User login
Study: Hospitalists Improve ED Throughput
Hospitalist-led, active-bed management can save hospitals millions of dollars a year by reducing ambulance diversions and trimming emergency department (ED) throughput times, according to a single-institution study.
The study in Annals of Internal Medicine (2008;149(11):804-810) found twice-daily bed management rounds in the ICU and regular visits to the ED to assess flow reduced ED throughput by 98 minutes. It also cut the number of ambulance diversions for overcrowding by 6% and reduced diversions caused by a lack of ICU beds by 27%. The study compared data from November 2005 to February 2006 (control period) and November 2006 to February 2007 (intervention period).
Lead author Eric Howell, MD, a hospitalist with Collaborative Inpatient Medical Service at Johns Hopkins Bayview Medical Center in Baltimore, estimated ambulance diversions cost hospitals $1,000 to $8,000 an hour. His study found a decrease of more than 2,000 hours in ambulance diversions, which could translate into $16 million in annual savings
"You've got to have the money up front," Dr. Howell says, acknowledging startup costs for a new or expanded hospitalist program. "You can't do it on the cheap. It falls apart."
Smaller hospitalist groups without the staffing for a full-time program could run a 12-hour, daytime version, or a trial run during specific hours.
"It's not an easy thing to do," he says, "but it adds tremendous value to a hospitalist group."
Hospitalist-led, active-bed management can save hospitals millions of dollars a year by reducing ambulance diversions and trimming emergency department (ED) throughput times, according to a single-institution study.
The study in Annals of Internal Medicine (2008;149(11):804-810) found twice-daily bed management rounds in the ICU and regular visits to the ED to assess flow reduced ED throughput by 98 minutes. It also cut the number of ambulance diversions for overcrowding by 6% and reduced diversions caused by a lack of ICU beds by 27%. The study compared data from November 2005 to February 2006 (control period) and November 2006 to February 2007 (intervention period).
Lead author Eric Howell, MD, a hospitalist with Collaborative Inpatient Medical Service at Johns Hopkins Bayview Medical Center in Baltimore, estimated ambulance diversions cost hospitals $1,000 to $8,000 an hour. His study found a decrease of more than 2,000 hours in ambulance diversions, which could translate into $16 million in annual savings
"You've got to have the money up front," Dr. Howell says, acknowledging startup costs for a new or expanded hospitalist program. "You can't do it on the cheap. It falls apart."
Smaller hospitalist groups without the staffing for a full-time program could run a 12-hour, daytime version, or a trial run during specific hours.
"It's not an easy thing to do," he says, "but it adds tremendous value to a hospitalist group."
Hospitalist-led, active-bed management can save hospitals millions of dollars a year by reducing ambulance diversions and trimming emergency department (ED) throughput times, according to a single-institution study.
The study in Annals of Internal Medicine (2008;149(11):804-810) found twice-daily bed management rounds in the ICU and regular visits to the ED to assess flow reduced ED throughput by 98 minutes. It also cut the number of ambulance diversions for overcrowding by 6% and reduced diversions caused by a lack of ICU beds by 27%. The study compared data from November 2005 to February 2006 (control period) and November 2006 to February 2007 (intervention period).
Lead author Eric Howell, MD, a hospitalist with Collaborative Inpatient Medical Service at Johns Hopkins Bayview Medical Center in Baltimore, estimated ambulance diversions cost hospitals $1,000 to $8,000 an hour. His study found a decrease of more than 2,000 hours in ambulance diversions, which could translate into $16 million in annual savings
"You've got to have the money up front," Dr. Howell says, acknowledging startup costs for a new or expanded hospitalist program. "You can't do it on the cheap. It falls apart."
Smaller hospitalist groups without the staffing for a full-time program could run a 12-hour, daytime version, or a trial run during specific hours.
"It's not an easy thing to do," he says, "but it adds tremendous value to a hospitalist group."
Research Roundup
Question: Can thrombolysis with alteplase improve stroke symptoms more than three hours after symptom onset?
Background: For acute ischemic stroke (AIS), trial evidence demonstrates tissue plasminogen activator (tPA) improves outcomes when given within three hours of symptom onset. Although the benefit of alteplase (a recombinant tPA therapeutic) decreases as the time from stroke symptom onset increases, some observational studies have suggested this drug may improve long-term neurologic outcomes even after the three-hour time limit.
Study Design: A multicenter, double blind, randomized placebo-controlled study with intention-to-treat analysis.
Setting: Multicenter, multinational study in Europe.
Synopsis: More than 800 patients presenting with stroke symptoms were diagnosed with ischemic stroke. Stroke severity was somewhat lower than prior tPA trials, and patients were randomized to receive either 0.9 mg per kg of alteplase by IV (up to 90 mg) or placebo.
Based on validated patient symptom scores, outcome was favorable in 52.4% of the alteplase group and 45.2% in the placebo group, leading to an absolute benefit of 7.2% and a number-needed-to-treat of 14. Although risk of any intracranial hemorrhage (ICH) was higher in the alteplase group (2.4% vs. 0.3%), symptomatic ICH was only marginally different, and mortality was similar in both groups, (7.7% vs. 8.4%).
This trial suggests the use of alteplase can be extended to four and a half hours after the onset of AIS symptoms without significantly increasing the overall mortality of these patients. A premium should be placed on earlier treatment, as the efficacy of alteplase decreases exponentially with time.
Bottom Line: Alteplase shows a statistically significant and clinically important improvement of AIS symptoms up to four and a half hours after the onset of stroke, without increasing mortality.
Citation: NEJM. 2008;359:1317-1329.
—Reviewed for the e-wire by Elbert Chun, MD, John Vazquez, MD, Larry Beer, MD, Maged Doss, MD, Vana Bollineni, MD, Mohammed S. Singapuri, MD, Dan Dressler, MD, MsCR, Emory University Hospital, Atlanta
Question: Can thrombolysis with alteplase improve stroke symptoms more than three hours after symptom onset?
Background: For acute ischemic stroke (AIS), trial evidence demonstrates tissue plasminogen activator (tPA) improves outcomes when given within three hours of symptom onset. Although the benefit of alteplase (a recombinant tPA therapeutic) decreases as the time from stroke symptom onset increases, some observational studies have suggested this drug may improve long-term neurologic outcomes even after the three-hour time limit.
Study Design: A multicenter, double blind, randomized placebo-controlled study with intention-to-treat analysis.
Setting: Multicenter, multinational study in Europe.
Synopsis: More than 800 patients presenting with stroke symptoms were diagnosed with ischemic stroke. Stroke severity was somewhat lower than prior tPA trials, and patients were randomized to receive either 0.9 mg per kg of alteplase by IV (up to 90 mg) or placebo.
Based on validated patient symptom scores, outcome was favorable in 52.4% of the alteplase group and 45.2% in the placebo group, leading to an absolute benefit of 7.2% and a number-needed-to-treat of 14. Although risk of any intracranial hemorrhage (ICH) was higher in the alteplase group (2.4% vs. 0.3%), symptomatic ICH was only marginally different, and mortality was similar in both groups, (7.7% vs. 8.4%).
This trial suggests the use of alteplase can be extended to four and a half hours after the onset of AIS symptoms without significantly increasing the overall mortality of these patients. A premium should be placed on earlier treatment, as the efficacy of alteplase decreases exponentially with time.
Bottom Line: Alteplase shows a statistically significant and clinically important improvement of AIS symptoms up to four and a half hours after the onset of stroke, without increasing mortality.
Citation: NEJM. 2008;359:1317-1329.
—Reviewed for the e-wire by Elbert Chun, MD, John Vazquez, MD, Larry Beer, MD, Maged Doss, MD, Vana Bollineni, MD, Mohammed S. Singapuri, MD, Dan Dressler, MD, MsCR, Emory University Hospital, Atlanta
Question: Can thrombolysis with alteplase improve stroke symptoms more than three hours after symptom onset?
Background: For acute ischemic stroke (AIS), trial evidence demonstrates tissue plasminogen activator (tPA) improves outcomes when given within three hours of symptom onset. Although the benefit of alteplase (a recombinant tPA therapeutic) decreases as the time from stroke symptom onset increases, some observational studies have suggested this drug may improve long-term neurologic outcomes even after the three-hour time limit.
Study Design: A multicenter, double blind, randomized placebo-controlled study with intention-to-treat analysis.
Setting: Multicenter, multinational study in Europe.
Synopsis: More than 800 patients presenting with stroke symptoms were diagnosed with ischemic stroke. Stroke severity was somewhat lower than prior tPA trials, and patients were randomized to receive either 0.9 mg per kg of alteplase by IV (up to 90 mg) or placebo.
Based on validated patient symptom scores, outcome was favorable in 52.4% of the alteplase group and 45.2% in the placebo group, leading to an absolute benefit of 7.2% and a number-needed-to-treat of 14. Although risk of any intracranial hemorrhage (ICH) was higher in the alteplase group (2.4% vs. 0.3%), symptomatic ICH was only marginally different, and mortality was similar in both groups, (7.7% vs. 8.4%).
This trial suggests the use of alteplase can be extended to four and a half hours after the onset of AIS symptoms without significantly increasing the overall mortality of these patients. A premium should be placed on earlier treatment, as the efficacy of alteplase decreases exponentially with time.
Bottom Line: Alteplase shows a statistically significant and clinically important improvement of AIS symptoms up to four and a half hours after the onset of stroke, without increasing mortality.
Citation: NEJM. 2008;359:1317-1329.
—Reviewed for the e-wire by Elbert Chun, MD, John Vazquez, MD, Larry Beer, MD, Maged Doss, MD, Vana Bollineni, MD, Mohammed S. Singapuri, MD, Dan Dressler, MD, MsCR, Emory University Hospital, Atlanta
At Work in Washington

SHM’s 20-member Public Policy Committee, headed by Eric Siegal, MD, had a busy 2008 addressing the many legislative and regulatory issues affecting hospitalists. As a result of our efforts, SHM’s visibility with key lawmakers, the Medicare Payment Advisory Commission (MedPAC), and the Centers for Medicare and Medicaid Services (CMS) has increased significantly, and policymakers now routinely turn to us for advice and expertise. Here is an update on how the committee represents hospital medicine—and your interests—in our nation’s capital.
Medicare Payment Cuts Prevented
Earlier this year, SHM’s Public Policy Committee (PPC) worked tirelessly to secure passage of H.R. 6331, the Medicare Improvements for Patients and Providers Act (MIPPA). It blocked the 10.6% physician payment cut, which was scheduled to go into effect July 1, 2008, and extended current payment rates through Dec. 31, 2009 Thanks in part to concerted advocacy by SHM and its members, Congress voted overwhelmingly July 15 to override President Bush’s veto of this measure. The new law (P.L. 110-275) includes 18 months of relief from scheduled cuts in Part B Medicare payments, other increases in reimbursement for inpatient evaluation and management services, and several SHM-supported quality improvement initiatives.
Beginning Jan. 1, hospitalists will see a 1.1% increase in Part B payments, instead of a projected 5.4% cut. SHM strongly supported provisions of the bill, which requires CMS to apply budget-neutral adjustments for 2007 and 2008 to the conversion factor. This change will result in an estimated average gain of another 3% in total Medicare payments for hospitalists.
SHM also supported continued funding of the Physician Quality Reporting Initiative (PQRI) and other quality provisions contained in MIPPA. Under the new Medicare act, hospitalists who successfully report quality measures are eligible for a bonus payment in 2009 and 2010 of 2% (up from 1.5%) of their total Medicare allowed charges. The new law also requires the U.S. Secretary of Health and Human Services (HHS) to develop a plan to transition to a value-based purchasing program for physician services.
SHM took multiple steps to influence MIPPA’s successful passage. Here are a few of those steps:
- When Sen. Max Baucus (D-Montana), chair of the Senate Finance Committee, announced he was working on legislation to reverse the pending cuts in Medicare payments, the PPC immediately commended his efforts and offered our assistance with the bill, specifically focusing on quality improvement provisions.
- Sen. Baucus introduced S. 3101, which formed the basis of the final Medicare bill, H.R. 6331, in June. On June 9, the PPC sent a letter to Sen. Baucus, formally supporting his efforts to address the pending payment reductions and praising other provisions in the bill promoting quality reporting, including continuation of the PQRI program; a requirement that measures be endorsed by a consensus-based, standard-setting entity such as the National Quality Forum, which CMS posts on its Web site the list of providers who participate in the PQRI; and that HHS provide confidential feedback to providers regarding their resource use.
- In a July 10 letter, we urged President Bush to immediately sign H.R. 6331 into law, stating, “This legislation, passed by bipartisan margins in both the House and Senate, contains many positive elements for hospitalists and their patients, and deserves your support.”
- As part of our advocacy efforts, the PPC launched a comprehensive, grassroots campaign to pass MIPPA, sending an unprecedented number of communications to inform and mobilize SHM members. In the month leading up to final passage of H.R. 6331, we sent 12 legislative e-mail updates to SHM members, urging each member to contact his or her lawmakers via our legislation action center. As a result of this outreach, hospitalists generated a total of 1,269 messages to members of Congress, urging policy makers to stop the Medicare cuts to physician payments. We thank all of you who visited the action center and contacted your lawmakers. Your efforts were vital to the success of SHM’s campaign to stop the cuts and passage of H.R. 6331.
Payment Advisory Commission Interaction
The PPC has made SHM’s engagement with the influential Medicare Payment Advisory Commission a top priority. An independent Congressional agency established by the Balanced Budget Act of 1997, MedPAC advises Congress on issues affecting the Medicare program. Our efforts to educate the commission and staff about hospital medicine are paying off. Through attendance at MedPAC meetings, as well as conference calls and face-to-face meetings with staff, SHM has educated the commission about the positive contributions hospitalists are making throughout the country.
PPC members attended MedPAC’s March 5, 2008, meeting and addressed the commission during the public comment period. The PPC offered to further educate the commission regarding the role hospital medicine can play in Medicare reform. In June, key SHM leaders met with MedPAC staff, including executive director Mark Miller, in Washington to discuss hospital medicine and SHM’s quality improvement initiatives, including Project BOOST. At MedPAC’s request, the PPC has worked to develop a “starter set” of metrics to define high-performing hospitalist programs. It might form a basis for future value-based purchasing initiatives.
Also in June, MedPAC released its report to Congress on “Reforming the Delivery System,” which contained extensive information and feedback from SHM.
The PPC continues to monitor MedPAC’s work, particularly its recommendations for changes in Medicare payment for care provided around a hospitalization to encourage care coordination and efficiency. To reduce hospital readmissions, the commission’s June report to Congress recommended, among other things, that CMS conduct a voluntary pilot program to test bundled payment for all services around a hospitalization for select conditions.
Value-Based Purchasing
Together with SHM’s Performance and Standards Committee, the PPC continues to monitor and comment on CMS’s value-based purchasing (VBP) initiatives, as well as educate SHM members on what the initiatives mean for hospitalists. On June 11, SHM hosted a teleconference on VBP. It featured Thomas Valuck, MD, JD, medical officer and senior adviser, Center for Medicare Management, CMS. In his presentation, Dr. Valuck acknowledged the unique role hospitalists play in VBP programs, and he commended SHM for its proactive stance and constructive engagement with CMS.
On Aug. 29, the PPC submitted comments on CMS’s proposed FY 2009 physician payment rule. This rule proposed additional improvements to the PQRI; discussed CMS’s interest in developing a “Physician Compare” Web site to report quality of care and value for services provided by physicians; solicited comments on CMS’s proposed preventable hospital-acquired conditions; and proposed a new, targeted exception to the physician self-referral statute for programs using economic incentives to foster high quality, cost-effective care. Visit http://www.hospital medicine.org/Content/Navigation Menu/AdvocacyPolicy/LegislativeRegulatoryUpdates/Legislative_Regulato.htm for a summary of the final rule.
Increased AHRQ Appropriations
Through its participation in the Friends of Agency for Healthcare Research and Quality (AHRQ) coalition, visits to Congressional offices by members and staff, and grassroots advocacy via our legislative action center, SHM continues to advocate for increased funding for this important agency. Last fall, President Bush signed a continuing resolution, P.L. 110-329, which funds government agencies, including AHRQ, at current levels through March 6. The resolution was necessary because none of the 12 individual FY 2009 appropriations bills, including the Labor Department-Health and Human Services-Education Department measure, which contains funding for AHRQ and the National Institutes of Health, had been enacted into law. Early this year lawmakers are expected to attempt to pass the remaining appropriations bills and forward them to President-elect Obama.
What’s Ahead?
This year promises to be busy on the healthcare policy front. SHM is poised to make major contributions to the debate, given its advocacy on key issues over the past year and the goodwill it has generated among policymakers. The PPC will devote considerable time to crafting hospital medicine-specific recommendations on health reform, including bundling and its implications for hospitalists; and providing input to CMS’s value-based purchasing initiatives, including the agency’s report to Congress, which is due May 2010. We also will continue to pursue a separate CMS specialty billing code for hospitalists.
The PPC strives to keep SHM members informed about legislative and regulatory activities through monthly updates posted to the advocacy section of the SHM Web site, articles in The Hospitalist, and items in the new SHM e-Wire. Letters to Congress and CMS are located on the Web site, as well.
When an important issue arises, you likely will receive an e-mail urging you to visit our legislative action center (www.hospitalmedicine.org/beheard) and contact your members of Congress. We depend on your involvement in the legislative process in order to be effective in Washington. TH
Laura Allendorf is senior advisor for advocacy and government affairs for the Society of Hospital Medicine. Contact her at [email protected].
SHM’s 20-member Public Policy Committee, headed by Eric Siegal, MD, had a busy 2008 addressing the many legislative and regulatory issues affecting hospitalists. As a result of our efforts, SHM’s visibility with key lawmakers, the Medicare Payment Advisory Commission (MedPAC), and the Centers for Medicare and Medicaid Services (CMS) has increased significantly, and policymakers now routinely turn to us for advice and expertise. Here is an update on how the committee represents hospital medicine—and your interests—in our nation’s capital.
Medicare Payment Cuts Prevented
Earlier this year, SHM’s Public Policy Committee (PPC) worked tirelessly to secure passage of H.R. 6331, the Medicare Improvements for Patients and Providers Act (MIPPA). It blocked the 10.6% physician payment cut, which was scheduled to go into effect July 1, 2008, and extended current payment rates through Dec. 31, 2009 Thanks in part to concerted advocacy by SHM and its members, Congress voted overwhelmingly July 15 to override President Bush’s veto of this measure. The new law (P.L. 110-275) includes 18 months of relief from scheduled cuts in Part B Medicare payments, other increases in reimbursement for inpatient evaluation and management services, and several SHM-supported quality improvement initiatives.
Beginning Jan. 1, hospitalists will see a 1.1% increase in Part B payments, instead of a projected 5.4% cut. SHM strongly supported provisions of the bill, which requires CMS to apply budget-neutral adjustments for 2007 and 2008 to the conversion factor. This change will result in an estimated average gain of another 3% in total Medicare payments for hospitalists.
SHM also supported continued funding of the Physician Quality Reporting Initiative (PQRI) and other quality provisions contained in MIPPA. Under the new Medicare act, hospitalists who successfully report quality measures are eligible for a bonus payment in 2009 and 2010 of 2% (up from 1.5%) of their total Medicare allowed charges. The new law also requires the U.S. Secretary of Health and Human Services (HHS) to develop a plan to transition to a value-based purchasing program for physician services.
SHM took multiple steps to influence MIPPA’s successful passage. Here are a few of those steps:
- When Sen. Max Baucus (D-Montana), chair of the Senate Finance Committee, announced he was working on legislation to reverse the pending cuts in Medicare payments, the PPC immediately commended his efforts and offered our assistance with the bill, specifically focusing on quality improvement provisions.
- Sen. Baucus introduced S. 3101, which formed the basis of the final Medicare bill, H.R. 6331, in June. On June 9, the PPC sent a letter to Sen. Baucus, formally supporting his efforts to address the pending payment reductions and praising other provisions in the bill promoting quality reporting, including continuation of the PQRI program; a requirement that measures be endorsed by a consensus-based, standard-setting entity such as the National Quality Forum, which CMS posts on its Web site the list of providers who participate in the PQRI; and that HHS provide confidential feedback to providers regarding their resource use.
- In a July 10 letter, we urged President Bush to immediately sign H.R. 6331 into law, stating, “This legislation, passed by bipartisan margins in both the House and Senate, contains many positive elements for hospitalists and their patients, and deserves your support.”
- As part of our advocacy efforts, the PPC launched a comprehensive, grassroots campaign to pass MIPPA, sending an unprecedented number of communications to inform and mobilize SHM members. In the month leading up to final passage of H.R. 6331, we sent 12 legislative e-mail updates to SHM members, urging each member to contact his or her lawmakers via our legislation action center. As a result of this outreach, hospitalists generated a total of 1,269 messages to members of Congress, urging policy makers to stop the Medicare cuts to physician payments. We thank all of you who visited the action center and contacted your lawmakers. Your efforts were vital to the success of SHM’s campaign to stop the cuts and passage of H.R. 6331.
Payment Advisory Commission Interaction
The PPC has made SHM’s engagement with the influential Medicare Payment Advisory Commission a top priority. An independent Congressional agency established by the Balanced Budget Act of 1997, MedPAC advises Congress on issues affecting the Medicare program. Our efforts to educate the commission and staff about hospital medicine are paying off. Through attendance at MedPAC meetings, as well as conference calls and face-to-face meetings with staff, SHM has educated the commission about the positive contributions hospitalists are making throughout the country.
PPC members attended MedPAC’s March 5, 2008, meeting and addressed the commission during the public comment period. The PPC offered to further educate the commission regarding the role hospital medicine can play in Medicare reform. In June, key SHM leaders met with MedPAC staff, including executive director Mark Miller, in Washington to discuss hospital medicine and SHM’s quality improvement initiatives, including Project BOOST. At MedPAC’s request, the PPC has worked to develop a “starter set” of metrics to define high-performing hospitalist programs. It might form a basis for future value-based purchasing initiatives.
Also in June, MedPAC released its report to Congress on “Reforming the Delivery System,” which contained extensive information and feedback from SHM.
The PPC continues to monitor MedPAC’s work, particularly its recommendations for changes in Medicare payment for care provided around a hospitalization to encourage care coordination and efficiency. To reduce hospital readmissions, the commission’s June report to Congress recommended, among other things, that CMS conduct a voluntary pilot program to test bundled payment for all services around a hospitalization for select conditions.
Value-Based Purchasing
Together with SHM’s Performance and Standards Committee, the PPC continues to monitor and comment on CMS’s value-based purchasing (VBP) initiatives, as well as educate SHM members on what the initiatives mean for hospitalists. On June 11, SHM hosted a teleconference on VBP. It featured Thomas Valuck, MD, JD, medical officer and senior adviser, Center for Medicare Management, CMS. In his presentation, Dr. Valuck acknowledged the unique role hospitalists play in VBP programs, and he commended SHM for its proactive stance and constructive engagement with CMS.
On Aug. 29, the PPC submitted comments on CMS’s proposed FY 2009 physician payment rule. This rule proposed additional improvements to the PQRI; discussed CMS’s interest in developing a “Physician Compare” Web site to report quality of care and value for services provided by physicians; solicited comments on CMS’s proposed preventable hospital-acquired conditions; and proposed a new, targeted exception to the physician self-referral statute for programs using economic incentives to foster high quality, cost-effective care. Visit http://www.hospital medicine.org/Content/Navigation Menu/AdvocacyPolicy/LegislativeRegulatoryUpdates/Legislative_Regulato.htm for a summary of the final rule.
Increased AHRQ Appropriations
Through its participation in the Friends of Agency for Healthcare Research and Quality (AHRQ) coalition, visits to Congressional offices by members and staff, and grassroots advocacy via our legislative action center, SHM continues to advocate for increased funding for this important agency. Last fall, President Bush signed a continuing resolution, P.L. 110-329, which funds government agencies, including AHRQ, at current levels through March 6. The resolution was necessary because none of the 12 individual FY 2009 appropriations bills, including the Labor Department-Health and Human Services-Education Department measure, which contains funding for AHRQ and the National Institutes of Health, had been enacted into law. Early this year lawmakers are expected to attempt to pass the remaining appropriations bills and forward them to President-elect Obama.
What’s Ahead?
This year promises to be busy on the healthcare policy front. SHM is poised to make major contributions to the debate, given its advocacy on key issues over the past year and the goodwill it has generated among policymakers. The PPC will devote considerable time to crafting hospital medicine-specific recommendations on health reform, including bundling and its implications for hospitalists; and providing input to CMS’s value-based purchasing initiatives, including the agency’s report to Congress, which is due May 2010. We also will continue to pursue a separate CMS specialty billing code for hospitalists.
The PPC strives to keep SHM members informed about legislative and regulatory activities through monthly updates posted to the advocacy section of the SHM Web site, articles in The Hospitalist, and items in the new SHM e-Wire. Letters to Congress and CMS are located on the Web site, as well.
When an important issue arises, you likely will receive an e-mail urging you to visit our legislative action center (www.hospitalmedicine.org/beheard) and contact your members of Congress. We depend on your involvement in the legislative process in order to be effective in Washington. TH
Laura Allendorf is senior advisor for advocacy and government affairs for the Society of Hospital Medicine. Contact her at [email protected].
SHM’s 20-member Public Policy Committee, headed by Eric Siegal, MD, had a busy 2008 addressing the many legislative and regulatory issues affecting hospitalists. As a result of our efforts, SHM’s visibility with key lawmakers, the Medicare Payment Advisory Commission (MedPAC), and the Centers for Medicare and Medicaid Services (CMS) has increased significantly, and policymakers now routinely turn to us for advice and expertise. Here is an update on how the committee represents hospital medicine—and your interests—in our nation’s capital.
Medicare Payment Cuts Prevented
Earlier this year, SHM’s Public Policy Committee (PPC) worked tirelessly to secure passage of H.R. 6331, the Medicare Improvements for Patients and Providers Act (MIPPA). It blocked the 10.6% physician payment cut, which was scheduled to go into effect July 1, 2008, and extended current payment rates through Dec. 31, 2009 Thanks in part to concerted advocacy by SHM and its members, Congress voted overwhelmingly July 15 to override President Bush’s veto of this measure. The new law (P.L. 110-275) includes 18 months of relief from scheduled cuts in Part B Medicare payments, other increases in reimbursement for inpatient evaluation and management services, and several SHM-supported quality improvement initiatives.
Beginning Jan. 1, hospitalists will see a 1.1% increase in Part B payments, instead of a projected 5.4% cut. SHM strongly supported provisions of the bill, which requires CMS to apply budget-neutral adjustments for 2007 and 2008 to the conversion factor. This change will result in an estimated average gain of another 3% in total Medicare payments for hospitalists.
SHM also supported continued funding of the Physician Quality Reporting Initiative (PQRI) and other quality provisions contained in MIPPA. Under the new Medicare act, hospitalists who successfully report quality measures are eligible for a bonus payment in 2009 and 2010 of 2% (up from 1.5%) of their total Medicare allowed charges. The new law also requires the U.S. Secretary of Health and Human Services (HHS) to develop a plan to transition to a value-based purchasing program for physician services.
SHM took multiple steps to influence MIPPA’s successful passage. Here are a few of those steps:
- When Sen. Max Baucus (D-Montana), chair of the Senate Finance Committee, announced he was working on legislation to reverse the pending cuts in Medicare payments, the PPC immediately commended his efforts and offered our assistance with the bill, specifically focusing on quality improvement provisions.
- Sen. Baucus introduced S. 3101, which formed the basis of the final Medicare bill, H.R. 6331, in June. On June 9, the PPC sent a letter to Sen. Baucus, formally supporting his efforts to address the pending payment reductions and praising other provisions in the bill promoting quality reporting, including continuation of the PQRI program; a requirement that measures be endorsed by a consensus-based, standard-setting entity such as the National Quality Forum, which CMS posts on its Web site the list of providers who participate in the PQRI; and that HHS provide confidential feedback to providers regarding their resource use.
- In a July 10 letter, we urged President Bush to immediately sign H.R. 6331 into law, stating, “This legislation, passed by bipartisan margins in both the House and Senate, contains many positive elements for hospitalists and their patients, and deserves your support.”
- As part of our advocacy efforts, the PPC launched a comprehensive, grassroots campaign to pass MIPPA, sending an unprecedented number of communications to inform and mobilize SHM members. In the month leading up to final passage of H.R. 6331, we sent 12 legislative e-mail updates to SHM members, urging each member to contact his or her lawmakers via our legislation action center. As a result of this outreach, hospitalists generated a total of 1,269 messages to members of Congress, urging policy makers to stop the Medicare cuts to physician payments. We thank all of you who visited the action center and contacted your lawmakers. Your efforts were vital to the success of SHM’s campaign to stop the cuts and passage of H.R. 6331.
Payment Advisory Commission Interaction
The PPC has made SHM’s engagement with the influential Medicare Payment Advisory Commission a top priority. An independent Congressional agency established by the Balanced Budget Act of 1997, MedPAC advises Congress on issues affecting the Medicare program. Our efforts to educate the commission and staff about hospital medicine are paying off. Through attendance at MedPAC meetings, as well as conference calls and face-to-face meetings with staff, SHM has educated the commission about the positive contributions hospitalists are making throughout the country.
PPC members attended MedPAC’s March 5, 2008, meeting and addressed the commission during the public comment period. The PPC offered to further educate the commission regarding the role hospital medicine can play in Medicare reform. In June, key SHM leaders met with MedPAC staff, including executive director Mark Miller, in Washington to discuss hospital medicine and SHM’s quality improvement initiatives, including Project BOOST. At MedPAC’s request, the PPC has worked to develop a “starter set” of metrics to define high-performing hospitalist programs. It might form a basis for future value-based purchasing initiatives.
Also in June, MedPAC released its report to Congress on “Reforming the Delivery System,” which contained extensive information and feedback from SHM.
The PPC continues to monitor MedPAC’s work, particularly its recommendations for changes in Medicare payment for care provided around a hospitalization to encourage care coordination and efficiency. To reduce hospital readmissions, the commission’s June report to Congress recommended, among other things, that CMS conduct a voluntary pilot program to test bundled payment for all services around a hospitalization for select conditions.
Value-Based Purchasing
Together with SHM’s Performance and Standards Committee, the PPC continues to monitor and comment on CMS’s value-based purchasing (VBP) initiatives, as well as educate SHM members on what the initiatives mean for hospitalists. On June 11, SHM hosted a teleconference on VBP. It featured Thomas Valuck, MD, JD, medical officer and senior adviser, Center for Medicare Management, CMS. In his presentation, Dr. Valuck acknowledged the unique role hospitalists play in VBP programs, and he commended SHM for its proactive stance and constructive engagement with CMS.
On Aug. 29, the PPC submitted comments on CMS’s proposed FY 2009 physician payment rule. This rule proposed additional improvements to the PQRI; discussed CMS’s interest in developing a “Physician Compare” Web site to report quality of care and value for services provided by physicians; solicited comments on CMS’s proposed preventable hospital-acquired conditions; and proposed a new, targeted exception to the physician self-referral statute for programs using economic incentives to foster high quality, cost-effective care. Visit http://www.hospital medicine.org/Content/Navigation Menu/AdvocacyPolicy/LegislativeRegulatoryUpdates/Legislative_Regulato.htm for a summary of the final rule.
Increased AHRQ Appropriations
Through its participation in the Friends of Agency for Healthcare Research and Quality (AHRQ) coalition, visits to Congressional offices by members and staff, and grassroots advocacy via our legislative action center, SHM continues to advocate for increased funding for this important agency. Last fall, President Bush signed a continuing resolution, P.L. 110-329, which funds government agencies, including AHRQ, at current levels through March 6. The resolution was necessary because none of the 12 individual FY 2009 appropriations bills, including the Labor Department-Health and Human Services-Education Department measure, which contains funding for AHRQ and the National Institutes of Health, had been enacted into law. Early this year lawmakers are expected to attempt to pass the remaining appropriations bills and forward them to President-elect Obama.
What’s Ahead?
This year promises to be busy on the healthcare policy front. SHM is poised to make major contributions to the debate, given its advocacy on key issues over the past year and the goodwill it has generated among policymakers. The PPC will devote considerable time to crafting hospital medicine-specific recommendations on health reform, including bundling and its implications for hospitalists; and providing input to CMS’s value-based purchasing initiatives, including the agency’s report to Congress, which is due May 2010. We also will continue to pursue a separate CMS specialty billing code for hospitalists.
The PPC strives to keep SHM members informed about legislative and regulatory activities through monthly updates posted to the advocacy section of the SHM Web site, articles in The Hospitalist, and items in the new SHM e-Wire. Letters to Congress and CMS are located on the Web site, as well.
When an important issue arises, you likely will receive an e-mail urging you to visit our legislative action center (www.hospitalmedicine.org/beheard) and contact your members of Congress. We depend on your involvement in the legislative process in order to be effective in Washington. TH
Laura Allendorf is senior advisor for advocacy and government affairs for the Society of Hospital Medicine. Contact her at [email protected].
Non-Physician Providers Critical to Hospital Medicine Practices
Non-Physician Providers Critical to Hospital Medicine Practices
In “Maximizing NPPs in Hospitalist Practices” (Practice Management, October 2008, p. 69), John Nelson, MD, implies a financial advantage to hiring nurse practitioners (NPs) rather than physician assistants (PAs):
“A PA’s work will nearly always require a physician being physically present during some portion of the patient visit and co-signing chart notes and orders. Nurse practitioners, on the other hand, may be able to perform certain patient-care activities independently. Medicare and other payers typically reimburse at 85% of the rate customarily paid to MDs for the same service.”
Dr. Nelson: Do you mean to imply NPs providing hospitalist coverage should function independently, without any physician supervision, oversight, or input? I do not know of any hospitals that allow NPs to admit and manage patients independently, despite their independent practice status. In my opinion, it would be detrimental to patient care to allow non-physician providers to provide completely independent, unsupervised hospital care.
In addition, I know of no hospital practice setting that requires a physician to be physically present during a PA’s exam. In all hospital practice settings that I am aware of, a supervising or attending physician (not necessarily the one on the PA’s license), must see the patient at some time each day, not necessarily at the same time as the PA.
In that scenario, the physician pays a brief visit to the patient to corroborate the PA’s exam and plan. The physician then writes a brief, seen-and-agreed note, and can bill 100% of the Medicare reimbursement, not the 85% for a NP visit. How this compares economically depends, of course, on relative salaries. But it provides physician oversight of the NPP, which assumedly will improve quality of care (though I have no studies to cite to that effect).
Do you have some other reason for your anti-PA, pro-NP bias? Is your personal experience colored by working with more competent NPs, in your opinion, and less competent PAs? Perhaps that would make for a more interesting, better-defined column.
Richard Buckberg, PA-C, Maine Medical Center, Portland, Maine
SHM’s Non-Physician Provider (NPP) Committee was delighted to see the recent article by Dr. Nelson, MD, regarding the use of NPPs in hospital medicine. These columns spotlighted HM’s increasing utilization of NPPs, a role that only is expected to increase in number and breadth, based on the increasing need for competent, comprehensive care of the acutely ill.
Dr. Nelson made many excellent points that may benefit from some clarification or expansion. Dr. Nelson states a PA “will nearly always require a physician being physically present during some portion of the patient visit and co-signing chart notes and orders.” This is incorrect, and seems to imply PAs are more difficult to assimilate into practice than NPs. Depending upon variability in legal limitations and hospital bylaws, both PAs and NPs may see a patient without a physician present. In order to bill for a shared visit and receive 100% reimbursement, the attending physician needs to have a face-to-face encounter with that patient at some point in the day and document a portion of the evaluation and management (e.g., history, physical exam, or medical decision-making).
Alternatively, in most states, both NPs and PAs can see patients independently and bill for their services at 85% of the physician’s Medicare rate. Obtaining 100% reimbursement for NPP services when using the shared billing model is efficient and simple. Though variability exists, more specifically in hospital culture, utilization of these roles can positively impact the bottom line of reimbursement. Therefore, it is essential to understand all of the regulations before developing a business model incorporating NPPs.
Dr. Nelson also points out that using NPPs in a clerical fashion, such as discharge planning, is not optimizing the NPPs capabilities. This point is well taken. If clerical assistance is needed, hire the appropriate discipline.
He also says in order to integrate and maximize the benefit of the NPP to a particular practice, a careful analysis of the needs of the practice should be performed prior to hiring. This will allow both physicians and the NPP to have clear expectations. An emphasis should be placed on the importance of recognizing the level of experience will likely impact the role. For example, utilizing a new graduate in a short-stay observation unit with limited diagnoses and treatments may make sense, but utilizing an NPP as a nocturnist, cross-covering and independently admitting patients, would require an NPP with years of experience. It is crucial to hire the right NPP with the right qualifications and experience for the job.
Dr. Nelson states patient satisfaction may decline by adding NPPs to the practice. Rather than focusing on possible dilution of a patient experience with the addition of another healthcare provider, one should instead consider the potential of adding another perspective to the team. Two providers with unique educational backgrounds and insight may indeed be better than one. Utilization of NPPs can increase face time with patients, subsequently increasing patient satisfaction. Additionally, research in outpatient settings shows no difference in patient satisfaction between physicians and NPPs; more research in inpatient settings needs to be performed.
We encourage HMG directors to refer to our expanded section on SHM’s Web site, “Practice Resources,” which has a wealth of information regarding NPP utilization. The NPP committee will be hosting two courses at the annual meeting, “The Basics: Can NPs/PAs Meet Our Needs,” and “Advanced Concepts: Three Different Practice Models.”
Jina Saltzman, PA-C, University of Chicago Medical Center
Dear readers: My main goal in writing the column was to indicate that NPs and PAs could be valuable contributors to many hospitalist practices. Yet, many practices fail to create the optimal role for them, one that contributes to patient outcomes, efficiency and economic health, and provides a rewarding role for the NPP.
I do not have an anti-PA, pro-NP bias. My intention was not to take sides regarding whether PAs or NPs are more skilled or better for patients. I think that is a function of the individual, much more than their training certificate (same with MDs). I did not intend to imply “nurse practitioners providing hospitalist coverage should function independently, without any physician supervision, oversight, or input.”
Lastly, I did make a factual error regarding the differences in physician supervision required for NPs and PAs. Saltzman’s letter above addresses the error. TH
Non-Physician Providers Critical to Hospital Medicine Practices
In “Maximizing NPPs in Hospitalist Practices” (Practice Management, October 2008, p. 69), John Nelson, MD, implies a financial advantage to hiring nurse practitioners (NPs) rather than physician assistants (PAs):
“A PA’s work will nearly always require a physician being physically present during some portion of the patient visit and co-signing chart notes and orders. Nurse practitioners, on the other hand, may be able to perform certain patient-care activities independently. Medicare and other payers typically reimburse at 85% of the rate customarily paid to MDs for the same service.”
Dr. Nelson: Do you mean to imply NPs providing hospitalist coverage should function independently, without any physician supervision, oversight, or input? I do not know of any hospitals that allow NPs to admit and manage patients independently, despite their independent practice status. In my opinion, it would be detrimental to patient care to allow non-physician providers to provide completely independent, unsupervised hospital care.
In addition, I know of no hospital practice setting that requires a physician to be physically present during a PA’s exam. In all hospital practice settings that I am aware of, a supervising or attending physician (not necessarily the one on the PA’s license), must see the patient at some time each day, not necessarily at the same time as the PA.
In that scenario, the physician pays a brief visit to the patient to corroborate the PA’s exam and plan. The physician then writes a brief, seen-and-agreed note, and can bill 100% of the Medicare reimbursement, not the 85% for a NP visit. How this compares economically depends, of course, on relative salaries. But it provides physician oversight of the NPP, which assumedly will improve quality of care (though I have no studies to cite to that effect).
Do you have some other reason for your anti-PA, pro-NP bias? Is your personal experience colored by working with more competent NPs, in your opinion, and less competent PAs? Perhaps that would make for a more interesting, better-defined column.
Richard Buckberg, PA-C, Maine Medical Center, Portland, Maine
SHM’s Non-Physician Provider (NPP) Committee was delighted to see the recent article by Dr. Nelson, MD, regarding the use of NPPs in hospital medicine. These columns spotlighted HM’s increasing utilization of NPPs, a role that only is expected to increase in number and breadth, based on the increasing need for competent, comprehensive care of the acutely ill.
Dr. Nelson made many excellent points that may benefit from some clarification or expansion. Dr. Nelson states a PA “will nearly always require a physician being physically present during some portion of the patient visit and co-signing chart notes and orders.” This is incorrect, and seems to imply PAs are more difficult to assimilate into practice than NPs. Depending upon variability in legal limitations and hospital bylaws, both PAs and NPs may see a patient without a physician present. In order to bill for a shared visit and receive 100% reimbursement, the attending physician needs to have a face-to-face encounter with that patient at some point in the day and document a portion of the evaluation and management (e.g., history, physical exam, or medical decision-making).
Alternatively, in most states, both NPs and PAs can see patients independently and bill for their services at 85% of the physician’s Medicare rate. Obtaining 100% reimbursement for NPP services when using the shared billing model is efficient and simple. Though variability exists, more specifically in hospital culture, utilization of these roles can positively impact the bottom line of reimbursement. Therefore, it is essential to understand all of the regulations before developing a business model incorporating NPPs.
Dr. Nelson also points out that using NPPs in a clerical fashion, such as discharge planning, is not optimizing the NPPs capabilities. This point is well taken. If clerical assistance is needed, hire the appropriate discipline.
He also says in order to integrate and maximize the benefit of the NPP to a particular practice, a careful analysis of the needs of the practice should be performed prior to hiring. This will allow both physicians and the NPP to have clear expectations. An emphasis should be placed on the importance of recognizing the level of experience will likely impact the role. For example, utilizing a new graduate in a short-stay observation unit with limited diagnoses and treatments may make sense, but utilizing an NPP as a nocturnist, cross-covering and independently admitting patients, would require an NPP with years of experience. It is crucial to hire the right NPP with the right qualifications and experience for the job.
Dr. Nelson states patient satisfaction may decline by adding NPPs to the practice. Rather than focusing on possible dilution of a patient experience with the addition of another healthcare provider, one should instead consider the potential of adding another perspective to the team. Two providers with unique educational backgrounds and insight may indeed be better than one. Utilization of NPPs can increase face time with patients, subsequently increasing patient satisfaction. Additionally, research in outpatient settings shows no difference in patient satisfaction between physicians and NPPs; more research in inpatient settings needs to be performed.
We encourage HMG directors to refer to our expanded section on SHM’s Web site, “Practice Resources,” which has a wealth of information regarding NPP utilization. The NPP committee will be hosting two courses at the annual meeting, “The Basics: Can NPs/PAs Meet Our Needs,” and “Advanced Concepts: Three Different Practice Models.”
Jina Saltzman, PA-C, University of Chicago Medical Center
Dear readers: My main goal in writing the column was to indicate that NPs and PAs could be valuable contributors to many hospitalist practices. Yet, many practices fail to create the optimal role for them, one that contributes to patient outcomes, efficiency and economic health, and provides a rewarding role for the NPP.
I do not have an anti-PA, pro-NP bias. My intention was not to take sides regarding whether PAs or NPs are more skilled or better for patients. I think that is a function of the individual, much more than their training certificate (same with MDs). I did not intend to imply “nurse practitioners providing hospitalist coverage should function independently, without any physician supervision, oversight, or input.”
Lastly, I did make a factual error regarding the differences in physician supervision required for NPs and PAs. Saltzman’s letter above addresses the error. TH
Non-Physician Providers Critical to Hospital Medicine Practices
In “Maximizing NPPs in Hospitalist Practices” (Practice Management, October 2008, p. 69), John Nelson, MD, implies a financial advantage to hiring nurse practitioners (NPs) rather than physician assistants (PAs):
“A PA’s work will nearly always require a physician being physically present during some portion of the patient visit and co-signing chart notes and orders. Nurse practitioners, on the other hand, may be able to perform certain patient-care activities independently. Medicare and other payers typically reimburse at 85% of the rate customarily paid to MDs for the same service.”
Dr. Nelson: Do you mean to imply NPs providing hospitalist coverage should function independently, without any physician supervision, oversight, or input? I do not know of any hospitals that allow NPs to admit and manage patients independently, despite their independent practice status. In my opinion, it would be detrimental to patient care to allow non-physician providers to provide completely independent, unsupervised hospital care.
In addition, I know of no hospital practice setting that requires a physician to be physically present during a PA’s exam. In all hospital practice settings that I am aware of, a supervising or attending physician (not necessarily the one on the PA’s license), must see the patient at some time each day, not necessarily at the same time as the PA.
In that scenario, the physician pays a brief visit to the patient to corroborate the PA’s exam and plan. The physician then writes a brief, seen-and-agreed note, and can bill 100% of the Medicare reimbursement, not the 85% for a NP visit. How this compares economically depends, of course, on relative salaries. But it provides physician oversight of the NPP, which assumedly will improve quality of care (though I have no studies to cite to that effect).
Do you have some other reason for your anti-PA, pro-NP bias? Is your personal experience colored by working with more competent NPs, in your opinion, and less competent PAs? Perhaps that would make for a more interesting, better-defined column.
Richard Buckberg, PA-C, Maine Medical Center, Portland, Maine
SHM’s Non-Physician Provider (NPP) Committee was delighted to see the recent article by Dr. Nelson, MD, regarding the use of NPPs in hospital medicine. These columns spotlighted HM’s increasing utilization of NPPs, a role that only is expected to increase in number and breadth, based on the increasing need for competent, comprehensive care of the acutely ill.
Dr. Nelson made many excellent points that may benefit from some clarification or expansion. Dr. Nelson states a PA “will nearly always require a physician being physically present during some portion of the patient visit and co-signing chart notes and orders.” This is incorrect, and seems to imply PAs are more difficult to assimilate into practice than NPs. Depending upon variability in legal limitations and hospital bylaws, both PAs and NPs may see a patient without a physician present. In order to bill for a shared visit and receive 100% reimbursement, the attending physician needs to have a face-to-face encounter with that patient at some point in the day and document a portion of the evaluation and management (e.g., history, physical exam, or medical decision-making).
Alternatively, in most states, both NPs and PAs can see patients independently and bill for their services at 85% of the physician’s Medicare rate. Obtaining 100% reimbursement for NPP services when using the shared billing model is efficient and simple. Though variability exists, more specifically in hospital culture, utilization of these roles can positively impact the bottom line of reimbursement. Therefore, it is essential to understand all of the regulations before developing a business model incorporating NPPs.
Dr. Nelson also points out that using NPPs in a clerical fashion, such as discharge planning, is not optimizing the NPPs capabilities. This point is well taken. If clerical assistance is needed, hire the appropriate discipline.
He also says in order to integrate and maximize the benefit of the NPP to a particular practice, a careful analysis of the needs of the practice should be performed prior to hiring. This will allow both physicians and the NPP to have clear expectations. An emphasis should be placed on the importance of recognizing the level of experience will likely impact the role. For example, utilizing a new graduate in a short-stay observation unit with limited diagnoses and treatments may make sense, but utilizing an NPP as a nocturnist, cross-covering and independently admitting patients, would require an NPP with years of experience. It is crucial to hire the right NPP with the right qualifications and experience for the job.
Dr. Nelson states patient satisfaction may decline by adding NPPs to the practice. Rather than focusing on possible dilution of a patient experience with the addition of another healthcare provider, one should instead consider the potential of adding another perspective to the team. Two providers with unique educational backgrounds and insight may indeed be better than one. Utilization of NPPs can increase face time with patients, subsequently increasing patient satisfaction. Additionally, research in outpatient settings shows no difference in patient satisfaction between physicians and NPPs; more research in inpatient settings needs to be performed.
We encourage HMG directors to refer to our expanded section on SHM’s Web site, “Practice Resources,” which has a wealth of information regarding NPP utilization. The NPP committee will be hosting two courses at the annual meeting, “The Basics: Can NPs/PAs Meet Our Needs,” and “Advanced Concepts: Three Different Practice Models.”
Jina Saltzman, PA-C, University of Chicago Medical Center
Dear readers: My main goal in writing the column was to indicate that NPs and PAs could be valuable contributors to many hospitalist practices. Yet, many practices fail to create the optimal role for them, one that contributes to patient outcomes, efficiency and economic health, and provides a rewarding role for the NPP.
I do not have an anti-PA, pro-NP bias. My intention was not to take sides regarding whether PAs or NPs are more skilled or better for patients. I think that is a function of the individual, much more than their training certificate (same with MDs). I did not intend to imply “nurse practitioners providing hospitalist coverage should function independently, without any physician supervision, oversight, or input.”
Lastly, I did make a factual error regarding the differences in physician supervision required for NPs and PAs. Saltzman’s letter above addresses the error. TH
Perilous Intersection
When Amsterdam Airport Schiphol in the Netherlands revamped its men’s restrooms, the architects installed small, Euro-style urinals: a surefire way to throw urine off target. To solve this problem, the black outline of a fly was etched in the porcelain near each urinal’s drain. Users’ aim improved and spillage was reduced by 80%. “They try to power blast it away,” says Sanjay Saint, MD, hospitalist and professor of internal medicine at the Ann Arbor VA Medical Center, University of Michigan. “By the time they might realize that the fly isn’t going anywhere, the men are done and walking away.”
It’s a guy thing, sure. It also is an example of a human factors intervention. “Science teaches us that implementing a design for a machine or device that elicits an instinctive reaction from someone using it is a clear-cut way to avoid error,” Dr. Saint explains.
What It Is and Why It’s Important
Human factors (HF), or human factors engineering (HFE), also sometimes called usability engineering or systems-based practice, refers to the study of human abilities and characteristics as they affect the design and smooth operation of equipment, systems, and jobs.1 HF is the basic science underlying much of patient safety practice. For instance, the current recommendation that hospitals standardize equipment, such as ventilators, programmable IV pumps, and defibrillators, is an example of making tasks human friendly. The use of cognitive psychology and biomechanics to develop and improve software and hand tools are another example of HF principles.
In general, HF examines the component tasks of an activity in terms of three factorial domains: physical and environmental factors, cognitive factors (skill demands and mental workload), and organizational factors. Each task is assessed in terms of necessary interactions of the individual and work environment, the device/system design, and associated team dynamics.
HF use in healthcare is not new; for roughly four decades HF researchers have emphasized the key role of HF in safe medical design, healthcare facility operations, and patient safety processes. HF helps organizations deepen analyses of adverse events and develop effective solutions.2 HF is used in the design of labeling, warnings or alarms, software programs, information displays, paper forms, process and activity flow, workplace design, cognitive aids, decision support systems, and policies and protocols.
For hospitalists, human factors knowledge is most useful in process improvement, says John Gosbee, MD, MS, a human factors engineering and healthcare specialist at the University of Michigan. Dr. Gosbee, who has worked with hospitalists in Ann Arbor and around the country, originally studied aerospace medicine, pursued a subspecialty in occupational medicine, and from 1988 to 1992 worked at NASA designing space hospitals. In the dozens of lectures and workshops he has conducted, he has learned numerous physicians resist learning about HF. At first they protest, claiming they “didn’t go to medical school to become engineers” or “weren’t hired to have you tell us we need to be some kind of designer or computer-science software evaluator.”
Dr. Gosbee couldn’t agree more, but after the a-ha! moment, usually in an interactive experience when the hospitalist sees a poor system design is an obstacle to safety and process flow, they open up to adopting the HF mindset. Once on board with HF, hospitalists are quick to translate the theories to their own practices, identifying potential vulnerabilities and risks.
Manufacturers of healthcare equipment and systems don’t want to hear from “safety geeks,” Dr. Gosbee says; the companies want to hear from front-line providers who regularly use the products. “Hospitalists are in great position to provide that input because they see what happens across a broad swath of hospital settings,” he says, “and they could amalgamate the fact that everyone across specialties is having some trouble with this computer screen or new infusion device.”
Dr. Gosbee’s first-hand knowledge and experience solving hospitalist issues with HF techniques evolved into a teaching career. He says the university administration supports his belief in the practicality of HF lessons, and he now works as the lead instructor for a majority of the university’s medical residents.
“Human factors engineering is an efficient way to flip people’s brains around 180 degrees toward systems thinking,” Dr. Gosbee explains, “which is required if the organization wants to become a high-reliability organization.”3
Examples in Medicine
Russ Cucina, MD, MS, hospitalist at the University of California San Francisco Medical Center, describes a practical example of human factors engineering in a simple, widely used design. When cars ran on leaded gasoline, the design of the leaded gas pump nozzle precluded it from being inserted into an unleaded gas tank. “Even though one was clearly labeled leaded and other unleaded, human beings are bad at catching those things, especially when they’re in a hurry and under stress,” says Dr. Cucina, whose research includes clinical human-computer interaction science with an emphasis on human factors and patient safety.
A similar concept is what is missing from the Swan Ganz catheter design. The three ports (proximal, middle, and distal) connecting the catheter to the ICU monitor all have the same shape, making it easy to errantly connect one or more to the wrong port. “You’d think the manufacturers would shape the connectors in a way that would preclude incorrect connections,” Dr. Cucina says, “but that has not been done. We leave it to the vigilance of the bedside nurse or intensivist or hospitalist to hook these up correctly, rather than redesigning them so that cannot be done incorrectly.”
One way to think about human factors engineering is to think about forcing “a round peg into a square hole.” In the hospital setting, round pegs into square holes equate to errors. HF tries to solve the issue (round peg into a round hole, and vice versa). “Were you to apply human factors to the Swan Ganz catheter port connectors,” Dr. Cucina says, “you’d have round into the round hole, square into the square, and triangular into the triangular. You’d have no choice but to do the right thing.”
Efforts to implement systems that anticipate and minimize the chances of human error, such as computer physician order entry and patient bar coding, are attempts to overcome by design those instances where it is possible to place round pegs into square holes.
HF Projects in Motion
A number of hospitalists around the country have or are using HF as part of projects and studies to reduce human errors.
Culture change: In the early 2000s, Janet Nagamine, MD, a hospitalist with Kaiser Permanente in Santa Clara, Calif., and her colleagues took human factors concepts to front-line ICU staff. The human factors training provided a framework to reinforce three basic concepts: all humans make errors; processes can be designed to reduce the possibility of error; and processes can be designed so errors are detected and corrected before causing injury.4 “My colleagues and I knew that the punitive, ‘shame-and-blame’ culture around mistakes and errors were preventing us from identifying problems and moving forward with solutions,” Dr. Nagamine says.
A former ICU nurse and current chair of SHM’s Hospital Quality and Patient Safety (HQPS) Committee, Dr. Nagamine first became involved in HF when she realized how many patients suffered adverse events stemming from poorly designed medical systems. “Some of my most respected mentors were involved in these kinds of cases, and I knew eventually that would be me,” she says. It was a disturbing reality. During her medical training it was drilled into her head smart, diligent doctors would be successful. “But bad things happen in medicine; it’s part of what we do,” she says. “Rather than deny that things will inevitably go wrong, I wanted to study safety science and reliable system design.” She asked herself, how can we prevent the same mistakes from happening to competent people who practice in poorly designed systems? “The patterns are there,” she says. “You can train your eyes to look for vulnerabilities and patterns, then find the solutions.”
After she started looking at adverse events as system failures, rather than solely personal failures, she engaged the staff to redesign systems. She introduced HF concepts and provided an infrastructure to make it safe to report and discuss problems. The project included a new medication error reporting system and the creation of departmental patient safety teams. A palpable culture change developed when front-line staff and managers became empowered to find solutions working side-by-side with the quality and risk management departments.
The result? A dramatic increase in medication errors and near-miss reports: from eight faulty problems per quarter in 2000 to 200 reports per quarter by 2001.
To sum up the essence of Dr. Nagamine’s project, she invokes her favorite quotation from systems expert James Reason: “We can’t change the human condition, but we can change the conditions under which humans work.”1,5
Bar coding workarounds: Hospitalist Tosha Wetterneck, MD, and her colleagues at the University of Wisconsin School of Medicine and Dentistry focused their HF-trained eyes on medication errors.5 The team applied HF concepts as part of a study of bar-coded medication administration systems (BCMAs). Ideally, BCMAs help confirm the five rights of medication administration: the right patient, drug, dose, route, and time. The study authors identified the hospitalist staff had developed 46 workarounds in place of proper use of the BCMA. With each workaround, the researchers identified six potential errors. Furthermore, nurses were overriding the BCMA alerts for 4.2% of patients charted, and for 10.3% of total medication.
By creating an exhaustive template, the study authors broke down the use of BCMA workarounds to the finest detail of task component. They learned many workarounds were engendered by difficulties with the technology and by interactions between BCMA technologies and environmental, technical, process, workload, training, and policy concerns. Data shows BCMAs still have an important role in preventing error; during one year, almost 24,000 BCMA alerts led users to change their action, instead of overriding an alert. “These causes (and related workarounds) are neither rare nor secret,” the authors write. “They are hiding in plain sight.”1,5
Dr. Wetterneck is part of the Systems Engineering Initiative for Patient Safety (SEIPS), an interdisciplinary research group located within the Center for Quality and Productivity Improvement in the College of Engineering at the University of Wisconsin-Madison.6,7 SEIPS uses HF principles to study the safety and quality of healthcare systems.
Congestive heart failure order sets: Researchers in another study incorporated HF science in their review of clinical practice guideline use and application for congestive heart failure (CHF). Reingold and Kulstad studied the impact of HF design elements on order set utilization and recommendations compliance.8
Using retrospective medical record review of adult patients admitted from the emergency department with CHF, the study measured acuity and clinical practice guideline (CPG) parameters before and after introducing new orders. In 87 adult patients before and 84 patients after beginning the new order set, attention to HF design elements significantly improved utilization of the orders and CPG compliance.
Infusion device programming: In another instance, a multidisciplinary research team applied HF design principles to common nursing procedures: programming an insulin infusion and programming a heparin infusion.9,10 An HF usability checklist was developed, and it revealed systematic error-provoking conditions in both tasks.
The good news is the pitfalls were remedied easily.
Not only did researchers subsequently commit to modify training procedures and redesign preprinted orders, they took the bigger step of providing feedback to the manufacturer and committing to incorporate usability testing in future procurement of medical devices. TH
Andrea M. Sattinger is a medical writer based in North Carolina and a frequent contributor to The Hospitalist.
References
1. Gosbee JW. Conclusion: you need human factors engineering expertise to see design hazards that are hiding in “plain sight!” Jt Comm J Qual Saf. 2004;30(12):696-700.
2. Gosbee J. Introduction to the human factors engineering series. Jt Comm J Qual Saf. 2004;30(4): 215-219.
3. Reason J. Human error: models and management. BMJ. 2000; 320(7237):768-770.
4. Etchells E, Juurlink D, Levinson W. Medication errors: the human factor. CMAJ. 2008;178(1):63-64.
5. Koppel R, Wetterneck T, Telles JL, Karsh BT. Workarounds to barcode medication administration systems: their occurrences, causes, and threats to patient safety. J Am Med Inform Assoc. 2008;15(4):408-423.
6. SEIPS model. http://cqpi.engr.wisc.edu/seips_ home/. Accessed Dec. 20, 2008.
7. Carayon P, Schoofs Hundt A, Karsh BT, et al. Work system design for patient safety: the SEIPS model. Qual Saf Health Care. 2006;15 Suppl 1:850-858.
8. Reingold S, Kulstad E. Impact of human factor design on the use of order sets in the treatment of congestive heart failure. Acad Emerg Med. 2007;14(11):1097-1105.
9. Etchells E, Bailey C, Biason R, et al. Human factors in action: getting “pumped” at a nursing usability laboratory. Healthc Q. 2006;9 Spec No:69-74.
10. Carayon P, Wetterneck T, Schoofs Hundt A, et al. Observing nurse interaction with infusion pump technologies. In: Henriksen K, Battles J, Lewin D, eds. Advances in Patient Safety: From Research to Implementation. Rockville, Md.: Agency for Healthcare Research and Quality; Feb. 2005, AHRQ Publication No. 05-0021-2.
When Amsterdam Airport Schiphol in the Netherlands revamped its men’s restrooms, the architects installed small, Euro-style urinals: a surefire way to throw urine off target. To solve this problem, the black outline of a fly was etched in the porcelain near each urinal’s drain. Users’ aim improved and spillage was reduced by 80%. “They try to power blast it away,” says Sanjay Saint, MD, hospitalist and professor of internal medicine at the Ann Arbor VA Medical Center, University of Michigan. “By the time they might realize that the fly isn’t going anywhere, the men are done and walking away.”
It’s a guy thing, sure. It also is an example of a human factors intervention. “Science teaches us that implementing a design for a machine or device that elicits an instinctive reaction from someone using it is a clear-cut way to avoid error,” Dr. Saint explains.
What It Is and Why It’s Important
Human factors (HF), or human factors engineering (HFE), also sometimes called usability engineering or systems-based practice, refers to the study of human abilities and characteristics as they affect the design and smooth operation of equipment, systems, and jobs.1 HF is the basic science underlying much of patient safety practice. For instance, the current recommendation that hospitals standardize equipment, such as ventilators, programmable IV pumps, and defibrillators, is an example of making tasks human friendly. The use of cognitive psychology and biomechanics to develop and improve software and hand tools are another example of HF principles.
In general, HF examines the component tasks of an activity in terms of three factorial domains: physical and environmental factors, cognitive factors (skill demands and mental workload), and organizational factors. Each task is assessed in terms of necessary interactions of the individual and work environment, the device/system design, and associated team dynamics.
HF use in healthcare is not new; for roughly four decades HF researchers have emphasized the key role of HF in safe medical design, healthcare facility operations, and patient safety processes. HF helps organizations deepen analyses of adverse events and develop effective solutions.2 HF is used in the design of labeling, warnings or alarms, software programs, information displays, paper forms, process and activity flow, workplace design, cognitive aids, decision support systems, and policies and protocols.
For hospitalists, human factors knowledge is most useful in process improvement, says John Gosbee, MD, MS, a human factors engineering and healthcare specialist at the University of Michigan. Dr. Gosbee, who has worked with hospitalists in Ann Arbor and around the country, originally studied aerospace medicine, pursued a subspecialty in occupational medicine, and from 1988 to 1992 worked at NASA designing space hospitals. In the dozens of lectures and workshops he has conducted, he has learned numerous physicians resist learning about HF. At first they protest, claiming they “didn’t go to medical school to become engineers” or “weren’t hired to have you tell us we need to be some kind of designer or computer-science software evaluator.”
Dr. Gosbee couldn’t agree more, but after the a-ha! moment, usually in an interactive experience when the hospitalist sees a poor system design is an obstacle to safety and process flow, they open up to adopting the HF mindset. Once on board with HF, hospitalists are quick to translate the theories to their own practices, identifying potential vulnerabilities and risks.
Manufacturers of healthcare equipment and systems don’t want to hear from “safety geeks,” Dr. Gosbee says; the companies want to hear from front-line providers who regularly use the products. “Hospitalists are in great position to provide that input because they see what happens across a broad swath of hospital settings,” he says, “and they could amalgamate the fact that everyone across specialties is having some trouble with this computer screen or new infusion device.”
Dr. Gosbee’s first-hand knowledge and experience solving hospitalist issues with HF techniques evolved into a teaching career. He says the university administration supports his belief in the practicality of HF lessons, and he now works as the lead instructor for a majority of the university’s medical residents.
“Human factors engineering is an efficient way to flip people’s brains around 180 degrees toward systems thinking,” Dr. Gosbee explains, “which is required if the organization wants to become a high-reliability organization.”3
Examples in Medicine
Russ Cucina, MD, MS, hospitalist at the University of California San Francisco Medical Center, describes a practical example of human factors engineering in a simple, widely used design. When cars ran on leaded gasoline, the design of the leaded gas pump nozzle precluded it from being inserted into an unleaded gas tank. “Even though one was clearly labeled leaded and other unleaded, human beings are bad at catching those things, especially when they’re in a hurry and under stress,” says Dr. Cucina, whose research includes clinical human-computer interaction science with an emphasis on human factors and patient safety.
A similar concept is what is missing from the Swan Ganz catheter design. The three ports (proximal, middle, and distal) connecting the catheter to the ICU monitor all have the same shape, making it easy to errantly connect one or more to the wrong port. “You’d think the manufacturers would shape the connectors in a way that would preclude incorrect connections,” Dr. Cucina says, “but that has not been done. We leave it to the vigilance of the bedside nurse or intensivist or hospitalist to hook these up correctly, rather than redesigning them so that cannot be done incorrectly.”
One way to think about human factors engineering is to think about forcing “a round peg into a square hole.” In the hospital setting, round pegs into square holes equate to errors. HF tries to solve the issue (round peg into a round hole, and vice versa). “Were you to apply human factors to the Swan Ganz catheter port connectors,” Dr. Cucina says, “you’d have round into the round hole, square into the square, and triangular into the triangular. You’d have no choice but to do the right thing.”
Efforts to implement systems that anticipate and minimize the chances of human error, such as computer physician order entry and patient bar coding, are attempts to overcome by design those instances where it is possible to place round pegs into square holes.
HF Projects in Motion
A number of hospitalists around the country have or are using HF as part of projects and studies to reduce human errors.
Culture change: In the early 2000s, Janet Nagamine, MD, a hospitalist with Kaiser Permanente in Santa Clara, Calif., and her colleagues took human factors concepts to front-line ICU staff. The human factors training provided a framework to reinforce three basic concepts: all humans make errors; processes can be designed to reduce the possibility of error; and processes can be designed so errors are detected and corrected before causing injury.4 “My colleagues and I knew that the punitive, ‘shame-and-blame’ culture around mistakes and errors were preventing us from identifying problems and moving forward with solutions,” Dr. Nagamine says.
A former ICU nurse and current chair of SHM’s Hospital Quality and Patient Safety (HQPS) Committee, Dr. Nagamine first became involved in HF when she realized how many patients suffered adverse events stemming from poorly designed medical systems. “Some of my most respected mentors were involved in these kinds of cases, and I knew eventually that would be me,” she says. It was a disturbing reality. During her medical training it was drilled into her head smart, diligent doctors would be successful. “But bad things happen in medicine; it’s part of what we do,” she says. “Rather than deny that things will inevitably go wrong, I wanted to study safety science and reliable system design.” She asked herself, how can we prevent the same mistakes from happening to competent people who practice in poorly designed systems? “The patterns are there,” she says. “You can train your eyes to look for vulnerabilities and patterns, then find the solutions.”
After she started looking at adverse events as system failures, rather than solely personal failures, she engaged the staff to redesign systems. She introduced HF concepts and provided an infrastructure to make it safe to report and discuss problems. The project included a new medication error reporting system and the creation of departmental patient safety teams. A palpable culture change developed when front-line staff and managers became empowered to find solutions working side-by-side with the quality and risk management departments.
The result? A dramatic increase in medication errors and near-miss reports: from eight faulty problems per quarter in 2000 to 200 reports per quarter by 2001.
To sum up the essence of Dr. Nagamine’s project, she invokes her favorite quotation from systems expert James Reason: “We can’t change the human condition, but we can change the conditions under which humans work.”1,5
Bar coding workarounds: Hospitalist Tosha Wetterneck, MD, and her colleagues at the University of Wisconsin School of Medicine and Dentistry focused their HF-trained eyes on medication errors.5 The team applied HF concepts as part of a study of bar-coded medication administration systems (BCMAs). Ideally, BCMAs help confirm the five rights of medication administration: the right patient, drug, dose, route, and time. The study authors identified the hospitalist staff had developed 46 workarounds in place of proper use of the BCMA. With each workaround, the researchers identified six potential errors. Furthermore, nurses were overriding the BCMA alerts for 4.2% of patients charted, and for 10.3% of total medication.
By creating an exhaustive template, the study authors broke down the use of BCMA workarounds to the finest detail of task component. They learned many workarounds were engendered by difficulties with the technology and by interactions between BCMA technologies and environmental, technical, process, workload, training, and policy concerns. Data shows BCMAs still have an important role in preventing error; during one year, almost 24,000 BCMA alerts led users to change their action, instead of overriding an alert. “These causes (and related workarounds) are neither rare nor secret,” the authors write. “They are hiding in plain sight.”1,5
Dr. Wetterneck is part of the Systems Engineering Initiative for Patient Safety (SEIPS), an interdisciplinary research group located within the Center for Quality and Productivity Improvement in the College of Engineering at the University of Wisconsin-Madison.6,7 SEIPS uses HF principles to study the safety and quality of healthcare systems.
Congestive heart failure order sets: Researchers in another study incorporated HF science in their review of clinical practice guideline use and application for congestive heart failure (CHF). Reingold and Kulstad studied the impact of HF design elements on order set utilization and recommendations compliance.8
Using retrospective medical record review of adult patients admitted from the emergency department with CHF, the study measured acuity and clinical practice guideline (CPG) parameters before and after introducing new orders. In 87 adult patients before and 84 patients after beginning the new order set, attention to HF design elements significantly improved utilization of the orders and CPG compliance.
Infusion device programming: In another instance, a multidisciplinary research team applied HF design principles to common nursing procedures: programming an insulin infusion and programming a heparin infusion.9,10 An HF usability checklist was developed, and it revealed systematic error-provoking conditions in both tasks.
The good news is the pitfalls were remedied easily.
Not only did researchers subsequently commit to modify training procedures and redesign preprinted orders, they took the bigger step of providing feedback to the manufacturer and committing to incorporate usability testing in future procurement of medical devices. TH
Andrea M. Sattinger is a medical writer based in North Carolina and a frequent contributor to The Hospitalist.
References
1. Gosbee JW. Conclusion: you need human factors engineering expertise to see design hazards that are hiding in “plain sight!” Jt Comm J Qual Saf. 2004;30(12):696-700.
2. Gosbee J. Introduction to the human factors engineering series. Jt Comm J Qual Saf. 2004;30(4): 215-219.
3. Reason J. Human error: models and management. BMJ. 2000; 320(7237):768-770.
4. Etchells E, Juurlink D, Levinson W. Medication errors: the human factor. CMAJ. 2008;178(1):63-64.
5. Koppel R, Wetterneck T, Telles JL, Karsh BT. Workarounds to barcode medication administration systems: their occurrences, causes, and threats to patient safety. J Am Med Inform Assoc. 2008;15(4):408-423.
6. SEIPS model. http://cqpi.engr.wisc.edu/seips_ home/. Accessed Dec. 20, 2008.
7. Carayon P, Schoofs Hundt A, Karsh BT, et al. Work system design for patient safety: the SEIPS model. Qual Saf Health Care. 2006;15 Suppl 1:850-858.
8. Reingold S, Kulstad E. Impact of human factor design on the use of order sets in the treatment of congestive heart failure. Acad Emerg Med. 2007;14(11):1097-1105.
9. Etchells E, Bailey C, Biason R, et al. Human factors in action: getting “pumped” at a nursing usability laboratory. Healthc Q. 2006;9 Spec No:69-74.
10. Carayon P, Wetterneck T, Schoofs Hundt A, et al. Observing nurse interaction with infusion pump technologies. In: Henriksen K, Battles J, Lewin D, eds. Advances in Patient Safety: From Research to Implementation. Rockville, Md.: Agency for Healthcare Research and Quality; Feb. 2005, AHRQ Publication No. 05-0021-2.
When Amsterdam Airport Schiphol in the Netherlands revamped its men’s restrooms, the architects installed small, Euro-style urinals: a surefire way to throw urine off target. To solve this problem, the black outline of a fly was etched in the porcelain near each urinal’s drain. Users’ aim improved and spillage was reduced by 80%. “They try to power blast it away,” says Sanjay Saint, MD, hospitalist and professor of internal medicine at the Ann Arbor VA Medical Center, University of Michigan. “By the time they might realize that the fly isn’t going anywhere, the men are done and walking away.”
It’s a guy thing, sure. It also is an example of a human factors intervention. “Science teaches us that implementing a design for a machine or device that elicits an instinctive reaction from someone using it is a clear-cut way to avoid error,” Dr. Saint explains.
What It Is and Why It’s Important
Human factors (HF), or human factors engineering (HFE), also sometimes called usability engineering or systems-based practice, refers to the study of human abilities and characteristics as they affect the design and smooth operation of equipment, systems, and jobs.1 HF is the basic science underlying much of patient safety practice. For instance, the current recommendation that hospitals standardize equipment, such as ventilators, programmable IV pumps, and defibrillators, is an example of making tasks human friendly. The use of cognitive psychology and biomechanics to develop and improve software and hand tools are another example of HF principles.
In general, HF examines the component tasks of an activity in terms of three factorial domains: physical and environmental factors, cognitive factors (skill demands and mental workload), and organizational factors. Each task is assessed in terms of necessary interactions of the individual and work environment, the device/system design, and associated team dynamics.
HF use in healthcare is not new; for roughly four decades HF researchers have emphasized the key role of HF in safe medical design, healthcare facility operations, and patient safety processes. HF helps organizations deepen analyses of adverse events and develop effective solutions.2 HF is used in the design of labeling, warnings or alarms, software programs, information displays, paper forms, process and activity flow, workplace design, cognitive aids, decision support systems, and policies and protocols.
For hospitalists, human factors knowledge is most useful in process improvement, says John Gosbee, MD, MS, a human factors engineering and healthcare specialist at the University of Michigan. Dr. Gosbee, who has worked with hospitalists in Ann Arbor and around the country, originally studied aerospace medicine, pursued a subspecialty in occupational medicine, and from 1988 to 1992 worked at NASA designing space hospitals. In the dozens of lectures and workshops he has conducted, he has learned numerous physicians resist learning about HF. At first they protest, claiming they “didn’t go to medical school to become engineers” or “weren’t hired to have you tell us we need to be some kind of designer or computer-science software evaluator.”
Dr. Gosbee couldn’t agree more, but after the a-ha! moment, usually in an interactive experience when the hospitalist sees a poor system design is an obstacle to safety and process flow, they open up to adopting the HF mindset. Once on board with HF, hospitalists are quick to translate the theories to their own practices, identifying potential vulnerabilities and risks.
Manufacturers of healthcare equipment and systems don’t want to hear from “safety geeks,” Dr. Gosbee says; the companies want to hear from front-line providers who regularly use the products. “Hospitalists are in great position to provide that input because they see what happens across a broad swath of hospital settings,” he says, “and they could amalgamate the fact that everyone across specialties is having some trouble with this computer screen or new infusion device.”
Dr. Gosbee’s first-hand knowledge and experience solving hospitalist issues with HF techniques evolved into a teaching career. He says the university administration supports his belief in the practicality of HF lessons, and he now works as the lead instructor for a majority of the university’s medical residents.
“Human factors engineering is an efficient way to flip people’s brains around 180 degrees toward systems thinking,” Dr. Gosbee explains, “which is required if the organization wants to become a high-reliability organization.”3
Examples in Medicine
Russ Cucina, MD, MS, hospitalist at the University of California San Francisco Medical Center, describes a practical example of human factors engineering in a simple, widely used design. When cars ran on leaded gasoline, the design of the leaded gas pump nozzle precluded it from being inserted into an unleaded gas tank. “Even though one was clearly labeled leaded and other unleaded, human beings are bad at catching those things, especially when they’re in a hurry and under stress,” says Dr. Cucina, whose research includes clinical human-computer interaction science with an emphasis on human factors and patient safety.
A similar concept is what is missing from the Swan Ganz catheter design. The three ports (proximal, middle, and distal) connecting the catheter to the ICU monitor all have the same shape, making it easy to errantly connect one or more to the wrong port. “You’d think the manufacturers would shape the connectors in a way that would preclude incorrect connections,” Dr. Cucina says, “but that has not been done. We leave it to the vigilance of the bedside nurse or intensivist or hospitalist to hook these up correctly, rather than redesigning them so that cannot be done incorrectly.”
One way to think about human factors engineering is to think about forcing “a round peg into a square hole.” In the hospital setting, round pegs into square holes equate to errors. HF tries to solve the issue (round peg into a round hole, and vice versa). “Were you to apply human factors to the Swan Ganz catheter port connectors,” Dr. Cucina says, “you’d have round into the round hole, square into the square, and triangular into the triangular. You’d have no choice but to do the right thing.”
Efforts to implement systems that anticipate and minimize the chances of human error, such as computer physician order entry and patient bar coding, are attempts to overcome by design those instances where it is possible to place round pegs into square holes.
HF Projects in Motion
A number of hospitalists around the country have or are using HF as part of projects and studies to reduce human errors.
Culture change: In the early 2000s, Janet Nagamine, MD, a hospitalist with Kaiser Permanente in Santa Clara, Calif., and her colleagues took human factors concepts to front-line ICU staff. The human factors training provided a framework to reinforce three basic concepts: all humans make errors; processes can be designed to reduce the possibility of error; and processes can be designed so errors are detected and corrected before causing injury.4 “My colleagues and I knew that the punitive, ‘shame-and-blame’ culture around mistakes and errors were preventing us from identifying problems and moving forward with solutions,” Dr. Nagamine says.
A former ICU nurse and current chair of SHM’s Hospital Quality and Patient Safety (HQPS) Committee, Dr. Nagamine first became involved in HF when she realized how many patients suffered adverse events stemming from poorly designed medical systems. “Some of my most respected mentors were involved in these kinds of cases, and I knew eventually that would be me,” she says. It was a disturbing reality. During her medical training it was drilled into her head smart, diligent doctors would be successful. “But bad things happen in medicine; it’s part of what we do,” she says. “Rather than deny that things will inevitably go wrong, I wanted to study safety science and reliable system design.” She asked herself, how can we prevent the same mistakes from happening to competent people who practice in poorly designed systems? “The patterns are there,” she says. “You can train your eyes to look for vulnerabilities and patterns, then find the solutions.”
After she started looking at adverse events as system failures, rather than solely personal failures, she engaged the staff to redesign systems. She introduced HF concepts and provided an infrastructure to make it safe to report and discuss problems. The project included a new medication error reporting system and the creation of departmental patient safety teams. A palpable culture change developed when front-line staff and managers became empowered to find solutions working side-by-side with the quality and risk management departments.
The result? A dramatic increase in medication errors and near-miss reports: from eight faulty problems per quarter in 2000 to 200 reports per quarter by 2001.
To sum up the essence of Dr. Nagamine’s project, she invokes her favorite quotation from systems expert James Reason: “We can’t change the human condition, but we can change the conditions under which humans work.”1,5
Bar coding workarounds: Hospitalist Tosha Wetterneck, MD, and her colleagues at the University of Wisconsin School of Medicine and Dentistry focused their HF-trained eyes on medication errors.5 The team applied HF concepts as part of a study of bar-coded medication administration systems (BCMAs). Ideally, BCMAs help confirm the five rights of medication administration: the right patient, drug, dose, route, and time. The study authors identified the hospitalist staff had developed 46 workarounds in place of proper use of the BCMA. With each workaround, the researchers identified six potential errors. Furthermore, nurses were overriding the BCMA alerts for 4.2% of patients charted, and for 10.3% of total medication.
By creating an exhaustive template, the study authors broke down the use of BCMA workarounds to the finest detail of task component. They learned many workarounds were engendered by difficulties with the technology and by interactions between BCMA technologies and environmental, technical, process, workload, training, and policy concerns. Data shows BCMAs still have an important role in preventing error; during one year, almost 24,000 BCMA alerts led users to change their action, instead of overriding an alert. “These causes (and related workarounds) are neither rare nor secret,” the authors write. “They are hiding in plain sight.”1,5
Dr. Wetterneck is part of the Systems Engineering Initiative for Patient Safety (SEIPS), an interdisciplinary research group located within the Center for Quality and Productivity Improvement in the College of Engineering at the University of Wisconsin-Madison.6,7 SEIPS uses HF principles to study the safety and quality of healthcare systems.
Congestive heart failure order sets: Researchers in another study incorporated HF science in their review of clinical practice guideline use and application for congestive heart failure (CHF). Reingold and Kulstad studied the impact of HF design elements on order set utilization and recommendations compliance.8
Using retrospective medical record review of adult patients admitted from the emergency department with CHF, the study measured acuity and clinical practice guideline (CPG) parameters before and after introducing new orders. In 87 adult patients before and 84 patients after beginning the new order set, attention to HF design elements significantly improved utilization of the orders and CPG compliance.
Infusion device programming: In another instance, a multidisciplinary research team applied HF design principles to common nursing procedures: programming an insulin infusion and programming a heparin infusion.9,10 An HF usability checklist was developed, and it revealed systematic error-provoking conditions in both tasks.
The good news is the pitfalls were remedied easily.
Not only did researchers subsequently commit to modify training procedures and redesign preprinted orders, they took the bigger step of providing feedback to the manufacturer and committing to incorporate usability testing in future procurement of medical devices. TH
Andrea M. Sattinger is a medical writer based in North Carolina and a frequent contributor to The Hospitalist.
References
1. Gosbee JW. Conclusion: you need human factors engineering expertise to see design hazards that are hiding in “plain sight!” Jt Comm J Qual Saf. 2004;30(12):696-700.
2. Gosbee J. Introduction to the human factors engineering series. Jt Comm J Qual Saf. 2004;30(4): 215-219.
3. Reason J. Human error: models and management. BMJ. 2000; 320(7237):768-770.
4. Etchells E, Juurlink D, Levinson W. Medication errors: the human factor. CMAJ. 2008;178(1):63-64.
5. Koppel R, Wetterneck T, Telles JL, Karsh BT. Workarounds to barcode medication administration systems: their occurrences, causes, and threats to patient safety. J Am Med Inform Assoc. 2008;15(4):408-423.
6. SEIPS model. http://cqpi.engr.wisc.edu/seips_ home/. Accessed Dec. 20, 2008.
7. Carayon P, Schoofs Hundt A, Karsh BT, et al. Work system design for patient safety: the SEIPS model. Qual Saf Health Care. 2006;15 Suppl 1:850-858.
8. Reingold S, Kulstad E. Impact of human factor design on the use of order sets in the treatment of congestive heart failure. Acad Emerg Med. 2007;14(11):1097-1105.
9. Etchells E, Bailey C, Biason R, et al. Human factors in action: getting “pumped” at a nursing usability laboratory. Healthc Q. 2006;9 Spec No:69-74.
10. Carayon P, Wetterneck T, Schoofs Hundt A, et al. Observing nurse interaction with infusion pump technologies. In: Henriksen K, Battles J, Lewin D, eds. Advances in Patient Safety: From Research to Implementation. Rockville, Md.: Agency for Healthcare Research and Quality; Feb. 2005, AHRQ Publication No. 05-0021-2.
Resident Restrictions
Effective July 1, the Accreditation Council for Graduate Medical Education (ACGME) is adopting rules changes to further restrict the number of patients internal medicine residents follow. The impact of this change may reach beyond academic institutions and teaching services. Non-teaching services and institutions may see some fallout, as hospital administration shuffles caseloads of residents and hospitalist attendings. The potential results likely will impact resident training, hospitalist training, and hospitalist practice management, namely recruitment and hospitalist job satisfaction.
Why the Change?
With the 2003 restrictions on resident work-hours duty and now the capping of patient caseloads, the ACGME is attempting to ensure residency programs are not viewed as a source of cheap labor and excessive stress. Also, “the Residency Review Committee (RRC) is cognizant too much service can be a barrier to education,” says Lenny Feldman, MD, a hospitalist and associate program director at Johns Hopkins Medical Center in Baltimore. But there is a danger in the reverse: too little service may undersupply residents with the depth and breadth of cases they need under their belts to competently enter practice. “Education should be the foremost mission for residency programs, but trying to find that exact balance between service and education is tough,” Dr. Feldman says.
In a Nutshell
As leader of the 70-hospitalist Health Partners Medical Group in Minneapolis-St. Paul, a University of Minnesota affiliate working with internal medicine residents, Burke T. Kealey, MD, views the ACGME rule change on a professional and personal level. In the big picture, Dr. Kealey observes three main effects:
- Hospitalists will be seeing more patients and probably more patients at night;
- The cost of hospital care will increase for hospitals and hospital medicine groups (HMGs); and
- The experience level of new graduates applying to be hospitalists will diminish.
In essence, there are few ways to handle the looming cap on residents’ patient caseloads. (see Practical Approaches, p. 24) Given the financial constraints imposed by this new, unfunded mandate, and taking into account the fact most residency programs depend on federal funding, it generally is believed increasing the number of residents cannot be considered an option. “Given the looming physician shortage, there is pressure on the federal government to increase the amount of GME support and the number of residency spots,” Dr. Feldman says. “Medical schools have increased enrollment pretty significantly, but the bottleneck is the number of GME-supported residency positions.”
HM Crossroads
Leslie Flores, MHA, principal with Nelson Flores Hospital Medicine Consultants, and the director of SHM’s Practice Management Institute, believes the new rule dramatically will impact teaching hospitals and HMGs. “I think it is likely to be harder for academic hospitalists, who are working on teaching services, to generate reasonable productivity, which will place an even greater financial burden on academic practices,” she says. “But the larger effect will be that non-teaching services in teaching hospitals will be expected to pick up the slack and, subsequently, grow in order to accommodate the patient numbers.”
Asking staff physicians to increase their patient load, even incrementally, is a poor solution, at best, Dr. Kealey says. And it may be tough for some places to recruit more hospitalists, a function of the hospitalist labor shortage.
William Rifkin, MD, a hospitalist and associate director of clinical medicine at Albert Einstein College of Medicine, and director of the residency program at Jacobi Medical Center, Bronx, N.Y., estimates hospitalist jobs in teaching institutions will increasingly morph into non-teaching positions. “Where currently the ratio of teaching to nonteaching jobs is 50-50,” Dr. Rifkin says, “by 2009, 80% of internal medicine training programs will have to build or expand a new, non-teaching service, and more than half of hospitalist duties will be non-teaching.”
A recent recommendation from the Institute of Mecidine (IOM) reinforces the national movement to restructure resident work hours and duties. Released Dec. 2, 2008, the “Resident Duty Hours: Enhancing Sleep, Supervision, and Safety” report calls for a maximum shift length of 30 hours with admission of patients for up to 16 hours, plus a five-hour, uninterrupted sleep period between 10 p.m. and 8 a.m., with the remaining hours for transitional and educational activity.
The consensus is the ACGME rules changes likely will alter the hospitalist job description and produce an even greater shortage of qualified, experienced physicians. Leora Horwitz, MD, MHS, an assistant professor in internal medicine at Yale University School of Medicine in New Haven, Conn., says “hospitalists are really an amalgamation of two very distinct types: the short-term hospitalist who takes the job for a year or two right after residency and before fellowship, and the longer-term hospitalist who takes on the job as at least an intermediate-term career. It could be that recruitment and retention differ for these types.”
Dr. Rifkin isn’t alone when he asks, “Can a hospitalist last that long doing patient care alone? There are only so many people who will move up to be leaders in HMGs. So while this will probably be good for recruitment in the short term, in the long term, we don’t know.”
Immediate Consequences
Some ramifications of hospital medicine as a whole taking on more patients and more hospitalists will parallel the growing pains of individual HMGs. For instance, hospitalist group’s social bonds may not be as tight, says Dr. Feldman. But where many obstacles are surmountable, “what is not surmountable is if hospitals don’t choose to increase the size of their hospitalist programs. The deathblow to most hospitalist programs is if you ask the group, and each individual, to do more work that is not commensurate with the original expectations. And with the market already tight, most hospitals can’t afford to have unhappy hospitalists.”
Financially, the new rules will place a heavy burden on HMGs and hospital administrators. With no additional reimbursement under the GME system, most hospitals will have to get creative with existing budgets. “Part of the concern is that patients that hospitalists see on a teaching service tend to be the lower socioeconomic population of patients―Medicaid and self-pay patients―where there is inadequate reimbursement anyway,” Flores says. The answer likely will be sending those patients to a non-teaching service, which in essence transfers the financial burden. “Hospitals will have to find money from somewhere.”
Teaching hospitals not part of large academic medical centers contribute to hospitalists’ compensation when they help train family medicine and internal medicine residents. “Because they are not technically academic hospitalists,” Flores says, “they need to be alerted about how these rule changes may influence the way they manage and run the finances of their practice.”
Some of the solutions to the problems inherent in this change depend on the practice and scheduling model. In the aftermath of the work-hour restriction, many hospitalist programs changed their scheduling method to day float/night float, or the “drip” method of admission (taking admissions every day), versus the “bolus” method (every fourth or fifth day), Dr. Feldman says. The bolus method likely leads to scenarios where the new ACGME cap will come into play.
There is the possibility the rule change could turn out to be a boon to HMGs, Dr. Feldman says. Programs without hospitalists may hire them; small groups may expand, increasing job opportunities. Additionally, teaching opportunities for hospitalist attendings may improve with the decreased number of patients on a service residents follow. “Hopefully, this will increase opportunities for teaching residents and increase the satisfaction of those involved in teaching,” he says. “Ultimately, it may result in improved resident education while creating more job opportunities for hospitalists―a win-win for both groups.”
Will Training Suffer?
Dr. Kealey has concerns about the long-term effects on the training residents who become hospitalists. “First, they won’t get enough experience to be competent hospitalists on graduation. Second, the number of patients is being capped, but the number of ACGME-required outpatient clinic sessions is rising, increasing from about 108 to 130 over a 30-month period,” he says. “Residency programs will to have to figure out how to fit these sessions into training, and that may squeeze out inpatient time.”
Third, with the work hours and caseload restrictions on residents, educators are concerned residents will not receive an adequate level of training.
Kenneth P. Patrick, MD, director of the hospitalist program at Chestnut Hill Hospital in Philadelphia, is worried, too, especially when it comes to the educational implications. As a former residency program director, one who shares concerns about residents’ large workloads, Dr. Patrick believes strongly in medical education and is wary of the path it seems to be taking. “What a hospital medicine group can provide to residents is the opportunity to learn from a smaller patient load,” Dr. Patrick says, “and regulatory agencies should carefully address that. Cutting back on the number of service hours and patients can have both a positive and negative effect. Most people are only adjusting the numbers of hours and patients, and not viewing the whole picture.”
Another likely result of the rules change is the mindset residents could be developing, an issue that rings true with most HMG directors. “I worry that our residents will be sheltered during training and will emerge into a real world where there won’t be caps,” Dr. Kealey says. “They will be in systems where people have to cooperate with each other in order to handle patient surges and large patient volumes. Though they may graduate, join a group, and become acculturated, it concerns me that their initial primary training, rather than encouraging them to think as part of a system, may be training them to think of ‘my restrictions, my needs, my limitation.’ ”
Prepare for Change
What is the answer? Two hospitalists echoed the same, simple solutions: “Give us more money” and “We need more bodies.”
Simplicity aside, residency and hospital medicine programs will need to prepare for the change. “Instead of happening gradually, suddenly every [residency] program in the country will lose 20% of its capacity,” Dr. Rikfin says.
Michael Pistoria, DO, FACP, associate general division of internal medicine chief at Lehigh Valley Hospital in Allentown, Pa., believes institutions with closely aligned hospitalist and residency programs will benefit from “enlightenment on both sides. Residency programs are increasingly alert to the vital role that HMGs play in supporting residency programs,” he says. “They are more aware of the impact these types of decisions have on the staffing of HMGs.”
Mid-level providers are one possible solution. “Programs will increasingly look to supplement their existing group with advanced practice clinicians—physician assistants and non-physician providers―a less-expensive alternative,” Dr. Pistoria says
Does hiring mid-level practitioners pose a risk for unintended adverse events and delays to diagnosis? “There may be an extended growth curve for these providers,” Dr. Pistoria says, “due to less clinical exposure and experience than a new physician hospitalist just out of residency.”
However, these advanced practice clinicians often are quick to adapt to the hospitalist setting, learning the skills required to be an effective hospitalist through on-the-job training. “On-the-job training for physician hospitalists can focus on education, quality improvement, safety―some of the value-added pieces,” Dr. Pistoria points out.
Without a doubt, ACGME’s new cap on residency caseloads will impact hospital medicine, both at the national level and the individual group level. HMG efforts to recruit, schedule, train and pay hospitalists will be affected, as will the level of experience patients receive from recent residency graduates.
“It is incumbent on us to get involved in committees and process and performance improvement projects,” Dr. Pistoria says, “so that when leadership approaches administrators regarding residency caseload cutbacks, we can make a strong case for recruiting more hospitalists.” TH
Andrea M. Sattinger is a medical writer based in North Carolina and a frequent contributor to The Hospitalist.
Effective July 1, the Accreditation Council for Graduate Medical Education (ACGME) is adopting rules changes to further restrict the number of patients internal medicine residents follow. The impact of this change may reach beyond academic institutions and teaching services. Non-teaching services and institutions may see some fallout, as hospital administration shuffles caseloads of residents and hospitalist attendings. The potential results likely will impact resident training, hospitalist training, and hospitalist practice management, namely recruitment and hospitalist job satisfaction.
Why the Change?
With the 2003 restrictions on resident work-hours duty and now the capping of patient caseloads, the ACGME is attempting to ensure residency programs are not viewed as a source of cheap labor and excessive stress. Also, “the Residency Review Committee (RRC) is cognizant too much service can be a barrier to education,” says Lenny Feldman, MD, a hospitalist and associate program director at Johns Hopkins Medical Center in Baltimore. But there is a danger in the reverse: too little service may undersupply residents with the depth and breadth of cases they need under their belts to competently enter practice. “Education should be the foremost mission for residency programs, but trying to find that exact balance between service and education is tough,” Dr. Feldman says.
In a Nutshell
As leader of the 70-hospitalist Health Partners Medical Group in Minneapolis-St. Paul, a University of Minnesota affiliate working with internal medicine residents, Burke T. Kealey, MD, views the ACGME rule change on a professional and personal level. In the big picture, Dr. Kealey observes three main effects:
- Hospitalists will be seeing more patients and probably more patients at night;
- The cost of hospital care will increase for hospitals and hospital medicine groups (HMGs); and
- The experience level of new graduates applying to be hospitalists will diminish.
In essence, there are few ways to handle the looming cap on residents’ patient caseloads. (see Practical Approaches, p. 24) Given the financial constraints imposed by this new, unfunded mandate, and taking into account the fact most residency programs depend on federal funding, it generally is believed increasing the number of residents cannot be considered an option. “Given the looming physician shortage, there is pressure on the federal government to increase the amount of GME support and the number of residency spots,” Dr. Feldman says. “Medical schools have increased enrollment pretty significantly, but the bottleneck is the number of GME-supported residency positions.”
HM Crossroads
Leslie Flores, MHA, principal with Nelson Flores Hospital Medicine Consultants, and the director of SHM’s Practice Management Institute, believes the new rule dramatically will impact teaching hospitals and HMGs. “I think it is likely to be harder for academic hospitalists, who are working on teaching services, to generate reasonable productivity, which will place an even greater financial burden on academic practices,” she says. “But the larger effect will be that non-teaching services in teaching hospitals will be expected to pick up the slack and, subsequently, grow in order to accommodate the patient numbers.”
Asking staff physicians to increase their patient load, even incrementally, is a poor solution, at best, Dr. Kealey says. And it may be tough for some places to recruit more hospitalists, a function of the hospitalist labor shortage.
William Rifkin, MD, a hospitalist and associate director of clinical medicine at Albert Einstein College of Medicine, and director of the residency program at Jacobi Medical Center, Bronx, N.Y., estimates hospitalist jobs in teaching institutions will increasingly morph into non-teaching positions. “Where currently the ratio of teaching to nonteaching jobs is 50-50,” Dr. Rifkin says, “by 2009, 80% of internal medicine training programs will have to build or expand a new, non-teaching service, and more than half of hospitalist duties will be non-teaching.”
A recent recommendation from the Institute of Mecidine (IOM) reinforces the national movement to restructure resident work hours and duties. Released Dec. 2, 2008, the “Resident Duty Hours: Enhancing Sleep, Supervision, and Safety” report calls for a maximum shift length of 30 hours with admission of patients for up to 16 hours, plus a five-hour, uninterrupted sleep period between 10 p.m. and 8 a.m., with the remaining hours for transitional and educational activity.
The consensus is the ACGME rules changes likely will alter the hospitalist job description and produce an even greater shortage of qualified, experienced physicians. Leora Horwitz, MD, MHS, an assistant professor in internal medicine at Yale University School of Medicine in New Haven, Conn., says “hospitalists are really an amalgamation of two very distinct types: the short-term hospitalist who takes the job for a year or two right after residency and before fellowship, and the longer-term hospitalist who takes on the job as at least an intermediate-term career. It could be that recruitment and retention differ for these types.”
Dr. Rifkin isn’t alone when he asks, “Can a hospitalist last that long doing patient care alone? There are only so many people who will move up to be leaders in HMGs. So while this will probably be good for recruitment in the short term, in the long term, we don’t know.”
Immediate Consequences
Some ramifications of hospital medicine as a whole taking on more patients and more hospitalists will parallel the growing pains of individual HMGs. For instance, hospitalist group’s social bonds may not be as tight, says Dr. Feldman. But where many obstacles are surmountable, “what is not surmountable is if hospitals don’t choose to increase the size of their hospitalist programs. The deathblow to most hospitalist programs is if you ask the group, and each individual, to do more work that is not commensurate with the original expectations. And with the market already tight, most hospitals can’t afford to have unhappy hospitalists.”
Financially, the new rules will place a heavy burden on HMGs and hospital administrators. With no additional reimbursement under the GME system, most hospitals will have to get creative with existing budgets. “Part of the concern is that patients that hospitalists see on a teaching service tend to be the lower socioeconomic population of patients―Medicaid and self-pay patients―where there is inadequate reimbursement anyway,” Flores says. The answer likely will be sending those patients to a non-teaching service, which in essence transfers the financial burden. “Hospitals will have to find money from somewhere.”
Teaching hospitals not part of large academic medical centers contribute to hospitalists’ compensation when they help train family medicine and internal medicine residents. “Because they are not technically academic hospitalists,” Flores says, “they need to be alerted about how these rule changes may influence the way they manage and run the finances of their practice.”
Some of the solutions to the problems inherent in this change depend on the practice and scheduling model. In the aftermath of the work-hour restriction, many hospitalist programs changed their scheduling method to day float/night float, or the “drip” method of admission (taking admissions every day), versus the “bolus” method (every fourth or fifth day), Dr. Feldman says. The bolus method likely leads to scenarios where the new ACGME cap will come into play.
There is the possibility the rule change could turn out to be a boon to HMGs, Dr. Feldman says. Programs without hospitalists may hire them; small groups may expand, increasing job opportunities. Additionally, teaching opportunities for hospitalist attendings may improve with the decreased number of patients on a service residents follow. “Hopefully, this will increase opportunities for teaching residents and increase the satisfaction of those involved in teaching,” he says. “Ultimately, it may result in improved resident education while creating more job opportunities for hospitalists―a win-win for both groups.”
Will Training Suffer?
Dr. Kealey has concerns about the long-term effects on the training residents who become hospitalists. “First, they won’t get enough experience to be competent hospitalists on graduation. Second, the number of patients is being capped, but the number of ACGME-required outpatient clinic sessions is rising, increasing from about 108 to 130 over a 30-month period,” he says. “Residency programs will to have to figure out how to fit these sessions into training, and that may squeeze out inpatient time.”
Third, with the work hours and caseload restrictions on residents, educators are concerned residents will not receive an adequate level of training.
Kenneth P. Patrick, MD, director of the hospitalist program at Chestnut Hill Hospital in Philadelphia, is worried, too, especially when it comes to the educational implications. As a former residency program director, one who shares concerns about residents’ large workloads, Dr. Patrick believes strongly in medical education and is wary of the path it seems to be taking. “What a hospital medicine group can provide to residents is the opportunity to learn from a smaller patient load,” Dr. Patrick says, “and regulatory agencies should carefully address that. Cutting back on the number of service hours and patients can have both a positive and negative effect. Most people are only adjusting the numbers of hours and patients, and not viewing the whole picture.”
Another likely result of the rules change is the mindset residents could be developing, an issue that rings true with most HMG directors. “I worry that our residents will be sheltered during training and will emerge into a real world where there won’t be caps,” Dr. Kealey says. “They will be in systems where people have to cooperate with each other in order to handle patient surges and large patient volumes. Though they may graduate, join a group, and become acculturated, it concerns me that their initial primary training, rather than encouraging them to think as part of a system, may be training them to think of ‘my restrictions, my needs, my limitation.’ ”
Prepare for Change
What is the answer? Two hospitalists echoed the same, simple solutions: “Give us more money” and “We need more bodies.”
Simplicity aside, residency and hospital medicine programs will need to prepare for the change. “Instead of happening gradually, suddenly every [residency] program in the country will lose 20% of its capacity,” Dr. Rikfin says.
Michael Pistoria, DO, FACP, associate general division of internal medicine chief at Lehigh Valley Hospital in Allentown, Pa., believes institutions with closely aligned hospitalist and residency programs will benefit from “enlightenment on both sides. Residency programs are increasingly alert to the vital role that HMGs play in supporting residency programs,” he says. “They are more aware of the impact these types of decisions have on the staffing of HMGs.”
Mid-level providers are one possible solution. “Programs will increasingly look to supplement their existing group with advanced practice clinicians—physician assistants and non-physician providers―a less-expensive alternative,” Dr. Pistoria says
Does hiring mid-level practitioners pose a risk for unintended adverse events and delays to diagnosis? “There may be an extended growth curve for these providers,” Dr. Pistoria says, “due to less clinical exposure and experience than a new physician hospitalist just out of residency.”
However, these advanced practice clinicians often are quick to adapt to the hospitalist setting, learning the skills required to be an effective hospitalist through on-the-job training. “On-the-job training for physician hospitalists can focus on education, quality improvement, safety―some of the value-added pieces,” Dr. Pistoria points out.
Without a doubt, ACGME’s new cap on residency caseloads will impact hospital medicine, both at the national level and the individual group level. HMG efforts to recruit, schedule, train and pay hospitalists will be affected, as will the level of experience patients receive from recent residency graduates.
“It is incumbent on us to get involved in committees and process and performance improvement projects,” Dr. Pistoria says, “so that when leadership approaches administrators regarding residency caseload cutbacks, we can make a strong case for recruiting more hospitalists.” TH
Andrea M. Sattinger is a medical writer based in North Carolina and a frequent contributor to The Hospitalist.
Effective July 1, the Accreditation Council for Graduate Medical Education (ACGME) is adopting rules changes to further restrict the number of patients internal medicine residents follow. The impact of this change may reach beyond academic institutions and teaching services. Non-teaching services and institutions may see some fallout, as hospital administration shuffles caseloads of residents and hospitalist attendings. The potential results likely will impact resident training, hospitalist training, and hospitalist practice management, namely recruitment and hospitalist job satisfaction.
Why the Change?
With the 2003 restrictions on resident work-hours duty and now the capping of patient caseloads, the ACGME is attempting to ensure residency programs are not viewed as a source of cheap labor and excessive stress. Also, “the Residency Review Committee (RRC) is cognizant too much service can be a barrier to education,” says Lenny Feldman, MD, a hospitalist and associate program director at Johns Hopkins Medical Center in Baltimore. But there is a danger in the reverse: too little service may undersupply residents with the depth and breadth of cases they need under their belts to competently enter practice. “Education should be the foremost mission for residency programs, but trying to find that exact balance between service and education is tough,” Dr. Feldman says.
In a Nutshell
As leader of the 70-hospitalist Health Partners Medical Group in Minneapolis-St. Paul, a University of Minnesota affiliate working with internal medicine residents, Burke T. Kealey, MD, views the ACGME rule change on a professional and personal level. In the big picture, Dr. Kealey observes three main effects:
- Hospitalists will be seeing more patients and probably more patients at night;
- The cost of hospital care will increase for hospitals and hospital medicine groups (HMGs); and
- The experience level of new graduates applying to be hospitalists will diminish.
In essence, there are few ways to handle the looming cap on residents’ patient caseloads. (see Practical Approaches, p. 24) Given the financial constraints imposed by this new, unfunded mandate, and taking into account the fact most residency programs depend on federal funding, it generally is believed increasing the number of residents cannot be considered an option. “Given the looming physician shortage, there is pressure on the federal government to increase the amount of GME support and the number of residency spots,” Dr. Feldman says. “Medical schools have increased enrollment pretty significantly, but the bottleneck is the number of GME-supported residency positions.”
HM Crossroads
Leslie Flores, MHA, principal with Nelson Flores Hospital Medicine Consultants, and the director of SHM’s Practice Management Institute, believes the new rule dramatically will impact teaching hospitals and HMGs. “I think it is likely to be harder for academic hospitalists, who are working on teaching services, to generate reasonable productivity, which will place an even greater financial burden on academic practices,” she says. “But the larger effect will be that non-teaching services in teaching hospitals will be expected to pick up the slack and, subsequently, grow in order to accommodate the patient numbers.”
Asking staff physicians to increase their patient load, even incrementally, is a poor solution, at best, Dr. Kealey says. And it may be tough for some places to recruit more hospitalists, a function of the hospitalist labor shortage.
William Rifkin, MD, a hospitalist and associate director of clinical medicine at Albert Einstein College of Medicine, and director of the residency program at Jacobi Medical Center, Bronx, N.Y., estimates hospitalist jobs in teaching institutions will increasingly morph into non-teaching positions. “Where currently the ratio of teaching to nonteaching jobs is 50-50,” Dr. Rifkin says, “by 2009, 80% of internal medicine training programs will have to build or expand a new, non-teaching service, and more than half of hospitalist duties will be non-teaching.”
A recent recommendation from the Institute of Mecidine (IOM) reinforces the national movement to restructure resident work hours and duties. Released Dec. 2, 2008, the “Resident Duty Hours: Enhancing Sleep, Supervision, and Safety” report calls for a maximum shift length of 30 hours with admission of patients for up to 16 hours, plus a five-hour, uninterrupted sleep period between 10 p.m. and 8 a.m., with the remaining hours for transitional and educational activity.
The consensus is the ACGME rules changes likely will alter the hospitalist job description and produce an even greater shortage of qualified, experienced physicians. Leora Horwitz, MD, MHS, an assistant professor in internal medicine at Yale University School of Medicine in New Haven, Conn., says “hospitalists are really an amalgamation of two very distinct types: the short-term hospitalist who takes the job for a year or two right after residency and before fellowship, and the longer-term hospitalist who takes on the job as at least an intermediate-term career. It could be that recruitment and retention differ for these types.”
Dr. Rifkin isn’t alone when he asks, “Can a hospitalist last that long doing patient care alone? There are only so many people who will move up to be leaders in HMGs. So while this will probably be good for recruitment in the short term, in the long term, we don’t know.”
Immediate Consequences
Some ramifications of hospital medicine as a whole taking on more patients and more hospitalists will parallel the growing pains of individual HMGs. For instance, hospitalist group’s social bonds may not be as tight, says Dr. Feldman. But where many obstacles are surmountable, “what is not surmountable is if hospitals don’t choose to increase the size of their hospitalist programs. The deathblow to most hospitalist programs is if you ask the group, and each individual, to do more work that is not commensurate with the original expectations. And with the market already tight, most hospitals can’t afford to have unhappy hospitalists.”
Financially, the new rules will place a heavy burden on HMGs and hospital administrators. With no additional reimbursement under the GME system, most hospitals will have to get creative with existing budgets. “Part of the concern is that patients that hospitalists see on a teaching service tend to be the lower socioeconomic population of patients―Medicaid and self-pay patients―where there is inadequate reimbursement anyway,” Flores says. The answer likely will be sending those patients to a non-teaching service, which in essence transfers the financial burden. “Hospitals will have to find money from somewhere.”
Teaching hospitals not part of large academic medical centers contribute to hospitalists’ compensation when they help train family medicine and internal medicine residents. “Because they are not technically academic hospitalists,” Flores says, “they need to be alerted about how these rule changes may influence the way they manage and run the finances of their practice.”
Some of the solutions to the problems inherent in this change depend on the practice and scheduling model. In the aftermath of the work-hour restriction, many hospitalist programs changed their scheduling method to day float/night float, or the “drip” method of admission (taking admissions every day), versus the “bolus” method (every fourth or fifth day), Dr. Feldman says. The bolus method likely leads to scenarios where the new ACGME cap will come into play.
There is the possibility the rule change could turn out to be a boon to HMGs, Dr. Feldman says. Programs without hospitalists may hire them; small groups may expand, increasing job opportunities. Additionally, teaching opportunities for hospitalist attendings may improve with the decreased number of patients on a service residents follow. “Hopefully, this will increase opportunities for teaching residents and increase the satisfaction of those involved in teaching,” he says. “Ultimately, it may result in improved resident education while creating more job opportunities for hospitalists―a win-win for both groups.”
Will Training Suffer?
Dr. Kealey has concerns about the long-term effects on the training residents who become hospitalists. “First, they won’t get enough experience to be competent hospitalists on graduation. Second, the number of patients is being capped, but the number of ACGME-required outpatient clinic sessions is rising, increasing from about 108 to 130 over a 30-month period,” he says. “Residency programs will to have to figure out how to fit these sessions into training, and that may squeeze out inpatient time.”
Third, with the work hours and caseload restrictions on residents, educators are concerned residents will not receive an adequate level of training.
Kenneth P. Patrick, MD, director of the hospitalist program at Chestnut Hill Hospital in Philadelphia, is worried, too, especially when it comes to the educational implications. As a former residency program director, one who shares concerns about residents’ large workloads, Dr. Patrick believes strongly in medical education and is wary of the path it seems to be taking. “What a hospital medicine group can provide to residents is the opportunity to learn from a smaller patient load,” Dr. Patrick says, “and regulatory agencies should carefully address that. Cutting back on the number of service hours and patients can have both a positive and negative effect. Most people are only adjusting the numbers of hours and patients, and not viewing the whole picture.”
Another likely result of the rules change is the mindset residents could be developing, an issue that rings true with most HMG directors. “I worry that our residents will be sheltered during training and will emerge into a real world where there won’t be caps,” Dr. Kealey says. “They will be in systems where people have to cooperate with each other in order to handle patient surges and large patient volumes. Though they may graduate, join a group, and become acculturated, it concerns me that their initial primary training, rather than encouraging them to think as part of a system, may be training them to think of ‘my restrictions, my needs, my limitation.’ ”
Prepare for Change
What is the answer? Two hospitalists echoed the same, simple solutions: “Give us more money” and “We need more bodies.”
Simplicity aside, residency and hospital medicine programs will need to prepare for the change. “Instead of happening gradually, suddenly every [residency] program in the country will lose 20% of its capacity,” Dr. Rikfin says.
Michael Pistoria, DO, FACP, associate general division of internal medicine chief at Lehigh Valley Hospital in Allentown, Pa., believes institutions with closely aligned hospitalist and residency programs will benefit from “enlightenment on both sides. Residency programs are increasingly alert to the vital role that HMGs play in supporting residency programs,” he says. “They are more aware of the impact these types of decisions have on the staffing of HMGs.”
Mid-level providers are one possible solution. “Programs will increasingly look to supplement their existing group with advanced practice clinicians—physician assistants and non-physician providers―a less-expensive alternative,” Dr. Pistoria says
Does hiring mid-level practitioners pose a risk for unintended adverse events and delays to diagnosis? “There may be an extended growth curve for these providers,” Dr. Pistoria says, “due to less clinical exposure and experience than a new physician hospitalist just out of residency.”
However, these advanced practice clinicians often are quick to adapt to the hospitalist setting, learning the skills required to be an effective hospitalist through on-the-job training. “On-the-job training for physician hospitalists can focus on education, quality improvement, safety―some of the value-added pieces,” Dr. Pistoria points out.
Without a doubt, ACGME’s new cap on residency caseloads will impact hospital medicine, both at the national level and the individual group level. HMG efforts to recruit, schedule, train and pay hospitalists will be affected, as will the level of experience patients receive from recent residency graduates.
“It is incumbent on us to get involved in committees and process and performance improvement projects,” Dr. Pistoria says, “so that when leadership approaches administrators regarding residency caseload cutbacks, we can make a strong case for recruiting more hospitalists.” TH
Andrea M. Sattinger is a medical writer based in North Carolina and a frequent contributor to The Hospitalist.
Staffing Strategies

One of the most difficult challenges in staffing a hospitalist practice is handling the unpredictable daily fluctuations in patient volume. It isn’t difficult to decide how many hospitalists will work each day to handle the average number of daily visits (aka encounters), but the actual number of visits on any given day is almost always significantly different than the average. I think many groups could more effectively handle day-to-day variations in workload by eliminating predetermined lengths of the shifts that the doctors work. It isn’t a perfect strategy, but it is worth some consideration by nearly any practice. Let me explain.

First, think about how the workload for a typical day might be represented. For many or most practices it often looks something like the wavy line in Figure 1. (See Figure 1, p. 52.)
Of course, the line representing a day’s work will be different every day, but I’ve tried to draw it in a way that represents a typical day.
In Figure 2 (see p. 52), I’ve added horizontal bars to represent a common way that groups might schedule four daytime doctors who each work 7 a.m. to 7 p.m., and one night doctor working 7 p.m. to 7 a.m. The four horizontal bars represent the four day doctors, and the one horizontal bar at the bottom right represents the one night doctor. Ideally, the manpower (horizontal bars) should match the workload (wavy line) every hour of the day.
This graph shows that—at least for this particular day—there are many hours in the afternoon when there is excess manpower. The doctors may be sitting around waiting for their shift to end or waiting to see if it will suddenly get busy again. We all know that happens unpredictably. And from about 7 p.m. to about 11:30 p.m., the single night doctor has more work than he/she can reasonably handle.
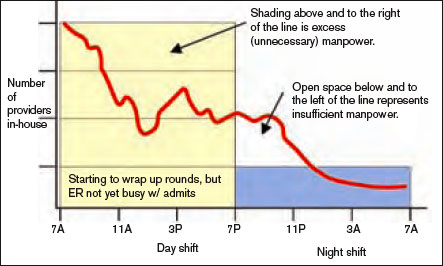
In fact, there probably isn’t ever a day when the work that needs to be done is just the right amount for all four doctors from 7 a.m. to 7 p.m. with a sudden drop at 7 p.m. that is just right for one doctor for the next 12 hours. Because the doctors have scheduled themselves to work 12-hour shifts, they know in advance that their manpower will quite regularly fail to match the workload for that day.
Groups have devised a number of strategies to try to get manpower to more closely match the unpredictable workload for a given day. These include having a member of the group available on standby (often called “jeopardy”) for that day; this physician comes in only if it is unusually busy. Some groups have a patient volume cap to prevent the practice from becoming too busy. I think a cap is a poor strategy that should be used only as a last resort, and I will discuss this in detail in a future column. Other groups have a swing shift from late in the afternoon until around 11 p.m. or so to help with evening admits and cross cover. And an often overlooked but potentially valuable strategy is to eliminate clearly specified start and stop times for the shifts that the doctors work. For an idea of what that might look like, see Figure 3 (p. 52).
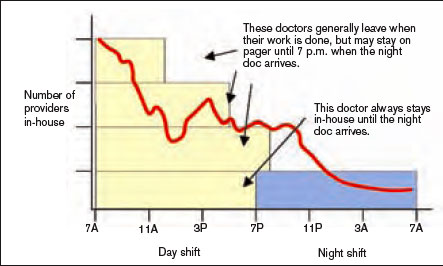
Notice that the right-hand side of each yellow bar in Figures 2 and 3 is indistinct. That is meant to show that the precise time that the doctor leaves varies, depending on the day’s workload. That way the manpower can be adjusted from one day to the next to more closely match the workload than if the doctors work fixed shifts of a specified duration. On some days, all of the doctors may stay 12 hours or more, but on many days at least some of the doctors will end up leaving in less than 12 hours. If all day doctors work a 12-hour shift, they have provided 48 hours—four doctors at 12 hours each—of physician manpower, but if there is some flexibility about when the doctors leave, the same four day doctors could provide between about 34 and 52 hours of manpower, depending on the day’s workload.
If your practice is contracted to keep a doctor in the hospital around the clock, you will probably need the night doctor and at least one day doctor to stay around—even if it is a slow day. But the other doctors might be able to leave when their work is done. And it is also reasonable for some groups to eliminate precise times that the doctors start working in the morning each day, though they might be required to be available by pager by a specified time in the morning.
One common concern about such a system is how to handle issues that arise with the patients cared for by a doctor who has left. I think it is best for the doctor to stay available by pager and handle simple issues by phone. For more complicated issues (e.g., a patient who needs attention at the bedside) the doctor could either come back to the hospital or phone another member of the practice (e.g., the doctor required to stay at least 12 hours that day) and see if he or she can handle the emergency.
All of the specifics of a system that allows doctors to leave when their work is done rather than according to shifts of a predetermined number of hours would be too long for this column. But they aren’t complicated, and given the variability that exists in the number of daily patient visits to any hospitalist practice, the application of this kind of approach is well worth considering. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is a co-founder and past-president of SHM. He is a principal in Nelson/Flores Associates, a national hospitalist practice management consulting firm. This column represents his views and is not intended to reflect an official position of SHM.
One of the most difficult challenges in staffing a hospitalist practice is handling the unpredictable daily fluctuations in patient volume. It isn’t difficult to decide how many hospitalists will work each day to handle the average number of daily visits (aka encounters), but the actual number of visits on any given day is almost always significantly different than the average. I think many groups could more effectively handle day-to-day variations in workload by eliminating predetermined lengths of the shifts that the doctors work. It isn’t a perfect strategy, but it is worth some consideration by nearly any practice. Let me explain.
First, think about how the workload for a typical day might be represented. For many or most practices it often looks something like the wavy line in Figure 1. (See Figure 1, p. 52.)
Of course, the line representing a day’s work will be different every day, but I’ve tried to draw it in a way that represents a typical day.
In Figure 2 (see p. 52), I’ve added horizontal bars to represent a common way that groups might schedule four daytime doctors who each work 7 a.m. to 7 p.m., and one night doctor working 7 p.m. to 7 a.m. The four horizontal bars represent the four day doctors, and the one horizontal bar at the bottom right represents the one night doctor. Ideally, the manpower (horizontal bars) should match the workload (wavy line) every hour of the day.
This graph shows that—at least for this particular day—there are many hours in the afternoon when there is excess manpower. The doctors may be sitting around waiting for their shift to end or waiting to see if it will suddenly get busy again. We all know that happens unpredictably. And from about 7 p.m. to about 11:30 p.m., the single night doctor has more work than he/she can reasonably handle.
In fact, there probably isn’t ever a day when the work that needs to be done is just the right amount for all four doctors from 7 a.m. to 7 p.m. with a sudden drop at 7 p.m. that is just right for one doctor for the next 12 hours. Because the doctors have scheduled themselves to work 12-hour shifts, they know in advance that their manpower will quite regularly fail to match the workload for that day.
Groups have devised a number of strategies to try to get manpower to more closely match the unpredictable workload for a given day. These include having a member of the group available on standby (often called “jeopardy”) for that day; this physician comes in only if it is unusually busy. Some groups have a patient volume cap to prevent the practice from becoming too busy. I think a cap is a poor strategy that should be used only as a last resort, and I will discuss this in detail in a future column. Other groups have a swing shift from late in the afternoon until around 11 p.m. or so to help with evening admits and cross cover. And an often overlooked but potentially valuable strategy is to eliminate clearly specified start and stop times for the shifts that the doctors work. For an idea of what that might look like, see Figure 3 (p. 52).
Notice that the right-hand side of each yellow bar in Figures 2 and 3 is indistinct. That is meant to show that the precise time that the doctor leaves varies, depending on the day’s workload. That way the manpower can be adjusted from one day to the next to more closely match the workload than if the doctors work fixed shifts of a specified duration. On some days, all of the doctors may stay 12 hours or more, but on many days at least some of the doctors will end up leaving in less than 12 hours. If all day doctors work a 12-hour shift, they have provided 48 hours—four doctors at 12 hours each—of physician manpower, but if there is some flexibility about when the doctors leave, the same four day doctors could provide between about 34 and 52 hours of manpower, depending on the day’s workload.
If your practice is contracted to keep a doctor in the hospital around the clock, you will probably need the night doctor and at least one day doctor to stay around—even if it is a slow day. But the other doctors might be able to leave when their work is done. And it is also reasonable for some groups to eliminate precise times that the doctors start working in the morning each day, though they might be required to be available by pager by a specified time in the morning.
One common concern about such a system is how to handle issues that arise with the patients cared for by a doctor who has left. I think it is best for the doctor to stay available by pager and handle simple issues by phone. For more complicated issues (e.g., a patient who needs attention at the bedside) the doctor could either come back to the hospital or phone another member of the practice (e.g., the doctor required to stay at least 12 hours that day) and see if he or she can handle the emergency.
All of the specifics of a system that allows doctors to leave when their work is done rather than according to shifts of a predetermined number of hours would be too long for this column. But they aren’t complicated, and given the variability that exists in the number of daily patient visits to any hospitalist practice, the application of this kind of approach is well worth considering. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is a co-founder and past-president of SHM. He is a principal in Nelson/Flores Associates, a national hospitalist practice management consulting firm. This column represents his views and is not intended to reflect an official position of SHM.
One of the most difficult challenges in staffing a hospitalist practice is handling the unpredictable daily fluctuations in patient volume. It isn’t difficult to decide how many hospitalists will work each day to handle the average number of daily visits (aka encounters), but the actual number of visits on any given day is almost always significantly different than the average. I think many groups could more effectively handle day-to-day variations in workload by eliminating predetermined lengths of the shifts that the doctors work. It isn’t a perfect strategy, but it is worth some consideration by nearly any practice. Let me explain.
First, think about how the workload for a typical day might be represented. For many or most practices it often looks something like the wavy line in Figure 1. (See Figure 1, p. 52.)
Of course, the line representing a day’s work will be different every day, but I’ve tried to draw it in a way that represents a typical day.
In Figure 2 (see p. 52), I’ve added horizontal bars to represent a common way that groups might schedule four daytime doctors who each work 7 a.m. to 7 p.m., and one night doctor working 7 p.m. to 7 a.m. The four horizontal bars represent the four day doctors, and the one horizontal bar at the bottom right represents the one night doctor. Ideally, the manpower (horizontal bars) should match the workload (wavy line) every hour of the day.
This graph shows that—at least for this particular day—there are many hours in the afternoon when there is excess manpower. The doctors may be sitting around waiting for their shift to end or waiting to see if it will suddenly get busy again. We all know that happens unpredictably. And from about 7 p.m. to about 11:30 p.m., the single night doctor has more work than he/she can reasonably handle.
In fact, there probably isn’t ever a day when the work that needs to be done is just the right amount for all four doctors from 7 a.m. to 7 p.m. with a sudden drop at 7 p.m. that is just right for one doctor for the next 12 hours. Because the doctors have scheduled themselves to work 12-hour shifts, they know in advance that their manpower will quite regularly fail to match the workload for that day.
Groups have devised a number of strategies to try to get manpower to more closely match the unpredictable workload for a given day. These include having a member of the group available on standby (often called “jeopardy”) for that day; this physician comes in only if it is unusually busy. Some groups have a patient volume cap to prevent the practice from becoming too busy. I think a cap is a poor strategy that should be used only as a last resort, and I will discuss this in detail in a future column. Other groups have a swing shift from late in the afternoon until around 11 p.m. or so to help with evening admits and cross cover. And an often overlooked but potentially valuable strategy is to eliminate clearly specified start and stop times for the shifts that the doctors work. For an idea of what that might look like, see Figure 3 (p. 52).
Notice that the right-hand side of each yellow bar in Figures 2 and 3 is indistinct. That is meant to show that the precise time that the doctor leaves varies, depending on the day’s workload. That way the manpower can be adjusted from one day to the next to more closely match the workload than if the doctors work fixed shifts of a specified duration. On some days, all of the doctors may stay 12 hours or more, but on many days at least some of the doctors will end up leaving in less than 12 hours. If all day doctors work a 12-hour shift, they have provided 48 hours—four doctors at 12 hours each—of physician manpower, but if there is some flexibility about when the doctors leave, the same four day doctors could provide between about 34 and 52 hours of manpower, depending on the day’s workload.
If your practice is contracted to keep a doctor in the hospital around the clock, you will probably need the night doctor and at least one day doctor to stay around—even if it is a slow day. But the other doctors might be able to leave when their work is done. And it is also reasonable for some groups to eliminate precise times that the doctors start working in the morning each day, though they might be required to be available by pager by a specified time in the morning.
One common concern about such a system is how to handle issues that arise with the patients cared for by a doctor who has left. I think it is best for the doctor to stay available by pager and handle simple issues by phone. For more complicated issues (e.g., a patient who needs attention at the bedside) the doctor could either come back to the hospital or phone another member of the practice (e.g., the doctor required to stay at least 12 hours that day) and see if he or she can handle the emergency.
All of the specifics of a system that allows doctors to leave when their work is done rather than according to shifts of a predetermined number of hours would be too long for this column. But they aren’t complicated, and given the variability that exists in the number of daily patient visits to any hospitalist practice, the application of this kind of approach is well worth considering. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is a co-founder and past-president of SHM. He is a principal in Nelson/Flores Associates, a national hospitalist practice management consulting firm. This column represents his views and is not intended to reflect an official position of SHM.
Deja Vu
We thought they were gone, but they’ve returned: diseases once considered “vintage bugs” that were common in as late as the mid-20th century. In the past these diseases killed one in three people younger than 20 who had survived an infancy during which many of their contemporaries died.1
“When you think about disease states, you think about some that are gone from the world,” says Erin Stucky, MD, a pediatric hospitalist at the University of California, San Diego, “but there are very few truly gone from the world.”
Some of the major infectious diseases that hospitalists may [still] see are pertussis (whooping cough), measles, and mumps, but scarlet fever and varicella (chicken pox) also endure—not to mention those occurrences of polio around the country that epidemiologists and infectious diseases specialists are monitoring closely. Rickets, a vitamin-D-deficiency-related disease also thought to be a relic of the 18th century, is showing up in certain patient populations—and not exclusively in infants and children.
This is a crossover clinical issue, our pediatric hospitalists say, and thus one to which their hospitalist partners who treat adult patients must also remain alert.
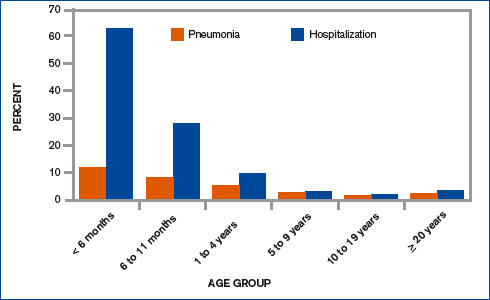
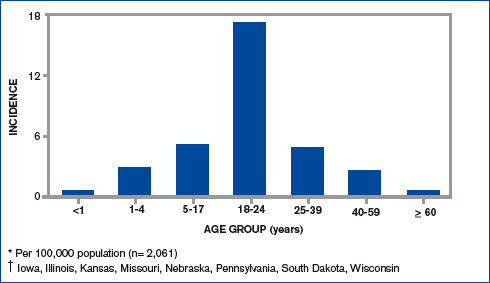
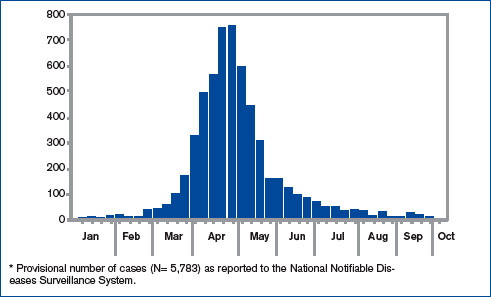
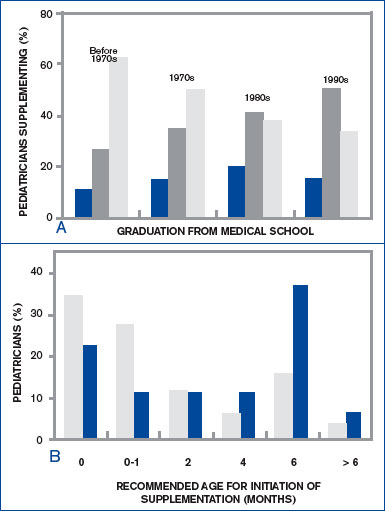
Pertussis (Whooping Cough)
Despite vaccination protocols, pediatric hospitalists continue to see whooping cough in young infants. (See Figure 1, p. 39.) Even with treatment, the damage can be severe, and the length of stay (LOS) is prolonged compared with those of most other patients with complex illnesses. “Vaccine fatigue” means that immunization lasts only until adolescence or early adulthood, at which time they need appropriate boosters. If the patient hasn’t receive boosters, the initial immunization loses its effectiveness; unprotected, they can be infected with the disease, though sometimes not badly enough for them to seek care. When they do, the diagnosis is often community-acquired mild pneumonia or a more traditional bronchitis. Either by accident or because the physician has given it thought, those illnesses are treated with a macrolide drug, which is also—coincidentally and serendipitously—the drug of choice for pertussis. But many remain carriers because they are not accurately diagnosed or never seek care.
“There is a huge reservoir of people carrying pertussis, particularly [in] the adolescent and adult population[s],” says Alison Holmes, MD, a pediatric hospitalist at Concord Hospital, N.H. “And the babies who get really sick from it are the under two- to three-month group who have not yet been immunized or have just been immunized. Because it is so rampant in the adolescent and adult community, those children can still get sick.”
“Unfortunately,” says Dr. Stucky, “what’s happening is that if physicians are not thinking pertussis, they don’t talk about pertussis to that adult patient who … is either around children or has children in the home. So they don’t know to tell that person to watch for these same signs and symptoms in that young infant, who then could have a much more severe outcome from getting [the infection].”
As with most patients who contract illnesses, these patients may never have heard of the disease and unless educated may not understand the implications of the diagnosis. They might realize their disease could spread to family members, “but most people don’t absorb that information and use that information thoughtfully,” says Dr. Stucky. The onus is, therefore, on the physician to warn adult patients specifically about the serious danger that exists for infants in the two- to three-month-old group, who may not have been vaccinated or whose single-vaccination immunity is not adequate protection against the disease.
While the numbers in babies appear to be what they have always been, the incidence has grown in the teen years and even later into adulthood. This is more likely the result of increased testing for pertussis, as opposed to being only due to a true resurgence. Data from studies of adults with prolonged cough revealed that 20% to 25% have serologic evidence of recent pertussis infection.2 Adults are the major reservoir of infection, and infection spreads quickly in a population in a closed environment where droplets spread easily person to person.5
For both teens and adults, testing and immunization with the newly recommended DTaP (diphtheria-tetanus-pertussis)—as opposed to the more limited Td—can help upgrade immunity. Although a patient can recover from pertussis on his or her own within one to two weeks following treatment, the intent of treatment is primarily to limit the spread of disease to others.4-7
The problem when adults get pertussis, says Dr. Holmes, who is also an assistant professor of community and family medicine at Dartmouth Medical School, Hanover, N.H., “is that they often don’t show up complaining about this horrible paroxysmal coughing until they’re about three or four weeks into the illness, and it hasn’t gone away. You go for hours and hours feeling completely fine and wonderful, and why would you bother going to the doctor?”
Babies are most at risk, however. “They often don’t have the energy or the muscle strength, so they just stop breathing instead,” she says.
Mark Dworkin, MD, MPH, TM, the state epidemiologist and team leader for the Rapid Response Team at the Illinois Department of Public Health, is active in outbreak investigation. He wrote a compelling argument for maintaining a high index of suspicion when physicians see adolescent and adult patients who have a cough that has lasted more than two weeks.4
It has been estimated that more than one million cases of pertussis occur in the United States each year; that number has continued to grow for 20 years. From 1990 to 2001, the incidence of pertussis in adults increased by 400%. But many physicians believe that pertussis is only a pediatric illness. A survey of internists in Washington state showed that only 38% of respondents knew about the risk of vaccine fatigue, and just 36% knew that the nasopharyngeal swab is the preferred method for sample collection. Public health professionals were also concerned with the finding that too many pediatricians and nonpediatricians (43% and 41%, respectively) were not able to define a reportable case.
The first challenge that faces internists, writes Dr. Dworkin, is recognizing pertussis, which in some cases presents with mild symptoms; some adults won’t even have a cough.4 But at the other end of the disease spectrum, symptoms may be as brutal as bilateral subconjunctival hemorrhage or rib fracture due to convulsive coughing. In any case, what goes unrecognized, undiagnosed, and untreated becomes a particularly serious risk for vulnerable infants. Once pertussis is identified, positive results on polymerase chain reaction or culture can help convince skeptical colleagues who may still believe pertussis is exclusively a childhood disease—and a vintage one at that.
“What we in pediatrics champion … is for [these immunizations] to help the young child; the less disease we have out there, the better off we’re going to eventually be,” says Dr. Stucky, who projects that, within just a few years, Tdap vaccinations for adolescents and adults up to age 64 might lead to a reduction of infection in the three-month-old group.6
Measles and Mumps
From January 1 to October 7, 2006, 45 states and the District of Columbia reported 5,783 confirmed or probable mumps cases to the Centers for Disease Control and Prevention (CDC). (See Figures 2 and 3, above.)8 The Advisory Committee on Immunization Practices (ACIP) announced that continuing data from surveillance reports meant that healthcare workers should remain alert to suspected cases, conduct appropriate laboratory testing, and use every opportunity to ensure adequate immunity, particularly among populations at high risk.7
In contrast to the circumstances with pertussis, with mumps “there have been pockets of people who have either chosen not to immunize their child[ren], or their child[ren] get exposed to it somehow,” says Dr. Stucky, “and although they might be immunized, they might not have had a good response.” In an environment such as a school, “where one child can cough on a few and then cough on a few [more],” there is an environment where the infection can spread rampantly.
With mumps and measles, these could be called true outbreaks, such as the classic example that occurred in Kansas 18 years ago or the epidemic that disseminated from a college campus in Iowa in the spring of 2006, which originated from only two airline passengers on nine different flights within one week.8
College dorms and cafeterias can be treacherous breeding grounds for pathogens, and this generation of college students is susceptible for a few reasons. For one, in the late 1980s, when they were infants, the vaccine schedule was changed; the measles/mumps/rubella vaccine was upgraded from one dose to two—and not all children received the two doses.
The unimmunized who are exposed to measles and mumps remain at highest risk for spreading the disease. Although in 2005, 76%-79% of children aged 19-35 months received the entire recommended series of shots against whooping cough, diphtheria, tetanus, polio, measles, mumps, rubella, chicken pox, hepatitis B, and Haemophilus influenza type B, that still means that 21%-24% of the children—or potentially one out of five kids—did not.9
Other factors causing low levels of immunization include parents’ Internet-fueled fears of links to autism; immigrants crossing U.S. borders from Mexico or other countries where immunization is not standardized; religious and philosophical reasons; and international travel.10
“When young adults travel internationally [to places] where they are exposed to young children and adults who have never been immunized,” that’s a big risk, says Dr. Stucky. “All it would take is one [infected] student coming into a dorm and passing it around [to others with lapsed coverage or no immunization for the disease].” And while providers may think of travelers being exposed to diseases such as malaria and typhoid fever in developing countries, “in reality, a lot of the common things we’re immunizing for in our country are not immunized for in other countries, and those can be brought back.”
Rickets
The incidence of rickets is increasing, especially in black and Hispanic children and particularly in the north.11,12 Epidemiologists trace the rise to an increase in breast-feeding (good for immunity, but breast milk lacks substantial vitamin D), overuse of sunscreen or lack of exposure to sunlight, and changes in physician recommendations for vitamin supplementation. The effects of rickets alone can be profound, but other long-term consequences of vitamin D deficiency may include type I diabetes, cancer (especially of the prostate), and osteoporosis.12
In the past few decades, physicians have been less likely to recommend vitamin D supplementation for babies, and an interesting study by Davenport and colleagues correlates the year of medical school completion to that decline as well as substantial variability as to the age at which supplement use is begun.12 (See Figures 4a and 4b, left.)
“Most of the cases I have run into have been in [recent] African immigrants, where the mothers stay covered and they are vitamin D deficient,” says Dr. Holmes. “It’s wonderful that they culturally breast-feed, but they come to the U.S., and they’re pretty afraid to go outside in a new society.”
Varicella (Chicken Pox)
Varicella was removed from the CDC’s national notifiable disease list in 1981, but in 1995 a varicella vaccine was recommended for routine childhood vaccination.13 Before the licensure of that vaccine, varicella was a universal childhood disease in the U.S., causing 4 million cases, 11,000 hospitalizations, and 100 deaths every year.14 In 2002, the Council of State and Territorial Epidemiologists recommended that varicella be included in the National Notifiable Surveillance System by 2003 and that case-based surveillance in all states be established by 2005.13 CDC’s ACIP recommended in 2006 that a routine second dose of varicella vaccine be given to children between the ages of four and six years old.
Contracting chicken pox as an adult is a much more morbid occurrence than catching it as a child. Although varicella is not life threatening (as are diphtheria, tetanus, and measles) or sterility-causing (as is mumps), when the vaccine was approved, some pediatricians, including Dr. Stucky, became concerned that “now we’re creating a population that has never seen the wild-type varicella virus, and what does that mean? Were we just delaying something into an age category where people will get sicker?” Recognizing varicella, therefore, is critical even for hospitalists who treat adults.
Conclusion
“I’ve seen mumps, measles, varicella, pertussis,” says Dr. Stucky, “but our adult [hospitalist] partners hadn’t.” She encourages her colleagues who treat adult populations “to read and be diligent. These diseases can exist in adults, or even in children who were once vaccinated, and all hospitalists need to know “what to do, how to treat them, and [that] the consequences in adults are hands down worse than in children.”
Dr. Stucky believes hospitalists who treat adults would do well to consult physicians who practiced in the 1950s because they understand the history as well as clinical signs and symptoms of these diseases; she says, “For the hospitalist who treats adults, these are the equivalent of emerging infectious diseases.” TH
Andrea Sattinger is a frequent contributor to The Hospitalist.
References
- Carmichael M. 'Vintage' bugs return. Newsweek. May 1, 2006:Vol. 147, p. 38. Available at: www.msnbc.msn.com/id/12440796/site/newsweek/. Accessed on November 29, 2006.
- Herwaldt LA. Pertussis in adults. What physicians need to know. Arch Intern Med. 1991;151:1510-1512.
- Schafer S, Gillette H, Hedberg K, et al. A community-wide pertussis outbreak: an argument for universal booster vaccination. Arch Intern Med. 2006 Jun 26;166(12):1317-1321.
- Dworkin MS. Adults are whooping, but are internists listening? Ann Intern Med. 2005 May 17;142(10):832-835. Available at: www.annals.org/cgi/reprint/142/10/832.pdf. Accessed on November 19, 2006.
- Gregory DS. Pertussis: a disease affecting all ages. Am Fam Physician. 2006 Aug 1;74(3):420-426.
- Finger R, Shoemaker J. Preventing pertussis in infants by vaccinating adults. Am Fam Physician. 2006 Aug 1;74(3):382.
- Broder KR, Cortese MM, Iskander JK, et al. Preventing tetanus, diphtheria, and pertussis among adolescents: use of tetanus toxoid, reduced diphtheria toxoid and acellular pertussis vaccines recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2006;55:1-34.
- MMWR. Brief report: update: mumps activity—United States, January 1-October 7, 2006. MMWR. 2006 Oct 27;55(42):1152-1153.
- National Briefing: Science and health: race gap closes in vaccinations, U.S. says. New York Times. September 15, 2006.
- Calandrillo SP. Vanishing vaccinations: why are so many Americans opting out of vaccinating their children? Univ Mich J Law Reform. 2004 Winter;37(2):353-440.
- Kreiter SR, Schwartz RP, Kirkman HN Jr, et al. Nutritional rickets in African American breast-fed infants. J Pediatr. 2000 Aug;137(2):153-157.
- Davenport ML, Uckun A, Calikoglu AS. Pediatrician patterns of prescribing vitamin supplementation for infants: do they contribute to rickets? Pediatrics. 2004 Jan;113(1 Pt 1):179-180.
- MMWR. Varicella surveillance practices—United States, 2004. MMWR. 2006 Oct 19;55:1126-1129.
- Seward JF, Watson BM, Peterson CL, et al. Varicella disease after introduction of varicella vaccine in the United States, 1995-2000. JAMA. 2002 Feb 6;287(5):606-611.
We thought they were gone, but they’ve returned: diseases once considered “vintage bugs” that were common in as late as the mid-20th century. In the past these diseases killed one in three people younger than 20 who had survived an infancy during which many of their contemporaries died.1
“When you think about disease states, you think about some that are gone from the world,” says Erin Stucky, MD, a pediatric hospitalist at the University of California, San Diego, “but there are very few truly gone from the world.”
Some of the major infectious diseases that hospitalists may [still] see are pertussis (whooping cough), measles, and mumps, but scarlet fever and varicella (chicken pox) also endure—not to mention those occurrences of polio around the country that epidemiologists and infectious diseases specialists are monitoring closely. Rickets, a vitamin-D-deficiency-related disease also thought to be a relic of the 18th century, is showing up in certain patient populations—and not exclusively in infants and children.
This is a crossover clinical issue, our pediatric hospitalists say, and thus one to which their hospitalist partners who treat adult patients must also remain alert.
Pertussis (Whooping Cough)
Despite vaccination protocols, pediatric hospitalists continue to see whooping cough in young infants. (See Figure 1, p. 39.) Even with treatment, the damage can be severe, and the length of stay (LOS) is prolonged compared with those of most other patients with complex illnesses. “Vaccine fatigue” means that immunization lasts only until adolescence or early adulthood, at which time they need appropriate boosters. If the patient hasn’t receive boosters, the initial immunization loses its effectiveness; unprotected, they can be infected with the disease, though sometimes not badly enough for them to seek care. When they do, the diagnosis is often community-acquired mild pneumonia or a more traditional bronchitis. Either by accident or because the physician has given it thought, those illnesses are treated with a macrolide drug, which is also—coincidentally and serendipitously—the drug of choice for pertussis. But many remain carriers because they are not accurately diagnosed or never seek care.
“There is a huge reservoir of people carrying pertussis, particularly [in] the adolescent and adult population[s],” says Alison Holmes, MD, a pediatric hospitalist at Concord Hospital, N.H. “And the babies who get really sick from it are the under two- to three-month group who have not yet been immunized or have just been immunized. Because it is so rampant in the adolescent and adult community, those children can still get sick.”
“Unfortunately,” says Dr. Stucky, “what’s happening is that if physicians are not thinking pertussis, they don’t talk about pertussis to that adult patient who … is either around children or has children in the home. So they don’t know to tell that person to watch for these same signs and symptoms in that young infant, who then could have a much more severe outcome from getting [the infection].”
As with most patients who contract illnesses, these patients may never have heard of the disease and unless educated may not understand the implications of the diagnosis. They might realize their disease could spread to family members, “but most people don’t absorb that information and use that information thoughtfully,” says Dr. Stucky. The onus is, therefore, on the physician to warn adult patients specifically about the serious danger that exists for infants in the two- to three-month-old group, who may not have been vaccinated or whose single-vaccination immunity is not adequate protection against the disease.
While the numbers in babies appear to be what they have always been, the incidence has grown in the teen years and even later into adulthood. This is more likely the result of increased testing for pertussis, as opposed to being only due to a true resurgence. Data from studies of adults with prolonged cough revealed that 20% to 25% have serologic evidence of recent pertussis infection.2 Adults are the major reservoir of infection, and infection spreads quickly in a population in a closed environment where droplets spread easily person to person.5
For both teens and adults, testing and immunization with the newly recommended DTaP (diphtheria-tetanus-pertussis)—as opposed to the more limited Td—can help upgrade immunity. Although a patient can recover from pertussis on his or her own within one to two weeks following treatment, the intent of treatment is primarily to limit the spread of disease to others.4-7
The problem when adults get pertussis, says Dr. Holmes, who is also an assistant professor of community and family medicine at Dartmouth Medical School, Hanover, N.H., “is that they often don’t show up complaining about this horrible paroxysmal coughing until they’re about three or four weeks into the illness, and it hasn’t gone away. You go for hours and hours feeling completely fine and wonderful, and why would you bother going to the doctor?”
Babies are most at risk, however. “They often don’t have the energy or the muscle strength, so they just stop breathing instead,” she says.
Mark Dworkin, MD, MPH, TM, the state epidemiologist and team leader for the Rapid Response Team at the Illinois Department of Public Health, is active in outbreak investigation. He wrote a compelling argument for maintaining a high index of suspicion when physicians see adolescent and adult patients who have a cough that has lasted more than two weeks.4
It has been estimated that more than one million cases of pertussis occur in the United States each year; that number has continued to grow for 20 years. From 1990 to 2001, the incidence of pertussis in adults increased by 400%. But many physicians believe that pertussis is only a pediatric illness. A survey of internists in Washington state showed that only 38% of respondents knew about the risk of vaccine fatigue, and just 36% knew that the nasopharyngeal swab is the preferred method for sample collection. Public health professionals were also concerned with the finding that too many pediatricians and nonpediatricians (43% and 41%, respectively) were not able to define a reportable case.
The first challenge that faces internists, writes Dr. Dworkin, is recognizing pertussis, which in some cases presents with mild symptoms; some adults won’t even have a cough.4 But at the other end of the disease spectrum, symptoms may be as brutal as bilateral subconjunctival hemorrhage or rib fracture due to convulsive coughing. In any case, what goes unrecognized, undiagnosed, and untreated becomes a particularly serious risk for vulnerable infants. Once pertussis is identified, positive results on polymerase chain reaction or culture can help convince skeptical colleagues who may still believe pertussis is exclusively a childhood disease—and a vintage one at that.
“What we in pediatrics champion … is for [these immunizations] to help the young child; the less disease we have out there, the better off we’re going to eventually be,” says Dr. Stucky, who projects that, within just a few years, Tdap vaccinations for adolescents and adults up to age 64 might lead to a reduction of infection in the three-month-old group.6
Measles and Mumps
From January 1 to October 7, 2006, 45 states and the District of Columbia reported 5,783 confirmed or probable mumps cases to the Centers for Disease Control and Prevention (CDC). (See Figures 2 and 3, above.)8 The Advisory Committee on Immunization Practices (ACIP) announced that continuing data from surveillance reports meant that healthcare workers should remain alert to suspected cases, conduct appropriate laboratory testing, and use every opportunity to ensure adequate immunity, particularly among populations at high risk.7
In contrast to the circumstances with pertussis, with mumps “there have been pockets of people who have either chosen not to immunize their child[ren], or their child[ren] get exposed to it somehow,” says Dr. Stucky, “and although they might be immunized, they might not have had a good response.” In an environment such as a school, “where one child can cough on a few and then cough on a few [more],” there is an environment where the infection can spread rampantly.
With mumps and measles, these could be called true outbreaks, such as the classic example that occurred in Kansas 18 years ago or the epidemic that disseminated from a college campus in Iowa in the spring of 2006, which originated from only two airline passengers on nine different flights within one week.8
College dorms and cafeterias can be treacherous breeding grounds for pathogens, and this generation of college students is susceptible for a few reasons. For one, in the late 1980s, when they were infants, the vaccine schedule was changed; the measles/mumps/rubella vaccine was upgraded from one dose to two—and not all children received the two doses.
The unimmunized who are exposed to measles and mumps remain at highest risk for spreading the disease. Although in 2005, 76%-79% of children aged 19-35 months received the entire recommended series of shots against whooping cough, diphtheria, tetanus, polio, measles, mumps, rubella, chicken pox, hepatitis B, and Haemophilus influenza type B, that still means that 21%-24% of the children—or potentially one out of five kids—did not.9
Other factors causing low levels of immunization include parents’ Internet-fueled fears of links to autism; immigrants crossing U.S. borders from Mexico or other countries where immunization is not standardized; religious and philosophical reasons; and international travel.10
“When young adults travel internationally [to places] where they are exposed to young children and adults who have never been immunized,” that’s a big risk, says Dr. Stucky. “All it would take is one [infected] student coming into a dorm and passing it around [to others with lapsed coverage or no immunization for the disease].” And while providers may think of travelers being exposed to diseases such as malaria and typhoid fever in developing countries, “in reality, a lot of the common things we’re immunizing for in our country are not immunized for in other countries, and those can be brought back.”
Rickets
The incidence of rickets is increasing, especially in black and Hispanic children and particularly in the north.11,12 Epidemiologists trace the rise to an increase in breast-feeding (good for immunity, but breast milk lacks substantial vitamin D), overuse of sunscreen or lack of exposure to sunlight, and changes in physician recommendations for vitamin supplementation. The effects of rickets alone can be profound, but other long-term consequences of vitamin D deficiency may include type I diabetes, cancer (especially of the prostate), and osteoporosis.12
In the past few decades, physicians have been less likely to recommend vitamin D supplementation for babies, and an interesting study by Davenport and colleagues correlates the year of medical school completion to that decline as well as substantial variability as to the age at which supplement use is begun.12 (See Figures 4a and 4b, left.)
“Most of the cases I have run into have been in [recent] African immigrants, where the mothers stay covered and they are vitamin D deficient,” says Dr. Holmes. “It’s wonderful that they culturally breast-feed, but they come to the U.S., and they’re pretty afraid to go outside in a new society.”
Varicella (Chicken Pox)
Varicella was removed from the CDC’s national notifiable disease list in 1981, but in 1995 a varicella vaccine was recommended for routine childhood vaccination.13 Before the licensure of that vaccine, varicella was a universal childhood disease in the U.S., causing 4 million cases, 11,000 hospitalizations, and 100 deaths every year.14 In 2002, the Council of State and Territorial Epidemiologists recommended that varicella be included in the National Notifiable Surveillance System by 2003 and that case-based surveillance in all states be established by 2005.13 CDC’s ACIP recommended in 2006 that a routine second dose of varicella vaccine be given to children between the ages of four and six years old.
Contracting chicken pox as an adult is a much more morbid occurrence than catching it as a child. Although varicella is not life threatening (as are diphtheria, tetanus, and measles) or sterility-causing (as is mumps), when the vaccine was approved, some pediatricians, including Dr. Stucky, became concerned that “now we’re creating a population that has never seen the wild-type varicella virus, and what does that mean? Were we just delaying something into an age category where people will get sicker?” Recognizing varicella, therefore, is critical even for hospitalists who treat adults.
Conclusion
“I’ve seen mumps, measles, varicella, pertussis,” says Dr. Stucky, “but our adult [hospitalist] partners hadn’t.” She encourages her colleagues who treat adult populations “to read and be diligent. These diseases can exist in adults, or even in children who were once vaccinated, and all hospitalists need to know “what to do, how to treat them, and [that] the consequences in adults are hands down worse than in children.”
Dr. Stucky believes hospitalists who treat adults would do well to consult physicians who practiced in the 1950s because they understand the history as well as clinical signs and symptoms of these diseases; she says, “For the hospitalist who treats adults, these are the equivalent of emerging infectious diseases.” TH
Andrea Sattinger is a frequent contributor to The Hospitalist.
References
- Carmichael M. 'Vintage' bugs return. Newsweek. May 1, 2006:Vol. 147, p. 38. Available at: www.msnbc.msn.com/id/12440796/site/newsweek/. Accessed on November 29, 2006.
- Herwaldt LA. Pertussis in adults. What physicians need to know. Arch Intern Med. 1991;151:1510-1512.
- Schafer S, Gillette H, Hedberg K, et al. A community-wide pertussis outbreak: an argument for universal booster vaccination. Arch Intern Med. 2006 Jun 26;166(12):1317-1321.
- Dworkin MS. Adults are whooping, but are internists listening? Ann Intern Med. 2005 May 17;142(10):832-835. Available at: www.annals.org/cgi/reprint/142/10/832.pdf. Accessed on November 19, 2006.
- Gregory DS. Pertussis: a disease affecting all ages. Am Fam Physician. 2006 Aug 1;74(3):420-426.
- Finger R, Shoemaker J. Preventing pertussis in infants by vaccinating adults. Am Fam Physician. 2006 Aug 1;74(3):382.
- Broder KR, Cortese MM, Iskander JK, et al. Preventing tetanus, diphtheria, and pertussis among adolescents: use of tetanus toxoid, reduced diphtheria toxoid and acellular pertussis vaccines recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2006;55:1-34.
- MMWR. Brief report: update: mumps activity—United States, January 1-October 7, 2006. MMWR. 2006 Oct 27;55(42):1152-1153.
- National Briefing: Science and health: race gap closes in vaccinations, U.S. says. New York Times. September 15, 2006.
- Calandrillo SP. Vanishing vaccinations: why are so many Americans opting out of vaccinating their children? Univ Mich J Law Reform. 2004 Winter;37(2):353-440.
- Kreiter SR, Schwartz RP, Kirkman HN Jr, et al. Nutritional rickets in African American breast-fed infants. J Pediatr. 2000 Aug;137(2):153-157.
- Davenport ML, Uckun A, Calikoglu AS. Pediatrician patterns of prescribing vitamin supplementation for infants: do they contribute to rickets? Pediatrics. 2004 Jan;113(1 Pt 1):179-180.
- MMWR. Varicella surveillance practices—United States, 2004. MMWR. 2006 Oct 19;55:1126-1129.
- Seward JF, Watson BM, Peterson CL, et al. Varicella disease after introduction of varicella vaccine in the United States, 1995-2000. JAMA. 2002 Feb 6;287(5):606-611.
We thought they were gone, but they’ve returned: diseases once considered “vintage bugs” that were common in as late as the mid-20th century. In the past these diseases killed one in three people younger than 20 who had survived an infancy during which many of their contemporaries died.1
“When you think about disease states, you think about some that are gone from the world,” says Erin Stucky, MD, a pediatric hospitalist at the University of California, San Diego, “but there are very few truly gone from the world.”
Some of the major infectious diseases that hospitalists may [still] see are pertussis (whooping cough), measles, and mumps, but scarlet fever and varicella (chicken pox) also endure—not to mention those occurrences of polio around the country that epidemiologists and infectious diseases specialists are monitoring closely. Rickets, a vitamin-D-deficiency-related disease also thought to be a relic of the 18th century, is showing up in certain patient populations—and not exclusively in infants and children.
This is a crossover clinical issue, our pediatric hospitalists say, and thus one to which their hospitalist partners who treat adult patients must also remain alert.
Pertussis (Whooping Cough)
Despite vaccination protocols, pediatric hospitalists continue to see whooping cough in young infants. (See Figure 1, p. 39.) Even with treatment, the damage can be severe, and the length of stay (LOS) is prolonged compared with those of most other patients with complex illnesses. “Vaccine fatigue” means that immunization lasts only until adolescence or early adulthood, at which time they need appropriate boosters. If the patient hasn’t receive boosters, the initial immunization loses its effectiveness; unprotected, they can be infected with the disease, though sometimes not badly enough for them to seek care. When they do, the diagnosis is often community-acquired mild pneumonia or a more traditional bronchitis. Either by accident or because the physician has given it thought, those illnesses are treated with a macrolide drug, which is also—coincidentally and serendipitously—the drug of choice for pertussis. But many remain carriers because they are not accurately diagnosed or never seek care.
“There is a huge reservoir of people carrying pertussis, particularly [in] the adolescent and adult population[s],” says Alison Holmes, MD, a pediatric hospitalist at Concord Hospital, N.H. “And the babies who get really sick from it are the under two- to three-month group who have not yet been immunized or have just been immunized. Because it is so rampant in the adolescent and adult community, those children can still get sick.”
“Unfortunately,” says Dr. Stucky, “what’s happening is that if physicians are not thinking pertussis, they don’t talk about pertussis to that adult patient who … is either around children or has children in the home. So they don’t know to tell that person to watch for these same signs and symptoms in that young infant, who then could have a much more severe outcome from getting [the infection].”
As with most patients who contract illnesses, these patients may never have heard of the disease and unless educated may not understand the implications of the diagnosis. They might realize their disease could spread to family members, “but most people don’t absorb that information and use that information thoughtfully,” says Dr. Stucky. The onus is, therefore, on the physician to warn adult patients specifically about the serious danger that exists for infants in the two- to three-month-old group, who may not have been vaccinated or whose single-vaccination immunity is not adequate protection against the disease.
While the numbers in babies appear to be what they have always been, the incidence has grown in the teen years and even later into adulthood. This is more likely the result of increased testing for pertussis, as opposed to being only due to a true resurgence. Data from studies of adults with prolonged cough revealed that 20% to 25% have serologic evidence of recent pertussis infection.2 Adults are the major reservoir of infection, and infection spreads quickly in a population in a closed environment where droplets spread easily person to person.5
For both teens and adults, testing and immunization with the newly recommended DTaP (diphtheria-tetanus-pertussis)—as opposed to the more limited Td—can help upgrade immunity. Although a patient can recover from pertussis on his or her own within one to two weeks following treatment, the intent of treatment is primarily to limit the spread of disease to others.4-7
The problem when adults get pertussis, says Dr. Holmes, who is also an assistant professor of community and family medicine at Dartmouth Medical School, Hanover, N.H., “is that they often don’t show up complaining about this horrible paroxysmal coughing until they’re about three or four weeks into the illness, and it hasn’t gone away. You go for hours and hours feeling completely fine and wonderful, and why would you bother going to the doctor?”
Babies are most at risk, however. “They often don’t have the energy or the muscle strength, so they just stop breathing instead,” she says.
Mark Dworkin, MD, MPH, TM, the state epidemiologist and team leader for the Rapid Response Team at the Illinois Department of Public Health, is active in outbreak investigation. He wrote a compelling argument for maintaining a high index of suspicion when physicians see adolescent and adult patients who have a cough that has lasted more than two weeks.4
It has been estimated that more than one million cases of pertussis occur in the United States each year; that number has continued to grow for 20 years. From 1990 to 2001, the incidence of pertussis in adults increased by 400%. But many physicians believe that pertussis is only a pediatric illness. A survey of internists in Washington state showed that only 38% of respondents knew about the risk of vaccine fatigue, and just 36% knew that the nasopharyngeal swab is the preferred method for sample collection. Public health professionals were also concerned with the finding that too many pediatricians and nonpediatricians (43% and 41%, respectively) were not able to define a reportable case.
The first challenge that faces internists, writes Dr. Dworkin, is recognizing pertussis, which in some cases presents with mild symptoms; some adults won’t even have a cough.4 But at the other end of the disease spectrum, symptoms may be as brutal as bilateral subconjunctival hemorrhage or rib fracture due to convulsive coughing. In any case, what goes unrecognized, undiagnosed, and untreated becomes a particularly serious risk for vulnerable infants. Once pertussis is identified, positive results on polymerase chain reaction or culture can help convince skeptical colleagues who may still believe pertussis is exclusively a childhood disease—and a vintage one at that.
“What we in pediatrics champion … is for [these immunizations] to help the young child; the less disease we have out there, the better off we’re going to eventually be,” says Dr. Stucky, who projects that, within just a few years, Tdap vaccinations for adolescents and adults up to age 64 might lead to a reduction of infection in the three-month-old group.6
Measles and Mumps
From January 1 to October 7, 2006, 45 states and the District of Columbia reported 5,783 confirmed or probable mumps cases to the Centers for Disease Control and Prevention (CDC). (See Figures 2 and 3, above.)8 The Advisory Committee on Immunization Practices (ACIP) announced that continuing data from surveillance reports meant that healthcare workers should remain alert to suspected cases, conduct appropriate laboratory testing, and use every opportunity to ensure adequate immunity, particularly among populations at high risk.7
In contrast to the circumstances with pertussis, with mumps “there have been pockets of people who have either chosen not to immunize their child[ren], or their child[ren] get exposed to it somehow,” says Dr. Stucky, “and although they might be immunized, they might not have had a good response.” In an environment such as a school, “where one child can cough on a few and then cough on a few [more],” there is an environment where the infection can spread rampantly.
With mumps and measles, these could be called true outbreaks, such as the classic example that occurred in Kansas 18 years ago or the epidemic that disseminated from a college campus in Iowa in the spring of 2006, which originated from only two airline passengers on nine different flights within one week.8
College dorms and cafeterias can be treacherous breeding grounds for pathogens, and this generation of college students is susceptible for a few reasons. For one, in the late 1980s, when they were infants, the vaccine schedule was changed; the measles/mumps/rubella vaccine was upgraded from one dose to two—and not all children received the two doses.
The unimmunized who are exposed to measles and mumps remain at highest risk for spreading the disease. Although in 2005, 76%-79% of children aged 19-35 months received the entire recommended series of shots against whooping cough, diphtheria, tetanus, polio, measles, mumps, rubella, chicken pox, hepatitis B, and Haemophilus influenza type B, that still means that 21%-24% of the children—or potentially one out of five kids—did not.9
Other factors causing low levels of immunization include parents’ Internet-fueled fears of links to autism; immigrants crossing U.S. borders from Mexico or other countries where immunization is not standardized; religious and philosophical reasons; and international travel.10
“When young adults travel internationally [to places] where they are exposed to young children and adults who have never been immunized,” that’s a big risk, says Dr. Stucky. “All it would take is one [infected] student coming into a dorm and passing it around [to others with lapsed coverage or no immunization for the disease].” And while providers may think of travelers being exposed to diseases such as malaria and typhoid fever in developing countries, “in reality, a lot of the common things we’re immunizing for in our country are not immunized for in other countries, and those can be brought back.”
Rickets
The incidence of rickets is increasing, especially in black and Hispanic children and particularly in the north.11,12 Epidemiologists trace the rise to an increase in breast-feeding (good for immunity, but breast milk lacks substantial vitamin D), overuse of sunscreen or lack of exposure to sunlight, and changes in physician recommendations for vitamin supplementation. The effects of rickets alone can be profound, but other long-term consequences of vitamin D deficiency may include type I diabetes, cancer (especially of the prostate), and osteoporosis.12
In the past few decades, physicians have been less likely to recommend vitamin D supplementation for babies, and an interesting study by Davenport and colleagues correlates the year of medical school completion to that decline as well as substantial variability as to the age at which supplement use is begun.12 (See Figures 4a and 4b, left.)
“Most of the cases I have run into have been in [recent] African immigrants, where the mothers stay covered and they are vitamin D deficient,” says Dr. Holmes. “It’s wonderful that they culturally breast-feed, but they come to the U.S., and they’re pretty afraid to go outside in a new society.”
Varicella (Chicken Pox)
Varicella was removed from the CDC’s national notifiable disease list in 1981, but in 1995 a varicella vaccine was recommended for routine childhood vaccination.13 Before the licensure of that vaccine, varicella was a universal childhood disease in the U.S., causing 4 million cases, 11,000 hospitalizations, and 100 deaths every year.14 In 2002, the Council of State and Territorial Epidemiologists recommended that varicella be included in the National Notifiable Surveillance System by 2003 and that case-based surveillance in all states be established by 2005.13 CDC’s ACIP recommended in 2006 that a routine second dose of varicella vaccine be given to children between the ages of four and six years old.
Contracting chicken pox as an adult is a much more morbid occurrence than catching it as a child. Although varicella is not life threatening (as are diphtheria, tetanus, and measles) or sterility-causing (as is mumps), when the vaccine was approved, some pediatricians, including Dr. Stucky, became concerned that “now we’re creating a population that has never seen the wild-type varicella virus, and what does that mean? Were we just delaying something into an age category where people will get sicker?” Recognizing varicella, therefore, is critical even for hospitalists who treat adults.
Conclusion
“I’ve seen mumps, measles, varicella, pertussis,” says Dr. Stucky, “but our adult [hospitalist] partners hadn’t.” She encourages her colleagues who treat adult populations “to read and be diligent. These diseases can exist in adults, or even in children who were once vaccinated, and all hospitalists need to know “what to do, how to treat them, and [that] the consequences in adults are hands down worse than in children.”
Dr. Stucky believes hospitalists who treat adults would do well to consult physicians who practiced in the 1950s because they understand the history as well as clinical signs and symptoms of these diseases; she says, “For the hospitalist who treats adults, these are the equivalent of emerging infectious diseases.” TH
Andrea Sattinger is a frequent contributor to The Hospitalist.
References
- Carmichael M. 'Vintage' bugs return. Newsweek. May 1, 2006:Vol. 147, p. 38. Available at: www.msnbc.msn.com/id/12440796/site/newsweek/. Accessed on November 29, 2006.
- Herwaldt LA. Pertussis in adults. What physicians need to know. Arch Intern Med. 1991;151:1510-1512.
- Schafer S, Gillette H, Hedberg K, et al. A community-wide pertussis outbreak: an argument for universal booster vaccination. Arch Intern Med. 2006 Jun 26;166(12):1317-1321.
- Dworkin MS. Adults are whooping, but are internists listening? Ann Intern Med. 2005 May 17;142(10):832-835. Available at: www.annals.org/cgi/reprint/142/10/832.pdf. Accessed on November 19, 2006.
- Gregory DS. Pertussis: a disease affecting all ages. Am Fam Physician. 2006 Aug 1;74(3):420-426.
- Finger R, Shoemaker J. Preventing pertussis in infants by vaccinating adults. Am Fam Physician. 2006 Aug 1;74(3):382.
- Broder KR, Cortese MM, Iskander JK, et al. Preventing tetanus, diphtheria, and pertussis among adolescents: use of tetanus toxoid, reduced diphtheria toxoid and acellular pertussis vaccines recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2006;55:1-34.
- MMWR. Brief report: update: mumps activity—United States, January 1-October 7, 2006. MMWR. 2006 Oct 27;55(42):1152-1153.
- National Briefing: Science and health: race gap closes in vaccinations, U.S. says. New York Times. September 15, 2006.
- Calandrillo SP. Vanishing vaccinations: why are so many Americans opting out of vaccinating their children? Univ Mich J Law Reform. 2004 Winter;37(2):353-440.
- Kreiter SR, Schwartz RP, Kirkman HN Jr, et al. Nutritional rickets in African American breast-fed infants. J Pediatr. 2000 Aug;137(2):153-157.
- Davenport ML, Uckun A, Calikoglu AS. Pediatrician patterns of prescribing vitamin supplementation for infants: do they contribute to rickets? Pediatrics. 2004 Jan;113(1 Pt 1):179-180.
- MMWR. Varicella surveillance practices—United States, 2004. MMWR. 2006 Oct 19;55:1126-1129.
- Seward JF, Watson BM, Peterson CL, et al. Varicella disease after introduction of varicella vaccine in the United States, 1995-2000. JAMA. 2002 Feb 6;287(5):606-611.
Coming to a Hospital Near You: E-prescribe

Coming to a Hospital Near You: E-prescribe
Will hospitalists and hospital medicine groups get to participate in the Centers for Medicare and Medicaid e-prescribe program?
Jettie Eddleman, BSN, RN, Quality Initiatives Program Director North Texas Specialty Physicians (NTSP), Fort Worth, Texas
Dr. Hospitalist responds: According to a December 2007 study by SureScripts, only 6% of U.S. physicians prescribe medications electronically. Medicare would like more physicians to electronically prescribe prescriptions because “e-prescribing is more efficient and convenient for consumers, improves the quality of care, lowers administrative costs, and its widespread use would eliminate thousands of medication errors every year.”
Medicare is not the only organization encouraging e-prescribing. Blue Cross Blue Shield of Massachusetts (BCBSMA) recently announced e-prescribing will be required for any physician to participate in any of the BCBSMA physician incentive programs, effective January, 2011. To speed the adoption of e-prescribing, Medicare will provide financial incentives to physicians who e-prescribe. Starting in this year, Medicare will pay a 2% bonus to physicians who prescribe under Part D. This incentive bonus will decrease to 1% in 2011 and 0.5% in 2013. Starting in 2012, physicians who are not e-prescribing will lose 1% of their Medicare payment. This penalty will increase to 1.5% in 2013 and 2% in 2014. In other words, you can e-prescribe sooner or later, but hospital medicine groups will increase revenue if they start sooner.
Do you have a problem or concern that you’d like Dr. Hospitalist to address? E-mail your questions to [email protected].
E-prescribing is, however, not without barriers. For example, the Drug Enforcement Agency (DEA) presently prohibits e-prescription of controlled substances. Medicare is working with the DEA to address this issue. In the meantime, providers who implement e-prescription will need to continue a separate system for prescription of controlled substances.
Some physicians have electronic medical record (EMR) systems, which send prescriptions to pharmacies via facsimile machines. For clarification, this is not considered e-prescribing. In fact, under Medicare statute, most EMR-faxed prescriptions no longer are allowed for Part D. As the plan currently stands, this incentive is tied to specific Current Procedural Terminology (CPT) codes, used primarily by primary care providers and not by hospitalists. Unless the hospitalists in your group also provide outpatient care, it is unlikely any will be able to participate in this Medicare e-prescribing incentive plan.
Test Results Post-Discharge
Some of my patients have laboratory test results pending at the time of discharge from the hospital. Most of my patients do not have a routine outpatient provider. At the time of discharge, I always set them up with outpatient follow-up with a new primary care provider, but I have no way of knowing if the patients are showing up at their appointments. My concern is if they don’t show up, they won’t know about their test results. Is this something I need to address? If so, how do you suggest I go about doing it?
Z. Taylor, Durant, Okla.
Dr. Hospitalist responds: This definitely is an issue that needs to be addressed. It is not only a quality of care issue, but also potentially a medical/legal issue. Transitions in care are risky for patients because these are periods with increased risk for medical error. For example, this is why the Joint Commission mandates medication reconciliation each time a patient sees a provider.
When patients are discharged from the hospital, which provider is responsible for notifying the patient of pending laboratory test results? Is it the primary care provider (PCP) or the hospitalist? As you described in your question, what if the patient does not have a regular PCP? Does the responsibility then rest with the hospitalist who discharged the patient? One could argue the physician who ordered the test is responsible. What if the hospitalist who ordered the test is not the same hospitalist who discharged the patient? It would seem under that circumstance, the hospitalist who discharged the patient bears more responsibility than the hospitalist who ordered the study.
To further complicate matters, what if the physician who ordered the test was a consultant? I am not aware of any rules specifying provider responsibility for notifying the patient. I typically recommend the hospitalist in charge of discharging the patient from the hospital make a practice of looking for studies whose results are pending at the time of discharge. The hospitalist should inform the patient the results are pending and discuss a plan of action for the patient to get the study results. Then I suggest the hospitalist document this discussion and plan/forward this documentation to the provider who is scheduled to see the patient in follow up. It is typically easier for hospitalists to include this information as part of the discharge summary sent to the PCP.
As you suggested, these steps may be insufficient when the patient does not follow up with the designated PCP. For that reason, it is necessary for the hospitalist who discharged the patient to follow up on these pending results. The hospitalist must notify the patient if the results are abnormal. To do this, prior to hospital discharge, one needs to know how to contact a patient post-discharge. Always document the fact you have notified the patient of the abnormal result. I recognize this type of follow up is not easy after a patient is discharged, especially when most results will return as normal studies. The volume-to-noise ratio is not great. But it is that one out of 100 abnormal result that will end up hurting the patient and potentially result in litigation.
One important piece of advice: Only order necessary tests. The fewer tests you order, the less it is likely you will have test results pending at discharge. If a test is not likely to change how you manage a patient during their inpatient stay, consider not ordering the test. Such practice is not only more cost-effective care, but also simplifies the system and minimizes the risk of error associated with notifying patients of abnormal test results.
A Little Common Courtesy, Please
I find it incredibly annoying when we are holding a staff meeting and some of my colleagues are checking e-mail on their Blackberry. At the risk of sounding like a codger, is it too much to ask for some common courtesy?
K. Moore, Austin, Texas
Dr. Hospitalist responds: You are, of course, correct at pointing out it is rude for people to check messages during meetings, not to mention anytime a supervisor or colleague is speaking. Do I condone the behavior? No. Do I understand the behavior? Yes. (In the spirit of full disclosure, I am addicted to my Blackberry and, occasionally, am guilty of checking for messages when I should be paying attention).
We live in an information age and the expectation for communication is greater than ever. As hospitalists, we know this all too well. For many of us, the Blackberry affords us the opportunity to multitask, shaving minutes or hours off our workday. I agree it is not an unreasonable request to ask everyone to turn off their cell phones and put down their Blackberrys during meetings.
That said, doing without the Blackberry for much longer than an hour or two is not an option for many of us.
Please note President-elect Obama will have to ditch his Blackberry this month, if not sooner, due to concerns surrounding e-mail privacy. He also is subject to the Presidential Records Act, which eliminates any privacy regarding this correspondence. (Memo to self, another reason not to run for president.) TH
Coming to a Hospital Near You: E-prescribe
Will hospitalists and hospital medicine groups get to participate in the Centers for Medicare and Medicaid e-prescribe program?
Jettie Eddleman, BSN, RN, Quality Initiatives Program Director North Texas Specialty Physicians (NTSP), Fort Worth, Texas
Dr. Hospitalist responds: According to a December 2007 study by SureScripts, only 6% of U.S. physicians prescribe medications electronically. Medicare would like more physicians to electronically prescribe prescriptions because “e-prescribing is more efficient and convenient for consumers, improves the quality of care, lowers administrative costs, and its widespread use would eliminate thousands of medication errors every year.”
Medicare is not the only organization encouraging e-prescribing. Blue Cross Blue Shield of Massachusetts (BCBSMA) recently announced e-prescribing will be required for any physician to participate in any of the BCBSMA physician incentive programs, effective January, 2011. To speed the adoption of e-prescribing, Medicare will provide financial incentives to physicians who e-prescribe. Starting in this year, Medicare will pay a 2% bonus to physicians who prescribe under Part D. This incentive bonus will decrease to 1% in 2011 and 0.5% in 2013. Starting in 2012, physicians who are not e-prescribing will lose 1% of their Medicare payment. This penalty will increase to 1.5% in 2013 and 2% in 2014. In other words, you can e-prescribe sooner or later, but hospital medicine groups will increase revenue if they start sooner.
Do you have a problem or concern that you’d like Dr. Hospitalist to address? E-mail your questions to [email protected].
E-prescribing is, however, not without barriers. For example, the Drug Enforcement Agency (DEA) presently prohibits e-prescription of controlled substances. Medicare is working with the DEA to address this issue. In the meantime, providers who implement e-prescription will need to continue a separate system for prescription of controlled substances.
Some physicians have electronic medical record (EMR) systems, which send prescriptions to pharmacies via facsimile machines. For clarification, this is not considered e-prescribing. In fact, under Medicare statute, most EMR-faxed prescriptions no longer are allowed for Part D. As the plan currently stands, this incentive is tied to specific Current Procedural Terminology (CPT) codes, used primarily by primary care providers and not by hospitalists. Unless the hospitalists in your group also provide outpatient care, it is unlikely any will be able to participate in this Medicare e-prescribing incentive plan.
Test Results Post-Discharge
Some of my patients have laboratory test results pending at the time of discharge from the hospital. Most of my patients do not have a routine outpatient provider. At the time of discharge, I always set them up with outpatient follow-up with a new primary care provider, but I have no way of knowing if the patients are showing up at their appointments. My concern is if they don’t show up, they won’t know about their test results. Is this something I need to address? If so, how do you suggest I go about doing it?
Z. Taylor, Durant, Okla.
Dr. Hospitalist responds: This definitely is an issue that needs to be addressed. It is not only a quality of care issue, but also potentially a medical/legal issue. Transitions in care are risky for patients because these are periods with increased risk for medical error. For example, this is why the Joint Commission mandates medication reconciliation each time a patient sees a provider.
When patients are discharged from the hospital, which provider is responsible for notifying the patient of pending laboratory test results? Is it the primary care provider (PCP) or the hospitalist? As you described in your question, what if the patient does not have a regular PCP? Does the responsibility then rest with the hospitalist who discharged the patient? One could argue the physician who ordered the test is responsible. What if the hospitalist who ordered the test is not the same hospitalist who discharged the patient? It would seem under that circumstance, the hospitalist who discharged the patient bears more responsibility than the hospitalist who ordered the study.
To further complicate matters, what if the physician who ordered the test was a consultant? I am not aware of any rules specifying provider responsibility for notifying the patient. I typically recommend the hospitalist in charge of discharging the patient from the hospital make a practice of looking for studies whose results are pending at the time of discharge. The hospitalist should inform the patient the results are pending and discuss a plan of action for the patient to get the study results. Then I suggest the hospitalist document this discussion and plan/forward this documentation to the provider who is scheduled to see the patient in follow up. It is typically easier for hospitalists to include this information as part of the discharge summary sent to the PCP.
As you suggested, these steps may be insufficient when the patient does not follow up with the designated PCP. For that reason, it is necessary for the hospitalist who discharged the patient to follow up on these pending results. The hospitalist must notify the patient if the results are abnormal. To do this, prior to hospital discharge, one needs to know how to contact a patient post-discharge. Always document the fact you have notified the patient of the abnormal result. I recognize this type of follow up is not easy after a patient is discharged, especially when most results will return as normal studies. The volume-to-noise ratio is not great. But it is that one out of 100 abnormal result that will end up hurting the patient and potentially result in litigation.
One important piece of advice: Only order necessary tests. The fewer tests you order, the less it is likely you will have test results pending at discharge. If a test is not likely to change how you manage a patient during their inpatient stay, consider not ordering the test. Such practice is not only more cost-effective care, but also simplifies the system and minimizes the risk of error associated with notifying patients of abnormal test results.
A Little Common Courtesy, Please
I find it incredibly annoying when we are holding a staff meeting and some of my colleagues are checking e-mail on their Blackberry. At the risk of sounding like a codger, is it too much to ask for some common courtesy?
K. Moore, Austin, Texas
Dr. Hospitalist responds: You are, of course, correct at pointing out it is rude for people to check messages during meetings, not to mention anytime a supervisor or colleague is speaking. Do I condone the behavior? No. Do I understand the behavior? Yes. (In the spirit of full disclosure, I am addicted to my Blackberry and, occasionally, am guilty of checking for messages when I should be paying attention).
We live in an information age and the expectation for communication is greater than ever. As hospitalists, we know this all too well. For many of us, the Blackberry affords us the opportunity to multitask, shaving minutes or hours off our workday. I agree it is not an unreasonable request to ask everyone to turn off their cell phones and put down their Blackberrys during meetings.
That said, doing without the Blackberry for much longer than an hour or two is not an option for many of us.
Please note President-elect Obama will have to ditch his Blackberry this month, if not sooner, due to concerns surrounding e-mail privacy. He also is subject to the Presidential Records Act, which eliminates any privacy regarding this correspondence. (Memo to self, another reason not to run for president.) TH
Coming to a Hospital Near You: E-prescribe
Will hospitalists and hospital medicine groups get to participate in the Centers for Medicare and Medicaid e-prescribe program?
Jettie Eddleman, BSN, RN, Quality Initiatives Program Director North Texas Specialty Physicians (NTSP), Fort Worth, Texas
Dr. Hospitalist responds: According to a December 2007 study by SureScripts, only 6% of U.S. physicians prescribe medications electronically. Medicare would like more physicians to electronically prescribe prescriptions because “e-prescribing is more efficient and convenient for consumers, improves the quality of care, lowers administrative costs, and its widespread use would eliminate thousands of medication errors every year.”
Medicare is not the only organization encouraging e-prescribing. Blue Cross Blue Shield of Massachusetts (BCBSMA) recently announced e-prescribing will be required for any physician to participate in any of the BCBSMA physician incentive programs, effective January, 2011. To speed the adoption of e-prescribing, Medicare will provide financial incentives to physicians who e-prescribe. Starting in this year, Medicare will pay a 2% bonus to physicians who prescribe under Part D. This incentive bonus will decrease to 1% in 2011 and 0.5% in 2013. Starting in 2012, physicians who are not e-prescribing will lose 1% of their Medicare payment. This penalty will increase to 1.5% in 2013 and 2% in 2014. In other words, you can e-prescribe sooner or later, but hospital medicine groups will increase revenue if they start sooner.
Do you have a problem or concern that you’d like Dr. Hospitalist to address? E-mail your questions to [email protected].
E-prescribing is, however, not without barriers. For example, the Drug Enforcement Agency (DEA) presently prohibits e-prescription of controlled substances. Medicare is working with the DEA to address this issue. In the meantime, providers who implement e-prescription will need to continue a separate system for prescription of controlled substances.
Some physicians have electronic medical record (EMR) systems, which send prescriptions to pharmacies via facsimile machines. For clarification, this is not considered e-prescribing. In fact, under Medicare statute, most EMR-faxed prescriptions no longer are allowed for Part D. As the plan currently stands, this incentive is tied to specific Current Procedural Terminology (CPT) codes, used primarily by primary care providers and not by hospitalists. Unless the hospitalists in your group also provide outpatient care, it is unlikely any will be able to participate in this Medicare e-prescribing incentive plan.
Test Results Post-Discharge
Some of my patients have laboratory test results pending at the time of discharge from the hospital. Most of my patients do not have a routine outpatient provider. At the time of discharge, I always set them up with outpatient follow-up with a new primary care provider, but I have no way of knowing if the patients are showing up at their appointments. My concern is if they don’t show up, they won’t know about their test results. Is this something I need to address? If so, how do you suggest I go about doing it?
Z. Taylor, Durant, Okla.
Dr. Hospitalist responds: This definitely is an issue that needs to be addressed. It is not only a quality of care issue, but also potentially a medical/legal issue. Transitions in care are risky for patients because these are periods with increased risk for medical error. For example, this is why the Joint Commission mandates medication reconciliation each time a patient sees a provider.
When patients are discharged from the hospital, which provider is responsible for notifying the patient of pending laboratory test results? Is it the primary care provider (PCP) or the hospitalist? As you described in your question, what if the patient does not have a regular PCP? Does the responsibility then rest with the hospitalist who discharged the patient? One could argue the physician who ordered the test is responsible. What if the hospitalist who ordered the test is not the same hospitalist who discharged the patient? It would seem under that circumstance, the hospitalist who discharged the patient bears more responsibility than the hospitalist who ordered the study.
To further complicate matters, what if the physician who ordered the test was a consultant? I am not aware of any rules specifying provider responsibility for notifying the patient. I typically recommend the hospitalist in charge of discharging the patient from the hospital make a practice of looking for studies whose results are pending at the time of discharge. The hospitalist should inform the patient the results are pending and discuss a plan of action for the patient to get the study results. Then I suggest the hospitalist document this discussion and plan/forward this documentation to the provider who is scheduled to see the patient in follow up. It is typically easier for hospitalists to include this information as part of the discharge summary sent to the PCP.
As you suggested, these steps may be insufficient when the patient does not follow up with the designated PCP. For that reason, it is necessary for the hospitalist who discharged the patient to follow up on these pending results. The hospitalist must notify the patient if the results are abnormal. To do this, prior to hospital discharge, one needs to know how to contact a patient post-discharge. Always document the fact you have notified the patient of the abnormal result. I recognize this type of follow up is not easy after a patient is discharged, especially when most results will return as normal studies. The volume-to-noise ratio is not great. But it is that one out of 100 abnormal result that will end up hurting the patient and potentially result in litigation.
One important piece of advice: Only order necessary tests. The fewer tests you order, the less it is likely you will have test results pending at discharge. If a test is not likely to change how you manage a patient during their inpatient stay, consider not ordering the test. Such practice is not only more cost-effective care, but also simplifies the system and minimizes the risk of error associated with notifying patients of abnormal test results.
A Little Common Courtesy, Please
I find it incredibly annoying when we are holding a staff meeting and some of my colleagues are checking e-mail on their Blackberry. At the risk of sounding like a codger, is it too much to ask for some common courtesy?
K. Moore, Austin, Texas
Dr. Hospitalist responds: You are, of course, correct at pointing out it is rude for people to check messages during meetings, not to mention anytime a supervisor or colleague is speaking. Do I condone the behavior? No. Do I understand the behavior? Yes. (In the spirit of full disclosure, I am addicted to my Blackberry and, occasionally, am guilty of checking for messages when I should be paying attention).
We live in an information age and the expectation for communication is greater than ever. As hospitalists, we know this all too well. For many of us, the Blackberry affords us the opportunity to multitask, shaving minutes or hours off our workday. I agree it is not an unreasonable request to ask everyone to turn off their cell phones and put down their Blackberrys during meetings.
That said, doing without the Blackberry for much longer than an hour or two is not an option for many of us.
Please note President-elect Obama will have to ditch his Blackberry this month, if not sooner, due to concerns surrounding e-mail privacy. He also is subject to the Presidential Records Act, which eliminates any privacy regarding this correspondence. (Memo to self, another reason not to run for president.) TH
Satisfaction Scorecard

Patient satisfaction became a much more important issue earlier in 2008 when hospitals began reporting their performance on the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey. The 18-item survey was developed by the Centers for Medicare and Medicaid Services (CMS) and the Agency for Healthcare Research and Quality, and is completed by patients to record their level of satisfaction with the hospital and the care they received.
If you don’t know your hospital’s publicly reported HCAHPS scores, you should make it your business to become familiar with them. Survey results from participating hospitals are updated periodically and available at www.hospitalcompare.hhs.gov.
I regularly hear hospitalists say they could more effectively direct their energy and resources in ways consistent with their hospital CEO’s goals, if they only knew what those goals were. If you find yourself in the same category, then you should know there is a really good chance the upper-level hospital leadership has a salary bonus in place for achieving certain patient safety and satisfaction goals. A recent survey in the Journal of Patient Safety showed this is the case for more than half of the nation’s hospital executives.1 Therefore, learning more about your hospital’s patient satisfaction scores, and your group’s contribution to the score, is worth the effort.
The publicly reported HCAHPS data does not address hospitalists’ effect on patient satisfaction separately from other doctors and hospital attributes. As a result, some hospitalist groups conduct their own survey. This may range from a very brief series of written questions, or a more-involved survey administered via phone. Since it is difficult to drill down to patient satisfaction with individual hospitalists, data from these surveys often times are collected for the entire hospitalist practice in aggregate, rather than by the individual doctor.
Incorporate Information
Investigate both internal and external resources, if you believe your group could benefit from an intervention (e.g., education or training to improve patient satisfaction). Many hospitals have someone on staff to provide employee training in this area; the trainer probably would be impressed and pleased if you initiate contact to learn more about available training. Resources external to your hospital include survey companies (e.g., Press Ganey, NCR Picker, among others). These companies can provide training and guidance, or put you in contact with firms with whom they work. I have seen something as simple as a one- or two-hour online or DVD course be a valuable tool for some practices.
I know some very efficient hospitalists who maintain incredibly high levels of patient satisfaction despite very high workloads. I think these doctors probably are outliers, and most of us will see satisfaction scores decline as workloads become unreasonably high. Each group should challenge itself continually to find the optimal point between patient volume and important outcomes like patient satisfaction.
Here are some strategies hospitalist groups could use to improve patient satisfaction:
- Call patients after discharge. This is a potent way to improve patient satisfaction. It is a valuable clinical encounter, since it offers a chance to reinforce discharge instructions (e.g, the value of smoking cessation, or the need to have ongoing INR monitoring), and to address any issues arising in the interim. The calls are most effective if done by the doctor who discharged the patient, but can be of some value if made by a nurse or other person connected to the practice. Note, it is best not to inform patients you will be making this call, since it will reduce the surprise and pleasure when the call comes, and could lead to frustration if a patient never gets the call they were told to expect.
- Provide referring doctors, emergency room doctors, and nurses at the hospital with a “script” to use when introducing or describing the hospitalist practice to patients. Without coaching, most of these people likely will say to the patient something like “your doctor [primary care physician] doesn’t come to the hospital anymore, so the hospitalist will see you.” Such a description may make the patient feel like they’re getting a second-class doctor. Instead, encourage these workers to say something like “Your doctor has decided to focus her practice on the office, to be more available to you there. As a result, she has decided to refer you to Dr. McCartney, a doctor who specializes in the care of hospitalized patients with problems like yours. Dr. McCartney will communicate with your primary doctor, and you should plan to follow up with her when you are discharged.”
- Communicate regularly, even daily, with family members of patients who have dementia or other cognitive impairment. This usually means calling the family.
- During initial and subsequent visits with a patient, shake hands or gently touch the patient in some way. Sit down and, while still sitting, conclude the conversation by asking if the patient has any questions and asking “Is there anything special I can do for you today?”
- Provide patients with a copy of their discharge summary. It can serve as a valuable education tool for the patient, their loved ones, visiting nurses, etc. Ideally, the summary should be transcribed on a stat basis, so it can be available for the nurse to give to the patient on the way out of the hospital. Alternatively, it could be mailed to the patient later.
- Track your ongoing satisfaction performance by regularly including available data from HCAHPS and/or other sources in the practice dashboard or report card.
- Ensure patients and their families are provided a copy of your group’s brochure. Brochures delivered to primary care offices rarely find their way into the hands of the patients, and often times are forgotten or misplaced by the time hospital care is needed. It usually is more effective to ensure hospital staff provides the brochure. This could be done via a standing protocol stating that a clerical person will provide all patients with a hospitalist as attending or consultant will get a copy. Or, ensure an order to this effect is written on every one of your patients. It’s best to do this via pre-printed order sets.
If you don’t have a group brochure, develop one. It should include the name of the group with a one- or two-line biography and photograph for each hospitalist (e.g., where they attended medical school and residency), and other key information about the practice. If you’d like a sample brochure, visit www.hospitalmedicine.org and search “sample brochure for hospitalist program.”
Each hospitalist in your group could carry business cards with the doctor’s picture next to their name and key info. One small study showed this enhanced patients’ ability to correctly identify their hospitalist and presumably increased their satisfaction, as well.
- Maximize hospitalist-patient continuity. Each group should adjust their work schedule to maximize continuity between patient and hospitalist while still providing a sustainable lifestyle. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is co-founder and past president of SHM. He is a principal in Nelson/Flores Associates, a national hospitalist practice management consulting firm. He is part of the faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official SHM position..
Reference
1. Vaughn T, Koepke M, Kroch E, et al. Engagement of leadership in quality improvement initiatives: executive quality improvement survey results. J Patient Saf. 2006;2(1):2-9.
Patient satisfaction became a much more important issue earlier in 2008 when hospitals began reporting their performance on the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey. The 18-item survey was developed by the Centers for Medicare and Medicaid Services (CMS) and the Agency for Healthcare Research and Quality, and is completed by patients to record their level of satisfaction with the hospital and the care they received.
If you don’t know your hospital’s publicly reported HCAHPS scores, you should make it your business to become familiar with them. Survey results from participating hospitals are updated periodically and available at www.hospitalcompare.hhs.gov.
I regularly hear hospitalists say they could more effectively direct their energy and resources in ways consistent with their hospital CEO’s goals, if they only knew what those goals were. If you find yourself in the same category, then you should know there is a really good chance the upper-level hospital leadership has a salary bonus in place for achieving certain patient safety and satisfaction goals. A recent survey in the Journal of Patient Safety showed this is the case for more than half of the nation’s hospital executives.1 Therefore, learning more about your hospital’s patient satisfaction scores, and your group’s contribution to the score, is worth the effort.
The publicly reported HCAHPS data does not address hospitalists’ effect on patient satisfaction separately from other doctors and hospital attributes. As a result, some hospitalist groups conduct their own survey. This may range from a very brief series of written questions, or a more-involved survey administered via phone. Since it is difficult to drill down to patient satisfaction with individual hospitalists, data from these surveys often times are collected for the entire hospitalist practice in aggregate, rather than by the individual doctor.
Incorporate Information
Investigate both internal and external resources, if you believe your group could benefit from an intervention (e.g., education or training to improve patient satisfaction). Many hospitals have someone on staff to provide employee training in this area; the trainer probably would be impressed and pleased if you initiate contact to learn more about available training. Resources external to your hospital include survey companies (e.g., Press Ganey, NCR Picker, among others). These companies can provide training and guidance, or put you in contact with firms with whom they work. I have seen something as simple as a one- or two-hour online or DVD course be a valuable tool for some practices.
I know some very efficient hospitalists who maintain incredibly high levels of patient satisfaction despite very high workloads. I think these doctors probably are outliers, and most of us will see satisfaction scores decline as workloads become unreasonably high. Each group should challenge itself continually to find the optimal point between patient volume and important outcomes like patient satisfaction.
Here are some strategies hospitalist groups could use to improve patient satisfaction:
- Call patients after discharge. This is a potent way to improve patient satisfaction. It is a valuable clinical encounter, since it offers a chance to reinforce discharge instructions (e.g, the value of smoking cessation, or the need to have ongoing INR monitoring), and to address any issues arising in the interim. The calls are most effective if done by the doctor who discharged the patient, but can be of some value if made by a nurse or other person connected to the practice. Note, it is best not to inform patients you will be making this call, since it will reduce the surprise and pleasure when the call comes, and could lead to frustration if a patient never gets the call they were told to expect.
- Provide referring doctors, emergency room doctors, and nurses at the hospital with a “script” to use when introducing or describing the hospitalist practice to patients. Without coaching, most of these people likely will say to the patient something like “your doctor [primary care physician] doesn’t come to the hospital anymore, so the hospitalist will see you.” Such a description may make the patient feel like they’re getting a second-class doctor. Instead, encourage these workers to say something like “Your doctor has decided to focus her practice on the office, to be more available to you there. As a result, she has decided to refer you to Dr. McCartney, a doctor who specializes in the care of hospitalized patients with problems like yours. Dr. McCartney will communicate with your primary doctor, and you should plan to follow up with her when you are discharged.”
- Communicate regularly, even daily, with family members of patients who have dementia or other cognitive impairment. This usually means calling the family.
- During initial and subsequent visits with a patient, shake hands or gently touch the patient in some way. Sit down and, while still sitting, conclude the conversation by asking if the patient has any questions and asking “Is there anything special I can do for you today?”
- Provide patients with a copy of their discharge summary. It can serve as a valuable education tool for the patient, their loved ones, visiting nurses, etc. Ideally, the summary should be transcribed on a stat basis, so it can be available for the nurse to give to the patient on the way out of the hospital. Alternatively, it could be mailed to the patient later.
- Track your ongoing satisfaction performance by regularly including available data from HCAHPS and/or other sources in the practice dashboard or report card.
- Ensure patients and their families are provided a copy of your group’s brochure. Brochures delivered to primary care offices rarely find their way into the hands of the patients, and often times are forgotten or misplaced by the time hospital care is needed. It usually is more effective to ensure hospital staff provides the brochure. This could be done via a standing protocol stating that a clerical person will provide all patients with a hospitalist as attending or consultant will get a copy. Or, ensure an order to this effect is written on every one of your patients. It’s best to do this via pre-printed order sets.
If you don’t have a group brochure, develop one. It should include the name of the group with a one- or two-line biography and photograph for each hospitalist (e.g., where they attended medical school and residency), and other key information about the practice. If you’d like a sample brochure, visit www.hospitalmedicine.org and search “sample brochure for hospitalist program.”
Each hospitalist in your group could carry business cards with the doctor’s picture next to their name and key info. One small study showed this enhanced patients’ ability to correctly identify their hospitalist and presumably increased their satisfaction, as well.
- Maximize hospitalist-patient continuity. Each group should adjust their work schedule to maximize continuity between patient and hospitalist while still providing a sustainable lifestyle. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is co-founder and past president of SHM. He is a principal in Nelson/Flores Associates, a national hospitalist practice management consulting firm. He is part of the faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official SHM position..
Reference
1. Vaughn T, Koepke M, Kroch E, et al. Engagement of leadership in quality improvement initiatives: executive quality improvement survey results. J Patient Saf. 2006;2(1):2-9.
Patient satisfaction became a much more important issue earlier in 2008 when hospitals began reporting their performance on the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey. The 18-item survey was developed by the Centers for Medicare and Medicaid Services (CMS) and the Agency for Healthcare Research and Quality, and is completed by patients to record their level of satisfaction with the hospital and the care they received.
If you don’t know your hospital’s publicly reported HCAHPS scores, you should make it your business to become familiar with them. Survey results from participating hospitals are updated periodically and available at www.hospitalcompare.hhs.gov.
I regularly hear hospitalists say they could more effectively direct their energy and resources in ways consistent with their hospital CEO’s goals, if they only knew what those goals were. If you find yourself in the same category, then you should know there is a really good chance the upper-level hospital leadership has a salary bonus in place for achieving certain patient safety and satisfaction goals. A recent survey in the Journal of Patient Safety showed this is the case for more than half of the nation’s hospital executives.1 Therefore, learning more about your hospital’s patient satisfaction scores, and your group’s contribution to the score, is worth the effort.
The publicly reported HCAHPS data does not address hospitalists’ effect on patient satisfaction separately from other doctors and hospital attributes. As a result, some hospitalist groups conduct their own survey. This may range from a very brief series of written questions, or a more-involved survey administered via phone. Since it is difficult to drill down to patient satisfaction with individual hospitalists, data from these surveys often times are collected for the entire hospitalist practice in aggregate, rather than by the individual doctor.
Incorporate Information
Investigate both internal and external resources, if you believe your group could benefit from an intervention (e.g., education or training to improve patient satisfaction). Many hospitals have someone on staff to provide employee training in this area; the trainer probably would be impressed and pleased if you initiate contact to learn more about available training. Resources external to your hospital include survey companies (e.g., Press Ganey, NCR Picker, among others). These companies can provide training and guidance, or put you in contact with firms with whom they work. I have seen something as simple as a one- or two-hour online or DVD course be a valuable tool for some practices.
I know some very efficient hospitalists who maintain incredibly high levels of patient satisfaction despite very high workloads. I think these doctors probably are outliers, and most of us will see satisfaction scores decline as workloads become unreasonably high. Each group should challenge itself continually to find the optimal point between patient volume and important outcomes like patient satisfaction.
Here are some strategies hospitalist groups could use to improve patient satisfaction:
- Call patients after discharge. This is a potent way to improve patient satisfaction. It is a valuable clinical encounter, since it offers a chance to reinforce discharge instructions (e.g, the value of smoking cessation, or the need to have ongoing INR monitoring), and to address any issues arising in the interim. The calls are most effective if done by the doctor who discharged the patient, but can be of some value if made by a nurse or other person connected to the practice. Note, it is best not to inform patients you will be making this call, since it will reduce the surprise and pleasure when the call comes, and could lead to frustration if a patient never gets the call they were told to expect.
- Provide referring doctors, emergency room doctors, and nurses at the hospital with a “script” to use when introducing or describing the hospitalist practice to patients. Without coaching, most of these people likely will say to the patient something like “your doctor [primary care physician] doesn’t come to the hospital anymore, so the hospitalist will see you.” Such a description may make the patient feel like they’re getting a second-class doctor. Instead, encourage these workers to say something like “Your doctor has decided to focus her practice on the office, to be more available to you there. As a result, she has decided to refer you to Dr. McCartney, a doctor who specializes in the care of hospitalized patients with problems like yours. Dr. McCartney will communicate with your primary doctor, and you should plan to follow up with her when you are discharged.”
- Communicate regularly, even daily, with family members of patients who have dementia or other cognitive impairment. This usually means calling the family.
- During initial and subsequent visits with a patient, shake hands or gently touch the patient in some way. Sit down and, while still sitting, conclude the conversation by asking if the patient has any questions and asking “Is there anything special I can do for you today?”
- Provide patients with a copy of their discharge summary. It can serve as a valuable education tool for the patient, their loved ones, visiting nurses, etc. Ideally, the summary should be transcribed on a stat basis, so it can be available for the nurse to give to the patient on the way out of the hospital. Alternatively, it could be mailed to the patient later.
- Track your ongoing satisfaction performance by regularly including available data from HCAHPS and/or other sources in the practice dashboard or report card.
- Ensure patients and their families are provided a copy of your group’s brochure. Brochures delivered to primary care offices rarely find their way into the hands of the patients, and often times are forgotten or misplaced by the time hospital care is needed. It usually is more effective to ensure hospital staff provides the brochure. This could be done via a standing protocol stating that a clerical person will provide all patients with a hospitalist as attending or consultant will get a copy. Or, ensure an order to this effect is written on every one of your patients. It’s best to do this via pre-printed order sets.
If you don’t have a group brochure, develop one. It should include the name of the group with a one- or two-line biography and photograph for each hospitalist (e.g., where they attended medical school and residency), and other key information about the practice. If you’d like a sample brochure, visit www.hospitalmedicine.org and search “sample brochure for hospitalist program.”
Each hospitalist in your group could carry business cards with the doctor’s picture next to their name and key info. One small study showed this enhanced patients’ ability to correctly identify their hospitalist and presumably increased their satisfaction, as well.
- Maximize hospitalist-patient continuity. Each group should adjust their work schedule to maximize continuity between patient and hospitalist while still providing a sustainable lifestyle. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is co-founder and past president of SHM. He is a principal in Nelson/Flores Associates, a national hospitalist practice management consulting firm. He is part of the faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official SHM position..
Reference
1. Vaughn T, Koepke M, Kroch E, et al. Engagement of leadership in quality improvement initiatives: executive quality improvement survey results. J Patient Saf. 2006;2(1):2-9.