User login
A Plea to Reconsider the Diagnosis
An eight-month-old unvaccinated boy presented to an emergency department (ED) with fever, neck pain, and lethargy. Examination of the cerebrospinal fluid (CSF) demonstrated hazy fluid with a white blood cell count of 3,906 cells/uL (90% polymorphonuclear cells, 6% lymphocytes, and 4% monocytes), 0 red blood cells/uL, protein of 40 mg/dL, and glucose of 56 mg/dL. No organisms were seen on Gram stain. Ceftriaxone and vancomycin were administered. CSF, blood, and urine cultures remained sterile; arbovirus serology was nonreactive, and polymerase chain reactions (PCRs) for enterovirus, Herpes simplex virus (HSV), Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenza were negative. His irritability improved, but his fevers continued. The antibiotics were stopped after 10 days of empiric treatment, and his fever resolved within 36 hours of cessation of antibiotics. He was diagnosed with aseptic meningitis and possible drug fever, attributed to either ceftriaxone or vancomycin.
There are many possibilities to consider in an unimmunized child with signs and symptoms of meningitis. The vaccine-preventable infections are ruled out in the setting of negative cultures and PCRs. While the most common etiology of aseptic meningitis is secondary to viral infections, the considerations of drug fever and aseptic meningitis deserve more attention. A thorough medication history should be taken as nonsteroidal anti-inflammatory drugs (NSAIDs) are relatively commonly linked to aseptic meningitis. Evaluation should focus on family history, medications, and exposures.
There was no family history of meningitis nor known exposures to mosquitos or ticks. The patient did not have a history of atypical or severe infections. He had one episode of acute otitis media that resolved without antibiotics. He had a history of delayed speech and was more irritable than his siblings.
Sixteen months later, at two years of age, he presented to his primary care physician in Wisconsin for evaluation of one day of fever and fussiness without rhinorrhea or cough. Examination showed enlarged tonsils without exudate or pharyngeal erythema. His tympanic membranes were normal, and the lung fields were clear. Two of his older siblings and his father had been diagnosed with streptococcal pharyngitis and were receiving antibiotic treatment. A rapid streptococcal antigen test was positive, and amoxicillin started.
Group A streptococcal (GAS) pharyngitis is an acute infection of the oropharynx or nasopharynx caused by Streptococcus pyogenes and is most common in school-aged children. GAS pharyngitis is less common at age two years unless there is definite exposure. The most frequent presentations in this age group (<3 years of age) include protracted nasal symptoms (congestion and rhinorrhea) and cough instead of a well-localized episode of pharyngitis.
The amoxicillin was continued for four days without improvement in fever or fussiness. His oral intake decreased, and he developed nonbilious, nonbloody emesis without diarrhea. He followed up with his pediatrician for the presumed streptococcal pharyngitis. Because of the previous concern for drug fever related to his ceftriaxone exposure, the amoxicillin was discontinued. Supportive care was recommended.
While viral infections remain the most likely etiology, noninfectious etiologies, such as vasculitis, should be considered. Kawasaki disease should be considered in any child with prolonged fever. Kawasaki disease can also cause aseptic meningitis that could provide an explanation for his original episode at eight months of age; nevertheless, it is rare for Kawasaki disease to recur.
Over the next three days, his temperature was as high as 38.8°C (101.8°F), he became more irritable, and his vomiting worsened; his family believed he had a headache. He was again seen by his pediatrician, now with eight days of fever. On examination, his oropharynx was mildly erythematous with palatal petechiae and 2+ tonsillar enlargement; shotty anterior cervical lymphadenopathy was present. Concern for incompletely treated streptococcal pharyngitis prompted prescription of azithromycin for five days.
This information does not change the differential diagnosis significantly. Azithromycin is as effective as beta lactams for the treatment of GAS pharyngitis if the GAS is susceptible to macrolides. Macrolide resistance rates vary between communities and have been as high as 15% in Wisconsin; knowledge of local resistance patterns is important.1
Despite the azithromycin, his symptoms worsened, and he became lethargic. The family believed the symptoms were similar to those during his previous episode of meningitis. They presented to an ED where he was febrile to 39.4°C (102.9°F) with a heart rate of 159 beats per minute and blood pressure of 113/84 mm Hg. His head circumference was 50.5 cm (97th percentile) compared with his weight of 10.8 kg (23.81 lbs; 22nd percentile). He was listless when undisturbed and irritable during the examination; his neck was supple and strong, and reflexes were normal. The remainder of his examination, including joints and skin, was normal. His white blood cell count was 18.6 K/uL, hemoglobin 11.8 g/dL, and platelets 401 K/uL. A chest radiograph was normal.
The patient is presenting on the 10th day of fever—a long time for any patient to remain febrile. Although most typically due to infectious etiologies, rheumatologic and oncologic diseases must be considered. It is important to characterize the pattern of fevers during the past 10 days and whether the patient has had similar febrile illnesses in the past. In this case, his past medical history substantially alters the differential diagnosis. The positive rapid strep test and history of recent strep pharyngitis are of uncertain importance, and the patient’s nonresponsiveness to antibiotics should raise concern for a second disease process (other than streptococcal infection) causing the fever. His unimmunized status changes the pretest probability of serious conditions such as bacterial meningitis caused by S. pneumoniae. A lumbar puncture should be performed, including an opening pressure; if the CSF again shows pleocytosis, but no infectious etiology is identified, then imaging of the brain (magnetic resonance imaging [MRI] or magnetic resonance angiogram) should be performed to evaluate for anatomic abnormalities.
CSF examination revealed 9,327 white blood cells/uL (82% polymorphonuclear cells, 1% lymphocytes, and 17% monocytes), 114 red blood cells/uL, protein of 87 mg/dL, and glucose of 63 mg/dL. Gram stain revealed no organisms. Ceftriaxone, vancomycin, and acyclovir were started, and he was transferred to a children’s hospital.
This history must be viewed through two alternate lenses: that the two episodes of meningitis are related or that they are unrelated. The finding of a neutrophil predominance in the CSF in the setting of aseptic (or nonbacterial) meningitis is less common than a lymphocytic predominance. Most commonly, aseptic meningitis is due to viral infection and is typically associated with a lymphocyte predominance, although a moderate neutrophil predominance can be seen in patients with enterovirus meningitis. Neutrophil-predominant aseptic meningitis can also accompany genetic auto-inflammatory syndromes (eg, familial Mediterranean fever and cryopyrin-associated periodic syndrome). This finding can also be seen in other noninfectious conditions such as neurosarcoidosis, Behçet’s disease, Cogan syndrome, and other vasculitides. Drug-induced aseptic meningitis can also cause neutrophil predominance. Additionally, the apparent neutrophil predominance could be explained if the patient had lymphopenia associated with primary or acquired immunodeficiency; therefore, the peripheral leukocyte differential obtained at the same time as the CSF should be evaluated. However, immunodeficiency is less likely given the patient’s lack of history of recurrent infections.
The main objective information added here is that the patient now has his second episode of likely aseptic meningitis with neutrophilic predominance, although it is possible that antibiotic therapy may have led to a false-negative CSF culture. However, this possible partial treatment was not a consideration in the first episode of meningitis. Having two similar episodes increases the likelihood that the patient has an underlying inflammatory/immune disorder, likely genetic (now termed “inborn errors of immunity”), or that there is a common exposure not yet revealed in the history (eg, drug-induced meningitis). Primary immunodeficiency is less likely than an autoinflammatory disease, considering the patient’s course of recovery with the first episode.
Further evaluation of the CSF did not reveal a pathogen. Bacterial CSF culture was sterile, and PCRs for HSV and enterovirus were negative.
The differential diagnosis is narrowing to include causes of recurrent, aseptic, neutrophilic meningitis. The incongruous head circumference and weight could be due to a relatively large head, a relatively low weight, or both. To interpret these data properly, one also needs to know the patient’s length, the trajectory of his growth parameters over time, and the parents’ heights and head circumferences. One possible scenario, considering the rest of the history, is that the patient has a chronic inflammatory condition of the central nervous system (CNS), leading to hydrocephalus and macrocephaly. It is possible that systemic inflammation could also lead to poor weight gain.
When considering chronic causes of aseptic meningitis associated with neutrophil predominance in the CSF, autoinflammatory disorders (cryopyrin-associated periodic syndrome, Muckle–Wells syndrome, neonatal-onset multisystem inflammatory disease [NOMID], and chronic infantile neurological cutaneous articular syndrome [CINCA]) should be considered. The patient lacks the typical deforming arthropathy of the most severe NOMID/CINCA phenotype. If the brain imaging does not reveal another etiology, then genetic testing of the patient is indicated.
Because of the history of recurrent meningitis with marked neutrophilic pleocytosis, yet no evidence of infection given normal glucose, only mildly elevated protein, and no culture growth, an MRI of the brain was obtained. MRI revealed a sharply circumscribed, homogeneous, nonenhancing 2.6 cm diameter cystic suprasellar mass with a thin rim of capsular enhancement (Figure). The appearance was most consistent with an epidermoid cyst, a dermoid, Rathke’s cleft cyst (RCC), or, less likely, a craniopharyngioma. The recurrent aseptic meningitis was attributed to chemical meningitis secondary to episodic discharging of the tumor. There was no hydrocephalus on imaging, and the enlarged head circumference was attributed to large parental head circumference.
Antibiotics were discontinued and supportive care continued. A CSF cholesterol level of 4 mg/dL was found (normal range 0.6 ± 0.2 mg/dL) on the CSF from admission. Fevers and symptoms ultimately improved with 72 hours of admission. He was discharged with neurosurgical follow-up, and within a year, he developed a third episode of aseptic meningitis. He eventually underwent a craniotomy with near-total resection of the cyst. Histopathological analysis indicated the presence of an underlying RCC, despite initial clinical and radiographic suspicion of an epidermoid cyst. He recovered well with follow-up imaging demonstrating stable resolution of the RCC and no further incidents of aseptic meningitis in the 12 months since the surgery.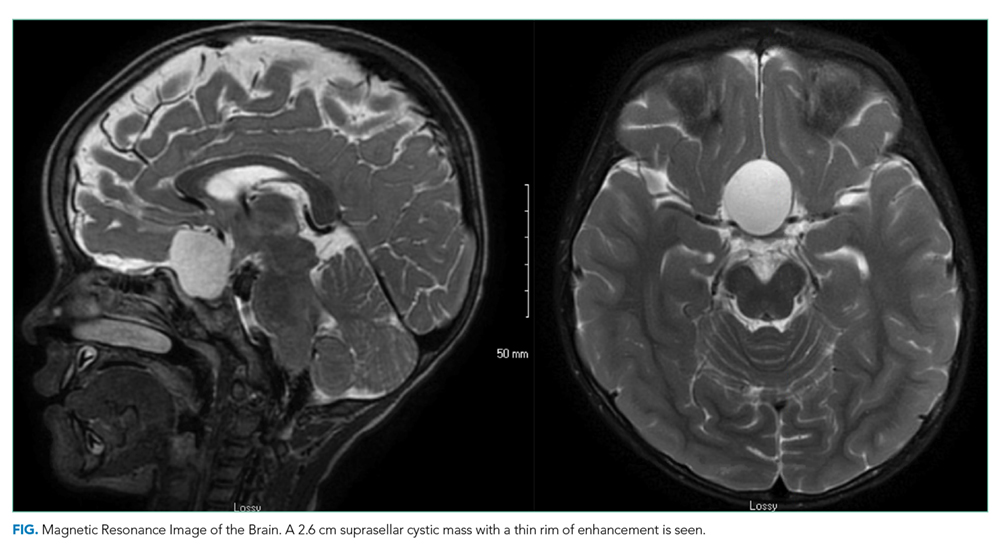
DISCUSSION
Aseptic meningitis is defined as meningitis with negative bacterial cultures from CSF and is habitually equated with viral meningitis.2 This erroneous equivalence may curb critical thinking about alternative diagnoses, as aseptic meningitis may also be associated with a wide range of both infectious and noninfectious etiologies (Table). A thorough history and physical examination are the essential first steps in determining the etiology of aseptic meningitis, as many of the listed etiologies can be effectively eliminated through the evaluation of risk factors and exposures. Laboratory evaluation of CSF including cell count with differential, glucose, and protein levels is required. Gram stain and culture should be obtained to evaluate for bacterial meningitis even if suspicion is low. Multiplex and dedicated PCRs to viral agents as well as a serologic test for arboviruses, are widely available. Multiple episodes of aseptic meningitis with HSV, known as Mollaret’s meningitis, or enterovirus, which is more common in males with X-linked agammaglobulinemia, should be considered in patients with recurrent disease. Imaging is not indicated for every patient with aseptic meningitis; however, if anatomic abnormalities or malignancy are suspected, or in the evaluation of recurrent disease, then an MRI of the brain should be considered.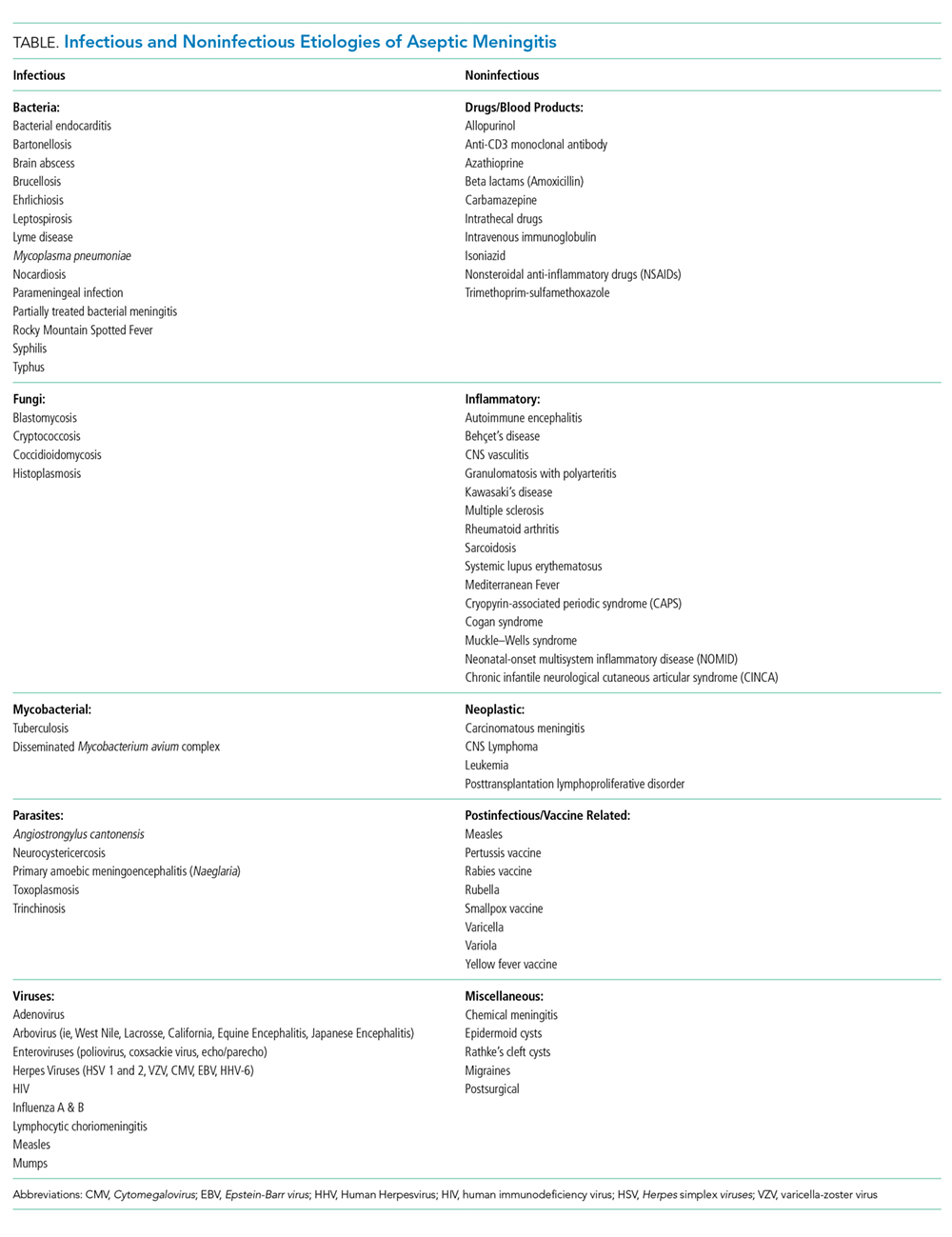
This case highlights how the analysis of CSF pleocytosis is not always predictive of a specific underlying etiology. The classic teaching that viral meningitis is associated with lymphocytic pleocytosis is based on studies of mumps meningitis.3 It is important to recognize that a neutrophilic pleocytosis is also described in viral infections including those caused by an enterovirus, herpes simplex, and arboviruses.4,5 Moreover, while the magnitude of the neutrophilic pleocytosis should always raise suspicion of bacterial meningitis, it should also be associated with hypoglycorrhachia and elevated CSF protein levels. Antibiotic pretreatment of bacterial meningitis can alter CSF chemistries (raise CSF glucose levels and lower CSF protein levels), but chemistries are unlikely to return completely to normal.6 In this case, one clue that hinted toward a noninfectious etiology was the recurrence of relatively normal CSF glucose and protein levels in the setting of such a highly inflammatory pleocytosis on multiple occasions.
There is a wide spectrum of CNS mass lesions known for causing chemical meningitis including epidermoid, dermoid, craniopharyngiomas, and RCCs. While imaging can be suggestive, histological examination is often required to make a specific diagnosis. In this patient, the diagnosis of chemical meningitis secondary to a ruptured brain tumor was confirmed by MRI. CNS tumors that may cause aseptic meningitis are typically slow-growing lesions that cause symptoms due both to local growth and regional neurovascular compression. These masses can rupture and disseminate inflammatory contents into the subarachnoid space giving rise to chemical aseptic meningitis. Their contents may include materials rich in keratin, cholesterol, and lipids, which cause an intense sterile inflammatory reaction when discharged, possibly via cholesterol activation of the inflammasome.7,8 The subsequent inflammatory response produces a neutrophilic pleocytosis, often suggestive of bacterial meningitis, while simultaneously maintaining the near normalcy of the CSF glucose and protein levels. Elevated levels of CSF cholesterol may raise suspicion of the diagnosis. Not all discharging tumors result in purely chemical meningitis, as secondary bacterial meningitis with S. pneumoniae or other respiratory flora can coexist if cysts communicate with the respiratory tract.9
Rathke’s cleft is formed during the development of the pituitary gland by the evagination of oral ectoderm through the precursor of the oral cavity.10 The cleft gives rise to the endocrine cells of the anterior pituitary. It subsequently disconnects from the oral cavity and develops into the pars intermedia between the anterior and posterior pituitary. Cystic enlargement of Rathke’s cleft through epithelial proliferation and secondary secretions leads to the development of an RCC. RCCs are nonneoplastic lesions, and the majority are diagnosed incidentally. Asymptomatic RCCs are common and are detected in 13%-22% of routine autopsies.11 Symptomatic lesions may present with headaches due to mechanical effects on pain-sensitive dura or cranial nerves. Severe acute onset headaches may arise secondary to pituitary hemorrhage. RCCs can also cause ophthalmic or endocrinological impairment due to sellar compression. As in the present case, rarely cystic rupture and subarachnoid extravasation of epithelial-derived contents lead to a chemical aseptic meningitis.12
Surgical resection is indicated for symptomatic RCCs that exert neurologic, ophthalmic, or endocrinological symptoms.13 The surgical goal is the removal of the lesion and complete excision of the capsule unless it is extremely adherent to neurovascular structures. Surgical morbidity is related to the risk of hypopituitarism, visual decline, incomplete resection with lesion regrowth, and aseptic meningitis. Surgical approaches to this region are potentially complicated by proximity to optic nerves, pituitary glands, major arteries, and perforating vessels belonging to the circle of Willis. In addition, potential dehiscence of the skull base floor due to progressive cyst growth can give rise to a delayed risk of CSF leak and complicate surgical recovery. Surgery was indicated for this patient because of the parasellar location of his cyst placing him at risk for visual decline due to compression of the optic chiasm as well as pituitary dysfunction or obstructive hydrocephalus from ventricular compression.
This case is illustrative for learning because, at the outset, there were many possibilities to explore in an unimmunized child with meningitis. This patient’s neutrophilic cell count and partial antibiotic treatment only compounded the certainty of a bacterial etiology. However, further scrutiny of the history and laboratory parameters revealed the true underlying diagnosis of RCC. Ultimately, the plea to reconsider the pleocytosis was heard.
KEY LEARNING POINTS
- The CSF cell count and differential should be used in conjunction with CSF chemistries (glucose and protein) to raise or lower suspicion of bacterial meningitis.
- Aseptic meningitis is a syndrome and not a specific diagnosis. Clinicians should be alert to key aspects of the history and physical examination, which prompt consideration of noninfectious etiologies.
- Aseptic chemical meningitis secondary to discharging CNS tumors, including RCCs, should be considered in episodes of recurrent culture-negative meningitis.
1. DeMuri GP, Sterkel AK, Kubica PA, Duster MN, Reed KD, Wald ER. Macrolide and clindamycin resistance in group a streptococci isolated from children with pharyngitis. Pediatr Infect Dis J. 2017;36(3):342-344. https://doi.org/10.1097/INF.0000000000001442.
2. Lee BE, Davies HD. Aseptic meningitis. Curr Opin Infect Dis. 2007;20(3):272-277. https://doi.org/10.1097/QCO.0b013e3280ad4672.
3. Ritter BS. Mumps meningoencephalitis in children. J Pediatr. 1958;52(4):424-433. https://doi.org/10.1016/S0022-3476(58)80063-3.
4. Miller SA, Wald ER, Bergman I, DeBiasio R. Enteroviral meningitis in January with marked cerebrospinal fluid pleocytosis. Pediatr Infect Dis. 1986;5(6):706-707. https://doi.org/10.1097/00006454-198611000-00024.
5. Jaijakul S, Salazar L, Wootton SH, Aguilera E, Hasbun R. The clinical significance of neutrophilic pleocytosis in cerebrospinal fluid in patients with viral central nervous system infections. Int J Infect Dis. 2017;59:77-81. https://doi.org/10.1016/j.ijid.2017.04.010.
6. Nigrovic LE, Malley R, Macias CG, et al. Effect of antibiotic pretreatment on cerebrospinal fluid profiles of children with bacterial meningitis. Pediatrics. 2008;122(4):726-730. https://doi.org/10.1542/peds.2007-3275.
7. Cherian A, Baheti NN, Easwar HV, Nair DS, Iype T. Recurrent meningitis due to epidermoid. J Pediatr Neurosci. 2012;7(1):47-48. https://doi.org/10.4103/1817-1745.97624.
8. Grebe A, Latz E. Cholesterol crystals and inflammation. Curr Rheumatol Rep. 2013;15(3):313. https://doi.org/10.1007/s11926-012-0313-z.
9. Kriss TC, Kriss VM, Warf BC. Recurrent meningitis: the search for the dermoid or epidermoid tumor. Pediatr Infect Dis J. 1995;14(8):697-700.
10. Bresson D, Herman P, Polivka M, Froelich S. Sellar lesions/pathology. Otolaryngol Clin North Am. 2016;49(1):63-93. https://doi.org/10.1016/j.otc.2015.09.004.
11. Billeci D, Marton E, Tripodi M, Orvieto E, Longatti P. Symptomatic Rathke’s cleft cysts: a radiological, surgical and pathological review. Pituitary. 2004;7(3):131-137. https://doi.org/10.1007/s11102-005-1755-3.
12. Steinberg GK, Koenig GH, Golden JB. Symptomatic Rathke’s cleft cysts. Report of two cases. J Neurosurg. 1982;56(2):290-295. https://doi.org/10.3171/jns.1982.56.2.0290.
13. Zada G. Rathke cleft cysts: a review of clinical and surgical management. Neurosurg Focus. 2011;31(1):E1. https://doi.org/10.3171/2011.5.FOCUS1183.
An eight-month-old unvaccinated boy presented to an emergency department (ED) with fever, neck pain, and lethargy. Examination of the cerebrospinal fluid (CSF) demonstrated hazy fluid with a white blood cell count of 3,906 cells/uL (90% polymorphonuclear cells, 6% lymphocytes, and 4% monocytes), 0 red blood cells/uL, protein of 40 mg/dL, and glucose of 56 mg/dL. No organisms were seen on Gram stain. Ceftriaxone and vancomycin were administered. CSF, blood, and urine cultures remained sterile; arbovirus serology was nonreactive, and polymerase chain reactions (PCRs) for enterovirus, Herpes simplex virus (HSV), Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenza were negative. His irritability improved, but his fevers continued. The antibiotics were stopped after 10 days of empiric treatment, and his fever resolved within 36 hours of cessation of antibiotics. He was diagnosed with aseptic meningitis and possible drug fever, attributed to either ceftriaxone or vancomycin.
There are many possibilities to consider in an unimmunized child with signs and symptoms of meningitis. The vaccine-preventable infections are ruled out in the setting of negative cultures and PCRs. While the most common etiology of aseptic meningitis is secondary to viral infections, the considerations of drug fever and aseptic meningitis deserve more attention. A thorough medication history should be taken as nonsteroidal anti-inflammatory drugs (NSAIDs) are relatively commonly linked to aseptic meningitis. Evaluation should focus on family history, medications, and exposures.
There was no family history of meningitis nor known exposures to mosquitos or ticks. The patient did not have a history of atypical or severe infections. He had one episode of acute otitis media that resolved without antibiotics. He had a history of delayed speech and was more irritable than his siblings.
Sixteen months later, at two years of age, he presented to his primary care physician in Wisconsin for evaluation of one day of fever and fussiness without rhinorrhea or cough. Examination showed enlarged tonsils without exudate or pharyngeal erythema. His tympanic membranes were normal, and the lung fields were clear. Two of his older siblings and his father had been diagnosed with streptococcal pharyngitis and were receiving antibiotic treatment. A rapid streptococcal antigen test was positive, and amoxicillin started.
Group A streptococcal (GAS) pharyngitis is an acute infection of the oropharynx or nasopharynx caused by Streptococcus pyogenes and is most common in school-aged children. GAS pharyngitis is less common at age two years unless there is definite exposure. The most frequent presentations in this age group (<3 years of age) include protracted nasal symptoms (congestion and rhinorrhea) and cough instead of a well-localized episode of pharyngitis.
The amoxicillin was continued for four days without improvement in fever or fussiness. His oral intake decreased, and he developed nonbilious, nonbloody emesis without diarrhea. He followed up with his pediatrician for the presumed streptococcal pharyngitis. Because of the previous concern for drug fever related to his ceftriaxone exposure, the amoxicillin was discontinued. Supportive care was recommended.
While viral infections remain the most likely etiology, noninfectious etiologies, such as vasculitis, should be considered. Kawasaki disease should be considered in any child with prolonged fever. Kawasaki disease can also cause aseptic meningitis that could provide an explanation for his original episode at eight months of age; nevertheless, it is rare for Kawasaki disease to recur.
Over the next three days, his temperature was as high as 38.8°C (101.8°F), he became more irritable, and his vomiting worsened; his family believed he had a headache. He was again seen by his pediatrician, now with eight days of fever. On examination, his oropharynx was mildly erythematous with palatal petechiae and 2+ tonsillar enlargement; shotty anterior cervical lymphadenopathy was present. Concern for incompletely treated streptococcal pharyngitis prompted prescription of azithromycin for five days.
This information does not change the differential diagnosis significantly. Azithromycin is as effective as beta lactams for the treatment of GAS pharyngitis if the GAS is susceptible to macrolides. Macrolide resistance rates vary between communities and have been as high as 15% in Wisconsin; knowledge of local resistance patterns is important.1
Despite the azithromycin, his symptoms worsened, and he became lethargic. The family believed the symptoms were similar to those during his previous episode of meningitis. They presented to an ED where he was febrile to 39.4°C (102.9°F) with a heart rate of 159 beats per minute and blood pressure of 113/84 mm Hg. His head circumference was 50.5 cm (97th percentile) compared with his weight of 10.8 kg (23.81 lbs; 22nd percentile). He was listless when undisturbed and irritable during the examination; his neck was supple and strong, and reflexes were normal. The remainder of his examination, including joints and skin, was normal. His white blood cell count was 18.6 K/uL, hemoglobin 11.8 g/dL, and platelets 401 K/uL. A chest radiograph was normal.
The patient is presenting on the 10th day of fever—a long time for any patient to remain febrile. Although most typically due to infectious etiologies, rheumatologic and oncologic diseases must be considered. It is important to characterize the pattern of fevers during the past 10 days and whether the patient has had similar febrile illnesses in the past. In this case, his past medical history substantially alters the differential diagnosis. The positive rapid strep test and history of recent strep pharyngitis are of uncertain importance, and the patient’s nonresponsiveness to antibiotics should raise concern for a second disease process (other than streptococcal infection) causing the fever. His unimmunized status changes the pretest probability of serious conditions such as bacterial meningitis caused by S. pneumoniae. A lumbar puncture should be performed, including an opening pressure; if the CSF again shows pleocytosis, but no infectious etiology is identified, then imaging of the brain (magnetic resonance imaging [MRI] or magnetic resonance angiogram) should be performed to evaluate for anatomic abnormalities.
CSF examination revealed 9,327 white blood cells/uL (82% polymorphonuclear cells, 1% lymphocytes, and 17% monocytes), 114 red blood cells/uL, protein of 87 mg/dL, and glucose of 63 mg/dL. Gram stain revealed no organisms. Ceftriaxone, vancomycin, and acyclovir were started, and he was transferred to a children’s hospital.
This history must be viewed through two alternate lenses: that the two episodes of meningitis are related or that they are unrelated. The finding of a neutrophil predominance in the CSF in the setting of aseptic (or nonbacterial) meningitis is less common than a lymphocytic predominance. Most commonly, aseptic meningitis is due to viral infection and is typically associated with a lymphocyte predominance, although a moderate neutrophil predominance can be seen in patients with enterovirus meningitis. Neutrophil-predominant aseptic meningitis can also accompany genetic auto-inflammatory syndromes (eg, familial Mediterranean fever and cryopyrin-associated periodic syndrome). This finding can also be seen in other noninfectious conditions such as neurosarcoidosis, Behçet’s disease, Cogan syndrome, and other vasculitides. Drug-induced aseptic meningitis can also cause neutrophil predominance. Additionally, the apparent neutrophil predominance could be explained if the patient had lymphopenia associated with primary or acquired immunodeficiency; therefore, the peripheral leukocyte differential obtained at the same time as the CSF should be evaluated. However, immunodeficiency is less likely given the patient’s lack of history of recurrent infections.
The main objective information added here is that the patient now has his second episode of likely aseptic meningitis with neutrophilic predominance, although it is possible that antibiotic therapy may have led to a false-negative CSF culture. However, this possible partial treatment was not a consideration in the first episode of meningitis. Having two similar episodes increases the likelihood that the patient has an underlying inflammatory/immune disorder, likely genetic (now termed “inborn errors of immunity”), or that there is a common exposure not yet revealed in the history (eg, drug-induced meningitis). Primary immunodeficiency is less likely than an autoinflammatory disease, considering the patient’s course of recovery with the first episode.
Further evaluation of the CSF did not reveal a pathogen. Bacterial CSF culture was sterile, and PCRs for HSV and enterovirus were negative.
The differential diagnosis is narrowing to include causes of recurrent, aseptic, neutrophilic meningitis. The incongruous head circumference and weight could be due to a relatively large head, a relatively low weight, or both. To interpret these data properly, one also needs to know the patient’s length, the trajectory of his growth parameters over time, and the parents’ heights and head circumferences. One possible scenario, considering the rest of the history, is that the patient has a chronic inflammatory condition of the central nervous system (CNS), leading to hydrocephalus and macrocephaly. It is possible that systemic inflammation could also lead to poor weight gain.
When considering chronic causes of aseptic meningitis associated with neutrophil predominance in the CSF, autoinflammatory disorders (cryopyrin-associated periodic syndrome, Muckle–Wells syndrome, neonatal-onset multisystem inflammatory disease [NOMID], and chronic infantile neurological cutaneous articular syndrome [CINCA]) should be considered. The patient lacks the typical deforming arthropathy of the most severe NOMID/CINCA phenotype. If the brain imaging does not reveal another etiology, then genetic testing of the patient is indicated.
Because of the history of recurrent meningitis with marked neutrophilic pleocytosis, yet no evidence of infection given normal glucose, only mildly elevated protein, and no culture growth, an MRI of the brain was obtained. MRI revealed a sharply circumscribed, homogeneous, nonenhancing 2.6 cm diameter cystic suprasellar mass with a thin rim of capsular enhancement (Figure). The appearance was most consistent with an epidermoid cyst, a dermoid, Rathke’s cleft cyst (RCC), or, less likely, a craniopharyngioma. The recurrent aseptic meningitis was attributed to chemical meningitis secondary to episodic discharging of the tumor. There was no hydrocephalus on imaging, and the enlarged head circumference was attributed to large parental head circumference.
Antibiotics were discontinued and supportive care continued. A CSF cholesterol level of 4 mg/dL was found (normal range 0.6 ± 0.2 mg/dL) on the CSF from admission. Fevers and symptoms ultimately improved with 72 hours of admission. He was discharged with neurosurgical follow-up, and within a year, he developed a third episode of aseptic meningitis. He eventually underwent a craniotomy with near-total resection of the cyst. Histopathological analysis indicated the presence of an underlying RCC, despite initial clinical and radiographic suspicion of an epidermoid cyst. He recovered well with follow-up imaging demonstrating stable resolution of the RCC and no further incidents of aseptic meningitis in the 12 months since the surgery.
DISCUSSION
Aseptic meningitis is defined as meningitis with negative bacterial cultures from CSF and is habitually equated with viral meningitis.2 This erroneous equivalence may curb critical thinking about alternative diagnoses, as aseptic meningitis may also be associated with a wide range of both infectious and noninfectious etiologies (Table). A thorough history and physical examination are the essential first steps in determining the etiology of aseptic meningitis, as many of the listed etiologies can be effectively eliminated through the evaluation of risk factors and exposures. Laboratory evaluation of CSF including cell count with differential, glucose, and protein levels is required. Gram stain and culture should be obtained to evaluate for bacterial meningitis even if suspicion is low. Multiplex and dedicated PCRs to viral agents as well as a serologic test for arboviruses, are widely available. Multiple episodes of aseptic meningitis with HSV, known as Mollaret’s meningitis, or enterovirus, which is more common in males with X-linked agammaglobulinemia, should be considered in patients with recurrent disease. Imaging is not indicated for every patient with aseptic meningitis; however, if anatomic abnormalities or malignancy are suspected, or in the evaluation of recurrent disease, then an MRI of the brain should be considered.
This case highlights how the analysis of CSF pleocytosis is not always predictive of a specific underlying etiology. The classic teaching that viral meningitis is associated with lymphocytic pleocytosis is based on studies of mumps meningitis.3 It is important to recognize that a neutrophilic pleocytosis is also described in viral infections including those caused by an enterovirus, herpes simplex, and arboviruses.4,5 Moreover, while the magnitude of the neutrophilic pleocytosis should always raise suspicion of bacterial meningitis, it should also be associated with hypoglycorrhachia and elevated CSF protein levels. Antibiotic pretreatment of bacterial meningitis can alter CSF chemistries (raise CSF glucose levels and lower CSF protein levels), but chemistries are unlikely to return completely to normal.6 In this case, one clue that hinted toward a noninfectious etiology was the recurrence of relatively normal CSF glucose and protein levels in the setting of such a highly inflammatory pleocytosis on multiple occasions.
There is a wide spectrum of CNS mass lesions known for causing chemical meningitis including epidermoid, dermoid, craniopharyngiomas, and RCCs. While imaging can be suggestive, histological examination is often required to make a specific diagnosis. In this patient, the diagnosis of chemical meningitis secondary to a ruptured brain tumor was confirmed by MRI. CNS tumors that may cause aseptic meningitis are typically slow-growing lesions that cause symptoms due both to local growth and regional neurovascular compression. These masses can rupture and disseminate inflammatory contents into the subarachnoid space giving rise to chemical aseptic meningitis. Their contents may include materials rich in keratin, cholesterol, and lipids, which cause an intense sterile inflammatory reaction when discharged, possibly via cholesterol activation of the inflammasome.7,8 The subsequent inflammatory response produces a neutrophilic pleocytosis, often suggestive of bacterial meningitis, while simultaneously maintaining the near normalcy of the CSF glucose and protein levels. Elevated levels of CSF cholesterol may raise suspicion of the diagnosis. Not all discharging tumors result in purely chemical meningitis, as secondary bacterial meningitis with S. pneumoniae or other respiratory flora can coexist if cysts communicate with the respiratory tract.9
Rathke’s cleft is formed during the development of the pituitary gland by the evagination of oral ectoderm through the precursor of the oral cavity.10 The cleft gives rise to the endocrine cells of the anterior pituitary. It subsequently disconnects from the oral cavity and develops into the pars intermedia between the anterior and posterior pituitary. Cystic enlargement of Rathke’s cleft through epithelial proliferation and secondary secretions leads to the development of an RCC. RCCs are nonneoplastic lesions, and the majority are diagnosed incidentally. Asymptomatic RCCs are common and are detected in 13%-22% of routine autopsies.11 Symptomatic lesions may present with headaches due to mechanical effects on pain-sensitive dura or cranial nerves. Severe acute onset headaches may arise secondary to pituitary hemorrhage. RCCs can also cause ophthalmic or endocrinological impairment due to sellar compression. As in the present case, rarely cystic rupture and subarachnoid extravasation of epithelial-derived contents lead to a chemical aseptic meningitis.12
Surgical resection is indicated for symptomatic RCCs that exert neurologic, ophthalmic, or endocrinological symptoms.13 The surgical goal is the removal of the lesion and complete excision of the capsule unless it is extremely adherent to neurovascular structures. Surgical morbidity is related to the risk of hypopituitarism, visual decline, incomplete resection with lesion regrowth, and aseptic meningitis. Surgical approaches to this region are potentially complicated by proximity to optic nerves, pituitary glands, major arteries, and perforating vessels belonging to the circle of Willis. In addition, potential dehiscence of the skull base floor due to progressive cyst growth can give rise to a delayed risk of CSF leak and complicate surgical recovery. Surgery was indicated for this patient because of the parasellar location of his cyst placing him at risk for visual decline due to compression of the optic chiasm as well as pituitary dysfunction or obstructive hydrocephalus from ventricular compression.
This case is illustrative for learning because, at the outset, there were many possibilities to explore in an unimmunized child with meningitis. This patient’s neutrophilic cell count and partial antibiotic treatment only compounded the certainty of a bacterial etiology. However, further scrutiny of the history and laboratory parameters revealed the true underlying diagnosis of RCC. Ultimately, the plea to reconsider the pleocytosis was heard.
KEY LEARNING POINTS
- The CSF cell count and differential should be used in conjunction with CSF chemistries (glucose and protein) to raise or lower suspicion of bacterial meningitis.
- Aseptic meningitis is a syndrome and not a specific diagnosis. Clinicians should be alert to key aspects of the history and physical examination, which prompt consideration of noninfectious etiologies.
- Aseptic chemical meningitis secondary to discharging CNS tumors, including RCCs, should be considered in episodes of recurrent culture-negative meningitis.
An eight-month-old unvaccinated boy presented to an emergency department (ED) with fever, neck pain, and lethargy. Examination of the cerebrospinal fluid (CSF) demonstrated hazy fluid with a white blood cell count of 3,906 cells/uL (90% polymorphonuclear cells, 6% lymphocytes, and 4% monocytes), 0 red blood cells/uL, protein of 40 mg/dL, and glucose of 56 mg/dL. No organisms were seen on Gram stain. Ceftriaxone and vancomycin were administered. CSF, blood, and urine cultures remained sterile; arbovirus serology was nonreactive, and polymerase chain reactions (PCRs) for enterovirus, Herpes simplex virus (HSV), Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenza were negative. His irritability improved, but his fevers continued. The antibiotics were stopped after 10 days of empiric treatment, and his fever resolved within 36 hours of cessation of antibiotics. He was diagnosed with aseptic meningitis and possible drug fever, attributed to either ceftriaxone or vancomycin.
There are many possibilities to consider in an unimmunized child with signs and symptoms of meningitis. The vaccine-preventable infections are ruled out in the setting of negative cultures and PCRs. While the most common etiology of aseptic meningitis is secondary to viral infections, the considerations of drug fever and aseptic meningitis deserve more attention. A thorough medication history should be taken as nonsteroidal anti-inflammatory drugs (NSAIDs) are relatively commonly linked to aseptic meningitis. Evaluation should focus on family history, medications, and exposures.
There was no family history of meningitis nor known exposures to mosquitos or ticks. The patient did not have a history of atypical or severe infections. He had one episode of acute otitis media that resolved without antibiotics. He had a history of delayed speech and was more irritable than his siblings.
Sixteen months later, at two years of age, he presented to his primary care physician in Wisconsin for evaluation of one day of fever and fussiness without rhinorrhea or cough. Examination showed enlarged tonsils without exudate or pharyngeal erythema. His tympanic membranes were normal, and the lung fields were clear. Two of his older siblings and his father had been diagnosed with streptococcal pharyngitis and were receiving antibiotic treatment. A rapid streptococcal antigen test was positive, and amoxicillin started.
Group A streptococcal (GAS) pharyngitis is an acute infection of the oropharynx or nasopharynx caused by Streptococcus pyogenes and is most common in school-aged children. GAS pharyngitis is less common at age two years unless there is definite exposure. The most frequent presentations in this age group (<3 years of age) include protracted nasal symptoms (congestion and rhinorrhea) and cough instead of a well-localized episode of pharyngitis.
The amoxicillin was continued for four days without improvement in fever or fussiness. His oral intake decreased, and he developed nonbilious, nonbloody emesis without diarrhea. He followed up with his pediatrician for the presumed streptococcal pharyngitis. Because of the previous concern for drug fever related to his ceftriaxone exposure, the amoxicillin was discontinued. Supportive care was recommended.
While viral infections remain the most likely etiology, noninfectious etiologies, such as vasculitis, should be considered. Kawasaki disease should be considered in any child with prolonged fever. Kawasaki disease can also cause aseptic meningitis that could provide an explanation for his original episode at eight months of age; nevertheless, it is rare for Kawasaki disease to recur.
Over the next three days, his temperature was as high as 38.8°C (101.8°F), he became more irritable, and his vomiting worsened; his family believed he had a headache. He was again seen by his pediatrician, now with eight days of fever. On examination, his oropharynx was mildly erythematous with palatal petechiae and 2+ tonsillar enlargement; shotty anterior cervical lymphadenopathy was present. Concern for incompletely treated streptococcal pharyngitis prompted prescription of azithromycin for five days.
This information does not change the differential diagnosis significantly. Azithromycin is as effective as beta lactams for the treatment of GAS pharyngitis if the GAS is susceptible to macrolides. Macrolide resistance rates vary between communities and have been as high as 15% in Wisconsin; knowledge of local resistance patterns is important.1
Despite the azithromycin, his symptoms worsened, and he became lethargic. The family believed the symptoms were similar to those during his previous episode of meningitis. They presented to an ED where he was febrile to 39.4°C (102.9°F) with a heart rate of 159 beats per minute and blood pressure of 113/84 mm Hg. His head circumference was 50.5 cm (97th percentile) compared with his weight of 10.8 kg (23.81 lbs; 22nd percentile). He was listless when undisturbed and irritable during the examination; his neck was supple and strong, and reflexes were normal. The remainder of his examination, including joints and skin, was normal. His white blood cell count was 18.6 K/uL, hemoglobin 11.8 g/dL, and platelets 401 K/uL. A chest radiograph was normal.
The patient is presenting on the 10th day of fever—a long time for any patient to remain febrile. Although most typically due to infectious etiologies, rheumatologic and oncologic diseases must be considered. It is important to characterize the pattern of fevers during the past 10 days and whether the patient has had similar febrile illnesses in the past. In this case, his past medical history substantially alters the differential diagnosis. The positive rapid strep test and history of recent strep pharyngitis are of uncertain importance, and the patient’s nonresponsiveness to antibiotics should raise concern for a second disease process (other than streptococcal infection) causing the fever. His unimmunized status changes the pretest probability of serious conditions such as bacterial meningitis caused by S. pneumoniae. A lumbar puncture should be performed, including an opening pressure; if the CSF again shows pleocytosis, but no infectious etiology is identified, then imaging of the brain (magnetic resonance imaging [MRI] or magnetic resonance angiogram) should be performed to evaluate for anatomic abnormalities.
CSF examination revealed 9,327 white blood cells/uL (82% polymorphonuclear cells, 1% lymphocytes, and 17% monocytes), 114 red blood cells/uL, protein of 87 mg/dL, and glucose of 63 mg/dL. Gram stain revealed no organisms. Ceftriaxone, vancomycin, and acyclovir were started, and he was transferred to a children’s hospital.
This history must be viewed through two alternate lenses: that the two episodes of meningitis are related or that they are unrelated. The finding of a neutrophil predominance in the CSF in the setting of aseptic (or nonbacterial) meningitis is less common than a lymphocytic predominance. Most commonly, aseptic meningitis is due to viral infection and is typically associated with a lymphocyte predominance, although a moderate neutrophil predominance can be seen in patients with enterovirus meningitis. Neutrophil-predominant aseptic meningitis can also accompany genetic auto-inflammatory syndromes (eg, familial Mediterranean fever and cryopyrin-associated periodic syndrome). This finding can also be seen in other noninfectious conditions such as neurosarcoidosis, Behçet’s disease, Cogan syndrome, and other vasculitides. Drug-induced aseptic meningitis can also cause neutrophil predominance. Additionally, the apparent neutrophil predominance could be explained if the patient had lymphopenia associated with primary or acquired immunodeficiency; therefore, the peripheral leukocyte differential obtained at the same time as the CSF should be evaluated. However, immunodeficiency is less likely given the patient’s lack of history of recurrent infections.
The main objective information added here is that the patient now has his second episode of likely aseptic meningitis with neutrophilic predominance, although it is possible that antibiotic therapy may have led to a false-negative CSF culture. However, this possible partial treatment was not a consideration in the first episode of meningitis. Having two similar episodes increases the likelihood that the patient has an underlying inflammatory/immune disorder, likely genetic (now termed “inborn errors of immunity”), or that there is a common exposure not yet revealed in the history (eg, drug-induced meningitis). Primary immunodeficiency is less likely than an autoinflammatory disease, considering the patient’s course of recovery with the first episode.
Further evaluation of the CSF did not reveal a pathogen. Bacterial CSF culture was sterile, and PCRs for HSV and enterovirus were negative.
The differential diagnosis is narrowing to include causes of recurrent, aseptic, neutrophilic meningitis. The incongruous head circumference and weight could be due to a relatively large head, a relatively low weight, or both. To interpret these data properly, one also needs to know the patient’s length, the trajectory of his growth parameters over time, and the parents’ heights and head circumferences. One possible scenario, considering the rest of the history, is that the patient has a chronic inflammatory condition of the central nervous system (CNS), leading to hydrocephalus and macrocephaly. It is possible that systemic inflammation could also lead to poor weight gain.
When considering chronic causes of aseptic meningitis associated with neutrophil predominance in the CSF, autoinflammatory disorders (cryopyrin-associated periodic syndrome, Muckle–Wells syndrome, neonatal-onset multisystem inflammatory disease [NOMID], and chronic infantile neurological cutaneous articular syndrome [CINCA]) should be considered. The patient lacks the typical deforming arthropathy of the most severe NOMID/CINCA phenotype. If the brain imaging does not reveal another etiology, then genetic testing of the patient is indicated.
Because of the history of recurrent meningitis with marked neutrophilic pleocytosis, yet no evidence of infection given normal glucose, only mildly elevated protein, and no culture growth, an MRI of the brain was obtained. MRI revealed a sharply circumscribed, homogeneous, nonenhancing 2.6 cm diameter cystic suprasellar mass with a thin rim of capsular enhancement (Figure). The appearance was most consistent with an epidermoid cyst, a dermoid, Rathke’s cleft cyst (RCC), or, less likely, a craniopharyngioma. The recurrent aseptic meningitis was attributed to chemical meningitis secondary to episodic discharging of the tumor. There was no hydrocephalus on imaging, and the enlarged head circumference was attributed to large parental head circumference.
Antibiotics were discontinued and supportive care continued. A CSF cholesterol level of 4 mg/dL was found (normal range 0.6 ± 0.2 mg/dL) on the CSF from admission. Fevers and symptoms ultimately improved with 72 hours of admission. He was discharged with neurosurgical follow-up, and within a year, he developed a third episode of aseptic meningitis. He eventually underwent a craniotomy with near-total resection of the cyst. Histopathological analysis indicated the presence of an underlying RCC, despite initial clinical and radiographic suspicion of an epidermoid cyst. He recovered well with follow-up imaging demonstrating stable resolution of the RCC and no further incidents of aseptic meningitis in the 12 months since the surgery.
DISCUSSION
Aseptic meningitis is defined as meningitis with negative bacterial cultures from CSF and is habitually equated with viral meningitis.2 This erroneous equivalence may curb critical thinking about alternative diagnoses, as aseptic meningitis may also be associated with a wide range of both infectious and noninfectious etiologies (Table). A thorough history and physical examination are the essential first steps in determining the etiology of aseptic meningitis, as many of the listed etiologies can be effectively eliminated through the evaluation of risk factors and exposures. Laboratory evaluation of CSF including cell count with differential, glucose, and protein levels is required. Gram stain and culture should be obtained to evaluate for bacterial meningitis even if suspicion is low. Multiplex and dedicated PCRs to viral agents as well as a serologic test for arboviruses, are widely available. Multiple episodes of aseptic meningitis with HSV, known as Mollaret’s meningitis, or enterovirus, which is more common in males with X-linked agammaglobulinemia, should be considered in patients with recurrent disease. Imaging is not indicated for every patient with aseptic meningitis; however, if anatomic abnormalities or malignancy are suspected, or in the evaluation of recurrent disease, then an MRI of the brain should be considered.
This case highlights how the analysis of CSF pleocytosis is not always predictive of a specific underlying etiology. The classic teaching that viral meningitis is associated with lymphocytic pleocytosis is based on studies of mumps meningitis.3 It is important to recognize that a neutrophilic pleocytosis is also described in viral infections including those caused by an enterovirus, herpes simplex, and arboviruses.4,5 Moreover, while the magnitude of the neutrophilic pleocytosis should always raise suspicion of bacterial meningitis, it should also be associated with hypoglycorrhachia and elevated CSF protein levels. Antibiotic pretreatment of bacterial meningitis can alter CSF chemistries (raise CSF glucose levels and lower CSF protein levels), but chemistries are unlikely to return completely to normal.6 In this case, one clue that hinted toward a noninfectious etiology was the recurrence of relatively normal CSF glucose and protein levels in the setting of such a highly inflammatory pleocytosis on multiple occasions.
There is a wide spectrum of CNS mass lesions known for causing chemical meningitis including epidermoid, dermoid, craniopharyngiomas, and RCCs. While imaging can be suggestive, histological examination is often required to make a specific diagnosis. In this patient, the diagnosis of chemical meningitis secondary to a ruptured brain tumor was confirmed by MRI. CNS tumors that may cause aseptic meningitis are typically slow-growing lesions that cause symptoms due both to local growth and regional neurovascular compression. These masses can rupture and disseminate inflammatory contents into the subarachnoid space giving rise to chemical aseptic meningitis. Their contents may include materials rich in keratin, cholesterol, and lipids, which cause an intense sterile inflammatory reaction when discharged, possibly via cholesterol activation of the inflammasome.7,8 The subsequent inflammatory response produces a neutrophilic pleocytosis, often suggestive of bacterial meningitis, while simultaneously maintaining the near normalcy of the CSF glucose and protein levels. Elevated levels of CSF cholesterol may raise suspicion of the diagnosis. Not all discharging tumors result in purely chemical meningitis, as secondary bacterial meningitis with S. pneumoniae or other respiratory flora can coexist if cysts communicate with the respiratory tract.9
Rathke’s cleft is formed during the development of the pituitary gland by the evagination of oral ectoderm through the precursor of the oral cavity.10 The cleft gives rise to the endocrine cells of the anterior pituitary. It subsequently disconnects from the oral cavity and develops into the pars intermedia between the anterior and posterior pituitary. Cystic enlargement of Rathke’s cleft through epithelial proliferation and secondary secretions leads to the development of an RCC. RCCs are nonneoplastic lesions, and the majority are diagnosed incidentally. Asymptomatic RCCs are common and are detected in 13%-22% of routine autopsies.11 Symptomatic lesions may present with headaches due to mechanical effects on pain-sensitive dura or cranial nerves. Severe acute onset headaches may arise secondary to pituitary hemorrhage. RCCs can also cause ophthalmic or endocrinological impairment due to sellar compression. As in the present case, rarely cystic rupture and subarachnoid extravasation of epithelial-derived contents lead to a chemical aseptic meningitis.12
Surgical resection is indicated for symptomatic RCCs that exert neurologic, ophthalmic, or endocrinological symptoms.13 The surgical goal is the removal of the lesion and complete excision of the capsule unless it is extremely adherent to neurovascular structures. Surgical morbidity is related to the risk of hypopituitarism, visual decline, incomplete resection with lesion regrowth, and aseptic meningitis. Surgical approaches to this region are potentially complicated by proximity to optic nerves, pituitary glands, major arteries, and perforating vessels belonging to the circle of Willis. In addition, potential dehiscence of the skull base floor due to progressive cyst growth can give rise to a delayed risk of CSF leak and complicate surgical recovery. Surgery was indicated for this patient because of the parasellar location of his cyst placing him at risk for visual decline due to compression of the optic chiasm as well as pituitary dysfunction or obstructive hydrocephalus from ventricular compression.
This case is illustrative for learning because, at the outset, there were many possibilities to explore in an unimmunized child with meningitis. This patient’s neutrophilic cell count and partial antibiotic treatment only compounded the certainty of a bacterial etiology. However, further scrutiny of the history and laboratory parameters revealed the true underlying diagnosis of RCC. Ultimately, the plea to reconsider the pleocytosis was heard.
KEY LEARNING POINTS
- The CSF cell count and differential should be used in conjunction with CSF chemistries (glucose and protein) to raise or lower suspicion of bacterial meningitis.
- Aseptic meningitis is a syndrome and not a specific diagnosis. Clinicians should be alert to key aspects of the history and physical examination, which prompt consideration of noninfectious etiologies.
- Aseptic chemical meningitis secondary to discharging CNS tumors, including RCCs, should be considered in episodes of recurrent culture-negative meningitis.
1. DeMuri GP, Sterkel AK, Kubica PA, Duster MN, Reed KD, Wald ER. Macrolide and clindamycin resistance in group a streptococci isolated from children with pharyngitis. Pediatr Infect Dis J. 2017;36(3):342-344. https://doi.org/10.1097/INF.0000000000001442.
2. Lee BE, Davies HD. Aseptic meningitis. Curr Opin Infect Dis. 2007;20(3):272-277. https://doi.org/10.1097/QCO.0b013e3280ad4672.
3. Ritter BS. Mumps meningoencephalitis in children. J Pediatr. 1958;52(4):424-433. https://doi.org/10.1016/S0022-3476(58)80063-3.
4. Miller SA, Wald ER, Bergman I, DeBiasio R. Enteroviral meningitis in January with marked cerebrospinal fluid pleocytosis. Pediatr Infect Dis. 1986;5(6):706-707. https://doi.org/10.1097/00006454-198611000-00024.
5. Jaijakul S, Salazar L, Wootton SH, Aguilera E, Hasbun R. The clinical significance of neutrophilic pleocytosis in cerebrospinal fluid in patients with viral central nervous system infections. Int J Infect Dis. 2017;59:77-81. https://doi.org/10.1016/j.ijid.2017.04.010.
6. Nigrovic LE, Malley R, Macias CG, et al. Effect of antibiotic pretreatment on cerebrospinal fluid profiles of children with bacterial meningitis. Pediatrics. 2008;122(4):726-730. https://doi.org/10.1542/peds.2007-3275.
7. Cherian A, Baheti NN, Easwar HV, Nair DS, Iype T. Recurrent meningitis due to epidermoid. J Pediatr Neurosci. 2012;7(1):47-48. https://doi.org/10.4103/1817-1745.97624.
8. Grebe A, Latz E. Cholesterol crystals and inflammation. Curr Rheumatol Rep. 2013;15(3):313. https://doi.org/10.1007/s11926-012-0313-z.
9. Kriss TC, Kriss VM, Warf BC. Recurrent meningitis: the search for the dermoid or epidermoid tumor. Pediatr Infect Dis J. 1995;14(8):697-700.
10. Bresson D, Herman P, Polivka M, Froelich S. Sellar lesions/pathology. Otolaryngol Clin North Am. 2016;49(1):63-93. https://doi.org/10.1016/j.otc.2015.09.004.
11. Billeci D, Marton E, Tripodi M, Orvieto E, Longatti P. Symptomatic Rathke’s cleft cysts: a radiological, surgical and pathological review. Pituitary. 2004;7(3):131-137. https://doi.org/10.1007/s11102-005-1755-3.
12. Steinberg GK, Koenig GH, Golden JB. Symptomatic Rathke’s cleft cysts. Report of two cases. J Neurosurg. 1982;56(2):290-295. https://doi.org/10.3171/jns.1982.56.2.0290.
13. Zada G. Rathke cleft cysts: a review of clinical and surgical management. Neurosurg Focus. 2011;31(1):E1. https://doi.org/10.3171/2011.5.FOCUS1183.
1. DeMuri GP, Sterkel AK, Kubica PA, Duster MN, Reed KD, Wald ER. Macrolide and clindamycin resistance in group a streptococci isolated from children with pharyngitis. Pediatr Infect Dis J. 2017;36(3):342-344. https://doi.org/10.1097/INF.0000000000001442.
2. Lee BE, Davies HD. Aseptic meningitis. Curr Opin Infect Dis. 2007;20(3):272-277. https://doi.org/10.1097/QCO.0b013e3280ad4672.
3. Ritter BS. Mumps meningoencephalitis in children. J Pediatr. 1958;52(4):424-433. https://doi.org/10.1016/S0022-3476(58)80063-3.
4. Miller SA, Wald ER, Bergman I, DeBiasio R. Enteroviral meningitis in January with marked cerebrospinal fluid pleocytosis. Pediatr Infect Dis. 1986;5(6):706-707. https://doi.org/10.1097/00006454-198611000-00024.
5. Jaijakul S, Salazar L, Wootton SH, Aguilera E, Hasbun R. The clinical significance of neutrophilic pleocytosis in cerebrospinal fluid in patients with viral central nervous system infections. Int J Infect Dis. 2017;59:77-81. https://doi.org/10.1016/j.ijid.2017.04.010.
6. Nigrovic LE, Malley R, Macias CG, et al. Effect of antibiotic pretreatment on cerebrospinal fluid profiles of children with bacterial meningitis. Pediatrics. 2008;122(4):726-730. https://doi.org/10.1542/peds.2007-3275.
7. Cherian A, Baheti NN, Easwar HV, Nair DS, Iype T. Recurrent meningitis due to epidermoid. J Pediatr Neurosci. 2012;7(1):47-48. https://doi.org/10.4103/1817-1745.97624.
8. Grebe A, Latz E. Cholesterol crystals and inflammation. Curr Rheumatol Rep. 2013;15(3):313. https://doi.org/10.1007/s11926-012-0313-z.
9. Kriss TC, Kriss VM, Warf BC. Recurrent meningitis: the search for the dermoid or epidermoid tumor. Pediatr Infect Dis J. 1995;14(8):697-700.
10. Bresson D, Herman P, Polivka M, Froelich S. Sellar lesions/pathology. Otolaryngol Clin North Am. 2016;49(1):63-93. https://doi.org/10.1016/j.otc.2015.09.004.
11. Billeci D, Marton E, Tripodi M, Orvieto E, Longatti P. Symptomatic Rathke’s cleft cysts: a radiological, surgical and pathological review. Pituitary. 2004;7(3):131-137. https://doi.org/10.1007/s11102-005-1755-3.
12. Steinberg GK, Koenig GH, Golden JB. Symptomatic Rathke’s cleft cysts. Report of two cases. J Neurosurg. 1982;56(2):290-295. https://doi.org/10.3171/jns.1982.56.2.0290.
13. Zada G. Rathke cleft cysts: a review of clinical and surgical management. Neurosurg Focus. 2011;31(1):E1. https://doi.org/10.3171/2011.5.FOCUS1183.
© 2019 Society of Hospital Medicine
A Branching Algorithm
A 21-year-old man with a history of hypertension presented to the emergency department with four days of generalized abdominal pain, nausea, and vomiting as well as one month of loose stools. He also had a headache (not further specified) for one day. Due to his nausea, he had been unable to take his medications for two days. Home blood pressure measurements over the preceding two days revealed systolic pressures exceeding 200 mm Hg. He did not experience fever, dyspnea, chest pain, vision changes, numbness, weakness, diaphoresis, or palpitations.
Abdominal pain with vomiting and diarrhea is often caused by a self-limited gastroenteritis. However, the priority initially is to exclude serious intraabdominal processes including arterial insufficiency, bowel obstruction, organ perforation, or organ-based infection or inflammation (eg, appendicitis, cholecystitis, pancreatitis). Essential hypertension accounts for 95% of cases of hypertension in the United States, but given this patient’s young age, secondary causes should be evaluated. These include primary aldosteronism (the most common endocrine cause for hypertension in young patients), chronic kidney disease, fibromuscular dysplasia, illicit drug use, hypercortisolism, pheochromocytoma, and coarctation of the aorta. Thyrotoxicosis can elevate blood pressure (although usually not to this extent) and cause hyperdefecation. While the etiology of the chronic hypertension is uncertain, the proximate cause of the acute rise in blood pressure is likely the stress of his acute illness and the inability to take his prescribed antihypertensive medications. In the setting of severe hypertension, his headache may reflect an intracranial hemorrhage and his abdominal pain could signal an aortic dissection.
His medical history included hypertension diagnosed at age 16 as well as anxiety diagnosed following a panic attack at age 19. Over the past year, he had also developed persistent nausea, which was attributed to gastroesophageal reflux disease. His medications included metoprolol 50 mg daily, amlodipine 5 mg daily, hydrochlorothiazide 12.5 mg daily, escitalopram 20 mg daily, and omeprazole 20 mg daily. His father and 15-year-old brother also had hypertension. He was a part-time student while working at a car dealership. He did not smoke or use drugs and he rarely drank alcohol.
The need for three antihypertensive medications (albeit at submaximal doses) reflects the severity of his hypertension (provided challenges with medication adherence have been excluded). His family history, especially that of his brother who was diagnosed with hypertension at an early age, and the patient’s own early onset hypertension point toward an inherited form of hypertension. Autosomal dominant polycystic kidney disease often results in hypertension before chronic kidney disease develops. Rare inherited forms of hypertension include familial hyperaldosteronism, apparent mineralocorticoid excess, Liddle syndrome, or a hereditary endocrine tumor syndrome predisposing to pheochromocytoma. Even among patients who report classic pheochromocytoma symptoms, such as headache and anxiety, the diagnosis remains unlikely as these symptoms are nonspecific and highly prevalent in the general population. However, once secondary hypertension is plausible or suspected, testing for hyperadrenergic states, which can also cause nausea and vomiting during times of catecholamine excess, should be pursued.
His temperature was 97.5°F, heart rate 95 beats per minute and regular, respiratory rate 18 breaths per minute, blood pressure 181/118 mm Hg (systolic and diastolic pressures in each arm were within 10 mm Hg), and oxygen saturation 100% on room air. Systolic and diastolic pressures did not decrease by more than 20 mm Hg and 10 mm Hg, respectively, after he stood for two minutes. His body mass index was 24 kg/m2. He was alert and appeared slightly anxious. There was a bounding point of maximal impulse in the fifth intercostal space at the midclavicular line and a 3/6 systolic murmur at the left upper sternal border with radiation to the carotid arteries. His abdomen was soft with generalized tenderness to palpation and without rebound tenderness, masses, organomegaly, or bruits. There was no costovertebral angle tenderness. No lymphadenopathy was present. His fundoscopic, pulmonary, skin and neurologic examinations were normal.
Laboratory studies revealed a white blood cell count of 13.3 × 103/uL with a normal differential, hemoglobin 13.9 g/dL, platelet count 373 × 103/uL, sodium 142 mmol/L, potassium 3.8 mmol/L, chloride 103 mmol/L,bicarbonate 25 mmol/L, blood urea nitrogen 12 mg/dL, creatinine 1.3 mg/dL (a baseline creatinine level was not available), glucose 88 mg/dL, calcium 10.6 mg/dL, albumin 4.9 g/dL, aspartate aminotransferase 27 IU/L, alanine aminotransferase 37 IU/L, and lipase 40 IU/L. Urinalysis revealed 5-10 white blood cells per high power field without casts and 10 mg/dL protein. Urine toxicology was not performed. Electrocardiogram (ECG) showed left ventricular hypertrophy (LVH). Chest radiography was normal.
The abdominal examination does not suggest peritonitis. The laboratory tests do not suggest inflammation of the liver, pancreas, or biliary tree as the cause of his abdominal pain or diarrhea. The murmur may indicate hypertrophic cardiomyopathy or a congenital anomaly such as bicuspid aortic valve; but neither would explain hypertension unless they were associated with another developmental abnormality, such as coarctation of the aorta. Tricuspid regurgitation is conceivable and if confirmed, might raise concern for carcinoid syndrome, which can cause diarrhea. The normal neurologic examination, including the absence of papilledema, lowers suspicion of intracranial hemorrhage as a cause of his headache.
The albumin of 4.9 g/dL likely reflects hypovolemia resulting from vomiting and diarrhea. Vasoconstriction associated with pheochromocytoma can cause pressure diuresis and resultant hypovolemia. Hyperaldosteronism arising from bilateral adrenal hyperplasia or adrenal adenoma commonly causes hypokalemia, although this is not a universal feature.
The duration of his mildly decreased glomerular filtration rate is uncertain. He may have chronic kidney disease from sustained hypertension, or acute kidney injury from hypovolemia. The mild pyuria could indicate infection or renal calculi, either of which could account for generalized abdominal pain or could reflect an acute renal injury from acute interstitial nephritis from his proton pump inhibitor or hydrochlorothiazide.
LVH on the ECG indicates longstanding hypertension. The chest radiograph does not reveal clues to the etiology of or sequelae from hypertension. In particular, there is no widened aorta to suggest aortic dissection, no pulmonary edema to indicate heart failure, and no rib notching that points toward aortic coarctation. A transthoracic echocardiogram to assess for valvular and other structural abnormalities is warranted.
Tests for secondary hypertension should be sent, including serum aldosterone and renin levels to assess for primary aldosteronism and plasma or 24-hour urine normetanephrine and metanephrine levels to assess for pheochromocytoma. Biochemical evaluation is the mainstay for endocrine hypertension evaluation and should be followed by imaging if abnormal results are found.
Intact parathyroid hormone (PTH) was 78 pg/mL (normal, 10-65 pg/mL), thyroid stimulating hormone 3.6 mIU/L (normal, 0.30-5.50 mIU/L), and morning cortisol 4.1 ug/dL (normal, >7.0 ug/dL). Plasma aldosterone was 14.6 ng/dL (normal, 1-16 ng/dL), plasma renin activity 3.6 ng/mL/hr (normal, 0.5-3.5 ng/mL/hr), and aldosterone-renin ratio 4.1 (normal, <20). Transthoracic echocardiogram showed LVH with normal valves, wall motion, and proximal aorta; the left ventricular ejection fraction was 70%. Magnetic resonance angiography of the renal vessels demonstrated no abnormalities.
Computed tomography (CT) of the abdomen and pelvis with oral and intravenous contrast revealed a 5 cm heterogeneous enhancing mass associated with the prostate gland extending into the base of the bladder. The mass obstructed the right renal collecting system and ureter causing severe right-sided ureterectasis and hydronephrosis. There was also 2.8 cm right-sided paracaval lymph node enlargement and 2.1 cm right-sided and 1.5 cm left-sided external iliac lymph node enlargement (Figure 1). There were no adrenal masses.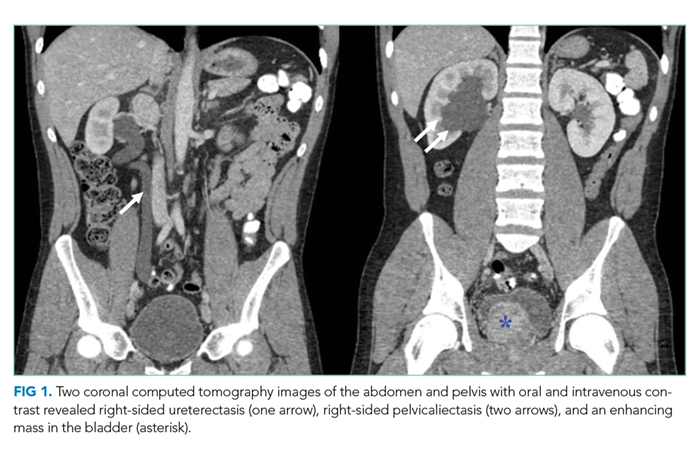
He is young for prostate, bladder, or colorectal cancer, but early onset variations of these tumors, along with metastatic testicular cancer, must be considered for the pelvic mass and associated lymphadenopathy. Prostatic masses can be infectious (eg, abscess) or malignant (eg, adenocarcinoma, small cell carcinoma). Additional considerations for abdominopelvic cancer are sarcomas, germ cell tumors, or lymphoma. A low aldosterone-renin ratio coupled with a normal potassium level makes primary aldosteronism unlikely. The normal angiography excludes renovascular hypertension.
His abdominal pain and gastrointestinal symptoms could arise from irritation of the bowel, distension of the right-sided urinary collecting system, or products secreted from the mass (eg, catecholamines). The hyperdynamic precordium, elevated ejection fraction, and murmur may reflect augmented blood flow from a hyperadrenergic state. A unifying diagnosis would be a pheochromocytoma. However, given the normal appearance of the adrenal glands on CT imaging, catecholamines arising from a paraganglioma, a tumor of the autonomic nervous system, is more likely. These tumors often secrete catecholamines and can be metastatic (suggested here by the lymphadenopathy). Functional imaging or biopsy of either the mass or an adjacent lymph node is indicated. However, because of the possibility of a catecholamine-secreting tumor, he should be treated with an alpha-adrenergic receptor antagonist before undergoing a biopsy to prevent unopposed vasoconstriction from catecholamine leakage.
Scrotal ultrasound revealed no evidence of a testicular tumor. Lactate dehydrogenase (LDH) was 179 IU/L (normal, 120-240 IU/L) and prostate specific antigen (PSA) was 0.7 ng/mL (normal, <2.5 ng/mL). The patient was given amlodipine and labetalol with improvement of blood pressures to 160s/100s. His creatinine decreased to 1.1 mg/dL. He underwent CT-guided biopsy of a pelvic lymph node. CT of the head without intravenous contrast demonstrated no intracranial abnormalities. His headache resolved with improvement in blood pressure, and he had minimal gastrointestinal symptoms during his hospitalization. No stool studies were sent. A right-sided percutaneous nephrostomy was placed which yielded >15 L of urine from the tube over the next four days.
Upon the first episode of micturition through the urethra four days after percutaneous nephrostomy placement, he experienced severe lightheadedness, diaphoresis, and palpitations. These symptoms prompted him to recall similar episodes following micturition for several months prior to his hospitalization.
It is likely that contraction of the bladder during episodes of urination caused irritation of the pelvic mass, leading to catecholamine secretion. Another explanation for his recurrent lightheadedness would be a neurocardiogenic reflex with micturition (which when it culminates with loss of consciousness is called micturition syncope), but this would not explain his hypertension or bladder mass.
Biochemical tests that were ordered on admission but sent to a reference lab then returned. Plasma metanephrine was 0.2 nmol/L (normal, <0.5 nmol/L) and plasma normetanephrine 34.6 nmol/L (normal, <0.9 nmol/L). His 24-hour urine metanephrine was 72 ug/24 hr (normal, 0-300 ug/24 hr) and normetanephrine 8,511 ug/24 hr (normal, 50-800 ug/24 hr).
The markedly elevated plasma and urine normetanephrine levels confirm a diagnosis of a catecholamine-secreting tumor (paraganglioma). The tissue obtained from the CT-guided lymph node biopsy should be sent for markers of neuroendocrine tumors including chromogranin.
Lymph node biopsy revealed metastatic paraganglioma that was chromogranin A and synaptophysin positive (Figure 2). A fluorodeoxyglucose positron emission tomography (FDG-PET) scan disclosed skull metastases. He was treated with phenoxybenzamine, amlodipine, and labetalol. Surgical resection of the pelvic mass was discussed, but the patient elected to defer surgery as the location of the primary tumor made it challenging to resect and would have required an ileal conduit.
After the diagnosis was made, the patient’s family recalled that a maternal uncle had been diagnosed with a paraganglioma of the carotid body. Genetic testing of the patient identified a succinate dehydrogenase complex subunit B (SDHB) pathogenic variant and confirmed hereditary paraganglioma syndrome (HPGL). One year after the diagnosis, liver and lung metastases developed. He was treated with lanreotide (somatostatin analogue), capecitabine, and temozolomide, as well as a craniotomy and radiotherapy for palliation of bony metastases. The patient died less than two years after diagnosis.
DISCUSSION
Most patients with hypertension (defined as blood pressure >130/80 mm Hg1) do not have an identifiable etiology (primary hypertension). Many components of this patient’s history, however, including his young age of onset, a teenage sibling with hypertension, lack of obesity, hypertension refractory to multiple medications, and LVH suggested secondary hypertension. Hypertension onset at an age less than 30 years, resistance to three or more medications,1,2 and/or acute onset hypertension at any age should prompt an evaluation for secondary causes.1 The prevalence of secondary hypertension is approximately 30% in hypertensive patients ages 18 to 40 years compared with 5%-10% in the overall adult population with hypertension.3 Among children and adolescents ages 0 to 19 years with hypertension, the prevalence of secondary hypertension may be as high as 57%.4
The most common etiology of secondary hypertension is primary aldosteronism.5,6 However, in young adults (ages 19 to 39 years), common etiologies also include renovascular disease and renal parenchymal disease.7 Other causes include obstructive sleep apnea, medications, stimulants (cocaine and amphetamines),8 and endocrinopathies such as thyrotoxicosis, Cushing syndrome, and catecholamine-secreting tumors.7 Less than 1% of secondary hypertension in all adults is due to catecholamine-secreting tumors, and the minority of those catecholamine-secreting tumors are paragangliomas.9
Paragangliomas are tumors of the peripheral autonomic nervous system. These neoplasms arise in the sympathetic and parasympathetic chains along the paravertebral and paraaortic axes. They are closely related to pheochromocytomas, which arise in the adrenal medulla.9 Most head and neck paragangliomas are biochemically silent and are generally discovered due to mass effect.10 The subset of paragangliomas that secrete catecholamines most often arise in the abdomen and pelvis, and their clinical presentation mimics that of pheochromocytomas, including episodic hypertension, palpitations, pallor, and diaphoresis.
This patient had persistent, nonepisodic hypertension, while palpitations and diaphoresis only manifested following micturition. Other cases of urinary bladder paragangliomas have described micturition-associated symptoms and hypertensive crises. Three-fold increases of catecholamine secretion after micturition have been observed in these patients, likely due to muscle contraction and pressure changes in the bladder leading to the systemic release of catecholamines.11
Epinephrine and norepinephrine are monoamine neurotransmitters that activate alpha-adrenergic and beta-adrenergic receptors. Adrenergic receptors are present in all tissues of the body but have prominent effects on the smooth muscle in the vasculature, gastrointestinal tract, urinary tract, and airways.12 Alpha-adrenergic vasoconstriction causes hypertension, which is commonly observed in patients with catecholamine-secreting tumors.10 Catecholamine excess due to secretion from these tumors causes headache in 60%-80% of patients, tachycardia/palpitations in 50%-70%, anxiety in 20%-40%, and nausea in 20%-25%.10 Other symptoms include sweating, pallor, dyspnea, and vertigo.9,10 This patient’s chronic nausea, which was attributed to gastroesophageal reflux, and his anxiety, attributed to generalized anxiety disorder, were likely symptoms of catecholamine excess.13
The best test for the diagnosis of paragangliomas and pheochromocytomas is the measurement of plasma free or 24-hour urinary fractionated metanephrines (test sensitivity of >90% and >90%, respectively).14 Screening for pheochromocytoma should be considered in hypertensive patients who have symptoms of catecholamine excess, refractory or paroxysmal hypertension, and/or familial pheochromocytoma/paraganglioma syndromes.15 Screening for pheochromocytoma should also be performed in children and adolescents with systolic or diastolic blood pressure that is greater than the 95th percentile for their age plus 5 mm Hg.16
While a typical tumor location and elevated metanephrine levels are sufficient to make the diagnosis of a pheochromocytoma or catecholamine-secreting paraganglioma, functional imaging with FDG-PET, Ga-DOTATATE-PET, or 123I-meta-iodobenzylguanidine (123I-MIBG) can further confirm the diagnosis and detect distant metastases. However, imaging has low sensitivity for these tumors and thus should only be considered for patients in whom metastatic disease is suspected.14 Biopsy is rarely needed and should be reserved for unusual metastatic locations. Treatment with an alpha-adrenergic receptor antagonist often reduces symptoms and lowers blood pressure. Definitive management typically involves surgical resection for benign disease. Surgery, radionuclide therapy, or chemotherapy is used for malignant disease.
While most pheochromocytomas are sporadic, up to 40% of paragangliomas are due to germline pathogenic variants.17 Mutations in the succinate dehydrogenase (SDH) group of genes are the most common germline pathogenic variants in the autosomal dominant hereditary paraganglioma syndrome (HPGL). Most paragangliomas and pheochromocytomas are localized and benign, but 10%-15% are metastatic.18 SDHB mutations are associated with a high risk of metastasis.19 Thus, genetic testing for patients and subsequent cascade testing to identify at-risk family members is advised in all patients with pheochromocytomas or paragangliomas.20 This patient’s younger brother and mother were both found to carry the same pathogenic SDHB variant, but neither was found to have paragangliomas. Annual metanephrine levels (urine or plasma) and every other year whole-body magnetic resonance imaging (MRI) scans were recommended for tumor surveillance.
The clinician team followed a logical branching algorithm for the diagnosis of severe hypertension with biochemical testing, advanced imaging, histology, and genetic testing to arrive at the final diagnosis of hereditary paraganglioma syndrome. Although this patient presented for urgent care because of the acute effects of catecholamine excess, he suffered from chronic effects (nausea, anxiety, and hypertension) for years. Each symptom had been diagnosed and treated in isolation, but the combination and severity in a young patient suggested a unifying diagnosis. The family history of hypertension (brother and father) suggested an inherited diagnosis from the father’s family, but the final answer rested on the other branch (maternal uncle) of the family tree.
KEY TEACHING POINTS
- Hypertension in a young adult is due to a secondary cause in up to 30% of patients.
- Pathologic catecholamine excess leads to hypertension, tachycardia, pallor, sweating, anxiety, and nausea. A sustained and unexplained combination of these symptoms should prompt a biochemical evaluation for pheochromocytoma or paraganglioma.
- Paragangliomas are tumors of the autonomic nervous system. The frequency of catecholamine secretion depends on their location in the body, and they are commonly caused by germline pathogenic variants.
Acknowledgments
This conundrum was presented during a live Grand Rounds with the expert clinician’s responses recorded and edited for space and clarity.
Disclosures
Dr. Dhaliwal reports speaking honoraria from ISMIE Mutual Insurance Company and GE Healthcare. All other authors have nothing to disclose.
Funding
No sources of funding.
1. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):e13-e115. https://doi.org/10.1161/HYP.0000000000000065.
2. Acelajado MC, Calhoun DA. Resistant hypertension, secondary hypertension, and hypertensive crises: diagnostic evaluation and treatment. Cardiol Clin. 2010;28(4):639-654. https://doi.org/10.1016/j.ccl.2010.07.002.
3. Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension. 2003;42(6):1206-1252. https://doi.org/10.1161/01.HYP.0000107251.49515.c2.
4. Gupta-Malhotra M, Banker A, Shete S, et al. Essential hypertension vs. secondary hypertension among children. Am J Hypertens. 2015;28(1):73-80. https://doi.org/10.1093/ajh/hpu083.
5. Mosso L, Carvajal C, Gonzalez A, et al. Primary aldosteronism and hypertensive disease. Hypertension. 2003;42(2):161-165. https://doi.org/10.1161/01.HYP.0000079505.25750.11.
6. Kayser SC, Dekkers T, Groenewoud HJ, et al. Study heterogeneity and estimation of prevalence of primary aldosteronism: a systematic review and meta-regression analysis. J Clin Endocrinol Metab. 2016;101(7):2826-2835. https://doi.org/10.1210/jc.2016-1472.
7. Charles L, Triscott J, Dobbs B. Secondary hypertension: discovering the underlying cause. Am Fam Physician. 2017;96(7):453-461.
8. Aronow WS. Drug-induced causes of secondary hypertension. Ann Transl Med. 2017;5(17):349. https://doi.org/10.21037/atm.2017.06.16.
9. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665-675. https://doi.org/10.1016/S0140-6736(05)67139-5.
10. Mannelli M, Lenders JW, Pacak K, Parenti G, Eisenhofer G. Subclinical phaeochromocytoma. Best Pract Res Clin Endocrinol Metab. 2012;26(4):507-515. https://doi.org/10.1016/j.beem.2011.10.008.
11. Kappers MH, van den Meiracker AH, Alwani RA, Kats E, Baggen MG. Paraganglioma of the urinary bladder. Neth J Med. 2008;66(4):163-165.
12. Paravati S, Warrington SJ. Physiology, Catecholamines. In: StatPearls. Treasure Island, FL: StatPearls Publishing LLC; 2019.
13. King KS, Darmani NA, Hughes MS, Adams KT, Pacak K. Exercise-induced nausea and vomiting: another sign and symptom of pheochromocytoma and paraganglioma. Endocrine. 2010;37(3):403-407. https://doi.org/10.1007/s12020-010-9319-3.
14. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942. https://doi.org/10.1210/jc.2014-1498.
15. Lenders JWM, Eisenhofer G. Update on modern management of pheochromocytoma and paraganglioma. Endocrinol Metab (Seoul). 2017;32(2):152-161. https://doi.org/10.3803/EnM.2017.32.2.152.
16. National High Blood Pressure Education Program Working Group. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics. 2004;114(2):555-576.
17. Else T, Greenberg S, Fishbein L. Hereditary Paraganglioma-Pheochromocytoma Syndromes. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. Gene Reviews. Seattle, WA: University of Washington; 1993.
18. Goldstein RE, O’Neill JA, Jr., Holcomb GW, 3rd, et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg. 1999;229(6):755-764; discussion 764-756. https://doi.org/10.1097/00000658-199906000-00001.
19. Amar L, Baudin E, Burnichon N, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92(10):3822-3828. https://doi.org/10.1210/jc.2007-0709.
20. Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11(2):101-111. https://doi.org/10.1038/nrendo.2014.188.
A 21-year-old man with a history of hypertension presented to the emergency department with four days of generalized abdominal pain, nausea, and vomiting as well as one month of loose stools. He also had a headache (not further specified) for one day. Due to his nausea, he had been unable to take his medications for two days. Home blood pressure measurements over the preceding two days revealed systolic pressures exceeding 200 mm Hg. He did not experience fever, dyspnea, chest pain, vision changes, numbness, weakness, diaphoresis, or palpitations.
Abdominal pain with vomiting and diarrhea is often caused by a self-limited gastroenteritis. However, the priority initially is to exclude serious intraabdominal processes including arterial insufficiency, bowel obstruction, organ perforation, or organ-based infection or inflammation (eg, appendicitis, cholecystitis, pancreatitis). Essential hypertension accounts for 95% of cases of hypertension in the United States, but given this patient’s young age, secondary causes should be evaluated. These include primary aldosteronism (the most common endocrine cause for hypertension in young patients), chronic kidney disease, fibromuscular dysplasia, illicit drug use, hypercortisolism, pheochromocytoma, and coarctation of the aorta. Thyrotoxicosis can elevate blood pressure (although usually not to this extent) and cause hyperdefecation. While the etiology of the chronic hypertension is uncertain, the proximate cause of the acute rise in blood pressure is likely the stress of his acute illness and the inability to take his prescribed antihypertensive medications. In the setting of severe hypertension, his headache may reflect an intracranial hemorrhage and his abdominal pain could signal an aortic dissection.
His medical history included hypertension diagnosed at age 16 as well as anxiety diagnosed following a panic attack at age 19. Over the past year, he had also developed persistent nausea, which was attributed to gastroesophageal reflux disease. His medications included metoprolol 50 mg daily, amlodipine 5 mg daily, hydrochlorothiazide 12.5 mg daily, escitalopram 20 mg daily, and omeprazole 20 mg daily. His father and 15-year-old brother also had hypertension. He was a part-time student while working at a car dealership. He did not smoke or use drugs and he rarely drank alcohol.
The need for three antihypertensive medications (albeit at submaximal doses) reflects the severity of his hypertension (provided challenges with medication adherence have been excluded). His family history, especially that of his brother who was diagnosed with hypertension at an early age, and the patient’s own early onset hypertension point toward an inherited form of hypertension. Autosomal dominant polycystic kidney disease often results in hypertension before chronic kidney disease develops. Rare inherited forms of hypertension include familial hyperaldosteronism, apparent mineralocorticoid excess, Liddle syndrome, or a hereditary endocrine tumor syndrome predisposing to pheochromocytoma. Even among patients who report classic pheochromocytoma symptoms, such as headache and anxiety, the diagnosis remains unlikely as these symptoms are nonspecific and highly prevalent in the general population. However, once secondary hypertension is plausible or suspected, testing for hyperadrenergic states, which can also cause nausea and vomiting during times of catecholamine excess, should be pursued.
His temperature was 97.5°F, heart rate 95 beats per minute and regular, respiratory rate 18 breaths per minute, blood pressure 181/118 mm Hg (systolic and diastolic pressures in each arm were within 10 mm Hg), and oxygen saturation 100% on room air. Systolic and diastolic pressures did not decrease by more than 20 mm Hg and 10 mm Hg, respectively, after he stood for two minutes. His body mass index was 24 kg/m2. He was alert and appeared slightly anxious. There was a bounding point of maximal impulse in the fifth intercostal space at the midclavicular line and a 3/6 systolic murmur at the left upper sternal border with radiation to the carotid arteries. His abdomen was soft with generalized tenderness to palpation and without rebound tenderness, masses, organomegaly, or bruits. There was no costovertebral angle tenderness. No lymphadenopathy was present. His fundoscopic, pulmonary, skin and neurologic examinations were normal.
Laboratory studies revealed a white blood cell count of 13.3 × 103/uL with a normal differential, hemoglobin 13.9 g/dL, platelet count 373 × 103/uL, sodium 142 mmol/L, potassium 3.8 mmol/L, chloride 103 mmol/L,bicarbonate 25 mmol/L, blood urea nitrogen 12 mg/dL, creatinine 1.3 mg/dL (a baseline creatinine level was not available), glucose 88 mg/dL, calcium 10.6 mg/dL, albumin 4.9 g/dL, aspartate aminotransferase 27 IU/L, alanine aminotransferase 37 IU/L, and lipase 40 IU/L. Urinalysis revealed 5-10 white blood cells per high power field without casts and 10 mg/dL protein. Urine toxicology was not performed. Electrocardiogram (ECG) showed left ventricular hypertrophy (LVH). Chest radiography was normal.
The abdominal examination does not suggest peritonitis. The laboratory tests do not suggest inflammation of the liver, pancreas, or biliary tree as the cause of his abdominal pain or diarrhea. The murmur may indicate hypertrophic cardiomyopathy or a congenital anomaly such as bicuspid aortic valve; but neither would explain hypertension unless they were associated with another developmental abnormality, such as coarctation of the aorta. Tricuspid regurgitation is conceivable and if confirmed, might raise concern for carcinoid syndrome, which can cause diarrhea. The normal neurologic examination, including the absence of papilledema, lowers suspicion of intracranial hemorrhage as a cause of his headache.
The albumin of 4.9 g/dL likely reflects hypovolemia resulting from vomiting and diarrhea. Vasoconstriction associated with pheochromocytoma can cause pressure diuresis and resultant hypovolemia. Hyperaldosteronism arising from bilateral adrenal hyperplasia or adrenal adenoma commonly causes hypokalemia, although this is not a universal feature.
The duration of his mildly decreased glomerular filtration rate is uncertain. He may have chronic kidney disease from sustained hypertension, or acute kidney injury from hypovolemia. The mild pyuria could indicate infection or renal calculi, either of which could account for generalized abdominal pain or could reflect an acute renal injury from acute interstitial nephritis from his proton pump inhibitor or hydrochlorothiazide.
LVH on the ECG indicates longstanding hypertension. The chest radiograph does not reveal clues to the etiology of or sequelae from hypertension. In particular, there is no widened aorta to suggest aortic dissection, no pulmonary edema to indicate heart failure, and no rib notching that points toward aortic coarctation. A transthoracic echocardiogram to assess for valvular and other structural abnormalities is warranted.
Tests for secondary hypertension should be sent, including serum aldosterone and renin levels to assess for primary aldosteronism and plasma or 24-hour urine normetanephrine and metanephrine levels to assess for pheochromocytoma. Biochemical evaluation is the mainstay for endocrine hypertension evaluation and should be followed by imaging if abnormal results are found.
Intact parathyroid hormone (PTH) was 78 pg/mL (normal, 10-65 pg/mL), thyroid stimulating hormone 3.6 mIU/L (normal, 0.30-5.50 mIU/L), and morning cortisol 4.1 ug/dL (normal, >7.0 ug/dL). Plasma aldosterone was 14.6 ng/dL (normal, 1-16 ng/dL), plasma renin activity 3.6 ng/mL/hr (normal, 0.5-3.5 ng/mL/hr), and aldosterone-renin ratio 4.1 (normal, <20). Transthoracic echocardiogram showed LVH with normal valves, wall motion, and proximal aorta; the left ventricular ejection fraction was 70%. Magnetic resonance angiography of the renal vessels demonstrated no abnormalities.
Computed tomography (CT) of the abdomen and pelvis with oral and intravenous contrast revealed a 5 cm heterogeneous enhancing mass associated with the prostate gland extending into the base of the bladder. The mass obstructed the right renal collecting system and ureter causing severe right-sided ureterectasis and hydronephrosis. There was also 2.8 cm right-sided paracaval lymph node enlargement and 2.1 cm right-sided and 1.5 cm left-sided external iliac lymph node enlargement (Figure 1). There were no adrenal masses.
He is young for prostate, bladder, or colorectal cancer, but early onset variations of these tumors, along with metastatic testicular cancer, must be considered for the pelvic mass and associated lymphadenopathy. Prostatic masses can be infectious (eg, abscess) or malignant (eg, adenocarcinoma, small cell carcinoma). Additional considerations for abdominopelvic cancer are sarcomas, germ cell tumors, or lymphoma. A low aldosterone-renin ratio coupled with a normal potassium level makes primary aldosteronism unlikely. The normal angiography excludes renovascular hypertension.
His abdominal pain and gastrointestinal symptoms could arise from irritation of the bowel, distension of the right-sided urinary collecting system, or products secreted from the mass (eg, catecholamines). The hyperdynamic precordium, elevated ejection fraction, and murmur may reflect augmented blood flow from a hyperadrenergic state. A unifying diagnosis would be a pheochromocytoma. However, given the normal appearance of the adrenal glands on CT imaging, catecholamines arising from a paraganglioma, a tumor of the autonomic nervous system, is more likely. These tumors often secrete catecholamines and can be metastatic (suggested here by the lymphadenopathy). Functional imaging or biopsy of either the mass or an adjacent lymph node is indicated. However, because of the possibility of a catecholamine-secreting tumor, he should be treated with an alpha-adrenergic receptor antagonist before undergoing a biopsy to prevent unopposed vasoconstriction from catecholamine leakage.
Scrotal ultrasound revealed no evidence of a testicular tumor. Lactate dehydrogenase (LDH) was 179 IU/L (normal, 120-240 IU/L) and prostate specific antigen (PSA) was 0.7 ng/mL (normal, <2.5 ng/mL). The patient was given amlodipine and labetalol with improvement of blood pressures to 160s/100s. His creatinine decreased to 1.1 mg/dL. He underwent CT-guided biopsy of a pelvic lymph node. CT of the head without intravenous contrast demonstrated no intracranial abnormalities. His headache resolved with improvement in blood pressure, and he had minimal gastrointestinal symptoms during his hospitalization. No stool studies were sent. A right-sided percutaneous nephrostomy was placed which yielded >15 L of urine from the tube over the next four days.
Upon the first episode of micturition through the urethra four days after percutaneous nephrostomy placement, he experienced severe lightheadedness, diaphoresis, and palpitations. These symptoms prompted him to recall similar episodes following micturition for several months prior to his hospitalization.
It is likely that contraction of the bladder during episodes of urination caused irritation of the pelvic mass, leading to catecholamine secretion. Another explanation for his recurrent lightheadedness would be a neurocardiogenic reflex with micturition (which when it culminates with loss of consciousness is called micturition syncope), but this would not explain his hypertension or bladder mass.
Biochemical tests that were ordered on admission but sent to a reference lab then returned. Plasma metanephrine was 0.2 nmol/L (normal, <0.5 nmol/L) and plasma normetanephrine 34.6 nmol/L (normal, <0.9 nmol/L). His 24-hour urine metanephrine was 72 ug/24 hr (normal, 0-300 ug/24 hr) and normetanephrine 8,511 ug/24 hr (normal, 50-800 ug/24 hr).
The markedly elevated plasma and urine normetanephrine levels confirm a diagnosis of a catecholamine-secreting tumor (paraganglioma). The tissue obtained from the CT-guided lymph node biopsy should be sent for markers of neuroendocrine tumors including chromogranin.
Lymph node biopsy revealed metastatic paraganglioma that was chromogranin A and synaptophysin positive (Figure 2). A fluorodeoxyglucose positron emission tomography (FDG-PET) scan disclosed skull metastases. He was treated with phenoxybenzamine, amlodipine, and labetalol. Surgical resection of the pelvic mass was discussed, but the patient elected to defer surgery as the location of the primary tumor made it challenging to resect and would have required an ileal conduit.
After the diagnosis was made, the patient’s family recalled that a maternal uncle had been diagnosed with a paraganglioma of the carotid body. Genetic testing of the patient identified a succinate dehydrogenase complex subunit B (SDHB) pathogenic variant and confirmed hereditary paraganglioma syndrome (HPGL). One year after the diagnosis, liver and lung metastases developed. He was treated with lanreotide (somatostatin analogue), capecitabine, and temozolomide, as well as a craniotomy and radiotherapy for palliation of bony metastases. The patient died less than two years after diagnosis.
DISCUSSION
Most patients with hypertension (defined as blood pressure >130/80 mm Hg1) do not have an identifiable etiology (primary hypertension). Many components of this patient’s history, however, including his young age of onset, a teenage sibling with hypertension, lack of obesity, hypertension refractory to multiple medications, and LVH suggested secondary hypertension. Hypertension onset at an age less than 30 years, resistance to three or more medications,1,2 and/or acute onset hypertension at any age should prompt an evaluation for secondary causes.1 The prevalence of secondary hypertension is approximately 30% in hypertensive patients ages 18 to 40 years compared with 5%-10% in the overall adult population with hypertension.3 Among children and adolescents ages 0 to 19 years with hypertension, the prevalence of secondary hypertension may be as high as 57%.4
The most common etiology of secondary hypertension is primary aldosteronism.5,6 However, in young adults (ages 19 to 39 years), common etiologies also include renovascular disease and renal parenchymal disease.7 Other causes include obstructive sleep apnea, medications, stimulants (cocaine and amphetamines),8 and endocrinopathies such as thyrotoxicosis, Cushing syndrome, and catecholamine-secreting tumors.7 Less than 1% of secondary hypertension in all adults is due to catecholamine-secreting tumors, and the minority of those catecholamine-secreting tumors are paragangliomas.9
Paragangliomas are tumors of the peripheral autonomic nervous system. These neoplasms arise in the sympathetic and parasympathetic chains along the paravertebral and paraaortic axes. They are closely related to pheochromocytomas, which arise in the adrenal medulla.9 Most head and neck paragangliomas are biochemically silent and are generally discovered due to mass effect.10 The subset of paragangliomas that secrete catecholamines most often arise in the abdomen and pelvis, and their clinical presentation mimics that of pheochromocytomas, including episodic hypertension, palpitations, pallor, and diaphoresis.
This patient had persistent, nonepisodic hypertension, while palpitations and diaphoresis only manifested following micturition. Other cases of urinary bladder paragangliomas have described micturition-associated symptoms and hypertensive crises. Three-fold increases of catecholamine secretion after micturition have been observed in these patients, likely due to muscle contraction and pressure changes in the bladder leading to the systemic release of catecholamines.11
Epinephrine and norepinephrine are monoamine neurotransmitters that activate alpha-adrenergic and beta-adrenergic receptors. Adrenergic receptors are present in all tissues of the body but have prominent effects on the smooth muscle in the vasculature, gastrointestinal tract, urinary tract, and airways.12 Alpha-adrenergic vasoconstriction causes hypertension, which is commonly observed in patients with catecholamine-secreting tumors.10 Catecholamine excess due to secretion from these tumors causes headache in 60%-80% of patients, tachycardia/palpitations in 50%-70%, anxiety in 20%-40%, and nausea in 20%-25%.10 Other symptoms include sweating, pallor, dyspnea, and vertigo.9,10 This patient’s chronic nausea, which was attributed to gastroesophageal reflux, and his anxiety, attributed to generalized anxiety disorder, were likely symptoms of catecholamine excess.13
The best test for the diagnosis of paragangliomas and pheochromocytomas is the measurement of plasma free or 24-hour urinary fractionated metanephrines (test sensitivity of >90% and >90%, respectively).14 Screening for pheochromocytoma should be considered in hypertensive patients who have symptoms of catecholamine excess, refractory or paroxysmal hypertension, and/or familial pheochromocytoma/paraganglioma syndromes.15 Screening for pheochromocytoma should also be performed in children and adolescents with systolic or diastolic blood pressure that is greater than the 95th percentile for their age plus 5 mm Hg.16
While a typical tumor location and elevated metanephrine levels are sufficient to make the diagnosis of a pheochromocytoma or catecholamine-secreting paraganglioma, functional imaging with FDG-PET, Ga-DOTATATE-PET, or 123I-meta-iodobenzylguanidine (123I-MIBG) can further confirm the diagnosis and detect distant metastases. However, imaging has low sensitivity for these tumors and thus should only be considered for patients in whom metastatic disease is suspected.14 Biopsy is rarely needed and should be reserved for unusual metastatic locations. Treatment with an alpha-adrenergic receptor antagonist often reduces symptoms and lowers blood pressure. Definitive management typically involves surgical resection for benign disease. Surgery, radionuclide therapy, or chemotherapy is used for malignant disease.
While most pheochromocytomas are sporadic, up to 40% of paragangliomas are due to germline pathogenic variants.17 Mutations in the succinate dehydrogenase (SDH) group of genes are the most common germline pathogenic variants in the autosomal dominant hereditary paraganglioma syndrome (HPGL). Most paragangliomas and pheochromocytomas are localized and benign, but 10%-15% are metastatic.18 SDHB mutations are associated with a high risk of metastasis.19 Thus, genetic testing for patients and subsequent cascade testing to identify at-risk family members is advised in all patients with pheochromocytomas or paragangliomas.20 This patient’s younger brother and mother were both found to carry the same pathogenic SDHB variant, but neither was found to have paragangliomas. Annual metanephrine levels (urine or plasma) and every other year whole-body magnetic resonance imaging (MRI) scans were recommended for tumor surveillance.
The clinician team followed a logical branching algorithm for the diagnosis of severe hypertension with biochemical testing, advanced imaging, histology, and genetic testing to arrive at the final diagnosis of hereditary paraganglioma syndrome. Although this patient presented for urgent care because of the acute effects of catecholamine excess, he suffered from chronic effects (nausea, anxiety, and hypertension) for years. Each symptom had been diagnosed and treated in isolation, but the combination and severity in a young patient suggested a unifying diagnosis. The family history of hypertension (brother and father) suggested an inherited diagnosis from the father’s family, but the final answer rested on the other branch (maternal uncle) of the family tree.
KEY TEACHING POINTS
- Hypertension in a young adult is due to a secondary cause in up to 30% of patients.
- Pathologic catecholamine excess leads to hypertension, tachycardia, pallor, sweating, anxiety, and nausea. A sustained and unexplained combination of these symptoms should prompt a biochemical evaluation for pheochromocytoma or paraganglioma.
- Paragangliomas are tumors of the autonomic nervous system. The frequency of catecholamine secretion depends on their location in the body, and they are commonly caused by germline pathogenic variants.
Acknowledgments
This conundrum was presented during a live Grand Rounds with the expert clinician’s responses recorded and edited for space and clarity.
Disclosures
Dr. Dhaliwal reports speaking honoraria from ISMIE Mutual Insurance Company and GE Healthcare. All other authors have nothing to disclose.
Funding
No sources of funding.
A 21-year-old man with a history of hypertension presented to the emergency department with four days of generalized abdominal pain, nausea, and vomiting as well as one month of loose stools. He also had a headache (not further specified) for one day. Due to his nausea, he had been unable to take his medications for two days. Home blood pressure measurements over the preceding two days revealed systolic pressures exceeding 200 mm Hg. He did not experience fever, dyspnea, chest pain, vision changes, numbness, weakness, diaphoresis, or palpitations.
Abdominal pain with vomiting and diarrhea is often caused by a self-limited gastroenteritis. However, the priority initially is to exclude serious intraabdominal processes including arterial insufficiency, bowel obstruction, organ perforation, or organ-based infection or inflammation (eg, appendicitis, cholecystitis, pancreatitis). Essential hypertension accounts for 95% of cases of hypertension in the United States, but given this patient’s young age, secondary causes should be evaluated. These include primary aldosteronism (the most common endocrine cause for hypertension in young patients), chronic kidney disease, fibromuscular dysplasia, illicit drug use, hypercortisolism, pheochromocytoma, and coarctation of the aorta. Thyrotoxicosis can elevate blood pressure (although usually not to this extent) and cause hyperdefecation. While the etiology of the chronic hypertension is uncertain, the proximate cause of the acute rise in blood pressure is likely the stress of his acute illness and the inability to take his prescribed antihypertensive medications. In the setting of severe hypertension, his headache may reflect an intracranial hemorrhage and his abdominal pain could signal an aortic dissection.
His medical history included hypertension diagnosed at age 16 as well as anxiety diagnosed following a panic attack at age 19. Over the past year, he had also developed persistent nausea, which was attributed to gastroesophageal reflux disease. His medications included metoprolol 50 mg daily, amlodipine 5 mg daily, hydrochlorothiazide 12.5 mg daily, escitalopram 20 mg daily, and omeprazole 20 mg daily. His father and 15-year-old brother also had hypertension. He was a part-time student while working at a car dealership. He did not smoke or use drugs and he rarely drank alcohol.
The need for three antihypertensive medications (albeit at submaximal doses) reflects the severity of his hypertension (provided challenges with medication adherence have been excluded). His family history, especially that of his brother who was diagnosed with hypertension at an early age, and the patient’s own early onset hypertension point toward an inherited form of hypertension. Autosomal dominant polycystic kidney disease often results in hypertension before chronic kidney disease develops. Rare inherited forms of hypertension include familial hyperaldosteronism, apparent mineralocorticoid excess, Liddle syndrome, or a hereditary endocrine tumor syndrome predisposing to pheochromocytoma. Even among patients who report classic pheochromocytoma symptoms, such as headache and anxiety, the diagnosis remains unlikely as these symptoms are nonspecific and highly prevalent in the general population. However, once secondary hypertension is plausible or suspected, testing for hyperadrenergic states, which can also cause nausea and vomiting during times of catecholamine excess, should be pursued.
His temperature was 97.5°F, heart rate 95 beats per minute and regular, respiratory rate 18 breaths per minute, blood pressure 181/118 mm Hg (systolic and diastolic pressures in each arm were within 10 mm Hg), and oxygen saturation 100% on room air. Systolic and diastolic pressures did not decrease by more than 20 mm Hg and 10 mm Hg, respectively, after he stood for two minutes. His body mass index was 24 kg/m2. He was alert and appeared slightly anxious. There was a bounding point of maximal impulse in the fifth intercostal space at the midclavicular line and a 3/6 systolic murmur at the left upper sternal border with radiation to the carotid arteries. His abdomen was soft with generalized tenderness to palpation and without rebound tenderness, masses, organomegaly, or bruits. There was no costovertebral angle tenderness. No lymphadenopathy was present. His fundoscopic, pulmonary, skin and neurologic examinations were normal.
Laboratory studies revealed a white blood cell count of 13.3 × 103/uL with a normal differential, hemoglobin 13.9 g/dL, platelet count 373 × 103/uL, sodium 142 mmol/L, potassium 3.8 mmol/L, chloride 103 mmol/L,bicarbonate 25 mmol/L, blood urea nitrogen 12 mg/dL, creatinine 1.3 mg/dL (a baseline creatinine level was not available), glucose 88 mg/dL, calcium 10.6 mg/dL, albumin 4.9 g/dL, aspartate aminotransferase 27 IU/L, alanine aminotransferase 37 IU/L, and lipase 40 IU/L. Urinalysis revealed 5-10 white blood cells per high power field without casts and 10 mg/dL protein. Urine toxicology was not performed. Electrocardiogram (ECG) showed left ventricular hypertrophy (LVH). Chest radiography was normal.
The abdominal examination does not suggest peritonitis. The laboratory tests do not suggest inflammation of the liver, pancreas, or biliary tree as the cause of his abdominal pain or diarrhea. The murmur may indicate hypertrophic cardiomyopathy or a congenital anomaly such as bicuspid aortic valve; but neither would explain hypertension unless they were associated with another developmental abnormality, such as coarctation of the aorta. Tricuspid regurgitation is conceivable and if confirmed, might raise concern for carcinoid syndrome, which can cause diarrhea. The normal neurologic examination, including the absence of papilledema, lowers suspicion of intracranial hemorrhage as a cause of his headache.
The albumin of 4.9 g/dL likely reflects hypovolemia resulting from vomiting and diarrhea. Vasoconstriction associated with pheochromocytoma can cause pressure diuresis and resultant hypovolemia. Hyperaldosteronism arising from bilateral adrenal hyperplasia or adrenal adenoma commonly causes hypokalemia, although this is not a universal feature.
The duration of his mildly decreased glomerular filtration rate is uncertain. He may have chronic kidney disease from sustained hypertension, or acute kidney injury from hypovolemia. The mild pyuria could indicate infection or renal calculi, either of which could account for generalized abdominal pain or could reflect an acute renal injury from acute interstitial nephritis from his proton pump inhibitor or hydrochlorothiazide.
LVH on the ECG indicates longstanding hypertension. The chest radiograph does not reveal clues to the etiology of or sequelae from hypertension. In particular, there is no widened aorta to suggest aortic dissection, no pulmonary edema to indicate heart failure, and no rib notching that points toward aortic coarctation. A transthoracic echocardiogram to assess for valvular and other structural abnormalities is warranted.
Tests for secondary hypertension should be sent, including serum aldosterone and renin levels to assess for primary aldosteronism and plasma or 24-hour urine normetanephrine and metanephrine levels to assess for pheochromocytoma. Biochemical evaluation is the mainstay for endocrine hypertension evaluation and should be followed by imaging if abnormal results are found.
Intact parathyroid hormone (PTH) was 78 pg/mL (normal, 10-65 pg/mL), thyroid stimulating hormone 3.6 mIU/L (normal, 0.30-5.50 mIU/L), and morning cortisol 4.1 ug/dL (normal, >7.0 ug/dL). Plasma aldosterone was 14.6 ng/dL (normal, 1-16 ng/dL), plasma renin activity 3.6 ng/mL/hr (normal, 0.5-3.5 ng/mL/hr), and aldosterone-renin ratio 4.1 (normal, <20). Transthoracic echocardiogram showed LVH with normal valves, wall motion, and proximal aorta; the left ventricular ejection fraction was 70%. Magnetic resonance angiography of the renal vessels demonstrated no abnormalities.
Computed tomography (CT) of the abdomen and pelvis with oral and intravenous contrast revealed a 5 cm heterogeneous enhancing mass associated with the prostate gland extending into the base of the bladder. The mass obstructed the right renal collecting system and ureter causing severe right-sided ureterectasis and hydronephrosis. There was also 2.8 cm right-sided paracaval lymph node enlargement and 2.1 cm right-sided and 1.5 cm left-sided external iliac lymph node enlargement (Figure 1). There were no adrenal masses.
He is young for prostate, bladder, or colorectal cancer, but early onset variations of these tumors, along with metastatic testicular cancer, must be considered for the pelvic mass and associated lymphadenopathy. Prostatic masses can be infectious (eg, abscess) or malignant (eg, adenocarcinoma, small cell carcinoma). Additional considerations for abdominopelvic cancer are sarcomas, germ cell tumors, or lymphoma. A low aldosterone-renin ratio coupled with a normal potassium level makes primary aldosteronism unlikely. The normal angiography excludes renovascular hypertension.
His abdominal pain and gastrointestinal symptoms could arise from irritation of the bowel, distension of the right-sided urinary collecting system, or products secreted from the mass (eg, catecholamines). The hyperdynamic precordium, elevated ejection fraction, and murmur may reflect augmented blood flow from a hyperadrenergic state. A unifying diagnosis would be a pheochromocytoma. However, given the normal appearance of the adrenal glands on CT imaging, catecholamines arising from a paraganglioma, a tumor of the autonomic nervous system, is more likely. These tumors often secrete catecholamines and can be metastatic (suggested here by the lymphadenopathy). Functional imaging or biopsy of either the mass or an adjacent lymph node is indicated. However, because of the possibility of a catecholamine-secreting tumor, he should be treated with an alpha-adrenergic receptor antagonist before undergoing a biopsy to prevent unopposed vasoconstriction from catecholamine leakage.
Scrotal ultrasound revealed no evidence of a testicular tumor. Lactate dehydrogenase (LDH) was 179 IU/L (normal, 120-240 IU/L) and prostate specific antigen (PSA) was 0.7 ng/mL (normal, <2.5 ng/mL). The patient was given amlodipine and labetalol with improvement of blood pressures to 160s/100s. His creatinine decreased to 1.1 mg/dL. He underwent CT-guided biopsy of a pelvic lymph node. CT of the head without intravenous contrast demonstrated no intracranial abnormalities. His headache resolved with improvement in blood pressure, and he had minimal gastrointestinal symptoms during his hospitalization. No stool studies were sent. A right-sided percutaneous nephrostomy was placed which yielded >15 L of urine from the tube over the next four days.
Upon the first episode of micturition through the urethra four days after percutaneous nephrostomy placement, he experienced severe lightheadedness, diaphoresis, and palpitations. These symptoms prompted him to recall similar episodes following micturition for several months prior to his hospitalization.
It is likely that contraction of the bladder during episodes of urination caused irritation of the pelvic mass, leading to catecholamine secretion. Another explanation for his recurrent lightheadedness would be a neurocardiogenic reflex with micturition (which when it culminates with loss of consciousness is called micturition syncope), but this would not explain his hypertension or bladder mass.
Biochemical tests that were ordered on admission but sent to a reference lab then returned. Plasma metanephrine was 0.2 nmol/L (normal, <0.5 nmol/L) and plasma normetanephrine 34.6 nmol/L (normal, <0.9 nmol/L). His 24-hour urine metanephrine was 72 ug/24 hr (normal, 0-300 ug/24 hr) and normetanephrine 8,511 ug/24 hr (normal, 50-800 ug/24 hr).
The markedly elevated plasma and urine normetanephrine levels confirm a diagnosis of a catecholamine-secreting tumor (paraganglioma). The tissue obtained from the CT-guided lymph node biopsy should be sent for markers of neuroendocrine tumors including chromogranin.
Lymph node biopsy revealed metastatic paraganglioma that was chromogranin A and synaptophysin positive (Figure 2). A fluorodeoxyglucose positron emission tomography (FDG-PET) scan disclosed skull metastases. He was treated with phenoxybenzamine, amlodipine, and labetalol. Surgical resection of the pelvic mass was discussed, but the patient elected to defer surgery as the location of the primary tumor made it challenging to resect and would have required an ileal conduit.
After the diagnosis was made, the patient’s family recalled that a maternal uncle had been diagnosed with a paraganglioma of the carotid body. Genetic testing of the patient identified a succinate dehydrogenase complex subunit B (SDHB) pathogenic variant and confirmed hereditary paraganglioma syndrome (HPGL). One year after the diagnosis, liver and lung metastases developed. He was treated with lanreotide (somatostatin analogue), capecitabine, and temozolomide, as well as a craniotomy and radiotherapy for palliation of bony metastases. The patient died less than two years after diagnosis.
DISCUSSION
Most patients with hypertension (defined as blood pressure >130/80 mm Hg1) do not have an identifiable etiology (primary hypertension). Many components of this patient’s history, however, including his young age of onset, a teenage sibling with hypertension, lack of obesity, hypertension refractory to multiple medications, and LVH suggested secondary hypertension. Hypertension onset at an age less than 30 years, resistance to three or more medications,1,2 and/or acute onset hypertension at any age should prompt an evaluation for secondary causes.1 The prevalence of secondary hypertension is approximately 30% in hypertensive patients ages 18 to 40 years compared with 5%-10% in the overall adult population with hypertension.3 Among children and adolescents ages 0 to 19 years with hypertension, the prevalence of secondary hypertension may be as high as 57%.4
The most common etiology of secondary hypertension is primary aldosteronism.5,6 However, in young adults (ages 19 to 39 years), common etiologies also include renovascular disease and renal parenchymal disease.7 Other causes include obstructive sleep apnea, medications, stimulants (cocaine and amphetamines),8 and endocrinopathies such as thyrotoxicosis, Cushing syndrome, and catecholamine-secreting tumors.7 Less than 1% of secondary hypertension in all adults is due to catecholamine-secreting tumors, and the minority of those catecholamine-secreting tumors are paragangliomas.9
Paragangliomas are tumors of the peripheral autonomic nervous system. These neoplasms arise in the sympathetic and parasympathetic chains along the paravertebral and paraaortic axes. They are closely related to pheochromocytomas, which arise in the adrenal medulla.9 Most head and neck paragangliomas are biochemically silent and are generally discovered due to mass effect.10 The subset of paragangliomas that secrete catecholamines most often arise in the abdomen and pelvis, and their clinical presentation mimics that of pheochromocytomas, including episodic hypertension, palpitations, pallor, and diaphoresis.
This patient had persistent, nonepisodic hypertension, while palpitations and diaphoresis only manifested following micturition. Other cases of urinary bladder paragangliomas have described micturition-associated symptoms and hypertensive crises. Three-fold increases of catecholamine secretion after micturition have been observed in these patients, likely due to muscle contraction and pressure changes in the bladder leading to the systemic release of catecholamines.11
Epinephrine and norepinephrine are monoamine neurotransmitters that activate alpha-adrenergic and beta-adrenergic receptors. Adrenergic receptors are present in all tissues of the body but have prominent effects on the smooth muscle in the vasculature, gastrointestinal tract, urinary tract, and airways.12 Alpha-adrenergic vasoconstriction causes hypertension, which is commonly observed in patients with catecholamine-secreting tumors.10 Catecholamine excess due to secretion from these tumors causes headache in 60%-80% of patients, tachycardia/palpitations in 50%-70%, anxiety in 20%-40%, and nausea in 20%-25%.10 Other symptoms include sweating, pallor, dyspnea, and vertigo.9,10 This patient’s chronic nausea, which was attributed to gastroesophageal reflux, and his anxiety, attributed to generalized anxiety disorder, were likely symptoms of catecholamine excess.13
The best test for the diagnosis of paragangliomas and pheochromocytomas is the measurement of plasma free or 24-hour urinary fractionated metanephrines (test sensitivity of >90% and >90%, respectively).14 Screening for pheochromocytoma should be considered in hypertensive patients who have symptoms of catecholamine excess, refractory or paroxysmal hypertension, and/or familial pheochromocytoma/paraganglioma syndromes.15 Screening for pheochromocytoma should also be performed in children and adolescents with systolic or diastolic blood pressure that is greater than the 95th percentile for their age plus 5 mm Hg.16
While a typical tumor location and elevated metanephrine levels are sufficient to make the diagnosis of a pheochromocytoma or catecholamine-secreting paraganglioma, functional imaging with FDG-PET, Ga-DOTATATE-PET, or 123I-meta-iodobenzylguanidine (123I-MIBG) can further confirm the diagnosis and detect distant metastases. However, imaging has low sensitivity for these tumors and thus should only be considered for patients in whom metastatic disease is suspected.14 Biopsy is rarely needed and should be reserved for unusual metastatic locations. Treatment with an alpha-adrenergic receptor antagonist often reduces symptoms and lowers blood pressure. Definitive management typically involves surgical resection for benign disease. Surgery, radionuclide therapy, or chemotherapy is used for malignant disease.
While most pheochromocytomas are sporadic, up to 40% of paragangliomas are due to germline pathogenic variants.17 Mutations in the succinate dehydrogenase (SDH) group of genes are the most common germline pathogenic variants in the autosomal dominant hereditary paraganglioma syndrome (HPGL). Most paragangliomas and pheochromocytomas are localized and benign, but 10%-15% are metastatic.18 SDHB mutations are associated with a high risk of metastasis.19 Thus, genetic testing for patients and subsequent cascade testing to identify at-risk family members is advised in all patients with pheochromocytomas or paragangliomas.20 This patient’s younger brother and mother were both found to carry the same pathogenic SDHB variant, but neither was found to have paragangliomas. Annual metanephrine levels (urine or plasma) and every other year whole-body magnetic resonance imaging (MRI) scans were recommended for tumor surveillance.
The clinician team followed a logical branching algorithm for the diagnosis of severe hypertension with biochemical testing, advanced imaging, histology, and genetic testing to arrive at the final diagnosis of hereditary paraganglioma syndrome. Although this patient presented for urgent care because of the acute effects of catecholamine excess, he suffered from chronic effects (nausea, anxiety, and hypertension) for years. Each symptom had been diagnosed and treated in isolation, but the combination and severity in a young patient suggested a unifying diagnosis. The family history of hypertension (brother and father) suggested an inherited diagnosis from the father’s family, but the final answer rested on the other branch (maternal uncle) of the family tree.
KEY TEACHING POINTS
- Hypertension in a young adult is due to a secondary cause in up to 30% of patients.
- Pathologic catecholamine excess leads to hypertension, tachycardia, pallor, sweating, anxiety, and nausea. A sustained and unexplained combination of these symptoms should prompt a biochemical evaluation for pheochromocytoma or paraganglioma.
- Paragangliomas are tumors of the autonomic nervous system. The frequency of catecholamine secretion depends on their location in the body, and they are commonly caused by germline pathogenic variants.
Acknowledgments
This conundrum was presented during a live Grand Rounds with the expert clinician’s responses recorded and edited for space and clarity.
Disclosures
Dr. Dhaliwal reports speaking honoraria from ISMIE Mutual Insurance Company and GE Healthcare. All other authors have nothing to disclose.
Funding
No sources of funding.
1. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):e13-e115. https://doi.org/10.1161/HYP.0000000000000065.
2. Acelajado MC, Calhoun DA. Resistant hypertension, secondary hypertension, and hypertensive crises: diagnostic evaluation and treatment. Cardiol Clin. 2010;28(4):639-654. https://doi.org/10.1016/j.ccl.2010.07.002.
3. Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension. 2003;42(6):1206-1252. https://doi.org/10.1161/01.HYP.0000107251.49515.c2.
4. Gupta-Malhotra M, Banker A, Shete S, et al. Essential hypertension vs. secondary hypertension among children. Am J Hypertens. 2015;28(1):73-80. https://doi.org/10.1093/ajh/hpu083.
5. Mosso L, Carvajal C, Gonzalez A, et al. Primary aldosteronism and hypertensive disease. Hypertension. 2003;42(2):161-165. https://doi.org/10.1161/01.HYP.0000079505.25750.11.
6. Kayser SC, Dekkers T, Groenewoud HJ, et al. Study heterogeneity and estimation of prevalence of primary aldosteronism: a systematic review and meta-regression analysis. J Clin Endocrinol Metab. 2016;101(7):2826-2835. https://doi.org/10.1210/jc.2016-1472.
7. Charles L, Triscott J, Dobbs B. Secondary hypertension: discovering the underlying cause. Am Fam Physician. 2017;96(7):453-461.
8. Aronow WS. Drug-induced causes of secondary hypertension. Ann Transl Med. 2017;5(17):349. https://doi.org/10.21037/atm.2017.06.16.
9. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665-675. https://doi.org/10.1016/S0140-6736(05)67139-5.
10. Mannelli M, Lenders JW, Pacak K, Parenti G, Eisenhofer G. Subclinical phaeochromocytoma. Best Pract Res Clin Endocrinol Metab. 2012;26(4):507-515. https://doi.org/10.1016/j.beem.2011.10.008.
11. Kappers MH, van den Meiracker AH, Alwani RA, Kats E, Baggen MG. Paraganglioma of the urinary bladder. Neth J Med. 2008;66(4):163-165.
12. Paravati S, Warrington SJ. Physiology, Catecholamines. In: StatPearls. Treasure Island, FL: StatPearls Publishing LLC; 2019.
13. King KS, Darmani NA, Hughes MS, Adams KT, Pacak K. Exercise-induced nausea and vomiting: another sign and symptom of pheochromocytoma and paraganglioma. Endocrine. 2010;37(3):403-407. https://doi.org/10.1007/s12020-010-9319-3.
14. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942. https://doi.org/10.1210/jc.2014-1498.
15. Lenders JWM, Eisenhofer G. Update on modern management of pheochromocytoma and paraganglioma. Endocrinol Metab (Seoul). 2017;32(2):152-161. https://doi.org/10.3803/EnM.2017.32.2.152.
16. National High Blood Pressure Education Program Working Group. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics. 2004;114(2):555-576.
17. Else T, Greenberg S, Fishbein L. Hereditary Paraganglioma-Pheochromocytoma Syndromes. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. Gene Reviews. Seattle, WA: University of Washington; 1993.
18. Goldstein RE, O’Neill JA, Jr., Holcomb GW, 3rd, et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg. 1999;229(6):755-764; discussion 764-756. https://doi.org/10.1097/00000658-199906000-00001.
19. Amar L, Baudin E, Burnichon N, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92(10):3822-3828. https://doi.org/10.1210/jc.2007-0709.
20. Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11(2):101-111. https://doi.org/10.1038/nrendo.2014.188.
1. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):e13-e115. https://doi.org/10.1161/HYP.0000000000000065.
2. Acelajado MC, Calhoun DA. Resistant hypertension, secondary hypertension, and hypertensive crises: diagnostic evaluation and treatment. Cardiol Clin. 2010;28(4):639-654. https://doi.org/10.1016/j.ccl.2010.07.002.
3. Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension. 2003;42(6):1206-1252. https://doi.org/10.1161/01.HYP.0000107251.49515.c2.
4. Gupta-Malhotra M, Banker A, Shete S, et al. Essential hypertension vs. secondary hypertension among children. Am J Hypertens. 2015;28(1):73-80. https://doi.org/10.1093/ajh/hpu083.
5. Mosso L, Carvajal C, Gonzalez A, et al. Primary aldosteronism and hypertensive disease. Hypertension. 2003;42(2):161-165. https://doi.org/10.1161/01.HYP.0000079505.25750.11.
6. Kayser SC, Dekkers T, Groenewoud HJ, et al. Study heterogeneity and estimation of prevalence of primary aldosteronism: a systematic review and meta-regression analysis. J Clin Endocrinol Metab. 2016;101(7):2826-2835. https://doi.org/10.1210/jc.2016-1472.
7. Charles L, Triscott J, Dobbs B. Secondary hypertension: discovering the underlying cause. Am Fam Physician. 2017;96(7):453-461.
8. Aronow WS. Drug-induced causes of secondary hypertension. Ann Transl Med. 2017;5(17):349. https://doi.org/10.21037/atm.2017.06.16.
9. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665-675. https://doi.org/10.1016/S0140-6736(05)67139-5.
10. Mannelli M, Lenders JW, Pacak K, Parenti G, Eisenhofer G. Subclinical phaeochromocytoma. Best Pract Res Clin Endocrinol Metab. 2012;26(4):507-515. https://doi.org/10.1016/j.beem.2011.10.008.
11. Kappers MH, van den Meiracker AH, Alwani RA, Kats E, Baggen MG. Paraganglioma of the urinary bladder. Neth J Med. 2008;66(4):163-165.
12. Paravati S, Warrington SJ. Physiology, Catecholamines. In: StatPearls. Treasure Island, FL: StatPearls Publishing LLC; 2019.
13. King KS, Darmani NA, Hughes MS, Adams KT, Pacak K. Exercise-induced nausea and vomiting: another sign and symptom of pheochromocytoma and paraganglioma. Endocrine. 2010;37(3):403-407. https://doi.org/10.1007/s12020-010-9319-3.
14. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942. https://doi.org/10.1210/jc.2014-1498.
15. Lenders JWM, Eisenhofer G. Update on modern management of pheochromocytoma and paraganglioma. Endocrinol Metab (Seoul). 2017;32(2):152-161. https://doi.org/10.3803/EnM.2017.32.2.152.
16. National High Blood Pressure Education Program Working Group. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics. 2004;114(2):555-576.
17. Else T, Greenberg S, Fishbein L. Hereditary Paraganglioma-Pheochromocytoma Syndromes. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. Gene Reviews. Seattle, WA: University of Washington; 1993.
18. Goldstein RE, O’Neill JA, Jr., Holcomb GW, 3rd, et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg. 1999;229(6):755-764; discussion 764-756. https://doi.org/10.1097/00000658-199906000-00001.
19. Amar L, Baudin E, Burnichon N, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92(10):3822-3828. https://doi.org/10.1210/jc.2007-0709.
20. Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11(2):101-111. https://doi.org/10.1038/nrendo.2014.188.
© 2019 Society of Hospital Medicine
All in the Stream
A 67-year-old man presented to the emergency department with four days of nausea, vomiting, and chills. He was originally from the Philippines but lived in the United States for six years. His past medical history was notable for nephrolithiasis for which a ureteral stent had been placed and was subsequently removed three years prior. He reported no abdominal pain, diarrhea, dysuria, urinary frequency, hematuria, cough, headache, or fever. He was a retired high school teacher and a lifelong nonsmoker.
This patient presents with a nonspecific constellation of constitutional and gastrointestinal (GI) symptoms. A system-based approach may be helpful when considering the differential diagnosis. Chills most often suggest infection, especially in older patients. With regard to GI causes, acute gastroenteritis and other food-borne infections can cause nausea, vomiting, and chills, but these are typically accompanied by abdominal pain and diarrhea and often resolve in less than four days. Abdominal pain would also be expected with cholecystitis as well as more life-threatening causes of nausea such as acute pancreatitis, mesenteric ischemia, and bowel obstruction. In contrast, abdominal pain would not be expected with a central nervous system (CNS) infection such as a brain abscess, which may cause nausea from increased intracranial pressure. Headaches occur in a majority of these cases, making CNS etiologies of nausea less likely. Cardiovascular causes, including myocardial ischemia and infarction, may lead to nausea and vomiting, but these are less likely given the absence of chest pain. Genitourinary causes must be considered, especially given his history of both nephrolithiasis and instrumentation. A stricture or recurrence of nephrolithiasis could lead to acute pyelonephritis or perinephric abscess, although both commonly present with urinary tract symptoms. Uremia, possibly from obstructive uropathy given the patient’s history of nephrolithiasis, could also lead to this constellation of symptoms.
On examination, temperature was 101.6 °F, heart rate 126 beats per minute, respiratory rate 18 breaths per minute, blood pressure 120/76 mm Hg, and oxygen saturation 98% on room air. The oral mucosa was moist, heart sounds were normal without murmurs, lungs were clear to auscultation, and abdomen was soft, nontender, and nondistended. There was no flank tenderness, and penile, testicular, and prostate examination findings were normal.
Laboratory studies revealed a serum sodium of 126 mEq/L, potassium 5.0 mEq/L, chloride 98 mEq/L, bicarbonate 15 mEq/L, blood urea nitrogen 88 mg/dL, creatinine 9.0 mg/dL, calcium 8 mg/dL, glucose 110 mg/dL, and albumin 3.3 g/dL. One year prior, serum creatinine was 1.4 mg/dL. His white blood cell (WBC) count was 8.0 k/uL with normal differential, hemoglobin 11.4 g/dL with normal MCV, and platelet count was normal. Serum osmolality was 286 mOsm/kg and serum parathyroid hormone (PTH) level 63 pg/mL (normal, 10-65). The urinalysis revealed cloudy urine with a specific gravity 1.009, 54 red blood cells (RBC), 236 WBC, large leukocyte esterase, negative nitrite, trace protein, and no casts or dysmorphic RBCs. A random urine specimen revealed sodium of 86 mEq/L, potassium 16 mEq/L, chloride 80 mEq/L, and creatinine 70 mg/dL.
Fever and tachycardia support an infectious cause of his symptoms. Absent flank tenderness and a normal genitourinary examination have only moderate negative predictive values for acute pyelonephritis and prostatitis, respectively. The most striking laboratory finding is his azotemia. Acute kidney injury (AKI) is more likely than chronic kidney disease (CKD) given that the PTH level is normal and the serum creatinine from a year ago was near normal. The most useful finding to differentiate AKI from CKD is the presence of atrophic kidneys on imaging. The low bicarbonate level indicates a metabolic acidosis. His serum anion gap is 13 mEq/L, which falls above most normal ranges. A mildly elevated serum anion gap together with a “delta serum anion gap/delta serum bicarbonate” ratio less than one suggest concomitant anion gap metabolic acidosis and non anion gap metabolic acidosis. The latter, coupled with a history of nephrolithiasis, may point to the possibility of renal tubular acidosis and AKI caused by urinary tract obstruction. This could also account for the marked hyponatremia. Moreover, his high fractional excretion of sodium (9%) is not suggestive of prerenal injury, the most common acute renal injury among patients who present to the emergency department. Hematuria carries a broad differential diagnosis, but most common causes include nephrolithiasis, urinary tract infection (UTI), prostatitis, neoplasm, and glomerulonephritis (GN). The lack of casts and dysmorphic RBCs makes GN unlikely. Taken together, his vital signs, examination, and laboratory studies suggest a high likelihood of an upper UTI (acute obstructive pyelonephritis) in the context of AKI due to obstructive uropathy.
Despite both a normal serum WBC count (which has only a moderate negative predictive value) and his low risk of developing life-threating organ dysfunction from sepsis based on a quick Sequential Organ Failure Assessment (qSOFA) score of zero, it is appropriate to start antibiotics after drawing blood and doing urine cultures. The next step should include administration of a broad-spectrum regimen that is appropriately dose-adjusted for renal dysfunction, such as an antipseudomonal carbapenem and vancomycin to cover extended-spectrum beta-lactamase (ESBL)-producing organisms, Pseudomonas aeruginosa, and methicillin-resistant Staphylococcus aureus (MRSA). This broad coverage is indicated for empiric treatment of complicated obstructive pyelonephritis, a condition that may arise from significant urinary obstruction and that carries a high risk of rapid clinical deterioration. He should undergo a rapid bedside test to assess for urethral or bladder outlet obstruction: either a bladder ultrasound or temporary insertion of a bladder catheter. He should also have an urgent computed tomography (CT) of his abdomen and pelvis without intravenous (IV) contrast, looking for evidence of urinary tract obstruction. A CT is preferred over ultrasound of the kidneys and bladder as CT has higher sensitivity and specificity for nephrolithiasis and neoplasm.
A CT of the abdomen and pelvis without IV contrast revealed bilateral hydroureter and hydronephrosis with multiple punctate calcified stones within the right calyces and the distal right ureter (Figure 1, Figure 2). However, these appeared too small to cause the degree of obstruction visualized. There were no stones noted in the left ureter to account for the obstruction, though small stones were noted in the left calyces. The bladder appeared normal.
Rarely are both ureters obstructed proximal to the ureterovesical junctions in the retroperitoneum. When they are, CT scans usually reveal culprit lesions that are extrinsic to the urinary tract, such as masses or retroperitoneal fibrosis, the latter of which can be associated with IgG4-related disease. Intrinsic causes of urinary tract obstructions include ureteral strictures (from infections, nephrolithiasis, instrumentation, prior radiotherapy, or rarely urothelial cancer), blood clots, metastatic ureteral deposits, or nephrolithiasis. While most intrinsic causes are unilateral, the patient is predisposed to strictures given his history of ureteral instrumentation. A preexisting unilateral obstruction due to a stricture may now, therefore, be unmasked by a second intrinsic obstruction in the contralateral ureter. Alternatively, given his remote history of living in the Philippines, a site where Schistosoma haematobium is endemic, chronic genitourinary schistosomiasis may have caused ureteral strictures due to granulomas, fibrosis, or pseudopolyps.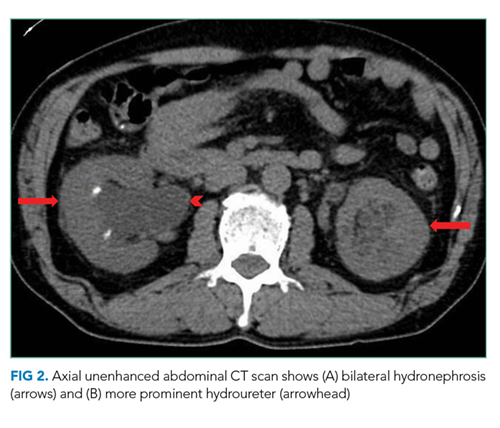
More commonly, bilateral hydroureter with bilateral hydronephrosis is caused by an obstruction of the bladder or urethra. CT scans can reveal prostatic hyperplasia (occasionally with protrusion into the bladder) and increased bladder wall thickness as a result of chronic bladder outlet obstruction, but the negative predictive value of either finding is modest. More revealing is that the patient reported neither an inability to pass urine (in fact, a “random” urine sample was obtained) nor suprapubic discomfort. Both symptoms would be pronounced with acute bladder obstruction but may be minimal with slowly progressive obstruction. In either case, a distended bladder would have been seen on the CT scan.
Regardless of the cause or whether the obstruction is in the upper or lower urinary tract, emergency intervention is needed to relieve the obstruction when AKI presents with bilateral hydronephrosis. A urology consultation should be sought urgently to determine the best strategy to relieve the obstruction. This may include bilateral percutaneous nephrostomy tubes given that the obstruction appears to be above the level of the bladder. Their findings will also direct additional diagnostic workup.
The patient received ceftriaxone and underwent cystoscopy, which revealed a stricture of the distal bulbar urethra. The ureters and bladder could not be completely visualized due to hematuria. The urethral stricture was dilated, and a Foley catheter was placed. In the operating room, the patient had a fever of 103 °F and developed severe hypotension unresponsive to 3 L of intravenous normal saline. Norepinephrine infusion was initiated for refractory hypotension.
Except for transurethral prostate resections, endoscopic urologic procedures rarely lead to a stricture of any segment of the urethra, suggesting that the previous ureteral stent placement and retrieval were not causal. Longstanding Mycobacterium tuberculosis or Schistosoma haematobium infection occasionally causes urethral strictures presenting as bilateral hydronephrosis. The multiple punctate calcified “stones” demonstrated on CT may suggest either diagnosis if they were actually calcified granulomas.
Regardless of the cause, most patients with a urethral stricture have chronic lower urinary tract symptoms such as decreased stream and the feeling of incomplete bladder emptying. Since this patient does not report these symptoms, it is important to consider if the stricture might be merely incidental. The absence of pain is more telling than the absence of chronic or recurrent symptoms. Lack of pain argues strongly against a pure de novo acute obstruction because abrupt stretching of the renal capsules and the walls of the collecting system is usually painful. Slow stretching caused by a progressive stricture may mask the pain of a superimposed acute obstruction. A blood clot, for example, may have precipitated an acute-on-chronic obstruction upon lodging at the urethral stricture.
The worsening systemic response to the procedure may be due to increased intravesical pressure by dilation and cystoscopy, which may have caused subsequent backflow of bacteria from the renal parenchyma into the circulation (pyelorenal backflow). The broad-spectrum antibiotic regimen suggested above and IV crystalloid infusion should be continued with close hemodynamic monitoring.
Treatment for severe sepsis was initiated with empiric piperacillin-tazobactam, ceftriaxone was discontinued, and the patient was transferred to the intensive care unit (ICU). Norepinephrine was discontinued after 24 hours. Despite the indwelling Foley catheter, his kidney function worsened (creatinine increased to 10.5 over the next 36 hours, and he remained oliguric). Therefore, bilateral percutaneous nephrostomy tubes were placed to relieve the ongoing obstruction. In the ICU, he remained febrile, despite receiving piperacillin-tazobactam, through hospital day 7. Serial blood and urine cultures all remained negative. HIV testing was negative. His chest radiograph was unremarkable, and transthoracic echocardiogram was normal. His creatinine improved but plateaued at 2.5 mg/dL by day 7.
Worsening renal function (alongside oliguria or anuria) despite a functioning Foley catheter suggests either intrinsic renal disease or bilateral ureteral obstructions. The initial attempt at relieving the obstruction with a Foley catheter did not take into consideration the bilateral ureteral strictures. As a result, soon thereafter, the insertion of percutaneous nephrostomy tubes was necessary. Given the severity of his illness, underlying obstructive uropathy, and persistent fever, suggesting an ongoing infection, one strategy would be to continue antibiotics with broader coverage than piperacillin-tazobactam. This approach may be reasonable, given the emergence of ESBL organisms and the possibility of MRSA due to instrumentation. However, it is important to note that only sterile pyuria has been identified to date, which raises the possibility of nonbacterial infections. Although chronic infection with Schistosoma haematobium can cause bilateral ureteral strictures, associated fever is limited to the acute phase of infection and not the chronic obstructive phase, unless there is a superimposed infection. Genitourinary Mycobacterium tuberculosis remains a likely possibility, regardless of the unrevealing chest radiograph. Urine nucleic acid amplification and acid-fast bacilli (AFB) smear and culture, the best initial diagnostic test, should be sent. Although less definitive, a tuberculin skin test and an interferon-gamma release assay should also be conducted. Histopathology of the ureters obtained by repeat cystoscopy may be diagnostic, but given the limited visualization during the last cystoscopy and the recent dilation of the urethra, this option should be kept in reserve for now.
Antibiotics were discontinued on day 7, but the patient continued to experience ongoing fever. Urine Histoplasma and serum cryptococcal antigens were negative. His urine AFB smear was 1+ positive. Liver function tests revealed a total protein of 7.0 g/dL, albumin 3.0 g/dL, total bilirubin 1.2 mg/dL, direct bilirubin 0.3 mg/dL, alkaline phosphatase 418 U/L, aspartate aminotransferase 65 U/L, alanine aminotransferase 88 U/L, gamma-glutamyltransferase (GGT) 609 U/L (normal, 3-60), and lactate dehydrogenase 284 U/L (normal, 85-210).
Acid-fast bacillus in the urine strongly suggests Mycobacterium tuberculosis (MTB) with several reports of likelihood ratios greater than 10. Nevertheless, confirmation is needed to rule out nontuberculous mycobacteria because of potential hepatotoxicity from treatment. Up to six urine samples should be sent for mycobacterium culture. However, false negative rates of up to 90% are reported, and final test results can take up to two months, so other methods of confirmation should be simultaneously sought. A nucleic acid amplification test of urine could rule in a pathogenic species within 24 hours. Alternatively, the probability of a nonpathogenic colonizing species would be negligible if a caseating granuloma was found. Biopsy could be obtained from the ureters, as suggested above. Liver biopsy should also be considered, especially if the moderate elevations in alkaline phosphatase and GGT (the most common liver enzyme abnormalities in hepatic tuberculosis) did not merely wax and wane with sepsis.
A CT of the thorax without IV contrast was done to evaluate for evidence of pulmonary disease given the positive urine AFB. This demonstrated bilateral fibrotic upper lobe opacities suggestive of prior granulomatous disease but no cavitary lung lesions (Figure 3). Three sputum smears were negative for AFB, but one sample showed Mycobacterium tuberculosis detected by a polymerase chain reaction (PCR) probe.
Given the concern for genitourinary tuberculosis (GUTB), it is appropriate to place the patient in respiratory isolation to exclude concomitant pulmonary tuberculosis (TB). AFB smears were negative, but the sputum PCR probe was positive, confirming pathogenic MTB. However, the negative AFB smears make the likelihood of pulmonary infectivity low. As a result, contact tracing is often deemed unnecessary by hospital infection control teams. Though his chest radiograph was normal, CT showed bilateral upper lobe fibrotic disease suggestive of prior pulmonary TB, thus making it likely that the current GUTB represents reactivation.
The two-month initiation phase of treatment with four antituberculosis drugs should begin while drug susceptibility tests are pending. Potential hepatotoxicity should be closely monitored, ideally by a clinician with experience treating tuberculosis in patients with existing liver disease. As a general precaution, alcohol should be avoided as should medications such as acetaminophen that are known to be hepatotoxic. Urology follow-up is also needed because about one-third of tuberculous ureteral strictures treated initially with percutaneous nephrostomy do not resolve with antituberculosis therapy.
The patient was started on weight-based antituberculosis treatment with four antimicrobial agents (rifampin, ethambutol, pyrazinamide, and isoniazid). He was seen in the infectious disease clinic two weeks later; his fever had resolved, and his liver function tests showed normalization of AST and LDH as well as a 45% reduction in his GGT and alkaline phosphatase levels. Two months following discharge, a nuclear medicine radionuclide angiogram renal flow scan showed normal right kidney function. The right nephrostomy tube was subsequently removed. He continued to have left kidney outflow obstruction due to a residual ureteral stricture (Figure 4). Repeat cystoscopy and attempted left ureteral stenting was unsuccessful. The left nephrostomy tube remained in place.
DISCUSSION
According to the Centers for Disease Control, in 2017, 10 million people became sick with TB, and there were 1.3 million TB-related deaths worldwide with 9,150 cases reported in the United States. Extrapulmonary TB (EPTB) constitutes 10% of all TB cases globally.1-4 GUTB is the second most common form of EPTB after lymph node TB, and it occurs in up to 20% of all pulmonary TB cases.2,3
Mycobacteria reach the genitourinary (GU) tract via hematogenous spread during primary infection or reactivation of TB. This leads to cortical and medullary lesions, which can heal spontaneously or eventually (average of 22 years) rupture into the tubules and onto the collecting system, ureters, and bladder.5,6 Spread to the ureter and bladder leads to multiple ureteral strictures and contracture of the bladder with disruption of the ureterovesical junction (UVJ), which causes hydroureter and hydronephrosis.7 Unilateral kidney involvement is most common, but bilateral involvement can occur following exacerbated hematogenous spread, particularly in immune deficient patients. Bilateral kidney involvement is also possible from retrograde spread to the good kidney due to bladder contracture and UVJ disruption.8,9 Distal infection can involve all aspects of the male and female genital tracts, but urethral strictures are extremely rare.10,11
GUTB affects males more than females (2:1) and presents insidiously at 40 to 60 years of age.12 Other risk factors for TB include birth in TB endemic areas, prior TB infection, immunosuppression, malnutrition, severe systemic disease, diabetes, and cirrhosis. It is crucial to assess these risk factors when creating and refining differential diagnoses. Many patients have hematuria and sterile pyuria as incidental initial findings. The most common symptoms arise from bladder involvement, including frequency, urgency, and dysuria. Low back pain and gross hematuria are also common, but fever and constitutional symptoms are uncommon.10 Bilateral ureteral strictures can lead to obstructive renal failure, and involvement of the genital tracts can lead to pelvic or scrotal pain, swelling, and fistula formation.10
Diagnosis involves the demonstration of TB bacilli in urine or GU tissue. The urinalysis reveals hematuria and sterile pyuria.13 Urine AFB stains are positive in 52% of cases but are not diagnostic as nontuberculous mycobacteria may also cause a positive test result.13,14 Urine cultures for Mycobacterium tuberculosis are positive in up to 90% of cases after six to eight weeks. As many as three to six morning urine samples are required to achieve diagnostic accuracy.10,14 Urine PCR for Mycobacterium tuberculosis has 96% sensitivity and up to 98% specificity,14 while PCR on GU tissue has a sensitivity of 88% and specificity of 87%.15 The rapid nucleic acid amplification assay Xpert MTB/RIF in urine has a sensitivity of 83%, and specificity of 98%.16 Imaging is required to evaluate for obstruction, and the CT scan is abnormal in up to 90% of cases, showing multiple ureteral stenoses, hydroureter and hydronephrosis, and a contracted bladder.10,17
GUTB is treated with standard antituberculosis regimens.18 Patients with urinary obstruction benefit from ureteral stenting or percutaneous nephrostomy, bladder diversion, or ureteral reconstruction surgery. Unilateral nephrectomy for a nonfunctioning kidney with extensive disease is occasionally required.19 Following treatment, relapse occurs in up to 6% of patients over five years, and long-term follow-up with urine cultures and PCR every six months is recommended.10,20 Appropriate screening and treatment for latent tuberculosis infection greatly reduces the risk of reactivation GUTB.
This patient presented with features of an infection, which, combined with his history of renal stones and his urinalysis, led to an appropriate suspicion of and empiric treatment for an upper UTI. Given the AKI and nephrolithiasis, imaging was done to exclude obstruction. The CT finding of bilateral hydroureters and hydronephrosis absent an obstructing stone or mass or abnormal bladder was the initial clue that this was not a typical bacterial infection and that there was likely an underlying infectious pathologic process such as TB involving the GU tract diffusely. The care team treated the patient as an individual with fever and sterile pyuria in the context of multiple urinary tract strictures and an initial unrevealing infectious diagnostic workup. They recognized that the clues to the ultimate diagnosis of GUTB were all in the stream.
KEY TEACHING POINTS
- GUTB is a significant cause of sterile pyuria.
- In the presence of bilateral hydronephrosis, it is vital to determine the level of obstruction. If the bladder is not distended or contracted, then obstruction is likely at the level of the ureters and initial use of percutaneous nephrostomy tubes to relieve obstruction may be preferred.
- Imaging abnormalities such as multiple ureteral strictures, hydroureter and hydronephrosis (absent an obstructing stone or mass), and the finding of a contracted bladder are highly suggestive of GUTB.
- The mainstay of treatment for GUTB is standard antituberculosis treatment regimens in combination with the relief of urinary obstruction by ureteral stenting, percutaneous nephrostomy or open surgery.
- GUTB can relapse in up to 6% of treated cases over five years, and long-term follow-up and surveillance with urine culture and PCR every six months are recommended.
Disclosures
Benjamin Mba, Nathan Houchens, Marie Jennifer Seares, and Udit Joshi have no financial conflicts of interest and no disclosures.
Funding
Brian P. Lucas receives funding from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development and Dartmouth SYNERGY, National Institutes of Health, and National Center for Translational Science (UL1TR001086).
1. Forssbohm M, Zwahlen M, Loddenkemper R, Rieder HL. Demographic characteristics of patients with extrapulmonary tuberculosis in Germany. Eur Resp J. 2008;31(1):99-105. https://doi.org/10.1183/09031936.00020607.
2. French CE, Antoine D, Gelb D, Jones JA, Gilbert RL, Watson JM. Tuberculosis in non-UK-born persons, England and Wales, 2001-2003. Int J Tuberc Lung Dis. 2007;11(5):577-584.
3. Peto HM, Pratt RH, Harrington TA, LoBue PA, Armstrong LR. Epidemiology of extrapulmonary tuberculosis in the United States, 1993-2006. Clin Infect Dis. 2009;49(9):1350-1357. https://doi.org/10.1086/605559.
4. Alvarez S, McCabe WR. Extrapulmonary tuberculosis revisited: a review of experience at Boston City and other hospitals. Medicine. 1984;63(1):25-55.
5. Simon HB, Weinstein AJ, Pasternak MS, Swartz MN, Kunz LJ. Genitourinary tuberculosis. Clinical features in a general hospital population. Am J Med. 1977;63(3):410-420. https://doi.org/10.1016/0002-9343(77)90279-0.
6. Christensen WI. Genitourinary tuberculosis: review of 102 cases. Medicine. 1974;53(5):377-390. https://doi.org/10.1016/0002-9343(77)90279-0.
7. Eastwood JB, Corbishley CM, Grange JM. Tuberculosis and the kidney. J Am Soc Nephrol. 2001;12(6):1307-1314.
8. Garcia-Rodriguez JA, Garcia Sanchez JE, Munoz Bellido JL, et al. Genitourinary tuberculosis in Spain: review of 81 cases. Clin Infect Dis.1994;18(4):557-561. https://doi.org/10.1093/clinids/18.4.557.
9. de Figueiredo AA, Lucon AM, Srougi M. Bladder augmentation for the treatment of chronic tuberculous cystitis. Clinical and urodynamic evaluation of 25 patients after long term follow-up. Neurourol Urodyn. 2006;25(5):433-440. https://doi.org/10.1002/nau.20264.
10. Figueiredo AA, Lucon AM, Srougi M. Urogenital Tuberculosis. Microbiol Spectr. 2017;5. https://doi.org/10.1128/microbiolspec.TNMI7-0015-2016.
11. Gupta N, Mandal AK, Singh SK. Tuberculosis of the prostate and urethra: A review. Indian J Urol. 2008;24(3):388-391. https://doi.org/10.4103/0970-1591.42623.
12. Figueiredo AA, Lucon AM, Junior RF, Srougi M. Epidemiology of urogenital tuberculosis worldwide. Int J Urol. 2008;15(9):827-832. https://doi.org/10.1111/j.1442-2042.2008.02099.x.
13. Mortier E, Pouchot J, Girard L, Boussougant Y, Vinceneux P. Assessment of urine analysis for the diagnosis of tuberculosis. BMJ (Clinical research ed). 1996;312:27-28. https://doi.org/10.1136/bmj.312.7022.27.
14. Moussa OM, Eraky I, El-Far MA, et al. Rapid diagnosis of genitourinary tuberculosis by polymerase chain reaction and non-radioactive DNA hybridization. J Urol. 2000;164(2):584-588. https://doi.org/10.1016/S0022-5347(05)67427-7.
15. Chawla A, Chawla K, Reddy S, et al. Can tissue PCR augment the diagnostic accuracy in genitourinary tract tuberculosis? Urol Int. 2012;88(1):34-38. https://doi.org/10.1159/000327039.
16. Kohli M, Schiller I, Dendukuri N, et al. Xpert((R)) MTB/RIF assay for extrapulmonary tuberculosis and rifampicin resistance. Cochrane Database Syst Rev. 2018;8:Cd012768. https://doi.org/10.1002/14651858.CD012768.pub2.
17. Figueiredo AA, Lucon AM, Arvellos AN, et al. A better understanding of urogenital tuberculosis pathophysiology based on radiological findings. Eur J Radiol. 2010;76(2):246-257. https://doi.org/10.1016/j.ejrad.2009.05.049.
18. Treatment of Tuberculosis: Guidelines. 4th edition. Geneva: World Health Organization. 2010.
19. O’Flynn D. Surgical treatment of genito-urinary tuberculosis. A report on 762 cases. Br J Urol. 1970;42(6):667-671. https://doi.org/10.1111/j.1464-410X.1970.tb06789.x.
20. Butler MR, O’Flynn JD. Reactivation of genito-urinary tuberculosis: a retrospective review of 838 cases. Eur Urol. 1975;1:14-17. https://doi.org/10.1159/000455566.
A 67-year-old man presented to the emergency department with four days of nausea, vomiting, and chills. He was originally from the Philippines but lived in the United States for six years. His past medical history was notable for nephrolithiasis for which a ureteral stent had been placed and was subsequently removed three years prior. He reported no abdominal pain, diarrhea, dysuria, urinary frequency, hematuria, cough, headache, or fever. He was a retired high school teacher and a lifelong nonsmoker.
This patient presents with a nonspecific constellation of constitutional and gastrointestinal (GI) symptoms. A system-based approach may be helpful when considering the differential diagnosis. Chills most often suggest infection, especially in older patients. With regard to GI causes, acute gastroenteritis and other food-borne infections can cause nausea, vomiting, and chills, but these are typically accompanied by abdominal pain and diarrhea and often resolve in less than four days. Abdominal pain would also be expected with cholecystitis as well as more life-threatening causes of nausea such as acute pancreatitis, mesenteric ischemia, and bowel obstruction. In contrast, abdominal pain would not be expected with a central nervous system (CNS) infection such as a brain abscess, which may cause nausea from increased intracranial pressure. Headaches occur in a majority of these cases, making CNS etiologies of nausea less likely. Cardiovascular causes, including myocardial ischemia and infarction, may lead to nausea and vomiting, but these are less likely given the absence of chest pain. Genitourinary causes must be considered, especially given his history of both nephrolithiasis and instrumentation. A stricture or recurrence of nephrolithiasis could lead to acute pyelonephritis or perinephric abscess, although both commonly present with urinary tract symptoms. Uremia, possibly from obstructive uropathy given the patient’s history of nephrolithiasis, could also lead to this constellation of symptoms.
On examination, temperature was 101.6 °F, heart rate 126 beats per minute, respiratory rate 18 breaths per minute, blood pressure 120/76 mm Hg, and oxygen saturation 98% on room air. The oral mucosa was moist, heart sounds were normal without murmurs, lungs were clear to auscultation, and abdomen was soft, nontender, and nondistended. There was no flank tenderness, and penile, testicular, and prostate examination findings were normal.
Laboratory studies revealed a serum sodium of 126 mEq/L, potassium 5.0 mEq/L, chloride 98 mEq/L, bicarbonate 15 mEq/L, blood urea nitrogen 88 mg/dL, creatinine 9.0 mg/dL, calcium 8 mg/dL, glucose 110 mg/dL, and albumin 3.3 g/dL. One year prior, serum creatinine was 1.4 mg/dL. His white blood cell (WBC) count was 8.0 k/uL with normal differential, hemoglobin 11.4 g/dL with normal MCV, and platelet count was normal. Serum osmolality was 286 mOsm/kg and serum parathyroid hormone (PTH) level 63 pg/mL (normal, 10-65). The urinalysis revealed cloudy urine with a specific gravity 1.009, 54 red blood cells (RBC), 236 WBC, large leukocyte esterase, negative nitrite, trace protein, and no casts or dysmorphic RBCs. A random urine specimen revealed sodium of 86 mEq/L, potassium 16 mEq/L, chloride 80 mEq/L, and creatinine 70 mg/dL.
Fever and tachycardia support an infectious cause of his symptoms. Absent flank tenderness and a normal genitourinary examination have only moderate negative predictive values for acute pyelonephritis and prostatitis, respectively. The most striking laboratory finding is his azotemia. Acute kidney injury (AKI) is more likely than chronic kidney disease (CKD) given that the PTH level is normal and the serum creatinine from a year ago was near normal. The most useful finding to differentiate AKI from CKD is the presence of atrophic kidneys on imaging. The low bicarbonate level indicates a metabolic acidosis. His serum anion gap is 13 mEq/L, which falls above most normal ranges. A mildly elevated serum anion gap together with a “delta serum anion gap/delta serum bicarbonate” ratio less than one suggest concomitant anion gap metabolic acidosis and non anion gap metabolic acidosis. The latter, coupled with a history of nephrolithiasis, may point to the possibility of renal tubular acidosis and AKI caused by urinary tract obstruction. This could also account for the marked hyponatremia. Moreover, his high fractional excretion of sodium (9%) is not suggestive of prerenal injury, the most common acute renal injury among patients who present to the emergency department. Hematuria carries a broad differential diagnosis, but most common causes include nephrolithiasis, urinary tract infection (UTI), prostatitis, neoplasm, and glomerulonephritis (GN). The lack of casts and dysmorphic RBCs makes GN unlikely. Taken together, his vital signs, examination, and laboratory studies suggest a high likelihood of an upper UTI (acute obstructive pyelonephritis) in the context of AKI due to obstructive uropathy.
Despite both a normal serum WBC count (which has only a moderate negative predictive value) and his low risk of developing life-threating organ dysfunction from sepsis based on a quick Sequential Organ Failure Assessment (qSOFA) score of zero, it is appropriate to start antibiotics after drawing blood and doing urine cultures. The next step should include administration of a broad-spectrum regimen that is appropriately dose-adjusted for renal dysfunction, such as an antipseudomonal carbapenem and vancomycin to cover extended-spectrum beta-lactamase (ESBL)-producing organisms, Pseudomonas aeruginosa, and methicillin-resistant Staphylococcus aureus (MRSA). This broad coverage is indicated for empiric treatment of complicated obstructive pyelonephritis, a condition that may arise from significant urinary obstruction and that carries a high risk of rapid clinical deterioration. He should undergo a rapid bedside test to assess for urethral or bladder outlet obstruction: either a bladder ultrasound or temporary insertion of a bladder catheter. He should also have an urgent computed tomography (CT) of his abdomen and pelvis without intravenous (IV) contrast, looking for evidence of urinary tract obstruction. A CT is preferred over ultrasound of the kidneys and bladder as CT has higher sensitivity and specificity for nephrolithiasis and neoplasm.
A CT of the abdomen and pelvis without IV contrast revealed bilateral hydroureter and hydronephrosis with multiple punctate calcified stones within the right calyces and the distal right ureter (Figure 1, Figure 2). However, these appeared too small to cause the degree of obstruction visualized. There were no stones noted in the left ureter to account for the obstruction, though small stones were noted in the left calyces. The bladder appeared normal.
Rarely are both ureters obstructed proximal to the ureterovesical junctions in the retroperitoneum. When they are, CT scans usually reveal culprit lesions that are extrinsic to the urinary tract, such as masses or retroperitoneal fibrosis, the latter of which can be associated with IgG4-related disease. Intrinsic causes of urinary tract obstructions include ureteral strictures (from infections, nephrolithiasis, instrumentation, prior radiotherapy, or rarely urothelial cancer), blood clots, metastatic ureteral deposits, or nephrolithiasis. While most intrinsic causes are unilateral, the patient is predisposed to strictures given his history of ureteral instrumentation. A preexisting unilateral obstruction due to a stricture may now, therefore, be unmasked by a second intrinsic obstruction in the contralateral ureter. Alternatively, given his remote history of living in the Philippines, a site where Schistosoma haematobium is endemic, chronic genitourinary schistosomiasis may have caused ureteral strictures due to granulomas, fibrosis, or pseudopolyps.
More commonly, bilateral hydroureter with bilateral hydronephrosis is caused by an obstruction of the bladder or urethra. CT scans can reveal prostatic hyperplasia (occasionally with protrusion into the bladder) and increased bladder wall thickness as a result of chronic bladder outlet obstruction, but the negative predictive value of either finding is modest. More revealing is that the patient reported neither an inability to pass urine (in fact, a “random” urine sample was obtained) nor suprapubic discomfort. Both symptoms would be pronounced with acute bladder obstruction but may be minimal with slowly progressive obstruction. In either case, a distended bladder would have been seen on the CT scan.
Regardless of the cause or whether the obstruction is in the upper or lower urinary tract, emergency intervention is needed to relieve the obstruction when AKI presents with bilateral hydronephrosis. A urology consultation should be sought urgently to determine the best strategy to relieve the obstruction. This may include bilateral percutaneous nephrostomy tubes given that the obstruction appears to be above the level of the bladder. Their findings will also direct additional diagnostic workup.
The patient received ceftriaxone and underwent cystoscopy, which revealed a stricture of the distal bulbar urethra. The ureters and bladder could not be completely visualized due to hematuria. The urethral stricture was dilated, and a Foley catheter was placed. In the operating room, the patient had a fever of 103 °F and developed severe hypotension unresponsive to 3 L of intravenous normal saline. Norepinephrine infusion was initiated for refractory hypotension.
Except for transurethral prostate resections, endoscopic urologic procedures rarely lead to a stricture of any segment of the urethra, suggesting that the previous ureteral stent placement and retrieval were not causal. Longstanding Mycobacterium tuberculosis or Schistosoma haematobium infection occasionally causes urethral strictures presenting as bilateral hydronephrosis. The multiple punctate calcified “stones” demonstrated on CT may suggest either diagnosis if they were actually calcified granulomas.
Regardless of the cause, most patients with a urethral stricture have chronic lower urinary tract symptoms such as decreased stream and the feeling of incomplete bladder emptying. Since this patient does not report these symptoms, it is important to consider if the stricture might be merely incidental. The absence of pain is more telling than the absence of chronic or recurrent symptoms. Lack of pain argues strongly against a pure de novo acute obstruction because abrupt stretching of the renal capsules and the walls of the collecting system is usually painful. Slow stretching caused by a progressive stricture may mask the pain of a superimposed acute obstruction. A blood clot, for example, may have precipitated an acute-on-chronic obstruction upon lodging at the urethral stricture.
The worsening systemic response to the procedure may be due to increased intravesical pressure by dilation and cystoscopy, which may have caused subsequent backflow of bacteria from the renal parenchyma into the circulation (pyelorenal backflow). The broad-spectrum antibiotic regimen suggested above and IV crystalloid infusion should be continued with close hemodynamic monitoring.
Treatment for severe sepsis was initiated with empiric piperacillin-tazobactam, ceftriaxone was discontinued, and the patient was transferred to the intensive care unit (ICU). Norepinephrine was discontinued after 24 hours. Despite the indwelling Foley catheter, his kidney function worsened (creatinine increased to 10.5 over the next 36 hours, and he remained oliguric). Therefore, bilateral percutaneous nephrostomy tubes were placed to relieve the ongoing obstruction. In the ICU, he remained febrile, despite receiving piperacillin-tazobactam, through hospital day 7. Serial blood and urine cultures all remained negative. HIV testing was negative. His chest radiograph was unremarkable, and transthoracic echocardiogram was normal. His creatinine improved but plateaued at 2.5 mg/dL by day 7.
Worsening renal function (alongside oliguria or anuria) despite a functioning Foley catheter suggests either intrinsic renal disease or bilateral ureteral obstructions. The initial attempt at relieving the obstruction with a Foley catheter did not take into consideration the bilateral ureteral strictures. As a result, soon thereafter, the insertion of percutaneous nephrostomy tubes was necessary. Given the severity of his illness, underlying obstructive uropathy, and persistent fever, suggesting an ongoing infection, one strategy would be to continue antibiotics with broader coverage than piperacillin-tazobactam. This approach may be reasonable, given the emergence of ESBL organisms and the possibility of MRSA due to instrumentation. However, it is important to note that only sterile pyuria has been identified to date, which raises the possibility of nonbacterial infections. Although chronic infection with Schistosoma haematobium can cause bilateral ureteral strictures, associated fever is limited to the acute phase of infection and not the chronic obstructive phase, unless there is a superimposed infection. Genitourinary Mycobacterium tuberculosis remains a likely possibility, regardless of the unrevealing chest radiograph. Urine nucleic acid amplification and acid-fast bacilli (AFB) smear and culture, the best initial diagnostic test, should be sent. Although less definitive, a tuberculin skin test and an interferon-gamma release assay should also be conducted. Histopathology of the ureters obtained by repeat cystoscopy may be diagnostic, but given the limited visualization during the last cystoscopy and the recent dilation of the urethra, this option should be kept in reserve for now.
Antibiotics were discontinued on day 7, but the patient continued to experience ongoing fever. Urine Histoplasma and serum cryptococcal antigens were negative. His urine AFB smear was 1+ positive. Liver function tests revealed a total protein of 7.0 g/dL, albumin 3.0 g/dL, total bilirubin 1.2 mg/dL, direct bilirubin 0.3 mg/dL, alkaline phosphatase 418 U/L, aspartate aminotransferase 65 U/L, alanine aminotransferase 88 U/L, gamma-glutamyltransferase (GGT) 609 U/L (normal, 3-60), and lactate dehydrogenase 284 U/L (normal, 85-210).
Acid-fast bacillus in the urine strongly suggests Mycobacterium tuberculosis (MTB) with several reports of likelihood ratios greater than 10. Nevertheless, confirmation is needed to rule out nontuberculous mycobacteria because of potential hepatotoxicity from treatment. Up to six urine samples should be sent for mycobacterium culture. However, false negative rates of up to 90% are reported, and final test results can take up to two months, so other methods of confirmation should be simultaneously sought. A nucleic acid amplification test of urine could rule in a pathogenic species within 24 hours. Alternatively, the probability of a nonpathogenic colonizing species would be negligible if a caseating granuloma was found. Biopsy could be obtained from the ureters, as suggested above. Liver biopsy should also be considered, especially if the moderate elevations in alkaline phosphatase and GGT (the most common liver enzyme abnormalities in hepatic tuberculosis) did not merely wax and wane with sepsis.
A CT of the thorax without IV contrast was done to evaluate for evidence of pulmonary disease given the positive urine AFB. This demonstrated bilateral fibrotic upper lobe opacities suggestive of prior granulomatous disease but no cavitary lung lesions (Figure 3). Three sputum smears were negative for AFB, but one sample showed Mycobacterium tuberculosis detected by a polymerase chain reaction (PCR) probe.
Given the concern for genitourinary tuberculosis (GUTB), it is appropriate to place the patient in respiratory isolation to exclude concomitant pulmonary tuberculosis (TB). AFB smears were negative, but the sputum PCR probe was positive, confirming pathogenic MTB. However, the negative AFB smears make the likelihood of pulmonary infectivity low. As a result, contact tracing is often deemed unnecessary by hospital infection control teams. Though his chest radiograph was normal, CT showed bilateral upper lobe fibrotic disease suggestive of prior pulmonary TB, thus making it likely that the current GUTB represents reactivation.
The two-month initiation phase of treatment with four antituberculosis drugs should begin while drug susceptibility tests are pending. Potential hepatotoxicity should be closely monitored, ideally by a clinician with experience treating tuberculosis in patients with existing liver disease. As a general precaution, alcohol should be avoided as should medications such as acetaminophen that are known to be hepatotoxic. Urology follow-up is also needed because about one-third of tuberculous ureteral strictures treated initially with percutaneous nephrostomy do not resolve with antituberculosis therapy.
The patient was started on weight-based antituberculosis treatment with four antimicrobial agents (rifampin, ethambutol, pyrazinamide, and isoniazid). He was seen in the infectious disease clinic two weeks later; his fever had resolved, and his liver function tests showed normalization of AST and LDH as well as a 45% reduction in his GGT and alkaline phosphatase levels. Two months following discharge, a nuclear medicine radionuclide angiogram renal flow scan showed normal right kidney function. The right nephrostomy tube was subsequently removed. He continued to have left kidney outflow obstruction due to a residual ureteral stricture (Figure 4). Repeat cystoscopy and attempted left ureteral stenting was unsuccessful. The left nephrostomy tube remained in place.
DISCUSSION
According to the Centers for Disease Control, in 2017, 10 million people became sick with TB, and there were 1.3 million TB-related deaths worldwide with 9,150 cases reported in the United States. Extrapulmonary TB (EPTB) constitutes 10% of all TB cases globally.1-4 GUTB is the second most common form of EPTB after lymph node TB, and it occurs in up to 20% of all pulmonary TB cases.2,3
Mycobacteria reach the genitourinary (GU) tract via hematogenous spread during primary infection or reactivation of TB. This leads to cortical and medullary lesions, which can heal spontaneously or eventually (average of 22 years) rupture into the tubules and onto the collecting system, ureters, and bladder.5,6 Spread to the ureter and bladder leads to multiple ureteral strictures and contracture of the bladder with disruption of the ureterovesical junction (UVJ), which causes hydroureter and hydronephrosis.7 Unilateral kidney involvement is most common, but bilateral involvement can occur following exacerbated hematogenous spread, particularly in immune deficient patients. Bilateral kidney involvement is also possible from retrograde spread to the good kidney due to bladder contracture and UVJ disruption.8,9 Distal infection can involve all aspects of the male and female genital tracts, but urethral strictures are extremely rare.10,11
GUTB affects males more than females (2:1) and presents insidiously at 40 to 60 years of age.12 Other risk factors for TB include birth in TB endemic areas, prior TB infection, immunosuppression, malnutrition, severe systemic disease, diabetes, and cirrhosis. It is crucial to assess these risk factors when creating and refining differential diagnoses. Many patients have hematuria and sterile pyuria as incidental initial findings. The most common symptoms arise from bladder involvement, including frequency, urgency, and dysuria. Low back pain and gross hematuria are also common, but fever and constitutional symptoms are uncommon.10 Bilateral ureteral strictures can lead to obstructive renal failure, and involvement of the genital tracts can lead to pelvic or scrotal pain, swelling, and fistula formation.10
Diagnosis involves the demonstration of TB bacilli in urine or GU tissue. The urinalysis reveals hematuria and sterile pyuria.13 Urine AFB stains are positive in 52% of cases but are not diagnostic as nontuberculous mycobacteria may also cause a positive test result.13,14 Urine cultures for Mycobacterium tuberculosis are positive in up to 90% of cases after six to eight weeks. As many as three to six morning urine samples are required to achieve diagnostic accuracy.10,14 Urine PCR for Mycobacterium tuberculosis has 96% sensitivity and up to 98% specificity,14 while PCR on GU tissue has a sensitivity of 88% and specificity of 87%.15 The rapid nucleic acid amplification assay Xpert MTB/RIF in urine has a sensitivity of 83%, and specificity of 98%.16 Imaging is required to evaluate for obstruction, and the CT scan is abnormal in up to 90% of cases, showing multiple ureteral stenoses, hydroureter and hydronephrosis, and a contracted bladder.10,17
GUTB is treated with standard antituberculosis regimens.18 Patients with urinary obstruction benefit from ureteral stenting or percutaneous nephrostomy, bladder diversion, or ureteral reconstruction surgery. Unilateral nephrectomy for a nonfunctioning kidney with extensive disease is occasionally required.19 Following treatment, relapse occurs in up to 6% of patients over five years, and long-term follow-up with urine cultures and PCR every six months is recommended.10,20 Appropriate screening and treatment for latent tuberculosis infection greatly reduces the risk of reactivation GUTB.
This patient presented with features of an infection, which, combined with his history of renal stones and his urinalysis, led to an appropriate suspicion of and empiric treatment for an upper UTI. Given the AKI and nephrolithiasis, imaging was done to exclude obstruction. The CT finding of bilateral hydroureters and hydronephrosis absent an obstructing stone or mass or abnormal bladder was the initial clue that this was not a typical bacterial infection and that there was likely an underlying infectious pathologic process such as TB involving the GU tract diffusely. The care team treated the patient as an individual with fever and sterile pyuria in the context of multiple urinary tract strictures and an initial unrevealing infectious diagnostic workup. They recognized that the clues to the ultimate diagnosis of GUTB were all in the stream.
KEY TEACHING POINTS
- GUTB is a significant cause of sterile pyuria.
- In the presence of bilateral hydronephrosis, it is vital to determine the level of obstruction. If the bladder is not distended or contracted, then obstruction is likely at the level of the ureters and initial use of percutaneous nephrostomy tubes to relieve obstruction may be preferred.
- Imaging abnormalities such as multiple ureteral strictures, hydroureter and hydronephrosis (absent an obstructing stone or mass), and the finding of a contracted bladder are highly suggestive of GUTB.
- The mainstay of treatment for GUTB is standard antituberculosis treatment regimens in combination with the relief of urinary obstruction by ureteral stenting, percutaneous nephrostomy or open surgery.
- GUTB can relapse in up to 6% of treated cases over five years, and long-term follow-up and surveillance with urine culture and PCR every six months are recommended.
Disclosures
Benjamin Mba, Nathan Houchens, Marie Jennifer Seares, and Udit Joshi have no financial conflicts of interest and no disclosures.
Funding
Brian P. Lucas receives funding from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development and Dartmouth SYNERGY, National Institutes of Health, and National Center for Translational Science (UL1TR001086).
A 67-year-old man presented to the emergency department with four days of nausea, vomiting, and chills. He was originally from the Philippines but lived in the United States for six years. His past medical history was notable for nephrolithiasis for which a ureteral stent had been placed and was subsequently removed three years prior. He reported no abdominal pain, diarrhea, dysuria, urinary frequency, hematuria, cough, headache, or fever. He was a retired high school teacher and a lifelong nonsmoker.
This patient presents with a nonspecific constellation of constitutional and gastrointestinal (GI) symptoms. A system-based approach may be helpful when considering the differential diagnosis. Chills most often suggest infection, especially in older patients. With regard to GI causes, acute gastroenteritis and other food-borne infections can cause nausea, vomiting, and chills, but these are typically accompanied by abdominal pain and diarrhea and often resolve in less than four days. Abdominal pain would also be expected with cholecystitis as well as more life-threatening causes of nausea such as acute pancreatitis, mesenteric ischemia, and bowel obstruction. In contrast, abdominal pain would not be expected with a central nervous system (CNS) infection such as a brain abscess, which may cause nausea from increased intracranial pressure. Headaches occur in a majority of these cases, making CNS etiologies of nausea less likely. Cardiovascular causes, including myocardial ischemia and infarction, may lead to nausea and vomiting, but these are less likely given the absence of chest pain. Genitourinary causes must be considered, especially given his history of both nephrolithiasis and instrumentation. A stricture or recurrence of nephrolithiasis could lead to acute pyelonephritis or perinephric abscess, although both commonly present with urinary tract symptoms. Uremia, possibly from obstructive uropathy given the patient’s history of nephrolithiasis, could also lead to this constellation of symptoms.
On examination, temperature was 101.6 °F, heart rate 126 beats per minute, respiratory rate 18 breaths per minute, blood pressure 120/76 mm Hg, and oxygen saturation 98% on room air. The oral mucosa was moist, heart sounds were normal without murmurs, lungs were clear to auscultation, and abdomen was soft, nontender, and nondistended. There was no flank tenderness, and penile, testicular, and prostate examination findings were normal.
Laboratory studies revealed a serum sodium of 126 mEq/L, potassium 5.0 mEq/L, chloride 98 mEq/L, bicarbonate 15 mEq/L, blood urea nitrogen 88 mg/dL, creatinine 9.0 mg/dL, calcium 8 mg/dL, glucose 110 mg/dL, and albumin 3.3 g/dL. One year prior, serum creatinine was 1.4 mg/dL. His white blood cell (WBC) count was 8.0 k/uL with normal differential, hemoglobin 11.4 g/dL with normal MCV, and platelet count was normal. Serum osmolality was 286 mOsm/kg and serum parathyroid hormone (PTH) level 63 pg/mL (normal, 10-65). The urinalysis revealed cloudy urine with a specific gravity 1.009, 54 red blood cells (RBC), 236 WBC, large leukocyte esterase, negative nitrite, trace protein, and no casts or dysmorphic RBCs. A random urine specimen revealed sodium of 86 mEq/L, potassium 16 mEq/L, chloride 80 mEq/L, and creatinine 70 mg/dL.
Fever and tachycardia support an infectious cause of his symptoms. Absent flank tenderness and a normal genitourinary examination have only moderate negative predictive values for acute pyelonephritis and prostatitis, respectively. The most striking laboratory finding is his azotemia. Acute kidney injury (AKI) is more likely than chronic kidney disease (CKD) given that the PTH level is normal and the serum creatinine from a year ago was near normal. The most useful finding to differentiate AKI from CKD is the presence of atrophic kidneys on imaging. The low bicarbonate level indicates a metabolic acidosis. His serum anion gap is 13 mEq/L, which falls above most normal ranges. A mildly elevated serum anion gap together with a “delta serum anion gap/delta serum bicarbonate” ratio less than one suggest concomitant anion gap metabolic acidosis and non anion gap metabolic acidosis. The latter, coupled with a history of nephrolithiasis, may point to the possibility of renal tubular acidosis and AKI caused by urinary tract obstruction. This could also account for the marked hyponatremia. Moreover, his high fractional excretion of sodium (9%) is not suggestive of prerenal injury, the most common acute renal injury among patients who present to the emergency department. Hematuria carries a broad differential diagnosis, but most common causes include nephrolithiasis, urinary tract infection (UTI), prostatitis, neoplasm, and glomerulonephritis (GN). The lack of casts and dysmorphic RBCs makes GN unlikely. Taken together, his vital signs, examination, and laboratory studies suggest a high likelihood of an upper UTI (acute obstructive pyelonephritis) in the context of AKI due to obstructive uropathy.
Despite both a normal serum WBC count (which has only a moderate negative predictive value) and his low risk of developing life-threating organ dysfunction from sepsis based on a quick Sequential Organ Failure Assessment (qSOFA) score of zero, it is appropriate to start antibiotics after drawing blood and doing urine cultures. The next step should include administration of a broad-spectrum regimen that is appropriately dose-adjusted for renal dysfunction, such as an antipseudomonal carbapenem and vancomycin to cover extended-spectrum beta-lactamase (ESBL)-producing organisms, Pseudomonas aeruginosa, and methicillin-resistant Staphylococcus aureus (MRSA). This broad coverage is indicated for empiric treatment of complicated obstructive pyelonephritis, a condition that may arise from significant urinary obstruction and that carries a high risk of rapid clinical deterioration. He should undergo a rapid bedside test to assess for urethral or bladder outlet obstruction: either a bladder ultrasound or temporary insertion of a bladder catheter. He should also have an urgent computed tomography (CT) of his abdomen and pelvis without intravenous (IV) contrast, looking for evidence of urinary tract obstruction. A CT is preferred over ultrasound of the kidneys and bladder as CT has higher sensitivity and specificity for nephrolithiasis and neoplasm.
A CT of the abdomen and pelvis without IV contrast revealed bilateral hydroureter and hydronephrosis with multiple punctate calcified stones within the right calyces and the distal right ureter (Figure 1, Figure 2). However, these appeared too small to cause the degree of obstruction visualized. There were no stones noted in the left ureter to account for the obstruction, though small stones were noted in the left calyces. The bladder appeared normal.
Rarely are both ureters obstructed proximal to the ureterovesical junctions in the retroperitoneum. When they are, CT scans usually reveal culprit lesions that are extrinsic to the urinary tract, such as masses or retroperitoneal fibrosis, the latter of which can be associated with IgG4-related disease. Intrinsic causes of urinary tract obstructions include ureteral strictures (from infections, nephrolithiasis, instrumentation, prior radiotherapy, or rarely urothelial cancer), blood clots, metastatic ureteral deposits, or nephrolithiasis. While most intrinsic causes are unilateral, the patient is predisposed to strictures given his history of ureteral instrumentation. A preexisting unilateral obstruction due to a stricture may now, therefore, be unmasked by a second intrinsic obstruction in the contralateral ureter. Alternatively, given his remote history of living in the Philippines, a site where Schistosoma haematobium is endemic, chronic genitourinary schistosomiasis may have caused ureteral strictures due to granulomas, fibrosis, or pseudopolyps.
More commonly, bilateral hydroureter with bilateral hydronephrosis is caused by an obstruction of the bladder or urethra. CT scans can reveal prostatic hyperplasia (occasionally with protrusion into the bladder) and increased bladder wall thickness as a result of chronic bladder outlet obstruction, but the negative predictive value of either finding is modest. More revealing is that the patient reported neither an inability to pass urine (in fact, a “random” urine sample was obtained) nor suprapubic discomfort. Both symptoms would be pronounced with acute bladder obstruction but may be minimal with slowly progressive obstruction. In either case, a distended bladder would have been seen on the CT scan.
Regardless of the cause or whether the obstruction is in the upper or lower urinary tract, emergency intervention is needed to relieve the obstruction when AKI presents with bilateral hydronephrosis. A urology consultation should be sought urgently to determine the best strategy to relieve the obstruction. This may include bilateral percutaneous nephrostomy tubes given that the obstruction appears to be above the level of the bladder. Their findings will also direct additional diagnostic workup.
The patient received ceftriaxone and underwent cystoscopy, which revealed a stricture of the distal bulbar urethra. The ureters and bladder could not be completely visualized due to hematuria. The urethral stricture was dilated, and a Foley catheter was placed. In the operating room, the patient had a fever of 103 °F and developed severe hypotension unresponsive to 3 L of intravenous normal saline. Norepinephrine infusion was initiated for refractory hypotension.
Except for transurethral prostate resections, endoscopic urologic procedures rarely lead to a stricture of any segment of the urethra, suggesting that the previous ureteral stent placement and retrieval were not causal. Longstanding Mycobacterium tuberculosis or Schistosoma haematobium infection occasionally causes urethral strictures presenting as bilateral hydronephrosis. The multiple punctate calcified “stones” demonstrated on CT may suggest either diagnosis if they were actually calcified granulomas.
Regardless of the cause, most patients with a urethral stricture have chronic lower urinary tract symptoms such as decreased stream and the feeling of incomplete bladder emptying. Since this patient does not report these symptoms, it is important to consider if the stricture might be merely incidental. The absence of pain is more telling than the absence of chronic or recurrent symptoms. Lack of pain argues strongly against a pure de novo acute obstruction because abrupt stretching of the renal capsules and the walls of the collecting system is usually painful. Slow stretching caused by a progressive stricture may mask the pain of a superimposed acute obstruction. A blood clot, for example, may have precipitated an acute-on-chronic obstruction upon lodging at the urethral stricture.
The worsening systemic response to the procedure may be due to increased intravesical pressure by dilation and cystoscopy, which may have caused subsequent backflow of bacteria from the renal parenchyma into the circulation (pyelorenal backflow). The broad-spectrum antibiotic regimen suggested above and IV crystalloid infusion should be continued with close hemodynamic monitoring.
Treatment for severe sepsis was initiated with empiric piperacillin-tazobactam, ceftriaxone was discontinued, and the patient was transferred to the intensive care unit (ICU). Norepinephrine was discontinued after 24 hours. Despite the indwelling Foley catheter, his kidney function worsened (creatinine increased to 10.5 over the next 36 hours, and he remained oliguric). Therefore, bilateral percutaneous nephrostomy tubes were placed to relieve the ongoing obstruction. In the ICU, he remained febrile, despite receiving piperacillin-tazobactam, through hospital day 7. Serial blood and urine cultures all remained negative. HIV testing was negative. His chest radiograph was unremarkable, and transthoracic echocardiogram was normal. His creatinine improved but plateaued at 2.5 mg/dL by day 7.
Worsening renal function (alongside oliguria or anuria) despite a functioning Foley catheter suggests either intrinsic renal disease or bilateral ureteral obstructions. The initial attempt at relieving the obstruction with a Foley catheter did not take into consideration the bilateral ureteral strictures. As a result, soon thereafter, the insertion of percutaneous nephrostomy tubes was necessary. Given the severity of his illness, underlying obstructive uropathy, and persistent fever, suggesting an ongoing infection, one strategy would be to continue antibiotics with broader coverage than piperacillin-tazobactam. This approach may be reasonable, given the emergence of ESBL organisms and the possibility of MRSA due to instrumentation. However, it is important to note that only sterile pyuria has been identified to date, which raises the possibility of nonbacterial infections. Although chronic infection with Schistosoma haematobium can cause bilateral ureteral strictures, associated fever is limited to the acute phase of infection and not the chronic obstructive phase, unless there is a superimposed infection. Genitourinary Mycobacterium tuberculosis remains a likely possibility, regardless of the unrevealing chest radiograph. Urine nucleic acid amplification and acid-fast bacilli (AFB) smear and culture, the best initial diagnostic test, should be sent. Although less definitive, a tuberculin skin test and an interferon-gamma release assay should also be conducted. Histopathology of the ureters obtained by repeat cystoscopy may be diagnostic, but given the limited visualization during the last cystoscopy and the recent dilation of the urethra, this option should be kept in reserve for now.
Antibiotics were discontinued on day 7, but the patient continued to experience ongoing fever. Urine Histoplasma and serum cryptococcal antigens were negative. His urine AFB smear was 1+ positive. Liver function tests revealed a total protein of 7.0 g/dL, albumin 3.0 g/dL, total bilirubin 1.2 mg/dL, direct bilirubin 0.3 mg/dL, alkaline phosphatase 418 U/L, aspartate aminotransferase 65 U/L, alanine aminotransferase 88 U/L, gamma-glutamyltransferase (GGT) 609 U/L (normal, 3-60), and lactate dehydrogenase 284 U/L (normal, 85-210).
Acid-fast bacillus in the urine strongly suggests Mycobacterium tuberculosis (MTB) with several reports of likelihood ratios greater than 10. Nevertheless, confirmation is needed to rule out nontuberculous mycobacteria because of potential hepatotoxicity from treatment. Up to six urine samples should be sent for mycobacterium culture. However, false negative rates of up to 90% are reported, and final test results can take up to two months, so other methods of confirmation should be simultaneously sought. A nucleic acid amplification test of urine could rule in a pathogenic species within 24 hours. Alternatively, the probability of a nonpathogenic colonizing species would be negligible if a caseating granuloma was found. Biopsy could be obtained from the ureters, as suggested above. Liver biopsy should also be considered, especially if the moderate elevations in alkaline phosphatase and GGT (the most common liver enzyme abnormalities in hepatic tuberculosis) did not merely wax and wane with sepsis.
A CT of the thorax without IV contrast was done to evaluate for evidence of pulmonary disease given the positive urine AFB. This demonstrated bilateral fibrotic upper lobe opacities suggestive of prior granulomatous disease but no cavitary lung lesions (Figure 3). Three sputum smears were negative for AFB, but one sample showed Mycobacterium tuberculosis detected by a polymerase chain reaction (PCR) probe.
Given the concern for genitourinary tuberculosis (GUTB), it is appropriate to place the patient in respiratory isolation to exclude concomitant pulmonary tuberculosis (TB). AFB smears were negative, but the sputum PCR probe was positive, confirming pathogenic MTB. However, the negative AFB smears make the likelihood of pulmonary infectivity low. As a result, contact tracing is often deemed unnecessary by hospital infection control teams. Though his chest radiograph was normal, CT showed bilateral upper lobe fibrotic disease suggestive of prior pulmonary TB, thus making it likely that the current GUTB represents reactivation.
The two-month initiation phase of treatment with four antituberculosis drugs should begin while drug susceptibility tests are pending. Potential hepatotoxicity should be closely monitored, ideally by a clinician with experience treating tuberculosis in patients with existing liver disease. As a general precaution, alcohol should be avoided as should medications such as acetaminophen that are known to be hepatotoxic. Urology follow-up is also needed because about one-third of tuberculous ureteral strictures treated initially with percutaneous nephrostomy do not resolve with antituberculosis therapy.
The patient was started on weight-based antituberculosis treatment with four antimicrobial agents (rifampin, ethambutol, pyrazinamide, and isoniazid). He was seen in the infectious disease clinic two weeks later; his fever had resolved, and his liver function tests showed normalization of AST and LDH as well as a 45% reduction in his GGT and alkaline phosphatase levels. Two months following discharge, a nuclear medicine radionuclide angiogram renal flow scan showed normal right kidney function. The right nephrostomy tube was subsequently removed. He continued to have left kidney outflow obstruction due to a residual ureteral stricture (Figure 4). Repeat cystoscopy and attempted left ureteral stenting was unsuccessful. The left nephrostomy tube remained in place.
DISCUSSION
According to the Centers for Disease Control, in 2017, 10 million people became sick with TB, and there were 1.3 million TB-related deaths worldwide with 9,150 cases reported in the United States. Extrapulmonary TB (EPTB) constitutes 10% of all TB cases globally.1-4 GUTB is the second most common form of EPTB after lymph node TB, and it occurs in up to 20% of all pulmonary TB cases.2,3
Mycobacteria reach the genitourinary (GU) tract via hematogenous spread during primary infection or reactivation of TB. This leads to cortical and medullary lesions, which can heal spontaneously or eventually (average of 22 years) rupture into the tubules and onto the collecting system, ureters, and bladder.5,6 Spread to the ureter and bladder leads to multiple ureteral strictures and contracture of the bladder with disruption of the ureterovesical junction (UVJ), which causes hydroureter and hydronephrosis.7 Unilateral kidney involvement is most common, but bilateral involvement can occur following exacerbated hematogenous spread, particularly in immune deficient patients. Bilateral kidney involvement is also possible from retrograde spread to the good kidney due to bladder contracture and UVJ disruption.8,9 Distal infection can involve all aspects of the male and female genital tracts, but urethral strictures are extremely rare.10,11
GUTB affects males more than females (2:1) and presents insidiously at 40 to 60 years of age.12 Other risk factors for TB include birth in TB endemic areas, prior TB infection, immunosuppression, malnutrition, severe systemic disease, diabetes, and cirrhosis. It is crucial to assess these risk factors when creating and refining differential diagnoses. Many patients have hematuria and sterile pyuria as incidental initial findings. The most common symptoms arise from bladder involvement, including frequency, urgency, and dysuria. Low back pain and gross hematuria are also common, but fever and constitutional symptoms are uncommon.10 Bilateral ureteral strictures can lead to obstructive renal failure, and involvement of the genital tracts can lead to pelvic or scrotal pain, swelling, and fistula formation.10
Diagnosis involves the demonstration of TB bacilli in urine or GU tissue. The urinalysis reveals hematuria and sterile pyuria.13 Urine AFB stains are positive in 52% of cases but are not diagnostic as nontuberculous mycobacteria may also cause a positive test result.13,14 Urine cultures for Mycobacterium tuberculosis are positive in up to 90% of cases after six to eight weeks. As many as three to six morning urine samples are required to achieve diagnostic accuracy.10,14 Urine PCR for Mycobacterium tuberculosis has 96% sensitivity and up to 98% specificity,14 while PCR on GU tissue has a sensitivity of 88% and specificity of 87%.15 The rapid nucleic acid amplification assay Xpert MTB/RIF in urine has a sensitivity of 83%, and specificity of 98%.16 Imaging is required to evaluate for obstruction, and the CT scan is abnormal in up to 90% of cases, showing multiple ureteral stenoses, hydroureter and hydronephrosis, and a contracted bladder.10,17
GUTB is treated with standard antituberculosis regimens.18 Patients with urinary obstruction benefit from ureteral stenting or percutaneous nephrostomy, bladder diversion, or ureteral reconstruction surgery. Unilateral nephrectomy for a nonfunctioning kidney with extensive disease is occasionally required.19 Following treatment, relapse occurs in up to 6% of patients over five years, and long-term follow-up with urine cultures and PCR every six months is recommended.10,20 Appropriate screening and treatment for latent tuberculosis infection greatly reduces the risk of reactivation GUTB.
This patient presented with features of an infection, which, combined with his history of renal stones and his urinalysis, led to an appropriate suspicion of and empiric treatment for an upper UTI. Given the AKI and nephrolithiasis, imaging was done to exclude obstruction. The CT finding of bilateral hydroureters and hydronephrosis absent an obstructing stone or mass or abnormal bladder was the initial clue that this was not a typical bacterial infection and that there was likely an underlying infectious pathologic process such as TB involving the GU tract diffusely. The care team treated the patient as an individual with fever and sterile pyuria in the context of multiple urinary tract strictures and an initial unrevealing infectious diagnostic workup. They recognized that the clues to the ultimate diagnosis of GUTB were all in the stream.
KEY TEACHING POINTS
- GUTB is a significant cause of sterile pyuria.
- In the presence of bilateral hydronephrosis, it is vital to determine the level of obstruction. If the bladder is not distended or contracted, then obstruction is likely at the level of the ureters and initial use of percutaneous nephrostomy tubes to relieve obstruction may be preferred.
- Imaging abnormalities such as multiple ureteral strictures, hydroureter and hydronephrosis (absent an obstructing stone or mass), and the finding of a contracted bladder are highly suggestive of GUTB.
- The mainstay of treatment for GUTB is standard antituberculosis treatment regimens in combination with the relief of urinary obstruction by ureteral stenting, percutaneous nephrostomy or open surgery.
- GUTB can relapse in up to 6% of treated cases over five years, and long-term follow-up and surveillance with urine culture and PCR every six months are recommended.
Disclosures
Benjamin Mba, Nathan Houchens, Marie Jennifer Seares, and Udit Joshi have no financial conflicts of interest and no disclosures.
Funding
Brian P. Lucas receives funding from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development and Dartmouth SYNERGY, National Institutes of Health, and National Center for Translational Science (UL1TR001086).
1. Forssbohm M, Zwahlen M, Loddenkemper R, Rieder HL. Demographic characteristics of patients with extrapulmonary tuberculosis in Germany. Eur Resp J. 2008;31(1):99-105. https://doi.org/10.1183/09031936.00020607.
2. French CE, Antoine D, Gelb D, Jones JA, Gilbert RL, Watson JM. Tuberculosis in non-UK-born persons, England and Wales, 2001-2003. Int J Tuberc Lung Dis. 2007;11(5):577-584.
3. Peto HM, Pratt RH, Harrington TA, LoBue PA, Armstrong LR. Epidemiology of extrapulmonary tuberculosis in the United States, 1993-2006. Clin Infect Dis. 2009;49(9):1350-1357. https://doi.org/10.1086/605559.
4. Alvarez S, McCabe WR. Extrapulmonary tuberculosis revisited: a review of experience at Boston City and other hospitals. Medicine. 1984;63(1):25-55.
5. Simon HB, Weinstein AJ, Pasternak MS, Swartz MN, Kunz LJ. Genitourinary tuberculosis. Clinical features in a general hospital population. Am J Med. 1977;63(3):410-420. https://doi.org/10.1016/0002-9343(77)90279-0.
6. Christensen WI. Genitourinary tuberculosis: review of 102 cases. Medicine. 1974;53(5):377-390. https://doi.org/10.1016/0002-9343(77)90279-0.
7. Eastwood JB, Corbishley CM, Grange JM. Tuberculosis and the kidney. J Am Soc Nephrol. 2001;12(6):1307-1314.
8. Garcia-Rodriguez JA, Garcia Sanchez JE, Munoz Bellido JL, et al. Genitourinary tuberculosis in Spain: review of 81 cases. Clin Infect Dis.1994;18(4):557-561. https://doi.org/10.1093/clinids/18.4.557.
9. de Figueiredo AA, Lucon AM, Srougi M. Bladder augmentation for the treatment of chronic tuberculous cystitis. Clinical and urodynamic evaluation of 25 patients after long term follow-up. Neurourol Urodyn. 2006;25(5):433-440. https://doi.org/10.1002/nau.20264.
10. Figueiredo AA, Lucon AM, Srougi M. Urogenital Tuberculosis. Microbiol Spectr. 2017;5. https://doi.org/10.1128/microbiolspec.TNMI7-0015-2016.
11. Gupta N, Mandal AK, Singh SK. Tuberculosis of the prostate and urethra: A review. Indian J Urol. 2008;24(3):388-391. https://doi.org/10.4103/0970-1591.42623.
12. Figueiredo AA, Lucon AM, Junior RF, Srougi M. Epidemiology of urogenital tuberculosis worldwide. Int J Urol. 2008;15(9):827-832. https://doi.org/10.1111/j.1442-2042.2008.02099.x.
13. Mortier E, Pouchot J, Girard L, Boussougant Y, Vinceneux P. Assessment of urine analysis for the diagnosis of tuberculosis. BMJ (Clinical research ed). 1996;312:27-28. https://doi.org/10.1136/bmj.312.7022.27.
14. Moussa OM, Eraky I, El-Far MA, et al. Rapid diagnosis of genitourinary tuberculosis by polymerase chain reaction and non-radioactive DNA hybridization. J Urol. 2000;164(2):584-588. https://doi.org/10.1016/S0022-5347(05)67427-7.
15. Chawla A, Chawla K, Reddy S, et al. Can tissue PCR augment the diagnostic accuracy in genitourinary tract tuberculosis? Urol Int. 2012;88(1):34-38. https://doi.org/10.1159/000327039.
16. Kohli M, Schiller I, Dendukuri N, et al. Xpert((R)) MTB/RIF assay for extrapulmonary tuberculosis and rifampicin resistance. Cochrane Database Syst Rev. 2018;8:Cd012768. https://doi.org/10.1002/14651858.CD012768.pub2.
17. Figueiredo AA, Lucon AM, Arvellos AN, et al. A better understanding of urogenital tuberculosis pathophysiology based on radiological findings. Eur J Radiol. 2010;76(2):246-257. https://doi.org/10.1016/j.ejrad.2009.05.049.
18. Treatment of Tuberculosis: Guidelines. 4th edition. Geneva: World Health Organization. 2010.
19. O’Flynn D. Surgical treatment of genito-urinary tuberculosis. A report on 762 cases. Br J Urol. 1970;42(6):667-671. https://doi.org/10.1111/j.1464-410X.1970.tb06789.x.
20. Butler MR, O’Flynn JD. Reactivation of genito-urinary tuberculosis: a retrospective review of 838 cases. Eur Urol. 1975;1:14-17. https://doi.org/10.1159/000455566.
1. Forssbohm M, Zwahlen M, Loddenkemper R, Rieder HL. Demographic characteristics of patients with extrapulmonary tuberculosis in Germany. Eur Resp J. 2008;31(1):99-105. https://doi.org/10.1183/09031936.00020607.
2. French CE, Antoine D, Gelb D, Jones JA, Gilbert RL, Watson JM. Tuberculosis in non-UK-born persons, England and Wales, 2001-2003. Int J Tuberc Lung Dis. 2007;11(5):577-584.
3. Peto HM, Pratt RH, Harrington TA, LoBue PA, Armstrong LR. Epidemiology of extrapulmonary tuberculosis in the United States, 1993-2006. Clin Infect Dis. 2009;49(9):1350-1357. https://doi.org/10.1086/605559.
4. Alvarez S, McCabe WR. Extrapulmonary tuberculosis revisited: a review of experience at Boston City and other hospitals. Medicine. 1984;63(1):25-55.
5. Simon HB, Weinstein AJ, Pasternak MS, Swartz MN, Kunz LJ. Genitourinary tuberculosis. Clinical features in a general hospital population. Am J Med. 1977;63(3):410-420. https://doi.org/10.1016/0002-9343(77)90279-0.
6. Christensen WI. Genitourinary tuberculosis: review of 102 cases. Medicine. 1974;53(5):377-390. https://doi.org/10.1016/0002-9343(77)90279-0.
7. Eastwood JB, Corbishley CM, Grange JM. Tuberculosis and the kidney. J Am Soc Nephrol. 2001;12(6):1307-1314.
8. Garcia-Rodriguez JA, Garcia Sanchez JE, Munoz Bellido JL, et al. Genitourinary tuberculosis in Spain: review of 81 cases. Clin Infect Dis.1994;18(4):557-561. https://doi.org/10.1093/clinids/18.4.557.
9. de Figueiredo AA, Lucon AM, Srougi M. Bladder augmentation for the treatment of chronic tuberculous cystitis. Clinical and urodynamic evaluation of 25 patients after long term follow-up. Neurourol Urodyn. 2006;25(5):433-440. https://doi.org/10.1002/nau.20264.
10. Figueiredo AA, Lucon AM, Srougi M. Urogenital Tuberculosis. Microbiol Spectr. 2017;5. https://doi.org/10.1128/microbiolspec.TNMI7-0015-2016.
11. Gupta N, Mandal AK, Singh SK. Tuberculosis of the prostate and urethra: A review. Indian J Urol. 2008;24(3):388-391. https://doi.org/10.4103/0970-1591.42623.
12. Figueiredo AA, Lucon AM, Junior RF, Srougi M. Epidemiology of urogenital tuberculosis worldwide. Int J Urol. 2008;15(9):827-832. https://doi.org/10.1111/j.1442-2042.2008.02099.x.
13. Mortier E, Pouchot J, Girard L, Boussougant Y, Vinceneux P. Assessment of urine analysis for the diagnosis of tuberculosis. BMJ (Clinical research ed). 1996;312:27-28. https://doi.org/10.1136/bmj.312.7022.27.
14. Moussa OM, Eraky I, El-Far MA, et al. Rapid diagnosis of genitourinary tuberculosis by polymerase chain reaction and non-radioactive DNA hybridization. J Urol. 2000;164(2):584-588. https://doi.org/10.1016/S0022-5347(05)67427-7.
15. Chawla A, Chawla K, Reddy S, et al. Can tissue PCR augment the diagnostic accuracy in genitourinary tract tuberculosis? Urol Int. 2012;88(1):34-38. https://doi.org/10.1159/000327039.
16. Kohli M, Schiller I, Dendukuri N, et al. Xpert((R)) MTB/RIF assay for extrapulmonary tuberculosis and rifampicin resistance. Cochrane Database Syst Rev. 2018;8:Cd012768. https://doi.org/10.1002/14651858.CD012768.pub2.
17. Figueiredo AA, Lucon AM, Arvellos AN, et al. A better understanding of urogenital tuberculosis pathophysiology based on radiological findings. Eur J Radiol. 2010;76(2):246-257. https://doi.org/10.1016/j.ejrad.2009.05.049.
18. Treatment of Tuberculosis: Guidelines. 4th edition. Geneva: World Health Organization. 2010.
19. O’Flynn D. Surgical treatment of genito-urinary tuberculosis. A report on 762 cases. Br J Urol. 1970;42(6):667-671. https://doi.org/10.1111/j.1464-410X.1970.tb06789.x.
20. Butler MR, O’Flynn JD. Reactivation of genito-urinary tuberculosis: a retrospective review of 838 cases. Eur Urol. 1975;1:14-17. https://doi.org/10.1159/000455566.
© 2019 Society of Hospital Medicine
Last Resort
A 22-year-old man presented to a Canadian community hospital emergency department complaining of 2-3 weeks of abdominal pain and bloating associated with early satiety. He also noted weight loss of 20 pounds over the preceding months, leg and abdominal swelling with increased girth, and 1-2 loose, nonbloody stools per day.
Early satiety and bloating are nonspecific symptoms that can be due to gastroesophageal reflux disease, peptic ulcer disease, gastrointestinal obstruction, or gastroparesis. Weight loss in a young person, particularly if >5% of body weight, is concerning for a serious underlying medical issue. It could reflect reduced intake due to anorexia, odynophagia, or dysphagia or increased energy expenditure due to an inflammatory state such as infection or rheumatic disease. The etiology of the swelling needs to be elucidated. It may be due to increased hydrostatic forces as in heart failure, venous or lymphatic obstruction, or from lowered oncotic pressure resulting from hepatic disease, nephrotic syndrome, severe malnutrition (nonbloody loose stools), or a protein losing enteropathy.
The patient was transferred to a tertiary care center for closer access to specialty consultation. He described generalized abdominal pain increasing in intensity over three weeks; bilateral lower extremity, scrotal, abdominal wall, and sacral edema; and mild dyspnea on exertion. The early satiety was not associated with dysphagia, odynophagia, nausea, or vomiting. He denied fevers, chills, night sweats, nausea, vomiting, jaundice, easy bruising, orthopnea, paroxysmal nocturnal dyspnea (PND), or chest pain. His past medical history included asthma treated with fluticasone/salmeterol and albuterol. He was a Canadian of East Asian descent working as a plumber. He previously smoked three to four cigarettes per day for six years. He stopped smoking one month before presentation. He had one alcoholic beverage per week and smoked marijuana weekly. He denied any family history of similar symptoms or malignancy.The differential diagnosis for weight loss and anasarca is broad and includes malignancies, infectious diseases, rheumatic or inflammatory disorders, malabsorption, and advanced cardiac, renal, or liver disease. His history does not classically point in one direction. The mild dyspnea on exertion may be due to cardiac disease, but it is unlikely in the absence of orthopnea and PND. The dyspnea could be due to increased abdominal pressure if ascites are present, his underlying asthma, or another etiology such as anemia. Fevers, chills, and/or night sweats can be expected in infections and some malignancies, but their absence does not exclude infections and malignancies from the differential diagnoses. Particular attention should be paid to lymphadenopathy on the physical examination. The presence of an umbilical nodule (Sister Mary Joseph sign) could indicate a malignancy (gastrointestinal or lymphoma).
The differential diagnosis for weight loss and anasarca is broad and includes malignancies, infectious diseases, rheumatic or inflammatory disorders, malabsorption, and advanced cardiac, renal, or liver disease. His history does not classically point in one direction. The mild dyspnea on exertion may be due to cardiac disease, but it is unlikely in the absence of orthopnea and PND. The dyspnea could be due to increased abdominal pressure if ascites are present, his underlying asthma, or another etiology such as anemia. Fevers, chills, and/or night sweats can be expected in infections and some malignancies, but their absence does not exclude infections and malignancies from the differential diagnoses. Particular attention should be paid to lymphadenopathy on the physical examination. The presence of an umbilical nodule (Sister Mary Joseph sign) could indicate a malignancy (gastrointestinal or lymphoma).
On physical examination, his temperature was 38.1°C, heart rate was 138 beats per minute, blood pressure was 123/86 mm Hg, respiratory rate was 20 breaths per minute, and oxygen saturation was 97% on room air. He appeared uncomfortable and diaphoretic. No scleral icterus or jaundice was appreciated. There were no palpable cervical, axillary, or inguinal lymph nodes. Cardiac examination revealed tachycardia and no murmurs, rubs, gallops, or jugular venous distention. Abdominal examination revealed abdominal distention, diffuse tenderness to deep palpation, bulging flanks, and a positive fluid wave. Liver and spleen could not be palpated or percussed secondary to abdominal distention. He had pitting bilateral lower extremity edema that extended to and included the scrotum. Neurologic and pulmonary examinations were unremarkable.
His examination reveals low-grade fever, tachycardia, and diaphoresis. Whether this represents progression of his primary disease or he has acutely developed a superimposed infection is uncertain at this point. He has notable anasarca but no jugular venous distention, crackles, or S3 gallop. The lack of evidence of pulmonary edema or increased central venous pressure on physical examination increases the likelihood of cirrhosis, hypoalbuminemia, or obstruction (lymphatic or venous) and decreases the likelihood of heart failure as the etiology of his peripheral edema and likely ascites. Despite the prominence of gastrointestinal symptoms, he has neither jaundice nor stigmata of chronic liver disease. Periorbital edema, which may be present in nephrotic syndrome, is also absent. Although he has no palpable peripheral lymphadenopathy, malignancy remains a concern.
Testing should include urinalysis for proteinuria and coagulation studies to assess synthetic function of the liver. Abdominal ultrasound is indicated to confirm ascites. If present, a diagnostic paracentesis should be performed to rule out spontaneous bacterial peritonitis and determine whether the ascites is from portal hypertension, hypoalbuminemia, or peritoneal disease. If the transaminases are elevated or if the ascitic fluid is concerning for malignancy, he will need a computed tomography (CT) of the abdomen and pelvis. A protein losing enteropathy due to malignancies (gastric cancer or lymphoma), rheumatic disease (systemic lupus erythematosus [SLE]), or infiltrative disease (amyloid) is also a possibility. If the other studies are unrevealing, stool should be sent for alpha-1 antitrypsin.
Laboratory studies revealed hemoglobin 7.8 g/dL, platelets 53 k/mm3, white blood cell count (WBC) 10.6 k/mm3, alkaline phosphatase (ALP) 217 U/L, albumin 2.7 g/dL, reticulocyte count 3 k/mm3 (reference range, 30-110 k/mm3), and ferritin 1,310 ng/mL (reference range, 20-400 ng/L). Serum aminotransferase levels, bilirubin, coagulation panel, electrolytes, and creatinine were normal. Urinalysis was negative for blood, leukocytes, and protein. Diagnostic paracentesis demonstrated a serum-ascites-albumin gradient (SAAG) of two and macrophage predominance (WBC 250 U/L). Ascitic fluid cytology and culture were negative. Blood cultures, human immunodeficiency virus (HIV)-1 and 2, cytomegalovirus (CMV), and Epstein–Barr virus (EBV) serologies were negative. Viral serologies for hepatitis A, B, and C were negative. Antinuclear antibody (ANA), anti-ds DNA, antineutrophilic cytoplasmic antibody (ANCA), serum angiotensin-converting enzyme (ACE) level, and quantitative immunoglobulin levels were all within the normal range. Chest, abdomen, and pelvis CT with contrast revealed large-volume abdominal and pelvic ascites, diffuse subcutaneous edema (Figure 1), modest hepatosplenomegaly, small bilateral pleural effusions, and mediastinal, axillary, mesenteric, periportal, peripancreatic, and retroperitoneal lymphadenopathy (Figure 2).
Malignancy is highest on the differential. In the absence of evidence of a primary tumor, a lymphoma would be the most likely diagnosis. Multicentric Castleman disease (MCD), a rare lymphoproliferative disorder with a clinical picture similar to lymphoma, should be considered.
Some of the more common viral etiologies of generalized lymphadenopathy and cytopenias are unlikely because serologies for HIV, hepatitis B and C, EBV, and CMV are negative. Tuberculosis fits with the insidious nature of his presentation and remains on the differential although a low SAAG would be expected. From a rheumatologic standpoint, the lack of characteristic findings on history and physical examination and the negative ANA and anti-ds DNA results make SLE unlikely. Although elevated in the majority of untreated sarcoid patients, a normal ACE level is not sufficient to rule out this diagnosis. IgG, IgA, and IgM levels would be low if there was significant gastrointestinal protein loss and elevated in MCD. The markedly increased ferritin level, an acute-phase reactant often elevated in the setting of inflammation or malignancy, raises suspicion for adult Still’s disease (despite the lack of characteristic arthralgias and/or rash) and hemophagocytic lymphohistiocytosis (HLH).
A SAAG greater than or equal to 1.1 indicates the presence of portal hypertension. Portal hypertension most often results from cirrhosis for which this patient has no apparent clinical findings. Etiologies of noncirrhotic portal hypertension are classified as prehepatic, intrahepatic, and posthepatic. There is no clinical or radiologic evidence of portal or splenic vein thrombosis (prehepatic) or heart failure (posthepatic). Possible intrahepatic etiologies include malignancy and sarcoid. Although uncommon, patients with malignancy-related ascites may have a high SAAG without coexisting cirrhosis. This occurs if there is portal hypertension due to extensive metastases in the liver or involvement of the portal venous system. The cytology of the ascitic fluid is negative. However, cytology is <80% sensitive in the absence of peritoneal carcinomatosis.
The most likely diagnosis at this point is lymphoma. Bone marrow biopsy is indicated to further assess his thrombocytopenia and hypoproliferative anemia and may be diagnostic for malignancy. Pathologic examination of a lymph node should be performed. Due to concern for lymphoproliferative disease, excisional biopsy is preferred to preserve tissue architecture.
Hematology was consulted for evaluation of the lymphadenopathy, anemia, and thrombocytopenia and recommended bone marrow and excisional lymph node biopsies. Bone marrow biopsy showed trilineage hypercellularity (Figure 3A) with reduced erythropoiesis and reticulin fibrosis (Figure 3B). An axillary lymph node biopsy with flow cytometry was nondiagnostic for a lymphoproliferative disorder or malignancy.
Both biopsies fail to provide a definitive diagnosis. Hypercellularity in the marrow (>70% cellularity) and reticulin fibrosis are nonspecific and could be from a malignant or reactive disease process. Lymphoma remains the most likely diagnosis. Peripheral blood for flow cytometry, lactate dehydrogenase (LDH), and uric acid should be sent. A repeat excisional biopsy of another lymph node should be performed.
Gastroenterology was consulted to evaluate the loose stools, anasarca, and hepatomegaly, and esophagogastroduodenoscopy, enteroscopy, and colonoscopy with biopsies were performed. Gastric biopsy revealed mild gastropathy. Duodenal, jejunal, and right and left colon biopsies were all normal. A liver biopsy was performed and revealed periportal inflammation. Rheumatology and infectious disease consultations did not suspect that the patient had a rheumatologic or infectious disease.
After appropriate workup and no definitive diagnosis, it is important to reassess the patient for overall stability and the presence of any new or changing symptoms (worsening symptoms, persistent fevers) that could direct further evaluation. Lymphoma remains on the differential despite multiple negative biopsies, but other less common diseases that mimic lymphoma and cause multisystem disease should be investigated. Review of the previous lymph node and tissue biopsies with the pathologist and hematologist should focus on features of adult Still’s disease (paracortical immunoblastic hyperplasia), MCD (histopathology of angiofollicular lymph node hyperplasia and presence of human herpes virus-8 (HHV-8), and HLH (hemophagocytosis). A positron emission tomography scan may not distinguish between malignancy and other fluorodeoxyglucose avid inflammatory processes but is recommended to determine the site of a future excisional lymph node biopsy.
A 10-day trial of prednisone 50 mg daily was initiated for presumed lymphoma. He experienced symptomatic improvement with decreased peripheral edema and ascites and resolution of his fevers. He was discharged home seven days after completing steroids with follow-up.
Five days after discharge, he was readmitted with worsening anasarca, massive ascites, and acute kidney injury. Admission laboratory studies revealed creatinine 1.66 mg/dL, hemoglobin 11.5 g/dL, and platelets 94 k/mm3. In addition, his ferritin level was 1,907 ng/L (reference range, 20-400 ng/L), erythrocyte sedimentation rate (ESR) was 50 mm/h (reference range, 0-20 mm/h), and C-reactive protein concentration (CRP) was 12.1 mg/dL (reference range, 0-0.5 mg/dL).
Steroids are used to treat a wide variety of illnesses, some of which are still under consideration in this patient including lymphoma, MCD, adult Still’s disease, and HLH. His symptoms recurred quickly after discontinuation of steroids in the setting of elevated ferritin, ESR, and CRP levels reflecting marked ongoing inflammation. Serologic testing for soluble IL-2 receptor, often elevated in MCD and HLH, should be performed. Excisional biopsy of an accessible node should be performed urgently.
His acute kidney injury resolved; however, he continued to have intermittent fevers, anemia, thromobocytopenia, lymphadenopathy, and hepatosplenomegaly. A hematology case-conference recommended testing for HLH, including soluble IL-2 receptor (CD25), soluble CD163, and natural killer cell degranulation assay, all of which were negative. A right inguinal lymph node biopsy revealed reactive lymphoid tissue and stained negative for HHV-8. Based on the lack of an alternative diagnosis (particularly lymphoma), the presence of multiple areas of lymphadenopathy, anemia, fevers, organomegaly, weight loss, reactive lymphoid tissue on lymph node biopsy, and elevated CRP and ESR, a working diagnosis of MCD was made. The negative HHV-8 testing was consistent with idiopathic MCD (iMCD); however, features inconsistent with iMCD included lack of polyclonal hypergammaglobulinemia and the presence of significant anasarca and thrombocytopenia. Therefore, an internet search was performed using the patient’s salient symptoms and findings. The search revealed a few recently published case reports of a rare variant of iMCD, TAFRO syndrome. TAFRO syndrome, characterized by thrombocytopenia, anasarca, fever, reticulin fibrosis and/or renal insufficiency, and organomegaly, fully explained the patient’s presentation. He was started on prednisone, rituximab (anti-CD20 antibody), and furosemide. After one month of treatment, he showed complete resolution of cytopenias, lymphadenopathy, organomegaly, anasarca, and ascites. Therapy continued for approximately three months, and he has remained symptom-free.
COMMENTARY
Castleman’s disease (CD) is a rare lymphoproliferative disorder divided into unicentric (solitary enlarged lymph node) and multicentric (multifocal enlarged lymph nodes).1 MCD typically presents with systemic inflammation, reactive proliferation of benign lymphocytes, multifocal lymphadenopathy, elevated inflammatory markers, anemia, hypoalbuminemia, and polyclonal gammaglobulinemia.1 It is hypothesized that HHV-8 drives the systemic inflammation of MCD via high levels of interleukin-6 (IL-6) activity.1 iMCD is an HHV-8-negative variant of MCD.1
TAFRO syndrome was first described in 2010 in three Japanese patients demonstrating high fever, anasarca, hepatosplenomegaly, lymphadenopathy, severe thrombocytopenia, and reticulin fibrosis.2 In 2015, the All Japan TAFRO Syndrome Research Group recognized TAFRO syndrome as a variant of iMCD and created diagnostic criteria and a severity classification system.3 Major criteria consist of anasarca, including pleural effusion and/or ascites identified on CT scan and general edema, thrombocytopenia (platelet count <100 k/mm3), and systemic inflammation (fever >37.5°C and/or serum CRP greater than or equal to 2 mg/dL).3 Two of four minor criteria must be met, which include (1) lymph node histology consistent with CD, (2) reticulin myelofibrosis and/or increased number of megakaryocytes in bone marrow, (3) mild organomegaly, including hepatomegaly, splenomegaly, and lymphadenopathy <1.5 cm in diameter identified on CT scan, and (4) progressive renal insufficiency (serum creatinine >1.2 mg/dL in males or >1.0 mg/dL in females).3 In addition, several patients with TAFRO syndrome demonstrate elevated ALP, low-normal LDH, elevated vascular endothelial growth factor, elevated IL-6, microcytic anemia, and slight polyclonal hypergammopathy.3 Malignancies such as lymphoma and myeloma, autoimmune diseases such as SLE and ANCA-associated vasculitis, infectious diseases such as those caused by mycobacteria, and POEMS (polyneuropathy, organomegaly, endocrine diseases, M-protein, and skin lesions) syndrome must be excluded to diagnose TAFRO syndrome.3,4
The pathophysiology of TAFRO syndrome is unknown, and it is unclear whether the syndrome is truly a variant of iMCD or a distinct entity.3 IL-6 is typically only mildly elevated in TAFRO syndrome, without the consequent thrombocytosis and polyclonal hypergammaglobulinemia seen in MCD, which is associated with higher levels of IL-6.1 Multiple non-HHV-8 mechanisms for TAFRO syndrome have been proposed, including (1) systemic inflammation, autoimmune/autoinflammatory mechanisms, (2) neoplastic, ectopic cytokine secretion by malignant or benign tumor cells, and/or (3) infectious, such as non-HHV-8 virus.5
Immunosuppression is the mainstay of treatment for TAFRO syndrome based on recommendations from the 2015 TAFRO Research Group.3 Glucocorticoids are considered first-line therapy.3 Cyclosporin A is recommended for individuals refractory to glucocorticoids.3 In patients with a contraindication to cyclosporin A, anti-IL-6 receptor antibodies such as tocilizumab (approved for treatment of iMCD in Japan) and siltuximab (approved for treatment of iMCD in North America and Europe) or the anti-CD20 antibody rituximab should be prescribed.3 There is evidence for the thrombopoietin receptor agonists romiplostim and eltrombopag to treat persistent thrombocytopenia.3 Additional treatments for refractory TAFRO syndrome include IVIG and plasma exchange, chemotherapy (cyclophosphamide, doxorubicin, vincristine, prednisolone), and thalidomide.3,6
Little is known about the epidemiologic characterization of TAFRO syndrome as less than 40 cases of TAFRO syndrome have been reported in the United States, Asia, and Europe.
1,4,7-9 TAFRO syndrome occurs primarily in the fourth and fifth decades of life, with case reports ranging from 14 to 78 years of age.1,3,10,11 Gender distribution varies but is likely equal for males and females.3 Mortality in TAFRO syndrome is estimated at 11%-12%.1,3 Over the past several years, a North American and European patient registry and natural history study for CD, ACCELERATE, has been initiated.4 In addition, the international Castleman Disease Collaborative Network, a Japanese multicenter retrospective study for MCD, and a nationwide Japanese research team for CD have been created.3,4 Previously, CD did not have an International Classification of Diseases (ICD) code and was likely under-recognized. An ICD-10 for CD was added, making CD and its variants easier to research for prevalence, characterization, mortality, and treatment.
After prolonged hospitalizations and extensive workup with no diagnosis, the patient’s clinical picture was most consistent with the lymphoproliferative disorder iMCD. However, iMCD is notable for polyclonal hypergammaglobulinemia, thrombocytosis, and mild anasarca. This patient had normal gammaglobulins, significant thrombocyotopenia, and profound, difficult-to-treat anasarca and ascites. Recognizing that the patient’s presentation did not fit neatly into a known clinical syndrome, an internet search was conducted based on his clinical features. This revealed TAFRO syndrome, which was at the time a newly described clinical syndrome with only a few published case reports. It was an internet search undertaken as a last resort that ultimately led to the patient’s diagnosis and successful treatment.
TEACHING POINTS
- Key clinical and pathologic features of TAFRO syndrome include thrombocytopenia, anasarca, fever, reticulin fibrosis and/or renal insufficiency, and organomegaly.
- TAFRO syndrome may be under-recognized due to very recent characterization and no previous ICD code for CD.
- TAFRO syndrome experts recommend immunosuppression for treatment of TAFRO syndrome, including glucocorticoids as first-line treatment.
- Internet searches can be helpful in the diagnosis of challenging cases, particularly with rare, unusual, and emerging diseases that have not yet been described in reference texts and only infrequently reported in the medical literature.
Disclosures
Jonathan S. Zipursky, Keri T. Holmes-Maybank, Steven L. Shumak, and Ashley A. Ducketthave none to declare.
1. Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol. 2016;91(2):220-226. PubMed
2. Takai K, Nikkuni K, Shibuya H, Hashidate H. Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly. Rinsho Ketsueki. 2010;51(5):320-325. PubMed
3. Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol. 2016;103:686-692. https://doi.org/10.1007/s12185-016-1979-1.
4. Liu AY, Nabel CS, Finkelman BS, et al. Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol. 2016;3:e163-e175. https://doi.org/10.1016/S2352-3026(16)00006-5.
5. Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924-2933. https://doi.org/10.1182/blood-2013-12-545087.
6. Sakashita K, Murata K, Takamori M. TAFRO syndrome: Current perspectives. J Blood Med. 2018;9:15-23. doi: 10.2147/JBM.S127822.
7. Louis C, Vijgen S, Samii K, et al. TAFRO syndrome in caucasians: A case report and review of the literature. Front Med. 2017;4(149):1-8. https://doi.org/10.3389/fmed.2017.00149.
8. Courtier F, Ruault NM, Crepin T, et al. A comparison of TAFRO syndrome between Japanese and non-Japanese cases: a case report and literature review. Ann Hematol. 2018;97:401-407. https://doi.org/10.1007/s00277-017-3138-z.
9. Jain P, Verstovsek S, Loghavi S, et al. Durable remission with rituximab in a patient with an unusual variant of Castleman’s disease with myelofibrosis-TAFRO syndrome. Am J Hematol. 2015;90(11):1091-1092. https://doi.org/10.1002/ajh.24015.
10. Igawa T, Sato Y. TAFRO syndeome. Hematol Oncol Clin N Am. 2018;32(1):107-118. https://doi.org/10.1016/j.hoc.2017.09.009.
11. Hawkins JM, Pillai V. TAFRO syndrome or Castleman-Kojima syndrome: a variant of multicentric Castleman disease. Blood. 2015;126(18):2163. https://doi.org/10.1182/blood-2015-07-662122.
A 22-year-old man presented to a Canadian community hospital emergency department complaining of 2-3 weeks of abdominal pain and bloating associated with early satiety. He also noted weight loss of 20 pounds over the preceding months, leg and abdominal swelling with increased girth, and 1-2 loose, nonbloody stools per day.
Early satiety and bloating are nonspecific symptoms that can be due to gastroesophageal reflux disease, peptic ulcer disease, gastrointestinal obstruction, or gastroparesis. Weight loss in a young person, particularly if >5% of body weight, is concerning for a serious underlying medical issue. It could reflect reduced intake due to anorexia, odynophagia, or dysphagia or increased energy expenditure due to an inflammatory state such as infection or rheumatic disease. The etiology of the swelling needs to be elucidated. It may be due to increased hydrostatic forces as in heart failure, venous or lymphatic obstruction, or from lowered oncotic pressure resulting from hepatic disease, nephrotic syndrome, severe malnutrition (nonbloody loose stools), or a protein losing enteropathy.
The patient was transferred to a tertiary care center for closer access to specialty consultation. He described generalized abdominal pain increasing in intensity over three weeks; bilateral lower extremity, scrotal, abdominal wall, and sacral edema; and mild dyspnea on exertion. The early satiety was not associated with dysphagia, odynophagia, nausea, or vomiting. He denied fevers, chills, night sweats, nausea, vomiting, jaundice, easy bruising, orthopnea, paroxysmal nocturnal dyspnea (PND), or chest pain. His past medical history included asthma treated with fluticasone/salmeterol and albuterol. He was a Canadian of East Asian descent working as a plumber. He previously smoked three to four cigarettes per day for six years. He stopped smoking one month before presentation. He had one alcoholic beverage per week and smoked marijuana weekly. He denied any family history of similar symptoms or malignancy.The differential diagnosis for weight loss and anasarca is broad and includes malignancies, infectious diseases, rheumatic or inflammatory disorders, malabsorption, and advanced cardiac, renal, or liver disease. His history does not classically point in one direction. The mild dyspnea on exertion may be due to cardiac disease, but it is unlikely in the absence of orthopnea and PND. The dyspnea could be due to increased abdominal pressure if ascites are present, his underlying asthma, or another etiology such as anemia. Fevers, chills, and/or night sweats can be expected in infections and some malignancies, but their absence does not exclude infections and malignancies from the differential diagnoses. Particular attention should be paid to lymphadenopathy on the physical examination. The presence of an umbilical nodule (Sister Mary Joseph sign) could indicate a malignancy (gastrointestinal or lymphoma).
The differential diagnosis for weight loss and anasarca is broad and includes malignancies, infectious diseases, rheumatic or inflammatory disorders, malabsorption, and advanced cardiac, renal, or liver disease. His history does not classically point in one direction. The mild dyspnea on exertion may be due to cardiac disease, but it is unlikely in the absence of orthopnea and PND. The dyspnea could be due to increased abdominal pressure if ascites are present, his underlying asthma, or another etiology such as anemia. Fevers, chills, and/or night sweats can be expected in infections and some malignancies, but their absence does not exclude infections and malignancies from the differential diagnoses. Particular attention should be paid to lymphadenopathy on the physical examination. The presence of an umbilical nodule (Sister Mary Joseph sign) could indicate a malignancy (gastrointestinal or lymphoma).
On physical examination, his temperature was 38.1°C, heart rate was 138 beats per minute, blood pressure was 123/86 mm Hg, respiratory rate was 20 breaths per minute, and oxygen saturation was 97% on room air. He appeared uncomfortable and diaphoretic. No scleral icterus or jaundice was appreciated. There were no palpable cervical, axillary, or inguinal lymph nodes. Cardiac examination revealed tachycardia and no murmurs, rubs, gallops, or jugular venous distention. Abdominal examination revealed abdominal distention, diffuse tenderness to deep palpation, bulging flanks, and a positive fluid wave. Liver and spleen could not be palpated or percussed secondary to abdominal distention. He had pitting bilateral lower extremity edema that extended to and included the scrotum. Neurologic and pulmonary examinations were unremarkable.
His examination reveals low-grade fever, tachycardia, and diaphoresis. Whether this represents progression of his primary disease or he has acutely developed a superimposed infection is uncertain at this point. He has notable anasarca but no jugular venous distention, crackles, or S3 gallop. The lack of evidence of pulmonary edema or increased central venous pressure on physical examination increases the likelihood of cirrhosis, hypoalbuminemia, or obstruction (lymphatic or venous) and decreases the likelihood of heart failure as the etiology of his peripheral edema and likely ascites. Despite the prominence of gastrointestinal symptoms, he has neither jaundice nor stigmata of chronic liver disease. Periorbital edema, which may be present in nephrotic syndrome, is also absent. Although he has no palpable peripheral lymphadenopathy, malignancy remains a concern.
Testing should include urinalysis for proteinuria and coagulation studies to assess synthetic function of the liver. Abdominal ultrasound is indicated to confirm ascites. If present, a diagnostic paracentesis should be performed to rule out spontaneous bacterial peritonitis and determine whether the ascites is from portal hypertension, hypoalbuminemia, or peritoneal disease. If the transaminases are elevated or if the ascitic fluid is concerning for malignancy, he will need a computed tomography (CT) of the abdomen and pelvis. A protein losing enteropathy due to malignancies (gastric cancer or lymphoma), rheumatic disease (systemic lupus erythematosus [SLE]), or infiltrative disease (amyloid) is also a possibility. If the other studies are unrevealing, stool should be sent for alpha-1 antitrypsin.
Laboratory studies revealed hemoglobin 7.8 g/dL, platelets 53 k/mm3, white blood cell count (WBC) 10.6 k/mm3, alkaline phosphatase (ALP) 217 U/L, albumin 2.7 g/dL, reticulocyte count 3 k/mm3 (reference range, 30-110 k/mm3), and ferritin 1,310 ng/mL (reference range, 20-400 ng/L). Serum aminotransferase levels, bilirubin, coagulation panel, electrolytes, and creatinine were normal. Urinalysis was negative for blood, leukocytes, and protein. Diagnostic paracentesis demonstrated a serum-ascites-albumin gradient (SAAG) of two and macrophage predominance (WBC 250 U/L). Ascitic fluid cytology and culture were negative. Blood cultures, human immunodeficiency virus (HIV)-1 and 2, cytomegalovirus (CMV), and Epstein–Barr virus (EBV) serologies were negative. Viral serologies for hepatitis A, B, and C were negative. Antinuclear antibody (ANA), anti-ds DNA, antineutrophilic cytoplasmic antibody (ANCA), serum angiotensin-converting enzyme (ACE) level, and quantitative immunoglobulin levels were all within the normal range. Chest, abdomen, and pelvis CT with contrast revealed large-volume abdominal and pelvic ascites, diffuse subcutaneous edema (Figure 1), modest hepatosplenomegaly, small bilateral pleural effusions, and mediastinal, axillary, mesenteric, periportal, peripancreatic, and retroperitoneal lymphadenopathy (Figure 2).
Malignancy is highest on the differential. In the absence of evidence of a primary tumor, a lymphoma would be the most likely diagnosis. Multicentric Castleman disease (MCD), a rare lymphoproliferative disorder with a clinical picture similar to lymphoma, should be considered.
Some of the more common viral etiologies of generalized lymphadenopathy and cytopenias are unlikely because serologies for HIV, hepatitis B and C, EBV, and CMV are negative. Tuberculosis fits with the insidious nature of his presentation and remains on the differential although a low SAAG would be expected. From a rheumatologic standpoint, the lack of characteristic findings on history and physical examination and the negative ANA and anti-ds DNA results make SLE unlikely. Although elevated in the majority of untreated sarcoid patients, a normal ACE level is not sufficient to rule out this diagnosis. IgG, IgA, and IgM levels would be low if there was significant gastrointestinal protein loss and elevated in MCD. The markedly increased ferritin level, an acute-phase reactant often elevated in the setting of inflammation or malignancy, raises suspicion for adult Still’s disease (despite the lack of characteristic arthralgias and/or rash) and hemophagocytic lymphohistiocytosis (HLH).
A SAAG greater than or equal to 1.1 indicates the presence of portal hypertension. Portal hypertension most often results from cirrhosis for which this patient has no apparent clinical findings. Etiologies of noncirrhotic portal hypertension are classified as prehepatic, intrahepatic, and posthepatic. There is no clinical or radiologic evidence of portal or splenic vein thrombosis (prehepatic) or heart failure (posthepatic). Possible intrahepatic etiologies include malignancy and sarcoid. Although uncommon, patients with malignancy-related ascites may have a high SAAG without coexisting cirrhosis. This occurs if there is portal hypertension due to extensive metastases in the liver or involvement of the portal venous system. The cytology of the ascitic fluid is negative. However, cytology is <80% sensitive in the absence of peritoneal carcinomatosis.
The most likely diagnosis at this point is lymphoma. Bone marrow biopsy is indicated to further assess his thrombocytopenia and hypoproliferative anemia and may be diagnostic for malignancy. Pathologic examination of a lymph node should be performed. Due to concern for lymphoproliferative disease, excisional biopsy is preferred to preserve tissue architecture.
Hematology was consulted for evaluation of the lymphadenopathy, anemia, and thrombocytopenia and recommended bone marrow and excisional lymph node biopsies. Bone marrow biopsy showed trilineage hypercellularity (Figure 3A) with reduced erythropoiesis and reticulin fibrosis (Figure 3B). An axillary lymph node biopsy with flow cytometry was nondiagnostic for a lymphoproliferative disorder or malignancy.
Both biopsies fail to provide a definitive diagnosis. Hypercellularity in the marrow (>70% cellularity) and reticulin fibrosis are nonspecific and could be from a malignant or reactive disease process. Lymphoma remains the most likely diagnosis. Peripheral blood for flow cytometry, lactate dehydrogenase (LDH), and uric acid should be sent. A repeat excisional biopsy of another lymph node should be performed.
Gastroenterology was consulted to evaluate the loose stools, anasarca, and hepatomegaly, and esophagogastroduodenoscopy, enteroscopy, and colonoscopy with biopsies were performed. Gastric biopsy revealed mild gastropathy. Duodenal, jejunal, and right and left colon biopsies were all normal. A liver biopsy was performed and revealed periportal inflammation. Rheumatology and infectious disease consultations did not suspect that the patient had a rheumatologic or infectious disease.
After appropriate workup and no definitive diagnosis, it is important to reassess the patient for overall stability and the presence of any new or changing symptoms (worsening symptoms, persistent fevers) that could direct further evaluation. Lymphoma remains on the differential despite multiple negative biopsies, but other less common diseases that mimic lymphoma and cause multisystem disease should be investigated. Review of the previous lymph node and tissue biopsies with the pathologist and hematologist should focus on features of adult Still’s disease (paracortical immunoblastic hyperplasia), MCD (histopathology of angiofollicular lymph node hyperplasia and presence of human herpes virus-8 (HHV-8), and HLH (hemophagocytosis). A positron emission tomography scan may not distinguish between malignancy and other fluorodeoxyglucose avid inflammatory processes but is recommended to determine the site of a future excisional lymph node biopsy.
A 10-day trial of prednisone 50 mg daily was initiated for presumed lymphoma. He experienced symptomatic improvement with decreased peripheral edema and ascites and resolution of his fevers. He was discharged home seven days after completing steroids with follow-up.
Five days after discharge, he was readmitted with worsening anasarca, massive ascites, and acute kidney injury. Admission laboratory studies revealed creatinine 1.66 mg/dL, hemoglobin 11.5 g/dL, and platelets 94 k/mm3. In addition, his ferritin level was 1,907 ng/L (reference range, 20-400 ng/L), erythrocyte sedimentation rate (ESR) was 50 mm/h (reference range, 0-20 mm/h), and C-reactive protein concentration (CRP) was 12.1 mg/dL (reference range, 0-0.5 mg/dL).
Steroids are used to treat a wide variety of illnesses, some of which are still under consideration in this patient including lymphoma, MCD, adult Still’s disease, and HLH. His symptoms recurred quickly after discontinuation of steroids in the setting of elevated ferritin, ESR, and CRP levels reflecting marked ongoing inflammation. Serologic testing for soluble IL-2 receptor, often elevated in MCD and HLH, should be performed. Excisional biopsy of an accessible node should be performed urgently.
His acute kidney injury resolved; however, he continued to have intermittent fevers, anemia, thromobocytopenia, lymphadenopathy, and hepatosplenomegaly. A hematology case-conference recommended testing for HLH, including soluble IL-2 receptor (CD25), soluble CD163, and natural killer cell degranulation assay, all of which were negative. A right inguinal lymph node biopsy revealed reactive lymphoid tissue and stained negative for HHV-8. Based on the lack of an alternative diagnosis (particularly lymphoma), the presence of multiple areas of lymphadenopathy, anemia, fevers, organomegaly, weight loss, reactive lymphoid tissue on lymph node biopsy, and elevated CRP and ESR, a working diagnosis of MCD was made. The negative HHV-8 testing was consistent with idiopathic MCD (iMCD); however, features inconsistent with iMCD included lack of polyclonal hypergammaglobulinemia and the presence of significant anasarca and thrombocytopenia. Therefore, an internet search was performed using the patient’s salient symptoms and findings. The search revealed a few recently published case reports of a rare variant of iMCD, TAFRO syndrome. TAFRO syndrome, characterized by thrombocytopenia, anasarca, fever, reticulin fibrosis and/or renal insufficiency, and organomegaly, fully explained the patient’s presentation. He was started on prednisone, rituximab (anti-CD20 antibody), and furosemide. After one month of treatment, he showed complete resolution of cytopenias, lymphadenopathy, organomegaly, anasarca, and ascites. Therapy continued for approximately three months, and he has remained symptom-free.
COMMENTARY
Castleman’s disease (CD) is a rare lymphoproliferative disorder divided into unicentric (solitary enlarged lymph node) and multicentric (multifocal enlarged lymph nodes).1 MCD typically presents with systemic inflammation, reactive proliferation of benign lymphocytes, multifocal lymphadenopathy, elevated inflammatory markers, anemia, hypoalbuminemia, and polyclonal gammaglobulinemia.1 It is hypothesized that HHV-8 drives the systemic inflammation of MCD via high levels of interleukin-6 (IL-6) activity.1 iMCD is an HHV-8-negative variant of MCD.1
TAFRO syndrome was first described in 2010 in three Japanese patients demonstrating high fever, anasarca, hepatosplenomegaly, lymphadenopathy, severe thrombocytopenia, and reticulin fibrosis.2 In 2015, the All Japan TAFRO Syndrome Research Group recognized TAFRO syndrome as a variant of iMCD and created diagnostic criteria and a severity classification system.3 Major criteria consist of anasarca, including pleural effusion and/or ascites identified on CT scan and general edema, thrombocytopenia (platelet count <100 k/mm3), and systemic inflammation (fever >37.5°C and/or serum CRP greater than or equal to 2 mg/dL).3 Two of four minor criteria must be met, which include (1) lymph node histology consistent with CD, (2) reticulin myelofibrosis and/or increased number of megakaryocytes in bone marrow, (3) mild organomegaly, including hepatomegaly, splenomegaly, and lymphadenopathy <1.5 cm in diameter identified on CT scan, and (4) progressive renal insufficiency (serum creatinine >1.2 mg/dL in males or >1.0 mg/dL in females).3 In addition, several patients with TAFRO syndrome demonstrate elevated ALP, low-normal LDH, elevated vascular endothelial growth factor, elevated IL-6, microcytic anemia, and slight polyclonal hypergammopathy.3 Malignancies such as lymphoma and myeloma, autoimmune diseases such as SLE and ANCA-associated vasculitis, infectious diseases such as those caused by mycobacteria, and POEMS (polyneuropathy, organomegaly, endocrine diseases, M-protein, and skin lesions) syndrome must be excluded to diagnose TAFRO syndrome.3,4
The pathophysiology of TAFRO syndrome is unknown, and it is unclear whether the syndrome is truly a variant of iMCD or a distinct entity.3 IL-6 is typically only mildly elevated in TAFRO syndrome, without the consequent thrombocytosis and polyclonal hypergammaglobulinemia seen in MCD, which is associated with higher levels of IL-6.1 Multiple non-HHV-8 mechanisms for TAFRO syndrome have been proposed, including (1) systemic inflammation, autoimmune/autoinflammatory mechanisms, (2) neoplastic, ectopic cytokine secretion by malignant or benign tumor cells, and/or (3) infectious, such as non-HHV-8 virus.5
Immunosuppression is the mainstay of treatment for TAFRO syndrome based on recommendations from the 2015 TAFRO Research Group.3 Glucocorticoids are considered first-line therapy.3 Cyclosporin A is recommended for individuals refractory to glucocorticoids.3 In patients with a contraindication to cyclosporin A, anti-IL-6 receptor antibodies such as tocilizumab (approved for treatment of iMCD in Japan) and siltuximab (approved for treatment of iMCD in North America and Europe) or the anti-CD20 antibody rituximab should be prescribed.3 There is evidence for the thrombopoietin receptor agonists romiplostim and eltrombopag to treat persistent thrombocytopenia.3 Additional treatments for refractory TAFRO syndrome include IVIG and plasma exchange, chemotherapy (cyclophosphamide, doxorubicin, vincristine, prednisolone), and thalidomide.3,6
Little is known about the epidemiologic characterization of TAFRO syndrome as less than 40 cases of TAFRO syndrome have been reported in the United States, Asia, and Europe.
1,4,7-9 TAFRO syndrome occurs primarily in the fourth and fifth decades of life, with case reports ranging from 14 to 78 years of age.1,3,10,11 Gender distribution varies but is likely equal for males and females.3 Mortality in TAFRO syndrome is estimated at 11%-12%.1,3 Over the past several years, a North American and European patient registry and natural history study for CD, ACCELERATE, has been initiated.4 In addition, the international Castleman Disease Collaborative Network, a Japanese multicenter retrospective study for MCD, and a nationwide Japanese research team for CD have been created.3,4 Previously, CD did not have an International Classification of Diseases (ICD) code and was likely under-recognized. An ICD-10 for CD was added, making CD and its variants easier to research for prevalence, characterization, mortality, and treatment.
After prolonged hospitalizations and extensive workup with no diagnosis, the patient’s clinical picture was most consistent with the lymphoproliferative disorder iMCD. However, iMCD is notable for polyclonal hypergammaglobulinemia, thrombocytosis, and mild anasarca. This patient had normal gammaglobulins, significant thrombocyotopenia, and profound, difficult-to-treat anasarca and ascites. Recognizing that the patient’s presentation did not fit neatly into a known clinical syndrome, an internet search was conducted based on his clinical features. This revealed TAFRO syndrome, which was at the time a newly described clinical syndrome with only a few published case reports. It was an internet search undertaken as a last resort that ultimately led to the patient’s diagnosis and successful treatment.
TEACHING POINTS
- Key clinical and pathologic features of TAFRO syndrome include thrombocytopenia, anasarca, fever, reticulin fibrosis and/or renal insufficiency, and organomegaly.
- TAFRO syndrome may be under-recognized due to very recent characterization and no previous ICD code for CD.
- TAFRO syndrome experts recommend immunosuppression for treatment of TAFRO syndrome, including glucocorticoids as first-line treatment.
- Internet searches can be helpful in the diagnosis of challenging cases, particularly with rare, unusual, and emerging diseases that have not yet been described in reference texts and only infrequently reported in the medical literature.
Disclosures
Jonathan S. Zipursky, Keri T. Holmes-Maybank, Steven L. Shumak, and Ashley A. Ducketthave none to declare.
A 22-year-old man presented to a Canadian community hospital emergency department complaining of 2-3 weeks of abdominal pain and bloating associated with early satiety. He also noted weight loss of 20 pounds over the preceding months, leg and abdominal swelling with increased girth, and 1-2 loose, nonbloody stools per day.
Early satiety and bloating are nonspecific symptoms that can be due to gastroesophageal reflux disease, peptic ulcer disease, gastrointestinal obstruction, or gastroparesis. Weight loss in a young person, particularly if >5% of body weight, is concerning for a serious underlying medical issue. It could reflect reduced intake due to anorexia, odynophagia, or dysphagia or increased energy expenditure due to an inflammatory state such as infection or rheumatic disease. The etiology of the swelling needs to be elucidated. It may be due to increased hydrostatic forces as in heart failure, venous or lymphatic obstruction, or from lowered oncotic pressure resulting from hepatic disease, nephrotic syndrome, severe malnutrition (nonbloody loose stools), or a protein losing enteropathy.
The patient was transferred to a tertiary care center for closer access to specialty consultation. He described generalized abdominal pain increasing in intensity over three weeks; bilateral lower extremity, scrotal, abdominal wall, and sacral edema; and mild dyspnea on exertion. The early satiety was not associated with dysphagia, odynophagia, nausea, or vomiting. He denied fevers, chills, night sweats, nausea, vomiting, jaundice, easy bruising, orthopnea, paroxysmal nocturnal dyspnea (PND), or chest pain. His past medical history included asthma treated with fluticasone/salmeterol and albuterol. He was a Canadian of East Asian descent working as a plumber. He previously smoked three to four cigarettes per day for six years. He stopped smoking one month before presentation. He had one alcoholic beverage per week and smoked marijuana weekly. He denied any family history of similar symptoms or malignancy.The differential diagnosis for weight loss and anasarca is broad and includes malignancies, infectious diseases, rheumatic or inflammatory disorders, malabsorption, and advanced cardiac, renal, or liver disease. His history does not classically point in one direction. The mild dyspnea on exertion may be due to cardiac disease, but it is unlikely in the absence of orthopnea and PND. The dyspnea could be due to increased abdominal pressure if ascites are present, his underlying asthma, or another etiology such as anemia. Fevers, chills, and/or night sweats can be expected in infections and some malignancies, but their absence does not exclude infections and malignancies from the differential diagnoses. Particular attention should be paid to lymphadenopathy on the physical examination. The presence of an umbilical nodule (Sister Mary Joseph sign) could indicate a malignancy (gastrointestinal or lymphoma).
The differential diagnosis for weight loss and anasarca is broad and includes malignancies, infectious diseases, rheumatic or inflammatory disorders, malabsorption, and advanced cardiac, renal, or liver disease. His history does not classically point in one direction. The mild dyspnea on exertion may be due to cardiac disease, but it is unlikely in the absence of orthopnea and PND. The dyspnea could be due to increased abdominal pressure if ascites are present, his underlying asthma, or another etiology such as anemia. Fevers, chills, and/or night sweats can be expected in infections and some malignancies, but their absence does not exclude infections and malignancies from the differential diagnoses. Particular attention should be paid to lymphadenopathy on the physical examination. The presence of an umbilical nodule (Sister Mary Joseph sign) could indicate a malignancy (gastrointestinal or lymphoma).
On physical examination, his temperature was 38.1°C, heart rate was 138 beats per minute, blood pressure was 123/86 mm Hg, respiratory rate was 20 breaths per minute, and oxygen saturation was 97% on room air. He appeared uncomfortable and diaphoretic. No scleral icterus or jaundice was appreciated. There were no palpable cervical, axillary, or inguinal lymph nodes. Cardiac examination revealed tachycardia and no murmurs, rubs, gallops, or jugular venous distention. Abdominal examination revealed abdominal distention, diffuse tenderness to deep palpation, bulging flanks, and a positive fluid wave. Liver and spleen could not be palpated or percussed secondary to abdominal distention. He had pitting bilateral lower extremity edema that extended to and included the scrotum. Neurologic and pulmonary examinations were unremarkable.
His examination reveals low-grade fever, tachycardia, and diaphoresis. Whether this represents progression of his primary disease or he has acutely developed a superimposed infection is uncertain at this point. He has notable anasarca but no jugular venous distention, crackles, or S3 gallop. The lack of evidence of pulmonary edema or increased central venous pressure on physical examination increases the likelihood of cirrhosis, hypoalbuminemia, or obstruction (lymphatic or venous) and decreases the likelihood of heart failure as the etiology of his peripheral edema and likely ascites. Despite the prominence of gastrointestinal symptoms, he has neither jaundice nor stigmata of chronic liver disease. Periorbital edema, which may be present in nephrotic syndrome, is also absent. Although he has no palpable peripheral lymphadenopathy, malignancy remains a concern.
Testing should include urinalysis for proteinuria and coagulation studies to assess synthetic function of the liver. Abdominal ultrasound is indicated to confirm ascites. If present, a diagnostic paracentesis should be performed to rule out spontaneous bacterial peritonitis and determine whether the ascites is from portal hypertension, hypoalbuminemia, or peritoneal disease. If the transaminases are elevated or if the ascitic fluid is concerning for malignancy, he will need a computed tomography (CT) of the abdomen and pelvis. A protein losing enteropathy due to malignancies (gastric cancer or lymphoma), rheumatic disease (systemic lupus erythematosus [SLE]), or infiltrative disease (amyloid) is also a possibility. If the other studies are unrevealing, stool should be sent for alpha-1 antitrypsin.
Laboratory studies revealed hemoglobin 7.8 g/dL, platelets 53 k/mm3, white blood cell count (WBC) 10.6 k/mm3, alkaline phosphatase (ALP) 217 U/L, albumin 2.7 g/dL, reticulocyte count 3 k/mm3 (reference range, 30-110 k/mm3), and ferritin 1,310 ng/mL (reference range, 20-400 ng/L). Serum aminotransferase levels, bilirubin, coagulation panel, electrolytes, and creatinine were normal. Urinalysis was negative for blood, leukocytes, and protein. Diagnostic paracentesis demonstrated a serum-ascites-albumin gradient (SAAG) of two and macrophage predominance (WBC 250 U/L). Ascitic fluid cytology and culture were negative. Blood cultures, human immunodeficiency virus (HIV)-1 and 2, cytomegalovirus (CMV), and Epstein–Barr virus (EBV) serologies were negative. Viral serologies for hepatitis A, B, and C were negative. Antinuclear antibody (ANA), anti-ds DNA, antineutrophilic cytoplasmic antibody (ANCA), serum angiotensin-converting enzyme (ACE) level, and quantitative immunoglobulin levels were all within the normal range. Chest, abdomen, and pelvis CT with contrast revealed large-volume abdominal and pelvic ascites, diffuse subcutaneous edema (Figure 1), modest hepatosplenomegaly, small bilateral pleural effusions, and mediastinal, axillary, mesenteric, periportal, peripancreatic, and retroperitoneal lymphadenopathy (Figure 2).
Malignancy is highest on the differential. In the absence of evidence of a primary tumor, a lymphoma would be the most likely diagnosis. Multicentric Castleman disease (MCD), a rare lymphoproliferative disorder with a clinical picture similar to lymphoma, should be considered.
Some of the more common viral etiologies of generalized lymphadenopathy and cytopenias are unlikely because serologies for HIV, hepatitis B and C, EBV, and CMV are negative. Tuberculosis fits with the insidious nature of his presentation and remains on the differential although a low SAAG would be expected. From a rheumatologic standpoint, the lack of characteristic findings on history and physical examination and the negative ANA and anti-ds DNA results make SLE unlikely. Although elevated in the majority of untreated sarcoid patients, a normal ACE level is not sufficient to rule out this diagnosis. IgG, IgA, and IgM levels would be low if there was significant gastrointestinal protein loss and elevated in MCD. The markedly increased ferritin level, an acute-phase reactant often elevated in the setting of inflammation or malignancy, raises suspicion for adult Still’s disease (despite the lack of characteristic arthralgias and/or rash) and hemophagocytic lymphohistiocytosis (HLH).
A SAAG greater than or equal to 1.1 indicates the presence of portal hypertension. Portal hypertension most often results from cirrhosis for which this patient has no apparent clinical findings. Etiologies of noncirrhotic portal hypertension are classified as prehepatic, intrahepatic, and posthepatic. There is no clinical or radiologic evidence of portal or splenic vein thrombosis (prehepatic) or heart failure (posthepatic). Possible intrahepatic etiologies include malignancy and sarcoid. Although uncommon, patients with malignancy-related ascites may have a high SAAG without coexisting cirrhosis. This occurs if there is portal hypertension due to extensive metastases in the liver or involvement of the portal venous system. The cytology of the ascitic fluid is negative. However, cytology is <80% sensitive in the absence of peritoneal carcinomatosis.
The most likely diagnosis at this point is lymphoma. Bone marrow biopsy is indicated to further assess his thrombocytopenia and hypoproliferative anemia and may be diagnostic for malignancy. Pathologic examination of a lymph node should be performed. Due to concern for lymphoproliferative disease, excisional biopsy is preferred to preserve tissue architecture.
Hematology was consulted for evaluation of the lymphadenopathy, anemia, and thrombocytopenia and recommended bone marrow and excisional lymph node biopsies. Bone marrow biopsy showed trilineage hypercellularity (Figure 3A) with reduced erythropoiesis and reticulin fibrosis (Figure 3B). An axillary lymph node biopsy with flow cytometry was nondiagnostic for a lymphoproliferative disorder or malignancy.
Both biopsies fail to provide a definitive diagnosis. Hypercellularity in the marrow (>70% cellularity) and reticulin fibrosis are nonspecific and could be from a malignant or reactive disease process. Lymphoma remains the most likely diagnosis. Peripheral blood for flow cytometry, lactate dehydrogenase (LDH), and uric acid should be sent. A repeat excisional biopsy of another lymph node should be performed.
Gastroenterology was consulted to evaluate the loose stools, anasarca, and hepatomegaly, and esophagogastroduodenoscopy, enteroscopy, and colonoscopy with biopsies were performed. Gastric biopsy revealed mild gastropathy. Duodenal, jejunal, and right and left colon biopsies were all normal. A liver biopsy was performed and revealed periportal inflammation. Rheumatology and infectious disease consultations did not suspect that the patient had a rheumatologic or infectious disease.
After appropriate workup and no definitive diagnosis, it is important to reassess the patient for overall stability and the presence of any new or changing symptoms (worsening symptoms, persistent fevers) that could direct further evaluation. Lymphoma remains on the differential despite multiple negative biopsies, but other less common diseases that mimic lymphoma and cause multisystem disease should be investigated. Review of the previous lymph node and tissue biopsies with the pathologist and hematologist should focus on features of adult Still’s disease (paracortical immunoblastic hyperplasia), MCD (histopathology of angiofollicular lymph node hyperplasia and presence of human herpes virus-8 (HHV-8), and HLH (hemophagocytosis). A positron emission tomography scan may not distinguish between malignancy and other fluorodeoxyglucose avid inflammatory processes but is recommended to determine the site of a future excisional lymph node biopsy.
A 10-day trial of prednisone 50 mg daily was initiated for presumed lymphoma. He experienced symptomatic improvement with decreased peripheral edema and ascites and resolution of his fevers. He was discharged home seven days after completing steroids with follow-up.
Five days after discharge, he was readmitted with worsening anasarca, massive ascites, and acute kidney injury. Admission laboratory studies revealed creatinine 1.66 mg/dL, hemoglobin 11.5 g/dL, and platelets 94 k/mm3. In addition, his ferritin level was 1,907 ng/L (reference range, 20-400 ng/L), erythrocyte sedimentation rate (ESR) was 50 mm/h (reference range, 0-20 mm/h), and C-reactive protein concentration (CRP) was 12.1 mg/dL (reference range, 0-0.5 mg/dL).
Steroids are used to treat a wide variety of illnesses, some of which are still under consideration in this patient including lymphoma, MCD, adult Still’s disease, and HLH. His symptoms recurred quickly after discontinuation of steroids in the setting of elevated ferritin, ESR, and CRP levels reflecting marked ongoing inflammation. Serologic testing for soluble IL-2 receptor, often elevated in MCD and HLH, should be performed. Excisional biopsy of an accessible node should be performed urgently.
His acute kidney injury resolved; however, he continued to have intermittent fevers, anemia, thromobocytopenia, lymphadenopathy, and hepatosplenomegaly. A hematology case-conference recommended testing for HLH, including soluble IL-2 receptor (CD25), soluble CD163, and natural killer cell degranulation assay, all of which were negative. A right inguinal lymph node biopsy revealed reactive lymphoid tissue and stained negative for HHV-8. Based on the lack of an alternative diagnosis (particularly lymphoma), the presence of multiple areas of lymphadenopathy, anemia, fevers, organomegaly, weight loss, reactive lymphoid tissue on lymph node biopsy, and elevated CRP and ESR, a working diagnosis of MCD was made. The negative HHV-8 testing was consistent with idiopathic MCD (iMCD); however, features inconsistent with iMCD included lack of polyclonal hypergammaglobulinemia and the presence of significant anasarca and thrombocytopenia. Therefore, an internet search was performed using the patient’s salient symptoms and findings. The search revealed a few recently published case reports of a rare variant of iMCD, TAFRO syndrome. TAFRO syndrome, characterized by thrombocytopenia, anasarca, fever, reticulin fibrosis and/or renal insufficiency, and organomegaly, fully explained the patient’s presentation. He was started on prednisone, rituximab (anti-CD20 antibody), and furosemide. After one month of treatment, he showed complete resolution of cytopenias, lymphadenopathy, organomegaly, anasarca, and ascites. Therapy continued for approximately three months, and he has remained symptom-free.
COMMENTARY
Castleman’s disease (CD) is a rare lymphoproliferative disorder divided into unicentric (solitary enlarged lymph node) and multicentric (multifocal enlarged lymph nodes).1 MCD typically presents with systemic inflammation, reactive proliferation of benign lymphocytes, multifocal lymphadenopathy, elevated inflammatory markers, anemia, hypoalbuminemia, and polyclonal gammaglobulinemia.1 It is hypothesized that HHV-8 drives the systemic inflammation of MCD via high levels of interleukin-6 (IL-6) activity.1 iMCD is an HHV-8-negative variant of MCD.1
TAFRO syndrome was first described in 2010 in three Japanese patients demonstrating high fever, anasarca, hepatosplenomegaly, lymphadenopathy, severe thrombocytopenia, and reticulin fibrosis.2 In 2015, the All Japan TAFRO Syndrome Research Group recognized TAFRO syndrome as a variant of iMCD and created diagnostic criteria and a severity classification system.3 Major criteria consist of anasarca, including pleural effusion and/or ascites identified on CT scan and general edema, thrombocytopenia (platelet count <100 k/mm3), and systemic inflammation (fever >37.5°C and/or serum CRP greater than or equal to 2 mg/dL).3 Two of four minor criteria must be met, which include (1) lymph node histology consistent with CD, (2) reticulin myelofibrosis and/or increased number of megakaryocytes in bone marrow, (3) mild organomegaly, including hepatomegaly, splenomegaly, and lymphadenopathy <1.5 cm in diameter identified on CT scan, and (4) progressive renal insufficiency (serum creatinine >1.2 mg/dL in males or >1.0 mg/dL in females).3 In addition, several patients with TAFRO syndrome demonstrate elevated ALP, low-normal LDH, elevated vascular endothelial growth factor, elevated IL-6, microcytic anemia, and slight polyclonal hypergammopathy.3 Malignancies such as lymphoma and myeloma, autoimmune diseases such as SLE and ANCA-associated vasculitis, infectious diseases such as those caused by mycobacteria, and POEMS (polyneuropathy, organomegaly, endocrine diseases, M-protein, and skin lesions) syndrome must be excluded to diagnose TAFRO syndrome.3,4
The pathophysiology of TAFRO syndrome is unknown, and it is unclear whether the syndrome is truly a variant of iMCD or a distinct entity.3 IL-6 is typically only mildly elevated in TAFRO syndrome, without the consequent thrombocytosis and polyclonal hypergammaglobulinemia seen in MCD, which is associated with higher levels of IL-6.1 Multiple non-HHV-8 mechanisms for TAFRO syndrome have been proposed, including (1) systemic inflammation, autoimmune/autoinflammatory mechanisms, (2) neoplastic, ectopic cytokine secretion by malignant or benign tumor cells, and/or (3) infectious, such as non-HHV-8 virus.5
Immunosuppression is the mainstay of treatment for TAFRO syndrome based on recommendations from the 2015 TAFRO Research Group.3 Glucocorticoids are considered first-line therapy.3 Cyclosporin A is recommended for individuals refractory to glucocorticoids.3 In patients with a contraindication to cyclosporin A, anti-IL-6 receptor antibodies such as tocilizumab (approved for treatment of iMCD in Japan) and siltuximab (approved for treatment of iMCD in North America and Europe) or the anti-CD20 antibody rituximab should be prescribed.3 There is evidence for the thrombopoietin receptor agonists romiplostim and eltrombopag to treat persistent thrombocytopenia.3 Additional treatments for refractory TAFRO syndrome include IVIG and plasma exchange, chemotherapy (cyclophosphamide, doxorubicin, vincristine, prednisolone), and thalidomide.3,6
Little is known about the epidemiologic characterization of TAFRO syndrome as less than 40 cases of TAFRO syndrome have been reported in the United States, Asia, and Europe.
1,4,7-9 TAFRO syndrome occurs primarily in the fourth and fifth decades of life, with case reports ranging from 14 to 78 years of age.1,3,10,11 Gender distribution varies but is likely equal for males and females.3 Mortality in TAFRO syndrome is estimated at 11%-12%.1,3 Over the past several years, a North American and European patient registry and natural history study for CD, ACCELERATE, has been initiated.4 In addition, the international Castleman Disease Collaborative Network, a Japanese multicenter retrospective study for MCD, and a nationwide Japanese research team for CD have been created.3,4 Previously, CD did not have an International Classification of Diseases (ICD) code and was likely under-recognized. An ICD-10 for CD was added, making CD and its variants easier to research for prevalence, characterization, mortality, and treatment.
After prolonged hospitalizations and extensive workup with no diagnosis, the patient’s clinical picture was most consistent with the lymphoproliferative disorder iMCD. However, iMCD is notable for polyclonal hypergammaglobulinemia, thrombocytosis, and mild anasarca. This patient had normal gammaglobulins, significant thrombocyotopenia, and profound, difficult-to-treat anasarca and ascites. Recognizing that the patient’s presentation did not fit neatly into a known clinical syndrome, an internet search was conducted based on his clinical features. This revealed TAFRO syndrome, which was at the time a newly described clinical syndrome with only a few published case reports. It was an internet search undertaken as a last resort that ultimately led to the patient’s diagnosis and successful treatment.
TEACHING POINTS
- Key clinical and pathologic features of TAFRO syndrome include thrombocytopenia, anasarca, fever, reticulin fibrosis and/or renal insufficiency, and organomegaly.
- TAFRO syndrome may be under-recognized due to very recent characterization and no previous ICD code for CD.
- TAFRO syndrome experts recommend immunosuppression for treatment of TAFRO syndrome, including glucocorticoids as first-line treatment.
- Internet searches can be helpful in the diagnosis of challenging cases, particularly with rare, unusual, and emerging diseases that have not yet been described in reference texts and only infrequently reported in the medical literature.
Disclosures
Jonathan S. Zipursky, Keri T. Holmes-Maybank, Steven L. Shumak, and Ashley A. Ducketthave none to declare.
1. Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol. 2016;91(2):220-226. PubMed
2. Takai K, Nikkuni K, Shibuya H, Hashidate H. Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly. Rinsho Ketsueki. 2010;51(5):320-325. PubMed
3. Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol. 2016;103:686-692. https://doi.org/10.1007/s12185-016-1979-1.
4. Liu AY, Nabel CS, Finkelman BS, et al. Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol. 2016;3:e163-e175. https://doi.org/10.1016/S2352-3026(16)00006-5.
5. Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924-2933. https://doi.org/10.1182/blood-2013-12-545087.
6. Sakashita K, Murata K, Takamori M. TAFRO syndrome: Current perspectives. J Blood Med. 2018;9:15-23. doi: 10.2147/JBM.S127822.
7. Louis C, Vijgen S, Samii K, et al. TAFRO syndrome in caucasians: A case report and review of the literature. Front Med. 2017;4(149):1-8. https://doi.org/10.3389/fmed.2017.00149.
8. Courtier F, Ruault NM, Crepin T, et al. A comparison of TAFRO syndrome between Japanese and non-Japanese cases: a case report and literature review. Ann Hematol. 2018;97:401-407. https://doi.org/10.1007/s00277-017-3138-z.
9. Jain P, Verstovsek S, Loghavi S, et al. Durable remission with rituximab in a patient with an unusual variant of Castleman’s disease with myelofibrosis-TAFRO syndrome. Am J Hematol. 2015;90(11):1091-1092. https://doi.org/10.1002/ajh.24015.
10. Igawa T, Sato Y. TAFRO syndeome. Hematol Oncol Clin N Am. 2018;32(1):107-118. https://doi.org/10.1016/j.hoc.2017.09.009.
11. Hawkins JM, Pillai V. TAFRO syndrome or Castleman-Kojima syndrome: a variant of multicentric Castleman disease. Blood. 2015;126(18):2163. https://doi.org/10.1182/blood-2015-07-662122.
1. Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol. 2016;91(2):220-226. PubMed
2. Takai K, Nikkuni K, Shibuya H, Hashidate H. Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly. Rinsho Ketsueki. 2010;51(5):320-325. PubMed
3. Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol. 2016;103:686-692. https://doi.org/10.1007/s12185-016-1979-1.
4. Liu AY, Nabel CS, Finkelman BS, et al. Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol. 2016;3:e163-e175. https://doi.org/10.1016/S2352-3026(16)00006-5.
5. Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924-2933. https://doi.org/10.1182/blood-2013-12-545087.
6. Sakashita K, Murata K, Takamori M. TAFRO syndrome: Current perspectives. J Blood Med. 2018;9:15-23. doi: 10.2147/JBM.S127822.
7. Louis C, Vijgen S, Samii K, et al. TAFRO syndrome in caucasians: A case report and review of the literature. Front Med. 2017;4(149):1-8. https://doi.org/10.3389/fmed.2017.00149.
8. Courtier F, Ruault NM, Crepin T, et al. A comparison of TAFRO syndrome between Japanese and non-Japanese cases: a case report and literature review. Ann Hematol. 2018;97:401-407. https://doi.org/10.1007/s00277-017-3138-z.
9. Jain P, Verstovsek S, Loghavi S, et al. Durable remission with rituximab in a patient with an unusual variant of Castleman’s disease with myelofibrosis-TAFRO syndrome. Am J Hematol. 2015;90(11):1091-1092. https://doi.org/10.1002/ajh.24015.
10. Igawa T, Sato Y. TAFRO syndeome. Hematol Oncol Clin N Am. 2018;32(1):107-118. https://doi.org/10.1016/j.hoc.2017.09.009.
11. Hawkins JM, Pillai V. TAFRO syndrome or Castleman-Kojima syndrome: a variant of multicentric Castleman disease. Blood. 2015;126(18):2163. https://doi.org/10.1182/blood-2015-07-662122.
© 2019 Society of Hospital Medicine
Past is Prologue
A 56-year-old Japanese man with a history of renal transplantation 20 years prior presented to the emergency department (ED) with two months of dyspnea on exertion and one day of fever and chills. The patient was in his usual state of health until two months prior to presentation, when he gradually noticed shortness of breath after sustained or effortful physical activities. The dyspnea improved with rest. Over the following two months, he noticed that the shortness of breath came on with lesser degrees of exertion, such as walking 100 meters. One day before presentation, he developed a fever of 39°C and chills at home, which prompted him to seek ED care. He denied chest pain, cough, leg swelling, or paroxysmal nocturnal dyspnea.
The differential diagnosis of exertional dyspnea progressing over several months includes cardiac, pulmonary, hematologic, and neuromuscular conditions. The patient’s history of renal transplantation prompts consideration of worsening indolent pneumonia (eg, Aspergillus, cytomegalovirus [CMV], or Pneumocystis pneumonia), allograft dysfunction with volume overload, recrudescence of the underlying disease that incited renal failure earlier in life (eg, vasculitis), or a late-onset posttransplantation lymphoproliferative disorder (PTLD). Additionally, acute fever in an immunocompromised patient immediately raises suspicion for infection (eg, pneumonia, enteritis, or urinary tract infection). At this point, it is difficult to know whether the subacute-to-chronic exertional dyspnea and the acute fever are consequences of the same disease or separate, potentially overlapping, processes.
His past medical history was significant for end-stage renal disease due to membranoproliferative glomerular nephropathy (MPGN), for which living, related-donor kidney transplantation was performed 20 years earlier. He also had type 2 diabetes mellitus, hypertension, and basal cell carcinoma of the face, which had been resected three years prior without spread or recurrence. He had no known allergies. Medications included prednisolone 15 mg daily, azathioprine 100 mg daily, and cyclosporine 100 mg daily, as well as amlodipine and candesartan. He lived in Japan with his wife and children. He denied any animal or environmental exposures. He did not smoke cigarettes or drink alcohol and had not traveled recently. His father had diabetes mellitus.
Recrudescence of an underlying autoimmune condition that may have incited MPGN earlier in life is unlikely while taking an immunosuppressive regimen consisting of prednisolone, azathioprine, and cyclosporine. However, these medications do increase susceptibility to infections, lymphoma, and skin cancers. Though he is immunocompromised, the patient is not on prophylaxis for Pneumocystis pneumonia (PCP). PCP in HIV-negative patients is associated with recent glucocorticoid exposure and typically follows an acute-to-subacute course with hypoxemia and respiratory distress. Though the risk of PCP infection is considered highest in the early posttransplantation period (when immunosuppression is most intense), many cases are diagnosed years after transplantation among patients no longer on prophylaxis. The patient has type 2 diabetes mellitus and hypertension, which are known complications of calcineurin inhibitor and steroid therapy and increase the risk of cardiovascular disease. Cardiovascular disease is a major cause of death among renal transplant recipients. Exertional dyspnea may be the presenting symptom of coronary artery disease.
On physical examination, the patient was alert, oriented, and in no acute distress. His temperature was 38.5°C, blood pressure 120/60 mm Hg, heart rate 146 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 93% while breathing ambient air. The conjunctiva were normal without pallor or icterus. There was no cervical lymphadenopathy. Cardiac examination revealed tachycardia with a regular rhythm, normal S1 and S2, and no murmurs, rubs, or gallops. Jugular venous pressure was not elevated, and there was no lower extremity edema. Lungs were clear to auscultation bilaterally. The abdomen was soft, nontender, and nondistended. There was no tenderness over the transplanted kidney and no hepatosplenomegaly.
Dyspnea, fever, and tachycardia may be the sole manifestations of pneumonia in solid organ transplant recipients. The absence of cough or adventitious breath sounds does not eliminate concern for pneumonia. Pathogens that cause indolent pneumonia in immunocompromised patients include viruses (such as typical respiratory viruses and CMV), bacteria (typical organisms, Nocardia, Rhodococcus), and mycobacteria. Fungal causes include Aspergillus, Candida, Cryptococcus, Pneumocystis, and endemic mycoses. A detailed environmental history should be taken, and providers should ascertain which fungal diseases are endemic in the patient’s region of residence. There are no examination features suggesting hypervolemia or anemia. Although there is no hepatosplenomegaly or lymphadenopathy, PTLD often involves extranodal tissues, including the lungs. The incidence of PTLD is highest in the 12 months following transplantation, but it may occur at any time in the posttransplantation course. A complete blood count, comprehensive metabolic panel, lactate dehydrogenase (LDH), and blood and sputum cultures are indicated, along with computed tomography (CT) of the chest.
The leukocyte count was 3,500 cells/mm3, the hemoglobin level 9.0 g/dL, mean corpuscular volume 102 fL, and the platelet count 137,000 cells/mm3. The sodium level was 130 mEq/L, potassium 4.6 mEq/L, blood urea nitrogen 41 mg/dL, and creatinine 3.5 mg/dL. These complete blood count and serum electrolyte results were unchanged from the patient’s baseline values. The serum LDH level was 1,895 IU/L (normal range, 115-245 IU/L). The serum ferritin was 2,114 ng/mL (normal range, 13-277 ng/mL). A chest radiograph revealed diffuse, airspace-filling opacities in the bilateral lung bases. The urinalysis was normal. The patient was admitted and started empirically on intravenous ceftriaxone for potential bacterial pneumonia.
Chronic pancytopenia may result from azathioprine or cyclosporine use, marrow suppression or infiltration by a multisystem disease, or nutritional deficiency. Hemophagocytic lymphohistiocytosis (HLH) triggered by infection, a rheumatologic condition, acquired immunodeficiency, or malignancy can present with fevers, pancytopenia, and elevated ferritin, while splenomegaly may be absent. The euvolemic state, baseline creatinine level, and normal urinalysis argue against allograft dysfunction. The elevated serum ferritin nonspecifically confirms systemic inflammation. LDH, an intracellular enzyme involved in the bidirectional conversion of lactate to pyruvate, is expressed across tissue types. Elevated serum LDH attests to cell destruction, in this case potentially from lung infection (such as PCP) or malignancy (such as PTLD). At this point, the differential diagnosis of fever and pulmonary infiltrates in this patient remains broad.
Azathioprine and cyclosporine were stopped. The patient remained febrile despite the administration of intravenous antibiotics. His hypoxia worsened with an oxygen saturation of 90%-93% on 5 L/min of supplemental oxygen administered by nasal cannula. Noncontrast chest CT obtained on the second hospital day revealed ground-glass opacities in the bilateral lung bases. Blood, sputum, and urine cultures were sterile. As empiric therapies, ganciclovir was started for CMV infection, ciprofloxacin added for atypical pneumonia, and trimethoprim-sulfamethoxazole added for Pneumocystis infection.
These chest imaging findings help prioritize the differential diagnosis. Bibasilar ground-glass opacities are evident, while pulmonary masses, nodules, cavitation, adenopathy, and pleural effusions are absent. The differential diagnosis of multifocal ground-glass opacities on chest imaging includes infectious pneumonia, chronic interstitial lung disease, acute alveolar conditions (eg, cardiogenic pulmonary edema, acute respiratory distress syndrome, diffuse alveolar hemorrhage), or other pathologies (eg, drug toxicity, bronchoalveolar carcinoma, cryptogenic organizing pneumonia).
Infectious pneumonia is the principal concern. A diagnosis of PCP could be unifying, given dyspnea, progressive respiratory failure with hypoxia, and elevated LDH in an immunocompromised patient who is not prescribed PCP prophylaxis. The bilateral lung infiltrates and the absence of thoracic adenopathy or pleural effusions are characteristic of PCP as well. However, caution should be exercised in making specific infectious diagnoses in immunocompromised hosts on the basis of clinical and imaging findings alone. There can be overlap in the radiologic appearance of various infections (eg, CMV pneumonia can also present with bilateral ground-glass infiltrates, with concurrent fever, hypoxia, and pancytopenia). Additionally, more than one pneumonic pathogen may be implicated (eg, acute viral pneumonia superimposed on indolent fungal pneumonia). Polymerase chain reaction (PCR) analysis of respiratory secretions for viruses, serum PCR and serologic testing for herpes viruses, and serum beta-D-glucan and galactomannan assays are indicated. Serum serologic testing for fungi and bacteria such as Nocardia can be helpful, though the negative predictive values of these tests may be reduced in patients with impaired humoral immunity. Timely bronchoalveolar lavage (BAL) with microbiologic and PCR analysis and cytology is advised.
Fever, elevated LDH, cytopenias, and pulmonary infiltrates also raise suspicion for an underlying hematologic malignancy, such as PTLD. However, pulmonary PTLD is seen more often in lung transplant recipients than in patients who have undergone transplantation of other solid organs. In kidney transplant recipients, PTLD most commonly manifests in the allograft itself, gastrointestinal tract, central nervous system, or lymph nodes; lung involvement is less common. Chest imaging in affected patients may reveal nodular or reticulonodular infiltrates of basilar predominance, solitary or multiple masses, cavitating or necrotic lesions, and/or lymphadenopathy. In this patient who has undergone renal transplantation, late-onset PTLD with isolated pulmonary involvement, with only ground-glass opacities on lung imaging, would be an atypical presentation of an uncommon syndrome.
Despite empiric treatment with antibiotics and antiviral agents, the patient’s fever persisted. His respiratory rate increased to 30 breaths per minute. His hypoxia worsened, and he required nasal cannula high-flow oxygen supplementation at 30 L/min with a fraction of inspired oxygen (FiO2) of 40%. On the fifth hospital day, contrast CT scan of the chest and abdomen showed new infiltrates in the bilateral upper lung fields as well as an area of low density in the tail of the pancreas without a focal mass (Figure 1). At this point, BAL was performed, and fluid PCR analysis returned positive for Pneumocystis jirovecii. CMV direct immunoperoxidase staining of leukocytes with peroxidase-labeled monoclonal antibody (C7-HRP test) was positive at five cells per 7.35 × 104 peripheral blood leukocytes. The serum Epstein-Barr virus (EBV) viral capsid antigen (VCA) IgG was positive, while VCA IgM and EBV nuclear antigen IgG were negative. A bone marrow biopsy revealed mild hemophagocytosis. His serum soluble interleukin-2 (sIL2R) level was elevated at 5,254 U/mL (normal range, 122-496 U/mL). Given the BAL Pneumocystis PCR result, the dose of prednisolone was increased to 30 mg/day, and the patient’s fever subsided. Supplemental oxygen was weaned to an FiO2 of 35%.
These studies should be interpreted carefully considering the biphasic clinical course. After two months of exertional dyspnea, the patient acutely developed persistent fever and progressive lung infiltrates. His clinical course, the positive PCR assay for Pneumocystis jirovecii in BAL fluid, and the compatible lung imaging findings make Pneumocystis jirovecii a likely pathogen. But PCP may only explain the second phase of this patient’s illness, considering its often-fulminant course in HIV-negative patients. To explain the two months of exertional dyspnea, marrow hemophagocytosis, pancreatic abnormality, and perhaps even the patient’s heightened susceptibility to PCP infection, an index of suspicion should be maintained for a separate, antecedent process. This could be either an indolent infection (eg, CMV or Aspergillus pneumonia) or a malignancy (eg, lymphoma or PTLD). Completion of serum serologic testing for viruses, bacteria, and fungi and comprehensive BAL fluid analysis (culture, viral PCR, and cytology) is recommended.
A CMV antigenemia assay returned positive, suggesting prior CMV infection. However, to diagnose CMV pneumonia, the virus must be detected in BAL fluid by PCR or cytologic analysis. CMV infection has been associated with cytopenias, HLH, pancreatic infiltration, and an increased risk for fungal infections and EBV-related PTLD. CMV infection could explain the first phase of this patient’s illness. Serum and BAL PCR for CMV are advised. Meanwhile, EBV testing indicates prior infection but does not distinguish between recent or more distant infection. EBV has been implicated in the pathophysiology of PTLD, as EBV-infected lymphoid tissue may proliferate in a variety of organs under reduced T-cell surveillance. EBV infection or PTLD with resulting immunomodulation may pose other explanations for this patient’s development of PCP infection. Cytologic analysis of the BAL fluid and marrow aspirate for evidence of PTLD is warranted. Finally, CMV, EBV, and PTLD have each been reported to trigger HLH. Though this patient has fevers, mild marrow hemophagocytosis, elevated serum ferritin, and elevated serum IL-2 receptor levels, he does not meet other diagnostic criteria for HLH (such as more pronounced cytopenias, splenomegaly, hypertriglyceridemia, hypofibrinogenemia, and low or absent natural killer T-cell activity). However, HLH may be muted in this patient because he was prescribed cyclosporine, which has been used in HLH treatment protocols.
On the 11th hospital day, the patient developed hemorrhagic shock due to massive hematemesis and was transferred to the intensive care unit. His hemoglobin level was 5.9 g/dL. A total of 18 units of packed red blood cells were transfused over the following week for ongoing gastrointestinal bleeding. The serum LDH level increased to 4,139 IU/L, and the ferritin level rose to 7,855 ng/mL. The EBV copy level by serum PCR returned at 1 × 106 copies/mL (normal range, less than 2 x 102 copies/mL). The patient was started on methylprednisolone (1 g/day for three days) and transitioned to dexamethasone and cyclosporine for possible EBV-related HLH. Ceftazidime, vancomycin, trimethoprim-sulfamethoxazole, and ciprofloxacin were administered. Amphotericin-B was initiated empirically for potential fungal pneumonia. Ganciclovir was continued. However, the patient remained in shock despite vasopressors and transfusions and died on the 22nd hospital day.
The patient deteriorated despite broad antimicrobial therapy. Laboratory studies revealed EBV viremia and rising serum LDH. Recent EBV infection may have induced PTLD in the gastrointestinal tract, which is a commonly involved site among affected renal transplant patients. Corticosteroids and stress from critical illness can contribute to intestinal mucosal erosion and bleeding from a luminal PTLD lesion. Overall, the patient’s condition was likely explained by EBV infection, which triggered HLH and gastrointestinal PTLD. The resulting immunomodulation increased his risk for PCP infection beyond that conferred by chronic immunosuppression. It is still possible that he had concomitant CMV pneumonia, Aspergillus pneumonia, or even pulmonary PTLD, in addition to the proven PCP diagnosis.
An autopsy was performed. Atypical lymphocytic infiltration and diffuse alveolar damage were shown in the right upper lobe (Figure 2). EBV RNA-positive atypical lymphocytes coexpressing CD20 were demonstrated in multiple organs including the bone marrow, lungs, heart, stomach, adrenal glands, duodenum, ileum, and mesentery (Figure 3). This confirmed the diagnosis of an underlying EBV-positive posttransplant lymphoproliferative disorder. Serum and BAL CMV PCR assays returned negative. Neither CMV nor Aspergillus was identified in autopsy specimens.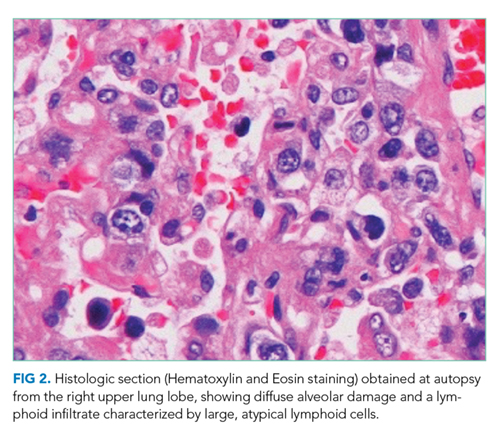
COMMENTARY
A broad differential diagnosis should be considered when acute fever develops in a patient who has undergone solid organ transplantation. Causes may include opportunistic and nonopportunistic infections as well as noninfectious etiologies such as malignancy, organ rejection, inflammatory conditions, and medication toxicity.1,2 As the discussant noted, more than one infection, or both infection and malignancy, can coexist in immunocompromised patients. For example, while viral pathogens such as EBV, CMV, and respiratory syncytial virus can cause illness due to direct tissue infection, they can also exert indirect effects in transplant recipients: acting as cofactors for and enabling other infections by causing immunosuppression (eg, Aspergillus or PCP developing after CMV infection), triggering graft rejection by upregulating proinflammatory cytokines, and inducing oncogenesis (eg, EBV-related PTLD).1,3-5
PTLD is a rare but serious complication of solid organ transplantation and immunosuppression. Most cases are driven by EBV infection and subsequent transformation of infected lymphoid tissue in a variety of organs in the context of reduced T-cell surveillance.6 The incidence of PTLD varies based on the organ transplanted, ranging from 0.8%-2.5% in those who have undergone renal transplantation to 1.0%-5.5% in liver transplant recipients and 3.0%-10% in lung transplant recipients.3 The incidence has increased over the past decade. This may be due to a greater number of solid organ transplantations being performed, aging of the transplant donor/recipient population, new immunosuppressive regimens, and improved PTLD diagnosis due to superior diagnostic tools and clinician awareness.3 However, the mortality rate among solid organ transplant recipients with PTLD remains high, ranging from 40% to 70%.6
Risk factors for PTLD include a greater intensity of T-cell immunosuppression,7 history of pretransplant malignancy, recipient EBV seronegativity and donor seropositivity, and younger age at the time of transplantation.8-10 EBV-related PTLD incidence in solid organ transplant recipients is greatest in the early posttransplantation course (the period of most intense immunosuppression) with over 80% of cases occurring in the first posttransplant year.11
A high index of suspicion for PTLD is warranted in any solid organ transplant recipient who presents with constitutional symptoms, adenopathy, or cytopenias. Clinical suspicion of PTLD can be informed by risk factors, constitutional symptoms, elevated serum LDH, a detectable or rising serum EBV viral load, and radiologic adenopathy or visceral tissue infiltration.12 The clinical presentation of PTLD is heterogeneous and varies in accordance with the organs affected. Extranodal involvement, such as pulmonary, gastrointestinal, and bone marrow involvement, is more common in PTLD than in other types of lymphoma.13 In this patient, the cytopenias, elevated serum LDH level, lung infiltrates, and radiologic pancreatic tail abnormality served as early clues to the presence of underlying PTLD.
The standard approach to diagnosing PTLD is biopsy of a suspicious lesion (adenopathy or an infiltrated visceral organ) with histopathological examination. Pathology may demonstrate distorted tissue architecture, clonal lymphocytes, or EBV-positive lymphocytes.14 Conventional CT is the most commonly used imaging modality to detect adenopathy or tissue infiltration related to PTLD,3 though 18F-fluorodeoxyglucose position-emission tomography (FDG-PET) is also used. Although FDG-PET has high diagnostic accuracy, with an overall sensitivity of 89% and specificity of 89%, false-negative results have been reported, particularly in cases of early PTLD lesions and diffuse large B-cell lymphoma.15 The majority of patients with EBV-associated PTLD demonstrate significant elevations in the serum EBV viral load compared with immunosuppressed controls without PTLD.16 An elevated EBV viral load can support a diagnosis of PTLD, though the absence of EBV viremia does not rule it out.17 Some transplant centers perform posttransplantation monitoring of the serum EBV viral load to aid in PTLD risk assessment and early diagnosis.
Management of PTLD is patient-specific and may involve reduction of immunosuppressive therapy, rituximab, chemotherapy, surgical excision, and/or radiation.13 Reduction of immunosuppression is the cornerstone of treatment.18 In patients who do not respond to the reduction of immunosuppression, rituximab and immunochemotherapy are second-line treatment options. A prospective, multicenter phase 2 trial (the PTLD-1 trial) demonstrated a complete response rate of 40% among patients with PTLD managed with rituximab.19
In summary, this case illustrates the importance of maintaining a broad differential diagnosis when acute fever develops in a patient who has undergone solid organ transplantation. The presence of more than one condition should be considered when the clinical presentation cannot be explained by a single diagnosis, as infections and malignancies can coexist in immunocompromised hosts. This case also highlights an unusual clinical presentation of PTLD, which was heralded mainly by its immunomodulatory effects rather than by compatible symptoms or obvious mass lesions.
Carefully reviewing the patient’s medical history and understanding how it sets the stage for the present illness is an essential step in clinical problem solving, because what is past is prologue.
TEACHING POINTS
- Fever in solid organ transplant recipients should prompt consideration of a broad differential diagnosis, including infection, malignancy, organ graft rejection, autoimmune disease, and medication toxicity.
- PTLD is a rare but serious complication of organ transplantation. Most cases are driven by EBV infection and transformation of infected lymphocytes in a variety of organs in the context of reduced T-cell surveillance. The clinical presentation can be heterogeneous and varies depending on the organs and tissues involved.
- More than one infection, or both infection and malignancy, can coexist in organ transplant recipients. Viral pathogens can exert direct pathologic effects on tissue but can also exert indirect effects, such as contributing to opportunistic infection susceptibility, graft rejection, and oncogenesis.
Disclosures
The authors have nothing to disclose.
Previous Publication
This case was originally reported in the 121st Okinawa Association of Medical Sciences in 2015 in Okinawa, Japan, and the conference abstracts were covered in The Okinawa Medical Journal. The publication did not provide any detailed, step-by-step analysis of clinical decision-making.
1. Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med. 2007;357(25):2601-2614. https://doi.org/10.1056/NEJMra064928.
2. Bouza E, Loeches B, Muñoz P. Fever of unknown origin in solid organ transplant recipients. Infect Dis Clin North Am. 2007;21(4):1033-1054, ix-x. https://doi.org/10.1016/j.idc.2007.09.001,
3. Kotton CN, Fishman JA. Viral infection in the renal transplant recipient. J Am Soc Nephrol. 2005;16(6):1758-1774. https://doi.org/10.1681/ASN.2004121113.
4. Arend SM, Westendorp RG, Kroon FP, et al. Rejection treatment and cytomegalovirus infection as risk factors for Pneumocystis carinii pneumonia in renal transplant recipients. Clin Infect Dis. 1996;22(6):920-925. https://doi.org/10.1093/clinids/22.6.920.
5. Reinke P, Fietze E, Ode-Hakim S, et al. Late-acute renal allograft rejection and symptomless cytomegalovirus infection. Lancet. 1994;344(8939-8940):1737-1738. https://doi.org/10.1016/S0140-6736(94)92887-8.
6. Tsai DE, Douglas L, Andreadis C, et al. EBV PCR in the diagnosis and monitoring of posttransplant lymphoproliferative disorder: results of a two-arm prospective trial. Am J Transplant. 2008;8(5):1016-1024. https://doi.org/10.1111/j.1600-6143.2008.02183.x.
7. Penn I. Cancers complicating organ transplantation. N Engl J Med. 1990;323(25):1767-1769. https://doi.org/10.1056/NEJM199012203232510
8. Walker RC, Marshall WF, Strickler JG, et al. Pretransplantation assessment of the risk of lymphoproliferative disorder. Clin Infect Dis. 1995;20(5):1346-1353. https://doi.org/10.1093/clinids/20.5.1346.
9. Opelz G, Döhler B. Lymphomas after solid organ transplantation: a collaborative transplant study report. Am J Transplant. 2004;4(2):222-230. https://doi.org/10.1046/j.1600-6143.2003.00325.x.
10. Caillard S, Dharnidharka V, Agodoa L, Bohen E, Abbott K. Posttransplant lymphoproliferative disorders after renal transplantation in the United States in era of modern immunosuppression. Transplantation. 2005;80(9):1233-1243. doi: 10.1097/01.tp.0000179639.98338.39.
11. Opelz G, Henderson R. Incidence of non-Hodgkin lymphoma in kidney and heart transplant recipients. Lancet. 1993;342(8886-8887):1514-1516. https://doi.org/10.1016/S0140-6736(05)80084-4.
12. Samant H, Kothadia JP. Transplantation Posttransplantation Lymphoproliferative Disorders. Treasure Island, FL: StatPearls Publishing; 2018. PubMed
13. Dierickx D, Habermann TM. Post-transplantation lymphoproliferative disorders in adults. N Engl J Med. 2018;378(6):549-562. https://doi.org/10.1056/NEJMra1702693.
14. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390. https://doi.org/10.1182/blood-2016-01-643569.
15. Dierickx D, Tousseyn T, Requilé A, et al. The accuracy of positron emission tomography in the detection of posttransplant lymphoproliferative disorder. Haematologica. 2013;98(5):771-775. https://doi.org/10.3324/haematol.2012.074500.
16. Wagner HJ, Wessel M, Jabs W, et al. Patients at risk for development of posttransplant lymphoproliferative disorder: plasma versus peripheral blood mononuclear cells as material for quantification of Epstein-Barr viral load by using real-time quantitative polymerase chain reaction. Transplantation. 2001;72(6):1012-1019. PubMed
17. Baldanti F, Rognoni V, Cascina A, Oggionni T, Tinelli C, Meloni F. Post-transplant lymphoproliferative disorders and Epstein-Barr virus DNAemia in a cohort of lung transplant recipients. Virol J. 2011;8:421. https://doi.org/10.1186/1743-422X-8-421.
18. Parker A, Bowles K, Bradley JA, et al. Management of post-transplant lymphoproliferative disorder in adult solid organ transplant recipients - BCSH and BTS Guidelines. Br J Haematol. 2010;149(5):693-705. https://doi.org/10.1111/j.1365-2141.2010.08160.x.
19. Trappe R, Oertel S, Leblond V, et al. Sequential treatment with rituximab followed by CHOP chemotherapy in adult B-cell post-transplant lymphoproliferative disorder (PTLD): the prospective international multicentre phase 2 PTLD-1 trial. Lancet Oncol. 2012;13(2):196-206. https://doi.org/10.1016/S1470-2045(11)70300-X.
A 56-year-old Japanese man with a history of renal transplantation 20 years prior presented to the emergency department (ED) with two months of dyspnea on exertion and one day of fever and chills. The patient was in his usual state of health until two months prior to presentation, when he gradually noticed shortness of breath after sustained or effortful physical activities. The dyspnea improved with rest. Over the following two months, he noticed that the shortness of breath came on with lesser degrees of exertion, such as walking 100 meters. One day before presentation, he developed a fever of 39°C and chills at home, which prompted him to seek ED care. He denied chest pain, cough, leg swelling, or paroxysmal nocturnal dyspnea.
The differential diagnosis of exertional dyspnea progressing over several months includes cardiac, pulmonary, hematologic, and neuromuscular conditions. The patient’s history of renal transplantation prompts consideration of worsening indolent pneumonia (eg, Aspergillus, cytomegalovirus [CMV], or Pneumocystis pneumonia), allograft dysfunction with volume overload, recrudescence of the underlying disease that incited renal failure earlier in life (eg, vasculitis), or a late-onset posttransplantation lymphoproliferative disorder (PTLD). Additionally, acute fever in an immunocompromised patient immediately raises suspicion for infection (eg, pneumonia, enteritis, or urinary tract infection). At this point, it is difficult to know whether the subacute-to-chronic exertional dyspnea and the acute fever are consequences of the same disease or separate, potentially overlapping, processes.
His past medical history was significant for end-stage renal disease due to membranoproliferative glomerular nephropathy (MPGN), for which living, related-donor kidney transplantation was performed 20 years earlier. He also had type 2 diabetes mellitus, hypertension, and basal cell carcinoma of the face, which had been resected three years prior without spread or recurrence. He had no known allergies. Medications included prednisolone 15 mg daily, azathioprine 100 mg daily, and cyclosporine 100 mg daily, as well as amlodipine and candesartan. He lived in Japan with his wife and children. He denied any animal or environmental exposures. He did not smoke cigarettes or drink alcohol and had not traveled recently. His father had diabetes mellitus.
Recrudescence of an underlying autoimmune condition that may have incited MPGN earlier in life is unlikely while taking an immunosuppressive regimen consisting of prednisolone, azathioprine, and cyclosporine. However, these medications do increase susceptibility to infections, lymphoma, and skin cancers. Though he is immunocompromised, the patient is not on prophylaxis for Pneumocystis pneumonia (PCP). PCP in HIV-negative patients is associated with recent glucocorticoid exposure and typically follows an acute-to-subacute course with hypoxemia and respiratory distress. Though the risk of PCP infection is considered highest in the early posttransplantation period (when immunosuppression is most intense), many cases are diagnosed years after transplantation among patients no longer on prophylaxis. The patient has type 2 diabetes mellitus and hypertension, which are known complications of calcineurin inhibitor and steroid therapy and increase the risk of cardiovascular disease. Cardiovascular disease is a major cause of death among renal transplant recipients. Exertional dyspnea may be the presenting symptom of coronary artery disease.
On physical examination, the patient was alert, oriented, and in no acute distress. His temperature was 38.5°C, blood pressure 120/60 mm Hg, heart rate 146 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 93% while breathing ambient air. The conjunctiva were normal without pallor or icterus. There was no cervical lymphadenopathy. Cardiac examination revealed tachycardia with a regular rhythm, normal S1 and S2, and no murmurs, rubs, or gallops. Jugular venous pressure was not elevated, and there was no lower extremity edema. Lungs were clear to auscultation bilaterally. The abdomen was soft, nontender, and nondistended. There was no tenderness over the transplanted kidney and no hepatosplenomegaly.
Dyspnea, fever, and tachycardia may be the sole manifestations of pneumonia in solid organ transplant recipients. The absence of cough or adventitious breath sounds does not eliminate concern for pneumonia. Pathogens that cause indolent pneumonia in immunocompromised patients include viruses (such as typical respiratory viruses and CMV), bacteria (typical organisms, Nocardia, Rhodococcus), and mycobacteria. Fungal causes include Aspergillus, Candida, Cryptococcus, Pneumocystis, and endemic mycoses. A detailed environmental history should be taken, and providers should ascertain which fungal diseases are endemic in the patient’s region of residence. There are no examination features suggesting hypervolemia or anemia. Although there is no hepatosplenomegaly or lymphadenopathy, PTLD often involves extranodal tissues, including the lungs. The incidence of PTLD is highest in the 12 months following transplantation, but it may occur at any time in the posttransplantation course. A complete blood count, comprehensive metabolic panel, lactate dehydrogenase (LDH), and blood and sputum cultures are indicated, along with computed tomography (CT) of the chest.
The leukocyte count was 3,500 cells/mm3, the hemoglobin level 9.0 g/dL, mean corpuscular volume 102 fL, and the platelet count 137,000 cells/mm3. The sodium level was 130 mEq/L, potassium 4.6 mEq/L, blood urea nitrogen 41 mg/dL, and creatinine 3.5 mg/dL. These complete blood count and serum electrolyte results were unchanged from the patient’s baseline values. The serum LDH level was 1,895 IU/L (normal range, 115-245 IU/L). The serum ferritin was 2,114 ng/mL (normal range, 13-277 ng/mL). A chest radiograph revealed diffuse, airspace-filling opacities in the bilateral lung bases. The urinalysis was normal. The patient was admitted and started empirically on intravenous ceftriaxone for potential bacterial pneumonia.
Chronic pancytopenia may result from azathioprine or cyclosporine use, marrow suppression or infiltration by a multisystem disease, or nutritional deficiency. Hemophagocytic lymphohistiocytosis (HLH) triggered by infection, a rheumatologic condition, acquired immunodeficiency, or malignancy can present with fevers, pancytopenia, and elevated ferritin, while splenomegaly may be absent. The euvolemic state, baseline creatinine level, and normal urinalysis argue against allograft dysfunction. The elevated serum ferritin nonspecifically confirms systemic inflammation. LDH, an intracellular enzyme involved in the bidirectional conversion of lactate to pyruvate, is expressed across tissue types. Elevated serum LDH attests to cell destruction, in this case potentially from lung infection (such as PCP) or malignancy (such as PTLD). At this point, the differential diagnosis of fever and pulmonary infiltrates in this patient remains broad.
Azathioprine and cyclosporine were stopped. The patient remained febrile despite the administration of intravenous antibiotics. His hypoxia worsened with an oxygen saturation of 90%-93% on 5 L/min of supplemental oxygen administered by nasal cannula. Noncontrast chest CT obtained on the second hospital day revealed ground-glass opacities in the bilateral lung bases. Blood, sputum, and urine cultures were sterile. As empiric therapies, ganciclovir was started for CMV infection, ciprofloxacin added for atypical pneumonia, and trimethoprim-sulfamethoxazole added for Pneumocystis infection.
These chest imaging findings help prioritize the differential diagnosis. Bibasilar ground-glass opacities are evident, while pulmonary masses, nodules, cavitation, adenopathy, and pleural effusions are absent. The differential diagnosis of multifocal ground-glass opacities on chest imaging includes infectious pneumonia, chronic interstitial lung disease, acute alveolar conditions (eg, cardiogenic pulmonary edema, acute respiratory distress syndrome, diffuse alveolar hemorrhage), or other pathologies (eg, drug toxicity, bronchoalveolar carcinoma, cryptogenic organizing pneumonia).
Infectious pneumonia is the principal concern. A diagnosis of PCP could be unifying, given dyspnea, progressive respiratory failure with hypoxia, and elevated LDH in an immunocompromised patient who is not prescribed PCP prophylaxis. The bilateral lung infiltrates and the absence of thoracic adenopathy or pleural effusions are characteristic of PCP as well. However, caution should be exercised in making specific infectious diagnoses in immunocompromised hosts on the basis of clinical and imaging findings alone. There can be overlap in the radiologic appearance of various infections (eg, CMV pneumonia can also present with bilateral ground-glass infiltrates, with concurrent fever, hypoxia, and pancytopenia). Additionally, more than one pneumonic pathogen may be implicated (eg, acute viral pneumonia superimposed on indolent fungal pneumonia). Polymerase chain reaction (PCR) analysis of respiratory secretions for viruses, serum PCR and serologic testing for herpes viruses, and serum beta-D-glucan and galactomannan assays are indicated. Serum serologic testing for fungi and bacteria such as Nocardia can be helpful, though the negative predictive values of these tests may be reduced in patients with impaired humoral immunity. Timely bronchoalveolar lavage (BAL) with microbiologic and PCR analysis and cytology is advised.
Fever, elevated LDH, cytopenias, and pulmonary infiltrates also raise suspicion for an underlying hematologic malignancy, such as PTLD. However, pulmonary PTLD is seen more often in lung transplant recipients than in patients who have undergone transplantation of other solid organs. In kidney transplant recipients, PTLD most commonly manifests in the allograft itself, gastrointestinal tract, central nervous system, or lymph nodes; lung involvement is less common. Chest imaging in affected patients may reveal nodular or reticulonodular infiltrates of basilar predominance, solitary or multiple masses, cavitating or necrotic lesions, and/or lymphadenopathy. In this patient who has undergone renal transplantation, late-onset PTLD with isolated pulmonary involvement, with only ground-glass opacities on lung imaging, would be an atypical presentation of an uncommon syndrome.
Despite empiric treatment with antibiotics and antiviral agents, the patient’s fever persisted. His respiratory rate increased to 30 breaths per minute. His hypoxia worsened, and he required nasal cannula high-flow oxygen supplementation at 30 L/min with a fraction of inspired oxygen (FiO2) of 40%. On the fifth hospital day, contrast CT scan of the chest and abdomen showed new infiltrates in the bilateral upper lung fields as well as an area of low density in the tail of the pancreas without a focal mass (Figure 1). At this point, BAL was performed, and fluid PCR analysis returned positive for Pneumocystis jirovecii. CMV direct immunoperoxidase staining of leukocytes with peroxidase-labeled monoclonal antibody (C7-HRP test) was positive at five cells per 7.35 × 104 peripheral blood leukocytes. The serum Epstein-Barr virus (EBV) viral capsid antigen (VCA) IgG was positive, while VCA IgM and EBV nuclear antigen IgG were negative. A bone marrow biopsy revealed mild hemophagocytosis. His serum soluble interleukin-2 (sIL2R) level was elevated at 5,254 U/mL (normal range, 122-496 U/mL). Given the BAL Pneumocystis PCR result, the dose of prednisolone was increased to 30 mg/day, and the patient’s fever subsided. Supplemental oxygen was weaned to an FiO2 of 35%.
These studies should be interpreted carefully considering the biphasic clinical course. After two months of exertional dyspnea, the patient acutely developed persistent fever and progressive lung infiltrates. His clinical course, the positive PCR assay for Pneumocystis jirovecii in BAL fluid, and the compatible lung imaging findings make Pneumocystis jirovecii a likely pathogen. But PCP may only explain the second phase of this patient’s illness, considering its often-fulminant course in HIV-negative patients. To explain the two months of exertional dyspnea, marrow hemophagocytosis, pancreatic abnormality, and perhaps even the patient’s heightened susceptibility to PCP infection, an index of suspicion should be maintained for a separate, antecedent process. This could be either an indolent infection (eg, CMV or Aspergillus pneumonia) or a malignancy (eg, lymphoma or PTLD). Completion of serum serologic testing for viruses, bacteria, and fungi and comprehensive BAL fluid analysis (culture, viral PCR, and cytology) is recommended.
A CMV antigenemia assay returned positive, suggesting prior CMV infection. However, to diagnose CMV pneumonia, the virus must be detected in BAL fluid by PCR or cytologic analysis. CMV infection has been associated with cytopenias, HLH, pancreatic infiltration, and an increased risk for fungal infections and EBV-related PTLD. CMV infection could explain the first phase of this patient’s illness. Serum and BAL PCR for CMV are advised. Meanwhile, EBV testing indicates prior infection but does not distinguish between recent or more distant infection. EBV has been implicated in the pathophysiology of PTLD, as EBV-infected lymphoid tissue may proliferate in a variety of organs under reduced T-cell surveillance. EBV infection or PTLD with resulting immunomodulation may pose other explanations for this patient’s development of PCP infection. Cytologic analysis of the BAL fluid and marrow aspirate for evidence of PTLD is warranted. Finally, CMV, EBV, and PTLD have each been reported to trigger HLH. Though this patient has fevers, mild marrow hemophagocytosis, elevated serum ferritin, and elevated serum IL-2 receptor levels, he does not meet other diagnostic criteria for HLH (such as more pronounced cytopenias, splenomegaly, hypertriglyceridemia, hypofibrinogenemia, and low or absent natural killer T-cell activity). However, HLH may be muted in this patient because he was prescribed cyclosporine, which has been used in HLH treatment protocols.
On the 11th hospital day, the patient developed hemorrhagic shock due to massive hematemesis and was transferred to the intensive care unit. His hemoglobin level was 5.9 g/dL. A total of 18 units of packed red blood cells were transfused over the following week for ongoing gastrointestinal bleeding. The serum LDH level increased to 4,139 IU/L, and the ferritin level rose to 7,855 ng/mL. The EBV copy level by serum PCR returned at 1 × 106 copies/mL (normal range, less than 2 x 102 copies/mL). The patient was started on methylprednisolone (1 g/day for three days) and transitioned to dexamethasone and cyclosporine for possible EBV-related HLH. Ceftazidime, vancomycin, trimethoprim-sulfamethoxazole, and ciprofloxacin were administered. Amphotericin-B was initiated empirically for potential fungal pneumonia. Ganciclovir was continued. However, the patient remained in shock despite vasopressors and transfusions and died on the 22nd hospital day.
The patient deteriorated despite broad antimicrobial therapy. Laboratory studies revealed EBV viremia and rising serum LDH. Recent EBV infection may have induced PTLD in the gastrointestinal tract, which is a commonly involved site among affected renal transplant patients. Corticosteroids and stress from critical illness can contribute to intestinal mucosal erosion and bleeding from a luminal PTLD lesion. Overall, the patient’s condition was likely explained by EBV infection, which triggered HLH and gastrointestinal PTLD. The resulting immunomodulation increased his risk for PCP infection beyond that conferred by chronic immunosuppression. It is still possible that he had concomitant CMV pneumonia, Aspergillus pneumonia, or even pulmonary PTLD, in addition to the proven PCP diagnosis.
An autopsy was performed. Atypical lymphocytic infiltration and diffuse alveolar damage were shown in the right upper lobe (Figure 2). EBV RNA-positive atypical lymphocytes coexpressing CD20 were demonstrated in multiple organs including the bone marrow, lungs, heart, stomach, adrenal glands, duodenum, ileum, and mesentery (Figure 3). This confirmed the diagnosis of an underlying EBV-positive posttransplant lymphoproliferative disorder. Serum and BAL CMV PCR assays returned negative. Neither CMV nor Aspergillus was identified in autopsy specimens.
COMMENTARY
A broad differential diagnosis should be considered when acute fever develops in a patient who has undergone solid organ transplantation. Causes may include opportunistic and nonopportunistic infections as well as noninfectious etiologies such as malignancy, organ rejection, inflammatory conditions, and medication toxicity.1,2 As the discussant noted, more than one infection, or both infection and malignancy, can coexist in immunocompromised patients. For example, while viral pathogens such as EBV, CMV, and respiratory syncytial virus can cause illness due to direct tissue infection, they can also exert indirect effects in transplant recipients: acting as cofactors for and enabling other infections by causing immunosuppression (eg, Aspergillus or PCP developing after CMV infection), triggering graft rejection by upregulating proinflammatory cytokines, and inducing oncogenesis (eg, EBV-related PTLD).1,3-5
PTLD is a rare but serious complication of solid organ transplantation and immunosuppression. Most cases are driven by EBV infection and subsequent transformation of infected lymphoid tissue in a variety of organs in the context of reduced T-cell surveillance.6 The incidence of PTLD varies based on the organ transplanted, ranging from 0.8%-2.5% in those who have undergone renal transplantation to 1.0%-5.5% in liver transplant recipients and 3.0%-10% in lung transplant recipients.3 The incidence has increased over the past decade. This may be due to a greater number of solid organ transplantations being performed, aging of the transplant donor/recipient population, new immunosuppressive regimens, and improved PTLD diagnosis due to superior diagnostic tools and clinician awareness.3 However, the mortality rate among solid organ transplant recipients with PTLD remains high, ranging from 40% to 70%.6
Risk factors for PTLD include a greater intensity of T-cell immunosuppression,7 history of pretransplant malignancy, recipient EBV seronegativity and donor seropositivity, and younger age at the time of transplantation.8-10 EBV-related PTLD incidence in solid organ transplant recipients is greatest in the early posttransplantation course (the period of most intense immunosuppression) with over 80% of cases occurring in the first posttransplant year.11
A high index of suspicion for PTLD is warranted in any solid organ transplant recipient who presents with constitutional symptoms, adenopathy, or cytopenias. Clinical suspicion of PTLD can be informed by risk factors, constitutional symptoms, elevated serum LDH, a detectable or rising serum EBV viral load, and radiologic adenopathy or visceral tissue infiltration.12 The clinical presentation of PTLD is heterogeneous and varies in accordance with the organs affected. Extranodal involvement, such as pulmonary, gastrointestinal, and bone marrow involvement, is more common in PTLD than in other types of lymphoma.13 In this patient, the cytopenias, elevated serum LDH level, lung infiltrates, and radiologic pancreatic tail abnormality served as early clues to the presence of underlying PTLD.
The standard approach to diagnosing PTLD is biopsy of a suspicious lesion (adenopathy or an infiltrated visceral organ) with histopathological examination. Pathology may demonstrate distorted tissue architecture, clonal lymphocytes, or EBV-positive lymphocytes.14 Conventional CT is the most commonly used imaging modality to detect adenopathy or tissue infiltration related to PTLD,3 though 18F-fluorodeoxyglucose position-emission tomography (FDG-PET) is also used. Although FDG-PET has high diagnostic accuracy, with an overall sensitivity of 89% and specificity of 89%, false-negative results have been reported, particularly in cases of early PTLD lesions and diffuse large B-cell lymphoma.15 The majority of patients with EBV-associated PTLD demonstrate significant elevations in the serum EBV viral load compared with immunosuppressed controls without PTLD.16 An elevated EBV viral load can support a diagnosis of PTLD, though the absence of EBV viremia does not rule it out.17 Some transplant centers perform posttransplantation monitoring of the serum EBV viral load to aid in PTLD risk assessment and early diagnosis.
Management of PTLD is patient-specific and may involve reduction of immunosuppressive therapy, rituximab, chemotherapy, surgical excision, and/or radiation.13 Reduction of immunosuppression is the cornerstone of treatment.18 In patients who do not respond to the reduction of immunosuppression, rituximab and immunochemotherapy are second-line treatment options. A prospective, multicenter phase 2 trial (the PTLD-1 trial) demonstrated a complete response rate of 40% among patients with PTLD managed with rituximab.19
In summary, this case illustrates the importance of maintaining a broad differential diagnosis when acute fever develops in a patient who has undergone solid organ transplantation. The presence of more than one condition should be considered when the clinical presentation cannot be explained by a single diagnosis, as infections and malignancies can coexist in immunocompromised hosts. This case also highlights an unusual clinical presentation of PTLD, which was heralded mainly by its immunomodulatory effects rather than by compatible symptoms or obvious mass lesions.
Carefully reviewing the patient’s medical history and understanding how it sets the stage for the present illness is an essential step in clinical problem solving, because what is past is prologue.
TEACHING POINTS
- Fever in solid organ transplant recipients should prompt consideration of a broad differential diagnosis, including infection, malignancy, organ graft rejection, autoimmune disease, and medication toxicity.
- PTLD is a rare but serious complication of organ transplantation. Most cases are driven by EBV infection and transformation of infected lymphocytes in a variety of organs in the context of reduced T-cell surveillance. The clinical presentation can be heterogeneous and varies depending on the organs and tissues involved.
- More than one infection, or both infection and malignancy, can coexist in organ transplant recipients. Viral pathogens can exert direct pathologic effects on tissue but can also exert indirect effects, such as contributing to opportunistic infection susceptibility, graft rejection, and oncogenesis.
Disclosures
The authors have nothing to disclose.
Previous Publication
This case was originally reported in the 121st Okinawa Association of Medical Sciences in 2015 in Okinawa, Japan, and the conference abstracts were covered in The Okinawa Medical Journal. The publication did not provide any detailed, step-by-step analysis of clinical decision-making.
A 56-year-old Japanese man with a history of renal transplantation 20 years prior presented to the emergency department (ED) with two months of dyspnea on exertion and one day of fever and chills. The patient was in his usual state of health until two months prior to presentation, when he gradually noticed shortness of breath after sustained or effortful physical activities. The dyspnea improved with rest. Over the following two months, he noticed that the shortness of breath came on with lesser degrees of exertion, such as walking 100 meters. One day before presentation, he developed a fever of 39°C and chills at home, which prompted him to seek ED care. He denied chest pain, cough, leg swelling, or paroxysmal nocturnal dyspnea.
The differential diagnosis of exertional dyspnea progressing over several months includes cardiac, pulmonary, hematologic, and neuromuscular conditions. The patient’s history of renal transplantation prompts consideration of worsening indolent pneumonia (eg, Aspergillus, cytomegalovirus [CMV], or Pneumocystis pneumonia), allograft dysfunction with volume overload, recrudescence of the underlying disease that incited renal failure earlier in life (eg, vasculitis), or a late-onset posttransplantation lymphoproliferative disorder (PTLD). Additionally, acute fever in an immunocompromised patient immediately raises suspicion for infection (eg, pneumonia, enteritis, or urinary tract infection). At this point, it is difficult to know whether the subacute-to-chronic exertional dyspnea and the acute fever are consequences of the same disease or separate, potentially overlapping, processes.
His past medical history was significant for end-stage renal disease due to membranoproliferative glomerular nephropathy (MPGN), for which living, related-donor kidney transplantation was performed 20 years earlier. He also had type 2 diabetes mellitus, hypertension, and basal cell carcinoma of the face, which had been resected three years prior without spread or recurrence. He had no known allergies. Medications included prednisolone 15 mg daily, azathioprine 100 mg daily, and cyclosporine 100 mg daily, as well as amlodipine and candesartan. He lived in Japan with his wife and children. He denied any animal or environmental exposures. He did not smoke cigarettes or drink alcohol and had not traveled recently. His father had diabetes mellitus.
Recrudescence of an underlying autoimmune condition that may have incited MPGN earlier in life is unlikely while taking an immunosuppressive regimen consisting of prednisolone, azathioprine, and cyclosporine. However, these medications do increase susceptibility to infections, lymphoma, and skin cancers. Though he is immunocompromised, the patient is not on prophylaxis for Pneumocystis pneumonia (PCP). PCP in HIV-negative patients is associated with recent glucocorticoid exposure and typically follows an acute-to-subacute course with hypoxemia and respiratory distress. Though the risk of PCP infection is considered highest in the early posttransplantation period (when immunosuppression is most intense), many cases are diagnosed years after transplantation among patients no longer on prophylaxis. The patient has type 2 diabetes mellitus and hypertension, which are known complications of calcineurin inhibitor and steroid therapy and increase the risk of cardiovascular disease. Cardiovascular disease is a major cause of death among renal transplant recipients. Exertional dyspnea may be the presenting symptom of coronary artery disease.
On physical examination, the patient was alert, oriented, and in no acute distress. His temperature was 38.5°C, blood pressure 120/60 mm Hg, heart rate 146 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 93% while breathing ambient air. The conjunctiva were normal without pallor or icterus. There was no cervical lymphadenopathy. Cardiac examination revealed tachycardia with a regular rhythm, normal S1 and S2, and no murmurs, rubs, or gallops. Jugular venous pressure was not elevated, and there was no lower extremity edema. Lungs were clear to auscultation bilaterally. The abdomen was soft, nontender, and nondistended. There was no tenderness over the transplanted kidney and no hepatosplenomegaly.
Dyspnea, fever, and tachycardia may be the sole manifestations of pneumonia in solid organ transplant recipients. The absence of cough or adventitious breath sounds does not eliminate concern for pneumonia. Pathogens that cause indolent pneumonia in immunocompromised patients include viruses (such as typical respiratory viruses and CMV), bacteria (typical organisms, Nocardia, Rhodococcus), and mycobacteria. Fungal causes include Aspergillus, Candida, Cryptococcus, Pneumocystis, and endemic mycoses. A detailed environmental history should be taken, and providers should ascertain which fungal diseases are endemic in the patient’s region of residence. There are no examination features suggesting hypervolemia or anemia. Although there is no hepatosplenomegaly or lymphadenopathy, PTLD often involves extranodal tissues, including the lungs. The incidence of PTLD is highest in the 12 months following transplantation, but it may occur at any time in the posttransplantation course. A complete blood count, comprehensive metabolic panel, lactate dehydrogenase (LDH), and blood and sputum cultures are indicated, along with computed tomography (CT) of the chest.
The leukocyte count was 3,500 cells/mm3, the hemoglobin level 9.0 g/dL, mean corpuscular volume 102 fL, and the platelet count 137,000 cells/mm3. The sodium level was 130 mEq/L, potassium 4.6 mEq/L, blood urea nitrogen 41 mg/dL, and creatinine 3.5 mg/dL. These complete blood count and serum electrolyte results were unchanged from the patient’s baseline values. The serum LDH level was 1,895 IU/L (normal range, 115-245 IU/L). The serum ferritin was 2,114 ng/mL (normal range, 13-277 ng/mL). A chest radiograph revealed diffuse, airspace-filling opacities in the bilateral lung bases. The urinalysis was normal. The patient was admitted and started empirically on intravenous ceftriaxone for potential bacterial pneumonia.
Chronic pancytopenia may result from azathioprine or cyclosporine use, marrow suppression or infiltration by a multisystem disease, or nutritional deficiency. Hemophagocytic lymphohistiocytosis (HLH) triggered by infection, a rheumatologic condition, acquired immunodeficiency, or malignancy can present with fevers, pancytopenia, and elevated ferritin, while splenomegaly may be absent. The euvolemic state, baseline creatinine level, and normal urinalysis argue against allograft dysfunction. The elevated serum ferritin nonspecifically confirms systemic inflammation. LDH, an intracellular enzyme involved in the bidirectional conversion of lactate to pyruvate, is expressed across tissue types. Elevated serum LDH attests to cell destruction, in this case potentially from lung infection (such as PCP) or malignancy (such as PTLD). At this point, the differential diagnosis of fever and pulmonary infiltrates in this patient remains broad.
Azathioprine and cyclosporine were stopped. The patient remained febrile despite the administration of intravenous antibiotics. His hypoxia worsened with an oxygen saturation of 90%-93% on 5 L/min of supplemental oxygen administered by nasal cannula. Noncontrast chest CT obtained on the second hospital day revealed ground-glass opacities in the bilateral lung bases. Blood, sputum, and urine cultures were sterile. As empiric therapies, ganciclovir was started for CMV infection, ciprofloxacin added for atypical pneumonia, and trimethoprim-sulfamethoxazole added for Pneumocystis infection.
These chest imaging findings help prioritize the differential diagnosis. Bibasilar ground-glass opacities are evident, while pulmonary masses, nodules, cavitation, adenopathy, and pleural effusions are absent. The differential diagnosis of multifocal ground-glass opacities on chest imaging includes infectious pneumonia, chronic interstitial lung disease, acute alveolar conditions (eg, cardiogenic pulmonary edema, acute respiratory distress syndrome, diffuse alveolar hemorrhage), or other pathologies (eg, drug toxicity, bronchoalveolar carcinoma, cryptogenic organizing pneumonia).
Infectious pneumonia is the principal concern. A diagnosis of PCP could be unifying, given dyspnea, progressive respiratory failure with hypoxia, and elevated LDH in an immunocompromised patient who is not prescribed PCP prophylaxis. The bilateral lung infiltrates and the absence of thoracic adenopathy or pleural effusions are characteristic of PCP as well. However, caution should be exercised in making specific infectious diagnoses in immunocompromised hosts on the basis of clinical and imaging findings alone. There can be overlap in the radiologic appearance of various infections (eg, CMV pneumonia can also present with bilateral ground-glass infiltrates, with concurrent fever, hypoxia, and pancytopenia). Additionally, more than one pneumonic pathogen may be implicated (eg, acute viral pneumonia superimposed on indolent fungal pneumonia). Polymerase chain reaction (PCR) analysis of respiratory secretions for viruses, serum PCR and serologic testing for herpes viruses, and serum beta-D-glucan and galactomannan assays are indicated. Serum serologic testing for fungi and bacteria such as Nocardia can be helpful, though the negative predictive values of these tests may be reduced in patients with impaired humoral immunity. Timely bronchoalveolar lavage (BAL) with microbiologic and PCR analysis and cytology is advised.
Fever, elevated LDH, cytopenias, and pulmonary infiltrates also raise suspicion for an underlying hematologic malignancy, such as PTLD. However, pulmonary PTLD is seen more often in lung transplant recipients than in patients who have undergone transplantation of other solid organs. In kidney transplant recipients, PTLD most commonly manifests in the allograft itself, gastrointestinal tract, central nervous system, or lymph nodes; lung involvement is less common. Chest imaging in affected patients may reveal nodular or reticulonodular infiltrates of basilar predominance, solitary or multiple masses, cavitating or necrotic lesions, and/or lymphadenopathy. In this patient who has undergone renal transplantation, late-onset PTLD with isolated pulmonary involvement, with only ground-glass opacities on lung imaging, would be an atypical presentation of an uncommon syndrome.
Despite empiric treatment with antibiotics and antiviral agents, the patient’s fever persisted. His respiratory rate increased to 30 breaths per minute. His hypoxia worsened, and he required nasal cannula high-flow oxygen supplementation at 30 L/min with a fraction of inspired oxygen (FiO2) of 40%. On the fifth hospital day, contrast CT scan of the chest and abdomen showed new infiltrates in the bilateral upper lung fields as well as an area of low density in the tail of the pancreas without a focal mass (Figure 1). At this point, BAL was performed, and fluid PCR analysis returned positive for Pneumocystis jirovecii. CMV direct immunoperoxidase staining of leukocytes with peroxidase-labeled monoclonal antibody (C7-HRP test) was positive at five cells per 7.35 × 104 peripheral blood leukocytes. The serum Epstein-Barr virus (EBV) viral capsid antigen (VCA) IgG was positive, while VCA IgM and EBV nuclear antigen IgG were negative. A bone marrow biopsy revealed mild hemophagocytosis. His serum soluble interleukin-2 (sIL2R) level was elevated at 5,254 U/mL (normal range, 122-496 U/mL). Given the BAL Pneumocystis PCR result, the dose of prednisolone was increased to 30 mg/day, and the patient’s fever subsided. Supplemental oxygen was weaned to an FiO2 of 35%.
These studies should be interpreted carefully considering the biphasic clinical course. After two months of exertional dyspnea, the patient acutely developed persistent fever and progressive lung infiltrates. His clinical course, the positive PCR assay for Pneumocystis jirovecii in BAL fluid, and the compatible lung imaging findings make Pneumocystis jirovecii a likely pathogen. But PCP may only explain the second phase of this patient’s illness, considering its often-fulminant course in HIV-negative patients. To explain the two months of exertional dyspnea, marrow hemophagocytosis, pancreatic abnormality, and perhaps even the patient’s heightened susceptibility to PCP infection, an index of suspicion should be maintained for a separate, antecedent process. This could be either an indolent infection (eg, CMV or Aspergillus pneumonia) or a malignancy (eg, lymphoma or PTLD). Completion of serum serologic testing for viruses, bacteria, and fungi and comprehensive BAL fluid analysis (culture, viral PCR, and cytology) is recommended.
A CMV antigenemia assay returned positive, suggesting prior CMV infection. However, to diagnose CMV pneumonia, the virus must be detected in BAL fluid by PCR or cytologic analysis. CMV infection has been associated with cytopenias, HLH, pancreatic infiltration, and an increased risk for fungal infections and EBV-related PTLD. CMV infection could explain the first phase of this patient’s illness. Serum and BAL PCR for CMV are advised. Meanwhile, EBV testing indicates prior infection but does not distinguish between recent or more distant infection. EBV has been implicated in the pathophysiology of PTLD, as EBV-infected lymphoid tissue may proliferate in a variety of organs under reduced T-cell surveillance. EBV infection or PTLD with resulting immunomodulation may pose other explanations for this patient’s development of PCP infection. Cytologic analysis of the BAL fluid and marrow aspirate for evidence of PTLD is warranted. Finally, CMV, EBV, and PTLD have each been reported to trigger HLH. Though this patient has fevers, mild marrow hemophagocytosis, elevated serum ferritin, and elevated serum IL-2 receptor levels, he does not meet other diagnostic criteria for HLH (such as more pronounced cytopenias, splenomegaly, hypertriglyceridemia, hypofibrinogenemia, and low or absent natural killer T-cell activity). However, HLH may be muted in this patient because he was prescribed cyclosporine, which has been used in HLH treatment protocols.
On the 11th hospital day, the patient developed hemorrhagic shock due to massive hematemesis and was transferred to the intensive care unit. His hemoglobin level was 5.9 g/dL. A total of 18 units of packed red blood cells were transfused over the following week for ongoing gastrointestinal bleeding. The serum LDH level increased to 4,139 IU/L, and the ferritin level rose to 7,855 ng/mL. The EBV copy level by serum PCR returned at 1 × 106 copies/mL (normal range, less than 2 x 102 copies/mL). The patient was started on methylprednisolone (1 g/day for three days) and transitioned to dexamethasone and cyclosporine for possible EBV-related HLH. Ceftazidime, vancomycin, trimethoprim-sulfamethoxazole, and ciprofloxacin were administered. Amphotericin-B was initiated empirically for potential fungal pneumonia. Ganciclovir was continued. However, the patient remained in shock despite vasopressors and transfusions and died on the 22nd hospital day.
The patient deteriorated despite broad antimicrobial therapy. Laboratory studies revealed EBV viremia and rising serum LDH. Recent EBV infection may have induced PTLD in the gastrointestinal tract, which is a commonly involved site among affected renal transplant patients. Corticosteroids and stress from critical illness can contribute to intestinal mucosal erosion and bleeding from a luminal PTLD lesion. Overall, the patient’s condition was likely explained by EBV infection, which triggered HLH and gastrointestinal PTLD. The resulting immunomodulation increased his risk for PCP infection beyond that conferred by chronic immunosuppression. It is still possible that he had concomitant CMV pneumonia, Aspergillus pneumonia, or even pulmonary PTLD, in addition to the proven PCP diagnosis.
An autopsy was performed. Atypical lymphocytic infiltration and diffuse alveolar damage were shown in the right upper lobe (Figure 2). EBV RNA-positive atypical lymphocytes coexpressing CD20 were demonstrated in multiple organs including the bone marrow, lungs, heart, stomach, adrenal glands, duodenum, ileum, and mesentery (Figure 3). This confirmed the diagnosis of an underlying EBV-positive posttransplant lymphoproliferative disorder. Serum and BAL CMV PCR assays returned negative. Neither CMV nor Aspergillus was identified in autopsy specimens.
COMMENTARY
A broad differential diagnosis should be considered when acute fever develops in a patient who has undergone solid organ transplantation. Causes may include opportunistic and nonopportunistic infections as well as noninfectious etiologies such as malignancy, organ rejection, inflammatory conditions, and medication toxicity.1,2 As the discussant noted, more than one infection, or both infection and malignancy, can coexist in immunocompromised patients. For example, while viral pathogens such as EBV, CMV, and respiratory syncytial virus can cause illness due to direct tissue infection, they can also exert indirect effects in transplant recipients: acting as cofactors for and enabling other infections by causing immunosuppression (eg, Aspergillus or PCP developing after CMV infection), triggering graft rejection by upregulating proinflammatory cytokines, and inducing oncogenesis (eg, EBV-related PTLD).1,3-5
PTLD is a rare but serious complication of solid organ transplantation and immunosuppression. Most cases are driven by EBV infection and subsequent transformation of infected lymphoid tissue in a variety of organs in the context of reduced T-cell surveillance.6 The incidence of PTLD varies based on the organ transplanted, ranging from 0.8%-2.5% in those who have undergone renal transplantation to 1.0%-5.5% in liver transplant recipients and 3.0%-10% in lung transplant recipients.3 The incidence has increased over the past decade. This may be due to a greater number of solid organ transplantations being performed, aging of the transplant donor/recipient population, new immunosuppressive regimens, and improved PTLD diagnosis due to superior diagnostic tools and clinician awareness.3 However, the mortality rate among solid organ transplant recipients with PTLD remains high, ranging from 40% to 70%.6
Risk factors for PTLD include a greater intensity of T-cell immunosuppression,7 history of pretransplant malignancy, recipient EBV seronegativity and donor seropositivity, and younger age at the time of transplantation.8-10 EBV-related PTLD incidence in solid organ transplant recipients is greatest in the early posttransplantation course (the period of most intense immunosuppression) with over 80% of cases occurring in the first posttransplant year.11
A high index of suspicion for PTLD is warranted in any solid organ transplant recipient who presents with constitutional symptoms, adenopathy, or cytopenias. Clinical suspicion of PTLD can be informed by risk factors, constitutional symptoms, elevated serum LDH, a detectable or rising serum EBV viral load, and radiologic adenopathy or visceral tissue infiltration.12 The clinical presentation of PTLD is heterogeneous and varies in accordance with the organs affected. Extranodal involvement, such as pulmonary, gastrointestinal, and bone marrow involvement, is more common in PTLD than in other types of lymphoma.13 In this patient, the cytopenias, elevated serum LDH level, lung infiltrates, and radiologic pancreatic tail abnormality served as early clues to the presence of underlying PTLD.
The standard approach to diagnosing PTLD is biopsy of a suspicious lesion (adenopathy or an infiltrated visceral organ) with histopathological examination. Pathology may demonstrate distorted tissue architecture, clonal lymphocytes, or EBV-positive lymphocytes.14 Conventional CT is the most commonly used imaging modality to detect adenopathy or tissue infiltration related to PTLD,3 though 18F-fluorodeoxyglucose position-emission tomography (FDG-PET) is also used. Although FDG-PET has high diagnostic accuracy, with an overall sensitivity of 89% and specificity of 89%, false-negative results have been reported, particularly in cases of early PTLD lesions and diffuse large B-cell lymphoma.15 The majority of patients with EBV-associated PTLD demonstrate significant elevations in the serum EBV viral load compared with immunosuppressed controls without PTLD.16 An elevated EBV viral load can support a diagnosis of PTLD, though the absence of EBV viremia does not rule it out.17 Some transplant centers perform posttransplantation monitoring of the serum EBV viral load to aid in PTLD risk assessment and early diagnosis.
Management of PTLD is patient-specific and may involve reduction of immunosuppressive therapy, rituximab, chemotherapy, surgical excision, and/or radiation.13 Reduction of immunosuppression is the cornerstone of treatment.18 In patients who do not respond to the reduction of immunosuppression, rituximab and immunochemotherapy are second-line treatment options. A prospective, multicenter phase 2 trial (the PTLD-1 trial) demonstrated a complete response rate of 40% among patients with PTLD managed with rituximab.19
In summary, this case illustrates the importance of maintaining a broad differential diagnosis when acute fever develops in a patient who has undergone solid organ transplantation. The presence of more than one condition should be considered when the clinical presentation cannot be explained by a single diagnosis, as infections and malignancies can coexist in immunocompromised hosts. This case also highlights an unusual clinical presentation of PTLD, which was heralded mainly by its immunomodulatory effects rather than by compatible symptoms or obvious mass lesions.
Carefully reviewing the patient’s medical history and understanding how it sets the stage for the present illness is an essential step in clinical problem solving, because what is past is prologue.
TEACHING POINTS
- Fever in solid organ transplant recipients should prompt consideration of a broad differential diagnosis, including infection, malignancy, organ graft rejection, autoimmune disease, and medication toxicity.
- PTLD is a rare but serious complication of organ transplantation. Most cases are driven by EBV infection and transformation of infected lymphocytes in a variety of organs in the context of reduced T-cell surveillance. The clinical presentation can be heterogeneous and varies depending on the organs and tissues involved.
- More than one infection, or both infection and malignancy, can coexist in organ transplant recipients. Viral pathogens can exert direct pathologic effects on tissue but can also exert indirect effects, such as contributing to opportunistic infection susceptibility, graft rejection, and oncogenesis.
Disclosures
The authors have nothing to disclose.
Previous Publication
This case was originally reported in the 121st Okinawa Association of Medical Sciences in 2015 in Okinawa, Japan, and the conference abstracts were covered in The Okinawa Medical Journal. The publication did not provide any detailed, step-by-step analysis of clinical decision-making.
1. Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med. 2007;357(25):2601-2614. https://doi.org/10.1056/NEJMra064928.
2. Bouza E, Loeches B, Muñoz P. Fever of unknown origin in solid organ transplant recipients. Infect Dis Clin North Am. 2007;21(4):1033-1054, ix-x. https://doi.org/10.1016/j.idc.2007.09.001,
3. Kotton CN, Fishman JA. Viral infection in the renal transplant recipient. J Am Soc Nephrol. 2005;16(6):1758-1774. https://doi.org/10.1681/ASN.2004121113.
4. Arend SM, Westendorp RG, Kroon FP, et al. Rejection treatment and cytomegalovirus infection as risk factors for Pneumocystis carinii pneumonia in renal transplant recipients. Clin Infect Dis. 1996;22(6):920-925. https://doi.org/10.1093/clinids/22.6.920.
5. Reinke P, Fietze E, Ode-Hakim S, et al. Late-acute renal allograft rejection and symptomless cytomegalovirus infection. Lancet. 1994;344(8939-8940):1737-1738. https://doi.org/10.1016/S0140-6736(94)92887-8.
6. Tsai DE, Douglas L, Andreadis C, et al. EBV PCR in the diagnosis and monitoring of posttransplant lymphoproliferative disorder: results of a two-arm prospective trial. Am J Transplant. 2008;8(5):1016-1024. https://doi.org/10.1111/j.1600-6143.2008.02183.x.
7. Penn I. Cancers complicating organ transplantation. N Engl J Med. 1990;323(25):1767-1769. https://doi.org/10.1056/NEJM199012203232510
8. Walker RC, Marshall WF, Strickler JG, et al. Pretransplantation assessment of the risk of lymphoproliferative disorder. Clin Infect Dis. 1995;20(5):1346-1353. https://doi.org/10.1093/clinids/20.5.1346.
9. Opelz G, Döhler B. Lymphomas after solid organ transplantation: a collaborative transplant study report. Am J Transplant. 2004;4(2):222-230. https://doi.org/10.1046/j.1600-6143.2003.00325.x.
10. Caillard S, Dharnidharka V, Agodoa L, Bohen E, Abbott K. Posttransplant lymphoproliferative disorders after renal transplantation in the United States in era of modern immunosuppression. Transplantation. 2005;80(9):1233-1243. doi: 10.1097/01.tp.0000179639.98338.39.
11. Opelz G, Henderson R. Incidence of non-Hodgkin lymphoma in kidney and heart transplant recipients. Lancet. 1993;342(8886-8887):1514-1516. https://doi.org/10.1016/S0140-6736(05)80084-4.
12. Samant H, Kothadia JP. Transplantation Posttransplantation Lymphoproliferative Disorders. Treasure Island, FL: StatPearls Publishing; 2018. PubMed
13. Dierickx D, Habermann TM. Post-transplantation lymphoproliferative disorders in adults. N Engl J Med. 2018;378(6):549-562. https://doi.org/10.1056/NEJMra1702693.
14. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390. https://doi.org/10.1182/blood-2016-01-643569.
15. Dierickx D, Tousseyn T, Requilé A, et al. The accuracy of positron emission tomography in the detection of posttransplant lymphoproliferative disorder. Haematologica. 2013;98(5):771-775. https://doi.org/10.3324/haematol.2012.074500.
16. Wagner HJ, Wessel M, Jabs W, et al. Patients at risk for development of posttransplant lymphoproliferative disorder: plasma versus peripheral blood mononuclear cells as material for quantification of Epstein-Barr viral load by using real-time quantitative polymerase chain reaction. Transplantation. 2001;72(6):1012-1019. PubMed
17. Baldanti F, Rognoni V, Cascina A, Oggionni T, Tinelli C, Meloni F. Post-transplant lymphoproliferative disorders and Epstein-Barr virus DNAemia in a cohort of lung transplant recipients. Virol J. 2011;8:421. https://doi.org/10.1186/1743-422X-8-421.
18. Parker A, Bowles K, Bradley JA, et al. Management of post-transplant lymphoproliferative disorder in adult solid organ transplant recipients - BCSH and BTS Guidelines. Br J Haematol. 2010;149(5):693-705. https://doi.org/10.1111/j.1365-2141.2010.08160.x.
19. Trappe R, Oertel S, Leblond V, et al. Sequential treatment with rituximab followed by CHOP chemotherapy in adult B-cell post-transplant lymphoproliferative disorder (PTLD): the prospective international multicentre phase 2 PTLD-1 trial. Lancet Oncol. 2012;13(2):196-206. https://doi.org/10.1016/S1470-2045(11)70300-X.
1. Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med. 2007;357(25):2601-2614. https://doi.org/10.1056/NEJMra064928.
2. Bouza E, Loeches B, Muñoz P. Fever of unknown origin in solid organ transplant recipients. Infect Dis Clin North Am. 2007;21(4):1033-1054, ix-x. https://doi.org/10.1016/j.idc.2007.09.001,
3. Kotton CN, Fishman JA. Viral infection in the renal transplant recipient. J Am Soc Nephrol. 2005;16(6):1758-1774. https://doi.org/10.1681/ASN.2004121113.
4. Arend SM, Westendorp RG, Kroon FP, et al. Rejection treatment and cytomegalovirus infection as risk factors for Pneumocystis carinii pneumonia in renal transplant recipients. Clin Infect Dis. 1996;22(6):920-925. https://doi.org/10.1093/clinids/22.6.920.
5. Reinke P, Fietze E, Ode-Hakim S, et al. Late-acute renal allograft rejection and symptomless cytomegalovirus infection. Lancet. 1994;344(8939-8940):1737-1738. https://doi.org/10.1016/S0140-6736(94)92887-8.
6. Tsai DE, Douglas L, Andreadis C, et al. EBV PCR in the diagnosis and monitoring of posttransplant lymphoproliferative disorder: results of a two-arm prospective trial. Am J Transplant. 2008;8(5):1016-1024. https://doi.org/10.1111/j.1600-6143.2008.02183.x.
7. Penn I. Cancers complicating organ transplantation. N Engl J Med. 1990;323(25):1767-1769. https://doi.org/10.1056/NEJM199012203232510
8. Walker RC, Marshall WF, Strickler JG, et al. Pretransplantation assessment of the risk of lymphoproliferative disorder. Clin Infect Dis. 1995;20(5):1346-1353. https://doi.org/10.1093/clinids/20.5.1346.
9. Opelz G, Döhler B. Lymphomas after solid organ transplantation: a collaborative transplant study report. Am J Transplant. 2004;4(2):222-230. https://doi.org/10.1046/j.1600-6143.2003.00325.x.
10. Caillard S, Dharnidharka V, Agodoa L, Bohen E, Abbott K. Posttransplant lymphoproliferative disorders after renal transplantation in the United States in era of modern immunosuppression. Transplantation. 2005;80(9):1233-1243. doi: 10.1097/01.tp.0000179639.98338.39.
11. Opelz G, Henderson R. Incidence of non-Hodgkin lymphoma in kidney and heart transplant recipients. Lancet. 1993;342(8886-8887):1514-1516. https://doi.org/10.1016/S0140-6736(05)80084-4.
12. Samant H, Kothadia JP. Transplantation Posttransplantation Lymphoproliferative Disorders. Treasure Island, FL: StatPearls Publishing; 2018. PubMed
13. Dierickx D, Habermann TM. Post-transplantation lymphoproliferative disorders in adults. N Engl J Med. 2018;378(6):549-562. https://doi.org/10.1056/NEJMra1702693.
14. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390. https://doi.org/10.1182/blood-2016-01-643569.
15. Dierickx D, Tousseyn T, Requilé A, et al. The accuracy of positron emission tomography in the detection of posttransplant lymphoproliferative disorder. Haematologica. 2013;98(5):771-775. https://doi.org/10.3324/haematol.2012.074500.
16. Wagner HJ, Wessel M, Jabs W, et al. Patients at risk for development of posttransplant lymphoproliferative disorder: plasma versus peripheral blood mononuclear cells as material for quantification of Epstein-Barr viral load by using real-time quantitative polymerase chain reaction. Transplantation. 2001;72(6):1012-1019. PubMed
17. Baldanti F, Rognoni V, Cascina A, Oggionni T, Tinelli C, Meloni F. Post-transplant lymphoproliferative disorders and Epstein-Barr virus DNAemia in a cohort of lung transplant recipients. Virol J. 2011;8:421. https://doi.org/10.1186/1743-422X-8-421.
18. Parker A, Bowles K, Bradley JA, et al. Management of post-transplant lymphoproliferative disorder in adult solid organ transplant recipients - BCSH and BTS Guidelines. Br J Haematol. 2010;149(5):693-705. https://doi.org/10.1111/j.1365-2141.2010.08160.x.
19. Trappe R, Oertel S, Leblond V, et al. Sequential treatment with rituximab followed by CHOP chemotherapy in adult B-cell post-transplant lymphoproliferative disorder (PTLD): the prospective international multicentre phase 2 PTLD-1 trial. Lancet Oncol. 2012;13(2):196-206. https://doi.org/10.1016/S1470-2045(11)70300-X.
© 2019 Society of Hospital Medicine
The Right Frame
A 65-year-old man was transferred to a tertiary academic medical center with one week of progressive shortness of breath, dry cough, and fevers. He reported no weight loss or night sweats but had experienced mild right upper quadrant pain and anorexia for the preceding three weeks. Several years had passed since he had consulted a physician, and he did not take any medications. He immigrated to the United States from Mexico four decades prior. He traveled back frequently to visit his family, most recently one month before his presentation. He worked as a farming supervisor in the Central Valley of California. He smoked tobacco and had a 30 pack-year history. He drank alcohol occasionally and denied any drug use.
Causes of subacute cough and dyspnea include bronchitis, pneumonia, heart failure, and asthma. Pneumonia and heart failure might cause right upper quadrant pain from diaphragmatic irritation and hepatic congestion, respectively. Metastatic cancer or infection may lead to synchronous pulmonary and hepatic involvement. The patient is at increased risk of lung cancer, given his extensive smoking history.
The patient’s place of residence in the Southwestern United States places him at risk of respiratory illness from coccidioidomycosis. His exact involvement with animals and their products should be further explored. For example, consumption of unpasteurized milk might result in pneumonia, hepatitis, or both from M. bovis, Brucella species, or C. burnetii. His travel to Mexico prompts consideration of tuberculosis, histoplasmosis, and paracoccidiomycosis as causes of respiratory and possible hepatic illness.
Two weeks prior, the patient had initially presented to another hospital with one week of intermittent right upper quadrant pain unrelated to eating. An abdominal ultrasound and hepatobiliary iminodiacetic acid (HIDA) scan were normal. Computed tomography (CT) of the chest, abdomen, and pelvis with contrast demonstrated a left upper lobe lung mass measuring 5.5 × 4.4 × 3.7 cm3 and scattered right-sided pulmonary nodules (Figure 1). He underwent CT-guided biopsy of the mass and was discharged with a presumed diagnosis of primary pulmonary malignancy with plans for outpatient follow-up.
Over the next four days, the patient developed progressive dyspnea with cough and subjective fevers. The patient was readmitted with a diagnosis of postobstructive pneumonia and acute kidney injury (creatinine increased from 0.7 mg/dL to 2.9 mg/dL between admissions), and this finding was attributed to contrast-induced nephropathy from his recent CT scan. He was treated with vancomycin and piperacillin/tazobactam for two days but wished to transfer to a tertiary care hospital for a second opinion.
Postobstructive pneumonia, pulmonary embolism, and pleural effusion are common causes of dyspnea in patients with lung cancer. The patient’s travel and occupational history, lung nodules, acute renal insufficiency, and rapidly progressive respiratory symptoms prompt consideration for radiographic mimickers of lung cancer. Tuberculosis might present as a lung mass (pulmonary tuberculoma) during primary infection or reactivation. Noninfectious causes of pulmonary masses and nodules include metastatic cancer (eg, colon cancer), sarcoidosis, IgG4-related disease, and granulomatous polyangiitis (GPA).
Contrast-induced nephropathy is unusual in patients with normal renal function. More probable explanations include hypovolemia or acute tubular necrosis (ATN) from underlying inflammation. The patient’s CT-negative right upper quadrant pain may be a distinct process or represent another facet of a disseminated illness such as hepatic infiltration from lymphoma.
Upon arrival, the patient’s temperature was 38°C, heart rate (HR) 107 beats per minute, blood pressure (BP) 159/89 mm Hg, respiratory rate 25 breaths per minute, and oxygen saturation 92% on 2 L of oxygen per minute. He showed no signs of distress. Mild scleral icterus was noted. The cardiac exam was normal. Auscultation revealed scattered wheezes and crackles in the left upper lobe. Mild right upper quadrant tenderness without hepatosplenomegaly was noted on the abdominal exam. The patient’s lower extremities exhibited bilateral trace edema. No rash was observed, and his neurologic exam was normal.
The white blood cell (WBC) count was 28,300 per cubic millimeter (87% neutrophils, 3.6% lymphocytes, and 0.03% eosinophils), hemoglobin 11.1 g per deciliter, and platelet count 789,000 per cubic millimeter. Sodium was 127 mmol per liter, potassium 4.6 mmol per liter, chloride 101 mmol per liter, bicarbonate 13 mmol per liter, blood urea nitrogen 60 mg per deciliter, and creatinine 3.4 mg per deciliter. Aspartate aminotransferase and alanine aminotransferase levels were normal. Alkaline phosphatase was 283 units per liter (normal range, 31-95), and total bilirubin was 4.5 mg per deciliter (normal range, 0.2-1.3) with a direct bilirubin of 2.7 mg per deciliter. Urinalysis demonstrated urine protein of 30 mg/dL, specific gravity of 1.013, negative nitrites, 10-21 white cells per high-powered field (normal, < 5), and 21-50 red cells per high-powered field (normal, < 3). Urine microscopy revealed muddy brown casts but no cellular casts or dysmorphic red cells. A chest radiograph (CXR) showed patchy consolidations in the bilateral upper lobes and left lower lobe along with Kerley B lines, a small left pleural effusion, and thickened right horizontal fissure; the left upper lobe mass was re-demonstrated. Vancomycin, piperacillin-tazobactam, and azithromycin were administered.
At this point, the most likely source of sepsis is multifocal pneumonia. The patient is at risk for S. aureus and P. aeruginosa given his recent hospitalization. A severe form of leptospirosis (Weil’s disease) is associated with pulmonary disease, hyperbilirubinemia, and renal failure. Repeat abdominal imaging is necessary to evaluate for cholangitis given the patient’s right upper quadrant pain, fever, and jaundice. It would also help categorize his cholestatic pattern of liver injury as intrahepatic or extrahepatic (eg, stricture). An infiltrative disease such as sarcoidosis may cause both intrahepatic cholestasis and parenchymal lung disease, although the pleural pathology is uncommon.
His normal cardiac exam does not exclude cardiogenic pulmonary edema, a common cause of interstitial edema and pleural effusion. In this setting of systemic inflammation (neutrophilia, thrombocytosis, and hypoalbuminemia), the thickened right horizontal fissure and interlobular septa might represent an infiltrative process, such as lymphangitic carcinomatosis, lymphoma, or sarcoidosis.
Muddy brown casts are characteristic of ATN. The patient’s risk factors for ATN include sepsis and previously administered iodinated contrast. Fluid retention from oliguric renal failure is likely contributing to his hyponatremia and lower extremity edema. Pathology isolated to the tubules, however, would not cause hematuria and pyuria and suggests glomerular or interstitial disease. The lack of cellular casts on a single urinary specimen does not eliminate the likelihood of either disease. Hematuria and diffuse parenchymal lung disease prompt consideration of pulmonary-renal syndromes, such as anti-glomerular basement membrane disease, GPA, and systemic lupus erythematosus, which can all be triggered by infection.
On the night of transfer, the patient experienced acute respiratory distress. Heart rate was 130 beats per minute, BP 170/95 mm Hg, respiratory rate 40 breaths per minute, and oxygen saturation 88% on six liters of supplemental oxygen by nasal cannula. His arterial blood gas demonstrated a pH of 7.23, PaCO2 of 32 mm Hg, and PaO2 of 65 mm Hg. He was emergently intubated for progressive hypoxemic respiratory failure. A small amount of blood was noted in the endotracheal tube. A noncontrast CT of the chest demonstrated multifocal airspace opacities and bilateral pleural effusions. The previously noted left upper lobe mass was unchanged.
Rapid respiratory decline and diffuse alveolar disease commonly result from aspiration, flash pulmonary edema, and acute respiratory distress syndrome (ARDS). Necrotizing pneumonia (eg, S. aureus) and trauma during intubation are possible causes of blood in his endotracheal tube. However, in the setting of multifocal airspace opacity, renal insufficiency, hematuria, and rapid respiratory decline, the blood might represent diffuse alveolar hemorrhage (DAH). Bronchoscopy with bronchioalveolar lavage to evaluate for pulmonary edema, infection, and hemorrhage would be indicated.
The patient subsequently developed oliguria, requiring continuous renal replacement therapy. An echocardiogram demonstrated impaired left ventricular relaxation and a reduced ejection fraction of 45% without segmental wall motion abnormalities or valvular disease, and a right ventricular systolic pressure of 36 mm Hg. Over the next 12 hours, his respiratory status improved, and he was extubated to 15 L per minute of supplemental oxygen by high-flow nasal cannula (HFNC).
The pathology report of the lung biopsy from the other hospital disclosed chronic inflammation and fibrosis with ill-defined areas of necrosis and myxoid degeneration surrounded by nuclear palisading suggestive of granulomatous inflammation. Staining for acid-fast bacilli (AFB) and fungal organisms was negative.
The rapid pulmonary recovery is inconsistent with multifocal pneumonia or ARDS. Flash pulmonary edema might result in sudden hypoxemic respiratory failure that resolves with positive pressure ventilation and ultrafiltration. However, this condition would not explain the biopsy results. Granulomatous lung pathology often results from mycobacterial or fungal disease. Tuberculosis and fungal pneumonia are not excluded with negative staining alone. However, neither would cause self-limited respiratory failure. Histologic evidence of necrosis lessens the likelihood of sarcoidosis, which rarely causes fulminant pulmonary disease. Lymphoma can result in granulomatous inflammation but would not cause transient pulmonary disease. GPA, a cause of necrotizing granulomatous lung disease, might result in a lung mass and worsened hypoxemia through DAH.
The patient continued to require 15 L of oxygen per minute by HFNC. He had persistent bilateral perihilar alveolar and interstitial opacities on CXR. Repeat WBC count was 29,200 per cubic millimeter, hemoglobin 7.8 g per deciliter, and platelets 656,000 per cubic millimeter. The C-reactive protein was 300 mg per L (normal range, <6.3) and erythrocyte sedimentation rate 100 mm per hour (normal range, <10). Legionella urinary antigen, serum immunodiffusion for Coccidiodes imitus, human immunodeficiency virus antibody, respiratory viral panel, and beta-D glucan were negative. Rare acid-fast bacilli were visualized in one out of three concentrated AFB sputum smears. He was started on empiric antituberculous therapy with rifampin, isoniazid, pyrazinamide, and ethambutol.
The sputum sample is suggestive of pulmonary tuberculosis. The salient features of this case include systemic inflammation, pulmonary nodules and mass, necrotizing granulomatous lung pathology, renal insufficiency, and hematuria. Disseminated tuberculosis might explain all these findings. However, a positive AFB smear may signal the presence of a nontuberculous mycobacteria, which is less likely to cause this clinical syndrome.
M. tuberculosis complex polymerase chain reaction (MTB PCR) assay returned negative for M. tuberculosis. Antiproteinase 3 antibody was 1,930 units (normal range, <20). Antimyeloperoxidase and antiglomerular basement membrane antibodies were negative.
Tuberculosis and GPA share several overlapping features, such as necrotizing lung pathology and less commonly antineutrophil cytoplasmic autoantibody (ANCA)-associated antibodies. However, the lung mass, acute renal and respiratory failure, hematuria, and the degree of anti-proteinase 3 level elevation are highly suggestive of GPA. The negative MTB PCR raises the possibility that a nontuberculous mycobacterium was detected on the sputum smear. Nevertheless, continued treatment until finalization of culture results is appropriate given that tuberculosis is endemic in Mexico.
The patient’s presenting features of right upper quadrant tenderness, jaundice, and cholestatic hepatitis remain poorly explained by either of these diagnoses. Neither tuberculosis nor GPA commonly presents with accompanying hepatic involvement, though both have been occasionally described as causing hepatitis. As the greatest concern in this patient remains his progressive renal failure and accompanying pulmonary hemorrhage, a renal biopsy to assess for glomerulonephritis associated with GPA is warranted before further investigation into the cause of his cholestatic hepatitis.
A core renal biopsy demonstrated pauci-immune focal crescentic and necrotizing glomerulonephritis with mixed tubulointerstitial inflammation (Figure 2). In conjunction with the pulmonary syndrome and positive antiproteinase 3 serology, a diagnosis of granulomatosis with polyangiitis was made. The patient was treated with pulse dose steroids, rituximab, and plasma exchange. Two weeks later, the sputum mycobacterial culture returned positive for Mycobacterium llatzerense and anti-tuberculous treatment was discontinued.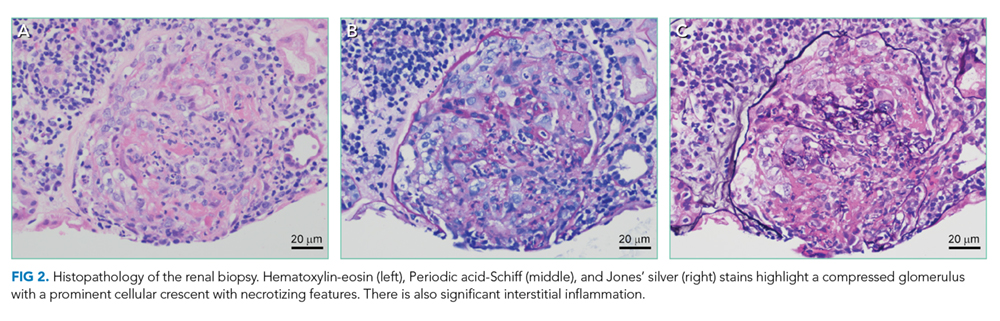
Over the following weeks, the patient improved and was transitioned off dialysis prior to hospital discharge. By six months later, he had resolution of his hemoptysis, shortness of breath, liver biochemical test abnormalities, and significant improvement in his renal function. Repeat sputum mycobacterial cultures were negative.
DISCUSSION
A 65-year-old man from Mexico with a significant smoking history presented with an apical lung mass and cough, prioritizing tuberculosis and pulmonary malignancy. As the case unfolded, renal failure, multifocal lung opacities, conflicting tuberculosis test results, positive anti-proteinase 3 antibody, and ultimately a renal biopsy led to the diagnosis of granulomatosis with polyangiitis (GPA).
The correct interpretation of occasionally conflicting mycobacterial testing is crucial. Mycobacterial cultures remain the gold standard for diagnosing tuberculosis. However, results take weeks to return. Rapid tests include acid-fast bacilli (AFB) smear microscopy and nucleic acid-amplification tests (NAAT) of sputum or bronchoalveolar samples.1 When three sputum smears are performed, the sensitivity of AFB smear microscopy for tuberculosis in immunocompetent hosts is 70%.1 The AFB smear does not distinguish between different mycobacterial organisms. Thus, a positive result must be interpreted with the relative prevalence of tuberculosis and nontuberculous mycobacteria (NTM) in mind. The addition of NAAT-based assays has allowed for enhanced sensitivity and specificity in the diagnosis of tuberculosis, such that a negative NAAT in a patient with a positive AFB smear strongly argues for the presence of a NTM.2-4
NTM are widely prevalent environmental microbes, with over 140 species described, and careful consideration is required to determine if an isolate is pathogenic.5 Given their ubiquitous nature, a high rate of asymptomatic respiratory and cutaneous colonization occurs. Correspondingly, the diagnosis of NTM disease requires multiple positive cultures or pathologic features on tissue biopsy, compatible clinical findings, and diligent exclusion of other causes.5 A retrospective study of all NTM isolates in Oregon from 2005-2006 revealed that only 47% of patients met the guideline criteria for having symptomatic NTM disease.6 In our case, the patient’s sputum grew M. llatzerense, an aerobic, nonfermenting mycobacterium found in water sources that has only infrequently been implicated as a human pathogen.7,8 Subsequent AFB sputum cultures were negative, and serial imaging showed resolution of the pulmonary findings without additional antimycobacterial therapy, suggesting that this organism was not responsible for the disease process.
Along with microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA), GPA is an antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis that predominantly affects small to medium sized vessels. Although it can occur at any age, GPA most commonly afflicts older adults, with men and women being diagnosed at roughly equal rates.9 GPA is a multisystem disease with a wide array of clinical manifestations. The most frequently involved sites of disease are the respiratory tract and kidneys, although virtually any organ can be affected. Sino-nasal disease, such as destructive sinusitis, or ear involvement are nearly universal. Lower respiratory manifestations occur in 60% of patients, but are highly diverse and reflect the inherent difficulty in diagnosing this condition.9-11 Additionally, GPA is a frequent cause of the pulmonary-renal syndromes, with glomerulonephritis occurring in 80% of patients.9
The diagnosis of GPA in this case was delayed, in part, due to features suggestive of malignancy and pulmonary tuberculosis. While sino-nasal disease was not noted during this hospitalization, the patient had many different respiratory manifestations, including a dominant pulmonary mass, diffuse nodules, and hypoxemic respiratory failure due to suspected diffuse alveolar hemorrhage (DAH), all of which have been reported in GPA.12 Dysmorphic red cells and red blood cell casts are not sensitive for renal involvement in GPA; their absence does not exclude the possibility of an ANCA-associated vasculitis.13 Hematuria and rapid progression to oliguric renal failure are characteristic of a vasculitic process and should sway clinicians away from a working diagnosis of ATN.
The diagnosis of GPA involves the synthesis of clinical data, radiographic findings, serologic testing, and histopathology. ANCA testing is an essential step in the diagnosis of GPA but has limitations. Patients with GPA more commonly have ANCAs targeting the enzyme proteinase-3 (PR3-ANCA), with MPA being more closely associated with myeloperoxidase (MPO-ANCA), although cross-reactivity and antibody-negative disease can occur.14 Although 90% of patients with GPA with multiorgan involvement will have a positive ANCA, a negative test is more common in localized upper airway disease, where only 50% have a positive ANCA.15 A number of drugs, medications, infections, and nonvasculitic autoimmune diseases have been associated with positive ANCA serologies in the absence of systemic vasculitis.14,16,17 As such, pathologic demonstration of vasculitis is necessary for establishing the diagnosis. Typical sites for biopsy include the kidneys and lungs.9
This case illustrates how clinicians often find themselves at a diagnostic crossroads—being forced to choose which clinical elements to prioritize. At various points, our patient’s presentation could have been framed as “a man from a Tb-endemic country with hemoptysis and an apical opacity,” “an elderly man with extensive smoking history and lung mass,” or “a patient with elevated inflammatory markers and pulmonary-renal syndrome”. In such situations, it is incumbent on the clinician to evaluate how well a given problem representation encompasses or highlights the salient features of a case. As with painting or photography, an essential aspect of appreciating the whole picture involves carefully selecting the right frame.
KEY TEACHING POINTS
- The diagnosis of tuberculosis relies on smear microscopy, nucleic-acid amplification testing (NAAT), and cultures. A positive AFB smear with negative NAAT suggests the presence of a nontuberculous mycobacteria (NTM).
- NTM are common environmental organisms and often exist as nonpathogenic colonizers.6 The diagnosis of NTM disease requires exclusion of other causes and careful clinical, microbiologic, and radiographic correlation.
- Granulomatosis with polyangiitis is a multisystem disease often involving the respiratory track and kidney. Pulmonary disease can present with airway involvement, parenchymal nodules, opacities, pleural findings, and diffuse alveolar hemorrhage.12
Disclosures
Drs. Minter, Geha, Boslett, Chung, and Ramani have no disclosures. Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME).
1. Lewinsohn DM, Leonard MK, LoBue PA, et al. Official American Thoracic Society/Infectious Diseases Society of America/Centers for Disease Control and Prevention clinical practice guidelines: diagnosis of tuberculosis in adults and children. Clin Infect Dis. 2017;64(2):e1-e33. PubMed
2. Steingart KR, Sohn H, Schiller I, et al. Xpert(R) MTB/RIF assay for pulmonary tuberculosis and rifampicin resistance in adults. Cochrane Database Syst Rev. 2013;(1):Cd009593. PubMed
3. Luetkemeyer AF, Firnhaber C, Kendall MA, et al. Evaluation of Xpert MTB/RIF versus afb smear and culture to identify pulmonary tuberculosis in patients with suspected tuberculosis from low and higher prevalence settings. Clin Infect Dis. 2016;62(9):1081-1088. PubMed
4. Boehme CC, Nabeta P, Hillemann D, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med. 2010;363(11):1005-1015. PubMed
5. Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175(4):367-416. PubMed
6. Winthrop KL, McNelley E, Kendall B, et al. Pulmonary nontuberculous mycobacterial disease prevalence and clinical features: an emerging public health disease. Am J Respir Crit Care Med. 2010;182(7):977-982. PubMed
7. Teixeira L, Avery RK, Iseman M, et al. Mycobacterium llatzerense lung infection in a liver transplant recipient: case report and review of the literature. Am J Transplant. 2013;13(8):2198-2200. PubMed
8. Cárdenas AM, Gomila M, Lalucat J, Edelstein PH. Abdominal abscess caused by Mycobacterium llatzerense. J Clin Microbiol. 2014;52(4):1287-1289. PubMed
9. Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med. 1997;337(21):1512-1523. PubMed
10. Mahr A, Katsahian S, Varet H, et al. Revisiting the classification of clinical phenotypes of anti-neutrophil cytoplasmic antibody-associated vasculitis: a cluster analysis. Ann Rheum Dis. 2013;72(6):1003-1010. PubMed
11. Holle JU, Gross WL, Latza U, et al. Improved outcome in 445 patients with Wegener’s granulomatosis in a German vasculitis center over four decades. Arthritis Rheum. 2011;63(1):257-266. PubMed
12. Cordier JF, Valeyre D, Guillevin L, Loire R, Brechot JM. Pulmonary Wegener’s granulomatosis. A clinical and imaging study of 77 cases. Chest. 1990;97(4):906-912. PubMed
13. Hamadah AM, Gharaibeh K, Mara KC, et al. Urinalysis for the diagnosis of glomerulonephritis: role of dysmorphic red blood cells. Nephrol Dial Transplant. 2018;33(8):1397-1403. PubMed
14. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol. 2014;10(8):463-473. PubMed
15. Borner U, Landis BN, Banz Y, et al. Diagnostic value of biopsies in identifying cytoplasmic antineutrophil cytoplasmic antibody-negative localized Wegener’s granulomatosis presenting primarily with sinonasal disease. Am J Rhinol Allergy. 2012;26(6):475-480. PubMed
16. Mahr A, Batteux F, Tubiana S, et al. Brief report: prevalence of antineutrophil cytoplasmic antibodies in infective endocarditis. Arthritis Rheumatol. 2014;66(6):1672-1677. PubMed
17. Sherkat R, Mostafavizadeh K, Zeydabadi L, Shoaei P, Rostami S. Antineutrophil cytoplasmic antibodies in patients with pulmonary tuberculosis. Iran J Immunol. 2011;8(1):52-57. PubMed
A 65-year-old man was transferred to a tertiary academic medical center with one week of progressive shortness of breath, dry cough, and fevers. He reported no weight loss or night sweats but had experienced mild right upper quadrant pain and anorexia for the preceding three weeks. Several years had passed since he had consulted a physician, and he did not take any medications. He immigrated to the United States from Mexico four decades prior. He traveled back frequently to visit his family, most recently one month before his presentation. He worked as a farming supervisor in the Central Valley of California. He smoked tobacco and had a 30 pack-year history. He drank alcohol occasionally and denied any drug use.
Causes of subacute cough and dyspnea include bronchitis, pneumonia, heart failure, and asthma. Pneumonia and heart failure might cause right upper quadrant pain from diaphragmatic irritation and hepatic congestion, respectively. Metastatic cancer or infection may lead to synchronous pulmonary and hepatic involvement. The patient is at increased risk of lung cancer, given his extensive smoking history.
The patient’s place of residence in the Southwestern United States places him at risk of respiratory illness from coccidioidomycosis. His exact involvement with animals and their products should be further explored. For example, consumption of unpasteurized milk might result in pneumonia, hepatitis, or both from M. bovis, Brucella species, or C. burnetii. His travel to Mexico prompts consideration of tuberculosis, histoplasmosis, and paracoccidiomycosis as causes of respiratory and possible hepatic illness.
Two weeks prior, the patient had initially presented to another hospital with one week of intermittent right upper quadrant pain unrelated to eating. An abdominal ultrasound and hepatobiliary iminodiacetic acid (HIDA) scan were normal. Computed tomography (CT) of the chest, abdomen, and pelvis with contrast demonstrated a left upper lobe lung mass measuring 5.5 × 4.4 × 3.7 cm3 and scattered right-sided pulmonary nodules (Figure 1). He underwent CT-guided biopsy of the mass and was discharged with a presumed diagnosis of primary pulmonary malignancy with plans for outpatient follow-up.
Over the next four days, the patient developed progressive dyspnea with cough and subjective fevers. The patient was readmitted with a diagnosis of postobstructive pneumonia and acute kidney injury (creatinine increased from 0.7 mg/dL to 2.9 mg/dL between admissions), and this finding was attributed to contrast-induced nephropathy from his recent CT scan. He was treated with vancomycin and piperacillin/tazobactam for two days but wished to transfer to a tertiary care hospital for a second opinion.
Postobstructive pneumonia, pulmonary embolism, and pleural effusion are common causes of dyspnea in patients with lung cancer. The patient’s travel and occupational history, lung nodules, acute renal insufficiency, and rapidly progressive respiratory symptoms prompt consideration for radiographic mimickers of lung cancer. Tuberculosis might present as a lung mass (pulmonary tuberculoma) during primary infection or reactivation. Noninfectious causes of pulmonary masses and nodules include metastatic cancer (eg, colon cancer), sarcoidosis, IgG4-related disease, and granulomatous polyangiitis (GPA).
Contrast-induced nephropathy is unusual in patients with normal renal function. More probable explanations include hypovolemia or acute tubular necrosis (ATN) from underlying inflammation. The patient’s CT-negative right upper quadrant pain may be a distinct process or represent another facet of a disseminated illness such as hepatic infiltration from lymphoma.
Upon arrival, the patient’s temperature was 38°C, heart rate (HR) 107 beats per minute, blood pressure (BP) 159/89 mm Hg, respiratory rate 25 breaths per minute, and oxygen saturation 92% on 2 L of oxygen per minute. He showed no signs of distress. Mild scleral icterus was noted. The cardiac exam was normal. Auscultation revealed scattered wheezes and crackles in the left upper lobe. Mild right upper quadrant tenderness without hepatosplenomegaly was noted on the abdominal exam. The patient’s lower extremities exhibited bilateral trace edema. No rash was observed, and his neurologic exam was normal.
The white blood cell (WBC) count was 28,300 per cubic millimeter (87% neutrophils, 3.6% lymphocytes, and 0.03% eosinophils), hemoglobin 11.1 g per deciliter, and platelet count 789,000 per cubic millimeter. Sodium was 127 mmol per liter, potassium 4.6 mmol per liter, chloride 101 mmol per liter, bicarbonate 13 mmol per liter, blood urea nitrogen 60 mg per deciliter, and creatinine 3.4 mg per deciliter. Aspartate aminotransferase and alanine aminotransferase levels were normal. Alkaline phosphatase was 283 units per liter (normal range, 31-95), and total bilirubin was 4.5 mg per deciliter (normal range, 0.2-1.3) with a direct bilirubin of 2.7 mg per deciliter. Urinalysis demonstrated urine protein of 30 mg/dL, specific gravity of 1.013, negative nitrites, 10-21 white cells per high-powered field (normal, < 5), and 21-50 red cells per high-powered field (normal, < 3). Urine microscopy revealed muddy brown casts but no cellular casts or dysmorphic red cells. A chest radiograph (CXR) showed patchy consolidations in the bilateral upper lobes and left lower lobe along with Kerley B lines, a small left pleural effusion, and thickened right horizontal fissure; the left upper lobe mass was re-demonstrated. Vancomycin, piperacillin-tazobactam, and azithromycin were administered.
At this point, the most likely source of sepsis is multifocal pneumonia. The patient is at risk for S. aureus and P. aeruginosa given his recent hospitalization. A severe form of leptospirosis (Weil’s disease) is associated with pulmonary disease, hyperbilirubinemia, and renal failure. Repeat abdominal imaging is necessary to evaluate for cholangitis given the patient’s right upper quadrant pain, fever, and jaundice. It would also help categorize his cholestatic pattern of liver injury as intrahepatic or extrahepatic (eg, stricture). An infiltrative disease such as sarcoidosis may cause both intrahepatic cholestasis and parenchymal lung disease, although the pleural pathology is uncommon.
His normal cardiac exam does not exclude cardiogenic pulmonary edema, a common cause of interstitial edema and pleural effusion. In this setting of systemic inflammation (neutrophilia, thrombocytosis, and hypoalbuminemia), the thickened right horizontal fissure and interlobular septa might represent an infiltrative process, such as lymphangitic carcinomatosis, lymphoma, or sarcoidosis.
Muddy brown casts are characteristic of ATN. The patient’s risk factors for ATN include sepsis and previously administered iodinated contrast. Fluid retention from oliguric renal failure is likely contributing to his hyponatremia and lower extremity edema. Pathology isolated to the tubules, however, would not cause hematuria and pyuria and suggests glomerular or interstitial disease. The lack of cellular casts on a single urinary specimen does not eliminate the likelihood of either disease. Hematuria and diffuse parenchymal lung disease prompt consideration of pulmonary-renal syndromes, such as anti-glomerular basement membrane disease, GPA, and systemic lupus erythematosus, which can all be triggered by infection.
On the night of transfer, the patient experienced acute respiratory distress. Heart rate was 130 beats per minute, BP 170/95 mm Hg, respiratory rate 40 breaths per minute, and oxygen saturation 88% on six liters of supplemental oxygen by nasal cannula. His arterial blood gas demonstrated a pH of 7.23, PaCO2 of 32 mm Hg, and PaO2 of 65 mm Hg. He was emergently intubated for progressive hypoxemic respiratory failure. A small amount of blood was noted in the endotracheal tube. A noncontrast CT of the chest demonstrated multifocal airspace opacities and bilateral pleural effusions. The previously noted left upper lobe mass was unchanged.
Rapid respiratory decline and diffuse alveolar disease commonly result from aspiration, flash pulmonary edema, and acute respiratory distress syndrome (ARDS). Necrotizing pneumonia (eg, S. aureus) and trauma during intubation are possible causes of blood in his endotracheal tube. However, in the setting of multifocal airspace opacity, renal insufficiency, hematuria, and rapid respiratory decline, the blood might represent diffuse alveolar hemorrhage (DAH). Bronchoscopy with bronchioalveolar lavage to evaluate for pulmonary edema, infection, and hemorrhage would be indicated.
The patient subsequently developed oliguria, requiring continuous renal replacement therapy. An echocardiogram demonstrated impaired left ventricular relaxation and a reduced ejection fraction of 45% without segmental wall motion abnormalities or valvular disease, and a right ventricular systolic pressure of 36 mm Hg. Over the next 12 hours, his respiratory status improved, and he was extubated to 15 L per minute of supplemental oxygen by high-flow nasal cannula (HFNC).
The pathology report of the lung biopsy from the other hospital disclosed chronic inflammation and fibrosis with ill-defined areas of necrosis and myxoid degeneration surrounded by nuclear palisading suggestive of granulomatous inflammation. Staining for acid-fast bacilli (AFB) and fungal organisms was negative.
The rapid pulmonary recovery is inconsistent with multifocal pneumonia or ARDS. Flash pulmonary edema might result in sudden hypoxemic respiratory failure that resolves with positive pressure ventilation and ultrafiltration. However, this condition would not explain the biopsy results. Granulomatous lung pathology often results from mycobacterial or fungal disease. Tuberculosis and fungal pneumonia are not excluded with negative staining alone. However, neither would cause self-limited respiratory failure. Histologic evidence of necrosis lessens the likelihood of sarcoidosis, which rarely causes fulminant pulmonary disease. Lymphoma can result in granulomatous inflammation but would not cause transient pulmonary disease. GPA, a cause of necrotizing granulomatous lung disease, might result in a lung mass and worsened hypoxemia through DAH.
The patient continued to require 15 L of oxygen per minute by HFNC. He had persistent bilateral perihilar alveolar and interstitial opacities on CXR. Repeat WBC count was 29,200 per cubic millimeter, hemoglobin 7.8 g per deciliter, and platelets 656,000 per cubic millimeter. The C-reactive protein was 300 mg per L (normal range, <6.3) and erythrocyte sedimentation rate 100 mm per hour (normal range, <10). Legionella urinary antigen, serum immunodiffusion for Coccidiodes imitus, human immunodeficiency virus antibody, respiratory viral panel, and beta-D glucan were negative. Rare acid-fast bacilli were visualized in one out of three concentrated AFB sputum smears. He was started on empiric antituberculous therapy with rifampin, isoniazid, pyrazinamide, and ethambutol.
The sputum sample is suggestive of pulmonary tuberculosis. The salient features of this case include systemic inflammation, pulmonary nodules and mass, necrotizing granulomatous lung pathology, renal insufficiency, and hematuria. Disseminated tuberculosis might explain all these findings. However, a positive AFB smear may signal the presence of a nontuberculous mycobacteria, which is less likely to cause this clinical syndrome.
M. tuberculosis complex polymerase chain reaction (MTB PCR) assay returned negative for M. tuberculosis. Antiproteinase 3 antibody was 1,930 units (normal range, <20). Antimyeloperoxidase and antiglomerular basement membrane antibodies were negative.
Tuberculosis and GPA share several overlapping features, such as necrotizing lung pathology and less commonly antineutrophil cytoplasmic autoantibody (ANCA)-associated antibodies. However, the lung mass, acute renal and respiratory failure, hematuria, and the degree of anti-proteinase 3 level elevation are highly suggestive of GPA. The negative MTB PCR raises the possibility that a nontuberculous mycobacterium was detected on the sputum smear. Nevertheless, continued treatment until finalization of culture results is appropriate given that tuberculosis is endemic in Mexico.
The patient’s presenting features of right upper quadrant tenderness, jaundice, and cholestatic hepatitis remain poorly explained by either of these diagnoses. Neither tuberculosis nor GPA commonly presents with accompanying hepatic involvement, though both have been occasionally described as causing hepatitis. As the greatest concern in this patient remains his progressive renal failure and accompanying pulmonary hemorrhage, a renal biopsy to assess for glomerulonephritis associated with GPA is warranted before further investigation into the cause of his cholestatic hepatitis.
A core renal biopsy demonstrated pauci-immune focal crescentic and necrotizing glomerulonephritis with mixed tubulointerstitial inflammation (Figure 2). In conjunction with the pulmonary syndrome and positive antiproteinase 3 serology, a diagnosis of granulomatosis with polyangiitis was made. The patient was treated with pulse dose steroids, rituximab, and plasma exchange. Two weeks later, the sputum mycobacterial culture returned positive for Mycobacterium llatzerense and anti-tuberculous treatment was discontinued.
Over the following weeks, the patient improved and was transitioned off dialysis prior to hospital discharge. By six months later, he had resolution of his hemoptysis, shortness of breath, liver biochemical test abnormalities, and significant improvement in his renal function. Repeat sputum mycobacterial cultures were negative.
DISCUSSION
A 65-year-old man from Mexico with a significant smoking history presented with an apical lung mass and cough, prioritizing tuberculosis and pulmonary malignancy. As the case unfolded, renal failure, multifocal lung opacities, conflicting tuberculosis test results, positive anti-proteinase 3 antibody, and ultimately a renal biopsy led to the diagnosis of granulomatosis with polyangiitis (GPA).
The correct interpretation of occasionally conflicting mycobacterial testing is crucial. Mycobacterial cultures remain the gold standard for diagnosing tuberculosis. However, results take weeks to return. Rapid tests include acid-fast bacilli (AFB) smear microscopy and nucleic acid-amplification tests (NAAT) of sputum or bronchoalveolar samples.1 When three sputum smears are performed, the sensitivity of AFB smear microscopy for tuberculosis in immunocompetent hosts is 70%.1 The AFB smear does not distinguish between different mycobacterial organisms. Thus, a positive result must be interpreted with the relative prevalence of tuberculosis and nontuberculous mycobacteria (NTM) in mind. The addition of NAAT-based assays has allowed for enhanced sensitivity and specificity in the diagnosis of tuberculosis, such that a negative NAAT in a patient with a positive AFB smear strongly argues for the presence of a NTM.2-4
NTM are widely prevalent environmental microbes, with over 140 species described, and careful consideration is required to determine if an isolate is pathogenic.5 Given their ubiquitous nature, a high rate of asymptomatic respiratory and cutaneous colonization occurs. Correspondingly, the diagnosis of NTM disease requires multiple positive cultures or pathologic features on tissue biopsy, compatible clinical findings, and diligent exclusion of other causes.5 A retrospective study of all NTM isolates in Oregon from 2005-2006 revealed that only 47% of patients met the guideline criteria for having symptomatic NTM disease.6 In our case, the patient’s sputum grew M. llatzerense, an aerobic, nonfermenting mycobacterium found in water sources that has only infrequently been implicated as a human pathogen.7,8 Subsequent AFB sputum cultures were negative, and serial imaging showed resolution of the pulmonary findings without additional antimycobacterial therapy, suggesting that this organism was not responsible for the disease process.
Along with microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA), GPA is an antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis that predominantly affects small to medium sized vessels. Although it can occur at any age, GPA most commonly afflicts older adults, with men and women being diagnosed at roughly equal rates.9 GPA is a multisystem disease with a wide array of clinical manifestations. The most frequently involved sites of disease are the respiratory tract and kidneys, although virtually any organ can be affected. Sino-nasal disease, such as destructive sinusitis, or ear involvement are nearly universal. Lower respiratory manifestations occur in 60% of patients, but are highly diverse and reflect the inherent difficulty in diagnosing this condition.9-11 Additionally, GPA is a frequent cause of the pulmonary-renal syndromes, with glomerulonephritis occurring in 80% of patients.9
The diagnosis of GPA in this case was delayed, in part, due to features suggestive of malignancy and pulmonary tuberculosis. While sino-nasal disease was not noted during this hospitalization, the patient had many different respiratory manifestations, including a dominant pulmonary mass, diffuse nodules, and hypoxemic respiratory failure due to suspected diffuse alveolar hemorrhage (DAH), all of which have been reported in GPA.12 Dysmorphic red cells and red blood cell casts are not sensitive for renal involvement in GPA; their absence does not exclude the possibility of an ANCA-associated vasculitis.13 Hematuria and rapid progression to oliguric renal failure are characteristic of a vasculitic process and should sway clinicians away from a working diagnosis of ATN.
The diagnosis of GPA involves the synthesis of clinical data, radiographic findings, serologic testing, and histopathology. ANCA testing is an essential step in the diagnosis of GPA but has limitations. Patients with GPA more commonly have ANCAs targeting the enzyme proteinase-3 (PR3-ANCA), with MPA being more closely associated with myeloperoxidase (MPO-ANCA), although cross-reactivity and antibody-negative disease can occur.14 Although 90% of patients with GPA with multiorgan involvement will have a positive ANCA, a negative test is more common in localized upper airway disease, where only 50% have a positive ANCA.15 A number of drugs, medications, infections, and nonvasculitic autoimmune diseases have been associated with positive ANCA serologies in the absence of systemic vasculitis.14,16,17 As such, pathologic demonstration of vasculitis is necessary for establishing the diagnosis. Typical sites for biopsy include the kidneys and lungs.9
This case illustrates how clinicians often find themselves at a diagnostic crossroads—being forced to choose which clinical elements to prioritize. At various points, our patient’s presentation could have been framed as “a man from a Tb-endemic country with hemoptysis and an apical opacity,” “an elderly man with extensive smoking history and lung mass,” or “a patient with elevated inflammatory markers and pulmonary-renal syndrome”. In such situations, it is incumbent on the clinician to evaluate how well a given problem representation encompasses or highlights the salient features of a case. As with painting or photography, an essential aspect of appreciating the whole picture involves carefully selecting the right frame.
KEY TEACHING POINTS
- The diagnosis of tuberculosis relies on smear microscopy, nucleic-acid amplification testing (NAAT), and cultures. A positive AFB smear with negative NAAT suggests the presence of a nontuberculous mycobacteria (NTM).
- NTM are common environmental organisms and often exist as nonpathogenic colonizers.6 The diagnosis of NTM disease requires exclusion of other causes and careful clinical, microbiologic, and radiographic correlation.
- Granulomatosis with polyangiitis is a multisystem disease often involving the respiratory track and kidney. Pulmonary disease can present with airway involvement, parenchymal nodules, opacities, pleural findings, and diffuse alveolar hemorrhage.12
Disclosures
Drs. Minter, Geha, Boslett, Chung, and Ramani have no disclosures. Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME).
A 65-year-old man was transferred to a tertiary academic medical center with one week of progressive shortness of breath, dry cough, and fevers. He reported no weight loss or night sweats but had experienced mild right upper quadrant pain and anorexia for the preceding three weeks. Several years had passed since he had consulted a physician, and he did not take any medications. He immigrated to the United States from Mexico four decades prior. He traveled back frequently to visit his family, most recently one month before his presentation. He worked as a farming supervisor in the Central Valley of California. He smoked tobacco and had a 30 pack-year history. He drank alcohol occasionally and denied any drug use.
Causes of subacute cough and dyspnea include bronchitis, pneumonia, heart failure, and asthma. Pneumonia and heart failure might cause right upper quadrant pain from diaphragmatic irritation and hepatic congestion, respectively. Metastatic cancer or infection may lead to synchronous pulmonary and hepatic involvement. The patient is at increased risk of lung cancer, given his extensive smoking history.
The patient’s place of residence in the Southwestern United States places him at risk of respiratory illness from coccidioidomycosis. His exact involvement with animals and their products should be further explored. For example, consumption of unpasteurized milk might result in pneumonia, hepatitis, or both from M. bovis, Brucella species, or C. burnetii. His travel to Mexico prompts consideration of tuberculosis, histoplasmosis, and paracoccidiomycosis as causes of respiratory and possible hepatic illness.
Two weeks prior, the patient had initially presented to another hospital with one week of intermittent right upper quadrant pain unrelated to eating. An abdominal ultrasound and hepatobiliary iminodiacetic acid (HIDA) scan were normal. Computed tomography (CT) of the chest, abdomen, and pelvis with contrast demonstrated a left upper lobe lung mass measuring 5.5 × 4.4 × 3.7 cm3 and scattered right-sided pulmonary nodules (Figure 1). He underwent CT-guided biopsy of the mass and was discharged with a presumed diagnosis of primary pulmonary malignancy with plans for outpatient follow-up.
Over the next four days, the patient developed progressive dyspnea with cough and subjective fevers. The patient was readmitted with a diagnosis of postobstructive pneumonia and acute kidney injury (creatinine increased from 0.7 mg/dL to 2.9 mg/dL between admissions), and this finding was attributed to contrast-induced nephropathy from his recent CT scan. He was treated with vancomycin and piperacillin/tazobactam for two days but wished to transfer to a tertiary care hospital for a second opinion.
Postobstructive pneumonia, pulmonary embolism, and pleural effusion are common causes of dyspnea in patients with lung cancer. The patient’s travel and occupational history, lung nodules, acute renal insufficiency, and rapidly progressive respiratory symptoms prompt consideration for radiographic mimickers of lung cancer. Tuberculosis might present as a lung mass (pulmonary tuberculoma) during primary infection or reactivation. Noninfectious causes of pulmonary masses and nodules include metastatic cancer (eg, colon cancer), sarcoidosis, IgG4-related disease, and granulomatous polyangiitis (GPA).
Contrast-induced nephropathy is unusual in patients with normal renal function. More probable explanations include hypovolemia or acute tubular necrosis (ATN) from underlying inflammation. The patient’s CT-negative right upper quadrant pain may be a distinct process or represent another facet of a disseminated illness such as hepatic infiltration from lymphoma.
Upon arrival, the patient’s temperature was 38°C, heart rate (HR) 107 beats per minute, blood pressure (BP) 159/89 mm Hg, respiratory rate 25 breaths per minute, and oxygen saturation 92% on 2 L of oxygen per minute. He showed no signs of distress. Mild scleral icterus was noted. The cardiac exam was normal. Auscultation revealed scattered wheezes and crackles in the left upper lobe. Mild right upper quadrant tenderness without hepatosplenomegaly was noted on the abdominal exam. The patient’s lower extremities exhibited bilateral trace edema. No rash was observed, and his neurologic exam was normal.
The white blood cell (WBC) count was 28,300 per cubic millimeter (87% neutrophils, 3.6% lymphocytes, and 0.03% eosinophils), hemoglobin 11.1 g per deciliter, and platelet count 789,000 per cubic millimeter. Sodium was 127 mmol per liter, potassium 4.6 mmol per liter, chloride 101 mmol per liter, bicarbonate 13 mmol per liter, blood urea nitrogen 60 mg per deciliter, and creatinine 3.4 mg per deciliter. Aspartate aminotransferase and alanine aminotransferase levels were normal. Alkaline phosphatase was 283 units per liter (normal range, 31-95), and total bilirubin was 4.5 mg per deciliter (normal range, 0.2-1.3) with a direct bilirubin of 2.7 mg per deciliter. Urinalysis demonstrated urine protein of 30 mg/dL, specific gravity of 1.013, negative nitrites, 10-21 white cells per high-powered field (normal, < 5), and 21-50 red cells per high-powered field (normal, < 3). Urine microscopy revealed muddy brown casts but no cellular casts or dysmorphic red cells. A chest radiograph (CXR) showed patchy consolidations in the bilateral upper lobes and left lower lobe along with Kerley B lines, a small left pleural effusion, and thickened right horizontal fissure; the left upper lobe mass was re-demonstrated. Vancomycin, piperacillin-tazobactam, and azithromycin were administered.
At this point, the most likely source of sepsis is multifocal pneumonia. The patient is at risk for S. aureus and P. aeruginosa given his recent hospitalization. A severe form of leptospirosis (Weil’s disease) is associated with pulmonary disease, hyperbilirubinemia, and renal failure. Repeat abdominal imaging is necessary to evaluate for cholangitis given the patient’s right upper quadrant pain, fever, and jaundice. It would also help categorize his cholestatic pattern of liver injury as intrahepatic or extrahepatic (eg, stricture). An infiltrative disease such as sarcoidosis may cause both intrahepatic cholestasis and parenchymal lung disease, although the pleural pathology is uncommon.
His normal cardiac exam does not exclude cardiogenic pulmonary edema, a common cause of interstitial edema and pleural effusion. In this setting of systemic inflammation (neutrophilia, thrombocytosis, and hypoalbuminemia), the thickened right horizontal fissure and interlobular septa might represent an infiltrative process, such as lymphangitic carcinomatosis, lymphoma, or sarcoidosis.
Muddy brown casts are characteristic of ATN. The patient’s risk factors for ATN include sepsis and previously administered iodinated contrast. Fluid retention from oliguric renal failure is likely contributing to his hyponatremia and lower extremity edema. Pathology isolated to the tubules, however, would not cause hematuria and pyuria and suggests glomerular or interstitial disease. The lack of cellular casts on a single urinary specimen does not eliminate the likelihood of either disease. Hematuria and diffuse parenchymal lung disease prompt consideration of pulmonary-renal syndromes, such as anti-glomerular basement membrane disease, GPA, and systemic lupus erythematosus, which can all be triggered by infection.
On the night of transfer, the patient experienced acute respiratory distress. Heart rate was 130 beats per minute, BP 170/95 mm Hg, respiratory rate 40 breaths per minute, and oxygen saturation 88% on six liters of supplemental oxygen by nasal cannula. His arterial blood gas demonstrated a pH of 7.23, PaCO2 of 32 mm Hg, and PaO2 of 65 mm Hg. He was emergently intubated for progressive hypoxemic respiratory failure. A small amount of blood was noted in the endotracheal tube. A noncontrast CT of the chest demonstrated multifocal airspace opacities and bilateral pleural effusions. The previously noted left upper lobe mass was unchanged.
Rapid respiratory decline and diffuse alveolar disease commonly result from aspiration, flash pulmonary edema, and acute respiratory distress syndrome (ARDS). Necrotizing pneumonia (eg, S. aureus) and trauma during intubation are possible causes of blood in his endotracheal tube. However, in the setting of multifocal airspace opacity, renal insufficiency, hematuria, and rapid respiratory decline, the blood might represent diffuse alveolar hemorrhage (DAH). Bronchoscopy with bronchioalveolar lavage to evaluate for pulmonary edema, infection, and hemorrhage would be indicated.
The patient subsequently developed oliguria, requiring continuous renal replacement therapy. An echocardiogram demonstrated impaired left ventricular relaxation and a reduced ejection fraction of 45% without segmental wall motion abnormalities or valvular disease, and a right ventricular systolic pressure of 36 mm Hg. Over the next 12 hours, his respiratory status improved, and he was extubated to 15 L per minute of supplemental oxygen by high-flow nasal cannula (HFNC).
The pathology report of the lung biopsy from the other hospital disclosed chronic inflammation and fibrosis with ill-defined areas of necrosis and myxoid degeneration surrounded by nuclear palisading suggestive of granulomatous inflammation. Staining for acid-fast bacilli (AFB) and fungal organisms was negative.
The rapid pulmonary recovery is inconsistent with multifocal pneumonia or ARDS. Flash pulmonary edema might result in sudden hypoxemic respiratory failure that resolves with positive pressure ventilation and ultrafiltration. However, this condition would not explain the biopsy results. Granulomatous lung pathology often results from mycobacterial or fungal disease. Tuberculosis and fungal pneumonia are not excluded with negative staining alone. However, neither would cause self-limited respiratory failure. Histologic evidence of necrosis lessens the likelihood of sarcoidosis, which rarely causes fulminant pulmonary disease. Lymphoma can result in granulomatous inflammation but would not cause transient pulmonary disease. GPA, a cause of necrotizing granulomatous lung disease, might result in a lung mass and worsened hypoxemia through DAH.
The patient continued to require 15 L of oxygen per minute by HFNC. He had persistent bilateral perihilar alveolar and interstitial opacities on CXR. Repeat WBC count was 29,200 per cubic millimeter, hemoglobin 7.8 g per deciliter, and platelets 656,000 per cubic millimeter. The C-reactive protein was 300 mg per L (normal range, <6.3) and erythrocyte sedimentation rate 100 mm per hour (normal range, <10). Legionella urinary antigen, serum immunodiffusion for Coccidiodes imitus, human immunodeficiency virus antibody, respiratory viral panel, and beta-D glucan were negative. Rare acid-fast bacilli were visualized in one out of three concentrated AFB sputum smears. He was started on empiric antituberculous therapy with rifampin, isoniazid, pyrazinamide, and ethambutol.
The sputum sample is suggestive of pulmonary tuberculosis. The salient features of this case include systemic inflammation, pulmonary nodules and mass, necrotizing granulomatous lung pathology, renal insufficiency, and hematuria. Disseminated tuberculosis might explain all these findings. However, a positive AFB smear may signal the presence of a nontuberculous mycobacteria, which is less likely to cause this clinical syndrome.
M. tuberculosis complex polymerase chain reaction (MTB PCR) assay returned negative for M. tuberculosis. Antiproteinase 3 antibody was 1,930 units (normal range, <20). Antimyeloperoxidase and antiglomerular basement membrane antibodies were negative.
Tuberculosis and GPA share several overlapping features, such as necrotizing lung pathology and less commonly antineutrophil cytoplasmic autoantibody (ANCA)-associated antibodies. However, the lung mass, acute renal and respiratory failure, hematuria, and the degree of anti-proteinase 3 level elevation are highly suggestive of GPA. The negative MTB PCR raises the possibility that a nontuberculous mycobacterium was detected on the sputum smear. Nevertheless, continued treatment until finalization of culture results is appropriate given that tuberculosis is endemic in Mexico.
The patient’s presenting features of right upper quadrant tenderness, jaundice, and cholestatic hepatitis remain poorly explained by either of these diagnoses. Neither tuberculosis nor GPA commonly presents with accompanying hepatic involvement, though both have been occasionally described as causing hepatitis. As the greatest concern in this patient remains his progressive renal failure and accompanying pulmonary hemorrhage, a renal biopsy to assess for glomerulonephritis associated with GPA is warranted before further investigation into the cause of his cholestatic hepatitis.
A core renal biopsy demonstrated pauci-immune focal crescentic and necrotizing glomerulonephritis with mixed tubulointerstitial inflammation (Figure 2). In conjunction with the pulmonary syndrome and positive antiproteinase 3 serology, a diagnosis of granulomatosis with polyangiitis was made. The patient was treated with pulse dose steroids, rituximab, and plasma exchange. Two weeks later, the sputum mycobacterial culture returned positive for Mycobacterium llatzerense and anti-tuberculous treatment was discontinued.
Over the following weeks, the patient improved and was transitioned off dialysis prior to hospital discharge. By six months later, he had resolution of his hemoptysis, shortness of breath, liver biochemical test abnormalities, and significant improvement in his renal function. Repeat sputum mycobacterial cultures were negative.
DISCUSSION
A 65-year-old man from Mexico with a significant smoking history presented with an apical lung mass and cough, prioritizing tuberculosis and pulmonary malignancy. As the case unfolded, renal failure, multifocal lung opacities, conflicting tuberculosis test results, positive anti-proteinase 3 antibody, and ultimately a renal biopsy led to the diagnosis of granulomatosis with polyangiitis (GPA).
The correct interpretation of occasionally conflicting mycobacterial testing is crucial. Mycobacterial cultures remain the gold standard for diagnosing tuberculosis. However, results take weeks to return. Rapid tests include acid-fast bacilli (AFB) smear microscopy and nucleic acid-amplification tests (NAAT) of sputum or bronchoalveolar samples.1 When three sputum smears are performed, the sensitivity of AFB smear microscopy for tuberculosis in immunocompetent hosts is 70%.1 The AFB smear does not distinguish between different mycobacterial organisms. Thus, a positive result must be interpreted with the relative prevalence of tuberculosis and nontuberculous mycobacteria (NTM) in mind. The addition of NAAT-based assays has allowed for enhanced sensitivity and specificity in the diagnosis of tuberculosis, such that a negative NAAT in a patient with a positive AFB smear strongly argues for the presence of a NTM.2-4
NTM are widely prevalent environmental microbes, with over 140 species described, and careful consideration is required to determine if an isolate is pathogenic.5 Given their ubiquitous nature, a high rate of asymptomatic respiratory and cutaneous colonization occurs. Correspondingly, the diagnosis of NTM disease requires multiple positive cultures or pathologic features on tissue biopsy, compatible clinical findings, and diligent exclusion of other causes.5 A retrospective study of all NTM isolates in Oregon from 2005-2006 revealed that only 47% of patients met the guideline criteria for having symptomatic NTM disease.6 In our case, the patient’s sputum grew M. llatzerense, an aerobic, nonfermenting mycobacterium found in water sources that has only infrequently been implicated as a human pathogen.7,8 Subsequent AFB sputum cultures were negative, and serial imaging showed resolution of the pulmonary findings without additional antimycobacterial therapy, suggesting that this organism was not responsible for the disease process.
Along with microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA), GPA is an antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis that predominantly affects small to medium sized vessels. Although it can occur at any age, GPA most commonly afflicts older adults, with men and women being diagnosed at roughly equal rates.9 GPA is a multisystem disease with a wide array of clinical manifestations. The most frequently involved sites of disease are the respiratory tract and kidneys, although virtually any organ can be affected. Sino-nasal disease, such as destructive sinusitis, or ear involvement are nearly universal. Lower respiratory manifestations occur in 60% of patients, but are highly diverse and reflect the inherent difficulty in diagnosing this condition.9-11 Additionally, GPA is a frequent cause of the pulmonary-renal syndromes, with glomerulonephritis occurring in 80% of patients.9
The diagnosis of GPA in this case was delayed, in part, due to features suggestive of malignancy and pulmonary tuberculosis. While sino-nasal disease was not noted during this hospitalization, the patient had many different respiratory manifestations, including a dominant pulmonary mass, diffuse nodules, and hypoxemic respiratory failure due to suspected diffuse alveolar hemorrhage (DAH), all of which have been reported in GPA.12 Dysmorphic red cells and red blood cell casts are not sensitive for renal involvement in GPA; their absence does not exclude the possibility of an ANCA-associated vasculitis.13 Hematuria and rapid progression to oliguric renal failure are characteristic of a vasculitic process and should sway clinicians away from a working diagnosis of ATN.
The diagnosis of GPA involves the synthesis of clinical data, radiographic findings, serologic testing, and histopathology. ANCA testing is an essential step in the diagnosis of GPA but has limitations. Patients with GPA more commonly have ANCAs targeting the enzyme proteinase-3 (PR3-ANCA), with MPA being more closely associated with myeloperoxidase (MPO-ANCA), although cross-reactivity and antibody-negative disease can occur.14 Although 90% of patients with GPA with multiorgan involvement will have a positive ANCA, a negative test is more common in localized upper airway disease, where only 50% have a positive ANCA.15 A number of drugs, medications, infections, and nonvasculitic autoimmune diseases have been associated with positive ANCA serologies in the absence of systemic vasculitis.14,16,17 As such, pathologic demonstration of vasculitis is necessary for establishing the diagnosis. Typical sites for biopsy include the kidneys and lungs.9
This case illustrates how clinicians often find themselves at a diagnostic crossroads—being forced to choose which clinical elements to prioritize. At various points, our patient’s presentation could have been framed as “a man from a Tb-endemic country with hemoptysis and an apical opacity,” “an elderly man with extensive smoking history and lung mass,” or “a patient with elevated inflammatory markers and pulmonary-renal syndrome”. In such situations, it is incumbent on the clinician to evaluate how well a given problem representation encompasses or highlights the salient features of a case. As with painting or photography, an essential aspect of appreciating the whole picture involves carefully selecting the right frame.
KEY TEACHING POINTS
- The diagnosis of tuberculosis relies on smear microscopy, nucleic-acid amplification testing (NAAT), and cultures. A positive AFB smear with negative NAAT suggests the presence of a nontuberculous mycobacteria (NTM).
- NTM are common environmental organisms and often exist as nonpathogenic colonizers.6 The diagnosis of NTM disease requires exclusion of other causes and careful clinical, microbiologic, and radiographic correlation.
- Granulomatosis with polyangiitis is a multisystem disease often involving the respiratory track and kidney. Pulmonary disease can present with airway involvement, parenchymal nodules, opacities, pleural findings, and diffuse alveolar hemorrhage.12
Disclosures
Drs. Minter, Geha, Boslett, Chung, and Ramani have no disclosures. Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME).
1. Lewinsohn DM, Leonard MK, LoBue PA, et al. Official American Thoracic Society/Infectious Diseases Society of America/Centers for Disease Control and Prevention clinical practice guidelines: diagnosis of tuberculosis in adults and children. Clin Infect Dis. 2017;64(2):e1-e33. PubMed
2. Steingart KR, Sohn H, Schiller I, et al. Xpert(R) MTB/RIF assay for pulmonary tuberculosis and rifampicin resistance in adults. Cochrane Database Syst Rev. 2013;(1):Cd009593. PubMed
3. Luetkemeyer AF, Firnhaber C, Kendall MA, et al. Evaluation of Xpert MTB/RIF versus afb smear and culture to identify pulmonary tuberculosis in patients with suspected tuberculosis from low and higher prevalence settings. Clin Infect Dis. 2016;62(9):1081-1088. PubMed
4. Boehme CC, Nabeta P, Hillemann D, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med. 2010;363(11):1005-1015. PubMed
5. Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175(4):367-416. PubMed
6. Winthrop KL, McNelley E, Kendall B, et al. Pulmonary nontuberculous mycobacterial disease prevalence and clinical features: an emerging public health disease. Am J Respir Crit Care Med. 2010;182(7):977-982. PubMed
7. Teixeira L, Avery RK, Iseman M, et al. Mycobacterium llatzerense lung infection in a liver transplant recipient: case report and review of the literature. Am J Transplant. 2013;13(8):2198-2200. PubMed
8. Cárdenas AM, Gomila M, Lalucat J, Edelstein PH. Abdominal abscess caused by Mycobacterium llatzerense. J Clin Microbiol. 2014;52(4):1287-1289. PubMed
9. Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med. 1997;337(21):1512-1523. PubMed
10. Mahr A, Katsahian S, Varet H, et al. Revisiting the classification of clinical phenotypes of anti-neutrophil cytoplasmic antibody-associated vasculitis: a cluster analysis. Ann Rheum Dis. 2013;72(6):1003-1010. PubMed
11. Holle JU, Gross WL, Latza U, et al. Improved outcome in 445 patients with Wegener’s granulomatosis in a German vasculitis center over four decades. Arthritis Rheum. 2011;63(1):257-266. PubMed
12. Cordier JF, Valeyre D, Guillevin L, Loire R, Brechot JM. Pulmonary Wegener’s granulomatosis. A clinical and imaging study of 77 cases. Chest. 1990;97(4):906-912. PubMed
13. Hamadah AM, Gharaibeh K, Mara KC, et al. Urinalysis for the diagnosis of glomerulonephritis: role of dysmorphic red blood cells. Nephrol Dial Transplant. 2018;33(8):1397-1403. PubMed
14. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol. 2014;10(8):463-473. PubMed
15. Borner U, Landis BN, Banz Y, et al. Diagnostic value of biopsies in identifying cytoplasmic antineutrophil cytoplasmic antibody-negative localized Wegener’s granulomatosis presenting primarily with sinonasal disease. Am J Rhinol Allergy. 2012;26(6):475-480. PubMed
16. Mahr A, Batteux F, Tubiana S, et al. Brief report: prevalence of antineutrophil cytoplasmic antibodies in infective endocarditis. Arthritis Rheumatol. 2014;66(6):1672-1677. PubMed
17. Sherkat R, Mostafavizadeh K, Zeydabadi L, Shoaei P, Rostami S. Antineutrophil cytoplasmic antibodies in patients with pulmonary tuberculosis. Iran J Immunol. 2011;8(1):52-57. PubMed
1. Lewinsohn DM, Leonard MK, LoBue PA, et al. Official American Thoracic Society/Infectious Diseases Society of America/Centers for Disease Control and Prevention clinical practice guidelines: diagnosis of tuberculosis in adults and children. Clin Infect Dis. 2017;64(2):e1-e33. PubMed
2. Steingart KR, Sohn H, Schiller I, et al. Xpert(R) MTB/RIF assay for pulmonary tuberculosis and rifampicin resistance in adults. Cochrane Database Syst Rev. 2013;(1):Cd009593. PubMed
3. Luetkemeyer AF, Firnhaber C, Kendall MA, et al. Evaluation of Xpert MTB/RIF versus afb smear and culture to identify pulmonary tuberculosis in patients with suspected tuberculosis from low and higher prevalence settings. Clin Infect Dis. 2016;62(9):1081-1088. PubMed
4. Boehme CC, Nabeta P, Hillemann D, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med. 2010;363(11):1005-1015. PubMed
5. Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175(4):367-416. PubMed
6. Winthrop KL, McNelley E, Kendall B, et al. Pulmonary nontuberculous mycobacterial disease prevalence and clinical features: an emerging public health disease. Am J Respir Crit Care Med. 2010;182(7):977-982. PubMed
7. Teixeira L, Avery RK, Iseman M, et al. Mycobacterium llatzerense lung infection in a liver transplant recipient: case report and review of the literature. Am J Transplant. 2013;13(8):2198-2200. PubMed
8. Cárdenas AM, Gomila M, Lalucat J, Edelstein PH. Abdominal abscess caused by Mycobacterium llatzerense. J Clin Microbiol. 2014;52(4):1287-1289. PubMed
9. Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med. 1997;337(21):1512-1523. PubMed
10. Mahr A, Katsahian S, Varet H, et al. Revisiting the classification of clinical phenotypes of anti-neutrophil cytoplasmic antibody-associated vasculitis: a cluster analysis. Ann Rheum Dis. 2013;72(6):1003-1010. PubMed
11. Holle JU, Gross WL, Latza U, et al. Improved outcome in 445 patients with Wegener’s granulomatosis in a German vasculitis center over four decades. Arthritis Rheum. 2011;63(1):257-266. PubMed
12. Cordier JF, Valeyre D, Guillevin L, Loire R, Brechot JM. Pulmonary Wegener’s granulomatosis. A clinical and imaging study of 77 cases. Chest. 1990;97(4):906-912. PubMed
13. Hamadah AM, Gharaibeh K, Mara KC, et al. Urinalysis for the diagnosis of glomerulonephritis: role of dysmorphic red blood cells. Nephrol Dial Transplant. 2018;33(8):1397-1403. PubMed
14. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol. 2014;10(8):463-473. PubMed
15. Borner U, Landis BN, Banz Y, et al. Diagnostic value of biopsies in identifying cytoplasmic antineutrophil cytoplasmic antibody-negative localized Wegener’s granulomatosis presenting primarily with sinonasal disease. Am J Rhinol Allergy. 2012;26(6):475-480. PubMed
16. Mahr A, Batteux F, Tubiana S, et al. Brief report: prevalence of antineutrophil cytoplasmic antibodies in infective endocarditis. Arthritis Rheumatol. 2014;66(6):1672-1677. PubMed
17. Sherkat R, Mostafavizadeh K, Zeydabadi L, Shoaei P, Rostami S. Antineutrophil cytoplasmic antibodies in patients with pulmonary tuberculosis. Iran J Immunol. 2011;8(1):52-57. PubMed
© 2019 Society of Hospital Medicine