User login
A 22-year-old man presented to a Canadian community hospital emergency department complaining of 2-3 weeks of abdominal pain and bloating associated with early satiety. He also noted weight loss of 20 pounds over the preceding months, leg and abdominal swelling with increased girth, and 1-2 loose, nonbloody stools per day.
Early satiety and bloating are nonspecific symptoms that can be due to gastroesophageal reflux disease, peptic ulcer disease, gastrointestinal obstruction, or gastroparesis. Weight loss in a young person, particularly if >5% of body weight, is concerning for a serious underlying medical issue. It could reflect reduced intake due to anorexia, odynophagia, or dysphagia or increased energy expenditure due to an inflammatory state such as infection or rheumatic disease. The etiology of the swelling needs to be elucidated. It may be due to increased hydrostatic forces as in heart failure, venous or lymphatic obstruction, or from lowered oncotic pressure resulting from hepatic disease, nephrotic syndrome, severe malnutrition (nonbloody loose stools), or a protein losing enteropathy.
The patient was transferred to a tertiary care center for closer access to specialty consultation. He described generalized abdominal pain increasing in intensity over three weeks; bilateral lower extremity, scrotal, abdominal wall, and sacral edema; and mild dyspnea on exertion. The early satiety was not associated with dysphagia, odynophagia, nausea, or vomiting. He denied fevers, chills, night sweats, nausea, vomiting, jaundice, easy bruising, orthopnea, paroxysmal nocturnal dyspnea (PND), or chest pain. His past medical history included asthma treated with fluticasone/salmeterol and albuterol. He was a Canadian of East Asian descent working as a plumber. He previously smoked three to four cigarettes per day for six years. He stopped smoking one month before presentation. He had one alcoholic beverage per week and smoked marijuana weekly. He denied any family history of similar symptoms or malignancy.The differential diagnosis for weight loss and anasarca is broad and includes malignancies, infectious diseases, rheumatic or inflammatory disorders, malabsorption, and advanced cardiac, renal, or liver disease. His history does not classically point in one direction. The mild dyspnea on exertion may be due to cardiac disease, but it is unlikely in the absence of orthopnea and PND. The dyspnea could be due to increased abdominal pressure if ascites are present, his underlying asthma, or another etiology such as anemia. Fevers, chills, and/or night sweats can be expected in infections and some malignancies, but their absence does not exclude infections and malignancies from the differential diagnoses. Particular attention should be paid to lymphadenopathy on the physical examination. The presence of an umbilical nodule (Sister Mary Joseph sign) could indicate a malignancy (gastrointestinal or lymphoma).
The differential diagnosis for weight loss and anasarca is broad and includes malignancies, infectious diseases, rheumatic or inflammatory disorders, malabsorption, and advanced cardiac, renal, or liver disease. His history does not classically point in one direction. The mild dyspnea on exertion may be due to cardiac disease, but it is unlikely in the absence of orthopnea and PND. The dyspnea could be due to increased abdominal pressure if ascites are present, his underlying asthma, or another etiology such as anemia. Fevers, chills, and/or night sweats can be expected in infections and some malignancies, but their absence does not exclude infections and malignancies from the differential diagnoses. Particular attention should be paid to lymphadenopathy on the physical examination. The presence of an umbilical nodule (Sister Mary Joseph sign) could indicate a malignancy (gastrointestinal or lymphoma).
On physical examination, his temperature was 38.1°C, heart rate was 138 beats per minute, blood pressure was 123/86 mm Hg, respiratory rate was 20 breaths per minute, and oxygen saturation was 97% on room air. He appeared uncomfortable and diaphoretic. No scleral icterus or jaundice was appreciated. There were no palpable cervical, axillary, or inguinal lymph nodes. Cardiac examination revealed tachycardia and no murmurs, rubs, gallops, or jugular venous distention. Abdominal examination revealed abdominal distention, diffuse tenderness to deep palpation, bulging flanks, and a positive fluid wave. Liver and spleen could not be palpated or percussed secondary to abdominal distention. He had pitting bilateral lower extremity edema that extended to and included the scrotum. Neurologic and pulmonary examinations were unremarkable.
His examination reveals low-grade fever, tachycardia, and diaphoresis. Whether this represents progression of his primary disease or he has acutely developed a superimposed infection is uncertain at this point. He has notable anasarca but no jugular venous distention, crackles, or S3 gallop. The lack of evidence of pulmonary edema or increased central venous pressure on physical examination increases the likelihood of cirrhosis, hypoalbuminemia, or obstruction (lymphatic or venous) and decreases the likelihood of heart failure as the etiology of his peripheral edema and likely ascites. Despite the prominence of gastrointestinal symptoms, he has neither jaundice nor stigmata of chronic liver disease. Periorbital edema, which may be present in nephrotic syndrome, is also absent. Although he has no palpable peripheral lymphadenopathy, malignancy remains a concern.
Testing should include urinalysis for proteinuria and coagulation studies to assess synthetic function of the liver. Abdominal ultrasound is indicated to confirm ascites. If present, a diagnostic paracentesis should be performed to rule out spontaneous bacterial peritonitis and determine whether the ascites is from portal hypertension, hypoalbuminemia, or peritoneal disease. If the transaminases are elevated or if the ascitic fluid is concerning for malignancy, he will need a computed tomography (CT) of the abdomen and pelvis. A protein losing enteropathy due to malignancies (gastric cancer or lymphoma), rheumatic disease (systemic lupus erythematosus [SLE]), or infiltrative disease (amyloid) is also a possibility. If the other studies are unrevealing, stool should be sent for alpha-1 antitrypsin.
Laboratory studies revealed hemoglobin 7.8 g/dL, platelets 53 k/mm3, white blood cell count (WBC) 10.6 k/mm3, alkaline phosphatase (ALP) 217 U/L, albumin 2.7 g/dL, reticulocyte count 3 k/mm3 (reference range, 30-110 k/mm3), and ferritin 1,310 ng/mL (reference range, 20-400 ng/L). Serum aminotransferase levels, bilirubin, coagulation panel, electrolytes, and creatinine were normal. Urinalysis was negative for blood, leukocytes, and protein. Diagnostic paracentesis demonstrated a serum-ascites-albumin gradient (SAAG) of two and macrophage predominance (WBC 250 U/L). Ascitic fluid cytology and culture were negative. Blood cultures, human immunodeficiency virus (HIV)-1 and 2, cytomegalovirus (CMV), and Epstein–Barr virus (EBV) serologies were negative. Viral serologies for hepatitis A, B, and C were negative. Antinuclear antibody (ANA), anti-ds DNA, antineutrophilic cytoplasmic antibody (ANCA), serum angiotensin-converting enzyme (ACE) level, and quantitative immunoglobulin levels were all within the normal range. Chest, abdomen, and pelvis CT with contrast revealed large-volume abdominal and pelvic ascites, diffuse subcutaneous edema (Figure 1), modest hepatosplenomegaly, small bilateral pleural effusions, and mediastinal, axillary, mesenteric, periportal, peripancreatic, and retroperitoneal lymphadenopathy (Figure 2).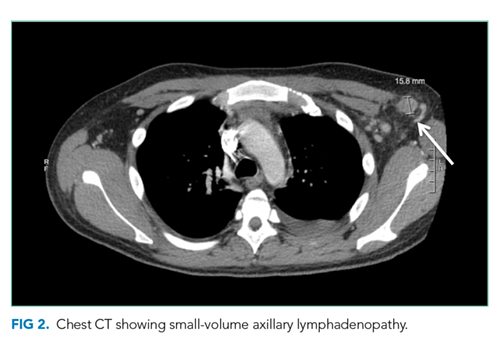
Malignancy is highest on the differential. In the absence of evidence of a primary tumor, a lymphoma would be the most likely diagnosis. Multicentric Castleman disease (MCD), a rare lymphoproliferative disorder with a clinical picture similar to lymphoma, should be considered.
Some of the more common viral etiologies of generalized lymphadenopathy and cytopenias are unlikely because serologies for HIV, hepatitis B and C, EBV, and CMV are negative. Tuberculosis fits with the insidious nature of his presentation and remains on the differential although a low SAAG would be expected. From a rheumatologic standpoint, the lack of characteristic findings on history and physical examination and the negative ANA and anti-ds DNA results make SLE unlikely. Although elevated in the majority of untreated sarcoid patients, a normal ACE level is not sufficient to rule out this diagnosis. IgG, IgA, and IgM levels would be low if there was significant gastrointestinal protein loss and elevated in MCD. The markedly increased ferritin level, an acute-phase reactant often elevated in the setting of inflammation or malignancy, raises suspicion for adult Still’s disease (despite the lack of characteristic arthralgias and/or rash) and hemophagocytic lymphohistiocytosis (HLH).
A SAAG greater than or equal to 1.1 indicates the presence of portal hypertension. Portal hypertension most often results from cirrhosis for which this patient has no apparent clinical findings. Etiologies of noncirrhotic portal hypertension are classified as prehepatic, intrahepatic, and posthepatic. There is no clinical or radiologic evidence of portal or splenic vein thrombosis (prehepatic) or heart failure (posthepatic). Possible intrahepatic etiologies include malignancy and sarcoid. Although uncommon, patients with malignancy-related ascites may have a high SAAG without coexisting cirrhosis. This occurs if there is portal hypertension due to extensive metastases in the liver or involvement of the portal venous system. The cytology of the ascitic fluid is negative. However, cytology is <80% sensitive in the absence of peritoneal carcinomatosis.
The most likely diagnosis at this point is lymphoma. Bone marrow biopsy is indicated to further assess his thrombocytopenia and hypoproliferative anemia and may be diagnostic for malignancy. Pathologic examination of a lymph node should be performed. Due to concern for lymphoproliferative disease, excisional biopsy is preferred to preserve tissue architecture.
Hematology was consulted for evaluation of the lymphadenopathy, anemia, and thrombocytopenia and recommended bone marrow and excisional lymph node biopsies. Bone marrow biopsy showed trilineage hypercellularity (Figure 3A) with reduced erythropoiesis and reticulin fibrosis (Figure 3B). An axillary lymph node biopsy with flow cytometry was nondiagnostic for a lymphoproliferative disorder or malignancy.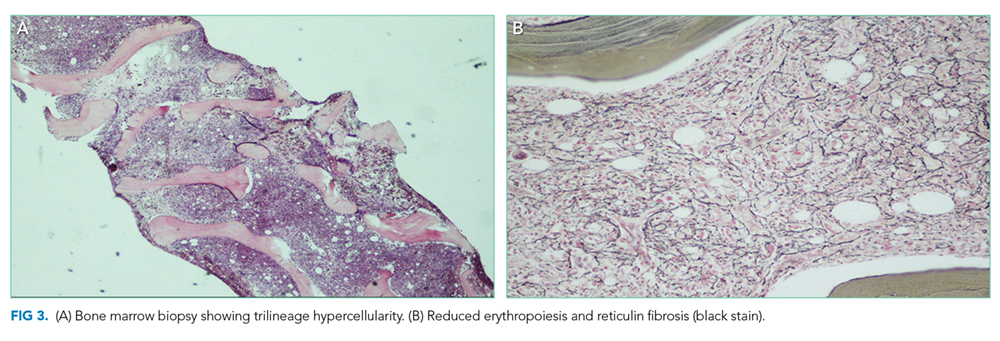
Both biopsies fail to provide a definitive diagnosis. Hypercellularity in the marrow (>70% cellularity) and reticulin fibrosis are nonspecific and could be from a malignant or reactive disease process. Lymphoma remains the most likely diagnosis. Peripheral blood for flow cytometry, lactate dehydrogenase (LDH), and uric acid should be sent. A repeat excisional biopsy of another lymph node should be performed.
Gastroenterology was consulted to evaluate the loose stools, anasarca, and hepatomegaly, and esophagogastroduodenoscopy, enteroscopy, and colonoscopy with biopsies were performed. Gastric biopsy revealed mild gastropathy. Duodenal, jejunal, and right and left colon biopsies were all normal. A liver biopsy was performed and revealed periportal inflammation. Rheumatology and infectious disease consultations did not suspect that the patient had a rheumatologic or infectious disease.
After appropriate workup and no definitive diagnosis, it is important to reassess the patient for overall stability and the presence of any new or changing symptoms (worsening symptoms, persistent fevers) that could direct further evaluation. Lymphoma remains on the differential despite multiple negative biopsies, but other less common diseases that mimic lymphoma and cause multisystem disease should be investigated. Review of the previous lymph node and tissue biopsies with the pathologist and hematologist should focus on features of adult Still’s disease (paracortical immunoblastic hyperplasia), MCD (histopathology of angiofollicular lymph node hyperplasia and presence of human herpes virus-8 (HHV-8), and HLH (hemophagocytosis). A positron emission tomography scan may not distinguish between malignancy and other fluorodeoxyglucose avid inflammatory processes but is recommended to determine the site of a future excisional lymph node biopsy.
A 10-day trial of prednisone 50 mg daily was initiated for presumed lymphoma. He experienced symptomatic improvement with decreased peripheral edema and ascites and resolution of his fevers. He was discharged home seven days after completing steroids with follow-up.
Five days after discharge, he was readmitted with worsening anasarca, massive ascites, and acute kidney injury. Admission laboratory studies revealed creatinine 1.66 mg/dL, hemoglobin 11.5 g/dL, and platelets 94 k/mm3. In addition, his ferritin level was 1,907 ng/L (reference range, 20-400 ng/L), erythrocyte sedimentation rate (ESR) was 50 mm/h (reference range, 0-20 mm/h), and C-reactive protein concentration (CRP) was 12.1 mg/dL (reference range, 0-0.5 mg/dL).
Steroids are used to treat a wide variety of illnesses, some of which are still under consideration in this patient including lymphoma, MCD, adult Still’s disease, and HLH. His symptoms recurred quickly after discontinuation of steroids in the setting of elevated ferritin, ESR, and CRP levels reflecting marked ongoing inflammation. Serologic testing for soluble IL-2 receptor, often elevated in MCD and HLH, should be performed. Excisional biopsy of an accessible node should be performed urgently.
His acute kidney injury resolved; however, he continued to have intermittent fevers, anemia, thromobocytopenia, lymphadenopathy, and hepatosplenomegaly. A hematology case-conference recommended testing for HLH, including soluble IL-2 receptor (CD25), soluble CD163, and natural killer cell degranulation assay, all of which were negative. A right inguinal lymph node biopsy revealed reactive lymphoid tissue and stained negative for HHV-8. Based on the lack of an alternative diagnosis (particularly lymphoma), the presence of multiple areas of lymphadenopathy, anemia, fevers, organomegaly, weight loss, reactive lymphoid tissue on lymph node biopsy, and elevated CRP and ESR, a working diagnosis of MCD was made. The negative HHV-8 testing was consistent with idiopathic MCD (iMCD); however, features inconsistent with iMCD included lack of polyclonal hypergammaglobulinemia and the presence of significant anasarca and thrombocytopenia. Therefore, an internet search was performed using the patient’s salient symptoms and findings. The search revealed a few recently published case reports of a rare variant of iMCD, TAFRO syndrome. TAFRO syndrome, characterized by thrombocytopenia, anasarca, fever, reticulin fibrosis and/or renal insufficiency, and organomegaly, fully explained the patient’s presentation. He was started on prednisone, rituximab (anti-CD20 antibody), and furosemide. After one month of treatment, he showed complete resolution of cytopenias, lymphadenopathy, organomegaly, anasarca, and ascites. Therapy continued for approximately three months, and he has remained symptom-free.
COMMENTARY
Castleman’s disease (CD) is a rare lymphoproliferative disorder divided into unicentric (solitary enlarged lymph node) and multicentric (multifocal enlarged lymph nodes).1 MCD typically presents with systemic inflammation, reactive proliferation of benign lymphocytes, multifocal lymphadenopathy, elevated inflammatory markers, anemia, hypoalbuminemia, and polyclonal gammaglobulinemia.1 It is hypothesized that HHV-8 drives the systemic inflammation of MCD via high levels of interleukin-6 (IL-6) activity.1 iMCD is an HHV-8-negative variant of MCD.1
TAFRO syndrome was first described in 2010 in three Japanese patients demonstrating high fever, anasarca, hepatosplenomegaly, lymphadenopathy, severe thrombocytopenia, and reticulin fibrosis.2 In 2015, the All Japan TAFRO Syndrome Research Group recognized TAFRO syndrome as a variant of iMCD and created diagnostic criteria and a severity classification system.3 Major criteria consist of anasarca, including pleural effusion and/or ascites identified on CT scan and general edema, thrombocytopenia (platelet count <100 k/mm3), and systemic inflammation (fever >37.5°C and/or serum CRP greater than or equal to 2 mg/dL).3 Two of four minor criteria must be met, which include (1) lymph node histology consistent with CD, (2) reticulin myelofibrosis and/or increased number of megakaryocytes in bone marrow, (3) mild organomegaly, including hepatomegaly, splenomegaly, and lymphadenopathy <1.5 cm in diameter identified on CT scan, and (4) progressive renal insufficiency (serum creatinine >1.2 mg/dL in males or >1.0 mg/dL in females).3 In addition, several patients with TAFRO syndrome demonstrate elevated ALP, low-normal LDH, elevated vascular endothelial growth factor, elevated IL-6, microcytic anemia, and slight polyclonal hypergammopathy.3 Malignancies such as lymphoma and myeloma, autoimmune diseases such as SLE and ANCA-associated vasculitis, infectious diseases such as those caused by mycobacteria, and POEMS (polyneuropathy, organomegaly, endocrine diseases, M-protein, and skin lesions) syndrome must be excluded to diagnose TAFRO syndrome.3,4
The pathophysiology of TAFRO syndrome is unknown, and it is unclear whether the syndrome is truly a variant of iMCD or a distinct entity.3 IL-6 is typically only mildly elevated in TAFRO syndrome, without the consequent thrombocytosis and polyclonal hypergammaglobulinemia seen in MCD, which is associated with higher levels of IL-6.1 Multiple non-HHV-8 mechanisms for TAFRO syndrome have been proposed, including (1) systemic inflammation, autoimmune/autoinflammatory mechanisms, (2) neoplastic, ectopic cytokine secretion by malignant or benign tumor cells, and/or (3) infectious, such as non-HHV-8 virus.5
Immunosuppression is the mainstay of treatment for TAFRO syndrome based on recommendations from the 2015 TAFRO Research Group.3 Glucocorticoids are considered first-line therapy.3 Cyclosporin A is recommended for individuals refractory to glucocorticoids.3 In patients with a contraindication to cyclosporin A, anti-IL-6 receptor antibodies such as tocilizumab (approved for treatment of iMCD in Japan) and siltuximab (approved for treatment of iMCD in North America and Europe) or the anti-CD20 antibody rituximab should be prescribed.3 There is evidence for the thrombopoietin receptor agonists romiplostim and eltrombopag to treat persistent thrombocytopenia.3 Additional treatments for refractory TAFRO syndrome include IVIG and plasma exchange, chemotherapy (cyclophosphamide, doxorubicin, vincristine, prednisolone), and thalidomide.3,6
Little is known about the epidemiologic characterization of TAFRO syndrome as less than 40 cases of TAFRO syndrome have been reported in the United States, Asia, and Europe.
1,4,7-9 TAFRO syndrome occurs primarily in the fourth and fifth decades of life, with case reports ranging from 14 to 78 years of age.1,3,10,11 Gender distribution varies but is likely equal for males and females.3 Mortality in TAFRO syndrome is estimated at 11%-12%.1,3 Over the past several years, a North American and European patient registry and natural history study for CD, ACCELERATE, has been initiated.4 In addition, the international Castleman Disease Collaborative Network, a Japanese multicenter retrospective study for MCD, and a nationwide Japanese research team for CD have been created.3,4 Previously, CD did not have an International Classification of Diseases (ICD) code and was likely under-recognized. An ICD-10 for CD was added, making CD and its variants easier to research for prevalence, characterization, mortality, and treatment.
After prolonged hospitalizations and extensive workup with no diagnosis, the patient’s clinical picture was most consistent with the lymphoproliferative disorder iMCD. However, iMCD is notable for polyclonal hypergammaglobulinemia, thrombocytosis, and mild anasarca. This patient had normal gammaglobulins, significant thrombocyotopenia, and profound, difficult-to-treat anasarca and ascites. Recognizing that the patient’s presentation did not fit neatly into a known clinical syndrome, an internet search was conducted based on his clinical features. This revealed TAFRO syndrome, which was at the time a newly described clinical syndrome with only a few published case reports. It was an internet search undertaken as a last resort that ultimately led to the patient’s diagnosis and successful treatment.
TEACHING POINTS
- Key clinical and pathologic features of TAFRO syndrome include thrombocytopenia, anasarca, fever, reticulin fibrosis and/or renal insufficiency, and organomegaly.
- TAFRO syndrome may be under-recognized due to very recent characterization and no previous ICD code for CD.
- TAFRO syndrome experts recommend immunosuppression for treatment of TAFRO syndrome, including glucocorticoids as first-line treatment.
- Internet searches can be helpful in the diagnosis of challenging cases, particularly with rare, unusual, and emerging diseases that have not yet been described in reference texts and only infrequently reported in the medical literature.
Disclosures
Jonathan S. Zipursky, Keri T. Holmes-Maybank, Steven L. Shumak, and Ashley A. Ducketthave none to declare.
1. Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol. 2016;91(2):220-226. PubMed
2. Takai K, Nikkuni K, Shibuya H, Hashidate H. Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly. Rinsho Ketsueki. 2010;51(5):320-325. PubMed
3. Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol. 2016;103:686-692. https://doi.org/10.1007/s12185-016-1979-1.
4. Liu AY, Nabel CS, Finkelman BS, et al. Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol. 2016;3:e163-e175. https://doi.org/10.1016/S2352-3026(16)00006-5.
5. Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924-2933. https://doi.org/10.1182/blood-2013-12-545087.
6. Sakashita K, Murata K, Takamori M. TAFRO syndrome: Current perspectives. J Blood Med. 2018;9:15-23. doi: 10.2147/JBM.S127822.
7. Louis C, Vijgen S, Samii K, et al. TAFRO syndrome in caucasians: A case report and review of the literature. Front Med. 2017;4(149):1-8. https://doi.org/10.3389/fmed.2017.00149.
8. Courtier F, Ruault NM, Crepin T, et al. A comparison of TAFRO syndrome between Japanese and non-Japanese cases: a case report and literature review. Ann Hematol. 2018;97:401-407. https://doi.org/10.1007/s00277-017-3138-z.
9. Jain P, Verstovsek S, Loghavi S, et al. Durable remission with rituximab in a patient with an unusual variant of Castleman’s disease with myelofibrosis-TAFRO syndrome. Am J Hematol. 2015;90(11):1091-1092. https://doi.org/10.1002/ajh.24015.
10. Igawa T, Sato Y. TAFRO syndeome. Hematol Oncol Clin N Am. 2018;32(1):107-118. https://doi.org/10.1016/j.hoc.2017.09.009.
11. Hawkins JM, Pillai V. TAFRO syndrome or Castleman-Kojima syndrome: a variant of multicentric Castleman disease. Blood. 2015;126(18):2163. https://doi.org/10.1182/blood-2015-07-662122.
A 22-year-old man presented to a Canadian community hospital emergency department complaining of 2-3 weeks of abdominal pain and bloating associated with early satiety. He also noted weight loss of 20 pounds over the preceding months, leg and abdominal swelling with increased girth, and 1-2 loose, nonbloody stools per day.
Early satiety and bloating are nonspecific symptoms that can be due to gastroesophageal reflux disease, peptic ulcer disease, gastrointestinal obstruction, or gastroparesis. Weight loss in a young person, particularly if >5% of body weight, is concerning for a serious underlying medical issue. It could reflect reduced intake due to anorexia, odynophagia, or dysphagia or increased energy expenditure due to an inflammatory state such as infection or rheumatic disease. The etiology of the swelling needs to be elucidated. It may be due to increased hydrostatic forces as in heart failure, venous or lymphatic obstruction, or from lowered oncotic pressure resulting from hepatic disease, nephrotic syndrome, severe malnutrition (nonbloody loose stools), or a protein losing enteropathy.
The patient was transferred to a tertiary care center for closer access to specialty consultation. He described generalized abdominal pain increasing in intensity over three weeks; bilateral lower extremity, scrotal, abdominal wall, and sacral edema; and mild dyspnea on exertion. The early satiety was not associated with dysphagia, odynophagia, nausea, or vomiting. He denied fevers, chills, night sweats, nausea, vomiting, jaundice, easy bruising, orthopnea, paroxysmal nocturnal dyspnea (PND), or chest pain. His past medical history included asthma treated with fluticasone/salmeterol and albuterol. He was a Canadian of East Asian descent working as a plumber. He previously smoked three to four cigarettes per day for six years. He stopped smoking one month before presentation. He had one alcoholic beverage per week and smoked marijuana weekly. He denied any family history of similar symptoms or malignancy.The differential diagnosis for weight loss and anasarca is broad and includes malignancies, infectious diseases, rheumatic or inflammatory disorders, malabsorption, and advanced cardiac, renal, or liver disease. His history does not classically point in one direction. The mild dyspnea on exertion may be due to cardiac disease, but it is unlikely in the absence of orthopnea and PND. The dyspnea could be due to increased abdominal pressure if ascites are present, his underlying asthma, or another etiology such as anemia. Fevers, chills, and/or night sweats can be expected in infections and some malignancies, but their absence does not exclude infections and malignancies from the differential diagnoses. Particular attention should be paid to lymphadenopathy on the physical examination. The presence of an umbilical nodule (Sister Mary Joseph sign) could indicate a malignancy (gastrointestinal or lymphoma).
The differential diagnosis for weight loss and anasarca is broad and includes malignancies, infectious diseases, rheumatic or inflammatory disorders, malabsorption, and advanced cardiac, renal, or liver disease. His history does not classically point in one direction. The mild dyspnea on exertion may be due to cardiac disease, but it is unlikely in the absence of orthopnea and PND. The dyspnea could be due to increased abdominal pressure if ascites are present, his underlying asthma, or another etiology such as anemia. Fevers, chills, and/or night sweats can be expected in infections and some malignancies, but their absence does not exclude infections and malignancies from the differential diagnoses. Particular attention should be paid to lymphadenopathy on the physical examination. The presence of an umbilical nodule (Sister Mary Joseph sign) could indicate a malignancy (gastrointestinal or lymphoma).
On physical examination, his temperature was 38.1°C, heart rate was 138 beats per minute, blood pressure was 123/86 mm Hg, respiratory rate was 20 breaths per minute, and oxygen saturation was 97% on room air. He appeared uncomfortable and diaphoretic. No scleral icterus or jaundice was appreciated. There were no palpable cervical, axillary, or inguinal lymph nodes. Cardiac examination revealed tachycardia and no murmurs, rubs, gallops, or jugular venous distention. Abdominal examination revealed abdominal distention, diffuse tenderness to deep palpation, bulging flanks, and a positive fluid wave. Liver and spleen could not be palpated or percussed secondary to abdominal distention. He had pitting bilateral lower extremity edema that extended to and included the scrotum. Neurologic and pulmonary examinations were unremarkable.
His examination reveals low-grade fever, tachycardia, and diaphoresis. Whether this represents progression of his primary disease or he has acutely developed a superimposed infection is uncertain at this point. He has notable anasarca but no jugular venous distention, crackles, or S3 gallop. The lack of evidence of pulmonary edema or increased central venous pressure on physical examination increases the likelihood of cirrhosis, hypoalbuminemia, or obstruction (lymphatic or venous) and decreases the likelihood of heart failure as the etiology of his peripheral edema and likely ascites. Despite the prominence of gastrointestinal symptoms, he has neither jaundice nor stigmata of chronic liver disease. Periorbital edema, which may be present in nephrotic syndrome, is also absent. Although he has no palpable peripheral lymphadenopathy, malignancy remains a concern.
Testing should include urinalysis for proteinuria and coagulation studies to assess synthetic function of the liver. Abdominal ultrasound is indicated to confirm ascites. If present, a diagnostic paracentesis should be performed to rule out spontaneous bacterial peritonitis and determine whether the ascites is from portal hypertension, hypoalbuminemia, or peritoneal disease. If the transaminases are elevated or if the ascitic fluid is concerning for malignancy, he will need a computed tomography (CT) of the abdomen and pelvis. A protein losing enteropathy due to malignancies (gastric cancer or lymphoma), rheumatic disease (systemic lupus erythematosus [SLE]), or infiltrative disease (amyloid) is also a possibility. If the other studies are unrevealing, stool should be sent for alpha-1 antitrypsin.
Laboratory studies revealed hemoglobin 7.8 g/dL, platelets 53 k/mm3, white blood cell count (WBC) 10.6 k/mm3, alkaline phosphatase (ALP) 217 U/L, albumin 2.7 g/dL, reticulocyte count 3 k/mm3 (reference range, 30-110 k/mm3), and ferritin 1,310 ng/mL (reference range, 20-400 ng/L). Serum aminotransferase levels, bilirubin, coagulation panel, electrolytes, and creatinine were normal. Urinalysis was negative for blood, leukocytes, and protein. Diagnostic paracentesis demonstrated a serum-ascites-albumin gradient (SAAG) of two and macrophage predominance (WBC 250 U/L). Ascitic fluid cytology and culture were negative. Blood cultures, human immunodeficiency virus (HIV)-1 and 2, cytomegalovirus (CMV), and Epstein–Barr virus (EBV) serologies were negative. Viral serologies for hepatitis A, B, and C were negative. Antinuclear antibody (ANA), anti-ds DNA, antineutrophilic cytoplasmic antibody (ANCA), serum angiotensin-converting enzyme (ACE) level, and quantitative immunoglobulin levels were all within the normal range. Chest, abdomen, and pelvis CT with contrast revealed large-volume abdominal and pelvic ascites, diffuse subcutaneous edema (Figure 1), modest hepatosplenomegaly, small bilateral pleural effusions, and mediastinal, axillary, mesenteric, periportal, peripancreatic, and retroperitoneal lymphadenopathy (Figure 2).
Malignancy is highest on the differential. In the absence of evidence of a primary tumor, a lymphoma would be the most likely diagnosis. Multicentric Castleman disease (MCD), a rare lymphoproliferative disorder with a clinical picture similar to lymphoma, should be considered.
Some of the more common viral etiologies of generalized lymphadenopathy and cytopenias are unlikely because serologies for HIV, hepatitis B and C, EBV, and CMV are negative. Tuberculosis fits with the insidious nature of his presentation and remains on the differential although a low SAAG would be expected. From a rheumatologic standpoint, the lack of characteristic findings on history and physical examination and the negative ANA and anti-ds DNA results make SLE unlikely. Although elevated in the majority of untreated sarcoid patients, a normal ACE level is not sufficient to rule out this diagnosis. IgG, IgA, and IgM levels would be low if there was significant gastrointestinal protein loss and elevated in MCD. The markedly increased ferritin level, an acute-phase reactant often elevated in the setting of inflammation or malignancy, raises suspicion for adult Still’s disease (despite the lack of characteristic arthralgias and/or rash) and hemophagocytic lymphohistiocytosis (HLH).
A SAAG greater than or equal to 1.1 indicates the presence of portal hypertension. Portal hypertension most often results from cirrhosis for which this patient has no apparent clinical findings. Etiologies of noncirrhotic portal hypertension are classified as prehepatic, intrahepatic, and posthepatic. There is no clinical or radiologic evidence of portal or splenic vein thrombosis (prehepatic) or heart failure (posthepatic). Possible intrahepatic etiologies include malignancy and sarcoid. Although uncommon, patients with malignancy-related ascites may have a high SAAG without coexisting cirrhosis. This occurs if there is portal hypertension due to extensive metastases in the liver or involvement of the portal venous system. The cytology of the ascitic fluid is negative. However, cytology is <80% sensitive in the absence of peritoneal carcinomatosis.
The most likely diagnosis at this point is lymphoma. Bone marrow biopsy is indicated to further assess his thrombocytopenia and hypoproliferative anemia and may be diagnostic for malignancy. Pathologic examination of a lymph node should be performed. Due to concern for lymphoproliferative disease, excisional biopsy is preferred to preserve tissue architecture.
Hematology was consulted for evaluation of the lymphadenopathy, anemia, and thrombocytopenia and recommended bone marrow and excisional lymph node biopsies. Bone marrow biopsy showed trilineage hypercellularity (Figure 3A) with reduced erythropoiesis and reticulin fibrosis (Figure 3B). An axillary lymph node biopsy with flow cytometry was nondiagnostic for a lymphoproliferative disorder or malignancy.
Both biopsies fail to provide a definitive diagnosis. Hypercellularity in the marrow (>70% cellularity) and reticulin fibrosis are nonspecific and could be from a malignant or reactive disease process. Lymphoma remains the most likely diagnosis. Peripheral blood for flow cytometry, lactate dehydrogenase (LDH), and uric acid should be sent. A repeat excisional biopsy of another lymph node should be performed.
Gastroenterology was consulted to evaluate the loose stools, anasarca, and hepatomegaly, and esophagogastroduodenoscopy, enteroscopy, and colonoscopy with biopsies were performed. Gastric biopsy revealed mild gastropathy. Duodenal, jejunal, and right and left colon biopsies were all normal. A liver biopsy was performed and revealed periportal inflammation. Rheumatology and infectious disease consultations did not suspect that the patient had a rheumatologic or infectious disease.
After appropriate workup and no definitive diagnosis, it is important to reassess the patient for overall stability and the presence of any new or changing symptoms (worsening symptoms, persistent fevers) that could direct further evaluation. Lymphoma remains on the differential despite multiple negative biopsies, but other less common diseases that mimic lymphoma and cause multisystem disease should be investigated. Review of the previous lymph node and tissue biopsies with the pathologist and hematologist should focus on features of adult Still’s disease (paracortical immunoblastic hyperplasia), MCD (histopathology of angiofollicular lymph node hyperplasia and presence of human herpes virus-8 (HHV-8), and HLH (hemophagocytosis). A positron emission tomography scan may not distinguish between malignancy and other fluorodeoxyglucose avid inflammatory processes but is recommended to determine the site of a future excisional lymph node biopsy.
A 10-day trial of prednisone 50 mg daily was initiated for presumed lymphoma. He experienced symptomatic improvement with decreased peripheral edema and ascites and resolution of his fevers. He was discharged home seven days after completing steroids with follow-up.
Five days after discharge, he was readmitted with worsening anasarca, massive ascites, and acute kidney injury. Admission laboratory studies revealed creatinine 1.66 mg/dL, hemoglobin 11.5 g/dL, and platelets 94 k/mm3. In addition, his ferritin level was 1,907 ng/L (reference range, 20-400 ng/L), erythrocyte sedimentation rate (ESR) was 50 mm/h (reference range, 0-20 mm/h), and C-reactive protein concentration (CRP) was 12.1 mg/dL (reference range, 0-0.5 mg/dL).
Steroids are used to treat a wide variety of illnesses, some of which are still under consideration in this patient including lymphoma, MCD, adult Still’s disease, and HLH. His symptoms recurred quickly after discontinuation of steroids in the setting of elevated ferritin, ESR, and CRP levels reflecting marked ongoing inflammation. Serologic testing for soluble IL-2 receptor, often elevated in MCD and HLH, should be performed. Excisional biopsy of an accessible node should be performed urgently.
His acute kidney injury resolved; however, he continued to have intermittent fevers, anemia, thromobocytopenia, lymphadenopathy, and hepatosplenomegaly. A hematology case-conference recommended testing for HLH, including soluble IL-2 receptor (CD25), soluble CD163, and natural killer cell degranulation assay, all of which were negative. A right inguinal lymph node biopsy revealed reactive lymphoid tissue and stained negative for HHV-8. Based on the lack of an alternative diagnosis (particularly lymphoma), the presence of multiple areas of lymphadenopathy, anemia, fevers, organomegaly, weight loss, reactive lymphoid tissue on lymph node biopsy, and elevated CRP and ESR, a working diagnosis of MCD was made. The negative HHV-8 testing was consistent with idiopathic MCD (iMCD); however, features inconsistent with iMCD included lack of polyclonal hypergammaglobulinemia and the presence of significant anasarca and thrombocytopenia. Therefore, an internet search was performed using the patient’s salient symptoms and findings. The search revealed a few recently published case reports of a rare variant of iMCD, TAFRO syndrome. TAFRO syndrome, characterized by thrombocytopenia, anasarca, fever, reticulin fibrosis and/or renal insufficiency, and organomegaly, fully explained the patient’s presentation. He was started on prednisone, rituximab (anti-CD20 antibody), and furosemide. After one month of treatment, he showed complete resolution of cytopenias, lymphadenopathy, organomegaly, anasarca, and ascites. Therapy continued for approximately three months, and he has remained symptom-free.
COMMENTARY
Castleman’s disease (CD) is a rare lymphoproliferative disorder divided into unicentric (solitary enlarged lymph node) and multicentric (multifocal enlarged lymph nodes).1 MCD typically presents with systemic inflammation, reactive proliferation of benign lymphocytes, multifocal lymphadenopathy, elevated inflammatory markers, anemia, hypoalbuminemia, and polyclonal gammaglobulinemia.1 It is hypothesized that HHV-8 drives the systemic inflammation of MCD via high levels of interleukin-6 (IL-6) activity.1 iMCD is an HHV-8-negative variant of MCD.1
TAFRO syndrome was first described in 2010 in three Japanese patients demonstrating high fever, anasarca, hepatosplenomegaly, lymphadenopathy, severe thrombocytopenia, and reticulin fibrosis.2 In 2015, the All Japan TAFRO Syndrome Research Group recognized TAFRO syndrome as a variant of iMCD and created diagnostic criteria and a severity classification system.3 Major criteria consist of anasarca, including pleural effusion and/or ascites identified on CT scan and general edema, thrombocytopenia (platelet count <100 k/mm3), and systemic inflammation (fever >37.5°C and/or serum CRP greater than or equal to 2 mg/dL).3 Two of four minor criteria must be met, which include (1) lymph node histology consistent with CD, (2) reticulin myelofibrosis and/or increased number of megakaryocytes in bone marrow, (3) mild organomegaly, including hepatomegaly, splenomegaly, and lymphadenopathy <1.5 cm in diameter identified on CT scan, and (4) progressive renal insufficiency (serum creatinine >1.2 mg/dL in males or >1.0 mg/dL in females).3 In addition, several patients with TAFRO syndrome demonstrate elevated ALP, low-normal LDH, elevated vascular endothelial growth factor, elevated IL-6, microcytic anemia, and slight polyclonal hypergammopathy.3 Malignancies such as lymphoma and myeloma, autoimmune diseases such as SLE and ANCA-associated vasculitis, infectious diseases such as those caused by mycobacteria, and POEMS (polyneuropathy, organomegaly, endocrine diseases, M-protein, and skin lesions) syndrome must be excluded to diagnose TAFRO syndrome.3,4
The pathophysiology of TAFRO syndrome is unknown, and it is unclear whether the syndrome is truly a variant of iMCD or a distinct entity.3 IL-6 is typically only mildly elevated in TAFRO syndrome, without the consequent thrombocytosis and polyclonal hypergammaglobulinemia seen in MCD, which is associated with higher levels of IL-6.1 Multiple non-HHV-8 mechanisms for TAFRO syndrome have been proposed, including (1) systemic inflammation, autoimmune/autoinflammatory mechanisms, (2) neoplastic, ectopic cytokine secretion by malignant or benign tumor cells, and/or (3) infectious, such as non-HHV-8 virus.5
Immunosuppression is the mainstay of treatment for TAFRO syndrome based on recommendations from the 2015 TAFRO Research Group.3 Glucocorticoids are considered first-line therapy.3 Cyclosporin A is recommended for individuals refractory to glucocorticoids.3 In patients with a contraindication to cyclosporin A, anti-IL-6 receptor antibodies such as tocilizumab (approved for treatment of iMCD in Japan) and siltuximab (approved for treatment of iMCD in North America and Europe) or the anti-CD20 antibody rituximab should be prescribed.3 There is evidence for the thrombopoietin receptor agonists romiplostim and eltrombopag to treat persistent thrombocytopenia.3 Additional treatments for refractory TAFRO syndrome include IVIG and plasma exchange, chemotherapy (cyclophosphamide, doxorubicin, vincristine, prednisolone), and thalidomide.3,6
Little is known about the epidemiologic characterization of TAFRO syndrome as less than 40 cases of TAFRO syndrome have been reported in the United States, Asia, and Europe.
1,4,7-9 TAFRO syndrome occurs primarily in the fourth and fifth decades of life, with case reports ranging from 14 to 78 years of age.1,3,10,11 Gender distribution varies but is likely equal for males and females.3 Mortality in TAFRO syndrome is estimated at 11%-12%.1,3 Over the past several years, a North American and European patient registry and natural history study for CD, ACCELERATE, has been initiated.4 In addition, the international Castleman Disease Collaborative Network, a Japanese multicenter retrospective study for MCD, and a nationwide Japanese research team for CD have been created.3,4 Previously, CD did not have an International Classification of Diseases (ICD) code and was likely under-recognized. An ICD-10 for CD was added, making CD and its variants easier to research for prevalence, characterization, mortality, and treatment.
After prolonged hospitalizations and extensive workup with no diagnosis, the patient’s clinical picture was most consistent with the lymphoproliferative disorder iMCD. However, iMCD is notable for polyclonal hypergammaglobulinemia, thrombocytosis, and mild anasarca. This patient had normal gammaglobulins, significant thrombocyotopenia, and profound, difficult-to-treat anasarca and ascites. Recognizing that the patient’s presentation did not fit neatly into a known clinical syndrome, an internet search was conducted based on his clinical features. This revealed TAFRO syndrome, which was at the time a newly described clinical syndrome with only a few published case reports. It was an internet search undertaken as a last resort that ultimately led to the patient’s diagnosis and successful treatment.
TEACHING POINTS
- Key clinical and pathologic features of TAFRO syndrome include thrombocytopenia, anasarca, fever, reticulin fibrosis and/or renal insufficiency, and organomegaly.
- TAFRO syndrome may be under-recognized due to very recent characterization and no previous ICD code for CD.
- TAFRO syndrome experts recommend immunosuppression for treatment of TAFRO syndrome, including glucocorticoids as first-line treatment.
- Internet searches can be helpful in the diagnosis of challenging cases, particularly with rare, unusual, and emerging diseases that have not yet been described in reference texts and only infrequently reported in the medical literature.
Disclosures
Jonathan S. Zipursky, Keri T. Holmes-Maybank, Steven L. Shumak, and Ashley A. Ducketthave none to declare.
A 22-year-old man presented to a Canadian community hospital emergency department complaining of 2-3 weeks of abdominal pain and bloating associated with early satiety. He also noted weight loss of 20 pounds over the preceding months, leg and abdominal swelling with increased girth, and 1-2 loose, nonbloody stools per day.
Early satiety and bloating are nonspecific symptoms that can be due to gastroesophageal reflux disease, peptic ulcer disease, gastrointestinal obstruction, or gastroparesis. Weight loss in a young person, particularly if >5% of body weight, is concerning for a serious underlying medical issue. It could reflect reduced intake due to anorexia, odynophagia, or dysphagia or increased energy expenditure due to an inflammatory state such as infection or rheumatic disease. The etiology of the swelling needs to be elucidated. It may be due to increased hydrostatic forces as in heart failure, venous or lymphatic obstruction, or from lowered oncotic pressure resulting from hepatic disease, nephrotic syndrome, severe malnutrition (nonbloody loose stools), or a protein losing enteropathy.
The patient was transferred to a tertiary care center for closer access to specialty consultation. He described generalized abdominal pain increasing in intensity over three weeks; bilateral lower extremity, scrotal, abdominal wall, and sacral edema; and mild dyspnea on exertion. The early satiety was not associated with dysphagia, odynophagia, nausea, or vomiting. He denied fevers, chills, night sweats, nausea, vomiting, jaundice, easy bruising, orthopnea, paroxysmal nocturnal dyspnea (PND), or chest pain. His past medical history included asthma treated with fluticasone/salmeterol and albuterol. He was a Canadian of East Asian descent working as a plumber. He previously smoked three to four cigarettes per day for six years. He stopped smoking one month before presentation. He had one alcoholic beverage per week and smoked marijuana weekly. He denied any family history of similar symptoms or malignancy.The differential diagnosis for weight loss and anasarca is broad and includes malignancies, infectious diseases, rheumatic or inflammatory disorders, malabsorption, and advanced cardiac, renal, or liver disease. His history does not classically point in one direction. The mild dyspnea on exertion may be due to cardiac disease, but it is unlikely in the absence of orthopnea and PND. The dyspnea could be due to increased abdominal pressure if ascites are present, his underlying asthma, or another etiology such as anemia. Fevers, chills, and/or night sweats can be expected in infections and some malignancies, but their absence does not exclude infections and malignancies from the differential diagnoses. Particular attention should be paid to lymphadenopathy on the physical examination. The presence of an umbilical nodule (Sister Mary Joseph sign) could indicate a malignancy (gastrointestinal or lymphoma).
The differential diagnosis for weight loss and anasarca is broad and includes malignancies, infectious diseases, rheumatic or inflammatory disorders, malabsorption, and advanced cardiac, renal, or liver disease. His history does not classically point in one direction. The mild dyspnea on exertion may be due to cardiac disease, but it is unlikely in the absence of orthopnea and PND. The dyspnea could be due to increased abdominal pressure if ascites are present, his underlying asthma, or another etiology such as anemia. Fevers, chills, and/or night sweats can be expected in infections and some malignancies, but their absence does not exclude infections and malignancies from the differential diagnoses. Particular attention should be paid to lymphadenopathy on the physical examination. The presence of an umbilical nodule (Sister Mary Joseph sign) could indicate a malignancy (gastrointestinal or lymphoma).
On physical examination, his temperature was 38.1°C, heart rate was 138 beats per minute, blood pressure was 123/86 mm Hg, respiratory rate was 20 breaths per minute, and oxygen saturation was 97% on room air. He appeared uncomfortable and diaphoretic. No scleral icterus or jaundice was appreciated. There were no palpable cervical, axillary, or inguinal lymph nodes. Cardiac examination revealed tachycardia and no murmurs, rubs, gallops, or jugular venous distention. Abdominal examination revealed abdominal distention, diffuse tenderness to deep palpation, bulging flanks, and a positive fluid wave. Liver and spleen could not be palpated or percussed secondary to abdominal distention. He had pitting bilateral lower extremity edema that extended to and included the scrotum. Neurologic and pulmonary examinations were unremarkable.
His examination reveals low-grade fever, tachycardia, and diaphoresis. Whether this represents progression of his primary disease or he has acutely developed a superimposed infection is uncertain at this point. He has notable anasarca but no jugular venous distention, crackles, or S3 gallop. The lack of evidence of pulmonary edema or increased central venous pressure on physical examination increases the likelihood of cirrhosis, hypoalbuminemia, or obstruction (lymphatic or venous) and decreases the likelihood of heart failure as the etiology of his peripheral edema and likely ascites. Despite the prominence of gastrointestinal symptoms, he has neither jaundice nor stigmata of chronic liver disease. Periorbital edema, which may be present in nephrotic syndrome, is also absent. Although he has no palpable peripheral lymphadenopathy, malignancy remains a concern.
Testing should include urinalysis for proteinuria and coagulation studies to assess synthetic function of the liver. Abdominal ultrasound is indicated to confirm ascites. If present, a diagnostic paracentesis should be performed to rule out spontaneous bacterial peritonitis and determine whether the ascites is from portal hypertension, hypoalbuminemia, or peritoneal disease. If the transaminases are elevated or if the ascitic fluid is concerning for malignancy, he will need a computed tomography (CT) of the abdomen and pelvis. A protein losing enteropathy due to malignancies (gastric cancer or lymphoma), rheumatic disease (systemic lupus erythematosus [SLE]), or infiltrative disease (amyloid) is also a possibility. If the other studies are unrevealing, stool should be sent for alpha-1 antitrypsin.
Laboratory studies revealed hemoglobin 7.8 g/dL, platelets 53 k/mm3, white blood cell count (WBC) 10.6 k/mm3, alkaline phosphatase (ALP) 217 U/L, albumin 2.7 g/dL, reticulocyte count 3 k/mm3 (reference range, 30-110 k/mm3), and ferritin 1,310 ng/mL (reference range, 20-400 ng/L). Serum aminotransferase levels, bilirubin, coagulation panel, electrolytes, and creatinine were normal. Urinalysis was negative for blood, leukocytes, and protein. Diagnostic paracentesis demonstrated a serum-ascites-albumin gradient (SAAG) of two and macrophage predominance (WBC 250 U/L). Ascitic fluid cytology and culture were negative. Blood cultures, human immunodeficiency virus (HIV)-1 and 2, cytomegalovirus (CMV), and Epstein–Barr virus (EBV) serologies were negative. Viral serologies for hepatitis A, B, and C were negative. Antinuclear antibody (ANA), anti-ds DNA, antineutrophilic cytoplasmic antibody (ANCA), serum angiotensin-converting enzyme (ACE) level, and quantitative immunoglobulin levels were all within the normal range. Chest, abdomen, and pelvis CT with contrast revealed large-volume abdominal and pelvic ascites, diffuse subcutaneous edema (Figure 1), modest hepatosplenomegaly, small bilateral pleural effusions, and mediastinal, axillary, mesenteric, periportal, peripancreatic, and retroperitoneal lymphadenopathy (Figure 2).
Malignancy is highest on the differential. In the absence of evidence of a primary tumor, a lymphoma would be the most likely diagnosis. Multicentric Castleman disease (MCD), a rare lymphoproliferative disorder with a clinical picture similar to lymphoma, should be considered.
Some of the more common viral etiologies of generalized lymphadenopathy and cytopenias are unlikely because serologies for HIV, hepatitis B and C, EBV, and CMV are negative. Tuberculosis fits with the insidious nature of his presentation and remains on the differential although a low SAAG would be expected. From a rheumatologic standpoint, the lack of characteristic findings on history and physical examination and the negative ANA and anti-ds DNA results make SLE unlikely. Although elevated in the majority of untreated sarcoid patients, a normal ACE level is not sufficient to rule out this diagnosis. IgG, IgA, and IgM levels would be low if there was significant gastrointestinal protein loss and elevated in MCD. The markedly increased ferritin level, an acute-phase reactant often elevated in the setting of inflammation or malignancy, raises suspicion for adult Still’s disease (despite the lack of characteristic arthralgias and/or rash) and hemophagocytic lymphohistiocytosis (HLH).
A SAAG greater than or equal to 1.1 indicates the presence of portal hypertension. Portal hypertension most often results from cirrhosis for which this patient has no apparent clinical findings. Etiologies of noncirrhotic portal hypertension are classified as prehepatic, intrahepatic, and posthepatic. There is no clinical or radiologic evidence of portal or splenic vein thrombosis (prehepatic) or heart failure (posthepatic). Possible intrahepatic etiologies include malignancy and sarcoid. Although uncommon, patients with malignancy-related ascites may have a high SAAG without coexisting cirrhosis. This occurs if there is portal hypertension due to extensive metastases in the liver or involvement of the portal venous system. The cytology of the ascitic fluid is negative. However, cytology is <80% sensitive in the absence of peritoneal carcinomatosis.
The most likely diagnosis at this point is lymphoma. Bone marrow biopsy is indicated to further assess his thrombocytopenia and hypoproliferative anemia and may be diagnostic for malignancy. Pathologic examination of a lymph node should be performed. Due to concern for lymphoproliferative disease, excisional biopsy is preferred to preserve tissue architecture.
Hematology was consulted for evaluation of the lymphadenopathy, anemia, and thrombocytopenia and recommended bone marrow and excisional lymph node biopsies. Bone marrow biopsy showed trilineage hypercellularity (Figure 3A) with reduced erythropoiesis and reticulin fibrosis (Figure 3B). An axillary lymph node biopsy with flow cytometry was nondiagnostic for a lymphoproliferative disorder or malignancy.
Both biopsies fail to provide a definitive diagnosis. Hypercellularity in the marrow (>70% cellularity) and reticulin fibrosis are nonspecific and could be from a malignant or reactive disease process. Lymphoma remains the most likely diagnosis. Peripheral blood for flow cytometry, lactate dehydrogenase (LDH), and uric acid should be sent. A repeat excisional biopsy of another lymph node should be performed.
Gastroenterology was consulted to evaluate the loose stools, anasarca, and hepatomegaly, and esophagogastroduodenoscopy, enteroscopy, and colonoscopy with biopsies were performed. Gastric biopsy revealed mild gastropathy. Duodenal, jejunal, and right and left colon biopsies were all normal. A liver biopsy was performed and revealed periportal inflammation. Rheumatology and infectious disease consultations did not suspect that the patient had a rheumatologic or infectious disease.
After appropriate workup and no definitive diagnosis, it is important to reassess the patient for overall stability and the presence of any new or changing symptoms (worsening symptoms, persistent fevers) that could direct further evaluation. Lymphoma remains on the differential despite multiple negative biopsies, but other less common diseases that mimic lymphoma and cause multisystem disease should be investigated. Review of the previous lymph node and tissue biopsies with the pathologist and hematologist should focus on features of adult Still’s disease (paracortical immunoblastic hyperplasia), MCD (histopathology of angiofollicular lymph node hyperplasia and presence of human herpes virus-8 (HHV-8), and HLH (hemophagocytosis). A positron emission tomography scan may not distinguish between malignancy and other fluorodeoxyglucose avid inflammatory processes but is recommended to determine the site of a future excisional lymph node biopsy.
A 10-day trial of prednisone 50 mg daily was initiated for presumed lymphoma. He experienced symptomatic improvement with decreased peripheral edema and ascites and resolution of his fevers. He was discharged home seven days after completing steroids with follow-up.
Five days after discharge, he was readmitted with worsening anasarca, massive ascites, and acute kidney injury. Admission laboratory studies revealed creatinine 1.66 mg/dL, hemoglobin 11.5 g/dL, and platelets 94 k/mm3. In addition, his ferritin level was 1,907 ng/L (reference range, 20-400 ng/L), erythrocyte sedimentation rate (ESR) was 50 mm/h (reference range, 0-20 mm/h), and C-reactive protein concentration (CRP) was 12.1 mg/dL (reference range, 0-0.5 mg/dL).
Steroids are used to treat a wide variety of illnesses, some of which are still under consideration in this patient including lymphoma, MCD, adult Still’s disease, and HLH. His symptoms recurred quickly after discontinuation of steroids in the setting of elevated ferritin, ESR, and CRP levels reflecting marked ongoing inflammation. Serologic testing for soluble IL-2 receptor, often elevated in MCD and HLH, should be performed. Excisional biopsy of an accessible node should be performed urgently.
His acute kidney injury resolved; however, he continued to have intermittent fevers, anemia, thromobocytopenia, lymphadenopathy, and hepatosplenomegaly. A hematology case-conference recommended testing for HLH, including soluble IL-2 receptor (CD25), soluble CD163, and natural killer cell degranulation assay, all of which were negative. A right inguinal lymph node biopsy revealed reactive lymphoid tissue and stained negative for HHV-8. Based on the lack of an alternative diagnosis (particularly lymphoma), the presence of multiple areas of lymphadenopathy, anemia, fevers, organomegaly, weight loss, reactive lymphoid tissue on lymph node biopsy, and elevated CRP and ESR, a working diagnosis of MCD was made. The negative HHV-8 testing was consistent with idiopathic MCD (iMCD); however, features inconsistent with iMCD included lack of polyclonal hypergammaglobulinemia and the presence of significant anasarca and thrombocytopenia. Therefore, an internet search was performed using the patient’s salient symptoms and findings. The search revealed a few recently published case reports of a rare variant of iMCD, TAFRO syndrome. TAFRO syndrome, characterized by thrombocytopenia, anasarca, fever, reticulin fibrosis and/or renal insufficiency, and organomegaly, fully explained the patient’s presentation. He was started on prednisone, rituximab (anti-CD20 antibody), and furosemide. After one month of treatment, he showed complete resolution of cytopenias, lymphadenopathy, organomegaly, anasarca, and ascites. Therapy continued for approximately three months, and he has remained symptom-free.
COMMENTARY
Castleman’s disease (CD) is a rare lymphoproliferative disorder divided into unicentric (solitary enlarged lymph node) and multicentric (multifocal enlarged lymph nodes).1 MCD typically presents with systemic inflammation, reactive proliferation of benign lymphocytes, multifocal lymphadenopathy, elevated inflammatory markers, anemia, hypoalbuminemia, and polyclonal gammaglobulinemia.1 It is hypothesized that HHV-8 drives the systemic inflammation of MCD via high levels of interleukin-6 (IL-6) activity.1 iMCD is an HHV-8-negative variant of MCD.1
TAFRO syndrome was first described in 2010 in three Japanese patients demonstrating high fever, anasarca, hepatosplenomegaly, lymphadenopathy, severe thrombocytopenia, and reticulin fibrosis.2 In 2015, the All Japan TAFRO Syndrome Research Group recognized TAFRO syndrome as a variant of iMCD and created diagnostic criteria and a severity classification system.3 Major criteria consist of anasarca, including pleural effusion and/or ascites identified on CT scan and general edema, thrombocytopenia (platelet count <100 k/mm3), and systemic inflammation (fever >37.5°C and/or serum CRP greater than or equal to 2 mg/dL).3 Two of four minor criteria must be met, which include (1) lymph node histology consistent with CD, (2) reticulin myelofibrosis and/or increased number of megakaryocytes in bone marrow, (3) mild organomegaly, including hepatomegaly, splenomegaly, and lymphadenopathy <1.5 cm in diameter identified on CT scan, and (4) progressive renal insufficiency (serum creatinine >1.2 mg/dL in males or >1.0 mg/dL in females).3 In addition, several patients with TAFRO syndrome demonstrate elevated ALP, low-normal LDH, elevated vascular endothelial growth factor, elevated IL-6, microcytic anemia, and slight polyclonal hypergammopathy.3 Malignancies such as lymphoma and myeloma, autoimmune diseases such as SLE and ANCA-associated vasculitis, infectious diseases such as those caused by mycobacteria, and POEMS (polyneuropathy, organomegaly, endocrine diseases, M-protein, and skin lesions) syndrome must be excluded to diagnose TAFRO syndrome.3,4
The pathophysiology of TAFRO syndrome is unknown, and it is unclear whether the syndrome is truly a variant of iMCD or a distinct entity.3 IL-6 is typically only mildly elevated in TAFRO syndrome, without the consequent thrombocytosis and polyclonal hypergammaglobulinemia seen in MCD, which is associated with higher levels of IL-6.1 Multiple non-HHV-8 mechanisms for TAFRO syndrome have been proposed, including (1) systemic inflammation, autoimmune/autoinflammatory mechanisms, (2) neoplastic, ectopic cytokine secretion by malignant or benign tumor cells, and/or (3) infectious, such as non-HHV-8 virus.5
Immunosuppression is the mainstay of treatment for TAFRO syndrome based on recommendations from the 2015 TAFRO Research Group.3 Glucocorticoids are considered first-line therapy.3 Cyclosporin A is recommended for individuals refractory to glucocorticoids.3 In patients with a contraindication to cyclosporin A, anti-IL-6 receptor antibodies such as tocilizumab (approved for treatment of iMCD in Japan) and siltuximab (approved for treatment of iMCD in North America and Europe) or the anti-CD20 antibody rituximab should be prescribed.3 There is evidence for the thrombopoietin receptor agonists romiplostim and eltrombopag to treat persistent thrombocytopenia.3 Additional treatments for refractory TAFRO syndrome include IVIG and plasma exchange, chemotherapy (cyclophosphamide, doxorubicin, vincristine, prednisolone), and thalidomide.3,6
Little is known about the epidemiologic characterization of TAFRO syndrome as less than 40 cases of TAFRO syndrome have been reported in the United States, Asia, and Europe.
1,4,7-9 TAFRO syndrome occurs primarily in the fourth and fifth decades of life, with case reports ranging from 14 to 78 years of age.1,3,10,11 Gender distribution varies but is likely equal for males and females.3 Mortality in TAFRO syndrome is estimated at 11%-12%.1,3 Over the past several years, a North American and European patient registry and natural history study for CD, ACCELERATE, has been initiated.4 In addition, the international Castleman Disease Collaborative Network, a Japanese multicenter retrospective study for MCD, and a nationwide Japanese research team for CD have been created.3,4 Previously, CD did not have an International Classification of Diseases (ICD) code and was likely under-recognized. An ICD-10 for CD was added, making CD and its variants easier to research for prevalence, characterization, mortality, and treatment.
After prolonged hospitalizations and extensive workup with no diagnosis, the patient’s clinical picture was most consistent with the lymphoproliferative disorder iMCD. However, iMCD is notable for polyclonal hypergammaglobulinemia, thrombocytosis, and mild anasarca. This patient had normal gammaglobulins, significant thrombocyotopenia, and profound, difficult-to-treat anasarca and ascites. Recognizing that the patient’s presentation did not fit neatly into a known clinical syndrome, an internet search was conducted based on his clinical features. This revealed TAFRO syndrome, which was at the time a newly described clinical syndrome with only a few published case reports. It was an internet search undertaken as a last resort that ultimately led to the patient’s diagnosis and successful treatment.
TEACHING POINTS
- Key clinical and pathologic features of TAFRO syndrome include thrombocytopenia, anasarca, fever, reticulin fibrosis and/or renal insufficiency, and organomegaly.
- TAFRO syndrome may be under-recognized due to very recent characterization and no previous ICD code for CD.
- TAFRO syndrome experts recommend immunosuppression for treatment of TAFRO syndrome, including glucocorticoids as first-line treatment.
- Internet searches can be helpful in the diagnosis of challenging cases, particularly with rare, unusual, and emerging diseases that have not yet been described in reference texts and only infrequently reported in the medical literature.
Disclosures
Jonathan S. Zipursky, Keri T. Holmes-Maybank, Steven L. Shumak, and Ashley A. Ducketthave none to declare.
1. Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol. 2016;91(2):220-226. PubMed
2. Takai K, Nikkuni K, Shibuya H, Hashidate H. Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly. Rinsho Ketsueki. 2010;51(5):320-325. PubMed
3. Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol. 2016;103:686-692. https://doi.org/10.1007/s12185-016-1979-1.
4. Liu AY, Nabel CS, Finkelman BS, et al. Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol. 2016;3:e163-e175. https://doi.org/10.1016/S2352-3026(16)00006-5.
5. Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924-2933. https://doi.org/10.1182/blood-2013-12-545087.
6. Sakashita K, Murata K, Takamori M. TAFRO syndrome: Current perspectives. J Blood Med. 2018;9:15-23. doi: 10.2147/JBM.S127822.
7. Louis C, Vijgen S, Samii K, et al. TAFRO syndrome in caucasians: A case report and review of the literature. Front Med. 2017;4(149):1-8. https://doi.org/10.3389/fmed.2017.00149.
8. Courtier F, Ruault NM, Crepin T, et al. A comparison of TAFRO syndrome between Japanese and non-Japanese cases: a case report and literature review. Ann Hematol. 2018;97:401-407. https://doi.org/10.1007/s00277-017-3138-z.
9. Jain P, Verstovsek S, Loghavi S, et al. Durable remission with rituximab in a patient with an unusual variant of Castleman’s disease with myelofibrosis-TAFRO syndrome. Am J Hematol. 2015;90(11):1091-1092. https://doi.org/10.1002/ajh.24015.
10. Igawa T, Sato Y. TAFRO syndeome. Hematol Oncol Clin N Am. 2018;32(1):107-118. https://doi.org/10.1016/j.hoc.2017.09.009.
11. Hawkins JM, Pillai V. TAFRO syndrome or Castleman-Kojima syndrome: a variant of multicentric Castleman disease. Blood. 2015;126(18):2163. https://doi.org/10.1182/blood-2015-07-662122.
1. Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol. 2016;91(2):220-226. PubMed
2. Takai K, Nikkuni K, Shibuya H, Hashidate H. Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly. Rinsho Ketsueki. 2010;51(5):320-325. PubMed
3. Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol. 2016;103:686-692. https://doi.org/10.1007/s12185-016-1979-1.
4. Liu AY, Nabel CS, Finkelman BS, et al. Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol. 2016;3:e163-e175. https://doi.org/10.1016/S2352-3026(16)00006-5.
5. Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924-2933. https://doi.org/10.1182/blood-2013-12-545087.
6. Sakashita K, Murata K, Takamori M. TAFRO syndrome: Current perspectives. J Blood Med. 2018;9:15-23. doi: 10.2147/JBM.S127822.
7. Louis C, Vijgen S, Samii K, et al. TAFRO syndrome in caucasians: A case report and review of the literature. Front Med. 2017;4(149):1-8. https://doi.org/10.3389/fmed.2017.00149.
8. Courtier F, Ruault NM, Crepin T, et al. A comparison of TAFRO syndrome between Japanese and non-Japanese cases: a case report and literature review. Ann Hematol. 2018;97:401-407. https://doi.org/10.1007/s00277-017-3138-z.
9. Jain P, Verstovsek S, Loghavi S, et al. Durable remission with rituximab in a patient with an unusual variant of Castleman’s disease with myelofibrosis-TAFRO syndrome. Am J Hematol. 2015;90(11):1091-1092. https://doi.org/10.1002/ajh.24015.
10. Igawa T, Sato Y. TAFRO syndeome. Hematol Oncol Clin N Am. 2018;32(1):107-118. https://doi.org/10.1016/j.hoc.2017.09.009.
11. Hawkins JM, Pillai V. TAFRO syndrome or Castleman-Kojima syndrome: a variant of multicentric Castleman disease. Blood. 2015;126(18):2163. https://doi.org/10.1182/blood-2015-07-662122.
© 2019 Society of Hospital Medicine