User login
Fever, tachycardia, and tachypnea during a psychotic exacerbation
CASE Posing a threat to his family
Mr. C, age 23, who was diagnosed with schizophrenia with daily auditory hallucinations 4 years earlier, is transferred from an outside psychiatric hospital to our emergency department (ED) after developing fever, tachycardia, headache, and nasal congestion for the past day. He had been admitted to the psychiatric hospital 3 weeks ago due to concerns he was experiencing increased hallucinations and delusions and posed a threat to his sister and her children, with whom he had been living.
Mr. C tells us that while at the psychiatric hospital, he had been started on clozapine, 250 mg/d. He said that prior to clozapine, he had been taking risperidone. We are unable to confirm past treatment information with the psychiatric hospital, including exactly when the clozapine had been started or how fast it had been titrated. We also were not able to obtain information on his prior medication regimen.
In the ED, Mr. C is febrile (39.4°C; 102.9°F), tachycardic (160 beats per minute; reference range 60 to 100), and tachypneic (24 breaths per minute; reference range 12 to 20). His blood pressure is 130/68 mm Hg, and his lactate level is 2.3 mmol/L (reference range <1.9 mmol/L). After he receives 3 liters of fluid, Mr. C’s heart rate decreases to 117 and his lactate level to 1.1 mmol/L. His white blood cell count is 10.6 × 103/mm3 (reference range 4.0 to 10.0 × 103/mm3); a differential can be found in the Table. His electrocardiogram (ECG) demonstrates sinus tachycardia and a QTc of 510 ms (reference range <430 ms), but is otherwise unremarkable. His creatinine kinase (CK) level is within normal limits at 76 U/L (reference range 52 to 336 U/L). A C-reactive protein (CRP) level was not drawn at this time. Other than marijuana and cocaine use, Mr. C’s medical history is unremarkable.

Mr. C is admitted to the hospital and is started on treatment for sepsis. On the evening of Day 1, Mr. C experiences worsening tachycardia (140 beats per minute) and tachypnea (≥40 breaths per minute). His temperature increases to 103.3°F, and his blood pressure drops to 97/55 mm Hg. His troponin level is 19.0 ng/mL (reference range <0.01 ng/mL) and CK level is 491 U/L.
As Mr. C continues to deteriorate, a rapid response is called and he is placed on non-rebreather oxygen and transferred to the medical intensive care unit (MICU).
[polldaddy:10226034]
The authors’ observations
With Mr. C’s presenting symptoms, multiple conditions were included in the differential diagnosis. The initial concern was for sepsis. Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.1 Organ dysfunction is defined by a quick Sepsis-Related Organ Failure Assessment (qSOFA) score ≥2 and is associated with an increased probability of mortality (>10%). Although no infection had been identified in Mr. C, the combination of fever, altered vital signs, and elevated lactate level in the setting of a qSOFA score of 2 (for respiratory rate and blood pressure) raised suspicion enough to start empiric treatment.
With Mr. C’s subsequent deterioration on the evening of Day 1, we considered cardiopulmonary etiologies. His symptoms of dyspnea, hypotension, tachycardia, tachypnea, and fever were nonspecific and thus required consideration of multiple life-threatening etiologies. Thygesen et al2 published an expert consensus of the definition of myocardial infarction, which was of concern given our patient’s elevated troponin level. Because there was already concern for sepsis, the addition of cardiac symptoms required us to consider infectious endocarditis.3 Sudden onset of dyspnea and a drop in blood pressure were concerning for pulmonary embolism, although our patient did not have the usual risk factors (cancer, immobilization, recent surgery, etc.).4 Additionally, in light of Mr. C’s psychiatric history and recent stressors of being moved from his sister’s house and admitted to a psychiatric hospital, coupled with dyspnea and hypotension, we included Takotsubo cardiomyopathy in the differential.5,6 This disease often occurs in response to an emotional or physical stressor and is characterized by transient systolic dysfunction in the setting of ventricular wall-motion abnormalities reaching beyond the distribution of a single coronary artery. Acute ECG and biomarker findings mimic those of myocardial infarction.6
Continue to: Finally, we needed to consider...
Finally, we needed to consider the potential adverse effects of clozapine. Clozapine is a second-generation antipsychotic (SGA) used to treat patients with schizophrenia for whom other antipsychotic medications are ineffective. Clozapine has been shown to be more effective than first-generation antipsychotics (FGA) in reducing symptoms of schizophrenia.7 It has also been shown to be more effective than several SGAs, including quetiapine, risperidone, and olanzapine.7 In fact, in patients with an insufficient therapeutic response to an SGA, clozapine proves to be more effective than switching to a different SGA. As a result of more than 20 years of research, clozapine is the gold-standard for treatment-resistant schizophrenia.7 Yet despite this strong evidence supporting its use in patients with treatment-resistant schizophrenia, the medication continues to be underutilized, especially in patients at risk for suicide.7
It appears that clozapine remains a third-choice medication in the treatment of schizophrenia largely due to its serious adverse effect profile.7 The medication includes several black-box warnings, including severe neutropenia, orthostatic hypotension, bradycardia, syncope, seizures, myocarditis, cardiomyopathy, and mitral valve incompetence.8 Tachycardia, bradycardia, and orthostatic hypotension are all clozapine-related adverse effects associated with autonomic dysfunction, which can result in serious long-term cardiac complications.9 With regards to the drug’s neutropenia risk, the establishment of the Clozapine Risk Evaluation and Mitigation Strategy (REMS) program has allowed for safer use of clozapine and reduced deaths due to clozapine-induced agranulocytosis. Clinicians and pharmacists must be certified in order to prescribe clozapine, and patients must be registered and undergo frequent absolute neutrophil count (ANC) monitoring.
Clozapine-induced myocarditis, a condition observed in up to 3% of patients started on the medication,9 is more likely to develop early on during treatment, with a median time of detection of 16 days following drug initiation.10 Myocarditis often presents with nonspecific signs and symptoms that include chest pain, tachycardia, palpitations, dyspnea, fever, flu-like symptoms, and/or hypotension.
[polldaddy:10226036]
The authors’ observations
Initial workup in the MICU for Mr. C included an ABG analysis, ECG, and cardiology consult. The ABG analysis demonstrated metabolic alkalosis; his ECG demonstrated sinus tachycardia and nonspecific ST elevation in the lateral leads (Figure). The cardiology consult team started Mr. C on treatment for a non-ST-elevation myocardial infarction (NSTEMI), which it believed to be most likely due to myocarditis with secondary demand ischemia, and less likely acute coronary syndrome. The cardiology consult team also recommended performing a workup for pulmonary emboli and infectious endocarditis if Mr. C’s symptoms persist or the infectious source could not be identified.

EVALUATION Gradual improvement
Mr. C demonstrates gradual improvement as his workup continues, and clozapine is held on the recommendation of the cardiac consult team. By Day 2, he stops complaining of auditory hallucinations, and does not report their return during the rest of his stay. His troponin level decreases to 8.6 ng/mL and lactate level to 1.4 mmol/L; trending is stopped for both. The erythrocyte sedimentation rate (ESR) is elevated at 59 mm/hr (reference range 0 to 22 mm/hr), along with a CRP level of 21 mg/L (reference range <8.0 mg/L). An echocardiogram demonstrates a 40% ejection fraction (reference range 55% to 75%) and moderate global hypokinesis. The cardiology consult team is concerned for Takotsubo cardiomyopathy with sepsis as a source of adrenergic surge vs myopericarditis of viral etiology. The cardiology team also suggests continued stoppage of clozapine, because the medication can cause hypotension and tachycardia.
Continue to: On Day 3...
On Day 3, Mr. C’s ST elevation resolves on ECG, and his CK level decreases to 70 U/L, at which point trending is stopped. On Day 5, Mr. C undergoes MRI, which demonstrates an ejection fraction of 55% and confirms myocarditis. No infectious source is identified.
By Day 6, with all other sources ruled out, clozapine is confirmed as the source of myocarditis for Mr. C.
The authors’ observations
Close cardiovascular monitoring should occur during the first 4 weeks after starting clozapine because 80% of cases of clozapine-induced myocarditis occur within 4 weeks of clozapine initiation.10 Baseline CRP, troponin I/T, and vital signs should be obtained before starting clozapine.11 Vital signs must be monitored to assess for fever, tachycardia, and deviations from baseline blood pressures.11 Although eosinophil counts and percentages can also be considered in addition to a baseline CRP value, they have not proven to be sensitive or specific for clozapine-induced myocarditis.12 A baseline echocardiogram can also be obtained, but is not necessary, especially given that it may not be readily available in all clinics, and could therefore delay initiation of clozapine and limit its use. C-reactive protein and troponin levels should be assessed weekly during the first 6 weeks of clozapine therapy.11 For symptomatic patients presenting with concern for clozapine-induced myocarditis, a CRP level >100 mg/L has 100% sensitivity in detecting clozapine-induced myocarditis.13 Clozapine should also be stopped if troponins levels reach twice the upper limit of normal. More mild elevations of CRP and troponins in the setting of persistent tachycardia or signs of an infectious process should be followed by daily CRP and troponins levels until these features resolve.11
Mr. C’s case highlights clinical features that clinicians should consider when screening for myocarditis. The development of myocarditis is associated with quick titrations of clozapine during Days 1 to 9. In this case, Mr. C had recently been titrated at an outside hospital, and the time frame during which this titration occurred was unknown. Given this lack of information, the potential for a rapid titration should alert the clinician to the risk of developing myocarditis. Increased age is also associated with an increased risk of myocarditis, with a 31% increase for each decade. Further, the concomitant use of valproate sodium during the titration period also increases the risk of myocarditis 2.5-fold.14
When evaluating a patient such as Mr. C, an important clinical sign that must not be overlooked is that an elevation of body temperature of 1°C is expected to give rise to a 10-beats-per-minute increase in heart rate when the fever is the result of an infection.15 During Day 1 of his hospitalization, Mr. C was tachycardic to 160 beats per minute, with a fever of 39.4°C. Thus, his heart rate was elevated well beyond what would be expected from a fever secondary to an infectious process. This further illustrates the need to consider adverse effects caused by medication, such as clozapine-induced tachycardia.
Continue to: While clozapine had already been stopped...
While clozapine had already been stopped in Mr. C, it is conceivable that other patients would potentially continue receiving it because of the medication’s demonstrated efficacy in reducing hallucinations; however, this would result in worsening and potentially serious cardiac symptoms.
[polldaddy:10226037]
The authors’ observations
A diagnosis of clozapine-induced myocarditis should be followed by a prompt discontinuation of clozapine. Discontinuation of the drug should lead to spontaneous resolution of the myocarditis, with significantly improved left ventricular function observed within 5 days.13 Historically, rechallenging a patient with clozapine was not recommended, due to fear of recurrence of myocarditis. However, recent case studies indicate that myocarditis need not be an absolute contraindication to restarting clozapine.16 Rather, the risks must be balanced against demonstrated efficacy in patients who had a limited response to other antipsychotics, as was the case with Mr. C. For these patients, the decision to rechallenge should be made with the patient’s informed consent and involve slow dose titration and increased monitoring.17 Should this rechallenge fail, another antipsychotic plus augmentation with a mood stabilizer or ECT may be more efficacious than an antipsychotic alone.18,19
OUTCOME Return to the psychiatric hospital
On Day 8, Mr. C is medically cleared; he had not reported auditory hallucinations since Day 2. He is discharged back to the psychiatric hospital for additional medication management of his schizophrenia.
Bottom Line
Clozapine-induced myocarditis should be included in the differential diagnosis for patients who present with nonspecific complaints and have an incomplete history pertaining to clozapine use. After discontinuing clozapine, and after myocarditis symptoms resolve, consider restarting clozapine in patients who have limited response to other treatments. If rechallenging fails, another antipsychotic plus augmentation with a mood stabilizer or electroconvulsive therapy may be more efficacious than an antipsychotic alone.
Related Resources
- Clozapine Risk Evaluation and Mitigation Strategy [REMS] Program. What is the Clozapine REMS Program? https://www.clozapinerems.com.
- Keating D, McWilliams S, Schneider I, et al. Pharmacological guidelines for schizophrenia: a systematic review and comparison of recommendations for the first episode. BMJ Open. 2017;7(1):e013881.
- Curto M, Girardi N, Lionetto L, et al. Systematic review of clozapine cardiotoxicity. Curr Psychiatry Rep. 2016;18(7):68.
Drug Brand Names
Clozapine • Clozaril
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Valproate • Depacon
1. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-810.
2. Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551-2567.
3. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893.
4. Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest. 1991;100(3):598-603.
5. Summers MR, Lennon RJ, Prasad A. Pre-morbid psychiatric and cardiovascular diseases in apical ballooning syndrome (tako-tsubo/stress-induced cardiomyopathy): potential pre-disposing factors? J Am Coll Cardiol. 2010;55(7):700-701.
6. Templin C, Ghadri JR, Diekmann J, et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N Engl J Med. 2015;373(10):929-938.
7. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
8. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc.; 2016.
9. Ronaldson KJ. Cardiovascular disease in clozapine-treated Patients: evidence, mechanisms and management. CNS Drugs. 2017;31(9):777-795.
10. Haas SJ, Hill R, Krum H, et al. Clozapine-associated myocarditis: a review of 116 cases of suspected myocarditis associated with the use of clozapine in Australia during 1993-2003. Drug Saf. 2007;30(1):47-57.
11. Goldsmith DR, Cotes RO. An unmet need: a clozapine-induced myocarditis screening protocol. Prim Care Companion CNS Disord. 2017;19(4): doi: 10.4088/PCC.16l02083.
12. Ronaldson KJ, Fitzgerald PB, McNeil JJ. Evolution of troponin, C-reactive protein and eosinophil count with the onset of clozapine-induced myocarditis. Aust N Z J Psychiatry. 2015;49(5):486-487.
13. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
14. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: a case-control study. Schizophr Res. 2012;141(2-3):173-178.
15. Davies P, Maconochie I. The relationship between body temperature, heart rate and respiratory rate in children. Emerg Med J. 2009;26(9):641-643.
16. Cook SC, Ferguson BA, Cotes RO, et al. Clozapine-induced myocarditis: prevention and considerations in rechallenge. Psychosomatics. 2015;56(6):685-690.
17. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Observations from 8 cases of clozapine rechallenge after development of myocarditis. J Clin Psychiatry. 2012;73(2):252-254.
18. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
19. Wenzheng W, Chengcheng PU, Jiangling Jiang, et al. Efficacy and safety of treating patients with refractory schizophrenia with antipsychotic medication and adjunctive electroconvulsive therapy: a systematic review and meta-analysis. Shanghai Arch Psychiatry. 2015;27(4):206-219.
CASE Posing a threat to his family
Mr. C, age 23, who was diagnosed with schizophrenia with daily auditory hallucinations 4 years earlier, is transferred from an outside psychiatric hospital to our emergency department (ED) after developing fever, tachycardia, headache, and nasal congestion for the past day. He had been admitted to the psychiatric hospital 3 weeks ago due to concerns he was experiencing increased hallucinations and delusions and posed a threat to his sister and her children, with whom he had been living.
Mr. C tells us that while at the psychiatric hospital, he had been started on clozapine, 250 mg/d. He said that prior to clozapine, he had been taking risperidone. We are unable to confirm past treatment information with the psychiatric hospital, including exactly when the clozapine had been started or how fast it had been titrated. We also were not able to obtain information on his prior medication regimen.
In the ED, Mr. C is febrile (39.4°C; 102.9°F), tachycardic (160 beats per minute; reference range 60 to 100), and tachypneic (24 breaths per minute; reference range 12 to 20). His blood pressure is 130/68 mm Hg, and his lactate level is 2.3 mmol/L (reference range <1.9 mmol/L). After he receives 3 liters of fluid, Mr. C’s heart rate decreases to 117 and his lactate level to 1.1 mmol/L. His white blood cell count is 10.6 × 103/mm3 (reference range 4.0 to 10.0 × 103/mm3); a differential can be found in the Table. His electrocardiogram (ECG) demonstrates sinus tachycardia and a QTc of 510 ms (reference range <430 ms), but is otherwise unremarkable. His creatinine kinase (CK) level is within normal limits at 76 U/L (reference range 52 to 336 U/L). A C-reactive protein (CRP) level was not drawn at this time. Other than marijuana and cocaine use, Mr. C’s medical history is unremarkable.
Mr. C is admitted to the hospital and is started on treatment for sepsis. On the evening of Day 1, Mr. C experiences worsening tachycardia (140 beats per minute) and tachypnea (≥40 breaths per minute). His temperature increases to 103.3°F, and his blood pressure drops to 97/55 mm Hg. His troponin level is 19.0 ng/mL (reference range <0.01 ng/mL) and CK level is 491 U/L.
As Mr. C continues to deteriorate, a rapid response is called and he is placed on non-rebreather oxygen and transferred to the medical intensive care unit (MICU).
[polldaddy:10226034]
The authors’ observations
With Mr. C’s presenting symptoms, multiple conditions were included in the differential diagnosis. The initial concern was for sepsis. Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.1 Organ dysfunction is defined by a quick Sepsis-Related Organ Failure Assessment (qSOFA) score ≥2 and is associated with an increased probability of mortality (>10%). Although no infection had been identified in Mr. C, the combination of fever, altered vital signs, and elevated lactate level in the setting of a qSOFA score of 2 (for respiratory rate and blood pressure) raised suspicion enough to start empiric treatment.
With Mr. C’s subsequent deterioration on the evening of Day 1, we considered cardiopulmonary etiologies. His symptoms of dyspnea, hypotension, tachycardia, tachypnea, and fever were nonspecific and thus required consideration of multiple life-threatening etiologies. Thygesen et al2 published an expert consensus of the definition of myocardial infarction, which was of concern given our patient’s elevated troponin level. Because there was already concern for sepsis, the addition of cardiac symptoms required us to consider infectious endocarditis.3 Sudden onset of dyspnea and a drop in blood pressure were concerning for pulmonary embolism, although our patient did not have the usual risk factors (cancer, immobilization, recent surgery, etc.).4 Additionally, in light of Mr. C’s psychiatric history and recent stressors of being moved from his sister’s house and admitted to a psychiatric hospital, coupled with dyspnea and hypotension, we included Takotsubo cardiomyopathy in the differential.5,6 This disease often occurs in response to an emotional or physical stressor and is characterized by transient systolic dysfunction in the setting of ventricular wall-motion abnormalities reaching beyond the distribution of a single coronary artery. Acute ECG and biomarker findings mimic those of myocardial infarction.6
Continue to: Finally, we needed to consider...
Finally, we needed to consider the potential adverse effects of clozapine. Clozapine is a second-generation antipsychotic (SGA) used to treat patients with schizophrenia for whom other antipsychotic medications are ineffective. Clozapine has been shown to be more effective than first-generation antipsychotics (FGA) in reducing symptoms of schizophrenia.7 It has also been shown to be more effective than several SGAs, including quetiapine, risperidone, and olanzapine.7 In fact, in patients with an insufficient therapeutic response to an SGA, clozapine proves to be more effective than switching to a different SGA. As a result of more than 20 years of research, clozapine is the gold-standard for treatment-resistant schizophrenia.7 Yet despite this strong evidence supporting its use in patients with treatment-resistant schizophrenia, the medication continues to be underutilized, especially in patients at risk for suicide.7
It appears that clozapine remains a third-choice medication in the treatment of schizophrenia largely due to its serious adverse effect profile.7 The medication includes several black-box warnings, including severe neutropenia, orthostatic hypotension, bradycardia, syncope, seizures, myocarditis, cardiomyopathy, and mitral valve incompetence.8 Tachycardia, bradycardia, and orthostatic hypotension are all clozapine-related adverse effects associated with autonomic dysfunction, which can result in serious long-term cardiac complications.9 With regards to the drug’s neutropenia risk, the establishment of the Clozapine Risk Evaluation and Mitigation Strategy (REMS) program has allowed for safer use of clozapine and reduced deaths due to clozapine-induced agranulocytosis. Clinicians and pharmacists must be certified in order to prescribe clozapine, and patients must be registered and undergo frequent absolute neutrophil count (ANC) monitoring.
Clozapine-induced myocarditis, a condition observed in up to 3% of patients started on the medication,9 is more likely to develop early on during treatment, with a median time of detection of 16 days following drug initiation.10 Myocarditis often presents with nonspecific signs and symptoms that include chest pain, tachycardia, palpitations, dyspnea, fever, flu-like symptoms, and/or hypotension.
[polldaddy:10226036]
The authors’ observations
Initial workup in the MICU for Mr. C included an ABG analysis, ECG, and cardiology consult. The ABG analysis demonstrated metabolic alkalosis; his ECG demonstrated sinus tachycardia and nonspecific ST elevation in the lateral leads (Figure). The cardiology consult team started Mr. C on treatment for a non-ST-elevation myocardial infarction (NSTEMI), which it believed to be most likely due to myocarditis with secondary demand ischemia, and less likely acute coronary syndrome. The cardiology consult team also recommended performing a workup for pulmonary emboli and infectious endocarditis if Mr. C’s symptoms persist or the infectious source could not be identified.
EVALUATION Gradual improvement
Mr. C demonstrates gradual improvement as his workup continues, and clozapine is held on the recommendation of the cardiac consult team. By Day 2, he stops complaining of auditory hallucinations, and does not report their return during the rest of his stay. His troponin level decreases to 8.6 ng/mL and lactate level to 1.4 mmol/L; trending is stopped for both. The erythrocyte sedimentation rate (ESR) is elevated at 59 mm/hr (reference range 0 to 22 mm/hr), along with a CRP level of 21 mg/L (reference range <8.0 mg/L). An echocardiogram demonstrates a 40% ejection fraction (reference range 55% to 75%) and moderate global hypokinesis. The cardiology consult team is concerned for Takotsubo cardiomyopathy with sepsis as a source of adrenergic surge vs myopericarditis of viral etiology. The cardiology team also suggests continued stoppage of clozapine, because the medication can cause hypotension and tachycardia.
Continue to: On Day 3...
On Day 3, Mr. C’s ST elevation resolves on ECG, and his CK level decreases to 70 U/L, at which point trending is stopped. On Day 5, Mr. C undergoes MRI, which demonstrates an ejection fraction of 55% and confirms myocarditis. No infectious source is identified.
By Day 6, with all other sources ruled out, clozapine is confirmed as the source of myocarditis for Mr. C.
The authors’ observations
Close cardiovascular monitoring should occur during the first 4 weeks after starting clozapine because 80% of cases of clozapine-induced myocarditis occur within 4 weeks of clozapine initiation.10 Baseline CRP, troponin I/T, and vital signs should be obtained before starting clozapine.11 Vital signs must be monitored to assess for fever, tachycardia, and deviations from baseline blood pressures.11 Although eosinophil counts and percentages can also be considered in addition to a baseline CRP value, they have not proven to be sensitive or specific for clozapine-induced myocarditis.12 A baseline echocardiogram can also be obtained, but is not necessary, especially given that it may not be readily available in all clinics, and could therefore delay initiation of clozapine and limit its use. C-reactive protein and troponin levels should be assessed weekly during the first 6 weeks of clozapine therapy.11 For symptomatic patients presenting with concern for clozapine-induced myocarditis, a CRP level >100 mg/L has 100% sensitivity in detecting clozapine-induced myocarditis.13 Clozapine should also be stopped if troponins levels reach twice the upper limit of normal. More mild elevations of CRP and troponins in the setting of persistent tachycardia or signs of an infectious process should be followed by daily CRP and troponins levels until these features resolve.11
Mr. C’s case highlights clinical features that clinicians should consider when screening for myocarditis. The development of myocarditis is associated with quick titrations of clozapine during Days 1 to 9. In this case, Mr. C had recently been titrated at an outside hospital, and the time frame during which this titration occurred was unknown. Given this lack of information, the potential for a rapid titration should alert the clinician to the risk of developing myocarditis. Increased age is also associated with an increased risk of myocarditis, with a 31% increase for each decade. Further, the concomitant use of valproate sodium during the titration period also increases the risk of myocarditis 2.5-fold.14
When evaluating a patient such as Mr. C, an important clinical sign that must not be overlooked is that an elevation of body temperature of 1°C is expected to give rise to a 10-beats-per-minute increase in heart rate when the fever is the result of an infection.15 During Day 1 of his hospitalization, Mr. C was tachycardic to 160 beats per minute, with a fever of 39.4°C. Thus, his heart rate was elevated well beyond what would be expected from a fever secondary to an infectious process. This further illustrates the need to consider adverse effects caused by medication, such as clozapine-induced tachycardia.
Continue to: While clozapine had already been stopped...
While clozapine had already been stopped in Mr. C, it is conceivable that other patients would potentially continue receiving it because of the medication’s demonstrated efficacy in reducing hallucinations; however, this would result in worsening and potentially serious cardiac symptoms.
[polldaddy:10226037]
The authors’ observations
A diagnosis of clozapine-induced myocarditis should be followed by a prompt discontinuation of clozapine. Discontinuation of the drug should lead to spontaneous resolution of the myocarditis, with significantly improved left ventricular function observed within 5 days.13 Historically, rechallenging a patient with clozapine was not recommended, due to fear of recurrence of myocarditis. However, recent case studies indicate that myocarditis need not be an absolute contraindication to restarting clozapine.16 Rather, the risks must be balanced against demonstrated efficacy in patients who had a limited response to other antipsychotics, as was the case with Mr. C. For these patients, the decision to rechallenge should be made with the patient’s informed consent and involve slow dose titration and increased monitoring.17 Should this rechallenge fail, another antipsychotic plus augmentation with a mood stabilizer or ECT may be more efficacious than an antipsychotic alone.18,19
OUTCOME Return to the psychiatric hospital
On Day 8, Mr. C is medically cleared; he had not reported auditory hallucinations since Day 2. He is discharged back to the psychiatric hospital for additional medication management of his schizophrenia.
Bottom Line
Clozapine-induced myocarditis should be included in the differential diagnosis for patients who present with nonspecific complaints and have an incomplete history pertaining to clozapine use. After discontinuing clozapine, and after myocarditis symptoms resolve, consider restarting clozapine in patients who have limited response to other treatments. If rechallenging fails, another antipsychotic plus augmentation with a mood stabilizer or electroconvulsive therapy may be more efficacious than an antipsychotic alone.
Related Resources
- Clozapine Risk Evaluation and Mitigation Strategy [REMS] Program. What is the Clozapine REMS Program? https://www.clozapinerems.com.
- Keating D, McWilliams S, Schneider I, et al. Pharmacological guidelines for schizophrenia: a systematic review and comparison of recommendations for the first episode. BMJ Open. 2017;7(1):e013881.
- Curto M, Girardi N, Lionetto L, et al. Systematic review of clozapine cardiotoxicity. Curr Psychiatry Rep. 2016;18(7):68.
Drug Brand Names
Clozapine • Clozaril
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Valproate • Depacon
CASE Posing a threat to his family
Mr. C, age 23, who was diagnosed with schizophrenia with daily auditory hallucinations 4 years earlier, is transferred from an outside psychiatric hospital to our emergency department (ED) after developing fever, tachycardia, headache, and nasal congestion for the past day. He had been admitted to the psychiatric hospital 3 weeks ago due to concerns he was experiencing increased hallucinations and delusions and posed a threat to his sister and her children, with whom he had been living.
Mr. C tells us that while at the psychiatric hospital, he had been started on clozapine, 250 mg/d. He said that prior to clozapine, he had been taking risperidone. We are unable to confirm past treatment information with the psychiatric hospital, including exactly when the clozapine had been started or how fast it had been titrated. We also were not able to obtain information on his prior medication regimen.
In the ED, Mr. C is febrile (39.4°C; 102.9°F), tachycardic (160 beats per minute; reference range 60 to 100), and tachypneic (24 breaths per minute; reference range 12 to 20). His blood pressure is 130/68 mm Hg, and his lactate level is 2.3 mmol/L (reference range <1.9 mmol/L). After he receives 3 liters of fluid, Mr. C’s heart rate decreases to 117 and his lactate level to 1.1 mmol/L. His white blood cell count is 10.6 × 103/mm3 (reference range 4.0 to 10.0 × 103/mm3); a differential can be found in the Table. His electrocardiogram (ECG) demonstrates sinus tachycardia and a QTc of 510 ms (reference range <430 ms), but is otherwise unremarkable. His creatinine kinase (CK) level is within normal limits at 76 U/L (reference range 52 to 336 U/L). A C-reactive protein (CRP) level was not drawn at this time. Other than marijuana and cocaine use, Mr. C’s medical history is unremarkable.
Mr. C is admitted to the hospital and is started on treatment for sepsis. On the evening of Day 1, Mr. C experiences worsening tachycardia (140 beats per minute) and tachypnea (≥40 breaths per minute). His temperature increases to 103.3°F, and his blood pressure drops to 97/55 mm Hg. His troponin level is 19.0 ng/mL (reference range <0.01 ng/mL) and CK level is 491 U/L.
As Mr. C continues to deteriorate, a rapid response is called and he is placed on non-rebreather oxygen and transferred to the medical intensive care unit (MICU).
[polldaddy:10226034]
The authors’ observations
With Mr. C’s presenting symptoms, multiple conditions were included in the differential diagnosis. The initial concern was for sepsis. Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.1 Organ dysfunction is defined by a quick Sepsis-Related Organ Failure Assessment (qSOFA) score ≥2 and is associated with an increased probability of mortality (>10%). Although no infection had been identified in Mr. C, the combination of fever, altered vital signs, and elevated lactate level in the setting of a qSOFA score of 2 (for respiratory rate and blood pressure) raised suspicion enough to start empiric treatment.
With Mr. C’s subsequent deterioration on the evening of Day 1, we considered cardiopulmonary etiologies. His symptoms of dyspnea, hypotension, tachycardia, tachypnea, and fever were nonspecific and thus required consideration of multiple life-threatening etiologies. Thygesen et al2 published an expert consensus of the definition of myocardial infarction, which was of concern given our patient’s elevated troponin level. Because there was already concern for sepsis, the addition of cardiac symptoms required us to consider infectious endocarditis.3 Sudden onset of dyspnea and a drop in blood pressure were concerning for pulmonary embolism, although our patient did not have the usual risk factors (cancer, immobilization, recent surgery, etc.).4 Additionally, in light of Mr. C’s psychiatric history and recent stressors of being moved from his sister’s house and admitted to a psychiatric hospital, coupled with dyspnea and hypotension, we included Takotsubo cardiomyopathy in the differential.5,6 This disease often occurs in response to an emotional or physical stressor and is characterized by transient systolic dysfunction in the setting of ventricular wall-motion abnormalities reaching beyond the distribution of a single coronary artery. Acute ECG and biomarker findings mimic those of myocardial infarction.6
Continue to: Finally, we needed to consider...
Finally, we needed to consider the potential adverse effects of clozapine. Clozapine is a second-generation antipsychotic (SGA) used to treat patients with schizophrenia for whom other antipsychotic medications are ineffective. Clozapine has been shown to be more effective than first-generation antipsychotics (FGA) in reducing symptoms of schizophrenia.7 It has also been shown to be more effective than several SGAs, including quetiapine, risperidone, and olanzapine.7 In fact, in patients with an insufficient therapeutic response to an SGA, clozapine proves to be more effective than switching to a different SGA. As a result of more than 20 years of research, clozapine is the gold-standard for treatment-resistant schizophrenia.7 Yet despite this strong evidence supporting its use in patients with treatment-resistant schizophrenia, the medication continues to be underutilized, especially in patients at risk for suicide.7
It appears that clozapine remains a third-choice medication in the treatment of schizophrenia largely due to its serious adverse effect profile.7 The medication includes several black-box warnings, including severe neutropenia, orthostatic hypotension, bradycardia, syncope, seizures, myocarditis, cardiomyopathy, and mitral valve incompetence.8 Tachycardia, bradycardia, and orthostatic hypotension are all clozapine-related adverse effects associated with autonomic dysfunction, which can result in serious long-term cardiac complications.9 With regards to the drug’s neutropenia risk, the establishment of the Clozapine Risk Evaluation and Mitigation Strategy (REMS) program has allowed for safer use of clozapine and reduced deaths due to clozapine-induced agranulocytosis. Clinicians and pharmacists must be certified in order to prescribe clozapine, and patients must be registered and undergo frequent absolute neutrophil count (ANC) monitoring.
Clozapine-induced myocarditis, a condition observed in up to 3% of patients started on the medication,9 is more likely to develop early on during treatment, with a median time of detection of 16 days following drug initiation.10 Myocarditis often presents with nonspecific signs and symptoms that include chest pain, tachycardia, palpitations, dyspnea, fever, flu-like symptoms, and/or hypotension.
[polldaddy:10226036]
The authors’ observations
Initial workup in the MICU for Mr. C included an ABG analysis, ECG, and cardiology consult. The ABG analysis demonstrated metabolic alkalosis; his ECG demonstrated sinus tachycardia and nonspecific ST elevation in the lateral leads (Figure). The cardiology consult team started Mr. C on treatment for a non-ST-elevation myocardial infarction (NSTEMI), which it believed to be most likely due to myocarditis with secondary demand ischemia, and less likely acute coronary syndrome. The cardiology consult team also recommended performing a workup for pulmonary emboli and infectious endocarditis if Mr. C’s symptoms persist or the infectious source could not be identified.
EVALUATION Gradual improvement
Mr. C demonstrates gradual improvement as his workup continues, and clozapine is held on the recommendation of the cardiac consult team. By Day 2, he stops complaining of auditory hallucinations, and does not report their return during the rest of his stay. His troponin level decreases to 8.6 ng/mL and lactate level to 1.4 mmol/L; trending is stopped for both. The erythrocyte sedimentation rate (ESR) is elevated at 59 mm/hr (reference range 0 to 22 mm/hr), along with a CRP level of 21 mg/L (reference range <8.0 mg/L). An echocardiogram demonstrates a 40% ejection fraction (reference range 55% to 75%) and moderate global hypokinesis. The cardiology consult team is concerned for Takotsubo cardiomyopathy with sepsis as a source of adrenergic surge vs myopericarditis of viral etiology. The cardiology team also suggests continued stoppage of clozapine, because the medication can cause hypotension and tachycardia.
Continue to: On Day 3...
On Day 3, Mr. C’s ST elevation resolves on ECG, and his CK level decreases to 70 U/L, at which point trending is stopped. On Day 5, Mr. C undergoes MRI, which demonstrates an ejection fraction of 55% and confirms myocarditis. No infectious source is identified.
By Day 6, with all other sources ruled out, clozapine is confirmed as the source of myocarditis for Mr. C.
The authors’ observations
Close cardiovascular monitoring should occur during the first 4 weeks after starting clozapine because 80% of cases of clozapine-induced myocarditis occur within 4 weeks of clozapine initiation.10 Baseline CRP, troponin I/T, and vital signs should be obtained before starting clozapine.11 Vital signs must be monitored to assess for fever, tachycardia, and deviations from baseline blood pressures.11 Although eosinophil counts and percentages can also be considered in addition to a baseline CRP value, they have not proven to be sensitive or specific for clozapine-induced myocarditis.12 A baseline echocardiogram can also be obtained, but is not necessary, especially given that it may not be readily available in all clinics, and could therefore delay initiation of clozapine and limit its use. C-reactive protein and troponin levels should be assessed weekly during the first 6 weeks of clozapine therapy.11 For symptomatic patients presenting with concern for clozapine-induced myocarditis, a CRP level >100 mg/L has 100% sensitivity in detecting clozapine-induced myocarditis.13 Clozapine should also be stopped if troponins levels reach twice the upper limit of normal. More mild elevations of CRP and troponins in the setting of persistent tachycardia or signs of an infectious process should be followed by daily CRP and troponins levels until these features resolve.11
Mr. C’s case highlights clinical features that clinicians should consider when screening for myocarditis. The development of myocarditis is associated with quick titrations of clozapine during Days 1 to 9. In this case, Mr. C had recently been titrated at an outside hospital, and the time frame during which this titration occurred was unknown. Given this lack of information, the potential for a rapid titration should alert the clinician to the risk of developing myocarditis. Increased age is also associated with an increased risk of myocarditis, with a 31% increase for each decade. Further, the concomitant use of valproate sodium during the titration period also increases the risk of myocarditis 2.5-fold.14
When evaluating a patient such as Mr. C, an important clinical sign that must not be overlooked is that an elevation of body temperature of 1°C is expected to give rise to a 10-beats-per-minute increase in heart rate when the fever is the result of an infection.15 During Day 1 of his hospitalization, Mr. C was tachycardic to 160 beats per minute, with a fever of 39.4°C. Thus, his heart rate was elevated well beyond what would be expected from a fever secondary to an infectious process. This further illustrates the need to consider adverse effects caused by medication, such as clozapine-induced tachycardia.
Continue to: While clozapine had already been stopped...
While clozapine had already been stopped in Mr. C, it is conceivable that other patients would potentially continue receiving it because of the medication’s demonstrated efficacy in reducing hallucinations; however, this would result in worsening and potentially serious cardiac symptoms.
[polldaddy:10226037]
The authors’ observations
A diagnosis of clozapine-induced myocarditis should be followed by a prompt discontinuation of clozapine. Discontinuation of the drug should lead to spontaneous resolution of the myocarditis, with significantly improved left ventricular function observed within 5 days.13 Historically, rechallenging a patient with clozapine was not recommended, due to fear of recurrence of myocarditis. However, recent case studies indicate that myocarditis need not be an absolute contraindication to restarting clozapine.16 Rather, the risks must be balanced against demonstrated efficacy in patients who had a limited response to other antipsychotics, as was the case with Mr. C. For these patients, the decision to rechallenge should be made with the patient’s informed consent and involve slow dose titration and increased monitoring.17 Should this rechallenge fail, another antipsychotic plus augmentation with a mood stabilizer or ECT may be more efficacious than an antipsychotic alone.18,19
OUTCOME Return to the psychiatric hospital
On Day 8, Mr. C is medically cleared; he had not reported auditory hallucinations since Day 2. He is discharged back to the psychiatric hospital for additional medication management of his schizophrenia.
Bottom Line
Clozapine-induced myocarditis should be included in the differential diagnosis for patients who present with nonspecific complaints and have an incomplete history pertaining to clozapine use. After discontinuing clozapine, and after myocarditis symptoms resolve, consider restarting clozapine in patients who have limited response to other treatments. If rechallenging fails, another antipsychotic plus augmentation with a mood stabilizer or electroconvulsive therapy may be more efficacious than an antipsychotic alone.
Related Resources
- Clozapine Risk Evaluation and Mitigation Strategy [REMS] Program. What is the Clozapine REMS Program? https://www.clozapinerems.com.
- Keating D, McWilliams S, Schneider I, et al. Pharmacological guidelines for schizophrenia: a systematic review and comparison of recommendations for the first episode. BMJ Open. 2017;7(1):e013881.
- Curto M, Girardi N, Lionetto L, et al. Systematic review of clozapine cardiotoxicity. Curr Psychiatry Rep. 2016;18(7):68.
Drug Brand Names
Clozapine • Clozaril
Olanzapine • Zyprexa
Quetiapine • Seroquel
Risperidone • Risperdal
Valproate • Depacon
1. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-810.
2. Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551-2567.
3. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893.
4. Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest. 1991;100(3):598-603.
5. Summers MR, Lennon RJ, Prasad A. Pre-morbid psychiatric and cardiovascular diseases in apical ballooning syndrome (tako-tsubo/stress-induced cardiomyopathy): potential pre-disposing factors? J Am Coll Cardiol. 2010;55(7):700-701.
6. Templin C, Ghadri JR, Diekmann J, et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N Engl J Med. 2015;373(10):929-938.
7. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
8. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc.; 2016.
9. Ronaldson KJ. Cardiovascular disease in clozapine-treated Patients: evidence, mechanisms and management. CNS Drugs. 2017;31(9):777-795.
10. Haas SJ, Hill R, Krum H, et al. Clozapine-associated myocarditis: a review of 116 cases of suspected myocarditis associated with the use of clozapine in Australia during 1993-2003. Drug Saf. 2007;30(1):47-57.
11. Goldsmith DR, Cotes RO. An unmet need: a clozapine-induced myocarditis screening protocol. Prim Care Companion CNS Disord. 2017;19(4): doi: 10.4088/PCC.16l02083.
12. Ronaldson KJ, Fitzgerald PB, McNeil JJ. Evolution of troponin, C-reactive protein and eosinophil count with the onset of clozapine-induced myocarditis. Aust N Z J Psychiatry. 2015;49(5):486-487.
13. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
14. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: a case-control study. Schizophr Res. 2012;141(2-3):173-178.
15. Davies P, Maconochie I. The relationship between body temperature, heart rate and respiratory rate in children. Emerg Med J. 2009;26(9):641-643.
16. Cook SC, Ferguson BA, Cotes RO, et al. Clozapine-induced myocarditis: prevention and considerations in rechallenge. Psychosomatics. 2015;56(6):685-690.
17. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Observations from 8 cases of clozapine rechallenge after development of myocarditis. J Clin Psychiatry. 2012;73(2):252-254.
18. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
19. Wenzheng W, Chengcheng PU, Jiangling Jiang, et al. Efficacy and safety of treating patients with refractory schizophrenia with antipsychotic medication and adjunctive electroconvulsive therapy: a systematic review and meta-analysis. Shanghai Arch Psychiatry. 2015;27(4):206-219.
1. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-810.
2. Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551-2567.
3. Cahill TJ, Prendergast BD. Infective endocarditis. Lancet. 2016;387(10021):882-893.
4. Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest. 1991;100(3):598-603.
5. Summers MR, Lennon RJ, Prasad A. Pre-morbid psychiatric and cardiovascular diseases in apical ballooning syndrome (tako-tsubo/stress-induced cardiomyopathy): potential pre-disposing factors? J Am Coll Cardiol. 2010;55(7):700-701.
6. Templin C, Ghadri JR, Diekmann J, et al. Clinical features and outcomes of Takotsubo (stress) cardiomyopathy. N Engl J Med. 2015;373(10):929-938.
7. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
8. Clozaril [package insert]. Rosemont, PA: HLS Therapeutics (USA), Inc.; 2016.
9. Ronaldson KJ. Cardiovascular disease in clozapine-treated Patients: evidence, mechanisms and management. CNS Drugs. 2017;31(9):777-795.
10. Haas SJ, Hill R, Krum H, et al. Clozapine-associated myocarditis: a review of 116 cases of suspected myocarditis associated with the use of clozapine in Australia during 1993-2003. Drug Saf. 2007;30(1):47-57.
11. Goldsmith DR, Cotes RO. An unmet need: a clozapine-induced myocarditis screening protocol. Prim Care Companion CNS Disord. 2017;19(4): doi: 10.4088/PCC.16l02083.
12. Ronaldson KJ, Fitzgerald PB, McNeil JJ. Evolution of troponin, C-reactive protein and eosinophil count with the onset of clozapine-induced myocarditis. Aust N Z J Psychiatry. 2015;49(5):486-487.
13. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
14. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: a case-control study. Schizophr Res. 2012;141(2-3):173-178.
15. Davies P, Maconochie I. The relationship between body temperature, heart rate and respiratory rate in children. Emerg Med J. 2009;26(9):641-643.
16. Cook SC, Ferguson BA, Cotes RO, et al. Clozapine-induced myocarditis: prevention and considerations in rechallenge. Psychosomatics. 2015;56(6):685-690.
17. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. Observations from 8 cases of clozapine rechallenge after development of myocarditis. J Clin Psychiatry. 2012;73(2):252-254.
18. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
19. Wenzheng W, Chengcheng PU, Jiangling Jiang, et al. Efficacy and safety of treating patients with refractory schizophrenia with antipsychotic medication and adjunctive electroconvulsive therapy: a systematic review and meta-analysis. Shanghai Arch Psychiatry. 2015;27(4):206-219.
Psychotropic-induced hyponatremia

Hyponatremia is a common, multifactorial clinical condition. Hyponatremia is usually defined as a plasma sodium level <135 mmol/L; however, some studies define it as a level <130 mmol/L. Hyponatremia results from the inability of the kidney to excrete a sufficient amount of fluid, or is due to excessive fluid intake. Increases in osmolality stimulate thirst and result in increased fluid intake. This increase in osmolality is recognized by the osmoreceptors located in the hypothalamus, which release antidiuretic hormone (ADH). Antidiuretic hormone works on the collecting ducts within the kidneys, triggering increased fluid reabsorption resulting in decreased fluid loss and a reduction in thirst.
The syndrome of inappropriate antidiuretic hormone (SIADH) occurs when there is persistent ADH stimulation resulting in hyponatremia. SIADH commonly presents as euvolemic hyponatremia. Common diagnostic criteria for SIADH are listed in Table 1.1

Medications are a major cause of SIADH, and psychotropics are a primary offender. Most of the data for drug-induced SIADH come from case reports and small case series, such as those described in Table 2.2-4 The extent to which each psychotropic class causes SIADH remains unknown. In this article, we focus on 3 classes of psychotropics, and their role in causing SIADH.

Antidepressants
There is a fair amount of data associating antidepressants with SIADH. The incidence of SIADH with selective serotonin reuptake inhibitors (SSRIs) varies greatly among studies, from .06% to 40%.5-12 This wide variation is due to the way each study defined hyponatremia. A higher incidence was found when hyponatremia was defined as <135 mmol/L as opposed to <130 mmol/L. A large cohort study of SSRIs found that there was an increased risk with fluoxetine, escitalopram, and citalopram (.078% to .085%) vs paroxetine and sertraline (.033% to .053%).13 Studies comparing the incidence of SIADH with SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs) found that the rates were equal or slightly higher with the SNRI venlafaxine.13 SNRIs as a group have an estimated incidence of .08% to 4%, based on studies that defined hyponatremia as <130 mmol/L.13,14 Tricyclic antidepressants have an estimated incidence of .005% to 16.7%, based on a retrospective study that reviewed 15 studies and 100 case reports.15 Mirtazapine and bupropion do not have enough evidence to obtain a true definition of incidence; case reports for these drugs suggest a causal link for hyponatremia. Table 37,9,12-15 provides an overview of the incidence rate of hyponatremia for select antidepressants. It is clear that a more stringent cutoff for hyponatremia (<130 mmol/L) reduces the incidence rates. More evidence is needed to identify the true incidence and prevalence of SIADH with these agents.

Antipsychotics
Compared with antidepressants, there’s less evidence linking SIADH with antipsychotics; this data come mainly from case reports and observational studies. Serrano et al16 reported on a cross-sectional study that included 88 patients receiving clozapine, 61 patients receiving other atypical antipsychotics, 23 patients receiving typical antipsychotics, and 11 patients receiving both typical and atypical antipsychotics. They reported incidence rates of 3.4% for clozapine, 4.9% for atypical antipsychotics, 26.1% for typical antipsychotics, and 9.1% for the group receiving both typical and atypical antipsychotics.16 The primary theory for the decreased incidence of SIADH with use of atypical antipsychotics is related to decreased rates of psychogenic polydipsia leading to lower incidence of hyponatremia.
Mood stabilizers
Several studies have associated carbamazepine/oxcarbazepine, valproic acid, and lamotrigine with SIADH.17-23 Studies show incidence rates ranging from 4.8% to 41.5% for these medications. Carbamazepine appears to have the highest incidence of SIADH. A limitation of these studies is the small sample sizes, which ranged from 12 to 60 participants.
Pathophysiology
The kidneys are responsible for maintaining homeostasis between bodily fluids and serum sodium levels. ADH, which is produced by the hypothalamus, plays a significant role in fluid balance, thirst, and fluid retention. Inappropriate and continuous secretion of ADH, despite normal or high fluid status, results in hyposmolality and hyponatremia. The specific mechanisms by which psychotropic medications cause SIADH are listed in Table 4.24

Diagnosis
Diagnosis of SIADH can be complex because there are many clinical reasons a patient may have hyponatremia. For example, SIADH and psychogenic polydipsia both result in hyponatremia, and sometimes the 2 conditions can be difficult to distinguish. Hyponatremia is typically discovered by routine blood testing if the patient is asymptomatic. Table 525 highlights the major laboratory markers that distinguish SIADH and psychogenic polydipsia.

Continue to: Treatment
Treatment
The primary treatment for SIADH is cessation of the offending agent. Based on the patient’s clinical presentation, free water restriction (.5 to 1 L/d) can be implemented to increase serum sodium levels. If the patient is having neurologic complications due to the severity of hyponatremia, correction with hypertonic saline is indicated. Upon resolution, the recommended course of action is to switch to a medication in a different class. Re-challenging the patient with the same medication is not recommended unless there is no other alternative class of medication.24 Table 626 highlights other causes of hyponatremia, what laboratory markers to assess, and how to treat high-risk individuals.

Hyponatremia is a complex medical complication that can be life-threatening. Psychotropics are a relatively common cause of hyponatremia, specifically SIADH. Older adults appear to be at highest risk, as most case reports are in patients age ≥65. Patients who are prescribed psychotropics should be treated with the lowest effective dose and monitored for signs and symptoms of hyponatremia throughout therapy.
Related Resources
- Spasovski G, Vanholder R, Allolio B, et al. Clinical practice guidelines on diagnosis and treatment of hyponatremia. Eur J Endocrinol. 2014;170(3):G1-G47.
- Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013;126(10 Suppl 1):S1-S42.
Drug Brand Names
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Citalopram • Celexa
Clozapine • Clozaril
Escitalopram • Lexapro
Fluoxetine • Prozac
Haloperidol • Haldol
Lamotrigine • Lamictal
Levathyroxine • Levothroid
Mirtazapine • Remeron
Oxcarbazepine • Trileptal
Paroxetine • Paxil
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Valproic acid • Depakote
Venlafaxine • Effexor
1. Sahay M, Sahay R. Hyponatremia: a practical approach. Indian J Endocrinol Metab. 2014;18(6):760-771.
2. Kenes MT, Hamblin S, Tumuluri SS, et al. Syndrome of inappropriate antidiuretic hormone in a patient receiving high-dose haloperidol and quetiapine therapy. J Neuropsychiatry Clin Neurosci. 2016;28(2):e29-e30. doi: 10.1176/appi.neuropsych.15110392.
3. Twardowschy CA, Bertolucci CB, Gracia Cde M, et al. Severe hyponatremia and the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) associated with fluoxetine: case report. Arq Neuropsiquiatr. 2006;64(1):142-145.
4. Patel KR, Meesala A, Stanilla JK. Sodium valproate–induced hyponatremia: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(5):PCC.09100941. doi: 10.4088/PCC.09100941.
5. Pillans PI, Coulter DM. Fluoxetine and hyponatraemia—a potential hazard in the elderly. N Z Med J. 1994;107(973):85‑86.
6. Strachan J, Shepherd J. Hyponatraemia associated with the use of selective serotonin reuptake inhibitors. Aust N Z J Psychiatry. 1998;32(2):295‑298.
7. Bouman WP, Pinner G, Johnson H. Incidence of selective serotonin reuptake inhibitor (SSRI) induced hyponatraemia due to the syndrome of inappropriate antidiuretic hormone (SIADH) secretion in the elderly. Int J Geriatr Psychiatry. 1998;13(1):12‑15.
8. Wilkinson TJ, Begg EJ, Winter AC, et al. Incidence and risk factors for hyponatraemia following treatment with fluoxetine or paroxetine in elderly people. Br J Clin Pharmacol. 1999;47(2):211‑217.
9. Kirby D, Harrigan S, Ames D. Hyponatraemia in elderly psychiatric patients treated with selective serotonin reuptake inhibitors and venlafaxine: a retrospective controlled study in an inpatient unit. Int J Geriatr Psychiatry. 2002;17(3):231‑237.
10. Wee R, Lim WK. Selective serotonin re‑uptake inhibitors (SSRIs) and hyponatraemia in the elderly. Int J Geriatr Psychiatry. 2004;19(6):590‑591.
11. Jung YE, Jun TY, Kim KS, et al. Hyponatremia associated with selective serotonin reuptake inhibitors, mirtazapine, and venlafaxine in Korean patients with major depressive disorder. Int J Clin Pharmacol Ther. 2011;49(7):437‑443.
12. Letmaier M, Painold A, Holl AK, et al. Hyponatremia during psychopharmacological treatment: Results of a drug surveillance program. Int J Neuropsychopharmacol. 2012;15(6):739‑748.
13. Coupland CA, Dhiman P, Barton G, et al. A study of the safety and harms of antidepressant drugs for older people: a cohort study using a large primary care database. Health Technol Assess. 2011;15(28):1‑202, iii‑iv.
14. Leah-Møller KB, Hansen AH, Torstensson M, et al. Antidepressants and the risk of hyponatremia: a Danish register-based population study. BMJ Open. 2016;6(5):e011200. doi: 10.1136/bmjopen-2016-011200.
15. De Picker LD, Van Den Eede F, Dumont G, et al. Antidepressants and the risk of hyponatremia: a class by class review of literature. Psychosomatics. 2014;55(6):536-547.
16. Serrano A, Rangel N, Carrizo E, et al. Safety of long-term clozapine administration. Frequency of cardiomyopathy and hyponatraemia: two cross-sectional, naturalistic studies. Aust N Z J Psychiatry. 2014;48(2):183‑192.
17. Uhde TW, Post RM. Effects of carbamazepine on serum electrolytes: clinical and theoretical implications. J Clin Psychopharmacol. 1983;3(2):103‑106.
18. Lahr MB. Hyponatremia during carbamazepine therapy. Clin Pharmacol Ther. 1985;37(6):693‑696.
19. Joffe RT, Post RM, Uhde TW. Effects of carbamazepine on serum electrolytes in affectively ill patients. Psychol Med. 1986;16(2):331‑335.
20. Vieweg V, Glick JL, Herring S, et al. Absence of carbamazepine‑induced hyponatremia among patients also given lithium. Am J Psychiatry. 1987;144(7):943‑947.
21. Yassa R, Iskandar H, Nastase C, et al. Carbamazepine and hyponatremia in patients with affective disorder. Am J Psychiatry. 1988;145(3):339‑342.
22. Kastner T, Friedman DL, Pond WS. Carbamazepine‑induced hyponatremia in patients with mental retardation. Am J Ment Retard. 1992;96(5):536‑540.
23. Kelly BD, Hillery J. Hyponatremia during carbamazepine therapy in patients with intellectual disability. J Intellect Disabil Res. 2001;45(Pt 2):152‑156.
24. Sahoo S, Grover S. Hyponatremia and psychotropics. J Geriatr Ment Health. 2016;3(2):108-122.
25. Siragy HM. Hyponatremia, fluid-electrolyte disorders and the syndrome of inappropriate antidiuretic hormone secretion: diagnosis and treatment options. Endocr Pract. 2006;12(4):446-457.
26. Braun M, Barstow CH, Pyzocha NJ. Diagnosis and management of sodium disorders: hyponatremia and hypernatremia. Am Fam Physician. 2015;91(5):299-307.
Hyponatremia is a common, multifactorial clinical condition. Hyponatremia is usually defined as a plasma sodium level <135 mmol/L; however, some studies define it as a level <130 mmol/L. Hyponatremia results from the inability of the kidney to excrete a sufficient amount of fluid, or is due to excessive fluid intake. Increases in osmolality stimulate thirst and result in increased fluid intake. This increase in osmolality is recognized by the osmoreceptors located in the hypothalamus, which release antidiuretic hormone (ADH). Antidiuretic hormone works on the collecting ducts within the kidneys, triggering increased fluid reabsorption resulting in decreased fluid loss and a reduction in thirst.
The syndrome of inappropriate antidiuretic hormone (SIADH) occurs when there is persistent ADH stimulation resulting in hyponatremia. SIADH commonly presents as euvolemic hyponatremia. Common diagnostic criteria for SIADH are listed in Table 1.1
Medications are a major cause of SIADH, and psychotropics are a primary offender. Most of the data for drug-induced SIADH come from case reports and small case series, such as those described in Table 2.2-4 The extent to which each psychotropic class causes SIADH remains unknown. In this article, we focus on 3 classes of psychotropics, and their role in causing SIADH.
Antidepressants
There is a fair amount of data associating antidepressants with SIADH. The incidence of SIADH with selective serotonin reuptake inhibitors (SSRIs) varies greatly among studies, from .06% to 40%.5-12 This wide variation is due to the way each study defined hyponatremia. A higher incidence was found when hyponatremia was defined as <135 mmol/L as opposed to <130 mmol/L. A large cohort study of SSRIs found that there was an increased risk with fluoxetine, escitalopram, and citalopram (.078% to .085%) vs paroxetine and sertraline (.033% to .053%).13 Studies comparing the incidence of SIADH with SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs) found that the rates were equal or slightly higher with the SNRI venlafaxine.13 SNRIs as a group have an estimated incidence of .08% to 4%, based on studies that defined hyponatremia as <130 mmol/L.13,14 Tricyclic antidepressants have an estimated incidence of .005% to 16.7%, based on a retrospective study that reviewed 15 studies and 100 case reports.15 Mirtazapine and bupropion do not have enough evidence to obtain a true definition of incidence; case reports for these drugs suggest a causal link for hyponatremia. Table 37,9,12-15 provides an overview of the incidence rate of hyponatremia for select antidepressants. It is clear that a more stringent cutoff for hyponatremia (<130 mmol/L) reduces the incidence rates. More evidence is needed to identify the true incidence and prevalence of SIADH with these agents.
Antipsychotics
Compared with antidepressants, there’s less evidence linking SIADH with antipsychotics; this data come mainly from case reports and observational studies. Serrano et al16 reported on a cross-sectional study that included 88 patients receiving clozapine, 61 patients receiving other atypical antipsychotics, 23 patients receiving typical antipsychotics, and 11 patients receiving both typical and atypical antipsychotics. They reported incidence rates of 3.4% for clozapine, 4.9% for atypical antipsychotics, 26.1% for typical antipsychotics, and 9.1% for the group receiving both typical and atypical antipsychotics.16 The primary theory for the decreased incidence of SIADH with use of atypical antipsychotics is related to decreased rates of psychogenic polydipsia leading to lower incidence of hyponatremia.
Mood stabilizers
Several studies have associated carbamazepine/oxcarbazepine, valproic acid, and lamotrigine with SIADH.17-23 Studies show incidence rates ranging from 4.8% to 41.5% for these medications. Carbamazepine appears to have the highest incidence of SIADH. A limitation of these studies is the small sample sizes, which ranged from 12 to 60 participants.
Pathophysiology
The kidneys are responsible for maintaining homeostasis between bodily fluids and serum sodium levels. ADH, which is produced by the hypothalamus, plays a significant role in fluid balance, thirst, and fluid retention. Inappropriate and continuous secretion of ADH, despite normal or high fluid status, results in hyposmolality and hyponatremia. The specific mechanisms by which psychotropic medications cause SIADH are listed in Table 4.24
Diagnosis
Diagnosis of SIADH can be complex because there are many clinical reasons a patient may have hyponatremia. For example, SIADH and psychogenic polydipsia both result in hyponatremia, and sometimes the 2 conditions can be difficult to distinguish. Hyponatremia is typically discovered by routine blood testing if the patient is asymptomatic. Table 525 highlights the major laboratory markers that distinguish SIADH and psychogenic polydipsia.
Continue to: Treatment
Treatment
The primary treatment for SIADH is cessation of the offending agent. Based on the patient’s clinical presentation, free water restriction (.5 to 1 L/d) can be implemented to increase serum sodium levels. If the patient is having neurologic complications due to the severity of hyponatremia, correction with hypertonic saline is indicated. Upon resolution, the recommended course of action is to switch to a medication in a different class. Re-challenging the patient with the same medication is not recommended unless there is no other alternative class of medication.24 Table 626 highlights other causes of hyponatremia, what laboratory markers to assess, and how to treat high-risk individuals.
Hyponatremia is a complex medical complication that can be life-threatening. Psychotropics are a relatively common cause of hyponatremia, specifically SIADH. Older adults appear to be at highest risk, as most case reports are in patients age ≥65. Patients who are prescribed psychotropics should be treated with the lowest effective dose and monitored for signs and symptoms of hyponatremia throughout therapy.
Related Resources
- Spasovski G, Vanholder R, Allolio B, et al. Clinical practice guidelines on diagnosis and treatment of hyponatremia. Eur J Endocrinol. 2014;170(3):G1-G47.
- Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013;126(10 Suppl 1):S1-S42.
Drug Brand Names
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Citalopram • Celexa
Clozapine • Clozaril
Escitalopram • Lexapro
Fluoxetine • Prozac
Haloperidol • Haldol
Lamotrigine • Lamictal
Levathyroxine • Levothroid
Mirtazapine • Remeron
Oxcarbazepine • Trileptal
Paroxetine • Paxil
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Valproic acid • Depakote
Venlafaxine • Effexor
Hyponatremia is a common, multifactorial clinical condition. Hyponatremia is usually defined as a plasma sodium level <135 mmol/L; however, some studies define it as a level <130 mmol/L. Hyponatremia results from the inability of the kidney to excrete a sufficient amount of fluid, or is due to excessive fluid intake. Increases in osmolality stimulate thirst and result in increased fluid intake. This increase in osmolality is recognized by the osmoreceptors located in the hypothalamus, which release antidiuretic hormone (ADH). Antidiuretic hormone works on the collecting ducts within the kidneys, triggering increased fluid reabsorption resulting in decreased fluid loss and a reduction in thirst.
The syndrome of inappropriate antidiuretic hormone (SIADH) occurs when there is persistent ADH stimulation resulting in hyponatremia. SIADH commonly presents as euvolemic hyponatremia. Common diagnostic criteria for SIADH are listed in Table 1.1
Medications are a major cause of SIADH, and psychotropics are a primary offender. Most of the data for drug-induced SIADH come from case reports and small case series, such as those described in Table 2.2-4 The extent to which each psychotropic class causes SIADH remains unknown. In this article, we focus on 3 classes of psychotropics, and their role in causing SIADH.
Antidepressants
There is a fair amount of data associating antidepressants with SIADH. The incidence of SIADH with selective serotonin reuptake inhibitors (SSRIs) varies greatly among studies, from .06% to 40%.5-12 This wide variation is due to the way each study defined hyponatremia. A higher incidence was found when hyponatremia was defined as <135 mmol/L as opposed to <130 mmol/L. A large cohort study of SSRIs found that there was an increased risk with fluoxetine, escitalopram, and citalopram (.078% to .085%) vs paroxetine and sertraline (.033% to .053%).13 Studies comparing the incidence of SIADH with SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs) found that the rates were equal or slightly higher with the SNRI venlafaxine.13 SNRIs as a group have an estimated incidence of .08% to 4%, based on studies that defined hyponatremia as <130 mmol/L.13,14 Tricyclic antidepressants have an estimated incidence of .005% to 16.7%, based on a retrospective study that reviewed 15 studies and 100 case reports.15 Mirtazapine and bupropion do not have enough evidence to obtain a true definition of incidence; case reports for these drugs suggest a causal link for hyponatremia. Table 37,9,12-15 provides an overview of the incidence rate of hyponatremia for select antidepressants. It is clear that a more stringent cutoff for hyponatremia (<130 mmol/L) reduces the incidence rates. More evidence is needed to identify the true incidence and prevalence of SIADH with these agents.
Antipsychotics
Compared with antidepressants, there’s less evidence linking SIADH with antipsychotics; this data come mainly from case reports and observational studies. Serrano et al16 reported on a cross-sectional study that included 88 patients receiving clozapine, 61 patients receiving other atypical antipsychotics, 23 patients receiving typical antipsychotics, and 11 patients receiving both typical and atypical antipsychotics. They reported incidence rates of 3.4% for clozapine, 4.9% for atypical antipsychotics, 26.1% for typical antipsychotics, and 9.1% for the group receiving both typical and atypical antipsychotics.16 The primary theory for the decreased incidence of SIADH with use of atypical antipsychotics is related to decreased rates of psychogenic polydipsia leading to lower incidence of hyponatremia.
Mood stabilizers
Several studies have associated carbamazepine/oxcarbazepine, valproic acid, and lamotrigine with SIADH.17-23 Studies show incidence rates ranging from 4.8% to 41.5% for these medications. Carbamazepine appears to have the highest incidence of SIADH. A limitation of these studies is the small sample sizes, which ranged from 12 to 60 participants.
Pathophysiology
The kidneys are responsible for maintaining homeostasis between bodily fluids and serum sodium levels. ADH, which is produced by the hypothalamus, plays a significant role in fluid balance, thirst, and fluid retention. Inappropriate and continuous secretion of ADH, despite normal or high fluid status, results in hyposmolality and hyponatremia. The specific mechanisms by which psychotropic medications cause SIADH are listed in Table 4.24
Diagnosis
Diagnosis of SIADH can be complex because there are many clinical reasons a patient may have hyponatremia. For example, SIADH and psychogenic polydipsia both result in hyponatremia, and sometimes the 2 conditions can be difficult to distinguish. Hyponatremia is typically discovered by routine blood testing if the patient is asymptomatic. Table 525 highlights the major laboratory markers that distinguish SIADH and psychogenic polydipsia.
Continue to: Treatment
Treatment
The primary treatment for SIADH is cessation of the offending agent. Based on the patient’s clinical presentation, free water restriction (.5 to 1 L/d) can be implemented to increase serum sodium levels. If the patient is having neurologic complications due to the severity of hyponatremia, correction with hypertonic saline is indicated. Upon resolution, the recommended course of action is to switch to a medication in a different class. Re-challenging the patient with the same medication is not recommended unless there is no other alternative class of medication.24 Table 626 highlights other causes of hyponatremia, what laboratory markers to assess, and how to treat high-risk individuals.
Hyponatremia is a complex medical complication that can be life-threatening. Psychotropics are a relatively common cause of hyponatremia, specifically SIADH. Older adults appear to be at highest risk, as most case reports are in patients age ≥65. Patients who are prescribed psychotropics should be treated with the lowest effective dose and monitored for signs and symptoms of hyponatremia throughout therapy.
Related Resources
- Spasovski G, Vanholder R, Allolio B, et al. Clinical practice guidelines on diagnosis and treatment of hyponatremia. Eur J Endocrinol. 2014;170(3):G1-G47.
- Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013;126(10 Suppl 1):S1-S42.
Drug Brand Names
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Citalopram • Celexa
Clozapine • Clozaril
Escitalopram • Lexapro
Fluoxetine • Prozac
Haloperidol • Haldol
Lamotrigine • Lamictal
Levathyroxine • Levothroid
Mirtazapine • Remeron
Oxcarbazepine • Trileptal
Paroxetine • Paxil
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Valproic acid • Depakote
Venlafaxine • Effexor
1. Sahay M, Sahay R. Hyponatremia: a practical approach. Indian J Endocrinol Metab. 2014;18(6):760-771.
2. Kenes MT, Hamblin S, Tumuluri SS, et al. Syndrome of inappropriate antidiuretic hormone in a patient receiving high-dose haloperidol and quetiapine therapy. J Neuropsychiatry Clin Neurosci. 2016;28(2):e29-e30. doi: 10.1176/appi.neuropsych.15110392.
3. Twardowschy CA, Bertolucci CB, Gracia Cde M, et al. Severe hyponatremia and the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) associated with fluoxetine: case report. Arq Neuropsiquiatr. 2006;64(1):142-145.
4. Patel KR, Meesala A, Stanilla JK. Sodium valproate–induced hyponatremia: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(5):PCC.09100941. doi: 10.4088/PCC.09100941.
5. Pillans PI, Coulter DM. Fluoxetine and hyponatraemia—a potential hazard in the elderly. N Z Med J. 1994;107(973):85‑86.
6. Strachan J, Shepherd J. Hyponatraemia associated with the use of selective serotonin reuptake inhibitors. Aust N Z J Psychiatry. 1998;32(2):295‑298.
7. Bouman WP, Pinner G, Johnson H. Incidence of selective serotonin reuptake inhibitor (SSRI) induced hyponatraemia due to the syndrome of inappropriate antidiuretic hormone (SIADH) secretion in the elderly. Int J Geriatr Psychiatry. 1998;13(1):12‑15.
8. Wilkinson TJ, Begg EJ, Winter AC, et al. Incidence and risk factors for hyponatraemia following treatment with fluoxetine or paroxetine in elderly people. Br J Clin Pharmacol. 1999;47(2):211‑217.
9. Kirby D, Harrigan S, Ames D. Hyponatraemia in elderly psychiatric patients treated with selective serotonin reuptake inhibitors and venlafaxine: a retrospective controlled study in an inpatient unit. Int J Geriatr Psychiatry. 2002;17(3):231‑237.
10. Wee R, Lim WK. Selective serotonin re‑uptake inhibitors (SSRIs) and hyponatraemia in the elderly. Int J Geriatr Psychiatry. 2004;19(6):590‑591.
11. Jung YE, Jun TY, Kim KS, et al. Hyponatremia associated with selective serotonin reuptake inhibitors, mirtazapine, and venlafaxine in Korean patients with major depressive disorder. Int J Clin Pharmacol Ther. 2011;49(7):437‑443.
12. Letmaier M, Painold A, Holl AK, et al. Hyponatremia during psychopharmacological treatment: Results of a drug surveillance program. Int J Neuropsychopharmacol. 2012;15(6):739‑748.
13. Coupland CA, Dhiman P, Barton G, et al. A study of the safety and harms of antidepressant drugs for older people: a cohort study using a large primary care database. Health Technol Assess. 2011;15(28):1‑202, iii‑iv.
14. Leah-Møller KB, Hansen AH, Torstensson M, et al. Antidepressants and the risk of hyponatremia: a Danish register-based population study. BMJ Open. 2016;6(5):e011200. doi: 10.1136/bmjopen-2016-011200.
15. De Picker LD, Van Den Eede F, Dumont G, et al. Antidepressants and the risk of hyponatremia: a class by class review of literature. Psychosomatics. 2014;55(6):536-547.
16. Serrano A, Rangel N, Carrizo E, et al. Safety of long-term clozapine administration. Frequency of cardiomyopathy and hyponatraemia: two cross-sectional, naturalistic studies. Aust N Z J Psychiatry. 2014;48(2):183‑192.
17. Uhde TW, Post RM. Effects of carbamazepine on serum electrolytes: clinical and theoretical implications. J Clin Psychopharmacol. 1983;3(2):103‑106.
18. Lahr MB. Hyponatremia during carbamazepine therapy. Clin Pharmacol Ther. 1985;37(6):693‑696.
19. Joffe RT, Post RM, Uhde TW. Effects of carbamazepine on serum electrolytes in affectively ill patients. Psychol Med. 1986;16(2):331‑335.
20. Vieweg V, Glick JL, Herring S, et al. Absence of carbamazepine‑induced hyponatremia among patients also given lithium. Am J Psychiatry. 1987;144(7):943‑947.
21. Yassa R, Iskandar H, Nastase C, et al. Carbamazepine and hyponatremia in patients with affective disorder. Am J Psychiatry. 1988;145(3):339‑342.
22. Kastner T, Friedman DL, Pond WS. Carbamazepine‑induced hyponatremia in patients with mental retardation. Am J Ment Retard. 1992;96(5):536‑540.
23. Kelly BD, Hillery J. Hyponatremia during carbamazepine therapy in patients with intellectual disability. J Intellect Disabil Res. 2001;45(Pt 2):152‑156.
24. Sahoo S, Grover S. Hyponatremia and psychotropics. J Geriatr Ment Health. 2016;3(2):108-122.
25. Siragy HM. Hyponatremia, fluid-electrolyte disorders and the syndrome of inappropriate antidiuretic hormone secretion: diagnosis and treatment options. Endocr Pract. 2006;12(4):446-457.
26. Braun M, Barstow CH, Pyzocha NJ. Diagnosis and management of sodium disorders: hyponatremia and hypernatremia. Am Fam Physician. 2015;91(5):299-307.
1. Sahay M, Sahay R. Hyponatremia: a practical approach. Indian J Endocrinol Metab. 2014;18(6):760-771.
2. Kenes MT, Hamblin S, Tumuluri SS, et al. Syndrome of inappropriate antidiuretic hormone in a patient receiving high-dose haloperidol and quetiapine therapy. J Neuropsychiatry Clin Neurosci. 2016;28(2):e29-e30. doi: 10.1176/appi.neuropsych.15110392.
3. Twardowschy CA, Bertolucci CB, Gracia Cde M, et al. Severe hyponatremia and the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) associated with fluoxetine: case report. Arq Neuropsiquiatr. 2006;64(1):142-145.
4. Patel KR, Meesala A, Stanilla JK. Sodium valproate–induced hyponatremia: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(5):PCC.09100941. doi: 10.4088/PCC.09100941.
5. Pillans PI, Coulter DM. Fluoxetine and hyponatraemia—a potential hazard in the elderly. N Z Med J. 1994;107(973):85‑86.
6. Strachan J, Shepherd J. Hyponatraemia associated with the use of selective serotonin reuptake inhibitors. Aust N Z J Psychiatry. 1998;32(2):295‑298.
7. Bouman WP, Pinner G, Johnson H. Incidence of selective serotonin reuptake inhibitor (SSRI) induced hyponatraemia due to the syndrome of inappropriate antidiuretic hormone (SIADH) secretion in the elderly. Int J Geriatr Psychiatry. 1998;13(1):12‑15.
8. Wilkinson TJ, Begg EJ, Winter AC, et al. Incidence and risk factors for hyponatraemia following treatment with fluoxetine or paroxetine in elderly people. Br J Clin Pharmacol. 1999;47(2):211‑217.
9. Kirby D, Harrigan S, Ames D. Hyponatraemia in elderly psychiatric patients treated with selective serotonin reuptake inhibitors and venlafaxine: a retrospective controlled study in an inpatient unit. Int J Geriatr Psychiatry. 2002;17(3):231‑237.
10. Wee R, Lim WK. Selective serotonin re‑uptake inhibitors (SSRIs) and hyponatraemia in the elderly. Int J Geriatr Psychiatry. 2004;19(6):590‑591.
11. Jung YE, Jun TY, Kim KS, et al. Hyponatremia associated with selective serotonin reuptake inhibitors, mirtazapine, and venlafaxine in Korean patients with major depressive disorder. Int J Clin Pharmacol Ther. 2011;49(7):437‑443.
12. Letmaier M, Painold A, Holl AK, et al. Hyponatremia during psychopharmacological treatment: Results of a drug surveillance program. Int J Neuropsychopharmacol. 2012;15(6):739‑748.
13. Coupland CA, Dhiman P, Barton G, et al. A study of the safety and harms of antidepressant drugs for older people: a cohort study using a large primary care database. Health Technol Assess. 2011;15(28):1‑202, iii‑iv.
14. Leah-Møller KB, Hansen AH, Torstensson M, et al. Antidepressants and the risk of hyponatremia: a Danish register-based population study. BMJ Open. 2016;6(5):e011200. doi: 10.1136/bmjopen-2016-011200.
15. De Picker LD, Van Den Eede F, Dumont G, et al. Antidepressants and the risk of hyponatremia: a class by class review of literature. Psychosomatics. 2014;55(6):536-547.
16. Serrano A, Rangel N, Carrizo E, et al. Safety of long-term clozapine administration. Frequency of cardiomyopathy and hyponatraemia: two cross-sectional, naturalistic studies. Aust N Z J Psychiatry. 2014;48(2):183‑192.
17. Uhde TW, Post RM. Effects of carbamazepine on serum electrolytes: clinical and theoretical implications. J Clin Psychopharmacol. 1983;3(2):103‑106.
18. Lahr MB. Hyponatremia during carbamazepine therapy. Clin Pharmacol Ther. 1985;37(6):693‑696.
19. Joffe RT, Post RM, Uhde TW. Effects of carbamazepine on serum electrolytes in affectively ill patients. Psychol Med. 1986;16(2):331‑335.
20. Vieweg V, Glick JL, Herring S, et al. Absence of carbamazepine‑induced hyponatremia among patients also given lithium. Am J Psychiatry. 1987;144(7):943‑947.
21. Yassa R, Iskandar H, Nastase C, et al. Carbamazepine and hyponatremia in patients with affective disorder. Am J Psychiatry. 1988;145(3):339‑342.
22. Kastner T, Friedman DL, Pond WS. Carbamazepine‑induced hyponatremia in patients with mental retardation. Am J Ment Retard. 1992;96(5):536‑540.
23. Kelly BD, Hillery J. Hyponatremia during carbamazepine therapy in patients with intellectual disability. J Intellect Disabil Res. 2001;45(Pt 2):152‑156.
24. Sahoo S, Grover S. Hyponatremia and psychotropics. J Geriatr Ment Health. 2016;3(2):108-122.
25. Siragy HM. Hyponatremia, fluid-electrolyte disorders and the syndrome of inappropriate antidiuretic hormone secretion: diagnosis and treatment options. Endocr Pract. 2006;12(4):446-457.
26. Braun M, Barstow CH, Pyzocha NJ. Diagnosis and management of sodium disorders: hyponatremia and hypernatremia. Am Fam Physician. 2015;91(5):299-307.
Older-age bipolar disorder: A case series
Although the peak age of onset of bipolar disorder (BD) is between 20 and 40 years,1 some patients develop BD later in life. The International Society for Bipolar Disorders Task Force has classified the illness into 3 categories:
- early-onset bipolar disorder (EOBD), in which the first manic episode occurs before age 40
- late-onset bipolar disorder (LOBD), in which the initial manic/hypomanic episode occurs after age 50
- older-age bipolar disorder (OABD), in which the first manic/hypomanic episode occurs after age 60.2
OABD represents 25% of the population with BD.3 OABD differs from EOBD in its clinical presentation, biological factors, and psychiatric and somatic comorbidities.4 Studies suggest OABD warrants a more extensive workup to rule out organic causes because symptoms are often attributable to a variety of organic etiologies.
This article describes 3 cases of OABD, including treatments and outcomes. We discuss general treatment recommendations for patients with OABD as cited in the literature. Further research is needed to expand our ability to better care for this unique population.
CASE 1
Mr. D was a 66-year-old African American male with no psychiatric history. His medical history was significant for hypertension, poorly controlled diabetes mellitus, and chronic kidney disease. One year ago, he was diagnosed with cholangiocarcinoma, and underwent uncomplicated right trisegmentectomy, resection of extrahepatic biliary tree, and complete portal lymphadenectomy, with Roux-en-Y hepaticojejunostomy to 2 intrahepatic ducts. He presented to the emergency department (ED) with disorganized behavior for 3 weeks. During that time, Mr. D reported increased distractibility, irritability, hyper-religiosity, racing thoughts, decreased appetite, and decreased need for sleep. There was no pertinent family history.
On mental status examination, Mr. D was agitated, noncooperative, and guarded. His speech was loud and pressured. Mr. D was distractible, tangential, and goal-directed. His Young Mania Rating Scale (YMRS) score was 31, which is highly indicative of mania.5 Computed tomography (CT) scan of the head (Figure 1)
")
CASE 2
Mr. M was a 63-year-old African American male with no psychiatric history and a medical history significant for hypertension and hypercholesterolemia. He presented to the ED with behavioral changes for 2 weeks. During this time, he experienced decreased need for sleep, agitation, excessive spending, self-conversing, hypersexuality, and paranoia. His family history was significant for schizoaffective disorder, bipolar type.

A mental status examination revealed pressured speech, grandiose delusions, hyper-religiosity, flight of ideas, looseness of association, auditory hallucinations, and tangential thought processes. Mr. M’s initial YMRS score was 56. A CT scan of the head revealed no acute abnormality, but MRI of the brain (Figure 2) showed chronic microvascular ischemic change. Mr. M was diagnosed with bipolar I disorder and admitted. He was started on quetiapine extended release, which was titrated to 600 mg nightly. Divalproex sodium extended release was titrated to 1,500 mg nightly, with subsequent improvement. At discharge, his YMRS score was 15.
Continue to: CASE 3
CASE 3
Ms. F was a 69-year-old White female with no psychiatric history. Her medical history was significant for hypertension, osteoarthritis, and stage III-C ovarian adenocarcinoma with a debulking surgical procedure 5 years earlier. After that, she received adjuvant therapy with paclitaxel and carboplatin, which resulted in a 10-month disease-free interval. Subsequent progression led to cycles of doxorubicin liposomal and gemcitabine. She was in remission until 1 week earlier, when a CT scan of the abdomen/pelvis showed recurrence. She presented to the hospital after disrobing in the street due to hyper-religiosity and divine instruction. She endorsed elevated mood and increased energy despite sleeping only 2 hours daily. Her family psychiatric history was significant for her daughter’s suicide attempt.
A mental status examination revealed disorganized behavior and agitation. Her speech was loud and pressured. She described a “great” mood with congruent affect. Her thought process was circumstantial and illogical. She displayed flight of ideas, grandiose delusions, and paranoia. Ms. F’s initial YMRS score was 38. Vital signs were significant for an elevated blood pressure of 153/113 mm Hg. A CT scan of the head (Figure 3) showed age-related change with no acute findings. Ms. F was admitted with a diagnosis of bipolar I disorder and prescribed olanzapine, 2.5 mg nightly. Due to continued manic symptoms, olanzapine was discontinued, and Ms. F was started on quetiapine, 300 mg nightly, with subsequent improvement. At discharge, her YMRS score was 10.
")
Differences between EOBD and OABD
BD has always been considered a multi-system illness; however, comorbidity is much more common in OABD than in EOBD. Comorbid conditions are 3 to 4 times more common in patients with OABD.2 Common comorbidities include metabolic syndrome, allergic rhinitis, arthritis, asthma, and cardiovascular disease.
Compared with younger individuals, older patients with BD score lower on the YMRS in the areas of increased activity-energy, language-thought disorder, and sexual interest.6 Psychotic symptoms are less common or less severe in OABD. Although symptom severity is lower, the prevalence of rapid cycling illness is 20% higher in patients with OABD.6 OABD is less commonly associated with a family history.7 This may suggest a difference from the popular genetic component typically found in patients with EOBD.
Cognitive impairment is more commonly found in OABD. Patients with OABD suffer from neuropsychological deficits even during euthymic phases.8 While these deficits may also be found in patients with EOBD, compared with younger patients, older adults are more susceptible to accelerated decline in cognition. OABD can first present within the context of cardiovascular or neuropsychological impairment. It has also been linked to a greater prevalence of white matter hyperintensities compared with EOBD.9,10
Continue to: Treatment is not specific to OABD
Treatment is not specific to OABD
No established treatment guidelines specifically address OABD. It has been treated similarly to EOBD, with antipsychotics, mood stabilizers, antidepressants, and electroconvulsive therapy (ECT). Although lithium is effective, special precautions should be taken when prescribing it to older adults because these patients may be more sensitive to adverse events.11 Drug–drug interactions may also be more likely due to concomitant use of medications for common medical issues such as hypertension.
Treatment with antipsychotics in older patients carries risks. Use of antipsychotics may result in higher rates of morbidity and mortality related to cardiovascular, metabolic, and infectious etiologies. Some literature recommends the use of antipsychotics for OABD; however, the potential benefits must outweigh the risks.6 Monotherapy followed by combination therapy has demonstrated effectiveness in OABD.11 Because symptoms of OABD are often less severe, it may be best to avoid maintenance antipsychotic therapy when possible. With a higher prevalence of depressed mood following manic episodes, use of antidepressant therapy is common in OABD.6 ECT should be considered for patients with treatment-refractory BD.11
Lessons from our case series
Our case series included 3 patients with OABD. These patients’ comorbid conditions included hypertension, hypercholesteremia, and diabetes mellitus. Two patients had a history of cancer, but there was no metastasis to the brain in either case. However, we considered the possibility of structural changes in the brain or cognitive impairment secondary to cancer or its treatment. A literature review confirmed that adult patients treated for noncentral nervous system cancer experienced cancer-related cognitive impairment (CRCI).12 New research suggests that CRCI could be related to altered neuronal integrity along with a disturbance of brain structure networks that process and integrate information.13
We used the YMRS to compare symptom severity and treatment response (Figure 4). Two patients were treated with atypical antipsychotics with a mood stabilizer, and the third patient was prescribed an antipsychotic only. We avoided lithium and carbamazepine as mood stabilizers due to their adverse effect profiles and potential for drug–drug interactions. Each patient responded well to treatment without adverse events.

Future studies are needed to clearly define the safest and most effective treatment guidelines in patients with OABD. We believe that OABD may require the development of a unique treatment algorithm due to the high likelihood of medical comorbidity and age-related variations in treatment response.
Continue to: Etiology of OABD may be different
Etiology of OABD may be different
OABD may be associated with manic presentations and vascular risk factors. MRI imaging that found more white matter hyperintensities and cerebrovascular lesions in patients with OABD compared with younger patients provides evidence of possible differing etiologies.14 Cassidy and Carroll15 found a higher incidence of smoking, hypertension, diabetes mellitus, coronary heart disease, and atrial fibrillation in patients in the older onset group. Bellivier et al16 proposed 3 subgroups of bipolar I disorder; the late-onset subgroup’s etiology was multifactorial. EOBD and OABD subgroups have similar gender ratios,17 first-episode descriptions, and alcohol use rates; however, OABD subgroups have more neurological comorbidity, lesser severe psychosis, and less genetic predisposition.
Although 25% of BD cases are late onset,3 there is still little consensus regarding subgroups and etiological causes. Therefore, additional research specifically focusing on vascular risks may provide much-needed information. Controlling and mitigating vascular risks in OABD may affect its development and course. Despite debated etiologies, the treatment of BD remains consistent, with anticonvulsants preferred over lithium in older individuals.

The Table summarizes clinical pearls about the features and treatment of OABD.
Bottom Line
Compared with younger patients with bipolar disorder (BD), those who develop BD later in life may be more likely to have rapid cycling, medical comorbidities, and cognitive impairment. Older patients with BD also may be more likely to experience adverse effects of the medications commonly used to treat BD, including antipsychotics, lithium, and carbamazepine.
Related Resources
- Carlino AR, Stinnett JL, Kim DR. New onset of bipolar disorder in late life. Psychosomatics. 2013;54(1):94-97.
- Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: Focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
Drug Brand Names
Carbamazepine • Carbatrol, Tegretol
Carboplatin • Paraplatin
Divalproex sodium • Depakote
Doxorubicin liposome injection • Doxil
Gemcitabine injection • Gemzar
Lithium • Eskalith, Lithobid
Olanzapine • Zyprexa
Paclitaxel injection • Abraxane
Quetiapine • Seroquel
1. Prabhakar D, Balon R. Late-onset bipolar disorder: a case for careful appraisal. Psychiatry (Edgmont). 2010;7(1):34-37.
2. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
3. Arciniegas DB. New-onset bipolar disorder in late life: a case of mistaken identity. Am J Psychiatry. 2006;163(2):198-203.
4. Chou P-H, Tseng W-J, Chen L-M, et al. Late onset bipolar disorder: a case report and review of the literature. Journal of Clinical Gerontology and Geriatrics. 2015;6(1):27-29.
5. Lukasiewicz M, Gerard S, Besnard A, et al; Emblem Study Group. Young Mania Rating Scale: how to interpret the numbers? Determination of a severity threshold and of the minimal clinically significant difference in the EMBLEM cohort. Int J Methods Psychiatr Res. 2013;22(1):46-58.
6. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
7. Depp CA, Jeste D V. Bipolar disorder in older adults: A critical review. Bipolar Disord. 2004;6(5):343-367.8.
8. Gildengers AG, Butters MA, et al. Cognitive functioning in late-life bipolar disorder. Am J Psychiatry. 2004. doi:10.1176/appi.ajp.161.4.736
9. Steffens DC, Krishnan KR. Structural neuroimaging and mood disorders: Recent findings, implications for classification, and future directions. Biological Psychiatry. 1998;43(10):705-712.
10. Tamashiro JH, Zung S, Zanetti MV, et al. Increased rates of white matter hyperintensities in late-onset bipolar disorder. Bipolar Disord. 2008;10(7):765-775.
11. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
12. Wefel JS, Kesler SR, Noll KR, et al. Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. CA Cancer J Clin. 2015;65(2):123-138.
13. Amidi A, Hosseini SMH, Leemans A, et al. Changes in brain structural networks and cognitive functions in testicular cancer patients receiving cisplatin-based chemotherapy. J Natl Cancer Inst. 2017;109(12). doi: 10.1093/jnci/djx085.
14. Torrence C, Jackson J. New onset mania in late life: case report and literature review. J Mississippi Acad Sci. 2016;61(1):159.
15. Cassidy F, Carroll BJ. Vascular risk factors in late onset mania. Psychol Med. 2002;32(2):359-362.
16. Bellivier F, Golmard JL, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160(5):999-1001.
17. Almeida OP, Fenner S. Bipolar disorder: similarities and differences between patients with illness onset before and after 65 years of age. Int Psychogeriatr. 2002;14(3):311-322.
18. Schürhoff F, Bellivier F, Jouvent R, et al. Early and late onset bipolar disorders: two different forms of manic-depressive illness? J Affect Disord. 2000;58(3):215-21.
Although the peak age of onset of bipolar disorder (BD) is between 20 and 40 years,1 some patients develop BD later in life. The International Society for Bipolar Disorders Task Force has classified the illness into 3 categories:
- early-onset bipolar disorder (EOBD), in which the first manic episode occurs before age 40
- late-onset bipolar disorder (LOBD), in which the initial manic/hypomanic episode occurs after age 50
- older-age bipolar disorder (OABD), in which the first manic/hypomanic episode occurs after age 60.2
OABD represents 25% of the population with BD.3 OABD differs from EOBD in its clinical presentation, biological factors, and psychiatric and somatic comorbidities.4 Studies suggest OABD warrants a more extensive workup to rule out organic causes because symptoms are often attributable to a variety of organic etiologies.
This article describes 3 cases of OABD, including treatments and outcomes. We discuss general treatment recommendations for patients with OABD as cited in the literature. Further research is needed to expand our ability to better care for this unique population.
CASE 1
Mr. D was a 66-year-old African American male with no psychiatric history. His medical history was significant for hypertension, poorly controlled diabetes mellitus, and chronic kidney disease. One year ago, he was diagnosed with cholangiocarcinoma, and underwent uncomplicated right trisegmentectomy, resection of extrahepatic biliary tree, and complete portal lymphadenectomy, with Roux-en-Y hepaticojejunostomy to 2 intrahepatic ducts. He presented to the emergency department (ED) with disorganized behavior for 3 weeks. During that time, Mr. D reported increased distractibility, irritability, hyper-religiosity, racing thoughts, decreased appetite, and decreased need for sleep. There was no pertinent family history.
On mental status examination, Mr. D was agitated, noncooperative, and guarded. His speech was loud and pressured. Mr. D was distractible, tangential, and goal-directed. His Young Mania Rating Scale (YMRS) score was 31, which is highly indicative of mania.5 Computed tomography (CT) scan of the head (Figure 1)
CASE 2
Mr. M was a 63-year-old African American male with no psychiatric history and a medical history significant for hypertension and hypercholesterolemia. He presented to the ED with behavioral changes for 2 weeks. During this time, he experienced decreased need for sleep, agitation, excessive spending, self-conversing, hypersexuality, and paranoia. His family history was significant for schizoaffective disorder, bipolar type.
A mental status examination revealed pressured speech, grandiose delusions, hyper-religiosity, flight of ideas, looseness of association, auditory hallucinations, and tangential thought processes. Mr. M’s initial YMRS score was 56. A CT scan of the head revealed no acute abnormality, but MRI of the brain (Figure 2) showed chronic microvascular ischemic change. Mr. M was diagnosed with bipolar I disorder and admitted. He was started on quetiapine extended release, which was titrated to 600 mg nightly. Divalproex sodium extended release was titrated to 1,500 mg nightly, with subsequent improvement. At discharge, his YMRS score was 15.
Continue to: CASE 3
CASE 3
Ms. F was a 69-year-old White female with no psychiatric history. Her medical history was significant for hypertension, osteoarthritis, and stage III-C ovarian adenocarcinoma with a debulking surgical procedure 5 years earlier. After that, she received adjuvant therapy with paclitaxel and carboplatin, which resulted in a 10-month disease-free interval. Subsequent progression led to cycles of doxorubicin liposomal and gemcitabine. She was in remission until 1 week earlier, when a CT scan of the abdomen/pelvis showed recurrence. She presented to the hospital after disrobing in the street due to hyper-religiosity and divine instruction. She endorsed elevated mood and increased energy despite sleeping only 2 hours daily. Her family psychiatric history was significant for her daughter’s suicide attempt.
A mental status examination revealed disorganized behavior and agitation. Her speech was loud and pressured. She described a “great” mood with congruent affect. Her thought process was circumstantial and illogical. She displayed flight of ideas, grandiose delusions, and paranoia. Ms. F’s initial YMRS score was 38. Vital signs were significant for an elevated blood pressure of 153/113 mm Hg. A CT scan of the head (Figure 3) showed age-related change with no acute findings. Ms. F was admitted with a diagnosis of bipolar I disorder and prescribed olanzapine, 2.5 mg nightly. Due to continued manic symptoms, olanzapine was discontinued, and Ms. F was started on quetiapine, 300 mg nightly, with subsequent improvement. At discharge, her YMRS score was 10.
Differences between EOBD and OABD
BD has always been considered a multi-system illness; however, comorbidity is much more common in OABD than in EOBD. Comorbid conditions are 3 to 4 times more common in patients with OABD.2 Common comorbidities include metabolic syndrome, allergic rhinitis, arthritis, asthma, and cardiovascular disease.
Compared with younger individuals, older patients with BD score lower on the YMRS in the areas of increased activity-energy, language-thought disorder, and sexual interest.6 Psychotic symptoms are less common or less severe in OABD. Although symptom severity is lower, the prevalence of rapid cycling illness is 20% higher in patients with OABD.6 OABD is less commonly associated with a family history.7 This may suggest a difference from the popular genetic component typically found in patients with EOBD.
Cognitive impairment is more commonly found in OABD. Patients with OABD suffer from neuropsychological deficits even during euthymic phases.8 While these deficits may also be found in patients with EOBD, compared with younger patients, older adults are more susceptible to accelerated decline in cognition. OABD can first present within the context of cardiovascular or neuropsychological impairment. It has also been linked to a greater prevalence of white matter hyperintensities compared with EOBD.9,10
Continue to: Treatment is not specific to OABD
Treatment is not specific to OABD
No established treatment guidelines specifically address OABD. It has been treated similarly to EOBD, with antipsychotics, mood stabilizers, antidepressants, and electroconvulsive therapy (ECT). Although lithium is effective, special precautions should be taken when prescribing it to older adults because these patients may be more sensitive to adverse events.11 Drug–drug interactions may also be more likely due to concomitant use of medications for common medical issues such as hypertension.
Treatment with antipsychotics in older patients carries risks. Use of antipsychotics may result in higher rates of morbidity and mortality related to cardiovascular, metabolic, and infectious etiologies. Some literature recommends the use of antipsychotics for OABD; however, the potential benefits must outweigh the risks.6 Monotherapy followed by combination therapy has demonstrated effectiveness in OABD.11 Because symptoms of OABD are often less severe, it may be best to avoid maintenance antipsychotic therapy when possible. With a higher prevalence of depressed mood following manic episodes, use of antidepressant therapy is common in OABD.6 ECT should be considered for patients with treatment-refractory BD.11
Lessons from our case series
Our case series included 3 patients with OABD. These patients’ comorbid conditions included hypertension, hypercholesteremia, and diabetes mellitus. Two patients had a history of cancer, but there was no metastasis to the brain in either case. However, we considered the possibility of structural changes in the brain or cognitive impairment secondary to cancer or its treatment. A literature review confirmed that adult patients treated for noncentral nervous system cancer experienced cancer-related cognitive impairment (CRCI).12 New research suggests that CRCI could be related to altered neuronal integrity along with a disturbance of brain structure networks that process and integrate information.13
We used the YMRS to compare symptom severity and treatment response (Figure 4). Two patients were treated with atypical antipsychotics with a mood stabilizer, and the third patient was prescribed an antipsychotic only. We avoided lithium and carbamazepine as mood stabilizers due to their adverse effect profiles and potential for drug–drug interactions. Each patient responded well to treatment without adverse events.
Future studies are needed to clearly define the safest and most effective treatment guidelines in patients with OABD. We believe that OABD may require the development of a unique treatment algorithm due to the high likelihood of medical comorbidity and age-related variations in treatment response.
Continue to: Etiology of OABD may be different
Etiology of OABD may be different
OABD may be associated with manic presentations and vascular risk factors. MRI imaging that found more white matter hyperintensities and cerebrovascular lesions in patients with OABD compared with younger patients provides evidence of possible differing etiologies.14 Cassidy and Carroll15 found a higher incidence of smoking, hypertension, diabetes mellitus, coronary heart disease, and atrial fibrillation in patients in the older onset group. Bellivier et al16 proposed 3 subgroups of bipolar I disorder; the late-onset subgroup’s etiology was multifactorial. EOBD and OABD subgroups have similar gender ratios,17 first-episode descriptions, and alcohol use rates; however, OABD subgroups have more neurological comorbidity, lesser severe psychosis, and less genetic predisposition.
Although 25% of BD cases are late onset,3 there is still little consensus regarding subgroups and etiological causes. Therefore, additional research specifically focusing on vascular risks may provide much-needed information. Controlling and mitigating vascular risks in OABD may affect its development and course. Despite debated etiologies, the treatment of BD remains consistent, with anticonvulsants preferred over lithium in older individuals.
The Table summarizes clinical pearls about the features and treatment of OABD.
Bottom Line
Compared with younger patients with bipolar disorder (BD), those who develop BD later in life may be more likely to have rapid cycling, medical comorbidities, and cognitive impairment. Older patients with BD also may be more likely to experience adverse effects of the medications commonly used to treat BD, including antipsychotics, lithium, and carbamazepine.
Related Resources
- Carlino AR, Stinnett JL, Kim DR. New onset of bipolar disorder in late life. Psychosomatics. 2013;54(1):94-97.
- Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: Focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
Drug Brand Names
Carbamazepine • Carbatrol, Tegretol
Carboplatin • Paraplatin
Divalproex sodium • Depakote
Doxorubicin liposome injection • Doxil
Gemcitabine injection • Gemzar
Lithium • Eskalith, Lithobid
Olanzapine • Zyprexa
Paclitaxel injection • Abraxane
Quetiapine • Seroquel
Although the peak age of onset of bipolar disorder (BD) is between 20 and 40 years,1 some patients develop BD later in life. The International Society for Bipolar Disorders Task Force has classified the illness into 3 categories:
- early-onset bipolar disorder (EOBD), in which the first manic episode occurs before age 40
- late-onset bipolar disorder (LOBD), in which the initial manic/hypomanic episode occurs after age 50
- older-age bipolar disorder (OABD), in which the first manic/hypomanic episode occurs after age 60.2
OABD represents 25% of the population with BD.3 OABD differs from EOBD in its clinical presentation, biological factors, and psychiatric and somatic comorbidities.4 Studies suggest OABD warrants a more extensive workup to rule out organic causes because symptoms are often attributable to a variety of organic etiologies.
This article describes 3 cases of OABD, including treatments and outcomes. We discuss general treatment recommendations for patients with OABD as cited in the literature. Further research is needed to expand our ability to better care for this unique population.
CASE 1
Mr. D was a 66-year-old African American male with no psychiatric history. His medical history was significant for hypertension, poorly controlled diabetes mellitus, and chronic kidney disease. One year ago, he was diagnosed with cholangiocarcinoma, and underwent uncomplicated right trisegmentectomy, resection of extrahepatic biliary tree, and complete portal lymphadenectomy, with Roux-en-Y hepaticojejunostomy to 2 intrahepatic ducts. He presented to the emergency department (ED) with disorganized behavior for 3 weeks. During that time, Mr. D reported increased distractibility, irritability, hyper-religiosity, racing thoughts, decreased appetite, and decreased need for sleep. There was no pertinent family history.
On mental status examination, Mr. D was agitated, noncooperative, and guarded. His speech was loud and pressured. Mr. D was distractible, tangential, and goal-directed. His Young Mania Rating Scale (YMRS) score was 31, which is highly indicative of mania.5 Computed tomography (CT) scan of the head (Figure 1)
CASE 2
Mr. M was a 63-year-old African American male with no psychiatric history and a medical history significant for hypertension and hypercholesterolemia. He presented to the ED with behavioral changes for 2 weeks. During this time, he experienced decreased need for sleep, agitation, excessive spending, self-conversing, hypersexuality, and paranoia. His family history was significant for schizoaffective disorder, bipolar type.
A mental status examination revealed pressured speech, grandiose delusions, hyper-religiosity, flight of ideas, looseness of association, auditory hallucinations, and tangential thought processes. Mr. M’s initial YMRS score was 56. A CT scan of the head revealed no acute abnormality, but MRI of the brain (Figure 2) showed chronic microvascular ischemic change. Mr. M was diagnosed with bipolar I disorder and admitted. He was started on quetiapine extended release, which was titrated to 600 mg nightly. Divalproex sodium extended release was titrated to 1,500 mg nightly, with subsequent improvement. At discharge, his YMRS score was 15.
Continue to: CASE 3
CASE 3
Ms. F was a 69-year-old White female with no psychiatric history. Her medical history was significant for hypertension, osteoarthritis, and stage III-C ovarian adenocarcinoma with a debulking surgical procedure 5 years earlier. After that, she received adjuvant therapy with paclitaxel and carboplatin, which resulted in a 10-month disease-free interval. Subsequent progression led to cycles of doxorubicin liposomal and gemcitabine. She was in remission until 1 week earlier, when a CT scan of the abdomen/pelvis showed recurrence. She presented to the hospital after disrobing in the street due to hyper-religiosity and divine instruction. She endorsed elevated mood and increased energy despite sleeping only 2 hours daily. Her family psychiatric history was significant for her daughter’s suicide attempt.
A mental status examination revealed disorganized behavior and agitation. Her speech was loud and pressured. She described a “great” mood with congruent affect. Her thought process was circumstantial and illogical. She displayed flight of ideas, grandiose delusions, and paranoia. Ms. F’s initial YMRS score was 38. Vital signs were significant for an elevated blood pressure of 153/113 mm Hg. A CT scan of the head (Figure 3) showed age-related change with no acute findings. Ms. F was admitted with a diagnosis of bipolar I disorder and prescribed olanzapine, 2.5 mg nightly. Due to continued manic symptoms, olanzapine was discontinued, and Ms. F was started on quetiapine, 300 mg nightly, with subsequent improvement. At discharge, her YMRS score was 10.
Differences between EOBD and OABD
BD has always been considered a multi-system illness; however, comorbidity is much more common in OABD than in EOBD. Comorbid conditions are 3 to 4 times more common in patients with OABD.2 Common comorbidities include metabolic syndrome, allergic rhinitis, arthritis, asthma, and cardiovascular disease.
Compared with younger individuals, older patients with BD score lower on the YMRS in the areas of increased activity-energy, language-thought disorder, and sexual interest.6 Psychotic symptoms are less common or less severe in OABD. Although symptom severity is lower, the prevalence of rapid cycling illness is 20% higher in patients with OABD.6 OABD is less commonly associated with a family history.7 This may suggest a difference from the popular genetic component typically found in patients with EOBD.
Cognitive impairment is more commonly found in OABD. Patients with OABD suffer from neuropsychological deficits even during euthymic phases.8 While these deficits may also be found in patients with EOBD, compared with younger patients, older adults are more susceptible to accelerated decline in cognition. OABD can first present within the context of cardiovascular or neuropsychological impairment. It has also been linked to a greater prevalence of white matter hyperintensities compared with EOBD.9,10
Continue to: Treatment is not specific to OABD
Treatment is not specific to OABD
No established treatment guidelines specifically address OABD. It has been treated similarly to EOBD, with antipsychotics, mood stabilizers, antidepressants, and electroconvulsive therapy (ECT). Although lithium is effective, special precautions should be taken when prescribing it to older adults because these patients may be more sensitive to adverse events.11 Drug–drug interactions may also be more likely due to concomitant use of medications for common medical issues such as hypertension.
Treatment with antipsychotics in older patients carries risks. Use of antipsychotics may result in higher rates of morbidity and mortality related to cardiovascular, metabolic, and infectious etiologies. Some literature recommends the use of antipsychotics for OABD; however, the potential benefits must outweigh the risks.6 Monotherapy followed by combination therapy has demonstrated effectiveness in OABD.11 Because symptoms of OABD are often less severe, it may be best to avoid maintenance antipsychotic therapy when possible. With a higher prevalence of depressed mood following manic episodes, use of antidepressant therapy is common in OABD.6 ECT should be considered for patients with treatment-refractory BD.11
Lessons from our case series
Our case series included 3 patients with OABD. These patients’ comorbid conditions included hypertension, hypercholesteremia, and diabetes mellitus. Two patients had a history of cancer, but there was no metastasis to the brain in either case. However, we considered the possibility of structural changes in the brain or cognitive impairment secondary to cancer or its treatment. A literature review confirmed that adult patients treated for noncentral nervous system cancer experienced cancer-related cognitive impairment (CRCI).12 New research suggests that CRCI could be related to altered neuronal integrity along with a disturbance of brain structure networks that process and integrate information.13
We used the YMRS to compare symptom severity and treatment response (Figure 4). Two patients were treated with atypical antipsychotics with a mood stabilizer, and the third patient was prescribed an antipsychotic only. We avoided lithium and carbamazepine as mood stabilizers due to their adverse effect profiles and potential for drug–drug interactions. Each patient responded well to treatment without adverse events.
Future studies are needed to clearly define the safest and most effective treatment guidelines in patients with OABD. We believe that OABD may require the development of a unique treatment algorithm due to the high likelihood of medical comorbidity and age-related variations in treatment response.
Continue to: Etiology of OABD may be different
Etiology of OABD may be different
OABD may be associated with manic presentations and vascular risk factors. MRI imaging that found more white matter hyperintensities and cerebrovascular lesions in patients with OABD compared with younger patients provides evidence of possible differing etiologies.14 Cassidy and Carroll15 found a higher incidence of smoking, hypertension, diabetes mellitus, coronary heart disease, and atrial fibrillation in patients in the older onset group. Bellivier et al16 proposed 3 subgroups of bipolar I disorder; the late-onset subgroup’s etiology was multifactorial. EOBD and OABD subgroups have similar gender ratios,17 first-episode descriptions, and alcohol use rates; however, OABD subgroups have more neurological comorbidity, lesser severe psychosis, and less genetic predisposition.
Although 25% of BD cases are late onset,3 there is still little consensus regarding subgroups and etiological causes. Therefore, additional research specifically focusing on vascular risks may provide much-needed information. Controlling and mitigating vascular risks in OABD may affect its development and course. Despite debated etiologies, the treatment of BD remains consistent, with anticonvulsants preferred over lithium in older individuals.
The Table summarizes clinical pearls about the features and treatment of OABD.
Bottom Line
Compared with younger patients with bipolar disorder (BD), those who develop BD later in life may be more likely to have rapid cycling, medical comorbidities, and cognitive impairment. Older patients with BD also may be more likely to experience adverse effects of the medications commonly used to treat BD, including antipsychotics, lithium, and carbamazepine.
Related Resources
- Carlino AR, Stinnett JL, Kim DR. New onset of bipolar disorder in late life. Psychosomatics. 2013;54(1):94-97.
- Sajatovic M, Kales HC, Mulsant BH. Prescribing antipsychotics in geriatric patients: Focus on schizophrenia and bipolar disorder. Current Psychiatry. 2017;16(10):20-26,28.
Drug Brand Names
Carbamazepine • Carbatrol, Tegretol
Carboplatin • Paraplatin
Divalproex sodium • Depakote
Doxorubicin liposome injection • Doxil
Gemcitabine injection • Gemzar
Lithium • Eskalith, Lithobid
Olanzapine • Zyprexa
Paclitaxel injection • Abraxane
Quetiapine • Seroquel
1. Prabhakar D, Balon R. Late-onset bipolar disorder: a case for careful appraisal. Psychiatry (Edgmont). 2010;7(1):34-37.
2. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
3. Arciniegas DB. New-onset bipolar disorder in late life: a case of mistaken identity. Am J Psychiatry. 2006;163(2):198-203.
4. Chou P-H, Tseng W-J, Chen L-M, et al. Late onset bipolar disorder: a case report and review of the literature. Journal of Clinical Gerontology and Geriatrics. 2015;6(1):27-29.
5. Lukasiewicz M, Gerard S, Besnard A, et al; Emblem Study Group. Young Mania Rating Scale: how to interpret the numbers? Determination of a severity threshold and of the minimal clinically significant difference in the EMBLEM cohort. Int J Methods Psychiatr Res. 2013;22(1):46-58.
6. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
7. Depp CA, Jeste D V. Bipolar disorder in older adults: A critical review. Bipolar Disord. 2004;6(5):343-367.8.
8. Gildengers AG, Butters MA, et al. Cognitive functioning in late-life bipolar disorder. Am J Psychiatry. 2004. doi:10.1176/appi.ajp.161.4.736
9. Steffens DC, Krishnan KR. Structural neuroimaging and mood disorders: Recent findings, implications for classification, and future directions. Biological Psychiatry. 1998;43(10):705-712.
10. Tamashiro JH, Zung S, Zanetti MV, et al. Increased rates of white matter hyperintensities in late-onset bipolar disorder. Bipolar Disord. 2008;10(7):765-775.
11. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
12. Wefel JS, Kesler SR, Noll KR, et al. Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. CA Cancer J Clin. 2015;65(2):123-138.
13. Amidi A, Hosseini SMH, Leemans A, et al. Changes in brain structural networks and cognitive functions in testicular cancer patients receiving cisplatin-based chemotherapy. J Natl Cancer Inst. 2017;109(12). doi: 10.1093/jnci/djx085.
14. Torrence C, Jackson J. New onset mania in late life: case report and literature review. J Mississippi Acad Sci. 2016;61(1):159.
15. Cassidy F, Carroll BJ. Vascular risk factors in late onset mania. Psychol Med. 2002;32(2):359-362.
16. Bellivier F, Golmard JL, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160(5):999-1001.
17. Almeida OP, Fenner S. Bipolar disorder: similarities and differences between patients with illness onset before and after 65 years of age. Int Psychogeriatr. 2002;14(3):311-322.
18. Schürhoff F, Bellivier F, Jouvent R, et al. Early and late onset bipolar disorders: two different forms of manic-depressive illness? J Affect Disord. 2000;58(3):215-21.
1. Prabhakar D, Balon R. Late-onset bipolar disorder: a case for careful appraisal. Psychiatry (Edgmont). 2010;7(1):34-37.
2. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
3. Arciniegas DB. New-onset bipolar disorder in late life: a case of mistaken identity. Am J Psychiatry. 2006;163(2):198-203.
4. Chou P-H, Tseng W-J, Chen L-M, et al. Late onset bipolar disorder: a case report and review of the literature. Journal of Clinical Gerontology and Geriatrics. 2015;6(1):27-29.
5. Lukasiewicz M, Gerard S, Besnard A, et al; Emblem Study Group. Young Mania Rating Scale: how to interpret the numbers? Determination of a severity threshold and of the minimal clinically significant difference in the EMBLEM cohort. Int J Methods Psychiatr Res. 2013;22(1):46-58.
6. Oostervink F, Boomsma MM, Nolen WA; EMBLEM Advisory Board. Bipolar disorder in the elderly; different effects of age and of age of onset. J Affect Disord. 2009;116(3):176-183.
7. Depp CA, Jeste D V. Bipolar disorder in older adults: A critical review. Bipolar Disord. 2004;6(5):343-367.8.
8. Gildengers AG, Butters MA, et al. Cognitive functioning in late-life bipolar disorder. Am J Psychiatry. 2004. doi:10.1176/appi.ajp.161.4.736
9. Steffens DC, Krishnan KR. Structural neuroimaging and mood disorders: Recent findings, implications for classification, and future directions. Biological Psychiatry. 1998;43(10):705-712.
10. Tamashiro JH, Zung S, Zanetti MV, et al. Increased rates of white matter hyperintensities in late-onset bipolar disorder. Bipolar Disord. 2008;10(7):765-775.
11. Aziz R, Lorberg B, Tampi RR. Treatments for late-life bipolar disorder. Am J Geriatr Pharmacother. 2006;4(4):347-364.
12. Wefel JS, Kesler SR, Noll KR, et al. Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. CA Cancer J Clin. 2015;65(2):123-138.
13. Amidi A, Hosseini SMH, Leemans A, et al. Changes in brain structural networks and cognitive functions in testicular cancer patients receiving cisplatin-based chemotherapy. J Natl Cancer Inst. 2017;109(12). doi: 10.1093/jnci/djx085.
14. Torrence C, Jackson J. New onset mania in late life: case report and literature review. J Mississippi Acad Sci. 2016;61(1):159.
15. Cassidy F, Carroll BJ. Vascular risk factors in late onset mania. Psychol Med. 2002;32(2):359-362.
16. Bellivier F, Golmard JL, Rietschel M, et al. Age at onset in bipolar I affective disorder: further evidence for three subgroups. Am J Psychiatry. 2003;160(5):999-1001.
17. Almeida OP, Fenner S. Bipolar disorder: similarities and differences between patients with illness onset before and after 65 years of age. Int Psychogeriatr. 2002;14(3):311-322.
18. Schürhoff F, Bellivier F, Jouvent R, et al. Early and late onset bipolar disorders: two different forms of manic-depressive illness? J Affect Disord. 2000;58(3):215-21.

Catatonia: Recognition, management, and prevention of complications
Mr. W, age 50, who has been diagnosed with hypertension and catatonia associated with schizophrenia, is brought to the emergency department by his case manager for evaluation of increasing disorganization, inability to function, and nonadherence to medications. He has not been bathing, eating, or drinking. During the admission interview, he is mute, and is noted to have purposeless activity, alternating between rocking from leg to leg to pacing in circles. At times Mr. W holds a rigid, prayer-type posture with his arms. Negativism is present, primarily opposition to interviewer requests.
Previously stable on
On the inpatient psychiatry unit, Mr. W continues to be mute, staying in bed except to use the bathroom. He refuses all food and fluids. The team initiates subcutaneous

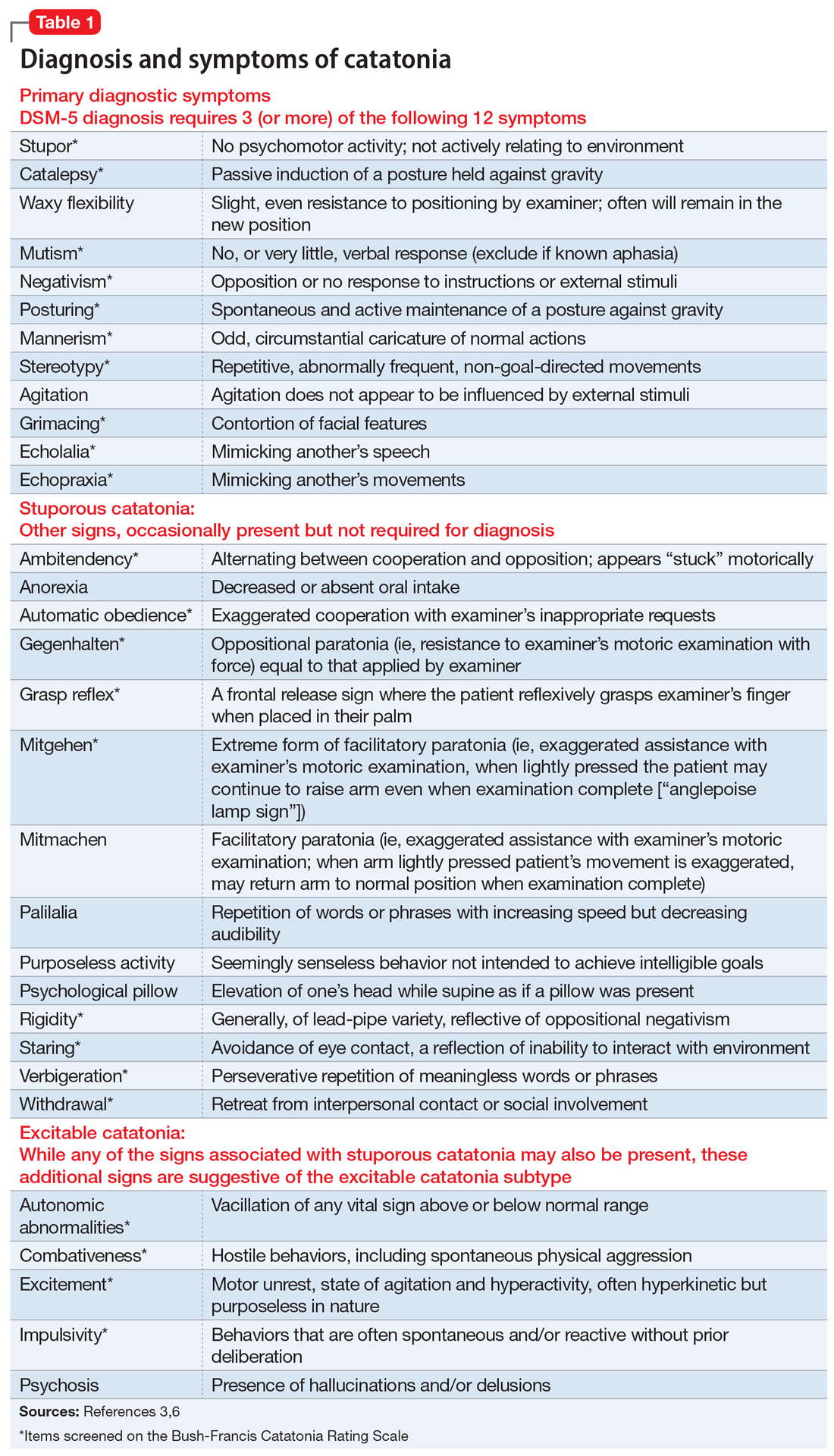
Continue to: Medical complications can be fatal
Medical complications can be fatal

Treatment usually starts with lorazepam
Benzodiazepines are a first-line option for the management of catatonia.2,5 Controversy exists as to effectiveness of different routes of administration. Generally, IV lorazepam is preferred due to its ease of administration, fast onset, and longer duration of action.1 Some inpatient psychiatric units are unable to administer IV benzodiazepines; in these scenarios, IM administration is preferred to oral benzodiazepines.
The initial lorazepam challenge dose should be 2 mg. A positive response to the lorazepam challenge often confirms the catatonia diagnosis.2,7 This challenge should be followed by maintenance doses ranging from 6 to 8 mg/d in divided doses (3 or 4 times a day). Higher doses (up to 24 mg/d) are sometimes used.2,5,8 A recent case report described catatonia remission using lorazepam, 28 mg/d, after unsuccessful ECT.9 The lorazepam dose prior to ECT was 8 mg/d.9 Response is usually seen within 3 to 7 days of an adequate dose.2,8 Parenteral lorazepam typically is continued for several days before converting to oral lorazepam.1 Approximately 70% to 80% of patients with catatonia will show improvement in symptoms with lorazepam.2,7,8
The optimal duration of benzodiazepine treatment is unclear.2 In some cases, once remission of the underlying illness is achieved, benzodiazepines are discontinued.2 However, in other cases, symptoms of catatonia may emerge when lorazepam is tapered, therefore suggesting the need for a longer duration of treatment.2 Despite this high rate of improvement, many patients ultimately receive ECT due to unsustained response or to prevent future episodes of catatonia.
A recent review of 60 Turkish patients with catatonia found 91.7% (n = 55) received oral lorazepam (up to 15 mg/d) as the first-line therapy.7 Improvement was seen in 23.7% (n = 13) of patients treated with lorazepam, yet 70% (n = 42) showed either no response or partial response, and ultimately received ECT in combination with lorazepam.7 The lower improvement rate seen in this review may be secondary to the use of oral lorazepam instead of parenteral, or may highlight the frequency in which patients ultimately go on to receive ECT.
Continue to: ECT
ECT. If high doses of benzodiazepines are not effective within 48 to 72 hours, ECT should be considered.1,7 ECT should be considered sooner for patients with life-threatening catatonia or those who present with excited features or malignant catatonia.1,2,7 In patients with catatonia, ECT response rates range from 80% to 100%.2,7 Unal et al7 reported a 100% response rate if ECT was used as the first-line treatment (n = 5), and a 92.9% (n = 39) response rate after adding ECT to lorazepam. Lorazepam may interfere with the seizure threshold, but if indicated, this medication can be continued.2 A minimum of 6 ECT treatments are suggested; however, as many as 20 treatments have been needed.1 Mr. W required a total of 18 ECT treatments. In some cases, maintenance ECT may be required.2
Antipsychotics. Discontinuation of antipsychotics is generally encouraged in patients presenting with catatonia.2,7,8 Antipsychotics carry a risk of potentially worsening catatonia, conversion to malignant catatonia, or precipitation of NMS; therefore, carefully weigh the risks vs benefits.1,2 If catatonia is secondary to psychosis, as in Mr. W’s case, antipsychotics may be considered once catatonia improves.2 If an antipsychotic is warranted, consider aripiprazole (because of its D2 partial agonist activity) or low-dose olanzapine.1,2 If catatonia is secondary to clozapine withdrawal, the initial therapy should be clozapine re-initiation.1 Although high-potency agents, such as haloperidol and risperidone, typically are not preferred, risperidone was restarted for Mr. W because of his history of response to and tolerability of this medication during a previous catatonic episode.
Other treatments. In a recent review, Beach et al1 described the use of additional agents, mostly in a small number of positive case reports, for managing catatonia. These included:
- zolpidem (zolpidem 10 mg as a challenge test, and doses of ≤40 mg/d)
- the N-methyl-
D -aspartic acid antagonists amantadine (100 to 600 mg/d) or memantine (5 to 20 mg/d) - carbidopa/levodopa
- methylphenidate
- antiepileptics (eg, carbamazepine, topiramate, and divalproex sodium)
- anticholinergics.1,2
Lithium has been used in attempts to prevent recurrent catatonia with limited success.2 There are also a few reports of using transcranial magnetic stimulation (TMS) to manage catatonia.1
Beach et al1 proposed a treatment algorithm in which IV lorazepam (Step 1) and ECT (Step 2) remain the preferred treatments. Next, for Step 3 consider a glutamate antagonist (amantadine or memantine), followed by an antiepileptic (Step 4), and lastly an atypical antipsychotic (aripiprazole, olanzapine, or clozapine) in combination with lorazepam (Step 5).
When indicated, don’t delay ECT
Initial management of catatonia is with a benzodiazepine challenge. Ultimately, the gold-standard treatment of catatonia that does not improve with benzodiazepines is ECT, and ECT should be implemented as soon as it is clear that pharmacotherapy is less than fully effective. Consider ECT initially in life-threatening cases and for patients with malignant catatonia. Although additional agents and TMS have been explored, these should be reserved for patients who fail to respond to, or who are not candidates for, benzodiazepines or ECT.
CASE CONTINUED
After 5 ECT treatments, Mr. W says a few words, but he communicates primarily with gestures (primarily waving people away). After 10 to 12 ECT treatments, Mr. W becomes more interactive and conversant, and his nutrition improves; however, he still exhibits symptoms of catatonia and is not at baseline. He undergoes a total of 18 ECT treatments. Antipsychotics were initially discontinued; however, given Mr. W’s improvement with ECT and the presence of auditory hallucinations, oral risperidone is restarted and titrated to 2 mg, 2 times a day, and he is transitioned back to paliperidone palmitate before he is discharged. Lorazepam is tapered and discontinued. Mr. W is discharged back to his nursing home and is interactive (laughing and joking with family) and attending to his activities of daily living. Unfortunately, Mr. W did not followup with the recommendation for maintenance ECT, and adherence to paliperidone palmitate injections is unknown. Mr. W presented to our facility again 6 months later with symptoms of catatonia and ultimately transferred to a state hospital.
Related Resources
- Fink M, Taylor MA. Catatonia: A clinician’s guide to diagnosis and treatment. New York, NY: Cambridge University Press; 2006. • Carroll BT, Spiegel DR. Catatonia on the consultation liaison service and other clinical settings. Hauppauge, NY: Nova Science Pub Inc.; 2016.
- Benarous X, Raffin M, Ferrafiat V, et al. Catatonia in children and adolescents: new perspectives. Schizophr Res. 2018;200:56-67.
- Malignant Hyperthermia Association of the United States. What is NMSIS? http://www.mhaus.org/nmsis/about-us/ what-is-nmsis/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Asenapine • Saphris
Carbamazepine • Carbatrol, Tegretol
Carbidopa/Levodopa • Sinemet
Citalopram • Celexa
Clozapine • Clozaril
Divalproex Sodium • Depakote
Enoxaparin • Lovenox
Fluoxetine • Prozac
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Lurasidone • Latuda
Memantine • Namenda
Methylphenidate • Concerta, Ritalin
Mirtazapine • Remeron
Olanzapine • Zyprexa
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone long-acting injection • Risperdal Consta
Topiramate • Topamax
Zolpidem • Ambien
1. Beach SR, Gomez-Bernal F, Huffman JC, et al. Alternative treatment strategies for catatonia: a systematic review. Gen Hosp Psychiatry. 2017;48:1-19.
2. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:1-6.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome: focus on treatment and rechallenge. Ann Pharmacother. 2016;50(11):973-981.
5. Ohi K, Kuwata A, Shimada T, et al. Response to benzodiazepines and clinical course in malignant catatonia associated with schizophrenia: a case report. Medicine (Baltimore). 2017;96(16):e6566. doi: 10.1097/MD.0000000000006566.
6. Bush G, Fink M, Petrides G, et al. Catatonia I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
7. Unal A, Altindag A, Demir B, et al. The use of lorazepam and electroconvulsive therapy in the treatment of catatonia: treatment characteristics and outcomes in 60 patients. J ECT. 2017;33(4):290-293.
8. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1182-1183.
9. van der Markt A, Heller HM, van Exel E. A woman with catatonia, what to do after ECT fails: a case report. J ECT. 2016;32(3):e6-7. doi: 10.1097/YCT.0000000000000290.
Mr. W, age 50, who has been diagnosed with hypertension and catatonia associated with schizophrenia, is brought to the emergency department by his case manager for evaluation of increasing disorganization, inability to function, and nonadherence to medications. He has not been bathing, eating, or drinking. During the admission interview, he is mute, and is noted to have purposeless activity, alternating between rocking from leg to leg to pacing in circles. At times Mr. W holds a rigid, prayer-type posture with his arms. Negativism is present, primarily opposition to interviewer requests.
Previously stable on
On the inpatient psychiatry unit, Mr. W continues to be mute, staying in bed except to use the bathroom. He refuses all food and fluids. The team initiates subcutaneous
Continue to: Medical complications can be fatal
Medical complications can be fatal
Treatment usually starts with lorazepam
Benzodiazepines are a first-line option for the management of catatonia.2,5 Controversy exists as to effectiveness of different routes of administration. Generally, IV lorazepam is preferred due to its ease of administration, fast onset, and longer duration of action.1 Some inpatient psychiatric units are unable to administer IV benzodiazepines; in these scenarios, IM administration is preferred to oral benzodiazepines.
The initial lorazepam challenge dose should be 2 mg. A positive response to the lorazepam challenge often confirms the catatonia diagnosis.2,7 This challenge should be followed by maintenance doses ranging from 6 to 8 mg/d in divided doses (3 or 4 times a day). Higher doses (up to 24 mg/d) are sometimes used.2,5,8 A recent case report described catatonia remission using lorazepam, 28 mg/d, after unsuccessful ECT.9 The lorazepam dose prior to ECT was 8 mg/d.9 Response is usually seen within 3 to 7 days of an adequate dose.2,8 Parenteral lorazepam typically is continued for several days before converting to oral lorazepam.1 Approximately 70% to 80% of patients with catatonia will show improvement in symptoms with lorazepam.2,7,8
The optimal duration of benzodiazepine treatment is unclear.2 In some cases, once remission of the underlying illness is achieved, benzodiazepines are discontinued.2 However, in other cases, symptoms of catatonia may emerge when lorazepam is tapered, therefore suggesting the need for a longer duration of treatment.2 Despite this high rate of improvement, many patients ultimately receive ECT due to unsustained response or to prevent future episodes of catatonia.
A recent review of 60 Turkish patients with catatonia found 91.7% (n = 55) received oral lorazepam (up to 15 mg/d) as the first-line therapy.7 Improvement was seen in 23.7% (n = 13) of patients treated with lorazepam, yet 70% (n = 42) showed either no response or partial response, and ultimately received ECT in combination with lorazepam.7 The lower improvement rate seen in this review may be secondary to the use of oral lorazepam instead of parenteral, or may highlight the frequency in which patients ultimately go on to receive ECT.
Continue to: ECT
ECT. If high doses of benzodiazepines are not effective within 48 to 72 hours, ECT should be considered.1,7 ECT should be considered sooner for patients with life-threatening catatonia or those who present with excited features or malignant catatonia.1,2,7 In patients with catatonia, ECT response rates range from 80% to 100%.2,7 Unal et al7 reported a 100% response rate if ECT was used as the first-line treatment (n = 5), and a 92.9% (n = 39) response rate after adding ECT to lorazepam. Lorazepam may interfere with the seizure threshold, but if indicated, this medication can be continued.2 A minimum of 6 ECT treatments are suggested; however, as many as 20 treatments have been needed.1 Mr. W required a total of 18 ECT treatments. In some cases, maintenance ECT may be required.2
Antipsychotics. Discontinuation of antipsychotics is generally encouraged in patients presenting with catatonia.2,7,8 Antipsychotics carry a risk of potentially worsening catatonia, conversion to malignant catatonia, or precipitation of NMS; therefore, carefully weigh the risks vs benefits.1,2 If catatonia is secondary to psychosis, as in Mr. W’s case, antipsychotics may be considered once catatonia improves.2 If an antipsychotic is warranted, consider aripiprazole (because of its D2 partial agonist activity) or low-dose olanzapine.1,2 If catatonia is secondary to clozapine withdrawal, the initial therapy should be clozapine re-initiation.1 Although high-potency agents, such as haloperidol and risperidone, typically are not preferred, risperidone was restarted for Mr. W because of his history of response to and tolerability of this medication during a previous catatonic episode.
Other treatments. In a recent review, Beach et al1 described the use of additional agents, mostly in a small number of positive case reports, for managing catatonia. These included:
- zolpidem (zolpidem 10 mg as a challenge test, and doses of ≤40 mg/d)
- the N-methyl-
D -aspartic acid antagonists amantadine (100 to 600 mg/d) or memantine (5 to 20 mg/d) - carbidopa/levodopa
- methylphenidate
- antiepileptics (eg, carbamazepine, topiramate, and divalproex sodium)
- anticholinergics.1,2
Lithium has been used in attempts to prevent recurrent catatonia with limited success.2 There are also a few reports of using transcranial magnetic stimulation (TMS) to manage catatonia.1
Beach et al1 proposed a treatment algorithm in which IV lorazepam (Step 1) and ECT (Step 2) remain the preferred treatments. Next, for Step 3 consider a glutamate antagonist (amantadine or memantine), followed by an antiepileptic (Step 4), and lastly an atypical antipsychotic (aripiprazole, olanzapine, or clozapine) in combination with lorazepam (Step 5).
When indicated, don’t delay ECT
Initial management of catatonia is with a benzodiazepine challenge. Ultimately, the gold-standard treatment of catatonia that does not improve with benzodiazepines is ECT, and ECT should be implemented as soon as it is clear that pharmacotherapy is less than fully effective. Consider ECT initially in life-threatening cases and for patients with malignant catatonia. Although additional agents and TMS have been explored, these should be reserved for patients who fail to respond to, or who are not candidates for, benzodiazepines or ECT.
CASE CONTINUED
After 5 ECT treatments, Mr. W says a few words, but he communicates primarily with gestures (primarily waving people away). After 10 to 12 ECT treatments, Mr. W becomes more interactive and conversant, and his nutrition improves; however, he still exhibits symptoms of catatonia and is not at baseline. He undergoes a total of 18 ECT treatments. Antipsychotics were initially discontinued; however, given Mr. W’s improvement with ECT and the presence of auditory hallucinations, oral risperidone is restarted and titrated to 2 mg, 2 times a day, and he is transitioned back to paliperidone palmitate before he is discharged. Lorazepam is tapered and discontinued. Mr. W is discharged back to his nursing home and is interactive (laughing and joking with family) and attending to his activities of daily living. Unfortunately, Mr. W did not followup with the recommendation for maintenance ECT, and adherence to paliperidone palmitate injections is unknown. Mr. W presented to our facility again 6 months later with symptoms of catatonia and ultimately transferred to a state hospital.
Related Resources
- Fink M, Taylor MA. Catatonia: A clinician’s guide to diagnosis and treatment. New York, NY: Cambridge University Press; 2006. • Carroll BT, Spiegel DR. Catatonia on the consultation liaison service and other clinical settings. Hauppauge, NY: Nova Science Pub Inc.; 2016.
- Benarous X, Raffin M, Ferrafiat V, et al. Catatonia in children and adolescents: new perspectives. Schizophr Res. 2018;200:56-67.
- Malignant Hyperthermia Association of the United States. What is NMSIS? http://www.mhaus.org/nmsis/about-us/ what-is-nmsis/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Asenapine • Saphris
Carbamazepine • Carbatrol, Tegretol
Carbidopa/Levodopa • Sinemet
Citalopram • Celexa
Clozapine • Clozaril
Divalproex Sodium • Depakote
Enoxaparin • Lovenox
Fluoxetine • Prozac
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Lurasidone • Latuda
Memantine • Namenda
Methylphenidate • Concerta, Ritalin
Mirtazapine • Remeron
Olanzapine • Zyprexa
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone long-acting injection • Risperdal Consta
Topiramate • Topamax
Zolpidem • Ambien
Mr. W, age 50, who has been diagnosed with hypertension and catatonia associated with schizophrenia, is brought to the emergency department by his case manager for evaluation of increasing disorganization, inability to function, and nonadherence to medications. He has not been bathing, eating, or drinking. During the admission interview, he is mute, and is noted to have purposeless activity, alternating between rocking from leg to leg to pacing in circles. At times Mr. W holds a rigid, prayer-type posture with his arms. Negativism is present, primarily opposition to interviewer requests.
Previously stable on
On the inpatient psychiatry unit, Mr. W continues to be mute, staying in bed except to use the bathroom. He refuses all food and fluids. The team initiates subcutaneous
Continue to: Medical complications can be fatal
Medical complications can be fatal
Treatment usually starts with lorazepam
Benzodiazepines are a first-line option for the management of catatonia.2,5 Controversy exists as to effectiveness of different routes of administration. Generally, IV lorazepam is preferred due to its ease of administration, fast onset, and longer duration of action.1 Some inpatient psychiatric units are unable to administer IV benzodiazepines; in these scenarios, IM administration is preferred to oral benzodiazepines.
The initial lorazepam challenge dose should be 2 mg. A positive response to the lorazepam challenge often confirms the catatonia diagnosis.2,7 This challenge should be followed by maintenance doses ranging from 6 to 8 mg/d in divided doses (3 or 4 times a day). Higher doses (up to 24 mg/d) are sometimes used.2,5,8 A recent case report described catatonia remission using lorazepam, 28 mg/d, after unsuccessful ECT.9 The lorazepam dose prior to ECT was 8 mg/d.9 Response is usually seen within 3 to 7 days of an adequate dose.2,8 Parenteral lorazepam typically is continued for several days before converting to oral lorazepam.1 Approximately 70% to 80% of patients with catatonia will show improvement in symptoms with lorazepam.2,7,8
The optimal duration of benzodiazepine treatment is unclear.2 In some cases, once remission of the underlying illness is achieved, benzodiazepines are discontinued.2 However, in other cases, symptoms of catatonia may emerge when lorazepam is tapered, therefore suggesting the need for a longer duration of treatment.2 Despite this high rate of improvement, many patients ultimately receive ECT due to unsustained response or to prevent future episodes of catatonia.
A recent review of 60 Turkish patients with catatonia found 91.7% (n = 55) received oral lorazepam (up to 15 mg/d) as the first-line therapy.7 Improvement was seen in 23.7% (n = 13) of patients treated with lorazepam, yet 70% (n = 42) showed either no response or partial response, and ultimately received ECT in combination with lorazepam.7 The lower improvement rate seen in this review may be secondary to the use of oral lorazepam instead of parenteral, or may highlight the frequency in which patients ultimately go on to receive ECT.
Continue to: ECT
ECT. If high doses of benzodiazepines are not effective within 48 to 72 hours, ECT should be considered.1,7 ECT should be considered sooner for patients with life-threatening catatonia or those who present with excited features or malignant catatonia.1,2,7 In patients with catatonia, ECT response rates range from 80% to 100%.2,7 Unal et al7 reported a 100% response rate if ECT was used as the first-line treatment (n = 5), and a 92.9% (n = 39) response rate after adding ECT to lorazepam. Lorazepam may interfere with the seizure threshold, but if indicated, this medication can be continued.2 A minimum of 6 ECT treatments are suggested; however, as many as 20 treatments have been needed.1 Mr. W required a total of 18 ECT treatments. In some cases, maintenance ECT may be required.2
Antipsychotics. Discontinuation of antipsychotics is generally encouraged in patients presenting with catatonia.2,7,8 Antipsychotics carry a risk of potentially worsening catatonia, conversion to malignant catatonia, or precipitation of NMS; therefore, carefully weigh the risks vs benefits.1,2 If catatonia is secondary to psychosis, as in Mr. W’s case, antipsychotics may be considered once catatonia improves.2 If an antipsychotic is warranted, consider aripiprazole (because of its D2 partial agonist activity) or low-dose olanzapine.1,2 If catatonia is secondary to clozapine withdrawal, the initial therapy should be clozapine re-initiation.1 Although high-potency agents, such as haloperidol and risperidone, typically are not preferred, risperidone was restarted for Mr. W because of his history of response to and tolerability of this medication during a previous catatonic episode.
Other treatments. In a recent review, Beach et al1 described the use of additional agents, mostly in a small number of positive case reports, for managing catatonia. These included:
- zolpidem (zolpidem 10 mg as a challenge test, and doses of ≤40 mg/d)
- the N-methyl-
D -aspartic acid antagonists amantadine (100 to 600 mg/d) or memantine (5 to 20 mg/d) - carbidopa/levodopa
- methylphenidate
- antiepileptics (eg, carbamazepine, topiramate, and divalproex sodium)
- anticholinergics.1,2
Lithium has been used in attempts to prevent recurrent catatonia with limited success.2 There are also a few reports of using transcranial magnetic stimulation (TMS) to manage catatonia.1
Beach et al1 proposed a treatment algorithm in which IV lorazepam (Step 1) and ECT (Step 2) remain the preferred treatments. Next, for Step 3 consider a glutamate antagonist (amantadine or memantine), followed by an antiepileptic (Step 4), and lastly an atypical antipsychotic (aripiprazole, olanzapine, or clozapine) in combination with lorazepam (Step 5).
When indicated, don’t delay ECT
Initial management of catatonia is with a benzodiazepine challenge. Ultimately, the gold-standard treatment of catatonia that does not improve with benzodiazepines is ECT, and ECT should be implemented as soon as it is clear that pharmacotherapy is less than fully effective. Consider ECT initially in life-threatening cases and for patients with malignant catatonia. Although additional agents and TMS have been explored, these should be reserved for patients who fail to respond to, or who are not candidates for, benzodiazepines or ECT.
CASE CONTINUED
After 5 ECT treatments, Mr. W says a few words, but he communicates primarily with gestures (primarily waving people away). After 10 to 12 ECT treatments, Mr. W becomes more interactive and conversant, and his nutrition improves; however, he still exhibits symptoms of catatonia and is not at baseline. He undergoes a total of 18 ECT treatments. Antipsychotics were initially discontinued; however, given Mr. W’s improvement with ECT and the presence of auditory hallucinations, oral risperidone is restarted and titrated to 2 mg, 2 times a day, and he is transitioned back to paliperidone palmitate before he is discharged. Lorazepam is tapered and discontinued. Mr. W is discharged back to his nursing home and is interactive (laughing and joking with family) and attending to his activities of daily living. Unfortunately, Mr. W did not followup with the recommendation for maintenance ECT, and adherence to paliperidone palmitate injections is unknown. Mr. W presented to our facility again 6 months later with symptoms of catatonia and ultimately transferred to a state hospital.
Related Resources
- Fink M, Taylor MA. Catatonia: A clinician’s guide to diagnosis and treatment. New York, NY: Cambridge University Press; 2006. • Carroll BT, Spiegel DR. Catatonia on the consultation liaison service and other clinical settings. Hauppauge, NY: Nova Science Pub Inc.; 2016.
- Benarous X, Raffin M, Ferrafiat V, et al. Catatonia in children and adolescents: new perspectives. Schizophr Res. 2018;200:56-67.
- Malignant Hyperthermia Association of the United States. What is NMSIS? http://www.mhaus.org/nmsis/about-us/ what-is-nmsis/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Asenapine • Saphris
Carbamazepine • Carbatrol, Tegretol
Carbidopa/Levodopa • Sinemet
Citalopram • Celexa
Clozapine • Clozaril
Divalproex Sodium • Depakote
Enoxaparin • Lovenox
Fluoxetine • Prozac
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Lurasidone • Latuda
Memantine • Namenda
Methylphenidate • Concerta, Ritalin
Mirtazapine • Remeron
Olanzapine • Zyprexa
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone long-acting injection • Risperdal Consta
Topiramate • Topamax
Zolpidem • Ambien
1. Beach SR, Gomez-Bernal F, Huffman JC, et al. Alternative treatment strategies for catatonia: a systematic review. Gen Hosp Psychiatry. 2017;48:1-19.
2. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:1-6.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome: focus on treatment and rechallenge. Ann Pharmacother. 2016;50(11):973-981.
5. Ohi K, Kuwata A, Shimada T, et al. Response to benzodiazepines and clinical course in malignant catatonia associated with schizophrenia: a case report. Medicine (Baltimore). 2017;96(16):e6566. doi: 10.1097/MD.0000000000006566.
6. Bush G, Fink M, Petrides G, et al. Catatonia I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
7. Unal A, Altindag A, Demir B, et al. The use of lorazepam and electroconvulsive therapy in the treatment of catatonia: treatment characteristics and outcomes in 60 patients. J ECT. 2017;33(4):290-293.
8. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1182-1183.
9. van der Markt A, Heller HM, van Exel E. A woman with catatonia, what to do after ECT fails: a case report. J ECT. 2016;32(3):e6-7. doi: 10.1097/YCT.0000000000000290.
1. Beach SR, Gomez-Bernal F, Huffman JC, et al. Alternative treatment strategies for catatonia: a systematic review. Gen Hosp Psychiatry. 2017;48:1-19.
2. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:1-6.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome: focus on treatment and rechallenge. Ann Pharmacother. 2016;50(11):973-981.
5. Ohi K, Kuwata A, Shimada T, et al. Response to benzodiazepines and clinical course in malignant catatonia associated with schizophrenia: a case report. Medicine (Baltimore). 2017;96(16):e6566. doi: 10.1097/MD.0000000000006566.
6. Bush G, Fink M, Petrides G, et al. Catatonia I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
7. Unal A, Altindag A, Demir B, et al. The use of lorazepam and electroconvulsive therapy in the treatment of catatonia: treatment characteristics and outcomes in 60 patients. J ECT. 2017;33(4):290-293.
8. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1182-1183.
9. van der Markt A, Heller HM, van Exel E. A woman with catatonia, what to do after ECT fails: a case report. J ECT. 2016;32(3):e6-7. doi: 10.1097/YCT.0000000000000290.
A mood disorder complicated by multiple sclerosis
CASE Depression, or something else?
Ms. A, age 56, presents to the emergency department (ED) with depressed mood, poor sleep, anhedonia, irritability, agitation, and recent self-injurious behavior; she had superficially cut her wrists. She also has a longstanding history of multiple sclerosis (MS), depression, and anxiety. She is admitted voluntarily to an inpatient psychiatric unit.
According to medical records, at age 32, Ms. A was diagnosed with relapsing-remitting MS, which initially presented with facial numbness, and later with optic neuritis with transient loss of vision. As her disease progressed to the secondary progressive type, she experienced spasticity and vertigo. In the past few years, she also had experienced cognitive difficulties, particularly with memory and focus.
Ms. A has a history of recurrent depressive symptoms that began at an unspecified time after being diagnosed with MS. In the past few years, she had greatly increased her alcohol use in response to multiple psychosocial stressors and as an attempt to self-medicate MS-related pain. Several years ago, Ms. A had been admitted to a rehabilitation facility to address her alcohol use.
In the past, Ms. A’s depressive symptoms had been treated with various antidepressants, including fluoxetine (unspecified dose), which for a time was effective. The most recently prescribed antidepressant was duloxetine, 60 mg/d, which was discontinued because Ms. A felt it activated her mood lability. A few years before this current hospitalization, Ms. A had been started on a trial of dextromethorphan/quinidine (20 mg/10 mg, twice daily), which was discontinued due to concomitant use of an unspecified serotonin-norepinephrine reuptake inhibitor (SNRI) and subsequent precipitation of serotonin syndrome.
At the time of this current admission to the psychiatric unit, Ms. A is being treated for MS with rituximab (10 mg/mL IV, every 6 months). Additionally, just before her admission, she was taking alprazolam (.25 mg, 3 times per day) for anxiety. She denies experiencing any spasticity or vision impairment.
[polldaddy:10175070]
The authors’ observations
We initially considered a diagnosis of MDD due to Ms. A’s past history of depressive episodes, her recent increase in tearfulness and anhedonia, and her self-injurious behaviors. However, diagnosis of a mood disorder was complicated by her complex history of longstanding MS and other psychosocial factors.

Continue to: Several factors contribute to the neuropsychiatric course of patients with MS...
Several factors contribute to the neuropsychiatric course of patients with MS, including the impact of the patient accepting a chronic and incurable diagnosis, the toll of progressive neurologic/physical disability and subsequent decline in functioning, and the availability of a support system.2 As opposed to disorders such as Parkinson’s disease, where disease progression is relatively more predictable, the culture of MS involves the obscurity of symptom fluctuation, both from the patient’s and/or clinician’s viewpoint. Psychiatric and neurologic symptoms may be difficult to predict, leading to speculation and projection as to the progression of the disease. The diagnosis of psychiatric conditions, such as depression, can be complicated by the fact that MS and psychiatric disorders share presenting symptoms; for example, disturbances in sleep and concentration may be seen in both conditions.
While studies have examined the neurobiology of MS lesions and their effects on mood symptoms, there has been no clear consensus of specific lesion distributions, although lesions in the superior frontal lobe and right temporal lobe regions have been identified in depressed MS patients.8 Lesions in the left frontal lobe may also have some contribution; studies have shown hyperintense lesion load in this area, which was found to be an independent predictor of MDD in MS.9 This, in turn, coincides with the association of left frontal cortex involvement in modulating affective depression, evidenced by studies that have associated depression severity with left frontal lobe damage in post-stroke patients10 as well as the use of transcranial magnetic stimulation of the left prefrontal cortex for treatment-resistant MDD.11 Lesions along the orbitofrontal prefrontal cortex have similarly been connected to mood lability and impulsivity, which are characteristics of bipolar disorder.8 Within the general population, bipolar disorder is associated with areas of hyperintensity on MRI, particularly in the frontal and parietal white matter, which may provide clues as to the role of MS demyelinating lesions in similar locations, although research concerning the relationship between MS and bipolar disorder remains limited.12
EVALUATION No exacerbation of MS
Upon admission, Ms. A’s lability of affect is apparent as she quickly switches from being tearful to bright depending on the topic of discussion. She smiles when talking about the hobbies she enjoys and becomes tearful when speaking of personal problems within her family. She denies suicidal ideation/intent, shows no evidence of psychosis, and denies any history of bipolar disorder or recollection of hypomanic/manic symptoms. Overall, she exhibits low energy and difficulty sleeping, and reiterates her various psychosocial stressors, including her family history of depression and ongoing marital conflicts. Ms. A denies experiencing any acute exacerbations of clinical neurologic features of MS immediately before or during her admission. Laboratory values are normal, except for an elevated thyroid stimulating hormone (TSH) value of 11.136 uIU/mL, which is expected given her history of hypothyroidism. Results of the most recent brain MRI scans for Ms. A are pending.
The authors’ observations
Although we considered a diagnosis of bipolar disorder–mixed subtype, this was less likely to be the diagnosis considering her lack of any frank manic/hypomanic symptoms or history of such symptoms. Additionally, while we also considered a diagnosis of pseudobulbar affect due to her current mood swings and past trial of dextromethorphan/quinidine, this diagnosis was also less likely because Ms. A’s affect was not characterized by uncontrollable outbursts of emotion but was congruent with her experiences and surroundings. For example, Ms. A smiled when talking about her hobbies and became tearful when speaking of conflicts within her family.
Given Ms. A’s mood dysregulation and lability and her history of depressive episodes that began to manifest after her diagnosis of MS was established, and after ruling out other etiologic psychiatric disorders, a diagnosis of mood disorder secondary to MS was made.
[polldaddy:10175136]
Continue to: TREATMENT Mood stabilization
TREATMENT Mood stabilization
We start Ms. A on divalproex sodium, 250 mg 2 times a day, which is eventually titrated to 250 mg every morning with an additional daily 750 mg (total daily dose of 1,000 mg) for mood stabilization. Additionally, quetiapine, 50 mg nightly, is added and eventually titrated to 300 mg to augment mood stabilization and to aid sleep. Before being admitted, Ms. A had been prescribed
The authors’ observations
Definitive treatments for psychiatric conditions in patients with MS have been lacking, and current recommendations are based on regimens used to treat general psychiatric populations. For example, selective serotonin reuptake inhibitors are frequently considered for treatment of MDD in patients with MS, whereas SNRIs are considered for patients with concomitant neuropathic pain.13 Similarly,
OUTCOME Improved mood, energy
After 2 weeks of inpatient treatment, Ms. A shows improvement in mood lability and energy levels, and she is able to tolerate titration of divalproex sodium and quetiapine to therapeutic levels. She is referred to an outpatient psychiatrist after discharge, as well as a follow-up appointment with her neurologist. On discharge, Ms. A expresses a commitment to treatment and hope for the future.
1. National Multiple Sclerosis Society. Signs and symptoms consistent with demyelinating disease (for professionals). https://www.nationalmssociety.org/For-Professionals/Clinical-Care/Diagnosing-MS/Signs-and-Symptoms-Consistent-with-Demyelinating-D. Accessed October 29, 2018.
2. Politte LC, Huffman JC, Stern TA. Neuropsychiatric manifestations of multiple sclerosis. Prim Care Companion J Clin Psychiatry. 2008;10(4):318-324.
3. Siegert RJ, Abernethy D. Depression in multiple sclerosis: a review. J Neurol Neurosurg Psychiatry. 2005;76(4):469-475.
4. Scalfari A, Knappertz V, Cutter G, et al. Mortality in patients with multiple sclerosis. Neurology. 2013;81(2):184-192.
5. Ghaffar O, Feinstein A. The neuropsychiatry of multiple sclerosis: a review of recent developments. Curr Opin Psychiatry. 2007;20(3):278-285.
6. Duncan A, Malcolm-Smith S, Ameen O, et al. The incidence of euphoria in multiple sclerosis: artefact of measure. Mult Scler Int. 2016;2016:1-8.
7. Paparrigopoulos T, Ferentinos P, Kouzoupis A, et al. The neuropsychiatry of multiple sclerosis: focus on disorders of mood, affect and behaviour. Int Rev Psychiatry. 2010;22(1):14-21.
8. Bakshi R, Czarnecki D, Shaikh ZA, et al. Brain MRI lesions and atrophy are related to depression in multiple sclerosis. Neuroreport. 2000;11(6):1153-1158.
9. Feinstein A, Roy P, Lobaugh N, et al. Structural brain abnormalities in multiple sclerosis patients with major depression. Neurology. 2004;62(4):586-590.
10. Hama S, Yamashita H, Shigenobu M, et al. Post-stroke affective or apathetic depression and lesion location: left frontal lobe and bilateral basal ganglia. Eur Arch Psychiatry Clin Neurosci. 2007;257(3):149-152.
11. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
12. Beyer JL, Young R, Kuchibhatla M, et al. Hyperintense MRI lesions in bipolar disorder: a meta-analysis and review. Int Rev Psychiatry. 2009;21(4):394-409.
13. Feinstein A. Neuropsychiatric syndromes associated with multiple sclerosis. J Neurol. 2007;254(S2):1173-1176.
14. Thomas PW, Thomas S, Hillier C, et al. Psychological interventions for multiple sclerosis. Cochrane Database Syst Rev. 2006;(1):CD004431. doi: 10.1002/14651858.cd004431.pub2.
CASE Depression, or something else?
Ms. A, age 56, presents to the emergency department (ED) with depressed mood, poor sleep, anhedonia, irritability, agitation, and recent self-injurious behavior; she had superficially cut her wrists. She also has a longstanding history of multiple sclerosis (MS), depression, and anxiety. She is admitted voluntarily to an inpatient psychiatric unit.
According to medical records, at age 32, Ms. A was diagnosed with relapsing-remitting MS, which initially presented with facial numbness, and later with optic neuritis with transient loss of vision. As her disease progressed to the secondary progressive type, she experienced spasticity and vertigo. In the past few years, she also had experienced cognitive difficulties, particularly with memory and focus.
Ms. A has a history of recurrent depressive symptoms that began at an unspecified time after being diagnosed with MS. In the past few years, she had greatly increased her alcohol use in response to multiple psychosocial stressors and as an attempt to self-medicate MS-related pain. Several years ago, Ms. A had been admitted to a rehabilitation facility to address her alcohol use.
In the past, Ms. A’s depressive symptoms had been treated with various antidepressants, including fluoxetine (unspecified dose), which for a time was effective. The most recently prescribed antidepressant was duloxetine, 60 mg/d, which was discontinued because Ms. A felt it activated her mood lability. A few years before this current hospitalization, Ms. A had been started on a trial of dextromethorphan/quinidine (20 mg/10 mg, twice daily), which was discontinued due to concomitant use of an unspecified serotonin-norepinephrine reuptake inhibitor (SNRI) and subsequent precipitation of serotonin syndrome.
At the time of this current admission to the psychiatric unit, Ms. A is being treated for MS with rituximab (10 mg/mL IV, every 6 months). Additionally, just before her admission, she was taking alprazolam (.25 mg, 3 times per day) for anxiety. She denies experiencing any spasticity or vision impairment.
[polldaddy:10175070]
The authors’ observations
We initially considered a diagnosis of MDD due to Ms. A’s past history of depressive episodes, her recent increase in tearfulness and anhedonia, and her self-injurious behaviors. However, diagnosis of a mood disorder was complicated by her complex history of longstanding MS and other psychosocial factors.
Continue to: Several factors contribute to the neuropsychiatric course of patients with MS...
Several factors contribute to the neuropsychiatric course of patients with MS, including the impact of the patient accepting a chronic and incurable diagnosis, the toll of progressive neurologic/physical disability and subsequent decline in functioning, and the availability of a support system.2 As opposed to disorders such as Parkinson’s disease, where disease progression is relatively more predictable, the culture of MS involves the obscurity of symptom fluctuation, both from the patient’s and/or clinician’s viewpoint. Psychiatric and neurologic symptoms may be difficult to predict, leading to speculation and projection as to the progression of the disease. The diagnosis of psychiatric conditions, such as depression, can be complicated by the fact that MS and psychiatric disorders share presenting symptoms; for example, disturbances in sleep and concentration may be seen in both conditions.
While studies have examined the neurobiology of MS lesions and their effects on mood symptoms, there has been no clear consensus of specific lesion distributions, although lesions in the superior frontal lobe and right temporal lobe regions have been identified in depressed MS patients.8 Lesions in the left frontal lobe may also have some contribution; studies have shown hyperintense lesion load in this area, which was found to be an independent predictor of MDD in MS.9 This, in turn, coincides with the association of left frontal cortex involvement in modulating affective depression, evidenced by studies that have associated depression severity with left frontal lobe damage in post-stroke patients10 as well as the use of transcranial magnetic stimulation of the left prefrontal cortex for treatment-resistant MDD.11 Lesions along the orbitofrontal prefrontal cortex have similarly been connected to mood lability and impulsivity, which are characteristics of bipolar disorder.8 Within the general population, bipolar disorder is associated with areas of hyperintensity on MRI, particularly in the frontal and parietal white matter, which may provide clues as to the role of MS demyelinating lesions in similar locations, although research concerning the relationship between MS and bipolar disorder remains limited.12
EVALUATION No exacerbation of MS
Upon admission, Ms. A’s lability of affect is apparent as she quickly switches from being tearful to bright depending on the topic of discussion. She smiles when talking about the hobbies she enjoys and becomes tearful when speaking of personal problems within her family. She denies suicidal ideation/intent, shows no evidence of psychosis, and denies any history of bipolar disorder or recollection of hypomanic/manic symptoms. Overall, she exhibits low energy and difficulty sleeping, and reiterates her various psychosocial stressors, including her family history of depression and ongoing marital conflicts. Ms. A denies experiencing any acute exacerbations of clinical neurologic features of MS immediately before or during her admission. Laboratory values are normal, except for an elevated thyroid stimulating hormone (TSH) value of 11.136 uIU/mL, which is expected given her history of hypothyroidism. Results of the most recent brain MRI scans for Ms. A are pending.
The authors’ observations
Although we considered a diagnosis of bipolar disorder–mixed subtype, this was less likely to be the diagnosis considering her lack of any frank manic/hypomanic symptoms or history of such symptoms. Additionally, while we also considered a diagnosis of pseudobulbar affect due to her current mood swings and past trial of dextromethorphan/quinidine, this diagnosis was also less likely because Ms. A’s affect was not characterized by uncontrollable outbursts of emotion but was congruent with her experiences and surroundings. For example, Ms. A smiled when talking about her hobbies and became tearful when speaking of conflicts within her family.
Given Ms. A’s mood dysregulation and lability and her history of depressive episodes that began to manifest after her diagnosis of MS was established, and after ruling out other etiologic psychiatric disorders, a diagnosis of mood disorder secondary to MS was made.
[polldaddy:10175136]
Continue to: TREATMENT Mood stabilization
TREATMENT Mood stabilization
We start Ms. A on divalproex sodium, 250 mg 2 times a day, which is eventually titrated to 250 mg every morning with an additional daily 750 mg (total daily dose of 1,000 mg) for mood stabilization. Additionally, quetiapine, 50 mg nightly, is added and eventually titrated to 300 mg to augment mood stabilization and to aid sleep. Before being admitted, Ms. A had been prescribed
The authors’ observations
Definitive treatments for psychiatric conditions in patients with MS have been lacking, and current recommendations are based on regimens used to treat general psychiatric populations. For example, selective serotonin reuptake inhibitors are frequently considered for treatment of MDD in patients with MS, whereas SNRIs are considered for patients with concomitant neuropathic pain.13 Similarly,
OUTCOME Improved mood, energy
After 2 weeks of inpatient treatment, Ms. A shows improvement in mood lability and energy levels, and she is able to tolerate titration of divalproex sodium and quetiapine to therapeutic levels. She is referred to an outpatient psychiatrist after discharge, as well as a follow-up appointment with her neurologist. On discharge, Ms. A expresses a commitment to treatment and hope for the future.
CASE Depression, or something else?
Ms. A, age 56, presents to the emergency department (ED) with depressed mood, poor sleep, anhedonia, irritability, agitation, and recent self-injurious behavior; she had superficially cut her wrists. She also has a longstanding history of multiple sclerosis (MS), depression, and anxiety. She is admitted voluntarily to an inpatient psychiatric unit.
According to medical records, at age 32, Ms. A was diagnosed with relapsing-remitting MS, which initially presented with facial numbness, and later with optic neuritis with transient loss of vision. As her disease progressed to the secondary progressive type, she experienced spasticity and vertigo. In the past few years, she also had experienced cognitive difficulties, particularly with memory and focus.
Ms. A has a history of recurrent depressive symptoms that began at an unspecified time after being diagnosed with MS. In the past few years, she had greatly increased her alcohol use in response to multiple psychosocial stressors and as an attempt to self-medicate MS-related pain. Several years ago, Ms. A had been admitted to a rehabilitation facility to address her alcohol use.
In the past, Ms. A’s depressive symptoms had been treated with various antidepressants, including fluoxetine (unspecified dose), which for a time was effective. The most recently prescribed antidepressant was duloxetine, 60 mg/d, which was discontinued because Ms. A felt it activated her mood lability. A few years before this current hospitalization, Ms. A had been started on a trial of dextromethorphan/quinidine (20 mg/10 mg, twice daily), which was discontinued due to concomitant use of an unspecified serotonin-norepinephrine reuptake inhibitor (SNRI) and subsequent precipitation of serotonin syndrome.
At the time of this current admission to the psychiatric unit, Ms. A is being treated for MS with rituximab (10 mg/mL IV, every 6 months). Additionally, just before her admission, she was taking alprazolam (.25 mg, 3 times per day) for anxiety. She denies experiencing any spasticity or vision impairment.
[polldaddy:10175070]
The authors’ observations
We initially considered a diagnosis of MDD due to Ms. A’s past history of depressive episodes, her recent increase in tearfulness and anhedonia, and her self-injurious behaviors. However, diagnosis of a mood disorder was complicated by her complex history of longstanding MS and other psychosocial factors.
Continue to: Several factors contribute to the neuropsychiatric course of patients with MS...
Several factors contribute to the neuropsychiatric course of patients with MS, including the impact of the patient accepting a chronic and incurable diagnosis, the toll of progressive neurologic/physical disability and subsequent decline in functioning, and the availability of a support system.2 As opposed to disorders such as Parkinson’s disease, where disease progression is relatively more predictable, the culture of MS involves the obscurity of symptom fluctuation, both from the patient’s and/or clinician’s viewpoint. Psychiatric and neurologic symptoms may be difficult to predict, leading to speculation and projection as to the progression of the disease. The diagnosis of psychiatric conditions, such as depression, can be complicated by the fact that MS and psychiatric disorders share presenting symptoms; for example, disturbances in sleep and concentration may be seen in both conditions.
While studies have examined the neurobiology of MS lesions and their effects on mood symptoms, there has been no clear consensus of specific lesion distributions, although lesions in the superior frontal lobe and right temporal lobe regions have been identified in depressed MS patients.8 Lesions in the left frontal lobe may also have some contribution; studies have shown hyperintense lesion load in this area, which was found to be an independent predictor of MDD in MS.9 This, in turn, coincides with the association of left frontal cortex involvement in modulating affective depression, evidenced by studies that have associated depression severity with left frontal lobe damage in post-stroke patients10 as well as the use of transcranial magnetic stimulation of the left prefrontal cortex for treatment-resistant MDD.11 Lesions along the orbitofrontal prefrontal cortex have similarly been connected to mood lability and impulsivity, which are characteristics of bipolar disorder.8 Within the general population, bipolar disorder is associated with areas of hyperintensity on MRI, particularly in the frontal and parietal white matter, which may provide clues as to the role of MS demyelinating lesions in similar locations, although research concerning the relationship between MS and bipolar disorder remains limited.12
EVALUATION No exacerbation of MS
Upon admission, Ms. A’s lability of affect is apparent as she quickly switches from being tearful to bright depending on the topic of discussion. She smiles when talking about the hobbies she enjoys and becomes tearful when speaking of personal problems within her family. She denies suicidal ideation/intent, shows no evidence of psychosis, and denies any history of bipolar disorder or recollection of hypomanic/manic symptoms. Overall, she exhibits low energy and difficulty sleeping, and reiterates her various psychosocial stressors, including her family history of depression and ongoing marital conflicts. Ms. A denies experiencing any acute exacerbations of clinical neurologic features of MS immediately before or during her admission. Laboratory values are normal, except for an elevated thyroid stimulating hormone (TSH) value of 11.136 uIU/mL, which is expected given her history of hypothyroidism. Results of the most recent brain MRI scans for Ms. A are pending.
The authors’ observations
Although we considered a diagnosis of bipolar disorder–mixed subtype, this was less likely to be the diagnosis considering her lack of any frank manic/hypomanic symptoms or history of such symptoms. Additionally, while we also considered a diagnosis of pseudobulbar affect due to her current mood swings and past trial of dextromethorphan/quinidine, this diagnosis was also less likely because Ms. A’s affect was not characterized by uncontrollable outbursts of emotion but was congruent with her experiences and surroundings. For example, Ms. A smiled when talking about her hobbies and became tearful when speaking of conflicts within her family.
Given Ms. A’s mood dysregulation and lability and her history of depressive episodes that began to manifest after her diagnosis of MS was established, and after ruling out other etiologic psychiatric disorders, a diagnosis of mood disorder secondary to MS was made.
[polldaddy:10175136]
Continue to: TREATMENT Mood stabilization
TREATMENT Mood stabilization
We start Ms. A on divalproex sodium, 250 mg 2 times a day, which is eventually titrated to 250 mg every morning with an additional daily 750 mg (total daily dose of 1,000 mg) for mood stabilization. Additionally, quetiapine, 50 mg nightly, is added and eventually titrated to 300 mg to augment mood stabilization and to aid sleep. Before being admitted, Ms. A had been prescribed
The authors’ observations
Definitive treatments for psychiatric conditions in patients with MS have been lacking, and current recommendations are based on regimens used to treat general psychiatric populations. For example, selective serotonin reuptake inhibitors are frequently considered for treatment of MDD in patients with MS, whereas SNRIs are considered for patients with concomitant neuropathic pain.13 Similarly,
OUTCOME Improved mood, energy
After 2 weeks of inpatient treatment, Ms. A shows improvement in mood lability and energy levels, and she is able to tolerate titration of divalproex sodium and quetiapine to therapeutic levels. She is referred to an outpatient psychiatrist after discharge, as well as a follow-up appointment with her neurologist. On discharge, Ms. A expresses a commitment to treatment and hope for the future.
1. National Multiple Sclerosis Society. Signs and symptoms consistent with demyelinating disease (for professionals). https://www.nationalmssociety.org/For-Professionals/Clinical-Care/Diagnosing-MS/Signs-and-Symptoms-Consistent-with-Demyelinating-D. Accessed October 29, 2018.
2. Politte LC, Huffman JC, Stern TA. Neuropsychiatric manifestations of multiple sclerosis. Prim Care Companion J Clin Psychiatry. 2008;10(4):318-324.
3. Siegert RJ, Abernethy D. Depression in multiple sclerosis: a review. J Neurol Neurosurg Psychiatry. 2005;76(4):469-475.
4. Scalfari A, Knappertz V, Cutter G, et al. Mortality in patients with multiple sclerosis. Neurology. 2013;81(2):184-192.
5. Ghaffar O, Feinstein A. The neuropsychiatry of multiple sclerosis: a review of recent developments. Curr Opin Psychiatry. 2007;20(3):278-285.
6. Duncan A, Malcolm-Smith S, Ameen O, et al. The incidence of euphoria in multiple sclerosis: artefact of measure. Mult Scler Int. 2016;2016:1-8.
7. Paparrigopoulos T, Ferentinos P, Kouzoupis A, et al. The neuropsychiatry of multiple sclerosis: focus on disorders of mood, affect and behaviour. Int Rev Psychiatry. 2010;22(1):14-21.
8. Bakshi R, Czarnecki D, Shaikh ZA, et al. Brain MRI lesions and atrophy are related to depression in multiple sclerosis. Neuroreport. 2000;11(6):1153-1158.
9. Feinstein A, Roy P, Lobaugh N, et al. Structural brain abnormalities in multiple sclerosis patients with major depression. Neurology. 2004;62(4):586-590.
10. Hama S, Yamashita H, Shigenobu M, et al. Post-stroke affective or apathetic depression and lesion location: left frontal lobe and bilateral basal ganglia. Eur Arch Psychiatry Clin Neurosci. 2007;257(3):149-152.
11. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
12. Beyer JL, Young R, Kuchibhatla M, et al. Hyperintense MRI lesions in bipolar disorder: a meta-analysis and review. Int Rev Psychiatry. 2009;21(4):394-409.
13. Feinstein A. Neuropsychiatric syndromes associated with multiple sclerosis. J Neurol. 2007;254(S2):1173-1176.
14. Thomas PW, Thomas S, Hillier C, et al. Psychological interventions for multiple sclerosis. Cochrane Database Syst Rev. 2006;(1):CD004431. doi: 10.1002/14651858.cd004431.pub2.
1. National Multiple Sclerosis Society. Signs and symptoms consistent with demyelinating disease (for professionals). https://www.nationalmssociety.org/For-Professionals/Clinical-Care/Diagnosing-MS/Signs-and-Symptoms-Consistent-with-Demyelinating-D. Accessed October 29, 2018.
2. Politte LC, Huffman JC, Stern TA. Neuropsychiatric manifestations of multiple sclerosis. Prim Care Companion J Clin Psychiatry. 2008;10(4):318-324.
3. Siegert RJ, Abernethy D. Depression in multiple sclerosis: a review. J Neurol Neurosurg Psychiatry. 2005;76(4):469-475.
4. Scalfari A, Knappertz V, Cutter G, et al. Mortality in patients with multiple sclerosis. Neurology. 2013;81(2):184-192.
5. Ghaffar O, Feinstein A. The neuropsychiatry of multiple sclerosis: a review of recent developments. Curr Opin Psychiatry. 2007;20(3):278-285.
6. Duncan A, Malcolm-Smith S, Ameen O, et al. The incidence of euphoria in multiple sclerosis: artefact of measure. Mult Scler Int. 2016;2016:1-8.
7. Paparrigopoulos T, Ferentinos P, Kouzoupis A, et al. The neuropsychiatry of multiple sclerosis: focus on disorders of mood, affect and behaviour. Int Rev Psychiatry. 2010;22(1):14-21.
8. Bakshi R, Czarnecki D, Shaikh ZA, et al. Brain MRI lesions and atrophy are related to depression in multiple sclerosis. Neuroreport. 2000;11(6):1153-1158.
9. Feinstein A, Roy P, Lobaugh N, et al. Structural brain abnormalities in multiple sclerosis patients with major depression. Neurology. 2004;62(4):586-590.
10. Hama S, Yamashita H, Shigenobu M, et al. Post-stroke affective or apathetic depression and lesion location: left frontal lobe and bilateral basal ganglia. Eur Arch Psychiatry Clin Neurosci. 2007;257(3):149-152.
11. Carpenter LL, Janicak PG, Aaronson ST, et al. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587-596.
12. Beyer JL, Young R, Kuchibhatla M, et al. Hyperintense MRI lesions in bipolar disorder: a meta-analysis and review. Int Rev Psychiatry. 2009;21(4):394-409.
13. Feinstein A. Neuropsychiatric syndromes associated with multiple sclerosis. J Neurol. 2007;254(S2):1173-1176.
14. Thomas PW, Thomas S, Hillier C, et al. Psychological interventions for multiple sclerosis. Cochrane Database Syst Rev. 2006;(1):CD004431. doi: 10.1002/14651858.cd004431.pub2.
The teenager who couldn’t stay awake
CASE Somnolent, confused, and hungry
Mr. G, age 14, presents to the emergency department (ED) for acute-onset hypersomnia that has gradually worsened over the course of a few days. Mr. G now sleeps most of the day, has altered mental status, and is experiencing emotional dysregulation with no clear etiology. His mother, who accompanies him to the ED, says that prior to the onset of these symptoms her son had been healthy. She notes that he has been eating more than usual, which she assumes is due to a growth spurt.
Mr. G’s symptoms began 4 days ago when he became increasingly fatigued, sleeping for 11 to 12 hours per day, with intermittent episodes of staring and unresponsiveness from which he rapidly returned to baseline. During the next 3 days, he became more confused and somnolent, and began to display bizarre behavior, including eating food out of the trash and attempting to microwave a full metal pot. He exhibited unexplained crying spells, calling out for his “mommy,” and saying he was “afraid I’m dying.”
During the 2 days before he came to our ED, Mr. G was seen at 2 other hospitals. Following extensive imaging and laboratory work-up, clinicians at these facilities attributed his symptoms to intoxication from an unknown substance. Mr. G has a history of marijuana use, and his mother reports that his friends had recently been using synthetic marijuana. However, no intoxicant was identified on urine or gas chromatography drug screening.
Mr. G’s history includes oppositional behavior, and a brief psychiatric hospitalization at age 5 for aggression. He has otherwise been healthy. His family history is significant for maternal substance use and anxiety disorders. In addition to sporadic cannabis use, Mr. G’s social history includes multiple recent family losses, previous foster care placement, and recent declining academic performance.
[polldaddy:10148168]
EVALUATION No red flags
On admission, Mr. G appears somnolent and displays disorganized speech, impulsivity, frequent disorientation, and intermittent agitation/anxiety; he sleeps 16 to 18 hours per day. Mr. G is admitted with a presumptive diagnosis of substance intoxication and transferred to the general pediatric inpatient unit. Upon arrival, he is found to be bradycardic (42 beats per minute), although afebrile with otherwise age-appropriate vitals. On exam, he is somnolent but arousable and follows simple commands.
Continue to: Mr. G undergoes a Monospot test...
Mr. G undergoes a Monospot test, which is positive, with subsequent evidence of a prior, but not active, Epstein-Barr virus (EBV) infection. He also has a mildly elevated CSF protein level, but subsequent CSF labs are negative for both infectious and non-infectious processes. An EEG reveals focal neuronal slowing.
During brief periods of wakefulness, Mr. G calls out to his mother and says, “I’m not going to make it to my birthday,” and “You’re going to have to let me go.” He occasionally becomes combative, pulling at IV lines and swearing at staff. His bradycardia resolves without intervention during his admission. On Day 8 of his hospitalization, Mr. G displays hypersexuality and makes sexually suggestive comments toward female staff members. He also experiences recurrence of hyperphagia.
On Day 10 of his stay on the pediatric unit, because of the emergence of hypersexuality and hyperphagia, along with a largely negative medical evaluation, Mr. G is transferred to the pediatric psychiatric unit for ongoing evaluation and management.
[polldaddy:10148172]
The authors’ observations
Given Mr. G’s rapid onset of confusion, hypersomnia, and emotional dysregulation, our differential diagnosis included delirium of unclear etiology, substance intoxication, autoimmune encephalitis, viral meningitis, heavy metal intoxication, primary psychotic disorder, and KLS. Mr. G underwent an extensive diagnostic evaluation, which was largely unremarkable (Table). He had a mildly elevated CSF protein level, but subsequent CSF labs were negative for both infectious and non-infectious processes. When Mr. G was transferred to the pediatric inpatient psychiatric unit on Day 10, the presumptive diagnosis was KLS.
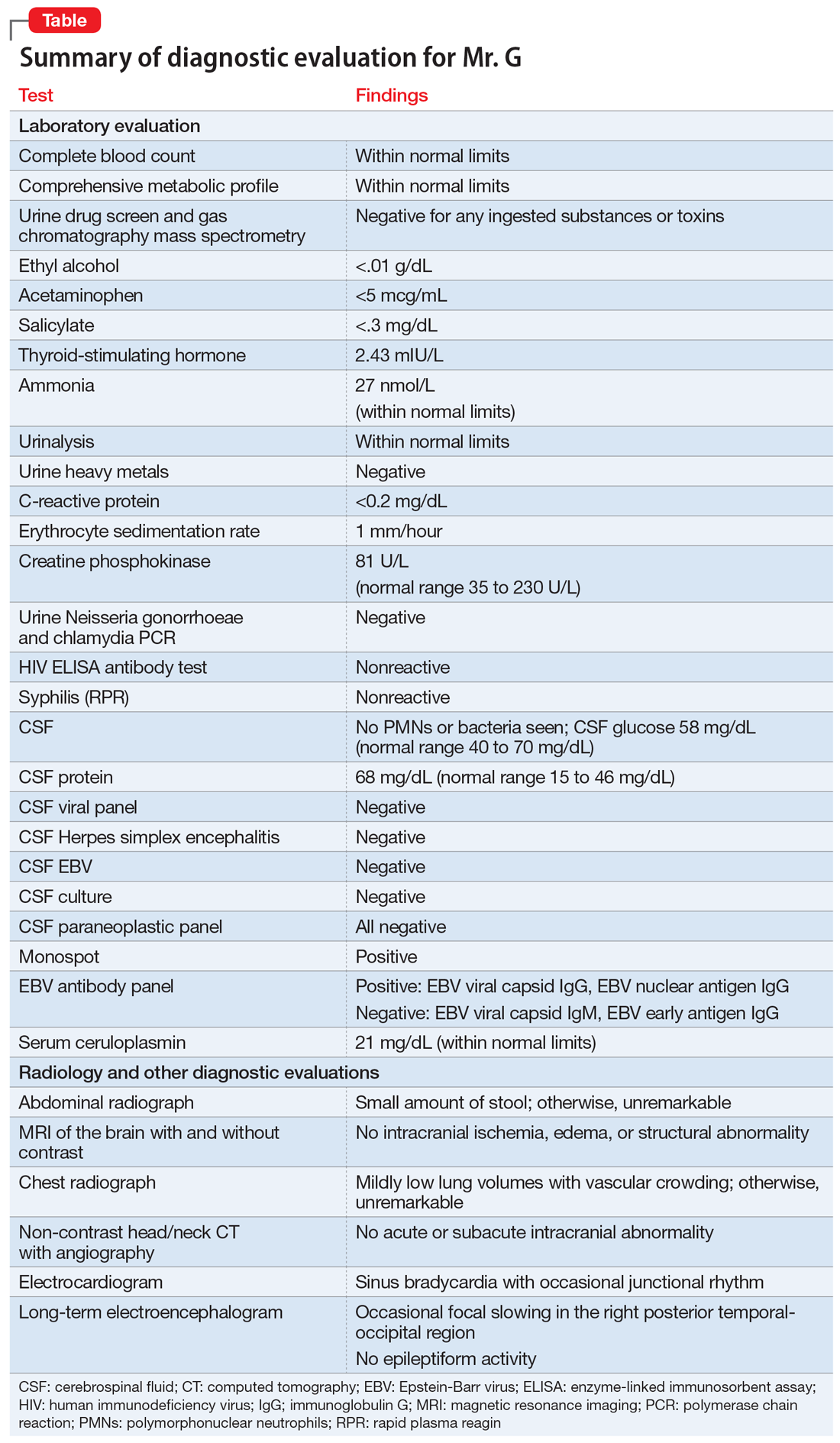
KLS is a rare neurologic disorder, with an incidence of 1 to 5 in 1 million and a 4:1 male-to-female predominance.1 It poses a diagnostic challenge due to its low prevalence and broad differential. The disorder typically presents in early adolescence and is characterized by episodes of severe hypersomnia with associated cognitive and/or behavioral disturbance2 (Box2-4). Bradycardia, as seen in Mr. G, and other forms of autonomic dysregulation have been reported in the literature, as has the focal neuronal slowing noted on Mr. G’s EEG.3

[polldaddy:10148174]
Continue to: TREATMENT Methylphenidate and a safety plan
TREATMENT Methylphenidate and a safety plan
On Day 11 of hospitalization, Mr. G is started on methylphenidate, 10 mg in the morning and 5 mg in the afternoon. After starting methylphenidate, he sustains more regular wakefulness, with improved thought organization, engagement, and fewer disruptive behaviors. He receives infrequent, as-needed doses of olanzapine, and by Day 14, he returns to his baseline behavior and cognition.
A safety plan is created for the family to address worsening symptoms or future episodes. The safety plan is developed with Mr. G and input from his family. It is to be administered in all settings and we particularly emphasized using it in the school setting, where staff may not be familiar with KLS. The safety plan involves a description of KLS, its symptoms, the risks for hypersomnolence, hypersexuality, and psychotic symptoms or behavioral dysregulation. It stresses close supervision of Mr. G, not allowing him to be unsupervised or unchaperoned on school trips or other outings, and lethal means restriction. It outlines a detailed plan if Mr. G’s behavior decompensates or escalates, including a step-wise approach to engaging psychological interventions and mental health resources, and securing crisis services as needed.
On Day 15, he is discharged to home in stable condition with outpatient mental health follow-up and continues to take the prescribed methylphenidate.
The authors’ observations
Management of KLS is primarily supportive. Stimulants may help reduce hypersomnia, impulsivity, and inattention early in the disease course.1 However, in a systematic review, 89% of patients with KLS who received methylphenidate experienced worsening or no improvement, and 11% showed only partial improvement.2 Amantadine was more promising, with 29% of patients with KLS showing partial benefit and 12% showing significant benefit.2 Multiple other pharmacologic agents have been described with varying efficacy, including lithium, valproate, risperidone, bupropion, and immunoglobulins.2 Furthermore, lithium and valproate have been suggested to be helpful in preventing recurrences in some cases.6
The circumstances surrounding Mr. G’s symptom onset are unclear and may have been multifactorial. It is possible that his prior EBV infection was a trigger for this KLS-associated episode, as EBV is a known precipitant for KLS episodes.3 Mr. G’s history of cannabis use may also have served as an early trigger for KLS.3
This case highlights the importance of multidisciplinary collaboration in a diagnostically challenging case. It emphasizes the need for a broad differential and the importance of challenging a previous diagnosis in the face of mounting evidence to the contrary. In this case, the patient’s history of irritability, aggression, and cannabis use resulted in multiple clinicians misattributing his symptoms to substance use or a primary psychiatric disorder. However, given his symptom acuity, progression, and the lack of findings on diagnostic evaluation to explain his presentation, these initial diagnoses did not explain the severity, nature, or duration of his symptoms. Keeping KLS in the differential is particularly important for patients with a prior history of psychiatric illness or substance use, because these patients are at higher risk for misattribution of symptoms to pre-existing psychiatric illness. Evolution of symptoms, a negative diagnostic evaluation, and maintaining a broad differential resulted in eventually reaching the final diagnosis of KLS and development of a longitudinal management plan.
While further work must be done to clearly define the pharmacologic approach to acute management of KLS episodes, nonpharmacologic aspects of care must not be neglected. Behavioral planning, adjustment of the environment, engagement with schools/community supports, and family education are valuable tools for facilitating the patient’s de-escalation, avoiding unneeded polypharmacy, reducing anxieties, and safeguarding the patient from unnecessary harm.7 Clinicians can support their patients’ transitions back into the community by ensuring careful outpatient follow-up for symptom monitoring and by communicating with patients’ schools and employers.
OUTCOME Asymptomatic; no recurrence of symptoms
Forty-six days after his symptoms began and 31 days after hospital discharge, Mr. G is asymptomatic with no recurrence of symptoms.
Bottom Line
Kleine-Levin syndrome (KLS) is a rare, often-overlooked condition that should be considered in the differential diagnosis for patients who present with hypersomnolence and altered mental status without a clear etiology. Rapid recognition of KLS can prevent misattribution of symptoms, unnecessary treatment, and missed opportunities for care.
1. Billiard M, Jaussent I, Dauvilliers Y, et al. Recurrent hypersomnia: a review of 339 cases. Sleep Med. 2011;15(4):247-257.
2. Arnulf I, Lin L, Gadoth N, et al. Kleine-Levin syndrome: a systematic study of 108 patients. Ann Neurol. 2008;63(4):482-493.
3. Arnulf I. Kleine-Levin syndrome: a systematic review of 186 cases in the literature. Brain. 2005;128(12):2763-2776.
4. Lisk R. Kleine-Levin syndrome. Pract Neurol. 2009;9(1);42-45.
5. de Araújo Lima TF, da Silva Behrens NS, Lopes E, et al. Kleine–Levin Syndrome: a case report. Sleep Sci. 2014;7(2):122-125.
6. Goldberg MA. The treatment of Kleine-Levin syndrome with lithium. Can J Psychiatry. 1983;28:491-493.
7. Gadoth N, Kesler A, Vainstein G, et al. Clinical and polysomnographic characteristics of 34 patients with Kleine-Levin syndrome. J Sleep Res. 2001;10(4):337-341.
CASE Somnolent, confused, and hungry
Mr. G, age 14, presents to the emergency department (ED) for acute-onset hypersomnia that has gradually worsened over the course of a few days. Mr. G now sleeps most of the day, has altered mental status, and is experiencing emotional dysregulation with no clear etiology. His mother, who accompanies him to the ED, says that prior to the onset of these symptoms her son had been healthy. She notes that he has been eating more than usual, which she assumes is due to a growth spurt.
Mr. G’s symptoms began 4 days ago when he became increasingly fatigued, sleeping for 11 to 12 hours per day, with intermittent episodes of staring and unresponsiveness from which he rapidly returned to baseline. During the next 3 days, he became more confused and somnolent, and began to display bizarre behavior, including eating food out of the trash and attempting to microwave a full metal pot. He exhibited unexplained crying spells, calling out for his “mommy,” and saying he was “afraid I’m dying.”
During the 2 days before he came to our ED, Mr. G was seen at 2 other hospitals. Following extensive imaging and laboratory work-up, clinicians at these facilities attributed his symptoms to intoxication from an unknown substance. Mr. G has a history of marijuana use, and his mother reports that his friends had recently been using synthetic marijuana. However, no intoxicant was identified on urine or gas chromatography drug screening.
Mr. G’s history includes oppositional behavior, and a brief psychiatric hospitalization at age 5 for aggression. He has otherwise been healthy. His family history is significant for maternal substance use and anxiety disorders. In addition to sporadic cannabis use, Mr. G’s social history includes multiple recent family losses, previous foster care placement, and recent declining academic performance.
[polldaddy:10148168]
EVALUATION No red flags
On admission, Mr. G appears somnolent and displays disorganized speech, impulsivity, frequent disorientation, and intermittent agitation/anxiety; he sleeps 16 to 18 hours per day. Mr. G is admitted with a presumptive diagnosis of substance intoxication and transferred to the general pediatric inpatient unit. Upon arrival, he is found to be bradycardic (42 beats per minute), although afebrile with otherwise age-appropriate vitals. On exam, he is somnolent but arousable and follows simple commands.
Continue to: Mr. G undergoes a Monospot test...
Mr. G undergoes a Monospot test, which is positive, with subsequent evidence of a prior, but not active, Epstein-Barr virus (EBV) infection. He also has a mildly elevated CSF protein level, but subsequent CSF labs are negative for both infectious and non-infectious processes. An EEG reveals focal neuronal slowing.
During brief periods of wakefulness, Mr. G calls out to his mother and says, “I’m not going to make it to my birthday,” and “You’re going to have to let me go.” He occasionally becomes combative, pulling at IV lines and swearing at staff. His bradycardia resolves without intervention during his admission. On Day 8 of his hospitalization, Mr. G displays hypersexuality and makes sexually suggestive comments toward female staff members. He also experiences recurrence of hyperphagia.
On Day 10 of his stay on the pediatric unit, because of the emergence of hypersexuality and hyperphagia, along with a largely negative medical evaluation, Mr. G is transferred to the pediatric psychiatric unit for ongoing evaluation and management.
[polldaddy:10148172]
The authors’ observations
Given Mr. G’s rapid onset of confusion, hypersomnia, and emotional dysregulation, our differential diagnosis included delirium of unclear etiology, substance intoxication, autoimmune encephalitis, viral meningitis, heavy metal intoxication, primary psychotic disorder, and KLS. Mr. G underwent an extensive diagnostic evaluation, which was largely unremarkable (Table). He had a mildly elevated CSF protein level, but subsequent CSF labs were negative for both infectious and non-infectious processes. When Mr. G was transferred to the pediatric inpatient psychiatric unit on Day 10, the presumptive diagnosis was KLS.
KLS is a rare neurologic disorder, with an incidence of 1 to 5 in 1 million and a 4:1 male-to-female predominance.1 It poses a diagnostic challenge due to its low prevalence and broad differential. The disorder typically presents in early adolescence and is characterized by episodes of severe hypersomnia with associated cognitive and/or behavioral disturbance2 (Box2-4). Bradycardia, as seen in Mr. G, and other forms of autonomic dysregulation have been reported in the literature, as has the focal neuronal slowing noted on Mr. G’s EEG.3
[polldaddy:10148174]
Continue to: TREATMENT Methylphenidate and a safety plan
TREATMENT Methylphenidate and a safety plan
On Day 11 of hospitalization, Mr. G is started on methylphenidate, 10 mg in the morning and 5 mg in the afternoon. After starting methylphenidate, he sustains more regular wakefulness, with improved thought organization, engagement, and fewer disruptive behaviors. He receives infrequent, as-needed doses of olanzapine, and by Day 14, he returns to his baseline behavior and cognition.
A safety plan is created for the family to address worsening symptoms or future episodes. The safety plan is developed with Mr. G and input from his family. It is to be administered in all settings and we particularly emphasized using it in the school setting, where staff may not be familiar with KLS. The safety plan involves a description of KLS, its symptoms, the risks for hypersomnolence, hypersexuality, and psychotic symptoms or behavioral dysregulation. It stresses close supervision of Mr. G, not allowing him to be unsupervised or unchaperoned on school trips or other outings, and lethal means restriction. It outlines a detailed plan if Mr. G’s behavior decompensates or escalates, including a step-wise approach to engaging psychological interventions and mental health resources, and securing crisis services as needed.
On Day 15, he is discharged to home in stable condition with outpatient mental health follow-up and continues to take the prescribed methylphenidate.
The authors’ observations
Management of KLS is primarily supportive. Stimulants may help reduce hypersomnia, impulsivity, and inattention early in the disease course.1 However, in a systematic review, 89% of patients with KLS who received methylphenidate experienced worsening or no improvement, and 11% showed only partial improvement.2 Amantadine was more promising, with 29% of patients with KLS showing partial benefit and 12% showing significant benefit.2 Multiple other pharmacologic agents have been described with varying efficacy, including lithium, valproate, risperidone, bupropion, and immunoglobulins.2 Furthermore, lithium and valproate have been suggested to be helpful in preventing recurrences in some cases.6
The circumstances surrounding Mr. G’s symptom onset are unclear and may have been multifactorial. It is possible that his prior EBV infection was a trigger for this KLS-associated episode, as EBV is a known precipitant for KLS episodes.3 Mr. G’s history of cannabis use may also have served as an early trigger for KLS.3
This case highlights the importance of multidisciplinary collaboration in a diagnostically challenging case. It emphasizes the need for a broad differential and the importance of challenging a previous diagnosis in the face of mounting evidence to the contrary. In this case, the patient’s history of irritability, aggression, and cannabis use resulted in multiple clinicians misattributing his symptoms to substance use or a primary psychiatric disorder. However, given his symptom acuity, progression, and the lack of findings on diagnostic evaluation to explain his presentation, these initial diagnoses did not explain the severity, nature, or duration of his symptoms. Keeping KLS in the differential is particularly important for patients with a prior history of psychiatric illness or substance use, because these patients are at higher risk for misattribution of symptoms to pre-existing psychiatric illness. Evolution of symptoms, a negative diagnostic evaluation, and maintaining a broad differential resulted in eventually reaching the final diagnosis of KLS and development of a longitudinal management plan.
While further work must be done to clearly define the pharmacologic approach to acute management of KLS episodes, nonpharmacologic aspects of care must not be neglected. Behavioral planning, adjustment of the environment, engagement with schools/community supports, and family education are valuable tools for facilitating the patient’s de-escalation, avoiding unneeded polypharmacy, reducing anxieties, and safeguarding the patient from unnecessary harm.7 Clinicians can support their patients’ transitions back into the community by ensuring careful outpatient follow-up for symptom monitoring and by communicating with patients’ schools and employers.
OUTCOME Asymptomatic; no recurrence of symptoms
Forty-six days after his symptoms began and 31 days after hospital discharge, Mr. G is asymptomatic with no recurrence of symptoms.
Bottom Line
Kleine-Levin syndrome (KLS) is a rare, often-overlooked condition that should be considered in the differential diagnosis for patients who present with hypersomnolence and altered mental status without a clear etiology. Rapid recognition of KLS can prevent misattribution of symptoms, unnecessary treatment, and missed opportunities for care.
CASE Somnolent, confused, and hungry
Mr. G, age 14, presents to the emergency department (ED) for acute-onset hypersomnia that has gradually worsened over the course of a few days. Mr. G now sleeps most of the day, has altered mental status, and is experiencing emotional dysregulation with no clear etiology. His mother, who accompanies him to the ED, says that prior to the onset of these symptoms her son had been healthy. She notes that he has been eating more than usual, which she assumes is due to a growth spurt.
Mr. G’s symptoms began 4 days ago when he became increasingly fatigued, sleeping for 11 to 12 hours per day, with intermittent episodes of staring and unresponsiveness from which he rapidly returned to baseline. During the next 3 days, he became more confused and somnolent, and began to display bizarre behavior, including eating food out of the trash and attempting to microwave a full metal pot. He exhibited unexplained crying spells, calling out for his “mommy,” and saying he was “afraid I’m dying.”
During the 2 days before he came to our ED, Mr. G was seen at 2 other hospitals. Following extensive imaging and laboratory work-up, clinicians at these facilities attributed his symptoms to intoxication from an unknown substance. Mr. G has a history of marijuana use, and his mother reports that his friends had recently been using synthetic marijuana. However, no intoxicant was identified on urine or gas chromatography drug screening.
Mr. G’s history includes oppositional behavior, and a brief psychiatric hospitalization at age 5 for aggression. He has otherwise been healthy. His family history is significant for maternal substance use and anxiety disorders. In addition to sporadic cannabis use, Mr. G’s social history includes multiple recent family losses, previous foster care placement, and recent declining academic performance.
[polldaddy:10148168]
EVALUATION No red flags
On admission, Mr. G appears somnolent and displays disorganized speech, impulsivity, frequent disorientation, and intermittent agitation/anxiety; he sleeps 16 to 18 hours per day. Mr. G is admitted with a presumptive diagnosis of substance intoxication and transferred to the general pediatric inpatient unit. Upon arrival, he is found to be bradycardic (42 beats per minute), although afebrile with otherwise age-appropriate vitals. On exam, he is somnolent but arousable and follows simple commands.
Continue to: Mr. G undergoes a Monospot test...
Mr. G undergoes a Monospot test, which is positive, with subsequent evidence of a prior, but not active, Epstein-Barr virus (EBV) infection. He also has a mildly elevated CSF protein level, but subsequent CSF labs are negative for both infectious and non-infectious processes. An EEG reveals focal neuronal slowing.
During brief periods of wakefulness, Mr. G calls out to his mother and says, “I’m not going to make it to my birthday,” and “You’re going to have to let me go.” He occasionally becomes combative, pulling at IV lines and swearing at staff. His bradycardia resolves without intervention during his admission. On Day 8 of his hospitalization, Mr. G displays hypersexuality and makes sexually suggestive comments toward female staff members. He also experiences recurrence of hyperphagia.
On Day 10 of his stay on the pediatric unit, because of the emergence of hypersexuality and hyperphagia, along with a largely negative medical evaluation, Mr. G is transferred to the pediatric psychiatric unit for ongoing evaluation and management.
[polldaddy:10148172]
The authors’ observations
Given Mr. G’s rapid onset of confusion, hypersomnia, and emotional dysregulation, our differential diagnosis included delirium of unclear etiology, substance intoxication, autoimmune encephalitis, viral meningitis, heavy metal intoxication, primary psychotic disorder, and KLS. Mr. G underwent an extensive diagnostic evaluation, which was largely unremarkable (Table). He had a mildly elevated CSF protein level, but subsequent CSF labs were negative for both infectious and non-infectious processes. When Mr. G was transferred to the pediatric inpatient psychiatric unit on Day 10, the presumptive diagnosis was KLS.
KLS is a rare neurologic disorder, with an incidence of 1 to 5 in 1 million and a 4:1 male-to-female predominance.1 It poses a diagnostic challenge due to its low prevalence and broad differential. The disorder typically presents in early adolescence and is characterized by episodes of severe hypersomnia with associated cognitive and/or behavioral disturbance2 (Box2-4). Bradycardia, as seen in Mr. G, and other forms of autonomic dysregulation have been reported in the literature, as has the focal neuronal slowing noted on Mr. G’s EEG.3
[polldaddy:10148174]
Continue to: TREATMENT Methylphenidate and a safety plan
TREATMENT Methylphenidate and a safety plan
On Day 11 of hospitalization, Mr. G is started on methylphenidate, 10 mg in the morning and 5 mg in the afternoon. After starting methylphenidate, he sustains more regular wakefulness, with improved thought organization, engagement, and fewer disruptive behaviors. He receives infrequent, as-needed doses of olanzapine, and by Day 14, he returns to his baseline behavior and cognition.
A safety plan is created for the family to address worsening symptoms or future episodes. The safety plan is developed with Mr. G and input from his family. It is to be administered in all settings and we particularly emphasized using it in the school setting, where staff may not be familiar with KLS. The safety plan involves a description of KLS, its symptoms, the risks for hypersomnolence, hypersexuality, and psychotic symptoms or behavioral dysregulation. It stresses close supervision of Mr. G, not allowing him to be unsupervised or unchaperoned on school trips or other outings, and lethal means restriction. It outlines a detailed plan if Mr. G’s behavior decompensates or escalates, including a step-wise approach to engaging psychological interventions and mental health resources, and securing crisis services as needed.
On Day 15, he is discharged to home in stable condition with outpatient mental health follow-up and continues to take the prescribed methylphenidate.
The authors’ observations
Management of KLS is primarily supportive. Stimulants may help reduce hypersomnia, impulsivity, and inattention early in the disease course.1 However, in a systematic review, 89% of patients with KLS who received methylphenidate experienced worsening or no improvement, and 11% showed only partial improvement.2 Amantadine was more promising, with 29% of patients with KLS showing partial benefit and 12% showing significant benefit.2 Multiple other pharmacologic agents have been described with varying efficacy, including lithium, valproate, risperidone, bupropion, and immunoglobulins.2 Furthermore, lithium and valproate have been suggested to be helpful in preventing recurrences in some cases.6
The circumstances surrounding Mr. G’s symptom onset are unclear and may have been multifactorial. It is possible that his prior EBV infection was a trigger for this KLS-associated episode, as EBV is a known precipitant for KLS episodes.3 Mr. G’s history of cannabis use may also have served as an early trigger for KLS.3
This case highlights the importance of multidisciplinary collaboration in a diagnostically challenging case. It emphasizes the need for a broad differential and the importance of challenging a previous diagnosis in the face of mounting evidence to the contrary. In this case, the patient’s history of irritability, aggression, and cannabis use resulted in multiple clinicians misattributing his symptoms to substance use or a primary psychiatric disorder. However, given his symptom acuity, progression, and the lack of findings on diagnostic evaluation to explain his presentation, these initial diagnoses did not explain the severity, nature, or duration of his symptoms. Keeping KLS in the differential is particularly important for patients with a prior history of psychiatric illness or substance use, because these patients are at higher risk for misattribution of symptoms to pre-existing psychiatric illness. Evolution of symptoms, a negative diagnostic evaluation, and maintaining a broad differential resulted in eventually reaching the final diagnosis of KLS and development of a longitudinal management plan.
While further work must be done to clearly define the pharmacologic approach to acute management of KLS episodes, nonpharmacologic aspects of care must not be neglected. Behavioral planning, adjustment of the environment, engagement with schools/community supports, and family education are valuable tools for facilitating the patient’s de-escalation, avoiding unneeded polypharmacy, reducing anxieties, and safeguarding the patient from unnecessary harm.7 Clinicians can support their patients’ transitions back into the community by ensuring careful outpatient follow-up for symptom monitoring and by communicating with patients’ schools and employers.
OUTCOME Asymptomatic; no recurrence of symptoms
Forty-six days after his symptoms began and 31 days after hospital discharge, Mr. G is asymptomatic with no recurrence of symptoms.
Bottom Line
Kleine-Levin syndrome (KLS) is a rare, often-overlooked condition that should be considered in the differential diagnosis for patients who present with hypersomnolence and altered mental status without a clear etiology. Rapid recognition of KLS can prevent misattribution of symptoms, unnecessary treatment, and missed opportunities for care.
1. Billiard M, Jaussent I, Dauvilliers Y, et al. Recurrent hypersomnia: a review of 339 cases. Sleep Med. 2011;15(4):247-257.
2. Arnulf I, Lin L, Gadoth N, et al. Kleine-Levin syndrome: a systematic study of 108 patients. Ann Neurol. 2008;63(4):482-493.
3. Arnulf I. Kleine-Levin syndrome: a systematic review of 186 cases in the literature. Brain. 2005;128(12):2763-2776.
4. Lisk R. Kleine-Levin syndrome. Pract Neurol. 2009;9(1);42-45.
5. de Araújo Lima TF, da Silva Behrens NS, Lopes E, et al. Kleine–Levin Syndrome: a case report. Sleep Sci. 2014;7(2):122-125.
6. Goldberg MA. The treatment of Kleine-Levin syndrome with lithium. Can J Psychiatry. 1983;28:491-493.
7. Gadoth N, Kesler A, Vainstein G, et al. Clinical and polysomnographic characteristics of 34 patients with Kleine-Levin syndrome. J Sleep Res. 2001;10(4):337-341.
1. Billiard M, Jaussent I, Dauvilliers Y, et al. Recurrent hypersomnia: a review of 339 cases. Sleep Med. 2011;15(4):247-257.
2. Arnulf I, Lin L, Gadoth N, et al. Kleine-Levin syndrome: a systematic study of 108 patients. Ann Neurol. 2008;63(4):482-493.
3. Arnulf I. Kleine-Levin syndrome: a systematic review of 186 cases in the literature. Brain. 2005;128(12):2763-2776.
4. Lisk R. Kleine-Levin syndrome. Pract Neurol. 2009;9(1);42-45.
5. de Araújo Lima TF, da Silva Behrens NS, Lopes E, et al. Kleine–Levin Syndrome: a case report. Sleep Sci. 2014;7(2):122-125.
6. Goldberg MA. The treatment of Kleine-Levin syndrome with lithium. Can J Psychiatry. 1983;28:491-493.
7. Gadoth N, Kesler A, Vainstein G, et al. Clinical and polysomnographic characteristics of 34 patients with Kleine-Levin syndrome. J Sleep Res. 2001;10(4):337-341.
Childhood adversity & lifelong health: From research to action
The rising prevalence of obesity, widespread community violence, and the opioid epidemic are urgent health crises that we have, so far, failed to solve. Physicians must therefore ask: Are we employing the right framework to effectively understand and address these complex problems?
Careful review of the literature reveals that these problems and many others begin with, and are profoundly affected by, childhood adversity. Compounding this, studies over the past 20 years that have focused on abuse and neglect without including community, structural, and historical adversity demonstrate that our definitions of adversity and trauma have been too narrow. The prevalence and diversity of factors affecting development and health is much greater than our medical model anticipates.1,2
CASE
Eileen W, a 55-year-old married, self-employed woman with a 20-year history of autoimmune thyroiditis, longstanding insomnia, and anxiety presents with intense episodes of terror related to public speaking, which are compromising her work performance. Her history is significant for tobacco and alcohol use beginning in early adolescence and continuing into young adulthood, as well as 2 unplanned pregnancies in her 20s. Additional adversities included the murder of her maternal aunt while Ms. W was in utero, resulting in her parents having fostered 2 young cousins; bullying; and the premature death of a special-needs sibling.
What treatment strategies might have been undertaken to manage consequences of the adversities of Ms. W’s childhood—both on her own initiative and as interventions by her health care providers?

Our medical model must be updated to be effective
Because at least 60% of Americans have had 1 or more experiences of childhood adversity, family physicians care for affected patients every day—a reality incompletely addressed by our conventional theories and practices.1,3 Consequently, updating our medical model to incorporate research that confirms the critical and widespread impact of childhood experience on health and illness is an essential task for family medicine.
Core values of family medicine integrate biological, clinical, and behavioral sciences. They include comprehensive and compassionate care that is provided within the context of family and community across the lifespan.4,5 Family medicine is therefore the ideal specialty to lead a movement that will translate scientific evidence of the effects of childhood adversity on health into training, delivery of care, and research—transforming clinical practice and patient health across the lifespan.
This article describes the dramatic impact of childhood adversity on health and well-being and calls on family physicians to play a crucial role in preventing, mitigating, and treating the consequences of childhood adversity, an important root cause of disease.
Continue to: Childhood adversity makes us sick
Childhood adversity makes us sick
The first paper about the landmark Adverse Childhood Experiences (ACE) Study, published 20 years ago, is 1 of more than 90 on this topic.3 This study explored the relationship of physical, emotional, and social health in adulthood and self-reported childhood adversity, and comprised 10 categories of abuse, neglect, and household distress between birth and 18 years of age. One of the largest epidemiological studies of its kind, the ACE Study surveyed more than 17,000 mostly white, middle-aged, educated, and insured participants. Study researchers developed an “ACE Score”—the total number of ACEs faced by a person before her (his) 18th birthday—and found that 64% of respondents endorsed 1 or more ACEs; 27% reported 3 or more ACEs; and 5% experienced 6 or more.
The ACE Study revealed a dose–response relationship between ACEs and more than 40 health-compromising behaviors, negative health conditions, and poor social outcomes. Examples include cardiac, autoimmune disease, obesity, intravenous drug abuse, depression and anxiety, adolescent pregnancy, and worker absenteeism. Tragically, an ACE score of ≥6 conferred a significant risk for premature death.1
ACE data have been collected in diverse populations in 32 states and many countries through the Behavioral Risk Factor Surveillance Survey conducted by the Centers for Disease Control and Prevention3; the Child & Adolescent Health Measurement Initiative’s National Survey of Children’s Health6; and The World Health Organization’s ACE International Questionnaire7—underscoring the pervasiveness of childhood adversity. Evaluation of ACEs in special populations, such as people experiencing homelessness,8 incarcerated youth,9 people struggling with addiction,10 and even health care workers,11 uncovers notably higher rates of ACEs in these populations than in the general population.
Is childhood adversity a true cause of bad outcomes?
Or is the relationship between the 2 entities merely an association? To help answer this question, researchers evaluated the ACE Study using Bradford Hill criteria—9 epidemiological principles employed to infer causation. Their findings strongly support the hypothesis that not only are ACEs associated with myriad negative outcomes, they are their root cause12 and therefore a powerful determinant of our most pressing and expensive health and social problems.Nevertheless, strategies to prevent and address childhood adversity, which are critical to meeting national health goals of successful prevention and treatment of myriad conditions, are absent from the paradigm and practice of most physicians.
The body of research about the health impact of additional adverse experiences is growing to include community violence, poverty, longstanding discrimination,2 and other experiences that we describe as social determinants of health. Furthermore, social determinants of health, or adverse community experiences, appear to maintain a dose–response relationship with health and social outcomes.2 ,13 Along with adverse collective historical experiences (historical trauma),14 these community experiences are forcing further re-examination of existing paradigms of health.
Continue to: The biological pathway from experience to illness
The biological pathway from experience to illness
Neuroscience supports the epidemiology of ACEs.12 The brain develops from the bottom up, in a use-dependent fashion, contingent on genetic potential and, most importantly, on our experiences, which also influence genetic expression. Although present across the lifespan, the brain’s capacity to change—neuroplasticity—is most robust from the prenatal period until about 3 years of age.15 The autonomic nervous system receives information from the body about our internal world and from sensory organs about our external environment and sends it to the brain for processing and interpretation, resulting in micro- and macro-adaptations in structure and function, both within the brain and in the rest of the body.16

Neuroscience demonstrates that adverse experiences, in the context of insufficient protective factors and depending on their timing, severity, and frequency, cause overactivation or prolonged activation, or both, of the stress response system, thus derailing optimal growth and development of the brain and disrupting healthy signaling in all body systems. The dysregulated stress response drives inflammation and subsequent chronic disease (FIGURE17,18), and may influence genetic expression in this, and future, generations.12,14,19 Using neuroimaging and assessment of biomarkers, researchers can see the harm caused by inadequately buffered adversity on overall anatomy and physiology. Protective factors such as a safe environment and positive relationships provide hope that normal biological responses to adverse circumstances can be prevented or reversed, leading to clinical, cognitive, and functional improvement11 (TABLE 120-22).

Evidence-based primary prevention of childhood adversity succeeds
Primary prevention of childhood adversity offers significant benefits across the lifespan and, likely, into the next generation. It ensures that every infant has at least 1 nurturing, attuned caregiver with whom to develop a secure attachment relationship that is essential for optimal growth and development of brain and body.
Primary prevention is most effective when it focuses on supporting caregivers during the perinatal and early childhood periods of their families, before children’s brains are fully organized. Primary prevention involves evidence-based program implementation; collaboration among multiple sectors, including early childhood education, child welfare, criminal justice, business, faith, and health care; and, ultimately, policy change. It incorporates individual, family, and community-based strategies to meet basic needs, ensure safety, fortify a sense of love and belonging in families, and support parents in developing optimal parenting skills. This allows caregivers to devote attention to their children, thus strengthening attunement and attachment, reducing toxic stress, and building protective factors and resilience. Evidence-based and -informed prevention programs include the Nurse–Family Partnership (NFP), Positive Parenting Program (Triple P), and the Family-Centered Medical Home.
NFP. Randomized controlled trials of the NFP, a perinatal home visiting program for low-income, first-time pregnant women and their offspring, showed a reduction in the incidence of domestic violence, child maltreatment, and maternal smoking, with improvement in maternal financial stability, cognitive and socioemotional outcomes, and rates of substance abuse and incarceration in children and/or youth.23
Continue to: Triple P
Triple P. A randomized controlled trial of Triple P, an evidence-based, multilevel, population-based preventive intervention system that was designed to support parents and enhance parenting practices for families with at least 1 child (birth to 12 years old), demonstrated a statistically significant reduction in substantiated child maltreatment cases, out-of-home placements, and emergency room visits and hospitalizations for childhood injuries that were the result of child maltreatment.24
The Family-Centered Medical Home, a primary care strategy to reduce premature and low-birth-weight deliveries, used Medicaid dollars for services not traditionally considered “medical” to address all physical and emotional needs of mothers and families as part of the medical relationship. This program eliminated premature delivery and low birth weight,25 both considered evidence of in utero toxic stress.26
Screening can be brief: In some cases, a single question
The prevalence and impact of childhood adversity, along with the opportunity for significant health improvements and savings, inspires providers to explore screening. Existing screening programs have consistent goals27,28:
- identify unique experiences shaping our patients’ health
- reframe “What’s wrong with you?” as “What happened to you?” “What’s right with you?” and “What matters to you?”
- facilitate health education and neuro-education, particularly meaning-making and self-regulation
- prevent and mitigate the sequelae of exposure to ACEs
- promote health in this and subsequent generations.
The ACE Study screened patients in the context of a comprehensive periodic health assessment. Study participants completed an at-home questionnaire and reviewed it with their physician.1 The Urban ACE Survey added important community stressors such as neighborhood violence, bullying, and food insecurity to the original ACE questionnaire.2
Primary care tool. Wade developed a short, 2-question ACE pre-screener for primary care29 and is exploring screening for childhood adversity in pediatric practice, as are primary care clinicians around the country.
Continue to: Single-question screener
Single-question screener. A Chicago internist interviewed more than 500 patients using a single-question screener that asked whether growing up was “mostly okay or pretty difficult.” This tool accurately confirmed childhood adversity in patients with complex chronic illness, prevented re-traumatization by allowing patients control over disclosure, and opened the door to collaborative healing work over time.30
The Hague Protocol, now mandated in the Netherlands for health and justice professionals, focuses its efforts upstream by offering early detection of children at risk for adverse experiences. The protocol requires asking adults who present with intimate partner violence, suicidality, psychiatric disturbance, or severe substance abuse whether they care for children in any capacity. Those who are so identified are referred to a center at which support services are offered.31
Uncertainty about the utility of existing tools. Many screening tools appear to be promising in terms of identification of the risk for, or actual, childhood adversity, patient and provider satisfaction, and their “fit” in the clinical workflow. Even so, no best practice guidelines exist in primary care to steer screening efforts. Questions remain about27-29:
- broad implementation of a specific tool
- how, when, and where screening should take place
- whether to screen adults, parents, or children—or all 3
- how best to use the content and pacing of screening questions to promote self-regulation and prevent re-traumatization
- best strategies for training and supporting health care workers around screening activities
- how to optimally manage a positive screen.
How best to approach treatment
Treatment includes trauma-informed care, an organizational transformation process (described in TABLE 232; in “The lexicon of childhood adversity: Concepts and tools for care”33-45; and in the subsection, “Lessons from neuroscience”), and individual treatment strategies. The Substance Abuse and Mental Health Services Administration (SAMHSA) of the US Department of Health and Human Services is advocating for implementation of trauma-informed approaches in health systems.

Continue to: The lexicon of childhood adversity...
SIDEBAR
The lexicon of childhood adversity: Concepts and tools for care33-45
Adversity A state or instance of serious or continued difficulty or misfortune. 33
Attachment A special, enduring form of emotional relationship with a specific person involving soothing, pleasure, and comfort.34
Attunement The ability to read and respond to the cues of another.35
Eye-movement desensitization and reprocessing (EMDR) An evidence-based psychotherapy for posttraumatic stress disorder and other psychiatric disorders, mental health problems, and somatic symptoms. EMDR facilitates resumption of normal information processing and integration; the patient attends to emotionally disturbing material in brief sequential doses while simultaneously focusing on an external stimulus. EMDR targets past experience, current triggers, and future potential challenges, and results in alleviation of presenting symptoms; a decrease or elimination of distress from the disturbing memory; improved view of the self; relief from bodily disturbance; and resolution of present and future anticipated triggers.36
Historical trauma Cumulative emotional and psychological wounding, resulting from group traumatic experiences, transmitted across generations within a community.37
Neurofeedback Electroencephalographic biofeedback is a method for retraining brainwave patterns through operant conditioning; it is used to treat posttraumatic stress disorder, various mental health conditions, addiction, chronic pain, epilepsy, and other disorders.38
Neuromodulatory Having the capacity to alter nerve activity through targeted delivery of a stimulus, such as electrical stimulation or chemical agents, to specific neurological sites in the body to help restore function or relieve symptoms.39
Social determinants of health/adverse community experiences Conditions in which people are born, grow, live, work, and age and that are shaped by distribution of money, power, and resources at all levels.40,41
Trauma An event or circumstance experienced or observed by a person as physically or emotionally harmful or threatening and having lasting adverse effects on that person's functioning and well-being.42
Trauma-focused cognitive behavioral therapy An evidence-based trauma treatment for children 3 to 18 years and their parents comprising the elements of the acronym PRACTICE: Psychoeducation and parenting; Relaxation methods; Affective expression and regulation skills; Cognitive coping skills and processing; Trauma narrative and processing; In vivo exposure; Conjoint parent-child therapy sessions; and Enhancing personal safety and growth.43
Trauma-informed approach This "4-R" approach can be implemented in any type of service setting, organization, or program that: Realizes the widespread impact of trauma and understands potential paths for recovery; recognizes signs and symptoms of trauma in clients, families, staff, and others involved with the system; responds by fully integrating knowledge about trauma into policies, procedures, and practices; and seeks to actively resist re-traumatization.44
Use-dependent The organization and function of neurons, the neural system, and the brain depends on repetitive, patterned stimulation.45
Continue to: Trauma-informed care is a model...
Trauma-informed care is a model intended to promote healing and reduce the risk for re-traumatization of patients by staff—significant concerns in clinical settings, where the dynamics of loss of power, control, and safety that are inherent in traumatic experience can be replicated.46 To operationalize trauma-informed care more formally, the Center for Health Care Strategies, Inc., and the National Council for Behavioral Health are developing recommendations for 1) standardized screening and assessment tools, evidence-based clinical interventions, implementation processes, and relevant and replicable outcome measures, and 2) policy changes to improve patient and staff engagement, enhance health outcomes, and reduce avoidable care and excess costs.47,48
Lessons from neuroscience guide effective treatment.16 Treatment begins with bottom-up strategies that are focused on decreasing suboptimal excitatory input from the survival brainstem to create safety, connect patients to resources to meet basic needs, teach self-regulation skills, and improve relational health in and outside of the office. Later-stage top-down methods, such as education and other cognitive activities, focus on strengthening the regulatory capacity of the thinking cortex.16 In many ways, treatment mirrors prevention: It emphasizes first helping patients feel safe and loved.
In a follow-up to the ACE Study, 100,000 patients had a primary care visit in which their practitioner reviewed the ACE questionnaire with them; said “I see that you have________. Tell me how that has affected you later in your life” for every “Yes” response; and listened to the answers without passing judgment. This simple intervention profoundly decreased health resource utilization by these patients during the following year: a reduction of 35% in office visits, 11% in emergency room visits, and 3% in hospitalizations.1
The neurosequential model of therapeutics assesses neurodevelopment in the context of childhood adversity and relational health to evaluate consequences of childhood adversity and direct treatment. Adopted domestically and internationally, this model has had statistically significant success facilitating improvement in patients’ physical, emotional, and social health status.16,49
Trauma-specific treatment modalities such as trauma-focused cognitive behavioral therapy and eye-movement desensitization and reprocessing (EMDR),50 a trauma-specific treatment effective in resolving painful childhood memories, are evidence-based treatments that reduce trauma-related symptoms; evidence is also emerging about the efficacy of yoga51 and neurofeedback.52 These therapies have been best studied as treatment for posttraumatic stress disorder and other mental health disorders and also hold promise for addressing physical and social consequences of adversity. They present a low risk for harm, appear to be cost-effective, and improve outcomes.
Continue to: Best regimens involve a multifaceted approach that combines...
Best regimens involve a multifaceted approach that combines health-system resources with referral to other community practitioners and agencies. An excellent example is a current collaboration between health systems and affordable housing programs to reduce and, ultimately, eliminate chronic homelessness. Positive outcomes of this collaboration include both improved health and life satisfaction for participants and cost savings to the health system.53
CASE
Beginning in adulthood, Ms. W began long-term psychotherapy and had a therapeutic trial of antidepressants, without significant improvement. None of her medical or mental-health providers educated her about the connection between childhood adversity and illness to help her make sense of her health history and autoimmune disease, or to guide treatment. She learned from a friend about the relationship between childhood adversity and poor health and self-administered the ACE questionnaire, scoring 5 points out of a possible 10.
Ms. W enjoyed loving relationships with her mother, sisters, and friends. She had long-standing personal practices of individual and group physical activity, journaling, and spending time in nature.
About 10 years ago, Ms. W committed to regular yoga practice and later saw a functional medicine provider, who focused on nutrition and restorative sleep. She noticed improvement in all signs and symptoms; however, the terror of public speaking remained. Through friends, she found a practitioner who offered EMDR. Over the past 2 years, her terror has resolved and general anxiety and insomnia have continued to improve; she is now able to speak with fluency and comfort in any arena.
Addressing childhood adversity: Our “natural domain”
Experiences, positive and negative, shape our psychology and biology; they are powerful determinants of health—or illness. Prevention of, and response to, childhood adversity demand a systems approach to the whole person in context—the natural domain of family medicine.
Continue to: Although clinical translation is still unfolding...
Although clinical translation is still unfolding, the risks of implementing promising prevention and treatment strategies are low, the stakes are high, and the potential benefits are vast. Therefore, we as family physicians can—must—learn and incorporate the science of childhood adversity, neurobiology, and life course into our training, research, and clinical paradigm and practice; we can do that by embedding this framework throughout our training and continuing education in formal didactics, case discussions, hands-on skill-building, scientific investigation, and patient care.
We must make our offices and hospitals trauma-informed; connect patients with resources to meet basic needs and with home-visiting and parent education programs; educate patients about the impact of protective and adverse factors on health; provide and practice self-regulation training in our offices or by referral; and advocate for equity.
Using these strategies, family physicians will play a crucial role in the prevention, mitigation, and treatment of the root cause of disease and society’s deepest individual and collective suffering.
CORRESPONDENCE
Audrey Stillerman, MD, ABFM, ABIHM, ABOIM, Office of Community Engagement and Neighborhood Health Partnerships, 808 South Wolcott Street, Room 809, Chicago, IL 60612; [email protected].
ACKNOWLEDGMENT
Patricia Rush, MD, MBA, and Adrienne Williams, PhD, reviewed the manuscript of this article.
1. Felitti V, Anda R. The relationship of adverse childhood experiences to adult medical disease, psychiatric disorders and sexual behavior: implications for healthcare. In: Lanius RA, Vermetten E, Pain C, eds. The Impact of Early Life Trauma on Health and Disease: The Hidden Epidemic. Cambridge, UK: Cambridge University Press; 2011:77-87.
2. Wade R Jr, Shea JA, Rubin D, et al. Adverse childhood experiences of low-income urban youth. Pediatrics. 2014;134:e13-e20.
3. Centers for Disease Control and Prevention. Child abuse and neglect prevention. April 10, 2018. www.cdc.gov/violenceprevention/childabuseandneglect/index.html. Accessed September 20, 2018.
4. American Academy of Family Physicians. Definition of family medicine. www.aafp.org/about/policies/all/family-medicine-definition.html. Accessed March 5, 2018.
5. Martin JC, Avant RF, Bowman MA, et al; The Future of Family Medicine Project Leadership Committee. The Future of Family Medicine: a collaborative project of the family medicine community. Ann Fam Med. 2004;2 Suppl 1:S3-S32.
6. Child & Adolescent Health Measurement Initiative (CAHMI). A national and across-state profile on Adverse Childhood Experiences among U.S. children and possibilities to heal and thrive. Issue Brief. October 2017. www.cahmi.org/wp-content/uploads/2018/05/aces_brief_final.pdf. Accessed September 20, 2018.
7. World Health Organization. Adverse Childhood Experiences International Questionnaire (ACE-IQ). www.who.int/violence_injury_prevention/violence/activities/adverse_childhood_experiences/en/. Accessed September 20, 2018.
8. Roos LE, Mota N, Afifi TO, et al. Relationship between adverse childhood experiences and homelessness and the impact of axis I and II disorders. Am J Public Health. 2013;103(Suppl 2):S275-S281.
9. Baglivio MT. Wolff KT. Piquero AR, et al. The relationship between adverse childhood experiences (ACE) and juvenile offending trajectories in a juvenile offender sample. J Crim Justice. 2015;43:229-241.
10. Dube SR. Felitti VF. Dong M, et al. Childhood abuse, neglect, and household dysfunction and the risk of illicit drug use: the adverse childhood experiences study. Pediatrics. 2003;111:564-572.
11. Maunder RG, Peladeau N, Savage D, et al. The prevalence of childhood adversity among healthcare workers and its relationship to adult life events, distress and impairment. Child Abuse Negl. 2010;34:114-123.
12. Anda RF, Felitti VJ, Bremner JD, et al. The enduring effects of abuse and related adverse experiences in childhood: a convergence of evidence from neurobiology and epidemiology. Eur Arch Psychiatry Clin Neurosci. 2006;256:174-186.
13. Braveman PA, Cubbin C, Egerter S, et al. Socioeconomic disparities in health in the United States: what the patterns tell us. Am J Public Health. 2010;100(Suppl 1):S186-S196.
14. Bowers ME, Yehuda R. Intergenerational transmission of stress in humans. Neuropsychopharmacology. 2016;41:232-244.
15. Perry BD. Memories of fears: How the brain stores and retrieves traumatic experiences. In: Goodwin J, Attias R, eds. Splintered Reflections: Images of the Body in Trauma. New York, NY: Basic Books; 1999:9-38.
16. Perry BD. Examining child maltreatment through a neurodevelopmental lens: clinical application of the Neurosequential Model of Therapeutics. J Loss Trauma. 2009;14:240-255.
17.
18. Adding layers to the ACEs pyramid—What do you think? Trauma and social location. ACES Connection, RYSE Center. 2015. www.acesconnection.com/blog/adding-layers-to-the-aces-pyramid-what-do-you-think. Accessed October 10, 2018.
19. Berens AE, Jensen SKG, Nelson CA 3rd. Biological embedding of childhood adversity: from physiological mechanisms to clinical implications. BMC Med. 2017;15:135.
20. Rostad WL, Basile KC, Clayton HB. Association among television and computer/video game use, victimization, and suicide risk among U.S. high school students. J Interpers Violence. 2018 Mar 1:886260518760020.
21. Coyne SM, Nelson DA, Graham-Kevan N, et al. Media depictions of physical and relational aggression: connections with aggression in young adults’ romantic relationships. Aggress Behav. 2011;37:56-62.
22. Centers for Disease Control and Prevention. Violence prevention: Child abuse and neglect: risk and protective factors. April 10, 2018. www.cdc.gov/violenceprevention/childabuseandneglect/riskprotectivefactors.html. Accessed October 10, 2018.
23. Miller TR. Projected outcomes of nurse-family partnership home visitation during 1996-2013, United States. Prev Sci. 2015;16:765-777.
24. Prinz RJ, Sanders MR, Shapiro CJ, et al. Population-based prevention of child maltreatment: the U.S. Triple P system population trial. Prev Sci. 2009;10:1-12.
25. Kraft C. Building capacity & support for two generation primary care. 2015 Midwest Regional Summit on Adverse Childhood Experiences. March 13, 2015. www.hmprg.org/assets/root/PDFs/2015/Summit%20Notes%20for%20Day%20Two.pdf. Accessed September 20, 2018.
26. Smith MV, Gotman N, Yonkers KA. Early childhood adversity and pregnancy outcomes. Matern Child Health J. 2016;20:790-798.
27. Leitch L. Action steps using ACEs and trauma-informed care: a resilience model. Health & Justice. 2017;5:1-10.
28. Bethell CD, Carle A, Hudziak J, et al. Methods to assess adverse childhood experiences of children and families: toward approaches to promote child well-being in policy and practice. Acad Pediatr. 2017;17:S51-S69.
29. Wade R Jr, Becker BD, Bevans KB, et al. Development and evaluation of a short adverse childhood experiences measure. Am J Prev Med. 2017;52:163-172.
30. Rush P. How learning about emotional trauma led me to a new understanding of chronic illness and health disparity. Becoming trauma-informed: Perspectives from public health, faith communities, education and medicine. Presented at 2016 Advocate Symposium, “Becoming a Trauma-Informed Children’s Hospital and Community: Building Foundations of Care, Collaboration and Practice.” Oaklawn, IL: Advocate Children’s Hospital; November 16, 2016.
31. Diderich HM, Fekkes M, Verkerk PH, et al. A new protocol for screening adults presenting with their own medical problems at the Emergency Department to identify children at high risk for maltreatment. Child Abuse Negl. 2013;37:1122-1131.
32. Fact Sheet: Key ingredients for trauma-informed care. Center for Health Care Strategies, Inc. August 2017. www.chcs.org/media/ATC-Key-Ingredients-Fact-Sheet_081417.pdf. Accessed September 22, 2018.
33. Adversity. In: Merriam-Webster Online Dictionary. Springfield, MA: Merriam-Webster, Inc. www.merriam-webster.com/dictionary/adversity. Accessed September 21, 2018.
34. Perry BD. Understanding traumatized and maltreated children: the core concepts. Child Trauma Academy Video Training Series, Video 4;2004:12. Child Trauma Academy (http://childtrauma.org/).
35. Perry BD. Understanding traumatized and maltreated children: the core concepts. Child Trauma Academy Video Training Series, Video 4;2004:19. Child Trauma Academy (http://childtrauma.org/).
36. EMDRIA’s definition of EMDR (eye movement desensitization and reprocessing). Austin, TX: EMDRIA: EMDR International Association. http://c.ymcdn.com/sites/www.emdria.org/resource/resmgr/imported/EMDRIA%20Definition%20of%20EMDR.pdf. Revised February 25 2012. Accessed September 21, 2018.
37. Types of trauma and violence: Historical trauma. Washington, DC: Substance Abuse and Mental Health Services Administration. www.samhsa.gov/trauma-violence/types. Accessed September 21, 2018.
38. Hammond DC. What is neurofeedback? An update. J Neurotherapy. 2011;15:305-336.
39. International Neuromodulation Society. Neuromodulation, or neuromodulatory effect. www.neuromodulation.com/neuromodulation-defined. November 9, 2017. Accessed September 21, 2018.
40. World Health Organization. Social determinants of health. www.who.int/social_determinants/sdh_definition/en/. Accessed September 21, 2018.
41. Davis R, Pinderhughes H, Williams M. Adverse community experiences and resilience: a framework for addressing and preventing community trauma. Oakland, CA: Prevention Institute; 2015:4-5. www.preventioninstitute.org/publications/adverse-community-experiences-and-resilience-framework-addressing-and-preventing. Accessed September 30, 2018.
42. SAMHSA-HRSA Center for Integrated Health Solutions. Trauma. Rockville, MD: Substance Abuse and Mental Health Services Administration and Health Resources and Services Administration, US Department of Health and Human Services. www.integration.samhsa.gov/clinical-practice/trauma. Accessed September 21, 2018.
43. Cohen JA, Mandarino AP. Trauma-focused cognitive behavioural therapy for children and parents. Child Adolesc Ment Health. 2008;13:158-162.
44. Trauma-informed approach and trauma-specific interventions: Trauma-informed approach. Washington, DC: National Center for Trauma Informed Care and Alternatives to Seclusion and Restraints; Substance Abuse and Mental Health Services Administration. www.samhsa.gov/nctic/trauma-interventions. Accessed September 21, 2018.
45. Perry BD. How the brain develops: the importance of early childhood. Child Trauma Academy Video Training Series, Video 1;2004:21. Child Trauma Academy (http://childtrauma.org/).
46. Huang LN, Sharp CS, Gunther T. It’s just good medicine: trauma-informed primary care. (SAMHSA-HRSA Center for Integrated Health Solutions webinar); August 6, 2013. www.integration.samhsa.gov/about-us/CIHS_TIC_Webinar_PDF.pdf. Accessed September 20, 2018.
47. CHCS: Center for Health Care Strategies, Inc. Fact sheet: Key ingredients for trauma-informed care. August 2017. www.chcs.org/media/ATC-Key-Ingredients-Fact-Sheet_081417.pdf. Accessed September 20, 2018.
48. National Council for Behavioral Health. Trauma-informed primary care: fostering resilience and recovery. www.thenationalcouncil.org/consulting-areas-of-expertise/trauma-informed-primary-care/. Accessed September 20, 2018.
49. Child Trauma Academy. The Neurosequential Model of Therapeutics as evidence-based practice. https://childtrauma.org/wp-content/uploads/2015/05/NMT_EvidenceBasedPract_5_2_15.pdf. Accessed September 30, 2018.
50. Bisson JI, Ehlers A, Matthews R, et al. Psychological treatments for chronic post-traumatic stress disorder. Systematic review and meta-analysis. Br J Psychiatry. 2007;190:97-104.
51. Metcalf O, Varker T, Forbes D, et al. Efficacy of fifteen emerging interventions for the treatment of posttraumatic stress disorder: a systematic review. J Trauma Stress. 2016;29:88-92.
52. van der Kolk BA, Hodgdon H, Gapen M, et al. A randomized controlled study of neurofeedback for chronic PTSD. 2016; PLoS One. 2016;11:e0166752.
53. Bryan M. A hospital offers frequent ER patients an out—free housing. “All Things Considered.” National Public Radio. June 29, 2016. www.npr.org/sections/health-shots/2016/06/29/482994000/a-hospital-offers-frequent-er-patients-an-out-free-housing. Acces-sed September 20, 2018.
The rising prevalence of obesity, widespread community violence, and the opioid epidemic are urgent health crises that we have, so far, failed to solve. Physicians must therefore ask: Are we employing the right framework to effectively understand and address these complex problems?
Careful review of the literature reveals that these problems and many others begin with, and are profoundly affected by, childhood adversity. Compounding this, studies over the past 20 years that have focused on abuse and neglect without including community, structural, and historical adversity demonstrate that our definitions of adversity and trauma have been too narrow. The prevalence and diversity of factors affecting development and health is much greater than our medical model anticipates.1,2
CASE
Eileen W, a 55-year-old married, self-employed woman with a 20-year history of autoimmune thyroiditis, longstanding insomnia, and anxiety presents with intense episodes of terror related to public speaking, which are compromising her work performance. Her history is significant for tobacco and alcohol use beginning in early adolescence and continuing into young adulthood, as well as 2 unplanned pregnancies in her 20s. Additional adversities included the murder of her maternal aunt while Ms. W was in utero, resulting in her parents having fostered 2 young cousins; bullying; and the premature death of a special-needs sibling.
What treatment strategies might have been undertaken to manage consequences of the adversities of Ms. W’s childhood—both on her own initiative and as interventions by her health care providers?
Our medical model must be updated to be effective
Because at least 60% of Americans have had 1 or more experiences of childhood adversity, family physicians care for affected patients every day—a reality incompletely addressed by our conventional theories and practices.1,3 Consequently, updating our medical model to incorporate research that confirms the critical and widespread impact of childhood experience on health and illness is an essential task for family medicine.
Core values of family medicine integrate biological, clinical, and behavioral sciences. They include comprehensive and compassionate care that is provided within the context of family and community across the lifespan.4,5 Family medicine is therefore the ideal specialty to lead a movement that will translate scientific evidence of the effects of childhood adversity on health into training, delivery of care, and research—transforming clinical practice and patient health across the lifespan.
This article describes the dramatic impact of childhood adversity on health and well-being and calls on family physicians to play a crucial role in preventing, mitigating, and treating the consequences of childhood adversity, an important root cause of disease.
Continue to: Childhood adversity makes us sick
Childhood adversity makes us sick
The first paper about the landmark Adverse Childhood Experiences (ACE) Study, published 20 years ago, is 1 of more than 90 on this topic.3 This study explored the relationship of physical, emotional, and social health in adulthood and self-reported childhood adversity, and comprised 10 categories of abuse, neglect, and household distress between birth and 18 years of age. One of the largest epidemiological studies of its kind, the ACE Study surveyed more than 17,000 mostly white, middle-aged, educated, and insured participants. Study researchers developed an “ACE Score”—the total number of ACEs faced by a person before her (his) 18th birthday—and found that 64% of respondents endorsed 1 or more ACEs; 27% reported 3 or more ACEs; and 5% experienced 6 or more.
The ACE Study revealed a dose–response relationship between ACEs and more than 40 health-compromising behaviors, negative health conditions, and poor social outcomes. Examples include cardiac, autoimmune disease, obesity, intravenous drug abuse, depression and anxiety, adolescent pregnancy, and worker absenteeism. Tragically, an ACE score of ≥6 conferred a significant risk for premature death.1
ACE data have been collected in diverse populations in 32 states and many countries through the Behavioral Risk Factor Surveillance Survey conducted by the Centers for Disease Control and Prevention3; the Child & Adolescent Health Measurement Initiative’s National Survey of Children’s Health6; and The World Health Organization’s ACE International Questionnaire7—underscoring the pervasiveness of childhood adversity. Evaluation of ACEs in special populations, such as people experiencing homelessness,8 incarcerated youth,9 people struggling with addiction,10 and even health care workers,11 uncovers notably higher rates of ACEs in these populations than in the general population.
Is childhood adversity a true cause of bad outcomes?
Or is the relationship between the 2 entities merely an association? To help answer this question, researchers evaluated the ACE Study using Bradford Hill criteria—9 epidemiological principles employed to infer causation. Their findings strongly support the hypothesis that not only are ACEs associated with myriad negative outcomes, they are their root cause12 and therefore a powerful determinant of our most pressing and expensive health and social problems.Nevertheless, strategies to prevent and address childhood adversity, which are critical to meeting national health goals of successful prevention and treatment of myriad conditions, are absent from the paradigm and practice of most physicians.
The body of research about the health impact of additional adverse experiences is growing to include community violence, poverty, longstanding discrimination,2 and other experiences that we describe as social determinants of health. Furthermore, social determinants of health, or adverse community experiences, appear to maintain a dose–response relationship with health and social outcomes.2 ,13 Along with adverse collective historical experiences (historical trauma),14 these community experiences are forcing further re-examination of existing paradigms of health.
Continue to: The biological pathway from experience to illness
The biological pathway from experience to illness
Neuroscience supports the epidemiology of ACEs.12 The brain develops from the bottom up, in a use-dependent fashion, contingent on genetic potential and, most importantly, on our experiences, which also influence genetic expression. Although present across the lifespan, the brain’s capacity to change—neuroplasticity—is most robust from the prenatal period until about 3 years of age.15 The autonomic nervous system receives information from the body about our internal world and from sensory organs about our external environment and sends it to the brain for processing and interpretation, resulting in micro- and macro-adaptations in structure and function, both within the brain and in the rest of the body.16
Neuroscience demonstrates that adverse experiences, in the context of insufficient protective factors and depending on their timing, severity, and frequency, cause overactivation or prolonged activation, or both, of the stress response system, thus derailing optimal growth and development of the brain and disrupting healthy signaling in all body systems. The dysregulated stress response drives inflammation and subsequent chronic disease (FIGURE17,18), and may influence genetic expression in this, and future, generations.12,14,19 Using neuroimaging and assessment of biomarkers, researchers can see the harm caused by inadequately buffered adversity on overall anatomy and physiology. Protective factors such as a safe environment and positive relationships provide hope that normal biological responses to adverse circumstances can be prevented or reversed, leading to clinical, cognitive, and functional improvement11 (TABLE 120-22).
Evidence-based primary prevention of childhood adversity succeeds
Primary prevention of childhood adversity offers significant benefits across the lifespan and, likely, into the next generation. It ensures that every infant has at least 1 nurturing, attuned caregiver with whom to develop a secure attachment relationship that is essential for optimal growth and development of brain and body.
Primary prevention is most effective when it focuses on supporting caregivers during the perinatal and early childhood periods of their families, before children’s brains are fully organized. Primary prevention involves evidence-based program implementation; collaboration among multiple sectors, including early childhood education, child welfare, criminal justice, business, faith, and health care; and, ultimately, policy change. It incorporates individual, family, and community-based strategies to meet basic needs, ensure safety, fortify a sense of love and belonging in families, and support parents in developing optimal parenting skills. This allows caregivers to devote attention to their children, thus strengthening attunement and attachment, reducing toxic stress, and building protective factors and resilience. Evidence-based and -informed prevention programs include the Nurse–Family Partnership (NFP), Positive Parenting Program (Triple P), and the Family-Centered Medical Home.
NFP. Randomized controlled trials of the NFP, a perinatal home visiting program for low-income, first-time pregnant women and their offspring, showed a reduction in the incidence of domestic violence, child maltreatment, and maternal smoking, with improvement in maternal financial stability, cognitive and socioemotional outcomes, and rates of substance abuse and incarceration in children and/or youth.23
Continue to: Triple P
Triple P. A randomized controlled trial of Triple P, an evidence-based, multilevel, population-based preventive intervention system that was designed to support parents and enhance parenting practices for families with at least 1 child (birth to 12 years old), demonstrated a statistically significant reduction in substantiated child maltreatment cases, out-of-home placements, and emergency room visits and hospitalizations for childhood injuries that were the result of child maltreatment.24
The Family-Centered Medical Home, a primary care strategy to reduce premature and low-birth-weight deliveries, used Medicaid dollars for services not traditionally considered “medical” to address all physical and emotional needs of mothers and families as part of the medical relationship. This program eliminated premature delivery and low birth weight,25 both considered evidence of in utero toxic stress.26
Screening can be brief: In some cases, a single question
The prevalence and impact of childhood adversity, along with the opportunity for significant health improvements and savings, inspires providers to explore screening. Existing screening programs have consistent goals27,28:
- identify unique experiences shaping our patients’ health
- reframe “What’s wrong with you?” as “What happened to you?” “What’s right with you?” and “What matters to you?”
- facilitate health education and neuro-education, particularly meaning-making and self-regulation
- prevent and mitigate the sequelae of exposure to ACEs
- promote health in this and subsequent generations.
The ACE Study screened patients in the context of a comprehensive periodic health assessment. Study participants completed an at-home questionnaire and reviewed it with their physician.1 The Urban ACE Survey added important community stressors such as neighborhood violence, bullying, and food insecurity to the original ACE questionnaire.2
Primary care tool. Wade developed a short, 2-question ACE pre-screener for primary care29 and is exploring screening for childhood adversity in pediatric practice, as are primary care clinicians around the country.
Continue to: Single-question screener
Single-question screener. A Chicago internist interviewed more than 500 patients using a single-question screener that asked whether growing up was “mostly okay or pretty difficult.” This tool accurately confirmed childhood adversity in patients with complex chronic illness, prevented re-traumatization by allowing patients control over disclosure, and opened the door to collaborative healing work over time.30
The Hague Protocol, now mandated in the Netherlands for health and justice professionals, focuses its efforts upstream by offering early detection of children at risk for adverse experiences. The protocol requires asking adults who present with intimate partner violence, suicidality, psychiatric disturbance, or severe substance abuse whether they care for children in any capacity. Those who are so identified are referred to a center at which support services are offered.31
Uncertainty about the utility of existing tools. Many screening tools appear to be promising in terms of identification of the risk for, or actual, childhood adversity, patient and provider satisfaction, and their “fit” in the clinical workflow. Even so, no best practice guidelines exist in primary care to steer screening efforts. Questions remain about27-29:
- broad implementation of a specific tool
- how, when, and where screening should take place
- whether to screen adults, parents, or children—or all 3
- how best to use the content and pacing of screening questions to promote self-regulation and prevent re-traumatization
- best strategies for training and supporting health care workers around screening activities
- how to optimally manage a positive screen.
How best to approach treatment
Treatment includes trauma-informed care, an organizational transformation process (described in TABLE 232; in “The lexicon of childhood adversity: Concepts and tools for care”33-45; and in the subsection, “Lessons from neuroscience”), and individual treatment strategies. The Substance Abuse and Mental Health Services Administration (SAMHSA) of the US Department of Health and Human Services is advocating for implementation of trauma-informed approaches in health systems.
Continue to: The lexicon of childhood adversity...
SIDEBAR
The lexicon of childhood adversity: Concepts and tools for care33-45
Adversity A state or instance of serious or continued difficulty or misfortune. 33
Attachment A special, enduring form of emotional relationship with a specific person involving soothing, pleasure, and comfort.34
Attunement The ability to read and respond to the cues of another.35
Eye-movement desensitization and reprocessing (EMDR) An evidence-based psychotherapy for posttraumatic stress disorder and other psychiatric disorders, mental health problems, and somatic symptoms. EMDR facilitates resumption of normal information processing and integration; the patient attends to emotionally disturbing material in brief sequential doses while simultaneously focusing on an external stimulus. EMDR targets past experience, current triggers, and future potential challenges, and results in alleviation of presenting symptoms; a decrease or elimination of distress from the disturbing memory; improved view of the self; relief from bodily disturbance; and resolution of present and future anticipated triggers.36
Historical trauma Cumulative emotional and psychological wounding, resulting from group traumatic experiences, transmitted across generations within a community.37
Neurofeedback Electroencephalographic biofeedback is a method for retraining brainwave patterns through operant conditioning; it is used to treat posttraumatic stress disorder, various mental health conditions, addiction, chronic pain, epilepsy, and other disorders.38
Neuromodulatory Having the capacity to alter nerve activity through targeted delivery of a stimulus, such as electrical stimulation or chemical agents, to specific neurological sites in the body to help restore function or relieve symptoms.39
Social determinants of health/adverse community experiences Conditions in which people are born, grow, live, work, and age and that are shaped by distribution of money, power, and resources at all levels.40,41
Trauma An event or circumstance experienced or observed by a person as physically or emotionally harmful or threatening and having lasting adverse effects on that person's functioning and well-being.42
Trauma-focused cognitive behavioral therapy An evidence-based trauma treatment for children 3 to 18 years and their parents comprising the elements of the acronym PRACTICE: Psychoeducation and parenting; Relaxation methods; Affective expression and regulation skills; Cognitive coping skills and processing; Trauma narrative and processing; In vivo exposure; Conjoint parent-child therapy sessions; and Enhancing personal safety and growth.43
Trauma-informed approach This "4-R" approach can be implemented in any type of service setting, organization, or program that: Realizes the widespread impact of trauma and understands potential paths for recovery; recognizes signs and symptoms of trauma in clients, families, staff, and others involved with the system; responds by fully integrating knowledge about trauma into policies, procedures, and practices; and seeks to actively resist re-traumatization.44
Use-dependent The organization and function of neurons, the neural system, and the brain depends on repetitive, patterned stimulation.45
Continue to: Trauma-informed care is a model...
Trauma-informed care is a model intended to promote healing and reduce the risk for re-traumatization of patients by staff—significant concerns in clinical settings, where the dynamics of loss of power, control, and safety that are inherent in traumatic experience can be replicated.46 To operationalize trauma-informed care more formally, the Center for Health Care Strategies, Inc., and the National Council for Behavioral Health are developing recommendations for 1) standardized screening and assessment tools, evidence-based clinical interventions, implementation processes, and relevant and replicable outcome measures, and 2) policy changes to improve patient and staff engagement, enhance health outcomes, and reduce avoidable care and excess costs.47,48
Lessons from neuroscience guide effective treatment.16 Treatment begins with bottom-up strategies that are focused on decreasing suboptimal excitatory input from the survival brainstem to create safety, connect patients to resources to meet basic needs, teach self-regulation skills, and improve relational health in and outside of the office. Later-stage top-down methods, such as education and other cognitive activities, focus on strengthening the regulatory capacity of the thinking cortex.16 In many ways, treatment mirrors prevention: It emphasizes first helping patients feel safe and loved.
In a follow-up to the ACE Study, 100,000 patients had a primary care visit in which their practitioner reviewed the ACE questionnaire with them; said “I see that you have________. Tell me how that has affected you later in your life” for every “Yes” response; and listened to the answers without passing judgment. This simple intervention profoundly decreased health resource utilization by these patients during the following year: a reduction of 35% in office visits, 11% in emergency room visits, and 3% in hospitalizations.1
The neurosequential model of therapeutics assesses neurodevelopment in the context of childhood adversity and relational health to evaluate consequences of childhood adversity and direct treatment. Adopted domestically and internationally, this model has had statistically significant success facilitating improvement in patients’ physical, emotional, and social health status.16,49
Trauma-specific treatment modalities such as trauma-focused cognitive behavioral therapy and eye-movement desensitization and reprocessing (EMDR),50 a trauma-specific treatment effective in resolving painful childhood memories, are evidence-based treatments that reduce trauma-related symptoms; evidence is also emerging about the efficacy of yoga51 and neurofeedback.52 These therapies have been best studied as treatment for posttraumatic stress disorder and other mental health disorders and also hold promise for addressing physical and social consequences of adversity. They present a low risk for harm, appear to be cost-effective, and improve outcomes.
Continue to: Best regimens involve a multifaceted approach that combines...
Best regimens involve a multifaceted approach that combines health-system resources with referral to other community practitioners and agencies. An excellent example is a current collaboration between health systems and affordable housing programs to reduce and, ultimately, eliminate chronic homelessness. Positive outcomes of this collaboration include both improved health and life satisfaction for participants and cost savings to the health system.53
CASE
Beginning in adulthood, Ms. W began long-term psychotherapy and had a therapeutic trial of antidepressants, without significant improvement. None of her medical or mental-health providers educated her about the connection between childhood adversity and illness to help her make sense of her health history and autoimmune disease, or to guide treatment. She learned from a friend about the relationship between childhood adversity and poor health and self-administered the ACE questionnaire, scoring 5 points out of a possible 10.
Ms. W enjoyed loving relationships with her mother, sisters, and friends. She had long-standing personal practices of individual and group physical activity, journaling, and spending time in nature.
About 10 years ago, Ms. W committed to regular yoga practice and later saw a functional medicine provider, who focused on nutrition and restorative sleep. She noticed improvement in all signs and symptoms; however, the terror of public speaking remained. Through friends, she found a practitioner who offered EMDR. Over the past 2 years, her terror has resolved and general anxiety and insomnia have continued to improve; she is now able to speak with fluency and comfort in any arena.
Addressing childhood adversity: Our “natural domain”
Experiences, positive and negative, shape our psychology and biology; they are powerful determinants of health—or illness. Prevention of, and response to, childhood adversity demand a systems approach to the whole person in context—the natural domain of family medicine.
Continue to: Although clinical translation is still unfolding...
Although clinical translation is still unfolding, the risks of implementing promising prevention and treatment strategies are low, the stakes are high, and the potential benefits are vast. Therefore, we as family physicians can—must—learn and incorporate the science of childhood adversity, neurobiology, and life course into our training, research, and clinical paradigm and practice; we can do that by embedding this framework throughout our training and continuing education in formal didactics, case discussions, hands-on skill-building, scientific investigation, and patient care.
We must make our offices and hospitals trauma-informed; connect patients with resources to meet basic needs and with home-visiting and parent education programs; educate patients about the impact of protective and adverse factors on health; provide and practice self-regulation training in our offices or by referral; and advocate for equity.
Using these strategies, family physicians will play a crucial role in the prevention, mitigation, and treatment of the root cause of disease and society’s deepest individual and collective suffering.
CORRESPONDENCE
Audrey Stillerman, MD, ABFM, ABIHM, ABOIM, Office of Community Engagement and Neighborhood Health Partnerships, 808 South Wolcott Street, Room 809, Chicago, IL 60612; [email protected].
ACKNOWLEDGMENT
Patricia Rush, MD, MBA, and Adrienne Williams, PhD, reviewed the manuscript of this article.
The rising prevalence of obesity, widespread community violence, and the opioid epidemic are urgent health crises that we have, so far, failed to solve. Physicians must therefore ask: Are we employing the right framework to effectively understand and address these complex problems?
Careful review of the literature reveals that these problems and many others begin with, and are profoundly affected by, childhood adversity. Compounding this, studies over the past 20 years that have focused on abuse and neglect without including community, structural, and historical adversity demonstrate that our definitions of adversity and trauma have been too narrow. The prevalence and diversity of factors affecting development and health is much greater than our medical model anticipates.1,2
CASE
Eileen W, a 55-year-old married, self-employed woman with a 20-year history of autoimmune thyroiditis, longstanding insomnia, and anxiety presents with intense episodes of terror related to public speaking, which are compromising her work performance. Her history is significant for tobacco and alcohol use beginning in early adolescence and continuing into young adulthood, as well as 2 unplanned pregnancies in her 20s. Additional adversities included the murder of her maternal aunt while Ms. W was in utero, resulting in her parents having fostered 2 young cousins; bullying; and the premature death of a special-needs sibling.
What treatment strategies might have been undertaken to manage consequences of the adversities of Ms. W’s childhood—both on her own initiative and as interventions by her health care providers?
Our medical model must be updated to be effective
Because at least 60% of Americans have had 1 or more experiences of childhood adversity, family physicians care for affected patients every day—a reality incompletely addressed by our conventional theories and practices.1,3 Consequently, updating our medical model to incorporate research that confirms the critical and widespread impact of childhood experience on health and illness is an essential task for family medicine.
Core values of family medicine integrate biological, clinical, and behavioral sciences. They include comprehensive and compassionate care that is provided within the context of family and community across the lifespan.4,5 Family medicine is therefore the ideal specialty to lead a movement that will translate scientific evidence of the effects of childhood adversity on health into training, delivery of care, and research—transforming clinical practice and patient health across the lifespan.
This article describes the dramatic impact of childhood adversity on health and well-being and calls on family physicians to play a crucial role in preventing, mitigating, and treating the consequences of childhood adversity, an important root cause of disease.
Continue to: Childhood adversity makes us sick
Childhood adversity makes us sick
The first paper about the landmark Adverse Childhood Experiences (ACE) Study, published 20 years ago, is 1 of more than 90 on this topic.3 This study explored the relationship of physical, emotional, and social health in adulthood and self-reported childhood adversity, and comprised 10 categories of abuse, neglect, and household distress between birth and 18 years of age. One of the largest epidemiological studies of its kind, the ACE Study surveyed more than 17,000 mostly white, middle-aged, educated, and insured participants. Study researchers developed an “ACE Score”—the total number of ACEs faced by a person before her (his) 18th birthday—and found that 64% of respondents endorsed 1 or more ACEs; 27% reported 3 or more ACEs; and 5% experienced 6 or more.
The ACE Study revealed a dose–response relationship between ACEs and more than 40 health-compromising behaviors, negative health conditions, and poor social outcomes. Examples include cardiac, autoimmune disease, obesity, intravenous drug abuse, depression and anxiety, adolescent pregnancy, and worker absenteeism. Tragically, an ACE score of ≥6 conferred a significant risk for premature death.1
ACE data have been collected in diverse populations in 32 states and many countries through the Behavioral Risk Factor Surveillance Survey conducted by the Centers for Disease Control and Prevention3; the Child & Adolescent Health Measurement Initiative’s National Survey of Children’s Health6; and The World Health Organization’s ACE International Questionnaire7—underscoring the pervasiveness of childhood adversity. Evaluation of ACEs in special populations, such as people experiencing homelessness,8 incarcerated youth,9 people struggling with addiction,10 and even health care workers,11 uncovers notably higher rates of ACEs in these populations than in the general population.
Is childhood adversity a true cause of bad outcomes?
Or is the relationship between the 2 entities merely an association? To help answer this question, researchers evaluated the ACE Study using Bradford Hill criteria—9 epidemiological principles employed to infer causation. Their findings strongly support the hypothesis that not only are ACEs associated with myriad negative outcomes, they are their root cause12 and therefore a powerful determinant of our most pressing and expensive health and social problems.Nevertheless, strategies to prevent and address childhood adversity, which are critical to meeting national health goals of successful prevention and treatment of myriad conditions, are absent from the paradigm and practice of most physicians.
The body of research about the health impact of additional adverse experiences is growing to include community violence, poverty, longstanding discrimination,2 and other experiences that we describe as social determinants of health. Furthermore, social determinants of health, or adverse community experiences, appear to maintain a dose–response relationship with health and social outcomes.2 ,13 Along with adverse collective historical experiences (historical trauma),14 these community experiences are forcing further re-examination of existing paradigms of health.
Continue to: The biological pathway from experience to illness
The biological pathway from experience to illness
Neuroscience supports the epidemiology of ACEs.12 The brain develops from the bottom up, in a use-dependent fashion, contingent on genetic potential and, most importantly, on our experiences, which also influence genetic expression. Although present across the lifespan, the brain’s capacity to change—neuroplasticity—is most robust from the prenatal period until about 3 years of age.15 The autonomic nervous system receives information from the body about our internal world and from sensory organs about our external environment and sends it to the brain for processing and interpretation, resulting in micro- and macro-adaptations in structure and function, both within the brain and in the rest of the body.16
Neuroscience demonstrates that adverse experiences, in the context of insufficient protective factors and depending on their timing, severity, and frequency, cause overactivation or prolonged activation, or both, of the stress response system, thus derailing optimal growth and development of the brain and disrupting healthy signaling in all body systems. The dysregulated stress response drives inflammation and subsequent chronic disease (FIGURE17,18), and may influence genetic expression in this, and future, generations.12,14,19 Using neuroimaging and assessment of biomarkers, researchers can see the harm caused by inadequately buffered adversity on overall anatomy and physiology. Protective factors such as a safe environment and positive relationships provide hope that normal biological responses to adverse circumstances can be prevented or reversed, leading to clinical, cognitive, and functional improvement11 (TABLE 120-22).
Evidence-based primary prevention of childhood adversity succeeds
Primary prevention of childhood adversity offers significant benefits across the lifespan and, likely, into the next generation. It ensures that every infant has at least 1 nurturing, attuned caregiver with whom to develop a secure attachment relationship that is essential for optimal growth and development of brain and body.
Primary prevention is most effective when it focuses on supporting caregivers during the perinatal and early childhood periods of their families, before children’s brains are fully organized. Primary prevention involves evidence-based program implementation; collaboration among multiple sectors, including early childhood education, child welfare, criminal justice, business, faith, and health care; and, ultimately, policy change. It incorporates individual, family, and community-based strategies to meet basic needs, ensure safety, fortify a sense of love and belonging in families, and support parents in developing optimal parenting skills. This allows caregivers to devote attention to their children, thus strengthening attunement and attachment, reducing toxic stress, and building protective factors and resilience. Evidence-based and -informed prevention programs include the Nurse–Family Partnership (NFP), Positive Parenting Program (Triple P), and the Family-Centered Medical Home.
NFP. Randomized controlled trials of the NFP, a perinatal home visiting program for low-income, first-time pregnant women and their offspring, showed a reduction in the incidence of domestic violence, child maltreatment, and maternal smoking, with improvement in maternal financial stability, cognitive and socioemotional outcomes, and rates of substance abuse and incarceration in children and/or youth.23
Continue to: Triple P
Triple P. A randomized controlled trial of Triple P, an evidence-based, multilevel, population-based preventive intervention system that was designed to support parents and enhance parenting practices for families with at least 1 child (birth to 12 years old), demonstrated a statistically significant reduction in substantiated child maltreatment cases, out-of-home placements, and emergency room visits and hospitalizations for childhood injuries that were the result of child maltreatment.24
The Family-Centered Medical Home, a primary care strategy to reduce premature and low-birth-weight deliveries, used Medicaid dollars for services not traditionally considered “medical” to address all physical and emotional needs of mothers and families as part of the medical relationship. This program eliminated premature delivery and low birth weight,25 both considered evidence of in utero toxic stress.26
Screening can be brief: In some cases, a single question
The prevalence and impact of childhood adversity, along with the opportunity for significant health improvements and savings, inspires providers to explore screening. Existing screening programs have consistent goals27,28:
- identify unique experiences shaping our patients’ health
- reframe “What’s wrong with you?” as “What happened to you?” “What’s right with you?” and “What matters to you?”
- facilitate health education and neuro-education, particularly meaning-making and self-regulation
- prevent and mitigate the sequelae of exposure to ACEs
- promote health in this and subsequent generations.
The ACE Study screened patients in the context of a comprehensive periodic health assessment. Study participants completed an at-home questionnaire and reviewed it with their physician.1 The Urban ACE Survey added important community stressors such as neighborhood violence, bullying, and food insecurity to the original ACE questionnaire.2
Primary care tool. Wade developed a short, 2-question ACE pre-screener for primary care29 and is exploring screening for childhood adversity in pediatric practice, as are primary care clinicians around the country.
Continue to: Single-question screener
Single-question screener. A Chicago internist interviewed more than 500 patients using a single-question screener that asked whether growing up was “mostly okay or pretty difficult.” This tool accurately confirmed childhood adversity in patients with complex chronic illness, prevented re-traumatization by allowing patients control over disclosure, and opened the door to collaborative healing work over time.30
The Hague Protocol, now mandated in the Netherlands for health and justice professionals, focuses its efforts upstream by offering early detection of children at risk for adverse experiences. The protocol requires asking adults who present with intimate partner violence, suicidality, psychiatric disturbance, or severe substance abuse whether they care for children in any capacity. Those who are so identified are referred to a center at which support services are offered.31
Uncertainty about the utility of existing tools. Many screening tools appear to be promising in terms of identification of the risk for, or actual, childhood adversity, patient and provider satisfaction, and their “fit” in the clinical workflow. Even so, no best practice guidelines exist in primary care to steer screening efforts. Questions remain about27-29:
- broad implementation of a specific tool
- how, when, and where screening should take place
- whether to screen adults, parents, or children—or all 3
- how best to use the content and pacing of screening questions to promote self-regulation and prevent re-traumatization
- best strategies for training and supporting health care workers around screening activities
- how to optimally manage a positive screen.
How best to approach treatment
Treatment includes trauma-informed care, an organizational transformation process (described in TABLE 232; in “The lexicon of childhood adversity: Concepts and tools for care”33-45; and in the subsection, “Lessons from neuroscience”), and individual treatment strategies. The Substance Abuse and Mental Health Services Administration (SAMHSA) of the US Department of Health and Human Services is advocating for implementation of trauma-informed approaches in health systems.
Continue to: The lexicon of childhood adversity...
SIDEBAR
The lexicon of childhood adversity: Concepts and tools for care33-45
Adversity A state or instance of serious or continued difficulty or misfortune. 33
Attachment A special, enduring form of emotional relationship with a specific person involving soothing, pleasure, and comfort.34
Attunement The ability to read and respond to the cues of another.35
Eye-movement desensitization and reprocessing (EMDR) An evidence-based psychotherapy for posttraumatic stress disorder and other psychiatric disorders, mental health problems, and somatic symptoms. EMDR facilitates resumption of normal information processing and integration; the patient attends to emotionally disturbing material in brief sequential doses while simultaneously focusing on an external stimulus. EMDR targets past experience, current triggers, and future potential challenges, and results in alleviation of presenting symptoms; a decrease or elimination of distress from the disturbing memory; improved view of the self; relief from bodily disturbance; and resolution of present and future anticipated triggers.36
Historical trauma Cumulative emotional and psychological wounding, resulting from group traumatic experiences, transmitted across generations within a community.37
Neurofeedback Electroencephalographic biofeedback is a method for retraining brainwave patterns through operant conditioning; it is used to treat posttraumatic stress disorder, various mental health conditions, addiction, chronic pain, epilepsy, and other disorders.38
Neuromodulatory Having the capacity to alter nerve activity through targeted delivery of a stimulus, such as electrical stimulation or chemical agents, to specific neurological sites in the body to help restore function or relieve symptoms.39
Social determinants of health/adverse community experiences Conditions in which people are born, grow, live, work, and age and that are shaped by distribution of money, power, and resources at all levels.40,41
Trauma An event or circumstance experienced or observed by a person as physically or emotionally harmful or threatening and having lasting adverse effects on that person's functioning and well-being.42
Trauma-focused cognitive behavioral therapy An evidence-based trauma treatment for children 3 to 18 years and their parents comprising the elements of the acronym PRACTICE: Psychoeducation and parenting; Relaxation methods; Affective expression and regulation skills; Cognitive coping skills and processing; Trauma narrative and processing; In vivo exposure; Conjoint parent-child therapy sessions; and Enhancing personal safety and growth.43
Trauma-informed approach This "4-R" approach can be implemented in any type of service setting, organization, or program that: Realizes the widespread impact of trauma and understands potential paths for recovery; recognizes signs and symptoms of trauma in clients, families, staff, and others involved with the system; responds by fully integrating knowledge about trauma into policies, procedures, and practices; and seeks to actively resist re-traumatization.44
Use-dependent The organization and function of neurons, the neural system, and the brain depends on repetitive, patterned stimulation.45
Continue to: Trauma-informed care is a model...
Trauma-informed care is a model intended to promote healing and reduce the risk for re-traumatization of patients by staff—significant concerns in clinical settings, where the dynamics of loss of power, control, and safety that are inherent in traumatic experience can be replicated.46 To operationalize trauma-informed care more formally, the Center for Health Care Strategies, Inc., and the National Council for Behavioral Health are developing recommendations for 1) standardized screening and assessment tools, evidence-based clinical interventions, implementation processes, and relevant and replicable outcome measures, and 2) policy changes to improve patient and staff engagement, enhance health outcomes, and reduce avoidable care and excess costs.47,48
Lessons from neuroscience guide effective treatment.16 Treatment begins with bottom-up strategies that are focused on decreasing suboptimal excitatory input from the survival brainstem to create safety, connect patients to resources to meet basic needs, teach self-regulation skills, and improve relational health in and outside of the office. Later-stage top-down methods, such as education and other cognitive activities, focus on strengthening the regulatory capacity of the thinking cortex.16 In many ways, treatment mirrors prevention: It emphasizes first helping patients feel safe and loved.
In a follow-up to the ACE Study, 100,000 patients had a primary care visit in which their practitioner reviewed the ACE questionnaire with them; said “I see that you have________. Tell me how that has affected you later in your life” for every “Yes” response; and listened to the answers without passing judgment. This simple intervention profoundly decreased health resource utilization by these patients during the following year: a reduction of 35% in office visits, 11% in emergency room visits, and 3% in hospitalizations.1
The neurosequential model of therapeutics assesses neurodevelopment in the context of childhood adversity and relational health to evaluate consequences of childhood adversity and direct treatment. Adopted domestically and internationally, this model has had statistically significant success facilitating improvement in patients’ physical, emotional, and social health status.16,49
Trauma-specific treatment modalities such as trauma-focused cognitive behavioral therapy and eye-movement desensitization and reprocessing (EMDR),50 a trauma-specific treatment effective in resolving painful childhood memories, are evidence-based treatments that reduce trauma-related symptoms; evidence is also emerging about the efficacy of yoga51 and neurofeedback.52 These therapies have been best studied as treatment for posttraumatic stress disorder and other mental health disorders and also hold promise for addressing physical and social consequences of adversity. They present a low risk for harm, appear to be cost-effective, and improve outcomes.
Continue to: Best regimens involve a multifaceted approach that combines...
Best regimens involve a multifaceted approach that combines health-system resources with referral to other community practitioners and agencies. An excellent example is a current collaboration between health systems and affordable housing programs to reduce and, ultimately, eliminate chronic homelessness. Positive outcomes of this collaboration include both improved health and life satisfaction for participants and cost savings to the health system.53
CASE
Beginning in adulthood, Ms. W began long-term psychotherapy and had a therapeutic trial of antidepressants, without significant improvement. None of her medical or mental-health providers educated her about the connection between childhood adversity and illness to help her make sense of her health history and autoimmune disease, or to guide treatment. She learned from a friend about the relationship between childhood adversity and poor health and self-administered the ACE questionnaire, scoring 5 points out of a possible 10.
Ms. W enjoyed loving relationships with her mother, sisters, and friends. She had long-standing personal practices of individual and group physical activity, journaling, and spending time in nature.
About 10 years ago, Ms. W committed to regular yoga practice and later saw a functional medicine provider, who focused on nutrition and restorative sleep. She noticed improvement in all signs and symptoms; however, the terror of public speaking remained. Through friends, she found a practitioner who offered EMDR. Over the past 2 years, her terror has resolved and general anxiety and insomnia have continued to improve; she is now able to speak with fluency and comfort in any arena.
Addressing childhood adversity: Our “natural domain”
Experiences, positive and negative, shape our psychology and biology; they are powerful determinants of health—or illness. Prevention of, and response to, childhood adversity demand a systems approach to the whole person in context—the natural domain of family medicine.
Continue to: Although clinical translation is still unfolding...
Although clinical translation is still unfolding, the risks of implementing promising prevention and treatment strategies are low, the stakes are high, and the potential benefits are vast. Therefore, we as family physicians can—must—learn and incorporate the science of childhood adversity, neurobiology, and life course into our training, research, and clinical paradigm and practice; we can do that by embedding this framework throughout our training and continuing education in formal didactics, case discussions, hands-on skill-building, scientific investigation, and patient care.
We must make our offices and hospitals trauma-informed; connect patients with resources to meet basic needs and with home-visiting and parent education programs; educate patients about the impact of protective and adverse factors on health; provide and practice self-regulation training in our offices or by referral; and advocate for equity.
Using these strategies, family physicians will play a crucial role in the prevention, mitigation, and treatment of the root cause of disease and society’s deepest individual and collective suffering.
CORRESPONDENCE
Audrey Stillerman, MD, ABFM, ABIHM, ABOIM, Office of Community Engagement and Neighborhood Health Partnerships, 808 South Wolcott Street, Room 809, Chicago, IL 60612; [email protected].
ACKNOWLEDGMENT
Patricia Rush, MD, MBA, and Adrienne Williams, PhD, reviewed the manuscript of this article.
1. Felitti V, Anda R. The relationship of adverse childhood experiences to adult medical disease, psychiatric disorders and sexual behavior: implications for healthcare. In: Lanius RA, Vermetten E, Pain C, eds. The Impact of Early Life Trauma on Health and Disease: The Hidden Epidemic. Cambridge, UK: Cambridge University Press; 2011:77-87.
2. Wade R Jr, Shea JA, Rubin D, et al. Adverse childhood experiences of low-income urban youth. Pediatrics. 2014;134:e13-e20.
3. Centers for Disease Control and Prevention. Child abuse and neglect prevention. April 10, 2018. www.cdc.gov/violenceprevention/childabuseandneglect/index.html. Accessed September 20, 2018.
4. American Academy of Family Physicians. Definition of family medicine. www.aafp.org/about/policies/all/family-medicine-definition.html. Accessed March 5, 2018.
5. Martin JC, Avant RF, Bowman MA, et al; The Future of Family Medicine Project Leadership Committee. The Future of Family Medicine: a collaborative project of the family medicine community. Ann Fam Med. 2004;2 Suppl 1:S3-S32.
6. Child & Adolescent Health Measurement Initiative (CAHMI). A national and across-state profile on Adverse Childhood Experiences among U.S. children and possibilities to heal and thrive. Issue Brief. October 2017. www.cahmi.org/wp-content/uploads/2018/05/aces_brief_final.pdf. Accessed September 20, 2018.
7. World Health Organization. Adverse Childhood Experiences International Questionnaire (ACE-IQ). www.who.int/violence_injury_prevention/violence/activities/adverse_childhood_experiences/en/. Accessed September 20, 2018.
8. Roos LE, Mota N, Afifi TO, et al. Relationship between adverse childhood experiences and homelessness and the impact of axis I and II disorders. Am J Public Health. 2013;103(Suppl 2):S275-S281.
9. Baglivio MT. Wolff KT. Piquero AR, et al. The relationship between adverse childhood experiences (ACE) and juvenile offending trajectories in a juvenile offender sample. J Crim Justice. 2015;43:229-241.
10. Dube SR. Felitti VF. Dong M, et al. Childhood abuse, neglect, and household dysfunction and the risk of illicit drug use: the adverse childhood experiences study. Pediatrics. 2003;111:564-572.
11. Maunder RG, Peladeau N, Savage D, et al. The prevalence of childhood adversity among healthcare workers and its relationship to adult life events, distress and impairment. Child Abuse Negl. 2010;34:114-123.
12. Anda RF, Felitti VJ, Bremner JD, et al. The enduring effects of abuse and related adverse experiences in childhood: a convergence of evidence from neurobiology and epidemiology. Eur Arch Psychiatry Clin Neurosci. 2006;256:174-186.
13. Braveman PA, Cubbin C, Egerter S, et al. Socioeconomic disparities in health in the United States: what the patterns tell us. Am J Public Health. 2010;100(Suppl 1):S186-S196.
14. Bowers ME, Yehuda R. Intergenerational transmission of stress in humans. Neuropsychopharmacology. 2016;41:232-244.
15. Perry BD. Memories of fears: How the brain stores and retrieves traumatic experiences. In: Goodwin J, Attias R, eds. Splintered Reflections: Images of the Body in Trauma. New York, NY: Basic Books; 1999:9-38.
16. Perry BD. Examining child maltreatment through a neurodevelopmental lens: clinical application of the Neurosequential Model of Therapeutics. J Loss Trauma. 2009;14:240-255.
17.
18. Adding layers to the ACEs pyramid—What do you think? Trauma and social location. ACES Connection, RYSE Center. 2015. www.acesconnection.com/blog/adding-layers-to-the-aces-pyramid-what-do-you-think. Accessed October 10, 2018.
19. Berens AE, Jensen SKG, Nelson CA 3rd. Biological embedding of childhood adversity: from physiological mechanisms to clinical implications. BMC Med. 2017;15:135.
20. Rostad WL, Basile KC, Clayton HB. Association among television and computer/video game use, victimization, and suicide risk among U.S. high school students. J Interpers Violence. 2018 Mar 1:886260518760020.
21. Coyne SM, Nelson DA, Graham-Kevan N, et al. Media depictions of physical and relational aggression: connections with aggression in young adults’ romantic relationships. Aggress Behav. 2011;37:56-62.
22. Centers for Disease Control and Prevention. Violence prevention: Child abuse and neglect: risk and protective factors. April 10, 2018. www.cdc.gov/violenceprevention/childabuseandneglect/riskprotectivefactors.html. Accessed October 10, 2018.
23. Miller TR. Projected outcomes of nurse-family partnership home visitation during 1996-2013, United States. Prev Sci. 2015;16:765-777.
24. Prinz RJ, Sanders MR, Shapiro CJ, et al. Population-based prevention of child maltreatment: the U.S. Triple P system population trial. Prev Sci. 2009;10:1-12.
25. Kraft C. Building capacity & support for two generation primary care. 2015 Midwest Regional Summit on Adverse Childhood Experiences. March 13, 2015. www.hmprg.org/assets/root/PDFs/2015/Summit%20Notes%20for%20Day%20Two.pdf. Accessed September 20, 2018.
26. Smith MV, Gotman N, Yonkers KA. Early childhood adversity and pregnancy outcomes. Matern Child Health J. 2016;20:790-798.
27. Leitch L. Action steps using ACEs and trauma-informed care: a resilience model. Health & Justice. 2017;5:1-10.
28. Bethell CD, Carle A, Hudziak J, et al. Methods to assess adverse childhood experiences of children and families: toward approaches to promote child well-being in policy and practice. Acad Pediatr. 2017;17:S51-S69.
29. Wade R Jr, Becker BD, Bevans KB, et al. Development and evaluation of a short adverse childhood experiences measure. Am J Prev Med. 2017;52:163-172.
30. Rush P. How learning about emotional trauma led me to a new understanding of chronic illness and health disparity. Becoming trauma-informed: Perspectives from public health, faith communities, education and medicine. Presented at 2016 Advocate Symposium, “Becoming a Trauma-Informed Children’s Hospital and Community: Building Foundations of Care, Collaboration and Practice.” Oaklawn, IL: Advocate Children’s Hospital; November 16, 2016.
31. Diderich HM, Fekkes M, Verkerk PH, et al. A new protocol for screening adults presenting with their own medical problems at the Emergency Department to identify children at high risk for maltreatment. Child Abuse Negl. 2013;37:1122-1131.
32. Fact Sheet: Key ingredients for trauma-informed care. Center for Health Care Strategies, Inc. August 2017. www.chcs.org/media/ATC-Key-Ingredients-Fact-Sheet_081417.pdf. Accessed September 22, 2018.
33. Adversity. In: Merriam-Webster Online Dictionary. Springfield, MA: Merriam-Webster, Inc. www.merriam-webster.com/dictionary/adversity. Accessed September 21, 2018.
34. Perry BD. Understanding traumatized and maltreated children: the core concepts. Child Trauma Academy Video Training Series, Video 4;2004:12. Child Trauma Academy (http://childtrauma.org/).
35. Perry BD. Understanding traumatized and maltreated children: the core concepts. Child Trauma Academy Video Training Series, Video 4;2004:19. Child Trauma Academy (http://childtrauma.org/).
36. EMDRIA’s definition of EMDR (eye movement desensitization and reprocessing). Austin, TX: EMDRIA: EMDR International Association. http://c.ymcdn.com/sites/www.emdria.org/resource/resmgr/imported/EMDRIA%20Definition%20of%20EMDR.pdf. Revised February 25 2012. Accessed September 21, 2018.
37. Types of trauma and violence: Historical trauma. Washington, DC: Substance Abuse and Mental Health Services Administration. www.samhsa.gov/trauma-violence/types. Accessed September 21, 2018.
38. Hammond DC. What is neurofeedback? An update. J Neurotherapy. 2011;15:305-336.
39. International Neuromodulation Society. Neuromodulation, or neuromodulatory effect. www.neuromodulation.com/neuromodulation-defined. November 9, 2017. Accessed September 21, 2018.
40. World Health Organization. Social determinants of health. www.who.int/social_determinants/sdh_definition/en/. Accessed September 21, 2018.
41. Davis R, Pinderhughes H, Williams M. Adverse community experiences and resilience: a framework for addressing and preventing community trauma. Oakland, CA: Prevention Institute; 2015:4-5. www.preventioninstitute.org/publications/adverse-community-experiences-and-resilience-framework-addressing-and-preventing. Accessed September 30, 2018.
42. SAMHSA-HRSA Center for Integrated Health Solutions. Trauma. Rockville, MD: Substance Abuse and Mental Health Services Administration and Health Resources and Services Administration, US Department of Health and Human Services. www.integration.samhsa.gov/clinical-practice/trauma. Accessed September 21, 2018.
43. Cohen JA, Mandarino AP. Trauma-focused cognitive behavioural therapy for children and parents. Child Adolesc Ment Health. 2008;13:158-162.
44. Trauma-informed approach and trauma-specific interventions: Trauma-informed approach. Washington, DC: National Center for Trauma Informed Care and Alternatives to Seclusion and Restraints; Substance Abuse and Mental Health Services Administration. www.samhsa.gov/nctic/trauma-interventions. Accessed September 21, 2018.
45. Perry BD. How the brain develops: the importance of early childhood. Child Trauma Academy Video Training Series, Video 1;2004:21. Child Trauma Academy (http://childtrauma.org/).
46. Huang LN, Sharp CS, Gunther T. It’s just good medicine: trauma-informed primary care. (SAMHSA-HRSA Center for Integrated Health Solutions webinar); August 6, 2013. www.integration.samhsa.gov/about-us/CIHS_TIC_Webinar_PDF.pdf. Accessed September 20, 2018.
47. CHCS: Center for Health Care Strategies, Inc. Fact sheet: Key ingredients for trauma-informed care. August 2017. www.chcs.org/media/ATC-Key-Ingredients-Fact-Sheet_081417.pdf. Accessed September 20, 2018.
48. National Council for Behavioral Health. Trauma-informed primary care: fostering resilience and recovery. www.thenationalcouncil.org/consulting-areas-of-expertise/trauma-informed-primary-care/. Accessed September 20, 2018.
49. Child Trauma Academy. The Neurosequential Model of Therapeutics as evidence-based practice. https://childtrauma.org/wp-content/uploads/2015/05/NMT_EvidenceBasedPract_5_2_15.pdf. Accessed September 30, 2018.
50. Bisson JI, Ehlers A, Matthews R, et al. Psychological treatments for chronic post-traumatic stress disorder. Systematic review and meta-analysis. Br J Psychiatry. 2007;190:97-104.
51. Metcalf O, Varker T, Forbes D, et al. Efficacy of fifteen emerging interventions for the treatment of posttraumatic stress disorder: a systematic review. J Trauma Stress. 2016;29:88-92.
52. van der Kolk BA, Hodgdon H, Gapen M, et al. A randomized controlled study of neurofeedback for chronic PTSD. 2016; PLoS One. 2016;11:e0166752.
53. Bryan M. A hospital offers frequent ER patients an out—free housing. “All Things Considered.” National Public Radio. June 29, 2016. www.npr.org/sections/health-shots/2016/06/29/482994000/a-hospital-offers-frequent-er-patients-an-out-free-housing. Acces-sed September 20, 2018.
1. Felitti V, Anda R. The relationship of adverse childhood experiences to adult medical disease, psychiatric disorders and sexual behavior: implications for healthcare. In: Lanius RA, Vermetten E, Pain C, eds. The Impact of Early Life Trauma on Health and Disease: The Hidden Epidemic. Cambridge, UK: Cambridge University Press; 2011:77-87.
2. Wade R Jr, Shea JA, Rubin D, et al. Adverse childhood experiences of low-income urban youth. Pediatrics. 2014;134:e13-e20.
3. Centers for Disease Control and Prevention. Child abuse and neglect prevention. April 10, 2018. www.cdc.gov/violenceprevention/childabuseandneglect/index.html. Accessed September 20, 2018.
4. American Academy of Family Physicians. Definition of family medicine. www.aafp.org/about/policies/all/family-medicine-definition.html. Accessed March 5, 2018.
5. Martin JC, Avant RF, Bowman MA, et al; The Future of Family Medicine Project Leadership Committee. The Future of Family Medicine: a collaborative project of the family medicine community. Ann Fam Med. 2004;2 Suppl 1:S3-S32.
6. Child & Adolescent Health Measurement Initiative (CAHMI). A national and across-state profile on Adverse Childhood Experiences among U.S. children and possibilities to heal and thrive. Issue Brief. October 2017. www.cahmi.org/wp-content/uploads/2018/05/aces_brief_final.pdf. Accessed September 20, 2018.
7. World Health Organization. Adverse Childhood Experiences International Questionnaire (ACE-IQ). www.who.int/violence_injury_prevention/violence/activities/adverse_childhood_experiences/en/. Accessed September 20, 2018.
8. Roos LE, Mota N, Afifi TO, et al. Relationship between adverse childhood experiences and homelessness and the impact of axis I and II disorders. Am J Public Health. 2013;103(Suppl 2):S275-S281.
9. Baglivio MT. Wolff KT. Piquero AR, et al. The relationship between adverse childhood experiences (ACE) and juvenile offending trajectories in a juvenile offender sample. J Crim Justice. 2015;43:229-241.
10. Dube SR. Felitti VF. Dong M, et al. Childhood abuse, neglect, and household dysfunction and the risk of illicit drug use: the adverse childhood experiences study. Pediatrics. 2003;111:564-572.
11. Maunder RG, Peladeau N, Savage D, et al. The prevalence of childhood adversity among healthcare workers and its relationship to adult life events, distress and impairment. Child Abuse Negl. 2010;34:114-123.
12. Anda RF, Felitti VJ, Bremner JD, et al. The enduring effects of abuse and related adverse experiences in childhood: a convergence of evidence from neurobiology and epidemiology. Eur Arch Psychiatry Clin Neurosci. 2006;256:174-186.
13. Braveman PA, Cubbin C, Egerter S, et al. Socioeconomic disparities in health in the United States: what the patterns tell us. Am J Public Health. 2010;100(Suppl 1):S186-S196.
14. Bowers ME, Yehuda R. Intergenerational transmission of stress in humans. Neuropsychopharmacology. 2016;41:232-244.
15. Perry BD. Memories of fears: How the brain stores and retrieves traumatic experiences. In: Goodwin J, Attias R, eds. Splintered Reflections: Images of the Body in Trauma. New York, NY: Basic Books; 1999:9-38.
16. Perry BD. Examining child maltreatment through a neurodevelopmental lens: clinical application of the Neurosequential Model of Therapeutics. J Loss Trauma. 2009;14:240-255.
17.
18. Adding layers to the ACEs pyramid—What do you think? Trauma and social location. ACES Connection, RYSE Center. 2015. www.acesconnection.com/blog/adding-layers-to-the-aces-pyramid-what-do-you-think. Accessed October 10, 2018.
19. Berens AE, Jensen SKG, Nelson CA 3rd. Biological embedding of childhood adversity: from physiological mechanisms to clinical implications. BMC Med. 2017;15:135.
20. Rostad WL, Basile KC, Clayton HB. Association among television and computer/video game use, victimization, and suicide risk among U.S. high school students. J Interpers Violence. 2018 Mar 1:886260518760020.
21. Coyne SM, Nelson DA, Graham-Kevan N, et al. Media depictions of physical and relational aggression: connections with aggression in young adults’ romantic relationships. Aggress Behav. 2011;37:56-62.
22. Centers for Disease Control and Prevention. Violence prevention: Child abuse and neglect: risk and protective factors. April 10, 2018. www.cdc.gov/violenceprevention/childabuseandneglect/riskprotectivefactors.html. Accessed October 10, 2018.
23. Miller TR. Projected outcomes of nurse-family partnership home visitation during 1996-2013, United States. Prev Sci. 2015;16:765-777.
24. Prinz RJ, Sanders MR, Shapiro CJ, et al. Population-based prevention of child maltreatment: the U.S. Triple P system population trial. Prev Sci. 2009;10:1-12.
25. Kraft C. Building capacity & support for two generation primary care. 2015 Midwest Regional Summit on Adverse Childhood Experiences. March 13, 2015. www.hmprg.org/assets/root/PDFs/2015/Summit%20Notes%20for%20Day%20Two.pdf. Accessed September 20, 2018.
26. Smith MV, Gotman N, Yonkers KA. Early childhood adversity and pregnancy outcomes. Matern Child Health J. 2016;20:790-798.
27. Leitch L. Action steps using ACEs and trauma-informed care: a resilience model. Health & Justice. 2017;5:1-10.
28. Bethell CD, Carle A, Hudziak J, et al. Methods to assess adverse childhood experiences of children and families: toward approaches to promote child well-being in policy and practice. Acad Pediatr. 2017;17:S51-S69.
29. Wade R Jr, Becker BD, Bevans KB, et al. Development and evaluation of a short adverse childhood experiences measure. Am J Prev Med. 2017;52:163-172.
30. Rush P. How learning about emotional trauma led me to a new understanding of chronic illness and health disparity. Becoming trauma-informed: Perspectives from public health, faith communities, education and medicine. Presented at 2016 Advocate Symposium, “Becoming a Trauma-Informed Children’s Hospital and Community: Building Foundations of Care, Collaboration and Practice.” Oaklawn, IL: Advocate Children’s Hospital; November 16, 2016.
31. Diderich HM, Fekkes M, Verkerk PH, et al. A new protocol for screening adults presenting with their own medical problems at the Emergency Department to identify children at high risk for maltreatment. Child Abuse Negl. 2013;37:1122-1131.
32. Fact Sheet: Key ingredients for trauma-informed care. Center for Health Care Strategies, Inc. August 2017. www.chcs.org/media/ATC-Key-Ingredients-Fact-Sheet_081417.pdf. Accessed September 22, 2018.
33. Adversity. In: Merriam-Webster Online Dictionary. Springfield, MA: Merriam-Webster, Inc. www.merriam-webster.com/dictionary/adversity. Accessed September 21, 2018.
34. Perry BD. Understanding traumatized and maltreated children: the core concepts. Child Trauma Academy Video Training Series, Video 4;2004:12. Child Trauma Academy (http://childtrauma.org/).
35. Perry BD. Understanding traumatized and maltreated children: the core concepts. Child Trauma Academy Video Training Series, Video 4;2004:19. Child Trauma Academy (http://childtrauma.org/).
36. EMDRIA’s definition of EMDR (eye movement desensitization and reprocessing). Austin, TX: EMDRIA: EMDR International Association. http://c.ymcdn.com/sites/www.emdria.org/resource/resmgr/imported/EMDRIA%20Definition%20of%20EMDR.pdf. Revised February 25 2012. Accessed September 21, 2018.
37. Types of trauma and violence: Historical trauma. Washington, DC: Substance Abuse and Mental Health Services Administration. www.samhsa.gov/trauma-violence/types. Accessed September 21, 2018.
38. Hammond DC. What is neurofeedback? An update. J Neurotherapy. 2011;15:305-336.
39. International Neuromodulation Society. Neuromodulation, or neuromodulatory effect. www.neuromodulation.com/neuromodulation-defined. November 9, 2017. Accessed September 21, 2018.
40. World Health Organization. Social determinants of health. www.who.int/social_determinants/sdh_definition/en/. Accessed September 21, 2018.
41. Davis R, Pinderhughes H, Williams M. Adverse community experiences and resilience: a framework for addressing and preventing community trauma. Oakland, CA: Prevention Institute; 2015:4-5. www.preventioninstitute.org/publications/adverse-community-experiences-and-resilience-framework-addressing-and-preventing. Accessed September 30, 2018.
42. SAMHSA-HRSA Center for Integrated Health Solutions. Trauma. Rockville, MD: Substance Abuse and Mental Health Services Administration and Health Resources and Services Administration, US Department of Health and Human Services. www.integration.samhsa.gov/clinical-practice/trauma. Accessed September 21, 2018.
43. Cohen JA, Mandarino AP. Trauma-focused cognitive behavioural therapy for children and parents. Child Adolesc Ment Health. 2008;13:158-162.
44. Trauma-informed approach and trauma-specific interventions: Trauma-informed approach. Washington, DC: National Center for Trauma Informed Care and Alternatives to Seclusion and Restraints; Substance Abuse and Mental Health Services Administration. www.samhsa.gov/nctic/trauma-interventions. Accessed September 21, 2018.
45. Perry BD. How the brain develops: the importance of early childhood. Child Trauma Academy Video Training Series, Video 1;2004:21. Child Trauma Academy (http://childtrauma.org/).
46. Huang LN, Sharp CS, Gunther T. It’s just good medicine: trauma-informed primary care. (SAMHSA-HRSA Center for Integrated Health Solutions webinar); August 6, 2013. www.integration.samhsa.gov/about-us/CIHS_TIC_Webinar_PDF.pdf. Accessed September 20, 2018.
47. CHCS: Center for Health Care Strategies, Inc. Fact sheet: Key ingredients for trauma-informed care. August 2017. www.chcs.org/media/ATC-Key-Ingredients-Fact-Sheet_081417.pdf. Accessed September 20, 2018.
48. National Council for Behavioral Health. Trauma-informed primary care: fostering resilience and recovery. www.thenationalcouncil.org/consulting-areas-of-expertise/trauma-informed-primary-care/. Accessed September 20, 2018.
49. Child Trauma Academy. The Neurosequential Model of Therapeutics as evidence-based practice. https://childtrauma.org/wp-content/uploads/2015/05/NMT_EvidenceBasedPract_5_2_15.pdf. Accessed September 30, 2018.
50. Bisson JI, Ehlers A, Matthews R, et al. Psychological treatments for chronic post-traumatic stress disorder. Systematic review and meta-analysis. Br J Psychiatry. 2007;190:97-104.
51. Metcalf O, Varker T, Forbes D, et al. Efficacy of fifteen emerging interventions for the treatment of posttraumatic stress disorder: a systematic review. J Trauma Stress. 2016;29:88-92.
52. van der Kolk BA, Hodgdon H, Gapen M, et al. A randomized controlled study of neurofeedback for chronic PTSD. 2016; PLoS One. 2016;11:e0166752.
53. Bryan M. A hospital offers frequent ER patients an out—free housing. “All Things Considered.” National Public Radio. June 29, 2016. www.npr.org/sections/health-shots/2016/06/29/482994000/a-hospital-offers-frequent-er-patients-an-out-free-housing. Acces-sed September 20, 2018.
PRACTICE RECOMMENDATIONS
› Refer eligible patients to an evidence-based perinatal home-visiting program and all parents to an evidence-based parenting program to prevent childhood adversity. A
› Consider screening adult patients and parents for their own history (and their children’s history) of childhood adversity. B
› Recommend trauma-focused cognitive behavioral therapy and eye-movement desensitization and reprocessing as first-line treatments for adversity and trauma. A
› Consider prescribing yoga, neurofeedback, and other neuromodulatory modalities to treat the consequences of childhood adversity and trauma. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

Protein binding changes and drug interactions: What do we know?
Mr. S, age 47, weighs 209 lb and has a history of seizure disorder, bipolar disorder not otherwise specified, hypertension, and type 2 diabetes mellitus. He presents to the emergency department after not taking his medications for 2 days while on vacation. He has increased energy, decreased sleep, and pressured speech, and insists on walking for up to 10 hours per day “in preparation for a marathon,” even though he has a 4-cm foot ulcer. His family reports that he had been compliant with his medications until the present incident.
Mr. S has no known drug allergies. His medications include oral divalproex sodium delayed release (valproic acid [VPA]), 1,000 mg twice a day, oral lisinopril, 20 mg every morning, and insulin glargine, 22 units subcutaneously every evening.
A complete blood count, basic metabolic panel, creatine kinase level, VPA level, and urine drug screen are ordered. Relevant results include a serum creatinine level of 1.4 mg/dL (normal range: 0.6 to 1.2 mg/dL), a glucose serum level of 188 mg/dL (normal range: 70 to 100 mg/dL), and a VPA level of 23 mcg/mL (therapeutic range: 50 to 125 mcg/mL). A liver function panel is within normal limits: albumin level of 3.9 g/dL, aspartate aminotransferase level of 18 IU/L, and alanine aminotransferase level of 14 IU/L. In light of Mr. S’s seizure history, neurology is consulted and the decision is made to continue treating him with VPA because he has been seizure-free for 4.5 years and this medication has also helped with his bipolar disorder.
Mr. S is admitted to the hospital and his home medications are resumed at the current doses. On hospital Day 3, Mr. S’s VPA level is 62 mcg/mL, his obsession with a marathon has remitted, and his sleep pattern has normalized. Infectious disease and podiatry services are consulted for his diabetic foot infection, which has ulcerated down to the bone. IV ertapenem, 1,000 mg/d, is initiated with plans for debridement the following week. Two days later, Mr. S has a witnessed seizure; his VPA level is 9 mcg/mL.

A common question asked of pharmacists is, “Will protein binding changes affect drug dosages?” In this article, I describe how protein binding changes may occur, and the complexity of the dynamic. Being highly bound to a protein typically does not mean all medications will interact, but some interactions can be important. This article does not cover medications that bind to hormones.
Why is protein binding important? When a medication is bound to plasma protein, it is not free to act. There can be a delay in therapeutic effect (because no drug is available to react), delayed elimination, or possibly displacement of another protein-bound medication. Additionally, medications tend not to cross the blood-brain barrier or be eliminated when bound. For example, if a drug is 99% bound (leaving 1% free) and displacement now leaves 2% of the drug free, this event has doubled the amount of free drug. As the unbound medication is eliminated, the drug that is bound to the protein can act as a reservoir. A dynamic relationship exists between bound drug, unbound drug, and rate of elimination.
Which proteins do drugs commonly bind to? The proteins often associated with binding include albumin, alpha-1-acid glycoprotein (AAG), and lipoproteins. Albumin comprises 60% of total plasma protein in the plasma. Lipoproteins include very high-density lipoprotein (VHDL), high-density lipoprotein (HDL), very low-density lipoprotein (VLDL), and low-density lipoprotein (LDL).1 Medications that bind to lipoproteins include cyclosporine, tacrolimus, and propofol.2
Continued to: What common disease states can cause hypoalbuminemia?
What common disease states can cause hypoalbuminemia? Many disease states can result in low albumin levels. The most common ones are malnutrition, malignancies, stress, injury, burns, pregnancy, and diabetes.3 When there is less albumin to bind to, free drug levels may be increased.
Can AAG levels change with disease states as well? Because AAG accounts for a lower percentage of total plasma protein than albumin, there may be less clinical concern regarding AAG. AAG levels usually do not drop, but instead can become elevated during times of trauma, inflammation, and acute myocardial infarction. This could result in increased binding of the free drug.4Which medications bind to red blood cells (RBCs)? There are several locations for drugs to bind to RBCs, including to hemoglobin and the plasma membrane. Medications that commonly bind to RBCs include barbiturates, chlorpromazine, imipramine, and phenytoin.5
What are common highly-bound medications? The Table1 provides examples of medications that are >90% protein-bound. However, this information may be misleading because many medications are highly bound. Zhang et al1 compiled binding data for 222 drugs, half of which bind 90% to 100%. However, the literature does not indicate that they all have clinically significant interactions. Benet and Hoener6 discuss how factors other than protein binding affect potential drug interactions, and the complexity of the body’s ability to compensate for increased free drug. Medication characteristics that may contribute to producing a significant interaction include, but are not limited to:
- free vs protein-bound drug in the plasma or tissue
- volume of distribution
- organs affected
- hepatic bioavailability
- drug clearance.
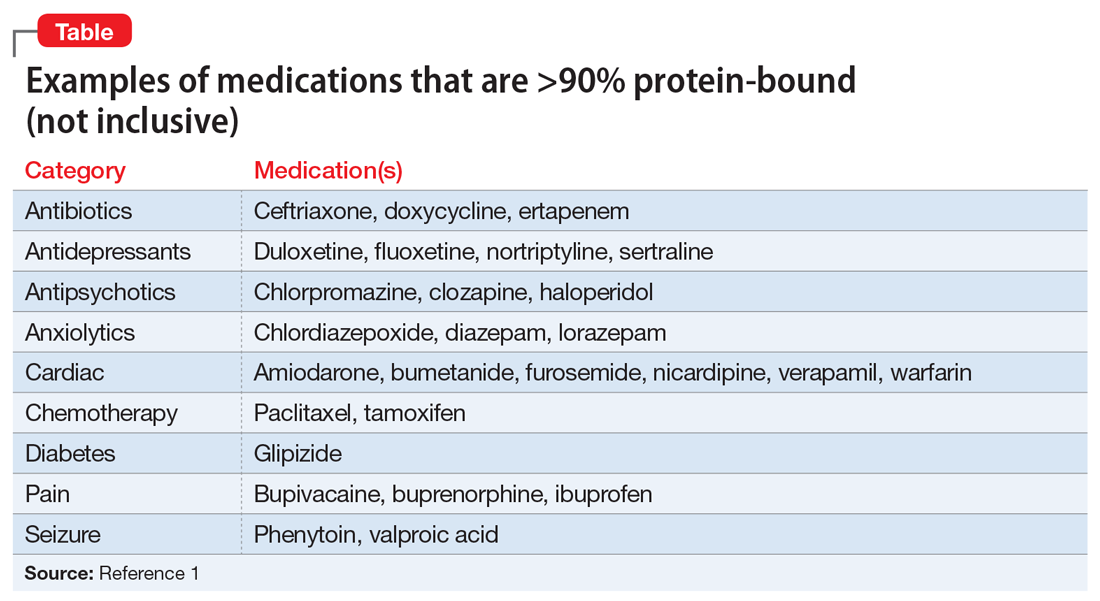
For example, VPA is 93% protein-bound and phenytoin is 91% protein-bound.1 However, this interaction is affected by more than just protein binding. VPA not only displaces the protein-bound phenytoin, but also inhibits its metabolism, which together result in increased free phenytoin levels.
Continued to: Another area of concern is a critically ill patient...
Another area of concern is a critically ill patient who has a change in his or her pH. Medications that are highly bound and have high clearance rates may be affected. This is of particular concern when prescribing antibiotics that are time-dependent, such as beta-lactams.3
What happened to Mr. S? Mr. S likely experienced a drug–drug interaction that resulted in a subtherapeutic VPA level and subsequent seizure. Case reports have shown evidence that the carbapenem class of antibiotics, which includes ertapenem, interacts with VPA.7 Proposed mechanisms include a lowering of VPA serum levels due to a redistribution of the VPA onto the RBCs due to carbapenem. Other theories include the possibility that carbapenems may limit oral VPA absorption, decrease VPA enterohepatic recirculation, and increase VPA metabolism.7 Using VPA and ertapenem together is discouraged because seizures have been reported among patients receiving this combination. If it is medically necessary to administer VPA and ertapenem, closely monitor VPA levels. In Mr. S’s case, another broad-spectrum antibiotic, such as piperacillin-tazobactam, could have been used, for his diabetic foot infection.
While many medications may have high protein binding, there are few clinically important known interactions. However, our understanding of the relationship between protein binding and drug interactions may improve with additional research.
CASE CONTINUED
Under neurology’s care, lacosamide is added for treatment of Mr. S’s seizures. No more seizures are noted during the remainder of his hospitalization. Infectious disease services change his antibiotic to piperacillin-tazobactam. Mr. S continues to progress well and is discharged to a rehabilitation center 2 days later.
Related Resource
- DrugBank. www.drugbank.ca. Canadian Institutes of Health Research.
Drug Brand Names
Amiodarone • Cordarone, Pacerone
Bumetanide • Bumex
Bupivacaine • Marcaine, Sensorcaine
Buprenorphine • Belbuca, Subutex
Ceftriaxone • Rocephin
Chlordiazepoxide • Librium
Chlorpromazine • Thorazine
Clozapine • Clozaril
Cyclosporine • Gengraf, Neoral
Diazepam • Valium
Doxycycline • Acticlate, Doryx
Duloxetine • Cymbalta
Ertapenem • Invanz
Fluoxetine • Prozac, Sarafem
Furosemide • Lasix
Glargine (Insulin) • Lantus, Toujeo
Glipizide • Glucotrol
Haloperidol • Haldol
Ibuprofen • Advil, Motrin
Imipramine • Tofranil
Lacosamide • Vimpat
Lisinopril • Prinivil, Zestril
Lorazepam • Ativan
Nicardipine • Cardene
Nortriptyline • Pamelor
Paclitaxel • Abraxane, Taxol
Phenytoin • Dilantin, Phenytek
Piperacillin-tazobactam • Zosyn
Propofol • Diprivan
Sertraline • Zoloft
Tacrolimus • Prograf
Tamoxifen • Soltamox
Valproic acid • Depakene, Depakote
Verapamil • Calan, Verelan
Warfarin • Coumadin, Jantoven
1. Zhang F, Xue J, Shao J, et al. Compilation of 222 drugs’ plasma protein binding data and guidance for study designs. Drug Discov Today. 2012;17(9-10):475-485.
2. Mehvar R. Role of protein binding in pharmacokinetics. Am J Pharm Edu. 2005;69(5): Article 103;1-8.
3. Roberts JA, Pea F, Lipman J. The clinical relevance of plasma protein binding changes. Clin Pharmacokinet. 2013;52(1):1-8.
4. Schmidt S, Gonzalez D, Derendork H. Significance of protein binding in pharmacokinetics and pharmacodynamics. J Pharm Sci. 2010;99(3):1107-1122.
5. Hinderling P. Red blood cells: a neglected compartment in pharmacokinetics and pharmacodynamics. Pharmacol Rev. 1997;49(3):279-295.
6. Benet LZ, Hoener B. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther. 2002;71(3):115-121.
7. Park MK, Lim KS, Kim T, et al. Reduced valproic acid serum concentrations due to drug interactions with carbapenem antibiotics: overview of 6 cases. Ther Drug Monit. 2012;34(5):599-603.
Mr. S, age 47, weighs 209 lb and has a history of seizure disorder, bipolar disorder not otherwise specified, hypertension, and type 2 diabetes mellitus. He presents to the emergency department after not taking his medications for 2 days while on vacation. He has increased energy, decreased sleep, and pressured speech, and insists on walking for up to 10 hours per day “in preparation for a marathon,” even though he has a 4-cm foot ulcer. His family reports that he had been compliant with his medications until the present incident.
Mr. S has no known drug allergies. His medications include oral divalproex sodium delayed release (valproic acid [VPA]), 1,000 mg twice a day, oral lisinopril, 20 mg every morning, and insulin glargine, 22 units subcutaneously every evening.
A complete blood count, basic metabolic panel, creatine kinase level, VPA level, and urine drug screen are ordered. Relevant results include a serum creatinine level of 1.4 mg/dL (normal range: 0.6 to 1.2 mg/dL), a glucose serum level of 188 mg/dL (normal range: 70 to 100 mg/dL), and a VPA level of 23 mcg/mL (therapeutic range: 50 to 125 mcg/mL). A liver function panel is within normal limits: albumin level of 3.9 g/dL, aspartate aminotransferase level of 18 IU/L, and alanine aminotransferase level of 14 IU/L. In light of Mr. S’s seizure history, neurology is consulted and the decision is made to continue treating him with VPA because he has been seizure-free for 4.5 years and this medication has also helped with his bipolar disorder.
Mr. S is admitted to the hospital and his home medications are resumed at the current doses. On hospital Day 3, Mr. S’s VPA level is 62 mcg/mL, his obsession with a marathon has remitted, and his sleep pattern has normalized. Infectious disease and podiatry services are consulted for his diabetic foot infection, which has ulcerated down to the bone. IV ertapenem, 1,000 mg/d, is initiated with plans for debridement the following week. Two days later, Mr. S has a witnessed seizure; his VPA level is 9 mcg/mL.
A common question asked of pharmacists is, “Will protein binding changes affect drug dosages?” In this article, I describe how protein binding changes may occur, and the complexity of the dynamic. Being highly bound to a protein typically does not mean all medications will interact, but some interactions can be important. This article does not cover medications that bind to hormones.
Why is protein binding important? When a medication is bound to plasma protein, it is not free to act. There can be a delay in therapeutic effect (because no drug is available to react), delayed elimination, or possibly displacement of another protein-bound medication. Additionally, medications tend not to cross the blood-brain barrier or be eliminated when bound. For example, if a drug is 99% bound (leaving 1% free) and displacement now leaves 2% of the drug free, this event has doubled the amount of free drug. As the unbound medication is eliminated, the drug that is bound to the protein can act as a reservoir. A dynamic relationship exists between bound drug, unbound drug, and rate of elimination.
Which proteins do drugs commonly bind to? The proteins often associated with binding include albumin, alpha-1-acid glycoprotein (AAG), and lipoproteins. Albumin comprises 60% of total plasma protein in the plasma. Lipoproteins include very high-density lipoprotein (VHDL), high-density lipoprotein (HDL), very low-density lipoprotein (VLDL), and low-density lipoprotein (LDL).1 Medications that bind to lipoproteins include cyclosporine, tacrolimus, and propofol.2
Continued to: What common disease states can cause hypoalbuminemia?
What common disease states can cause hypoalbuminemia? Many disease states can result in low albumin levels. The most common ones are malnutrition, malignancies, stress, injury, burns, pregnancy, and diabetes.3 When there is less albumin to bind to, free drug levels may be increased.
Can AAG levels change with disease states as well? Because AAG accounts for a lower percentage of total plasma protein than albumin, there may be less clinical concern regarding AAG. AAG levels usually do not drop, but instead can become elevated during times of trauma, inflammation, and acute myocardial infarction. This could result in increased binding of the free drug.4Which medications bind to red blood cells (RBCs)? There are several locations for drugs to bind to RBCs, including to hemoglobin and the plasma membrane. Medications that commonly bind to RBCs include barbiturates, chlorpromazine, imipramine, and phenytoin.5
What are common highly-bound medications? The Table1 provides examples of medications that are >90% protein-bound. However, this information may be misleading because many medications are highly bound. Zhang et al1 compiled binding data for 222 drugs, half of which bind 90% to 100%. However, the literature does not indicate that they all have clinically significant interactions. Benet and Hoener6 discuss how factors other than protein binding affect potential drug interactions, and the complexity of the body’s ability to compensate for increased free drug. Medication characteristics that may contribute to producing a significant interaction include, but are not limited to:
- free vs protein-bound drug in the plasma or tissue
- volume of distribution
- organs affected
- hepatic bioavailability
- drug clearance.
For example, VPA is 93% protein-bound and phenytoin is 91% protein-bound.1 However, this interaction is affected by more than just protein binding. VPA not only displaces the protein-bound phenytoin, but also inhibits its metabolism, which together result in increased free phenytoin levels.
Continued to: Another area of concern is a critically ill patient...
Another area of concern is a critically ill patient who has a change in his or her pH. Medications that are highly bound and have high clearance rates may be affected. This is of particular concern when prescribing antibiotics that are time-dependent, such as beta-lactams.3
What happened to Mr. S? Mr. S likely experienced a drug–drug interaction that resulted in a subtherapeutic VPA level and subsequent seizure. Case reports have shown evidence that the carbapenem class of antibiotics, which includes ertapenem, interacts with VPA.7 Proposed mechanisms include a lowering of VPA serum levels due to a redistribution of the VPA onto the RBCs due to carbapenem. Other theories include the possibility that carbapenems may limit oral VPA absorption, decrease VPA enterohepatic recirculation, and increase VPA metabolism.7 Using VPA and ertapenem together is discouraged because seizures have been reported among patients receiving this combination. If it is medically necessary to administer VPA and ertapenem, closely monitor VPA levels. In Mr. S’s case, another broad-spectrum antibiotic, such as piperacillin-tazobactam, could have been used, for his diabetic foot infection.
While many medications may have high protein binding, there are few clinically important known interactions. However, our understanding of the relationship between protein binding and drug interactions may improve with additional research.
CASE CONTINUED
Under neurology’s care, lacosamide is added for treatment of Mr. S’s seizures. No more seizures are noted during the remainder of his hospitalization. Infectious disease services change his antibiotic to piperacillin-tazobactam. Mr. S continues to progress well and is discharged to a rehabilitation center 2 days later.
Related Resource
- DrugBank. www.drugbank.ca. Canadian Institutes of Health Research.
Drug Brand Names
Amiodarone • Cordarone, Pacerone
Bumetanide • Bumex
Bupivacaine • Marcaine, Sensorcaine
Buprenorphine • Belbuca, Subutex
Ceftriaxone • Rocephin
Chlordiazepoxide • Librium
Chlorpromazine • Thorazine
Clozapine • Clozaril
Cyclosporine • Gengraf, Neoral
Diazepam • Valium
Doxycycline • Acticlate, Doryx
Duloxetine • Cymbalta
Ertapenem • Invanz
Fluoxetine • Prozac, Sarafem
Furosemide • Lasix
Glargine (Insulin) • Lantus, Toujeo
Glipizide • Glucotrol
Haloperidol • Haldol
Ibuprofen • Advil, Motrin
Imipramine • Tofranil
Lacosamide • Vimpat
Lisinopril • Prinivil, Zestril
Lorazepam • Ativan
Nicardipine • Cardene
Nortriptyline • Pamelor
Paclitaxel • Abraxane, Taxol
Phenytoin • Dilantin, Phenytek
Piperacillin-tazobactam • Zosyn
Propofol • Diprivan
Sertraline • Zoloft
Tacrolimus • Prograf
Tamoxifen • Soltamox
Valproic acid • Depakene, Depakote
Verapamil • Calan, Verelan
Warfarin • Coumadin, Jantoven
Mr. S, age 47, weighs 209 lb and has a history of seizure disorder, bipolar disorder not otherwise specified, hypertension, and type 2 diabetes mellitus. He presents to the emergency department after not taking his medications for 2 days while on vacation. He has increased energy, decreased sleep, and pressured speech, and insists on walking for up to 10 hours per day “in preparation for a marathon,” even though he has a 4-cm foot ulcer. His family reports that he had been compliant with his medications until the present incident.
Mr. S has no known drug allergies. His medications include oral divalproex sodium delayed release (valproic acid [VPA]), 1,000 mg twice a day, oral lisinopril, 20 mg every morning, and insulin glargine, 22 units subcutaneously every evening.
A complete blood count, basic metabolic panel, creatine kinase level, VPA level, and urine drug screen are ordered. Relevant results include a serum creatinine level of 1.4 mg/dL (normal range: 0.6 to 1.2 mg/dL), a glucose serum level of 188 mg/dL (normal range: 70 to 100 mg/dL), and a VPA level of 23 mcg/mL (therapeutic range: 50 to 125 mcg/mL). A liver function panel is within normal limits: albumin level of 3.9 g/dL, aspartate aminotransferase level of 18 IU/L, and alanine aminotransferase level of 14 IU/L. In light of Mr. S’s seizure history, neurology is consulted and the decision is made to continue treating him with VPA because he has been seizure-free for 4.5 years and this medication has also helped with his bipolar disorder.
Mr. S is admitted to the hospital and his home medications are resumed at the current doses. On hospital Day 3, Mr. S’s VPA level is 62 mcg/mL, his obsession with a marathon has remitted, and his sleep pattern has normalized. Infectious disease and podiatry services are consulted for his diabetic foot infection, which has ulcerated down to the bone. IV ertapenem, 1,000 mg/d, is initiated with plans for debridement the following week. Two days later, Mr. S has a witnessed seizure; his VPA level is 9 mcg/mL.
A common question asked of pharmacists is, “Will protein binding changes affect drug dosages?” In this article, I describe how protein binding changes may occur, and the complexity of the dynamic. Being highly bound to a protein typically does not mean all medications will interact, but some interactions can be important. This article does not cover medications that bind to hormones.
Why is protein binding important? When a medication is bound to plasma protein, it is not free to act. There can be a delay in therapeutic effect (because no drug is available to react), delayed elimination, or possibly displacement of another protein-bound medication. Additionally, medications tend not to cross the blood-brain barrier or be eliminated when bound. For example, if a drug is 99% bound (leaving 1% free) and displacement now leaves 2% of the drug free, this event has doubled the amount of free drug. As the unbound medication is eliminated, the drug that is bound to the protein can act as a reservoir. A dynamic relationship exists between bound drug, unbound drug, and rate of elimination.
Which proteins do drugs commonly bind to? The proteins often associated with binding include albumin, alpha-1-acid glycoprotein (AAG), and lipoproteins. Albumin comprises 60% of total plasma protein in the plasma. Lipoproteins include very high-density lipoprotein (VHDL), high-density lipoprotein (HDL), very low-density lipoprotein (VLDL), and low-density lipoprotein (LDL).1 Medications that bind to lipoproteins include cyclosporine, tacrolimus, and propofol.2
Continued to: What common disease states can cause hypoalbuminemia?
What common disease states can cause hypoalbuminemia? Many disease states can result in low albumin levels. The most common ones are malnutrition, malignancies, stress, injury, burns, pregnancy, and diabetes.3 When there is less albumin to bind to, free drug levels may be increased.
Can AAG levels change with disease states as well? Because AAG accounts for a lower percentage of total plasma protein than albumin, there may be less clinical concern regarding AAG. AAG levels usually do not drop, but instead can become elevated during times of trauma, inflammation, and acute myocardial infarction. This could result in increased binding of the free drug.4Which medications bind to red blood cells (RBCs)? There are several locations for drugs to bind to RBCs, including to hemoglobin and the plasma membrane. Medications that commonly bind to RBCs include barbiturates, chlorpromazine, imipramine, and phenytoin.5
What are common highly-bound medications? The Table1 provides examples of medications that are >90% protein-bound. However, this information may be misleading because many medications are highly bound. Zhang et al1 compiled binding data for 222 drugs, half of which bind 90% to 100%. However, the literature does not indicate that they all have clinically significant interactions. Benet and Hoener6 discuss how factors other than protein binding affect potential drug interactions, and the complexity of the body’s ability to compensate for increased free drug. Medication characteristics that may contribute to producing a significant interaction include, but are not limited to:
- free vs protein-bound drug in the plasma or tissue
- volume of distribution
- organs affected
- hepatic bioavailability
- drug clearance.
For example, VPA is 93% protein-bound and phenytoin is 91% protein-bound.1 However, this interaction is affected by more than just protein binding. VPA not only displaces the protein-bound phenytoin, but also inhibits its metabolism, which together result in increased free phenytoin levels.
Continued to: Another area of concern is a critically ill patient...
Another area of concern is a critically ill patient who has a change in his or her pH. Medications that are highly bound and have high clearance rates may be affected. This is of particular concern when prescribing antibiotics that are time-dependent, such as beta-lactams.3
What happened to Mr. S? Mr. S likely experienced a drug–drug interaction that resulted in a subtherapeutic VPA level and subsequent seizure. Case reports have shown evidence that the carbapenem class of antibiotics, which includes ertapenem, interacts with VPA.7 Proposed mechanisms include a lowering of VPA serum levels due to a redistribution of the VPA onto the RBCs due to carbapenem. Other theories include the possibility that carbapenems may limit oral VPA absorption, decrease VPA enterohepatic recirculation, and increase VPA metabolism.7 Using VPA and ertapenem together is discouraged because seizures have been reported among patients receiving this combination. If it is medically necessary to administer VPA and ertapenem, closely monitor VPA levels. In Mr. S’s case, another broad-spectrum antibiotic, such as piperacillin-tazobactam, could have been used, for his diabetic foot infection.
While many medications may have high protein binding, there are few clinically important known interactions. However, our understanding of the relationship between protein binding and drug interactions may improve with additional research.
CASE CONTINUED
Under neurology’s care, lacosamide is added for treatment of Mr. S’s seizures. No more seizures are noted during the remainder of his hospitalization. Infectious disease services change his antibiotic to piperacillin-tazobactam. Mr. S continues to progress well and is discharged to a rehabilitation center 2 days later.
Related Resource
- DrugBank. www.drugbank.ca. Canadian Institutes of Health Research.
Drug Brand Names
Amiodarone • Cordarone, Pacerone
Bumetanide • Bumex
Bupivacaine • Marcaine, Sensorcaine
Buprenorphine • Belbuca, Subutex
Ceftriaxone • Rocephin
Chlordiazepoxide • Librium
Chlorpromazine • Thorazine
Clozapine • Clozaril
Cyclosporine • Gengraf, Neoral
Diazepam • Valium
Doxycycline • Acticlate, Doryx
Duloxetine • Cymbalta
Ertapenem • Invanz
Fluoxetine • Prozac, Sarafem
Furosemide • Lasix
Glargine (Insulin) • Lantus, Toujeo
Glipizide • Glucotrol
Haloperidol • Haldol
Ibuprofen • Advil, Motrin
Imipramine • Tofranil
Lacosamide • Vimpat
Lisinopril • Prinivil, Zestril
Lorazepam • Ativan
Nicardipine • Cardene
Nortriptyline • Pamelor
Paclitaxel • Abraxane, Taxol
Phenytoin • Dilantin, Phenytek
Piperacillin-tazobactam • Zosyn
Propofol • Diprivan
Sertraline • Zoloft
Tacrolimus • Prograf
Tamoxifen • Soltamox
Valproic acid • Depakene, Depakote
Verapamil • Calan, Verelan
Warfarin • Coumadin, Jantoven
1. Zhang F, Xue J, Shao J, et al. Compilation of 222 drugs’ plasma protein binding data and guidance for study designs. Drug Discov Today. 2012;17(9-10):475-485.
2. Mehvar R. Role of protein binding in pharmacokinetics. Am J Pharm Edu. 2005;69(5): Article 103;1-8.
3. Roberts JA, Pea F, Lipman J. The clinical relevance of plasma protein binding changes. Clin Pharmacokinet. 2013;52(1):1-8.
4. Schmidt S, Gonzalez D, Derendork H. Significance of protein binding in pharmacokinetics and pharmacodynamics. J Pharm Sci. 2010;99(3):1107-1122.
5. Hinderling P. Red blood cells: a neglected compartment in pharmacokinetics and pharmacodynamics. Pharmacol Rev. 1997;49(3):279-295.
6. Benet LZ, Hoener B. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther. 2002;71(3):115-121.
7. Park MK, Lim KS, Kim T, et al. Reduced valproic acid serum concentrations due to drug interactions with carbapenem antibiotics: overview of 6 cases. Ther Drug Monit. 2012;34(5):599-603.
1. Zhang F, Xue J, Shao J, et al. Compilation of 222 drugs’ plasma protein binding data and guidance for study designs. Drug Discov Today. 2012;17(9-10):475-485.
2. Mehvar R. Role of protein binding in pharmacokinetics. Am J Pharm Edu. 2005;69(5): Article 103;1-8.
3. Roberts JA, Pea F, Lipman J. The clinical relevance of plasma protein binding changes. Clin Pharmacokinet. 2013;52(1):1-8.
4. Schmidt S, Gonzalez D, Derendork H. Significance of protein binding in pharmacokinetics and pharmacodynamics. J Pharm Sci. 2010;99(3):1107-1122.
5. Hinderling P. Red blood cells: a neglected compartment in pharmacokinetics and pharmacodynamics. Pharmacol Rev. 1997;49(3):279-295.
6. Benet LZ, Hoener B. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther. 2002;71(3):115-121.
7. Park MK, Lim KS, Kim T, et al. Reduced valproic acid serum concentrations due to drug interactions with carbapenem antibiotics: overview of 6 cases. Ther Drug Monit. 2012;34(5):599-603.
Unrelenting depression: ‘I would rather be dead than feel this way’
CASE Suicidal ideation, flare-up of ulcerative colitis
Mr. J, age 56, who has a history of major depressive disorder (MDD), generalized anxiety disorder (GAD), and ulcerative colitis (UC), presents to the emergency department (ED) with suicidal ideation and a plan to overdose on his medications. He reports no current emotional or financial stressors in his personal life. Home medications documented at the time of his arrival to the ED include sertraline, 100 mg/d, bupropion, 150 mg/d, buspirone, 10 mg 3 times daily, diazepam 10 mg 3 times daily, as needed, adalimumab, 40 mg IM every 2 weeks, and diphenhydramine, 50 mg every night.
A recent flare-up of UC resulted in Mr. J being placed on a 15-week prednisone taper, beginning at 80 mg/d and decreasing by 5 mg weekly, which was completed 2 weeks before he presented to the ED. After completing the prednisone taper, Mr. J went to his primary care physician (PCP) on 3 separate occasions due to episodes of severe depression. Although the PCP prescribed multiple medications to target Mr. J’s depressive symptoms, he continued to decline.
Subsequently, Mr. J came to the ED and is admitted to the psychiatric unit for safety and stabilization. Upon admission, Mr. J becomes bedridden, and reports that his current depressive episode is the most severe that he has ever experienced in his more than 30 years of having MDD. He says that neither bupropion nor buspirone are helping with his depression, anxiety, or any related symptom.
[polldaddy:10120537]
The authors’ observations
At admission, all of Mr. J’s home medications, except sertraline and adalimumab, which had been prescribed to treat UC (Box1,2), were discontinued. His diazepam was discontinued because the clinician felt it may have been contributing to Mr. J’s inability to walk or get out of bed. Diazepam was not tapered because it was initiated 7 days prior to admission and was thought to be exacerbating his depression and suicidal ideation. Bupropion and buspirone, which were initiated 2 weeks prior, were discontinued because Mr. J reported that neither medication was helping with his depression, anxiety, or any related symptom.
Box
Ulcerative colitis and depressive episodes
Ulcerative colitis (UC) is a chronic condition associated with inflammation in the colon causing extreme abdominal discomfort during acute flare-ups. Moderate to severe UC flare-ups are commonly treated with corticosteroids due to these medications’ anti-inflammatory properties. Although rare, corticosteroid withdrawal has been documented to induce episodes of depression. The pathophysiology of corticosteroid withdrawal inducing neuropsychiatric sequelae remains unclear; however, it is thought to be due to hypothalamic-pituitary-adrenocortical suppression.1 Fardet et al2 concluded that incident rates per 100 person-years at risk during the withdrawal period were 11.1 (95% confidence interval, 10.0, 12.3) for depression.
EVALUATION Poor appetite, anxiety, and continued suicidality
During evaluation, vital signs, laboratory findings, and diagnostic testing are found to be unremarkable. Mr. J’s presentation and complaints are entirely subjective, and include poor appetite, fatigue, difficulty sleeping, sorrow, anxiety, and continued suicidality. Mr. J reports that he feels miserable, which is reflected by his poor eye contact, soft speech, and body language.
Continued to: The authors' observations
The authors’ observations
MDD is a mood disorder characterized by depressed mood and/or loss of interest or pleasure for more than 2 weeks.3 First-line pharmacotherapy for MDD includes monotherapy with a selective serotonin reuptake inhibitor (SSRI), serotonin-norepinephrine reuptake inhibitor (SNRI), mirtazapine, or bupropion.4 Medication selection is typically based on patient-specific factors, adverse effect profile, drug–drug interactions, and cost. Other treatments include electroconvulsive therapy (ECT) or cognitive-behavioral therapy (CBT).4,5 Augmentation agents, such as second-generation antipsychotics, lithium, thyroid hormone supplementation, buspirone, anticonvulsants, and combinations of antidepressants, may also be considered.4
TREATMENT Condition worsens
On Day 2 of hospitalization, Mr. J is started on aripiprazole, 5 mg/d, clonazepam, 1 mg twice daily, and melatonin, 5 mg, each night for sleep. Aripiprazole, 5 mg/d, is initiated as an adjunct to sertraline for MDD because Mr. J reports feeling much worse and continues to report that he would “rather die than feel this way.” Mr. J begins to believe that his current state is his new baseline, and that feeling better is no longer possible.
On Day 3 of hospitalization, records are obtained from a clinician at an outside facility who previously treated Mr
By Day 8 of hospitalization, there is no notable change in Mr. J’s depressive symptoms. On Day 9, sertraline is increased to 200 mg/d, with little improvement from Mr. J’s perspective. The multidisciplinary team evaluates him, and when directly asked, Mr. J cites his 4 greatest complaints to be poor sleep, fatigue, no appetite, and depressed mood. Once again, he states, “I would rather be dead than go on feeling this way.”
[polldaddy:10120587]
The authors’ observations
Due to Mr. J’s severe, unrelenting depressive episode, the treatment team obtained his informed consent to undergo ECT. On Day 9, before initiating ECT, the pharmacist recommended mirtazapine, even though the patient weighed almost 89 kg (196.21 lb) and had a body mass index of 27.8 kg/m2. The treatment team thought that mirtazapine augmentation could potentially help the sertraline work more quickly while targeting Mr. J’s 4 greatest complaints.
Mirtazapine is a central alpha-2 antagonist or noradrenergic and specific serotonergic antidepressant (NaSSA) that works through antagonism of the presynaptic alpha-2 adrenergic receptors to indirectly regulate release of monoamines and increase the release of serotonin and norepinephrine.6 Additionally, mirtazapine has antagonist actions at 5HT2A, 5HT2C, 5HT3, and histamine-1 receptors.6 Potential adverse effects include drowsiness and increased appetite leading to weight gain.7 Mirtazapine’s therapeutic efficacy is similar to SSRIs for treating depression.4 Mirtazapine in combination with an SNRI has been referred to as “California rocket fuel” due to the theoretical pharmacologic synergy and resulting strong antidepressant action.6 It was hypothesized that similar effects could be seen by augmenting the SSRI sertraline with mirtazapine.
Continued to: The time to efficacy with mirtazapine...
The time to efficacy with mirtazapine is approximately 2 to 4 weeks, but anxiety symptoms and poor sleep or insomnia may improve in the first week.8 Studies have suggested the possibility of a more rapid onset of efficacy with mirtazapine than with SSRIs, as well as potential response acceleration in MDD and other psychiatric illnesses such as anxiety disorders or obsessive-compulsive disorder (OCD).9,10 A review that included several double-blind studies and compared mirtazapine with SSRIs found the amount of responders with persistent improvement with onset in Week 1 was more pronounced with mirtazapine.9
Augmenting an SSRI with mirtazapine is a potential therapeutic option because it can help boost the efficacy of the prescribed SSRI while enhancing appetite and blunting the activating or anxiety-like effects of some SSRIs, which may help with relaxation and sleep.4 The combination of an SSRI plus mirtazapine has been studied in patients with MDD, posttraumatic stress disorder, and OCD; it was found to improve symptoms of those conditions due to the medications’ complementary mechanisms of action.4,11-13 Also, mirtazapine has been shown to decrease the rates of relapse after an acute phase of depression.4,14
OUTCOME Rapid improvement
On Day 9, Mr. J receives the first dose of mirtazapine, 7.5 mg at bedtime. On Day 10, when Mr. J wakes, his mood is notably improved. He is more interactive (sitting up in bed reading and making eye contact with the staff during an interview), and he reports improved sleep and eats most of his breakfast.
After receiving 3 doses of mirtazapine, Mr. J reports that he feels back to his normal self; he is interactive, alert, and eating well. Due to the rapid improvement in mood, ECT is discontinued, and he does not receive any ECT treatment during the remainder of his hospitalization.
On Day 11, divalproex is discontinued. Because Mr. J receives only 5 days of therapy with this agent, his divalproex level is not checked. At this point, the treatment team feels confident in ruling out bipolar disorder.
On Day 15, Mr. J is discharged with sertraline, 200 mg/d, mirtazapine, 7.5 mg/d at 7
Ten months after his depressive episode, Mr. J has had no further admissions at the hospital where he received the treatment described here.
Bottom Line
Evidence for the treatment of major depressive disorder induced by corticosteroid withdrawal is limited. Despite trials of agents from multiple medication classes, the depressive episode may not improve. Adding mirtazapine to a selective serotonin reuptake inhibitor or serotonin-norepinephrine reuptake inhibitor may prove successful.
Related Resources
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev. 2011;(12):CD006528.
- Kenna HA, Poon AW, de los Angeles CP, et al. Psychiatric complications of treatment with corticosteroids: review with case report. Psychiatry Clin Neurosci. 2011;65(6):549-560.
Drug Brand Names
Adalimumab • Humira
Aripiprazole • Abilify
Bupropion • Wellbutrin, Zyban
Buspirone • Buspar
Clonazepam • Klonopin
Diazepam • Valium
Diphenhydramine • Benadryl
Divalproex • Depakote, Depakote ER
Lithium • Eskalith, Lithobid
Mirtazapine • Remeron
Prednisone • Deltasone
Sertraline • Zoloft
1. Dixon R, Christy N. On the various forms of corticosteroid withdrawal syndrome. Am J Med. 1980;68(2):224-30.
2. Fardet L, Petersen I, Nazareth I. Suicidal behavior and severe neuropsychiatric disorders following glucocorticoid therapy in primary care. Am J Psychiatry. 2012;169(5):491-497.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder, 3rd ed. Arlington Virginia: American Psychiatric Association. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Published October 2010. Accessed March 15, 2017.
5. National Institute for Health and Clinical Excellence (NICE) Clinical Guideline 90. Depression in adults: recognition and management. https://www.nice.org.uk/guidance/cg90. Accessed March 15, 2017.
6. Stahl SM. Stahl’s essential psychopharmacology: neuroscientific basis and practical applications, 4th ed. Cambridge, United Kingdom: Cambridge University Press; 2013;317-322; 363-364.
7. Remeron [package insert]. Whitehouse Station, NJ: Merck & Co., Inc.; 2018.
8. Gorman JM. Mirtazapine: clinical overview. J Clin Psychiatry. 1999;60(suppl 17):9-13; discussion 46-48.
9. Quitkin FM, Taylor BP, Kremer C. Does mirtazapine have a more rapid onset than SSRIs? J Clin Psychiatry. 2001;62(5):358-361.
10. Pallanti S, Quercioli L, Bruscoli M. Response acceleration with mirtazapine augmentation of citalopram in obsessive-compulsive disorder patients without comorbid depression: a pilot study. J Clin Psychiatry. 2004;65(10):1394-1399.
11. Blier P, Gobbi G, Turcotte JE, et al. Mirtazapine and paroxetine in major depression: a comparison of monotherapy versus their combination from treatment initiation. Eur Neuropsychopharmacol. 2009;19(7):457-465.
12. Blier P, Ward HE, Tremblay P, et al. Combination of antidepressant medications from treatment initiation for major depressive disorder: a double-blind randomized study. Am J Psychiatry. 2010;167(3):281-288.
13. Carpenter LL, Yasmin S, Price LH. A double-blind, placebo-controlled study of antidepressant augmentation with mirtazapine. Biol Psychiatry. 2002;51(2):183-188.
14. Schneier FR, Campeas R, Carcamo J, et al. Combined mirtazapine and SSRI treatment of PTSD: a placebo-controlled trial. Depress Anxiety. 2015;32(8):570-579.
CASE Suicidal ideation, flare-up of ulcerative colitis
Mr. J, age 56, who has a history of major depressive disorder (MDD), generalized anxiety disorder (GAD), and ulcerative colitis (UC), presents to the emergency department (ED) with suicidal ideation and a plan to overdose on his medications. He reports no current emotional or financial stressors in his personal life. Home medications documented at the time of his arrival to the ED include sertraline, 100 mg/d, bupropion, 150 mg/d, buspirone, 10 mg 3 times daily, diazepam 10 mg 3 times daily, as needed, adalimumab, 40 mg IM every 2 weeks, and diphenhydramine, 50 mg every night.
A recent flare-up of UC resulted in Mr. J being placed on a 15-week prednisone taper, beginning at 80 mg/d and decreasing by 5 mg weekly, which was completed 2 weeks before he presented to the ED. After completing the prednisone taper, Mr. J went to his primary care physician (PCP) on 3 separate occasions due to episodes of severe depression. Although the PCP prescribed multiple medications to target Mr. J’s depressive symptoms, he continued to decline.
Subsequently, Mr. J came to the ED and is admitted to the psychiatric unit for safety and stabilization. Upon admission, Mr. J becomes bedridden, and reports that his current depressive episode is the most severe that he has ever experienced in his more than 30 years of having MDD. He says that neither bupropion nor buspirone are helping with his depression, anxiety, or any related symptom.
[polldaddy:10120537]
The authors’ observations
At admission, all of Mr. J’s home medications, except sertraline and adalimumab, which had been prescribed to treat UC (Box1,2), were discontinued. His diazepam was discontinued because the clinician felt it may have been contributing to Mr. J’s inability to walk or get out of bed. Diazepam was not tapered because it was initiated 7 days prior to admission and was thought to be exacerbating his depression and suicidal ideation. Bupropion and buspirone, which were initiated 2 weeks prior, were discontinued because Mr. J reported that neither medication was helping with his depression, anxiety, or any related symptom.
Box
Ulcerative colitis and depressive episodes
Ulcerative colitis (UC) is a chronic condition associated with inflammation in the colon causing extreme abdominal discomfort during acute flare-ups. Moderate to severe UC flare-ups are commonly treated with corticosteroids due to these medications’ anti-inflammatory properties. Although rare, corticosteroid withdrawal has been documented to induce episodes of depression. The pathophysiology of corticosteroid withdrawal inducing neuropsychiatric sequelae remains unclear; however, it is thought to be due to hypothalamic-pituitary-adrenocortical suppression.1 Fardet et al2 concluded that incident rates per 100 person-years at risk during the withdrawal period were 11.1 (95% confidence interval, 10.0, 12.3) for depression.
EVALUATION Poor appetite, anxiety, and continued suicidality
During evaluation, vital signs, laboratory findings, and diagnostic testing are found to be unremarkable. Mr. J’s presentation and complaints are entirely subjective, and include poor appetite, fatigue, difficulty sleeping, sorrow, anxiety, and continued suicidality. Mr. J reports that he feels miserable, which is reflected by his poor eye contact, soft speech, and body language.
Continued to: The authors' observations
The authors’ observations
MDD is a mood disorder characterized by depressed mood and/or loss of interest or pleasure for more than 2 weeks.3 First-line pharmacotherapy for MDD includes monotherapy with a selective serotonin reuptake inhibitor (SSRI), serotonin-norepinephrine reuptake inhibitor (SNRI), mirtazapine, or bupropion.4 Medication selection is typically based on patient-specific factors, adverse effect profile, drug–drug interactions, and cost. Other treatments include electroconvulsive therapy (ECT) or cognitive-behavioral therapy (CBT).4,5 Augmentation agents, such as second-generation antipsychotics, lithium, thyroid hormone supplementation, buspirone, anticonvulsants, and combinations of antidepressants, may also be considered.4
TREATMENT Condition worsens
On Day 2 of hospitalization, Mr. J is started on aripiprazole, 5 mg/d, clonazepam, 1 mg twice daily, and melatonin, 5 mg, each night for sleep. Aripiprazole, 5 mg/d, is initiated as an adjunct to sertraline for MDD because Mr. J reports feeling much worse and continues to report that he would “rather die than feel this way.” Mr. J begins to believe that his current state is his new baseline, and that feeling better is no longer possible.
On Day 3 of hospitalization, records are obtained from a clinician at an outside facility who previously treated Mr
By Day 8 of hospitalization, there is no notable change in Mr. J’s depressive symptoms. On Day 9, sertraline is increased to 200 mg/d, with little improvement from Mr. J’s perspective. The multidisciplinary team evaluates him, and when directly asked, Mr. J cites his 4 greatest complaints to be poor sleep, fatigue, no appetite, and depressed mood. Once again, he states, “I would rather be dead than go on feeling this way.”
[polldaddy:10120587]
The authors’ observations
Due to Mr. J’s severe, unrelenting depressive episode, the treatment team obtained his informed consent to undergo ECT. On Day 9, before initiating ECT, the pharmacist recommended mirtazapine, even though the patient weighed almost 89 kg (196.21 lb) and had a body mass index of 27.8 kg/m2. The treatment team thought that mirtazapine augmentation could potentially help the sertraline work more quickly while targeting Mr. J’s 4 greatest complaints.
Mirtazapine is a central alpha-2 antagonist or noradrenergic and specific serotonergic antidepressant (NaSSA) that works through antagonism of the presynaptic alpha-2 adrenergic receptors to indirectly regulate release of monoamines and increase the release of serotonin and norepinephrine.6 Additionally, mirtazapine has antagonist actions at 5HT2A, 5HT2C, 5HT3, and histamine-1 receptors.6 Potential adverse effects include drowsiness and increased appetite leading to weight gain.7 Mirtazapine’s therapeutic efficacy is similar to SSRIs for treating depression.4 Mirtazapine in combination with an SNRI has been referred to as “California rocket fuel” due to the theoretical pharmacologic synergy and resulting strong antidepressant action.6 It was hypothesized that similar effects could be seen by augmenting the SSRI sertraline with mirtazapine.
Continued to: The time to efficacy with mirtazapine...
The time to efficacy with mirtazapine is approximately 2 to 4 weeks, but anxiety symptoms and poor sleep or insomnia may improve in the first week.8 Studies have suggested the possibility of a more rapid onset of efficacy with mirtazapine than with SSRIs, as well as potential response acceleration in MDD and other psychiatric illnesses such as anxiety disorders or obsessive-compulsive disorder (OCD).9,10 A review that included several double-blind studies and compared mirtazapine with SSRIs found the amount of responders with persistent improvement with onset in Week 1 was more pronounced with mirtazapine.9
Augmenting an SSRI with mirtazapine is a potential therapeutic option because it can help boost the efficacy of the prescribed SSRI while enhancing appetite and blunting the activating or anxiety-like effects of some SSRIs, which may help with relaxation and sleep.4 The combination of an SSRI plus mirtazapine has been studied in patients with MDD, posttraumatic stress disorder, and OCD; it was found to improve symptoms of those conditions due to the medications’ complementary mechanisms of action.4,11-13 Also, mirtazapine has been shown to decrease the rates of relapse after an acute phase of depression.4,14
OUTCOME Rapid improvement
On Day 9, Mr. J receives the first dose of mirtazapine, 7.5 mg at bedtime. On Day 10, when Mr. J wakes, his mood is notably improved. He is more interactive (sitting up in bed reading and making eye contact with the staff during an interview), and he reports improved sleep and eats most of his breakfast.
After receiving 3 doses of mirtazapine, Mr. J reports that he feels back to his normal self; he is interactive, alert, and eating well. Due to the rapid improvement in mood, ECT is discontinued, and he does not receive any ECT treatment during the remainder of his hospitalization.
On Day 11, divalproex is discontinued. Because Mr. J receives only 5 days of therapy with this agent, his divalproex level is not checked. At this point, the treatment team feels confident in ruling out bipolar disorder.
On Day 15, Mr. J is discharged with sertraline, 200 mg/d, mirtazapine, 7.5 mg/d at 7
Ten months after his depressive episode, Mr. J has had no further admissions at the hospital where he received the treatment described here.
Bottom Line
Evidence for the treatment of major depressive disorder induced by corticosteroid withdrawal is limited. Despite trials of agents from multiple medication classes, the depressive episode may not improve. Adding mirtazapine to a selective serotonin reuptake inhibitor or serotonin-norepinephrine reuptake inhibitor may prove successful.
Related Resources
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev. 2011;(12):CD006528.
- Kenna HA, Poon AW, de los Angeles CP, et al. Psychiatric complications of treatment with corticosteroids: review with case report. Psychiatry Clin Neurosci. 2011;65(6):549-560.
Drug Brand Names
Adalimumab • Humira
Aripiprazole • Abilify
Bupropion • Wellbutrin, Zyban
Buspirone • Buspar
Clonazepam • Klonopin
Diazepam • Valium
Diphenhydramine • Benadryl
Divalproex • Depakote, Depakote ER
Lithium • Eskalith, Lithobid
Mirtazapine • Remeron
Prednisone • Deltasone
Sertraline • Zoloft
CASE Suicidal ideation, flare-up of ulcerative colitis
Mr. J, age 56, who has a history of major depressive disorder (MDD), generalized anxiety disorder (GAD), and ulcerative colitis (UC), presents to the emergency department (ED) with suicidal ideation and a plan to overdose on his medications. He reports no current emotional or financial stressors in his personal life. Home medications documented at the time of his arrival to the ED include sertraline, 100 mg/d, bupropion, 150 mg/d, buspirone, 10 mg 3 times daily, diazepam 10 mg 3 times daily, as needed, adalimumab, 40 mg IM every 2 weeks, and diphenhydramine, 50 mg every night.
A recent flare-up of UC resulted in Mr. J being placed on a 15-week prednisone taper, beginning at 80 mg/d and decreasing by 5 mg weekly, which was completed 2 weeks before he presented to the ED. After completing the prednisone taper, Mr. J went to his primary care physician (PCP) on 3 separate occasions due to episodes of severe depression. Although the PCP prescribed multiple medications to target Mr. J’s depressive symptoms, he continued to decline.
Subsequently, Mr. J came to the ED and is admitted to the psychiatric unit for safety and stabilization. Upon admission, Mr. J becomes bedridden, and reports that his current depressive episode is the most severe that he has ever experienced in his more than 30 years of having MDD. He says that neither bupropion nor buspirone are helping with his depression, anxiety, or any related symptom.
[polldaddy:10120537]
The authors’ observations
At admission, all of Mr. J’s home medications, except sertraline and adalimumab, which had been prescribed to treat UC (Box1,2), were discontinued. His diazepam was discontinued because the clinician felt it may have been contributing to Mr. J’s inability to walk or get out of bed. Diazepam was not tapered because it was initiated 7 days prior to admission and was thought to be exacerbating his depression and suicidal ideation. Bupropion and buspirone, which were initiated 2 weeks prior, were discontinued because Mr. J reported that neither medication was helping with his depression, anxiety, or any related symptom.
Box
Ulcerative colitis and depressive episodes
Ulcerative colitis (UC) is a chronic condition associated with inflammation in the colon causing extreme abdominal discomfort during acute flare-ups. Moderate to severe UC flare-ups are commonly treated with corticosteroids due to these medications’ anti-inflammatory properties. Although rare, corticosteroid withdrawal has been documented to induce episodes of depression. The pathophysiology of corticosteroid withdrawal inducing neuropsychiatric sequelae remains unclear; however, it is thought to be due to hypothalamic-pituitary-adrenocortical suppression.1 Fardet et al2 concluded that incident rates per 100 person-years at risk during the withdrawal period were 11.1 (95% confidence interval, 10.0, 12.3) for depression.
EVALUATION Poor appetite, anxiety, and continued suicidality
During evaluation, vital signs, laboratory findings, and diagnostic testing are found to be unremarkable. Mr. J’s presentation and complaints are entirely subjective, and include poor appetite, fatigue, difficulty sleeping, sorrow, anxiety, and continued suicidality. Mr. J reports that he feels miserable, which is reflected by his poor eye contact, soft speech, and body language.
Continued to: The authors' observations
The authors’ observations
MDD is a mood disorder characterized by depressed mood and/or loss of interest or pleasure for more than 2 weeks.3 First-line pharmacotherapy for MDD includes monotherapy with a selective serotonin reuptake inhibitor (SSRI), serotonin-norepinephrine reuptake inhibitor (SNRI), mirtazapine, or bupropion.4 Medication selection is typically based on patient-specific factors, adverse effect profile, drug–drug interactions, and cost. Other treatments include electroconvulsive therapy (ECT) or cognitive-behavioral therapy (CBT).4,5 Augmentation agents, such as second-generation antipsychotics, lithium, thyroid hormone supplementation, buspirone, anticonvulsants, and combinations of antidepressants, may also be considered.4
TREATMENT Condition worsens
On Day 2 of hospitalization, Mr. J is started on aripiprazole, 5 mg/d, clonazepam, 1 mg twice daily, and melatonin, 5 mg, each night for sleep. Aripiprazole, 5 mg/d, is initiated as an adjunct to sertraline for MDD because Mr. J reports feeling much worse and continues to report that he would “rather die than feel this way.” Mr. J begins to believe that his current state is his new baseline, and that feeling better is no longer possible.
On Day 3 of hospitalization, records are obtained from a clinician at an outside facility who previously treated Mr
By Day 8 of hospitalization, there is no notable change in Mr. J’s depressive symptoms. On Day 9, sertraline is increased to 200 mg/d, with little improvement from Mr. J’s perspective. The multidisciplinary team evaluates him, and when directly asked, Mr. J cites his 4 greatest complaints to be poor sleep, fatigue, no appetite, and depressed mood. Once again, he states, “I would rather be dead than go on feeling this way.”
[polldaddy:10120587]
The authors’ observations
Due to Mr. J’s severe, unrelenting depressive episode, the treatment team obtained his informed consent to undergo ECT. On Day 9, before initiating ECT, the pharmacist recommended mirtazapine, even though the patient weighed almost 89 kg (196.21 lb) and had a body mass index of 27.8 kg/m2. The treatment team thought that mirtazapine augmentation could potentially help the sertraline work more quickly while targeting Mr. J’s 4 greatest complaints.
Mirtazapine is a central alpha-2 antagonist or noradrenergic and specific serotonergic antidepressant (NaSSA) that works through antagonism of the presynaptic alpha-2 adrenergic receptors to indirectly regulate release of monoamines and increase the release of serotonin and norepinephrine.6 Additionally, mirtazapine has antagonist actions at 5HT2A, 5HT2C, 5HT3, and histamine-1 receptors.6 Potential adverse effects include drowsiness and increased appetite leading to weight gain.7 Mirtazapine’s therapeutic efficacy is similar to SSRIs for treating depression.4 Mirtazapine in combination with an SNRI has been referred to as “California rocket fuel” due to the theoretical pharmacologic synergy and resulting strong antidepressant action.6 It was hypothesized that similar effects could be seen by augmenting the SSRI sertraline with mirtazapine.
Continued to: The time to efficacy with mirtazapine...
The time to efficacy with mirtazapine is approximately 2 to 4 weeks, but anxiety symptoms and poor sleep or insomnia may improve in the first week.8 Studies have suggested the possibility of a more rapid onset of efficacy with mirtazapine than with SSRIs, as well as potential response acceleration in MDD and other psychiatric illnesses such as anxiety disorders or obsessive-compulsive disorder (OCD).9,10 A review that included several double-blind studies and compared mirtazapine with SSRIs found the amount of responders with persistent improvement with onset in Week 1 was more pronounced with mirtazapine.9
Augmenting an SSRI with mirtazapine is a potential therapeutic option because it can help boost the efficacy of the prescribed SSRI while enhancing appetite and blunting the activating or anxiety-like effects of some SSRIs, which may help with relaxation and sleep.4 The combination of an SSRI plus mirtazapine has been studied in patients with MDD, posttraumatic stress disorder, and OCD; it was found to improve symptoms of those conditions due to the medications’ complementary mechanisms of action.4,11-13 Also, mirtazapine has been shown to decrease the rates of relapse after an acute phase of depression.4,14
OUTCOME Rapid improvement
On Day 9, Mr. J receives the first dose of mirtazapine, 7.5 mg at bedtime. On Day 10, when Mr. J wakes, his mood is notably improved. He is more interactive (sitting up in bed reading and making eye contact with the staff during an interview), and he reports improved sleep and eats most of his breakfast.
After receiving 3 doses of mirtazapine, Mr. J reports that he feels back to his normal self; he is interactive, alert, and eating well. Due to the rapid improvement in mood, ECT is discontinued, and he does not receive any ECT treatment during the remainder of his hospitalization.
On Day 11, divalproex is discontinued. Because Mr. J receives only 5 days of therapy with this agent, his divalproex level is not checked. At this point, the treatment team feels confident in ruling out bipolar disorder.
On Day 15, Mr. J is discharged with sertraline, 200 mg/d, mirtazapine, 7.5 mg/d at 7
Ten months after his depressive episode, Mr. J has had no further admissions at the hospital where he received the treatment described here.
Bottom Line
Evidence for the treatment of major depressive disorder induced by corticosteroid withdrawal is limited. Despite trials of agents from multiple medication classes, the depressive episode may not improve. Adding mirtazapine to a selective serotonin reuptake inhibitor or serotonin-norepinephrine reuptake inhibitor may prove successful.
Related Resources
- Watanabe N, Omori IM, Nakagawa A, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database Syst Rev. 2011;(12):CD006528.
- Kenna HA, Poon AW, de los Angeles CP, et al. Psychiatric complications of treatment with corticosteroids: review with case report. Psychiatry Clin Neurosci. 2011;65(6):549-560.
Drug Brand Names
Adalimumab • Humira
Aripiprazole • Abilify
Bupropion • Wellbutrin, Zyban
Buspirone • Buspar
Clonazepam • Klonopin
Diazepam • Valium
Diphenhydramine • Benadryl
Divalproex • Depakote, Depakote ER
Lithium • Eskalith, Lithobid
Mirtazapine • Remeron
Prednisone • Deltasone
Sertraline • Zoloft
1. Dixon R, Christy N. On the various forms of corticosteroid withdrawal syndrome. Am J Med. 1980;68(2):224-30.
2. Fardet L, Petersen I, Nazareth I. Suicidal behavior and severe neuropsychiatric disorders following glucocorticoid therapy in primary care. Am J Psychiatry. 2012;169(5):491-497.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder, 3rd ed. Arlington Virginia: American Psychiatric Association. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Published October 2010. Accessed March 15, 2017.
5. National Institute for Health and Clinical Excellence (NICE) Clinical Guideline 90. Depression in adults: recognition and management. https://www.nice.org.uk/guidance/cg90. Accessed March 15, 2017.
6. Stahl SM. Stahl’s essential psychopharmacology: neuroscientific basis and practical applications, 4th ed. Cambridge, United Kingdom: Cambridge University Press; 2013;317-322; 363-364.
7. Remeron [package insert]. Whitehouse Station, NJ: Merck & Co., Inc.; 2018.
8. Gorman JM. Mirtazapine: clinical overview. J Clin Psychiatry. 1999;60(suppl 17):9-13; discussion 46-48.
9. Quitkin FM, Taylor BP, Kremer C. Does mirtazapine have a more rapid onset than SSRIs? J Clin Psychiatry. 2001;62(5):358-361.
10. Pallanti S, Quercioli L, Bruscoli M. Response acceleration with mirtazapine augmentation of citalopram in obsessive-compulsive disorder patients without comorbid depression: a pilot study. J Clin Psychiatry. 2004;65(10):1394-1399.
11. Blier P, Gobbi G, Turcotte JE, et al. Mirtazapine and paroxetine in major depression: a comparison of monotherapy versus their combination from treatment initiation. Eur Neuropsychopharmacol. 2009;19(7):457-465.
12. Blier P, Ward HE, Tremblay P, et al. Combination of antidepressant medications from treatment initiation for major depressive disorder: a double-blind randomized study. Am J Psychiatry. 2010;167(3):281-288.
13. Carpenter LL, Yasmin S, Price LH. A double-blind, placebo-controlled study of antidepressant augmentation with mirtazapine. Biol Psychiatry. 2002;51(2):183-188.
14. Schneier FR, Campeas R, Carcamo J, et al. Combined mirtazapine and SSRI treatment of PTSD: a placebo-controlled trial. Depress Anxiety. 2015;32(8):570-579.
1. Dixon R, Christy N. On the various forms of corticosteroid withdrawal syndrome. Am J Med. 1980;68(2):224-30.
2. Fardet L, Petersen I, Nazareth I. Suicidal behavior and severe neuropsychiatric disorders following glucocorticoid therapy in primary care. Am J Psychiatry. 2012;169(5):491-497.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder, 3rd ed. Arlington Virginia: American Psychiatric Association. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Published October 2010. Accessed March 15, 2017.
5. National Institute for Health and Clinical Excellence (NICE) Clinical Guideline 90. Depression in adults: recognition and management. https://www.nice.org.uk/guidance/cg90. Accessed March 15, 2017.
6. Stahl SM. Stahl’s essential psychopharmacology: neuroscientific basis and practical applications, 4th ed. Cambridge, United Kingdom: Cambridge University Press; 2013;317-322; 363-364.
7. Remeron [package insert]. Whitehouse Station, NJ: Merck & Co., Inc.; 2018.
8. Gorman JM. Mirtazapine: clinical overview. J Clin Psychiatry. 1999;60(suppl 17):9-13; discussion 46-48.
9. Quitkin FM, Taylor BP, Kremer C. Does mirtazapine have a more rapid onset than SSRIs? J Clin Psychiatry. 2001;62(5):358-361.
10. Pallanti S, Quercioli L, Bruscoli M. Response acceleration with mirtazapine augmentation of citalopram in obsessive-compulsive disorder patients without comorbid depression: a pilot study. J Clin Psychiatry. 2004;65(10):1394-1399.
11. Blier P, Gobbi G, Turcotte JE, et al. Mirtazapine and paroxetine in major depression: a comparison of monotherapy versus their combination from treatment initiation. Eur Neuropsychopharmacol. 2009;19(7):457-465.
12. Blier P, Ward HE, Tremblay P, et al. Combination of antidepressant medications from treatment initiation for major depressive disorder: a double-blind randomized study. Am J Psychiatry. 2010;167(3):281-288.
13. Carpenter LL, Yasmin S, Price LH. A double-blind, placebo-controlled study of antidepressant augmentation with mirtazapine. Biol Psychiatry. 2002;51(2):183-188.
14. Schneier FR, Campeas R, Carcamo J, et al. Combined mirtazapine and SSRI treatment of PTSD: a placebo-controlled trial. Depress Anxiety. 2015;32(8):570-579.
Small Cell Lung Cancer
INTRODUCTION
Small cell lung cancer (SCLC) is an aggressive cancer of neuroendocrine origin that accounts for approximately 15% of all lung cancer cases, with approximately 33,000 patients diagnosed annually.1 The incidence of SCLC in the United States has steadily declined over the past 30 years, presumably because of a decrease in the number of smokers and a change to low-tar filter cigarettes.2 Although the overall incidence of SCLC has been decreasing, the incidence in women is increasing and the male-to-female incidence ratio is now 1:1.3 Nearly all cases of SCLC are associated with heavy tobacco exposure, making it a heterogeneous disease with a complex genomic landscape consisting of thousands of mutations.4,5 Despite recent advances in the treatment of non-small cell lung cancer, the therapeutic options for SCLC remain limited, with a median overall survival (OS) of 9 months in patients with advanced disease.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 61-year-old man presents to the emergency department with progressive shortness of breath and cough over the past 6 weeks. He also reports a 20-lb weight loss over the same period. He is a current smoker and has been smoking 1 pack of cigarettes per day since the age of 18 years. A chest radiograph obtained in the emergency department shows a right hilar mass. Computed tomography (CT) scan confirms the presence of a 4.5-cm right hilar mass and enlarged mediastinal lymph nodes bilaterally.
• What are the next steps in diagnosis?
SCLC is characterized by rapid growth and early hematogenous metastasis. Consequently, only 25% of patients have limited-stage disease at the time of diagnosis. According to the Veterans Administration Lung Study Group (VALSG) staging system, limited-stage disease is defined as tumor that is confined to 1 hemithorax and can be encompassed within 1 radiation field. This typically includes mediastinal lymph nodes and ipsilateral supraclavicular lymph nodes. Approximately 75% of patients present with extensive-stage disease, which is defined as disease that cannot be classified as limited, including disease that extends beyond 1 hemithorax. Extensive-stage disease includes the presence of malignant pleural effusion and/or distant metastasis.6 The VALSG classification and staging system is more commonly used in clinical practice than the American Joint Committee on Cancer TNM staging system because it is less complex and directs treatment decisions, as most of the literature on SCLC classifies patients based on the VALSG system.7
Given SCLC’s propensity to metastasize quickly, none of the currently available screening methods have proven successful in early detection of SCLC. In the National Lung Cancer Screening Trial, 86% of the 125 patients who were diagnosed with SCLC while undergoing annual low-dose chest CT scans had advanced disease at diagnosis.8,9 These results highlight the fact that most cases of SCLC develop in the interval between annual screening imaging.
SCLC frequently presents with a large hilar mass that is symptomatic. Common symptoms include shortness of breath and cough. In addition, patients with SCLC usually have bulky mediastinal adenopathy at presentation. SCLC is commonly located submucosally in the bronchus, and therefore hemoptysis is not a very common symptom at the time of presentation. Patients may present with superior vena cava syndrome from local compression by the tumor. Not infrequently, SCLC is associated with paraneoplastic syndromes that arise due to ectopic secretion of hormones or antibodies by the tumor cells. The paraneoplastic syndromes can be broadly categorized as endocrine or neurologic (Table 1). The presence of a paraneoplastic syndrome is often a clue to the potential diagnosis of SCLC in the presence of a hilar mass. Additionally, some paraneoplastic syndromes, more specifically endocrine paraneoplastic syndromes, follow the pattern of disease response and relapse, and therefore can sometimes serve as an early marker of disease relapse or progression.

The common sites of metastases include brain, liver, and bone. Therefore, the staging workup should include fluorodeoxyglucose (FDG) positron emission tomography (PET)/CT scan. Contrast-enhanced CT scan of the chest and abdomen and bone scan can be obtained for staging in lieu of PET scan. Due to the physiologic FDG uptake, cerebral metastases cannot be assessed with sufficient certainty using PET-CT.10 Therefore, brain imaging with contrast-enhanced CT or magnetic resonance imaging (MRI) is also necessary. Although the incidence of metastasis to bone marrow is less than 10%, bone marrow aspiration and biopsy are warranted in patients with unexplained cytopenias, especially when the cytopenia is associated with teardrop-shaped red cells or nucleated red cells on peripheral blood smear, findings indicative of a marrow infiltrative process.7 The tissue diagnosis is established by obtaining a biopsy of the primary tumor or 1 of the metastatic sites. In localized disease, bronchoscopy (with endobronchial ultrasound, if necessary) with biopsy of the centrally located tumor and/or lymph node is required. Histologically, SCLC consists of monomorphic cells, a high nuclear-cytoplasmic ratio, and confluent necrosis. The tumor cells are positive for chromogranin, synaptophysin, and CD56 by immunohistochemistry, and very frequently are also positive for thyroid transcription factor 1.11 Although serum tumor markers, including neuron-specific enolase and progastrin-releasing peptide, are frequently elevated in patients with SCLC, these markers are of limited value in clinical practice because they lack sensitivity and specificity.12
MANAGEMENT OF LIMITED-STAGE DISEASE
CASE CONTINUED
The patient undergoes FDG PET scan, which shows the presence of a hypermetabolic right hilar mass in addition to enlarged and hypermetabolic bilateral mediastinal lymph nodes. There are no other areas of FDG avidity. Brain MRI does not show any evidence of brain metastasis. Thus, the patient is confirmed to have limited-stage SCLC.
• What is the standard of care treatment for limited-stage SCLC?
SCLC is exquisitely sensitive to both chemotherapy and radiation, especially at the time of initial presentation. The standard of care treatment of limited-stage SCLC is 4 cycles of platinum-based chemotherapy in combination with thoracic radiation started within the first 2 cycles of chemotherapy (Figure 1). .")
CHOICE OF CHEMOTHERAPY
Etoposide and cisplatin is the most commonly used initial combination chemotherapy regimen in limited-stage SCLC.14 This combination has largely replaced anthracycline-based regimens given its favorable efficacy and toxicity profile.15–17 Several small randomized trials have shown comparable efficacy of carboplatin and etoposide in extensive-stage SCLC.18–20 A meta-analysis of 4 randomized trials comparing cisplatin-based versus carboplatin-based regimens in 663 patients with SCLC (32% had limited-stage disease and 68% had extensive-stage disease) showed no statistically significant difference in response rate, progression-free survival (PFS), or OS between the 2 regimens.21 Therefore, in clinical practice carboplatin is frequently used instead of cisplatin in patients with extensive-stage disease. In patients with limited-stage disease, cisplatin is still the drug of choice. However, the toxicity profile of the 2 regimens is different. Cisplatin-based regimens are more commonly associated with neuropathy, nephrotoxicity, and chemotherapy-induced nausea/vomiting,18 while carboplatin-based regimens are more myelosuppressive.22 In addition, the combination of thoracic radiation with either of these regimens is associated with a higher risk of esophagitis, pneumonitis, and myelosuppression.23 The use of myeloid growth factors is not recommended in patients undergoing concurrent chemoradiation.24 Of note, intravenous etoposide is always preferred over oral etoposide, especially in the curative setting given the unreliable absorption and bioavailability of oral formulations.
THORACIC RADIOTHERAPY
Adding thoracic radiotherapy to platinum-etoposide chemotherapy improves local control and OS. Two meta-analyses of 13 trials including more than 2000 patients have shown a 25% to 30% decrease in local failure and a 5% to 7% increase in 2-year OS with chemoradiation compared to chemotherapy alone in limited-stage SCLC.25,26 Early (within the first 2 cycles) concurrent thoracic radiation is superior to delayed and/or sequential radiation in terms of local control and OS.23,27,28 The dose and fractionation of thoracic radiation in limited-stage SCLC has remained a controversial issue. The Eastern Cooperative Oncology Group/Radiation Therapy Oncology Group randomized trial compared 45 Gy of radiotherapy delivered twice daily over a period of 3 weeks to 45 Gy once daily over 5 weeks concurrently with chemotherapy. The twice daily regimen led to a 10% improvement in 5-year OS (26% versus 16%), but a higher incidence of grade 3 and 4 adverse events.13 Despite the survival advantage demonstrated by hyperfractionated radiotherapy, the results need to be interpreted with caution because the radiation doses are not biologically equivalent. In addition, the difficult logistics of patients receiving radiation twice a day has limited the routine implementation of this strategy. Subsequently, another randomized phase 3 trial (CONVERT) compared 45 Gy radiotherapy twice daily with 66 Gy radiotherapy once daily in limited-stage SCLC.29 This trial did not show any difference in OS. The patients in the twice daily arm had a higher incidence of grade 4 neutropenia. Considering the results of these trials, both strategies—45 Gy fractionated twice daily or 60 Gy fractionated once daily, delivered concurrently with chemotherapy—are acceptable in the setting of limited-stage SCLC. However, quite often a hyperfractionated regimen is not feasible for patients and many radiation oncology centers. Hopefully, the ongoing CALGB 30610 study will clarify the optimal radiation schedule for limited-stage disease.
PROPHYLACTIC CRANIAL IRRADIATION
Approximately 75% of patients with limited-stage disease experience disease recurrence, and brain is the site of recurrence in approximately half of these patients.30 Prophylactic cranial irradiation (PCI) consisting of 25 Gy radiotherapy delivered in 10 fractions has been shown to be effective in decreasing the incidence of cerebral metastases.30–32 Although individual small studies have not shown a survival benefit of PCI because of small sample size and limited power, a meta-analysis of these studies has shown a 25% decrease in the 3-year incidence of brain metastasis and 5.4% increase in 3-year OS.30 Most patients included in these studies had limited-stage disease. Therefore, PCI is the standard of care for patients with limited-stage disease who attain a partial or complete response to chemoradiation.
ROLE OF SURGERY
Surgical resection may be an acceptable choice in a very limited subset of patients with peripherally located small (< 5 cm) tumors where mediastinal lymph nodes have been confirmed to be uninvolved with complete mediastinal staging.33,34 Most of the data in this setting are derived from retrospective studies.35,36 A 5-year OS between 40% and 60% has been reported with this strategy in patients with clinical stage I disease. In general, when surgery is considered, lobectomy with mediastinal lymph node dissection followed by chemotherapy (if there is no nodal involvement) or chemoradiation (if nodal involvement) is recommended.37,38 Wedge or segmental resections are not considered to be optimal surgical options.
MANAGEMENT OF EXTENSIVE-STAGE DISEASE
CASE CONTINUED
The patient receives 4 cycles of cisplatin and etoposide along with 70 Gy radiotherapy concurrently with the first 2 cycles of chemotherapy. His post-treatment CT scans show a partial response. He undergoes PCI 6 weeks after completion of treatment. At routine follow-up 18 months later, he is doing generally well except for mildly decreased appetite and an unintentional weight loss of 5 lb. CT scans demonstrate multiple hypodense liver lesions ranging from 7 mm to 2 cm in size and a 2-cm left adrenal gland lesion highly concerning for metastasis. FDG PET scan confirms that the adrenal and liver lesions are hypermetabolic. In addition, the PET scan shows multiple FDG-avid bone lesions throughout the spine. Brain MRI is negative for brain metastasis.
• What is the standard of care for treatment of extensive-stage disease?
.")
Chemotherapy is the mainstay of treatment for extensive-stage SCLC; the goals of treatment are prolongation of survival, prevention or alleviation of cancer-related symptoms, and improvement in quality of life. The combination of etoposide with a platinum agent (carboplatin or cisplatin) is the preferred first-line treatment option. Carboplatin is more commonly used in clinical practice in this setting because of its comparable efficacy and better tolerability compared to cisplatin (Figure 2).21 A Japanese phase 3 trial comparing cisplatin plus irinotecan with cisplatin plus etoposide in the first-line setting in extensive-stage SCLC showed improvement in median and 2-year OS with the cisplatin/irinotecan regimen; however, 2 subsequent phase 3 trials conducted in the United States comparing these 2 regimens did not show any difference in OS. In addition, the cisplatin/irinotecan regimen was more toxic than the etoposide-based regimen.39,40 Therefore, 4 to 6 cycles of platinum/etoposide remains the standard of care first-line treatment for extensive-stage SCLC in the United States. The combination yields a 60% to 70% response rate, but the majority of patients invariably experience disease progression, with a median OS of 9 to 11 months.41 Maintenance chemotherapy beyond the initial 4 to 6 cycles does not improve survival and is associated with higher cumulative toxicity.42
Multiple attempts at improving first-line chemotherapy in extensive-stage disease have failed to show any meaningful difference in OS. For example, the addition of ifosfamide, palifosfamide, cyclophosphamide, taxane, or anthracycline to platinum doublet failed to show improvement in OS and led to more toxicity.43–46 Additionally, the use of alternating or cyclic chemotherapies in an attempt to curb drug resistance has also failed to show survival benefit.47–49 The addition of the antiangiogenic agent bevacizumab to standard platinum-based doublet has not prolonged OS in SCLC and has led to an unacceptably higher rate of tracheoesophageal fistula when used in conjunction with chemoradiation in limited-stage disease.50–55 Finally, the immune checkpoint inhibitor ipilimumab in combination with platinum plus etoposide failed to improve PFS or OS compared to platinum plus etoposide alone in a recent phase 3 trial, and maintenance pembrolizumab after completion of platinum-based chemotherapy did not improve PFS.56,57
More recently, a phase 2 study of pembrolizumab in extensive-stage SCLC (KEYNOTE 158) reported an overall response rate of 35.7%, median PFS of 2.1 months, and median OS of 14.6 months in patients who tested positive for programmed death ligand-1 (PD-L1) expression (which was defined as a PD-L1 Combined Positive Score ≥ 1).58 The median duration of response has not been reached in this study, indicating that pembrolizumab may be a promising approach in patients with extensive-stage SCLC, especially for those with PD-L1–positive tumors.
Patients with extensive-stage disease who have brain metastasis at the time of diagnosis can be treated with systemic chemotherapy first if the brain metastases are asymptomatic and there is significant extracranial disease burden. In that case, whole brain radiotherapy should be given after completion of systemic therapy.
SECOND-LINE CHEMOTHERAPY
Despite being exquisitely chemosensitive, SCLC is associated with a very poor prognosis largely because of invariable disease progression following first-line therapy and lack of effective second-line treatment options that can lead to appreciable disease control. The choice of second-line treatment is predominantly determined by the time of disease relapse after first-line platinum-based therapy. If this interval is 6 months or longer, re-treatment utilizing the same platinum doublet is appropriate. However, if the interval is 6 months or less, second-line systemic therapy options should be explored. Unfortunately, the response rate tends to be less than 10% with most of the second-line therapies in platinum-resistant disease (defined as disease progression within 3 months of receiving platinum-based therapy). If disease progression occurs between 3 and 6 months after completion of platinum-based therapy, the response rate with second-line chemotherapy is in the range of 25%.59,60

A number of second-line chemotherapy options have been explored in small studies, including topotecan, irinotecan, paclitaxel, docetaxel, temozolomide, vinorelbine, oral etoposide, gemcitabine, bendamustine, and CAV (
IMMUNOTHERAPY
The role of immune checkpoint inhibitors in the treatment of SCLC is evolving, and currently there are no FDA-approved immunotherapy agents for treating SCLC. A recently conducted phase 1/2 trial (CheckMate 032) studied the anti-programmed death(PD)-1 antibody nivolumab with or without the anti-cytotoxic T-lymphocyte–associated antigen (CTLA) -4 antibody ipilimumab in patients with relapsed SCLC. The authors reported response rates of 10% with nivolumab 3 mg/kg and 21% with nivolumab 1 mg/kg plus ipilimumab 3 mg/kg.78,79 The 2-year OS was 26% with the combination and 14% with single-agent nivolumab. Only 18% of patients had PD-L1 expression of ≥ 1%, and the response rate did not correlate with PD-L1 status. The rate of grade 3 or 4 adverse events was approximately 20%, and only 10% of patients discontinued treatment because of toxicity. Based on these data, nivolumab plus ipilimumab is now included in the National Comprehensive Cancer Network guidelines as an option for patients with SCLC who experience disease relapse within 6 months of receiving platinum-based therapy;7 however, it is questionable whether routine use of this combination is justified based on currently available data. The evidence for the combination of nivolumab and ipilimumab remains limited. The efficacy and toxicity data from both randomized and nonrandomized cohorts were presented together, making it hard to interpret the results.
Another phase 1b study (KEYNOTE-028) evaluated the anti-PD-1 antibody pembrolizumab (10 mg/kg intravenously every 2 weeks) in patients with relapsed SCLC who had received 1 or more prior lines of therapy and had PD-L1 expression of ≥ 1%. This study showed a response rate of 33%, with a median duration of response of 19 months and 1-year OS of 38%.80 Although only 28% of screened patients had PD-L1 expression of ≥ 1%, these results indicated that at least a subset of SCLC patients are able to achieve durable responses with immune checkpoint inhibition. A number of clinical trials utilizing immune checkpoint inhibitors in various combinations and settings are currently underway.
ROLE OF PROPHYLACTIC CRANIAL IRRADIATION
The role of PCI in extensive-stage SCLC is not clearly defined. A randomized phase 3 trial conducted by the European Organization for Research and Treatment of Cancer (EORTC) comparing PCI with no PCI in patients with extensive-stage SCLC who had a partial or complete response to initial platinum-based chemotherapy showed a decrease in the incidence of symptomatic brain metastasis and improvement in 1-year OS with PCI.81 However, this trial did not require mandatory brain imaging prior to PCI, and thus it is unclear if some patients in the PCI group had asymptomatic brain metastasis prior to enrollment and therefore received therapeutic benefit from brain radiation. Additionally, the dose and fractionation of PCI was not standardized across patient groups.
A more recent phase 3 study conducted in Japan that compared PCI (25 Gy in 10 fractions) with no PCI reported no difference in survival between the 2 groups.82 As opposed to the EORTC study, the Japanese study did require baseline brain imaging to confirm the absence of brain metastasis prior to enrollment. In addition, the control patients underwent periodic brain MRI to allow early detection of brain metastasis. Given the emergence of the new data, the impact of PCI on survival in patients with extensive-stage SCLC is unproven, and PCI likely has a role in a highly selected small group of patients with extensive-stage SCLC. PCI is not recommended for patients with poor performance status (ECOG performance score of 3 or 4) or underlying neurocognitive disorders.34,83
The NMDA-receptor antagonist memantine can be used in patients undergoing PCI to delay the occurrence of cognitive dysfunction.61 Memantine 20 mg daily delayed time to cognitive decline and reduced the rate of decline in memory, executive function, and processing speed compared to placebo in patients receiving whole brain radiotherapy.84
ROLE OF RADIOTHERAPY
A subset of patients with extensive-stage SCLC may benefit from consolidative thoracic radiotherapy after completion of platinum-based chemotherapy. A randomized trial that enrolled patients who achieved complete or near complete response after 3 cycles of cisplatin plus etoposide compared thoracic radiotherapy in combination with continued chemotherapy versus chemotherapy alone.85 The median OS was longer with the addition of thoracic radiotherapy compared to chemotherapy alone. Another phase 3 trial did not show improvement in 1-year OS with consolidative thoracic radiotherapy, but 2-year OS and 6-month PFS were longer.86 In general, consolidative thoracic radiotherapy benefits patients who have residual thoracic disease and low-bulk extrathoracic disease that has responded to systemic therapy.87 In addition, patients who initially presented with bulky symptomatic thoracic disease should also be considered for consolidative radiation.
Similar to other solid tumors, radiotherapy should be utilized for palliative purposes in patients with painful bone metastasis, spinal cord compression, or brain metastasis. Surgery is generally not recommended for spinal cord compression given the short life expectancy of patients with extensive-stage disease. Whole brain radiotherapy is preferred over stereotactic radiosurgery because micrometastasis is frequently present even in the setting of 1 or 2 radiographically evident brain metastasis.
NOVEL THERAPIES
The very complex genetic landscape of SCLC accounts for its resistance to conventional therapy and high recurrence rate; however, at the same time this complexity can form the basis for effective targeted therapy for the disease. One of the major factors hindering the development of targeted therapies in SCLC is limited availability of tissue due to small tissue samples and the frequent presence of significant necrosis in the samples. In recent years, several different therapeutic strategies and targeted agents have been investigated for their potential role in SCLC. Several of them, including EGFR tyrosine kinase inhibitors (TKIs), BCR-ABL TKIs, mTOR inhibitors, and VEGF inhibitors, have not been shown to provide a survival advantage in this disease. Several others, including PARP inhibitors, cellular developmental pathway inhibitors, and antibody-drug conjugates, are being tested. A phase 1 study of veliparib combined with cisplatin and etoposide in patients with previously untreated extensive-stage SCLC demonstrated a complete response in 14.3%, a partial response in 57.1%, and stable disease in 28.6% of patients with an acceptable safety profile.88 So far, none of these agents are approved for use in SCLC, and the majority are in early- phase clinical trials.89
One of the emerging targets in the treatment of SCLC is delta-like protein 3 (DLL3). DLL3 is expressed on more than 80% of SCLC tumor cells and cancer stem cells. Rovalpituzumab tesirine is an antibody-drug conjugate consisting of humanized anti-DLL3 monoclonal antibody linked to SC-DR002, a DNA-crosslinking agent. A phase 1 trial of rovalpituzumab in patients with relapsed SCLC after 1 or 2 prior lines of therapy reported a response rate of 31% in patients with DLL3 expression of ≥ 50%. The median duration of response and median PFS were both 4.6 months.90 Rovalpituzumab is currently in later phases of clinical trials and has a potential to serve as an option for patients with extensive-stage disease after disease progression on platinum-based therapy.
SUMMARY
Four to 6 cycles of carboplatin and etoposide remain the standard of care first-line treatment for patients with extensive stage SCLC. The only FDA-approved second-line treatment option is topotecan. Re-treatment with the original platinum doublet is a reasonable option for patients who have disease progression 6 months or longer after completion of platinum-based therapy. The immune checkpoint inhibitors pembrolizumab and combination nivolumab and ipilimumab have shown promising results in the second-line setting and beyond. The role of PCI has become more controversial in recent years, and periodic brain MRI in lieu of PCI is now an acceptable approach.
RESPONSE ASSESSMENT/SURVEILLANCE
For patients undergoing treatment for limited-stage SCLC, response assessment with contrast-enhanced CT of the chest/abdomen should be performed after completion of 4 cycles of chemotherapy and thoracic radiation.7 The surveillance guidelines consist of history, physical exam, and imaging every 3 months during the first 2 years, every 6 months during the third year, and annually thereafter. If PCI is not performed, brain MRI or contrast-enhanced CT scan should be performed every 3 or 4 months during the first 2 years of follow up. For extensive-stage disease, response assessment should be performed after every 2 cycles of therapy. After completion of therapy, history, physical exam, and imaging should be done every 2 months during the first year, every 3 or 4 months during years 2 and 3, every 6 months during years 4 and 5, and annually thereafter. Routine use of PET scan for surveillance is not recommended. Any new pulmonary nodule should prompt evaluation for a second primary lung malignancy. Finally, smoking cessation counseling is an integral part of management of any patient with SCLC and should be included with every clinic visit.
CONCLUSION
SCLC is a heterogeneous and genetically complex disease with a very high mortality rate. The current standard of care includes concurrent chemoradiation with cisplatin and etoposide for limited-stage SCLC and the combination of platinum and etoposide for extensive SCLC. A number of novel treatment approaches, including immune checkpoint inhibitors and antibody-drug conjugates, have had promising results in early clinical trials. Given the limited treatment options and large unmet need for new treatment options, enrollment in clinical trials is strongly recommended for patients with SCLC.
1. American Cancer Society. Cancer Facts & Figures 2017. American Cancer Society website. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2017/cancer-facts-and-figures-2017.pdf. Published 2017. Accessed July 11, 2018.
2. Govindan R, Page N, Morgensztern D, et al. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol 2006;24:4539–44.
3. Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2014. National Cancer Institute website. https://seer.cancer.gov/csr/1975_2014/. Updated April 2, 2018. Accessed July 11, 2018.
4. Varghese AM, Zakowski MF, Yu HA, et al. Small-cell lung cancers in patients who never smoked cigarettes. J Thorac Oncol 2014;9:892–6.
5. Pleasance ED, Stephens PJ, O’Meara S, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010;463:184–90.
6. Green RA, Humphrey E, Close H, Patno ME. Alkylating agents in bronchogenic carcinoma. Am J Med 1969;46:516–25.
7. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology for small cell lung cancer (Version 2.2018). www.nccn.org/professionals/physician_gls/pdf/sclc.pdf. Accessed August 12, 2018.
8. National Lung Screening Trial Research Team, Aberle DR, Adams AM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011;365:395–409.
9. Aberle DR, DeMello S, Berg CD, et al. Results of the two incidence screenings in the National Lung Screening Trial. N Engl J Med 2013;369:920–31.
10. Kitajima K, Nakamoto Y, Okizuka H, et al. Accuracy of whole-body FDG-PET/CT for detecting brain metastases from non-central nervous system tumors. Ann Nucl Med 2008;22:595–602.
11. Ordonez NG. Value of thyroid transcription factor-1 immunostaining in distinguishing small cell lung carcinomas from other small cell carcinomas. Am J Surg Pathol 2000;24:1217–23.
12. Karnak D, Beder S, Kayacan O, et al. Neuron-specific enolase and lung cancer. Am J Clin Oncol 2005;28:586–90.
13. Turrisi AT 3rd, Kim K, Blum R, et al. Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. N Engl J Med 1999;340:265–71.
14. Evans WK, Shepherd FA, Feld R, et al. VP-16 and cisplatin as first-line therapy for small-cell lung cancer. J Clin Oncol 1985;3:1471–7.
15. Pujol JL, Carestia L, Daurés JP. Is there a case for cisplatin in the treatment of small-cell lung cancer? A meta-analysis of randomized trials of a cisplatin-containing regimen versus a regimen without this alkylating agent. Br J Cancer 2000;83:8–15.
16. Mascaux C, Paesmans M, Berghmans T, et al; European Lung Cancer Working Party (ELCWP). A systematic review of the role of etoposide and cisplatin in the chemotherapy of small cell lung cancer with methodology assessment and meta-analysis. Lung Cancer 2000;30:23–36.
17. Sundstrøm S, Bremnes RM, Kaasa S, et al; Norwegian Lung Cancer Study Group. Cisplatin and etoposide regimen is superior to cyclophosphamide, epirubicin, and vincristine regimen in small-cell lung cancer: results from a randomized phase III trial with 5 years’ follow-up. J Clin Oncol 2002;20:4665–72.
18. Hatfield LA, Huskamp HA, Lamont EB. Survival and toxicity after cisplatin plus etoposide versus carboplatin plus etoposide for extensive-stage small-cell lung cancer in elderly patients. J Oncol Pract 2016;12:666–73.
19. Okamoto H, Watanabe K, Kunikane H, et al. Randomised phase III trial of carboplatin plus etoposide vs split doses of cisplatin plus etoposide in elderly or poor-risk patients with extensive disease small-cell lung cancer: JCOG 9702. Br J Cancer 2007;97:162–9.
20. Skarlos DV, Samantas E, Kosmidis P, et al. Randomized comparison of etoposide-cisplatin vs. etoposide-carboplatin and irradiation in small-cell lung cancer. A Hellenic Co-operative Oncology Group study. Ann Oncol 1994;5:601–7.
21. Rossi A, Di Maio M, Chiodini P, et al. Carboplatin- or cisplatin-based chemotherapy in first-line treatment of small-cell lung cancer: the COCIS meta-analysis of individual patient data J Clin Oncol 2012;30:1692–8.
22. Bishop JF, Raghavan D, Stuart-Harris R, et al. Carboplatin (CBDCA, JM-8) and VP-16-213 in previously untreated patients with small-cell lung cancer. J Clin Oncol 1987;5:1574–8.
23. Takada M, Fukuoka M, Kawahara M, et al. Phase III study of concurrent versus sequential thoracic radiotherapy in combination with cisplatin and etoposide for limited-stage small-cell lung cancer: results of the Japan Clinical Oncology Group Study 9104. J Clin Oncol 2002;20:3054–60.
24. Bunn PA Jr, Crowley J, Kelly K, et al. Chemoradiotherapy with or without granulocyte-macrophage colony-stimulating factor in the treatment of limited-stage small-cell lung cancer: a prospective phase III randomized study of the Southwest Oncology Group. J Clin Oncol 1995;13:1632–41.
25. Pignon JP, Arriagada R, Ihde DC, et al. A meta-analysis of thoracic radiotherapy for small-cell lung cancer. N Engl J Med 1992;327:1618–24.
26. Warde P, Payne D. Does thoracic irradiation improve survival and local control in limited-stage small-cell carcinoma of the lung? A meta-analysis. J Clin Oncol 1992;10:890–5.
27. Murray N, Coy P, Pater JL, et al. Importance of timing for thoracic irradiation in the combined modality treatment of limited-stage small-cell lung cancer. The National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 1993;11:336–44.
28. De Ruysscher D, Lueza B, Le Péchoux C, et al. Impact of thoracic radiotherapy timing in limited-stage small-cell lung cancer: usefulness of the individual patient data meta-analysis. Ann Oncol 2016;27:1818–28.
29. Faivre-Finn C, Snee M, Ashcroft L, et al. Concurrent once-daily versus twice-daily chemoradiotherapy in patients with limited-stage small-cell lung cancer (CONVERT): an open-label, phase 3, randomised, superiority trial. Lancet Oncol 2017;18:1116–25.
30. Aupérin A, Arriagada R, Pignon JP, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. Prophylactic Cranial Irradiation Overview Collaborative Group. N Engl J Med 1999;341:476–84.
31. Arriagada R, Le Chevalier T, Borie F, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. J Natl Cancer Inst 1995;87:183–90.
32. Le Péchoux C, Dunant A, Senan S, et al; Prophylactic Cranial Irradiation (PCI) Collaborative Group. Standard-dose versus higher-dose prophylactic cranial irradiation (PCI) in patients with limited-stage small-cell lung cancer in complete remission after chemotherapy and thoracic radiotherapy (PCI 99-01, EORTC 22003-08004, RTOG 0212, and IFCT 99-01): a randomised clinical trial. Lancet Oncol 2009;10:467–74.
33. Schneider BJ, Saxena A, Downey RJ. Surgery for early-stage small cell lung cancer. J Natl Compr Canc Netw 2011;9:1132–9.
34. Inoue M, Nakagawa K, Fujiwara K, et al. Results of preoperative mediastinoscopy for small cell lung cancer. Ann Thorac Surg 2000;70:1620–3.
35. Lim E, Belcher E, Yap YK, et al. The role of surgery in the treatment of limited disease small cell lung cancer: time to reevaluate. J Thorac Oncol 2008;3:1267–71.
36. Inoue M, Miyoshi S, Yasumitsu T, et al. Surgical results for small cell lung cancer based on the new TNM staging system. Thoracic Surgery Study Group of Osaka University, Osaka, Japan. Ann Thorac Surg 2000;70:1615–9.
37. Yang CF, Chan DY, Speicher PJ, et al. Role of adjuvant therapy in a population-based cohort of patients with early-stage small-cell lung cancer. J Clin Oncol 2016;34:1057–64.
38. Shepherd FA, Evans WK, Feld R, et al. Adjuvant chemotherapy following surgical resection for small-cell carcinoma of the lung. J Clin Oncol 1988;6:832–8.
39. Noda K, Nishiwaki Y, Kawahara M, et al; Japan Clinical Oncology Group. Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small-cell lung cancer. N Engl J Med 2002;346:85–91.
40. Lara PN Jr, Natale R, Crowley J, et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol 2009;27:2530–5.
41. Chute JP, Chen T, Feigal E, et al. Twenty years of phase III trials for patients with extensive-stage small-cell lung cancer: perceptible progress. J Clin Oncol 1999;17:1794–801.
42. Zhou H, Zeng C, Wei Y, et al. Duration of chemotherapy for small cell lung cancer: a meta-analysis. PloS One 2013;8:e73805.
43. Loehrer PJ Sr, Ansari R, Gonin R, et al. Cisplatin plus etoposide with and without ifosfamide in extensive small-cell lung cancer: a Hoosier Oncology Group study. J Clin Oncol;13:2594–9.
44. Pujol JL, Daurés JP, Riviére A, et al. Etoposide plus cisplatin with or without the combination of 4’-epidoxorubicin plus cyclophosphamide in treatment of extensive small-cell lung cancer: a French Federation of Cancer Institutes multicenter phase III randomized study. J Natl Cancer Inst 2001;93:300–8.
45. Berghmans T, Scherpereel A, Meert AP, et al; European Lung Cancer Working Party (ELCWP). A phase III randomized study comparing a chemotherapy with cisplatin and etoposide to a etoposide regimen without cisplatin for patients with extensive small-cell lung cancer. Front Oncol 2017;7:217.
46. Jalal SI, Lavin P, Lo G, et al. Carboplatin and etoposide with or without palifosfamide in untreated extensive-stage small-cell lung cancer: a Multicenter, Adaptive, Randomized Phase III Study (MATISSE). J Clin Oncol 2017;35:2619–23.
47. Fukuoka M, Furuse K, Saijo N, et al. Randomized trial of cyclophosphamide, doxorubicin, and vincristine versus cisplatin and etoposide versus alternation of these regimens in small-cell lung cancer. J Natl Cancer Inst 1991;83:855–61.
48. Roth BJ, Johnson DH, Einhorn LH, et al. Randomized study of cyclophosphamide, doxorubicin, and vincristine versus etoposide and cisplatin versus alternation of these two regimens in extensive small-cell lung cancer: a phase III trial of the Southeastern Cancer Study Group. J Clin Oncol 1992;10:282–91.
49. Miles DW, Earl HM, Souhami RL, et al. Intensive weekly chemotherapy for good-prognosis patients with small-cell lung cancer. J Clin Oncol 1991;9:280–5.
50. Petrioli R, Roviello G, Laera L, et al. Cisplatin, etoposide, and bevacizumab regimen followed by oral etoposide and bevacizumab maintenance treatment in patients with extensive-stage small cell lung cancer: a single-institution experience. Clin Lung Cancer 2015;16:e229–34.
51. Spigel DR, Greco FA, Zubkus JD, et al. Phase II trial of irinotecan, carboplatin, and bevacizumab in the treatment of patients with extensive-stage small-cell lung cancer. J Thorac Oncol 2009;4:1555–60.
52. Spigel DR, Townley PM, Waterhouse DM, et al. Randomized phase II study of bevacizumab in combination with chemotherapy in previously untreated extensive-stage small-cell lung cancer: results from the SALUTE trial. J Clin Oncol 2011;29:2215–22.
53. Horn L, Dahlberg SE, Sandler AB, et al. Phase II study of cisplatin plus etoposide and bevacizumab for previously untreated, extensive-stage small-cell lung cancer: Eastern Cooperative Oncology Group Study E3501. J Clin Oncol 2009;27:6006–11.
54. Tiseo M, Boni L, Ambrosio F, et al. Italian, multicenter, phase III, randomized study of cisplatin plus etoposide with or without bevacizumab as first-line treatment in extensive-disease small-cell lung cancer: the GOIRC-AIFA FARM6PMFJM trial. J Clin Oncol 2017;35:1281–7.
55. Pujol JL, Lavole A, Quoix E, et al. Randomized phase II-III study of bevacizumab in combination with chemotherapy in previously untreated extensive small-cell lung cancer: results from the IFCT-0802 trial. Ann Oncol 2015;26:908–14.
56. Gadgeel SM, Ventimiglia J, Kalemkerian GP, et al. Phase II study of maintenance pembrolizumab (pembro) in extensive stage small cell lung cancer (ES-SCLC) patients (pts) [abstract]. J Clin Oncol 2017;35(15_suppl):8504.
57. Reck M, Luft A, Szczesna A, et al. Phase III randomized trial of ipilimumab plus etoposide and platinum versus placebo plus etoposide and platinum in extensive-stage small-cell lung cancer. J Clin Oncol 2016;34:3740–8.
58. Chung HC, Lopez-Martin JA, Kao SC, et al. Phase 2 study of pembrolizumab in advanced small-cell lung cancer (SCLC): KEYNOTE-158 [abstract]. J Clin Oncol 2018;36(suppl):8506.
59. Owonikoko TK, Behera M, Chen Z, et al. A systematic analysis of efficacy of second-line chemotherapy in sensitive and refractory small-cell lung cancer. J Thorac Oncol 2012;7:866–72.
60. Postmus PE, Berendsen HH, van Zandwijk N, et al. Retreatment with the induction regimen in small cell lung cancer relapsing after an initial response to short term chemotherapy. Eur J Cancer Clin Oncol 1987;23:1409–11.
61. von Pawel J, Schiller JH, Shepherd FA, et al. Topotecan versus cyclophosphamide, doxorubicin, and vincristine for the treatment of recurrent small-cell lung cancer. J Clin Oncol 1999;17:658–667.
62. O’Brien ME, Ciuleanu TE, Tsekov H, et al. Phase III trial comparing supportive care alone with supportive care with oral topotecan in patients with relapsed small-cell lung cancer. J Clin Oncol 2006;24:5441–7.
63. Eckardt JR, von Pawel J, Pujol JL, et al. Phase III study of oral compared with intravenous topotecan as second-line therapy in small-cell lung cancer. J Clin Oncol 2007;25:2086–92.
64. Masuda N, Fukuoka M, Kusunoki Y, et al. CPT-11: a new derivative of camptothecin for the treatment of refractory or relapsed small-cell lung cancer. J Clin Oncol 1992;10:1225–9.
65. Smit EF, Fokkema E, Biesma B, et al. A phase II study of paclitaxel in heavily pretreated patients with small-cell lung cancer. Br J Cancer 1998;77:347–51.
66. Yamamoto N, Tsurutani J, Yoshimura N, et al. Phase II study of weekly paclitaxel for relapsed and refractory small cell lung cancer. Anticancer Res 2006;26:777–81.
67. Smyth JF, Smith IE, Sessa C, et al. Activity of docetaxel (Taxotere) in small cell lung cancer. Eur J Cancer 1994;30A:1058–60.
68. Pietanza MC, Kadota K, Huberman K, et al. Phase II trial of temozolomide with relapsed sensitive or refractory small cell lung cancer, with assessment of methylguanine-DNA methyltransferase as a potential biomarker. Clin Cancer Res 2012;18:1138–45.
69. Zauderer MG, Drilon A, Kadota K, et al. Trial of a 5-day dosing regimen of temozolomide in patients with relapsed small cell lung cancers with assessment of methylguanine-DNA methyltransferase. Lung Cancer 2014;86:237–40.
70. Jassem J, Karnicka-Mlodkowska H, van Pottelsberghe C, et al. Phase II study of vinorelbine (Navelbine) in previously treated small cell lung cancer patients. Eur J Cancer 1993;29A:1720–2.
71. Furuse K, Kuboa K, Kawahara M, et al. Phase II study of vinorelbine in heavily previously treated small cell lung cancer. Oncology 1996;53:169–72.
72. Einhorn LH, Pennington K, McClean J. Phase II trial of daily oral VP-16 in refractory small cell lung cancer. Semin Oncol 1990;17:32–5.
73. Johnson DH, Greco FA, Strupp J, et al. Prolonged administration of oral etoposide in patients with relapsed or refractory small-cell lung cancer: a phase II trial. J Clin Oncol 1990;8:1613–7.
74. Van der Lee I, Smit EF, van Putten JW, et al. Single-agent gemcitabine in patients with resistant small-cell lung cancer. Ann Oncol 2001;12:557–61.
75. Masters GA, Declerck L, Blanke C, et al. Phase II trial of gemcitabine in refractory or relapsed small-cell lung cancer. J Clin Oncol 2003;21:1550–5.
76. von Pawel J, Schiller JH, Shepherd FA, et al. Topotecan versus cyclophosphamide, doxorubicin, and vincristine for the treatment of recurrent small-cell lung cancer. J Clin Oncol 1999;17:658–67.
77. Lammers PE, Shyr Y, Li CI, et al. Phase II study of bendamustine in relapsed chemotherapy sensitive or resistant small-cell lung cancer. J Thorac Oncol 2014;9:559–62.
78. Hellmann MD, Ott PA, Zugazagoitia J, et al. Nivolumab (nivo) ± ipilimumab (ipi) in advanced small-cell lung cancer (SCLC): First report of a randomized expansion cohort from CheckMate 032 [abstract]. J Clin Oncol 2017;35(15_suppl):8503.
79. Antonia SJ, López-Martin JA, Bendell J, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol 2016;17:883–95.
80. Ott PA, Elez E, Hiret S, et al. Pembrolizumab in patients with extensive-stage small-cell lung cancer: results from the Phase Ib KEYNOTE-028 study. J Clin Oncol 2017;35:3823–9.
81. Slotman B, Faivre-Finn C, Kramer G, et al. Prophylactic cranial irradiation in extensive small-cell lung cancer. N Engl J Med 2007;357:664–72.
82. Takahashi T, Yamanaka T, Seto T, et al. Prophylactic cranial irradiation versus observation in patients with extensive-disease small-cell lung cancer: a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2017;18:663–71.
83. Slotman BJ, Mauer ME, Bottomley A, et al. Prophylactic cranial irradiation in extensive disease small-cell lung cancer: short-term health-related quality of life and patient reported symptoms: results of an international Phase III randomized controlled trial by the EORTC Radiation Oncology and Lung Cancer Groups. J Clin Oncol 2009;27:78–84.
84. Brown PD, Pugh S, Laack NN, et al; Radiation Therapy Oncology Group (RTOG). Memantine for the prevention of cognitive dysfunction in patients receiving whole-brain radiotherapy: a randomized, double-blind, placebo-controlled trial. Neuro Oncol 2013;15:1429–37.
85. Jeremic B, Shibamoto Y, Nikolic N, et al. Role of radiation therapy in the combined-modality treatment of patients with extensive disease small-cell lung cancer: a randomized study. J Clin Oncol 1999;17:2092–9.
86. Slotman BJ, van Tinteren H, Praag JO, et al. Use of thoracic radiotherapy for extensive stage small-cell lung cancer: a phase 3 randomised controlled trial. Lancet 2015;385:36–42.
87. Slotman BJ, van Tinteren H, Praag JO, et al. Radiotherapy for extensive stage small-cell lung cancer - authors’ reply. Lancet 2015;385:1292–3.
88. Owonikoko TK, Dahlberg SE, Khan SA, et al. A phase 1 safety study of veliparib combined with cisplatin and etoposide in extensive stage small cell lung cancer: A trial of the ECOG-ACRIN Cancer Research Group (E2511). Lung Cancer 2015;89:66–70.
89. Mamdani H, Induru R, Jalal SI. Novel therapies in small cell lung cancer. Transl Lung Cancer Res 2015;4:533–44.
90. Rudin CM, Pietanza MC, Bauer TM, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol 2017;18:42–51.
INTRODUCTION
Small cell lung cancer (SCLC) is an aggressive cancer of neuroendocrine origin that accounts for approximately 15% of all lung cancer cases, with approximately 33,000 patients diagnosed annually.1 The incidence of SCLC in the United States has steadily declined over the past 30 years, presumably because of a decrease in the number of smokers and a change to low-tar filter cigarettes.2 Although the overall incidence of SCLC has been decreasing, the incidence in women is increasing and the male-to-female incidence ratio is now 1:1.3 Nearly all cases of SCLC are associated with heavy tobacco exposure, making it a heterogeneous disease with a complex genomic landscape consisting of thousands of mutations.4,5 Despite recent advances in the treatment of non-small cell lung cancer, the therapeutic options for SCLC remain limited, with a median overall survival (OS) of 9 months in patients with advanced disease.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 61-year-old man presents to the emergency department with progressive shortness of breath and cough over the past 6 weeks. He also reports a 20-lb weight loss over the same period. He is a current smoker and has been smoking 1 pack of cigarettes per day since the age of 18 years. A chest radiograph obtained in the emergency department shows a right hilar mass. Computed tomography (CT) scan confirms the presence of a 4.5-cm right hilar mass and enlarged mediastinal lymph nodes bilaterally.
• What are the next steps in diagnosis?
SCLC is characterized by rapid growth and early hematogenous metastasis. Consequently, only 25% of patients have limited-stage disease at the time of diagnosis. According to the Veterans Administration Lung Study Group (VALSG) staging system, limited-stage disease is defined as tumor that is confined to 1 hemithorax and can be encompassed within 1 radiation field. This typically includes mediastinal lymph nodes and ipsilateral supraclavicular lymph nodes. Approximately 75% of patients present with extensive-stage disease, which is defined as disease that cannot be classified as limited, including disease that extends beyond 1 hemithorax. Extensive-stage disease includes the presence of malignant pleural effusion and/or distant metastasis.6 The VALSG classification and staging system is more commonly used in clinical practice than the American Joint Committee on Cancer TNM staging system because it is less complex and directs treatment decisions, as most of the literature on SCLC classifies patients based on the VALSG system.7
Given SCLC’s propensity to metastasize quickly, none of the currently available screening methods have proven successful in early detection of SCLC. In the National Lung Cancer Screening Trial, 86% of the 125 patients who were diagnosed with SCLC while undergoing annual low-dose chest CT scans had advanced disease at diagnosis.8,9 These results highlight the fact that most cases of SCLC develop in the interval between annual screening imaging.
SCLC frequently presents with a large hilar mass that is symptomatic. Common symptoms include shortness of breath and cough. In addition, patients with SCLC usually have bulky mediastinal adenopathy at presentation. SCLC is commonly located submucosally in the bronchus, and therefore hemoptysis is not a very common symptom at the time of presentation. Patients may present with superior vena cava syndrome from local compression by the tumor. Not infrequently, SCLC is associated with paraneoplastic syndromes that arise due to ectopic secretion of hormones or antibodies by the tumor cells. The paraneoplastic syndromes can be broadly categorized as endocrine or neurologic (Table 1). The presence of a paraneoplastic syndrome is often a clue to the potential diagnosis of SCLC in the presence of a hilar mass. Additionally, some paraneoplastic syndromes, more specifically endocrine paraneoplastic syndromes, follow the pattern of disease response and relapse, and therefore can sometimes serve as an early marker of disease relapse or progression.
The common sites of metastases include brain, liver, and bone. Therefore, the staging workup should include fluorodeoxyglucose (FDG) positron emission tomography (PET)/CT scan. Contrast-enhanced CT scan of the chest and abdomen and bone scan can be obtained for staging in lieu of PET scan. Due to the physiologic FDG uptake, cerebral metastases cannot be assessed with sufficient certainty using PET-CT.10 Therefore, brain imaging with contrast-enhanced CT or magnetic resonance imaging (MRI) is also necessary. Although the incidence of metastasis to bone marrow is less than 10%, bone marrow aspiration and biopsy are warranted in patients with unexplained cytopenias, especially when the cytopenia is associated with teardrop-shaped red cells or nucleated red cells on peripheral blood smear, findings indicative of a marrow infiltrative process.7 The tissue diagnosis is established by obtaining a biopsy of the primary tumor or 1 of the metastatic sites. In localized disease, bronchoscopy (with endobronchial ultrasound, if necessary) with biopsy of the centrally located tumor and/or lymph node is required. Histologically, SCLC consists of monomorphic cells, a high nuclear-cytoplasmic ratio, and confluent necrosis. The tumor cells are positive for chromogranin, synaptophysin, and CD56 by immunohistochemistry, and very frequently are also positive for thyroid transcription factor 1.11 Although serum tumor markers, including neuron-specific enolase and progastrin-releasing peptide, are frequently elevated in patients with SCLC, these markers are of limited value in clinical practice because they lack sensitivity and specificity.12
MANAGEMENT OF LIMITED-STAGE DISEASE
CASE CONTINUED
The patient undergoes FDG PET scan, which shows the presence of a hypermetabolic right hilar mass in addition to enlarged and hypermetabolic bilateral mediastinal lymph nodes. There are no other areas of FDG avidity. Brain MRI does not show any evidence of brain metastasis. Thus, the patient is confirmed to have limited-stage SCLC.
• What is the standard of care treatment for limited-stage SCLC?
SCLC is exquisitely sensitive to both chemotherapy and radiation, especially at the time of initial presentation. The standard of care treatment of limited-stage SCLC is 4 cycles of platinum-based chemotherapy in combination with thoracic radiation started within the first 2 cycles of chemotherapy (Figure 1).
CHOICE OF CHEMOTHERAPY
Etoposide and cisplatin is the most commonly used initial combination chemotherapy regimen in limited-stage SCLC.14 This combination has largely replaced anthracycline-based regimens given its favorable efficacy and toxicity profile.15–17 Several small randomized trials have shown comparable efficacy of carboplatin and etoposide in extensive-stage SCLC.18–20 A meta-analysis of 4 randomized trials comparing cisplatin-based versus carboplatin-based regimens in 663 patients with SCLC (32% had limited-stage disease and 68% had extensive-stage disease) showed no statistically significant difference in response rate, progression-free survival (PFS), or OS between the 2 regimens.21 Therefore, in clinical practice carboplatin is frequently used instead of cisplatin in patients with extensive-stage disease. In patients with limited-stage disease, cisplatin is still the drug of choice. However, the toxicity profile of the 2 regimens is different. Cisplatin-based regimens are more commonly associated with neuropathy, nephrotoxicity, and chemotherapy-induced nausea/vomiting,18 while carboplatin-based regimens are more myelosuppressive.22 In addition, the combination of thoracic radiation with either of these regimens is associated with a higher risk of esophagitis, pneumonitis, and myelosuppression.23 The use of myeloid growth factors is not recommended in patients undergoing concurrent chemoradiation.24 Of note, intravenous etoposide is always preferred over oral etoposide, especially in the curative setting given the unreliable absorption and bioavailability of oral formulations.
THORACIC RADIOTHERAPY
Adding thoracic radiotherapy to platinum-etoposide chemotherapy improves local control and OS. Two meta-analyses of 13 trials including more than 2000 patients have shown a 25% to 30% decrease in local failure and a 5% to 7% increase in 2-year OS with chemoradiation compared to chemotherapy alone in limited-stage SCLC.25,26 Early (within the first 2 cycles) concurrent thoracic radiation is superior to delayed and/or sequential radiation in terms of local control and OS.23,27,28 The dose and fractionation of thoracic radiation in limited-stage SCLC has remained a controversial issue. The Eastern Cooperative Oncology Group/Radiation Therapy Oncology Group randomized trial compared 45 Gy of radiotherapy delivered twice daily over a period of 3 weeks to 45 Gy once daily over 5 weeks concurrently with chemotherapy. The twice daily regimen led to a 10% improvement in 5-year OS (26% versus 16%), but a higher incidence of grade 3 and 4 adverse events.13 Despite the survival advantage demonstrated by hyperfractionated radiotherapy, the results need to be interpreted with caution because the radiation doses are not biologically equivalent. In addition, the difficult logistics of patients receiving radiation twice a day has limited the routine implementation of this strategy. Subsequently, another randomized phase 3 trial (CONVERT) compared 45 Gy radiotherapy twice daily with 66 Gy radiotherapy once daily in limited-stage SCLC.29 This trial did not show any difference in OS. The patients in the twice daily arm had a higher incidence of grade 4 neutropenia. Considering the results of these trials, both strategies—45 Gy fractionated twice daily or 60 Gy fractionated once daily, delivered concurrently with chemotherapy—are acceptable in the setting of limited-stage SCLC. However, quite often a hyperfractionated regimen is not feasible for patients and many radiation oncology centers. Hopefully, the ongoing CALGB 30610 study will clarify the optimal radiation schedule for limited-stage disease.
PROPHYLACTIC CRANIAL IRRADIATION
Approximately 75% of patients with limited-stage disease experience disease recurrence, and brain is the site of recurrence in approximately half of these patients.30 Prophylactic cranial irradiation (PCI) consisting of 25 Gy radiotherapy delivered in 10 fractions has been shown to be effective in decreasing the incidence of cerebral metastases.30–32 Although individual small studies have not shown a survival benefit of PCI because of small sample size and limited power, a meta-analysis of these studies has shown a 25% decrease in the 3-year incidence of brain metastasis and 5.4% increase in 3-year OS.30 Most patients included in these studies had limited-stage disease. Therefore, PCI is the standard of care for patients with limited-stage disease who attain a partial or complete response to chemoradiation.
ROLE OF SURGERY
Surgical resection may be an acceptable choice in a very limited subset of patients with peripherally located small (< 5 cm) tumors where mediastinal lymph nodes have been confirmed to be uninvolved with complete mediastinal staging.33,34 Most of the data in this setting are derived from retrospective studies.35,36 A 5-year OS between 40% and 60% has been reported with this strategy in patients with clinical stage I disease. In general, when surgery is considered, lobectomy with mediastinal lymph node dissection followed by chemotherapy (if there is no nodal involvement) or chemoradiation (if nodal involvement) is recommended.37,38 Wedge or segmental resections are not considered to be optimal surgical options.
MANAGEMENT OF EXTENSIVE-STAGE DISEASE
CASE CONTINUED
The patient receives 4 cycles of cisplatin and etoposide along with 70 Gy radiotherapy concurrently with the first 2 cycles of chemotherapy. His post-treatment CT scans show a partial response. He undergoes PCI 6 weeks after completion of treatment. At routine follow-up 18 months later, he is doing generally well except for mildly decreased appetite and an unintentional weight loss of 5 lb. CT scans demonstrate multiple hypodense liver lesions ranging from 7 mm to 2 cm in size and a 2-cm left adrenal gland lesion highly concerning for metastasis. FDG PET scan confirms that the adrenal and liver lesions are hypermetabolic. In addition, the PET scan shows multiple FDG-avid bone lesions throughout the spine. Brain MRI is negative for brain metastasis.
• What is the standard of care for treatment of extensive-stage disease?
Chemotherapy is the mainstay of treatment for extensive-stage SCLC; the goals of treatment are prolongation of survival, prevention or alleviation of cancer-related symptoms, and improvement in quality of life. The combination of etoposide with a platinum agent (carboplatin or cisplatin) is the preferred first-line treatment option. Carboplatin is more commonly used in clinical practice in this setting because of its comparable efficacy and better tolerability compared to cisplatin (Figure 2).21 A Japanese phase 3 trial comparing cisplatin plus irinotecan with cisplatin plus etoposide in the first-line setting in extensive-stage SCLC showed improvement in median and 2-year OS with the cisplatin/irinotecan regimen; however, 2 subsequent phase 3 trials conducted in the United States comparing these 2 regimens did not show any difference in OS. In addition, the cisplatin/irinotecan regimen was more toxic than the etoposide-based regimen.39,40 Therefore, 4 to 6 cycles of platinum/etoposide remains the standard of care first-line treatment for extensive-stage SCLC in the United States. The combination yields a 60% to 70% response rate, but the majority of patients invariably experience disease progression, with a median OS of 9 to 11 months.41 Maintenance chemotherapy beyond the initial 4 to 6 cycles does not improve survival and is associated with higher cumulative toxicity.42
Multiple attempts at improving first-line chemotherapy in extensive-stage disease have failed to show any meaningful difference in OS. For example, the addition of ifosfamide, palifosfamide, cyclophosphamide, taxane, or anthracycline to platinum doublet failed to show improvement in OS and led to more toxicity.43–46 Additionally, the use of alternating or cyclic chemotherapies in an attempt to curb drug resistance has also failed to show survival benefit.47–49 The addition of the antiangiogenic agent bevacizumab to standard platinum-based doublet has not prolonged OS in SCLC and has led to an unacceptably higher rate of tracheoesophageal fistula when used in conjunction with chemoradiation in limited-stage disease.50–55 Finally, the immune checkpoint inhibitor ipilimumab in combination with platinum plus etoposide failed to improve PFS or OS compared to platinum plus etoposide alone in a recent phase 3 trial, and maintenance pembrolizumab after completion of platinum-based chemotherapy did not improve PFS.56,57
More recently, a phase 2 study of pembrolizumab in extensive-stage SCLC (KEYNOTE 158) reported an overall response rate of 35.7%, median PFS of 2.1 months, and median OS of 14.6 months in patients who tested positive for programmed death ligand-1 (PD-L1) expression (which was defined as a PD-L1 Combined Positive Score ≥ 1).58 The median duration of response has not been reached in this study, indicating that pembrolizumab may be a promising approach in patients with extensive-stage SCLC, especially for those with PD-L1–positive tumors.
Patients with extensive-stage disease who have brain metastasis at the time of diagnosis can be treated with systemic chemotherapy first if the brain metastases are asymptomatic and there is significant extracranial disease burden. In that case, whole brain radiotherapy should be given after completion of systemic therapy.
SECOND-LINE CHEMOTHERAPY
Despite being exquisitely chemosensitive, SCLC is associated with a very poor prognosis largely because of invariable disease progression following first-line therapy and lack of effective second-line treatment options that can lead to appreciable disease control. The choice of second-line treatment is predominantly determined by the time of disease relapse after first-line platinum-based therapy. If this interval is 6 months or longer, re-treatment utilizing the same platinum doublet is appropriate. However, if the interval is 6 months or less, second-line systemic therapy options should be explored. Unfortunately, the response rate tends to be less than 10% with most of the second-line therapies in platinum-resistant disease (defined as disease progression within 3 months of receiving platinum-based therapy). If disease progression occurs between 3 and 6 months after completion of platinum-based therapy, the response rate with second-line chemotherapy is in the range of 25%.59,60
A number of second-line chemotherapy options have been explored in small studies, including topotecan, irinotecan, paclitaxel, docetaxel, temozolomide, vinorelbine, oral etoposide, gemcitabine, bendamustine, and CAV (
IMMUNOTHERAPY
The role of immune checkpoint inhibitors in the treatment of SCLC is evolving, and currently there are no FDA-approved immunotherapy agents for treating SCLC. A recently conducted phase 1/2 trial (CheckMate 032) studied the anti-programmed death(PD)-1 antibody nivolumab with or without the anti-cytotoxic T-lymphocyte–associated antigen (CTLA) -4 antibody ipilimumab in patients with relapsed SCLC. The authors reported response rates of 10% with nivolumab 3 mg/kg and 21% with nivolumab 1 mg/kg plus ipilimumab 3 mg/kg.78,79 The 2-year OS was 26% with the combination and 14% with single-agent nivolumab. Only 18% of patients had PD-L1 expression of ≥ 1%, and the response rate did not correlate with PD-L1 status. The rate of grade 3 or 4 adverse events was approximately 20%, and only 10% of patients discontinued treatment because of toxicity. Based on these data, nivolumab plus ipilimumab is now included in the National Comprehensive Cancer Network guidelines as an option for patients with SCLC who experience disease relapse within 6 months of receiving platinum-based therapy;7 however, it is questionable whether routine use of this combination is justified based on currently available data. The evidence for the combination of nivolumab and ipilimumab remains limited. The efficacy and toxicity data from both randomized and nonrandomized cohorts were presented together, making it hard to interpret the results.
Another phase 1b study (KEYNOTE-028) evaluated the anti-PD-1 antibody pembrolizumab (10 mg/kg intravenously every 2 weeks) in patients with relapsed SCLC who had received 1 or more prior lines of therapy and had PD-L1 expression of ≥ 1%. This study showed a response rate of 33%, with a median duration of response of 19 months and 1-year OS of 38%.80 Although only 28% of screened patients had PD-L1 expression of ≥ 1%, these results indicated that at least a subset of SCLC patients are able to achieve durable responses with immune checkpoint inhibition. A number of clinical trials utilizing immune checkpoint inhibitors in various combinations and settings are currently underway.
ROLE OF PROPHYLACTIC CRANIAL IRRADIATION
The role of PCI in extensive-stage SCLC is not clearly defined. A randomized phase 3 trial conducted by the European Organization for Research and Treatment of Cancer (EORTC) comparing PCI with no PCI in patients with extensive-stage SCLC who had a partial or complete response to initial platinum-based chemotherapy showed a decrease in the incidence of symptomatic brain metastasis and improvement in 1-year OS with PCI.81 However, this trial did not require mandatory brain imaging prior to PCI, and thus it is unclear if some patients in the PCI group had asymptomatic brain metastasis prior to enrollment and therefore received therapeutic benefit from brain radiation. Additionally, the dose and fractionation of PCI was not standardized across patient groups.
A more recent phase 3 study conducted in Japan that compared PCI (25 Gy in 10 fractions) with no PCI reported no difference in survival between the 2 groups.82 As opposed to the EORTC study, the Japanese study did require baseline brain imaging to confirm the absence of brain metastasis prior to enrollment. In addition, the control patients underwent periodic brain MRI to allow early detection of brain metastasis. Given the emergence of the new data, the impact of PCI on survival in patients with extensive-stage SCLC is unproven, and PCI likely has a role in a highly selected small group of patients with extensive-stage SCLC. PCI is not recommended for patients with poor performance status (ECOG performance score of 3 or 4) or underlying neurocognitive disorders.34,83
The NMDA-receptor antagonist memantine can be used in patients undergoing PCI to delay the occurrence of cognitive dysfunction.61 Memantine 20 mg daily delayed time to cognitive decline and reduced the rate of decline in memory, executive function, and processing speed compared to placebo in patients receiving whole brain radiotherapy.84
ROLE OF RADIOTHERAPY
A subset of patients with extensive-stage SCLC may benefit from consolidative thoracic radiotherapy after completion of platinum-based chemotherapy. A randomized trial that enrolled patients who achieved complete or near complete response after 3 cycles of cisplatin plus etoposide compared thoracic radiotherapy in combination with continued chemotherapy versus chemotherapy alone.85 The median OS was longer with the addition of thoracic radiotherapy compared to chemotherapy alone. Another phase 3 trial did not show improvement in 1-year OS with consolidative thoracic radiotherapy, but 2-year OS and 6-month PFS were longer.86 In general, consolidative thoracic radiotherapy benefits patients who have residual thoracic disease and low-bulk extrathoracic disease that has responded to systemic therapy.87 In addition, patients who initially presented with bulky symptomatic thoracic disease should also be considered for consolidative radiation.
Similar to other solid tumors, radiotherapy should be utilized for palliative purposes in patients with painful bone metastasis, spinal cord compression, or brain metastasis. Surgery is generally not recommended for spinal cord compression given the short life expectancy of patients with extensive-stage disease. Whole brain radiotherapy is preferred over stereotactic radiosurgery because micrometastasis is frequently present even in the setting of 1 or 2 radiographically evident brain metastasis.
NOVEL THERAPIES
The very complex genetic landscape of SCLC accounts for its resistance to conventional therapy and high recurrence rate; however, at the same time this complexity can form the basis for effective targeted therapy for the disease. One of the major factors hindering the development of targeted therapies in SCLC is limited availability of tissue due to small tissue samples and the frequent presence of significant necrosis in the samples. In recent years, several different therapeutic strategies and targeted agents have been investigated for their potential role in SCLC. Several of them, including EGFR tyrosine kinase inhibitors (TKIs), BCR-ABL TKIs, mTOR inhibitors, and VEGF inhibitors, have not been shown to provide a survival advantage in this disease. Several others, including PARP inhibitors, cellular developmental pathway inhibitors, and antibody-drug conjugates, are being tested. A phase 1 study of veliparib combined with cisplatin and etoposide in patients with previously untreated extensive-stage SCLC demonstrated a complete response in 14.3%, a partial response in 57.1%, and stable disease in 28.6% of patients with an acceptable safety profile.88 So far, none of these agents are approved for use in SCLC, and the majority are in early- phase clinical trials.89
One of the emerging targets in the treatment of SCLC is delta-like protein 3 (DLL3). DLL3 is expressed on more than 80% of SCLC tumor cells and cancer stem cells. Rovalpituzumab tesirine is an antibody-drug conjugate consisting of humanized anti-DLL3 monoclonal antibody linked to SC-DR002, a DNA-crosslinking agent. A phase 1 trial of rovalpituzumab in patients with relapsed SCLC after 1 or 2 prior lines of therapy reported a response rate of 31% in patients with DLL3 expression of ≥ 50%. The median duration of response and median PFS were both 4.6 months.90 Rovalpituzumab is currently in later phases of clinical trials and has a potential to serve as an option for patients with extensive-stage disease after disease progression on platinum-based therapy.
SUMMARY
Four to 6 cycles of carboplatin and etoposide remain the standard of care first-line treatment for patients with extensive stage SCLC. The only FDA-approved second-line treatment option is topotecan. Re-treatment with the original platinum doublet is a reasonable option for patients who have disease progression 6 months or longer after completion of platinum-based therapy. The immune checkpoint inhibitors pembrolizumab and combination nivolumab and ipilimumab have shown promising results in the second-line setting and beyond. The role of PCI has become more controversial in recent years, and periodic brain MRI in lieu of PCI is now an acceptable approach.
RESPONSE ASSESSMENT/SURVEILLANCE
For patients undergoing treatment for limited-stage SCLC, response assessment with contrast-enhanced CT of the chest/abdomen should be performed after completion of 4 cycles of chemotherapy and thoracic radiation.7 The surveillance guidelines consist of history, physical exam, and imaging every 3 months during the first 2 years, every 6 months during the third year, and annually thereafter. If PCI is not performed, brain MRI or contrast-enhanced CT scan should be performed every 3 or 4 months during the first 2 years of follow up. For extensive-stage disease, response assessment should be performed after every 2 cycles of therapy. After completion of therapy, history, physical exam, and imaging should be done every 2 months during the first year, every 3 or 4 months during years 2 and 3, every 6 months during years 4 and 5, and annually thereafter. Routine use of PET scan for surveillance is not recommended. Any new pulmonary nodule should prompt evaluation for a second primary lung malignancy. Finally, smoking cessation counseling is an integral part of management of any patient with SCLC and should be included with every clinic visit.
CONCLUSION
SCLC is a heterogeneous and genetically complex disease with a very high mortality rate. The current standard of care includes concurrent chemoradiation with cisplatin and etoposide for limited-stage SCLC and the combination of platinum and etoposide for extensive SCLC. A number of novel treatment approaches, including immune checkpoint inhibitors and antibody-drug conjugates, have had promising results in early clinical trials. Given the limited treatment options and large unmet need for new treatment options, enrollment in clinical trials is strongly recommended for patients with SCLC.
INTRODUCTION
Small cell lung cancer (SCLC) is an aggressive cancer of neuroendocrine origin that accounts for approximately 15% of all lung cancer cases, with approximately 33,000 patients diagnosed annually.1 The incidence of SCLC in the United States has steadily declined over the past 30 years, presumably because of a decrease in the number of smokers and a change to low-tar filter cigarettes.2 Although the overall incidence of SCLC has been decreasing, the incidence in women is increasing and the male-to-female incidence ratio is now 1:1.3 Nearly all cases of SCLC are associated with heavy tobacco exposure, making it a heterogeneous disease with a complex genomic landscape consisting of thousands of mutations.4,5 Despite recent advances in the treatment of non-small cell lung cancer, the therapeutic options for SCLC remain limited, with a median overall survival (OS) of 9 months in patients with advanced disease.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 61-year-old man presents to the emergency department with progressive shortness of breath and cough over the past 6 weeks. He also reports a 20-lb weight loss over the same period. He is a current smoker and has been smoking 1 pack of cigarettes per day since the age of 18 years. A chest radiograph obtained in the emergency department shows a right hilar mass. Computed tomography (CT) scan confirms the presence of a 4.5-cm right hilar mass and enlarged mediastinal lymph nodes bilaterally.
• What are the next steps in diagnosis?
SCLC is characterized by rapid growth and early hematogenous metastasis. Consequently, only 25% of patients have limited-stage disease at the time of diagnosis. According to the Veterans Administration Lung Study Group (VALSG) staging system, limited-stage disease is defined as tumor that is confined to 1 hemithorax and can be encompassed within 1 radiation field. This typically includes mediastinal lymph nodes and ipsilateral supraclavicular lymph nodes. Approximately 75% of patients present with extensive-stage disease, which is defined as disease that cannot be classified as limited, including disease that extends beyond 1 hemithorax. Extensive-stage disease includes the presence of malignant pleural effusion and/or distant metastasis.6 The VALSG classification and staging system is more commonly used in clinical practice than the American Joint Committee on Cancer TNM staging system because it is less complex and directs treatment decisions, as most of the literature on SCLC classifies patients based on the VALSG system.7
Given SCLC’s propensity to metastasize quickly, none of the currently available screening methods have proven successful in early detection of SCLC. In the National Lung Cancer Screening Trial, 86% of the 125 patients who were diagnosed with SCLC while undergoing annual low-dose chest CT scans had advanced disease at diagnosis.8,9 These results highlight the fact that most cases of SCLC develop in the interval between annual screening imaging.
SCLC frequently presents with a large hilar mass that is symptomatic. Common symptoms include shortness of breath and cough. In addition, patients with SCLC usually have bulky mediastinal adenopathy at presentation. SCLC is commonly located submucosally in the bronchus, and therefore hemoptysis is not a very common symptom at the time of presentation. Patients may present with superior vena cava syndrome from local compression by the tumor. Not infrequently, SCLC is associated with paraneoplastic syndromes that arise due to ectopic secretion of hormones or antibodies by the tumor cells. The paraneoplastic syndromes can be broadly categorized as endocrine or neurologic (Table 1). The presence of a paraneoplastic syndrome is often a clue to the potential diagnosis of SCLC in the presence of a hilar mass. Additionally, some paraneoplastic syndromes, more specifically endocrine paraneoplastic syndromes, follow the pattern of disease response and relapse, and therefore can sometimes serve as an early marker of disease relapse or progression.
The common sites of metastases include brain, liver, and bone. Therefore, the staging workup should include fluorodeoxyglucose (FDG) positron emission tomography (PET)/CT scan. Contrast-enhanced CT scan of the chest and abdomen and bone scan can be obtained for staging in lieu of PET scan. Due to the physiologic FDG uptake, cerebral metastases cannot be assessed with sufficient certainty using PET-CT.10 Therefore, brain imaging with contrast-enhanced CT or magnetic resonance imaging (MRI) is also necessary. Although the incidence of metastasis to bone marrow is less than 10%, bone marrow aspiration and biopsy are warranted in patients with unexplained cytopenias, especially when the cytopenia is associated with teardrop-shaped red cells or nucleated red cells on peripheral blood smear, findings indicative of a marrow infiltrative process.7 The tissue diagnosis is established by obtaining a biopsy of the primary tumor or 1 of the metastatic sites. In localized disease, bronchoscopy (with endobronchial ultrasound, if necessary) with biopsy of the centrally located tumor and/or lymph node is required. Histologically, SCLC consists of monomorphic cells, a high nuclear-cytoplasmic ratio, and confluent necrosis. The tumor cells are positive for chromogranin, synaptophysin, and CD56 by immunohistochemistry, and very frequently are also positive for thyroid transcription factor 1.11 Although serum tumor markers, including neuron-specific enolase and progastrin-releasing peptide, are frequently elevated in patients with SCLC, these markers are of limited value in clinical practice because they lack sensitivity and specificity.12
MANAGEMENT OF LIMITED-STAGE DISEASE
CASE CONTINUED
The patient undergoes FDG PET scan, which shows the presence of a hypermetabolic right hilar mass in addition to enlarged and hypermetabolic bilateral mediastinal lymph nodes. There are no other areas of FDG avidity. Brain MRI does not show any evidence of brain metastasis. Thus, the patient is confirmed to have limited-stage SCLC.
• What is the standard of care treatment for limited-stage SCLC?
SCLC is exquisitely sensitive to both chemotherapy and radiation, especially at the time of initial presentation. The standard of care treatment of limited-stage SCLC is 4 cycles of platinum-based chemotherapy in combination with thoracic radiation started within the first 2 cycles of chemotherapy (Figure 1).
CHOICE OF CHEMOTHERAPY
Etoposide and cisplatin is the most commonly used initial combination chemotherapy regimen in limited-stage SCLC.14 This combination has largely replaced anthracycline-based regimens given its favorable efficacy and toxicity profile.15–17 Several small randomized trials have shown comparable efficacy of carboplatin and etoposide in extensive-stage SCLC.18–20 A meta-analysis of 4 randomized trials comparing cisplatin-based versus carboplatin-based regimens in 663 patients with SCLC (32% had limited-stage disease and 68% had extensive-stage disease) showed no statistically significant difference in response rate, progression-free survival (PFS), or OS between the 2 regimens.21 Therefore, in clinical practice carboplatin is frequently used instead of cisplatin in patients with extensive-stage disease. In patients with limited-stage disease, cisplatin is still the drug of choice. However, the toxicity profile of the 2 regimens is different. Cisplatin-based regimens are more commonly associated with neuropathy, nephrotoxicity, and chemotherapy-induced nausea/vomiting,18 while carboplatin-based regimens are more myelosuppressive.22 In addition, the combination of thoracic radiation with either of these regimens is associated with a higher risk of esophagitis, pneumonitis, and myelosuppression.23 The use of myeloid growth factors is not recommended in patients undergoing concurrent chemoradiation.24 Of note, intravenous etoposide is always preferred over oral etoposide, especially in the curative setting given the unreliable absorption and bioavailability of oral formulations.
THORACIC RADIOTHERAPY
Adding thoracic radiotherapy to platinum-etoposide chemotherapy improves local control and OS. Two meta-analyses of 13 trials including more than 2000 patients have shown a 25% to 30% decrease in local failure and a 5% to 7% increase in 2-year OS with chemoradiation compared to chemotherapy alone in limited-stage SCLC.25,26 Early (within the first 2 cycles) concurrent thoracic radiation is superior to delayed and/or sequential radiation in terms of local control and OS.23,27,28 The dose and fractionation of thoracic radiation in limited-stage SCLC has remained a controversial issue. The Eastern Cooperative Oncology Group/Radiation Therapy Oncology Group randomized trial compared 45 Gy of radiotherapy delivered twice daily over a period of 3 weeks to 45 Gy once daily over 5 weeks concurrently with chemotherapy. The twice daily regimen led to a 10% improvement in 5-year OS (26% versus 16%), but a higher incidence of grade 3 and 4 adverse events.13 Despite the survival advantage demonstrated by hyperfractionated radiotherapy, the results need to be interpreted with caution because the radiation doses are not biologically equivalent. In addition, the difficult logistics of patients receiving radiation twice a day has limited the routine implementation of this strategy. Subsequently, another randomized phase 3 trial (CONVERT) compared 45 Gy radiotherapy twice daily with 66 Gy radiotherapy once daily in limited-stage SCLC.29 This trial did not show any difference in OS. The patients in the twice daily arm had a higher incidence of grade 4 neutropenia. Considering the results of these trials, both strategies—45 Gy fractionated twice daily or 60 Gy fractionated once daily, delivered concurrently with chemotherapy—are acceptable in the setting of limited-stage SCLC. However, quite often a hyperfractionated regimen is not feasible for patients and many radiation oncology centers. Hopefully, the ongoing CALGB 30610 study will clarify the optimal radiation schedule for limited-stage disease.
PROPHYLACTIC CRANIAL IRRADIATION
Approximately 75% of patients with limited-stage disease experience disease recurrence, and brain is the site of recurrence in approximately half of these patients.30 Prophylactic cranial irradiation (PCI) consisting of 25 Gy radiotherapy delivered in 10 fractions has been shown to be effective in decreasing the incidence of cerebral metastases.30–32 Although individual small studies have not shown a survival benefit of PCI because of small sample size and limited power, a meta-analysis of these studies has shown a 25% decrease in the 3-year incidence of brain metastasis and 5.4% increase in 3-year OS.30 Most patients included in these studies had limited-stage disease. Therefore, PCI is the standard of care for patients with limited-stage disease who attain a partial or complete response to chemoradiation.
ROLE OF SURGERY
Surgical resection may be an acceptable choice in a very limited subset of patients with peripherally located small (< 5 cm) tumors where mediastinal lymph nodes have been confirmed to be uninvolved with complete mediastinal staging.33,34 Most of the data in this setting are derived from retrospective studies.35,36 A 5-year OS between 40% and 60% has been reported with this strategy in patients with clinical stage I disease. In general, when surgery is considered, lobectomy with mediastinal lymph node dissection followed by chemotherapy (if there is no nodal involvement) or chemoradiation (if nodal involvement) is recommended.37,38 Wedge or segmental resections are not considered to be optimal surgical options.
MANAGEMENT OF EXTENSIVE-STAGE DISEASE
CASE CONTINUED
The patient receives 4 cycles of cisplatin and etoposide along with 70 Gy radiotherapy concurrently with the first 2 cycles of chemotherapy. His post-treatment CT scans show a partial response. He undergoes PCI 6 weeks after completion of treatment. At routine follow-up 18 months later, he is doing generally well except for mildly decreased appetite and an unintentional weight loss of 5 lb. CT scans demonstrate multiple hypodense liver lesions ranging from 7 mm to 2 cm in size and a 2-cm left adrenal gland lesion highly concerning for metastasis. FDG PET scan confirms that the adrenal and liver lesions are hypermetabolic. In addition, the PET scan shows multiple FDG-avid bone lesions throughout the spine. Brain MRI is negative for brain metastasis.
• What is the standard of care for treatment of extensive-stage disease?
Chemotherapy is the mainstay of treatment for extensive-stage SCLC; the goals of treatment are prolongation of survival, prevention or alleviation of cancer-related symptoms, and improvement in quality of life. The combination of etoposide with a platinum agent (carboplatin or cisplatin) is the preferred first-line treatment option. Carboplatin is more commonly used in clinical practice in this setting because of its comparable efficacy and better tolerability compared to cisplatin (Figure 2).21 A Japanese phase 3 trial comparing cisplatin plus irinotecan with cisplatin plus etoposide in the first-line setting in extensive-stage SCLC showed improvement in median and 2-year OS with the cisplatin/irinotecan regimen; however, 2 subsequent phase 3 trials conducted in the United States comparing these 2 regimens did not show any difference in OS. In addition, the cisplatin/irinotecan regimen was more toxic than the etoposide-based regimen.39,40 Therefore, 4 to 6 cycles of platinum/etoposide remains the standard of care first-line treatment for extensive-stage SCLC in the United States. The combination yields a 60% to 70% response rate, but the majority of patients invariably experience disease progression, with a median OS of 9 to 11 months.41 Maintenance chemotherapy beyond the initial 4 to 6 cycles does not improve survival and is associated with higher cumulative toxicity.42
Multiple attempts at improving first-line chemotherapy in extensive-stage disease have failed to show any meaningful difference in OS. For example, the addition of ifosfamide, palifosfamide, cyclophosphamide, taxane, or anthracycline to platinum doublet failed to show improvement in OS and led to more toxicity.43–46 Additionally, the use of alternating or cyclic chemotherapies in an attempt to curb drug resistance has also failed to show survival benefit.47–49 The addition of the antiangiogenic agent bevacizumab to standard platinum-based doublet has not prolonged OS in SCLC and has led to an unacceptably higher rate of tracheoesophageal fistula when used in conjunction with chemoradiation in limited-stage disease.50–55 Finally, the immune checkpoint inhibitor ipilimumab in combination with platinum plus etoposide failed to improve PFS or OS compared to platinum plus etoposide alone in a recent phase 3 trial, and maintenance pembrolizumab after completion of platinum-based chemotherapy did not improve PFS.56,57
More recently, a phase 2 study of pembrolizumab in extensive-stage SCLC (KEYNOTE 158) reported an overall response rate of 35.7%, median PFS of 2.1 months, and median OS of 14.6 months in patients who tested positive for programmed death ligand-1 (PD-L1) expression (which was defined as a PD-L1 Combined Positive Score ≥ 1).58 The median duration of response has not been reached in this study, indicating that pembrolizumab may be a promising approach in patients with extensive-stage SCLC, especially for those with PD-L1–positive tumors.
Patients with extensive-stage disease who have brain metastasis at the time of diagnosis can be treated with systemic chemotherapy first if the brain metastases are asymptomatic and there is significant extracranial disease burden. In that case, whole brain radiotherapy should be given after completion of systemic therapy.
SECOND-LINE CHEMOTHERAPY
Despite being exquisitely chemosensitive, SCLC is associated with a very poor prognosis largely because of invariable disease progression following first-line therapy and lack of effective second-line treatment options that can lead to appreciable disease control. The choice of second-line treatment is predominantly determined by the time of disease relapse after first-line platinum-based therapy. If this interval is 6 months or longer, re-treatment utilizing the same platinum doublet is appropriate. However, if the interval is 6 months or less, second-line systemic therapy options should be explored. Unfortunately, the response rate tends to be less than 10% with most of the second-line therapies in platinum-resistant disease (defined as disease progression within 3 months of receiving platinum-based therapy). If disease progression occurs between 3 and 6 months after completion of platinum-based therapy, the response rate with second-line chemotherapy is in the range of 25%.59,60
A number of second-line chemotherapy options have been explored in small studies, including topotecan, irinotecan, paclitaxel, docetaxel, temozolomide, vinorelbine, oral etoposide, gemcitabine, bendamustine, and CAV (
IMMUNOTHERAPY
The role of immune checkpoint inhibitors in the treatment of SCLC is evolving, and currently there are no FDA-approved immunotherapy agents for treating SCLC. A recently conducted phase 1/2 trial (CheckMate 032) studied the anti-programmed death(PD)-1 antibody nivolumab with or without the anti-cytotoxic T-lymphocyte–associated antigen (CTLA) -4 antibody ipilimumab in patients with relapsed SCLC. The authors reported response rates of 10% with nivolumab 3 mg/kg and 21% with nivolumab 1 mg/kg plus ipilimumab 3 mg/kg.78,79 The 2-year OS was 26% with the combination and 14% with single-agent nivolumab. Only 18% of patients had PD-L1 expression of ≥ 1%, and the response rate did not correlate with PD-L1 status. The rate of grade 3 or 4 adverse events was approximately 20%, and only 10% of patients discontinued treatment because of toxicity. Based on these data, nivolumab plus ipilimumab is now included in the National Comprehensive Cancer Network guidelines as an option for patients with SCLC who experience disease relapse within 6 months of receiving platinum-based therapy;7 however, it is questionable whether routine use of this combination is justified based on currently available data. The evidence for the combination of nivolumab and ipilimumab remains limited. The efficacy and toxicity data from both randomized and nonrandomized cohorts were presented together, making it hard to interpret the results.
Another phase 1b study (KEYNOTE-028) evaluated the anti-PD-1 antibody pembrolizumab (10 mg/kg intravenously every 2 weeks) in patients with relapsed SCLC who had received 1 or more prior lines of therapy and had PD-L1 expression of ≥ 1%. This study showed a response rate of 33%, with a median duration of response of 19 months and 1-year OS of 38%.80 Although only 28% of screened patients had PD-L1 expression of ≥ 1%, these results indicated that at least a subset of SCLC patients are able to achieve durable responses with immune checkpoint inhibition. A number of clinical trials utilizing immune checkpoint inhibitors in various combinations and settings are currently underway.
ROLE OF PROPHYLACTIC CRANIAL IRRADIATION
The role of PCI in extensive-stage SCLC is not clearly defined. A randomized phase 3 trial conducted by the European Organization for Research and Treatment of Cancer (EORTC) comparing PCI with no PCI in patients with extensive-stage SCLC who had a partial or complete response to initial platinum-based chemotherapy showed a decrease in the incidence of symptomatic brain metastasis and improvement in 1-year OS with PCI.81 However, this trial did not require mandatory brain imaging prior to PCI, and thus it is unclear if some patients in the PCI group had asymptomatic brain metastasis prior to enrollment and therefore received therapeutic benefit from brain radiation. Additionally, the dose and fractionation of PCI was not standardized across patient groups.
A more recent phase 3 study conducted in Japan that compared PCI (25 Gy in 10 fractions) with no PCI reported no difference in survival between the 2 groups.82 As opposed to the EORTC study, the Japanese study did require baseline brain imaging to confirm the absence of brain metastasis prior to enrollment. In addition, the control patients underwent periodic brain MRI to allow early detection of brain metastasis. Given the emergence of the new data, the impact of PCI on survival in patients with extensive-stage SCLC is unproven, and PCI likely has a role in a highly selected small group of patients with extensive-stage SCLC. PCI is not recommended for patients with poor performance status (ECOG performance score of 3 or 4) or underlying neurocognitive disorders.34,83
The NMDA-receptor antagonist memantine can be used in patients undergoing PCI to delay the occurrence of cognitive dysfunction.61 Memantine 20 mg daily delayed time to cognitive decline and reduced the rate of decline in memory, executive function, and processing speed compared to placebo in patients receiving whole brain radiotherapy.84
ROLE OF RADIOTHERAPY
A subset of patients with extensive-stage SCLC may benefit from consolidative thoracic radiotherapy after completion of platinum-based chemotherapy. A randomized trial that enrolled patients who achieved complete or near complete response after 3 cycles of cisplatin plus etoposide compared thoracic radiotherapy in combination with continued chemotherapy versus chemotherapy alone.85 The median OS was longer with the addition of thoracic radiotherapy compared to chemotherapy alone. Another phase 3 trial did not show improvement in 1-year OS with consolidative thoracic radiotherapy, but 2-year OS and 6-month PFS were longer.86 In general, consolidative thoracic radiotherapy benefits patients who have residual thoracic disease and low-bulk extrathoracic disease that has responded to systemic therapy.87 In addition, patients who initially presented with bulky symptomatic thoracic disease should also be considered for consolidative radiation.
Similar to other solid tumors, radiotherapy should be utilized for palliative purposes in patients with painful bone metastasis, spinal cord compression, or brain metastasis. Surgery is generally not recommended for spinal cord compression given the short life expectancy of patients with extensive-stage disease. Whole brain radiotherapy is preferred over stereotactic radiosurgery because micrometastasis is frequently present even in the setting of 1 or 2 radiographically evident brain metastasis.
NOVEL THERAPIES
The very complex genetic landscape of SCLC accounts for its resistance to conventional therapy and high recurrence rate; however, at the same time this complexity can form the basis for effective targeted therapy for the disease. One of the major factors hindering the development of targeted therapies in SCLC is limited availability of tissue due to small tissue samples and the frequent presence of significant necrosis in the samples. In recent years, several different therapeutic strategies and targeted agents have been investigated for their potential role in SCLC. Several of them, including EGFR tyrosine kinase inhibitors (TKIs), BCR-ABL TKIs, mTOR inhibitors, and VEGF inhibitors, have not been shown to provide a survival advantage in this disease. Several others, including PARP inhibitors, cellular developmental pathway inhibitors, and antibody-drug conjugates, are being tested. A phase 1 study of veliparib combined with cisplatin and etoposide in patients with previously untreated extensive-stage SCLC demonstrated a complete response in 14.3%, a partial response in 57.1%, and stable disease in 28.6% of patients with an acceptable safety profile.88 So far, none of these agents are approved for use in SCLC, and the majority are in early- phase clinical trials.89
One of the emerging targets in the treatment of SCLC is delta-like protein 3 (DLL3). DLL3 is expressed on more than 80% of SCLC tumor cells and cancer stem cells. Rovalpituzumab tesirine is an antibody-drug conjugate consisting of humanized anti-DLL3 monoclonal antibody linked to SC-DR002, a DNA-crosslinking agent. A phase 1 trial of rovalpituzumab in patients with relapsed SCLC after 1 or 2 prior lines of therapy reported a response rate of 31% in patients with DLL3 expression of ≥ 50%. The median duration of response and median PFS were both 4.6 months.90 Rovalpituzumab is currently in later phases of clinical trials and has a potential to serve as an option for patients with extensive-stage disease after disease progression on platinum-based therapy.
SUMMARY
Four to 6 cycles of carboplatin and etoposide remain the standard of care first-line treatment for patients with extensive stage SCLC. The only FDA-approved second-line treatment option is topotecan. Re-treatment with the original platinum doublet is a reasonable option for patients who have disease progression 6 months or longer after completion of platinum-based therapy. The immune checkpoint inhibitors pembrolizumab and combination nivolumab and ipilimumab have shown promising results in the second-line setting and beyond. The role of PCI has become more controversial in recent years, and periodic brain MRI in lieu of PCI is now an acceptable approach.
RESPONSE ASSESSMENT/SURVEILLANCE
For patients undergoing treatment for limited-stage SCLC, response assessment with contrast-enhanced CT of the chest/abdomen should be performed after completion of 4 cycles of chemotherapy and thoracic radiation.7 The surveillance guidelines consist of history, physical exam, and imaging every 3 months during the first 2 years, every 6 months during the third year, and annually thereafter. If PCI is not performed, brain MRI or contrast-enhanced CT scan should be performed every 3 or 4 months during the first 2 years of follow up. For extensive-stage disease, response assessment should be performed after every 2 cycles of therapy. After completion of therapy, history, physical exam, and imaging should be done every 2 months during the first year, every 3 or 4 months during years 2 and 3, every 6 months during years 4 and 5, and annually thereafter. Routine use of PET scan for surveillance is not recommended. Any new pulmonary nodule should prompt evaluation for a second primary lung malignancy. Finally, smoking cessation counseling is an integral part of management of any patient with SCLC and should be included with every clinic visit.
CONCLUSION
SCLC is a heterogeneous and genetically complex disease with a very high mortality rate. The current standard of care includes concurrent chemoradiation with cisplatin and etoposide for limited-stage SCLC and the combination of platinum and etoposide for extensive SCLC. A number of novel treatment approaches, including immune checkpoint inhibitors and antibody-drug conjugates, have had promising results in early clinical trials. Given the limited treatment options and large unmet need for new treatment options, enrollment in clinical trials is strongly recommended for patients with SCLC.
1. American Cancer Society. Cancer Facts & Figures 2017. American Cancer Society website. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2017/cancer-facts-and-figures-2017.pdf. Published 2017. Accessed July 11, 2018.
2. Govindan R, Page N, Morgensztern D, et al. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol 2006;24:4539–44.
3. Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2014. National Cancer Institute website. https://seer.cancer.gov/csr/1975_2014/. Updated April 2, 2018. Accessed July 11, 2018.
4. Varghese AM, Zakowski MF, Yu HA, et al. Small-cell lung cancers in patients who never smoked cigarettes. J Thorac Oncol 2014;9:892–6.
5. Pleasance ED, Stephens PJ, O’Meara S, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010;463:184–90.
6. Green RA, Humphrey E, Close H, Patno ME. Alkylating agents in bronchogenic carcinoma. Am J Med 1969;46:516–25.
7. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology for small cell lung cancer (Version 2.2018). www.nccn.org/professionals/physician_gls/pdf/sclc.pdf. Accessed August 12, 2018.
8. National Lung Screening Trial Research Team, Aberle DR, Adams AM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011;365:395–409.
9. Aberle DR, DeMello S, Berg CD, et al. Results of the two incidence screenings in the National Lung Screening Trial. N Engl J Med 2013;369:920–31.
10. Kitajima K, Nakamoto Y, Okizuka H, et al. Accuracy of whole-body FDG-PET/CT for detecting brain metastases from non-central nervous system tumors. Ann Nucl Med 2008;22:595–602.
11. Ordonez NG. Value of thyroid transcription factor-1 immunostaining in distinguishing small cell lung carcinomas from other small cell carcinomas. Am J Surg Pathol 2000;24:1217–23.
12. Karnak D, Beder S, Kayacan O, et al. Neuron-specific enolase and lung cancer. Am J Clin Oncol 2005;28:586–90.
13. Turrisi AT 3rd, Kim K, Blum R, et al. Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. N Engl J Med 1999;340:265–71.
14. Evans WK, Shepherd FA, Feld R, et al. VP-16 and cisplatin as first-line therapy for small-cell lung cancer. J Clin Oncol 1985;3:1471–7.
15. Pujol JL, Carestia L, Daurés JP. Is there a case for cisplatin in the treatment of small-cell lung cancer? A meta-analysis of randomized trials of a cisplatin-containing regimen versus a regimen without this alkylating agent. Br J Cancer 2000;83:8–15.
16. Mascaux C, Paesmans M, Berghmans T, et al; European Lung Cancer Working Party (ELCWP). A systematic review of the role of etoposide and cisplatin in the chemotherapy of small cell lung cancer with methodology assessment and meta-analysis. Lung Cancer 2000;30:23–36.
17. Sundstrøm S, Bremnes RM, Kaasa S, et al; Norwegian Lung Cancer Study Group. Cisplatin and etoposide regimen is superior to cyclophosphamide, epirubicin, and vincristine regimen in small-cell lung cancer: results from a randomized phase III trial with 5 years’ follow-up. J Clin Oncol 2002;20:4665–72.
18. Hatfield LA, Huskamp HA, Lamont EB. Survival and toxicity after cisplatin plus etoposide versus carboplatin plus etoposide for extensive-stage small-cell lung cancer in elderly patients. J Oncol Pract 2016;12:666–73.
19. Okamoto H, Watanabe K, Kunikane H, et al. Randomised phase III trial of carboplatin plus etoposide vs split doses of cisplatin plus etoposide in elderly or poor-risk patients with extensive disease small-cell lung cancer: JCOG 9702. Br J Cancer 2007;97:162–9.
20. Skarlos DV, Samantas E, Kosmidis P, et al. Randomized comparison of etoposide-cisplatin vs. etoposide-carboplatin and irradiation in small-cell lung cancer. A Hellenic Co-operative Oncology Group study. Ann Oncol 1994;5:601–7.
21. Rossi A, Di Maio M, Chiodini P, et al. Carboplatin- or cisplatin-based chemotherapy in first-line treatment of small-cell lung cancer: the COCIS meta-analysis of individual patient data J Clin Oncol 2012;30:1692–8.
22. Bishop JF, Raghavan D, Stuart-Harris R, et al. Carboplatin (CBDCA, JM-8) and VP-16-213 in previously untreated patients with small-cell lung cancer. J Clin Oncol 1987;5:1574–8.
23. Takada M, Fukuoka M, Kawahara M, et al. Phase III study of concurrent versus sequential thoracic radiotherapy in combination with cisplatin and etoposide for limited-stage small-cell lung cancer: results of the Japan Clinical Oncology Group Study 9104. J Clin Oncol 2002;20:3054–60.
24. Bunn PA Jr, Crowley J, Kelly K, et al. Chemoradiotherapy with or without granulocyte-macrophage colony-stimulating factor in the treatment of limited-stage small-cell lung cancer: a prospective phase III randomized study of the Southwest Oncology Group. J Clin Oncol 1995;13:1632–41.
25. Pignon JP, Arriagada R, Ihde DC, et al. A meta-analysis of thoracic radiotherapy for small-cell lung cancer. N Engl J Med 1992;327:1618–24.
26. Warde P, Payne D. Does thoracic irradiation improve survival and local control in limited-stage small-cell carcinoma of the lung? A meta-analysis. J Clin Oncol 1992;10:890–5.
27. Murray N, Coy P, Pater JL, et al. Importance of timing for thoracic irradiation in the combined modality treatment of limited-stage small-cell lung cancer. The National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 1993;11:336–44.
28. De Ruysscher D, Lueza B, Le Péchoux C, et al. Impact of thoracic radiotherapy timing in limited-stage small-cell lung cancer: usefulness of the individual patient data meta-analysis. Ann Oncol 2016;27:1818–28.
29. Faivre-Finn C, Snee M, Ashcroft L, et al. Concurrent once-daily versus twice-daily chemoradiotherapy in patients with limited-stage small-cell lung cancer (CONVERT): an open-label, phase 3, randomised, superiority trial. Lancet Oncol 2017;18:1116–25.
30. Aupérin A, Arriagada R, Pignon JP, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. Prophylactic Cranial Irradiation Overview Collaborative Group. N Engl J Med 1999;341:476–84.
31. Arriagada R, Le Chevalier T, Borie F, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. J Natl Cancer Inst 1995;87:183–90.
32. Le Péchoux C, Dunant A, Senan S, et al; Prophylactic Cranial Irradiation (PCI) Collaborative Group. Standard-dose versus higher-dose prophylactic cranial irradiation (PCI) in patients with limited-stage small-cell lung cancer in complete remission after chemotherapy and thoracic radiotherapy (PCI 99-01, EORTC 22003-08004, RTOG 0212, and IFCT 99-01): a randomised clinical trial. Lancet Oncol 2009;10:467–74.
33. Schneider BJ, Saxena A, Downey RJ. Surgery for early-stage small cell lung cancer. J Natl Compr Canc Netw 2011;9:1132–9.
34. Inoue M, Nakagawa K, Fujiwara K, et al. Results of preoperative mediastinoscopy for small cell lung cancer. Ann Thorac Surg 2000;70:1620–3.
35. Lim E, Belcher E, Yap YK, et al. The role of surgery in the treatment of limited disease small cell lung cancer: time to reevaluate. J Thorac Oncol 2008;3:1267–71.
36. Inoue M, Miyoshi S, Yasumitsu T, et al. Surgical results for small cell lung cancer based on the new TNM staging system. Thoracic Surgery Study Group of Osaka University, Osaka, Japan. Ann Thorac Surg 2000;70:1615–9.
37. Yang CF, Chan DY, Speicher PJ, et al. Role of adjuvant therapy in a population-based cohort of patients with early-stage small-cell lung cancer. J Clin Oncol 2016;34:1057–64.
38. Shepherd FA, Evans WK, Feld R, et al. Adjuvant chemotherapy following surgical resection for small-cell carcinoma of the lung. J Clin Oncol 1988;6:832–8.
39. Noda K, Nishiwaki Y, Kawahara M, et al; Japan Clinical Oncology Group. Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small-cell lung cancer. N Engl J Med 2002;346:85–91.
40. Lara PN Jr, Natale R, Crowley J, et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol 2009;27:2530–5.
41. Chute JP, Chen T, Feigal E, et al. Twenty years of phase III trials for patients with extensive-stage small-cell lung cancer: perceptible progress. J Clin Oncol 1999;17:1794–801.
42. Zhou H, Zeng C, Wei Y, et al. Duration of chemotherapy for small cell lung cancer: a meta-analysis. PloS One 2013;8:e73805.
43. Loehrer PJ Sr, Ansari R, Gonin R, et al. Cisplatin plus etoposide with and without ifosfamide in extensive small-cell lung cancer: a Hoosier Oncology Group study. J Clin Oncol;13:2594–9.
44. Pujol JL, Daurés JP, Riviére A, et al. Etoposide plus cisplatin with or without the combination of 4’-epidoxorubicin plus cyclophosphamide in treatment of extensive small-cell lung cancer: a French Federation of Cancer Institutes multicenter phase III randomized study. J Natl Cancer Inst 2001;93:300–8.
45. Berghmans T, Scherpereel A, Meert AP, et al; European Lung Cancer Working Party (ELCWP). A phase III randomized study comparing a chemotherapy with cisplatin and etoposide to a etoposide regimen without cisplatin for patients with extensive small-cell lung cancer. Front Oncol 2017;7:217.
46. Jalal SI, Lavin P, Lo G, et al. Carboplatin and etoposide with or without palifosfamide in untreated extensive-stage small-cell lung cancer: a Multicenter, Adaptive, Randomized Phase III Study (MATISSE). J Clin Oncol 2017;35:2619–23.
47. Fukuoka M, Furuse K, Saijo N, et al. Randomized trial of cyclophosphamide, doxorubicin, and vincristine versus cisplatin and etoposide versus alternation of these regimens in small-cell lung cancer. J Natl Cancer Inst 1991;83:855–61.
48. Roth BJ, Johnson DH, Einhorn LH, et al. Randomized study of cyclophosphamide, doxorubicin, and vincristine versus etoposide and cisplatin versus alternation of these two regimens in extensive small-cell lung cancer: a phase III trial of the Southeastern Cancer Study Group. J Clin Oncol 1992;10:282–91.
49. Miles DW, Earl HM, Souhami RL, et al. Intensive weekly chemotherapy for good-prognosis patients with small-cell lung cancer. J Clin Oncol 1991;9:280–5.
50. Petrioli R, Roviello G, Laera L, et al. Cisplatin, etoposide, and bevacizumab regimen followed by oral etoposide and bevacizumab maintenance treatment in patients with extensive-stage small cell lung cancer: a single-institution experience. Clin Lung Cancer 2015;16:e229–34.
51. Spigel DR, Greco FA, Zubkus JD, et al. Phase II trial of irinotecan, carboplatin, and bevacizumab in the treatment of patients with extensive-stage small-cell lung cancer. J Thorac Oncol 2009;4:1555–60.
52. Spigel DR, Townley PM, Waterhouse DM, et al. Randomized phase II study of bevacizumab in combination with chemotherapy in previously untreated extensive-stage small-cell lung cancer: results from the SALUTE trial. J Clin Oncol 2011;29:2215–22.
53. Horn L, Dahlberg SE, Sandler AB, et al. Phase II study of cisplatin plus etoposide and bevacizumab for previously untreated, extensive-stage small-cell lung cancer: Eastern Cooperative Oncology Group Study E3501. J Clin Oncol 2009;27:6006–11.
54. Tiseo M, Boni L, Ambrosio F, et al. Italian, multicenter, phase III, randomized study of cisplatin plus etoposide with or without bevacizumab as first-line treatment in extensive-disease small-cell lung cancer: the GOIRC-AIFA FARM6PMFJM trial. J Clin Oncol 2017;35:1281–7.
55. Pujol JL, Lavole A, Quoix E, et al. Randomized phase II-III study of bevacizumab in combination with chemotherapy in previously untreated extensive small-cell lung cancer: results from the IFCT-0802 trial. Ann Oncol 2015;26:908–14.
56. Gadgeel SM, Ventimiglia J, Kalemkerian GP, et al. Phase II study of maintenance pembrolizumab (pembro) in extensive stage small cell lung cancer (ES-SCLC) patients (pts) [abstract]. J Clin Oncol 2017;35(15_suppl):8504.
57. Reck M, Luft A, Szczesna A, et al. Phase III randomized trial of ipilimumab plus etoposide and platinum versus placebo plus etoposide and platinum in extensive-stage small-cell lung cancer. J Clin Oncol 2016;34:3740–8.
58. Chung HC, Lopez-Martin JA, Kao SC, et al. Phase 2 study of pembrolizumab in advanced small-cell lung cancer (SCLC): KEYNOTE-158 [abstract]. J Clin Oncol 2018;36(suppl):8506.
59. Owonikoko TK, Behera M, Chen Z, et al. A systematic analysis of efficacy of second-line chemotherapy in sensitive and refractory small-cell lung cancer. J Thorac Oncol 2012;7:866–72.
60. Postmus PE, Berendsen HH, van Zandwijk N, et al. Retreatment with the induction regimen in small cell lung cancer relapsing after an initial response to short term chemotherapy. Eur J Cancer Clin Oncol 1987;23:1409–11.
61. von Pawel J, Schiller JH, Shepherd FA, et al. Topotecan versus cyclophosphamide, doxorubicin, and vincristine for the treatment of recurrent small-cell lung cancer. J Clin Oncol 1999;17:658–667.
62. O’Brien ME, Ciuleanu TE, Tsekov H, et al. Phase III trial comparing supportive care alone with supportive care with oral topotecan in patients with relapsed small-cell lung cancer. J Clin Oncol 2006;24:5441–7.
63. Eckardt JR, von Pawel J, Pujol JL, et al. Phase III study of oral compared with intravenous topotecan as second-line therapy in small-cell lung cancer. J Clin Oncol 2007;25:2086–92.
64. Masuda N, Fukuoka M, Kusunoki Y, et al. CPT-11: a new derivative of camptothecin for the treatment of refractory or relapsed small-cell lung cancer. J Clin Oncol 1992;10:1225–9.
65. Smit EF, Fokkema E, Biesma B, et al. A phase II study of paclitaxel in heavily pretreated patients with small-cell lung cancer. Br J Cancer 1998;77:347–51.
66. Yamamoto N, Tsurutani J, Yoshimura N, et al. Phase II study of weekly paclitaxel for relapsed and refractory small cell lung cancer. Anticancer Res 2006;26:777–81.
67. Smyth JF, Smith IE, Sessa C, et al. Activity of docetaxel (Taxotere) in small cell lung cancer. Eur J Cancer 1994;30A:1058–60.
68. Pietanza MC, Kadota K, Huberman K, et al. Phase II trial of temozolomide with relapsed sensitive or refractory small cell lung cancer, with assessment of methylguanine-DNA methyltransferase as a potential biomarker. Clin Cancer Res 2012;18:1138–45.
69. Zauderer MG, Drilon A, Kadota K, et al. Trial of a 5-day dosing regimen of temozolomide in patients with relapsed small cell lung cancers with assessment of methylguanine-DNA methyltransferase. Lung Cancer 2014;86:237–40.
70. Jassem J, Karnicka-Mlodkowska H, van Pottelsberghe C, et al. Phase II study of vinorelbine (Navelbine) in previously treated small cell lung cancer patients. Eur J Cancer 1993;29A:1720–2.
71. Furuse K, Kuboa K, Kawahara M, et al. Phase II study of vinorelbine in heavily previously treated small cell lung cancer. Oncology 1996;53:169–72.
72. Einhorn LH, Pennington K, McClean J. Phase II trial of daily oral VP-16 in refractory small cell lung cancer. Semin Oncol 1990;17:32–5.
73. Johnson DH, Greco FA, Strupp J, et al. Prolonged administration of oral etoposide in patients with relapsed or refractory small-cell lung cancer: a phase II trial. J Clin Oncol 1990;8:1613–7.
74. Van der Lee I, Smit EF, van Putten JW, et al. Single-agent gemcitabine in patients with resistant small-cell lung cancer. Ann Oncol 2001;12:557–61.
75. Masters GA, Declerck L, Blanke C, et al. Phase II trial of gemcitabine in refractory or relapsed small-cell lung cancer. J Clin Oncol 2003;21:1550–5.
76. von Pawel J, Schiller JH, Shepherd FA, et al. Topotecan versus cyclophosphamide, doxorubicin, and vincristine for the treatment of recurrent small-cell lung cancer. J Clin Oncol 1999;17:658–67.
77. Lammers PE, Shyr Y, Li CI, et al. Phase II study of bendamustine in relapsed chemotherapy sensitive or resistant small-cell lung cancer. J Thorac Oncol 2014;9:559–62.
78. Hellmann MD, Ott PA, Zugazagoitia J, et al. Nivolumab (nivo) ± ipilimumab (ipi) in advanced small-cell lung cancer (SCLC): First report of a randomized expansion cohort from CheckMate 032 [abstract]. J Clin Oncol 2017;35(15_suppl):8503.
79. Antonia SJ, López-Martin JA, Bendell J, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol 2016;17:883–95.
80. Ott PA, Elez E, Hiret S, et al. Pembrolizumab in patients with extensive-stage small-cell lung cancer: results from the Phase Ib KEYNOTE-028 study. J Clin Oncol 2017;35:3823–9.
81. Slotman B, Faivre-Finn C, Kramer G, et al. Prophylactic cranial irradiation in extensive small-cell lung cancer. N Engl J Med 2007;357:664–72.
82. Takahashi T, Yamanaka T, Seto T, et al. Prophylactic cranial irradiation versus observation in patients with extensive-disease small-cell lung cancer: a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2017;18:663–71.
83. Slotman BJ, Mauer ME, Bottomley A, et al. Prophylactic cranial irradiation in extensive disease small-cell lung cancer: short-term health-related quality of life and patient reported symptoms: results of an international Phase III randomized controlled trial by the EORTC Radiation Oncology and Lung Cancer Groups. J Clin Oncol 2009;27:78–84.
84. Brown PD, Pugh S, Laack NN, et al; Radiation Therapy Oncology Group (RTOG). Memantine for the prevention of cognitive dysfunction in patients receiving whole-brain radiotherapy: a randomized, double-blind, placebo-controlled trial. Neuro Oncol 2013;15:1429–37.
85. Jeremic B, Shibamoto Y, Nikolic N, et al. Role of radiation therapy in the combined-modality treatment of patients with extensive disease small-cell lung cancer: a randomized study. J Clin Oncol 1999;17:2092–9.
86. Slotman BJ, van Tinteren H, Praag JO, et al. Use of thoracic radiotherapy for extensive stage small-cell lung cancer: a phase 3 randomised controlled trial. Lancet 2015;385:36–42.
87. Slotman BJ, van Tinteren H, Praag JO, et al. Radiotherapy for extensive stage small-cell lung cancer - authors’ reply. Lancet 2015;385:1292–3.
88. Owonikoko TK, Dahlberg SE, Khan SA, et al. A phase 1 safety study of veliparib combined with cisplatin and etoposide in extensive stage small cell lung cancer: A trial of the ECOG-ACRIN Cancer Research Group (E2511). Lung Cancer 2015;89:66–70.
89. Mamdani H, Induru R, Jalal SI. Novel therapies in small cell lung cancer. Transl Lung Cancer Res 2015;4:533–44.
90. Rudin CM, Pietanza MC, Bauer TM, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol 2017;18:42–51.
1. American Cancer Society. Cancer Facts & Figures 2017. American Cancer Society website. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2017/cancer-facts-and-figures-2017.pdf. Published 2017. Accessed July 11, 2018.
2. Govindan R, Page N, Morgensztern D, et al. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol 2006;24:4539–44.
3. Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2014. National Cancer Institute website. https://seer.cancer.gov/csr/1975_2014/. Updated April 2, 2018. Accessed July 11, 2018.
4. Varghese AM, Zakowski MF, Yu HA, et al. Small-cell lung cancers in patients who never smoked cigarettes. J Thorac Oncol 2014;9:892–6.
5. Pleasance ED, Stephens PJ, O’Meara S, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010;463:184–90.
6. Green RA, Humphrey E, Close H, Patno ME. Alkylating agents in bronchogenic carcinoma. Am J Med 1969;46:516–25.
7. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology for small cell lung cancer (Version 2.2018). www.nccn.org/professionals/physician_gls/pdf/sclc.pdf. Accessed August 12, 2018.
8. National Lung Screening Trial Research Team, Aberle DR, Adams AM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011;365:395–409.
9. Aberle DR, DeMello S, Berg CD, et al. Results of the two incidence screenings in the National Lung Screening Trial. N Engl J Med 2013;369:920–31.
10. Kitajima K, Nakamoto Y, Okizuka H, et al. Accuracy of whole-body FDG-PET/CT for detecting brain metastases from non-central nervous system tumors. Ann Nucl Med 2008;22:595–602.
11. Ordonez NG. Value of thyroid transcription factor-1 immunostaining in distinguishing small cell lung carcinomas from other small cell carcinomas. Am J Surg Pathol 2000;24:1217–23.
12. Karnak D, Beder S, Kayacan O, et al. Neuron-specific enolase and lung cancer. Am J Clin Oncol 2005;28:586–90.
13. Turrisi AT 3rd, Kim K, Blum R, et al. Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. N Engl J Med 1999;340:265–71.
14. Evans WK, Shepherd FA, Feld R, et al. VP-16 and cisplatin as first-line therapy for small-cell lung cancer. J Clin Oncol 1985;3:1471–7.
15. Pujol JL, Carestia L, Daurés JP. Is there a case for cisplatin in the treatment of small-cell lung cancer? A meta-analysis of randomized trials of a cisplatin-containing regimen versus a regimen without this alkylating agent. Br J Cancer 2000;83:8–15.
16. Mascaux C, Paesmans M, Berghmans T, et al; European Lung Cancer Working Party (ELCWP). A systematic review of the role of etoposide and cisplatin in the chemotherapy of small cell lung cancer with methodology assessment and meta-analysis. Lung Cancer 2000;30:23–36.
17. Sundstrøm S, Bremnes RM, Kaasa S, et al; Norwegian Lung Cancer Study Group. Cisplatin and etoposide regimen is superior to cyclophosphamide, epirubicin, and vincristine regimen in small-cell lung cancer: results from a randomized phase III trial with 5 years’ follow-up. J Clin Oncol 2002;20:4665–72.
18. Hatfield LA, Huskamp HA, Lamont EB. Survival and toxicity after cisplatin plus etoposide versus carboplatin plus etoposide for extensive-stage small-cell lung cancer in elderly patients. J Oncol Pract 2016;12:666–73.
19. Okamoto H, Watanabe K, Kunikane H, et al. Randomised phase III trial of carboplatin plus etoposide vs split doses of cisplatin plus etoposide in elderly or poor-risk patients with extensive disease small-cell lung cancer: JCOG 9702. Br J Cancer 2007;97:162–9.
20. Skarlos DV, Samantas E, Kosmidis P, et al. Randomized comparison of etoposide-cisplatin vs. etoposide-carboplatin and irradiation in small-cell lung cancer. A Hellenic Co-operative Oncology Group study. Ann Oncol 1994;5:601–7.
21. Rossi A, Di Maio M, Chiodini P, et al. Carboplatin- or cisplatin-based chemotherapy in first-line treatment of small-cell lung cancer: the COCIS meta-analysis of individual patient data J Clin Oncol 2012;30:1692–8.
22. Bishop JF, Raghavan D, Stuart-Harris R, et al. Carboplatin (CBDCA, JM-8) and VP-16-213 in previously untreated patients with small-cell lung cancer. J Clin Oncol 1987;5:1574–8.
23. Takada M, Fukuoka M, Kawahara M, et al. Phase III study of concurrent versus sequential thoracic radiotherapy in combination with cisplatin and etoposide for limited-stage small-cell lung cancer: results of the Japan Clinical Oncology Group Study 9104. J Clin Oncol 2002;20:3054–60.
24. Bunn PA Jr, Crowley J, Kelly K, et al. Chemoradiotherapy with or without granulocyte-macrophage colony-stimulating factor in the treatment of limited-stage small-cell lung cancer: a prospective phase III randomized study of the Southwest Oncology Group. J Clin Oncol 1995;13:1632–41.
25. Pignon JP, Arriagada R, Ihde DC, et al. A meta-analysis of thoracic radiotherapy for small-cell lung cancer. N Engl J Med 1992;327:1618–24.
26. Warde P, Payne D. Does thoracic irradiation improve survival and local control in limited-stage small-cell carcinoma of the lung? A meta-analysis. J Clin Oncol 1992;10:890–5.
27. Murray N, Coy P, Pater JL, et al. Importance of timing for thoracic irradiation in the combined modality treatment of limited-stage small-cell lung cancer. The National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 1993;11:336–44.
28. De Ruysscher D, Lueza B, Le Péchoux C, et al. Impact of thoracic radiotherapy timing in limited-stage small-cell lung cancer: usefulness of the individual patient data meta-analysis. Ann Oncol 2016;27:1818–28.
29. Faivre-Finn C, Snee M, Ashcroft L, et al. Concurrent once-daily versus twice-daily chemoradiotherapy in patients with limited-stage small-cell lung cancer (CONVERT): an open-label, phase 3, randomised, superiority trial. Lancet Oncol 2017;18:1116–25.
30. Aupérin A, Arriagada R, Pignon JP, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. Prophylactic Cranial Irradiation Overview Collaborative Group. N Engl J Med 1999;341:476–84.
31. Arriagada R, Le Chevalier T, Borie F, et al. Prophylactic cranial irradiation for patients with small-cell lung cancer in complete remission. J Natl Cancer Inst 1995;87:183–90.
32. Le Péchoux C, Dunant A, Senan S, et al; Prophylactic Cranial Irradiation (PCI) Collaborative Group. Standard-dose versus higher-dose prophylactic cranial irradiation (PCI) in patients with limited-stage small-cell lung cancer in complete remission after chemotherapy and thoracic radiotherapy (PCI 99-01, EORTC 22003-08004, RTOG 0212, and IFCT 99-01): a randomised clinical trial. Lancet Oncol 2009;10:467–74.
33. Schneider BJ, Saxena A, Downey RJ. Surgery for early-stage small cell lung cancer. J Natl Compr Canc Netw 2011;9:1132–9.
34. Inoue M, Nakagawa K, Fujiwara K, et al. Results of preoperative mediastinoscopy for small cell lung cancer. Ann Thorac Surg 2000;70:1620–3.
35. Lim E, Belcher E, Yap YK, et al. The role of surgery in the treatment of limited disease small cell lung cancer: time to reevaluate. J Thorac Oncol 2008;3:1267–71.
36. Inoue M, Miyoshi S, Yasumitsu T, et al. Surgical results for small cell lung cancer based on the new TNM staging system. Thoracic Surgery Study Group of Osaka University, Osaka, Japan. Ann Thorac Surg 2000;70:1615–9.
37. Yang CF, Chan DY, Speicher PJ, et al. Role of adjuvant therapy in a population-based cohort of patients with early-stage small-cell lung cancer. J Clin Oncol 2016;34:1057–64.
38. Shepherd FA, Evans WK, Feld R, et al. Adjuvant chemotherapy following surgical resection for small-cell carcinoma of the lung. J Clin Oncol 1988;6:832–8.
39. Noda K, Nishiwaki Y, Kawahara M, et al; Japan Clinical Oncology Group. Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small-cell lung cancer. N Engl J Med 2002;346:85–91.
40. Lara PN Jr, Natale R, Crowley J, et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol 2009;27:2530–5.
41. Chute JP, Chen T, Feigal E, et al. Twenty years of phase III trials for patients with extensive-stage small-cell lung cancer: perceptible progress. J Clin Oncol 1999;17:1794–801.
42. Zhou H, Zeng C, Wei Y, et al. Duration of chemotherapy for small cell lung cancer: a meta-analysis. PloS One 2013;8:e73805.
43. Loehrer PJ Sr, Ansari R, Gonin R, et al. Cisplatin plus etoposide with and without ifosfamide in extensive small-cell lung cancer: a Hoosier Oncology Group study. J Clin Oncol;13:2594–9.
44. Pujol JL, Daurés JP, Riviére A, et al. Etoposide plus cisplatin with or without the combination of 4’-epidoxorubicin plus cyclophosphamide in treatment of extensive small-cell lung cancer: a French Federation of Cancer Institutes multicenter phase III randomized study. J Natl Cancer Inst 2001;93:300–8.
45. Berghmans T, Scherpereel A, Meert AP, et al; European Lung Cancer Working Party (ELCWP). A phase III randomized study comparing a chemotherapy with cisplatin and etoposide to a etoposide regimen without cisplatin for patients with extensive small-cell lung cancer. Front Oncol 2017;7:217.
46. Jalal SI, Lavin P, Lo G, et al. Carboplatin and etoposide with or without palifosfamide in untreated extensive-stage small-cell lung cancer: a Multicenter, Adaptive, Randomized Phase III Study (MATISSE). J Clin Oncol 2017;35:2619–23.
47. Fukuoka M, Furuse K, Saijo N, et al. Randomized trial of cyclophosphamide, doxorubicin, and vincristine versus cisplatin and etoposide versus alternation of these regimens in small-cell lung cancer. J Natl Cancer Inst 1991;83:855–61.
48. Roth BJ, Johnson DH, Einhorn LH, et al. Randomized study of cyclophosphamide, doxorubicin, and vincristine versus etoposide and cisplatin versus alternation of these two regimens in extensive small-cell lung cancer: a phase III trial of the Southeastern Cancer Study Group. J Clin Oncol 1992;10:282–91.
49. Miles DW, Earl HM, Souhami RL, et al. Intensive weekly chemotherapy for good-prognosis patients with small-cell lung cancer. J Clin Oncol 1991;9:280–5.
50. Petrioli R, Roviello G, Laera L, et al. Cisplatin, etoposide, and bevacizumab regimen followed by oral etoposide and bevacizumab maintenance treatment in patients with extensive-stage small cell lung cancer: a single-institution experience. Clin Lung Cancer 2015;16:e229–34.
51. Spigel DR, Greco FA, Zubkus JD, et al. Phase II trial of irinotecan, carboplatin, and bevacizumab in the treatment of patients with extensive-stage small-cell lung cancer. J Thorac Oncol 2009;4:1555–60.
52. Spigel DR, Townley PM, Waterhouse DM, et al. Randomized phase II study of bevacizumab in combination with chemotherapy in previously untreated extensive-stage small-cell lung cancer: results from the SALUTE trial. J Clin Oncol 2011;29:2215–22.
53. Horn L, Dahlberg SE, Sandler AB, et al. Phase II study of cisplatin plus etoposide and bevacizumab for previously untreated, extensive-stage small-cell lung cancer: Eastern Cooperative Oncology Group Study E3501. J Clin Oncol 2009;27:6006–11.
54. Tiseo M, Boni L, Ambrosio F, et al. Italian, multicenter, phase III, randomized study of cisplatin plus etoposide with or without bevacizumab as first-line treatment in extensive-disease small-cell lung cancer: the GOIRC-AIFA FARM6PMFJM trial. J Clin Oncol 2017;35:1281–7.
55. Pujol JL, Lavole A, Quoix E, et al. Randomized phase II-III study of bevacizumab in combination with chemotherapy in previously untreated extensive small-cell lung cancer: results from the IFCT-0802 trial. Ann Oncol 2015;26:908–14.
56. Gadgeel SM, Ventimiglia J, Kalemkerian GP, et al. Phase II study of maintenance pembrolizumab (pembro) in extensive stage small cell lung cancer (ES-SCLC) patients (pts) [abstract]. J Clin Oncol 2017;35(15_suppl):8504.
57. Reck M, Luft A, Szczesna A, et al. Phase III randomized trial of ipilimumab plus etoposide and platinum versus placebo plus etoposide and platinum in extensive-stage small-cell lung cancer. J Clin Oncol 2016;34:3740–8.
58. Chung HC, Lopez-Martin JA, Kao SC, et al. Phase 2 study of pembrolizumab in advanced small-cell lung cancer (SCLC): KEYNOTE-158 [abstract]. J Clin Oncol 2018;36(suppl):8506.
59. Owonikoko TK, Behera M, Chen Z, et al. A systematic analysis of efficacy of second-line chemotherapy in sensitive and refractory small-cell lung cancer. J Thorac Oncol 2012;7:866–72.
60. Postmus PE, Berendsen HH, van Zandwijk N, et al. Retreatment with the induction regimen in small cell lung cancer relapsing after an initial response to short term chemotherapy. Eur J Cancer Clin Oncol 1987;23:1409–11.
61. von Pawel J, Schiller JH, Shepherd FA, et al. Topotecan versus cyclophosphamide, doxorubicin, and vincristine for the treatment of recurrent small-cell lung cancer. J Clin Oncol 1999;17:658–667.
62. O’Brien ME, Ciuleanu TE, Tsekov H, et al. Phase III trial comparing supportive care alone with supportive care with oral topotecan in patients with relapsed small-cell lung cancer. J Clin Oncol 2006;24:5441–7.
63. Eckardt JR, von Pawel J, Pujol JL, et al. Phase III study of oral compared with intravenous topotecan as second-line therapy in small-cell lung cancer. J Clin Oncol 2007;25:2086–92.
64. Masuda N, Fukuoka M, Kusunoki Y, et al. CPT-11: a new derivative of camptothecin for the treatment of refractory or relapsed small-cell lung cancer. J Clin Oncol 1992;10:1225–9.
65. Smit EF, Fokkema E, Biesma B, et al. A phase II study of paclitaxel in heavily pretreated patients with small-cell lung cancer. Br J Cancer 1998;77:347–51.
66. Yamamoto N, Tsurutani J, Yoshimura N, et al. Phase II study of weekly paclitaxel for relapsed and refractory small cell lung cancer. Anticancer Res 2006;26:777–81.
67. Smyth JF, Smith IE, Sessa C, et al. Activity of docetaxel (Taxotere) in small cell lung cancer. Eur J Cancer 1994;30A:1058–60.
68. Pietanza MC, Kadota K, Huberman K, et al. Phase II trial of temozolomide with relapsed sensitive or refractory small cell lung cancer, with assessment of methylguanine-DNA methyltransferase as a potential biomarker. Clin Cancer Res 2012;18:1138–45.
69. Zauderer MG, Drilon A, Kadota K, et al. Trial of a 5-day dosing regimen of temozolomide in patients with relapsed small cell lung cancers with assessment of methylguanine-DNA methyltransferase. Lung Cancer 2014;86:237–40.
70. Jassem J, Karnicka-Mlodkowska H, van Pottelsberghe C, et al. Phase II study of vinorelbine (Navelbine) in previously treated small cell lung cancer patients. Eur J Cancer 1993;29A:1720–2.
71. Furuse K, Kuboa K, Kawahara M, et al. Phase II study of vinorelbine in heavily previously treated small cell lung cancer. Oncology 1996;53:169–72.
72. Einhorn LH, Pennington K, McClean J. Phase II trial of daily oral VP-16 in refractory small cell lung cancer. Semin Oncol 1990;17:32–5.
73. Johnson DH, Greco FA, Strupp J, et al. Prolonged administration of oral etoposide in patients with relapsed or refractory small-cell lung cancer: a phase II trial. J Clin Oncol 1990;8:1613–7.
74. Van der Lee I, Smit EF, van Putten JW, et al. Single-agent gemcitabine in patients with resistant small-cell lung cancer. Ann Oncol 2001;12:557–61.
75. Masters GA, Declerck L, Blanke C, et al. Phase II trial of gemcitabine in refractory or relapsed small-cell lung cancer. J Clin Oncol 2003;21:1550–5.
76. von Pawel J, Schiller JH, Shepherd FA, et al. Topotecan versus cyclophosphamide, doxorubicin, and vincristine for the treatment of recurrent small-cell lung cancer. J Clin Oncol 1999;17:658–67.
77. Lammers PE, Shyr Y, Li CI, et al. Phase II study of bendamustine in relapsed chemotherapy sensitive or resistant small-cell lung cancer. J Thorac Oncol 2014;9:559–62.
78. Hellmann MD, Ott PA, Zugazagoitia J, et al. Nivolumab (nivo) ± ipilimumab (ipi) in advanced small-cell lung cancer (SCLC): First report of a randomized expansion cohort from CheckMate 032 [abstract]. J Clin Oncol 2017;35(15_suppl):8503.
79. Antonia SJ, López-Martin JA, Bendell J, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol 2016;17:883–95.
80. Ott PA, Elez E, Hiret S, et al. Pembrolizumab in patients with extensive-stage small-cell lung cancer: results from the Phase Ib KEYNOTE-028 study. J Clin Oncol 2017;35:3823–9.
81. Slotman B, Faivre-Finn C, Kramer G, et al. Prophylactic cranial irradiation in extensive small-cell lung cancer. N Engl J Med 2007;357:664–72.
82. Takahashi T, Yamanaka T, Seto T, et al. Prophylactic cranial irradiation versus observation in patients with extensive-disease small-cell lung cancer: a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2017;18:663–71.
83. Slotman BJ, Mauer ME, Bottomley A, et al. Prophylactic cranial irradiation in extensive disease small-cell lung cancer: short-term health-related quality of life and patient reported symptoms: results of an international Phase III randomized controlled trial by the EORTC Radiation Oncology and Lung Cancer Groups. J Clin Oncol 2009;27:78–84.
84. Brown PD, Pugh S, Laack NN, et al; Radiation Therapy Oncology Group (RTOG). Memantine for the prevention of cognitive dysfunction in patients receiving whole-brain radiotherapy: a randomized, double-blind, placebo-controlled trial. Neuro Oncol 2013;15:1429–37.
85. Jeremic B, Shibamoto Y, Nikolic N, et al. Role of radiation therapy in the combined-modality treatment of patients with extensive disease small-cell lung cancer: a randomized study. J Clin Oncol 1999;17:2092–9.
86. Slotman BJ, van Tinteren H, Praag JO, et al. Use of thoracic radiotherapy for extensive stage small-cell lung cancer: a phase 3 randomised controlled trial. Lancet 2015;385:36–42.
87. Slotman BJ, van Tinteren H, Praag JO, et al. Radiotherapy for extensive stage small-cell lung cancer - authors’ reply. Lancet 2015;385:1292–3.
88. Owonikoko TK, Dahlberg SE, Khan SA, et al. A phase 1 safety study of veliparib combined with cisplatin and etoposide in extensive stage small cell lung cancer: A trial of the ECOG-ACRIN Cancer Research Group (E2511). Lung Cancer 2015;89:66–70.
89. Mamdani H, Induru R, Jalal SI. Novel therapies in small cell lung cancer. Transl Lung Cancer Res 2015;4:533–44.
90. Rudin CM, Pietanza MC, Bauer TM, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol 2017;18:42–51.