User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'main-prefix')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
div[contains(@class, 'view-medstat-quiz-listing-panes')]
div[contains(@class, 'pane-article-sidebar-latest-news')]
div[contains(@class, 'medstat-accordion-set article-series')]
Pregnancy studies on psoriasis, PsA medications pick up
Christina Chambers, PhD, MPH, who runs the MotherToBaby Pregnancy Studies research center at the University of California, San Diego, has found most pregnant women to be “entirely altruistic” about sharing their experiences with drug treatment during pregnancy.

– but dermatologists, rheumatologists, and their female patients with psoriasis and psoriatic arthritis (PsA) want much more.
And women’s participation in the MotherToBaby studies conducted by the nonprofit Organization of Teratology Information Specialists (OTIS) is key, say physicians who are treating women of reproductive age. OTIS is now listed in drug labeling as the “pregnancy registry” contact for many of the medications they may be discussing with patients.
Dr. Chambers said that most women appreciate “that participating in a study may not help her with her pregnancy, but it can help her sister or her friend or someone else who has these same questions in planning a pregnancy of ‘Can I stay on my treatment?’ or, in the case of an unplanned pregnancy, ‘Should I be concerned?’ ”
OTIS has enrolled women with psoriasis and/or PsA in studies of nine medications, most of them biologics (both TNF-alpha blockers and newer anti-interleukin agents).
Four of the studies – those evaluating etanercept (Enbrel), adalimumab (Humira), abatacept (Orencia), and ustekinumab (Stelara) – are now closed to enrollment with analyses either underway or completed. The other five are currently enrolling patients and involve treatment with certolizumab pegol (Cimzia), tildrakizumab (Ilumya), apremilast (Otezla), guselkumab (Tremfya), and tofacitinib (Xeljanz).

Lisa R. Sammaritano, MD, a rheumatologist at the Hospital for Special Surgery, New York, who led the development of the American College of Rheumatology’s first guideline for the management of reproductive health in rheumatic and musculoskeletal diseases, recommends to some of her patients that they contact OTIS. “Their pregnancy registry studies have added important information to the field over the years,” she said.
Most recently, a study of the anti–TNF-alpha medication adalimumab that began in 2004 in pregnant patients with RA and Crohn’s disease culminated in a 2019 PLOS ONE paper reporting no associations between exposure to the medication and an increased risk of adverse outcomes. The outcomes studied were major structural birth defects, minor defects, spontaneous abortion, preterm delivery, prenatal and postnatal growth deficiency, serious or opportunistic infections, and malignancies.
An analysis is underway of adalimumab exposure in women with PsA – a patient subset that was added after the study started. But in the meantime, Dr. Chambers said, the 2019 research article is relevant to questions of drug safety across indications.
OTIS’s MothertoBaby studies are structured as prospective cohort studies. Dr. Chambers, a perinatal epidemiologist, is president of OTIS, which recruits women who have an exposure to the medication under study – at least one dose, for any length of time. And in most cases, it also recruits women with the underlying condition but no exposure and healthy women without the condition to represent the general population.
It’s the disease-matched comparison group that makes OTIS’s studies different from traditional pregnancy registries involving “a simple exposure series and outcomes that are described in the context of what you’d expect in the general population,” said Dr. Chambers, professor in the department of pediatrics, as well as family and preventative medicine, at UCSD and codirector of the Center for Better Beginnings at that university. “Many maternal conditions themselves [or their comorbidities] carry some risk of adverse outcomes in pregnancy.”
The OTIS studies typically involve at least 100 exposed pregnancies and a similar number of unexposed pregnancies; some have cohorts of 200-300.
The recently published study of adalimumab, for instance, included 257 women with exposure to the drug and 120 women in a disease comparison group with no exposure. In addition to finding no associations between drug exposure and adverse outcomes, the study found that women with RA or Crohn’s were at increased risk of preterm delivery, irrespective of adalimumab exposure.
“There’s insufficient [power with any of these numbers] to come to the conclusion that a drug is safe,” she said. “But what we have been able to say [through our studies] is that we’ve looked carefully at the whole array of outcomes ... and we don’t see anything unusual. That early view can be reassuring” until large population-based studies or claims analyses become possible.
Dr. Sammaritano, also with Weill Cornell Medicine, New York, said that she does not recommend registry participation for patients who stop biologics at the diagnosis of pregnancy. Since “the start of IgG antibody transfer during pregnancy is about 16 weeks,” she worries that including these patients might lead to falsely reassuring findings. “We are most interested in [knowing the outcomes of] patients who must continue the drugs through pregnancy,” she said.
Dr. Chambers, however, said that in her view, placental transfer is not a requirement for a medication to have some effect on the outcome of pregnancy. “The outcome could be influenced by an effect of the medication that doesn’t require placental transfer or require placental transfer in large amounts,” she said. “So it’s relevant to examine exposures that have occurred only in the first trimester, and this is especially true for the outcome of major birth defects, most of which are initiated in the first trimester.”
The MotherToBaby studies typically include both early, short exposures and longer exposures, she said. “And certainly, duration of use is a factor that we do consider in looking at specific outcomes such as growth, preterm delivery, and risk of serious or opportunistic infections.”
(In the published study of adalimumab, 65.3% of women in the medication-exposed cohort used the medication in all three trimesters, 10.5% in the first and second trimesters, and 22.4% in the first trimester only.)
Women participating in the MotherToBaby studies complete two to four interviews during pregnancy and may be interviewed again after delivery. They are asked for their permission to share a copy of their medical records – and their baby’s medical records – and their babies receive a follow-up pediatric exam by a pediatrician with expertise in dysmorphology/genetics (who is blinded to exposure status), most commonly in the participant’s home. Providers are not asked to enter any data.

Eliza Chakravarty, MD, a rheumatologist with the Oklahoma Medical Research Foundation in Oklahoma City who treats patients with PsA who are pregnant or considering pregnancy, said that her referrals for research participation “have been mostly to MothertoBaby.”
“Most drug companies [in the autoimmune space] are now contracting with them [for their pregnancy exposure research],” she said. “I really like that it’s become so centralized.”
She tells patients that many questions can be answered through research, that their experience matters, and that “there are benefits” to the extra pediatric examination. “I give them the information and let them decide whether or not they want to call [MotherToBaby],” she said. “I don’t want to impose. I want to make them aware.”
Dr. Chambers emphasizes to patients and physicians that the studies are strictly observational and do not require any changes in personal or medical regimens. “When people hear the word ‘research’ they think of clinical trials. We’re saying, you and your provider do everything you normally would do, just let us observe what happens during your pregnancy.”
Physicians should assure patients, moreover, that “just because the drug is being studied doesn’t mean there’s a known risk or even a suspected risk,” she said.
The MotherToBaby studies receive funding from the pharmaceutical companies, which are required by the Food and Drug Administration to conduct pregnancy exposure registries for medications used during pregnancy or in women of reproductive age. OTIS has an independent advisory board, however, and independently analyzes and publishes its findings. Progress reports are shared with the pharmaceutical companies, and in turn, the FDA, Dr. Chambers said.
To refer patients for MotherToBaby studies, physicians can use an online referral form found on the MothertoBaby web site, a service of OTIS, or call the pregnancy studies team at 877-311-8972 to provide them with the patient’s name or number. Patients may also be given the number and advised to consider calling. MotherToBaby offers medication fact sheets that answer questions about exposures during pregnancy and breastfeeding, and runs a free and confidential teratogen counseling service: 866-626-6847.
Christina Chambers, PhD, MPH, who runs the MotherToBaby Pregnancy Studies research center at the University of California, San Diego, has found most pregnant women to be “entirely altruistic” about sharing their experiences with drug treatment during pregnancy.
– but dermatologists, rheumatologists, and their female patients with psoriasis and psoriatic arthritis (PsA) want much more.
And women’s participation in the MotherToBaby studies conducted by the nonprofit Organization of Teratology Information Specialists (OTIS) is key, say physicians who are treating women of reproductive age. OTIS is now listed in drug labeling as the “pregnancy registry” contact for many of the medications they may be discussing with patients.
Dr. Chambers said that most women appreciate “that participating in a study may not help her with her pregnancy, but it can help her sister or her friend or someone else who has these same questions in planning a pregnancy of ‘Can I stay on my treatment?’ or, in the case of an unplanned pregnancy, ‘Should I be concerned?’ ”
OTIS has enrolled women with psoriasis and/or PsA in studies of nine medications, most of them biologics (both TNF-alpha blockers and newer anti-interleukin agents).
Four of the studies – those evaluating etanercept (Enbrel), adalimumab (Humira), abatacept (Orencia), and ustekinumab (Stelara) – are now closed to enrollment with analyses either underway or completed. The other five are currently enrolling patients and involve treatment with certolizumab pegol (Cimzia), tildrakizumab (Ilumya), apremilast (Otezla), guselkumab (Tremfya), and tofacitinib (Xeljanz).
Lisa R. Sammaritano, MD, a rheumatologist at the Hospital for Special Surgery, New York, who led the development of the American College of Rheumatology’s first guideline for the management of reproductive health in rheumatic and musculoskeletal diseases, recommends to some of her patients that they contact OTIS. “Their pregnancy registry studies have added important information to the field over the years,” she said.
Most recently, a study of the anti–TNF-alpha medication adalimumab that began in 2004 in pregnant patients with RA and Crohn’s disease culminated in a 2019 PLOS ONE paper reporting no associations between exposure to the medication and an increased risk of adverse outcomes. The outcomes studied were major structural birth defects, minor defects, spontaneous abortion, preterm delivery, prenatal and postnatal growth deficiency, serious or opportunistic infections, and malignancies.
An analysis is underway of adalimumab exposure in women with PsA – a patient subset that was added after the study started. But in the meantime, Dr. Chambers said, the 2019 research article is relevant to questions of drug safety across indications.
OTIS’s MothertoBaby studies are structured as prospective cohort studies. Dr. Chambers, a perinatal epidemiologist, is president of OTIS, which recruits women who have an exposure to the medication under study – at least one dose, for any length of time. And in most cases, it also recruits women with the underlying condition but no exposure and healthy women without the condition to represent the general population.
It’s the disease-matched comparison group that makes OTIS’s studies different from traditional pregnancy registries involving “a simple exposure series and outcomes that are described in the context of what you’d expect in the general population,” said Dr. Chambers, professor in the department of pediatrics, as well as family and preventative medicine, at UCSD and codirector of the Center for Better Beginnings at that university. “Many maternal conditions themselves [or their comorbidities] carry some risk of adverse outcomes in pregnancy.”
The OTIS studies typically involve at least 100 exposed pregnancies and a similar number of unexposed pregnancies; some have cohorts of 200-300.
The recently published study of adalimumab, for instance, included 257 women with exposure to the drug and 120 women in a disease comparison group with no exposure. In addition to finding no associations between drug exposure and adverse outcomes, the study found that women with RA or Crohn’s were at increased risk of preterm delivery, irrespective of adalimumab exposure.
“There’s insufficient [power with any of these numbers] to come to the conclusion that a drug is safe,” she said. “But what we have been able to say [through our studies] is that we’ve looked carefully at the whole array of outcomes ... and we don’t see anything unusual. That early view can be reassuring” until large population-based studies or claims analyses become possible.
Dr. Sammaritano, also with Weill Cornell Medicine, New York, said that she does not recommend registry participation for patients who stop biologics at the diagnosis of pregnancy. Since “the start of IgG antibody transfer during pregnancy is about 16 weeks,” she worries that including these patients might lead to falsely reassuring findings. “We are most interested in [knowing the outcomes of] patients who must continue the drugs through pregnancy,” she said.
Dr. Chambers, however, said that in her view, placental transfer is not a requirement for a medication to have some effect on the outcome of pregnancy. “The outcome could be influenced by an effect of the medication that doesn’t require placental transfer or require placental transfer in large amounts,” she said. “So it’s relevant to examine exposures that have occurred only in the first trimester, and this is especially true for the outcome of major birth defects, most of which are initiated in the first trimester.”
The MotherToBaby studies typically include both early, short exposures and longer exposures, she said. “And certainly, duration of use is a factor that we do consider in looking at specific outcomes such as growth, preterm delivery, and risk of serious or opportunistic infections.”
(In the published study of adalimumab, 65.3% of women in the medication-exposed cohort used the medication in all three trimesters, 10.5% in the first and second trimesters, and 22.4% in the first trimester only.)
Women participating in the MotherToBaby studies complete two to four interviews during pregnancy and may be interviewed again after delivery. They are asked for their permission to share a copy of their medical records – and their baby’s medical records – and their babies receive a follow-up pediatric exam by a pediatrician with expertise in dysmorphology/genetics (who is blinded to exposure status), most commonly in the participant’s home. Providers are not asked to enter any data.
Eliza Chakravarty, MD, a rheumatologist with the Oklahoma Medical Research Foundation in Oklahoma City who treats patients with PsA who are pregnant or considering pregnancy, said that her referrals for research participation “have been mostly to MothertoBaby.”
“Most drug companies [in the autoimmune space] are now contracting with them [for their pregnancy exposure research],” she said. “I really like that it’s become so centralized.”
She tells patients that many questions can be answered through research, that their experience matters, and that “there are benefits” to the extra pediatric examination. “I give them the information and let them decide whether or not they want to call [MotherToBaby],” she said. “I don’t want to impose. I want to make them aware.”
Dr. Chambers emphasizes to patients and physicians that the studies are strictly observational and do not require any changes in personal or medical regimens. “When people hear the word ‘research’ they think of clinical trials. We’re saying, you and your provider do everything you normally would do, just let us observe what happens during your pregnancy.”
Physicians should assure patients, moreover, that “just because the drug is being studied doesn’t mean there’s a known risk or even a suspected risk,” she said.
The MotherToBaby studies receive funding from the pharmaceutical companies, which are required by the Food and Drug Administration to conduct pregnancy exposure registries for medications used during pregnancy or in women of reproductive age. OTIS has an independent advisory board, however, and independently analyzes and publishes its findings. Progress reports are shared with the pharmaceutical companies, and in turn, the FDA, Dr. Chambers said.
To refer patients for MotherToBaby studies, physicians can use an online referral form found on the MothertoBaby web site, a service of OTIS, or call the pregnancy studies team at 877-311-8972 to provide them with the patient’s name or number. Patients may also be given the number and advised to consider calling. MotherToBaby offers medication fact sheets that answer questions about exposures during pregnancy and breastfeeding, and runs a free and confidential teratogen counseling service: 866-626-6847.
Christina Chambers, PhD, MPH, who runs the MotherToBaby Pregnancy Studies research center at the University of California, San Diego, has found most pregnant women to be “entirely altruistic” about sharing their experiences with drug treatment during pregnancy.
– but dermatologists, rheumatologists, and their female patients with psoriasis and psoriatic arthritis (PsA) want much more.
And women’s participation in the MotherToBaby studies conducted by the nonprofit Organization of Teratology Information Specialists (OTIS) is key, say physicians who are treating women of reproductive age. OTIS is now listed in drug labeling as the “pregnancy registry” contact for many of the medications they may be discussing with patients.
Dr. Chambers said that most women appreciate “that participating in a study may not help her with her pregnancy, but it can help her sister or her friend or someone else who has these same questions in planning a pregnancy of ‘Can I stay on my treatment?’ or, in the case of an unplanned pregnancy, ‘Should I be concerned?’ ”
OTIS has enrolled women with psoriasis and/or PsA in studies of nine medications, most of them biologics (both TNF-alpha blockers and newer anti-interleukin agents).
Four of the studies – those evaluating etanercept (Enbrel), adalimumab (Humira), abatacept (Orencia), and ustekinumab (Stelara) – are now closed to enrollment with analyses either underway or completed. The other five are currently enrolling patients and involve treatment with certolizumab pegol (Cimzia), tildrakizumab (Ilumya), apremilast (Otezla), guselkumab (Tremfya), and tofacitinib (Xeljanz).
Lisa R. Sammaritano, MD, a rheumatologist at the Hospital for Special Surgery, New York, who led the development of the American College of Rheumatology’s first guideline for the management of reproductive health in rheumatic and musculoskeletal diseases, recommends to some of her patients that they contact OTIS. “Their pregnancy registry studies have added important information to the field over the years,” she said.
Most recently, a study of the anti–TNF-alpha medication adalimumab that began in 2004 in pregnant patients with RA and Crohn’s disease culminated in a 2019 PLOS ONE paper reporting no associations between exposure to the medication and an increased risk of adverse outcomes. The outcomes studied were major structural birth defects, minor defects, spontaneous abortion, preterm delivery, prenatal and postnatal growth deficiency, serious or opportunistic infections, and malignancies.
An analysis is underway of adalimumab exposure in women with PsA – a patient subset that was added after the study started. But in the meantime, Dr. Chambers said, the 2019 research article is relevant to questions of drug safety across indications.
OTIS’s MothertoBaby studies are structured as prospective cohort studies. Dr. Chambers, a perinatal epidemiologist, is president of OTIS, which recruits women who have an exposure to the medication under study – at least one dose, for any length of time. And in most cases, it also recruits women with the underlying condition but no exposure and healthy women without the condition to represent the general population.
It’s the disease-matched comparison group that makes OTIS’s studies different from traditional pregnancy registries involving “a simple exposure series and outcomes that are described in the context of what you’d expect in the general population,” said Dr. Chambers, professor in the department of pediatrics, as well as family and preventative medicine, at UCSD and codirector of the Center for Better Beginnings at that university. “Many maternal conditions themselves [or their comorbidities] carry some risk of adverse outcomes in pregnancy.”
The OTIS studies typically involve at least 100 exposed pregnancies and a similar number of unexposed pregnancies; some have cohorts of 200-300.
The recently published study of adalimumab, for instance, included 257 women with exposure to the drug and 120 women in a disease comparison group with no exposure. In addition to finding no associations between drug exposure and adverse outcomes, the study found that women with RA or Crohn’s were at increased risk of preterm delivery, irrespective of adalimumab exposure.
“There’s insufficient [power with any of these numbers] to come to the conclusion that a drug is safe,” she said. “But what we have been able to say [through our studies] is that we’ve looked carefully at the whole array of outcomes ... and we don’t see anything unusual. That early view can be reassuring” until large population-based studies or claims analyses become possible.
Dr. Sammaritano, also with Weill Cornell Medicine, New York, said that she does not recommend registry participation for patients who stop biologics at the diagnosis of pregnancy. Since “the start of IgG antibody transfer during pregnancy is about 16 weeks,” she worries that including these patients might lead to falsely reassuring findings. “We are most interested in [knowing the outcomes of] patients who must continue the drugs through pregnancy,” she said.
Dr. Chambers, however, said that in her view, placental transfer is not a requirement for a medication to have some effect on the outcome of pregnancy. “The outcome could be influenced by an effect of the medication that doesn’t require placental transfer or require placental transfer in large amounts,” she said. “So it’s relevant to examine exposures that have occurred only in the first trimester, and this is especially true for the outcome of major birth defects, most of which are initiated in the first trimester.”
The MotherToBaby studies typically include both early, short exposures and longer exposures, she said. “And certainly, duration of use is a factor that we do consider in looking at specific outcomes such as growth, preterm delivery, and risk of serious or opportunistic infections.”
(In the published study of adalimumab, 65.3% of women in the medication-exposed cohort used the medication in all three trimesters, 10.5% in the first and second trimesters, and 22.4% in the first trimester only.)
Women participating in the MotherToBaby studies complete two to four interviews during pregnancy and may be interviewed again after delivery. They are asked for their permission to share a copy of their medical records – and their baby’s medical records – and their babies receive a follow-up pediatric exam by a pediatrician with expertise in dysmorphology/genetics (who is blinded to exposure status), most commonly in the participant’s home. Providers are not asked to enter any data.
Eliza Chakravarty, MD, a rheumatologist with the Oklahoma Medical Research Foundation in Oklahoma City who treats patients with PsA who are pregnant or considering pregnancy, said that her referrals for research participation “have been mostly to MothertoBaby.”
“Most drug companies [in the autoimmune space] are now contracting with them [for their pregnancy exposure research],” she said. “I really like that it’s become so centralized.”
She tells patients that many questions can be answered through research, that their experience matters, and that “there are benefits” to the extra pediatric examination. “I give them the information and let them decide whether or not they want to call [MotherToBaby],” she said. “I don’t want to impose. I want to make them aware.”
Dr. Chambers emphasizes to patients and physicians that the studies are strictly observational and do not require any changes in personal or medical regimens. “When people hear the word ‘research’ they think of clinical trials. We’re saying, you and your provider do everything you normally would do, just let us observe what happens during your pregnancy.”
Physicians should assure patients, moreover, that “just because the drug is being studied doesn’t mean there’s a known risk or even a suspected risk,” she said.
The MotherToBaby studies receive funding from the pharmaceutical companies, which are required by the Food and Drug Administration to conduct pregnancy exposure registries for medications used during pregnancy or in women of reproductive age. OTIS has an independent advisory board, however, and independently analyzes and publishes its findings. Progress reports are shared with the pharmaceutical companies, and in turn, the FDA, Dr. Chambers said.
To refer patients for MotherToBaby studies, physicians can use an online referral form found on the MothertoBaby web site, a service of OTIS, or call the pregnancy studies team at 877-311-8972 to provide them with the patient’s name or number. Patients may also be given the number and advised to consider calling. MotherToBaby offers medication fact sheets that answer questions about exposures during pregnancy and breastfeeding, and runs a free and confidential teratogen counseling service: 866-626-6847.
‘Overwhelming evidence’ FDA’s opioid approval process is shoddy
Despite the ongoing epidemic of misuse, overuse, and diversion of opioids, the Food and Drug Administration has set a low bar for approval of these medications over the past 20 years, new research suggests.
The study results also show that the FDA did not require manufacturers to collect safety data on tolerance, withdrawal, overdose, misuse, and diversion in any rigorous fashion.
In addition, during the study period, 17 of the 39 new drug applications (NDAs) (only one was an innovator product, known as a new molecular entity) for chronic pain were approved with an “enriched enrollment randomized withdrawal” (EERW) trial design. Such a design, in this case, allowed manufacturers to exclude 32%-43% of the initially enrolled patients from the double-blind treatment phase.
“The question for regulators, policy makers, and others is: How did we get to a point where these approvals took place based on trials that were by design unlikely to yield some of the most important information about safety and efficacy that patients and clinicians would care about?” study investigator G. Caleb Alexander, MD, Johns Hopkins University, Baltimore, said in an interview.
The study was published online Sept. 29 in the Annals of Internal Medicine.
‘Cooking the books’
Little is known about the evidence required by the FDA for new approvals of opioid analgesics.
To characterize the quality of safety and efficacy data in NDAs for opioid analgesics approved by the FDA between 1997 and 2018, the investigators conducted the cross-sectional analysis using data from ClinicalTrials.gov, FDA reviews, and peer-reviewed publications regarding phase 3 pivotal trials.
The investigators examined the key characteristics of each NDA, including the number, size, and duration of pivotal trials, trial control groups, use of EERW, and systematically measured safety outcomes.
Results showed that most of the 48 NDAs evaluated were for new dosage forms (52.1%) or new formulations (18.8%). Only one (2.1%) was for a new molecular entity.
Of 39 NDAs approved for the treatment of chronic pain, only 21 products were supported by at least one pivotal trial. The mean duration of these 28 trials was 84 days, and they enrolled a median of 299 patients.
Results showed that, for 17 of the 39 opioids approved for chronic pain, pivotal trials had an EERW design. For the latest period – 2012-2018 – trials of all eight of the approved opioids used the EERW method.
This EERW design allows the manufacturer to assess efficacy “among a subset of patients most likely to respond and least likely to have adverse effects, reducing generalizability to real-world settings,” the investigators noted.
They called on the FDA to stop relying on this type of trial to assess opioid efficacy.
In an August 2020 article, Andrew Kolodny, MD, pointed out the pitfalls of the EERW approach. In such a study, all participants are made physiologically dependent on the opioid in a 4- to 6-week open-label phase. Only those who tolerate the drug and find it helpful are included in the randomized study. Dr. Kolodny is codirector of opioid policy research at Brandeis University, Waltham, Mass.
“Critics of EERW have correctly described this methodology as ‘cooking the books,’ ” Dr. Kolodny writes.
He noted that the agency’s decision to rely on EERW trials for opioids was “based on discussions at private meetings between FDA officials and pharmaceutical company executives hosted by an organization called Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials.” The 2013 meetings were reported in an article published in the Washington Post.
Little sign of change
Among NDAs for chronic pain, the investigators found that eight (20.5%) included pooled safety reviews that reported systematic assessment of diversion. Seven (17.9%) reported systematic measurement of nonmedical use, and 15 (38.5%) assessed incident tolerance.
The study revealed that eight of nine products that were approved for acute pain were supported by at least one pivotal trial. The median duration of these 19 trials was 1 day, and they enrolled a median of 329 patients.
The investigators noted that the findings “underscore the evidence gaps that have limited clinicians’ and patients’ understanding and appreciation of the inherent risks of prescription opioid analgesics.”
Dr. Alexander, who has been an FDA advisory committee chairman and currently serves as a consultant to plaintiffs who are suing opioid manufacturers in federal multidistrict litigation, said the study “is a story about missed opportunities to improve the safety and to improve the regulatory review of these products.”
Coinvestigator Peter Lurie, MD, who was an official at the FDA from 2009 to 2017, said that “there’s not a lot of signs that things are changing” at the agency.
The study shows that the FDA has “accepted what the companies have been presenting,” said Dr. Lurie, who is president of the Center for Science in the Public Interest.
The FDA “absolutely has the authority” to require manufacturers to undertake more rigorous trials, but agency culture keeps it from making such demands, especially if doing so means a new applicant might have to conduct trials that weren’t previously required, Dr. Lurie said in an interview.
“FDA is pretty rigorous about trying to establish a level playing field. That’s a virtuous thing, but it becomes problematic when that prevents change,” said Dr. Lurie.
The most recent FDA guidance to manufacturers, issued in 2019, does not provide advice on criteria for endpoints, study duration, or which populations are most likely to benefit from opioid treatment. The agency also does not require drug manufacturers to formally collect data on safety, tolerance, overdose symptoms, or constipation.
The guidance does suggest that the agency would likely take into account public health considerations when evaluating opioids, such as the risk to the overall population for overdose and diversion.
‘Overwhelming evidence’
Dr. Kolodny said that, as far as he is aware, “this is the first scientific publication in a peer-reviewed journal demonstrating clearly the problems with FDA’s opioid approval process.”
The article offers “overwhelming evidence that they are improperly approving the most dangerous medications – medications that killed more people than any other medication on the market,” added Dr. Kolodny, who is also president of Physicians for Responsible Opioid Prescribing.
Asked to respond to the study findings, FDA spokesperson Charles Kohler said the agency “does not comment on specific studies but evaluates them as part of the body of evidence to further our understanding about a particular issue and assist in our mission to protect public health.”
A version of this article originally appeared on Medscape.com.
Despite the ongoing epidemic of misuse, overuse, and diversion of opioids, the Food and Drug Administration has set a low bar for approval of these medications over the past 20 years, new research suggests.
The study results also show that the FDA did not require manufacturers to collect safety data on tolerance, withdrawal, overdose, misuse, and diversion in any rigorous fashion.
In addition, during the study period, 17 of the 39 new drug applications (NDAs) (only one was an innovator product, known as a new molecular entity) for chronic pain were approved with an “enriched enrollment randomized withdrawal” (EERW) trial design. Such a design, in this case, allowed manufacturers to exclude 32%-43% of the initially enrolled patients from the double-blind treatment phase.
“The question for regulators, policy makers, and others is: How did we get to a point where these approvals took place based on trials that were by design unlikely to yield some of the most important information about safety and efficacy that patients and clinicians would care about?” study investigator G. Caleb Alexander, MD, Johns Hopkins University, Baltimore, said in an interview.
The study was published online Sept. 29 in the Annals of Internal Medicine.
‘Cooking the books’
Little is known about the evidence required by the FDA for new approvals of opioid analgesics.
To characterize the quality of safety and efficacy data in NDAs for opioid analgesics approved by the FDA between 1997 and 2018, the investigators conducted the cross-sectional analysis using data from ClinicalTrials.gov, FDA reviews, and peer-reviewed publications regarding phase 3 pivotal trials.
The investigators examined the key characteristics of each NDA, including the number, size, and duration of pivotal trials, trial control groups, use of EERW, and systematically measured safety outcomes.
Results showed that most of the 48 NDAs evaluated were for new dosage forms (52.1%) or new formulations (18.8%). Only one (2.1%) was for a new molecular entity.
Of 39 NDAs approved for the treatment of chronic pain, only 21 products were supported by at least one pivotal trial. The mean duration of these 28 trials was 84 days, and they enrolled a median of 299 patients.
Results showed that, for 17 of the 39 opioids approved for chronic pain, pivotal trials had an EERW design. For the latest period – 2012-2018 – trials of all eight of the approved opioids used the EERW method.
This EERW design allows the manufacturer to assess efficacy “among a subset of patients most likely to respond and least likely to have adverse effects, reducing generalizability to real-world settings,” the investigators noted.
They called on the FDA to stop relying on this type of trial to assess opioid efficacy.
In an August 2020 article, Andrew Kolodny, MD, pointed out the pitfalls of the EERW approach. In such a study, all participants are made physiologically dependent on the opioid in a 4- to 6-week open-label phase. Only those who tolerate the drug and find it helpful are included in the randomized study. Dr. Kolodny is codirector of opioid policy research at Brandeis University, Waltham, Mass.
“Critics of EERW have correctly described this methodology as ‘cooking the books,’ ” Dr. Kolodny writes.
He noted that the agency’s decision to rely on EERW trials for opioids was “based on discussions at private meetings between FDA officials and pharmaceutical company executives hosted by an organization called Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials.” The 2013 meetings were reported in an article published in the Washington Post.
Little sign of change
Among NDAs for chronic pain, the investigators found that eight (20.5%) included pooled safety reviews that reported systematic assessment of diversion. Seven (17.9%) reported systematic measurement of nonmedical use, and 15 (38.5%) assessed incident tolerance.
The study revealed that eight of nine products that were approved for acute pain were supported by at least one pivotal trial. The median duration of these 19 trials was 1 day, and they enrolled a median of 329 patients.
The investigators noted that the findings “underscore the evidence gaps that have limited clinicians’ and patients’ understanding and appreciation of the inherent risks of prescription opioid analgesics.”
Dr. Alexander, who has been an FDA advisory committee chairman and currently serves as a consultant to plaintiffs who are suing opioid manufacturers in federal multidistrict litigation, said the study “is a story about missed opportunities to improve the safety and to improve the regulatory review of these products.”
Coinvestigator Peter Lurie, MD, who was an official at the FDA from 2009 to 2017, said that “there’s not a lot of signs that things are changing” at the agency.
The study shows that the FDA has “accepted what the companies have been presenting,” said Dr. Lurie, who is president of the Center for Science in the Public Interest.
The FDA “absolutely has the authority” to require manufacturers to undertake more rigorous trials, but agency culture keeps it from making such demands, especially if doing so means a new applicant might have to conduct trials that weren’t previously required, Dr. Lurie said in an interview.
“FDA is pretty rigorous about trying to establish a level playing field. That’s a virtuous thing, but it becomes problematic when that prevents change,” said Dr. Lurie.
The most recent FDA guidance to manufacturers, issued in 2019, does not provide advice on criteria for endpoints, study duration, or which populations are most likely to benefit from opioid treatment. The agency also does not require drug manufacturers to formally collect data on safety, tolerance, overdose symptoms, or constipation.
The guidance does suggest that the agency would likely take into account public health considerations when evaluating opioids, such as the risk to the overall population for overdose and diversion.
‘Overwhelming evidence’
Dr. Kolodny said that, as far as he is aware, “this is the first scientific publication in a peer-reviewed journal demonstrating clearly the problems with FDA’s opioid approval process.”
The article offers “overwhelming evidence that they are improperly approving the most dangerous medications – medications that killed more people than any other medication on the market,” added Dr. Kolodny, who is also president of Physicians for Responsible Opioid Prescribing.
Asked to respond to the study findings, FDA spokesperson Charles Kohler said the agency “does not comment on specific studies but evaluates them as part of the body of evidence to further our understanding about a particular issue and assist in our mission to protect public health.”
A version of this article originally appeared on Medscape.com.
Despite the ongoing epidemic of misuse, overuse, and diversion of opioids, the Food and Drug Administration has set a low bar for approval of these medications over the past 20 years, new research suggests.
The study results also show that the FDA did not require manufacturers to collect safety data on tolerance, withdrawal, overdose, misuse, and diversion in any rigorous fashion.
In addition, during the study period, 17 of the 39 new drug applications (NDAs) (only one was an innovator product, known as a new molecular entity) for chronic pain were approved with an “enriched enrollment randomized withdrawal” (EERW) trial design. Such a design, in this case, allowed manufacturers to exclude 32%-43% of the initially enrolled patients from the double-blind treatment phase.
“The question for regulators, policy makers, and others is: How did we get to a point where these approvals took place based on trials that were by design unlikely to yield some of the most important information about safety and efficacy that patients and clinicians would care about?” study investigator G. Caleb Alexander, MD, Johns Hopkins University, Baltimore, said in an interview.
The study was published online Sept. 29 in the Annals of Internal Medicine.
‘Cooking the books’
Little is known about the evidence required by the FDA for new approvals of opioid analgesics.
To characterize the quality of safety and efficacy data in NDAs for opioid analgesics approved by the FDA between 1997 and 2018, the investigators conducted the cross-sectional analysis using data from ClinicalTrials.gov, FDA reviews, and peer-reviewed publications regarding phase 3 pivotal trials.
The investigators examined the key characteristics of each NDA, including the number, size, and duration of pivotal trials, trial control groups, use of EERW, and systematically measured safety outcomes.
Results showed that most of the 48 NDAs evaluated were for new dosage forms (52.1%) or new formulations (18.8%). Only one (2.1%) was for a new molecular entity.
Of 39 NDAs approved for the treatment of chronic pain, only 21 products were supported by at least one pivotal trial. The mean duration of these 28 trials was 84 days, and they enrolled a median of 299 patients.
Results showed that, for 17 of the 39 opioids approved for chronic pain, pivotal trials had an EERW design. For the latest period – 2012-2018 – trials of all eight of the approved opioids used the EERW method.
This EERW design allows the manufacturer to assess efficacy “among a subset of patients most likely to respond and least likely to have adverse effects, reducing generalizability to real-world settings,” the investigators noted.
They called on the FDA to stop relying on this type of trial to assess opioid efficacy.
In an August 2020 article, Andrew Kolodny, MD, pointed out the pitfalls of the EERW approach. In such a study, all participants are made physiologically dependent on the opioid in a 4- to 6-week open-label phase. Only those who tolerate the drug and find it helpful are included in the randomized study. Dr. Kolodny is codirector of opioid policy research at Brandeis University, Waltham, Mass.
“Critics of EERW have correctly described this methodology as ‘cooking the books,’ ” Dr. Kolodny writes.
He noted that the agency’s decision to rely on EERW trials for opioids was “based on discussions at private meetings between FDA officials and pharmaceutical company executives hosted by an organization called Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials.” The 2013 meetings were reported in an article published in the Washington Post.
Little sign of change
Among NDAs for chronic pain, the investigators found that eight (20.5%) included pooled safety reviews that reported systematic assessment of diversion. Seven (17.9%) reported systematic measurement of nonmedical use, and 15 (38.5%) assessed incident tolerance.
The study revealed that eight of nine products that were approved for acute pain were supported by at least one pivotal trial. The median duration of these 19 trials was 1 day, and they enrolled a median of 329 patients.
The investigators noted that the findings “underscore the evidence gaps that have limited clinicians’ and patients’ understanding and appreciation of the inherent risks of prescription opioid analgesics.”
Dr. Alexander, who has been an FDA advisory committee chairman and currently serves as a consultant to plaintiffs who are suing opioid manufacturers in federal multidistrict litigation, said the study “is a story about missed opportunities to improve the safety and to improve the regulatory review of these products.”
Coinvestigator Peter Lurie, MD, who was an official at the FDA from 2009 to 2017, said that “there’s not a lot of signs that things are changing” at the agency.
The study shows that the FDA has “accepted what the companies have been presenting,” said Dr. Lurie, who is president of the Center for Science in the Public Interest.
The FDA “absolutely has the authority” to require manufacturers to undertake more rigorous trials, but agency culture keeps it from making such demands, especially if doing so means a new applicant might have to conduct trials that weren’t previously required, Dr. Lurie said in an interview.
“FDA is pretty rigorous about trying to establish a level playing field. That’s a virtuous thing, but it becomes problematic when that prevents change,” said Dr. Lurie.
The most recent FDA guidance to manufacturers, issued in 2019, does not provide advice on criteria for endpoints, study duration, or which populations are most likely to benefit from opioid treatment. The agency also does not require drug manufacturers to formally collect data on safety, tolerance, overdose symptoms, or constipation.
The guidance does suggest that the agency would likely take into account public health considerations when evaluating opioids, such as the risk to the overall population for overdose and diversion.
‘Overwhelming evidence’
Dr. Kolodny said that, as far as he is aware, “this is the first scientific publication in a peer-reviewed journal demonstrating clearly the problems with FDA’s opioid approval process.”
The article offers “overwhelming evidence that they are improperly approving the most dangerous medications – medications that killed more people than any other medication on the market,” added Dr. Kolodny, who is also president of Physicians for Responsible Opioid Prescribing.
Asked to respond to the study findings, FDA spokesperson Charles Kohler said the agency “does not comment on specific studies but evaluates them as part of the body of evidence to further our understanding about a particular issue and assist in our mission to protect public health.”
A version of this article originally appeared on Medscape.com.
Orthopedic problems in children can be the first indication of acute lymphoblastic leukemia
The diagnosis of acute lymphoblastic leukemia (ALL) can be delayed because of vague presentation and normal hematological results. Orthopedic manifestations may be the primary presentation of ALL to physicians, and such symptoms in children should be cause for suspicion, even in the absence of hematological abnormalities, according to a report published in the Journal of Orthopaedics.
The study retrospectively assessed 250 consecutive ALL patients at a single institution to identify the frequency of ALL cases presented to the orthopedic department and to determine the number of these patients presenting with normal hematological results, according to Amrath Raj BK, MD, and colleagues at the Manipal (India) Academy of Higher Education.
Suspicion warranted
Twenty-two of the 250 patients (8.8%) presented primarily to the orthopedic department (4 with vertebral compression fractures, 12 with joint pain, and 6 with bone pain), but were subsequently diagnosed with ALL. These results were comparable to previous studies. The mean patient age at the first visit was 5.6 years; 13 patients were boys, and 9 were girls. Six of these 22 patients (27.3%) had a normal peripheral blood smear, according to the researchers.
“Acute leukemia should be considered strongly as a differential diagnosis in children with severe osteoporosis and vertebral fractures. Initial orthopedic manifestations are not uncommon, and the primary physician should maintain a high index of suspicion as a peripheral smear is not diagnostic in all patients,” the researchers concluded.
The authors reported that there was no outside funding source and that they had no conflicts.
SOURCE: Raj BK A et al. Journal of Orthopaedics. 2020;22:326-330.
The diagnosis of acute lymphoblastic leukemia (ALL) can be delayed because of vague presentation and normal hematological results. Orthopedic manifestations may be the primary presentation of ALL to physicians, and such symptoms in children should be cause for suspicion, even in the absence of hematological abnormalities, according to a report published in the Journal of Orthopaedics.
The study retrospectively assessed 250 consecutive ALL patients at a single institution to identify the frequency of ALL cases presented to the orthopedic department and to determine the number of these patients presenting with normal hematological results, according to Amrath Raj BK, MD, and colleagues at the Manipal (India) Academy of Higher Education.
Suspicion warranted
Twenty-two of the 250 patients (8.8%) presented primarily to the orthopedic department (4 with vertebral compression fractures, 12 with joint pain, and 6 with bone pain), but were subsequently diagnosed with ALL. These results were comparable to previous studies. The mean patient age at the first visit was 5.6 years; 13 patients were boys, and 9 were girls. Six of these 22 patients (27.3%) had a normal peripheral blood smear, according to the researchers.
“Acute leukemia should be considered strongly as a differential diagnosis in children with severe osteoporosis and vertebral fractures. Initial orthopedic manifestations are not uncommon, and the primary physician should maintain a high index of suspicion as a peripheral smear is not diagnostic in all patients,” the researchers concluded.
The authors reported that there was no outside funding source and that they had no conflicts.
SOURCE: Raj BK A et al. Journal of Orthopaedics. 2020;22:326-330.
The diagnosis of acute lymphoblastic leukemia (ALL) can be delayed because of vague presentation and normal hematological results. Orthopedic manifestations may be the primary presentation of ALL to physicians, and such symptoms in children should be cause for suspicion, even in the absence of hematological abnormalities, according to a report published in the Journal of Orthopaedics.
The study retrospectively assessed 250 consecutive ALL patients at a single institution to identify the frequency of ALL cases presented to the orthopedic department and to determine the number of these patients presenting with normal hematological results, according to Amrath Raj BK, MD, and colleagues at the Manipal (India) Academy of Higher Education.
Suspicion warranted
Twenty-two of the 250 patients (8.8%) presented primarily to the orthopedic department (4 with vertebral compression fractures, 12 with joint pain, and 6 with bone pain), but were subsequently diagnosed with ALL. These results were comparable to previous studies. The mean patient age at the first visit was 5.6 years; 13 patients were boys, and 9 were girls. Six of these 22 patients (27.3%) had a normal peripheral blood smear, according to the researchers.
“Acute leukemia should be considered strongly as a differential diagnosis in children with severe osteoporosis and vertebral fractures. Initial orthopedic manifestations are not uncommon, and the primary physician should maintain a high index of suspicion as a peripheral smear is not diagnostic in all patients,” the researchers concluded.
The authors reported that there was no outside funding source and that they had no conflicts.
SOURCE: Raj BK A et al. Journal of Orthopaedics. 2020;22:326-330.
FROM THE JOURNAL OF ORTHOPAEDICS
Genetics and epigenetics could predict response to RA therapies
Machine-based learning of genetic and epigenetic characteristics of patients with rheumatoid arthritis could help to predict who is likely to benefit from the biologic drugs adalimumab and etanercept, according to results from a longitudinal, observational cohort study.
In the study, machine learning models created by researchers from Utrecht University in the Netherlands using different parameters predicted true-positive rates for response to adalimumab ranging from 76% to 90% and true-negative rates ranging from 70% to 89%, while for etanercept true-positive rates ranged from about 60% to 80% and true-negative rates ranged from about 82% to 98%.
“These results suggest that we can accurately predict the clinical response before adalimumab and etanercept treatment using molecular signatures-based machine learning models, although the prediction accuracy of these molecular signatures differs between cell types and treatments, underlining the need to study more than one drug, cell type, or epigenetic layers,” first author Weiyang Tao and colleagues wrote in Arthritis & Rheumatology. The ability to predict which tumor necrosis factor inhibitor (TNFi) is the first choice for treatment would be highly beneficial in reducing the time to effective treatment, which has been extensively proven to be a paramount factor for achieving long-sustained disease remission, they noted.
The researchers analyzed gene expression and epigenetic signatures in 80 patients with rheumatoid arthritis prior to treatment with adalimumab or etanercept and then examined patients’ response to treatment at 6 months. They then used that information to build a machine learning model to try to predict treatment response.
Overall, 47.5% of patients were treated with adalimumab, and 52.5% were treated with etanercept. Among the adalimumab group, 53% had a good or moderate response to treatment at 6 months, and among those treated with etanercept, 45% had a good or moderate response.
While there were no differences in baseline clinical parameters between responders and nonresponders, the study found significant genetic and epigenetic differences between patients.
They identified 549 genes that showed significantly different levels of expression between responders and nonresponders treated with adalimumab – in particular, genes involved in DNA and nucleotide binding – and 460 genes that were differentially expressed between etanercept responders and nonresponders, including genes involved in TNF-receptor signaling. However, only 2% of these differentially expressed genes were common in both the adalimumab and etanercept groups, suggesting treatment responses for these two medications have distinct gene signatures.
Looking at DNA methylation, researchers found 16,141 CpG positions – sites of DNA methylation – that were differentially methylated between adalimumab responders and nonresponders, 46% of which were hypermethylated among responders but not nonresponders. In the etanercept group, there were 17,026 differentially methylated sites in responders and nonresponders, 76.3% of which were hypermethylated among responders.
The researchers also noted that among the adalimumab responders, the hypermethylated sites were more likely to be found in the upstream and promoter regions of genes, and on CpG islands.
“Thus, on epigenetic level, we observed a distinct hypermethylation pattern between adalimumab and etanercept responders, suggesting the role of epigenetics in defining response towards adalimumab and to etanercept in PBMCs [peripheral blood mononuclear cells],” the authors wrote.
Given the differences in gene signatures seen in the adalimumab responders and etanercept responders, researchers speculated that different cell types might be involved in the responses to these two treatments. They undertook RNA sequencing on the variety of immune cell types known to be involved in rheumatoid arthritis, which revealed gene-expression differences between adalimumab responders and nonresponders in their CD4+ T cells but not in monocytes. However, the gene-expression differences between etanercept responders and nonresponders were seen in both CD4+ T cells and monocytes.
The study was supported by AbbVie, which manufactures adalimumab, and two authors were supported by the China Scholarship Council and the Netherlands Organization for Scientific Research. No conflicts of interest were declared.
SOURCE: Tao W et al. Arthritis Rheumatol. 2020 Sep 10. doi: 10.1002/art.41516.
Machine-based learning of genetic and epigenetic characteristics of patients with rheumatoid arthritis could help to predict who is likely to benefit from the biologic drugs adalimumab and etanercept, according to results from a longitudinal, observational cohort study.
In the study, machine learning models created by researchers from Utrecht University in the Netherlands using different parameters predicted true-positive rates for response to adalimumab ranging from 76% to 90% and true-negative rates ranging from 70% to 89%, while for etanercept true-positive rates ranged from about 60% to 80% and true-negative rates ranged from about 82% to 98%.
“These results suggest that we can accurately predict the clinical response before adalimumab and etanercept treatment using molecular signatures-based machine learning models, although the prediction accuracy of these molecular signatures differs between cell types and treatments, underlining the need to study more than one drug, cell type, or epigenetic layers,” first author Weiyang Tao and colleagues wrote in Arthritis & Rheumatology. The ability to predict which tumor necrosis factor inhibitor (TNFi) is the first choice for treatment would be highly beneficial in reducing the time to effective treatment, which has been extensively proven to be a paramount factor for achieving long-sustained disease remission, they noted.
The researchers analyzed gene expression and epigenetic signatures in 80 patients with rheumatoid arthritis prior to treatment with adalimumab or etanercept and then examined patients’ response to treatment at 6 months. They then used that information to build a machine learning model to try to predict treatment response.
Overall, 47.5% of patients were treated with adalimumab, and 52.5% were treated with etanercept. Among the adalimumab group, 53% had a good or moderate response to treatment at 6 months, and among those treated with etanercept, 45% had a good or moderate response.
While there were no differences in baseline clinical parameters between responders and nonresponders, the study found significant genetic and epigenetic differences between patients.
They identified 549 genes that showed significantly different levels of expression between responders and nonresponders treated with adalimumab – in particular, genes involved in DNA and nucleotide binding – and 460 genes that were differentially expressed between etanercept responders and nonresponders, including genes involved in TNF-receptor signaling. However, only 2% of these differentially expressed genes were common in both the adalimumab and etanercept groups, suggesting treatment responses for these two medications have distinct gene signatures.
Looking at DNA methylation, researchers found 16,141 CpG positions – sites of DNA methylation – that were differentially methylated between adalimumab responders and nonresponders, 46% of which were hypermethylated among responders but not nonresponders. In the etanercept group, there were 17,026 differentially methylated sites in responders and nonresponders, 76.3% of which were hypermethylated among responders.
The researchers also noted that among the adalimumab responders, the hypermethylated sites were more likely to be found in the upstream and promoter regions of genes, and on CpG islands.
“Thus, on epigenetic level, we observed a distinct hypermethylation pattern between adalimumab and etanercept responders, suggesting the role of epigenetics in defining response towards adalimumab and to etanercept in PBMCs [peripheral blood mononuclear cells],” the authors wrote.
Given the differences in gene signatures seen in the adalimumab responders and etanercept responders, researchers speculated that different cell types might be involved in the responses to these two treatments. They undertook RNA sequencing on the variety of immune cell types known to be involved in rheumatoid arthritis, which revealed gene-expression differences between adalimumab responders and nonresponders in their CD4+ T cells but not in monocytes. However, the gene-expression differences between etanercept responders and nonresponders were seen in both CD4+ T cells and monocytes.
The study was supported by AbbVie, which manufactures adalimumab, and two authors were supported by the China Scholarship Council and the Netherlands Organization for Scientific Research. No conflicts of interest were declared.
SOURCE: Tao W et al. Arthritis Rheumatol. 2020 Sep 10. doi: 10.1002/art.41516.
Machine-based learning of genetic and epigenetic characteristics of patients with rheumatoid arthritis could help to predict who is likely to benefit from the biologic drugs adalimumab and etanercept, according to results from a longitudinal, observational cohort study.
In the study, machine learning models created by researchers from Utrecht University in the Netherlands using different parameters predicted true-positive rates for response to adalimumab ranging from 76% to 90% and true-negative rates ranging from 70% to 89%, while for etanercept true-positive rates ranged from about 60% to 80% and true-negative rates ranged from about 82% to 98%.
“These results suggest that we can accurately predict the clinical response before adalimumab and etanercept treatment using molecular signatures-based machine learning models, although the prediction accuracy of these molecular signatures differs between cell types and treatments, underlining the need to study more than one drug, cell type, or epigenetic layers,” first author Weiyang Tao and colleagues wrote in Arthritis & Rheumatology. The ability to predict which tumor necrosis factor inhibitor (TNFi) is the first choice for treatment would be highly beneficial in reducing the time to effective treatment, which has been extensively proven to be a paramount factor for achieving long-sustained disease remission, they noted.
The researchers analyzed gene expression and epigenetic signatures in 80 patients with rheumatoid arthritis prior to treatment with adalimumab or etanercept and then examined patients’ response to treatment at 6 months. They then used that information to build a machine learning model to try to predict treatment response.
Overall, 47.5% of patients were treated with adalimumab, and 52.5% were treated with etanercept. Among the adalimumab group, 53% had a good or moderate response to treatment at 6 months, and among those treated with etanercept, 45% had a good or moderate response.
While there were no differences in baseline clinical parameters between responders and nonresponders, the study found significant genetic and epigenetic differences between patients.
They identified 549 genes that showed significantly different levels of expression between responders and nonresponders treated with adalimumab – in particular, genes involved in DNA and nucleotide binding – and 460 genes that were differentially expressed between etanercept responders and nonresponders, including genes involved in TNF-receptor signaling. However, only 2% of these differentially expressed genes were common in both the adalimumab and etanercept groups, suggesting treatment responses for these two medications have distinct gene signatures.
Looking at DNA methylation, researchers found 16,141 CpG positions – sites of DNA methylation – that were differentially methylated between adalimumab responders and nonresponders, 46% of which were hypermethylated among responders but not nonresponders. In the etanercept group, there were 17,026 differentially methylated sites in responders and nonresponders, 76.3% of which were hypermethylated among responders.
The researchers also noted that among the adalimumab responders, the hypermethylated sites were more likely to be found in the upstream and promoter regions of genes, and on CpG islands.
“Thus, on epigenetic level, we observed a distinct hypermethylation pattern between adalimumab and etanercept responders, suggesting the role of epigenetics in defining response towards adalimumab and to etanercept in PBMCs [peripheral blood mononuclear cells],” the authors wrote.
Given the differences in gene signatures seen in the adalimumab responders and etanercept responders, researchers speculated that different cell types might be involved in the responses to these two treatments. They undertook RNA sequencing on the variety of immune cell types known to be involved in rheumatoid arthritis, which revealed gene-expression differences between adalimumab responders and nonresponders in their CD4+ T cells but not in monocytes. However, the gene-expression differences between etanercept responders and nonresponders were seen in both CD4+ T cells and monocytes.
The study was supported by AbbVie, which manufactures adalimumab, and two authors were supported by the China Scholarship Council and the Netherlands Organization for Scientific Research. No conflicts of interest were declared.
SOURCE: Tao W et al. Arthritis Rheumatol. 2020 Sep 10. doi: 10.1002/art.41516.
FROM ARTHRITIS & RHEUMATOLOGY
FDA adds polyarticular-course JIA to approved indications for tofacitinib
The Food and Drug Administration has (pJIA).

The approval, announced Sept. 28 by tofacitinib’s manufacturer, Pfizer, marks the first JAK inhibitor to be approved for the condition in the United States and is the fourth indication to be approved for the drug after approvals in adult patients with moderate to severe rheumatoid arthritis following methotrexate failure, active psoriatic arthritis after disease-modifying antirheumatic drug failure, and moderate to severe ulcerative colitis after failure on a tumor necrosis factor inhibitor.
The agency based its approval on a phase 3, multinational, randomized, double-blind, controlled withdrawal study that had an 18-week, open-label, run-in phase involving 225 patients who twice daily took either a 5-mg tablet or, in patients under 40 kg, a weight-based lower dose in the form of a 1 mg/mL oral solution, according to the company press release. A total of 173 patients from this phase met JIA American College of Rheumatology 30 response criteria, defined as 30% or greater improvement in three of six JIA core set variables and worsening in no more than one of the core set variables; they were then randomized in part 2 of the study to continue the same dose of tofacitinib or receive placebo until 44 weeks. By the end of this period, 31% who received tofacitinib had a disease flare, compared with 55% on placebo (P = .0007). Disease flare was defined as a 30% or greater worsening in at least three of the six variables of the JIA core set, with no more than one of the remaining JIA core response variables improving by 30% or more after randomization.
The types of adverse drug reactions in patients with pJIA were consistent with those seen in adult rheumatoid arthritis patients, according to Pfizer. Serious adverse drug reactions have most commonly been serious infections that may lead to hospitalization or death, and most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids. Common adverse drug reactions reported in 2% or more of patients during the first 3 months in controlled clinical trials in patients with rheumatoid arthritis taking tofacitinib at 5 mg twice daily were upper respiratory tract infection, nasopharyngitis, diarrhea, headache, and hypertension.
While the 5-mg tablet formulation is already available, Pfizer said it expects the oral solution to be available by the end of the first quarter in 2021.
Prescribing information can be found on the FDA website.
The Food and Drug Administration has (pJIA).
The approval, announced Sept. 28 by tofacitinib’s manufacturer, Pfizer, marks the first JAK inhibitor to be approved for the condition in the United States and is the fourth indication to be approved for the drug after approvals in adult patients with moderate to severe rheumatoid arthritis following methotrexate failure, active psoriatic arthritis after disease-modifying antirheumatic drug failure, and moderate to severe ulcerative colitis after failure on a tumor necrosis factor inhibitor.
The agency based its approval on a phase 3, multinational, randomized, double-blind, controlled withdrawal study that had an 18-week, open-label, run-in phase involving 225 patients who twice daily took either a 5-mg tablet or, in patients under 40 kg, a weight-based lower dose in the form of a 1 mg/mL oral solution, according to the company press release. A total of 173 patients from this phase met JIA American College of Rheumatology 30 response criteria, defined as 30% or greater improvement in three of six JIA core set variables and worsening in no more than one of the core set variables; they were then randomized in part 2 of the study to continue the same dose of tofacitinib or receive placebo until 44 weeks. By the end of this period, 31% who received tofacitinib had a disease flare, compared with 55% on placebo (P = .0007). Disease flare was defined as a 30% or greater worsening in at least three of the six variables of the JIA core set, with no more than one of the remaining JIA core response variables improving by 30% or more after randomization.
The types of adverse drug reactions in patients with pJIA were consistent with those seen in adult rheumatoid arthritis patients, according to Pfizer. Serious adverse drug reactions have most commonly been serious infections that may lead to hospitalization or death, and most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids. Common adverse drug reactions reported in 2% or more of patients during the first 3 months in controlled clinical trials in patients with rheumatoid arthritis taking tofacitinib at 5 mg twice daily were upper respiratory tract infection, nasopharyngitis, diarrhea, headache, and hypertension.
While the 5-mg tablet formulation is already available, Pfizer said it expects the oral solution to be available by the end of the first quarter in 2021.
Prescribing information can be found on the FDA website.
The Food and Drug Administration has (pJIA).
The approval, announced Sept. 28 by tofacitinib’s manufacturer, Pfizer, marks the first JAK inhibitor to be approved for the condition in the United States and is the fourth indication to be approved for the drug after approvals in adult patients with moderate to severe rheumatoid arthritis following methotrexate failure, active psoriatic arthritis after disease-modifying antirheumatic drug failure, and moderate to severe ulcerative colitis after failure on a tumor necrosis factor inhibitor.
The agency based its approval on a phase 3, multinational, randomized, double-blind, controlled withdrawal study that had an 18-week, open-label, run-in phase involving 225 patients who twice daily took either a 5-mg tablet or, in patients under 40 kg, a weight-based lower dose in the form of a 1 mg/mL oral solution, according to the company press release. A total of 173 patients from this phase met JIA American College of Rheumatology 30 response criteria, defined as 30% or greater improvement in three of six JIA core set variables and worsening in no more than one of the core set variables; they were then randomized in part 2 of the study to continue the same dose of tofacitinib or receive placebo until 44 weeks. By the end of this period, 31% who received tofacitinib had a disease flare, compared with 55% on placebo (P = .0007). Disease flare was defined as a 30% or greater worsening in at least three of the six variables of the JIA core set, with no more than one of the remaining JIA core response variables improving by 30% or more after randomization.
The types of adverse drug reactions in patients with pJIA were consistent with those seen in adult rheumatoid arthritis patients, according to Pfizer. Serious adverse drug reactions have most commonly been serious infections that may lead to hospitalization or death, and most patients who developed these infections were taking concomitant immunosuppressants, such as methotrexate or corticosteroids. Common adverse drug reactions reported in 2% or more of patients during the first 3 months in controlled clinical trials in patients with rheumatoid arthritis taking tofacitinib at 5 mg twice daily were upper respiratory tract infection, nasopharyngitis, diarrhea, headache, and hypertension.
While the 5-mg tablet formulation is already available, Pfizer said it expects the oral solution to be available by the end of the first quarter in 2021.
Prescribing information can be found on the FDA website.
Children’s share of new COVID-19 cases is on the rise
The cumulative percentage of COVID-19 cases reported in children continues to climb, but “the history behind that cumulative number shows substantial change,” according to a new analysis of state health department data.
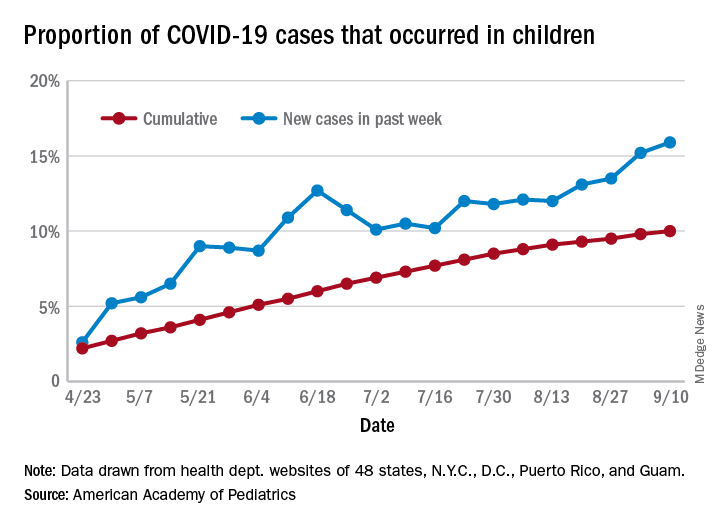
As of Sept. 10, the 549,432 cases in children represented 10.0% of all reported COVID-19 cases in the United States following a substantial rise over the course of the pandemic – the figure was 7.7% on July 16 and 3.2% on May 7, Blake Sisk, PhD, of the American Academy of Pediatrics and associates reported Sept. 29 in Pediatrics.
Unlike the cumulative number, the weekly proportion of cases in children fell early in the summer but then started climbing again in late July. Dr. Sisk and associates wrote.
Despite the increase, however, the proportion of pediatric COVID-19 cases is still well below children’s share of the overall population (22.6%). Also, “it is unclear how much of the increase in child cases is due to increased testing capacity, although CDC data from public and commercial laboratories show the share of all tests administered to children ages 0-17 has remained stable at 5%-7% since late April,” they said.
Data for the current report were drawn from 49 state health department websites (New York state does not report ages for COVID-19 cases), along with New York City, the District of Columbia, Puerto Rico, and Guam. Alabama changed its definition of a child case in August and was not included in the trend analysis (see graph), the investigators explained.
Those data show “substantial variation in case growth by region: in April, a preponderance of cases was in the Northeast. In June, cases surged in the South and West, followed by mid-July increases in the Midwest,” Dr. Sisk and associates said.
The increase among children in Midwest states is ongoing with the number of new cases reaching its highest level yet during the week ending Sept. 10, they reported.
SOURCE: Sisk B et al. Pediatrics. 2020 Sep 29. doi: 10.1542/peds.2020-027425.
The cumulative percentage of COVID-19 cases reported in children continues to climb, but “the history behind that cumulative number shows substantial change,” according to a new analysis of state health department data.
As of Sept. 10, the 549,432 cases in children represented 10.0% of all reported COVID-19 cases in the United States following a substantial rise over the course of the pandemic – the figure was 7.7% on July 16 and 3.2% on May 7, Blake Sisk, PhD, of the American Academy of Pediatrics and associates reported Sept. 29 in Pediatrics.
Unlike the cumulative number, the weekly proportion of cases in children fell early in the summer but then started climbing again in late July. Dr. Sisk and associates wrote.
Despite the increase, however, the proportion of pediatric COVID-19 cases is still well below children’s share of the overall population (22.6%). Also, “it is unclear how much of the increase in child cases is due to increased testing capacity, although CDC data from public and commercial laboratories show the share of all tests administered to children ages 0-17 has remained stable at 5%-7% since late April,” they said.
Data for the current report were drawn from 49 state health department websites (New York state does not report ages for COVID-19 cases), along with New York City, the District of Columbia, Puerto Rico, and Guam. Alabama changed its definition of a child case in August and was not included in the trend analysis (see graph), the investigators explained.
Those data show “substantial variation in case growth by region: in April, a preponderance of cases was in the Northeast. In June, cases surged in the South and West, followed by mid-July increases in the Midwest,” Dr. Sisk and associates said.
The increase among children in Midwest states is ongoing with the number of new cases reaching its highest level yet during the week ending Sept. 10, they reported.
SOURCE: Sisk B et al. Pediatrics. 2020 Sep 29. doi: 10.1542/peds.2020-027425.
The cumulative percentage of COVID-19 cases reported in children continues to climb, but “the history behind that cumulative number shows substantial change,” according to a new analysis of state health department data.
As of Sept. 10, the 549,432 cases in children represented 10.0% of all reported COVID-19 cases in the United States following a substantial rise over the course of the pandemic – the figure was 7.7% on July 16 and 3.2% on May 7, Blake Sisk, PhD, of the American Academy of Pediatrics and associates reported Sept. 29 in Pediatrics.
Unlike the cumulative number, the weekly proportion of cases in children fell early in the summer but then started climbing again in late July. Dr. Sisk and associates wrote.
Despite the increase, however, the proportion of pediatric COVID-19 cases is still well below children’s share of the overall population (22.6%). Also, “it is unclear how much of the increase in child cases is due to increased testing capacity, although CDC data from public and commercial laboratories show the share of all tests administered to children ages 0-17 has remained stable at 5%-7% since late April,” they said.
Data for the current report were drawn from 49 state health department websites (New York state does not report ages for COVID-19 cases), along with New York City, the District of Columbia, Puerto Rico, and Guam. Alabama changed its definition of a child case in August and was not included in the trend analysis (see graph), the investigators explained.
Those data show “substantial variation in case growth by region: in April, a preponderance of cases was in the Northeast. In June, cases surged in the South and West, followed by mid-July increases in the Midwest,” Dr. Sisk and associates said.
The increase among children in Midwest states is ongoing with the number of new cases reaching its highest level yet during the week ending Sept. 10, they reported.
SOURCE: Sisk B et al. Pediatrics. 2020 Sep 29. doi: 10.1542/peds.2020-027425.
FROM PEDIATRICS
Pandemic poses new challenges for rural doctors
These include struggling with seeing patients virtually and treating patients who have politicized the virus. Additionally, the pandemic has exposed rural practices to greater financial difficulties.

Before the pandemic some rurally based primary care physicians were already working through big challenges, such as having few local medical colleagues to consult and working in small practices with lean budgets. In fact, data gathered by the National Rural Health Association showed that there are only 40 primary care physicians per 100,000 patients in rural regions, compared with 53 in urban areas – and the number of physicians overall is 13 per 10,000 in rural areas, compared with 31 in cities.
In the prepandemic world, for some doctors, the challenges were balanced by the benefits of practicing in these sparsely populated communities with scenic, low-traffic roads. Some perks of practicing in rural areas touted by doctors included having a fast commute, being able to swim in a lake near the office before work, having a low cost of living, and feeling like they are making a difference in their communities as they treat generations of the families they see around town.
But today, new hurdles to practicing medicine in rural America created by the COVID-19 pandemic have caused the hardships to feel heavier than the joys at times for some physicians interviewed by MDedge.
Many independent rural practices in need of assistance were not able to get much from the federal Provider Relief Funds, said John M. Westfall, MD, who is director of the Robert Graham Center for Policy Studies in Family Medicine and Primary Care, in an interview.
“Rural primary care doctors function independently or in smaller critical access hospitals and community health centers,” said Dr. Westfall, who previously practiced family medicine in a small town in Colorado. “Many of these have much less financial reserves so are at risk of cutbacks and closure.”
Jacqueline W. Fincher, MD, an internist based in a tiny Georgia community along the highway between Atlanta and Augusta, said her small practice works on really thin margins and doesn’t have much cushion. At the beginning of the pandemic, all visits were down, and her practice operated at a loss. To help, Dr. Fincher and her colleagues applied for funding from the Small Business Administration’s Paycheck Protection Program (PPP) through the CARES Act.
“COVID-19 has had a tremendous impact especially on primary care practices. We live and die by volume. … Our volume in mid-March to mid-May really dropped dramatically,” explained Dr. Fincher, who is also president of the American College of Physicians. “The PPP sustained us for 2 months, enabling us to pay our staff and to remain open and get us up and running on telehealth.”
Starting up telemedicine
Experiencing spotty or no access to broadband Internet is nothing new to rural physicians, but having this problem interfere with their ability to provide care to patients is.
As much of the American health system rapidly embraced telehealth during the pandemic, obtaining access to high-speed Internet has been a major challenge for rural patients, noted Dr. Westfall.
“Some practices were able to quickly adopt some telehealth capacity with phone and video. Changes in payment for telehealth helped. But in some rural communities there was not adequate Internet bandwidth for quality video connections. And some patients did not have the means for high-speed video connections,” Dr. Westfall said.
Indeed, according to a 2019 Pew Research Center survey, 63% of rural Americans say they can access the Internet through a broadband connection at home, compared with 75% and 79% in suburban and urban areas, respectively.

In the Appalachian town of Zanesville, Ohio, for example, family physician Shelly L. Dunmyer, MD, and her colleagues discovered that many patients don’t have Internet access at home. Dr. Fincher has to go to the office to conduct telehealth visits because her own Internet access at home is unpredictable. As for patients, it may take 15 minutes for them to work out technical glitches and find good Internet reception, said Dr. Fincher. For internist Y. Ki Shin, MD, who practices in the coastal town of Montesano in Washington state, about 25% of his practice’s telehealth visits must be conducted by phone because of limitations on video, such as lack of high-speed access.
But telephone visits are often insufficient replacements for appointments via video, according to several rural physicians interviewed for this piece.
“Telehealth can be frustrating at times due to connectivity issues which can be difficult at times in the rural areas,” said Dr. Fincher. “In order for telehealth to be reasonably helpful to patients and physicians to care for people with chronic problems, the patients must have things like blood pressure monitors, glucometers, and scales to address problems like hypertension, diabetes myelitis, and congestive heart failure.”
“If you have the audio and video and the data from these devices, you’re good. If you don’t have these data, and/or don’t have the video you just can’t provide good care,” she explained.

Dr. Dunmyer and her colleagues at Medical Home Primary Care Center in Zanesville, Ohio, found a way to get around the problem of patients not being able to access Internet to participate in video visits from their homes. This involved having her patients drive into her practice’s parking lot to participate in modified telehealth visits. Staffers gave iPads to patients in their cars, and Dr. Dunmyer conducted visits from her office, about 50 yards away.
“We were even doing Medicare wellness visits: Instead of asking them to get up and move around the room, we would sit at the window and wave at them, ask them to get out, walk around the car. We were able to check mobility and all kinds of things that we’d normally do in the office,” Dr. Dunmyer explained in an interview.
The family physician noted that her practice is now conducting fewer parking lot visits since her office is allowing in-person appointments, but that they’re still an option for her patients.
Treating political adversaries
Some rural physicians have experienced strained relationships with patients for reasons other than technology – stark differences in opinion over the pandemic itself. Certain patients are following President Trump’s lead and questioning everything from the pandemic death toll to preventive measures recommended by scientists and medical experts, physicians interviewed by MDedge said.
Patients everywhere share these viewpoints, of course, but research and election results confirm that rural areas are more receptive to conservative viewpoints. In 2018, a Pew Research Center survey reported that rural and urban areas are “becoming more polarized politically,” and “rural areas tend to have a higher concentration of Republicans and Republican-leaning independents.” For example, 40% of rural respondents reported “very warm” or “somewhat warm” feelings toward Donald Trump, compared with just 19% in urban areas.
Dr. Shin has struggled to cope with patients who want to argue about pandemic safety precautions like wearing masks and seem to question whether systemic racism exists.
“We are seeing a lot more people who feel that this pandemic is not real, that it’s a political and not-true infection,” he said in an interview. “We’ve had patients who were angry at us because we made them wear masks, and some were demanding hydroxychloroquine and wanted to have an argument because we’re not going to prescribe it for them.”
In one situation, which he found especially disturbing, Dr. Shin had to leave the exam room because a patient wouldn’t stop challenging him regarding the pandemic. Things have gotten so bad that Dr. Shin has even questioned whether he wants to continue his long career in his small town because of local political attitudes such as opposition to mask-wearing and social distancing.
“Mr. Trump’s misinformation on this pandemic made my job much more difficult. As a minority, I feel less safe in my community than ever,” said Dr. Shin, who described himself as Asian American.
Despite these new stressors, Dr. Shin has experienced some joyful moments while practicing medicine in the pandemic.

He said a recent home visit to a patient who had been hospitalized for over 3 months and nearly died helped him put political disputes with his patients into perspective.
“He was discharged home but is bedbound. He had gangrene on his toes, and I could not fully examine him using video,” Dr. Shin recalled. “It was tricky to find the house, but a very large Trump sign was very helpful in locating it. It was a good visit: He was happy to see me, and I was happy to see that he was doing okay at home.”
“I need to remind myself that supporting Mr. Trump does not always mean that my patient supports Mr. Trump’s view on the pandemic and the race issues in our country,” Dr. Shin added.
The Washington-based internist said he also tells himself that, even if his patients refuse to follow his strong advice regarding pandemic precautions, it does not mean he has failed as a doctor.
“I need to continue to educate patients about the dangers of COVID infection but cannot be angry if they don’t choose to follow my recommendations,” he noted.
Dr. Fincher says her close connection with patients has allowed her to smooth over politically charged claims about the pandemic in the town of Thomson, Georgia, with a population 6,800.
“I have a sense that, even though we may differ in our understanding of some basic facts, they appreciate what I say since we have a long-term relationship built on trust,” she said. This kind of trust, Dr. Fincher suggested, may be more common than in urban areas where there’s a larger supply of physicians, and patients don’t see the same doctors for long periods of time.
“It’s more meaningful when it comes from me, rather than doctors who are [new to patients] every year when their employer changes their insurance,” she noted.
These include struggling with seeing patients virtually and treating patients who have politicized the virus. Additionally, the pandemic has exposed rural practices to greater financial difficulties.
Before the pandemic some rurally based primary care physicians were already working through big challenges, such as having few local medical colleagues to consult and working in small practices with lean budgets. In fact, data gathered by the National Rural Health Association showed that there are only 40 primary care physicians per 100,000 patients in rural regions, compared with 53 in urban areas – and the number of physicians overall is 13 per 10,000 in rural areas, compared with 31 in cities.
In the prepandemic world, for some doctors, the challenges were balanced by the benefits of practicing in these sparsely populated communities with scenic, low-traffic roads. Some perks of practicing in rural areas touted by doctors included having a fast commute, being able to swim in a lake near the office before work, having a low cost of living, and feeling like they are making a difference in their communities as they treat generations of the families they see around town.
But today, new hurdles to practicing medicine in rural America created by the COVID-19 pandemic have caused the hardships to feel heavier than the joys at times for some physicians interviewed by MDedge.
Many independent rural practices in need of assistance were not able to get much from the federal Provider Relief Funds, said John M. Westfall, MD, who is director of the Robert Graham Center for Policy Studies in Family Medicine and Primary Care, in an interview.
“Rural primary care doctors function independently or in smaller critical access hospitals and community health centers,” said Dr. Westfall, who previously practiced family medicine in a small town in Colorado. “Many of these have much less financial reserves so are at risk of cutbacks and closure.”
Jacqueline W. Fincher, MD, an internist based in a tiny Georgia community along the highway between Atlanta and Augusta, said her small practice works on really thin margins and doesn’t have much cushion. At the beginning of the pandemic, all visits were down, and her practice operated at a loss. To help, Dr. Fincher and her colleagues applied for funding from the Small Business Administration’s Paycheck Protection Program (PPP) through the CARES Act.
“COVID-19 has had a tremendous impact especially on primary care practices. We live and die by volume. … Our volume in mid-March to mid-May really dropped dramatically,” explained Dr. Fincher, who is also president of the American College of Physicians. “The PPP sustained us for 2 months, enabling us to pay our staff and to remain open and get us up and running on telehealth.”
Starting up telemedicine
Experiencing spotty or no access to broadband Internet is nothing new to rural physicians, but having this problem interfere with their ability to provide care to patients is.
As much of the American health system rapidly embraced telehealth during the pandemic, obtaining access to high-speed Internet has been a major challenge for rural patients, noted Dr. Westfall.
“Some practices were able to quickly adopt some telehealth capacity with phone and video. Changes in payment for telehealth helped. But in some rural communities there was not adequate Internet bandwidth for quality video connections. And some patients did not have the means for high-speed video connections,” Dr. Westfall said.
Indeed, according to a 2019 Pew Research Center survey, 63% of rural Americans say they can access the Internet through a broadband connection at home, compared with 75% and 79% in suburban and urban areas, respectively.
In the Appalachian town of Zanesville, Ohio, for example, family physician Shelly L. Dunmyer, MD, and her colleagues discovered that many patients don’t have Internet access at home. Dr. Fincher has to go to the office to conduct telehealth visits because her own Internet access at home is unpredictable. As for patients, it may take 15 minutes for them to work out technical glitches and find good Internet reception, said Dr. Fincher. For internist Y. Ki Shin, MD, who practices in the coastal town of Montesano in Washington state, about 25% of his practice’s telehealth visits must be conducted by phone because of limitations on video, such as lack of high-speed access.
But telephone visits are often insufficient replacements for appointments via video, according to several rural physicians interviewed for this piece.
“Telehealth can be frustrating at times due to connectivity issues which can be difficult at times in the rural areas,” said Dr. Fincher. “In order for telehealth to be reasonably helpful to patients and physicians to care for people with chronic problems, the patients must have things like blood pressure monitors, glucometers, and scales to address problems like hypertension, diabetes myelitis, and congestive heart failure.”
“If you have the audio and video and the data from these devices, you’re good. If you don’t have these data, and/or don’t have the video you just can’t provide good care,” she explained.
Dr. Dunmyer and her colleagues at Medical Home Primary Care Center in Zanesville, Ohio, found a way to get around the problem of patients not being able to access Internet to participate in video visits from their homes. This involved having her patients drive into her practice’s parking lot to participate in modified telehealth visits. Staffers gave iPads to patients in their cars, and Dr. Dunmyer conducted visits from her office, about 50 yards away.
“We were even doing Medicare wellness visits: Instead of asking them to get up and move around the room, we would sit at the window and wave at them, ask them to get out, walk around the car. We were able to check mobility and all kinds of things that we’d normally do in the office,” Dr. Dunmyer explained in an interview.
The family physician noted that her practice is now conducting fewer parking lot visits since her office is allowing in-person appointments, but that they’re still an option for her patients.
Treating political adversaries
Some rural physicians have experienced strained relationships with patients for reasons other than technology – stark differences in opinion over the pandemic itself. Certain patients are following President Trump’s lead and questioning everything from the pandemic death toll to preventive measures recommended by scientists and medical experts, physicians interviewed by MDedge said.
Patients everywhere share these viewpoints, of course, but research and election results confirm that rural areas are more receptive to conservative viewpoints. In 2018, a Pew Research Center survey reported that rural and urban areas are “becoming more polarized politically,” and “rural areas tend to have a higher concentration of Republicans and Republican-leaning independents.” For example, 40% of rural respondents reported “very warm” or “somewhat warm” feelings toward Donald Trump, compared with just 19% in urban areas.
Dr. Shin has struggled to cope with patients who want to argue about pandemic safety precautions like wearing masks and seem to question whether systemic racism exists.
“We are seeing a lot more people who feel that this pandemic is not real, that it’s a political and not-true infection,” he said in an interview. “We’ve had patients who were angry at us because we made them wear masks, and some were demanding hydroxychloroquine and wanted to have an argument because we’re not going to prescribe it for them.”
In one situation, which he found especially disturbing, Dr. Shin had to leave the exam room because a patient wouldn’t stop challenging him regarding the pandemic. Things have gotten so bad that Dr. Shin has even questioned whether he wants to continue his long career in his small town because of local political attitudes such as opposition to mask-wearing and social distancing.
“Mr. Trump’s misinformation on this pandemic made my job much more difficult. As a minority, I feel less safe in my community than ever,” said Dr. Shin, who described himself as Asian American.
Despite these new stressors, Dr. Shin has experienced some joyful moments while practicing medicine in the pandemic.
He said a recent home visit to a patient who had been hospitalized for over 3 months and nearly died helped him put political disputes with his patients into perspective.
“He was discharged home but is bedbound. He had gangrene on his toes, and I could not fully examine him using video,” Dr. Shin recalled. “It was tricky to find the house, but a very large Trump sign was very helpful in locating it. It was a good visit: He was happy to see me, and I was happy to see that he was doing okay at home.”
“I need to remind myself that supporting Mr. Trump does not always mean that my patient supports Mr. Trump’s view on the pandemic and the race issues in our country,” Dr. Shin added.
The Washington-based internist said he also tells himself that, even if his patients refuse to follow his strong advice regarding pandemic precautions, it does not mean he has failed as a doctor.
“I need to continue to educate patients about the dangers of COVID infection but cannot be angry if they don’t choose to follow my recommendations,” he noted.
Dr. Fincher says her close connection with patients has allowed her to smooth over politically charged claims about the pandemic in the town of Thomson, Georgia, with a population 6,800.
“I have a sense that, even though we may differ in our understanding of some basic facts, they appreciate what I say since we have a long-term relationship built on trust,” she said. This kind of trust, Dr. Fincher suggested, may be more common than in urban areas where there’s a larger supply of physicians, and patients don’t see the same doctors for long periods of time.
“It’s more meaningful when it comes from me, rather than doctors who are [new to patients] every year when their employer changes their insurance,” she noted.
These include struggling with seeing patients virtually and treating patients who have politicized the virus. Additionally, the pandemic has exposed rural practices to greater financial difficulties.
Before the pandemic some rurally based primary care physicians were already working through big challenges, such as having few local medical colleagues to consult and working in small practices with lean budgets. In fact, data gathered by the National Rural Health Association showed that there are only 40 primary care physicians per 100,000 patients in rural regions, compared with 53 in urban areas – and the number of physicians overall is 13 per 10,000 in rural areas, compared with 31 in cities.
In the prepandemic world, for some doctors, the challenges were balanced by the benefits of practicing in these sparsely populated communities with scenic, low-traffic roads. Some perks of practicing in rural areas touted by doctors included having a fast commute, being able to swim in a lake near the office before work, having a low cost of living, and feeling like they are making a difference in their communities as they treat generations of the families they see around town.
But today, new hurdles to practicing medicine in rural America created by the COVID-19 pandemic have caused the hardships to feel heavier than the joys at times for some physicians interviewed by MDedge.
Many independent rural practices in need of assistance were not able to get much from the federal Provider Relief Funds, said John M. Westfall, MD, who is director of the Robert Graham Center for Policy Studies in Family Medicine and Primary Care, in an interview.
“Rural primary care doctors function independently or in smaller critical access hospitals and community health centers,” said Dr. Westfall, who previously practiced family medicine in a small town in Colorado. “Many of these have much less financial reserves so are at risk of cutbacks and closure.”
Jacqueline W. Fincher, MD, an internist based in a tiny Georgia community along the highway between Atlanta and Augusta, said her small practice works on really thin margins and doesn’t have much cushion. At the beginning of the pandemic, all visits were down, and her practice operated at a loss. To help, Dr. Fincher and her colleagues applied for funding from the Small Business Administration’s Paycheck Protection Program (PPP) through the CARES Act.
“COVID-19 has had a tremendous impact especially on primary care practices. We live and die by volume. … Our volume in mid-March to mid-May really dropped dramatically,” explained Dr. Fincher, who is also president of the American College of Physicians. “The PPP sustained us for 2 months, enabling us to pay our staff and to remain open and get us up and running on telehealth.”
Starting up telemedicine
Experiencing spotty or no access to broadband Internet is nothing new to rural physicians, but having this problem interfere with their ability to provide care to patients is.
As much of the American health system rapidly embraced telehealth during the pandemic, obtaining access to high-speed Internet has been a major challenge for rural patients, noted Dr. Westfall.
“Some practices were able to quickly adopt some telehealth capacity with phone and video. Changes in payment for telehealth helped. But in some rural communities there was not adequate Internet bandwidth for quality video connections. And some patients did not have the means for high-speed video connections,” Dr. Westfall said.
Indeed, according to a 2019 Pew Research Center survey, 63% of rural Americans say they can access the Internet through a broadband connection at home, compared with 75% and 79% in suburban and urban areas, respectively.
In the Appalachian town of Zanesville, Ohio, for example, family physician Shelly L. Dunmyer, MD, and her colleagues discovered that many patients don’t have Internet access at home. Dr. Fincher has to go to the office to conduct telehealth visits because her own Internet access at home is unpredictable. As for patients, it may take 15 minutes for them to work out technical glitches and find good Internet reception, said Dr. Fincher. For internist Y. Ki Shin, MD, who practices in the coastal town of Montesano in Washington state, about 25% of his practice’s telehealth visits must be conducted by phone because of limitations on video, such as lack of high-speed access.
But telephone visits are often insufficient replacements for appointments via video, according to several rural physicians interviewed for this piece.
“Telehealth can be frustrating at times due to connectivity issues which can be difficult at times in the rural areas,” said Dr. Fincher. “In order for telehealth to be reasonably helpful to patients and physicians to care for people with chronic problems, the patients must have things like blood pressure monitors, glucometers, and scales to address problems like hypertension, diabetes myelitis, and congestive heart failure.”
“If you have the audio and video and the data from these devices, you’re good. If you don’t have these data, and/or don’t have the video you just can’t provide good care,” she explained.
Dr. Dunmyer and her colleagues at Medical Home Primary Care Center in Zanesville, Ohio, found a way to get around the problem of patients not being able to access Internet to participate in video visits from their homes. This involved having her patients drive into her practice’s parking lot to participate in modified telehealth visits. Staffers gave iPads to patients in their cars, and Dr. Dunmyer conducted visits from her office, about 50 yards away.
“We were even doing Medicare wellness visits: Instead of asking them to get up and move around the room, we would sit at the window and wave at them, ask them to get out, walk around the car. We were able to check mobility and all kinds of things that we’d normally do in the office,” Dr. Dunmyer explained in an interview.
The family physician noted that her practice is now conducting fewer parking lot visits since her office is allowing in-person appointments, but that they’re still an option for her patients.
Treating political adversaries
Some rural physicians have experienced strained relationships with patients for reasons other than technology – stark differences in opinion over the pandemic itself. Certain patients are following President Trump’s lead and questioning everything from the pandemic death toll to preventive measures recommended by scientists and medical experts, physicians interviewed by MDedge said.
Patients everywhere share these viewpoints, of course, but research and election results confirm that rural areas are more receptive to conservative viewpoints. In 2018, a Pew Research Center survey reported that rural and urban areas are “becoming more polarized politically,” and “rural areas tend to have a higher concentration of Republicans and Republican-leaning independents.” For example, 40% of rural respondents reported “very warm” or “somewhat warm” feelings toward Donald Trump, compared with just 19% in urban areas.
Dr. Shin has struggled to cope with patients who want to argue about pandemic safety precautions like wearing masks and seem to question whether systemic racism exists.
“We are seeing a lot more people who feel that this pandemic is not real, that it’s a political and not-true infection,” he said in an interview. “We’ve had patients who were angry at us because we made them wear masks, and some were demanding hydroxychloroquine and wanted to have an argument because we’re not going to prescribe it for them.”
In one situation, which he found especially disturbing, Dr. Shin had to leave the exam room because a patient wouldn’t stop challenging him regarding the pandemic. Things have gotten so bad that Dr. Shin has even questioned whether he wants to continue his long career in his small town because of local political attitudes such as opposition to mask-wearing and social distancing.
“Mr. Trump’s misinformation on this pandemic made my job much more difficult. As a minority, I feel less safe in my community than ever,” said Dr. Shin, who described himself as Asian American.
Despite these new stressors, Dr. Shin has experienced some joyful moments while practicing medicine in the pandemic.
He said a recent home visit to a patient who had been hospitalized for over 3 months and nearly died helped him put political disputes with his patients into perspective.
“He was discharged home but is bedbound. He had gangrene on his toes, and I could not fully examine him using video,” Dr. Shin recalled. “It was tricky to find the house, but a very large Trump sign was very helpful in locating it. It was a good visit: He was happy to see me, and I was happy to see that he was doing okay at home.”
“I need to remind myself that supporting Mr. Trump does not always mean that my patient supports Mr. Trump’s view on the pandemic and the race issues in our country,” Dr. Shin added.
The Washington-based internist said he also tells himself that, even if his patients refuse to follow his strong advice regarding pandemic precautions, it does not mean he has failed as a doctor.
“I need to continue to educate patients about the dangers of COVID infection but cannot be angry if they don’t choose to follow my recommendations,” he noted.
Dr. Fincher says her close connection with patients has allowed her to smooth over politically charged claims about the pandemic in the town of Thomson, Georgia, with a population 6,800.
“I have a sense that, even though we may differ in our understanding of some basic facts, they appreciate what I say since we have a long-term relationship built on trust,” she said. This kind of trust, Dr. Fincher suggested, may be more common than in urban areas where there’s a larger supply of physicians, and patients don’t see the same doctors for long periods of time.
“It’s more meaningful when it comes from me, rather than doctors who are [new to patients] every year when their employer changes their insurance,” she noted.
Dr. Brian Mandell gives his take on ACR’s newest gout guideline
Guidance on the initiation and use of urate-lowering therapies was among the strong recommendations in the updated gout guideline recently issued by the American College of Rheumatology, said Brian F. Mandell, MD, PhD, in a virtual presentation at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education.

The 2020 American College of Rheumatology Guideline for the Management of Gout is “intended to provide guidance for particular patterns of practice and not to dictate the care of a particular patient,” said Dr. Mandell, chair of academic medicine at the Cleveland Clinic in Ohio and cochair of the conference. However, “there was a hope that, with additional evidence since the previous guideline issued in 2012, the recommendations are more firmly based and will improve care,” he said.
Of 42 recommendations, 16 were strong, and these included guidance on several points: when to initiate urate-lowering therapy and using a treat-to-target strategy for lowering serum uric acid to less than 6 mg/dL; prophylaxis against attacks; the use of allopurinol as the first-choice drug and how to avoid hypersensitivity reactions; the use of pegloticase (Krystexxa); and treating flares.
Hyperuricemia does not automatically equal gout, Dr. Mandell said. A 2018 published analysis of data from several large cohorts including 18,889 adults who were gout-free at baseline showed that serum uric acid levels could not accurately predict an initial gout attack. Therefore, the guideline conditionally recommends against initiating any pharmacologic urate-lowering therapy in patients with asymptomatic hyperuricemia. The guideline authors intentionally did not include the presence of comorbidities or deposits of uric acid in making their recommendation. But when advising an individual patient, these factors plus the patient’s age and family history should be considered, he said. “Individualize the decision to use ULT [urate-lowering therapy] to prevent possible future flares,” he advised, with consideration of age, the effects of the flare on the patient’s life, and challenges in treating flares.
For patients who are being treated with urate-lowering therapy, a published study indicated that if treatment is discontinued, “gout attacks will recur, depending on the new serum urate level,” Dr. Mandell said. “Maintenance of low SUA [serum uric acid] must be lifelong to stop attacks,” he emphasized, noting that this is counter to a management guideline published by the American College of Physicians in 2017.
“For patients starting any ULT, we strongly recommend allopurinol over all other urate-lowering therapies as the preferred first-line agent for all patients, including those with CKD [chronic kidney disease] stage 3 or higher,” according to the new guideline, which also recommends starting at a low dose followed by dose titration to target versus starting at a higher dose.
Two reasons in support of a slow up-titration of urate-lowering therapy are a lower frequency of mobilization flares and a possibly lower chance of allopurinol hypersensitivity reactions, Dr. Mandell said.
Although the guideline recommends allopurinol over probenecid, “probenecid works well as monotherapy and effectively as add-on therapy to a xanthine oxidase inhibitor, and it is cheap,” Dr. Mandell said.
Allopurinol can be associated with life-threatening hypersensitivity reactions, but most of these have been associated with a higher-than-recommended starting dose, according to the literature, he noted. The new guideline suggests checking for the HLA-B*5801 haplotype in high-risk demographic groups, and if it is present, to use an alternative to allopurinol if possible
The updated guideline also carries a strong recommendation for the use of pegloticase for patients with frequent gout flares and nonresolving subcutaneous tophi, but it strongly recommends against switching to pegloticase for patients with infrequent gout flares and no tophi.
However, Dr. Mandell said that he will consider off-label treatment of gout with pegloticase “in patients where a shorter time to response really matters,” which is consistent with his belief that, within these treatment principles, the management of gout must be individualized to the specific patient.
For treating acute gout flares, the guideline recommendations strongly supports the use of oral colchicine, NSAIDs, or glucocorticoids as an appropriate first-line therapy, based on patient factors and preferences, instead of using interleukin-1 inhibitors or adrenocorticotropic hormone. However, the interleukin-1 inhibitor anakinra has shown relatively rapid and successful response in treating patients hospitalized with acute gout, Dr. Mandell said. No large, randomized, trials have been conducted, but he cited his experience at the Cleveland Clinic in Ohio, where anakinra is the most common treatment for acute gout on inpatient consults, and he cited a representative small study of 26 patients in which 73% showed “significant response” within 5 days of treatment, which meant that they were able to move and bear weight without pain. In addition, a more recent study of 100 hospitalized patients in the Journal of Rheumatology, found that 75% showed a rapid response to anakinra and improvement or resolution of flares within 4 days, Dr. Mandell said.
Dr. Mandell disclosed relationships with companies including Horizon, Ardea/AstraZeneca/Ironwood, and Takeda. He served as coauthor on the 2012 American College of Rheumatology gout guideline.
Global Academy for Medical Education and this news organization are owned by the same parent company.
Guidance on the initiation and use of urate-lowering therapies was among the strong recommendations in the updated gout guideline recently issued by the American College of Rheumatology, said Brian F. Mandell, MD, PhD, in a virtual presentation at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education.
The 2020 American College of Rheumatology Guideline for the Management of Gout is “intended to provide guidance for particular patterns of practice and not to dictate the care of a particular patient,” said Dr. Mandell, chair of academic medicine at the Cleveland Clinic in Ohio and cochair of the conference. However, “there was a hope that, with additional evidence since the previous guideline issued in 2012, the recommendations are more firmly based and will improve care,” he said.
Of 42 recommendations, 16 were strong, and these included guidance on several points: when to initiate urate-lowering therapy and using a treat-to-target strategy for lowering serum uric acid to less than 6 mg/dL; prophylaxis against attacks; the use of allopurinol as the first-choice drug and how to avoid hypersensitivity reactions; the use of pegloticase (Krystexxa); and treating flares.
Hyperuricemia does not automatically equal gout, Dr. Mandell said. A 2018 published analysis of data from several large cohorts including 18,889 adults who were gout-free at baseline showed that serum uric acid levels could not accurately predict an initial gout attack. Therefore, the guideline conditionally recommends against initiating any pharmacologic urate-lowering therapy in patients with asymptomatic hyperuricemia. The guideline authors intentionally did not include the presence of comorbidities or deposits of uric acid in making their recommendation. But when advising an individual patient, these factors plus the patient’s age and family history should be considered, he said. “Individualize the decision to use ULT [urate-lowering therapy] to prevent possible future flares,” he advised, with consideration of age, the effects of the flare on the patient’s life, and challenges in treating flares.
For patients who are being treated with urate-lowering therapy, a published study indicated that if treatment is discontinued, “gout attacks will recur, depending on the new serum urate level,” Dr. Mandell said. “Maintenance of low SUA [serum uric acid] must be lifelong to stop attacks,” he emphasized, noting that this is counter to a management guideline published by the American College of Physicians in 2017.
“For patients starting any ULT, we strongly recommend allopurinol over all other urate-lowering therapies as the preferred first-line agent for all patients, including those with CKD [chronic kidney disease] stage 3 or higher,” according to the new guideline, which also recommends starting at a low dose followed by dose titration to target versus starting at a higher dose.
Two reasons in support of a slow up-titration of urate-lowering therapy are a lower frequency of mobilization flares and a possibly lower chance of allopurinol hypersensitivity reactions, Dr. Mandell said.
Although the guideline recommends allopurinol over probenecid, “probenecid works well as monotherapy and effectively as add-on therapy to a xanthine oxidase inhibitor, and it is cheap,” Dr. Mandell said.
Allopurinol can be associated with life-threatening hypersensitivity reactions, but most of these have been associated with a higher-than-recommended starting dose, according to the literature, he noted. The new guideline suggests checking for the HLA-B*5801 haplotype in high-risk demographic groups, and if it is present, to use an alternative to allopurinol if possible
The updated guideline also carries a strong recommendation for the use of pegloticase for patients with frequent gout flares and nonresolving subcutaneous tophi, but it strongly recommends against switching to pegloticase for patients with infrequent gout flares and no tophi.
However, Dr. Mandell said that he will consider off-label treatment of gout with pegloticase “in patients where a shorter time to response really matters,” which is consistent with his belief that, within these treatment principles, the management of gout must be individualized to the specific patient.
For treating acute gout flares, the guideline recommendations strongly supports the use of oral colchicine, NSAIDs, or glucocorticoids as an appropriate first-line therapy, based on patient factors and preferences, instead of using interleukin-1 inhibitors or adrenocorticotropic hormone. However, the interleukin-1 inhibitor anakinra has shown relatively rapid and successful response in treating patients hospitalized with acute gout, Dr. Mandell said. No large, randomized, trials have been conducted, but he cited his experience at the Cleveland Clinic in Ohio, where anakinra is the most common treatment for acute gout on inpatient consults, and he cited a representative small study of 26 patients in which 73% showed “significant response” within 5 days of treatment, which meant that they were able to move and bear weight without pain. In addition, a more recent study of 100 hospitalized patients in the Journal of Rheumatology, found that 75% showed a rapid response to anakinra and improvement or resolution of flares within 4 days, Dr. Mandell said.
Dr. Mandell disclosed relationships with companies including Horizon, Ardea/AstraZeneca/Ironwood, and Takeda. He served as coauthor on the 2012 American College of Rheumatology gout guideline.
Global Academy for Medical Education and this news organization are owned by the same parent company.
Guidance on the initiation and use of urate-lowering therapies was among the strong recommendations in the updated gout guideline recently issued by the American College of Rheumatology, said Brian F. Mandell, MD, PhD, in a virtual presentation at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education.
The 2020 American College of Rheumatology Guideline for the Management of Gout is “intended to provide guidance for particular patterns of practice and not to dictate the care of a particular patient,” said Dr. Mandell, chair of academic medicine at the Cleveland Clinic in Ohio and cochair of the conference. However, “there was a hope that, with additional evidence since the previous guideline issued in 2012, the recommendations are more firmly based and will improve care,” he said.
Of 42 recommendations, 16 were strong, and these included guidance on several points: when to initiate urate-lowering therapy and using a treat-to-target strategy for lowering serum uric acid to less than 6 mg/dL; prophylaxis against attacks; the use of allopurinol as the first-choice drug and how to avoid hypersensitivity reactions; the use of pegloticase (Krystexxa); and treating flares.
Hyperuricemia does not automatically equal gout, Dr. Mandell said. A 2018 published analysis of data from several large cohorts including 18,889 adults who were gout-free at baseline showed that serum uric acid levels could not accurately predict an initial gout attack. Therefore, the guideline conditionally recommends against initiating any pharmacologic urate-lowering therapy in patients with asymptomatic hyperuricemia. The guideline authors intentionally did not include the presence of comorbidities or deposits of uric acid in making their recommendation. But when advising an individual patient, these factors plus the patient’s age and family history should be considered, he said. “Individualize the decision to use ULT [urate-lowering therapy] to prevent possible future flares,” he advised, with consideration of age, the effects of the flare on the patient’s life, and challenges in treating flares.
For patients who are being treated with urate-lowering therapy, a published study indicated that if treatment is discontinued, “gout attacks will recur, depending on the new serum urate level,” Dr. Mandell said. “Maintenance of low SUA [serum uric acid] must be lifelong to stop attacks,” he emphasized, noting that this is counter to a management guideline published by the American College of Physicians in 2017.
“For patients starting any ULT, we strongly recommend allopurinol over all other urate-lowering therapies as the preferred first-line agent for all patients, including those with CKD [chronic kidney disease] stage 3 or higher,” according to the new guideline, which also recommends starting at a low dose followed by dose titration to target versus starting at a higher dose.
Two reasons in support of a slow up-titration of urate-lowering therapy are a lower frequency of mobilization flares and a possibly lower chance of allopurinol hypersensitivity reactions, Dr. Mandell said.
Although the guideline recommends allopurinol over probenecid, “probenecid works well as monotherapy and effectively as add-on therapy to a xanthine oxidase inhibitor, and it is cheap,” Dr. Mandell said.
Allopurinol can be associated with life-threatening hypersensitivity reactions, but most of these have been associated with a higher-than-recommended starting dose, according to the literature, he noted. The new guideline suggests checking for the HLA-B*5801 haplotype in high-risk demographic groups, and if it is present, to use an alternative to allopurinol if possible
The updated guideline also carries a strong recommendation for the use of pegloticase for patients with frequent gout flares and nonresolving subcutaneous tophi, but it strongly recommends against switching to pegloticase for patients with infrequent gout flares and no tophi.
However, Dr. Mandell said that he will consider off-label treatment of gout with pegloticase “in patients where a shorter time to response really matters,” which is consistent with his belief that, within these treatment principles, the management of gout must be individualized to the specific patient.
For treating acute gout flares, the guideline recommendations strongly supports the use of oral colchicine, NSAIDs, or glucocorticoids as an appropriate first-line therapy, based on patient factors and preferences, instead of using interleukin-1 inhibitors or adrenocorticotropic hormone. However, the interleukin-1 inhibitor anakinra has shown relatively rapid and successful response in treating patients hospitalized with acute gout, Dr. Mandell said. No large, randomized, trials have been conducted, but he cited his experience at the Cleveland Clinic in Ohio, where anakinra is the most common treatment for acute gout on inpatient consults, and he cited a representative small study of 26 patients in which 73% showed “significant response” within 5 days of treatment, which meant that they were able to move and bear weight without pain. In addition, a more recent study of 100 hospitalized patients in the Journal of Rheumatology, found that 75% showed a rapid response to anakinra and improvement or resolution of flares within 4 days, Dr. Mandell said.
Dr. Mandell disclosed relationships with companies including Horizon, Ardea/AstraZeneca/Ironwood, and Takeda. He served as coauthor on the 2012 American College of Rheumatology gout guideline.
Global Academy for Medical Education and this news organization are owned by the same parent company.
FROM PRD 2020
Osteoporosis: September 2020

Romosozumab is a novel osteoporosis treatment agent that stimulates bone formation and inhibits bone resorption. Due to concerns for adverse cardiovascular effects, this medication is generally reserved for patients with severe osteoporosis despite multiple previous treatments. However, the influence of previous osteoporosis pharmacotherapy on romosozumab’s skeletal efficacy remains poorly studied, especially in real world clinical scenarios. In a prospective non-randomized study of 130 patients treated with romosozumab in Japan, the effects of romosozumab treatment on bone turnover markers and bone mineral density (BMD) was assessed based on previous treatment with either no osteoporosis medication, bisphosphonates, denosumab, or teriparatide. In general, treatment-naïve individuals experienced superior spine BMD and bone turnover marker gains versus patients who had previously received osteoporosis pharmacotherapy. Notably, prior treatment with the potent anti-resorptive agent denosumab was associated with suboptimal spine BMD response to romosozumab. These results highlight the potential impact of previous anti-resorptive treatment on romosozumab efficacy and indicate that careful consideration of treatment sequence is warranted when deciding amongst the multiple osteoporosis agents that are currently available.
Denosumab is a potent anti-resorptive osteoporosis agent that works by neutralizing RANKL, the key cytokine that drives osteoclast differentiation. While effective at suppressing bone resorption, increasing BMD, and preventing fragility fractures when administered every 6 months, rapid discontinuation of denosumab has been associated with an increased risk of vertebral compression fractures. In routine clinical practice, denosumab injection delays are common. However, risks of vertebral fractures associated with such injection delays remain unknown. In this study of a UK population-based cohort of 2,594 patients, a large electronic database was used to assess the relationship between denosumab dosing intervals (defined as ‘on time’, ‘short delay’, and ‘long delay’) and fractures in the 6 month window after the recommended treatment date. Compared with ‘on time’ denosumab treatment, individuals who received delayed denosumab injections showed an increased risk of interval vertebral fractures. Notably, the delay time was associated with fracture risk. This powerful study design further underscores the importance of timely (every 6 months) denosumab injections. Given current health care delivery challenges associated with the COVID-19 pandemic, these results should be considered when selecting initial osteoporosis treatment agents and in management of patients currently receiving denosumab.
Abaloparatide, a synthetic analog of parathyroid hormone related peptide (PTHrP) is a bone anabolic osteoporosis treatment agent. In the ACTIVE study, abaloparatide increased bone mineral density and reduced fracture risk compared to both placebo and teriparatide. Although abaloparatide is generally well-tolerated, non-serious adverse events have been reported such as dizziness and palpitations. PTHrP can act in a paracrine manner to cause vasodilation. Therefore, in this post hoc drug safety study, the effects of abaloparatide on blood pressure, pulse, and cardiovascular adverse events were assessed in detail in subjects from the ACTIVE study (one hour post treatment) plus an additional group of 55 subjects for more detailed hemodynamic assessment. Compared to placebo injection, both abaloparatide and teriparatide treatment caused mild and transient increases in pulse and decreases in systolic blood pressure. These mild hemodynamic changes were not associated with an increased risk of adverse cardiovascular events. In general, these data are reassuring and support the cardiac safety of PTH analogs for osteoporosis. However, the transient hemodynamic changes observed should be considered for patients with cardiovascular comorbidities or baseline problems with orthostasis.
Marc Wein, M.D., Ph.D
Assistant Professor of Medicine
Massachusetts General Hospital Endocrine Unit, Harvard Medical School
Romosozumab is a novel osteoporosis treatment agent that stimulates bone formation and inhibits bone resorption. Due to concerns for adverse cardiovascular effects, this medication is generally reserved for patients with severe osteoporosis despite multiple previous treatments. However, the influence of previous osteoporosis pharmacotherapy on romosozumab’s skeletal efficacy remains poorly studied, especially in real world clinical scenarios. In a prospective non-randomized study of 130 patients treated with romosozumab in Japan, the effects of romosozumab treatment on bone turnover markers and bone mineral density (BMD) was assessed based on previous treatment with either no osteoporosis medication, bisphosphonates, denosumab, or teriparatide. In general, treatment-naïve individuals experienced superior spine BMD and bone turnover marker gains versus patients who had previously received osteoporosis pharmacotherapy. Notably, prior treatment with the potent anti-resorptive agent denosumab was associated with suboptimal spine BMD response to romosozumab. These results highlight the potential impact of previous anti-resorptive treatment on romosozumab efficacy and indicate that careful consideration of treatment sequence is warranted when deciding amongst the multiple osteoporosis agents that are currently available.
Denosumab is a potent anti-resorptive osteoporosis agent that works by neutralizing RANKL, the key cytokine that drives osteoclast differentiation. While effective at suppressing bone resorption, increasing BMD, and preventing fragility fractures when administered every 6 months, rapid discontinuation of denosumab has been associated with an increased risk of vertebral compression fractures. In routine clinical practice, denosumab injection delays are common. However, risks of vertebral fractures associated with such injection delays remain unknown. In this study of a UK population-based cohort of 2,594 patients, a large electronic database was used to assess the relationship between denosumab dosing intervals (defined as ‘on time’, ‘short delay’, and ‘long delay’) and fractures in the 6 month window after the recommended treatment date. Compared with ‘on time’ denosumab treatment, individuals who received delayed denosumab injections showed an increased risk of interval vertebral fractures. Notably, the delay time was associated with fracture risk. This powerful study design further underscores the importance of timely (every 6 months) denosumab injections. Given current health care delivery challenges associated with the COVID-19 pandemic, these results should be considered when selecting initial osteoporosis treatment agents and in management of patients currently receiving denosumab.
Abaloparatide, a synthetic analog of parathyroid hormone related peptide (PTHrP) is a bone anabolic osteoporosis treatment agent. In the ACTIVE study, abaloparatide increased bone mineral density and reduced fracture risk compared to both placebo and teriparatide. Although abaloparatide is generally well-tolerated, non-serious adverse events have been reported such as dizziness and palpitations. PTHrP can act in a paracrine manner to cause vasodilation. Therefore, in this post hoc drug safety study, the effects of abaloparatide on blood pressure, pulse, and cardiovascular adverse events were assessed in detail in subjects from the ACTIVE study (one hour post treatment) plus an additional group of 55 subjects for more detailed hemodynamic assessment. Compared to placebo injection, both abaloparatide and teriparatide treatment caused mild and transient increases in pulse and decreases in systolic blood pressure. These mild hemodynamic changes were not associated with an increased risk of adverse cardiovascular events. In general, these data are reassuring and support the cardiac safety of PTH analogs for osteoporosis. However, the transient hemodynamic changes observed should be considered for patients with cardiovascular comorbidities or baseline problems with orthostasis.
Marc Wein, M.D., Ph.D
Assistant Professor of Medicine
Massachusetts General Hospital Endocrine Unit, Harvard Medical School
Romosozumab is a novel osteoporosis treatment agent that stimulates bone formation and inhibits bone resorption. Due to concerns for adverse cardiovascular effects, this medication is generally reserved for patients with severe osteoporosis despite multiple previous treatments. However, the influence of previous osteoporosis pharmacotherapy on romosozumab’s skeletal efficacy remains poorly studied, especially in real world clinical scenarios. In a prospective non-randomized study of 130 patients treated with romosozumab in Japan, the effects of romosozumab treatment on bone turnover markers and bone mineral density (BMD) was assessed based on previous treatment with either no osteoporosis medication, bisphosphonates, denosumab, or teriparatide. In general, treatment-naïve individuals experienced superior spine BMD and bone turnover marker gains versus patients who had previously received osteoporosis pharmacotherapy. Notably, prior treatment with the potent anti-resorptive agent denosumab was associated with suboptimal spine BMD response to romosozumab. These results highlight the potential impact of previous anti-resorptive treatment on romosozumab efficacy and indicate that careful consideration of treatment sequence is warranted when deciding amongst the multiple osteoporosis agents that are currently available.
Denosumab is a potent anti-resorptive osteoporosis agent that works by neutralizing RANKL, the key cytokine that drives osteoclast differentiation. While effective at suppressing bone resorption, increasing BMD, and preventing fragility fractures when administered every 6 months, rapid discontinuation of denosumab has been associated with an increased risk of vertebral compression fractures. In routine clinical practice, denosumab injection delays are common. However, risks of vertebral fractures associated with such injection delays remain unknown. In this study of a UK population-based cohort of 2,594 patients, a large electronic database was used to assess the relationship between denosumab dosing intervals (defined as ‘on time’, ‘short delay’, and ‘long delay’) and fractures in the 6 month window after the recommended treatment date. Compared with ‘on time’ denosumab treatment, individuals who received delayed denosumab injections showed an increased risk of interval vertebral fractures. Notably, the delay time was associated with fracture risk. This powerful study design further underscores the importance of timely (every 6 months) denosumab injections. Given current health care delivery challenges associated with the COVID-19 pandemic, these results should be considered when selecting initial osteoporosis treatment agents and in management of patients currently receiving denosumab.
Abaloparatide, a synthetic analog of parathyroid hormone related peptide (PTHrP) is a bone anabolic osteoporosis treatment agent. In the ACTIVE study, abaloparatide increased bone mineral density and reduced fracture risk compared to both placebo and teriparatide. Although abaloparatide is generally well-tolerated, non-serious adverse events have been reported such as dizziness and palpitations. PTHrP can act in a paracrine manner to cause vasodilation. Therefore, in this post hoc drug safety study, the effects of abaloparatide on blood pressure, pulse, and cardiovascular adverse events were assessed in detail in subjects from the ACTIVE study (one hour post treatment) plus an additional group of 55 subjects for more detailed hemodynamic assessment. Compared to placebo injection, both abaloparatide and teriparatide treatment caused mild and transient increases in pulse and decreases in systolic blood pressure. These mild hemodynamic changes were not associated with an increased risk of adverse cardiovascular events. In general, these data are reassuring and support the cardiac safety of PTH analogs for osteoporosis. However, the transient hemodynamic changes observed should be considered for patients with cardiovascular comorbidities or baseline problems with orthostasis.
Marc Wein, M.D., Ph.D
Assistant Professor of Medicine
Massachusetts General Hospital Endocrine Unit, Harvard Medical School
Glucocorticoid-induced osteoporosis: Teriparatide may be a good alternative to denosumab
Key clinical point: Teriparatide may have better therapeutic effects than denosumab in glucocorticoid-induced osteoporosis (GIO) patients previously treated with bisphosphonates.
Major finding: Teriparatide group showed a significantly greater improvement in the bone mineral density (BMD) of lumbar spine (P less than .01) and femoral neck (P less than .05) vs. denosumab group at 12 months, but not at 24 months. At 24 months, BMD of lumbar spine had significantly increased from baseline in both the groups, whereas BMD of femoral neck had increased only in the teriparatide group.
Study details: A prospective, open-label, nonrandomized, single-center study including 47 patients with GIO assigned to either teriparatide group (n = 23) or denosumab group (n = 24).
Disclosures: The study did not receive any specific funding. The authors declared no conflicts of interest.
Source: Hirooka Y et al. Bone Rep. 2020 Jul 4. doi: 10.1016/j.bonr.2020.100293.
Key clinical point: Teriparatide may have better therapeutic effects than denosumab in glucocorticoid-induced osteoporosis (GIO) patients previously treated with bisphosphonates.
Major finding: Teriparatide group showed a significantly greater improvement in the bone mineral density (BMD) of lumbar spine (P less than .01) and femoral neck (P less than .05) vs. denosumab group at 12 months, but not at 24 months. At 24 months, BMD of lumbar spine had significantly increased from baseline in both the groups, whereas BMD of femoral neck had increased only in the teriparatide group.
Study details: A prospective, open-label, nonrandomized, single-center study including 47 patients with GIO assigned to either teriparatide group (n = 23) or denosumab group (n = 24).
Disclosures: The study did not receive any specific funding. The authors declared no conflicts of interest.
Source: Hirooka Y et al. Bone Rep. 2020 Jul 4. doi: 10.1016/j.bonr.2020.100293.
Key clinical point: Teriparatide may have better therapeutic effects than denosumab in glucocorticoid-induced osteoporosis (GIO) patients previously treated with bisphosphonates.
Major finding: Teriparatide group showed a significantly greater improvement in the bone mineral density (BMD) of lumbar spine (P less than .01) and femoral neck (P less than .05) vs. denosumab group at 12 months, but not at 24 months. At 24 months, BMD of lumbar spine had significantly increased from baseline in both the groups, whereas BMD of femoral neck had increased only in the teriparatide group.
Study details: A prospective, open-label, nonrandomized, single-center study including 47 patients with GIO assigned to either teriparatide group (n = 23) or denosumab group (n = 24).
Disclosures: The study did not receive any specific funding. The authors declared no conflicts of interest.
Source: Hirooka Y et al. Bone Rep. 2020 Jul 4. doi: 10.1016/j.bonr.2020.100293.