User login
‘They’re out to get me!’: Evaluating rational fears and bizarre delusions in paranoia
Even among healthy individuals, feelings of paranoia are not unusual. In modern psychiatry, we consider paranoia to be a pattern of unfounded thinking, centered on the fearful experience of perceived victimization or threat of intentional harm. This means that a patient with paranoia is, by nature, difficult to engage in treatment. A patient might perceive the clinician as attempting to mislead or manipulate him. A therapeutic alliance could require patience on the part of the clinician, creativity,1 and abandoning attempts at rational “therapeutic” persuasion. The severity of symptoms determines the approach.
In this article, we review the nature of paranoia and the continuum of syndromes to which it is a central feature, as well as treatment approaches.
Categorization and etiology


Until recently, clinicians considered “paranoid” to be a subtype of schizophrenia (Box2-7); in DSM-5 the limited diagnostic stability and reliability of the categorization rendered the distinction obsolete.8 There are several levels of severity of paranoia; this thought process can present in simple variations of normal fears and concerns or in severe forms, with highly organized delusional systems.
The etiology of paranoia is not clear. Over the years, it has been attributed to defense mechanisms of the ego, habitual fears from repetitive exposure, or irregular activity of the amygdala. It is possible that various types of paranoia could have different causes. Functional MRIs indicate that the amygdala is involved in anxiety and threat perception in both primates and humans.9
Rational fear vs paranoia
Under the right circumstances, anyone could sense that he (she) is being threatened. Such feelings are normal in occupied countries and nations at war, and are not pathologic in such contexts. Anxiety about potential danger and harassment under truly oppressive circumstances might be biologically ingrained and have value for survival. It is important to employ cultural sensitivity when distinguishing pathological and nonpathological paranoia because some immigrant populations might have increased prevalence rates but without a true mental illness.10
Perhaps the key to separating realistic fear from paranoia is the recognition of whether the environment is truly safe or hostile; sometimes this is not initially evident to the clinician. The first author (J.A.W.) experienced this when discovering that a patient who was thought to be paranoid was indeed being stalked by another patient.
Rapid social change makes sweeping explanations about the range of threats experienced by any one person of limited value. Persons living with serious and persistent mental illness experience stigma—harassment, abuse, disgrace—and, similar to victims of repeated sexual abuse and other violence, are not necessarily unreasonable in their inner experience of omnipresent threat. In addition, advances in surveillance technology, as well as the media proliferation of depictions of vulnerability and threat, can plant generalized doubt of historically trusted individuals and systems. Under conditions of severe social discrimination or life under a totalitarian regime, constant fear for safety and worry about the intentions of others is reasonable. We must remember that during the Cold War many people in Eastern Europe had legitimate concerns that their phones were tapped. There are still many places in the world where the fear of government or of one’s neighbors exists.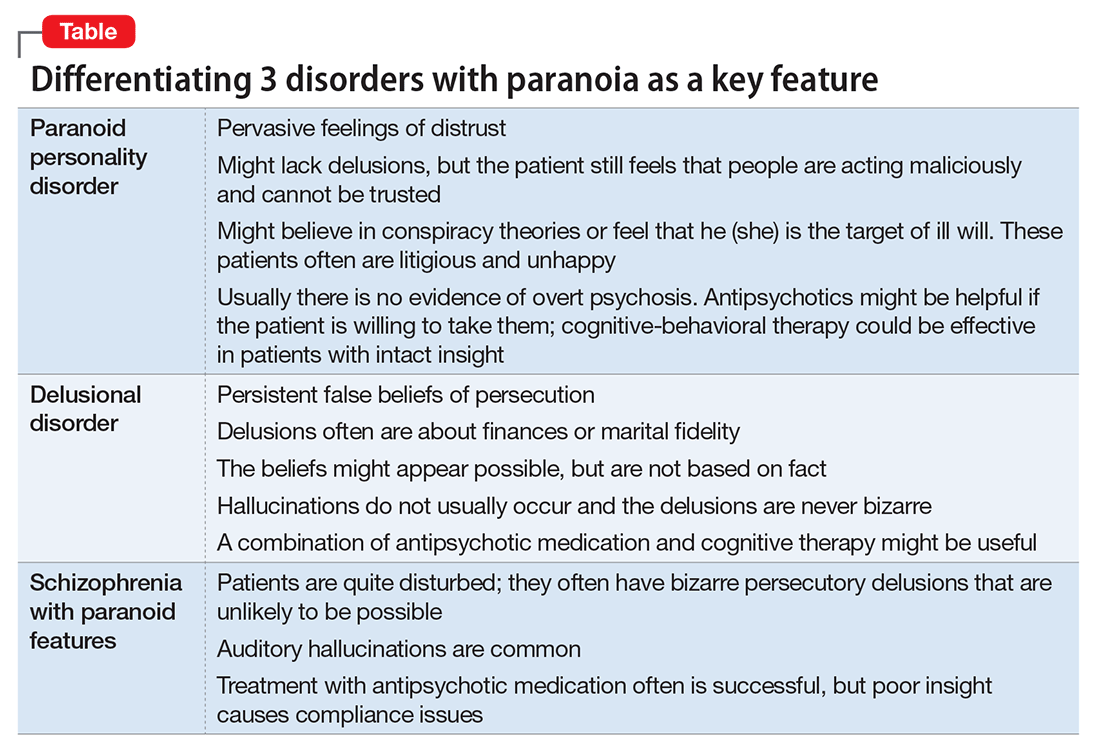
- paranoid personality disorder
- delusional disorder
- paranoia in schizophrenia (Table).
Paranoid personality disorder
The nature of any personality disorder is a long-standing psychological and behavioral pattern that differs significantly from the expectations of one’s culture. Such beliefs and behaviors typically are pervasive across most aspects of the individual’s interactions, and these enduring patterns of personality usually are evident by adolescence or young adulthood. Paranoid personality disorder is marked by pervasive distrust of others. Typical features include:
- suspicion about other people’s motives
- sensitivity to criticism
- keeping grudges against alleged offenders.8
The patient must have 4 of the following symptoms to confirm the diagnosis:
- suspicion of others and their motives
- reluctance to confide in others, due to lack of trust
- recurrent doubts about the fidelity of a significant other
- preoccupation with doubt regarding trusting others
- seeing threatening meanings behind benign remarks or events
- perception of attacks upon one’s character or reputation
- bears persistent grudges.8
Individuals with paranoid personality disorder tend to lead maladaptive lifestyles and might present as irritable, unpleasant, and emotionally guarded. Paranoid personality disorder is not a form of delusion, but is a pattern of habitual distrust of others.
The disorder generally is expressed verbally, and is seldom accompanied by hallucinations or unpredictable behavior. Distrust of others might result in social isolation and litigious behavior.8 Alternately, a patient with this disorder might not present for treatment until later in life after the loss of significant supporting factors, such as the death of parents or loss of steady employment. Examination of these older individuals is likely to reveal long-standing suspiciousness and distrust that previously was hidden by family members. For example, a 68-year-old woman might present saying that she can’t trust her daughter, but her recently deceased spouse would not let her discuss the topic outside of the home.
The etiology of paranoid personality disorder is unknown. Family studies suggest a possible a genetic connection to paranoia in schizophrenia.12 Others hypothesize that this dysfunction of personality might originate in early feelings of anxiety and low self-esteem, learned from a controlling, cruel, or sadistic parent; the patient then expects others to reject him (her) as the parent did.13,14 Such individuals might develop deep-seated distrust of others as a defense mechanism. Under stress, such as during a medical illness, patients could develop brief psychoses. Antipsychotic treatment might be useful in some cases of paranoid personality disorder, but should be limited.
Delusional disorder
Delusional disorder is a unique form of psychosis. Patients with delusional disorder might appear rational—as long as they are in independent roles—and their general functioning could go unnoticed. This could change when the delusions predominate their thoughts, or their delusional behavior is unacceptable in a structured environment. Such individuals often suffer from a highly specific delusion fixed on 1 topic. These delusions generally are the only psychotic feature. The most common theme is that of persecution. For example, a person firmly believes he is being followed by foreign agents or by a religious organization, which is blatantly untrue. Another common theme is infidelity.
Paranoia in delusional disorder is about something that is not actually occurring, but could.3 In other words, the delusion is not necessarily bizarre. The patient may have no evidence or could invent “evidence,” yet remain completely resistant to any logical argument against his belief system. In many situations, individuals with delusional disorder function normally in society, until the delusion becomes severe enough to prompt clinical attention.
Paranoia in schizophrenia
In patients with schizophrenia with paranoia, the typical symptoms of disorganization and disturbed affect are less prominent. The condition develops in young adulthood, but could start at any age. Its course typically is chronic and requires psychiatric treatment; the patient may require hospital care.
Although patients with delusional disorder and those with schizophrenia both have delusions, the delusions of the latter typically are bizarre and unlikely to be possible. For example, the patient might believe that her body has been replaced with the inner workings of an alien being or a robot. The paranoid delusions of persons with delusional disorder are much more mundane and could be plausible. Karl Jaspers, a clinician and researcher in the early 20th century, separated delusional disorder from paranoid schizophrenia by noting that the former could be “understandable, even if untrue” while the latter was “not within the realm of understandability.”5
A patient with schizophrenia with paranoid delusions usually experiences auditory hallucinations, such as voices threatening persecution or harm. When predominant, patients could be aroused by these fears and can be dangerous to others.2,4,5
Other presentations of paranoia
Paranoia can occur in affective disorders as well.13 Although the cause is only now being understood, clinicians have put forth theories for many years. A depressed person might suffer from excessive guilt and feel that he deserves to be persecuted, while a manic patient might think she is being persecuted for her greatness. In the past, response to electroconvulsive therapy was used to distinguish affective paranoia from other types.2
Paranoia in organic states
Substance use. Psychostimulants, which are known for their motor activity and arousal enhancing properties, as well as the potential for abuse and other negative consequences, could lead to acute paranoid states in susceptible individuals.15-17 In addition, tetrahydrocannabinol, the active chemical in Cannabis, can cause acute psychotic symptoms, such as paranoia,18,19 in a dose-dependent manner. A growing body of evidence suggests that a combination of Cannabis use with a genetic predisposition to psychosis may put some individuals at high risk of decompensation.19 Of growing concern is the evidence that synthetic cannabinoids, which are among the most commonly used new psychoactive substances, could be associated with psychosis, including paranoia.20
Dementia. Persons with dementia often are paranoid. In geriatric patients with dementia, a delusion of thievery is common. When a person has misplaced objects and can’t remember where, the “default” cognition is that someone has taken them. This confabulation may progress to a persistent paranoia and can be draining on caregivers.
Treating paranoia
A patient with paranoia usually has poor insight and cannot be reasoned with. Such individuals are quick to incorporate others into their delusional theories and easily develop notions of conspiracy. In acute psychosis, when the patient presents with fixed beliefs that are not amenable to reality orientation, and poses a threat to his well-being or that of others, alleviating underlying fear and anxiety is the first priority. Swift pharmacologic measures are required to decrease the patient’s underlying anxiety or anger, before you can try to earn his trust.
Psychopharmacologic interventions should be specific to the diagnosis. Antipsychotic medications generally will help decrease most paranoia, but affective syndromes usually require lithium or divalproex for best results.14,21
Develop a therapeutic relationship. The clinician must approach the patient in a practical and straightforward manner, and should not expect a quick therapeutic alliance. Transference and countertransference develop easily in the context of paranoia. Focus on behaviors that are problematic for the patient or the milieu, such as to ensure a safe environment. The patient needs to be aware of how he could come across to others. Clear feedback about behavior, such as “I cannot really listen to you when you’re yelling,” may be effective. It might be unwise to confront delusional paranoia in an agitated patient. Honesty and respect must continue in all communications to build trust. During assessment of a paranoid individual, evaluate the level of dangerousness. Ask your patient if he feels like acting on his beliefs or harming the people that are the targets of his paranoia.
As the patient begins to manage his anxiety and fear, you can develop a therapeutic alliance. The goals of treatment need be those of the patient—such as staying out of the hospital, or behaving in a manner that is required for employment. Over time, work toward growing the patient’s capacity for social interaction and productive activity. Insight might be elusive; however, some patients with paranoia can learn to take a detached view of their thoughts and emotions, and consider them impermanent events of the mind that make their lives difficult. Practice good judgment when aiming for recovery in a patient who does not have insight. For example, a patient can recognize that although there could be a microchip in his brain, he feels better when he takes medication.
In the case of paranoid personality disorder, treatment, as with most personality disorders, can be difficult. The patient might be unlikely to accept help and could distrust caregivers. Cognitive-behavioral therapy could be useful, if the patient can be engaged in the therapeutic process. Although it might be difficult to obtain enhanced insight, the patient could accept logical explanations for situations that provoke distrust. As long as anxiety and anger can be kept under control, the individual might learn the value of adopting the lessons of therapy. Pharmacological treatments are aimed at reducing the anxiety and anger experienced by the paranoid individual. Antipsychotics may be useful for short periods or during a crisis.14,21
The clinician must remain calm and reassuring when approaching an individual with paranoia, and not react to the projection of paranoid feelings from the patient. Respect for the patient can be conveyed without agreeing with delusions or bizarre thinking. The clinician must keep agreements and appointments with the client to prevent the erosion of trust. Paranoid conditions might respond slowly to pharmacological treatment, therefore establishing a consistent therapeutic relationship is essential.
1. Frank C. Delirium, consent to treatment, and Shakespeare. A geriatric experience. Can Fam Physician. 1999;45:875-876.
2. Hamilton M. Fish’s schizophrenia. Bristol, United Kingdom: John Wright and Sons; 1962.
3. Munro A. Delusional disorder. New York, NY: Cambridge University Press; 2000.
4. Kahlbaum K. Die gruppierung de psychischen krankheiten. Danzig, Germany: Verlag von A. W. Kafemann; 1853.
5. Kraepelin E. Manic depressive insanity and paranoia. Barclay RM, trans. New York, NY: Arno Press; 1976.
6. Bleuler E. Dementia praecox or the group of schizophrenias. Ainkia J, trans. New York, NY: International University Press; 1950.
7. Mayer W. Uber paraphrene psychosen. Zeitschrift fur die gesamte. Neurology und Psychiatrie. 1921;71:187-206.
8. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
9. Pinkham AE, Liu P, Lu H, et al. Amygdala hyperactivity at rest in paranoid individuals with schizophrenia. Am J Psychiatry. 2015;172(8):784-792.
10. Sen P, Chowdhury AN. Culture, ethnicity and paranoia. Curr Psychiatry Rep. 2006;8(3):174-178.
11. Szasz TS. The manufacture of madness: a comparative study of the inquisition and the mental health movement. New York, NY: Harper and Row; 1970.
12. Schanda H, Berner P, Gabriel E, et al. The genetics of delusional psychosis. Schizophr Bull. 1983;9(4):563-570.
13. Levy B, Tsoy E, Brodt T, et al. Stigma, social anxiety and illness severity in bipolar disorder: implications for treatment. Ann Clin Psychiatry. 2015;27(1):55-64.
14. Benjamin LS. Interpersonal diagnosis and treatment of personality disorders. New York, NY: Gilford Press; 1993.
15. Busardo FP, Kyriakou C, Cipilloni L, et al. From clinical application to cognitive enhancement. Curr Neuropharmacol. 2015;13(2):281-295.
16. McKetin R, Gardner J, Baker AL, et al. Correlates of transient versus persistent psychotic symptoms among dependent methylamphetamine users. Psychiatry Res. 2016;238:166-171.
17. Djamshidian A. The neurobehavioral sequelae of psychostimulant abuse. Int Rev Neurobiol. 2015;120:161-177.
18. Haney M, Evins AE. Does cannabis cause, exacerbate or ameliorate psychiatric disorders? An oversimplified debate discussed. Neuropsychopharmacology. 2016;41(2):393-401.
19. Bui QM, Simpson S, Nordstrom K. Psychiatric and medical management of marijuana intoxication in the emergency department. West J Emerg Med. 2015;16(3):414-417.
20. Seely KA, Lapoint J, Moran JH, et al. Spice drugs are more than harmless herbal blends: a review of the pharmacology and toxicology of synthetic cannabinoids. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(2):234-243.
21. Lake CR. Hypothesis: grandiosity and guilt cause paranoia; paranoid schizophrenia is a psychotic mood disorder: a review. Schizophr Bull. 2008;34(6):1151-1162.
Even among healthy individuals, feelings of paranoia are not unusual. In modern psychiatry, we consider paranoia to be a pattern of unfounded thinking, centered on the fearful experience of perceived victimization or threat of intentional harm. This means that a patient with paranoia is, by nature, difficult to engage in treatment. A patient might perceive the clinician as attempting to mislead or manipulate him. A therapeutic alliance could require patience on the part of the clinician, creativity,1 and abandoning attempts at rational “therapeutic” persuasion. The severity of symptoms determines the approach.
In this article, we review the nature of paranoia and the continuum of syndromes to which it is a central feature, as well as treatment approaches.
Categorization and etiology
Until recently, clinicians considered “paranoid” to be a subtype of schizophrenia (Box2-7); in DSM-5 the limited diagnostic stability and reliability of the categorization rendered the distinction obsolete.8 There are several levels of severity of paranoia; this thought process can present in simple variations of normal fears and concerns or in severe forms, with highly organized delusional systems.
The etiology of paranoia is not clear. Over the years, it has been attributed to defense mechanisms of the ego, habitual fears from repetitive exposure, or irregular activity of the amygdala. It is possible that various types of paranoia could have different causes. Functional MRIs indicate that the amygdala is involved in anxiety and threat perception in both primates and humans.9
Rational fear vs paranoia
Under the right circumstances, anyone could sense that he (she) is being threatened. Such feelings are normal in occupied countries and nations at war, and are not pathologic in such contexts. Anxiety about potential danger and harassment under truly oppressive circumstances might be biologically ingrained and have value for survival. It is important to employ cultural sensitivity when distinguishing pathological and nonpathological paranoia because some immigrant populations might have increased prevalence rates but without a true mental illness.10
Perhaps the key to separating realistic fear from paranoia is the recognition of whether the environment is truly safe or hostile; sometimes this is not initially evident to the clinician. The first author (J.A.W.) experienced this when discovering that a patient who was thought to be paranoid was indeed being stalked by another patient.
Rapid social change makes sweeping explanations about the range of threats experienced by any one person of limited value. Persons living with serious and persistent mental illness experience stigma—harassment, abuse, disgrace—and, similar to victims of repeated sexual abuse and other violence, are not necessarily unreasonable in their inner experience of omnipresent threat. In addition, advances in surveillance technology, as well as the media proliferation of depictions of vulnerability and threat, can plant generalized doubt of historically trusted individuals and systems. Under conditions of severe social discrimination or life under a totalitarian regime, constant fear for safety and worry about the intentions of others is reasonable. We must remember that during the Cold War many people in Eastern Europe had legitimate concerns that their phones were tapped. There are still many places in the world where the fear of government or of one’s neighbors exists.
- paranoid personality disorder
- delusional disorder
- paranoia in schizophrenia (Table).
Paranoid personality disorder
The nature of any personality disorder is a long-standing psychological and behavioral pattern that differs significantly from the expectations of one’s culture. Such beliefs and behaviors typically are pervasive across most aspects of the individual’s interactions, and these enduring patterns of personality usually are evident by adolescence or young adulthood. Paranoid personality disorder is marked by pervasive distrust of others. Typical features include:
- suspicion about other people’s motives
- sensitivity to criticism
- keeping grudges against alleged offenders.8
The patient must have 4 of the following symptoms to confirm the diagnosis:
- suspicion of others and their motives
- reluctance to confide in others, due to lack of trust
- recurrent doubts about the fidelity of a significant other
- preoccupation with doubt regarding trusting others
- seeing threatening meanings behind benign remarks or events
- perception of attacks upon one’s character or reputation
- bears persistent grudges.8
Individuals with paranoid personality disorder tend to lead maladaptive lifestyles and might present as irritable, unpleasant, and emotionally guarded. Paranoid personality disorder is not a form of delusion, but is a pattern of habitual distrust of others.
The disorder generally is expressed verbally, and is seldom accompanied by hallucinations or unpredictable behavior. Distrust of others might result in social isolation and litigious behavior.8 Alternately, a patient with this disorder might not present for treatment until later in life after the loss of significant supporting factors, such as the death of parents or loss of steady employment. Examination of these older individuals is likely to reveal long-standing suspiciousness and distrust that previously was hidden by family members. For example, a 68-year-old woman might present saying that she can’t trust her daughter, but her recently deceased spouse would not let her discuss the topic outside of the home.
The etiology of paranoid personality disorder is unknown. Family studies suggest a possible a genetic connection to paranoia in schizophrenia.12 Others hypothesize that this dysfunction of personality might originate in early feelings of anxiety and low self-esteem, learned from a controlling, cruel, or sadistic parent; the patient then expects others to reject him (her) as the parent did.13,14 Such individuals might develop deep-seated distrust of others as a defense mechanism. Under stress, such as during a medical illness, patients could develop brief psychoses. Antipsychotic treatment might be useful in some cases of paranoid personality disorder, but should be limited.
Delusional disorder
Delusional disorder is a unique form of psychosis. Patients with delusional disorder might appear rational—as long as they are in independent roles—and their general functioning could go unnoticed. This could change when the delusions predominate their thoughts, or their delusional behavior is unacceptable in a structured environment. Such individuals often suffer from a highly specific delusion fixed on 1 topic. These delusions generally are the only psychotic feature. The most common theme is that of persecution. For example, a person firmly believes he is being followed by foreign agents or by a religious organization, which is blatantly untrue. Another common theme is infidelity.
Paranoia in delusional disorder is about something that is not actually occurring, but could.3 In other words, the delusion is not necessarily bizarre. The patient may have no evidence or could invent “evidence,” yet remain completely resistant to any logical argument against his belief system. In many situations, individuals with delusional disorder function normally in society, until the delusion becomes severe enough to prompt clinical attention.
Paranoia in schizophrenia
In patients with schizophrenia with paranoia, the typical symptoms of disorganization and disturbed affect are less prominent. The condition develops in young adulthood, but could start at any age. Its course typically is chronic and requires psychiatric treatment; the patient may require hospital care.
Although patients with delusional disorder and those with schizophrenia both have delusions, the delusions of the latter typically are bizarre and unlikely to be possible. For example, the patient might believe that her body has been replaced with the inner workings of an alien being or a robot. The paranoid delusions of persons with delusional disorder are much more mundane and could be plausible. Karl Jaspers, a clinician and researcher in the early 20th century, separated delusional disorder from paranoid schizophrenia by noting that the former could be “understandable, even if untrue” while the latter was “not within the realm of understandability.”5
A patient with schizophrenia with paranoid delusions usually experiences auditory hallucinations, such as voices threatening persecution or harm. When predominant, patients could be aroused by these fears and can be dangerous to others.2,4,5
Other presentations of paranoia
Paranoia can occur in affective disorders as well.13 Although the cause is only now being understood, clinicians have put forth theories for many years. A depressed person might suffer from excessive guilt and feel that he deserves to be persecuted, while a manic patient might think she is being persecuted for her greatness. In the past, response to electroconvulsive therapy was used to distinguish affective paranoia from other types.2
Paranoia in organic states
Substance use. Psychostimulants, which are known for their motor activity and arousal enhancing properties, as well as the potential for abuse and other negative consequences, could lead to acute paranoid states in susceptible individuals.15-17 In addition, tetrahydrocannabinol, the active chemical in Cannabis, can cause acute psychotic symptoms, such as paranoia,18,19 in a dose-dependent manner. A growing body of evidence suggests that a combination of Cannabis use with a genetic predisposition to psychosis may put some individuals at high risk of decompensation.19 Of growing concern is the evidence that synthetic cannabinoids, which are among the most commonly used new psychoactive substances, could be associated with psychosis, including paranoia.20
Dementia. Persons with dementia often are paranoid. In geriatric patients with dementia, a delusion of thievery is common. When a person has misplaced objects and can’t remember where, the “default” cognition is that someone has taken them. This confabulation may progress to a persistent paranoia and can be draining on caregivers.
Treating paranoia
A patient with paranoia usually has poor insight and cannot be reasoned with. Such individuals are quick to incorporate others into their delusional theories and easily develop notions of conspiracy. In acute psychosis, when the patient presents with fixed beliefs that are not amenable to reality orientation, and poses a threat to his well-being or that of others, alleviating underlying fear and anxiety is the first priority. Swift pharmacologic measures are required to decrease the patient’s underlying anxiety or anger, before you can try to earn his trust.
Psychopharmacologic interventions should be specific to the diagnosis. Antipsychotic medications generally will help decrease most paranoia, but affective syndromes usually require lithium or divalproex for best results.14,21
Develop a therapeutic relationship. The clinician must approach the patient in a practical and straightforward manner, and should not expect a quick therapeutic alliance. Transference and countertransference develop easily in the context of paranoia. Focus on behaviors that are problematic for the patient or the milieu, such as to ensure a safe environment. The patient needs to be aware of how he could come across to others. Clear feedback about behavior, such as “I cannot really listen to you when you’re yelling,” may be effective. It might be unwise to confront delusional paranoia in an agitated patient. Honesty and respect must continue in all communications to build trust. During assessment of a paranoid individual, evaluate the level of dangerousness. Ask your patient if he feels like acting on his beliefs or harming the people that are the targets of his paranoia.
As the patient begins to manage his anxiety and fear, you can develop a therapeutic alliance. The goals of treatment need be those of the patient—such as staying out of the hospital, or behaving in a manner that is required for employment. Over time, work toward growing the patient’s capacity for social interaction and productive activity. Insight might be elusive; however, some patients with paranoia can learn to take a detached view of their thoughts and emotions, and consider them impermanent events of the mind that make their lives difficult. Practice good judgment when aiming for recovery in a patient who does not have insight. For example, a patient can recognize that although there could be a microchip in his brain, he feels better when he takes medication.
In the case of paranoid personality disorder, treatment, as with most personality disorders, can be difficult. The patient might be unlikely to accept help and could distrust caregivers. Cognitive-behavioral therapy could be useful, if the patient can be engaged in the therapeutic process. Although it might be difficult to obtain enhanced insight, the patient could accept logical explanations for situations that provoke distrust. As long as anxiety and anger can be kept under control, the individual might learn the value of adopting the lessons of therapy. Pharmacological treatments are aimed at reducing the anxiety and anger experienced by the paranoid individual. Antipsychotics may be useful for short periods or during a crisis.14,21
The clinician must remain calm and reassuring when approaching an individual with paranoia, and not react to the projection of paranoid feelings from the patient. Respect for the patient can be conveyed without agreeing with delusions or bizarre thinking. The clinician must keep agreements and appointments with the client to prevent the erosion of trust. Paranoid conditions might respond slowly to pharmacological treatment, therefore establishing a consistent therapeutic relationship is essential.
Even among healthy individuals, feelings of paranoia are not unusual. In modern psychiatry, we consider paranoia to be a pattern of unfounded thinking, centered on the fearful experience of perceived victimization or threat of intentional harm. This means that a patient with paranoia is, by nature, difficult to engage in treatment. A patient might perceive the clinician as attempting to mislead or manipulate him. A therapeutic alliance could require patience on the part of the clinician, creativity,1 and abandoning attempts at rational “therapeutic” persuasion. The severity of symptoms determines the approach.
In this article, we review the nature of paranoia and the continuum of syndromes to which it is a central feature, as well as treatment approaches.
Categorization and etiology
Until recently, clinicians considered “paranoid” to be a subtype of schizophrenia (Box2-7); in DSM-5 the limited diagnostic stability and reliability of the categorization rendered the distinction obsolete.8 There are several levels of severity of paranoia; this thought process can present in simple variations of normal fears and concerns or in severe forms, with highly organized delusional systems.
The etiology of paranoia is not clear. Over the years, it has been attributed to defense mechanisms of the ego, habitual fears from repetitive exposure, or irregular activity of the amygdala. It is possible that various types of paranoia could have different causes. Functional MRIs indicate that the amygdala is involved in anxiety and threat perception in both primates and humans.9
Rational fear vs paranoia
Under the right circumstances, anyone could sense that he (she) is being threatened. Such feelings are normal in occupied countries and nations at war, and are not pathologic in such contexts. Anxiety about potential danger and harassment under truly oppressive circumstances might be biologically ingrained and have value for survival. It is important to employ cultural sensitivity when distinguishing pathological and nonpathological paranoia because some immigrant populations might have increased prevalence rates but without a true mental illness.10
Perhaps the key to separating realistic fear from paranoia is the recognition of whether the environment is truly safe or hostile; sometimes this is not initially evident to the clinician. The first author (J.A.W.) experienced this when discovering that a patient who was thought to be paranoid was indeed being stalked by another patient.
Rapid social change makes sweeping explanations about the range of threats experienced by any one person of limited value. Persons living with serious and persistent mental illness experience stigma—harassment, abuse, disgrace—and, similar to victims of repeated sexual abuse and other violence, are not necessarily unreasonable in their inner experience of omnipresent threat. In addition, advances in surveillance technology, as well as the media proliferation of depictions of vulnerability and threat, can plant generalized doubt of historically trusted individuals and systems. Under conditions of severe social discrimination or life under a totalitarian regime, constant fear for safety and worry about the intentions of others is reasonable. We must remember that during the Cold War many people in Eastern Europe had legitimate concerns that their phones were tapped. There are still many places in the world where the fear of government or of one’s neighbors exists.
- paranoid personality disorder
- delusional disorder
- paranoia in schizophrenia (Table).
Paranoid personality disorder
The nature of any personality disorder is a long-standing psychological and behavioral pattern that differs significantly from the expectations of one’s culture. Such beliefs and behaviors typically are pervasive across most aspects of the individual’s interactions, and these enduring patterns of personality usually are evident by adolescence or young adulthood. Paranoid personality disorder is marked by pervasive distrust of others. Typical features include:
- suspicion about other people’s motives
- sensitivity to criticism
- keeping grudges against alleged offenders.8
The patient must have 4 of the following symptoms to confirm the diagnosis:
- suspicion of others and their motives
- reluctance to confide in others, due to lack of trust
- recurrent doubts about the fidelity of a significant other
- preoccupation with doubt regarding trusting others
- seeing threatening meanings behind benign remarks or events
- perception of attacks upon one’s character or reputation
- bears persistent grudges.8
Individuals with paranoid personality disorder tend to lead maladaptive lifestyles and might present as irritable, unpleasant, and emotionally guarded. Paranoid personality disorder is not a form of delusion, but is a pattern of habitual distrust of others.
The disorder generally is expressed verbally, and is seldom accompanied by hallucinations or unpredictable behavior. Distrust of others might result in social isolation and litigious behavior.8 Alternately, a patient with this disorder might not present for treatment until later in life after the loss of significant supporting factors, such as the death of parents or loss of steady employment. Examination of these older individuals is likely to reveal long-standing suspiciousness and distrust that previously was hidden by family members. For example, a 68-year-old woman might present saying that she can’t trust her daughter, but her recently deceased spouse would not let her discuss the topic outside of the home.
The etiology of paranoid personality disorder is unknown. Family studies suggest a possible a genetic connection to paranoia in schizophrenia.12 Others hypothesize that this dysfunction of personality might originate in early feelings of anxiety and low self-esteem, learned from a controlling, cruel, or sadistic parent; the patient then expects others to reject him (her) as the parent did.13,14 Such individuals might develop deep-seated distrust of others as a defense mechanism. Under stress, such as during a medical illness, patients could develop brief psychoses. Antipsychotic treatment might be useful in some cases of paranoid personality disorder, but should be limited.
Delusional disorder
Delusional disorder is a unique form of psychosis. Patients with delusional disorder might appear rational—as long as they are in independent roles—and their general functioning could go unnoticed. This could change when the delusions predominate their thoughts, or their delusional behavior is unacceptable in a structured environment. Such individuals often suffer from a highly specific delusion fixed on 1 topic. These delusions generally are the only psychotic feature. The most common theme is that of persecution. For example, a person firmly believes he is being followed by foreign agents or by a religious organization, which is blatantly untrue. Another common theme is infidelity.
Paranoia in delusional disorder is about something that is not actually occurring, but could.3 In other words, the delusion is not necessarily bizarre. The patient may have no evidence or could invent “evidence,” yet remain completely resistant to any logical argument against his belief system. In many situations, individuals with delusional disorder function normally in society, until the delusion becomes severe enough to prompt clinical attention.
Paranoia in schizophrenia
In patients with schizophrenia with paranoia, the typical symptoms of disorganization and disturbed affect are less prominent. The condition develops in young adulthood, but could start at any age. Its course typically is chronic and requires psychiatric treatment; the patient may require hospital care.
Although patients with delusional disorder and those with schizophrenia both have delusions, the delusions of the latter typically are bizarre and unlikely to be possible. For example, the patient might believe that her body has been replaced with the inner workings of an alien being or a robot. The paranoid delusions of persons with delusional disorder are much more mundane and could be plausible. Karl Jaspers, a clinician and researcher in the early 20th century, separated delusional disorder from paranoid schizophrenia by noting that the former could be “understandable, even if untrue” while the latter was “not within the realm of understandability.”5
A patient with schizophrenia with paranoid delusions usually experiences auditory hallucinations, such as voices threatening persecution or harm. When predominant, patients could be aroused by these fears and can be dangerous to others.2,4,5
Other presentations of paranoia
Paranoia can occur in affective disorders as well.13 Although the cause is only now being understood, clinicians have put forth theories for many years. A depressed person might suffer from excessive guilt and feel that he deserves to be persecuted, while a manic patient might think she is being persecuted for her greatness. In the past, response to electroconvulsive therapy was used to distinguish affective paranoia from other types.2
Paranoia in organic states
Substance use. Psychostimulants, which are known for their motor activity and arousal enhancing properties, as well as the potential for abuse and other negative consequences, could lead to acute paranoid states in susceptible individuals.15-17 In addition, tetrahydrocannabinol, the active chemical in Cannabis, can cause acute psychotic symptoms, such as paranoia,18,19 in a dose-dependent manner. A growing body of evidence suggests that a combination of Cannabis use with a genetic predisposition to psychosis may put some individuals at high risk of decompensation.19 Of growing concern is the evidence that synthetic cannabinoids, which are among the most commonly used new psychoactive substances, could be associated with psychosis, including paranoia.20
Dementia. Persons with dementia often are paranoid. In geriatric patients with dementia, a delusion of thievery is common. When a person has misplaced objects and can’t remember where, the “default” cognition is that someone has taken them. This confabulation may progress to a persistent paranoia and can be draining on caregivers.
Treating paranoia
A patient with paranoia usually has poor insight and cannot be reasoned with. Such individuals are quick to incorporate others into their delusional theories and easily develop notions of conspiracy. In acute psychosis, when the patient presents with fixed beliefs that are not amenable to reality orientation, and poses a threat to his well-being or that of others, alleviating underlying fear and anxiety is the first priority. Swift pharmacologic measures are required to decrease the patient’s underlying anxiety or anger, before you can try to earn his trust.
Psychopharmacologic interventions should be specific to the diagnosis. Antipsychotic medications generally will help decrease most paranoia, but affective syndromes usually require lithium or divalproex for best results.14,21
Develop a therapeutic relationship. The clinician must approach the patient in a practical and straightforward manner, and should not expect a quick therapeutic alliance. Transference and countertransference develop easily in the context of paranoia. Focus on behaviors that are problematic for the patient or the milieu, such as to ensure a safe environment. The patient needs to be aware of how he could come across to others. Clear feedback about behavior, such as “I cannot really listen to you when you’re yelling,” may be effective. It might be unwise to confront delusional paranoia in an agitated patient. Honesty and respect must continue in all communications to build trust. During assessment of a paranoid individual, evaluate the level of dangerousness. Ask your patient if he feels like acting on his beliefs or harming the people that are the targets of his paranoia.
As the patient begins to manage his anxiety and fear, you can develop a therapeutic alliance. The goals of treatment need be those of the patient—such as staying out of the hospital, or behaving in a manner that is required for employment. Over time, work toward growing the patient’s capacity for social interaction and productive activity. Insight might be elusive; however, some patients with paranoia can learn to take a detached view of their thoughts and emotions, and consider them impermanent events of the mind that make their lives difficult. Practice good judgment when aiming for recovery in a patient who does not have insight. For example, a patient can recognize that although there could be a microchip in his brain, he feels better when he takes medication.
In the case of paranoid personality disorder, treatment, as with most personality disorders, can be difficult. The patient might be unlikely to accept help and could distrust caregivers. Cognitive-behavioral therapy could be useful, if the patient can be engaged in the therapeutic process. Although it might be difficult to obtain enhanced insight, the patient could accept logical explanations for situations that provoke distrust. As long as anxiety and anger can be kept under control, the individual might learn the value of adopting the lessons of therapy. Pharmacological treatments are aimed at reducing the anxiety and anger experienced by the paranoid individual. Antipsychotics may be useful for short periods or during a crisis.14,21
The clinician must remain calm and reassuring when approaching an individual with paranoia, and not react to the projection of paranoid feelings from the patient. Respect for the patient can be conveyed without agreeing with delusions or bizarre thinking. The clinician must keep agreements and appointments with the client to prevent the erosion of trust. Paranoid conditions might respond slowly to pharmacological treatment, therefore establishing a consistent therapeutic relationship is essential.
1. Frank C. Delirium, consent to treatment, and Shakespeare. A geriatric experience. Can Fam Physician. 1999;45:875-876.
2. Hamilton M. Fish’s schizophrenia. Bristol, United Kingdom: John Wright and Sons; 1962.
3. Munro A. Delusional disorder. New York, NY: Cambridge University Press; 2000.
4. Kahlbaum K. Die gruppierung de psychischen krankheiten. Danzig, Germany: Verlag von A. W. Kafemann; 1853.
5. Kraepelin E. Manic depressive insanity and paranoia. Barclay RM, trans. New York, NY: Arno Press; 1976.
6. Bleuler E. Dementia praecox or the group of schizophrenias. Ainkia J, trans. New York, NY: International University Press; 1950.
7. Mayer W. Uber paraphrene psychosen. Zeitschrift fur die gesamte. Neurology und Psychiatrie. 1921;71:187-206.
8. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
9. Pinkham AE, Liu P, Lu H, et al. Amygdala hyperactivity at rest in paranoid individuals with schizophrenia. Am J Psychiatry. 2015;172(8):784-792.
10. Sen P, Chowdhury AN. Culture, ethnicity and paranoia. Curr Psychiatry Rep. 2006;8(3):174-178.
11. Szasz TS. The manufacture of madness: a comparative study of the inquisition and the mental health movement. New York, NY: Harper and Row; 1970.
12. Schanda H, Berner P, Gabriel E, et al. The genetics of delusional psychosis. Schizophr Bull. 1983;9(4):563-570.
13. Levy B, Tsoy E, Brodt T, et al. Stigma, social anxiety and illness severity in bipolar disorder: implications for treatment. Ann Clin Psychiatry. 2015;27(1):55-64.
14. Benjamin LS. Interpersonal diagnosis and treatment of personality disorders. New York, NY: Gilford Press; 1993.
15. Busardo FP, Kyriakou C, Cipilloni L, et al. From clinical application to cognitive enhancement. Curr Neuropharmacol. 2015;13(2):281-295.
16. McKetin R, Gardner J, Baker AL, et al. Correlates of transient versus persistent psychotic symptoms among dependent methylamphetamine users. Psychiatry Res. 2016;238:166-171.
17. Djamshidian A. The neurobehavioral sequelae of psychostimulant abuse. Int Rev Neurobiol. 2015;120:161-177.
18. Haney M, Evins AE. Does cannabis cause, exacerbate or ameliorate psychiatric disorders? An oversimplified debate discussed. Neuropsychopharmacology. 2016;41(2):393-401.
19. Bui QM, Simpson S, Nordstrom K. Psychiatric and medical management of marijuana intoxication in the emergency department. West J Emerg Med. 2015;16(3):414-417.
20. Seely KA, Lapoint J, Moran JH, et al. Spice drugs are more than harmless herbal blends: a review of the pharmacology and toxicology of synthetic cannabinoids. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(2):234-243.
21. Lake CR. Hypothesis: grandiosity and guilt cause paranoia; paranoid schizophrenia is a psychotic mood disorder: a review. Schizophr Bull. 2008;34(6):1151-1162.
1. Frank C. Delirium, consent to treatment, and Shakespeare. A geriatric experience. Can Fam Physician. 1999;45:875-876.
2. Hamilton M. Fish’s schizophrenia. Bristol, United Kingdom: John Wright and Sons; 1962.
3. Munro A. Delusional disorder. New York, NY: Cambridge University Press; 2000.
4. Kahlbaum K. Die gruppierung de psychischen krankheiten. Danzig, Germany: Verlag von A. W. Kafemann; 1853.
5. Kraepelin E. Manic depressive insanity and paranoia. Barclay RM, trans. New York, NY: Arno Press; 1976.
6. Bleuler E. Dementia praecox or the group of schizophrenias. Ainkia J, trans. New York, NY: International University Press; 1950.
7. Mayer W. Uber paraphrene psychosen. Zeitschrift fur die gesamte. Neurology und Psychiatrie. 1921;71:187-206.
8. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
9. Pinkham AE, Liu P, Lu H, et al. Amygdala hyperactivity at rest in paranoid individuals with schizophrenia. Am J Psychiatry. 2015;172(8):784-792.
10. Sen P, Chowdhury AN. Culture, ethnicity and paranoia. Curr Psychiatry Rep. 2006;8(3):174-178.
11. Szasz TS. The manufacture of madness: a comparative study of the inquisition and the mental health movement. New York, NY: Harper and Row; 1970.
12. Schanda H, Berner P, Gabriel E, et al. The genetics of delusional psychosis. Schizophr Bull. 1983;9(4):563-570.
13. Levy B, Tsoy E, Brodt T, et al. Stigma, social anxiety and illness severity in bipolar disorder: implications for treatment. Ann Clin Psychiatry. 2015;27(1):55-64.
14. Benjamin LS. Interpersonal diagnosis and treatment of personality disorders. New York, NY: Gilford Press; 1993.
15. Busardo FP, Kyriakou C, Cipilloni L, et al. From clinical application to cognitive enhancement. Curr Neuropharmacol. 2015;13(2):281-295.
16. McKetin R, Gardner J, Baker AL, et al. Correlates of transient versus persistent psychotic symptoms among dependent methylamphetamine users. Psychiatry Res. 2016;238:166-171.
17. Djamshidian A. The neurobehavioral sequelae of psychostimulant abuse. Int Rev Neurobiol. 2015;120:161-177.
18. Haney M, Evins AE. Does cannabis cause, exacerbate or ameliorate psychiatric disorders? An oversimplified debate discussed. Neuropsychopharmacology. 2016;41(2):393-401.
19. Bui QM, Simpson S, Nordstrom K. Psychiatric and medical management of marijuana intoxication in the emergency department. West J Emerg Med. 2015;16(3):414-417.
20. Seely KA, Lapoint J, Moran JH, et al. Spice drugs are more than harmless herbal blends: a review of the pharmacology and toxicology of synthetic cannabinoids. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(2):234-243.
21. Lake CR. Hypothesis: grandiosity and guilt cause paranoia; paranoid schizophrenia is a psychotic mood disorder: a review. Schizophr Bull. 2008;34(6):1151-1162.

An irritable, inattentive, and disruptive child: Is it ADHD or bipolar disorder?
Differentiating the irritable, oppositional child with attention-deficit/hyperactivity disorder (ADHD) from the child with bipolar disorder (BD) often is difficult. To make matters more complicated, 50% to 70% of patients with BD have comorbid ADHD.1,2 Accordingly, clinicians are often faced with the moody, irritable, disruptive child whose parents want to know if he (she) is “bipolar” to try to deal with oppositional and mood behaviors.
In this article, we present an approach that will help you distinguish these 2 disorders from each other.
Precision medicineThere is a lack of evidence-based methods for diagnosing psychiatric disorders in children and adolescents. DSM-5 provides clinicians with diagnostic checklists that rely on the clinician’s judgment and training in evaluating a patient.3 In The innovator’s prescription: a disruptive solution for health care, Christensen et al4 describe how medicine is moving from “intuitive medicine” to empirical medicine and toward “precision medicine.” Intuitive medicine depends on the clinician’s expertise, training, and exposure to different disorders, which is the traditional clinical model that predominates in child psychiatry. Empirical medicine relies on laboratory results, scans, scales, and other standardized tools.
Precision medicine occurs when a disorder can be precisely diagnosed and its cause understood, and when it can be treated with effective, evidence-based therapies. An example of this movement toward precision is Timothy syndrome (TS), a rare autosomal dominant disorder characterized by physical malformations, cardiac arrhythmias and structural heart defects, webbing of fingers and toes, and autism spectrum disorder. In the past, a child with TS would have been given a diagnosis of intellectual disability, or a specialist in developmental disorders might recognize the pattern of TS. It is now known that TS is caused by mutations in CACNA1C, the gene encoding the calcium channel Cav1.2α subunit, allowing precise diagnosis by genotyping.5
Although there are several tools that help clinicians assess symptoms of ADHD and BD, including rating scales such the Vanderbilt ADHD Diagnostic Rating Scale and Young Mania Rating Scale, none of these scales are diagnostic. Youngstrom et al6,7 have developed an evidence-based strategy to diagnose pediatric BD. This method uses a nomogram that takes into account the base rate of BD in a clinical setting and family history of BD.
We will describe and contrast the epidemiologic and clinical characteristics of pediatric BD from ADHD and use the Youngstrom nomogram to better define these patients. Although still far from precision medicine, the type of approach represents an ongoing effort in mental health care to increase diagnostic accuracy and improve treatment outcomes.
Pediatric bipolar disorder
Prevalence of pediatric BD is 1.8% (95% CI, 1.1% to 3.0%),8 which does not include sub-threshold cases of BD. ADHD and oppositional defiant disorder (ODD) are 8 to 10 times more prevalent. For the purposes of the nomogram, the “base rate” is the rate at which a disorder occurs in different clinical settings. In general outpatient clinics, BD might occur 6% to 8% of the time, whereas in a county-run child psychiatry inpatient facility the rate is 11%.6 A reasonable rate in an outpatient pediatric setting is 6%.
Family history. In the Bipolar Offspring Study,9 the rate of BD in children of parents with BD was 13 times greater than that of controls, and the rate of anxiety and behavior disorders was approximately twice that of children of parents without BD (Table 1).9 This study evaluated 388 children of 233 parents with BD and 251 children of 143 demographically matched controls.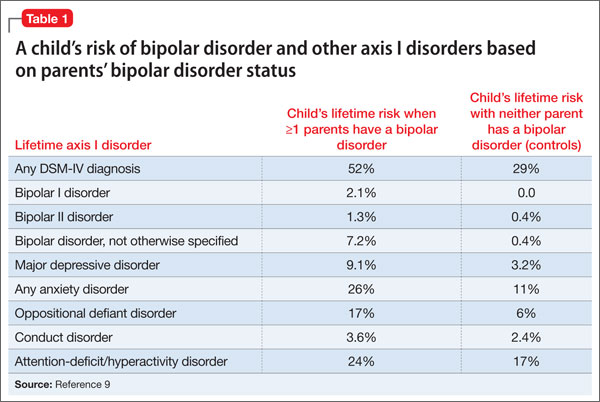
Clinical characteristics. Children and adolescents with BD typically manifest with what can be described as a “mood cycle”—a pronounced shift in mood and energy from one extreme to another. An example would be a child who wakes up with extreme silliness, high energy, and intrusive behavior that persists for several hours, then later becomes sad, depressed, and suicidal with no precipitant for either mood cycle.10 Pediatric patients with BD also exhibit other symptoms of mania during mood cycling periods.
Elevated or expansive mood. The child might have a mood that is inappropriately giddy, silly, elated, or euphoric. Often this mood will be present without reason and last for several hours. It may be distinguished from a transient cheerful mood by the intensity and duration of the episode. The child with BD may have little to no insight about the inappropriate nature of their elevated mood, when present.
Irritable mood. The child might become markedly belligerent or irritated with intense outbursts of anger, 2 to 3 times a day for several hours. An adolescent might appear extremely oppositional, belligerent, or hostile with parents and others.
Grandiosity or inflated self-esteem can be confused with brief childhood fantasies of increased capability. Typically, true grandiosity can manifest as assertion of great competency in all areas of life, which usually cannot be altered by contrary external evidence. Occasionally, this is bizarre and includes delusions of “super powers.” The child in a manic episode will not only assert that she can fly, but will jump off the garage roof to prove it.
Decreased need for sleep. The child may only require 4 to 5 hours of sleep a night during a manic episode without feeling fatigued or showing evidence of tiredness. Consider substance use in this differential diagnosis, especially in adolescents.
Increased talkativeness. Lack of inhibition to social norms may lead pediatric BD patients to blurt out answers during class or repeatedly be disciplined for talking to peers in class. Speech typically is rapid and pressured to the point where it might be continuous and seems to jump between loosely related subjects.
Flight of ideas or racing thoughts. The child or adolescent might report a subjective feeling that his thoughts are moving so rapidly that his speech cannot keep up. Often this is differentiated from rapid speech by the degree of rapidity the patient expresses loosely related topics that might seem completely unrelated to the listener.
Distractibility, short attention span. During a manic episode, the child or adolescent might report that it is impossible to pay attention to class or other outside events because of rapidly changing focus of their thoughts. This symptom must be carefully distinguished from the distractibility and inattention of ADHD, which typically is a more fixed and long-standing pattern rather than a brief episodic phenomenon in a manic or hypomanic episode.
Increase in goal-directed activity. During a mild manic episode, the child or adolescent may be capable of accomplishing a great deal of work. However, episodes that are more severe manifest as an individual starting numerous ambitious projects that she later is unable to complete.
Excessive risk-taking activities. The child or adolescent might become involved in forbidden, pleasurable activities that have a high risk of adverse consequences. This can manifest as hypersexual behavior, frequent fighting, increased recklessness, use of drugs and alcohol, shopping sprees, and reckless driving.
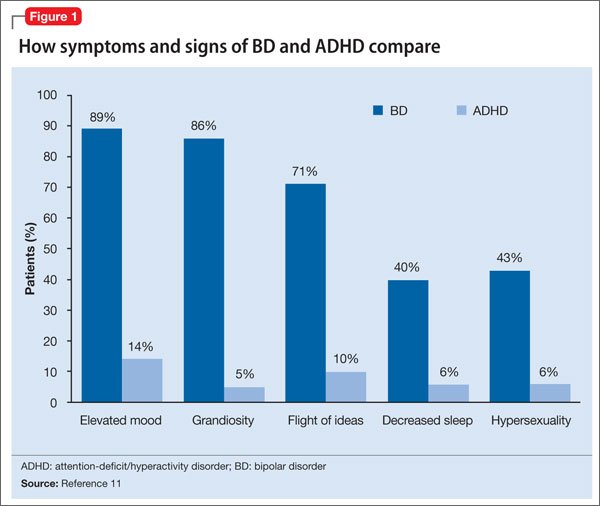
There are few studies comparing patients with comorbid BD and ADHD with patients with only ADHD. Geller et al11 compared 60 children with BD and ADHD (mean age, 10) to age- and sex-matched patients with ADHD and no mood disorder. Compared with children who had ADHD, those with BD exhibited significantly greater elevated mood, grandiosity, flight and/or racing of ideas, decreased need for sleep, and hypersexuality (Figure 1,11). Features common to both groups—and therefore not useful in differentiating the disorders—included irritability, hyperactivity, accelerated speech, and distractibility.
CASE REPORTIrritable and disruptiveBill, age 12, has been brought to see you by his mother because she is concerned about escalating behavior problems at home and school in the past several months. The school principal has called her about his obnoxious behavior with teachers and about other parents’ complaints that he has made unwanted sexual advances to girls who sit next to him in class.
Bill, who is in the 7th grade, is on the verge of being suspended for his inappropriate and disruptive behavior. His parents report that he is irritable around them and stays up all night, messaging his friends on the Internet from his iPad in his bedroom. They attribute his inappropriate sexual behavior to puberty and possibly to the Web sites he views.
Bill’s mother is concerned about his:
• increasing behavior problems during the last several months at home and school
• intensifying irritability and depressive symptoms
• staying up all night on the Internet, phoning friends, and doing projects
• frequent unprovoked, outbursts of rage occurring with increasing frequency and intensity (almost daily)
• moderate grandiosity, including telling the soccer coach and teachers how to do their jobs
• inappropriate sexual behavior, including kissing and touching female classmates.
During your history, you learn that Bill has been a bright and artistic child, with good academic performance. His peer relationships have been satisfactory, but not excellent—he tends to be “bossy” with his peers. He is medically healthy and not taking any medications. As part of your history, you also talk with Bill and his family about exposure to trauma or significant stressors, which they deny. You learn that Bill’s father was diagnosed with BD I at age 32.
Completing the nomogram developed by Youngstrom et al6,7 using these variables (see this article at CurrentPsychiatry.com for Figure 2)6,7 gives Bill a post-test probability of approximately 42%. The threshold for moving ahead with assessment and possible treatment, the “test-treatment threshold,” depends on your clinical setting.12,13 Our clinical experience is that, when the post-test probability exceeds 30%, further assessment for BD is warranted.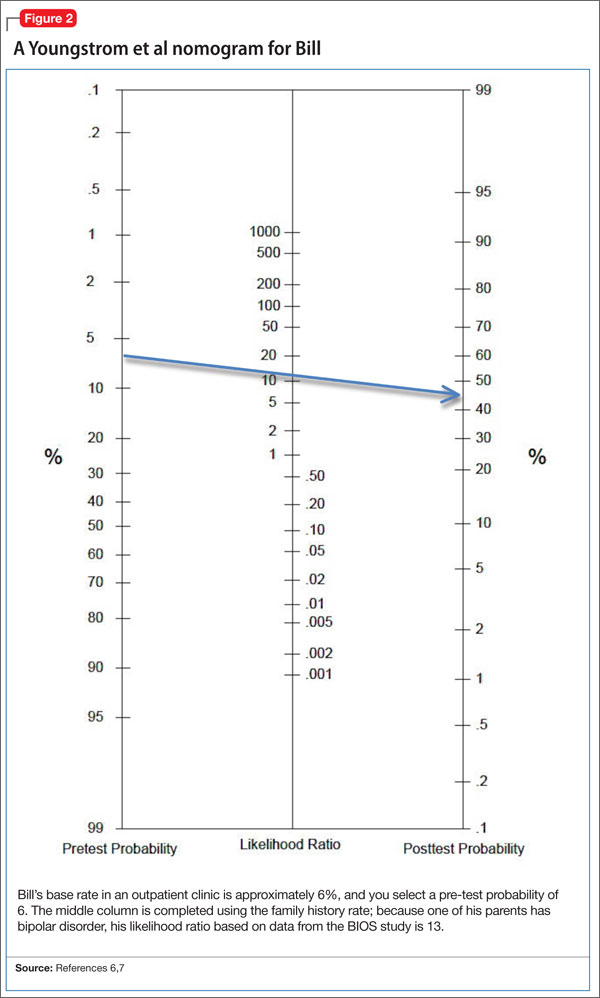
The next strategy is to look at Bill’s scores on externalizing behaviors using an instrument such as the Vanderbilt ADHD Diagnostic Parent Rating Scale. Few pediatric patients with BD will score low on externalizing behaviors.14 Bill scores in the clinically significant range for hyperactivity/impulsivity and positive on the screeners for ODD, conduct disorder (CD), and anxiety/depression.
You decide that Bill is at high risk of pediatric BD; he has a post-test probability of approximately 45%, and many externalizing behaviors on the Vanderbilt. You give Bill a diagnosis of BD I and ADHD and prescribe risperidone, 0.5 mg/d, which results in significant improvement in mood swings and other manic behaviors.
ADHD
Epidemiology. ADHD is one of the most common neurodevelopmental disorders in childhood, with prevalence estimates of 8% of U.S. children.15,16 Overall, boys are more likely to be assigned a diagnosis of ADHD than girls.15 Although ADHD often is diagnosed in early childhood, research is working to clarify the lifetime prevalence of ADHD into late adolescence and adulthood. Current estimates suggest that ADHD persists into adulthood in close to two-thirds of patients.17 However, the symptom presentation can change during adolescence and adulthood, with less overt hyperactivity and symptoms of impulsivity transitioning to risky behaviors involving trouble with the law, substance use, and sexual promiscuity.17
As in pediatric BD, comorbidity is common in ADHD, with uncomplicated ADHD being the exception rather than the rule. Recent studies have suggested that approximately two-thirds of children who have a diagnosis of ADHD have ≥1 comorbid diagnoses.15 Common comorbidities are similar to those seen in BD, including ODD, CD, anxiety disorders, depression, and learning disability. Several tools and resources are available to help clinicians navigate these issues within their practices.
Family history. Genetics appear to play a large role in ADHD, with twin studies suggesting inheritance of approximately 76%.18 Environmental factors contribute, either in the development of ADHD or in the exacerbation of an underlying familial predisposition. Interestingly, in children with BD, family history often is significant for several family members who have both ADHD and BD. However, in children with ADHD only, family history often reflects an absence of family members with BD.19 Although not diagnostic, this pattern can be helpful when considering a diagnosis of BD vs ADHD.
Clinical picture. ADHD often is recognized in childhood; DSM-5 criteria specify that symptoms be present before age 12 and persist for at least 6 months. This characterization of the timing of symptoms helps exclude behavioral disruptions related to external factors such as trauma (eg, death of a caregiver) or abuse. It also is important to note that symptoms might be present earlier but not come to attention clinically until a later age, perhaps because of increasing demands placed on the child by school, peer groups, and extracurricular activities. To make an ADHD diagnosis, symptoms must be present in >1 setting and interfere with functioning or development.
Core symptoms of ADHD include inattention, hyperactivity, and impulsivity that are out of proportion to the child’s developmental level (Table 2).20 When considering diagnosis of ADHD, 6 of 9 symptoms for inattention and/or hyperactivity-impulsivity must be present at a clinically significant level.
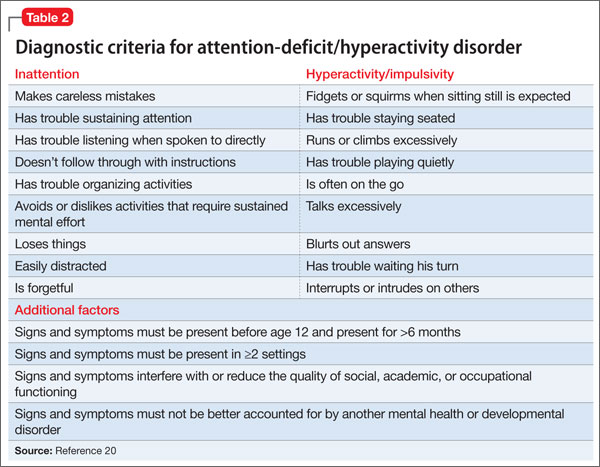
Three different ADHD presentations are recognized: combined, inattentive, and hyperactive impulsive. Children with predominant impulsive and hyperactive behaviors generally come to clinical attention at a younger age; inattentive symptoms often take longer to identify.
Children with ADHD have been noted to have lower tolerance for frustration, which might make anger outbursts and aggressive behavior more likely. Anger and aggression in ADHD often stem from impulsivity, rather than irritable mood seen with BD.18 Issues related to self-esteem, depression, substance use, and CD can contribute to symptoms of irritability, anger, and aggression that can occur in children with ADHD. Although these symptoms can overlap with those seen in children with BD, other core symptoms of ADHD will not be present.
ODD is one of the most common comorbidities among children with ADHD, and the combination of ODD and ADHD may be confused with BD. Children with ODD often are noted to exhibit a pattern of negative and defiant behavior that is out of proportion to what is seen in their peers and for their age and developmental level (Table 3).20 When considering an ODD diagnosis, 4 out of 8 symptoms must be present at a clinically significant level.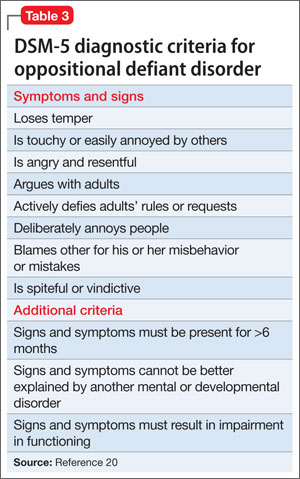
The following case highlights the potential similarities between ADHD/ODD and BD, with tips on how to distinguish them.
CASE REPORT
Angry and destructiveSam, age 7, has been given a diagnosis of ADHD, but his parents think that he isn’t improving with methylphenidate treatment. They are concerned that he has anger issues like his uncle, who has “bipolar disorder.”
Sam’s parents find that he gets frustrated easily and note that he has frequent short “meltdowns” and “mood swings.” During these episodes he yells, is aggressive towards others, and can be destructive. They are concerned because Sam will become angry quickly, then act as if nothing happened after the meltdown has blown over. Sam’s parents feel that he doesn’t listen to them and often argues when they make a request. His parents note that when they push harder, Sam digs in his heels, which can trigger his meltdowns.
Despite clearly disobeying his parents, Sam often says that things aren’t his fault and blames his parents or siblings instead. Sam seems to disagree with people often. His mother reports “if I say the water looks blue, he’ll say it’s green.” Often, Sam seems to argue or pester others to get a rise out of them. This is causing problems for Sam with his siblings and peers, and significant stress for his parents. Family history suggests that Sam’s uncle may have ADHD with CD or a substance use disorder, rather than true BD. Other than Sam’s uncle, there is no family history for BD.
Sam’s parents say that extended release methylphenidate, 20 mg/d, has helped with hyperactivity, but they are concerned that other symptoms have not improved. Aside from the symptoms listed above, Sam is described as a happy child. There is no history of trauma, and no symptoms of anxiety are noted. Sam sometimes gets “down” when things don’t go his way, but this lasts only for a few hours. Sam has a history of delayed sleep onset, which responded well to melatonin. No other symptoms that suggest mania are described.
You complete the pediatric bipolar nomogram (Figure 3)6,7 and Sam’s parents complete a Vanderbilt ADHD Diagnostic Parent Rating Scale. At first, Sam seems to have several factors that might indicate BD: aggressive behavior, mood swings, sleep problems, and, possibly, a family history of BD.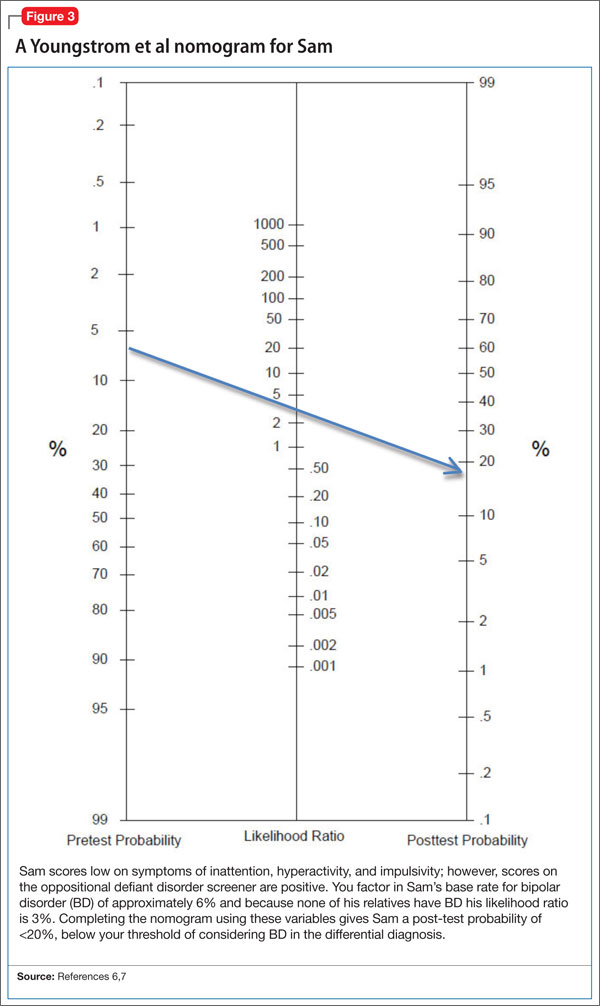
However, a careful history provides several clues that Sam has a comorbid diagnosis of ODD. Sam is exhibiting the classic pattern of negativist behavior seen in children with ODD. In contrast to the episodic pattern of BD, these symptoms are prevalent and persistent, and manifest as an overall pattern of functioning. Impulsivity seen in children with ADHD can complicate the picture, but again appears as a consistent pattern rather than bouts of irritability. Sam’s core symptoms of ADHD (hyperactivity) improved with methylphenidate, but the underlying symptoms of ODD persisted.
Sleep problems are common in children who have ADHD and BD, but Sam’s delayed sleep onset responded to melatonin, whereas the insomnia seen in BD often is refractory to lower-intensity interventions, such as melatonin. Taking a careful family history led you to believe that BD in the family is unlikely. Although this type of detail may not always be available, it can be helpful to ask about mental health symptoms that seem to “run in the family.”
Bottom Line
Distinguishing the child who has bipolar disorder from one who has attention-deficit/hyperactivity disorder can be challenging. A careful history helps ensure that you are on the path toward understanding the diagnostic possibilities. Tools such as the Vanderbilt Rating Scale can further clarify possible diagnoses, and the nomogram approach can provide even more predictive information when considering a diagnosis of bipolar disorder.
Related Resources
• Children and Adults with Attention Deficit/Hyperactivity Disorder (CHADD). www.chadd.org.
• American Academy of Child and Adolescent Psychiatry. Facts for Families. www.aacap.org/cs/root/facts_for_families/ facts_for_families.
• Froehlich TE, Delgado SV, Anixt JS. Expanding medication options for pediatric ADHD. Current Psychiatry. 2013;(12)12:20-29.
• Passarotti AM, Pavuluri MN. Brain functional domains inform therapeutic interventions in attention-deficit/hyperactivity disorder and pediatric bipolar disorder. Expert Rev Neurother. 2011;11(6):897-914.
Drug Brand Names
Methylphenidate • Ritalin, Methylin, Metadate CD, Metadate ER, Methylin ER, Ritalin LA, Ritalin SR, Concerta, Quillivant XR, Daytrana
Risperidone • Risperdal
1. Faraone SV, Biederman J, Wozniak J, et al. Is comorbidity with ADHD a marker for juvenile-onset mania? J Am Acad Child Adolesc Psychiatry. 1997;36(8):1046-1055.
2. West SA, McElroy SL, Strakowski SM, et al. Attention deficit hyperactivity disorder in adolescent mania. Am J Psychiatry. 1995;152(2):271-273.
3. McHugh PR, Slavney PR. Mental illness–comprehensive evaluation or checklist? N Engl J Med. 2012;366(20): 1853-1855.
4. Christensen CM, Grossman JH, Hwang J. The innovator’s prescription: a disruptive solution for health care. New York, NY: McGraw-Hill; 2009.
5. Yazawa M, Hsueh B, Jia X, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471(7337):230-234.
6. Youngstrom EA, Duax J. Evidence-based assessment of pediatric bipolar disorder, part I: base rate and family history. J Am Acad Child Adolesc Psychiatry. 2005;44(7): 712-717.
7. Youngstrom EA, Jenkins MM, Doss AJ, et al. Evidence-based assessment strategies for pediatric bipolar disorder. Isr J Psychiatry Relat Sci. 2012;49(1):15-27.
8. Van Meter AR, Moreira AL, Youngstrom EA. Meta-analysis of epidemiologic studies of pediatric bipolar disorder. J Clin Psychiatry. 2011;72(9):1250-1256.
9. Birmaher B, Axelson D, Monk K, et al. Lifetime psychiatric disorders in school-aged offspring of parents with bipolar disorder: the Pittsburgh Bipolar Offspring study. Arch Gen Psychiatry. 2009;66(3):287-296.
10. Youngstrom EA, Birmaher B, Findling RL. Pediatric bipolar disorder: validity, phenomenology, and recommendations for diagnosis. Bipolar Disord. 2008;10 (1 pt 2):194-214.
11. Geller B, Warner K, Williams M, et al. Prepubertal and young adolescent bipolarity versus ADHD: assessment and validity using the WASH-U-KSADS, CBCL and TRF. J Affect Disord. 1998;51(2):93-100.
12. Richardson WS, Wilson MC, Guyatt GH, et al. Users’ guides to the medical literature: XV. How to use an article about disease probability for differential diagnosis. Evidence-Based Medicine Working Group. JAMA. 1999;281(13):1214-1219.
13. Nease RF Jr, Owens DK, Sox HC Jr. Threshold analysis using diagnostic tests with multiple results. Med Decis Making. 1989;9(2):91-103.
14. Youngstrom EA, Youngstrom JK. Evidence-based assessment of pediatric bipolar disorder, Part II: incorporating information from behavior checklists. J Am Acad Child Adolesc Psychiatry. 2005;44(8):823-828.
15. Merikangas KR, He JP, Brody D, et al. Prevalence and treatment of mental disorders among US children in the 2001-2004 NHANES. Pediatrics. 2010;125(1):75-81.
16. Larson K, Russ SA, Kahn RS, et al. Patterns of comorbidity, functioning, and service use for US children with ADHD, 2007. Pediatrics. 2011;127(3):462-470.
17. Simon V, Czobor P, Bálint S, et al. Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br J Psychiatry. 2009;194(3):204-211.
18. Biederman J, Faraone SV. Attention-deficit hyperactivity disorder. Lancet. 2005;366(9481):237-248.
19. Sood AB, Razdan A, Weller EB, et al. How to differentiate bipolar disorder from attention deficit hyperactivity disorder and other common psychiatric disorders: a guide for clinicians. Curr Psychiatry Rep. 2005;7(2): 98-103.
20. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
Differentiating the irritable, oppositional child with attention-deficit/hyperactivity disorder (ADHD) from the child with bipolar disorder (BD) often is difficult. To make matters more complicated, 50% to 70% of patients with BD have comorbid ADHD.1,2 Accordingly, clinicians are often faced with the moody, irritable, disruptive child whose parents want to know if he (she) is “bipolar” to try to deal with oppositional and mood behaviors.
In this article, we present an approach that will help you distinguish these 2 disorders from each other.
Precision medicineThere is a lack of evidence-based methods for diagnosing psychiatric disorders in children and adolescents. DSM-5 provides clinicians with diagnostic checklists that rely on the clinician’s judgment and training in evaluating a patient.3 In The innovator’s prescription: a disruptive solution for health care, Christensen et al4 describe how medicine is moving from “intuitive medicine” to empirical medicine and toward “precision medicine.” Intuitive medicine depends on the clinician’s expertise, training, and exposure to different disorders, which is the traditional clinical model that predominates in child psychiatry. Empirical medicine relies on laboratory results, scans, scales, and other standardized tools.
Precision medicine occurs when a disorder can be precisely diagnosed and its cause understood, and when it can be treated with effective, evidence-based therapies. An example of this movement toward precision is Timothy syndrome (TS), a rare autosomal dominant disorder characterized by physical malformations, cardiac arrhythmias and structural heart defects, webbing of fingers and toes, and autism spectrum disorder. In the past, a child with TS would have been given a diagnosis of intellectual disability, or a specialist in developmental disorders might recognize the pattern of TS. It is now known that TS is caused by mutations in CACNA1C, the gene encoding the calcium channel Cav1.2α subunit, allowing precise diagnosis by genotyping.5
Although there are several tools that help clinicians assess symptoms of ADHD and BD, including rating scales such the Vanderbilt ADHD Diagnostic Rating Scale and Young Mania Rating Scale, none of these scales are diagnostic. Youngstrom et al6,7 have developed an evidence-based strategy to diagnose pediatric BD. This method uses a nomogram that takes into account the base rate of BD in a clinical setting and family history of BD.
We will describe and contrast the epidemiologic and clinical characteristics of pediatric BD from ADHD and use the Youngstrom nomogram to better define these patients. Although still far from precision medicine, the type of approach represents an ongoing effort in mental health care to increase diagnostic accuracy and improve treatment outcomes.
Pediatric bipolar disorder
Prevalence of pediatric BD is 1.8% (95% CI, 1.1% to 3.0%),8 which does not include sub-threshold cases of BD. ADHD and oppositional defiant disorder (ODD) are 8 to 10 times more prevalent. For the purposes of the nomogram, the “base rate” is the rate at which a disorder occurs in different clinical settings. In general outpatient clinics, BD might occur 6% to 8% of the time, whereas in a county-run child psychiatry inpatient facility the rate is 11%.6 A reasonable rate in an outpatient pediatric setting is 6%.
Family history. In the Bipolar Offspring Study,9 the rate of BD in children of parents with BD was 13 times greater than that of controls, and the rate of anxiety and behavior disorders was approximately twice that of children of parents without BD (Table 1).9 This study evaluated 388 children of 233 parents with BD and 251 children of 143 demographically matched controls.
Clinical characteristics. Children and adolescents with BD typically manifest with what can be described as a “mood cycle”—a pronounced shift in mood and energy from one extreme to another. An example would be a child who wakes up with extreme silliness, high energy, and intrusive behavior that persists for several hours, then later becomes sad, depressed, and suicidal with no precipitant for either mood cycle.10 Pediatric patients with BD also exhibit other symptoms of mania during mood cycling periods.
Elevated or expansive mood. The child might have a mood that is inappropriately giddy, silly, elated, or euphoric. Often this mood will be present without reason and last for several hours. It may be distinguished from a transient cheerful mood by the intensity and duration of the episode. The child with BD may have little to no insight about the inappropriate nature of their elevated mood, when present.
Irritable mood. The child might become markedly belligerent or irritated with intense outbursts of anger, 2 to 3 times a day for several hours. An adolescent might appear extremely oppositional, belligerent, or hostile with parents and others.
Grandiosity or inflated self-esteem can be confused with brief childhood fantasies of increased capability. Typically, true grandiosity can manifest as assertion of great competency in all areas of life, which usually cannot be altered by contrary external evidence. Occasionally, this is bizarre and includes delusions of “super powers.” The child in a manic episode will not only assert that she can fly, but will jump off the garage roof to prove it.
Decreased need for sleep. The child may only require 4 to 5 hours of sleep a night during a manic episode without feeling fatigued or showing evidence of tiredness. Consider substance use in this differential diagnosis, especially in adolescents.
Increased talkativeness. Lack of inhibition to social norms may lead pediatric BD patients to blurt out answers during class or repeatedly be disciplined for talking to peers in class. Speech typically is rapid and pressured to the point where it might be continuous and seems to jump between loosely related subjects.
Flight of ideas or racing thoughts. The child or adolescent might report a subjective feeling that his thoughts are moving so rapidly that his speech cannot keep up. Often this is differentiated from rapid speech by the degree of rapidity the patient expresses loosely related topics that might seem completely unrelated to the listener.
Distractibility, short attention span. During a manic episode, the child or adolescent might report that it is impossible to pay attention to class or other outside events because of rapidly changing focus of their thoughts. This symptom must be carefully distinguished from the distractibility and inattention of ADHD, which typically is a more fixed and long-standing pattern rather than a brief episodic phenomenon in a manic or hypomanic episode.
Increase in goal-directed activity. During a mild manic episode, the child or adolescent may be capable of accomplishing a great deal of work. However, episodes that are more severe manifest as an individual starting numerous ambitious projects that she later is unable to complete.
Excessive risk-taking activities. The child or adolescent might become involved in forbidden, pleasurable activities that have a high risk of adverse consequences. This can manifest as hypersexual behavior, frequent fighting, increased recklessness, use of drugs and alcohol, shopping sprees, and reckless driving.
There are few studies comparing patients with comorbid BD and ADHD with patients with only ADHD. Geller et al11 compared 60 children with BD and ADHD (mean age, 10) to age- and sex-matched patients with ADHD and no mood disorder. Compared with children who had ADHD, those with BD exhibited significantly greater elevated mood, grandiosity, flight and/or racing of ideas, decreased need for sleep, and hypersexuality (Figure 1,11). Features common to both groups—and therefore not useful in differentiating the disorders—included irritability, hyperactivity, accelerated speech, and distractibility.
CASE REPORTIrritable and disruptiveBill, age 12, has been brought to see you by his mother because she is concerned about escalating behavior problems at home and school in the past several months. The school principal has called her about his obnoxious behavior with teachers and about other parents’ complaints that he has made unwanted sexual advances to girls who sit next to him in class.
Bill, who is in the 7th grade, is on the verge of being suspended for his inappropriate and disruptive behavior. His parents report that he is irritable around them and stays up all night, messaging his friends on the Internet from his iPad in his bedroom. They attribute his inappropriate sexual behavior to puberty and possibly to the Web sites he views.
Bill’s mother is concerned about his:
• increasing behavior problems during the last several months at home and school
• intensifying irritability and depressive symptoms
• staying up all night on the Internet, phoning friends, and doing projects
• frequent unprovoked, outbursts of rage occurring with increasing frequency and intensity (almost daily)
• moderate grandiosity, including telling the soccer coach and teachers how to do their jobs
• inappropriate sexual behavior, including kissing and touching female classmates.
During your history, you learn that Bill has been a bright and artistic child, with good academic performance. His peer relationships have been satisfactory, but not excellent—he tends to be “bossy” with his peers. He is medically healthy and not taking any medications. As part of your history, you also talk with Bill and his family about exposure to trauma or significant stressors, which they deny. You learn that Bill’s father was diagnosed with BD I at age 32.
Completing the nomogram developed by Youngstrom et al6,7 using these variables (see this article at CurrentPsychiatry.com for Figure 2)6,7 gives Bill a post-test probability of approximately 42%. The threshold for moving ahead with assessment and possible treatment, the “test-treatment threshold,” depends on your clinical setting.12,13 Our clinical experience is that, when the post-test probability exceeds 30%, further assessment for BD is warranted.
The next strategy is to look at Bill’s scores on externalizing behaviors using an instrument such as the Vanderbilt ADHD Diagnostic Parent Rating Scale. Few pediatric patients with BD will score low on externalizing behaviors.14 Bill scores in the clinically significant range for hyperactivity/impulsivity and positive on the screeners for ODD, conduct disorder (CD), and anxiety/depression.
You decide that Bill is at high risk of pediatric BD; he has a post-test probability of approximately 45%, and many externalizing behaviors on the Vanderbilt. You give Bill a diagnosis of BD I and ADHD and prescribe risperidone, 0.5 mg/d, which results in significant improvement in mood swings and other manic behaviors.
ADHD
Epidemiology. ADHD is one of the most common neurodevelopmental disorders in childhood, with prevalence estimates of 8% of U.S. children.15,16 Overall, boys are more likely to be assigned a diagnosis of ADHD than girls.15 Although ADHD often is diagnosed in early childhood, research is working to clarify the lifetime prevalence of ADHD into late adolescence and adulthood. Current estimates suggest that ADHD persists into adulthood in close to two-thirds of patients.17 However, the symptom presentation can change during adolescence and adulthood, with less overt hyperactivity and symptoms of impulsivity transitioning to risky behaviors involving trouble with the law, substance use, and sexual promiscuity.17
As in pediatric BD, comorbidity is common in ADHD, with uncomplicated ADHD being the exception rather than the rule. Recent studies have suggested that approximately two-thirds of children who have a diagnosis of ADHD have ≥1 comorbid diagnoses.15 Common comorbidities are similar to those seen in BD, including ODD, CD, anxiety disorders, depression, and learning disability. Several tools and resources are available to help clinicians navigate these issues within their practices.
Family history. Genetics appear to play a large role in ADHD, with twin studies suggesting inheritance of approximately 76%.18 Environmental factors contribute, either in the development of ADHD or in the exacerbation of an underlying familial predisposition. Interestingly, in children with BD, family history often is significant for several family members who have both ADHD and BD. However, in children with ADHD only, family history often reflects an absence of family members with BD.19 Although not diagnostic, this pattern can be helpful when considering a diagnosis of BD vs ADHD.
Clinical picture. ADHD often is recognized in childhood; DSM-5 criteria specify that symptoms be present before age 12 and persist for at least 6 months. This characterization of the timing of symptoms helps exclude behavioral disruptions related to external factors such as trauma (eg, death of a caregiver) or abuse. It also is important to note that symptoms might be present earlier but not come to attention clinically until a later age, perhaps because of increasing demands placed on the child by school, peer groups, and extracurricular activities. To make an ADHD diagnosis, symptoms must be present in >1 setting and interfere with functioning or development.
Core symptoms of ADHD include inattention, hyperactivity, and impulsivity that are out of proportion to the child’s developmental level (Table 2).20 When considering diagnosis of ADHD, 6 of 9 symptoms for inattention and/or hyperactivity-impulsivity must be present at a clinically significant level.
Three different ADHD presentations are recognized: combined, inattentive, and hyperactive impulsive. Children with predominant impulsive and hyperactive behaviors generally come to clinical attention at a younger age; inattentive symptoms often take longer to identify.
Children with ADHD have been noted to have lower tolerance for frustration, which might make anger outbursts and aggressive behavior more likely. Anger and aggression in ADHD often stem from impulsivity, rather than irritable mood seen with BD.18 Issues related to self-esteem, depression, substance use, and CD can contribute to symptoms of irritability, anger, and aggression that can occur in children with ADHD. Although these symptoms can overlap with those seen in children with BD, other core symptoms of ADHD will not be present.
ODD is one of the most common comorbidities among children with ADHD, and the combination of ODD and ADHD may be confused with BD. Children with ODD often are noted to exhibit a pattern of negative and defiant behavior that is out of proportion to what is seen in their peers and for their age and developmental level (Table 3).20 When considering an ODD diagnosis, 4 out of 8 symptoms must be present at a clinically significant level.
The following case highlights the potential similarities between ADHD/ODD and BD, with tips on how to distinguish them.
CASE REPORT
Angry and destructiveSam, age 7, has been given a diagnosis of ADHD, but his parents think that he isn’t improving with methylphenidate treatment. They are concerned that he has anger issues like his uncle, who has “bipolar disorder.”
Sam’s parents find that he gets frustrated easily and note that he has frequent short “meltdowns” and “mood swings.” During these episodes he yells, is aggressive towards others, and can be destructive. They are concerned because Sam will become angry quickly, then act as if nothing happened after the meltdown has blown over. Sam’s parents feel that he doesn’t listen to them and often argues when they make a request. His parents note that when they push harder, Sam digs in his heels, which can trigger his meltdowns.
Despite clearly disobeying his parents, Sam often says that things aren’t his fault and blames his parents or siblings instead. Sam seems to disagree with people often. His mother reports “if I say the water looks blue, he’ll say it’s green.” Often, Sam seems to argue or pester others to get a rise out of them. This is causing problems for Sam with his siblings and peers, and significant stress for his parents. Family history suggests that Sam’s uncle may have ADHD with CD or a substance use disorder, rather than true BD. Other than Sam’s uncle, there is no family history for BD.
Sam’s parents say that extended release methylphenidate, 20 mg/d, has helped with hyperactivity, but they are concerned that other symptoms have not improved. Aside from the symptoms listed above, Sam is described as a happy child. There is no history of trauma, and no symptoms of anxiety are noted. Sam sometimes gets “down” when things don’t go his way, but this lasts only for a few hours. Sam has a history of delayed sleep onset, which responded well to melatonin. No other symptoms that suggest mania are described.
You complete the pediatric bipolar nomogram (Figure 3)6,7 and Sam’s parents complete a Vanderbilt ADHD Diagnostic Parent Rating Scale. At first, Sam seems to have several factors that might indicate BD: aggressive behavior, mood swings, sleep problems, and, possibly, a family history of BD.
However, a careful history provides several clues that Sam has a comorbid diagnosis of ODD. Sam is exhibiting the classic pattern of negativist behavior seen in children with ODD. In contrast to the episodic pattern of BD, these symptoms are prevalent and persistent, and manifest as an overall pattern of functioning. Impulsivity seen in children with ADHD can complicate the picture, but again appears as a consistent pattern rather than bouts of irritability. Sam’s core symptoms of ADHD (hyperactivity) improved with methylphenidate, but the underlying symptoms of ODD persisted.
Sleep problems are common in children who have ADHD and BD, but Sam’s delayed sleep onset responded to melatonin, whereas the insomnia seen in BD often is refractory to lower-intensity interventions, such as melatonin. Taking a careful family history led you to believe that BD in the family is unlikely. Although this type of detail may not always be available, it can be helpful to ask about mental health symptoms that seem to “run in the family.”
Bottom Line
Distinguishing the child who has bipolar disorder from one who has attention-deficit/hyperactivity disorder can be challenging. A careful history helps ensure that you are on the path toward understanding the diagnostic possibilities. Tools such as the Vanderbilt Rating Scale can further clarify possible diagnoses, and the nomogram approach can provide even more predictive information when considering a diagnosis of bipolar disorder.
Related Resources
• Children and Adults with Attention Deficit/Hyperactivity Disorder (CHADD). www.chadd.org.
• American Academy of Child and Adolescent Psychiatry. Facts for Families. www.aacap.org/cs/root/facts_for_families/ facts_for_families.
• Froehlich TE, Delgado SV, Anixt JS. Expanding medication options for pediatric ADHD. Current Psychiatry. 2013;(12)12:20-29.
• Passarotti AM, Pavuluri MN. Brain functional domains inform therapeutic interventions in attention-deficit/hyperactivity disorder and pediatric bipolar disorder. Expert Rev Neurother. 2011;11(6):897-914.
Drug Brand Names
Methylphenidate • Ritalin, Methylin, Metadate CD, Metadate ER, Methylin ER, Ritalin LA, Ritalin SR, Concerta, Quillivant XR, Daytrana
Risperidone • Risperdal
Differentiating the irritable, oppositional child with attention-deficit/hyperactivity disorder (ADHD) from the child with bipolar disorder (BD) often is difficult. To make matters more complicated, 50% to 70% of patients with BD have comorbid ADHD.1,2 Accordingly, clinicians are often faced with the moody, irritable, disruptive child whose parents want to know if he (she) is “bipolar” to try to deal with oppositional and mood behaviors.
In this article, we present an approach that will help you distinguish these 2 disorders from each other.
Precision medicineThere is a lack of evidence-based methods for diagnosing psychiatric disorders in children and adolescents. DSM-5 provides clinicians with diagnostic checklists that rely on the clinician’s judgment and training in evaluating a patient.3 In The innovator’s prescription: a disruptive solution for health care, Christensen et al4 describe how medicine is moving from “intuitive medicine” to empirical medicine and toward “precision medicine.” Intuitive medicine depends on the clinician’s expertise, training, and exposure to different disorders, which is the traditional clinical model that predominates in child psychiatry. Empirical medicine relies on laboratory results, scans, scales, and other standardized tools.
Precision medicine occurs when a disorder can be precisely diagnosed and its cause understood, and when it can be treated with effective, evidence-based therapies. An example of this movement toward precision is Timothy syndrome (TS), a rare autosomal dominant disorder characterized by physical malformations, cardiac arrhythmias and structural heart defects, webbing of fingers and toes, and autism spectrum disorder. In the past, a child with TS would have been given a diagnosis of intellectual disability, or a specialist in developmental disorders might recognize the pattern of TS. It is now known that TS is caused by mutations in CACNA1C, the gene encoding the calcium channel Cav1.2α subunit, allowing precise diagnosis by genotyping.5
Although there are several tools that help clinicians assess symptoms of ADHD and BD, including rating scales such the Vanderbilt ADHD Diagnostic Rating Scale and Young Mania Rating Scale, none of these scales are diagnostic. Youngstrom et al6,7 have developed an evidence-based strategy to diagnose pediatric BD. This method uses a nomogram that takes into account the base rate of BD in a clinical setting and family history of BD.
We will describe and contrast the epidemiologic and clinical characteristics of pediatric BD from ADHD and use the Youngstrom nomogram to better define these patients. Although still far from precision medicine, the type of approach represents an ongoing effort in mental health care to increase diagnostic accuracy and improve treatment outcomes.
Pediatric bipolar disorder
Prevalence of pediatric BD is 1.8% (95% CI, 1.1% to 3.0%),8 which does not include sub-threshold cases of BD. ADHD and oppositional defiant disorder (ODD) are 8 to 10 times more prevalent. For the purposes of the nomogram, the “base rate” is the rate at which a disorder occurs in different clinical settings. In general outpatient clinics, BD might occur 6% to 8% of the time, whereas in a county-run child psychiatry inpatient facility the rate is 11%.6 A reasonable rate in an outpatient pediatric setting is 6%.
Family history. In the Bipolar Offspring Study,9 the rate of BD in children of parents with BD was 13 times greater than that of controls, and the rate of anxiety and behavior disorders was approximately twice that of children of parents without BD (Table 1).9 This study evaluated 388 children of 233 parents with BD and 251 children of 143 demographically matched controls.
Clinical characteristics. Children and adolescents with BD typically manifest with what can be described as a “mood cycle”—a pronounced shift in mood and energy from one extreme to another. An example would be a child who wakes up with extreme silliness, high energy, and intrusive behavior that persists for several hours, then later becomes sad, depressed, and suicidal with no precipitant for either mood cycle.10 Pediatric patients with BD also exhibit other symptoms of mania during mood cycling periods.
Elevated or expansive mood. The child might have a mood that is inappropriately giddy, silly, elated, or euphoric. Often this mood will be present without reason and last for several hours. It may be distinguished from a transient cheerful mood by the intensity and duration of the episode. The child with BD may have little to no insight about the inappropriate nature of their elevated mood, when present.
Irritable mood. The child might become markedly belligerent or irritated with intense outbursts of anger, 2 to 3 times a day for several hours. An adolescent might appear extremely oppositional, belligerent, or hostile with parents and others.
Grandiosity or inflated self-esteem can be confused with brief childhood fantasies of increased capability. Typically, true grandiosity can manifest as assertion of great competency in all areas of life, which usually cannot be altered by contrary external evidence. Occasionally, this is bizarre and includes delusions of “super powers.” The child in a manic episode will not only assert that she can fly, but will jump off the garage roof to prove it.
Decreased need for sleep. The child may only require 4 to 5 hours of sleep a night during a manic episode without feeling fatigued or showing evidence of tiredness. Consider substance use in this differential diagnosis, especially in adolescents.
Increased talkativeness. Lack of inhibition to social norms may lead pediatric BD patients to blurt out answers during class or repeatedly be disciplined for talking to peers in class. Speech typically is rapid and pressured to the point where it might be continuous and seems to jump between loosely related subjects.
Flight of ideas or racing thoughts. The child or adolescent might report a subjective feeling that his thoughts are moving so rapidly that his speech cannot keep up. Often this is differentiated from rapid speech by the degree of rapidity the patient expresses loosely related topics that might seem completely unrelated to the listener.
Distractibility, short attention span. During a manic episode, the child or adolescent might report that it is impossible to pay attention to class or other outside events because of rapidly changing focus of their thoughts. This symptom must be carefully distinguished from the distractibility and inattention of ADHD, which typically is a more fixed and long-standing pattern rather than a brief episodic phenomenon in a manic or hypomanic episode.
Increase in goal-directed activity. During a mild manic episode, the child or adolescent may be capable of accomplishing a great deal of work. However, episodes that are more severe manifest as an individual starting numerous ambitious projects that she later is unable to complete.
Excessive risk-taking activities. The child or adolescent might become involved in forbidden, pleasurable activities that have a high risk of adverse consequences. This can manifest as hypersexual behavior, frequent fighting, increased recklessness, use of drugs and alcohol, shopping sprees, and reckless driving.
There are few studies comparing patients with comorbid BD and ADHD with patients with only ADHD. Geller et al11 compared 60 children with BD and ADHD (mean age, 10) to age- and sex-matched patients with ADHD and no mood disorder. Compared with children who had ADHD, those with BD exhibited significantly greater elevated mood, grandiosity, flight and/or racing of ideas, decreased need for sleep, and hypersexuality (Figure 1,11). Features common to both groups—and therefore not useful in differentiating the disorders—included irritability, hyperactivity, accelerated speech, and distractibility.
CASE REPORTIrritable and disruptiveBill, age 12, has been brought to see you by his mother because she is concerned about escalating behavior problems at home and school in the past several months. The school principal has called her about his obnoxious behavior with teachers and about other parents’ complaints that he has made unwanted sexual advances to girls who sit next to him in class.
Bill, who is in the 7th grade, is on the verge of being suspended for his inappropriate and disruptive behavior. His parents report that he is irritable around them and stays up all night, messaging his friends on the Internet from his iPad in his bedroom. They attribute his inappropriate sexual behavior to puberty and possibly to the Web sites he views.
Bill’s mother is concerned about his:
• increasing behavior problems during the last several months at home and school
• intensifying irritability and depressive symptoms
• staying up all night on the Internet, phoning friends, and doing projects
• frequent unprovoked, outbursts of rage occurring with increasing frequency and intensity (almost daily)
• moderate grandiosity, including telling the soccer coach and teachers how to do their jobs
• inappropriate sexual behavior, including kissing and touching female classmates.
During your history, you learn that Bill has been a bright and artistic child, with good academic performance. His peer relationships have been satisfactory, but not excellent—he tends to be “bossy” with his peers. He is medically healthy and not taking any medications. As part of your history, you also talk with Bill and his family about exposure to trauma or significant stressors, which they deny. You learn that Bill’s father was diagnosed with BD I at age 32.
Completing the nomogram developed by Youngstrom et al6,7 using these variables (see this article at CurrentPsychiatry.com for Figure 2)6,7 gives Bill a post-test probability of approximately 42%. The threshold for moving ahead with assessment and possible treatment, the “test-treatment threshold,” depends on your clinical setting.12,13 Our clinical experience is that, when the post-test probability exceeds 30%, further assessment for BD is warranted.
The next strategy is to look at Bill’s scores on externalizing behaviors using an instrument such as the Vanderbilt ADHD Diagnostic Parent Rating Scale. Few pediatric patients with BD will score low on externalizing behaviors.14 Bill scores in the clinically significant range for hyperactivity/impulsivity and positive on the screeners for ODD, conduct disorder (CD), and anxiety/depression.
You decide that Bill is at high risk of pediatric BD; he has a post-test probability of approximately 45%, and many externalizing behaviors on the Vanderbilt. You give Bill a diagnosis of BD I and ADHD and prescribe risperidone, 0.5 mg/d, which results in significant improvement in mood swings and other manic behaviors.
ADHD
Epidemiology. ADHD is one of the most common neurodevelopmental disorders in childhood, with prevalence estimates of 8% of U.S. children.15,16 Overall, boys are more likely to be assigned a diagnosis of ADHD than girls.15 Although ADHD often is diagnosed in early childhood, research is working to clarify the lifetime prevalence of ADHD into late adolescence and adulthood. Current estimates suggest that ADHD persists into adulthood in close to two-thirds of patients.17 However, the symptom presentation can change during adolescence and adulthood, with less overt hyperactivity and symptoms of impulsivity transitioning to risky behaviors involving trouble with the law, substance use, and sexual promiscuity.17
As in pediatric BD, comorbidity is common in ADHD, with uncomplicated ADHD being the exception rather than the rule. Recent studies have suggested that approximately two-thirds of children who have a diagnosis of ADHD have ≥1 comorbid diagnoses.15 Common comorbidities are similar to those seen in BD, including ODD, CD, anxiety disorders, depression, and learning disability. Several tools and resources are available to help clinicians navigate these issues within their practices.
Family history. Genetics appear to play a large role in ADHD, with twin studies suggesting inheritance of approximately 76%.18 Environmental factors contribute, either in the development of ADHD or in the exacerbation of an underlying familial predisposition. Interestingly, in children with BD, family history often is significant for several family members who have both ADHD and BD. However, in children with ADHD only, family history often reflects an absence of family members with BD.19 Although not diagnostic, this pattern can be helpful when considering a diagnosis of BD vs ADHD.
Clinical picture. ADHD often is recognized in childhood; DSM-5 criteria specify that symptoms be present before age 12 and persist for at least 6 months. This characterization of the timing of symptoms helps exclude behavioral disruptions related to external factors such as trauma (eg, death of a caregiver) or abuse. It also is important to note that symptoms might be present earlier but not come to attention clinically until a later age, perhaps because of increasing demands placed on the child by school, peer groups, and extracurricular activities. To make an ADHD diagnosis, symptoms must be present in >1 setting and interfere with functioning or development.
Core symptoms of ADHD include inattention, hyperactivity, and impulsivity that are out of proportion to the child’s developmental level (Table 2).20 When considering diagnosis of ADHD, 6 of 9 symptoms for inattention and/or hyperactivity-impulsivity must be present at a clinically significant level.
Three different ADHD presentations are recognized: combined, inattentive, and hyperactive impulsive. Children with predominant impulsive and hyperactive behaviors generally come to clinical attention at a younger age; inattentive symptoms often take longer to identify.
Children with ADHD have been noted to have lower tolerance for frustration, which might make anger outbursts and aggressive behavior more likely. Anger and aggression in ADHD often stem from impulsivity, rather than irritable mood seen with BD.18 Issues related to self-esteem, depression, substance use, and CD can contribute to symptoms of irritability, anger, and aggression that can occur in children with ADHD. Although these symptoms can overlap with those seen in children with BD, other core symptoms of ADHD will not be present.
ODD is one of the most common comorbidities among children with ADHD, and the combination of ODD and ADHD may be confused with BD. Children with ODD often are noted to exhibit a pattern of negative and defiant behavior that is out of proportion to what is seen in their peers and for their age and developmental level (Table 3).20 When considering an ODD diagnosis, 4 out of 8 symptoms must be present at a clinically significant level.
The following case highlights the potential similarities between ADHD/ODD and BD, with tips on how to distinguish them.
CASE REPORT
Angry and destructiveSam, age 7, has been given a diagnosis of ADHD, but his parents think that he isn’t improving with methylphenidate treatment. They are concerned that he has anger issues like his uncle, who has “bipolar disorder.”
Sam’s parents find that he gets frustrated easily and note that he has frequent short “meltdowns” and “mood swings.” During these episodes he yells, is aggressive towards others, and can be destructive. They are concerned because Sam will become angry quickly, then act as if nothing happened after the meltdown has blown over. Sam’s parents feel that he doesn’t listen to them and often argues when they make a request. His parents note that when they push harder, Sam digs in his heels, which can trigger his meltdowns.
Despite clearly disobeying his parents, Sam often says that things aren’t his fault and blames his parents or siblings instead. Sam seems to disagree with people often. His mother reports “if I say the water looks blue, he’ll say it’s green.” Often, Sam seems to argue or pester others to get a rise out of them. This is causing problems for Sam with his siblings and peers, and significant stress for his parents. Family history suggests that Sam’s uncle may have ADHD with CD or a substance use disorder, rather than true BD. Other than Sam’s uncle, there is no family history for BD.
Sam’s parents say that extended release methylphenidate, 20 mg/d, has helped with hyperactivity, but they are concerned that other symptoms have not improved. Aside from the symptoms listed above, Sam is described as a happy child. There is no history of trauma, and no symptoms of anxiety are noted. Sam sometimes gets “down” when things don’t go his way, but this lasts only for a few hours. Sam has a history of delayed sleep onset, which responded well to melatonin. No other symptoms that suggest mania are described.
You complete the pediatric bipolar nomogram (Figure 3)6,7 and Sam’s parents complete a Vanderbilt ADHD Diagnostic Parent Rating Scale. At first, Sam seems to have several factors that might indicate BD: aggressive behavior, mood swings, sleep problems, and, possibly, a family history of BD.
However, a careful history provides several clues that Sam has a comorbid diagnosis of ODD. Sam is exhibiting the classic pattern of negativist behavior seen in children with ODD. In contrast to the episodic pattern of BD, these symptoms are prevalent and persistent, and manifest as an overall pattern of functioning. Impulsivity seen in children with ADHD can complicate the picture, but again appears as a consistent pattern rather than bouts of irritability. Sam’s core symptoms of ADHD (hyperactivity) improved with methylphenidate, but the underlying symptoms of ODD persisted.
Sleep problems are common in children who have ADHD and BD, but Sam’s delayed sleep onset responded to melatonin, whereas the insomnia seen in BD often is refractory to lower-intensity interventions, such as melatonin. Taking a careful family history led you to believe that BD in the family is unlikely. Although this type of detail may not always be available, it can be helpful to ask about mental health symptoms that seem to “run in the family.”
Bottom Line
Distinguishing the child who has bipolar disorder from one who has attention-deficit/hyperactivity disorder can be challenging. A careful history helps ensure that you are on the path toward understanding the diagnostic possibilities. Tools such as the Vanderbilt Rating Scale can further clarify possible diagnoses, and the nomogram approach can provide even more predictive information when considering a diagnosis of bipolar disorder.
Related Resources
• Children and Adults with Attention Deficit/Hyperactivity Disorder (CHADD). www.chadd.org.
• American Academy of Child and Adolescent Psychiatry. Facts for Families. www.aacap.org/cs/root/facts_for_families/ facts_for_families.
• Froehlich TE, Delgado SV, Anixt JS. Expanding medication options for pediatric ADHD. Current Psychiatry. 2013;(12)12:20-29.
• Passarotti AM, Pavuluri MN. Brain functional domains inform therapeutic interventions in attention-deficit/hyperactivity disorder and pediatric bipolar disorder. Expert Rev Neurother. 2011;11(6):897-914.
Drug Brand Names
Methylphenidate • Ritalin, Methylin, Metadate CD, Metadate ER, Methylin ER, Ritalin LA, Ritalin SR, Concerta, Quillivant XR, Daytrana
Risperidone • Risperdal
1. Faraone SV, Biederman J, Wozniak J, et al. Is comorbidity with ADHD a marker for juvenile-onset mania? J Am Acad Child Adolesc Psychiatry. 1997;36(8):1046-1055.
2. West SA, McElroy SL, Strakowski SM, et al. Attention deficit hyperactivity disorder in adolescent mania. Am J Psychiatry. 1995;152(2):271-273.
3. McHugh PR, Slavney PR. Mental illness–comprehensive evaluation or checklist? N Engl J Med. 2012;366(20): 1853-1855.
4. Christensen CM, Grossman JH, Hwang J. The innovator’s prescription: a disruptive solution for health care. New York, NY: McGraw-Hill; 2009.
5. Yazawa M, Hsueh B, Jia X, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471(7337):230-234.
6. Youngstrom EA, Duax J. Evidence-based assessment of pediatric bipolar disorder, part I: base rate and family history. J Am Acad Child Adolesc Psychiatry. 2005;44(7): 712-717.
7. Youngstrom EA, Jenkins MM, Doss AJ, et al. Evidence-based assessment strategies for pediatric bipolar disorder. Isr J Psychiatry Relat Sci. 2012;49(1):15-27.
8. Van Meter AR, Moreira AL, Youngstrom EA. Meta-analysis of epidemiologic studies of pediatric bipolar disorder. J Clin Psychiatry. 2011;72(9):1250-1256.
9. Birmaher B, Axelson D, Monk K, et al. Lifetime psychiatric disorders in school-aged offspring of parents with bipolar disorder: the Pittsburgh Bipolar Offspring study. Arch Gen Psychiatry. 2009;66(3):287-296.
10. Youngstrom EA, Birmaher B, Findling RL. Pediatric bipolar disorder: validity, phenomenology, and recommendations for diagnosis. Bipolar Disord. 2008;10 (1 pt 2):194-214.
11. Geller B, Warner K, Williams M, et al. Prepubertal and young adolescent bipolarity versus ADHD: assessment and validity using the WASH-U-KSADS, CBCL and TRF. J Affect Disord. 1998;51(2):93-100.
12. Richardson WS, Wilson MC, Guyatt GH, et al. Users’ guides to the medical literature: XV. How to use an article about disease probability for differential diagnosis. Evidence-Based Medicine Working Group. JAMA. 1999;281(13):1214-1219.
13. Nease RF Jr, Owens DK, Sox HC Jr. Threshold analysis using diagnostic tests with multiple results. Med Decis Making. 1989;9(2):91-103.
14. Youngstrom EA, Youngstrom JK. Evidence-based assessment of pediatric bipolar disorder, Part II: incorporating information from behavior checklists. J Am Acad Child Adolesc Psychiatry. 2005;44(8):823-828.
15. Merikangas KR, He JP, Brody D, et al. Prevalence and treatment of mental disorders among US children in the 2001-2004 NHANES. Pediatrics. 2010;125(1):75-81.
16. Larson K, Russ SA, Kahn RS, et al. Patterns of comorbidity, functioning, and service use for US children with ADHD, 2007. Pediatrics. 2011;127(3):462-470.
17. Simon V, Czobor P, Bálint S, et al. Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br J Psychiatry. 2009;194(3):204-211.
18. Biederman J, Faraone SV. Attention-deficit hyperactivity disorder. Lancet. 2005;366(9481):237-248.
19. Sood AB, Razdan A, Weller EB, et al. How to differentiate bipolar disorder from attention deficit hyperactivity disorder and other common psychiatric disorders: a guide for clinicians. Curr Psychiatry Rep. 2005;7(2): 98-103.
20. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
1. Faraone SV, Biederman J, Wozniak J, et al. Is comorbidity with ADHD a marker for juvenile-onset mania? J Am Acad Child Adolesc Psychiatry. 1997;36(8):1046-1055.
2. West SA, McElroy SL, Strakowski SM, et al. Attention deficit hyperactivity disorder in adolescent mania. Am J Psychiatry. 1995;152(2):271-273.
3. McHugh PR, Slavney PR. Mental illness–comprehensive evaluation or checklist? N Engl J Med. 2012;366(20): 1853-1855.
4. Christensen CM, Grossman JH, Hwang J. The innovator’s prescription: a disruptive solution for health care. New York, NY: McGraw-Hill; 2009.
5. Yazawa M, Hsueh B, Jia X, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471(7337):230-234.
6. Youngstrom EA, Duax J. Evidence-based assessment of pediatric bipolar disorder, part I: base rate and family history. J Am Acad Child Adolesc Psychiatry. 2005;44(7): 712-717.
7. Youngstrom EA, Jenkins MM, Doss AJ, et al. Evidence-based assessment strategies for pediatric bipolar disorder. Isr J Psychiatry Relat Sci. 2012;49(1):15-27.
8. Van Meter AR, Moreira AL, Youngstrom EA. Meta-analysis of epidemiologic studies of pediatric bipolar disorder. J Clin Psychiatry. 2011;72(9):1250-1256.
9. Birmaher B, Axelson D, Monk K, et al. Lifetime psychiatric disorders in school-aged offspring of parents with bipolar disorder: the Pittsburgh Bipolar Offspring study. Arch Gen Psychiatry. 2009;66(3):287-296.
10. Youngstrom EA, Birmaher B, Findling RL. Pediatric bipolar disorder: validity, phenomenology, and recommendations for diagnosis. Bipolar Disord. 2008;10 (1 pt 2):194-214.
11. Geller B, Warner K, Williams M, et al. Prepubertal and young adolescent bipolarity versus ADHD: assessment and validity using the WASH-U-KSADS, CBCL and TRF. J Affect Disord. 1998;51(2):93-100.
12. Richardson WS, Wilson MC, Guyatt GH, et al. Users’ guides to the medical literature: XV. How to use an article about disease probability for differential diagnosis. Evidence-Based Medicine Working Group. JAMA. 1999;281(13):1214-1219.
13. Nease RF Jr, Owens DK, Sox HC Jr. Threshold analysis using diagnostic tests with multiple results. Med Decis Making. 1989;9(2):91-103.
14. Youngstrom EA, Youngstrom JK. Evidence-based assessment of pediatric bipolar disorder, Part II: incorporating information from behavior checklists. J Am Acad Child Adolesc Psychiatry. 2005;44(8):823-828.
15. Merikangas KR, He JP, Brody D, et al. Prevalence and treatment of mental disorders among US children in the 2001-2004 NHANES. Pediatrics. 2010;125(1):75-81.
16. Larson K, Russ SA, Kahn RS, et al. Patterns of comorbidity, functioning, and service use for US children with ADHD, 2007. Pediatrics. 2011;127(3):462-470.
17. Simon V, Czobor P, Bálint S, et al. Prevalence and correlates of adult attention-deficit hyperactivity disorder: meta-analysis. Br J Psychiatry. 2009;194(3):204-211.
18. Biederman J, Faraone SV. Attention-deficit hyperactivity disorder. Lancet. 2005;366(9481):237-248.
19. Sood AB, Razdan A, Weller EB, et al. How to differentiate bipolar disorder from attention deficit hyperactivity disorder and other common psychiatric disorders: a guide for clinicians. Curr Psychiatry Rep. 2005;7(2): 98-103.
20. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.

Neuroimaging in children and adolescents: When do you scan? With which modalities?
The first 15 years of the new millennium have seen a great increase in research on neuroimaging in children and adolescents who have a psychiatric disorder. In addition, imaging modalities continue to evolve, and are becoming increasingly accessible and informative. The literature is now replete with reports of neurostructural differences between patients and healthy subjects in a variety of common pediatric psychiatric conditions, including anxiety disorders, mood disorders, autism spectrum disorder (ASD), and attention-deficit/hyperactivity disorder (ADHD).
Historically, the clinical utility of neuroimaging was restricted to the identification of structural pathology. Today, accumulating data reveal novel roles for neuroimaging; these revelations are supported by studies demonstrating that treatment response for psychotherapeutic and psychopharmacotherapeutic interventions can be predicted by neurochemical and neurofunctional characteristics assessed by advanced imaging technologies, such as magnetic resonance spectroscopy (MRS) and functional MRI.
However, such advanced techniques are (at least at present) not ready for routine clinical use for this purpose. Instead, neuroimaging in the child and adolescent psychiatric clinic remains largely focused on ruling out neurostructural, neurologic, “nonpsychiatric” causes of our patients’ symptoms.
Understanding the role and limitations of major imaging modalities is key to guiding efficient and appropriate neuroimaging selection for pediatric patients. In this article, we describe and review:
- neuroimaging approaches for children and adolescents with psychiatric disorders
- the role of neuroimaging in (1) the differential diagnosis and workup of common psychiatric disorders and (2) urgent clinical situations
- how to determine what type of imaging to obtain.
Computed tomography
CT, which utilizes ionizing radiation, often is reserved, in the pediatric setting, for (1) emergency evaluation and (2) excluding potentially catastrophic neurologic injury resulting from:
- ischemic or hemorrhagic stroke
- herniation
- intracerebral hemorrhage
- subdural and epidural hematoma
- large intracranial mass with mass effect
- increased intracranial pressure
- acute skull fracture.
Although a CT scan is, typically, quick and has excellent sensitivity for acute bleeding and bony pathology, it exposes the patient to radiation and provides poor resolution compared with MRI.
In pediatrics, there has been practice-changing recognition of the importance of limiting lifetime radiation exposure incurred from medical procedures and imaging. As a result, most providers now agree that use of MRI in lieu of CT is appropriate in many, if not most, non-emergent situations. In an emergent situation, however, CT imaging is appropriate and should not be delayed. Moreover, in an emergent situation, you should not hesitate to use head CT in children, although timely discussion with the radiologist is recommended to review your differential diagnosis to better determine the preferred imaging modality.
Magnetic resonance imaging
Over the past several decades, MRI has been increasingly available in most pediatric health care facilities. The modality offers specific advantages for pediatric patients, including:
- better spatial resolution
- the ability to concurrently assess multiple pathologic processes
- lack of exposure to ionizing radiation.1
A number of MRI sequences, described below, can be used to assess vascular, inflammatory, structural, and metabolic processes.
A look inside. Comprehensive review of the physics that underlies MRI is beyond the scope of this article; several important principles are relevant to clinicians, however. Image contrast is dependent on intrinsic properties of tissue with regard to proton density, longitudinal relaxation time (T1), and transverse relaxation time (T2). Pulse sequences, which describe the strength and timing of the radiofrequency pulse and gradient pulses, define imaging acquisition parameters (eg, repetition time between the radio frequency pulse and echo time).
In turn, the intensity of the signal that is “seen” with various pulse sequences is differentially affected by intrinsic properties of tissue. At most pediatric institutions, the standard MRI-examination protocol includes: a T1-weighted image (Figure 1A); a T2-weighted scan (Figure 1B); fluid attenuated inversion recovery (FLAIR) (Figure 1C); and diffusion-weighted imaging (DWI) (Figure 1D).
Specific MRI sequences
T1 images. T1 sequences, or so-called anatomy sequences, are ideally suited for detailed neuroanatomic evaluations. They are generated in such a way that structures containing fluid are dark (hypo-intense), whereas other structures, with higher fat or protein content, are brighter (iso-intense, even hyper-intense). For this reason, CSF in the intracranial ventricles is dark, and white matter is brighter than the cortex because of lipid in myelin sheaths.
In addition, to view structural abnormalities that are characterized by altered vascular supply or flow, such as tumors and infections (abscesses), contrast imaging can be particularly helpful; such images generally are obtained as T1 sequences.
T2 images. By contrast to the T1-weighted sequence, the T2-weighted sequences emphasize fluid signal; structures such as the ventricles, which contain CSF, therefore will be bright (hyper-intense). Pathology that produces edema or fluid, such as edema surrounding demyelinating lesions or infections, also will show bright hyper-intense signal. In T2-weighted images of the brain, white matter shows lower signal intensity than the cortex because of the relatively lower water content in white matter tracts and myelin sheaths.
Fluid attenuation inversion recovery. FLAIR images are generated so that the baseline bright T2 signal seen in normal structures, such as the CSF, containing ventricles is cancelled out, or attenuated. In effect, this subtraction of typical background hyper-intense fluid signal leaves only abnormal T2 bright hyper-intense signal, such as vasogenic edema surrounding tumors, cytotoxic edema within an infarction, or extra-axial fluid collections such as a subarachnoid or subdural hemorrhage.
Diffusion-weighted imaging. DWI utilizes the random motion (ie, diffusion) of water molecules to generate contrast. In this regard, the diffusion of any molecule is influenced by its interaction with other molecules (eg, white-matter fibers and membranes, and macromolecules). Diffusion patterns therefore reflect details about tissue boundaries; as such, DWI is sensitive to a number of neurologic processes, such as ischemia, demyelinating disease, and some tumors, which restrict the free motion of water. DWI detects this so-called restricted diffusion and displays an area of bright signal.
Susceptibility-weighted imaging (SWI). In the pediatric population, SWI (Figure 2) utilizes a long-echo, 3-dimensional, velocity-compensated gradient recalled echo for image acquisition2 and, ultimately, leverages susceptibility differences across tissues by employing the phase image to identify these differences. SWI, which uses both magnitude and phase images and is remarkably sensitive to venous blood (and blood products), iron, and calcifications, therefore might be of increasing utility in pediatric patients with traumatic brain injury (TBI) (Figure 2B). As such, SWI has become a critical component of many pediatric MRI studies.3
Magnetic resonance angiography (MRA) (Figure 3A) is helpful for assessing intracranial arteries and may be employed in the evaluation of:
- vessel pathology and injury underlying stroke, such as vessel occlusion or injury
- patterns of vessel involvement suggestive of vasculitis
- developmental or acquired structural vascular abnormalities, such as aneurysm or vascular malformations
- determination of tumor blood supply.

MRA can be performed without or with contrast, although MRA with contrast might provide a higher quality study and therefore be of greater utility. Of note: The spatial resolution of MRA is not as good as CT angiography; abnormalities, such as a small aneurysm, might not be apparent.
Magnetic resonance venography (MRV) (Figure 3B) is most commonly performed when the possibility of thrombosis of the dural venous sinuses is being considered; it also is employed to evaluate vascular malformations, tumor drainage patterns, and other pathologic states. As with MRA, MRV can be performed without or with contrast, although post-contrast MRV is generally of higher quality and might be preferred when assessing for sinus thrombosis.
Magnetic resonance spectroscopy (MRS) resides at the border between research and clinical practice. In children and adolescents, MRS provides data on neuronal and axonal viability as well as energetics and cell membranes.4 Pediatric neurologists often use MRS to evaluate for congenital neurometabolic disease; this modality also can help distinguish between an active intracranial tumor from an abscess or gliosis.5
Neuroimaging in pediatric neuropsychiatric conditions: Evidence, guidance
Delirium (altered mental status). The acute neuropsychiatric syndrome characterized by impaired attention and sensorium might have a broad underlying etiology, but it is always associated with alteration of CNS neurophysiology. Children with neurostructural abnormalities might have increased vulnerability to CNS insult and therefore be at increased risk of delirium.6 Additionally, delirium can present subtly in children, with the precise signs dependent on the individual patient’s developmental stage.
Neuroimaging may be helpful when an infectious, inflammatory, toxic, or a metabolic basis for delirium is suspected, or when a patient has new focal neurologic findings. In this regard, focal neurologic findings suggest an underlying localizable lesion and warrant dedicated neuroimaging to localize the lesion.
In general, the differential diagnosis should guide consideration of neuroimaging. When considering the possibility of an unwitnessed seizure in a child who presents with altered mental status, neuroimaging certainly is an important component of the workup.
As another example, when underlying trauma, intracranial hemorrhage, or mass is a possibility in acute delirium, urgent head CT is appropriate. In non-emergent cases, MRI is the modality of choice. In immunocompromised patients presenting with delirium, maintain a low threshold for neuroimaging with contrast to rule out opportunistic intracranial infection.
Last, in children who have hydrocephalus with a shunt, delirium could be a harbinger of underlying shunt malfunction, warranting a “shunt series.”
ADHD. The diagnosis of ADHD remains a clinical one; for the typical pediatric patient with ADHD but who does not have focal neurologic deficits, neuroimaging is unnecessary. Some structural MRI studies of youth with ADHD suggest diminished volume of the globus pallidus, putamen, and caudate7; other studies reveal changes in gyrification and cortical thickness8 in regions subserving attentional processes. However, intra-individual and developmental-related variability preclude routine use of neuroimaging in the standard diagnostic work-up of ADHD.
Nevertheless, neuroimaging should be strongly considered in a child with progressive worsening of inattention, especially if combined with other psychiatric or neurologic findings. In such a case, MRI should be obtained to evaluate for a progressive neurodegenerative leukoencephalopathy (eg, adrenoleukodystrophy).9
Depressive and anxiety disorders. In pediatric patients who exhibit depressive or anxiety symptoms, abnormalities have been observed in cortical thickness10 and gray matter volume,11,12 and functional signatures13 have been identified in the circuitry of the prefrontal amygdala. No data suggest that, in an individual patient, neuroimaging can be of diagnostic utility—particularly in the absence of focal neurologic findings.
That being said, headache and other somatic symptoms are common in pediatric patients with a mood or anxiety disorder. Evidence for neuroimaging in the context of pediatric headache suggests that MRI should be considered when headache is associated with neurologic signs or symptoms, such as aura, or accompanied by focal neurologic deficit.14
Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) and pediatric acute-onset neuropsychiatric syndrome (PANS). Neuroimaging studies of patients with confirmed PANDAS or PANS are rare, but group analyses suggest a decreased average volume of the caudate, putamen, and globus pallidus in patients with PANDAS compared with healthy comparison subjects, although total cerebral volume does not appear to differ.15 Moreover, thalamic findings in patients with PANDAS have been noted to be similar to what is seen in patients with Sydenham’s chorea.
The most recent consensus statement regarding the treatment and assessment of PANDAS and PANS recommends ordering brain MRI when other conditions are suspected (eg, CNS, small vessel vasculitis, limbic encephalitis) or when the patient has severe headache, gait disturbance, cognitive deterioration, or psychosis.16 Furthermore, the consensus statement notes the potential utility of T2-weighted imaging with contrast to evaluate inflammatory changes in the basal ganglia.16
Autism spectrum disorder. Significant progress has been made during the past decade on the neuroanatomic characterization of ASD. Accumulating data indicate that, in pediatric patients with ASD, (1) development of white matter and gray matter is disrupted early in the course of the disorder and (2) cortical thickness is increased in regions subserving social cognition.17,18
Several studies have examined the presence of patient-level findings in samples of pediatric patients with ASD. Approximately 8% of pediatric patients with ASD were found to have some abnormality on routine brain MRI, the most common being white-matter signal abnormalities, dilated Virchow-Robin space, and temporal lobe abnormalities.19
Although abnormalities might be present in a large percentage of individual scans, routine screening MRI is unlikely to be of clinical utility in youth with ASD. In fact, no recommendation for routine MRI screening in patients with ASD has been made by the American Academy of Child & Adolescent Psychiatry, the Child Neurology Society of the American Academy of Neurology, or the American Academy of Pediatrics.
However, in patients with an underlying neurostructural disease that is phenotypically associated with ASD-like symptoms, imaging might be of use. In tuberous sclerosis, for example, MRI is especially important to classify intracranial lesions; determine burden and location; or identify treatment options (Figure 4). For patients with tuberous sclerosis—of whom more than one-third meet diagnostic criteria for ASD20—MRI study should include FLAIR, spin-echo, and gradient-echo sequences.

Movement disorders. Hyperkinetic movement disorders, including tic disorders and drug-induced movement disorders (eg, tremor) are common in pediatric patients. In pediatric patients with a tic disorder or Tourette’s disorder (TD), neuroimaging typically is unnecessary, despite the suggestion that the caudate nucleus volume is reduced in groups of patients with TD.
Many CNS-acting medications can exacerbate physiologic tremor; in pediatric patients with symptoms of a movement disorder, home medications should be carefully reviewed for potentially offending agents. When the patient is clinically and biochemically euthyroid and medication-induced movement disorder has been ruled out, or when the patient meets clinical diagnostic criteria for TD or a tic disorder, routine neuroimaging generally is unnecessary.
When tremor accompanies other cerebellar signs, such as ataxia or dysmetria, strongly consider MRI of the brain to evaluate pathology in the posterior fossa. In addition, neuroimaging should be considered for children with a new-onset abnormality on neurologic exam, including rapid onset of abnormal movements (other than common tics), continuous progressive worsening of symptoms, or any loss of developmental milestones.
Last, although tics and stereotypies often are transient and wane with age, other abnormal movements, such as dystonia, chorea, and parkinsonism (aside from those potentially associated with antipsychotic use), are never expected during typical development and warrant MRI.
Traumatic brain injury. Prompt evaluation and intervention for TBI can significantly affect overall outcome. Moreover, there has been increased enthusiasm around the pre-hospital assessment of TBI severity using (1) any of several proprietary testing systems (eg, Immediate Post-Concussion Assessment and Cognitive Testing [ImPACT]) plus (2) standard clinical staging, which is based on duration of loss of consciousness, persistence of memory loss, and the Glasgow Coma Scale score.
The goal for any TBI patient during the acute post-injury phase is to minimize continued neuronal injury from secondary effects of TBI, such as cerebral edema and herniation, and to optimize protection of surviving brain tissue; neuroimaging is a critical component of assessment during both acute and chronic recovery periods of TBI.21
The optimal imaging modality varies with the amount of time that has passed since initial injury.22 Urgent neuroimaging (the first 24 hours after brain injury) is typically obtained using head CT to assist decision-making in acute neurosurgical management. In this setting, head CT is fast and efficient; minimizes the amount of time that the patient is in the scanner; and provides valuable information on the acuity and extent of injury, degree of cerebral edema, and evidence or risk of pending herniation.
On the other hand, MRI is superior to CT during 48 to 72 hours after injury, given its higher resolution; superior imaging of the brainstem and deep gray nuclei; and ability to detect axonal injury, small contusions, and subtle neuronal damage. Specifically, SWI sequences can be particularly helpful in TBI for detecting diffuse axonal injury and micro-hemorrhages; several recent studies also suggest that SWI may be of particular value in pediatric patients with TBI.23
Additionally, given the increased sensitivity of MRI to detect subtle injuries, this modality can assist in identifying chronic sequelae of brain injury—thus contributing to determining of chronic therapy options and assisting with long-term prognosis. Gross structural changes resulting from TBI often are evident even in the acute post-injury phase; synaptic remodeling continues, however, for an indefinite period after injury, and this remodeling capacity is even more pronounced in the highly plastic brain of a young child.
Microstructural changes might not be detectable using traditional, readily available imaging sequences (CT, MRI). When those traditional modalities are used in concert with functional imaging techniques (eg, PET to evaluate cerebral metabolism and SPECT imaging which can detect abnormalities in cerebral blood flow), the combination of older and newer might provide a more complete picture of recovery after TBI.24
The important role of neuroimaging in severe TBI is intuitive. However, it is important to consider the role of neuroimaging in mild TBI in children, especially in the setting of repetitive mild injury.25 A growing body of evidence supports close, serial monitoring of children after even mild closed head injury for neurologic and psychiatric sequelae. Although it is rare that a child who is awake, interactive, and lacking focal neurologic deficits would need emergent (ie, CT) imaging after mild closed head injury, there might be a role for MRI later in the course of that child’s recovery—especially if recovery is complicated by clinical sequelae of mild TBI, such as cognitive impairment, headaches, or altered behavior.
When is additional neuroimaging needed?
It’s worthwhile briefly reviewing 4 scenarios that you might encounter, when you work with children and adolescents, in which urgent or emergent neuroimaging (often with consultation) should be obtained. The Table describes these situations and appropriate first- and second-line interventions.
1. When the presentation of your patient is consistent with an acute neurologic deficit, acute TBI, progressive neuropsychiatric decline, CNS infection, mass, demyelinating process, or toxic exposure, neuroimaging is likely critical.
2. In patients with progressive neurologic decline, including loss of developmental milestones, MRI, MRS, and referral to neurology should be part of the comprehensive evaluation.
3. In young children who exhibit a decrease in head circumference on growth curves, MRI is important to evaluate for underlying structural causes.
4. Pediatric patients with symptoms consistent with either stroke (ie, a new, persistent neurologic deficit) or a demyelinating process (eg, multiple episodes of variable transient focal neurologic symptoms), MRI should be obtained without compunction.

Consultation with pediatric neuroradiology
In deciding whether to obtain neuroimaging for a particular case, you should discuss your concerns with the pediatric radiologist or pediatric neuroradiologist, who will likely provide important guidance on key aspects of the study (eg, modifying slice thickness in a particular scan; recommending the use of contrast; including MRS in the order for imaging; performing appropriate vessel imaging). Consider asking 1 or more important questions when you discuss a patient’s presentation with the pediatric radiologist or pediatric neuroradiologist:
- “What neuroimaging studies are appropriate, based on my differential diagnosis?”
- “Are there specific imaging sequences that we should consider?”
- “Are there contraindications to the imaging modality for my patient?”
- “Is my patient likely to have difficulty tolerating the imaging procedure?”
- “Does my patient need sedation to tolerate this procedure?”
- “Should additional regions be included in the scan?” (Examples: In a child with stroke it might be important to include neck and chest vasculature and the heart. Other conditions might warrant imaging of the spinal cord.)
1. Abdelhalim AN, Alberico RA. Pediatric neuroimaging. Neurol Clin. 2009;27(1):285-301, x.
2. Sehgal V, Delproposto Z, Haacke EM, et al. Clinical applications of neuroimaging with susceptibility-weighted imaging. J Magn Reson Imaging. 2005;22(4):439-450.
3. Bosemani T, Poretti A, Huisman TA. Susceptibility-weighted imaging in pediatric neuroimaging. J Magn Reson Imaging. 2014;40(3):530-544.
4. Cecil KM. Proton magnetic resonance spectroscopy: technique for the neuroradiologist. Neuroimaging Clin N Am. 2013;23(3):381-392.
5. Panigrahy A, Nelson MD Jr, Blüml S. Magnetic resonance spectroscopy in pediatric neuroradiology: clinical and research applications. Pediatr Radiol. 2010;40(1):3-30.
6. Leentjens AF, Schieveld JN, Leonard M, et al. A comparison of the phenomenology of pediatric, adult, and geriatric delirium. J Psychosom Res. 2008;64(2):219-223.
7. Frodl T, Skokauskas N. Meta-analysis of structural MRI studies in children and adults with attention deficit hyperactivity disorder indicates treatment effects. Acta Psychiatr Scand. 2012;125(2):114-126.
8. Shaw P, Malek M, Watson B, et al. Development of cortical surface area and gyrification in attention-deficit/hyperactivity disorder. Biol Psychiatry. 2012;72(3):191-197.
9. Phelan JA, Lowe LH, Glasier CM. Pediatric neurodegenerative white matter processes: leukodystrophies and beyond. Pediatr Radiol. 2008;38(7):729-749.
10. Strawn JR, Wegman CJ, Dominick KC, et al. Cortical surface anatomy in pediatric patients with generalized anxiety disorder. J Anxiety Disord. 2014;28(7):717-723.
11. Mueller SC, Aouidad A, Gorodetsky E, et al. Gray matter volume in adolescent anxiety: an impact of the brain-derived neurotrophic factor Val(66)Met polymorphism [Erratum in J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195]? J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195.
12. Strawn JR, Hamm L, Fitzgerald DA, et al. Neurostructural abnormalities in pediatric anxiety disorders. J Anxiety Disord. 2015;32:81-88.
13. Strawn JR, Dominick KC, Patino LR, et al. Neurobiology of pediatric anxiety disorders. Curr Behav Neurosci Reports. 2014;1(3):154-160.
14. Alexiou GA, Argyropoulou MI. Neuroimaging in childhood headache: a systematic review. Pediatr Radiol. 2013;43(7):777-784.
15. Giedd JN, Rapoport JL, Garvey MA, et al. MRI assessment of children with obsessive-compulsive disorder or tics associated with streptococcal infection. Am J Psychiatry. 2000;157(2):281-283.
16. Chang K, Frankovich J, Cooperstock M, et al; PANS Collaborative Consortium. Clinical evaluation of youth with pediatric acute-onset neuropsychiatric syndrome (PANS): recommendations from the 2013 PANS Consensus Conference. J Child Adolesc Psychopharmacol. 2014;25(1):3-13.
17. Wallace GL, Robustelli B, Dankner N, et al. Increased gyrification, but comparable surface area in adolescents with autism spectrum disorders. Brain. 2013;136(pt 6):1956-1967.
18. Libero LE, DeRamus TP, Deshpande HD, et al. Surface-based morphometry of the cortical architecture of autism spectrum disorders: volume, thickness, area, and gyrification. Neuropsychologia. 2014;62:1-10.
19. Boddaert N, Zilbovicius M, Philipe A, et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS One. 2009;4:e445. doi: 10.1371/journal.pone.0004415.
20. Richards C, Jones C, Groves L, et al. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry. 2015;2(10):909-916.
21. Wilde EA, Hunter JV, Bigler ED. Pediatric traumatic brain injury: neuroimaging and neurorehabilitation outcome. NeuroRehabilitation. 2012;31(3):245-260.
22. Mechtler LL, Shastri KK, Crutchfield KE. Advanced neuroimaging of mild traumatic brain injury. Neurol Clin. 2014;32(1):31-58.
23. Ashwal S, Tong KA, Ghosh N, et al. Application of advanced neuroimaging modalities in pediatric traumatic brain injury. J Child Neurol. 2014;29(12):1704-1717.
24. Munson S, Schroth E, Ernst M. The role of functional neuroimaging in pediatric brain injury. Pediatrics. 2006;117(4):1372-1381.
25. Wozniak JR, Krach L, Ward E, et al. Neurocognitive and neuroimaging correlates of pediatric traumatic brain injury: a diffusion tensor imaging (DTI) study. Arch Clin Neuropsychol. 2007;22(5):555-568.
The first 15 years of the new millennium have seen a great increase in research on neuroimaging in children and adolescents who have a psychiatric disorder. In addition, imaging modalities continue to evolve, and are becoming increasingly accessible and informative. The literature is now replete with reports of neurostructural differences between patients and healthy subjects in a variety of common pediatric psychiatric conditions, including anxiety disorders, mood disorders, autism spectrum disorder (ASD), and attention-deficit/hyperactivity disorder (ADHD).
Historically, the clinical utility of neuroimaging was restricted to the identification of structural pathology. Today, accumulating data reveal novel roles for neuroimaging; these revelations are supported by studies demonstrating that treatment response for psychotherapeutic and psychopharmacotherapeutic interventions can be predicted by neurochemical and neurofunctional characteristics assessed by advanced imaging technologies, such as magnetic resonance spectroscopy (MRS) and functional MRI.
However, such advanced techniques are (at least at present) not ready for routine clinical use for this purpose. Instead, neuroimaging in the child and adolescent psychiatric clinic remains largely focused on ruling out neurostructural, neurologic, “nonpsychiatric” causes of our patients’ symptoms.
Understanding the role and limitations of major imaging modalities is key to guiding efficient and appropriate neuroimaging selection for pediatric patients. In this article, we describe and review:
- neuroimaging approaches for children and adolescents with psychiatric disorders
- the role of neuroimaging in (1) the differential diagnosis and workup of common psychiatric disorders and (2) urgent clinical situations
- how to determine what type of imaging to obtain.
Computed tomography
CT, which utilizes ionizing radiation, often is reserved, in the pediatric setting, for (1) emergency evaluation and (2) excluding potentially catastrophic neurologic injury resulting from:
- ischemic or hemorrhagic stroke
- herniation
- intracerebral hemorrhage
- subdural and epidural hematoma
- large intracranial mass with mass effect
- increased intracranial pressure
- acute skull fracture.
Although a CT scan is, typically, quick and has excellent sensitivity for acute bleeding and bony pathology, it exposes the patient to radiation and provides poor resolution compared with MRI.
In pediatrics, there has been practice-changing recognition of the importance of limiting lifetime radiation exposure incurred from medical procedures and imaging. As a result, most providers now agree that use of MRI in lieu of CT is appropriate in many, if not most, non-emergent situations. In an emergent situation, however, CT imaging is appropriate and should not be delayed. Moreover, in an emergent situation, you should not hesitate to use head CT in children, although timely discussion with the radiologist is recommended to review your differential diagnosis to better determine the preferred imaging modality.
Magnetic resonance imaging
Over the past several decades, MRI has been increasingly available in most pediatric health care facilities. The modality offers specific advantages for pediatric patients, including:
- better spatial resolution
- the ability to concurrently assess multiple pathologic processes
- lack of exposure to ionizing radiation.1
A number of MRI sequences, described below, can be used to assess vascular, inflammatory, structural, and metabolic processes.
A look inside. Comprehensive review of the physics that underlies MRI is beyond the scope of this article; several important principles are relevant to clinicians, however. Image contrast is dependent on intrinsic properties of tissue with regard to proton density, longitudinal relaxation time (T1), and transverse relaxation time (T2). Pulse sequences, which describe the strength and timing of the radiofrequency pulse and gradient pulses, define imaging acquisition parameters (eg, repetition time between the radio frequency pulse and echo time).
In turn, the intensity of the signal that is “seen” with various pulse sequences is differentially affected by intrinsic properties of tissue. At most pediatric institutions, the standard MRI-examination protocol includes: a T1-weighted image (Figure 1A); a T2-weighted scan (Figure 1B); fluid attenuated inversion recovery (FLAIR) (Figure 1C); and diffusion-weighted imaging (DWI) (Figure 1D).
Specific MRI sequences
T1 images. T1 sequences, or so-called anatomy sequences, are ideally suited for detailed neuroanatomic evaluations. They are generated in such a way that structures containing fluid are dark (hypo-intense), whereas other structures, with higher fat or protein content, are brighter (iso-intense, even hyper-intense). For this reason, CSF in the intracranial ventricles is dark, and white matter is brighter than the cortex because of lipid in myelin sheaths.
In addition, to view structural abnormalities that are characterized by altered vascular supply or flow, such as tumors and infections (abscesses), contrast imaging can be particularly helpful; such images generally are obtained as T1 sequences.
T2 images. By contrast to the T1-weighted sequence, the T2-weighted sequences emphasize fluid signal; structures such as the ventricles, which contain CSF, therefore will be bright (hyper-intense). Pathology that produces edema or fluid, such as edema surrounding demyelinating lesions or infections, also will show bright hyper-intense signal. In T2-weighted images of the brain, white matter shows lower signal intensity than the cortex because of the relatively lower water content in white matter tracts and myelin sheaths.
Fluid attenuation inversion recovery. FLAIR images are generated so that the baseline bright T2 signal seen in normal structures, such as the CSF, containing ventricles is cancelled out, or attenuated. In effect, this subtraction of typical background hyper-intense fluid signal leaves only abnormal T2 bright hyper-intense signal, such as vasogenic edema surrounding tumors, cytotoxic edema within an infarction, or extra-axial fluid collections such as a subarachnoid or subdural hemorrhage.
Diffusion-weighted imaging. DWI utilizes the random motion (ie, diffusion) of water molecules to generate contrast. In this regard, the diffusion of any molecule is influenced by its interaction with other molecules (eg, white-matter fibers and membranes, and macromolecules). Diffusion patterns therefore reflect details about tissue boundaries; as such, DWI is sensitive to a number of neurologic processes, such as ischemia, demyelinating disease, and some tumors, which restrict the free motion of water. DWI detects this so-called restricted diffusion and displays an area of bright signal.
Susceptibility-weighted imaging (SWI). In the pediatric population, SWI (Figure 2) utilizes a long-echo, 3-dimensional, velocity-compensated gradient recalled echo for image acquisition2 and, ultimately, leverages susceptibility differences across tissues by employing the phase image to identify these differences. SWI, which uses both magnitude and phase images and is remarkably sensitive to venous blood (and blood products), iron, and calcifications, therefore might be of increasing utility in pediatric patients with traumatic brain injury (TBI) (Figure 2B). As such, SWI has become a critical component of many pediatric MRI studies.3
Magnetic resonance angiography (MRA) (Figure 3A) is helpful for assessing intracranial arteries and may be employed in the evaluation of:
- vessel pathology and injury underlying stroke, such as vessel occlusion or injury
- patterns of vessel involvement suggestive of vasculitis
- developmental or acquired structural vascular abnormalities, such as aneurysm or vascular malformations
- determination of tumor blood supply.
MRA can be performed without or with contrast, although MRA with contrast might provide a higher quality study and therefore be of greater utility. Of note: The spatial resolution of MRA is not as good as CT angiography; abnormalities, such as a small aneurysm, might not be apparent.
Magnetic resonance venography (MRV) (Figure 3B) is most commonly performed when the possibility of thrombosis of the dural venous sinuses is being considered; it also is employed to evaluate vascular malformations, tumor drainage patterns, and other pathologic states. As with MRA, MRV can be performed without or with contrast, although post-contrast MRV is generally of higher quality and might be preferred when assessing for sinus thrombosis.
Magnetic resonance spectroscopy (MRS) resides at the border between research and clinical practice. In children and adolescents, MRS provides data on neuronal and axonal viability as well as energetics and cell membranes.4 Pediatric neurologists often use MRS to evaluate for congenital neurometabolic disease; this modality also can help distinguish between an active intracranial tumor from an abscess or gliosis.5
Neuroimaging in pediatric neuropsychiatric conditions: Evidence, guidance
Delirium (altered mental status). The acute neuropsychiatric syndrome characterized by impaired attention and sensorium might have a broad underlying etiology, but it is always associated with alteration of CNS neurophysiology. Children with neurostructural abnormalities might have increased vulnerability to CNS insult and therefore be at increased risk of delirium.6 Additionally, delirium can present subtly in children, with the precise signs dependent on the individual patient’s developmental stage.
Neuroimaging may be helpful when an infectious, inflammatory, toxic, or a metabolic basis for delirium is suspected, or when a patient has new focal neurologic findings. In this regard, focal neurologic findings suggest an underlying localizable lesion and warrant dedicated neuroimaging to localize the lesion.
In general, the differential diagnosis should guide consideration of neuroimaging. When considering the possibility of an unwitnessed seizure in a child who presents with altered mental status, neuroimaging certainly is an important component of the workup.
As another example, when underlying trauma, intracranial hemorrhage, or mass is a possibility in acute delirium, urgent head CT is appropriate. In non-emergent cases, MRI is the modality of choice. In immunocompromised patients presenting with delirium, maintain a low threshold for neuroimaging with contrast to rule out opportunistic intracranial infection.
Last, in children who have hydrocephalus with a shunt, delirium could be a harbinger of underlying shunt malfunction, warranting a “shunt series.”
ADHD. The diagnosis of ADHD remains a clinical one; for the typical pediatric patient with ADHD but who does not have focal neurologic deficits, neuroimaging is unnecessary. Some structural MRI studies of youth with ADHD suggest diminished volume of the globus pallidus, putamen, and caudate7; other studies reveal changes in gyrification and cortical thickness8 in regions subserving attentional processes. However, intra-individual and developmental-related variability preclude routine use of neuroimaging in the standard diagnostic work-up of ADHD.
Nevertheless, neuroimaging should be strongly considered in a child with progressive worsening of inattention, especially if combined with other psychiatric or neurologic findings. In such a case, MRI should be obtained to evaluate for a progressive neurodegenerative leukoencephalopathy (eg, adrenoleukodystrophy).9
Depressive and anxiety disorders. In pediatric patients who exhibit depressive or anxiety symptoms, abnormalities have been observed in cortical thickness10 and gray matter volume,11,12 and functional signatures13 have been identified in the circuitry of the prefrontal amygdala. No data suggest that, in an individual patient, neuroimaging can be of diagnostic utility—particularly in the absence of focal neurologic findings.
That being said, headache and other somatic symptoms are common in pediatric patients with a mood or anxiety disorder. Evidence for neuroimaging in the context of pediatric headache suggests that MRI should be considered when headache is associated with neurologic signs or symptoms, such as aura, or accompanied by focal neurologic deficit.14
Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) and pediatric acute-onset neuropsychiatric syndrome (PANS). Neuroimaging studies of patients with confirmed PANDAS or PANS are rare, but group analyses suggest a decreased average volume of the caudate, putamen, and globus pallidus in patients with PANDAS compared with healthy comparison subjects, although total cerebral volume does not appear to differ.15 Moreover, thalamic findings in patients with PANDAS have been noted to be similar to what is seen in patients with Sydenham’s chorea.
The most recent consensus statement regarding the treatment and assessment of PANDAS and PANS recommends ordering brain MRI when other conditions are suspected (eg, CNS, small vessel vasculitis, limbic encephalitis) or when the patient has severe headache, gait disturbance, cognitive deterioration, or psychosis.16 Furthermore, the consensus statement notes the potential utility of T2-weighted imaging with contrast to evaluate inflammatory changes in the basal ganglia.16
Autism spectrum disorder. Significant progress has been made during the past decade on the neuroanatomic characterization of ASD. Accumulating data indicate that, in pediatric patients with ASD, (1) development of white matter and gray matter is disrupted early in the course of the disorder and (2) cortical thickness is increased in regions subserving social cognition.17,18
Several studies have examined the presence of patient-level findings in samples of pediatric patients with ASD. Approximately 8% of pediatric patients with ASD were found to have some abnormality on routine brain MRI, the most common being white-matter signal abnormalities, dilated Virchow-Robin space, and temporal lobe abnormalities.19
Although abnormalities might be present in a large percentage of individual scans, routine screening MRI is unlikely to be of clinical utility in youth with ASD. In fact, no recommendation for routine MRI screening in patients with ASD has been made by the American Academy of Child & Adolescent Psychiatry, the Child Neurology Society of the American Academy of Neurology, or the American Academy of Pediatrics.
However, in patients with an underlying neurostructural disease that is phenotypically associated with ASD-like symptoms, imaging might be of use. In tuberous sclerosis, for example, MRI is especially important to classify intracranial lesions; determine burden and location; or identify treatment options (Figure 4). For patients with tuberous sclerosis—of whom more than one-third meet diagnostic criteria for ASD20—MRI study should include FLAIR, spin-echo, and gradient-echo sequences.
Movement disorders. Hyperkinetic movement disorders, including tic disorders and drug-induced movement disorders (eg, tremor) are common in pediatric patients. In pediatric patients with a tic disorder or Tourette’s disorder (TD), neuroimaging typically is unnecessary, despite the suggestion that the caudate nucleus volume is reduced in groups of patients with TD.
Many CNS-acting medications can exacerbate physiologic tremor; in pediatric patients with symptoms of a movement disorder, home medications should be carefully reviewed for potentially offending agents. When the patient is clinically and biochemically euthyroid and medication-induced movement disorder has been ruled out, or when the patient meets clinical diagnostic criteria for TD or a tic disorder, routine neuroimaging generally is unnecessary.
When tremor accompanies other cerebellar signs, such as ataxia or dysmetria, strongly consider MRI of the brain to evaluate pathology in the posterior fossa. In addition, neuroimaging should be considered for children with a new-onset abnormality on neurologic exam, including rapid onset of abnormal movements (other than common tics), continuous progressive worsening of symptoms, or any loss of developmental milestones.
Last, although tics and stereotypies often are transient and wane with age, other abnormal movements, such as dystonia, chorea, and parkinsonism (aside from those potentially associated with antipsychotic use), are never expected during typical development and warrant MRI.
Traumatic brain injury. Prompt evaluation and intervention for TBI can significantly affect overall outcome. Moreover, there has been increased enthusiasm around the pre-hospital assessment of TBI severity using (1) any of several proprietary testing systems (eg, Immediate Post-Concussion Assessment and Cognitive Testing [ImPACT]) plus (2) standard clinical staging, which is based on duration of loss of consciousness, persistence of memory loss, and the Glasgow Coma Scale score.
The goal for any TBI patient during the acute post-injury phase is to minimize continued neuronal injury from secondary effects of TBI, such as cerebral edema and herniation, and to optimize protection of surviving brain tissue; neuroimaging is a critical component of assessment during both acute and chronic recovery periods of TBI.21
The optimal imaging modality varies with the amount of time that has passed since initial injury.22 Urgent neuroimaging (the first 24 hours after brain injury) is typically obtained using head CT to assist decision-making in acute neurosurgical management. In this setting, head CT is fast and efficient; minimizes the amount of time that the patient is in the scanner; and provides valuable information on the acuity and extent of injury, degree of cerebral edema, and evidence or risk of pending herniation.
On the other hand, MRI is superior to CT during 48 to 72 hours after injury, given its higher resolution; superior imaging of the brainstem and deep gray nuclei; and ability to detect axonal injury, small contusions, and subtle neuronal damage. Specifically, SWI sequences can be particularly helpful in TBI for detecting diffuse axonal injury and micro-hemorrhages; several recent studies also suggest that SWI may be of particular value in pediatric patients with TBI.23
Additionally, given the increased sensitivity of MRI to detect subtle injuries, this modality can assist in identifying chronic sequelae of brain injury—thus contributing to determining of chronic therapy options and assisting with long-term prognosis. Gross structural changes resulting from TBI often are evident even in the acute post-injury phase; synaptic remodeling continues, however, for an indefinite period after injury, and this remodeling capacity is even more pronounced in the highly plastic brain of a young child.
Microstructural changes might not be detectable using traditional, readily available imaging sequences (CT, MRI). When those traditional modalities are used in concert with functional imaging techniques (eg, PET to evaluate cerebral metabolism and SPECT imaging which can detect abnormalities in cerebral blood flow), the combination of older and newer might provide a more complete picture of recovery after TBI.24
The important role of neuroimaging in severe TBI is intuitive. However, it is important to consider the role of neuroimaging in mild TBI in children, especially in the setting of repetitive mild injury.25 A growing body of evidence supports close, serial monitoring of children after even mild closed head injury for neurologic and psychiatric sequelae. Although it is rare that a child who is awake, interactive, and lacking focal neurologic deficits would need emergent (ie, CT) imaging after mild closed head injury, there might be a role for MRI later in the course of that child’s recovery—especially if recovery is complicated by clinical sequelae of mild TBI, such as cognitive impairment, headaches, or altered behavior.
When is additional neuroimaging needed?
It’s worthwhile briefly reviewing 4 scenarios that you might encounter, when you work with children and adolescents, in which urgent or emergent neuroimaging (often with consultation) should be obtained. The Table describes these situations and appropriate first- and second-line interventions.
1. When the presentation of your patient is consistent with an acute neurologic deficit, acute TBI, progressive neuropsychiatric decline, CNS infection, mass, demyelinating process, or toxic exposure, neuroimaging is likely critical.
2. In patients with progressive neurologic decline, including loss of developmental milestones, MRI, MRS, and referral to neurology should be part of the comprehensive evaluation.
3. In young children who exhibit a decrease in head circumference on growth curves, MRI is important to evaluate for underlying structural causes.
4. Pediatric patients with symptoms consistent with either stroke (ie, a new, persistent neurologic deficit) or a demyelinating process (eg, multiple episodes of variable transient focal neurologic symptoms), MRI should be obtained without compunction.
Consultation with pediatric neuroradiology
In deciding whether to obtain neuroimaging for a particular case, you should discuss your concerns with the pediatric radiologist or pediatric neuroradiologist, who will likely provide important guidance on key aspects of the study (eg, modifying slice thickness in a particular scan; recommending the use of contrast; including MRS in the order for imaging; performing appropriate vessel imaging). Consider asking 1 or more important questions when you discuss a patient’s presentation with the pediatric radiologist or pediatric neuroradiologist:
- “What neuroimaging studies are appropriate, based on my differential diagnosis?”
- “Are there specific imaging sequences that we should consider?”
- “Are there contraindications to the imaging modality for my patient?”
- “Is my patient likely to have difficulty tolerating the imaging procedure?”
- “Does my patient need sedation to tolerate this procedure?”
- “Should additional regions be included in the scan?” (Examples: In a child with stroke it might be important to include neck and chest vasculature and the heart. Other conditions might warrant imaging of the spinal cord.)
The first 15 years of the new millennium have seen a great increase in research on neuroimaging in children and adolescents who have a psychiatric disorder. In addition, imaging modalities continue to evolve, and are becoming increasingly accessible and informative. The literature is now replete with reports of neurostructural differences between patients and healthy subjects in a variety of common pediatric psychiatric conditions, including anxiety disorders, mood disorders, autism spectrum disorder (ASD), and attention-deficit/hyperactivity disorder (ADHD).
Historically, the clinical utility of neuroimaging was restricted to the identification of structural pathology. Today, accumulating data reveal novel roles for neuroimaging; these revelations are supported by studies demonstrating that treatment response for psychotherapeutic and psychopharmacotherapeutic interventions can be predicted by neurochemical and neurofunctional characteristics assessed by advanced imaging technologies, such as magnetic resonance spectroscopy (MRS) and functional MRI.
However, such advanced techniques are (at least at present) not ready for routine clinical use for this purpose. Instead, neuroimaging in the child and adolescent psychiatric clinic remains largely focused on ruling out neurostructural, neurologic, “nonpsychiatric” causes of our patients’ symptoms.
Understanding the role and limitations of major imaging modalities is key to guiding efficient and appropriate neuroimaging selection for pediatric patients. In this article, we describe and review:
- neuroimaging approaches for children and adolescents with psychiatric disorders
- the role of neuroimaging in (1) the differential diagnosis and workup of common psychiatric disorders and (2) urgent clinical situations
- how to determine what type of imaging to obtain.
Computed tomography
CT, which utilizes ionizing radiation, often is reserved, in the pediatric setting, for (1) emergency evaluation and (2) excluding potentially catastrophic neurologic injury resulting from:
- ischemic or hemorrhagic stroke
- herniation
- intracerebral hemorrhage
- subdural and epidural hematoma
- large intracranial mass with mass effect
- increased intracranial pressure
- acute skull fracture.
Although a CT scan is, typically, quick and has excellent sensitivity for acute bleeding and bony pathology, it exposes the patient to radiation and provides poor resolution compared with MRI.
In pediatrics, there has been practice-changing recognition of the importance of limiting lifetime radiation exposure incurred from medical procedures and imaging. As a result, most providers now agree that use of MRI in lieu of CT is appropriate in many, if not most, non-emergent situations. In an emergent situation, however, CT imaging is appropriate and should not be delayed. Moreover, in an emergent situation, you should not hesitate to use head CT in children, although timely discussion with the radiologist is recommended to review your differential diagnosis to better determine the preferred imaging modality.
Magnetic resonance imaging
Over the past several decades, MRI has been increasingly available in most pediatric health care facilities. The modality offers specific advantages for pediatric patients, including:
- better spatial resolution
- the ability to concurrently assess multiple pathologic processes
- lack of exposure to ionizing radiation.1
A number of MRI sequences, described below, can be used to assess vascular, inflammatory, structural, and metabolic processes.
A look inside. Comprehensive review of the physics that underlies MRI is beyond the scope of this article; several important principles are relevant to clinicians, however. Image contrast is dependent on intrinsic properties of tissue with regard to proton density, longitudinal relaxation time (T1), and transverse relaxation time (T2). Pulse sequences, which describe the strength and timing of the radiofrequency pulse and gradient pulses, define imaging acquisition parameters (eg, repetition time between the radio frequency pulse and echo time).
In turn, the intensity of the signal that is “seen” with various pulse sequences is differentially affected by intrinsic properties of tissue. At most pediatric institutions, the standard MRI-examination protocol includes: a T1-weighted image (Figure 1A); a T2-weighted scan (Figure 1B); fluid attenuated inversion recovery (FLAIR) (Figure 1C); and diffusion-weighted imaging (DWI) (Figure 1D).
Specific MRI sequences
T1 images. T1 sequences, or so-called anatomy sequences, are ideally suited for detailed neuroanatomic evaluations. They are generated in such a way that structures containing fluid are dark (hypo-intense), whereas other structures, with higher fat or protein content, are brighter (iso-intense, even hyper-intense). For this reason, CSF in the intracranial ventricles is dark, and white matter is brighter than the cortex because of lipid in myelin sheaths.
In addition, to view structural abnormalities that are characterized by altered vascular supply or flow, such as tumors and infections (abscesses), contrast imaging can be particularly helpful; such images generally are obtained as T1 sequences.
T2 images. By contrast to the T1-weighted sequence, the T2-weighted sequences emphasize fluid signal; structures such as the ventricles, which contain CSF, therefore will be bright (hyper-intense). Pathology that produces edema or fluid, such as edema surrounding demyelinating lesions or infections, also will show bright hyper-intense signal. In T2-weighted images of the brain, white matter shows lower signal intensity than the cortex because of the relatively lower water content in white matter tracts and myelin sheaths.
Fluid attenuation inversion recovery. FLAIR images are generated so that the baseline bright T2 signal seen in normal structures, such as the CSF, containing ventricles is cancelled out, or attenuated. In effect, this subtraction of typical background hyper-intense fluid signal leaves only abnormal T2 bright hyper-intense signal, such as vasogenic edema surrounding tumors, cytotoxic edema within an infarction, or extra-axial fluid collections such as a subarachnoid or subdural hemorrhage.
Diffusion-weighted imaging. DWI utilizes the random motion (ie, diffusion) of water molecules to generate contrast. In this regard, the diffusion of any molecule is influenced by its interaction with other molecules (eg, white-matter fibers and membranes, and macromolecules). Diffusion patterns therefore reflect details about tissue boundaries; as such, DWI is sensitive to a number of neurologic processes, such as ischemia, demyelinating disease, and some tumors, which restrict the free motion of water. DWI detects this so-called restricted diffusion and displays an area of bright signal.
Susceptibility-weighted imaging (SWI). In the pediatric population, SWI (Figure 2) utilizes a long-echo, 3-dimensional, velocity-compensated gradient recalled echo for image acquisition2 and, ultimately, leverages susceptibility differences across tissues by employing the phase image to identify these differences. SWI, which uses both magnitude and phase images and is remarkably sensitive to venous blood (and blood products), iron, and calcifications, therefore might be of increasing utility in pediatric patients with traumatic brain injury (TBI) (Figure 2B). As such, SWI has become a critical component of many pediatric MRI studies.3
Magnetic resonance angiography (MRA) (Figure 3A) is helpful for assessing intracranial arteries and may be employed in the evaluation of:
- vessel pathology and injury underlying stroke, such as vessel occlusion or injury
- patterns of vessel involvement suggestive of vasculitis
- developmental or acquired structural vascular abnormalities, such as aneurysm or vascular malformations
- determination of tumor blood supply.
MRA can be performed without or with contrast, although MRA with contrast might provide a higher quality study and therefore be of greater utility. Of note: The spatial resolution of MRA is not as good as CT angiography; abnormalities, such as a small aneurysm, might not be apparent.
Magnetic resonance venography (MRV) (Figure 3B) is most commonly performed when the possibility of thrombosis of the dural venous sinuses is being considered; it also is employed to evaluate vascular malformations, tumor drainage patterns, and other pathologic states. As with MRA, MRV can be performed without or with contrast, although post-contrast MRV is generally of higher quality and might be preferred when assessing for sinus thrombosis.
Magnetic resonance spectroscopy (MRS) resides at the border between research and clinical practice. In children and adolescents, MRS provides data on neuronal and axonal viability as well as energetics and cell membranes.4 Pediatric neurologists often use MRS to evaluate for congenital neurometabolic disease; this modality also can help distinguish between an active intracranial tumor from an abscess or gliosis.5
Neuroimaging in pediatric neuropsychiatric conditions: Evidence, guidance
Delirium (altered mental status). The acute neuropsychiatric syndrome characterized by impaired attention and sensorium might have a broad underlying etiology, but it is always associated with alteration of CNS neurophysiology. Children with neurostructural abnormalities might have increased vulnerability to CNS insult and therefore be at increased risk of delirium.6 Additionally, delirium can present subtly in children, with the precise signs dependent on the individual patient’s developmental stage.
Neuroimaging may be helpful when an infectious, inflammatory, toxic, or a metabolic basis for delirium is suspected, or when a patient has new focal neurologic findings. In this regard, focal neurologic findings suggest an underlying localizable lesion and warrant dedicated neuroimaging to localize the lesion.
In general, the differential diagnosis should guide consideration of neuroimaging. When considering the possibility of an unwitnessed seizure in a child who presents with altered mental status, neuroimaging certainly is an important component of the workup.
As another example, when underlying trauma, intracranial hemorrhage, or mass is a possibility in acute delirium, urgent head CT is appropriate. In non-emergent cases, MRI is the modality of choice. In immunocompromised patients presenting with delirium, maintain a low threshold for neuroimaging with contrast to rule out opportunistic intracranial infection.
Last, in children who have hydrocephalus with a shunt, delirium could be a harbinger of underlying shunt malfunction, warranting a “shunt series.”
ADHD. The diagnosis of ADHD remains a clinical one; for the typical pediatric patient with ADHD but who does not have focal neurologic deficits, neuroimaging is unnecessary. Some structural MRI studies of youth with ADHD suggest diminished volume of the globus pallidus, putamen, and caudate7; other studies reveal changes in gyrification and cortical thickness8 in regions subserving attentional processes. However, intra-individual and developmental-related variability preclude routine use of neuroimaging in the standard diagnostic work-up of ADHD.
Nevertheless, neuroimaging should be strongly considered in a child with progressive worsening of inattention, especially if combined with other psychiatric or neurologic findings. In such a case, MRI should be obtained to evaluate for a progressive neurodegenerative leukoencephalopathy (eg, adrenoleukodystrophy).9
Depressive and anxiety disorders. In pediatric patients who exhibit depressive or anxiety symptoms, abnormalities have been observed in cortical thickness10 and gray matter volume,11,12 and functional signatures13 have been identified in the circuitry of the prefrontal amygdala. No data suggest that, in an individual patient, neuroimaging can be of diagnostic utility—particularly in the absence of focal neurologic findings.
That being said, headache and other somatic symptoms are common in pediatric patients with a mood or anxiety disorder. Evidence for neuroimaging in the context of pediatric headache suggests that MRI should be considered when headache is associated with neurologic signs or symptoms, such as aura, or accompanied by focal neurologic deficit.14
Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) and pediatric acute-onset neuropsychiatric syndrome (PANS). Neuroimaging studies of patients with confirmed PANDAS or PANS are rare, but group analyses suggest a decreased average volume of the caudate, putamen, and globus pallidus in patients with PANDAS compared with healthy comparison subjects, although total cerebral volume does not appear to differ.15 Moreover, thalamic findings in patients with PANDAS have been noted to be similar to what is seen in patients with Sydenham’s chorea.
The most recent consensus statement regarding the treatment and assessment of PANDAS and PANS recommends ordering brain MRI when other conditions are suspected (eg, CNS, small vessel vasculitis, limbic encephalitis) or when the patient has severe headache, gait disturbance, cognitive deterioration, or psychosis.16 Furthermore, the consensus statement notes the potential utility of T2-weighted imaging with contrast to evaluate inflammatory changes in the basal ganglia.16
Autism spectrum disorder. Significant progress has been made during the past decade on the neuroanatomic characterization of ASD. Accumulating data indicate that, in pediatric patients with ASD, (1) development of white matter and gray matter is disrupted early in the course of the disorder and (2) cortical thickness is increased in regions subserving social cognition.17,18
Several studies have examined the presence of patient-level findings in samples of pediatric patients with ASD. Approximately 8% of pediatric patients with ASD were found to have some abnormality on routine brain MRI, the most common being white-matter signal abnormalities, dilated Virchow-Robin space, and temporal lobe abnormalities.19
Although abnormalities might be present in a large percentage of individual scans, routine screening MRI is unlikely to be of clinical utility in youth with ASD. In fact, no recommendation for routine MRI screening in patients with ASD has been made by the American Academy of Child & Adolescent Psychiatry, the Child Neurology Society of the American Academy of Neurology, or the American Academy of Pediatrics.
However, in patients with an underlying neurostructural disease that is phenotypically associated with ASD-like symptoms, imaging might be of use. In tuberous sclerosis, for example, MRI is especially important to classify intracranial lesions; determine burden and location; or identify treatment options (Figure 4). For patients with tuberous sclerosis—of whom more than one-third meet diagnostic criteria for ASD20—MRI study should include FLAIR, spin-echo, and gradient-echo sequences.
Movement disorders. Hyperkinetic movement disorders, including tic disorders and drug-induced movement disorders (eg, tremor) are common in pediatric patients. In pediatric patients with a tic disorder or Tourette’s disorder (TD), neuroimaging typically is unnecessary, despite the suggestion that the caudate nucleus volume is reduced in groups of patients with TD.
Many CNS-acting medications can exacerbate physiologic tremor; in pediatric patients with symptoms of a movement disorder, home medications should be carefully reviewed for potentially offending agents. When the patient is clinically and biochemically euthyroid and medication-induced movement disorder has been ruled out, or when the patient meets clinical diagnostic criteria for TD or a tic disorder, routine neuroimaging generally is unnecessary.
When tremor accompanies other cerebellar signs, such as ataxia or dysmetria, strongly consider MRI of the brain to evaluate pathology in the posterior fossa. In addition, neuroimaging should be considered for children with a new-onset abnormality on neurologic exam, including rapid onset of abnormal movements (other than common tics), continuous progressive worsening of symptoms, or any loss of developmental milestones.
Last, although tics and stereotypies often are transient and wane with age, other abnormal movements, such as dystonia, chorea, and parkinsonism (aside from those potentially associated with antipsychotic use), are never expected during typical development and warrant MRI.
Traumatic brain injury. Prompt evaluation and intervention for TBI can significantly affect overall outcome. Moreover, there has been increased enthusiasm around the pre-hospital assessment of TBI severity using (1) any of several proprietary testing systems (eg, Immediate Post-Concussion Assessment and Cognitive Testing [ImPACT]) plus (2) standard clinical staging, which is based on duration of loss of consciousness, persistence of memory loss, and the Glasgow Coma Scale score.
The goal for any TBI patient during the acute post-injury phase is to minimize continued neuronal injury from secondary effects of TBI, such as cerebral edema and herniation, and to optimize protection of surviving brain tissue; neuroimaging is a critical component of assessment during both acute and chronic recovery periods of TBI.21
The optimal imaging modality varies with the amount of time that has passed since initial injury.22 Urgent neuroimaging (the first 24 hours after brain injury) is typically obtained using head CT to assist decision-making in acute neurosurgical management. In this setting, head CT is fast and efficient; minimizes the amount of time that the patient is in the scanner; and provides valuable information on the acuity and extent of injury, degree of cerebral edema, and evidence or risk of pending herniation.
On the other hand, MRI is superior to CT during 48 to 72 hours after injury, given its higher resolution; superior imaging of the brainstem and deep gray nuclei; and ability to detect axonal injury, small contusions, and subtle neuronal damage. Specifically, SWI sequences can be particularly helpful in TBI for detecting diffuse axonal injury and micro-hemorrhages; several recent studies also suggest that SWI may be of particular value in pediatric patients with TBI.23
Additionally, given the increased sensitivity of MRI to detect subtle injuries, this modality can assist in identifying chronic sequelae of brain injury—thus contributing to determining of chronic therapy options and assisting with long-term prognosis. Gross structural changes resulting from TBI often are evident even in the acute post-injury phase; synaptic remodeling continues, however, for an indefinite period after injury, and this remodeling capacity is even more pronounced in the highly plastic brain of a young child.
Microstructural changes might not be detectable using traditional, readily available imaging sequences (CT, MRI). When those traditional modalities are used in concert with functional imaging techniques (eg, PET to evaluate cerebral metabolism and SPECT imaging which can detect abnormalities in cerebral blood flow), the combination of older and newer might provide a more complete picture of recovery after TBI.24
The important role of neuroimaging in severe TBI is intuitive. However, it is important to consider the role of neuroimaging in mild TBI in children, especially in the setting of repetitive mild injury.25 A growing body of evidence supports close, serial monitoring of children after even mild closed head injury for neurologic and psychiatric sequelae. Although it is rare that a child who is awake, interactive, and lacking focal neurologic deficits would need emergent (ie, CT) imaging after mild closed head injury, there might be a role for MRI later in the course of that child’s recovery—especially if recovery is complicated by clinical sequelae of mild TBI, such as cognitive impairment, headaches, or altered behavior.
When is additional neuroimaging needed?
It’s worthwhile briefly reviewing 4 scenarios that you might encounter, when you work with children and adolescents, in which urgent or emergent neuroimaging (often with consultation) should be obtained. The Table describes these situations and appropriate first- and second-line interventions.
1. When the presentation of your patient is consistent with an acute neurologic deficit, acute TBI, progressive neuropsychiatric decline, CNS infection, mass, demyelinating process, or toxic exposure, neuroimaging is likely critical.
2. In patients with progressive neurologic decline, including loss of developmental milestones, MRI, MRS, and referral to neurology should be part of the comprehensive evaluation.
3. In young children who exhibit a decrease in head circumference on growth curves, MRI is important to evaluate for underlying structural causes.
4. Pediatric patients with symptoms consistent with either stroke (ie, a new, persistent neurologic deficit) or a demyelinating process (eg, multiple episodes of variable transient focal neurologic symptoms), MRI should be obtained without compunction.
Consultation with pediatric neuroradiology
In deciding whether to obtain neuroimaging for a particular case, you should discuss your concerns with the pediatric radiologist or pediatric neuroradiologist, who will likely provide important guidance on key aspects of the study (eg, modifying slice thickness in a particular scan; recommending the use of contrast; including MRS in the order for imaging; performing appropriate vessel imaging). Consider asking 1 or more important questions when you discuss a patient’s presentation with the pediatric radiologist or pediatric neuroradiologist:
- “What neuroimaging studies are appropriate, based on my differential diagnosis?”
- “Are there specific imaging sequences that we should consider?”
- “Are there contraindications to the imaging modality for my patient?”
- “Is my patient likely to have difficulty tolerating the imaging procedure?”
- “Does my patient need sedation to tolerate this procedure?”
- “Should additional regions be included in the scan?” (Examples: In a child with stroke it might be important to include neck and chest vasculature and the heart. Other conditions might warrant imaging of the spinal cord.)
1. Abdelhalim AN, Alberico RA. Pediatric neuroimaging. Neurol Clin. 2009;27(1):285-301, x.
2. Sehgal V, Delproposto Z, Haacke EM, et al. Clinical applications of neuroimaging with susceptibility-weighted imaging. J Magn Reson Imaging. 2005;22(4):439-450.
3. Bosemani T, Poretti A, Huisman TA. Susceptibility-weighted imaging in pediatric neuroimaging. J Magn Reson Imaging. 2014;40(3):530-544.
4. Cecil KM. Proton magnetic resonance spectroscopy: technique for the neuroradiologist. Neuroimaging Clin N Am. 2013;23(3):381-392.
5. Panigrahy A, Nelson MD Jr, Blüml S. Magnetic resonance spectroscopy in pediatric neuroradiology: clinical and research applications. Pediatr Radiol. 2010;40(1):3-30.
6. Leentjens AF, Schieveld JN, Leonard M, et al. A comparison of the phenomenology of pediatric, adult, and geriatric delirium. J Psychosom Res. 2008;64(2):219-223.
7. Frodl T, Skokauskas N. Meta-analysis of structural MRI studies in children and adults with attention deficit hyperactivity disorder indicates treatment effects. Acta Psychiatr Scand. 2012;125(2):114-126.
8. Shaw P, Malek M, Watson B, et al. Development of cortical surface area and gyrification in attention-deficit/hyperactivity disorder. Biol Psychiatry. 2012;72(3):191-197.
9. Phelan JA, Lowe LH, Glasier CM. Pediatric neurodegenerative white matter processes: leukodystrophies and beyond. Pediatr Radiol. 2008;38(7):729-749.
10. Strawn JR, Wegman CJ, Dominick KC, et al. Cortical surface anatomy in pediatric patients with generalized anxiety disorder. J Anxiety Disord. 2014;28(7):717-723.
11. Mueller SC, Aouidad A, Gorodetsky E, et al. Gray matter volume in adolescent anxiety: an impact of the brain-derived neurotrophic factor Val(66)Met polymorphism [Erratum in J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195]? J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195.
12. Strawn JR, Hamm L, Fitzgerald DA, et al. Neurostructural abnormalities in pediatric anxiety disorders. J Anxiety Disord. 2015;32:81-88.
13. Strawn JR, Dominick KC, Patino LR, et al. Neurobiology of pediatric anxiety disorders. Curr Behav Neurosci Reports. 2014;1(3):154-160.
14. Alexiou GA, Argyropoulou MI. Neuroimaging in childhood headache: a systematic review. Pediatr Radiol. 2013;43(7):777-784.
15. Giedd JN, Rapoport JL, Garvey MA, et al. MRI assessment of children with obsessive-compulsive disorder or tics associated with streptococcal infection. Am J Psychiatry. 2000;157(2):281-283.
16. Chang K, Frankovich J, Cooperstock M, et al; PANS Collaborative Consortium. Clinical evaluation of youth with pediatric acute-onset neuropsychiatric syndrome (PANS): recommendations from the 2013 PANS Consensus Conference. J Child Adolesc Psychopharmacol. 2014;25(1):3-13.
17. Wallace GL, Robustelli B, Dankner N, et al. Increased gyrification, but comparable surface area in adolescents with autism spectrum disorders. Brain. 2013;136(pt 6):1956-1967.
18. Libero LE, DeRamus TP, Deshpande HD, et al. Surface-based morphometry of the cortical architecture of autism spectrum disorders: volume, thickness, area, and gyrification. Neuropsychologia. 2014;62:1-10.
19. Boddaert N, Zilbovicius M, Philipe A, et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS One. 2009;4:e445. doi: 10.1371/journal.pone.0004415.
20. Richards C, Jones C, Groves L, et al. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry. 2015;2(10):909-916.
21. Wilde EA, Hunter JV, Bigler ED. Pediatric traumatic brain injury: neuroimaging and neurorehabilitation outcome. NeuroRehabilitation. 2012;31(3):245-260.
22. Mechtler LL, Shastri KK, Crutchfield KE. Advanced neuroimaging of mild traumatic brain injury. Neurol Clin. 2014;32(1):31-58.
23. Ashwal S, Tong KA, Ghosh N, et al. Application of advanced neuroimaging modalities in pediatric traumatic brain injury. J Child Neurol. 2014;29(12):1704-1717.
24. Munson S, Schroth E, Ernst M. The role of functional neuroimaging in pediatric brain injury. Pediatrics. 2006;117(4):1372-1381.
25. Wozniak JR, Krach L, Ward E, et al. Neurocognitive and neuroimaging correlates of pediatric traumatic brain injury: a diffusion tensor imaging (DTI) study. Arch Clin Neuropsychol. 2007;22(5):555-568.
1. Abdelhalim AN, Alberico RA. Pediatric neuroimaging. Neurol Clin. 2009;27(1):285-301, x.
2. Sehgal V, Delproposto Z, Haacke EM, et al. Clinical applications of neuroimaging with susceptibility-weighted imaging. J Magn Reson Imaging. 2005;22(4):439-450.
3. Bosemani T, Poretti A, Huisman TA. Susceptibility-weighted imaging in pediatric neuroimaging. J Magn Reson Imaging. 2014;40(3):530-544.
4. Cecil KM. Proton magnetic resonance spectroscopy: technique for the neuroradiologist. Neuroimaging Clin N Am. 2013;23(3):381-392.
5. Panigrahy A, Nelson MD Jr, Blüml S. Magnetic resonance spectroscopy in pediatric neuroradiology: clinical and research applications. Pediatr Radiol. 2010;40(1):3-30.
6. Leentjens AF, Schieveld JN, Leonard M, et al. A comparison of the phenomenology of pediatric, adult, and geriatric delirium. J Psychosom Res. 2008;64(2):219-223.
7. Frodl T, Skokauskas N. Meta-analysis of structural MRI studies in children and adults with attention deficit hyperactivity disorder indicates treatment effects. Acta Psychiatr Scand. 2012;125(2):114-126.
8. Shaw P, Malek M, Watson B, et al. Development of cortical surface area and gyrification in attention-deficit/hyperactivity disorder. Biol Psychiatry. 2012;72(3):191-197.
9. Phelan JA, Lowe LH, Glasier CM. Pediatric neurodegenerative white matter processes: leukodystrophies and beyond. Pediatr Radiol. 2008;38(7):729-749.
10. Strawn JR, Wegman CJ, Dominick KC, et al. Cortical surface anatomy in pediatric patients with generalized anxiety disorder. J Anxiety Disord. 2014;28(7):717-723.
11. Mueller SC, Aouidad A, Gorodetsky E, et al. Gray matter volume in adolescent anxiety: an impact of the brain-derived neurotrophic factor Val(66)Met polymorphism [Erratum in J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195]? J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195.
12. Strawn JR, Hamm L, Fitzgerald DA, et al. Neurostructural abnormalities in pediatric anxiety disorders. J Anxiety Disord. 2015;32:81-88.
13. Strawn JR, Dominick KC, Patino LR, et al. Neurobiology of pediatric anxiety disorders. Curr Behav Neurosci Reports. 2014;1(3):154-160.
14. Alexiou GA, Argyropoulou MI. Neuroimaging in childhood headache: a systematic review. Pediatr Radiol. 2013;43(7):777-784.
15. Giedd JN, Rapoport JL, Garvey MA, et al. MRI assessment of children with obsessive-compulsive disorder or tics associated with streptococcal infection. Am J Psychiatry. 2000;157(2):281-283.
16. Chang K, Frankovich J, Cooperstock M, et al; PANS Collaborative Consortium. Clinical evaluation of youth with pediatric acute-onset neuropsychiatric syndrome (PANS): recommendations from the 2013 PANS Consensus Conference. J Child Adolesc Psychopharmacol. 2014;25(1):3-13.
17. Wallace GL, Robustelli B, Dankner N, et al. Increased gyrification, but comparable surface area in adolescents with autism spectrum disorders. Brain. 2013;136(pt 6):1956-1967.
18. Libero LE, DeRamus TP, Deshpande HD, et al. Surface-based morphometry of the cortical architecture of autism spectrum disorders: volume, thickness, area, and gyrification. Neuropsychologia. 2014;62:1-10.
19. Boddaert N, Zilbovicius M, Philipe A, et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS One. 2009;4:e445. doi: 10.1371/journal.pone.0004415.
20. Richards C, Jones C, Groves L, et al. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry. 2015;2(10):909-916.
21. Wilde EA, Hunter JV, Bigler ED. Pediatric traumatic brain injury: neuroimaging and neurorehabilitation outcome. NeuroRehabilitation. 2012;31(3):245-260.
22. Mechtler LL, Shastri KK, Crutchfield KE. Advanced neuroimaging of mild traumatic brain injury. Neurol Clin. 2014;32(1):31-58.
23. Ashwal S, Tong KA, Ghosh N, et al. Application of advanced neuroimaging modalities in pediatric traumatic brain injury. J Child Neurol. 2014;29(12):1704-1717.
24. Munson S, Schroth E, Ernst M. The role of functional neuroimaging in pediatric brain injury. Pediatrics. 2006;117(4):1372-1381.
25. Wozniak JR, Krach L, Ward E, et al. Neurocognitive and neuroimaging correlates of pediatric traumatic brain injury: a diffusion tensor imaging (DTI) study. Arch Clin Neuropsychol. 2007;22(5):555-568.

Pseudobulbar affect: When patients laugh or cry, but don’t know why
Pseudobulbar affect (PBA) is a disorder of affective expression that manifests as stereotyped and frequent outbursts of crying (not limited to lacrimation) or laughter. Symptoms are involuntary, uncontrolled, and exaggerated or incongruent with current mood. Episodes, lasting a few seconds to several minutes, may be unprovoked or occur in response to a mild stimulus, and patients typically display a normal affect between episodes.1 PBA is estimated to affect 1 to 2 million people in the United States, although some studies suggest as many as 7 million,1,2 depending on the evaluation method and threshold criteria used.3

Where to look for pseudobulbar affect
PBA has been most commonly described in 6 major neurologic disorders:
- Alzheimer’s disease
- amyotrophic lateral sclerosis (ALS)
- multiple sclerosis (MS)
- Parkinson’s disease
- stroke
- traumatic brain injury (TBI).

Of these disorders, most studies have found the highest PBA prevalence in patients with ALS and TBI, with lesser (although significant) prevalence in Parkinson’s disease (Table 2).1,12 These “big 6” diagnoses are not a comprehensive list, as many other disease states are associated with PBA (Table 3).12-14


2 Pathways: ‘Generator’ and ‘governor’
Despite the many and varied injuries and illnesses associated with PBA, Lauterbach et al10 noted patterns that suggest dysregulation of 2 distinct but interconnected brain pathways: an emotional pathway controlled by a separate volitional pathway. Lesions to the volitional pathway (or its associated feedback or processing circuits) are thought to cause PBA symptoms.
To borrow an analogy from engineering, the emotional pathway is the “generator” of affect, whereas the volitional pathway is the “governor” of affect. Thus, injury to the “governor” results in overspill, or overflow, of affect that usually would be suppressed.
The emotional pathway, which coordinates the motor aspect of reflex laughing or crying, originates at the frontotemporal cortex, relaying to the amygdala and hypothalamus, then projecting to the dorsal brainstem, which includes the midbrain-pontine periaqueductal gray (PAG), dorsal tegmentum, and related brainstem.
The volitional pathway, which regulates the emotional pathway, originates in the dorsal and lateral frontoparietal cortex, projects through the internal capsule and midbrain basis pedunculi, and continues on to the anteroventral basis pontis. The basis pontis then serves as an afferent relay center for cerebellar activity. Projections from the pons then regulate the emotional circuitry primarily at the level of the PAG.10
Lesions of the volitional pathway have been correlated with conditions of PBA, whereas direct activation of the emotional pathway tended to lead to emotional lability or the crying and laughing behaviors observed in dacrystic or gelastic epilepsy.10 The pivotal nature of the regulation occurring at the PAG has guided treatment options. Neurotransmitter receptors most closely associated with this region include glutamatergic N-methyl-
When to screen for PBA
Ask the right question. PBA as a disease state likely has been widely under-reported, under-recognized, and misdiagnosed (typically, as a primary mood disorder).9 Three factors underscore this problem:
- Patients do not specifically report symptoms of affective disturbance (perhaps because they lack a vocabulary to separate concepts of mood and affect)
- Physicians do not ask patients about separations of mood and affect
- Perhaps most importantly, PBA lacks a general awareness and understanding.
Co-occurring mood disorders also may thwart PBA detection. One study of PBA in Alzheimer’s dementia found that 53% of patients with symptoms consistent with PBA also had a distinct mood disorder.17 This suggests that a PBA-specific screening test is needed for accurate diagnosis.
A single question might best refine the likelihood that a patient has PBA: “Do you ever cry for no reason?” In primary psychiatric illness, crying typically is associated with a specific trigger (eg, depressed mood, despair, anxiety). A patient’s inability to identify a trigger for crying suggests the pathological separation of mood and affect—the core of PBA, and worthy of further investigation.
Clinical rating scales that correlate to disease severity appear to be the most effective in identifying PBA. The PRISM study, to date the largest clinic-based study of PBA symptoms, used the Center for Neurologic Study-Liability Scale (CNS-LS) to gauge the presence and severity of PBA symptoms.1 A 7-question, patient self-administered tool, the CNS-LS is graded on a 5-point Likert scale. A score ≥13 has high sensitivity and specificity for diagnosis of PBA, compared with physician diagnosis.
Another option, the 16-question Pathological Laughing and Crying Scale, is a clinician-administered screening tool. Again, a score ≥13 is consistent with symptoms required for a PBA diagnosis.
Treating PBA symptoms
Until recently, most pharmacotherapeutic interventions for PBA were based on off-label use of tricyclic antidepressants (TCAs) or selective serotonin reuptake inhibitors (SSRIs). From 1980 to 2010, only 7 of 22 case reports or trials of TCAs or SSRIs for PBA were randomized, double-blind, and placebo-controlled. Five had 12 to 28 patients, and 2 had 106 and 128 patients, respectively. Only 1 controlled trial included a validated symptom severity scale, and none included a scale validated for PBA.18
In particular, imipramine and nortriptyline were studied for managing PBA in patients with stroke; amitriptyline, in patients with MS; and various SSRIs, in patients with stroke.11 Response of PBA symptoms to antidepressant therapy was greater in all placebo-controlled trials than response to placebo.18 As seen in pharmacotherapy of depression, the lower burden of adverse effects and overall better tolerability of SSRIs resulted in their preferred use over TCAs. In some cases, the side effects of TCAs can be leveraged for therapeutic gain. If insomnia is a problem, a nighttime dose of a TCA could ameliorate this. Similarly, if a patient has sialorrhea, the anticholinergic effect of a TCA may show some benefit.19
Dextromethorphan plus quinidine. Dextromethorphan has long been of interest for a variety of neurodegenerative diseases. Studies of its efficacy were largely unsuccessful, however, because rapid metabolism by cytochrome P450 (CYP) 2D6 prevented CNS penetration.20 Quinidine is an avid inhibitor of CYP2D6, even at very low dosages. Adding quinidine to dextromethorphan limits metabolism, allowing dextromethorphan to accumulate to a plasma concentration sufficient to penetrate the CNS.12 In 2010, the combination agent dextromethorphan hydrobromide (20 mg)/quinidine (10 mg) (DM/Q) became the first treatment to receive FDA approval for managing PBA.11
Mechanism of action. The exact mechanism of DM/Q in PBA remains unknown. Dextromethorphan is an agonist of sigma-1 receptors and a relatively specific noncompetitive antagonist of NMDA receptors. It also has been shown to modulate glutamate and serotonin neurotransmission and ion channel function.20 Sigma-1 receptors are concentrated in the brainstem and parts of the cerebellum that are thought to coordinate motor emotional responses. Agonism of sigma-1 receptors on glutamatergic neurons has been proposed to limit release of glutamate from the presynaptic neuron while also limiting downstream transmission of glutamatergic signal in postsynaptic neurons.
Clinical trials. Two large trials have demonstrated efficacy of DM/Q in PBA. STAR was a 12-week, double-blind, placebo-controlled trial with 326 patients diagnosed with ALS or MS who showed PBA symptoms (CNS-LS score ≥13). Compared with placebo, DM/Q use was associated with significantly reduced (P < .01) daily episodes of PBA at 2, 4, 8, and 12 weeks.20 The effect was rapid, with 30% fewer PBA episodes after the first week (P < .0167). At 12 weeks, 51% of patients on DM/Q had been symptom-free for at least 2 weeks.
The PRISM II study examined the efficacy of DM/Q in managing PBA in 102 individuals with dementia, 92 with stroke, and 67 with TBI. After 30 and 90 days, CNL-LS scores were significantly reduced (P < .001) compared with baseline scores.20
Prescribing information. Dextromethorphan—typically in the form of cough syrup—has been implicated as a substance of abuse. A placebo-controlled trial demonstrated that co-administering quinidine with dextromethorphan limits measures of positive reinforcement, such as euphoria and drug liking. This suggests that quinidine may be used to reduce abuse of dextromethorphan.20 As such, the abuse potential of DM/Q appears to be low.
The most common adverse effects reported with DM/Q are diarrhea, dizziness, and cough.12 Notably, patients who received DM/Q in the STAR trial were more likely to report dizziness than those receiving placebo (10.3% vs 5.5%), but patients receiving placebo were more likely to fall.21,22
Package labeling warns that DM/Q causes dose-dependent QTc prolongation.21 Quinidine can be associated with significant QTc prolongation when dosed at antiarrhythmic levels, although mean plasma concentrations found with the 10 mg of quinidine in the approved DM/Q formulation are 1% to 3% of those associated with typical dosages used in antiarrhythmic therapy. Electrophysiology studies of quinidine 10 mg dosed every 12 hours have demonstrated a mean QTc increase at steady state of 6.8 milliseconds, compared with 9.1 milliseconds for a reference control (moxifloxacin).12,21
Although this would seem to indicate a relatively low risk of clinically significant QTc prolongation at these ultra-low dosages of quinidine, it may be advisable to obtain an initial pre-dose and post-dose ECG and longitudinally monitor the QTc interval in patients with conditions that predispose to cardiac arrhythmias. Because quinidine inhibits CYP2D6, use caution when prescribing and monitoring other medications metabolized by this pathway.
Bottom Line
1. Brooks BR, Crumpacker D, Fellus J, et al. PRISM: a novel research tool to assess the prevalence of pseudobulbar affect symptoms across neurological conditions. PLoS One. 2013;8(8):e72232. doi: 10.1371/journal.pone.0072232.
2. Cruz MP. Nuedexta for the treatment of pseudobulbar affect: a condition of involuntary laughing and crying. P T. 2013;38(6):325-328.
3. Work SS, Colamonico JA, Bradley WG, et al. Pseudobulbar affect: an under-recognized and under-treated neurological disorder. Adv Ther. 2011;28(7):586-601.
4. Arciniegas DB, Lauterbach EC, Anderson KE, et al. The differential diagnosis of pseudobulbar affect (PBA). Distinguishing PBA among disorders of mood and affect. Proceedings of a roundtable meeting. CNS Spectr. 2005;10(5):1-14; quiz 15-16.
5. Darwin C. The expression of the emotions in man and animals. London, United Kingdom: John Murray; 1872.
6. Oppenheim H, Siemerling E. Mitteilungen über Pseudobulbärparalyse und akute Bulbärparalyse. Berl Kli Woch. 1886;46.
7. Wilson SA. Original papers: some problems in neurology. J Neurol Psychopathol. 1924;4(16):299-333.
8. Poeck K, Risso M, Pilleri G. Contribution to the pathophysiology and clinical systematology of pathological laughing and crying [in German]. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1963;204:181-198.
9. Cummings JL, Gilbart J, Andersen G. Pseudobulbar affect - a disabling but under-recognised consequence of neurological disease and brain injury. Eur Neurol Rev. 2013;8(2):74-81.
10. Lauterbach EC, Cummings JL, Kuppuswamy PS. Toward a more precise, clinically–informed pathophysiology of pathological laughing and crying. Neurosci Biobehav Rev. 2013;37(8):1893-1916.
11. Pioro EP. Review of dextromethorphan 20 mg/quinidine 10 mg (Nuedexta(®)) for pseudobulbar affect. Neurol Ther. 2014;3(1):15-28.
12. Schoedel KA, Morrow SA, Sellers EM. Evaluating the safety and efficacy of dextromethorphan/quinidine in the treatment of pseudobulbar affect. Neuropsychiatr Dis Treat. 2014;10:1161-1174.
13. Li Z, Luo S, Ou J, et al. Persistent pseudobulbar affect secondary to acute disseminated encephalomyelitis. Socioaffect Neurosci Psychol. 2015;5:26210. doi: 10.3402/snp.v5.26210.
14. Pattee GL, Wymer JP, Lomen-Hoerth C, et al. An open-label multicenter study to assess the safety of dextromethorphan/quinidine in patients with pseudobulbar affect associated with a range of underlying neurological conditions. Curr Med Res Opin. 2014;30(11):2255-2265.
15. Strowd RE, Cartwright MS, Okun MS, et al. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257(8):1382-1387.
16. Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness in the USA. Adv Ther. 2012;29(9):775-798.
17. Starkstein SE, Migliorelli R, Tesón A, et al. Prevalence and clinical correlates of pathological affective display in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1995;59(1):55-60.
18. Pioro EP. Current concepts in the pharmacotherapy of pseudobulbar affect. Drugs. 2011;71(9):1193-1207.
19. Ahmed A, Simmons A. Pseudobulbar affect: prevalence and management. Ther Clin Risk Manag. 2013;9:483-489.
20. Yang LP, Deeks ED. Dextromethorphan/quinidine: a review of its use in adults with pseudobulbar affect. Drugs. 2015;75(1):83-90.
21. Nuedexta [package insert]. Aliso Viejo, CA: Avanir Pharmaceuticals, Inc.; 2015.
22. Pioro EP, Brooks BR, Cummings J, et al; Safety, Tolerability, and Efficacy trial of AVP-923 in PBA Investigators. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol. 2010;68(5):693-702.
Pseudobulbar affect (PBA) is a disorder of affective expression that manifests as stereotyped and frequent outbursts of crying (not limited to lacrimation) or laughter. Symptoms are involuntary, uncontrolled, and exaggerated or incongruent with current mood. Episodes, lasting a few seconds to several minutes, may be unprovoked or occur in response to a mild stimulus, and patients typically display a normal affect between episodes.1 PBA is estimated to affect 1 to 2 million people in the United States, although some studies suggest as many as 7 million,1,2 depending on the evaluation method and threshold criteria used.3
Where to look for pseudobulbar affect
PBA has been most commonly described in 6 major neurologic disorders:
- Alzheimer’s disease
- amyotrophic lateral sclerosis (ALS)
- multiple sclerosis (MS)
- Parkinson’s disease
- stroke
- traumatic brain injury (TBI).
Of these disorders, most studies have found the highest PBA prevalence in patients with ALS and TBI, with lesser (although significant) prevalence in Parkinson’s disease (Table 2).1,12 These “big 6” diagnoses are not a comprehensive list, as many other disease states are associated with PBA (Table 3).12-14
2 Pathways: ‘Generator’ and ‘governor’
Despite the many and varied injuries and illnesses associated with PBA, Lauterbach et al10 noted patterns that suggest dysregulation of 2 distinct but interconnected brain pathways: an emotional pathway controlled by a separate volitional pathway. Lesions to the volitional pathway (or its associated feedback or processing circuits) are thought to cause PBA symptoms.
To borrow an analogy from engineering, the emotional pathway is the “generator” of affect, whereas the volitional pathway is the “governor” of affect. Thus, injury to the “governor” results in overspill, or overflow, of affect that usually would be suppressed.
The emotional pathway, which coordinates the motor aspect of reflex laughing or crying, originates at the frontotemporal cortex, relaying to the amygdala and hypothalamus, then projecting to the dorsal brainstem, which includes the midbrain-pontine periaqueductal gray (PAG), dorsal tegmentum, and related brainstem.
The volitional pathway, which regulates the emotional pathway, originates in the dorsal and lateral frontoparietal cortex, projects through the internal capsule and midbrain basis pedunculi, and continues on to the anteroventral basis pontis. The basis pontis then serves as an afferent relay center for cerebellar activity. Projections from the pons then regulate the emotional circuitry primarily at the level of the PAG.10
Lesions of the volitional pathway have been correlated with conditions of PBA, whereas direct activation of the emotional pathway tended to lead to emotional lability or the crying and laughing behaviors observed in dacrystic or gelastic epilepsy.10 The pivotal nature of the regulation occurring at the PAG has guided treatment options. Neurotransmitter receptors most closely associated with this region include glutamatergic N-methyl-
When to screen for PBA
Ask the right question. PBA as a disease state likely has been widely under-reported, under-recognized, and misdiagnosed (typically, as a primary mood disorder).9 Three factors underscore this problem:
- Patients do not specifically report symptoms of affective disturbance (perhaps because they lack a vocabulary to separate concepts of mood and affect)
- Physicians do not ask patients about separations of mood and affect
- Perhaps most importantly, PBA lacks a general awareness and understanding.
Co-occurring mood disorders also may thwart PBA detection. One study of PBA in Alzheimer’s dementia found that 53% of patients with symptoms consistent with PBA also had a distinct mood disorder.17 This suggests that a PBA-specific screening test is needed for accurate diagnosis.
A single question might best refine the likelihood that a patient has PBA: “Do you ever cry for no reason?” In primary psychiatric illness, crying typically is associated with a specific trigger (eg, depressed mood, despair, anxiety). A patient’s inability to identify a trigger for crying suggests the pathological separation of mood and affect—the core of PBA, and worthy of further investigation.
Clinical rating scales that correlate to disease severity appear to be the most effective in identifying PBA. The PRISM study, to date the largest clinic-based study of PBA symptoms, used the Center for Neurologic Study-Liability Scale (CNS-LS) to gauge the presence and severity of PBA symptoms.1 A 7-question, patient self-administered tool, the CNS-LS is graded on a 5-point Likert scale. A score ≥13 has high sensitivity and specificity for diagnosis of PBA, compared with physician diagnosis.
Another option, the 16-question Pathological Laughing and Crying Scale, is a clinician-administered screening tool. Again, a score ≥13 is consistent with symptoms required for a PBA diagnosis.
Treating PBA symptoms
Until recently, most pharmacotherapeutic interventions for PBA were based on off-label use of tricyclic antidepressants (TCAs) or selective serotonin reuptake inhibitors (SSRIs). From 1980 to 2010, only 7 of 22 case reports or trials of TCAs or SSRIs for PBA were randomized, double-blind, and placebo-controlled. Five had 12 to 28 patients, and 2 had 106 and 128 patients, respectively. Only 1 controlled trial included a validated symptom severity scale, and none included a scale validated for PBA.18
In particular, imipramine and nortriptyline were studied for managing PBA in patients with stroke; amitriptyline, in patients with MS; and various SSRIs, in patients with stroke.11 Response of PBA symptoms to antidepressant therapy was greater in all placebo-controlled trials than response to placebo.18 As seen in pharmacotherapy of depression, the lower burden of adverse effects and overall better tolerability of SSRIs resulted in their preferred use over TCAs. In some cases, the side effects of TCAs can be leveraged for therapeutic gain. If insomnia is a problem, a nighttime dose of a TCA could ameliorate this. Similarly, if a patient has sialorrhea, the anticholinergic effect of a TCA may show some benefit.19
Dextromethorphan plus quinidine. Dextromethorphan has long been of interest for a variety of neurodegenerative diseases. Studies of its efficacy were largely unsuccessful, however, because rapid metabolism by cytochrome P450 (CYP) 2D6 prevented CNS penetration.20 Quinidine is an avid inhibitor of CYP2D6, even at very low dosages. Adding quinidine to dextromethorphan limits metabolism, allowing dextromethorphan to accumulate to a plasma concentration sufficient to penetrate the CNS.12 In 2010, the combination agent dextromethorphan hydrobromide (20 mg)/quinidine (10 mg) (DM/Q) became the first treatment to receive FDA approval for managing PBA.11
Mechanism of action. The exact mechanism of DM/Q in PBA remains unknown. Dextromethorphan is an agonist of sigma-1 receptors and a relatively specific noncompetitive antagonist of NMDA receptors. It also has been shown to modulate glutamate and serotonin neurotransmission and ion channel function.20 Sigma-1 receptors are concentrated in the brainstem and parts of the cerebellum that are thought to coordinate motor emotional responses. Agonism of sigma-1 receptors on glutamatergic neurons has been proposed to limit release of glutamate from the presynaptic neuron while also limiting downstream transmission of glutamatergic signal in postsynaptic neurons.
Clinical trials. Two large trials have demonstrated efficacy of DM/Q in PBA. STAR was a 12-week, double-blind, placebo-controlled trial with 326 patients diagnosed with ALS or MS who showed PBA symptoms (CNS-LS score ≥13). Compared with placebo, DM/Q use was associated with significantly reduced (P < .01) daily episodes of PBA at 2, 4, 8, and 12 weeks.20 The effect was rapid, with 30% fewer PBA episodes after the first week (P < .0167). At 12 weeks, 51% of patients on DM/Q had been symptom-free for at least 2 weeks.
The PRISM II study examined the efficacy of DM/Q in managing PBA in 102 individuals with dementia, 92 with stroke, and 67 with TBI. After 30 and 90 days, CNL-LS scores were significantly reduced (P < .001) compared with baseline scores.20
Prescribing information. Dextromethorphan—typically in the form of cough syrup—has been implicated as a substance of abuse. A placebo-controlled trial demonstrated that co-administering quinidine with dextromethorphan limits measures of positive reinforcement, such as euphoria and drug liking. This suggests that quinidine may be used to reduce abuse of dextromethorphan.20 As such, the abuse potential of DM/Q appears to be low.
The most common adverse effects reported with DM/Q are diarrhea, dizziness, and cough.12 Notably, patients who received DM/Q in the STAR trial were more likely to report dizziness than those receiving placebo (10.3% vs 5.5%), but patients receiving placebo were more likely to fall.21,22
Package labeling warns that DM/Q causes dose-dependent QTc prolongation.21 Quinidine can be associated with significant QTc prolongation when dosed at antiarrhythmic levels, although mean plasma concentrations found with the 10 mg of quinidine in the approved DM/Q formulation are 1% to 3% of those associated with typical dosages used in antiarrhythmic therapy. Electrophysiology studies of quinidine 10 mg dosed every 12 hours have demonstrated a mean QTc increase at steady state of 6.8 milliseconds, compared with 9.1 milliseconds for a reference control (moxifloxacin).12,21
Although this would seem to indicate a relatively low risk of clinically significant QTc prolongation at these ultra-low dosages of quinidine, it may be advisable to obtain an initial pre-dose and post-dose ECG and longitudinally monitor the QTc interval in patients with conditions that predispose to cardiac arrhythmias. Because quinidine inhibits CYP2D6, use caution when prescribing and monitoring other medications metabolized by this pathway.
Bottom Line
Pseudobulbar affect (PBA) is a disorder of affective expression that manifests as stereotyped and frequent outbursts of crying (not limited to lacrimation) or laughter. Symptoms are involuntary, uncontrolled, and exaggerated or incongruent with current mood. Episodes, lasting a few seconds to several minutes, may be unprovoked or occur in response to a mild stimulus, and patients typically display a normal affect between episodes.1 PBA is estimated to affect 1 to 2 million people in the United States, although some studies suggest as many as 7 million,1,2 depending on the evaluation method and threshold criteria used.3
Where to look for pseudobulbar affect
PBA has been most commonly described in 6 major neurologic disorders:
- Alzheimer’s disease
- amyotrophic lateral sclerosis (ALS)
- multiple sclerosis (MS)
- Parkinson’s disease
- stroke
- traumatic brain injury (TBI).
Of these disorders, most studies have found the highest PBA prevalence in patients with ALS and TBI, with lesser (although significant) prevalence in Parkinson’s disease (Table 2).1,12 These “big 6” diagnoses are not a comprehensive list, as many other disease states are associated with PBA (Table 3).12-14
2 Pathways: ‘Generator’ and ‘governor’
Despite the many and varied injuries and illnesses associated with PBA, Lauterbach et al10 noted patterns that suggest dysregulation of 2 distinct but interconnected brain pathways: an emotional pathway controlled by a separate volitional pathway. Lesions to the volitional pathway (or its associated feedback or processing circuits) are thought to cause PBA symptoms.
To borrow an analogy from engineering, the emotional pathway is the “generator” of affect, whereas the volitional pathway is the “governor” of affect. Thus, injury to the “governor” results in overspill, or overflow, of affect that usually would be suppressed.
The emotional pathway, which coordinates the motor aspect of reflex laughing or crying, originates at the frontotemporal cortex, relaying to the amygdala and hypothalamus, then projecting to the dorsal brainstem, which includes the midbrain-pontine periaqueductal gray (PAG), dorsal tegmentum, and related brainstem.
The volitional pathway, which regulates the emotional pathway, originates in the dorsal and lateral frontoparietal cortex, projects through the internal capsule and midbrain basis pedunculi, and continues on to the anteroventral basis pontis. The basis pontis then serves as an afferent relay center for cerebellar activity. Projections from the pons then regulate the emotional circuitry primarily at the level of the PAG.10
Lesions of the volitional pathway have been correlated with conditions of PBA, whereas direct activation of the emotional pathway tended to lead to emotional lability or the crying and laughing behaviors observed in dacrystic or gelastic epilepsy.10 The pivotal nature of the regulation occurring at the PAG has guided treatment options. Neurotransmitter receptors most closely associated with this region include glutamatergic N-methyl-
When to screen for PBA
Ask the right question. PBA as a disease state likely has been widely under-reported, under-recognized, and misdiagnosed (typically, as a primary mood disorder).9 Three factors underscore this problem:
- Patients do not specifically report symptoms of affective disturbance (perhaps because they lack a vocabulary to separate concepts of mood and affect)
- Physicians do not ask patients about separations of mood and affect
- Perhaps most importantly, PBA lacks a general awareness and understanding.
Co-occurring mood disorders also may thwart PBA detection. One study of PBA in Alzheimer’s dementia found that 53% of patients with symptoms consistent with PBA also had a distinct mood disorder.17 This suggests that a PBA-specific screening test is needed for accurate diagnosis.
A single question might best refine the likelihood that a patient has PBA: “Do you ever cry for no reason?” In primary psychiatric illness, crying typically is associated with a specific trigger (eg, depressed mood, despair, anxiety). A patient’s inability to identify a trigger for crying suggests the pathological separation of mood and affect—the core of PBA, and worthy of further investigation.
Clinical rating scales that correlate to disease severity appear to be the most effective in identifying PBA. The PRISM study, to date the largest clinic-based study of PBA symptoms, used the Center for Neurologic Study-Liability Scale (CNS-LS) to gauge the presence and severity of PBA symptoms.1 A 7-question, patient self-administered tool, the CNS-LS is graded on a 5-point Likert scale. A score ≥13 has high sensitivity and specificity for diagnosis of PBA, compared with physician diagnosis.
Another option, the 16-question Pathological Laughing and Crying Scale, is a clinician-administered screening tool. Again, a score ≥13 is consistent with symptoms required for a PBA diagnosis.
Treating PBA symptoms
Until recently, most pharmacotherapeutic interventions for PBA were based on off-label use of tricyclic antidepressants (TCAs) or selective serotonin reuptake inhibitors (SSRIs). From 1980 to 2010, only 7 of 22 case reports or trials of TCAs or SSRIs for PBA were randomized, double-blind, and placebo-controlled. Five had 12 to 28 patients, and 2 had 106 and 128 patients, respectively. Only 1 controlled trial included a validated symptom severity scale, and none included a scale validated for PBA.18
In particular, imipramine and nortriptyline were studied for managing PBA in patients with stroke; amitriptyline, in patients with MS; and various SSRIs, in patients with stroke.11 Response of PBA symptoms to antidepressant therapy was greater in all placebo-controlled trials than response to placebo.18 As seen in pharmacotherapy of depression, the lower burden of adverse effects and overall better tolerability of SSRIs resulted in their preferred use over TCAs. In some cases, the side effects of TCAs can be leveraged for therapeutic gain. If insomnia is a problem, a nighttime dose of a TCA could ameliorate this. Similarly, if a patient has sialorrhea, the anticholinergic effect of a TCA may show some benefit.19
Dextromethorphan plus quinidine. Dextromethorphan has long been of interest for a variety of neurodegenerative diseases. Studies of its efficacy were largely unsuccessful, however, because rapid metabolism by cytochrome P450 (CYP) 2D6 prevented CNS penetration.20 Quinidine is an avid inhibitor of CYP2D6, even at very low dosages. Adding quinidine to dextromethorphan limits metabolism, allowing dextromethorphan to accumulate to a plasma concentration sufficient to penetrate the CNS.12 In 2010, the combination agent dextromethorphan hydrobromide (20 mg)/quinidine (10 mg) (DM/Q) became the first treatment to receive FDA approval for managing PBA.11
Mechanism of action. The exact mechanism of DM/Q in PBA remains unknown. Dextromethorphan is an agonist of sigma-1 receptors and a relatively specific noncompetitive antagonist of NMDA receptors. It also has been shown to modulate glutamate and serotonin neurotransmission and ion channel function.20 Sigma-1 receptors are concentrated in the brainstem and parts of the cerebellum that are thought to coordinate motor emotional responses. Agonism of sigma-1 receptors on glutamatergic neurons has been proposed to limit release of glutamate from the presynaptic neuron while also limiting downstream transmission of glutamatergic signal in postsynaptic neurons.
Clinical trials. Two large trials have demonstrated efficacy of DM/Q in PBA. STAR was a 12-week, double-blind, placebo-controlled trial with 326 patients diagnosed with ALS or MS who showed PBA symptoms (CNS-LS score ≥13). Compared with placebo, DM/Q use was associated with significantly reduced (P < .01) daily episodes of PBA at 2, 4, 8, and 12 weeks.20 The effect was rapid, with 30% fewer PBA episodes after the first week (P < .0167). At 12 weeks, 51% of patients on DM/Q had been symptom-free for at least 2 weeks.
The PRISM II study examined the efficacy of DM/Q in managing PBA in 102 individuals with dementia, 92 with stroke, and 67 with TBI. After 30 and 90 days, CNL-LS scores were significantly reduced (P < .001) compared with baseline scores.20
Prescribing information. Dextromethorphan—typically in the form of cough syrup—has been implicated as a substance of abuse. A placebo-controlled trial demonstrated that co-administering quinidine with dextromethorphan limits measures of positive reinforcement, such as euphoria and drug liking. This suggests that quinidine may be used to reduce abuse of dextromethorphan.20 As such, the abuse potential of DM/Q appears to be low.
The most common adverse effects reported with DM/Q are diarrhea, dizziness, and cough.12 Notably, patients who received DM/Q in the STAR trial were more likely to report dizziness than those receiving placebo (10.3% vs 5.5%), but patients receiving placebo were more likely to fall.21,22
Package labeling warns that DM/Q causes dose-dependent QTc prolongation.21 Quinidine can be associated with significant QTc prolongation when dosed at antiarrhythmic levels, although mean plasma concentrations found with the 10 mg of quinidine in the approved DM/Q formulation are 1% to 3% of those associated with typical dosages used in antiarrhythmic therapy. Electrophysiology studies of quinidine 10 mg dosed every 12 hours have demonstrated a mean QTc increase at steady state of 6.8 milliseconds, compared with 9.1 milliseconds for a reference control (moxifloxacin).12,21
Although this would seem to indicate a relatively low risk of clinically significant QTc prolongation at these ultra-low dosages of quinidine, it may be advisable to obtain an initial pre-dose and post-dose ECG and longitudinally monitor the QTc interval in patients with conditions that predispose to cardiac arrhythmias. Because quinidine inhibits CYP2D6, use caution when prescribing and monitoring other medications metabolized by this pathway.
Bottom Line
1. Brooks BR, Crumpacker D, Fellus J, et al. PRISM: a novel research tool to assess the prevalence of pseudobulbar affect symptoms across neurological conditions. PLoS One. 2013;8(8):e72232. doi: 10.1371/journal.pone.0072232.
2. Cruz MP. Nuedexta for the treatment of pseudobulbar affect: a condition of involuntary laughing and crying. P T. 2013;38(6):325-328.
3. Work SS, Colamonico JA, Bradley WG, et al. Pseudobulbar affect: an under-recognized and under-treated neurological disorder. Adv Ther. 2011;28(7):586-601.
4. Arciniegas DB, Lauterbach EC, Anderson KE, et al. The differential diagnosis of pseudobulbar affect (PBA). Distinguishing PBA among disorders of mood and affect. Proceedings of a roundtable meeting. CNS Spectr. 2005;10(5):1-14; quiz 15-16.
5. Darwin C. The expression of the emotions in man and animals. London, United Kingdom: John Murray; 1872.
6. Oppenheim H, Siemerling E. Mitteilungen über Pseudobulbärparalyse und akute Bulbärparalyse. Berl Kli Woch. 1886;46.
7. Wilson SA. Original papers: some problems in neurology. J Neurol Psychopathol. 1924;4(16):299-333.
8. Poeck K, Risso M, Pilleri G. Contribution to the pathophysiology and clinical systematology of pathological laughing and crying [in German]. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1963;204:181-198.
9. Cummings JL, Gilbart J, Andersen G. Pseudobulbar affect - a disabling but under-recognised consequence of neurological disease and brain injury. Eur Neurol Rev. 2013;8(2):74-81.
10. Lauterbach EC, Cummings JL, Kuppuswamy PS. Toward a more precise, clinically–informed pathophysiology of pathological laughing and crying. Neurosci Biobehav Rev. 2013;37(8):1893-1916.
11. Pioro EP. Review of dextromethorphan 20 mg/quinidine 10 mg (Nuedexta(®)) for pseudobulbar affect. Neurol Ther. 2014;3(1):15-28.
12. Schoedel KA, Morrow SA, Sellers EM. Evaluating the safety and efficacy of dextromethorphan/quinidine in the treatment of pseudobulbar affect. Neuropsychiatr Dis Treat. 2014;10:1161-1174.
13. Li Z, Luo S, Ou J, et al. Persistent pseudobulbar affect secondary to acute disseminated encephalomyelitis. Socioaffect Neurosci Psychol. 2015;5:26210. doi: 10.3402/snp.v5.26210.
14. Pattee GL, Wymer JP, Lomen-Hoerth C, et al. An open-label multicenter study to assess the safety of dextromethorphan/quinidine in patients with pseudobulbar affect associated with a range of underlying neurological conditions. Curr Med Res Opin. 2014;30(11):2255-2265.
15. Strowd RE, Cartwright MS, Okun MS, et al. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257(8):1382-1387.
16. Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness in the USA. Adv Ther. 2012;29(9):775-798.
17. Starkstein SE, Migliorelli R, Tesón A, et al. Prevalence and clinical correlates of pathological affective display in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1995;59(1):55-60.
18. Pioro EP. Current concepts in the pharmacotherapy of pseudobulbar affect. Drugs. 2011;71(9):1193-1207.
19. Ahmed A, Simmons A. Pseudobulbar affect: prevalence and management. Ther Clin Risk Manag. 2013;9:483-489.
20. Yang LP, Deeks ED. Dextromethorphan/quinidine: a review of its use in adults with pseudobulbar affect. Drugs. 2015;75(1):83-90.
21. Nuedexta [package insert]. Aliso Viejo, CA: Avanir Pharmaceuticals, Inc.; 2015.
22. Pioro EP, Brooks BR, Cummings J, et al; Safety, Tolerability, and Efficacy trial of AVP-923 in PBA Investigators. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol. 2010;68(5):693-702.
1. Brooks BR, Crumpacker D, Fellus J, et al. PRISM: a novel research tool to assess the prevalence of pseudobulbar affect symptoms across neurological conditions. PLoS One. 2013;8(8):e72232. doi: 10.1371/journal.pone.0072232.
2. Cruz MP. Nuedexta for the treatment of pseudobulbar affect: a condition of involuntary laughing and crying. P T. 2013;38(6):325-328.
3. Work SS, Colamonico JA, Bradley WG, et al. Pseudobulbar affect: an under-recognized and under-treated neurological disorder. Adv Ther. 2011;28(7):586-601.
4. Arciniegas DB, Lauterbach EC, Anderson KE, et al. The differential diagnosis of pseudobulbar affect (PBA). Distinguishing PBA among disorders of mood and affect. Proceedings of a roundtable meeting. CNS Spectr. 2005;10(5):1-14; quiz 15-16.
5. Darwin C. The expression of the emotions in man and animals. London, United Kingdom: John Murray; 1872.
6. Oppenheim H, Siemerling E. Mitteilungen über Pseudobulbärparalyse und akute Bulbärparalyse. Berl Kli Woch. 1886;46.
7. Wilson SA. Original papers: some problems in neurology. J Neurol Psychopathol. 1924;4(16):299-333.
8. Poeck K, Risso M, Pilleri G. Contribution to the pathophysiology and clinical systematology of pathological laughing and crying [in German]. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1963;204:181-198.
9. Cummings JL, Gilbart J, Andersen G. Pseudobulbar affect - a disabling but under-recognised consequence of neurological disease and brain injury. Eur Neurol Rev. 2013;8(2):74-81.
10. Lauterbach EC, Cummings JL, Kuppuswamy PS. Toward a more precise, clinically–informed pathophysiology of pathological laughing and crying. Neurosci Biobehav Rev. 2013;37(8):1893-1916.
11. Pioro EP. Review of dextromethorphan 20 mg/quinidine 10 mg (Nuedexta(®)) for pseudobulbar affect. Neurol Ther. 2014;3(1):15-28.
12. Schoedel KA, Morrow SA, Sellers EM. Evaluating the safety and efficacy of dextromethorphan/quinidine in the treatment of pseudobulbar affect. Neuropsychiatr Dis Treat. 2014;10:1161-1174.
13. Li Z, Luo S, Ou J, et al. Persistent pseudobulbar affect secondary to acute disseminated encephalomyelitis. Socioaffect Neurosci Psychol. 2015;5:26210. doi: 10.3402/snp.v5.26210.
14. Pattee GL, Wymer JP, Lomen-Hoerth C, et al. An open-label multicenter study to assess the safety of dextromethorphan/quinidine in patients with pseudobulbar affect associated with a range of underlying neurological conditions. Curr Med Res Opin. 2014;30(11):2255-2265.
15. Strowd RE, Cartwright MS, Okun MS, et al. Pseudobulbar affect: prevalence and quality of life impact in movement disorders. J Neurol. 2010;257(8):1382-1387.
16. Colamonico J, Formella A, Bradley W. Pseudobulbar affect: burden of illness in the USA. Adv Ther. 2012;29(9):775-798.
17. Starkstein SE, Migliorelli R, Tesón A, et al. Prevalence and clinical correlates of pathological affective display in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1995;59(1):55-60.
18. Pioro EP. Current concepts in the pharmacotherapy of pseudobulbar affect. Drugs. 2011;71(9):1193-1207.
19. Ahmed A, Simmons A. Pseudobulbar affect: prevalence and management. Ther Clin Risk Manag. 2013;9:483-489.
20. Yang LP, Deeks ED. Dextromethorphan/quinidine: a review of its use in adults with pseudobulbar affect. Drugs. 2015;75(1):83-90.
21. Nuedexta [package insert]. Aliso Viejo, CA: Avanir Pharmaceuticals, Inc.; 2015.
22. Pioro EP, Brooks BR, Cummings J, et al; Safety, Tolerability, and Efficacy trial of AVP-923 in PBA Investigators. Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol. 2010;68(5):693-702.

Rediscovering clozapine: Adverse effects develop—what should you do now?
Clozapine is a highly effective antipsychotic with superior efficacy in treatment-resistant schizophrenia, but its side effect profile is daunting (Figure 1).1 Adverse reactions lead to approximately 17% of patients who take clozapine eventually discontinuing the medication.1 As we noted in Part 1 of this 3-part series,2 clozapine remains the most efficacious, but most tedious, antipsychotic available to psychiatrists because of its monitoring requirements and potential side effects.
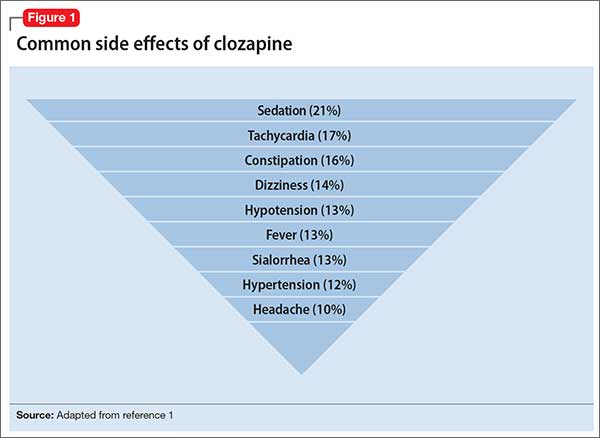
A powerful rationale for prescribing clozapine, despite its drawbacks, is its association with a reduced risk of all-cause mortality.3,4 People with serious mental illness, including schizophrenia, have a median 10-year shorter life expectancy than the general population.5
A recent cohort study6 examined electronic health records to test whether intensive monitoring or lower suicide risk might account for the reduced mortality with clozapine. The authors found that the reduced mortality rate was not directly related to clozapine’s clinical monitoring or other confounding factors. They did find an association between clozapine use and reduced risk of death from both natural and unnatural causes.
This second article in our series examines clozapine’s adverse effects from a systems perspective. Severe neutropenia, myocarditis, sedation, weight gain, orthostatic hypotension, and sialorrhea appear to be the most studied adverse effects, but myriad others can occur.7 We offer guidance to help the astute clinician continue this effective antipsychotic by monitoring carefully, treating side effects early, and managing potential drug interactions (Table 1).8
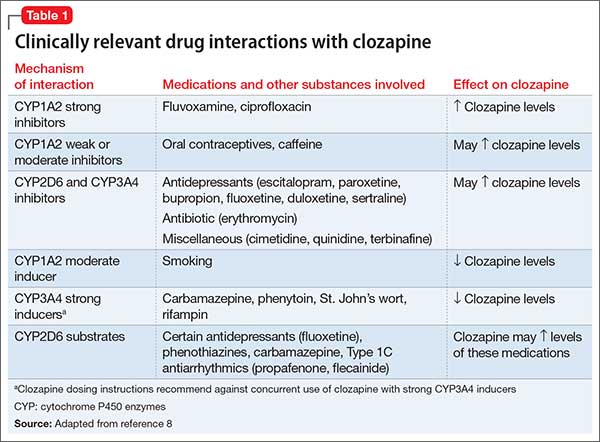
Hematologic eventsSevere neutropenia, defined as absolute neutrophil count (ANC) <500/µL, is a well-known adverse effect of clozapine that requires specific clinical monitoring, a requirement that was updated by the FDA in 2015.2 The incidence of severe neutropenia peaks in the first 2 months of clozapine therapy and tapers after 6 months, but some risk always remains.
Older efficacy studies in the United States gauged the 1-year cumulative incidence of severe clozapine-induced neutropenia to be 2%.9 A 1998 study of the effects of using a clozapine registry reported a lower incidence—0.38%—than the 2% noted above.10 Early recognition and recommended interventions can improve clinical outcomes.2
Drug interactions and neutropenia. A retrospective study of mental health inpatients taking clozapine concurrently with oseltamivir during an influenza outbreak found a statistically significant—but not clinically significant—change in ANC values.11 The authors noted that viral infection might lead to blood dyscrasia early in illness, and that oseltamivir has been associated with a small incidence of blood dyscrasia.11-13 This information might be useful when treating influenza in patients taking clozapine, although no specific change in management is recommended.
Similarly, concomitant treatment with clozapine and lithium can affect both white blood cell and ANC values.14,15 Lithium-treated patients often demonstrate increased circulating neutrophils via enhancement of granulocyte-colony stimulating factor.16 Case studies describe how initiating lithium treatment enabled some patients to continue clozapine after developing neutropenia.14,17 Leukocytosis can affect blood monitoring, possibly masking other blood dyscrasias, when lithium is used concurrently with clozapine.
Eosinophilia (blood eosinophil count >700/µL) occurs in approximately 1% of clozapine users, usually in the first 4 weeks of treatment.18 It can be benign and transient or a harbinger of a more rare adverse reaction such as myocarditis, pancreatitis, hepatitis, colitis, or nephritis.19 If a patient taking clozapine develops eosinophilia, clozapine’s package insert recommends that you:
- evaluate promptly for other systemic involvement (rash, other evidence of allergic reaction, myocarditis, other organ-specific disease)
- stop clozapine immediately if any of these are found.
If other causes of eosinophilia are identified (asthma, allergies, collagen vascular disease, parasitic infection, neoplasm), treat these and continue clozapine.
The manufacturer also mentions the occurrence of clozapine-related eosinophilia without organ involvement that can resolve without intervention, with careful monitoring over several weeks.8 In this scenario, there is flexibility to judge whether clozapine should be stopped or re-challenged, or if close monitoring is adequate. Consulting with an internal medicine or hematology specialist might be helpful.
Cardiovascular side effectsMost common events. Three of the 10 most common clozapine side effects are cardiac: tachycardia, hypotension, and hypertension (Figure 1).1 Orthostatic hypotension, bradycardia, and syncope also can occur, especially with rapid clozapine titration. Baseline electrocardiogram (ECG) can help differentiate whether abnormalities are clozapine-induced or related to a preexisting condition.
Reducing the dosage of clozapine or slowing titration could reverse cardiac side effects.8 If dosage reduction is not an option or is ineffective, first consider treating the side effect rather than discontinuing clozapine.20
Sinus tachycardia is one of the most common side effects of clozapine. First, rule out serious conditions—myocarditis, cardiomyopathy, neuroleptic malignant syndrome (NMS)—then consider waiting and monitoring for the first few months of clozapine treatment. If tachycardia continues, consider dosage reduction. Slower titration, or treatment with a cardio-selective beta blocker such as atenolol.21,22 Note that a recent Cochrane Review concluded that there is not enough randomized evidence to support any particular treatment for clozapine-induced tachycardia; the prescriber must therefore make a case-by-case clinical judgment.22
Similarly, orthostatic hypotension can be managed with a reduced dosage of clozapine or slower titration. Increased fluid intake, compression stockings, and, if necessary, fludrocortisone also can be initiated.20
Rare, potentially fatal events. Myocarditis, pericarditis, and cardiomyopathy are among the rare but potentially fatal adverse effects of clozapine. A recent study reported the incidence of myocarditis with clozapine at a range of 0.015% to 1.3%; cardiomyopathy was even more rare.23 Pulmonary embolism and deep venous thrombosis also are very rare possibilities; keep them in mind, however, when patients taking clozapine report new cardiovascular symptoms.
Patients with clozapine-induced cardiovascular effects most commonly report shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).7,24
Clozapine’s “black-box” warning specifically recommends discontinuing clozapine and consulting cardiology when myocarditis or cardiomyopathy is suspected. In 50% of cases, myocarditis symptoms present in the first few weeks of clozapine treatment.23 The manufacturer states that myocarditis usually presents in the first 2 months, and cardiomyopathy after 8 weeks of treatment; however, either can present at any time.8Figure 2 provides a clinical reference for monitoring a clozapine patient for cardiomyopathy.24
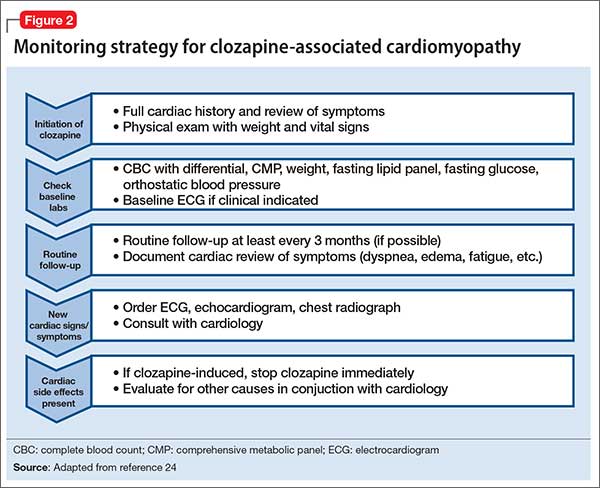
Laboratory findings that support a diagnosis of clozapine-related myocarditis include:
- elevated C-reactive protein
- elevated troponin I or T
- elevated creatine kinase-MB
- peripheral eosinophilia.8,25
ECG, echocardiography, and cardiac MRI can be helpful in diagnosis, in consultation with a cardiologist.
Neurologic side effectsSeizures are listed in the “black-box” warning for clozapine. Seizure incidence with clozapine is 5% per year, with higher incidence at dosages ≥600 mg/d.8 Because clozapine-induced seizures are dosage-dependent, slow titration can mitigate this risk. Tonic-clonic seizures are the most common type associated with clozapine.
The manufacturer recommends caution when using clozapine in patients with a known seizure disorder, alcohol use disorder, or other CNS pathology.8 Patients with a seizure disorder may be at increased risk of experiencing clozapine-induced seizures, but this is not an absolute contraindication.26 Smoking cessation increases clozapine blood levels by an average of 57.4%, further increasing seizure risk.26,27
Discontinuing clozapine is unnecessary when a patient experiences a seizure. Instead, you can:
- halve the dosage prescribed at the time of the seizure (or at least reduce to the last seizure-free dosage)
- consider any medications or medical problems that might have contributed to a lower seizure threshold
- consider prophylaxis with an antiepileptic medication (eg, valproic acid has efficacy for both myoclonic and tonic-clonic seizures).20,26
Sedation is the most common side effect of clozapine.1 Patients experiencing severe sedation should not drive or operate heavy machinery. To reduce sedation, consider instructing the patient to take all or most of the clozapine dosage at bedtime. A critical review of modafinil for sedation caused by antipsychotics in schizophrenia found only 1 open-label study that showed any positive effects; the authors concluded that further study is needed.28
Cognitive and motor slowing are possible neurologic side effects of clozapine. Caution patients about the risk of participating in activities that require cognitive or motor performance until the individual effects of clozapine are known.8
Tardive dyskinesia. Clozapine carries some risk of tardive dyskinesia, although that risk is lower than with other antipsychotics. Similarly, all antipsychotics including clozapine are associated with a risk of NMS. In the rare case of clozapine-induced NMS, stop clozapine immediately and initiate supportive therapy. Clozapine-induced NMS is not an absolute contraindication to re-challenging a patient with clozapine, however, if doing so is clinically appropriate.20
Cerebrovascular events. In older people with dementia, the use of antipsychotics—including clozapine—has been shown to increase the risk of cerebrovascular events. Because most antipsychotics are not FDA-approved for treating psychosis associated with dementia (only pimavanserin is FDA-approved for symptoms of psychosis in Parkinson’s disease), a risk-benefit analysis should be documented when prescribing any antipsychotic in this population. In practice, clozapine’s benefits may outweigh the mortality risks in specific situations.29,30
CASE Sialorrhea puts progress at risk
Ms. B, age 40, has a history of treatment-resistant schizophrenia and is starting clozapine because of residual psychosis during trials of other antipsychotics. She develops severe persistent drooling, mostly at night, during clozapine titration. Sugar-free candy, multiple bed pillows, and changing the dosing schedule do not significantly improve the sialorrhea.
As a result, Ms. B is embarrassed to continue her usual activities. She asks to stop clozapine, even though her psychotic symptoms have improved and she is functioning at her highest level in years.
Ms. B already is taking trihexyphenidyl, 5 mg, 3 times daily, to manage extrapyramidal symptoms related to haloperidol decanoate treatment. After discussing other medication options for sialorrhea, she agrees to a trial of glycopyrrolate, 1 mg, twice daily. She experiences significant improvement and continues taking clozapine.
Sialorrhea develops in 13% of patients taking clozapine.1 As in Ms. B’s case, this side effect can be embarrassing, can limit social or occupational functioning, and might lead patients to discontinue clozapine treatment despite efficacy. Nonpharmacotherapeutic options include covering the pillow with a towel, lowering the clozapine dosage or titrating slowly (or both), and using sugarless gum or candy to increase swallowing.
If the benefits of additional medications targeting side effects outweigh the risks, pharmacotherapeutic intervention may be appropriate. Options include the tricyclic antidepressant amitriptyline31; alpha-adrenergic agonists or antagonists (clonidine, terazosin); and anti-muscarinic medications (benztropine, atropine, trihexyphenidyl, glycopyrrolate) (Table 231). Scopolamine transdermal patch is another possible treatment strategy; however, the scopolamine patch was used for clozapine-induced sialorrhea in only a few case reports, and it is not considered a first-line treatment choice.30
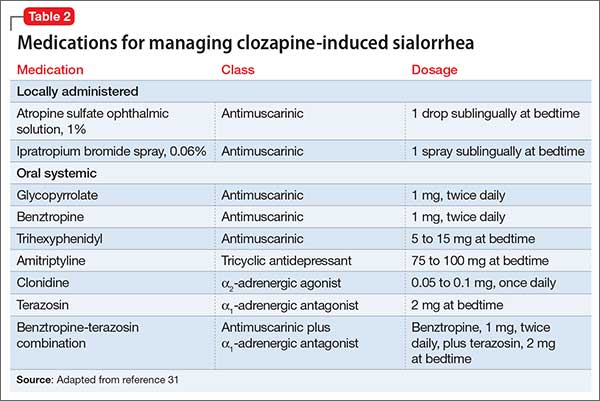
When prescribing, consider the possibility of combined side effects with clozapine and adjunct medications having antimuscarinic or alpha-adrenergic activity, or both. Even atropine ophthalmic drops, administered sublingually, are readily absorbed and cross the blood–brain barrier.31 Another antimuscarinic agent, glycopyrrolate, is less likely to cross the blood–brain barrier and therefore is less likely to cause cognitive side effects. Glycopyrrolate is 5 times more potent at blocking the muscarinic receptor than atropine.31,32 Ipratropium bromide, another nonselective muscarinic receptor antagonist, has less systemic absorption than atropine drops, with less anticholinergic side effects when administered sublingually.
Limited evidence supports the efficacy of alpha-adrenergic medications for managing clozapine-induced sialorrhea. Monitor blood pressure when prescribing terazosin or clonidine, which could potentiate clozapine’s hypotensive effects.
Endocrine side effectsAmong antipsychotics, clozapine is associated with the greatest weight gain—averaging nearly 10% of body weight.33,34 Similarly, the risk of new-onset diabetes mellitus is highest with clozapine in relation to other antipsychotics: 43% reported in a 10-year naturalistic study.35 The risk of hyperlipidemia also increases with clozapine treatment.36 These metabolic changes increase the risk of cardiovascular-related death, with a 10-year mortality rate from cardiovascular disease reported at 9% in clozapine-treated patients.35
Despite clozapine’s metabolic side effects, patients with schizophrenia who are treated with clozapine show a significant reduction in overall mortality compared with patients not treated with clozapine.6 Effective identification and management of metabolic side effects can prevent the need to discontinue clozapine.
Behavioral weight management and exercise are recommended as initial therapy.20 If, based on clinical judgment, these alone are insufficient, data support the use of pharmacotherapeutic interventions. Metformin demonstrates a positive effect on body weight, insulin resistance, and lipids, making it the first choice for adjunctive treatment of clozapine-induced metabolic side effects.37-39
Gastrointestinal side effectsClozapine’s anticholinergic activity can lead to serious gastrointestinal (GI) side effects, including constipation, intestinal obstruction, fecal impaction, and paralytic ileus.8 Ileus has produced more fatal adverse reactions with clozapine than has severe neutropenia.20,40 Co-administered anticholinergic medications could increase the risk of ileus. Obtaining a GI review of systems and monitoring bowel movements (in inpatient or residential facilities) can aid in early identification and limit morbidity and mortality from GI adverse events. A high-fiber diet, adequate hydration, bulk laxatives in patients who can reliably maintain hydration, and GI consultation (if needed) may help manage GI side effects.20
Constitutional side effectsFever can occur with clozapine, most often in the first month of treatment, but the incidence is quite variable (0.5% to 55%).20,41 Although benign fever is common, agranulocytosis with infection, NMS, and other systemic illness must be ruled out. The recommended workup when a patient develops fever while taking clozapine includes physical examination and relevant testing (urinalysis, measurement of ANC and serum creatine kinase, chest radiograph, ECG, and, possibly, blood cultures).41
If evidence supports a serious adverse reaction, stop clozapine immediately.20 If benign clozapine-related fever is suspected, acetaminophen or another antipyretic might provide symptomatic relief; discontinuing clozapine is then unnecessary.41
Pregnancy. When a patient with schizophrenia requires clozapine treatment during pregnancy, reliable clinical guidance is limited. The American College of Obstetricians and Gynecologists Practice Bulletin on the use of psychiatric medications during pregnancy and lactation can be a useful resource.42
Be aware that the FDA very recently made major changes to the format and content of pregnancy and lactation labeling, removing the letter categories that have been used for medications approved on or after June 30, 2001. The manufacturers of medications (such as clozapine) that were approved before June 30, 2001, have 3 years to comply with new requirements.43
The FDA had rated clozapine a pregnancy risk category B medication, meaning no evidence of risk in humans. In 2011, the FDA issued a general warning that antipsychotic use in pregnancy can cause extrapyramidal symptoms and discontinuation symptoms in newborns.44,45
A 2015 review of psychotropic medications and pregnancy noted that approximately 60% of women with schizophrenia became pregnant, with an increased incidence of unplanned pregnancy. A high risk of psychotic relapse (65%) during pregnancy and in the postpartum period may lead to insufficient prenatal care, drug use, and obstetric complications.45 Some data suggest low fetal birth weight and an increased rate of therapeutic abortions in women with schizophrenia.42,46
When treating a pregnant patient, weigh the benefits of clozapine against the risks of adverse events, and clearly document the analysis. Clozapine treatment is not recommended during breast-feeding because of the risk of side effects for newborns.8
We highly recommend keeping updated on the literature regarding pregnancy and lactation information with antipsychotics, including clozapine, because prescribing information will likely be updated in the near future to comply with recent FDA labeling changes.
Final installment: Using clozapine off-labelClozapine is FDA-approved for refractory schizophrenia and for reducing the risk of recurrent suicidal behavior in schizophrenia or schizoaffective disorder. In Part 3 of this series, we review off-label uses—such as managing bipolar disorder, borderline personality disorder, and aggressive behavior—that have varying degrees of scientific support.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Newman WJ, Newman BM. Rediscovering clozapine: after a turbulent history, current guidance on initiating and monitoring. Current Psychiatry. 2016;15(7):42-46,48-49.
3. Walker AM, Lanza LL, Arellano F, et al. Mortality in current and former users of clozapine. Epidemiology. 1997;8(6):671-677.
4. Tiihonen J, Lönnqvist J, Wahlbeck K, et al. 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort study (FIN11 study). Lancet. 2009;374(9690):620-627.
5. Walker E, McGee RE, Druss BG. Mortality in mental disorders and global disease burden Implications: a systematic review and meta-analysis. JAMA Psychiatry. 2015;72(4):334-341.
6. Hayes RD, Downs J, Chang CK, et al. The effect of clozapine on premature mortality: an assessment of clinical monitoring and other potential confounders. Schizophr Bull. 2015;41(3):644-655.
7. De Fazio P, Gaetano R, Caroleo M, et al. Rare and very rare adverse effects of clozapine. Neuropsychiatr Dis Treat. 2015;11:1995-2003.
8. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 29, 2016.
9. Lieberman JA, Johns CA, Kane JM, et al. Clozapine-induced agranulocytosis: non-cross-reactivity with other psychotropic drugs. J Clin Psychiatry. 1988;49(7):271-277.
10. Honigfeld G, Arellano F, Sethi J, et al. Reducing clozapine-related morbidity and mortality: 5 years of experience with the Clozaril National Registry. J Clin Psychiatry. 1998;59(suppl 3):3-7.
11. Demler TL, Trigoboff E. Are clozapine patients at risk for blood dyscrasias with concomitant tamiflu use? Psychiatry (Edgmont). 2009;6(11):29-33.
12. Karalakulasingam R, Schacht RA, Lansing AM, et al. Influenza virus pneumonia after renal transplant. Postgrad Med. 1977;62(2):164-167.
13. Hoffman-La Roche Limited. Product monograph: Tamiflu. http://www.rochecanada.com/content/dam/roche_canada/en_CA/documents/Research/ClinicalTrialsForms/Products/ConsumerInformation/MonographsandPublicAdvisories/Tamiflu/Tamiflu_PM_E.pdf. Updated January 26, 2015. Accessed November 28, 2015.
14. Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
15. Palominao A, Kukoyi O, Xiong GL. Leukocytosis after lithium and clozapine combination therapy. Ann Clin Psychiatry. 2010;22(3):205-206.
16. Focosi D, Azzarà A, Kast RE, et al. Lithium and hematology: established and proposed uses. J Leukoc Biol. 2009;85(1):20-28.
17. Papetti F, Darcourt G, Giordana JY, et al. Treatment of clozapine-induced granulocytopenia with lithium (two observations) [in French]. Encephale. 2004;30(6):578-582.
18. Hummer M, Sperner-Unterweger B, Kemmler G, et al. Does eosinophilia predict clozapine induced neutropenia? Psychopharmacology (Berl). 1996;124(1-2):201-204.
19. Aneja J, Sharma N, Mahajan S, et al. Eosinophilia induced by clozapine: a report of two cases and review of the literature. J Family Med Prim Care. 2015;4(1):127-129.
20. Nielsen J, Correll CU, Manu P, et al. Termination of clozapine treatment due to medical reasons: when is it warranted and how can it be avoided? J Clin Psychiatry. 2013;74(6):603-613.
21. Stryjer R, Timinsky I, Reznik, I, et al. Beta-adrenergic antagonists for the treatment of clozapine-induced sinus tachycardia: a retrospective study. Clin Neuropharmacol. 2009;32(5):290-292.
22. Lally J, Docherty MJ, MacCabe JH. Pharmacological interventions for clozapine-induced sinus tachycardia. Cochrane Database Syst Rev. 2016;9(6):CD011566.
23. Kamphuis H, Arends J, Timmerman L, et al. Myocarditis and cardiomyopathy: underestimated complications resulting from clozapine therapy [in Dutch]. Tijdschr Psychiatr. 2010;52(4):223-233.
24. Alawami M, Wasywich C, Cicovic A, et al. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
25. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
26. Williams AM, Park SH. Seizure associated with clozapine: incidence, etiology, and management. CNS Drugs. 2015;29(2):101-111.
27. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
28. Saavedra-Velez C, Yusim A, Anbarasan D, et al. Modafinil as an adjunctive treatment of sedation, negative symptoms, and cognition in schizophrenia: a critical review. J Clin Psychiatry. 2009;70(1):104-112.
29. Klein C, Gordon J, Pollak L, et al. Clozapine in Parkinson’s disease psychosis: 5-year follow-up review. Clin Neuropharmacol. 2003;26(1):8-11.
30. Lutz UC, Sirfy A, Wiatr G, et al. Clozapine serum concentrations in dopamimetic psychosis in Parkinson’s disease and related disorders. Eur J Clin Pharmacol. 2014;70(12):1471-1476.
31. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
32. Duggal HS. Glycopyrrolate for clozapine-induced sialorrhea. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(7):1546-1547.
33. Leadbetter R, Shutty M, Pavalonis D, et al. Clozapine-induced weight gain: prevalence and clinical relevance. Am J Psychiatry. 1992;149(1):68-72.
34. Lundblad W, Azzam PN, Gopalan, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;10(8):537-543.
35. Henderson DC, Nguyen DD, Copeland PM, et al. Clozapine, diabetes mellitus, hyperlipidemia, and cardiovascular risks and mortality: results of a 10-year naturalistic study. J Clin Psychiatry. 2005;66(9):1116-1121.
36. Stroup TS, Gerhard T, Crystal S, et al. Comparative effectiveness of clozapine and standard antipsychotic treatment in adults with schizophrenia. Am J Psychiatry. 2016;173(2):166-173.
37. Carrizo E, Fernández V, Connell L, et al. Extended release metformin for metabolic control assistance during prolonged clozapine administration: a 14 week, double-blind, parallel group, placebo-controlled study. Schizophr Res. 2009;113(1):19-26.
38. Chen CH, Huang MC, Kao CF, et al. Effects of adjunctive metformin on metabolic traits in nondiabetic clozapine-treated patients with schizophrenia and the effect of metformin discontinuation on body weight: a 24-week, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(5):e424-e430.
39. Mizuno Y, Suzuki T, Nakagawa A, et al. Pharmacological strategies to counteract antipsychotic-induced weight gain and metabolic adverse effects in schizophrenia: a systematic review and meta-analysis. Schizophr Bull. 2014;40(6):1385-1403.
40. Nielsen J, Meyer JM. Risk factors for ileus in patients with schizophrenia. Schizophr Bull. 2012;38(3):592-598.
41. Lowe CM, Grube RR, Scates AC. Characterization and clinical management of clozapine-induced fever. Ann Pharmacother. 2007;41(10):1700-1704.
42. ACOG Committee on Practice Bulletins–Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces practice bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol. 2008;111(4):1001-1020.
43. U.S. Food and Drug Administration. Pregnancy and Lactation Labeling (Drugs) Final Rule. https://s3.amazonaws.com/public-inspection.federalregister.gov/2014-28241.pdf. Published December 4, 2014. Accessed July 6, 2016.
44. Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: a reference guide to fetal and neonatal risk. 9th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2011.
45. Larsen ER, Damkier P, Pedersen LH, et al; Danish Psychiatric Society; Danish Society of Obstetrics and Gynecology; Danish Paediatric Society; Danish Society of Clinical Pharmacology. Use of psychotropic drugs during pregnancy and breast-feeding. Acta Psychiatr Scand Suppl. 2015;(445):1-28.
46. McKenna K, Koren G, Tetelbaum M, et al. Pregnancy outcome of women using atypical antipsychotic drugs: a prospective comparative study. J Clin Psychiatry. 2005;66(4):444-449.
Clozapine is a highly effective antipsychotic with superior efficacy in treatment-resistant schizophrenia, but its side effect profile is daunting (Figure 1).1 Adverse reactions lead to approximately 17% of patients who take clozapine eventually discontinuing the medication.1 As we noted in Part 1 of this 3-part series,2 clozapine remains the most efficacious, but most tedious, antipsychotic available to psychiatrists because of its monitoring requirements and potential side effects.
A powerful rationale for prescribing clozapine, despite its drawbacks, is its association with a reduced risk of all-cause mortality.3,4 People with serious mental illness, including schizophrenia, have a median 10-year shorter life expectancy than the general population.5
A recent cohort study6 examined electronic health records to test whether intensive monitoring or lower suicide risk might account for the reduced mortality with clozapine. The authors found that the reduced mortality rate was not directly related to clozapine’s clinical monitoring or other confounding factors. They did find an association between clozapine use and reduced risk of death from both natural and unnatural causes.
This second article in our series examines clozapine’s adverse effects from a systems perspective. Severe neutropenia, myocarditis, sedation, weight gain, orthostatic hypotension, and sialorrhea appear to be the most studied adverse effects, but myriad others can occur.7 We offer guidance to help the astute clinician continue this effective antipsychotic by monitoring carefully, treating side effects early, and managing potential drug interactions (Table 1).8
Hematologic eventsSevere neutropenia, defined as absolute neutrophil count (ANC) <500/µL, is a well-known adverse effect of clozapine that requires specific clinical monitoring, a requirement that was updated by the FDA in 2015.2 The incidence of severe neutropenia peaks in the first 2 months of clozapine therapy and tapers after 6 months, but some risk always remains.
Older efficacy studies in the United States gauged the 1-year cumulative incidence of severe clozapine-induced neutropenia to be 2%.9 A 1998 study of the effects of using a clozapine registry reported a lower incidence—0.38%—than the 2% noted above.10 Early recognition and recommended interventions can improve clinical outcomes.2
Drug interactions and neutropenia. A retrospective study of mental health inpatients taking clozapine concurrently with oseltamivir during an influenza outbreak found a statistically significant—but not clinically significant—change in ANC values.11 The authors noted that viral infection might lead to blood dyscrasia early in illness, and that oseltamivir has been associated with a small incidence of blood dyscrasia.11-13 This information might be useful when treating influenza in patients taking clozapine, although no specific change in management is recommended.
Similarly, concomitant treatment with clozapine and lithium can affect both white blood cell and ANC values.14,15 Lithium-treated patients often demonstrate increased circulating neutrophils via enhancement of granulocyte-colony stimulating factor.16 Case studies describe how initiating lithium treatment enabled some patients to continue clozapine after developing neutropenia.14,17 Leukocytosis can affect blood monitoring, possibly masking other blood dyscrasias, when lithium is used concurrently with clozapine.
Eosinophilia (blood eosinophil count >700/µL) occurs in approximately 1% of clozapine users, usually in the first 4 weeks of treatment.18 It can be benign and transient or a harbinger of a more rare adverse reaction such as myocarditis, pancreatitis, hepatitis, colitis, or nephritis.19 If a patient taking clozapine develops eosinophilia, clozapine’s package insert recommends that you:
- evaluate promptly for other systemic involvement (rash, other evidence of allergic reaction, myocarditis, other organ-specific disease)
- stop clozapine immediately if any of these are found.
If other causes of eosinophilia are identified (asthma, allergies, collagen vascular disease, parasitic infection, neoplasm), treat these and continue clozapine.
The manufacturer also mentions the occurrence of clozapine-related eosinophilia without organ involvement that can resolve without intervention, with careful monitoring over several weeks.8 In this scenario, there is flexibility to judge whether clozapine should be stopped or re-challenged, or if close monitoring is adequate. Consulting with an internal medicine or hematology specialist might be helpful.
Cardiovascular side effectsMost common events. Three of the 10 most common clozapine side effects are cardiac: tachycardia, hypotension, and hypertension (Figure 1).1 Orthostatic hypotension, bradycardia, and syncope also can occur, especially with rapid clozapine titration. Baseline electrocardiogram (ECG) can help differentiate whether abnormalities are clozapine-induced or related to a preexisting condition.
Reducing the dosage of clozapine or slowing titration could reverse cardiac side effects.8 If dosage reduction is not an option or is ineffective, first consider treating the side effect rather than discontinuing clozapine.20
Sinus tachycardia is one of the most common side effects of clozapine. First, rule out serious conditions—myocarditis, cardiomyopathy, neuroleptic malignant syndrome (NMS)—then consider waiting and monitoring for the first few months of clozapine treatment. If tachycardia continues, consider dosage reduction. Slower titration, or treatment with a cardio-selective beta blocker such as atenolol.21,22 Note that a recent Cochrane Review concluded that there is not enough randomized evidence to support any particular treatment for clozapine-induced tachycardia; the prescriber must therefore make a case-by-case clinical judgment.22
Similarly, orthostatic hypotension can be managed with a reduced dosage of clozapine or slower titration. Increased fluid intake, compression stockings, and, if necessary, fludrocortisone also can be initiated.20
Rare, potentially fatal events. Myocarditis, pericarditis, and cardiomyopathy are among the rare but potentially fatal adverse effects of clozapine. A recent study reported the incidence of myocarditis with clozapine at a range of 0.015% to 1.3%; cardiomyopathy was even more rare.23 Pulmonary embolism and deep venous thrombosis also are very rare possibilities; keep them in mind, however, when patients taking clozapine report new cardiovascular symptoms.
Patients with clozapine-induced cardiovascular effects most commonly report shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).7,24
Clozapine’s “black-box” warning specifically recommends discontinuing clozapine and consulting cardiology when myocarditis or cardiomyopathy is suspected. In 50% of cases, myocarditis symptoms present in the first few weeks of clozapine treatment.23 The manufacturer states that myocarditis usually presents in the first 2 months, and cardiomyopathy after 8 weeks of treatment; however, either can present at any time.8Figure 2 provides a clinical reference for monitoring a clozapine patient for cardiomyopathy.24
Laboratory findings that support a diagnosis of clozapine-related myocarditis include:
- elevated C-reactive protein
- elevated troponin I or T
- elevated creatine kinase-MB
- peripheral eosinophilia.8,25
ECG, echocardiography, and cardiac MRI can be helpful in diagnosis, in consultation with a cardiologist.
Neurologic side effectsSeizures are listed in the “black-box” warning for clozapine. Seizure incidence with clozapine is 5% per year, with higher incidence at dosages ≥600 mg/d.8 Because clozapine-induced seizures are dosage-dependent, slow titration can mitigate this risk. Tonic-clonic seizures are the most common type associated with clozapine.
The manufacturer recommends caution when using clozapine in patients with a known seizure disorder, alcohol use disorder, or other CNS pathology.8 Patients with a seizure disorder may be at increased risk of experiencing clozapine-induced seizures, but this is not an absolute contraindication.26 Smoking cessation increases clozapine blood levels by an average of 57.4%, further increasing seizure risk.26,27
Discontinuing clozapine is unnecessary when a patient experiences a seizure. Instead, you can:
- halve the dosage prescribed at the time of the seizure (or at least reduce to the last seizure-free dosage)
- consider any medications or medical problems that might have contributed to a lower seizure threshold
- consider prophylaxis with an antiepileptic medication (eg, valproic acid has efficacy for both myoclonic and tonic-clonic seizures).20,26
Sedation is the most common side effect of clozapine.1 Patients experiencing severe sedation should not drive or operate heavy machinery. To reduce sedation, consider instructing the patient to take all or most of the clozapine dosage at bedtime. A critical review of modafinil for sedation caused by antipsychotics in schizophrenia found only 1 open-label study that showed any positive effects; the authors concluded that further study is needed.28
Cognitive and motor slowing are possible neurologic side effects of clozapine. Caution patients about the risk of participating in activities that require cognitive or motor performance until the individual effects of clozapine are known.8
Tardive dyskinesia. Clozapine carries some risk of tardive dyskinesia, although that risk is lower than with other antipsychotics. Similarly, all antipsychotics including clozapine are associated with a risk of NMS. In the rare case of clozapine-induced NMS, stop clozapine immediately and initiate supportive therapy. Clozapine-induced NMS is not an absolute contraindication to re-challenging a patient with clozapine, however, if doing so is clinically appropriate.20
Cerebrovascular events. In older people with dementia, the use of antipsychotics—including clozapine—has been shown to increase the risk of cerebrovascular events. Because most antipsychotics are not FDA-approved for treating psychosis associated with dementia (only pimavanserin is FDA-approved for symptoms of psychosis in Parkinson’s disease), a risk-benefit analysis should be documented when prescribing any antipsychotic in this population. In practice, clozapine’s benefits may outweigh the mortality risks in specific situations.29,30
CASE Sialorrhea puts progress at risk
Ms. B, age 40, has a history of treatment-resistant schizophrenia and is starting clozapine because of residual psychosis during trials of other antipsychotics. She develops severe persistent drooling, mostly at night, during clozapine titration. Sugar-free candy, multiple bed pillows, and changing the dosing schedule do not significantly improve the sialorrhea.
As a result, Ms. B is embarrassed to continue her usual activities. She asks to stop clozapine, even though her psychotic symptoms have improved and she is functioning at her highest level in years.
Ms. B already is taking trihexyphenidyl, 5 mg, 3 times daily, to manage extrapyramidal symptoms related to haloperidol decanoate treatment. After discussing other medication options for sialorrhea, she agrees to a trial of glycopyrrolate, 1 mg, twice daily. She experiences significant improvement and continues taking clozapine.
Sialorrhea develops in 13% of patients taking clozapine.1 As in Ms. B’s case, this side effect can be embarrassing, can limit social or occupational functioning, and might lead patients to discontinue clozapine treatment despite efficacy. Nonpharmacotherapeutic options include covering the pillow with a towel, lowering the clozapine dosage or titrating slowly (or both), and using sugarless gum or candy to increase swallowing.
If the benefits of additional medications targeting side effects outweigh the risks, pharmacotherapeutic intervention may be appropriate. Options include the tricyclic antidepressant amitriptyline31; alpha-adrenergic agonists or antagonists (clonidine, terazosin); and anti-muscarinic medications (benztropine, atropine, trihexyphenidyl, glycopyrrolate) (Table 231). Scopolamine transdermal patch is another possible treatment strategy; however, the scopolamine patch was used for clozapine-induced sialorrhea in only a few case reports, and it is not considered a first-line treatment choice.30
When prescribing, consider the possibility of combined side effects with clozapine and adjunct medications having antimuscarinic or alpha-adrenergic activity, or both. Even atropine ophthalmic drops, administered sublingually, are readily absorbed and cross the blood–brain barrier.31 Another antimuscarinic agent, glycopyrrolate, is less likely to cross the blood–brain barrier and therefore is less likely to cause cognitive side effects. Glycopyrrolate is 5 times more potent at blocking the muscarinic receptor than atropine.31,32 Ipratropium bromide, another nonselective muscarinic receptor antagonist, has less systemic absorption than atropine drops, with less anticholinergic side effects when administered sublingually.
Limited evidence supports the efficacy of alpha-adrenergic medications for managing clozapine-induced sialorrhea. Monitor blood pressure when prescribing terazosin or clonidine, which could potentiate clozapine’s hypotensive effects.
Endocrine side effectsAmong antipsychotics, clozapine is associated with the greatest weight gain—averaging nearly 10% of body weight.33,34 Similarly, the risk of new-onset diabetes mellitus is highest with clozapine in relation to other antipsychotics: 43% reported in a 10-year naturalistic study.35 The risk of hyperlipidemia also increases with clozapine treatment.36 These metabolic changes increase the risk of cardiovascular-related death, with a 10-year mortality rate from cardiovascular disease reported at 9% in clozapine-treated patients.35
Despite clozapine’s metabolic side effects, patients with schizophrenia who are treated with clozapine show a significant reduction in overall mortality compared with patients not treated with clozapine.6 Effective identification and management of metabolic side effects can prevent the need to discontinue clozapine.
Behavioral weight management and exercise are recommended as initial therapy.20 If, based on clinical judgment, these alone are insufficient, data support the use of pharmacotherapeutic interventions. Metformin demonstrates a positive effect on body weight, insulin resistance, and lipids, making it the first choice for adjunctive treatment of clozapine-induced metabolic side effects.37-39
Gastrointestinal side effectsClozapine’s anticholinergic activity can lead to serious gastrointestinal (GI) side effects, including constipation, intestinal obstruction, fecal impaction, and paralytic ileus.8 Ileus has produced more fatal adverse reactions with clozapine than has severe neutropenia.20,40 Co-administered anticholinergic medications could increase the risk of ileus. Obtaining a GI review of systems and monitoring bowel movements (in inpatient or residential facilities) can aid in early identification and limit morbidity and mortality from GI adverse events. A high-fiber diet, adequate hydration, bulk laxatives in patients who can reliably maintain hydration, and GI consultation (if needed) may help manage GI side effects.20
Constitutional side effectsFever can occur with clozapine, most often in the first month of treatment, but the incidence is quite variable (0.5% to 55%).20,41 Although benign fever is common, agranulocytosis with infection, NMS, and other systemic illness must be ruled out. The recommended workup when a patient develops fever while taking clozapine includes physical examination and relevant testing (urinalysis, measurement of ANC and serum creatine kinase, chest radiograph, ECG, and, possibly, blood cultures).41
If evidence supports a serious adverse reaction, stop clozapine immediately.20 If benign clozapine-related fever is suspected, acetaminophen or another antipyretic might provide symptomatic relief; discontinuing clozapine is then unnecessary.41
Pregnancy. When a patient with schizophrenia requires clozapine treatment during pregnancy, reliable clinical guidance is limited. The American College of Obstetricians and Gynecologists Practice Bulletin on the use of psychiatric medications during pregnancy and lactation can be a useful resource.42
Be aware that the FDA very recently made major changes to the format and content of pregnancy and lactation labeling, removing the letter categories that have been used for medications approved on or after June 30, 2001. The manufacturers of medications (such as clozapine) that were approved before June 30, 2001, have 3 years to comply with new requirements.43
The FDA had rated clozapine a pregnancy risk category B medication, meaning no evidence of risk in humans. In 2011, the FDA issued a general warning that antipsychotic use in pregnancy can cause extrapyramidal symptoms and discontinuation symptoms in newborns.44,45
A 2015 review of psychotropic medications and pregnancy noted that approximately 60% of women with schizophrenia became pregnant, with an increased incidence of unplanned pregnancy. A high risk of psychotic relapse (65%) during pregnancy and in the postpartum period may lead to insufficient prenatal care, drug use, and obstetric complications.45 Some data suggest low fetal birth weight and an increased rate of therapeutic abortions in women with schizophrenia.42,46
When treating a pregnant patient, weigh the benefits of clozapine against the risks of adverse events, and clearly document the analysis. Clozapine treatment is not recommended during breast-feeding because of the risk of side effects for newborns.8
We highly recommend keeping updated on the literature regarding pregnancy and lactation information with antipsychotics, including clozapine, because prescribing information will likely be updated in the near future to comply with recent FDA labeling changes.
Final installment: Using clozapine off-labelClozapine is FDA-approved for refractory schizophrenia and for reducing the risk of recurrent suicidal behavior in schizophrenia or schizoaffective disorder. In Part 3 of this series, we review off-label uses—such as managing bipolar disorder, borderline personality disorder, and aggressive behavior—that have varying degrees of scientific support.
Clozapine is a highly effective antipsychotic with superior efficacy in treatment-resistant schizophrenia, but its side effect profile is daunting (Figure 1).1 Adverse reactions lead to approximately 17% of patients who take clozapine eventually discontinuing the medication.1 As we noted in Part 1 of this 3-part series,2 clozapine remains the most efficacious, but most tedious, antipsychotic available to psychiatrists because of its monitoring requirements and potential side effects.
A powerful rationale for prescribing clozapine, despite its drawbacks, is its association with a reduced risk of all-cause mortality.3,4 People with serious mental illness, including schizophrenia, have a median 10-year shorter life expectancy than the general population.5
A recent cohort study6 examined electronic health records to test whether intensive monitoring or lower suicide risk might account for the reduced mortality with clozapine. The authors found that the reduced mortality rate was not directly related to clozapine’s clinical monitoring or other confounding factors. They did find an association between clozapine use and reduced risk of death from both natural and unnatural causes.
This second article in our series examines clozapine’s adverse effects from a systems perspective. Severe neutropenia, myocarditis, sedation, weight gain, orthostatic hypotension, and sialorrhea appear to be the most studied adverse effects, but myriad others can occur.7 We offer guidance to help the astute clinician continue this effective antipsychotic by monitoring carefully, treating side effects early, and managing potential drug interactions (Table 1).8
Hematologic eventsSevere neutropenia, defined as absolute neutrophil count (ANC) <500/µL, is a well-known adverse effect of clozapine that requires specific clinical monitoring, a requirement that was updated by the FDA in 2015.2 The incidence of severe neutropenia peaks in the first 2 months of clozapine therapy and tapers after 6 months, but some risk always remains.
Older efficacy studies in the United States gauged the 1-year cumulative incidence of severe clozapine-induced neutropenia to be 2%.9 A 1998 study of the effects of using a clozapine registry reported a lower incidence—0.38%—than the 2% noted above.10 Early recognition and recommended interventions can improve clinical outcomes.2
Drug interactions and neutropenia. A retrospective study of mental health inpatients taking clozapine concurrently with oseltamivir during an influenza outbreak found a statistically significant—but not clinically significant—change in ANC values.11 The authors noted that viral infection might lead to blood dyscrasia early in illness, and that oseltamivir has been associated with a small incidence of blood dyscrasia.11-13 This information might be useful when treating influenza in patients taking clozapine, although no specific change in management is recommended.
Similarly, concomitant treatment with clozapine and lithium can affect both white blood cell and ANC values.14,15 Lithium-treated patients often demonstrate increased circulating neutrophils via enhancement of granulocyte-colony stimulating factor.16 Case studies describe how initiating lithium treatment enabled some patients to continue clozapine after developing neutropenia.14,17 Leukocytosis can affect blood monitoring, possibly masking other blood dyscrasias, when lithium is used concurrently with clozapine.
Eosinophilia (blood eosinophil count >700/µL) occurs in approximately 1% of clozapine users, usually in the first 4 weeks of treatment.18 It can be benign and transient or a harbinger of a more rare adverse reaction such as myocarditis, pancreatitis, hepatitis, colitis, or nephritis.19 If a patient taking clozapine develops eosinophilia, clozapine’s package insert recommends that you:
- evaluate promptly for other systemic involvement (rash, other evidence of allergic reaction, myocarditis, other organ-specific disease)
- stop clozapine immediately if any of these are found.
If other causes of eosinophilia are identified (asthma, allergies, collagen vascular disease, parasitic infection, neoplasm), treat these and continue clozapine.
The manufacturer also mentions the occurrence of clozapine-related eosinophilia without organ involvement that can resolve without intervention, with careful monitoring over several weeks.8 In this scenario, there is flexibility to judge whether clozapine should be stopped or re-challenged, or if close monitoring is adequate. Consulting with an internal medicine or hematology specialist might be helpful.
Cardiovascular side effectsMost common events. Three of the 10 most common clozapine side effects are cardiac: tachycardia, hypotension, and hypertension (Figure 1).1 Orthostatic hypotension, bradycardia, and syncope also can occur, especially with rapid clozapine titration. Baseline electrocardiogram (ECG) can help differentiate whether abnormalities are clozapine-induced or related to a preexisting condition.
Reducing the dosage of clozapine or slowing titration could reverse cardiac side effects.8 If dosage reduction is not an option or is ineffective, first consider treating the side effect rather than discontinuing clozapine.20
Sinus tachycardia is one of the most common side effects of clozapine. First, rule out serious conditions—myocarditis, cardiomyopathy, neuroleptic malignant syndrome (NMS)—then consider waiting and monitoring for the first few months of clozapine treatment. If tachycardia continues, consider dosage reduction. Slower titration, or treatment with a cardio-selective beta blocker such as atenolol.21,22 Note that a recent Cochrane Review concluded that there is not enough randomized evidence to support any particular treatment for clozapine-induced tachycardia; the prescriber must therefore make a case-by-case clinical judgment.22
Similarly, orthostatic hypotension can be managed with a reduced dosage of clozapine or slower titration. Increased fluid intake, compression stockings, and, if necessary, fludrocortisone also can be initiated.20
Rare, potentially fatal events. Myocarditis, pericarditis, and cardiomyopathy are among the rare but potentially fatal adverse effects of clozapine. A recent study reported the incidence of myocarditis with clozapine at a range of 0.015% to 1.3%; cardiomyopathy was even more rare.23 Pulmonary embolism and deep venous thrombosis also are very rare possibilities; keep them in mind, however, when patients taking clozapine report new cardiovascular symptoms.
Patients with clozapine-induced cardiovascular effects most commonly report shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).7,24
Clozapine’s “black-box” warning specifically recommends discontinuing clozapine and consulting cardiology when myocarditis or cardiomyopathy is suspected. In 50% of cases, myocarditis symptoms present in the first few weeks of clozapine treatment.23 The manufacturer states that myocarditis usually presents in the first 2 months, and cardiomyopathy after 8 weeks of treatment; however, either can present at any time.8Figure 2 provides a clinical reference for monitoring a clozapine patient for cardiomyopathy.24
Laboratory findings that support a diagnosis of clozapine-related myocarditis include:
- elevated C-reactive protein
- elevated troponin I or T
- elevated creatine kinase-MB
- peripheral eosinophilia.8,25
ECG, echocardiography, and cardiac MRI can be helpful in diagnosis, in consultation with a cardiologist.
Neurologic side effectsSeizures are listed in the “black-box” warning for clozapine. Seizure incidence with clozapine is 5% per year, with higher incidence at dosages ≥600 mg/d.8 Because clozapine-induced seizures are dosage-dependent, slow titration can mitigate this risk. Tonic-clonic seizures are the most common type associated with clozapine.
The manufacturer recommends caution when using clozapine in patients with a known seizure disorder, alcohol use disorder, or other CNS pathology.8 Patients with a seizure disorder may be at increased risk of experiencing clozapine-induced seizures, but this is not an absolute contraindication.26 Smoking cessation increases clozapine blood levels by an average of 57.4%, further increasing seizure risk.26,27
Discontinuing clozapine is unnecessary when a patient experiences a seizure. Instead, you can:
- halve the dosage prescribed at the time of the seizure (or at least reduce to the last seizure-free dosage)
- consider any medications or medical problems that might have contributed to a lower seizure threshold
- consider prophylaxis with an antiepileptic medication (eg, valproic acid has efficacy for both myoclonic and tonic-clonic seizures).20,26
Sedation is the most common side effect of clozapine.1 Patients experiencing severe sedation should not drive or operate heavy machinery. To reduce sedation, consider instructing the patient to take all or most of the clozapine dosage at bedtime. A critical review of modafinil for sedation caused by antipsychotics in schizophrenia found only 1 open-label study that showed any positive effects; the authors concluded that further study is needed.28
Cognitive and motor slowing are possible neurologic side effects of clozapine. Caution patients about the risk of participating in activities that require cognitive or motor performance until the individual effects of clozapine are known.8
Tardive dyskinesia. Clozapine carries some risk of tardive dyskinesia, although that risk is lower than with other antipsychotics. Similarly, all antipsychotics including clozapine are associated with a risk of NMS. In the rare case of clozapine-induced NMS, stop clozapine immediately and initiate supportive therapy. Clozapine-induced NMS is not an absolute contraindication to re-challenging a patient with clozapine, however, if doing so is clinically appropriate.20
Cerebrovascular events. In older people with dementia, the use of antipsychotics—including clozapine—has been shown to increase the risk of cerebrovascular events. Because most antipsychotics are not FDA-approved for treating psychosis associated with dementia (only pimavanserin is FDA-approved for symptoms of psychosis in Parkinson’s disease), a risk-benefit analysis should be documented when prescribing any antipsychotic in this population. In practice, clozapine’s benefits may outweigh the mortality risks in specific situations.29,30
CASE Sialorrhea puts progress at risk
Ms. B, age 40, has a history of treatment-resistant schizophrenia and is starting clozapine because of residual psychosis during trials of other antipsychotics. She develops severe persistent drooling, mostly at night, during clozapine titration. Sugar-free candy, multiple bed pillows, and changing the dosing schedule do not significantly improve the sialorrhea.
As a result, Ms. B is embarrassed to continue her usual activities. She asks to stop clozapine, even though her psychotic symptoms have improved and she is functioning at her highest level in years.
Ms. B already is taking trihexyphenidyl, 5 mg, 3 times daily, to manage extrapyramidal symptoms related to haloperidol decanoate treatment. After discussing other medication options for sialorrhea, she agrees to a trial of glycopyrrolate, 1 mg, twice daily. She experiences significant improvement and continues taking clozapine.
Sialorrhea develops in 13% of patients taking clozapine.1 As in Ms. B’s case, this side effect can be embarrassing, can limit social or occupational functioning, and might lead patients to discontinue clozapine treatment despite efficacy. Nonpharmacotherapeutic options include covering the pillow with a towel, lowering the clozapine dosage or titrating slowly (or both), and using sugarless gum or candy to increase swallowing.
If the benefits of additional medications targeting side effects outweigh the risks, pharmacotherapeutic intervention may be appropriate. Options include the tricyclic antidepressant amitriptyline31; alpha-adrenergic agonists or antagonists (clonidine, terazosin); and anti-muscarinic medications (benztropine, atropine, trihexyphenidyl, glycopyrrolate) (Table 231). Scopolamine transdermal patch is another possible treatment strategy; however, the scopolamine patch was used for clozapine-induced sialorrhea in only a few case reports, and it is not considered a first-line treatment choice.30
When prescribing, consider the possibility of combined side effects with clozapine and adjunct medications having antimuscarinic or alpha-adrenergic activity, or both. Even atropine ophthalmic drops, administered sublingually, are readily absorbed and cross the blood–brain barrier.31 Another antimuscarinic agent, glycopyrrolate, is less likely to cross the blood–brain barrier and therefore is less likely to cause cognitive side effects. Glycopyrrolate is 5 times more potent at blocking the muscarinic receptor than atropine.31,32 Ipratropium bromide, another nonselective muscarinic receptor antagonist, has less systemic absorption than atropine drops, with less anticholinergic side effects when administered sublingually.
Limited evidence supports the efficacy of alpha-adrenergic medications for managing clozapine-induced sialorrhea. Monitor blood pressure when prescribing terazosin or clonidine, which could potentiate clozapine’s hypotensive effects.
Endocrine side effectsAmong antipsychotics, clozapine is associated with the greatest weight gain—averaging nearly 10% of body weight.33,34 Similarly, the risk of new-onset diabetes mellitus is highest with clozapine in relation to other antipsychotics: 43% reported in a 10-year naturalistic study.35 The risk of hyperlipidemia also increases with clozapine treatment.36 These metabolic changes increase the risk of cardiovascular-related death, with a 10-year mortality rate from cardiovascular disease reported at 9% in clozapine-treated patients.35
Despite clozapine’s metabolic side effects, patients with schizophrenia who are treated with clozapine show a significant reduction in overall mortality compared with patients not treated with clozapine.6 Effective identification and management of metabolic side effects can prevent the need to discontinue clozapine.
Behavioral weight management and exercise are recommended as initial therapy.20 If, based on clinical judgment, these alone are insufficient, data support the use of pharmacotherapeutic interventions. Metformin demonstrates a positive effect on body weight, insulin resistance, and lipids, making it the first choice for adjunctive treatment of clozapine-induced metabolic side effects.37-39
Gastrointestinal side effectsClozapine’s anticholinergic activity can lead to serious gastrointestinal (GI) side effects, including constipation, intestinal obstruction, fecal impaction, and paralytic ileus.8 Ileus has produced more fatal adverse reactions with clozapine than has severe neutropenia.20,40 Co-administered anticholinergic medications could increase the risk of ileus. Obtaining a GI review of systems and monitoring bowel movements (in inpatient or residential facilities) can aid in early identification and limit morbidity and mortality from GI adverse events. A high-fiber diet, adequate hydration, bulk laxatives in patients who can reliably maintain hydration, and GI consultation (if needed) may help manage GI side effects.20
Constitutional side effectsFever can occur with clozapine, most often in the first month of treatment, but the incidence is quite variable (0.5% to 55%).20,41 Although benign fever is common, agranulocytosis with infection, NMS, and other systemic illness must be ruled out. The recommended workup when a patient develops fever while taking clozapine includes physical examination and relevant testing (urinalysis, measurement of ANC and serum creatine kinase, chest radiograph, ECG, and, possibly, blood cultures).41
If evidence supports a serious adverse reaction, stop clozapine immediately.20 If benign clozapine-related fever is suspected, acetaminophen or another antipyretic might provide symptomatic relief; discontinuing clozapine is then unnecessary.41
Pregnancy. When a patient with schizophrenia requires clozapine treatment during pregnancy, reliable clinical guidance is limited. The American College of Obstetricians and Gynecologists Practice Bulletin on the use of psychiatric medications during pregnancy and lactation can be a useful resource.42
Be aware that the FDA very recently made major changes to the format and content of pregnancy and lactation labeling, removing the letter categories that have been used for medications approved on or after June 30, 2001. The manufacturers of medications (such as clozapine) that were approved before June 30, 2001, have 3 years to comply with new requirements.43
The FDA had rated clozapine a pregnancy risk category B medication, meaning no evidence of risk in humans. In 2011, the FDA issued a general warning that antipsychotic use in pregnancy can cause extrapyramidal symptoms and discontinuation symptoms in newborns.44,45
A 2015 review of psychotropic medications and pregnancy noted that approximately 60% of women with schizophrenia became pregnant, with an increased incidence of unplanned pregnancy. A high risk of psychotic relapse (65%) during pregnancy and in the postpartum period may lead to insufficient prenatal care, drug use, and obstetric complications.45 Some data suggest low fetal birth weight and an increased rate of therapeutic abortions in women with schizophrenia.42,46
When treating a pregnant patient, weigh the benefits of clozapine against the risks of adverse events, and clearly document the analysis. Clozapine treatment is not recommended during breast-feeding because of the risk of side effects for newborns.8
We highly recommend keeping updated on the literature regarding pregnancy and lactation information with antipsychotics, including clozapine, because prescribing information will likely be updated in the near future to comply with recent FDA labeling changes.
Final installment: Using clozapine off-labelClozapine is FDA-approved for refractory schizophrenia and for reducing the risk of recurrent suicidal behavior in schizophrenia or schizoaffective disorder. In Part 3 of this series, we review off-label uses—such as managing bipolar disorder, borderline personality disorder, and aggressive behavior—that have varying degrees of scientific support.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Newman WJ, Newman BM. Rediscovering clozapine: after a turbulent history, current guidance on initiating and monitoring. Current Psychiatry. 2016;15(7):42-46,48-49.
3. Walker AM, Lanza LL, Arellano F, et al. Mortality in current and former users of clozapine. Epidemiology. 1997;8(6):671-677.
4. Tiihonen J, Lönnqvist J, Wahlbeck K, et al. 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort study (FIN11 study). Lancet. 2009;374(9690):620-627.
5. Walker E, McGee RE, Druss BG. Mortality in mental disorders and global disease burden Implications: a systematic review and meta-analysis. JAMA Psychiatry. 2015;72(4):334-341.
6. Hayes RD, Downs J, Chang CK, et al. The effect of clozapine on premature mortality: an assessment of clinical monitoring and other potential confounders. Schizophr Bull. 2015;41(3):644-655.
7. De Fazio P, Gaetano R, Caroleo M, et al. Rare and very rare adverse effects of clozapine. Neuropsychiatr Dis Treat. 2015;11:1995-2003.
8. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 29, 2016.
9. Lieberman JA, Johns CA, Kane JM, et al. Clozapine-induced agranulocytosis: non-cross-reactivity with other psychotropic drugs. J Clin Psychiatry. 1988;49(7):271-277.
10. Honigfeld G, Arellano F, Sethi J, et al. Reducing clozapine-related morbidity and mortality: 5 years of experience with the Clozaril National Registry. J Clin Psychiatry. 1998;59(suppl 3):3-7.
11. Demler TL, Trigoboff E. Are clozapine patients at risk for blood dyscrasias with concomitant tamiflu use? Psychiatry (Edgmont). 2009;6(11):29-33.
12. Karalakulasingam R, Schacht RA, Lansing AM, et al. Influenza virus pneumonia after renal transplant. Postgrad Med. 1977;62(2):164-167.
13. Hoffman-La Roche Limited. Product monograph: Tamiflu. http://www.rochecanada.com/content/dam/roche_canada/en_CA/documents/Research/ClinicalTrialsForms/Products/ConsumerInformation/MonographsandPublicAdvisories/Tamiflu/Tamiflu_PM_E.pdf. Updated January 26, 2015. Accessed November 28, 2015.
14. Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
15. Palominao A, Kukoyi O, Xiong GL. Leukocytosis after lithium and clozapine combination therapy. Ann Clin Psychiatry. 2010;22(3):205-206.
16. Focosi D, Azzarà A, Kast RE, et al. Lithium and hematology: established and proposed uses. J Leukoc Biol. 2009;85(1):20-28.
17. Papetti F, Darcourt G, Giordana JY, et al. Treatment of clozapine-induced granulocytopenia with lithium (two observations) [in French]. Encephale. 2004;30(6):578-582.
18. Hummer M, Sperner-Unterweger B, Kemmler G, et al. Does eosinophilia predict clozapine induced neutropenia? Psychopharmacology (Berl). 1996;124(1-2):201-204.
19. Aneja J, Sharma N, Mahajan S, et al. Eosinophilia induced by clozapine: a report of two cases and review of the literature. J Family Med Prim Care. 2015;4(1):127-129.
20. Nielsen J, Correll CU, Manu P, et al. Termination of clozapine treatment due to medical reasons: when is it warranted and how can it be avoided? J Clin Psychiatry. 2013;74(6):603-613.
21. Stryjer R, Timinsky I, Reznik, I, et al. Beta-adrenergic antagonists for the treatment of clozapine-induced sinus tachycardia: a retrospective study. Clin Neuropharmacol. 2009;32(5):290-292.
22. Lally J, Docherty MJ, MacCabe JH. Pharmacological interventions for clozapine-induced sinus tachycardia. Cochrane Database Syst Rev. 2016;9(6):CD011566.
23. Kamphuis H, Arends J, Timmerman L, et al. Myocarditis and cardiomyopathy: underestimated complications resulting from clozapine therapy [in Dutch]. Tijdschr Psychiatr. 2010;52(4):223-233.
24. Alawami M, Wasywich C, Cicovic A, et al. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
25. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
26. Williams AM, Park SH. Seizure associated with clozapine: incidence, etiology, and management. CNS Drugs. 2015;29(2):101-111.
27. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
28. Saavedra-Velez C, Yusim A, Anbarasan D, et al. Modafinil as an adjunctive treatment of sedation, negative symptoms, and cognition in schizophrenia: a critical review. J Clin Psychiatry. 2009;70(1):104-112.
29. Klein C, Gordon J, Pollak L, et al. Clozapine in Parkinson’s disease psychosis: 5-year follow-up review. Clin Neuropharmacol. 2003;26(1):8-11.
30. Lutz UC, Sirfy A, Wiatr G, et al. Clozapine serum concentrations in dopamimetic psychosis in Parkinson’s disease and related disorders. Eur J Clin Pharmacol. 2014;70(12):1471-1476.
31. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
32. Duggal HS. Glycopyrrolate for clozapine-induced sialorrhea. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(7):1546-1547.
33. Leadbetter R, Shutty M, Pavalonis D, et al. Clozapine-induced weight gain: prevalence and clinical relevance. Am J Psychiatry. 1992;149(1):68-72.
34. Lundblad W, Azzam PN, Gopalan, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;10(8):537-543.
35. Henderson DC, Nguyen DD, Copeland PM, et al. Clozapine, diabetes mellitus, hyperlipidemia, and cardiovascular risks and mortality: results of a 10-year naturalistic study. J Clin Psychiatry. 2005;66(9):1116-1121.
36. Stroup TS, Gerhard T, Crystal S, et al. Comparative effectiveness of clozapine and standard antipsychotic treatment in adults with schizophrenia. Am J Psychiatry. 2016;173(2):166-173.
37. Carrizo E, Fernández V, Connell L, et al. Extended release metformin for metabolic control assistance during prolonged clozapine administration: a 14 week, double-blind, parallel group, placebo-controlled study. Schizophr Res. 2009;113(1):19-26.
38. Chen CH, Huang MC, Kao CF, et al. Effects of adjunctive metformin on metabolic traits in nondiabetic clozapine-treated patients with schizophrenia and the effect of metformin discontinuation on body weight: a 24-week, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(5):e424-e430.
39. Mizuno Y, Suzuki T, Nakagawa A, et al. Pharmacological strategies to counteract antipsychotic-induced weight gain and metabolic adverse effects in schizophrenia: a systematic review and meta-analysis. Schizophr Bull. 2014;40(6):1385-1403.
40. Nielsen J, Meyer JM. Risk factors for ileus in patients with schizophrenia. Schizophr Bull. 2012;38(3):592-598.
41. Lowe CM, Grube RR, Scates AC. Characterization and clinical management of clozapine-induced fever. Ann Pharmacother. 2007;41(10):1700-1704.
42. ACOG Committee on Practice Bulletins–Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces practice bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol. 2008;111(4):1001-1020.
43. U.S. Food and Drug Administration. Pregnancy and Lactation Labeling (Drugs) Final Rule. https://s3.amazonaws.com/public-inspection.federalregister.gov/2014-28241.pdf. Published December 4, 2014. Accessed July 6, 2016.
44. Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: a reference guide to fetal and neonatal risk. 9th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2011.
45. Larsen ER, Damkier P, Pedersen LH, et al; Danish Psychiatric Society; Danish Society of Obstetrics and Gynecology; Danish Paediatric Society; Danish Society of Clinical Pharmacology. Use of psychotropic drugs during pregnancy and breast-feeding. Acta Psychiatr Scand Suppl. 2015;(445):1-28.
46. McKenna K, Koren G, Tetelbaum M, et al. Pregnancy outcome of women using atypical antipsychotic drugs: a prospective comparative study. J Clin Psychiatry. 2005;66(4):444-449.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Newman WJ, Newman BM. Rediscovering clozapine: after a turbulent history, current guidance on initiating and monitoring. Current Psychiatry. 2016;15(7):42-46,48-49.
3. Walker AM, Lanza LL, Arellano F, et al. Mortality in current and former users of clozapine. Epidemiology. 1997;8(6):671-677.
4. Tiihonen J, Lönnqvist J, Wahlbeck K, et al. 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort study (FIN11 study). Lancet. 2009;374(9690):620-627.
5. Walker E, McGee RE, Druss BG. Mortality in mental disorders and global disease burden Implications: a systematic review and meta-analysis. JAMA Psychiatry. 2015;72(4):334-341.
6. Hayes RD, Downs J, Chang CK, et al. The effect of clozapine on premature mortality: an assessment of clinical monitoring and other potential confounders. Schizophr Bull. 2015;41(3):644-655.
7. De Fazio P, Gaetano R, Caroleo M, et al. Rare and very rare adverse effects of clozapine. Neuropsychiatr Dis Treat. 2015;11:1995-2003.
8. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 29, 2016.
9. Lieberman JA, Johns CA, Kane JM, et al. Clozapine-induced agranulocytosis: non-cross-reactivity with other psychotropic drugs. J Clin Psychiatry. 1988;49(7):271-277.
10. Honigfeld G, Arellano F, Sethi J, et al. Reducing clozapine-related morbidity and mortality: 5 years of experience with the Clozaril National Registry. J Clin Psychiatry. 1998;59(suppl 3):3-7.
11. Demler TL, Trigoboff E. Are clozapine patients at risk for blood dyscrasias with concomitant tamiflu use? Psychiatry (Edgmont). 2009;6(11):29-33.
12. Karalakulasingam R, Schacht RA, Lansing AM, et al. Influenza virus pneumonia after renal transplant. Postgrad Med. 1977;62(2):164-167.
13. Hoffman-La Roche Limited. Product monograph: Tamiflu. http://www.rochecanada.com/content/dam/roche_canada/en_CA/documents/Research/ClinicalTrialsForms/Products/ConsumerInformation/MonographsandPublicAdvisories/Tamiflu/Tamiflu_PM_E.pdf. Updated January 26, 2015. Accessed November 28, 2015.
14. Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
15. Palominao A, Kukoyi O, Xiong GL. Leukocytosis after lithium and clozapine combination therapy. Ann Clin Psychiatry. 2010;22(3):205-206.
16. Focosi D, Azzarà A, Kast RE, et al. Lithium and hematology: established and proposed uses. J Leukoc Biol. 2009;85(1):20-28.
17. Papetti F, Darcourt G, Giordana JY, et al. Treatment of clozapine-induced granulocytopenia with lithium (two observations) [in French]. Encephale. 2004;30(6):578-582.
18. Hummer M, Sperner-Unterweger B, Kemmler G, et al. Does eosinophilia predict clozapine induced neutropenia? Psychopharmacology (Berl). 1996;124(1-2):201-204.
19. Aneja J, Sharma N, Mahajan S, et al. Eosinophilia induced by clozapine: a report of two cases and review of the literature. J Family Med Prim Care. 2015;4(1):127-129.
20. Nielsen J, Correll CU, Manu P, et al. Termination of clozapine treatment due to medical reasons: when is it warranted and how can it be avoided? J Clin Psychiatry. 2013;74(6):603-613.
21. Stryjer R, Timinsky I, Reznik, I, et al. Beta-adrenergic antagonists for the treatment of clozapine-induced sinus tachycardia: a retrospective study. Clin Neuropharmacol. 2009;32(5):290-292.
22. Lally J, Docherty MJ, MacCabe JH. Pharmacological interventions for clozapine-induced sinus tachycardia. Cochrane Database Syst Rev. 2016;9(6):CD011566.
23. Kamphuis H, Arends J, Timmerman L, et al. Myocarditis and cardiomyopathy: underestimated complications resulting from clozapine therapy [in Dutch]. Tijdschr Psychiatr. 2010;52(4):223-233.
24. Alawami M, Wasywich C, Cicovic A, et al. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
25. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
26. Williams AM, Park SH. Seizure associated with clozapine: incidence, etiology, and management. CNS Drugs. 2015;29(2):101-111.
27. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
28. Saavedra-Velez C, Yusim A, Anbarasan D, et al. Modafinil as an adjunctive treatment of sedation, negative symptoms, and cognition in schizophrenia: a critical review. J Clin Psychiatry. 2009;70(1):104-112.
29. Klein C, Gordon J, Pollak L, et al. Clozapine in Parkinson’s disease psychosis: 5-year follow-up review. Clin Neuropharmacol. 2003;26(1):8-11.
30. Lutz UC, Sirfy A, Wiatr G, et al. Clozapine serum concentrations in dopamimetic psychosis in Parkinson’s disease and related disorders. Eur J Clin Pharmacol. 2014;70(12):1471-1476.
31. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
32. Duggal HS. Glycopyrrolate for clozapine-induced sialorrhea. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(7):1546-1547.
33. Leadbetter R, Shutty M, Pavalonis D, et al. Clozapine-induced weight gain: prevalence and clinical relevance. Am J Psychiatry. 1992;149(1):68-72.
34. Lundblad W, Azzam PN, Gopalan, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;10(8):537-543.
35. Henderson DC, Nguyen DD, Copeland PM, et al. Clozapine, diabetes mellitus, hyperlipidemia, and cardiovascular risks and mortality: results of a 10-year naturalistic study. J Clin Psychiatry. 2005;66(9):1116-1121.
36. Stroup TS, Gerhard T, Crystal S, et al. Comparative effectiveness of clozapine and standard antipsychotic treatment in adults with schizophrenia. Am J Psychiatry. 2016;173(2):166-173.
37. Carrizo E, Fernández V, Connell L, et al. Extended release metformin for metabolic control assistance during prolonged clozapine administration: a 14 week, double-blind, parallel group, placebo-controlled study. Schizophr Res. 2009;113(1):19-26.
38. Chen CH, Huang MC, Kao CF, et al. Effects of adjunctive metformin on metabolic traits in nondiabetic clozapine-treated patients with schizophrenia and the effect of metformin discontinuation on body weight: a 24-week, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(5):e424-e430.
39. Mizuno Y, Suzuki T, Nakagawa A, et al. Pharmacological strategies to counteract antipsychotic-induced weight gain and metabolic adverse effects in schizophrenia: a systematic review and meta-analysis. Schizophr Bull. 2014;40(6):1385-1403.
40. Nielsen J, Meyer JM. Risk factors for ileus in patients with schizophrenia. Schizophr Bull. 2012;38(3):592-598.
41. Lowe CM, Grube RR, Scates AC. Characterization and clinical management of clozapine-induced fever. Ann Pharmacother. 2007;41(10):1700-1704.
42. ACOG Committee on Practice Bulletins–Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces practice bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol. 2008;111(4):1001-1020.
43. U.S. Food and Drug Administration. Pregnancy and Lactation Labeling (Drugs) Final Rule. https://s3.amazonaws.com/public-inspection.federalregister.gov/2014-28241.pdf. Published December 4, 2014. Accessed July 6, 2016.
44. Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: a reference guide to fetal and neonatal risk. 9th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2011.
45. Larsen ER, Damkier P, Pedersen LH, et al; Danish Psychiatric Society; Danish Society of Obstetrics and Gynecology; Danish Paediatric Society; Danish Society of Clinical Pharmacology. Use of psychotropic drugs during pregnancy and breast-feeding. Acta Psychiatr Scand Suppl. 2015;(445):1-28.
46. McKenna K, Koren G, Tetelbaum M, et al. Pregnancy outcome of women using atypical antipsychotic drugs: a prospective comparative study. J Clin Psychiatry. 2005;66(4):444-449.

What clinicians need to know about treating opioid use disorder
Communities across the United States have experienced a near-epidemic of opioid abuse. Deaths from opioid overdose have doubled since 2000 and increased 14% from 2013 to 2014.1 Treatment strategies for opioid use disorder (OUD) target individual well-being with the goal of preventing relapse. Most treatment approaches help patients gain self-confidence and have been described by patients as “giving them their life back.”
Broadly, the major phases of treatment for OUD are similar to those for other substance use disorders, and involve acute detoxification followed by long-term maintenance of sobriety. There are 3 main phases of treatment for OUD:
- treatment engagement
- stabilization and harm reduction
- sustained abstinence.
Long-term maintenance of sobriety in OUD could involve medications that are FDA-approved for this indication: methadone, buprenorphine, and naltrexone. Psychosocial interventions can be used individually and in combination with pharmacotherapy. Because OUD is a chronic disorder typically characterized by intermittent relapse, patients could move back and forth between the different phases of treatment.
In this article, we highlight the medication and non-medication treatment options for the long-term management of OUD.
Defining abuseExogenous opioids are synthetic substances used for their analgesic and morphine-like properties. Opioids are indicated for the treatment of certain pain conditions and exist in varying potencies and delivery systems, which are tailored for specific types of pain.2 Opioids work through activity at opioid receptors found in the brain, spinal cord, gut, and other organs. Agonism of specific opioid receptors results in a decreased perception of pain and contributes to their abuse potential.
Because of their abuse potential, prescription of opioids is governed by the Controlled Substances Act3 of the United States. Despite these regulations, approximately 4.5% of U.S. adults from a nationally representative sample were found to be misusing prescription opioids.4 Another study used data from the National Survey on Drug Use and Health and showed an increasing prevalence of OUD involving prescription drugs and resulting in increased mortality.5 The mortality rate from prescription opioids was found to be higher than for all other illicit drugs combined in 2013.6 Recently, Congress passed the Comprehensive Addiction and Recovery Act, which is intended to address the opioid crisis by expanding access to treatments for opioid overdose and addiction treatment services.7
The term “opioid use disorder” is found in DSM-58 and replaces the previous diagnoses of opioid abuse and dependence in DSM-IV-TR.9 OUD is characterized by a strong desire to continue using opioids despite problems associated with their use. Patients with OUD often experience cravings for opioids, tolerance, repeated failures at cutting down or limiting use, and decreased involvement in social activities.
Maladaptive use of opioids can result in physiologic dependence and lead to withdrawal symptoms, including anxiety, drug craving, insomnia, rhinorrhea, lacrimation, diarrhea, and piloerection. However, physiologic dependence might not necessarily lead to development of OUD, which can be diagnosed when enough other diagnostic criteria present in the absence of physiologic dependence.
Misuse of prescription opioids is more prevalent among patients who meet criteria for other substance use disorders than among patients who do not.10 Misuse of prescription opioids often results in greater health care utilization, including the need for emergency services, hospitalization, and detoxification.
Assessing for OUDThe Addiction Severity Index was designed to classify and monitor misuse of opioids. The Index has poor sensitivity; it detects recent opioid use but fails to differentiate patients using prescribed opioids from those who are abusing opioids.11 By comparison, the Current Opioid Misuse Measure (COMM) was developed to monitor opioid use in chemical dependency treatment settings.12 The COMM has reliable internal consistency13 and validity and can be used to assess use of opioids outside of routine medical care. To gauge the severity of withdrawal symptoms, the Objective Opioid Withdrawal Scale or Clinical Opiate Withdrawal Scale14 can be used. Table 1 summarizes some standardized tools used to assess OUD.
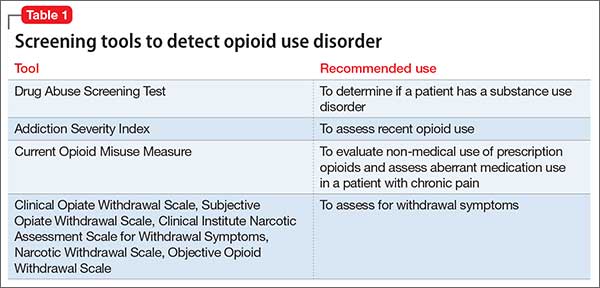
Neurobiological considerationsOpioids work through activity at mu, kappa, and delta opioid receptors. These receptors are present in both the peripheral and the central nervous system. Exogenous opioids work through activity at G protein-coupled receptors and by activating specific neurotransmitter systems. The effect of a given opioid drug is dependent on the type and location of receptors it modulates and can range from CNS depression to euphoria.
For example, mu receptor activation produces sedation, euphoria, or analgesia depending upon the location, frequency, and duration of receptor occupancy. Activation of CNS mu receptors can cause miosis and respiratory depression, whereas mu receptor activation in the peripheral nervous system can cause constipation and cough suppression. Mu receptor stimulation through various G-proteins triggers the second messenger cascade, generating enzymes such as cyclic adenosine monophosphate.
Clinical considerationsTreating OUD is challenging because of the ease with which patients can obtain opioids and because sometimes OUD occurs iatrogenically. Engaging patients in treatment is an important step in recovery, but it does not necessarily lead to reduction in opioid use. The engagement stage can involve outreach workers to encourage further treatment. Developing a therapeutic alliance and appropriately incentivizing patients also promotes entry into treatment. Motivational interviewing is used often in substance use treatment programs and can help engage patients in treatment and evaluate their willingness to change problematic behaviors.
Managing acute withdrawal symptoms. Withdrawal symptoms usually are not life threatening, but can be in the context of other medical conditions, such as autonomic instability, hypertension, cardiovascular disease, and dehydration. Withdrawal symptoms also can be life-threatening during in utero exposure to a fetus. Pharmacotherapeutic options to treat opioid withdrawal symptoms include long-acting opioids, such as methadone and buprenorphine,15 which can be administered in an ambulatory setting. The combination of buprenorphine and naloxone also can be used to treat opioid withdrawal symptoms.
The alpha-2 agonist16 clonidine, although not FDA-approved for OUD or opioid withdrawal, could be used to shorten the duration of withdrawal symptoms. Clonidine also decreases methadone withdrawal and can be combined with naloxone to target naloxone-induced opioid withdrawal symptoms.17,18 Nalbuphine and butorphanol should be avoided during opioid withdrawal because they antagonize opioid receptors and can precipitate withdrawal symptoms.19,20
Maintenance phase involves long-term stabilization and relapse prevention. Treatment options include medication and non-medication interventions.21
Non-pharmacologic treatment options,22 principally psychosocial interventions, can be used on their own or in combination with medications for maintenance treatment of OUD. Psychosocial interventions include structured, professionally administered interventions such as cognitive-behavioral therapy (CBT), aversion therapy, and day-treatment programs. Interventions such as peer counseling and self-help groups also are considered psychosocial interventions, but do not require the same type of professional training.
Peer support groups such as Narcotics Anonymous (NA) help members achieve and maintain sobriety and often focus on a traditional 12-step format or on the more recent Matrix Model,23,24 which is an intensive outpatient treatment program based on components of relapse prevention, motivational interviewing, CBT, and psychoeducation.
In these peer support models, group members discuss patterns of substance use and help one another recognize and overcome problematic behaviors. Groups may vary in terms of their specific approach. For example, NA encourages group members to focus on addiction itself while Methadone Anonymous prefers participants also discuss pharmacologic treatment experiences. Additional services for finding housing and assisting with job placement are also part of some relapse prevention strategies.
Although studies on the use of abstinence-based treatments are limited, abstinence-based therapy is an option for patients wishing to undergo chemical dependency treatment without taking prescription medications to address cravings or withdrawal symptoms.25 However, abstinence-based treatments have been shown to be less effective in improving outcomes than medication-assisted treatment (MAT).26 MAT combines medications and behavioral therapies for treating substance use disorders.
Pharmacologically, OUD can be treated with opioid agonist and antagonist medications. As summarized in Tables 2-4,27-31 these medications differ based on their pharmacokinetic and pharmacodynamic profiles and intrinsic activity at mu opioid receptors. Opioid system agonists, such as methadone and buprenorphine, decrease cravings by mimicking the activity of exogenous opioids. The opioid antagonists naloxone and naltrexone reinforce abstinence by inhibiting the euphoric effects associated with opioid use. The medication of choice for a given patient depends on:
- treatment adherence
- clinical setting
- degree of withdrawal symptoms
- motivation.26
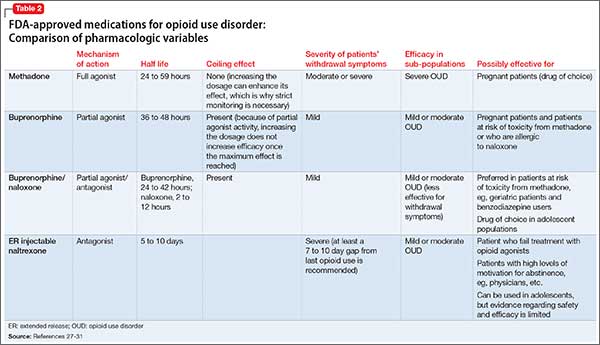
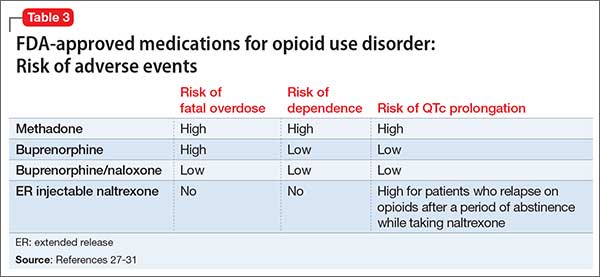
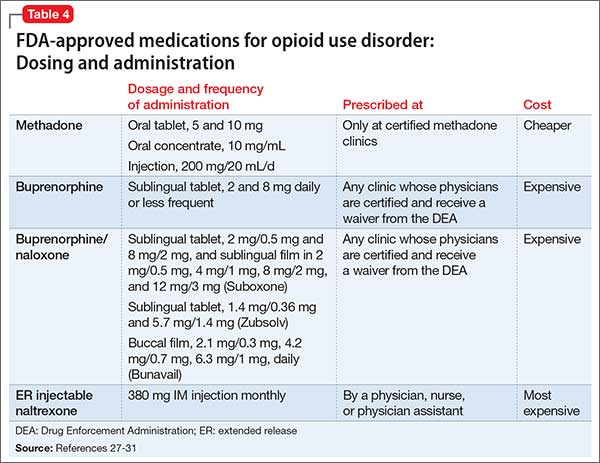
If a patient is actively seeking abstinence from opioids, either agonist or antagonist treatment can be used. In cases where a patient is not seeking abstinence, then preference should be given to opioid agonists to prevent overdose.26
Evidence suggests MAT can improve outcomes with OUD when compared with abstinence treatment alone. Several randomized, controlled, trials showed methadone and buprenorphine were more effective at treating OUD compared with treatment without medication. To date, 3 medications have been FDA-approved for treating OUD: methadone, buprenorphine, and naltrexone.26 All 3 medications differ in their pharmacokinetic and pharmacodynamic profiles and intrinsic activities at central mu-opioid receptor, as summarized in Tables 2-4.27-31
MethadoneMethadone reduces the euphoric effects of opioid use because it binds to and blocks opioid receptors. Methadone is an opioid replacement strategy; higher dosages are used for maintenance treatment to prevent additional dosages of opioids from causing euphoria. Methadone typically is administered once daily. However, in certain circumstances, such as rapid metabolism or pregnancy, it can be given as a twice-daily dosing regimen. Specific ABCB1 variants and DRD2 genetic polymorphisms (simultaneous occurrence of ≥2 genetically determined phenotypes) might determine the dosage requirements of methadone.32
Methadone during pregnancy. Methadone is the treatment of choice for opioid-dependent women during pregnancy33 and is listed as pregnancy category C because it can result in physiologic dependence of the newborn, although there are no documented controlled studies in humans to assess this risk. Methadone can be used while breast-feeding as long as patients are HIV-negative and not abusing other drugs.34,35 Because the methadone concentration in breast milk generally is low, the medication can be administered to nursing mothers after a careful consideration of risks and benefits.
Methadone administration. There are stringent eligibility criteria for methadone administration; not all physicians are authorized to prescribe methadone. Its use is federally regulated and only licensed treatment programs and licensed inpatient detoxification units can prescribe and dispense methadone in controlled settings and under the direct supervision of clinical personnel (Table 5).36 Patients meeting eligibility criteria can attend a specialized methadone clinic.

One of the challenges when using methadone for long-term management of OUD is tapering the dosage and attempting to discontinue the medication. Discontinuation of methadone leads to withdrawal symptoms and requires a carefully tailored tapering schedule. The literature on methadone tapering is limited. Tapering schedules could differ from practice to practice and, in many cases, are highly individualized based on the need and response of specific patients.
Dosage reduction schedules can last from 2 to 3 weeks to 6 months. Studies indicate rapid reduction worsens treatment outcomes and protracted tapering is associated with better outcomes. A suggested tapering schedule could involve decreasing the dosage by 20% to 25% until reaching a dosage of 30 mg/d, then decreasing by 5 mg/d every 3 to 5 days until reaching a dosage of 10 mg/d, before finally decreasing by 2.5 mg/d every 3 to 5 days.
Some randomized trials have shown better outcomes with long-term treatment. The goal of many programs is transitioning from maintenance treatment to abstinence. However, programs targeting maintenance rather than abstinence have been shown to be more effective.
The FDA has no defined limits for treatment duration with either methadone or buprenorphine. Therefore, the decision to taper or discontinue either medication should be made carefully case by case, using sound clinical judgment. Studies show that methadone treatment could reduce the spread of HIV,37,38 decrease criminal behaviors,39 and reduce overall mortality rates.40 A follow-up study comparing individuals randomly assigned to receive methadone or buprenorphine for OUD showed reduced risk of mortality overall40 in both groups.
Adverse events reported during treatment with methadone include decreased libido, erectile dysfunction, constipation, drowsiness, QTc prolongation, and torsade de pointes.41 Therefore, the FDA recommends obtaining a detailed medical history and baseline electrocardiogram (ECG), with a repeat ECG within the first month of treatment and then annually. Informing patients about the possibility of arrhythmias is part of the informed consent process before starting methadone.
Clinicians also should be vigilant when using methadone in combination with other medications that can prolong the QTc interval (eg, some antipsychotics). Methadone has a greater risk of fatal overdose then buprenorphine. A large-scale study of >16,000 patients reported a 4-fold increase in mortality resulting from methadone overdose compared with buprenorphine.42
BuprenorphineBuprenorphine is a partial opioid agonist at the mu opioid receptor. A full opioid agonist binds and fully activates the opioid receptors; an antagonist blocks the same. An opioid receptor partial agonist partially activates the receptor. Therefore, an opioid system partial agonist is a functional antagonist and, at lower dosages, has weak agonist effects; at higher dosages, a partial agonist antagonizes other endogenous and exogenous opioids that compete for binding at the same receptor.43 Because of the partial agonist effect, buprenorphine could result in less physical dependence and less withdrawal symptoms.
Administration. In contrast to methadone, buprenorphine can be prescribed by physicians for long-term management of OUD in the United States. Buprenorphine is available in 2 formulations: a sublingual form for daily use and a long-acting form that causes less withdrawal symptoms and cravings. In May 2016 the FDA approved the first buprenorphine implant for use in opioid dependence.44,45
To prevent withdrawal symptoms, a 24-hour period of opioid abstinence is recommended before starting buprenorphine or buprenorphine/naloxone treatment.46 Although lacking empirical evidence, catechol-O-methyltransferase (COMT) inhibitors, such as entacapone, have an anti-craving affect and are used by some clinicians to improve adherence with buprenorphine. This is because of their ability to balance dopamine, which is central to the reward pathway responsible for cravings. Although use of COMT inhibitors might make sense intuitively, such use is off-label and should be based on clinical judgment and a review of the available literature. A study showed that tapering buprenorphine for 4 weeks in combination with naltrexone improved the abstinence rate.47
Adverse effects. Some of the adverse events reported during treatment with buprenorphine include fever, back pain, nausea, cough, sedation, difficulty with urination, and constipation. Respiratory depression is a less common effect of buprenorphine, compared with full opioid agonists, because of the medication’s mechanism of action as a partial agonist.48 As a result, buprenorphine has been shown to have a lower risk of fatal overdose than methadone.49 Studies have shown buprenorphine to be more likely than methadone to reduce neonatal abstinence syndromes.50
NaltrexoneNaltrexone is an opioid antagonist and is an option to promote relapse prevention. Because of its antagonist properties, naltrexone treatment should always start after opioid detoxification because it can potentiate immediate withdrawal symptoms. Naltrexone is available in oral and long-acting formulations, the latter of which may be considered in patients who have difficulty with adherence.37
Oral naltrexone is taken as a single 50 mg-tablet once daily, whereas dosing for long-acting naltrexone in injectable and implantable formulations varies. These long-acting naltrexone formulations typically are administered monthly. Some of the adverse events reported during treatment with naltrexone are nausea, liver damage, and injection site pain.51
Buprenorphine/naloxoneBecause of buprenorphine’s agonist effects, it has a relatively high abuse potential compared with other opioids.52 Naloxone, on the other hand, is an opioid antagonist and is poorly absorbed when given orally and is associated with withdrawal symptoms if used intravenously. Therefore, naloxone is added to buprenorphine to decrease the likelihood of abuse when both are used as a combination product.53
Buprenorphine is combined with naloxone in a ratio of 4:1. Induction begins by using a 2 mg/0.5 mg tablet with dosage titration until symptoms abate. A combination of buprenorphine and naloxone also is available in film and tablet formulations. Patients must abstain from other opioids for at least 24 hours before initiating buprenorphine/naloxone treatment to prevent the precipitation of withdrawal symptoms.
1. Rudd RA, Aleshire N, Zibbell JE, et al. Increases in drug and opioid overdose deaths—United States, 2000-2014. MMWR Morb Mortal Wkly Rep. 2016;64(50-51):1378-1382.
2. Portenoy RK, Lesage P. Management of cancer pain. Lancet. 1999;353(9165):1695-1700.
3. Passik SD, Weinreb HJ. Managing chronic nonmalignant pain: overcoming obstacles to the use of opioids. Adv Ther. 2000;17(2):70-83.
4. Becker WC, Sullivan LE, Tetrault JM, et al. Non-medical use, abuse and dependence on prescription opioids among U.S. adults: psychiatric, medical and substance use correlates. Drug Alcohol Depend. 2008;94(1-3):38-47.
5. Han B, Compton WM, Jones CM, et al. Nonmedical prescription opioid use and use disorders among adults aged 18 through 64 years in the United States, 2003-2013. JAMA. 2015;314(14):1468-1478.
6. Centers for Disease Control and Prevention. National Center for Health Statistics, 2014. Multiple cause of death data. http://wonder.cdc.gov/mcd.html.
7. Twachtman G. Congress sends opioid legislation to the President. Clinical Psychiatry News. http://www.clinicalpsychiatrynews.com/?id=2407&tx_ttnews[tt_news]=524025&cHash=e93d5d1f86d20e53d3e2d8b07e9562b2. Published July 15, 2016. Accessed July 18, 2016.
8. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
9. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
10. McCabe SE, Cranford JA, West BT. Trends in prescription drug abuse and dependence, co-occurrence with other substance use disorders, and treatment utilization: results from two national surveys. Addict Behav. 2008;33(10):1297-1305.
11. Butler SF, Villapiano A, Malinow A. The effect of computer-mediated administration on self-disclosure of problems on the Addiction Severity Index. J Addict Med. 2009;3(4):194-203.
12. Meltzer EC, Rybin D, Saitz R, et al. Identifying prescription opioid use disorder in primary care: diagnostic characteristics of the Current Opioid Misuse Measure (COMM). Pain. 2011;152(2):397-402.
13. Butler SF, Budman SH, Fanciullo GJ, et al. Cross validation of the current opioid misuse measure to monitor chronic pain patients on opioid therapy. Clin J Pain. 2010;26(9):770-776.
14. Wesson DR, Ling W. The clinical opiate withdrawal scale (COWS). J Psychoactive Drugs. 2003;35(2):253-259.
15. Fiellin DA, O’Connor PG. Clinical practice. Office-based treatment of opioid-dependent patients. N Engl J Med. 2002;347(11):817-823.
16. Gowing LR, Farrell M, Ali RL, et al. α2‐Adrenergic agonists in opioid withdrawal. Addiction. 2002;97(1):49-58.
17. Loimer N, Hofmann P, Chaudhry H. Ultrashort noninvasive opiate detoxification. Am J Psychiatry. 1993;150(5):839.
18. Charney DS, Sternberg DE, Kleber HD, et al. The clinical use of clonidine in abrupt withdrawal from methadone. Effects on blood pressure and specific signs and symptoms. Arch Gen Psychiatry. 1981;38(11):1273-1277.
19. Preston KL, Bigelow GE, Liebson IA. Antagonist effects of nalbuphine in opioid-dependent human volunteers. J Pharmacol Exp Ther. 1989;248(3):929-937.
20. Preston KL, Bigelow GE, Liebson IA. Discrimination of butorphanol and nalbuphine in opioid-dependent humans. Pharmacol Biochem Behav. 1990;37(3):511-522.
21. Effective medical treatment of opiate addiction. National Consensus Development Panel on Effective Medical Treatment of Opiate Addiction. JAMA. 1998;280(22):1936-1943.
22. Amato L, Minozzi S, Davoli, M, et al. Psychosocial combined with agonist maintenance treatments versus agonist maintenance treatments alone for treatment of opioid dependence. Cochrane Database Syst Rev. 2011;(10):CD004147. doi: 10.1002/14651858.CD004147.pub4.
23. Obert JL, McCann MJ, Marinelli-Casey P, et al. The matrix model of outpatient stimulant abuse treatment: history and description. J Psychoactive Drugs. 2000;32(2):157-164.
24. Mayet S, Farrell M, Ferri M, et al. Psychosocial treatment for opiate abuse and dependence. Cochrane Database Syst Rev. 2005:CD004330.
25. McAuliffe WE. A randomized controlled trial of recovery training and self-help for opioid addicts in New England and Hong Kong. J Psychoactive Drugs. 1990;22(2):197-209.
26. Connery HS. Medication-assisted treatment of opioid use disorder: review of the evidence and future directions. Harv Rev Psychiatry. 2015;23(2):63-75.
27. Gibson AE, Degenhardt LJ. Mortality related to pharmacotherapies for opioid dependence: a comparative analysis of coronial records. Drug Alcohol Rev. 2007;26:405-410.
28. Clark L, Haram E, Johnson K, et al. Getting started with medication-assisted treatment with lessons from advancing recovery. Madison, WI: University of Wisconsin-Madison; 2010.
29. U.S. Food and Drug Administration. Vivitrol (naltrexone for extended-release injectable suspension): NDA 21-897C—Briefing document/background package. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharmacologic DrugsAdvisoryCommittee/UCM225664.pdf. Published September 16, 2010. Accessed July 11, 2016.
30. Providers Clinical Support System. PCSS Guidance. Buprenorphine induction. http://pcssmat.org/wp-content/uploads/2014/02/PCSS-MATGuidanceBuprenorphineInduction.Casadonte.pdf. Updated November 27, 2013. Accessed July 18, 2016.
31. An introduction to extended-release injectable naltrexone for the treatment of people with opioid dependence. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2012. HHS Publication No. (SMA) 12-4682.
32. Doehring A, von Hentig N, Graff J, et al. Genetic variants altering dopamine D2 receptor expression or function modulate the risk of opiate addiction and the dosage requirements of methadone substitution. Pharmacogenet Genomics. 2009;19(6):407-414.
33. Mitchell JL. Pregnant, substance-abusing women: treatment improvement protocol (TIP) Series 2. Rockville, MD: Substance Abuse and Mental Health Services Administration; 1993. DHHS Publication No. (SMA) 95-3056.
34. McCarthy JJ, Posey BL. Methadone levels in human milk. J Hum Lact. 2000;16(2):115-120.
35. Geraghty B, Graham EA, Logan B, et al. Methadone levels in breast milk. J Hum Lact. 1997;13(3):227-230.
36. Krambeer LL, von McKnelly W Jr, Gabrielli WF Jr, et al. Methadone therapy for opioid dependence. Am Fam Physician. 2001;63(12):2404-2410.
37. Novick DM, Joseph H, Croxson TS, et al. Absence of antibody to human immunodeficiency virus in long-term, socially rehabilitated methadone maintenance patients. Arch Intern Med. 1990;150(1):97-99.
38. Gowing LR, Farrell M, Bornemann R, et al. Brief report: methadone treatment of injecting opioid users for prevention of HIV infection. J Gen Intern Med. 2006;21(2):193-195.
39. Nurco DN, Ball JC, Shaffer JW, et al. The criminality of narcotic addicts. J Nerv Ment Dis. 1985;173(2):94-102.
40. Gibson A, Degenhardt L, Mattick RP, et al. Exposure to opioid maintenance treatment reduces long-term mortality. Addiction. 2008;103(3):462-468.
41. Pearson EC, Woosley RL. QT prolongation and torsades de pointes among methadone users: reports to the FDA spontaneous reporting system. Pharmacoepidemiol Drug Saf. 2005;14(11):747-753.
42. Bell JR, Butler B, Lawrance A, et al. Comparing overdose mortality associated with methadone and buprenorphine treatment. Drug Alcohol Depend. 2009;104(1-2):73-77.
43. Bickel WK, Amass L. Buprenorphine treatment of opioid dependence: a review. Experimental and Clinical Psychopharmacology. 1995;3(4):477-489.
44. U.S. Food and Drug Administration. FDA approves first buprenorphine implant for treatment of opioid dependence. http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm503719.htm. Published May 26, 2016. Accessed July 18, 2016.
45. The National Alliance of Advocates for Buprenorphine Treatment. https://www.naabt.org/index.cfm. Accessed July 18, 2016.
46. Buprenorphine: an alternative to methadone. Med Lett Drugs Ther. 2003;45(1150):13-15.
47. Sigmon SC, Dunn KE, Saulsgiver K, et al. A randomized, double-blind evaluation of buprenorphine taper duration in primary prescription opioid abusers. JAMA Psychiatry. 2013;70(12):1347-1354.
48. Dahan A, Yassen A, Bijl H, et al. Comparison of the respiratory effects of intravenous buprenorphine and fentanyl in humans and rats. Br J Anaesth. 2005;94(6):825-834.
49. Bell JR, Butler B, Lawrance A, et al. Comparing overdose mortality associated with methadone and buprenorphine treatment. Drug Alcohol Depend. 2009;104(1-2):73-77.
50. Kakko J, Heilig M, Sarman I. Buprenorphine and methadone treatment of opiate dependence during pregnancy: comparison of fetal growth and neonatal outcomes in two consecutive case series. Drug Alcohol Depend. 2008;96(1-2):69-78.
51. Stotts AL, Dodrill CL, Kosten TR. Opioid dependence treatment: options in pharmacotherapy. Expert Opin Pharmacother. 2009;10(11):1727-1740.
52. Robinson GM, Dukes PD, Robinson BJ, et al. The misuse of buprenorphine and a buprenorphine-naloxone combination in Wellington, New Zealand. Drug Alcohol Depend. 1993;33(1):81-86.
53. Fudala PJ, Bridge TP, Herbert S, et al; Buprenorphine/Naloxone Collaborative Study Group. Office-based treatment of opiate addiction with a sublingual-tablet formulation of buprenorphine and naloxone. N Engl J Med. 2003;349(10):949-958.
Communities across the United States have experienced a near-epidemic of opioid abuse. Deaths from opioid overdose have doubled since 2000 and increased 14% from 2013 to 2014.1 Treatment strategies for opioid use disorder (OUD) target individual well-being with the goal of preventing relapse. Most treatment approaches help patients gain self-confidence and have been described by patients as “giving them their life back.”
Broadly, the major phases of treatment for OUD are similar to those for other substance use disorders, and involve acute detoxification followed by long-term maintenance of sobriety. There are 3 main phases of treatment for OUD:
- treatment engagement
- stabilization and harm reduction
- sustained abstinence.
Long-term maintenance of sobriety in OUD could involve medications that are FDA-approved for this indication: methadone, buprenorphine, and naltrexone. Psychosocial interventions can be used individually and in combination with pharmacotherapy. Because OUD is a chronic disorder typically characterized by intermittent relapse, patients could move back and forth between the different phases of treatment.
In this article, we highlight the medication and non-medication treatment options for the long-term management of OUD.
Defining abuseExogenous opioids are synthetic substances used for their analgesic and morphine-like properties. Opioids are indicated for the treatment of certain pain conditions and exist in varying potencies and delivery systems, which are tailored for specific types of pain.2 Opioids work through activity at opioid receptors found in the brain, spinal cord, gut, and other organs. Agonism of specific opioid receptors results in a decreased perception of pain and contributes to their abuse potential.
Because of their abuse potential, prescription of opioids is governed by the Controlled Substances Act3 of the United States. Despite these regulations, approximately 4.5% of U.S. adults from a nationally representative sample were found to be misusing prescription opioids.4 Another study used data from the National Survey on Drug Use and Health and showed an increasing prevalence of OUD involving prescription drugs and resulting in increased mortality.5 The mortality rate from prescription opioids was found to be higher than for all other illicit drugs combined in 2013.6 Recently, Congress passed the Comprehensive Addiction and Recovery Act, which is intended to address the opioid crisis by expanding access to treatments for opioid overdose and addiction treatment services.7
The term “opioid use disorder” is found in DSM-58 and replaces the previous diagnoses of opioid abuse and dependence in DSM-IV-TR.9 OUD is characterized by a strong desire to continue using opioids despite problems associated with their use. Patients with OUD often experience cravings for opioids, tolerance, repeated failures at cutting down or limiting use, and decreased involvement in social activities.
Maladaptive use of opioids can result in physiologic dependence and lead to withdrawal symptoms, including anxiety, drug craving, insomnia, rhinorrhea, lacrimation, diarrhea, and piloerection. However, physiologic dependence might not necessarily lead to development of OUD, which can be diagnosed when enough other diagnostic criteria present in the absence of physiologic dependence.
Misuse of prescription opioids is more prevalent among patients who meet criteria for other substance use disorders than among patients who do not.10 Misuse of prescription opioids often results in greater health care utilization, including the need for emergency services, hospitalization, and detoxification.
Assessing for OUDThe Addiction Severity Index was designed to classify and monitor misuse of opioids. The Index has poor sensitivity; it detects recent opioid use but fails to differentiate patients using prescribed opioids from those who are abusing opioids.11 By comparison, the Current Opioid Misuse Measure (COMM) was developed to monitor opioid use in chemical dependency treatment settings.12 The COMM has reliable internal consistency13 and validity and can be used to assess use of opioids outside of routine medical care. To gauge the severity of withdrawal symptoms, the Objective Opioid Withdrawal Scale or Clinical Opiate Withdrawal Scale14 can be used. Table 1 summarizes some standardized tools used to assess OUD.
Neurobiological considerationsOpioids work through activity at mu, kappa, and delta opioid receptors. These receptors are present in both the peripheral and the central nervous system. Exogenous opioids work through activity at G protein-coupled receptors and by activating specific neurotransmitter systems. The effect of a given opioid drug is dependent on the type and location of receptors it modulates and can range from CNS depression to euphoria.
For example, mu receptor activation produces sedation, euphoria, or analgesia depending upon the location, frequency, and duration of receptor occupancy. Activation of CNS mu receptors can cause miosis and respiratory depression, whereas mu receptor activation in the peripheral nervous system can cause constipation and cough suppression. Mu receptor stimulation through various G-proteins triggers the second messenger cascade, generating enzymes such as cyclic adenosine monophosphate.
Clinical considerationsTreating OUD is challenging because of the ease with which patients can obtain opioids and because sometimes OUD occurs iatrogenically. Engaging patients in treatment is an important step in recovery, but it does not necessarily lead to reduction in opioid use. The engagement stage can involve outreach workers to encourage further treatment. Developing a therapeutic alliance and appropriately incentivizing patients also promotes entry into treatment. Motivational interviewing is used often in substance use treatment programs and can help engage patients in treatment and evaluate their willingness to change problematic behaviors.
Managing acute withdrawal symptoms. Withdrawal symptoms usually are not life threatening, but can be in the context of other medical conditions, such as autonomic instability, hypertension, cardiovascular disease, and dehydration. Withdrawal symptoms also can be life-threatening during in utero exposure to a fetus. Pharmacotherapeutic options to treat opioid withdrawal symptoms include long-acting opioids, such as methadone and buprenorphine,15 which can be administered in an ambulatory setting. The combination of buprenorphine and naloxone also can be used to treat opioid withdrawal symptoms.
The alpha-2 agonist16 clonidine, although not FDA-approved for OUD or opioid withdrawal, could be used to shorten the duration of withdrawal symptoms. Clonidine also decreases methadone withdrawal and can be combined with naloxone to target naloxone-induced opioid withdrawal symptoms.17,18 Nalbuphine and butorphanol should be avoided during opioid withdrawal because they antagonize opioid receptors and can precipitate withdrawal symptoms.19,20
Maintenance phase involves long-term stabilization and relapse prevention. Treatment options include medication and non-medication interventions.21
Non-pharmacologic treatment options,22 principally psychosocial interventions, can be used on their own or in combination with medications for maintenance treatment of OUD. Psychosocial interventions include structured, professionally administered interventions such as cognitive-behavioral therapy (CBT), aversion therapy, and day-treatment programs. Interventions such as peer counseling and self-help groups also are considered psychosocial interventions, but do not require the same type of professional training.
Peer support groups such as Narcotics Anonymous (NA) help members achieve and maintain sobriety and often focus on a traditional 12-step format or on the more recent Matrix Model,23,24 which is an intensive outpatient treatment program based on components of relapse prevention, motivational interviewing, CBT, and psychoeducation.
In these peer support models, group members discuss patterns of substance use and help one another recognize and overcome problematic behaviors. Groups may vary in terms of their specific approach. For example, NA encourages group members to focus on addiction itself while Methadone Anonymous prefers participants also discuss pharmacologic treatment experiences. Additional services for finding housing and assisting with job placement are also part of some relapse prevention strategies.
Although studies on the use of abstinence-based treatments are limited, abstinence-based therapy is an option for patients wishing to undergo chemical dependency treatment without taking prescription medications to address cravings or withdrawal symptoms.25 However, abstinence-based treatments have been shown to be less effective in improving outcomes than medication-assisted treatment (MAT).26 MAT combines medications and behavioral therapies for treating substance use disorders.
Pharmacologically, OUD can be treated with opioid agonist and antagonist medications. As summarized in Tables 2-4,27-31 these medications differ based on their pharmacokinetic and pharmacodynamic profiles and intrinsic activity at mu opioid receptors. Opioid system agonists, such as methadone and buprenorphine, decrease cravings by mimicking the activity of exogenous opioids. The opioid antagonists naloxone and naltrexone reinforce abstinence by inhibiting the euphoric effects associated with opioid use. The medication of choice for a given patient depends on:
- treatment adherence
- clinical setting
- degree of withdrawal symptoms
- motivation.26
If a patient is actively seeking abstinence from opioids, either agonist or antagonist treatment can be used. In cases where a patient is not seeking abstinence, then preference should be given to opioid agonists to prevent overdose.26
Evidence suggests MAT can improve outcomes with OUD when compared with abstinence treatment alone. Several randomized, controlled, trials showed methadone and buprenorphine were more effective at treating OUD compared with treatment without medication. To date, 3 medications have been FDA-approved for treating OUD: methadone, buprenorphine, and naltrexone.26 All 3 medications differ in their pharmacokinetic and pharmacodynamic profiles and intrinsic activities at central mu-opioid receptor, as summarized in Tables 2-4.27-31
MethadoneMethadone reduces the euphoric effects of opioid use because it binds to and blocks opioid receptors. Methadone is an opioid replacement strategy; higher dosages are used for maintenance treatment to prevent additional dosages of opioids from causing euphoria. Methadone typically is administered once daily. However, in certain circumstances, such as rapid metabolism or pregnancy, it can be given as a twice-daily dosing regimen. Specific ABCB1 variants and DRD2 genetic polymorphisms (simultaneous occurrence of ≥2 genetically determined phenotypes) might determine the dosage requirements of methadone.32
Methadone during pregnancy. Methadone is the treatment of choice for opioid-dependent women during pregnancy33 and is listed as pregnancy category C because it can result in physiologic dependence of the newborn, although there are no documented controlled studies in humans to assess this risk. Methadone can be used while breast-feeding as long as patients are HIV-negative and not abusing other drugs.34,35 Because the methadone concentration in breast milk generally is low, the medication can be administered to nursing mothers after a careful consideration of risks and benefits.
Methadone administration. There are stringent eligibility criteria for methadone administration; not all physicians are authorized to prescribe methadone. Its use is federally regulated and only licensed treatment programs and licensed inpatient detoxification units can prescribe and dispense methadone in controlled settings and under the direct supervision of clinical personnel (Table 5).36 Patients meeting eligibility criteria can attend a specialized methadone clinic.
One of the challenges when using methadone for long-term management of OUD is tapering the dosage and attempting to discontinue the medication. Discontinuation of methadone leads to withdrawal symptoms and requires a carefully tailored tapering schedule. The literature on methadone tapering is limited. Tapering schedules could differ from practice to practice and, in many cases, are highly individualized based on the need and response of specific patients.
Dosage reduction schedules can last from 2 to 3 weeks to 6 months. Studies indicate rapid reduction worsens treatment outcomes and protracted tapering is associated with better outcomes. A suggested tapering schedule could involve decreasing the dosage by 20% to 25% until reaching a dosage of 30 mg/d, then decreasing by 5 mg/d every 3 to 5 days until reaching a dosage of 10 mg/d, before finally decreasing by 2.5 mg/d every 3 to 5 days.
Some randomized trials have shown better outcomes with long-term treatment. The goal of many programs is transitioning from maintenance treatment to abstinence. However, programs targeting maintenance rather than abstinence have been shown to be more effective.
The FDA has no defined limits for treatment duration with either methadone or buprenorphine. Therefore, the decision to taper or discontinue either medication should be made carefully case by case, using sound clinical judgment. Studies show that methadone treatment could reduce the spread of HIV,37,38 decrease criminal behaviors,39 and reduce overall mortality rates.40 A follow-up study comparing individuals randomly assigned to receive methadone or buprenorphine for OUD showed reduced risk of mortality overall40 in both groups.
Adverse events reported during treatment with methadone include decreased libido, erectile dysfunction, constipation, drowsiness, QTc prolongation, and torsade de pointes.41 Therefore, the FDA recommends obtaining a detailed medical history and baseline electrocardiogram (ECG), with a repeat ECG within the first month of treatment and then annually. Informing patients about the possibility of arrhythmias is part of the informed consent process before starting methadone.
Clinicians also should be vigilant when using methadone in combination with other medications that can prolong the QTc interval (eg, some antipsychotics). Methadone has a greater risk of fatal overdose then buprenorphine. A large-scale study of >16,000 patients reported a 4-fold increase in mortality resulting from methadone overdose compared with buprenorphine.42
BuprenorphineBuprenorphine is a partial opioid agonist at the mu opioid receptor. A full opioid agonist binds and fully activates the opioid receptors; an antagonist blocks the same. An opioid receptor partial agonist partially activates the receptor. Therefore, an opioid system partial agonist is a functional antagonist and, at lower dosages, has weak agonist effects; at higher dosages, a partial agonist antagonizes other endogenous and exogenous opioids that compete for binding at the same receptor.43 Because of the partial agonist effect, buprenorphine could result in less physical dependence and less withdrawal symptoms.
Administration. In contrast to methadone, buprenorphine can be prescribed by physicians for long-term management of OUD in the United States. Buprenorphine is available in 2 formulations: a sublingual form for daily use and a long-acting form that causes less withdrawal symptoms and cravings. In May 2016 the FDA approved the first buprenorphine implant for use in opioid dependence.44,45
To prevent withdrawal symptoms, a 24-hour period of opioid abstinence is recommended before starting buprenorphine or buprenorphine/naloxone treatment.46 Although lacking empirical evidence, catechol-O-methyltransferase (COMT) inhibitors, such as entacapone, have an anti-craving affect and are used by some clinicians to improve adherence with buprenorphine. This is because of their ability to balance dopamine, which is central to the reward pathway responsible for cravings. Although use of COMT inhibitors might make sense intuitively, such use is off-label and should be based on clinical judgment and a review of the available literature. A study showed that tapering buprenorphine for 4 weeks in combination with naltrexone improved the abstinence rate.47
Adverse effects. Some of the adverse events reported during treatment with buprenorphine include fever, back pain, nausea, cough, sedation, difficulty with urination, and constipation. Respiratory depression is a less common effect of buprenorphine, compared with full opioid agonists, because of the medication’s mechanism of action as a partial agonist.48 As a result, buprenorphine has been shown to have a lower risk of fatal overdose than methadone.49 Studies have shown buprenorphine to be more likely than methadone to reduce neonatal abstinence syndromes.50
NaltrexoneNaltrexone is an opioid antagonist and is an option to promote relapse prevention. Because of its antagonist properties, naltrexone treatment should always start after opioid detoxification because it can potentiate immediate withdrawal symptoms. Naltrexone is available in oral and long-acting formulations, the latter of which may be considered in patients who have difficulty with adherence.37
Oral naltrexone is taken as a single 50 mg-tablet once daily, whereas dosing for long-acting naltrexone in injectable and implantable formulations varies. These long-acting naltrexone formulations typically are administered monthly. Some of the adverse events reported during treatment with naltrexone are nausea, liver damage, and injection site pain.51
Buprenorphine/naloxoneBecause of buprenorphine’s agonist effects, it has a relatively high abuse potential compared with other opioids.52 Naloxone, on the other hand, is an opioid antagonist and is poorly absorbed when given orally and is associated with withdrawal symptoms if used intravenously. Therefore, naloxone is added to buprenorphine to decrease the likelihood of abuse when both are used as a combination product.53
Buprenorphine is combined with naloxone in a ratio of 4:1. Induction begins by using a 2 mg/0.5 mg tablet with dosage titration until symptoms abate. A combination of buprenorphine and naloxone also is available in film and tablet formulations. Patients must abstain from other opioids for at least 24 hours before initiating buprenorphine/naloxone treatment to prevent the precipitation of withdrawal symptoms.
Communities across the United States have experienced a near-epidemic of opioid abuse. Deaths from opioid overdose have doubled since 2000 and increased 14% from 2013 to 2014.1 Treatment strategies for opioid use disorder (OUD) target individual well-being with the goal of preventing relapse. Most treatment approaches help patients gain self-confidence and have been described by patients as “giving them their life back.”
Broadly, the major phases of treatment for OUD are similar to those for other substance use disorders, and involve acute detoxification followed by long-term maintenance of sobriety. There are 3 main phases of treatment for OUD:
- treatment engagement
- stabilization and harm reduction
- sustained abstinence.
Long-term maintenance of sobriety in OUD could involve medications that are FDA-approved for this indication: methadone, buprenorphine, and naltrexone. Psychosocial interventions can be used individually and in combination with pharmacotherapy. Because OUD is a chronic disorder typically characterized by intermittent relapse, patients could move back and forth between the different phases of treatment.
In this article, we highlight the medication and non-medication treatment options for the long-term management of OUD.
Defining abuseExogenous opioids are synthetic substances used for their analgesic and morphine-like properties. Opioids are indicated for the treatment of certain pain conditions and exist in varying potencies and delivery systems, which are tailored for specific types of pain.2 Opioids work through activity at opioid receptors found in the brain, spinal cord, gut, and other organs. Agonism of specific opioid receptors results in a decreased perception of pain and contributes to their abuse potential.
Because of their abuse potential, prescription of opioids is governed by the Controlled Substances Act3 of the United States. Despite these regulations, approximately 4.5% of U.S. adults from a nationally representative sample were found to be misusing prescription opioids.4 Another study used data from the National Survey on Drug Use and Health and showed an increasing prevalence of OUD involving prescription drugs and resulting in increased mortality.5 The mortality rate from prescription opioids was found to be higher than for all other illicit drugs combined in 2013.6 Recently, Congress passed the Comprehensive Addiction and Recovery Act, which is intended to address the opioid crisis by expanding access to treatments for opioid overdose and addiction treatment services.7
The term “opioid use disorder” is found in DSM-58 and replaces the previous diagnoses of opioid abuse and dependence in DSM-IV-TR.9 OUD is characterized by a strong desire to continue using opioids despite problems associated with their use. Patients with OUD often experience cravings for opioids, tolerance, repeated failures at cutting down or limiting use, and decreased involvement in social activities.
Maladaptive use of opioids can result in physiologic dependence and lead to withdrawal symptoms, including anxiety, drug craving, insomnia, rhinorrhea, lacrimation, diarrhea, and piloerection. However, physiologic dependence might not necessarily lead to development of OUD, which can be diagnosed when enough other diagnostic criteria present in the absence of physiologic dependence.
Misuse of prescription opioids is more prevalent among patients who meet criteria for other substance use disorders than among patients who do not.10 Misuse of prescription opioids often results in greater health care utilization, including the need for emergency services, hospitalization, and detoxification.
Assessing for OUDThe Addiction Severity Index was designed to classify and monitor misuse of opioids. The Index has poor sensitivity; it detects recent opioid use but fails to differentiate patients using prescribed opioids from those who are abusing opioids.11 By comparison, the Current Opioid Misuse Measure (COMM) was developed to monitor opioid use in chemical dependency treatment settings.12 The COMM has reliable internal consistency13 and validity and can be used to assess use of opioids outside of routine medical care. To gauge the severity of withdrawal symptoms, the Objective Opioid Withdrawal Scale or Clinical Opiate Withdrawal Scale14 can be used. Table 1 summarizes some standardized tools used to assess OUD.
Neurobiological considerationsOpioids work through activity at mu, kappa, and delta opioid receptors. These receptors are present in both the peripheral and the central nervous system. Exogenous opioids work through activity at G protein-coupled receptors and by activating specific neurotransmitter systems. The effect of a given opioid drug is dependent on the type and location of receptors it modulates and can range from CNS depression to euphoria.
For example, mu receptor activation produces sedation, euphoria, or analgesia depending upon the location, frequency, and duration of receptor occupancy. Activation of CNS mu receptors can cause miosis and respiratory depression, whereas mu receptor activation in the peripheral nervous system can cause constipation and cough suppression. Mu receptor stimulation through various G-proteins triggers the second messenger cascade, generating enzymes such as cyclic adenosine monophosphate.
Clinical considerationsTreating OUD is challenging because of the ease with which patients can obtain opioids and because sometimes OUD occurs iatrogenically. Engaging patients in treatment is an important step in recovery, but it does not necessarily lead to reduction in opioid use. The engagement stage can involve outreach workers to encourage further treatment. Developing a therapeutic alliance and appropriately incentivizing patients also promotes entry into treatment. Motivational interviewing is used often in substance use treatment programs and can help engage patients in treatment and evaluate their willingness to change problematic behaviors.
Managing acute withdrawal symptoms. Withdrawal symptoms usually are not life threatening, but can be in the context of other medical conditions, such as autonomic instability, hypertension, cardiovascular disease, and dehydration. Withdrawal symptoms also can be life-threatening during in utero exposure to a fetus. Pharmacotherapeutic options to treat opioid withdrawal symptoms include long-acting opioids, such as methadone and buprenorphine,15 which can be administered in an ambulatory setting. The combination of buprenorphine and naloxone also can be used to treat opioid withdrawal symptoms.
The alpha-2 agonist16 clonidine, although not FDA-approved for OUD or opioid withdrawal, could be used to shorten the duration of withdrawal symptoms. Clonidine also decreases methadone withdrawal and can be combined with naloxone to target naloxone-induced opioid withdrawal symptoms.17,18 Nalbuphine and butorphanol should be avoided during opioid withdrawal because they antagonize opioid receptors and can precipitate withdrawal symptoms.19,20
Maintenance phase involves long-term stabilization and relapse prevention. Treatment options include medication and non-medication interventions.21
Non-pharmacologic treatment options,22 principally psychosocial interventions, can be used on their own or in combination with medications for maintenance treatment of OUD. Psychosocial interventions include structured, professionally administered interventions such as cognitive-behavioral therapy (CBT), aversion therapy, and day-treatment programs. Interventions such as peer counseling and self-help groups also are considered psychosocial interventions, but do not require the same type of professional training.
Peer support groups such as Narcotics Anonymous (NA) help members achieve and maintain sobriety and often focus on a traditional 12-step format or on the more recent Matrix Model,23,24 which is an intensive outpatient treatment program based on components of relapse prevention, motivational interviewing, CBT, and psychoeducation.
In these peer support models, group members discuss patterns of substance use and help one another recognize and overcome problematic behaviors. Groups may vary in terms of their specific approach. For example, NA encourages group members to focus on addiction itself while Methadone Anonymous prefers participants also discuss pharmacologic treatment experiences. Additional services for finding housing and assisting with job placement are also part of some relapse prevention strategies.
Although studies on the use of abstinence-based treatments are limited, abstinence-based therapy is an option for patients wishing to undergo chemical dependency treatment without taking prescription medications to address cravings or withdrawal symptoms.25 However, abstinence-based treatments have been shown to be less effective in improving outcomes than medication-assisted treatment (MAT).26 MAT combines medications and behavioral therapies for treating substance use disorders.
Pharmacologically, OUD can be treated with opioid agonist and antagonist medications. As summarized in Tables 2-4,27-31 these medications differ based on their pharmacokinetic and pharmacodynamic profiles and intrinsic activity at mu opioid receptors. Opioid system agonists, such as methadone and buprenorphine, decrease cravings by mimicking the activity of exogenous opioids. The opioid antagonists naloxone and naltrexone reinforce abstinence by inhibiting the euphoric effects associated with opioid use. The medication of choice for a given patient depends on:
- treatment adherence
- clinical setting
- degree of withdrawal symptoms
- motivation.26
If a patient is actively seeking abstinence from opioids, either agonist or antagonist treatment can be used. In cases where a patient is not seeking abstinence, then preference should be given to opioid agonists to prevent overdose.26
Evidence suggests MAT can improve outcomes with OUD when compared with abstinence treatment alone. Several randomized, controlled, trials showed methadone and buprenorphine were more effective at treating OUD compared with treatment without medication. To date, 3 medications have been FDA-approved for treating OUD: methadone, buprenorphine, and naltrexone.26 All 3 medications differ in their pharmacokinetic and pharmacodynamic profiles and intrinsic activities at central mu-opioid receptor, as summarized in Tables 2-4.27-31
MethadoneMethadone reduces the euphoric effects of opioid use because it binds to and blocks opioid receptors. Methadone is an opioid replacement strategy; higher dosages are used for maintenance treatment to prevent additional dosages of opioids from causing euphoria. Methadone typically is administered once daily. However, in certain circumstances, such as rapid metabolism or pregnancy, it can be given as a twice-daily dosing regimen. Specific ABCB1 variants and DRD2 genetic polymorphisms (simultaneous occurrence of ≥2 genetically determined phenotypes) might determine the dosage requirements of methadone.32
Methadone during pregnancy. Methadone is the treatment of choice for opioid-dependent women during pregnancy33 and is listed as pregnancy category C because it can result in physiologic dependence of the newborn, although there are no documented controlled studies in humans to assess this risk. Methadone can be used while breast-feeding as long as patients are HIV-negative and not abusing other drugs.34,35 Because the methadone concentration in breast milk generally is low, the medication can be administered to nursing mothers after a careful consideration of risks and benefits.
Methadone administration. There are stringent eligibility criteria for methadone administration; not all physicians are authorized to prescribe methadone. Its use is federally regulated and only licensed treatment programs and licensed inpatient detoxification units can prescribe and dispense methadone in controlled settings and under the direct supervision of clinical personnel (Table 5).36 Patients meeting eligibility criteria can attend a specialized methadone clinic.
One of the challenges when using methadone for long-term management of OUD is tapering the dosage and attempting to discontinue the medication. Discontinuation of methadone leads to withdrawal symptoms and requires a carefully tailored tapering schedule. The literature on methadone tapering is limited. Tapering schedules could differ from practice to practice and, in many cases, are highly individualized based on the need and response of specific patients.
Dosage reduction schedules can last from 2 to 3 weeks to 6 months. Studies indicate rapid reduction worsens treatment outcomes and protracted tapering is associated with better outcomes. A suggested tapering schedule could involve decreasing the dosage by 20% to 25% until reaching a dosage of 30 mg/d, then decreasing by 5 mg/d every 3 to 5 days until reaching a dosage of 10 mg/d, before finally decreasing by 2.5 mg/d every 3 to 5 days.
Some randomized trials have shown better outcomes with long-term treatment. The goal of many programs is transitioning from maintenance treatment to abstinence. However, programs targeting maintenance rather than abstinence have been shown to be more effective.
The FDA has no defined limits for treatment duration with either methadone or buprenorphine. Therefore, the decision to taper or discontinue either medication should be made carefully case by case, using sound clinical judgment. Studies show that methadone treatment could reduce the spread of HIV,37,38 decrease criminal behaviors,39 and reduce overall mortality rates.40 A follow-up study comparing individuals randomly assigned to receive methadone or buprenorphine for OUD showed reduced risk of mortality overall40 in both groups.
Adverse events reported during treatment with methadone include decreased libido, erectile dysfunction, constipation, drowsiness, QTc prolongation, and torsade de pointes.41 Therefore, the FDA recommends obtaining a detailed medical history and baseline electrocardiogram (ECG), with a repeat ECG within the first month of treatment and then annually. Informing patients about the possibility of arrhythmias is part of the informed consent process before starting methadone.
Clinicians also should be vigilant when using methadone in combination with other medications that can prolong the QTc interval (eg, some antipsychotics). Methadone has a greater risk of fatal overdose then buprenorphine. A large-scale study of >16,000 patients reported a 4-fold increase in mortality resulting from methadone overdose compared with buprenorphine.42
BuprenorphineBuprenorphine is a partial opioid agonist at the mu opioid receptor. A full opioid agonist binds and fully activates the opioid receptors; an antagonist blocks the same. An opioid receptor partial agonist partially activates the receptor. Therefore, an opioid system partial agonist is a functional antagonist and, at lower dosages, has weak agonist effects; at higher dosages, a partial agonist antagonizes other endogenous and exogenous opioids that compete for binding at the same receptor.43 Because of the partial agonist effect, buprenorphine could result in less physical dependence and less withdrawal symptoms.
Administration. In contrast to methadone, buprenorphine can be prescribed by physicians for long-term management of OUD in the United States. Buprenorphine is available in 2 formulations: a sublingual form for daily use and a long-acting form that causes less withdrawal symptoms and cravings. In May 2016 the FDA approved the first buprenorphine implant for use in opioid dependence.44,45
To prevent withdrawal symptoms, a 24-hour period of opioid abstinence is recommended before starting buprenorphine or buprenorphine/naloxone treatment.46 Although lacking empirical evidence, catechol-O-methyltransferase (COMT) inhibitors, such as entacapone, have an anti-craving affect and are used by some clinicians to improve adherence with buprenorphine. This is because of their ability to balance dopamine, which is central to the reward pathway responsible for cravings. Although use of COMT inhibitors might make sense intuitively, such use is off-label and should be based on clinical judgment and a review of the available literature. A study showed that tapering buprenorphine for 4 weeks in combination with naltrexone improved the abstinence rate.47
Adverse effects. Some of the adverse events reported during treatment with buprenorphine include fever, back pain, nausea, cough, sedation, difficulty with urination, and constipation. Respiratory depression is a less common effect of buprenorphine, compared with full opioid agonists, because of the medication’s mechanism of action as a partial agonist.48 As a result, buprenorphine has been shown to have a lower risk of fatal overdose than methadone.49 Studies have shown buprenorphine to be more likely than methadone to reduce neonatal abstinence syndromes.50
NaltrexoneNaltrexone is an opioid antagonist and is an option to promote relapse prevention. Because of its antagonist properties, naltrexone treatment should always start after opioid detoxification because it can potentiate immediate withdrawal symptoms. Naltrexone is available in oral and long-acting formulations, the latter of which may be considered in patients who have difficulty with adherence.37
Oral naltrexone is taken as a single 50 mg-tablet once daily, whereas dosing for long-acting naltrexone in injectable and implantable formulations varies. These long-acting naltrexone formulations typically are administered monthly. Some of the adverse events reported during treatment with naltrexone are nausea, liver damage, and injection site pain.51
Buprenorphine/naloxoneBecause of buprenorphine’s agonist effects, it has a relatively high abuse potential compared with other opioids.52 Naloxone, on the other hand, is an opioid antagonist and is poorly absorbed when given orally and is associated with withdrawal symptoms if used intravenously. Therefore, naloxone is added to buprenorphine to decrease the likelihood of abuse when both are used as a combination product.53
Buprenorphine is combined with naloxone in a ratio of 4:1. Induction begins by using a 2 mg/0.5 mg tablet with dosage titration until symptoms abate. A combination of buprenorphine and naloxone also is available in film and tablet formulations. Patients must abstain from other opioids for at least 24 hours before initiating buprenorphine/naloxone treatment to prevent the precipitation of withdrawal symptoms.
1. Rudd RA, Aleshire N, Zibbell JE, et al. Increases in drug and opioid overdose deaths—United States, 2000-2014. MMWR Morb Mortal Wkly Rep. 2016;64(50-51):1378-1382.
2. Portenoy RK, Lesage P. Management of cancer pain. Lancet. 1999;353(9165):1695-1700.
3. Passik SD, Weinreb HJ. Managing chronic nonmalignant pain: overcoming obstacles to the use of opioids. Adv Ther. 2000;17(2):70-83.
4. Becker WC, Sullivan LE, Tetrault JM, et al. Non-medical use, abuse and dependence on prescription opioids among U.S. adults: psychiatric, medical and substance use correlates. Drug Alcohol Depend. 2008;94(1-3):38-47.
5. Han B, Compton WM, Jones CM, et al. Nonmedical prescription opioid use and use disorders among adults aged 18 through 64 years in the United States, 2003-2013. JAMA. 2015;314(14):1468-1478.
6. Centers for Disease Control and Prevention. National Center for Health Statistics, 2014. Multiple cause of death data. http://wonder.cdc.gov/mcd.html.
7. Twachtman G. Congress sends opioid legislation to the President. Clinical Psychiatry News. http://www.clinicalpsychiatrynews.com/?id=2407&tx_ttnews[tt_news]=524025&cHash=e93d5d1f86d20e53d3e2d8b07e9562b2. Published July 15, 2016. Accessed July 18, 2016.
8. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
9. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
10. McCabe SE, Cranford JA, West BT. Trends in prescription drug abuse and dependence, co-occurrence with other substance use disorders, and treatment utilization: results from two national surveys. Addict Behav. 2008;33(10):1297-1305.
11. Butler SF, Villapiano A, Malinow A. The effect of computer-mediated administration on self-disclosure of problems on the Addiction Severity Index. J Addict Med. 2009;3(4):194-203.
12. Meltzer EC, Rybin D, Saitz R, et al. Identifying prescription opioid use disorder in primary care: diagnostic characteristics of the Current Opioid Misuse Measure (COMM). Pain. 2011;152(2):397-402.
13. Butler SF, Budman SH, Fanciullo GJ, et al. Cross validation of the current opioid misuse measure to monitor chronic pain patients on opioid therapy. Clin J Pain. 2010;26(9):770-776.
14. Wesson DR, Ling W. The clinical opiate withdrawal scale (COWS). J Psychoactive Drugs. 2003;35(2):253-259.
15. Fiellin DA, O’Connor PG. Clinical practice. Office-based treatment of opioid-dependent patients. N Engl J Med. 2002;347(11):817-823.
16. Gowing LR, Farrell M, Ali RL, et al. α2‐Adrenergic agonists in opioid withdrawal. Addiction. 2002;97(1):49-58.
17. Loimer N, Hofmann P, Chaudhry H. Ultrashort noninvasive opiate detoxification. Am J Psychiatry. 1993;150(5):839.
18. Charney DS, Sternberg DE, Kleber HD, et al. The clinical use of clonidine in abrupt withdrawal from methadone. Effects on blood pressure and specific signs and symptoms. Arch Gen Psychiatry. 1981;38(11):1273-1277.
19. Preston KL, Bigelow GE, Liebson IA. Antagonist effects of nalbuphine in opioid-dependent human volunteers. J Pharmacol Exp Ther. 1989;248(3):929-937.
20. Preston KL, Bigelow GE, Liebson IA. Discrimination of butorphanol and nalbuphine in opioid-dependent humans. Pharmacol Biochem Behav. 1990;37(3):511-522.
21. Effective medical treatment of opiate addiction. National Consensus Development Panel on Effective Medical Treatment of Opiate Addiction. JAMA. 1998;280(22):1936-1943.
22. Amato L, Minozzi S, Davoli, M, et al. Psychosocial combined with agonist maintenance treatments versus agonist maintenance treatments alone for treatment of opioid dependence. Cochrane Database Syst Rev. 2011;(10):CD004147. doi: 10.1002/14651858.CD004147.pub4.
23. Obert JL, McCann MJ, Marinelli-Casey P, et al. The matrix model of outpatient stimulant abuse treatment: history and description. J Psychoactive Drugs. 2000;32(2):157-164.
24. Mayet S, Farrell M, Ferri M, et al. Psychosocial treatment for opiate abuse and dependence. Cochrane Database Syst Rev. 2005:CD004330.
25. McAuliffe WE. A randomized controlled trial of recovery training and self-help for opioid addicts in New England and Hong Kong. J Psychoactive Drugs. 1990;22(2):197-209.
26. Connery HS. Medication-assisted treatment of opioid use disorder: review of the evidence and future directions. Harv Rev Psychiatry. 2015;23(2):63-75.
27. Gibson AE, Degenhardt LJ. Mortality related to pharmacotherapies for opioid dependence: a comparative analysis of coronial records. Drug Alcohol Rev. 2007;26:405-410.
28. Clark L, Haram E, Johnson K, et al. Getting started with medication-assisted treatment with lessons from advancing recovery. Madison, WI: University of Wisconsin-Madison; 2010.
29. U.S. Food and Drug Administration. Vivitrol (naltrexone for extended-release injectable suspension): NDA 21-897C—Briefing document/background package. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharmacologic DrugsAdvisoryCommittee/UCM225664.pdf. Published September 16, 2010. Accessed July 11, 2016.
30. Providers Clinical Support System. PCSS Guidance. Buprenorphine induction. http://pcssmat.org/wp-content/uploads/2014/02/PCSS-MATGuidanceBuprenorphineInduction.Casadonte.pdf. Updated November 27, 2013. Accessed July 18, 2016.
31. An introduction to extended-release injectable naltrexone for the treatment of people with opioid dependence. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2012. HHS Publication No. (SMA) 12-4682.
32. Doehring A, von Hentig N, Graff J, et al. Genetic variants altering dopamine D2 receptor expression or function modulate the risk of opiate addiction and the dosage requirements of methadone substitution. Pharmacogenet Genomics. 2009;19(6):407-414.
33. Mitchell JL. Pregnant, substance-abusing women: treatment improvement protocol (TIP) Series 2. Rockville, MD: Substance Abuse and Mental Health Services Administration; 1993. DHHS Publication No. (SMA) 95-3056.
34. McCarthy JJ, Posey BL. Methadone levels in human milk. J Hum Lact. 2000;16(2):115-120.
35. Geraghty B, Graham EA, Logan B, et al. Methadone levels in breast milk. J Hum Lact. 1997;13(3):227-230.
36. Krambeer LL, von McKnelly W Jr, Gabrielli WF Jr, et al. Methadone therapy for opioid dependence. Am Fam Physician. 2001;63(12):2404-2410.
37. Novick DM, Joseph H, Croxson TS, et al. Absence of antibody to human immunodeficiency virus in long-term, socially rehabilitated methadone maintenance patients. Arch Intern Med. 1990;150(1):97-99.
38. Gowing LR, Farrell M, Bornemann R, et al. Brief report: methadone treatment of injecting opioid users for prevention of HIV infection. J Gen Intern Med. 2006;21(2):193-195.
39. Nurco DN, Ball JC, Shaffer JW, et al. The criminality of narcotic addicts. J Nerv Ment Dis. 1985;173(2):94-102.
40. Gibson A, Degenhardt L, Mattick RP, et al. Exposure to opioid maintenance treatment reduces long-term mortality. Addiction. 2008;103(3):462-468.
41. Pearson EC, Woosley RL. QT prolongation and torsades de pointes among methadone users: reports to the FDA spontaneous reporting system. Pharmacoepidemiol Drug Saf. 2005;14(11):747-753.
42. Bell JR, Butler B, Lawrance A, et al. Comparing overdose mortality associated with methadone and buprenorphine treatment. Drug Alcohol Depend. 2009;104(1-2):73-77.
43. Bickel WK, Amass L. Buprenorphine treatment of opioid dependence: a review. Experimental and Clinical Psychopharmacology. 1995;3(4):477-489.
44. U.S. Food and Drug Administration. FDA approves first buprenorphine implant for treatment of opioid dependence. http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm503719.htm. Published May 26, 2016. Accessed July 18, 2016.
45. The National Alliance of Advocates for Buprenorphine Treatment. https://www.naabt.org/index.cfm. Accessed July 18, 2016.
46. Buprenorphine: an alternative to methadone. Med Lett Drugs Ther. 2003;45(1150):13-15.
47. Sigmon SC, Dunn KE, Saulsgiver K, et al. A randomized, double-blind evaluation of buprenorphine taper duration in primary prescription opioid abusers. JAMA Psychiatry. 2013;70(12):1347-1354.
48. Dahan A, Yassen A, Bijl H, et al. Comparison of the respiratory effects of intravenous buprenorphine and fentanyl in humans and rats. Br J Anaesth. 2005;94(6):825-834.
49. Bell JR, Butler B, Lawrance A, et al. Comparing overdose mortality associated with methadone and buprenorphine treatment. Drug Alcohol Depend. 2009;104(1-2):73-77.
50. Kakko J, Heilig M, Sarman I. Buprenorphine and methadone treatment of opiate dependence during pregnancy: comparison of fetal growth and neonatal outcomes in two consecutive case series. Drug Alcohol Depend. 2008;96(1-2):69-78.
51. Stotts AL, Dodrill CL, Kosten TR. Opioid dependence treatment: options in pharmacotherapy. Expert Opin Pharmacother. 2009;10(11):1727-1740.
52. Robinson GM, Dukes PD, Robinson BJ, et al. The misuse of buprenorphine and a buprenorphine-naloxone combination in Wellington, New Zealand. Drug Alcohol Depend. 1993;33(1):81-86.
53. Fudala PJ, Bridge TP, Herbert S, et al; Buprenorphine/Naloxone Collaborative Study Group. Office-based treatment of opiate addiction with a sublingual-tablet formulation of buprenorphine and naloxone. N Engl J Med. 2003;349(10):949-958.
1. Rudd RA, Aleshire N, Zibbell JE, et al. Increases in drug and opioid overdose deaths—United States, 2000-2014. MMWR Morb Mortal Wkly Rep. 2016;64(50-51):1378-1382.
2. Portenoy RK, Lesage P. Management of cancer pain. Lancet. 1999;353(9165):1695-1700.
3. Passik SD, Weinreb HJ. Managing chronic nonmalignant pain: overcoming obstacles to the use of opioids. Adv Ther. 2000;17(2):70-83.
4. Becker WC, Sullivan LE, Tetrault JM, et al. Non-medical use, abuse and dependence on prescription opioids among U.S. adults: psychiatric, medical and substance use correlates. Drug Alcohol Depend. 2008;94(1-3):38-47.
5. Han B, Compton WM, Jones CM, et al. Nonmedical prescription opioid use and use disorders among adults aged 18 through 64 years in the United States, 2003-2013. JAMA. 2015;314(14):1468-1478.
6. Centers for Disease Control and Prevention. National Center for Health Statistics, 2014. Multiple cause of death data. http://wonder.cdc.gov/mcd.html.
7. Twachtman G. Congress sends opioid legislation to the President. Clinical Psychiatry News. http://www.clinicalpsychiatrynews.com/?id=2407&tx_ttnews[tt_news]=524025&cHash=e93d5d1f86d20e53d3e2d8b07e9562b2. Published July 15, 2016. Accessed July 18, 2016.
8. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
9. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
10. McCabe SE, Cranford JA, West BT. Trends in prescription drug abuse and dependence, co-occurrence with other substance use disorders, and treatment utilization: results from two national surveys. Addict Behav. 2008;33(10):1297-1305.
11. Butler SF, Villapiano A, Malinow A. The effect of computer-mediated administration on self-disclosure of problems on the Addiction Severity Index. J Addict Med. 2009;3(4):194-203.
12. Meltzer EC, Rybin D, Saitz R, et al. Identifying prescription opioid use disorder in primary care: diagnostic characteristics of the Current Opioid Misuse Measure (COMM). Pain. 2011;152(2):397-402.
13. Butler SF, Budman SH, Fanciullo GJ, et al. Cross validation of the current opioid misuse measure to monitor chronic pain patients on opioid therapy. Clin J Pain. 2010;26(9):770-776.
14. Wesson DR, Ling W. The clinical opiate withdrawal scale (COWS). J Psychoactive Drugs. 2003;35(2):253-259.
15. Fiellin DA, O’Connor PG. Clinical practice. Office-based treatment of opioid-dependent patients. N Engl J Med. 2002;347(11):817-823.
16. Gowing LR, Farrell M, Ali RL, et al. α2‐Adrenergic agonists in opioid withdrawal. Addiction. 2002;97(1):49-58.
17. Loimer N, Hofmann P, Chaudhry H. Ultrashort noninvasive opiate detoxification. Am J Psychiatry. 1993;150(5):839.
18. Charney DS, Sternberg DE, Kleber HD, et al. The clinical use of clonidine in abrupt withdrawal from methadone. Effects on blood pressure and specific signs and symptoms. Arch Gen Psychiatry. 1981;38(11):1273-1277.
19. Preston KL, Bigelow GE, Liebson IA. Antagonist effects of nalbuphine in opioid-dependent human volunteers. J Pharmacol Exp Ther. 1989;248(3):929-937.
20. Preston KL, Bigelow GE, Liebson IA. Discrimination of butorphanol and nalbuphine in opioid-dependent humans. Pharmacol Biochem Behav. 1990;37(3):511-522.
21. Effective medical treatment of opiate addiction. National Consensus Development Panel on Effective Medical Treatment of Opiate Addiction. JAMA. 1998;280(22):1936-1943.
22. Amato L, Minozzi S, Davoli, M, et al. Psychosocial combined with agonist maintenance treatments versus agonist maintenance treatments alone for treatment of opioid dependence. Cochrane Database Syst Rev. 2011;(10):CD004147. doi: 10.1002/14651858.CD004147.pub4.
23. Obert JL, McCann MJ, Marinelli-Casey P, et al. The matrix model of outpatient stimulant abuse treatment: history and description. J Psychoactive Drugs. 2000;32(2):157-164.
24. Mayet S, Farrell M, Ferri M, et al. Psychosocial treatment for opiate abuse and dependence. Cochrane Database Syst Rev. 2005:CD004330.
25. McAuliffe WE. A randomized controlled trial of recovery training and self-help for opioid addicts in New England and Hong Kong. J Psychoactive Drugs. 1990;22(2):197-209.
26. Connery HS. Medication-assisted treatment of opioid use disorder: review of the evidence and future directions. Harv Rev Psychiatry. 2015;23(2):63-75.
27. Gibson AE, Degenhardt LJ. Mortality related to pharmacotherapies for opioid dependence: a comparative analysis of coronial records. Drug Alcohol Rev. 2007;26:405-410.
28. Clark L, Haram E, Johnson K, et al. Getting started with medication-assisted treatment with lessons from advancing recovery. Madison, WI: University of Wisconsin-Madison; 2010.
29. U.S. Food and Drug Administration. Vivitrol (naltrexone for extended-release injectable suspension): NDA 21-897C—Briefing document/background package. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharmacologic DrugsAdvisoryCommittee/UCM225664.pdf. Published September 16, 2010. Accessed July 11, 2016.
30. Providers Clinical Support System. PCSS Guidance. Buprenorphine induction. http://pcssmat.org/wp-content/uploads/2014/02/PCSS-MATGuidanceBuprenorphineInduction.Casadonte.pdf. Updated November 27, 2013. Accessed July 18, 2016.
31. An introduction to extended-release injectable naltrexone for the treatment of people with opioid dependence. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2012. HHS Publication No. (SMA) 12-4682.
32. Doehring A, von Hentig N, Graff J, et al. Genetic variants altering dopamine D2 receptor expression or function modulate the risk of opiate addiction and the dosage requirements of methadone substitution. Pharmacogenet Genomics. 2009;19(6):407-414.
33. Mitchell JL. Pregnant, substance-abusing women: treatment improvement protocol (TIP) Series 2. Rockville, MD: Substance Abuse and Mental Health Services Administration; 1993. DHHS Publication No. (SMA) 95-3056.
34. McCarthy JJ, Posey BL. Methadone levels in human milk. J Hum Lact. 2000;16(2):115-120.
35. Geraghty B, Graham EA, Logan B, et al. Methadone levels in breast milk. J Hum Lact. 1997;13(3):227-230.
36. Krambeer LL, von McKnelly W Jr, Gabrielli WF Jr, et al. Methadone therapy for opioid dependence. Am Fam Physician. 2001;63(12):2404-2410.
37. Novick DM, Joseph H, Croxson TS, et al. Absence of antibody to human immunodeficiency virus in long-term, socially rehabilitated methadone maintenance patients. Arch Intern Med. 1990;150(1):97-99.
38. Gowing LR, Farrell M, Bornemann R, et al. Brief report: methadone treatment of injecting opioid users for prevention of HIV infection. J Gen Intern Med. 2006;21(2):193-195.
39. Nurco DN, Ball JC, Shaffer JW, et al. The criminality of narcotic addicts. J Nerv Ment Dis. 1985;173(2):94-102.
40. Gibson A, Degenhardt L, Mattick RP, et al. Exposure to opioid maintenance treatment reduces long-term mortality. Addiction. 2008;103(3):462-468.
41. Pearson EC, Woosley RL. QT prolongation and torsades de pointes among methadone users: reports to the FDA spontaneous reporting system. Pharmacoepidemiol Drug Saf. 2005;14(11):747-753.
42. Bell JR, Butler B, Lawrance A, et al. Comparing overdose mortality associated with methadone and buprenorphine treatment. Drug Alcohol Depend. 2009;104(1-2):73-77.
43. Bickel WK, Amass L. Buprenorphine treatment of opioid dependence: a review. Experimental and Clinical Psychopharmacology. 1995;3(4):477-489.
44. U.S. Food and Drug Administration. FDA approves first buprenorphine implant for treatment of opioid dependence. http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm503719.htm. Published May 26, 2016. Accessed July 18, 2016.
45. The National Alliance of Advocates for Buprenorphine Treatment. https://www.naabt.org/index.cfm. Accessed July 18, 2016.
46. Buprenorphine: an alternative to methadone. Med Lett Drugs Ther. 2003;45(1150):13-15.
47. Sigmon SC, Dunn KE, Saulsgiver K, et al. A randomized, double-blind evaluation of buprenorphine taper duration in primary prescription opioid abusers. JAMA Psychiatry. 2013;70(12):1347-1354.
48. Dahan A, Yassen A, Bijl H, et al. Comparison of the respiratory effects of intravenous buprenorphine and fentanyl in humans and rats. Br J Anaesth. 2005;94(6):825-834.
49. Bell JR, Butler B, Lawrance A, et al. Comparing overdose mortality associated with methadone and buprenorphine treatment. Drug Alcohol Depend. 2009;104(1-2):73-77.
50. Kakko J, Heilig M, Sarman I. Buprenorphine and methadone treatment of opiate dependence during pregnancy: comparison of fetal growth and neonatal outcomes in two consecutive case series. Drug Alcohol Depend. 2008;96(1-2):69-78.
51. Stotts AL, Dodrill CL, Kosten TR. Opioid dependence treatment: options in pharmacotherapy. Expert Opin Pharmacother. 2009;10(11):1727-1740.
52. Robinson GM, Dukes PD, Robinson BJ, et al. The misuse of buprenorphine and a buprenorphine-naloxone combination in Wellington, New Zealand. Drug Alcohol Depend. 1993;33(1):81-86.
53. Fudala PJ, Bridge TP, Herbert S, et al; Buprenorphine/Naloxone Collaborative Study Group. Office-based treatment of opiate addiction with a sublingual-tablet formulation of buprenorphine and naloxone. N Engl J Med. 2003;349(10):949-958.

Rediscovering clozapine: After a turbulent history, current guidance on initiating and monitoring
Although clozapine is the medication with the clearest benefits in treatment-resistant schizophrenia, many eligible patients never receive it. In the United States, 20% to 30% of patients with schizophrenia can be classified as treatment resistant, but clozapine accounts for <5% of antipsychotics prescribed.1,2 Clinicians worldwide tend to under-prescribe clozapine3—a reluctance one author coined as “clozaphobia.”4
Admittedly, clozapine has had a turbulent history—both lauded as a near-miracle drug and condemned as a deadly agent. The FDA has overhauled its prescribing and monitoring guidelines, however, offering psychiatrists a perfect opportunity to reacquaint themselves with this potentially life-changing intervention.
We begin this article with clozapine’s story, then spotlight new terrain the FDA created in 2015 when the agency introduced the Clozapine Risk Evaluation and Mitigation Strategy (REMS). Our goal in the 3 articles of this series is to deepen your appreciation for this tricyclic antipsychotic and provide practical clinical guidance for using it safely and effectively.
Setbacks, but the drug has an enduring presenceThe 1950s was an exciting era of exploration for new psychotropic medications. While searching for tricyclic antidepressants, Wander Laboratories discovered neuroleptic tricyclics, with clozapine identified in 1959 (Figure 1). Haloperidol’s development and release in the 1960s reinforced the prevailing dogma of the time that effective neuroleptics correlated with extrapyramidal symptoms, thus limiting interest in the newly discovered, but pharmacologically unique, clozapine. Throughout the 1960s, most research on clozapine was published in German, with less of an international presence.5
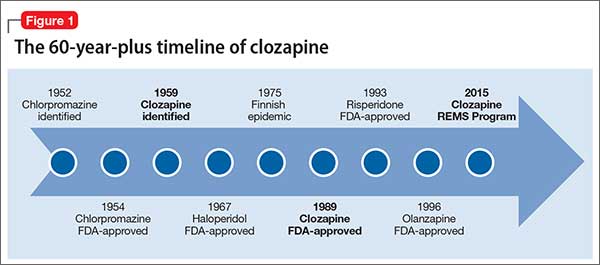
Agranulocytosis deaths. Clozapine earned its scarlet letter in 1975, when 8 patients in Finland died of agranulocytosis.6 Sandoz, its manufacturer, withdrew clozapine from the market and halted all clinical trials. The Finnish epidemic triggered detailed investigations into blood dyscrasias and early identification of agranulocytosis associated with clozapine and other antipsychotics.7
Clozapine endured only because of its unique efficacy. When psychiatrists witnessed relapses in patients who had to discontinue clozapine, some countries allowed its use with strict monitoring.5 The FDA kept clozapine minimally available in the United States by allowing so-called “compassionate need programs” to continue.7
New data, FDA approval. Two studies in 1987 and 1988 that compared clozapine with chlorpromazine for treatment-refractory schizophrenia demonstrated clozapine’s superior effect on both negative and positive symptoms.8,9 The FDA approved clozapine for refractory schizophrenia in 1989, and clozapine became clinically available in 1990.
Initially, the high annual cost of clozapine’s required “bundle” ($8,900 per patient for medication and monitoring) led to political outcry. As patients and their family struggled to afford the newly released medication, multiple states filed antitrust lawsuits. A federal court found both the manufacturer and individual states at fault and required expanded access to clozapine and its necessary monitoring. National clozapine registries were formed, and bundling was eliminated.7
The clozapine REMS programSix clozapine registries operated independently, each managed by a different manufacturer,10 until the FDA introduced REMS in September 2015. The REMS program created a centralized registry to monitor all U.S. patients treated with clozapine and made important changes to prescribing and monitoring guidelines.11,12 It also incorporated the National Non-Rechallenge Master File (NNRMF).
Initially, the REMS program was scheduled for rollout October 12, 2015, the closing date of the 6 registries. Since November 2015, pharmacies have been required to register with the program to dispense clozapine. A similar registration deadline for clozapine prescribers was extended indefinitely, however, because of technical problems. Once the deadline is finalized, all clozapine prescribers must complete 3 steps to be certified in the REMS program (Table 1).11

New requirements. Certified clozapine prescribers will have new responsibilities: enrolling patients and submitting lab results. They can designate someone else to perform these tasks on their behalf, but designees must enroll in the REMS program and the prescriber must confirm the designee. Pharmacists can no longer enroll patients for clozapine therapy unless they are confirmed as a prescriber designee. For outpatients, the absolute neutrophil count (ANC) must be reported before the pharmacy can dispense clozapine. For inpatients, the ANC must be reported within 7 days of the patient’s most recent blood draw.
Once the system is fully operational, Social Security numbers will no longer be used as patient identification for dispensing clozapine. Instead, outpatient pharmacies will obtain a predispense authorization, or PDA, from the REMS program. A person initiated on clozapine as an inpatient must be re-enrolled after discharge by their outpatient prescriber.
The REMS program includes information about clozapine patients who were maintained through the 6 registries, and these patients have been allowed to continue clozapine treatment. Data pertaining to patients last prescribed clozapine before October 1, 2012, did not transfer into the new system unless their name was on the NNRMF.
CASE
Is Mr. A a candidate for clozapine?Age 28, with schizophrenia, Mr. A is highly disorganized and psychotic when brought to the emergency room by police for inappropriate behavior. His family arrives and reports that similar events have occurred several times over the past few years. Mr. A’s outpatient psychiatrist has prescribed 3 different antipsychotic medications at adequate dosages, including 1 long-acting injectable, but Mr. A has remained consistently symptomatic.
Although disorganized and psychotic, Mr. A does not meet criteria for long-term involuntary hospitalization. His family wants to take him home, and the treatment team discusses clozapine as an antipsychotic option. Mr. A and his family agree to a trial of clozapine during voluntary hospitalization, but they would like him home within a week to attend his sister’s birthday party.
The treatment team decides to initiate clozapine and monitor his response in a controlled setting for a few days before transitioning him to outpatient care.
Initiating clozapine therapyThe case of Mr. A exemplifies a situation in which initiating clozapine is a reasonable clinical consideration. As the first step, we recommend checking baseline lab values and vital signs (Table 2), keeping in mind that the REMS program requires a baseline ANC within 7 days of initiating clozapine. When working with a highly disorganized or agitated patient, balance benefits of testing against the risk of harm to staff and patient.

REMS guidelines recommend a baseline ANC ≥1,500/µL for a new patient starting clozapine, except when benign ethnic neutropenia (BEN) has been confirmed. (Initiation guidelines for BEN are discussed later in this article.)
Dosing alternatives. We recommend following the manufacturer’s dosing guidelines when initiating clozapine (Figure 2).13,14 Three oral forms are available: tablet, disintegrating tablet, and suspension. All can be titrated using the schedule suggested with tablets. The disintegrating tablets or suspension might be beneficial for a patient with either:
- a history of “cheeking” or otherwise disposing of tablets
- a medical condition that affects swallowing or absorption.
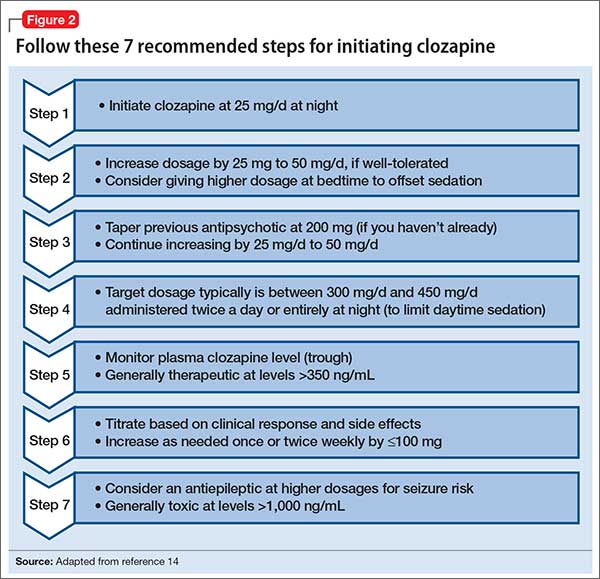
The disintegrating tablet is available in 12.5-mg, 25-mg, 100-mg, 150-mg, and 200-mg doses. It dissolves without requiring additional liquids. Each mL of the suspension contains 50 mg of clozapine.
Rapid titration? One group, working in Romania, examined the safety and efficacy of rapid titration of clozapine in 111 inpatients with schizophrenia.15 In the absence of additional studies, we do not recommend routine rapid titration of clozapine.
Monitoring: Greater flexibilityUnder the REMS program, laboratory monitoring of clozapine treatment must continue indefinitely. If not, pharmacies cannot dispense clozapine. Fortunately, the ANC is the only lab value tracked by the registry, and the frequency of required blood draws decreases over time (Figure 3).

Other guideline changes provide clinicians with greater flexibility to make patient-specific treatment decisions; for example, the allowable ANC to continue clozapine therapy has decreased. Usually, clozapine therapy should be interrupted for an ANC <1,000/µL if the prescriber suspects clozapine-induced neutropenia. Even when the ANC drops below 1,000/µL, however, prescribers can now continue clozapine treatment if they consider the benefits to outweigh risks for a given patient.
Separate guidelines now exist for patients with BEN, most commonly observed in persons of certain ethnic groups. BEN typically is diagnosed based on repeated ANC values <1,500/µL over several months. Patients with BEN do not have an increased risk of oral or systemic infections, as occur with other congenital neutropenias.16 In patients with BEN, clozapine therapy:
- can be initiated only after at least 2 baseline ANC measurements ≥1,000/µL
- should be interrupted for an ANC <500/µL if the prescriber suspects clozapine-induced neutropenia.
Substantial drops in ANC no longer require action (repeat lab draws) unless the drop causes neutropenia. Prescribers will receive an automated notification any time a patient experiences neutropenia that is considered mild (ANC 1,000 to 1,499/µL), moderate (ANC 500 to 999/µL), or severe (ANC <500/µL).
The NNRMF list is no longer definitive. All patients are now eligible for rechallenge, assuming they meet the new clozapine initiation criteria.
Next, when rediscovering clozapine: Adverse effectsDespite an intimidating list of side effects and interactions, clozapine is associated with a significant reduction in patients’ risk of overall mortality. In Part 2 of this series in the August 2016 issue, we discuss early identification of clozapine’s adverse effects and provide guidance for management.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stroup TS, Gerhard T, Crystal S, et al. Geographic and clinical variation in clozapine use in the United States. Psychiatr Serv. 2014;65(2):186-192.
2. Olfson M, Gerhard T, Crystal S, et al. Clozapine for schizophrenia: state variation in evidence-based practice. Psychiatr Serv. 2016;67(2):152.
3. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
4. Cetin M. Clozaphobia: fear of prescribers of clozapine for treatment of schizophrenia. Klinik Psikofarmakol Bulteni. 2014;24(4):295-301.
5. Hippius H. A historical perspective of clozapine. J Clin Psychiatry. 1999;60(suppl 12):22-23.
6. Amsler HA, Teerenhovi L, Barth E, et al. Agranulocytosis in patients treated with clozapine. A study of the Finnish epidemic. Acta Psychiatr Scand. 1977;56(4):241-248.
7. Crilly J. The history of clozapine and its emergence in the U.S. market: a review and analysis. Hist Psychiatry. 2007;18(1):39-60.
8. Claghorn J, Honigfeld G, Abuzzahab FS, et al. The risks and benefits of clozapine versus chlorpromazine. J Clin Psychopharmacol. 1987;7(6):377-384.
9. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
10. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA modified monitoring for neutropenia associated with schizophrenia medicine clozapine; approves new shared REMS program for all clozapine medicines. http://www.fda.gov/Drugs/DrugSafety/ucm461853.htm. Published September 15, 2015. Accessed November 23, 2015.
11. Clozapine REMS Program. What’s new with clozapine: an overview. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/WhatsNEWwithClozapine_An%20Overview.pdf. Published September 2015. Accessed November 23, 2015.
12. Clozapine REMS Program. Clozapine and the risk of neutropenia: a guide for healthcare providers. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/resources/Clozapine_REMS_HCP_Guide.pdf. Published September 2015. Accessed November 23, 2015.
13. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 16, 2016.
14. Newman WJ. Psychopharmacologic management of aggression. Psychiatr Clin North Am. 2012;35(4):957-972.
15. Ifteni P, Nielsen J, Burtea V, et al. Effectiveness and safety of rapid clozapine titration in schizophrenia. Acta Psychiatr Scand. 2014;130(1):25-29.
16. Hsieh MM, Tisdale JF, Rodgers GP, et al. Neutrophil count in African Americans: lowering the target cutoff to initiate or resume chemotherapy? J Clin Oncol. 2010;28(10):1633-1637.
Although clozapine is the medication with the clearest benefits in treatment-resistant schizophrenia, many eligible patients never receive it. In the United States, 20% to 30% of patients with schizophrenia can be classified as treatment resistant, but clozapine accounts for <5% of antipsychotics prescribed.1,2 Clinicians worldwide tend to under-prescribe clozapine3—a reluctance one author coined as “clozaphobia.”4
Admittedly, clozapine has had a turbulent history—both lauded as a near-miracle drug and condemned as a deadly agent. The FDA has overhauled its prescribing and monitoring guidelines, however, offering psychiatrists a perfect opportunity to reacquaint themselves with this potentially life-changing intervention.
We begin this article with clozapine’s story, then spotlight new terrain the FDA created in 2015 when the agency introduced the Clozapine Risk Evaluation and Mitigation Strategy (REMS). Our goal in the 3 articles of this series is to deepen your appreciation for this tricyclic antipsychotic and provide practical clinical guidance for using it safely and effectively.
Setbacks, but the drug has an enduring presenceThe 1950s was an exciting era of exploration for new psychotropic medications. While searching for tricyclic antidepressants, Wander Laboratories discovered neuroleptic tricyclics, with clozapine identified in 1959 (Figure 1). Haloperidol’s development and release in the 1960s reinforced the prevailing dogma of the time that effective neuroleptics correlated with extrapyramidal symptoms, thus limiting interest in the newly discovered, but pharmacologically unique, clozapine. Throughout the 1960s, most research on clozapine was published in German, with less of an international presence.5
Agranulocytosis deaths. Clozapine earned its scarlet letter in 1975, when 8 patients in Finland died of agranulocytosis.6 Sandoz, its manufacturer, withdrew clozapine from the market and halted all clinical trials. The Finnish epidemic triggered detailed investigations into blood dyscrasias and early identification of agranulocytosis associated with clozapine and other antipsychotics.7
Clozapine endured only because of its unique efficacy. When psychiatrists witnessed relapses in patients who had to discontinue clozapine, some countries allowed its use with strict monitoring.5 The FDA kept clozapine minimally available in the United States by allowing so-called “compassionate need programs” to continue.7
New data, FDA approval. Two studies in 1987 and 1988 that compared clozapine with chlorpromazine for treatment-refractory schizophrenia demonstrated clozapine’s superior effect on both negative and positive symptoms.8,9 The FDA approved clozapine for refractory schizophrenia in 1989, and clozapine became clinically available in 1990.
Initially, the high annual cost of clozapine’s required “bundle” ($8,900 per patient for medication and monitoring) led to political outcry. As patients and their family struggled to afford the newly released medication, multiple states filed antitrust lawsuits. A federal court found both the manufacturer and individual states at fault and required expanded access to clozapine and its necessary monitoring. National clozapine registries were formed, and bundling was eliminated.7
The clozapine REMS programSix clozapine registries operated independently, each managed by a different manufacturer,10 until the FDA introduced REMS in September 2015. The REMS program created a centralized registry to monitor all U.S. patients treated with clozapine and made important changes to prescribing and monitoring guidelines.11,12 It also incorporated the National Non-Rechallenge Master File (NNRMF).
Initially, the REMS program was scheduled for rollout October 12, 2015, the closing date of the 6 registries. Since November 2015, pharmacies have been required to register with the program to dispense clozapine. A similar registration deadline for clozapine prescribers was extended indefinitely, however, because of technical problems. Once the deadline is finalized, all clozapine prescribers must complete 3 steps to be certified in the REMS program (Table 1).11
New requirements. Certified clozapine prescribers will have new responsibilities: enrolling patients and submitting lab results. They can designate someone else to perform these tasks on their behalf, but designees must enroll in the REMS program and the prescriber must confirm the designee. Pharmacists can no longer enroll patients for clozapine therapy unless they are confirmed as a prescriber designee. For outpatients, the absolute neutrophil count (ANC) must be reported before the pharmacy can dispense clozapine. For inpatients, the ANC must be reported within 7 days of the patient’s most recent blood draw.
Once the system is fully operational, Social Security numbers will no longer be used as patient identification for dispensing clozapine. Instead, outpatient pharmacies will obtain a predispense authorization, or PDA, from the REMS program. A person initiated on clozapine as an inpatient must be re-enrolled after discharge by their outpatient prescriber.
The REMS program includes information about clozapine patients who were maintained through the 6 registries, and these patients have been allowed to continue clozapine treatment. Data pertaining to patients last prescribed clozapine before October 1, 2012, did not transfer into the new system unless their name was on the NNRMF.
CASE
Is Mr. A a candidate for clozapine?Age 28, with schizophrenia, Mr. A is highly disorganized and psychotic when brought to the emergency room by police for inappropriate behavior. His family arrives and reports that similar events have occurred several times over the past few years. Mr. A’s outpatient psychiatrist has prescribed 3 different antipsychotic medications at adequate dosages, including 1 long-acting injectable, but Mr. A has remained consistently symptomatic.
Although disorganized and psychotic, Mr. A does not meet criteria for long-term involuntary hospitalization. His family wants to take him home, and the treatment team discusses clozapine as an antipsychotic option. Mr. A and his family agree to a trial of clozapine during voluntary hospitalization, but they would like him home within a week to attend his sister’s birthday party.
The treatment team decides to initiate clozapine and monitor his response in a controlled setting for a few days before transitioning him to outpatient care.
Initiating clozapine therapyThe case of Mr. A exemplifies a situation in which initiating clozapine is a reasonable clinical consideration. As the first step, we recommend checking baseline lab values and vital signs (Table 2), keeping in mind that the REMS program requires a baseline ANC within 7 days of initiating clozapine. When working with a highly disorganized or agitated patient, balance benefits of testing against the risk of harm to staff and patient.
REMS guidelines recommend a baseline ANC ≥1,500/µL for a new patient starting clozapine, except when benign ethnic neutropenia (BEN) has been confirmed. (Initiation guidelines for BEN are discussed later in this article.)
Dosing alternatives. We recommend following the manufacturer’s dosing guidelines when initiating clozapine (Figure 2).13,14 Three oral forms are available: tablet, disintegrating tablet, and suspension. All can be titrated using the schedule suggested with tablets. The disintegrating tablets or suspension might be beneficial for a patient with either:
- a history of “cheeking” or otherwise disposing of tablets
- a medical condition that affects swallowing or absorption.
The disintegrating tablet is available in 12.5-mg, 25-mg, 100-mg, 150-mg, and 200-mg doses. It dissolves without requiring additional liquids. Each mL of the suspension contains 50 mg of clozapine.
Rapid titration? One group, working in Romania, examined the safety and efficacy of rapid titration of clozapine in 111 inpatients with schizophrenia.15 In the absence of additional studies, we do not recommend routine rapid titration of clozapine.
Monitoring: Greater flexibilityUnder the REMS program, laboratory monitoring of clozapine treatment must continue indefinitely. If not, pharmacies cannot dispense clozapine. Fortunately, the ANC is the only lab value tracked by the registry, and the frequency of required blood draws decreases over time (Figure 3).
Other guideline changes provide clinicians with greater flexibility to make patient-specific treatment decisions; for example, the allowable ANC to continue clozapine therapy has decreased. Usually, clozapine therapy should be interrupted for an ANC <1,000/µL if the prescriber suspects clozapine-induced neutropenia. Even when the ANC drops below 1,000/µL, however, prescribers can now continue clozapine treatment if they consider the benefits to outweigh risks for a given patient.
Separate guidelines now exist for patients with BEN, most commonly observed in persons of certain ethnic groups. BEN typically is diagnosed based on repeated ANC values <1,500/µL over several months. Patients with BEN do not have an increased risk of oral or systemic infections, as occur with other congenital neutropenias.16 In patients with BEN, clozapine therapy:
- can be initiated only after at least 2 baseline ANC measurements ≥1,000/µL
- should be interrupted for an ANC <500/µL if the prescriber suspects clozapine-induced neutropenia.
Substantial drops in ANC no longer require action (repeat lab draws) unless the drop causes neutropenia. Prescribers will receive an automated notification any time a patient experiences neutropenia that is considered mild (ANC 1,000 to 1,499/µL), moderate (ANC 500 to 999/µL), or severe (ANC <500/µL).
The NNRMF list is no longer definitive. All patients are now eligible for rechallenge, assuming they meet the new clozapine initiation criteria.
Next, when rediscovering clozapine: Adverse effectsDespite an intimidating list of side effects and interactions, clozapine is associated with a significant reduction in patients’ risk of overall mortality. In Part 2 of this series in the August 2016 issue, we discuss early identification of clozapine’s adverse effects and provide guidance for management.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Although clozapine is the medication with the clearest benefits in treatment-resistant schizophrenia, many eligible patients never receive it. In the United States, 20% to 30% of patients with schizophrenia can be classified as treatment resistant, but clozapine accounts for <5% of antipsychotics prescribed.1,2 Clinicians worldwide tend to under-prescribe clozapine3—a reluctance one author coined as “clozaphobia.”4
Admittedly, clozapine has had a turbulent history—both lauded as a near-miracle drug and condemned as a deadly agent. The FDA has overhauled its prescribing and monitoring guidelines, however, offering psychiatrists a perfect opportunity to reacquaint themselves with this potentially life-changing intervention.
We begin this article with clozapine’s story, then spotlight new terrain the FDA created in 2015 when the agency introduced the Clozapine Risk Evaluation and Mitigation Strategy (REMS). Our goal in the 3 articles of this series is to deepen your appreciation for this tricyclic antipsychotic and provide practical clinical guidance for using it safely and effectively.
Setbacks, but the drug has an enduring presenceThe 1950s was an exciting era of exploration for new psychotropic medications. While searching for tricyclic antidepressants, Wander Laboratories discovered neuroleptic tricyclics, with clozapine identified in 1959 (Figure 1). Haloperidol’s development and release in the 1960s reinforced the prevailing dogma of the time that effective neuroleptics correlated with extrapyramidal symptoms, thus limiting interest in the newly discovered, but pharmacologically unique, clozapine. Throughout the 1960s, most research on clozapine was published in German, with less of an international presence.5
Agranulocytosis deaths. Clozapine earned its scarlet letter in 1975, when 8 patients in Finland died of agranulocytosis.6 Sandoz, its manufacturer, withdrew clozapine from the market and halted all clinical trials. The Finnish epidemic triggered detailed investigations into blood dyscrasias and early identification of agranulocytosis associated with clozapine and other antipsychotics.7
Clozapine endured only because of its unique efficacy. When psychiatrists witnessed relapses in patients who had to discontinue clozapine, some countries allowed its use with strict monitoring.5 The FDA kept clozapine minimally available in the United States by allowing so-called “compassionate need programs” to continue.7
New data, FDA approval. Two studies in 1987 and 1988 that compared clozapine with chlorpromazine for treatment-refractory schizophrenia demonstrated clozapine’s superior effect on both negative and positive symptoms.8,9 The FDA approved clozapine for refractory schizophrenia in 1989, and clozapine became clinically available in 1990.
Initially, the high annual cost of clozapine’s required “bundle” ($8,900 per patient for medication and monitoring) led to political outcry. As patients and their family struggled to afford the newly released medication, multiple states filed antitrust lawsuits. A federal court found both the manufacturer and individual states at fault and required expanded access to clozapine and its necessary monitoring. National clozapine registries were formed, and bundling was eliminated.7
The clozapine REMS programSix clozapine registries operated independently, each managed by a different manufacturer,10 until the FDA introduced REMS in September 2015. The REMS program created a centralized registry to monitor all U.S. patients treated with clozapine and made important changes to prescribing and monitoring guidelines.11,12 It also incorporated the National Non-Rechallenge Master File (NNRMF).
Initially, the REMS program was scheduled for rollout October 12, 2015, the closing date of the 6 registries. Since November 2015, pharmacies have been required to register with the program to dispense clozapine. A similar registration deadline for clozapine prescribers was extended indefinitely, however, because of technical problems. Once the deadline is finalized, all clozapine prescribers must complete 3 steps to be certified in the REMS program (Table 1).11
New requirements. Certified clozapine prescribers will have new responsibilities: enrolling patients and submitting lab results. They can designate someone else to perform these tasks on their behalf, but designees must enroll in the REMS program and the prescriber must confirm the designee. Pharmacists can no longer enroll patients for clozapine therapy unless they are confirmed as a prescriber designee. For outpatients, the absolute neutrophil count (ANC) must be reported before the pharmacy can dispense clozapine. For inpatients, the ANC must be reported within 7 days of the patient’s most recent blood draw.
Once the system is fully operational, Social Security numbers will no longer be used as patient identification for dispensing clozapine. Instead, outpatient pharmacies will obtain a predispense authorization, or PDA, from the REMS program. A person initiated on clozapine as an inpatient must be re-enrolled after discharge by their outpatient prescriber.
The REMS program includes information about clozapine patients who were maintained through the 6 registries, and these patients have been allowed to continue clozapine treatment. Data pertaining to patients last prescribed clozapine before October 1, 2012, did not transfer into the new system unless their name was on the NNRMF.
CASE
Is Mr. A a candidate for clozapine?Age 28, with schizophrenia, Mr. A is highly disorganized and psychotic when brought to the emergency room by police for inappropriate behavior. His family arrives and reports that similar events have occurred several times over the past few years. Mr. A’s outpatient psychiatrist has prescribed 3 different antipsychotic medications at adequate dosages, including 1 long-acting injectable, but Mr. A has remained consistently symptomatic.
Although disorganized and psychotic, Mr. A does not meet criteria for long-term involuntary hospitalization. His family wants to take him home, and the treatment team discusses clozapine as an antipsychotic option. Mr. A and his family agree to a trial of clozapine during voluntary hospitalization, but they would like him home within a week to attend his sister’s birthday party.
The treatment team decides to initiate clozapine and monitor his response in a controlled setting for a few days before transitioning him to outpatient care.
Initiating clozapine therapyThe case of Mr. A exemplifies a situation in which initiating clozapine is a reasonable clinical consideration. As the first step, we recommend checking baseline lab values and vital signs (Table 2), keeping in mind that the REMS program requires a baseline ANC within 7 days of initiating clozapine. When working with a highly disorganized or agitated patient, balance benefits of testing against the risk of harm to staff and patient.
REMS guidelines recommend a baseline ANC ≥1,500/µL for a new patient starting clozapine, except when benign ethnic neutropenia (BEN) has been confirmed. (Initiation guidelines for BEN are discussed later in this article.)
Dosing alternatives. We recommend following the manufacturer’s dosing guidelines when initiating clozapine (Figure 2).13,14 Three oral forms are available: tablet, disintegrating tablet, and suspension. All can be titrated using the schedule suggested with tablets. The disintegrating tablets or suspension might be beneficial for a patient with either:
- a history of “cheeking” or otherwise disposing of tablets
- a medical condition that affects swallowing or absorption.
The disintegrating tablet is available in 12.5-mg, 25-mg, 100-mg, 150-mg, and 200-mg doses. It dissolves without requiring additional liquids. Each mL of the suspension contains 50 mg of clozapine.
Rapid titration? One group, working in Romania, examined the safety and efficacy of rapid titration of clozapine in 111 inpatients with schizophrenia.15 In the absence of additional studies, we do not recommend routine rapid titration of clozapine.
Monitoring: Greater flexibilityUnder the REMS program, laboratory monitoring of clozapine treatment must continue indefinitely. If not, pharmacies cannot dispense clozapine. Fortunately, the ANC is the only lab value tracked by the registry, and the frequency of required blood draws decreases over time (Figure 3).
Other guideline changes provide clinicians with greater flexibility to make patient-specific treatment decisions; for example, the allowable ANC to continue clozapine therapy has decreased. Usually, clozapine therapy should be interrupted for an ANC <1,000/µL if the prescriber suspects clozapine-induced neutropenia. Even when the ANC drops below 1,000/µL, however, prescribers can now continue clozapine treatment if they consider the benefits to outweigh risks for a given patient.
Separate guidelines now exist for patients with BEN, most commonly observed in persons of certain ethnic groups. BEN typically is diagnosed based on repeated ANC values <1,500/µL over several months. Patients with BEN do not have an increased risk of oral or systemic infections, as occur with other congenital neutropenias.16 In patients with BEN, clozapine therapy:
- can be initiated only after at least 2 baseline ANC measurements ≥1,000/µL
- should be interrupted for an ANC <500/µL if the prescriber suspects clozapine-induced neutropenia.
Substantial drops in ANC no longer require action (repeat lab draws) unless the drop causes neutropenia. Prescribers will receive an automated notification any time a patient experiences neutropenia that is considered mild (ANC 1,000 to 1,499/µL), moderate (ANC 500 to 999/µL), or severe (ANC <500/µL).
The NNRMF list is no longer definitive. All patients are now eligible for rechallenge, assuming they meet the new clozapine initiation criteria.
Next, when rediscovering clozapine: Adverse effectsDespite an intimidating list of side effects and interactions, clozapine is associated with a significant reduction in patients’ risk of overall mortality. In Part 2 of this series in the August 2016 issue, we discuss early identification of clozapine’s adverse effects and provide guidance for management.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stroup TS, Gerhard T, Crystal S, et al. Geographic and clinical variation in clozapine use in the United States. Psychiatr Serv. 2014;65(2):186-192.
2. Olfson M, Gerhard T, Crystal S, et al. Clozapine for schizophrenia: state variation in evidence-based practice. Psychiatr Serv. 2016;67(2):152.
3. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
4. Cetin M. Clozaphobia: fear of prescribers of clozapine for treatment of schizophrenia. Klinik Psikofarmakol Bulteni. 2014;24(4):295-301.
5. Hippius H. A historical perspective of clozapine. J Clin Psychiatry. 1999;60(suppl 12):22-23.
6. Amsler HA, Teerenhovi L, Barth E, et al. Agranulocytosis in patients treated with clozapine. A study of the Finnish epidemic. Acta Psychiatr Scand. 1977;56(4):241-248.
7. Crilly J. The history of clozapine and its emergence in the U.S. market: a review and analysis. Hist Psychiatry. 2007;18(1):39-60.
8. Claghorn J, Honigfeld G, Abuzzahab FS, et al. The risks and benefits of clozapine versus chlorpromazine. J Clin Psychopharmacol. 1987;7(6):377-384.
9. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
10. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA modified monitoring for neutropenia associated with schizophrenia medicine clozapine; approves new shared REMS program for all clozapine medicines. http://www.fda.gov/Drugs/DrugSafety/ucm461853.htm. Published September 15, 2015. Accessed November 23, 2015.
11. Clozapine REMS Program. What’s new with clozapine: an overview. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/WhatsNEWwithClozapine_An%20Overview.pdf. Published September 2015. Accessed November 23, 2015.
12. Clozapine REMS Program. Clozapine and the risk of neutropenia: a guide for healthcare providers. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/resources/Clozapine_REMS_HCP_Guide.pdf. Published September 2015. Accessed November 23, 2015.
13. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 16, 2016.
14. Newman WJ. Psychopharmacologic management of aggression. Psychiatr Clin North Am. 2012;35(4):957-972.
15. Ifteni P, Nielsen J, Burtea V, et al. Effectiveness and safety of rapid clozapine titration in schizophrenia. Acta Psychiatr Scand. 2014;130(1):25-29.
16. Hsieh MM, Tisdale JF, Rodgers GP, et al. Neutrophil count in African Americans: lowering the target cutoff to initiate or resume chemotherapy? J Clin Oncol. 2010;28(10):1633-1637.
1. Stroup TS, Gerhard T, Crystal S, et al. Geographic and clinical variation in clozapine use in the United States. Psychiatr Serv. 2014;65(2):186-192.
2. Olfson M, Gerhard T, Crystal S, et al. Clozapine for schizophrenia: state variation in evidence-based practice. Psychiatr Serv. 2016;67(2):152.
3. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
4. Cetin M. Clozaphobia: fear of prescribers of clozapine for treatment of schizophrenia. Klinik Psikofarmakol Bulteni. 2014;24(4):295-301.
5. Hippius H. A historical perspective of clozapine. J Clin Psychiatry. 1999;60(suppl 12):22-23.
6. Amsler HA, Teerenhovi L, Barth E, et al. Agranulocytosis in patients treated with clozapine. A study of the Finnish epidemic. Acta Psychiatr Scand. 1977;56(4):241-248.
7. Crilly J. The history of clozapine and its emergence in the U.S. market: a review and analysis. Hist Psychiatry. 2007;18(1):39-60.
8. Claghorn J, Honigfeld G, Abuzzahab FS, et al. The risks and benefits of clozapine versus chlorpromazine. J Clin Psychopharmacol. 1987;7(6):377-384.
9. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
10. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA modified monitoring for neutropenia associated with schizophrenia medicine clozapine; approves new shared REMS program for all clozapine medicines. http://www.fda.gov/Drugs/DrugSafety/ucm461853.htm. Published September 15, 2015. Accessed November 23, 2015.
11. Clozapine REMS Program. What’s new with clozapine: an overview. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/WhatsNEWwithClozapine_An%20Overview.pdf. Published September 2015. Accessed November 23, 2015.
12. Clozapine REMS Program. Clozapine and the risk of neutropenia: a guide for healthcare providers. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/resources/Clozapine_REMS_HCP_Guide.pdf. Published September 2015. Accessed November 23, 2015.
13. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 16, 2016.
14. Newman WJ. Psychopharmacologic management of aggression. Psychiatr Clin North Am. 2012;35(4):957-972.
15. Ifteni P, Nielsen J, Burtea V, et al. Effectiveness and safety of rapid clozapine titration in schizophrenia. Acta Psychiatr Scand. 2014;130(1):25-29.
16. Hsieh MM, Tisdale JF, Rodgers GP, et al. Neutrophil count in African Americans: lowering the target cutoff to initiate or resume chemotherapy? J Clin Oncol. 2010;28(10):1633-1637.

Pregnant and nursing patients benefit from ‘ambitious’ changes to drug labeling for safety
In December 2014, the FDA issued draft guidance for sweeping changes to labeling of pharmaceutical treatments in regard to pregnancy and lactation information. These changes are now in effect for use in practice.1 The undertaking has been years in the making, and is truly ambitious.
The outdated system of letter categories (A, B, C, D, X) falls short of clinical needs in several ways:
- the quality and volume of data can be lacking
- comparative risk is not described
- using letters can led to oversimplification or, in some cases, exaggeration of risk and safety (Box).

Other drawbacks include infrequent updating of information and omission of information about baseline rates of reproductive-related adverse events, to provide a more meaningful context for risk assessment.
A note before we continue discussion of labeling: Recognize that pregnancy itself is inherently risky; poor outcomes are, regrettably, not uncommon. The rate of birth defects in the United States is approximately 3%, and obstetric complications, such as prematurity, are common.2,3
New system described
The new labeling content has been described in the FDA’s Pregnancy and Lactation Labeling Rule (also called the “final rule”), issued in December 2014. For each medication, there will be subsections in the labeling:
- Pregnancy
- Lactation
- Females and Males of Reproductive Potential.
In addition, FDA instructions now state that labeling:
- must be updated when new information becomes available
- needs to include evaluation of human data that becomes available mainly after the drug is approved
- needs to include information about the background rates of adverse events related to reproduction.
Labeling in pregnancy. As an example, the “Pregnancy” section of every label contains 3 subsections, all of great clinical importance. First is information about pregnancy exposure registries, with a listing of scientifically acceptable registries (if a registry is available for that drug) and contact information; this section focuses on the high value of data that are systematically and prospectively collected. The second summarizes risk associated with the drug during pregnancy, based on available human, animal, and pharmacologic data. Third is a discussion of clinical considerations.
Need for appropriate controls. Psychiatric disorders increase the risk of pregnancy complications, and often are associated with variables that might increase the risk of a poor pregnancy outcome. For example, a patient who has a psychiatric disorder might be less likely to seek prenatal care, take a prenatal vitamin, and sleep and eat well; she also might use alcohol, tobacco, or other substances of abuse.
The medical literature on the reproductive safety of psychotropic medications is fraught with confounding variables other than the medications themselves. These include variables that, taken alone, might confer a poorer outcome on the fetus or newborn of a pregnant or lactating woman who has a psychiatric illness (to the extent that she uses psychotropics during a pregnancy), compared with what would be seen in (1) a healthy woman who is not taking such medication or (2) the general population.
On the new labels, detailed statements on human data include information from clinical trials, pregnancy exposure registries, and epidemiologic studies. Labels are also to include:
- incidence of adverse events
- effect of dosage
- effect of duration of exposure
- effect of gestational timing of exposure.
The labels emphasize quantifying risk relative to the risk of the same outcome in infants born to women who have not been exposed to the particular drug, but who have the disease or condition for which the drug is indicated (ie, appropriate controls).
Clinical considerations are to include information on the following related to the specific medication (when that information is known):
- more information for prescribers, to further risk-benefit counseling
- disease-associated maternal-fetal risks
- dosage adjustments during pregnancy and postpartum
- maternal adverse reactions
- fetal and neonatal adverse reactions
- labor and delivery.
Clearly, this overdue shift in providing information regarding reproductive safety has the potential to inform clinicians and patients in a meaningful way about the risks and benefits of specific treatments during pregnancy and lactation. Translating that information into practice is daunting, however.
Important aspects of implementation
Pregnancy exposure registries will play a crucial role. For most medications, no systematic registry has been established; to do so, rigorous methodology is required to acquire prospective data and account for confounding variables.4 Appropriate control groups also are required to yield data that are useful and interpretable. Primary outcomes require verification, such as review of medical records. Last, registries must be well-conducted and therefore adequately funded, yet labeling changes have not been accompanied by funding requirements set forth by regulators to pharmaceutical manufacturers.
Labeling must be updated continually. Furthermore, it is unclear who will review data for precision and comprehensiveness.
Data need to be understandable to health care providers across disciplines and to patients with varying levels of education for the label to have a meaningful impact on clinical care.
As noted, there is no mandate for funding the meticulous pharmacovigilance required to provide definitive data for labeling. It is unclear if the potential benefits of the new labeling can be reaped without adequate financing of the pharmacovigilance mechanisms required to inform patients adequately.
Role of pregnancy registries
Over the past 2 decades, pregnancy registries have emerged as a rapid, systematic means of collecting important reproductive safety data on the risk for major malformations after prenatal exposure to a medication or a class of medications.5,6 Such registries enhance the rigor of available cohort studies and other analyses of reproductive safety data that have been derived from large administrative databases.
NPRAA and NPRAD. Recently, the National Pregnancy Registry for Atypical Antipsychotics (NPRAA) and the National Pregnancy Registry for Antidepressants (NPRAD) were established in an effort to obtain reproductive safety data about fetal exposure to second-generation antipsychotics (SGAs) and to newer antidepressants.7 Based at Massachusetts General Hospital in Boston, NPRAA and NPRAD systematically and prospectively evaluate the risk of malformations among infants who have been exposed in utero to an SGA or an antidepressant.
The structure of both registries are the same, modeled after the North American Antiepileptic Drug Registry.5,8 Data are collected prospectively from pregnant women, age 18 to 45, by means of 3 telephone interviews conducted proximate to enrollment, at 7 months’ gestation, and at 2 or 3 months’ postpartum.
Participants include (1) pregnant women who have a history of fetal exposure to an SGA or an antidepressant, or both, and (2) a comparison group of non-exposed pregnant women who have a history of a psychiatric illness. Authorization for release of medical records is obtained for obstetric care, labor and delivery, and neonatal care (≤6 months of age).
Information on the presence of major malformations is abstracted from the medical record, along with other data on neonatal and maternal health outcomes. Identified cases of a congenital malformation are sent to a dysmorphologist, who has been blinded to drug exposure, for final adjudication. Release of findings is dictated by a governing Scientific Advisory Board.
Results so far. Results are available from the NPRAA.9 As of December 2014, 487 women were enrolled: 353 who used an SGA and 134 comparison women. Medical records were obtained for 82.2% of participants. A total of 303 women completed the study and were eligible for inclusion in the analysis. Findings include:
- Of 214 live births with first-trimester exposure to an SGA, 3 major malformations were confirmed. In the control group (n = 89), 1 major malformation was confirmed
- The absolute risk of a major malformation was 1.4% for an exposed infant and 1.1% for an unexposed infant
- The odds ratio for a major malformation, comparing exposed infants with unexposed infants, was 1.25 (95% CI, 0.13–12.19).
It is reasonable, therefore, to conclude that, as a class, SGAs are not major teratogens. Although the confidence intervals around the odds ratio estimate remain wide, with the probability for change over the course of the study, it is unlikely that risk will rise to the level of known major teratogens, such as valproate and thalidomide.10,11
Help with decision-making
Given recent FDA guidance about the importance of pregnancy registries (www.fda.gov/pregnancyregistries), such carefully collected data might help clinicians and patients make informed choices about treatment. Future efforts of NPRAA and NPRAD will focus on sustaining growth in enrollment of participants so that the reproductive safety of SGAs and newer antidepressants can be delineated more clearly.
Last, you can refer potential participants to NPRAA and NPRAD by calling 1-866-961-2388. More information is available at www.womensmentalhealth.org.
1. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Pregnancy, lactation, and reproductive potential: labeling for human prescription drug and biological products—content and format: guidance for industry. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM425398.pdf. Published December 2014. Accessed June 7, 2016.
2. Centers for Disease Control and Prevention. Birth defects. http://www.cdc.gov/ncbddd/birthdefects/facts.html. Updated September 21, 2005. Accessed June 7, 2016.
3. Centers for Disease Control and Prevention. Preterm birth. http://www.cdc.gov/reproductivehealth/maternalinfanthealth/pretermbirth.htm. Updated December 4, 2015. Accessed June 7, 2016.
4. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Guidance for industry: establishing pregnancy exposure registries. http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133332.pdf. Published August 2002. Accessed June 7, 2016.
5. Holmes LB, Wyszynski DF. North American antiepileptic drug pregnancy registry. Epilepsia. 2004;45(11):1465.
6. Tomson T, Battino D, Craig J, et al; ILAE Commission on Therapeutic Strategies. Pregnancy registries: differences, similarities, and possible harmonization. Epilepsia. 2010;51(5):909-915.
7. Cohen LS, Viguera AC, McInerney KA, et al. Establishment of the National Pregnancy Registry for Atypical Antipsychotics. J Clin Psychiatry. 2015;76(7):986-989.
8. Holmes LB, Wyszynski DF, Lieberman E. The AED (antiepileptic drug) pregnancy registry: a 6-year experience. Arch Neurol. 2004;61(5):673-678.
9. Cohen LS, Viguera AC, McInerney KA, et al. Reproductive safety of second-generation antipsychotics: current data from the Massachusetts General Hospital National Pregnancy Registry for Atypical Antipsychotics. Am J Psychiatry. 2016;173(3):263-270.
10. McBride WG. Thalidomide and congenital abnormalities. Lancet. 1961;2(7216):1358.
11. Wyszynski DF, Nambisan M, Surve T, et al; Antiepileptic Drug Pregnancy Registry. Increased rate of major malformations in offspring exposed to valproate during pregnancy. Neurology. 2005;64(6):961-965.
In December 2014, the FDA issued draft guidance for sweeping changes to labeling of pharmaceutical treatments in regard to pregnancy and lactation information. These changes are now in effect for use in practice.1 The undertaking has been years in the making, and is truly ambitious.
The outdated system of letter categories (A, B, C, D, X) falls short of clinical needs in several ways:
- the quality and volume of data can be lacking
- comparative risk is not described
- using letters can led to oversimplification or, in some cases, exaggeration of risk and safety (Box).
Other drawbacks include infrequent updating of information and omission of information about baseline rates of reproductive-related adverse events, to provide a more meaningful context for risk assessment.
A note before we continue discussion of labeling: Recognize that pregnancy itself is inherently risky; poor outcomes are, regrettably, not uncommon. The rate of birth defects in the United States is approximately 3%, and obstetric complications, such as prematurity, are common.2,3
New system described
The new labeling content has been described in the FDA’s Pregnancy and Lactation Labeling Rule (also called the “final rule”), issued in December 2014. For each medication, there will be subsections in the labeling:
- Pregnancy
- Lactation
- Females and Males of Reproductive Potential.
In addition, FDA instructions now state that labeling:
- must be updated when new information becomes available
- needs to include evaluation of human data that becomes available mainly after the drug is approved
- needs to include information about the background rates of adverse events related to reproduction.
Labeling in pregnancy. As an example, the “Pregnancy” section of every label contains 3 subsections, all of great clinical importance. First is information about pregnancy exposure registries, with a listing of scientifically acceptable registries (if a registry is available for that drug) and contact information; this section focuses on the high value of data that are systematically and prospectively collected. The second summarizes risk associated with the drug during pregnancy, based on available human, animal, and pharmacologic data. Third is a discussion of clinical considerations.
Need for appropriate controls. Psychiatric disorders increase the risk of pregnancy complications, and often are associated with variables that might increase the risk of a poor pregnancy outcome. For example, a patient who has a psychiatric disorder might be less likely to seek prenatal care, take a prenatal vitamin, and sleep and eat well; she also might use alcohol, tobacco, or other substances of abuse.
The medical literature on the reproductive safety of psychotropic medications is fraught with confounding variables other than the medications themselves. These include variables that, taken alone, might confer a poorer outcome on the fetus or newborn of a pregnant or lactating woman who has a psychiatric illness (to the extent that she uses psychotropics during a pregnancy), compared with what would be seen in (1) a healthy woman who is not taking such medication or (2) the general population.
On the new labels, detailed statements on human data include information from clinical trials, pregnancy exposure registries, and epidemiologic studies. Labels are also to include:
- incidence of adverse events
- effect of dosage
- effect of duration of exposure
- effect of gestational timing of exposure.
The labels emphasize quantifying risk relative to the risk of the same outcome in infants born to women who have not been exposed to the particular drug, but who have the disease or condition for which the drug is indicated (ie, appropriate controls).
Clinical considerations are to include information on the following related to the specific medication (when that information is known):
- more information for prescribers, to further risk-benefit counseling
- disease-associated maternal-fetal risks
- dosage adjustments during pregnancy and postpartum
- maternal adverse reactions
- fetal and neonatal adverse reactions
- labor and delivery.
Clearly, this overdue shift in providing information regarding reproductive safety has the potential to inform clinicians and patients in a meaningful way about the risks and benefits of specific treatments during pregnancy and lactation. Translating that information into practice is daunting, however.
Important aspects of implementation
Pregnancy exposure registries will play a crucial role. For most medications, no systematic registry has been established; to do so, rigorous methodology is required to acquire prospective data and account for confounding variables.4 Appropriate control groups also are required to yield data that are useful and interpretable. Primary outcomes require verification, such as review of medical records. Last, registries must be well-conducted and therefore adequately funded, yet labeling changes have not been accompanied by funding requirements set forth by regulators to pharmaceutical manufacturers.
Labeling must be updated continually. Furthermore, it is unclear who will review data for precision and comprehensiveness.
Data need to be understandable to health care providers across disciplines and to patients with varying levels of education for the label to have a meaningful impact on clinical care.
As noted, there is no mandate for funding the meticulous pharmacovigilance required to provide definitive data for labeling. It is unclear if the potential benefits of the new labeling can be reaped without adequate financing of the pharmacovigilance mechanisms required to inform patients adequately.
Role of pregnancy registries
Over the past 2 decades, pregnancy registries have emerged as a rapid, systematic means of collecting important reproductive safety data on the risk for major malformations after prenatal exposure to a medication or a class of medications.5,6 Such registries enhance the rigor of available cohort studies and other analyses of reproductive safety data that have been derived from large administrative databases.
NPRAA and NPRAD. Recently, the National Pregnancy Registry for Atypical Antipsychotics (NPRAA) and the National Pregnancy Registry for Antidepressants (NPRAD) were established in an effort to obtain reproductive safety data about fetal exposure to second-generation antipsychotics (SGAs) and to newer antidepressants.7 Based at Massachusetts General Hospital in Boston, NPRAA and NPRAD systematically and prospectively evaluate the risk of malformations among infants who have been exposed in utero to an SGA or an antidepressant.
The structure of both registries are the same, modeled after the North American Antiepileptic Drug Registry.5,8 Data are collected prospectively from pregnant women, age 18 to 45, by means of 3 telephone interviews conducted proximate to enrollment, at 7 months’ gestation, and at 2 or 3 months’ postpartum.
Participants include (1) pregnant women who have a history of fetal exposure to an SGA or an antidepressant, or both, and (2) a comparison group of non-exposed pregnant women who have a history of a psychiatric illness. Authorization for release of medical records is obtained for obstetric care, labor and delivery, and neonatal care (≤6 months of age).
Information on the presence of major malformations is abstracted from the medical record, along with other data on neonatal and maternal health outcomes. Identified cases of a congenital malformation are sent to a dysmorphologist, who has been blinded to drug exposure, for final adjudication. Release of findings is dictated by a governing Scientific Advisory Board.
Results so far. Results are available from the NPRAA.9 As of December 2014, 487 women were enrolled: 353 who used an SGA and 134 comparison women. Medical records were obtained for 82.2% of participants. A total of 303 women completed the study and were eligible for inclusion in the analysis. Findings include:
- Of 214 live births with first-trimester exposure to an SGA, 3 major malformations were confirmed. In the control group (n = 89), 1 major malformation was confirmed
- The absolute risk of a major malformation was 1.4% for an exposed infant and 1.1% for an unexposed infant
- The odds ratio for a major malformation, comparing exposed infants with unexposed infants, was 1.25 (95% CI, 0.13–12.19).
It is reasonable, therefore, to conclude that, as a class, SGAs are not major teratogens. Although the confidence intervals around the odds ratio estimate remain wide, with the probability for change over the course of the study, it is unlikely that risk will rise to the level of known major teratogens, such as valproate and thalidomide.10,11
Help with decision-making
Given recent FDA guidance about the importance of pregnancy registries (www.fda.gov/pregnancyregistries), such carefully collected data might help clinicians and patients make informed choices about treatment. Future efforts of NPRAA and NPRAD will focus on sustaining growth in enrollment of participants so that the reproductive safety of SGAs and newer antidepressants can be delineated more clearly.
Last, you can refer potential participants to NPRAA and NPRAD by calling 1-866-961-2388. More information is available at www.womensmentalhealth.org.
In December 2014, the FDA issued draft guidance for sweeping changes to labeling of pharmaceutical treatments in regard to pregnancy and lactation information. These changes are now in effect for use in practice.1 The undertaking has been years in the making, and is truly ambitious.
The outdated system of letter categories (A, B, C, D, X) falls short of clinical needs in several ways:
- the quality and volume of data can be lacking
- comparative risk is not described
- using letters can led to oversimplification or, in some cases, exaggeration of risk and safety (Box).
Other drawbacks include infrequent updating of information and omission of information about baseline rates of reproductive-related adverse events, to provide a more meaningful context for risk assessment.
A note before we continue discussion of labeling: Recognize that pregnancy itself is inherently risky; poor outcomes are, regrettably, not uncommon. The rate of birth defects in the United States is approximately 3%, and obstetric complications, such as prematurity, are common.2,3
New system described
The new labeling content has been described in the FDA’s Pregnancy and Lactation Labeling Rule (also called the “final rule”), issued in December 2014. For each medication, there will be subsections in the labeling:
- Pregnancy
- Lactation
- Females and Males of Reproductive Potential.
In addition, FDA instructions now state that labeling:
- must be updated when new information becomes available
- needs to include evaluation of human data that becomes available mainly after the drug is approved
- needs to include information about the background rates of adverse events related to reproduction.
Labeling in pregnancy. As an example, the “Pregnancy” section of every label contains 3 subsections, all of great clinical importance. First is information about pregnancy exposure registries, with a listing of scientifically acceptable registries (if a registry is available for that drug) and contact information; this section focuses on the high value of data that are systematically and prospectively collected. The second summarizes risk associated with the drug during pregnancy, based on available human, animal, and pharmacologic data. Third is a discussion of clinical considerations.
Need for appropriate controls. Psychiatric disorders increase the risk of pregnancy complications, and often are associated with variables that might increase the risk of a poor pregnancy outcome. For example, a patient who has a psychiatric disorder might be less likely to seek prenatal care, take a prenatal vitamin, and sleep and eat well; she also might use alcohol, tobacco, or other substances of abuse.
The medical literature on the reproductive safety of psychotropic medications is fraught with confounding variables other than the medications themselves. These include variables that, taken alone, might confer a poorer outcome on the fetus or newborn of a pregnant or lactating woman who has a psychiatric illness (to the extent that she uses psychotropics during a pregnancy), compared with what would be seen in (1) a healthy woman who is not taking such medication or (2) the general population.
On the new labels, detailed statements on human data include information from clinical trials, pregnancy exposure registries, and epidemiologic studies. Labels are also to include:
- incidence of adverse events
- effect of dosage
- effect of duration of exposure
- effect of gestational timing of exposure.
The labels emphasize quantifying risk relative to the risk of the same outcome in infants born to women who have not been exposed to the particular drug, but who have the disease or condition for which the drug is indicated (ie, appropriate controls).
Clinical considerations are to include information on the following related to the specific medication (when that information is known):
- more information for prescribers, to further risk-benefit counseling
- disease-associated maternal-fetal risks
- dosage adjustments during pregnancy and postpartum
- maternal adverse reactions
- fetal and neonatal adverse reactions
- labor and delivery.
Clearly, this overdue shift in providing information regarding reproductive safety has the potential to inform clinicians and patients in a meaningful way about the risks and benefits of specific treatments during pregnancy and lactation. Translating that information into practice is daunting, however.
Important aspects of implementation
Pregnancy exposure registries will play a crucial role. For most medications, no systematic registry has been established; to do so, rigorous methodology is required to acquire prospective data and account for confounding variables.4 Appropriate control groups also are required to yield data that are useful and interpretable. Primary outcomes require verification, such as review of medical records. Last, registries must be well-conducted and therefore adequately funded, yet labeling changes have not been accompanied by funding requirements set forth by regulators to pharmaceutical manufacturers.
Labeling must be updated continually. Furthermore, it is unclear who will review data for precision and comprehensiveness.
Data need to be understandable to health care providers across disciplines and to patients with varying levels of education for the label to have a meaningful impact on clinical care.
As noted, there is no mandate for funding the meticulous pharmacovigilance required to provide definitive data for labeling. It is unclear if the potential benefits of the new labeling can be reaped without adequate financing of the pharmacovigilance mechanisms required to inform patients adequately.
Role of pregnancy registries
Over the past 2 decades, pregnancy registries have emerged as a rapid, systematic means of collecting important reproductive safety data on the risk for major malformations after prenatal exposure to a medication or a class of medications.5,6 Such registries enhance the rigor of available cohort studies and other analyses of reproductive safety data that have been derived from large administrative databases.
NPRAA and NPRAD. Recently, the National Pregnancy Registry for Atypical Antipsychotics (NPRAA) and the National Pregnancy Registry for Antidepressants (NPRAD) were established in an effort to obtain reproductive safety data about fetal exposure to second-generation antipsychotics (SGAs) and to newer antidepressants.7 Based at Massachusetts General Hospital in Boston, NPRAA and NPRAD systematically and prospectively evaluate the risk of malformations among infants who have been exposed in utero to an SGA or an antidepressant.
The structure of both registries are the same, modeled after the North American Antiepileptic Drug Registry.5,8 Data are collected prospectively from pregnant women, age 18 to 45, by means of 3 telephone interviews conducted proximate to enrollment, at 7 months’ gestation, and at 2 or 3 months’ postpartum.
Participants include (1) pregnant women who have a history of fetal exposure to an SGA or an antidepressant, or both, and (2) a comparison group of non-exposed pregnant women who have a history of a psychiatric illness. Authorization for release of medical records is obtained for obstetric care, labor and delivery, and neonatal care (≤6 months of age).
Information on the presence of major malformations is abstracted from the medical record, along with other data on neonatal and maternal health outcomes. Identified cases of a congenital malformation are sent to a dysmorphologist, who has been blinded to drug exposure, for final adjudication. Release of findings is dictated by a governing Scientific Advisory Board.
Results so far. Results are available from the NPRAA.9 As of December 2014, 487 women were enrolled: 353 who used an SGA and 134 comparison women. Medical records were obtained for 82.2% of participants. A total of 303 women completed the study and were eligible for inclusion in the analysis. Findings include:
- Of 214 live births with first-trimester exposure to an SGA, 3 major malformations were confirmed. In the control group (n = 89), 1 major malformation was confirmed
- The absolute risk of a major malformation was 1.4% for an exposed infant and 1.1% for an unexposed infant
- The odds ratio for a major malformation, comparing exposed infants with unexposed infants, was 1.25 (95% CI, 0.13–12.19).
It is reasonable, therefore, to conclude that, as a class, SGAs are not major teratogens. Although the confidence intervals around the odds ratio estimate remain wide, with the probability for change over the course of the study, it is unlikely that risk will rise to the level of known major teratogens, such as valproate and thalidomide.10,11
Help with decision-making
Given recent FDA guidance about the importance of pregnancy registries (www.fda.gov/pregnancyregistries), such carefully collected data might help clinicians and patients make informed choices about treatment. Future efforts of NPRAA and NPRAD will focus on sustaining growth in enrollment of participants so that the reproductive safety of SGAs and newer antidepressants can be delineated more clearly.
Last, you can refer potential participants to NPRAA and NPRAD by calling 1-866-961-2388. More information is available at www.womensmentalhealth.org.
1. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Pregnancy, lactation, and reproductive potential: labeling for human prescription drug and biological products—content and format: guidance for industry. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM425398.pdf. Published December 2014. Accessed June 7, 2016.
2. Centers for Disease Control and Prevention. Birth defects. http://www.cdc.gov/ncbddd/birthdefects/facts.html. Updated September 21, 2005. Accessed June 7, 2016.
3. Centers for Disease Control and Prevention. Preterm birth. http://www.cdc.gov/reproductivehealth/maternalinfanthealth/pretermbirth.htm. Updated December 4, 2015. Accessed June 7, 2016.
4. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Guidance for industry: establishing pregnancy exposure registries. http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133332.pdf. Published August 2002. Accessed June 7, 2016.
5. Holmes LB, Wyszynski DF. North American antiepileptic drug pregnancy registry. Epilepsia. 2004;45(11):1465.
6. Tomson T, Battino D, Craig J, et al; ILAE Commission on Therapeutic Strategies. Pregnancy registries: differences, similarities, and possible harmonization. Epilepsia. 2010;51(5):909-915.
7. Cohen LS, Viguera AC, McInerney KA, et al. Establishment of the National Pregnancy Registry for Atypical Antipsychotics. J Clin Psychiatry. 2015;76(7):986-989.
8. Holmes LB, Wyszynski DF, Lieberman E. The AED (antiepileptic drug) pregnancy registry: a 6-year experience. Arch Neurol. 2004;61(5):673-678.
9. Cohen LS, Viguera AC, McInerney KA, et al. Reproductive safety of second-generation antipsychotics: current data from the Massachusetts General Hospital National Pregnancy Registry for Atypical Antipsychotics. Am J Psychiatry. 2016;173(3):263-270.
10. McBride WG. Thalidomide and congenital abnormalities. Lancet. 1961;2(7216):1358.
11. Wyszynski DF, Nambisan M, Surve T, et al; Antiepileptic Drug Pregnancy Registry. Increased rate of major malformations in offspring exposed to valproate during pregnancy. Neurology. 2005;64(6):961-965.
1. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Pregnancy, lactation, and reproductive potential: labeling for human prescription drug and biological products—content and format: guidance for industry. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM425398.pdf. Published December 2014. Accessed June 7, 2016.
2. Centers for Disease Control and Prevention. Birth defects. http://www.cdc.gov/ncbddd/birthdefects/facts.html. Updated September 21, 2005. Accessed June 7, 2016.
3. Centers for Disease Control and Prevention. Preterm birth. http://www.cdc.gov/reproductivehealth/maternalinfanthealth/pretermbirth.htm. Updated December 4, 2015. Accessed June 7, 2016.
4. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Guidance for industry: establishing pregnancy exposure registries. http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133332.pdf. Published August 2002. Accessed June 7, 2016.
5. Holmes LB, Wyszynski DF. North American antiepileptic drug pregnancy registry. Epilepsia. 2004;45(11):1465.
6. Tomson T, Battino D, Craig J, et al; ILAE Commission on Therapeutic Strategies. Pregnancy registries: differences, similarities, and possible harmonization. Epilepsia. 2010;51(5):909-915.
7. Cohen LS, Viguera AC, McInerney KA, et al. Establishment of the National Pregnancy Registry for Atypical Antipsychotics. J Clin Psychiatry. 2015;76(7):986-989.
8. Holmes LB, Wyszynski DF, Lieberman E. The AED (antiepileptic drug) pregnancy registry: a 6-year experience. Arch Neurol. 2004;61(5):673-678.
9. Cohen LS, Viguera AC, McInerney KA, et al. Reproductive safety of second-generation antipsychotics: current data from the Massachusetts General Hospital National Pregnancy Registry for Atypical Antipsychotics. Am J Psychiatry. 2016;173(3):263-270.
10. McBride WG. Thalidomide and congenital abnormalities. Lancet. 1961;2(7216):1358.
11. Wyszynski DF, Nambisan M, Surve T, et al; Antiepileptic Drug Pregnancy Registry. Increased rate of major malformations in offspring exposed to valproate during pregnancy. Neurology. 2005;64(6):961-965.

Advances in transcranial magnetic stimulation for managing major depressive disorders
Since 2008, the FDA has cleared 4 transcranial magnetic stimulation (TMS) devices for treating depression (Related Resources). In that time, the availability of TMS has steadily grown within and outside the United States.
Parallel with increasing clinical utilization of this technology, research continues into the benefit of TMS for treatment-resistant depression; such research includes additional, supportive, acute, sham-controlled trials; comparison trials with electroconvulsive therapy (ECT) for more severe episodes of depression; short- and long-term real-world outcome studies; exploration of alternative treatment parameters to further enhance its efficacy; and the development of other TMS approaches. In this article, we review recent developments in the application of TMS to treat major depressive disorder—in particular, treatment-resistant depression (Box).

Therapeutic neuromodulation
The underlying premise of neuromodulation is that the brain is an electrochemical organ that can be modulated by pharmacotherapy or device-based approaches, or their combination.1 ECT is the prototypic device-based neuromodulation approach, and remains one of the most effective treatments for severe depression.
More recently, however, other methods have been, and continue to be, developed to treat patients who do not achieve adequate benefit from psychotherapy or medical therapy, or both, and who might not be an ideal candidate for ECT (Table,1). In addition to the potential therapeutic benefit of these alternative strategies, some could avoid safety and tolerability concerns associated with medication (weight gain, sexual dysfunction) and ECT (eg, cognitive deficits).
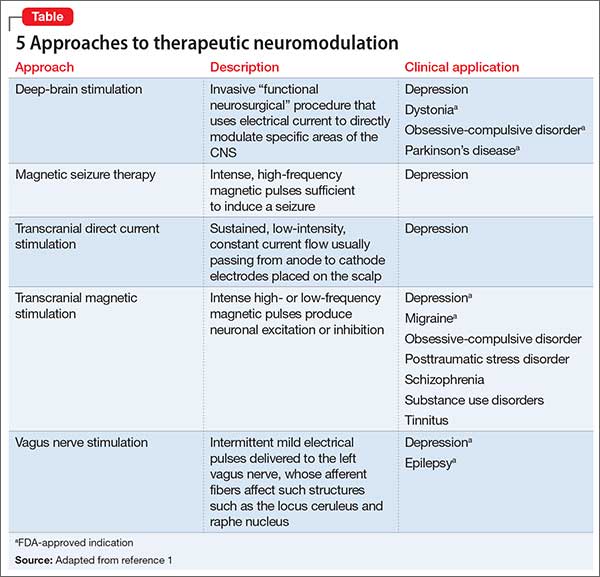
TMS, which utilizes intense, localized magnetic fields to alter activity in neural circuits implicated in the pathophysiology of depression, represents an important example of this initiative.2
TMS has established efficacy for depression
Sham-controlled trials. Several randomized, sham-controlled acute trials have demonstrated the efficacy of TMS for treatment-resistant depression.
A recent meta-analysis considered 18 studies (N = 1,970) that met the authors’ criteria for inclusion.3 They found that TMS monotherapy was statistically and clinically more effective than a sham procedure based on:
- improvement in depressive symptoms (mean decrease in baseline Hamilton Depression Rating Scale [HDRS] score, −4.53 [95% CI, −6.11 to −2.96])
- response rate; response was 3 times more likely with TMS (relative risk 3.38 [95% CI, 2.24 to 5.10])
- remission rate; remission was 5 times more likely with TMS (relative risk, 5.07 [95% CI, 2.50 to 10.30]).
Another meta-analysis (7 studies, N = 279) considered TMS as an augmentation strategy to standard medication for treatment-resistant depression.4 The authors reported that, based on change in HDRS scores, the pooled standardized mean difference between active and sham TMS augmentation was 0.86 (P < .00001). Furthermore, the pooled response rate with TMS augmentation was 46.6%, compared with 22.1% with the sham procedure (P < .0003).
Acute naturalistic TMS studies. The efficacy of TMS is supported by a large, naturalistic study of 307 patients with treatment-resistant depression who were assessed at baseline and during a standard course of TMS.5 Considering change score in the Clinician Global Impressions-Severity (CGI-S) scale, significant improvement was seen from baseline to end of treatment (−1.9 ± 1.4; P < .0001), with a clinician-assessed response rate of 58.0% and remission rate of 37.1%. Of note: Self-reported quality-of-life measures (on the Medical Outcomes Study 36-Item Short-Form Health Survey and EuroQol 5-Dimensions) also significantly improved during this relatively brief period.6
Maintenance strategies after acute TMS response. Most patients referred for TMS have a depressive illness characterized by a chronic, relapsing course and inadequate response to pharmacotherapy or psychotherapy, or their combination. An effective maintenance strategy after acute response to TMS is paramount. This includes:
- prolonged tapering schedule after an acute TMS course is completed
- maintenance medication or psychotherapy, or both
- scheduled periodic maintenance TMS sessions (usually as an augmentation strategy)
- reintroduction of TMS as needed with early signs of relapse. In this context, several trials have assessed the durability of acute TMS benefit.
A semi-controlled maintenance study followed 99 patients who had at least a 25% decrease in baseline HDRS score after acute TMS treatment.7 They were then tapered from their TMS sessions over 3 weeks while an antidepressant was titrated up. If, at any time during the subsequent 6 months, early signs of depression relapse were noted (ie, change of at least 1 point on the CGI-S for 2 consecutive weeks), TMS was reintroduced. At the end of the trial, 10 patients (13%) had relapsed and 38 (38%) had an exacerbation of symptoms sufficient to warrant reintroduction of TMS. Of those, 32 (84%) re-achieved mood stability.
In another study, 50 patients who had achieved remission during an acute course of TMS were followed for 3 months.8 After TMS taper and continued pharmacotherapy or naturalistic follow-up, 29 (58%) remained in remission; 2 (4%) maintained partial response; and 1 (2%) relapsed.
In a controlled, pilot, maintenance trial, 67 unmedicated patients with treatment-resistant depression received an acute course of TMS.9 Forty-seven of the responders were then randomized to a 1-year follow-up trial with or without a scheduled monthly TMS session. All patients could receive reintroduction TMS if they met criteria for symptom worsening.
Both groups had a similar outcome. The number of patients who did not require TMS reintroduction was 9 of 23 (39%) in the scheduled TMS group vs 9 of 26 (35%) in the no-scheduled TMS group (P < .1). Although no difference was noted between groups, the authors commented that these preliminary results will help inform larger, more definitive trials. They concluded that both acute and maintenance TMS monotherapy might be an option—for some patients.
A long-term, naturalistic outcomes study followed 257 treatment-resistant depressed patients for 1 year after they responded to an acute course of TMS.10 In addition to most patients receiving ongoing maintenance medication, they also could receive reintroduction of TMS if symptoms became worse.
Compared with pre-TMS baseline, there was a statistically significant reduction in the mean total score on the CGI-S scale (primary outcome, P < .0001) at the end of acute treatment that was sustained at follow-up. Ninety-six patients (36.2%) required reintroduction of TMS and 75 of 120 (62.5%) who initially met response or remission criteria after acute treatment continued to meet response criteria after 1 year. The authors concluded that TMS demonstrated both a statistically and clinically meaningful durability of acute benefit during this time frame.
TMS and electroconvulsive therapy
For more than 75 years, ECT has consistently proved to be an effective treatment for major depressive disorder. Although the use of ECT has fluctuated over this period, one practice survey estimated that 100,000 patients receive ECT annually.11
ECT has limitations, however, including cost, the need for general anesthesia, and cognitive deficits that range from short-term confusion to anterograde and retrograde amnesia, which can persist for weeks beyond active treatment.12 Despite increasing awareness of mental illness, stigma also remains a significant barrier to receiving ECT.
TMS vs ECT. Several trials have directly compared ECT and TMS:
- A recent meta-analysis of 9 trials included 384 patients with depression who were considered clinically appropriate for ECT and were randomized to one or the other treatment.13 Both modalities produced a significant reduction in baseline HDRS score, but ECT (15.4 point reduction) was superior to TMS (9.3 point reduction) in the degree of improvement (P < .01).
- Another meta-analysis of 9 trials (N = 425) found ECT superior to TMS in terms of response (P < .03) and remission (P < .006) rates, based on improvement in the HDRS score.14 When psychotic depressed patients were excluded, however, TMS produced effects equivalent to ECT.
In contrast to what was seen with ECT, cognitive testing of patients who received TMS revealed no deterioration in any domain. Furthermore, one of the comparison studies observed a modest, but statistically significant, improvement in patient’s working memory-executive function, objective memory, and fine-motor speed over the course of TMS treatment.15
TMS plus ECT. A 2-week, randomized, single-blind, controlled pilot study (N = 22) examined the combination of TMS and ECT as acute treatment of depression.16 Patients were assigned to receive either unilateral non-dominant (UND) ECT 3 days a week or a combination of 1 UND ECT treatment followed by 4 days of TMS. At the conclusion of treatment, UND ECT plus TMS group produced comparable efficacy and fewer adverse effects compared with the UND ECT-only group.
TMS maintenance after acute ECT response. Most patients who are referred for ECT have a depressive illness characterized by repeated episodes and incomplete response to pharmacotherapy or psychotherapy, or both. The need for an effective maintenance strategy after the acute response is therefore critical. Medication or ECT, or both, are commonly used to maintain acute benefit but, regrettably, a recent systematic review of the durability of benefit with such strategies found a substantial percentage (approximately 50%) of patients relapsed within the first year.17
- In this context, a case series report found that 1 or 2 weekly, sequential, bilateral TMS treatments after a successful acute course of ECT maintained response in 5 of 6 patients over 6 to 12 months.18
- Another case series (N = 6) transitioned stable patients from maintenance ECT to maintenance TMS, primarily because of adverse effects with ECT.19 With a mean frequency of 1 TMS treatment every 3.5 weeks, all 6 patients remained stable for as long as 6 months. Subsequently, 2 patients relapsed—1 at 8 months and 1 at 9 months.
Advantages of maintenance TMS over maintenance ECT include lower cost, fewer adverse effects (particularly cognitive deficits), and the ability to remain independent during the period of the treatment sessions.
TMS as an assessment tool for ECT response. TMS can be used to study excitability in cortical circuits. In a study, EEG potentials evoked by TMS before and after a course of ECT in 8 severely depressed patients revealed an increase in frontal cortical excitability, compared with baseline.20 Such findings support the ability of ECT to produce synaptic potentiation in humans. Furthermore, to the extent that depression presents with alterations in frontal cortical excitability, serial EEG-TMS measurements might be an effective tool to guide and monitor treatment progress with ECT, as well as other forms of therapeutic modulation.
Summing up: TMS and ECT. Although a definitive comparative study is needed, available evidence suggests that TMS might be an alternative treatment in a subgroup of patients who are referred for ECT. Factors that might warrant considering TMS over ECT include:
- patient preference
- fear of anesthesia
- concern about cognitive deficits
- stigma.
Although TMS might offer a workable alternative to ECT for acute and maintenance treatment of depression in selected patients, further refinement of the delivery of TMS is also needed to (1) enhance its efficacy and (2) identify clinical and biological markers to better define this select population.
Standard TMS treatment parameters
Superficial TMS. Superficial TMS for depression typically involves a single coil placed over the left dorsolateral prefrontal cortex. The standard, FDA-approved protocol includes stimulating at 110% of motor threshold with 75, 4-second trains at 10 Hz (ie, 40 stimulations) interspersed by 26-second intertrain intervals. Without interruption, a standard treatment session takes 37.5 minutes and delivers a total of 3,000 pulses. Most patients require 20 to 30 sessions, on a Monday-through-Friday schedule, to achieve optimal benefit.
This approach stimulates to a depth of approximately 2 or 3 cm. The coil usually is placed over the left dorsolateral prefrontal cortex because earlier studies indicated that decreased activity in this part of the brain correlates with symptoms of depression. When TMS is administered in a rapid repetitive fashion (at >1 Hz; typically, at 10 Hz), blood flow and metabolism in that area of the brain are increased. In addition, imaging studies indicate that trans-synaptic connections with deeper parts of the brain also allow modulation of other relevant neural circuits.
An alternate approach, less well-studied, involves low-frequency stimulation over the right dorsolateral prefrontal cortex. Parameters differ from what is used in left high-frequency dorsolateral prefrontal cortex TMS: frequency <1 Hz; train durations as long as 15 minutes; an intertrain interval of 25 to 180 seconds; 120 to 900 stimulations per train; and 2,400 to 18,000 total stimulations.
One hypothesis is that this low-frequency approach selectively stimulates inhibitory interneurons, decreases local neuronal activity and diminishes blood flow to deeper structures, such as the amygdala. Although right low-frequency TMS, compared with left high-frequency TMS, has potential advantages of better tolerability and decreased risk for seizures, its relative efficacy is unclear.
Deep TMS. Studies also are pursuing different coil configurations that allow for more direct stimulation of relevant structures (eg, prefrontal neuronal pathways associated with the reward system).
One of these coil designs (ie, the H-coil), coupled to a Magstim TMS stimulator, recently received FDA clearance for treatment-resistant depression. In the pivotal, sham-controlled study, patients received 20 treatment sessions over 4 weeks.21 The treatment protocol consisted of a helmet-like coil placed over the medial and lateral prefrontal cortex. Stimulation parameters included an 18-Hz frequency; stimulation intensity of 120% motor threshold; stimulation train duration of 2 seconds; and an intertrain interval of 20 seconds. The treatment sessions lasted 20.2 minutes and delivered a total of 1,980 stimulations.
Based on the 21-item HDRS, the active treatment coil group achieved a significantly greater decrease in baseline score (6.39 vs 3.28; P < .008); a greater response rate (37% vs 27.8%; P < .03); and a greater remission rate (30.4% vs 15.8%; P < .016) compared with the sham coil group.
Next, in what is the only randomized, controlled maintenance assessment to date, the same patients were followed for an additional 12 weeks, continuing blinded treatments twice weekly. At the end of the second phase, the active treatment group also demonstrated greater benefit than the sham group (P < .03). One seizure did occur, possibly related to excessive alcohol use; but this raises the question of whether treating at a higher frequency (18 Hz) with greater depth and less focality might increase the risk of seizure.
To assess the potential advantages, as well as the relative safety, of this approach over standard TMS delivery, an adequately designed and powered trial comparing the H-coil and a single-coil device is needed.
Alternate TMS approaches
Efforts to improve the clinical effectiveness of TMS for treating depression include several approaches.
Theta burst stimulation (TBS) is a patterned form of TMS pulse delivery that utilizes high and low frequencies in the same stimulus train (eg, three 50-Hz bursts delivered 5 times a second). Such a pulse sequence can modulate long-term depression and long-term potentiation mechanisms that induce plasticity in areas such as the hippocampus.22
Intermittent TBS (iTBS) administers stimulations over a relatively brief duration (eg, 2 seconds) or intermittently (eg, every 10 seconds) for a specific period (eg, 190 seconds [600 pulses in total]) over the left dorsolateral prefrontal cortex. This technique induces long-term potentiation and produces effects similar to those of high-frequency TMS.
In contrast, continuous TBS (cTBS) administers a continuous train (eg, 40 seconds [600 total pulses]) over the right dorsolateral prefrontal cortex. This induces long-term depression and produces effects similar to low-frequency TMS.
Recent studies using different delivery paradigms have generated mixed results:
Study 1: Fifty-six patients with depression received active treatment; 17 others, a sham procedure.23 This study used 3 different conditions:
- a combination of low-frequency and high-frequency TMS stimulation, administered over the right and left dorsolateral prefrontal cortices, respectively
- a combination of iTBS over the left dorsolateral prefrontal cortex and cTBS over the right dorsolateral prefrontal cortex
- a sham procedure, in which no magnetic field was created.
Neither active treatment arm separated from the sham procedure based on change scores in the 21-item HDRS (P = not significant).
Study 2: Sixty treatment-resistant depression patients were assigned to cTBS, iTBS, a combination of the 2 procedures, or a sham procedure.24 After 2 weeks, the active treatment arms produced the greatest benefit, based on change in scores on the 17-item HDRS, which differed significantly among the 4 groups (F value = 6.166; P < .001); the iTBS and combination arms demonstrated the most robust effect.
There were also significantly more responders in the iTBS (40.0%) and combination groups (66.7%) than in the cTBS (25.0%) and sham groups (13.3%) (P < .010). A lower level of treatment refractoriness predicted a better outcome.
Study 3: Twenty-nine depressed patients were randomized to cTBS over the right dorsolateral prefrontal cortex or a sham procedure.25 Overall, there was no difference between groups; however, actively treated patients who were unmedicated (n = 3) or remained on a stable dosage of medication during treatment (n = 8) did experience a significantly greater reduction in the HDRS score.
Study 4: In a pilot trial, 32 depressed patients were randomized to 30 sessions of adjunctive combined iTBS plus cTBS or bilateral sham TBS.26 Based on reduction from the baseline Montgomery-Åsberg Depression Rating Scale score, 9 patients in the active treatment group and 4 in the sham group achieved response (odds ratio, 3.86; P < .048).
If at least comparable efficacy can be clearly demonstrated, advantages of TBS over standard TMS include a significantly reduced administration time, which might allow for more patients to be treated and reduce associated costs of treatment.27
Magnetic low-field synchronized stimulation is produced by rotating spherical rare-earth magnets that are synchronized to an individual’s alpha frequency. A recent 6-week, double-blind, sham-controlled trial (N = 202) reported that, in the intention-to-treat population, there was no difference in outcome between treatment arms. In patients who completed the study according to protocol (120 of 202), however, active treatment was significantly better in decreasing baseline HDRS score (P < .033).28
Magnetic seizure therapy (MST) is an experimental approach to treating patients with more severe depression that is resistant to medical therapy. The primary aim is to use TMS to induce a seizure, thus achieving the same efficacy as provided by ECT but without the adverse cognitive effects of ECT. With MST, the TMS device uses much higher stimulation settings to produce a seizure—the goal being to avoid direct electrical current to the brain’s memory centers.29
A pilot study considered the clinical and cognitive effects of MST in a group of 26 treatment-resistant depression patients (10 randomized; 16 open-label).30 Based on reduction in baseline HDRS scores at the end of the trial, 69% of patients achieved response and 46% met remission criteria; however, one-half of patients relapsed within 6 months.
Importantly, no cognitive adverse effects were observed. Furthermore, the antidepressant and anti-anxiety effects of MST were associated with localized metabolic changes in brain areas implicated in the pathophysiology of depression.
The investigators concluded that MST might constitute an effective, well-tolerated, and safe treatment for patients unable to benefit from available medical therapies for depression. In addition to confirmation of acute benefit in more definitive trials, the issue of durability of effect needs further clarification.
TMS is a key component of neuropsychiatric practice
It has been 3 decades since Barker et al31 developed the technology to deliver intense, localized magnetic pulses to specific areas of the nervous system. During this period, the role of TMS as a probe of the central and peripheral nervous systems has expanded to include various therapeutic applications, primarily focusing on treatment-resistant major depressive disorder.
Now, increasing sophistication in the choice of stimulation parameters and other ongoing efforts to optimize the benefits of TMS are yielding improved clinical outcomes. Research is still needed to better define the place of TMS in the management of subtypes of depression that are particularly difficult to treat and that do not benefit adequately from medications or psychotherapy or their combination.
Growing support from controlled trials, systematic reviews, meta-analyses, naturalistic outcome studies, and professional guidelines indicate that TMS has an increasingly important role in clinical practice.
1. Janicak PG, Dowd SM, Rado JT, et al. The re-emerging role of therapeutic neuromodulation. Current Psychiatry. 2010;9(11):66-70,72-74.
2. Janicak PG, Dokucu ME. Transcranial magnetic stimulation for the treatment of major depression. Neuropsychiatr Dis Treat. 2015;11:1549-1560.
3. Gaynes BN, Lloyd SW, Lux L, et al. Repetitive transcranial magnetic stimulation for treatment-resistant depression: a systematic review and meta-analysis. J Clin Psychiatry. 2014;75(5):477-489; quiz 489.
4. Liu B, Zhang Y, Zhang L, et al. Repetitive transcranial magnetic stimulation as an augmentative strategy for treatment-resistant depression, a meta-analysis of randomized, double-blind and sham-controlled study. BMC Psychiatry. 2014;14:342.
5. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
6. Janicak PG, Dunner DL, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of quality of life outcome measures in clinical practice. CNS Spectr. 2013;18(6):322-332.
7. Janicak PG, Nahas Z, Lisanby SH, et al. Durability of clinical benefit with transcranial magnetic stimulation (TMS) in the treatment of pharmacoresistant major depression: assessment of relapse during a 6-month, multisite, open-label study. Brain Stimul. 2010;3(4):187-199.
8. Mantovani A, Pavlicova M, Avery D, et al. Long-term efficacy of repeated daily prefrontal transcranial magnetic stimulation (TMS) in treatment-resistant depression. Depress Anxiety. 2012;29(10):883-890.
9. Philip NS, Dunner DL, Dowd SM, et al. Can medication free, treatment-resistant, depressed patients who initially respond to TMS be maintained off medications? A prospective, 12-month multisite randomized pilot study. Brain Stimul. 2016;9(2):251-257.
10. Dunner DL, Aaronson ST, Sackeim HA, et al. A multisite, naturalistic, observational study of transcranial magnetic stimulation for patients with pharmacoresistant major depressive disorder: durability of benefit over a 1-year follow-up period. J Clin Psychiatry. 2014;75(12):1394-1401.
11. Hermann RC, Dorwart RA, Hoover CW. Variation in ECT use in the United States. Am J Psychiatry. 1995;152(6):869-875.
12. Sackeim HA. Memory and ECT: from polarization to reconciliation. J ECT. 2000;16(2):87-96.
13. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
14. Ren J, Li H, Palaniyappan L, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depression: a systematic review and meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2014;51:181-189.
15. Martis B, Alam D, Dowd SM, et al. Neurocognitive effects of repetitive transcranial magnetic stimulation in severe major depression. Clin Neurophysiol. 2003;114(6):1125-1132.
16. Pridmore S, Rybak M, Turnier-Shea Y, et al. Comparison of transcranial magnetic stimulation and electroconvulsive therapy in depression. In: Miyoshi K, Shapiro CM, Gaviria M, et al, eds. Contemporary neuropsychiatry. Tokyo, Japan: Springer; 2001:237-241.
17. Jelovac A, Kolshus E, McLoughlin DM. Relapse following successful electroconvulsive therapy for major depression: a meta-analysis. Neuropsychopharmacology. 2013;38(12):2467-2474.
18. Noda Y, Daskalakis Z, Ramos C, et al. Repetitive transcranial magnetic stimulation to maintain treatment response to electroconvulsive therapy in depression: a case series. Front Psychiatry. 2013;4:73.
19. Cristancho MA, Helmer A, Connolly R, et al. Transcranial magnetic stimulation maintenance as a substitute for maintenance electroconvulsive therapy: a case series. J ECT. 2013;29(2):106-108.
20. Casarotto S, Canali P, Rosanova M, et al. Assessing the effects of electroconvulsive therapy on cortical excitability by means of transcranial magnetic stimulation and electroencephalography. Brain Topogr. 2013;26(2):326-337.
21. Levkovitz Y, Isserles M, Padberg F, et al. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective multicenter randomized controlled trial. World Psychiatry. 2015;14(1):64-73.
22. Daskalakis ZJ. Theta-burst transcranial magnetic stimulation in depression: when less may be more. Brain. 2014;137(pt 7):1860-1862.
23. Prasser J, Schecklmann M, Poeppl TB, et al. Bilateral prefrontal rTMS and theta burst TMS as an add-on treatment for depression: a randomized placebo controlled trial. World J Biol Psychiatry. 2015;16(1):57-65.
24. Li CT, Chen MH, Juan CH, et al. Efficacy of prefrontal theta-burst stimulation in refractory depression: a randomized sham-controlled study. Brain. 2014;137(pt 7):2088-2098.
25. Chistyakov A, Kreinin B, Marmor S, et al. Preliminary assessment of the therapeutic efficacy of continuous theta-burst magnetic stimulation (cTBS) in major depression: a double-blind sham-controlled study. J Affect Disord. 2015;170:225-229.
26. Plewnia C, Pasqualetti P, Große S, et al. Treatment of major depression with bilateral theta burst stimulation: a randomized controlled pilot trial. J Affect Disord. 2014;156:219-223.
27. Chung SW, Hoy KE, Fitzgerald PB. Theta-burst stimulation: a new form of TMS treatment for depression? Depress Anxiety. 2015;32(3):182-192.
28. Leuchter AF, Cook IA, Feifel D, et al. Efficacy and safety of low-field synchronized transcranial magnetic stimulation (sTMS) for treatment of major depression. Brain Stimul. 2015;8(4):787-794.
29. Cretaz E, Brunoni AR, Lafer B. Magnetic seizure therapy for unipolar and bipolar depression: a systematic review. Neural Plast. 2015;2015:521398. doi: 10.1155/2015/521398.
30. Kayser S, Bewernick BH, Matusch A, et al. Magnetic seizure therapy in treatment-resistant depression: clinical, neuropsychological and metabolic effects. Psychol Med. 2015;45(5):1073-1092.
31. Barker AT, Jalinous R, Freeston IL. Non-invasive magnetic stimulation of human motor cortex. Lancet. 1985;1(8437):1106-1107.
Since 2008, the FDA has cleared 4 transcranial magnetic stimulation (TMS) devices for treating depression (Related Resources). In that time, the availability of TMS has steadily grown within and outside the United States.
Parallel with increasing clinical utilization of this technology, research continues into the benefit of TMS for treatment-resistant depression; such research includes additional, supportive, acute, sham-controlled trials; comparison trials with electroconvulsive therapy (ECT) for more severe episodes of depression; short- and long-term real-world outcome studies; exploration of alternative treatment parameters to further enhance its efficacy; and the development of other TMS approaches. In this article, we review recent developments in the application of TMS to treat major depressive disorder—in particular, treatment-resistant depression (Box).
Therapeutic neuromodulation
The underlying premise of neuromodulation is that the brain is an electrochemical organ that can be modulated by pharmacotherapy or device-based approaches, or their combination.1 ECT is the prototypic device-based neuromodulation approach, and remains one of the most effective treatments for severe depression.
More recently, however, other methods have been, and continue to be, developed to treat patients who do not achieve adequate benefit from psychotherapy or medical therapy, or both, and who might not be an ideal candidate for ECT (Table,1). In addition to the potential therapeutic benefit of these alternative strategies, some could avoid safety and tolerability concerns associated with medication (weight gain, sexual dysfunction) and ECT (eg, cognitive deficits).
TMS, which utilizes intense, localized magnetic fields to alter activity in neural circuits implicated in the pathophysiology of depression, represents an important example of this initiative.2
TMS has established efficacy for depression
Sham-controlled trials. Several randomized, sham-controlled acute trials have demonstrated the efficacy of TMS for treatment-resistant depression.
A recent meta-analysis considered 18 studies (N = 1,970) that met the authors’ criteria for inclusion.3 They found that TMS monotherapy was statistically and clinically more effective than a sham procedure based on:
- improvement in depressive symptoms (mean decrease in baseline Hamilton Depression Rating Scale [HDRS] score, −4.53 [95% CI, −6.11 to −2.96])
- response rate; response was 3 times more likely with TMS (relative risk 3.38 [95% CI, 2.24 to 5.10])
- remission rate; remission was 5 times more likely with TMS (relative risk, 5.07 [95% CI, 2.50 to 10.30]).
Another meta-analysis (7 studies, N = 279) considered TMS as an augmentation strategy to standard medication for treatment-resistant depression.4 The authors reported that, based on change in HDRS scores, the pooled standardized mean difference between active and sham TMS augmentation was 0.86 (P < .00001). Furthermore, the pooled response rate with TMS augmentation was 46.6%, compared with 22.1% with the sham procedure (P < .0003).
Acute naturalistic TMS studies. The efficacy of TMS is supported by a large, naturalistic study of 307 patients with treatment-resistant depression who were assessed at baseline and during a standard course of TMS.5 Considering change score in the Clinician Global Impressions-Severity (CGI-S) scale, significant improvement was seen from baseline to end of treatment (−1.9 ± 1.4; P < .0001), with a clinician-assessed response rate of 58.0% and remission rate of 37.1%. Of note: Self-reported quality-of-life measures (on the Medical Outcomes Study 36-Item Short-Form Health Survey and EuroQol 5-Dimensions) also significantly improved during this relatively brief period.6
Maintenance strategies after acute TMS response. Most patients referred for TMS have a depressive illness characterized by a chronic, relapsing course and inadequate response to pharmacotherapy or psychotherapy, or their combination. An effective maintenance strategy after acute response to TMS is paramount. This includes:
- prolonged tapering schedule after an acute TMS course is completed
- maintenance medication or psychotherapy, or both
- scheduled periodic maintenance TMS sessions (usually as an augmentation strategy)
- reintroduction of TMS as needed with early signs of relapse. In this context, several trials have assessed the durability of acute TMS benefit.
A semi-controlled maintenance study followed 99 patients who had at least a 25% decrease in baseline HDRS score after acute TMS treatment.7 They were then tapered from their TMS sessions over 3 weeks while an antidepressant was titrated up. If, at any time during the subsequent 6 months, early signs of depression relapse were noted (ie, change of at least 1 point on the CGI-S for 2 consecutive weeks), TMS was reintroduced. At the end of the trial, 10 patients (13%) had relapsed and 38 (38%) had an exacerbation of symptoms sufficient to warrant reintroduction of TMS. Of those, 32 (84%) re-achieved mood stability.
In another study, 50 patients who had achieved remission during an acute course of TMS were followed for 3 months.8 After TMS taper and continued pharmacotherapy or naturalistic follow-up, 29 (58%) remained in remission; 2 (4%) maintained partial response; and 1 (2%) relapsed.
In a controlled, pilot, maintenance trial, 67 unmedicated patients with treatment-resistant depression received an acute course of TMS.9 Forty-seven of the responders were then randomized to a 1-year follow-up trial with or without a scheduled monthly TMS session. All patients could receive reintroduction TMS if they met criteria for symptom worsening.
Both groups had a similar outcome. The number of patients who did not require TMS reintroduction was 9 of 23 (39%) in the scheduled TMS group vs 9 of 26 (35%) in the no-scheduled TMS group (P < .1). Although no difference was noted between groups, the authors commented that these preliminary results will help inform larger, more definitive trials. They concluded that both acute and maintenance TMS monotherapy might be an option—for some patients.
A long-term, naturalistic outcomes study followed 257 treatment-resistant depressed patients for 1 year after they responded to an acute course of TMS.10 In addition to most patients receiving ongoing maintenance medication, they also could receive reintroduction of TMS if symptoms became worse.
Compared with pre-TMS baseline, there was a statistically significant reduction in the mean total score on the CGI-S scale (primary outcome, P < .0001) at the end of acute treatment that was sustained at follow-up. Ninety-six patients (36.2%) required reintroduction of TMS and 75 of 120 (62.5%) who initially met response or remission criteria after acute treatment continued to meet response criteria after 1 year. The authors concluded that TMS demonstrated both a statistically and clinically meaningful durability of acute benefit during this time frame.
TMS and electroconvulsive therapy
For more than 75 years, ECT has consistently proved to be an effective treatment for major depressive disorder. Although the use of ECT has fluctuated over this period, one practice survey estimated that 100,000 patients receive ECT annually.11
ECT has limitations, however, including cost, the need for general anesthesia, and cognitive deficits that range from short-term confusion to anterograde and retrograde amnesia, which can persist for weeks beyond active treatment.12 Despite increasing awareness of mental illness, stigma also remains a significant barrier to receiving ECT.
TMS vs ECT. Several trials have directly compared ECT and TMS:
- A recent meta-analysis of 9 trials included 384 patients with depression who were considered clinically appropriate for ECT and were randomized to one or the other treatment.13 Both modalities produced a significant reduction in baseline HDRS score, but ECT (15.4 point reduction) was superior to TMS (9.3 point reduction) in the degree of improvement (P < .01).
- Another meta-analysis of 9 trials (N = 425) found ECT superior to TMS in terms of response (P < .03) and remission (P < .006) rates, based on improvement in the HDRS score.14 When psychotic depressed patients were excluded, however, TMS produced effects equivalent to ECT.
In contrast to what was seen with ECT, cognitive testing of patients who received TMS revealed no deterioration in any domain. Furthermore, one of the comparison studies observed a modest, but statistically significant, improvement in patient’s working memory-executive function, objective memory, and fine-motor speed over the course of TMS treatment.15
TMS plus ECT. A 2-week, randomized, single-blind, controlled pilot study (N = 22) examined the combination of TMS and ECT as acute treatment of depression.16 Patients were assigned to receive either unilateral non-dominant (UND) ECT 3 days a week or a combination of 1 UND ECT treatment followed by 4 days of TMS. At the conclusion of treatment, UND ECT plus TMS group produced comparable efficacy and fewer adverse effects compared with the UND ECT-only group.
TMS maintenance after acute ECT response. Most patients who are referred for ECT have a depressive illness characterized by repeated episodes and incomplete response to pharmacotherapy or psychotherapy, or both. The need for an effective maintenance strategy after the acute response is therefore critical. Medication or ECT, or both, are commonly used to maintain acute benefit but, regrettably, a recent systematic review of the durability of benefit with such strategies found a substantial percentage (approximately 50%) of patients relapsed within the first year.17
- In this context, a case series report found that 1 or 2 weekly, sequential, bilateral TMS treatments after a successful acute course of ECT maintained response in 5 of 6 patients over 6 to 12 months.18
- Another case series (N = 6) transitioned stable patients from maintenance ECT to maintenance TMS, primarily because of adverse effects with ECT.19 With a mean frequency of 1 TMS treatment every 3.5 weeks, all 6 patients remained stable for as long as 6 months. Subsequently, 2 patients relapsed—1 at 8 months and 1 at 9 months.
Advantages of maintenance TMS over maintenance ECT include lower cost, fewer adverse effects (particularly cognitive deficits), and the ability to remain independent during the period of the treatment sessions.
TMS as an assessment tool for ECT response. TMS can be used to study excitability in cortical circuits. In a study, EEG potentials evoked by TMS before and after a course of ECT in 8 severely depressed patients revealed an increase in frontal cortical excitability, compared with baseline.20 Such findings support the ability of ECT to produce synaptic potentiation in humans. Furthermore, to the extent that depression presents with alterations in frontal cortical excitability, serial EEG-TMS measurements might be an effective tool to guide and monitor treatment progress with ECT, as well as other forms of therapeutic modulation.
Summing up: TMS and ECT. Although a definitive comparative study is needed, available evidence suggests that TMS might be an alternative treatment in a subgroup of patients who are referred for ECT. Factors that might warrant considering TMS over ECT include:
- patient preference
- fear of anesthesia
- concern about cognitive deficits
- stigma.
Although TMS might offer a workable alternative to ECT for acute and maintenance treatment of depression in selected patients, further refinement of the delivery of TMS is also needed to (1) enhance its efficacy and (2) identify clinical and biological markers to better define this select population.
Standard TMS treatment parameters
Superficial TMS. Superficial TMS for depression typically involves a single coil placed over the left dorsolateral prefrontal cortex. The standard, FDA-approved protocol includes stimulating at 110% of motor threshold with 75, 4-second trains at 10 Hz (ie, 40 stimulations) interspersed by 26-second intertrain intervals. Without interruption, a standard treatment session takes 37.5 minutes and delivers a total of 3,000 pulses. Most patients require 20 to 30 sessions, on a Monday-through-Friday schedule, to achieve optimal benefit.
This approach stimulates to a depth of approximately 2 or 3 cm. The coil usually is placed over the left dorsolateral prefrontal cortex because earlier studies indicated that decreased activity in this part of the brain correlates with symptoms of depression. When TMS is administered in a rapid repetitive fashion (at >1 Hz; typically, at 10 Hz), blood flow and metabolism in that area of the brain are increased. In addition, imaging studies indicate that trans-synaptic connections with deeper parts of the brain also allow modulation of other relevant neural circuits.
An alternate approach, less well-studied, involves low-frequency stimulation over the right dorsolateral prefrontal cortex. Parameters differ from what is used in left high-frequency dorsolateral prefrontal cortex TMS: frequency <1 Hz; train durations as long as 15 minutes; an intertrain interval of 25 to 180 seconds; 120 to 900 stimulations per train; and 2,400 to 18,000 total stimulations.
One hypothesis is that this low-frequency approach selectively stimulates inhibitory interneurons, decreases local neuronal activity and diminishes blood flow to deeper structures, such as the amygdala. Although right low-frequency TMS, compared with left high-frequency TMS, has potential advantages of better tolerability and decreased risk for seizures, its relative efficacy is unclear.
Deep TMS. Studies also are pursuing different coil configurations that allow for more direct stimulation of relevant structures (eg, prefrontal neuronal pathways associated with the reward system).
One of these coil designs (ie, the H-coil), coupled to a Magstim TMS stimulator, recently received FDA clearance for treatment-resistant depression. In the pivotal, sham-controlled study, patients received 20 treatment sessions over 4 weeks.21 The treatment protocol consisted of a helmet-like coil placed over the medial and lateral prefrontal cortex. Stimulation parameters included an 18-Hz frequency; stimulation intensity of 120% motor threshold; stimulation train duration of 2 seconds; and an intertrain interval of 20 seconds. The treatment sessions lasted 20.2 minutes and delivered a total of 1,980 stimulations.
Based on the 21-item HDRS, the active treatment coil group achieved a significantly greater decrease in baseline score (6.39 vs 3.28; P < .008); a greater response rate (37% vs 27.8%; P < .03); and a greater remission rate (30.4% vs 15.8%; P < .016) compared with the sham coil group.
Next, in what is the only randomized, controlled maintenance assessment to date, the same patients were followed for an additional 12 weeks, continuing blinded treatments twice weekly. At the end of the second phase, the active treatment group also demonstrated greater benefit than the sham group (P < .03). One seizure did occur, possibly related to excessive alcohol use; but this raises the question of whether treating at a higher frequency (18 Hz) with greater depth and less focality might increase the risk of seizure.
To assess the potential advantages, as well as the relative safety, of this approach over standard TMS delivery, an adequately designed and powered trial comparing the H-coil and a single-coil device is needed.
Alternate TMS approaches
Efforts to improve the clinical effectiveness of TMS for treating depression include several approaches.
Theta burst stimulation (TBS) is a patterned form of TMS pulse delivery that utilizes high and low frequencies in the same stimulus train (eg, three 50-Hz bursts delivered 5 times a second). Such a pulse sequence can modulate long-term depression and long-term potentiation mechanisms that induce plasticity in areas such as the hippocampus.22
Intermittent TBS (iTBS) administers stimulations over a relatively brief duration (eg, 2 seconds) or intermittently (eg, every 10 seconds) for a specific period (eg, 190 seconds [600 pulses in total]) over the left dorsolateral prefrontal cortex. This technique induces long-term potentiation and produces effects similar to those of high-frequency TMS.
In contrast, continuous TBS (cTBS) administers a continuous train (eg, 40 seconds [600 total pulses]) over the right dorsolateral prefrontal cortex. This induces long-term depression and produces effects similar to low-frequency TMS.
Recent studies using different delivery paradigms have generated mixed results:
Study 1: Fifty-six patients with depression received active treatment; 17 others, a sham procedure.23 This study used 3 different conditions:
- a combination of low-frequency and high-frequency TMS stimulation, administered over the right and left dorsolateral prefrontal cortices, respectively
- a combination of iTBS over the left dorsolateral prefrontal cortex and cTBS over the right dorsolateral prefrontal cortex
- a sham procedure, in which no magnetic field was created.
Neither active treatment arm separated from the sham procedure based on change scores in the 21-item HDRS (P = not significant).
Study 2: Sixty treatment-resistant depression patients were assigned to cTBS, iTBS, a combination of the 2 procedures, or a sham procedure.24 After 2 weeks, the active treatment arms produced the greatest benefit, based on change in scores on the 17-item HDRS, which differed significantly among the 4 groups (F value = 6.166; P < .001); the iTBS and combination arms demonstrated the most robust effect.
There were also significantly more responders in the iTBS (40.0%) and combination groups (66.7%) than in the cTBS (25.0%) and sham groups (13.3%) (P < .010). A lower level of treatment refractoriness predicted a better outcome.
Study 3: Twenty-nine depressed patients were randomized to cTBS over the right dorsolateral prefrontal cortex or a sham procedure.25 Overall, there was no difference between groups; however, actively treated patients who were unmedicated (n = 3) or remained on a stable dosage of medication during treatment (n = 8) did experience a significantly greater reduction in the HDRS score.
Study 4: In a pilot trial, 32 depressed patients were randomized to 30 sessions of adjunctive combined iTBS plus cTBS or bilateral sham TBS.26 Based on reduction from the baseline Montgomery-Åsberg Depression Rating Scale score, 9 patients in the active treatment group and 4 in the sham group achieved response (odds ratio, 3.86; P < .048).
If at least comparable efficacy can be clearly demonstrated, advantages of TBS over standard TMS include a significantly reduced administration time, which might allow for more patients to be treated and reduce associated costs of treatment.27
Magnetic low-field synchronized stimulation is produced by rotating spherical rare-earth magnets that are synchronized to an individual’s alpha frequency. A recent 6-week, double-blind, sham-controlled trial (N = 202) reported that, in the intention-to-treat population, there was no difference in outcome between treatment arms. In patients who completed the study according to protocol (120 of 202), however, active treatment was significantly better in decreasing baseline HDRS score (P < .033).28
Magnetic seizure therapy (MST) is an experimental approach to treating patients with more severe depression that is resistant to medical therapy. The primary aim is to use TMS to induce a seizure, thus achieving the same efficacy as provided by ECT but without the adverse cognitive effects of ECT. With MST, the TMS device uses much higher stimulation settings to produce a seizure—the goal being to avoid direct electrical current to the brain’s memory centers.29
A pilot study considered the clinical and cognitive effects of MST in a group of 26 treatment-resistant depression patients (10 randomized; 16 open-label).30 Based on reduction in baseline HDRS scores at the end of the trial, 69% of patients achieved response and 46% met remission criteria; however, one-half of patients relapsed within 6 months.
Importantly, no cognitive adverse effects were observed. Furthermore, the antidepressant and anti-anxiety effects of MST were associated with localized metabolic changes in brain areas implicated in the pathophysiology of depression.
The investigators concluded that MST might constitute an effective, well-tolerated, and safe treatment for patients unable to benefit from available medical therapies for depression. In addition to confirmation of acute benefit in more definitive trials, the issue of durability of effect needs further clarification.
TMS is a key component of neuropsychiatric practice
It has been 3 decades since Barker et al31 developed the technology to deliver intense, localized magnetic pulses to specific areas of the nervous system. During this period, the role of TMS as a probe of the central and peripheral nervous systems has expanded to include various therapeutic applications, primarily focusing on treatment-resistant major depressive disorder.
Now, increasing sophistication in the choice of stimulation parameters and other ongoing efforts to optimize the benefits of TMS are yielding improved clinical outcomes. Research is still needed to better define the place of TMS in the management of subtypes of depression that are particularly difficult to treat and that do not benefit adequately from medications or psychotherapy or their combination.
Growing support from controlled trials, systematic reviews, meta-analyses, naturalistic outcome studies, and professional guidelines indicate that TMS has an increasingly important role in clinical practice.
Since 2008, the FDA has cleared 4 transcranial magnetic stimulation (TMS) devices for treating depression (Related Resources). In that time, the availability of TMS has steadily grown within and outside the United States.
Parallel with increasing clinical utilization of this technology, research continues into the benefit of TMS for treatment-resistant depression; such research includes additional, supportive, acute, sham-controlled trials; comparison trials with electroconvulsive therapy (ECT) for more severe episodes of depression; short- and long-term real-world outcome studies; exploration of alternative treatment parameters to further enhance its efficacy; and the development of other TMS approaches. In this article, we review recent developments in the application of TMS to treat major depressive disorder—in particular, treatment-resistant depression (Box).
Therapeutic neuromodulation
The underlying premise of neuromodulation is that the brain is an electrochemical organ that can be modulated by pharmacotherapy or device-based approaches, or their combination.1 ECT is the prototypic device-based neuromodulation approach, and remains one of the most effective treatments for severe depression.
More recently, however, other methods have been, and continue to be, developed to treat patients who do not achieve adequate benefit from psychotherapy or medical therapy, or both, and who might not be an ideal candidate for ECT (Table,1). In addition to the potential therapeutic benefit of these alternative strategies, some could avoid safety and tolerability concerns associated with medication (weight gain, sexual dysfunction) and ECT (eg, cognitive deficits).
TMS, which utilizes intense, localized magnetic fields to alter activity in neural circuits implicated in the pathophysiology of depression, represents an important example of this initiative.2
TMS has established efficacy for depression
Sham-controlled trials. Several randomized, sham-controlled acute trials have demonstrated the efficacy of TMS for treatment-resistant depression.
A recent meta-analysis considered 18 studies (N = 1,970) that met the authors’ criteria for inclusion.3 They found that TMS monotherapy was statistically and clinically more effective than a sham procedure based on:
- improvement in depressive symptoms (mean decrease in baseline Hamilton Depression Rating Scale [HDRS] score, −4.53 [95% CI, −6.11 to −2.96])
- response rate; response was 3 times more likely with TMS (relative risk 3.38 [95% CI, 2.24 to 5.10])
- remission rate; remission was 5 times more likely with TMS (relative risk, 5.07 [95% CI, 2.50 to 10.30]).
Another meta-analysis (7 studies, N = 279) considered TMS as an augmentation strategy to standard medication for treatment-resistant depression.4 The authors reported that, based on change in HDRS scores, the pooled standardized mean difference between active and sham TMS augmentation was 0.86 (P < .00001). Furthermore, the pooled response rate with TMS augmentation was 46.6%, compared with 22.1% with the sham procedure (P < .0003).
Acute naturalistic TMS studies. The efficacy of TMS is supported by a large, naturalistic study of 307 patients with treatment-resistant depression who were assessed at baseline and during a standard course of TMS.5 Considering change score in the Clinician Global Impressions-Severity (CGI-S) scale, significant improvement was seen from baseline to end of treatment (−1.9 ± 1.4; P < .0001), with a clinician-assessed response rate of 58.0% and remission rate of 37.1%. Of note: Self-reported quality-of-life measures (on the Medical Outcomes Study 36-Item Short-Form Health Survey and EuroQol 5-Dimensions) also significantly improved during this relatively brief period.6
Maintenance strategies after acute TMS response. Most patients referred for TMS have a depressive illness characterized by a chronic, relapsing course and inadequate response to pharmacotherapy or psychotherapy, or their combination. An effective maintenance strategy after acute response to TMS is paramount. This includes:
- prolonged tapering schedule after an acute TMS course is completed
- maintenance medication or psychotherapy, or both
- scheduled periodic maintenance TMS sessions (usually as an augmentation strategy)
- reintroduction of TMS as needed with early signs of relapse. In this context, several trials have assessed the durability of acute TMS benefit.
A semi-controlled maintenance study followed 99 patients who had at least a 25% decrease in baseline HDRS score after acute TMS treatment.7 They were then tapered from their TMS sessions over 3 weeks while an antidepressant was titrated up. If, at any time during the subsequent 6 months, early signs of depression relapse were noted (ie, change of at least 1 point on the CGI-S for 2 consecutive weeks), TMS was reintroduced. At the end of the trial, 10 patients (13%) had relapsed and 38 (38%) had an exacerbation of symptoms sufficient to warrant reintroduction of TMS. Of those, 32 (84%) re-achieved mood stability.
In another study, 50 patients who had achieved remission during an acute course of TMS were followed for 3 months.8 After TMS taper and continued pharmacotherapy or naturalistic follow-up, 29 (58%) remained in remission; 2 (4%) maintained partial response; and 1 (2%) relapsed.
In a controlled, pilot, maintenance trial, 67 unmedicated patients with treatment-resistant depression received an acute course of TMS.9 Forty-seven of the responders were then randomized to a 1-year follow-up trial with or without a scheduled monthly TMS session. All patients could receive reintroduction TMS if they met criteria for symptom worsening.
Both groups had a similar outcome. The number of patients who did not require TMS reintroduction was 9 of 23 (39%) in the scheduled TMS group vs 9 of 26 (35%) in the no-scheduled TMS group (P < .1). Although no difference was noted between groups, the authors commented that these preliminary results will help inform larger, more definitive trials. They concluded that both acute and maintenance TMS monotherapy might be an option—for some patients.
A long-term, naturalistic outcomes study followed 257 treatment-resistant depressed patients for 1 year after they responded to an acute course of TMS.10 In addition to most patients receiving ongoing maintenance medication, they also could receive reintroduction of TMS if symptoms became worse.
Compared with pre-TMS baseline, there was a statistically significant reduction in the mean total score on the CGI-S scale (primary outcome, P < .0001) at the end of acute treatment that was sustained at follow-up. Ninety-six patients (36.2%) required reintroduction of TMS and 75 of 120 (62.5%) who initially met response or remission criteria after acute treatment continued to meet response criteria after 1 year. The authors concluded that TMS demonstrated both a statistically and clinically meaningful durability of acute benefit during this time frame.
TMS and electroconvulsive therapy
For more than 75 years, ECT has consistently proved to be an effective treatment for major depressive disorder. Although the use of ECT has fluctuated over this period, one practice survey estimated that 100,000 patients receive ECT annually.11
ECT has limitations, however, including cost, the need for general anesthesia, and cognitive deficits that range from short-term confusion to anterograde and retrograde amnesia, which can persist for weeks beyond active treatment.12 Despite increasing awareness of mental illness, stigma also remains a significant barrier to receiving ECT.
TMS vs ECT. Several trials have directly compared ECT and TMS:
- A recent meta-analysis of 9 trials included 384 patients with depression who were considered clinically appropriate for ECT and were randomized to one or the other treatment.13 Both modalities produced a significant reduction in baseline HDRS score, but ECT (15.4 point reduction) was superior to TMS (9.3 point reduction) in the degree of improvement (P < .01).
- Another meta-analysis of 9 trials (N = 425) found ECT superior to TMS in terms of response (P < .03) and remission (P < .006) rates, based on improvement in the HDRS score.14 When psychotic depressed patients were excluded, however, TMS produced effects equivalent to ECT.
In contrast to what was seen with ECT, cognitive testing of patients who received TMS revealed no deterioration in any domain. Furthermore, one of the comparison studies observed a modest, but statistically significant, improvement in patient’s working memory-executive function, objective memory, and fine-motor speed over the course of TMS treatment.15
TMS plus ECT. A 2-week, randomized, single-blind, controlled pilot study (N = 22) examined the combination of TMS and ECT as acute treatment of depression.16 Patients were assigned to receive either unilateral non-dominant (UND) ECT 3 days a week or a combination of 1 UND ECT treatment followed by 4 days of TMS. At the conclusion of treatment, UND ECT plus TMS group produced comparable efficacy and fewer adverse effects compared with the UND ECT-only group.
TMS maintenance after acute ECT response. Most patients who are referred for ECT have a depressive illness characterized by repeated episodes and incomplete response to pharmacotherapy or psychotherapy, or both. The need for an effective maintenance strategy after the acute response is therefore critical. Medication or ECT, or both, are commonly used to maintain acute benefit but, regrettably, a recent systematic review of the durability of benefit with such strategies found a substantial percentage (approximately 50%) of patients relapsed within the first year.17
- In this context, a case series report found that 1 or 2 weekly, sequential, bilateral TMS treatments after a successful acute course of ECT maintained response in 5 of 6 patients over 6 to 12 months.18
- Another case series (N = 6) transitioned stable patients from maintenance ECT to maintenance TMS, primarily because of adverse effects with ECT.19 With a mean frequency of 1 TMS treatment every 3.5 weeks, all 6 patients remained stable for as long as 6 months. Subsequently, 2 patients relapsed—1 at 8 months and 1 at 9 months.
Advantages of maintenance TMS over maintenance ECT include lower cost, fewer adverse effects (particularly cognitive deficits), and the ability to remain independent during the period of the treatment sessions.
TMS as an assessment tool for ECT response. TMS can be used to study excitability in cortical circuits. In a study, EEG potentials evoked by TMS before and after a course of ECT in 8 severely depressed patients revealed an increase in frontal cortical excitability, compared with baseline.20 Such findings support the ability of ECT to produce synaptic potentiation in humans. Furthermore, to the extent that depression presents with alterations in frontal cortical excitability, serial EEG-TMS measurements might be an effective tool to guide and monitor treatment progress with ECT, as well as other forms of therapeutic modulation.
Summing up: TMS and ECT. Although a definitive comparative study is needed, available evidence suggests that TMS might be an alternative treatment in a subgroup of patients who are referred for ECT. Factors that might warrant considering TMS over ECT include:
- patient preference
- fear of anesthesia
- concern about cognitive deficits
- stigma.
Although TMS might offer a workable alternative to ECT for acute and maintenance treatment of depression in selected patients, further refinement of the delivery of TMS is also needed to (1) enhance its efficacy and (2) identify clinical and biological markers to better define this select population.
Standard TMS treatment parameters
Superficial TMS. Superficial TMS for depression typically involves a single coil placed over the left dorsolateral prefrontal cortex. The standard, FDA-approved protocol includes stimulating at 110% of motor threshold with 75, 4-second trains at 10 Hz (ie, 40 stimulations) interspersed by 26-second intertrain intervals. Without interruption, a standard treatment session takes 37.5 minutes and delivers a total of 3,000 pulses. Most patients require 20 to 30 sessions, on a Monday-through-Friday schedule, to achieve optimal benefit.
This approach stimulates to a depth of approximately 2 or 3 cm. The coil usually is placed over the left dorsolateral prefrontal cortex because earlier studies indicated that decreased activity in this part of the brain correlates with symptoms of depression. When TMS is administered in a rapid repetitive fashion (at >1 Hz; typically, at 10 Hz), blood flow and metabolism in that area of the brain are increased. In addition, imaging studies indicate that trans-synaptic connections with deeper parts of the brain also allow modulation of other relevant neural circuits.
An alternate approach, less well-studied, involves low-frequency stimulation over the right dorsolateral prefrontal cortex. Parameters differ from what is used in left high-frequency dorsolateral prefrontal cortex TMS: frequency <1 Hz; train durations as long as 15 minutes; an intertrain interval of 25 to 180 seconds; 120 to 900 stimulations per train; and 2,400 to 18,000 total stimulations.
One hypothesis is that this low-frequency approach selectively stimulates inhibitory interneurons, decreases local neuronal activity and diminishes blood flow to deeper structures, such as the amygdala. Although right low-frequency TMS, compared with left high-frequency TMS, has potential advantages of better tolerability and decreased risk for seizures, its relative efficacy is unclear.
Deep TMS. Studies also are pursuing different coil configurations that allow for more direct stimulation of relevant structures (eg, prefrontal neuronal pathways associated with the reward system).
One of these coil designs (ie, the H-coil), coupled to a Magstim TMS stimulator, recently received FDA clearance for treatment-resistant depression. In the pivotal, sham-controlled study, patients received 20 treatment sessions over 4 weeks.21 The treatment protocol consisted of a helmet-like coil placed over the medial and lateral prefrontal cortex. Stimulation parameters included an 18-Hz frequency; stimulation intensity of 120% motor threshold; stimulation train duration of 2 seconds; and an intertrain interval of 20 seconds. The treatment sessions lasted 20.2 minutes and delivered a total of 1,980 stimulations.
Based on the 21-item HDRS, the active treatment coil group achieved a significantly greater decrease in baseline score (6.39 vs 3.28; P < .008); a greater response rate (37% vs 27.8%; P < .03); and a greater remission rate (30.4% vs 15.8%; P < .016) compared with the sham coil group.
Next, in what is the only randomized, controlled maintenance assessment to date, the same patients were followed for an additional 12 weeks, continuing blinded treatments twice weekly. At the end of the second phase, the active treatment group also demonstrated greater benefit than the sham group (P < .03). One seizure did occur, possibly related to excessive alcohol use; but this raises the question of whether treating at a higher frequency (18 Hz) with greater depth and less focality might increase the risk of seizure.
To assess the potential advantages, as well as the relative safety, of this approach over standard TMS delivery, an adequately designed and powered trial comparing the H-coil and a single-coil device is needed.
Alternate TMS approaches
Efforts to improve the clinical effectiveness of TMS for treating depression include several approaches.
Theta burst stimulation (TBS) is a patterned form of TMS pulse delivery that utilizes high and low frequencies in the same stimulus train (eg, three 50-Hz bursts delivered 5 times a second). Such a pulse sequence can modulate long-term depression and long-term potentiation mechanisms that induce plasticity in areas such as the hippocampus.22
Intermittent TBS (iTBS) administers stimulations over a relatively brief duration (eg, 2 seconds) or intermittently (eg, every 10 seconds) for a specific period (eg, 190 seconds [600 pulses in total]) over the left dorsolateral prefrontal cortex. This technique induces long-term potentiation and produces effects similar to those of high-frequency TMS.
In contrast, continuous TBS (cTBS) administers a continuous train (eg, 40 seconds [600 total pulses]) over the right dorsolateral prefrontal cortex. This induces long-term depression and produces effects similar to low-frequency TMS.
Recent studies using different delivery paradigms have generated mixed results:
Study 1: Fifty-six patients with depression received active treatment; 17 others, a sham procedure.23 This study used 3 different conditions:
- a combination of low-frequency and high-frequency TMS stimulation, administered over the right and left dorsolateral prefrontal cortices, respectively
- a combination of iTBS over the left dorsolateral prefrontal cortex and cTBS over the right dorsolateral prefrontal cortex
- a sham procedure, in which no magnetic field was created.
Neither active treatment arm separated from the sham procedure based on change scores in the 21-item HDRS (P = not significant).
Study 2: Sixty treatment-resistant depression patients were assigned to cTBS, iTBS, a combination of the 2 procedures, or a sham procedure.24 After 2 weeks, the active treatment arms produced the greatest benefit, based on change in scores on the 17-item HDRS, which differed significantly among the 4 groups (F value = 6.166; P < .001); the iTBS and combination arms demonstrated the most robust effect.
There were also significantly more responders in the iTBS (40.0%) and combination groups (66.7%) than in the cTBS (25.0%) and sham groups (13.3%) (P < .010). A lower level of treatment refractoriness predicted a better outcome.
Study 3: Twenty-nine depressed patients were randomized to cTBS over the right dorsolateral prefrontal cortex or a sham procedure.25 Overall, there was no difference between groups; however, actively treated patients who were unmedicated (n = 3) or remained on a stable dosage of medication during treatment (n = 8) did experience a significantly greater reduction in the HDRS score.
Study 4: In a pilot trial, 32 depressed patients were randomized to 30 sessions of adjunctive combined iTBS plus cTBS or bilateral sham TBS.26 Based on reduction from the baseline Montgomery-Åsberg Depression Rating Scale score, 9 patients in the active treatment group and 4 in the sham group achieved response (odds ratio, 3.86; P < .048).
If at least comparable efficacy can be clearly demonstrated, advantages of TBS over standard TMS include a significantly reduced administration time, which might allow for more patients to be treated and reduce associated costs of treatment.27
Magnetic low-field synchronized stimulation is produced by rotating spherical rare-earth magnets that are synchronized to an individual’s alpha frequency. A recent 6-week, double-blind, sham-controlled trial (N = 202) reported that, in the intention-to-treat population, there was no difference in outcome between treatment arms. In patients who completed the study according to protocol (120 of 202), however, active treatment was significantly better in decreasing baseline HDRS score (P < .033).28
Magnetic seizure therapy (MST) is an experimental approach to treating patients with more severe depression that is resistant to medical therapy. The primary aim is to use TMS to induce a seizure, thus achieving the same efficacy as provided by ECT but without the adverse cognitive effects of ECT. With MST, the TMS device uses much higher stimulation settings to produce a seizure—the goal being to avoid direct electrical current to the brain’s memory centers.29
A pilot study considered the clinical and cognitive effects of MST in a group of 26 treatment-resistant depression patients (10 randomized; 16 open-label).30 Based on reduction in baseline HDRS scores at the end of the trial, 69% of patients achieved response and 46% met remission criteria; however, one-half of patients relapsed within 6 months.
Importantly, no cognitive adverse effects were observed. Furthermore, the antidepressant and anti-anxiety effects of MST were associated with localized metabolic changes in brain areas implicated in the pathophysiology of depression.
The investigators concluded that MST might constitute an effective, well-tolerated, and safe treatment for patients unable to benefit from available medical therapies for depression. In addition to confirmation of acute benefit in more definitive trials, the issue of durability of effect needs further clarification.
TMS is a key component of neuropsychiatric practice
It has been 3 decades since Barker et al31 developed the technology to deliver intense, localized magnetic pulses to specific areas of the nervous system. During this period, the role of TMS as a probe of the central and peripheral nervous systems has expanded to include various therapeutic applications, primarily focusing on treatment-resistant major depressive disorder.
Now, increasing sophistication in the choice of stimulation parameters and other ongoing efforts to optimize the benefits of TMS are yielding improved clinical outcomes. Research is still needed to better define the place of TMS in the management of subtypes of depression that are particularly difficult to treat and that do not benefit adequately from medications or psychotherapy or their combination.
Growing support from controlled trials, systematic reviews, meta-analyses, naturalistic outcome studies, and professional guidelines indicate that TMS has an increasingly important role in clinical practice.
1. Janicak PG, Dowd SM, Rado JT, et al. The re-emerging role of therapeutic neuromodulation. Current Psychiatry. 2010;9(11):66-70,72-74.
2. Janicak PG, Dokucu ME. Transcranial magnetic stimulation for the treatment of major depression. Neuropsychiatr Dis Treat. 2015;11:1549-1560.
3. Gaynes BN, Lloyd SW, Lux L, et al. Repetitive transcranial magnetic stimulation for treatment-resistant depression: a systematic review and meta-analysis. J Clin Psychiatry. 2014;75(5):477-489; quiz 489.
4. Liu B, Zhang Y, Zhang L, et al. Repetitive transcranial magnetic stimulation as an augmentative strategy for treatment-resistant depression, a meta-analysis of randomized, double-blind and sham-controlled study. BMC Psychiatry. 2014;14:342.
5. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
6. Janicak PG, Dunner DL, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of quality of life outcome measures in clinical practice. CNS Spectr. 2013;18(6):322-332.
7. Janicak PG, Nahas Z, Lisanby SH, et al. Durability of clinical benefit with transcranial magnetic stimulation (TMS) in the treatment of pharmacoresistant major depression: assessment of relapse during a 6-month, multisite, open-label study. Brain Stimul. 2010;3(4):187-199.
8. Mantovani A, Pavlicova M, Avery D, et al. Long-term efficacy of repeated daily prefrontal transcranial magnetic stimulation (TMS) in treatment-resistant depression. Depress Anxiety. 2012;29(10):883-890.
9. Philip NS, Dunner DL, Dowd SM, et al. Can medication free, treatment-resistant, depressed patients who initially respond to TMS be maintained off medications? A prospective, 12-month multisite randomized pilot study. Brain Stimul. 2016;9(2):251-257.
10. Dunner DL, Aaronson ST, Sackeim HA, et al. A multisite, naturalistic, observational study of transcranial magnetic stimulation for patients with pharmacoresistant major depressive disorder: durability of benefit over a 1-year follow-up period. J Clin Psychiatry. 2014;75(12):1394-1401.
11. Hermann RC, Dorwart RA, Hoover CW. Variation in ECT use in the United States. Am J Psychiatry. 1995;152(6):869-875.
12. Sackeim HA. Memory and ECT: from polarization to reconciliation. J ECT. 2000;16(2):87-96.
13. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
14. Ren J, Li H, Palaniyappan L, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depression: a systematic review and meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2014;51:181-189.
15. Martis B, Alam D, Dowd SM, et al. Neurocognitive effects of repetitive transcranial magnetic stimulation in severe major depression. Clin Neurophysiol. 2003;114(6):1125-1132.
16. Pridmore S, Rybak M, Turnier-Shea Y, et al. Comparison of transcranial magnetic stimulation and electroconvulsive therapy in depression. In: Miyoshi K, Shapiro CM, Gaviria M, et al, eds. Contemporary neuropsychiatry. Tokyo, Japan: Springer; 2001:237-241.
17. Jelovac A, Kolshus E, McLoughlin DM. Relapse following successful electroconvulsive therapy for major depression: a meta-analysis. Neuropsychopharmacology. 2013;38(12):2467-2474.
18. Noda Y, Daskalakis Z, Ramos C, et al. Repetitive transcranial magnetic stimulation to maintain treatment response to electroconvulsive therapy in depression: a case series. Front Psychiatry. 2013;4:73.
19. Cristancho MA, Helmer A, Connolly R, et al. Transcranial magnetic stimulation maintenance as a substitute for maintenance electroconvulsive therapy: a case series. J ECT. 2013;29(2):106-108.
20. Casarotto S, Canali P, Rosanova M, et al. Assessing the effects of electroconvulsive therapy on cortical excitability by means of transcranial magnetic stimulation and electroencephalography. Brain Topogr. 2013;26(2):326-337.
21. Levkovitz Y, Isserles M, Padberg F, et al. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective multicenter randomized controlled trial. World Psychiatry. 2015;14(1):64-73.
22. Daskalakis ZJ. Theta-burst transcranial magnetic stimulation in depression: when less may be more. Brain. 2014;137(pt 7):1860-1862.
23. Prasser J, Schecklmann M, Poeppl TB, et al. Bilateral prefrontal rTMS and theta burst TMS as an add-on treatment for depression: a randomized placebo controlled trial. World J Biol Psychiatry. 2015;16(1):57-65.
24. Li CT, Chen MH, Juan CH, et al. Efficacy of prefrontal theta-burst stimulation in refractory depression: a randomized sham-controlled study. Brain. 2014;137(pt 7):2088-2098.
25. Chistyakov A, Kreinin B, Marmor S, et al. Preliminary assessment of the therapeutic efficacy of continuous theta-burst magnetic stimulation (cTBS) in major depression: a double-blind sham-controlled study. J Affect Disord. 2015;170:225-229.
26. Plewnia C, Pasqualetti P, Große S, et al. Treatment of major depression with bilateral theta burst stimulation: a randomized controlled pilot trial. J Affect Disord. 2014;156:219-223.
27. Chung SW, Hoy KE, Fitzgerald PB. Theta-burst stimulation: a new form of TMS treatment for depression? Depress Anxiety. 2015;32(3):182-192.
28. Leuchter AF, Cook IA, Feifel D, et al. Efficacy and safety of low-field synchronized transcranial magnetic stimulation (sTMS) for treatment of major depression. Brain Stimul. 2015;8(4):787-794.
29. Cretaz E, Brunoni AR, Lafer B. Magnetic seizure therapy for unipolar and bipolar depression: a systematic review. Neural Plast. 2015;2015:521398. doi: 10.1155/2015/521398.
30. Kayser S, Bewernick BH, Matusch A, et al. Magnetic seizure therapy in treatment-resistant depression: clinical, neuropsychological and metabolic effects. Psychol Med. 2015;45(5):1073-1092.
31. Barker AT, Jalinous R, Freeston IL. Non-invasive magnetic stimulation of human motor cortex. Lancet. 1985;1(8437):1106-1107.
1. Janicak PG, Dowd SM, Rado JT, et al. The re-emerging role of therapeutic neuromodulation. Current Psychiatry. 2010;9(11):66-70,72-74.
2. Janicak PG, Dokucu ME. Transcranial magnetic stimulation for the treatment of major depression. Neuropsychiatr Dis Treat. 2015;11:1549-1560.
3. Gaynes BN, Lloyd SW, Lux L, et al. Repetitive transcranial magnetic stimulation for treatment-resistant depression: a systematic review and meta-analysis. J Clin Psychiatry. 2014;75(5):477-489; quiz 489.
4. Liu B, Zhang Y, Zhang L, et al. Repetitive transcranial magnetic stimulation as an augmentative strategy for treatment-resistant depression, a meta-analysis of randomized, double-blind and sham-controlled study. BMC Psychiatry. 2014;14:342.
5. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
6. Janicak PG, Dunner DL, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of quality of life outcome measures in clinical practice. CNS Spectr. 2013;18(6):322-332.
7. Janicak PG, Nahas Z, Lisanby SH, et al. Durability of clinical benefit with transcranial magnetic stimulation (TMS) in the treatment of pharmacoresistant major depression: assessment of relapse during a 6-month, multisite, open-label study. Brain Stimul. 2010;3(4):187-199.
8. Mantovani A, Pavlicova M, Avery D, et al. Long-term efficacy of repeated daily prefrontal transcranial magnetic stimulation (TMS) in treatment-resistant depression. Depress Anxiety. 2012;29(10):883-890.
9. Philip NS, Dunner DL, Dowd SM, et al. Can medication free, treatment-resistant, depressed patients who initially respond to TMS be maintained off medications? A prospective, 12-month multisite randomized pilot study. Brain Stimul. 2016;9(2):251-257.
10. Dunner DL, Aaronson ST, Sackeim HA, et al. A multisite, naturalistic, observational study of transcranial magnetic stimulation for patients with pharmacoresistant major depressive disorder: durability of benefit over a 1-year follow-up period. J Clin Psychiatry. 2014;75(12):1394-1401.
11. Hermann RC, Dorwart RA, Hoover CW. Variation in ECT use in the United States. Am J Psychiatry. 1995;152(6):869-875.
12. Sackeim HA. Memory and ECT: from polarization to reconciliation. J ECT. 2000;16(2):87-96.
13. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
14. Ren J, Li H, Palaniyappan L, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depression: a systematic review and meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2014;51:181-189.
15. Martis B, Alam D, Dowd SM, et al. Neurocognitive effects of repetitive transcranial magnetic stimulation in severe major depression. Clin Neurophysiol. 2003;114(6):1125-1132.
16. Pridmore S, Rybak M, Turnier-Shea Y, et al. Comparison of transcranial magnetic stimulation and electroconvulsive therapy in depression. In: Miyoshi K, Shapiro CM, Gaviria M, et al, eds. Contemporary neuropsychiatry. Tokyo, Japan: Springer; 2001:237-241.
17. Jelovac A, Kolshus E, McLoughlin DM. Relapse following successful electroconvulsive therapy for major depression: a meta-analysis. Neuropsychopharmacology. 2013;38(12):2467-2474.
18. Noda Y, Daskalakis Z, Ramos C, et al. Repetitive transcranial magnetic stimulation to maintain treatment response to electroconvulsive therapy in depression: a case series. Front Psychiatry. 2013;4:73.
19. Cristancho MA, Helmer A, Connolly R, et al. Transcranial magnetic stimulation maintenance as a substitute for maintenance electroconvulsive therapy: a case series. J ECT. 2013;29(2):106-108.
20. Casarotto S, Canali P, Rosanova M, et al. Assessing the effects of electroconvulsive therapy on cortical excitability by means of transcranial magnetic stimulation and electroencephalography. Brain Topogr. 2013;26(2):326-337.
21. Levkovitz Y, Isserles M, Padberg F, et al. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective multicenter randomized controlled trial. World Psychiatry. 2015;14(1):64-73.
22. Daskalakis ZJ. Theta-burst transcranial magnetic stimulation in depression: when less may be more. Brain. 2014;137(pt 7):1860-1862.
23. Prasser J, Schecklmann M, Poeppl TB, et al. Bilateral prefrontal rTMS and theta burst TMS as an add-on treatment for depression: a randomized placebo controlled trial. World J Biol Psychiatry. 2015;16(1):57-65.
24. Li CT, Chen MH, Juan CH, et al. Efficacy of prefrontal theta-burst stimulation in refractory depression: a randomized sham-controlled study. Brain. 2014;137(pt 7):2088-2098.
25. Chistyakov A, Kreinin B, Marmor S, et al. Preliminary assessment of the therapeutic efficacy of continuous theta-burst magnetic stimulation (cTBS) in major depression: a double-blind sham-controlled study. J Affect Disord. 2015;170:225-229.
26. Plewnia C, Pasqualetti P, Große S, et al. Treatment of major depression with bilateral theta burst stimulation: a randomized controlled pilot trial. J Affect Disord. 2014;156:219-223.
27. Chung SW, Hoy KE, Fitzgerald PB. Theta-burst stimulation: a new form of TMS treatment for depression? Depress Anxiety. 2015;32(3):182-192.
28. Leuchter AF, Cook IA, Feifel D, et al. Efficacy and safety of low-field synchronized transcranial magnetic stimulation (sTMS) for treatment of major depression. Brain Stimul. 2015;8(4):787-794.
29. Cretaz E, Brunoni AR, Lafer B. Magnetic seizure therapy for unipolar and bipolar depression: a systematic review. Neural Plast. 2015;2015:521398. doi: 10.1155/2015/521398.
30. Kayser S, Bewernick BH, Matusch A, et al. Magnetic seizure therapy in treatment-resistant depression: clinical, neuropsychological and metabolic effects. Psychol Med. 2015;45(5):1073-1092.
31. Barker AT, Jalinous R, Freeston IL. Non-invasive magnetic stimulation of human motor cortex. Lancet. 1985;1(8437):1106-1107.

How to control weight gain when prescribing antidepressants
The prevalence of undesired weight gain in the United States has reached an all-time high, with 68.5% of adults identified as overweight (body mass index [BMI] ≥25) or obese (BMI ≥30), 34.5% considered obese, and 6.4% considered extremely obese (BMI ≥40).1 Reasons for weight gain include various physical and nutritional factors in a patient’s life, but sometimes weight gain is iatrogenic. Many medications we prescribe are associated with weight gain, including most antidepressants and atypical antipsychotics. Clinicians might minimize or overlook the risk of weight gain when prescribing antidepressants.
Patients with major depression often have associated weight loss. Regaining weight can be seen as sign of successful treatment of depressive symptoms. If weight gain after treatment exceeds the amount of weight loss attributed to depression, however, medication could have caused the excessive gain. This is considered a side effect, or iatrogenic weight gain, and should not be considered normal or clinically acceptable.
Patients who are overweight or obese when beginning antidepressant treatment might be at greater medical risk when placed on a medication that can cause additional weight gain. The time to onset of weight gain during treatment can predict weight gain patterns; those affected in the first month are most at risk of future excessive weight gain.2
In this article, we discuss:
- considerations when prescribing antidepressants
- ways to approach weight gain
- medications available to assist in weight loss.
Our general recommendations
Screen. The United States Preventive Services Task Force maintains a Class-B recommendation for screening all patients for obesity. This means that the Task Force’s review panel determined that such screening is at least moderately or substantially beneficial.3 Screening is important in a setting of potential weight gain in patients taking an antidepressant.
Educate and treat. Provide at least some education and encouragement about eating a healthy diet and exercising, or refer the patient to a nutritionist or dietician. Next, initiate psychotherapy (motivational interviewing, cognitive-behavioral therapy [CBT]) as needed. Reserve anti-obesity medications for those who do not respond to weight loss efforts or who might be taking an antidepressant for the long term.
The need for medical management of weight gain has given rise to specialists who treat this complicated, multifactorial condition. Whether psychiatrists should be seen as a substitution for their specialty is not the purpose of this review; rather, how we might more effectively (1) work on our patients’ behalf to mitigate potential weight gain from the treatments that we prescribe and (2) participate in consultations that we’ve provided on their behalf.
BMI is not an absolute marker of healthBMI likely should not be viewed as a marker with absolute prognostic certainty of overall health of an overweight or obese person: An overweight person considered healthy from a cardiovascular and metabolic perspective could still benefit from preventing further weight gain.
Tomiyama et al4 concluded that BMI itself was insufficient to stratify health in a meaningful way—and that such a focus would lead to overweight and obese people in otherwise good health being penalized unfairly through higher health insurance premiums, and would divert focus on those with less optimal health but a normal BMI. The researchers’ goal was to use blood pressure, lipid levels, and glycemic markers as surrogate markers of health, and then statistically compare results with patients’ corresponding BMI. Their findings showed that approximately one-half of people who are overweight and 29% of obese people can be considered healthy.4
Potential causes of weight gainThere may be more than one reason for weight gain during depression treatment, so a multifactorial management approach might be necessary, depending on the patient’s medication regimen. Appetite might be influenced by physical (chemical, metabolic) and psychological (cultural, familial) factors. The following sections focus on specific antidepressant classes and their proclivity for weight gain.
Serotonergic antidepressantsMany patients with depression are treated with medications that alter serotonin levels in the body, such as selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs). This neurotransmitter often is affected through depression treatment, and therefore might be a factor contributing to unintended weight gain. In mice bred to lack serotonin 5-HT2c receptors in proopiomelanocortin (POMC) neurons, the expected anorectic reaction to serotonergic agents often is reversed, causing a robust increase in hyperphagia and obesity.5 This effect indicates that 5-HT2c receptor stimulation might control appetite and feeding.
After SSRI or SNRI treatment, accumulation of serotonin over time in the synaptic cleft is thought to result in down-regulation of 5-HT2c receptors. This may cause a relative absence of 5-HT2c receptors, similar to what is seen in mice who lack them biologically. The loss of these receptors or their activity often will result in excessive weight gain. Some sedating antidepressants (mirtazapine) and some second-generation antipsychotics (SGAs) (olanzapine, quetiapine) directly block 5-HT2c receptors and might cause more rapid weight gain. Lorcaserin, a selective 5-HT2c receptor agonist, theoretically could reverse this proposed weight gain mechanism and suppress appetite by activating the POMC pathway in the hypothalamus.
Continue to: Among SSRIs and SNRIs
Among SSRIs and SNRIs, paroxetine might be one of the worst for provoking long-term weight gain; a study showed an average increase of 2.73 kg over a 4-month period.6
Theoretically, SNRIs have the ability to increase noradrenergic tone. This might be associated with nausea and a decline in appetite or it might generally curb appetite. These agents likely will cause less future weight gain. SNRIs typically induce more noradrenergic tone at increasingly higher dosages. There may be a dose-response curve in this manner. Levomilnacipran likely is the most noradrenergic of the SNRIs; recent regulatory studies suggest no statistically significant weight gain over the long term.7
Sedating antidepressants
Mirtazapine has receptor-blocking effects on noradrenergic α-2 and serotonergic 5-HT2a and 5-HT2c receptors. Additionally, histamine blocking of H1 receptors can contribute to additional weight gain, similar to what is seen with some SGAs. H1 antagonism dampens satiety response, resulting in increased caloric intake. In that case, or when specific SGAs are used for managing depression, appetite increases (H1 antagonism) and metabolism slows (possibly 5-HT2c antagonism, muscarinic receptor antagonism, etc.), thus allowing for greater adipose tissue growth and leptin insensitivity.
In a meta-analysis, mean weight increased by 1.74 kg (P < .0001) in the first 4 to 12 weeks of mirtazapine treatment, with greater variability in periods >4 months.6 Among the more novel antidepressants released since the era of tricyclic antidepressants (TCAs) or monoamine oxidase inhibitor, mirtazapine might have the greatest weight gain potential.
Trazodone and nefazodone block 5-HT2a and 5-HT2c receptors, as well as serotonin reuptake transporters. Compared with trazodone, nefazodone has a more potent effect on 5-HT2a receptor antagonism and a less potent effect on 5-HT2c receptors, and also mildly inhibits uptake of norepinephrine—meaning that this drug might have less weight gain potential. These medications are not used frequently for treating depression, but trazodone is used as an adjunctive agent for insomnia. Used even at off-label low dosages, trazodone exerts H1-histaminic and α-1 adrenergic antagonistic properties, decreasing the level of consciousness and allowing sedation and somnolence. Because of its fast onset and relatively short duration of action, it can improve depression symptoms by promoting restful sleep as well as by facilitating monoamine neurotransmission. It also might add to weight gain because of its pharmacodynamic receptor profile.
Tricyclic antidepressants
Amitriptyline can be associated with release of tumor necrosis factor-alpha, which is implicated in causing weight gain. Many TCAs block H1 (amitriptyline, imipramine, clomipramine), likely causing weight gain. Most TCAs antagonize muscarinic receptors as well. The more noradrenergic TCAs could curb appetite (nortriptyline, desipramine, protriptyline) similar to SNRIs, therefore countering some of the weight gain drive.
As an example, in a meta-analysis examining weight gain with antidepressants, amitriptyline was associated with weight gain of 1.52 kg above baseline in the acute period (4 to 12 weeks) and 2.24 kg above baseline at 4 to 7 months.6 These results of the acute phase should be viewed cautiously because the authors reported high heterogeneity among these studies, and the possibility of publication bias (Egger test, P < .0001). In the same meta-analysis, the even the more noradrenergic nortriptyline was associated with an increase of 2.0 kg on average over baseline during acute treatment, with that number dropping to 1.24 kg over baseline at ≥4 months.6
Newer antidepressants
Vilazodone is a weak SSRI that aggressively partially agonizes pre-synaptic and post-synaptic 5-HT1a receptors in the CNS. This dual site 5-HT1a action is somewhat unique among antidepressants. This type of agent sometimes is called multimodal,8 or could be considered an “SSRI +” antidepressant. These drugs are SSRIs at the core but have additional 5-HT receptor modulating capabilities. Vilazodone has a favorable weight gain profile, as suggested in a 52-week trial reporting 1.7 kg gain over 52 weeks, compared with an average of 6.8 to 10 kg for long-term SSRI therapy.9
Vortioxetine is a stronger SSRI that also partially agonizes presynaptic 5-HT1a receptors. In addition, it antagonizes 5-HT1d, 5-HT3, and 5-HT7 receptors, giving it a unique pharmacodynamic profile.10 Vortioxetine also had minimal impact on drug-induced weight gain in 52-week studies, with data from 2 trials indicating either minimal weight gain in 6.1% of patients (mean increase of 0.41 kg over 52 weeks)11 or gain that was not statistically significant.12
Levomilnacipran is unique in that it has the most aggressive norepinephrine reuptake inhibition of all SNRIs.13 Again, increased noradrenergic tone might curb appetite and caloric intake. Many SNRIs cause low-grade nausea, which could account for decreased appetite. Long-term, 52-week data for this drug also shows minimal proclivity for weight gain, with the trial participants reporting a slight decrease of 4.34 kg on average from baseline.14
Continue to: Addressing weight gain
Addressing weight gain
Lifestyle modification. Eating smaller portions, combined with restricting foods high in calories and fat, should be the first step. A simple suggestion to a patient to eat the same foods, but remove 20% of the portion, is a simple intervention akin to that of suggesting sleep hygiene practices for insomnia management. Under medical supervision or with referral to a dietician or nutritionist, more rigid caloric restrictions could be employed.
Commercial weight-loss programs, such as Weight Watchers or Curves, can be helpful; some insurers will only cover medications for weight loss if one of these programs have been tried or is used in combination with medication. Some patients might ask about extreme weight-loss measures, such as low-calorie diets combined with intense exercise programs that have been popularized in the media. Although the motivation to initiate and maintain meaningful weight loss should be encouraged, doing so in a more gradual manner should be the goal.
Addressing portion size is a good approach in the early stages of managing obesity. Restaurants often serve portions that have more calories than should be consumed in one meal. Visual cues can influence this trend; using smaller plates can help reduce caloric intake.15
Exercise, sustained for at least 45 minutes, can have long-lasting effects, with a small study showing an increase in metabolic rate of 190 ± 71.4 kcal (P < .001) above baseline for 14 hours after exercise.16 Endurance exercise training is associated with a significant decrease in total cholesterol, triglycerides, and low-density lipoprotein cholesterol, as well as an increase in the high-density lipoprotein level over a 24-week period.17
Encouraging an exercise regimen that is appropriate for your patient can help maintain weight loss. In small trials,18,19 high-intensity exercise was shown to help suppress appetite and decrease 24-hour caloric consumption by 6% to 11%.18
Psychotherapy can become an important intervention for initiating and maintaining weight loss. CBT can help patients recognize and modify lifestyle components, and reinforce behaviors that promote weight loss. This can come from setting realistic weight loss goals; preventing triggering factors that lead to overeating; encouraging portion control during meals; and promoting exercise habits.
In a small, randomized controlled trial (RCT) examining weight loss in obese women, those who underwent CBT and psychoeducation for 2 hours a week for 10 weeks in addition to dietary changes and exercise showed an average weight loss of 10.4 kg at 18-month follow-up, compared with weight gain of 2.3 kg in the control group.20 The short duration of treatment in this study might be desirable to reduce cost and utilization of services. Group formats also could be employed.
Motivational interviewing is a useful tool in addiction psychiatry and shows promise for treating obesity and overeating as well. The approach may differ slightly because weight-loss therapy involves behavior modification rather than behavior cessation. In a meta-analysis of data from RCTs exploring motivational interviewing and its use as an intervention for weight loss, those in the intervention groups experienced significant weight loss as indicated by BMI decreasing a standardized mean difference of −0.51, compared with control groups.21
Medical management considerations
Diagnostics. Recognition and early intervention are instrumental in successfully treating medication-associated weight gain. It is important to obtain any family history of obesity, diabetes, hypertension, and hyperlipidemia. This will likely indicate a patient’s risk for weight gain before initiating medication.
Obtain vital signs at every visit, including blood pressure. Monitoring weight at every clinical visit can be used to calculate and monitor BMI, while also asking the patient to maintain a log of weight measurements obtained at home. Measuring abdominal girth is important to watch for metabolic syndrome, although often this is the least measured variable.
Laboratory testing is helpful. Obtaining a baseline lipid panel and a fasting glucose level (consider measuring hemoglobin A1c in patients with diabetes) is warranted. Including thyroid markers, such as thyroid-stimulating hormone and thyroxine (free T4), might be important considerations, because inadequate management of hypothyroidism can complicate the clinical picture.
Follow-up testing should be ordered every 3 to 12 months to monitor progress if your patient is showing signs of rapid weight gain, or if BMI nears ≥30 kg/m². These guidelines generally are assigned for prescribing of SGAs, but can be applied when using any psychotropic with weight gain potential.
Medications to considerWhen considering the medication regimen as an intervention point, consider changing the antidepressant to one that is not associated with significant weight gain. Although not specifically indicated as a monotherapy for weight loss, switching to or augmenting therapy with bupropion could aid weight loss through appetite suppression.20 Some newer antidepressants, such as vilazodone, vortioxetine, and levomilnacipran, might have less propensity to cause weight gain. In patients with severe depression, augmenting with medications containing amphetamine or methylphenidate could cause some weight loss, but greater care should be taken because of cardiovascular effects and dependency issues.
Continue to: Discussing with your patient...
Discussing with your patient the possibility of changing or worsening depressive symptoms when adding or switching medications allows them to be aware and engaged in the process and can encourage them to notice and report changes. Developing a sensible schedule to taper an existing medication slowly over several weeks and allowing a new one to build up gradually to a therapeutic level can help minimize adverse effects or a discontinuation syndrome.
If switching antidepressants is not possible, or is ineffective, an anti-obesity medication (Table 122-29) can be considered. These medications should not be considered first-line in weight loss management, but reserved for more difficult or refractory weight loss challenges and in patients who are not able to participate in weight loss or dieting programs because of cognitive disorders, a history of nonadherance, financial or travel limitations, or in those with poor social support systems such as homelessness.
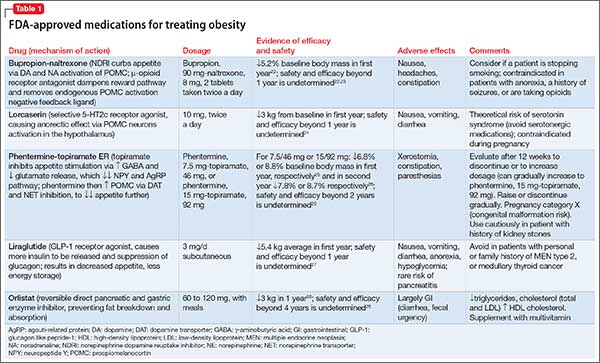
Some of these medications are not reimbursed by insurance companies; therefore, consider the financial burden to the patient and their capacity for adherence to therapy, and discuss this challenge before initiating treatment. There is some evidence for using medications off-label to treat obesity (Table 2,30-34).
Anti-obesity medications typically are considered for patients with a BMI >30 or in any overweight patient with diabetes, hyperlipidemia, or cardiovascular disease. As always, discuss with patients and their primary care provider the potential benefits and risks of adding any of these or other medications to an existing treatment regimen.
If weight loss goals are not met, consider discontinuing anti-obesity therapy. Patients and physicians should be cognizant of the need to continue long-term maintenance on these medications after successful treatment—perhaps indefinitely, because patients frequently regain weight after medication is discontinued.
Bottom Line
Many antidepressants are known to increase the risk of excessive weight gain, although risk of weigh gain varies among antidepressant classes. First, advise changes in diet and exercise; next, initiate psychotherapy as indicated, and then consider referral to a nutritionist. Consider switching to an antidepressant with less potential for causing weight gain or adding bupropion, which could lead to weight loss, if your patient can tolerate it. If these strategies are unsuccessful, consider an anti-obesity medication.
Related Resource
1. Ogden CL, Carroll MD, Kit BK, et al. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA. 2014;311(8):806-814.
2. Vandenberghe F, Gholam-Rezaee M, Saigí-Morgui N, et al. Importance of early weight changes to predict long-term weight gain during psychotropic drug treatment. J Clin Psychiatry. 2015;76(11):e1417-e1423.
3. Grade Definitions. Electronic Preventive Services Selector (ePSS). http://epss.ahrq.gov/ePSS/gradedef.jsp. Accessed May 2, 2016.
4. Tomiyama AJ, Hunger JM, Nguyen-Cuu J, et al. Misclassification of cardiometabolic health when using body mass index categories in NHANES 2005-2012 [published online February 4, 2016]. Int J Obes (Lond). doi: 10.1038/ijo.2016.17.
5. Berglund ED, Liu C, Sohn J, et al. Serotonin 2C receptors in pro-opiomelanocortin neurons regulate energy and glucose homeostasis. J Clin Invest. 2013;123(12):5061-5070.
6. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-anaylsis. J Clin Psychiatry. 2010;71(10):1259-1272.
7. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
8. Schwartz TL, Siddiqui UA, Stahl SM. Vilazodone: a brief pharmacological and clinical review of the novel serotonin partial agonist and reuptake inhibitor. Ther Adv Psychopharmacol. 2011;1(3):81-87.
9. Robinson DS, Kajdasz DK, Gallipoli S, et al. A 1-year, open-label study assessing the safety and tolerability of vilazodone in patients with major depressive disorder. J Clin Psychopharmacol. 2011;31(5):643-646.
10. Stahl SM. Modes and nodes explain the mechanism of action of vortioxetine, a multimodal agent (MMA): actions at serotonin receptors may enhance downstream release of four pro-cognitive neurotransmitters. CNS Spectr. 2015;20(6):515-519.
11. Jacobsen PL, Harper L, Chrones L, et al. Safety and tolerability of vortioxetine (15 and 20 mg) in patients with major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2015;30(5):255-264.
12. Boulenger JP, Loft H, Olsen CK. Efficacy and safety of vortioxetine (Lu AA21004), 15 and 20 mg/day: a randomized, double-blind, placebo-controlled, duloxetine-referenced study in the acute treatment of adult patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(3):138-149.
13. Grady MM, Stahl SM. Novel agents in development for the treatment of depression. CNS Spectr. 2013;18(suppl 1):37-40; quiz 41.
14. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
15. Hollands GJ, Shemilt I, Marteau TM, et al. Portion, package or tableware size for changing selection and consumption of food, alcohol and tobacco. Cochrane Database Syst Rev. 2015;9:CD011045.
16. Knab AM, Shanely RA, Corbin KD, et al. A 45-minute vigorous exercise bout increases metabolic rate for 14 hours. Med Sci Sports Exerc. 2011;43(9):1643-1648.
17. Halverstadt A, Phares DA, Wilund KR, et al. Endurance exercise training raises high-density lipoprotein cholesterol and lowers small low-density lipoprotein and very low-density lipoprotein independent of body fat phenotypes in older men and women. Metabolism. 2007;56(4):444-450.
18. Thivel D, Isacco L, Montaurier C, et al. The 24-h energy intake of obese adolescents is spontaneously reduced after intensive exercise: a randomized controlled trial in calorimetric chambers [published online January 17, 2012]. PLoS One. 2012;7(1):e29840. doi: 10.1371/journal.pone.0029840.
19. Sim AY, Wallman KE, Fairchild TJ, et al. High-intensity intermittent exercise attenuates ad-libitum energy intake. Int J Obes (Lond). 2014;38(3):417-422.
20. Stahre L, Tärnell B, Håkanson CE, et al. A randomized controlled trial of two weight-reducing short-term group treatment programs for obesity with an 18-month follow-up. Int J Behav Med. 2007;14(1):48-55.
21. Armstrong MJ, Mottershead TA, Ronksley PE, et al. Motivational interviewing to improve weight loss in overweight and/or obese patients: a systematic review and meta-analysis of randomized controlled trials. Obes Rev. 2011;12(9):709-723.
22. Apovian CM, Aronne L, Rubino D, et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Springs). 2013;21(5):935-943.
23. Greenway FL, Fujioka K, Plodkowski RA, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605.
24. Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96(10):3067-3077.
25. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial [Erratum in Lancet. 2011;377(9776):1494]. Lancet. 2011;377(9774):1341-1352.
26. Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr. 2012;95(2):297-308.
27. Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373(1):11-22.
28. Leblanc ES, O’Connor E, Whitlock EP, et al. Effectiveness of primary care-relevant treatments for obesity in adults: a systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med. 2011;155(7):434-447.
29. Torgerson JS, Hauptman J, Boldrin MN, et al. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27(1):155-161.
30. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393-403.
31. McDonagh MS, Selph S, Ozpinar A, et al. Systematic review of the benefits and risks of metformin in treating obesity in children aged 18 years and younger. JAMA Pediatr. 2014;168(2):178-184.
32. Dushay J, Gao C, Gopalakrishnan GS, et al. Short-term exenatide treatment leads to significant weight loss in a subset of obese women without diabetes. Diabetes Care. 2012;35(1):4-11.
33. Gadde KM, Kopping MF, Wagner HR 2nd, et al. Zonisamide for weight reduction in obese adults: a 1-year randomized controlled trial. Arch Intern Med. 2012;172(20):1557-1564.
34. Singh-Franco D, Perez A, Harrington C. The effect of pramlintide acetate on glycemic control and weight in patients with type 2 diabetes mellitus and in obese patients without diabetes: a systematic review and meta-analysis. Diabetes Obes Metab. 2011;13(2):169-180.
The prevalence of undesired weight gain in the United States has reached an all-time high, with 68.5% of adults identified as overweight (body mass index [BMI] ≥25) or obese (BMI ≥30), 34.5% considered obese, and 6.4% considered extremely obese (BMI ≥40).1 Reasons for weight gain include various physical and nutritional factors in a patient’s life, but sometimes weight gain is iatrogenic. Many medications we prescribe are associated with weight gain, including most antidepressants and atypical antipsychotics. Clinicians might minimize or overlook the risk of weight gain when prescribing antidepressants.
Patients with major depression often have associated weight loss. Regaining weight can be seen as sign of successful treatment of depressive symptoms. If weight gain after treatment exceeds the amount of weight loss attributed to depression, however, medication could have caused the excessive gain. This is considered a side effect, or iatrogenic weight gain, and should not be considered normal or clinically acceptable.
Patients who are overweight or obese when beginning antidepressant treatment might be at greater medical risk when placed on a medication that can cause additional weight gain. The time to onset of weight gain during treatment can predict weight gain patterns; those affected in the first month are most at risk of future excessive weight gain.2
In this article, we discuss:
- considerations when prescribing antidepressants
- ways to approach weight gain
- medications available to assist in weight loss.
Our general recommendations
Screen. The United States Preventive Services Task Force maintains a Class-B recommendation for screening all patients for obesity. This means that the Task Force’s review panel determined that such screening is at least moderately or substantially beneficial.3 Screening is important in a setting of potential weight gain in patients taking an antidepressant.
Educate and treat. Provide at least some education and encouragement about eating a healthy diet and exercising, or refer the patient to a nutritionist or dietician. Next, initiate psychotherapy (motivational interviewing, cognitive-behavioral therapy [CBT]) as needed. Reserve anti-obesity medications for those who do not respond to weight loss efforts or who might be taking an antidepressant for the long term.
The need for medical management of weight gain has given rise to specialists who treat this complicated, multifactorial condition. Whether psychiatrists should be seen as a substitution for their specialty is not the purpose of this review; rather, how we might more effectively (1) work on our patients’ behalf to mitigate potential weight gain from the treatments that we prescribe and (2) participate in consultations that we’ve provided on their behalf.
BMI is not an absolute marker of healthBMI likely should not be viewed as a marker with absolute prognostic certainty of overall health of an overweight or obese person: An overweight person considered healthy from a cardiovascular and metabolic perspective could still benefit from preventing further weight gain.
Tomiyama et al4 concluded that BMI itself was insufficient to stratify health in a meaningful way—and that such a focus would lead to overweight and obese people in otherwise good health being penalized unfairly through higher health insurance premiums, and would divert focus on those with less optimal health but a normal BMI. The researchers’ goal was to use blood pressure, lipid levels, and glycemic markers as surrogate markers of health, and then statistically compare results with patients’ corresponding BMI. Their findings showed that approximately one-half of people who are overweight and 29% of obese people can be considered healthy.4
Potential causes of weight gainThere may be more than one reason for weight gain during depression treatment, so a multifactorial management approach might be necessary, depending on the patient’s medication regimen. Appetite might be influenced by physical (chemical, metabolic) and psychological (cultural, familial) factors. The following sections focus on specific antidepressant classes and their proclivity for weight gain.
Serotonergic antidepressantsMany patients with depression are treated with medications that alter serotonin levels in the body, such as selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs). This neurotransmitter often is affected through depression treatment, and therefore might be a factor contributing to unintended weight gain. In mice bred to lack serotonin 5-HT2c receptors in proopiomelanocortin (POMC) neurons, the expected anorectic reaction to serotonergic agents often is reversed, causing a robust increase in hyperphagia and obesity.5 This effect indicates that 5-HT2c receptor stimulation might control appetite and feeding.
After SSRI or SNRI treatment, accumulation of serotonin over time in the synaptic cleft is thought to result in down-regulation of 5-HT2c receptors. This may cause a relative absence of 5-HT2c receptors, similar to what is seen in mice who lack them biologically. The loss of these receptors or their activity often will result in excessive weight gain. Some sedating antidepressants (mirtazapine) and some second-generation antipsychotics (SGAs) (olanzapine, quetiapine) directly block 5-HT2c receptors and might cause more rapid weight gain. Lorcaserin, a selective 5-HT2c receptor agonist, theoretically could reverse this proposed weight gain mechanism and suppress appetite by activating the POMC pathway in the hypothalamus.
Continue to: Among SSRIs and SNRIs
Among SSRIs and SNRIs, paroxetine might be one of the worst for provoking long-term weight gain; a study showed an average increase of 2.73 kg over a 4-month period.6
Theoretically, SNRIs have the ability to increase noradrenergic tone. This might be associated with nausea and a decline in appetite or it might generally curb appetite. These agents likely will cause less future weight gain. SNRIs typically induce more noradrenergic tone at increasingly higher dosages. There may be a dose-response curve in this manner. Levomilnacipran likely is the most noradrenergic of the SNRIs; recent regulatory studies suggest no statistically significant weight gain over the long term.7
Sedating antidepressants
Mirtazapine has receptor-blocking effects on noradrenergic α-2 and serotonergic 5-HT2a and 5-HT2c receptors. Additionally, histamine blocking of H1 receptors can contribute to additional weight gain, similar to what is seen with some SGAs. H1 antagonism dampens satiety response, resulting in increased caloric intake. In that case, or when specific SGAs are used for managing depression, appetite increases (H1 antagonism) and metabolism slows (possibly 5-HT2c antagonism, muscarinic receptor antagonism, etc.), thus allowing for greater adipose tissue growth and leptin insensitivity.
In a meta-analysis, mean weight increased by 1.74 kg (P < .0001) in the first 4 to 12 weeks of mirtazapine treatment, with greater variability in periods >4 months.6 Among the more novel antidepressants released since the era of tricyclic antidepressants (TCAs) or monoamine oxidase inhibitor, mirtazapine might have the greatest weight gain potential.
Trazodone and nefazodone block 5-HT2a and 5-HT2c receptors, as well as serotonin reuptake transporters. Compared with trazodone, nefazodone has a more potent effect on 5-HT2a receptor antagonism and a less potent effect on 5-HT2c receptors, and also mildly inhibits uptake of norepinephrine—meaning that this drug might have less weight gain potential. These medications are not used frequently for treating depression, but trazodone is used as an adjunctive agent for insomnia. Used even at off-label low dosages, trazodone exerts H1-histaminic and α-1 adrenergic antagonistic properties, decreasing the level of consciousness and allowing sedation and somnolence. Because of its fast onset and relatively short duration of action, it can improve depression symptoms by promoting restful sleep as well as by facilitating monoamine neurotransmission. It also might add to weight gain because of its pharmacodynamic receptor profile.
Tricyclic antidepressants
Amitriptyline can be associated with release of tumor necrosis factor-alpha, which is implicated in causing weight gain. Many TCAs block H1 (amitriptyline, imipramine, clomipramine), likely causing weight gain. Most TCAs antagonize muscarinic receptors as well. The more noradrenergic TCAs could curb appetite (nortriptyline, desipramine, protriptyline) similar to SNRIs, therefore countering some of the weight gain drive.
As an example, in a meta-analysis examining weight gain with antidepressants, amitriptyline was associated with weight gain of 1.52 kg above baseline in the acute period (4 to 12 weeks) and 2.24 kg above baseline at 4 to 7 months.6 These results of the acute phase should be viewed cautiously because the authors reported high heterogeneity among these studies, and the possibility of publication bias (Egger test, P < .0001). In the same meta-analysis, the even the more noradrenergic nortriptyline was associated with an increase of 2.0 kg on average over baseline during acute treatment, with that number dropping to 1.24 kg over baseline at ≥4 months.6
Newer antidepressants
Vilazodone is a weak SSRI that aggressively partially agonizes pre-synaptic and post-synaptic 5-HT1a receptors in the CNS. This dual site 5-HT1a action is somewhat unique among antidepressants. This type of agent sometimes is called multimodal,8 or could be considered an “SSRI +” antidepressant. These drugs are SSRIs at the core but have additional 5-HT receptor modulating capabilities. Vilazodone has a favorable weight gain profile, as suggested in a 52-week trial reporting 1.7 kg gain over 52 weeks, compared with an average of 6.8 to 10 kg for long-term SSRI therapy.9
Vortioxetine is a stronger SSRI that also partially agonizes presynaptic 5-HT1a receptors. In addition, it antagonizes 5-HT1d, 5-HT3, and 5-HT7 receptors, giving it a unique pharmacodynamic profile.10 Vortioxetine also had minimal impact on drug-induced weight gain in 52-week studies, with data from 2 trials indicating either minimal weight gain in 6.1% of patients (mean increase of 0.41 kg over 52 weeks)11 or gain that was not statistically significant.12
Levomilnacipran is unique in that it has the most aggressive norepinephrine reuptake inhibition of all SNRIs.13 Again, increased noradrenergic tone might curb appetite and caloric intake. Many SNRIs cause low-grade nausea, which could account for decreased appetite. Long-term, 52-week data for this drug also shows minimal proclivity for weight gain, with the trial participants reporting a slight decrease of 4.34 kg on average from baseline.14
Continue to: Addressing weight gain
Addressing weight gain
Lifestyle modification. Eating smaller portions, combined with restricting foods high in calories and fat, should be the first step. A simple suggestion to a patient to eat the same foods, but remove 20% of the portion, is a simple intervention akin to that of suggesting sleep hygiene practices for insomnia management. Under medical supervision or with referral to a dietician or nutritionist, more rigid caloric restrictions could be employed.
Commercial weight-loss programs, such as Weight Watchers or Curves, can be helpful; some insurers will only cover medications for weight loss if one of these programs have been tried or is used in combination with medication. Some patients might ask about extreme weight-loss measures, such as low-calorie diets combined with intense exercise programs that have been popularized in the media. Although the motivation to initiate and maintain meaningful weight loss should be encouraged, doing so in a more gradual manner should be the goal.
Addressing portion size is a good approach in the early stages of managing obesity. Restaurants often serve portions that have more calories than should be consumed in one meal. Visual cues can influence this trend; using smaller plates can help reduce caloric intake.15
Exercise, sustained for at least 45 minutes, can have long-lasting effects, with a small study showing an increase in metabolic rate of 190 ± 71.4 kcal (P < .001) above baseline for 14 hours after exercise.16 Endurance exercise training is associated with a significant decrease in total cholesterol, triglycerides, and low-density lipoprotein cholesterol, as well as an increase in the high-density lipoprotein level over a 24-week period.17
Encouraging an exercise regimen that is appropriate for your patient can help maintain weight loss. In small trials,18,19 high-intensity exercise was shown to help suppress appetite and decrease 24-hour caloric consumption by 6% to 11%.18
Psychotherapy can become an important intervention for initiating and maintaining weight loss. CBT can help patients recognize and modify lifestyle components, and reinforce behaviors that promote weight loss. This can come from setting realistic weight loss goals; preventing triggering factors that lead to overeating; encouraging portion control during meals; and promoting exercise habits.
In a small, randomized controlled trial (RCT) examining weight loss in obese women, those who underwent CBT and psychoeducation for 2 hours a week for 10 weeks in addition to dietary changes and exercise showed an average weight loss of 10.4 kg at 18-month follow-up, compared with weight gain of 2.3 kg in the control group.20 The short duration of treatment in this study might be desirable to reduce cost and utilization of services. Group formats also could be employed.
Motivational interviewing is a useful tool in addiction psychiatry and shows promise for treating obesity and overeating as well. The approach may differ slightly because weight-loss therapy involves behavior modification rather than behavior cessation. In a meta-analysis of data from RCTs exploring motivational interviewing and its use as an intervention for weight loss, those in the intervention groups experienced significant weight loss as indicated by BMI decreasing a standardized mean difference of −0.51, compared with control groups.21
Medical management considerations
Diagnostics. Recognition and early intervention are instrumental in successfully treating medication-associated weight gain. It is important to obtain any family history of obesity, diabetes, hypertension, and hyperlipidemia. This will likely indicate a patient’s risk for weight gain before initiating medication.
Obtain vital signs at every visit, including blood pressure. Monitoring weight at every clinical visit can be used to calculate and monitor BMI, while also asking the patient to maintain a log of weight measurements obtained at home. Measuring abdominal girth is important to watch for metabolic syndrome, although often this is the least measured variable.
Laboratory testing is helpful. Obtaining a baseline lipid panel and a fasting glucose level (consider measuring hemoglobin A1c in patients with diabetes) is warranted. Including thyroid markers, such as thyroid-stimulating hormone and thyroxine (free T4), might be important considerations, because inadequate management of hypothyroidism can complicate the clinical picture.
Follow-up testing should be ordered every 3 to 12 months to monitor progress if your patient is showing signs of rapid weight gain, or if BMI nears ≥30 kg/m². These guidelines generally are assigned for prescribing of SGAs, but can be applied when using any psychotropic with weight gain potential.
Medications to considerWhen considering the medication regimen as an intervention point, consider changing the antidepressant to one that is not associated with significant weight gain. Although not specifically indicated as a monotherapy for weight loss, switching to or augmenting therapy with bupropion could aid weight loss through appetite suppression.20 Some newer antidepressants, such as vilazodone, vortioxetine, and levomilnacipran, might have less propensity to cause weight gain. In patients with severe depression, augmenting with medications containing amphetamine or methylphenidate could cause some weight loss, but greater care should be taken because of cardiovascular effects and dependency issues.
Continue to: Discussing with your patient...
Discussing with your patient the possibility of changing or worsening depressive symptoms when adding or switching medications allows them to be aware and engaged in the process and can encourage them to notice and report changes. Developing a sensible schedule to taper an existing medication slowly over several weeks and allowing a new one to build up gradually to a therapeutic level can help minimize adverse effects or a discontinuation syndrome.
If switching antidepressants is not possible, or is ineffective, an anti-obesity medication (Table 122-29) can be considered. These medications should not be considered first-line in weight loss management, but reserved for more difficult or refractory weight loss challenges and in patients who are not able to participate in weight loss or dieting programs because of cognitive disorders, a history of nonadherance, financial or travel limitations, or in those with poor social support systems such as homelessness.
Some of these medications are not reimbursed by insurance companies; therefore, consider the financial burden to the patient and their capacity for adherence to therapy, and discuss this challenge before initiating treatment. There is some evidence for using medications off-label to treat obesity (Table 2,30-34).
Anti-obesity medications typically are considered for patients with a BMI >30 or in any overweight patient with diabetes, hyperlipidemia, or cardiovascular disease. As always, discuss with patients and their primary care provider the potential benefits and risks of adding any of these or other medications to an existing treatment regimen.
If weight loss goals are not met, consider discontinuing anti-obesity therapy. Patients and physicians should be cognizant of the need to continue long-term maintenance on these medications after successful treatment—perhaps indefinitely, because patients frequently regain weight after medication is discontinued.
Bottom Line
Many antidepressants are known to increase the risk of excessive weight gain, although risk of weigh gain varies among antidepressant classes. First, advise changes in diet and exercise; next, initiate psychotherapy as indicated, and then consider referral to a nutritionist. Consider switching to an antidepressant with less potential for causing weight gain or adding bupropion, which could lead to weight loss, if your patient can tolerate it. If these strategies are unsuccessful, consider an anti-obesity medication.
Related Resource
The prevalence of undesired weight gain in the United States has reached an all-time high, with 68.5% of adults identified as overweight (body mass index [BMI] ≥25) or obese (BMI ≥30), 34.5% considered obese, and 6.4% considered extremely obese (BMI ≥40).1 Reasons for weight gain include various physical and nutritional factors in a patient’s life, but sometimes weight gain is iatrogenic. Many medications we prescribe are associated with weight gain, including most antidepressants and atypical antipsychotics. Clinicians might minimize or overlook the risk of weight gain when prescribing antidepressants.
Patients with major depression often have associated weight loss. Regaining weight can be seen as sign of successful treatment of depressive symptoms. If weight gain after treatment exceeds the amount of weight loss attributed to depression, however, medication could have caused the excessive gain. This is considered a side effect, or iatrogenic weight gain, and should not be considered normal or clinically acceptable.
Patients who are overweight or obese when beginning antidepressant treatment might be at greater medical risk when placed on a medication that can cause additional weight gain. The time to onset of weight gain during treatment can predict weight gain patterns; those affected in the first month are most at risk of future excessive weight gain.2
In this article, we discuss:
- considerations when prescribing antidepressants
- ways to approach weight gain
- medications available to assist in weight loss.
Our general recommendations
Screen. The United States Preventive Services Task Force maintains a Class-B recommendation for screening all patients for obesity. This means that the Task Force’s review panel determined that such screening is at least moderately or substantially beneficial.3 Screening is important in a setting of potential weight gain in patients taking an antidepressant.
Educate and treat. Provide at least some education and encouragement about eating a healthy diet and exercising, or refer the patient to a nutritionist or dietician. Next, initiate psychotherapy (motivational interviewing, cognitive-behavioral therapy [CBT]) as needed. Reserve anti-obesity medications for those who do not respond to weight loss efforts or who might be taking an antidepressant for the long term.
The need for medical management of weight gain has given rise to specialists who treat this complicated, multifactorial condition. Whether psychiatrists should be seen as a substitution for their specialty is not the purpose of this review; rather, how we might more effectively (1) work on our patients’ behalf to mitigate potential weight gain from the treatments that we prescribe and (2) participate in consultations that we’ve provided on their behalf.
BMI is not an absolute marker of healthBMI likely should not be viewed as a marker with absolute prognostic certainty of overall health of an overweight or obese person: An overweight person considered healthy from a cardiovascular and metabolic perspective could still benefit from preventing further weight gain.
Tomiyama et al4 concluded that BMI itself was insufficient to stratify health in a meaningful way—and that such a focus would lead to overweight and obese people in otherwise good health being penalized unfairly through higher health insurance premiums, and would divert focus on those with less optimal health but a normal BMI. The researchers’ goal was to use blood pressure, lipid levels, and glycemic markers as surrogate markers of health, and then statistically compare results with patients’ corresponding BMI. Their findings showed that approximately one-half of people who are overweight and 29% of obese people can be considered healthy.4
Potential causes of weight gainThere may be more than one reason for weight gain during depression treatment, so a multifactorial management approach might be necessary, depending on the patient’s medication regimen. Appetite might be influenced by physical (chemical, metabolic) and psychological (cultural, familial) factors. The following sections focus on specific antidepressant classes and their proclivity for weight gain.
Serotonergic antidepressantsMany patients with depression are treated with medications that alter serotonin levels in the body, such as selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs). This neurotransmitter often is affected through depression treatment, and therefore might be a factor contributing to unintended weight gain. In mice bred to lack serotonin 5-HT2c receptors in proopiomelanocortin (POMC) neurons, the expected anorectic reaction to serotonergic agents often is reversed, causing a robust increase in hyperphagia and obesity.5 This effect indicates that 5-HT2c receptor stimulation might control appetite and feeding.
After SSRI or SNRI treatment, accumulation of serotonin over time in the synaptic cleft is thought to result in down-regulation of 5-HT2c receptors. This may cause a relative absence of 5-HT2c receptors, similar to what is seen in mice who lack them biologically. The loss of these receptors or their activity often will result in excessive weight gain. Some sedating antidepressants (mirtazapine) and some second-generation antipsychotics (SGAs) (olanzapine, quetiapine) directly block 5-HT2c receptors and might cause more rapid weight gain. Lorcaserin, a selective 5-HT2c receptor agonist, theoretically could reverse this proposed weight gain mechanism and suppress appetite by activating the POMC pathway in the hypothalamus.
Continue to: Among SSRIs and SNRIs
Among SSRIs and SNRIs, paroxetine might be one of the worst for provoking long-term weight gain; a study showed an average increase of 2.73 kg over a 4-month period.6
Theoretically, SNRIs have the ability to increase noradrenergic tone. This might be associated with nausea and a decline in appetite or it might generally curb appetite. These agents likely will cause less future weight gain. SNRIs typically induce more noradrenergic tone at increasingly higher dosages. There may be a dose-response curve in this manner. Levomilnacipran likely is the most noradrenergic of the SNRIs; recent regulatory studies suggest no statistically significant weight gain over the long term.7
Sedating antidepressants
Mirtazapine has receptor-blocking effects on noradrenergic α-2 and serotonergic 5-HT2a and 5-HT2c receptors. Additionally, histamine blocking of H1 receptors can contribute to additional weight gain, similar to what is seen with some SGAs. H1 antagonism dampens satiety response, resulting in increased caloric intake. In that case, or when specific SGAs are used for managing depression, appetite increases (H1 antagonism) and metabolism slows (possibly 5-HT2c antagonism, muscarinic receptor antagonism, etc.), thus allowing for greater adipose tissue growth and leptin insensitivity.
In a meta-analysis, mean weight increased by 1.74 kg (P < .0001) in the first 4 to 12 weeks of mirtazapine treatment, with greater variability in periods >4 months.6 Among the more novel antidepressants released since the era of tricyclic antidepressants (TCAs) or monoamine oxidase inhibitor, mirtazapine might have the greatest weight gain potential.
Trazodone and nefazodone block 5-HT2a and 5-HT2c receptors, as well as serotonin reuptake transporters. Compared with trazodone, nefazodone has a more potent effect on 5-HT2a receptor antagonism and a less potent effect on 5-HT2c receptors, and also mildly inhibits uptake of norepinephrine—meaning that this drug might have less weight gain potential. These medications are not used frequently for treating depression, but trazodone is used as an adjunctive agent for insomnia. Used even at off-label low dosages, trazodone exerts H1-histaminic and α-1 adrenergic antagonistic properties, decreasing the level of consciousness and allowing sedation and somnolence. Because of its fast onset and relatively short duration of action, it can improve depression symptoms by promoting restful sleep as well as by facilitating monoamine neurotransmission. It also might add to weight gain because of its pharmacodynamic receptor profile.
Tricyclic antidepressants
Amitriptyline can be associated with release of tumor necrosis factor-alpha, which is implicated in causing weight gain. Many TCAs block H1 (amitriptyline, imipramine, clomipramine), likely causing weight gain. Most TCAs antagonize muscarinic receptors as well. The more noradrenergic TCAs could curb appetite (nortriptyline, desipramine, protriptyline) similar to SNRIs, therefore countering some of the weight gain drive.
As an example, in a meta-analysis examining weight gain with antidepressants, amitriptyline was associated with weight gain of 1.52 kg above baseline in the acute period (4 to 12 weeks) and 2.24 kg above baseline at 4 to 7 months.6 These results of the acute phase should be viewed cautiously because the authors reported high heterogeneity among these studies, and the possibility of publication bias (Egger test, P < .0001). In the same meta-analysis, the even the more noradrenergic nortriptyline was associated with an increase of 2.0 kg on average over baseline during acute treatment, with that number dropping to 1.24 kg over baseline at ≥4 months.6
Newer antidepressants
Vilazodone is a weak SSRI that aggressively partially agonizes pre-synaptic and post-synaptic 5-HT1a receptors in the CNS. This dual site 5-HT1a action is somewhat unique among antidepressants. This type of agent sometimes is called multimodal,8 or could be considered an “SSRI +” antidepressant. These drugs are SSRIs at the core but have additional 5-HT receptor modulating capabilities. Vilazodone has a favorable weight gain profile, as suggested in a 52-week trial reporting 1.7 kg gain over 52 weeks, compared with an average of 6.8 to 10 kg for long-term SSRI therapy.9
Vortioxetine is a stronger SSRI that also partially agonizes presynaptic 5-HT1a receptors. In addition, it antagonizes 5-HT1d, 5-HT3, and 5-HT7 receptors, giving it a unique pharmacodynamic profile.10 Vortioxetine also had minimal impact on drug-induced weight gain in 52-week studies, with data from 2 trials indicating either minimal weight gain in 6.1% of patients (mean increase of 0.41 kg over 52 weeks)11 or gain that was not statistically significant.12
Levomilnacipran is unique in that it has the most aggressive norepinephrine reuptake inhibition of all SNRIs.13 Again, increased noradrenergic tone might curb appetite and caloric intake. Many SNRIs cause low-grade nausea, which could account for decreased appetite. Long-term, 52-week data for this drug also shows minimal proclivity for weight gain, with the trial participants reporting a slight decrease of 4.34 kg on average from baseline.14
Continue to: Addressing weight gain
Addressing weight gain
Lifestyle modification. Eating smaller portions, combined with restricting foods high in calories and fat, should be the first step. A simple suggestion to a patient to eat the same foods, but remove 20% of the portion, is a simple intervention akin to that of suggesting sleep hygiene practices for insomnia management. Under medical supervision or with referral to a dietician or nutritionist, more rigid caloric restrictions could be employed.
Commercial weight-loss programs, such as Weight Watchers or Curves, can be helpful; some insurers will only cover medications for weight loss if one of these programs have been tried or is used in combination with medication. Some patients might ask about extreme weight-loss measures, such as low-calorie diets combined with intense exercise programs that have been popularized in the media. Although the motivation to initiate and maintain meaningful weight loss should be encouraged, doing so in a more gradual manner should be the goal.
Addressing portion size is a good approach in the early stages of managing obesity. Restaurants often serve portions that have more calories than should be consumed in one meal. Visual cues can influence this trend; using smaller plates can help reduce caloric intake.15
Exercise, sustained for at least 45 minutes, can have long-lasting effects, with a small study showing an increase in metabolic rate of 190 ± 71.4 kcal (P < .001) above baseline for 14 hours after exercise.16 Endurance exercise training is associated with a significant decrease in total cholesterol, triglycerides, and low-density lipoprotein cholesterol, as well as an increase in the high-density lipoprotein level over a 24-week period.17
Encouraging an exercise regimen that is appropriate for your patient can help maintain weight loss. In small trials,18,19 high-intensity exercise was shown to help suppress appetite and decrease 24-hour caloric consumption by 6% to 11%.18
Psychotherapy can become an important intervention for initiating and maintaining weight loss. CBT can help patients recognize and modify lifestyle components, and reinforce behaviors that promote weight loss. This can come from setting realistic weight loss goals; preventing triggering factors that lead to overeating; encouraging portion control during meals; and promoting exercise habits.
In a small, randomized controlled trial (RCT) examining weight loss in obese women, those who underwent CBT and psychoeducation for 2 hours a week for 10 weeks in addition to dietary changes and exercise showed an average weight loss of 10.4 kg at 18-month follow-up, compared with weight gain of 2.3 kg in the control group.20 The short duration of treatment in this study might be desirable to reduce cost and utilization of services. Group formats also could be employed.
Motivational interviewing is a useful tool in addiction psychiatry and shows promise for treating obesity and overeating as well. The approach may differ slightly because weight-loss therapy involves behavior modification rather than behavior cessation. In a meta-analysis of data from RCTs exploring motivational interviewing and its use as an intervention for weight loss, those in the intervention groups experienced significant weight loss as indicated by BMI decreasing a standardized mean difference of −0.51, compared with control groups.21
Medical management considerations
Diagnostics. Recognition and early intervention are instrumental in successfully treating medication-associated weight gain. It is important to obtain any family history of obesity, diabetes, hypertension, and hyperlipidemia. This will likely indicate a patient’s risk for weight gain before initiating medication.
Obtain vital signs at every visit, including blood pressure. Monitoring weight at every clinical visit can be used to calculate and monitor BMI, while also asking the patient to maintain a log of weight measurements obtained at home. Measuring abdominal girth is important to watch for metabolic syndrome, although often this is the least measured variable.
Laboratory testing is helpful. Obtaining a baseline lipid panel and a fasting glucose level (consider measuring hemoglobin A1c in patients with diabetes) is warranted. Including thyroid markers, such as thyroid-stimulating hormone and thyroxine (free T4), might be important considerations, because inadequate management of hypothyroidism can complicate the clinical picture.
Follow-up testing should be ordered every 3 to 12 months to monitor progress if your patient is showing signs of rapid weight gain, or if BMI nears ≥30 kg/m². These guidelines generally are assigned for prescribing of SGAs, but can be applied when using any psychotropic with weight gain potential.
Medications to considerWhen considering the medication regimen as an intervention point, consider changing the antidepressant to one that is not associated with significant weight gain. Although not specifically indicated as a monotherapy for weight loss, switching to or augmenting therapy with bupropion could aid weight loss through appetite suppression.20 Some newer antidepressants, such as vilazodone, vortioxetine, and levomilnacipran, might have less propensity to cause weight gain. In patients with severe depression, augmenting with medications containing amphetamine or methylphenidate could cause some weight loss, but greater care should be taken because of cardiovascular effects and dependency issues.
Continue to: Discussing with your patient...
Discussing with your patient the possibility of changing or worsening depressive symptoms when adding or switching medications allows them to be aware and engaged in the process and can encourage them to notice and report changes. Developing a sensible schedule to taper an existing medication slowly over several weeks and allowing a new one to build up gradually to a therapeutic level can help minimize adverse effects or a discontinuation syndrome.
If switching antidepressants is not possible, or is ineffective, an anti-obesity medication (Table 122-29) can be considered. These medications should not be considered first-line in weight loss management, but reserved for more difficult or refractory weight loss challenges and in patients who are not able to participate in weight loss or dieting programs because of cognitive disorders, a history of nonadherance, financial or travel limitations, or in those with poor social support systems such as homelessness.
Some of these medications are not reimbursed by insurance companies; therefore, consider the financial burden to the patient and their capacity for adherence to therapy, and discuss this challenge before initiating treatment. There is some evidence for using medications off-label to treat obesity (Table 2,30-34).
Anti-obesity medications typically are considered for patients with a BMI >30 or in any overweight patient with diabetes, hyperlipidemia, or cardiovascular disease. As always, discuss with patients and their primary care provider the potential benefits and risks of adding any of these or other medications to an existing treatment regimen.
If weight loss goals are not met, consider discontinuing anti-obesity therapy. Patients and physicians should be cognizant of the need to continue long-term maintenance on these medications after successful treatment—perhaps indefinitely, because patients frequently regain weight after medication is discontinued.
Bottom Line
Many antidepressants are known to increase the risk of excessive weight gain, although risk of weigh gain varies among antidepressant classes. First, advise changes in diet and exercise; next, initiate psychotherapy as indicated, and then consider referral to a nutritionist. Consider switching to an antidepressant with less potential for causing weight gain or adding bupropion, which could lead to weight loss, if your patient can tolerate it. If these strategies are unsuccessful, consider an anti-obesity medication.
Related Resource
1. Ogden CL, Carroll MD, Kit BK, et al. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA. 2014;311(8):806-814.
2. Vandenberghe F, Gholam-Rezaee M, Saigí-Morgui N, et al. Importance of early weight changes to predict long-term weight gain during psychotropic drug treatment. J Clin Psychiatry. 2015;76(11):e1417-e1423.
3. Grade Definitions. Electronic Preventive Services Selector (ePSS). http://epss.ahrq.gov/ePSS/gradedef.jsp. Accessed May 2, 2016.
4. Tomiyama AJ, Hunger JM, Nguyen-Cuu J, et al. Misclassification of cardiometabolic health when using body mass index categories in NHANES 2005-2012 [published online February 4, 2016]. Int J Obes (Lond). doi: 10.1038/ijo.2016.17.
5. Berglund ED, Liu C, Sohn J, et al. Serotonin 2C receptors in pro-opiomelanocortin neurons regulate energy and glucose homeostasis. J Clin Invest. 2013;123(12):5061-5070.
6. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-anaylsis. J Clin Psychiatry. 2010;71(10):1259-1272.
7. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
8. Schwartz TL, Siddiqui UA, Stahl SM. Vilazodone: a brief pharmacological and clinical review of the novel serotonin partial agonist and reuptake inhibitor. Ther Adv Psychopharmacol. 2011;1(3):81-87.
9. Robinson DS, Kajdasz DK, Gallipoli S, et al. A 1-year, open-label study assessing the safety and tolerability of vilazodone in patients with major depressive disorder. J Clin Psychopharmacol. 2011;31(5):643-646.
10. Stahl SM. Modes and nodes explain the mechanism of action of vortioxetine, a multimodal agent (MMA): actions at serotonin receptors may enhance downstream release of four pro-cognitive neurotransmitters. CNS Spectr. 2015;20(6):515-519.
11. Jacobsen PL, Harper L, Chrones L, et al. Safety and tolerability of vortioxetine (15 and 20 mg) in patients with major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2015;30(5):255-264.
12. Boulenger JP, Loft H, Olsen CK. Efficacy and safety of vortioxetine (Lu AA21004), 15 and 20 mg/day: a randomized, double-blind, placebo-controlled, duloxetine-referenced study in the acute treatment of adult patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(3):138-149.
13. Grady MM, Stahl SM. Novel agents in development for the treatment of depression. CNS Spectr. 2013;18(suppl 1):37-40; quiz 41.
14. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
15. Hollands GJ, Shemilt I, Marteau TM, et al. Portion, package or tableware size for changing selection and consumption of food, alcohol and tobacco. Cochrane Database Syst Rev. 2015;9:CD011045.
16. Knab AM, Shanely RA, Corbin KD, et al. A 45-minute vigorous exercise bout increases metabolic rate for 14 hours. Med Sci Sports Exerc. 2011;43(9):1643-1648.
17. Halverstadt A, Phares DA, Wilund KR, et al. Endurance exercise training raises high-density lipoprotein cholesterol and lowers small low-density lipoprotein and very low-density lipoprotein independent of body fat phenotypes in older men and women. Metabolism. 2007;56(4):444-450.
18. Thivel D, Isacco L, Montaurier C, et al. The 24-h energy intake of obese adolescents is spontaneously reduced after intensive exercise: a randomized controlled trial in calorimetric chambers [published online January 17, 2012]. PLoS One. 2012;7(1):e29840. doi: 10.1371/journal.pone.0029840.
19. Sim AY, Wallman KE, Fairchild TJ, et al. High-intensity intermittent exercise attenuates ad-libitum energy intake. Int J Obes (Lond). 2014;38(3):417-422.
20. Stahre L, Tärnell B, Håkanson CE, et al. A randomized controlled trial of two weight-reducing short-term group treatment programs for obesity with an 18-month follow-up. Int J Behav Med. 2007;14(1):48-55.
21. Armstrong MJ, Mottershead TA, Ronksley PE, et al. Motivational interviewing to improve weight loss in overweight and/or obese patients: a systematic review and meta-analysis of randomized controlled trials. Obes Rev. 2011;12(9):709-723.
22. Apovian CM, Aronne L, Rubino D, et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Springs). 2013;21(5):935-943.
23. Greenway FL, Fujioka K, Plodkowski RA, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605.
24. Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96(10):3067-3077.
25. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial [Erratum in Lancet. 2011;377(9776):1494]. Lancet. 2011;377(9774):1341-1352.
26. Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr. 2012;95(2):297-308.
27. Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373(1):11-22.
28. Leblanc ES, O’Connor E, Whitlock EP, et al. Effectiveness of primary care-relevant treatments for obesity in adults: a systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med. 2011;155(7):434-447.
29. Torgerson JS, Hauptman J, Boldrin MN, et al. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27(1):155-161.
30. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393-403.
31. McDonagh MS, Selph S, Ozpinar A, et al. Systematic review of the benefits and risks of metformin in treating obesity in children aged 18 years and younger. JAMA Pediatr. 2014;168(2):178-184.
32. Dushay J, Gao C, Gopalakrishnan GS, et al. Short-term exenatide treatment leads to significant weight loss in a subset of obese women without diabetes. Diabetes Care. 2012;35(1):4-11.
33. Gadde KM, Kopping MF, Wagner HR 2nd, et al. Zonisamide for weight reduction in obese adults: a 1-year randomized controlled trial. Arch Intern Med. 2012;172(20):1557-1564.
34. Singh-Franco D, Perez A, Harrington C. The effect of pramlintide acetate on glycemic control and weight in patients with type 2 diabetes mellitus and in obese patients without diabetes: a systematic review and meta-analysis. Diabetes Obes Metab. 2011;13(2):169-180.
1. Ogden CL, Carroll MD, Kit BK, et al. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA. 2014;311(8):806-814.
2. Vandenberghe F, Gholam-Rezaee M, Saigí-Morgui N, et al. Importance of early weight changes to predict long-term weight gain during psychotropic drug treatment. J Clin Psychiatry. 2015;76(11):e1417-e1423.
3. Grade Definitions. Electronic Preventive Services Selector (ePSS). http://epss.ahrq.gov/ePSS/gradedef.jsp. Accessed May 2, 2016.
4. Tomiyama AJ, Hunger JM, Nguyen-Cuu J, et al. Misclassification of cardiometabolic health when using body mass index categories in NHANES 2005-2012 [published online February 4, 2016]. Int J Obes (Lond). doi: 10.1038/ijo.2016.17.
5. Berglund ED, Liu C, Sohn J, et al. Serotonin 2C receptors in pro-opiomelanocortin neurons regulate energy and glucose homeostasis. J Clin Invest. 2013;123(12):5061-5070.
6. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-anaylsis. J Clin Psychiatry. 2010;71(10):1259-1272.
7. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
8. Schwartz TL, Siddiqui UA, Stahl SM. Vilazodone: a brief pharmacological and clinical review of the novel serotonin partial agonist and reuptake inhibitor. Ther Adv Psychopharmacol. 2011;1(3):81-87.
9. Robinson DS, Kajdasz DK, Gallipoli S, et al. A 1-year, open-label study assessing the safety and tolerability of vilazodone in patients with major depressive disorder. J Clin Psychopharmacol. 2011;31(5):643-646.
10. Stahl SM. Modes and nodes explain the mechanism of action of vortioxetine, a multimodal agent (MMA): actions at serotonin receptors may enhance downstream release of four pro-cognitive neurotransmitters. CNS Spectr. 2015;20(6):515-519.
11. Jacobsen PL, Harper L, Chrones L, et al. Safety and tolerability of vortioxetine (15 and 20 mg) in patients with major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2015;30(5):255-264.
12. Boulenger JP, Loft H, Olsen CK. Efficacy and safety of vortioxetine (Lu AA21004), 15 and 20 mg/day: a randomized, double-blind, placebo-controlled, duloxetine-referenced study in the acute treatment of adult patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(3):138-149.
13. Grady MM, Stahl SM. Novel agents in development for the treatment of depression. CNS Spectr. 2013;18(suppl 1):37-40; quiz 41.
14. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
15. Hollands GJ, Shemilt I, Marteau TM, et al. Portion, package or tableware size for changing selection and consumption of food, alcohol and tobacco. Cochrane Database Syst Rev. 2015;9:CD011045.
16. Knab AM, Shanely RA, Corbin KD, et al. A 45-minute vigorous exercise bout increases metabolic rate for 14 hours. Med Sci Sports Exerc. 2011;43(9):1643-1648.
17. Halverstadt A, Phares DA, Wilund KR, et al. Endurance exercise training raises high-density lipoprotein cholesterol and lowers small low-density lipoprotein and very low-density lipoprotein independent of body fat phenotypes in older men and women. Metabolism. 2007;56(4):444-450.
18. Thivel D, Isacco L, Montaurier C, et al. The 24-h energy intake of obese adolescents is spontaneously reduced after intensive exercise: a randomized controlled trial in calorimetric chambers [published online January 17, 2012]. PLoS One. 2012;7(1):e29840. doi: 10.1371/journal.pone.0029840.
19. Sim AY, Wallman KE, Fairchild TJ, et al. High-intensity intermittent exercise attenuates ad-libitum energy intake. Int J Obes (Lond). 2014;38(3):417-422.
20. Stahre L, Tärnell B, Håkanson CE, et al. A randomized controlled trial of two weight-reducing short-term group treatment programs for obesity with an 18-month follow-up. Int J Behav Med. 2007;14(1):48-55.
21. Armstrong MJ, Mottershead TA, Ronksley PE, et al. Motivational interviewing to improve weight loss in overweight and/or obese patients: a systematic review and meta-analysis of randomized controlled trials. Obes Rev. 2011;12(9):709-723.
22. Apovian CM, Aronne L, Rubino D, et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Springs). 2013;21(5):935-943.
23. Greenway FL, Fujioka K, Plodkowski RA, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605.
24. Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96(10):3067-3077.
25. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial [Erratum in Lancet. 2011;377(9776):1494]. Lancet. 2011;377(9774):1341-1352.
26. Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr. 2012;95(2):297-308.
27. Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373(1):11-22.
28. Leblanc ES, O’Connor E, Whitlock EP, et al. Effectiveness of primary care-relevant treatments for obesity in adults: a systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med. 2011;155(7):434-447.
29. Torgerson JS, Hauptman J, Boldrin MN, et al. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27(1):155-161.
30. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393-403.
31. McDonagh MS, Selph S, Ozpinar A, et al. Systematic review of the benefits and risks of metformin in treating obesity in children aged 18 years and younger. JAMA Pediatr. 2014;168(2):178-184.
32. Dushay J, Gao C, Gopalakrishnan GS, et al. Short-term exenatide treatment leads to significant weight loss in a subset of obese women without diabetes. Diabetes Care. 2012;35(1):4-11.
33. Gadde KM, Kopping MF, Wagner HR 2nd, et al. Zonisamide for weight reduction in obese adults: a 1-year randomized controlled trial. Arch Intern Med. 2012;172(20):1557-1564.
34. Singh-Franco D, Perez A, Harrington C. The effect of pramlintide acetate on glycemic control and weight in patients with type 2 diabetes mellitus and in obese patients without diabetes: a systematic review and meta-analysis. Diabetes Obes Metab. 2011;13(2):169-180.
