User login
Ceftaroline fosamil: A super-cephalosporin?
Ceftaroline fosamil (Teflaro), introduced to the US market in October 2010, is the first beta-lactam agent with clinically useful activity against methicillin-resistant Staphylococcus aureus (MRSA). Currently, it is approved by the US Food and Drug Administration (FDA) to treat acute bacterial skin and skin-structure infections and community-acquired bacterial pneumonia caused by susceptible microorganisms.
In an era of increasing drug resistance and limited numbers of antimicrobials in the drug-production pipeline, ceftaroline is a step forward in fulfilling the Infectious Diseases Society of America’s “10 × ’20 Initiative” to increase support for drug research and manufacturing, with the goal of producing 10 new antimicrobial drugs by the year 2020.1 Ceftaroline was the first of several antibiotics to receive FDA approval in response to this initiative. It was followed by dalbavancin (May 2014), tedizolid phosphate (June 2014), oritavancin (August 2014), ceftolozane-tazobactam (December 2014), and ceftazidime-avibactam (February 2015). These antibiotic agents are aimed at treating infections caused by drug-resistant gram-positive and gram-negative microorganisms. It is important to understand and optimize the use of these new antibiotic agents in order to decrease the risk of emerging antibiotic resistance and superinfections (eg, Clostridium difficile infection) caused by antibiotic overuse or misuse.
This article provides an overview of ceftaroline’s mechanisms of action and resistance, spectrum of activity, pharmacokinetic properties, adverse effects, and current place in therapy.
AN ERA OF MULTIDRUG-RESISTANT MICROORGANISMS
Increasing rates of antimicrobial resistance threaten the efficacy of antimicrobial drugs in the daily practice of medicine. The World Health Organization has labeled antimicrobial resistance one of the three greatest threats to human health. Global efforts are under way to stimulate development of new antimicrobial agents and to decrease rates of antimicrobial resistance.
Staphylococcus aureus: A threat, even with vancomycin
Between 1998 and 2005, S aureus was one of the most common inpatient and outpatient isolates reported by clinical laboratories throughout the United States.2
Treatment of S aureus infection is complicated by a variety of resistance mechanisms that have evolved over time. In fact, the first resistant isolate of S aureus emerged not long after penicillin’s debut into clinical practice, and now the majority of strains are resistant to penicillin.
Methicillin was designed to overcome this beta-lactamase resistance and became the treatment of choice for penicillin-resistant S aureus isolates. However, MRSA isolates soon emerged because of the organism’s acquisition of penicillin-binding protein PBP2a via the mecA gene, leading to decreased binding affinity of methicillin.3
Since then, several agents active against MRSA (vancomycin, daptomycin, linezolid, tigecycline) have been introduced and continue to be widely used. While vancomycin is considered the first-line option for a variety of MRSA infections, its use has been threatened because of the emergence of vancomycin-intermediate-resistant S aureus (VISA), S aureus strains displaying vancomycin heteroresistance (hVISA), and vancomycin-resistant S aureus (VRSA) strains.4
VISA and hVISA isolates emerged through sequential mutations that lead to autolytic activity and cell-wall thickening. In contrast, the mechanism of resistance in VRSA is by acquisition of the vanA resistance gene, which alters the binding site of vancomycin from d-alanine-d-alanine to d-alanine-d-lactate.5
Streptococcus pneumoniae resistance: A continuing problem
The prevalence of drug resistance in S pneumoniae has risen since the late 1990s. A 2013 report from the SENTRY Antimicrobial Surveillance Program stated that almost 20% of S pneumoniae isolates were resistant to amoxicillin-clavulanate, and similar trends have been observed for penicillin (14.8%) and ceftriaxone (11.7%).6
S pneumoniae resistance is acquired through modifications of the penicillin-binding proteins, namely PBP1a, PBP2b, PBP2x, and, less frequently, PBP2a. These modifications lead to decreased binding affinity for most beta-lactams.7
Clinical impact of multidrug-resistant S aureus and S pneumoniae
In 2011, the US Centers for Disease Control and Prevention reported an estimated 80,000 severe MRSA infections and 11,000 MRSA-related deaths in the United States.8 In the same report, drug-resistant S pneumoniae was estimated to be responsible for almost 1.2 million illnesses and 7,000 deaths per year, leading to upwards of $96 million in related medical costs.
While invasive drug-resistant S pneumoniae infections usually affect patients at the extremes of age (under age 5 and over age 65), they have had a serious impact on patients of all ages.8
In light of the increasing prevalence of multidrug-resistant organisms, newer antimicrobial agents with novel mechanisms of action are needed.
CEFTAROLINE: A BETA-LACTAM WITH ANTI-MRSA ACTIVITY
The cephalosporins, a class of beta-lactam antibiotics, were originally derived from the fungus Cephalosporium (now called Acremonium). There are now many agents in this class, each containing a nucleus consisting of a beta-lactam ring fused to a six-member dihydrothiazine ring, and two side chains that can be modified to affect antibacterial activity and pharmacokinetic properties.
Cephalosporins are typically categorized into “generations.” With some exceptions, the first- and second-generation agents have good activity against gram-positive microorganisms, including methicillin-susceptible S aureus—but not against MRSA. The third- and fourth-generation cephalosporins have better gram-negative activity, with many agents having activity against the gram-negative bacterium Pseudomonas aeruginosa.
Enterococcal isolates are intrinsically resistant to cephalosporins. Additionally, cephalosporins are not active against anaerobic bacteria, except for a subset of structurally unique second-generation cephalosporins, ie, cefotetan and cefoxitin.
Ceftaroline was synthesized with specific manipulations of the side chains to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae isolates, making it the first available beta-lactam with this ability.
Mechanism of action
Ceftaroline binds to penicillin-binding proteins, inhibiting transpeptidation. This interaction blocks the final stage of peptidoglycan synthesis and inhibits bacterial cell wall formation, ultimately leading to cellular autolysis and microorganism death. Ceftaroline binds with high affinity to PBP2a and PBP2x, expanding its activity to encompass MRSA and penicillin-resistant S pneumoniae isolates.9
Spectrum of activity
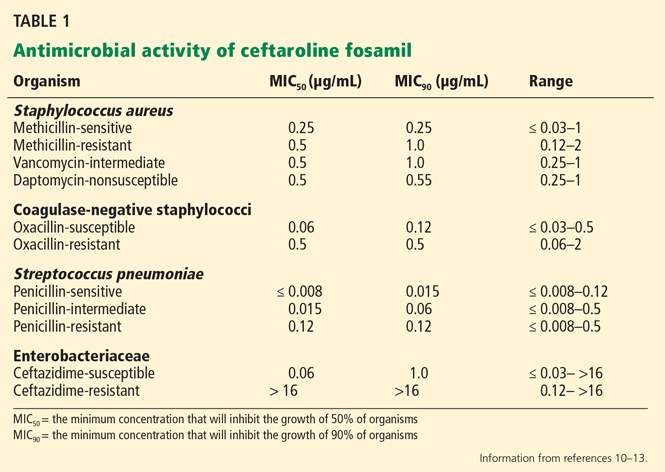
Ceftaroline has in vitro activity against many gram-positive and gram-negative bacteria,10–13 including (Table 1):
- Methicillin-susceptible and methicillin-resistant staphylococci
- VISA, VRSA, and hVISA
- Daptomycin-nonsusceptible S aureus
- Streptococcal species, including penicillin-resistant S pneumoniae
- Enterobacteriaceae, including Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Citrobacter koseri, Citrobacter freundii, Enterobacter cloacae, Enterobacter aerogenes, Moraxella catarrhalis, Morganella morganii, and Proteus mirabilis.
Of note, ceftaroline is not active against Pseudomonas species, Enterococcus species, or Bacteroides fragilis. In addition, it is not active against the “atypical” respiratory pathogens Mycoplasma pneumoniae, Chlamydophila pneumoniae, and Legionella pneumophila.
Ceftaroline resistance
Gram-negative organisms appear to develop resistance to ceftaroline at rates similar to those observed with the other oxyimino-cephalosporins (eg, ceftriaxone). Ceftaroline is inactive against gram-negative organisms producing extended-spectrum beta-lactamases, including K pneumoniae carbapenemase and metallo-beta-lactamases.14 In addition, it induces the expression of AmpC beta-lactamases.
Although currently uncommon, resistance to ceftaroline has also been reported in S aureus strains.15 The mechanism of resistance is decreased binding affinity for PBP2a due to amino acid substitutions on the nonpenicillin-binding domains.15
Pharmacokinetic profile
An understanding of pharmacokinetics is key in optimizing the dose of antimicrobials so that the drugs are used most effectively and pathogens do not develop resistance to them.

Ceftaroline fosamil is a prodrug that, upon intravenous administration, is rapidly converted by phosphatase enzymes to its active moiety, ceftaroline. Its pharmacokinetic profile is summarized in Table 2.16,17 Its volume of distribution is similar to that of the fourth-generation cephalosporin cefepime.
Ceftaroline is then hydrolyzed into its inactive metabolite, ceftaroline M-1. It undergoes little hepatic metabolism and lacks properties to make it a substrate, inhibitor, or inducer of the CYP450 enzyme system and therefore is not likely to cause notable CYP450-related drug-drug interactions.
Like most other beta-lactams, ceftaroline is primarily excreted by the kidneys. Furthermore, an estimated 21% of a dose is eliminated with each intermittent hemodialysis session. Therefore, renal and intermittent hemodialysis dose adjustments are necessary. The estimated elimination half-life is 2.6 hours, necessitating dosing two to three times daily, depending on the indication and infectious inoculum.
Ceftaroline dosing
Ceftaroline is available only in a parenteral preparation and is typically given at a dose of 600 mg every 12 hours.10 The intravenous infusion is given over 1 hour.
The current stability data require reconstituted ceftaroline to be used within 6 hours at room temperature and within 24 hours if refrigerated.10
Ceftaroline requires dosing adjustments for patients with renal insufficiency. Per the manufacturer, renal dosing adjustments are based on the creatinine clearance rate, as estimated by the Cockroft-Gault formula:
- Creatinine clearance > 50 mL/min: no dosage adjustment necessary
- Creatinine clearance > 30 to ≤ 50 mL/min: give 400 mg every 12 hours
- Creatinine clearance ≥ 15 to ≤ 30 mL/min: give 300 mg every 12 hours
- Creatinine clearance < 15 mL/min or on intermittent dialysis: give 200 mg every 12 hours.
Ongoing clinical trials are investigating a higher-dosing strategy of 600 mg every 8 hours for patients with community-acquired bacterial pneumonia at risk of MRSA bacteremia.18
CLINICAL TRIALS LEADING TO CEFTAROLINE’S APPROVAL
Ceftaroline was approved for the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections due to susceptible pathogens on the basis of phase 3 comparator trials.
Community-acquired bacterial pneumonia: The FOCUS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of community-acquired bacterial pneumonia was studied in two randomized, double-blind, noninferiority trials, known as Ceftaroline Community-acquired Pneumonia vs Ceftriaxone (FOCUS) 1 and FOCUS 2.19,20
Patients were adults and not critically ill, as was reflected by their being in Pneumonia Outcomes Research Team (PORT) risk class III or IV (with class V indicating the highest risk of death). Therefore, the results may not be completely applicable to critically ill patients or those not admitted to the hospital. Of note, patients were excluded from the trials if they had infections known or thought to be due to MRSA or to atypical organisms.21 Baseline characteristics and patient demographics were similar between study groups in both trials.
A bacterial pathogen was identified in 26.1% of the patients included in the modified intent-to-treat analysis of the pooled data of the trials; the most common pathogens were S pneumoniae, methicillin-sensitive S aureus, Haemophilus influenzae, K pneumoniae, and E coli.21
Treatment. Patients received either ceftaroline 600 mg every 12 hours (or a lower dose based on renal function) or ceftriaxone 1 g every 24 hours. In addition, in the FOCUS 1 trial, patients in both treatment groups received clarithromycin 500 mg every 12 hours for the first day.19

Results. In both trials and in the integrated analysis, ceftaroline was noninferior to ceftriaxone (Table 3).22 In the integrated analysis of both trials, compared with the ceftriaxone group, the ceftaroline group had a higher clinical cure rate among patients classified as PORT risk class III (86.8% vs 79.2%, weighted treatment difference 12.6%, 95% confidence interval [CI] 1.3–13.8) and among patients who had not received prior antibiotic treatment (85.5% vs 74.9%, weighted treatment difference 11.2%, 95% CI 4.5–18.0).21
Acute bacterial skin and skin-structure infections: The CANVAS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of complicated acute bacterial skin and skin-structure infections was studied in two randomized, double-blind trials: Ceftaroline Versus Vancomycin in Skin and Skin Structure Infections (CANVAS) 1 and CANVAS 2.23,24
Patients. Adult patients with a diagnosis of community-acquired skin and skin-structure infections warranting at least 5 days of intravenous antimicrobial therapy were included in the trials. Important protocol exclusions were patients with diabetic foot ulcers, decubitus ulcers, burns, ulcers associated with peripheral vascular disease accompanied by osteomyelitis, and suspected P aeruginosa infections.25 This limits the external validity of ceftaroline use in the aforementioned excluded patient populations.
Patients in each treatment group of the trials had similar demographic characteristics. The most common infections were cellulitis, major abscess requiring surgical intervention, wound infection, and infected ulcer. Bacteremia was present in 4.2% of patients in the ceftaroline group and in 3.8% of patients in the vancomycin-aztreonam group. The most common pathogen was S aureus. Methicillin resistance was present in 40% of the ceftaroline group and 34% of the control group.
Treatment. Patients received either ceftaroline 600 mg every 12 hours or the combination of vancomycin 1 g plus aztreonam 1 g given 12 hours, for 5 to 14 days.

Results. As assessed at a “test-of-cure” visit 8 to 15 days after the last dose of study medication, the efficacy of ceftaroline was similar to that of vancomycin-aztreonam, meeting the set noninferiority goal (Table 4).25 Moreover, if assessed on day 2 or 3 (a new end point recommended by the FDA), the rate of cessation of erythema spread and absence of fever was higher in the ceftaroline group than in the vancomycin-aztreonam group.26 However, this end point was not in the original trial protocol.
CEFTAROLINE FOR OTHER INDICATIONS
As noted, ceftaroline has been approved for treating community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections. In addition, it has been used in several studies in animals, and case reports of non-FDA approved indications including endocarditis and osteomyelitis have been published. Clinical trials are evaluating its use in pediatric patients, as well as for community-acquired bacterial pneumonia with risk for MRSA and for MRSA bacteremia.
Endocarditis
Animal studies have demonstrated ceftaroline to have bactericidal activity against MRSA and hVISA in endocarditis.27
A few case series have been published describing ceftaroline’s use as salvage therapy for persistent MRSA bacteremia and endocarditis. For example, Ho et al28 reported using it in three patients who had endocarditis as a source of their persistent bacteremia. All three patients had resolution of their MRSA bloodstream infection following ceftaroline therapy. The dosage was 600 mg every 8 hours, which is higher than in the manufacturer’s prescribing information.
Lin et al29 reported using ceftaroline in five patients with either possible or probable endocarditis. Three of the five patients had clinical cure as defined by resolution or improvement of all signs and symptoms of infection, and not requiring further antimicrobial therapy.29
More data from clinical trials would be beneficial in defining ceftaroline’s role in treating endocarditis caused by susceptible microorganisms.
Osteomyelitis
In animal studies of osteomyelitis, ceftaroline exhibited activity against MRSA in infected bone and joint fluid. Compared with vancomycin and linezolid, ceftaroline was associated with more significant decreases in bacterial load in the infected joint fluid, bone marrow, and bone.30
Lin et al29 gave ceftaroline to two patients with bone and joint infections, both of whom had received other therapies that had failed. The doses of ceftaroline were higher than those recommended in the prescribing information; clinical cure was noted in both cases following the switch.
These data come from case series, and more study of ceftaroline’s role in the treatment of osteomyelitis infections is warranted.
Meningitis
The use of ceftaroline in meningitis has been studied in rabbits. While ceftaroline penetrated into the cerebrospinal fluid in only negligible amounts in healthy rabbits (3% penetration), its penetration improved to 15% in animals with inflamed meninges. Ceftaroline cerebrospinal fluid levels in inflamed meninges were sufficient to provide bactericidal activity against penicillin-sensitive and resistant S pneumoniae strains as well as K pneumoniae and E coli strains.31,32
REPORTED ADVERSE EFFECTS OF CEFTAROLINE
Overall, ceftaroline was well tolerated in clinical trials, and its safety profile was similar to those of the comparator agents (ceftriaxone and vancomycin-aztreonam).
As with the other cephalosporins, hypersensitivity reactions have been reported with ceftaroline. In the clinical trials, 3% of patients developed a rash with ceftaroline.33,34 Patients with a history of beta-lactam allergy were excluded from the trials, so the rate of cross-reactivity with penicillins and with other cephalosporins is unknown.
In the phase 3 clinical trials, gastrointestinal side effects including diarrhea (5%), nausea (4%), and vomiting (2%) were reported with ceftaroline. C difficile-associated diarrhea has also been reported.33
As with other cephalosporins, ceftaroline can cause a false-positive result on the Coombs test. Approximately 11% of ceftaroline-treated patients in phase 3 clinical trials had a positive Coombs test, but hemolytic anemia did not occur in any patients.33,34
Discontinuation of ceftaroline due to an adverse reaction was reported in 2.7% of patients receiving the drug during phase 3 trials, compared with 3.7% with comparator agents.
WHEN SHOULD CEFTAROLINE BE USED IN DAILY PRACTICE?
Ceftaroline has been shown to be at least as effective as ceftriaxone in treating community-acquired bacterial pneumonia, and at least as effective as vancomycin-aztreonam in treating acute bacterial skin and skin-structure infections. The 2014 Infectious Diseases Society of America’s guidelines for the diagnosis and management of skin and soft-tissue infections recommend ceftaroline as an option for empiric therapy for purulent skin and soft-tissue infections.35
The guidelines on community-acquired pneumonia have not been updated since 2007, which was before ceftaroline was approved. However, these guidelines are currently undergoing revision and may provide insight on ceftaroline’s place in the treatment of community-acquired bacterial pneumonia.36
Currently, ceftaroline’s routine use for these indications should be balanced by its higher cost ($150 for a 600-mg dose) compared with ceftriaxone ($5 for a 1-g dose) or vancomycin ($25 for a 1-g dose). The drug’s in vitro activity against drug-resistant pneumococci and S aureus, including MRSA, hVISA, and VISA may help fill an unmet need or provide a safer and more tolerable alternative to currently available therapies.
However, ceftaroline’s lack of activity against P aeruginosa and carbapenem-resistant Enterobacteriaceae does not meet the public health threat needs stemming from these multidrug-resistant microorganisms. Ongoing clinical trials in patients with more serious MRSA infections will provide important information about ceftaroline’s role as an anti-MRSA agent.
While the discovery of antimicrobials has had one of the greatest impacts on medicine, continued antibiotic use is threatened by the emergence of drug-resistant pathogens. Therefore, it is as important as ever to be good stewards of our currently available antimicrobials. Developing usage and dosing criteria for antimicrobials based on available data and literature is a step forward in optimizing the use of antibiotics—a precious medical resource.
- Infectious Diseases Society of America. The 10 x ‘20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 2010; 50:1081–1083.
- Styers D, Sheehan DJ, Hogan P, Sahm DF. Laboratory-based surveillance of current antimicrobial resistance patterns and trends among Staphylococcus aureus: 2005 status in the United States. Ann Clin Microbiol Antimicrob 2006; 5:2.
- Farrell DJ, Castanheira M, Mendes RE, Sader HS, Jones RN. In vitro activity of ceftaroline against multidrug-resistant Staphylococcus aureus and Streptococcus pneumoniae: a review of published studies and the AWARE Surveillance Program (2008-2010). Clin Infect Dis 2012; 55(suppl 3):S206–S214.
- Holmes NE, Johnson PD, Howden BP. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J Clin Microbiol 2012; 50:2548–2552.
- Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 2003; 111:1265–1273.
- Jones RN, Sader HS, Mendes RE, Flamm RK. Update on antimicrobial susceptibility trends among Streptococcus pneumoniae in the United States: report of ceftaroline activity from the SENTRY Antimicrobial Surveillance Program (1998-2011). Diag Microbiol Infect Dis 2013; 75:107–109.
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 2008; 32:361–385.
- Centers for Disease Control and Prevention (CDC). Antibiotic resistance threats in the United States 2013. cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. Accessed June 1, 2015.
- Moisan H, Pruneau M, Malouin F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J Antimicrob Chemother 2010; 65:713–716.
- Forest Laboratories, Inc. Teflaro® (ceftaroline fosamil): prescribing information. www.frx.com/pi/teflaro_pi.pdf. Accessed June 1, 2015.
- Richter SS, Heilmann KP, Dohrn CL, et al. Activity of ceftaroline and epidemiologic trends in Staphylococcus aureus isolates collected from 43 medical centers in the United States in 2009. Antimicrob Agents Chemother 2011; 55:4154–4160.
- Ge Y, Biek D, Talbot GH, Sahm DF. In vitro profiling of ceftaroline against a collection of recent bacterial clinical isolates from across the United States. Antimicrob Agents Chemother 2008; 52:3398–3407.
- Saravolatz L, Pawlak J, Johnson L. In vitro activity of ceftaroline against community-associated methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, and daptomycin-nonsusceptible Staphylococcus aureus isolates. Antimicrob Agents Chemother 2010; 54:3027–3030.
- Mushtaq S, Livermore DM. AmpC induction by ceftaroline. J Antimicrob Chemother 2010; 65:586–588.
- Mendes RE, Tsakris A, Sader HS, et al. Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J Antimicrob Chemother 2012; 67:1321–1324.
- Lodise TP, Low DE. Ceftaroline fosamil in the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections. Drugs 2012; 72:1473–1493.
- Riccobene TA, Su SF, Rank D. Single- and multiple-dose study to determine the safety, tolerability, and pharmacokinetics of ceftaroline fosamil in combination with avibactam in healthy subjects. Antimicrob Agents Chemother 2013; 57:1496–1504.
- US National Institutes of Health. ClinicalTrials.gov. Evaluation of ceftaroline fosamil versus a comparator in adult subjects with community-acquired bacterial pneumonia (CABP) with risk for methicillin-resistant Staphylococcus aureus. http://clinicaltrials.gov/ct2/show/NCT01645735. Accessed June 1, 2015.
- File TM Jr, Low DE, Eckburg PB, et al; FOCUS 1 investigators. FOCUS 1: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii19–iii32.
- Low DE, File TM Jr, Eckburg PB, et al; FOCUS 2 investigators. FOCUS 2: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii33–iii44.
- File TM Jr, Low DE, Eckburg PB, et al. Integrated analysis of FOCUS 1 and FOCUS 2: randomized, doubled-blinded, multicenter phase 3 trials of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in patients with community-acquired pneumonia. Clin Infect Dis 2010; 51:1395–1405.
- Food and Drug Administration (FDA). Ceftaroline fosamil for the treatment of community-acquired bacterial pneumonia and complicated skin and skin structure infections. www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/anti-infectivedrugsadvisorycommittee/ucm224656.pdf. Accessed June 1, 2015.
- Corey GR, Wilcox MH, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 1 investigators. CANVAS 1: the first phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv41–iv51.
- Wilcox MH, Corey GR, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 2 investigators. CANVAS 2: the second phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv53-iv65.
- Corey GR, Wilcox M, Talbot GH, et al. Integrated analysis of CANVAS 1 and 2: phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin Infect Dis 2010; 51:641–650.
- Friedland HD, O’Neal T, Biek D, et al. CANVAS 1 and 2: analysis of clinical response at day 3 in two phase 3 trials of ceftaroline fosamil versus vancomycin plus aztreonam in treatment of acute bacterial skin and skin structure infections. Antimicrob Agents Chemother 2012; 56:2231–2236.
- Jacqueline C, Caillon J, Le Mabecque V, et al. In vivo efficacy of ceftaroline (PPI-0903), a new broad-spectrum cephalosporin, compared with linezolid and vancomycin against methicillin-resistant and vancomycin-intermediate Staphylococcus aureus in a rabbit endocarditis model. Antimicrob Agents Chemother 2007; 51:3397–3400.
- Ho TT, Cadena J, Childs LM, Gonzalez-Velez M, Lewis JS 2nd. Methicillin-resistant Staphylococcus aureus bacteraemia and endocarditis treated with ceftaroline salvage therapy. J Antimicrob Chemother 2012; 67:1267–1270.
- Lin JC, Aung G, Thomas A, Jahng M, Johns S, Fierer J. The use of ceftaroline fosamil in methicillin-resistant Staphylococcus aureus endocarditis and deep-seated MRSA infections: a retrospective case series of 10 patients. J Infect Chemother 2013; 19:42–49.
- Jacqueline C, Amador G, Caillon J, et al. Efficacy of the new cephalosporin ceftaroline in the treatment of experimental methicillin-resistant Staphylococcus aureus acute osteomyelitis. J Antimicrob Chemother 2010; 65:1749–1752.
- Stucki A, Acosta F, Cottagnoud M, Cottagnoud P. Efficacy of ceftaroline fosamil against Escherichia coli and Klebsiella pneumoniae strains in a rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:5808–5810.
- Cottagnoud P, Cottagnoud M, Acosta F, Stucki A. Efficacy of ceftaroline fosamil against penicillin-sensitive and -resistant Streptococcus pneumoniae in an experimental rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:4653–4655.
- Corrado ML. Integrated safety summary of CANVAS 1 and 2 trials: phase III, randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv67–iv71.
- Rank DR, Friedland HD, Laudano JB. Integrated safety summary of FOCUS 1 and FOCUS 2 trials: phase III randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii53–iii59.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis 2014; 59:147–159.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44:S27–S72.
Ceftaroline fosamil (Teflaro), introduced to the US market in October 2010, is the first beta-lactam agent with clinically useful activity against methicillin-resistant Staphylococcus aureus (MRSA). Currently, it is approved by the US Food and Drug Administration (FDA) to treat acute bacterial skin and skin-structure infections and community-acquired bacterial pneumonia caused by susceptible microorganisms.
In an era of increasing drug resistance and limited numbers of antimicrobials in the drug-production pipeline, ceftaroline is a step forward in fulfilling the Infectious Diseases Society of America’s “10 × ’20 Initiative” to increase support for drug research and manufacturing, with the goal of producing 10 new antimicrobial drugs by the year 2020.1 Ceftaroline was the first of several antibiotics to receive FDA approval in response to this initiative. It was followed by dalbavancin (May 2014), tedizolid phosphate (June 2014), oritavancin (August 2014), ceftolozane-tazobactam (December 2014), and ceftazidime-avibactam (February 2015). These antibiotic agents are aimed at treating infections caused by drug-resistant gram-positive and gram-negative microorganisms. It is important to understand and optimize the use of these new antibiotic agents in order to decrease the risk of emerging antibiotic resistance and superinfections (eg, Clostridium difficile infection) caused by antibiotic overuse or misuse.
This article provides an overview of ceftaroline’s mechanisms of action and resistance, spectrum of activity, pharmacokinetic properties, adverse effects, and current place in therapy.
AN ERA OF MULTIDRUG-RESISTANT MICROORGANISMS
Increasing rates of antimicrobial resistance threaten the efficacy of antimicrobial drugs in the daily practice of medicine. The World Health Organization has labeled antimicrobial resistance one of the three greatest threats to human health. Global efforts are under way to stimulate development of new antimicrobial agents and to decrease rates of antimicrobial resistance.
Staphylococcus aureus: A threat, even with vancomycin
Between 1998 and 2005, S aureus was one of the most common inpatient and outpatient isolates reported by clinical laboratories throughout the United States.2
Treatment of S aureus infection is complicated by a variety of resistance mechanisms that have evolved over time. In fact, the first resistant isolate of S aureus emerged not long after penicillin’s debut into clinical practice, and now the majority of strains are resistant to penicillin.
Methicillin was designed to overcome this beta-lactamase resistance and became the treatment of choice for penicillin-resistant S aureus isolates. However, MRSA isolates soon emerged because of the organism’s acquisition of penicillin-binding protein PBP2a via the mecA gene, leading to decreased binding affinity of methicillin.3
Since then, several agents active against MRSA (vancomycin, daptomycin, linezolid, tigecycline) have been introduced and continue to be widely used. While vancomycin is considered the first-line option for a variety of MRSA infections, its use has been threatened because of the emergence of vancomycin-intermediate-resistant S aureus (VISA), S aureus strains displaying vancomycin heteroresistance (hVISA), and vancomycin-resistant S aureus (VRSA) strains.4
VISA and hVISA isolates emerged through sequential mutations that lead to autolytic activity and cell-wall thickening. In contrast, the mechanism of resistance in VRSA is by acquisition of the vanA resistance gene, which alters the binding site of vancomycin from d-alanine-d-alanine to d-alanine-d-lactate.5
Streptococcus pneumoniae resistance: A continuing problem
The prevalence of drug resistance in S pneumoniae has risen since the late 1990s. A 2013 report from the SENTRY Antimicrobial Surveillance Program stated that almost 20% of S pneumoniae isolates were resistant to amoxicillin-clavulanate, and similar trends have been observed for penicillin (14.8%) and ceftriaxone (11.7%).6
S pneumoniae resistance is acquired through modifications of the penicillin-binding proteins, namely PBP1a, PBP2b, PBP2x, and, less frequently, PBP2a. These modifications lead to decreased binding affinity for most beta-lactams.7
Clinical impact of multidrug-resistant S aureus and S pneumoniae
In 2011, the US Centers for Disease Control and Prevention reported an estimated 80,000 severe MRSA infections and 11,000 MRSA-related deaths in the United States.8 In the same report, drug-resistant S pneumoniae was estimated to be responsible for almost 1.2 million illnesses and 7,000 deaths per year, leading to upwards of $96 million in related medical costs.
While invasive drug-resistant S pneumoniae infections usually affect patients at the extremes of age (under age 5 and over age 65), they have had a serious impact on patients of all ages.8
In light of the increasing prevalence of multidrug-resistant organisms, newer antimicrobial agents with novel mechanisms of action are needed.
CEFTAROLINE: A BETA-LACTAM WITH ANTI-MRSA ACTIVITY
The cephalosporins, a class of beta-lactam antibiotics, were originally derived from the fungus Cephalosporium (now called Acremonium). There are now many agents in this class, each containing a nucleus consisting of a beta-lactam ring fused to a six-member dihydrothiazine ring, and two side chains that can be modified to affect antibacterial activity and pharmacokinetic properties.
Cephalosporins are typically categorized into “generations.” With some exceptions, the first- and second-generation agents have good activity against gram-positive microorganisms, including methicillin-susceptible S aureus—but not against MRSA. The third- and fourth-generation cephalosporins have better gram-negative activity, with many agents having activity against the gram-negative bacterium Pseudomonas aeruginosa.
Enterococcal isolates are intrinsically resistant to cephalosporins. Additionally, cephalosporins are not active against anaerobic bacteria, except for a subset of structurally unique second-generation cephalosporins, ie, cefotetan and cefoxitin.
Ceftaroline was synthesized with specific manipulations of the side chains to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae isolates, making it the first available beta-lactam with this ability.
Mechanism of action
Ceftaroline binds to penicillin-binding proteins, inhibiting transpeptidation. This interaction blocks the final stage of peptidoglycan synthesis and inhibits bacterial cell wall formation, ultimately leading to cellular autolysis and microorganism death. Ceftaroline binds with high affinity to PBP2a and PBP2x, expanding its activity to encompass MRSA and penicillin-resistant S pneumoniae isolates.9
Spectrum of activity
Ceftaroline has in vitro activity against many gram-positive and gram-negative bacteria,10–13 including (Table 1):
- Methicillin-susceptible and methicillin-resistant staphylococci
- VISA, VRSA, and hVISA
- Daptomycin-nonsusceptible S aureus
- Streptococcal species, including penicillin-resistant S pneumoniae
- Enterobacteriaceae, including Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Citrobacter koseri, Citrobacter freundii, Enterobacter cloacae, Enterobacter aerogenes, Moraxella catarrhalis, Morganella morganii, and Proteus mirabilis.
Of note, ceftaroline is not active against Pseudomonas species, Enterococcus species, or Bacteroides fragilis. In addition, it is not active against the “atypical” respiratory pathogens Mycoplasma pneumoniae, Chlamydophila pneumoniae, and Legionella pneumophila.
Ceftaroline resistance
Gram-negative organisms appear to develop resistance to ceftaroline at rates similar to those observed with the other oxyimino-cephalosporins (eg, ceftriaxone). Ceftaroline is inactive against gram-negative organisms producing extended-spectrum beta-lactamases, including K pneumoniae carbapenemase and metallo-beta-lactamases.14 In addition, it induces the expression of AmpC beta-lactamases.
Although currently uncommon, resistance to ceftaroline has also been reported in S aureus strains.15 The mechanism of resistance is decreased binding affinity for PBP2a due to amino acid substitutions on the nonpenicillin-binding domains.15
Pharmacokinetic profile
An understanding of pharmacokinetics is key in optimizing the dose of antimicrobials so that the drugs are used most effectively and pathogens do not develop resistance to them.
Ceftaroline fosamil is a prodrug that, upon intravenous administration, is rapidly converted by phosphatase enzymes to its active moiety, ceftaroline. Its pharmacokinetic profile is summarized in Table 2.16,17 Its volume of distribution is similar to that of the fourth-generation cephalosporin cefepime.
Ceftaroline is then hydrolyzed into its inactive metabolite, ceftaroline M-1. It undergoes little hepatic metabolism and lacks properties to make it a substrate, inhibitor, or inducer of the CYP450 enzyme system and therefore is not likely to cause notable CYP450-related drug-drug interactions.
Like most other beta-lactams, ceftaroline is primarily excreted by the kidneys. Furthermore, an estimated 21% of a dose is eliminated with each intermittent hemodialysis session. Therefore, renal and intermittent hemodialysis dose adjustments are necessary. The estimated elimination half-life is 2.6 hours, necessitating dosing two to three times daily, depending on the indication and infectious inoculum.
Ceftaroline dosing
Ceftaroline is available only in a parenteral preparation and is typically given at a dose of 600 mg every 12 hours.10 The intravenous infusion is given over 1 hour.
The current stability data require reconstituted ceftaroline to be used within 6 hours at room temperature and within 24 hours if refrigerated.10
Ceftaroline requires dosing adjustments for patients with renal insufficiency. Per the manufacturer, renal dosing adjustments are based on the creatinine clearance rate, as estimated by the Cockroft-Gault formula:
- Creatinine clearance > 50 mL/min: no dosage adjustment necessary
- Creatinine clearance > 30 to ≤ 50 mL/min: give 400 mg every 12 hours
- Creatinine clearance ≥ 15 to ≤ 30 mL/min: give 300 mg every 12 hours
- Creatinine clearance < 15 mL/min or on intermittent dialysis: give 200 mg every 12 hours.
Ongoing clinical trials are investigating a higher-dosing strategy of 600 mg every 8 hours for patients with community-acquired bacterial pneumonia at risk of MRSA bacteremia.18
CLINICAL TRIALS LEADING TO CEFTAROLINE’S APPROVAL
Ceftaroline was approved for the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections due to susceptible pathogens on the basis of phase 3 comparator trials.
Community-acquired bacterial pneumonia: The FOCUS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of community-acquired bacterial pneumonia was studied in two randomized, double-blind, noninferiority trials, known as Ceftaroline Community-acquired Pneumonia vs Ceftriaxone (FOCUS) 1 and FOCUS 2.19,20
Patients were adults and not critically ill, as was reflected by their being in Pneumonia Outcomes Research Team (PORT) risk class III or IV (with class V indicating the highest risk of death). Therefore, the results may not be completely applicable to critically ill patients or those not admitted to the hospital. Of note, patients were excluded from the trials if they had infections known or thought to be due to MRSA or to atypical organisms.21 Baseline characteristics and patient demographics were similar between study groups in both trials.
A bacterial pathogen was identified in 26.1% of the patients included in the modified intent-to-treat analysis of the pooled data of the trials; the most common pathogens were S pneumoniae, methicillin-sensitive S aureus, Haemophilus influenzae, K pneumoniae, and E coli.21
Treatment. Patients received either ceftaroline 600 mg every 12 hours (or a lower dose based on renal function) or ceftriaxone 1 g every 24 hours. In addition, in the FOCUS 1 trial, patients in both treatment groups received clarithromycin 500 mg every 12 hours for the first day.19
Results. In both trials and in the integrated analysis, ceftaroline was noninferior to ceftriaxone (Table 3).22 In the integrated analysis of both trials, compared with the ceftriaxone group, the ceftaroline group had a higher clinical cure rate among patients classified as PORT risk class III (86.8% vs 79.2%, weighted treatment difference 12.6%, 95% confidence interval [CI] 1.3–13.8) and among patients who had not received prior antibiotic treatment (85.5% vs 74.9%, weighted treatment difference 11.2%, 95% CI 4.5–18.0).21
Acute bacterial skin and skin-structure infections: The CANVAS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of complicated acute bacterial skin and skin-structure infections was studied in two randomized, double-blind trials: Ceftaroline Versus Vancomycin in Skin and Skin Structure Infections (CANVAS) 1 and CANVAS 2.23,24
Patients. Adult patients with a diagnosis of community-acquired skin and skin-structure infections warranting at least 5 days of intravenous antimicrobial therapy were included in the trials. Important protocol exclusions were patients with diabetic foot ulcers, decubitus ulcers, burns, ulcers associated with peripheral vascular disease accompanied by osteomyelitis, and suspected P aeruginosa infections.25 This limits the external validity of ceftaroline use in the aforementioned excluded patient populations.
Patients in each treatment group of the trials had similar demographic characteristics. The most common infections were cellulitis, major abscess requiring surgical intervention, wound infection, and infected ulcer. Bacteremia was present in 4.2% of patients in the ceftaroline group and in 3.8% of patients in the vancomycin-aztreonam group. The most common pathogen was S aureus. Methicillin resistance was present in 40% of the ceftaroline group and 34% of the control group.
Treatment. Patients received either ceftaroline 600 mg every 12 hours or the combination of vancomycin 1 g plus aztreonam 1 g given 12 hours, for 5 to 14 days.
Results. As assessed at a “test-of-cure” visit 8 to 15 days after the last dose of study medication, the efficacy of ceftaroline was similar to that of vancomycin-aztreonam, meeting the set noninferiority goal (Table 4).25 Moreover, if assessed on day 2 or 3 (a new end point recommended by the FDA), the rate of cessation of erythema spread and absence of fever was higher in the ceftaroline group than in the vancomycin-aztreonam group.26 However, this end point was not in the original trial protocol.
CEFTAROLINE FOR OTHER INDICATIONS
As noted, ceftaroline has been approved for treating community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections. In addition, it has been used in several studies in animals, and case reports of non-FDA approved indications including endocarditis and osteomyelitis have been published. Clinical trials are evaluating its use in pediatric patients, as well as for community-acquired bacterial pneumonia with risk for MRSA and for MRSA bacteremia.
Endocarditis
Animal studies have demonstrated ceftaroline to have bactericidal activity against MRSA and hVISA in endocarditis.27
A few case series have been published describing ceftaroline’s use as salvage therapy for persistent MRSA bacteremia and endocarditis. For example, Ho et al28 reported using it in three patients who had endocarditis as a source of their persistent bacteremia. All three patients had resolution of their MRSA bloodstream infection following ceftaroline therapy. The dosage was 600 mg every 8 hours, which is higher than in the manufacturer’s prescribing information.
Lin et al29 reported using ceftaroline in five patients with either possible or probable endocarditis. Three of the five patients had clinical cure as defined by resolution or improvement of all signs and symptoms of infection, and not requiring further antimicrobial therapy.29
More data from clinical trials would be beneficial in defining ceftaroline’s role in treating endocarditis caused by susceptible microorganisms.
Osteomyelitis
In animal studies of osteomyelitis, ceftaroline exhibited activity against MRSA in infected bone and joint fluid. Compared with vancomycin and linezolid, ceftaroline was associated with more significant decreases in bacterial load in the infected joint fluid, bone marrow, and bone.30
Lin et al29 gave ceftaroline to two patients with bone and joint infections, both of whom had received other therapies that had failed. The doses of ceftaroline were higher than those recommended in the prescribing information; clinical cure was noted in both cases following the switch.
These data come from case series, and more study of ceftaroline’s role in the treatment of osteomyelitis infections is warranted.
Meningitis
The use of ceftaroline in meningitis has been studied in rabbits. While ceftaroline penetrated into the cerebrospinal fluid in only negligible amounts in healthy rabbits (3% penetration), its penetration improved to 15% in animals with inflamed meninges. Ceftaroline cerebrospinal fluid levels in inflamed meninges were sufficient to provide bactericidal activity against penicillin-sensitive and resistant S pneumoniae strains as well as K pneumoniae and E coli strains.31,32
REPORTED ADVERSE EFFECTS OF CEFTAROLINE
Overall, ceftaroline was well tolerated in clinical trials, and its safety profile was similar to those of the comparator agents (ceftriaxone and vancomycin-aztreonam).
As with the other cephalosporins, hypersensitivity reactions have been reported with ceftaroline. In the clinical trials, 3% of patients developed a rash with ceftaroline.33,34 Patients with a history of beta-lactam allergy were excluded from the trials, so the rate of cross-reactivity with penicillins and with other cephalosporins is unknown.
In the phase 3 clinical trials, gastrointestinal side effects including diarrhea (5%), nausea (4%), and vomiting (2%) were reported with ceftaroline. C difficile-associated diarrhea has also been reported.33
As with other cephalosporins, ceftaroline can cause a false-positive result on the Coombs test. Approximately 11% of ceftaroline-treated patients in phase 3 clinical trials had a positive Coombs test, but hemolytic anemia did not occur in any patients.33,34
Discontinuation of ceftaroline due to an adverse reaction was reported in 2.7% of patients receiving the drug during phase 3 trials, compared with 3.7% with comparator agents.
WHEN SHOULD CEFTAROLINE BE USED IN DAILY PRACTICE?
Ceftaroline has been shown to be at least as effective as ceftriaxone in treating community-acquired bacterial pneumonia, and at least as effective as vancomycin-aztreonam in treating acute bacterial skin and skin-structure infections. The 2014 Infectious Diseases Society of America’s guidelines for the diagnosis and management of skin and soft-tissue infections recommend ceftaroline as an option for empiric therapy for purulent skin and soft-tissue infections.35
The guidelines on community-acquired pneumonia have not been updated since 2007, which was before ceftaroline was approved. However, these guidelines are currently undergoing revision and may provide insight on ceftaroline’s place in the treatment of community-acquired bacterial pneumonia.36
Currently, ceftaroline’s routine use for these indications should be balanced by its higher cost ($150 for a 600-mg dose) compared with ceftriaxone ($5 for a 1-g dose) or vancomycin ($25 for a 1-g dose). The drug’s in vitro activity against drug-resistant pneumococci and S aureus, including MRSA, hVISA, and VISA may help fill an unmet need or provide a safer and more tolerable alternative to currently available therapies.
However, ceftaroline’s lack of activity against P aeruginosa and carbapenem-resistant Enterobacteriaceae does not meet the public health threat needs stemming from these multidrug-resistant microorganisms. Ongoing clinical trials in patients with more serious MRSA infections will provide important information about ceftaroline’s role as an anti-MRSA agent.
While the discovery of antimicrobials has had one of the greatest impacts on medicine, continued antibiotic use is threatened by the emergence of drug-resistant pathogens. Therefore, it is as important as ever to be good stewards of our currently available antimicrobials. Developing usage and dosing criteria for antimicrobials based on available data and literature is a step forward in optimizing the use of antibiotics—a precious medical resource.
Ceftaroline fosamil (Teflaro), introduced to the US market in October 2010, is the first beta-lactam agent with clinically useful activity against methicillin-resistant Staphylococcus aureus (MRSA). Currently, it is approved by the US Food and Drug Administration (FDA) to treat acute bacterial skin and skin-structure infections and community-acquired bacterial pneumonia caused by susceptible microorganisms.
In an era of increasing drug resistance and limited numbers of antimicrobials in the drug-production pipeline, ceftaroline is a step forward in fulfilling the Infectious Diseases Society of America’s “10 × ’20 Initiative” to increase support for drug research and manufacturing, with the goal of producing 10 new antimicrobial drugs by the year 2020.1 Ceftaroline was the first of several antibiotics to receive FDA approval in response to this initiative. It was followed by dalbavancin (May 2014), tedizolid phosphate (June 2014), oritavancin (August 2014), ceftolozane-tazobactam (December 2014), and ceftazidime-avibactam (February 2015). These antibiotic agents are aimed at treating infections caused by drug-resistant gram-positive and gram-negative microorganisms. It is important to understand and optimize the use of these new antibiotic agents in order to decrease the risk of emerging antibiotic resistance and superinfections (eg, Clostridium difficile infection) caused by antibiotic overuse or misuse.
This article provides an overview of ceftaroline’s mechanisms of action and resistance, spectrum of activity, pharmacokinetic properties, adverse effects, and current place in therapy.
AN ERA OF MULTIDRUG-RESISTANT MICROORGANISMS
Increasing rates of antimicrobial resistance threaten the efficacy of antimicrobial drugs in the daily practice of medicine. The World Health Organization has labeled antimicrobial resistance one of the three greatest threats to human health. Global efforts are under way to stimulate development of new antimicrobial agents and to decrease rates of antimicrobial resistance.
Staphylococcus aureus: A threat, even with vancomycin
Between 1998 and 2005, S aureus was one of the most common inpatient and outpatient isolates reported by clinical laboratories throughout the United States.2
Treatment of S aureus infection is complicated by a variety of resistance mechanisms that have evolved over time. In fact, the first resistant isolate of S aureus emerged not long after penicillin’s debut into clinical practice, and now the majority of strains are resistant to penicillin.
Methicillin was designed to overcome this beta-lactamase resistance and became the treatment of choice for penicillin-resistant S aureus isolates. However, MRSA isolates soon emerged because of the organism’s acquisition of penicillin-binding protein PBP2a via the mecA gene, leading to decreased binding affinity of methicillin.3
Since then, several agents active against MRSA (vancomycin, daptomycin, linezolid, tigecycline) have been introduced and continue to be widely used. While vancomycin is considered the first-line option for a variety of MRSA infections, its use has been threatened because of the emergence of vancomycin-intermediate-resistant S aureus (VISA), S aureus strains displaying vancomycin heteroresistance (hVISA), and vancomycin-resistant S aureus (VRSA) strains.4
VISA and hVISA isolates emerged through sequential mutations that lead to autolytic activity and cell-wall thickening. In contrast, the mechanism of resistance in VRSA is by acquisition of the vanA resistance gene, which alters the binding site of vancomycin from d-alanine-d-alanine to d-alanine-d-lactate.5
Streptococcus pneumoniae resistance: A continuing problem
The prevalence of drug resistance in S pneumoniae has risen since the late 1990s. A 2013 report from the SENTRY Antimicrobial Surveillance Program stated that almost 20% of S pneumoniae isolates were resistant to amoxicillin-clavulanate, and similar trends have been observed for penicillin (14.8%) and ceftriaxone (11.7%).6
S pneumoniae resistance is acquired through modifications of the penicillin-binding proteins, namely PBP1a, PBP2b, PBP2x, and, less frequently, PBP2a. These modifications lead to decreased binding affinity for most beta-lactams.7
Clinical impact of multidrug-resistant S aureus and S pneumoniae
In 2011, the US Centers for Disease Control and Prevention reported an estimated 80,000 severe MRSA infections and 11,000 MRSA-related deaths in the United States.8 In the same report, drug-resistant S pneumoniae was estimated to be responsible for almost 1.2 million illnesses and 7,000 deaths per year, leading to upwards of $96 million in related medical costs.
While invasive drug-resistant S pneumoniae infections usually affect patients at the extremes of age (under age 5 and over age 65), they have had a serious impact on patients of all ages.8
In light of the increasing prevalence of multidrug-resistant organisms, newer antimicrobial agents with novel mechanisms of action are needed.
CEFTAROLINE: A BETA-LACTAM WITH ANTI-MRSA ACTIVITY
The cephalosporins, a class of beta-lactam antibiotics, were originally derived from the fungus Cephalosporium (now called Acremonium). There are now many agents in this class, each containing a nucleus consisting of a beta-lactam ring fused to a six-member dihydrothiazine ring, and two side chains that can be modified to affect antibacterial activity and pharmacokinetic properties.
Cephalosporins are typically categorized into “generations.” With some exceptions, the first- and second-generation agents have good activity against gram-positive microorganisms, including methicillin-susceptible S aureus—but not against MRSA. The third- and fourth-generation cephalosporins have better gram-negative activity, with many agents having activity against the gram-negative bacterium Pseudomonas aeruginosa.
Enterococcal isolates are intrinsically resistant to cephalosporins. Additionally, cephalosporins are not active against anaerobic bacteria, except for a subset of structurally unique second-generation cephalosporins, ie, cefotetan and cefoxitin.
Ceftaroline was synthesized with specific manipulations of the side chains to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae isolates, making it the first available beta-lactam with this ability.
Mechanism of action
Ceftaroline binds to penicillin-binding proteins, inhibiting transpeptidation. This interaction blocks the final stage of peptidoglycan synthesis and inhibits bacterial cell wall formation, ultimately leading to cellular autolysis and microorganism death. Ceftaroline binds with high affinity to PBP2a and PBP2x, expanding its activity to encompass MRSA and penicillin-resistant S pneumoniae isolates.9
Spectrum of activity
Ceftaroline has in vitro activity against many gram-positive and gram-negative bacteria,10–13 including (Table 1):
- Methicillin-susceptible and methicillin-resistant staphylococci
- VISA, VRSA, and hVISA
- Daptomycin-nonsusceptible S aureus
- Streptococcal species, including penicillin-resistant S pneumoniae
- Enterobacteriaceae, including Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Citrobacter koseri, Citrobacter freundii, Enterobacter cloacae, Enterobacter aerogenes, Moraxella catarrhalis, Morganella morganii, and Proteus mirabilis.
Of note, ceftaroline is not active against Pseudomonas species, Enterococcus species, or Bacteroides fragilis. In addition, it is not active against the “atypical” respiratory pathogens Mycoplasma pneumoniae, Chlamydophila pneumoniae, and Legionella pneumophila.
Ceftaroline resistance
Gram-negative organisms appear to develop resistance to ceftaroline at rates similar to those observed with the other oxyimino-cephalosporins (eg, ceftriaxone). Ceftaroline is inactive against gram-negative organisms producing extended-spectrum beta-lactamases, including K pneumoniae carbapenemase and metallo-beta-lactamases.14 In addition, it induces the expression of AmpC beta-lactamases.
Although currently uncommon, resistance to ceftaroline has also been reported in S aureus strains.15 The mechanism of resistance is decreased binding affinity for PBP2a due to amino acid substitutions on the nonpenicillin-binding domains.15
Pharmacokinetic profile
An understanding of pharmacokinetics is key in optimizing the dose of antimicrobials so that the drugs are used most effectively and pathogens do not develop resistance to them.
Ceftaroline fosamil is a prodrug that, upon intravenous administration, is rapidly converted by phosphatase enzymes to its active moiety, ceftaroline. Its pharmacokinetic profile is summarized in Table 2.16,17 Its volume of distribution is similar to that of the fourth-generation cephalosporin cefepime.
Ceftaroline is then hydrolyzed into its inactive metabolite, ceftaroline M-1. It undergoes little hepatic metabolism and lacks properties to make it a substrate, inhibitor, or inducer of the CYP450 enzyme system and therefore is not likely to cause notable CYP450-related drug-drug interactions.
Like most other beta-lactams, ceftaroline is primarily excreted by the kidneys. Furthermore, an estimated 21% of a dose is eliminated with each intermittent hemodialysis session. Therefore, renal and intermittent hemodialysis dose adjustments are necessary. The estimated elimination half-life is 2.6 hours, necessitating dosing two to three times daily, depending on the indication and infectious inoculum.
Ceftaroline dosing
Ceftaroline is available only in a parenteral preparation and is typically given at a dose of 600 mg every 12 hours.10 The intravenous infusion is given over 1 hour.
The current stability data require reconstituted ceftaroline to be used within 6 hours at room temperature and within 24 hours if refrigerated.10
Ceftaroline requires dosing adjustments for patients with renal insufficiency. Per the manufacturer, renal dosing adjustments are based on the creatinine clearance rate, as estimated by the Cockroft-Gault formula:
- Creatinine clearance > 50 mL/min: no dosage adjustment necessary
- Creatinine clearance > 30 to ≤ 50 mL/min: give 400 mg every 12 hours
- Creatinine clearance ≥ 15 to ≤ 30 mL/min: give 300 mg every 12 hours
- Creatinine clearance < 15 mL/min or on intermittent dialysis: give 200 mg every 12 hours.
Ongoing clinical trials are investigating a higher-dosing strategy of 600 mg every 8 hours for patients with community-acquired bacterial pneumonia at risk of MRSA bacteremia.18
CLINICAL TRIALS LEADING TO CEFTAROLINE’S APPROVAL
Ceftaroline was approved for the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections due to susceptible pathogens on the basis of phase 3 comparator trials.
Community-acquired bacterial pneumonia: The FOCUS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of community-acquired bacterial pneumonia was studied in two randomized, double-blind, noninferiority trials, known as Ceftaroline Community-acquired Pneumonia vs Ceftriaxone (FOCUS) 1 and FOCUS 2.19,20
Patients were adults and not critically ill, as was reflected by their being in Pneumonia Outcomes Research Team (PORT) risk class III or IV (with class V indicating the highest risk of death). Therefore, the results may not be completely applicable to critically ill patients or those not admitted to the hospital. Of note, patients were excluded from the trials if they had infections known or thought to be due to MRSA or to atypical organisms.21 Baseline characteristics and patient demographics were similar between study groups in both trials.
A bacterial pathogen was identified in 26.1% of the patients included in the modified intent-to-treat analysis of the pooled data of the trials; the most common pathogens were S pneumoniae, methicillin-sensitive S aureus, Haemophilus influenzae, K pneumoniae, and E coli.21
Treatment. Patients received either ceftaroline 600 mg every 12 hours (or a lower dose based on renal function) or ceftriaxone 1 g every 24 hours. In addition, in the FOCUS 1 trial, patients in both treatment groups received clarithromycin 500 mg every 12 hours for the first day.19
Results. In both trials and in the integrated analysis, ceftaroline was noninferior to ceftriaxone (Table 3).22 In the integrated analysis of both trials, compared with the ceftriaxone group, the ceftaroline group had a higher clinical cure rate among patients classified as PORT risk class III (86.8% vs 79.2%, weighted treatment difference 12.6%, 95% confidence interval [CI] 1.3–13.8) and among patients who had not received prior antibiotic treatment (85.5% vs 74.9%, weighted treatment difference 11.2%, 95% CI 4.5–18.0).21
Acute bacterial skin and skin-structure infections: The CANVAS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of complicated acute bacterial skin and skin-structure infections was studied in two randomized, double-blind trials: Ceftaroline Versus Vancomycin in Skin and Skin Structure Infections (CANVAS) 1 and CANVAS 2.23,24
Patients. Adult patients with a diagnosis of community-acquired skin and skin-structure infections warranting at least 5 days of intravenous antimicrobial therapy were included in the trials. Important protocol exclusions were patients with diabetic foot ulcers, decubitus ulcers, burns, ulcers associated with peripheral vascular disease accompanied by osteomyelitis, and suspected P aeruginosa infections.25 This limits the external validity of ceftaroline use in the aforementioned excluded patient populations.
Patients in each treatment group of the trials had similar demographic characteristics. The most common infections were cellulitis, major abscess requiring surgical intervention, wound infection, and infected ulcer. Bacteremia was present in 4.2% of patients in the ceftaroline group and in 3.8% of patients in the vancomycin-aztreonam group. The most common pathogen was S aureus. Methicillin resistance was present in 40% of the ceftaroline group and 34% of the control group.
Treatment. Patients received either ceftaroline 600 mg every 12 hours or the combination of vancomycin 1 g plus aztreonam 1 g given 12 hours, for 5 to 14 days.
Results. As assessed at a “test-of-cure” visit 8 to 15 days after the last dose of study medication, the efficacy of ceftaroline was similar to that of vancomycin-aztreonam, meeting the set noninferiority goal (Table 4).25 Moreover, if assessed on day 2 or 3 (a new end point recommended by the FDA), the rate of cessation of erythema spread and absence of fever was higher in the ceftaroline group than in the vancomycin-aztreonam group.26 However, this end point was not in the original trial protocol.
CEFTAROLINE FOR OTHER INDICATIONS
As noted, ceftaroline has been approved for treating community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections. In addition, it has been used in several studies in animals, and case reports of non-FDA approved indications including endocarditis and osteomyelitis have been published. Clinical trials are evaluating its use in pediatric patients, as well as for community-acquired bacterial pneumonia with risk for MRSA and for MRSA bacteremia.
Endocarditis
Animal studies have demonstrated ceftaroline to have bactericidal activity against MRSA and hVISA in endocarditis.27
A few case series have been published describing ceftaroline’s use as salvage therapy for persistent MRSA bacteremia and endocarditis. For example, Ho et al28 reported using it in three patients who had endocarditis as a source of their persistent bacteremia. All three patients had resolution of their MRSA bloodstream infection following ceftaroline therapy. The dosage was 600 mg every 8 hours, which is higher than in the manufacturer’s prescribing information.
Lin et al29 reported using ceftaroline in five patients with either possible or probable endocarditis. Three of the five patients had clinical cure as defined by resolution or improvement of all signs and symptoms of infection, and not requiring further antimicrobial therapy.29
More data from clinical trials would be beneficial in defining ceftaroline’s role in treating endocarditis caused by susceptible microorganisms.
Osteomyelitis
In animal studies of osteomyelitis, ceftaroline exhibited activity against MRSA in infected bone and joint fluid. Compared with vancomycin and linezolid, ceftaroline was associated with more significant decreases in bacterial load in the infected joint fluid, bone marrow, and bone.30
Lin et al29 gave ceftaroline to two patients with bone and joint infections, both of whom had received other therapies that had failed. The doses of ceftaroline were higher than those recommended in the prescribing information; clinical cure was noted in both cases following the switch.
These data come from case series, and more study of ceftaroline’s role in the treatment of osteomyelitis infections is warranted.
Meningitis
The use of ceftaroline in meningitis has been studied in rabbits. While ceftaroline penetrated into the cerebrospinal fluid in only negligible amounts in healthy rabbits (3% penetration), its penetration improved to 15% in animals with inflamed meninges. Ceftaroline cerebrospinal fluid levels in inflamed meninges were sufficient to provide bactericidal activity against penicillin-sensitive and resistant S pneumoniae strains as well as K pneumoniae and E coli strains.31,32
REPORTED ADVERSE EFFECTS OF CEFTAROLINE
Overall, ceftaroline was well tolerated in clinical trials, and its safety profile was similar to those of the comparator agents (ceftriaxone and vancomycin-aztreonam).
As with the other cephalosporins, hypersensitivity reactions have been reported with ceftaroline. In the clinical trials, 3% of patients developed a rash with ceftaroline.33,34 Patients with a history of beta-lactam allergy were excluded from the trials, so the rate of cross-reactivity with penicillins and with other cephalosporins is unknown.
In the phase 3 clinical trials, gastrointestinal side effects including diarrhea (5%), nausea (4%), and vomiting (2%) were reported with ceftaroline. C difficile-associated diarrhea has also been reported.33
As with other cephalosporins, ceftaroline can cause a false-positive result on the Coombs test. Approximately 11% of ceftaroline-treated patients in phase 3 clinical trials had a positive Coombs test, but hemolytic anemia did not occur in any patients.33,34
Discontinuation of ceftaroline due to an adverse reaction was reported in 2.7% of patients receiving the drug during phase 3 trials, compared with 3.7% with comparator agents.
WHEN SHOULD CEFTAROLINE BE USED IN DAILY PRACTICE?
Ceftaroline has been shown to be at least as effective as ceftriaxone in treating community-acquired bacterial pneumonia, and at least as effective as vancomycin-aztreonam in treating acute bacterial skin and skin-structure infections. The 2014 Infectious Diseases Society of America’s guidelines for the diagnosis and management of skin and soft-tissue infections recommend ceftaroline as an option for empiric therapy for purulent skin and soft-tissue infections.35
The guidelines on community-acquired pneumonia have not been updated since 2007, which was before ceftaroline was approved. However, these guidelines are currently undergoing revision and may provide insight on ceftaroline’s place in the treatment of community-acquired bacterial pneumonia.36
Currently, ceftaroline’s routine use for these indications should be balanced by its higher cost ($150 for a 600-mg dose) compared with ceftriaxone ($5 for a 1-g dose) or vancomycin ($25 for a 1-g dose). The drug’s in vitro activity against drug-resistant pneumococci and S aureus, including MRSA, hVISA, and VISA may help fill an unmet need or provide a safer and more tolerable alternative to currently available therapies.
However, ceftaroline’s lack of activity against P aeruginosa and carbapenem-resistant Enterobacteriaceae does not meet the public health threat needs stemming from these multidrug-resistant microorganisms. Ongoing clinical trials in patients with more serious MRSA infections will provide important information about ceftaroline’s role as an anti-MRSA agent.
While the discovery of antimicrobials has had one of the greatest impacts on medicine, continued antibiotic use is threatened by the emergence of drug-resistant pathogens. Therefore, it is as important as ever to be good stewards of our currently available antimicrobials. Developing usage and dosing criteria for antimicrobials based on available data and literature is a step forward in optimizing the use of antibiotics—a precious medical resource.
- Infectious Diseases Society of America. The 10 x ‘20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 2010; 50:1081–1083.
- Styers D, Sheehan DJ, Hogan P, Sahm DF. Laboratory-based surveillance of current antimicrobial resistance patterns and trends among Staphylococcus aureus: 2005 status in the United States. Ann Clin Microbiol Antimicrob 2006; 5:2.
- Farrell DJ, Castanheira M, Mendes RE, Sader HS, Jones RN. In vitro activity of ceftaroline against multidrug-resistant Staphylococcus aureus and Streptococcus pneumoniae: a review of published studies and the AWARE Surveillance Program (2008-2010). Clin Infect Dis 2012; 55(suppl 3):S206–S214.
- Holmes NE, Johnson PD, Howden BP. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J Clin Microbiol 2012; 50:2548–2552.
- Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 2003; 111:1265–1273.
- Jones RN, Sader HS, Mendes RE, Flamm RK. Update on antimicrobial susceptibility trends among Streptococcus pneumoniae in the United States: report of ceftaroline activity from the SENTRY Antimicrobial Surveillance Program (1998-2011). Diag Microbiol Infect Dis 2013; 75:107–109.
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 2008; 32:361–385.
- Centers for Disease Control and Prevention (CDC). Antibiotic resistance threats in the United States 2013. cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. Accessed June 1, 2015.
- Moisan H, Pruneau M, Malouin F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J Antimicrob Chemother 2010; 65:713–716.
- Forest Laboratories, Inc. Teflaro® (ceftaroline fosamil): prescribing information. www.frx.com/pi/teflaro_pi.pdf. Accessed June 1, 2015.
- Richter SS, Heilmann KP, Dohrn CL, et al. Activity of ceftaroline and epidemiologic trends in Staphylococcus aureus isolates collected from 43 medical centers in the United States in 2009. Antimicrob Agents Chemother 2011; 55:4154–4160.
- Ge Y, Biek D, Talbot GH, Sahm DF. In vitro profiling of ceftaroline against a collection of recent bacterial clinical isolates from across the United States. Antimicrob Agents Chemother 2008; 52:3398–3407.
- Saravolatz L, Pawlak J, Johnson L. In vitro activity of ceftaroline against community-associated methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, and daptomycin-nonsusceptible Staphylococcus aureus isolates. Antimicrob Agents Chemother 2010; 54:3027–3030.
- Mushtaq S, Livermore DM. AmpC induction by ceftaroline. J Antimicrob Chemother 2010; 65:586–588.
- Mendes RE, Tsakris A, Sader HS, et al. Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J Antimicrob Chemother 2012; 67:1321–1324.
- Lodise TP, Low DE. Ceftaroline fosamil in the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections. Drugs 2012; 72:1473–1493.
- Riccobene TA, Su SF, Rank D. Single- and multiple-dose study to determine the safety, tolerability, and pharmacokinetics of ceftaroline fosamil in combination with avibactam in healthy subjects. Antimicrob Agents Chemother 2013; 57:1496–1504.
- US National Institutes of Health. ClinicalTrials.gov. Evaluation of ceftaroline fosamil versus a comparator in adult subjects with community-acquired bacterial pneumonia (CABP) with risk for methicillin-resistant Staphylococcus aureus. http://clinicaltrials.gov/ct2/show/NCT01645735. Accessed June 1, 2015.
- File TM Jr, Low DE, Eckburg PB, et al; FOCUS 1 investigators. FOCUS 1: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii19–iii32.
- Low DE, File TM Jr, Eckburg PB, et al; FOCUS 2 investigators. FOCUS 2: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii33–iii44.
- File TM Jr, Low DE, Eckburg PB, et al. Integrated analysis of FOCUS 1 and FOCUS 2: randomized, doubled-blinded, multicenter phase 3 trials of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in patients with community-acquired pneumonia. Clin Infect Dis 2010; 51:1395–1405.
- Food and Drug Administration (FDA). Ceftaroline fosamil for the treatment of community-acquired bacterial pneumonia and complicated skin and skin structure infections. www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/anti-infectivedrugsadvisorycommittee/ucm224656.pdf. Accessed June 1, 2015.
- Corey GR, Wilcox MH, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 1 investigators. CANVAS 1: the first phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv41–iv51.
- Wilcox MH, Corey GR, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 2 investigators. CANVAS 2: the second phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv53-iv65.
- Corey GR, Wilcox M, Talbot GH, et al. Integrated analysis of CANVAS 1 and 2: phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin Infect Dis 2010; 51:641–650.
- Friedland HD, O’Neal T, Biek D, et al. CANVAS 1 and 2: analysis of clinical response at day 3 in two phase 3 trials of ceftaroline fosamil versus vancomycin plus aztreonam in treatment of acute bacterial skin and skin structure infections. Antimicrob Agents Chemother 2012; 56:2231–2236.
- Jacqueline C, Caillon J, Le Mabecque V, et al. In vivo efficacy of ceftaroline (PPI-0903), a new broad-spectrum cephalosporin, compared with linezolid and vancomycin against methicillin-resistant and vancomycin-intermediate Staphylococcus aureus in a rabbit endocarditis model. Antimicrob Agents Chemother 2007; 51:3397–3400.
- Ho TT, Cadena J, Childs LM, Gonzalez-Velez M, Lewis JS 2nd. Methicillin-resistant Staphylococcus aureus bacteraemia and endocarditis treated with ceftaroline salvage therapy. J Antimicrob Chemother 2012; 67:1267–1270.
- Lin JC, Aung G, Thomas A, Jahng M, Johns S, Fierer J. The use of ceftaroline fosamil in methicillin-resistant Staphylococcus aureus endocarditis and deep-seated MRSA infections: a retrospective case series of 10 patients. J Infect Chemother 2013; 19:42–49.
- Jacqueline C, Amador G, Caillon J, et al. Efficacy of the new cephalosporin ceftaroline in the treatment of experimental methicillin-resistant Staphylococcus aureus acute osteomyelitis. J Antimicrob Chemother 2010; 65:1749–1752.
- Stucki A, Acosta F, Cottagnoud M, Cottagnoud P. Efficacy of ceftaroline fosamil against Escherichia coli and Klebsiella pneumoniae strains in a rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:5808–5810.
- Cottagnoud P, Cottagnoud M, Acosta F, Stucki A. Efficacy of ceftaroline fosamil against penicillin-sensitive and -resistant Streptococcus pneumoniae in an experimental rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:4653–4655.
- Corrado ML. Integrated safety summary of CANVAS 1 and 2 trials: phase III, randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv67–iv71.
- Rank DR, Friedland HD, Laudano JB. Integrated safety summary of FOCUS 1 and FOCUS 2 trials: phase III randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii53–iii59.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis 2014; 59:147–159.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44:S27–S72.
- Infectious Diseases Society of America. The 10 x ‘20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 2010; 50:1081–1083.
- Styers D, Sheehan DJ, Hogan P, Sahm DF. Laboratory-based surveillance of current antimicrobial resistance patterns and trends among Staphylococcus aureus: 2005 status in the United States. Ann Clin Microbiol Antimicrob 2006; 5:2.
- Farrell DJ, Castanheira M, Mendes RE, Sader HS, Jones RN. In vitro activity of ceftaroline against multidrug-resistant Staphylococcus aureus and Streptococcus pneumoniae: a review of published studies and the AWARE Surveillance Program (2008-2010). Clin Infect Dis 2012; 55(suppl 3):S206–S214.
- Holmes NE, Johnson PD, Howden BP. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J Clin Microbiol 2012; 50:2548–2552.
- Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 2003; 111:1265–1273.
- Jones RN, Sader HS, Mendes RE, Flamm RK. Update on antimicrobial susceptibility trends among Streptococcus pneumoniae in the United States: report of ceftaroline activity from the SENTRY Antimicrobial Surveillance Program (1998-2011). Diag Microbiol Infect Dis 2013; 75:107–109.
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 2008; 32:361–385.
- Centers for Disease Control and Prevention (CDC). Antibiotic resistance threats in the United States 2013. cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. Accessed June 1, 2015.
- Moisan H, Pruneau M, Malouin F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J Antimicrob Chemother 2010; 65:713–716.
- Forest Laboratories, Inc. Teflaro® (ceftaroline fosamil): prescribing information. www.frx.com/pi/teflaro_pi.pdf. Accessed June 1, 2015.
- Richter SS, Heilmann KP, Dohrn CL, et al. Activity of ceftaroline and epidemiologic trends in Staphylococcus aureus isolates collected from 43 medical centers in the United States in 2009. Antimicrob Agents Chemother 2011; 55:4154–4160.
- Ge Y, Biek D, Talbot GH, Sahm DF. In vitro profiling of ceftaroline against a collection of recent bacterial clinical isolates from across the United States. Antimicrob Agents Chemother 2008; 52:3398–3407.
- Saravolatz L, Pawlak J, Johnson L. In vitro activity of ceftaroline against community-associated methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, and daptomycin-nonsusceptible Staphylococcus aureus isolates. Antimicrob Agents Chemother 2010; 54:3027–3030.
- Mushtaq S, Livermore DM. AmpC induction by ceftaroline. J Antimicrob Chemother 2010; 65:586–588.
- Mendes RE, Tsakris A, Sader HS, et al. Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J Antimicrob Chemother 2012; 67:1321–1324.
- Lodise TP, Low DE. Ceftaroline fosamil in the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections. Drugs 2012; 72:1473–1493.
- Riccobene TA, Su SF, Rank D. Single- and multiple-dose study to determine the safety, tolerability, and pharmacokinetics of ceftaroline fosamil in combination with avibactam in healthy subjects. Antimicrob Agents Chemother 2013; 57:1496–1504.
- US National Institutes of Health. ClinicalTrials.gov. Evaluation of ceftaroline fosamil versus a comparator in adult subjects with community-acquired bacterial pneumonia (CABP) with risk for methicillin-resistant Staphylococcus aureus. http://clinicaltrials.gov/ct2/show/NCT01645735. Accessed June 1, 2015.
- File TM Jr, Low DE, Eckburg PB, et al; FOCUS 1 investigators. FOCUS 1: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii19–iii32.
- Low DE, File TM Jr, Eckburg PB, et al; FOCUS 2 investigators. FOCUS 2: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii33–iii44.
- File TM Jr, Low DE, Eckburg PB, et al. Integrated analysis of FOCUS 1 and FOCUS 2: randomized, doubled-blinded, multicenter phase 3 trials of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in patients with community-acquired pneumonia. Clin Infect Dis 2010; 51:1395–1405.
- Food and Drug Administration (FDA). Ceftaroline fosamil for the treatment of community-acquired bacterial pneumonia and complicated skin and skin structure infections. www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/anti-infectivedrugsadvisorycommittee/ucm224656.pdf. Accessed June 1, 2015.
- Corey GR, Wilcox MH, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 1 investigators. CANVAS 1: the first phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv41–iv51.
- Wilcox MH, Corey GR, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 2 investigators. CANVAS 2: the second phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv53-iv65.
- Corey GR, Wilcox M, Talbot GH, et al. Integrated analysis of CANVAS 1 and 2: phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin Infect Dis 2010; 51:641–650.
- Friedland HD, O’Neal T, Biek D, et al. CANVAS 1 and 2: analysis of clinical response at day 3 in two phase 3 trials of ceftaroline fosamil versus vancomycin plus aztreonam in treatment of acute bacterial skin and skin structure infections. Antimicrob Agents Chemother 2012; 56:2231–2236.
- Jacqueline C, Caillon J, Le Mabecque V, et al. In vivo efficacy of ceftaroline (PPI-0903), a new broad-spectrum cephalosporin, compared with linezolid and vancomycin against methicillin-resistant and vancomycin-intermediate Staphylococcus aureus in a rabbit endocarditis model. Antimicrob Agents Chemother 2007; 51:3397–3400.
- Ho TT, Cadena J, Childs LM, Gonzalez-Velez M, Lewis JS 2nd. Methicillin-resistant Staphylococcus aureus bacteraemia and endocarditis treated with ceftaroline salvage therapy. J Antimicrob Chemother 2012; 67:1267–1270.
- Lin JC, Aung G, Thomas A, Jahng M, Johns S, Fierer J. The use of ceftaroline fosamil in methicillin-resistant Staphylococcus aureus endocarditis and deep-seated MRSA infections: a retrospective case series of 10 patients. J Infect Chemother 2013; 19:42–49.
- Jacqueline C, Amador G, Caillon J, et al. Efficacy of the new cephalosporin ceftaroline in the treatment of experimental methicillin-resistant Staphylococcus aureus acute osteomyelitis. J Antimicrob Chemother 2010; 65:1749–1752.
- Stucki A, Acosta F, Cottagnoud M, Cottagnoud P. Efficacy of ceftaroline fosamil against Escherichia coli and Klebsiella pneumoniae strains in a rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:5808–5810.
- Cottagnoud P, Cottagnoud M, Acosta F, Stucki A. Efficacy of ceftaroline fosamil against penicillin-sensitive and -resistant Streptococcus pneumoniae in an experimental rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:4653–4655.
- Corrado ML. Integrated safety summary of CANVAS 1 and 2 trials: phase III, randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv67–iv71.
- Rank DR, Friedland HD, Laudano JB. Integrated safety summary of FOCUS 1 and FOCUS 2 trials: phase III randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii53–iii59.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis 2014; 59:147–159.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44:S27–S72.
KEY POINTS
- Resistance of S aureus and S pneumoniae to multiple antimicrobial drugs is on the rise, and new agents are urgently needed.
- Ceftaroline’s molecular structure was designed to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae.
- In clinical trials leading to its approval, ceftaroline was found to be at least as effective as ceftriaxone in treating community-acquired pneumonia and at least as effective as vancomycin plus aztreonam in treating acute bacterial skin and skin-structure infections.
- The routine use of ceftaroline for these indications should be balanced by its higher cost compared with ceftriaxone or vancomycin. Ongoing studies should shed more light on its role in treatment.
Denosumab: A novel antiresorptive drug for osteoporosis
A 68-year-old white woman presents with mid- thoracic back pain. Plain radiographs reveal a compression fracture of the 10th thoracic vertebra. She is diagnosed with osteoporosis on the basis of dual energy x-ray absorptiometry (DXA) scans that show T scores of –2.9 in her lumbar spine and –2.6 in her left femoral neck. Her 10-year probability of fracture is estimated as 23% for major osteoporotic fracture and 5.9% for hip fracture (based on the World Health Organization’s absolute fracture risk assessment tool, adapted for the United States, and available at www.shef.ac.uk/FRAX).
After excluding common secondary causes of osteoporosis, her physician recommends a bisphosphonate to reduce her risk of fracture, but she develops upper-gastrointestinal adverse effects with both alendronate and risedronate despite correctly following the instructions for oral administration.
What should her physician consider next?
OSTEOPOROSIS IS A MAJOR PROBLEM
Osteoporosis is a systemic skeletal disease characterized by low bone mass and microarchitectural deterioration of bone tissue, predisposing to an increased risk of fragility fractures, particularly of the spine, hip, and wrist.
It is a major public health problem, affecting 200 million people throughout the world, with 9 million osteoporotic fractures reported in the year 2000.1 The incidence of hip fracture alone is predicted to rise to 2.6 million by the year 2025, and to 4.5 million by the year 2050.2 In the United States, the total burden was estimated to be about 2 million incident fractures in the year 2005, projected to rise by another 50% by the year 2025,3 primarily because of the aging of the population. Population studies have indeed suggested that about 40% of white women and 13% of white men over the age of 50 are at risk of sustaining an osteoporotic fracture during the remainder of their lifetime.4
The consequences of osteoporotic fractures can be devastating. Hip fractures are associated with a risk of death ranging from 8.4% to 36% during the first year after fracture.5 One-fifth of patients who sustain a hip fracture require long-term nursing home care, and more than half of the survivors do not regain their previous level of independence.
Patients with vertebral fractures are also at increased risk of death, although the results of some studies suggest that this could be the result of comorbid factors.6–9 Vertebral fractures can result in chronic back pain, loss of height from spinal deformity, reduced mobility, loss of self-esteem, and in severe cases, respiratory and digestive problems because of contact between the lower ribs and pelvis.
A person with one vertebral compression fracture is five times more likely to have another vertebral fracture,10 and a person with two or more compression fractures is 12 times more likely.11
The costs of treating osteoporotic fractures are greater than those of treating myocardial infarction or stroke12,13; they include not only direct costs incurred in treating the fracture, but also indirect societal costs owing to the long-term morbidity associated with the fracture. In the United States, the total cost of treating osteoporotic fractures was estimated at $19 billion in the year 2005.3 By 2025, the annual costs are projected to rise by almost 50%.3
A NEED FOR MORE OPTIONS
Until fairly recently, bisphosphonates were the only drugs of first choice, but adherence to oral bisphosphonate therapy is generally poor (< 50% at 1 year),14 most commonly because of dyspepsia,15 and poor adherence has been shown to be associated with increased fracture risk.16,17 Hence the need for additional therapeutic options.
In this review, we discuss denosumab, an antiresorptive drug approved by the US Food and Drug Administration (FDA) in 2010. First, we discuss its mechanism of action, efficacy, and safety, and then we offer recommendations for its use in clinical practice.
WHAT IS DENOSUMAB AND HOW DOES IT WORK?
Bone remodeling is a dynamic process involving a balance between bone resorption by osteoclasts on the one hand and new bone formation by osteoblasts on the other. A net gain in bone occurs when the activity of osteoblasts exceeds that of osteoclasts, and bone loss occurs when there is increased osteoclast activity or reduced osteoblast activity, or both. The activities of osteoblasts and osteoclasts are tightly coupled because of the opposing effects of two sets of proteins, namely, receptor activator of nuclear factor kappa b ligand (RANKL) and osteoprotegerin.
Both RANKL and osteoprotegerin are produced by osteoblasts. RANKL binds to its receptor (RANK) on preosteoclasts and osteoclasts and induces their differentiation and activation, respectively. Osteoprotegerin is the decoy receptor and natural antagonist for RANKL. By binding with RANKL, it blocks its interaction with RANK.18 In healthy individuals, a fine balance between RANKL and osteoprotegerin ensures that bone remodeling is regulated.
In postmenopausal women, estrogen deficiency leads to an imbalance between RANKL and osteoprotegerin (increased RANKL and reduced osteoprotegerin), resulting in net bone loss. This imbalance is also a feature of rheumatoid arthritis, myeloma bone disease, and osteolytic metastatic bone disease; it also occurs in those receiving androgen deprivation therapy for prostate cancer or aromatase inhibitors for breast cancer.
Denosumab is a fully human monoclonal antibody that targets RANKL.19 By binding to RANKL, this drug prevents the maturation and differentiation of preosteoclasts and promotes apoptosis of osteoclasts. Bone resorption is therefore slowed. It was parenteral osteoprotegerin that was initially developed by denosumab’s manufacturer,20 but this approach failed because neutralizing antibodies developed to osteoprotegerin, rendering it ineffective. Development of neutralizing antibodies has thus far not been a problem with denosumab.
Denosumab, with its property of RANKL inhibition, has also been used to prevent skeletal events in patients with bone metastases from solid tumors and to treat unresectable giant cell tumors of the bone (both FDA-approved indications) and hypercalcemia of malignancy. There is limited clinical experience in Paget disease of the bone as well.21–23 These other potential uses of denosumab are beyond the scope of this review.
HOW WELL DOES DENOSUMAB WORK FOR OSTEOPOROSIS?
Several phase 2 and phase 3 randomized controlled trials have evaluated the efficacy of denosumab, but only one, the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) trial, included fracture reduction as the primary outcome measure. The rest evaluated changes in bone mineral density or in markers of bone turnover, or both.
FREEDOM was a double-blind, randomized controlled trial in 7,808 postmenopausal women with T scores between –2.5 and –4.0 at the lumbar spine or hip.24 Twenty-four percent of the patients had vertebral fractures at baseline. Patients were randomized to receive either denosumab 60 mg (n = 3,902) or placebo (n = 3,906) every 6 months for up to 36 months. All patients also received adequate calcium and vitamin D supplementation.
At 36 months, compared with those who were randomized to receive placebo, those who were randomized to denosumab had lower incidence rates of:
- New vertebral fracture
(2.3% vs 7.2%, risk ratio 0.32,
95% CI 0.26–0.41, P < .001) - Nonvertebral fracture
(6.5% vs 8.0%, risk ratio 0.80,
95% CI 0.67–0.95, P = .01) - Hip fracture
(0.7% vs 1.2%, risk ratio 0.60,
95% CI 0.37–0.97, P = .04).
Increases in bone mineral density at the lumbar spine and hip, and decreases in bone turnover markers were also significantly greater in the denosumab group. The number needed to treat to prevent one new fracture over 3 years was 21 for vertebral fracture, 67 for nonvertebral fracture, and 200 for hip fracture, reflecting the relatively low event rate in the study.
In an open-label extension of the FREEDOM trial, the fracture incidence rates among participants who continued to receive denosumab for an additional 5 years remained low, and still below those projected for a “virtual placebo cohort” (total duration of exposure of 8 years). The rates among participants who switched from placebo to denosumab were similar to those of the denosumab group from the parent trial.25,26
A subgroup analysis of the FREEDOM trial suggested that denosumab reduced the risk of new vertebral fractures irrespective of age, body mass index, femoral neck bone mineral density, prevalent vertebral fractures, or prior nonvertebral fractures (risk ratio 0.32; 95% CI 0.26–0.41, P < .001), whereas the risk of nonvertebral fractures was only reduced in those women with body mass indices less than 25 kg/m2, femoral neck bone mineral density T scores less than –2.5, and in those without a prevalent vertebral fracture.27
A post hoc analysis revealed that denosumab significantly reduced the risk of new vertebral and hip fractures even in subgroups of women at higher risk of fracture.28 At 10% fracture probability (as estimated by the FRAX risk calculator), denosumab reduced the fracture risk by 11% (P = .629), whereas at 30% probability (moderate to high risk), the reduction was 50% (P = .001).29
Other phase 2 and phase 3 trials, in postmenopausal women with low bone mineral density, demonstrated that compared with placebo, denosumab significantly increased bone mineral density at all skeletal sites, increased volumetric bone mineral density at the distal radius, improved hip structural analysis parameters, and reduced bone turnover markers.30–33 Increases in bone mineral density and reductions in bone turnover markers with denosumab have been shown in men as well.34
In a randomized controlled trial,35 improvement in bone mineral density was better in those who received the combination of denosumab and teriparatide than in those who received either drug on its own.
Denosumab has also been shown to reduce the incidence of new vertebral fractures and improve bone mineral density in men receiving androgen-deprivation therapy for nonmetastatic prostate cancer,36 and to improve bone mineral density in women with metastatic breast cancer and low bone mass who were receiving adjuvant aromatase inhibitor therapy.37
HOW DOES DENOSUMAB COMPARE WITH OTHER OSTEOPOROSIS DRUGS?
A double-blind randomized controlled trial in postmenopausal women with low bone mass demonstrated that denosumab was superior to alendronate in improving bone mineral density at all skeletal sites (3.5% vs 2.6% for total hip bone mineral density, P < .0001).38
Another double-blind trial demonstrated that in patients previously treated with alendronate, switching to denosumab resulted in significantly greater increases in bone mineral density at all skeletal sites compared with continuing with alendronate (P < .0001).39
Denosumab has also been shown to be superior to alendronate in improving cortical bone mineral density, as measured by quantitative computed tomography.40
No trial has directly compared the efficacy of denosumab with other osteoporosis drugs in reducing fracture risk, but a systematic literature review of multiple databases,41 comparing the antifracture efficacy of nine osteoporosis drugs, concluded that teriparatide, zoledronic acid, and denosumab had the highest probabilities of being most efficacious for nonvertebral and vertebral fractures, with the greatest effect sizes. Indirect comparisons of the relative risk of fracture with denosumab (based on the results of FREEDOM), alendronate, risedronate, raloxifene, and strontium (based on a meta-analysis of randomized controlled trials) are presented in Table 1.42
A 2-year randomized, open-label, crossover study43 randomized patients to receive either denosumab followed by alendronate or alendronate followed by denosumab over successive 12-month periods. The results suggested that postmenopausal women with osteoporosis were more adherent, compliant, and persistent with denosumab therapy (a subcutaneous injection every 6 months) than with alendronate therapy in the form of oral tablets, self-administered weekly (7.5% nonadherence vs 36.5% at the end of 2 years). After receiving both treatments, women reported greater satisfaction with denosumab, with 92.4% preferring it over oral alendronate. Bone mineral density remained stable when patients were switched from denosumab to alendronate, but improved further when they were switched from alendronate to denosumab.
HOW SAFE IS DENOSUMAB?
The most frequent adverse events with denosumab reported in the long-term extension of one phase 2 study were upper respiratory tract infections (13.5%), arthralgia (11.5%), and back pain (9.0%).30
Increased risk of infection, cancer, and dermatologic reactions has been a concern, as RANKL and RANK are expressed by a wide variety of cells, including T lymphocytes, B cells, and dendritic cells.44 However, there were no significant differences in the overall incidences of adverse events between patients who received denosumab and those who received placebo or alendronate in any of the phase 2, phase 3, or extension studies.
In the FREEDOM trial,24 there was no significant difference between the two groups in the overall incidence of infection (52.9% with denosumab vs 54.4% with placebo, P = .17), or serious infection (4.1% with denosumab vs 3.4% with placebo, P = .14), although the incidence of “serious” cellulitis requiring hospitalization was higher in the denosumab group (0.3% vs < 0.1%, P = .002). There were more serious infections involving the gastrointestinal system, urinary tract, and ear and cases of endocarditis in the denosumab group, but the number of events was small, and there was no relationship with the timing of administration or duration of exposure to denosumab.45 Eczema was more common in the denosumab than in the placebo group (3.0% vs 1.7%, P < .001), but the extension data from the first 3 years did not provide any evidence for an increased risk of cellulitis or eczema with denosumab.26
Although randomized controlled trials reported more cases of neoplasms in the denosumab than in the placebo groups, meta-analyses have failed to detect a statistically significant difference (risk ratio 1.11, 95% CI 0.91–1.36).46 The overall incidence of adverse and serious adverse events reported in the 8-year extension of FREEDOM were consistent with data reported in the previous extension studies.25
In the FREEDOM extension trial, four events in the long-term group (n = 2,343), and two in the crossover group (n = 2,207) were adjudicated as being consistent with osteonecrosis of the jaw.26 One mid-shaft fracture in the crossover group was adjudicated as an atypical femoral fracture. There were, however, no reports of osteonecrosis of the jaw or atypical femoral fracture in the long-term phase 2 trial after 8 years of follow-up.30 By September 2013, postmarketing safety surveillance data for denosumab (estimated exposure of 1.2 million patient-years) had recorded four cases of atypical femoral fracture. All four patients had previously been on bisphosphonates. There were also 32 reports of osteonecrosis of the jaw.47
Denosumab’s manufacturer aims to communicate the risks of treatment to health care professionals and patients. Information is available online at www.proliahcp.com/risk-evaluation-mitigation-strategy/.
WHAT ARE THE PRECAUTIONS?
Several precautions need to be taken when considering treatment with denosumab.
Antiresorptives can aggravate hypocalcemia by inhibiting bone turnover. Serum calcium should therefore be checked and preexisting hypocalcemia should be corrected before starting denosumab.48
Denosumab is contraindicated in women who are pregnant or are planning to become pregnant, as fetal loss and teratogenicity have been reported in animal experiments. (Denosumab is unlikely to be used in premenopausal women, as it is not approved for use in this group.)
There are no data on excretion of denosumab in human milk, so it should not be given to nursing mothers.
Renal impairment is not a contraindication, and no dose adjustment is necessary (even for patients on renal replacement therapy), as denosumab, being an antibody, is eliminated through the reticuloendothelial system.49,50 However, in practice, any antiresorptive agent should be used with caution in patients with severe renal impairment because of the possible presence of adynamic bone disease. Further reduction of bone turnover would be detrimental in such patients. Also, severe hypocalcemia has been reported in patients with a creatinine clearance rate less than 30 mL/min and in those receiving dialysis.51,52 Postmarketing surveillance data have reported eight cases of severe symptomatic hypocalcemia, of which seven were in patients with chronic kidney disease.47
The manufacturer suggests that patients receive a dental examination with appropriate preventive dentistry before starting denosumab to reduce the incidence of osteonecrosis of the jaw, despite the lack of evidence in support of this strategy. The American Dental Association recommends regular dental visits and maintenance of good oral hygiene for patients already established on antiresorptive therapy.53,54
SHOULD PATIENTS ON DENOSUMAB BE OFFERED A DRUG HOLIDAY?
A drug holiday (temporary discontinuation of the drug after a certain duration of treatment) has been proposed for patients receiving bisphosphonates because of the risk of atypical femoral fracture and osteonecrosis of the jaw (although small) consequent to long-term continuous suppression of bone turnover.55 The antifracture efficacy of bisphosphonates is likely to persist for an unknown length of time after discontinuation because of their long skeletal half-life, while the risks gradually diminish.
By contrast, denosumab targets RANKL in the extracellular fluid and does not become embedded within the bone tissue.56 Pharmacokinetic studies have shown that denosumab has a rapid offset of action, with a half-life of only 26 days and biological activity lasting only 6 months.57 The results of a phase 2 extension study suggest that bone mineral density starts to decline and bone turnover markers start to rise within 12 months of discontinuing denosumab.58
Although fracture risk did not increase in those who were randomized to stopping the treatment and bone mineral density increased further when treatment was restarted, a drug holiday cannot presently be recommended for patients receiving denosumab because of the lack of supportive data.
HOW COST-EFFECTIVE IS DENOSUMAB?
The wholesale acquisition cost is $825 per 60-mg prefilled syringe of denosumab, although this may vary depending on where the drug is obtained. This does not include physician-related service costs associated with administration of denosumab.
Cost-effectiveness analyses conducted in the United States, the United Kingdom, and Sweden have all concluded that denosumab would offer a cost-effective alternative to other osteoporosis medications for primary prevention and secondary prevention of fractures.59–61
The Swedish study also incorporated adherence in the cost-effectiveness model and showed that denosumab was a cost-effective alternative to oral bisphosphonates, particularly for patients who were not expected to adhere well to oral treatments.61
WHICH OSTEOPOROSIS PATIENTS ARE CANDIDATES FOR DENOSUMAB?
The FDA has approved denosumab for the treatment of postmenopausal women and men at high risk of fracture (defined as having a history of osteoporotic fracture or multiple risk factors for fracture), or in those who cannot tolerate other osteoporosis medications or for whom other medications have failed.
Denosumab is also approved for men at high risk of fracture receiving androgen deprivation therapy for nonmetastatic prostate cancer, and for women at high risk of fracture receiving adjuvant aromatase inhibitor therapy for breast cancer.
WHAT DO THE GUIDELINES RECOMMEND?
The National Osteoporosis Foundation guidelines recommend pharmacologic treatment for patients with hip or vertebral fractures (clinical or asymptomatic); T scores lower than –2.5 at the femoral neck, total hip, or lumbar spine; and those with a 10-year probability of hip fracture of more than 3% or of a major osteoporotic fracture more than 20% based on the US-adapted FRAX calculator.62 The American College of Endocrinology guidelines have proposed similar thresholds for pharmacologic treatment, and they recommend alendronate, risedronate, zoledronate, and denosumab as first-line agents.63
The 2010 Osteoporosis Canada guidelines recommend denosumab, alendronate, risedronate, and zoledronate as first-line therapies for preventing hip, nonvertebral, and vertebral fractures in postmenopausal women (grade A recommendation).64 The National Institute of Health and Clinical Excellence in England and Wales, on the other hand, recommends denosumab only for patients who are unable to take a bisphosphonate.65
PRACTICAL PRESCRIBING TIPS
The patient described at the beginning of this article has already sustained a vertebral compression fracture, and her DXA scan shows T scores in the osteoporotic range. She is therefore at increased risk of another fragility fracture (with a fivefold higher risk of another vertebral fracture). Pharmacologic therapy should be considered. In addition, she should be encouraged to adhere to lifestyle measures such as a healthy diet and regular weight-bearing exercise, her risk of falling should be assessed, and adequate calcium and vitamin D supplementation should be given.
Secondary causes of osteoporosis are present in about 30% of women and 55% of men who have vertebral fractures.66 A complete blood count, erythrocyte sedimentation rate, bone biochemistry, 25-hydroxyvitamin D, thyroid-stimulating hormone, and renal and liver function tests should be requested in all patients. Further tests should be considered depending on the clinical evaluation and results of initial investigations.
Because this patient cannot tolerate oral bisphosphonates, she could be offered the option of annual intravenous zoledronic acid infusions or 6-monthly subcutaneous denosumab injections. In clinical trials, gastrointestinal adverse effects were noted with intravenous bisphosphonates as well, but the adverse effects reported were no different than those with placebo. The potential advantages with denosumab include better bone mineral density gains, adherence and patient satisfaction compared with oral bisphosphonates, convenient twice-yearly administration, safety in patients with renal impairment, and absence of gastrointestinal effects.
Raloxifene, a selective estrogen receptor modulator, has estrogen-like action on the bone and antiestrogen actions on the breast and uterus. Unlike standard hormone replacement therapy, raloxifene can therefore increase bone mineral density without increasing the risk of breast and endometrial cancers. However, it has only been shown to reduce the risk of vertebral fracture, not hip fracture. Hence, it would be a more appropriate choice for younger postmenopausal women. Moreover, it may cause troublesome menopausal symptoms.
Teriparatide, the recombinant parathyroid hormone, is an anabolic agent. It is very expensive, and because of this, guidelines in several countries restrict its use to women with severe osteoporosis and multiple fractures who fail to respond to standard treatments. It cannot be used for longer than 2 years because of its association with osteosarcoma in rats.
If our patient prefers denosumab, therapy should be initiated after appropriate counseling (see precautions above). The dose is 60 mg, given subcutaneously, once every 6 months.
Monitoring
There is no consensus regarding the optimal frequency for monitoring patients on treatment, owing to the lack of prospective trial data. The National Osteoporosis Foundation recommends repeating the bone mineral density measurements about 2 years after starting therapy, and about every 2 years thereafter.62 Some studies suggest that changes in bone mineral density correlate with reduction in fracture risk.67,68 A change in bone mineral density is considered significant when it is greater than the range of error of the densitometer (also known as the least significant change).69 If the bone mineral density is stable or improving, therapy could be continued, but if it is declining and the decline is greater than the least significant change, a change in therapy should be considered if no secondary causes for bone loss are evident (but see What are the areas of uncertainty? below).
The National Osteoporosis Foundation also recommends measuring a bone turnover marker at baseline and then 3 to 6 months later, as its suppression predicts greater bone mineral density responses and fracture risk reduction.70 If there is a decrease of more than 30% in serum carboxy-terminal collagen crosslinks (CTX) or more than 50% in urinary N-telopeptide (NTX),71 the patient can be reassured that the next bone mineral density measurement will be stable or improved. In patients on oral bisphosphonates, measurement of bone turnover markers also provides evidence of compliance.
Clinical trials suggest that a numerical increase in bone mineral density can be expected in most patients on treatment, though this depends on the measurement site and the length of time between examinations. In one phase 3 trial of denosumab in postmenopausal women, only 5% of the participants had unchanged or diminished bone mineral density at the lumbar spine, and 8% at the hip, after 36 months of treatment.72 However, the CTX levels fell to below the lower limit of the reference interval as early as 1 month after commencing treatment in all denosumab-treated patients.68
Hence, bone turnover markers may be a more sensitive indicator of treatment effect than bone mineral density, but this would ultimately need to be evaluated against fracture rates in a real-world setting.
WHAT ARE THE AREAS OF UNCERTAINTY?
There are currently no guidelines for long-term management of patients on denosumab, and also no data to suggest whether patients should be switched to a weaker antiresorptive drug after a certain number of years in order to reduce the possible risk of atypical femoral fracture or osteonecrosis of the jaw.
No head-to-head trials have directly compared the antifracture efficacy of denosumab with that of other standard osteoporosis therapies. The antifracture efficacy and safety of combination therapies involving denosumab are also uncertain. For adherent patients who have a suboptimal response, there is no evidence to guide the further course of action. The International Osteoporosis Foundation guidelines suggest replacing a stronger antiresorptive with an anabolic agent, but acknowledge that this is only based on expert opinion.71
The very-long-term effects (beyond 8 years) of continuous denosumab administration on increasing the risk of atypical femoral fracture, osteonecrosis of the jaw, malignancy, or infection or the duration after which risks would start to outweigh benefits is not known. However, postmarketing safety data continue to be collected through the voluntary Post-marketing Active Safety Surveillance Program (for prespecified adverse events) in addition to the FDA’s MedWatch program.
CASE PROGRESSION
The patient described in the vignette is presented with two options—zoledronate and denosumab. She chooses denosumab. Her renal function and serum calcium are checked and are found to be satisfactory. She undergoes a dental examination, which is also satisfactory. She is counseled about the possible increased risk of infection, and then she is started on 60 mg of denosumab subcutaneously, once every 6 months.
When reviewed after 2 years, she reports no further fractures. Her bone mineral density remains stable compared with the values obtained before starting treatment. She reports no adverse effects and is happy to continue with denosumab.
- Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int 2006; 17:1726–1733.
- Gullberg B, Johnell O, Kanis JA. World-wide projections for hip fracture. Osteoporos Int 1997; 7:407–413.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res 2007; 22:465–475.
- Melton LJ 3rd, Chrischilles EA, Cooper C, Lane AW, Riggs BL. Perspective. How many women have osteoporosis? J Bone Miner Res 1992; 7:1005–1010.
- Abrahamsen B, van Staa T, Ariely R, Olson M, Cooper C. Excess mortality following hip fracture: a systematic epidemiological review. Osteoporos Int 2009; 20:1633–1650.
- Jalava T, Sarna S, Pylkkänen L, et al. Association between vertebral fracture and increased mortality in osteoporotic patients. J Bone Miner Res 2003; 18:1254–1260.
- Ismail AA, O’Neill TW, Cooper C, et al. Mortality associated with vertebral deformity in men and women: results from the European Prospective Osteoporosis Study (EPOS). Osteoporos Int 1998; 8:291–297.
- Ensrud KE, Thompson DE, Cauley JA, et al. Prevalent vertebral deformities predict mortality and hospitalization in older women with low bone mass. Fracture Intervention Trial Research Group. J Am Geriatr Soc 2000; 48:241–249.
- Kado DM, Browner WS, Palermo L, Nevitt MC, Genant HK, Cummings SR. Vertebral fractures and mortality in older women: a prospective study. Study of Osteoporotic Fractures Research Group. Arch Intern Med 1999; 159:1215–1220.
- Lindsay R, Silverman SL, Cooper C, et al. Risk of new vertebral fracture in the year following a fracture. JAMA 2001; 285:320–323.
- Ross PD, Davis JW, Epstein RS, Wasnich RD. Pre-existing fractures and bone mass predict vertebral fracture incidence in women. Ann Intern Med 1991; 114:919–923.
- Piscitelli P, Iolascon G, Argentiero A, et al. Incidence and costs of hip fractures vs strokes and acute myocardial infarction in Italy: comparative analysis based on national hospitalization records. Clin Interv Aging 2012; 7:575–583.
- Johnell O, Kanis JA, Jonsson B, Oden A, Johansson H, De Laet C. The burden of hospitalised fractures in Sweden. Osteoporos Int 2005; 16:222–228.
- Confavreux CB, Canoui-Poitrine F, Schott AM, Ambrosi V, Tainturier V, Chapurlat RD. Persistence at 1 year of oral antiosteoporotic drugs: a prospective study in a comprehensive health insurance database. Eur J Endocrinol 2012; 166:735–741.
- Biswas PN, Wilton LV, Shakir SA. Pharmacovigilance study of alendronate in England. Osteoporos Int 2003; 14:507–514.
- Landfeldt E, Ström O, Robbins S, Borgström F. Adherence to treatment of primary osteoporosis and its association to fractures—the Swedish Adherence Register Analysis (SARA). Osteoporos Int 2012; 23:433–443.
- Sampalis JS, Adachi JD, Rampakakis E, Vaillancourt J, Karellis A, Kindundu C. Long-term impact of adherence to oral bisphosphonates on osteoporotic fracture incidence. J Bone Miner Res 2012; 27:202–210.
- Schwarz EM, Ritchlin CT. Clinical development of anti-RANKL therapy. Arthritis Res Ther 2007; 9(suppl 1):S7.
- Hanley DA, Adachi JD, Bell A, Brown V. Denosumab: mechanism of action and clinical outcomes. Int J Clin Pract 2012; 66:1139–1146.
- Bekker PJ, Holloway D, Nakanishi A, Arrighi M, Leese PT, Dunstan CR. The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res 2001; 16:348–360.
- Rizzoli R, Body JJ, Brandi ML, et al; International Osteoporosis Foundation Committee of Scientific Advisors Working Group on Cancer-Induced Bone Disease. Cancer-associated bone disease. Osteoporos Int 2013; 24:2929–2953.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Hu MI, Glezerman IG, Leboulleux S, et al. Denosumab for treatment of hypercalcemia of malignancy. J Clin Endocrinol Metab 2014; Jun 10 [Epub ahead of print].
- Cummings SR, San Martin J, McClung MR, et al; FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Papapoulos S, Lippuner K, Roux C, et al. Eight years of denosumab treatment in postmenopausal women with osteoporosis: results from the first five years of the FREEDOM extension [abstract]. Presented at the 2013 annual meeting of the American Society for Bone and Mineral Research, Baltimore, MD, October 4–7, 2013.
- Bone HG, Chapurlat R, Brandi ML, et al. The effect of three or six years of denosumab exposure in women with postmenopausal osteoporosis: results from the FREEDOM extension. J Clin Endocrinol Metab 2013; 98:4483–4492.
- McClung MR, Boonen S, Törring O, et al. Effect of denosumab treatment on the risk of fractures in subgroups of women with postmenopausal osteoporosis. J Bone Miner Res 2012; 27:211–218.
- Boonen S, Adachi JD, Man Z, et al. Treatment with denosumab reduces the incidence of new vertebral and hip fractures in postmenopausal women at high risk. J Clin Endocrinol Metab 2011; 96:1727–1736.
- McCloskey EV, Johansson H, Oden A, et al. Denosumab reduces the risk of osteoporotic fractures in postmenopausal women, particularly in those with moderate to high fracture risk as assessed with FRAX. J Bone Miner Res 2012; 27:1480–1486.
- McClung MR, Lewiecki EM, Geller ML, et al. Effect of denosumab on bone mineral density and biochemical markers of bone turnover: 8-year results of a phase 2 clinical trial. Osteoporos Int 2013; 24:227–235.
- McClung MR, Lewiecki EM, Cohen SB, et al; AMG 162 Bone Loss Study Group. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med 2006; 354:821–831.
- Bone HG, Bolognese MA, Yuen CK, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab 2008; 93:2149–2157.
- Genant HK, Engelke K, Hanley DA, et al. Denosumab improves density and strength parameters as measured by QCT of the radius in postmenopausal women with low bone mineral density. Bone 2010; 47:131–139.
- Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab 2012; 97:3161–3169.
- Tsai JN, Uihlein AV, Lee H, et al. Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: the DATA study randomised trial. Lancet 2013; 382:50–56.
- Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med 2009; 361:745–755.
- Ellis GK, Bone HG, Chlebowski R, et al. Randomized trial of denosumab in patients receiving adjuvant aromatase inhibitors for nonmetastatic breast cancer. J Clin Oncol 2008; 26:4875–4882.
- Brown JP, Prince RL, Deal C, et al. Comparison of the effect of denosumab and alendronate on BMD and biochemical markers of bone turnover in postmenopausal women with low bone mass: a randomized, blinded, phase 3 trial. J Bone Miner Res 2009; 24:153–161.
- Kendler DL, Roux C, Benhamou CL, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res 2010; 25:72–81.
- Seeman E, Delmas PD, Hanley DA, et al. Microarchitectural deterioration of cortical and trabecular bone: differing effects of denosumab and alendronate. J Bone Miner Res 2010; 25:1886–1894.
- Hopkins RB, Goeree R, Pullenayegum E, et al. The relative efficacy of nine osteoporosis medications for reducing the rate of fractures in post-menopausal women. BMC Musculoskelet Disord 2011; 12:209.
- National Institute for Health and Care Excellence (NICE). NICE technology appraisal guidance: TA161. Alendronate, etidronate, risedronate, raloxifene, strontium ranelate and teriparatide for the secondary prevention of osteoporotic fragility fractures in postmenopausal women (amended). http://publications.nice.org.uk/alendronate-etidronate-risedronate-raloxifene-strontium-ranelate-and-teriparatide-for-ta161. Accessed January 9, 2015.
- Freemantle N, Satram-Hoang S, Tang ET, et al; DAPS Investigators. Final results of the DAPS (Denosumab Adherence Preference Satisfaction) study: a 24-month, randomized, crossover comparison with alendronate in postmenopausal women. Osteoporos Int 2012; 23:317–326.
- Lewiecki EM. Safety and tolerability of denosumab for the treatment of postmenopausal osteoporosis. Drug Healthc Patient Saf 2011; 3:79–91.
- Watts NB, Roux C, Modlin JF, et al. Infections in postmenopausal women with osteoporosis treated with denosumab or placebo: coincidence or causal association? Osteoporos Int 2012; 23:327–337.
- von Keyserlingk C, Hopkins R, Anastasilakis A, et al. Clinical efficacy and safety of denosumab in postmenopausal women with low bone mineral density and osteoporosis: a meta-analysis. Semin Arthritis Rheum 2011; 41:178–186.
- Geller M, Wagman RB, Ho PR, et al. Early findings from Prolia postmarketing safety surveillance for atypical femoral fracture, osteonecrosis of the jaw, severe symptomatic hypocalcemia, and anaphylaxis (abstract). Osteoporos Int 2014; 25(suppl 2). OC40; www.wco-iof-esceo.org/sites/ecceo14/docs/wco14-abstractbook.pdf. Accessed January 9, 2015.
- McCormick BB, Davis J, Burns KD. Severe hypocalcemia following denosumab injection in a hemodialysis patient. Am J Kidney Dis 2012; 60:626–628.
- Jamal SA, Ljunggren O, Stehman-Breen C, et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J Bone Miner Res 2011; 26:1829–1835.
- Block GA, Bone HG, Fang L, Lee E, Padhi D. A single-dose study of denosumab in patients with various degrees of renal impairment. J Bone Miner Res 2012; 27:1471–1479.
- Ungprasert P, Cheungpasitporn W, Srivali N, Kittanamongkolchai W, Bischof EF. Life-threatening hypocalcemia associated with denosumab in a patient with moderate renal insufficiency. Am J Emerg Med 2013; 31:756.e1–e2.
- McCormick BB, Davis J, Burns KD. Severe hypocalcemia following denosumab injection in a hemodialysis patient. Am J Kidney Dis 2012; 60:626–628.
- Rachner TD, Platzbecker U, Felsenberg D, Hofbauer LC. Osteonecrosis of the jaw after osteoporosis therapy with denosumab following long-term bisphosphonate therapy. Mayo Clin Proc 2013; 88:418–419.
- Epstein MS, Ephros HD, Epstein JB. Review of current literature and implications of RANKL inhibitors for oral health care providers. Oral Surg Oral Med Oral Pathol Oral Radiol 2013; 116:e437–e442.
- McClung M, Harris ST, Miller PD, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med 2013; 126:13–20.
- Baron R, Ferrari S, Russell RG. Denosumab and bisphosphonates: different mechanisms of action and effects. Bone 2011; 48:677–692.
- Bekker PJ, Holloway DL, Rasmussen AS, et al. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res 2004; 19:1059–1066.
- Miller PD, Bolognese MA, Lewiecki EM, et al; Amg Bone Loss Study Group. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone 2008; 43:222–229.
- Parthan A, Kruse M, Yurgin N, Huang J, Viswanathan HN, Taylor D. Cost effectiveness of denosumab versus oral bisphosphonates for postmenopausal osteoporosis in the US. Appl Health Econ Health Policy 2013; 11:485–497.
- Scotland G, Waugh N, Royle P, McNamee P, Henderson R, Hollick R. Denosumab for the prevention of osteoporotic fractures in post-menopausal women: a NICE single technology appraisal. Pharmacoeconomics 2011; 29:951–961.
- Jönsson B, Ström O, Eisman JA, et al. Cost-effectiveness of denosumab for the treatment of postmenopausal osteoporosis. Osteoporos Int 2011; 22:967–982.
- Clinician’s guide to prevention and treatment of osteoporosis. Washington DC: National Osteoporosis Foundation, 2013.
- Watts NB, Bilezikian JP, Camacho PM, et al; AACE Osteoporosis Task Force. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the diagnosis and treatment of postmenopausal osteoporosis. Endocr Pract 2010;16(suppl 3):1–37.
- Papaioannou A, Morin S, Cheung AM, et al; Scientific Advisory Council of Osteoporosis Canada. 2010 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada: summary. CMAJ 2010; 182:1864–1673.
- National Institute for Health and Care Excellence (NICE). NICE technology appraisal: TA204. Denosumab for the prevention of osteoporotic fractures in postmenopausal women. http://guidance.nice.org.uk/TA204. Accessed January 9, 2015.
- Premaor MO, Compston JE. Testing for secondary causes of osteoporosis. BMJ 2010; 341:c6959.
- Hochberg MC, Ross PD, Black D, et al. Larger increases in bone mineral density during alendronate therapy are associated with a lower risk of new vertebral fractures in women with postmenopausal osteoporosis. Fracture Intervention Trial Research Group. Arthritis Rheum 1999; 42:1246–1254.
- Eastell R, Vrijens B, Cahall DL, Ringe JD, Garnero P, Watts NB. Bone turnover markers and bone mineral density response with risedronate therapy: relationship with fracture risk and patient adherence. J Bone Miner Res 2011; 26:1662–1669.
- Diez-Perez A, Adachi JD, Agnusdei D, et al; IOF CSA Inadequate Responders Working Group. Treatment failure in osteoporosis. Osteoporos Int 2012; 23:2769–2774.
- Eastell R, Barton I, Hannon RA, Chines A, Garnero P, Delmas PD. Relationship of early changes in bone resorption to the reduction in fracture risk with risedronate. J Bone Miner Res 2003; 18:1051–1056.
- Rosen HN, Moses AC, Garber J, Ross DS, Lee SL, Greenspan SL. Utility of biochemical markers of bone turnover in the follow-up of patients treated with bisphosphonates. Calcif Tissue Int 1998; 63:363–368.
- Bolognese MA, Teglbjærg CS, Zanchetta JR, et al. Denosumab significantly increases DXA BMD at both trabecular and cortical sites: results from the FREEDOM study. J Clin Densitom 2013; 16:147–153.
A 68-year-old white woman presents with mid- thoracic back pain. Plain radiographs reveal a compression fracture of the 10th thoracic vertebra. She is diagnosed with osteoporosis on the basis of dual energy x-ray absorptiometry (DXA) scans that show T scores of –2.9 in her lumbar spine and –2.6 in her left femoral neck. Her 10-year probability of fracture is estimated as 23% for major osteoporotic fracture and 5.9% for hip fracture (based on the World Health Organization’s absolute fracture risk assessment tool, adapted for the United States, and available at www.shef.ac.uk/FRAX).
After excluding common secondary causes of osteoporosis, her physician recommends a bisphosphonate to reduce her risk of fracture, but she develops upper-gastrointestinal adverse effects with both alendronate and risedronate despite correctly following the instructions for oral administration.
What should her physician consider next?
OSTEOPOROSIS IS A MAJOR PROBLEM
Osteoporosis is a systemic skeletal disease characterized by low bone mass and microarchitectural deterioration of bone tissue, predisposing to an increased risk of fragility fractures, particularly of the spine, hip, and wrist.
It is a major public health problem, affecting 200 million people throughout the world, with 9 million osteoporotic fractures reported in the year 2000.1 The incidence of hip fracture alone is predicted to rise to 2.6 million by the year 2025, and to 4.5 million by the year 2050.2 In the United States, the total burden was estimated to be about 2 million incident fractures in the year 2005, projected to rise by another 50% by the year 2025,3 primarily because of the aging of the population. Population studies have indeed suggested that about 40% of white women and 13% of white men over the age of 50 are at risk of sustaining an osteoporotic fracture during the remainder of their lifetime.4
The consequences of osteoporotic fractures can be devastating. Hip fractures are associated with a risk of death ranging from 8.4% to 36% during the first year after fracture.5 One-fifth of patients who sustain a hip fracture require long-term nursing home care, and more than half of the survivors do not regain their previous level of independence.
Patients with vertebral fractures are also at increased risk of death, although the results of some studies suggest that this could be the result of comorbid factors.6–9 Vertebral fractures can result in chronic back pain, loss of height from spinal deformity, reduced mobility, loss of self-esteem, and in severe cases, respiratory and digestive problems because of contact between the lower ribs and pelvis.
A person with one vertebral compression fracture is five times more likely to have another vertebral fracture,10 and a person with two or more compression fractures is 12 times more likely.11
The costs of treating osteoporotic fractures are greater than those of treating myocardial infarction or stroke12,13; they include not only direct costs incurred in treating the fracture, but also indirect societal costs owing to the long-term morbidity associated with the fracture. In the United States, the total cost of treating osteoporotic fractures was estimated at $19 billion in the year 2005.3 By 2025, the annual costs are projected to rise by almost 50%.3
A NEED FOR MORE OPTIONS
Until fairly recently, bisphosphonates were the only drugs of first choice, but adherence to oral bisphosphonate therapy is generally poor (< 50% at 1 year),14 most commonly because of dyspepsia,15 and poor adherence has been shown to be associated with increased fracture risk.16,17 Hence the need for additional therapeutic options.
In this review, we discuss denosumab, an antiresorptive drug approved by the US Food and Drug Administration (FDA) in 2010. First, we discuss its mechanism of action, efficacy, and safety, and then we offer recommendations for its use in clinical practice.
WHAT IS DENOSUMAB AND HOW DOES IT WORK?
Bone remodeling is a dynamic process involving a balance between bone resorption by osteoclasts on the one hand and new bone formation by osteoblasts on the other. A net gain in bone occurs when the activity of osteoblasts exceeds that of osteoclasts, and bone loss occurs when there is increased osteoclast activity or reduced osteoblast activity, or both. The activities of osteoblasts and osteoclasts are tightly coupled because of the opposing effects of two sets of proteins, namely, receptor activator of nuclear factor kappa b ligand (RANKL) and osteoprotegerin.
Both RANKL and osteoprotegerin are produced by osteoblasts. RANKL binds to its receptor (RANK) on preosteoclasts and osteoclasts and induces their differentiation and activation, respectively. Osteoprotegerin is the decoy receptor and natural antagonist for RANKL. By binding with RANKL, it blocks its interaction with RANK.18 In healthy individuals, a fine balance between RANKL and osteoprotegerin ensures that bone remodeling is regulated.
In postmenopausal women, estrogen deficiency leads to an imbalance between RANKL and osteoprotegerin (increased RANKL and reduced osteoprotegerin), resulting in net bone loss. This imbalance is also a feature of rheumatoid arthritis, myeloma bone disease, and osteolytic metastatic bone disease; it also occurs in those receiving androgen deprivation therapy for prostate cancer or aromatase inhibitors for breast cancer.
Denosumab is a fully human monoclonal antibody that targets RANKL.19 By binding to RANKL, this drug prevents the maturation and differentiation of preosteoclasts and promotes apoptosis of osteoclasts. Bone resorption is therefore slowed. It was parenteral osteoprotegerin that was initially developed by denosumab’s manufacturer,20 but this approach failed because neutralizing antibodies developed to osteoprotegerin, rendering it ineffective. Development of neutralizing antibodies has thus far not been a problem with denosumab.
Denosumab, with its property of RANKL inhibition, has also been used to prevent skeletal events in patients with bone metastases from solid tumors and to treat unresectable giant cell tumors of the bone (both FDA-approved indications) and hypercalcemia of malignancy. There is limited clinical experience in Paget disease of the bone as well.21–23 These other potential uses of denosumab are beyond the scope of this review.
HOW WELL DOES DENOSUMAB WORK FOR OSTEOPOROSIS?
Several phase 2 and phase 3 randomized controlled trials have evaluated the efficacy of denosumab, but only one, the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) trial, included fracture reduction as the primary outcome measure. The rest evaluated changes in bone mineral density or in markers of bone turnover, or both.
FREEDOM was a double-blind, randomized controlled trial in 7,808 postmenopausal women with T scores between –2.5 and –4.0 at the lumbar spine or hip.24 Twenty-four percent of the patients had vertebral fractures at baseline. Patients were randomized to receive either denosumab 60 mg (n = 3,902) or placebo (n = 3,906) every 6 months for up to 36 months. All patients also received adequate calcium and vitamin D supplementation.
At 36 months, compared with those who were randomized to receive placebo, those who were randomized to denosumab had lower incidence rates of:
- New vertebral fracture
(2.3% vs 7.2%, risk ratio 0.32,
95% CI 0.26–0.41, P < .001) - Nonvertebral fracture
(6.5% vs 8.0%, risk ratio 0.80,
95% CI 0.67–0.95, P = .01) - Hip fracture
(0.7% vs 1.2%, risk ratio 0.60,
95% CI 0.37–0.97, P = .04).
Increases in bone mineral density at the lumbar spine and hip, and decreases in bone turnover markers were also significantly greater in the denosumab group. The number needed to treat to prevent one new fracture over 3 years was 21 for vertebral fracture, 67 for nonvertebral fracture, and 200 for hip fracture, reflecting the relatively low event rate in the study.
In an open-label extension of the FREEDOM trial, the fracture incidence rates among participants who continued to receive denosumab for an additional 5 years remained low, and still below those projected for a “virtual placebo cohort” (total duration of exposure of 8 years). The rates among participants who switched from placebo to denosumab were similar to those of the denosumab group from the parent trial.25,26
A subgroup analysis of the FREEDOM trial suggested that denosumab reduced the risk of new vertebral fractures irrespective of age, body mass index, femoral neck bone mineral density, prevalent vertebral fractures, or prior nonvertebral fractures (risk ratio 0.32; 95% CI 0.26–0.41, P < .001), whereas the risk of nonvertebral fractures was only reduced in those women with body mass indices less than 25 kg/m2, femoral neck bone mineral density T scores less than –2.5, and in those without a prevalent vertebral fracture.27
A post hoc analysis revealed that denosumab significantly reduced the risk of new vertebral and hip fractures even in subgroups of women at higher risk of fracture.28 At 10% fracture probability (as estimated by the FRAX risk calculator), denosumab reduced the fracture risk by 11% (P = .629), whereas at 30% probability (moderate to high risk), the reduction was 50% (P = .001).29
Other phase 2 and phase 3 trials, in postmenopausal women with low bone mineral density, demonstrated that compared with placebo, denosumab significantly increased bone mineral density at all skeletal sites, increased volumetric bone mineral density at the distal radius, improved hip structural analysis parameters, and reduced bone turnover markers.30–33 Increases in bone mineral density and reductions in bone turnover markers with denosumab have been shown in men as well.34
In a randomized controlled trial,35 improvement in bone mineral density was better in those who received the combination of denosumab and teriparatide than in those who received either drug on its own.
Denosumab has also been shown to reduce the incidence of new vertebral fractures and improve bone mineral density in men receiving androgen-deprivation therapy for nonmetastatic prostate cancer,36 and to improve bone mineral density in women with metastatic breast cancer and low bone mass who were receiving adjuvant aromatase inhibitor therapy.37
HOW DOES DENOSUMAB COMPARE WITH OTHER OSTEOPOROSIS DRUGS?
A double-blind randomized controlled trial in postmenopausal women with low bone mass demonstrated that denosumab was superior to alendronate in improving bone mineral density at all skeletal sites (3.5% vs 2.6% for total hip bone mineral density, P < .0001).38
Another double-blind trial demonstrated that in patients previously treated with alendronate, switching to denosumab resulted in significantly greater increases in bone mineral density at all skeletal sites compared with continuing with alendronate (P < .0001).39
Denosumab has also been shown to be superior to alendronate in improving cortical bone mineral density, as measured by quantitative computed tomography.40
No trial has directly compared the efficacy of denosumab with other osteoporosis drugs in reducing fracture risk, but a systematic literature review of multiple databases,41 comparing the antifracture efficacy of nine osteoporosis drugs, concluded that teriparatide, zoledronic acid, and denosumab had the highest probabilities of being most efficacious for nonvertebral and vertebral fractures, with the greatest effect sizes. Indirect comparisons of the relative risk of fracture with denosumab (based on the results of FREEDOM), alendronate, risedronate, raloxifene, and strontium (based on a meta-analysis of randomized controlled trials) are presented in Table 1.42
A 2-year randomized, open-label, crossover study43 randomized patients to receive either denosumab followed by alendronate or alendronate followed by denosumab over successive 12-month periods. The results suggested that postmenopausal women with osteoporosis were more adherent, compliant, and persistent with denosumab therapy (a subcutaneous injection every 6 months) than with alendronate therapy in the form of oral tablets, self-administered weekly (7.5% nonadherence vs 36.5% at the end of 2 years). After receiving both treatments, women reported greater satisfaction with denosumab, with 92.4% preferring it over oral alendronate. Bone mineral density remained stable when patients were switched from denosumab to alendronate, but improved further when they were switched from alendronate to denosumab.
HOW SAFE IS DENOSUMAB?
The most frequent adverse events with denosumab reported in the long-term extension of one phase 2 study were upper respiratory tract infections (13.5%), arthralgia (11.5%), and back pain (9.0%).30
Increased risk of infection, cancer, and dermatologic reactions has been a concern, as RANKL and RANK are expressed by a wide variety of cells, including T lymphocytes, B cells, and dendritic cells.44 However, there were no significant differences in the overall incidences of adverse events between patients who received denosumab and those who received placebo or alendronate in any of the phase 2, phase 3, or extension studies.
In the FREEDOM trial,24 there was no significant difference between the two groups in the overall incidence of infection (52.9% with denosumab vs 54.4% with placebo, P = .17), or serious infection (4.1% with denosumab vs 3.4% with placebo, P = .14), although the incidence of “serious” cellulitis requiring hospitalization was higher in the denosumab group (0.3% vs < 0.1%, P = .002). There were more serious infections involving the gastrointestinal system, urinary tract, and ear and cases of endocarditis in the denosumab group, but the number of events was small, and there was no relationship with the timing of administration or duration of exposure to denosumab.45 Eczema was more common in the denosumab than in the placebo group (3.0% vs 1.7%, P < .001), but the extension data from the first 3 years did not provide any evidence for an increased risk of cellulitis or eczema with denosumab.26
Although randomized controlled trials reported more cases of neoplasms in the denosumab than in the placebo groups, meta-analyses have failed to detect a statistically significant difference (risk ratio 1.11, 95% CI 0.91–1.36).46 The overall incidence of adverse and serious adverse events reported in the 8-year extension of FREEDOM were consistent with data reported in the previous extension studies.25
In the FREEDOM extension trial, four events in the long-term group (n = 2,343), and two in the crossover group (n = 2,207) were adjudicated as being consistent with osteonecrosis of the jaw.26 One mid-shaft fracture in the crossover group was adjudicated as an atypical femoral fracture. There were, however, no reports of osteonecrosis of the jaw or atypical femoral fracture in the long-term phase 2 trial after 8 years of follow-up.30 By September 2013, postmarketing safety surveillance data for denosumab (estimated exposure of 1.2 million patient-years) had recorded four cases of atypical femoral fracture. All four patients had previously been on bisphosphonates. There were also 32 reports of osteonecrosis of the jaw.47
Denosumab’s manufacturer aims to communicate the risks of treatment to health care professionals and patients. Information is available online at www.proliahcp.com/risk-evaluation-mitigation-strategy/.
WHAT ARE THE PRECAUTIONS?
Several precautions need to be taken when considering treatment with denosumab.
Antiresorptives can aggravate hypocalcemia by inhibiting bone turnover. Serum calcium should therefore be checked and preexisting hypocalcemia should be corrected before starting denosumab.48
Denosumab is contraindicated in women who are pregnant or are planning to become pregnant, as fetal loss and teratogenicity have been reported in animal experiments. (Denosumab is unlikely to be used in premenopausal women, as it is not approved for use in this group.)
There are no data on excretion of denosumab in human milk, so it should not be given to nursing mothers.
Renal impairment is not a contraindication, and no dose adjustment is necessary (even for patients on renal replacement therapy), as denosumab, being an antibody, is eliminated through the reticuloendothelial system.49,50 However, in practice, any antiresorptive agent should be used with caution in patients with severe renal impairment because of the possible presence of adynamic bone disease. Further reduction of bone turnover would be detrimental in such patients. Also, severe hypocalcemia has been reported in patients with a creatinine clearance rate less than 30 mL/min and in those receiving dialysis.51,52 Postmarketing surveillance data have reported eight cases of severe symptomatic hypocalcemia, of which seven were in patients with chronic kidney disease.47
The manufacturer suggests that patients receive a dental examination with appropriate preventive dentistry before starting denosumab to reduce the incidence of osteonecrosis of the jaw, despite the lack of evidence in support of this strategy. The American Dental Association recommends regular dental visits and maintenance of good oral hygiene for patients already established on antiresorptive therapy.53,54
SHOULD PATIENTS ON DENOSUMAB BE OFFERED A DRUG HOLIDAY?
A drug holiday (temporary discontinuation of the drug after a certain duration of treatment) has been proposed for patients receiving bisphosphonates because of the risk of atypical femoral fracture and osteonecrosis of the jaw (although small) consequent to long-term continuous suppression of bone turnover.55 The antifracture efficacy of bisphosphonates is likely to persist for an unknown length of time after discontinuation because of their long skeletal half-life, while the risks gradually diminish.
By contrast, denosumab targets RANKL in the extracellular fluid and does not become embedded within the bone tissue.56 Pharmacokinetic studies have shown that denosumab has a rapid offset of action, with a half-life of only 26 days and biological activity lasting only 6 months.57 The results of a phase 2 extension study suggest that bone mineral density starts to decline and bone turnover markers start to rise within 12 months of discontinuing denosumab.58
Although fracture risk did not increase in those who were randomized to stopping the treatment and bone mineral density increased further when treatment was restarted, a drug holiday cannot presently be recommended for patients receiving denosumab because of the lack of supportive data.
HOW COST-EFFECTIVE IS DENOSUMAB?
The wholesale acquisition cost is $825 per 60-mg prefilled syringe of denosumab, although this may vary depending on where the drug is obtained. This does not include physician-related service costs associated with administration of denosumab.
Cost-effectiveness analyses conducted in the United States, the United Kingdom, and Sweden have all concluded that denosumab would offer a cost-effective alternative to other osteoporosis medications for primary prevention and secondary prevention of fractures.59–61
The Swedish study also incorporated adherence in the cost-effectiveness model and showed that denosumab was a cost-effective alternative to oral bisphosphonates, particularly for patients who were not expected to adhere well to oral treatments.61
WHICH OSTEOPOROSIS PATIENTS ARE CANDIDATES FOR DENOSUMAB?
The FDA has approved denosumab for the treatment of postmenopausal women and men at high risk of fracture (defined as having a history of osteoporotic fracture or multiple risk factors for fracture), or in those who cannot tolerate other osteoporosis medications or for whom other medications have failed.
Denosumab is also approved for men at high risk of fracture receiving androgen deprivation therapy for nonmetastatic prostate cancer, and for women at high risk of fracture receiving adjuvant aromatase inhibitor therapy for breast cancer.
WHAT DO THE GUIDELINES RECOMMEND?
The National Osteoporosis Foundation guidelines recommend pharmacologic treatment for patients with hip or vertebral fractures (clinical or asymptomatic); T scores lower than –2.5 at the femoral neck, total hip, or lumbar spine; and those with a 10-year probability of hip fracture of more than 3% or of a major osteoporotic fracture more than 20% based on the US-adapted FRAX calculator.62 The American College of Endocrinology guidelines have proposed similar thresholds for pharmacologic treatment, and they recommend alendronate, risedronate, zoledronate, and denosumab as first-line agents.63
The 2010 Osteoporosis Canada guidelines recommend denosumab, alendronate, risedronate, and zoledronate as first-line therapies for preventing hip, nonvertebral, and vertebral fractures in postmenopausal women (grade A recommendation).64 The National Institute of Health and Clinical Excellence in England and Wales, on the other hand, recommends denosumab only for patients who are unable to take a bisphosphonate.65
PRACTICAL PRESCRIBING TIPS
The patient described at the beginning of this article has already sustained a vertebral compression fracture, and her DXA scan shows T scores in the osteoporotic range. She is therefore at increased risk of another fragility fracture (with a fivefold higher risk of another vertebral fracture). Pharmacologic therapy should be considered. In addition, she should be encouraged to adhere to lifestyle measures such as a healthy diet and regular weight-bearing exercise, her risk of falling should be assessed, and adequate calcium and vitamin D supplementation should be given.
Secondary causes of osteoporosis are present in about 30% of women and 55% of men who have vertebral fractures.66 A complete blood count, erythrocyte sedimentation rate, bone biochemistry, 25-hydroxyvitamin D, thyroid-stimulating hormone, and renal and liver function tests should be requested in all patients. Further tests should be considered depending on the clinical evaluation and results of initial investigations.
Because this patient cannot tolerate oral bisphosphonates, she could be offered the option of annual intravenous zoledronic acid infusions or 6-monthly subcutaneous denosumab injections. In clinical trials, gastrointestinal adverse effects were noted with intravenous bisphosphonates as well, but the adverse effects reported were no different than those with placebo. The potential advantages with denosumab include better bone mineral density gains, adherence and patient satisfaction compared with oral bisphosphonates, convenient twice-yearly administration, safety in patients with renal impairment, and absence of gastrointestinal effects.
Raloxifene, a selective estrogen receptor modulator, has estrogen-like action on the bone and antiestrogen actions on the breast and uterus. Unlike standard hormone replacement therapy, raloxifene can therefore increase bone mineral density without increasing the risk of breast and endometrial cancers. However, it has only been shown to reduce the risk of vertebral fracture, not hip fracture. Hence, it would be a more appropriate choice for younger postmenopausal women. Moreover, it may cause troublesome menopausal symptoms.
Teriparatide, the recombinant parathyroid hormone, is an anabolic agent. It is very expensive, and because of this, guidelines in several countries restrict its use to women with severe osteoporosis and multiple fractures who fail to respond to standard treatments. It cannot be used for longer than 2 years because of its association with osteosarcoma in rats.
If our patient prefers denosumab, therapy should be initiated after appropriate counseling (see precautions above). The dose is 60 mg, given subcutaneously, once every 6 months.
Monitoring
There is no consensus regarding the optimal frequency for monitoring patients on treatment, owing to the lack of prospective trial data. The National Osteoporosis Foundation recommends repeating the bone mineral density measurements about 2 years after starting therapy, and about every 2 years thereafter.62 Some studies suggest that changes in bone mineral density correlate with reduction in fracture risk.67,68 A change in bone mineral density is considered significant when it is greater than the range of error of the densitometer (also known as the least significant change).69 If the bone mineral density is stable or improving, therapy could be continued, but if it is declining and the decline is greater than the least significant change, a change in therapy should be considered if no secondary causes for bone loss are evident (but see What are the areas of uncertainty? below).
The National Osteoporosis Foundation also recommends measuring a bone turnover marker at baseline and then 3 to 6 months later, as its suppression predicts greater bone mineral density responses and fracture risk reduction.70 If there is a decrease of more than 30% in serum carboxy-terminal collagen crosslinks (CTX) or more than 50% in urinary N-telopeptide (NTX),71 the patient can be reassured that the next bone mineral density measurement will be stable or improved. In patients on oral bisphosphonates, measurement of bone turnover markers also provides evidence of compliance.
Clinical trials suggest that a numerical increase in bone mineral density can be expected in most patients on treatment, though this depends on the measurement site and the length of time between examinations. In one phase 3 trial of denosumab in postmenopausal women, only 5% of the participants had unchanged or diminished bone mineral density at the lumbar spine, and 8% at the hip, after 36 months of treatment.72 However, the CTX levels fell to below the lower limit of the reference interval as early as 1 month after commencing treatment in all denosumab-treated patients.68
Hence, bone turnover markers may be a more sensitive indicator of treatment effect than bone mineral density, but this would ultimately need to be evaluated against fracture rates in a real-world setting.
WHAT ARE THE AREAS OF UNCERTAINTY?
There are currently no guidelines for long-term management of patients on denosumab, and also no data to suggest whether patients should be switched to a weaker antiresorptive drug after a certain number of years in order to reduce the possible risk of atypical femoral fracture or osteonecrosis of the jaw.
No head-to-head trials have directly compared the antifracture efficacy of denosumab with that of other standard osteoporosis therapies. The antifracture efficacy and safety of combination therapies involving denosumab are also uncertain. For adherent patients who have a suboptimal response, there is no evidence to guide the further course of action. The International Osteoporosis Foundation guidelines suggest replacing a stronger antiresorptive with an anabolic agent, but acknowledge that this is only based on expert opinion.71
The very-long-term effects (beyond 8 years) of continuous denosumab administration on increasing the risk of atypical femoral fracture, osteonecrosis of the jaw, malignancy, or infection or the duration after which risks would start to outweigh benefits is not known. However, postmarketing safety data continue to be collected through the voluntary Post-marketing Active Safety Surveillance Program (for prespecified adverse events) in addition to the FDA’s MedWatch program.
CASE PROGRESSION
The patient described in the vignette is presented with two options—zoledronate and denosumab. She chooses denosumab. Her renal function and serum calcium are checked and are found to be satisfactory. She undergoes a dental examination, which is also satisfactory. She is counseled about the possible increased risk of infection, and then she is started on 60 mg of denosumab subcutaneously, once every 6 months.
When reviewed after 2 years, she reports no further fractures. Her bone mineral density remains stable compared with the values obtained before starting treatment. She reports no adverse effects and is happy to continue with denosumab.
A 68-year-old white woman presents with mid- thoracic back pain. Plain radiographs reveal a compression fracture of the 10th thoracic vertebra. She is diagnosed with osteoporosis on the basis of dual energy x-ray absorptiometry (DXA) scans that show T scores of –2.9 in her lumbar spine and –2.6 in her left femoral neck. Her 10-year probability of fracture is estimated as 23% for major osteoporotic fracture and 5.9% for hip fracture (based on the World Health Organization’s absolute fracture risk assessment tool, adapted for the United States, and available at www.shef.ac.uk/FRAX).
After excluding common secondary causes of osteoporosis, her physician recommends a bisphosphonate to reduce her risk of fracture, but she develops upper-gastrointestinal adverse effects with both alendronate and risedronate despite correctly following the instructions for oral administration.
What should her physician consider next?
OSTEOPOROSIS IS A MAJOR PROBLEM
Osteoporosis is a systemic skeletal disease characterized by low bone mass and microarchitectural deterioration of bone tissue, predisposing to an increased risk of fragility fractures, particularly of the spine, hip, and wrist.
It is a major public health problem, affecting 200 million people throughout the world, with 9 million osteoporotic fractures reported in the year 2000.1 The incidence of hip fracture alone is predicted to rise to 2.6 million by the year 2025, and to 4.5 million by the year 2050.2 In the United States, the total burden was estimated to be about 2 million incident fractures in the year 2005, projected to rise by another 50% by the year 2025,3 primarily because of the aging of the population. Population studies have indeed suggested that about 40% of white women and 13% of white men over the age of 50 are at risk of sustaining an osteoporotic fracture during the remainder of their lifetime.4
The consequences of osteoporotic fractures can be devastating. Hip fractures are associated with a risk of death ranging from 8.4% to 36% during the first year after fracture.5 One-fifth of patients who sustain a hip fracture require long-term nursing home care, and more than half of the survivors do not regain their previous level of independence.
Patients with vertebral fractures are also at increased risk of death, although the results of some studies suggest that this could be the result of comorbid factors.6–9 Vertebral fractures can result in chronic back pain, loss of height from spinal deformity, reduced mobility, loss of self-esteem, and in severe cases, respiratory and digestive problems because of contact between the lower ribs and pelvis.
A person with one vertebral compression fracture is five times more likely to have another vertebral fracture,10 and a person with two or more compression fractures is 12 times more likely.11
The costs of treating osteoporotic fractures are greater than those of treating myocardial infarction or stroke12,13; they include not only direct costs incurred in treating the fracture, but also indirect societal costs owing to the long-term morbidity associated with the fracture. In the United States, the total cost of treating osteoporotic fractures was estimated at $19 billion in the year 2005.3 By 2025, the annual costs are projected to rise by almost 50%.3
A NEED FOR MORE OPTIONS
Until fairly recently, bisphosphonates were the only drugs of first choice, but adherence to oral bisphosphonate therapy is generally poor (< 50% at 1 year),14 most commonly because of dyspepsia,15 and poor adherence has been shown to be associated with increased fracture risk.16,17 Hence the need for additional therapeutic options.
In this review, we discuss denosumab, an antiresorptive drug approved by the US Food and Drug Administration (FDA) in 2010. First, we discuss its mechanism of action, efficacy, and safety, and then we offer recommendations for its use in clinical practice.
WHAT IS DENOSUMAB AND HOW DOES IT WORK?
Bone remodeling is a dynamic process involving a balance between bone resorption by osteoclasts on the one hand and new bone formation by osteoblasts on the other. A net gain in bone occurs when the activity of osteoblasts exceeds that of osteoclasts, and bone loss occurs when there is increased osteoclast activity or reduced osteoblast activity, or both. The activities of osteoblasts and osteoclasts are tightly coupled because of the opposing effects of two sets of proteins, namely, receptor activator of nuclear factor kappa b ligand (RANKL) and osteoprotegerin.
Both RANKL and osteoprotegerin are produced by osteoblasts. RANKL binds to its receptor (RANK) on preosteoclasts and osteoclasts and induces their differentiation and activation, respectively. Osteoprotegerin is the decoy receptor and natural antagonist for RANKL. By binding with RANKL, it blocks its interaction with RANK.18 In healthy individuals, a fine balance between RANKL and osteoprotegerin ensures that bone remodeling is regulated.
In postmenopausal women, estrogen deficiency leads to an imbalance between RANKL and osteoprotegerin (increased RANKL and reduced osteoprotegerin), resulting in net bone loss. This imbalance is also a feature of rheumatoid arthritis, myeloma bone disease, and osteolytic metastatic bone disease; it also occurs in those receiving androgen deprivation therapy for prostate cancer or aromatase inhibitors for breast cancer.
Denosumab is a fully human monoclonal antibody that targets RANKL.19 By binding to RANKL, this drug prevents the maturation and differentiation of preosteoclasts and promotes apoptosis of osteoclasts. Bone resorption is therefore slowed. It was parenteral osteoprotegerin that was initially developed by denosumab’s manufacturer,20 but this approach failed because neutralizing antibodies developed to osteoprotegerin, rendering it ineffective. Development of neutralizing antibodies has thus far not been a problem with denosumab.
Denosumab, with its property of RANKL inhibition, has also been used to prevent skeletal events in patients with bone metastases from solid tumors and to treat unresectable giant cell tumors of the bone (both FDA-approved indications) and hypercalcemia of malignancy. There is limited clinical experience in Paget disease of the bone as well.21–23 These other potential uses of denosumab are beyond the scope of this review.
HOW WELL DOES DENOSUMAB WORK FOR OSTEOPOROSIS?
Several phase 2 and phase 3 randomized controlled trials have evaluated the efficacy of denosumab, but only one, the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) trial, included fracture reduction as the primary outcome measure. The rest evaluated changes in bone mineral density or in markers of bone turnover, or both.
FREEDOM was a double-blind, randomized controlled trial in 7,808 postmenopausal women with T scores between –2.5 and –4.0 at the lumbar spine or hip.24 Twenty-four percent of the patients had vertebral fractures at baseline. Patients were randomized to receive either denosumab 60 mg (n = 3,902) or placebo (n = 3,906) every 6 months for up to 36 months. All patients also received adequate calcium and vitamin D supplementation.
At 36 months, compared with those who were randomized to receive placebo, those who were randomized to denosumab had lower incidence rates of:
- New vertebral fracture
(2.3% vs 7.2%, risk ratio 0.32,
95% CI 0.26–0.41, P < .001) - Nonvertebral fracture
(6.5% vs 8.0%, risk ratio 0.80,
95% CI 0.67–0.95, P = .01) - Hip fracture
(0.7% vs 1.2%, risk ratio 0.60,
95% CI 0.37–0.97, P = .04).
Increases in bone mineral density at the lumbar spine and hip, and decreases in bone turnover markers were also significantly greater in the denosumab group. The number needed to treat to prevent one new fracture over 3 years was 21 for vertebral fracture, 67 for nonvertebral fracture, and 200 for hip fracture, reflecting the relatively low event rate in the study.
In an open-label extension of the FREEDOM trial, the fracture incidence rates among participants who continued to receive denosumab for an additional 5 years remained low, and still below those projected for a “virtual placebo cohort” (total duration of exposure of 8 years). The rates among participants who switched from placebo to denosumab were similar to those of the denosumab group from the parent trial.25,26
A subgroup analysis of the FREEDOM trial suggested that denosumab reduced the risk of new vertebral fractures irrespective of age, body mass index, femoral neck bone mineral density, prevalent vertebral fractures, or prior nonvertebral fractures (risk ratio 0.32; 95% CI 0.26–0.41, P < .001), whereas the risk of nonvertebral fractures was only reduced in those women with body mass indices less than 25 kg/m2, femoral neck bone mineral density T scores less than –2.5, and in those without a prevalent vertebral fracture.27
A post hoc analysis revealed that denosumab significantly reduced the risk of new vertebral and hip fractures even in subgroups of women at higher risk of fracture.28 At 10% fracture probability (as estimated by the FRAX risk calculator), denosumab reduced the fracture risk by 11% (P = .629), whereas at 30% probability (moderate to high risk), the reduction was 50% (P = .001).29
Other phase 2 and phase 3 trials, in postmenopausal women with low bone mineral density, demonstrated that compared with placebo, denosumab significantly increased bone mineral density at all skeletal sites, increased volumetric bone mineral density at the distal radius, improved hip structural analysis parameters, and reduced bone turnover markers.30–33 Increases in bone mineral density and reductions in bone turnover markers with denosumab have been shown in men as well.34
In a randomized controlled trial,35 improvement in bone mineral density was better in those who received the combination of denosumab and teriparatide than in those who received either drug on its own.
Denosumab has also been shown to reduce the incidence of new vertebral fractures and improve bone mineral density in men receiving androgen-deprivation therapy for nonmetastatic prostate cancer,36 and to improve bone mineral density in women with metastatic breast cancer and low bone mass who were receiving adjuvant aromatase inhibitor therapy.37
HOW DOES DENOSUMAB COMPARE WITH OTHER OSTEOPOROSIS DRUGS?
A double-blind randomized controlled trial in postmenopausal women with low bone mass demonstrated that denosumab was superior to alendronate in improving bone mineral density at all skeletal sites (3.5% vs 2.6% for total hip bone mineral density, P < .0001).38
Another double-blind trial demonstrated that in patients previously treated with alendronate, switching to denosumab resulted in significantly greater increases in bone mineral density at all skeletal sites compared with continuing with alendronate (P < .0001).39
Denosumab has also been shown to be superior to alendronate in improving cortical bone mineral density, as measured by quantitative computed tomography.40
No trial has directly compared the efficacy of denosumab with other osteoporosis drugs in reducing fracture risk, but a systematic literature review of multiple databases,41 comparing the antifracture efficacy of nine osteoporosis drugs, concluded that teriparatide, zoledronic acid, and denosumab had the highest probabilities of being most efficacious for nonvertebral and vertebral fractures, with the greatest effect sizes. Indirect comparisons of the relative risk of fracture with denosumab (based on the results of FREEDOM), alendronate, risedronate, raloxifene, and strontium (based on a meta-analysis of randomized controlled trials) are presented in Table 1.42
A 2-year randomized, open-label, crossover study43 randomized patients to receive either denosumab followed by alendronate or alendronate followed by denosumab over successive 12-month periods. The results suggested that postmenopausal women with osteoporosis were more adherent, compliant, and persistent with denosumab therapy (a subcutaneous injection every 6 months) than with alendronate therapy in the form of oral tablets, self-administered weekly (7.5% nonadherence vs 36.5% at the end of 2 years). After receiving both treatments, women reported greater satisfaction with denosumab, with 92.4% preferring it over oral alendronate. Bone mineral density remained stable when patients were switched from denosumab to alendronate, but improved further when they were switched from alendronate to denosumab.
HOW SAFE IS DENOSUMAB?
The most frequent adverse events with denosumab reported in the long-term extension of one phase 2 study were upper respiratory tract infections (13.5%), arthralgia (11.5%), and back pain (9.0%).30
Increased risk of infection, cancer, and dermatologic reactions has been a concern, as RANKL and RANK are expressed by a wide variety of cells, including T lymphocytes, B cells, and dendritic cells.44 However, there were no significant differences in the overall incidences of adverse events between patients who received denosumab and those who received placebo or alendronate in any of the phase 2, phase 3, or extension studies.
In the FREEDOM trial,24 there was no significant difference between the two groups in the overall incidence of infection (52.9% with denosumab vs 54.4% with placebo, P = .17), or serious infection (4.1% with denosumab vs 3.4% with placebo, P = .14), although the incidence of “serious” cellulitis requiring hospitalization was higher in the denosumab group (0.3% vs < 0.1%, P = .002). There were more serious infections involving the gastrointestinal system, urinary tract, and ear and cases of endocarditis in the denosumab group, but the number of events was small, and there was no relationship with the timing of administration or duration of exposure to denosumab.45 Eczema was more common in the denosumab than in the placebo group (3.0% vs 1.7%, P < .001), but the extension data from the first 3 years did not provide any evidence for an increased risk of cellulitis or eczema with denosumab.26
Although randomized controlled trials reported more cases of neoplasms in the denosumab than in the placebo groups, meta-analyses have failed to detect a statistically significant difference (risk ratio 1.11, 95% CI 0.91–1.36).46 The overall incidence of adverse and serious adverse events reported in the 8-year extension of FREEDOM were consistent with data reported in the previous extension studies.25
In the FREEDOM extension trial, four events in the long-term group (n = 2,343), and two in the crossover group (n = 2,207) were adjudicated as being consistent with osteonecrosis of the jaw.26 One mid-shaft fracture in the crossover group was adjudicated as an atypical femoral fracture. There were, however, no reports of osteonecrosis of the jaw or atypical femoral fracture in the long-term phase 2 trial after 8 years of follow-up.30 By September 2013, postmarketing safety surveillance data for denosumab (estimated exposure of 1.2 million patient-years) had recorded four cases of atypical femoral fracture. All four patients had previously been on bisphosphonates. There were also 32 reports of osteonecrosis of the jaw.47
Denosumab’s manufacturer aims to communicate the risks of treatment to health care professionals and patients. Information is available online at www.proliahcp.com/risk-evaluation-mitigation-strategy/.
WHAT ARE THE PRECAUTIONS?
Several precautions need to be taken when considering treatment with denosumab.
Antiresorptives can aggravate hypocalcemia by inhibiting bone turnover. Serum calcium should therefore be checked and preexisting hypocalcemia should be corrected before starting denosumab.48
Denosumab is contraindicated in women who are pregnant or are planning to become pregnant, as fetal loss and teratogenicity have been reported in animal experiments. (Denosumab is unlikely to be used in premenopausal women, as it is not approved for use in this group.)
There are no data on excretion of denosumab in human milk, so it should not be given to nursing mothers.
Renal impairment is not a contraindication, and no dose adjustment is necessary (even for patients on renal replacement therapy), as denosumab, being an antibody, is eliminated through the reticuloendothelial system.49,50 However, in practice, any antiresorptive agent should be used with caution in patients with severe renal impairment because of the possible presence of adynamic bone disease. Further reduction of bone turnover would be detrimental in such patients. Also, severe hypocalcemia has been reported in patients with a creatinine clearance rate less than 30 mL/min and in those receiving dialysis.51,52 Postmarketing surveillance data have reported eight cases of severe symptomatic hypocalcemia, of which seven were in patients with chronic kidney disease.47
The manufacturer suggests that patients receive a dental examination with appropriate preventive dentistry before starting denosumab to reduce the incidence of osteonecrosis of the jaw, despite the lack of evidence in support of this strategy. The American Dental Association recommends regular dental visits and maintenance of good oral hygiene for patients already established on antiresorptive therapy.53,54
SHOULD PATIENTS ON DENOSUMAB BE OFFERED A DRUG HOLIDAY?
A drug holiday (temporary discontinuation of the drug after a certain duration of treatment) has been proposed for patients receiving bisphosphonates because of the risk of atypical femoral fracture and osteonecrosis of the jaw (although small) consequent to long-term continuous suppression of bone turnover.55 The antifracture efficacy of bisphosphonates is likely to persist for an unknown length of time after discontinuation because of their long skeletal half-life, while the risks gradually diminish.
By contrast, denosumab targets RANKL in the extracellular fluid and does not become embedded within the bone tissue.56 Pharmacokinetic studies have shown that denosumab has a rapid offset of action, with a half-life of only 26 days and biological activity lasting only 6 months.57 The results of a phase 2 extension study suggest that bone mineral density starts to decline and bone turnover markers start to rise within 12 months of discontinuing denosumab.58
Although fracture risk did not increase in those who were randomized to stopping the treatment and bone mineral density increased further when treatment was restarted, a drug holiday cannot presently be recommended for patients receiving denosumab because of the lack of supportive data.
HOW COST-EFFECTIVE IS DENOSUMAB?
The wholesale acquisition cost is $825 per 60-mg prefilled syringe of denosumab, although this may vary depending on where the drug is obtained. This does not include physician-related service costs associated with administration of denosumab.
Cost-effectiveness analyses conducted in the United States, the United Kingdom, and Sweden have all concluded that denosumab would offer a cost-effective alternative to other osteoporosis medications for primary prevention and secondary prevention of fractures.59–61
The Swedish study also incorporated adherence in the cost-effectiveness model and showed that denosumab was a cost-effective alternative to oral bisphosphonates, particularly for patients who were not expected to adhere well to oral treatments.61
WHICH OSTEOPOROSIS PATIENTS ARE CANDIDATES FOR DENOSUMAB?
The FDA has approved denosumab for the treatment of postmenopausal women and men at high risk of fracture (defined as having a history of osteoporotic fracture or multiple risk factors for fracture), or in those who cannot tolerate other osteoporosis medications or for whom other medications have failed.
Denosumab is also approved for men at high risk of fracture receiving androgen deprivation therapy for nonmetastatic prostate cancer, and for women at high risk of fracture receiving adjuvant aromatase inhibitor therapy for breast cancer.
WHAT DO THE GUIDELINES RECOMMEND?
The National Osteoporosis Foundation guidelines recommend pharmacologic treatment for patients with hip or vertebral fractures (clinical or asymptomatic); T scores lower than –2.5 at the femoral neck, total hip, or lumbar spine; and those with a 10-year probability of hip fracture of more than 3% or of a major osteoporotic fracture more than 20% based on the US-adapted FRAX calculator.62 The American College of Endocrinology guidelines have proposed similar thresholds for pharmacologic treatment, and they recommend alendronate, risedronate, zoledronate, and denosumab as first-line agents.63
The 2010 Osteoporosis Canada guidelines recommend denosumab, alendronate, risedronate, and zoledronate as first-line therapies for preventing hip, nonvertebral, and vertebral fractures in postmenopausal women (grade A recommendation).64 The National Institute of Health and Clinical Excellence in England and Wales, on the other hand, recommends denosumab only for patients who are unable to take a bisphosphonate.65
PRACTICAL PRESCRIBING TIPS
The patient described at the beginning of this article has already sustained a vertebral compression fracture, and her DXA scan shows T scores in the osteoporotic range. She is therefore at increased risk of another fragility fracture (with a fivefold higher risk of another vertebral fracture). Pharmacologic therapy should be considered. In addition, she should be encouraged to adhere to lifestyle measures such as a healthy diet and regular weight-bearing exercise, her risk of falling should be assessed, and adequate calcium and vitamin D supplementation should be given.
Secondary causes of osteoporosis are present in about 30% of women and 55% of men who have vertebral fractures.66 A complete blood count, erythrocyte sedimentation rate, bone biochemistry, 25-hydroxyvitamin D, thyroid-stimulating hormone, and renal and liver function tests should be requested in all patients. Further tests should be considered depending on the clinical evaluation and results of initial investigations.
Because this patient cannot tolerate oral bisphosphonates, she could be offered the option of annual intravenous zoledronic acid infusions or 6-monthly subcutaneous denosumab injections. In clinical trials, gastrointestinal adverse effects were noted with intravenous bisphosphonates as well, but the adverse effects reported were no different than those with placebo. The potential advantages with denosumab include better bone mineral density gains, adherence and patient satisfaction compared with oral bisphosphonates, convenient twice-yearly administration, safety in patients with renal impairment, and absence of gastrointestinal effects.
Raloxifene, a selective estrogen receptor modulator, has estrogen-like action on the bone and antiestrogen actions on the breast and uterus. Unlike standard hormone replacement therapy, raloxifene can therefore increase bone mineral density without increasing the risk of breast and endometrial cancers. However, it has only been shown to reduce the risk of vertebral fracture, not hip fracture. Hence, it would be a more appropriate choice for younger postmenopausal women. Moreover, it may cause troublesome menopausal symptoms.
Teriparatide, the recombinant parathyroid hormone, is an anabolic agent. It is very expensive, and because of this, guidelines in several countries restrict its use to women with severe osteoporosis and multiple fractures who fail to respond to standard treatments. It cannot be used for longer than 2 years because of its association with osteosarcoma in rats.
If our patient prefers denosumab, therapy should be initiated after appropriate counseling (see precautions above). The dose is 60 mg, given subcutaneously, once every 6 months.
Monitoring
There is no consensus regarding the optimal frequency for monitoring patients on treatment, owing to the lack of prospective trial data. The National Osteoporosis Foundation recommends repeating the bone mineral density measurements about 2 years after starting therapy, and about every 2 years thereafter.62 Some studies suggest that changes in bone mineral density correlate with reduction in fracture risk.67,68 A change in bone mineral density is considered significant when it is greater than the range of error of the densitometer (also known as the least significant change).69 If the bone mineral density is stable or improving, therapy could be continued, but if it is declining and the decline is greater than the least significant change, a change in therapy should be considered if no secondary causes for bone loss are evident (but see What are the areas of uncertainty? below).
The National Osteoporosis Foundation also recommends measuring a bone turnover marker at baseline and then 3 to 6 months later, as its suppression predicts greater bone mineral density responses and fracture risk reduction.70 If there is a decrease of more than 30% in serum carboxy-terminal collagen crosslinks (CTX) or more than 50% in urinary N-telopeptide (NTX),71 the patient can be reassured that the next bone mineral density measurement will be stable or improved. In patients on oral bisphosphonates, measurement of bone turnover markers also provides evidence of compliance.
Clinical trials suggest that a numerical increase in bone mineral density can be expected in most patients on treatment, though this depends on the measurement site and the length of time between examinations. In one phase 3 trial of denosumab in postmenopausal women, only 5% of the participants had unchanged or diminished bone mineral density at the lumbar spine, and 8% at the hip, after 36 months of treatment.72 However, the CTX levels fell to below the lower limit of the reference interval as early as 1 month after commencing treatment in all denosumab-treated patients.68
Hence, bone turnover markers may be a more sensitive indicator of treatment effect than bone mineral density, but this would ultimately need to be evaluated against fracture rates in a real-world setting.
WHAT ARE THE AREAS OF UNCERTAINTY?
There are currently no guidelines for long-term management of patients on denosumab, and also no data to suggest whether patients should be switched to a weaker antiresorptive drug after a certain number of years in order to reduce the possible risk of atypical femoral fracture or osteonecrosis of the jaw.
No head-to-head trials have directly compared the antifracture efficacy of denosumab with that of other standard osteoporosis therapies. The antifracture efficacy and safety of combination therapies involving denosumab are also uncertain. For adherent patients who have a suboptimal response, there is no evidence to guide the further course of action. The International Osteoporosis Foundation guidelines suggest replacing a stronger antiresorptive with an anabolic agent, but acknowledge that this is only based on expert opinion.71
The very-long-term effects (beyond 8 years) of continuous denosumab administration on increasing the risk of atypical femoral fracture, osteonecrosis of the jaw, malignancy, or infection or the duration after which risks would start to outweigh benefits is not known. However, postmarketing safety data continue to be collected through the voluntary Post-marketing Active Safety Surveillance Program (for prespecified adverse events) in addition to the FDA’s MedWatch program.
CASE PROGRESSION
The patient described in the vignette is presented with two options—zoledronate and denosumab. She chooses denosumab. Her renal function and serum calcium are checked and are found to be satisfactory. She undergoes a dental examination, which is also satisfactory. She is counseled about the possible increased risk of infection, and then she is started on 60 mg of denosumab subcutaneously, once every 6 months.
When reviewed after 2 years, she reports no further fractures. Her bone mineral density remains stable compared with the values obtained before starting treatment. She reports no adverse effects and is happy to continue with denosumab.
- Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int 2006; 17:1726–1733.
- Gullberg B, Johnell O, Kanis JA. World-wide projections for hip fracture. Osteoporos Int 1997; 7:407–413.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res 2007; 22:465–475.
- Melton LJ 3rd, Chrischilles EA, Cooper C, Lane AW, Riggs BL. Perspective. How many women have osteoporosis? J Bone Miner Res 1992; 7:1005–1010.
- Abrahamsen B, van Staa T, Ariely R, Olson M, Cooper C. Excess mortality following hip fracture: a systematic epidemiological review. Osteoporos Int 2009; 20:1633–1650.
- Jalava T, Sarna S, Pylkkänen L, et al. Association between vertebral fracture and increased mortality in osteoporotic patients. J Bone Miner Res 2003; 18:1254–1260.
- Ismail AA, O’Neill TW, Cooper C, et al. Mortality associated with vertebral deformity in men and women: results from the European Prospective Osteoporosis Study (EPOS). Osteoporos Int 1998; 8:291–297.
- Ensrud KE, Thompson DE, Cauley JA, et al. Prevalent vertebral deformities predict mortality and hospitalization in older women with low bone mass. Fracture Intervention Trial Research Group. J Am Geriatr Soc 2000; 48:241–249.
- Kado DM, Browner WS, Palermo L, Nevitt MC, Genant HK, Cummings SR. Vertebral fractures and mortality in older women: a prospective study. Study of Osteoporotic Fractures Research Group. Arch Intern Med 1999; 159:1215–1220.
- Lindsay R, Silverman SL, Cooper C, et al. Risk of new vertebral fracture in the year following a fracture. JAMA 2001; 285:320–323.
- Ross PD, Davis JW, Epstein RS, Wasnich RD. Pre-existing fractures and bone mass predict vertebral fracture incidence in women. Ann Intern Med 1991; 114:919–923.
- Piscitelli P, Iolascon G, Argentiero A, et al. Incidence and costs of hip fractures vs strokes and acute myocardial infarction in Italy: comparative analysis based on national hospitalization records. Clin Interv Aging 2012; 7:575–583.
- Johnell O, Kanis JA, Jonsson B, Oden A, Johansson H, De Laet C. The burden of hospitalised fractures in Sweden. Osteoporos Int 2005; 16:222–228.
- Confavreux CB, Canoui-Poitrine F, Schott AM, Ambrosi V, Tainturier V, Chapurlat RD. Persistence at 1 year of oral antiosteoporotic drugs: a prospective study in a comprehensive health insurance database. Eur J Endocrinol 2012; 166:735–741.
- Biswas PN, Wilton LV, Shakir SA. Pharmacovigilance study of alendronate in England. Osteoporos Int 2003; 14:507–514.
- Landfeldt E, Ström O, Robbins S, Borgström F. Adherence to treatment of primary osteoporosis and its association to fractures—the Swedish Adherence Register Analysis (SARA). Osteoporos Int 2012; 23:433–443.
- Sampalis JS, Adachi JD, Rampakakis E, Vaillancourt J, Karellis A, Kindundu C. Long-term impact of adherence to oral bisphosphonates on osteoporotic fracture incidence. J Bone Miner Res 2012; 27:202–210.
- Schwarz EM, Ritchlin CT. Clinical development of anti-RANKL therapy. Arthritis Res Ther 2007; 9(suppl 1):S7.
- Hanley DA, Adachi JD, Bell A, Brown V. Denosumab: mechanism of action and clinical outcomes. Int J Clin Pract 2012; 66:1139–1146.
- Bekker PJ, Holloway D, Nakanishi A, Arrighi M, Leese PT, Dunstan CR. The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res 2001; 16:348–360.
- Rizzoli R, Body JJ, Brandi ML, et al; International Osteoporosis Foundation Committee of Scientific Advisors Working Group on Cancer-Induced Bone Disease. Cancer-associated bone disease. Osteoporos Int 2013; 24:2929–2953.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Hu MI, Glezerman IG, Leboulleux S, et al. Denosumab for treatment of hypercalcemia of malignancy. J Clin Endocrinol Metab 2014; Jun 10 [Epub ahead of print].
- Cummings SR, San Martin J, McClung MR, et al; FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Papapoulos S, Lippuner K, Roux C, et al. Eight years of denosumab treatment in postmenopausal women with osteoporosis: results from the first five years of the FREEDOM extension [abstract]. Presented at the 2013 annual meeting of the American Society for Bone and Mineral Research, Baltimore, MD, October 4–7, 2013.
- Bone HG, Chapurlat R, Brandi ML, et al. The effect of three or six years of denosumab exposure in women with postmenopausal osteoporosis: results from the FREEDOM extension. J Clin Endocrinol Metab 2013; 98:4483–4492.
- McClung MR, Boonen S, Törring O, et al. Effect of denosumab treatment on the risk of fractures in subgroups of women with postmenopausal osteoporosis. J Bone Miner Res 2012; 27:211–218.
- Boonen S, Adachi JD, Man Z, et al. Treatment with denosumab reduces the incidence of new vertebral and hip fractures in postmenopausal women at high risk. J Clin Endocrinol Metab 2011; 96:1727–1736.
- McCloskey EV, Johansson H, Oden A, et al. Denosumab reduces the risk of osteoporotic fractures in postmenopausal women, particularly in those with moderate to high fracture risk as assessed with FRAX. J Bone Miner Res 2012; 27:1480–1486.
- McClung MR, Lewiecki EM, Geller ML, et al. Effect of denosumab on bone mineral density and biochemical markers of bone turnover: 8-year results of a phase 2 clinical trial. Osteoporos Int 2013; 24:227–235.
- McClung MR, Lewiecki EM, Cohen SB, et al; AMG 162 Bone Loss Study Group. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med 2006; 354:821–831.
- Bone HG, Bolognese MA, Yuen CK, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab 2008; 93:2149–2157.
- Genant HK, Engelke K, Hanley DA, et al. Denosumab improves density and strength parameters as measured by QCT of the radius in postmenopausal women with low bone mineral density. Bone 2010; 47:131–139.
- Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab 2012; 97:3161–3169.
- Tsai JN, Uihlein AV, Lee H, et al. Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: the DATA study randomised trial. Lancet 2013; 382:50–56.
- Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med 2009; 361:745–755.
- Ellis GK, Bone HG, Chlebowski R, et al. Randomized trial of denosumab in patients receiving adjuvant aromatase inhibitors for nonmetastatic breast cancer. J Clin Oncol 2008; 26:4875–4882.
- Brown JP, Prince RL, Deal C, et al. Comparison of the effect of denosumab and alendronate on BMD and biochemical markers of bone turnover in postmenopausal women with low bone mass: a randomized, blinded, phase 3 trial. J Bone Miner Res 2009; 24:153–161.
- Kendler DL, Roux C, Benhamou CL, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res 2010; 25:72–81.
- Seeman E, Delmas PD, Hanley DA, et al. Microarchitectural deterioration of cortical and trabecular bone: differing effects of denosumab and alendronate. J Bone Miner Res 2010; 25:1886–1894.
- Hopkins RB, Goeree R, Pullenayegum E, et al. The relative efficacy of nine osteoporosis medications for reducing the rate of fractures in post-menopausal women. BMC Musculoskelet Disord 2011; 12:209.
- National Institute for Health and Care Excellence (NICE). NICE technology appraisal guidance: TA161. Alendronate, etidronate, risedronate, raloxifene, strontium ranelate and teriparatide for the secondary prevention of osteoporotic fragility fractures in postmenopausal women (amended). http://publications.nice.org.uk/alendronate-etidronate-risedronate-raloxifene-strontium-ranelate-and-teriparatide-for-ta161. Accessed January 9, 2015.
- Freemantle N, Satram-Hoang S, Tang ET, et al; DAPS Investigators. Final results of the DAPS (Denosumab Adherence Preference Satisfaction) study: a 24-month, randomized, crossover comparison with alendronate in postmenopausal women. Osteoporos Int 2012; 23:317–326.
- Lewiecki EM. Safety and tolerability of denosumab for the treatment of postmenopausal osteoporosis. Drug Healthc Patient Saf 2011; 3:79–91.
- Watts NB, Roux C, Modlin JF, et al. Infections in postmenopausal women with osteoporosis treated with denosumab or placebo: coincidence or causal association? Osteoporos Int 2012; 23:327–337.
- von Keyserlingk C, Hopkins R, Anastasilakis A, et al. Clinical efficacy and safety of denosumab in postmenopausal women with low bone mineral density and osteoporosis: a meta-analysis. Semin Arthritis Rheum 2011; 41:178–186.
- Geller M, Wagman RB, Ho PR, et al. Early findings from Prolia postmarketing safety surveillance for atypical femoral fracture, osteonecrosis of the jaw, severe symptomatic hypocalcemia, and anaphylaxis (abstract). Osteoporos Int 2014; 25(suppl 2). OC40; www.wco-iof-esceo.org/sites/ecceo14/docs/wco14-abstractbook.pdf. Accessed January 9, 2015.
- McCormick BB, Davis J, Burns KD. Severe hypocalcemia following denosumab injection in a hemodialysis patient. Am J Kidney Dis 2012; 60:626–628.
- Jamal SA, Ljunggren O, Stehman-Breen C, et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J Bone Miner Res 2011; 26:1829–1835.
- Block GA, Bone HG, Fang L, Lee E, Padhi D. A single-dose study of denosumab in patients with various degrees of renal impairment. J Bone Miner Res 2012; 27:1471–1479.
- Ungprasert P, Cheungpasitporn W, Srivali N, Kittanamongkolchai W, Bischof EF. Life-threatening hypocalcemia associated with denosumab in a patient with moderate renal insufficiency. Am J Emerg Med 2013; 31:756.e1–e2.
- McCormick BB, Davis J, Burns KD. Severe hypocalcemia following denosumab injection in a hemodialysis patient. Am J Kidney Dis 2012; 60:626–628.
- Rachner TD, Platzbecker U, Felsenberg D, Hofbauer LC. Osteonecrosis of the jaw after osteoporosis therapy with denosumab following long-term bisphosphonate therapy. Mayo Clin Proc 2013; 88:418–419.
- Epstein MS, Ephros HD, Epstein JB. Review of current literature and implications of RANKL inhibitors for oral health care providers. Oral Surg Oral Med Oral Pathol Oral Radiol 2013; 116:e437–e442.
- McClung M, Harris ST, Miller PD, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med 2013; 126:13–20.
- Baron R, Ferrari S, Russell RG. Denosumab and bisphosphonates: different mechanisms of action and effects. Bone 2011; 48:677–692.
- Bekker PJ, Holloway DL, Rasmussen AS, et al. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res 2004; 19:1059–1066.
- Miller PD, Bolognese MA, Lewiecki EM, et al; Amg Bone Loss Study Group. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone 2008; 43:222–229.
- Parthan A, Kruse M, Yurgin N, Huang J, Viswanathan HN, Taylor D. Cost effectiveness of denosumab versus oral bisphosphonates for postmenopausal osteoporosis in the US. Appl Health Econ Health Policy 2013; 11:485–497.
- Scotland G, Waugh N, Royle P, McNamee P, Henderson R, Hollick R. Denosumab for the prevention of osteoporotic fractures in post-menopausal women: a NICE single technology appraisal. Pharmacoeconomics 2011; 29:951–961.
- Jönsson B, Ström O, Eisman JA, et al. Cost-effectiveness of denosumab for the treatment of postmenopausal osteoporosis. Osteoporos Int 2011; 22:967–982.
- Clinician’s guide to prevention and treatment of osteoporosis. Washington DC: National Osteoporosis Foundation, 2013.
- Watts NB, Bilezikian JP, Camacho PM, et al; AACE Osteoporosis Task Force. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the diagnosis and treatment of postmenopausal osteoporosis. Endocr Pract 2010;16(suppl 3):1–37.
- Papaioannou A, Morin S, Cheung AM, et al; Scientific Advisory Council of Osteoporosis Canada. 2010 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada: summary. CMAJ 2010; 182:1864–1673.
- National Institute for Health and Care Excellence (NICE). NICE technology appraisal: TA204. Denosumab for the prevention of osteoporotic fractures in postmenopausal women. http://guidance.nice.org.uk/TA204. Accessed January 9, 2015.
- Premaor MO, Compston JE. Testing for secondary causes of osteoporosis. BMJ 2010; 341:c6959.
- Hochberg MC, Ross PD, Black D, et al. Larger increases in bone mineral density during alendronate therapy are associated with a lower risk of new vertebral fractures in women with postmenopausal osteoporosis. Fracture Intervention Trial Research Group. Arthritis Rheum 1999; 42:1246–1254.
- Eastell R, Vrijens B, Cahall DL, Ringe JD, Garnero P, Watts NB. Bone turnover markers and bone mineral density response with risedronate therapy: relationship with fracture risk and patient adherence. J Bone Miner Res 2011; 26:1662–1669.
- Diez-Perez A, Adachi JD, Agnusdei D, et al; IOF CSA Inadequate Responders Working Group. Treatment failure in osteoporosis. Osteoporos Int 2012; 23:2769–2774.
- Eastell R, Barton I, Hannon RA, Chines A, Garnero P, Delmas PD. Relationship of early changes in bone resorption to the reduction in fracture risk with risedronate. J Bone Miner Res 2003; 18:1051–1056.
- Rosen HN, Moses AC, Garber J, Ross DS, Lee SL, Greenspan SL. Utility of biochemical markers of bone turnover in the follow-up of patients treated with bisphosphonates. Calcif Tissue Int 1998; 63:363–368.
- Bolognese MA, Teglbjærg CS, Zanchetta JR, et al. Denosumab significantly increases DXA BMD at both trabecular and cortical sites: results from the FREEDOM study. J Clin Densitom 2013; 16:147–153.
- Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int 2006; 17:1726–1733.
- Gullberg B, Johnell O, Kanis JA. World-wide projections for hip fracture. Osteoporos Int 1997; 7:407–413.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res 2007; 22:465–475.
- Melton LJ 3rd, Chrischilles EA, Cooper C, Lane AW, Riggs BL. Perspective. How many women have osteoporosis? J Bone Miner Res 1992; 7:1005–1010.
- Abrahamsen B, van Staa T, Ariely R, Olson M, Cooper C. Excess mortality following hip fracture: a systematic epidemiological review. Osteoporos Int 2009; 20:1633–1650.
- Jalava T, Sarna S, Pylkkänen L, et al. Association between vertebral fracture and increased mortality in osteoporotic patients. J Bone Miner Res 2003; 18:1254–1260.
- Ismail AA, O’Neill TW, Cooper C, et al. Mortality associated with vertebral deformity in men and women: results from the European Prospective Osteoporosis Study (EPOS). Osteoporos Int 1998; 8:291–297.
- Ensrud KE, Thompson DE, Cauley JA, et al. Prevalent vertebral deformities predict mortality and hospitalization in older women with low bone mass. Fracture Intervention Trial Research Group. J Am Geriatr Soc 2000; 48:241–249.
- Kado DM, Browner WS, Palermo L, Nevitt MC, Genant HK, Cummings SR. Vertebral fractures and mortality in older women: a prospective study. Study of Osteoporotic Fractures Research Group. Arch Intern Med 1999; 159:1215–1220.
- Lindsay R, Silverman SL, Cooper C, et al. Risk of new vertebral fracture in the year following a fracture. JAMA 2001; 285:320–323.
- Ross PD, Davis JW, Epstein RS, Wasnich RD. Pre-existing fractures and bone mass predict vertebral fracture incidence in women. Ann Intern Med 1991; 114:919–923.
- Piscitelli P, Iolascon G, Argentiero A, et al. Incidence and costs of hip fractures vs strokes and acute myocardial infarction in Italy: comparative analysis based on national hospitalization records. Clin Interv Aging 2012; 7:575–583.
- Johnell O, Kanis JA, Jonsson B, Oden A, Johansson H, De Laet C. The burden of hospitalised fractures in Sweden. Osteoporos Int 2005; 16:222–228.
- Confavreux CB, Canoui-Poitrine F, Schott AM, Ambrosi V, Tainturier V, Chapurlat RD. Persistence at 1 year of oral antiosteoporotic drugs: a prospective study in a comprehensive health insurance database. Eur J Endocrinol 2012; 166:735–741.
- Biswas PN, Wilton LV, Shakir SA. Pharmacovigilance study of alendronate in England. Osteoporos Int 2003; 14:507–514.
- Landfeldt E, Ström O, Robbins S, Borgström F. Adherence to treatment of primary osteoporosis and its association to fractures—the Swedish Adherence Register Analysis (SARA). Osteoporos Int 2012; 23:433–443.
- Sampalis JS, Adachi JD, Rampakakis E, Vaillancourt J, Karellis A, Kindundu C. Long-term impact of adherence to oral bisphosphonates on osteoporotic fracture incidence. J Bone Miner Res 2012; 27:202–210.
- Schwarz EM, Ritchlin CT. Clinical development of anti-RANKL therapy. Arthritis Res Ther 2007; 9(suppl 1):S7.
- Hanley DA, Adachi JD, Bell A, Brown V. Denosumab: mechanism of action and clinical outcomes. Int J Clin Pract 2012; 66:1139–1146.
- Bekker PJ, Holloway D, Nakanishi A, Arrighi M, Leese PT, Dunstan CR. The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res 2001; 16:348–360.
- Rizzoli R, Body JJ, Brandi ML, et al; International Osteoporosis Foundation Committee of Scientific Advisors Working Group on Cancer-Induced Bone Disease. Cancer-associated bone disease. Osteoporos Int 2013; 24:2929–2953.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Hu MI, Glezerman IG, Leboulleux S, et al. Denosumab for treatment of hypercalcemia of malignancy. J Clin Endocrinol Metab 2014; Jun 10 [Epub ahead of print].
- Cummings SR, San Martin J, McClung MR, et al; FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361:756–765.
- Papapoulos S, Lippuner K, Roux C, et al. Eight years of denosumab treatment in postmenopausal women with osteoporosis: results from the first five years of the FREEDOM extension [abstract]. Presented at the 2013 annual meeting of the American Society for Bone and Mineral Research, Baltimore, MD, October 4–7, 2013.
- Bone HG, Chapurlat R, Brandi ML, et al. The effect of three or six years of denosumab exposure in women with postmenopausal osteoporosis: results from the FREEDOM extension. J Clin Endocrinol Metab 2013; 98:4483–4492.
- McClung MR, Boonen S, Törring O, et al. Effect of denosumab treatment on the risk of fractures in subgroups of women with postmenopausal osteoporosis. J Bone Miner Res 2012; 27:211–218.
- Boonen S, Adachi JD, Man Z, et al. Treatment with denosumab reduces the incidence of new vertebral and hip fractures in postmenopausal women at high risk. J Clin Endocrinol Metab 2011; 96:1727–1736.
- McCloskey EV, Johansson H, Oden A, et al. Denosumab reduces the risk of osteoporotic fractures in postmenopausal women, particularly in those with moderate to high fracture risk as assessed with FRAX. J Bone Miner Res 2012; 27:1480–1486.
- McClung MR, Lewiecki EM, Geller ML, et al. Effect of denosumab on bone mineral density and biochemical markers of bone turnover: 8-year results of a phase 2 clinical trial. Osteoporos Int 2013; 24:227–235.
- McClung MR, Lewiecki EM, Cohen SB, et al; AMG 162 Bone Loss Study Group. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med 2006; 354:821–831.
- Bone HG, Bolognese MA, Yuen CK, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab 2008; 93:2149–2157.
- Genant HK, Engelke K, Hanley DA, et al. Denosumab improves density and strength parameters as measured by QCT of the radius in postmenopausal women with low bone mineral density. Bone 2010; 47:131–139.
- Orwoll E, Teglbjærg CS, Langdahl BL, et al. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab 2012; 97:3161–3169.
- Tsai JN, Uihlein AV, Lee H, et al. Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: the DATA study randomised trial. Lancet 2013; 382:50–56.
- Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med 2009; 361:745–755.
- Ellis GK, Bone HG, Chlebowski R, et al. Randomized trial of denosumab in patients receiving adjuvant aromatase inhibitors for nonmetastatic breast cancer. J Clin Oncol 2008; 26:4875–4882.
- Brown JP, Prince RL, Deal C, et al. Comparison of the effect of denosumab and alendronate on BMD and biochemical markers of bone turnover in postmenopausal women with low bone mass: a randomized, blinded, phase 3 trial. J Bone Miner Res 2009; 24:153–161.
- Kendler DL, Roux C, Benhamou CL, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res 2010; 25:72–81.
- Seeman E, Delmas PD, Hanley DA, et al. Microarchitectural deterioration of cortical and trabecular bone: differing effects of denosumab and alendronate. J Bone Miner Res 2010; 25:1886–1894.
- Hopkins RB, Goeree R, Pullenayegum E, et al. The relative efficacy of nine osteoporosis medications for reducing the rate of fractures in post-menopausal women. BMC Musculoskelet Disord 2011; 12:209.
- National Institute for Health and Care Excellence (NICE). NICE technology appraisal guidance: TA161. Alendronate, etidronate, risedronate, raloxifene, strontium ranelate and teriparatide for the secondary prevention of osteoporotic fragility fractures in postmenopausal women (amended). http://publications.nice.org.uk/alendronate-etidronate-risedronate-raloxifene-strontium-ranelate-and-teriparatide-for-ta161. Accessed January 9, 2015.
- Freemantle N, Satram-Hoang S, Tang ET, et al; DAPS Investigators. Final results of the DAPS (Denosumab Adherence Preference Satisfaction) study: a 24-month, randomized, crossover comparison with alendronate in postmenopausal women. Osteoporos Int 2012; 23:317–326.
- Lewiecki EM. Safety and tolerability of denosumab for the treatment of postmenopausal osteoporosis. Drug Healthc Patient Saf 2011; 3:79–91.
- Watts NB, Roux C, Modlin JF, et al. Infections in postmenopausal women with osteoporosis treated with denosumab or placebo: coincidence or causal association? Osteoporos Int 2012; 23:327–337.
- von Keyserlingk C, Hopkins R, Anastasilakis A, et al. Clinical efficacy and safety of denosumab in postmenopausal women with low bone mineral density and osteoporosis: a meta-analysis. Semin Arthritis Rheum 2011; 41:178–186.
- Geller M, Wagman RB, Ho PR, et al. Early findings from Prolia postmarketing safety surveillance for atypical femoral fracture, osteonecrosis of the jaw, severe symptomatic hypocalcemia, and anaphylaxis (abstract). Osteoporos Int 2014; 25(suppl 2). OC40; www.wco-iof-esceo.org/sites/ecceo14/docs/wco14-abstractbook.pdf. Accessed January 9, 2015.
- McCormick BB, Davis J, Burns KD. Severe hypocalcemia following denosumab injection in a hemodialysis patient. Am J Kidney Dis 2012; 60:626–628.
- Jamal SA, Ljunggren O, Stehman-Breen C, et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J Bone Miner Res 2011; 26:1829–1835.
- Block GA, Bone HG, Fang L, Lee E, Padhi D. A single-dose study of denosumab in patients with various degrees of renal impairment. J Bone Miner Res 2012; 27:1471–1479.
- Ungprasert P, Cheungpasitporn W, Srivali N, Kittanamongkolchai W, Bischof EF. Life-threatening hypocalcemia associated with denosumab in a patient with moderate renal insufficiency. Am J Emerg Med 2013; 31:756.e1–e2.
- McCormick BB, Davis J, Burns KD. Severe hypocalcemia following denosumab injection in a hemodialysis patient. Am J Kidney Dis 2012; 60:626–628.
- Rachner TD, Platzbecker U, Felsenberg D, Hofbauer LC. Osteonecrosis of the jaw after osteoporosis therapy with denosumab following long-term bisphosphonate therapy. Mayo Clin Proc 2013; 88:418–419.
- Epstein MS, Ephros HD, Epstein JB. Review of current literature and implications of RANKL inhibitors for oral health care providers. Oral Surg Oral Med Oral Pathol Oral Radiol 2013; 116:e437–e442.
- McClung M, Harris ST, Miller PD, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med 2013; 126:13–20.
- Baron R, Ferrari S, Russell RG. Denosumab and bisphosphonates: different mechanisms of action and effects. Bone 2011; 48:677–692.
- Bekker PJ, Holloway DL, Rasmussen AS, et al. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res 2004; 19:1059–1066.
- Miller PD, Bolognese MA, Lewiecki EM, et al; Amg Bone Loss Study Group. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone 2008; 43:222–229.
- Parthan A, Kruse M, Yurgin N, Huang J, Viswanathan HN, Taylor D. Cost effectiveness of denosumab versus oral bisphosphonates for postmenopausal osteoporosis in the US. Appl Health Econ Health Policy 2013; 11:485–497.
- Scotland G, Waugh N, Royle P, McNamee P, Henderson R, Hollick R. Denosumab for the prevention of osteoporotic fractures in post-menopausal women: a NICE single technology appraisal. Pharmacoeconomics 2011; 29:951–961.
- Jönsson B, Ström O, Eisman JA, et al. Cost-effectiveness of denosumab for the treatment of postmenopausal osteoporosis. Osteoporos Int 2011; 22:967–982.
- Clinician’s guide to prevention and treatment of osteoporosis. Washington DC: National Osteoporosis Foundation, 2013.
- Watts NB, Bilezikian JP, Camacho PM, et al; AACE Osteoporosis Task Force. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the diagnosis and treatment of postmenopausal osteoporosis. Endocr Pract 2010;16(suppl 3):1–37.
- Papaioannou A, Morin S, Cheung AM, et al; Scientific Advisory Council of Osteoporosis Canada. 2010 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada: summary. CMAJ 2010; 182:1864–1673.
- National Institute for Health and Care Excellence (NICE). NICE technology appraisal: TA204. Denosumab for the prevention of osteoporotic fractures in postmenopausal women. http://guidance.nice.org.uk/TA204. Accessed January 9, 2015.
- Premaor MO, Compston JE. Testing for secondary causes of osteoporosis. BMJ 2010; 341:c6959.
- Hochberg MC, Ross PD, Black D, et al. Larger increases in bone mineral density during alendronate therapy are associated with a lower risk of new vertebral fractures in women with postmenopausal osteoporosis. Fracture Intervention Trial Research Group. Arthritis Rheum 1999; 42:1246–1254.
- Eastell R, Vrijens B, Cahall DL, Ringe JD, Garnero P, Watts NB. Bone turnover markers and bone mineral density response with risedronate therapy: relationship with fracture risk and patient adherence. J Bone Miner Res 2011; 26:1662–1669.
- Diez-Perez A, Adachi JD, Agnusdei D, et al; IOF CSA Inadequate Responders Working Group. Treatment failure in osteoporosis. Osteoporos Int 2012; 23:2769–2774.
- Eastell R, Barton I, Hannon RA, Chines A, Garnero P, Delmas PD. Relationship of early changes in bone resorption to the reduction in fracture risk with risedronate. J Bone Miner Res 2003; 18:1051–1056.
- Rosen HN, Moses AC, Garber J, Ross DS, Lee SL, Greenspan SL. Utility of biochemical markers of bone turnover in the follow-up of patients treated with bisphosphonates. Calcif Tissue Int 1998; 63:363–368.
- Bolognese MA, Teglbjærg CS, Zanchetta JR, et al. Denosumab significantly increases DXA BMD at both trabecular and cortical sites: results from the FREEDOM study. J Clin Densitom 2013; 16:147–153.
KEY POINTS
- Denosumab is a fully human monoclonal antibody that targets the receptor activator of nuclear factor kappa b ligand, a key mediator of osteoclastic bone resorption.
- Commpared with placebo, denosumab has been shown to significantly reduce the risk of vertebral, nonvertebral, and hip fractures in postmenopausal women with osteoporosis.
- Patients taking denosumab are more adherent, compliant, and persistent with therapy than those taking alendronate. Denosumab is also superior to alendronate in improving bone mineral density at all skeletal sites.
- Denosumab is safe, with safety data now available for up to 8 years of exposure.
Hepatitis C virus: Here comes all-oral treatment
In late 2013, the US Food and Drug Administration (FDA) approved sofosbuvir and simeprevir, the newest direct-acting antiviral agents for treating chronic hepatitis C virus (HCV) infection. Multiple clinical trials have demonstrated dramatically improved treatment outcomes with these agents, opening the door to all-oral regimens or interferon-free regimens as the future standard of care for HCV.
In this article, we discuss the results of the trials that established the efficacy and safety of sofosbuvir and simeprevir and led to their FDA approval. We also summarize the importance of these agents and evaluate other direct-acting antivirals currently in the pipeline for HCV treatment.
HCV IS A RISING PROBLEM
Chronic HCV infection is a major clinical and public health problem, with the estimated number of people infected exceeding 170 million worldwide, including 3.2 million in the United States.1 It is a leading cause of cirrhosis, and its complications include hepatocellular carcinoma and liver failure. Cirrhosis due to HCV remains the leading indication for liver transplantation in the United States, accounting for nearly 40% of liver transplants in adults.2

The clinical impact of HCV will only continue to escalate, and in parallel, so will the cost to society. Models suggest that HCV-related deaths will double between 2010 and 2019, and considering only direct medical costs, the projected financial burden of treating HCV-related disease during this interval is estimated at between $6.5 and $13.6 billion.3
AN RNA VIRUS WITH SIX GENOTYPES

HCV, first identified in 1989, is an enveloped, single-stranded RNA flavivirus of the Hepacivirus genus measuring 50 to 60 nm in diameter.4 There are six viral genotypes, with genotype 1 being the most common in the United States and traditionally the most difficult to treat.
Once inside the host cell, the virus releases its RNA strand, which is translated into a single polyprotein of about 3,000 amino acids. This large molecule is then cleaved by proteases into several domains: three structural proteins (C, E1, and E2), a small protein called p7, and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Figure 1).5 These nonstructural proteins enable the virus to replicate.
GOAL OF TREATING HCV: A SUSTAINED VIROLOGIC RESPONSE
The aim of HCV treatment is to achieve a sustained virologic response, defined as having no detectable viral RNA after completion of antiviral therapy. This is associated with substantially better clinical outcomes, lower rates of liver-related morbidity and all-cause mortality, and stabilization of or even improvement in liver histology.6,7 This end point has traditionally been assessed at 6 months after the end of therapy, but recent data suggest the rates at 12 weeks are essentially equivalent.

Table 1 summarizes the patterns of virologic response in treating HCV infection.
Interferon plus ribavirin: The standard of care for many years
HCV treatment has evolved over the past 20 years. Before 2011, the standard of care was a combination of interferon alfa-polyethylene glycol (peg-interferon), given as a weekly injection, and oral ribavirin. Neither drug has specific antiviral activity, and when they are used together they result in a sustained virologic response in fewer than 50% of patients with HCV genotype 1 and, at best, in 70% to 80% of patients with other genotypes.8
Nearly all patients receiving interferon experience side effects, which can be serious. Fatigue and flu-like symptoms are common, and the drug can also cause psychiatric symptoms (including depression or psychosis), weight loss, seizures, peripheral neuropathy, and bone marrow suppression. Ribavirin causes hemolysis and skin complications and is teratogenic.9
An important bit of information to know when using interferon is the patient’s IL28B genotype. This refers to a single-nucleotide polymorphism (C or T) on chromosome 19q13 (rs12979860) upstream of the IL28B gene encoding for interferon lambda-3. It is strongly associated with responsiveness to interferon: patients with the IL28B CC genotype have a much better chance of a sustained virologic response with interferon than do patients with CT or TT.
Boceprevir and telaprevir: First-generation protease inhibitors
In May 2011, the FDA approved the NS3/4A protease inhibitors boceprevir and telaprevir for treating HCV genotype 1, marking the beginning of the era of direct-acting antiviral agents.10 When these drugs are used in combination with peg-interferon alfa and ribavirin, up to 75% of patients with HCV genotype 1 who have had no previous treatment achieve a sustained virologic response.
But despite greatly improving the response rate, these first-generation protease inhibitors have substantial limitations. Twenty-five percent of patients with HCV genotype 1 who have received no previous treatment and 71% of patients who did not respond to previous treatment will not achieve a sustained virologic response with these agents.11 Further, they are effective only against HCV genotype 1, being highly specific for the amino acid target sequence of the NS3 region.
Also, they must be used in combination with interferon alfa and ribavirin because the virus needs to mutate only a little—a few amino-acid substitutions—to gain resistance to them.12 Therefore, patients are still exposed to interferon and ribavirin, with their toxicity. In addition, dysgeusia is seen with boceprevir, rash with telaprevir, and anemia with both.13,14
Finally, serious drug-drug interactions prompted the FDA to impose warnings for the use of these agents with other medications that interact with CYP3A4, the principal enzyme responsible for their metabolism. Thus, these significant adverse effects dampen the enthusiasm of patients contemplating a long course of treatment with these agents.
The need to improve the rate of sustained virologic response, shorten the duration of treatment, avoid serious side effects, improve efficacy in treating patients infected with genotypes other than 1, and, importantly, eliminate the need for interferon alfa and its serious adverse effects have driven the development of new direct-acting antiviral agents, including the two newly FDA-approved drugs, sofosbuvir and simeprevir.
SOFOSBUVIR: A POLYMERASE INHIBITOR
Sofosbuvir is a uridine nucleotide analogue that selectively inhibits the HCV NS5B RNA-dependent RNA polymerase (Figure 1). It targets the highly conserved nucleotide-binding pocket of this enzyme and functions as a chain terminator.15 While the protease inhibitors are genotype-dependent, inhibition of the highly conserved viral polymerase has an impact that spans genotypes.
Early clinical trials of sofosbuvir

Sofosbuvir has been tested in combination with interferon alfa and ribavirin, as well as in interferon-free regimens (Table 2).16–20
Rodriguez-Torres et al,15
- 56% with sofosbuvir 100 mg, peg-interferon, and ribavirin
- 83% with sofosbuvir 200 mg, peg-interferon, and ribavirin
- 80% with sofosbuvir 400 mg, peg-interferon, and ribavirin
- 43% with peg-interferon and ribavirin alone.
The ATOMIC trial16 tested the efficacy and safety of sofosbuvir in combination with peg-interferon and ribavirin in patients with HCV genotype 1, 4, or 6, without cirrhosis, who had not received any previous treatment. Patients with HCV genotype 1 were randomized to three treatments:
- Sofosbuvir 400 mg orally once daily plus peg-interferon and ribavirin for 12 weeks
- The same regimen, but for 24 weeks
- Sofosbuvir plus peg-interferon and ribavirin for 12 weeks, followed by 12 weeks of either sofosbuvir monotherapy or sofosbuvir plus ribavirin.
The rates of sustained virologic response were very high and were not significantly different among the three groups: 89%, 89%, and 87%, respectively. Patients who were able to complete a full course of therapy achieved even higher rates of sustained virologic response, ranging from 96% to 98%. The likelihood of response was not adversely affected by the usual markers of a poorer prognosis, such as a high viral load (≥ 800,000 IU/mL) or a non-CC IL28B genotype. Although patients with cirrhosis (another predictor of no response) were excluded from this study, the presence of bridging fibrosis did not seem to affect the rate of sustained virologic response. The results in patients with genotypes other than 1 were very encouraging, but the small number of patients enrolled precluded drawing firm conclusions in this group.
Important implications of the ATOMIC trial include the following:
There is no benefit in prolonging treatment with sofosbuvir beyond 12 weeks, since adverse events increased without any improvement in the rate of sustained virologic response.
There is a very low likelihood of developing viral resistance or mutation when using sofosbuvir.
There is no role for response-guided therapy, a concept used with protease inhibitor-based regimens in which patients who have complete clearance of the virus within the first 4 weeks of treatment (a rapid virologic response) and remain clear through 12 weeks of treatment (an extended rapid viral response) can be treated for a shorter duration without decreasing the likelihood of a sustained virologic response.
Lawitz et al17 conducted a randomized double-blind phase 2 trial to evaluate the effect of sofosbuvir dosing on response in noncirrhotic, previously untreated patients with HCV genotype 1, 2, or 3. Patients with HCV genotype 1 were randomized to one of three treatment groups in a 2:2:1 ratio: sofosbuvir 200 mg orally once daily, sofosbuvir 400 mg orally once daily, or placebo, all for 12 weeks in combination with peg-interferon (180 μg weekly) and ribavirin in a dosage based on weight. Depending on the viral response, patients continued peg-interferon and ribavirin for an additional 12 weeks if they achieved an extended rapid viral response, or 36 weeks if they did not achieve an extended rapid virologic response, and in all patients who received placebo. Patients with HCV genotype 2 or 3 were given sofosbuvir 400 mg once daily in combination with interferon and ribavirin for 12 weeks.
As in the ATOMIC trial, all patients treated with sofosbuvir had a very rapid reduction in viral load: 98% of patients with genotype 1 developed a rapid virologic response, and therefore almost all were eligible for the shorter treatment course of 24 weeks.17 The latter finding again suggested that response-guided treatment is not relevant with sofosbuvir-based regimens.
Very high rates of sustained virologic response were seen: 90% in patients with genotype 1 treated with sofosbuvir 200 mg, 91% in those with genotype 1 treated with 400 mg, and 92% in those with genotype 2 or 3. Although 6% of patients in the 200-mg group had virologic breakthrough after completing sofosbuvir treatment, no virologic breakthrough was observed in the 400-mg group, suggesting that the 400-mg dose might suppress the virus more effectively.17
The ELECTRON trial18 was a phase 2 study designed to evaluate the efficacy and safety of sofosbuvir and ribavirin in interferon-sparing and interferon-free regimens in patients with HCV genotype 1, 2, or 3 infection. Sofosbuvir was tested with peg-interferon and ribavirin, with ribavirin alone, and as monotherapy in previously untreated patients with genotype 2 or 3. A small number of patients with genotype 1 who were previously untreated and who were previously nonresponders were also treated with sofosbuvir and ribavirin.
All patients had a rapid virologic response, and viral suppression was sustained through the end of treatment. All patients with genotype 2 or 3 treated with double therapy (sofosbuvir and ribavirin) or triple therapy (sofosbuvir, peg-interferon, and ribavirin) achieved a sustained virologic response, compared with only 60% of patients treated with sofosbuvir monotherapy. The monotherapy group had an equal number of relapsers among those with genotype 2 or 3. Of the genotype 1 patients treated with sofosbuvir and ribavirin, 84% of those previously untreated developed a sustained virologic response, whereas only 10% of the previous nonresponders did.
Phase 3 clinical trials of sofosbuvir
The NEUTRINO trial19 studied the efficacy and safety of sofosbuvir in previously untreated patients with HCV genotype 1, 4, 5, or 6. In this phase 3 open-label study, all patients received sofosbuvir plus peg-interferon and weight-based ribavirin therapy for 12 weeks. Of the patients enrolled, 89% had genotype 1, while 9% had genotype 4 and 2% had genotype 5 or 6. Overall, 17% of the patients had cirrhosis.
The viral load rapidly decreased in all patients treated with sofosbuvir irrespective of the HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Ninety-nine percent of patients with genotype 1, 4, 5, or 6 achieved a rapid virologic response, and 90% achieved a sustained virologic response at 12 weeks after completion of treatment with sofosbuvir and ribavirin. Patients with cirrhosis had a slightly lower rate of sustained virologic response (80%, compared with 92% in patients without cirrhosis). Also, patients with non-CC IL28B genotypes had a lower rate of sustained virologic response (87% in non-CC allele vs 98% in patients with the favorable CC allele).
The FISSION trial19 recruited previously untreated patients with genotype 2 or 3 and randomized them to therapy with either sofosbuvir plus ribavirin in a weight-based dose for 12 weeks, or 24 weeks of interferon and ribavirin. In this study, 20% of patients in each treatment group had cirrhosis.
As in the NEUTRINO trial, the viral load rapidly decreased in all patients treated with sofosbuvir irrespective of HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Here, 100% of patients with genotype 2 or 3 who were treated with sofosbuvir and ribavirin achieved a rapid virologic response. Differences in outcome emerged based on genotype: 97% of those with genotype 2 and 56% of those with genotype 3 achieved a sustained virologic response. The overall rate was 67%, which was not different from patients treated with peg-interferon and ribavirin. In the subgroup of patients with cirrhosis, 47% of those treated with sofosbuvir and ribavirin achieved a sustained virologic response, vs 38% of those who received peg-interferon plus ribavirin.
In both the NEUTRINO and FISSION trials, few patients discontinued treatment, with higher rates of most adverse events occurring in patients treated with peg-interferon and ribavirin.
POSITRON,20 a phase 3 clinical trial, tested sofosbuvir in patients with HCV genotype 2 or 3 who were ineligible for peg-interferon, unwilling to take peg-interferon, or unable to tolerate peg-interferon (mainly because of clinically significant psychiatric disorders). Patients were randomized to two treatment groups for 12 weeks: sofosbuvir plus ribavirin, or placebo. About 50% of patients had HCV genotype 3, and 16% had cirrhosis.
The overall rate of sustained virologic response at 12 weeks after treatment was 78% in the sofosbuvir-and-ribavirin group (93% in genotype 2 patients and 61% in genotype 3 patients). Again, cirrhosis was associated with a lower rate of sustained virologic response (61% of patients with cirrhosis achieved a sustained virologic response vs 81% of patients without cirrhosis). None of the sofosbuvir-treated patients had virologic failure while on treatment.
FUSION,20 another phase 3 trial, evaluated sofosbuvir in patients infected with HCV genotype 2 or 3 for whom interferon-based treatment had failed. They were randomized to either 12 weeks or 16 weeks of sofosbuvir and weight-based ribavirin treatment. About 60% of patients had HCV genotype 3, and 34% had cirrhosis.
The overall sustained virologic response rate was 50% in the patients treated for 12 weeks and 73% in those treated for 16 weeks: specifically, 86% of patients with genotype 2 achieved a sustained virologic response at 12 weeks and 94% at 16 weeks, whereas in those with genotype 3 the rates were 30% at 12 weeks and 62% at 16 weeks.
Cirrhosis was again a predictor of lack of response to sofosbuvir. In the group treated for 12 weeks, 31% of those with cirrhosis achieved a sustained virologic response compared with 61% in those without cirrhosis. In the group treated for 16 weeks, 61% of those with cirrhosis achieved a sustained virologic response compared with 76% in those without cirrhosis.
In both the POSITRON and FUSION trials, relapse accounted for all treatment failures, and no virologic resistance was detected in patients who did not have a sustained virologic response. The investigators concluded that 12 weeks of treatment with sofosbuvir and ribavirin can be effective for HCV genotype 2 infection, but extending the treatment to 16 weeks may be beneficial for genotype 3. This may be especially important in patients with cirrhosis or those who did not have a response to peg-interferon-based treatment.
VALENCE,21 an ongoing phase 3 trial in Europe, is assessing the safety and efficacy of sofosbuvir 400 mg once daily and weight-based ribavirin in patients with HCV genotype 2 or 3. Eighty-five percent of the trial participants have received previous treatment, and 21% have cirrhosis. Patients were originally randomized in a 4:1 ratio to receive sofosbuvir plus ribavirin for 12 weeks or matching placebo, but as a result of emerging data suggesting that patients with genotype 3 would benefit from more than 12 weeks of treatment, the study was subsequently amended to extend treatment to 24 weeks for patients with genotype 3.
Overall rates of sustained virologic response were 93% in patients with genotype 2 and 85% in patients with genotype 3. In previously treated patients with genotype 2 who were treated for 12 weeks, the rates of sustained virologic response were 91% in those without cirrhosis vs 88% in those with cirrhosis. In previously treated patients with genotype 3, the rates in those treated for 24 weeks were 87% in patients without cirrhosis vs 60% with cirrhosis. The safety profile was consistent with that of ribavirin.
Side effects of sofosbuvir
In clinical trials, side effects occurred most often when sofosbuvir was combined with interferon and ribavirin and were consistent with the known side effects of the latter two agents. The most frequently reported side effects included fatigue, insomnia, nausea, rash, anemia, headache, and arthralgia, with most of these adverse events rated by treating clinicians as being mild in severity.15,20
In the ATOMIC trial, the most common events leading to drug discontinuation were anemia and neutropenia, both associated with interferon and ribavirin. Patients receiving sofosbuvir monotherapy after 12 weeks of triple therapy showed rapid improvement in hemoglobin levels and neutrophil counts, indicating that hematologic abnormalities attributed solely to sofosbuvir are minimal. In the FISSION trial, the incidence of adverse events was consistently lower in those receiving sofosbuvir-ribavirin than in patients receiving interferon-ribavirin without sofosbuvir.19
In the POSITRON trial, discontinuation of sofosbuvir because of adverse events was uncommon, and there were no differences in the incidence of adverse events and laboratory abnormalities between patients with and without cirrhosis when they received sofosbuvir and ribavirin.20
Sofosbuvir dosage and indications

Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3 and in combination with ribavirin and interferon alfa in patients infected with HCV genotype 1 or 4 (Table 3). It could be considered for HCV genotype 1 in combination with ribavirin alone for 24 weeks in patients who are ineligible for interferon.
Sofosbuvir is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation, for up to 48 weeks or until they receive a transplant, to prevent posttransplant reinfection with HCV.
Sofosbuvir is expensive
A course of therapy is expected to cost about $84,000, which is significantly more than the cost of previous triple therapy (peg-interferon, ribavirin, and either boceprevir or telaprevir).22 This high cost will undoubtedly lead to less widespread use in developing countries, and potentially even in the United States. As newer direct-acting antiviral agents become available, the price will likely come down, enhancing access to these drugs.
SIMEPREVIR: A SECOND-GENERATION PROTEASE INHIBITOR
Telaprevir and boceprevir are NS3/A4 protease inhibitors that belong to the alfa-ketoamid derivative class. Simeprevir belongs to the macrocyclic class and has a different way of binding to the target enzyme.23 Like sofosbuvir, simeprevir was recently approved by the FDA for the treatment of HCV genotype 1.
The therapeutic efficacy of simeprevir has been tested in several clinical trials (Table 4), including QUEST-124 and QUEST-225 (in previously untreated patients), PROMISE26 (in prior relapsers), and ASPIRE27 (in prior partial and null responders). Results from these trials showed high overall rates of sustained virologic response with triple therapy (ie, simeprevir combined with peg-interferon and ribavirin). It was generally well tolerated, and most adverse events reported during 12 weeks of treatment were of mild to moderate severity.
In QUEST-1 and QUEST-2, both double-blind phase 3 clinical trials, previously untreated patients infected with HCV genotype 1 were randomized in a 2:1 ratio to receive either simeprevir 150 mg daily or placebo for 12 weeks; both groups also received peg-interferon and ribavirin. Patients then received peg-interferon and ribavirin alone for 12 or 36 weeks in the simeprevir group (based on response) and for 36 weeks in the placebo group.
The overall rate of sustained virologic response at 12 weeks was 80% in the simeprevir group (75% in those with genotype 1a and 85% in those with genotype 1b) vs 50% in the placebo group (receiving peg-interferon and ribavirin alone).24,25
PROMISE,26 another double-blind randomized phase 3 clinical trial, evaluated simeprevir in patients with HCV genotype 1 who relapsed after previous interferon-based therapy. It had a similar design to QUEST-1 and QUEST-2, and 15% of all patients had cirrhosis.
The overall sustained virologic response rate at 12 weeks after treatment was 79% in the simeprevir group (70% in patients with genotype 1a and 86% in those with genotype 1b) vs 37% in the placebo group. Rates were similar in patients with absent to moderate fibrosis (82%), advanced fibrosis (73%), or cirrhosis (74%).
ASPIRE.27 Simeprevir efficacy in patients with HCV genotype 1 for whom previous therapy with peg-interferon and ribavirin had failed was tested in ASPIRE, a double-blind randomized phase 2 clinical trial. Patients were randomized to receive simeprevir (either 100 mg or 150 mg daily) for 12, 24, or 48 weeks in combination with 48 weeks of peg-interferon and ribavirin, or placebo plus peg-interferon and ribavirin for 48 weeks.
The primary end point was the rate of sustained virologic response at 24 weeks. Overall, rates were 61% to 80% for the simeprevir treatment groups compared with 23% with placebo, regardless of prior response to peg-interferon and ribavirin. By subgroup, rates were:
- 77% to 89% with simeprevir vs 37% with placebo in prior relapsers
- 48% to 86% with simeprevir vs 9% with placebo in prior partial responders
- 38% to 59% with placebo vs 19% for prior nonresponders.
The best rates of sustained viral response at 24 weeks were in the groups that received simeprevir 150 mg daily: 85% in prior relapsers, 75% in prior partial responders, and 51% in prior nonresponders.
Simeprevir vs other direct-acting antiviral drugs
Advantages of simeprevir over the earlier protease inhibitors include once-daily dosing, a lower rate of adverse events (the most common being fatigue, headache, rash, photosensitivity, and pruritus), a lower likelihood of discontinuation because of adverse events, and fewer drug-drug interactions (since it is a weak inhibitor of the CYP3A4 enzyme).
Unlike sofosbuvir, simeprevir was FDA-approved only for HCV genotype 1 and in combination with interferon alfa and ribavirin. Compared with sofosbuvir, the treatment duration with simeprevir regimens is longer overall (interferon alfa and ribavirin are given for 12 weeks in sofosbuvir-based regimens vs 24 to 48 weeks with simeprevir). As with sofosbuvir, the estimated cost of simeprevir is high, about $66,000 for a 12-week course.
Simeprevir dosage and indications
Simeprevir was approved at an oral dose of 150 mg once daily in combination with ribavirin and interferon alfa in patients with HCV genotype 1 (Table 5).
The approved regimens for simeprevir are fixed in total duration based on the patient’s treatment history. Specifically, all patients receive the drug in combination with peg-interferon and ribavirin for 12 weeks. Then, previously untreated patients and prior relapsers continue to receive peg-interferon and ribavirin alone for another 12 weeks, and those with a partial or null response continue with these drugs for another 36 weeks.
Patients infected with HCV genotype 1a should be screened for the NS3 Q80K polymorphism at baseline, as it has been associated with substantially reduced response to simeprevir.
Sofosbuvir and simeprevir in combination
The COSMOS trial.28 Given their differences in mechanism of action, sofosbuvir and simeprevir are being tested in combination. The COSMOS trial is an ongoing phase 2 randomized open-label study investigating the efficacy and safety of simeprevir and sofosbuvir in combination with and without ribavirin in patients with HCV genotype 1, including nonresponders and those with cirrhosis. Early results are promising, with very high rates of sustained virologic response with the sofosbuvir-simeprevir combination (93% to 100%) and indicate that the addition of ribavirin might not be needed to achieve sustained virologic response in this patient population.
THE FUTURE
The emergence of all-oral regimens for HCV treatment with increasingly sophisticated agents such as sofosbuvir and simeprevir will dramatically alter the management of HCV patients. In view of the improvement in sustained virologic response rates with these treatments, and since most HCV-infected persons have no symptoms, the US Centers for Disease Control and Prevention29 recently recommended one-time testing of the cohort in which the prevalence of HCV infection is highest: all persons born between 1945 and 1965. This undoubtedly will increase the detection of this infection—and the number of new patients expecting treatment.

Future drugs promise further improvements (Table 6).30–35 Advances in knowledge of the HCV molecular structure have led to the development of numerous direct-acting antiviral agents with very specific viral targets. A second wave of protease inhibitors and of nucleoside and nonnucleoside polymerase inhibitors will soon be available. Inhibitors of NS5A (a protein important in the assembly of the viral replication complex) such as daclatasvir and ledipasvir, are currently in phase 3 clinical trials. Other viral proteins involved in assembly of the virus, including the core protein and p7, are being explored as drug targets. In addition, inhibiting host targets such as cyclophilin A and miR122 has gained traction recently, with specific agents currently in phase 2 and 3 clinical trials.
Factors that previously were major determinants of response to treatment, such as IL28B genotype, viral load, race, age, extent of fibrosis, and genotype 1 subtypes, will become much less important with the introduction of highly potent direct-acting antiviral agents.
Many all-oral combinations are being evaluated in clinical trials. For example, the open-label, phase 2 LONESTAR trial tested the utility of combining sofosbuvir and ledipasvir (an NS5A inhibitor) with and without ribavirin for 8 or 12 weeks in previously untreated patients with HCV genotype 1, and for 12 weeks in patients with HCV genotype 1 who did not achieve a sustained virologic response after receiving a protease inhibitor-based regimen (half of whom had compensated cirrhosis).36 Sustained virologic response rates were very high (95% to 100%) in both previously treated and previously untreated patients, including those with cirrhosis. Similar rates were achieved by the 8-week and 12-week groups in noncirrhotic patients who had not been previously treated for HCV. The typical hematologic abnormalities associated with interferon were not observed except for mild anemia in patients who received ribavirin. These results suggest that the combination of sofosbuvir and ledipasvir could offer a very effective, short, all-oral treatment for patients with HCV genotype 1, including those with cirrhosis, who up to now have been difficult to treat.
Challenges remaining

The success of sofosbuvir and simeprevir paves the way for interferon-free regimens.37 For a long time, the treatment of HCV infection required close monitoring of patients while managing the side effects of interferon, but the current and emerging direct-acting antiviral agents will soon change this practice. Given the synergistic effects of combination therapy—targeting the virus at multiple locations, decreasing the likelihood of drug resistance, and improving efficacy—combination regimens seem to be the optimal solution to the HCV epidemic. Lower risk of side effects and shorter treatment duration will definitely improve the acceptance of any new regimen. New agents that act against conserved viral targets, thereby yielding activity across multiple genotypes, will be advantageous as well. Table 7 compares the rates of sustained virologic response of the different currently approved HCV treatment regimens.
Clinical challenges remain, including the management of special patient populations for whom data are still limited. These include patients with cirrhosis, chronic kidney disease, renal failure, and concurrent infection with human immunodeficiency virus, and patients who have undergone solid organ transplantation. Clinical trials are under way to evaluate the treatment options for these patients, who will likely need to wait for the emergence of additional agents before dramatic improvement in sustained virologic response rates may be expected.38
As the treatment of HCV becomes simpler, safer, and more effective, primary care physicians will increasingly be expected to manage it. Difficult-to-treat patients, including the special populations above, will require specialist management and individualized treatment regimens, at least until better therapies are available. The high projected cost of the new agents may limit access, at least initially. However, the dramatic improvement in sustained virologic response rates and all that that implies in terms of decreased risk of advanced liver disease and its complications will undoubtedly make these therapies cost-effective.39
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the united states. Clin Infect Dis 2012; 55(suppl 1):S10–S15.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first international liver transplantation society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transplant 2003; 9:S1–S9.
- Wong JB, McQuillan GM, McHutchison JG, Poynard T. Estimating future hepatitis C morbidity, mortality, and costs in the United States. Am J Public Health 2000; 90:1562–1569.
- Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132:1979–1998.
- Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol 2000; 81:1631–1648.
- Singal AG, Volk ML, Jensen D, Di Bisceglie AM, Schoenfeld PS. A sustained viral response is associated with reduced liver-related morbidity and mortality in patients with hepatitis C virus. Clin Gastroenterol Hepatol 2010; 8:280–288,288.e1.
- Camma C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Paeshuyse J, Dallmeier K, Neyts J. Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol 2011; 1:590–598.
- Thomas E, Ghany MG, Liang TJ. The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir Chem Chemother 2012; 23:1–12.
- Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB; American Association for Study of Liver Diseases. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54:1433–1444.
- Soriano V, Vispo E, Poveda E, Labarga P, Barreiro P. Treatment failure with new hepatitis C drugs. Expert Opin Pharmacother 2012; 13:313–323.
- Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int 2013; 33(suppl 1):93–104.
- Manns MP, McCone J, Davis MN, et al. Overall safety profile of boceprevir plus peginterferon alfa-2b and ribavirin in patients with chronic hepatitis C genotype 1: a combined analysis of 3 phase 2/3 clinical trials. Liver Int 2013; Aug 2. doi: 10.1111/liv.12300. [Epub ahead of print]
- Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Rodriguez-Torres M, Lawitz E, Kowdley KV, et al. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: a randomized, 28-day, dose-ranging trial. J Hepatol 2013; 58:663–668.
- Kowdley KV, Lawitz E, Crespo I, et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 2013; 381:2100–2107.
- Lawitz E, Lalezari JP, Hassanein T, et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis 2013; 13:401–408.
- Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 2013; 368:34–44.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Jacobson IM, Gordon SC, Kowdley KV, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 2013; 368:1867–1877.
- Zeuzem S, Dusheiko G, Salupere R, et al. Sofosbuvir + ribavirin for 12 or 24 weeks for patients with HCV genotype 2 or 3: the VALENCE trial [abstract no.1085]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- You DM, Pockros PJ. Simeprevir for the treatment of chronic hepatitis C. Expert Opin Pharmacother 2013; 14:2581–2589.
- Jacobson IM, Dore GJ, Foster G, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive patients: results from Quest-1, a phase III trial [abstract no. 1425]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, Netherlands.
- Manns M, Marcellin P, Poordad FP, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naïve patients: results from QUEST-2, a phase III trial [abstract no. 1413]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, The Netherlands.
- Lawitz E, Forns X, Zeuzem S, et al. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in patients who relapsed after previous interferon-based therapy: results from promise, a phase III trial [abstract no. 869b]. Digestive Disease Week; May 18–21, 2013; Orlando, FL.
- Zeuzem S, Berg T, Gane E, et al. Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology epub Oct 31, 2013.
- Jacobson IM, Ghalib RM, Rodriguez-Torres M, et al. SVR results of a once-daily regimen of simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naive and prior null responder patients: the COSMOS study [abstract LB-3]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Smith BD, Morgan RL, Beckett GA, et al. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm Rep 2012; 61( RR-4):1–32.
- Sulkowski MS, Kang M, Matining R, et al. Safety and antiviral activity of the HCV entry inhibitor ITX5061 in treatment-naive HCV-infected adults: a randomized, double-blind, phase 1b study. J Infect Dis 2013 Oct 9. [Epub ahead of print]
- Pawlotsky JM. NS5A inhibitors in the treatment of hepatitis C. J Hepatol 2013; 59:375–382.
- Yu M, Corsa AC, Xu S, et al. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antiviral Res 2013; 100:439–445.
- Aghemo A, De Francesco R. New horizons in hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 2013; 58:428–438.
- Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med 2013; 368:1907–1917.
- Flisiak R, Jaroszewicz J, Parfieniuk-Kowerda A. Emerging treatments for hepatitis C. Expert Opin Emerg Drugs 2013; 18:461–475.
- Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 2013 Nov 1. doi: 10.1016/S0140-6736(13)62121-2 [Epub ahead of print]
- Drenth JP. HCV treatment—no more room for interferonologists? N Engl J Med 2013; 368:1931–1932.
- Casey LC, Lee WM. Hepatitis C virus therapy update 2013. Curr Opin Gastroenterol 2013; 29:243–249.
- Afdhal NH, Zeuzem S, Schooley RT, et al. The new paradigm of hepatitis C therapy: integration of oral therapies into best practices. J Viral Hepatol 2013; 20:745–760.
In late 2013, the US Food and Drug Administration (FDA) approved sofosbuvir and simeprevir, the newest direct-acting antiviral agents for treating chronic hepatitis C virus (HCV) infection. Multiple clinical trials have demonstrated dramatically improved treatment outcomes with these agents, opening the door to all-oral regimens or interferon-free regimens as the future standard of care for HCV.
In this article, we discuss the results of the trials that established the efficacy and safety of sofosbuvir and simeprevir and led to their FDA approval. We also summarize the importance of these agents and evaluate other direct-acting antivirals currently in the pipeline for HCV treatment.
HCV IS A RISING PROBLEM
Chronic HCV infection is a major clinical and public health problem, with the estimated number of people infected exceeding 170 million worldwide, including 3.2 million in the United States.1 It is a leading cause of cirrhosis, and its complications include hepatocellular carcinoma and liver failure. Cirrhosis due to HCV remains the leading indication for liver transplantation in the United States, accounting for nearly 40% of liver transplants in adults.2
The clinical impact of HCV will only continue to escalate, and in parallel, so will the cost to society. Models suggest that HCV-related deaths will double between 2010 and 2019, and considering only direct medical costs, the projected financial burden of treating HCV-related disease during this interval is estimated at between $6.5 and $13.6 billion.3
AN RNA VIRUS WITH SIX GENOTYPES
HCV, first identified in 1989, is an enveloped, single-stranded RNA flavivirus of the Hepacivirus genus measuring 50 to 60 nm in diameter.4 There are six viral genotypes, with genotype 1 being the most common in the United States and traditionally the most difficult to treat.
Once inside the host cell, the virus releases its RNA strand, which is translated into a single polyprotein of about 3,000 amino acids. This large molecule is then cleaved by proteases into several domains: three structural proteins (C, E1, and E2), a small protein called p7, and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Figure 1).5 These nonstructural proteins enable the virus to replicate.
GOAL OF TREATING HCV: A SUSTAINED VIROLOGIC RESPONSE
The aim of HCV treatment is to achieve a sustained virologic response, defined as having no detectable viral RNA after completion of antiviral therapy. This is associated with substantially better clinical outcomes, lower rates of liver-related morbidity and all-cause mortality, and stabilization of or even improvement in liver histology.6,7 This end point has traditionally been assessed at 6 months after the end of therapy, but recent data suggest the rates at 12 weeks are essentially equivalent.
Table 1 summarizes the patterns of virologic response in treating HCV infection.
Interferon plus ribavirin: The standard of care for many years
HCV treatment has evolved over the past 20 years. Before 2011, the standard of care was a combination of interferon alfa-polyethylene glycol (peg-interferon), given as a weekly injection, and oral ribavirin. Neither drug has specific antiviral activity, and when they are used together they result in a sustained virologic response in fewer than 50% of patients with HCV genotype 1 and, at best, in 70% to 80% of patients with other genotypes.8
Nearly all patients receiving interferon experience side effects, which can be serious. Fatigue and flu-like symptoms are common, and the drug can also cause psychiatric symptoms (including depression or psychosis), weight loss, seizures, peripheral neuropathy, and bone marrow suppression. Ribavirin causes hemolysis and skin complications and is teratogenic.9
An important bit of information to know when using interferon is the patient’s IL28B genotype. This refers to a single-nucleotide polymorphism (C or T) on chromosome 19q13 (rs12979860) upstream of the IL28B gene encoding for interferon lambda-3. It is strongly associated with responsiveness to interferon: patients with the IL28B CC genotype have a much better chance of a sustained virologic response with interferon than do patients with CT or TT.
Boceprevir and telaprevir: First-generation protease inhibitors
In May 2011, the FDA approved the NS3/4A protease inhibitors boceprevir and telaprevir for treating HCV genotype 1, marking the beginning of the era of direct-acting antiviral agents.10 When these drugs are used in combination with peg-interferon alfa and ribavirin, up to 75% of patients with HCV genotype 1 who have had no previous treatment achieve a sustained virologic response.
But despite greatly improving the response rate, these first-generation protease inhibitors have substantial limitations. Twenty-five percent of patients with HCV genotype 1 who have received no previous treatment and 71% of patients who did not respond to previous treatment will not achieve a sustained virologic response with these agents.11 Further, they are effective only against HCV genotype 1, being highly specific for the amino acid target sequence of the NS3 region.
Also, they must be used in combination with interferon alfa and ribavirin because the virus needs to mutate only a little—a few amino-acid substitutions—to gain resistance to them.12 Therefore, patients are still exposed to interferon and ribavirin, with their toxicity. In addition, dysgeusia is seen with boceprevir, rash with telaprevir, and anemia with both.13,14
Finally, serious drug-drug interactions prompted the FDA to impose warnings for the use of these agents with other medications that interact with CYP3A4, the principal enzyme responsible for their metabolism. Thus, these significant adverse effects dampen the enthusiasm of patients contemplating a long course of treatment with these agents.
The need to improve the rate of sustained virologic response, shorten the duration of treatment, avoid serious side effects, improve efficacy in treating patients infected with genotypes other than 1, and, importantly, eliminate the need for interferon alfa and its serious adverse effects have driven the development of new direct-acting antiviral agents, including the two newly FDA-approved drugs, sofosbuvir and simeprevir.
SOFOSBUVIR: A POLYMERASE INHIBITOR
Sofosbuvir is a uridine nucleotide analogue that selectively inhibits the HCV NS5B RNA-dependent RNA polymerase (Figure 1). It targets the highly conserved nucleotide-binding pocket of this enzyme and functions as a chain terminator.15 While the protease inhibitors are genotype-dependent, inhibition of the highly conserved viral polymerase has an impact that spans genotypes.
Early clinical trials of sofosbuvir
Sofosbuvir has been tested in combination with interferon alfa and ribavirin, as well as in interferon-free regimens (Table 2).16–20
Rodriguez-Torres et al,15
- 56% with sofosbuvir 100 mg, peg-interferon, and ribavirin
- 83% with sofosbuvir 200 mg, peg-interferon, and ribavirin
- 80% with sofosbuvir 400 mg, peg-interferon, and ribavirin
- 43% with peg-interferon and ribavirin alone.
The ATOMIC trial16 tested the efficacy and safety of sofosbuvir in combination with peg-interferon and ribavirin in patients with HCV genotype 1, 4, or 6, without cirrhosis, who had not received any previous treatment. Patients with HCV genotype 1 were randomized to three treatments:
- Sofosbuvir 400 mg orally once daily plus peg-interferon and ribavirin for 12 weeks
- The same regimen, but for 24 weeks
- Sofosbuvir plus peg-interferon and ribavirin for 12 weeks, followed by 12 weeks of either sofosbuvir monotherapy or sofosbuvir plus ribavirin.
The rates of sustained virologic response were very high and were not significantly different among the three groups: 89%, 89%, and 87%, respectively. Patients who were able to complete a full course of therapy achieved even higher rates of sustained virologic response, ranging from 96% to 98%. The likelihood of response was not adversely affected by the usual markers of a poorer prognosis, such as a high viral load (≥ 800,000 IU/mL) or a non-CC IL28B genotype. Although patients with cirrhosis (another predictor of no response) were excluded from this study, the presence of bridging fibrosis did not seem to affect the rate of sustained virologic response. The results in patients with genotypes other than 1 were very encouraging, but the small number of patients enrolled precluded drawing firm conclusions in this group.
Important implications of the ATOMIC trial include the following:
There is no benefit in prolonging treatment with sofosbuvir beyond 12 weeks, since adverse events increased without any improvement in the rate of sustained virologic response.
There is a very low likelihood of developing viral resistance or mutation when using sofosbuvir.
There is no role for response-guided therapy, a concept used with protease inhibitor-based regimens in which patients who have complete clearance of the virus within the first 4 weeks of treatment (a rapid virologic response) and remain clear through 12 weeks of treatment (an extended rapid viral response) can be treated for a shorter duration without decreasing the likelihood of a sustained virologic response.
Lawitz et al17 conducted a randomized double-blind phase 2 trial to evaluate the effect of sofosbuvir dosing on response in noncirrhotic, previously untreated patients with HCV genotype 1, 2, or 3. Patients with HCV genotype 1 were randomized to one of three treatment groups in a 2:2:1 ratio: sofosbuvir 200 mg orally once daily, sofosbuvir 400 mg orally once daily, or placebo, all for 12 weeks in combination with peg-interferon (180 μg weekly) and ribavirin in a dosage based on weight. Depending on the viral response, patients continued peg-interferon and ribavirin for an additional 12 weeks if they achieved an extended rapid viral response, or 36 weeks if they did not achieve an extended rapid virologic response, and in all patients who received placebo. Patients with HCV genotype 2 or 3 were given sofosbuvir 400 mg once daily in combination with interferon and ribavirin for 12 weeks.
As in the ATOMIC trial, all patients treated with sofosbuvir had a very rapid reduction in viral load: 98% of patients with genotype 1 developed a rapid virologic response, and therefore almost all were eligible for the shorter treatment course of 24 weeks.17 The latter finding again suggested that response-guided treatment is not relevant with sofosbuvir-based regimens.
Very high rates of sustained virologic response were seen: 90% in patients with genotype 1 treated with sofosbuvir 200 mg, 91% in those with genotype 1 treated with 400 mg, and 92% in those with genotype 2 or 3. Although 6% of patients in the 200-mg group had virologic breakthrough after completing sofosbuvir treatment, no virologic breakthrough was observed in the 400-mg group, suggesting that the 400-mg dose might suppress the virus more effectively.17
The ELECTRON trial18 was a phase 2 study designed to evaluate the efficacy and safety of sofosbuvir and ribavirin in interferon-sparing and interferon-free regimens in patients with HCV genotype 1, 2, or 3 infection. Sofosbuvir was tested with peg-interferon and ribavirin, with ribavirin alone, and as monotherapy in previously untreated patients with genotype 2 or 3. A small number of patients with genotype 1 who were previously untreated and who were previously nonresponders were also treated with sofosbuvir and ribavirin.
All patients had a rapid virologic response, and viral suppression was sustained through the end of treatment. All patients with genotype 2 or 3 treated with double therapy (sofosbuvir and ribavirin) or triple therapy (sofosbuvir, peg-interferon, and ribavirin) achieved a sustained virologic response, compared with only 60% of patients treated with sofosbuvir monotherapy. The monotherapy group had an equal number of relapsers among those with genotype 2 or 3. Of the genotype 1 patients treated with sofosbuvir and ribavirin, 84% of those previously untreated developed a sustained virologic response, whereas only 10% of the previous nonresponders did.
Phase 3 clinical trials of sofosbuvir
The NEUTRINO trial19 studied the efficacy and safety of sofosbuvir in previously untreated patients with HCV genotype 1, 4, 5, or 6. In this phase 3 open-label study, all patients received sofosbuvir plus peg-interferon and weight-based ribavirin therapy for 12 weeks. Of the patients enrolled, 89% had genotype 1, while 9% had genotype 4 and 2% had genotype 5 or 6. Overall, 17% of the patients had cirrhosis.
The viral load rapidly decreased in all patients treated with sofosbuvir irrespective of the HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Ninety-nine percent of patients with genotype 1, 4, 5, or 6 achieved a rapid virologic response, and 90% achieved a sustained virologic response at 12 weeks after completion of treatment with sofosbuvir and ribavirin. Patients with cirrhosis had a slightly lower rate of sustained virologic response (80%, compared with 92% in patients without cirrhosis). Also, patients with non-CC IL28B genotypes had a lower rate of sustained virologic response (87% in non-CC allele vs 98% in patients with the favorable CC allele).
The FISSION trial19 recruited previously untreated patients with genotype 2 or 3 and randomized them to therapy with either sofosbuvir plus ribavirin in a weight-based dose for 12 weeks, or 24 weeks of interferon and ribavirin. In this study, 20% of patients in each treatment group had cirrhosis.
As in the NEUTRINO trial, the viral load rapidly decreased in all patients treated with sofosbuvir irrespective of HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Here, 100% of patients with genotype 2 or 3 who were treated with sofosbuvir and ribavirin achieved a rapid virologic response. Differences in outcome emerged based on genotype: 97% of those with genotype 2 and 56% of those with genotype 3 achieved a sustained virologic response. The overall rate was 67%, which was not different from patients treated with peg-interferon and ribavirin. In the subgroup of patients with cirrhosis, 47% of those treated with sofosbuvir and ribavirin achieved a sustained virologic response, vs 38% of those who received peg-interferon plus ribavirin.
In both the NEUTRINO and FISSION trials, few patients discontinued treatment, with higher rates of most adverse events occurring in patients treated with peg-interferon and ribavirin.
POSITRON,20 a phase 3 clinical trial, tested sofosbuvir in patients with HCV genotype 2 or 3 who were ineligible for peg-interferon, unwilling to take peg-interferon, or unable to tolerate peg-interferon (mainly because of clinically significant psychiatric disorders). Patients were randomized to two treatment groups for 12 weeks: sofosbuvir plus ribavirin, or placebo. About 50% of patients had HCV genotype 3, and 16% had cirrhosis.
The overall rate of sustained virologic response at 12 weeks after treatment was 78% in the sofosbuvir-and-ribavirin group (93% in genotype 2 patients and 61% in genotype 3 patients). Again, cirrhosis was associated with a lower rate of sustained virologic response (61% of patients with cirrhosis achieved a sustained virologic response vs 81% of patients without cirrhosis). None of the sofosbuvir-treated patients had virologic failure while on treatment.
FUSION,20 another phase 3 trial, evaluated sofosbuvir in patients infected with HCV genotype 2 or 3 for whom interferon-based treatment had failed. They were randomized to either 12 weeks or 16 weeks of sofosbuvir and weight-based ribavirin treatment. About 60% of patients had HCV genotype 3, and 34% had cirrhosis.
The overall sustained virologic response rate was 50% in the patients treated for 12 weeks and 73% in those treated for 16 weeks: specifically, 86% of patients with genotype 2 achieved a sustained virologic response at 12 weeks and 94% at 16 weeks, whereas in those with genotype 3 the rates were 30% at 12 weeks and 62% at 16 weeks.
Cirrhosis was again a predictor of lack of response to sofosbuvir. In the group treated for 12 weeks, 31% of those with cirrhosis achieved a sustained virologic response compared with 61% in those without cirrhosis. In the group treated for 16 weeks, 61% of those with cirrhosis achieved a sustained virologic response compared with 76% in those without cirrhosis.
In both the POSITRON and FUSION trials, relapse accounted for all treatment failures, and no virologic resistance was detected in patients who did not have a sustained virologic response. The investigators concluded that 12 weeks of treatment with sofosbuvir and ribavirin can be effective for HCV genotype 2 infection, but extending the treatment to 16 weeks may be beneficial for genotype 3. This may be especially important in patients with cirrhosis or those who did not have a response to peg-interferon-based treatment.
VALENCE,21 an ongoing phase 3 trial in Europe, is assessing the safety and efficacy of sofosbuvir 400 mg once daily and weight-based ribavirin in patients with HCV genotype 2 or 3. Eighty-five percent of the trial participants have received previous treatment, and 21% have cirrhosis. Patients were originally randomized in a 4:1 ratio to receive sofosbuvir plus ribavirin for 12 weeks or matching placebo, but as a result of emerging data suggesting that patients with genotype 3 would benefit from more than 12 weeks of treatment, the study was subsequently amended to extend treatment to 24 weeks for patients with genotype 3.
Overall rates of sustained virologic response were 93% in patients with genotype 2 and 85% in patients with genotype 3. In previously treated patients with genotype 2 who were treated for 12 weeks, the rates of sustained virologic response were 91% in those without cirrhosis vs 88% in those with cirrhosis. In previously treated patients with genotype 3, the rates in those treated for 24 weeks were 87% in patients without cirrhosis vs 60% with cirrhosis. The safety profile was consistent with that of ribavirin.
Side effects of sofosbuvir
In clinical trials, side effects occurred most often when sofosbuvir was combined with interferon and ribavirin and were consistent with the known side effects of the latter two agents. The most frequently reported side effects included fatigue, insomnia, nausea, rash, anemia, headache, and arthralgia, with most of these adverse events rated by treating clinicians as being mild in severity.15,20
In the ATOMIC trial, the most common events leading to drug discontinuation were anemia and neutropenia, both associated with interferon and ribavirin. Patients receiving sofosbuvir monotherapy after 12 weeks of triple therapy showed rapid improvement in hemoglobin levels and neutrophil counts, indicating that hematologic abnormalities attributed solely to sofosbuvir are minimal. In the FISSION trial, the incidence of adverse events was consistently lower in those receiving sofosbuvir-ribavirin than in patients receiving interferon-ribavirin without sofosbuvir.19
In the POSITRON trial, discontinuation of sofosbuvir because of adverse events was uncommon, and there were no differences in the incidence of adverse events and laboratory abnormalities between patients with and without cirrhosis when they received sofosbuvir and ribavirin.20
Sofosbuvir dosage and indications
Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3 and in combination with ribavirin and interferon alfa in patients infected with HCV genotype 1 or 4 (Table 3). It could be considered for HCV genotype 1 in combination with ribavirin alone for 24 weeks in patients who are ineligible for interferon.
Sofosbuvir is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation, for up to 48 weeks or until they receive a transplant, to prevent posttransplant reinfection with HCV.
Sofosbuvir is expensive
A course of therapy is expected to cost about $84,000, which is significantly more than the cost of previous triple therapy (peg-interferon, ribavirin, and either boceprevir or telaprevir).22 This high cost will undoubtedly lead to less widespread use in developing countries, and potentially even in the United States. As newer direct-acting antiviral agents become available, the price will likely come down, enhancing access to these drugs.
SIMEPREVIR: A SECOND-GENERATION PROTEASE INHIBITOR
Telaprevir and boceprevir are NS3/A4 protease inhibitors that belong to the alfa-ketoamid derivative class. Simeprevir belongs to the macrocyclic class and has a different way of binding to the target enzyme.23 Like sofosbuvir, simeprevir was recently approved by the FDA for the treatment of HCV genotype 1.
The therapeutic efficacy of simeprevir has been tested in several clinical trials (Table 4), including QUEST-124 and QUEST-225 (in previously untreated patients), PROMISE26 (in prior relapsers), and ASPIRE27 (in prior partial and null responders). Results from these trials showed high overall rates of sustained virologic response with triple therapy (ie, simeprevir combined with peg-interferon and ribavirin). It was generally well tolerated, and most adverse events reported during 12 weeks of treatment were of mild to moderate severity.
In QUEST-1 and QUEST-2, both double-blind phase 3 clinical trials, previously untreated patients infected with HCV genotype 1 were randomized in a 2:1 ratio to receive either simeprevir 150 mg daily or placebo for 12 weeks; both groups also received peg-interferon and ribavirin. Patients then received peg-interferon and ribavirin alone for 12 or 36 weeks in the simeprevir group (based on response) and for 36 weeks in the placebo group.
The overall rate of sustained virologic response at 12 weeks was 80% in the simeprevir group (75% in those with genotype 1a and 85% in those with genotype 1b) vs 50% in the placebo group (receiving peg-interferon and ribavirin alone).24,25
PROMISE,26 another double-blind randomized phase 3 clinical trial, evaluated simeprevir in patients with HCV genotype 1 who relapsed after previous interferon-based therapy. It had a similar design to QUEST-1 and QUEST-2, and 15% of all patients had cirrhosis.
The overall sustained virologic response rate at 12 weeks after treatment was 79% in the simeprevir group (70% in patients with genotype 1a and 86% in those with genotype 1b) vs 37% in the placebo group. Rates were similar in patients with absent to moderate fibrosis (82%), advanced fibrosis (73%), or cirrhosis (74%).
ASPIRE.27 Simeprevir efficacy in patients with HCV genotype 1 for whom previous therapy with peg-interferon and ribavirin had failed was tested in ASPIRE, a double-blind randomized phase 2 clinical trial. Patients were randomized to receive simeprevir (either 100 mg or 150 mg daily) for 12, 24, or 48 weeks in combination with 48 weeks of peg-interferon and ribavirin, or placebo plus peg-interferon and ribavirin for 48 weeks.
The primary end point was the rate of sustained virologic response at 24 weeks. Overall, rates were 61% to 80% for the simeprevir treatment groups compared with 23% with placebo, regardless of prior response to peg-interferon and ribavirin. By subgroup, rates were:
- 77% to 89% with simeprevir vs 37% with placebo in prior relapsers
- 48% to 86% with simeprevir vs 9% with placebo in prior partial responders
- 38% to 59% with placebo vs 19% for prior nonresponders.
The best rates of sustained viral response at 24 weeks were in the groups that received simeprevir 150 mg daily: 85% in prior relapsers, 75% in prior partial responders, and 51% in prior nonresponders.
Simeprevir vs other direct-acting antiviral drugs
Advantages of simeprevir over the earlier protease inhibitors include once-daily dosing, a lower rate of adverse events (the most common being fatigue, headache, rash, photosensitivity, and pruritus), a lower likelihood of discontinuation because of adverse events, and fewer drug-drug interactions (since it is a weak inhibitor of the CYP3A4 enzyme).
Unlike sofosbuvir, simeprevir was FDA-approved only for HCV genotype 1 and in combination with interferon alfa and ribavirin. Compared with sofosbuvir, the treatment duration with simeprevir regimens is longer overall (interferon alfa and ribavirin are given for 12 weeks in sofosbuvir-based regimens vs 24 to 48 weeks with simeprevir). As with sofosbuvir, the estimated cost of simeprevir is high, about $66,000 for a 12-week course.
Simeprevir dosage and indications
Simeprevir was approved at an oral dose of 150 mg once daily in combination with ribavirin and interferon alfa in patients with HCV genotype 1 (Table 5).
The approved regimens for simeprevir are fixed in total duration based on the patient’s treatment history. Specifically, all patients receive the drug in combination with peg-interferon and ribavirin for 12 weeks. Then, previously untreated patients and prior relapsers continue to receive peg-interferon and ribavirin alone for another 12 weeks, and those with a partial or null response continue with these drugs for another 36 weeks.
Patients infected with HCV genotype 1a should be screened for the NS3 Q80K polymorphism at baseline, as it has been associated with substantially reduced response to simeprevir.
Sofosbuvir and simeprevir in combination
The COSMOS trial.28 Given their differences in mechanism of action, sofosbuvir and simeprevir are being tested in combination. The COSMOS trial is an ongoing phase 2 randomized open-label study investigating the efficacy and safety of simeprevir and sofosbuvir in combination with and without ribavirin in patients with HCV genotype 1, including nonresponders and those with cirrhosis. Early results are promising, with very high rates of sustained virologic response with the sofosbuvir-simeprevir combination (93% to 100%) and indicate that the addition of ribavirin might not be needed to achieve sustained virologic response in this patient population.
THE FUTURE
The emergence of all-oral regimens for HCV treatment with increasingly sophisticated agents such as sofosbuvir and simeprevir will dramatically alter the management of HCV patients. In view of the improvement in sustained virologic response rates with these treatments, and since most HCV-infected persons have no symptoms, the US Centers for Disease Control and Prevention29 recently recommended one-time testing of the cohort in which the prevalence of HCV infection is highest: all persons born between 1945 and 1965. This undoubtedly will increase the detection of this infection—and the number of new patients expecting treatment.
Future drugs promise further improvements (Table 6).30–35 Advances in knowledge of the HCV molecular structure have led to the development of numerous direct-acting antiviral agents with very specific viral targets. A second wave of protease inhibitors and of nucleoside and nonnucleoside polymerase inhibitors will soon be available. Inhibitors of NS5A (a protein important in the assembly of the viral replication complex) such as daclatasvir and ledipasvir, are currently in phase 3 clinical trials. Other viral proteins involved in assembly of the virus, including the core protein and p7, are being explored as drug targets. In addition, inhibiting host targets such as cyclophilin A and miR122 has gained traction recently, with specific agents currently in phase 2 and 3 clinical trials.
Factors that previously were major determinants of response to treatment, such as IL28B genotype, viral load, race, age, extent of fibrosis, and genotype 1 subtypes, will become much less important with the introduction of highly potent direct-acting antiviral agents.
Many all-oral combinations are being evaluated in clinical trials. For example, the open-label, phase 2 LONESTAR trial tested the utility of combining sofosbuvir and ledipasvir (an NS5A inhibitor) with and without ribavirin for 8 or 12 weeks in previously untreated patients with HCV genotype 1, and for 12 weeks in patients with HCV genotype 1 who did not achieve a sustained virologic response after receiving a protease inhibitor-based regimen (half of whom had compensated cirrhosis).36 Sustained virologic response rates were very high (95% to 100%) in both previously treated and previously untreated patients, including those with cirrhosis. Similar rates were achieved by the 8-week and 12-week groups in noncirrhotic patients who had not been previously treated for HCV. The typical hematologic abnormalities associated with interferon were not observed except for mild anemia in patients who received ribavirin. These results suggest that the combination of sofosbuvir and ledipasvir could offer a very effective, short, all-oral treatment for patients with HCV genotype 1, including those with cirrhosis, who up to now have been difficult to treat.
Challenges remaining
The success of sofosbuvir and simeprevir paves the way for interferon-free regimens.37 For a long time, the treatment of HCV infection required close monitoring of patients while managing the side effects of interferon, but the current and emerging direct-acting antiviral agents will soon change this practice. Given the synergistic effects of combination therapy—targeting the virus at multiple locations, decreasing the likelihood of drug resistance, and improving efficacy—combination regimens seem to be the optimal solution to the HCV epidemic. Lower risk of side effects and shorter treatment duration will definitely improve the acceptance of any new regimen. New agents that act against conserved viral targets, thereby yielding activity across multiple genotypes, will be advantageous as well. Table 7 compares the rates of sustained virologic response of the different currently approved HCV treatment regimens.
Clinical challenges remain, including the management of special patient populations for whom data are still limited. These include patients with cirrhosis, chronic kidney disease, renal failure, and concurrent infection with human immunodeficiency virus, and patients who have undergone solid organ transplantation. Clinical trials are under way to evaluate the treatment options for these patients, who will likely need to wait for the emergence of additional agents before dramatic improvement in sustained virologic response rates may be expected.38
As the treatment of HCV becomes simpler, safer, and more effective, primary care physicians will increasingly be expected to manage it. Difficult-to-treat patients, including the special populations above, will require specialist management and individualized treatment regimens, at least until better therapies are available. The high projected cost of the new agents may limit access, at least initially. However, the dramatic improvement in sustained virologic response rates and all that that implies in terms of decreased risk of advanced liver disease and its complications will undoubtedly make these therapies cost-effective.39
In late 2013, the US Food and Drug Administration (FDA) approved sofosbuvir and simeprevir, the newest direct-acting antiviral agents for treating chronic hepatitis C virus (HCV) infection. Multiple clinical trials have demonstrated dramatically improved treatment outcomes with these agents, opening the door to all-oral regimens or interferon-free regimens as the future standard of care for HCV.
In this article, we discuss the results of the trials that established the efficacy and safety of sofosbuvir and simeprevir and led to their FDA approval. We also summarize the importance of these agents and evaluate other direct-acting antivirals currently in the pipeline for HCV treatment.
HCV IS A RISING PROBLEM
Chronic HCV infection is a major clinical and public health problem, with the estimated number of people infected exceeding 170 million worldwide, including 3.2 million in the United States.1 It is a leading cause of cirrhosis, and its complications include hepatocellular carcinoma and liver failure. Cirrhosis due to HCV remains the leading indication for liver transplantation in the United States, accounting for nearly 40% of liver transplants in adults.2
The clinical impact of HCV will only continue to escalate, and in parallel, so will the cost to society. Models suggest that HCV-related deaths will double between 2010 and 2019, and considering only direct medical costs, the projected financial burden of treating HCV-related disease during this interval is estimated at between $6.5 and $13.6 billion.3
AN RNA VIRUS WITH SIX GENOTYPES
HCV, first identified in 1989, is an enveloped, single-stranded RNA flavivirus of the Hepacivirus genus measuring 50 to 60 nm in diameter.4 There are six viral genotypes, with genotype 1 being the most common in the United States and traditionally the most difficult to treat.
Once inside the host cell, the virus releases its RNA strand, which is translated into a single polyprotein of about 3,000 amino acids. This large molecule is then cleaved by proteases into several domains: three structural proteins (C, E1, and E2), a small protein called p7, and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Figure 1).5 These nonstructural proteins enable the virus to replicate.
GOAL OF TREATING HCV: A SUSTAINED VIROLOGIC RESPONSE
The aim of HCV treatment is to achieve a sustained virologic response, defined as having no detectable viral RNA after completion of antiviral therapy. This is associated with substantially better clinical outcomes, lower rates of liver-related morbidity and all-cause mortality, and stabilization of or even improvement in liver histology.6,7 This end point has traditionally been assessed at 6 months after the end of therapy, but recent data suggest the rates at 12 weeks are essentially equivalent.
Table 1 summarizes the patterns of virologic response in treating HCV infection.
Interferon plus ribavirin: The standard of care for many years
HCV treatment has evolved over the past 20 years. Before 2011, the standard of care was a combination of interferon alfa-polyethylene glycol (peg-interferon), given as a weekly injection, and oral ribavirin. Neither drug has specific antiviral activity, and when they are used together they result in a sustained virologic response in fewer than 50% of patients with HCV genotype 1 and, at best, in 70% to 80% of patients with other genotypes.8
Nearly all patients receiving interferon experience side effects, which can be serious. Fatigue and flu-like symptoms are common, and the drug can also cause psychiatric symptoms (including depression or psychosis), weight loss, seizures, peripheral neuropathy, and bone marrow suppression. Ribavirin causes hemolysis and skin complications and is teratogenic.9
An important bit of information to know when using interferon is the patient’s IL28B genotype. This refers to a single-nucleotide polymorphism (C or T) on chromosome 19q13 (rs12979860) upstream of the IL28B gene encoding for interferon lambda-3. It is strongly associated with responsiveness to interferon: patients with the IL28B CC genotype have a much better chance of a sustained virologic response with interferon than do patients with CT or TT.
Boceprevir and telaprevir: First-generation protease inhibitors
In May 2011, the FDA approved the NS3/4A protease inhibitors boceprevir and telaprevir for treating HCV genotype 1, marking the beginning of the era of direct-acting antiviral agents.10 When these drugs are used in combination with peg-interferon alfa and ribavirin, up to 75% of patients with HCV genotype 1 who have had no previous treatment achieve a sustained virologic response.
But despite greatly improving the response rate, these first-generation protease inhibitors have substantial limitations. Twenty-five percent of patients with HCV genotype 1 who have received no previous treatment and 71% of patients who did not respond to previous treatment will not achieve a sustained virologic response with these agents.11 Further, they are effective only against HCV genotype 1, being highly specific for the amino acid target sequence of the NS3 region.
Also, they must be used in combination with interferon alfa and ribavirin because the virus needs to mutate only a little—a few amino-acid substitutions—to gain resistance to them.12 Therefore, patients are still exposed to interferon and ribavirin, with their toxicity. In addition, dysgeusia is seen with boceprevir, rash with telaprevir, and anemia with both.13,14
Finally, serious drug-drug interactions prompted the FDA to impose warnings for the use of these agents with other medications that interact with CYP3A4, the principal enzyme responsible for their metabolism. Thus, these significant adverse effects dampen the enthusiasm of patients contemplating a long course of treatment with these agents.
The need to improve the rate of sustained virologic response, shorten the duration of treatment, avoid serious side effects, improve efficacy in treating patients infected with genotypes other than 1, and, importantly, eliminate the need for interferon alfa and its serious adverse effects have driven the development of new direct-acting antiviral agents, including the two newly FDA-approved drugs, sofosbuvir and simeprevir.
SOFOSBUVIR: A POLYMERASE INHIBITOR
Sofosbuvir is a uridine nucleotide analogue that selectively inhibits the HCV NS5B RNA-dependent RNA polymerase (Figure 1). It targets the highly conserved nucleotide-binding pocket of this enzyme and functions as a chain terminator.15 While the protease inhibitors are genotype-dependent, inhibition of the highly conserved viral polymerase has an impact that spans genotypes.
Early clinical trials of sofosbuvir
Sofosbuvir has been tested in combination with interferon alfa and ribavirin, as well as in interferon-free regimens (Table 2).16–20
Rodriguez-Torres et al,15
- 56% with sofosbuvir 100 mg, peg-interferon, and ribavirin
- 83% with sofosbuvir 200 mg, peg-interferon, and ribavirin
- 80% with sofosbuvir 400 mg, peg-interferon, and ribavirin
- 43% with peg-interferon and ribavirin alone.
The ATOMIC trial16 tested the efficacy and safety of sofosbuvir in combination with peg-interferon and ribavirin in patients with HCV genotype 1, 4, or 6, without cirrhosis, who had not received any previous treatment. Patients with HCV genotype 1 were randomized to three treatments:
- Sofosbuvir 400 mg orally once daily plus peg-interferon and ribavirin for 12 weeks
- The same regimen, but for 24 weeks
- Sofosbuvir plus peg-interferon and ribavirin for 12 weeks, followed by 12 weeks of either sofosbuvir monotherapy or sofosbuvir plus ribavirin.
The rates of sustained virologic response were very high and were not significantly different among the three groups: 89%, 89%, and 87%, respectively. Patients who were able to complete a full course of therapy achieved even higher rates of sustained virologic response, ranging from 96% to 98%. The likelihood of response was not adversely affected by the usual markers of a poorer prognosis, such as a high viral load (≥ 800,000 IU/mL) or a non-CC IL28B genotype. Although patients with cirrhosis (another predictor of no response) were excluded from this study, the presence of bridging fibrosis did not seem to affect the rate of sustained virologic response. The results in patients with genotypes other than 1 were very encouraging, but the small number of patients enrolled precluded drawing firm conclusions in this group.
Important implications of the ATOMIC trial include the following:
There is no benefit in prolonging treatment with sofosbuvir beyond 12 weeks, since adverse events increased without any improvement in the rate of sustained virologic response.
There is a very low likelihood of developing viral resistance or mutation when using sofosbuvir.
There is no role for response-guided therapy, a concept used with protease inhibitor-based regimens in which patients who have complete clearance of the virus within the first 4 weeks of treatment (a rapid virologic response) and remain clear through 12 weeks of treatment (an extended rapid viral response) can be treated for a shorter duration without decreasing the likelihood of a sustained virologic response.
Lawitz et al17 conducted a randomized double-blind phase 2 trial to evaluate the effect of sofosbuvir dosing on response in noncirrhotic, previously untreated patients with HCV genotype 1, 2, or 3. Patients with HCV genotype 1 were randomized to one of three treatment groups in a 2:2:1 ratio: sofosbuvir 200 mg orally once daily, sofosbuvir 400 mg orally once daily, or placebo, all for 12 weeks in combination with peg-interferon (180 μg weekly) and ribavirin in a dosage based on weight. Depending on the viral response, patients continued peg-interferon and ribavirin for an additional 12 weeks if they achieved an extended rapid viral response, or 36 weeks if they did not achieve an extended rapid virologic response, and in all patients who received placebo. Patients with HCV genotype 2 or 3 were given sofosbuvir 400 mg once daily in combination with interferon and ribavirin for 12 weeks.
As in the ATOMIC trial, all patients treated with sofosbuvir had a very rapid reduction in viral load: 98% of patients with genotype 1 developed a rapid virologic response, and therefore almost all were eligible for the shorter treatment course of 24 weeks.17 The latter finding again suggested that response-guided treatment is not relevant with sofosbuvir-based regimens.
Very high rates of sustained virologic response were seen: 90% in patients with genotype 1 treated with sofosbuvir 200 mg, 91% in those with genotype 1 treated with 400 mg, and 92% in those with genotype 2 or 3. Although 6% of patients in the 200-mg group had virologic breakthrough after completing sofosbuvir treatment, no virologic breakthrough was observed in the 400-mg group, suggesting that the 400-mg dose might suppress the virus more effectively.17
The ELECTRON trial18 was a phase 2 study designed to evaluate the efficacy and safety of sofosbuvir and ribavirin in interferon-sparing and interferon-free regimens in patients with HCV genotype 1, 2, or 3 infection. Sofosbuvir was tested with peg-interferon and ribavirin, with ribavirin alone, and as monotherapy in previously untreated patients with genotype 2 or 3. A small number of patients with genotype 1 who were previously untreated and who were previously nonresponders were also treated with sofosbuvir and ribavirin.
All patients had a rapid virologic response, and viral suppression was sustained through the end of treatment. All patients with genotype 2 or 3 treated with double therapy (sofosbuvir and ribavirin) or triple therapy (sofosbuvir, peg-interferon, and ribavirin) achieved a sustained virologic response, compared with only 60% of patients treated with sofosbuvir monotherapy. The monotherapy group had an equal number of relapsers among those with genotype 2 or 3. Of the genotype 1 patients treated with sofosbuvir and ribavirin, 84% of those previously untreated developed a sustained virologic response, whereas only 10% of the previous nonresponders did.
Phase 3 clinical trials of sofosbuvir
The NEUTRINO trial19 studied the efficacy and safety of sofosbuvir in previously untreated patients with HCV genotype 1, 4, 5, or 6. In this phase 3 open-label study, all patients received sofosbuvir plus peg-interferon and weight-based ribavirin therapy for 12 weeks. Of the patients enrolled, 89% had genotype 1, while 9% had genotype 4 and 2% had genotype 5 or 6. Overall, 17% of the patients had cirrhosis.
The viral load rapidly decreased in all patients treated with sofosbuvir irrespective of the HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Ninety-nine percent of patients with genotype 1, 4, 5, or 6 achieved a rapid virologic response, and 90% achieved a sustained virologic response at 12 weeks after completion of treatment with sofosbuvir and ribavirin. Patients with cirrhosis had a slightly lower rate of sustained virologic response (80%, compared with 92% in patients without cirrhosis). Also, patients with non-CC IL28B genotypes had a lower rate of sustained virologic response (87% in non-CC allele vs 98% in patients with the favorable CC allele).
The FISSION trial19 recruited previously untreated patients with genotype 2 or 3 and randomized them to therapy with either sofosbuvir plus ribavirin in a weight-based dose for 12 weeks, or 24 weeks of interferon and ribavirin. In this study, 20% of patients in each treatment group had cirrhosis.
As in the NEUTRINO trial, the viral load rapidly decreased in all patients treated with sofosbuvir irrespective of HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Here, 100% of patients with genotype 2 or 3 who were treated with sofosbuvir and ribavirin achieved a rapid virologic response. Differences in outcome emerged based on genotype: 97% of those with genotype 2 and 56% of those with genotype 3 achieved a sustained virologic response. The overall rate was 67%, which was not different from patients treated with peg-interferon and ribavirin. In the subgroup of patients with cirrhosis, 47% of those treated with sofosbuvir and ribavirin achieved a sustained virologic response, vs 38% of those who received peg-interferon plus ribavirin.
In both the NEUTRINO and FISSION trials, few patients discontinued treatment, with higher rates of most adverse events occurring in patients treated with peg-interferon and ribavirin.
POSITRON,20 a phase 3 clinical trial, tested sofosbuvir in patients with HCV genotype 2 or 3 who were ineligible for peg-interferon, unwilling to take peg-interferon, or unable to tolerate peg-interferon (mainly because of clinically significant psychiatric disorders). Patients were randomized to two treatment groups for 12 weeks: sofosbuvir plus ribavirin, or placebo. About 50% of patients had HCV genotype 3, and 16% had cirrhosis.
The overall rate of sustained virologic response at 12 weeks after treatment was 78% in the sofosbuvir-and-ribavirin group (93% in genotype 2 patients and 61% in genotype 3 patients). Again, cirrhosis was associated with a lower rate of sustained virologic response (61% of patients with cirrhosis achieved a sustained virologic response vs 81% of patients without cirrhosis). None of the sofosbuvir-treated patients had virologic failure while on treatment.
FUSION,20 another phase 3 trial, evaluated sofosbuvir in patients infected with HCV genotype 2 or 3 for whom interferon-based treatment had failed. They were randomized to either 12 weeks or 16 weeks of sofosbuvir and weight-based ribavirin treatment. About 60% of patients had HCV genotype 3, and 34% had cirrhosis.
The overall sustained virologic response rate was 50% in the patients treated for 12 weeks and 73% in those treated for 16 weeks: specifically, 86% of patients with genotype 2 achieved a sustained virologic response at 12 weeks and 94% at 16 weeks, whereas in those with genotype 3 the rates were 30% at 12 weeks and 62% at 16 weeks.
Cirrhosis was again a predictor of lack of response to sofosbuvir. In the group treated for 12 weeks, 31% of those with cirrhosis achieved a sustained virologic response compared with 61% in those without cirrhosis. In the group treated for 16 weeks, 61% of those with cirrhosis achieved a sustained virologic response compared with 76% in those without cirrhosis.
In both the POSITRON and FUSION trials, relapse accounted for all treatment failures, and no virologic resistance was detected in patients who did not have a sustained virologic response. The investigators concluded that 12 weeks of treatment with sofosbuvir and ribavirin can be effective for HCV genotype 2 infection, but extending the treatment to 16 weeks may be beneficial for genotype 3. This may be especially important in patients with cirrhosis or those who did not have a response to peg-interferon-based treatment.
VALENCE,21 an ongoing phase 3 trial in Europe, is assessing the safety and efficacy of sofosbuvir 400 mg once daily and weight-based ribavirin in patients with HCV genotype 2 or 3. Eighty-five percent of the trial participants have received previous treatment, and 21% have cirrhosis. Patients were originally randomized in a 4:1 ratio to receive sofosbuvir plus ribavirin for 12 weeks or matching placebo, but as a result of emerging data suggesting that patients with genotype 3 would benefit from more than 12 weeks of treatment, the study was subsequently amended to extend treatment to 24 weeks for patients with genotype 3.
Overall rates of sustained virologic response were 93% in patients with genotype 2 and 85% in patients with genotype 3. In previously treated patients with genotype 2 who were treated for 12 weeks, the rates of sustained virologic response were 91% in those without cirrhosis vs 88% in those with cirrhosis. In previously treated patients with genotype 3, the rates in those treated for 24 weeks were 87% in patients without cirrhosis vs 60% with cirrhosis. The safety profile was consistent with that of ribavirin.
Side effects of sofosbuvir
In clinical trials, side effects occurred most often when sofosbuvir was combined with interferon and ribavirin and were consistent with the known side effects of the latter two agents. The most frequently reported side effects included fatigue, insomnia, nausea, rash, anemia, headache, and arthralgia, with most of these adverse events rated by treating clinicians as being mild in severity.15,20
In the ATOMIC trial, the most common events leading to drug discontinuation were anemia and neutropenia, both associated with interferon and ribavirin. Patients receiving sofosbuvir monotherapy after 12 weeks of triple therapy showed rapid improvement in hemoglobin levels and neutrophil counts, indicating that hematologic abnormalities attributed solely to sofosbuvir are minimal. In the FISSION trial, the incidence of adverse events was consistently lower in those receiving sofosbuvir-ribavirin than in patients receiving interferon-ribavirin without sofosbuvir.19
In the POSITRON trial, discontinuation of sofosbuvir because of adverse events was uncommon, and there were no differences in the incidence of adverse events and laboratory abnormalities between patients with and without cirrhosis when they received sofosbuvir and ribavirin.20
Sofosbuvir dosage and indications
Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3 and in combination with ribavirin and interferon alfa in patients infected with HCV genotype 1 or 4 (Table 3). It could be considered for HCV genotype 1 in combination with ribavirin alone for 24 weeks in patients who are ineligible for interferon.
Sofosbuvir is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation, for up to 48 weeks or until they receive a transplant, to prevent posttransplant reinfection with HCV.
Sofosbuvir is expensive
A course of therapy is expected to cost about $84,000, which is significantly more than the cost of previous triple therapy (peg-interferon, ribavirin, and either boceprevir or telaprevir).22 This high cost will undoubtedly lead to less widespread use in developing countries, and potentially even in the United States. As newer direct-acting antiviral agents become available, the price will likely come down, enhancing access to these drugs.
SIMEPREVIR: A SECOND-GENERATION PROTEASE INHIBITOR
Telaprevir and boceprevir are NS3/A4 protease inhibitors that belong to the alfa-ketoamid derivative class. Simeprevir belongs to the macrocyclic class and has a different way of binding to the target enzyme.23 Like sofosbuvir, simeprevir was recently approved by the FDA for the treatment of HCV genotype 1.
The therapeutic efficacy of simeprevir has been tested in several clinical trials (Table 4), including QUEST-124 and QUEST-225 (in previously untreated patients), PROMISE26 (in prior relapsers), and ASPIRE27 (in prior partial and null responders). Results from these trials showed high overall rates of sustained virologic response with triple therapy (ie, simeprevir combined with peg-interferon and ribavirin). It was generally well tolerated, and most adverse events reported during 12 weeks of treatment were of mild to moderate severity.
In QUEST-1 and QUEST-2, both double-blind phase 3 clinical trials, previously untreated patients infected with HCV genotype 1 were randomized in a 2:1 ratio to receive either simeprevir 150 mg daily or placebo for 12 weeks; both groups also received peg-interferon and ribavirin. Patients then received peg-interferon and ribavirin alone for 12 or 36 weeks in the simeprevir group (based on response) and for 36 weeks in the placebo group.
The overall rate of sustained virologic response at 12 weeks was 80% in the simeprevir group (75% in those with genotype 1a and 85% in those with genotype 1b) vs 50% in the placebo group (receiving peg-interferon and ribavirin alone).24,25
PROMISE,26 another double-blind randomized phase 3 clinical trial, evaluated simeprevir in patients with HCV genotype 1 who relapsed after previous interferon-based therapy. It had a similar design to QUEST-1 and QUEST-2, and 15% of all patients had cirrhosis.
The overall sustained virologic response rate at 12 weeks after treatment was 79% in the simeprevir group (70% in patients with genotype 1a and 86% in those with genotype 1b) vs 37% in the placebo group. Rates were similar in patients with absent to moderate fibrosis (82%), advanced fibrosis (73%), or cirrhosis (74%).
ASPIRE.27 Simeprevir efficacy in patients with HCV genotype 1 for whom previous therapy with peg-interferon and ribavirin had failed was tested in ASPIRE, a double-blind randomized phase 2 clinical trial. Patients were randomized to receive simeprevir (either 100 mg or 150 mg daily) for 12, 24, or 48 weeks in combination with 48 weeks of peg-interferon and ribavirin, or placebo plus peg-interferon and ribavirin for 48 weeks.
The primary end point was the rate of sustained virologic response at 24 weeks. Overall, rates were 61% to 80% for the simeprevir treatment groups compared with 23% with placebo, regardless of prior response to peg-interferon and ribavirin. By subgroup, rates were:
- 77% to 89% with simeprevir vs 37% with placebo in prior relapsers
- 48% to 86% with simeprevir vs 9% with placebo in prior partial responders
- 38% to 59% with placebo vs 19% for prior nonresponders.
The best rates of sustained viral response at 24 weeks were in the groups that received simeprevir 150 mg daily: 85% in prior relapsers, 75% in prior partial responders, and 51% in prior nonresponders.
Simeprevir vs other direct-acting antiviral drugs
Advantages of simeprevir over the earlier protease inhibitors include once-daily dosing, a lower rate of adverse events (the most common being fatigue, headache, rash, photosensitivity, and pruritus), a lower likelihood of discontinuation because of adverse events, and fewer drug-drug interactions (since it is a weak inhibitor of the CYP3A4 enzyme).
Unlike sofosbuvir, simeprevir was FDA-approved only for HCV genotype 1 and in combination with interferon alfa and ribavirin. Compared with sofosbuvir, the treatment duration with simeprevir regimens is longer overall (interferon alfa and ribavirin are given for 12 weeks in sofosbuvir-based regimens vs 24 to 48 weeks with simeprevir). As with sofosbuvir, the estimated cost of simeprevir is high, about $66,000 for a 12-week course.
Simeprevir dosage and indications
Simeprevir was approved at an oral dose of 150 mg once daily in combination with ribavirin and interferon alfa in patients with HCV genotype 1 (Table 5).
The approved regimens for simeprevir are fixed in total duration based on the patient’s treatment history. Specifically, all patients receive the drug in combination with peg-interferon and ribavirin for 12 weeks. Then, previously untreated patients and prior relapsers continue to receive peg-interferon and ribavirin alone for another 12 weeks, and those with a partial or null response continue with these drugs for another 36 weeks.
Patients infected with HCV genotype 1a should be screened for the NS3 Q80K polymorphism at baseline, as it has been associated with substantially reduced response to simeprevir.
Sofosbuvir and simeprevir in combination
The COSMOS trial.28 Given their differences in mechanism of action, sofosbuvir and simeprevir are being tested in combination. The COSMOS trial is an ongoing phase 2 randomized open-label study investigating the efficacy and safety of simeprevir and sofosbuvir in combination with and without ribavirin in patients with HCV genotype 1, including nonresponders and those with cirrhosis. Early results are promising, with very high rates of sustained virologic response with the sofosbuvir-simeprevir combination (93% to 100%) and indicate that the addition of ribavirin might not be needed to achieve sustained virologic response in this patient population.
THE FUTURE
The emergence of all-oral regimens for HCV treatment with increasingly sophisticated agents such as sofosbuvir and simeprevir will dramatically alter the management of HCV patients. In view of the improvement in sustained virologic response rates with these treatments, and since most HCV-infected persons have no symptoms, the US Centers for Disease Control and Prevention29 recently recommended one-time testing of the cohort in which the prevalence of HCV infection is highest: all persons born between 1945 and 1965. This undoubtedly will increase the detection of this infection—and the number of new patients expecting treatment.
Future drugs promise further improvements (Table 6).30–35 Advances in knowledge of the HCV molecular structure have led to the development of numerous direct-acting antiviral agents with very specific viral targets. A second wave of protease inhibitors and of nucleoside and nonnucleoside polymerase inhibitors will soon be available. Inhibitors of NS5A (a protein important in the assembly of the viral replication complex) such as daclatasvir and ledipasvir, are currently in phase 3 clinical trials. Other viral proteins involved in assembly of the virus, including the core protein and p7, are being explored as drug targets. In addition, inhibiting host targets such as cyclophilin A and miR122 has gained traction recently, with specific agents currently in phase 2 and 3 clinical trials.
Factors that previously were major determinants of response to treatment, such as IL28B genotype, viral load, race, age, extent of fibrosis, and genotype 1 subtypes, will become much less important with the introduction of highly potent direct-acting antiviral agents.
Many all-oral combinations are being evaluated in clinical trials. For example, the open-label, phase 2 LONESTAR trial tested the utility of combining sofosbuvir and ledipasvir (an NS5A inhibitor) with and without ribavirin for 8 or 12 weeks in previously untreated patients with HCV genotype 1, and for 12 weeks in patients with HCV genotype 1 who did not achieve a sustained virologic response after receiving a protease inhibitor-based regimen (half of whom had compensated cirrhosis).36 Sustained virologic response rates were very high (95% to 100%) in both previously treated and previously untreated patients, including those with cirrhosis. Similar rates were achieved by the 8-week and 12-week groups in noncirrhotic patients who had not been previously treated for HCV. The typical hematologic abnormalities associated with interferon were not observed except for mild anemia in patients who received ribavirin. These results suggest that the combination of sofosbuvir and ledipasvir could offer a very effective, short, all-oral treatment for patients with HCV genotype 1, including those with cirrhosis, who up to now have been difficult to treat.
Challenges remaining
The success of sofosbuvir and simeprevir paves the way for interferon-free regimens.37 For a long time, the treatment of HCV infection required close monitoring of patients while managing the side effects of interferon, but the current and emerging direct-acting antiviral agents will soon change this practice. Given the synergistic effects of combination therapy—targeting the virus at multiple locations, decreasing the likelihood of drug resistance, and improving efficacy—combination regimens seem to be the optimal solution to the HCV epidemic. Lower risk of side effects and shorter treatment duration will definitely improve the acceptance of any new regimen. New agents that act against conserved viral targets, thereby yielding activity across multiple genotypes, will be advantageous as well. Table 7 compares the rates of sustained virologic response of the different currently approved HCV treatment regimens.
Clinical challenges remain, including the management of special patient populations for whom data are still limited. These include patients with cirrhosis, chronic kidney disease, renal failure, and concurrent infection with human immunodeficiency virus, and patients who have undergone solid organ transplantation. Clinical trials are under way to evaluate the treatment options for these patients, who will likely need to wait for the emergence of additional agents before dramatic improvement in sustained virologic response rates may be expected.38
As the treatment of HCV becomes simpler, safer, and more effective, primary care physicians will increasingly be expected to manage it. Difficult-to-treat patients, including the special populations above, will require specialist management and individualized treatment regimens, at least until better therapies are available. The high projected cost of the new agents may limit access, at least initially. However, the dramatic improvement in sustained virologic response rates and all that that implies in terms of decreased risk of advanced liver disease and its complications will undoubtedly make these therapies cost-effective.39
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the united states. Clin Infect Dis 2012; 55(suppl 1):S10–S15.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first international liver transplantation society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transplant 2003; 9:S1–S9.
- Wong JB, McQuillan GM, McHutchison JG, Poynard T. Estimating future hepatitis C morbidity, mortality, and costs in the United States. Am J Public Health 2000; 90:1562–1569.
- Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132:1979–1998.
- Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol 2000; 81:1631–1648.
- Singal AG, Volk ML, Jensen D, Di Bisceglie AM, Schoenfeld PS. A sustained viral response is associated with reduced liver-related morbidity and mortality in patients with hepatitis C virus. Clin Gastroenterol Hepatol 2010; 8:280–288,288.e1.
- Camma C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Paeshuyse J, Dallmeier K, Neyts J. Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol 2011; 1:590–598.
- Thomas E, Ghany MG, Liang TJ. The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir Chem Chemother 2012; 23:1–12.
- Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB; American Association for Study of Liver Diseases. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54:1433–1444.
- Soriano V, Vispo E, Poveda E, Labarga P, Barreiro P. Treatment failure with new hepatitis C drugs. Expert Opin Pharmacother 2012; 13:313–323.
- Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int 2013; 33(suppl 1):93–104.
- Manns MP, McCone J, Davis MN, et al. Overall safety profile of boceprevir plus peginterferon alfa-2b and ribavirin in patients with chronic hepatitis C genotype 1: a combined analysis of 3 phase 2/3 clinical trials. Liver Int 2013; Aug 2. doi: 10.1111/liv.12300. [Epub ahead of print]
- Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Rodriguez-Torres M, Lawitz E, Kowdley KV, et al. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: a randomized, 28-day, dose-ranging trial. J Hepatol 2013; 58:663–668.
- Kowdley KV, Lawitz E, Crespo I, et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 2013; 381:2100–2107.
- Lawitz E, Lalezari JP, Hassanein T, et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis 2013; 13:401–408.
- Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 2013; 368:34–44.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Jacobson IM, Gordon SC, Kowdley KV, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 2013; 368:1867–1877.
- Zeuzem S, Dusheiko G, Salupere R, et al. Sofosbuvir + ribavirin for 12 or 24 weeks for patients with HCV genotype 2 or 3: the VALENCE trial [abstract no.1085]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- You DM, Pockros PJ. Simeprevir for the treatment of chronic hepatitis C. Expert Opin Pharmacother 2013; 14:2581–2589.
- Jacobson IM, Dore GJ, Foster G, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive patients: results from Quest-1, a phase III trial [abstract no. 1425]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, Netherlands.
- Manns M, Marcellin P, Poordad FP, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naïve patients: results from QUEST-2, a phase III trial [abstract no. 1413]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, The Netherlands.
- Lawitz E, Forns X, Zeuzem S, et al. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in patients who relapsed after previous interferon-based therapy: results from promise, a phase III trial [abstract no. 869b]. Digestive Disease Week; May 18–21, 2013; Orlando, FL.
- Zeuzem S, Berg T, Gane E, et al. Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology epub Oct 31, 2013.
- Jacobson IM, Ghalib RM, Rodriguez-Torres M, et al. SVR results of a once-daily regimen of simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naive and prior null responder patients: the COSMOS study [abstract LB-3]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Smith BD, Morgan RL, Beckett GA, et al. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm Rep 2012; 61( RR-4):1–32.
- Sulkowski MS, Kang M, Matining R, et al. Safety and antiviral activity of the HCV entry inhibitor ITX5061 in treatment-naive HCV-infected adults: a randomized, double-blind, phase 1b study. J Infect Dis 2013 Oct 9. [Epub ahead of print]
- Pawlotsky JM. NS5A inhibitors in the treatment of hepatitis C. J Hepatol 2013; 59:375–382.
- Yu M, Corsa AC, Xu S, et al. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antiviral Res 2013; 100:439–445.
- Aghemo A, De Francesco R. New horizons in hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 2013; 58:428–438.
- Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med 2013; 368:1907–1917.
- Flisiak R, Jaroszewicz J, Parfieniuk-Kowerda A. Emerging treatments for hepatitis C. Expert Opin Emerg Drugs 2013; 18:461–475.
- Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 2013 Nov 1. doi: 10.1016/S0140-6736(13)62121-2 [Epub ahead of print]
- Drenth JP. HCV treatment—no more room for interferonologists? N Engl J Med 2013; 368:1931–1932.
- Casey LC, Lee WM. Hepatitis C virus therapy update 2013. Curr Opin Gastroenterol 2013; 29:243–249.
- Afdhal NH, Zeuzem S, Schooley RT, et al. The new paradigm of hepatitis C therapy: integration of oral therapies into best practices. J Viral Hepatol 2013; 20:745–760.
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the united states. Clin Infect Dis 2012; 55(suppl 1):S10–S15.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first international liver transplantation society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transplant 2003; 9:S1–S9.
- Wong JB, McQuillan GM, McHutchison JG, Poynard T. Estimating future hepatitis C morbidity, mortality, and costs in the United States. Am J Public Health 2000; 90:1562–1569.
- Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132:1979–1998.
- Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol 2000; 81:1631–1648.
- Singal AG, Volk ML, Jensen D, Di Bisceglie AM, Schoenfeld PS. A sustained viral response is associated with reduced liver-related morbidity and mortality in patients with hepatitis C virus. Clin Gastroenterol Hepatol 2010; 8:280–288,288.e1.
- Camma C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Paeshuyse J, Dallmeier K, Neyts J. Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol 2011; 1:590–598.
- Thomas E, Ghany MG, Liang TJ. The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir Chem Chemother 2012; 23:1–12.
- Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB; American Association for Study of Liver Diseases. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54:1433–1444.
- Soriano V, Vispo E, Poveda E, Labarga P, Barreiro P. Treatment failure with new hepatitis C drugs. Expert Opin Pharmacother 2012; 13:313–323.
- Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int 2013; 33(suppl 1):93–104.
- Manns MP, McCone J, Davis MN, et al. Overall safety profile of boceprevir plus peginterferon alfa-2b and ribavirin in patients with chronic hepatitis C genotype 1: a combined analysis of 3 phase 2/3 clinical trials. Liver Int 2013; Aug 2. doi: 10.1111/liv.12300. [Epub ahead of print]
- Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Rodriguez-Torres M, Lawitz E, Kowdley KV, et al. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: a randomized, 28-day, dose-ranging trial. J Hepatol 2013; 58:663–668.
- Kowdley KV, Lawitz E, Crespo I, et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 2013; 381:2100–2107.
- Lawitz E, Lalezari JP, Hassanein T, et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis 2013; 13:401–408.
- Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 2013; 368:34–44.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Jacobson IM, Gordon SC, Kowdley KV, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 2013; 368:1867–1877.
- Zeuzem S, Dusheiko G, Salupere R, et al. Sofosbuvir + ribavirin for 12 or 24 weeks for patients with HCV genotype 2 or 3: the VALENCE trial [abstract no.1085]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- You DM, Pockros PJ. Simeprevir for the treatment of chronic hepatitis C. Expert Opin Pharmacother 2013; 14:2581–2589.
- Jacobson IM, Dore GJ, Foster G, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive patients: results from Quest-1, a phase III trial [abstract no. 1425]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, Netherlands.
- Manns M, Marcellin P, Poordad FP, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naïve patients: results from QUEST-2, a phase III trial [abstract no. 1413]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, The Netherlands.
- Lawitz E, Forns X, Zeuzem S, et al. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in patients who relapsed after previous interferon-based therapy: results from promise, a phase III trial [abstract no. 869b]. Digestive Disease Week; May 18–21, 2013; Orlando, FL.
- Zeuzem S, Berg T, Gane E, et al. Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology epub Oct 31, 2013.
- Jacobson IM, Ghalib RM, Rodriguez-Torres M, et al. SVR results of a once-daily regimen of simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naive and prior null responder patients: the COSMOS study [abstract LB-3]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Smith BD, Morgan RL, Beckett GA, et al. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm Rep 2012; 61( RR-4):1–32.
- Sulkowski MS, Kang M, Matining R, et al. Safety and antiviral activity of the HCV entry inhibitor ITX5061 in treatment-naive HCV-infected adults: a randomized, double-blind, phase 1b study. J Infect Dis 2013 Oct 9. [Epub ahead of print]
- Pawlotsky JM. NS5A inhibitors in the treatment of hepatitis C. J Hepatol 2013; 59:375–382.
- Yu M, Corsa AC, Xu S, et al. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antiviral Res 2013; 100:439–445.
- Aghemo A, De Francesco R. New horizons in hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 2013; 58:428–438.
- Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med 2013; 368:1907–1917.
- Flisiak R, Jaroszewicz J, Parfieniuk-Kowerda A. Emerging treatments for hepatitis C. Expert Opin Emerg Drugs 2013; 18:461–475.
- Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 2013 Nov 1. doi: 10.1016/S0140-6736(13)62121-2 [Epub ahead of print]
- Drenth JP. HCV treatment—no more room for interferonologists? N Engl J Med 2013; 368:1931–1932.
- Casey LC, Lee WM. Hepatitis C virus therapy update 2013. Curr Opin Gastroenterol 2013; 29:243–249.
- Afdhal NH, Zeuzem S, Schooley RT, et al. The new paradigm of hepatitis C therapy: integration of oral therapies into best practices. J Viral Hepatol 2013; 20:745–760.
KEY POINTS
- In clinical trials of treatment for chronic HCV infection, regimens that included a direct-acting antiviral agent were more effective than ones that did not.
- Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3, and in combination with ribavirin and interferon in patients infected with HCV genotype 1 or 4. It is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation.
- Simeprevir is approved in an oral dose of 150 mg once daily in combination with ribavirin and interferon for patients with HCV genotype 1.
- The new drugs are expensive, a potential barrier for many patients. As more direct-acting antiviral agents become available, their cost will likely decrease.
- Combinations of direct-acting antiviral agents of different classes may prove even more effective and could eliminate the need for interferon entirely.
Canagliflozin: Improving diabetes by making urine sweet
Glycosuria used to be a sign of uncontrolled diabetes and was something to be corrected, not a therapeutic mechanism. But now we have a new class of drugs that lower plasma glucose levels by increasing the renal excretion of glucose.
Here, we will review canagliflozin, the first in a new class of drugs for type 2 diabetes: how it works, who is a candidate for it, and what to watch out for.
THE NEED FOR NEW DIABETES DRUGS
Diabetes mellitus affects more than 25.8 million people in the United States—8.3% of the population—and this staggering number is rising.1 Among US residents age 65 and older, more than 10.9 million (26.9%) have diabetes.1 People with uncontrolled diabetes are at risk of microvascular complications such as retinopathy, nephropathy, and neuropathy, as well as cardiovascular disease. Diabetes is the leading cause of blindness, chronic kidney disease, and nontraumatic lower-limb amputation in the United States.1
Type 2 diabetes accounts for more than 90% of cases of diabetes in the United States, Europe, and Canada.2 It is characterized by insulin resistance, decreased beta-cell function, and progressive beta-cell decline.3
Current American Diabetes Association guidelines for the treatment of diabetes recommend a hemoglobin A1c target of less than 7.0%.4 Initial management includes lifestyle modifications such as changes in diet and an increase in exercise, as well as consideration of metformin treatment at the same time. If glucose levels remain uncontrolled despite these efforts, other drugs should be added.

A number of oral and injectable antihyperglycemic drugs are available to help achieve this goal, though none is without risk of adverse effects. Those available up to now include metformin, sulfonylureas, meglitinides, alpha-glucosidase inhibitors, thiazolidinediones, gliptins, glucagon-like peptide-1 agonists, amylin analogues, colesevelam, dopamine agonists, and insulin.5 Most of the available antihyperglycemics target the liver, pancreas, gut, and muscle to improve insulin sensitivity, reduce insulin resistance, or stimulate insulin secretion.
Despite the abundance of agents, type 2 diabetes remains uncontrolled in many patients. Only 57.1% of participants with previously diagnosed diabetes in the 2003–2006 National Health and Nutrition Examination Survey were at the hemoglobin A1c goal of less than 7.0%.6 Possible reasons for failure include adverse effects such as hypoglycemia, weight gain, and gastrointestinal symptoms resulting in discontinued use, nonadherence to the prescribed regimen, and failure to increase the dosage or to add additional agents, including insulin, to optimize glycemic control as beta-cell function declines over time.
HOW THE KIDNEYS HANDLE GLUCOSE
In the kidney, glucose is filtered in the glomerulus and then is reabsorbed in the proximal tubule. Normally, the filtered glucose is all reabsorbed unless the glucose load exceeds the kidney’s absorptive capacity. Membrane proteins called sodium-glucose cotransporters reabsorb glucose at the proximal tubule and return it into the peripheral circulation. Glucose enters the tubular epithelial cell with sodium by passive cotransport via the sodiumglucose cotransporters, and then exits on the other side via the glucose transporter GLUT in the basolateral membrane.
Two sodium-glucose transporters that act in the proximal tubule of the kidney have been identified: SGLT1 and SGLT2. SGLT2 reabsorbs most of the glucose in the early segment of the proximal tubule, while SLGT1 reabsorbs the remaining glucose at the distal end.7 SGLT2 is responsible for more than 90% of renal tubular reabsorption of glucose and is found only in the proximal tubule, whereas SGLT1 is found mainly in the gastrointestinal tract.8
Patients with type 2 diabetes have a higher capacity for glucose reabsorption in the proximal tubule as a result of the up-regulation of SGLT2.9
SGLT2 INHIBITORS AND TYPE 2 DIABETES
Drugs that inhibit SGLT2 block reabsorption of glucose in the proximal tubule, lowering the renal threshold for glucose and thereby increasing urinary glucose excretion and lowering the serum glucose level in patients with hyperglycemia. This mechanism of action is insulin-independent.
On March 29, 2013, canagliflozin became the first SGLT2 inhibitor to be approved in the United States for the treatment of type 2 diabetes.10 However, it is not the first of its class to be introduced.
Dapagliflozin was the first SGLT2 inhibitor approved in Europe and has been available there since November 2012. However, the US Food and Drug Administration withheld its approval in the United States in January 2012 because of concerns of a possible association with cancer, specifically breast and bladder cancers, as well as possible liver injury.10 Canagliflozin does not appear to share this risk.
Several other SGLT2 inhibitors may soon be available. Empagliflozin is in phase III trials, and the manufacturer has filed for approval in the United States. Ipragliflozin is awaiting approval in Japan.
CANAGLIFLOZIN: PHARMACOKINETICS AND THERAPEUTIC EFFICACY
Canagliflozin reaches its peak plasma concentration within 1 to 2 hours of oral administration.11 Its half-life is 10.6 hours with a 100-mg dose and 13.1 hours with a 300-mg dose. A steady state is typically achieved in 4 to 5 days.11
Canagliflozin lowers fasting plasma glucose and hemoglobin A1c levels in a dose-dependent manner.10,11 These effects are independent of age, sex, body mass index, and race.12 Postprandial glucose levels are also lowered.
Other potential benefits of canagliflozin include lowering of the systolic blood pressure and, especially important in obese people with type 2 diabetes, weight loss.12 Aside from metformin, which occasionally results in modest weight loss, other oral drugs used in treating type 2 diabetes are weight-neutral or can cause weight gain.
Trials of canagliflozin
Nine phase III trials of canagliflozin have enrolled 10,285 patients, in one of the largest clinical trial programs in type 2 diabetes to date.10 Several of these trials evaluated canagliflozin as monotherapy, whereas others assessed its effect as an add-on therapy in combination with another antihyperglycemic agent such as a sulfonylurea, metformin, pioglitazone, or insulin. There has not yet been a trial directly comparing canagliflozin with metformin.
Four of the placebo-controlled trials evaluated canagliflozin as monotherapy, canagliflozin added to metformin alone, canagliflozin added to metformin plus glimepiride, and canagliflozin added to metformin plus pioglitazone.
When canagliflozin was used as monotherapy, hemoglobin A1c levels at 26 weeks were an absolute 0.91% lower in the canagliflozin 100 mg/day group than in the placebo group, and an absolute 1.16% lower in the canagliflozin 300 mg/day group than in the placebo group (P < .001 for both).12 Patients lost 2.8% of their body weight with canagliflozin 100 mg and 3.3% with canagliflozin 300 mg, compared with 0.6% with placebo. Systolic blood pressure fell by a mean of 3.7 mm Hg with the 100-mg dose and by a mean of 5.4 mm Hg with the 300-mg dose compared with placebo (P < .001 for both dose groups).12
When canagliflozin was added to metformin, with glimepiride as the comparator drug, there was a 5.2% weight reduction with the 100-mg dose, a 5.7% reduction with 300 mg, and a 1% gain with glimepiride. Hemoglobin A1c fell about equally in the three groups.11
When canagliflozin was added to metformin and a sulfonylurea, with sitagliptin as the comparator third drug, the 300-mg canagliflozin dosage group had a 2.8% weight reduction.11
WHAT ARE THE ADVERSE EFFECTS?
Overall, canagliflozin seems to be well tolerated. The most common adverse effects reported in the clinical trials were genital yeast infections, urinary tract infections, and increased urination.
Genital yeast infections were more common in women than in men, occurring in 10.4% of women who received canagliflozin 100 mg and in 11.4% of women who received 300 mg, compared with only 3.2% in the placebo group.11
Urinary tract infections occurred in 5.9% of the 100-mg group and in 4.3% of the 300-mg group, compared with 4.0% of the placebo group.11
Postural hypotension. Lowering of blood pressure and symptoms of postural hypotension were also reported, and these may be attributed to the drug’s mild osmotic diuretic effect. The risk of adverse effects of volume depletion was dose-dependent; in patients over age 75, they occurred in 4.9% of those taking 100 mg and in 8.7% of those taking 300 mg, compared with 2.6% of those in the placebo or active-comparator groups.11 Therefore, one should exercise particular caution when starting this drug in the elderly or in patients taking diuretics or multiple antihypertensive drugs.
Hypoglycemia. When canagliflozin was used as monotherapy, the incidence of hypoglycemia over 26 weeks was similar to that with placebo, occurring in 3.6% of the 100-mg group, 3.0% of the 300-mg group, and 2.6% of the placebo group.12 Canagliflozin was associated with fewer episodes of hypoglycemia than were sulfonylureas, and the number of episodes was similar to that in patients taking gliptins. There was a higher overall incidence of hypoglycemia when canagliflozin was used in combination with a sulfonylurea or with insulin than when it was used as monotherapy.11
Hyperkalemia. Patients with moderate renal impairment or who are on potassiumsparing drugs or drugs that interfere with the renin-angiotensin-aldosterone system may be at higher risk of hyperkalemia, so close monitoring of potassium is recommended. There was also a dose-dependent increase in serum phosphate and magnesium levels, more notably in patients with moderate renal impairment within the first 3 weeks of starting the drug.11
Patients on canagliflozin who are also taking digoxin, ritonavir, phenytoin, phenobarbital, or rifampin should be closely monitored because of the risk of drug-drug interactions.11 Specifically, there was an increase in mean peak digoxin concentrations when used with canagliflozin 300 mg, and the use of phenytoin, phenobarbital, and ritonavir decreased the efficacy of canagliflozin.
WHAT ARE THE CARDIOVASCULAR RISKS OR LONG-TERM CONCERNS?
Dose-dependent increases in low-density lipoprotein cholesterol (LDL-C) may be seen with canagliflozin. Mean changes from baseline compared with placebo were 4.4 mg/dL (4.5%) with canagliflozin 100 mg and 8.3 mg/dL (8%) with canagliflozin 300 mg.11
There was also an increase in non-high-density lipoprotein cholesterol (non-HDL-C).12 Compared with placebo, mean non-HDL-C levels rose by 2.1 mg/dL (1.5%) with canagliflozin 100 mg and 5.1 mg/dL (3.6%) with 300 mg.11
In the 26-week canagliflozin monotherapy trial, archived blood samples in a small subgroup of patients (n = 349) were measured for apolipoprotein-B, which was found to increase by 1.2% with canagliflozin 100 mg and 3.5% with canagliflozin 300 mg, compared with 0.9% in the placebo group.12
Although small, the increase in LDL-C seen with this drug could be a concern, as diabetic patients are already at higher risk of cardiovascular events. The mechanism of this increase is not yet known, though it may be related to metabolic changes from urinary glucose excretion.12
The Canagliflozin Cardiovascular Assessment Study (CANVAS) is a randomized placebo-controlled trial in more than 4,000 patients with type 2 diabetes who have a history of or are at high risk of cardiovascular events. Currently under way, it is evaluating the occurrence of major adverse cardiovascular events (the primary end point) in patients randomized to receive canagliflozin 100 mg, canagliflozin 300 mg, or placebo once daily for up to 4 years. Secondary end points will be the drug’s effects on fasting plasma insulin and glucose, progression of albuminuria, body weight, blood pressure, HDL-C, LDL-C, bone mineral density, markers of bone turnover, and body composition.10 This trial will run for 9 years, to be completed in 2018.13
The CANVAS investigators have already reported that within the first month of treatment, 13 patients taking canagliflozin suffered a major cardiovascular event, including stroke (one of which was fatal) compared with just one patient taking placebo. These events were not seen after the first month. The hazard ratio for major adverse cardiovascular events within the first 30 days was 6.49, but this dropped to 0.89 after the first 30 days.10
Additional issues that should be addressed in long-term postmarketing studies include possible relationships with cancers and pancreatitis and the safety of the drug in pregnancy and in children with diabetes.10
WHO IS A CANDIDATE FOR THIS DRUG?
Canagliflozin is approved for use as monotherapy in addition to lifestyle modifications. It is also approved for use with other antihyperglycemic drugs, including metformin.
Obese patients with type 2 diabetes and normal kidney function may have the greatest benefit. Because of canagliflozin’s insulinin-dependent mechanism of action, patients with both early and late type 2 diabetes may benefit from its ability to lower hemoglobin A1c and blood glucose.14
Although it can be used in patients with moderate (but not severe) kidney disease, canagliflozin does not appear to be as effective in these patients, who had higher rates of adverse effects.11 It is not indicated for patients with type 1 diabetes, type 2 diabetes with ketonuria, or end-stage renal disease (estimated glomerular filtration rate < 45 mL/min or receiving dialysis).11 It also is not yet recommended for use in pregnant women or patients under age 18.
The recommended starting dose of canagliflozin is 100 mg once daily, taken with breakfast. This can be increased to 300 mg once daily if tolerated. However, patients with an estimated glomerular filtration rate of 45 to 60 mL/min should not exceed the 100- mg dose. No dose adjustment is required in patients with mild to moderate hepatic impairment. It is not recommended, however, in patients with severe hepatic impairment.11
Comment. Although canagliflozin is approved as monotherapy, metformin remains my choice for first-line oral therapy. Because canagliflozin is more expensive and its long-term affects are still relatively unknown, I prefer to use it as an adjunct, and believe it will be a useful addition, especially in obese patients who are seeking to lose weight.
WHAT IS THE COST OF THIS DRUG?
The suggested price is $10.53 per tablet (AmerisourceBergen), which is comparable to that of other newer drugs for type 2 diabetes.
THE BOTTOM LINE
The availability of canagliflozin as an additional oral antihyperglycemic option may prove helpful in managing patients with type 2 diabetes who experience adverse effects with other antihyperglycemic drugs.
As with any new drug, questions remain about the long-term risks of canagliflozin. However, it seems to be well tolerated, especially in patients with normal kidney function, and poses a low risk of hypoglycemia. The slight increase in LDL-C may prompt more aggressive lipid management. Whether blood pressure-lowering and weight loss will offset this increase in LDL-C is yet to be determined. Ongoing studies will help to further elucidate whether there is an increased risk of cardiovascular events.
Finally, canagliflozin distinguishes itself from other oral diabetes drugs by its added benefit of weight loss, an appealing side effect, especially in the growing population of obese individuals with type 2 diabetes mellitus.
- Centers for Disease Control and Prevention (CDC). Diabetes data and trends. www.cdc.gov/diabetes/statistics/. Accessed September 6, 2013.
- National Diabetes Information Clearinghouse (NDIC), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). National diabetes statistics, 2011. www.diabetes.niddk.nih.gov/dm/pubs/statistics/. Accessed September 6, 2013.
- Campbell RK. Fate of the beta-cell in the pathophysiology of type 2 diabetes. J Am Pharm Assoc (2003). 2009; 49(suppl 1):S10–S15.
- American Diabetes Association. Executive summary: standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S4–S10.
- Blonde L. Current antihyperglycemic treatment strategies for patients with type 2 diabetes mellitus. Cleve Clin J Med 2009; 76(suppl 5):S4–S11.
- Cheung BM, Ong KL, Cherny SS, Sham PC, Tso AW, Lam KS. Diabetes prevalence and therapeutic target achievement in the United States, 1999 to 2006. Am J Med 2009; 122:443–453.
- Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 2011; 300:R1009–R1022.
- DeFronzo RA, Davidson JA, Del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab 2012; 14:5–14.
- Pfister M, Whaley JM, Zhang L, List JF. Inhibition of SGLT2: a novel strategy for treatment of type 2 diabetes mellitus. Clin Pharmacol Ther 2011; 89:621–625.
- Food and Drug Administration (FDA). FDA Briefing Document. NDA 204042. Invokana (canagliflozin) Tablets. www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM334550.pdf. Accessed September 6, 2013.
- INVOKANA (canagliflozin) tablets, for oral use. Prescribing Information. Janssen Pharmaceuticals, Inc. www.janssenpharmaceuticalsinc.com/assets/invokana_prescribing_info.pdf. Accessed September 6, 2013.
- Stenlöf K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab 2013; 15:372–382.
- US National Institutes of Health. ClinicalTrials.gov. CANVAS—CA Nagliflozin cardio Vascular Assessment Study. http://clinicaltrials.gov/show/NCT01032629. Accessed September 6, 2013.
- Devineni D, Morrow L, Hompesch M, et al. Canagliflozin improves glycaemic control over 28 days in subjects with type 2 diabetes not optimally controlled on insulin. Diabetes Obes Metab 2012; 14:539–545.
Glycosuria used to be a sign of uncontrolled diabetes and was something to be corrected, not a therapeutic mechanism. But now we have a new class of drugs that lower plasma glucose levels by increasing the renal excretion of glucose.
Here, we will review canagliflozin, the first in a new class of drugs for type 2 diabetes: how it works, who is a candidate for it, and what to watch out for.
THE NEED FOR NEW DIABETES DRUGS
Diabetes mellitus affects more than 25.8 million people in the United States—8.3% of the population—and this staggering number is rising.1 Among US residents age 65 and older, more than 10.9 million (26.9%) have diabetes.1 People with uncontrolled diabetes are at risk of microvascular complications such as retinopathy, nephropathy, and neuropathy, as well as cardiovascular disease. Diabetes is the leading cause of blindness, chronic kidney disease, and nontraumatic lower-limb amputation in the United States.1
Type 2 diabetes accounts for more than 90% of cases of diabetes in the United States, Europe, and Canada.2 It is characterized by insulin resistance, decreased beta-cell function, and progressive beta-cell decline.3
Current American Diabetes Association guidelines for the treatment of diabetes recommend a hemoglobin A1c target of less than 7.0%.4 Initial management includes lifestyle modifications such as changes in diet and an increase in exercise, as well as consideration of metformin treatment at the same time. If glucose levels remain uncontrolled despite these efforts, other drugs should be added.
A number of oral and injectable antihyperglycemic drugs are available to help achieve this goal, though none is without risk of adverse effects. Those available up to now include metformin, sulfonylureas, meglitinides, alpha-glucosidase inhibitors, thiazolidinediones, gliptins, glucagon-like peptide-1 agonists, amylin analogues, colesevelam, dopamine agonists, and insulin.5 Most of the available antihyperglycemics target the liver, pancreas, gut, and muscle to improve insulin sensitivity, reduce insulin resistance, or stimulate insulin secretion.
Despite the abundance of agents, type 2 diabetes remains uncontrolled in many patients. Only 57.1% of participants with previously diagnosed diabetes in the 2003–2006 National Health and Nutrition Examination Survey were at the hemoglobin A1c goal of less than 7.0%.6 Possible reasons for failure include adverse effects such as hypoglycemia, weight gain, and gastrointestinal symptoms resulting in discontinued use, nonadherence to the prescribed regimen, and failure to increase the dosage or to add additional agents, including insulin, to optimize glycemic control as beta-cell function declines over time.
HOW THE KIDNEYS HANDLE GLUCOSE
In the kidney, glucose is filtered in the glomerulus and then is reabsorbed in the proximal tubule. Normally, the filtered glucose is all reabsorbed unless the glucose load exceeds the kidney’s absorptive capacity. Membrane proteins called sodium-glucose cotransporters reabsorb glucose at the proximal tubule and return it into the peripheral circulation. Glucose enters the tubular epithelial cell with sodium by passive cotransport via the sodiumglucose cotransporters, and then exits on the other side via the glucose transporter GLUT in the basolateral membrane.
Two sodium-glucose transporters that act in the proximal tubule of the kidney have been identified: SGLT1 and SGLT2. SGLT2 reabsorbs most of the glucose in the early segment of the proximal tubule, while SLGT1 reabsorbs the remaining glucose at the distal end.7 SGLT2 is responsible for more than 90% of renal tubular reabsorption of glucose and is found only in the proximal tubule, whereas SGLT1 is found mainly in the gastrointestinal tract.8
Patients with type 2 diabetes have a higher capacity for glucose reabsorption in the proximal tubule as a result of the up-regulation of SGLT2.9
SGLT2 INHIBITORS AND TYPE 2 DIABETES
Drugs that inhibit SGLT2 block reabsorption of glucose in the proximal tubule, lowering the renal threshold for glucose and thereby increasing urinary glucose excretion and lowering the serum glucose level in patients with hyperglycemia. This mechanism of action is insulin-independent.
On March 29, 2013, canagliflozin became the first SGLT2 inhibitor to be approved in the United States for the treatment of type 2 diabetes.10 However, it is not the first of its class to be introduced.
Dapagliflozin was the first SGLT2 inhibitor approved in Europe and has been available there since November 2012. However, the US Food and Drug Administration withheld its approval in the United States in January 2012 because of concerns of a possible association with cancer, specifically breast and bladder cancers, as well as possible liver injury.10 Canagliflozin does not appear to share this risk.
Several other SGLT2 inhibitors may soon be available. Empagliflozin is in phase III trials, and the manufacturer has filed for approval in the United States. Ipragliflozin is awaiting approval in Japan.
CANAGLIFLOZIN: PHARMACOKINETICS AND THERAPEUTIC EFFICACY
Canagliflozin reaches its peak plasma concentration within 1 to 2 hours of oral administration.11 Its half-life is 10.6 hours with a 100-mg dose and 13.1 hours with a 300-mg dose. A steady state is typically achieved in 4 to 5 days.11
Canagliflozin lowers fasting plasma glucose and hemoglobin A1c levels in a dose-dependent manner.10,11 These effects are independent of age, sex, body mass index, and race.12 Postprandial glucose levels are also lowered.
Other potential benefits of canagliflozin include lowering of the systolic blood pressure and, especially important in obese people with type 2 diabetes, weight loss.12 Aside from metformin, which occasionally results in modest weight loss, other oral drugs used in treating type 2 diabetes are weight-neutral or can cause weight gain.
Trials of canagliflozin
Nine phase III trials of canagliflozin have enrolled 10,285 patients, in one of the largest clinical trial programs in type 2 diabetes to date.10 Several of these trials evaluated canagliflozin as monotherapy, whereas others assessed its effect as an add-on therapy in combination with another antihyperglycemic agent such as a sulfonylurea, metformin, pioglitazone, or insulin. There has not yet been a trial directly comparing canagliflozin with metformin.
Four of the placebo-controlled trials evaluated canagliflozin as monotherapy, canagliflozin added to metformin alone, canagliflozin added to metformin plus glimepiride, and canagliflozin added to metformin plus pioglitazone.
When canagliflozin was used as monotherapy, hemoglobin A1c levels at 26 weeks were an absolute 0.91% lower in the canagliflozin 100 mg/day group than in the placebo group, and an absolute 1.16% lower in the canagliflozin 300 mg/day group than in the placebo group (P < .001 for both).12 Patients lost 2.8% of their body weight with canagliflozin 100 mg and 3.3% with canagliflozin 300 mg, compared with 0.6% with placebo. Systolic blood pressure fell by a mean of 3.7 mm Hg with the 100-mg dose and by a mean of 5.4 mm Hg with the 300-mg dose compared with placebo (P < .001 for both dose groups).12
When canagliflozin was added to metformin, with glimepiride as the comparator drug, there was a 5.2% weight reduction with the 100-mg dose, a 5.7% reduction with 300 mg, and a 1% gain with glimepiride. Hemoglobin A1c fell about equally in the three groups.11
When canagliflozin was added to metformin and a sulfonylurea, with sitagliptin as the comparator third drug, the 300-mg canagliflozin dosage group had a 2.8% weight reduction.11
WHAT ARE THE ADVERSE EFFECTS?
Overall, canagliflozin seems to be well tolerated. The most common adverse effects reported in the clinical trials were genital yeast infections, urinary tract infections, and increased urination.
Genital yeast infections were more common in women than in men, occurring in 10.4% of women who received canagliflozin 100 mg and in 11.4% of women who received 300 mg, compared with only 3.2% in the placebo group.11
Urinary tract infections occurred in 5.9% of the 100-mg group and in 4.3% of the 300-mg group, compared with 4.0% of the placebo group.11
Postural hypotension. Lowering of blood pressure and symptoms of postural hypotension were also reported, and these may be attributed to the drug’s mild osmotic diuretic effect. The risk of adverse effects of volume depletion was dose-dependent; in patients over age 75, they occurred in 4.9% of those taking 100 mg and in 8.7% of those taking 300 mg, compared with 2.6% of those in the placebo or active-comparator groups.11 Therefore, one should exercise particular caution when starting this drug in the elderly or in patients taking diuretics or multiple antihypertensive drugs.
Hypoglycemia. When canagliflozin was used as monotherapy, the incidence of hypoglycemia over 26 weeks was similar to that with placebo, occurring in 3.6% of the 100-mg group, 3.0% of the 300-mg group, and 2.6% of the placebo group.12 Canagliflozin was associated with fewer episodes of hypoglycemia than were sulfonylureas, and the number of episodes was similar to that in patients taking gliptins. There was a higher overall incidence of hypoglycemia when canagliflozin was used in combination with a sulfonylurea or with insulin than when it was used as monotherapy.11
Hyperkalemia. Patients with moderate renal impairment or who are on potassiumsparing drugs or drugs that interfere with the renin-angiotensin-aldosterone system may be at higher risk of hyperkalemia, so close monitoring of potassium is recommended. There was also a dose-dependent increase in serum phosphate and magnesium levels, more notably in patients with moderate renal impairment within the first 3 weeks of starting the drug.11
Patients on canagliflozin who are also taking digoxin, ritonavir, phenytoin, phenobarbital, or rifampin should be closely monitored because of the risk of drug-drug interactions.11 Specifically, there was an increase in mean peak digoxin concentrations when used with canagliflozin 300 mg, and the use of phenytoin, phenobarbital, and ritonavir decreased the efficacy of canagliflozin.
WHAT ARE THE CARDIOVASCULAR RISKS OR LONG-TERM CONCERNS?
Dose-dependent increases in low-density lipoprotein cholesterol (LDL-C) may be seen with canagliflozin. Mean changes from baseline compared with placebo were 4.4 mg/dL (4.5%) with canagliflozin 100 mg and 8.3 mg/dL (8%) with canagliflozin 300 mg.11
There was also an increase in non-high-density lipoprotein cholesterol (non-HDL-C).12 Compared with placebo, mean non-HDL-C levels rose by 2.1 mg/dL (1.5%) with canagliflozin 100 mg and 5.1 mg/dL (3.6%) with 300 mg.11
In the 26-week canagliflozin monotherapy trial, archived blood samples in a small subgroup of patients (n = 349) were measured for apolipoprotein-B, which was found to increase by 1.2% with canagliflozin 100 mg and 3.5% with canagliflozin 300 mg, compared with 0.9% in the placebo group.12
Although small, the increase in LDL-C seen with this drug could be a concern, as diabetic patients are already at higher risk of cardiovascular events. The mechanism of this increase is not yet known, though it may be related to metabolic changes from urinary glucose excretion.12
The Canagliflozin Cardiovascular Assessment Study (CANVAS) is a randomized placebo-controlled trial in more than 4,000 patients with type 2 diabetes who have a history of or are at high risk of cardiovascular events. Currently under way, it is evaluating the occurrence of major adverse cardiovascular events (the primary end point) in patients randomized to receive canagliflozin 100 mg, canagliflozin 300 mg, or placebo once daily for up to 4 years. Secondary end points will be the drug’s effects on fasting plasma insulin and glucose, progression of albuminuria, body weight, blood pressure, HDL-C, LDL-C, bone mineral density, markers of bone turnover, and body composition.10 This trial will run for 9 years, to be completed in 2018.13
The CANVAS investigators have already reported that within the first month of treatment, 13 patients taking canagliflozin suffered a major cardiovascular event, including stroke (one of which was fatal) compared with just one patient taking placebo. These events were not seen after the first month. The hazard ratio for major adverse cardiovascular events within the first 30 days was 6.49, but this dropped to 0.89 after the first 30 days.10
Additional issues that should be addressed in long-term postmarketing studies include possible relationships with cancers and pancreatitis and the safety of the drug in pregnancy and in children with diabetes.10
WHO IS A CANDIDATE FOR THIS DRUG?
Canagliflozin is approved for use as monotherapy in addition to lifestyle modifications. It is also approved for use with other antihyperglycemic drugs, including metformin.
Obese patients with type 2 diabetes and normal kidney function may have the greatest benefit. Because of canagliflozin’s insulinin-dependent mechanism of action, patients with both early and late type 2 diabetes may benefit from its ability to lower hemoglobin A1c and blood glucose.14
Although it can be used in patients with moderate (but not severe) kidney disease, canagliflozin does not appear to be as effective in these patients, who had higher rates of adverse effects.11 It is not indicated for patients with type 1 diabetes, type 2 diabetes with ketonuria, or end-stage renal disease (estimated glomerular filtration rate < 45 mL/min or receiving dialysis).11 It also is not yet recommended for use in pregnant women or patients under age 18.
The recommended starting dose of canagliflozin is 100 mg once daily, taken with breakfast. This can be increased to 300 mg once daily if tolerated. However, patients with an estimated glomerular filtration rate of 45 to 60 mL/min should not exceed the 100- mg dose. No dose adjustment is required in patients with mild to moderate hepatic impairment. It is not recommended, however, in patients with severe hepatic impairment.11
Comment. Although canagliflozin is approved as monotherapy, metformin remains my choice for first-line oral therapy. Because canagliflozin is more expensive and its long-term affects are still relatively unknown, I prefer to use it as an adjunct, and believe it will be a useful addition, especially in obese patients who are seeking to lose weight.
WHAT IS THE COST OF THIS DRUG?
The suggested price is $10.53 per tablet (AmerisourceBergen), which is comparable to that of other newer drugs for type 2 diabetes.
THE BOTTOM LINE
The availability of canagliflozin as an additional oral antihyperglycemic option may prove helpful in managing patients with type 2 diabetes who experience adverse effects with other antihyperglycemic drugs.
As with any new drug, questions remain about the long-term risks of canagliflozin. However, it seems to be well tolerated, especially in patients with normal kidney function, and poses a low risk of hypoglycemia. The slight increase in LDL-C may prompt more aggressive lipid management. Whether blood pressure-lowering and weight loss will offset this increase in LDL-C is yet to be determined. Ongoing studies will help to further elucidate whether there is an increased risk of cardiovascular events.
Finally, canagliflozin distinguishes itself from other oral diabetes drugs by its added benefit of weight loss, an appealing side effect, especially in the growing population of obese individuals with type 2 diabetes mellitus.
Glycosuria used to be a sign of uncontrolled diabetes and was something to be corrected, not a therapeutic mechanism. But now we have a new class of drugs that lower plasma glucose levels by increasing the renal excretion of glucose.
Here, we will review canagliflozin, the first in a new class of drugs for type 2 diabetes: how it works, who is a candidate for it, and what to watch out for.
THE NEED FOR NEW DIABETES DRUGS
Diabetes mellitus affects more than 25.8 million people in the United States—8.3% of the population—and this staggering number is rising.1 Among US residents age 65 and older, more than 10.9 million (26.9%) have diabetes.1 People with uncontrolled diabetes are at risk of microvascular complications such as retinopathy, nephropathy, and neuropathy, as well as cardiovascular disease. Diabetes is the leading cause of blindness, chronic kidney disease, and nontraumatic lower-limb amputation in the United States.1
Type 2 diabetes accounts for more than 90% of cases of diabetes in the United States, Europe, and Canada.2 It is characterized by insulin resistance, decreased beta-cell function, and progressive beta-cell decline.3
Current American Diabetes Association guidelines for the treatment of diabetes recommend a hemoglobin A1c target of less than 7.0%.4 Initial management includes lifestyle modifications such as changes in diet and an increase in exercise, as well as consideration of metformin treatment at the same time. If glucose levels remain uncontrolled despite these efforts, other drugs should be added.
A number of oral and injectable antihyperglycemic drugs are available to help achieve this goal, though none is without risk of adverse effects. Those available up to now include metformin, sulfonylureas, meglitinides, alpha-glucosidase inhibitors, thiazolidinediones, gliptins, glucagon-like peptide-1 agonists, amylin analogues, colesevelam, dopamine agonists, and insulin.5 Most of the available antihyperglycemics target the liver, pancreas, gut, and muscle to improve insulin sensitivity, reduce insulin resistance, or stimulate insulin secretion.
Despite the abundance of agents, type 2 diabetes remains uncontrolled in many patients. Only 57.1% of participants with previously diagnosed diabetes in the 2003–2006 National Health and Nutrition Examination Survey were at the hemoglobin A1c goal of less than 7.0%.6 Possible reasons for failure include adverse effects such as hypoglycemia, weight gain, and gastrointestinal symptoms resulting in discontinued use, nonadherence to the prescribed regimen, and failure to increase the dosage or to add additional agents, including insulin, to optimize glycemic control as beta-cell function declines over time.
HOW THE KIDNEYS HANDLE GLUCOSE
In the kidney, glucose is filtered in the glomerulus and then is reabsorbed in the proximal tubule. Normally, the filtered glucose is all reabsorbed unless the glucose load exceeds the kidney’s absorptive capacity. Membrane proteins called sodium-glucose cotransporters reabsorb glucose at the proximal tubule and return it into the peripheral circulation. Glucose enters the tubular epithelial cell with sodium by passive cotransport via the sodiumglucose cotransporters, and then exits on the other side via the glucose transporter GLUT in the basolateral membrane.
Two sodium-glucose transporters that act in the proximal tubule of the kidney have been identified: SGLT1 and SGLT2. SGLT2 reabsorbs most of the glucose in the early segment of the proximal tubule, while SLGT1 reabsorbs the remaining glucose at the distal end.7 SGLT2 is responsible for more than 90% of renal tubular reabsorption of glucose and is found only in the proximal tubule, whereas SGLT1 is found mainly in the gastrointestinal tract.8
Patients with type 2 diabetes have a higher capacity for glucose reabsorption in the proximal tubule as a result of the up-regulation of SGLT2.9
SGLT2 INHIBITORS AND TYPE 2 DIABETES
Drugs that inhibit SGLT2 block reabsorption of glucose in the proximal tubule, lowering the renal threshold for glucose and thereby increasing urinary glucose excretion and lowering the serum glucose level in patients with hyperglycemia. This mechanism of action is insulin-independent.
On March 29, 2013, canagliflozin became the first SGLT2 inhibitor to be approved in the United States for the treatment of type 2 diabetes.10 However, it is not the first of its class to be introduced.
Dapagliflozin was the first SGLT2 inhibitor approved in Europe and has been available there since November 2012. However, the US Food and Drug Administration withheld its approval in the United States in January 2012 because of concerns of a possible association with cancer, specifically breast and bladder cancers, as well as possible liver injury.10 Canagliflozin does not appear to share this risk.
Several other SGLT2 inhibitors may soon be available. Empagliflozin is in phase III trials, and the manufacturer has filed for approval in the United States. Ipragliflozin is awaiting approval in Japan.
CANAGLIFLOZIN: PHARMACOKINETICS AND THERAPEUTIC EFFICACY
Canagliflozin reaches its peak plasma concentration within 1 to 2 hours of oral administration.11 Its half-life is 10.6 hours with a 100-mg dose and 13.1 hours with a 300-mg dose. A steady state is typically achieved in 4 to 5 days.11
Canagliflozin lowers fasting plasma glucose and hemoglobin A1c levels in a dose-dependent manner.10,11 These effects are independent of age, sex, body mass index, and race.12 Postprandial glucose levels are also lowered.
Other potential benefits of canagliflozin include lowering of the systolic blood pressure and, especially important in obese people with type 2 diabetes, weight loss.12 Aside from metformin, which occasionally results in modest weight loss, other oral drugs used in treating type 2 diabetes are weight-neutral or can cause weight gain.
Trials of canagliflozin
Nine phase III trials of canagliflozin have enrolled 10,285 patients, in one of the largest clinical trial programs in type 2 diabetes to date.10 Several of these trials evaluated canagliflozin as monotherapy, whereas others assessed its effect as an add-on therapy in combination with another antihyperglycemic agent such as a sulfonylurea, metformin, pioglitazone, or insulin. There has not yet been a trial directly comparing canagliflozin with metformin.
Four of the placebo-controlled trials evaluated canagliflozin as monotherapy, canagliflozin added to metformin alone, canagliflozin added to metformin plus glimepiride, and canagliflozin added to metformin plus pioglitazone.
When canagliflozin was used as monotherapy, hemoglobin A1c levels at 26 weeks were an absolute 0.91% lower in the canagliflozin 100 mg/day group than in the placebo group, and an absolute 1.16% lower in the canagliflozin 300 mg/day group than in the placebo group (P < .001 for both).12 Patients lost 2.8% of their body weight with canagliflozin 100 mg and 3.3% with canagliflozin 300 mg, compared with 0.6% with placebo. Systolic blood pressure fell by a mean of 3.7 mm Hg with the 100-mg dose and by a mean of 5.4 mm Hg with the 300-mg dose compared with placebo (P < .001 for both dose groups).12
When canagliflozin was added to metformin, with glimepiride as the comparator drug, there was a 5.2% weight reduction with the 100-mg dose, a 5.7% reduction with 300 mg, and a 1% gain with glimepiride. Hemoglobin A1c fell about equally in the three groups.11
When canagliflozin was added to metformin and a sulfonylurea, with sitagliptin as the comparator third drug, the 300-mg canagliflozin dosage group had a 2.8% weight reduction.11
WHAT ARE THE ADVERSE EFFECTS?
Overall, canagliflozin seems to be well tolerated. The most common adverse effects reported in the clinical trials were genital yeast infections, urinary tract infections, and increased urination.
Genital yeast infections were more common in women than in men, occurring in 10.4% of women who received canagliflozin 100 mg and in 11.4% of women who received 300 mg, compared with only 3.2% in the placebo group.11
Urinary tract infections occurred in 5.9% of the 100-mg group and in 4.3% of the 300-mg group, compared with 4.0% of the placebo group.11
Postural hypotension. Lowering of blood pressure and symptoms of postural hypotension were also reported, and these may be attributed to the drug’s mild osmotic diuretic effect. The risk of adverse effects of volume depletion was dose-dependent; in patients over age 75, they occurred in 4.9% of those taking 100 mg and in 8.7% of those taking 300 mg, compared with 2.6% of those in the placebo or active-comparator groups.11 Therefore, one should exercise particular caution when starting this drug in the elderly or in patients taking diuretics or multiple antihypertensive drugs.
Hypoglycemia. When canagliflozin was used as monotherapy, the incidence of hypoglycemia over 26 weeks was similar to that with placebo, occurring in 3.6% of the 100-mg group, 3.0% of the 300-mg group, and 2.6% of the placebo group.12 Canagliflozin was associated with fewer episodes of hypoglycemia than were sulfonylureas, and the number of episodes was similar to that in patients taking gliptins. There was a higher overall incidence of hypoglycemia when canagliflozin was used in combination with a sulfonylurea or with insulin than when it was used as monotherapy.11
Hyperkalemia. Patients with moderate renal impairment or who are on potassiumsparing drugs or drugs that interfere with the renin-angiotensin-aldosterone system may be at higher risk of hyperkalemia, so close monitoring of potassium is recommended. There was also a dose-dependent increase in serum phosphate and magnesium levels, more notably in patients with moderate renal impairment within the first 3 weeks of starting the drug.11
Patients on canagliflozin who are also taking digoxin, ritonavir, phenytoin, phenobarbital, or rifampin should be closely monitored because of the risk of drug-drug interactions.11 Specifically, there was an increase in mean peak digoxin concentrations when used with canagliflozin 300 mg, and the use of phenytoin, phenobarbital, and ritonavir decreased the efficacy of canagliflozin.
WHAT ARE THE CARDIOVASCULAR RISKS OR LONG-TERM CONCERNS?
Dose-dependent increases in low-density lipoprotein cholesterol (LDL-C) may be seen with canagliflozin. Mean changes from baseline compared with placebo were 4.4 mg/dL (4.5%) with canagliflozin 100 mg and 8.3 mg/dL (8%) with canagliflozin 300 mg.11
There was also an increase in non-high-density lipoprotein cholesterol (non-HDL-C).12 Compared with placebo, mean non-HDL-C levels rose by 2.1 mg/dL (1.5%) with canagliflozin 100 mg and 5.1 mg/dL (3.6%) with 300 mg.11
In the 26-week canagliflozin monotherapy trial, archived blood samples in a small subgroup of patients (n = 349) were measured for apolipoprotein-B, which was found to increase by 1.2% with canagliflozin 100 mg and 3.5% with canagliflozin 300 mg, compared with 0.9% in the placebo group.12
Although small, the increase in LDL-C seen with this drug could be a concern, as diabetic patients are already at higher risk of cardiovascular events. The mechanism of this increase is not yet known, though it may be related to metabolic changes from urinary glucose excretion.12
The Canagliflozin Cardiovascular Assessment Study (CANVAS) is a randomized placebo-controlled trial in more than 4,000 patients with type 2 diabetes who have a history of or are at high risk of cardiovascular events. Currently under way, it is evaluating the occurrence of major adverse cardiovascular events (the primary end point) in patients randomized to receive canagliflozin 100 mg, canagliflozin 300 mg, or placebo once daily for up to 4 years. Secondary end points will be the drug’s effects on fasting plasma insulin and glucose, progression of albuminuria, body weight, blood pressure, HDL-C, LDL-C, bone mineral density, markers of bone turnover, and body composition.10 This trial will run for 9 years, to be completed in 2018.13
The CANVAS investigators have already reported that within the first month of treatment, 13 patients taking canagliflozin suffered a major cardiovascular event, including stroke (one of which was fatal) compared with just one patient taking placebo. These events were not seen after the first month. The hazard ratio for major adverse cardiovascular events within the first 30 days was 6.49, but this dropped to 0.89 after the first 30 days.10
Additional issues that should be addressed in long-term postmarketing studies include possible relationships with cancers and pancreatitis and the safety of the drug in pregnancy and in children with diabetes.10
WHO IS A CANDIDATE FOR THIS DRUG?
Canagliflozin is approved for use as monotherapy in addition to lifestyle modifications. It is also approved for use with other antihyperglycemic drugs, including metformin.
Obese patients with type 2 diabetes and normal kidney function may have the greatest benefit. Because of canagliflozin’s insulinin-dependent mechanism of action, patients with both early and late type 2 diabetes may benefit from its ability to lower hemoglobin A1c and blood glucose.14
Although it can be used in patients with moderate (but not severe) kidney disease, canagliflozin does not appear to be as effective in these patients, who had higher rates of adverse effects.11 It is not indicated for patients with type 1 diabetes, type 2 diabetes with ketonuria, or end-stage renal disease (estimated glomerular filtration rate < 45 mL/min or receiving dialysis).11 It also is not yet recommended for use in pregnant women or patients under age 18.
The recommended starting dose of canagliflozin is 100 mg once daily, taken with breakfast. This can be increased to 300 mg once daily if tolerated. However, patients with an estimated glomerular filtration rate of 45 to 60 mL/min should not exceed the 100- mg dose. No dose adjustment is required in patients with mild to moderate hepatic impairment. It is not recommended, however, in patients with severe hepatic impairment.11
Comment. Although canagliflozin is approved as monotherapy, metformin remains my choice for first-line oral therapy. Because canagliflozin is more expensive and its long-term affects are still relatively unknown, I prefer to use it as an adjunct, and believe it will be a useful addition, especially in obese patients who are seeking to lose weight.
WHAT IS THE COST OF THIS DRUG?
The suggested price is $10.53 per tablet (AmerisourceBergen), which is comparable to that of other newer drugs for type 2 diabetes.
THE BOTTOM LINE
The availability of canagliflozin as an additional oral antihyperglycemic option may prove helpful in managing patients with type 2 diabetes who experience adverse effects with other antihyperglycemic drugs.
As with any new drug, questions remain about the long-term risks of canagliflozin. However, it seems to be well tolerated, especially in patients with normal kidney function, and poses a low risk of hypoglycemia. The slight increase in LDL-C may prompt more aggressive lipid management. Whether blood pressure-lowering and weight loss will offset this increase in LDL-C is yet to be determined. Ongoing studies will help to further elucidate whether there is an increased risk of cardiovascular events.
Finally, canagliflozin distinguishes itself from other oral diabetes drugs by its added benefit of weight loss, an appealing side effect, especially in the growing population of obese individuals with type 2 diabetes mellitus.
- Centers for Disease Control and Prevention (CDC). Diabetes data and trends. www.cdc.gov/diabetes/statistics/. Accessed September 6, 2013.
- National Diabetes Information Clearinghouse (NDIC), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). National diabetes statistics, 2011. www.diabetes.niddk.nih.gov/dm/pubs/statistics/. Accessed September 6, 2013.
- Campbell RK. Fate of the beta-cell in the pathophysiology of type 2 diabetes. J Am Pharm Assoc (2003). 2009; 49(suppl 1):S10–S15.
- American Diabetes Association. Executive summary: standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S4–S10.
- Blonde L. Current antihyperglycemic treatment strategies for patients with type 2 diabetes mellitus. Cleve Clin J Med 2009; 76(suppl 5):S4–S11.
- Cheung BM, Ong KL, Cherny SS, Sham PC, Tso AW, Lam KS. Diabetes prevalence and therapeutic target achievement in the United States, 1999 to 2006. Am J Med 2009; 122:443–453.
- Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 2011; 300:R1009–R1022.
- DeFronzo RA, Davidson JA, Del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab 2012; 14:5–14.
- Pfister M, Whaley JM, Zhang L, List JF. Inhibition of SGLT2: a novel strategy for treatment of type 2 diabetes mellitus. Clin Pharmacol Ther 2011; 89:621–625.
- Food and Drug Administration (FDA). FDA Briefing Document. NDA 204042. Invokana (canagliflozin) Tablets. www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM334550.pdf. Accessed September 6, 2013.
- INVOKANA (canagliflozin) tablets, for oral use. Prescribing Information. Janssen Pharmaceuticals, Inc. www.janssenpharmaceuticalsinc.com/assets/invokana_prescribing_info.pdf. Accessed September 6, 2013.
- Stenlöf K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab 2013; 15:372–382.
- US National Institutes of Health. ClinicalTrials.gov. CANVAS—CA Nagliflozin cardio Vascular Assessment Study. http://clinicaltrials.gov/show/NCT01032629. Accessed September 6, 2013.
- Devineni D, Morrow L, Hompesch M, et al. Canagliflozin improves glycaemic control over 28 days in subjects with type 2 diabetes not optimally controlled on insulin. Diabetes Obes Metab 2012; 14:539–545.
- Centers for Disease Control and Prevention (CDC). Diabetes data and trends. www.cdc.gov/diabetes/statistics/. Accessed September 6, 2013.
- National Diabetes Information Clearinghouse (NDIC), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). National diabetes statistics, 2011. www.diabetes.niddk.nih.gov/dm/pubs/statistics/. Accessed September 6, 2013.
- Campbell RK. Fate of the beta-cell in the pathophysiology of type 2 diabetes. J Am Pharm Assoc (2003). 2009; 49(suppl 1):S10–S15.
- American Diabetes Association. Executive summary: standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S4–S10.
- Blonde L. Current antihyperglycemic treatment strategies for patients with type 2 diabetes mellitus. Cleve Clin J Med 2009; 76(suppl 5):S4–S11.
- Cheung BM, Ong KL, Cherny SS, Sham PC, Tso AW, Lam KS. Diabetes prevalence and therapeutic target achievement in the United States, 1999 to 2006. Am J Med 2009; 122:443–453.
- Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 2011; 300:R1009–R1022.
- DeFronzo RA, Davidson JA, Del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab 2012; 14:5–14.
- Pfister M, Whaley JM, Zhang L, List JF. Inhibition of SGLT2: a novel strategy for treatment of type 2 diabetes mellitus. Clin Pharmacol Ther 2011; 89:621–625.
- Food and Drug Administration (FDA). FDA Briefing Document. NDA 204042. Invokana (canagliflozin) Tablets. www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM334550.pdf. Accessed September 6, 2013.
- INVOKANA (canagliflozin) tablets, for oral use. Prescribing Information. Janssen Pharmaceuticals, Inc. www.janssenpharmaceuticalsinc.com/assets/invokana_prescribing_info.pdf. Accessed September 6, 2013.
- Stenlöf K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab 2013; 15:372–382.
- US National Institutes of Health. ClinicalTrials.gov. CANVAS—CA Nagliflozin cardio Vascular Assessment Study. http://clinicaltrials.gov/show/NCT01032629. Accessed September 6, 2013.
- Devineni D, Morrow L, Hompesch M, et al. Canagliflozin improves glycaemic control over 28 days in subjects with type 2 diabetes not optimally controlled on insulin. Diabetes Obes Metab 2012; 14:539–545.
KEY POINTS
- Type 2 diabetes is ubiquitous and, despite an abundance of agents, often remains uncontrolled.
- Canagliflozin and other drugs of its class cause glucose to be spilled in the urine by reducing the amount reabsorbed by the kidney.
- In clinical trials, canagliflozin lowered hemoglobin A1c levels by approximately 1 absolute percentage point.
- Beyond the adverse effects to be expected from the mechanism of action of the drug (ie, genital yeast infections, urinary tract infections, and hypotension caused by osmotic diuresis), canagliflozin seems to increase plasma levels of low-density lipoprotein cholesterol. This may be worrisome, as diabetic patients are already at increased risk of cardiovascular disease.
A practical approach to prescribing antidepressants
With the variety of drugs available for treating depression, choosing one can be daunting. Different agents have characteristics that may make them a better choice for different types of patients, but even so, treating any kind of mental illness often requires an element of trial and error.
Primary care providers are on the frontline of treating mental illness, often evaluating patients before they are seen by a psychiatrist. The purpose of this article is to provide insight into the art of prescribing antidepressants in the primary care setting. We will discuss common patient presentations, including depressed patients without other medical comorbidities as well as those with common comorbidities, with our recommendations for first-line treatment.
We hope our recommendations will help you to navigate the uncertainty more confidently, resulting in more efficient and tailored treatment for your patients.
BASELINE TESTING
When starting a patient on antidepressant drug therapy, we recommend obtaining a set of baseline laboratory tests to rule out underlying medical conditions that may be contributing to the patient’s depression or that may preclude the use of a given drug. (For example, elevation of liver enzymes may preclude the use of duloxetine.) Tests should include:
- A complete blood cell count
- A complete metabolic panel
- A thyroid-stimulating hormone level.
Electrocardiography may also be useful, as some antidepressants can prolong the QT interval or elevate the blood levels of other drugs with this effect.
GENERAL TREATMENT CONSIDERATIONS
There are several classes of antidepressants, and each class has a number of agents. Research has found little difference in efficacy among agents. So to simplify choosing which one to use, we recommend becoming comfortable with an agent from each class, ie:
- A selective serotonin reuptake inhibitor (SSRI)
- A selective serotonin-norepinephrine reuptake inhibitor (SNRI)
- A tricyclic antidepressant (TCA)
- A monoamine oxidase (MAO) inhibitor.
Each class includes generic agents, many of which are on the discount lists of retail pharmacies. Table 1 shows representative drugs from each class, with their relative costs.
Start low and go slow. In general, when starting an antidepressant, consider starting at half the normal dose, titrating upward as tolerated about every 14 days. This approach can minimize side effects. For example, if prescribing fluoxetine, start with 10 mg and titrate every 2 weeks based on tolerance and patient response. That said, each patient may respond differently, requiring perhaps a lower starting dose or a longer titration schedule.
Anticipate side effects. Most of the side effects of an antidepressant drug can be explained by its mechanism of action. Although side effects should certainly be considered when choosing an agent, patients can be reassured that most are transient and benign. A detailed discussion of side effects of antidepressant drugs is beyond the scope of this article, but a review by Khawam et al1 was published earlier in this journal.
Reassess. If after 4 to 6 weeks the patient has had little or no response, it is reasonable to switch agents. For a patient who was on an SSRI, the change can be to another SSRI or to an SNRI. However, if two SSRIs have already failed, then choose an SNRI. Agents are commonly cross-tapered during the switch to avoid abrupt cessation of one drug or the increased risk of adverse events such as cytochrome P450 interactions, serotonin syndrome, or hypertensive crisis (when switching to an MAO inhibitor).
Beware of interactions. All SSRIs and SNRIs are metabolized through the P450 system in the liver and therefore have the potential for drug-drug interactions. Care must be taken when giving these agents together with drugs whose metabolism can be altered by P450 inhibition. For TCAs, blood levels can be checked if there is concern about toxicity; however, dosing is not strictly based on this level. Great care should be taken if a TCA is given together with an SNRI or an SSRI, as the TCA blood level can become significantly elevated. This may result in QT interval prolongation, as mentioned earlier.
Refer. Referral to a psychiatrist is appropriate for patients for whom multiple classes have failed, for patients who have another psychiatric comorbidity (such as psychosis, hypomania, or mania), or for patients who may need hospitalization. Referral is also appropriate if the physician is concerned about suicide risk.
PATIENTS WITH MAJOR DEPRESSION ONLY
For a patient presenting with depression but no other significant medical comorbidity, the first-line therapy is often an SSRI. Several generic SSRIs are available, and some are on the discount lists at retail pharmacies.
Symptoms should start to improve in about 2 weeks, and the optimal response should be achieved in 4 to 6 weeks of treatment. If this does not occur, consider either adding an augmenting agent or switching to a different antidepressant.
PATIENTS WITH CHRONIC PAIN

Chronic pain and depression often go hand in hand and can potentiate each other. When considering an antidepressant in a patient who has both conditions, the SNRIs and TCAs are typically preferred. Some SNRIs, namely duloxetine and milnacipran, are approved for certain chronic pain conditions, such as fibromyalgia. SNRIs are frequently used off-label for other chronic pain conditions such as headache and neuropathic pain.2
TCAs such as amitriptyline, nortriptyline, and doxepin are also often used in patients with chronic pain. These agents, like the SNRIs, inhibit the reuptake of serotonin and norepinephrine and are used off-label for neuropathic pain,3,4 migraine, interstitial cystitis,5 and other pain conditions.6–9
For TCAs and SNRIs, the effective dose range for chronic pain overlaps that for depression. However, TCAs are often given at lower doses to patients without depression. We recommend starting at a low dose and slowly titrating upward to an effective dose. SNRIs are often preferred over TCAs because they do not have anticholinergic side effects and because an overdose is much less likely to be lethal.
PATIENTS WITH SEXUAL DYSFUNCTION
One of the more commonly reported side effects of antidepressants is sexual dysfunction, generally in the form of delayed orgasm or decreased libido.10 Typically, these complaints are attributed to SSRIs and SNRIs; however, TCAs and MAO inhibitors have also been associated wth sexual dysfunction.
Both erectile dysfunction and priapism have been linked to certain antidepressants. In particular, trazodone is a known cause of priapism. Even if using low doses for sleep, male patients should be made aware of this adverse effect.
Switching from one agent to another in the same class is not likely to improve sexual side effects. In particular, all the SSRIs are similar in their likelihood of causing sexual dysfunction. In a patient taking an SSRI who experiences this side effect, switching to bupropion11 or mirtazapine12 can be quite useful. Bupropion acts primarily on dopamine and norepinephrine, whereas mirtazapine acts on serotonin and norepinephrine but in a different manner from SSRIs and SNRIs.
Adjunctive treatment such as a cholinergic agonist, yohimbine (contraindicated with MAO inhibitors), a serotonergic agent (eg, buspirone), or a drug that acts on nitric oxide (eg, sildenafil, tadalafil) may have some utility but is often ineffective. Dose reduction, if possible, can be of value.
PATIENTS WITH ANXIETY
Many antidepressants are also approved for anxiety disorders, and still more are used off-label for this purpose. Anxiety and depression often occur together, so being able to treat both conditions with one drug can be quite useful.13 In general, the antidepressant effects are seen at lower doses of SSRIs and SNRIs, whereas more of the anxiolytic effects are seen at higher doses, particularly for obsessive-compulsive disorder.14
First-line treatment would be an SSRI or SNRI. Most anxiety disorders respond to either class, but there are some more-specific recommendations. SSRIs are best studied in panic disorder, generalized anxiety disorder, social anxiety disorder, posttraumatic stress disorder, and obsessive-compulsive disorder. Fluoxetine, citalopram, escitalopram, and sertraline15 can all be effective in both major depressive disorder and generalized anxiety disorder. Panic disorder also tends to respond well to SSRIs. SNRIs have been evaluated primarily in generalized anxiety disorder but may also be useful in many of the other conditions.
Additionally, mirtazapine (used off-label)12 and the TCAs16–18 can help treat anxiety. Clomipramine is used to treat obsessive-compulsive disorder.19 These drugs are especially useful for nighttime anxiety, as they can aid sleep. Of note, the anxiolytic effect of mirtazapine may be greater at higher doses.
MAO inhibitors often go unused because of the dietary and medication restrictions involved. However, very refractory cases of certain anxiety disorders may respond preferentially to these agents.
Bupropion tends to be more activating than other antidepressants, so is often avoided in anxious patients. However, some research suggests this is not always necessary.20 If the anxiety is secondary to depression, it will often improve significantly with this agent.
When starting or increasing the dose of an antidepressant, patients may experience increased anxiety or feel “jittery.” This feeling usually passes within the first week of treatment, and it is important to inform patients about this effect. “Start low and go slow” in patients with significant comorbid anxiety. Temporarily using a benzodiazepine such as clonazepam may make the transition more tolerable.
PATIENTS WITH CHRONIC FATIGUE SYNDROME OR FIBROMYALGIA
Increasing recognition of both chronic fatigue syndrome and fibromyalgia has led to more proactive treatment for these disorders. Depression can go hand in hand with these disorders, and certain antidepressants, namely the SNRIs, can be useful in this population.
More data exist for the treatment of fibromyalgia. Both duloxetine and milnacipran are approved by the US Food and Drug Administration (FDA) for the treatment of fibromyalgia.21 Venlafaxine is also used off-label for this purpose. SSRIs such as fluoxetine and citalopram have had mixed results.21–23 TCAs have been used with some success; however, their side effects and lethal potential are often limiting.21,24,25 A recent study in Spain also suggested there may be benefit from using MAO inhibitors for fibromyalgia, but data are quite limited.26
The data for treating chronic fatigue syndrome with SSRIs, SNRIs, or MAO inhibitors are conflicting.27–29 However, managing the co-existing depression may provide some relief in and of itself.
PATIENTS WITH FREQUENT INSOMNIA
Insomnia can be a symptom of depression, but it can also be a side effect of certain antidepressants. The SSRIs and SNRIs can disrupt sleep patterns in some patients by shortening the rapid-eye-movement (REM) stage.30,31
In patients with severe insomnia, it may be best to first recommend taking the antidepressant in the morning if they notice worsening sleep after initiating treatment. Patients can be told with any antidepressant, “If it makes you tired, take it at night, and if it wakes you up, take it in the morning.” Of note, a recent South African study suggested that escitalopram may be able to improve sleep.32
If that does not solve the problem, there are other options. For instance, mirtazapine, particularly in doses of 15 mg or 30 mg, aids depression and insomnia. At higher doses (45 mg), the sleep-aiding effect may be reduced. Low doses of TCAs, particularly doxepin, maprotiline (technically speaking, a tetracyclic antidepressant), amitriptyline, and nortriptyline can be effective sleep aids. These agents may be used as an adjunct to another antidepressant to enhance sleep and mood. However, the TCAs also shorten the REM stage of sleep.33
The previously mentioned drug interactions with SSRIs and SNRIs also need to be considered. Caution should be used when discontinuing these medications, as patients may experience rebound symptoms in the form of much more vivid dreams. MAO inhibitors may worsen insomnia because they suppress REM sleep.34
Trazodone is another agent that at lower doses (25–150 mg) can be an effective, nonaddicting sleep aid. When used as an antidepressant, it is generally prescribed at higher doses (300–400 mg), but its sedating effects can be quite limiting at these levels. It is important to remember the possibility of priapism in male patients.
GERIATRIC PATIENTS
Old age brings its own set of concerns when treating depression. Elderly patients are more susceptible to potential bradycardia caused by SSRIs. The TCAs have the more worrisome cardiac side effect of QTc prolongation. TCAs can slow cognitive function, whereas the SSRIs, bupropion, and the SNRIs tend not to affect cognition. Escitalopram and duloxetine have been suggested to be particularly effective in the elderly.35,36 A study from the Netherlands linked SSRIs with increased risk of falling in geriatric patients with dementia.37 Constipation, which could lead to ileus, is increased with TCAs and certain other agents (ie, paroxetine) in the geriatric population.
Mirtazapine is often very useful in elderly patients for many reasons: it treats both anxiety and depression, stimulates appetite and weight gain, can help with nausea, and is an effective sleep aid. Concerns about weight, appetite, and sleep are particularly common in the elderly, whereas younger patients can be less tolerant of drugs that make them gain weight and sleep more. Normal age-related changes to the sleep cycle contribute to decreased satisfaction with sleep as we age. In addition, depression often further impairs sleep. So, in the elderly, optimizing sleep is key. Research has also shown mirtazapine to be effective in patients with both Alzheimer dementia and depression.38
DIABETIC PATIENTS
One of the more worrisome side effects of psychiatric medications in diabetic patients is weight gain. Certain antidepressants have a greater propensity for weight gain and should likely be avoided as first-line treatments in this population.12 Typically, these agents include those that have more antihistamine action such as paroxetine and the TCAs. These agents also may lead to constipation, which could potentially worsen gastroparesis. Mirtazapine and the MAO inhibitors are also known to cause weight gain.
Bupropion and nefazodone are the most weight-neutral of all antidepressants. Nefazodone has fallen out of favor because of its potential to cause fulminant liver failure in rare cases. However, it remains a reasonable option for patients with comorbid anxiety and depression who have significant weight gain with other agents.
SSRIs and MAO inhibitors may improve or be neutral toward glucose metabolism, and some data suggest that SNRIs may impair this process.39
PATIENTS WITH CARDIAC CONDITIONS
Major depression often coexists with cardiac conditions. In particular, many patients develop depression after suffering a myocardial infarction, and increasingly they are being treated for it.40 Treatment in this situation is appropriate, since depression, if untreated, can increase the risk of recurrence of myocardial infarction.41
However, there are many concerns that accompany treating depression in cardiac patients. Therefore, a baseline electrocardiogram should be obtained before starting an antidepressant.
TCAs and tetracyclic agents have a tendency to prolong the QTc interval and potentiate ventricular arrhythmias,42 so it may be prudent to avoid these in patients at risk. These agents can also significantly increase the pulse rate. This tachycardia increases the risk of angina or myocardial infarction from the anticholinergic effects of these drugs.
In February 2013, the FDA issued a warning about possible arrhythmias with citalopram at doses greater than 40 mg in adult patients43; however, research has suggested citalopram is effective in treating depression in cardiac patients.44 Research has not shown an increase in efficacy at doses greater than 40 mg daily, so we recommend following the black-box warning.
TCAs and MAO inhibitors can also cause orthostatic hypotension. On the other hand, consuming large amounts of tyramine, in foods such as aged cheese, can precipitate a hypertensive crisis in patients taking MAO inhibitors.
Which antidepressants tend to be safer in cardiac patients? Sertraline has been shown to be safe in congestive heart failure and coronary artery disease,45–47 but the SSRIs are typically safe. Fluoxetine has shown efficacy in patients who have had a myocardial infarction.48 Mirtazapine has also been shown to be efficacious in cardiac patients.49 Nefazodone, mirtazapine, bupropion, SSRIs, and SNRIs have little or no tendency toward orthostatic hypotension.
- Khawam EA, Laurencic G, Malone DA. Side effects of antidepressants: an overview. Cleve Clin J Med 2006; 73:351–361.
- Ziegler D. Painful diabetic neuropathy: treatment and future aspects. Diabetes Metab Res Rev 2008; 24(suppl 1):S52–S57.
- Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry 2010; 81:1372–1373.
- Tanenberg RJ, Irving GA, Risser RC, et al. Duloxetine, pregabalin, and duloxetine plus gabapentin for diabetic peripheral neuropathic pain management in patients with inadequate pain response to gabapentin: an open-label, randomized, noninferiority comparison. Mayo Clin Proc 2011; 86:615–626.
- Hertle L, van Ophoven A. Long-term results of amitriptyline treatment for interstitial cystitis. Aktuelle Urol 2010; 41(suppl 1):S61–S65.
- Nguyen TM, Eslick GD. Systematic review: the treatment of noncardiac chest pain with antidepressants. Aliment Pharmacol Ther 2012; 35:493–500.
- Lee H, Kim JH, Min BH, et al. Efficacy of venlafaxine for symptomatic relief in young adult patients with functional chest pain: a randomized, double-blind, placebo-controlled, crossover trial. Am J Gastroenterol 2010; 105:1504–1512.
- Varia I, Logue E, O’Connor C, et al. Randomized trial of sertraline in patients with unexplained chest pain of noncardiac origin. Am Heart J 2000; 140:367–372.
- Doraiswamy PM, Varia I, Hellegers C, et al. A randomized controlled trial of paroxetine for noncardiac chest pain. Psychopharmacol Bull 2006; 39:15–24.
- Clayton AH. Understanding antidepressant mechanism of action and its effect on efficacy and safety. J Clin Psychiatry 2012; 73:e11.
- Gartlehner G, Hansen RA, Morgan LC, et al. Second-generation antidepressants in the pharmacologic treatment of adult depression: an update of the 2007 comparative effectiveness review (Internet). Rockville (MD): Agency for Healthcare Research and Quality (US); 2011 Dec. Comparative Effectiveness Reviews, No. 46. http://www.ncbi.nlm.nih.gov/books/NBK83442/. Accessed February 27, 2013.
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev 2011; 12:CD006528.
- Hofmeijer-Sevink MK, Batelaan NM, van Megen HJ, et al. Clinical relevance of comorbidity in anxiety disorders: a report from the Netherlands Study of Depression and Anxiety (NESDA). J Affect Disord 2012; 137:106–112.
- Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci 2011; 13:423–437.
- Sheehan DV, Kamijima K. An evidence-based review of the clinical use of sertraline in mood and anxiety disorders. Int Clin Psychopharmacol 2009; 24:43–60.
- Huh J, Goebert D, Takeshita J, Lu BY, Kang M. Treatment of generalized anxiety disorder: a comprehensive review of the literature for psychopharmacologic alternatives to newer antidepressants and benzodiazepines. Prim Care Companion CNS Disord 2011; 13: 4088/PCC.08r00709blu.
- Rickels K, Downing R, Schweizer E, Hassman H. Antidepressants for the treatment of generalized anxiety disorder. A placebo-controlled comparison of imipramine, trazodone, and diazepam. Arch Gen Psychiatry 1993; 50:884–895.
- Uher R, Maier W, Hauser J, et al. Differential efficacy of escitalopram and nortriptyline on dimensional measures of depression. Br J Psychiatry 2009; 194:252–259.
- Kellner M. Drug treatment of obsessive-compulsive disorder. Dialogues Clin Neurosci 2010; 12:187–197.
- Rush AJ, Trivedi MH, Carmody TJ, et al. Response in relation to baseline anxiety levels in major depressive disorder treated with bupropion sustained release or sertraline. Neuropsychopharmacology 2001; 25:131–138.
- Mease PJ, Dundon K, Sarzi-Puttini P. Pharmacotherapy of fibromyalgia. Best Pract Res Clin Rheumatol 2011; 25:285–297.
- Wolfe F, Cathey MA, Hawley DJ. A double-blind placebo controlled trial of fluoxetine in fibromyalgia. Scand J Rheumatol 1994; 23:255–259.
- Arnold LM, Hess EV, Hudson JI, Welge JA, Berno SE, Keck PE. A randomized, placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med 2002; 112:191–197.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics 2000; 41:104–113.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Tort S, Urrútia G, Nishishinya MB, Walitt B. Monoamine oxidase inhibitors (MAOIs) for fibromyalgia syndrome. Cochrane Database Syst Rev 2012; 4:CD009807.
- Vercoulen JH, Swanink CM, Zitman FG, et al. Randomised, double-blind, placebo-controlled study of fluoxetine in chronic fatigue syndrome. Lancet 1996; 347:858–861.
- Natelson BH, Cheu J, Pareja J, Ellis SP, Policastro T, Findley TW. Randomized, double blind, controlled placebo-phase in trial of low dose phenelzine in the chronic fatigue syndrome. Psychopharmacology (Berl) 1996; 124:226–230.
- Reid S, Chalder T, Cleare A, Hotopf M, Wessely S. Chronic fatigue syndrome. BMJ 2000; 320:292–296.
- Kupfer DJ, Spiker DG, Coble PA, Neil JF, Ulrich R, Shaw DH. Sleep and treatment prediction in endogenous depression. Am J Psychiatry 1981; 138:429–434.
- Argyropoulos SV, Hicks JA, Nash JR, et al. Redistribution of slow wave activity of sleep during pharmacological treatment of depression with paroxetine but not with nefazodone. J Sleep Res 2009; 18:342–348.
- Stein DJ, Lopez AG. Effects of escitalopram on sleep problems in patients with major depression or generalized anxiety disorder. Adv Ther 2011; 28:1021–1037.
- Ehlers CL, Havstad JW, Kupfer DJ. Estimation of the time course of slow-wave sleep over the night in depressed patients: effects of clomipramine and clinical response. Biol Psychiatry 1996; 39:171–181.
- Landolt HP, Raimo EB, Schnierow BJ, Kelsoe JR, Rapaport MH, Gillin JC. Sleep and sleep electroencephalogram in depressed patients treated with phenelzine. Arch Gen Psychiatry 2001; 58:268–276.
- Chen YM, Huang XM, Thompson R, Zhao YB. Clinical features and efficacy of escitalopram treatment for geriatric depression. J Int Med Res 2011; 39:1946–1953.
- Dolder C, Nelson M, Stump A. Pharmacological and clinical profile of newer antidepressants: implications for the treatment of elderly patients. Drugs Aging 2010; 27:625–640.
- Sterke CS, Ziere G, van Beeck EF, Looman CW, van der Cammen TJ. Dose-response relationship between selective serotonin re-uptake inhibitors and injurious falls: a study in nursing home residents with dementia. Br J Clin Pharmacol 2012; 73:812–820.
- Raji MA, Brady SR. Mirtazapine for treatment of depression and comorbidities in Alzheimer disease. Ann Pharmacother 2001; 35:1024–1027.
- Hennings JM, Schaaf L, Fulda S. Glucose metabolism and antidepressant medication. Curr Pharm Des 2012; 18:5900–5919.
- Czarny MJ, Arthurs E, Coffie DF, et al. Prevalence of antidepressant prescription or use in patients with acute coronary syndrome: a systematic review. PLoS One 2011; 6:e27671.
- Zuidersma M, Ormel J, Conradi HJ, de Jonge P. An increase in depressive symptoms after myocardial infarction predicts new cardiac events irrespective of depressive symptoms before myocardial infarction. Psychol Med 2012; 42:683–693.
- van Noord C, Straus SM, Sturkenboom MC, et al. Psychotropic drugs associated with corrected QT interval prolongation. J Clin Psychopharmacol 2009; 29:9–15.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: abnormal heart rhythms associated with high doses of Celexa (citalopram hydrobromide). http://www.fda.gov/Drugs/DrugSafety/ucm269086.htm. Accessed August 25, 2013.
- Lespérance F, Frasure-Smith N, Koszycki D, et al; CREATE Investigators. Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA 2007; 297:367–379.
- O’Connor CM, Jiang W, Kuchibhatla M, et al; SADHART-CHF Investigators. Safety and efficacy of sertraline for depression in patients with heart failure: results of the SADHART-CHF (Sertraline Against Depression and Heart Disease in Chronic Heart Failure) trial. J Am Coll Cardiol 2010; 56:692–699.
- Glassman AH, O’Connor CM, Califf RM, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHEART) Group. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Swenson JR, O’Connor CM, Barton D, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHART) Group. Influence of depression and effect of treatment with sertraline on quality of life after hospitalization for acute coronary syndrome. Am J Cardiol 2003; 92:1271–1276.
- Strik JJ, Honig A, Lousberg R, et al. Efficacy and safety of fluoxetine in the treatment of patients with major depression after first myocardial infarction: findings from a double-blind, placebo-controlled trial. Psychosom Med 2000; 62:783–789.
- Honig A, Kuyper AM, Schene AH, et al; MIND-IT investigators. Treatment of post-myocardial infarction depressive disorder: a randomized, placebo-controlled trial with mirtazapine. Psychosom Med 2007; 69:606–613.
With the variety of drugs available for treating depression, choosing one can be daunting. Different agents have characteristics that may make them a better choice for different types of patients, but even so, treating any kind of mental illness often requires an element of trial and error.
Primary care providers are on the frontline of treating mental illness, often evaluating patients before they are seen by a psychiatrist. The purpose of this article is to provide insight into the art of prescribing antidepressants in the primary care setting. We will discuss common patient presentations, including depressed patients without other medical comorbidities as well as those with common comorbidities, with our recommendations for first-line treatment.
We hope our recommendations will help you to navigate the uncertainty more confidently, resulting in more efficient and tailored treatment for your patients.
BASELINE TESTING
When starting a patient on antidepressant drug therapy, we recommend obtaining a set of baseline laboratory tests to rule out underlying medical conditions that may be contributing to the patient’s depression or that may preclude the use of a given drug. (For example, elevation of liver enzymes may preclude the use of duloxetine.) Tests should include:
- A complete blood cell count
- A complete metabolic panel
- A thyroid-stimulating hormone level.
Electrocardiography may also be useful, as some antidepressants can prolong the QT interval or elevate the blood levels of other drugs with this effect.
GENERAL TREATMENT CONSIDERATIONS
There are several classes of antidepressants, and each class has a number of agents. Research has found little difference in efficacy among agents. So to simplify choosing which one to use, we recommend becoming comfortable with an agent from each class, ie:
- A selective serotonin reuptake inhibitor (SSRI)
- A selective serotonin-norepinephrine reuptake inhibitor (SNRI)
- A tricyclic antidepressant (TCA)
- A monoamine oxidase (MAO) inhibitor.
Each class includes generic agents, many of which are on the discount lists of retail pharmacies. Table 1 shows representative drugs from each class, with their relative costs.
Start low and go slow. In general, when starting an antidepressant, consider starting at half the normal dose, titrating upward as tolerated about every 14 days. This approach can minimize side effects. For example, if prescribing fluoxetine, start with 10 mg and titrate every 2 weeks based on tolerance and patient response. That said, each patient may respond differently, requiring perhaps a lower starting dose or a longer titration schedule.
Anticipate side effects. Most of the side effects of an antidepressant drug can be explained by its mechanism of action. Although side effects should certainly be considered when choosing an agent, patients can be reassured that most are transient and benign. A detailed discussion of side effects of antidepressant drugs is beyond the scope of this article, but a review by Khawam et al1 was published earlier in this journal.
Reassess. If after 4 to 6 weeks the patient has had little or no response, it is reasonable to switch agents. For a patient who was on an SSRI, the change can be to another SSRI or to an SNRI. However, if two SSRIs have already failed, then choose an SNRI. Agents are commonly cross-tapered during the switch to avoid abrupt cessation of one drug or the increased risk of adverse events such as cytochrome P450 interactions, serotonin syndrome, or hypertensive crisis (when switching to an MAO inhibitor).
Beware of interactions. All SSRIs and SNRIs are metabolized through the P450 system in the liver and therefore have the potential for drug-drug interactions. Care must be taken when giving these agents together with drugs whose metabolism can be altered by P450 inhibition. For TCAs, blood levels can be checked if there is concern about toxicity; however, dosing is not strictly based on this level. Great care should be taken if a TCA is given together with an SNRI or an SSRI, as the TCA blood level can become significantly elevated. This may result in QT interval prolongation, as mentioned earlier.
Refer. Referral to a psychiatrist is appropriate for patients for whom multiple classes have failed, for patients who have another psychiatric comorbidity (such as psychosis, hypomania, or mania), or for patients who may need hospitalization. Referral is also appropriate if the physician is concerned about suicide risk.
PATIENTS WITH MAJOR DEPRESSION ONLY
For a patient presenting with depression but no other significant medical comorbidity, the first-line therapy is often an SSRI. Several generic SSRIs are available, and some are on the discount lists at retail pharmacies.
Symptoms should start to improve in about 2 weeks, and the optimal response should be achieved in 4 to 6 weeks of treatment. If this does not occur, consider either adding an augmenting agent or switching to a different antidepressant.
PATIENTS WITH CHRONIC PAIN
Chronic pain and depression often go hand in hand and can potentiate each other. When considering an antidepressant in a patient who has both conditions, the SNRIs and TCAs are typically preferred. Some SNRIs, namely duloxetine and milnacipran, are approved for certain chronic pain conditions, such as fibromyalgia. SNRIs are frequently used off-label for other chronic pain conditions such as headache and neuropathic pain.2
TCAs such as amitriptyline, nortriptyline, and doxepin are also often used in patients with chronic pain. These agents, like the SNRIs, inhibit the reuptake of serotonin and norepinephrine and are used off-label for neuropathic pain,3,4 migraine, interstitial cystitis,5 and other pain conditions.6–9
For TCAs and SNRIs, the effective dose range for chronic pain overlaps that for depression. However, TCAs are often given at lower doses to patients without depression. We recommend starting at a low dose and slowly titrating upward to an effective dose. SNRIs are often preferred over TCAs because they do not have anticholinergic side effects and because an overdose is much less likely to be lethal.
PATIENTS WITH SEXUAL DYSFUNCTION
One of the more commonly reported side effects of antidepressants is sexual dysfunction, generally in the form of delayed orgasm or decreased libido.10 Typically, these complaints are attributed to SSRIs and SNRIs; however, TCAs and MAO inhibitors have also been associated wth sexual dysfunction.
Both erectile dysfunction and priapism have been linked to certain antidepressants. In particular, trazodone is a known cause of priapism. Even if using low doses for sleep, male patients should be made aware of this adverse effect.
Switching from one agent to another in the same class is not likely to improve sexual side effects. In particular, all the SSRIs are similar in their likelihood of causing sexual dysfunction. In a patient taking an SSRI who experiences this side effect, switching to bupropion11 or mirtazapine12 can be quite useful. Bupropion acts primarily on dopamine and norepinephrine, whereas mirtazapine acts on serotonin and norepinephrine but in a different manner from SSRIs and SNRIs.
Adjunctive treatment such as a cholinergic agonist, yohimbine (contraindicated with MAO inhibitors), a serotonergic agent (eg, buspirone), or a drug that acts on nitric oxide (eg, sildenafil, tadalafil) may have some utility but is often ineffective. Dose reduction, if possible, can be of value.
PATIENTS WITH ANXIETY
Many antidepressants are also approved for anxiety disorders, and still more are used off-label for this purpose. Anxiety and depression often occur together, so being able to treat both conditions with one drug can be quite useful.13 In general, the antidepressant effects are seen at lower doses of SSRIs and SNRIs, whereas more of the anxiolytic effects are seen at higher doses, particularly for obsessive-compulsive disorder.14
First-line treatment would be an SSRI or SNRI. Most anxiety disorders respond to either class, but there are some more-specific recommendations. SSRIs are best studied in panic disorder, generalized anxiety disorder, social anxiety disorder, posttraumatic stress disorder, and obsessive-compulsive disorder. Fluoxetine, citalopram, escitalopram, and sertraline15 can all be effective in both major depressive disorder and generalized anxiety disorder. Panic disorder also tends to respond well to SSRIs. SNRIs have been evaluated primarily in generalized anxiety disorder but may also be useful in many of the other conditions.
Additionally, mirtazapine (used off-label)12 and the TCAs16–18 can help treat anxiety. Clomipramine is used to treat obsessive-compulsive disorder.19 These drugs are especially useful for nighttime anxiety, as they can aid sleep. Of note, the anxiolytic effect of mirtazapine may be greater at higher doses.
MAO inhibitors often go unused because of the dietary and medication restrictions involved. However, very refractory cases of certain anxiety disorders may respond preferentially to these agents.
Bupropion tends to be more activating than other antidepressants, so is often avoided in anxious patients. However, some research suggests this is not always necessary.20 If the anxiety is secondary to depression, it will often improve significantly with this agent.
When starting or increasing the dose of an antidepressant, patients may experience increased anxiety or feel “jittery.” This feeling usually passes within the first week of treatment, and it is important to inform patients about this effect. “Start low and go slow” in patients with significant comorbid anxiety. Temporarily using a benzodiazepine such as clonazepam may make the transition more tolerable.
PATIENTS WITH CHRONIC FATIGUE SYNDROME OR FIBROMYALGIA
Increasing recognition of both chronic fatigue syndrome and fibromyalgia has led to more proactive treatment for these disorders. Depression can go hand in hand with these disorders, and certain antidepressants, namely the SNRIs, can be useful in this population.
More data exist for the treatment of fibromyalgia. Both duloxetine and milnacipran are approved by the US Food and Drug Administration (FDA) for the treatment of fibromyalgia.21 Venlafaxine is also used off-label for this purpose. SSRIs such as fluoxetine and citalopram have had mixed results.21–23 TCAs have been used with some success; however, their side effects and lethal potential are often limiting.21,24,25 A recent study in Spain also suggested there may be benefit from using MAO inhibitors for fibromyalgia, but data are quite limited.26
The data for treating chronic fatigue syndrome with SSRIs, SNRIs, or MAO inhibitors are conflicting.27–29 However, managing the co-existing depression may provide some relief in and of itself.
PATIENTS WITH FREQUENT INSOMNIA
Insomnia can be a symptom of depression, but it can also be a side effect of certain antidepressants. The SSRIs and SNRIs can disrupt sleep patterns in some patients by shortening the rapid-eye-movement (REM) stage.30,31
In patients with severe insomnia, it may be best to first recommend taking the antidepressant in the morning if they notice worsening sleep after initiating treatment. Patients can be told with any antidepressant, “If it makes you tired, take it at night, and if it wakes you up, take it in the morning.” Of note, a recent South African study suggested that escitalopram may be able to improve sleep.32
If that does not solve the problem, there are other options. For instance, mirtazapine, particularly in doses of 15 mg or 30 mg, aids depression and insomnia. At higher doses (45 mg), the sleep-aiding effect may be reduced. Low doses of TCAs, particularly doxepin, maprotiline (technically speaking, a tetracyclic antidepressant), amitriptyline, and nortriptyline can be effective sleep aids. These agents may be used as an adjunct to another antidepressant to enhance sleep and mood. However, the TCAs also shorten the REM stage of sleep.33
The previously mentioned drug interactions with SSRIs and SNRIs also need to be considered. Caution should be used when discontinuing these medications, as patients may experience rebound symptoms in the form of much more vivid dreams. MAO inhibitors may worsen insomnia because they suppress REM sleep.34
Trazodone is another agent that at lower doses (25–150 mg) can be an effective, nonaddicting sleep aid. When used as an antidepressant, it is generally prescribed at higher doses (300–400 mg), but its sedating effects can be quite limiting at these levels. It is important to remember the possibility of priapism in male patients.
GERIATRIC PATIENTS
Old age brings its own set of concerns when treating depression. Elderly patients are more susceptible to potential bradycardia caused by SSRIs. The TCAs have the more worrisome cardiac side effect of QTc prolongation. TCAs can slow cognitive function, whereas the SSRIs, bupropion, and the SNRIs tend not to affect cognition. Escitalopram and duloxetine have been suggested to be particularly effective in the elderly.35,36 A study from the Netherlands linked SSRIs with increased risk of falling in geriatric patients with dementia.37 Constipation, which could lead to ileus, is increased with TCAs and certain other agents (ie, paroxetine) in the geriatric population.
Mirtazapine is often very useful in elderly patients for many reasons: it treats both anxiety and depression, stimulates appetite and weight gain, can help with nausea, and is an effective sleep aid. Concerns about weight, appetite, and sleep are particularly common in the elderly, whereas younger patients can be less tolerant of drugs that make them gain weight and sleep more. Normal age-related changes to the sleep cycle contribute to decreased satisfaction with sleep as we age. In addition, depression often further impairs sleep. So, in the elderly, optimizing sleep is key. Research has also shown mirtazapine to be effective in patients with both Alzheimer dementia and depression.38
DIABETIC PATIENTS
One of the more worrisome side effects of psychiatric medications in diabetic patients is weight gain. Certain antidepressants have a greater propensity for weight gain and should likely be avoided as first-line treatments in this population.12 Typically, these agents include those that have more antihistamine action such as paroxetine and the TCAs. These agents also may lead to constipation, which could potentially worsen gastroparesis. Mirtazapine and the MAO inhibitors are also known to cause weight gain.
Bupropion and nefazodone are the most weight-neutral of all antidepressants. Nefazodone has fallen out of favor because of its potential to cause fulminant liver failure in rare cases. However, it remains a reasonable option for patients with comorbid anxiety and depression who have significant weight gain with other agents.
SSRIs and MAO inhibitors may improve or be neutral toward glucose metabolism, and some data suggest that SNRIs may impair this process.39
PATIENTS WITH CARDIAC CONDITIONS
Major depression often coexists with cardiac conditions. In particular, many patients develop depression after suffering a myocardial infarction, and increasingly they are being treated for it.40 Treatment in this situation is appropriate, since depression, if untreated, can increase the risk of recurrence of myocardial infarction.41
However, there are many concerns that accompany treating depression in cardiac patients. Therefore, a baseline electrocardiogram should be obtained before starting an antidepressant.
TCAs and tetracyclic agents have a tendency to prolong the QTc interval and potentiate ventricular arrhythmias,42 so it may be prudent to avoid these in patients at risk. These agents can also significantly increase the pulse rate. This tachycardia increases the risk of angina or myocardial infarction from the anticholinergic effects of these drugs.
In February 2013, the FDA issued a warning about possible arrhythmias with citalopram at doses greater than 40 mg in adult patients43; however, research has suggested citalopram is effective in treating depression in cardiac patients.44 Research has not shown an increase in efficacy at doses greater than 40 mg daily, so we recommend following the black-box warning.
TCAs and MAO inhibitors can also cause orthostatic hypotension. On the other hand, consuming large amounts of tyramine, in foods such as aged cheese, can precipitate a hypertensive crisis in patients taking MAO inhibitors.
Which antidepressants tend to be safer in cardiac patients? Sertraline has been shown to be safe in congestive heart failure and coronary artery disease,45–47 but the SSRIs are typically safe. Fluoxetine has shown efficacy in patients who have had a myocardial infarction.48 Mirtazapine has also been shown to be efficacious in cardiac patients.49 Nefazodone, mirtazapine, bupropion, SSRIs, and SNRIs have little or no tendency toward orthostatic hypotension.
With the variety of drugs available for treating depression, choosing one can be daunting. Different agents have characteristics that may make them a better choice for different types of patients, but even so, treating any kind of mental illness often requires an element of trial and error.
Primary care providers are on the frontline of treating mental illness, often evaluating patients before they are seen by a psychiatrist. The purpose of this article is to provide insight into the art of prescribing antidepressants in the primary care setting. We will discuss common patient presentations, including depressed patients without other medical comorbidities as well as those with common comorbidities, with our recommendations for first-line treatment.
We hope our recommendations will help you to navigate the uncertainty more confidently, resulting in more efficient and tailored treatment for your patients.
BASELINE TESTING
When starting a patient on antidepressant drug therapy, we recommend obtaining a set of baseline laboratory tests to rule out underlying medical conditions that may be contributing to the patient’s depression or that may preclude the use of a given drug. (For example, elevation of liver enzymes may preclude the use of duloxetine.) Tests should include:
- A complete blood cell count
- A complete metabolic panel
- A thyroid-stimulating hormone level.
Electrocardiography may also be useful, as some antidepressants can prolong the QT interval or elevate the blood levels of other drugs with this effect.
GENERAL TREATMENT CONSIDERATIONS
There are several classes of antidepressants, and each class has a number of agents. Research has found little difference in efficacy among agents. So to simplify choosing which one to use, we recommend becoming comfortable with an agent from each class, ie:
- A selective serotonin reuptake inhibitor (SSRI)
- A selective serotonin-norepinephrine reuptake inhibitor (SNRI)
- A tricyclic antidepressant (TCA)
- A monoamine oxidase (MAO) inhibitor.
Each class includes generic agents, many of which are on the discount lists of retail pharmacies. Table 1 shows representative drugs from each class, with their relative costs.
Start low and go slow. In general, when starting an antidepressant, consider starting at half the normal dose, titrating upward as tolerated about every 14 days. This approach can minimize side effects. For example, if prescribing fluoxetine, start with 10 mg and titrate every 2 weeks based on tolerance and patient response. That said, each patient may respond differently, requiring perhaps a lower starting dose or a longer titration schedule.
Anticipate side effects. Most of the side effects of an antidepressant drug can be explained by its mechanism of action. Although side effects should certainly be considered when choosing an agent, patients can be reassured that most are transient and benign. A detailed discussion of side effects of antidepressant drugs is beyond the scope of this article, but a review by Khawam et al1 was published earlier in this journal.
Reassess. If after 4 to 6 weeks the patient has had little or no response, it is reasonable to switch agents. For a patient who was on an SSRI, the change can be to another SSRI or to an SNRI. However, if two SSRIs have already failed, then choose an SNRI. Agents are commonly cross-tapered during the switch to avoid abrupt cessation of one drug or the increased risk of adverse events such as cytochrome P450 interactions, serotonin syndrome, or hypertensive crisis (when switching to an MAO inhibitor).
Beware of interactions. All SSRIs and SNRIs are metabolized through the P450 system in the liver and therefore have the potential for drug-drug interactions. Care must be taken when giving these agents together with drugs whose metabolism can be altered by P450 inhibition. For TCAs, blood levels can be checked if there is concern about toxicity; however, dosing is not strictly based on this level. Great care should be taken if a TCA is given together with an SNRI or an SSRI, as the TCA blood level can become significantly elevated. This may result in QT interval prolongation, as mentioned earlier.
Refer. Referral to a psychiatrist is appropriate for patients for whom multiple classes have failed, for patients who have another psychiatric comorbidity (such as psychosis, hypomania, or mania), or for patients who may need hospitalization. Referral is also appropriate if the physician is concerned about suicide risk.
PATIENTS WITH MAJOR DEPRESSION ONLY
For a patient presenting with depression but no other significant medical comorbidity, the first-line therapy is often an SSRI. Several generic SSRIs are available, and some are on the discount lists at retail pharmacies.
Symptoms should start to improve in about 2 weeks, and the optimal response should be achieved in 4 to 6 weeks of treatment. If this does not occur, consider either adding an augmenting agent or switching to a different antidepressant.
PATIENTS WITH CHRONIC PAIN
Chronic pain and depression often go hand in hand and can potentiate each other. When considering an antidepressant in a patient who has both conditions, the SNRIs and TCAs are typically preferred. Some SNRIs, namely duloxetine and milnacipran, are approved for certain chronic pain conditions, such as fibromyalgia. SNRIs are frequently used off-label for other chronic pain conditions such as headache and neuropathic pain.2
TCAs such as amitriptyline, nortriptyline, and doxepin are also often used in patients with chronic pain. These agents, like the SNRIs, inhibit the reuptake of serotonin and norepinephrine and are used off-label for neuropathic pain,3,4 migraine, interstitial cystitis,5 and other pain conditions.6–9
For TCAs and SNRIs, the effective dose range for chronic pain overlaps that for depression. However, TCAs are often given at lower doses to patients without depression. We recommend starting at a low dose and slowly titrating upward to an effective dose. SNRIs are often preferred over TCAs because they do not have anticholinergic side effects and because an overdose is much less likely to be lethal.
PATIENTS WITH SEXUAL DYSFUNCTION
One of the more commonly reported side effects of antidepressants is sexual dysfunction, generally in the form of delayed orgasm or decreased libido.10 Typically, these complaints are attributed to SSRIs and SNRIs; however, TCAs and MAO inhibitors have also been associated wth sexual dysfunction.
Both erectile dysfunction and priapism have been linked to certain antidepressants. In particular, trazodone is a known cause of priapism. Even if using low doses for sleep, male patients should be made aware of this adverse effect.
Switching from one agent to another in the same class is not likely to improve sexual side effects. In particular, all the SSRIs are similar in their likelihood of causing sexual dysfunction. In a patient taking an SSRI who experiences this side effect, switching to bupropion11 or mirtazapine12 can be quite useful. Bupropion acts primarily on dopamine and norepinephrine, whereas mirtazapine acts on serotonin and norepinephrine but in a different manner from SSRIs and SNRIs.
Adjunctive treatment such as a cholinergic agonist, yohimbine (contraindicated with MAO inhibitors), a serotonergic agent (eg, buspirone), or a drug that acts on nitric oxide (eg, sildenafil, tadalafil) may have some utility but is often ineffective. Dose reduction, if possible, can be of value.
PATIENTS WITH ANXIETY
Many antidepressants are also approved for anxiety disorders, and still more are used off-label for this purpose. Anxiety and depression often occur together, so being able to treat both conditions with one drug can be quite useful.13 In general, the antidepressant effects are seen at lower doses of SSRIs and SNRIs, whereas more of the anxiolytic effects are seen at higher doses, particularly for obsessive-compulsive disorder.14
First-line treatment would be an SSRI or SNRI. Most anxiety disorders respond to either class, but there are some more-specific recommendations. SSRIs are best studied in panic disorder, generalized anxiety disorder, social anxiety disorder, posttraumatic stress disorder, and obsessive-compulsive disorder. Fluoxetine, citalopram, escitalopram, and sertraline15 can all be effective in both major depressive disorder and generalized anxiety disorder. Panic disorder also tends to respond well to SSRIs. SNRIs have been evaluated primarily in generalized anxiety disorder but may also be useful in many of the other conditions.
Additionally, mirtazapine (used off-label)12 and the TCAs16–18 can help treat anxiety. Clomipramine is used to treat obsessive-compulsive disorder.19 These drugs are especially useful for nighttime anxiety, as they can aid sleep. Of note, the anxiolytic effect of mirtazapine may be greater at higher doses.
MAO inhibitors often go unused because of the dietary and medication restrictions involved. However, very refractory cases of certain anxiety disorders may respond preferentially to these agents.
Bupropion tends to be more activating than other antidepressants, so is often avoided in anxious patients. However, some research suggests this is not always necessary.20 If the anxiety is secondary to depression, it will often improve significantly with this agent.
When starting or increasing the dose of an antidepressant, patients may experience increased anxiety or feel “jittery.” This feeling usually passes within the first week of treatment, and it is important to inform patients about this effect. “Start low and go slow” in patients with significant comorbid anxiety. Temporarily using a benzodiazepine such as clonazepam may make the transition more tolerable.
PATIENTS WITH CHRONIC FATIGUE SYNDROME OR FIBROMYALGIA
Increasing recognition of both chronic fatigue syndrome and fibromyalgia has led to more proactive treatment for these disorders. Depression can go hand in hand with these disorders, and certain antidepressants, namely the SNRIs, can be useful in this population.
More data exist for the treatment of fibromyalgia. Both duloxetine and milnacipran are approved by the US Food and Drug Administration (FDA) for the treatment of fibromyalgia.21 Venlafaxine is also used off-label for this purpose. SSRIs such as fluoxetine and citalopram have had mixed results.21–23 TCAs have been used with some success; however, their side effects and lethal potential are often limiting.21,24,25 A recent study in Spain also suggested there may be benefit from using MAO inhibitors for fibromyalgia, but data are quite limited.26
The data for treating chronic fatigue syndrome with SSRIs, SNRIs, or MAO inhibitors are conflicting.27–29 However, managing the co-existing depression may provide some relief in and of itself.
PATIENTS WITH FREQUENT INSOMNIA
Insomnia can be a symptom of depression, but it can also be a side effect of certain antidepressants. The SSRIs and SNRIs can disrupt sleep patterns in some patients by shortening the rapid-eye-movement (REM) stage.30,31
In patients with severe insomnia, it may be best to first recommend taking the antidepressant in the morning if they notice worsening sleep after initiating treatment. Patients can be told with any antidepressant, “If it makes you tired, take it at night, and if it wakes you up, take it in the morning.” Of note, a recent South African study suggested that escitalopram may be able to improve sleep.32
If that does not solve the problem, there are other options. For instance, mirtazapine, particularly in doses of 15 mg or 30 mg, aids depression and insomnia. At higher doses (45 mg), the sleep-aiding effect may be reduced. Low doses of TCAs, particularly doxepin, maprotiline (technically speaking, a tetracyclic antidepressant), amitriptyline, and nortriptyline can be effective sleep aids. These agents may be used as an adjunct to another antidepressant to enhance sleep and mood. However, the TCAs also shorten the REM stage of sleep.33
The previously mentioned drug interactions with SSRIs and SNRIs also need to be considered. Caution should be used when discontinuing these medications, as patients may experience rebound symptoms in the form of much more vivid dreams. MAO inhibitors may worsen insomnia because they suppress REM sleep.34
Trazodone is another agent that at lower doses (25–150 mg) can be an effective, nonaddicting sleep aid. When used as an antidepressant, it is generally prescribed at higher doses (300–400 mg), but its sedating effects can be quite limiting at these levels. It is important to remember the possibility of priapism in male patients.
GERIATRIC PATIENTS
Old age brings its own set of concerns when treating depression. Elderly patients are more susceptible to potential bradycardia caused by SSRIs. The TCAs have the more worrisome cardiac side effect of QTc prolongation. TCAs can slow cognitive function, whereas the SSRIs, bupropion, and the SNRIs tend not to affect cognition. Escitalopram and duloxetine have been suggested to be particularly effective in the elderly.35,36 A study from the Netherlands linked SSRIs with increased risk of falling in geriatric patients with dementia.37 Constipation, which could lead to ileus, is increased with TCAs and certain other agents (ie, paroxetine) in the geriatric population.
Mirtazapine is often very useful in elderly patients for many reasons: it treats both anxiety and depression, stimulates appetite and weight gain, can help with nausea, and is an effective sleep aid. Concerns about weight, appetite, and sleep are particularly common in the elderly, whereas younger patients can be less tolerant of drugs that make them gain weight and sleep more. Normal age-related changes to the sleep cycle contribute to decreased satisfaction with sleep as we age. In addition, depression often further impairs sleep. So, in the elderly, optimizing sleep is key. Research has also shown mirtazapine to be effective in patients with both Alzheimer dementia and depression.38
DIABETIC PATIENTS
One of the more worrisome side effects of psychiatric medications in diabetic patients is weight gain. Certain antidepressants have a greater propensity for weight gain and should likely be avoided as first-line treatments in this population.12 Typically, these agents include those that have more antihistamine action such as paroxetine and the TCAs. These agents also may lead to constipation, which could potentially worsen gastroparesis. Mirtazapine and the MAO inhibitors are also known to cause weight gain.
Bupropion and nefazodone are the most weight-neutral of all antidepressants. Nefazodone has fallen out of favor because of its potential to cause fulminant liver failure in rare cases. However, it remains a reasonable option for patients with comorbid anxiety and depression who have significant weight gain with other agents.
SSRIs and MAO inhibitors may improve or be neutral toward glucose metabolism, and some data suggest that SNRIs may impair this process.39
PATIENTS WITH CARDIAC CONDITIONS
Major depression often coexists with cardiac conditions. In particular, many patients develop depression after suffering a myocardial infarction, and increasingly they are being treated for it.40 Treatment in this situation is appropriate, since depression, if untreated, can increase the risk of recurrence of myocardial infarction.41
However, there are many concerns that accompany treating depression in cardiac patients. Therefore, a baseline electrocardiogram should be obtained before starting an antidepressant.
TCAs and tetracyclic agents have a tendency to prolong the QTc interval and potentiate ventricular arrhythmias,42 so it may be prudent to avoid these in patients at risk. These agents can also significantly increase the pulse rate. This tachycardia increases the risk of angina or myocardial infarction from the anticholinergic effects of these drugs.
In February 2013, the FDA issued a warning about possible arrhythmias with citalopram at doses greater than 40 mg in adult patients43; however, research has suggested citalopram is effective in treating depression in cardiac patients.44 Research has not shown an increase in efficacy at doses greater than 40 mg daily, so we recommend following the black-box warning.
TCAs and MAO inhibitors can also cause orthostatic hypotension. On the other hand, consuming large amounts of tyramine, in foods such as aged cheese, can precipitate a hypertensive crisis in patients taking MAO inhibitors.
Which antidepressants tend to be safer in cardiac patients? Sertraline has been shown to be safe in congestive heart failure and coronary artery disease,45–47 but the SSRIs are typically safe. Fluoxetine has shown efficacy in patients who have had a myocardial infarction.48 Mirtazapine has also been shown to be efficacious in cardiac patients.49 Nefazodone, mirtazapine, bupropion, SSRIs, and SNRIs have little or no tendency toward orthostatic hypotension.
- Khawam EA, Laurencic G, Malone DA. Side effects of antidepressants: an overview. Cleve Clin J Med 2006; 73:351–361.
- Ziegler D. Painful diabetic neuropathy: treatment and future aspects. Diabetes Metab Res Rev 2008; 24(suppl 1):S52–S57.
- Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry 2010; 81:1372–1373.
- Tanenberg RJ, Irving GA, Risser RC, et al. Duloxetine, pregabalin, and duloxetine plus gabapentin for diabetic peripheral neuropathic pain management in patients with inadequate pain response to gabapentin: an open-label, randomized, noninferiority comparison. Mayo Clin Proc 2011; 86:615–626.
- Hertle L, van Ophoven A. Long-term results of amitriptyline treatment for interstitial cystitis. Aktuelle Urol 2010; 41(suppl 1):S61–S65.
- Nguyen TM, Eslick GD. Systematic review: the treatment of noncardiac chest pain with antidepressants. Aliment Pharmacol Ther 2012; 35:493–500.
- Lee H, Kim JH, Min BH, et al. Efficacy of venlafaxine for symptomatic relief in young adult patients with functional chest pain: a randomized, double-blind, placebo-controlled, crossover trial. Am J Gastroenterol 2010; 105:1504–1512.
- Varia I, Logue E, O’Connor C, et al. Randomized trial of sertraline in patients with unexplained chest pain of noncardiac origin. Am Heart J 2000; 140:367–372.
- Doraiswamy PM, Varia I, Hellegers C, et al. A randomized controlled trial of paroxetine for noncardiac chest pain. Psychopharmacol Bull 2006; 39:15–24.
- Clayton AH. Understanding antidepressant mechanism of action and its effect on efficacy and safety. J Clin Psychiatry 2012; 73:e11.
- Gartlehner G, Hansen RA, Morgan LC, et al. Second-generation antidepressants in the pharmacologic treatment of adult depression: an update of the 2007 comparative effectiveness review (Internet). Rockville (MD): Agency for Healthcare Research and Quality (US); 2011 Dec. Comparative Effectiveness Reviews, No. 46. http://www.ncbi.nlm.nih.gov/books/NBK83442/. Accessed February 27, 2013.
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev 2011; 12:CD006528.
- Hofmeijer-Sevink MK, Batelaan NM, van Megen HJ, et al. Clinical relevance of comorbidity in anxiety disorders: a report from the Netherlands Study of Depression and Anxiety (NESDA). J Affect Disord 2012; 137:106–112.
- Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci 2011; 13:423–437.
- Sheehan DV, Kamijima K. An evidence-based review of the clinical use of sertraline in mood and anxiety disorders. Int Clin Psychopharmacol 2009; 24:43–60.
- Huh J, Goebert D, Takeshita J, Lu BY, Kang M. Treatment of generalized anxiety disorder: a comprehensive review of the literature for psychopharmacologic alternatives to newer antidepressants and benzodiazepines. Prim Care Companion CNS Disord 2011; 13: 4088/PCC.08r00709blu.
- Rickels K, Downing R, Schweizer E, Hassman H. Antidepressants for the treatment of generalized anxiety disorder. A placebo-controlled comparison of imipramine, trazodone, and diazepam. Arch Gen Psychiatry 1993; 50:884–895.
- Uher R, Maier W, Hauser J, et al. Differential efficacy of escitalopram and nortriptyline on dimensional measures of depression. Br J Psychiatry 2009; 194:252–259.
- Kellner M. Drug treatment of obsessive-compulsive disorder. Dialogues Clin Neurosci 2010; 12:187–197.
- Rush AJ, Trivedi MH, Carmody TJ, et al. Response in relation to baseline anxiety levels in major depressive disorder treated with bupropion sustained release or sertraline. Neuropsychopharmacology 2001; 25:131–138.
- Mease PJ, Dundon K, Sarzi-Puttini P. Pharmacotherapy of fibromyalgia. Best Pract Res Clin Rheumatol 2011; 25:285–297.
- Wolfe F, Cathey MA, Hawley DJ. A double-blind placebo controlled trial of fluoxetine in fibromyalgia. Scand J Rheumatol 1994; 23:255–259.
- Arnold LM, Hess EV, Hudson JI, Welge JA, Berno SE, Keck PE. A randomized, placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med 2002; 112:191–197.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics 2000; 41:104–113.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Tort S, Urrútia G, Nishishinya MB, Walitt B. Monoamine oxidase inhibitors (MAOIs) for fibromyalgia syndrome. Cochrane Database Syst Rev 2012; 4:CD009807.
- Vercoulen JH, Swanink CM, Zitman FG, et al. Randomised, double-blind, placebo-controlled study of fluoxetine in chronic fatigue syndrome. Lancet 1996; 347:858–861.
- Natelson BH, Cheu J, Pareja J, Ellis SP, Policastro T, Findley TW. Randomized, double blind, controlled placebo-phase in trial of low dose phenelzine in the chronic fatigue syndrome. Psychopharmacology (Berl) 1996; 124:226–230.
- Reid S, Chalder T, Cleare A, Hotopf M, Wessely S. Chronic fatigue syndrome. BMJ 2000; 320:292–296.
- Kupfer DJ, Spiker DG, Coble PA, Neil JF, Ulrich R, Shaw DH. Sleep and treatment prediction in endogenous depression. Am J Psychiatry 1981; 138:429–434.
- Argyropoulos SV, Hicks JA, Nash JR, et al. Redistribution of slow wave activity of sleep during pharmacological treatment of depression with paroxetine but not with nefazodone. J Sleep Res 2009; 18:342–348.
- Stein DJ, Lopez AG. Effects of escitalopram on sleep problems in patients with major depression or generalized anxiety disorder. Adv Ther 2011; 28:1021–1037.
- Ehlers CL, Havstad JW, Kupfer DJ. Estimation of the time course of slow-wave sleep over the night in depressed patients: effects of clomipramine and clinical response. Biol Psychiatry 1996; 39:171–181.
- Landolt HP, Raimo EB, Schnierow BJ, Kelsoe JR, Rapaport MH, Gillin JC. Sleep and sleep electroencephalogram in depressed patients treated with phenelzine. Arch Gen Psychiatry 2001; 58:268–276.
- Chen YM, Huang XM, Thompson R, Zhao YB. Clinical features and efficacy of escitalopram treatment for geriatric depression. J Int Med Res 2011; 39:1946–1953.
- Dolder C, Nelson M, Stump A. Pharmacological and clinical profile of newer antidepressants: implications for the treatment of elderly patients. Drugs Aging 2010; 27:625–640.
- Sterke CS, Ziere G, van Beeck EF, Looman CW, van der Cammen TJ. Dose-response relationship between selective serotonin re-uptake inhibitors and injurious falls: a study in nursing home residents with dementia. Br J Clin Pharmacol 2012; 73:812–820.
- Raji MA, Brady SR. Mirtazapine for treatment of depression and comorbidities in Alzheimer disease. Ann Pharmacother 2001; 35:1024–1027.
- Hennings JM, Schaaf L, Fulda S. Glucose metabolism and antidepressant medication. Curr Pharm Des 2012; 18:5900–5919.
- Czarny MJ, Arthurs E, Coffie DF, et al. Prevalence of antidepressant prescription or use in patients with acute coronary syndrome: a systematic review. PLoS One 2011; 6:e27671.
- Zuidersma M, Ormel J, Conradi HJ, de Jonge P. An increase in depressive symptoms after myocardial infarction predicts new cardiac events irrespective of depressive symptoms before myocardial infarction. Psychol Med 2012; 42:683–693.
- van Noord C, Straus SM, Sturkenboom MC, et al. Psychotropic drugs associated with corrected QT interval prolongation. J Clin Psychopharmacol 2009; 29:9–15.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: abnormal heart rhythms associated with high doses of Celexa (citalopram hydrobromide). http://www.fda.gov/Drugs/DrugSafety/ucm269086.htm. Accessed August 25, 2013.
- Lespérance F, Frasure-Smith N, Koszycki D, et al; CREATE Investigators. Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA 2007; 297:367–379.
- O’Connor CM, Jiang W, Kuchibhatla M, et al; SADHART-CHF Investigators. Safety and efficacy of sertraline for depression in patients with heart failure: results of the SADHART-CHF (Sertraline Against Depression and Heart Disease in Chronic Heart Failure) trial. J Am Coll Cardiol 2010; 56:692–699.
- Glassman AH, O’Connor CM, Califf RM, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHEART) Group. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Swenson JR, O’Connor CM, Barton D, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHART) Group. Influence of depression and effect of treatment with sertraline on quality of life after hospitalization for acute coronary syndrome. Am J Cardiol 2003; 92:1271–1276.
- Strik JJ, Honig A, Lousberg R, et al. Efficacy and safety of fluoxetine in the treatment of patients with major depression after first myocardial infarction: findings from a double-blind, placebo-controlled trial. Psychosom Med 2000; 62:783–789.
- Honig A, Kuyper AM, Schene AH, et al; MIND-IT investigators. Treatment of post-myocardial infarction depressive disorder: a randomized, placebo-controlled trial with mirtazapine. Psychosom Med 2007; 69:606–613.
- Khawam EA, Laurencic G, Malone DA. Side effects of antidepressants: an overview. Cleve Clin J Med 2006; 73:351–361.
- Ziegler D. Painful diabetic neuropathy: treatment and future aspects. Diabetes Metab Res Rev 2008; 24(suppl 1):S52–S57.
- Saarto T, Wiffen PJ. Antidepressants for neuropathic pain: a Cochrane review. J Neurol Neurosurg Psychiatry 2010; 81:1372–1373.
- Tanenberg RJ, Irving GA, Risser RC, et al. Duloxetine, pregabalin, and duloxetine plus gabapentin for diabetic peripheral neuropathic pain management in patients with inadequate pain response to gabapentin: an open-label, randomized, noninferiority comparison. Mayo Clin Proc 2011; 86:615–626.
- Hertle L, van Ophoven A. Long-term results of amitriptyline treatment for interstitial cystitis. Aktuelle Urol 2010; 41(suppl 1):S61–S65.
- Nguyen TM, Eslick GD. Systematic review: the treatment of noncardiac chest pain with antidepressants. Aliment Pharmacol Ther 2012; 35:493–500.
- Lee H, Kim JH, Min BH, et al. Efficacy of venlafaxine for symptomatic relief in young adult patients with functional chest pain: a randomized, double-blind, placebo-controlled, crossover trial. Am J Gastroenterol 2010; 105:1504–1512.
- Varia I, Logue E, O’Connor C, et al. Randomized trial of sertraline in patients with unexplained chest pain of noncardiac origin. Am Heart J 2000; 140:367–372.
- Doraiswamy PM, Varia I, Hellegers C, et al. A randomized controlled trial of paroxetine for noncardiac chest pain. Psychopharmacol Bull 2006; 39:15–24.
- Clayton AH. Understanding antidepressant mechanism of action and its effect on efficacy and safety. J Clin Psychiatry 2012; 73:e11.
- Gartlehner G, Hansen RA, Morgan LC, et al. Second-generation antidepressants in the pharmacologic treatment of adult depression: an update of the 2007 comparative effectiveness review (Internet). Rockville (MD): Agency for Healthcare Research and Quality (US); 2011 Dec. Comparative Effectiveness Reviews, No. 46. http://www.ncbi.nlm.nih.gov/books/NBK83442/. Accessed February 27, 2013.
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev 2011; 12:CD006528.
- Hofmeijer-Sevink MK, Batelaan NM, van Megen HJ, et al. Clinical relevance of comorbidity in anxiety disorders: a report from the Netherlands Study of Depression and Anxiety (NESDA). J Affect Disord 2012; 137:106–112.
- Koen N, Stein DJ. Pharmacotherapy of anxiety disorders: a critical review. Dialogues Clin Neurosci 2011; 13:423–437.
- Sheehan DV, Kamijima K. An evidence-based review of the clinical use of sertraline in mood and anxiety disorders. Int Clin Psychopharmacol 2009; 24:43–60.
- Huh J, Goebert D, Takeshita J, Lu BY, Kang M. Treatment of generalized anxiety disorder: a comprehensive review of the literature for psychopharmacologic alternatives to newer antidepressants and benzodiazepines. Prim Care Companion CNS Disord 2011; 13: 4088/PCC.08r00709blu.
- Rickels K, Downing R, Schweizer E, Hassman H. Antidepressants for the treatment of generalized anxiety disorder. A placebo-controlled comparison of imipramine, trazodone, and diazepam. Arch Gen Psychiatry 1993; 50:884–895.
- Uher R, Maier W, Hauser J, et al. Differential efficacy of escitalopram and nortriptyline on dimensional measures of depression. Br J Psychiatry 2009; 194:252–259.
- Kellner M. Drug treatment of obsessive-compulsive disorder. Dialogues Clin Neurosci 2010; 12:187–197.
- Rush AJ, Trivedi MH, Carmody TJ, et al. Response in relation to baseline anxiety levels in major depressive disorder treated with bupropion sustained release or sertraline. Neuropsychopharmacology 2001; 25:131–138.
- Mease PJ, Dundon K, Sarzi-Puttini P. Pharmacotherapy of fibromyalgia. Best Pract Res Clin Rheumatol 2011; 25:285–297.
- Wolfe F, Cathey MA, Hawley DJ. A double-blind placebo controlled trial of fluoxetine in fibromyalgia. Scand J Rheumatol 1994; 23:255–259.
- Arnold LM, Hess EV, Hudson JI, Welge JA, Berno SE, Keck PE. A randomized, placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med 2002; 112:191–197.
- Arnold LM, Keck PE, Welge JA. Antidepressant treatment of fibromyalgia. A meta-analysis and review. Psychosomatics 2000; 41:104–113.
- Goldenberg DL, Burckhardt C, Crofford L. Management of fibromyalgia syndrome. JAMA 2004; 292:2388–2395.
- Tort S, Urrútia G, Nishishinya MB, Walitt B. Monoamine oxidase inhibitors (MAOIs) for fibromyalgia syndrome. Cochrane Database Syst Rev 2012; 4:CD009807.
- Vercoulen JH, Swanink CM, Zitman FG, et al. Randomised, double-blind, placebo-controlled study of fluoxetine in chronic fatigue syndrome. Lancet 1996; 347:858–861.
- Natelson BH, Cheu J, Pareja J, Ellis SP, Policastro T, Findley TW. Randomized, double blind, controlled placebo-phase in trial of low dose phenelzine in the chronic fatigue syndrome. Psychopharmacology (Berl) 1996; 124:226–230.
- Reid S, Chalder T, Cleare A, Hotopf M, Wessely S. Chronic fatigue syndrome. BMJ 2000; 320:292–296.
- Kupfer DJ, Spiker DG, Coble PA, Neil JF, Ulrich R, Shaw DH. Sleep and treatment prediction in endogenous depression. Am J Psychiatry 1981; 138:429–434.
- Argyropoulos SV, Hicks JA, Nash JR, et al. Redistribution of slow wave activity of sleep during pharmacological treatment of depression with paroxetine but not with nefazodone. J Sleep Res 2009; 18:342–348.
- Stein DJ, Lopez AG. Effects of escitalopram on sleep problems in patients with major depression or generalized anxiety disorder. Adv Ther 2011; 28:1021–1037.
- Ehlers CL, Havstad JW, Kupfer DJ. Estimation of the time course of slow-wave sleep over the night in depressed patients: effects of clomipramine and clinical response. Biol Psychiatry 1996; 39:171–181.
- Landolt HP, Raimo EB, Schnierow BJ, Kelsoe JR, Rapaport MH, Gillin JC. Sleep and sleep electroencephalogram in depressed patients treated with phenelzine. Arch Gen Psychiatry 2001; 58:268–276.
- Chen YM, Huang XM, Thompson R, Zhao YB. Clinical features and efficacy of escitalopram treatment for geriatric depression. J Int Med Res 2011; 39:1946–1953.
- Dolder C, Nelson M, Stump A. Pharmacological and clinical profile of newer antidepressants: implications for the treatment of elderly patients. Drugs Aging 2010; 27:625–640.
- Sterke CS, Ziere G, van Beeck EF, Looman CW, van der Cammen TJ. Dose-response relationship between selective serotonin re-uptake inhibitors and injurious falls: a study in nursing home residents with dementia. Br J Clin Pharmacol 2012; 73:812–820.
- Raji MA, Brady SR. Mirtazapine for treatment of depression and comorbidities in Alzheimer disease. Ann Pharmacother 2001; 35:1024–1027.
- Hennings JM, Schaaf L, Fulda S. Glucose metabolism and antidepressant medication. Curr Pharm Des 2012; 18:5900–5919.
- Czarny MJ, Arthurs E, Coffie DF, et al. Prevalence of antidepressant prescription or use in patients with acute coronary syndrome: a systematic review. PLoS One 2011; 6:e27671.
- Zuidersma M, Ormel J, Conradi HJ, de Jonge P. An increase in depressive symptoms after myocardial infarction predicts new cardiac events irrespective of depressive symptoms before myocardial infarction. Psychol Med 2012; 42:683–693.
- van Noord C, Straus SM, Sturkenboom MC, et al. Psychotropic drugs associated with corrected QT interval prolongation. J Clin Psychopharmacol 2009; 29:9–15.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: abnormal heart rhythms associated with high doses of Celexa (citalopram hydrobromide). http://www.fda.gov/Drugs/DrugSafety/ucm269086.htm. Accessed August 25, 2013.
- Lespérance F, Frasure-Smith N, Koszycki D, et al; CREATE Investigators. Effects of citalopram and interpersonal psychotherapy on depression in patients with coronary artery disease: the Canadian Cardiac Randomized Evaluation of Antidepressant and Psychotherapy Efficacy (CREATE) trial. JAMA 2007; 297:367–379.
- O’Connor CM, Jiang W, Kuchibhatla M, et al; SADHART-CHF Investigators. Safety and efficacy of sertraline for depression in patients with heart failure: results of the SADHART-CHF (Sertraline Against Depression and Heart Disease in Chronic Heart Failure) trial. J Am Coll Cardiol 2010; 56:692–699.
- Glassman AH, O’Connor CM, Califf RM, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHEART) Group. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Swenson JR, O’Connor CM, Barton D, et al; Sertraline Antidepressant Heart Attack Randomized Trial (SADHART) Group. Influence of depression and effect of treatment with sertraline on quality of life after hospitalization for acute coronary syndrome. Am J Cardiol 2003; 92:1271–1276.
- Strik JJ, Honig A, Lousberg R, et al. Efficacy and safety of fluoxetine in the treatment of patients with major depression after first myocardial infarction: findings from a double-blind, placebo-controlled trial. Psychosom Med 2000; 62:783–789.
- Honig A, Kuyper AM, Schene AH, et al; MIND-IT investigators. Treatment of post-myocardial infarction depressive disorder: a randomized, placebo-controlled trial with mirtazapine. Psychosom Med 2007; 69:606–613.
KEY POINTS
- We suggest that clinicians become familiar with one drug from each class of antidepressants.
- Many antidepressants are also approved for conditions other than depression, and for patients who have both depression and one or more of these comcomitant conditions, these drugs can have a “two-for-one” benefit.
- Adverse effects of an antidepressant are usually predictable on the basis of the drug’s mechanism of action.
Protease inhibitors: Silver bullets for chronic hepatitis C infection?
The treatment of hepatitis c virus (HCV) infection is on the brink of major changes with the recent approval of the first direct-acting antiviral agents, the protease inhibitors boceprevir (Victrelis) and telaprevir (Incivek).
Both drugs were approved by the US Food and Drug Administration (FDA) Advisory Panel for Chronic Hepatitis C in May 2011 and are believed to significantly improve treatment outcomes for patients with HCV genotype 1 infection.
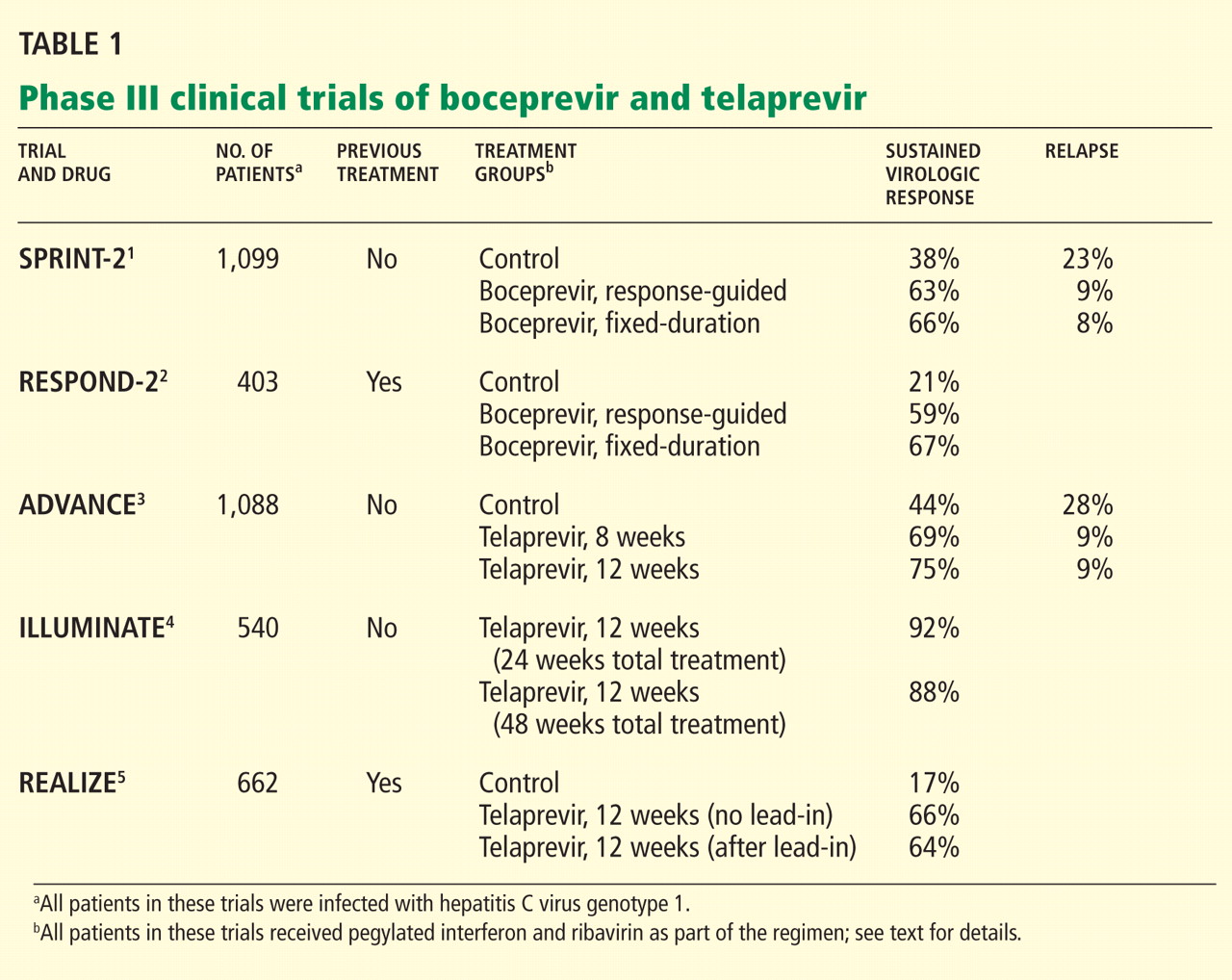
A MAJOR PUBLIC HEALTH PROBLEM
HCV infection is a major public health problem. Nearly 4 million people in the United States are infected.6,7 Most patients with acute HCV infection become chronically infected, and up to 25% eventually develop cirrhosis and its complications, making HCV infection the leading indication for liver transplantation.8–10
Chronic HCV infection has a large global impact, with 180 million people affected across all economic and social groups.11 The highest prevalence of HCV has been reported in Egypt (14%), in part due to the use of inadequately sterilized needles in mass programs to treat endemic schistosomiasis. In developed countries, hepatocellular carcinoma associated with HCV has the fastest growing cancer-related death rate.12
CURRENTLY, FEWER THAN 50% OF PATIENTS ARE CURED
The goal of HCV treatment is to eradicate the virus. However, most infected patients (especially in the United States and Europe) are infected with HCV genotype 1, which is the most difficult genotype to treat.
Successful treatment of HCV is defined as achieving a sustained virologic response—ie, the absence of detectable HCV RNA in the serum 24 weeks after completion of therapy. Once a sustained virologic response is achieved, lifetime “cure” of HCV infection is expected in more than 99% of patients.13
The current standard therapy for HCV, pegylated interferon plus ribavirin for 48 weeks, is effective in only 40% to 50% of patients with genotype 1 infection.14 Therefore, assessing predictors of response before starting treatment can help select patients who are most likely to benefit from therapy.
Viral factors associated with a sustained virologic response include HCV genotypes other than genotype 1 and a low baseline viral load.
Beneficial patient-related factors include younger age, nonblack ethnicity, low body weight (≤ 75 kg), low body mass index, absence of insulin resistance, and absence of advanced fibrosis or cirrhosis.
More recently, a single-nucleotide polymorphism near the interleukin 28B (IL28B) gene, coding for interferon lambda 3, was found to be associated with a twofold difference in the rates of sustained virologic response: patients with the favorable genotype CC were two times more likely to achieve a sustained virologic response than patients with the CT or TT genotypes.15–17
PROTEASE INHIBITORS: MECHANISM OF ACTION
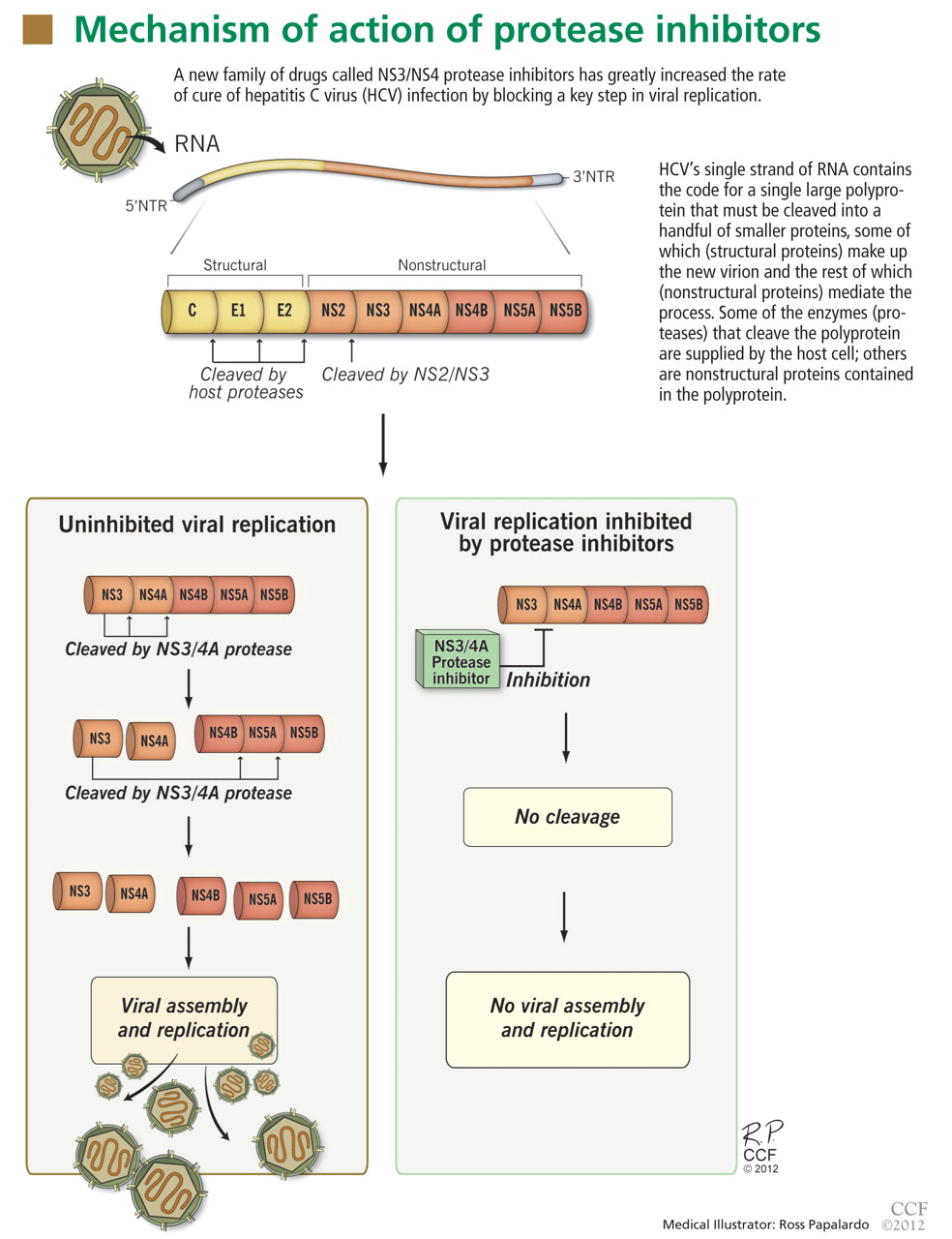
NS3/4A protease inhibitors rely on the principle of end-product inhibition, in which the cleavage product of the protease (a peptide) acts to inhibit the enzyme activity; this is why they are called peptidomimetics. The active site of the NS3/4A protease is a shallow groove composed of three highly conserved amino acid residues, which may explain why protease inhibitors display high antiviral efficacy but pose a low barrier to the development of resistance.20
Protease inhibitors are prone to resistance
The development of viral resistance to protease inhibitors has been a major drawback to their use in patients with chronic HCV infection.21
HCV is a highly variable virus with many genetically distinct but closely related quasispecies circulating in the blood at any given time. Drug-resistant, mutated variants preexist within the patient’s quasispecies, but only in small quantities because of their lesser replication fitness compared with the wild-type virus.22 When direct-acting antiviral therapy is started, the quantity of the wild-type virus decreases and the mutated virus gains replication fitness. Using protease inhibitors as monotherapy selects resistant viral populations rapidly within a few days or weeks.
HCV subtypes 1a and 1b may have different resistance profiles. With genotype 1a, some resistance-associated amino acid substitutions require only one nucleotide change, but with genotype 1b, two nucleotide changes are needed, making resistance less frequent in patients with HCV genotype 1b.23
BOCEPREVIR
Boceprevir is a specific inhibitor of the HCV viral protease NS3/4A.
In phase 3 clinical trials, boceprevir 800 mg three times a day was used with pegylated interferon alfa-2b (PegIntron) 1.5 μg/kg/week and ribavirin (Rebetol) 600 to 1,400 mg daily according to body weight.
Before patients started taking boceprevir, they went through a 4-week lead-in phase, during which they received pegylated interferon and ribavirin. This schedule appeared to reduce the incidence of viral breakthrough in phase 2 trials, and it produced higher rates of sustained virologic response and lower relapse rates compared with triple therapy without a lead-in phase.
Rapid virologic response was defined as undetectable HCV RNA at week 4 of boceprevir therapy (week 8 of the whole regimen).
Boceprevir in previously untreated patients with HCV genotype 1: The SPRINT-2 trial
The Serine Protease Inhibitor Therapy 2 (SPRINT-2) trial1 included more than 1,000 previously untreated adults with HCV genotype 1 infection (938 nonblack patients and 159 black patients; two other nonblack patients did not receive any study drug and were not included in the analysis). In this double-blind trial, patients were randomized into three groups:
- The control group received the standard of care with pegylated interferon and ribavirin for 48 weeks
- The response-guided therapy group received boceprevir plus pegylated interferon and ribavirin for 24 weeks after the 4-week lead-in phase; if HCV RNA was undetectable from week 8 to week 24, treatment was considered complete, but if HCV RNA was detectable at any point from week 8 to week 24, pegylated interferon and ribavirin were continued for a total of 48 weeks.
- The fixed-duration therapy group received boceprevir, pegylated interferon, and ribavirin for 44 weeks after the lead-in period.
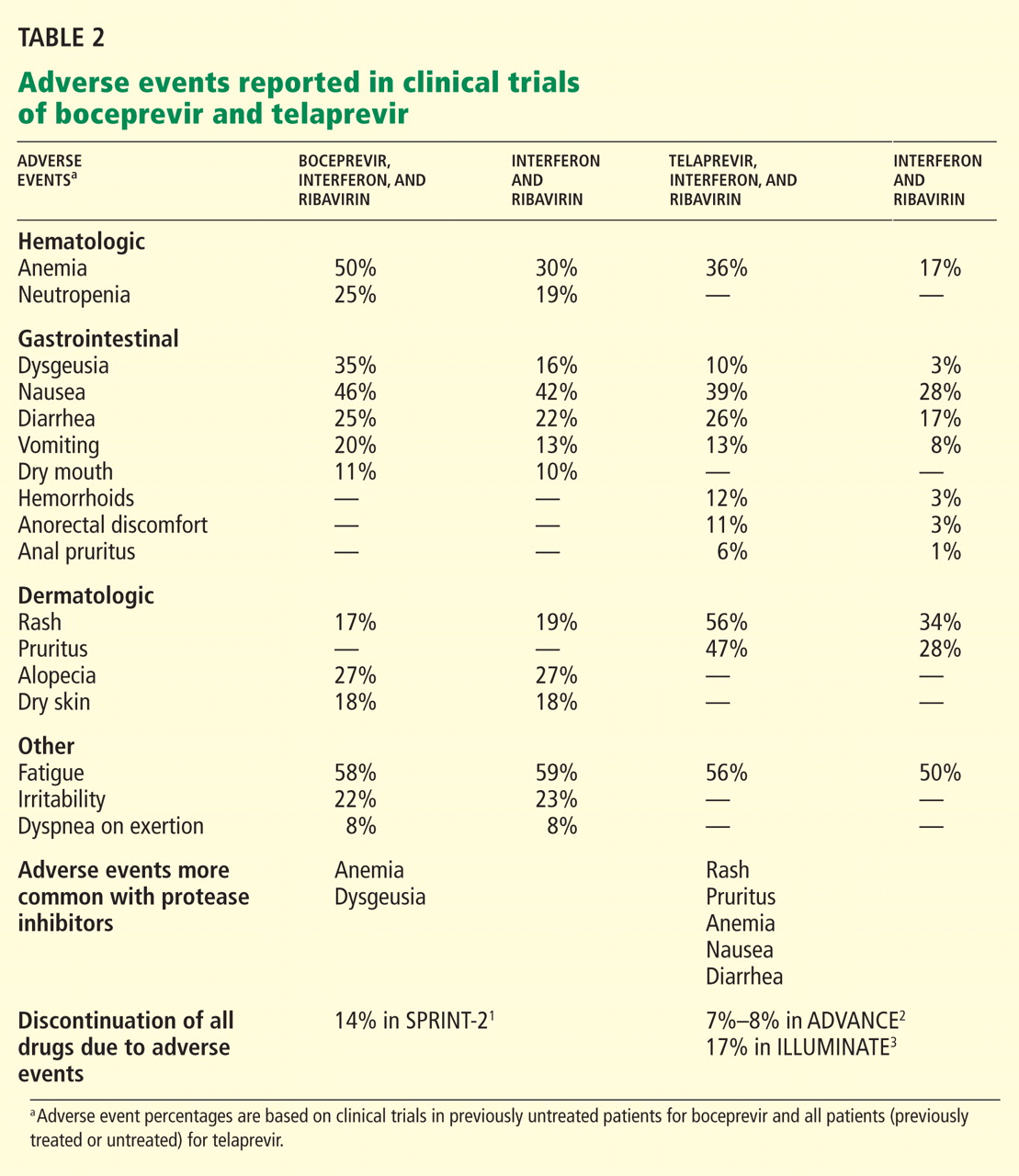
The rate of relapse was 8% and 9% in the boceprevir groups vs 23% in the control group. Patients in the boceprevir groups who had a decrease in HCV RNA of less than 1 log10 during the lead-in phase were found to have a significantly higher rate of boceprevirresistant variants than those who achieved a decrease of HCV RNA of 1 log10 or more.
Boceprevir in previously treated patients with HCV genotype 1: The RESPOND-2 trial
The Retreatment With HCV Serine Protease Inhibitor Boceprevir and PegIntron/Rebetol 2) (RESPOND-2) trial2 was designed to assess the efficacy of combined boceprevir, pegylated interferon, and ribavirin for repeat treatment of patients with HCV genotype 1. These patients had previously undergone standard treatment and had a reduction of 2 log10 or more in HCV RNA after 12 weeks of therapy but with detectable HCV RNA during the therapy period or had had a relapse (defined as undetectable HCV RNA at the end of a previous course of therapy with HCV RNA positivity thereafter). Importantly, null-responders (those who had a reduction of less than 2 log10 in HCV RNA after 12 weeks of therapy) were excluded from this trial.
After a lead-in period of interferon-ribavirin treatment for 4 weeks, 403 patients were assigned to one of three treatment groups:
- Pegylated interferon and ribavirin for 44 weeks (the control group)
- Boceprevir, pegylated interferon, and ribavirin in a response-guided regimen
- Boceprevir, pegylated interferon, and ribavirin for 44 weeks (the fixed-duration group).
Sustained virologic response was achieved in only 21% of patients in the control group. Adding boceprevir increased the rate to 59% in the response-guided therapy group and to 67% in the fixed-duration group. Previous relapsers had better rates than partial responders (69%–75% vs 40%–52%).
Importantly, patients who had a poor response to pegylated interferon and ribavirin during the lead-in phase (defined as having less than a 1-log decrease in the virus before starting boceprevir) had significantly lower rates of sustained virologic response and higher rates of resistance-associated virus variants.
Side effects of boceprevir
Overall, boceprevir is well tolerated. The most common side effects of triple therapy are those usually seen with pegylated interferon and ribavirin, such as flulike symptoms and fatigue (Table 2). However, anemia was more frequent in the boceprevir groups in both SPRINT-2 and RESPOND-2 (45%–50% compared with 20%–29% in the control groups). Erythropoietin was allowed in these studies and was used in about 40% of patients.
The other common side effect associated with boceprevir was dysgeusia (alteration of taste). Dysgeusia was reported by approximately 40% of patients; however, most dysgeusia events were mild to moderate in intensity and did not lead to treatment cessation.
In the SPRINT-2 trial,1 the study drugs had to be discontinued in 12% to 16% of patients in the boceprevir groups because of adverse events, which was similar to the rate (16%) in the control group. Erythropoietin was allowed in this trial, and it was used in 43% of patients in the boceprevir groups compared with 24% in the control group, with discontinuation owing to anemia occurring in 2% and 1% of cases, respectively.
TELAPREVIR
Telaprevir, the other protease NS3/4A inhibitor, has also shown efficacy over current standard therapy in phase 3 clinical trials. It was used in a dose of 750 mg three times a day with pegylated interferon alfa-2a (Pegasys) 180 μg per week and ribavirin (Copegus) 1,000 to 1,200 mg daily according to body weight. A lead-in phase with pegylated interferon and ribavirin was not applied with telaprevir, as it was in the boceprevir trials. Extended rapid virologic response was defined as an undetectable HCV RNA at weeks 4 and 12 of therapy.
Telaprevir in previously untreated patients with HCV genotype 1
The ADVANCE study3 was a double-blind randomized trial assessing the efficacy and safety of telaprevir in combination with pegylated interferon and ribavirin in more than 1,000 previously untreated patients. The three treatment groups received:
- Telaprevir, pegylated interferon, and ribavirin for 8 weeks, followed by pegylated interferon and ribavirin alone for 16 weeks in patients who achieved an extended rapid virologic response (total duration of 24 weeks) or 40 weeks in patients who did not (total duration of 48 weeks)
- Telaprevir, pegylated interferon, and ribavirin for 12 weeks, followed by pegylated interferon-ribavirin alone for 12 (total of 24 weeks) or 36 weeks (total of 48 weeks) according to extended rapid virologic response
- Standard care with pegylated interferon and ribavirin for 48 weeks.
The rate of sustained virologic response was 69% in the group that received telaprevir for 8 weeks and 75% in the group that received it for 12 weeks compared with 44% in the control group (P < .0001 for both) (Table 2). Patients infected with HCV genotype 1b had a higher sustained virologic response rate (79%) than those infected with HCV genotype 1a (71%).
Sustained virologic response rates were lower in black patients and patients with bridging fibrosis or cirrhosis, but were still significantly higher in the telaprevir groups than in the control group. The results of this subset analysis were limited by small numbers of patients in each category.
In total, 57% of those who received telaprevir for 8 weeks and 58% of those who received it for 12 weeks achieved an extended rapid virologic response and were able to cut the duration of their therapy in half (from 48 weeks to 24 weeks).
The relapse rates were 9% in the telaprevir groups and 28% in the control group.
The rate of virologic failure was lower in patients who received triple therapy than in those who received interferon-ribavirin alone (8% in the group that got telaprevir for 12 weeks and 13% in the group that got it for 8 weeks, vs 32% in the control group). The failure rate was also lower in patients with HCV genotype 1b infection than in those with genotype 1a.
The ILLUMINATE study4 (Illustrating the Effects of Combination Therapy With Telaprevir) investigated whether longer duration of treatment than that given in the ADVANCE trial increased the rate of sustained virologic response. Previously untreated patients received telaprevir, interferon, and ribavirin for 12 weeks, and those who achieved an extended rapid virologic response were randomized at week 20 to continue interferonribavirin treatment for 24 or 48 weeks of total treatment.
The sustained virologic response rates in patients who achieved an extended rapid virologic response were 92% in the group that received pegylated interferon and ribavirin for 12 weeks, and 88% in those who received it for 48 weeks. Thus, the results of this study support the use of response-guided therapy for telaprevir-based regimens.
Telaprevir in previously treated patients with HCV genotype 1: The REALIZE trial
In this phase 3 placebo-controlled trial,5 622 patients with prior relapse, partial response, or null response were randomly allocated into one of three groups:
- Telaprevir for 12 weeks plus pegylated interferon and ribavirin for 48 weeks
- Lead-in for 4 weeks followed by 12 weeks of triple therapy and another 32 weeks of pegylated interferon and ribavirin
- Pegylated interferon and ribavirin for 48 weeks (the control group).
The overall sustained virologic response rates were 66% and 64%, respectively, in the telaprevir groups vs 17% in the control group (P < .0001). The sustained virologic response rates in the telaprevir groups were 83% to 88% in prior relapsers, 54% to 59% in partial responders, and 29% to 33% in null-responders. Of note, patients did not benefit from the lead-in phase.
This was the only trial to investigate the response to triple therapy in null-responders, a group in which treatment has been considered hopeless. A response rate of approximately 31% was encouraging, especially if we compare it with the 5% response rate achieved with the current standard of care with pegylated interferon and ribavirin.
Telaprevir side effects
As with boceprevir-based triple therapy, the most common adverse events were related to pegylated interferon (Table 2).
Nearly 50% of patients who receive telaprevir develop a skin rash that is primarily eczematous, can be managed with topical steroids, and usually resolves when telaprevir is discontinued. Severe rashes occurred in 3% to 6% of patients in the ADVANCE trial,3 and three suspected cases of Stevens-Johnson syndrome have been reported to the FDA.
Other side effects that were more frequent with telaprevir included pruritus, nausea, diarrhea, and anemia. On average, the hemoglobin level decreased by an additional 1 g/dL in the telaprevir treatment groups compared with the groups that received only pegylated interferon-ribavirin. Erythropoietin use was not allowed in the phase 3 telaprevir studies, and anemia was managed by ribavirin dose reduction.
In the ADVANCE trial,3 study drugs were discontinued owing to adverse events in 7% to 8% of the patients in the telaprevir groups compared with 4% in the control group. In the ILLUMINATE trial,4 17% of patients had to permanently discontinue all study drugs due to adverse events.
FDA-APPROVED TREATMENT REGIMENS FOR BOCEPREVIR AND TELAPREVIR
For treatment algorithms, see the eFigures that accompany this article online.
Boceprevir in previously untreated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 28—Stop all treatment if HCV RNA was undetectable at weeks 8 and 24
- Week 36—Measure HCV RNA; stop boceprevir
- Week 48—Stop all treatment (eFigure 1).
Boceprevir in previously treated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 36—if HCV RNA was not detectable at week 8, stop all treatment now; if HCV RNA was detectable at week 8, stop boceprevir now but continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 2).
Telaprevir in previously untreated patients and prior relapsers
- Week 0—start telaprevir, pegylated interferon, and ribavirin
- Week 4—measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL
- Week 12—Stop telaprevir; measure HCV RNA; stop all treatment if HCV RNA is more than 1,000 IU/mL
- Week 24—Stop pegylated interferon and ribavirin if HCV RNA was undetectable at week 12; measure HCV RNA and stop treatment if it is detectable; otherwise, continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 3).
Telaprevir in patients who previously achieved a partial or null response
- Week 0—Start telaprevir, pegylated interferon, and ribavirin
- Week 4—Measure HCV RNA; stop treatment if it is more than 1,000 IU/mL
- Week 12—Measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL; if less than 1,000 IU/mL then stop telaprevir but continue pegylated interferon and ribavirin
- Week 24—Measure HCV RNA; stop treatment if HCV RNA is detectable
- Week 48—Stop all treatment (eFigure 4).
Drug interactions with boceprevir and telaprevir
Both boceprevir and telaprevir inhibit cytochrome P450 3A (CYP3A) and thus are contraindicated in combination with drugs highly dependent on CYP3A for clearance and with drugs for which elevated plasma concentrations are associated with serious adverse events, such as atorvastatin (Lipitor), simvastatin (Zocor), sildenafil (Viagra), midazolam (Versed), and St. John’s wort. Giving potent inducers of CYP3A with boceprevir or telaprevir may lead to lower exposure and loss of efficacy of both protease inhibitors.
EMERGING THERAPIES FOR HCV
Thanks to a better understanding of the biology of HCV infection, the effort to develop new therapeutic agents started to focus on targeting specific steps of the viral life cycle, including attachment, entry into cells, replication, and release.24
Currently, more than 50 clinical trials are evaluating new direct-acting antivirals to treat HCV infection.25 Monoclonal and polyclonal antibodies that target the molecular process involved in HCV attachment and entry are being developed.26 The nonstructural protein NS5B (RNA polymerase) is intimately involved in viral replication and represents a promising target.27 Several nucleosides and nonnucleoside protease inhibitors have already entered clinical trials.
The low fidelity of the HCV replication machinery leads to a very high mutation rate, thus enabling the virus to quickly develop mutations that resist agents targeting viral enzymes.28 Therefore, a novel approach is to target host cofactors that are essential for HCV replication. An intriguing study by Lanford et al29 demonstrated that antagonizing microRNA-122 (the most abundant microRNA in the liver and an essential cofactor for viral RNA replication) by the oligonucleotide SPC3649 caused marked and prolonged reduction of HCV viremia in chronically infected chimpanzees.29
Although we are still in the early stages of drug development, the future holds great promise for newer drugs to improve the sustained virologic response, shorten the duration of treatment, improve tolerability with interferon-sparing regimens, and decrease viral resistance.
FUTURE PERSPECTIVES
With the introduction of the first direct-acting antiviral medications for HCV (boceprevir and telaprevir), 2011 will be marked as the year that changed hepatitis C treatment for the better. Triple therapy with pegylated interferon, ribavirin, and either boceprevir or telaprevir has the potential for increasing the rate of sustained virologic response to around 70% in previously untreated patients and 65% in previously treated patients who are infected with HCV genotype 1. The IL28B polymorphisms appear to play a role in the rate of sustained virologic response achieved with triple therapy, with preliminary data showing a better response rate in patients who have the CC genotype.17
These drugs will add up to $50,000 to the cost of treating hepatitis C virus infection, depending on the drug used and the length of treatment. However, they may be well worth it if they prevent liver failure and the need for transplantation.
Many questions remain, such as how to use these new regimens to treat special patient populations—for example, those with a recurrence of HCV infection after liver transplantation, those co-infected with HCV and human immunodeficiency virus, and those infected with HCV genotypes other than genotype 1.
Other direct-acting antiviral agents that specifically target the replication cycle of HCV are currently in clinical development. In fact, the future has already started with the release of the Interferon-Free Regimen for the Management of HCV (INFORM-1) study results.30 This was the first trial to evaluate an interferon-free regimen for patients with chronic HCV infection using two direct-acting antiviral drugs (the protease inhibitor danoprevir and the polymerase inhibitor RG7128), with promising results.
- Poordad F, McCone J, Bacon BR, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1195–1206.
- Bacon BR, Gordon SC, Lawitz E, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1207–1217.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; for the ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Sherman KE, Flamm SL, Afdhal NH, et al; for the ILLUMINATE Study Team. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med 2011; 365:1014–1024.
- Zeuzem S, Andreone P, Pol S, et al; for the REALIZE Study Team. Telaprevir for retreatment of HCV infection. N Engl J Med 2011; 364:2417–2428.
- Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 2006; 144:705–714.
- Mitchell AE, Colvin HM, Palmer Beasley R. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology 2010; 51:729–733.
- Kim WR. The burden of hepatitis C in the United States. Hepatology 2002; 36:S30–S34.
- Marcellin P, Asselah T, Boyer N. Fibrosis and disease progression in hepatitis C. Hepatology 2002; 36:S47–S56.
- Seeff LB. Natural history of chronic hepatitis C. Hepatology 2002; 36:S35–S46.
- Lavanchy D. The global burden of hepatitis C. Liver Int 2009; 29(suppl 1):74–81.
- National Institutes of Health Consensus Development Conference Statement: Management of hepatitis C: 2002—June 10–12, 2002. Hepatology 2002; 36:S3–S20.
- Pearlman BL, Traub N. Sustained virologic response to antiviral therapy for chronic hepatitis C virus infection: a cure and so much more. Clin Infect Dis 2011; 52:889–900.
- Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med 2006; 355:2444–2451.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009; 461:399–401.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e118.
- Nielsen SU, Bassendine MF, Burt AD, Bevitt DJ, Toms GL. Characterization of the genome and structural proteins of hepatitis C virus resolved from infected human liver. J Gen Virol 2004; 85:1497–1507.
- Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology 2004; 39:5–19.
- Nelson DR. The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1 naive patients. Liver Int 2011; 31(suppl 1):53–57.
- Pawlotsky JM. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 2011; 53:1742–1751.
- Monto A, Schooley RT, Lai JC, et al. Lessons from HIV therapy applied to viral hepatitis therapy: summary of a workshop. Am J Gastroenterol 2010; 105:989–1004.
- McCown MF, Rajyaguru S, Kular S, Cammack N, Najera I. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob Agents Chemother 2009; 53:2129–2132.
- Cholongitas E, Papatheodoridis GV. Review article: novel therapeutic options for chronic hepatitis C. Aliment Pharmacol Ther 2008; 27:866–884.
- Naggie S, Patel K, McHutchison J. Hepatitis C virus directly acting antivirals: current developments with NS3/4A HCV serine protease inhibitors. J Antimicrob Chemother 2010; 65:2063–2069.
- Mir HM, Birerdinc A, Younossi ZM. Monoclonal and polyclonal antibodies against the HCV envelope proteins. Clin Liver Dis 2009; 13:477–486.
- Birerdinc A, Younossi ZM. Emerging therapies for hepatitis C virus. Expert Opin Emerg Drugs 2010; 15:535–544.
- Khattab MA. Targeting host factors: a novel rationale for the management of hepatitis C virus. World J Gastroenterol 2009; 15:3472–3479.
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010; 327:198–201.
- Gane EJ, Roberts SK, Stedman CA, et al. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2010; 376:1467–1475.
The treatment of hepatitis c virus (HCV) infection is on the brink of major changes with the recent approval of the first direct-acting antiviral agents, the protease inhibitors boceprevir (Victrelis) and telaprevir (Incivek).
Both drugs were approved by the US Food and Drug Administration (FDA) Advisory Panel for Chronic Hepatitis C in May 2011 and are believed to significantly improve treatment outcomes for patients with HCV genotype 1 infection.
A MAJOR PUBLIC HEALTH PROBLEM
HCV infection is a major public health problem. Nearly 4 million people in the United States are infected.6,7 Most patients with acute HCV infection become chronically infected, and up to 25% eventually develop cirrhosis and its complications, making HCV infection the leading indication for liver transplantation.8–10
Chronic HCV infection has a large global impact, with 180 million people affected across all economic and social groups.11 The highest prevalence of HCV has been reported in Egypt (14%), in part due to the use of inadequately sterilized needles in mass programs to treat endemic schistosomiasis. In developed countries, hepatocellular carcinoma associated with HCV has the fastest growing cancer-related death rate.12
CURRENTLY, FEWER THAN 50% OF PATIENTS ARE CURED
The goal of HCV treatment is to eradicate the virus. However, most infected patients (especially in the United States and Europe) are infected with HCV genotype 1, which is the most difficult genotype to treat.
Successful treatment of HCV is defined as achieving a sustained virologic response—ie, the absence of detectable HCV RNA in the serum 24 weeks after completion of therapy. Once a sustained virologic response is achieved, lifetime “cure” of HCV infection is expected in more than 99% of patients.13
The current standard therapy for HCV, pegylated interferon plus ribavirin for 48 weeks, is effective in only 40% to 50% of patients with genotype 1 infection.14 Therefore, assessing predictors of response before starting treatment can help select patients who are most likely to benefit from therapy.
Viral factors associated with a sustained virologic response include HCV genotypes other than genotype 1 and a low baseline viral load.
Beneficial patient-related factors include younger age, nonblack ethnicity, low body weight (≤ 75 kg), low body mass index, absence of insulin resistance, and absence of advanced fibrosis or cirrhosis.
More recently, a single-nucleotide polymorphism near the interleukin 28B (IL28B) gene, coding for interferon lambda 3, was found to be associated with a twofold difference in the rates of sustained virologic response: patients with the favorable genotype CC were two times more likely to achieve a sustained virologic response than patients with the CT or TT genotypes.15–17
PROTEASE INHIBITORS: MECHANISM OF ACTION
NS3/4A protease inhibitors rely on the principle of end-product inhibition, in which the cleavage product of the protease (a peptide) acts to inhibit the enzyme activity; this is why they are called peptidomimetics. The active site of the NS3/4A protease is a shallow groove composed of three highly conserved amino acid residues, which may explain why protease inhibitors display high antiviral efficacy but pose a low barrier to the development of resistance.20
Protease inhibitors are prone to resistance
The development of viral resistance to protease inhibitors has been a major drawback to their use in patients with chronic HCV infection.21
HCV is a highly variable virus with many genetically distinct but closely related quasispecies circulating in the blood at any given time. Drug-resistant, mutated variants preexist within the patient’s quasispecies, but only in small quantities because of their lesser replication fitness compared with the wild-type virus.22 When direct-acting antiviral therapy is started, the quantity of the wild-type virus decreases and the mutated virus gains replication fitness. Using protease inhibitors as monotherapy selects resistant viral populations rapidly within a few days or weeks.
HCV subtypes 1a and 1b may have different resistance profiles. With genotype 1a, some resistance-associated amino acid substitutions require only one nucleotide change, but with genotype 1b, two nucleotide changes are needed, making resistance less frequent in patients with HCV genotype 1b.23
BOCEPREVIR
Boceprevir is a specific inhibitor of the HCV viral protease NS3/4A.
In phase 3 clinical trials, boceprevir 800 mg three times a day was used with pegylated interferon alfa-2b (PegIntron) 1.5 μg/kg/week and ribavirin (Rebetol) 600 to 1,400 mg daily according to body weight.
Before patients started taking boceprevir, they went through a 4-week lead-in phase, during which they received pegylated interferon and ribavirin. This schedule appeared to reduce the incidence of viral breakthrough in phase 2 trials, and it produced higher rates of sustained virologic response and lower relapse rates compared with triple therapy without a lead-in phase.
Rapid virologic response was defined as undetectable HCV RNA at week 4 of boceprevir therapy (week 8 of the whole regimen).
Boceprevir in previously untreated patients with HCV genotype 1: The SPRINT-2 trial
The Serine Protease Inhibitor Therapy 2 (SPRINT-2) trial1 included more than 1,000 previously untreated adults with HCV genotype 1 infection (938 nonblack patients and 159 black patients; two other nonblack patients did not receive any study drug and were not included in the analysis). In this double-blind trial, patients were randomized into three groups:
- The control group received the standard of care with pegylated interferon and ribavirin for 48 weeks
- The response-guided therapy group received boceprevir plus pegylated interferon and ribavirin for 24 weeks after the 4-week lead-in phase; if HCV RNA was undetectable from week 8 to week 24, treatment was considered complete, but if HCV RNA was detectable at any point from week 8 to week 24, pegylated interferon and ribavirin were continued for a total of 48 weeks.
- The fixed-duration therapy group received boceprevir, pegylated interferon, and ribavirin for 44 weeks after the lead-in period.
The rate of relapse was 8% and 9% in the boceprevir groups vs 23% in the control group. Patients in the boceprevir groups who had a decrease in HCV RNA of less than 1 log10 during the lead-in phase were found to have a significantly higher rate of boceprevirresistant variants than those who achieved a decrease of HCV RNA of 1 log10 or more.
Boceprevir in previously treated patients with HCV genotype 1: The RESPOND-2 trial
The Retreatment With HCV Serine Protease Inhibitor Boceprevir and PegIntron/Rebetol 2) (RESPOND-2) trial2 was designed to assess the efficacy of combined boceprevir, pegylated interferon, and ribavirin for repeat treatment of patients with HCV genotype 1. These patients had previously undergone standard treatment and had a reduction of 2 log10 or more in HCV RNA after 12 weeks of therapy but with detectable HCV RNA during the therapy period or had had a relapse (defined as undetectable HCV RNA at the end of a previous course of therapy with HCV RNA positivity thereafter). Importantly, null-responders (those who had a reduction of less than 2 log10 in HCV RNA after 12 weeks of therapy) were excluded from this trial.
After a lead-in period of interferon-ribavirin treatment for 4 weeks, 403 patients were assigned to one of three treatment groups:
- Pegylated interferon and ribavirin for 44 weeks (the control group)
- Boceprevir, pegylated interferon, and ribavirin in a response-guided regimen
- Boceprevir, pegylated interferon, and ribavirin for 44 weeks (the fixed-duration group).
Sustained virologic response was achieved in only 21% of patients in the control group. Adding boceprevir increased the rate to 59% in the response-guided therapy group and to 67% in the fixed-duration group. Previous relapsers had better rates than partial responders (69%–75% vs 40%–52%).
Importantly, patients who had a poor response to pegylated interferon and ribavirin during the lead-in phase (defined as having less than a 1-log decrease in the virus before starting boceprevir) had significantly lower rates of sustained virologic response and higher rates of resistance-associated virus variants.
Side effects of boceprevir
Overall, boceprevir is well tolerated. The most common side effects of triple therapy are those usually seen with pegylated interferon and ribavirin, such as flulike symptoms and fatigue (Table 2). However, anemia was more frequent in the boceprevir groups in both SPRINT-2 and RESPOND-2 (45%–50% compared with 20%–29% in the control groups). Erythropoietin was allowed in these studies and was used in about 40% of patients.
The other common side effect associated with boceprevir was dysgeusia (alteration of taste). Dysgeusia was reported by approximately 40% of patients; however, most dysgeusia events were mild to moderate in intensity and did not lead to treatment cessation.
In the SPRINT-2 trial,1 the study drugs had to be discontinued in 12% to 16% of patients in the boceprevir groups because of adverse events, which was similar to the rate (16%) in the control group. Erythropoietin was allowed in this trial, and it was used in 43% of patients in the boceprevir groups compared with 24% in the control group, with discontinuation owing to anemia occurring in 2% and 1% of cases, respectively.
TELAPREVIR
Telaprevir, the other protease NS3/4A inhibitor, has also shown efficacy over current standard therapy in phase 3 clinical trials. It was used in a dose of 750 mg three times a day with pegylated interferon alfa-2a (Pegasys) 180 μg per week and ribavirin (Copegus) 1,000 to 1,200 mg daily according to body weight. A lead-in phase with pegylated interferon and ribavirin was not applied with telaprevir, as it was in the boceprevir trials. Extended rapid virologic response was defined as an undetectable HCV RNA at weeks 4 and 12 of therapy.
Telaprevir in previously untreated patients with HCV genotype 1
The ADVANCE study3 was a double-blind randomized trial assessing the efficacy and safety of telaprevir in combination with pegylated interferon and ribavirin in more than 1,000 previously untreated patients. The three treatment groups received:
- Telaprevir, pegylated interferon, and ribavirin for 8 weeks, followed by pegylated interferon and ribavirin alone for 16 weeks in patients who achieved an extended rapid virologic response (total duration of 24 weeks) or 40 weeks in patients who did not (total duration of 48 weeks)
- Telaprevir, pegylated interferon, and ribavirin for 12 weeks, followed by pegylated interferon-ribavirin alone for 12 (total of 24 weeks) or 36 weeks (total of 48 weeks) according to extended rapid virologic response
- Standard care with pegylated interferon and ribavirin for 48 weeks.
The rate of sustained virologic response was 69% in the group that received telaprevir for 8 weeks and 75% in the group that received it for 12 weeks compared with 44% in the control group (P < .0001 for both) (Table 2). Patients infected with HCV genotype 1b had a higher sustained virologic response rate (79%) than those infected with HCV genotype 1a (71%).
Sustained virologic response rates were lower in black patients and patients with bridging fibrosis or cirrhosis, but were still significantly higher in the telaprevir groups than in the control group. The results of this subset analysis were limited by small numbers of patients in each category.
In total, 57% of those who received telaprevir for 8 weeks and 58% of those who received it for 12 weeks achieved an extended rapid virologic response and were able to cut the duration of their therapy in half (from 48 weeks to 24 weeks).
The relapse rates were 9% in the telaprevir groups and 28% in the control group.
The rate of virologic failure was lower in patients who received triple therapy than in those who received interferon-ribavirin alone (8% in the group that got telaprevir for 12 weeks and 13% in the group that got it for 8 weeks, vs 32% in the control group). The failure rate was also lower in patients with HCV genotype 1b infection than in those with genotype 1a.
The ILLUMINATE study4 (Illustrating the Effects of Combination Therapy With Telaprevir) investigated whether longer duration of treatment than that given in the ADVANCE trial increased the rate of sustained virologic response. Previously untreated patients received telaprevir, interferon, and ribavirin for 12 weeks, and those who achieved an extended rapid virologic response were randomized at week 20 to continue interferonribavirin treatment for 24 or 48 weeks of total treatment.
The sustained virologic response rates in patients who achieved an extended rapid virologic response were 92% in the group that received pegylated interferon and ribavirin for 12 weeks, and 88% in those who received it for 48 weeks. Thus, the results of this study support the use of response-guided therapy for telaprevir-based regimens.
Telaprevir in previously treated patients with HCV genotype 1: The REALIZE trial
In this phase 3 placebo-controlled trial,5 622 patients with prior relapse, partial response, or null response were randomly allocated into one of three groups:
- Telaprevir for 12 weeks plus pegylated interferon and ribavirin for 48 weeks
- Lead-in for 4 weeks followed by 12 weeks of triple therapy and another 32 weeks of pegylated interferon and ribavirin
- Pegylated interferon and ribavirin for 48 weeks (the control group).
The overall sustained virologic response rates were 66% and 64%, respectively, in the telaprevir groups vs 17% in the control group (P < .0001). The sustained virologic response rates in the telaprevir groups were 83% to 88% in prior relapsers, 54% to 59% in partial responders, and 29% to 33% in null-responders. Of note, patients did not benefit from the lead-in phase.
This was the only trial to investigate the response to triple therapy in null-responders, a group in which treatment has been considered hopeless. A response rate of approximately 31% was encouraging, especially if we compare it with the 5% response rate achieved with the current standard of care with pegylated interferon and ribavirin.
Telaprevir side effects
As with boceprevir-based triple therapy, the most common adverse events were related to pegylated interferon (Table 2).
Nearly 50% of patients who receive telaprevir develop a skin rash that is primarily eczematous, can be managed with topical steroids, and usually resolves when telaprevir is discontinued. Severe rashes occurred in 3% to 6% of patients in the ADVANCE trial,3 and three suspected cases of Stevens-Johnson syndrome have been reported to the FDA.
Other side effects that were more frequent with telaprevir included pruritus, nausea, diarrhea, and anemia. On average, the hemoglobin level decreased by an additional 1 g/dL in the telaprevir treatment groups compared with the groups that received only pegylated interferon-ribavirin. Erythropoietin use was not allowed in the phase 3 telaprevir studies, and anemia was managed by ribavirin dose reduction.
In the ADVANCE trial,3 study drugs were discontinued owing to adverse events in 7% to 8% of the patients in the telaprevir groups compared with 4% in the control group. In the ILLUMINATE trial,4 17% of patients had to permanently discontinue all study drugs due to adverse events.
FDA-APPROVED TREATMENT REGIMENS FOR BOCEPREVIR AND TELAPREVIR
For treatment algorithms, see the eFigures that accompany this article online.
Boceprevir in previously untreated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 28—Stop all treatment if HCV RNA was undetectable at weeks 8 and 24
- Week 36—Measure HCV RNA; stop boceprevir
- Week 48—Stop all treatment (eFigure 1).
Boceprevir in previously treated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 36—if HCV RNA was not detectable at week 8, stop all treatment now; if HCV RNA was detectable at week 8, stop boceprevir now but continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 2).
Telaprevir in previously untreated patients and prior relapsers
- Week 0—start telaprevir, pegylated interferon, and ribavirin
- Week 4—measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL
- Week 12—Stop telaprevir; measure HCV RNA; stop all treatment if HCV RNA is more than 1,000 IU/mL
- Week 24—Stop pegylated interferon and ribavirin if HCV RNA was undetectable at week 12; measure HCV RNA and stop treatment if it is detectable; otherwise, continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 3).
Telaprevir in patients who previously achieved a partial or null response
- Week 0—Start telaprevir, pegylated interferon, and ribavirin
- Week 4—Measure HCV RNA; stop treatment if it is more than 1,000 IU/mL
- Week 12—Measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL; if less than 1,000 IU/mL then stop telaprevir but continue pegylated interferon and ribavirin
- Week 24—Measure HCV RNA; stop treatment if HCV RNA is detectable
- Week 48—Stop all treatment (eFigure 4).
Drug interactions with boceprevir and telaprevir
Both boceprevir and telaprevir inhibit cytochrome P450 3A (CYP3A) and thus are contraindicated in combination with drugs highly dependent on CYP3A for clearance and with drugs for which elevated plasma concentrations are associated with serious adverse events, such as atorvastatin (Lipitor), simvastatin (Zocor), sildenafil (Viagra), midazolam (Versed), and St. John’s wort. Giving potent inducers of CYP3A with boceprevir or telaprevir may lead to lower exposure and loss of efficacy of both protease inhibitors.
EMERGING THERAPIES FOR HCV
Thanks to a better understanding of the biology of HCV infection, the effort to develop new therapeutic agents started to focus on targeting specific steps of the viral life cycle, including attachment, entry into cells, replication, and release.24
Currently, more than 50 clinical trials are evaluating new direct-acting antivirals to treat HCV infection.25 Monoclonal and polyclonal antibodies that target the molecular process involved in HCV attachment and entry are being developed.26 The nonstructural protein NS5B (RNA polymerase) is intimately involved in viral replication and represents a promising target.27 Several nucleosides and nonnucleoside protease inhibitors have already entered clinical trials.
The low fidelity of the HCV replication machinery leads to a very high mutation rate, thus enabling the virus to quickly develop mutations that resist agents targeting viral enzymes.28 Therefore, a novel approach is to target host cofactors that are essential for HCV replication. An intriguing study by Lanford et al29 demonstrated that antagonizing microRNA-122 (the most abundant microRNA in the liver and an essential cofactor for viral RNA replication) by the oligonucleotide SPC3649 caused marked and prolonged reduction of HCV viremia in chronically infected chimpanzees.29
Although we are still in the early stages of drug development, the future holds great promise for newer drugs to improve the sustained virologic response, shorten the duration of treatment, improve tolerability with interferon-sparing regimens, and decrease viral resistance.
FUTURE PERSPECTIVES
With the introduction of the first direct-acting antiviral medications for HCV (boceprevir and telaprevir), 2011 will be marked as the year that changed hepatitis C treatment for the better. Triple therapy with pegylated interferon, ribavirin, and either boceprevir or telaprevir has the potential for increasing the rate of sustained virologic response to around 70% in previously untreated patients and 65% in previously treated patients who are infected with HCV genotype 1. The IL28B polymorphisms appear to play a role in the rate of sustained virologic response achieved with triple therapy, with preliminary data showing a better response rate in patients who have the CC genotype.17
These drugs will add up to $50,000 to the cost of treating hepatitis C virus infection, depending on the drug used and the length of treatment. However, they may be well worth it if they prevent liver failure and the need for transplantation.
Many questions remain, such as how to use these new regimens to treat special patient populations—for example, those with a recurrence of HCV infection after liver transplantation, those co-infected with HCV and human immunodeficiency virus, and those infected with HCV genotypes other than genotype 1.
Other direct-acting antiviral agents that specifically target the replication cycle of HCV are currently in clinical development. In fact, the future has already started with the release of the Interferon-Free Regimen for the Management of HCV (INFORM-1) study results.30 This was the first trial to evaluate an interferon-free regimen for patients with chronic HCV infection using two direct-acting antiviral drugs (the protease inhibitor danoprevir and the polymerase inhibitor RG7128), with promising results.
The treatment of hepatitis c virus (HCV) infection is on the brink of major changes with the recent approval of the first direct-acting antiviral agents, the protease inhibitors boceprevir (Victrelis) and telaprevir (Incivek).
Both drugs were approved by the US Food and Drug Administration (FDA) Advisory Panel for Chronic Hepatitis C in May 2011 and are believed to significantly improve treatment outcomes for patients with HCV genotype 1 infection.
A MAJOR PUBLIC HEALTH PROBLEM
HCV infection is a major public health problem. Nearly 4 million people in the United States are infected.6,7 Most patients with acute HCV infection become chronically infected, and up to 25% eventually develop cirrhosis and its complications, making HCV infection the leading indication for liver transplantation.8–10
Chronic HCV infection has a large global impact, with 180 million people affected across all economic and social groups.11 The highest prevalence of HCV has been reported in Egypt (14%), in part due to the use of inadequately sterilized needles in mass programs to treat endemic schistosomiasis. In developed countries, hepatocellular carcinoma associated with HCV has the fastest growing cancer-related death rate.12
CURRENTLY, FEWER THAN 50% OF PATIENTS ARE CURED
The goal of HCV treatment is to eradicate the virus. However, most infected patients (especially in the United States and Europe) are infected with HCV genotype 1, which is the most difficult genotype to treat.
Successful treatment of HCV is defined as achieving a sustained virologic response—ie, the absence of detectable HCV RNA in the serum 24 weeks after completion of therapy. Once a sustained virologic response is achieved, lifetime “cure” of HCV infection is expected in more than 99% of patients.13
The current standard therapy for HCV, pegylated interferon plus ribavirin for 48 weeks, is effective in only 40% to 50% of patients with genotype 1 infection.14 Therefore, assessing predictors of response before starting treatment can help select patients who are most likely to benefit from therapy.
Viral factors associated with a sustained virologic response include HCV genotypes other than genotype 1 and a low baseline viral load.
Beneficial patient-related factors include younger age, nonblack ethnicity, low body weight (≤ 75 kg), low body mass index, absence of insulin resistance, and absence of advanced fibrosis or cirrhosis.
More recently, a single-nucleotide polymorphism near the interleukin 28B (IL28B) gene, coding for interferon lambda 3, was found to be associated with a twofold difference in the rates of sustained virologic response: patients with the favorable genotype CC were two times more likely to achieve a sustained virologic response than patients with the CT or TT genotypes.15–17
PROTEASE INHIBITORS: MECHANISM OF ACTION
NS3/4A protease inhibitors rely on the principle of end-product inhibition, in which the cleavage product of the protease (a peptide) acts to inhibit the enzyme activity; this is why they are called peptidomimetics. The active site of the NS3/4A protease is a shallow groove composed of three highly conserved amino acid residues, which may explain why protease inhibitors display high antiviral efficacy but pose a low barrier to the development of resistance.20
Protease inhibitors are prone to resistance
The development of viral resistance to protease inhibitors has been a major drawback to their use in patients with chronic HCV infection.21
HCV is a highly variable virus with many genetically distinct but closely related quasispecies circulating in the blood at any given time. Drug-resistant, mutated variants preexist within the patient’s quasispecies, but only in small quantities because of their lesser replication fitness compared with the wild-type virus.22 When direct-acting antiviral therapy is started, the quantity of the wild-type virus decreases and the mutated virus gains replication fitness. Using protease inhibitors as monotherapy selects resistant viral populations rapidly within a few days or weeks.
HCV subtypes 1a and 1b may have different resistance profiles. With genotype 1a, some resistance-associated amino acid substitutions require only one nucleotide change, but with genotype 1b, two nucleotide changes are needed, making resistance less frequent in patients with HCV genotype 1b.23
BOCEPREVIR
Boceprevir is a specific inhibitor of the HCV viral protease NS3/4A.
In phase 3 clinical trials, boceprevir 800 mg three times a day was used with pegylated interferon alfa-2b (PegIntron) 1.5 μg/kg/week and ribavirin (Rebetol) 600 to 1,400 mg daily according to body weight.
Before patients started taking boceprevir, they went through a 4-week lead-in phase, during which they received pegylated interferon and ribavirin. This schedule appeared to reduce the incidence of viral breakthrough in phase 2 trials, and it produced higher rates of sustained virologic response and lower relapse rates compared with triple therapy without a lead-in phase.
Rapid virologic response was defined as undetectable HCV RNA at week 4 of boceprevir therapy (week 8 of the whole regimen).
Boceprevir in previously untreated patients with HCV genotype 1: The SPRINT-2 trial
The Serine Protease Inhibitor Therapy 2 (SPRINT-2) trial1 included more than 1,000 previously untreated adults with HCV genotype 1 infection (938 nonblack patients and 159 black patients; two other nonblack patients did not receive any study drug and were not included in the analysis). In this double-blind trial, patients were randomized into three groups:
- The control group received the standard of care with pegylated interferon and ribavirin for 48 weeks
- The response-guided therapy group received boceprevir plus pegylated interferon and ribavirin for 24 weeks after the 4-week lead-in phase; if HCV RNA was undetectable from week 8 to week 24, treatment was considered complete, but if HCV RNA was detectable at any point from week 8 to week 24, pegylated interferon and ribavirin were continued for a total of 48 weeks.
- The fixed-duration therapy group received boceprevir, pegylated interferon, and ribavirin for 44 weeks after the lead-in period.
The rate of relapse was 8% and 9% in the boceprevir groups vs 23% in the control group. Patients in the boceprevir groups who had a decrease in HCV RNA of less than 1 log10 during the lead-in phase were found to have a significantly higher rate of boceprevirresistant variants than those who achieved a decrease of HCV RNA of 1 log10 or more.
Boceprevir in previously treated patients with HCV genotype 1: The RESPOND-2 trial
The Retreatment With HCV Serine Protease Inhibitor Boceprevir and PegIntron/Rebetol 2) (RESPOND-2) trial2 was designed to assess the efficacy of combined boceprevir, pegylated interferon, and ribavirin for repeat treatment of patients with HCV genotype 1. These patients had previously undergone standard treatment and had a reduction of 2 log10 or more in HCV RNA after 12 weeks of therapy but with detectable HCV RNA during the therapy period or had had a relapse (defined as undetectable HCV RNA at the end of a previous course of therapy with HCV RNA positivity thereafter). Importantly, null-responders (those who had a reduction of less than 2 log10 in HCV RNA after 12 weeks of therapy) were excluded from this trial.
After a lead-in period of interferon-ribavirin treatment for 4 weeks, 403 patients were assigned to one of three treatment groups:
- Pegylated interferon and ribavirin for 44 weeks (the control group)
- Boceprevir, pegylated interferon, and ribavirin in a response-guided regimen
- Boceprevir, pegylated interferon, and ribavirin for 44 weeks (the fixed-duration group).
Sustained virologic response was achieved in only 21% of patients in the control group. Adding boceprevir increased the rate to 59% in the response-guided therapy group and to 67% in the fixed-duration group. Previous relapsers had better rates than partial responders (69%–75% vs 40%–52%).
Importantly, patients who had a poor response to pegylated interferon and ribavirin during the lead-in phase (defined as having less than a 1-log decrease in the virus before starting boceprevir) had significantly lower rates of sustained virologic response and higher rates of resistance-associated virus variants.
Side effects of boceprevir
Overall, boceprevir is well tolerated. The most common side effects of triple therapy are those usually seen with pegylated interferon and ribavirin, such as flulike symptoms and fatigue (Table 2). However, anemia was more frequent in the boceprevir groups in both SPRINT-2 and RESPOND-2 (45%–50% compared with 20%–29% in the control groups). Erythropoietin was allowed in these studies and was used in about 40% of patients.
The other common side effect associated with boceprevir was dysgeusia (alteration of taste). Dysgeusia was reported by approximately 40% of patients; however, most dysgeusia events were mild to moderate in intensity and did not lead to treatment cessation.
In the SPRINT-2 trial,1 the study drugs had to be discontinued in 12% to 16% of patients in the boceprevir groups because of adverse events, which was similar to the rate (16%) in the control group. Erythropoietin was allowed in this trial, and it was used in 43% of patients in the boceprevir groups compared with 24% in the control group, with discontinuation owing to anemia occurring in 2% and 1% of cases, respectively.
TELAPREVIR
Telaprevir, the other protease NS3/4A inhibitor, has also shown efficacy over current standard therapy in phase 3 clinical trials. It was used in a dose of 750 mg three times a day with pegylated interferon alfa-2a (Pegasys) 180 μg per week and ribavirin (Copegus) 1,000 to 1,200 mg daily according to body weight. A lead-in phase with pegylated interferon and ribavirin was not applied with telaprevir, as it was in the boceprevir trials. Extended rapid virologic response was defined as an undetectable HCV RNA at weeks 4 and 12 of therapy.
Telaprevir in previously untreated patients with HCV genotype 1
The ADVANCE study3 was a double-blind randomized trial assessing the efficacy and safety of telaprevir in combination with pegylated interferon and ribavirin in more than 1,000 previously untreated patients. The three treatment groups received:
- Telaprevir, pegylated interferon, and ribavirin for 8 weeks, followed by pegylated interferon and ribavirin alone for 16 weeks in patients who achieved an extended rapid virologic response (total duration of 24 weeks) or 40 weeks in patients who did not (total duration of 48 weeks)
- Telaprevir, pegylated interferon, and ribavirin for 12 weeks, followed by pegylated interferon-ribavirin alone for 12 (total of 24 weeks) or 36 weeks (total of 48 weeks) according to extended rapid virologic response
- Standard care with pegylated interferon and ribavirin for 48 weeks.
The rate of sustained virologic response was 69% in the group that received telaprevir for 8 weeks and 75% in the group that received it for 12 weeks compared with 44% in the control group (P < .0001 for both) (Table 2). Patients infected with HCV genotype 1b had a higher sustained virologic response rate (79%) than those infected with HCV genotype 1a (71%).
Sustained virologic response rates were lower in black patients and patients with bridging fibrosis or cirrhosis, but were still significantly higher in the telaprevir groups than in the control group. The results of this subset analysis were limited by small numbers of patients in each category.
In total, 57% of those who received telaprevir for 8 weeks and 58% of those who received it for 12 weeks achieved an extended rapid virologic response and were able to cut the duration of their therapy in half (from 48 weeks to 24 weeks).
The relapse rates were 9% in the telaprevir groups and 28% in the control group.
The rate of virologic failure was lower in patients who received triple therapy than in those who received interferon-ribavirin alone (8% in the group that got telaprevir for 12 weeks and 13% in the group that got it for 8 weeks, vs 32% in the control group). The failure rate was also lower in patients with HCV genotype 1b infection than in those with genotype 1a.
The ILLUMINATE study4 (Illustrating the Effects of Combination Therapy With Telaprevir) investigated whether longer duration of treatment than that given in the ADVANCE trial increased the rate of sustained virologic response. Previously untreated patients received telaprevir, interferon, and ribavirin for 12 weeks, and those who achieved an extended rapid virologic response were randomized at week 20 to continue interferonribavirin treatment for 24 or 48 weeks of total treatment.
The sustained virologic response rates in patients who achieved an extended rapid virologic response were 92% in the group that received pegylated interferon and ribavirin for 12 weeks, and 88% in those who received it for 48 weeks. Thus, the results of this study support the use of response-guided therapy for telaprevir-based regimens.
Telaprevir in previously treated patients with HCV genotype 1: The REALIZE trial
In this phase 3 placebo-controlled trial,5 622 patients with prior relapse, partial response, or null response were randomly allocated into one of three groups:
- Telaprevir for 12 weeks plus pegylated interferon and ribavirin for 48 weeks
- Lead-in for 4 weeks followed by 12 weeks of triple therapy and another 32 weeks of pegylated interferon and ribavirin
- Pegylated interferon and ribavirin for 48 weeks (the control group).
The overall sustained virologic response rates were 66% and 64%, respectively, in the telaprevir groups vs 17% in the control group (P < .0001). The sustained virologic response rates in the telaprevir groups were 83% to 88% in prior relapsers, 54% to 59% in partial responders, and 29% to 33% in null-responders. Of note, patients did not benefit from the lead-in phase.
This was the only trial to investigate the response to triple therapy in null-responders, a group in which treatment has been considered hopeless. A response rate of approximately 31% was encouraging, especially if we compare it with the 5% response rate achieved with the current standard of care with pegylated interferon and ribavirin.
Telaprevir side effects
As with boceprevir-based triple therapy, the most common adverse events were related to pegylated interferon (Table 2).
Nearly 50% of patients who receive telaprevir develop a skin rash that is primarily eczematous, can be managed with topical steroids, and usually resolves when telaprevir is discontinued. Severe rashes occurred in 3% to 6% of patients in the ADVANCE trial,3 and three suspected cases of Stevens-Johnson syndrome have been reported to the FDA.
Other side effects that were more frequent with telaprevir included pruritus, nausea, diarrhea, and anemia. On average, the hemoglobin level decreased by an additional 1 g/dL in the telaprevir treatment groups compared with the groups that received only pegylated interferon-ribavirin. Erythropoietin use was not allowed in the phase 3 telaprevir studies, and anemia was managed by ribavirin dose reduction.
In the ADVANCE trial,3 study drugs were discontinued owing to adverse events in 7% to 8% of the patients in the telaprevir groups compared with 4% in the control group. In the ILLUMINATE trial,4 17% of patients had to permanently discontinue all study drugs due to adverse events.
FDA-APPROVED TREATMENT REGIMENS FOR BOCEPREVIR AND TELAPREVIR
For treatment algorithms, see the eFigures that accompany this article online.
Boceprevir in previously untreated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 28—Stop all treatment if HCV RNA was undetectable at weeks 8 and 24
- Week 36—Measure HCV RNA; stop boceprevir
- Week 48—Stop all treatment (eFigure 1).
Boceprevir in previously treated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 36—if HCV RNA was not detectable at week 8, stop all treatment now; if HCV RNA was detectable at week 8, stop boceprevir now but continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 2).
Telaprevir in previously untreated patients and prior relapsers
- Week 0—start telaprevir, pegylated interferon, and ribavirin
- Week 4—measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL
- Week 12—Stop telaprevir; measure HCV RNA; stop all treatment if HCV RNA is more than 1,000 IU/mL
- Week 24—Stop pegylated interferon and ribavirin if HCV RNA was undetectable at week 12; measure HCV RNA and stop treatment if it is detectable; otherwise, continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 3).
Telaprevir in patients who previously achieved a partial or null response
- Week 0—Start telaprevir, pegylated interferon, and ribavirin
- Week 4—Measure HCV RNA; stop treatment if it is more than 1,000 IU/mL
- Week 12—Measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL; if less than 1,000 IU/mL then stop telaprevir but continue pegylated interferon and ribavirin
- Week 24—Measure HCV RNA; stop treatment if HCV RNA is detectable
- Week 48—Stop all treatment (eFigure 4).
Drug interactions with boceprevir and telaprevir
Both boceprevir and telaprevir inhibit cytochrome P450 3A (CYP3A) and thus are contraindicated in combination with drugs highly dependent on CYP3A for clearance and with drugs for which elevated plasma concentrations are associated with serious adverse events, such as atorvastatin (Lipitor), simvastatin (Zocor), sildenafil (Viagra), midazolam (Versed), and St. John’s wort. Giving potent inducers of CYP3A with boceprevir or telaprevir may lead to lower exposure and loss of efficacy of both protease inhibitors.
EMERGING THERAPIES FOR HCV
Thanks to a better understanding of the biology of HCV infection, the effort to develop new therapeutic agents started to focus on targeting specific steps of the viral life cycle, including attachment, entry into cells, replication, and release.24
Currently, more than 50 clinical trials are evaluating new direct-acting antivirals to treat HCV infection.25 Monoclonal and polyclonal antibodies that target the molecular process involved in HCV attachment and entry are being developed.26 The nonstructural protein NS5B (RNA polymerase) is intimately involved in viral replication and represents a promising target.27 Several nucleosides and nonnucleoside protease inhibitors have already entered clinical trials.
The low fidelity of the HCV replication machinery leads to a very high mutation rate, thus enabling the virus to quickly develop mutations that resist agents targeting viral enzymes.28 Therefore, a novel approach is to target host cofactors that are essential for HCV replication. An intriguing study by Lanford et al29 demonstrated that antagonizing microRNA-122 (the most abundant microRNA in the liver and an essential cofactor for viral RNA replication) by the oligonucleotide SPC3649 caused marked and prolonged reduction of HCV viremia in chronically infected chimpanzees.29
Although we are still in the early stages of drug development, the future holds great promise for newer drugs to improve the sustained virologic response, shorten the duration of treatment, improve tolerability with interferon-sparing regimens, and decrease viral resistance.
FUTURE PERSPECTIVES
With the introduction of the first direct-acting antiviral medications for HCV (boceprevir and telaprevir), 2011 will be marked as the year that changed hepatitis C treatment for the better. Triple therapy with pegylated interferon, ribavirin, and either boceprevir or telaprevir has the potential for increasing the rate of sustained virologic response to around 70% in previously untreated patients and 65% in previously treated patients who are infected with HCV genotype 1. The IL28B polymorphisms appear to play a role in the rate of sustained virologic response achieved with triple therapy, with preliminary data showing a better response rate in patients who have the CC genotype.17
These drugs will add up to $50,000 to the cost of treating hepatitis C virus infection, depending on the drug used and the length of treatment. However, they may be well worth it if they prevent liver failure and the need for transplantation.
Many questions remain, such as how to use these new regimens to treat special patient populations—for example, those with a recurrence of HCV infection after liver transplantation, those co-infected with HCV and human immunodeficiency virus, and those infected with HCV genotypes other than genotype 1.
Other direct-acting antiviral agents that specifically target the replication cycle of HCV are currently in clinical development. In fact, the future has already started with the release of the Interferon-Free Regimen for the Management of HCV (INFORM-1) study results.30 This was the first trial to evaluate an interferon-free regimen for patients with chronic HCV infection using two direct-acting antiviral drugs (the protease inhibitor danoprevir and the polymerase inhibitor RG7128), with promising results.
- Poordad F, McCone J, Bacon BR, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1195–1206.
- Bacon BR, Gordon SC, Lawitz E, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1207–1217.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; for the ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Sherman KE, Flamm SL, Afdhal NH, et al; for the ILLUMINATE Study Team. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med 2011; 365:1014–1024.
- Zeuzem S, Andreone P, Pol S, et al; for the REALIZE Study Team. Telaprevir for retreatment of HCV infection. N Engl J Med 2011; 364:2417–2428.
- Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 2006; 144:705–714.
- Mitchell AE, Colvin HM, Palmer Beasley R. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology 2010; 51:729–733.
- Kim WR. The burden of hepatitis C in the United States. Hepatology 2002; 36:S30–S34.
- Marcellin P, Asselah T, Boyer N. Fibrosis and disease progression in hepatitis C. Hepatology 2002; 36:S47–S56.
- Seeff LB. Natural history of chronic hepatitis C. Hepatology 2002; 36:S35–S46.
- Lavanchy D. The global burden of hepatitis C. Liver Int 2009; 29(suppl 1):74–81.
- National Institutes of Health Consensus Development Conference Statement: Management of hepatitis C: 2002—June 10–12, 2002. Hepatology 2002; 36:S3–S20.
- Pearlman BL, Traub N. Sustained virologic response to antiviral therapy for chronic hepatitis C virus infection: a cure and so much more. Clin Infect Dis 2011; 52:889–900.
- Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med 2006; 355:2444–2451.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009; 461:399–401.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e118.
- Nielsen SU, Bassendine MF, Burt AD, Bevitt DJ, Toms GL. Characterization of the genome and structural proteins of hepatitis C virus resolved from infected human liver. J Gen Virol 2004; 85:1497–1507.
- Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology 2004; 39:5–19.
- Nelson DR. The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1 naive patients. Liver Int 2011; 31(suppl 1):53–57.
- Pawlotsky JM. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 2011; 53:1742–1751.
- Monto A, Schooley RT, Lai JC, et al. Lessons from HIV therapy applied to viral hepatitis therapy: summary of a workshop. Am J Gastroenterol 2010; 105:989–1004.
- McCown MF, Rajyaguru S, Kular S, Cammack N, Najera I. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob Agents Chemother 2009; 53:2129–2132.
- Cholongitas E, Papatheodoridis GV. Review article: novel therapeutic options for chronic hepatitis C. Aliment Pharmacol Ther 2008; 27:866–884.
- Naggie S, Patel K, McHutchison J. Hepatitis C virus directly acting antivirals: current developments with NS3/4A HCV serine protease inhibitors. J Antimicrob Chemother 2010; 65:2063–2069.
- Mir HM, Birerdinc A, Younossi ZM. Monoclonal and polyclonal antibodies against the HCV envelope proteins. Clin Liver Dis 2009; 13:477–486.
- Birerdinc A, Younossi ZM. Emerging therapies for hepatitis C virus. Expert Opin Emerg Drugs 2010; 15:535–544.
- Khattab MA. Targeting host factors: a novel rationale for the management of hepatitis C virus. World J Gastroenterol 2009; 15:3472–3479.
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010; 327:198–201.
- Gane EJ, Roberts SK, Stedman CA, et al. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2010; 376:1467–1475.
- Poordad F, McCone J, Bacon BR, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1195–1206.
- Bacon BR, Gordon SC, Lawitz E, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1207–1217.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; for the ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Sherman KE, Flamm SL, Afdhal NH, et al; for the ILLUMINATE Study Team. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med 2011; 365:1014–1024.
- Zeuzem S, Andreone P, Pol S, et al; for the REALIZE Study Team. Telaprevir for retreatment of HCV infection. N Engl J Med 2011; 364:2417–2428.
- Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 2006; 144:705–714.
- Mitchell AE, Colvin HM, Palmer Beasley R. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology 2010; 51:729–733.
- Kim WR. The burden of hepatitis C in the United States. Hepatology 2002; 36:S30–S34.
- Marcellin P, Asselah T, Boyer N. Fibrosis and disease progression in hepatitis C. Hepatology 2002; 36:S47–S56.
- Seeff LB. Natural history of chronic hepatitis C. Hepatology 2002; 36:S35–S46.
- Lavanchy D. The global burden of hepatitis C. Liver Int 2009; 29(suppl 1):74–81.
- National Institutes of Health Consensus Development Conference Statement: Management of hepatitis C: 2002—June 10–12, 2002. Hepatology 2002; 36:S3–S20.
- Pearlman BL, Traub N. Sustained virologic response to antiviral therapy for chronic hepatitis C virus infection: a cure and so much more. Clin Infect Dis 2011; 52:889–900.
- Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med 2006; 355:2444–2451.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009; 461:399–401.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e118.
- Nielsen SU, Bassendine MF, Burt AD, Bevitt DJ, Toms GL. Characterization of the genome and structural proteins of hepatitis C virus resolved from infected human liver. J Gen Virol 2004; 85:1497–1507.
- Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology 2004; 39:5–19.
- Nelson DR. The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1 naive patients. Liver Int 2011; 31(suppl 1):53–57.
- Pawlotsky JM. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 2011; 53:1742–1751.
- Monto A, Schooley RT, Lai JC, et al. Lessons from HIV therapy applied to viral hepatitis therapy: summary of a workshop. Am J Gastroenterol 2010; 105:989–1004.
- McCown MF, Rajyaguru S, Kular S, Cammack N, Najera I. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob Agents Chemother 2009; 53:2129–2132.
- Cholongitas E, Papatheodoridis GV. Review article: novel therapeutic options for chronic hepatitis C. Aliment Pharmacol Ther 2008; 27:866–884.
- Naggie S, Patel K, McHutchison J. Hepatitis C virus directly acting antivirals: current developments with NS3/4A HCV serine protease inhibitors. J Antimicrob Chemother 2010; 65:2063–2069.
- Mir HM, Birerdinc A, Younossi ZM. Monoclonal and polyclonal antibodies against the HCV envelope proteins. Clin Liver Dis 2009; 13:477–486.
- Birerdinc A, Younossi ZM. Emerging therapies for hepatitis C virus. Expert Opin Emerg Drugs 2010; 15:535–544.
- Khattab MA. Targeting host factors: a novel rationale for the management of hepatitis C virus. World J Gastroenterol 2009; 15:3472–3479.
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010; 327:198–201.
- Gane EJ, Roberts SK, Stedman CA, et al. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2010; 376:1467–1475.
KEY POINTS
- Standard care with the combination of pegylated interferon and ribavirin produces a sustained virologic response in about 40% of patients infected with HCV genotype 1, the most prevalent genotype in North America.
- New phase 3 trials showed that the addition of an oral protease inhibitor (boceprevir or telaprevir) increased the sustained virologic response rates to 70% in patients infected with HCV genotype 1.
- Boceprevir and telaprevir must be used in combination with pegylated interferon and ribavirin; they should not be used as monotherapy because of concern about the development of drug-resistant mutations.
- The main side effects of boceprevir were anemia and dysgeusia. Adverse events associated with telaprevir included rash, pruritus, anemia, and diarrhea.
Bioidentical hormone therapy: Clarifying the misconceptions
Recent product endorsements from celebrities on television have brought a new term into the vocabulary of many American women: bioidentical hormone therapy—treatment with hormone products that are identical in molecular structure to those in the human body.
Since 2002, when results of the Women’s Health Initiative1 raised questions about the safety of hormone replacement therapy, women have been inundated by commercials, talk shows, and self-help books that promote bioidentical hormone therapy as a safe and natural way to treat menopausal symptoms—and more.
Although this publicity has helped promote discussion about menopause, it has also perpetuated confusion and misinformation among the lay public and the general medical community concerning menopausal hormone therapy.
Many postmenopausal women suffering from vasomotor symptoms, vaginal dryness, and vaginal atrophy are apprehensive about seeking therapy, owing to concerns resulting from misinterpreted information derived from the Women’s Health Initiative trial.1 (See “What are the known risks of FDA-approved hormone therapy.”2–8) Many others are told to suffer through their symptoms, which may eventually pass. It is not surprising, then, that women turn to unconventional treatments that are claimed to be safer. This unfortunate situation has driven the business of many compounding pharmacies into the multibillion dollar level.
In this paper, we hope to clarify some of the misconceptions surrounding this issue. But first we need to define some terms in what has become a confusing area.

WHAT ARE BIOIDENTICAL HORMONES?
“Bioidentical” means identical in molecular structure to endogenous hormones. However, as we will see, a better distinction should be made between products that are approved and regulated by the US Food and Drug Administration (FDA) and those that are not.
Endogenous reproductive hormones
Women produce various reproductive hormones, including three estrogens—estradiol, estrone, and estriol—as well as progesterone and testosterone.9
17-beta estradiol (E2) is the most bioactive endogenous estrogen. It is primarily produced by the dominant ovarian follicle and the corpus luteum and is synthesized intracellularly through aromatase activity.10,11 The rest of the circulating estradiol is derived from peripheral conversion of estrone to estradiol, and this is the primary source in postmenopausal women not on hormone therapy.11
In postmenopausal women, serum estradiol levels are often below 15 pg/mL. Many physiologic effects of the cellular compartmentalized estradiol contribute to an over-riding force in certain tissues even after menopause.10 With the loss of estradiol, many tissues in postmenopausal women can be affected, particularly resulting in genitourinary atrophy and bone loss.
Estrone (E1), the second dominant human estrogen, is primarily derived from the metabolism of estradiol and from the aromatization of androstenedione in adipose tissue, with a small quantity being secreted directly by the ovary and the adrenal glands.9 In postmenopausal women, mean estrone levels are about 30 pg/mL.11
Estriol (E3), the least active of the endogenous estrogens, is very short-acting.
Progesterone is a 21-carbon steroid secreted by the human ovary.9 It is formed during the transformation of cholesterol to estrogens and androgens and is no longer produced after menopause.9
Testosterone. In premenopausal women, the androgen testosterone is synthesized by the ovary, the adrenal cortex, and the peripheral conversion of circulating androstenedione and dehydroepiandrosterone (DHEA).9 Over a woman’s life span, her androgen levels decline progressively.10 The rate of decline has not been shown to be appreciably affected by the onset of menopause.10
All these hormone therapy products are synthesized
Many nonmedical women’s health books erroneously classify the forms of estrogen used in hormone therapy as either bioidentical or synthetic. In fact, they are all man-made.
Bioidentical hormones are synthesized by chemically extracting diosgenin from plants such as yams and soy.12 Diosgenin is chemically modified to yield the precursor progesterone, which is then used to synthesize bioidentical estrogens and androgens.10
Nonbioidentical estrogen products include conjugated equine estrogens (CEE), which is extracted from the urine of pregnant mares. The two predominant estrogens found in CEE are equilin sulfate (native to horses) and estrone sulfate.10
Other nonbioidentical products include ethinyl estradiol, which is used in most combined oral contraceptives. It is formed after a minor chemical modification of estradiol that makes it one of the most potent estrogens. The ethinyl group at carbon 17 of ring D of the steroid nucleus greatly slows the hepatic and enzymatic degradation of the molecule and, thereby, makes oral ethinyl estradiol 15 to 20 times more active than oral estradiol.
Mestranol is an inactive prodrug that is converted in the body to ethinyl estradiol.
While many women may find the idea of natural bioidentical hormones derived from sweet potatoes or soybeans more acceptable than taking one made from horse’s urine, all the products undergo extensive chemical processing and modification.
Misconception: FDA-regulated products are not bioidentical
WHAT IS CUSTOMIZED COMPOUNDED HORMONAL THERAPY?
There is often confusion between the terms “bioidentical hormones” and “customized compounded therapy,” which are often used interchangeably. Compounded therapy combines ratios of bioidentical hormones into a particular recipe or mixture. Customized compounding can be done by local compounding pharmacies.2

Compounded bioidentical estrogen products
There are several commonly marketed compounded products.
Tri-estrogen (tri-est) is a compounded hormone preparation made up of a mixture of 80% estriol, 10% estrone, and 10% estradiol.12
Bi-estrogen (bi-est) contains estriol and estradiol in a ratio of 8:1 or 9:1.
Although both tri-est and bi-est are largely composed of estriol, given the low potency of estriol, the effects of these products may be solely mediated by their major bioactive component, estradiol.10,12 No large prospective, well-controlled clinical trial has investigated the compounded ratios of these mixtures of estrogens.10
Tri-est and bi-est are frequently promoted as posing less risk of breast or endometrial cancer than FDA-approved agents, although there is no research to back up this claim.12 In fact, estriol may have a stimulatory effect on the breast and endometrium.9
In addition to these “standard” compounded preparations, women can receive more customized compounds.
Valid uses for customized compounded formulations
Some clinical providers use customized compounded formulations when prescribing hormone therapy to women who have allergies to certain ingredients, such as peanut oil (found in the FDA-regulated oral product Prometrium). Customized compounded formulations have also been used when prescribing hormones currently not FDA-approved for women, such as testosterone and DHEA.12 Before oral micronized progesterone was marketed in the United States as Prometrium, it was frequently prescribed as a compounded hormone.
HORMONE THERAPY COMES IN VARIOUS FORMS
Both FDA-regulated hormone therapy and unregulated compounded hormone therapy come in various doses and dosage forms administered by different routes, allowing for individualization for each woman’s specific characteristics.
Estrogens: Oral, transdermal, others
Estrogen therapy can be given orally, transvaginally (as creams, tablets, and rings), transdermally (as patches, gels, and creams), subcutaneously in pellets, intranasally (in Europe), and by injection.11
Most oral contraceptives contain the synthetic estrogen ethinyl estradiol. Ethinyl estradiol is more potent than human estrogens,11 specifically in increasing the production of hepatic proteins (sex-hormone-binding globulin, renin substrate, corticosteroid-binding globulin, and thyroid-binding globulin).11
Bioidentical estradiol, taken orally in tablet form, is first processed through the liver and converted into estrone.12 This stimulates proteins such as C-reactive protein, activated protein C, and clotting factors, which may increase the risk of clotting.12 Estradiol given transdermally by patch or gel or vaginally bypasses the liver and enters the bloodstream as 17-beta estradiol, therefore avoiding stimulation of these proteins.12 Case-control data have shown an associated lower risk of deep venous thromboembolism with transdermal therapy.3
Subcutaneous pellet therapy is a less common, non-FDA-approved method of hormone therapy to relieve postmenopausal symptoms.10 In an outpatient procedure, the pellet is inserted into the subcutaneous fat of the abdomen.10 The crystalline pellet is biodegradable and contains a mixture of testosterone and 17-beta estradiol.10 It is important to remember that endometrial stimulation may be prolonged with this form of therapy and levels may be supraphysiologic.
Progestogens can also be given by different routes
Oral progesterone has poor gastrointestinal absorption and a short half-life.10 Therefore, it is micronized with oil for better absorption. Reported side effects include sedative and anesthetic effects; therefore, it is recommended that oral progesterone be taken at bedtime.9 Medroxyprogesterone acetate may interfere more with estrogen’s positive effects on cholesterol than micronized progesterone does.13
Topical progesterone preparations vary widely in dosage and formulation. Over-the-counter progesterone creams vary in concentration from no active ingredient to 450 mg or more of progesterone per ounce. Application sites for progesterone cream include the inner arm, chest, and inner thigh. No transdermal hormone should be applied to areas of the body that may allow possible contact and transference to others.
Progestogen products
Progestogen products include “natural” progesterone and synthetic progestins. They should be given concurrently with estrogen therapy in women who have an intact uterus to prevent endometrial hyperplasia.9
Bioidentical progesterone is micronized in the laboratory for better absorption in the gut.2
Misconception: Transdermal progesterone protects the endometrium
In general, transdermal progesterone should be avoided, as it does not protect against endometrial cancer.
Many forms of progesterone are available by prescription at compounding pharmacies as lotions, gels, creams, capsules, trochees, and suppositories.9 Transdermal progesterone creams are also available over the counter at health stores. Some of these creams contain only diosgenin, a progesterone precursor derived from wild yams.10 Diosgenin cannot be converted into progesterone within the body and thus does not provide an adequate amount of absorbable progesterone.9 Therefore, progesterone cream that contains only diosgenin is not effective in preventing endometrial hyperplasia and cancer.
To achieve a physiologic response, progesterone levels must be at least in the nanogram range.10 Transdermal progesterone cream has not been shown to reach this level and may not significantly improve vasomotor symptoms.12 Some practitioners prescribe cream that contains more than 400 mg progesterone per ounce. This may achieve physiologic levels of progesterone, but no improvement has been proven for bone mineral density or endometrial protection. In general, no transdermal progesterone cream can be assumed to protect the endometrium against the stimulatory effects of estrogen.
CUSTOM COMPOUNDING AND SALIVA TESTING TO INDIVIDUALIZE THERAPY
Some clinicians who prescribe compounded hormones order saliva tests. They argue the tests help them to establish which hormones are deficient and therefore to customize therapy.12 The basis for this is that saliva is similar to an ultrafiltrate of blood and, theoretically, hormone levels in saliva should represent the bioavailable hormone in serum.10
Unfortunately, this testing is often unreliable due to poor stability of samples in storage and large interassay variability.10 Many factors may alter hormone levels in saliva and make test results unreproducible, including the time of day the sample is collected and dietary habits.10 The FDA states that there is no scientific basis for using salivary testing to adjust hormone levels.2
Levels of drugs with clearance that varies depending on hepatic enzyme activity and plasma binding (capacity-limited metabolism) such as estradiol and testosterone can be monitored with total blood serum concentrations.10 However, many physiologic effects of estrogens are determined intracellularly at the level of tissues.10 Therefore, although levels during therapy with bioidentical estrogens can be monitored more precisely, the FDA states that hormone therapy should be guided by symptom response and findings on physical examination and not by hormone levels alone.2,12 It may be reasonable to order serum levels of estradiol in women being treated with therapeutic doses of bioidentical estrogen but still not achieving symptom relief. If women are being treated with conjugated equine estrogens, serum levels cannot be monitored. Total estrogen can be monitored as a send-out laboratory test.
MISCONCEPTION: HORMONE THERAPY IS A FOUNTAIN OF YOUTH
Customized compounded hormonal therapy is marketed as being able to help with rejuvenation, improve memory, sexual function, and reverse the aging process, essentially promising to be an elixir or fountain of youth.
These claims are not substantiated. However, the actual benefits of hormone therapy in women who have menopausal symptoms include alleviation of moderate to severe vasomotor symptoms and vaginal atrophy that can result in dyspareunia. By alleviating their symptoms, hormone therapy improves women’s quality of life. It also reduces the incidence of postmenopausal osteoporotic fractures.
A research finding that is often overlooked is that postmenopausal women younger than 60 years who started estrogen or estrogenprogestin therapy soon after menopause had a 30% lower rate of death from all causes.2,14 This difference was statistically significant when the estrogen and estrogen-progestin therapy groups were combined. No reduction in the mortality rate was seen if therapy was started after age 60.
MISCONCEPTION: COMPOUNDED THERAPY IS SAFER
Compounded hormone therapy is often marketed as a safer or more effective alternative to government-regulated and approved therapy. Unfortunately, these claims are often false and misleading, and safety information is not consistently provided to patients as is required with FDA-regulated hormone therapy.2
Since these compounds have not been approved by the FDA, there is no guarantee that the ingredients have been tested for purity, potency, and efficacy. There is no batch standardization. These unregulated therapies may use unapproved ingredients, routes of administration, and mixtures with contaminants such as dyes and preservatives.2
Also, custom-compounded prescriptions are considered experimental. Therefore, they are often not covered by insurance, and many women must pay for them out of pocket.11
The North American Menopause Society does not recommend custom-mixed products over well-tested, government-approved commercial products for most women.2 All bioidentical hormone prescriptions should include a patient package insert,11 identical to that required of FDA-approved products.2
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- North American Menopause Society. Estrogen and progestogen use in postmenopausal women: 2010 position statement of the North American Menopause Society. Menopause 2010; 17:242–255.
- Canonico M, Oger E, Plu-Bureau G; Estrogen and Thromboembolism Risk (ESTHER) Study Group. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation 2007; 115:840–845.
- Risks of postmenopausal hormone replacement (letters). JAMA 2002; 288:2819–2825.
- Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA 2007; 297:1465–1477.
- Grodstein F, Manson JE, Colditz GA, Willett WC, Speizer FE, Stampfer MJ. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann Intern Med 2000; 133:933–941.
- Shumaker SA, Legault C, Rapp SR, et al; WHIMS Investigators. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 2003; 289:2651–2662.
- Chlebowski RT, Anderson GL, Gass M, et al; WHI Investigators. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 2010; 304:1684–1692.
- Lobo RA. Treatment of the Postmenopausal Woman: Basic and Clinical Aspects. 3rd ed. Burlington, MA: Academic Press; 2007.
- Cirigliano M. Bioidentical hormone therapy: a review of the evidence. J Womens Health (Larchmt) 2007; 16:600–631.
- Menopause Practice: A Clinician’s Guide. 4th ed. Cleveland, OH: The North American Menopause Society; 2010.
- What are bioidentical hormones? Natural. Bioidentical. Compounded. Confusion about these terms is only adding to the confusion over hormone therapy. Harv Womens Health Watch 2006; 13:1–3.
- The Writing Group for the PEPI Trial. Effects of estrogen or estrogen/progestin regimens on heart disease risk factors in postmenopausal women. The Postmenopausal Estrogen/Progestin Interventions (PEPI) Trial. JAMA 1995; 273:199–208.
- Hodis HN, Mack WJ. Postmenopausal hormone therapy in clinical perspective. Menopause 2007; 14:944–957.
Recent product endorsements from celebrities on television have brought a new term into the vocabulary of many American women: bioidentical hormone therapy—treatment with hormone products that are identical in molecular structure to those in the human body.
Since 2002, when results of the Women’s Health Initiative1 raised questions about the safety of hormone replacement therapy, women have been inundated by commercials, talk shows, and self-help books that promote bioidentical hormone therapy as a safe and natural way to treat menopausal symptoms—and more.
Although this publicity has helped promote discussion about menopause, it has also perpetuated confusion and misinformation among the lay public and the general medical community concerning menopausal hormone therapy.
Many postmenopausal women suffering from vasomotor symptoms, vaginal dryness, and vaginal atrophy are apprehensive about seeking therapy, owing to concerns resulting from misinterpreted information derived from the Women’s Health Initiative trial.1 (See “What are the known risks of FDA-approved hormone therapy.”2–8) Many others are told to suffer through their symptoms, which may eventually pass. It is not surprising, then, that women turn to unconventional treatments that are claimed to be safer. This unfortunate situation has driven the business of many compounding pharmacies into the multibillion dollar level.
In this paper, we hope to clarify some of the misconceptions surrounding this issue. But first we need to define some terms in what has become a confusing area.
WHAT ARE BIOIDENTICAL HORMONES?
“Bioidentical” means identical in molecular structure to endogenous hormones. However, as we will see, a better distinction should be made between products that are approved and regulated by the US Food and Drug Administration (FDA) and those that are not.
Endogenous reproductive hormones
Women produce various reproductive hormones, including three estrogens—estradiol, estrone, and estriol—as well as progesterone and testosterone.9
17-beta estradiol (E2) is the most bioactive endogenous estrogen. It is primarily produced by the dominant ovarian follicle and the corpus luteum and is synthesized intracellularly through aromatase activity.10,11 The rest of the circulating estradiol is derived from peripheral conversion of estrone to estradiol, and this is the primary source in postmenopausal women not on hormone therapy.11
In postmenopausal women, serum estradiol levels are often below 15 pg/mL. Many physiologic effects of the cellular compartmentalized estradiol contribute to an over-riding force in certain tissues even after menopause.10 With the loss of estradiol, many tissues in postmenopausal women can be affected, particularly resulting in genitourinary atrophy and bone loss.
Estrone (E1), the second dominant human estrogen, is primarily derived from the metabolism of estradiol and from the aromatization of androstenedione in adipose tissue, with a small quantity being secreted directly by the ovary and the adrenal glands.9 In postmenopausal women, mean estrone levels are about 30 pg/mL.11
Estriol (E3), the least active of the endogenous estrogens, is very short-acting.
Progesterone is a 21-carbon steroid secreted by the human ovary.9 It is formed during the transformation of cholesterol to estrogens and androgens and is no longer produced after menopause.9
Testosterone. In premenopausal women, the androgen testosterone is synthesized by the ovary, the adrenal cortex, and the peripheral conversion of circulating androstenedione and dehydroepiandrosterone (DHEA).9 Over a woman’s life span, her androgen levels decline progressively.10 The rate of decline has not been shown to be appreciably affected by the onset of menopause.10
All these hormone therapy products are synthesized
Many nonmedical women’s health books erroneously classify the forms of estrogen used in hormone therapy as either bioidentical or synthetic. In fact, they are all man-made.
Bioidentical hormones are synthesized by chemically extracting diosgenin from plants such as yams and soy.12 Diosgenin is chemically modified to yield the precursor progesterone, which is then used to synthesize bioidentical estrogens and androgens.10
Nonbioidentical estrogen products include conjugated equine estrogens (CEE), which is extracted from the urine of pregnant mares. The two predominant estrogens found in CEE are equilin sulfate (native to horses) and estrone sulfate.10
Other nonbioidentical products include ethinyl estradiol, which is used in most combined oral contraceptives. It is formed after a minor chemical modification of estradiol that makes it one of the most potent estrogens. The ethinyl group at carbon 17 of ring D of the steroid nucleus greatly slows the hepatic and enzymatic degradation of the molecule and, thereby, makes oral ethinyl estradiol 15 to 20 times more active than oral estradiol.
Mestranol is an inactive prodrug that is converted in the body to ethinyl estradiol.
While many women may find the idea of natural bioidentical hormones derived from sweet potatoes or soybeans more acceptable than taking one made from horse’s urine, all the products undergo extensive chemical processing and modification.
Misconception: FDA-regulated products are not bioidentical
WHAT IS CUSTOMIZED COMPOUNDED HORMONAL THERAPY?
There is often confusion between the terms “bioidentical hormones” and “customized compounded therapy,” which are often used interchangeably. Compounded therapy combines ratios of bioidentical hormones into a particular recipe or mixture. Customized compounding can be done by local compounding pharmacies.2
Compounded bioidentical estrogen products
There are several commonly marketed compounded products.
Tri-estrogen (tri-est) is a compounded hormone preparation made up of a mixture of 80% estriol, 10% estrone, and 10% estradiol.12
Bi-estrogen (bi-est) contains estriol and estradiol in a ratio of 8:1 or 9:1.
Although both tri-est and bi-est are largely composed of estriol, given the low potency of estriol, the effects of these products may be solely mediated by their major bioactive component, estradiol.10,12 No large prospective, well-controlled clinical trial has investigated the compounded ratios of these mixtures of estrogens.10
Tri-est and bi-est are frequently promoted as posing less risk of breast or endometrial cancer than FDA-approved agents, although there is no research to back up this claim.12 In fact, estriol may have a stimulatory effect on the breast and endometrium.9
In addition to these “standard” compounded preparations, women can receive more customized compounds.
Valid uses for customized compounded formulations
Some clinical providers use customized compounded formulations when prescribing hormone therapy to women who have allergies to certain ingredients, such as peanut oil (found in the FDA-regulated oral product Prometrium). Customized compounded formulations have also been used when prescribing hormones currently not FDA-approved for women, such as testosterone and DHEA.12 Before oral micronized progesterone was marketed in the United States as Prometrium, it was frequently prescribed as a compounded hormone.
HORMONE THERAPY COMES IN VARIOUS FORMS
Both FDA-regulated hormone therapy and unregulated compounded hormone therapy come in various doses and dosage forms administered by different routes, allowing for individualization for each woman’s specific characteristics.
Estrogens: Oral, transdermal, others
Estrogen therapy can be given orally, transvaginally (as creams, tablets, and rings), transdermally (as patches, gels, and creams), subcutaneously in pellets, intranasally (in Europe), and by injection.11
Most oral contraceptives contain the synthetic estrogen ethinyl estradiol. Ethinyl estradiol is more potent than human estrogens,11 specifically in increasing the production of hepatic proteins (sex-hormone-binding globulin, renin substrate, corticosteroid-binding globulin, and thyroid-binding globulin).11
Bioidentical estradiol, taken orally in tablet form, is first processed through the liver and converted into estrone.12 This stimulates proteins such as C-reactive protein, activated protein C, and clotting factors, which may increase the risk of clotting.12 Estradiol given transdermally by patch or gel or vaginally bypasses the liver and enters the bloodstream as 17-beta estradiol, therefore avoiding stimulation of these proteins.12 Case-control data have shown an associated lower risk of deep venous thromboembolism with transdermal therapy.3
Subcutaneous pellet therapy is a less common, non-FDA-approved method of hormone therapy to relieve postmenopausal symptoms.10 In an outpatient procedure, the pellet is inserted into the subcutaneous fat of the abdomen.10 The crystalline pellet is biodegradable and contains a mixture of testosterone and 17-beta estradiol.10 It is important to remember that endometrial stimulation may be prolonged with this form of therapy and levels may be supraphysiologic.
Progestogens can also be given by different routes
Oral progesterone has poor gastrointestinal absorption and a short half-life.10 Therefore, it is micronized with oil for better absorption. Reported side effects include sedative and anesthetic effects; therefore, it is recommended that oral progesterone be taken at bedtime.9 Medroxyprogesterone acetate may interfere more with estrogen’s positive effects on cholesterol than micronized progesterone does.13
Topical progesterone preparations vary widely in dosage and formulation. Over-the-counter progesterone creams vary in concentration from no active ingredient to 450 mg or more of progesterone per ounce. Application sites for progesterone cream include the inner arm, chest, and inner thigh. No transdermal hormone should be applied to areas of the body that may allow possible contact and transference to others.
Progestogen products
Progestogen products include “natural” progesterone and synthetic progestins. They should be given concurrently with estrogen therapy in women who have an intact uterus to prevent endometrial hyperplasia.9
Bioidentical progesterone is micronized in the laboratory for better absorption in the gut.2
Misconception: Transdermal progesterone protects the endometrium
In general, transdermal progesterone should be avoided, as it does not protect against endometrial cancer.
Many forms of progesterone are available by prescription at compounding pharmacies as lotions, gels, creams, capsules, trochees, and suppositories.9 Transdermal progesterone creams are also available over the counter at health stores. Some of these creams contain only diosgenin, a progesterone precursor derived from wild yams.10 Diosgenin cannot be converted into progesterone within the body and thus does not provide an adequate amount of absorbable progesterone.9 Therefore, progesterone cream that contains only diosgenin is not effective in preventing endometrial hyperplasia and cancer.
To achieve a physiologic response, progesterone levels must be at least in the nanogram range.10 Transdermal progesterone cream has not been shown to reach this level and may not significantly improve vasomotor symptoms.12 Some practitioners prescribe cream that contains more than 400 mg progesterone per ounce. This may achieve physiologic levels of progesterone, but no improvement has been proven for bone mineral density or endometrial protection. In general, no transdermal progesterone cream can be assumed to protect the endometrium against the stimulatory effects of estrogen.
CUSTOM COMPOUNDING AND SALIVA TESTING TO INDIVIDUALIZE THERAPY
Some clinicians who prescribe compounded hormones order saliva tests. They argue the tests help them to establish which hormones are deficient and therefore to customize therapy.12 The basis for this is that saliva is similar to an ultrafiltrate of blood and, theoretically, hormone levels in saliva should represent the bioavailable hormone in serum.10
Unfortunately, this testing is often unreliable due to poor stability of samples in storage and large interassay variability.10 Many factors may alter hormone levels in saliva and make test results unreproducible, including the time of day the sample is collected and dietary habits.10 The FDA states that there is no scientific basis for using salivary testing to adjust hormone levels.2
Levels of drugs with clearance that varies depending on hepatic enzyme activity and plasma binding (capacity-limited metabolism) such as estradiol and testosterone can be monitored with total blood serum concentrations.10 However, many physiologic effects of estrogens are determined intracellularly at the level of tissues.10 Therefore, although levels during therapy with bioidentical estrogens can be monitored more precisely, the FDA states that hormone therapy should be guided by symptom response and findings on physical examination and not by hormone levels alone.2,12 It may be reasonable to order serum levels of estradiol in women being treated with therapeutic doses of bioidentical estrogen but still not achieving symptom relief. If women are being treated with conjugated equine estrogens, serum levels cannot be monitored. Total estrogen can be monitored as a send-out laboratory test.
MISCONCEPTION: HORMONE THERAPY IS A FOUNTAIN OF YOUTH
Customized compounded hormonal therapy is marketed as being able to help with rejuvenation, improve memory, sexual function, and reverse the aging process, essentially promising to be an elixir or fountain of youth.
These claims are not substantiated. However, the actual benefits of hormone therapy in women who have menopausal symptoms include alleviation of moderate to severe vasomotor symptoms and vaginal atrophy that can result in dyspareunia. By alleviating their symptoms, hormone therapy improves women’s quality of life. It also reduces the incidence of postmenopausal osteoporotic fractures.
A research finding that is often overlooked is that postmenopausal women younger than 60 years who started estrogen or estrogenprogestin therapy soon after menopause had a 30% lower rate of death from all causes.2,14 This difference was statistically significant when the estrogen and estrogen-progestin therapy groups were combined. No reduction in the mortality rate was seen if therapy was started after age 60.
MISCONCEPTION: COMPOUNDED THERAPY IS SAFER
Compounded hormone therapy is often marketed as a safer or more effective alternative to government-regulated and approved therapy. Unfortunately, these claims are often false and misleading, and safety information is not consistently provided to patients as is required with FDA-regulated hormone therapy.2
Since these compounds have not been approved by the FDA, there is no guarantee that the ingredients have been tested for purity, potency, and efficacy. There is no batch standardization. These unregulated therapies may use unapproved ingredients, routes of administration, and mixtures with contaminants such as dyes and preservatives.2
Also, custom-compounded prescriptions are considered experimental. Therefore, they are often not covered by insurance, and many women must pay for them out of pocket.11
The North American Menopause Society does not recommend custom-mixed products over well-tested, government-approved commercial products for most women.2 All bioidentical hormone prescriptions should include a patient package insert,11 identical to that required of FDA-approved products.2
Recent product endorsements from celebrities on television have brought a new term into the vocabulary of many American women: bioidentical hormone therapy—treatment with hormone products that are identical in molecular structure to those in the human body.
Since 2002, when results of the Women’s Health Initiative1 raised questions about the safety of hormone replacement therapy, women have been inundated by commercials, talk shows, and self-help books that promote bioidentical hormone therapy as a safe and natural way to treat menopausal symptoms—and more.
Although this publicity has helped promote discussion about menopause, it has also perpetuated confusion and misinformation among the lay public and the general medical community concerning menopausal hormone therapy.
Many postmenopausal women suffering from vasomotor symptoms, vaginal dryness, and vaginal atrophy are apprehensive about seeking therapy, owing to concerns resulting from misinterpreted information derived from the Women’s Health Initiative trial.1 (See “What are the known risks of FDA-approved hormone therapy.”2–8) Many others are told to suffer through their symptoms, which may eventually pass. It is not surprising, then, that women turn to unconventional treatments that are claimed to be safer. This unfortunate situation has driven the business of many compounding pharmacies into the multibillion dollar level.
In this paper, we hope to clarify some of the misconceptions surrounding this issue. But first we need to define some terms in what has become a confusing area.
WHAT ARE BIOIDENTICAL HORMONES?
“Bioidentical” means identical in molecular structure to endogenous hormones. However, as we will see, a better distinction should be made between products that are approved and regulated by the US Food and Drug Administration (FDA) and those that are not.
Endogenous reproductive hormones
Women produce various reproductive hormones, including three estrogens—estradiol, estrone, and estriol—as well as progesterone and testosterone.9
17-beta estradiol (E2) is the most bioactive endogenous estrogen. It is primarily produced by the dominant ovarian follicle and the corpus luteum and is synthesized intracellularly through aromatase activity.10,11 The rest of the circulating estradiol is derived from peripheral conversion of estrone to estradiol, and this is the primary source in postmenopausal women not on hormone therapy.11
In postmenopausal women, serum estradiol levels are often below 15 pg/mL. Many physiologic effects of the cellular compartmentalized estradiol contribute to an over-riding force in certain tissues even after menopause.10 With the loss of estradiol, many tissues in postmenopausal women can be affected, particularly resulting in genitourinary atrophy and bone loss.
Estrone (E1), the second dominant human estrogen, is primarily derived from the metabolism of estradiol and from the aromatization of androstenedione in adipose tissue, with a small quantity being secreted directly by the ovary and the adrenal glands.9 In postmenopausal women, mean estrone levels are about 30 pg/mL.11
Estriol (E3), the least active of the endogenous estrogens, is very short-acting.
Progesterone is a 21-carbon steroid secreted by the human ovary.9 It is formed during the transformation of cholesterol to estrogens and androgens and is no longer produced after menopause.9
Testosterone. In premenopausal women, the androgen testosterone is synthesized by the ovary, the adrenal cortex, and the peripheral conversion of circulating androstenedione and dehydroepiandrosterone (DHEA).9 Over a woman’s life span, her androgen levels decline progressively.10 The rate of decline has not been shown to be appreciably affected by the onset of menopause.10
All these hormone therapy products are synthesized
Many nonmedical women’s health books erroneously classify the forms of estrogen used in hormone therapy as either bioidentical or synthetic. In fact, they are all man-made.
Bioidentical hormones are synthesized by chemically extracting diosgenin from plants such as yams and soy.12 Diosgenin is chemically modified to yield the precursor progesterone, which is then used to synthesize bioidentical estrogens and androgens.10
Nonbioidentical estrogen products include conjugated equine estrogens (CEE), which is extracted from the urine of pregnant mares. The two predominant estrogens found in CEE are equilin sulfate (native to horses) and estrone sulfate.10
Other nonbioidentical products include ethinyl estradiol, which is used in most combined oral contraceptives. It is formed after a minor chemical modification of estradiol that makes it one of the most potent estrogens. The ethinyl group at carbon 17 of ring D of the steroid nucleus greatly slows the hepatic and enzymatic degradation of the molecule and, thereby, makes oral ethinyl estradiol 15 to 20 times more active than oral estradiol.
Mestranol is an inactive prodrug that is converted in the body to ethinyl estradiol.
While many women may find the idea of natural bioidentical hormones derived from sweet potatoes or soybeans more acceptable than taking one made from horse’s urine, all the products undergo extensive chemical processing and modification.
Misconception: FDA-regulated products are not bioidentical
WHAT IS CUSTOMIZED COMPOUNDED HORMONAL THERAPY?
There is often confusion between the terms “bioidentical hormones” and “customized compounded therapy,” which are often used interchangeably. Compounded therapy combines ratios of bioidentical hormones into a particular recipe or mixture. Customized compounding can be done by local compounding pharmacies.2
Compounded bioidentical estrogen products
There are several commonly marketed compounded products.
Tri-estrogen (tri-est) is a compounded hormone preparation made up of a mixture of 80% estriol, 10% estrone, and 10% estradiol.12
Bi-estrogen (bi-est) contains estriol and estradiol in a ratio of 8:1 or 9:1.
Although both tri-est and bi-est are largely composed of estriol, given the low potency of estriol, the effects of these products may be solely mediated by their major bioactive component, estradiol.10,12 No large prospective, well-controlled clinical trial has investigated the compounded ratios of these mixtures of estrogens.10
Tri-est and bi-est are frequently promoted as posing less risk of breast or endometrial cancer than FDA-approved agents, although there is no research to back up this claim.12 In fact, estriol may have a stimulatory effect on the breast and endometrium.9
In addition to these “standard” compounded preparations, women can receive more customized compounds.
Valid uses for customized compounded formulations
Some clinical providers use customized compounded formulations when prescribing hormone therapy to women who have allergies to certain ingredients, such as peanut oil (found in the FDA-regulated oral product Prometrium). Customized compounded formulations have also been used when prescribing hormones currently not FDA-approved for women, such as testosterone and DHEA.12 Before oral micronized progesterone was marketed in the United States as Prometrium, it was frequently prescribed as a compounded hormone.
HORMONE THERAPY COMES IN VARIOUS FORMS
Both FDA-regulated hormone therapy and unregulated compounded hormone therapy come in various doses and dosage forms administered by different routes, allowing for individualization for each woman’s specific characteristics.
Estrogens: Oral, transdermal, others
Estrogen therapy can be given orally, transvaginally (as creams, tablets, and rings), transdermally (as patches, gels, and creams), subcutaneously in pellets, intranasally (in Europe), and by injection.11
Most oral contraceptives contain the synthetic estrogen ethinyl estradiol. Ethinyl estradiol is more potent than human estrogens,11 specifically in increasing the production of hepatic proteins (sex-hormone-binding globulin, renin substrate, corticosteroid-binding globulin, and thyroid-binding globulin).11
Bioidentical estradiol, taken orally in tablet form, is first processed through the liver and converted into estrone.12 This stimulates proteins such as C-reactive protein, activated protein C, and clotting factors, which may increase the risk of clotting.12 Estradiol given transdermally by patch or gel or vaginally bypasses the liver and enters the bloodstream as 17-beta estradiol, therefore avoiding stimulation of these proteins.12 Case-control data have shown an associated lower risk of deep venous thromboembolism with transdermal therapy.3
Subcutaneous pellet therapy is a less common, non-FDA-approved method of hormone therapy to relieve postmenopausal symptoms.10 In an outpatient procedure, the pellet is inserted into the subcutaneous fat of the abdomen.10 The crystalline pellet is biodegradable and contains a mixture of testosterone and 17-beta estradiol.10 It is important to remember that endometrial stimulation may be prolonged with this form of therapy and levels may be supraphysiologic.
Progestogens can also be given by different routes
Oral progesterone has poor gastrointestinal absorption and a short half-life.10 Therefore, it is micronized with oil for better absorption. Reported side effects include sedative and anesthetic effects; therefore, it is recommended that oral progesterone be taken at bedtime.9 Medroxyprogesterone acetate may interfere more with estrogen’s positive effects on cholesterol than micronized progesterone does.13
Topical progesterone preparations vary widely in dosage and formulation. Over-the-counter progesterone creams vary in concentration from no active ingredient to 450 mg or more of progesterone per ounce. Application sites for progesterone cream include the inner arm, chest, and inner thigh. No transdermal hormone should be applied to areas of the body that may allow possible contact and transference to others.
Progestogen products
Progestogen products include “natural” progesterone and synthetic progestins. They should be given concurrently with estrogen therapy in women who have an intact uterus to prevent endometrial hyperplasia.9
Bioidentical progesterone is micronized in the laboratory for better absorption in the gut.2
Misconception: Transdermal progesterone protects the endometrium
In general, transdermal progesterone should be avoided, as it does not protect against endometrial cancer.
Many forms of progesterone are available by prescription at compounding pharmacies as lotions, gels, creams, capsules, trochees, and suppositories.9 Transdermal progesterone creams are also available over the counter at health stores. Some of these creams contain only diosgenin, a progesterone precursor derived from wild yams.10 Diosgenin cannot be converted into progesterone within the body and thus does not provide an adequate amount of absorbable progesterone.9 Therefore, progesterone cream that contains only diosgenin is not effective in preventing endometrial hyperplasia and cancer.
To achieve a physiologic response, progesterone levels must be at least in the nanogram range.10 Transdermal progesterone cream has not been shown to reach this level and may not significantly improve vasomotor symptoms.12 Some practitioners prescribe cream that contains more than 400 mg progesterone per ounce. This may achieve physiologic levels of progesterone, but no improvement has been proven for bone mineral density or endometrial protection. In general, no transdermal progesterone cream can be assumed to protect the endometrium against the stimulatory effects of estrogen.
CUSTOM COMPOUNDING AND SALIVA TESTING TO INDIVIDUALIZE THERAPY
Some clinicians who prescribe compounded hormones order saliva tests. They argue the tests help them to establish which hormones are deficient and therefore to customize therapy.12 The basis for this is that saliva is similar to an ultrafiltrate of blood and, theoretically, hormone levels in saliva should represent the bioavailable hormone in serum.10
Unfortunately, this testing is often unreliable due to poor stability of samples in storage and large interassay variability.10 Many factors may alter hormone levels in saliva and make test results unreproducible, including the time of day the sample is collected and dietary habits.10 The FDA states that there is no scientific basis for using salivary testing to adjust hormone levels.2
Levels of drugs with clearance that varies depending on hepatic enzyme activity and plasma binding (capacity-limited metabolism) such as estradiol and testosterone can be monitored with total blood serum concentrations.10 However, many physiologic effects of estrogens are determined intracellularly at the level of tissues.10 Therefore, although levels during therapy with bioidentical estrogens can be monitored more precisely, the FDA states that hormone therapy should be guided by symptom response and findings on physical examination and not by hormone levels alone.2,12 It may be reasonable to order serum levels of estradiol in women being treated with therapeutic doses of bioidentical estrogen but still not achieving symptom relief. If women are being treated with conjugated equine estrogens, serum levels cannot be monitored. Total estrogen can be monitored as a send-out laboratory test.
MISCONCEPTION: HORMONE THERAPY IS A FOUNTAIN OF YOUTH
Customized compounded hormonal therapy is marketed as being able to help with rejuvenation, improve memory, sexual function, and reverse the aging process, essentially promising to be an elixir or fountain of youth.
These claims are not substantiated. However, the actual benefits of hormone therapy in women who have menopausal symptoms include alleviation of moderate to severe vasomotor symptoms and vaginal atrophy that can result in dyspareunia. By alleviating their symptoms, hormone therapy improves women’s quality of life. It also reduces the incidence of postmenopausal osteoporotic fractures.
A research finding that is often overlooked is that postmenopausal women younger than 60 years who started estrogen or estrogenprogestin therapy soon after menopause had a 30% lower rate of death from all causes.2,14 This difference was statistically significant when the estrogen and estrogen-progestin therapy groups were combined. No reduction in the mortality rate was seen if therapy was started after age 60.
MISCONCEPTION: COMPOUNDED THERAPY IS SAFER
Compounded hormone therapy is often marketed as a safer or more effective alternative to government-regulated and approved therapy. Unfortunately, these claims are often false and misleading, and safety information is not consistently provided to patients as is required with FDA-regulated hormone therapy.2
Since these compounds have not been approved by the FDA, there is no guarantee that the ingredients have been tested for purity, potency, and efficacy. There is no batch standardization. These unregulated therapies may use unapproved ingredients, routes of administration, and mixtures with contaminants such as dyes and preservatives.2
Also, custom-compounded prescriptions are considered experimental. Therefore, they are often not covered by insurance, and many women must pay for them out of pocket.11
The North American Menopause Society does not recommend custom-mixed products over well-tested, government-approved commercial products for most women.2 All bioidentical hormone prescriptions should include a patient package insert,11 identical to that required of FDA-approved products.2
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- North American Menopause Society. Estrogen and progestogen use in postmenopausal women: 2010 position statement of the North American Menopause Society. Menopause 2010; 17:242–255.
- Canonico M, Oger E, Plu-Bureau G; Estrogen and Thromboembolism Risk (ESTHER) Study Group. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation 2007; 115:840–845.
- Risks of postmenopausal hormone replacement (letters). JAMA 2002; 288:2819–2825.
- Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA 2007; 297:1465–1477.
- Grodstein F, Manson JE, Colditz GA, Willett WC, Speizer FE, Stampfer MJ. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann Intern Med 2000; 133:933–941.
- Shumaker SA, Legault C, Rapp SR, et al; WHIMS Investigators. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 2003; 289:2651–2662.
- Chlebowski RT, Anderson GL, Gass M, et al; WHI Investigators. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 2010; 304:1684–1692.
- Lobo RA. Treatment of the Postmenopausal Woman: Basic and Clinical Aspects. 3rd ed. Burlington, MA: Academic Press; 2007.
- Cirigliano M. Bioidentical hormone therapy: a review of the evidence. J Womens Health (Larchmt) 2007; 16:600–631.
- Menopause Practice: A Clinician’s Guide. 4th ed. Cleveland, OH: The North American Menopause Society; 2010.
- What are bioidentical hormones? Natural. Bioidentical. Compounded. Confusion about these terms is only adding to the confusion over hormone therapy. Harv Womens Health Watch 2006; 13:1–3.
- The Writing Group for the PEPI Trial. Effects of estrogen or estrogen/progestin regimens on heart disease risk factors in postmenopausal women. The Postmenopausal Estrogen/Progestin Interventions (PEPI) Trial. JAMA 1995; 273:199–208.
- Hodis HN, Mack WJ. Postmenopausal hormone therapy in clinical perspective. Menopause 2007; 14:944–957.
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- North American Menopause Society. Estrogen and progestogen use in postmenopausal women: 2010 position statement of the North American Menopause Society. Menopause 2010; 17:242–255.
- Canonico M, Oger E, Plu-Bureau G; Estrogen and Thromboembolism Risk (ESTHER) Study Group. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation 2007; 115:840–845.
- Risks of postmenopausal hormone replacement (letters). JAMA 2002; 288:2819–2825.
- Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA 2007; 297:1465–1477.
- Grodstein F, Manson JE, Colditz GA, Willett WC, Speizer FE, Stampfer MJ. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann Intern Med 2000; 133:933–941.
- Shumaker SA, Legault C, Rapp SR, et al; WHIMS Investigators. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 2003; 289:2651–2662.
- Chlebowski RT, Anderson GL, Gass M, et al; WHI Investigators. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 2010; 304:1684–1692.
- Lobo RA. Treatment of the Postmenopausal Woman: Basic and Clinical Aspects. 3rd ed. Burlington, MA: Academic Press; 2007.
- Cirigliano M. Bioidentical hormone therapy: a review of the evidence. J Womens Health (Larchmt) 2007; 16:600–631.
- Menopause Practice: A Clinician’s Guide. 4th ed. Cleveland, OH: The North American Menopause Society; 2010.
- What are bioidentical hormones? Natural. Bioidentical. Compounded. Confusion about these terms is only adding to the confusion over hormone therapy. Harv Womens Health Watch 2006; 13:1–3.
- The Writing Group for the PEPI Trial. Effects of estrogen or estrogen/progestin regimens on heart disease risk factors in postmenopausal women. The Postmenopausal Estrogen/Progestin Interventions (PEPI) Trial. JAMA 1995; 273:199–208.
- Hodis HN, Mack WJ. Postmenopausal hormone therapy in clinical perspective. Menopause 2007; 14:944–957.
KEY POINTS
- Hormone therapy is indicated for relief of menopausal symptoms; claims of reversal of the aging process are unsubstantiated.
- Products that are custom-compounded are not regulated by the US Food and Drug Administration and therefore carry no assurance of purity, safety, or efficacy.
- Transdermal progesterone creams do not achieve high enough serum levels to protect the endometrium.
- Hormone therapy is titrated on the basis of symptom response. Measuring hormone levels in saliva is not called for and is probably not reliable.