User login
Androgen Deficiency Syndrome: A Rational Approach to Male Hypogonadism
During a routine physical examination, a 65-year-old man wants to find out if he has “Low T.” He complains of fatigue, decreased libido, and erectile dysfunction (ED) for the past five years. He has a history of type 2 diabetes, hypertension, hyperlipidemia, obstructive sleep apnea, and chronic low back pain. His current medications include metformin, glipizide, lisinopril, atorvastatin, and hydrocodone for back pain. Given these clinical features, the next step will be to find out if he has hypogonadism (androgen deficiency).
The Endocrine Society defines hypogonadism as a clinical syndrome in which the testes produce insufficient testosterone as a consequence of an interruption of the hypothalamic-pituitary-testicular axis. Although prevalence is high in older men, the Endocrine Society does not recommend screening the general population for hypogonadism.1 Rather, screening should be limited to patients with clinical conditions associated with high prevalence of hypogonadism. Of note, approximately 30% of adults with type 2 diabetes have a subnormal testosterone concentration.2
Q: What is pertinent in the history?
The first step in evaluation of hypogonadism is a detailed history. Signs and symptoms such as decreased libido, hot flashes, decreased shaving frequency, breast enlargement/tenderness, and decreased testicular size are highly suggestive of hypogonadism. Other, less specific signs and symptoms include dysthymia, poor concentration, sleep disturbances, fatigue, reduction in muscle strength, and diminished work performance.
If these signs and symptoms are present, the likelihood of hypogonadism is high and further evaluation is needed.1,3 Note any history of alcoholism, liver problems, and testicular trauma or surgery.
A detailed medication history is also important. Some medications, such as opiates, can affect the release of gonadotropins. Among men taking long-term opiates for chronic noncancer pain, the prevalence of hypogonadism is 75%.4 Other drugs, such as spironolactone, can block the androgen effect and lead to hypogonadism.1
Recent reports have suggested an association between testosterone replacement therapy and increased cardiovascular events, making a detailed cardiovascular history essential.5,6 One study found that men ages 75 and older with limited mobility and other comorbidities who used testosterone gel had an increased risk for cardiovascular events.7 Therefore, clinicians need to be cognizant of this risk when considering testosterone therapy for their patients.
On the next page: Physical exam, lab tests, and treatments >>
Q: What does the physical exam reveal?
In hypogonadotropic hy pogonadism, physical examination does not usually provide much information, as compared to congenital hypogonadal syndromes (eg, Klinefelter and Kallmann syndromes). However, small testicular volume and/or gynecomastia would indicate hypogonadism.
Q: What lab tests should be ordered?
Serum total and free testosterone should be measured, preferably by liquid gas chromatography. The sample should be drawn before 10 am to limit the effects of diurnal variation. If the total testosterone is less than
300 ng/dL, a second morning sample should be drawn and tested. Serum prolactin, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), complete blood count, prostate-specific antigen (PSA), comprehensive metabolic panel, and ferritin should also be measured.
There is generally little benefit to testosterone therapy when total testosterone is greater than 350 ng/dL.8 The level of testosterone at which hypogonadal symptoms manifest and testosterone replacement provides improvement is yet to be determined. Buvat et al suggest that men with total testosterone levels less than 230 ng/dL usually benefit from therapy.8 If the total testosterone level is less than 150 ng/dL in the setting of secondary hypogonadism (low to low-normal LH/FSH) or if prolactin is elevated, MRI of the sella is recommended to rule out pituitary adenoma.1
Q: Once the diagnosis is confirmed, what treatment should you recommend?
The goal of therapy for confirmed hypogonadism is to normalize the testosterone level. Testosterone replacement therapy may help to improve libido, fatigue, muscle strength, and bone density. However, in the elderly (particularly those older than 70), these therapeutic benefits have not been proven. Therefore, before initiating therapy, the clinician should discuss in detail the risks versus the benefits of testosterone replacement for a particular patient.
Simple lifestyle modifications, such as weight loss and exercise, have been shown to increase total and free testosterone levels.3,8 For patients with obstructive sleep apnea (OSA), a known risk factor for hypogonadism, compliance with CPAP therapy has been associated with modest improvement in testosterone level. If it is appropriate for the patient to discontinue use of certain medications, such as opiates, he or she may experience an improvement in testosterone level as a result.
If the patient’s testosterone levels remain low after these changes have been implemented, consider testosterone therapy. Testosterone products currently available in the United States include transdermal preparations (gel, patch), intramuscular injection, and subcutaneous pellets.
On the next page: Contraindications, adverse effects, and follow-up >>
Q: What are the contraindications to testosterone therapy?
Testosterone therapy is contraindicated in patients with metastatic prostate cancer and breast cancer. An unevaluated prostate nodule, indurated prostate, PSA greater than 4 ng/mL, elevated hematocrit (>50%), severe lower urinary tract symptoms, poorly controlled congestive heart failure, and untreated severe OSA are associated with moderate to high risk for adverse outcomes; the Endocrine Society has recommended against using testosterone in affected patients.1
Q: What are the adverse effects of testosterone replacement therapy?
Testosterone replacement may worsen symptoms of benign prostatic hyperplasia (ie, urinary urgency, hesitancy, and frequency). Also, testosterone replacement can lead to marked elevation of hemoglobin and hematocrit levels.
Increased cardiovascular events have been associated with androgen replacement, especially in men with prior coronary artery disease. A positive cardiovascular history necessitates discussion with the patient regarding the risks versus the benefits of testosterone replacement therapy.5 In a recent study of obese, hypogonadal men with severe OSA, testosterone therapy was associated with transient worsening of sleep apnea.9
Q: What does monitoring/ follow-up entail?
In patients with long-standing hypogonadism, a lower starting dose of testosterone is recommended, which can be gradually increased. After starting testosterone therapy, patients should be monitored in the first three to six months for total testosterone, PSA, and hematocrit and for improvement of symptoms (ie, fatigue, ED, decreased libido) or worsening of benign prostatic hyperplasia signs/symptoms.
For men ages 40 and older, if the baseline PSA is greater than 0.6 ng/mL, a digital rectal exam (DRE) is recommended prior to initiation of therapy and should be followed in accordance with prostate cancer screening guidelines.1
Patients placed on testosterone cypionate/enanthate IM injections should have their testosterone checked at a midpoint between their injections, with the target testosterone level between 400 and 700 ng/dL.1 For those using gel or transdermal preparations, a morning total testosterone level should be measured.
Urology consultation is recommended if the PSA concentration rises by 1.4 ng/dL within 12 months, if the American Urological Association/International Prostate Symptom Score is greater than 19, or if there is an abnormal DRE.1,8 Treatment with testosterone should be postponed or withheld if the patient’s hematocrit is greater than 54% but may be resumed when it has decreased to normal levels.1
On the next page: References >>
REFERENCES
1. Bhasin S, Cunningham GR, Hayes FJ, et al. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95(6):2536-2559.
2. Dandona P, Dhindsa S. Update: hypogonadotropic hypogonadism in type 2 diabetes and obesity. J Clin Endocrinol Metab. 2011;96(9): 2643-2651.
3. Tajar A, Forti G, O’Neill TW, et al. Characteristics of secondary, primary, and compensated hypogonadism in aging men: evidence from the European Male Ageing Study. J Clin Endocrinol Metab. 2010;95(4):1810-1818.
4. Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes. 2009;117(1):38-43.
5. Vigen R, O’Donnell CI, Baron AE, et al. Association of testosterone therapy with mortality, myocardial infarction, and stroke in men with low testosterone levels. JAMA. 2013;310(17): 1829-1836.
6. Finkle WD, Greenland S, Ridgeway GK, et al. Increased risk of non-fatal myocardial infarction following testosterone therapy prescription in men. PloS One. 2014;9(1): e85805.
7. Basaria S, Coviello AD, Travison TG, et al. Adverse events associated with testosterone administration. N Engl J Med. 2010;363(2):109-122.
8. Buvat J, Maggi M, Guay A, Torres LO. Testosterone deficiency in men: systematic review and standard operating procedures for diagnosis and treatment. J Sex Med. 2013;10(1): 245-284.
9. Hoyos CM, Killick R, Yee BJ, et al. Effects of testosterone therapy on sleep and breathing in obese men with severe obstructive sleep apnoea: a randomized placebo-controlled trial. Clin Endocrinol (Oxf). 2012;77(4):
599-607.
| Clinician Reviews in partnership with |
Sheila B. Pinkson practices at Audie L. Murphy VA Hospital in San Antonio and is an Adjunct Assistant Professor in Endocrinology at the University of Texas Health Science Center at San Antonio (UTHSCSA). Devjit Tripathy is a staff physician at Audie L. Murphy VA Hospital in San Antonio and an Associate Professor in the Endocrinology/Diabetes Division at UTHSCSA.
| Clinician Reviews in partnership with |
Sheila B. Pinkson practices at Audie L. Murphy VA Hospital in San Antonio and is an Adjunct Assistant Professor in Endocrinology at the University of Texas Health Science Center at San Antonio (UTHSCSA). Devjit Tripathy is a staff physician at Audie L. Murphy VA Hospital in San Antonio and an Associate Professor in the Endocrinology/Diabetes Division at UTHSCSA.
| Clinician Reviews in partnership with |
Sheila B. Pinkson practices at Audie L. Murphy VA Hospital in San Antonio and is an Adjunct Assistant Professor in Endocrinology at the University of Texas Health Science Center at San Antonio (UTHSCSA). Devjit Tripathy is a staff physician at Audie L. Murphy VA Hospital in San Antonio and an Associate Professor in the Endocrinology/Diabetes Division at UTHSCSA.
During a routine physical examination, a 65-year-old man wants to find out if he has “Low T.” He complains of fatigue, decreased libido, and erectile dysfunction (ED) for the past five years. He has a history of type 2 diabetes, hypertension, hyperlipidemia, obstructive sleep apnea, and chronic low back pain. His current medications include metformin, glipizide, lisinopril, atorvastatin, and hydrocodone for back pain. Given these clinical features, the next step will be to find out if he has hypogonadism (androgen deficiency).
The Endocrine Society defines hypogonadism as a clinical syndrome in which the testes produce insufficient testosterone as a consequence of an interruption of the hypothalamic-pituitary-testicular axis. Although prevalence is high in older men, the Endocrine Society does not recommend screening the general population for hypogonadism.1 Rather, screening should be limited to patients with clinical conditions associated with high prevalence of hypogonadism. Of note, approximately 30% of adults with type 2 diabetes have a subnormal testosterone concentration.2
Q: What is pertinent in the history?
The first step in evaluation of hypogonadism is a detailed history. Signs and symptoms such as decreased libido, hot flashes, decreased shaving frequency, breast enlargement/tenderness, and decreased testicular size are highly suggestive of hypogonadism. Other, less specific signs and symptoms include dysthymia, poor concentration, sleep disturbances, fatigue, reduction in muscle strength, and diminished work performance.
If these signs and symptoms are present, the likelihood of hypogonadism is high and further evaluation is needed.1,3 Note any history of alcoholism, liver problems, and testicular trauma or surgery.
A detailed medication history is also important. Some medications, such as opiates, can affect the release of gonadotropins. Among men taking long-term opiates for chronic noncancer pain, the prevalence of hypogonadism is 75%.4 Other drugs, such as spironolactone, can block the androgen effect and lead to hypogonadism.1
Recent reports have suggested an association between testosterone replacement therapy and increased cardiovascular events, making a detailed cardiovascular history essential.5,6 One study found that men ages 75 and older with limited mobility and other comorbidities who used testosterone gel had an increased risk for cardiovascular events.7 Therefore, clinicians need to be cognizant of this risk when considering testosterone therapy for their patients.
On the next page: Physical exam, lab tests, and treatments >>
Q: What does the physical exam reveal?
In hypogonadotropic hy pogonadism, physical examination does not usually provide much information, as compared to congenital hypogonadal syndromes (eg, Klinefelter and Kallmann syndromes). However, small testicular volume and/or gynecomastia would indicate hypogonadism.
Q: What lab tests should be ordered?
Serum total and free testosterone should be measured, preferably by liquid gas chromatography. The sample should be drawn before 10 am to limit the effects of diurnal variation. If the total testosterone is less than
300 ng/dL, a second morning sample should be drawn and tested. Serum prolactin, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), complete blood count, prostate-specific antigen (PSA), comprehensive metabolic panel, and ferritin should also be measured.
There is generally little benefit to testosterone therapy when total testosterone is greater than 350 ng/dL.8 The level of testosterone at which hypogonadal symptoms manifest and testosterone replacement provides improvement is yet to be determined. Buvat et al suggest that men with total testosterone levels less than 230 ng/dL usually benefit from therapy.8 If the total testosterone level is less than 150 ng/dL in the setting of secondary hypogonadism (low to low-normal LH/FSH) or if prolactin is elevated, MRI of the sella is recommended to rule out pituitary adenoma.1
Q: Once the diagnosis is confirmed, what treatment should you recommend?
The goal of therapy for confirmed hypogonadism is to normalize the testosterone level. Testosterone replacement therapy may help to improve libido, fatigue, muscle strength, and bone density. However, in the elderly (particularly those older than 70), these therapeutic benefits have not been proven. Therefore, before initiating therapy, the clinician should discuss in detail the risks versus the benefits of testosterone replacement for a particular patient.
Simple lifestyle modifications, such as weight loss and exercise, have been shown to increase total and free testosterone levels.3,8 For patients with obstructive sleep apnea (OSA), a known risk factor for hypogonadism, compliance with CPAP therapy has been associated with modest improvement in testosterone level. If it is appropriate for the patient to discontinue use of certain medications, such as opiates, he or she may experience an improvement in testosterone level as a result.
If the patient’s testosterone levels remain low after these changes have been implemented, consider testosterone therapy. Testosterone products currently available in the United States include transdermal preparations (gel, patch), intramuscular injection, and subcutaneous pellets.
On the next page: Contraindications, adverse effects, and follow-up >>
Q: What are the contraindications to testosterone therapy?
Testosterone therapy is contraindicated in patients with metastatic prostate cancer and breast cancer. An unevaluated prostate nodule, indurated prostate, PSA greater than 4 ng/mL, elevated hematocrit (>50%), severe lower urinary tract symptoms, poorly controlled congestive heart failure, and untreated severe OSA are associated with moderate to high risk for adverse outcomes; the Endocrine Society has recommended against using testosterone in affected patients.1
Q: What are the adverse effects of testosterone replacement therapy?
Testosterone replacement may worsen symptoms of benign prostatic hyperplasia (ie, urinary urgency, hesitancy, and frequency). Also, testosterone replacement can lead to marked elevation of hemoglobin and hematocrit levels.
Increased cardiovascular events have been associated with androgen replacement, especially in men with prior coronary artery disease. A positive cardiovascular history necessitates discussion with the patient regarding the risks versus the benefits of testosterone replacement therapy.5 In a recent study of obese, hypogonadal men with severe OSA, testosterone therapy was associated with transient worsening of sleep apnea.9
Q: What does monitoring/ follow-up entail?
In patients with long-standing hypogonadism, a lower starting dose of testosterone is recommended, which can be gradually increased. After starting testosterone therapy, patients should be monitored in the first three to six months for total testosterone, PSA, and hematocrit and for improvement of symptoms (ie, fatigue, ED, decreased libido) or worsening of benign prostatic hyperplasia signs/symptoms.
For men ages 40 and older, if the baseline PSA is greater than 0.6 ng/mL, a digital rectal exam (DRE) is recommended prior to initiation of therapy and should be followed in accordance with prostate cancer screening guidelines.1
Patients placed on testosterone cypionate/enanthate IM injections should have their testosterone checked at a midpoint between their injections, with the target testosterone level between 400 and 700 ng/dL.1 For those using gel or transdermal preparations, a morning total testosterone level should be measured.
Urology consultation is recommended if the PSA concentration rises by 1.4 ng/dL within 12 months, if the American Urological Association/International Prostate Symptom Score is greater than 19, or if there is an abnormal DRE.1,8 Treatment with testosterone should be postponed or withheld if the patient’s hematocrit is greater than 54% but may be resumed when it has decreased to normal levels.1
On the next page: References >>
REFERENCES
1. Bhasin S, Cunningham GR, Hayes FJ, et al. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95(6):2536-2559.
2. Dandona P, Dhindsa S. Update: hypogonadotropic hypogonadism in type 2 diabetes and obesity. J Clin Endocrinol Metab. 2011;96(9): 2643-2651.
3. Tajar A, Forti G, O’Neill TW, et al. Characteristics of secondary, primary, and compensated hypogonadism in aging men: evidence from the European Male Ageing Study. J Clin Endocrinol Metab. 2010;95(4):1810-1818.
4. Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes. 2009;117(1):38-43.
5. Vigen R, O’Donnell CI, Baron AE, et al. Association of testosterone therapy with mortality, myocardial infarction, and stroke in men with low testosterone levels. JAMA. 2013;310(17): 1829-1836.
6. Finkle WD, Greenland S, Ridgeway GK, et al. Increased risk of non-fatal myocardial infarction following testosterone therapy prescription in men. PloS One. 2014;9(1): e85805.
7. Basaria S, Coviello AD, Travison TG, et al. Adverse events associated with testosterone administration. N Engl J Med. 2010;363(2):109-122.
8. Buvat J, Maggi M, Guay A, Torres LO. Testosterone deficiency in men: systematic review and standard operating procedures for diagnosis and treatment. J Sex Med. 2013;10(1): 245-284.
9. Hoyos CM, Killick R, Yee BJ, et al. Effects of testosterone therapy on sleep and breathing in obese men with severe obstructive sleep apnoea: a randomized placebo-controlled trial. Clin Endocrinol (Oxf). 2012;77(4):
599-607.
During a routine physical examination, a 65-year-old man wants to find out if he has “Low T.” He complains of fatigue, decreased libido, and erectile dysfunction (ED) for the past five years. He has a history of type 2 diabetes, hypertension, hyperlipidemia, obstructive sleep apnea, and chronic low back pain. His current medications include metformin, glipizide, lisinopril, atorvastatin, and hydrocodone for back pain. Given these clinical features, the next step will be to find out if he has hypogonadism (androgen deficiency).
The Endocrine Society defines hypogonadism as a clinical syndrome in which the testes produce insufficient testosterone as a consequence of an interruption of the hypothalamic-pituitary-testicular axis. Although prevalence is high in older men, the Endocrine Society does not recommend screening the general population for hypogonadism.1 Rather, screening should be limited to patients with clinical conditions associated with high prevalence of hypogonadism. Of note, approximately 30% of adults with type 2 diabetes have a subnormal testosterone concentration.2
Q: What is pertinent in the history?
The first step in evaluation of hypogonadism is a detailed history. Signs and symptoms such as decreased libido, hot flashes, decreased shaving frequency, breast enlargement/tenderness, and decreased testicular size are highly suggestive of hypogonadism. Other, less specific signs and symptoms include dysthymia, poor concentration, sleep disturbances, fatigue, reduction in muscle strength, and diminished work performance.
If these signs and symptoms are present, the likelihood of hypogonadism is high and further evaluation is needed.1,3 Note any history of alcoholism, liver problems, and testicular trauma or surgery.
A detailed medication history is also important. Some medications, such as opiates, can affect the release of gonadotropins. Among men taking long-term opiates for chronic noncancer pain, the prevalence of hypogonadism is 75%.4 Other drugs, such as spironolactone, can block the androgen effect and lead to hypogonadism.1
Recent reports have suggested an association between testosterone replacement therapy and increased cardiovascular events, making a detailed cardiovascular history essential.5,6 One study found that men ages 75 and older with limited mobility and other comorbidities who used testosterone gel had an increased risk for cardiovascular events.7 Therefore, clinicians need to be cognizant of this risk when considering testosterone therapy for their patients.
On the next page: Physical exam, lab tests, and treatments >>
Q: What does the physical exam reveal?
In hypogonadotropic hy pogonadism, physical examination does not usually provide much information, as compared to congenital hypogonadal syndromes (eg, Klinefelter and Kallmann syndromes). However, small testicular volume and/or gynecomastia would indicate hypogonadism.
Q: What lab tests should be ordered?
Serum total and free testosterone should be measured, preferably by liquid gas chromatography. The sample should be drawn before 10 am to limit the effects of diurnal variation. If the total testosterone is less than
300 ng/dL, a second morning sample should be drawn and tested. Serum prolactin, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), complete blood count, prostate-specific antigen (PSA), comprehensive metabolic panel, and ferritin should also be measured.
There is generally little benefit to testosterone therapy when total testosterone is greater than 350 ng/dL.8 The level of testosterone at which hypogonadal symptoms manifest and testosterone replacement provides improvement is yet to be determined. Buvat et al suggest that men with total testosterone levels less than 230 ng/dL usually benefit from therapy.8 If the total testosterone level is less than 150 ng/dL in the setting of secondary hypogonadism (low to low-normal LH/FSH) or if prolactin is elevated, MRI of the sella is recommended to rule out pituitary adenoma.1
Q: Once the diagnosis is confirmed, what treatment should you recommend?
The goal of therapy for confirmed hypogonadism is to normalize the testosterone level. Testosterone replacement therapy may help to improve libido, fatigue, muscle strength, and bone density. However, in the elderly (particularly those older than 70), these therapeutic benefits have not been proven. Therefore, before initiating therapy, the clinician should discuss in detail the risks versus the benefits of testosterone replacement for a particular patient.
Simple lifestyle modifications, such as weight loss and exercise, have been shown to increase total and free testosterone levels.3,8 For patients with obstructive sleep apnea (OSA), a known risk factor for hypogonadism, compliance with CPAP therapy has been associated with modest improvement in testosterone level. If it is appropriate for the patient to discontinue use of certain medications, such as opiates, he or she may experience an improvement in testosterone level as a result.
If the patient’s testosterone levels remain low after these changes have been implemented, consider testosterone therapy. Testosterone products currently available in the United States include transdermal preparations (gel, patch), intramuscular injection, and subcutaneous pellets.
On the next page: Contraindications, adverse effects, and follow-up >>
Q: What are the contraindications to testosterone therapy?
Testosterone therapy is contraindicated in patients with metastatic prostate cancer and breast cancer. An unevaluated prostate nodule, indurated prostate, PSA greater than 4 ng/mL, elevated hematocrit (>50%), severe lower urinary tract symptoms, poorly controlled congestive heart failure, and untreated severe OSA are associated with moderate to high risk for adverse outcomes; the Endocrine Society has recommended against using testosterone in affected patients.1
Q: What are the adverse effects of testosterone replacement therapy?
Testosterone replacement may worsen symptoms of benign prostatic hyperplasia (ie, urinary urgency, hesitancy, and frequency). Also, testosterone replacement can lead to marked elevation of hemoglobin and hematocrit levels.
Increased cardiovascular events have been associated with androgen replacement, especially in men with prior coronary artery disease. A positive cardiovascular history necessitates discussion with the patient regarding the risks versus the benefits of testosterone replacement therapy.5 In a recent study of obese, hypogonadal men with severe OSA, testosterone therapy was associated with transient worsening of sleep apnea.9
Q: What does monitoring/ follow-up entail?
In patients with long-standing hypogonadism, a lower starting dose of testosterone is recommended, which can be gradually increased. After starting testosterone therapy, patients should be monitored in the first three to six months for total testosterone, PSA, and hematocrit and for improvement of symptoms (ie, fatigue, ED, decreased libido) or worsening of benign prostatic hyperplasia signs/symptoms.
For men ages 40 and older, if the baseline PSA is greater than 0.6 ng/mL, a digital rectal exam (DRE) is recommended prior to initiation of therapy and should be followed in accordance with prostate cancer screening guidelines.1
Patients placed on testosterone cypionate/enanthate IM injections should have their testosterone checked at a midpoint between their injections, with the target testosterone level between 400 and 700 ng/dL.1 For those using gel or transdermal preparations, a morning total testosterone level should be measured.
Urology consultation is recommended if the PSA concentration rises by 1.4 ng/dL within 12 months, if the American Urological Association/International Prostate Symptom Score is greater than 19, or if there is an abnormal DRE.1,8 Treatment with testosterone should be postponed or withheld if the patient’s hematocrit is greater than 54% but may be resumed when it has decreased to normal levels.1
On the next page: References >>
REFERENCES
1. Bhasin S, Cunningham GR, Hayes FJ, et al. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95(6):2536-2559.
2. Dandona P, Dhindsa S. Update: hypogonadotropic hypogonadism in type 2 diabetes and obesity. J Clin Endocrinol Metab. 2011;96(9): 2643-2651.
3. Tajar A, Forti G, O’Neill TW, et al. Characteristics of secondary, primary, and compensated hypogonadism in aging men: evidence from the European Male Ageing Study. J Clin Endocrinol Metab. 2010;95(4):1810-1818.
4. Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes. 2009;117(1):38-43.
5. Vigen R, O’Donnell CI, Baron AE, et al. Association of testosterone therapy with mortality, myocardial infarction, and stroke in men with low testosterone levels. JAMA. 2013;310(17): 1829-1836.
6. Finkle WD, Greenland S, Ridgeway GK, et al. Increased risk of non-fatal myocardial infarction following testosterone therapy prescription in men. PloS One. 2014;9(1): e85805.
7. Basaria S, Coviello AD, Travison TG, et al. Adverse events associated with testosterone administration. N Engl J Med. 2010;363(2):109-122.
8. Buvat J, Maggi M, Guay A, Torres LO. Testosterone deficiency in men: systematic review and standard operating procedures for diagnosis and treatment. J Sex Med. 2013;10(1): 245-284.
9. Hoyos CM, Killick R, Yee BJ, et al. Effects of testosterone therapy on sleep and breathing in obese men with severe obstructive sleep apnoea: a randomized placebo-controlled trial. Clin Endocrinol (Oxf). 2012;77(4):
599-607.
Diabetic Amyotrophy: A Rare but Striking Neuropathy
A 45-year-old man, RT, with a six-month history of poorly controlled type 2 diabetes presents for evaluation of increased weakness and pain in the left lower extremity. The symptoms developed in the past three weeks. Previously able to ambulate without assistance, he purchased a cane yesterday due to concerns about falling.
RT reports poor adherence to his diabetes medications. His fingerstick blood sugars have ranged from 200 to 380 mg/dL over the past month. His weight has been stable; his BMI is 34. Review of other systems is negative. Vital signs include a blood pressure of 125/82 mm Hg; pulse, 74 beats/min; and respiratory rate, 16 breaths/min.
Physical examination is notable for muscle atrophy and tenderness to compression in the left quadriceps. Straight leg raise does not elicit pain bilaterally. Muscle strength is 4-/5 in the left hip with pain elicited on hip flexion, 4-/5 in the left knee, and 5/5 in the left ankle. Muscle strength is 4+/5 in the right hip, 5/5 in the right knee, and 5/5 in the right ankle. Muscle strength in both upper extremities is 5/5. Patellar deep tendon reflexes (DTRs) and ankle DTRs are absent bilaterally. Biceps and triceps DTRs are each 2+ bilaterally. Gait is slow and unsteady with use of the cane. Cranial nerves I-XII are intact. Sensation to sharp and dull testing is normal in both the upper and lower extremities.
Labwork reveals an A1C of 10.8%. The patient’s thyroid function studies, creatine kinase, and vitamin B12 level are all in normal range. The serum creatinine is 1.2 mg/dL, and eGFR (estimated glomerular filtration rate) is 58 mL/min/1.73 m2. Liver enzymes are normal, and complete blood count and other chemistry panels are unremarkable.
RT is referred to neurology. MRI of the thoracic and lumbar spine shows no mass lesions or disc disease. Electromyography reveals findings consistent with denervation and axonal damage in the proximal muscles in both lower extremities (left > right).
RT is diagnosed with diabetic amyotrophy and begins physical therapy three days a week. He achieves aggressive improvement in blood sugar control, and after three months, his A1C has improved to 7%. Although still using a cane, he reports improved muscle strength in the lower extremities and better gait stability.
Continued on next page >>
PREVALENCE AND TYPES OF DIABETIC PERIPHERAL NEUROPATHY
According to the CDC, 25.8 million children and adults in the United States (8.3% of the population) have diabetes. Approximately 60% to 70% of them have mild to severe neuropathy.1
Distal symmetric neuropathy is the most common form of diabetic peripheral neuropathy, accounting for more than 50% of cases. It is characterized by distal onset, predominately sensory polyneuropathy, and slow proximal progression.2
In contrast, diabetic amyotrophy is very rare, accounting for only 1% of all cases of neuropathy in diabetes. Prevalence is higher in those with type 2 versus type 1 diabetes (1.1% and 0.3%, respectively).3,4 The most commonly misdiagnosed of the asymmetric diabetic neuropathies, diabetic amyotrophy is characterized by acute, progressive, asymmetrical weakness and pain in the muscles of the proximal lower extremities.5 It is also been referred to as proximal diabetic neuropathy, ischemic mononeuropathy multiplex, diabetic femoral neuropathy, Bruns-Garland syndrome, and diabetic lumbosacral polyradiculopathy.5
LOCALIZATION AND PATHOGENESIS
The site of the lesion in diabetic amyotrophy remains controversial; it is theorized that diabetic amyotrophy may result from involvement of multiple sites, such as lumbosacral anterior horn cells, motor roots, plexus, or motor axons to the muscles of the proximal lower limbs.4
The pathogenesis remains unknown. One theory is that hyperglycemia may cause metabolic derangements in nerve conduction. Another is that there is ischemic damage followed by axonal degeneration. Immune-mediated inflammatory processes, such as microvasculitis, have also been proposed as causes.4,6
CLINICAL FEATURES
Diabetic amyotrophy is characterized by relatively rapid, progressive asymmetrical weakness and pain in the muscles in the proximal lower extremities; it develops over weeks to months and may continue for more than one year.2,6 It typically begins unilaterally and can progress bilaterally—normally without impairment in sensation. Patients commonly experience pain in the hip, buttock, or thigh, as well as difficulty walking, standing, or climbing stairs. Occasionally, the condition is painless and can be associated with weight loss. It causes significant acute disability, with the degree of recovery variable.2,4
Diabetic amyotrophy often presents either at diagnosis of diabetes or shortly thereafter. It most commonly affects men ages 40 to 50 and older, with higher incidence in type 2 diabetes.2,5
Physical exam findings include proximal muscle weakness and atrophy in the quadriceps, hamstring, gluteal, hip adductors/abductors, and iliopsoas muscles.4,5 Typically, there is no sensory impairment; however, mild sensory loss may be observed in patients with coexisting chronic distal sensorimotor polyneuropathy.2,4 The patellar tendon reflexes are typically diminished or absent, and the ankle reflexes may be normal or diminished.4
Continued on next page >>
DIAGNOSTIC WORK-UP AND DIFFERENTIAL DIAGNOSIS
Although the diagnosis of diabetic amyotrophy is made primarily through detailed history taking and neurologic examination, other studies—electromyography, nerve conduction, imaging and labs, and nerve biopsy—may provide confirmation. Referral to neurology should also be considered.
The differential diagnosis is extensive and includes myopathies, muscular dystrophies, intervertebral disc disease, spinal stenosis, polyradiculopathies due to porphyria, amyloid, heavy metal poisoning, anterior horn cell diseases (eg, poliomyelitis), neoplasms, chronic inflammatory demyelinating polyneuropathy, Guillain-Barré syndrome, monoclonal gammopathy, inflammatory vasculitis, hypothyroidism, vitamin B6 or B12 deficiencies, syphilis, AIDS, Lyme disease, and Charcot-Marie-Tooth disease.2,5-7 Diabetic neuropathic cachexia should also be considered in the differential, as it presents with weight loss and lower limb pain but no weakness.5
Lab evaluation should begin with analysis of fasting plasma glucose, complete blood count, comprehensive metabolic profile, A1C, erythrocyte sedimentation rate (ESR), creatine kinase, vitamin B12, and thyroid-stimulating hormone levels.7 Elevations in ESR and positive rheumatoid factor and antinuclear antibody can occur in patients with diabetic amyotrophy and are suggestive of a coexisting autoimmune disorder.6 Serum creatine kinase and thyroid function studies are normal.4 Additional lab tests, if clinically indicated, include paraneoplastic panel to evaluate for occult malignancy, antimyelin-associated glycoprotein antibodies, antiganglioside antibodies, cryoglobulins, cerebrospinal fluid analysis, porphyrin titers, and testing for heavy metals.7
Electrodiagnostic studies are recommended if the diagnosis of diabetic amyotrophy remains unclear following history taking, physical examination, and preliminary testing. Electromyography and nerve conduction studies typically reveal findings consistent with denervation and axonal damage in proximal muscles of the lower extremities.4 If demyelination is observed, a diagnosis of chronic demyelinating polyneuropathy should be considered.5
Nerve biopsy is considered if the diagnosis remains unclear after laboratory and electrodiagnostic testing or when confirmation of the diagnosis is needed before starting aggressive treatment. The sural and superficial peroneal nerves are preferred for biopsy. In cases of diabetic amyotrophy, sural nerve biopsy reveals significant fiber loss in an asymmetric fashion, resembling focal ischemia.5
MRI or CT scan of the lumbosacral spine is employed to exclude mass lesions and structural disorders such as spinal stenosis and disc disease.4 Cerebrospinal fluid is typically acellular, with a mildly elevated protein level of 60 to 100 mg/dL (but occasionally as high as 400 mg/dL).5
Continued on next page >>
PROGNOSIS AND MANAGEMENT
The course of diabetic amyotrophy is variable. There is often gradual but incomplete restoration in muscle strength in correlation with aggressive glycemic control and physical therapy.2 The majority of patients have residual muscle weakness, absent patellar and/or ankle DTRs, exercise-related pain, stiffness, and difficulty walking or climbing stairs. Full recovery of strength only occurs in 10% to 20% of patients.6
Treatment with IV immunoglobulin or other immunosuppressive drugs is controversial. According to a Cochrane review of immunotherapy for diabetic amyotrophy, only one completed controlled trial using IV methylprednisolone was found. There is currently no evidence to support use of immunoglobulins to halt progression and improve symptoms.8
Neuropathic pain may be difficult to control. The severe pain associated with diabetic amyotrophy begins to diminish several months after onset, but residual pain may persist for several years. Pregabalin, duloxetine, tricyclic antidepressants, antiepileptic drugs, and narcotic analgesics can be helpful.2,4 High doses of corticosteroids may lead to improvement of severe pain in some patients with diabetic amyotrophy.5
References >>
REFERENCES
1. CDC. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention, 2011.
2. Nagsayi S, Somasekhar C, James CM. Diagnosis and management of diabetic amyotrophy. Geriatric Med. 2010;40:327-329.
3. Pasnoor M, Dimachkie MM, Kluding P, Barohn RJ. Diabetic neuropathy part 1: overview and symmetric phenotypes. Neurol Clin. 2013;31(2):425-445.
4. Sander HW, Chokroverty S. Diabetic amyotrophy: current concepts. Semin Neurol. 1996;16(2):173-177.
5. Pasnoor M, Dimachkie MM, Barohn RJ. Diabetic neuropathy part 2: proximal and asymmetric phenotypes. Neurol Clin. 2013;31(2): 447-462.
6. Idiculla J, Shirazi N, Opacka-Juffry J, Ganapathi. Diabetic amyotrophy: a brief review. Natl Med J India. 2004;17(4):
200-202.
7. Azhary H, Farooq M, Bhanushali M, Majid A. Peripheral neuropathy: differential diagnosis and management. Am Fam Physician. 2010;81(7):887-892.
8. Chan YC, Lo YL, Chan ES. Immunotherapy for diabetic amyotrophy. Cochrane Database Syst Rev. 2012;13(6):2-6.
A 45-year-old man, RT, with a six-month history of poorly controlled type 2 diabetes presents for evaluation of increased weakness and pain in the left lower extremity. The symptoms developed in the past three weeks. Previously able to ambulate without assistance, he purchased a cane yesterday due to concerns about falling.
RT reports poor adherence to his diabetes medications. His fingerstick blood sugars have ranged from 200 to 380 mg/dL over the past month. His weight has been stable; his BMI is 34. Review of other systems is negative. Vital signs include a blood pressure of 125/82 mm Hg; pulse, 74 beats/min; and respiratory rate, 16 breaths/min.
Physical examination is notable for muscle atrophy and tenderness to compression in the left quadriceps. Straight leg raise does not elicit pain bilaterally. Muscle strength is 4-/5 in the left hip with pain elicited on hip flexion, 4-/5 in the left knee, and 5/5 in the left ankle. Muscle strength is 4+/5 in the right hip, 5/5 in the right knee, and 5/5 in the right ankle. Muscle strength in both upper extremities is 5/5. Patellar deep tendon reflexes (DTRs) and ankle DTRs are absent bilaterally. Biceps and triceps DTRs are each 2+ bilaterally. Gait is slow and unsteady with use of the cane. Cranial nerves I-XII are intact. Sensation to sharp and dull testing is normal in both the upper and lower extremities.
Labwork reveals an A1C of 10.8%. The patient’s thyroid function studies, creatine kinase, and vitamin B12 level are all in normal range. The serum creatinine is 1.2 mg/dL, and eGFR (estimated glomerular filtration rate) is 58 mL/min/1.73 m2. Liver enzymes are normal, and complete blood count and other chemistry panels are unremarkable.
RT is referred to neurology. MRI of the thoracic and lumbar spine shows no mass lesions or disc disease. Electromyography reveals findings consistent with denervation and axonal damage in the proximal muscles in both lower extremities (left > right).
RT is diagnosed with diabetic amyotrophy and begins physical therapy three days a week. He achieves aggressive improvement in blood sugar control, and after three months, his A1C has improved to 7%. Although still using a cane, he reports improved muscle strength in the lower extremities and better gait stability.
Continued on next page >>
PREVALENCE AND TYPES OF DIABETIC PERIPHERAL NEUROPATHY
According to the CDC, 25.8 million children and adults in the United States (8.3% of the population) have diabetes. Approximately 60% to 70% of them have mild to severe neuropathy.1
Distal symmetric neuropathy is the most common form of diabetic peripheral neuropathy, accounting for more than 50% of cases. It is characterized by distal onset, predominately sensory polyneuropathy, and slow proximal progression.2
In contrast, diabetic amyotrophy is very rare, accounting for only 1% of all cases of neuropathy in diabetes. Prevalence is higher in those with type 2 versus type 1 diabetes (1.1% and 0.3%, respectively).3,4 The most commonly misdiagnosed of the asymmetric diabetic neuropathies, diabetic amyotrophy is characterized by acute, progressive, asymmetrical weakness and pain in the muscles of the proximal lower extremities.5 It is also been referred to as proximal diabetic neuropathy, ischemic mononeuropathy multiplex, diabetic femoral neuropathy, Bruns-Garland syndrome, and diabetic lumbosacral polyradiculopathy.5
LOCALIZATION AND PATHOGENESIS
The site of the lesion in diabetic amyotrophy remains controversial; it is theorized that diabetic amyotrophy may result from involvement of multiple sites, such as lumbosacral anterior horn cells, motor roots, plexus, or motor axons to the muscles of the proximal lower limbs.4
The pathogenesis remains unknown. One theory is that hyperglycemia may cause metabolic derangements in nerve conduction. Another is that there is ischemic damage followed by axonal degeneration. Immune-mediated inflammatory processes, such as microvasculitis, have also been proposed as causes.4,6
CLINICAL FEATURES
Diabetic amyotrophy is characterized by relatively rapid, progressive asymmetrical weakness and pain in the muscles in the proximal lower extremities; it develops over weeks to months and may continue for more than one year.2,6 It typically begins unilaterally and can progress bilaterally—normally without impairment in sensation. Patients commonly experience pain in the hip, buttock, or thigh, as well as difficulty walking, standing, or climbing stairs. Occasionally, the condition is painless and can be associated with weight loss. It causes significant acute disability, with the degree of recovery variable.2,4
Diabetic amyotrophy often presents either at diagnosis of diabetes or shortly thereafter. It most commonly affects men ages 40 to 50 and older, with higher incidence in type 2 diabetes.2,5
Physical exam findings include proximal muscle weakness and atrophy in the quadriceps, hamstring, gluteal, hip adductors/abductors, and iliopsoas muscles.4,5 Typically, there is no sensory impairment; however, mild sensory loss may be observed in patients with coexisting chronic distal sensorimotor polyneuropathy.2,4 The patellar tendon reflexes are typically diminished or absent, and the ankle reflexes may be normal or diminished.4
Continued on next page >>
DIAGNOSTIC WORK-UP AND DIFFERENTIAL DIAGNOSIS
Although the diagnosis of diabetic amyotrophy is made primarily through detailed history taking and neurologic examination, other studies—electromyography, nerve conduction, imaging and labs, and nerve biopsy—may provide confirmation. Referral to neurology should also be considered.
The differential diagnosis is extensive and includes myopathies, muscular dystrophies, intervertebral disc disease, spinal stenosis, polyradiculopathies due to porphyria, amyloid, heavy metal poisoning, anterior horn cell diseases (eg, poliomyelitis), neoplasms, chronic inflammatory demyelinating polyneuropathy, Guillain-Barré syndrome, monoclonal gammopathy, inflammatory vasculitis, hypothyroidism, vitamin B6 or B12 deficiencies, syphilis, AIDS, Lyme disease, and Charcot-Marie-Tooth disease.2,5-7 Diabetic neuropathic cachexia should also be considered in the differential, as it presents with weight loss and lower limb pain but no weakness.5
Lab evaluation should begin with analysis of fasting plasma glucose, complete blood count, comprehensive metabolic profile, A1C, erythrocyte sedimentation rate (ESR), creatine kinase, vitamin B12, and thyroid-stimulating hormone levels.7 Elevations in ESR and positive rheumatoid factor and antinuclear antibody can occur in patients with diabetic amyotrophy and are suggestive of a coexisting autoimmune disorder.6 Serum creatine kinase and thyroid function studies are normal.4 Additional lab tests, if clinically indicated, include paraneoplastic panel to evaluate for occult malignancy, antimyelin-associated glycoprotein antibodies, antiganglioside antibodies, cryoglobulins, cerebrospinal fluid analysis, porphyrin titers, and testing for heavy metals.7
Electrodiagnostic studies are recommended if the diagnosis of diabetic amyotrophy remains unclear following history taking, physical examination, and preliminary testing. Electromyography and nerve conduction studies typically reveal findings consistent with denervation and axonal damage in proximal muscles of the lower extremities.4 If demyelination is observed, a diagnosis of chronic demyelinating polyneuropathy should be considered.5
Nerve biopsy is considered if the diagnosis remains unclear after laboratory and electrodiagnostic testing or when confirmation of the diagnosis is needed before starting aggressive treatment. The sural and superficial peroneal nerves are preferred for biopsy. In cases of diabetic amyotrophy, sural nerve biopsy reveals significant fiber loss in an asymmetric fashion, resembling focal ischemia.5
MRI or CT scan of the lumbosacral spine is employed to exclude mass lesions and structural disorders such as spinal stenosis and disc disease.4 Cerebrospinal fluid is typically acellular, with a mildly elevated protein level of 60 to 100 mg/dL (but occasionally as high as 400 mg/dL).5
Continued on next page >>
PROGNOSIS AND MANAGEMENT
The course of diabetic amyotrophy is variable. There is often gradual but incomplete restoration in muscle strength in correlation with aggressive glycemic control and physical therapy.2 The majority of patients have residual muscle weakness, absent patellar and/or ankle DTRs, exercise-related pain, stiffness, and difficulty walking or climbing stairs. Full recovery of strength only occurs in 10% to 20% of patients.6
Treatment with IV immunoglobulin or other immunosuppressive drugs is controversial. According to a Cochrane review of immunotherapy for diabetic amyotrophy, only one completed controlled trial using IV methylprednisolone was found. There is currently no evidence to support use of immunoglobulins to halt progression and improve symptoms.8
Neuropathic pain may be difficult to control. The severe pain associated with diabetic amyotrophy begins to diminish several months after onset, but residual pain may persist for several years. Pregabalin, duloxetine, tricyclic antidepressants, antiepileptic drugs, and narcotic analgesics can be helpful.2,4 High doses of corticosteroids may lead to improvement of severe pain in some patients with diabetic amyotrophy.5
References >>
REFERENCES
1. CDC. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention, 2011.
2. Nagsayi S, Somasekhar C, James CM. Diagnosis and management of diabetic amyotrophy. Geriatric Med. 2010;40:327-329.
3. Pasnoor M, Dimachkie MM, Kluding P, Barohn RJ. Diabetic neuropathy part 1: overview and symmetric phenotypes. Neurol Clin. 2013;31(2):425-445.
4. Sander HW, Chokroverty S. Diabetic amyotrophy: current concepts. Semin Neurol. 1996;16(2):173-177.
5. Pasnoor M, Dimachkie MM, Barohn RJ. Diabetic neuropathy part 2: proximal and asymmetric phenotypes. Neurol Clin. 2013;31(2): 447-462.
6. Idiculla J, Shirazi N, Opacka-Juffry J, Ganapathi. Diabetic amyotrophy: a brief review. Natl Med J India. 2004;17(4):
200-202.
7. Azhary H, Farooq M, Bhanushali M, Majid A. Peripheral neuropathy: differential diagnosis and management. Am Fam Physician. 2010;81(7):887-892.
8. Chan YC, Lo YL, Chan ES. Immunotherapy for diabetic amyotrophy. Cochrane Database Syst Rev. 2012;13(6):2-6.
A 45-year-old man, RT, with a six-month history of poorly controlled type 2 diabetes presents for evaluation of increased weakness and pain in the left lower extremity. The symptoms developed in the past three weeks. Previously able to ambulate without assistance, he purchased a cane yesterday due to concerns about falling.
RT reports poor adherence to his diabetes medications. His fingerstick blood sugars have ranged from 200 to 380 mg/dL over the past month. His weight has been stable; his BMI is 34. Review of other systems is negative. Vital signs include a blood pressure of 125/82 mm Hg; pulse, 74 beats/min; and respiratory rate, 16 breaths/min.
Physical examination is notable for muscle atrophy and tenderness to compression in the left quadriceps. Straight leg raise does not elicit pain bilaterally. Muscle strength is 4-/5 in the left hip with pain elicited on hip flexion, 4-/5 in the left knee, and 5/5 in the left ankle. Muscle strength is 4+/5 in the right hip, 5/5 in the right knee, and 5/5 in the right ankle. Muscle strength in both upper extremities is 5/5. Patellar deep tendon reflexes (DTRs) and ankle DTRs are absent bilaterally. Biceps and triceps DTRs are each 2+ bilaterally. Gait is slow and unsteady with use of the cane. Cranial nerves I-XII are intact. Sensation to sharp and dull testing is normal in both the upper and lower extremities.
Labwork reveals an A1C of 10.8%. The patient’s thyroid function studies, creatine kinase, and vitamin B12 level are all in normal range. The serum creatinine is 1.2 mg/dL, and eGFR (estimated glomerular filtration rate) is 58 mL/min/1.73 m2. Liver enzymes are normal, and complete blood count and other chemistry panels are unremarkable.
RT is referred to neurology. MRI of the thoracic and lumbar spine shows no mass lesions or disc disease. Electromyography reveals findings consistent with denervation and axonal damage in the proximal muscles in both lower extremities (left > right).
RT is diagnosed with diabetic amyotrophy and begins physical therapy three days a week. He achieves aggressive improvement in blood sugar control, and after three months, his A1C has improved to 7%. Although still using a cane, he reports improved muscle strength in the lower extremities and better gait stability.
Continued on next page >>
PREVALENCE AND TYPES OF DIABETIC PERIPHERAL NEUROPATHY
According to the CDC, 25.8 million children and adults in the United States (8.3% of the population) have diabetes. Approximately 60% to 70% of them have mild to severe neuropathy.1
Distal symmetric neuropathy is the most common form of diabetic peripheral neuropathy, accounting for more than 50% of cases. It is characterized by distal onset, predominately sensory polyneuropathy, and slow proximal progression.2
In contrast, diabetic amyotrophy is very rare, accounting for only 1% of all cases of neuropathy in diabetes. Prevalence is higher in those with type 2 versus type 1 diabetes (1.1% and 0.3%, respectively).3,4 The most commonly misdiagnosed of the asymmetric diabetic neuropathies, diabetic amyotrophy is characterized by acute, progressive, asymmetrical weakness and pain in the muscles of the proximal lower extremities.5 It is also been referred to as proximal diabetic neuropathy, ischemic mononeuropathy multiplex, diabetic femoral neuropathy, Bruns-Garland syndrome, and diabetic lumbosacral polyradiculopathy.5
LOCALIZATION AND PATHOGENESIS
The site of the lesion in diabetic amyotrophy remains controversial; it is theorized that diabetic amyotrophy may result from involvement of multiple sites, such as lumbosacral anterior horn cells, motor roots, plexus, or motor axons to the muscles of the proximal lower limbs.4
The pathogenesis remains unknown. One theory is that hyperglycemia may cause metabolic derangements in nerve conduction. Another is that there is ischemic damage followed by axonal degeneration. Immune-mediated inflammatory processes, such as microvasculitis, have also been proposed as causes.4,6
CLINICAL FEATURES
Diabetic amyotrophy is characterized by relatively rapid, progressive asymmetrical weakness and pain in the muscles in the proximal lower extremities; it develops over weeks to months and may continue for more than one year.2,6 It typically begins unilaterally and can progress bilaterally—normally without impairment in sensation. Patients commonly experience pain in the hip, buttock, or thigh, as well as difficulty walking, standing, or climbing stairs. Occasionally, the condition is painless and can be associated with weight loss. It causes significant acute disability, with the degree of recovery variable.2,4
Diabetic amyotrophy often presents either at diagnosis of diabetes or shortly thereafter. It most commonly affects men ages 40 to 50 and older, with higher incidence in type 2 diabetes.2,5
Physical exam findings include proximal muscle weakness and atrophy in the quadriceps, hamstring, gluteal, hip adductors/abductors, and iliopsoas muscles.4,5 Typically, there is no sensory impairment; however, mild sensory loss may be observed in patients with coexisting chronic distal sensorimotor polyneuropathy.2,4 The patellar tendon reflexes are typically diminished or absent, and the ankle reflexes may be normal or diminished.4
Continued on next page >>
DIAGNOSTIC WORK-UP AND DIFFERENTIAL DIAGNOSIS
Although the diagnosis of diabetic amyotrophy is made primarily through detailed history taking and neurologic examination, other studies—electromyography, nerve conduction, imaging and labs, and nerve biopsy—may provide confirmation. Referral to neurology should also be considered.
The differential diagnosis is extensive and includes myopathies, muscular dystrophies, intervertebral disc disease, spinal stenosis, polyradiculopathies due to porphyria, amyloid, heavy metal poisoning, anterior horn cell diseases (eg, poliomyelitis), neoplasms, chronic inflammatory demyelinating polyneuropathy, Guillain-Barré syndrome, monoclonal gammopathy, inflammatory vasculitis, hypothyroidism, vitamin B6 or B12 deficiencies, syphilis, AIDS, Lyme disease, and Charcot-Marie-Tooth disease.2,5-7 Diabetic neuropathic cachexia should also be considered in the differential, as it presents with weight loss and lower limb pain but no weakness.5
Lab evaluation should begin with analysis of fasting plasma glucose, complete blood count, comprehensive metabolic profile, A1C, erythrocyte sedimentation rate (ESR), creatine kinase, vitamin B12, and thyroid-stimulating hormone levels.7 Elevations in ESR and positive rheumatoid factor and antinuclear antibody can occur in patients with diabetic amyotrophy and are suggestive of a coexisting autoimmune disorder.6 Serum creatine kinase and thyroid function studies are normal.4 Additional lab tests, if clinically indicated, include paraneoplastic panel to evaluate for occult malignancy, antimyelin-associated glycoprotein antibodies, antiganglioside antibodies, cryoglobulins, cerebrospinal fluid analysis, porphyrin titers, and testing for heavy metals.7
Electrodiagnostic studies are recommended if the diagnosis of diabetic amyotrophy remains unclear following history taking, physical examination, and preliminary testing. Electromyography and nerve conduction studies typically reveal findings consistent with denervation and axonal damage in proximal muscles of the lower extremities.4 If demyelination is observed, a diagnosis of chronic demyelinating polyneuropathy should be considered.5
Nerve biopsy is considered if the diagnosis remains unclear after laboratory and electrodiagnostic testing or when confirmation of the diagnosis is needed before starting aggressive treatment. The sural and superficial peroneal nerves are preferred for biopsy. In cases of diabetic amyotrophy, sural nerve biopsy reveals significant fiber loss in an asymmetric fashion, resembling focal ischemia.5
MRI or CT scan of the lumbosacral spine is employed to exclude mass lesions and structural disorders such as spinal stenosis and disc disease.4 Cerebrospinal fluid is typically acellular, with a mildly elevated protein level of 60 to 100 mg/dL (but occasionally as high as 400 mg/dL).5
Continued on next page >>
PROGNOSIS AND MANAGEMENT
The course of diabetic amyotrophy is variable. There is often gradual but incomplete restoration in muscle strength in correlation with aggressive glycemic control and physical therapy.2 The majority of patients have residual muscle weakness, absent patellar and/or ankle DTRs, exercise-related pain, stiffness, and difficulty walking or climbing stairs. Full recovery of strength only occurs in 10% to 20% of patients.6
Treatment with IV immunoglobulin or other immunosuppressive drugs is controversial. According to a Cochrane review of immunotherapy for diabetic amyotrophy, only one completed controlled trial using IV methylprednisolone was found. There is currently no evidence to support use of immunoglobulins to halt progression and improve symptoms.8
Neuropathic pain may be difficult to control. The severe pain associated with diabetic amyotrophy begins to diminish several months after onset, but residual pain may persist for several years. Pregabalin, duloxetine, tricyclic antidepressants, antiepileptic drugs, and narcotic analgesics can be helpful.2,4 High doses of corticosteroids may lead to improvement of severe pain in some patients with diabetic amyotrophy.5
References >>
REFERENCES
1. CDC. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention, 2011.
2. Nagsayi S, Somasekhar C, James CM. Diagnosis and management of diabetic amyotrophy. Geriatric Med. 2010;40:327-329.
3. Pasnoor M, Dimachkie MM, Kluding P, Barohn RJ. Diabetic neuropathy part 1: overview and symmetric phenotypes. Neurol Clin. 2013;31(2):425-445.
4. Sander HW, Chokroverty S. Diabetic amyotrophy: current concepts. Semin Neurol. 1996;16(2):173-177.
5. Pasnoor M, Dimachkie MM, Barohn RJ. Diabetic neuropathy part 2: proximal and asymmetric phenotypes. Neurol Clin. 2013;31(2): 447-462.
6. Idiculla J, Shirazi N, Opacka-Juffry J, Ganapathi. Diabetic amyotrophy: a brief review. Natl Med J India. 2004;17(4):
200-202.
7. Azhary H, Farooq M, Bhanushali M, Majid A. Peripheral neuropathy: differential diagnosis and management. Am Fam Physician. 2010;81(7):887-892.
8. Chan YC, Lo YL, Chan ES. Immunotherapy for diabetic amyotrophy. Cochrane Database Syst Rev. 2012;13(6):2-6.
Hyperprolactinemia: Causes and Treatments
A 31-year-old woman is referred by her Ob-Gyn for elevated prolactin. She initially presented with a three-month history of amenorrhea, a negative home pregnancy test, and 100% compliance with condom use. She denies hirsutism and acne but admits to thin milky nipple discharge upon squeezing (but not spontaneous).
Two weeks ago, her Ob-Gyn ordered labs; results were negative for serum beta human chorionic gonadotropin and within normal ranges for thyroid-stimulating hormone (TSH), luteinizing hormone, follicle-stimulating hormone, estradiol, free and total testosterone, dehydroepiandrosterone sulfate (DHEAs), complete chemistry panel, and complete blood count. Her serum prolactin level was 110 ng/mL (normal, 3 to 27 ng/mL).
Q: How is prolactin physiologically regulated?
The primary role of prolactin, which is produced by lactotroph cells in the anterior pituitary gland, is to stimulate lactation and breast development. Prolactin is regulated by dopamine (also known as prolactin inhibitory hormone), which is secreted from the hypothalamus via an inhibitory pathway unique to the hypothalamus-pituitary hormone system. Dopamine essentially suppresses prolactin.
Other hormones can have a stimulatory effect on the anterior pituitary gland and thus increase prolactin levels. Estrogen can induce lactotroph hyperplasia and elevated prolactin; however, this is only clinically relevant in the context of estrogen surge during pregnancy. (Estrogen therapy, such as oral contraception or hormone replacement therapy, on the other hand, is targeted to “normal” estrogen levels.) Thyrotropin-releasing hormone (TRH) from the hypothalamus also stimulates the anterior pituitary gland, so patients with inadequately treated or untreated primary hypothyroidism will have mildly elevated prolactin.
Neurogenic stimuli of the chest wall, through nipple suckling or varicella zoster infection (shingles), can also increase prolactin secretion. And since prolactin is eliminated by the liver (75%) and the kidney (25%), significant liver disease and/or renal insufficiency can raise prolactin levels, due to decreased clearance.
What are the possible etiologies for elevated prolactin? See answer on the next page...
Q: What are the possible etiologies for elevated prolactin?
The causes of hyperprolactinemia fall into three categories: physiologic, pharmacologic, and pathologic.2 The table provides examples from each category.
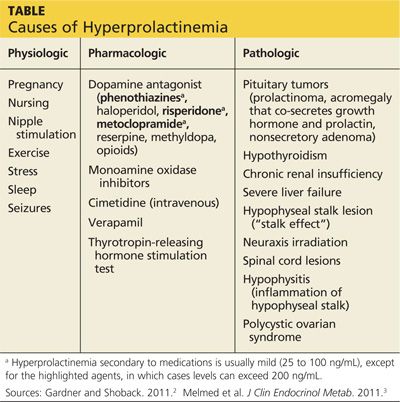
A nonsecretory pituitary adenoma or any lesion in the brain that would disrupt the hypophyseal stalk may interfere with dopamine’s inhibitory control and thereby increase prolactin. This is called the stalk effect. It is important to note that not all MRI-proven pituitary adenomas are prolactin secreting, even in the presence of hyperprolactinemia. According to an autopsy series, about 12% of the general population had pituitary microadenoma.3
There is rough correlation between prolactinoma size and level of prolactin. Large nonsecretory pituitary adenomas have prolactin levels less than 150 ng/mL. Microprolactinomas (< 1 cm) are usually in the range of 100 to 250 ng/mL, while macroprolactinomas (> 1 cm) are generally
≥ 250 ng/mL. If the tumor is very large and invades the cavernous sinus, prolactin can measure in the 1,000s.3
Polycystic ovarian syndrome (PCOS) is a common disorder affecting women of reproductive age and the most common cause of underlying ovulatory problems. Patients with PCOS can have mildly elevated prolactin; the exact mechanism of hyperprolactinemia in PCOS is unknown. One theory is that constant high levels of estrogen experienced in PCOS would stimulate prolactin production. It is important to rule out other causes of hyperprolactinemia before making the diagnosis of PCOS.
What is the clinical significance of elevated prolactin? Why do we have to work up and treat it? See answer on the next page...
Q: What is the clinical significance of elevated prolactin? Why do we have to work up and treat it?
By physiologic mechanisms not completely understood, hyperprolactinemia can interrupt the gonadal axis, leading to hypogonadism. In women, it can cause irregular menstrual cycles, oligomenorrhea, amenorrhea, and infertility. In men, it can lower testosterone levels. Long-term effects include declining bone mineral density due to insufficient estrogen in women or testosterone in men.
With macroadenoma, the size of the tumor can have a mass effect such as headache and visual defect by compressing the optic chiasm (bitemporal hemianopsia), which may lead to permanent vision loss if left untreated. Referral to an ophthalmologist may be necessary for formal visual field examination.
How is hyperprolactinemia treated? See answer on the next page...
Q: How is hyperprolactinemia treated?
There are three options for treatment: medication, surgery, and radiation.
Dopamine agonists (bromocriptine, cabergoline) are effective in normalizing prolactin and reducing the size of the tumor in the majority of cases. However, some patients may require long-term treatment. Bromocriptine has been used since the late 1970s, but, due to better tolerance and less frequent dosing, cabergoline is the preferred agent.3
Transsphenoidal surgery is indicated for patients who are intolerant to medication, who have a medication-resistant tumor or significant mass effect, or who prefer definitive treatment. Women of childbearing age with a macroadenoma might consider surgery due to the risk for tumor expansion during pregnancy (estrogen effect) and risk for pituitary apoplexy (hemorrhage or infarct of the pituitary gland). Surgical risk is usually low with a neurosurgeon who has extensive experience.
Radiation can be considered for large tumors that are resistant to medication. It can be used as adjunctive therapy to surgery, since reducing the size of the tumor can make the surgical field smaller. In some medication-resistant tumors, radiation can raise sensitivity to medication.
What does follow-up entail? See next page for answer...
Q: What does follow-up entail?
Once medication is initiated or dosage is adjusted, have the patient follow up in one month and recheck the prolactin level to assess responsiveness to medication (as well as medication adherence). When a therapeutic prolactin level is achieved, recheck the prolactin and have the patient follow up at three and six months and then every six months thereafter.3
MRI of the pituitary gland should be performed at baseline, then in six months to assess tumor response to medication, and then at 12 and 24 months.3 If tumor regression has stabilized or if the tumor has shrunk to a nondetectable size, consider discontinuing the dopamine agonist. If medication is discontinued, recheck prolactin every three months for the first year; if it remains in normal reference range, simply check serum prolactin annually.3
See next page for summary.

See next page for references.
REFERENCES
1. Jameson JL. Harrison’s Endocrinology. 18th ed. China: McGraw-Hill; 2010.
2. Gardner D, Shoback D. Greenspan’s Basic & Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.
A 31-year-old woman is referred by her Ob-Gyn for elevated prolactin. She initially presented with a three-month history of amenorrhea, a negative home pregnancy test, and 100% compliance with condom use. She denies hirsutism and acne but admits to thin milky nipple discharge upon squeezing (but not spontaneous).
Two weeks ago, her Ob-Gyn ordered labs; results were negative for serum beta human chorionic gonadotropin and within normal ranges for thyroid-stimulating hormone (TSH), luteinizing hormone, follicle-stimulating hormone, estradiol, free and total testosterone, dehydroepiandrosterone sulfate (DHEAs), complete chemistry panel, and complete blood count. Her serum prolactin level was 110 ng/mL (normal, 3 to 27 ng/mL).
Q: How is prolactin physiologically regulated?
The primary role of prolactin, which is produced by lactotroph cells in the anterior pituitary gland, is to stimulate lactation and breast development. Prolactin is regulated by dopamine (also known as prolactin inhibitory hormone), which is secreted from the hypothalamus via an inhibitory pathway unique to the hypothalamus-pituitary hormone system. Dopamine essentially suppresses prolactin.
Other hormones can have a stimulatory effect on the anterior pituitary gland and thus increase prolactin levels. Estrogen can induce lactotroph hyperplasia and elevated prolactin; however, this is only clinically relevant in the context of estrogen surge during pregnancy. (Estrogen therapy, such as oral contraception or hormone replacement therapy, on the other hand, is targeted to “normal” estrogen levels.) Thyrotropin-releasing hormone (TRH) from the hypothalamus also stimulates the anterior pituitary gland, so patients with inadequately treated or untreated primary hypothyroidism will have mildly elevated prolactin.
Neurogenic stimuli of the chest wall, through nipple suckling or varicella zoster infection (shingles), can also increase prolactin secretion. And since prolactin is eliminated by the liver (75%) and the kidney (25%), significant liver disease and/or renal insufficiency can raise prolactin levels, due to decreased clearance.
What are the possible etiologies for elevated prolactin? See answer on the next page...
Q: What are the possible etiologies for elevated prolactin?
The causes of hyperprolactinemia fall into three categories: physiologic, pharmacologic, and pathologic.2 The table provides examples from each category.
A nonsecretory pituitary adenoma or any lesion in the brain that would disrupt the hypophyseal stalk may interfere with dopamine’s inhibitory control and thereby increase prolactin. This is called the stalk effect. It is important to note that not all MRI-proven pituitary adenomas are prolactin secreting, even in the presence of hyperprolactinemia. According to an autopsy series, about 12% of the general population had pituitary microadenoma.3
There is rough correlation between prolactinoma size and level of prolactin. Large nonsecretory pituitary adenomas have prolactin levels less than 150 ng/mL. Microprolactinomas (< 1 cm) are usually in the range of 100 to 250 ng/mL, while macroprolactinomas (> 1 cm) are generally
≥ 250 ng/mL. If the tumor is very large and invades the cavernous sinus, prolactin can measure in the 1,000s.3
Polycystic ovarian syndrome (PCOS) is a common disorder affecting women of reproductive age and the most common cause of underlying ovulatory problems. Patients with PCOS can have mildly elevated prolactin; the exact mechanism of hyperprolactinemia in PCOS is unknown. One theory is that constant high levels of estrogen experienced in PCOS would stimulate prolactin production. It is important to rule out other causes of hyperprolactinemia before making the diagnosis of PCOS.
What is the clinical significance of elevated prolactin? Why do we have to work up and treat it? See answer on the next page...
Q: What is the clinical significance of elevated prolactin? Why do we have to work up and treat it?
By physiologic mechanisms not completely understood, hyperprolactinemia can interrupt the gonadal axis, leading to hypogonadism. In women, it can cause irregular menstrual cycles, oligomenorrhea, amenorrhea, and infertility. In men, it can lower testosterone levels. Long-term effects include declining bone mineral density due to insufficient estrogen in women or testosterone in men.
With macroadenoma, the size of the tumor can have a mass effect such as headache and visual defect by compressing the optic chiasm (bitemporal hemianopsia), which may lead to permanent vision loss if left untreated. Referral to an ophthalmologist may be necessary for formal visual field examination.
How is hyperprolactinemia treated? See answer on the next page...
Q: How is hyperprolactinemia treated?
There are three options for treatment: medication, surgery, and radiation.
Dopamine agonists (bromocriptine, cabergoline) are effective in normalizing prolactin and reducing the size of the tumor in the majority of cases. However, some patients may require long-term treatment. Bromocriptine has been used since the late 1970s, but, due to better tolerance and less frequent dosing, cabergoline is the preferred agent.3
Transsphenoidal surgery is indicated for patients who are intolerant to medication, who have a medication-resistant tumor or significant mass effect, or who prefer definitive treatment. Women of childbearing age with a macroadenoma might consider surgery due to the risk for tumor expansion during pregnancy (estrogen effect) and risk for pituitary apoplexy (hemorrhage or infarct of the pituitary gland). Surgical risk is usually low with a neurosurgeon who has extensive experience.
Radiation can be considered for large tumors that are resistant to medication. It can be used as adjunctive therapy to surgery, since reducing the size of the tumor can make the surgical field smaller. In some medication-resistant tumors, radiation can raise sensitivity to medication.
What does follow-up entail? See next page for answer...
Q: What does follow-up entail?
Once medication is initiated or dosage is adjusted, have the patient follow up in one month and recheck the prolactin level to assess responsiveness to medication (as well as medication adherence). When a therapeutic prolactin level is achieved, recheck the prolactin and have the patient follow up at three and six months and then every six months thereafter.3
MRI of the pituitary gland should be performed at baseline, then in six months to assess tumor response to medication, and then at 12 and 24 months.3 If tumor regression has stabilized or if the tumor has shrunk to a nondetectable size, consider discontinuing the dopamine agonist. If medication is discontinued, recheck prolactin every three months for the first year; if it remains in normal reference range, simply check serum prolactin annually.3
See next page for summary.
See next page for references.
REFERENCES
1. Jameson JL. Harrison’s Endocrinology. 18th ed. China: McGraw-Hill; 2010.
2. Gardner D, Shoback D. Greenspan’s Basic & Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.
A 31-year-old woman is referred by her Ob-Gyn for elevated prolactin. She initially presented with a three-month history of amenorrhea, a negative home pregnancy test, and 100% compliance with condom use. She denies hirsutism and acne but admits to thin milky nipple discharge upon squeezing (but not spontaneous).
Two weeks ago, her Ob-Gyn ordered labs; results were negative for serum beta human chorionic gonadotropin and within normal ranges for thyroid-stimulating hormone (TSH), luteinizing hormone, follicle-stimulating hormone, estradiol, free and total testosterone, dehydroepiandrosterone sulfate (DHEAs), complete chemistry panel, and complete blood count. Her serum prolactin level was 110 ng/mL (normal, 3 to 27 ng/mL).
Q: How is prolactin physiologically regulated?
The primary role of prolactin, which is produced by lactotroph cells in the anterior pituitary gland, is to stimulate lactation and breast development. Prolactin is regulated by dopamine (also known as prolactin inhibitory hormone), which is secreted from the hypothalamus via an inhibitory pathway unique to the hypothalamus-pituitary hormone system. Dopamine essentially suppresses prolactin.
Other hormones can have a stimulatory effect on the anterior pituitary gland and thus increase prolactin levels. Estrogen can induce lactotroph hyperplasia and elevated prolactin; however, this is only clinically relevant in the context of estrogen surge during pregnancy. (Estrogen therapy, such as oral contraception or hormone replacement therapy, on the other hand, is targeted to “normal” estrogen levels.) Thyrotropin-releasing hormone (TRH) from the hypothalamus also stimulates the anterior pituitary gland, so patients with inadequately treated or untreated primary hypothyroidism will have mildly elevated prolactin.
Neurogenic stimuli of the chest wall, through nipple suckling or varicella zoster infection (shingles), can also increase prolactin secretion. And since prolactin is eliminated by the liver (75%) and the kidney (25%), significant liver disease and/or renal insufficiency can raise prolactin levels, due to decreased clearance.
What are the possible etiologies for elevated prolactin? See answer on the next page...
Q: What are the possible etiologies for elevated prolactin?
The causes of hyperprolactinemia fall into three categories: physiologic, pharmacologic, and pathologic.2 The table provides examples from each category.
A nonsecretory pituitary adenoma or any lesion in the brain that would disrupt the hypophyseal stalk may interfere with dopamine’s inhibitory control and thereby increase prolactin. This is called the stalk effect. It is important to note that not all MRI-proven pituitary adenomas are prolactin secreting, even in the presence of hyperprolactinemia. According to an autopsy series, about 12% of the general population had pituitary microadenoma.3
There is rough correlation between prolactinoma size and level of prolactin. Large nonsecretory pituitary adenomas have prolactin levels less than 150 ng/mL. Microprolactinomas (< 1 cm) are usually in the range of 100 to 250 ng/mL, while macroprolactinomas (> 1 cm) are generally
≥ 250 ng/mL. If the tumor is very large and invades the cavernous sinus, prolactin can measure in the 1,000s.3
Polycystic ovarian syndrome (PCOS) is a common disorder affecting women of reproductive age and the most common cause of underlying ovulatory problems. Patients with PCOS can have mildly elevated prolactin; the exact mechanism of hyperprolactinemia in PCOS is unknown. One theory is that constant high levels of estrogen experienced in PCOS would stimulate prolactin production. It is important to rule out other causes of hyperprolactinemia before making the diagnosis of PCOS.
What is the clinical significance of elevated prolactin? Why do we have to work up and treat it? See answer on the next page...
Q: What is the clinical significance of elevated prolactin? Why do we have to work up and treat it?
By physiologic mechanisms not completely understood, hyperprolactinemia can interrupt the gonadal axis, leading to hypogonadism. In women, it can cause irregular menstrual cycles, oligomenorrhea, amenorrhea, and infertility. In men, it can lower testosterone levels. Long-term effects include declining bone mineral density due to insufficient estrogen in women or testosterone in men.
With macroadenoma, the size of the tumor can have a mass effect such as headache and visual defect by compressing the optic chiasm (bitemporal hemianopsia), which may lead to permanent vision loss if left untreated. Referral to an ophthalmologist may be necessary for formal visual field examination.
How is hyperprolactinemia treated? See answer on the next page...
Q: How is hyperprolactinemia treated?
There are three options for treatment: medication, surgery, and radiation.
Dopamine agonists (bromocriptine, cabergoline) are effective in normalizing prolactin and reducing the size of the tumor in the majority of cases. However, some patients may require long-term treatment. Bromocriptine has been used since the late 1970s, but, due to better tolerance and less frequent dosing, cabergoline is the preferred agent.3
Transsphenoidal surgery is indicated for patients who are intolerant to medication, who have a medication-resistant tumor or significant mass effect, or who prefer definitive treatment. Women of childbearing age with a macroadenoma might consider surgery due to the risk for tumor expansion during pregnancy (estrogen effect) and risk for pituitary apoplexy (hemorrhage or infarct of the pituitary gland). Surgical risk is usually low with a neurosurgeon who has extensive experience.
Radiation can be considered for large tumors that are resistant to medication. It can be used as adjunctive therapy to surgery, since reducing the size of the tumor can make the surgical field smaller. In some medication-resistant tumors, radiation can raise sensitivity to medication.
What does follow-up entail? See next page for answer...
Q: What does follow-up entail?
Once medication is initiated or dosage is adjusted, have the patient follow up in one month and recheck the prolactin level to assess responsiveness to medication (as well as medication adherence). When a therapeutic prolactin level is achieved, recheck the prolactin and have the patient follow up at three and six months and then every six months thereafter.3
MRI of the pituitary gland should be performed at baseline, then in six months to assess tumor response to medication, and then at 12 and 24 months.3 If tumor regression has stabilized or if the tumor has shrunk to a nondetectable size, consider discontinuing the dopamine agonist. If medication is discontinued, recheck prolactin every three months for the first year; if it remains in normal reference range, simply check serum prolactin annually.3
See next page for summary.
See next page for references.
REFERENCES
1. Jameson JL. Harrison’s Endocrinology. 18th ed. China: McGraw-Hill; 2010.
2. Gardner D, Shoback D. Greenspan’s Basic & Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.
Hereditary Hemochromatosis as a Cause of Hypogonadism
JR, a 34-year-old Caucasian man, was in his normal state of good health until several months ago, when he developed fatigue, low libido, and insomnia. He reports normal erectile function, adding that he fathered a child at age 24. His medical history and remaining review of systems are negative. Physical exam is unremarkable. His BMI is 23.
Labwork reveals low free and total testosterone levels with low FSH and LH levels. Thyroid-stimulating hormone, free T4, and prolactin levels are within normal range, comprehensive metabolic panel is unremarkable, and pituitary MRI is negative. The complete blood count reveals slightly elevated hemoglobin and hematocrit, prompting ordering of iron studies that reveal elevated ferritin and serum iron levels and elevated percent transferrin saturation. Lab values are shown in the table.
Based on his elevated ferritin and transferrin saturation levels, JR undergoes genetic testing for hereditary hemochromatosis (HH) with C282Y and H63D mutation analysis. He is found to have the homozygous C282Y genotype (C282Y/C282Y) for HH.
JR establishes care with a hematologist and is advised to receive therapeutic phlebotomy until his ferritin level is between 10 and 50 ng/mL. An abdominal ultrasound, ordered to screen for hepatomegaly, yields normal results. JR elects not to receive testosterone replacement therapy.
Three months later, labwork reveals a free testosterone level of 120 pg/mL with normal hemoglobin and hematocrit levels, normal transaminases, a ferritin level of 28 ng/mL, and a percent transferrin saturation of 31%. Additional values are shown in the table.
BACKGROUND AND GENETICS
Hereditary hemochromatosis is an autosomal recessive iron storage disorder in which intestinal iron absorption is markedly increased. This results in iron overload and excessive iron deposition in numerous tissues, glands, and organs.1
In patients with HH, a genetic defect causes abnormal expression of the HFE protein that regulates hepcidin production. Hepcidin is an iron regulatory hormone, secreted by hepatocytes, that decreases intestinal iron absorption in response to excess iron.2 Ninety percent of individuals affected by HH are homozygous for mutation at amino acid position 282 on the HFE gene, which causes an inappropriate decrease in hepcidin expression in response to elevated iron levels.2,3 Only 10% of individuals homozygous for the C282Y mutation actually develop clinically apparent end-organ damage.2
Being a carrier (heterozygous) for the C282Y mutation confers significantly lower risk for iron overload. The second most common mutation in the HFE gene, H63D, is associated with a milder phenotype. Those with compound heterozygosity for C282Y/H63D or homozygosity for H63D typically experience either mild or no detectable symptoms.1,3
There can be mutations in other genes involved in iron metabolism, but these represent more rare forms of hemochromatosis. Conditions such as thalassemia, sideroblastic anemia, porphyria cutanea tarda, and chronic liver disease may also be associated with iron overload.1,3
HH is most common in white populations of northern European descent. Multiple factors—including dietary iron intake, alcohol consumption, blood donation, blood loss associated with menstruation, and pregnancy—affect the expression of clinical features of hemochromatosis. Men are 24 times more likely than women to express clinical features of hemochromatosis.2 Approximately 70% of affected patients develop symptoms between ages 40 and 60.1
CLINICAL MANIFESTATIONS
The liver is typically the first organ affected, and hepatomegaly is present in 95% of symptomatic patients, even in the presence of normal transaminase levels.1 A bronzed, metallic, or slate gray skin coloration can occur due to increased iron deposition in the dermis. Arthralgias in the hands, wrists, hips, knees, and ankles are present in up to 50% of patients with hemochromatosis. Cardiac manifestations include restrictive cardiomyopathy, congestive heart failure, and arrhythmias.1
Iron deposition in the beta cells of the pancreas causes diabetes1 and in the pituitary causes hypogonadotropic hypogonadism in both men and women, resulting in decreased libido, amenorrhea, testicular atrophy, gynecomastia, and reduced body hair. Primary testicular dysfunction may occur due to iron deposition in the testicles.1,4 In the thyroid gland, iron deposition can lead to abnormal function. Secondary hypothyroidism is rare in the setting of iron overload, although iron deposition occasionally occurs in pituitary thyrotrophs (usually only to a mild degree). Adrenal insufficiency and hypoparathyroidism may also result from iron overload.5
CLINICAL STUDIES TO ASSESS IRON STORES
When assessing tissue iron stores, it is important to measure the serum iron level, total iron-binding capacity, and ferritin in the fasting state.2 This information can be used to calculate the percent transferrin saturation. If the serum ferritin is elevated (> 300 ng/mL in men and > 200 ng/mL in women) and/or the transferrin saturation is greater than 45%, referral to hematology or hepatology is recommended, along with genetic testing for hemochromatosis.1,2
Once the diagnosis of hemochromatosis has been confirmed, CT or MRI can be used to assess for increased density of the liver.1 Liver biopsy can determine the degree of fibrosis and is often considered in patients with more extreme elevations of serum ferritin levels and/or hepatomegaly. Liver biopsy is the only reliable method for determining whether hepatic cirrhosis, which increases risk for hepatocellular carcinoma, is present.1
TREATMENT
All patients with homozygous HH and evidence of iron overload require treatment, regardless of symptoms. Phlebotomy is the standard of care, due to its low cost and relative safety. Chelating agents are a second-line option when contraindications to phlebotomy (eg, anemia) exist.1,2
Alcohol consumption, especially in the presence of iron overload or liver disease, should be avoided, as it can increase risk for cirrhosis by nearly tenfold.1 Dietary modification is typically unnecessary, aside from the avoidance of iron and vitamin C supplementation.2 Patients should also eliminate raw shellfish from their diet, as they may carry bacteria that can cause potentially fatal infection (since high iron levels impair hepcidin bactericidal activity).2
The management of hepatic failure, cardiac failure, and diabetes in patients with HH is similar to conventional management of these conditions.1 With phlebotomy, the liver and spleen often decrease in size, liver function improves, skin pigmentation lightens, cardiac failure may be reversed, and diabetes control often improves.1,2 Testosterone levels may normalize after phlebotomy, especially if HH is diagnosed in the early stages. In more advanced cases, testosterone replacement therapy in combination with aggressive phlebotomy may be necessary.4
CONCLUSION
A high index of suspicion is required to diagnose hemochromatosis early. HH should be considered in the differential diagnosis for patients with hypogonadotropic hypogonadism, abnormal iron studies, elevated transaminase levels, and a family history of hemochromatosis.
Once the diagnosis is established, all first-degree relatives should be screened.1 Early therapy is crucial to prevent complications from iron overload.
REFERENCES
1. Powell LW. Hemochromatosis. In: Fauci AS, Braunwald E, Kasper DL, et al (eds). Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:2429-2433.
2. Crownover BK, Carlton JC. Hereditary hemochromatosis. Am Fam Phys. 2013;87(3):183-190.
3. Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717.
4. McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab. 2005;90(4):2451-2455.
5. Hudec M, Grigerova M, Walsh CH. Secondary hypothyroidism in hereditary hemochromatosis: recovery after iron depletion. Thyroid. 2008;18(2):255-257.
JR, a 34-year-old Caucasian man, was in his normal state of good health until several months ago, when he developed fatigue, low libido, and insomnia. He reports normal erectile function, adding that he fathered a child at age 24. His medical history and remaining review of systems are negative. Physical exam is unremarkable. His BMI is 23.
Labwork reveals low free and total testosterone levels with low FSH and LH levels. Thyroid-stimulating hormone, free T4, and prolactin levels are within normal range, comprehensive metabolic panel is unremarkable, and pituitary MRI is negative. The complete blood count reveals slightly elevated hemoglobin and hematocrit, prompting ordering of iron studies that reveal elevated ferritin and serum iron levels and elevated percent transferrin saturation. Lab values are shown in the table.
Based on his elevated ferritin and transferrin saturation levels, JR undergoes genetic testing for hereditary hemochromatosis (HH) with C282Y and H63D mutation analysis. He is found to have the homozygous C282Y genotype (C282Y/C282Y) for HH.
JR establishes care with a hematologist and is advised to receive therapeutic phlebotomy until his ferritin level is between 10 and 50 ng/mL. An abdominal ultrasound, ordered to screen for hepatomegaly, yields normal results. JR elects not to receive testosterone replacement therapy.
Three months later, labwork reveals a free testosterone level of 120 pg/mL with normal hemoglobin and hematocrit levels, normal transaminases, a ferritin level of 28 ng/mL, and a percent transferrin saturation of 31%. Additional values are shown in the table.
BACKGROUND AND GENETICS
Hereditary hemochromatosis is an autosomal recessive iron storage disorder in which intestinal iron absorption is markedly increased. This results in iron overload and excessive iron deposition in numerous tissues, glands, and organs.1
In patients with HH, a genetic defect causes abnormal expression of the HFE protein that regulates hepcidin production. Hepcidin is an iron regulatory hormone, secreted by hepatocytes, that decreases intestinal iron absorption in response to excess iron.2 Ninety percent of individuals affected by HH are homozygous for mutation at amino acid position 282 on the HFE gene, which causes an inappropriate decrease in hepcidin expression in response to elevated iron levels.2,3 Only 10% of individuals homozygous for the C282Y mutation actually develop clinically apparent end-organ damage.2
Being a carrier (heterozygous) for the C282Y mutation confers significantly lower risk for iron overload. The second most common mutation in the HFE gene, H63D, is associated with a milder phenotype. Those with compound heterozygosity for C282Y/H63D or homozygosity for H63D typically experience either mild or no detectable symptoms.1,3
There can be mutations in other genes involved in iron metabolism, but these represent more rare forms of hemochromatosis. Conditions such as thalassemia, sideroblastic anemia, porphyria cutanea tarda, and chronic liver disease may also be associated with iron overload.1,3
HH is most common in white populations of northern European descent. Multiple factors—including dietary iron intake, alcohol consumption, blood donation, blood loss associated with menstruation, and pregnancy—affect the expression of clinical features of hemochromatosis. Men are 24 times more likely than women to express clinical features of hemochromatosis.2 Approximately 70% of affected patients develop symptoms between ages 40 and 60.1
CLINICAL MANIFESTATIONS
The liver is typically the first organ affected, and hepatomegaly is present in 95% of symptomatic patients, even in the presence of normal transaminase levels.1 A bronzed, metallic, or slate gray skin coloration can occur due to increased iron deposition in the dermis. Arthralgias in the hands, wrists, hips, knees, and ankles are present in up to 50% of patients with hemochromatosis. Cardiac manifestations include restrictive cardiomyopathy, congestive heart failure, and arrhythmias.1
Iron deposition in the beta cells of the pancreas causes diabetes1 and in the pituitary causes hypogonadotropic hypogonadism in both men and women, resulting in decreased libido, amenorrhea, testicular atrophy, gynecomastia, and reduced body hair. Primary testicular dysfunction may occur due to iron deposition in the testicles.1,4 In the thyroid gland, iron deposition can lead to abnormal function. Secondary hypothyroidism is rare in the setting of iron overload, although iron deposition occasionally occurs in pituitary thyrotrophs (usually only to a mild degree). Adrenal insufficiency and hypoparathyroidism may also result from iron overload.5
CLINICAL STUDIES TO ASSESS IRON STORES
When assessing tissue iron stores, it is important to measure the serum iron level, total iron-binding capacity, and ferritin in the fasting state.2 This information can be used to calculate the percent transferrin saturation. If the serum ferritin is elevated (> 300 ng/mL in men and > 200 ng/mL in women) and/or the transferrin saturation is greater than 45%, referral to hematology or hepatology is recommended, along with genetic testing for hemochromatosis.1,2
Once the diagnosis of hemochromatosis has been confirmed, CT or MRI can be used to assess for increased density of the liver.1 Liver biopsy can determine the degree of fibrosis and is often considered in patients with more extreme elevations of serum ferritin levels and/or hepatomegaly. Liver biopsy is the only reliable method for determining whether hepatic cirrhosis, which increases risk for hepatocellular carcinoma, is present.1
TREATMENT
All patients with homozygous HH and evidence of iron overload require treatment, regardless of symptoms. Phlebotomy is the standard of care, due to its low cost and relative safety. Chelating agents are a second-line option when contraindications to phlebotomy (eg, anemia) exist.1,2
Alcohol consumption, especially in the presence of iron overload or liver disease, should be avoided, as it can increase risk for cirrhosis by nearly tenfold.1 Dietary modification is typically unnecessary, aside from the avoidance of iron and vitamin C supplementation.2 Patients should also eliminate raw shellfish from their diet, as they may carry bacteria that can cause potentially fatal infection (since high iron levels impair hepcidin bactericidal activity).2
The management of hepatic failure, cardiac failure, and diabetes in patients with HH is similar to conventional management of these conditions.1 With phlebotomy, the liver and spleen often decrease in size, liver function improves, skin pigmentation lightens, cardiac failure may be reversed, and diabetes control often improves.1,2 Testosterone levels may normalize after phlebotomy, especially if HH is diagnosed in the early stages. In more advanced cases, testosterone replacement therapy in combination with aggressive phlebotomy may be necessary.4
CONCLUSION
A high index of suspicion is required to diagnose hemochromatosis early. HH should be considered in the differential diagnosis for patients with hypogonadotropic hypogonadism, abnormal iron studies, elevated transaminase levels, and a family history of hemochromatosis.
Once the diagnosis is established, all first-degree relatives should be screened.1 Early therapy is crucial to prevent complications from iron overload.
REFERENCES
1. Powell LW. Hemochromatosis. In: Fauci AS, Braunwald E, Kasper DL, et al (eds). Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:2429-2433.
2. Crownover BK, Carlton JC. Hereditary hemochromatosis. Am Fam Phys. 2013;87(3):183-190.
3. Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717.
4. McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab. 2005;90(4):2451-2455.
5. Hudec M, Grigerova M, Walsh CH. Secondary hypothyroidism in hereditary hemochromatosis: recovery after iron depletion. Thyroid. 2008;18(2):255-257.
JR, a 34-year-old Caucasian man, was in his normal state of good health until several months ago, when he developed fatigue, low libido, and insomnia. He reports normal erectile function, adding that he fathered a child at age 24. His medical history and remaining review of systems are negative. Physical exam is unremarkable. His BMI is 23.
Labwork reveals low free and total testosterone levels with low FSH and LH levels. Thyroid-stimulating hormone, free T4, and prolactin levels are within normal range, comprehensive metabolic panel is unremarkable, and pituitary MRI is negative. The complete blood count reveals slightly elevated hemoglobin and hematocrit, prompting ordering of iron studies that reveal elevated ferritin and serum iron levels and elevated percent transferrin saturation. Lab values are shown in the table.
Based on his elevated ferritin and transferrin saturation levels, JR undergoes genetic testing for hereditary hemochromatosis (HH) with C282Y and H63D mutation analysis. He is found to have the homozygous C282Y genotype (C282Y/C282Y) for HH.
JR establishes care with a hematologist and is advised to receive therapeutic phlebotomy until his ferritin level is between 10 and 50 ng/mL. An abdominal ultrasound, ordered to screen for hepatomegaly, yields normal results. JR elects not to receive testosterone replacement therapy.
Three months later, labwork reveals a free testosterone level of 120 pg/mL with normal hemoglobin and hematocrit levels, normal transaminases, a ferritin level of 28 ng/mL, and a percent transferrin saturation of 31%. Additional values are shown in the table.
BACKGROUND AND GENETICS
Hereditary hemochromatosis is an autosomal recessive iron storage disorder in which intestinal iron absorption is markedly increased. This results in iron overload and excessive iron deposition in numerous tissues, glands, and organs.1
In patients with HH, a genetic defect causes abnormal expression of the HFE protein that regulates hepcidin production. Hepcidin is an iron regulatory hormone, secreted by hepatocytes, that decreases intestinal iron absorption in response to excess iron.2 Ninety percent of individuals affected by HH are homozygous for mutation at amino acid position 282 on the HFE gene, which causes an inappropriate decrease in hepcidin expression in response to elevated iron levels.2,3 Only 10% of individuals homozygous for the C282Y mutation actually develop clinically apparent end-organ damage.2
Being a carrier (heterozygous) for the C282Y mutation confers significantly lower risk for iron overload. The second most common mutation in the HFE gene, H63D, is associated with a milder phenotype. Those with compound heterozygosity for C282Y/H63D or homozygosity for H63D typically experience either mild or no detectable symptoms.1,3
There can be mutations in other genes involved in iron metabolism, but these represent more rare forms of hemochromatosis. Conditions such as thalassemia, sideroblastic anemia, porphyria cutanea tarda, and chronic liver disease may also be associated with iron overload.1,3
HH is most common in white populations of northern European descent. Multiple factors—including dietary iron intake, alcohol consumption, blood donation, blood loss associated with menstruation, and pregnancy—affect the expression of clinical features of hemochromatosis. Men are 24 times more likely than women to express clinical features of hemochromatosis.2 Approximately 70% of affected patients develop symptoms between ages 40 and 60.1
CLINICAL MANIFESTATIONS
The liver is typically the first organ affected, and hepatomegaly is present in 95% of symptomatic patients, even in the presence of normal transaminase levels.1 A bronzed, metallic, or slate gray skin coloration can occur due to increased iron deposition in the dermis. Arthralgias in the hands, wrists, hips, knees, and ankles are present in up to 50% of patients with hemochromatosis. Cardiac manifestations include restrictive cardiomyopathy, congestive heart failure, and arrhythmias.1
Iron deposition in the beta cells of the pancreas causes diabetes1 and in the pituitary causes hypogonadotropic hypogonadism in both men and women, resulting in decreased libido, amenorrhea, testicular atrophy, gynecomastia, and reduced body hair. Primary testicular dysfunction may occur due to iron deposition in the testicles.1,4 In the thyroid gland, iron deposition can lead to abnormal function. Secondary hypothyroidism is rare in the setting of iron overload, although iron deposition occasionally occurs in pituitary thyrotrophs (usually only to a mild degree). Adrenal insufficiency and hypoparathyroidism may also result from iron overload.5
CLINICAL STUDIES TO ASSESS IRON STORES
When assessing tissue iron stores, it is important to measure the serum iron level, total iron-binding capacity, and ferritin in the fasting state.2 This information can be used to calculate the percent transferrin saturation. If the serum ferritin is elevated (> 300 ng/mL in men and > 200 ng/mL in women) and/or the transferrin saturation is greater than 45%, referral to hematology or hepatology is recommended, along with genetic testing for hemochromatosis.1,2
Once the diagnosis of hemochromatosis has been confirmed, CT or MRI can be used to assess for increased density of the liver.1 Liver biopsy can determine the degree of fibrosis and is often considered in patients with more extreme elevations of serum ferritin levels and/or hepatomegaly. Liver biopsy is the only reliable method for determining whether hepatic cirrhosis, which increases risk for hepatocellular carcinoma, is present.1
TREATMENT
All patients with homozygous HH and evidence of iron overload require treatment, regardless of symptoms. Phlebotomy is the standard of care, due to its low cost and relative safety. Chelating agents are a second-line option when contraindications to phlebotomy (eg, anemia) exist.1,2
Alcohol consumption, especially in the presence of iron overload or liver disease, should be avoided, as it can increase risk for cirrhosis by nearly tenfold.1 Dietary modification is typically unnecessary, aside from the avoidance of iron and vitamin C supplementation.2 Patients should also eliminate raw shellfish from their diet, as they may carry bacteria that can cause potentially fatal infection (since high iron levels impair hepcidin bactericidal activity).2
The management of hepatic failure, cardiac failure, and diabetes in patients with HH is similar to conventional management of these conditions.1 With phlebotomy, the liver and spleen often decrease in size, liver function improves, skin pigmentation lightens, cardiac failure may be reversed, and diabetes control often improves.1,2 Testosterone levels may normalize after phlebotomy, especially if HH is diagnosed in the early stages. In more advanced cases, testosterone replacement therapy in combination with aggressive phlebotomy may be necessary.4
CONCLUSION
A high index of suspicion is required to diagnose hemochromatosis early. HH should be considered in the differential diagnosis for patients with hypogonadotropic hypogonadism, abnormal iron studies, elevated transaminase levels, and a family history of hemochromatosis.
Once the diagnosis is established, all first-degree relatives should be screened.1 Early therapy is crucial to prevent complications from iron overload.
REFERENCES
1. Powell LW. Hemochromatosis. In: Fauci AS, Braunwald E, Kasper DL, et al (eds). Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:2429-2433.
2. Crownover BK, Carlton JC. Hereditary hemochromatosis. Am Fam Phys. 2013;87(3):183-190.
3. Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106(12):3710-3717.
4. McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab. 2005;90(4):2451-2455.
5. Hudec M, Grigerova M, Walsh CH. Secondary hypothyroidism in hereditary hemochromatosis: recovery after iron depletion. Thyroid. 2008;18(2):255-257.
Managing Gestational Diabetes: Let’s Nip It in The Bud
One of the most common complications of pregnancy is gestational diabetes mellitus (GDM). It is defined as glucose intolerance with first onset during pregnancy.1 In 2011, the incidence of GDM in the United States was between 2% and 10% of all pregnancies. Potential complications associated with GDM include macrosomia, pre-eclampsia, preterm birth, increased risk for cesarean section, neonatal hypoglycemia, shoulder dystocia, and polyhydramnios. Women with a history of gestational diabetes have a 35% to 60% likelihood of developing type 2 diabetes over the following 10 to 20 years.2
Q: When should screening for GDM occur?
According to the American Diabetes Association’s (ADA) 2012 Clinical Practice Recommendations, a pregnant woman should be screened for undiagnosed type 2 diabetes at her first prenatal visit if she has certain risk factors.3 These include, but are not limited to, family history of diabetes, overweight/obesity, sedentary lifestyle, elevated blood pressure and/or cholesterol, impaired fasting glucose or impaired glucose tolerance, or certain ethnic backgrounds (eg, Hispanic, Native American, and non-Hispanic black).4 In 2011, the ADA revised its recommendations for GDM screening and diagnosis to be in accordance with those from the International Association of Diabetes and Pregnancy Study Groups (IADPSG), an international consensus group with representatives from multiple obstetric and diabetes organizations, including ADA.
Q: How is GDM diagnosed?
Current recommendations stipulate that women with no previous history of diabetes or prediabetes undergo one-step testing: a 75-g glucose tolerance test (GTT) at 24 to 28 weeks’ gestation.5,6 For women with a prior history of GDM, screening is recommended earlier in the pregnancy. The GTT should be performed after an overnight fast of at least eight hours.3 An elevation of any one of the values above normal reference range is consistent with the diagnosis of GDM. (Previously, the diagnostic criteria required two abnormal values.) Multiple international studies using the new criteria have estimated an increased incidence of gestational diabetes in up to 18% of pregnancies.5,6
Some organizations have not endorsed the IADPSG/ADA diagnostic criteria at this time; as a result, many practitioners continue to use two-step testing for diagnosing GDM. To do the two-step testing, a 50-g glucose load is given, followed by a blood glucose reading one hour later. If the one-hour reading is within normal range, no further testing is warranted and the patient does not have gestational diabetes. If the test is abnormal, she must undergo a fasting three-hour GTT using a 100-g glucose load.
Q: What advice should a woman get once she’s diagnosed with GDM?
As soon as a woman is diagnosed with GDM, she should be referred for a gestational diabetes education class and nutrition counseling. Specifically, she should learn what it means for her to have GDM, implications for her and her baby, and the importance of eating a healthy diet (not the proverbial concept of “eating for two”), physical activity, self-monitoring blood glucose, and adherence to any prescribed medications.
Probably the most important aspect of education is nutrition counseling. It is known that smaller meals consumed more frequently throughout the day reduce spikes in blood glucose levels. One suggestion is to eat three small meals and three low-carbohydrate (15 g) snacks each day. Meals and snacks are generally established based on fixed carbohydrate amounts. A certified diabetes educator or registered dietitian (RD) can recommend healthy meal and snack ideas that are tasty, promote satiety, and minimize spikes in glucose levels.
Q: What are the current treatment options for GDM?
During the process of receiving GDM education, the patient should be prescribed a glucometer, along with specific glucose targets. Blood glucose should be checked multiple times a day, preferably fasting and postprandial measurements. Medical practices vary in their preferred glucose targets; some individuals require tighter control than others. The ADA suggests the following targets:
• Before a meal (preprandial):
95 mg/dL or less.
• One hour after a meal (postprandial): 140 mg/dL or less.
• Two hours after a meal (postprandial): 120 mg/dL or less. 7
If blood glucose levels remain within normal range, it is possible to control gestational diabetes with dietary modification and physical activity. If readings are consistently elevated, then the patient must be started on medication. There are currently no FDA-approved oral medications to treat gestational diabetes. Glyburide is commonly used, although it is not FDA approved for this indication. More studies to establish its safety are likely needed for FDA approval.8
If pharmaceutical treatment is warranted, insulin is the safest and most effective agent. It is the only medication that is FDA approved for treatment of GDM. Levemir (insulin detemir [rDNA origin] injection) gained FDA approval for use in pregnancy in 2012, so it has become more widespread than NPH for basal insulin usage.9
Although it is usually managed by an endocrinologist or perinatologist, an experienced obstetrician could also manage GDM. Often, the patient is referred to an endocrinologist. The endocrine provider, along with the diabetes educator and RD, focus on nutrition counseling and diabetes management so the obstetrician can focus on maternal and fetal health.
Q: What is the recommended follow-up?
Since embryonic and fetal development occurs at such a rapid rate, time is of the essence for getting a patient’s blood glucose to goal. While treating diabetes in general can be challenging, this is usually not the case with GDM. Most women with GDM are motivated to take care of themselves for the well-being of their developing baby. The influence of a baby developing inside a mother is so strong that diabetic women who become pregnant often take better care of themselves than they do when they are not pregnant.
The patient’s daily responsibilities should include eating a healthy and diet checking her blood glucose levels throughout the day. These readings must be recorded. Clinic visits should occur often, with emailing of glucose readings between visits as needed. The frequency of visits varies among practices, depending on the patient’s level of glucose control and intensity of the treatment regimen.
Q: Why is postpartum testing important?
After delivery, most cases of GDM usually resolve. However, approximately 5% to 10% of women with gestational diabetes are found to have diabetes immediately after pregnancy.2 To evaluate for persistent diabetes, a two-hour GTT should be done at six weeks’ postpartum. Although an A1C can now be used to diagnose diabetes, the ADA does not recommend checking it for this purpose.3
If the two-hour GTT result is normal, a woman should be screened for diabetes every three years for the rest of her life.3 If a diagnosis of impaired fasting glucose or impaired glucose tolerance is made, then she should be tested for diabetes on an annual basis or in the interim if she develops classic symptoms of hyperglycemia.3 If diabetes is diagnosed, she should be treated accordingly as a type 2 diabetic patient.
At this time, the patient should be counseled on lifestyle interventions and consider starting metformin therapy if appropriate. Diabetes education classes are available for prediabetes. To maintain good health and prevent/delay onset of type 2 diabetes, here are some tips to follow:
• The same diet as during pregnancy does not have to be followed, although healthy eating habits are always a good idea.
• Physical activity (approximately 30 min five times a week) will help shed weight gained during pregnancy.
• Breastfeeding promotes weight loss.10
• Patients should aim for weight loss of 7% of body weight.3
• Continue annual physical exams, keeping an eye on blood pressure, weight, and cholesterol levels.
It’s reasonable for the patient to check glucose levels occasionally after delivery. If elevated readings occur, the patient can make an appointment with her primary care provider or endocrinologist.
References
1. American Association for Clinical Chemistry. A New Definition of Gestational Diabetes. www.aacc.org/publications/cln/2010/may/Pages/CoverStory2May2010.aspx. Accessed June 30, 2013.
2. National Diabetes Statistics, 2011. www.diabetes.niddk.nih.gov/dm/pubs/statistics/#Gestational. Accessed July 22, 2013.
3. American Diabetes Association. 2012 Clinical Practice Recommendations. Diabetes Care. 2012;35(suppl 1). http://professional.diabetes.org/SlideLibrary/media/4839/ADA%20Standards%20of%20Medical%20Care%202012%20FINAL.ppt. Accessed June 24, 2013.
4. American Diabetes Association. Diabetes basics: your risk. www.diabetes.org/diabetes-basics/prevention/risk-factors. Accessed August 13, 2013.
5. American Diabetes Association. Diabetes Basics: What is Gestational Diabetes? www.diabetes.org/diabetes-basics/gestational/what-is-gestational-diabetes.html. Accessed August 13, 2013.
6. Johnson K. New criteria for gestational diabetes increase diagnoses (December 5, 2011). www.medscape.com/viewarticle/754733. Accessed August 13, 2013.
7. American Diabetes Association. Diabetes basics: how to treat gestational diabetes. www.diabetes.org/diabetes-basics/gestational/how-to-treat-gestational.html. Accessed August 13, 2013.
8. Moore TR. Glyburide for the treatment of gestational diabetes: a critical appraisal. Diabetes Care. 2007;30(suppl 2). http://care.diabetesjournals.org/content/30/Supplement_2/S209.full. Accessed August 13, 2013.
9. Lowes R. Levemir assigned more reassuring pregnancy risk category (April 2, 2012). www.medscape.com/viewarticle/761349. Accessed August 13, 2013.
10. Buchanan TA, Xiang AH, Page KA. Gestational diabetes mellitus: risks and management during and after pregnancy. Nat Rev Endocrinol. 2012;8(11):639-649.
One of the most common complications of pregnancy is gestational diabetes mellitus (GDM). It is defined as glucose intolerance with first onset during pregnancy.1 In 2011, the incidence of GDM in the United States was between 2% and 10% of all pregnancies. Potential complications associated with GDM include macrosomia, pre-eclampsia, preterm birth, increased risk for cesarean section, neonatal hypoglycemia, shoulder dystocia, and polyhydramnios. Women with a history of gestational diabetes have a 35% to 60% likelihood of developing type 2 diabetes over the following 10 to 20 years.2
Q: When should screening for GDM occur?
According to the American Diabetes Association’s (ADA) 2012 Clinical Practice Recommendations, a pregnant woman should be screened for undiagnosed type 2 diabetes at her first prenatal visit if she has certain risk factors.3 These include, but are not limited to, family history of diabetes, overweight/obesity, sedentary lifestyle, elevated blood pressure and/or cholesterol, impaired fasting glucose or impaired glucose tolerance, or certain ethnic backgrounds (eg, Hispanic, Native American, and non-Hispanic black).4 In 2011, the ADA revised its recommendations for GDM screening and diagnosis to be in accordance with those from the International Association of Diabetes and Pregnancy Study Groups (IADPSG), an international consensus group with representatives from multiple obstetric and diabetes organizations, including ADA.
Q: How is GDM diagnosed?
Current recommendations stipulate that women with no previous history of diabetes or prediabetes undergo one-step testing: a 75-g glucose tolerance test (GTT) at 24 to 28 weeks’ gestation.5,6 For women with a prior history of GDM, screening is recommended earlier in the pregnancy. The GTT should be performed after an overnight fast of at least eight hours.3 An elevation of any one of the values above normal reference range is consistent with the diagnosis of GDM. (Previously, the diagnostic criteria required two abnormal values.) Multiple international studies using the new criteria have estimated an increased incidence of gestational diabetes in up to 18% of pregnancies.5,6
Some organizations have not endorsed the IADPSG/ADA diagnostic criteria at this time; as a result, many practitioners continue to use two-step testing for diagnosing GDM. To do the two-step testing, a 50-g glucose load is given, followed by a blood glucose reading one hour later. If the one-hour reading is within normal range, no further testing is warranted and the patient does not have gestational diabetes. If the test is abnormal, she must undergo a fasting three-hour GTT using a 100-g glucose load.
Q: What advice should a woman get once she’s diagnosed with GDM?
As soon as a woman is diagnosed with GDM, she should be referred for a gestational diabetes education class and nutrition counseling. Specifically, she should learn what it means for her to have GDM, implications for her and her baby, and the importance of eating a healthy diet (not the proverbial concept of “eating for two”), physical activity, self-monitoring blood glucose, and adherence to any prescribed medications.
Probably the most important aspect of education is nutrition counseling. It is known that smaller meals consumed more frequently throughout the day reduce spikes in blood glucose levels. One suggestion is to eat three small meals and three low-carbohydrate (15 g) snacks each day. Meals and snacks are generally established based on fixed carbohydrate amounts. A certified diabetes educator or registered dietitian (RD) can recommend healthy meal and snack ideas that are tasty, promote satiety, and minimize spikes in glucose levels.
Q: What are the current treatment options for GDM?
During the process of receiving GDM education, the patient should be prescribed a glucometer, along with specific glucose targets. Blood glucose should be checked multiple times a day, preferably fasting and postprandial measurements. Medical practices vary in their preferred glucose targets; some individuals require tighter control than others. The ADA suggests the following targets:
• Before a meal (preprandial):
95 mg/dL or less.
• One hour after a meal (postprandial): 140 mg/dL or less.
• Two hours after a meal (postprandial): 120 mg/dL or less. 7
If blood glucose levels remain within normal range, it is possible to control gestational diabetes with dietary modification and physical activity. If readings are consistently elevated, then the patient must be started on medication. There are currently no FDA-approved oral medications to treat gestational diabetes. Glyburide is commonly used, although it is not FDA approved for this indication. More studies to establish its safety are likely needed for FDA approval.8
If pharmaceutical treatment is warranted, insulin is the safest and most effective agent. It is the only medication that is FDA approved for treatment of GDM. Levemir (insulin detemir [rDNA origin] injection) gained FDA approval for use in pregnancy in 2012, so it has become more widespread than NPH for basal insulin usage.9
Although it is usually managed by an endocrinologist or perinatologist, an experienced obstetrician could also manage GDM. Often, the patient is referred to an endocrinologist. The endocrine provider, along with the diabetes educator and RD, focus on nutrition counseling and diabetes management so the obstetrician can focus on maternal and fetal health.
Q: What is the recommended follow-up?
Since embryonic and fetal development occurs at such a rapid rate, time is of the essence for getting a patient’s blood glucose to goal. While treating diabetes in general can be challenging, this is usually not the case with GDM. Most women with GDM are motivated to take care of themselves for the well-being of their developing baby. The influence of a baby developing inside a mother is so strong that diabetic women who become pregnant often take better care of themselves than they do when they are not pregnant.
The patient’s daily responsibilities should include eating a healthy and diet checking her blood glucose levels throughout the day. These readings must be recorded. Clinic visits should occur often, with emailing of glucose readings between visits as needed. The frequency of visits varies among practices, depending on the patient’s level of glucose control and intensity of the treatment regimen.
Q: Why is postpartum testing important?
After delivery, most cases of GDM usually resolve. However, approximately 5% to 10% of women with gestational diabetes are found to have diabetes immediately after pregnancy.2 To evaluate for persistent diabetes, a two-hour GTT should be done at six weeks’ postpartum. Although an A1C can now be used to diagnose diabetes, the ADA does not recommend checking it for this purpose.3
If the two-hour GTT result is normal, a woman should be screened for diabetes every three years for the rest of her life.3 If a diagnosis of impaired fasting glucose or impaired glucose tolerance is made, then she should be tested for diabetes on an annual basis or in the interim if she develops classic symptoms of hyperglycemia.3 If diabetes is diagnosed, she should be treated accordingly as a type 2 diabetic patient.
At this time, the patient should be counseled on lifestyle interventions and consider starting metformin therapy if appropriate. Diabetes education classes are available for prediabetes. To maintain good health and prevent/delay onset of type 2 diabetes, here are some tips to follow:
• The same diet as during pregnancy does not have to be followed, although healthy eating habits are always a good idea.
• Physical activity (approximately 30 min five times a week) will help shed weight gained during pregnancy.
• Breastfeeding promotes weight loss.10
• Patients should aim for weight loss of 7% of body weight.3
• Continue annual physical exams, keeping an eye on blood pressure, weight, and cholesterol levels.
It’s reasonable for the patient to check glucose levels occasionally after delivery. If elevated readings occur, the patient can make an appointment with her primary care provider or endocrinologist.
References
1. American Association for Clinical Chemistry. A New Definition of Gestational Diabetes. www.aacc.org/publications/cln/2010/may/Pages/CoverStory2May2010.aspx. Accessed June 30, 2013.
2. National Diabetes Statistics, 2011. www.diabetes.niddk.nih.gov/dm/pubs/statistics/#Gestational. Accessed July 22, 2013.
3. American Diabetes Association. 2012 Clinical Practice Recommendations. Diabetes Care. 2012;35(suppl 1). http://professional.diabetes.org/SlideLibrary/media/4839/ADA%20Standards%20of%20Medical%20Care%202012%20FINAL.ppt. Accessed June 24, 2013.
4. American Diabetes Association. Diabetes basics: your risk. www.diabetes.org/diabetes-basics/prevention/risk-factors. Accessed August 13, 2013.
5. American Diabetes Association. Diabetes Basics: What is Gestational Diabetes? www.diabetes.org/diabetes-basics/gestational/what-is-gestational-diabetes.html. Accessed August 13, 2013.
6. Johnson K. New criteria for gestational diabetes increase diagnoses (December 5, 2011). www.medscape.com/viewarticle/754733. Accessed August 13, 2013.
7. American Diabetes Association. Diabetes basics: how to treat gestational diabetes. www.diabetes.org/diabetes-basics/gestational/how-to-treat-gestational.html. Accessed August 13, 2013.
8. Moore TR. Glyburide for the treatment of gestational diabetes: a critical appraisal. Diabetes Care. 2007;30(suppl 2). http://care.diabetesjournals.org/content/30/Supplement_2/S209.full. Accessed August 13, 2013.
9. Lowes R. Levemir assigned more reassuring pregnancy risk category (April 2, 2012). www.medscape.com/viewarticle/761349. Accessed August 13, 2013.
10. Buchanan TA, Xiang AH, Page KA. Gestational diabetes mellitus: risks and management during and after pregnancy. Nat Rev Endocrinol. 2012;8(11):639-649.
One of the most common complications of pregnancy is gestational diabetes mellitus (GDM). It is defined as glucose intolerance with first onset during pregnancy.1 In 2011, the incidence of GDM in the United States was between 2% and 10% of all pregnancies. Potential complications associated with GDM include macrosomia, pre-eclampsia, preterm birth, increased risk for cesarean section, neonatal hypoglycemia, shoulder dystocia, and polyhydramnios. Women with a history of gestational diabetes have a 35% to 60% likelihood of developing type 2 diabetes over the following 10 to 20 years.2
Q: When should screening for GDM occur?
According to the American Diabetes Association’s (ADA) 2012 Clinical Practice Recommendations, a pregnant woman should be screened for undiagnosed type 2 diabetes at her first prenatal visit if she has certain risk factors.3 These include, but are not limited to, family history of diabetes, overweight/obesity, sedentary lifestyle, elevated blood pressure and/or cholesterol, impaired fasting glucose or impaired glucose tolerance, or certain ethnic backgrounds (eg, Hispanic, Native American, and non-Hispanic black).4 In 2011, the ADA revised its recommendations for GDM screening and diagnosis to be in accordance with those from the International Association of Diabetes and Pregnancy Study Groups (IADPSG), an international consensus group with representatives from multiple obstetric and diabetes organizations, including ADA.
Q: How is GDM diagnosed?
Current recommendations stipulate that women with no previous history of diabetes or prediabetes undergo one-step testing: a 75-g glucose tolerance test (GTT) at 24 to 28 weeks’ gestation.5,6 For women with a prior history of GDM, screening is recommended earlier in the pregnancy. The GTT should be performed after an overnight fast of at least eight hours.3 An elevation of any one of the values above normal reference range is consistent with the diagnosis of GDM. (Previously, the diagnostic criteria required two abnormal values.) Multiple international studies using the new criteria have estimated an increased incidence of gestational diabetes in up to 18% of pregnancies.5,6
Some organizations have not endorsed the IADPSG/ADA diagnostic criteria at this time; as a result, many practitioners continue to use two-step testing for diagnosing GDM. To do the two-step testing, a 50-g glucose load is given, followed by a blood glucose reading one hour later. If the one-hour reading is within normal range, no further testing is warranted and the patient does not have gestational diabetes. If the test is abnormal, she must undergo a fasting three-hour GTT using a 100-g glucose load.
Q: What advice should a woman get once she’s diagnosed with GDM?
As soon as a woman is diagnosed with GDM, she should be referred for a gestational diabetes education class and nutrition counseling. Specifically, she should learn what it means for her to have GDM, implications for her and her baby, and the importance of eating a healthy diet (not the proverbial concept of “eating for two”), physical activity, self-monitoring blood glucose, and adherence to any prescribed medications.
Probably the most important aspect of education is nutrition counseling. It is known that smaller meals consumed more frequently throughout the day reduce spikes in blood glucose levels. One suggestion is to eat three small meals and three low-carbohydrate (15 g) snacks each day. Meals and snacks are generally established based on fixed carbohydrate amounts. A certified diabetes educator or registered dietitian (RD) can recommend healthy meal and snack ideas that are tasty, promote satiety, and minimize spikes in glucose levels.
Q: What are the current treatment options for GDM?
During the process of receiving GDM education, the patient should be prescribed a glucometer, along with specific glucose targets. Blood glucose should be checked multiple times a day, preferably fasting and postprandial measurements. Medical practices vary in their preferred glucose targets; some individuals require tighter control than others. The ADA suggests the following targets:
• Before a meal (preprandial):
95 mg/dL or less.
• One hour after a meal (postprandial): 140 mg/dL or less.
• Two hours after a meal (postprandial): 120 mg/dL or less. 7
If blood glucose levels remain within normal range, it is possible to control gestational diabetes with dietary modification and physical activity. If readings are consistently elevated, then the patient must be started on medication. There are currently no FDA-approved oral medications to treat gestational diabetes. Glyburide is commonly used, although it is not FDA approved for this indication. More studies to establish its safety are likely needed for FDA approval.8
If pharmaceutical treatment is warranted, insulin is the safest and most effective agent. It is the only medication that is FDA approved for treatment of GDM. Levemir (insulin detemir [rDNA origin] injection) gained FDA approval for use in pregnancy in 2012, so it has become more widespread than NPH for basal insulin usage.9
Although it is usually managed by an endocrinologist or perinatologist, an experienced obstetrician could also manage GDM. Often, the patient is referred to an endocrinologist. The endocrine provider, along with the diabetes educator and RD, focus on nutrition counseling and diabetes management so the obstetrician can focus on maternal and fetal health.
Q: What is the recommended follow-up?
Since embryonic and fetal development occurs at such a rapid rate, time is of the essence for getting a patient’s blood glucose to goal. While treating diabetes in general can be challenging, this is usually not the case with GDM. Most women with GDM are motivated to take care of themselves for the well-being of their developing baby. The influence of a baby developing inside a mother is so strong that diabetic women who become pregnant often take better care of themselves than they do when they are not pregnant.
The patient’s daily responsibilities should include eating a healthy and diet checking her blood glucose levels throughout the day. These readings must be recorded. Clinic visits should occur often, with emailing of glucose readings between visits as needed. The frequency of visits varies among practices, depending on the patient’s level of glucose control and intensity of the treatment regimen.
Q: Why is postpartum testing important?
After delivery, most cases of GDM usually resolve. However, approximately 5% to 10% of women with gestational diabetes are found to have diabetes immediately after pregnancy.2 To evaluate for persistent diabetes, a two-hour GTT should be done at six weeks’ postpartum. Although an A1C can now be used to diagnose diabetes, the ADA does not recommend checking it for this purpose.3
If the two-hour GTT result is normal, a woman should be screened for diabetes every three years for the rest of her life.3 If a diagnosis of impaired fasting glucose or impaired glucose tolerance is made, then she should be tested for diabetes on an annual basis or in the interim if she develops classic symptoms of hyperglycemia.3 If diabetes is diagnosed, she should be treated accordingly as a type 2 diabetic patient.
At this time, the patient should be counseled on lifestyle interventions and consider starting metformin therapy if appropriate. Diabetes education classes are available for prediabetes. To maintain good health and prevent/delay onset of type 2 diabetes, here are some tips to follow:
• The same diet as during pregnancy does not have to be followed, although healthy eating habits are always a good idea.
• Physical activity (approximately 30 min five times a week) will help shed weight gained during pregnancy.
• Breastfeeding promotes weight loss.10
• Patients should aim for weight loss of 7% of body weight.3
• Continue annual physical exams, keeping an eye on blood pressure, weight, and cholesterol levels.
It’s reasonable for the patient to check glucose levels occasionally after delivery. If elevated readings occur, the patient can make an appointment with her primary care provider or endocrinologist.
References
1. American Association for Clinical Chemistry. A New Definition of Gestational Diabetes. www.aacc.org/publications/cln/2010/may/Pages/CoverStory2May2010.aspx. Accessed June 30, 2013.
2. National Diabetes Statistics, 2011. www.diabetes.niddk.nih.gov/dm/pubs/statistics/#Gestational. Accessed July 22, 2013.
3. American Diabetes Association. 2012 Clinical Practice Recommendations. Diabetes Care. 2012;35(suppl 1). http://professional.diabetes.org/SlideLibrary/media/4839/ADA%20Standards%20of%20Medical%20Care%202012%20FINAL.ppt. Accessed June 24, 2013.
4. American Diabetes Association. Diabetes basics: your risk. www.diabetes.org/diabetes-basics/prevention/risk-factors. Accessed August 13, 2013.
5. American Diabetes Association. Diabetes Basics: What is Gestational Diabetes? www.diabetes.org/diabetes-basics/gestational/what-is-gestational-diabetes.html. Accessed August 13, 2013.
6. Johnson K. New criteria for gestational diabetes increase diagnoses (December 5, 2011). www.medscape.com/viewarticle/754733. Accessed August 13, 2013.
7. American Diabetes Association. Diabetes basics: how to treat gestational diabetes. www.diabetes.org/diabetes-basics/gestational/how-to-treat-gestational.html. Accessed August 13, 2013.
8. Moore TR. Glyburide for the treatment of gestational diabetes: a critical appraisal. Diabetes Care. 2007;30(suppl 2). http://care.diabetesjournals.org/content/30/Supplement_2/S209.full. Accessed August 13, 2013.
9. Lowes R. Levemir assigned more reassuring pregnancy risk category (April 2, 2012). www.medscape.com/viewarticle/761349. Accessed August 13, 2013.
10. Buchanan TA, Xiang AH, Page KA. Gestational diabetes mellitus: risks and management during and after pregnancy. Nat Rev Endocrinol. 2012;8(11):639-649.
Underlying Factors Influence Insulin's Effect
Q: Help! How do you proceed if, after you’ve continually increased a patient’s insulin dose, his/her blood glucose levels do not improve?
This is a common scenario in diabetes management. Here are nine things to consider when a patient’s situation just doesn’t make sense clinically:
1. Noncompliance with the prescribed dose. This is the most common scenario. Ask the patient, “How many injections do you miss in a typical week?” Assure that he or she is actually taking the currently prescribed amount of insulin before you further increase the dose.
2. Inaccurate insulin dosing. This problem can be due to impaired vision, poor technique, dexterity issues, or dementia. Ask the patient to demonstrate for you how he/she draws up and takes the insulin at home. You might just be surprised at what you see, even in patients who have been giving themselves insulin for years. Consider prescribing an insulin pen or having a family member or significant other dose the insulin if the patient is no longer reliable to accurately dose it for him- or herself.
3. “Bad insulin.” What this actually means is loss of potency. This can be caused by improper storage, exposure to heat or cold, or use of an insulin delivery device (ie, vial or pen) past the 28- to 45-day period recommended, depending on the type of insulin. Replace the vial or pen and re-assess for improvement in diabetes control.
4. Lipohypertrophy of injection sites due to overuse. Palpate and visually inspect injection sites to look for firm or hypertrophied tissue. Advise the patient to avoid these areas for future injection, as absorption from these sites can be poor and unpredictable.
5. Dietary issues. The patient may be increasing his/her food intake along with the increased insulin doses. One clue that should raise suspicion for this occurrence is rapidly increasing body weight. Consider referring the patient to a dietitian for nutrition counseling.
6. New medication. Sometimes a new treatment is added to a patient’s regimen by another provider, and the medication might have an adverse effect on blood glucose control. Common examples include steroids (typically a cortisone injection) or methylprednisolone dose-packs taken during an asthma flare.
7. Occult infection. Urinary tract infections, pneumonia, and the like can impact blood glucose control. Consider ordering a urinalysis and complete blood count if infection seems a likely cause.
8. Major life stressors. Inquire as to what is happening in the patient’s life that might impact his/her body’s response to insulin. They might be in the middle of a divorce or other family crisis or experiencing severe stress at work.
9. Technique and equipment issues. Inaccurate glucose monitoring technique or use of expired strips can lead to “false high” readings. Also, patients with a continuous glucose monitor may record false high results when they are taking acetaminophen. If this is the case, increasing the insulin dose will often result in hypoglycemia.
It may be helpful to keep this clinical checklist handy and add to it any other issues that you come across when the clinical picture doesn’t make sense. You may also want to consider referral to a diabetes educator; patients will often confide what is really going on to an educator in a longer visit, rather than in the typically shorter visits with their health care provider.
SUGGESTED READING
Sadler C, Einhorn D. Tailoring insulin regimens for type 2 diabetes mellitus. JAAPA. 1998;11(4):55-71.
Q: Help! How do you proceed if, after you’ve continually increased a patient’s insulin dose, his/her blood glucose levels do not improve?
This is a common scenario in diabetes management. Here are nine things to consider when a patient’s situation just doesn’t make sense clinically:
1. Noncompliance with the prescribed dose. This is the most common scenario. Ask the patient, “How many injections do you miss in a typical week?” Assure that he or she is actually taking the currently prescribed amount of insulin before you further increase the dose.
2. Inaccurate insulin dosing. This problem can be due to impaired vision, poor technique, dexterity issues, or dementia. Ask the patient to demonstrate for you how he/she draws up and takes the insulin at home. You might just be surprised at what you see, even in patients who have been giving themselves insulin for years. Consider prescribing an insulin pen or having a family member or significant other dose the insulin if the patient is no longer reliable to accurately dose it for him- or herself.
3. “Bad insulin.” What this actually means is loss of potency. This can be caused by improper storage, exposure to heat or cold, or use of an insulin delivery device (ie, vial or pen) past the 28- to 45-day period recommended, depending on the type of insulin. Replace the vial or pen and re-assess for improvement in diabetes control.
4. Lipohypertrophy of injection sites due to overuse. Palpate and visually inspect injection sites to look for firm or hypertrophied tissue. Advise the patient to avoid these areas for future injection, as absorption from these sites can be poor and unpredictable.
5. Dietary issues. The patient may be increasing his/her food intake along with the increased insulin doses. One clue that should raise suspicion for this occurrence is rapidly increasing body weight. Consider referring the patient to a dietitian for nutrition counseling.
6. New medication. Sometimes a new treatment is added to a patient’s regimen by another provider, and the medication might have an adverse effect on blood glucose control. Common examples include steroids (typically a cortisone injection) or methylprednisolone dose-packs taken during an asthma flare.
7. Occult infection. Urinary tract infections, pneumonia, and the like can impact blood glucose control. Consider ordering a urinalysis and complete blood count if infection seems a likely cause.
8. Major life stressors. Inquire as to what is happening in the patient’s life that might impact his/her body’s response to insulin. They might be in the middle of a divorce or other family crisis or experiencing severe stress at work.
9. Technique and equipment issues. Inaccurate glucose monitoring technique or use of expired strips can lead to “false high” readings. Also, patients with a continuous glucose monitor may record false high results when they are taking acetaminophen. If this is the case, increasing the insulin dose will often result in hypoglycemia.
It may be helpful to keep this clinical checklist handy and add to it any other issues that you come across when the clinical picture doesn’t make sense. You may also want to consider referral to a diabetes educator; patients will often confide what is really going on to an educator in a longer visit, rather than in the typically shorter visits with their health care provider.
SUGGESTED READING
Sadler C, Einhorn D. Tailoring insulin regimens for type 2 diabetes mellitus. JAAPA. 1998;11(4):55-71.
Q: Help! How do you proceed if, after you’ve continually increased a patient’s insulin dose, his/her blood glucose levels do not improve?
This is a common scenario in diabetes management. Here are nine things to consider when a patient’s situation just doesn’t make sense clinically:
1. Noncompliance with the prescribed dose. This is the most common scenario. Ask the patient, “How many injections do you miss in a typical week?” Assure that he or she is actually taking the currently prescribed amount of insulin before you further increase the dose.
2. Inaccurate insulin dosing. This problem can be due to impaired vision, poor technique, dexterity issues, or dementia. Ask the patient to demonstrate for you how he/she draws up and takes the insulin at home. You might just be surprised at what you see, even in patients who have been giving themselves insulin for years. Consider prescribing an insulin pen or having a family member or significant other dose the insulin if the patient is no longer reliable to accurately dose it for him- or herself.
3. “Bad insulin.” What this actually means is loss of potency. This can be caused by improper storage, exposure to heat or cold, or use of an insulin delivery device (ie, vial or pen) past the 28- to 45-day period recommended, depending on the type of insulin. Replace the vial or pen and re-assess for improvement in diabetes control.
4. Lipohypertrophy of injection sites due to overuse. Palpate and visually inspect injection sites to look for firm or hypertrophied tissue. Advise the patient to avoid these areas for future injection, as absorption from these sites can be poor and unpredictable.
5. Dietary issues. The patient may be increasing his/her food intake along with the increased insulin doses. One clue that should raise suspicion for this occurrence is rapidly increasing body weight. Consider referring the patient to a dietitian for nutrition counseling.
6. New medication. Sometimes a new treatment is added to a patient’s regimen by another provider, and the medication might have an adverse effect on blood glucose control. Common examples include steroids (typically a cortisone injection) or methylprednisolone dose-packs taken during an asthma flare.
7. Occult infection. Urinary tract infections, pneumonia, and the like can impact blood glucose control. Consider ordering a urinalysis and complete blood count if infection seems a likely cause.
8. Major life stressors. Inquire as to what is happening in the patient’s life that might impact his/her body’s response to insulin. They might be in the middle of a divorce or other family crisis or experiencing severe stress at work.
9. Technique and equipment issues. Inaccurate glucose monitoring technique or use of expired strips can lead to “false high” readings. Also, patients with a continuous glucose monitor may record false high results when they are taking acetaminophen. If this is the case, increasing the insulin dose will often result in hypoglycemia.
It may be helpful to keep this clinical checklist handy and add to it any other issues that you come across when the clinical picture doesn’t make sense. You may also want to consider referral to a diabetes educator; patients will often confide what is really going on to an educator in a longer visit, rather than in the typically shorter visits with their health care provider.
SUGGESTED READING
Sadler C, Einhorn D. Tailoring insulin regimens for type 2 diabetes mellitus. JAAPA. 1998;11(4):55-71.
How to Handle "Incidentalomas"
Maggie, 42, presents to the emergency department with chronic intermittent abdominal pain and bloating with constipation and occasional diarrhea. She denies fever, chills, nausea, vomiting, melana, bright red blood per rectum, or changes in stool caliper, and she says she otherwise feels well.
Relevant lab and study results include: a comprehensive metabolic panel, complete blood count with differential, beta hCG (human chorionic gonadotropin), urinalysis, and amylase and lipase, all within normal limits; pregnancy test, negative; abdominal x-ray, within normal limits except increased stool in distal colon; and abdominal CT, 2.3-cm right adrenal mass and a Hounsfield measurement of 4 units.
Maggie has a right adrenal incidentaloma (incidentally discovered adenoma that was not in the differential diagnosis). Such findings have become all too often the case, due to the immediate access to and overutilization of high-resolution CT, MRI, and ultrasound. We are now seeing a significantly increased number of incidental adrenal lesions/masses discovered on images not intended to look for adrenal-related diseases (eg, Cushing syndrome, pheochromocytomas, and aldosterone-producing adenomas).
Q: How common are adrenal adenomas, and what must I consider?
Incidental adrenal adenomas are found on 4.4% of abdominal CTs, and in one autopsy series were discovered in 8.7%. Prevalence increases with age, with occurrence of < 1% in patients younger than 30 and about 7% for patients 70 or older.
Evaluation is based on two concerns: First, is the adrenal mass benign or malignant? Second, is the mass secretory or nonsecretory (non-hormone secreting) in nature?
The fortunate news about adrenal incidentalomas is that 80% are benign and nonsecretory, which provides immediate reassuring news to the patient. Examples of benign adrenal masses are: adenoma, lipoma, cyst, ganglioneuroma, hematoma, and infection (eg, tuberculosis, fungal).
The other encouraging statistic is that only 1:4,000 adrenal incidentalomas are malignant. Examples of malignant adrenal masses are: adrenocortical carcinoma, metastatic neoplasm, lymphoma, and malignant pheochromocytoma.
Q: Does adrenal adenoma size matter?
Yes, the larger the size of the adenoma, the higher the association with malignancy. The guide below (based on CT findings) shows not only malignancy potential as it relates to size, but also the importance of Hounsfield units and when surgical intervention is recommended.
Imaging (CT scan)
< 4 cm: homogeneous mass with smooth borders and < 10 Hounsfield units; suggests benign mass (likelihood of malignancy, about 2%)
4 to 6 cm: follow closely, consider surgery (likelihood of malignancy, about 6%)
> 6 cm: surgery indicated (likelihood of malignancy, about 25%)
Some providers and patients inquire whether it is helpful or necessary to biopsy. It is generally not advisable to biopsy, especially if the findings are favorable for benign nonsecretory masses, since there is a high false-negative rate. An indication for biopsy is if the patient has a history of extra-adrenal malignancy; this will distinguish recurrence or metastatic disease from a benign mass. A final proviso: If biopsy is performed, make sure the adrenal mass is not a pheochromocytoma, as biopsy of a hormone-secreting neoplasm can lead to a hypertensive emergency.
Q: How do I determine whether the mass is hormone-secreting?
Although 80% are nonsecretory, you must still maintain a high index of suspicion so as not to miss a potentially problematic and fully treatable adenoma. A thorough history is essential in screening for hormonal excess arising from adrenal adenomas, since the signs and symptoms can be insidious. The three hormones secreted by adrenal adenomas are cortisol, aldosterone, and catecholamines (seen in Cushing syndrome, aldosterone-producing adenoma [APA], and pheochromocytoma, respectively).
It is important to note that Cushing syndrome has an insidious onset and can be easily missed. Hyperaldosteronism presents with hypertension (requiring several medications) and commonly hypokalemia. And pheochromocytoma can be “written off as” anxiety disorder, panic attack, or even hypoglycemia symptoms (especially if patients are treated for diabetes with agents that cause hypoglycemia). To help in your differential diagnosis of secretory adenomas, know that APA accounts for only 1%, and therefore the majority will secrete cortisol and (far less likely) catecholamines.
Q: What is the appropriate laboratory work-up?
The best simple screening test for hypercortisolemia is a 1-mg overnight dexamethasone suppression test. If this value is increased to ≥ 3 µg/dL, it should be followed up with a more sensitive test (a 24-hour urine for creatinine and free cortisol) to further assess for hypercortisolemia.
Patients thought to have a potential pheochromocytoma should undergo measurement of plasma fractionated metanephrines and normetanephrines or 24-hour urine for total metanephrines and fractionated catecholamines.
Finally, for patients with hypokalemia and hypertension or refractory hypertension requiring multiple (> 3) antihypertensive medications, plasma renin activity (PRA) and plasma aldosterone concentration (PAC) should be obtained. A low PRA and a PAC > 15 ng/dL, along with a PAC/PRA ratio of > 20, is highly suggestive of an APA.
Q: What is the treatment and follow-up?
Here is a quick reference guide regarding surgical treatment and medical follow-up and surveillance:
• Adrenalectomy (pheochromocytoma, APA, Cushing syndrome): for masses 4 to 6 cm, consider surgery, especially if > 10 Hounsfield units; for masses > 6 cm, there is an increased risk for malignancy and surgery is required.
• Follow-up for low-suspicion, nonsecretory masses: abdominal CT 3 to 6 months after the initial scan, then annually for 1 to 2 years; hormonal evaluation and follow-up annually for 5 years, to evaluate for signs and symptoms of hormonal excess.
SUGGESTED READING
American Association of Clinical Endocrinologists/American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas. Endocr Pract. 2009;15(Suppl 1).
Management of the Clinically Inapparent Adrenal Mass (Incidentaloma). NIH State-of-the-Science Conference Statement; February 4-6, 2002.
Slawik M, Reincke M. Adrenal incidentalomas (Chapter 20). EndoText.com. www.endotext.org/adrenal/adrenal20/adrenal20.htm. Accessed October 12, 2012.
Fitzgerald PA, Goldfien A. Adrenal medulla. In: Greenspan F, Gardner D, eds. Basic and Clinical Endocrinology. 7th ed. McGraw-Hill: 2003;453-473.
The Washington Manual Endocrinology Specialty Consult. 2005;57-61, 71-84.
Endocrine Secrets. 4th ed. 2005;197-204, 241-252, 257-265.
Cleveland Clinic Endocrine & Metabolism Board Review. www.clevelandclinicmeded.com/live/courses/ann/endoreview/default.asp. Accessed October 12, 2012.
| Clinician Reviews in partnership with |
| Clinician Reviews in partnership with |
Maggie, 42, presents to the emergency department with chronic intermittent abdominal pain and bloating with constipation and occasional diarrhea. She denies fever, chills, nausea, vomiting, melana, bright red blood per rectum, or changes in stool caliper, and she says she otherwise feels well.
Relevant lab and study results include: a comprehensive metabolic panel, complete blood count with differential, beta hCG (human chorionic gonadotropin), urinalysis, and amylase and lipase, all within normal limits; pregnancy test, negative; abdominal x-ray, within normal limits except increased stool in distal colon; and abdominal CT, 2.3-cm right adrenal mass and a Hounsfield measurement of 4 units.
Maggie has a right adrenal incidentaloma (incidentally discovered adenoma that was not in the differential diagnosis). Such findings have become all too often the case, due to the immediate access to and overutilization of high-resolution CT, MRI, and ultrasound. We are now seeing a significantly increased number of incidental adrenal lesions/masses discovered on images not intended to look for adrenal-related diseases (eg, Cushing syndrome, pheochromocytomas, and aldosterone-producing adenomas).
Q: How common are adrenal adenomas, and what must I consider?
Incidental adrenal adenomas are found on 4.4% of abdominal CTs, and in one autopsy series were discovered in 8.7%. Prevalence increases with age, with occurrence of < 1% in patients younger than 30 and about 7% for patients 70 or older.
Evaluation is based on two concerns: First, is the adrenal mass benign or malignant? Second, is the mass secretory or nonsecretory (non-hormone secreting) in nature?
The fortunate news about adrenal incidentalomas is that 80% are benign and nonsecretory, which provides immediate reassuring news to the patient. Examples of benign adrenal masses are: adenoma, lipoma, cyst, ganglioneuroma, hematoma, and infection (eg, tuberculosis, fungal).
The other encouraging statistic is that only 1:4,000 adrenal incidentalomas are malignant. Examples of malignant adrenal masses are: adrenocortical carcinoma, metastatic neoplasm, lymphoma, and malignant pheochromocytoma.
Q: Does adrenal adenoma size matter?
Yes, the larger the size of the adenoma, the higher the association with malignancy. The guide below (based on CT findings) shows not only malignancy potential as it relates to size, but also the importance of Hounsfield units and when surgical intervention is recommended.
Imaging (CT scan)
< 4 cm: homogeneous mass with smooth borders and < 10 Hounsfield units; suggests benign mass (likelihood of malignancy, about 2%)
4 to 6 cm: follow closely, consider surgery (likelihood of malignancy, about 6%)
> 6 cm: surgery indicated (likelihood of malignancy, about 25%)
Some providers and patients inquire whether it is helpful or necessary to biopsy. It is generally not advisable to biopsy, especially if the findings are favorable for benign nonsecretory masses, since there is a high false-negative rate. An indication for biopsy is if the patient has a history of extra-adrenal malignancy; this will distinguish recurrence or metastatic disease from a benign mass. A final proviso: If biopsy is performed, make sure the adrenal mass is not a pheochromocytoma, as biopsy of a hormone-secreting neoplasm can lead to a hypertensive emergency.
Q: How do I determine whether the mass is hormone-secreting?
Although 80% are nonsecretory, you must still maintain a high index of suspicion so as not to miss a potentially problematic and fully treatable adenoma. A thorough history is essential in screening for hormonal excess arising from adrenal adenomas, since the signs and symptoms can be insidious. The three hormones secreted by adrenal adenomas are cortisol, aldosterone, and catecholamines (seen in Cushing syndrome, aldosterone-producing adenoma [APA], and pheochromocytoma, respectively).
It is important to note that Cushing syndrome has an insidious onset and can be easily missed. Hyperaldosteronism presents with hypertension (requiring several medications) and commonly hypokalemia. And pheochromocytoma can be “written off as” anxiety disorder, panic attack, or even hypoglycemia symptoms (especially if patients are treated for diabetes with agents that cause hypoglycemia). To help in your differential diagnosis of secretory adenomas, know that APA accounts for only 1%, and therefore the majority will secrete cortisol and (far less likely) catecholamines.
Q: What is the appropriate laboratory work-up?
The best simple screening test for hypercortisolemia is a 1-mg overnight dexamethasone suppression test. If this value is increased to ≥ 3 µg/dL, it should be followed up with a more sensitive test (a 24-hour urine for creatinine and free cortisol) to further assess for hypercortisolemia.
Patients thought to have a potential pheochromocytoma should undergo measurement of plasma fractionated metanephrines and normetanephrines or 24-hour urine for total metanephrines and fractionated catecholamines.
Finally, for patients with hypokalemia and hypertension or refractory hypertension requiring multiple (> 3) antihypertensive medications, plasma renin activity (PRA) and plasma aldosterone concentration (PAC) should be obtained. A low PRA and a PAC > 15 ng/dL, along with a PAC/PRA ratio of > 20, is highly suggestive of an APA.
Q: What is the treatment and follow-up?
Here is a quick reference guide regarding surgical treatment and medical follow-up and surveillance:
• Adrenalectomy (pheochromocytoma, APA, Cushing syndrome): for masses 4 to 6 cm, consider surgery, especially if > 10 Hounsfield units; for masses > 6 cm, there is an increased risk for malignancy and surgery is required.
• Follow-up for low-suspicion, nonsecretory masses: abdominal CT 3 to 6 months after the initial scan, then annually for 1 to 2 years; hormonal evaluation and follow-up annually for 5 years, to evaluate for signs and symptoms of hormonal excess.
SUGGESTED READING
American Association of Clinical Endocrinologists/American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas. Endocr Pract. 2009;15(Suppl 1).
Management of the Clinically Inapparent Adrenal Mass (Incidentaloma). NIH State-of-the-Science Conference Statement; February 4-6, 2002.
Slawik M, Reincke M. Adrenal incidentalomas (Chapter 20). EndoText.com. www.endotext.org/adrenal/adrenal20/adrenal20.htm. Accessed October 12, 2012.
Fitzgerald PA, Goldfien A. Adrenal medulla. In: Greenspan F, Gardner D, eds. Basic and Clinical Endocrinology. 7th ed. McGraw-Hill: 2003;453-473.
The Washington Manual Endocrinology Specialty Consult. 2005;57-61, 71-84.
Endocrine Secrets. 4th ed. 2005;197-204, 241-252, 257-265.
Cleveland Clinic Endocrine & Metabolism Board Review. www.clevelandclinicmeded.com/live/courses/ann/endoreview/default.asp. Accessed October 12, 2012.
Maggie, 42, presents to the emergency department with chronic intermittent abdominal pain and bloating with constipation and occasional diarrhea. She denies fever, chills, nausea, vomiting, melana, bright red blood per rectum, or changes in stool caliper, and she says she otherwise feels well.
Relevant lab and study results include: a comprehensive metabolic panel, complete blood count with differential, beta hCG (human chorionic gonadotropin), urinalysis, and amylase and lipase, all within normal limits; pregnancy test, negative; abdominal x-ray, within normal limits except increased stool in distal colon; and abdominal CT, 2.3-cm right adrenal mass and a Hounsfield measurement of 4 units.
Maggie has a right adrenal incidentaloma (incidentally discovered adenoma that was not in the differential diagnosis). Such findings have become all too often the case, due to the immediate access to and overutilization of high-resolution CT, MRI, and ultrasound. We are now seeing a significantly increased number of incidental adrenal lesions/masses discovered on images not intended to look for adrenal-related diseases (eg, Cushing syndrome, pheochromocytomas, and aldosterone-producing adenomas).
Q: How common are adrenal adenomas, and what must I consider?
Incidental adrenal adenomas are found on 4.4% of abdominal CTs, and in one autopsy series were discovered in 8.7%. Prevalence increases with age, with occurrence of < 1% in patients younger than 30 and about 7% for patients 70 or older.
Evaluation is based on two concerns: First, is the adrenal mass benign or malignant? Second, is the mass secretory or nonsecretory (non-hormone secreting) in nature?
The fortunate news about adrenal incidentalomas is that 80% are benign and nonsecretory, which provides immediate reassuring news to the patient. Examples of benign adrenal masses are: adenoma, lipoma, cyst, ganglioneuroma, hematoma, and infection (eg, tuberculosis, fungal).
The other encouraging statistic is that only 1:4,000 adrenal incidentalomas are malignant. Examples of malignant adrenal masses are: adrenocortical carcinoma, metastatic neoplasm, lymphoma, and malignant pheochromocytoma.
Q: Does adrenal adenoma size matter?
Yes, the larger the size of the adenoma, the higher the association with malignancy. The guide below (based on CT findings) shows not only malignancy potential as it relates to size, but also the importance of Hounsfield units and when surgical intervention is recommended.
Imaging (CT scan)
< 4 cm: homogeneous mass with smooth borders and < 10 Hounsfield units; suggests benign mass (likelihood of malignancy, about 2%)
4 to 6 cm: follow closely, consider surgery (likelihood of malignancy, about 6%)
> 6 cm: surgery indicated (likelihood of malignancy, about 25%)
Some providers and patients inquire whether it is helpful or necessary to biopsy. It is generally not advisable to biopsy, especially if the findings are favorable for benign nonsecretory masses, since there is a high false-negative rate. An indication for biopsy is if the patient has a history of extra-adrenal malignancy; this will distinguish recurrence or metastatic disease from a benign mass. A final proviso: If biopsy is performed, make sure the adrenal mass is not a pheochromocytoma, as biopsy of a hormone-secreting neoplasm can lead to a hypertensive emergency.
Q: How do I determine whether the mass is hormone-secreting?
Although 80% are nonsecretory, you must still maintain a high index of suspicion so as not to miss a potentially problematic and fully treatable adenoma. A thorough history is essential in screening for hormonal excess arising from adrenal adenomas, since the signs and symptoms can be insidious. The three hormones secreted by adrenal adenomas are cortisol, aldosterone, and catecholamines (seen in Cushing syndrome, aldosterone-producing adenoma [APA], and pheochromocytoma, respectively).
It is important to note that Cushing syndrome has an insidious onset and can be easily missed. Hyperaldosteronism presents with hypertension (requiring several medications) and commonly hypokalemia. And pheochromocytoma can be “written off as” anxiety disorder, panic attack, or even hypoglycemia symptoms (especially if patients are treated for diabetes with agents that cause hypoglycemia). To help in your differential diagnosis of secretory adenomas, know that APA accounts for only 1%, and therefore the majority will secrete cortisol and (far less likely) catecholamines.
Q: What is the appropriate laboratory work-up?
The best simple screening test for hypercortisolemia is a 1-mg overnight dexamethasone suppression test. If this value is increased to ≥ 3 µg/dL, it should be followed up with a more sensitive test (a 24-hour urine for creatinine and free cortisol) to further assess for hypercortisolemia.
Patients thought to have a potential pheochromocytoma should undergo measurement of plasma fractionated metanephrines and normetanephrines or 24-hour urine for total metanephrines and fractionated catecholamines.
Finally, for patients with hypokalemia and hypertension or refractory hypertension requiring multiple (> 3) antihypertensive medications, plasma renin activity (PRA) and plasma aldosterone concentration (PAC) should be obtained. A low PRA and a PAC > 15 ng/dL, along with a PAC/PRA ratio of > 20, is highly suggestive of an APA.
Q: What is the treatment and follow-up?
Here is a quick reference guide regarding surgical treatment and medical follow-up and surveillance:
• Adrenalectomy (pheochromocytoma, APA, Cushing syndrome): for masses 4 to 6 cm, consider surgery, especially if > 10 Hounsfield units; for masses > 6 cm, there is an increased risk for malignancy and surgery is required.
• Follow-up for low-suspicion, nonsecretory masses: abdominal CT 3 to 6 months after the initial scan, then annually for 1 to 2 years; hormonal evaluation and follow-up annually for 5 years, to evaluate for signs and symptoms of hormonal excess.
SUGGESTED READING
American Association of Clinical Endocrinologists/American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas. Endocr Pract. 2009;15(Suppl 1).
Management of the Clinically Inapparent Adrenal Mass (Incidentaloma). NIH State-of-the-Science Conference Statement; February 4-6, 2002.
Slawik M, Reincke M. Adrenal incidentalomas (Chapter 20). EndoText.com. www.endotext.org/adrenal/adrenal20/adrenal20.htm. Accessed October 12, 2012.
Fitzgerald PA, Goldfien A. Adrenal medulla. In: Greenspan F, Gardner D, eds. Basic and Clinical Endocrinology. 7th ed. McGraw-Hill: 2003;453-473.
The Washington Manual Endocrinology Specialty Consult. 2005;57-61, 71-84.
Endocrine Secrets. 4th ed. 2005;197-204, 241-252, 257-265.
Cleveland Clinic Endocrine & Metabolism Board Review. www.clevelandclinicmeded.com/live/courses/ann/endoreview/default.asp. Accessed October 12, 2012.
Glucose Control and Avoidance of Hypoglycemia
Q: I am frustrated by the “always bring the blood sugars down slowly” philosophy, which I know is intended to avoid hypoglycemic symptoms. However, it often seems to be done at the expense of prolonged hyperglycemia, which is dangerous for patients’ long-term health and may cause more rapid beta-cell destruction. What’s the deal? There is evidence that rapid achievement of tight glucose control using intensive insulin therapy with multiple daily injections or insulin pumps in patients with newly diagnosed type 2 diabetes has favorable outcomes on recovery and maintenance of beta-cell function and prolonged glycemic remission, compared with treatment with oral hypoglycemic agents.1 However, this approach is time consuming and not practical in most primary care settings. Overcoming “clinical inertia” (the failure to initiate or intensify therapy when indicated) has been identified as a major barrier to achieving rapid glycemic control, to the detriment of the patient’s health. One recent study showed that more frequent follow-up with a multidisciplinary team and regular use of a computer-analyzed 7-point glucose profile resulted in more rapid and significantly better glycemic control with a lower A1C, compared to standard care.2 This approach is much more practical in a primary care setting. Additionally, we always treat our patients as individuals. There are very few maxims that are correct in all situations. Almost every answer to a clinical question begins with the qualifier “It depends….” The specifics of the individual case will clarify the appropriate answer. In regard to this particular question, the answer will vary by the clinical history of the patient. For example, for a pregnant patient with poor glycemic control, potential hospitalization and rapid titration of insulin would be the most judicious plan. In this case, quickly bringing glucose into tight control helps minimize risks to the developing fetus. However, if the patient is a frail 80-year-old with advanced cardiovascular disease, then slow and careful titration of medications would be the prudent course to meticulously avoid hypoglycemia. New guidelines from the American Diabetes Association and the European Association for the Study of Diabetes (ADA/EASD)3 are helpful in that they identify various clinical issues and give guidance on which medication regimens would be more appropriate for the specific clinical history. They categorize medications based on efficacy, weight gain, hypoglycemia, major side effects, and costs. Guidelines from the American Association of Clinical Endocrinologists/American College of Endocrinology (AACE/ACE)4 are also very useful, because they categorize treatment based on the A1C level, as well as potential for weight gain and hypoglycemia. For example, a patient with an A1C < 7.5% may be an appropriate candidate for monotherapy, while a symptomatic patient with an A1C > 9% would likely benefit from insulin therapy or triple oral agent therapy. First, it is helpful to set individual glycemic targets for your patient. The following factors can help you in determining A1C targets: • Psychosocial considerations (motivation, adherence to therapy, self-care capacity) • Resources or support systems (family support, community resources, living situation, etc) • Risk for hypoglycemia • Duration of diabetes • Life expectancy • Microvascular complications • Cardiovascular disease and coexisting conditions. For example, an older individual with poor motivation, lack of support systems, short life expectancy, and coexisting terminal cancer would have a less stringent A1C target of ≤ 8%, whereas a young, motivated individual with no complications or serious coexisting complications would have an A1C target of 6%. The new ADA/EASD guidelines list additional considerations for medication choices for various comorbidities, including coronary disease, heart failure, renal disease, liver dysfunction, and hypoglycemia. For each comorbidity listed, there are suggested medications that are preferred and those that should be avoided. If your goal is to avoid hypoglycemia, the ADA/EASD guidelines list medication choices that have low propensity to cause hypoglycemia (eg, metformin, pioglitazone, DPP-4 inhibitors, and GLP-1 receptor agonists). (Of note, special attention is given to medications that do not cause weight gain, such as GLP-1 receptor agonists, DPP-4 inhibitors, and metformin.) Finally, the consensus statement emphasizes the need for individualizing therapy. Many patients have multiple comorbidities and may have medication sensitivities, cost constraints, etc. All of these factors must be taken into consideration when making therapeutic choices. Keep in mind, “one size does not fit all” when it comes to diabetes therapy. The recent releases from both the ADA/EASD and AACE/ACE give us much more detailed guidance addressing medication choices in regard to efficacy, potential for hypoglycemia and weight gain, major side effects, and costs. As always, guidelines do not replace good clinical judgment, based on the patient sitting in front of you. REFERENCES 2. Pimazoni-Netto A, Rodbard D, Zanella MT; Diabetes Education and Control Group. Rapid improvement of glycemic control in type 2 diabetes using weekly intensive multifactorial interventions: structured glucose monitoring, patient education, and adjustment of therapy—a randomized controlled trial. Diabetes Technol Therapeutics. 2011;13(10):997-1004. 3. Inzucchi SE, Bergenstahl RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient centered approach. Diabetes Care. [Epub ahead of print; April 19, 2012]. 4. Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control. Endocr Pract. 2009;15(6):540-559. |
Q: I am frustrated by the “always bring the blood sugars down slowly” philosophy, which I know is intended to avoid hypoglycemic symptoms. However, it often seems to be done at the expense of prolonged hyperglycemia, which is dangerous for patients’ long-term health and may cause more rapid beta-cell destruction. What’s the deal? There is evidence that rapid achievement of tight glucose control using intensive insulin therapy with multiple daily injections or insulin pumps in patients with newly diagnosed type 2 diabetes has favorable outcomes on recovery and maintenance of beta-cell function and prolonged glycemic remission, compared with treatment with oral hypoglycemic agents.1 However, this approach is time consuming and not practical in most primary care settings. Overcoming “clinical inertia” (the failure to initiate or intensify therapy when indicated) has been identified as a major barrier to achieving rapid glycemic control, to the detriment of the patient’s health. One recent study showed that more frequent follow-up with a multidisciplinary team and regular use of a computer-analyzed 7-point glucose profile resulted in more rapid and significantly better glycemic control with a lower A1C, compared to standard care.2 This approach is much more practical in a primary care setting. Additionally, we always treat our patients as individuals. There are very few maxims that are correct in all situations. Almost every answer to a clinical question begins with the qualifier “It depends….” The specifics of the individual case will clarify the appropriate answer. In regard to this particular question, the answer will vary by the clinical history of the patient. For example, for a pregnant patient with poor glycemic control, potential hospitalization and rapid titration of insulin would be the most judicious plan. In this case, quickly bringing glucose into tight control helps minimize risks to the developing fetus. However, if the patient is a frail 80-year-old with advanced cardiovascular disease, then slow and careful titration of medications would be the prudent course to meticulously avoid hypoglycemia. New guidelines from the American Diabetes Association and the European Association for the Study of Diabetes (ADA/EASD)3 are helpful in that they identify various clinical issues and give guidance on which medication regimens would be more appropriate for the specific clinical history. They categorize medications based on efficacy, weight gain, hypoglycemia, major side effects, and costs. Guidelines from the American Association of Clinical Endocrinologists/American College of Endocrinology (AACE/ACE)4 are also very useful, because they categorize treatment based on the A1C level, as well as potential for weight gain and hypoglycemia. For example, a patient with an A1C < 7.5% may be an appropriate candidate for monotherapy, while a symptomatic patient with an A1C > 9% would likely benefit from insulin therapy or triple oral agent therapy. First, it is helpful to set individual glycemic targets for your patient. The following factors can help you in determining A1C targets: • Psychosocial considerations (motivation, adherence to therapy, self-care capacity) • Resources or support systems (family support, community resources, living situation, etc) • Risk for hypoglycemia • Duration of diabetes • Life expectancy • Microvascular complications • Cardiovascular disease and coexisting conditions. For example, an older individual with poor motivation, lack of support systems, short life expectancy, and coexisting terminal cancer would have a less stringent A1C target of ≤ 8%, whereas a young, motivated individual with no complications or serious coexisting complications would have an A1C target of 6%. The new ADA/EASD guidelines list additional considerations for medication choices for various comorbidities, including coronary disease, heart failure, renal disease, liver dysfunction, and hypoglycemia. For each comorbidity listed, there are suggested medications that are preferred and those that should be avoided. If your goal is to avoid hypoglycemia, the ADA/EASD guidelines list medication choices that have low propensity to cause hypoglycemia (eg, metformin, pioglitazone, DPP-4 inhibitors, and GLP-1 receptor agonists). (Of note, special attention is given to medications that do not cause weight gain, such as GLP-1 receptor agonists, DPP-4 inhibitors, and metformin.) Finally, the consensus statement emphasizes the need for individualizing therapy. Many patients have multiple comorbidities and may have medication sensitivities, cost constraints, etc. All of these factors must be taken into consideration when making therapeutic choices. Keep in mind, “one size does not fit all” when it comes to diabetes therapy. The recent releases from both the ADA/EASD and AACE/ACE give us much more detailed guidance addressing medication choices in regard to efficacy, potential for hypoglycemia and weight gain, major side effects, and costs. As always, guidelines do not replace good clinical judgment, based on the patient sitting in front of you. REFERENCES 2. Pimazoni-Netto A, Rodbard D, Zanella MT; Diabetes Education and Control Group. Rapid improvement of glycemic control in type 2 diabetes using weekly intensive multifactorial interventions: structured glucose monitoring, patient education, and adjustment of therapy—a randomized controlled trial. Diabetes Technol Therapeutics. 2011;13(10):997-1004. 3. Inzucchi SE, Bergenstahl RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient centered approach. Diabetes Care. [Epub ahead of print; April 19, 2012]. 4. Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control. Endocr Pract. 2009;15(6):540-559. |
Q: I am frustrated by the “always bring the blood sugars down slowly” philosophy, which I know is intended to avoid hypoglycemic symptoms. However, it often seems to be done at the expense of prolonged hyperglycemia, which is dangerous for patients’ long-term health and may cause more rapid beta-cell destruction. What’s the deal? There is evidence that rapid achievement of tight glucose control using intensive insulin therapy with multiple daily injections or insulin pumps in patients with newly diagnosed type 2 diabetes has favorable outcomes on recovery and maintenance of beta-cell function and prolonged glycemic remission, compared with treatment with oral hypoglycemic agents.1 However, this approach is time consuming and not practical in most primary care settings. Overcoming “clinical inertia” (the failure to initiate or intensify therapy when indicated) has been identified as a major barrier to achieving rapid glycemic control, to the detriment of the patient’s health. One recent study showed that more frequent follow-up with a multidisciplinary team and regular use of a computer-analyzed 7-point glucose profile resulted in more rapid and significantly better glycemic control with a lower A1C, compared to standard care.2 This approach is much more practical in a primary care setting. Additionally, we always treat our patients as individuals. There are very few maxims that are correct in all situations. Almost every answer to a clinical question begins with the qualifier “It depends….” The specifics of the individual case will clarify the appropriate answer. In regard to this particular question, the answer will vary by the clinical history of the patient. For example, for a pregnant patient with poor glycemic control, potential hospitalization and rapid titration of insulin would be the most judicious plan. In this case, quickly bringing glucose into tight control helps minimize risks to the developing fetus. However, if the patient is a frail 80-year-old with advanced cardiovascular disease, then slow and careful titration of medications would be the prudent course to meticulously avoid hypoglycemia. New guidelines from the American Diabetes Association and the European Association for the Study of Diabetes (ADA/EASD)3 are helpful in that they identify various clinical issues and give guidance on which medication regimens would be more appropriate for the specific clinical history. They categorize medications based on efficacy, weight gain, hypoglycemia, major side effects, and costs. Guidelines from the American Association of Clinical Endocrinologists/American College of Endocrinology (AACE/ACE)4 are also very useful, because they categorize treatment based on the A1C level, as well as potential for weight gain and hypoglycemia. For example, a patient with an A1C < 7.5% may be an appropriate candidate for monotherapy, while a symptomatic patient with an A1C > 9% would likely benefit from insulin therapy or triple oral agent therapy. First, it is helpful to set individual glycemic targets for your patient. The following factors can help you in determining A1C targets: • Psychosocial considerations (motivation, adherence to therapy, self-care capacity) • Resources or support systems (family support, community resources, living situation, etc) • Risk for hypoglycemia • Duration of diabetes • Life expectancy • Microvascular complications • Cardiovascular disease and coexisting conditions. For example, an older individual with poor motivation, lack of support systems, short life expectancy, and coexisting terminal cancer would have a less stringent A1C target of ≤ 8%, whereas a young, motivated individual with no complications or serious coexisting complications would have an A1C target of 6%. The new ADA/EASD guidelines list additional considerations for medication choices for various comorbidities, including coronary disease, heart failure, renal disease, liver dysfunction, and hypoglycemia. For each comorbidity listed, there are suggested medications that are preferred and those that should be avoided. If your goal is to avoid hypoglycemia, the ADA/EASD guidelines list medication choices that have low propensity to cause hypoglycemia (eg, metformin, pioglitazone, DPP-4 inhibitors, and GLP-1 receptor agonists). (Of note, special attention is given to medications that do not cause weight gain, such as GLP-1 receptor agonists, DPP-4 inhibitors, and metformin.) Finally, the consensus statement emphasizes the need for individualizing therapy. Many patients have multiple comorbidities and may have medication sensitivities, cost constraints, etc. All of these factors must be taken into consideration when making therapeutic choices. Keep in mind, “one size does not fit all” when it comes to diabetes therapy. The recent releases from both the ADA/EASD and AACE/ACE give us much more detailed guidance addressing medication choices in regard to efficacy, potential for hypoglycemia and weight gain, major side effects, and costs. As always, guidelines do not replace good clinical judgment, based on the patient sitting in front of you. REFERENCES 2. Pimazoni-Netto A, Rodbard D, Zanella MT; Diabetes Education and Control Group. Rapid improvement of glycemic control in type 2 diabetes using weekly intensive multifactorial interventions: structured glucose monitoring, patient education, and adjustment of therapy—a randomized controlled trial. Diabetes Technol Therapeutics. 2011;13(10):997-1004. 3. Inzucchi SE, Bergenstahl RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient centered approach. Diabetes Care. [Epub ahead of print; April 19, 2012]. 4. Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control. Endocr Pract. 2009;15(6):540-559. |
Thyroid Peroxidase Antibodies
Q: I have a patient with premature ovarian failure (diagnosed when she was 32) who is now in her late 40s. She is fatigued, and a blood test revealed a thyroid peroxidase antibodies level of 587 IU/mL. Would you supplement with thyroid replacement hormone, even though she has a TSH of 1.004?
The short answer is: No. Thyroid peroxidase (TPO) antibodies are a marker for the presence of autoimmune thyroid disease. Blood test results for TPO antibodies are positive in 95% of patients with chronic lymphocytic thyroiditis, also known as Hashimoto’s disease, and in 50% to 80% of patients with Graves’ disease.
Patients with high levels of TPO antibodies are at risk for future thyroid dysfunction. Not all patients with Hashimoto’s develop hypothyroidism, and if present, it may not persist. Patients with Hashimoto’s, although rarely, can experience a change from a hypothyroid to a euthyroid or even a hyperthyroid state, because of the development of coexisting TSH-receptor antibodies (TRAb), which include thyroid-stimulating immunoglobulin (TSI) and thyrotropin-binding inhibitory immunoglobulin (TBII), as seen in Graves’ disease.
Thyroid nodules are common with Hashimoto’s and are associated with a small risk (5% to 7%) for thyroid cancer. Sudden enlargement of the thyroid gland in a patient with Hashimoto’s should raise concern about thyroid lymphoma. Some endocrinologists will give supplemental thyroid hormone to a patient with Hashimoto’s, even if the TSH is normal, in an attempt to shrink the size of the gland. However, the closer the TSH is to < 1, the less room there is to further suppress it without making the patient overtly hyperthyroid, and the less likely it is that you will achieve much shrinkage of the gland.
Therefore, in the absence of a symptomatic goiter, there is no clinical reason to initiate any therapy. Even with mildly elevated TSH levels (5 to 10 mIU/L; ie, subclinical hypothyroidism) in an asymptomatic patient, there is considerable controversy about thyroid hormone initiation when the free T4 and T3 levels are normal. Most authorities agree that treatment should be initiated in most patients when the TSH rises above 10 mIU/L, regardless of symptoms. However, there are clearer indications to start thyroid hormone in women who want to become, or who are, pregnant, to maintain a TSH of < 2.5 mIU/L. Also, individuals with depression or hyperlipidemia warrant extra consideration for the use of thyroid hormone.
Since this particular patient had premature ovarian failure, which is often autoimmune in nature, she must be considered at risk for future development of hypothyroidism. This patient should be followed annually to ensure that her TSH does not rise. Should she develop symptoms suggestive of hypothyroidism and her TSH rises above 3, some endocrinologists would initiate a brief empiric trial of thyroid replacement to see if her symptoms respond when the TSH lowers again. If they do not, the thyroid hormone might be stopped, and the patient should continue to be followed.
Note: The definition of a “normal” TSH is evolving. Levels above 3.0 (suggested normal therapeutic range: 0.5 to 3.0) are considered possibly suspicious in symptomatic young people, while levels slightly above the normal reference range (5 to 7 mIU/L) may be deemed normal for the asymptomatic geriatric population.
The other point to remember is that when a clinician initiates any thyroid therapy, some patients fixate on the thyroid as the only source of their symptoms, such as fatigue, weight gain, and obesity, to the exclusion of any other etiologies. For example, sleep deprivation is a far more common cause of fatigue in our “open 24 hours” society, and lifestyle remains the major cause of obesity. Thus, there can be unintended consequences of a diagnosis of thyroid “disease.”
SUGGESTED READING
American Association of Clinical Endocrinologists medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. www.aace.com/publications/guidelines. Accessed March 5, 2012.
Bremner AP, Feddema P, Leedman PJ, et al. Age-related changes in thyroid function: a longitudinal study of a community-based cohort. J Clin Endocrinol Metab. 2012 Feb 16; [Epub ahead of print].
Hutfless S, Matos P, Talor MV, et al. Significance of prediagnostic thyroid antibodies in women with autoimmune thyroid disease. J Clin Endocrinol Metab. 2011;96(9):E1466-E1471.
Kauffman RP, Castracane VD. Premature ovarian failure associated with autoimmune polyglandular syndrome: pathophysiological mechanisms and future fertility. J Womens Health (Larchmt). 2003;12(5):513-520.
Q: I have a patient with premature ovarian failure (diagnosed when she was 32) who is now in her late 40s. She is fatigued, and a blood test revealed a thyroid peroxidase antibodies level of 587 IU/mL. Would you supplement with thyroid replacement hormone, even though she has a TSH of 1.004?
The short answer is: No. Thyroid peroxidase (TPO) antibodies are a marker for the presence of autoimmune thyroid disease. Blood test results for TPO antibodies are positive in 95% of patients with chronic lymphocytic thyroiditis, also known as Hashimoto’s disease, and in 50% to 80% of patients with Graves’ disease.
Patients with high levels of TPO antibodies are at risk for future thyroid dysfunction. Not all patients with Hashimoto’s develop hypothyroidism, and if present, it may not persist. Patients with Hashimoto’s, although rarely, can experience a change from a hypothyroid to a euthyroid or even a hyperthyroid state, because of the development of coexisting TSH-receptor antibodies (TRAb), which include thyroid-stimulating immunoglobulin (TSI) and thyrotropin-binding inhibitory immunoglobulin (TBII), as seen in Graves’ disease.
Thyroid nodules are common with Hashimoto’s and are associated with a small risk (5% to 7%) for thyroid cancer. Sudden enlargement of the thyroid gland in a patient with Hashimoto’s should raise concern about thyroid lymphoma. Some endocrinologists will give supplemental thyroid hormone to a patient with Hashimoto’s, even if the TSH is normal, in an attempt to shrink the size of the gland. However, the closer the TSH is to < 1, the less room there is to further suppress it without making the patient overtly hyperthyroid, and the less likely it is that you will achieve much shrinkage of the gland.
Therefore, in the absence of a symptomatic goiter, there is no clinical reason to initiate any therapy. Even with mildly elevated TSH levels (5 to 10 mIU/L; ie, subclinical hypothyroidism) in an asymptomatic patient, there is considerable controversy about thyroid hormone initiation when the free T4 and T3 levels are normal. Most authorities agree that treatment should be initiated in most patients when the TSH rises above 10 mIU/L, regardless of symptoms. However, there are clearer indications to start thyroid hormone in women who want to become, or who are, pregnant, to maintain a TSH of < 2.5 mIU/L. Also, individuals with depression or hyperlipidemia warrant extra consideration for the use of thyroid hormone.
Since this particular patient had premature ovarian failure, which is often autoimmune in nature, she must be considered at risk for future development of hypothyroidism. This patient should be followed annually to ensure that her TSH does not rise. Should she develop symptoms suggestive of hypothyroidism and her TSH rises above 3, some endocrinologists would initiate a brief empiric trial of thyroid replacement to see if her symptoms respond when the TSH lowers again. If they do not, the thyroid hormone might be stopped, and the patient should continue to be followed.
Note: The definition of a “normal” TSH is evolving. Levels above 3.0 (suggested normal therapeutic range: 0.5 to 3.0) are considered possibly suspicious in symptomatic young people, while levels slightly above the normal reference range (5 to 7 mIU/L) may be deemed normal for the asymptomatic geriatric population.
The other point to remember is that when a clinician initiates any thyroid therapy, some patients fixate on the thyroid as the only source of their symptoms, such as fatigue, weight gain, and obesity, to the exclusion of any other etiologies. For example, sleep deprivation is a far more common cause of fatigue in our “open 24 hours” society, and lifestyle remains the major cause of obesity. Thus, there can be unintended consequences of a diagnosis of thyroid “disease.”
SUGGESTED READING
American Association of Clinical Endocrinologists medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. www.aace.com/publications/guidelines. Accessed March 5, 2012.
Bremner AP, Feddema P, Leedman PJ, et al. Age-related changes in thyroid function: a longitudinal study of a community-based cohort. J Clin Endocrinol Metab. 2012 Feb 16; [Epub ahead of print].
Hutfless S, Matos P, Talor MV, et al. Significance of prediagnostic thyroid antibodies in women with autoimmune thyroid disease. J Clin Endocrinol Metab. 2011;96(9):E1466-E1471.
Kauffman RP, Castracane VD. Premature ovarian failure associated with autoimmune polyglandular syndrome: pathophysiological mechanisms and future fertility. J Womens Health (Larchmt). 2003;12(5):513-520.
Q: I have a patient with premature ovarian failure (diagnosed when she was 32) who is now in her late 40s. She is fatigued, and a blood test revealed a thyroid peroxidase antibodies level of 587 IU/mL. Would you supplement with thyroid replacement hormone, even though she has a TSH of 1.004?
The short answer is: No. Thyroid peroxidase (TPO) antibodies are a marker for the presence of autoimmune thyroid disease. Blood test results for TPO antibodies are positive in 95% of patients with chronic lymphocytic thyroiditis, also known as Hashimoto’s disease, and in 50% to 80% of patients with Graves’ disease.
Patients with high levels of TPO antibodies are at risk for future thyroid dysfunction. Not all patients with Hashimoto’s develop hypothyroidism, and if present, it may not persist. Patients with Hashimoto’s, although rarely, can experience a change from a hypothyroid to a euthyroid or even a hyperthyroid state, because of the development of coexisting TSH-receptor antibodies (TRAb), which include thyroid-stimulating immunoglobulin (TSI) and thyrotropin-binding inhibitory immunoglobulin (TBII), as seen in Graves’ disease.
Thyroid nodules are common with Hashimoto’s and are associated with a small risk (5% to 7%) for thyroid cancer. Sudden enlargement of the thyroid gland in a patient with Hashimoto’s should raise concern about thyroid lymphoma. Some endocrinologists will give supplemental thyroid hormone to a patient with Hashimoto’s, even if the TSH is normal, in an attempt to shrink the size of the gland. However, the closer the TSH is to < 1, the less room there is to further suppress it without making the patient overtly hyperthyroid, and the less likely it is that you will achieve much shrinkage of the gland.
Therefore, in the absence of a symptomatic goiter, there is no clinical reason to initiate any therapy. Even with mildly elevated TSH levels (5 to 10 mIU/L; ie, subclinical hypothyroidism) in an asymptomatic patient, there is considerable controversy about thyroid hormone initiation when the free T4 and T3 levels are normal. Most authorities agree that treatment should be initiated in most patients when the TSH rises above 10 mIU/L, regardless of symptoms. However, there are clearer indications to start thyroid hormone in women who want to become, or who are, pregnant, to maintain a TSH of < 2.5 mIU/L. Also, individuals with depression or hyperlipidemia warrant extra consideration for the use of thyroid hormone.
Since this particular patient had premature ovarian failure, which is often autoimmune in nature, she must be considered at risk for future development of hypothyroidism. This patient should be followed annually to ensure that her TSH does not rise. Should she develop symptoms suggestive of hypothyroidism and her TSH rises above 3, some endocrinologists would initiate a brief empiric trial of thyroid replacement to see if her symptoms respond when the TSH lowers again. If they do not, the thyroid hormone might be stopped, and the patient should continue to be followed.
Note: The definition of a “normal” TSH is evolving. Levels above 3.0 (suggested normal therapeutic range: 0.5 to 3.0) are considered possibly suspicious in symptomatic young people, while levels slightly above the normal reference range (5 to 7 mIU/L) may be deemed normal for the asymptomatic geriatric population.
The other point to remember is that when a clinician initiates any thyroid therapy, some patients fixate on the thyroid as the only source of their symptoms, such as fatigue, weight gain, and obesity, to the exclusion of any other etiologies. For example, sleep deprivation is a far more common cause of fatigue in our “open 24 hours” society, and lifestyle remains the major cause of obesity. Thus, there can be unintended consequences of a diagnosis of thyroid “disease.”
SUGGESTED READING
American Association of Clinical Endocrinologists medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. www.aace.com/publications/guidelines. Accessed March 5, 2012.
Bremner AP, Feddema P, Leedman PJ, et al. Age-related changes in thyroid function: a longitudinal study of a community-based cohort. J Clin Endocrinol Metab. 2012 Feb 16; [Epub ahead of print].
Hutfless S, Matos P, Talor MV, et al. Significance of prediagnostic thyroid antibodies in women with autoimmune thyroid disease. J Clin Endocrinol Metab. 2011;96(9):E1466-E1471.
Kauffman RP, Castracane VD. Premature ovarian failure associated with autoimmune polyglandular syndrome: pathophysiological mechanisms and future fertility. J Womens Health (Larchmt). 2003;12(5):513-520.
Hypercalcemia: Common Yet Challenging
A 21-year-old woman presents with a history of recurrent renal stones. Her serum calcium level is 11.5 mg/dL (normal, 8.6 to 10.5 mg/dL); serum phosphorus, 2.4 mg/dL (2.5 to 4.8 mg/dL); intact parathyroid hormone (PTH), 198 pg/mL (7 to 53 pg/mL); and serum 25-hydroxyvitamin D [25(OH)D], 12.6 ng/mL (30 to 60 ng/mL). After six weeks of therapy with vitamin D (50,000 IU three times/week), the serum calcium level is 11 mg/dL; PTH, 164 pg/mL; and 25(OH)D, 28 ng/mL. With all lab results improved but still abnormal, what other information would be helpful?
With this particular case, the striking history is recurrent renal stones. Analysis of one of the stones to determine if they are calcium oxalate would be beneficial; however, a 24-hour urine calcium measurement would provide useful information about the potential cause of the renal stones. Vitamin D deficiency can cause mild hypercalcemia but can also mask underlying primary hyperparathyroidism—as it did in this case. A Tc-99 sestamibi parathyroid scan will often localize a parathyroid adenoma.
This patient’s 24-hour urine calcium was high, and her parathyroid scan suggested an adenoma in the left lower lobe of the thyroid. An experienced parathyroid surgeon was consulted, and surgical excision of a 1.5-cm parathyroid adenoma followed. The intraoperative PTH went from 183 to 39 pg/mL, and the intraoperative calcium from 11.6 to 9.2 mg/dL. There was no postoperative hypocalcemia.
Q: What is the differential diagnosis for hypercalcemia?
• Parathyroid adenoma or carcinoma
• Hypercalcemia of malignancy (eg, breast, lung, pancreas)
• Multiple myeloma
• Multiple endocrine neoplasia types 1 and 2
• Familial hypocalciuric hypercalcemia
• Excess 1,25 dihydroxy vitamin D [1,25(OH)2D] production: sarcoid or other granulomatous disorders, lymphomas
• Miscellaneous: immobilization, milk-alkali syndrome, and parenteral nutrition
• Drug-related: vitamin D deficiency or intoxication; use of thiazide diuretics or lithium
• Nonparathyroid endocrine causes: hyperthyroidism, pheochromocytoma, Addison’s disease, islet cell tumors
Q: What are the clinical manifestations of hypercalcemia?
Mild hypercalcemia is usually asymptomatic, especially if serum calcium is 10.5 to 11.5 mg/dL. Polyuria and polydypsia, renal stones, constipation, nausea, and weight loss are nonspecific symptoms. Decreased mental alertness and depression can be seen, especially if calcium is higher than 12 mg/dL. Bone pain, arthralgias, and decreased bone density can occur with longstanding hypercalcemia. ECG changes, including bradycardia, atrioventricular block, and short QT interval, are sometimes noted.
Q: What is the significance of familial hypocalciuric hypercalcemia (FHH)?
Patients with this genetic disorder, which involves mutated calcium-sensor receptors, often have a mildly elevated PTH but may have a normal PTH in the presence of hypercalcemia. A 24-hour urine calcium level below 100 mg is indicative of FHH.
A calcium/creatinine clearance ratio (calculated as urine calcium/serum calcium divided by urine creatinine/serum creatinine) of < 0.01 is suggestive of FHH, particularly if there is a family history of mild hypercalcemia.
An important point is that parathyroid surgery is ineffective in these patients, and they seldom develop clinical symptoms or stones.
Q: Often, hypercalcemia is identified through routine labs. What diagnostic studies should be obtained with the initial work-up?
Since it is not uncommon to discover mild hypercalcemia on routine labs, it may be prudent to simply recheck serum calcium before launching into an extensive work-up. A comprehensive metabolic panel will give you the calcium, albumin, and serum protein.
When serum albumin is reduced, a corrected calcium level is calculated by adding 0.8 mg/dL to the total calcium for every decrement of 1 g/dL in serum albumin below the reference value of 4 g/dL. Serum phosphate is often low, except in secondary hyperparathyroidism due to renal failure, in which case phosphate is high. Urine calcium excretion may be high or normal.
A 25(OH)D level should also be obtained, as vitamin D deficiency is a common cause of hypercalcemia. Adequate vitamin D replacement will often correct the hypercalcemia; however, vitamin D deficiency may be masking underlying primary hyperparathyroidism.
The PTH level will be high in primary hyperparathyroidism, although it is possible to have a normal intact PTH in patients who have had long-standing mild primary hyperparathyroidism. Secondary hyperparathyroidism due to vitamin D deficiency will also result in an elevated PTH.
A suppressed PTH level in the presence of severe hypercalcemia suggests nonparathyroid-mediated hypercalcemia, often due to malignancy. Hypercalcemia of malignancy is usually symptomatic and severe (≥ 15 mg/dL).
Q: What other nonroutine studies should be considered in the work-up?
A 24-hour urine for calcium, phosphorus, and creatinine clearance, as well as a DXA bone density test, are important for making treatment decisions. A Tc-99 sestamibi parathyroid scan is important to localize a parathyroid adenoma.
Ultrasound of the neck may help to localize an enlarged parathyroid gland, especially if the scan is negative or equivocal.
Q: What are the complications of untreated hypercalcemia?
These include renal stones and urinary tract infections; peptic ulcer; altered mental status; pancreatitis; and during pregnancy, neonatal hypocalcemia.
Q: What is the medical treatment for hypercalcemia?
For acute hypercalcemia, use IV fluids at a high rate, such as normal saline 2,000 cc/hr, unless contraindicated.
Bisphosphonates, such as IV zoledronic acid, are potent inhibitors of bone resorption of calcium and can temporarily treat hypercalcemia, especially in cases of malignancy or severe hyperparathyroidism. It is important to know that oral bisphosphonates are not effective in treating hypercalcemia.
Avoid thiazide diuretics, as well as vitamin A, vitamin D, and calcium supplements. Another caveat: In the face of vitamin D deficiency, correct the vitamin D level to 40 to 60 ng/dL. Patients with 1,25(OH)2D-mediated hypercalcemia should be treated with glucocorticoids (prednisone or IV hydrocortisone), as they decrease 1,25(OH)2D.
Cinacalcet is approved for treatment of secondary hyperparathyroidism due to chronic renal failure, parathyroid carcinoma, and severe hypercalcemia in patients with primary hyperparathyroidism who are unable to undergo parathyroidectomy. The mode of action of cinacalcet is by binding to the parathyroid glands’ extracellular calcium-sensing receptors (CaSRs) to increase their affinity for extracellular calcium and decrease PTH secretion production.
Q: What are the indications for surgical intervention?
Surgery is recommended for patients with kidney stones or bone disease or with notable symptoms; those who have osteoporosis (identified on DXA scan); patients younger than 50; and those with a glomerular filtration rate below 60 mL/min and calcium 1.0 mg/dL or more above the upper limit of normal. Surgical removal of a parathyroid adenoma usually results in a cure.
Q: What causes secondary hyperparathyroidism?
Chronic renal failure is usually the cause. Hyperphosphatemia and decreased 1,25(OH)2D produce a decrease in ionized calcium. The parathyroid glands are thus stimulated and enlarge.
Vitamin D deficiency is another common cause; it is corrected with adequate vitamin D replacement. Once the vitamin D level is corrected, additional calcium supplementation should be given.
Q: What is the prognosis of hypercalcemia?
Primary hyperparathyroidism is usually chronic and progressive unless surgically cured or medically corrected. The prognosis of hypercalcemia is directly related to the degree of renal impairment or the underlying cause, such as malignancy. The presence of pancreatitis increases the mortality rate.
Regular monitoring and follow-up are important, especially if there is a trend of worsening hypercalcemia and etiology has not been identified. Monitor calcium and albumin at least every three months and renal function at least every six months.
Furthermore, check the 24-hour urine calcium and order DXA bone density testing annually.
SUGGESTED READING
American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons. AACE/AAES position statement on the diagnosis and management of primary hyperparathyroidism. Endocr Prac. 2005;11(1): 49-54.
McPhee SJ, Papadakis MA, eds. 2011 Current Medical Diagnosis and Treatment. McGraw Hill; 2011:1090-1097; 1098-1105; 1575-1579.
Jameson J, ed. Harrison’s Endocrinology. 2nd ed. McGraw Hill; 2010:367-378; 406-410; 411-442.
Brown SA. Hyperparathyroidism. In: Runge MS, Greganti MA, eds. Netter’s Internal Medicine. 2nd ed. Saunders; 2009:316-320.
Bilezikian JP, Khan AA, Potts JT Jr. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Third International Workshop. J Clin Endocrinol Metab. 2009;94(2):335-339.
A 21-year-old woman presents with a history of recurrent renal stones. Her serum calcium level is 11.5 mg/dL (normal, 8.6 to 10.5 mg/dL); serum phosphorus, 2.4 mg/dL (2.5 to 4.8 mg/dL); intact parathyroid hormone (PTH), 198 pg/mL (7 to 53 pg/mL); and serum 25-hydroxyvitamin D [25(OH)D], 12.6 ng/mL (30 to 60 ng/mL). After six weeks of therapy with vitamin D (50,000 IU three times/week), the serum calcium level is 11 mg/dL; PTH, 164 pg/mL; and 25(OH)D, 28 ng/mL. With all lab results improved but still abnormal, what other information would be helpful?
With this particular case, the striking history is recurrent renal stones. Analysis of one of the stones to determine if they are calcium oxalate would be beneficial; however, a 24-hour urine calcium measurement would provide useful information about the potential cause of the renal stones. Vitamin D deficiency can cause mild hypercalcemia but can also mask underlying primary hyperparathyroidism—as it did in this case. A Tc-99 sestamibi parathyroid scan will often localize a parathyroid adenoma.
This patient’s 24-hour urine calcium was high, and her parathyroid scan suggested an adenoma in the left lower lobe of the thyroid. An experienced parathyroid surgeon was consulted, and surgical excision of a 1.5-cm parathyroid adenoma followed. The intraoperative PTH went from 183 to 39 pg/mL, and the intraoperative calcium from 11.6 to 9.2 mg/dL. There was no postoperative hypocalcemia.
Q: What is the differential diagnosis for hypercalcemia?
• Parathyroid adenoma or carcinoma
• Hypercalcemia of malignancy (eg, breast, lung, pancreas)
• Multiple myeloma
• Multiple endocrine neoplasia types 1 and 2
• Familial hypocalciuric hypercalcemia
• Excess 1,25 dihydroxy vitamin D [1,25(OH)2D] production: sarcoid or other granulomatous disorders, lymphomas
• Miscellaneous: immobilization, milk-alkali syndrome, and parenteral nutrition
• Drug-related: vitamin D deficiency or intoxication; use of thiazide diuretics or lithium
• Nonparathyroid endocrine causes: hyperthyroidism, pheochromocytoma, Addison’s disease, islet cell tumors
Q: What are the clinical manifestations of hypercalcemia?
Mild hypercalcemia is usually asymptomatic, especially if serum calcium is 10.5 to 11.5 mg/dL. Polyuria and polydypsia, renal stones, constipation, nausea, and weight loss are nonspecific symptoms. Decreased mental alertness and depression can be seen, especially if calcium is higher than 12 mg/dL. Bone pain, arthralgias, and decreased bone density can occur with longstanding hypercalcemia. ECG changes, including bradycardia, atrioventricular block, and short QT interval, are sometimes noted.
Q: What is the significance of familial hypocalciuric hypercalcemia (FHH)?
Patients with this genetic disorder, which involves mutated calcium-sensor receptors, often have a mildly elevated PTH but may have a normal PTH in the presence of hypercalcemia. A 24-hour urine calcium level below 100 mg is indicative of FHH.
A calcium/creatinine clearance ratio (calculated as urine calcium/serum calcium divided by urine creatinine/serum creatinine) of < 0.01 is suggestive of FHH, particularly if there is a family history of mild hypercalcemia.
An important point is that parathyroid surgery is ineffective in these patients, and they seldom develop clinical symptoms or stones.
Q: Often, hypercalcemia is identified through routine labs. What diagnostic studies should be obtained with the initial work-up?
Since it is not uncommon to discover mild hypercalcemia on routine labs, it may be prudent to simply recheck serum calcium before launching into an extensive work-up. A comprehensive metabolic panel will give you the calcium, albumin, and serum protein.
When serum albumin is reduced, a corrected calcium level is calculated by adding 0.8 mg/dL to the total calcium for every decrement of 1 g/dL in serum albumin below the reference value of 4 g/dL. Serum phosphate is often low, except in secondary hyperparathyroidism due to renal failure, in which case phosphate is high. Urine calcium excretion may be high or normal.
A 25(OH)D level should also be obtained, as vitamin D deficiency is a common cause of hypercalcemia. Adequate vitamin D replacement will often correct the hypercalcemia; however, vitamin D deficiency may be masking underlying primary hyperparathyroidism.
The PTH level will be high in primary hyperparathyroidism, although it is possible to have a normal intact PTH in patients who have had long-standing mild primary hyperparathyroidism. Secondary hyperparathyroidism due to vitamin D deficiency will also result in an elevated PTH.
A suppressed PTH level in the presence of severe hypercalcemia suggests nonparathyroid-mediated hypercalcemia, often due to malignancy. Hypercalcemia of malignancy is usually symptomatic and severe (≥ 15 mg/dL).
Q: What other nonroutine studies should be considered in the work-up?
A 24-hour urine for calcium, phosphorus, and creatinine clearance, as well as a DXA bone density test, are important for making treatment decisions. A Tc-99 sestamibi parathyroid scan is important to localize a parathyroid adenoma.
Ultrasound of the neck may help to localize an enlarged parathyroid gland, especially if the scan is negative or equivocal.
Q: What are the complications of untreated hypercalcemia?
These include renal stones and urinary tract infections; peptic ulcer; altered mental status; pancreatitis; and during pregnancy, neonatal hypocalcemia.
Q: What is the medical treatment for hypercalcemia?
For acute hypercalcemia, use IV fluids at a high rate, such as normal saline 2,000 cc/hr, unless contraindicated.
Bisphosphonates, such as IV zoledronic acid, are potent inhibitors of bone resorption of calcium and can temporarily treat hypercalcemia, especially in cases of malignancy or severe hyperparathyroidism. It is important to know that oral bisphosphonates are not effective in treating hypercalcemia.
Avoid thiazide diuretics, as well as vitamin A, vitamin D, and calcium supplements. Another caveat: In the face of vitamin D deficiency, correct the vitamin D level to 40 to 60 ng/dL. Patients with 1,25(OH)2D-mediated hypercalcemia should be treated with glucocorticoids (prednisone or IV hydrocortisone), as they decrease 1,25(OH)2D.
Cinacalcet is approved for treatment of secondary hyperparathyroidism due to chronic renal failure, parathyroid carcinoma, and severe hypercalcemia in patients with primary hyperparathyroidism who are unable to undergo parathyroidectomy. The mode of action of cinacalcet is by binding to the parathyroid glands’ extracellular calcium-sensing receptors (CaSRs) to increase their affinity for extracellular calcium and decrease PTH secretion production.
Q: What are the indications for surgical intervention?
Surgery is recommended for patients with kidney stones or bone disease or with notable symptoms; those who have osteoporosis (identified on DXA scan); patients younger than 50; and those with a glomerular filtration rate below 60 mL/min and calcium 1.0 mg/dL or more above the upper limit of normal. Surgical removal of a parathyroid adenoma usually results in a cure.
Q: What causes secondary hyperparathyroidism?
Chronic renal failure is usually the cause. Hyperphosphatemia and decreased 1,25(OH)2D produce a decrease in ionized calcium. The parathyroid glands are thus stimulated and enlarge.
Vitamin D deficiency is another common cause; it is corrected with adequate vitamin D replacement. Once the vitamin D level is corrected, additional calcium supplementation should be given.
Q: What is the prognosis of hypercalcemia?
Primary hyperparathyroidism is usually chronic and progressive unless surgically cured or medically corrected. The prognosis of hypercalcemia is directly related to the degree of renal impairment or the underlying cause, such as malignancy. The presence of pancreatitis increases the mortality rate.
Regular monitoring and follow-up are important, especially if there is a trend of worsening hypercalcemia and etiology has not been identified. Monitor calcium and albumin at least every three months and renal function at least every six months.
Furthermore, check the 24-hour urine calcium and order DXA bone density testing annually.
SUGGESTED READING
American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons. AACE/AAES position statement on the diagnosis and management of primary hyperparathyroidism. Endocr Prac. 2005;11(1): 49-54.
McPhee SJ, Papadakis MA, eds. 2011 Current Medical Diagnosis and Treatment. McGraw Hill; 2011:1090-1097; 1098-1105; 1575-1579.
Jameson J, ed. Harrison’s Endocrinology. 2nd ed. McGraw Hill; 2010:367-378; 406-410; 411-442.
Brown SA. Hyperparathyroidism. In: Runge MS, Greganti MA, eds. Netter’s Internal Medicine. 2nd ed. Saunders; 2009:316-320.
Bilezikian JP, Khan AA, Potts JT Jr. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Third International Workshop. J Clin Endocrinol Metab. 2009;94(2):335-339.
A 21-year-old woman presents with a history of recurrent renal stones. Her serum calcium level is 11.5 mg/dL (normal, 8.6 to 10.5 mg/dL); serum phosphorus, 2.4 mg/dL (2.5 to 4.8 mg/dL); intact parathyroid hormone (PTH), 198 pg/mL (7 to 53 pg/mL); and serum 25-hydroxyvitamin D [25(OH)D], 12.6 ng/mL (30 to 60 ng/mL). After six weeks of therapy with vitamin D (50,000 IU three times/week), the serum calcium level is 11 mg/dL; PTH, 164 pg/mL; and 25(OH)D, 28 ng/mL. With all lab results improved but still abnormal, what other information would be helpful?
With this particular case, the striking history is recurrent renal stones. Analysis of one of the stones to determine if they are calcium oxalate would be beneficial; however, a 24-hour urine calcium measurement would provide useful information about the potential cause of the renal stones. Vitamin D deficiency can cause mild hypercalcemia but can also mask underlying primary hyperparathyroidism—as it did in this case. A Tc-99 sestamibi parathyroid scan will often localize a parathyroid adenoma.
This patient’s 24-hour urine calcium was high, and her parathyroid scan suggested an adenoma in the left lower lobe of the thyroid. An experienced parathyroid surgeon was consulted, and surgical excision of a 1.5-cm parathyroid adenoma followed. The intraoperative PTH went from 183 to 39 pg/mL, and the intraoperative calcium from 11.6 to 9.2 mg/dL. There was no postoperative hypocalcemia.
Q: What is the differential diagnosis for hypercalcemia?
• Parathyroid adenoma or carcinoma
• Hypercalcemia of malignancy (eg, breast, lung, pancreas)
• Multiple myeloma
• Multiple endocrine neoplasia types 1 and 2
• Familial hypocalciuric hypercalcemia
• Excess 1,25 dihydroxy vitamin D [1,25(OH)2D] production: sarcoid or other granulomatous disorders, lymphomas
• Miscellaneous: immobilization, milk-alkali syndrome, and parenteral nutrition
• Drug-related: vitamin D deficiency or intoxication; use of thiazide diuretics or lithium
• Nonparathyroid endocrine causes: hyperthyroidism, pheochromocytoma, Addison’s disease, islet cell tumors
Q: What are the clinical manifestations of hypercalcemia?
Mild hypercalcemia is usually asymptomatic, especially if serum calcium is 10.5 to 11.5 mg/dL. Polyuria and polydypsia, renal stones, constipation, nausea, and weight loss are nonspecific symptoms. Decreased mental alertness and depression can be seen, especially if calcium is higher than 12 mg/dL. Bone pain, arthralgias, and decreased bone density can occur with longstanding hypercalcemia. ECG changes, including bradycardia, atrioventricular block, and short QT interval, are sometimes noted.
Q: What is the significance of familial hypocalciuric hypercalcemia (FHH)?
Patients with this genetic disorder, which involves mutated calcium-sensor receptors, often have a mildly elevated PTH but may have a normal PTH in the presence of hypercalcemia. A 24-hour urine calcium level below 100 mg is indicative of FHH.
A calcium/creatinine clearance ratio (calculated as urine calcium/serum calcium divided by urine creatinine/serum creatinine) of < 0.01 is suggestive of FHH, particularly if there is a family history of mild hypercalcemia.
An important point is that parathyroid surgery is ineffective in these patients, and they seldom develop clinical symptoms or stones.
Q: Often, hypercalcemia is identified through routine labs. What diagnostic studies should be obtained with the initial work-up?
Since it is not uncommon to discover mild hypercalcemia on routine labs, it may be prudent to simply recheck serum calcium before launching into an extensive work-up. A comprehensive metabolic panel will give you the calcium, albumin, and serum protein.
When serum albumin is reduced, a corrected calcium level is calculated by adding 0.8 mg/dL to the total calcium for every decrement of 1 g/dL in serum albumin below the reference value of 4 g/dL. Serum phosphate is often low, except in secondary hyperparathyroidism due to renal failure, in which case phosphate is high. Urine calcium excretion may be high or normal.
A 25(OH)D level should also be obtained, as vitamin D deficiency is a common cause of hypercalcemia. Adequate vitamin D replacement will often correct the hypercalcemia; however, vitamin D deficiency may be masking underlying primary hyperparathyroidism.
The PTH level will be high in primary hyperparathyroidism, although it is possible to have a normal intact PTH in patients who have had long-standing mild primary hyperparathyroidism. Secondary hyperparathyroidism due to vitamin D deficiency will also result in an elevated PTH.
A suppressed PTH level in the presence of severe hypercalcemia suggests nonparathyroid-mediated hypercalcemia, often due to malignancy. Hypercalcemia of malignancy is usually symptomatic and severe (≥ 15 mg/dL).
Q: What other nonroutine studies should be considered in the work-up?
A 24-hour urine for calcium, phosphorus, and creatinine clearance, as well as a DXA bone density test, are important for making treatment decisions. A Tc-99 sestamibi parathyroid scan is important to localize a parathyroid adenoma.
Ultrasound of the neck may help to localize an enlarged parathyroid gland, especially if the scan is negative or equivocal.
Q: What are the complications of untreated hypercalcemia?
These include renal stones and urinary tract infections; peptic ulcer; altered mental status; pancreatitis; and during pregnancy, neonatal hypocalcemia.
Q: What is the medical treatment for hypercalcemia?
For acute hypercalcemia, use IV fluids at a high rate, such as normal saline 2,000 cc/hr, unless contraindicated.
Bisphosphonates, such as IV zoledronic acid, are potent inhibitors of bone resorption of calcium and can temporarily treat hypercalcemia, especially in cases of malignancy or severe hyperparathyroidism. It is important to know that oral bisphosphonates are not effective in treating hypercalcemia.
Avoid thiazide diuretics, as well as vitamin A, vitamin D, and calcium supplements. Another caveat: In the face of vitamin D deficiency, correct the vitamin D level to 40 to 60 ng/dL. Patients with 1,25(OH)2D-mediated hypercalcemia should be treated with glucocorticoids (prednisone or IV hydrocortisone), as they decrease 1,25(OH)2D.
Cinacalcet is approved for treatment of secondary hyperparathyroidism due to chronic renal failure, parathyroid carcinoma, and severe hypercalcemia in patients with primary hyperparathyroidism who are unable to undergo parathyroidectomy. The mode of action of cinacalcet is by binding to the parathyroid glands’ extracellular calcium-sensing receptors (CaSRs) to increase their affinity for extracellular calcium and decrease PTH secretion production.
Q: What are the indications for surgical intervention?
Surgery is recommended for patients with kidney stones or bone disease or with notable symptoms; those who have osteoporosis (identified on DXA scan); patients younger than 50; and those with a glomerular filtration rate below 60 mL/min and calcium 1.0 mg/dL or more above the upper limit of normal. Surgical removal of a parathyroid adenoma usually results in a cure.
Q: What causes secondary hyperparathyroidism?
Chronic renal failure is usually the cause. Hyperphosphatemia and decreased 1,25(OH)2D produce a decrease in ionized calcium. The parathyroid glands are thus stimulated and enlarge.
Vitamin D deficiency is another common cause; it is corrected with adequate vitamin D replacement. Once the vitamin D level is corrected, additional calcium supplementation should be given.
Q: What is the prognosis of hypercalcemia?
Primary hyperparathyroidism is usually chronic and progressive unless surgically cured or medically corrected. The prognosis of hypercalcemia is directly related to the degree of renal impairment or the underlying cause, such as malignancy. The presence of pancreatitis increases the mortality rate.
Regular monitoring and follow-up are important, especially if there is a trend of worsening hypercalcemia and etiology has not been identified. Monitor calcium and albumin at least every three months and renal function at least every six months.
Furthermore, check the 24-hour urine calcium and order DXA bone density testing annually.
SUGGESTED READING
American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons. AACE/AAES position statement on the diagnosis and management of primary hyperparathyroidism. Endocr Prac. 2005;11(1): 49-54.
McPhee SJ, Papadakis MA, eds. 2011 Current Medical Diagnosis and Treatment. McGraw Hill; 2011:1090-1097; 1098-1105; 1575-1579.
Jameson J, ed. Harrison’s Endocrinology. 2nd ed. McGraw Hill; 2010:367-378; 406-410; 411-442.
Brown SA. Hyperparathyroidism. In: Runge MS, Greganti MA, eds. Netter’s Internal Medicine. 2nd ed. Saunders; 2009:316-320.
Bilezikian JP, Khan AA, Potts JT Jr. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Third International Workshop. J Clin Endocrinol Metab. 2009;94(2):335-339.