User login
Every Nook and Cranny
A 46-year-old man presented to the emergency room in the postmonsoon month of September with a seven-day history of high fevers as well as a four-day history of a dry cough, dyspnea, and progressive rash. The patient reported no chest pain, hemoptysis, chest tightness, palpitations, wheezing, orthopnea, paroxysmal nocturnal dyspnea, or leg swelling. He lived and sought healthcare in Delhi, India.
Fever followed by a progressive but as yet uncharacterized rash and pulmonary symptoms in a middle-aged man suggests a host of possibilities. While it is tempting to ascribe his symptoms to an infectious process, especially a “tropical” infection based on his residence in Delhi, the location may simply represent a red herring. Potential infections can be divided into those endemic to the Indian subcontinent, and those encountered more globally. The former include diseases such as measles and dengue, while the latter include entities such as Mycoplasma pneumonia, varicella, and acute human immunodeficiency virus (HIV) infection. Noninfectious categories of diseases that should be considered include drug reactions and rheumatologic processes. Several rheumatologic diseases, including granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis, and systemic lupus erythematosus (SLE) may present with fever, rash, and pulmonary symptomatology.
A history of the patient’s exposures, both environmental and pharmaceutical, should be obtained. More information regarding his immunization history, rash characteristics (distribution and nature of the lesions), and other salient exam findings such as organomegaly and joint abnormalities will be helpful.
Fever reached a maximum of 103° Fahrenheit and was associated with chills but not rigors. There were several fever spikes daily, relieved completely with antipyretics. The patient’s dyspnea was predominantly noted on exertion, nonpleuritic, not temporally related to cough, and progressively worsening over three days. The skin lesions were first noticed on his trunk and were described as reddish, flat, and pinpoint size. However, the rash spread to the face and extremities sparing the palms and soles. There was no bleeding, nausea, vomiting, abdominal pain, change in bowel habits, dysuria, headache, photophobia, neck stiffness, or joint pain.
The patient reported no significant past medical history, took no medications, and had no recent travel outside of Delhi, India in the past year. He was married and monogamous. He had no pets nor did he report any contact with animals. He did not use tobacco, alcohol, or illicit substances. He did not remember being bitten by an insect. He worked as a software engineer. There was no history of similar illness in the patient’s family or at his workplace. He had no history of recent blood transfusion or immunization (including MMR and Tdap).
Several noninfectious and inflammatory conditions can explain his symptoms. Eosinophilic granulomatosis with polyangiitis is considerably less likely in the absence of asthma, and vasculitic processes, in general, are less likely given the nongravity dependent nature of the rash. SLE and sarcoidosis are possible causes of a systemic inflammatory illness presenting acutely with fever, rash, and pulmonary symptoms.
The patient’s expanded history makes several infections less likely. Although much of the presentation is consistent with measles, the initial appearance of the truncal rash is atypical, and there is no mention of coryza or conjunctivitis. Likewise, the description of the exanthem is not suggestive of varicella, and dengue and chikungunya are much less likely in the absence of a headache and arthralgias. Other infections including leptospirosis and scrub typhus are possible, and both might be contracted in greater Delhi. Typhoid is another infectious syndrome endemic to the Indian subcontinent that should be considered. The presence of rash involving the face and extremities would be highly atypical, however; and the presence of dyspnea and the absence of a headache argue against typhoid. Acute HIV infection and Mycoplasma pneumonia remain possible diagnoses. Toxic shock syndrome is possible, but a faster and fulminant course would be expected.
On physical examination, the temperature was 103° Fahrenheit, heart rate was 120 beats per minute and regular, respiratory rate was 24 breaths per minute, blood pressure was 100/60 mm Hg, and resting oxygen saturation was 93% while breathing ambient air. He appeared uncomfortable. Jugular venous pulse was elevated at 10 cm H2O. Mild icterus was present, but there was neither conjunctival congestion nor subconjunctival hemorrhage. S1 and S2 heart sounds were loud, but there were no murmurs. Chest auscultation revealed bilateral basal coarse crackles. The abdominal right upper quadrant was mildly tender to palpation, and the liver edge was palpable 2 cm below the subcostal margin. There was neither splenomegaly nor peripheral lymphadenopathy. Kernig and Brudzinski signs were negative, and there were no focal neurological deficits. A generalized, nonpalpable, maculopapular and petechial rash was present on the face, extremities, and trunk.
The patient’s presentation must now incorporate the additional findings of bibasilar chest crackles, maculopapular/petechial rash, icterus, modest hypoxia, and hepatomegaly. Some of the noninfectious entities already mentioned (SLE and sarcoidosis) remain possible explanations. Hemophagocytic lymphohistiocytosis (HLH) may also explain most of the patient’s presenting signs and symptoms, and several other infectious diseases account for his presentation. Scrub typhus (or a more uncommon rickettsia disease, Indian tick typhus), leptospirosis, and perhaps infective endocarditis seem most likely to provide a unifying diagnosis for the symptoms mentioned above. Leptospirosis presents in a minority of instances as a severe illness known as Weil disease, characterized by several of this patient’s findings including icterus, kidney injury, and pulmonary symptoms. However, the rash is relatively uncommon in leptospirosis and when present, is usually more localized. The patient’s rash as described is not typically expected in infective endocarditis, although high-grade Staphylococcus aureus bacteremia will occasionally present with a diffuse rash that may be confused with that of meningococcemia. The etiology of the patient’s elevated jugular venous pressure is not readily apparent, with the cardiac examination making acute valvular insufficiency much less likely. Myocarditis, however, is possible in the setting of several of the diseases listed above, including leptospirosis, scrub typhus, SLE, and dengue.
In addition to basic laboratory studies and a chest radiograph, multiple sets of blood cultures should be obtained, along with a transthoracic echocardiogram and a ferritin level. The evidence to support leptospirosis and scrub typhus is strong enough to justify empiric use of doxycycline once the blood cultures are obtained, especially given the difficulty in definitively diagnosing these diseases in a timely fashion.
Laboratory analysis revealed a total leukocyte count of 13,600/uL (85% neutrophils), hemoglobin 10 g/dL, and platelet count 35,000/uL. Absolute eosinophil count was 136/uL. Serum chemistry showed sodium of 145 meq/L, potassium 4.1 meq/L, blood urea nitrogen 80 mg/dL, creatinine 1.6 mg/dL, aspartate transaminase (AST) 44 U/L (normal, 0-40), alanine transaminase (ALT) 81 U/L (normal, 0-40), direct bilirubin 3 mg/dL, and indirect bilirubin 3 mg/dL. Lactate dehydrogenase, alkaline phosphatase, albumin, and coagulation studies were normal. Erythrocyte sedimentation rate (ESR) was 42 mm (normal, 0-25) and highly sensitive C-reactive protein was 42 mg/L (normal, 0-10). Arterial blood gas on ambient air revealed a pH of 7.52, PaCO2 24 mm Hg, PaO2 55 mm Hg, and bicarbonate 20 meq/L. Urinalysis was normal. Blood cultures were obtained. Electrocardiogram (ECG) showed regular narrow complex tachycardia with incomplete left bundle branch block. Old ECGs were not available for comparison. Chest radiograph showed bilateral air space opacities with evidence of vein cephalization. Abdominal and pelvis ultrasonography showed pericholecystic fluid and mild hepatomegaly, but no free fluid, pleural effusion, or evidence of cholecystitis. Point of care immunochromatographic rapid malarial antigen detection test (detects Plasmodium falciparum, Plasmodium vivax, Plasmodium malaria, and Plasmodium ovale) was negative.
Most of the findings described are commonly observed in both scrub typhus and leptospirosis, including cytopenias, parenchymal infiltrates, hepatomegaly, elevated transaminases and bilirubin, cardiac involvement, fever, and rash. The rash described is more consistent with scrub typhus than with leptospirosis. The absence of a headache and joint findings argue modestly against these diagnoses. Likewise, HLH provides an adequate explanation for most of the patient’s symptoms, signs, and test results. These include fever, lung involvement, rash, hepatomegaly, elevated bilirubin, and cytopenias; however, leukocytosis and cardiac involvement are less characteristic. SLE also provides a satisfactory explanation for much of the symptoms, although the rash characteristics, normal urinalysis, and leukocytosis make this diagnosis less likely.
Additional testing that should be performed includes serum antinuclear antibody (ANA) and ferritin, since the latter may be markedly elevated in the setting of HLH. Bone marrow aspirate and biopsy should be performed looking specifically for evidence of hemophagocytosis. Finally, a transthoracic echocardiogram (TTE) should be performed to assess evidence of myocardial dysfunction as it may alter the therapeutic approach, although the results will be unlikely to differentiate between the preceding considerations.
Troponin I was negative, but N-terminal probrain natriuretic peptide was elevated at 20,000 pg/mL (normal, 0-900). D-dimer was negative. TTE showed left ventricular ejection fraction (LVEF) of 35% with global left ventricular hypokinesis. On three separate examinations, the peripheral blood smear did not show malarial parasites, atypical lymphocytes, or schistocytes. Three sets of blood cultures, testing for bacteria and fungi, were sterile. A throat culture was sterile. Widal test, as well as Leptospira and Mycoplasma serologies, were negative. Serology for Legionella pneumophila was positive, but the urinary antigen testing was negative. Antibodies to HIV 1 and 2 and anti-hepatitis C virus (HCV) antibody were negative. Dengue IgM ELISA (qualitative) returned positive.
Despite the absence of arthralgias, myalgias, headache, and retro-orbital pain, a positive dengue IgM ELISA supports acute dengue infection, provided the patient did not experience an unexplained febrile illness in the previous months. Most of his presentation may be explained by dengue, including fever, rash, liver abnormalities, myocardial dysfunction, and thrombocytopenia. The bilateral airspace opacities seen on chest radiograph also fit reasonably provided these actually reflect pulmonary edema. Leukocytosis (as opposed to leukopenia) is highly unexpected in dengue, but its presence could be an outlier.
If dengue does indeed explain the entire presentation, defervescence should have occurred by the time the blood cultures and serologic studies returned. Also, by that time, the patient would be expected to demonstrate evidence of improvement, barring the appearance of the serious complications of dengue hemorrhagic fever/dengue shock syndrome. Should fever persist and signs of recovery fail to materialize, the possibility of a superimposed process will need to be considered. Of note, the sensitivity of Leptospira serology early in the course of illness is low, and leptospirosis is thus not yet excluded.
A presumptive diagnosis of severe dengue fever was made, based on evidence of pulmonary edema and sepsis. The patient was managed conservatively with oral fluid restriction, low dose of diuretics, and supplemental oxygenation. The patient was also given levofloxacin for possible legionellosis. Despite these therapies, the patient had no improvement in 24 hours. His tachypnea increased, and his measured PaO2 to FIO2 (P:F) ratio decreased to 230 from 285 on admission. This prompted the initiation of BiPAP at 10 cm H2O inspiration PAP and 5 cm H2O expiration PAP. However, he did not tolerate BiPAP, and his P:F ratio decreased to below 200.
The patient was transferred to the intensive care unit and underwent elective intubation with mechanical ventilation. Axial and coronal computed tomography of the thorax (Figure 1A and 1B, respectively) showed extensive ground-glass opacities and consolidation sparing the nondependent portions of the lungs. On physical inspection, a round, well-defined, painless black lesion surrounded by erythema was noticed in the right axilla (Figure 2). The rest of the examination findings were unchanged.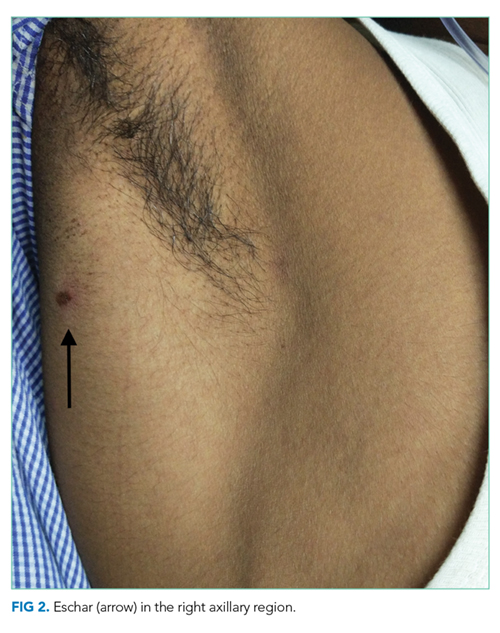
The discovery of eschar in the axilla provides a “pivot point” in determining the cause of the patient’s illness. This finding appears to point, with high specificity, toward rickettsia as the explanation of the patient’s disease, and this is most likely to be scrub typhus. The report of a positive dengue IgM may represent concurrent infection or may simply reflect a recent infection in an area that is highly endemic for dengue. Although most of the patient’s clinical presentation could be attributed to dengue, multiple features including the leukocytosis, myocarditis, and elevated bilirubin are more likely to be seen in scrub typhus. In any event, dengue cannot satisfactorily explain the eschar.
No mention has been made to the initiation of doxycycline thus far; this agent needs to be started promptly. Polymerase chain reaction (PCR) testing for scrub typhus should be ordered if available; if not, acute and convalescent serology may be obtained.
Given the finding of axillary eschar, the patient was diagnosed with scrub typhus. Doxycycline 100 mg by nasogastric tube twice a day was initiated. The patient began to show marked symptomatic improvement. His P:F ratio improved, and he was successfully weaned off and extubated after 24 hours. Postextubation, he was kept on BiPAP for 12 hours. He was transferred out of the ICU and monitored for 72 hours. With therapy, his cytopenias, liver and renal function, and ECG normalized. Indirect immunofluorescence assay for scrub typhus returned positive at a dilution of > 1:512. PCR assay targeting the 56 kDa region of Orientia tsutsugamushi was also positive. Repeated TTE showed an LVEF of 65%. He was subsequently discharged with oral doxycycline and a plan to complete a course of 14 days on an outpatient basis. The final diagnosis was scrub typhus with myocarditis leading to acutely decompensated heart failure with reduced ejection fraction.
DISCUSSION
Scrub typhus is a mite-borne tropical infection caused by the gram-negative intracellular parasite Orientia tsutsugamushi from the Rickettsiaceae family that is known to occur in certain parts of Asia and Australia. Although this entity is well known in the Sub Himalayan belt and southern part of India, very few cases have been described in Delhi, the capital state in North India. Scrub typhus, like most other tropical infections, is found most often during the postmonsoon season.1,2
Patients with scrub typhus present with fever in addition to a variety of nonspecific symptoms and findings. These often manifest within 10 days of being bitten by a mite. Malaise, headache, myalgias, lymphadenopathy, and maculopapular or petechial rash are common. If present, the rash manifests on the 3rd to 5th day of fever.3 Disseminated vasculitis due to scrub typhus can frequently result in multiorgan system involvement. Pulmonary involvement often leads to acute respiratory distress syndrome (ARDS) with an incidence of 8%-10%.1,4 Acute kidney injury, mostly mild and nonoliguric, has been reported in up to 2/3 cases.4-6 The cardiac myocyte is a known target cell affected by scrub typhus, and therefore patients commonly present with myocarditis.7 Liver involvement in scrub typhus is evident through elevated liver enzymes and can occur without other clinical evidence of the illness.4,6,8,9 As in dengue, patients often develop thrombocytopenia, but normal hemoglobin in scrub typhus differentiates it from dengue.6,8
Given the nonspecific presentation, it can be challenging to diagnose and treat scrub typhus. The gold standard for diagnosis is the detection of IgM antibodies to Orientia tsutsugamushi using an indirect immunofluorescence assay (IFA). For patients from endemic regions, it may be necessary to show a four-fold increase in titers two weeks apart to distinguish from background immunity. Presence of the characteristic eschar, as discussed below, is highly suggestive of scrub typhus. The treatment of choice is doxycycline or azithromycin for seven days.10,11 Early initiation of doxycycline when considering either scrub typhus or leptospirosis is appropriate and may be life-saving.
Medical decision making is fraught with uncertainty, and physicians must use their experience, evidence base, and cognitive heuristics wisely to care for patients effectively. For this patient, the region of Delhi experiences massive outbreaks of dengue every year during the time the patient presented to the hospital, whereas rickettsia infections are relatively uncommon. The clinical presentation was conceivably consistent with either dengue or scrub typhus, though somewhat more suggestive of the latter. Once the serological diagnosis of recent or concomitant dengue was obtained, however, scrub typhus was considered even less. The team called upon Occam’s razor or the heuristic that the simplest and most unifying explanation for any given problem is the one most likely to be correct and that other, less satisfactory explanations (in this case, scrub typhus) are “shaven off.” The patient was managed conservatively for dengue. Only when his condition worsened did the team recognize this conflicting information without dismissing it, consider alternative possibilities, and reexamined the patient.
An eschar can be an important clue in the diagnosis of scrub typhus, though it is not often obvious. The presence of this necrotic skin lesion with black crust is highly suggestive of scrub typhus, and in the right clinical context, it is virtually diagnostic. However, it is uncommon (9.5%-45%) in most of the studies from the Indian subcontinent (ie, high specificity but low sensitivity).1,12 An eschar is often found in obscure locations such as the axillae or groin, areas that may easily be missed or overlooked. Eschars may be seen in a variety of other infectious diseases, including rickettsia pox, Rocky Mountain spotted fever, other members of the spotted fever group, tularemia, and cutaneous anthrax. Given this patient’s lack of improvement, repeated examination revealed an eschar in the right axilla, a finding that was either missed or still evolving at the time of presentation.
This case illustrates the challenges in interpreting the significance of multiple positive serological tests in the context of an undifferentiated clinical syndrome. Possible reasons for a positive dengue serology could have been persistent antibodies from a previous infection, recent asymptomatic infection, concurrent infection, or cross-reactivity with flaviviruses such as West Nile Virus or Japanese Encephalitis.13 The patient also had positive IgM antibodies against Legionella pneumophila, but the urinary antigen was negative. In view of a negative antigen test, low specificity of the serologic test, low incidence of legionellosis in the Indian subcontinent, and absence of therapeutic response to a trial of fluoroquinolones, the diagnosis of legionellosis was considered unlikely in this patient.
With rapid advancements in technology, the importance of history taking and physical examination is at risk of being overshadowed. Approximately 80% of correct diagnoses in medicine can arrive through history and physical examination alone.14,15 In this case, Occam’s razor combined with multiple serological tests was relied on to create the likely list of diagnoses. However, recognition of the limitations of these heuristics and tests proved critical. The life-saving diagnosis was only made when the clinicians returned to basics, looked in every nook and cranny, and found the eschar on physical examination.
KEY TEACHING POINTS
- In patients living in endemic areas who present with an acute febrile illness, the differential diagnosis should include “tropical” infections such as dengue, chikungunya, enteric fever, leptospirosis, malaria, and scrub typhus.
- Serology is commonly employed for diagnosis of tropical infections, which may be misleading. These tests can be falsely positive from past asymptomatic infection or cross reactivity between antibodies, or falsely negative, as in the first few days of infection.
- Presence of eschar is a very useful clue in the diagnosis of scrub typhus, but this finding can be missed since it is often found in obscure locations. A thorough clinical history and physical examination are paramount.
Disclosures
The authors do not report any conflict of interest.
1. Gupta N, Chaudhry R, Kabra SK, et al. In search of scrub typhus: a prospective analysis of clinical and epidemiological profile of patients from a tertiary care hospital in New Delhi. Adv Infect Dis. 2015;5(4):140. doi: 10.4236/aid.2015.54017.
2. Kamarasu K, Malathi M, Rajagopal V, Subramani K, Jagadeeshramasamy D, Mathai E. Serological evidence for the wide distribution of spotted fevers & typhus fever in Tamil Nadu. Indian J Med Res. 2007;126(2):128-130. PubMed
3. Mahajan SK. Scrub typhus. J Assoc Physicians India. 2005;53:954-958. PubMed
4. Mahajan SK, Rolain JM, Kashyap R, et al. Scrub typhus in the Himalayas. Emerg Infect Dis. 2006;12(10):1590-1592. doi: 10.3201/eid1210.051697. PubMed
5. Attur RP, Kuppasamy S, Bairy M, et al. Acute kidney injury in scrub typhus. Clin Exp Nephrol. 2013;17(5):725-729. doi: 10.1007/s10157-012-0753-9. PubMed
6. Varghese GM, Trowbridge P, Janardhanan J, et al. Clinical profile and improving mortality trend of scrub typhus in South India. Int J Infect Dis. 2014;23:39-43. doi: 10.1016/j.ijid.2014.02.009. PubMed
7. Taylor AJ, Paris DH, Newton PN. A systematic review of mortality from untreated scrub typhus (Orientia tsutsugamushi). PLoS Negl Trop Dis. 2015;9(8):e0003971. doi.org/10.1371/journal.pntd.0003971 PubMed
8. Chrispal A, Boorugu H, Gopinath KG, et al. Scrub typhus: an unrecognized threat in South India-clinical profile and predictors of mortality. Trop Doct. 2010;40(3):129-133. doi: 10.1258/td.2010.090452. PubMed
9. Mathai E, Rolain JM, Verghese GM, et al. Outbreak of scrub typhus in southern India during the cooler months. Ann N Y Acad Sci. 2003;990:359-364. doi: 10.1111/j.1749-6632.2003.tb07391.x PubMed
10. Gupta N, Chaudhry R, Kabra SK, et al. Comparative evaluation of serological and molecular methods for the diagnosis of scrub typhus in Indian settings. Jpn J Infect Dis. 2017;70(2):221-222. doi: 10.7883/yoken.JJID.2016.139. PubMed
11. Rahi M, Gupte MD, Bhargava A, Varghese GM, Arora R. DHR-ICMR Guidelines for diagnosis & management of Rickettsial diseases in India. Indian J Med Res. 2015;141(4):417-422. doi: 10.4103/0971-5916.159279. PubMed
12. Sharma A, Mahajan S, Gupta ML, Kanga A, Sharma V. Investigation of an outbreak of scrub typhus in the Himalayan region of India. Jpn J Infect Dis. 2005;58(4):208-210. PubMed
13. Gupta N, Chaudhry R, Mirdha B, et al. Scrub typhus and leptospirosis: the fallacy of diagnosing with IgM enzyme-linked immunosorbent assay. J Microb Biochem Technol. 2016;8:71-75. doi: 10.4172/1948-5948.1000265.
14. Peterson MC, Holbrook JH, Von Hales D, Smith NL, Staker LV. Contributions of the history, physical examination, and laboratory investigation in making medical diagnoses. West J Med. 1992;156(2):163-165. doi: 10.1097/00006254-199210000-00013 PubMed
15. Roshan M, Rao AP. A study on relative contributions of the history, physical examination and investigations in making a medical diagnosis. J Assoc Physicians India. 2000;48(8):771-775. PubMed
A 46-year-old man presented to the emergency room in the postmonsoon month of September with a seven-day history of high fevers as well as a four-day history of a dry cough, dyspnea, and progressive rash. The patient reported no chest pain, hemoptysis, chest tightness, palpitations, wheezing, orthopnea, paroxysmal nocturnal dyspnea, or leg swelling. He lived and sought healthcare in Delhi, India.
Fever followed by a progressive but as yet uncharacterized rash and pulmonary symptoms in a middle-aged man suggests a host of possibilities. While it is tempting to ascribe his symptoms to an infectious process, especially a “tropical” infection based on his residence in Delhi, the location may simply represent a red herring. Potential infections can be divided into those endemic to the Indian subcontinent, and those encountered more globally. The former include diseases such as measles and dengue, while the latter include entities such as Mycoplasma pneumonia, varicella, and acute human immunodeficiency virus (HIV) infection. Noninfectious categories of diseases that should be considered include drug reactions and rheumatologic processes. Several rheumatologic diseases, including granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis, and systemic lupus erythematosus (SLE) may present with fever, rash, and pulmonary symptomatology.
A history of the patient’s exposures, both environmental and pharmaceutical, should be obtained. More information regarding his immunization history, rash characteristics (distribution and nature of the lesions), and other salient exam findings such as organomegaly and joint abnormalities will be helpful.
Fever reached a maximum of 103° Fahrenheit and was associated with chills but not rigors. There were several fever spikes daily, relieved completely with antipyretics. The patient’s dyspnea was predominantly noted on exertion, nonpleuritic, not temporally related to cough, and progressively worsening over three days. The skin lesions were first noticed on his trunk and were described as reddish, flat, and pinpoint size. However, the rash spread to the face and extremities sparing the palms and soles. There was no bleeding, nausea, vomiting, abdominal pain, change in bowel habits, dysuria, headache, photophobia, neck stiffness, or joint pain.
The patient reported no significant past medical history, took no medications, and had no recent travel outside of Delhi, India in the past year. He was married and monogamous. He had no pets nor did he report any contact with animals. He did not use tobacco, alcohol, or illicit substances. He did not remember being bitten by an insect. He worked as a software engineer. There was no history of similar illness in the patient’s family or at his workplace. He had no history of recent blood transfusion or immunization (including MMR and Tdap).
Several noninfectious and inflammatory conditions can explain his symptoms. Eosinophilic granulomatosis with polyangiitis is considerably less likely in the absence of asthma, and vasculitic processes, in general, are less likely given the nongravity dependent nature of the rash. SLE and sarcoidosis are possible causes of a systemic inflammatory illness presenting acutely with fever, rash, and pulmonary symptoms.
The patient’s expanded history makes several infections less likely. Although much of the presentation is consistent with measles, the initial appearance of the truncal rash is atypical, and there is no mention of coryza or conjunctivitis. Likewise, the description of the exanthem is not suggestive of varicella, and dengue and chikungunya are much less likely in the absence of a headache and arthralgias. Other infections including leptospirosis and scrub typhus are possible, and both might be contracted in greater Delhi. Typhoid is another infectious syndrome endemic to the Indian subcontinent that should be considered. The presence of rash involving the face and extremities would be highly atypical, however; and the presence of dyspnea and the absence of a headache argue against typhoid. Acute HIV infection and Mycoplasma pneumonia remain possible diagnoses. Toxic shock syndrome is possible, but a faster and fulminant course would be expected.
On physical examination, the temperature was 103° Fahrenheit, heart rate was 120 beats per minute and regular, respiratory rate was 24 breaths per minute, blood pressure was 100/60 mm Hg, and resting oxygen saturation was 93% while breathing ambient air. He appeared uncomfortable. Jugular venous pulse was elevated at 10 cm H2O. Mild icterus was present, but there was neither conjunctival congestion nor subconjunctival hemorrhage. S1 and S2 heart sounds were loud, but there were no murmurs. Chest auscultation revealed bilateral basal coarse crackles. The abdominal right upper quadrant was mildly tender to palpation, and the liver edge was palpable 2 cm below the subcostal margin. There was neither splenomegaly nor peripheral lymphadenopathy. Kernig and Brudzinski signs were negative, and there were no focal neurological deficits. A generalized, nonpalpable, maculopapular and petechial rash was present on the face, extremities, and trunk.
The patient’s presentation must now incorporate the additional findings of bibasilar chest crackles, maculopapular/petechial rash, icterus, modest hypoxia, and hepatomegaly. Some of the noninfectious entities already mentioned (SLE and sarcoidosis) remain possible explanations. Hemophagocytic lymphohistiocytosis (HLH) may also explain most of the patient’s presenting signs and symptoms, and several other infectious diseases account for his presentation. Scrub typhus (or a more uncommon rickettsia disease, Indian tick typhus), leptospirosis, and perhaps infective endocarditis seem most likely to provide a unifying diagnosis for the symptoms mentioned above. Leptospirosis presents in a minority of instances as a severe illness known as Weil disease, characterized by several of this patient’s findings including icterus, kidney injury, and pulmonary symptoms. However, the rash is relatively uncommon in leptospirosis and when present, is usually more localized. The patient’s rash as described is not typically expected in infective endocarditis, although high-grade Staphylococcus aureus bacteremia will occasionally present with a diffuse rash that may be confused with that of meningococcemia. The etiology of the patient’s elevated jugular venous pressure is not readily apparent, with the cardiac examination making acute valvular insufficiency much less likely. Myocarditis, however, is possible in the setting of several of the diseases listed above, including leptospirosis, scrub typhus, SLE, and dengue.
In addition to basic laboratory studies and a chest radiograph, multiple sets of blood cultures should be obtained, along with a transthoracic echocardiogram and a ferritin level. The evidence to support leptospirosis and scrub typhus is strong enough to justify empiric use of doxycycline once the blood cultures are obtained, especially given the difficulty in definitively diagnosing these diseases in a timely fashion.
Laboratory analysis revealed a total leukocyte count of 13,600/uL (85% neutrophils), hemoglobin 10 g/dL, and platelet count 35,000/uL. Absolute eosinophil count was 136/uL. Serum chemistry showed sodium of 145 meq/L, potassium 4.1 meq/L, blood urea nitrogen 80 mg/dL, creatinine 1.6 mg/dL, aspartate transaminase (AST) 44 U/L (normal, 0-40), alanine transaminase (ALT) 81 U/L (normal, 0-40), direct bilirubin 3 mg/dL, and indirect bilirubin 3 mg/dL. Lactate dehydrogenase, alkaline phosphatase, albumin, and coagulation studies were normal. Erythrocyte sedimentation rate (ESR) was 42 mm (normal, 0-25) and highly sensitive C-reactive protein was 42 mg/L (normal, 0-10). Arterial blood gas on ambient air revealed a pH of 7.52, PaCO2 24 mm Hg, PaO2 55 mm Hg, and bicarbonate 20 meq/L. Urinalysis was normal. Blood cultures were obtained. Electrocardiogram (ECG) showed regular narrow complex tachycardia with incomplete left bundle branch block. Old ECGs were not available for comparison. Chest radiograph showed bilateral air space opacities with evidence of vein cephalization. Abdominal and pelvis ultrasonography showed pericholecystic fluid and mild hepatomegaly, but no free fluid, pleural effusion, or evidence of cholecystitis. Point of care immunochromatographic rapid malarial antigen detection test (detects Plasmodium falciparum, Plasmodium vivax, Plasmodium malaria, and Plasmodium ovale) was negative.
Most of the findings described are commonly observed in both scrub typhus and leptospirosis, including cytopenias, parenchymal infiltrates, hepatomegaly, elevated transaminases and bilirubin, cardiac involvement, fever, and rash. The rash described is more consistent with scrub typhus than with leptospirosis. The absence of a headache and joint findings argue modestly against these diagnoses. Likewise, HLH provides an adequate explanation for most of the patient’s symptoms, signs, and test results. These include fever, lung involvement, rash, hepatomegaly, elevated bilirubin, and cytopenias; however, leukocytosis and cardiac involvement are less characteristic. SLE also provides a satisfactory explanation for much of the symptoms, although the rash characteristics, normal urinalysis, and leukocytosis make this diagnosis less likely.
Additional testing that should be performed includes serum antinuclear antibody (ANA) and ferritin, since the latter may be markedly elevated in the setting of HLH. Bone marrow aspirate and biopsy should be performed looking specifically for evidence of hemophagocytosis. Finally, a transthoracic echocardiogram (TTE) should be performed to assess evidence of myocardial dysfunction as it may alter the therapeutic approach, although the results will be unlikely to differentiate between the preceding considerations.
Troponin I was negative, but N-terminal probrain natriuretic peptide was elevated at 20,000 pg/mL (normal, 0-900). D-dimer was negative. TTE showed left ventricular ejection fraction (LVEF) of 35% with global left ventricular hypokinesis. On three separate examinations, the peripheral blood smear did not show malarial parasites, atypical lymphocytes, or schistocytes. Three sets of blood cultures, testing for bacteria and fungi, were sterile. A throat culture was sterile. Widal test, as well as Leptospira and Mycoplasma serologies, were negative. Serology for Legionella pneumophila was positive, but the urinary antigen testing was negative. Antibodies to HIV 1 and 2 and anti-hepatitis C virus (HCV) antibody were negative. Dengue IgM ELISA (qualitative) returned positive.
Despite the absence of arthralgias, myalgias, headache, and retro-orbital pain, a positive dengue IgM ELISA supports acute dengue infection, provided the patient did not experience an unexplained febrile illness in the previous months. Most of his presentation may be explained by dengue, including fever, rash, liver abnormalities, myocardial dysfunction, and thrombocytopenia. The bilateral airspace opacities seen on chest radiograph also fit reasonably provided these actually reflect pulmonary edema. Leukocytosis (as opposed to leukopenia) is highly unexpected in dengue, but its presence could be an outlier.
If dengue does indeed explain the entire presentation, defervescence should have occurred by the time the blood cultures and serologic studies returned. Also, by that time, the patient would be expected to demonstrate evidence of improvement, barring the appearance of the serious complications of dengue hemorrhagic fever/dengue shock syndrome. Should fever persist and signs of recovery fail to materialize, the possibility of a superimposed process will need to be considered. Of note, the sensitivity of Leptospira serology early in the course of illness is low, and leptospirosis is thus not yet excluded.
A presumptive diagnosis of severe dengue fever was made, based on evidence of pulmonary edema and sepsis. The patient was managed conservatively with oral fluid restriction, low dose of diuretics, and supplemental oxygenation. The patient was also given levofloxacin for possible legionellosis. Despite these therapies, the patient had no improvement in 24 hours. His tachypnea increased, and his measured PaO2 to FIO2 (P:F) ratio decreased to 230 from 285 on admission. This prompted the initiation of BiPAP at 10 cm H2O inspiration PAP and 5 cm H2O expiration PAP. However, he did not tolerate BiPAP, and his P:F ratio decreased to below 200.
The patient was transferred to the intensive care unit and underwent elective intubation with mechanical ventilation. Axial and coronal computed tomography of the thorax (Figure 1A and 1B, respectively) showed extensive ground-glass opacities and consolidation sparing the nondependent portions of the lungs. On physical inspection, a round, well-defined, painless black lesion surrounded by erythema was noticed in the right axilla (Figure 2). The rest of the examination findings were unchanged.
The discovery of eschar in the axilla provides a “pivot point” in determining the cause of the patient’s illness. This finding appears to point, with high specificity, toward rickettsia as the explanation of the patient’s disease, and this is most likely to be scrub typhus. The report of a positive dengue IgM may represent concurrent infection or may simply reflect a recent infection in an area that is highly endemic for dengue. Although most of the patient’s clinical presentation could be attributed to dengue, multiple features including the leukocytosis, myocarditis, and elevated bilirubin are more likely to be seen in scrub typhus. In any event, dengue cannot satisfactorily explain the eschar.
No mention has been made to the initiation of doxycycline thus far; this agent needs to be started promptly. Polymerase chain reaction (PCR) testing for scrub typhus should be ordered if available; if not, acute and convalescent serology may be obtained.
Given the finding of axillary eschar, the patient was diagnosed with scrub typhus. Doxycycline 100 mg by nasogastric tube twice a day was initiated. The patient began to show marked symptomatic improvement. His P:F ratio improved, and he was successfully weaned off and extubated after 24 hours. Postextubation, he was kept on BiPAP for 12 hours. He was transferred out of the ICU and monitored for 72 hours. With therapy, his cytopenias, liver and renal function, and ECG normalized. Indirect immunofluorescence assay for scrub typhus returned positive at a dilution of > 1:512. PCR assay targeting the 56 kDa region of Orientia tsutsugamushi was also positive. Repeated TTE showed an LVEF of 65%. He was subsequently discharged with oral doxycycline and a plan to complete a course of 14 days on an outpatient basis. The final diagnosis was scrub typhus with myocarditis leading to acutely decompensated heart failure with reduced ejection fraction.
DISCUSSION
Scrub typhus is a mite-borne tropical infection caused by the gram-negative intracellular parasite Orientia tsutsugamushi from the Rickettsiaceae family that is known to occur in certain parts of Asia and Australia. Although this entity is well known in the Sub Himalayan belt and southern part of India, very few cases have been described in Delhi, the capital state in North India. Scrub typhus, like most other tropical infections, is found most often during the postmonsoon season.1,2
Patients with scrub typhus present with fever in addition to a variety of nonspecific symptoms and findings. These often manifest within 10 days of being bitten by a mite. Malaise, headache, myalgias, lymphadenopathy, and maculopapular or petechial rash are common. If present, the rash manifests on the 3rd to 5th day of fever.3 Disseminated vasculitis due to scrub typhus can frequently result in multiorgan system involvement. Pulmonary involvement often leads to acute respiratory distress syndrome (ARDS) with an incidence of 8%-10%.1,4 Acute kidney injury, mostly mild and nonoliguric, has been reported in up to 2/3 cases.4-6 The cardiac myocyte is a known target cell affected by scrub typhus, and therefore patients commonly present with myocarditis.7 Liver involvement in scrub typhus is evident through elevated liver enzymes and can occur without other clinical evidence of the illness.4,6,8,9 As in dengue, patients often develop thrombocytopenia, but normal hemoglobin in scrub typhus differentiates it from dengue.6,8
Given the nonspecific presentation, it can be challenging to diagnose and treat scrub typhus. The gold standard for diagnosis is the detection of IgM antibodies to Orientia tsutsugamushi using an indirect immunofluorescence assay (IFA). For patients from endemic regions, it may be necessary to show a four-fold increase in titers two weeks apart to distinguish from background immunity. Presence of the characteristic eschar, as discussed below, is highly suggestive of scrub typhus. The treatment of choice is doxycycline or azithromycin for seven days.10,11 Early initiation of doxycycline when considering either scrub typhus or leptospirosis is appropriate and may be life-saving.
Medical decision making is fraught with uncertainty, and physicians must use their experience, evidence base, and cognitive heuristics wisely to care for patients effectively. For this patient, the region of Delhi experiences massive outbreaks of dengue every year during the time the patient presented to the hospital, whereas rickettsia infections are relatively uncommon. The clinical presentation was conceivably consistent with either dengue or scrub typhus, though somewhat more suggestive of the latter. Once the serological diagnosis of recent or concomitant dengue was obtained, however, scrub typhus was considered even less. The team called upon Occam’s razor or the heuristic that the simplest and most unifying explanation for any given problem is the one most likely to be correct and that other, less satisfactory explanations (in this case, scrub typhus) are “shaven off.” The patient was managed conservatively for dengue. Only when his condition worsened did the team recognize this conflicting information without dismissing it, consider alternative possibilities, and reexamined the patient.
An eschar can be an important clue in the diagnosis of scrub typhus, though it is not often obvious. The presence of this necrotic skin lesion with black crust is highly suggestive of scrub typhus, and in the right clinical context, it is virtually diagnostic. However, it is uncommon (9.5%-45%) in most of the studies from the Indian subcontinent (ie, high specificity but low sensitivity).1,12 An eschar is often found in obscure locations such as the axillae or groin, areas that may easily be missed or overlooked. Eschars may be seen in a variety of other infectious diseases, including rickettsia pox, Rocky Mountain spotted fever, other members of the spotted fever group, tularemia, and cutaneous anthrax. Given this patient’s lack of improvement, repeated examination revealed an eschar in the right axilla, a finding that was either missed or still evolving at the time of presentation.
This case illustrates the challenges in interpreting the significance of multiple positive serological tests in the context of an undifferentiated clinical syndrome. Possible reasons for a positive dengue serology could have been persistent antibodies from a previous infection, recent asymptomatic infection, concurrent infection, or cross-reactivity with flaviviruses such as West Nile Virus or Japanese Encephalitis.13 The patient also had positive IgM antibodies against Legionella pneumophila, but the urinary antigen was negative. In view of a negative antigen test, low specificity of the serologic test, low incidence of legionellosis in the Indian subcontinent, and absence of therapeutic response to a trial of fluoroquinolones, the diagnosis of legionellosis was considered unlikely in this patient.
With rapid advancements in technology, the importance of history taking and physical examination is at risk of being overshadowed. Approximately 80% of correct diagnoses in medicine can arrive through history and physical examination alone.14,15 In this case, Occam’s razor combined with multiple serological tests was relied on to create the likely list of diagnoses. However, recognition of the limitations of these heuristics and tests proved critical. The life-saving diagnosis was only made when the clinicians returned to basics, looked in every nook and cranny, and found the eschar on physical examination.
KEY TEACHING POINTS
- In patients living in endemic areas who present with an acute febrile illness, the differential diagnosis should include “tropical” infections such as dengue, chikungunya, enteric fever, leptospirosis, malaria, and scrub typhus.
- Serology is commonly employed for diagnosis of tropical infections, which may be misleading. These tests can be falsely positive from past asymptomatic infection or cross reactivity between antibodies, or falsely negative, as in the first few days of infection.
- Presence of eschar is a very useful clue in the diagnosis of scrub typhus, but this finding can be missed since it is often found in obscure locations. A thorough clinical history and physical examination are paramount.
Disclosures
The authors do not report any conflict of interest.
A 46-year-old man presented to the emergency room in the postmonsoon month of September with a seven-day history of high fevers as well as a four-day history of a dry cough, dyspnea, and progressive rash. The patient reported no chest pain, hemoptysis, chest tightness, palpitations, wheezing, orthopnea, paroxysmal nocturnal dyspnea, or leg swelling. He lived and sought healthcare in Delhi, India.
Fever followed by a progressive but as yet uncharacterized rash and pulmonary symptoms in a middle-aged man suggests a host of possibilities. While it is tempting to ascribe his symptoms to an infectious process, especially a “tropical” infection based on his residence in Delhi, the location may simply represent a red herring. Potential infections can be divided into those endemic to the Indian subcontinent, and those encountered more globally. The former include diseases such as measles and dengue, while the latter include entities such as Mycoplasma pneumonia, varicella, and acute human immunodeficiency virus (HIV) infection. Noninfectious categories of diseases that should be considered include drug reactions and rheumatologic processes. Several rheumatologic diseases, including granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis, and systemic lupus erythematosus (SLE) may present with fever, rash, and pulmonary symptomatology.
A history of the patient’s exposures, both environmental and pharmaceutical, should be obtained. More information regarding his immunization history, rash characteristics (distribution and nature of the lesions), and other salient exam findings such as organomegaly and joint abnormalities will be helpful.
Fever reached a maximum of 103° Fahrenheit and was associated with chills but not rigors. There were several fever spikes daily, relieved completely with antipyretics. The patient’s dyspnea was predominantly noted on exertion, nonpleuritic, not temporally related to cough, and progressively worsening over three days. The skin lesions were first noticed on his trunk and were described as reddish, flat, and pinpoint size. However, the rash spread to the face and extremities sparing the palms and soles. There was no bleeding, nausea, vomiting, abdominal pain, change in bowel habits, dysuria, headache, photophobia, neck stiffness, or joint pain.
The patient reported no significant past medical history, took no medications, and had no recent travel outside of Delhi, India in the past year. He was married and monogamous. He had no pets nor did he report any contact with animals. He did not use tobacco, alcohol, or illicit substances. He did not remember being bitten by an insect. He worked as a software engineer. There was no history of similar illness in the patient’s family or at his workplace. He had no history of recent blood transfusion or immunization (including MMR and Tdap).
Several noninfectious and inflammatory conditions can explain his symptoms. Eosinophilic granulomatosis with polyangiitis is considerably less likely in the absence of asthma, and vasculitic processes, in general, are less likely given the nongravity dependent nature of the rash. SLE and sarcoidosis are possible causes of a systemic inflammatory illness presenting acutely with fever, rash, and pulmonary symptoms.
The patient’s expanded history makes several infections less likely. Although much of the presentation is consistent with measles, the initial appearance of the truncal rash is atypical, and there is no mention of coryza or conjunctivitis. Likewise, the description of the exanthem is not suggestive of varicella, and dengue and chikungunya are much less likely in the absence of a headache and arthralgias. Other infections including leptospirosis and scrub typhus are possible, and both might be contracted in greater Delhi. Typhoid is another infectious syndrome endemic to the Indian subcontinent that should be considered. The presence of rash involving the face and extremities would be highly atypical, however; and the presence of dyspnea and the absence of a headache argue against typhoid. Acute HIV infection and Mycoplasma pneumonia remain possible diagnoses. Toxic shock syndrome is possible, but a faster and fulminant course would be expected.
On physical examination, the temperature was 103° Fahrenheit, heart rate was 120 beats per minute and regular, respiratory rate was 24 breaths per minute, blood pressure was 100/60 mm Hg, and resting oxygen saturation was 93% while breathing ambient air. He appeared uncomfortable. Jugular venous pulse was elevated at 10 cm H2O. Mild icterus was present, but there was neither conjunctival congestion nor subconjunctival hemorrhage. S1 and S2 heart sounds were loud, but there were no murmurs. Chest auscultation revealed bilateral basal coarse crackles. The abdominal right upper quadrant was mildly tender to palpation, and the liver edge was palpable 2 cm below the subcostal margin. There was neither splenomegaly nor peripheral lymphadenopathy. Kernig and Brudzinski signs were negative, and there were no focal neurological deficits. A generalized, nonpalpable, maculopapular and petechial rash was present on the face, extremities, and trunk.
The patient’s presentation must now incorporate the additional findings of bibasilar chest crackles, maculopapular/petechial rash, icterus, modest hypoxia, and hepatomegaly. Some of the noninfectious entities already mentioned (SLE and sarcoidosis) remain possible explanations. Hemophagocytic lymphohistiocytosis (HLH) may also explain most of the patient’s presenting signs and symptoms, and several other infectious diseases account for his presentation. Scrub typhus (or a more uncommon rickettsia disease, Indian tick typhus), leptospirosis, and perhaps infective endocarditis seem most likely to provide a unifying diagnosis for the symptoms mentioned above. Leptospirosis presents in a minority of instances as a severe illness known as Weil disease, characterized by several of this patient’s findings including icterus, kidney injury, and pulmonary symptoms. However, the rash is relatively uncommon in leptospirosis and when present, is usually more localized. The patient’s rash as described is not typically expected in infective endocarditis, although high-grade Staphylococcus aureus bacteremia will occasionally present with a diffuse rash that may be confused with that of meningococcemia. The etiology of the patient’s elevated jugular venous pressure is not readily apparent, with the cardiac examination making acute valvular insufficiency much less likely. Myocarditis, however, is possible in the setting of several of the diseases listed above, including leptospirosis, scrub typhus, SLE, and dengue.
In addition to basic laboratory studies and a chest radiograph, multiple sets of blood cultures should be obtained, along with a transthoracic echocardiogram and a ferritin level. The evidence to support leptospirosis and scrub typhus is strong enough to justify empiric use of doxycycline once the blood cultures are obtained, especially given the difficulty in definitively diagnosing these diseases in a timely fashion.
Laboratory analysis revealed a total leukocyte count of 13,600/uL (85% neutrophils), hemoglobin 10 g/dL, and platelet count 35,000/uL. Absolute eosinophil count was 136/uL. Serum chemistry showed sodium of 145 meq/L, potassium 4.1 meq/L, blood urea nitrogen 80 mg/dL, creatinine 1.6 mg/dL, aspartate transaminase (AST) 44 U/L (normal, 0-40), alanine transaminase (ALT) 81 U/L (normal, 0-40), direct bilirubin 3 mg/dL, and indirect bilirubin 3 mg/dL. Lactate dehydrogenase, alkaline phosphatase, albumin, and coagulation studies were normal. Erythrocyte sedimentation rate (ESR) was 42 mm (normal, 0-25) and highly sensitive C-reactive protein was 42 mg/L (normal, 0-10). Arterial blood gas on ambient air revealed a pH of 7.52, PaCO2 24 mm Hg, PaO2 55 mm Hg, and bicarbonate 20 meq/L. Urinalysis was normal. Blood cultures were obtained. Electrocardiogram (ECG) showed regular narrow complex tachycardia with incomplete left bundle branch block. Old ECGs were not available for comparison. Chest radiograph showed bilateral air space opacities with evidence of vein cephalization. Abdominal and pelvis ultrasonography showed pericholecystic fluid and mild hepatomegaly, but no free fluid, pleural effusion, or evidence of cholecystitis. Point of care immunochromatographic rapid malarial antigen detection test (detects Plasmodium falciparum, Plasmodium vivax, Plasmodium malaria, and Plasmodium ovale) was negative.
Most of the findings described are commonly observed in both scrub typhus and leptospirosis, including cytopenias, parenchymal infiltrates, hepatomegaly, elevated transaminases and bilirubin, cardiac involvement, fever, and rash. The rash described is more consistent with scrub typhus than with leptospirosis. The absence of a headache and joint findings argue modestly against these diagnoses. Likewise, HLH provides an adequate explanation for most of the patient’s symptoms, signs, and test results. These include fever, lung involvement, rash, hepatomegaly, elevated bilirubin, and cytopenias; however, leukocytosis and cardiac involvement are less characteristic. SLE also provides a satisfactory explanation for much of the symptoms, although the rash characteristics, normal urinalysis, and leukocytosis make this diagnosis less likely.
Additional testing that should be performed includes serum antinuclear antibody (ANA) and ferritin, since the latter may be markedly elevated in the setting of HLH. Bone marrow aspirate and biopsy should be performed looking specifically for evidence of hemophagocytosis. Finally, a transthoracic echocardiogram (TTE) should be performed to assess evidence of myocardial dysfunction as it may alter the therapeutic approach, although the results will be unlikely to differentiate between the preceding considerations.
Troponin I was negative, but N-terminal probrain natriuretic peptide was elevated at 20,000 pg/mL (normal, 0-900). D-dimer was negative. TTE showed left ventricular ejection fraction (LVEF) of 35% with global left ventricular hypokinesis. On three separate examinations, the peripheral blood smear did not show malarial parasites, atypical lymphocytes, or schistocytes. Three sets of blood cultures, testing for bacteria and fungi, were sterile. A throat culture was sterile. Widal test, as well as Leptospira and Mycoplasma serologies, were negative. Serology for Legionella pneumophila was positive, but the urinary antigen testing was negative. Antibodies to HIV 1 and 2 and anti-hepatitis C virus (HCV) antibody were negative. Dengue IgM ELISA (qualitative) returned positive.
Despite the absence of arthralgias, myalgias, headache, and retro-orbital pain, a positive dengue IgM ELISA supports acute dengue infection, provided the patient did not experience an unexplained febrile illness in the previous months. Most of his presentation may be explained by dengue, including fever, rash, liver abnormalities, myocardial dysfunction, and thrombocytopenia. The bilateral airspace opacities seen on chest radiograph also fit reasonably provided these actually reflect pulmonary edema. Leukocytosis (as opposed to leukopenia) is highly unexpected in dengue, but its presence could be an outlier.
If dengue does indeed explain the entire presentation, defervescence should have occurred by the time the blood cultures and serologic studies returned. Also, by that time, the patient would be expected to demonstrate evidence of improvement, barring the appearance of the serious complications of dengue hemorrhagic fever/dengue shock syndrome. Should fever persist and signs of recovery fail to materialize, the possibility of a superimposed process will need to be considered. Of note, the sensitivity of Leptospira serology early in the course of illness is low, and leptospirosis is thus not yet excluded.
A presumptive diagnosis of severe dengue fever was made, based on evidence of pulmonary edema and sepsis. The patient was managed conservatively with oral fluid restriction, low dose of diuretics, and supplemental oxygenation. The patient was also given levofloxacin for possible legionellosis. Despite these therapies, the patient had no improvement in 24 hours. His tachypnea increased, and his measured PaO2 to FIO2 (P:F) ratio decreased to 230 from 285 on admission. This prompted the initiation of BiPAP at 10 cm H2O inspiration PAP and 5 cm H2O expiration PAP. However, he did not tolerate BiPAP, and his P:F ratio decreased to below 200.
The patient was transferred to the intensive care unit and underwent elective intubation with mechanical ventilation. Axial and coronal computed tomography of the thorax (Figure 1A and 1B, respectively) showed extensive ground-glass opacities and consolidation sparing the nondependent portions of the lungs. On physical inspection, a round, well-defined, painless black lesion surrounded by erythema was noticed in the right axilla (Figure 2). The rest of the examination findings were unchanged.
The discovery of eschar in the axilla provides a “pivot point” in determining the cause of the patient’s illness. This finding appears to point, with high specificity, toward rickettsia as the explanation of the patient’s disease, and this is most likely to be scrub typhus. The report of a positive dengue IgM may represent concurrent infection or may simply reflect a recent infection in an area that is highly endemic for dengue. Although most of the patient’s clinical presentation could be attributed to dengue, multiple features including the leukocytosis, myocarditis, and elevated bilirubin are more likely to be seen in scrub typhus. In any event, dengue cannot satisfactorily explain the eschar.
No mention has been made to the initiation of doxycycline thus far; this agent needs to be started promptly. Polymerase chain reaction (PCR) testing for scrub typhus should be ordered if available; if not, acute and convalescent serology may be obtained.
Given the finding of axillary eschar, the patient was diagnosed with scrub typhus. Doxycycline 100 mg by nasogastric tube twice a day was initiated. The patient began to show marked symptomatic improvement. His P:F ratio improved, and he was successfully weaned off and extubated after 24 hours. Postextubation, he was kept on BiPAP for 12 hours. He was transferred out of the ICU and monitored for 72 hours. With therapy, his cytopenias, liver and renal function, and ECG normalized. Indirect immunofluorescence assay for scrub typhus returned positive at a dilution of > 1:512. PCR assay targeting the 56 kDa region of Orientia tsutsugamushi was also positive. Repeated TTE showed an LVEF of 65%. He was subsequently discharged with oral doxycycline and a plan to complete a course of 14 days on an outpatient basis. The final diagnosis was scrub typhus with myocarditis leading to acutely decompensated heart failure with reduced ejection fraction.
DISCUSSION
Scrub typhus is a mite-borne tropical infection caused by the gram-negative intracellular parasite Orientia tsutsugamushi from the Rickettsiaceae family that is known to occur in certain parts of Asia and Australia. Although this entity is well known in the Sub Himalayan belt and southern part of India, very few cases have been described in Delhi, the capital state in North India. Scrub typhus, like most other tropical infections, is found most often during the postmonsoon season.1,2
Patients with scrub typhus present with fever in addition to a variety of nonspecific symptoms and findings. These often manifest within 10 days of being bitten by a mite. Malaise, headache, myalgias, lymphadenopathy, and maculopapular or petechial rash are common. If present, the rash manifests on the 3rd to 5th day of fever.3 Disseminated vasculitis due to scrub typhus can frequently result in multiorgan system involvement. Pulmonary involvement often leads to acute respiratory distress syndrome (ARDS) with an incidence of 8%-10%.1,4 Acute kidney injury, mostly mild and nonoliguric, has been reported in up to 2/3 cases.4-6 The cardiac myocyte is a known target cell affected by scrub typhus, and therefore patients commonly present with myocarditis.7 Liver involvement in scrub typhus is evident through elevated liver enzymes and can occur without other clinical evidence of the illness.4,6,8,9 As in dengue, patients often develop thrombocytopenia, but normal hemoglobin in scrub typhus differentiates it from dengue.6,8
Given the nonspecific presentation, it can be challenging to diagnose and treat scrub typhus. The gold standard for diagnosis is the detection of IgM antibodies to Orientia tsutsugamushi using an indirect immunofluorescence assay (IFA). For patients from endemic regions, it may be necessary to show a four-fold increase in titers two weeks apart to distinguish from background immunity. Presence of the characteristic eschar, as discussed below, is highly suggestive of scrub typhus. The treatment of choice is doxycycline or azithromycin for seven days.10,11 Early initiation of doxycycline when considering either scrub typhus or leptospirosis is appropriate and may be life-saving.
Medical decision making is fraught with uncertainty, and physicians must use their experience, evidence base, and cognitive heuristics wisely to care for patients effectively. For this patient, the region of Delhi experiences massive outbreaks of dengue every year during the time the patient presented to the hospital, whereas rickettsia infections are relatively uncommon. The clinical presentation was conceivably consistent with either dengue or scrub typhus, though somewhat more suggestive of the latter. Once the serological diagnosis of recent or concomitant dengue was obtained, however, scrub typhus was considered even less. The team called upon Occam’s razor or the heuristic that the simplest and most unifying explanation for any given problem is the one most likely to be correct and that other, less satisfactory explanations (in this case, scrub typhus) are “shaven off.” The patient was managed conservatively for dengue. Only when his condition worsened did the team recognize this conflicting information without dismissing it, consider alternative possibilities, and reexamined the patient.
An eschar can be an important clue in the diagnosis of scrub typhus, though it is not often obvious. The presence of this necrotic skin lesion with black crust is highly suggestive of scrub typhus, and in the right clinical context, it is virtually diagnostic. However, it is uncommon (9.5%-45%) in most of the studies from the Indian subcontinent (ie, high specificity but low sensitivity).1,12 An eschar is often found in obscure locations such as the axillae or groin, areas that may easily be missed or overlooked. Eschars may be seen in a variety of other infectious diseases, including rickettsia pox, Rocky Mountain spotted fever, other members of the spotted fever group, tularemia, and cutaneous anthrax. Given this patient’s lack of improvement, repeated examination revealed an eschar in the right axilla, a finding that was either missed or still evolving at the time of presentation.
This case illustrates the challenges in interpreting the significance of multiple positive serological tests in the context of an undifferentiated clinical syndrome. Possible reasons for a positive dengue serology could have been persistent antibodies from a previous infection, recent asymptomatic infection, concurrent infection, or cross-reactivity with flaviviruses such as West Nile Virus or Japanese Encephalitis.13 The patient also had positive IgM antibodies against Legionella pneumophila, but the urinary antigen was negative. In view of a negative antigen test, low specificity of the serologic test, low incidence of legionellosis in the Indian subcontinent, and absence of therapeutic response to a trial of fluoroquinolones, the diagnosis of legionellosis was considered unlikely in this patient.
With rapid advancements in technology, the importance of history taking and physical examination is at risk of being overshadowed. Approximately 80% of correct diagnoses in medicine can arrive through history and physical examination alone.14,15 In this case, Occam’s razor combined with multiple serological tests was relied on to create the likely list of diagnoses. However, recognition of the limitations of these heuristics and tests proved critical. The life-saving diagnosis was only made when the clinicians returned to basics, looked in every nook and cranny, and found the eschar on physical examination.
KEY TEACHING POINTS
- In patients living in endemic areas who present with an acute febrile illness, the differential diagnosis should include “tropical” infections such as dengue, chikungunya, enteric fever, leptospirosis, malaria, and scrub typhus.
- Serology is commonly employed for diagnosis of tropical infections, which may be misleading. These tests can be falsely positive from past asymptomatic infection or cross reactivity between antibodies, or falsely negative, as in the first few days of infection.
- Presence of eschar is a very useful clue in the diagnosis of scrub typhus, but this finding can be missed since it is often found in obscure locations. A thorough clinical history and physical examination are paramount.
Disclosures
The authors do not report any conflict of interest.
1. Gupta N, Chaudhry R, Kabra SK, et al. In search of scrub typhus: a prospective analysis of clinical and epidemiological profile of patients from a tertiary care hospital in New Delhi. Adv Infect Dis. 2015;5(4):140. doi: 10.4236/aid.2015.54017.
2. Kamarasu K, Malathi M, Rajagopal V, Subramani K, Jagadeeshramasamy D, Mathai E. Serological evidence for the wide distribution of spotted fevers & typhus fever in Tamil Nadu. Indian J Med Res. 2007;126(2):128-130. PubMed
3. Mahajan SK. Scrub typhus. J Assoc Physicians India. 2005;53:954-958. PubMed
4. Mahajan SK, Rolain JM, Kashyap R, et al. Scrub typhus in the Himalayas. Emerg Infect Dis. 2006;12(10):1590-1592. doi: 10.3201/eid1210.051697. PubMed
5. Attur RP, Kuppasamy S, Bairy M, et al. Acute kidney injury in scrub typhus. Clin Exp Nephrol. 2013;17(5):725-729. doi: 10.1007/s10157-012-0753-9. PubMed
6. Varghese GM, Trowbridge P, Janardhanan J, et al. Clinical profile and improving mortality trend of scrub typhus in South India. Int J Infect Dis. 2014;23:39-43. doi: 10.1016/j.ijid.2014.02.009. PubMed
7. Taylor AJ, Paris DH, Newton PN. A systematic review of mortality from untreated scrub typhus (Orientia tsutsugamushi). PLoS Negl Trop Dis. 2015;9(8):e0003971. doi.org/10.1371/journal.pntd.0003971 PubMed
8. Chrispal A, Boorugu H, Gopinath KG, et al. Scrub typhus: an unrecognized threat in South India-clinical profile and predictors of mortality. Trop Doct. 2010;40(3):129-133. doi: 10.1258/td.2010.090452. PubMed
9. Mathai E, Rolain JM, Verghese GM, et al. Outbreak of scrub typhus in southern India during the cooler months. Ann N Y Acad Sci. 2003;990:359-364. doi: 10.1111/j.1749-6632.2003.tb07391.x PubMed
10. Gupta N, Chaudhry R, Kabra SK, et al. Comparative evaluation of serological and molecular methods for the diagnosis of scrub typhus in Indian settings. Jpn J Infect Dis. 2017;70(2):221-222. doi: 10.7883/yoken.JJID.2016.139. PubMed
11. Rahi M, Gupte MD, Bhargava A, Varghese GM, Arora R. DHR-ICMR Guidelines for diagnosis & management of Rickettsial diseases in India. Indian J Med Res. 2015;141(4):417-422. doi: 10.4103/0971-5916.159279. PubMed
12. Sharma A, Mahajan S, Gupta ML, Kanga A, Sharma V. Investigation of an outbreak of scrub typhus in the Himalayan region of India. Jpn J Infect Dis. 2005;58(4):208-210. PubMed
13. Gupta N, Chaudhry R, Mirdha B, et al. Scrub typhus and leptospirosis: the fallacy of diagnosing with IgM enzyme-linked immunosorbent assay. J Microb Biochem Technol. 2016;8:71-75. doi: 10.4172/1948-5948.1000265.
14. Peterson MC, Holbrook JH, Von Hales D, Smith NL, Staker LV. Contributions of the history, physical examination, and laboratory investigation in making medical diagnoses. West J Med. 1992;156(2):163-165. doi: 10.1097/00006254-199210000-00013 PubMed
15. Roshan M, Rao AP. A study on relative contributions of the history, physical examination and investigations in making a medical diagnosis. J Assoc Physicians India. 2000;48(8):771-775. PubMed
1. Gupta N, Chaudhry R, Kabra SK, et al. In search of scrub typhus: a prospective analysis of clinical and epidemiological profile of patients from a tertiary care hospital in New Delhi. Adv Infect Dis. 2015;5(4):140. doi: 10.4236/aid.2015.54017.
2. Kamarasu K, Malathi M, Rajagopal V, Subramani K, Jagadeeshramasamy D, Mathai E. Serological evidence for the wide distribution of spotted fevers & typhus fever in Tamil Nadu. Indian J Med Res. 2007;126(2):128-130. PubMed
3. Mahajan SK. Scrub typhus. J Assoc Physicians India. 2005;53:954-958. PubMed
4. Mahajan SK, Rolain JM, Kashyap R, et al. Scrub typhus in the Himalayas. Emerg Infect Dis. 2006;12(10):1590-1592. doi: 10.3201/eid1210.051697. PubMed
5. Attur RP, Kuppasamy S, Bairy M, et al. Acute kidney injury in scrub typhus. Clin Exp Nephrol. 2013;17(5):725-729. doi: 10.1007/s10157-012-0753-9. PubMed
6. Varghese GM, Trowbridge P, Janardhanan J, et al. Clinical profile and improving mortality trend of scrub typhus in South India. Int J Infect Dis. 2014;23:39-43. doi: 10.1016/j.ijid.2014.02.009. PubMed
7. Taylor AJ, Paris DH, Newton PN. A systematic review of mortality from untreated scrub typhus (Orientia tsutsugamushi). PLoS Negl Trop Dis. 2015;9(8):e0003971. doi.org/10.1371/journal.pntd.0003971 PubMed
8. Chrispal A, Boorugu H, Gopinath KG, et al. Scrub typhus: an unrecognized threat in South India-clinical profile and predictors of mortality. Trop Doct. 2010;40(3):129-133. doi: 10.1258/td.2010.090452. PubMed
9. Mathai E, Rolain JM, Verghese GM, et al. Outbreak of scrub typhus in southern India during the cooler months. Ann N Y Acad Sci. 2003;990:359-364. doi: 10.1111/j.1749-6632.2003.tb07391.x PubMed
10. Gupta N, Chaudhry R, Kabra SK, et al. Comparative evaluation of serological and molecular methods for the diagnosis of scrub typhus in Indian settings. Jpn J Infect Dis. 2017;70(2):221-222. doi: 10.7883/yoken.JJID.2016.139. PubMed
11. Rahi M, Gupte MD, Bhargava A, Varghese GM, Arora R. DHR-ICMR Guidelines for diagnosis & management of Rickettsial diseases in India. Indian J Med Res. 2015;141(4):417-422. doi: 10.4103/0971-5916.159279. PubMed
12. Sharma A, Mahajan S, Gupta ML, Kanga A, Sharma V. Investigation of an outbreak of scrub typhus in the Himalayan region of India. Jpn J Infect Dis. 2005;58(4):208-210. PubMed
13. Gupta N, Chaudhry R, Mirdha B, et al. Scrub typhus and leptospirosis: the fallacy of diagnosing with IgM enzyme-linked immunosorbent assay. J Microb Biochem Technol. 2016;8:71-75. doi: 10.4172/1948-5948.1000265.
14. Peterson MC, Holbrook JH, Von Hales D, Smith NL, Staker LV. Contributions of the history, physical examination, and laboratory investigation in making medical diagnoses. West J Med. 1992;156(2):163-165. doi: 10.1097/00006254-199210000-00013 PubMed
15. Roshan M, Rao AP. A study on relative contributions of the history, physical examination and investigations in making a medical diagnosis. J Assoc Physicians India. 2000;48(8):771-775. PubMed
© 2019 Society of Hospital Medicine
Egad!
A 69-year-old woman presented to the clinic with pain in the right great toe lasting several days. She was prescribed colchicine and indomethacin empirically for gout. She took one tablet of colchicine (0.6 mg) every hour until her stools became loose after the eighth tablet. Her toe pain resolved, but two days later she developed bilateral lower extremity pruritus and paresthesia and presented to the emergency department (ED). On physical examination, no rash, weakness, or sensory deficits were observed, and she was able to ambulate without assistance. Her patellar reflexes were normal. The complete blood count was notable for an absolute lymphocyte count of 6,120/µL (normal: 1,100-4,800), and the comprehensive metabolic panel was normal. Serum creatine kinase (CK) was 341 U/L (normal: 24-170) and uric acid 7.7 mg/dL (normal: 2.4-6.4). Her lower extremity symptoms were attributed to colchicine, which was discontinued. She was prescribed diphenhydramine and discharged home.
Monoarthritis of the hallux is the classic manifestation of gout, although other considerations include pseudogout, sesamoiditis, and trauma. The typical side effects of colchicine include diarrhea and myositis. Colchicine-induced muscle injury often results in a modest elevation of CK levels and is associated with myalgia.
Paresthesia is defined as abnormal sensory symptoms that most commonly localize to the peripheral nerves or spinal cord. Acute neuropathies or myelopathies might result from vasculitis, heavy metal toxicity, vitamin deficiencies, and paraneoplastic neurologic syndromes. The normal motor, sensory, and reflex examination, however, make these unlikely.
The neuro-anatomic localization of pruritus is poorly understood but is proposed to include peripheral nerves, spinothalamic tracts, and thalami. Acute pruritus (lasting <6 weeks) typically results from a primary dermatologic process such as a drug reaction, eczema, or xerosis. Less common causes include uremia, cholestasis, and thyroid disease. Pruritus can also be seen with malignancy, most commonly hematologic or paraneoplastic syndromes, or with connective tissue diseases. At this stage, it is unclear whether her pruritus and paresthesia are part of a unifying disease process.
Five days later she re-presented to the ED with nausea and emesis after eating at a restaurant. Her symptoms improved with intravenous fluids, and she was discharged. Four days later she returned with difficulty ambulating, bilateral leg cramping, and continued pruritus and paresthesia. The chemistry panel was normal except for a potassium level of 2.6 mmol/L and a bicarbonate level of 32 mmol/L. She was admitted to the hospital because of severe hypokalemia and impaired ability to ambulate. Her potassium was replenished. Her CK was elevated (3,551 U/L on hospital day 7). She was given cyclobenzaprine, gabapentin, oxycodone, acetaminophen, and prednisone (40 mg); her cramping only mildly improved, and she remained unable to walk. On hospital day five she had visual hallucinations and confusion, which did not resolve with administration of haloperidol; a head CT was unremarkable. On hospital day eight the patient, with her family’s support, left the hospital and presented to a different ED for a second opinion.
Difficulty ambulating often results from weakness, sensory impairment, cerebellar ataxia, extrapyramidal dysfunction (eg, parkinsonism), and pain. In this patient, leg cramping suggests pain or true weakness due to a myopathic process as a contributing factor. Symptoms of muscle disease include cramps, myalgia, and difficulty walking. Causes of elevated CK and myalgia include inflammatory myopathies, endocrinopathies, drugs, infections, and electrolyte abnormalities (eg, hypokalemia). Her age and acuity of presentation decrease the likelihood of a metabolic myopathy due to a disorder of glycogen storage, lipid metabolism, or mitochondrial function. Her hypokalemic metabolic alkalosis likely resulted from vomiting. Hypokalemic periodic paralysis is unlikely as exacerbations typically only last hours to days. As such, her difficulty ambulating, muscle cramps, and elevated CK strongly support a primary myopathic disorder, although additional information regarding the neurologic examination is still required.
Acute changes in mental status without corresponding changes in cranial nerve, motor, or sensory function are common in the hospital setting and frequently relate to delirium, which is the most likely explanation for her confusion. Her age and exposure to muscle relaxants, opiates, and corticosteroids increase her risk considerably. Other possible explanations for isolated changes in mental status include nonconvulsive seizures, central nervous system (CNS) infection, and strokes that involve the thalamus, nondominant parietal lobe, and reticular activating system. A shower of emboli resulting in small multifocal strokes can have the same effect.
She was re-evaluated by her new providers. Her only prior medical history was hypertension, which was treated at home with atenolol and amlodipine. She had emigrated from Nigeria to the US many years prior. She occasionally consumed alcohol and never smoked tobacco or used illicit drugs. She was unsure if she had received a tetanus booster in the past 10 years.
On physical examination, her temperature was 36°C, blood pressure 149/70 mm Hg, pulse 56 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 98% on ambient air. She was diaphoretic and appeared anxious, grabbing both bedrails out of fear of falling. Cardiovascular, pulmonary, abdominal, and skin examinations were normal. She was alert and oriented to her identity, her location, and the time. Cranial nerves II to XII were normal. Tone was normal in her upper extremities but markedly increased in her lower extremities and back. There were spontaneous and stimulus-induced painful spasms, predominantly involving her axial muscles and distal lower extremities. Muscle bulk was normal. Strength was normal in the upper extremities and could not be assessed in the lower extremities due to rigidity. Reflexes were 2+ and symmetric throughout with downgoing toes on Babinski testing. A sensory examination was normal. Gait could not be tested because of the severe muscle spasms. The patient was admitted to the hospital.
Localized muscle spasms may be caused by muscle overuse, but more generalized spasms are associated with systemic diseases such as electrolyte disturbances, toxidromes, tetanus, peripheral nerve hyperexcitability syndromes (including Isaacs syndrome and Morvan syndrome), or stiff person syndrome (SPS). Hypokalemia is unlikely the cause as its correction did not improve her symptoms. Although tetanus is rare in the United States, it remains endemic in the developing world and can cause focal as well as generalized stimulus-induced spasms. The patient should be asked about potential exposure to Clostridium tetani infection, such as incurring a puncture wound. It is also important to consider neuroleptic malignant syndrome and serotonin syndrome, which can cause confusion, elevated CK, and increased muscle tone. Her confusion, however, was transient and the elevated CK preceded the administration of haloperidol.
SPS and progressive encephalomyelitis with rigidity and myoclonus (PERM) provide better explanations for her presentation. Both diseases cause severe spasms, impaired ambulation, and stiffness. They differ in their acuity of onset, accompanying symptoms, antibody associations, and responses to treatment. The rapid onset, paresthesia, and confusion seen in this patient are atypical of SPS. SPS usually presents with subacute-to-chronic stiffness or soreness of muscles in the back and lower extremities, followed by the upper extremities. Rigidity, stimulation-provoked spasms, hyperlordosis, and difficulty ambulating are typically later-stage findings. Her rapid escalation of symptoms is more consistent with PERM, which is often more acute and progressive than typical SPS; however, unlike this patient, PERM commonly causes widespread CNS dysfunction, including persistent encephalopathy, cranial neuropathies, hyperreflexia, and autonomic instability. Both are rare diagnoses that can manifest as a paraneoplastic neurologic syndrome.
Blood tests showed a leukocyte count of 17,350/µL, neutrophils 8,720/µL (normal: 1,500–7,800), lymphocytes 6,130/µL, hemoglobin 11.3 g/dL, and platelets 231,000/µL. The basic metabolic panel was normal. Serum total protein was 6.7 g/dL with albumin 3.5 g/dL. Aspartate aminotransferase (AST) was 94 U/L (normal: 0-31), alanine aminotransferase (ALT) 56 U/L (normal: 0-31), alkaline phosphatase 45 U/L, and total bilirubin 1.1 mg/dL. Vitamin B12 was 868 pg/mL. Hemoglobin A1c and thyrotropin levels were normal. Creatine kinase was 3,757 U/L and lactate dehydrogenase (LDH) 435 U/L (normal: 122-220). The syphilis treponemal test and hepatitis B surface antigen were negative. HIV and hepatitis C antibodies were nonreactive. The anti-nuclear antibody screen was negative and complement C3 and C4 were normal.
Neutrophilia likely reflects glucocorticoid-induced demargination, as opposed to an infectious process, given the temporal association with steroid administration. Persistent mild lymphocytosis is nonspecific but more likely to reflect a reactive rather than a clonal process. Elevated LDH and CK, as well as a greater increase of AST relative to ALT, suggest muscle injury, although mild concomitant hepatic injury cannot be excluded. Normal or negative serum studies for TSH, HIV, ANA, peripheral blood smear, and creatinine eliminate many of the systemic causes of her pruritus, but malignancy and associated paraneoplastic etiologies remain considerations.
The initial work-up for SPS includes electromyography (EMG) which would show spontaneous muscle activity. Her poorly localized sensory abnormalities, transient vestibular symptoms, and confusion warrant an MRI of the brain and spine to evaluate for inflammation (eg, encephalomyelitis), which could be consistent with PERM.
An MRI of the brain and cervicothoracic spine without contrast was significantly limited by motion artifact but without obvious intracranial or cord signal abnormalities. Electromyography demonstrated spontaneous muscle activity in both lower extremities with co-contraction of agonist and antagonist muscles (hamstrings and quadriceps as well as medial gastrocnemius and tibialis anterior). Sensory and motor nerve conductions were normal. Cerebral spinal fluid (CSF) contained six leukocytes (96% lymphocytes) and three red blood cells per microliter; glucose was 67 mg/dL and protein 24 mg/dL. There were two oligoclonal bands unique to the CSF. Cytology was negative for malignant cells.
The EMG narrows the differential diagnosis considerably. Co-contraction of opposing flexor and extensor groups (with predominance of extensors) on EMG is a diagnostic criterion for SPS and explains the myalgia and elevated CK. Her normal MRI studies effectively ruled out any focal lesion and did not show signs of encephalitis. Oligoclonal bands in the CSF are a sensitive marker of intrathecal inflammation, although not specific to one diagnosis. The mildly elevated cell count also supports CNS inflammation. In the setting of a lymphocytic pleocytosis and unique oligoclonal bands, it is important to consider infectious, neoplastic, autoimmune, and paraneoplastic causes of neuroinflammatory disorders.
Serum analyses, including antiglutamic acid decarboxylase 65 (GAD65) antibody and anti-amphiphysin antibody, should be ordered. The anti-GAD65 antibody is most commonly elevated in the setting of autoimmune diabetes mellitus; the titer, however, is usually dramatically higher in SPS. The CSF titer of anti-GAD65 antibodies is more specific than the serum titer for SPS. Antibodies against amphiphysin are typically elevated in paraneoplastic SPS, and anti-glycine receptor antibodies are associated with PERM, which commonly does not have elevated anti-GAD65 antibodies.
The serum GAD65 antibody level was greater than 265,000 × 103 IU/µL (normal <5,000), and the CSF level was 11.2 nmol/L (normal: ≤0.02). Serum amphiphysin antibody testing was negative.
Significantly elevated serum and CSF anti-GAD65 antibody levels are highly suggestive of SPS. Stiff person syndrome with rapidly progressive clinical symptoms raises the concern of a paraneoplastic neurologic syndrome. Although anti-amphiphysin antibody – the antibody classically associated with breast cancer and SPS – was negative, anti-GAD65 antibody has been implicated in paraneoplastic SPS with thymoma, lymphoma, and thyroid carcinoma. Paraneoplastic neurologic syndrome can predate a detectable malignancy by several years. As SPS and lymphoma are associated with pruritus and lymphocytosis, imaging is indicated to search for malignancy. Antiglycine receptor antibody, associated with PERM, is not routinely available commercially.
Computed tomography of the chest, abdomen, and pelvis with intravenous contrast revealed a 3.9 × 8.0 × 7.0 cm anterior mediastinal mass (Figure 1, Panel A). Biopsy of the mass demonstrated a thymoma. Given that the patient exhibited no further signs of CNS involvement, her initial transiently altered mental status was attributed to opioids and steroids. As she did not meet the clinical criteria for PERM, testing of antiglycine antibodies was not pursued.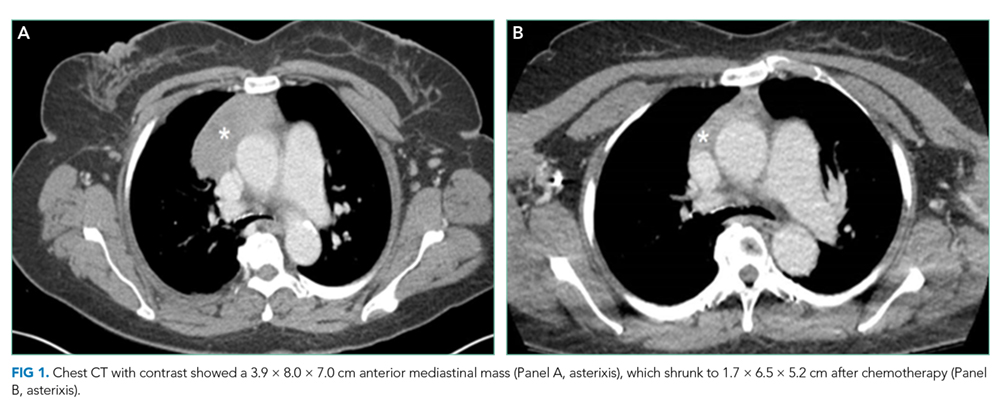
She received scheduled baclofen and diazepam with as needed cyclobenzaprine for continued muscle spasms. Over the next several days, her stiffness, spasms, and myoclonic jerks slowly improved, and she was able to attempt physical therapy (Appendix Video 1; https://youtu.be/d0gLpTgqaCs). She subsequently received intravenous immunoglobulin (IVIG) with further improvement. After five months of scheduled diazepam and baclofen, she was able to ambulate with minimal assistance (Appendix Video 2; https://youtu.be/I00i638u00o). Given the absence of safe tissue planes for resection, the patient received neoadjuvant chemotherapy with four cycles of cyclophosphamide, doxorubicin, and cisplatin. Tumor size decreased to 1.7 × 6.5 × 5.2 cm (Figure 1, Panel B), and she subsequently underwent resection (Figure 2). Pathological analysis demonstrated a type B1 thymoma.
COMMENTARY
SPS is a condition of muscle stiffness and spasticity. Diagnosis is difficult and often delayed due to its rarity, with an approximate prevalence of one to two cases per million people.1 SPS typically occurs in middle age, and women are diagnosed twice as often as men. Classic SPS is characterized by axial and limb muscle stiffness, episodic spasms precipitated by tactile or auditory stimuli, continuous motor unit activity in agonist and antagonist muscles on EMG, high-titer antibody to GAD65 or amphiphysin, and the absence of an alternate diagnosis.2 Variant syndromes have been described, including a milder variant limited to the limbs, a severe variant with brainstem and spinal cord involvement, and a paraneoplastic variant.3 This patient’s clinical presentation, EMG findings, and extraordinarily high anti-GAD titers in the serum and CSF were diagnostic of SPS.
The pathophysiology of SPS is associated with autoantibodies targeting proteins such as GAD65, amphiphysin, gephyrin, and GABAA receptor-associated protein (GABARAP). These proteins are critical to gamma-aminobutyric acid (GABA) signaling, the primary inhibitory neurotransmitter pathway in the CNS (Figure 3).4 The formation of GABA from glutamate is catalyzed by GAD65. Gamma-aminobutyric acid is loaded into secretory vesicles, and amphiphysin facilitates vesicle recycling from the synaptic space.5 In the postsynaptic neuron, GABA binds the GABAA receptor, leading to neuronal hyperpolarization and resistance to excitation. The GABAA receptor is clustered on the plasma membrane through a scaffold formed by gephyrin. GABARAP facilitates this clustering, in part by linking GABAA receptors and gephyrin.6 Autoantibodies to these proteins may be pathogenic; however, the direct effects on their targets are unclear. The end result is decreased GABAergic activity, leading to continuous activation of opposing muscle groups. The resulting stiffness is characteristic of this disorder. Colchicine is known to antagonize GABAA receptor signaling, and this may have brought the underlying diagnosis of SPS to clinical attention.7,8
Symptomatic treatment of SPS targets the GABAergic system. Typically, high doses of scheduled benzodiazepines9 and baclofen10 are necessary. When symptoms are not controlled by GABAergic drugs, immunosuppression with corticosteroids and IVIG has been used, as have plasmapheresis and rituximab.11 The efficacy of the latter, however, was not supported by a randomized, placebo-controlled trial.12 This patient experienced significant improvement with benzodiazepines, baclofen, IVIG, and neoadjuvant chemotherapy prior to thymoma resection. The pruritus, paresthesia, and lymphocytosis also resolved with medical therapy. Interestingly, GABA signaling suppresses itch, suggesting that loss of GABAA signaling may have contributed to the development of pruritus.
SPS occasionally occurs as a paraneoplastic neurologic syndrome. Breast cancer is the most commonly associated malignancy, although associations between thymomas and SPS13 with anti-GAD65 antibodies14 have also been described. The presentation of thymomas is variable, with approximately one-third discovered incidentally on imaging, one-third producing symptoms of local compression, and one-third identified in the setting of another syndrome, most commonly myasthenia gravis. In addition to myasthenia gravis, thymomas have been associated with conditions such as hypogammaglobulinemia, pure red cell aplasia, and agranulocytosis. Stiff person syndrome is a known, albeit infrequently associated, condition.15
A critical step in arriving at the relevant differential diagnosis requires correctly framing the patient’s case.16 The treatment team’s initial frame was “a 69-year-old woman with weakness and elevated CK,” which prioritized causes of weakness and myositis. Stiff person syndrome does not cause weakness, but rather impaired movement from marked stiffness and spasms. The patient’s elevated CK was a result of continual muscle contractions. The physical exam and lack of motor deficit on EMG led the treatment team to reframe as “a 69-year-old woman with severe stiffness and spasms.” Egad! This correct frame was the key to diagnosis and confirmed by EMG and GAD65 antibody testing.
KEY LEARNING POINTS
- Classic SPS is characterized by axial and limb muscle stiffness, episodic spasms precipitated by tactile or auditory stimuli, continuous motor unit activity in agonist and antagonist muscles on EMG, and high-titer antibody to GAD65 or amphiphysin.
- SPS typically occurs in middle age, and women are diagnosed twice as often as men.
- Symptomatic treatment of SPS targets the GABAergic system. Typically, high doses of scheduled benzodiazepines and baclofenare necessary.
- SPS occasionally occurs as a paraneoplastic neurologic syndrome, most commonly in association with breast cancer.
Acknowledgments
The authors wish to thank Jason Kern, MD for his preparation and interpretation of the pathologic image; and the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education, for supporting Reza Manesh, MD.
Disclosures
The authors have nothing to disclose.
Appendix Video 1: This video was taken during a physical therapy session after 1 week of scheduled benzodiazepine and 2 days of intravenous immunoglobuli
Appendix Video 2: This video was taken 5 months after scheduled diazepam and baclofen, and 1 week prior to thymectomy. (https://youtu.be/I00i638u00o)
1. Hadavi S, Noyce AJ, Leslie RD, Giovannoni G. Stiff person syndrome. Pract Neurol. 2011;11(5):272-282. doi: 10.1136/practneurol-2011-000071
2. Dalakas MC. Stiff person syndrome: advances in pathogenesis and therapeutic interventions. Curr Treat Options Neurol. 2009;11(2):102-110. doi: 10.1007/s11940-009-0013-9
3. Murinson BB. Stiff-person syndrome. Neurologist. 2004;10(3):131-137. doi: 10.1097/01.nrl.0000126587.37087.1a
4. Rakocevic G, Floeter MK. Autoimmune stiff person syndrome and related myelopathies: understanding of electrophysiological and immunological processes. Muscle Nerve. 2012;45(5):623-634. doi: 10.1002/mus.23234
5. Zhang B, Zelhof AC. Amphiphysins: raising the BAR for synaptic vesicle recycling and membrane dynamics. Bin-Amphiphysin-Rvsp. Traffic. 2002;3(7):452-460. doi: 10.1034/j.1600-0854.2002.30702.x
6. Tyagarajan SK, Fritschy JM. Gephyrin: a master regulator of neuronal function? Nat Rev Neurosci. 2014;15(3):141-156. doi: 10.1038/nrn3670
7. Bueno OF, Leidenheimer NJ. Colchicine inhibits GABA(A) receptors independently of microtubule depolymerization. Neuropharmacology. 1998;37(3):383-390. doi: 10.1016/S0028-3908(98)00020-3
8. Weiner JL, Buhler AV, Whatley VJ, Harris RA, Dunwiddie TV. Colchicine is a competitive antagonist at human recombinant γ-aminobutyric acidA receptors. J Pharmacol Exp Ther. 1998;284(1):95-102 . PubMed
9. Lorish TR, Thorsteinsson G, Howard FM Jr. Stiff-man syndrome updated. Mayo Clin Proc. 1989;64(6):629-636. doi: 10.1016/S0025-6196(12)65339-7
10. McKeon A, Robinson MT, McEvoy KM, et al. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol. 2012;69(2):230-238. doi: 10.1001/archneurol.2011.991
11. Dalakas MC, Li M, Fujii M, Jacobowitz DM. Stiff person syndrome: quantification, specificity, and intrathecal synthesis of GAD65 antibodies. Neurology. 2001;57(5):780-784. doi: 10.1212/WNL.57.5.780
12. Dalakas MC, Rakocevic G, Dambrosia JM, Alexopoulos H, McElroy B. A double-blind, placebo-controlled study of rituximab in patients with stiff person syndrome. Ann Neurol. 2017;82(2):271-277. doi: 10.1002/ana.25002
13. Hagiwara H, Enomoto-Nakatani S, Sakai K, et al. Stiff-person syndrome associated with invasive thymoma: a case report. J Neurol Sci. 2001;193(1):59-62. doi: 10.1016/S0022-510X(01)00602-5
14. Vernino S, Lennon VA. Autoantibody profiles and neurological correlations of thymoma. Clin Cancer Res. 2004;10(21):7270-7275. doi: 10.1158/1078-0432.CCR-04-0735 PubMed
15. Thomas CR, Wright CD, Loehrer PJ. Thymoma: state of the art. J Clin Oncol. 1999;17(7):2280-2289. doi: 10.1200/JCO.1999.17.7.2280 PubMed
16. Stuart S, Hartig JR, Willett L. The importance of framing. J Gen Intern Med. 2017;32(6):706-710. doi: 10.1007/s11606-016-3964-z PubMed
A 69-year-old woman presented to the clinic with pain in the right great toe lasting several days. She was prescribed colchicine and indomethacin empirically for gout. She took one tablet of colchicine (0.6 mg) every hour until her stools became loose after the eighth tablet. Her toe pain resolved, but two days later she developed bilateral lower extremity pruritus and paresthesia and presented to the emergency department (ED). On physical examination, no rash, weakness, or sensory deficits were observed, and she was able to ambulate without assistance. Her patellar reflexes were normal. The complete blood count was notable for an absolute lymphocyte count of 6,120/µL (normal: 1,100-4,800), and the comprehensive metabolic panel was normal. Serum creatine kinase (CK) was 341 U/L (normal: 24-170) and uric acid 7.7 mg/dL (normal: 2.4-6.4). Her lower extremity symptoms were attributed to colchicine, which was discontinued. She was prescribed diphenhydramine and discharged home.
Monoarthritis of the hallux is the classic manifestation of gout, although other considerations include pseudogout, sesamoiditis, and trauma. The typical side effects of colchicine include diarrhea and myositis. Colchicine-induced muscle injury often results in a modest elevation of CK levels and is associated with myalgia.
Paresthesia is defined as abnormal sensory symptoms that most commonly localize to the peripheral nerves or spinal cord. Acute neuropathies or myelopathies might result from vasculitis, heavy metal toxicity, vitamin deficiencies, and paraneoplastic neurologic syndromes. The normal motor, sensory, and reflex examination, however, make these unlikely.
The neuro-anatomic localization of pruritus is poorly understood but is proposed to include peripheral nerves, spinothalamic tracts, and thalami. Acute pruritus (lasting <6 weeks) typically results from a primary dermatologic process such as a drug reaction, eczema, or xerosis. Less common causes include uremia, cholestasis, and thyroid disease. Pruritus can also be seen with malignancy, most commonly hematologic or paraneoplastic syndromes, or with connective tissue diseases. At this stage, it is unclear whether her pruritus and paresthesia are part of a unifying disease process.
Five days later she re-presented to the ED with nausea and emesis after eating at a restaurant. Her symptoms improved with intravenous fluids, and she was discharged. Four days later she returned with difficulty ambulating, bilateral leg cramping, and continued pruritus and paresthesia. The chemistry panel was normal except for a potassium level of 2.6 mmol/L and a bicarbonate level of 32 mmol/L. She was admitted to the hospital because of severe hypokalemia and impaired ability to ambulate. Her potassium was replenished. Her CK was elevated (3,551 U/L on hospital day 7). She was given cyclobenzaprine, gabapentin, oxycodone, acetaminophen, and prednisone (40 mg); her cramping only mildly improved, and she remained unable to walk. On hospital day five she had visual hallucinations and confusion, which did not resolve with administration of haloperidol; a head CT was unremarkable. On hospital day eight the patient, with her family’s support, left the hospital and presented to a different ED for a second opinion.
Difficulty ambulating often results from weakness, sensory impairment, cerebellar ataxia, extrapyramidal dysfunction (eg, parkinsonism), and pain. In this patient, leg cramping suggests pain or true weakness due to a myopathic process as a contributing factor. Symptoms of muscle disease include cramps, myalgia, and difficulty walking. Causes of elevated CK and myalgia include inflammatory myopathies, endocrinopathies, drugs, infections, and electrolyte abnormalities (eg, hypokalemia). Her age and acuity of presentation decrease the likelihood of a metabolic myopathy due to a disorder of glycogen storage, lipid metabolism, or mitochondrial function. Her hypokalemic metabolic alkalosis likely resulted from vomiting. Hypokalemic periodic paralysis is unlikely as exacerbations typically only last hours to days. As such, her difficulty ambulating, muscle cramps, and elevated CK strongly support a primary myopathic disorder, although additional information regarding the neurologic examination is still required.
Acute changes in mental status without corresponding changes in cranial nerve, motor, or sensory function are common in the hospital setting and frequently relate to delirium, which is the most likely explanation for her confusion. Her age and exposure to muscle relaxants, opiates, and corticosteroids increase her risk considerably. Other possible explanations for isolated changes in mental status include nonconvulsive seizures, central nervous system (CNS) infection, and strokes that involve the thalamus, nondominant parietal lobe, and reticular activating system. A shower of emboli resulting in small multifocal strokes can have the same effect.
She was re-evaluated by her new providers. Her only prior medical history was hypertension, which was treated at home with atenolol and amlodipine. She had emigrated from Nigeria to the US many years prior. She occasionally consumed alcohol and never smoked tobacco or used illicit drugs. She was unsure if she had received a tetanus booster in the past 10 years.
On physical examination, her temperature was 36°C, blood pressure 149/70 mm Hg, pulse 56 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 98% on ambient air. She was diaphoretic and appeared anxious, grabbing both bedrails out of fear of falling. Cardiovascular, pulmonary, abdominal, and skin examinations were normal. She was alert and oriented to her identity, her location, and the time. Cranial nerves II to XII were normal. Tone was normal in her upper extremities but markedly increased in her lower extremities and back. There were spontaneous and stimulus-induced painful spasms, predominantly involving her axial muscles and distal lower extremities. Muscle bulk was normal. Strength was normal in the upper extremities and could not be assessed in the lower extremities due to rigidity. Reflexes were 2+ and symmetric throughout with downgoing toes on Babinski testing. A sensory examination was normal. Gait could not be tested because of the severe muscle spasms. The patient was admitted to the hospital.
Localized muscle spasms may be caused by muscle overuse, but more generalized spasms are associated with systemic diseases such as electrolyte disturbances, toxidromes, tetanus, peripheral nerve hyperexcitability syndromes (including Isaacs syndrome and Morvan syndrome), or stiff person syndrome (SPS). Hypokalemia is unlikely the cause as its correction did not improve her symptoms. Although tetanus is rare in the United States, it remains endemic in the developing world and can cause focal as well as generalized stimulus-induced spasms. The patient should be asked about potential exposure to Clostridium tetani infection, such as incurring a puncture wound. It is also important to consider neuroleptic malignant syndrome and serotonin syndrome, which can cause confusion, elevated CK, and increased muscle tone. Her confusion, however, was transient and the elevated CK preceded the administration of haloperidol.
SPS and progressive encephalomyelitis with rigidity and myoclonus (PERM) provide better explanations for her presentation. Both diseases cause severe spasms, impaired ambulation, and stiffness. They differ in their acuity of onset, accompanying symptoms, antibody associations, and responses to treatment. The rapid onset, paresthesia, and confusion seen in this patient are atypical of SPS. SPS usually presents with subacute-to-chronic stiffness or soreness of muscles in the back and lower extremities, followed by the upper extremities. Rigidity, stimulation-provoked spasms, hyperlordosis, and difficulty ambulating are typically later-stage findings. Her rapid escalation of symptoms is more consistent with PERM, which is often more acute and progressive than typical SPS; however, unlike this patient, PERM commonly causes widespread CNS dysfunction, including persistent encephalopathy, cranial neuropathies, hyperreflexia, and autonomic instability. Both are rare diagnoses that can manifest as a paraneoplastic neurologic syndrome.
Blood tests showed a leukocyte count of 17,350/µL, neutrophils 8,720/µL (normal: 1,500–7,800), lymphocytes 6,130/µL, hemoglobin 11.3 g/dL, and platelets 231,000/µL. The basic metabolic panel was normal. Serum total protein was 6.7 g/dL with albumin 3.5 g/dL. Aspartate aminotransferase (AST) was 94 U/L (normal: 0-31), alanine aminotransferase (ALT) 56 U/L (normal: 0-31), alkaline phosphatase 45 U/L, and total bilirubin 1.1 mg/dL. Vitamin B12 was 868 pg/mL. Hemoglobin A1c and thyrotropin levels were normal. Creatine kinase was 3,757 U/L and lactate dehydrogenase (LDH) 435 U/L (normal: 122-220). The syphilis treponemal test and hepatitis B surface antigen were negative. HIV and hepatitis C antibodies were nonreactive. The anti-nuclear antibody screen was negative and complement C3 and C4 were normal.
Neutrophilia likely reflects glucocorticoid-induced demargination, as opposed to an infectious process, given the temporal association with steroid administration. Persistent mild lymphocytosis is nonspecific but more likely to reflect a reactive rather than a clonal process. Elevated LDH and CK, as well as a greater increase of AST relative to ALT, suggest muscle injury, although mild concomitant hepatic injury cannot be excluded. Normal or negative serum studies for TSH, HIV, ANA, peripheral blood smear, and creatinine eliminate many of the systemic causes of her pruritus, but malignancy and associated paraneoplastic etiologies remain considerations.
The initial work-up for SPS includes electromyography (EMG) which would show spontaneous muscle activity. Her poorly localized sensory abnormalities, transient vestibular symptoms, and confusion warrant an MRI of the brain and spine to evaluate for inflammation (eg, encephalomyelitis), which could be consistent with PERM.
An MRI of the brain and cervicothoracic spine without contrast was significantly limited by motion artifact but without obvious intracranial or cord signal abnormalities. Electromyography demonstrated spontaneous muscle activity in both lower extremities with co-contraction of agonist and antagonist muscles (hamstrings and quadriceps as well as medial gastrocnemius and tibialis anterior). Sensory and motor nerve conductions were normal. Cerebral spinal fluid (CSF) contained six leukocytes (96% lymphocytes) and three red blood cells per microliter; glucose was 67 mg/dL and protein 24 mg/dL. There were two oligoclonal bands unique to the CSF. Cytology was negative for malignant cells.
The EMG narrows the differential diagnosis considerably. Co-contraction of opposing flexor and extensor groups (with predominance of extensors) on EMG is a diagnostic criterion for SPS and explains the myalgia and elevated CK. Her normal MRI studies effectively ruled out any focal lesion and did not show signs of encephalitis. Oligoclonal bands in the CSF are a sensitive marker of intrathecal inflammation, although not specific to one diagnosis. The mildly elevated cell count also supports CNS inflammation. In the setting of a lymphocytic pleocytosis and unique oligoclonal bands, it is important to consider infectious, neoplastic, autoimmune, and paraneoplastic causes of neuroinflammatory disorders.
Serum analyses, including antiglutamic acid decarboxylase 65 (GAD65) antibody and anti-amphiphysin antibody, should be ordered. The anti-GAD65 antibody is most commonly elevated in the setting of autoimmune diabetes mellitus; the titer, however, is usually dramatically higher in SPS. The CSF titer of anti-GAD65 antibodies is more specific than the serum titer for SPS. Antibodies against amphiphysin are typically elevated in paraneoplastic SPS, and anti-glycine receptor antibodies are associated with PERM, which commonly does not have elevated anti-GAD65 antibodies.
The serum GAD65 antibody level was greater than 265,000 × 103 IU/µL (normal <5,000), and the CSF level was 11.2 nmol/L (normal: ≤0.02). Serum amphiphysin antibody testing was negative.
Significantly elevated serum and CSF anti-GAD65 antibody levels are highly suggestive of SPS. Stiff person syndrome with rapidly progressive clinical symptoms raises the concern of a paraneoplastic neurologic syndrome. Although anti-amphiphysin antibody – the antibody classically associated with breast cancer and SPS – was negative, anti-GAD65 antibody has been implicated in paraneoplastic SPS with thymoma, lymphoma, and thyroid carcinoma. Paraneoplastic neurologic syndrome can predate a detectable malignancy by several years. As SPS and lymphoma are associated with pruritus and lymphocytosis, imaging is indicated to search for malignancy. Antiglycine receptor antibody, associated with PERM, is not routinely available commercially.
Computed tomography of the chest, abdomen, and pelvis with intravenous contrast revealed a 3.9 × 8.0 × 7.0 cm anterior mediastinal mass (Figure 1, Panel A). Biopsy of the mass demonstrated a thymoma. Given that the patient exhibited no further signs of CNS involvement, her initial transiently altered mental status was attributed to opioids and steroids. As she did not meet the clinical criteria for PERM, testing of antiglycine antibodies was not pursued.
She received scheduled baclofen and diazepam with as needed cyclobenzaprine for continued muscle spasms. Over the next several days, her stiffness, spasms, and myoclonic jerks slowly improved, and she was able to attempt physical therapy (Appendix Video 1; https://youtu.be/d0gLpTgqaCs). She subsequently received intravenous immunoglobulin (IVIG) with further improvement. After five months of scheduled diazepam and baclofen, she was able to ambulate with minimal assistance (Appendix Video 2; https://youtu.be/I00i638u00o). Given the absence of safe tissue planes for resection, the patient received neoadjuvant chemotherapy with four cycles of cyclophosphamide, doxorubicin, and cisplatin. Tumor size decreased to 1.7 × 6.5 × 5.2 cm (Figure 1, Panel B), and she subsequently underwent resection (Figure 2). Pathological analysis demonstrated a type B1 thymoma.
COMMENTARY
SPS is a condition of muscle stiffness and spasticity. Diagnosis is difficult and often delayed due to its rarity, with an approximate prevalence of one to two cases per million people.1 SPS typically occurs in middle age, and women are diagnosed twice as often as men. Classic SPS is characterized by axial and limb muscle stiffness, episodic spasms precipitated by tactile or auditory stimuli, continuous motor unit activity in agonist and antagonist muscles on EMG, high-titer antibody to GAD65 or amphiphysin, and the absence of an alternate diagnosis.2 Variant syndromes have been described, including a milder variant limited to the limbs, a severe variant with brainstem and spinal cord involvement, and a paraneoplastic variant.3 This patient’s clinical presentation, EMG findings, and extraordinarily high anti-GAD titers in the serum and CSF were diagnostic of SPS.
The pathophysiology of SPS is associated with autoantibodies targeting proteins such as GAD65, amphiphysin, gephyrin, and GABAA receptor-associated protein (GABARAP). These proteins are critical to gamma-aminobutyric acid (GABA) signaling, the primary inhibitory neurotransmitter pathway in the CNS (Figure 3).4 The formation of GABA from glutamate is catalyzed by GAD65. Gamma-aminobutyric acid is loaded into secretory vesicles, and amphiphysin facilitates vesicle recycling from the synaptic space.5 In the postsynaptic neuron, GABA binds the GABAA receptor, leading to neuronal hyperpolarization and resistance to excitation. The GABAA receptor is clustered on the plasma membrane through a scaffold formed by gephyrin. GABARAP facilitates this clustering, in part by linking GABAA receptors and gephyrin.6 Autoantibodies to these proteins may be pathogenic; however, the direct effects on their targets are unclear. The end result is decreased GABAergic activity, leading to continuous activation of opposing muscle groups. The resulting stiffness is characteristic of this disorder. Colchicine is known to antagonize GABAA receptor signaling, and this may have brought the underlying diagnosis of SPS to clinical attention.7,8
Symptomatic treatment of SPS targets the GABAergic system. Typically, high doses of scheduled benzodiazepines9 and baclofen10 are necessary. When symptoms are not controlled by GABAergic drugs, immunosuppression with corticosteroids and IVIG has been used, as have plasmapheresis and rituximab.11 The efficacy of the latter, however, was not supported by a randomized, placebo-controlled trial.12 This patient experienced significant improvement with benzodiazepines, baclofen, IVIG, and neoadjuvant chemotherapy prior to thymoma resection. The pruritus, paresthesia, and lymphocytosis also resolved with medical therapy. Interestingly, GABA signaling suppresses itch, suggesting that loss of GABAA signaling may have contributed to the development of pruritus.
SPS occasionally occurs as a paraneoplastic neurologic syndrome. Breast cancer is the most commonly associated malignancy, although associations between thymomas and SPS13 with anti-GAD65 antibodies14 have also been described. The presentation of thymomas is variable, with approximately one-third discovered incidentally on imaging, one-third producing symptoms of local compression, and one-third identified in the setting of another syndrome, most commonly myasthenia gravis. In addition to myasthenia gravis, thymomas have been associated with conditions such as hypogammaglobulinemia, pure red cell aplasia, and agranulocytosis. Stiff person syndrome is a known, albeit infrequently associated, condition.15
A critical step in arriving at the relevant differential diagnosis requires correctly framing the patient’s case.16 The treatment team’s initial frame was “a 69-year-old woman with weakness and elevated CK,” which prioritized causes of weakness and myositis. Stiff person syndrome does not cause weakness, but rather impaired movement from marked stiffness and spasms. The patient’s elevated CK was a result of continual muscle contractions. The physical exam and lack of motor deficit on EMG led the treatment team to reframe as “a 69-year-old woman with severe stiffness and spasms.” Egad! This correct frame was the key to diagnosis and confirmed by EMG and GAD65 antibody testing.
KEY LEARNING POINTS
- Classic SPS is characterized by axial and limb muscle stiffness, episodic spasms precipitated by tactile or auditory stimuli, continuous motor unit activity in agonist and antagonist muscles on EMG, and high-titer antibody to GAD65 or amphiphysin.
- SPS typically occurs in middle age, and women are diagnosed twice as often as men.
- Symptomatic treatment of SPS targets the GABAergic system. Typically, high doses of scheduled benzodiazepines and baclofenare necessary.
- SPS occasionally occurs as a paraneoplastic neurologic syndrome, most commonly in association with breast cancer.
Acknowledgments
The authors wish to thank Jason Kern, MD for his preparation and interpretation of the pathologic image; and the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education, for supporting Reza Manesh, MD.
Disclosures
The authors have nothing to disclose.
Appendix Video 1: This video was taken during a physical therapy session after 1 week of scheduled benzodiazepine and 2 days of intravenous immunoglobuli
Appendix Video 2: This video was taken 5 months after scheduled diazepam and baclofen, and 1 week prior to thymectomy. (https://youtu.be/I00i638u00o)
A 69-year-old woman presented to the clinic with pain in the right great toe lasting several days. She was prescribed colchicine and indomethacin empirically for gout. She took one tablet of colchicine (0.6 mg) every hour until her stools became loose after the eighth tablet. Her toe pain resolved, but two days later she developed bilateral lower extremity pruritus and paresthesia and presented to the emergency department (ED). On physical examination, no rash, weakness, or sensory deficits were observed, and she was able to ambulate without assistance. Her patellar reflexes were normal. The complete blood count was notable for an absolute lymphocyte count of 6,120/µL (normal: 1,100-4,800), and the comprehensive metabolic panel was normal. Serum creatine kinase (CK) was 341 U/L (normal: 24-170) and uric acid 7.7 mg/dL (normal: 2.4-6.4). Her lower extremity symptoms were attributed to colchicine, which was discontinued. She was prescribed diphenhydramine and discharged home.
Monoarthritis of the hallux is the classic manifestation of gout, although other considerations include pseudogout, sesamoiditis, and trauma. The typical side effects of colchicine include diarrhea and myositis. Colchicine-induced muscle injury often results in a modest elevation of CK levels and is associated with myalgia.
Paresthesia is defined as abnormal sensory symptoms that most commonly localize to the peripheral nerves or spinal cord. Acute neuropathies or myelopathies might result from vasculitis, heavy metal toxicity, vitamin deficiencies, and paraneoplastic neurologic syndromes. The normal motor, sensory, and reflex examination, however, make these unlikely.
The neuro-anatomic localization of pruritus is poorly understood but is proposed to include peripheral nerves, spinothalamic tracts, and thalami. Acute pruritus (lasting <6 weeks) typically results from a primary dermatologic process such as a drug reaction, eczema, or xerosis. Less common causes include uremia, cholestasis, and thyroid disease. Pruritus can also be seen with malignancy, most commonly hematologic or paraneoplastic syndromes, or with connective tissue diseases. At this stage, it is unclear whether her pruritus and paresthesia are part of a unifying disease process.
Five days later she re-presented to the ED with nausea and emesis after eating at a restaurant. Her symptoms improved with intravenous fluids, and she was discharged. Four days later she returned with difficulty ambulating, bilateral leg cramping, and continued pruritus and paresthesia. The chemistry panel was normal except for a potassium level of 2.6 mmol/L and a bicarbonate level of 32 mmol/L. She was admitted to the hospital because of severe hypokalemia and impaired ability to ambulate. Her potassium was replenished. Her CK was elevated (3,551 U/L on hospital day 7). She was given cyclobenzaprine, gabapentin, oxycodone, acetaminophen, and prednisone (40 mg); her cramping only mildly improved, and she remained unable to walk. On hospital day five she had visual hallucinations and confusion, which did not resolve with administration of haloperidol; a head CT was unremarkable. On hospital day eight the patient, with her family’s support, left the hospital and presented to a different ED for a second opinion.
Difficulty ambulating often results from weakness, sensory impairment, cerebellar ataxia, extrapyramidal dysfunction (eg, parkinsonism), and pain. In this patient, leg cramping suggests pain or true weakness due to a myopathic process as a contributing factor. Symptoms of muscle disease include cramps, myalgia, and difficulty walking. Causes of elevated CK and myalgia include inflammatory myopathies, endocrinopathies, drugs, infections, and electrolyte abnormalities (eg, hypokalemia). Her age and acuity of presentation decrease the likelihood of a metabolic myopathy due to a disorder of glycogen storage, lipid metabolism, or mitochondrial function. Her hypokalemic metabolic alkalosis likely resulted from vomiting. Hypokalemic periodic paralysis is unlikely as exacerbations typically only last hours to days. As such, her difficulty ambulating, muscle cramps, and elevated CK strongly support a primary myopathic disorder, although additional information regarding the neurologic examination is still required.
Acute changes in mental status without corresponding changes in cranial nerve, motor, or sensory function are common in the hospital setting and frequently relate to delirium, which is the most likely explanation for her confusion. Her age and exposure to muscle relaxants, opiates, and corticosteroids increase her risk considerably. Other possible explanations for isolated changes in mental status include nonconvulsive seizures, central nervous system (CNS) infection, and strokes that involve the thalamus, nondominant parietal lobe, and reticular activating system. A shower of emboli resulting in small multifocal strokes can have the same effect.
She was re-evaluated by her new providers. Her only prior medical history was hypertension, which was treated at home with atenolol and amlodipine. She had emigrated from Nigeria to the US many years prior. She occasionally consumed alcohol and never smoked tobacco or used illicit drugs. She was unsure if she had received a tetanus booster in the past 10 years.
On physical examination, her temperature was 36°C, blood pressure 149/70 mm Hg, pulse 56 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 98% on ambient air. She was diaphoretic and appeared anxious, grabbing both bedrails out of fear of falling. Cardiovascular, pulmonary, abdominal, and skin examinations were normal. She was alert and oriented to her identity, her location, and the time. Cranial nerves II to XII were normal. Tone was normal in her upper extremities but markedly increased in her lower extremities and back. There were spontaneous and stimulus-induced painful spasms, predominantly involving her axial muscles and distal lower extremities. Muscle bulk was normal. Strength was normal in the upper extremities and could not be assessed in the lower extremities due to rigidity. Reflexes were 2+ and symmetric throughout with downgoing toes on Babinski testing. A sensory examination was normal. Gait could not be tested because of the severe muscle spasms. The patient was admitted to the hospital.
Localized muscle spasms may be caused by muscle overuse, but more generalized spasms are associated with systemic diseases such as electrolyte disturbances, toxidromes, tetanus, peripheral nerve hyperexcitability syndromes (including Isaacs syndrome and Morvan syndrome), or stiff person syndrome (SPS). Hypokalemia is unlikely the cause as its correction did not improve her symptoms. Although tetanus is rare in the United States, it remains endemic in the developing world and can cause focal as well as generalized stimulus-induced spasms. The patient should be asked about potential exposure to Clostridium tetani infection, such as incurring a puncture wound. It is also important to consider neuroleptic malignant syndrome and serotonin syndrome, which can cause confusion, elevated CK, and increased muscle tone. Her confusion, however, was transient and the elevated CK preceded the administration of haloperidol.
SPS and progressive encephalomyelitis with rigidity and myoclonus (PERM) provide better explanations for her presentation. Both diseases cause severe spasms, impaired ambulation, and stiffness. They differ in their acuity of onset, accompanying symptoms, antibody associations, and responses to treatment. The rapid onset, paresthesia, and confusion seen in this patient are atypical of SPS. SPS usually presents with subacute-to-chronic stiffness or soreness of muscles in the back and lower extremities, followed by the upper extremities. Rigidity, stimulation-provoked spasms, hyperlordosis, and difficulty ambulating are typically later-stage findings. Her rapid escalation of symptoms is more consistent with PERM, which is often more acute and progressive than typical SPS; however, unlike this patient, PERM commonly causes widespread CNS dysfunction, including persistent encephalopathy, cranial neuropathies, hyperreflexia, and autonomic instability. Both are rare diagnoses that can manifest as a paraneoplastic neurologic syndrome.
Blood tests showed a leukocyte count of 17,350/µL, neutrophils 8,720/µL (normal: 1,500–7,800), lymphocytes 6,130/µL, hemoglobin 11.3 g/dL, and platelets 231,000/µL. The basic metabolic panel was normal. Serum total protein was 6.7 g/dL with albumin 3.5 g/dL. Aspartate aminotransferase (AST) was 94 U/L (normal: 0-31), alanine aminotransferase (ALT) 56 U/L (normal: 0-31), alkaline phosphatase 45 U/L, and total bilirubin 1.1 mg/dL. Vitamin B12 was 868 pg/mL. Hemoglobin A1c and thyrotropin levels were normal. Creatine kinase was 3,757 U/L and lactate dehydrogenase (LDH) 435 U/L (normal: 122-220). The syphilis treponemal test and hepatitis B surface antigen were negative. HIV and hepatitis C antibodies were nonreactive. The anti-nuclear antibody screen was negative and complement C3 and C4 were normal.
Neutrophilia likely reflects glucocorticoid-induced demargination, as opposed to an infectious process, given the temporal association with steroid administration. Persistent mild lymphocytosis is nonspecific but more likely to reflect a reactive rather than a clonal process. Elevated LDH and CK, as well as a greater increase of AST relative to ALT, suggest muscle injury, although mild concomitant hepatic injury cannot be excluded. Normal or negative serum studies for TSH, HIV, ANA, peripheral blood smear, and creatinine eliminate many of the systemic causes of her pruritus, but malignancy and associated paraneoplastic etiologies remain considerations.
The initial work-up for SPS includes electromyography (EMG) which would show spontaneous muscle activity. Her poorly localized sensory abnormalities, transient vestibular symptoms, and confusion warrant an MRI of the brain and spine to evaluate for inflammation (eg, encephalomyelitis), which could be consistent with PERM.
An MRI of the brain and cervicothoracic spine without contrast was significantly limited by motion artifact but without obvious intracranial or cord signal abnormalities. Electromyography demonstrated spontaneous muscle activity in both lower extremities with co-contraction of agonist and antagonist muscles (hamstrings and quadriceps as well as medial gastrocnemius and tibialis anterior). Sensory and motor nerve conductions were normal. Cerebral spinal fluid (CSF) contained six leukocytes (96% lymphocytes) and three red blood cells per microliter; glucose was 67 mg/dL and protein 24 mg/dL. There were two oligoclonal bands unique to the CSF. Cytology was negative for malignant cells.
The EMG narrows the differential diagnosis considerably. Co-contraction of opposing flexor and extensor groups (with predominance of extensors) on EMG is a diagnostic criterion for SPS and explains the myalgia and elevated CK. Her normal MRI studies effectively ruled out any focal lesion and did not show signs of encephalitis. Oligoclonal bands in the CSF are a sensitive marker of intrathecal inflammation, although not specific to one diagnosis. The mildly elevated cell count also supports CNS inflammation. In the setting of a lymphocytic pleocytosis and unique oligoclonal bands, it is important to consider infectious, neoplastic, autoimmune, and paraneoplastic causes of neuroinflammatory disorders.
Serum analyses, including antiglutamic acid decarboxylase 65 (GAD65) antibody and anti-amphiphysin antibody, should be ordered. The anti-GAD65 antibody is most commonly elevated in the setting of autoimmune diabetes mellitus; the titer, however, is usually dramatically higher in SPS. The CSF titer of anti-GAD65 antibodies is more specific than the serum titer for SPS. Antibodies against amphiphysin are typically elevated in paraneoplastic SPS, and anti-glycine receptor antibodies are associated with PERM, which commonly does not have elevated anti-GAD65 antibodies.
The serum GAD65 antibody level was greater than 265,000 × 103 IU/µL (normal <5,000), and the CSF level was 11.2 nmol/L (normal: ≤0.02). Serum amphiphysin antibody testing was negative.
Significantly elevated serum and CSF anti-GAD65 antibody levels are highly suggestive of SPS. Stiff person syndrome with rapidly progressive clinical symptoms raises the concern of a paraneoplastic neurologic syndrome. Although anti-amphiphysin antibody – the antibody classically associated with breast cancer and SPS – was negative, anti-GAD65 antibody has been implicated in paraneoplastic SPS with thymoma, lymphoma, and thyroid carcinoma. Paraneoplastic neurologic syndrome can predate a detectable malignancy by several years. As SPS and lymphoma are associated with pruritus and lymphocytosis, imaging is indicated to search for malignancy. Antiglycine receptor antibody, associated with PERM, is not routinely available commercially.
Computed tomography of the chest, abdomen, and pelvis with intravenous contrast revealed a 3.9 × 8.0 × 7.0 cm anterior mediastinal mass (Figure 1, Panel A). Biopsy of the mass demonstrated a thymoma. Given that the patient exhibited no further signs of CNS involvement, her initial transiently altered mental status was attributed to opioids and steroids. As she did not meet the clinical criteria for PERM, testing of antiglycine antibodies was not pursued.
She received scheduled baclofen and diazepam with as needed cyclobenzaprine for continued muscle spasms. Over the next several days, her stiffness, spasms, and myoclonic jerks slowly improved, and she was able to attempt physical therapy (Appendix Video 1; https://youtu.be/d0gLpTgqaCs). She subsequently received intravenous immunoglobulin (IVIG) with further improvement. After five months of scheduled diazepam and baclofen, she was able to ambulate with minimal assistance (Appendix Video 2; https://youtu.be/I00i638u00o). Given the absence of safe tissue planes for resection, the patient received neoadjuvant chemotherapy with four cycles of cyclophosphamide, doxorubicin, and cisplatin. Tumor size decreased to 1.7 × 6.5 × 5.2 cm (Figure 1, Panel B), and she subsequently underwent resection (Figure 2). Pathological analysis demonstrated a type B1 thymoma.
COMMENTARY
SPS is a condition of muscle stiffness and spasticity. Diagnosis is difficult and often delayed due to its rarity, with an approximate prevalence of one to two cases per million people.1 SPS typically occurs in middle age, and women are diagnosed twice as often as men. Classic SPS is characterized by axial and limb muscle stiffness, episodic spasms precipitated by tactile or auditory stimuli, continuous motor unit activity in agonist and antagonist muscles on EMG, high-titer antibody to GAD65 or amphiphysin, and the absence of an alternate diagnosis.2 Variant syndromes have been described, including a milder variant limited to the limbs, a severe variant with brainstem and spinal cord involvement, and a paraneoplastic variant.3 This patient’s clinical presentation, EMG findings, and extraordinarily high anti-GAD titers in the serum and CSF were diagnostic of SPS.
The pathophysiology of SPS is associated with autoantibodies targeting proteins such as GAD65, amphiphysin, gephyrin, and GABAA receptor-associated protein (GABARAP). These proteins are critical to gamma-aminobutyric acid (GABA) signaling, the primary inhibitory neurotransmitter pathway in the CNS (Figure 3).4 The formation of GABA from glutamate is catalyzed by GAD65. Gamma-aminobutyric acid is loaded into secretory vesicles, and amphiphysin facilitates vesicle recycling from the synaptic space.5 In the postsynaptic neuron, GABA binds the GABAA receptor, leading to neuronal hyperpolarization and resistance to excitation. The GABAA receptor is clustered on the plasma membrane through a scaffold formed by gephyrin. GABARAP facilitates this clustering, in part by linking GABAA receptors and gephyrin.6 Autoantibodies to these proteins may be pathogenic; however, the direct effects on their targets are unclear. The end result is decreased GABAergic activity, leading to continuous activation of opposing muscle groups. The resulting stiffness is characteristic of this disorder. Colchicine is known to antagonize GABAA receptor signaling, and this may have brought the underlying diagnosis of SPS to clinical attention.7,8
Symptomatic treatment of SPS targets the GABAergic system. Typically, high doses of scheduled benzodiazepines9 and baclofen10 are necessary. When symptoms are not controlled by GABAergic drugs, immunosuppression with corticosteroids and IVIG has been used, as have plasmapheresis and rituximab.11 The efficacy of the latter, however, was not supported by a randomized, placebo-controlled trial.12 This patient experienced significant improvement with benzodiazepines, baclofen, IVIG, and neoadjuvant chemotherapy prior to thymoma resection. The pruritus, paresthesia, and lymphocytosis also resolved with medical therapy. Interestingly, GABA signaling suppresses itch, suggesting that loss of GABAA signaling may have contributed to the development of pruritus.
SPS occasionally occurs as a paraneoplastic neurologic syndrome. Breast cancer is the most commonly associated malignancy, although associations between thymomas and SPS13 with anti-GAD65 antibodies14 have also been described. The presentation of thymomas is variable, with approximately one-third discovered incidentally on imaging, one-third producing symptoms of local compression, and one-third identified in the setting of another syndrome, most commonly myasthenia gravis. In addition to myasthenia gravis, thymomas have been associated with conditions such as hypogammaglobulinemia, pure red cell aplasia, and agranulocytosis. Stiff person syndrome is a known, albeit infrequently associated, condition.15
A critical step in arriving at the relevant differential diagnosis requires correctly framing the patient’s case.16 The treatment team’s initial frame was “a 69-year-old woman with weakness and elevated CK,” which prioritized causes of weakness and myositis. Stiff person syndrome does not cause weakness, but rather impaired movement from marked stiffness and spasms. The patient’s elevated CK was a result of continual muscle contractions. The physical exam and lack of motor deficit on EMG led the treatment team to reframe as “a 69-year-old woman with severe stiffness and spasms.” Egad! This correct frame was the key to diagnosis and confirmed by EMG and GAD65 antibody testing.
KEY LEARNING POINTS
- Classic SPS is characterized by axial and limb muscle stiffness, episodic spasms precipitated by tactile or auditory stimuli, continuous motor unit activity in agonist and antagonist muscles on EMG, and high-titer antibody to GAD65 or amphiphysin.
- SPS typically occurs in middle age, and women are diagnosed twice as often as men.
- Symptomatic treatment of SPS targets the GABAergic system. Typically, high doses of scheduled benzodiazepines and baclofenare necessary.
- SPS occasionally occurs as a paraneoplastic neurologic syndrome, most commonly in association with breast cancer.
Acknowledgments
The authors wish to thank Jason Kern, MD for his preparation and interpretation of the pathologic image; and the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education, for supporting Reza Manesh, MD.
Disclosures
The authors have nothing to disclose.
Appendix Video 1: This video was taken during a physical therapy session after 1 week of scheduled benzodiazepine and 2 days of intravenous immunoglobuli
Appendix Video 2: This video was taken 5 months after scheduled diazepam and baclofen, and 1 week prior to thymectomy. (https://youtu.be/I00i638u00o)
1. Hadavi S, Noyce AJ, Leslie RD, Giovannoni G. Stiff person syndrome. Pract Neurol. 2011;11(5):272-282. doi: 10.1136/practneurol-2011-000071
2. Dalakas MC. Stiff person syndrome: advances in pathogenesis and therapeutic interventions. Curr Treat Options Neurol. 2009;11(2):102-110. doi: 10.1007/s11940-009-0013-9
3. Murinson BB. Stiff-person syndrome. Neurologist. 2004;10(3):131-137. doi: 10.1097/01.nrl.0000126587.37087.1a
4. Rakocevic G, Floeter MK. Autoimmune stiff person syndrome and related myelopathies: understanding of electrophysiological and immunological processes. Muscle Nerve. 2012;45(5):623-634. doi: 10.1002/mus.23234
5. Zhang B, Zelhof AC. Amphiphysins: raising the BAR for synaptic vesicle recycling and membrane dynamics. Bin-Amphiphysin-Rvsp. Traffic. 2002;3(7):452-460. doi: 10.1034/j.1600-0854.2002.30702.x
6. Tyagarajan SK, Fritschy JM. Gephyrin: a master regulator of neuronal function? Nat Rev Neurosci. 2014;15(3):141-156. doi: 10.1038/nrn3670
7. Bueno OF, Leidenheimer NJ. Colchicine inhibits GABA(A) receptors independently of microtubule depolymerization. Neuropharmacology. 1998;37(3):383-390. doi: 10.1016/S0028-3908(98)00020-3
8. Weiner JL, Buhler AV, Whatley VJ, Harris RA, Dunwiddie TV. Colchicine is a competitive antagonist at human recombinant γ-aminobutyric acidA receptors. J Pharmacol Exp Ther. 1998;284(1):95-102 . PubMed
9. Lorish TR, Thorsteinsson G, Howard FM Jr. Stiff-man syndrome updated. Mayo Clin Proc. 1989;64(6):629-636. doi: 10.1016/S0025-6196(12)65339-7
10. McKeon A, Robinson MT, McEvoy KM, et al. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol. 2012;69(2):230-238. doi: 10.1001/archneurol.2011.991
11. Dalakas MC, Li M, Fujii M, Jacobowitz DM. Stiff person syndrome: quantification, specificity, and intrathecal synthesis of GAD65 antibodies. Neurology. 2001;57(5):780-784. doi: 10.1212/WNL.57.5.780
12. Dalakas MC, Rakocevic G, Dambrosia JM, Alexopoulos H, McElroy B. A double-blind, placebo-controlled study of rituximab in patients with stiff person syndrome. Ann Neurol. 2017;82(2):271-277. doi: 10.1002/ana.25002
13. Hagiwara H, Enomoto-Nakatani S, Sakai K, et al. Stiff-person syndrome associated with invasive thymoma: a case report. J Neurol Sci. 2001;193(1):59-62. doi: 10.1016/S0022-510X(01)00602-5
14. Vernino S, Lennon VA. Autoantibody profiles and neurological correlations of thymoma. Clin Cancer Res. 2004;10(21):7270-7275. doi: 10.1158/1078-0432.CCR-04-0735 PubMed
15. Thomas CR, Wright CD, Loehrer PJ. Thymoma: state of the art. J Clin Oncol. 1999;17(7):2280-2289. doi: 10.1200/JCO.1999.17.7.2280 PubMed
16. Stuart S, Hartig JR, Willett L. The importance of framing. J Gen Intern Med. 2017;32(6):706-710. doi: 10.1007/s11606-016-3964-z PubMed
1. Hadavi S, Noyce AJ, Leslie RD, Giovannoni G. Stiff person syndrome. Pract Neurol. 2011;11(5):272-282. doi: 10.1136/practneurol-2011-000071
2. Dalakas MC. Stiff person syndrome: advances in pathogenesis and therapeutic interventions. Curr Treat Options Neurol. 2009;11(2):102-110. doi: 10.1007/s11940-009-0013-9
3. Murinson BB. Stiff-person syndrome. Neurologist. 2004;10(3):131-137. doi: 10.1097/01.nrl.0000126587.37087.1a
4. Rakocevic G, Floeter MK. Autoimmune stiff person syndrome and related myelopathies: understanding of electrophysiological and immunological processes. Muscle Nerve. 2012;45(5):623-634. doi: 10.1002/mus.23234
5. Zhang B, Zelhof AC. Amphiphysins: raising the BAR for synaptic vesicle recycling and membrane dynamics. Bin-Amphiphysin-Rvsp. Traffic. 2002;3(7):452-460. doi: 10.1034/j.1600-0854.2002.30702.x
6. Tyagarajan SK, Fritschy JM. Gephyrin: a master regulator of neuronal function? Nat Rev Neurosci. 2014;15(3):141-156. doi: 10.1038/nrn3670
7. Bueno OF, Leidenheimer NJ. Colchicine inhibits GABA(A) receptors independently of microtubule depolymerization. Neuropharmacology. 1998;37(3):383-390. doi: 10.1016/S0028-3908(98)00020-3
8. Weiner JL, Buhler AV, Whatley VJ, Harris RA, Dunwiddie TV. Colchicine is a competitive antagonist at human recombinant γ-aminobutyric acidA receptors. J Pharmacol Exp Ther. 1998;284(1):95-102 . PubMed
9. Lorish TR, Thorsteinsson G, Howard FM Jr. Stiff-man syndrome updated. Mayo Clin Proc. 1989;64(6):629-636. doi: 10.1016/S0025-6196(12)65339-7
10. McKeon A, Robinson MT, McEvoy KM, et al. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol. 2012;69(2):230-238. doi: 10.1001/archneurol.2011.991
11. Dalakas MC, Li M, Fujii M, Jacobowitz DM. Stiff person syndrome: quantification, specificity, and intrathecal synthesis of GAD65 antibodies. Neurology. 2001;57(5):780-784. doi: 10.1212/WNL.57.5.780
12. Dalakas MC, Rakocevic G, Dambrosia JM, Alexopoulos H, McElroy B. A double-blind, placebo-controlled study of rituximab in patients with stiff person syndrome. Ann Neurol. 2017;82(2):271-277. doi: 10.1002/ana.25002
13. Hagiwara H, Enomoto-Nakatani S, Sakai K, et al. Stiff-person syndrome associated with invasive thymoma: a case report. J Neurol Sci. 2001;193(1):59-62. doi: 10.1016/S0022-510X(01)00602-5
14. Vernino S, Lennon VA. Autoantibody profiles and neurological correlations of thymoma. Clin Cancer Res. 2004;10(21):7270-7275. doi: 10.1158/1078-0432.CCR-04-0735 PubMed
15. Thomas CR, Wright CD, Loehrer PJ. Thymoma: state of the art. J Clin Oncol. 1999;17(7):2280-2289. doi: 10.1200/JCO.1999.17.7.2280 PubMed
16. Stuart S, Hartig JR, Willett L. The importance of framing. J Gen Intern Med. 2017;32(6):706-710. doi: 10.1007/s11606-016-3964-z PubMed
© 2019 Society of Hospital Medicine
A Protean Protein
A 39-year-old man presented to a neurologist with three weeks of progressive leg weakness associated with numbness in his feet and fingertips. His medical history included hypertriglyceridemia, hypogonadism, and gout. He was taking fenofibrate and colchicine as needed. There was no family history of neurologic issues. He did not smoke or drink alcohol.
The patient appeared well with a heart rate of 76 beats per minute, blood pressure 133/72 mm Hg, temperature 36.6°C, respiratory rate 16 breaths per minute, and oxygen saturation 100% on room air. His cardiopulmonary and abdominal examinations were normal. His skin was warm and dry without rashes. On neurologic examination, upper extremity strength and sensation was normal. Bilateral hip flexion, knee flexion, and knee extension strength was 4/5; bilateral ankle dorsiflexion and plantar flexion strength was 3/5. Reflexes were trace in the arms and absent at the patellae and ankles. He had symmetric, length-dependent reduction in vibration, pinprick, and light touch sensation in his legs.
Peripheral neuropathy presenting with ascending symmetric motor and sensory deficits progressing over three weeks raises the suspicion of an acquired inflammatory demyelinating polyneuropathy (AIDP), a variant of Guillain-Barre Syndrome. Alternative causes of acute polyneuropathy include thiamine (B1) deficiency, vasculitis, sarcoidosis, or malignancy, particularly lymphoma and multiple myeloma. Further evaluation should include electromyography, nerve conduction studies, lumbar puncture with cerebrospinal fluid (CSF) protein, glucose, and cell count differential. Follow-up laboratory testing based on results of the above may include serum protein electrophoresis (SPEP), serum free light chains (sFLC), vitamin B12, human immunodeficiency virus (HIV), hepatitis B and C testing, antinuclear antibody, and erythrocyte sedimentation rate.
Electromyography and nerve conduction studies revealed a sensorimotor mixed axonal/demyelinating polyneuropathy in all extremities. CSF analysis found one white cell per mm3, glucose of 93 mg/dL, and protein of 313 mg/dL. Magnetic resonance imaging (MRI) of the spine without contrast showed normal cord parenchyma. The vitamin B12 level was 441 pg/mL (normal >200 pg/mL). Antibodies to HIV-1, HIV-2, hepatitis C virus, and Borrelia burgdorferi were negative. Serum protein electrophoresis (SPEP) and immunofixation were normal.
The patient received two courses of intravenous immunoglobulin (IVIG) for suspected AIDP. His weakness progressed over the next several weeks to the point that he required a wheelchair.
Progression of symptoms beyond three weeks and lack of response to IVIG are atypical for AIDP. Alternate diagnoses for a sensorimotor polyneuropathy should be considered. Causes of subacute or chronic demyelinating polyneuropathy include inflammatory conditions (chronic inflammatory demyelinating polyneuropathy [CIDP], connective-tissue disorders), paraprotein disorders (myeloma, amyloidosis, lymphoplasmacytic lymphoma), paraneoplastic syndromes, infectious diseases (HIV, Lyme disease), infiltrative disorders (sarcoidosis), medications or toxins, and hereditary disorders. Of these etiologies, the first three seem the most likely given the history and clinical course, the negative HIV and Lyme testing, and the absence of exposures and family history. Normal SPEP and immunofixation make paraprotein disorders less likely, but sFLC testing should be sent to evaluate for a light chain-only paraprotein. A paraneoplastic antibody panel and a CT of the chest, abdomen, and pelvis should be ordered to evaluate for sarcoidosis, lymphoma, or other malignancies. Although a peripheral nerve biopsy would further classify the polyneuropathy, it is of low diagnostic yield in patients with subacute and chronic distal symmetric polyneuropathies and is associated with significant morbidity. In the absence of history or physical exam findings to narrow the differential diagnosis for polyneuropathy, testing for paraneoplastic antibodies and imaging is appropriate.
The patient tested negative for antiganglioside GM1 and antimyelin-associated glycoprotein antibodies. Urine arsenic, lead, and mercury levels were normal. Tests for serum antinuclear antibody, rapid plasmin reagin, and a paraneoplastic neuropathy panel including amphiphysin antibody, CV2 antibody, and Hu auto-antibody were negative. Repeat electrodiagnostic testing was consistent with CIDP. The patient received prednisone 60 mg daily for six weeks and was then tapered to 30 mg daily over six weeks. Concurrently, he underwent twelve cycles of plasma exchange. His strength improved, and he could walk with a cane; however, weakness recurred when steroids were further tapered.
He was maintained on prednisone 50 mg daily. Over the next year, the patient’s lower extremities became flaccid and severely atrophied. He developed hyperpigmented patches on his trunk, severe gastroesophageal reflux disease (GERD), dysphonia, and gynecomastia. He had lost 60 pounds since symptom onset. He was prescribed levothyroxine for subclinical hypothyroidism (thyroid stimulating hormone 12.63 µIU/mL [normal 0.10-5.50 µIU/mL], free thyroxine 0.8 ng/dL [0.8-1.7 ng/dL]).
At this point, the diagnosis of CIDP should be questioned, and additional investigation is warranted. Although improvement was initially observed with plasma exchange and steroids, subsequent progression of symptoms despite prednisone suggests a nonimmune-mediated etiology, such as a neoplastic or infiltrative process. Conversely, negative serologic testing for paraneoplastic antibodies may be due to an antibody that has not been well characterized.
While prednisone could explain GERD and gynecomastia, the weight loss, dysphonia, and subclinical hypothyroidism may offer clues to the diagnosis underlying the neurological symptoms. Weight loss raises suspicion of a hypercatabolic process such as cancer, cachexia, systemic inflammation, heart failure, or chronic obstructive pulmonary disease. Causes of dysphonia relevant to this presentation include neurologic dysfunction related to malignant invasion of the vagus nerve or demyelinating disease. Subclinical hypothyroidism due to chronic autoimmune thyroiditis seems most likely in the absence of a medication effect or thyroid injury, yet infiltrative disorders of the thyroid (eg, amyloidosis, sarcoidosis, lymphoma) should also be considered. A diagnosis that unifies the neurologic and nonneurologic findings would be desirable; lymphoma with paraneoplastic peripheral neuropathy manifesting as CIDP seems most likely. As of yet, CT of the chest, abdomen, and pelvis or an 18-Fluoro-deoxyglucose positron emission tomography (FDG-PET) scan have not been obtained and would be helpful to evaluate for underlying malignancy. Further evaluation for a paraprotein disorder that includes sFLC is also still indicated to rule out a paraneoplastic disorder that may be associated with polyneuropathy.
Repeat SPEP and serum immunofixation were normal. sFLC assay showed elevated levels of both kappa and lambda light chains with a ratio of 0.61 (reference range: 0.26-1.25). Urine protein electrophoresis (UPEP) from a 24-hour specimen showed a homogenous band in the gamma region, but urine immunofixation demonstrated polyclonal light chains. The plasma vascular endothelial growth factor (VEGF) level was 612 pg/mL (reference range, 31-86 pg/mL).
CT imaging of the chest, abdomen, and pelvis with contrast demonstrated an enlarged liver and spleen and possible splenic infarcts. A skeletal survey and whole-body FGD-PET scan were normal. The patient declined bone marrow biopsy.
Polyneuropathy secondary to a monoclonal protein was previously considered, and an SPEP was normal. Full evaluation for a monoclonal protein additionally requires sFLC testing. If clinical suspicion remains high after a negative result, 24-hour UPEP and urine immunofixation should be obtained. Normal results in this case argue against the presence of a monoclonal protein.
The presence of a monoclonal protein and polyneuropathy are mandatory diagnostic criteria for POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes), a plasma cell proliferative disorder. Major diagnostic criteria include osteosclerotic bone lesions, Castleman’s disease, and markedly elevated VEGF levels. Castleman’s disease is a lymphoproliferative disorder characterized by angiofollicular lymphoid hyperplasia that results in lymphadenopathy in one or multiple lymph node regions. Imaging studies reveal organomegaly, one of many minor criteria, but not bone lesions or lymphadenopathy. A diagnosis of POEMS syndrome requires the presence of both mandatory, one major, and one minor criteria. Since only one of two of the mandatory criteria are met at this point, a diagnosis of POEMS syndrome cannot be made.
Eighteen months after symptom onset, the patient presented to the emergency department with dyspnea, orthopnea, and lower extremity edema. B-type natriuretic peptide was 1564 pg/mL. Transthoracic echocardiography showed a severely dilated and hypertrophied left ventricle. Left ventricular ejection fraction was 20%. A furosemide infusion was initiated. Angiography of the coronary vessels was not performed. Congo red stain of an abdominal adipose biopsy was negative for amyloid.
On hospital day five, he developed gangrenous changes in his right first toe. CT angiography of the abdomen and lower extremities demonstrated patent three vessel runoff to the foot with an infrarenal aortic thrombus. Heparin infusion was started. On hospital day 10, the patient developed expressive aphasia and somnolence, prompting intubation for airway protection. MRI and MR angiography (MRA) of the brain and cerebral vessels revealed multiple bilateral acute ischemic strokes (Figure 1) without flow limiting stenosis in cerebral vessels.
These clinical developments lead to an important opportunity to rethink this patient’s working diagnosis. The new diagnosis of heart failure in this young patient with polyneuropathy raises suspicion for an infiltrative cardiomyopathy such as amyloidosis, sarcoidosis, or Fabry disease. Of these, Fabry disease is the least likely because it is typically characterized by a painful burning sensation in response to specific triggers. Although polyneuropathy and heart failure may be concurrently observed with both sarcoidosis and amyloidosis, the absence of an apparent arrhythmia make amyloidosis the more likely of these two diagnoses. The development of an arterial thrombus and multiple strokes may represent emboli from a cardiac thrombus.
Cardiac imaging and tissue biopsy of the heart or other affected organs would distinguish between these diagnostic possibilities. An abdominal adipose biopsy negative for amyloid does not rule out amyloidosis, as the test is approximately 80% sensitive when cardiac amyloidosis is present and varies depending on the etiology of the amyloid protein (ie, light chain vs transthyretin). Evaluation of cardiac amyloid in the setting of peripheral neuropathy should include echocardiography (as was performed here) and repeat testing for a monoclonal protein.
If clinical suspicion of a paraprotein-associated disorder remains high and both SPEP and sFLC are normal, it is important to obtain a 24-hour UPEP and immunofixation. A monoclonal protein can be overlooked by SPEP and serum immunofixation if the monoclonal protein is composed only of a light chain or if the monoclonal protein is IgD or IgE. In these rare circumstances, sFLC analysis or 24-hour UPEP and immunofixation should mitigate the potential for a falsely negative SPEP/IFE. These studies are normal in this case, which argues against the presence of a monoclonal protein.
Transesophageal echocardiography showed grade IV atheromatous plaque within the descending thoracic aorta with mobile elements suggesting a superimposed thrombus; there was no intracardiac shunt or thrombus. MRA of the neck and great vessels was normal.
Testing for heparin-induced thrombocytopenia (HIT) was sent due to thrombocytopenia and the presence of thrombosis. An immunoassay for antiheparin-platelet factor 4 (anti-PF4) antibodies was substantially positive (optical density 2.178); however, functional testing with a washed platelet heparin-induced platelet activation assay was negative. Anticoagulation was changed to argatroban due to concern for HIT. Dry gangrenous changes developed in all distal toes on the right foot and three toes on the left foot. A right radial artery thrombus formed at the site of a prior arterial line.
Thrombocytopenia that develops between the fifth and tenth day following heparin exposure in a patient with new thromboses is consistent with HIT. However, the patient’s infrarenal aortic thrombus preceded the initiation of heparin, and negative functional testing undermines the diagnosis of HIT in this case. Therefore, the arterial thromboses may be related to an underlying unifying diagnosis.
A third SPEP showed a 0.1 g/dL M-spike in the gamma region, but standard immunofixation did not reveal a monoclonal protein (Figure 2). However, a specific request for immunofixation testing using IgD antisera detected an IgD heavy chain. A lambda chain comprising 3% of urine protein was detected on 24-hour urine immunofixation but was not detectable by serum immunofixation. Bone marrow biopsy demonstrated plasma cells comprising 5% of bone marrow cellularity (Figure 3); flow cytometry of the aspirate demonstrated an abnormal lambda-restricted plasma cell population.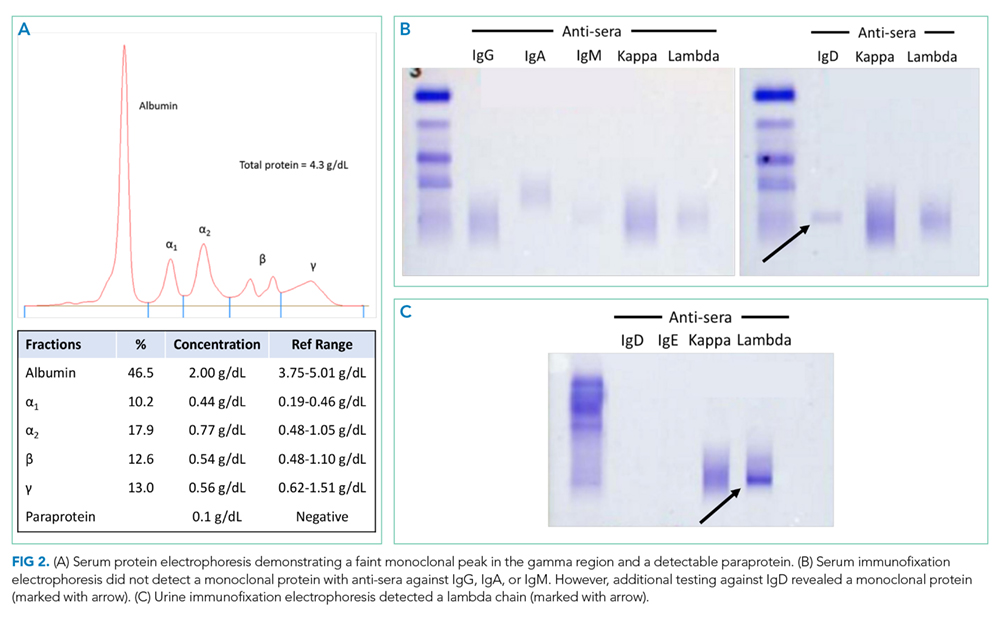
When a monoclonal protein is identified but does not react with standard antisera to detect IgG, IgM, and IgA, immunofixation with IgD and IgE antisera are necessary to rule out a monoclonal IgD or IgE protein. The underlying IgD isotype coupled with its low abundance made detection of this monoclonal protein especially challenging. With the discovery of a monoclonal protein in the context of polyneuropathy, the mandatory criteria of POEMS syndrome are met. The elevated VEGF level and hypothyroidism meet major and minor criteria, respectively. Arterial thromboses and heart failure are other features that may be observed in cases of POEMS syndrome.
POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes) was diagnosed. Prednisone was continued, and weekly cyclophosphamide was initiated. After six weeks, the VEGF level remained elevated, and a neurologic examination showed minimal improvement. Due to poor respiratory muscle strength and difficulty managing secretions, he underwent percutaneous tracheostomy and gastrostomy tube placement. Unfortunately, his condition further deteriorated and he subsequently died of sepsis from pneumonia.
An autopsy revealed acute bronchopneumonia and multiple acute and subacute cerebral infarctions. There was extensive peripheral mixed axonal/demyelinating neuropathy, hepatosplenomegaly, atrophy of the thyroid and adrenal glands, hyperpigmented patches and thickened integument, and severe aortic and coronary atherosclerotic disease with a healed myocardial infarction.
DISCUSSION
POEMS syndrome1 is a rare constellation of clinical and laboratory findings resulting from an underlying plasma cell proliferative disorder. This paraneoplastic syndrome is characterized by the chronic overproduction of proinflammatory and proangiogenic cytokines, including VEGF, which are postulated to drive its manifestations,2 though the exact pathogenesis is not understood. Some of the disease’s most common features are summarized by its name: polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes.3
The International Myeloma Working Group (IMWG) diagnostic criteria1 (Table) require the presence of both mandatory criteria (polyneuropathy and monoclonal plasma cell proliferation), plus at least one major and one minor criterion. Delayed diagnosis or misdiagnosis of this protean disorder is often driven by its rarity and clinical overlap with other paraprotein-associated polyneuropathies. These include amyloidosis, cryoglobulinemia, and monoclonal gammopathy of undetermined significance (MGUS), which can all produce antibodies directed against neural antigens. In addition, polyneuropathy is often the first and most striking manifestation of POEMS syndrome, fostering confusion with CIDP as both disorders are subacute, symmetric, motor-dominant, mixed axonal/demyelinating polyneuropathies.4
IgD and IgE monoclonal gammopathies are extremely rare. IgD myeloma, for instance, accounts for 2% of multiple myeloma cases, and IgE myeloma has been reported fewer than 50 times.5 IgD is secreted only in very small amounts, ordinarily representing 0.25% of the immunoglobulins in serum, while the majority is found in the plasma membranes of mature B-cells.6 These monoclonal gammopathies often escape detection for two reasons: (1) the very low paraprotein concentration produces undetectable or small M-protein levels on electrophoresis,5 and (2) immunofixation is routinely performed without antisera against IgD and IgE heavy chains.7
While this case depicts a rare manifestation of a rare disease, the principles underlying its elusive diagnosis are routinely encountered. Recognition of the specific limitations of the SPEP, UPEP, sFLC, and immunofixation tests, outlined below, can assist the hospitalist when suspicion for paraproteinemia is high.
First, low levels of monoclonal proteins may be associated with a normal SPEP. Accordingly, suspicion of a plasma cell dyscrasia should prompt serum immunofixation, even when the electrophoretic pattern appears normal.8
Second, laboratories routinely perform immunofixation with antisera against IgG, IgA, and IgM heavy chains and kappa and lambda light chains, whereas testing with IgD or IgE antisera must be specifically requested. Thus, clinicians should screen for the presence of IgD and IgE in patients with an apparently free monoclonal immunoglobulin light chain in the serum or with a monoclonal serum protein and negative immunofixation. In this case, the paraprotein was not detected on the first two serum electrophoreses, likely due to a low serum concentration, then missed on immunofixation due to a lack of IgD antiserum. On admission to the hospital, this patient had a very low paraprotein concentration (0.1 g/dL) on SPEP, and the lab initially reported negative immunofixation. When asked to test specifically for IgD and IgE, the lab ran a more comprehensive immunofixation revealing IgD heavy chain paraprotein.
Third, this case illustrates the limitations of the sFLC assay. IMWG guidelines specify that sFLC assay in combination with SPEP and serum immunofixation is sufficient to screen for monoclonal plasma cell proliferative disorders other than light chain amyloidosis (which requires all the serum tests as well as 24-hour urine immunofixation).9 Though the sFLC assay has been demonstrated to be more sensitive than urine analysis for detecting monoclonal free light chains,10 it is still subject to false negatives. Polyclonal gammopathy or reduced renal clearance with accumulation of free light chains in the serum may mask the presence of low levels of monoclonal sFLC,11 the latter of which likely explains why the sFLC ratio was repeatedly normal in this case. In these circumstances, monoclonal free light chains can be identified by urine studies.11 In this case, 24-hour urine immunofixation detected the excess light chain that was not evident on the sFLC assay. Even with these pitfalls in mind, there is still no evident explanation as to why the 24-hour urine studies done prior to the patient’s hospital admission did not reveal a monoclonal light chain.
This case also highlights the thrombotic diathesis in POEMS syndrome. Although the patient was treated with argatroban for suspected HIT, it is likely that the HIT antibody result was a false positive, and his thrombi were better explained by POEMS syndrome in and of itself. Coronary, limb, and cerebral artery thromboses have been linked to POEMS syndrome,12,13 all of which were present in this case. Laboratory testing for HIT involves an immunoassay to detect circulating HIT antibody and a functional assay to measure platelet activity in the presence of patient serum and heparin. The immunoassay binds anti-PF4/heparin complex irrespective of its ability to activate platelets. The presence of nonspecific antibodies may lead to cross-reactions with the immunoassay test components, which has been demonstrated in cases of MGUS.14 In this case, elevated production of monoclonal antibodies by plasma cells may have led to false-positive results. With moderate to high clinical suspicion of HIT, the combination of a positive immunoassay and negative functional assay (as in this case) make the diagnosis of HIT indeterminate.15
TEACHING POINTS
- If a monoclonal protein is suggested by SPEP but cannot be identified by standard immunofixation, request immunofixation for IgD or IgE. Screen patients for IgD and IgE paraproteins before making a diagnosis of light chain multiple myeloma.
- Polyclonal gammopathy or reduced renal clearance with accumulation of free light chains in the serum may mask the presence of low levels of monoclonal FLC and result in a normal sFLC ratio.
- Thrombosis is a less-recognized but documented feature of POEMS syndrome which may be mediated by the overproduction of proinflammatory and proangiogenic cytokines, though the precise pathogenesis is unknown.
Acknowledgment
The authors thank Dr. Theodore Kurtz and Dr. Anne Deucher from the department of laboratory medicine at the University of California, San Francisco for providing their respective expertise in clinical chemistry and hematopathology.
Disclosures
The authors have no conflicts of interests to disclose.
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-ee548. doi: 10.1016/S1470-2045(14)70442-5.
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-ee548. doi: 10.1016/S1470-2045(14)70442-5. PubMed
2. Watanabe O, Arimura K, Kitajima I, Osame M, Maruyama I. Greatly raised vascular endothelial growth factor (VEGF) in POEMS syndrome. Lancet. 1996;347(9002):702. doi: 10.1016/S0140-6736(96)91261-1. PubMed
3. Dispenzieri A. How I treat POEMS syndrome. Blood. 2012;119(24):5650-5658. doi: 10.1182/blood-2012-03-378992. PubMed
4. Nasu S, Misawa S, Sekiguchi Y, et al. Different neurological and physiological profiles in POEMS syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry. 2012;83(5):476-479. doi: 10.1136/jnnp-2011-301706. PubMed
5. Pandey S, Kyle RA. Unusual myelomas: a review of IgD and IgE variants. Oncology. 2013;27(8):798-803. PubMed
6. Vladutiu AO. Immunoglobulin D: properties, measurement, and clinical relevance. Clin Diagn Lab Immunol. 2000;7(2):131-140. doi: 10.1128/CDLI.7.2.131-140.2000. PubMed
7. Sinclair D, Cranfield T. IgD myeloma: A potential missed diagnosis. Ann Clin Biochem. 2001;38(5):564-565. doi: 10.1177/000456320103800517. PubMed
8. Dimopoulos M, Kyle R, Fermand JP, et al. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood. 2011;117(18):4701-4705. doi: 10.1182/blood-2010-10-299529. PubMed
9. Dispenzieri A, Kyle R, Merlini G, et al. International Myeloma Working Group. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia. 2009;23(2):215-224. doi: 10.1038/leu.2008.307. PubMed
10. Dejoie T, Attal M, Moreau P, Harousseau JL, Avet-Loiseau H. Comparison of serum free light chain and urine electrophoresis for the detection of the light chain component of monoclonal immunoglobulins in light chain and intact immunoglobulin multiple myeloma. Haematologica. 2016;101(3):356-362. doi: 10.3324/haematol.2015.126797. PubMed
11. Levinson SS. Polyclonal free light chain of Ig may interfere with interpretation of monoclonal free light chain κ/λ ratio. Ann Clin Lab Sci. 2010;40(4):348-353. PubMed
12. Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003;101(7):2496-2506. doi: 10.1182/blood-2002-07-2299. PubMed
13. Dupont SA, Dispenzieri A, Mauermann ML, Rabinstein AA, Brown RD. Cerebral infarction in POEMS syndrome: incidence, risk factors, and imaging characteristics. Neurology. 2009;73(16):1308-1312. doi: 10.1212/WNL.0b013e3181bd136b. PubMed
14. Markovic I, Debeljak Z, Bosnjak B, Marijanovic M. False positive immunoassay for heparin-induced thrombocytopenia in the presence of monoclonal gammopathy: a case report. Biochemia Medica. 2017;27(3):030801. doi: 10.11613/BM.2017.030801. PubMed
15. Cuker A, Cines DB. How I treat heparin-induced thrombocytopenia. Blood. 2012;119(10):2209-2218. doi: 10.1182/blood-2011-11-376293. PubMed
A 39-year-old man presented to a neurologist with three weeks of progressive leg weakness associated with numbness in his feet and fingertips. His medical history included hypertriglyceridemia, hypogonadism, and gout. He was taking fenofibrate and colchicine as needed. There was no family history of neurologic issues. He did not smoke or drink alcohol.
The patient appeared well with a heart rate of 76 beats per minute, blood pressure 133/72 mm Hg, temperature 36.6°C, respiratory rate 16 breaths per minute, and oxygen saturation 100% on room air. His cardiopulmonary and abdominal examinations were normal. His skin was warm and dry without rashes. On neurologic examination, upper extremity strength and sensation was normal. Bilateral hip flexion, knee flexion, and knee extension strength was 4/5; bilateral ankle dorsiflexion and plantar flexion strength was 3/5. Reflexes were trace in the arms and absent at the patellae and ankles. He had symmetric, length-dependent reduction in vibration, pinprick, and light touch sensation in his legs.
Peripheral neuropathy presenting with ascending symmetric motor and sensory deficits progressing over three weeks raises the suspicion of an acquired inflammatory demyelinating polyneuropathy (AIDP), a variant of Guillain-Barre Syndrome. Alternative causes of acute polyneuropathy include thiamine (B1) deficiency, vasculitis, sarcoidosis, or malignancy, particularly lymphoma and multiple myeloma. Further evaluation should include electromyography, nerve conduction studies, lumbar puncture with cerebrospinal fluid (CSF) protein, glucose, and cell count differential. Follow-up laboratory testing based on results of the above may include serum protein electrophoresis (SPEP), serum free light chains (sFLC), vitamin B12, human immunodeficiency virus (HIV), hepatitis B and C testing, antinuclear antibody, and erythrocyte sedimentation rate.
Electromyography and nerve conduction studies revealed a sensorimotor mixed axonal/demyelinating polyneuropathy in all extremities. CSF analysis found one white cell per mm3, glucose of 93 mg/dL, and protein of 313 mg/dL. Magnetic resonance imaging (MRI) of the spine without contrast showed normal cord parenchyma. The vitamin B12 level was 441 pg/mL (normal >200 pg/mL). Antibodies to HIV-1, HIV-2, hepatitis C virus, and Borrelia burgdorferi were negative. Serum protein electrophoresis (SPEP) and immunofixation were normal.
The patient received two courses of intravenous immunoglobulin (IVIG) for suspected AIDP. His weakness progressed over the next several weeks to the point that he required a wheelchair.
Progression of symptoms beyond three weeks and lack of response to IVIG are atypical for AIDP. Alternate diagnoses for a sensorimotor polyneuropathy should be considered. Causes of subacute or chronic demyelinating polyneuropathy include inflammatory conditions (chronic inflammatory demyelinating polyneuropathy [CIDP], connective-tissue disorders), paraprotein disorders (myeloma, amyloidosis, lymphoplasmacytic lymphoma), paraneoplastic syndromes, infectious diseases (HIV, Lyme disease), infiltrative disorders (sarcoidosis), medications or toxins, and hereditary disorders. Of these etiologies, the first three seem the most likely given the history and clinical course, the negative HIV and Lyme testing, and the absence of exposures and family history. Normal SPEP and immunofixation make paraprotein disorders less likely, but sFLC testing should be sent to evaluate for a light chain-only paraprotein. A paraneoplastic antibody panel and a CT of the chest, abdomen, and pelvis should be ordered to evaluate for sarcoidosis, lymphoma, or other malignancies. Although a peripheral nerve biopsy would further classify the polyneuropathy, it is of low diagnostic yield in patients with subacute and chronic distal symmetric polyneuropathies and is associated with significant morbidity. In the absence of history or physical exam findings to narrow the differential diagnosis for polyneuropathy, testing for paraneoplastic antibodies and imaging is appropriate.
The patient tested negative for antiganglioside GM1 and antimyelin-associated glycoprotein antibodies. Urine arsenic, lead, and mercury levels were normal. Tests for serum antinuclear antibody, rapid plasmin reagin, and a paraneoplastic neuropathy panel including amphiphysin antibody, CV2 antibody, and Hu auto-antibody were negative. Repeat electrodiagnostic testing was consistent with CIDP. The patient received prednisone 60 mg daily for six weeks and was then tapered to 30 mg daily over six weeks. Concurrently, he underwent twelve cycles of plasma exchange. His strength improved, and he could walk with a cane; however, weakness recurred when steroids were further tapered.
He was maintained on prednisone 50 mg daily. Over the next year, the patient’s lower extremities became flaccid and severely atrophied. He developed hyperpigmented patches on his trunk, severe gastroesophageal reflux disease (GERD), dysphonia, and gynecomastia. He had lost 60 pounds since symptom onset. He was prescribed levothyroxine for subclinical hypothyroidism (thyroid stimulating hormone 12.63 µIU/mL [normal 0.10-5.50 µIU/mL], free thyroxine 0.8 ng/dL [0.8-1.7 ng/dL]).
At this point, the diagnosis of CIDP should be questioned, and additional investigation is warranted. Although improvement was initially observed with plasma exchange and steroids, subsequent progression of symptoms despite prednisone suggests a nonimmune-mediated etiology, such as a neoplastic or infiltrative process. Conversely, negative serologic testing for paraneoplastic antibodies may be due to an antibody that has not been well characterized.
While prednisone could explain GERD and gynecomastia, the weight loss, dysphonia, and subclinical hypothyroidism may offer clues to the diagnosis underlying the neurological symptoms. Weight loss raises suspicion of a hypercatabolic process such as cancer, cachexia, systemic inflammation, heart failure, or chronic obstructive pulmonary disease. Causes of dysphonia relevant to this presentation include neurologic dysfunction related to malignant invasion of the vagus nerve or demyelinating disease. Subclinical hypothyroidism due to chronic autoimmune thyroiditis seems most likely in the absence of a medication effect or thyroid injury, yet infiltrative disorders of the thyroid (eg, amyloidosis, sarcoidosis, lymphoma) should also be considered. A diagnosis that unifies the neurologic and nonneurologic findings would be desirable; lymphoma with paraneoplastic peripheral neuropathy manifesting as CIDP seems most likely. As of yet, CT of the chest, abdomen, and pelvis or an 18-Fluoro-deoxyglucose positron emission tomography (FDG-PET) scan have not been obtained and would be helpful to evaluate for underlying malignancy. Further evaluation for a paraprotein disorder that includes sFLC is also still indicated to rule out a paraneoplastic disorder that may be associated with polyneuropathy.
Repeat SPEP and serum immunofixation were normal. sFLC assay showed elevated levels of both kappa and lambda light chains with a ratio of 0.61 (reference range: 0.26-1.25). Urine protein electrophoresis (UPEP) from a 24-hour specimen showed a homogenous band in the gamma region, but urine immunofixation demonstrated polyclonal light chains. The plasma vascular endothelial growth factor (VEGF) level was 612 pg/mL (reference range, 31-86 pg/mL).
CT imaging of the chest, abdomen, and pelvis with contrast demonstrated an enlarged liver and spleen and possible splenic infarcts. A skeletal survey and whole-body FGD-PET scan were normal. The patient declined bone marrow biopsy.
Polyneuropathy secondary to a monoclonal protein was previously considered, and an SPEP was normal. Full evaluation for a monoclonal protein additionally requires sFLC testing. If clinical suspicion remains high after a negative result, 24-hour UPEP and urine immunofixation should be obtained. Normal results in this case argue against the presence of a monoclonal protein.
The presence of a monoclonal protein and polyneuropathy are mandatory diagnostic criteria for POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes), a plasma cell proliferative disorder. Major diagnostic criteria include osteosclerotic bone lesions, Castleman’s disease, and markedly elevated VEGF levels. Castleman’s disease is a lymphoproliferative disorder characterized by angiofollicular lymphoid hyperplasia that results in lymphadenopathy in one or multiple lymph node regions. Imaging studies reveal organomegaly, one of many minor criteria, but not bone lesions or lymphadenopathy. A diagnosis of POEMS syndrome requires the presence of both mandatory, one major, and one minor criteria. Since only one of two of the mandatory criteria are met at this point, a diagnosis of POEMS syndrome cannot be made.
Eighteen months after symptom onset, the patient presented to the emergency department with dyspnea, orthopnea, and lower extremity edema. B-type natriuretic peptide was 1564 pg/mL. Transthoracic echocardiography showed a severely dilated and hypertrophied left ventricle. Left ventricular ejection fraction was 20%. A furosemide infusion was initiated. Angiography of the coronary vessels was not performed. Congo red stain of an abdominal adipose biopsy was negative for amyloid.
On hospital day five, he developed gangrenous changes in his right first toe. CT angiography of the abdomen and lower extremities demonstrated patent three vessel runoff to the foot with an infrarenal aortic thrombus. Heparin infusion was started. On hospital day 10, the patient developed expressive aphasia and somnolence, prompting intubation for airway protection. MRI and MR angiography (MRA) of the brain and cerebral vessels revealed multiple bilateral acute ischemic strokes (Figure 1) without flow limiting stenosis in cerebral vessels.
These clinical developments lead to an important opportunity to rethink this patient’s working diagnosis. The new diagnosis of heart failure in this young patient with polyneuropathy raises suspicion for an infiltrative cardiomyopathy such as amyloidosis, sarcoidosis, or Fabry disease. Of these, Fabry disease is the least likely because it is typically characterized by a painful burning sensation in response to specific triggers. Although polyneuropathy and heart failure may be concurrently observed with both sarcoidosis and amyloidosis, the absence of an apparent arrhythmia make amyloidosis the more likely of these two diagnoses. The development of an arterial thrombus and multiple strokes may represent emboli from a cardiac thrombus.
Cardiac imaging and tissue biopsy of the heart or other affected organs would distinguish between these diagnostic possibilities. An abdominal adipose biopsy negative for amyloid does not rule out amyloidosis, as the test is approximately 80% sensitive when cardiac amyloidosis is present and varies depending on the etiology of the amyloid protein (ie, light chain vs transthyretin). Evaluation of cardiac amyloid in the setting of peripheral neuropathy should include echocardiography (as was performed here) and repeat testing for a monoclonal protein.
If clinical suspicion of a paraprotein-associated disorder remains high and both SPEP and sFLC are normal, it is important to obtain a 24-hour UPEP and immunofixation. A monoclonal protein can be overlooked by SPEP and serum immunofixation if the monoclonal protein is composed only of a light chain or if the monoclonal protein is IgD or IgE. In these rare circumstances, sFLC analysis or 24-hour UPEP and immunofixation should mitigate the potential for a falsely negative SPEP/IFE. These studies are normal in this case, which argues against the presence of a monoclonal protein.
Transesophageal echocardiography showed grade IV atheromatous plaque within the descending thoracic aorta with mobile elements suggesting a superimposed thrombus; there was no intracardiac shunt or thrombus. MRA of the neck and great vessels was normal.
Testing for heparin-induced thrombocytopenia (HIT) was sent due to thrombocytopenia and the presence of thrombosis. An immunoassay for antiheparin-platelet factor 4 (anti-PF4) antibodies was substantially positive (optical density 2.178); however, functional testing with a washed platelet heparin-induced platelet activation assay was negative. Anticoagulation was changed to argatroban due to concern for HIT. Dry gangrenous changes developed in all distal toes on the right foot and three toes on the left foot. A right radial artery thrombus formed at the site of a prior arterial line.
Thrombocytopenia that develops between the fifth and tenth day following heparin exposure in a patient with new thromboses is consistent with HIT. However, the patient’s infrarenal aortic thrombus preceded the initiation of heparin, and negative functional testing undermines the diagnosis of HIT in this case. Therefore, the arterial thromboses may be related to an underlying unifying diagnosis.
A third SPEP showed a 0.1 g/dL M-spike in the gamma region, but standard immunofixation did not reveal a monoclonal protein (Figure 2). However, a specific request for immunofixation testing using IgD antisera detected an IgD heavy chain. A lambda chain comprising 3% of urine protein was detected on 24-hour urine immunofixation but was not detectable by serum immunofixation. Bone marrow biopsy demonstrated plasma cells comprising 5% of bone marrow cellularity (Figure 3); flow cytometry of the aspirate demonstrated an abnormal lambda-restricted plasma cell population.
When a monoclonal protein is identified but does not react with standard antisera to detect IgG, IgM, and IgA, immunofixation with IgD and IgE antisera are necessary to rule out a monoclonal IgD or IgE protein. The underlying IgD isotype coupled with its low abundance made detection of this monoclonal protein especially challenging. With the discovery of a monoclonal protein in the context of polyneuropathy, the mandatory criteria of POEMS syndrome are met. The elevated VEGF level and hypothyroidism meet major and minor criteria, respectively. Arterial thromboses and heart failure are other features that may be observed in cases of POEMS syndrome.
POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes) was diagnosed. Prednisone was continued, and weekly cyclophosphamide was initiated. After six weeks, the VEGF level remained elevated, and a neurologic examination showed minimal improvement. Due to poor respiratory muscle strength and difficulty managing secretions, he underwent percutaneous tracheostomy and gastrostomy tube placement. Unfortunately, his condition further deteriorated and he subsequently died of sepsis from pneumonia.
An autopsy revealed acute bronchopneumonia and multiple acute and subacute cerebral infarctions. There was extensive peripheral mixed axonal/demyelinating neuropathy, hepatosplenomegaly, atrophy of the thyroid and adrenal glands, hyperpigmented patches and thickened integument, and severe aortic and coronary atherosclerotic disease with a healed myocardial infarction.
DISCUSSION
POEMS syndrome1 is a rare constellation of clinical and laboratory findings resulting from an underlying plasma cell proliferative disorder. This paraneoplastic syndrome is characterized by the chronic overproduction of proinflammatory and proangiogenic cytokines, including VEGF, which are postulated to drive its manifestations,2 though the exact pathogenesis is not understood. Some of the disease’s most common features are summarized by its name: polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes.3
The International Myeloma Working Group (IMWG) diagnostic criteria1 (Table) require the presence of both mandatory criteria (polyneuropathy and monoclonal plasma cell proliferation), plus at least one major and one minor criterion. Delayed diagnosis or misdiagnosis of this protean disorder is often driven by its rarity and clinical overlap with other paraprotein-associated polyneuropathies. These include amyloidosis, cryoglobulinemia, and monoclonal gammopathy of undetermined significance (MGUS), which can all produce antibodies directed against neural antigens. In addition, polyneuropathy is often the first and most striking manifestation of POEMS syndrome, fostering confusion with CIDP as both disorders are subacute, symmetric, motor-dominant, mixed axonal/demyelinating polyneuropathies.4
IgD and IgE monoclonal gammopathies are extremely rare. IgD myeloma, for instance, accounts for 2% of multiple myeloma cases, and IgE myeloma has been reported fewer than 50 times.5 IgD is secreted only in very small amounts, ordinarily representing 0.25% of the immunoglobulins in serum, while the majority is found in the plasma membranes of mature B-cells.6 These monoclonal gammopathies often escape detection for two reasons: (1) the very low paraprotein concentration produces undetectable or small M-protein levels on electrophoresis,5 and (2) immunofixation is routinely performed without antisera against IgD and IgE heavy chains.7
While this case depicts a rare manifestation of a rare disease, the principles underlying its elusive diagnosis are routinely encountered. Recognition of the specific limitations of the SPEP, UPEP, sFLC, and immunofixation tests, outlined below, can assist the hospitalist when suspicion for paraproteinemia is high.
First, low levels of monoclonal proteins may be associated with a normal SPEP. Accordingly, suspicion of a plasma cell dyscrasia should prompt serum immunofixation, even when the electrophoretic pattern appears normal.8
Second, laboratories routinely perform immunofixation with antisera against IgG, IgA, and IgM heavy chains and kappa and lambda light chains, whereas testing with IgD or IgE antisera must be specifically requested. Thus, clinicians should screen for the presence of IgD and IgE in patients with an apparently free monoclonal immunoglobulin light chain in the serum or with a monoclonal serum protein and negative immunofixation. In this case, the paraprotein was not detected on the first two serum electrophoreses, likely due to a low serum concentration, then missed on immunofixation due to a lack of IgD antiserum. On admission to the hospital, this patient had a very low paraprotein concentration (0.1 g/dL) on SPEP, and the lab initially reported negative immunofixation. When asked to test specifically for IgD and IgE, the lab ran a more comprehensive immunofixation revealing IgD heavy chain paraprotein.
Third, this case illustrates the limitations of the sFLC assay. IMWG guidelines specify that sFLC assay in combination with SPEP and serum immunofixation is sufficient to screen for monoclonal plasma cell proliferative disorders other than light chain amyloidosis (which requires all the serum tests as well as 24-hour urine immunofixation).9 Though the sFLC assay has been demonstrated to be more sensitive than urine analysis for detecting monoclonal free light chains,10 it is still subject to false negatives. Polyclonal gammopathy or reduced renal clearance with accumulation of free light chains in the serum may mask the presence of low levels of monoclonal sFLC,11 the latter of which likely explains why the sFLC ratio was repeatedly normal in this case. In these circumstances, monoclonal free light chains can be identified by urine studies.11 In this case, 24-hour urine immunofixation detected the excess light chain that was not evident on the sFLC assay. Even with these pitfalls in mind, there is still no evident explanation as to why the 24-hour urine studies done prior to the patient’s hospital admission did not reveal a monoclonal light chain.
This case also highlights the thrombotic diathesis in POEMS syndrome. Although the patient was treated with argatroban for suspected HIT, it is likely that the HIT antibody result was a false positive, and his thrombi were better explained by POEMS syndrome in and of itself. Coronary, limb, and cerebral artery thromboses have been linked to POEMS syndrome,12,13 all of which were present in this case. Laboratory testing for HIT involves an immunoassay to detect circulating HIT antibody and a functional assay to measure platelet activity in the presence of patient serum and heparin. The immunoassay binds anti-PF4/heparin complex irrespective of its ability to activate platelets. The presence of nonspecific antibodies may lead to cross-reactions with the immunoassay test components, which has been demonstrated in cases of MGUS.14 In this case, elevated production of monoclonal antibodies by plasma cells may have led to false-positive results. With moderate to high clinical suspicion of HIT, the combination of a positive immunoassay and negative functional assay (as in this case) make the diagnosis of HIT indeterminate.15
TEACHING POINTS
- If a monoclonal protein is suggested by SPEP but cannot be identified by standard immunofixation, request immunofixation for IgD or IgE. Screen patients for IgD and IgE paraproteins before making a diagnosis of light chain multiple myeloma.
- Polyclonal gammopathy or reduced renal clearance with accumulation of free light chains in the serum may mask the presence of low levels of monoclonal FLC and result in a normal sFLC ratio.
- Thrombosis is a less-recognized but documented feature of POEMS syndrome which may be mediated by the overproduction of proinflammatory and proangiogenic cytokines, though the precise pathogenesis is unknown.
Acknowledgment
The authors thank Dr. Theodore Kurtz and Dr. Anne Deucher from the department of laboratory medicine at the University of California, San Francisco for providing their respective expertise in clinical chemistry and hematopathology.
Disclosures
The authors have no conflicts of interests to disclose.
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-ee548. doi: 10.1016/S1470-2045(14)70442-5.
A 39-year-old man presented to a neurologist with three weeks of progressive leg weakness associated with numbness in his feet and fingertips. His medical history included hypertriglyceridemia, hypogonadism, and gout. He was taking fenofibrate and colchicine as needed. There was no family history of neurologic issues. He did not smoke or drink alcohol.
The patient appeared well with a heart rate of 76 beats per minute, blood pressure 133/72 mm Hg, temperature 36.6°C, respiratory rate 16 breaths per minute, and oxygen saturation 100% on room air. His cardiopulmonary and abdominal examinations were normal. His skin was warm and dry without rashes. On neurologic examination, upper extremity strength and sensation was normal. Bilateral hip flexion, knee flexion, and knee extension strength was 4/5; bilateral ankle dorsiflexion and plantar flexion strength was 3/5. Reflexes were trace in the arms and absent at the patellae and ankles. He had symmetric, length-dependent reduction in vibration, pinprick, and light touch sensation in his legs.
Peripheral neuropathy presenting with ascending symmetric motor and sensory deficits progressing over three weeks raises the suspicion of an acquired inflammatory demyelinating polyneuropathy (AIDP), a variant of Guillain-Barre Syndrome. Alternative causes of acute polyneuropathy include thiamine (B1) deficiency, vasculitis, sarcoidosis, or malignancy, particularly lymphoma and multiple myeloma. Further evaluation should include electromyography, nerve conduction studies, lumbar puncture with cerebrospinal fluid (CSF) protein, glucose, and cell count differential. Follow-up laboratory testing based on results of the above may include serum protein electrophoresis (SPEP), serum free light chains (sFLC), vitamin B12, human immunodeficiency virus (HIV), hepatitis B and C testing, antinuclear antibody, and erythrocyte sedimentation rate.
Electromyography and nerve conduction studies revealed a sensorimotor mixed axonal/demyelinating polyneuropathy in all extremities. CSF analysis found one white cell per mm3, glucose of 93 mg/dL, and protein of 313 mg/dL. Magnetic resonance imaging (MRI) of the spine without contrast showed normal cord parenchyma. The vitamin B12 level was 441 pg/mL (normal >200 pg/mL). Antibodies to HIV-1, HIV-2, hepatitis C virus, and Borrelia burgdorferi were negative. Serum protein electrophoresis (SPEP) and immunofixation were normal.
The patient received two courses of intravenous immunoglobulin (IVIG) for suspected AIDP. His weakness progressed over the next several weeks to the point that he required a wheelchair.
Progression of symptoms beyond three weeks and lack of response to IVIG are atypical for AIDP. Alternate diagnoses for a sensorimotor polyneuropathy should be considered. Causes of subacute or chronic demyelinating polyneuropathy include inflammatory conditions (chronic inflammatory demyelinating polyneuropathy [CIDP], connective-tissue disorders), paraprotein disorders (myeloma, amyloidosis, lymphoplasmacytic lymphoma), paraneoplastic syndromes, infectious diseases (HIV, Lyme disease), infiltrative disorders (sarcoidosis), medications or toxins, and hereditary disorders. Of these etiologies, the first three seem the most likely given the history and clinical course, the negative HIV and Lyme testing, and the absence of exposures and family history. Normal SPEP and immunofixation make paraprotein disorders less likely, but sFLC testing should be sent to evaluate for a light chain-only paraprotein. A paraneoplastic antibody panel and a CT of the chest, abdomen, and pelvis should be ordered to evaluate for sarcoidosis, lymphoma, or other malignancies. Although a peripheral nerve biopsy would further classify the polyneuropathy, it is of low diagnostic yield in patients with subacute and chronic distal symmetric polyneuropathies and is associated with significant morbidity. In the absence of history or physical exam findings to narrow the differential diagnosis for polyneuropathy, testing for paraneoplastic antibodies and imaging is appropriate.
The patient tested negative for antiganglioside GM1 and antimyelin-associated glycoprotein antibodies. Urine arsenic, lead, and mercury levels were normal. Tests for serum antinuclear antibody, rapid plasmin reagin, and a paraneoplastic neuropathy panel including amphiphysin antibody, CV2 antibody, and Hu auto-antibody were negative. Repeat electrodiagnostic testing was consistent with CIDP. The patient received prednisone 60 mg daily for six weeks and was then tapered to 30 mg daily over six weeks. Concurrently, he underwent twelve cycles of plasma exchange. His strength improved, and he could walk with a cane; however, weakness recurred when steroids were further tapered.
He was maintained on prednisone 50 mg daily. Over the next year, the patient’s lower extremities became flaccid and severely atrophied. He developed hyperpigmented patches on his trunk, severe gastroesophageal reflux disease (GERD), dysphonia, and gynecomastia. He had lost 60 pounds since symptom onset. He was prescribed levothyroxine for subclinical hypothyroidism (thyroid stimulating hormone 12.63 µIU/mL [normal 0.10-5.50 µIU/mL], free thyroxine 0.8 ng/dL [0.8-1.7 ng/dL]).
At this point, the diagnosis of CIDP should be questioned, and additional investigation is warranted. Although improvement was initially observed with plasma exchange and steroids, subsequent progression of symptoms despite prednisone suggests a nonimmune-mediated etiology, such as a neoplastic or infiltrative process. Conversely, negative serologic testing for paraneoplastic antibodies may be due to an antibody that has not been well characterized.
While prednisone could explain GERD and gynecomastia, the weight loss, dysphonia, and subclinical hypothyroidism may offer clues to the diagnosis underlying the neurological symptoms. Weight loss raises suspicion of a hypercatabolic process such as cancer, cachexia, systemic inflammation, heart failure, or chronic obstructive pulmonary disease. Causes of dysphonia relevant to this presentation include neurologic dysfunction related to malignant invasion of the vagus nerve or demyelinating disease. Subclinical hypothyroidism due to chronic autoimmune thyroiditis seems most likely in the absence of a medication effect or thyroid injury, yet infiltrative disorders of the thyroid (eg, amyloidosis, sarcoidosis, lymphoma) should also be considered. A diagnosis that unifies the neurologic and nonneurologic findings would be desirable; lymphoma with paraneoplastic peripheral neuropathy manifesting as CIDP seems most likely. As of yet, CT of the chest, abdomen, and pelvis or an 18-Fluoro-deoxyglucose positron emission tomography (FDG-PET) scan have not been obtained and would be helpful to evaluate for underlying malignancy. Further evaluation for a paraprotein disorder that includes sFLC is also still indicated to rule out a paraneoplastic disorder that may be associated with polyneuropathy.
Repeat SPEP and serum immunofixation were normal. sFLC assay showed elevated levels of both kappa and lambda light chains with a ratio of 0.61 (reference range: 0.26-1.25). Urine protein electrophoresis (UPEP) from a 24-hour specimen showed a homogenous band in the gamma region, but urine immunofixation demonstrated polyclonal light chains. The plasma vascular endothelial growth factor (VEGF) level was 612 pg/mL (reference range, 31-86 pg/mL).
CT imaging of the chest, abdomen, and pelvis with contrast demonstrated an enlarged liver and spleen and possible splenic infarcts. A skeletal survey and whole-body FGD-PET scan were normal. The patient declined bone marrow biopsy.
Polyneuropathy secondary to a monoclonal protein was previously considered, and an SPEP was normal. Full evaluation for a monoclonal protein additionally requires sFLC testing. If clinical suspicion remains high after a negative result, 24-hour UPEP and urine immunofixation should be obtained. Normal results in this case argue against the presence of a monoclonal protein.
The presence of a monoclonal protein and polyneuropathy are mandatory diagnostic criteria for POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes), a plasma cell proliferative disorder. Major diagnostic criteria include osteosclerotic bone lesions, Castleman’s disease, and markedly elevated VEGF levels. Castleman’s disease is a lymphoproliferative disorder characterized by angiofollicular lymphoid hyperplasia that results in lymphadenopathy in one or multiple lymph node regions. Imaging studies reveal organomegaly, one of many minor criteria, but not bone lesions or lymphadenopathy. A diagnosis of POEMS syndrome requires the presence of both mandatory, one major, and one minor criteria. Since only one of two of the mandatory criteria are met at this point, a diagnosis of POEMS syndrome cannot be made.
Eighteen months after symptom onset, the patient presented to the emergency department with dyspnea, orthopnea, and lower extremity edema. B-type natriuretic peptide was 1564 pg/mL. Transthoracic echocardiography showed a severely dilated and hypertrophied left ventricle. Left ventricular ejection fraction was 20%. A furosemide infusion was initiated. Angiography of the coronary vessels was not performed. Congo red stain of an abdominal adipose biopsy was negative for amyloid.
On hospital day five, he developed gangrenous changes in his right first toe. CT angiography of the abdomen and lower extremities demonstrated patent three vessel runoff to the foot with an infrarenal aortic thrombus. Heparin infusion was started. On hospital day 10, the patient developed expressive aphasia and somnolence, prompting intubation for airway protection. MRI and MR angiography (MRA) of the brain and cerebral vessels revealed multiple bilateral acute ischemic strokes (Figure 1) without flow limiting stenosis in cerebral vessels.
These clinical developments lead to an important opportunity to rethink this patient’s working diagnosis. The new diagnosis of heart failure in this young patient with polyneuropathy raises suspicion for an infiltrative cardiomyopathy such as amyloidosis, sarcoidosis, or Fabry disease. Of these, Fabry disease is the least likely because it is typically characterized by a painful burning sensation in response to specific triggers. Although polyneuropathy and heart failure may be concurrently observed with both sarcoidosis and amyloidosis, the absence of an apparent arrhythmia make amyloidosis the more likely of these two diagnoses. The development of an arterial thrombus and multiple strokes may represent emboli from a cardiac thrombus.
Cardiac imaging and tissue biopsy of the heart or other affected organs would distinguish between these diagnostic possibilities. An abdominal adipose biopsy negative for amyloid does not rule out amyloidosis, as the test is approximately 80% sensitive when cardiac amyloidosis is present and varies depending on the etiology of the amyloid protein (ie, light chain vs transthyretin). Evaluation of cardiac amyloid in the setting of peripheral neuropathy should include echocardiography (as was performed here) and repeat testing for a monoclonal protein.
If clinical suspicion of a paraprotein-associated disorder remains high and both SPEP and sFLC are normal, it is important to obtain a 24-hour UPEP and immunofixation. A monoclonal protein can be overlooked by SPEP and serum immunofixation if the monoclonal protein is composed only of a light chain or if the monoclonal protein is IgD or IgE. In these rare circumstances, sFLC analysis or 24-hour UPEP and immunofixation should mitigate the potential for a falsely negative SPEP/IFE. These studies are normal in this case, which argues against the presence of a monoclonal protein.
Transesophageal echocardiography showed grade IV atheromatous plaque within the descending thoracic aorta with mobile elements suggesting a superimposed thrombus; there was no intracardiac shunt or thrombus. MRA of the neck and great vessels was normal.
Testing for heparin-induced thrombocytopenia (HIT) was sent due to thrombocytopenia and the presence of thrombosis. An immunoassay for antiheparin-platelet factor 4 (anti-PF4) antibodies was substantially positive (optical density 2.178); however, functional testing with a washed platelet heparin-induced platelet activation assay was negative. Anticoagulation was changed to argatroban due to concern for HIT. Dry gangrenous changes developed in all distal toes on the right foot and three toes on the left foot. A right radial artery thrombus formed at the site of a prior arterial line.
Thrombocytopenia that develops between the fifth and tenth day following heparin exposure in a patient with new thromboses is consistent with HIT. However, the patient’s infrarenal aortic thrombus preceded the initiation of heparin, and negative functional testing undermines the diagnosis of HIT in this case. Therefore, the arterial thromboses may be related to an underlying unifying diagnosis.
A third SPEP showed a 0.1 g/dL M-spike in the gamma region, but standard immunofixation did not reveal a monoclonal protein (Figure 2). However, a specific request for immunofixation testing using IgD antisera detected an IgD heavy chain. A lambda chain comprising 3% of urine protein was detected on 24-hour urine immunofixation but was not detectable by serum immunofixation. Bone marrow biopsy demonstrated plasma cells comprising 5% of bone marrow cellularity (Figure 3); flow cytometry of the aspirate demonstrated an abnormal lambda-restricted plasma cell population.
When a monoclonal protein is identified but does not react with standard antisera to detect IgG, IgM, and IgA, immunofixation with IgD and IgE antisera are necessary to rule out a monoclonal IgD or IgE protein. The underlying IgD isotype coupled with its low abundance made detection of this monoclonal protein especially challenging. With the discovery of a monoclonal protein in the context of polyneuropathy, the mandatory criteria of POEMS syndrome are met. The elevated VEGF level and hypothyroidism meet major and minor criteria, respectively. Arterial thromboses and heart failure are other features that may be observed in cases of POEMS syndrome.
POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes) was diagnosed. Prednisone was continued, and weekly cyclophosphamide was initiated. After six weeks, the VEGF level remained elevated, and a neurologic examination showed minimal improvement. Due to poor respiratory muscle strength and difficulty managing secretions, he underwent percutaneous tracheostomy and gastrostomy tube placement. Unfortunately, his condition further deteriorated and he subsequently died of sepsis from pneumonia.
An autopsy revealed acute bronchopneumonia and multiple acute and subacute cerebral infarctions. There was extensive peripheral mixed axonal/demyelinating neuropathy, hepatosplenomegaly, atrophy of the thyroid and adrenal glands, hyperpigmented patches and thickened integument, and severe aortic and coronary atherosclerotic disease with a healed myocardial infarction.
DISCUSSION
POEMS syndrome1 is a rare constellation of clinical and laboratory findings resulting from an underlying plasma cell proliferative disorder. This paraneoplastic syndrome is characterized by the chronic overproduction of proinflammatory and proangiogenic cytokines, including VEGF, which are postulated to drive its manifestations,2 though the exact pathogenesis is not understood. Some of the disease’s most common features are summarized by its name: polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes.3
The International Myeloma Working Group (IMWG) diagnostic criteria1 (Table) require the presence of both mandatory criteria (polyneuropathy and monoclonal plasma cell proliferation), plus at least one major and one minor criterion. Delayed diagnosis or misdiagnosis of this protean disorder is often driven by its rarity and clinical overlap with other paraprotein-associated polyneuropathies. These include amyloidosis, cryoglobulinemia, and monoclonal gammopathy of undetermined significance (MGUS), which can all produce antibodies directed against neural antigens. In addition, polyneuropathy is often the first and most striking manifestation of POEMS syndrome, fostering confusion with CIDP as both disorders are subacute, symmetric, motor-dominant, mixed axonal/demyelinating polyneuropathies.4
IgD and IgE monoclonal gammopathies are extremely rare. IgD myeloma, for instance, accounts for 2% of multiple myeloma cases, and IgE myeloma has been reported fewer than 50 times.5 IgD is secreted only in very small amounts, ordinarily representing 0.25% of the immunoglobulins in serum, while the majority is found in the plasma membranes of mature B-cells.6 These monoclonal gammopathies often escape detection for two reasons: (1) the very low paraprotein concentration produces undetectable or small M-protein levels on electrophoresis,5 and (2) immunofixation is routinely performed without antisera against IgD and IgE heavy chains.7
While this case depicts a rare manifestation of a rare disease, the principles underlying its elusive diagnosis are routinely encountered. Recognition of the specific limitations of the SPEP, UPEP, sFLC, and immunofixation tests, outlined below, can assist the hospitalist when suspicion for paraproteinemia is high.
First, low levels of monoclonal proteins may be associated with a normal SPEP. Accordingly, suspicion of a plasma cell dyscrasia should prompt serum immunofixation, even when the electrophoretic pattern appears normal.8
Second, laboratories routinely perform immunofixation with antisera against IgG, IgA, and IgM heavy chains and kappa and lambda light chains, whereas testing with IgD or IgE antisera must be specifically requested. Thus, clinicians should screen for the presence of IgD and IgE in patients with an apparently free monoclonal immunoglobulin light chain in the serum or with a monoclonal serum protein and negative immunofixation. In this case, the paraprotein was not detected on the first two serum electrophoreses, likely due to a low serum concentration, then missed on immunofixation due to a lack of IgD antiserum. On admission to the hospital, this patient had a very low paraprotein concentration (0.1 g/dL) on SPEP, and the lab initially reported negative immunofixation. When asked to test specifically for IgD and IgE, the lab ran a more comprehensive immunofixation revealing IgD heavy chain paraprotein.
Third, this case illustrates the limitations of the sFLC assay. IMWG guidelines specify that sFLC assay in combination with SPEP and serum immunofixation is sufficient to screen for monoclonal plasma cell proliferative disorders other than light chain amyloidosis (which requires all the serum tests as well as 24-hour urine immunofixation).9 Though the sFLC assay has been demonstrated to be more sensitive than urine analysis for detecting monoclonal free light chains,10 it is still subject to false negatives. Polyclonal gammopathy or reduced renal clearance with accumulation of free light chains in the serum may mask the presence of low levels of monoclonal sFLC,11 the latter of which likely explains why the sFLC ratio was repeatedly normal in this case. In these circumstances, monoclonal free light chains can be identified by urine studies.11 In this case, 24-hour urine immunofixation detected the excess light chain that was not evident on the sFLC assay. Even with these pitfalls in mind, there is still no evident explanation as to why the 24-hour urine studies done prior to the patient’s hospital admission did not reveal a monoclonal light chain.
This case also highlights the thrombotic diathesis in POEMS syndrome. Although the patient was treated with argatroban for suspected HIT, it is likely that the HIT antibody result was a false positive, and his thrombi were better explained by POEMS syndrome in and of itself. Coronary, limb, and cerebral artery thromboses have been linked to POEMS syndrome,12,13 all of which were present in this case. Laboratory testing for HIT involves an immunoassay to detect circulating HIT antibody and a functional assay to measure platelet activity in the presence of patient serum and heparin. The immunoassay binds anti-PF4/heparin complex irrespective of its ability to activate platelets. The presence of nonspecific antibodies may lead to cross-reactions with the immunoassay test components, which has been demonstrated in cases of MGUS.14 In this case, elevated production of monoclonal antibodies by plasma cells may have led to false-positive results. With moderate to high clinical suspicion of HIT, the combination of a positive immunoassay and negative functional assay (as in this case) make the diagnosis of HIT indeterminate.15
TEACHING POINTS
- If a monoclonal protein is suggested by SPEP but cannot be identified by standard immunofixation, request immunofixation for IgD or IgE. Screen patients for IgD and IgE paraproteins before making a diagnosis of light chain multiple myeloma.
- Polyclonal gammopathy or reduced renal clearance with accumulation of free light chains in the serum may mask the presence of low levels of monoclonal FLC and result in a normal sFLC ratio.
- Thrombosis is a less-recognized but documented feature of POEMS syndrome which may be mediated by the overproduction of proinflammatory and proangiogenic cytokines, though the precise pathogenesis is unknown.
Acknowledgment
The authors thank Dr. Theodore Kurtz and Dr. Anne Deucher from the department of laboratory medicine at the University of California, San Francisco for providing their respective expertise in clinical chemistry and hematopathology.
Disclosures
The authors have no conflicts of interests to disclose.
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-ee548. doi: 10.1016/S1470-2045(14)70442-5.
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-ee548. doi: 10.1016/S1470-2045(14)70442-5. PubMed
2. Watanabe O, Arimura K, Kitajima I, Osame M, Maruyama I. Greatly raised vascular endothelial growth factor (VEGF) in POEMS syndrome. Lancet. 1996;347(9002):702. doi: 10.1016/S0140-6736(96)91261-1. PubMed
3. Dispenzieri A. How I treat POEMS syndrome. Blood. 2012;119(24):5650-5658. doi: 10.1182/blood-2012-03-378992. PubMed
4. Nasu S, Misawa S, Sekiguchi Y, et al. Different neurological and physiological profiles in POEMS syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry. 2012;83(5):476-479. doi: 10.1136/jnnp-2011-301706. PubMed
5. Pandey S, Kyle RA. Unusual myelomas: a review of IgD and IgE variants. Oncology. 2013;27(8):798-803. PubMed
6. Vladutiu AO. Immunoglobulin D: properties, measurement, and clinical relevance. Clin Diagn Lab Immunol. 2000;7(2):131-140. doi: 10.1128/CDLI.7.2.131-140.2000. PubMed
7. Sinclair D, Cranfield T. IgD myeloma: A potential missed diagnosis. Ann Clin Biochem. 2001;38(5):564-565. doi: 10.1177/000456320103800517. PubMed
8. Dimopoulos M, Kyle R, Fermand JP, et al. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood. 2011;117(18):4701-4705. doi: 10.1182/blood-2010-10-299529. PubMed
9. Dispenzieri A, Kyle R, Merlini G, et al. International Myeloma Working Group. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia. 2009;23(2):215-224. doi: 10.1038/leu.2008.307. PubMed
10. Dejoie T, Attal M, Moreau P, Harousseau JL, Avet-Loiseau H. Comparison of serum free light chain and urine electrophoresis for the detection of the light chain component of monoclonal immunoglobulins in light chain and intact immunoglobulin multiple myeloma. Haematologica. 2016;101(3):356-362. doi: 10.3324/haematol.2015.126797. PubMed
11. Levinson SS. Polyclonal free light chain of Ig may interfere with interpretation of monoclonal free light chain κ/λ ratio. Ann Clin Lab Sci. 2010;40(4):348-353. PubMed
12. Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003;101(7):2496-2506. doi: 10.1182/blood-2002-07-2299. PubMed
13. Dupont SA, Dispenzieri A, Mauermann ML, Rabinstein AA, Brown RD. Cerebral infarction in POEMS syndrome: incidence, risk factors, and imaging characteristics. Neurology. 2009;73(16):1308-1312. doi: 10.1212/WNL.0b013e3181bd136b. PubMed
14. Markovic I, Debeljak Z, Bosnjak B, Marijanovic M. False positive immunoassay for heparin-induced thrombocytopenia in the presence of monoclonal gammopathy: a case report. Biochemia Medica. 2017;27(3):030801. doi: 10.11613/BM.2017.030801. PubMed
15. Cuker A, Cines DB. How I treat heparin-induced thrombocytopenia. Blood. 2012;119(10):2209-2218. doi: 10.1182/blood-2011-11-376293. PubMed
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-ee548. doi: 10.1016/S1470-2045(14)70442-5. PubMed
2. Watanabe O, Arimura K, Kitajima I, Osame M, Maruyama I. Greatly raised vascular endothelial growth factor (VEGF) in POEMS syndrome. Lancet. 1996;347(9002):702. doi: 10.1016/S0140-6736(96)91261-1. PubMed
3. Dispenzieri A. How I treat POEMS syndrome. Blood. 2012;119(24):5650-5658. doi: 10.1182/blood-2012-03-378992. PubMed
4. Nasu S, Misawa S, Sekiguchi Y, et al. Different neurological and physiological profiles in POEMS syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry. 2012;83(5):476-479. doi: 10.1136/jnnp-2011-301706. PubMed
5. Pandey S, Kyle RA. Unusual myelomas: a review of IgD and IgE variants. Oncology. 2013;27(8):798-803. PubMed
6. Vladutiu AO. Immunoglobulin D: properties, measurement, and clinical relevance. Clin Diagn Lab Immunol. 2000;7(2):131-140. doi: 10.1128/CDLI.7.2.131-140.2000. PubMed
7. Sinclair D, Cranfield T. IgD myeloma: A potential missed diagnosis. Ann Clin Biochem. 2001;38(5):564-565. doi: 10.1177/000456320103800517. PubMed
8. Dimopoulos M, Kyle R, Fermand JP, et al. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood. 2011;117(18):4701-4705. doi: 10.1182/blood-2010-10-299529. PubMed
9. Dispenzieri A, Kyle R, Merlini G, et al. International Myeloma Working Group. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia. 2009;23(2):215-224. doi: 10.1038/leu.2008.307. PubMed
10. Dejoie T, Attal M, Moreau P, Harousseau JL, Avet-Loiseau H. Comparison of serum free light chain and urine electrophoresis for the detection of the light chain component of monoclonal immunoglobulins in light chain and intact immunoglobulin multiple myeloma. Haematologica. 2016;101(3):356-362. doi: 10.3324/haematol.2015.126797. PubMed
11. Levinson SS. Polyclonal free light chain of Ig may interfere with interpretation of monoclonal free light chain κ/λ ratio. Ann Clin Lab Sci. 2010;40(4):348-353. PubMed
12. Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003;101(7):2496-2506. doi: 10.1182/blood-2002-07-2299. PubMed
13. Dupont SA, Dispenzieri A, Mauermann ML, Rabinstein AA, Brown RD. Cerebral infarction in POEMS syndrome: incidence, risk factors, and imaging characteristics. Neurology. 2009;73(16):1308-1312. doi: 10.1212/WNL.0b013e3181bd136b. PubMed
14. Markovic I, Debeljak Z, Bosnjak B, Marijanovic M. False positive immunoassay for heparin-induced thrombocytopenia in the presence of monoclonal gammopathy: a case report. Biochemia Medica. 2017;27(3):030801. doi: 10.11613/BM.2017.030801. PubMed
15. Cuker A, Cines DB. How I treat heparin-induced thrombocytopenia. Blood. 2012;119(10):2209-2218. doi: 10.1182/blood-2011-11-376293. PubMed
© 2019 Society of Hospital Medicine
The Basement Flight
A 14-year-old girl with a history of asthma presented to the Emergency Department (ED) with three months of persistent, nonproductive cough, and progressive shortness of breath. She reported fatigue, chest tightness, orthopnea, and dyspnea with exertion. She denied fever, rhinorrhea, congestion, hemoptysis, or paroxysmal nocturnal dyspnea.
Her age and past medical history of asthma are incongruent with her new symptoms, as asthma is typified by intermittent exacerbations, not progressive symptoms. Thus, another process, in addition to asthma, is most likely present; it is also important to question the accuracy of previous diagnoses in light of new information. Her symptoms may signify an underlying cardiopulmonary process, such as infiltrative diseases (eg, lymphoma or sarcoidosis), atypical infections, genetic conditions (eg, variant cystic fibrosis), autoimmune conditions, or cardiomyopathy. A detailed symptom history, family history, and careful physical examination will help expand and then refine the differential diagnosis. At this stage, typical infections are less likely.
She had presented two months prior with nonproductive cough and dyspnea. At that presentation, her temperature was 36.3°C, heart rate 110 beats per minute, blood pressure 119/63 mm Hg, respiratory rate 43 breaths per minute, and oxygen saturation 86% while breathing ambient air. A chest CT with contrast demonstrated diffuse patchy multifocal ground-glass opacities in the bilateral lungs as well as a mixture of atelectasis and lobular emphysema in the dependent lobes bilaterally (Figure 1). Her main pulmonary artery was dilated at 3.6 cm (mean of 2.42 cm with SD 0.22). She was diagnosed with atypical pneumonia. She was administered azithromycin, weaned off oxygen, and discharged after a seven-day hospitalization.
Two months prior, she had marked tachypnea, tachycardia, and hypoxemia, and imaging revealed diffuse ground-glass opacities. The differential diagnosis for this constellation of symptoms is extensive and includes many conditions that have an inflammatory component, such as atypical pneumonia caused by Mycoplasma or Chlamydia pneumoniae or a common respiratory virus such as rhinovirus or human metapneumovirus. However, two findings make an acute pneumonia unlikely to be the sole cause of her symptoms: underlying emphysema and an enlarged pulmonary artery. Emphysema is an uncommon finding in children and can be related to congenital or acquired causes; congenital lobar emphysema most often presents earlier in life and is focal, not diffuse. Alpha-1-anti-trypin deficiency and mutations in connective tissue genes such as those encoding for elastin and fibrillin can lead to pulmonary disease. While not diagnostic of pulmonary hypertension, her dilated pulmonary artery, coupled with her history, makes pulmonary hypertension a strong possibility. While her pulmonary hypertension is most likely secondary to chronic lung disease based on the emphysematous changes on CT, it could still be related to a cardiac etiology.
The patient had a history of seasonal allergies and well-controlled asthma. She was hospitalized at age six for an asthma exacerbation associated with a respiratory infection. She was discharged with an albuterol inhaler, but seldom used it. Her parents denied any regular coughing during the day or night. She was morbidly obese. Her tonsils and adenoids were removed to treat obstructive sleep apnea (OSA) at age seven, and a subsequent polysomnography was normal. Her medications included intranasal fluticasone propionate and oral iron supplementation. She had no known allergies or recent travels. She had never smoked. She had two pet cats and a dog. Her mother had a history of obesity, OSA, and eczema. Her father had diabetes and eczema.
The patient’s history prior to the recent few months sheds little light on the cause of her current symptoms. While it is possible that her current symptoms are related to the worsening of a process that had been present for many years which mimicked asthma, this seems implausible given the long period of time between her last asthma exacerbation and her present symptoms. Similarly, while tonsillar and adenoidal hypertrophy can be associated with infiltrative diseases (such as lymphoma), this is less common than the usual (and normal) disproportionate increase in size of the adenoids compared to other airway structures during growth in children.
She was admitted to the hospital. On initial examination, her temperature was 37.4°C, heart rate 125 beats per minute, blood pressure 143/69 mm Hg, respiratory rate 48 breaths per minute, and oxygen saturation 86% breathing ambient air. Her BMI was 58 kg/m2. Her exam demonstrated increased work of breathing with accessory muscle use, and decreased breath sounds at the bases. There were no wheezes or crackles. Cardiovascular, abdominal, and skin exams were normal except for tachycardia. At rest, later in the hospitalization, her oxygen saturation was 97% breathing ambient air and heart rate 110 bpm. After two minutes of walking, her oxygen saturation was 77% and heart rate 132 bpm. Two minutes after resting, her oxygen saturation increased to 91%.
Her white blood cell count was 11.9 x 10 9 /L (67% neutrophils, 24.2% lymphocytes, 6% monocytes, and 2% eosinophils), hemoglobin 11.2 g/dL, and platelet count 278,000/mm 3 . Her complete metabolic panel was normal. The C-reactive protein (CRP) was 24 mg/L (normal range, < 4.9) and erythrocyte sedimentation rate (ESR) 103 mm/hour (normal range, 0-32). A venous blood gas (VBG) showed a pH of 7.42 and pCO2 39. An EKG demonstrated sinus tachycardia.
The combination of the patient’s tachypnea, hypoxemia, respiratory distress, and obesity is striking. Her lack of adventitious lung sounds is surprising given her CT findings, but the sensitivity of chest auscultation may be limited in obese patients. Her laboratory findings help narrow the diagnostic frame: she has mild anemia and leukocytosis along with significant inflammation. The normal CO2 concentration on VBG is concerning given the degree of her tachypnea and reflects significant alveolar hypoventilation.
This marked inflammation with diffuse lung findings again raises the possibility of an inflammatory or, less likely, infectious disorder. Sjogren’s syndrome, systemic lupus erythematosus (SLE), and juvenile dermatomyositis can present in young women with interstitial lung disease. She does have exposure to pets and hypersensitivity pneumonitis can worsen rapidly with continued exposure. Another possibility is that she has an underlying immunodeficiency such as common variable immunodeficiency, although a history of recurrent infections such as pneumonia, bacteremia, or sinusitis is lacking.
An echocardiogram should be performed. In addition, laboratory evaluation for the aforementioned autoimmune causes of interstitial lung disease, immunoglobulin levels, pulmonary function testing (if available as an inpatient), and potentially a bronchoscopy with bronchoalveolar lavage (BAL), and biopsy should be pursued. The BAL and biopsy would be helpful in evaluating for infection and interstitial lung disease in an expeditious manner.
A chest CT without contrast was done and compared to the scan from two months prior. New diffuse, ill-defined centrilobular ground-glass opacities were evident throughout the lung fields; dilation of the main pulmonary artery was unchanged, and previously seen ground-glass opacities had resolved. There were patchy areas of air-trapping and mosaic attenuation in the lower lobes (Figure 2).
Transthoracic echocardiogram demonstrated a right ventricular systolic pressure of 58 mm Hg with flattened intraventricular septum during systole. Left and right ventricular systolic function were normal. The left ventricular diastolic function was normal. Pulmonary function testing demonstrated a FEV1/FVC ratio of 100 (112% predicted), FVC 1.07 L (35 % predicted) and FEV1 1.07 L (39% predicted), and total lung capacity was 2.7L (56% predicted) (Figure 3). Single-breath carbon monoxide uptake in the lung was not interpretable based on 2017 European Respiratory Society (ERS)/American Thoracic Society (ATS) technical standards.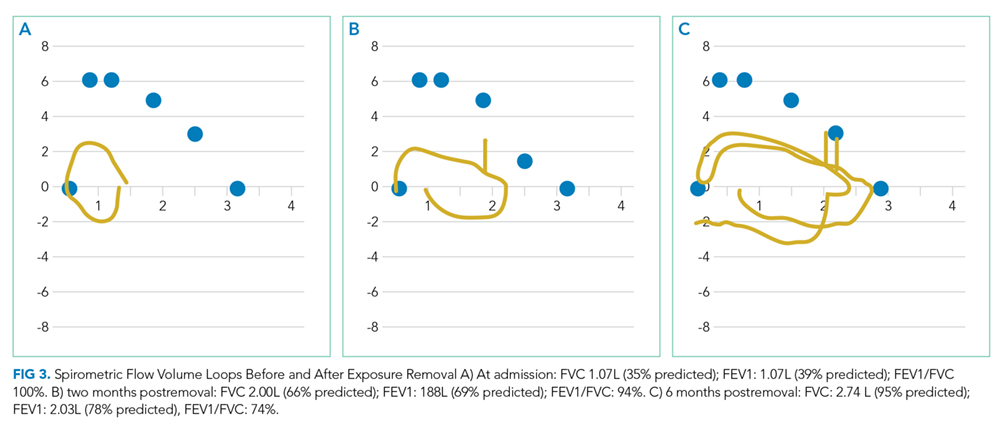
This information is helpful in classifying whether this patient’s primary condition is cardiac or pulmonary in nature. Her normal left ventricular systolic and diastolic function make a cardiac etiology for her pulmonary hypertension less likely. Further, the combination of pulmonary hypertension, a restrictive pattern on pulmonary function testing, and findings consistent with interstitial lung disease on cross-sectional imaging all suggest a primary pulmonary etiology rather than a cardiac, infectious, or thromboembolic condition. While chronic thromboembolic hypertension can result in nonspecific mosaic attenuation, it typically would not cause centrilobular ground-glass opacities nor restrictive lung disease. Thus, it seems most likely that this patient has a progressive pulmonary process resulting in hypoxia, pulmonary hypertension, centrilobular opacities, and lower-lobe mosaic attenuation. Considerations for this process can be broadly categorized as one of the childhood interstitial lung disease (chILD). While this differential diagnosis is broad, strong consideration should be given to hypersensitivity pneumonitis, chronic aspiration, sarcoidosis, and Sjogren’s syndrome. An intriguing possibility is that the patient’s “response to azithromycin” two months prior was due to the avoidance of an inhaled antigen while she was in the hospital; a detailed environmental history should be explored. The normal polysomnography after tonsilloadenoidectomy makes it unlikely that OSA is a major contributor to her current presentation. However, since the surgery was seven years ago, and her BMI is presently 58 kg/m2 she remains at risk for OSA and obesity-hypoventilation syndrome. Polysomnography should be done after her acute symptoms improve.
She was started on 5 mm Hg of continuous positive airway pressure (CPAP) at night after a sleep study on room air demonstrated severe OSA with a respiratory disturbance index of 13 events per hour. Antinuclear antibodies (ANA), anti-neutrophil cytoplasmic antibody (ANCA), anti-Jo-1 antibody, anti-RNP antibody, anti-Smith antibody, anti-Ro/SSA and anti-La/SSB antibody were negative as was the histoplasmin antibody. Serum angiotensin-converting enzyme (ACE) level was normal. Mycoplasma IgM and IgG were negative. IgE was 529 kU/L (normal range, <114).
This evaluation reduces the likelihood the patient has Sjogren’s syndrome, SLE, dermatomyositis, or ANCA-associated pulmonary disease. While many patients with dermatomyositis may have negative serologic evaluations, other findings usually present such as rash and myositis are lacking. The negative ANCA evaluation makes granulomatosis with polyangiitis and microscopic polyangiitis very unlikely given the high sensitivity of the ANCA assay for these conditions. ANCA assays are less sensitive for eosinophilic granulomatosis with polyangiitis (EGPA), but the lack of eosinophilia significantly decreases the likelihood of EGPA. ACE levels have relatively poor operating characteristics in the evaluation of sarcoidosis; however, sarcoidosis seems unlikely in this case, especially as patients with sarcoidosis tend to have low or normal IgE levels. Patients with asthma can have elevated IgE levels. However, very elevated IgE levels are more common in other conditions, including allergic bronchopulmonary aspergillosis (ABPA) and the Hyper-IgE syndrome. The latter manifests with recurrent infections and eczema, and is inherited in an autosomal dominant manner. However, both the Hyper-IgE syndrome and ABPA have much higher IgE levels than seen in this case. Allergen-specific IgE testing (including for antibodies to Aspergillus) should be sent. It seems that an interstitial lung disease is present; the waxing and waning pattern and clinical presentation, along with the lack of other systemic findings, make hypersensitivity pneumonitis most likely.
The family lived in an apartment building. Her symptoms started when the family’s neighbor recently moved his outdoor pigeon coop into his basement. The patient often smelled the pigeons and noted feathers coming through the holes in the wall.
One of the key diagnostic features of hypersensitivity pneumonitis (HP) is the history of exposure to a potential offending antigen—in this case likely bird feathers—along with worsening upon reexposure to that antigen. HP is primarily a clinical diagnosis, and testing for serum precipitants has limited value, given the high false negative rate and the frequent lack of clinical symptoms accompanying positive testing. Bronchoalveolar lavage fluid may reveal lymphocytosis and reduced CD4:CD8 ratio. Crackles are commonly heard on examination, but in this case were likely not auscultated due to her obese habitus. The most important treatment is withdrawal of the offending antigen. Limited data suggest that corticosteroid therapy may be helpful in certain HP cases, including subacute, chronic and severe cases as well as patients with hypoxemia, significant imaging findings, and those with significant abnormalities on pulmonary function testing (PFT).
A hypersensitivity pneumonitis precipitins panel was sent with positive antibodies to M. faeni, T. Vulgaris, A. Fumigatus 1 and 6, A. Flavus, and pigeon serum. Her symptoms gradually improved within five days of oral prednisone (60 mg). She was discharged home without dyspnea and normal oxygen saturation while breathing ambient air. A repeat echocardiogram after nighttime CPAP for 1 week demonstrated a right ventricular systolic pressure of 17 mm Hg consistent with improved pulmonary hypertension.
Three weeks later, she returned to clinic for follow up. She had re-experienced dyspnea, cough, and wheezing, which improved when she was outdoors. She was afebrile, tachypneic, tachycardic, and her oxygen saturation was 92% on ambient air.
Her steroid-responsive interstitial lung disease and rapid improvement upon avoidance of the offending antigen is consistent with HP. The positive serum precipitins assay lends further credence to the diagnosis of HP, although serologic analysis with such antibody assays is limited by false positives and false negatives; further, individuals exposed to pigeons often have antibodies present without evidence of HP. History taking at this visit should ask specifically about further pigeon exposure: were the pigeons removed from the home completely, were heating-cooling filters changed, carpets cleaned, and bedding laundered? An in-home evaluation may be helpful before conducting further diagnostic testing.
She was admitted for oxygen therapy and a bronchoscopy, which showed mucosal friability and cobblestoning, suggesting inflammation. BAL revealed a normal CD4:CD8 ratio of 3; BAL cultures were sterile. Her shortness of breath significantly improved following a prolonged course of systemic steroids and removal from the triggering environment. PFTs improved with a FEV1/FVC ratio of 94 (105% predicted), FVC of 2.00 L (66% predicted), FEV1 of 1.88L (69% predicted) (Figure 3B). Her presenting symptoms of persistent cough and progressive dyspnea on exertion, characteristic CT, sterile BAL cultures, positive serum precipitants against pigeon serum, and resolution of her symptoms with withdrawal of the offending antigen were diagnostic of hypersensitivity pneumonitis due to pigeon exposure, also known as bird fancier’s disease.
COMMENTARY
The patient’s original presentation of dyspnea, tachypnea, and hypoxia is commonly associated with pediatric pneumonia and asthma exacerbations.1 However, an alternative diagnosis was suggested by the lack of wheezing, absence of fever, and recurrent presentations with progressive symptoms.
Hypersensitivity pneumonitis (HP) represents an exaggerated T-cell meditated immune response to inhalation of an offending antigen that results in a restrictive ventilatory defect and interstitial infiltrates.2 Bird pneumonitis (also known as bird fancier’s disease) is a frequent cause of HP, accounting for approximately 65-70% of cases.3 HP, however, only manifests in a small number of subjects exposed to culprit antigens, suggesting an underlying genetic susceptibility.4 Prevalence estimates vary depending on bird species, county, climate, and other possible factors.
There are no standard criteria for the diagnosis of HP, though a combination of findings is suggestive. A recent prospective multicenter study created a scoring system for HP based on factors associated with the disease to aid in accurate diagnosis. The most relevant criteria included antigen exposure, recurrent symptoms noted within 4-8 hours after antigen exposure, weight loss, presence of specific IgG antibodies to avian antigens, and inspiratory crackles on exam. Using this rule, the probability that our patient has HP based on clinical characteristics was 93% with an area under the receiver operating curve of 0.93 (96% confidence interval: 0.90-0.95)5. Chest imaging (high resolution CT) often consists of a mosaic pattern of air trapping, as seen in this patient in combination with ground-glass opacities6. Bronchoalveolar lavage (BAL) is sensitive in detecting lung inflammation in a patient with suspected HP. On BAL, a lymphocytic alveolitis can be seen, but absence of this finding does not exclude HP.5,7,8 Pulmonary function tests (PFTs) may be normal in acute HP. When abnormal, PFTs may reveal a restrictive pattern and reduction in carbon monoxide diffusing capacity.7 However, BAL and PFT results are neither specific nor diagnostic of HP; it is important to consider results in the context of the clinical picture.
The respiratory response to inhalation of the avian antigen has traditionally been classified as acute, subacute, or chronic.9 The acute response occurs within hours of exposure to the offending agent and usually resolves within 24 hours after antigen withdrawal. The subacute presentation involves cough and dyspnea over several days to weeks, and can progress to chronic and permanent lung damage if unrecognized and untreated. In chronic presentations, lung abnormalities may persist despite antigen avoidance and pharmacologic interventions.4,10 The patient’s symptoms occurred over a six-month period which coincided with pigeon exposure and resolved during each hospitalization with steroid treatment and removal from the offending agent. Her presentation was consistent with a subacute time course of HP.
The dilated pulmonary artery, elevated right systolic ventricular pressure, and normal right ventricular function in our patient suggested pulmonary hypertension of chronic duration. Her risk factors for pulmonary hypertension included asthma, sleep apnea, possible obesity-hypoventilation syndrome, and HP-associated interstitial lung disease.11
The most important intervention in HP is avoidance of the causative antigen. Medical therapy without removal of antigen is inadequate. Systemic corticosteroids can help ameliorate acute symptoms though dosing and duration remains unclear. For chronic patients unresponsive to steroid therapy, lung transplantation can be considered.4
The key to diagnosis of HP in this patient—and to minimizing repeat testing upon the patient’s recrudescence of symptoms—was the clinician’s consideration that the major impetus for the patient’s improvement in the hospital was removal from the offending antigen in her home environment. As in this case, taking time to delve deeply into a patient’s environment—even by descending the basement stairs—may lead to the diagnosis.
LEARNING POINTS
- Consider hypersensitivity pneumonitis (HP) in patients with recurrent respiratory distress, offending exposure, and resolution of symptoms with removal of culprit antigen.
- The most important treatment of HP is removal of offending antigen; systemic and/or inhaled corticosteroids are indicated until the full resolution of respiratory symptoms.
- Prognosis is dependent on early diagnosis and removal of offending exposures.
- Failure to treat HP might result in end-stage lung disease from pulmonary fibrosis secondary to long-term inflammation.
Disclosures
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
1. Ebell MH. Clinical diagnosis of pneumonia in children. Am Fam Physician. 2010;82(2):192-193. PubMed
2. Cormier Y, Lacasse Y. Hypersensitivity pneumonitis and organic dust toxic syndrome. In: Malo J-L, Chan-Yeung M, Bernstein DI, eds. Asthma in the Workplace. Vol 32. Boca Raton, FL: Fourth Informa Healthcare; 2013:392-405.
3. Chan AL, Juarez MM, Leslie KO, Ismail HA, Albertson TE. Bird fancier’s lung: a state-of-the-art review. Clin Rev Allergy Immunol. 2012;43(1-2):69-83. doi: 10.1007/s12016-011-8282-y. PubMed
4. Camarena A, Juárez A, Mejía M, et al. Major histocompatibility complex and tumor necrosis factor-α polymorphisms in pigeon breeder’s disease. Am J Respir Crit Care Med. 2001;163(7):1528-1533. https:/doi.org/10.1164/ajrccm.163.7.2004023. PubMed
5. Lacasse Y, Selman M, Costabel U, et al. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2003;168(8):952-958. doi: 10.1164/rccm.200301-137OC. PubMed
6. Glazer CS, Rose CS, Lynch DA. Clinical and radiologic manifestations of hypersensitivity pneumonitis. J Thorac Imaging. 2002;17(4):261-272. PubMed
7. Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186(4):314-324. doi: 10.1164/rccm.201203-0513CI. PubMed
8. Calillad DM, Vergnon, JM, Madroszyk A, et al. Bronchoalveolar lavage in hypersensitivity pneumonitis: a series of 139 patients. Inflamm Allergy Drug Targets. 2012;11(1):15-19. doi: 10.2174/187152812798889330. PubMed
9. Richerson HB, Bernstein IL, Fink JN, et al. Guidelines for the clinical evaluation of hypersensitivity pneumonitis. Report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol. 1989;84(5 Pt 2):839-844. doi: 10.1016/0091-6749(89)90349-7. PubMed
10. Zacharisen MC, Schlueter DP, Kurup VP, Fink JN. The long-term outcome in acute, subacute, and chronic forms of pigeon breeder’s disease hypersensitivity pneumonitis. Ann Allergy Asthma Immunol. 2002;88(2):175-182. doi: 10.1016/S1081-1206(10)61993-X. PubMed
11. Raymond TE, Khabbaza JE, Yadav R, Tonelli AR. Significance of main pulmonary artery dilation on imaging studies. Ann Am Thorac Soc. 2014;11(10):1623-1632. doi: 10.1513/AnnalsATS.201406-253PP. PubMed
A 14-year-old girl with a history of asthma presented to the Emergency Department (ED) with three months of persistent, nonproductive cough, and progressive shortness of breath. She reported fatigue, chest tightness, orthopnea, and dyspnea with exertion. She denied fever, rhinorrhea, congestion, hemoptysis, or paroxysmal nocturnal dyspnea.
Her age and past medical history of asthma are incongruent with her new symptoms, as asthma is typified by intermittent exacerbations, not progressive symptoms. Thus, another process, in addition to asthma, is most likely present; it is also important to question the accuracy of previous diagnoses in light of new information. Her symptoms may signify an underlying cardiopulmonary process, such as infiltrative diseases (eg, lymphoma or sarcoidosis), atypical infections, genetic conditions (eg, variant cystic fibrosis), autoimmune conditions, or cardiomyopathy. A detailed symptom history, family history, and careful physical examination will help expand and then refine the differential diagnosis. At this stage, typical infections are less likely.
She had presented two months prior with nonproductive cough and dyspnea. At that presentation, her temperature was 36.3°C, heart rate 110 beats per minute, blood pressure 119/63 mm Hg, respiratory rate 43 breaths per minute, and oxygen saturation 86% while breathing ambient air. A chest CT with contrast demonstrated diffuse patchy multifocal ground-glass opacities in the bilateral lungs as well as a mixture of atelectasis and lobular emphysema in the dependent lobes bilaterally (Figure 1). Her main pulmonary artery was dilated at 3.6 cm (mean of 2.42 cm with SD 0.22). She was diagnosed with atypical pneumonia. She was administered azithromycin, weaned off oxygen, and discharged after a seven-day hospitalization.
Two months prior, she had marked tachypnea, tachycardia, and hypoxemia, and imaging revealed diffuse ground-glass opacities. The differential diagnosis for this constellation of symptoms is extensive and includes many conditions that have an inflammatory component, such as atypical pneumonia caused by Mycoplasma or Chlamydia pneumoniae or a common respiratory virus such as rhinovirus or human metapneumovirus. However, two findings make an acute pneumonia unlikely to be the sole cause of her symptoms: underlying emphysema and an enlarged pulmonary artery. Emphysema is an uncommon finding in children and can be related to congenital or acquired causes; congenital lobar emphysema most often presents earlier in life and is focal, not diffuse. Alpha-1-anti-trypin deficiency and mutations in connective tissue genes such as those encoding for elastin and fibrillin can lead to pulmonary disease. While not diagnostic of pulmonary hypertension, her dilated pulmonary artery, coupled with her history, makes pulmonary hypertension a strong possibility. While her pulmonary hypertension is most likely secondary to chronic lung disease based on the emphysematous changes on CT, it could still be related to a cardiac etiology.
The patient had a history of seasonal allergies and well-controlled asthma. She was hospitalized at age six for an asthma exacerbation associated with a respiratory infection. She was discharged with an albuterol inhaler, but seldom used it. Her parents denied any regular coughing during the day or night. She was morbidly obese. Her tonsils and adenoids were removed to treat obstructive sleep apnea (OSA) at age seven, and a subsequent polysomnography was normal. Her medications included intranasal fluticasone propionate and oral iron supplementation. She had no known allergies or recent travels. She had never smoked. She had two pet cats and a dog. Her mother had a history of obesity, OSA, and eczema. Her father had diabetes and eczema.
The patient’s history prior to the recent few months sheds little light on the cause of her current symptoms. While it is possible that her current symptoms are related to the worsening of a process that had been present for many years which mimicked asthma, this seems implausible given the long period of time between her last asthma exacerbation and her present symptoms. Similarly, while tonsillar and adenoidal hypertrophy can be associated with infiltrative diseases (such as lymphoma), this is less common than the usual (and normal) disproportionate increase in size of the adenoids compared to other airway structures during growth in children.
She was admitted to the hospital. On initial examination, her temperature was 37.4°C, heart rate 125 beats per minute, blood pressure 143/69 mm Hg, respiratory rate 48 breaths per minute, and oxygen saturation 86% breathing ambient air. Her BMI was 58 kg/m2. Her exam demonstrated increased work of breathing with accessory muscle use, and decreased breath sounds at the bases. There were no wheezes or crackles. Cardiovascular, abdominal, and skin exams were normal except for tachycardia. At rest, later in the hospitalization, her oxygen saturation was 97% breathing ambient air and heart rate 110 bpm. After two minutes of walking, her oxygen saturation was 77% and heart rate 132 bpm. Two minutes after resting, her oxygen saturation increased to 91%.
Her white blood cell count was 11.9 x 10 9 /L (67% neutrophils, 24.2% lymphocytes, 6% monocytes, and 2% eosinophils), hemoglobin 11.2 g/dL, and platelet count 278,000/mm 3 . Her complete metabolic panel was normal. The C-reactive protein (CRP) was 24 mg/L (normal range, < 4.9) and erythrocyte sedimentation rate (ESR) 103 mm/hour (normal range, 0-32). A venous blood gas (VBG) showed a pH of 7.42 and pCO2 39. An EKG demonstrated sinus tachycardia.
The combination of the patient’s tachypnea, hypoxemia, respiratory distress, and obesity is striking. Her lack of adventitious lung sounds is surprising given her CT findings, but the sensitivity of chest auscultation may be limited in obese patients. Her laboratory findings help narrow the diagnostic frame: she has mild anemia and leukocytosis along with significant inflammation. The normal CO2 concentration on VBG is concerning given the degree of her tachypnea and reflects significant alveolar hypoventilation.
This marked inflammation with diffuse lung findings again raises the possibility of an inflammatory or, less likely, infectious disorder. Sjogren’s syndrome, systemic lupus erythematosus (SLE), and juvenile dermatomyositis can present in young women with interstitial lung disease. She does have exposure to pets and hypersensitivity pneumonitis can worsen rapidly with continued exposure. Another possibility is that she has an underlying immunodeficiency such as common variable immunodeficiency, although a history of recurrent infections such as pneumonia, bacteremia, or sinusitis is lacking.
An echocardiogram should be performed. In addition, laboratory evaluation for the aforementioned autoimmune causes of interstitial lung disease, immunoglobulin levels, pulmonary function testing (if available as an inpatient), and potentially a bronchoscopy with bronchoalveolar lavage (BAL), and biopsy should be pursued. The BAL and biopsy would be helpful in evaluating for infection and interstitial lung disease in an expeditious manner.
A chest CT without contrast was done and compared to the scan from two months prior. New diffuse, ill-defined centrilobular ground-glass opacities were evident throughout the lung fields; dilation of the main pulmonary artery was unchanged, and previously seen ground-glass opacities had resolved. There were patchy areas of air-trapping and mosaic attenuation in the lower lobes (Figure 2).
Transthoracic echocardiogram demonstrated a right ventricular systolic pressure of 58 mm Hg with flattened intraventricular septum during systole. Left and right ventricular systolic function were normal. The left ventricular diastolic function was normal. Pulmonary function testing demonstrated a FEV1/FVC ratio of 100 (112% predicted), FVC 1.07 L (35 % predicted) and FEV1 1.07 L (39% predicted), and total lung capacity was 2.7L (56% predicted) (Figure 3). Single-breath carbon monoxide uptake in the lung was not interpretable based on 2017 European Respiratory Society (ERS)/American Thoracic Society (ATS) technical standards.
This information is helpful in classifying whether this patient’s primary condition is cardiac or pulmonary in nature. Her normal left ventricular systolic and diastolic function make a cardiac etiology for her pulmonary hypertension less likely. Further, the combination of pulmonary hypertension, a restrictive pattern on pulmonary function testing, and findings consistent with interstitial lung disease on cross-sectional imaging all suggest a primary pulmonary etiology rather than a cardiac, infectious, or thromboembolic condition. While chronic thromboembolic hypertension can result in nonspecific mosaic attenuation, it typically would not cause centrilobular ground-glass opacities nor restrictive lung disease. Thus, it seems most likely that this patient has a progressive pulmonary process resulting in hypoxia, pulmonary hypertension, centrilobular opacities, and lower-lobe mosaic attenuation. Considerations for this process can be broadly categorized as one of the childhood interstitial lung disease (chILD). While this differential diagnosis is broad, strong consideration should be given to hypersensitivity pneumonitis, chronic aspiration, sarcoidosis, and Sjogren’s syndrome. An intriguing possibility is that the patient’s “response to azithromycin” two months prior was due to the avoidance of an inhaled antigen while she was in the hospital; a detailed environmental history should be explored. The normal polysomnography after tonsilloadenoidectomy makes it unlikely that OSA is a major contributor to her current presentation. However, since the surgery was seven years ago, and her BMI is presently 58 kg/m2 she remains at risk for OSA and obesity-hypoventilation syndrome. Polysomnography should be done after her acute symptoms improve.
She was started on 5 mm Hg of continuous positive airway pressure (CPAP) at night after a sleep study on room air demonstrated severe OSA with a respiratory disturbance index of 13 events per hour. Antinuclear antibodies (ANA), anti-neutrophil cytoplasmic antibody (ANCA), anti-Jo-1 antibody, anti-RNP antibody, anti-Smith antibody, anti-Ro/SSA and anti-La/SSB antibody were negative as was the histoplasmin antibody. Serum angiotensin-converting enzyme (ACE) level was normal. Mycoplasma IgM and IgG were negative. IgE was 529 kU/L (normal range, <114).
This evaluation reduces the likelihood the patient has Sjogren’s syndrome, SLE, dermatomyositis, or ANCA-associated pulmonary disease. While many patients with dermatomyositis may have negative serologic evaluations, other findings usually present such as rash and myositis are lacking. The negative ANCA evaluation makes granulomatosis with polyangiitis and microscopic polyangiitis very unlikely given the high sensitivity of the ANCA assay for these conditions. ANCA assays are less sensitive for eosinophilic granulomatosis with polyangiitis (EGPA), but the lack of eosinophilia significantly decreases the likelihood of EGPA. ACE levels have relatively poor operating characteristics in the evaluation of sarcoidosis; however, sarcoidosis seems unlikely in this case, especially as patients with sarcoidosis tend to have low or normal IgE levels. Patients with asthma can have elevated IgE levels. However, very elevated IgE levels are more common in other conditions, including allergic bronchopulmonary aspergillosis (ABPA) and the Hyper-IgE syndrome. The latter manifests with recurrent infections and eczema, and is inherited in an autosomal dominant manner. However, both the Hyper-IgE syndrome and ABPA have much higher IgE levels than seen in this case. Allergen-specific IgE testing (including for antibodies to Aspergillus) should be sent. It seems that an interstitial lung disease is present; the waxing and waning pattern and clinical presentation, along with the lack of other systemic findings, make hypersensitivity pneumonitis most likely.
The family lived in an apartment building. Her symptoms started when the family’s neighbor recently moved his outdoor pigeon coop into his basement. The patient often smelled the pigeons and noted feathers coming through the holes in the wall.
One of the key diagnostic features of hypersensitivity pneumonitis (HP) is the history of exposure to a potential offending antigen—in this case likely bird feathers—along with worsening upon reexposure to that antigen. HP is primarily a clinical diagnosis, and testing for serum precipitants has limited value, given the high false negative rate and the frequent lack of clinical symptoms accompanying positive testing. Bronchoalveolar lavage fluid may reveal lymphocytosis and reduced CD4:CD8 ratio. Crackles are commonly heard on examination, but in this case were likely not auscultated due to her obese habitus. The most important treatment is withdrawal of the offending antigen. Limited data suggest that corticosteroid therapy may be helpful in certain HP cases, including subacute, chronic and severe cases as well as patients with hypoxemia, significant imaging findings, and those with significant abnormalities on pulmonary function testing (PFT).
A hypersensitivity pneumonitis precipitins panel was sent with positive antibodies to M. faeni, T. Vulgaris, A. Fumigatus 1 and 6, A. Flavus, and pigeon serum. Her symptoms gradually improved within five days of oral prednisone (60 mg). She was discharged home without dyspnea and normal oxygen saturation while breathing ambient air. A repeat echocardiogram after nighttime CPAP for 1 week demonstrated a right ventricular systolic pressure of 17 mm Hg consistent with improved pulmonary hypertension.
Three weeks later, she returned to clinic for follow up. She had re-experienced dyspnea, cough, and wheezing, which improved when she was outdoors. She was afebrile, tachypneic, tachycardic, and her oxygen saturation was 92% on ambient air.
Her steroid-responsive interstitial lung disease and rapid improvement upon avoidance of the offending antigen is consistent with HP. The positive serum precipitins assay lends further credence to the diagnosis of HP, although serologic analysis with such antibody assays is limited by false positives and false negatives; further, individuals exposed to pigeons often have antibodies present without evidence of HP. History taking at this visit should ask specifically about further pigeon exposure: were the pigeons removed from the home completely, were heating-cooling filters changed, carpets cleaned, and bedding laundered? An in-home evaluation may be helpful before conducting further diagnostic testing.
She was admitted for oxygen therapy and a bronchoscopy, which showed mucosal friability and cobblestoning, suggesting inflammation. BAL revealed a normal CD4:CD8 ratio of 3; BAL cultures were sterile. Her shortness of breath significantly improved following a prolonged course of systemic steroids and removal from the triggering environment. PFTs improved with a FEV1/FVC ratio of 94 (105% predicted), FVC of 2.00 L (66% predicted), FEV1 of 1.88L (69% predicted) (Figure 3B). Her presenting symptoms of persistent cough and progressive dyspnea on exertion, characteristic CT, sterile BAL cultures, positive serum precipitants against pigeon serum, and resolution of her symptoms with withdrawal of the offending antigen were diagnostic of hypersensitivity pneumonitis due to pigeon exposure, also known as bird fancier’s disease.
COMMENTARY
The patient’s original presentation of dyspnea, tachypnea, and hypoxia is commonly associated with pediatric pneumonia and asthma exacerbations.1 However, an alternative diagnosis was suggested by the lack of wheezing, absence of fever, and recurrent presentations with progressive symptoms.
Hypersensitivity pneumonitis (HP) represents an exaggerated T-cell meditated immune response to inhalation of an offending antigen that results in a restrictive ventilatory defect and interstitial infiltrates.2 Bird pneumonitis (also known as bird fancier’s disease) is a frequent cause of HP, accounting for approximately 65-70% of cases.3 HP, however, only manifests in a small number of subjects exposed to culprit antigens, suggesting an underlying genetic susceptibility.4 Prevalence estimates vary depending on bird species, county, climate, and other possible factors.
There are no standard criteria for the diagnosis of HP, though a combination of findings is suggestive. A recent prospective multicenter study created a scoring system for HP based on factors associated with the disease to aid in accurate diagnosis. The most relevant criteria included antigen exposure, recurrent symptoms noted within 4-8 hours after antigen exposure, weight loss, presence of specific IgG antibodies to avian antigens, and inspiratory crackles on exam. Using this rule, the probability that our patient has HP based on clinical characteristics was 93% with an area under the receiver operating curve of 0.93 (96% confidence interval: 0.90-0.95)5. Chest imaging (high resolution CT) often consists of a mosaic pattern of air trapping, as seen in this patient in combination with ground-glass opacities6. Bronchoalveolar lavage (BAL) is sensitive in detecting lung inflammation in a patient with suspected HP. On BAL, a lymphocytic alveolitis can be seen, but absence of this finding does not exclude HP.5,7,8 Pulmonary function tests (PFTs) may be normal in acute HP. When abnormal, PFTs may reveal a restrictive pattern and reduction in carbon monoxide diffusing capacity.7 However, BAL and PFT results are neither specific nor diagnostic of HP; it is important to consider results in the context of the clinical picture.
The respiratory response to inhalation of the avian antigen has traditionally been classified as acute, subacute, or chronic.9 The acute response occurs within hours of exposure to the offending agent and usually resolves within 24 hours after antigen withdrawal. The subacute presentation involves cough and dyspnea over several days to weeks, and can progress to chronic and permanent lung damage if unrecognized and untreated. In chronic presentations, lung abnormalities may persist despite antigen avoidance and pharmacologic interventions.4,10 The patient’s symptoms occurred over a six-month period which coincided with pigeon exposure and resolved during each hospitalization with steroid treatment and removal from the offending agent. Her presentation was consistent with a subacute time course of HP.
The dilated pulmonary artery, elevated right systolic ventricular pressure, and normal right ventricular function in our patient suggested pulmonary hypertension of chronic duration. Her risk factors for pulmonary hypertension included asthma, sleep apnea, possible obesity-hypoventilation syndrome, and HP-associated interstitial lung disease.11
The most important intervention in HP is avoidance of the causative antigen. Medical therapy without removal of antigen is inadequate. Systemic corticosteroids can help ameliorate acute symptoms though dosing and duration remains unclear. For chronic patients unresponsive to steroid therapy, lung transplantation can be considered.4
The key to diagnosis of HP in this patient—and to minimizing repeat testing upon the patient’s recrudescence of symptoms—was the clinician’s consideration that the major impetus for the patient’s improvement in the hospital was removal from the offending antigen in her home environment. As in this case, taking time to delve deeply into a patient’s environment—even by descending the basement stairs—may lead to the diagnosis.
LEARNING POINTS
- Consider hypersensitivity pneumonitis (HP) in patients with recurrent respiratory distress, offending exposure, and resolution of symptoms with removal of culprit antigen.
- The most important treatment of HP is removal of offending antigen; systemic and/or inhaled corticosteroids are indicated until the full resolution of respiratory symptoms.
- Prognosis is dependent on early diagnosis and removal of offending exposures.
- Failure to treat HP might result in end-stage lung disease from pulmonary fibrosis secondary to long-term inflammation.
Disclosures
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
A 14-year-old girl with a history of asthma presented to the Emergency Department (ED) with three months of persistent, nonproductive cough, and progressive shortness of breath. She reported fatigue, chest tightness, orthopnea, and dyspnea with exertion. She denied fever, rhinorrhea, congestion, hemoptysis, or paroxysmal nocturnal dyspnea.
Her age and past medical history of asthma are incongruent with her new symptoms, as asthma is typified by intermittent exacerbations, not progressive symptoms. Thus, another process, in addition to asthma, is most likely present; it is also important to question the accuracy of previous diagnoses in light of new information. Her symptoms may signify an underlying cardiopulmonary process, such as infiltrative diseases (eg, lymphoma or sarcoidosis), atypical infections, genetic conditions (eg, variant cystic fibrosis), autoimmune conditions, or cardiomyopathy. A detailed symptom history, family history, and careful physical examination will help expand and then refine the differential diagnosis. At this stage, typical infections are less likely.
She had presented two months prior with nonproductive cough and dyspnea. At that presentation, her temperature was 36.3°C, heart rate 110 beats per minute, blood pressure 119/63 mm Hg, respiratory rate 43 breaths per minute, and oxygen saturation 86% while breathing ambient air. A chest CT with contrast demonstrated diffuse patchy multifocal ground-glass opacities in the bilateral lungs as well as a mixture of atelectasis and lobular emphysema in the dependent lobes bilaterally (Figure 1). Her main pulmonary artery was dilated at 3.6 cm (mean of 2.42 cm with SD 0.22). She was diagnosed with atypical pneumonia. She was administered azithromycin, weaned off oxygen, and discharged after a seven-day hospitalization.
Two months prior, she had marked tachypnea, tachycardia, and hypoxemia, and imaging revealed diffuse ground-glass opacities. The differential diagnosis for this constellation of symptoms is extensive and includes many conditions that have an inflammatory component, such as atypical pneumonia caused by Mycoplasma or Chlamydia pneumoniae or a common respiratory virus such as rhinovirus or human metapneumovirus. However, two findings make an acute pneumonia unlikely to be the sole cause of her symptoms: underlying emphysema and an enlarged pulmonary artery. Emphysema is an uncommon finding in children and can be related to congenital or acquired causes; congenital lobar emphysema most often presents earlier in life and is focal, not diffuse. Alpha-1-anti-trypin deficiency and mutations in connective tissue genes such as those encoding for elastin and fibrillin can lead to pulmonary disease. While not diagnostic of pulmonary hypertension, her dilated pulmonary artery, coupled with her history, makes pulmonary hypertension a strong possibility. While her pulmonary hypertension is most likely secondary to chronic lung disease based on the emphysematous changes on CT, it could still be related to a cardiac etiology.
The patient had a history of seasonal allergies and well-controlled asthma. She was hospitalized at age six for an asthma exacerbation associated with a respiratory infection. She was discharged with an albuterol inhaler, but seldom used it. Her parents denied any regular coughing during the day or night. She was morbidly obese. Her tonsils and adenoids were removed to treat obstructive sleep apnea (OSA) at age seven, and a subsequent polysomnography was normal. Her medications included intranasal fluticasone propionate and oral iron supplementation. She had no known allergies or recent travels. She had never smoked. She had two pet cats and a dog. Her mother had a history of obesity, OSA, and eczema. Her father had diabetes and eczema.
The patient’s history prior to the recent few months sheds little light on the cause of her current symptoms. While it is possible that her current symptoms are related to the worsening of a process that had been present for many years which mimicked asthma, this seems implausible given the long period of time between her last asthma exacerbation and her present symptoms. Similarly, while tonsillar and adenoidal hypertrophy can be associated with infiltrative diseases (such as lymphoma), this is less common than the usual (and normal) disproportionate increase in size of the adenoids compared to other airway structures during growth in children.
She was admitted to the hospital. On initial examination, her temperature was 37.4°C, heart rate 125 beats per minute, blood pressure 143/69 mm Hg, respiratory rate 48 breaths per minute, and oxygen saturation 86% breathing ambient air. Her BMI was 58 kg/m2. Her exam demonstrated increased work of breathing with accessory muscle use, and decreased breath sounds at the bases. There were no wheezes or crackles. Cardiovascular, abdominal, and skin exams were normal except for tachycardia. At rest, later in the hospitalization, her oxygen saturation was 97% breathing ambient air and heart rate 110 bpm. After two minutes of walking, her oxygen saturation was 77% and heart rate 132 bpm. Two minutes after resting, her oxygen saturation increased to 91%.
Her white blood cell count was 11.9 x 10 9 /L (67% neutrophils, 24.2% lymphocytes, 6% monocytes, and 2% eosinophils), hemoglobin 11.2 g/dL, and platelet count 278,000/mm 3 . Her complete metabolic panel was normal. The C-reactive protein (CRP) was 24 mg/L (normal range, < 4.9) and erythrocyte sedimentation rate (ESR) 103 mm/hour (normal range, 0-32). A venous blood gas (VBG) showed a pH of 7.42 and pCO2 39. An EKG demonstrated sinus tachycardia.
The combination of the patient’s tachypnea, hypoxemia, respiratory distress, and obesity is striking. Her lack of adventitious lung sounds is surprising given her CT findings, but the sensitivity of chest auscultation may be limited in obese patients. Her laboratory findings help narrow the diagnostic frame: she has mild anemia and leukocytosis along with significant inflammation. The normal CO2 concentration on VBG is concerning given the degree of her tachypnea and reflects significant alveolar hypoventilation.
This marked inflammation with diffuse lung findings again raises the possibility of an inflammatory or, less likely, infectious disorder. Sjogren’s syndrome, systemic lupus erythematosus (SLE), and juvenile dermatomyositis can present in young women with interstitial lung disease. She does have exposure to pets and hypersensitivity pneumonitis can worsen rapidly with continued exposure. Another possibility is that she has an underlying immunodeficiency such as common variable immunodeficiency, although a history of recurrent infections such as pneumonia, bacteremia, or sinusitis is lacking.
An echocardiogram should be performed. In addition, laboratory evaluation for the aforementioned autoimmune causes of interstitial lung disease, immunoglobulin levels, pulmonary function testing (if available as an inpatient), and potentially a bronchoscopy with bronchoalveolar lavage (BAL), and biopsy should be pursued. The BAL and biopsy would be helpful in evaluating for infection and interstitial lung disease in an expeditious manner.
A chest CT without contrast was done and compared to the scan from two months prior. New diffuse, ill-defined centrilobular ground-glass opacities were evident throughout the lung fields; dilation of the main pulmonary artery was unchanged, and previously seen ground-glass opacities had resolved. There were patchy areas of air-trapping and mosaic attenuation in the lower lobes (Figure 2).
Transthoracic echocardiogram demonstrated a right ventricular systolic pressure of 58 mm Hg with flattened intraventricular septum during systole. Left and right ventricular systolic function were normal. The left ventricular diastolic function was normal. Pulmonary function testing demonstrated a FEV1/FVC ratio of 100 (112% predicted), FVC 1.07 L (35 % predicted) and FEV1 1.07 L (39% predicted), and total lung capacity was 2.7L (56% predicted) (Figure 3). Single-breath carbon monoxide uptake in the lung was not interpretable based on 2017 European Respiratory Society (ERS)/American Thoracic Society (ATS) technical standards.
This information is helpful in classifying whether this patient’s primary condition is cardiac or pulmonary in nature. Her normal left ventricular systolic and diastolic function make a cardiac etiology for her pulmonary hypertension less likely. Further, the combination of pulmonary hypertension, a restrictive pattern on pulmonary function testing, and findings consistent with interstitial lung disease on cross-sectional imaging all suggest a primary pulmonary etiology rather than a cardiac, infectious, or thromboembolic condition. While chronic thromboembolic hypertension can result in nonspecific mosaic attenuation, it typically would not cause centrilobular ground-glass opacities nor restrictive lung disease. Thus, it seems most likely that this patient has a progressive pulmonary process resulting in hypoxia, pulmonary hypertension, centrilobular opacities, and lower-lobe mosaic attenuation. Considerations for this process can be broadly categorized as one of the childhood interstitial lung disease (chILD). While this differential diagnosis is broad, strong consideration should be given to hypersensitivity pneumonitis, chronic aspiration, sarcoidosis, and Sjogren’s syndrome. An intriguing possibility is that the patient’s “response to azithromycin” two months prior was due to the avoidance of an inhaled antigen while she was in the hospital; a detailed environmental history should be explored. The normal polysomnography after tonsilloadenoidectomy makes it unlikely that OSA is a major contributor to her current presentation. However, since the surgery was seven years ago, and her BMI is presently 58 kg/m2 she remains at risk for OSA and obesity-hypoventilation syndrome. Polysomnography should be done after her acute symptoms improve.
She was started on 5 mm Hg of continuous positive airway pressure (CPAP) at night after a sleep study on room air demonstrated severe OSA with a respiratory disturbance index of 13 events per hour. Antinuclear antibodies (ANA), anti-neutrophil cytoplasmic antibody (ANCA), anti-Jo-1 antibody, anti-RNP antibody, anti-Smith antibody, anti-Ro/SSA and anti-La/SSB antibody were negative as was the histoplasmin antibody. Serum angiotensin-converting enzyme (ACE) level was normal. Mycoplasma IgM and IgG were negative. IgE was 529 kU/L (normal range, <114).
This evaluation reduces the likelihood the patient has Sjogren’s syndrome, SLE, dermatomyositis, or ANCA-associated pulmonary disease. While many patients with dermatomyositis may have negative serologic evaluations, other findings usually present such as rash and myositis are lacking. The negative ANCA evaluation makes granulomatosis with polyangiitis and microscopic polyangiitis very unlikely given the high sensitivity of the ANCA assay for these conditions. ANCA assays are less sensitive for eosinophilic granulomatosis with polyangiitis (EGPA), but the lack of eosinophilia significantly decreases the likelihood of EGPA. ACE levels have relatively poor operating characteristics in the evaluation of sarcoidosis; however, sarcoidosis seems unlikely in this case, especially as patients with sarcoidosis tend to have low or normal IgE levels. Patients with asthma can have elevated IgE levels. However, very elevated IgE levels are more common in other conditions, including allergic bronchopulmonary aspergillosis (ABPA) and the Hyper-IgE syndrome. The latter manifests with recurrent infections and eczema, and is inherited in an autosomal dominant manner. However, both the Hyper-IgE syndrome and ABPA have much higher IgE levels than seen in this case. Allergen-specific IgE testing (including for antibodies to Aspergillus) should be sent. It seems that an interstitial lung disease is present; the waxing and waning pattern and clinical presentation, along with the lack of other systemic findings, make hypersensitivity pneumonitis most likely.
The family lived in an apartment building. Her symptoms started when the family’s neighbor recently moved his outdoor pigeon coop into his basement. The patient often smelled the pigeons and noted feathers coming through the holes in the wall.
One of the key diagnostic features of hypersensitivity pneumonitis (HP) is the history of exposure to a potential offending antigen—in this case likely bird feathers—along with worsening upon reexposure to that antigen. HP is primarily a clinical diagnosis, and testing for serum precipitants has limited value, given the high false negative rate and the frequent lack of clinical symptoms accompanying positive testing. Bronchoalveolar lavage fluid may reveal lymphocytosis and reduced CD4:CD8 ratio. Crackles are commonly heard on examination, but in this case were likely not auscultated due to her obese habitus. The most important treatment is withdrawal of the offending antigen. Limited data suggest that corticosteroid therapy may be helpful in certain HP cases, including subacute, chronic and severe cases as well as patients with hypoxemia, significant imaging findings, and those with significant abnormalities on pulmonary function testing (PFT).
A hypersensitivity pneumonitis precipitins panel was sent with positive antibodies to M. faeni, T. Vulgaris, A. Fumigatus 1 and 6, A. Flavus, and pigeon serum. Her symptoms gradually improved within five days of oral prednisone (60 mg). She was discharged home without dyspnea and normal oxygen saturation while breathing ambient air. A repeat echocardiogram after nighttime CPAP for 1 week demonstrated a right ventricular systolic pressure of 17 mm Hg consistent with improved pulmonary hypertension.
Three weeks later, she returned to clinic for follow up. She had re-experienced dyspnea, cough, and wheezing, which improved when she was outdoors. She was afebrile, tachypneic, tachycardic, and her oxygen saturation was 92% on ambient air.
Her steroid-responsive interstitial lung disease and rapid improvement upon avoidance of the offending antigen is consistent with HP. The positive serum precipitins assay lends further credence to the diagnosis of HP, although serologic analysis with such antibody assays is limited by false positives and false negatives; further, individuals exposed to pigeons often have antibodies present without evidence of HP. History taking at this visit should ask specifically about further pigeon exposure: were the pigeons removed from the home completely, were heating-cooling filters changed, carpets cleaned, and bedding laundered? An in-home evaluation may be helpful before conducting further diagnostic testing.
She was admitted for oxygen therapy and a bronchoscopy, which showed mucosal friability and cobblestoning, suggesting inflammation. BAL revealed a normal CD4:CD8 ratio of 3; BAL cultures were sterile. Her shortness of breath significantly improved following a prolonged course of systemic steroids and removal from the triggering environment. PFTs improved with a FEV1/FVC ratio of 94 (105% predicted), FVC of 2.00 L (66% predicted), FEV1 of 1.88L (69% predicted) (Figure 3B). Her presenting symptoms of persistent cough and progressive dyspnea on exertion, characteristic CT, sterile BAL cultures, positive serum precipitants against pigeon serum, and resolution of her symptoms with withdrawal of the offending antigen were diagnostic of hypersensitivity pneumonitis due to pigeon exposure, also known as bird fancier’s disease.
COMMENTARY
The patient’s original presentation of dyspnea, tachypnea, and hypoxia is commonly associated with pediatric pneumonia and asthma exacerbations.1 However, an alternative diagnosis was suggested by the lack of wheezing, absence of fever, and recurrent presentations with progressive symptoms.
Hypersensitivity pneumonitis (HP) represents an exaggerated T-cell meditated immune response to inhalation of an offending antigen that results in a restrictive ventilatory defect and interstitial infiltrates.2 Bird pneumonitis (also known as bird fancier’s disease) is a frequent cause of HP, accounting for approximately 65-70% of cases.3 HP, however, only manifests in a small number of subjects exposed to culprit antigens, suggesting an underlying genetic susceptibility.4 Prevalence estimates vary depending on bird species, county, climate, and other possible factors.
There are no standard criteria for the diagnosis of HP, though a combination of findings is suggestive. A recent prospective multicenter study created a scoring system for HP based on factors associated with the disease to aid in accurate diagnosis. The most relevant criteria included antigen exposure, recurrent symptoms noted within 4-8 hours after antigen exposure, weight loss, presence of specific IgG antibodies to avian antigens, and inspiratory crackles on exam. Using this rule, the probability that our patient has HP based on clinical characteristics was 93% with an area under the receiver operating curve of 0.93 (96% confidence interval: 0.90-0.95)5. Chest imaging (high resolution CT) often consists of a mosaic pattern of air trapping, as seen in this patient in combination with ground-glass opacities6. Bronchoalveolar lavage (BAL) is sensitive in detecting lung inflammation in a patient with suspected HP. On BAL, a lymphocytic alveolitis can be seen, but absence of this finding does not exclude HP.5,7,8 Pulmonary function tests (PFTs) may be normal in acute HP. When abnormal, PFTs may reveal a restrictive pattern and reduction in carbon monoxide diffusing capacity.7 However, BAL and PFT results are neither specific nor diagnostic of HP; it is important to consider results in the context of the clinical picture.
The respiratory response to inhalation of the avian antigen has traditionally been classified as acute, subacute, or chronic.9 The acute response occurs within hours of exposure to the offending agent and usually resolves within 24 hours after antigen withdrawal. The subacute presentation involves cough and dyspnea over several days to weeks, and can progress to chronic and permanent lung damage if unrecognized and untreated. In chronic presentations, lung abnormalities may persist despite antigen avoidance and pharmacologic interventions.4,10 The patient’s symptoms occurred over a six-month period which coincided with pigeon exposure and resolved during each hospitalization with steroid treatment and removal from the offending agent. Her presentation was consistent with a subacute time course of HP.
The dilated pulmonary artery, elevated right systolic ventricular pressure, and normal right ventricular function in our patient suggested pulmonary hypertension of chronic duration. Her risk factors for pulmonary hypertension included asthma, sleep apnea, possible obesity-hypoventilation syndrome, and HP-associated interstitial lung disease.11
The most important intervention in HP is avoidance of the causative antigen. Medical therapy without removal of antigen is inadequate. Systemic corticosteroids can help ameliorate acute symptoms though dosing and duration remains unclear. For chronic patients unresponsive to steroid therapy, lung transplantation can be considered.4
The key to diagnosis of HP in this patient—and to minimizing repeat testing upon the patient’s recrudescence of symptoms—was the clinician’s consideration that the major impetus for the patient’s improvement in the hospital was removal from the offending antigen in her home environment. As in this case, taking time to delve deeply into a patient’s environment—even by descending the basement stairs—may lead to the diagnosis.
LEARNING POINTS
- Consider hypersensitivity pneumonitis (HP) in patients with recurrent respiratory distress, offending exposure, and resolution of symptoms with removal of culprit antigen.
- The most important treatment of HP is removal of offending antigen; systemic and/or inhaled corticosteroids are indicated until the full resolution of respiratory symptoms.
- Prognosis is dependent on early diagnosis and removal of offending exposures.
- Failure to treat HP might result in end-stage lung disease from pulmonary fibrosis secondary to long-term inflammation.
Disclosures
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
1. Ebell MH. Clinical diagnosis of pneumonia in children. Am Fam Physician. 2010;82(2):192-193. PubMed
2. Cormier Y, Lacasse Y. Hypersensitivity pneumonitis and organic dust toxic syndrome. In: Malo J-L, Chan-Yeung M, Bernstein DI, eds. Asthma in the Workplace. Vol 32. Boca Raton, FL: Fourth Informa Healthcare; 2013:392-405.
3. Chan AL, Juarez MM, Leslie KO, Ismail HA, Albertson TE. Bird fancier’s lung: a state-of-the-art review. Clin Rev Allergy Immunol. 2012;43(1-2):69-83. doi: 10.1007/s12016-011-8282-y. PubMed
4. Camarena A, Juárez A, Mejía M, et al. Major histocompatibility complex and tumor necrosis factor-α polymorphisms in pigeon breeder’s disease. Am J Respir Crit Care Med. 2001;163(7):1528-1533. https:/doi.org/10.1164/ajrccm.163.7.2004023. PubMed
5. Lacasse Y, Selman M, Costabel U, et al. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2003;168(8):952-958. doi: 10.1164/rccm.200301-137OC. PubMed
6. Glazer CS, Rose CS, Lynch DA. Clinical and radiologic manifestations of hypersensitivity pneumonitis. J Thorac Imaging. 2002;17(4):261-272. PubMed
7. Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186(4):314-324. doi: 10.1164/rccm.201203-0513CI. PubMed
8. Calillad DM, Vergnon, JM, Madroszyk A, et al. Bronchoalveolar lavage in hypersensitivity pneumonitis: a series of 139 patients. Inflamm Allergy Drug Targets. 2012;11(1):15-19. doi: 10.2174/187152812798889330. PubMed
9. Richerson HB, Bernstein IL, Fink JN, et al. Guidelines for the clinical evaluation of hypersensitivity pneumonitis. Report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol. 1989;84(5 Pt 2):839-844. doi: 10.1016/0091-6749(89)90349-7. PubMed
10. Zacharisen MC, Schlueter DP, Kurup VP, Fink JN. The long-term outcome in acute, subacute, and chronic forms of pigeon breeder’s disease hypersensitivity pneumonitis. Ann Allergy Asthma Immunol. 2002;88(2):175-182. doi: 10.1016/S1081-1206(10)61993-X. PubMed
11. Raymond TE, Khabbaza JE, Yadav R, Tonelli AR. Significance of main pulmonary artery dilation on imaging studies. Ann Am Thorac Soc. 2014;11(10):1623-1632. doi: 10.1513/AnnalsATS.201406-253PP. PubMed
1. Ebell MH. Clinical diagnosis of pneumonia in children. Am Fam Physician. 2010;82(2):192-193. PubMed
2. Cormier Y, Lacasse Y. Hypersensitivity pneumonitis and organic dust toxic syndrome. In: Malo J-L, Chan-Yeung M, Bernstein DI, eds. Asthma in the Workplace. Vol 32. Boca Raton, FL: Fourth Informa Healthcare; 2013:392-405.
3. Chan AL, Juarez MM, Leslie KO, Ismail HA, Albertson TE. Bird fancier’s lung: a state-of-the-art review. Clin Rev Allergy Immunol. 2012;43(1-2):69-83. doi: 10.1007/s12016-011-8282-y. PubMed
4. Camarena A, Juárez A, Mejía M, et al. Major histocompatibility complex and tumor necrosis factor-α polymorphisms in pigeon breeder’s disease. Am J Respir Crit Care Med. 2001;163(7):1528-1533. https:/doi.org/10.1164/ajrccm.163.7.2004023. PubMed
5. Lacasse Y, Selman M, Costabel U, et al. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2003;168(8):952-958. doi: 10.1164/rccm.200301-137OC. PubMed
6. Glazer CS, Rose CS, Lynch DA. Clinical and radiologic manifestations of hypersensitivity pneumonitis. J Thorac Imaging. 2002;17(4):261-272. PubMed
7. Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186(4):314-324. doi: 10.1164/rccm.201203-0513CI. PubMed
8. Calillad DM, Vergnon, JM, Madroszyk A, et al. Bronchoalveolar lavage in hypersensitivity pneumonitis: a series of 139 patients. Inflamm Allergy Drug Targets. 2012;11(1):15-19. doi: 10.2174/187152812798889330. PubMed
9. Richerson HB, Bernstein IL, Fink JN, et al. Guidelines for the clinical evaluation of hypersensitivity pneumonitis. Report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol. 1989;84(5 Pt 2):839-844. doi: 10.1016/0091-6749(89)90349-7. PubMed
10. Zacharisen MC, Schlueter DP, Kurup VP, Fink JN. The long-term outcome in acute, subacute, and chronic forms of pigeon breeder’s disease hypersensitivity pneumonitis. Ann Allergy Asthma Immunol. 2002;88(2):175-182. doi: 10.1016/S1081-1206(10)61993-X. PubMed
11. Raymond TE, Khabbaza JE, Yadav R, Tonelli AR. Significance of main pulmonary artery dilation on imaging studies. Ann Am Thorac Soc. 2014;11(10):1623-1632. doi: 10.1513/AnnalsATS.201406-253PP. PubMed
© 2019 Society of Hospital Medicine
So Much More than Bald and Bloated
A 44-year-old previously healthy semiprofessional male athlete presented with five days of nausea, vomiting, and abdominal pain. He had also experienced several months of decreased energy and new episodes of constipation three weeks prior to presentation.
At this point, we do not have sufficient information to completely determine the cause of his abdominal symptoms. Common causes of abdominal pain and vomiting in adults of his age group include peptic ulcer disease, pancreatic or hepatobiliary track disorders, small or large bowel processes, appendicitis, or even renal pathology. Further characterization may be possible by describing the location and quality of pain and factors that might relieve or exacerbate his pain. Despite the ambiguity, multiple clues might allow us to narrow the broad differential diagnosis of abdominal pain. In a previously healthy, vigorous, middle-aged man with subacute abdominal pain associated with constipation, the differential diagnosis should include disease states that may cause a bowel obstruction; these states include inflammatory bowel disease (IBD), gastrointestinal malignancy, or peptic ulcer disease. Mechanical obstruction due to volvulus or intussusception would be less likely in his age group. Given his history of several months of fatigue and several weeks of constipation, he should be evaluated for metabolic causes of abdominal pain and constipation, such as hypothyroidism or hypercalcemia. In addition to basic laboratory and imaging studies, obtaining additional history regarding prior abdominal surgeries, medication use, alcohol intake, and family and travel history will be the key in directing the evaluation.
Six months prior to admission, the patient began to feel more fatigue and exercise intolerance, reduced sweating, increased cold intolerance, and increased presyncopal episodes. He was diagnosed with hypothyroidism (TSH 6.69 μIU/mL; free T4 not done) and initiated on levothyroxine. One month prior to presentation, he developed constipation, loss of taste, reduced appetite, and weight loss of 30 pounds. He developed blurry vision and photophobia. He also complained of erectile dysfunction, urinary hesitancy and straining, which were diagnosed as benign prostatic hypertrophy.
Given the addition of numerous historical features in a previously healthy man, it is important to strive for a parsimonious diagnosis to unify his seemingly disparate features. His fatigue, constipation, and cold intolerance are consistent with his diagnosis of hypothyroidism but are nonspecific. Whether the degree of hypothyroidism caused his symptoms or signs is doubtful. The constellation of symptoms and signs are more likely to be representative of a nonthyroidal illness. His abdominal pain, unexplained weight loss, and presyncopal episodes should raise consideration of adrenal insufficiency. The combination of hypothyroidism and adrenal insufficiency suggest the possibility of an autoimmune polyendocrine syndrome or other pituitary pathology. In this case, history of headache, dysgeusia, and visual disturbances might support the diagnosis of pituitary adenoma. A cosyntropin stimulation test could establish the diagnosis of adrenal insufficiency. A low ACTH level would establish a diagnosis of pituitary or hypothalamic hypofunction. If pituitary hypofunction is documented, then a brain MRI would be needed to confirm the diagnosis of pituitary adenoma.
His newly reported erectile dysfunction suggests the possibility of a psychiatric, neurologic, hormonal, or vascular process and should be explored further. Sexual dysfunction is also associated with adrenal insufficiency and hypopituitarism. However, the presence of suspected prostatic hypertrophy in a male competitive athlete in his forties also raises the question of exogenous androgen use.
His past medical history was notable for a two-year history of alopecia totalis, seasonal allergies, asthma, and a repaired congenital aortic web with known aortic insufficiency. He was married with two children, worked an office job, and had no history of injection drug use, blood transfusions, or multiple sexual partners. His family history was notable for hypothyroidism and asthma in several family members in addition to Crohn disease, celiac disease, diabetes, cardiovascular disease, and cancers of the breast and lung.
His past medical, surgical, and family history supports a diagnosis of autoimmune disease. Although there is a personal and family history of atopic disorders, including allergic rhinitis and asthma, no association is found between atopy and autoimmunity. His family history of hypothyroidism, Crohn disease, and diabetes suggests a familial autoimmune genetic predisposition. His history of alopecia totalis in the setting of hypothyroidism and possible autoimmune adrenal insufficiency or autoimmune hypophysitis raises suspicion for the previously suggested diagnosis of polyglandular autoimmune syndrome, also known as autoimmune polyendocrine syndrome. Type I polyglandular autoimmune syndrome is associated with hypoparathyroidism and mucocutaneous candidiasis. In the absence of these symptoms, the patient more likely has type II polyglandular autoimmune syndrome. Type II syndrome is more prevalent and can occur in the setting of other nonendocrine autoimmune disorders, such as vitiligo, myasthenia gravis, or rheumatoid arthritis. Adrenal insufficiency can be the initial and most prominent manifestation of type II syndrome.
On physical exam, he was afebrile, with a heart rate of 68 beats per minute, respiratory rate of 16 breaths per minute, and normal oxygen saturation. His supine blood pressure and heart rate were 116/72 mm Hg and 66 beats per minute, respectively, and his standing blood pressure and heart rates were 80/48 mm Hg and 68 beats per minute respectively. He was thin, had diffuse scalp and body alopecia, and was ill-appearing with dry skin and dry mucous membranes. No evidence of Osler nodes, Janeway lesions, or splinter hemorrhages were found on cutaneous examination. No Roth spots or conjunctival hemorrhages were noted on ophthalmologic examination. He had both a 3/6 crescendo–decrescendo systolic murmur best heard at the right clavicle and radiated to the carotids and a 3/6 early diastolic decrescendo murmur best heard at the left sternal border. His abdomen was slightly protuberant, with reduced bowel sounds, hyperresonant to tympanitic on percussion, and a diffusely, moderately tender without peritoneal signs. Neurologic examination revealed 8 mm pupils with minimal response to light and accommodation. The remaining portions of his cranial nerve and complete neurologic examination were normal.
The presence of postural hypotension supports the previous suspicion of adrenal insufficiency, and the possibility of a pituitary or hypothalamic process remains. However, his dilated and minimally responsive pupils and potentially adynamic bowel are inconsistent with these diagnoses. Mydriasis and adynamic bowel in combination with orthostatic hypotension, dysgeusia, urinary retention, and erectile dysfunction are strongly suggestive of an autonomic process. Endocarditis is worth considering given his multisystem involvement, subacute decline, and known valve pathology. The absence of fever or stigmata of endocarditis make it difficult to explain his clinical syndrome. An echocardiogram would be reasonable for further assessment. At this point, it is prudent to explore his adrenal and pituitary function; if unrevealing, embark on an evaluation of his autonomic dysfunction.
Initial laboratory investigations were notable for mild normocytic anemia and hypoalbuminemia. His cosyntropin stimulation test was normal at 60 minutes. An abdominal CT scan demonstrated marked dilation in the small bowel loops (6 cm in caliber) with associated small bowel wall thickening and hyperemia. The echocardiogram was unrevealing and only confirmed the ongoing, progression of his known valve pathology without evidence of vegetation.
The above testing rules out primary adrenal insufficiency, but an appropriate response to the cosyntropin stimulation test does not rule out secondary, or pituitary, adrenal insufficiency. The echocardiogram and lack of other features make infective endocarditis unlikely. Thus, as mentioned, it is important now to commence a complete work-up of his probable dysautonomia to explain the majority of his features. Additionally, his hypothyroidism (if more than sick euthyroid syndrome), family history of autoimmune processes, and alopecia totalis all suggest the possibility of an immune-related syndrome. His CT scan revealed some thickened hyperemic bowel, which could suggest an IBD, such as Crohn disease; however, the absence of other signs, such as fever, diarrhea, or bloody stools, argues against this diagnosis. A syndrome that could unify his presentation is autoimmune autonomic ganglionopathy (AAG), a rare genetic autonomic system disorder that presents with pandysautonomia. The spectrum of autoimmunity was considered early in this case, but the differential diagnosis included more common conditions, such as adrenal insufficiency. Similarly, IBD remains a consideration. The serologic studies for IBD can be useful but they lack definitive diagnostic accuracy. Given that treatment for AAG differs from that for IBD, additional information will help guide the therapeutic approach. Anti-α3gnAChR antibodies, which are associated with AAG, should be checked.
His history of presyncope, anhidrosis, urinary retention, and ileus raised suspicion for pandysautonomia, as characterized by signs of sympathetic and parasympathetic dysfunction. The suspicion for pandysautonomia was confirmed via specialized autonomic testing, which included reduced heart rate variation on Valsalva and deep breathing maneuvers, orthostatic hypotension consistent autonomic insufficiency on Tilt table testing, and reduced sweat response to acetylcholine application (QSART test). The patient underwent further diagnostic serologic testing to differentiate causes of autonomic failure (Table 1). His personal and family history of autoimmunity led to the working diagnosis of AAG. Ultimate testing revealed high titers of autoantibodies, specifically anti-α3gnAChR (3.29 nmol/L, normal <0.02 nmol/L), directed against the ganglionic nicotinic acetylcholine receptor. This finding strongly supported the diagnosis of AAG.1,4-7 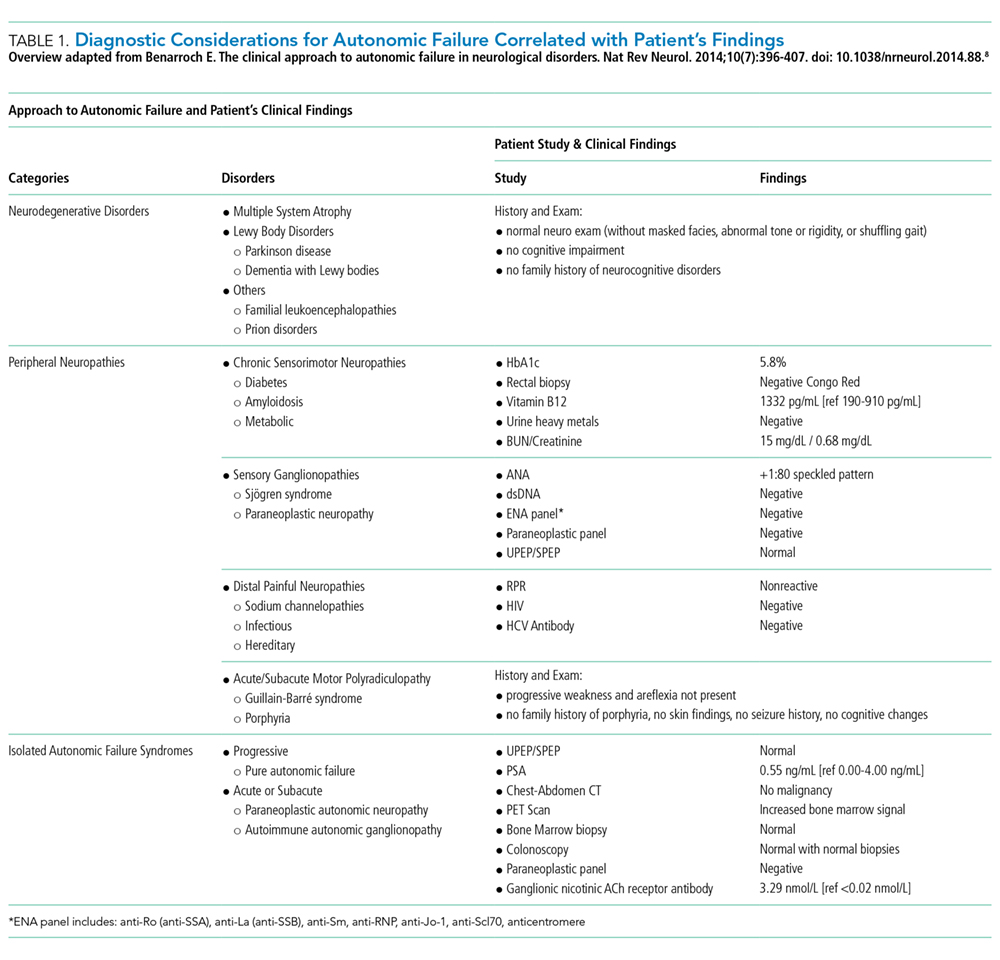
He was initially treated empirically with intravenous immunoglobulin (IVIG) with minimal improvement. He received additional immunomodulating therapies including methylprednisolone, plasmapheresis, and rituximab but did not tolerate a trial of mycophenolate. Six weeks after therapy initiation, his antibody titers decreased to 0.89 nmol/L with associated clinical improvement. Ultimately, he was discharged from the hospital on day 73 with a feeding tube and supplemental total parenteral nutrition. Four months postdischarge, he had returned to his prediagnosis weight, had eased back into his prior activities, and was off supplemental nutrition. Over a year later, he completed a 10-month prednisone taper and continued to receive monthly IVIG infusion
DISCUSSION
The clinical approach to dysautonomia is based on different etiologies: (1) those associated with neurodegenerative disorders; (2) those associated with peripheral neuropathies, and (3) isolated autonomic failure.2 Thus, clinical history and physical examination can assist greatly in guiding the evaluation of patients. Neurodegenerative disorders (such as Parkinson disease), combined disorders (such as multiple-system atrophy), and acquired or familial processes were considered. Our patient had neither a personal or family history nor physical examination supporting a neurodegenerative disorder. Disorders of the peripheral nerves were considered and can broadly be categorized as chronic sensorimotor neuropathies, sensory ganglionopathies, distal painful neuropathies, and acute or subacute motor polyradiculopathies. During evaluation, no historical, physical examination, or laboratory findings supported diabetes, amyloidosis, heavy metals, Sjögren syndrome, paraneoplastic neuropathy, sodium channel disorders, infectious etiologies, or porphyria (Table 1). Thus, in the absence of supportive evidence for primary neurodegenerative disorders or peripheral neuropathies, his syndrome appeared most compatible with an isolated autonomic failure syndrome. The principal differential for this syndrome is pure autonomic failure versus an immune-mediated autonomic disorder, including paraneoplastic autoimmune neuropathy and AAG. The diagnosis of pure autonomic failure is made after there is no clear unifying syndrome after more than five years of investigation. After exploration, no evidence of malignancy was discovered on body cross sectional imaging, PET scanning, bone marrow biopsy, colonoscopy, or laboratory testing. Thus, positive serologic testing in the absence of an underlying malignancy suggests a diagnosis of AAG.
AAG was first described in 1969 and is a rare, acquired disorder characterized by combined failure of the parasympathetic, sympathetic, and enteric nervous systems. This disorder typically presents in young-to-middle aged patients but has been described in all age groups. It is more commonly seen in patients with coexistent autoimmune diseases and/or a history of familial autoimmunity. The onset of clinical AAG may be subacute (less than three months) or insidious (more than three months). Patients present with signs or symptoms of pandysautonomia, such as severe orthostatic hypotension, syncope, constipation and gastrointestinal dysmotility, urinary retention, fixed and dilated pupils, and dry mouth and eyes (Table 2). Up to 40% of patients with AAG may also have significant cognitive impairment.3,4 Diagnosis relies on a combination of typical clinical features as discussed above and the exclusion of other diagnostic considerations. Diagnosis of AAG is aided by the presence of autoantibodies to ganglionic nicotinic acetylcholine receptors (gnAChR), particularly antiganglionic acetylcholine receptor α3 (anti-α3gAChR).1 Anti-gnAChR antibodies are only present in about half of patients with AAG. Antibody titers are highest in subacute AAG (40%-50%)3 compared with chronic AAG (30%-40%) or paraneoplastic AAG (10%-20%).5 Anti-gnAChR antibodies are not specific to AAG and have been identified in low levels in up to 20% of patients with thymomas, postural orthostatic tachycardia syndrome, chronic idiopathic anhidrosis, idiopathic gastrointestinal dysmotility, Lambert–Eaton syndrome, and myasthenia gravis without thymoma.1,5-7 These associations raise the question of shared pathology and perhaps a syndrome overlap. Individuals with seropositive AAG may also have other paraneoplastic antibodies, making it clinically indistinguishable from paraneoplastic autonomic neuropathy.5,8 Although the autoantibody lacks sensitivity and is imperfectly specific, its presence supports a diagnosis of AAG. Anti-gnAChR antibodies have been shown to be pathological in rabbit and mouse models.4 In patients with AAG, higher autoantibody titers correlate with increased disease severity.1,6,7 A decrease in autoantibody titers correlates with decreased disease severity.6 Case report series also described a distinct entity of seronegative AAG.2,3 Maintaining a high clinical suspicion for AAG even with negative antibodies is important.
Given the rarity of the disease, no standard therapeutic regimens are available. About one-third of individuals improve on their own, while other individuals require extensive immunomodulation and symptom management. Case series and observational trials currently make up the vast array of treatment data. Therapies include glucocorticoids, plasmapheresis, IVIG, and other immunosuppressive agents, such as rituximab.9-12 Patients with and without identified anti-gnAChRs antibodies may respond to therapy.12 The overall long-term prognosis of the disease is poorly characterized.9,10,13
Despite the rarity of the syndrome discussed, this case represents how diagnostic reasoning strategies, such as law of parsimony, shift how the case is framed. For example, a middle-aged man with several new, distinctly unrelated diagnoses versus a middle-aged man with signs and symptoms of autonomic failure alters the subsequent clinical reasoning and diagnostic approach. Many diseases, both common and rare, are associated with dysautonomia. Therefore, clinicians should have an approach to autonomic failure
TEACHING POINTS
- Recognize the following signs and symptoms suggesting a dysautonomic syndrome: orthostasis, syncope, anhidrosis, xerophthalmia, xerostomia, impaired pupillary constriction, blurry vision, photophobia, erectile dysfunction, urinary retention, gastroparesis, constipation, neurogenic bowel obstruction, and dysgeusia.
- Recognize the clinical features, diagnostic approach, and management of autoimmune autonomic ganglionopathy.
- When faced with a complex clinical presentation, early application of the “law of parsimony” may help identify a unifying syndrome.
Acknowledgments
The authors wish to thank our Blinded Expert, Anthony Montanaro, MD, for his expertise and guidance during this process.
Disclosures
There are no known conflicts of interest.
1. Gibbons C, Freeman R. Antibody titers predict clinical features of autoimmune autonomic ganglionopathy. Auton Neurosci. 2009;146(1-2):8-12. doi: 10.1016/j.autneu.2008.11.013. PubMed
2. Golden E, Bryarly M, Vernino S. Seronegative autoimmune autonomic neuropathy: a distinct clinical entity. Clin Auton Res. 2018;28(1):115-123. doi: 10.1007/s10286-017-0493-8. PubMed
3. Sandroni P, Vernino S, Klein CM, et al. Idiopathic autonomic neuropathy: comparison of cases seropositive and seronegative for ganglionic acetylcholine receptor antibody. Arch Neurol. 2004;61(1):44-48. doi: 10.1001/archneur.61.1.44. PubMed
4. Vernino S, Ermilov L, Sha L, Szurszewski J, Low P, Lennon V. Passive transfer of autoimmune autonomic neuropathy to mice. J Neurosci. 2004;24(32):7037-7042. doi: 10.1523/JNEUROSCI.1485-04.2004. PubMed
5. Vernino S, Hopkins S, Wang Z. Autonomic ganglia, acetylcholine receptor antibodies, and autoimmune ganglionopathy. Auton Neurosci. 2009;146(1-2):3-7. doi: 10.1016/j.autneu.2008.09.005. PubMed
6. Vernino S, Low P, Fealey R, Stewart J, Farrugia G, Lennon V. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343(12):847-855. doi: 10.1056/NEJM200009213431204. PubMed
7. Gibbons C, Vernino S, Freeman R. Autoimmune autonomic ganglionopathy – Symptom antibody correlations. Auton Neurosci. 2015;192:130. doi: 10.1016/j.autneu.2015.07.241 .
8. Benarroch E. The clinical approach to autonomic failure in neurological disorders. Nat Rev Neurol. 2014;10(7):396-407. doi: 10.1038/nrneurol.2014.88. PubMed
9. Baker SK, Morillo C, Vernino S. Autoimmune autonomic ganglionopathy with late-onset encephalopathy. Auton Neurosci. 2009;146(1-2):29-32. doi: 10.1016/j.autneu.2008.10.016. PubMed
10. Gibbons C, Centi J, Vernino S. Autoimmune autonomic ganglionoapthy with reversible cognitive impairment. Arch Neurol. 2012;69(4):461-466. doi: 10.1001/archneurol.2011.2372. PubMed
11. Boydston E, Muppidi S, Vernino S. Long-term outcomes in autoimmune autonomic ganglionopathy (P05.210). Neurology. 2012;78(1):P05.210. doi: 10.1212/WNL.78.1_MeetingAbstracts.P05.210.
12. Gehrking T, Sletten D, Fealey R, Low P, Singer W. 11-year follow-up of a case of autoimmune autonomic ganglionopathy (P03.024). Neurology. 2013;80(7):P03.024.
13. Imrich R, Vernino S, Eldadah BA, Holmes C, Goldstein DS. Autoimmune autonomic ganglionopathy: treatment by plasma exchanges and rituximab. Clin Auton Res. 2009;19(4):259-262. doi: 10.1007/s10286-009-0012-7. PubMed
14. Iodice V, Kimpinski K, Vernino S, Sandroni P, Fealey RD, Low PA. Efficacy of immunotherapy in seropositive and seronegative putative autoimmune autonomic ganglionopathy. Neurology. 2009;72(23):2002-8. doi: 10.1212/WNL.0b013e3181a92b52. PubMed
15. Hayashi M, Ishii Y. A Japanese case of autoimmune autonomic ganglionopathy (AAG) and a review of AAG cases in Japan. Auton Neurosci. 2009;146(1-2):26-8. doi: 10.1016/j.autneu.2008.12.013. PubMed
16. Baker, A. Simplicity. In: Baker A, Zalta E, eds. The Stanford Encyclopedia of Philosophy. Winter 2016 Edition. https://plato.stanford.edu/archives/win2016/entries/simplicity/. Accessed October 26, 2017.
A 44-year-old previously healthy semiprofessional male athlete presented with five days of nausea, vomiting, and abdominal pain. He had also experienced several months of decreased energy and new episodes of constipation three weeks prior to presentation.
At this point, we do not have sufficient information to completely determine the cause of his abdominal symptoms. Common causes of abdominal pain and vomiting in adults of his age group include peptic ulcer disease, pancreatic or hepatobiliary track disorders, small or large bowel processes, appendicitis, or even renal pathology. Further characterization may be possible by describing the location and quality of pain and factors that might relieve or exacerbate his pain. Despite the ambiguity, multiple clues might allow us to narrow the broad differential diagnosis of abdominal pain. In a previously healthy, vigorous, middle-aged man with subacute abdominal pain associated with constipation, the differential diagnosis should include disease states that may cause a bowel obstruction; these states include inflammatory bowel disease (IBD), gastrointestinal malignancy, or peptic ulcer disease. Mechanical obstruction due to volvulus or intussusception would be less likely in his age group. Given his history of several months of fatigue and several weeks of constipation, he should be evaluated for metabolic causes of abdominal pain and constipation, such as hypothyroidism or hypercalcemia. In addition to basic laboratory and imaging studies, obtaining additional history regarding prior abdominal surgeries, medication use, alcohol intake, and family and travel history will be the key in directing the evaluation.
Six months prior to admission, the patient began to feel more fatigue and exercise intolerance, reduced sweating, increased cold intolerance, and increased presyncopal episodes. He was diagnosed with hypothyroidism (TSH 6.69 μIU/mL; free T4 not done) and initiated on levothyroxine. One month prior to presentation, he developed constipation, loss of taste, reduced appetite, and weight loss of 30 pounds. He developed blurry vision and photophobia. He also complained of erectile dysfunction, urinary hesitancy and straining, which were diagnosed as benign prostatic hypertrophy.
Given the addition of numerous historical features in a previously healthy man, it is important to strive for a parsimonious diagnosis to unify his seemingly disparate features. His fatigue, constipation, and cold intolerance are consistent with his diagnosis of hypothyroidism but are nonspecific. Whether the degree of hypothyroidism caused his symptoms or signs is doubtful. The constellation of symptoms and signs are more likely to be representative of a nonthyroidal illness. His abdominal pain, unexplained weight loss, and presyncopal episodes should raise consideration of adrenal insufficiency. The combination of hypothyroidism and adrenal insufficiency suggest the possibility of an autoimmune polyendocrine syndrome or other pituitary pathology. In this case, history of headache, dysgeusia, and visual disturbances might support the diagnosis of pituitary adenoma. A cosyntropin stimulation test could establish the diagnosis of adrenal insufficiency. A low ACTH level would establish a diagnosis of pituitary or hypothalamic hypofunction. If pituitary hypofunction is documented, then a brain MRI would be needed to confirm the diagnosis of pituitary adenoma.
His newly reported erectile dysfunction suggests the possibility of a psychiatric, neurologic, hormonal, or vascular process and should be explored further. Sexual dysfunction is also associated with adrenal insufficiency and hypopituitarism. However, the presence of suspected prostatic hypertrophy in a male competitive athlete in his forties also raises the question of exogenous androgen use.
His past medical history was notable for a two-year history of alopecia totalis, seasonal allergies, asthma, and a repaired congenital aortic web with known aortic insufficiency. He was married with two children, worked an office job, and had no history of injection drug use, blood transfusions, or multiple sexual partners. His family history was notable for hypothyroidism and asthma in several family members in addition to Crohn disease, celiac disease, diabetes, cardiovascular disease, and cancers of the breast and lung.
His past medical, surgical, and family history supports a diagnosis of autoimmune disease. Although there is a personal and family history of atopic disorders, including allergic rhinitis and asthma, no association is found between atopy and autoimmunity. His family history of hypothyroidism, Crohn disease, and diabetes suggests a familial autoimmune genetic predisposition. His history of alopecia totalis in the setting of hypothyroidism and possible autoimmune adrenal insufficiency or autoimmune hypophysitis raises suspicion for the previously suggested diagnosis of polyglandular autoimmune syndrome, also known as autoimmune polyendocrine syndrome. Type I polyglandular autoimmune syndrome is associated with hypoparathyroidism and mucocutaneous candidiasis. In the absence of these symptoms, the patient more likely has type II polyglandular autoimmune syndrome. Type II syndrome is more prevalent and can occur in the setting of other nonendocrine autoimmune disorders, such as vitiligo, myasthenia gravis, or rheumatoid arthritis. Adrenal insufficiency can be the initial and most prominent manifestation of type II syndrome.
On physical exam, he was afebrile, with a heart rate of 68 beats per minute, respiratory rate of 16 breaths per minute, and normal oxygen saturation. His supine blood pressure and heart rate were 116/72 mm Hg and 66 beats per minute, respectively, and his standing blood pressure and heart rates were 80/48 mm Hg and 68 beats per minute respectively. He was thin, had diffuse scalp and body alopecia, and was ill-appearing with dry skin and dry mucous membranes. No evidence of Osler nodes, Janeway lesions, or splinter hemorrhages were found on cutaneous examination. No Roth spots or conjunctival hemorrhages were noted on ophthalmologic examination. He had both a 3/6 crescendo–decrescendo systolic murmur best heard at the right clavicle and radiated to the carotids and a 3/6 early diastolic decrescendo murmur best heard at the left sternal border. His abdomen was slightly protuberant, with reduced bowel sounds, hyperresonant to tympanitic on percussion, and a diffusely, moderately tender without peritoneal signs. Neurologic examination revealed 8 mm pupils with minimal response to light and accommodation. The remaining portions of his cranial nerve and complete neurologic examination were normal.
The presence of postural hypotension supports the previous suspicion of adrenal insufficiency, and the possibility of a pituitary or hypothalamic process remains. However, his dilated and minimally responsive pupils and potentially adynamic bowel are inconsistent with these diagnoses. Mydriasis and adynamic bowel in combination with orthostatic hypotension, dysgeusia, urinary retention, and erectile dysfunction are strongly suggestive of an autonomic process. Endocarditis is worth considering given his multisystem involvement, subacute decline, and known valve pathology. The absence of fever or stigmata of endocarditis make it difficult to explain his clinical syndrome. An echocardiogram would be reasonable for further assessment. At this point, it is prudent to explore his adrenal and pituitary function; if unrevealing, embark on an evaluation of his autonomic dysfunction.
Initial laboratory investigations were notable for mild normocytic anemia and hypoalbuminemia. His cosyntropin stimulation test was normal at 60 minutes. An abdominal CT scan demonstrated marked dilation in the small bowel loops (6 cm in caliber) with associated small bowel wall thickening and hyperemia. The echocardiogram was unrevealing and only confirmed the ongoing, progression of his known valve pathology without evidence of vegetation.
The above testing rules out primary adrenal insufficiency, but an appropriate response to the cosyntropin stimulation test does not rule out secondary, or pituitary, adrenal insufficiency. The echocardiogram and lack of other features make infective endocarditis unlikely. Thus, as mentioned, it is important now to commence a complete work-up of his probable dysautonomia to explain the majority of his features. Additionally, his hypothyroidism (if more than sick euthyroid syndrome), family history of autoimmune processes, and alopecia totalis all suggest the possibility of an immune-related syndrome. His CT scan revealed some thickened hyperemic bowel, which could suggest an IBD, such as Crohn disease; however, the absence of other signs, such as fever, diarrhea, or bloody stools, argues against this diagnosis. A syndrome that could unify his presentation is autoimmune autonomic ganglionopathy (AAG), a rare genetic autonomic system disorder that presents with pandysautonomia. The spectrum of autoimmunity was considered early in this case, but the differential diagnosis included more common conditions, such as adrenal insufficiency. Similarly, IBD remains a consideration. The serologic studies for IBD can be useful but they lack definitive diagnostic accuracy. Given that treatment for AAG differs from that for IBD, additional information will help guide the therapeutic approach. Anti-α3gnAChR antibodies, which are associated with AAG, should be checked.
His history of presyncope, anhidrosis, urinary retention, and ileus raised suspicion for pandysautonomia, as characterized by signs of sympathetic and parasympathetic dysfunction. The suspicion for pandysautonomia was confirmed via specialized autonomic testing, which included reduced heart rate variation on Valsalva and deep breathing maneuvers, orthostatic hypotension consistent autonomic insufficiency on Tilt table testing, and reduced sweat response to acetylcholine application (QSART test). The patient underwent further diagnostic serologic testing to differentiate causes of autonomic failure (Table 1). His personal and family history of autoimmunity led to the working diagnosis of AAG. Ultimate testing revealed high titers of autoantibodies, specifically anti-α3gnAChR (3.29 nmol/L, normal <0.02 nmol/L), directed against the ganglionic nicotinic acetylcholine receptor. This finding strongly supported the diagnosis of AAG.1,4-7
He was initially treated empirically with intravenous immunoglobulin (IVIG) with minimal improvement. He received additional immunomodulating therapies including methylprednisolone, plasmapheresis, and rituximab but did not tolerate a trial of mycophenolate. Six weeks after therapy initiation, his antibody titers decreased to 0.89 nmol/L with associated clinical improvement. Ultimately, he was discharged from the hospital on day 73 with a feeding tube and supplemental total parenteral nutrition. Four months postdischarge, he had returned to his prediagnosis weight, had eased back into his prior activities, and was off supplemental nutrition. Over a year later, he completed a 10-month prednisone taper and continued to receive monthly IVIG infusion
DISCUSSION
The clinical approach to dysautonomia is based on different etiologies: (1) those associated with neurodegenerative disorders; (2) those associated with peripheral neuropathies, and (3) isolated autonomic failure.2 Thus, clinical history and physical examination can assist greatly in guiding the evaluation of patients. Neurodegenerative disorders (such as Parkinson disease), combined disorders (such as multiple-system atrophy), and acquired or familial processes were considered. Our patient had neither a personal or family history nor physical examination supporting a neurodegenerative disorder. Disorders of the peripheral nerves were considered and can broadly be categorized as chronic sensorimotor neuropathies, sensory ganglionopathies, distal painful neuropathies, and acute or subacute motor polyradiculopathies. During evaluation, no historical, physical examination, or laboratory findings supported diabetes, amyloidosis, heavy metals, Sjögren syndrome, paraneoplastic neuropathy, sodium channel disorders, infectious etiologies, or porphyria (Table 1). Thus, in the absence of supportive evidence for primary neurodegenerative disorders or peripheral neuropathies, his syndrome appeared most compatible with an isolated autonomic failure syndrome. The principal differential for this syndrome is pure autonomic failure versus an immune-mediated autonomic disorder, including paraneoplastic autoimmune neuropathy and AAG. The diagnosis of pure autonomic failure is made after there is no clear unifying syndrome after more than five years of investigation. After exploration, no evidence of malignancy was discovered on body cross sectional imaging, PET scanning, bone marrow biopsy, colonoscopy, or laboratory testing. Thus, positive serologic testing in the absence of an underlying malignancy suggests a diagnosis of AAG.
AAG was first described in 1969 and is a rare, acquired disorder characterized by combined failure of the parasympathetic, sympathetic, and enteric nervous systems. This disorder typically presents in young-to-middle aged patients but has been described in all age groups. It is more commonly seen in patients with coexistent autoimmune diseases and/or a history of familial autoimmunity. The onset of clinical AAG may be subacute (less than three months) or insidious (more than three months). Patients present with signs or symptoms of pandysautonomia, such as severe orthostatic hypotension, syncope, constipation and gastrointestinal dysmotility, urinary retention, fixed and dilated pupils, and dry mouth and eyes (Table 2). Up to 40% of patients with AAG may also have significant cognitive impairment.3,4 Diagnosis relies on a combination of typical clinical features as discussed above and the exclusion of other diagnostic considerations. Diagnosis of AAG is aided by the presence of autoantibodies to ganglionic nicotinic acetylcholine receptors (gnAChR), particularly antiganglionic acetylcholine receptor α3 (anti-α3gAChR).1 Anti-gnAChR antibodies are only present in about half of patients with AAG. Antibody titers are highest in subacute AAG (40%-50%)3 compared with chronic AAG (30%-40%) or paraneoplastic AAG (10%-20%).5 Anti-gnAChR antibodies are not specific to AAG and have been identified in low levels in up to 20% of patients with thymomas, postural orthostatic tachycardia syndrome, chronic idiopathic anhidrosis, idiopathic gastrointestinal dysmotility, Lambert–Eaton syndrome, and myasthenia gravis without thymoma.1,5-7 These associations raise the question of shared pathology and perhaps a syndrome overlap. Individuals with seropositive AAG may also have other paraneoplastic antibodies, making it clinically indistinguishable from paraneoplastic autonomic neuropathy.5,8 Although the autoantibody lacks sensitivity and is imperfectly specific, its presence supports a diagnosis of AAG. Anti-gnAChR antibodies have been shown to be pathological in rabbit and mouse models.4 In patients with AAG, higher autoantibody titers correlate with increased disease severity.1,6,7 A decrease in autoantibody titers correlates with decreased disease severity.6 Case report series also described a distinct entity of seronegative AAG.2,3 Maintaining a high clinical suspicion for AAG even with negative antibodies is important.
Given the rarity of the disease, no standard therapeutic regimens are available. About one-third of individuals improve on their own, while other individuals require extensive immunomodulation and symptom management. Case series and observational trials currently make up the vast array of treatment data. Therapies include glucocorticoids, plasmapheresis, IVIG, and other immunosuppressive agents, such as rituximab.9-12 Patients with and without identified anti-gnAChRs antibodies may respond to therapy.12 The overall long-term prognosis of the disease is poorly characterized.9,10,13
Despite the rarity of the syndrome discussed, this case represents how diagnostic reasoning strategies, such as law of parsimony, shift how the case is framed. For example, a middle-aged man with several new, distinctly unrelated diagnoses versus a middle-aged man with signs and symptoms of autonomic failure alters the subsequent clinical reasoning and diagnostic approach. Many diseases, both common and rare, are associated with dysautonomia. Therefore, clinicians should have an approach to autonomic failure
TEACHING POINTS
- Recognize the following signs and symptoms suggesting a dysautonomic syndrome: orthostasis, syncope, anhidrosis, xerophthalmia, xerostomia, impaired pupillary constriction, blurry vision, photophobia, erectile dysfunction, urinary retention, gastroparesis, constipation, neurogenic bowel obstruction, and dysgeusia.
- Recognize the clinical features, diagnostic approach, and management of autoimmune autonomic ganglionopathy.
- When faced with a complex clinical presentation, early application of the “law of parsimony” may help identify a unifying syndrome.
Acknowledgments
The authors wish to thank our Blinded Expert, Anthony Montanaro, MD, for his expertise and guidance during this process.
Disclosures
There are no known conflicts of interest.
A 44-year-old previously healthy semiprofessional male athlete presented with five days of nausea, vomiting, and abdominal pain. He had also experienced several months of decreased energy and new episodes of constipation three weeks prior to presentation.
At this point, we do not have sufficient information to completely determine the cause of his abdominal symptoms. Common causes of abdominal pain and vomiting in adults of his age group include peptic ulcer disease, pancreatic or hepatobiliary track disorders, small or large bowel processes, appendicitis, or even renal pathology. Further characterization may be possible by describing the location and quality of pain and factors that might relieve or exacerbate his pain. Despite the ambiguity, multiple clues might allow us to narrow the broad differential diagnosis of abdominal pain. In a previously healthy, vigorous, middle-aged man with subacute abdominal pain associated with constipation, the differential diagnosis should include disease states that may cause a bowel obstruction; these states include inflammatory bowel disease (IBD), gastrointestinal malignancy, or peptic ulcer disease. Mechanical obstruction due to volvulus or intussusception would be less likely in his age group. Given his history of several months of fatigue and several weeks of constipation, he should be evaluated for metabolic causes of abdominal pain and constipation, such as hypothyroidism or hypercalcemia. In addition to basic laboratory and imaging studies, obtaining additional history regarding prior abdominal surgeries, medication use, alcohol intake, and family and travel history will be the key in directing the evaluation.
Six months prior to admission, the patient began to feel more fatigue and exercise intolerance, reduced sweating, increased cold intolerance, and increased presyncopal episodes. He was diagnosed with hypothyroidism (TSH 6.69 μIU/mL; free T4 not done) and initiated on levothyroxine. One month prior to presentation, he developed constipation, loss of taste, reduced appetite, and weight loss of 30 pounds. He developed blurry vision and photophobia. He also complained of erectile dysfunction, urinary hesitancy and straining, which were diagnosed as benign prostatic hypertrophy.
Given the addition of numerous historical features in a previously healthy man, it is important to strive for a parsimonious diagnosis to unify his seemingly disparate features. His fatigue, constipation, and cold intolerance are consistent with his diagnosis of hypothyroidism but are nonspecific. Whether the degree of hypothyroidism caused his symptoms or signs is doubtful. The constellation of symptoms and signs are more likely to be representative of a nonthyroidal illness. His abdominal pain, unexplained weight loss, and presyncopal episodes should raise consideration of adrenal insufficiency. The combination of hypothyroidism and adrenal insufficiency suggest the possibility of an autoimmune polyendocrine syndrome or other pituitary pathology. In this case, history of headache, dysgeusia, and visual disturbances might support the diagnosis of pituitary adenoma. A cosyntropin stimulation test could establish the diagnosis of adrenal insufficiency. A low ACTH level would establish a diagnosis of pituitary or hypothalamic hypofunction. If pituitary hypofunction is documented, then a brain MRI would be needed to confirm the diagnosis of pituitary adenoma.
His newly reported erectile dysfunction suggests the possibility of a psychiatric, neurologic, hormonal, or vascular process and should be explored further. Sexual dysfunction is also associated with adrenal insufficiency and hypopituitarism. However, the presence of suspected prostatic hypertrophy in a male competitive athlete in his forties also raises the question of exogenous androgen use.
His past medical history was notable for a two-year history of alopecia totalis, seasonal allergies, asthma, and a repaired congenital aortic web with known aortic insufficiency. He was married with two children, worked an office job, and had no history of injection drug use, blood transfusions, or multiple sexual partners. His family history was notable for hypothyroidism and asthma in several family members in addition to Crohn disease, celiac disease, diabetes, cardiovascular disease, and cancers of the breast and lung.
His past medical, surgical, and family history supports a diagnosis of autoimmune disease. Although there is a personal and family history of atopic disorders, including allergic rhinitis and asthma, no association is found between atopy and autoimmunity. His family history of hypothyroidism, Crohn disease, and diabetes suggests a familial autoimmune genetic predisposition. His history of alopecia totalis in the setting of hypothyroidism and possible autoimmune adrenal insufficiency or autoimmune hypophysitis raises suspicion for the previously suggested diagnosis of polyglandular autoimmune syndrome, also known as autoimmune polyendocrine syndrome. Type I polyglandular autoimmune syndrome is associated with hypoparathyroidism and mucocutaneous candidiasis. In the absence of these symptoms, the patient more likely has type II polyglandular autoimmune syndrome. Type II syndrome is more prevalent and can occur in the setting of other nonendocrine autoimmune disorders, such as vitiligo, myasthenia gravis, or rheumatoid arthritis. Adrenal insufficiency can be the initial and most prominent manifestation of type II syndrome.
On physical exam, he was afebrile, with a heart rate of 68 beats per minute, respiratory rate of 16 breaths per minute, and normal oxygen saturation. His supine blood pressure and heart rate were 116/72 mm Hg and 66 beats per minute, respectively, and his standing blood pressure and heart rates were 80/48 mm Hg and 68 beats per minute respectively. He was thin, had diffuse scalp and body alopecia, and was ill-appearing with dry skin and dry mucous membranes. No evidence of Osler nodes, Janeway lesions, or splinter hemorrhages were found on cutaneous examination. No Roth spots or conjunctival hemorrhages were noted on ophthalmologic examination. He had both a 3/6 crescendo–decrescendo systolic murmur best heard at the right clavicle and radiated to the carotids and a 3/6 early diastolic decrescendo murmur best heard at the left sternal border. His abdomen was slightly protuberant, with reduced bowel sounds, hyperresonant to tympanitic on percussion, and a diffusely, moderately tender without peritoneal signs. Neurologic examination revealed 8 mm pupils with minimal response to light and accommodation. The remaining portions of his cranial nerve and complete neurologic examination were normal.
The presence of postural hypotension supports the previous suspicion of adrenal insufficiency, and the possibility of a pituitary or hypothalamic process remains. However, his dilated and minimally responsive pupils and potentially adynamic bowel are inconsistent with these diagnoses. Mydriasis and adynamic bowel in combination with orthostatic hypotension, dysgeusia, urinary retention, and erectile dysfunction are strongly suggestive of an autonomic process. Endocarditis is worth considering given his multisystem involvement, subacute decline, and known valve pathology. The absence of fever or stigmata of endocarditis make it difficult to explain his clinical syndrome. An echocardiogram would be reasonable for further assessment. At this point, it is prudent to explore his adrenal and pituitary function; if unrevealing, embark on an evaluation of his autonomic dysfunction.
Initial laboratory investigations were notable for mild normocytic anemia and hypoalbuminemia. His cosyntropin stimulation test was normal at 60 minutes. An abdominal CT scan demonstrated marked dilation in the small bowel loops (6 cm in caliber) with associated small bowel wall thickening and hyperemia. The echocardiogram was unrevealing and only confirmed the ongoing, progression of his known valve pathology without evidence of vegetation.
The above testing rules out primary adrenal insufficiency, but an appropriate response to the cosyntropin stimulation test does not rule out secondary, or pituitary, adrenal insufficiency. The echocardiogram and lack of other features make infective endocarditis unlikely. Thus, as mentioned, it is important now to commence a complete work-up of his probable dysautonomia to explain the majority of his features. Additionally, his hypothyroidism (if more than sick euthyroid syndrome), family history of autoimmune processes, and alopecia totalis all suggest the possibility of an immune-related syndrome. His CT scan revealed some thickened hyperemic bowel, which could suggest an IBD, such as Crohn disease; however, the absence of other signs, such as fever, diarrhea, or bloody stools, argues against this diagnosis. A syndrome that could unify his presentation is autoimmune autonomic ganglionopathy (AAG), a rare genetic autonomic system disorder that presents with pandysautonomia. The spectrum of autoimmunity was considered early in this case, but the differential diagnosis included more common conditions, such as adrenal insufficiency. Similarly, IBD remains a consideration. The serologic studies for IBD can be useful but they lack definitive diagnostic accuracy. Given that treatment for AAG differs from that for IBD, additional information will help guide the therapeutic approach. Anti-α3gnAChR antibodies, which are associated with AAG, should be checked.
His history of presyncope, anhidrosis, urinary retention, and ileus raised suspicion for pandysautonomia, as characterized by signs of sympathetic and parasympathetic dysfunction. The suspicion for pandysautonomia was confirmed via specialized autonomic testing, which included reduced heart rate variation on Valsalva and deep breathing maneuvers, orthostatic hypotension consistent autonomic insufficiency on Tilt table testing, and reduced sweat response to acetylcholine application (QSART test). The patient underwent further diagnostic serologic testing to differentiate causes of autonomic failure (Table 1). His personal and family history of autoimmunity led to the working diagnosis of AAG. Ultimate testing revealed high titers of autoantibodies, specifically anti-α3gnAChR (3.29 nmol/L, normal <0.02 nmol/L), directed against the ganglionic nicotinic acetylcholine receptor. This finding strongly supported the diagnosis of AAG.1,4-7
He was initially treated empirically with intravenous immunoglobulin (IVIG) with minimal improvement. He received additional immunomodulating therapies including methylprednisolone, plasmapheresis, and rituximab but did not tolerate a trial of mycophenolate. Six weeks after therapy initiation, his antibody titers decreased to 0.89 nmol/L with associated clinical improvement. Ultimately, he was discharged from the hospital on day 73 with a feeding tube and supplemental total parenteral nutrition. Four months postdischarge, he had returned to his prediagnosis weight, had eased back into his prior activities, and was off supplemental nutrition. Over a year later, he completed a 10-month prednisone taper and continued to receive monthly IVIG infusion
DISCUSSION
The clinical approach to dysautonomia is based on different etiologies: (1) those associated with neurodegenerative disorders; (2) those associated with peripheral neuropathies, and (3) isolated autonomic failure.2 Thus, clinical history and physical examination can assist greatly in guiding the evaluation of patients. Neurodegenerative disorders (such as Parkinson disease), combined disorders (such as multiple-system atrophy), and acquired or familial processes were considered. Our patient had neither a personal or family history nor physical examination supporting a neurodegenerative disorder. Disorders of the peripheral nerves were considered and can broadly be categorized as chronic sensorimotor neuropathies, sensory ganglionopathies, distal painful neuropathies, and acute or subacute motor polyradiculopathies. During evaluation, no historical, physical examination, or laboratory findings supported diabetes, amyloidosis, heavy metals, Sjögren syndrome, paraneoplastic neuropathy, sodium channel disorders, infectious etiologies, or porphyria (Table 1). Thus, in the absence of supportive evidence for primary neurodegenerative disorders or peripheral neuropathies, his syndrome appeared most compatible with an isolated autonomic failure syndrome. The principal differential for this syndrome is pure autonomic failure versus an immune-mediated autonomic disorder, including paraneoplastic autoimmune neuropathy and AAG. The diagnosis of pure autonomic failure is made after there is no clear unifying syndrome after more than five years of investigation. After exploration, no evidence of malignancy was discovered on body cross sectional imaging, PET scanning, bone marrow biopsy, colonoscopy, or laboratory testing. Thus, positive serologic testing in the absence of an underlying malignancy suggests a diagnosis of AAG.
AAG was first described in 1969 and is a rare, acquired disorder characterized by combined failure of the parasympathetic, sympathetic, and enteric nervous systems. This disorder typically presents in young-to-middle aged patients but has been described in all age groups. It is more commonly seen in patients with coexistent autoimmune diseases and/or a history of familial autoimmunity. The onset of clinical AAG may be subacute (less than three months) or insidious (more than three months). Patients present with signs or symptoms of pandysautonomia, such as severe orthostatic hypotension, syncope, constipation and gastrointestinal dysmotility, urinary retention, fixed and dilated pupils, and dry mouth and eyes (Table 2). Up to 40% of patients with AAG may also have significant cognitive impairment.3,4 Diagnosis relies on a combination of typical clinical features as discussed above and the exclusion of other diagnostic considerations. Diagnosis of AAG is aided by the presence of autoantibodies to ganglionic nicotinic acetylcholine receptors (gnAChR), particularly antiganglionic acetylcholine receptor α3 (anti-α3gAChR).1 Anti-gnAChR antibodies are only present in about half of patients with AAG. Antibody titers are highest in subacute AAG (40%-50%)3 compared with chronic AAG (30%-40%) or paraneoplastic AAG (10%-20%).5 Anti-gnAChR antibodies are not specific to AAG and have been identified in low levels in up to 20% of patients with thymomas, postural orthostatic tachycardia syndrome, chronic idiopathic anhidrosis, idiopathic gastrointestinal dysmotility, Lambert–Eaton syndrome, and myasthenia gravis without thymoma.1,5-7 These associations raise the question of shared pathology and perhaps a syndrome overlap. Individuals with seropositive AAG may also have other paraneoplastic antibodies, making it clinically indistinguishable from paraneoplastic autonomic neuropathy.5,8 Although the autoantibody lacks sensitivity and is imperfectly specific, its presence supports a diagnosis of AAG. Anti-gnAChR antibodies have been shown to be pathological in rabbit and mouse models.4 In patients with AAG, higher autoantibody titers correlate with increased disease severity.1,6,7 A decrease in autoantibody titers correlates with decreased disease severity.6 Case report series also described a distinct entity of seronegative AAG.2,3 Maintaining a high clinical suspicion for AAG even with negative antibodies is important.
Given the rarity of the disease, no standard therapeutic regimens are available. About one-third of individuals improve on their own, while other individuals require extensive immunomodulation and symptom management. Case series and observational trials currently make up the vast array of treatment data. Therapies include glucocorticoids, plasmapheresis, IVIG, and other immunosuppressive agents, such as rituximab.9-12 Patients with and without identified anti-gnAChRs antibodies may respond to therapy.12 The overall long-term prognosis of the disease is poorly characterized.9,10,13
Despite the rarity of the syndrome discussed, this case represents how diagnostic reasoning strategies, such as law of parsimony, shift how the case is framed. For example, a middle-aged man with several new, distinctly unrelated diagnoses versus a middle-aged man with signs and symptoms of autonomic failure alters the subsequent clinical reasoning and diagnostic approach. Many diseases, both common and rare, are associated with dysautonomia. Therefore, clinicians should have an approach to autonomic failure
TEACHING POINTS
- Recognize the following signs and symptoms suggesting a dysautonomic syndrome: orthostasis, syncope, anhidrosis, xerophthalmia, xerostomia, impaired pupillary constriction, blurry vision, photophobia, erectile dysfunction, urinary retention, gastroparesis, constipation, neurogenic bowel obstruction, and dysgeusia.
- Recognize the clinical features, diagnostic approach, and management of autoimmune autonomic ganglionopathy.
- When faced with a complex clinical presentation, early application of the “law of parsimony” may help identify a unifying syndrome.
Acknowledgments
The authors wish to thank our Blinded Expert, Anthony Montanaro, MD, for his expertise and guidance during this process.
Disclosures
There are no known conflicts of interest.
1. Gibbons C, Freeman R. Antibody titers predict clinical features of autoimmune autonomic ganglionopathy. Auton Neurosci. 2009;146(1-2):8-12. doi: 10.1016/j.autneu.2008.11.013. PubMed
2. Golden E, Bryarly M, Vernino S. Seronegative autoimmune autonomic neuropathy: a distinct clinical entity. Clin Auton Res. 2018;28(1):115-123. doi: 10.1007/s10286-017-0493-8. PubMed
3. Sandroni P, Vernino S, Klein CM, et al. Idiopathic autonomic neuropathy: comparison of cases seropositive and seronegative for ganglionic acetylcholine receptor antibody. Arch Neurol. 2004;61(1):44-48. doi: 10.1001/archneur.61.1.44. PubMed
4. Vernino S, Ermilov L, Sha L, Szurszewski J, Low P, Lennon V. Passive transfer of autoimmune autonomic neuropathy to mice. J Neurosci. 2004;24(32):7037-7042. doi: 10.1523/JNEUROSCI.1485-04.2004. PubMed
5. Vernino S, Hopkins S, Wang Z. Autonomic ganglia, acetylcholine receptor antibodies, and autoimmune ganglionopathy. Auton Neurosci. 2009;146(1-2):3-7. doi: 10.1016/j.autneu.2008.09.005. PubMed
6. Vernino S, Low P, Fealey R, Stewart J, Farrugia G, Lennon V. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343(12):847-855. doi: 10.1056/NEJM200009213431204. PubMed
7. Gibbons C, Vernino S, Freeman R. Autoimmune autonomic ganglionopathy – Symptom antibody correlations. Auton Neurosci. 2015;192:130. doi: 10.1016/j.autneu.2015.07.241 .
8. Benarroch E. The clinical approach to autonomic failure in neurological disorders. Nat Rev Neurol. 2014;10(7):396-407. doi: 10.1038/nrneurol.2014.88. PubMed
9. Baker SK, Morillo C, Vernino S. Autoimmune autonomic ganglionopathy with late-onset encephalopathy. Auton Neurosci. 2009;146(1-2):29-32. doi: 10.1016/j.autneu.2008.10.016. PubMed
10. Gibbons C, Centi J, Vernino S. Autoimmune autonomic ganglionoapthy with reversible cognitive impairment. Arch Neurol. 2012;69(4):461-466. doi: 10.1001/archneurol.2011.2372. PubMed
11. Boydston E, Muppidi S, Vernino S. Long-term outcomes in autoimmune autonomic ganglionopathy (P05.210). Neurology. 2012;78(1):P05.210. doi: 10.1212/WNL.78.1_MeetingAbstracts.P05.210.
12. Gehrking T, Sletten D, Fealey R, Low P, Singer W. 11-year follow-up of a case of autoimmune autonomic ganglionopathy (P03.024). Neurology. 2013;80(7):P03.024.
13. Imrich R, Vernino S, Eldadah BA, Holmes C, Goldstein DS. Autoimmune autonomic ganglionopathy: treatment by plasma exchanges and rituximab. Clin Auton Res. 2009;19(4):259-262. doi: 10.1007/s10286-009-0012-7. PubMed
14. Iodice V, Kimpinski K, Vernino S, Sandroni P, Fealey RD, Low PA. Efficacy of immunotherapy in seropositive and seronegative putative autoimmune autonomic ganglionopathy. Neurology. 2009;72(23):2002-8. doi: 10.1212/WNL.0b013e3181a92b52. PubMed
15. Hayashi M, Ishii Y. A Japanese case of autoimmune autonomic ganglionopathy (AAG) and a review of AAG cases in Japan. Auton Neurosci. 2009;146(1-2):26-8. doi: 10.1016/j.autneu.2008.12.013. PubMed
16. Baker, A. Simplicity. In: Baker A, Zalta E, eds. The Stanford Encyclopedia of Philosophy. Winter 2016 Edition. https://plato.stanford.edu/archives/win2016/entries/simplicity/. Accessed October 26, 2017.
1. Gibbons C, Freeman R. Antibody titers predict clinical features of autoimmune autonomic ganglionopathy. Auton Neurosci. 2009;146(1-2):8-12. doi: 10.1016/j.autneu.2008.11.013. PubMed
2. Golden E, Bryarly M, Vernino S. Seronegative autoimmune autonomic neuropathy: a distinct clinical entity. Clin Auton Res. 2018;28(1):115-123. doi: 10.1007/s10286-017-0493-8. PubMed
3. Sandroni P, Vernino S, Klein CM, et al. Idiopathic autonomic neuropathy: comparison of cases seropositive and seronegative for ganglionic acetylcholine receptor antibody. Arch Neurol. 2004;61(1):44-48. doi: 10.1001/archneur.61.1.44. PubMed
4. Vernino S, Ermilov L, Sha L, Szurszewski J, Low P, Lennon V. Passive transfer of autoimmune autonomic neuropathy to mice. J Neurosci. 2004;24(32):7037-7042. doi: 10.1523/JNEUROSCI.1485-04.2004. PubMed
5. Vernino S, Hopkins S, Wang Z. Autonomic ganglia, acetylcholine receptor antibodies, and autoimmune ganglionopathy. Auton Neurosci. 2009;146(1-2):3-7. doi: 10.1016/j.autneu.2008.09.005. PubMed
6. Vernino S, Low P, Fealey R, Stewart J, Farrugia G, Lennon V. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343(12):847-855. doi: 10.1056/NEJM200009213431204. PubMed
7. Gibbons C, Vernino S, Freeman R. Autoimmune autonomic ganglionopathy – Symptom antibody correlations. Auton Neurosci. 2015;192:130. doi: 10.1016/j.autneu.2015.07.241 .
8. Benarroch E. The clinical approach to autonomic failure in neurological disorders. Nat Rev Neurol. 2014;10(7):396-407. doi: 10.1038/nrneurol.2014.88. PubMed
9. Baker SK, Morillo C, Vernino S. Autoimmune autonomic ganglionopathy with late-onset encephalopathy. Auton Neurosci. 2009;146(1-2):29-32. doi: 10.1016/j.autneu.2008.10.016. PubMed
10. Gibbons C, Centi J, Vernino S. Autoimmune autonomic ganglionoapthy with reversible cognitive impairment. Arch Neurol. 2012;69(4):461-466. doi: 10.1001/archneurol.2011.2372. PubMed
11. Boydston E, Muppidi S, Vernino S. Long-term outcomes in autoimmune autonomic ganglionopathy (P05.210). Neurology. 2012;78(1):P05.210. doi: 10.1212/WNL.78.1_MeetingAbstracts.P05.210.
12. Gehrking T, Sletten D, Fealey R, Low P, Singer W. 11-year follow-up of a case of autoimmune autonomic ganglionopathy (P03.024). Neurology. 2013;80(7):P03.024.
13. Imrich R, Vernino S, Eldadah BA, Holmes C, Goldstein DS. Autoimmune autonomic ganglionopathy: treatment by plasma exchanges and rituximab. Clin Auton Res. 2009;19(4):259-262. doi: 10.1007/s10286-009-0012-7. PubMed
14. Iodice V, Kimpinski K, Vernino S, Sandroni P, Fealey RD, Low PA. Efficacy of immunotherapy in seropositive and seronegative putative autoimmune autonomic ganglionopathy. Neurology. 2009;72(23):2002-8. doi: 10.1212/WNL.0b013e3181a92b52. PubMed
15. Hayashi M, Ishii Y. A Japanese case of autoimmune autonomic ganglionopathy (AAG) and a review of AAG cases in Japan. Auton Neurosci. 2009;146(1-2):26-8. doi: 10.1016/j.autneu.2008.12.013. PubMed
16. Baker, A. Simplicity. In: Baker A, Zalta E, eds. The Stanford Encyclopedia of Philosophy. Winter 2016 Edition. https://plato.stanford.edu/archives/win2016/entries/simplicity/. Accessed October 26, 2017.
© 2018 Society of Hospital Medicine
A Dark Horse Diagnosis
A 73-year-old man presented to the emergency department in late winter with fevers, myalgias, fatigue, low back pain, and poor oral intake. Four days earlier, he had fallen and hit his head. His partner also noticed a few episodes of confusion in the days leading up to presentation.
The patient’s symptoms are nonspecific. Fevers prompt the consideration of systemic infection, though fevers can also be seen in a broad range of noninfectious processes, including malignancy, vasculitis, autoimmune conditions, endocrinopathies, and drug reaction. The clinical picture warrants prompt and comprehensive evaluation, beginning with further detailed history (current illnesses, exposures, travel, vaccinations, medications, cancer screenings, weight change) and a careful physical examination, which will help guide laboratory testing and imaging.
His past medical history was notable for coronary artery disease for which he underwent coronary artery bypass grafting five years prior, hypertension, hyperlipidemia, diet-controlled type 2 diabetes mellitus, gastroesophageal reflux disease, osteoarthritis leading to chronic knee and hand pain, and a history of mildly low testosterone levels. His medications included hydrocodone and acetaminophen, metoprolol tartrate, omeprazole, topical testosterone gel (prescribed for daily use, used intermittently), and aspirin. He was retired and lived in rural Michigan with his female partner. He previously worked as a truck driver and used to train racehorses. He had quit smoking five years earlier. He denied alcohol or injection drug use.
The patient has significant underlying medical conditions. Considering infectious causes of his symptoms, it is notable that he has no reported immunodeficiency. It would be relevant to know if he has been tested for HIV. His rural residence and work with horses raise the possibility of zoonotic infections, including plague (Yersinia pestis), brucellosis (Brucella species), Q fever (Coxiella burnetti), Rhodococcus equi, or group C or G Streptococci. Information about tuberculosis risk factors, other geographic exposures, recent dental work, and ill contacts might be helpful to elucidate the causes of this nonspecific febrile illness with a possible CNS component. With regard to malignancy, it would be helpful to ask about recent weight loss, lymphadenopathy, and prior cancer screenings. Considering other etiologies, he does not report a history of autoimmune or endocrine conditions. However, it is important to consider a vasculitis, such as giant cell arteritis or polyarteritis nodosa, autoimmune conditions, and endocrinopathies such as thyrotoxicosis. The differential diagnosis for his clinical syndrome remains broad.
Vital signs were temperature 37.3°C, heart rate 88 beats per minute, respiratory rate 18 breaths per minute, blood pressure 105/64 mmHg, and oxygen saturation 93% on room air. Oral examination revealed poor dentition. The heart had a normal rate and regular rhythm with no murmurs, rubs, or gallops, and lungs were clear to auscultation bilaterally. The abdomen was unremarkable. Examination of the back was notable for mild tenderness to palpation over the sacrum. He was oriented to person, place, and time, with intact cranial nerves and a nonfocal neurologic examination. The remainder of his examination was normal. The white blood cell (WBC) count was 11.1 × 103/μL, with 84% neutrophils and 9% bands, hemoglobin 13.6 g/dL, platelet count 54 × 103/μL, sodium 122 mmol/L, potassium 3.3 mmol/L, chloride 89 mmol/L, bicarbonate 21 mmol/L, creatinine 1.64 mg/dL, albumin 2.7 g/dL, alkaline phosphatase 136 U/L, AST 60 U/L, ALT 37 U/L, and total bilirubin 2.1 mg/dL.
He had presented to the emergency department five days earlier with fever, flank pain, nausea, vomiting, and weakness. At that time, he had a temperature of 38.2°C, but vital signs otherwise had been normal. Laboratory studies had revealed WBC count 14.0 × 103/μL, hemoglobin 13.7 g/dL, platelet count 175 × 103/μL, sodium 129 mmol/L, chloride 97 mmol/dL, bicarbonate 23 mmol/L, creatinine 1.1 mg/dL, and total bilirubin 1.6 mg/dL. Urinalysis had been negative. He had received one liter of intravenous normal saline and ketorolac for pain and had been discharged with the diagnosis of a viral illness.
A picture of a progressive, subacute illness with multisystem involvement appears to be emerging, and there are several abnormalities consistent with infection, including fever, leukocytosis with bandemia, thrombocytopenia, renal dysfunction, and elevated bilirubin. His borderline hypotension may be due to uninterrupted use of his antihypertensive medication in the setting of poor oral intake or may indicate incipient sepsis. Focal sacral tenderness raises the possibility of vertebral osteomyelitis or epidural abscess, either from a contiguous focus of infection from the surrounding structures, or as a site of seeding from bacteremia. His prior confusion episodes might have been secondary to a systemic process; however, CNS imaging should be done, given the history of confusion and recent fall. Further diagnostic studies are warranted, including: blood cultures; peripheral blood smear; imaging of the spine, chest, abdomen, and pelvis; electrocardiogram; and possibly echocardiogram. Although noninfectious etiologies should not be discounted, the constellation of findings is more compatible with infection.
Two sets of blood cultures and a viral respiratory swab were obtained. Computed tomography (CT) of the head without contrast was negative for acute bleeding or other intracranial pathology. Lumbosacral radiography revealed degenerative changes with intact alignment of the sacrum. The patient was admitted with plans to pursue lumbar puncture if altered mental status recurred. The viral swab was negative. Within 24 hours, one set of blood cultures (both bottles) grew lactose-negative, oxidase-negative, gram-negative rods.
Gram-negative rods (GNRs) rarely are contaminants in blood cultures and should be considered significant until proven otherwise. Prompt empiric therapy and investigation to identify the primary source of bacteremia must be initiated. Although the most common GNRs isolated from blood cultures are enteric coliform organisms such as E. coli, Klebsiella, and Enterobacter, these typically are lactose-positive. Additional possibilities should be considered, including Salmonella species or other organisms comprising the “HACEK” group. This latter group is commonly associated with endocarditis, but the majority are oxidase-positive and have more fastidious growth requirements. Although there are other gram-negative organisms to consider, they have other distinguishing characteristics that have not been indicated in the microbiology results. Broad-spectrum antibiotic therapy is appropriate while awaiting the final identification of the GNR. A thorough search for a primary source and secondary sites of hematogenous seeding should be conducted. His only localizing symptom was tenderness over the sacrum, and this should be further assessed by sensitive imaging such as magnetic resonance imaging (MRI). The identity of the GNR would guide further diagnostic evaluation. For example, a respiratory organism such as Haemophilus influenzae would prompt a CT scan of the chest. Isolation of an enteric or a coliform GNR such as E. coli would prompt abdominal and pelvic imaging to assess for occult abscess. An “HACEK” group organism would prompt echocardiography to evaluate for endocarditis.
He was started on piperacillin–tazobactam. GNR bacteremia without a clear source prompted a CT of the chest, abdomen, and pelvis with and without contrast. The images were unremarkable, with the exception of a signal abnormality in the left psoas muscle concerning for abscess (Figure 1). MRI of the same region revealed L2-4 osteomyelitis and discitis with bilateral psoas abscesses but without epidural abscess (Figure 2).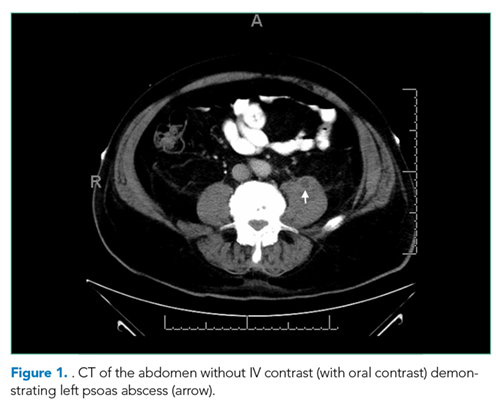
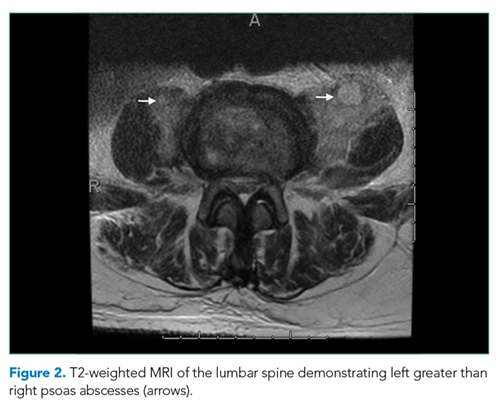
Because of increasing reports of antibiotic resistance in GNRs, even in community-acquired infections, it is appropriate to initially treat with a broad-spectrum antibiotic such as a fourth-generation cephalosporin or carbapenem while awaiting identification and susceptibility results to guide definitive therapy. In addition to antimicrobial therapy, treatment of psoas abscess usually requires drainage. Vertebral osteomyelitis from a hematogenous source can often be treated with antibiotics alone, as long as there are no associated complications such as epidural abscess and spine instability. Imaging should be reviewed for pathology of the surrounding structures, and surgical consultation should be obtained.
Neurosurgery, Interventional Radiology, and Infectious Disease services were consulted. Antibiotic coverage was expanded to vancomycin, cefepime, and metronidazole due to the possibility of polymicrobial infection. No surgical intervention was recommended since the abscesses were too small to drain.
The next day, the GNR was identified as Serratia marcescens.
S. marcescens is a widely distributed organism in the environment, but not a common component of endogenous human flora. Serratia is generally considered as an opportunistic nosocomial pathogen. Community-acquired infection with this organism is unusual and implies exogenous acquisition. A careful re-evaluation of exposures, including injection drug use or other parenteral exposures is important to identify the likely source of infection, as these have been previously linked to outbreaks of environmental organisms. Based on the presumed pathogenesis of infection and the initial microbiology suggesting monomicrobial Serratia infection, antibiotics should be narrowed based on the susceptibility results. There is concern that antibiotics might not adequately penetrate the abscesses and result in a lack of clinical improvement and/or lead to the emergence of antibiotic resistance during therapy. This is an important concern with Serratia, which typically harbors an AmpC beta-lactamase that can mediate resistance to broad-spectrum cephalosporins. If medical therapy alone without drainage is planned, short-interval re-imaging is warranted.
Blood cultures from days two and three of hospitalization also grew S. marcescens. No other organisms grew. Based on culture sensitivity data, antibiotics were narrowed to ceftriaxone.
This surprising culture result prompted the medical team to obtain screening laboratory tests for immunocompromising conditions and to revisit the patient’s history. His type 2 diabetes mellitus was well controlled with a hemoglobin A1c of 6.5%. HIV testing was negative. Further questioning of the patient revealed that he had fallen from a truck onto rocks four months prior, injuring his back and hip, but without puncture of the skin or loss of consciousness; he denied recent falls or other injuries but reported significant chronic knee pain. He had not been hospitalized recently. He had never taken corticosteroids or immunomodulatory medications. He continued to deny injection drug use. He did, however, clarify that his work with racehorses, which was originally understood to be a prior hobby, was ongoing, including recent work of cleaning the stables.
The following morning, he experienced confusion, rigors, and hypoxia, which prompted transfer to the intensive care unit (ICU).
Acute worsening during treatment is worrisome, and could be a potential complication of his infection or treatment – or even a separate process altogether. Knee pain in the setting of bacteremia raises the possibility of septic or crystal-induced arthritis and warrants imaging. Confusion and hypoxia might represent secondary sites of seeding from bacteremia (CNS infection and pneumonia, respectively) or manifestations of endocarditis, the latter being unusual for Serratia. An echocardiogram should be obtained. Other neurologic causes, including seizure, should also be considered. Further evaluation by chest imaging and repeat neurologic examination and imaging should be performed. Emergence of resistance during therapy is a theoretical concern with Serratia as an AmpC beta-lactamase-containing organism. While awaiting additional microbiology data, an empiric change to an AmpC beta-lactamase stable antibiotic such as a carbapenem should be made, especially since he has clinically deteriorated on therapy with a β-lactamase susceptible antibiotic, raising concerns of the emergence of resistance on initial therapy.
Antibiotics were changed to meropenem, vancomycin, and metronidazole given the clinical worsening and concerns that this represented infection unresponsive to prior antibiotics. The acute episode resolved spontaneously after one hour. His neurologic examination remained nonfocal. Chest radiography, urinalysis, urine culture, and right upper quadrant ultrasound were unremarkable. Transesophageal echocardiogram revealed no heart valve vegetations. MRI and bone scan of the lower extremities did not show any evidence of septic arthritis or other infection. He remained stable and was transferred out of the ICU the following day. Antibiotic coverage was switched to cefepime. On discussion with his significant other, this event was found to be similar to the intermittent confusion that occurred in the days prior to admission.
The acute onset and other features of these intermittent periods of deterioration are compatible with infection; intermittent seeding of the blood with microbes or their products (eg, lipopolysaccharides) from an abscess or vascular infection could explain these episodes. Some of the previous hypotheses to explain the episodes, such as a secondary infectious process, have not been supported by diagnostic testing or the clinical course. He needs close clinical monitoring and interval assessment of the known sites of infection.
Ten days after osteomyelitis and discitis were diagnosed, the patient developed worsening low back pain, prompting repeat spine MRI. This was significant for bilateral psoas abscess enlargement and extension of osteomyelitis and discitis (Figure 3). He was re-evaluated by Neurosurgery and Interventional Radiology and underwent psoas abscess drainage; abscess cultures grew S. marcescens.
He slowly improved over several weeks and was discharged to a subacute rehabilitation facility. He completed a 3.5-week course of intravenous antibiotics before leaving against medical advice. He completed eight weeks of oral trimethoprim-sulfamethoxazole and remains without long-term sequelae from the infection.
DISCUSSION
S. marcescens is a gram-negative rod in the Enterobacteriaceae family known for its red pigment. Primarily, S. marcescens causes nosocomial infections, most commonly of the respiratory and urinary tracts. However, a wide range of manifestations has been documented, including meningitis, ocular infections (conjunctivitis, keratitis, endophthalmitis), endocarditis, skin infections (cellulitis, necrotizing fasciitis), and osteomyelitis.1, 2 S. marcescens is often reported as the cause of outbreaks in ICUs;3-6 infection is thought to occur via contamination of water pipes, hospital equipment, and disinfectants.3, 7 Its natural environment includes soil, water, and GI tracts of animals,4 and there are published reports of S. marcescens infection in horses.8, 9 This patient was most likely exposed to S. marcescens through his work with horses and their environment.
S. marcescens has wide-ranging target organs, and successful treatment can be difficult. S. marcescens can infect the renal, respiratory, gastrointestinal, ocular, cardiovascular, and musculoskeletal systems. S. marcescens, like other “SPACE” organisms (Serratia, Pseudomonas, Acinetobacter, Citrobacter, Enterobacter), expresses inducible AmpC beta-lactamase.10 At baseline, AmpC beta-lactamase expression is repressed.11 Mutants with stably de-repressed (constitutively expressed) AmpC can be selected during therapy and lead to clinical failure, as has been best described during therapy for Enterobacter infections.12 Infectious Disease consultation may be helpful when caring for patients with S. marcescens bacteremia given these complexities.
This was an unusual case of S. marcescens infection. It most commonly infects immunocompromised hosts. Reported risk factors include solid organ or hematopoietic stem cell transplant, malignancy, HIV/AIDS, and receipt of immunosuppressive agents. The patient did not have these risk factors, but did have well-controlled type 2 diabetes mellitus. Although diabetes is associated with an increased risk of infection and more severe infections,13, 14 there is no evidence in the literature that well-controlled type 2 diabetes mellitus compromises the immune system. A few case reports document cutaneous S. marcescens infection in immunocompetent adults.15,16 A case report of S. marcescens septic arthritis and adjacent osteomyelitis has also been published, but the patient had poorly controlled diabetes.17 This case provides a report of systemic S. marcescens infection in an individual without clear risk factors.
S. marcescens osteomyelitis is rare, and there have been only a few prior case reports.2,18 The presentation of osteomyelitis, regardless of the causative organism, is subtle, often insidious, and can easily be missed. Hospitalists should have a high index of suspicion for the diagnosis as it requires prompt evaluation and treatment for complications, including epidural abscess. Risk factors include diabetes mellitus, rheumatoid arthritis, injection drug use, and other immunocompromising illnesses.19 Degenerative changes in the spine such as osteoarthritis may be risk factors as well,20 though not well studied or quantified. A hypothesized mechanism involves local inflammation and joint damage, leaving the area susceptible to bacterial seeding. Osteoarthritis and degenerative disc disease, along with exposure to racehorses, likely put this patient at risk for bacterial seeding in the vertebrae, ultimately leading to a “dark horse” diagnosis.
TEACHING POINTS
- Serratia marcescens is a gram-negative rod bacterium that most commonly infects immunocompromised individuals in hospital settings. This report demonstrates that S. marcescens can cause serious infection in immunocompetent, nonhospitalized adults.
- S. marcescens bacteremia or infection of organs outside of the urinary or respiratory systems is uncommon, and therapy can be complicated by emergence of resistance.
- The clinical presentation of vertebral osteomyelitis and discitis and psoas abscess can be subtle and may present without typical signs and symptoms of infection.
ACKNOWLEDGEMENTS
The authors thank the patient and his partner for their willingness to have his story published, Laura Petersen, MHSA, for providing assistance with references and manuscript editing, and Shadi Azar, MBBS, for assistance in selecting the cross-sectional images.
Disclosures
The authors have no conflicts of interest to disclose.
1. Hejazi A, Falkiner FR. Serratia marcescens. J Med Microbiol. 1997;46(11):903-912. doi: 10.1099/00222615-46-11-903. PubMed
2. Lau JX, Li JY, Yong TY. Non-contiguous multifocal vertebral osteomyelitis caused by erratia marcescens. Mod Rheumatol. 2015;25(2):303-306. doi: 10.3109/14397595.2013.874754. PubMed
3. Dessi A, Puddu M, Testa M, Marcialis MA, Pintus MC, Fanos V. Serratia marcescens infections and outbreaks in neonatal intensive care units. J Chemother. 2009;21(5):493-499. doi: 10.1179/joc.2009.21.5.493. PubMed
4. Mahlen SD. Serratia infections: from military experiments to current practice. Clin Microbiol Rev. 2011;24(4):755-791. doi: 10.1128/CMR.00017-11. PubMed
5. Montagnani C, Cocchi P, Lega L, et al. Serratia marcescens outbreak in a neonatal intensive care unit: crucial role of implementing hand hygiene among external consultants. BMC Infect Dis. 2015;15:11. doi: 10.1186/s12879-014-0734-6. PubMed
6. van Ogtrop ML, van Zoeren-Grobben D, Verbakel-Salomons EM, van Boven CP. Serratia marcescens infections in neonatal departments: description of an outbreak and review of the literature. J Hosp Infect. 1997;36(2):95-103. doi: 10.1016/S0195-6701(97)90115-8. PubMed
7. Weber DJ, Rutala WA, Sickbert-Bennett EE. Outbreaks associated with contaminated antiseptics and disinfectants. Antimicrob Agents Chemother. 2007;51(12):4217-4224. doi: 10.1128/AAC.00138-07. PubMed
8. Ewart S, Brown C, Derksen F, Kufuor-Mensa E. Serratia marcescens endocarditis in a horse. J Am Vet Med Assoc. 1992;200(7):961-963. PubMed
9. Jores J, Beutner G, Hirth-Schmidt I, Borchers K, Pitt TL, Lubke-Becker A. Isolation of Serratia marcescens from an equine abortion in Germany. Vet Rec. 2004;154(8):242-244. doi: 10.1136/vr.154.8.242. PubMed
10. Herra C, Falkiner FR. Serratia marcescens. http://www.antimicrobe.org/b26.asp. Accessed August 22, 2017.
11. Jacoby GA. AmpC beta-lactamases. Clin Microbiol Rev. 2009;22(1):161-182, Table of Contents. doi: 10.1128/CMR.00036-08. PubMed
12. Chow JW, Fine MJ, Shlaes DM, et al. Enterobacter bacteremia: clinical features and emergence of antibiotic resistance during therapy. Ann Intern Med. 1991;115(8):585-590. doi: 10.7326/0003-4819-115-8-585. PubMed
13. Goeijenbier M, van Sloten TT, Slobbe L, et al. Benefits of flu vaccination for persons with diabetes mellitus: A review. Vaccine. 2017;35(38):5095-5101. doi: 10.1016/j.vaccine.2017.07.095. PubMed
14. Gupta S, Koirala J, Khardori R, Khardori N. Infections in diabetes mellitus and hyperglycemia. Infect Dis Clin North Am. 2007;21(3):617-638, vii. doi: 10.1016/j.idc.2007.07.003. PubMed
15. Carlesimo M, Pennica A, Muscianese M, et al. Multiple skin ulcers due to Serratia marcescens in a immunocompetent patient. G Ital Dermatol Venereol. 2014;149(3):367-370. PubMed
16. Rallis E, Karanikola E, Papadakis P. Severe facial infection caused by Serratia marcescens in an immunocompetent soldier. J Am Acad Dermatol. 2008;58(5 Suppl 1):S109-S110. doi: 10.1016/j.jaad.2007.04.010. PubMed
17. Hadid H, Usman M, Thapa S. Severe osteomyelitis and septic arthritis due to Serratia marcescens in an immunocompetent patient. Case Rep Infect Dis. 2015;2015:347652. doi: 10.1155/2015/347652. PubMed
18. Berbari EF, Kanj SS, Kowalski TJ, et al. 2015 Infectious Diseases Society of America (IDSA) Clinical Practice Guidelines for the Diagnosis and Treatment of Native Vertebral Osteomyelitis in Adults. Clin Infect Dis. 2015;61(6):e26-e46. doi: 10.1093/cid/civ482. PubMed
19. Vertebral Osteomyelitis Guideline Team (Team Leader: Chenoweth CE; Team Members: Bassin BS HS, Mack MR, Kunapuli A, Park P, Quint DJ, Seagull FJ, Wesorick DH; Consultants: Patel RD, Riddell IV J, Lanava KM). Vertebral Osteomyelitis, Discitis, and Spinal Epidural Abscess in Adults. University of Michigan Guidelines for Clinical Care 2013; http://www.med.umich.edu/1info/FHP/practiceguides/vertebral/VO.pdf. Accessed October 26, 2017.
20. McDonald M. Vertebral osteomyelitis and discitis in adults. 2017; Available at: https://www.uptodate.com/contents/vertebral-osteomyelitis-and-discitis-in-adults. Accessed October 26, 2017.
A 73-year-old man presented to the emergency department in late winter with fevers, myalgias, fatigue, low back pain, and poor oral intake. Four days earlier, he had fallen and hit his head. His partner also noticed a few episodes of confusion in the days leading up to presentation.
The patient’s symptoms are nonspecific. Fevers prompt the consideration of systemic infection, though fevers can also be seen in a broad range of noninfectious processes, including malignancy, vasculitis, autoimmune conditions, endocrinopathies, and drug reaction. The clinical picture warrants prompt and comprehensive evaluation, beginning with further detailed history (current illnesses, exposures, travel, vaccinations, medications, cancer screenings, weight change) and a careful physical examination, which will help guide laboratory testing and imaging.
His past medical history was notable for coronary artery disease for which he underwent coronary artery bypass grafting five years prior, hypertension, hyperlipidemia, diet-controlled type 2 diabetes mellitus, gastroesophageal reflux disease, osteoarthritis leading to chronic knee and hand pain, and a history of mildly low testosterone levels. His medications included hydrocodone and acetaminophen, metoprolol tartrate, omeprazole, topical testosterone gel (prescribed for daily use, used intermittently), and aspirin. He was retired and lived in rural Michigan with his female partner. He previously worked as a truck driver and used to train racehorses. He had quit smoking five years earlier. He denied alcohol or injection drug use.
The patient has significant underlying medical conditions. Considering infectious causes of his symptoms, it is notable that he has no reported immunodeficiency. It would be relevant to know if he has been tested for HIV. His rural residence and work with horses raise the possibility of zoonotic infections, including plague (Yersinia pestis), brucellosis (Brucella species), Q fever (Coxiella burnetti), Rhodococcus equi, or group C or G Streptococci. Information about tuberculosis risk factors, other geographic exposures, recent dental work, and ill contacts might be helpful to elucidate the causes of this nonspecific febrile illness with a possible CNS component. With regard to malignancy, it would be helpful to ask about recent weight loss, lymphadenopathy, and prior cancer screenings. Considering other etiologies, he does not report a history of autoimmune or endocrine conditions. However, it is important to consider a vasculitis, such as giant cell arteritis or polyarteritis nodosa, autoimmune conditions, and endocrinopathies such as thyrotoxicosis. The differential diagnosis for his clinical syndrome remains broad.
Vital signs were temperature 37.3°C, heart rate 88 beats per minute, respiratory rate 18 breaths per minute, blood pressure 105/64 mmHg, and oxygen saturation 93% on room air. Oral examination revealed poor dentition. The heart had a normal rate and regular rhythm with no murmurs, rubs, or gallops, and lungs were clear to auscultation bilaterally. The abdomen was unremarkable. Examination of the back was notable for mild tenderness to palpation over the sacrum. He was oriented to person, place, and time, with intact cranial nerves and a nonfocal neurologic examination. The remainder of his examination was normal. The white blood cell (WBC) count was 11.1 × 103/μL, with 84% neutrophils and 9% bands, hemoglobin 13.6 g/dL, platelet count 54 × 103/μL, sodium 122 mmol/L, potassium 3.3 mmol/L, chloride 89 mmol/L, bicarbonate 21 mmol/L, creatinine 1.64 mg/dL, albumin 2.7 g/dL, alkaline phosphatase 136 U/L, AST 60 U/L, ALT 37 U/L, and total bilirubin 2.1 mg/dL.
He had presented to the emergency department five days earlier with fever, flank pain, nausea, vomiting, and weakness. At that time, he had a temperature of 38.2°C, but vital signs otherwise had been normal. Laboratory studies had revealed WBC count 14.0 × 103/μL, hemoglobin 13.7 g/dL, platelet count 175 × 103/μL, sodium 129 mmol/L, chloride 97 mmol/dL, bicarbonate 23 mmol/L, creatinine 1.1 mg/dL, and total bilirubin 1.6 mg/dL. Urinalysis had been negative. He had received one liter of intravenous normal saline and ketorolac for pain and had been discharged with the diagnosis of a viral illness.
A picture of a progressive, subacute illness with multisystem involvement appears to be emerging, and there are several abnormalities consistent with infection, including fever, leukocytosis with bandemia, thrombocytopenia, renal dysfunction, and elevated bilirubin. His borderline hypotension may be due to uninterrupted use of his antihypertensive medication in the setting of poor oral intake or may indicate incipient sepsis. Focal sacral tenderness raises the possibility of vertebral osteomyelitis or epidural abscess, either from a contiguous focus of infection from the surrounding structures, or as a site of seeding from bacteremia. His prior confusion episodes might have been secondary to a systemic process; however, CNS imaging should be done, given the history of confusion and recent fall. Further diagnostic studies are warranted, including: blood cultures; peripheral blood smear; imaging of the spine, chest, abdomen, and pelvis; electrocardiogram; and possibly echocardiogram. Although noninfectious etiologies should not be discounted, the constellation of findings is more compatible with infection.
Two sets of blood cultures and a viral respiratory swab were obtained. Computed tomography (CT) of the head without contrast was negative for acute bleeding or other intracranial pathology. Lumbosacral radiography revealed degenerative changes with intact alignment of the sacrum. The patient was admitted with plans to pursue lumbar puncture if altered mental status recurred. The viral swab was negative. Within 24 hours, one set of blood cultures (both bottles) grew lactose-negative, oxidase-negative, gram-negative rods.
Gram-negative rods (GNRs) rarely are contaminants in blood cultures and should be considered significant until proven otherwise. Prompt empiric therapy and investigation to identify the primary source of bacteremia must be initiated. Although the most common GNRs isolated from blood cultures are enteric coliform organisms such as E. coli, Klebsiella, and Enterobacter, these typically are lactose-positive. Additional possibilities should be considered, including Salmonella species or other organisms comprising the “HACEK” group. This latter group is commonly associated with endocarditis, but the majority are oxidase-positive and have more fastidious growth requirements. Although there are other gram-negative organisms to consider, they have other distinguishing characteristics that have not been indicated in the microbiology results. Broad-spectrum antibiotic therapy is appropriate while awaiting the final identification of the GNR. A thorough search for a primary source and secondary sites of hematogenous seeding should be conducted. His only localizing symptom was tenderness over the sacrum, and this should be further assessed by sensitive imaging such as magnetic resonance imaging (MRI). The identity of the GNR would guide further diagnostic evaluation. For example, a respiratory organism such as Haemophilus influenzae would prompt a CT scan of the chest. Isolation of an enteric or a coliform GNR such as E. coli would prompt abdominal and pelvic imaging to assess for occult abscess. An “HACEK” group organism would prompt echocardiography to evaluate for endocarditis.
He was started on piperacillin–tazobactam. GNR bacteremia without a clear source prompted a CT of the chest, abdomen, and pelvis with and without contrast. The images were unremarkable, with the exception of a signal abnormality in the left psoas muscle concerning for abscess (Figure 1). MRI of the same region revealed L2-4 osteomyelitis and discitis with bilateral psoas abscesses but without epidural abscess (Figure 2).
Because of increasing reports of antibiotic resistance in GNRs, even in community-acquired infections, it is appropriate to initially treat with a broad-spectrum antibiotic such as a fourth-generation cephalosporin or carbapenem while awaiting identification and susceptibility results to guide definitive therapy. In addition to antimicrobial therapy, treatment of psoas abscess usually requires drainage. Vertebral osteomyelitis from a hematogenous source can often be treated with antibiotics alone, as long as there are no associated complications such as epidural abscess and spine instability. Imaging should be reviewed for pathology of the surrounding structures, and surgical consultation should be obtained.
Neurosurgery, Interventional Radiology, and Infectious Disease services were consulted. Antibiotic coverage was expanded to vancomycin, cefepime, and metronidazole due to the possibility of polymicrobial infection. No surgical intervention was recommended since the abscesses were too small to drain.
The next day, the GNR was identified as Serratia marcescens.
S. marcescens is a widely distributed organism in the environment, but not a common component of endogenous human flora. Serratia is generally considered as an opportunistic nosocomial pathogen. Community-acquired infection with this organism is unusual and implies exogenous acquisition. A careful re-evaluation of exposures, including injection drug use or other parenteral exposures is important to identify the likely source of infection, as these have been previously linked to outbreaks of environmental organisms. Based on the presumed pathogenesis of infection and the initial microbiology suggesting monomicrobial Serratia infection, antibiotics should be narrowed based on the susceptibility results. There is concern that antibiotics might not adequately penetrate the abscesses and result in a lack of clinical improvement and/or lead to the emergence of antibiotic resistance during therapy. This is an important concern with Serratia, which typically harbors an AmpC beta-lactamase that can mediate resistance to broad-spectrum cephalosporins. If medical therapy alone without drainage is planned, short-interval re-imaging is warranted.
Blood cultures from days two and three of hospitalization also grew S. marcescens. No other organisms grew. Based on culture sensitivity data, antibiotics were narrowed to ceftriaxone.
This surprising culture result prompted the medical team to obtain screening laboratory tests for immunocompromising conditions and to revisit the patient’s history. His type 2 diabetes mellitus was well controlled with a hemoglobin A1c of 6.5%. HIV testing was negative. Further questioning of the patient revealed that he had fallen from a truck onto rocks four months prior, injuring his back and hip, but without puncture of the skin or loss of consciousness; he denied recent falls or other injuries but reported significant chronic knee pain. He had not been hospitalized recently. He had never taken corticosteroids or immunomodulatory medications. He continued to deny injection drug use. He did, however, clarify that his work with racehorses, which was originally understood to be a prior hobby, was ongoing, including recent work of cleaning the stables.
The following morning, he experienced confusion, rigors, and hypoxia, which prompted transfer to the intensive care unit (ICU).
Acute worsening during treatment is worrisome, and could be a potential complication of his infection or treatment – or even a separate process altogether. Knee pain in the setting of bacteremia raises the possibility of septic or crystal-induced arthritis and warrants imaging. Confusion and hypoxia might represent secondary sites of seeding from bacteremia (CNS infection and pneumonia, respectively) or manifestations of endocarditis, the latter being unusual for Serratia. An echocardiogram should be obtained. Other neurologic causes, including seizure, should also be considered. Further evaluation by chest imaging and repeat neurologic examination and imaging should be performed. Emergence of resistance during therapy is a theoretical concern with Serratia as an AmpC beta-lactamase-containing organism. While awaiting additional microbiology data, an empiric change to an AmpC beta-lactamase stable antibiotic such as a carbapenem should be made, especially since he has clinically deteriorated on therapy with a β-lactamase susceptible antibiotic, raising concerns of the emergence of resistance on initial therapy.
Antibiotics were changed to meropenem, vancomycin, and metronidazole given the clinical worsening and concerns that this represented infection unresponsive to prior antibiotics. The acute episode resolved spontaneously after one hour. His neurologic examination remained nonfocal. Chest radiography, urinalysis, urine culture, and right upper quadrant ultrasound were unremarkable. Transesophageal echocardiogram revealed no heart valve vegetations. MRI and bone scan of the lower extremities did not show any evidence of septic arthritis or other infection. He remained stable and was transferred out of the ICU the following day. Antibiotic coverage was switched to cefepime. On discussion with his significant other, this event was found to be similar to the intermittent confusion that occurred in the days prior to admission.
The acute onset and other features of these intermittent periods of deterioration are compatible with infection; intermittent seeding of the blood with microbes or their products (eg, lipopolysaccharides) from an abscess or vascular infection could explain these episodes. Some of the previous hypotheses to explain the episodes, such as a secondary infectious process, have not been supported by diagnostic testing or the clinical course. He needs close clinical monitoring and interval assessment of the known sites of infection.
Ten days after osteomyelitis and discitis were diagnosed, the patient developed worsening low back pain, prompting repeat spine MRI. This was significant for bilateral psoas abscess enlargement and extension of osteomyelitis and discitis (Figure 3). He was re-evaluated by Neurosurgery and Interventional Radiology and underwent psoas abscess drainage; abscess cultures grew S. marcescens.
He slowly improved over several weeks and was discharged to a subacute rehabilitation facility. He completed a 3.5-week course of intravenous antibiotics before leaving against medical advice. He completed eight weeks of oral trimethoprim-sulfamethoxazole and remains without long-term sequelae from the infection.
DISCUSSION
S. marcescens is a gram-negative rod in the Enterobacteriaceae family known for its red pigment. Primarily, S. marcescens causes nosocomial infections, most commonly of the respiratory and urinary tracts. However, a wide range of manifestations has been documented, including meningitis, ocular infections (conjunctivitis, keratitis, endophthalmitis), endocarditis, skin infections (cellulitis, necrotizing fasciitis), and osteomyelitis.1, 2 S. marcescens is often reported as the cause of outbreaks in ICUs;3-6 infection is thought to occur via contamination of water pipes, hospital equipment, and disinfectants.3, 7 Its natural environment includes soil, water, and GI tracts of animals,4 and there are published reports of S. marcescens infection in horses.8, 9 This patient was most likely exposed to S. marcescens through his work with horses and their environment.
S. marcescens has wide-ranging target organs, and successful treatment can be difficult. S. marcescens can infect the renal, respiratory, gastrointestinal, ocular, cardiovascular, and musculoskeletal systems. S. marcescens, like other “SPACE” organisms (Serratia, Pseudomonas, Acinetobacter, Citrobacter, Enterobacter), expresses inducible AmpC beta-lactamase.10 At baseline, AmpC beta-lactamase expression is repressed.11 Mutants with stably de-repressed (constitutively expressed) AmpC can be selected during therapy and lead to clinical failure, as has been best described during therapy for Enterobacter infections.12 Infectious Disease consultation may be helpful when caring for patients with S. marcescens bacteremia given these complexities.
This was an unusual case of S. marcescens infection. It most commonly infects immunocompromised hosts. Reported risk factors include solid organ or hematopoietic stem cell transplant, malignancy, HIV/AIDS, and receipt of immunosuppressive agents. The patient did not have these risk factors, but did have well-controlled type 2 diabetes mellitus. Although diabetes is associated with an increased risk of infection and more severe infections,13, 14 there is no evidence in the literature that well-controlled type 2 diabetes mellitus compromises the immune system. A few case reports document cutaneous S. marcescens infection in immunocompetent adults.15,16 A case report of S. marcescens septic arthritis and adjacent osteomyelitis has also been published, but the patient had poorly controlled diabetes.17 This case provides a report of systemic S. marcescens infection in an individual without clear risk factors.
S. marcescens osteomyelitis is rare, and there have been only a few prior case reports.2,18 The presentation of osteomyelitis, regardless of the causative organism, is subtle, often insidious, and can easily be missed. Hospitalists should have a high index of suspicion for the diagnosis as it requires prompt evaluation and treatment for complications, including epidural abscess. Risk factors include diabetes mellitus, rheumatoid arthritis, injection drug use, and other immunocompromising illnesses.19 Degenerative changes in the spine such as osteoarthritis may be risk factors as well,20 though not well studied or quantified. A hypothesized mechanism involves local inflammation and joint damage, leaving the area susceptible to bacterial seeding. Osteoarthritis and degenerative disc disease, along with exposure to racehorses, likely put this patient at risk for bacterial seeding in the vertebrae, ultimately leading to a “dark horse” diagnosis.
TEACHING POINTS
- Serratia marcescens is a gram-negative rod bacterium that most commonly infects immunocompromised individuals in hospital settings. This report demonstrates that S. marcescens can cause serious infection in immunocompetent, nonhospitalized adults.
- S. marcescens bacteremia or infection of organs outside of the urinary or respiratory systems is uncommon, and therapy can be complicated by emergence of resistance.
- The clinical presentation of vertebral osteomyelitis and discitis and psoas abscess can be subtle and may present without typical signs and symptoms of infection.
ACKNOWLEDGEMENTS
The authors thank the patient and his partner for their willingness to have his story published, Laura Petersen, MHSA, for providing assistance with references and manuscript editing, and Shadi Azar, MBBS, for assistance in selecting the cross-sectional images.
Disclosures
The authors have no conflicts of interest to disclose.
A 73-year-old man presented to the emergency department in late winter with fevers, myalgias, fatigue, low back pain, and poor oral intake. Four days earlier, he had fallen and hit his head. His partner also noticed a few episodes of confusion in the days leading up to presentation.
The patient’s symptoms are nonspecific. Fevers prompt the consideration of systemic infection, though fevers can also be seen in a broad range of noninfectious processes, including malignancy, vasculitis, autoimmune conditions, endocrinopathies, and drug reaction. The clinical picture warrants prompt and comprehensive evaluation, beginning with further detailed history (current illnesses, exposures, travel, vaccinations, medications, cancer screenings, weight change) and a careful physical examination, which will help guide laboratory testing and imaging.
His past medical history was notable for coronary artery disease for which he underwent coronary artery bypass grafting five years prior, hypertension, hyperlipidemia, diet-controlled type 2 diabetes mellitus, gastroesophageal reflux disease, osteoarthritis leading to chronic knee and hand pain, and a history of mildly low testosterone levels. His medications included hydrocodone and acetaminophen, metoprolol tartrate, omeprazole, topical testosterone gel (prescribed for daily use, used intermittently), and aspirin. He was retired and lived in rural Michigan with his female partner. He previously worked as a truck driver and used to train racehorses. He had quit smoking five years earlier. He denied alcohol or injection drug use.
The patient has significant underlying medical conditions. Considering infectious causes of his symptoms, it is notable that he has no reported immunodeficiency. It would be relevant to know if he has been tested for HIV. His rural residence and work with horses raise the possibility of zoonotic infections, including plague (Yersinia pestis), brucellosis (Brucella species), Q fever (Coxiella burnetti), Rhodococcus equi, or group C or G Streptococci. Information about tuberculosis risk factors, other geographic exposures, recent dental work, and ill contacts might be helpful to elucidate the causes of this nonspecific febrile illness with a possible CNS component. With regard to malignancy, it would be helpful to ask about recent weight loss, lymphadenopathy, and prior cancer screenings. Considering other etiologies, he does not report a history of autoimmune or endocrine conditions. However, it is important to consider a vasculitis, such as giant cell arteritis or polyarteritis nodosa, autoimmune conditions, and endocrinopathies such as thyrotoxicosis. The differential diagnosis for his clinical syndrome remains broad.
Vital signs were temperature 37.3°C, heart rate 88 beats per minute, respiratory rate 18 breaths per minute, blood pressure 105/64 mmHg, and oxygen saturation 93% on room air. Oral examination revealed poor dentition. The heart had a normal rate and regular rhythm with no murmurs, rubs, or gallops, and lungs were clear to auscultation bilaterally. The abdomen was unremarkable. Examination of the back was notable for mild tenderness to palpation over the sacrum. He was oriented to person, place, and time, with intact cranial nerves and a nonfocal neurologic examination. The remainder of his examination was normal. The white blood cell (WBC) count was 11.1 × 103/μL, with 84% neutrophils and 9% bands, hemoglobin 13.6 g/dL, platelet count 54 × 103/μL, sodium 122 mmol/L, potassium 3.3 mmol/L, chloride 89 mmol/L, bicarbonate 21 mmol/L, creatinine 1.64 mg/dL, albumin 2.7 g/dL, alkaline phosphatase 136 U/L, AST 60 U/L, ALT 37 U/L, and total bilirubin 2.1 mg/dL.
He had presented to the emergency department five days earlier with fever, flank pain, nausea, vomiting, and weakness. At that time, he had a temperature of 38.2°C, but vital signs otherwise had been normal. Laboratory studies had revealed WBC count 14.0 × 103/μL, hemoglobin 13.7 g/dL, platelet count 175 × 103/μL, sodium 129 mmol/L, chloride 97 mmol/dL, bicarbonate 23 mmol/L, creatinine 1.1 mg/dL, and total bilirubin 1.6 mg/dL. Urinalysis had been negative. He had received one liter of intravenous normal saline and ketorolac for pain and had been discharged with the diagnosis of a viral illness.
A picture of a progressive, subacute illness with multisystem involvement appears to be emerging, and there are several abnormalities consistent with infection, including fever, leukocytosis with bandemia, thrombocytopenia, renal dysfunction, and elevated bilirubin. His borderline hypotension may be due to uninterrupted use of his antihypertensive medication in the setting of poor oral intake or may indicate incipient sepsis. Focal sacral tenderness raises the possibility of vertebral osteomyelitis or epidural abscess, either from a contiguous focus of infection from the surrounding structures, or as a site of seeding from bacteremia. His prior confusion episodes might have been secondary to a systemic process; however, CNS imaging should be done, given the history of confusion and recent fall. Further diagnostic studies are warranted, including: blood cultures; peripheral blood smear; imaging of the spine, chest, abdomen, and pelvis; electrocardiogram; and possibly echocardiogram. Although noninfectious etiologies should not be discounted, the constellation of findings is more compatible with infection.
Two sets of blood cultures and a viral respiratory swab were obtained. Computed tomography (CT) of the head without contrast was negative for acute bleeding or other intracranial pathology. Lumbosacral radiography revealed degenerative changes with intact alignment of the sacrum. The patient was admitted with plans to pursue lumbar puncture if altered mental status recurred. The viral swab was negative. Within 24 hours, one set of blood cultures (both bottles) grew lactose-negative, oxidase-negative, gram-negative rods.
Gram-negative rods (GNRs) rarely are contaminants in blood cultures and should be considered significant until proven otherwise. Prompt empiric therapy and investigation to identify the primary source of bacteremia must be initiated. Although the most common GNRs isolated from blood cultures are enteric coliform organisms such as E. coli, Klebsiella, and Enterobacter, these typically are lactose-positive. Additional possibilities should be considered, including Salmonella species or other organisms comprising the “HACEK” group. This latter group is commonly associated with endocarditis, but the majority are oxidase-positive and have more fastidious growth requirements. Although there are other gram-negative organisms to consider, they have other distinguishing characteristics that have not been indicated in the microbiology results. Broad-spectrum antibiotic therapy is appropriate while awaiting the final identification of the GNR. A thorough search for a primary source and secondary sites of hematogenous seeding should be conducted. His only localizing symptom was tenderness over the sacrum, and this should be further assessed by sensitive imaging such as magnetic resonance imaging (MRI). The identity of the GNR would guide further diagnostic evaluation. For example, a respiratory organism such as Haemophilus influenzae would prompt a CT scan of the chest. Isolation of an enteric or a coliform GNR such as E. coli would prompt abdominal and pelvic imaging to assess for occult abscess. An “HACEK” group organism would prompt echocardiography to evaluate for endocarditis.
He was started on piperacillin–tazobactam. GNR bacteremia without a clear source prompted a CT of the chest, abdomen, and pelvis with and without contrast. The images were unremarkable, with the exception of a signal abnormality in the left psoas muscle concerning for abscess (Figure 1). MRI of the same region revealed L2-4 osteomyelitis and discitis with bilateral psoas abscesses but without epidural abscess (Figure 2).
Because of increasing reports of antibiotic resistance in GNRs, even in community-acquired infections, it is appropriate to initially treat with a broad-spectrum antibiotic such as a fourth-generation cephalosporin or carbapenem while awaiting identification and susceptibility results to guide definitive therapy. In addition to antimicrobial therapy, treatment of psoas abscess usually requires drainage. Vertebral osteomyelitis from a hematogenous source can often be treated with antibiotics alone, as long as there are no associated complications such as epidural abscess and spine instability. Imaging should be reviewed for pathology of the surrounding structures, and surgical consultation should be obtained.
Neurosurgery, Interventional Radiology, and Infectious Disease services were consulted. Antibiotic coverage was expanded to vancomycin, cefepime, and metronidazole due to the possibility of polymicrobial infection. No surgical intervention was recommended since the abscesses were too small to drain.
The next day, the GNR was identified as Serratia marcescens.
S. marcescens is a widely distributed organism in the environment, but not a common component of endogenous human flora. Serratia is generally considered as an opportunistic nosocomial pathogen. Community-acquired infection with this organism is unusual and implies exogenous acquisition. A careful re-evaluation of exposures, including injection drug use or other parenteral exposures is important to identify the likely source of infection, as these have been previously linked to outbreaks of environmental organisms. Based on the presumed pathogenesis of infection and the initial microbiology suggesting monomicrobial Serratia infection, antibiotics should be narrowed based on the susceptibility results. There is concern that antibiotics might not adequately penetrate the abscesses and result in a lack of clinical improvement and/or lead to the emergence of antibiotic resistance during therapy. This is an important concern with Serratia, which typically harbors an AmpC beta-lactamase that can mediate resistance to broad-spectrum cephalosporins. If medical therapy alone without drainage is planned, short-interval re-imaging is warranted.
Blood cultures from days two and three of hospitalization also grew S. marcescens. No other organisms grew. Based on culture sensitivity data, antibiotics were narrowed to ceftriaxone.
This surprising culture result prompted the medical team to obtain screening laboratory tests for immunocompromising conditions and to revisit the patient’s history. His type 2 diabetes mellitus was well controlled with a hemoglobin A1c of 6.5%. HIV testing was negative. Further questioning of the patient revealed that he had fallen from a truck onto rocks four months prior, injuring his back and hip, but without puncture of the skin or loss of consciousness; he denied recent falls or other injuries but reported significant chronic knee pain. He had not been hospitalized recently. He had never taken corticosteroids or immunomodulatory medications. He continued to deny injection drug use. He did, however, clarify that his work with racehorses, which was originally understood to be a prior hobby, was ongoing, including recent work of cleaning the stables.
The following morning, he experienced confusion, rigors, and hypoxia, which prompted transfer to the intensive care unit (ICU).
Acute worsening during treatment is worrisome, and could be a potential complication of his infection or treatment – or even a separate process altogether. Knee pain in the setting of bacteremia raises the possibility of septic or crystal-induced arthritis and warrants imaging. Confusion and hypoxia might represent secondary sites of seeding from bacteremia (CNS infection and pneumonia, respectively) or manifestations of endocarditis, the latter being unusual for Serratia. An echocardiogram should be obtained. Other neurologic causes, including seizure, should also be considered. Further evaluation by chest imaging and repeat neurologic examination and imaging should be performed. Emergence of resistance during therapy is a theoretical concern with Serratia as an AmpC beta-lactamase-containing organism. While awaiting additional microbiology data, an empiric change to an AmpC beta-lactamase stable antibiotic such as a carbapenem should be made, especially since he has clinically deteriorated on therapy with a β-lactamase susceptible antibiotic, raising concerns of the emergence of resistance on initial therapy.
Antibiotics were changed to meropenem, vancomycin, and metronidazole given the clinical worsening and concerns that this represented infection unresponsive to prior antibiotics. The acute episode resolved spontaneously after one hour. His neurologic examination remained nonfocal. Chest radiography, urinalysis, urine culture, and right upper quadrant ultrasound were unremarkable. Transesophageal echocardiogram revealed no heart valve vegetations. MRI and bone scan of the lower extremities did not show any evidence of septic arthritis or other infection. He remained stable and was transferred out of the ICU the following day. Antibiotic coverage was switched to cefepime. On discussion with his significant other, this event was found to be similar to the intermittent confusion that occurred in the days prior to admission.
The acute onset and other features of these intermittent periods of deterioration are compatible with infection; intermittent seeding of the blood with microbes or their products (eg, lipopolysaccharides) from an abscess or vascular infection could explain these episodes. Some of the previous hypotheses to explain the episodes, such as a secondary infectious process, have not been supported by diagnostic testing or the clinical course. He needs close clinical monitoring and interval assessment of the known sites of infection.
Ten days after osteomyelitis and discitis were diagnosed, the patient developed worsening low back pain, prompting repeat spine MRI. This was significant for bilateral psoas abscess enlargement and extension of osteomyelitis and discitis (Figure 3). He was re-evaluated by Neurosurgery and Interventional Radiology and underwent psoas abscess drainage; abscess cultures grew S. marcescens.
He slowly improved over several weeks and was discharged to a subacute rehabilitation facility. He completed a 3.5-week course of intravenous antibiotics before leaving against medical advice. He completed eight weeks of oral trimethoprim-sulfamethoxazole and remains without long-term sequelae from the infection.
DISCUSSION
S. marcescens is a gram-negative rod in the Enterobacteriaceae family known for its red pigment. Primarily, S. marcescens causes nosocomial infections, most commonly of the respiratory and urinary tracts. However, a wide range of manifestations has been documented, including meningitis, ocular infections (conjunctivitis, keratitis, endophthalmitis), endocarditis, skin infections (cellulitis, necrotizing fasciitis), and osteomyelitis.1, 2 S. marcescens is often reported as the cause of outbreaks in ICUs;3-6 infection is thought to occur via contamination of water pipes, hospital equipment, and disinfectants.3, 7 Its natural environment includes soil, water, and GI tracts of animals,4 and there are published reports of S. marcescens infection in horses.8, 9 This patient was most likely exposed to S. marcescens through his work with horses and their environment.
S. marcescens has wide-ranging target organs, and successful treatment can be difficult. S. marcescens can infect the renal, respiratory, gastrointestinal, ocular, cardiovascular, and musculoskeletal systems. S. marcescens, like other “SPACE” organisms (Serratia, Pseudomonas, Acinetobacter, Citrobacter, Enterobacter), expresses inducible AmpC beta-lactamase.10 At baseline, AmpC beta-lactamase expression is repressed.11 Mutants with stably de-repressed (constitutively expressed) AmpC can be selected during therapy and lead to clinical failure, as has been best described during therapy for Enterobacter infections.12 Infectious Disease consultation may be helpful when caring for patients with S. marcescens bacteremia given these complexities.
This was an unusual case of S. marcescens infection. It most commonly infects immunocompromised hosts. Reported risk factors include solid organ or hematopoietic stem cell transplant, malignancy, HIV/AIDS, and receipt of immunosuppressive agents. The patient did not have these risk factors, but did have well-controlled type 2 diabetes mellitus. Although diabetes is associated with an increased risk of infection and more severe infections,13, 14 there is no evidence in the literature that well-controlled type 2 diabetes mellitus compromises the immune system. A few case reports document cutaneous S. marcescens infection in immunocompetent adults.15,16 A case report of S. marcescens septic arthritis and adjacent osteomyelitis has also been published, but the patient had poorly controlled diabetes.17 This case provides a report of systemic S. marcescens infection in an individual without clear risk factors.
S. marcescens osteomyelitis is rare, and there have been only a few prior case reports.2,18 The presentation of osteomyelitis, regardless of the causative organism, is subtle, often insidious, and can easily be missed. Hospitalists should have a high index of suspicion for the diagnosis as it requires prompt evaluation and treatment for complications, including epidural abscess. Risk factors include diabetes mellitus, rheumatoid arthritis, injection drug use, and other immunocompromising illnesses.19 Degenerative changes in the spine such as osteoarthritis may be risk factors as well,20 though not well studied or quantified. A hypothesized mechanism involves local inflammation and joint damage, leaving the area susceptible to bacterial seeding. Osteoarthritis and degenerative disc disease, along with exposure to racehorses, likely put this patient at risk for bacterial seeding in the vertebrae, ultimately leading to a “dark horse” diagnosis.
TEACHING POINTS
- Serratia marcescens is a gram-negative rod bacterium that most commonly infects immunocompromised individuals in hospital settings. This report demonstrates that S. marcescens can cause serious infection in immunocompetent, nonhospitalized adults.
- S. marcescens bacteremia or infection of organs outside of the urinary or respiratory systems is uncommon, and therapy can be complicated by emergence of resistance.
- The clinical presentation of vertebral osteomyelitis and discitis and psoas abscess can be subtle and may present without typical signs and symptoms of infection.
ACKNOWLEDGEMENTS
The authors thank the patient and his partner for their willingness to have his story published, Laura Petersen, MHSA, for providing assistance with references and manuscript editing, and Shadi Azar, MBBS, for assistance in selecting the cross-sectional images.
Disclosures
The authors have no conflicts of interest to disclose.
1. Hejazi A, Falkiner FR. Serratia marcescens. J Med Microbiol. 1997;46(11):903-912. doi: 10.1099/00222615-46-11-903. PubMed
2. Lau JX, Li JY, Yong TY. Non-contiguous multifocal vertebral osteomyelitis caused by erratia marcescens. Mod Rheumatol. 2015;25(2):303-306. doi: 10.3109/14397595.2013.874754. PubMed
3. Dessi A, Puddu M, Testa M, Marcialis MA, Pintus MC, Fanos V. Serratia marcescens infections and outbreaks in neonatal intensive care units. J Chemother. 2009;21(5):493-499. doi: 10.1179/joc.2009.21.5.493. PubMed
4. Mahlen SD. Serratia infections: from military experiments to current practice. Clin Microbiol Rev. 2011;24(4):755-791. doi: 10.1128/CMR.00017-11. PubMed
5. Montagnani C, Cocchi P, Lega L, et al. Serratia marcescens outbreak in a neonatal intensive care unit: crucial role of implementing hand hygiene among external consultants. BMC Infect Dis. 2015;15:11. doi: 10.1186/s12879-014-0734-6. PubMed
6. van Ogtrop ML, van Zoeren-Grobben D, Verbakel-Salomons EM, van Boven CP. Serratia marcescens infections in neonatal departments: description of an outbreak and review of the literature. J Hosp Infect. 1997;36(2):95-103. doi: 10.1016/S0195-6701(97)90115-8. PubMed
7. Weber DJ, Rutala WA, Sickbert-Bennett EE. Outbreaks associated with contaminated antiseptics and disinfectants. Antimicrob Agents Chemother. 2007;51(12):4217-4224. doi: 10.1128/AAC.00138-07. PubMed
8. Ewart S, Brown C, Derksen F, Kufuor-Mensa E. Serratia marcescens endocarditis in a horse. J Am Vet Med Assoc. 1992;200(7):961-963. PubMed
9. Jores J, Beutner G, Hirth-Schmidt I, Borchers K, Pitt TL, Lubke-Becker A. Isolation of Serratia marcescens from an equine abortion in Germany. Vet Rec. 2004;154(8):242-244. doi: 10.1136/vr.154.8.242. PubMed
10. Herra C, Falkiner FR. Serratia marcescens. http://www.antimicrobe.org/b26.asp. Accessed August 22, 2017.
11. Jacoby GA. AmpC beta-lactamases. Clin Microbiol Rev. 2009;22(1):161-182, Table of Contents. doi: 10.1128/CMR.00036-08. PubMed
12. Chow JW, Fine MJ, Shlaes DM, et al. Enterobacter bacteremia: clinical features and emergence of antibiotic resistance during therapy. Ann Intern Med. 1991;115(8):585-590. doi: 10.7326/0003-4819-115-8-585. PubMed
13. Goeijenbier M, van Sloten TT, Slobbe L, et al. Benefits of flu vaccination for persons with diabetes mellitus: A review. Vaccine. 2017;35(38):5095-5101. doi: 10.1016/j.vaccine.2017.07.095. PubMed
14. Gupta S, Koirala J, Khardori R, Khardori N. Infections in diabetes mellitus and hyperglycemia. Infect Dis Clin North Am. 2007;21(3):617-638, vii. doi: 10.1016/j.idc.2007.07.003. PubMed
15. Carlesimo M, Pennica A, Muscianese M, et al. Multiple skin ulcers due to Serratia marcescens in a immunocompetent patient. G Ital Dermatol Venereol. 2014;149(3):367-370. PubMed
16. Rallis E, Karanikola E, Papadakis P. Severe facial infection caused by Serratia marcescens in an immunocompetent soldier. J Am Acad Dermatol. 2008;58(5 Suppl 1):S109-S110. doi: 10.1016/j.jaad.2007.04.010. PubMed
17. Hadid H, Usman M, Thapa S. Severe osteomyelitis and septic arthritis due to Serratia marcescens in an immunocompetent patient. Case Rep Infect Dis. 2015;2015:347652. doi: 10.1155/2015/347652. PubMed
18. Berbari EF, Kanj SS, Kowalski TJ, et al. 2015 Infectious Diseases Society of America (IDSA) Clinical Practice Guidelines for the Diagnosis and Treatment of Native Vertebral Osteomyelitis in Adults. Clin Infect Dis. 2015;61(6):e26-e46. doi: 10.1093/cid/civ482. PubMed
19. Vertebral Osteomyelitis Guideline Team (Team Leader: Chenoweth CE; Team Members: Bassin BS HS, Mack MR, Kunapuli A, Park P, Quint DJ, Seagull FJ, Wesorick DH; Consultants: Patel RD, Riddell IV J, Lanava KM). Vertebral Osteomyelitis, Discitis, and Spinal Epidural Abscess in Adults. University of Michigan Guidelines for Clinical Care 2013; http://www.med.umich.edu/1info/FHP/practiceguides/vertebral/VO.pdf. Accessed October 26, 2017.
20. McDonald M. Vertebral osteomyelitis and discitis in adults. 2017; Available at: https://www.uptodate.com/contents/vertebral-osteomyelitis-and-discitis-in-adults. Accessed October 26, 2017.
1. Hejazi A, Falkiner FR. Serratia marcescens. J Med Microbiol. 1997;46(11):903-912. doi: 10.1099/00222615-46-11-903. PubMed
2. Lau JX, Li JY, Yong TY. Non-contiguous multifocal vertebral osteomyelitis caused by erratia marcescens. Mod Rheumatol. 2015;25(2):303-306. doi: 10.3109/14397595.2013.874754. PubMed
3. Dessi A, Puddu M, Testa M, Marcialis MA, Pintus MC, Fanos V. Serratia marcescens infections and outbreaks in neonatal intensive care units. J Chemother. 2009;21(5):493-499. doi: 10.1179/joc.2009.21.5.493. PubMed
4. Mahlen SD. Serratia infections: from military experiments to current practice. Clin Microbiol Rev. 2011;24(4):755-791. doi: 10.1128/CMR.00017-11. PubMed
5. Montagnani C, Cocchi P, Lega L, et al. Serratia marcescens outbreak in a neonatal intensive care unit: crucial role of implementing hand hygiene among external consultants. BMC Infect Dis. 2015;15:11. doi: 10.1186/s12879-014-0734-6. PubMed
6. van Ogtrop ML, van Zoeren-Grobben D, Verbakel-Salomons EM, van Boven CP. Serratia marcescens infections in neonatal departments: description of an outbreak and review of the literature. J Hosp Infect. 1997;36(2):95-103. doi: 10.1016/S0195-6701(97)90115-8. PubMed
7. Weber DJ, Rutala WA, Sickbert-Bennett EE. Outbreaks associated with contaminated antiseptics and disinfectants. Antimicrob Agents Chemother. 2007;51(12):4217-4224. doi: 10.1128/AAC.00138-07. PubMed
8. Ewart S, Brown C, Derksen F, Kufuor-Mensa E. Serratia marcescens endocarditis in a horse. J Am Vet Med Assoc. 1992;200(7):961-963. PubMed
9. Jores J, Beutner G, Hirth-Schmidt I, Borchers K, Pitt TL, Lubke-Becker A. Isolation of Serratia marcescens from an equine abortion in Germany. Vet Rec. 2004;154(8):242-244. doi: 10.1136/vr.154.8.242. PubMed
10. Herra C, Falkiner FR. Serratia marcescens. http://www.antimicrobe.org/b26.asp. Accessed August 22, 2017.
11. Jacoby GA. AmpC beta-lactamases. Clin Microbiol Rev. 2009;22(1):161-182, Table of Contents. doi: 10.1128/CMR.00036-08. PubMed
12. Chow JW, Fine MJ, Shlaes DM, et al. Enterobacter bacteremia: clinical features and emergence of antibiotic resistance during therapy. Ann Intern Med. 1991;115(8):585-590. doi: 10.7326/0003-4819-115-8-585. PubMed
13. Goeijenbier M, van Sloten TT, Slobbe L, et al. Benefits of flu vaccination for persons with diabetes mellitus: A review. Vaccine. 2017;35(38):5095-5101. doi: 10.1016/j.vaccine.2017.07.095. PubMed
14. Gupta S, Koirala J, Khardori R, Khardori N. Infections in diabetes mellitus and hyperglycemia. Infect Dis Clin North Am. 2007;21(3):617-638, vii. doi: 10.1016/j.idc.2007.07.003. PubMed
15. Carlesimo M, Pennica A, Muscianese M, et al. Multiple skin ulcers due to Serratia marcescens in a immunocompetent patient. G Ital Dermatol Venereol. 2014;149(3):367-370. PubMed
16. Rallis E, Karanikola E, Papadakis P. Severe facial infection caused by Serratia marcescens in an immunocompetent soldier. J Am Acad Dermatol. 2008;58(5 Suppl 1):S109-S110. doi: 10.1016/j.jaad.2007.04.010. PubMed
17. Hadid H, Usman M, Thapa S. Severe osteomyelitis and septic arthritis due to Serratia marcescens in an immunocompetent patient. Case Rep Infect Dis. 2015;2015:347652. doi: 10.1155/2015/347652. PubMed
18. Berbari EF, Kanj SS, Kowalski TJ, et al. 2015 Infectious Diseases Society of America (IDSA) Clinical Practice Guidelines for the Diagnosis and Treatment of Native Vertebral Osteomyelitis in Adults. Clin Infect Dis. 2015;61(6):e26-e46. doi: 10.1093/cid/civ482. PubMed
19. Vertebral Osteomyelitis Guideline Team (Team Leader: Chenoweth CE; Team Members: Bassin BS HS, Mack MR, Kunapuli A, Park P, Quint DJ, Seagull FJ, Wesorick DH; Consultants: Patel RD, Riddell IV J, Lanava KM). Vertebral Osteomyelitis, Discitis, and Spinal Epidural Abscess in Adults. University of Michigan Guidelines for Clinical Care 2013; http://www.med.umich.edu/1info/FHP/practiceguides/vertebral/VO.pdf. Accessed October 26, 2017.
20. McDonald M. Vertebral osteomyelitis and discitis in adults. 2017; Available at: https://www.uptodate.com/contents/vertebral-osteomyelitis-and-discitis-in-adults. Accessed October 26, 2017.
© 2018 Society of Hospital Medicine