User login
RT of lymph nodes as good as dissection for the long-term
SAN ANTONIO – Both axillary radiation therapy and axillary lymph node dissection provide excellent, comparable locoregional control in patients with early-stage breast cancer who have a positive sentinel node, according to updated results of the European Organisation for Research and Treatment of Cancer’s AMAROS trial.
The 10-year cumulative incidence rate of axillary recurrence was 1.82% with radiation and 0.93% with lymph node dissection, a nonsignificant difference (hazard ratio, 1.71; P = .365). Distant metastasis–free survival and overall survival also were statistically on par. The findings reinforce the trial’s 5-year results, which additionally showed a markedly lower incidence of lymphedema with axillary radiation therapy. Lead investigator Emiel J. T. Rutgers, MD, PhD, reflected on hesitation in the uptake of axillary radiation therapy among oncologists and discussed the AMAROS results in the context of the ACOSOG Z11 trial. Dr. Rutgers, the principal investigator of the AMAROS trial and a surgical oncologist at the Netherlands Cancer Institute in Amsterdam, also described how the trial’s findings have altered practice at his institution.
Dr. Rutgers disclosed that he had no relevant conflicts of interest. The study was supported by the EORTC Charitable Trust.
SAN ANTONIO – Both axillary radiation therapy and axillary lymph node dissection provide excellent, comparable locoregional control in patients with early-stage breast cancer who have a positive sentinel node, according to updated results of the European Organisation for Research and Treatment of Cancer’s AMAROS trial.
The 10-year cumulative incidence rate of axillary recurrence was 1.82% with radiation and 0.93% with lymph node dissection, a nonsignificant difference (hazard ratio, 1.71; P = .365). Distant metastasis–free survival and overall survival also were statistically on par. The findings reinforce the trial’s 5-year results, which additionally showed a markedly lower incidence of lymphedema with axillary radiation therapy. Lead investigator Emiel J. T. Rutgers, MD, PhD, reflected on hesitation in the uptake of axillary radiation therapy among oncologists and discussed the AMAROS results in the context of the ACOSOG Z11 trial. Dr. Rutgers, the principal investigator of the AMAROS trial and a surgical oncologist at the Netherlands Cancer Institute in Amsterdam, also described how the trial’s findings have altered practice at his institution.
Dr. Rutgers disclosed that he had no relevant conflicts of interest. The study was supported by the EORTC Charitable Trust.
SAN ANTONIO – Both axillary radiation therapy and axillary lymph node dissection provide excellent, comparable locoregional control in patients with early-stage breast cancer who have a positive sentinel node, according to updated results of the European Organisation for Research and Treatment of Cancer’s AMAROS trial.
The 10-year cumulative incidence rate of axillary recurrence was 1.82% with radiation and 0.93% with lymph node dissection, a nonsignificant difference (hazard ratio, 1.71; P = .365). Distant metastasis–free survival and overall survival also were statistically on par. The findings reinforce the trial’s 5-year results, which additionally showed a markedly lower incidence of lymphedema with axillary radiation therapy. Lead investigator Emiel J. T. Rutgers, MD, PhD, reflected on hesitation in the uptake of axillary radiation therapy among oncologists and discussed the AMAROS results in the context of the ACOSOG Z11 trial. Dr. Rutgers, the principal investigator of the AMAROS trial and a surgical oncologist at the Netherlands Cancer Institute in Amsterdam, also described how the trial’s findings have altered practice at his institution.
Dr. Rutgers disclosed that he had no relevant conflicts of interest. The study was supported by the EORTC Charitable Trust.
REPORTING FROM SABCS 2018

Oral Bowenoid Papulosis
To the Editor:
A 22-year-old Somali woman presented to our institution with oral lesions of 2 years’ duration. The lesions started as small papules in the corners of the mouth that gradually continued to spread to the mucosal lips and gums. The lesions did not drain any material. The patient reported that they were not painful and had not regressed. She was concerned about the cosmetic appearance of the lesions. The patient believed the lesions had developed from working in a chicken factory and was concerned that they appeared possibly due to contact with a substance in the factory. Additionally, she noted that her voice had become hoarse. She was otherwise healthy and denied any sexual contact or ever having a blood transfusion.
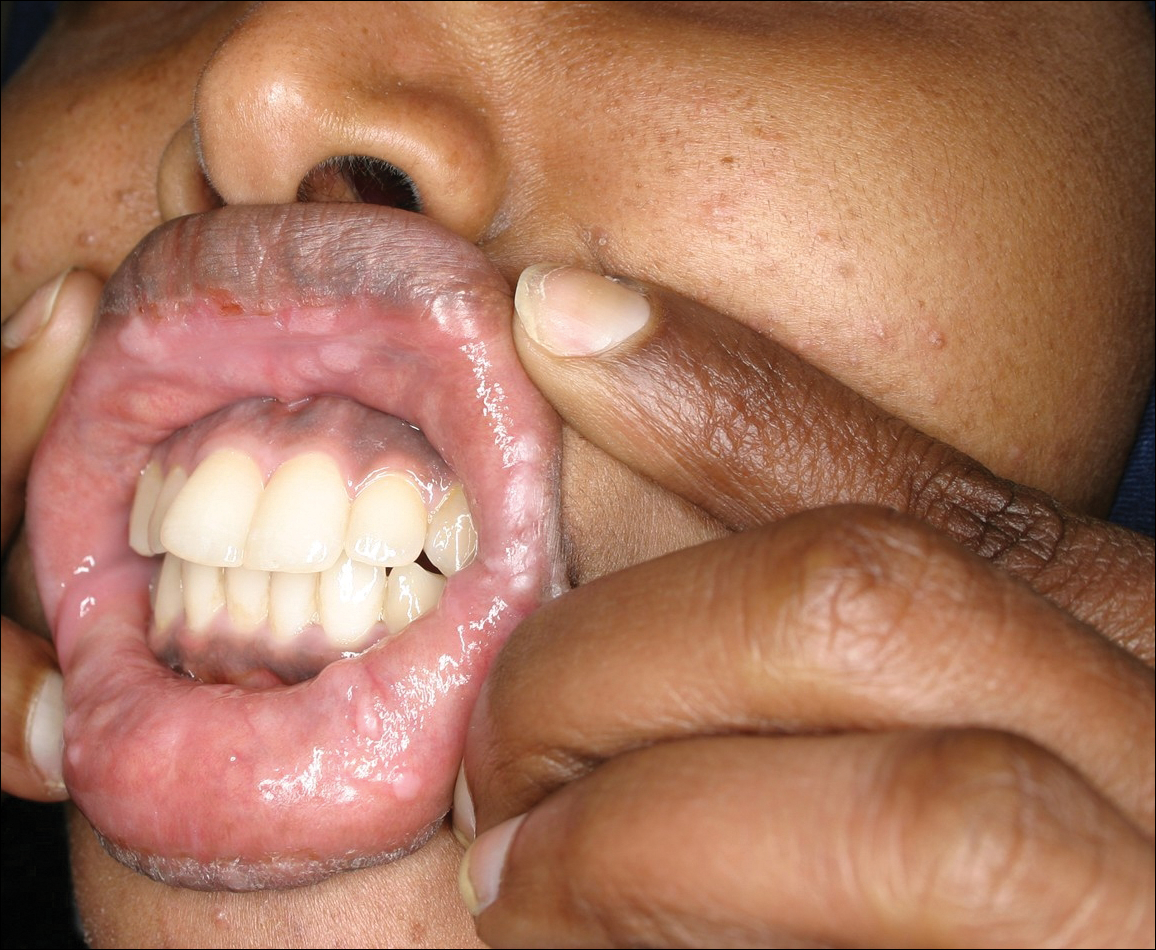
Physical examination revealed 10 to 15 flesh-colored papules measuring 2 to 3 mm in diameter on the vermilion, mucosal surfaces of the lips, and upper and lower gingivae (Figure 1). No lesions were seen on the hard and soft palate, tongue, buccal mucosa, or posterior pharynx.
Skin biopsy of the left lower mucosal lip revealed parakeratosis, acanthosis, superficial koilocytes, and atypical keratinocytes with frequent mitoses (Figures 2A–2C). In situ hybridization testing for human papillomavirus (HPV) was negative for low-risk types 6 and 11 but positive for high-risk types 16 and 18 (Figure 2D). Laboratory investigations including complete blood cell count, electrolyte panel, and liver function studies were normal, and serum was negative for syphilis and human immunodeficiency virus antibodies.
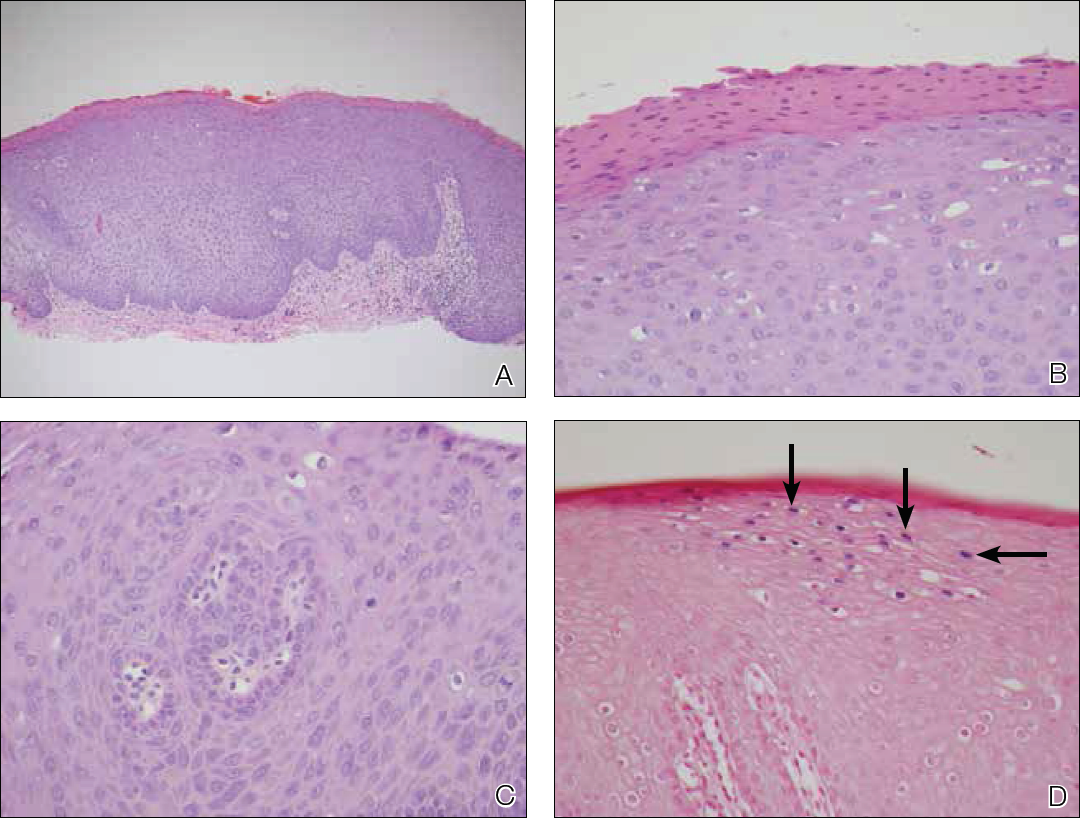
The combined clinical and histologic findings were diagnostic of oral bowenoid papulosis. Gynecologic evaluation showed that the patient had undergone female circumcision, and she had a normal Papanicolaou test. The patient was referred to both the ear, nose, and throat clinic as well as the dermatologic surgery department to discuss treatment options, but she was lost to follow-up.
Bowenoid papulosis is triggered by HPV infection and manifests clinically as solitary or multiple verrucous papules and plaques that are usually located on the genitalia.1 Only a few cases of bowenoid papulosis have been reported in the oral cavity.1-5 Because this disease is sexually transmitted, the mean age of onset of bowenoid papulosis is 31 years.2 There is a small risk (2%–3%) of developing invasive carcinoma in bowenoid papulosis.1-3,6 Most lesions are associated with HPV type 16; however, bowenoid papulosis also has been associated with HPV types 18, 31, 32, 35, and 39.2
Some investigators consider bowenoid papulosis and Bowen disease (a type of squamous cell carcinoma [SCC] in situ) to be histologically identical1,6; however, some histologic differences have been reported.1-3,6 Bowenoid papulosis has more dilated and tortuous dermal capillaries and less atypia and dyskeratosis than Bowen disease.1,6 In contrast to bowenoid papulosis, Bowen disease is characterized clinically as well-defined scaly plaques on sun-exposed areas of the skin in older adults. Invasive SCC can be seen in 5% of skin lesions and 30% of penile lesions associated with Bowen disease.2 Risk factors for Bowen disease include sun exposure; arsenic poisoning; and infection with HPV types 2, 16, 18, 31, 33, 52, and 67.1,6
Oral bowenoid papulosis is rare. A PubMed search of articles indexed for MEDLINE using the term oral bowenoid papulosis yielded 7 additional cases, which are summarized in the Table. In 1987 Lookingbill et al2 described one of the first reported cases of oral disease in a 33-year-old immunosuppressed man receiving prednisone therapy for systemic lupus erythematosus who had both mouth and genital lesions. All lesions were positive for HPV type 16. The patient subsequently developed SCC of the tongue.2

The risk for progression of oral bowenoid papulosis to invasive SCC is not known. Our search yielded only 1 case of this occurrence.2
Two of 3 cases of solitary lip lesions in oral bowenoid papulosis were treated with surgical excision.1 Other treatment options include CO2 laser therapy, cryotherapy, 5-fluorouracil, bleomycin, intralesional interferon alfa, and imiquimod.1-3,5,6
Our case represents a rare report of oral bowenoid papulosis. Recognition of this unusual presentation is important for the diagnosis and management of this disease.
- Daley T, Birek C, Wysocki GP. Oral bowenoid lesions: differential diagnosis and pathogenetic insights. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:466-473.
- Lookingbill DP, Kreider JW, Howett MK, et al. Human papillomavirus type 16 in bowenoid papulosis, intraoral papillomas, and squamous cell carcinoma of the tongue. Arch Dermatol. 1987;123:363-368.
- Kratochvil FJ, Cioffi GA, Auclair PL, et al. Virus-associated dysplasia (bowenoid papulosis?) of the oral cavity. Oral Surg Oral Med Oral Pathol. 1989;68:312-316.
- Degener AM, Latino L, Pierangeli A, et al. Human papilloma virus-32-positive extragenital bowenoid papulosis in a HIV patient with typical genital bowenoid papulosis localization. Sex Transm Dis. 2004;31:619-622.
- Rinaggio J, Glick M, Lambert WC. Oral bowenoid papulosis in an HIV-positive male [published online October 14, 2005]. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:328-332.
- Regezi JA, Dekker NP, Ramos DM, et al. Proliferation and invasion factors in HIV-associated dysplastic and nondysplastic oral warts and in oral squamous cell carcinoma: an immunohistochemical and RT-PCR evaluation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;94:724-731.
To the Editor:
A 22-year-old Somali woman presented to our institution with oral lesions of 2 years’ duration. The lesions started as small papules in the corners of the mouth that gradually continued to spread to the mucosal lips and gums. The lesions did not drain any material. The patient reported that they were not painful and had not regressed. She was concerned about the cosmetic appearance of the lesions. The patient believed the lesions had developed from working in a chicken factory and was concerned that they appeared possibly due to contact with a substance in the factory. Additionally, she noted that her voice had become hoarse. She was otherwise healthy and denied any sexual contact or ever having a blood transfusion.
Physical examination revealed 10 to 15 flesh-colored papules measuring 2 to 3 mm in diameter on the vermilion, mucosal surfaces of the lips, and upper and lower gingivae (Figure 1). No lesions were seen on the hard and soft palate, tongue, buccal mucosa, or posterior pharynx.
Skin biopsy of the left lower mucosal lip revealed parakeratosis, acanthosis, superficial koilocytes, and atypical keratinocytes with frequent mitoses (Figures 2A–2C). In situ hybridization testing for human papillomavirus (HPV) was negative for low-risk types 6 and 11 but positive for high-risk types 16 and 18 (Figure 2D). Laboratory investigations including complete blood cell count, electrolyte panel, and liver function studies were normal, and serum was negative for syphilis and human immunodeficiency virus antibodies.
The combined clinical and histologic findings were diagnostic of oral bowenoid papulosis. Gynecologic evaluation showed that the patient had undergone female circumcision, and she had a normal Papanicolaou test. The patient was referred to both the ear, nose, and throat clinic as well as the dermatologic surgery department to discuss treatment options, but she was lost to follow-up.
Bowenoid papulosis is triggered by HPV infection and manifests clinically as solitary or multiple verrucous papules and plaques that are usually located on the genitalia.1 Only a few cases of bowenoid papulosis have been reported in the oral cavity.1-5 Because this disease is sexually transmitted, the mean age of onset of bowenoid papulosis is 31 years.2 There is a small risk (2%–3%) of developing invasive carcinoma in bowenoid papulosis.1-3,6 Most lesions are associated with HPV type 16; however, bowenoid papulosis also has been associated with HPV types 18, 31, 32, 35, and 39.2
Some investigators consider bowenoid papulosis and Bowen disease (a type of squamous cell carcinoma [SCC] in situ) to be histologically identical1,6; however, some histologic differences have been reported.1-3,6 Bowenoid papulosis has more dilated and tortuous dermal capillaries and less atypia and dyskeratosis than Bowen disease.1,6 In contrast to bowenoid papulosis, Bowen disease is characterized clinically as well-defined scaly plaques on sun-exposed areas of the skin in older adults. Invasive SCC can be seen in 5% of skin lesions and 30% of penile lesions associated with Bowen disease.2 Risk factors for Bowen disease include sun exposure; arsenic poisoning; and infection with HPV types 2, 16, 18, 31, 33, 52, and 67.1,6
Oral bowenoid papulosis is rare. A PubMed search of articles indexed for MEDLINE using the term oral bowenoid papulosis yielded 7 additional cases, which are summarized in the Table. In 1987 Lookingbill et al2 described one of the first reported cases of oral disease in a 33-year-old immunosuppressed man receiving prednisone therapy for systemic lupus erythematosus who had both mouth and genital lesions. All lesions were positive for HPV type 16. The patient subsequently developed SCC of the tongue.2
The risk for progression of oral bowenoid papulosis to invasive SCC is not known. Our search yielded only 1 case of this occurrence.2
Two of 3 cases of solitary lip lesions in oral bowenoid papulosis were treated with surgical excision.1 Other treatment options include CO2 laser therapy, cryotherapy, 5-fluorouracil, bleomycin, intralesional interferon alfa, and imiquimod.1-3,5,6
Our case represents a rare report of oral bowenoid papulosis. Recognition of this unusual presentation is important for the diagnosis and management of this disease.
To the Editor:
A 22-year-old Somali woman presented to our institution with oral lesions of 2 years’ duration. The lesions started as small papules in the corners of the mouth that gradually continued to spread to the mucosal lips and gums. The lesions did not drain any material. The patient reported that they were not painful and had not regressed. She was concerned about the cosmetic appearance of the lesions. The patient believed the lesions had developed from working in a chicken factory and was concerned that they appeared possibly due to contact with a substance in the factory. Additionally, she noted that her voice had become hoarse. She was otherwise healthy and denied any sexual contact or ever having a blood transfusion.
Physical examination revealed 10 to 15 flesh-colored papules measuring 2 to 3 mm in diameter on the vermilion, mucosal surfaces of the lips, and upper and lower gingivae (Figure 1). No lesions were seen on the hard and soft palate, tongue, buccal mucosa, or posterior pharynx.
Skin biopsy of the left lower mucosal lip revealed parakeratosis, acanthosis, superficial koilocytes, and atypical keratinocytes with frequent mitoses (Figures 2A–2C). In situ hybridization testing for human papillomavirus (HPV) was negative for low-risk types 6 and 11 but positive for high-risk types 16 and 18 (Figure 2D). Laboratory investigations including complete blood cell count, electrolyte panel, and liver function studies were normal, and serum was negative for syphilis and human immunodeficiency virus antibodies.
The combined clinical and histologic findings were diagnostic of oral bowenoid papulosis. Gynecologic evaluation showed that the patient had undergone female circumcision, and she had a normal Papanicolaou test. The patient was referred to both the ear, nose, and throat clinic as well as the dermatologic surgery department to discuss treatment options, but she was lost to follow-up.
Bowenoid papulosis is triggered by HPV infection and manifests clinically as solitary or multiple verrucous papules and plaques that are usually located on the genitalia.1 Only a few cases of bowenoid papulosis have been reported in the oral cavity.1-5 Because this disease is sexually transmitted, the mean age of onset of bowenoid papulosis is 31 years.2 There is a small risk (2%–3%) of developing invasive carcinoma in bowenoid papulosis.1-3,6 Most lesions are associated with HPV type 16; however, bowenoid papulosis also has been associated with HPV types 18, 31, 32, 35, and 39.2
Some investigators consider bowenoid papulosis and Bowen disease (a type of squamous cell carcinoma [SCC] in situ) to be histologically identical1,6; however, some histologic differences have been reported.1-3,6 Bowenoid papulosis has more dilated and tortuous dermal capillaries and less atypia and dyskeratosis than Bowen disease.1,6 In contrast to bowenoid papulosis, Bowen disease is characterized clinically as well-defined scaly plaques on sun-exposed areas of the skin in older adults. Invasive SCC can be seen in 5% of skin lesions and 30% of penile lesions associated with Bowen disease.2 Risk factors for Bowen disease include sun exposure; arsenic poisoning; and infection with HPV types 2, 16, 18, 31, 33, 52, and 67.1,6
Oral bowenoid papulosis is rare. A PubMed search of articles indexed for MEDLINE using the term oral bowenoid papulosis yielded 7 additional cases, which are summarized in the Table. In 1987 Lookingbill et al2 described one of the first reported cases of oral disease in a 33-year-old immunosuppressed man receiving prednisone therapy for systemic lupus erythematosus who had both mouth and genital lesions. All lesions were positive for HPV type 16. The patient subsequently developed SCC of the tongue.2
The risk for progression of oral bowenoid papulosis to invasive SCC is not known. Our search yielded only 1 case of this occurrence.2
Two of 3 cases of solitary lip lesions in oral bowenoid papulosis were treated with surgical excision.1 Other treatment options include CO2 laser therapy, cryotherapy, 5-fluorouracil, bleomycin, intralesional interferon alfa, and imiquimod.1-3,5,6
Our case represents a rare report of oral bowenoid papulosis. Recognition of this unusual presentation is important for the diagnosis and management of this disease.
- Daley T, Birek C, Wysocki GP. Oral bowenoid lesions: differential diagnosis and pathogenetic insights. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:466-473.
- Lookingbill DP, Kreider JW, Howett MK, et al. Human papillomavirus type 16 in bowenoid papulosis, intraoral papillomas, and squamous cell carcinoma of the tongue. Arch Dermatol. 1987;123:363-368.
- Kratochvil FJ, Cioffi GA, Auclair PL, et al. Virus-associated dysplasia (bowenoid papulosis?) of the oral cavity. Oral Surg Oral Med Oral Pathol. 1989;68:312-316.
- Degener AM, Latino L, Pierangeli A, et al. Human papilloma virus-32-positive extragenital bowenoid papulosis in a HIV patient with typical genital bowenoid papulosis localization. Sex Transm Dis. 2004;31:619-622.
- Rinaggio J, Glick M, Lambert WC. Oral bowenoid papulosis in an HIV-positive male [published online October 14, 2005]. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:328-332.
- Regezi JA, Dekker NP, Ramos DM, et al. Proliferation and invasion factors in HIV-associated dysplastic and nondysplastic oral warts and in oral squamous cell carcinoma: an immunohistochemical and RT-PCR evaluation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;94:724-731.
- Daley T, Birek C, Wysocki GP. Oral bowenoid lesions: differential diagnosis and pathogenetic insights. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:466-473.
- Lookingbill DP, Kreider JW, Howett MK, et al. Human papillomavirus type 16 in bowenoid papulosis, intraoral papillomas, and squamous cell carcinoma of the tongue. Arch Dermatol. 1987;123:363-368.
- Kratochvil FJ, Cioffi GA, Auclair PL, et al. Virus-associated dysplasia (bowenoid papulosis?) of the oral cavity. Oral Surg Oral Med Oral Pathol. 1989;68:312-316.
- Degener AM, Latino L, Pierangeli A, et al. Human papilloma virus-32-positive extragenital bowenoid papulosis in a HIV patient with typical genital bowenoid papulosis localization. Sex Transm Dis. 2004;31:619-622.
- Rinaggio J, Glick M, Lambert WC. Oral bowenoid papulosis in an HIV-positive male [published online October 14, 2005]. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:328-332.
- Regezi JA, Dekker NP, Ramos DM, et al. Proliferation and invasion factors in HIV-associated dysplastic and nondysplastic oral warts and in oral squamous cell carcinoma: an immunohistochemical and RT-PCR evaluation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;94:724-731.
Practice Points
- Bowenoid papulosis is triggered by human papillomavirus infection and manifests clinically as solitary or multiple verrucous papules and plaques that usually are located on the genitalia.
- Oral bowenoid papulosis is rare, and recognition of this unusual presentation is important for the diagnosis and management of this disease.
Nothing to gain from chemo after pCR achieved
SAN ANTONIO – Women with localized breast cancer who achieve a pathological complete response (pCR) after neoadjuvant chemotherapy may have little to gain from subsequent adjuvant chemotherapy except toxicity, according to a patient-level meta-analysis of more than 27,000 women. The analysis, reported by lead investigator Laura M. Spring, MD, at the San Antonio Breast Cancer Symposium, confirmed that, compared with residual disease, pCR was associated with significantly reduced risks of event-free survival events (hazard ratio, 0.31) and death (HR, 0.22). Moreover, the EFS benefit of a pCR was similar whether women went on to receive adjuvant chemotherapy (HR, 0.36) or not (HR, 0.36) (P = .60 for difference). Dr. Spring discussed overall and subgroup findings, implications for use of neoadjuvant chemotherapy, and how these new data may inform escalation and de-escalation of adjuvant therapy.
Dr. Spring disclosed that she has a consulting or advisory role with Novartis and that she receives institutional research funding from Tesaro. The study was supported by grants from the National Cancer Institute and Susan G. Komen.
SAN ANTONIO – Women with localized breast cancer who achieve a pathological complete response (pCR) after neoadjuvant chemotherapy may have little to gain from subsequent adjuvant chemotherapy except toxicity, according to a patient-level meta-analysis of more than 27,000 women. The analysis, reported by lead investigator Laura M. Spring, MD, at the San Antonio Breast Cancer Symposium, confirmed that, compared with residual disease, pCR was associated with significantly reduced risks of event-free survival events (hazard ratio, 0.31) and death (HR, 0.22). Moreover, the EFS benefit of a pCR was similar whether women went on to receive adjuvant chemotherapy (HR, 0.36) or not (HR, 0.36) (P = .60 for difference). Dr. Spring discussed overall and subgroup findings, implications for use of neoadjuvant chemotherapy, and how these new data may inform escalation and de-escalation of adjuvant therapy.
Dr. Spring disclosed that she has a consulting or advisory role with Novartis and that she receives institutional research funding from Tesaro. The study was supported by grants from the National Cancer Institute and Susan G. Komen.
SAN ANTONIO – Women with localized breast cancer who achieve a pathological complete response (pCR) after neoadjuvant chemotherapy may have little to gain from subsequent adjuvant chemotherapy except toxicity, according to a patient-level meta-analysis of more than 27,000 women. The analysis, reported by lead investigator Laura M. Spring, MD, at the San Antonio Breast Cancer Symposium, confirmed that, compared with residual disease, pCR was associated with significantly reduced risks of event-free survival events (hazard ratio, 0.31) and death (HR, 0.22). Moreover, the EFS benefit of a pCR was similar whether women went on to receive adjuvant chemotherapy (HR, 0.36) or not (HR, 0.36) (P = .60 for difference). Dr. Spring discussed overall and subgroup findings, implications for use of neoadjuvant chemotherapy, and how these new data may inform escalation and de-escalation of adjuvant therapy.
Dr. Spring disclosed that she has a consulting or advisory role with Novartis and that she receives institutional research funding from Tesaro. The study was supported by grants from the National Cancer Institute and Susan G. Komen.
REPORTING FROM SABCS 2018

Uterine cancer incidence and mortality on the rise in the U.S.
Uterine cancer incidence and mortality in the United States increased significantly from 1999 to 2015, with the greatest increases among nonwhite women, according to data published in the Morbidity and Mortality Weekly Report.
Uterine cancer is the fourth most common cancer diagnosed and the seventh most common cause of cancer death among women in the United States, wrote S. Jane Henley and her colleagues from the division of cancer prevention and control at the National Center for Chronic Disease Prevention and Health Promotion. Its incidence is thought to be on the rise because of the increasing prevalence of overweight and obesity.
In this report, researchers analyzed data from population-based cancer registries to find new cases of invasive uterine cancer from 1999 to 2015.
Over that period, incidence rates of uterine cancer increased around 0.7% per year on average, with an overall 12% increase. However, among American Indian and Alaskan Native women, the incidence rate during that time increased by 53%, among black women it increased by 46%, among Asian/Pacific Island women the incidence rate increased by 38%, and among Hispanic women it increased by 32%.
Uterine cancer death rates also increased by 21% from 1999 to 2015, representing approximately a 1.1% average increase per year. Again, the greatest increases were seen in American Indian and Alaskan Native women (52%), Hispanic women (33%), and black women (29%). Death rates increased by 18% among white women but were stable among Asian/Pacific Island women.
The most common type of uterine cancer was the endometrioid carcinomas, which accounted for 68% of uterine cancers overall. However black women had a higher percentage of other carcinomas, carcinosarcomas, and sarcomas, compared with women from other ethnic groups.
Two-thirds of cancer overall were diagnosed at a localized stage, but this was less common in black women than women of other ethnicities, while the proportion of cancers diagnosed at distant stage was higher among black women.
“This report found that black women were more likely to receive a diagnosis at distant stage and with more aggressive histologic types than were other women, which might in part account for the higher death rate among black women,” the authors wrote.
Despite the increasing incidence and mortality, the authors wrote that population-based screening tests are not recommended for uterine cancer, partly because around 90% of women with uterine cancer report abnormal vaginal bleeding.
“Uterine cancer outcomes could be improved by increasing awareness among women that abnormal vaginal bleeding should be evaluated promptly by a health care provider,” they wrote.
No conflicts of interest were reported.
SOURCE: Henley SJ et al. MMWR Morb Mortal Wkly Rep. 2018 Dec 7;67:1333-8.
Uterine cancer incidence and mortality in the United States increased significantly from 1999 to 2015, with the greatest increases among nonwhite women, according to data published in the Morbidity and Mortality Weekly Report.
Uterine cancer is the fourth most common cancer diagnosed and the seventh most common cause of cancer death among women in the United States, wrote S. Jane Henley and her colleagues from the division of cancer prevention and control at the National Center for Chronic Disease Prevention and Health Promotion. Its incidence is thought to be on the rise because of the increasing prevalence of overweight and obesity.
In this report, researchers analyzed data from population-based cancer registries to find new cases of invasive uterine cancer from 1999 to 2015.
Over that period, incidence rates of uterine cancer increased around 0.7% per year on average, with an overall 12% increase. However, among American Indian and Alaskan Native women, the incidence rate during that time increased by 53%, among black women it increased by 46%, among Asian/Pacific Island women the incidence rate increased by 38%, and among Hispanic women it increased by 32%.
Uterine cancer death rates also increased by 21% from 1999 to 2015, representing approximately a 1.1% average increase per year. Again, the greatest increases were seen in American Indian and Alaskan Native women (52%), Hispanic women (33%), and black women (29%). Death rates increased by 18% among white women but were stable among Asian/Pacific Island women.
The most common type of uterine cancer was the endometrioid carcinomas, which accounted for 68% of uterine cancers overall. However black women had a higher percentage of other carcinomas, carcinosarcomas, and sarcomas, compared with women from other ethnic groups.
Two-thirds of cancer overall were diagnosed at a localized stage, but this was less common in black women than women of other ethnicities, while the proportion of cancers diagnosed at distant stage was higher among black women.
“This report found that black women were more likely to receive a diagnosis at distant stage and with more aggressive histologic types than were other women, which might in part account for the higher death rate among black women,” the authors wrote.
Despite the increasing incidence and mortality, the authors wrote that population-based screening tests are not recommended for uterine cancer, partly because around 90% of women with uterine cancer report abnormal vaginal bleeding.
“Uterine cancer outcomes could be improved by increasing awareness among women that abnormal vaginal bleeding should be evaluated promptly by a health care provider,” they wrote.
No conflicts of interest were reported.
SOURCE: Henley SJ et al. MMWR Morb Mortal Wkly Rep. 2018 Dec 7;67:1333-8.
Uterine cancer incidence and mortality in the United States increased significantly from 1999 to 2015, with the greatest increases among nonwhite women, according to data published in the Morbidity and Mortality Weekly Report.
Uterine cancer is the fourth most common cancer diagnosed and the seventh most common cause of cancer death among women in the United States, wrote S. Jane Henley and her colleagues from the division of cancer prevention and control at the National Center for Chronic Disease Prevention and Health Promotion. Its incidence is thought to be on the rise because of the increasing prevalence of overweight and obesity.
In this report, researchers analyzed data from population-based cancer registries to find new cases of invasive uterine cancer from 1999 to 2015.
Over that period, incidence rates of uterine cancer increased around 0.7% per year on average, with an overall 12% increase. However, among American Indian and Alaskan Native women, the incidence rate during that time increased by 53%, among black women it increased by 46%, among Asian/Pacific Island women the incidence rate increased by 38%, and among Hispanic women it increased by 32%.
Uterine cancer death rates also increased by 21% from 1999 to 2015, representing approximately a 1.1% average increase per year. Again, the greatest increases were seen in American Indian and Alaskan Native women (52%), Hispanic women (33%), and black women (29%). Death rates increased by 18% among white women but were stable among Asian/Pacific Island women.
The most common type of uterine cancer was the endometrioid carcinomas, which accounted for 68% of uterine cancers overall. However black women had a higher percentage of other carcinomas, carcinosarcomas, and sarcomas, compared with women from other ethnic groups.
Two-thirds of cancer overall were diagnosed at a localized stage, but this was less common in black women than women of other ethnicities, while the proportion of cancers diagnosed at distant stage was higher among black women.
“This report found that black women were more likely to receive a diagnosis at distant stage and with more aggressive histologic types than were other women, which might in part account for the higher death rate among black women,” the authors wrote.
Despite the increasing incidence and mortality, the authors wrote that population-based screening tests are not recommended for uterine cancer, partly because around 90% of women with uterine cancer report abnormal vaginal bleeding.
“Uterine cancer outcomes could be improved by increasing awareness among women that abnormal vaginal bleeding should be evaluated promptly by a health care provider,” they wrote.
No conflicts of interest were reported.
SOURCE: Henley SJ et al. MMWR Morb Mortal Wkly Rep. 2018 Dec 7;67:1333-8.
FROM MMWR
Key clinical point: Uterine cancer incidence and mortality rose from 1999 to 2015 in the United States.
Major finding: The incidence of uterine cancer in the United States increased 12% from 1999 to 2015.
Study details: An analysis of cancer registry data.
Disclosures: No conflicts of interest were reported.
Source: Henley SJ et al. MMWR Morb Mortal Wkly Rep. 2018 Dec 7;67:1333-8.
Small absolute difference between partial and whole breast irradiation
SAN ANTONIO – A phase 3 randomized NRG Oncology trial (NSABP B-39/RTOG 0413) was unable to rule out the possibility that, after lumpectomy, partial breast irradiation is inferior to whole breast irradiation when it comes to the ipsilateral breast tumor recurrences (invasive disease or ductal carcinoma in situ), reported Frank Vicini, MD, of MHP Radiation Oncology Institute, Pontiac, Mich.
The hazard ratio for this event with the former versus latter modality was 1.22, with the 90% confidence interval (0.94-1.58) falling just outside the predefined range to declare the two modalities equivalent (0.667-1.5). However, the absolute difference in the 10-year cumulative incidence of ipsilateral breast tumor recurrences was just 0.7% (4.6% vs. 3.9%). In a video interview, Dr. Vicini discussed whether this difference is clinically important, and the implications of the trial’s findings, taken together, for offering partial breast irradiation to patients.
Dr. Vicini disclosed that he is a research adviser for ImpediMed. The study was sponsored by the National Cancer Institute.
SAN ANTONIO – A phase 3 randomized NRG Oncology trial (NSABP B-39/RTOG 0413) was unable to rule out the possibility that, after lumpectomy, partial breast irradiation is inferior to whole breast irradiation when it comes to the ipsilateral breast tumor recurrences (invasive disease or ductal carcinoma in situ), reported Frank Vicini, MD, of MHP Radiation Oncology Institute, Pontiac, Mich.
The hazard ratio for this event with the former versus latter modality was 1.22, with the 90% confidence interval (0.94-1.58) falling just outside the predefined range to declare the two modalities equivalent (0.667-1.5). However, the absolute difference in the 10-year cumulative incidence of ipsilateral breast tumor recurrences was just 0.7% (4.6% vs. 3.9%). In a video interview, Dr. Vicini discussed whether this difference is clinically important, and the implications of the trial’s findings, taken together, for offering partial breast irradiation to patients.
Dr. Vicini disclosed that he is a research adviser for ImpediMed. The study was sponsored by the National Cancer Institute.
SAN ANTONIO – A phase 3 randomized NRG Oncology trial (NSABP B-39/RTOG 0413) was unable to rule out the possibility that, after lumpectomy, partial breast irradiation is inferior to whole breast irradiation when it comes to the ipsilateral breast tumor recurrences (invasive disease or ductal carcinoma in situ), reported Frank Vicini, MD, of MHP Radiation Oncology Institute, Pontiac, Mich.
The hazard ratio for this event with the former versus latter modality was 1.22, with the 90% confidence interval (0.94-1.58) falling just outside the predefined range to declare the two modalities equivalent (0.667-1.5). However, the absolute difference in the 10-year cumulative incidence of ipsilateral breast tumor recurrences was just 0.7% (4.6% vs. 3.9%). In a video interview, Dr. Vicini discussed whether this difference is clinically important, and the implications of the trial’s findings, taken together, for offering partial breast irradiation to patients.
Dr. Vicini disclosed that he is a research adviser for ImpediMed. The study was sponsored by the National Cancer Institute.
REPORTING FROM SABCS 2018

Low-dose tamoxifen halves recurrence of breast intraepithelial neoplasia
SAN ANTONIO – New life for old medicine: Women aged under 75 years with breast intraepithelial neoplasms (IEN) who took tamoxifen for 3 years at a dose of 5 mg per day – one-fourth the standard dose – had a 50% reduction in risk of IEN recurrence and an even more remarkable 75% reduction in the risk of contralateral breast cancer, compared with women who took placebos in the TAMO1 study.
Despite concerns about the known side effects of tamoxifen, there were no significant differences in either the rate of endometrial cancer or of deep vein thrombosis/pulmonary embolism between groups, and there was only a borderline increase in hot flashes among patients randomized to tamoxifen, reported Dr. Andrea De Censi, MD, from Ospedali Galliera in Genoa, Italy.
In a video interview, Dr. De Censi discusses how tamoxifen, a decades-old, inexpensive drug still offers real clinical benefit in day-to-day practice for patients with IEN.
The TAM01 study was supported by the Italian Ministry of Health, Italian Association for Cancer Research, and the Italian League Against Cancer. Dr. De Censi and his coauthors reported having no direct conflicts of interest.
SAN ANTONIO – New life for old medicine: Women aged under 75 years with breast intraepithelial neoplasms (IEN) who took tamoxifen for 3 years at a dose of 5 mg per day – one-fourth the standard dose – had a 50% reduction in risk of IEN recurrence and an even more remarkable 75% reduction in the risk of contralateral breast cancer, compared with women who took placebos in the TAMO1 study.
Despite concerns about the known side effects of tamoxifen, there were no significant differences in either the rate of endometrial cancer or of deep vein thrombosis/pulmonary embolism between groups, and there was only a borderline increase in hot flashes among patients randomized to tamoxifen, reported Dr. Andrea De Censi, MD, from Ospedali Galliera in Genoa, Italy.
In a video interview, Dr. De Censi discusses how tamoxifen, a decades-old, inexpensive drug still offers real clinical benefit in day-to-day practice for patients with IEN.
The TAM01 study was supported by the Italian Ministry of Health, Italian Association for Cancer Research, and the Italian League Against Cancer. Dr. De Censi and his coauthors reported having no direct conflicts of interest.
SAN ANTONIO – New life for old medicine: Women aged under 75 years with breast intraepithelial neoplasms (IEN) who took tamoxifen for 3 years at a dose of 5 mg per day – one-fourth the standard dose – had a 50% reduction in risk of IEN recurrence and an even more remarkable 75% reduction in the risk of contralateral breast cancer, compared with women who took placebos in the TAMO1 study.
Despite concerns about the known side effects of tamoxifen, there were no significant differences in either the rate of endometrial cancer or of deep vein thrombosis/pulmonary embolism between groups, and there was only a borderline increase in hot flashes among patients randomized to tamoxifen, reported Dr. Andrea De Censi, MD, from Ospedali Galliera in Genoa, Italy.
In a video interview, Dr. De Censi discusses how tamoxifen, a decades-old, inexpensive drug still offers real clinical benefit in day-to-day practice for patients with IEN.
The TAM01 study was supported by the Italian Ministry of Health, Italian Association for Cancer Research, and the Italian League Against Cancer. Dr. De Censi and his coauthors reported having no direct conflicts of interest.
REPORTING FROM SABCS 2018

Adjuvant capecitabine found disappointing in TNBC
SAN ANTONIO – Adjuvant capecitabine does not improve outcomes in women with early-stage triple-negative breast cancer (TNBC) who have undergone resection and received standard chemotherapy, finds a phase 3, randomized, controlled trial jointly conducted by the Spanish GEICAM group and the Central and South American CIBOMA group. But the story may not end there.

Findings reported in a session and press conference at the San Antonio Breast Cancer Symposium showed that, compared with observation, eight cycles of adjuvant capecitabine (Xeloda) reduced the 5-year risks of disease-free survival events and death by a nonsignificant relative 18% and 8%, respectively, among all 876 women randomized. However, the subgroup whose tumors had the nonbasal phenotype saw large significant benefits, with 47% and 58% relative reductions in these risks, respectively.
“The trial is formally negative,” commented lead investigator Miguel Martín, MD, PhD, head of the medical oncology service at Hospital Gregorio Marañón in Madrid. However, the observation arm fared better than expected. In addition, the trial had a very low–risk patient population, which may help explain why its findings differ somewhat from the more positive findings of the similar CREATE-X trial.
“Our data don’t speak against the CREATE-X study. My personal view is that capecitabine is useful for some TNBC patients,” Dr. Martin said. “Our study is not finished because we are going to look at the genomic characteristics of this group defined as non–basal-like because we want to know more about this subgroup. We are planning also to reproduce our subset in the CREATE-X trial to see if this is a real finding because we are in the era of personalized medicine.”
TNBC is a broad group defined only by negative findings for the main markers having available treatments, he elaborated. “So if we could define a subpopulation that actually benefited from capecitabine, this will be great for the patients.” Currently, conventional pathology does not routinely report on tumor basal phenotype, so all TNBC patients receive the same drugs. “This is a mistake. We should select the right drug for the right patient. Probably not all breast cancer patients are sensitive to the same drugs. But the fact is that we don’t have funding to run trials looking at that because this kind of trial is not interesting for pharma companies.”

“That’s an important message that triple-negative is really a big, very heterogeneous group,” agreed SABCS codirector and press conference moderator Carlos Arteaga, MD, director of the Harold C. Simmons Comprehensive Cancer Center and associate dean of oncology programs at the University of Texas, Dallas.
Patients and clinicians alike are largely unaware of the presence of TNBC basal and nonbasal subtypes and their potential importance, he said. “We all need some education on that, us included. It’s a very, very heterogeneous group, it is one that is very challenging. We need to start by educating all of us that there is a need to do research on that. ... We have a duty to define this phenotype better.”
Study details
“TNBC is sensitive to chemotherapy, but a significant proportion of patients will eventually relapse after conventional anthracycline and taxane combinations, so we need new approaches to this population,” Dr. Martín noted.
The trial, joint GEICAM/2003-11 and CIBOMA/2004-01, was designed in 2004. Although no information about capecitabine in breast cancer was available at the time, the investigators selected this drug because it is non–cross-resistant with anthracyclines and taxanes.
About 55% of the patients randomized had node-negative disease and roughly 80% received adjuvant chemotherapy alone because neoadjuvant chemotherapy was generally not used 14 years ago, Dr. Martín noted.
After a median follow-up of 7.34 years, the 5-year disease-free survival rate—the trial’s primary endpoint—was 79.6% with capecitabine and 76.8% with observation, a nonsignificant difference in both unadjusted analysis (hazard ratio, 0.82; P = .136) and adjusted analysis (HR, 0.79; P = .082). The 5-year overall survival rate was 86.2% with capecitabine and 85.9% with observation, another nonsignificant difference (HR, 0.92; P = .623).
However, in subgroup analyses among the 248 patients with nonbasal disease, defined as immunohistochemically negative for both EGFR and CK5/6, capecitabine conferred a significant disease-free survival advantage (HR, 0.53; P = .02) and overall survival advantage (HR, 0.420; P = .007) relative to observation.
Interaction of basal/nonbasal phenotype with treatment was marginal for disease-free survival (P = .0694) and significant for overall survival (P = .0052).
In the nonbasal subgroup, the disease-free survival benefit of capecitabine was mainly driven by a reduction in distant recurrences, particularly in the liver and the brain.
Adjuvant capecitabine had tolerability that was “exactly as expected,” according to Dr. Martín. The median dose intensity was 86.3%, and 75.2% of patients completed all of the planned eight cycles.
The trial was supported by Roche, which also provided capecitabine. Dr. Martín reported receiving speaker’s honoraria from Pfizer and Eli Lilly; honoraria for participation in advisory boards from AstraZeneca, Novartis, Roche-Genentech, Pfizer, GlaxoSmithKline, PharmaMar, Taiho Oncology, and Eli Lilly; and research grants from Novartis and Roche.
SOURCE: Martín M et al. SABCS 2018, Abstract GS2-04.
SAN ANTONIO – Adjuvant capecitabine does not improve outcomes in women with early-stage triple-negative breast cancer (TNBC) who have undergone resection and received standard chemotherapy, finds a phase 3, randomized, controlled trial jointly conducted by the Spanish GEICAM group and the Central and South American CIBOMA group. But the story may not end there.
Findings reported in a session and press conference at the San Antonio Breast Cancer Symposium showed that, compared with observation, eight cycles of adjuvant capecitabine (Xeloda) reduced the 5-year risks of disease-free survival events and death by a nonsignificant relative 18% and 8%, respectively, among all 876 women randomized. However, the subgroup whose tumors had the nonbasal phenotype saw large significant benefits, with 47% and 58% relative reductions in these risks, respectively.
“The trial is formally negative,” commented lead investigator Miguel Martín, MD, PhD, head of the medical oncology service at Hospital Gregorio Marañón in Madrid. However, the observation arm fared better than expected. In addition, the trial had a very low–risk patient population, which may help explain why its findings differ somewhat from the more positive findings of the similar CREATE-X trial.
“Our data don’t speak against the CREATE-X study. My personal view is that capecitabine is useful for some TNBC patients,” Dr. Martin said. “Our study is not finished because we are going to look at the genomic characteristics of this group defined as non–basal-like because we want to know more about this subgroup. We are planning also to reproduce our subset in the CREATE-X trial to see if this is a real finding because we are in the era of personalized medicine.”
TNBC is a broad group defined only by negative findings for the main markers having available treatments, he elaborated. “So if we could define a subpopulation that actually benefited from capecitabine, this will be great for the patients.” Currently, conventional pathology does not routinely report on tumor basal phenotype, so all TNBC patients receive the same drugs. “This is a mistake. We should select the right drug for the right patient. Probably not all breast cancer patients are sensitive to the same drugs. But the fact is that we don’t have funding to run trials looking at that because this kind of trial is not interesting for pharma companies.”
“That’s an important message that triple-negative is really a big, very heterogeneous group,” agreed SABCS codirector and press conference moderator Carlos Arteaga, MD, director of the Harold C. Simmons Comprehensive Cancer Center and associate dean of oncology programs at the University of Texas, Dallas.
Patients and clinicians alike are largely unaware of the presence of TNBC basal and nonbasal subtypes and their potential importance, he said. “We all need some education on that, us included. It’s a very, very heterogeneous group, it is one that is very challenging. We need to start by educating all of us that there is a need to do research on that. ... We have a duty to define this phenotype better.”
Study details
“TNBC is sensitive to chemotherapy, but a significant proportion of patients will eventually relapse after conventional anthracycline and taxane combinations, so we need new approaches to this population,” Dr. Martín noted.
The trial, joint GEICAM/2003-11 and CIBOMA/2004-01, was designed in 2004. Although no information about capecitabine in breast cancer was available at the time, the investigators selected this drug because it is non–cross-resistant with anthracyclines and taxanes.
About 55% of the patients randomized had node-negative disease and roughly 80% received adjuvant chemotherapy alone because neoadjuvant chemotherapy was generally not used 14 years ago, Dr. Martín noted.
After a median follow-up of 7.34 years, the 5-year disease-free survival rate—the trial’s primary endpoint—was 79.6% with capecitabine and 76.8% with observation, a nonsignificant difference in both unadjusted analysis (hazard ratio, 0.82; P = .136) and adjusted analysis (HR, 0.79; P = .082). The 5-year overall survival rate was 86.2% with capecitabine and 85.9% with observation, another nonsignificant difference (HR, 0.92; P = .623).
However, in subgroup analyses among the 248 patients with nonbasal disease, defined as immunohistochemically negative for both EGFR and CK5/6, capecitabine conferred a significant disease-free survival advantage (HR, 0.53; P = .02) and overall survival advantage (HR, 0.420; P = .007) relative to observation.
Interaction of basal/nonbasal phenotype with treatment was marginal for disease-free survival (P = .0694) and significant for overall survival (P = .0052).
In the nonbasal subgroup, the disease-free survival benefit of capecitabine was mainly driven by a reduction in distant recurrences, particularly in the liver and the brain.
Adjuvant capecitabine had tolerability that was “exactly as expected,” according to Dr. Martín. The median dose intensity was 86.3%, and 75.2% of patients completed all of the planned eight cycles.
The trial was supported by Roche, which also provided capecitabine. Dr. Martín reported receiving speaker’s honoraria from Pfizer and Eli Lilly; honoraria for participation in advisory boards from AstraZeneca, Novartis, Roche-Genentech, Pfizer, GlaxoSmithKline, PharmaMar, Taiho Oncology, and Eli Lilly; and research grants from Novartis and Roche.
SOURCE: Martín M et al. SABCS 2018, Abstract GS2-04.
SAN ANTONIO – Adjuvant capecitabine does not improve outcomes in women with early-stage triple-negative breast cancer (TNBC) who have undergone resection and received standard chemotherapy, finds a phase 3, randomized, controlled trial jointly conducted by the Spanish GEICAM group and the Central and South American CIBOMA group. But the story may not end there.
Findings reported in a session and press conference at the San Antonio Breast Cancer Symposium showed that, compared with observation, eight cycles of adjuvant capecitabine (Xeloda) reduced the 5-year risks of disease-free survival events and death by a nonsignificant relative 18% and 8%, respectively, among all 876 women randomized. However, the subgroup whose tumors had the nonbasal phenotype saw large significant benefits, with 47% and 58% relative reductions in these risks, respectively.
“The trial is formally negative,” commented lead investigator Miguel Martín, MD, PhD, head of the medical oncology service at Hospital Gregorio Marañón in Madrid. However, the observation arm fared better than expected. In addition, the trial had a very low–risk patient population, which may help explain why its findings differ somewhat from the more positive findings of the similar CREATE-X trial.
“Our data don’t speak against the CREATE-X study. My personal view is that capecitabine is useful for some TNBC patients,” Dr. Martin said. “Our study is not finished because we are going to look at the genomic characteristics of this group defined as non–basal-like because we want to know more about this subgroup. We are planning also to reproduce our subset in the CREATE-X trial to see if this is a real finding because we are in the era of personalized medicine.”
TNBC is a broad group defined only by negative findings for the main markers having available treatments, he elaborated. “So if we could define a subpopulation that actually benefited from capecitabine, this will be great for the patients.” Currently, conventional pathology does not routinely report on tumor basal phenotype, so all TNBC patients receive the same drugs. “This is a mistake. We should select the right drug for the right patient. Probably not all breast cancer patients are sensitive to the same drugs. But the fact is that we don’t have funding to run trials looking at that because this kind of trial is not interesting for pharma companies.”
“That’s an important message that triple-negative is really a big, very heterogeneous group,” agreed SABCS codirector and press conference moderator Carlos Arteaga, MD, director of the Harold C. Simmons Comprehensive Cancer Center and associate dean of oncology programs at the University of Texas, Dallas.
Patients and clinicians alike are largely unaware of the presence of TNBC basal and nonbasal subtypes and their potential importance, he said. “We all need some education on that, us included. It’s a very, very heterogeneous group, it is one that is very challenging. We need to start by educating all of us that there is a need to do research on that. ... We have a duty to define this phenotype better.”
Study details
“TNBC is sensitive to chemotherapy, but a significant proportion of patients will eventually relapse after conventional anthracycline and taxane combinations, so we need new approaches to this population,” Dr. Martín noted.
The trial, joint GEICAM/2003-11 and CIBOMA/2004-01, was designed in 2004. Although no information about capecitabine in breast cancer was available at the time, the investigators selected this drug because it is non–cross-resistant with anthracyclines and taxanes.
About 55% of the patients randomized had node-negative disease and roughly 80% received adjuvant chemotherapy alone because neoadjuvant chemotherapy was generally not used 14 years ago, Dr. Martín noted.
After a median follow-up of 7.34 years, the 5-year disease-free survival rate—the trial’s primary endpoint—was 79.6% with capecitabine and 76.8% with observation, a nonsignificant difference in both unadjusted analysis (hazard ratio, 0.82; P = .136) and adjusted analysis (HR, 0.79; P = .082). The 5-year overall survival rate was 86.2% with capecitabine and 85.9% with observation, another nonsignificant difference (HR, 0.92; P = .623).
However, in subgroup analyses among the 248 patients with nonbasal disease, defined as immunohistochemically negative for both EGFR and CK5/6, capecitabine conferred a significant disease-free survival advantage (HR, 0.53; P = .02) and overall survival advantage (HR, 0.420; P = .007) relative to observation.
Interaction of basal/nonbasal phenotype with treatment was marginal for disease-free survival (P = .0694) and significant for overall survival (P = .0052).
In the nonbasal subgroup, the disease-free survival benefit of capecitabine was mainly driven by a reduction in distant recurrences, particularly in the liver and the brain.
Adjuvant capecitabine had tolerability that was “exactly as expected,” according to Dr. Martín. The median dose intensity was 86.3%, and 75.2% of patients completed all of the planned eight cycles.
The trial was supported by Roche, which also provided capecitabine. Dr. Martín reported receiving speaker’s honoraria from Pfizer and Eli Lilly; honoraria for participation in advisory boards from AstraZeneca, Novartis, Roche-Genentech, Pfizer, GlaxoSmithKline, PharmaMar, Taiho Oncology, and Eli Lilly; and research grants from Novartis and Roche.
SOURCE: Martín M et al. SABCS 2018, Abstract GS2-04.
REPORTING FROM SABCS 2018
Key clinical point: Adjuvant capecitabine fails to improve outcomes in early-stage triple-negative breast cancer treated with surgery and standard chemotherapy.
Major finding: The 5-year disease-free survival rate was 79.6% with eight cycles of adjuvant capecitabine versus 76.8% with observation (adjusted hazard ratio, 0.79; P = .082).
Study details: A phase 3, randomized, controlled trial among 876 women with early-stage triple-negative breast cancer who had undergone surgery and received standard chemotherapy (joint GEICAM/2003-11 and CIBOMA/2004-01 trial).
Disclosures: The trial was supported by Roche, which also provided capecitabine. Dr. Martín reported receiving speaker’s honoraria from Pfizer and Eli Lilly; honoraria for participation in advisory boards from AstraZeneca, Novartis, Roche-Genentech, Pfizer, GlaxoSmithKline, PharmaMar, Taiho Oncology, and Eli Lilly; and research grants from Novartis and Roche.
Source: Martín M et al. SABCS 2018, Abstract GS2-04.
Cost-conscious minimally invasive hysterectomy: A case illustration
CASE Cost-conscious benign laparoscopic hysterectomy
A 43-year-old woman undergoes laparoscopic hysterectomy for treatment of presumed benign uterine fibroids and menorrhagia. Once she is prepped with ChloraPrep with tint, a RUMI II uterine manipulator is placed. Laparoscopic ports include a Kii Balloon Blunt Tip system, a Versaport Plus Pyramidal Bladed Trocar, and 2 Kii Fios First Entry trocars.
The surgeon uses the Harmonic ACE +7 device (a purely ultrasonic device) to perform most of the procedure. The uterus is morcellated and removed using the US Food and Drug Administration (FDA)-approved Olympus Contained Tissue Extraction System, and the vaginal cuff is closed using a series of 2-0 PDS II sutures. Skin incisions are closed using Dermabond skin adhesive.
Total cost of the products used in this case: $1,592.40. Could different product choices have reduced this figure?
Health-care costs continue to rise faster than inflation: Total health-care expenditures account for approximately 18% of gross domestic product in the United States. Physicians therefore face increasing pressure to take cost into account in their care of patients.1 Cost-effectiveness and outcome quality continue to increase in importance as measures in many clinical trials that compare standard and alternative therapies. And women’s health—specifically, minimally invasive gynecologic surgery—invites such comparisons.
Overall, conventional laparoscopic gynecologic procedures tend to cost less than laparotomy, a consequence of shorter hospital stays, faster recovery, and fewer complications.2-5 What is not fully appreciated, however, is how choice of laparoscopic instrumentation and associated products affects surgical costs. In this article, which revisits and updates a 2013 OBG Management examination of cost-consciousness in the selection of equipment and supplies for minimally invasive gynecologic surgery,6 we review these costs in 2018. Our goal is to raise awareness of the role of cost in care among minimally invasive gynecologic surgeons.
In the sections that follow, we highlight several aspects of laparoscopic gynecologic surgery that can affect your selection of instruments and products, describing differences in cost as well as some distinctive characteristics of products. Note that our comparisons focus solely on cost—not on ease of utility, effectiveness, surgical technique, risk of complications, or any other assessment. Note also that numerous other instruments and devices are commercially available besides those we list.
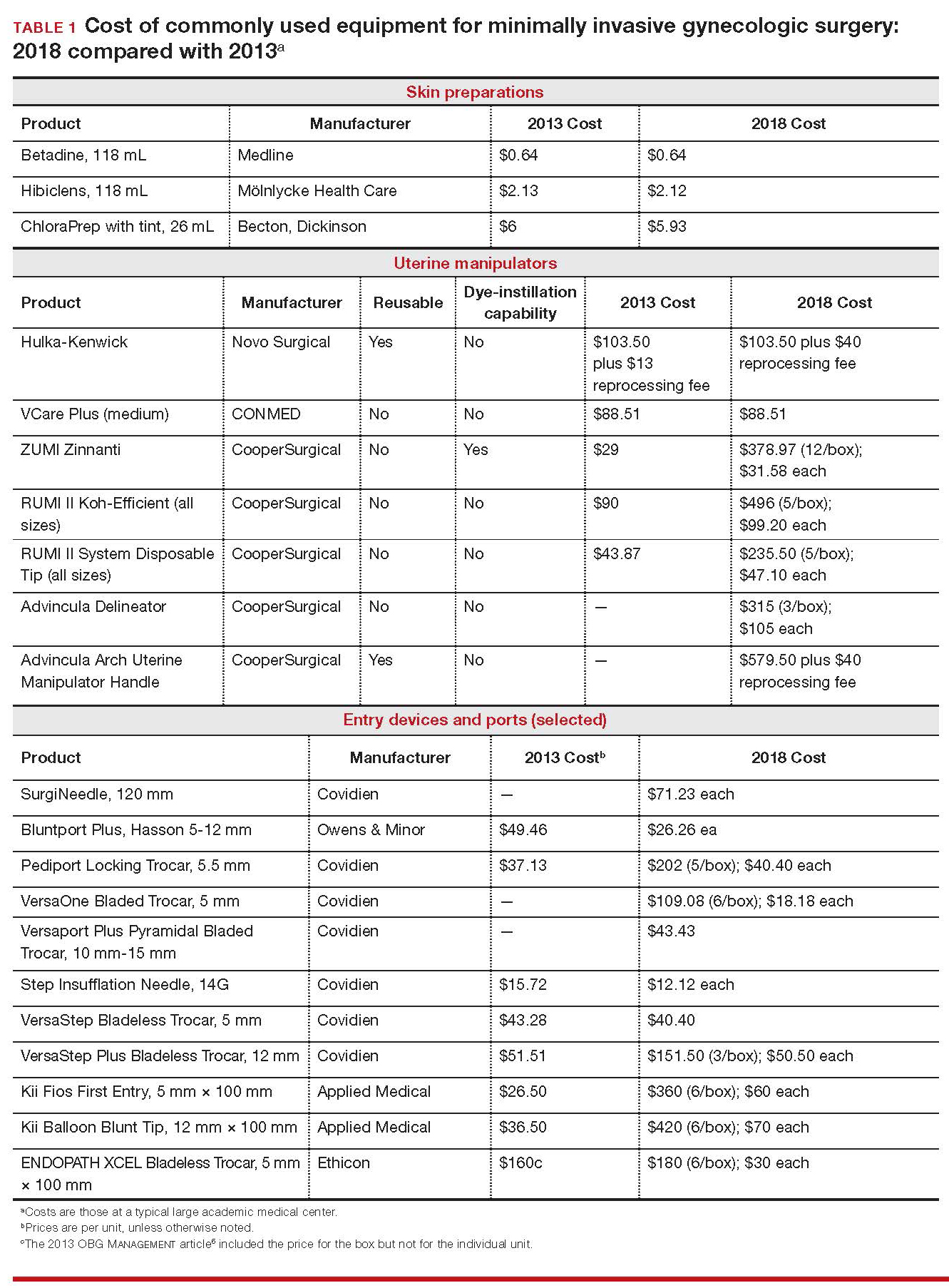
Importantly, 2013 and 2018 costs are included in TABLE 1. Unless otherwise noted, costs are per unit. Changes in manufacturers and material costs and technologic advances have contributed to some, but not all, of the changes in cost between 2013 and 2018.
Continue to: Variables to keep in mind
Variables to keep in mind
Even when taking cost into consideration, tailor your selection of instruments and supplies to your capabilities and comfort, as well as to the particular characteristics of the patient and the planned procedure. Also, remember that your institution might have arrangements with companies that supply minimally invasive instruments, and that such arrangements might limit your options, to some degree. Last, be aware that reprocessed ports and instruments are now available at a reduced cost. In short, we believe that it is crucial for surgeons to be cognizant of all products available to them prior to attending a surgical case.
Skin preparation and other preop considerations
Multiple preoperative skin preparations are available (TABLE 1). Traditionally, a povidone–iodine topical antiseptic, such as Betadine, has been used for skin and vaginal preparation prior to gynecologic surgery. Hibiclens and ChloraPrep are different combinations of chlorhexidine gluconate and isopropyl alcohol that act as broad-spectrum antiseptics.
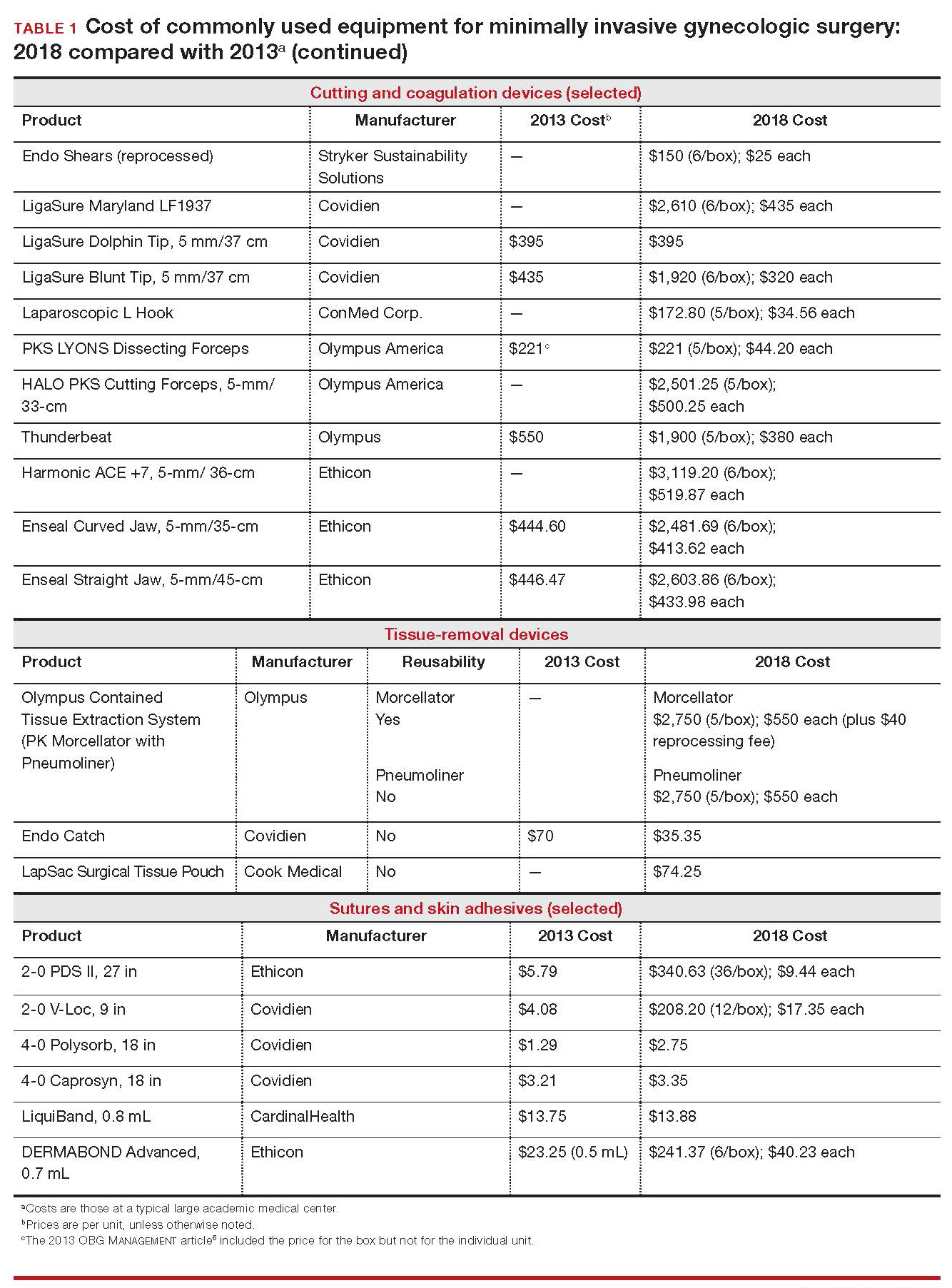
ChloraPrep is applied with a wand-like applicator and contains a much higher concentration of isopropyl alcohol than Hibiclens (70% and 4%, respectively), rendering it more flammable. It also requires longer drying time before surgery can be started. Clear and tinted ChloraPrep formulations are available.
Continue to: Uterine manipulators
Uterine manipulators
Cannulation of the cervical canal allows for uterine manipulation, increasing intraoperative traction and exposure as well as visualization of the adnexae and peritoneal surfaces.
The Hulka-Kenwick is a reusable uterine manipulator that is fairly standard and easy to apply. Specialized, single-use manipulators also are available, including the Advincula Delineator and VCare Plus uterine manipulator/elevator. The VCare Plus manipulator consists of 2 opposing cups: one cup (available in 4 sizes, small to extra-large) fits around the cervix and defines the site for colpotomy; the other helps maintain pneumoperitoneum once a colpotomy is created.
The ZUMI (Zinnanti Uterine Manipulator Injector) is a rigid, curved shaft with an intrauterine balloon to help prevent expulsion. It also has an integrated injection channel to allow for intraoperative chromotubation.
The RUMI II System fits individual patient anatomy with various tip lengths and colpotomy cup sizes. The Advincula Arch Uterine Manipulator Handle is a reusable alternative to the articulating RUMI II and works with the RUMI II System Disposable Tip (TABLE 1).
Continue to: Entry style and ports
Entry style and ports
The peritoneal cavity can be entered using either a closed (Veress needle) or open (Hasson) technique.7,8 Closed entry might allow for quicker access to the peritoneal cavity. A 2015 Cochrane review of 46 randomized, controlled trials of 7,389 patients undergoing laparoscopy compared outcomes between laparoscopic entry techniques and found no difference in major vascular or visceral injury between closed and open techniques at the umbilicus.9 However, open entry was associated with a greater likelihood of successful entry into the peritoneal cavity.9
Left upper-quadrant (Palmer’s point) entry is another option when adhesions are anticipated or abnormal anatomy is encountered at the umbilicus.
In general, complications related to laparoscopic entry are rare in gynecologic surgery, ranging from 0.18% to 0.5% of cases in studies.8,10,11 A minimally invasive surgeon might prefer one entry technique over another but should be able to perform both methods competently and recognize when a particular technique is warranted.
--
Choosing a port
Laparoscopic ports usually range from 5 mm to 12 mm and can be fixed or variable in size.
The primary port, usually placed through the umbilicus, can be a standard, blunt, 10-mm (Bluntport Plus Hasson) port, or it can be specialized to ease entry of the port or stabilize the port once it is introduced through the skin incision.
Optical trocars have a transparent tip that allows the surgeon to visualize the abdominal wall entry layer by layer using a 0° laparoscope, sometimes after pneumoperitoneum is created with a Veress needle. Other specialized ports include those that have balloons or foam collars, or both, to secure the port without traditional stay sutures on the fascia and to minimize leakage of pneumoperitoneum.
Continue to: Accessory ports
Accessory ports
When choosing an accessory port type and size, it is important to anticipate which instruments and devices, such as an Endo Catch bag, suture, or needle, will need to pass through it. Also, know whether 5-mm and 10-mm laparoscopes are available, and anticipate whether a second port with insufflation capabilities will be required.
The Pediport Locking Trocar is a user-friendly, 5-mm bladed port that deploys a mushroom-shaped stabilizer to prevent dislodgement. The Versaport bladed trocar has a spring-loaded entry shield, which slides over the blade to protect it once the peritoneal cavity is entered.
VersaStep Bladeless Trocars are introduced after a Step Insufflation Needle has been inserted. These trocars create a smaller fascial defect than conventional bladed trocars for an equivalent cannula size (TABLE 1).
Cutting and coagulating
Both monopolar and bipolar electrosurgical techniques are commonly employed in gynecologic laparoscopy. A wide variety of disposable and reusable instruments are available for monopolar energy, such as scissors, a hook, and a spatula.
Bipolar devices also can be disposable or reusable. Although bipolar electrosurgery minimizes injury to surrounding tissues by containing the current within the jaws of the forceps, it cannot cut or seal large vessels. As a result, several advanced bipolar devices with sealing and transecting capabilities have emerged (the LigaSure line of devices, Enseal). Ultrasonic devices, such as the Harmonic ACE, also can coagulate and cut at lower temperatures by converting electrical energy to mechanical energy (TABLE 1).
Continue to: Suture material
Suture material
Aspects of minimally invasive gynecologic surgery that require the use of suture include, but are not limited to, closure of the vaginal cuff, oophoropexy, and reapproximation of the ovarian cortex after cystectomy. Synthetic and delayed absorbable sutures, such as PDS II, are used frequently. The barbed suture also has gained popularity because it anchors to tissue without the need for intracorporeal or extracorporeal knots (TABLE 1).
Tissue removal
Adnexae and pathologic tissue, such as dermoid cysts, can be removed intact from the peritoneal cavity using an Endo Catch Single Use Specimen Pouch, a polyurethane sac. Careful use, with placement of the ovary with the cyst into the pouch prior to cystectomy, can contain or prevent spillage outside the bag.
A large uterus that cannot be extracted through a colpotomy can be manually morcellated. Appropriate candidates can undergo power morcellation using an FDA-approved device. (TABLE 1), allowing for the removal of smaller pieces through a small laparoscopic incision or the colpotomy.
Issues surrounding morcellation continue to require that gynecologic surgeons understand FDA recommendations. In 2014, the FDA issued a safety communication that morcellation is “contraindicated in gynecologic surgery if tissue is known or suspected to be malignant; it is contraindicated for uterine tissue removal with presumed benign fibroids in perimenopausal women.”12 A black-box warning was issued that uterine tissue might contain unsuspected cancer.
A task force created by AAGL addressed key issues in this controversy.
AAGL then provided guidelines related to morcellation13:
- Do not use morcellate in the setting of known malignancy.
- Provide appropriate preoperative evaluation with up-to-date Pap smear screening and image analysis.
- Increasing age significantly increases the risk of leiomyosarcoma, especially in a postmenopausal woman.
- Fibroid growth is not a reliable sign of malignancy.
- Do not use a morcellator if the patient is at high risk for malignancy.
- If leiomyosarcoma is the presumed pathology, await the final pathology report before proceeding with hysterectomy.
- Concomitant use of a bag might mitigate the risk of tissue spread.
- Obtain informed consent before proceeding with morcellation.
Continue to: Skin closure
Skin closure
Final subcuticular closure can be accomplished using sutures or skin adhesive. Sutures can be synthetic, absorbable monofilament (Caprosyn), or synthetic, absorbable, braided multifilament (Polysorb).
Skin adhesive closes incisions quickly, avoids inflammation related to foreign bodies, and can ease patients’ concerns that sometimes arise when absorbable suture persists postoperatively (TABLE 1).
The impact of physician experience
Physician experience has been shown to reduce cost while maintaining quality of care.14 That was the conclusion of researchers who undertook a retrospective study, addressing cost and clinical outcomes, of senior and junior attending physicians who performed laparoscopic-assisted vaginal hysterectomy on 120 patients. Studies such as these often lead to clinical pathways to facilitate cost-effective quality care.
--
CASE Same outcome at lower cost
The hypothetical 43-year-old patient in the opening case undergoes laparoscopic hysterectomy for treatment of uterine fibroids and menorrhagia. In this scenario, however, the surgeon makes the following product choices:
- The patient is prepped with Hibiclens.
- A VCare Plus uterine manipulator is placed.
- Laparoscopic ports include a VersaStep Plus Bladeless Trocar with Step Insufflation Needle; Versaport Plus Pyramidal Bladed Trocar; and 2 VersaOne Bladed trocars.
- The surgeon uses the PKS LYONS Dissecting Forceps and reprocessed Endo Shears to perform the hysterectomy.
- The uterus is enclosed in an Endo Catch bag and removed through the minilaparotomy site.
- The vaginal cuff is closed using 2-0 V-Loc barbed suture. Skin incisions are closed with 4-0 Polysorb, a polyglycolic acid absorbable suture.
The cost of this set of products? $360.44 or, roughly, $1,231.96 less than the set-up described in the case at the beginning of this article (TABLE 2).
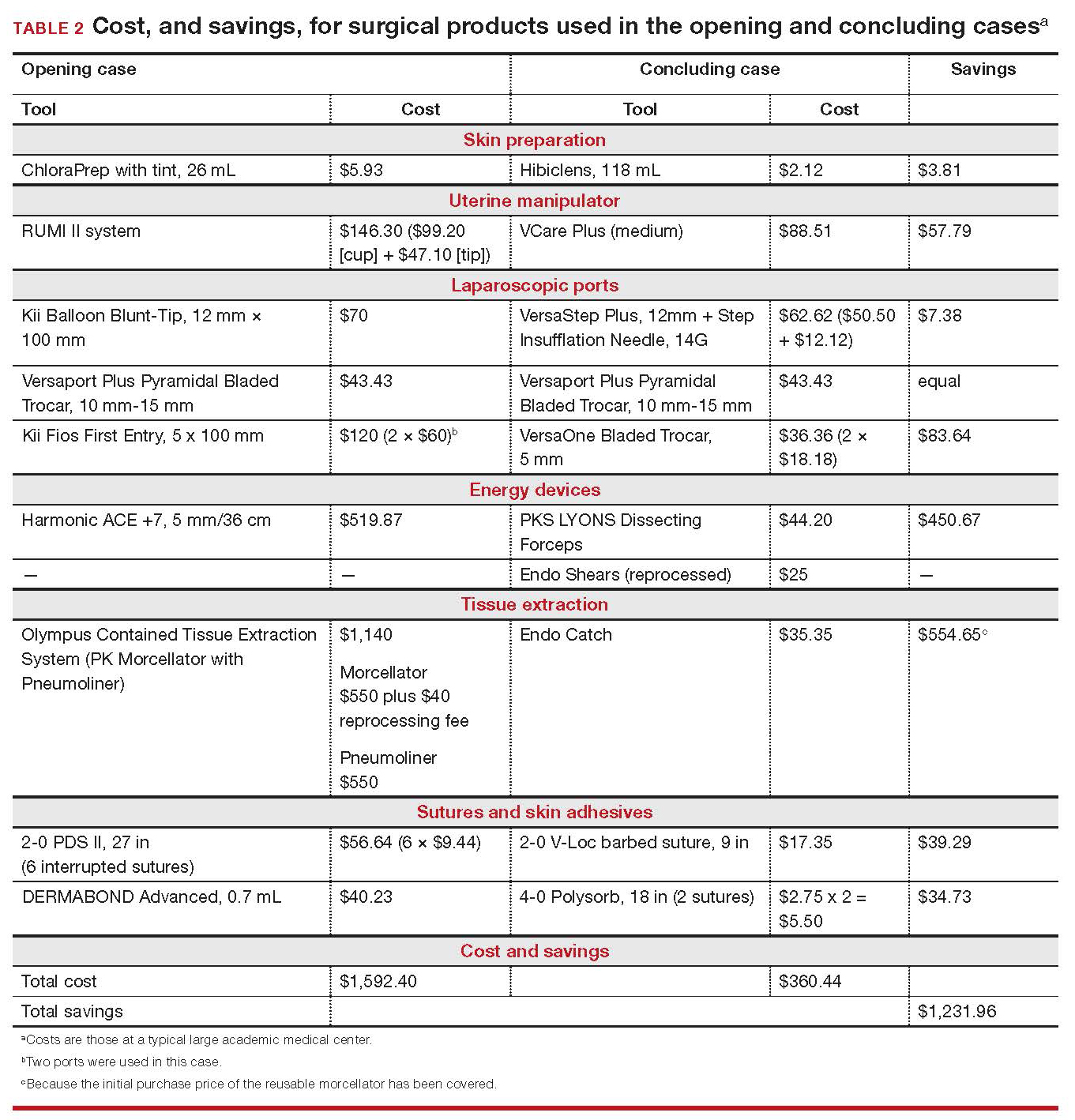
Continue to: Summing up
Summing up
Here are key points to take away from this analysis and discussion:
- As third-party payers and hospitals continue to evaluate surgeons individually and compare procedures from surgeon to surgeon, reimbursement might be stratified—thereby favoring physicians who demonstrate both quality outcomes and cost containment.
- There are many ways a minimally invasive surgeon can implement cost-conscious choices that have little or no impact on the quality of outcome.
- Surgeons who are familiar with surgical instruments and models available at their institution are better prepared to make wise cost-conscious decisions. (See “Caregivers should keep cost in mind: Here’s why,” in the Web version of this article at https://www.mdedge.com/obgyn.)
- Cost is not the only indicator of value: The surgeon must know how to apply tools correctly and be familiar with their limitations, and should choose instruments and products for their safety and ease of use. More often than not, a surgeon’s training and personal experience define—and sometimes restrict—the choice of devices.
- Last, it makes sense to have instruments and devices readily available in the operating room at the start of a case, to avoid unnecessary surgical delays. However, we recommend that you refrain from opening these tools until they are required intraoperatively. It is possible that the case will require conversion to laparotomy or that, after direct visualization of the pathology, different ports or instruments are required.
Acknowledgments
The authors would like to thank Meredith Snook, MD, who was coauthor of the original 2013 article6 and Kathleen Riordan, BSN, RN, for assistance in gathering specific cost-related information for this article.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Centers for Medicare & Medicaid Services. National health expenditure projections 2017-2026: Forecast summary. www.cms.gov/Research-Statistics-Data-and-Systems /Statistics-Trends-and-Reports/NationalHealthExpendData /Downloads/ForecastSummary.pdf. Accessed November 3, 2018.
- Vilos GA, Alshimmiri MM. Cost-benefit analysis of laparoscopic versus laparotomy salpingo-oophorectomy for benign tubo-ovarian disease. J Am Assoc Gynecol Laparosc. 1995;2(3):299-303.
- Gray DT, Thorburn J, Lundor P, et al. A cost-effectiveness study of a randomised trial of laparoscopy versus laparotomy for ectopic pregnancy. Lancet. 1995;345(8958):1139-1143.
- Chapron C, Fauconnier A, Goffinet F, et al. Laparoscopic surgery is not inherently dangerous for patients presenting with benign gynaecologic pathology. Results of a metaanalysis. Hum Reprod. 2002;17(5):1334-1342.
- Benezra V, Verma U, Whitted RW. Comparison of laparoscopy versus laparotomy for the surgical treatment of ovarian dermoid cysts. Obstet Gynecol Surv. 2006;61(1): 20-21.
- Sanfilippo JS, Snook ML. Cost-conscious choices for minimally invasive gynecologic surgery. OBG Manag. 2013;25(11):40-41,44,46-48,72.
- Hasson HM. A modified instrument and method for laparoscopy. Am J Obstet Gynecol. 1971;110(6):886-887.
- Ott J, Jaeger-Lansky A, Poschalko G, et al. Entry techniques in gynecologic laparoscopy—a review. Gynecol Surg. 2012;9(2):139-146.
- Ahmad G, Gent D, Henderson D, et al. Laparoscopic entry techniques. Cochrane Database Syst Rev. 2015;8:CD006583.
- Hasson HM, Rotman C, Rana N, et al. Open laparoscopy: 29-year experience. Obstet Gynecol. 2000;96(5 Pt 1):763-766.
- Schäfer M, Lauper M, Krähenbühl L. Trocar and Veress needle injuries during laparoscopy. Surg Endosc. 2001;15(3):275- 280.
- Immediately in effect guidance document: product labeling for laparoscopic power morcellators. Rockville, MD: US Department of Health and Human Services, Food and Drug Administration Center for Devices and Radiological Health; November 25, 2014. www.fda.gov/downloads /MedicalDevices/DeviceRegulationandGuidance /GuidanceDocuments/UCM424123.pdf. Accessed November 3, 2018.
- Tissue Extraction Task Force Members. Morcellation during uterine tissue extraction: an update. J Minim Invasive Gynecol. 2018;25(4):543-550.
- Chang WC, Li TC, Lin CC. The effect of physician experience on costs and clinical outcomes of laparoscopic-assisted vaginal hysterectomy: a multivariate analysis. J Am Assoc Gynecol Laparosc. 2003;10(3):356-359.

Dr. Kotha is Fellow, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh School of Medicine and Magee-Womens Hospital, Pittsburgh, Pennsylvania.
--

Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh School of Medicine, and Director, Reproductive Endocrinology and Infertility, at Magee-Womens Hospital. He also serves on the OBG Management Board of Editors.
--
The authors report no financial relationships relevant to this article.
Dr. Kotha is Fellow, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh School of Medicine and Magee-Womens Hospital, Pittsburgh, Pennsylvania.
--
Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh School of Medicine, and Director, Reproductive Endocrinology and Infertility, at Magee-Womens Hospital. He also serves on the OBG Management Board of Editors.
--
The authors report no financial relationships relevant to this article.
Dr. Kotha is Fellow, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh School of Medicine and Magee-Womens Hospital, Pittsburgh, Pennsylvania.
--
Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh School of Medicine, and Director, Reproductive Endocrinology and Infertility, at Magee-Womens Hospital. He also serves on the OBG Management Board of Editors.
--
The authors report no financial relationships relevant to this article.
CASE Cost-conscious benign laparoscopic hysterectomy
A 43-year-old woman undergoes laparoscopic hysterectomy for treatment of presumed benign uterine fibroids and menorrhagia. Once she is prepped with ChloraPrep with tint, a RUMI II uterine manipulator is placed. Laparoscopic ports include a Kii Balloon Blunt Tip system, a Versaport Plus Pyramidal Bladed Trocar, and 2 Kii Fios First Entry trocars.
The surgeon uses the Harmonic ACE +7 device (a purely ultrasonic device) to perform most of the procedure. The uterus is morcellated and removed using the US Food and Drug Administration (FDA)-approved Olympus Contained Tissue Extraction System, and the vaginal cuff is closed using a series of 2-0 PDS II sutures. Skin incisions are closed using Dermabond skin adhesive.
Total cost of the products used in this case: $1,592.40. Could different product choices have reduced this figure?
Health-care costs continue to rise faster than inflation: Total health-care expenditures account for approximately 18% of gross domestic product in the United States. Physicians therefore face increasing pressure to take cost into account in their care of patients.1 Cost-effectiveness and outcome quality continue to increase in importance as measures in many clinical trials that compare standard and alternative therapies. And women’s health—specifically, minimally invasive gynecologic surgery—invites such comparisons.
Overall, conventional laparoscopic gynecologic procedures tend to cost less than laparotomy, a consequence of shorter hospital stays, faster recovery, and fewer complications.2-5 What is not fully appreciated, however, is how choice of laparoscopic instrumentation and associated products affects surgical costs. In this article, which revisits and updates a 2013 OBG Management examination of cost-consciousness in the selection of equipment and supplies for minimally invasive gynecologic surgery,6 we review these costs in 2018. Our goal is to raise awareness of the role of cost in care among minimally invasive gynecologic surgeons.
In the sections that follow, we highlight several aspects of laparoscopic gynecologic surgery that can affect your selection of instruments and products, describing differences in cost as well as some distinctive characteristics of products. Note that our comparisons focus solely on cost—not on ease of utility, effectiveness, surgical technique, risk of complications, or any other assessment. Note also that numerous other instruments and devices are commercially available besides those we list.
Importantly, 2013 and 2018 costs are included in TABLE 1. Unless otherwise noted, costs are per unit. Changes in manufacturers and material costs and technologic advances have contributed to some, but not all, of the changes in cost between 2013 and 2018.
Continue to: Variables to keep in mind
Variables to keep in mind
Even when taking cost into consideration, tailor your selection of instruments and supplies to your capabilities and comfort, as well as to the particular characteristics of the patient and the planned procedure. Also, remember that your institution might have arrangements with companies that supply minimally invasive instruments, and that such arrangements might limit your options, to some degree. Last, be aware that reprocessed ports and instruments are now available at a reduced cost. In short, we believe that it is crucial for surgeons to be cognizant of all products available to them prior to attending a surgical case.
Skin preparation and other preop considerations
Multiple preoperative skin preparations are available (TABLE 1). Traditionally, a povidone–iodine topical antiseptic, such as Betadine, has been used for skin and vaginal preparation prior to gynecologic surgery. Hibiclens and ChloraPrep are different combinations of chlorhexidine gluconate and isopropyl alcohol that act as broad-spectrum antiseptics.
ChloraPrep is applied with a wand-like applicator and contains a much higher concentration of isopropyl alcohol than Hibiclens (70% and 4%, respectively), rendering it more flammable. It also requires longer drying time before surgery can be started. Clear and tinted ChloraPrep formulations are available.
Continue to: Uterine manipulators
Uterine manipulators
Cannulation of the cervical canal allows for uterine manipulation, increasing intraoperative traction and exposure as well as visualization of the adnexae and peritoneal surfaces.
The Hulka-Kenwick is a reusable uterine manipulator that is fairly standard and easy to apply. Specialized, single-use manipulators also are available, including the Advincula Delineator and VCare Plus uterine manipulator/elevator. The VCare Plus manipulator consists of 2 opposing cups: one cup (available in 4 sizes, small to extra-large) fits around the cervix and defines the site for colpotomy; the other helps maintain pneumoperitoneum once a colpotomy is created.
The ZUMI (Zinnanti Uterine Manipulator Injector) is a rigid, curved shaft with an intrauterine balloon to help prevent expulsion. It also has an integrated injection channel to allow for intraoperative chromotubation.
The RUMI II System fits individual patient anatomy with various tip lengths and colpotomy cup sizes. The Advincula Arch Uterine Manipulator Handle is a reusable alternative to the articulating RUMI II and works with the RUMI II System Disposable Tip (TABLE 1).
Continue to: Entry style and ports
Entry style and ports
The peritoneal cavity can be entered using either a closed (Veress needle) or open (Hasson) technique.7,8 Closed entry might allow for quicker access to the peritoneal cavity. A 2015 Cochrane review of 46 randomized, controlled trials of 7,389 patients undergoing laparoscopy compared outcomes between laparoscopic entry techniques and found no difference in major vascular or visceral injury between closed and open techniques at the umbilicus.9 However, open entry was associated with a greater likelihood of successful entry into the peritoneal cavity.9
Left upper-quadrant (Palmer’s point) entry is another option when adhesions are anticipated or abnormal anatomy is encountered at the umbilicus.
In general, complications related to laparoscopic entry are rare in gynecologic surgery, ranging from 0.18% to 0.5% of cases in studies.8,10,11 A minimally invasive surgeon might prefer one entry technique over another but should be able to perform both methods competently and recognize when a particular technique is warranted.
--
Choosing a port
Laparoscopic ports usually range from 5 mm to 12 mm and can be fixed or variable in size.
The primary port, usually placed through the umbilicus, can be a standard, blunt, 10-mm (Bluntport Plus Hasson) port, or it can be specialized to ease entry of the port or stabilize the port once it is introduced through the skin incision.
Optical trocars have a transparent tip that allows the surgeon to visualize the abdominal wall entry layer by layer using a 0° laparoscope, sometimes after pneumoperitoneum is created with a Veress needle. Other specialized ports include those that have balloons or foam collars, or both, to secure the port without traditional stay sutures on the fascia and to minimize leakage of pneumoperitoneum.
Continue to: Accessory ports
Accessory ports
When choosing an accessory port type and size, it is important to anticipate which instruments and devices, such as an Endo Catch bag, suture, or needle, will need to pass through it. Also, know whether 5-mm and 10-mm laparoscopes are available, and anticipate whether a second port with insufflation capabilities will be required.
The Pediport Locking Trocar is a user-friendly, 5-mm bladed port that deploys a mushroom-shaped stabilizer to prevent dislodgement. The Versaport bladed trocar has a spring-loaded entry shield, which slides over the blade to protect it once the peritoneal cavity is entered.
VersaStep Bladeless Trocars are introduced after a Step Insufflation Needle has been inserted. These trocars create a smaller fascial defect than conventional bladed trocars for an equivalent cannula size (TABLE 1).
Cutting and coagulating
Both monopolar and bipolar electrosurgical techniques are commonly employed in gynecologic laparoscopy. A wide variety of disposable and reusable instruments are available for monopolar energy, such as scissors, a hook, and a spatula.
Bipolar devices also can be disposable or reusable. Although bipolar electrosurgery minimizes injury to surrounding tissues by containing the current within the jaws of the forceps, it cannot cut or seal large vessels. As a result, several advanced bipolar devices with sealing and transecting capabilities have emerged (the LigaSure line of devices, Enseal). Ultrasonic devices, such as the Harmonic ACE, also can coagulate and cut at lower temperatures by converting electrical energy to mechanical energy (TABLE 1).
Continue to: Suture material
Suture material
Aspects of minimally invasive gynecologic surgery that require the use of suture include, but are not limited to, closure of the vaginal cuff, oophoropexy, and reapproximation of the ovarian cortex after cystectomy. Synthetic and delayed absorbable sutures, such as PDS II, are used frequently. The barbed suture also has gained popularity because it anchors to tissue without the need for intracorporeal or extracorporeal knots (TABLE 1).
Tissue removal
Adnexae and pathologic tissue, such as dermoid cysts, can be removed intact from the peritoneal cavity using an Endo Catch Single Use Specimen Pouch, a polyurethane sac. Careful use, with placement of the ovary with the cyst into the pouch prior to cystectomy, can contain or prevent spillage outside the bag.
A large uterus that cannot be extracted through a colpotomy can be manually morcellated. Appropriate candidates can undergo power morcellation using an FDA-approved device. (TABLE 1), allowing for the removal of smaller pieces through a small laparoscopic incision or the colpotomy.
Issues surrounding morcellation continue to require that gynecologic surgeons understand FDA recommendations. In 2014, the FDA issued a safety communication that morcellation is “contraindicated in gynecologic surgery if tissue is known or suspected to be malignant; it is contraindicated for uterine tissue removal with presumed benign fibroids in perimenopausal women.”12 A black-box warning was issued that uterine tissue might contain unsuspected cancer.
A task force created by AAGL addressed key issues in this controversy.
AAGL then provided guidelines related to morcellation13:
- Do not use morcellate in the setting of known malignancy.
- Provide appropriate preoperative evaluation with up-to-date Pap smear screening and image analysis.
- Increasing age significantly increases the risk of leiomyosarcoma, especially in a postmenopausal woman.
- Fibroid growth is not a reliable sign of malignancy.
- Do not use a morcellator if the patient is at high risk for malignancy.
- If leiomyosarcoma is the presumed pathology, await the final pathology report before proceeding with hysterectomy.
- Concomitant use of a bag might mitigate the risk of tissue spread.
- Obtain informed consent before proceeding with morcellation.
Continue to: Skin closure
Skin closure
Final subcuticular closure can be accomplished using sutures or skin adhesive. Sutures can be synthetic, absorbable monofilament (Caprosyn), or synthetic, absorbable, braided multifilament (Polysorb).
Skin adhesive closes incisions quickly, avoids inflammation related to foreign bodies, and can ease patients’ concerns that sometimes arise when absorbable suture persists postoperatively (TABLE 1).
The impact of physician experience
Physician experience has been shown to reduce cost while maintaining quality of care.14 That was the conclusion of researchers who undertook a retrospective study, addressing cost and clinical outcomes, of senior and junior attending physicians who performed laparoscopic-assisted vaginal hysterectomy on 120 patients. Studies such as these often lead to clinical pathways to facilitate cost-effective quality care.
--
CASE Same outcome at lower cost
The hypothetical 43-year-old patient in the opening case undergoes laparoscopic hysterectomy for treatment of uterine fibroids and menorrhagia. In this scenario, however, the surgeon makes the following product choices:
- The patient is prepped with Hibiclens.
- A VCare Plus uterine manipulator is placed.
- Laparoscopic ports include a VersaStep Plus Bladeless Trocar with Step Insufflation Needle; Versaport Plus Pyramidal Bladed Trocar; and 2 VersaOne Bladed trocars.
- The surgeon uses the PKS LYONS Dissecting Forceps and reprocessed Endo Shears to perform the hysterectomy.
- The uterus is enclosed in an Endo Catch bag and removed through the minilaparotomy site.
- The vaginal cuff is closed using 2-0 V-Loc barbed suture. Skin incisions are closed with 4-0 Polysorb, a polyglycolic acid absorbable suture.
The cost of this set of products? $360.44 or, roughly, $1,231.96 less than the set-up described in the case at the beginning of this article (TABLE 2).
Continue to: Summing up
Summing up
Here are key points to take away from this analysis and discussion:
- As third-party payers and hospitals continue to evaluate surgeons individually and compare procedures from surgeon to surgeon, reimbursement might be stratified—thereby favoring physicians who demonstrate both quality outcomes and cost containment.
- There are many ways a minimally invasive surgeon can implement cost-conscious choices that have little or no impact on the quality of outcome.
- Surgeons who are familiar with surgical instruments and models available at their institution are better prepared to make wise cost-conscious decisions. (See “Caregivers should keep cost in mind: Here’s why,” in the Web version of this article at https://www.mdedge.com/obgyn.)
- Cost is not the only indicator of value: The surgeon must know how to apply tools correctly and be familiar with their limitations, and should choose instruments and products for their safety and ease of use. More often than not, a surgeon’s training and personal experience define—and sometimes restrict—the choice of devices.
- Last, it makes sense to have instruments and devices readily available in the operating room at the start of a case, to avoid unnecessary surgical delays. However, we recommend that you refrain from opening these tools until they are required intraoperatively. It is possible that the case will require conversion to laparotomy or that, after direct visualization of the pathology, different ports or instruments are required.
Acknowledgments
The authors would like to thank Meredith Snook, MD, who was coauthor of the original 2013 article6 and Kathleen Riordan, BSN, RN, for assistance in gathering specific cost-related information for this article.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
CASE Cost-conscious benign laparoscopic hysterectomy
A 43-year-old woman undergoes laparoscopic hysterectomy for treatment of presumed benign uterine fibroids and menorrhagia. Once she is prepped with ChloraPrep with tint, a RUMI II uterine manipulator is placed. Laparoscopic ports include a Kii Balloon Blunt Tip system, a Versaport Plus Pyramidal Bladed Trocar, and 2 Kii Fios First Entry trocars.
The surgeon uses the Harmonic ACE +7 device (a purely ultrasonic device) to perform most of the procedure. The uterus is morcellated and removed using the US Food and Drug Administration (FDA)-approved Olympus Contained Tissue Extraction System, and the vaginal cuff is closed using a series of 2-0 PDS II sutures. Skin incisions are closed using Dermabond skin adhesive.
Total cost of the products used in this case: $1,592.40. Could different product choices have reduced this figure?
Health-care costs continue to rise faster than inflation: Total health-care expenditures account for approximately 18% of gross domestic product in the United States. Physicians therefore face increasing pressure to take cost into account in their care of patients.1 Cost-effectiveness and outcome quality continue to increase in importance as measures in many clinical trials that compare standard and alternative therapies. And women’s health—specifically, minimally invasive gynecologic surgery—invites such comparisons.
Overall, conventional laparoscopic gynecologic procedures tend to cost less than laparotomy, a consequence of shorter hospital stays, faster recovery, and fewer complications.2-5 What is not fully appreciated, however, is how choice of laparoscopic instrumentation and associated products affects surgical costs. In this article, which revisits and updates a 2013 OBG Management examination of cost-consciousness in the selection of equipment and supplies for minimally invasive gynecologic surgery,6 we review these costs in 2018. Our goal is to raise awareness of the role of cost in care among minimally invasive gynecologic surgeons.
In the sections that follow, we highlight several aspects of laparoscopic gynecologic surgery that can affect your selection of instruments and products, describing differences in cost as well as some distinctive characteristics of products. Note that our comparisons focus solely on cost—not on ease of utility, effectiveness, surgical technique, risk of complications, or any other assessment. Note also that numerous other instruments and devices are commercially available besides those we list.
Importantly, 2013 and 2018 costs are included in TABLE 1. Unless otherwise noted, costs are per unit. Changes in manufacturers and material costs and technologic advances have contributed to some, but not all, of the changes in cost between 2013 and 2018.
Continue to: Variables to keep in mind
Variables to keep in mind
Even when taking cost into consideration, tailor your selection of instruments and supplies to your capabilities and comfort, as well as to the particular characteristics of the patient and the planned procedure. Also, remember that your institution might have arrangements with companies that supply minimally invasive instruments, and that such arrangements might limit your options, to some degree. Last, be aware that reprocessed ports and instruments are now available at a reduced cost. In short, we believe that it is crucial for surgeons to be cognizant of all products available to them prior to attending a surgical case.
Skin preparation and other preop considerations
Multiple preoperative skin preparations are available (TABLE 1). Traditionally, a povidone–iodine topical antiseptic, such as Betadine, has been used for skin and vaginal preparation prior to gynecologic surgery. Hibiclens and ChloraPrep are different combinations of chlorhexidine gluconate and isopropyl alcohol that act as broad-spectrum antiseptics.
ChloraPrep is applied with a wand-like applicator and contains a much higher concentration of isopropyl alcohol than Hibiclens (70% and 4%, respectively), rendering it more flammable. It also requires longer drying time before surgery can be started. Clear and tinted ChloraPrep formulations are available.
Continue to: Uterine manipulators
Uterine manipulators
Cannulation of the cervical canal allows for uterine manipulation, increasing intraoperative traction and exposure as well as visualization of the adnexae and peritoneal surfaces.
The Hulka-Kenwick is a reusable uterine manipulator that is fairly standard and easy to apply. Specialized, single-use manipulators also are available, including the Advincula Delineator and VCare Plus uterine manipulator/elevator. The VCare Plus manipulator consists of 2 opposing cups: one cup (available in 4 sizes, small to extra-large) fits around the cervix and defines the site for colpotomy; the other helps maintain pneumoperitoneum once a colpotomy is created.
The ZUMI (Zinnanti Uterine Manipulator Injector) is a rigid, curved shaft with an intrauterine balloon to help prevent expulsion. It also has an integrated injection channel to allow for intraoperative chromotubation.
The RUMI II System fits individual patient anatomy with various tip lengths and colpotomy cup sizes. The Advincula Arch Uterine Manipulator Handle is a reusable alternative to the articulating RUMI II and works with the RUMI II System Disposable Tip (TABLE 1).
Continue to: Entry style and ports
Entry style and ports
The peritoneal cavity can be entered using either a closed (Veress needle) or open (Hasson) technique.7,8 Closed entry might allow for quicker access to the peritoneal cavity. A 2015 Cochrane review of 46 randomized, controlled trials of 7,389 patients undergoing laparoscopy compared outcomes between laparoscopic entry techniques and found no difference in major vascular or visceral injury between closed and open techniques at the umbilicus.9 However, open entry was associated with a greater likelihood of successful entry into the peritoneal cavity.9
Left upper-quadrant (Palmer’s point) entry is another option when adhesions are anticipated or abnormal anatomy is encountered at the umbilicus.
In general, complications related to laparoscopic entry are rare in gynecologic surgery, ranging from 0.18% to 0.5% of cases in studies.8,10,11 A minimally invasive surgeon might prefer one entry technique over another but should be able to perform both methods competently and recognize when a particular technique is warranted.
--
Choosing a port
Laparoscopic ports usually range from 5 mm to 12 mm and can be fixed or variable in size.
The primary port, usually placed through the umbilicus, can be a standard, blunt, 10-mm (Bluntport Plus Hasson) port, or it can be specialized to ease entry of the port or stabilize the port once it is introduced through the skin incision.
Optical trocars have a transparent tip that allows the surgeon to visualize the abdominal wall entry layer by layer using a 0° laparoscope, sometimes after pneumoperitoneum is created with a Veress needle. Other specialized ports include those that have balloons or foam collars, or both, to secure the port without traditional stay sutures on the fascia and to minimize leakage of pneumoperitoneum.
Continue to: Accessory ports
Accessory ports
When choosing an accessory port type and size, it is important to anticipate which instruments and devices, such as an Endo Catch bag, suture, or needle, will need to pass through it. Also, know whether 5-mm and 10-mm laparoscopes are available, and anticipate whether a second port with insufflation capabilities will be required.
The Pediport Locking Trocar is a user-friendly, 5-mm bladed port that deploys a mushroom-shaped stabilizer to prevent dislodgement. The Versaport bladed trocar has a spring-loaded entry shield, which slides over the blade to protect it once the peritoneal cavity is entered.
VersaStep Bladeless Trocars are introduced after a Step Insufflation Needle has been inserted. These trocars create a smaller fascial defect than conventional bladed trocars for an equivalent cannula size (TABLE 1).
Cutting and coagulating
Both monopolar and bipolar electrosurgical techniques are commonly employed in gynecologic laparoscopy. A wide variety of disposable and reusable instruments are available for monopolar energy, such as scissors, a hook, and a spatula.
Bipolar devices also can be disposable or reusable. Although bipolar electrosurgery minimizes injury to surrounding tissues by containing the current within the jaws of the forceps, it cannot cut or seal large vessels. As a result, several advanced bipolar devices with sealing and transecting capabilities have emerged (the LigaSure line of devices, Enseal). Ultrasonic devices, such as the Harmonic ACE, also can coagulate and cut at lower temperatures by converting electrical energy to mechanical energy (TABLE 1).
Continue to: Suture material
Suture material
Aspects of minimally invasive gynecologic surgery that require the use of suture include, but are not limited to, closure of the vaginal cuff, oophoropexy, and reapproximation of the ovarian cortex after cystectomy. Synthetic and delayed absorbable sutures, such as PDS II, are used frequently. The barbed suture also has gained popularity because it anchors to tissue without the need for intracorporeal or extracorporeal knots (TABLE 1).
Tissue removal
Adnexae and pathologic tissue, such as dermoid cysts, can be removed intact from the peritoneal cavity using an Endo Catch Single Use Specimen Pouch, a polyurethane sac. Careful use, with placement of the ovary with the cyst into the pouch prior to cystectomy, can contain or prevent spillage outside the bag.
A large uterus that cannot be extracted through a colpotomy can be manually morcellated. Appropriate candidates can undergo power morcellation using an FDA-approved device. (TABLE 1), allowing for the removal of smaller pieces through a small laparoscopic incision or the colpotomy.
Issues surrounding morcellation continue to require that gynecologic surgeons understand FDA recommendations. In 2014, the FDA issued a safety communication that morcellation is “contraindicated in gynecologic surgery if tissue is known or suspected to be malignant; it is contraindicated for uterine tissue removal with presumed benign fibroids in perimenopausal women.”12 A black-box warning was issued that uterine tissue might contain unsuspected cancer.
A task force created by AAGL addressed key issues in this controversy.
AAGL then provided guidelines related to morcellation13:
- Do not use morcellate in the setting of known malignancy.
- Provide appropriate preoperative evaluation with up-to-date Pap smear screening and image analysis.
- Increasing age significantly increases the risk of leiomyosarcoma, especially in a postmenopausal woman.
- Fibroid growth is not a reliable sign of malignancy.
- Do not use a morcellator if the patient is at high risk for malignancy.
- If leiomyosarcoma is the presumed pathology, await the final pathology report before proceeding with hysterectomy.
- Concomitant use of a bag might mitigate the risk of tissue spread.
- Obtain informed consent before proceeding with morcellation.
Continue to: Skin closure
Skin closure
Final subcuticular closure can be accomplished using sutures or skin adhesive. Sutures can be synthetic, absorbable monofilament (Caprosyn), or synthetic, absorbable, braided multifilament (Polysorb).
Skin adhesive closes incisions quickly, avoids inflammation related to foreign bodies, and can ease patients’ concerns that sometimes arise when absorbable suture persists postoperatively (TABLE 1).
The impact of physician experience
Physician experience has been shown to reduce cost while maintaining quality of care.14 That was the conclusion of researchers who undertook a retrospective study, addressing cost and clinical outcomes, of senior and junior attending physicians who performed laparoscopic-assisted vaginal hysterectomy on 120 patients. Studies such as these often lead to clinical pathways to facilitate cost-effective quality care.
--
CASE Same outcome at lower cost
The hypothetical 43-year-old patient in the opening case undergoes laparoscopic hysterectomy for treatment of uterine fibroids and menorrhagia. In this scenario, however, the surgeon makes the following product choices:
- The patient is prepped with Hibiclens.
- A VCare Plus uterine manipulator is placed.
- Laparoscopic ports include a VersaStep Plus Bladeless Trocar with Step Insufflation Needle; Versaport Plus Pyramidal Bladed Trocar; and 2 VersaOne Bladed trocars.
- The surgeon uses the PKS LYONS Dissecting Forceps and reprocessed Endo Shears to perform the hysterectomy.
- The uterus is enclosed in an Endo Catch bag and removed through the minilaparotomy site.
- The vaginal cuff is closed using 2-0 V-Loc barbed suture. Skin incisions are closed with 4-0 Polysorb, a polyglycolic acid absorbable suture.
The cost of this set of products? $360.44 or, roughly, $1,231.96 less than the set-up described in the case at the beginning of this article (TABLE 2).
Continue to: Summing up
Summing up
Here are key points to take away from this analysis and discussion:
- As third-party payers and hospitals continue to evaluate surgeons individually and compare procedures from surgeon to surgeon, reimbursement might be stratified—thereby favoring physicians who demonstrate both quality outcomes and cost containment.
- There are many ways a minimally invasive surgeon can implement cost-conscious choices that have little or no impact on the quality of outcome.
- Surgeons who are familiar with surgical instruments and models available at their institution are better prepared to make wise cost-conscious decisions. (See “Caregivers should keep cost in mind: Here’s why,” in the Web version of this article at https://www.mdedge.com/obgyn.)
- Cost is not the only indicator of value: The surgeon must know how to apply tools correctly and be familiar with their limitations, and should choose instruments and products for their safety and ease of use. More often than not, a surgeon’s training and personal experience define—and sometimes restrict—the choice of devices.
- Last, it makes sense to have instruments and devices readily available in the operating room at the start of a case, to avoid unnecessary surgical delays. However, we recommend that you refrain from opening these tools until they are required intraoperatively. It is possible that the case will require conversion to laparotomy or that, after direct visualization of the pathology, different ports or instruments are required.
Acknowledgments
The authors would like to thank Meredith Snook, MD, who was coauthor of the original 2013 article6 and Kathleen Riordan, BSN, RN, for assistance in gathering specific cost-related information for this article.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Centers for Medicare & Medicaid Services. National health expenditure projections 2017-2026: Forecast summary. www.cms.gov/Research-Statistics-Data-and-Systems /Statistics-Trends-and-Reports/NationalHealthExpendData /Downloads/ForecastSummary.pdf. Accessed November 3, 2018.
- Vilos GA, Alshimmiri MM. Cost-benefit analysis of laparoscopic versus laparotomy salpingo-oophorectomy for benign tubo-ovarian disease. J Am Assoc Gynecol Laparosc. 1995;2(3):299-303.
- Gray DT, Thorburn J, Lundor P, et al. A cost-effectiveness study of a randomised trial of laparoscopy versus laparotomy for ectopic pregnancy. Lancet. 1995;345(8958):1139-1143.
- Chapron C, Fauconnier A, Goffinet F, et al. Laparoscopic surgery is not inherently dangerous for patients presenting with benign gynaecologic pathology. Results of a metaanalysis. Hum Reprod. 2002;17(5):1334-1342.
- Benezra V, Verma U, Whitted RW. Comparison of laparoscopy versus laparotomy for the surgical treatment of ovarian dermoid cysts. Obstet Gynecol Surv. 2006;61(1): 20-21.
- Sanfilippo JS, Snook ML. Cost-conscious choices for minimally invasive gynecologic surgery. OBG Manag. 2013;25(11):40-41,44,46-48,72.
- Hasson HM. A modified instrument and method for laparoscopy. Am J Obstet Gynecol. 1971;110(6):886-887.
- Ott J, Jaeger-Lansky A, Poschalko G, et al. Entry techniques in gynecologic laparoscopy—a review. Gynecol Surg. 2012;9(2):139-146.
- Ahmad G, Gent D, Henderson D, et al. Laparoscopic entry techniques. Cochrane Database Syst Rev. 2015;8:CD006583.
- Hasson HM, Rotman C, Rana N, et al. Open laparoscopy: 29-year experience. Obstet Gynecol. 2000;96(5 Pt 1):763-766.
- Schäfer M, Lauper M, Krähenbühl L. Trocar and Veress needle injuries during laparoscopy. Surg Endosc. 2001;15(3):275- 280.
- Immediately in effect guidance document: product labeling for laparoscopic power morcellators. Rockville, MD: US Department of Health and Human Services, Food and Drug Administration Center for Devices and Radiological Health; November 25, 2014. www.fda.gov/downloads /MedicalDevices/DeviceRegulationandGuidance /GuidanceDocuments/UCM424123.pdf. Accessed November 3, 2018.
- Tissue Extraction Task Force Members. Morcellation during uterine tissue extraction: an update. J Minim Invasive Gynecol. 2018;25(4):543-550.
- Chang WC, Li TC, Lin CC. The effect of physician experience on costs and clinical outcomes of laparoscopic-assisted vaginal hysterectomy: a multivariate analysis. J Am Assoc Gynecol Laparosc. 2003;10(3):356-359.
- Centers for Medicare & Medicaid Services. National health expenditure projections 2017-2026: Forecast summary. www.cms.gov/Research-Statistics-Data-and-Systems /Statistics-Trends-and-Reports/NationalHealthExpendData /Downloads/ForecastSummary.pdf. Accessed November 3, 2018.
- Vilos GA, Alshimmiri MM. Cost-benefit analysis of laparoscopic versus laparotomy salpingo-oophorectomy for benign tubo-ovarian disease. J Am Assoc Gynecol Laparosc. 1995;2(3):299-303.
- Gray DT, Thorburn J, Lundor P, et al. A cost-effectiveness study of a randomised trial of laparoscopy versus laparotomy for ectopic pregnancy. Lancet. 1995;345(8958):1139-1143.
- Chapron C, Fauconnier A, Goffinet F, et al. Laparoscopic surgery is not inherently dangerous for patients presenting with benign gynaecologic pathology. Results of a metaanalysis. Hum Reprod. 2002;17(5):1334-1342.
- Benezra V, Verma U, Whitted RW. Comparison of laparoscopy versus laparotomy for the surgical treatment of ovarian dermoid cysts. Obstet Gynecol Surv. 2006;61(1): 20-21.
- Sanfilippo JS, Snook ML. Cost-conscious choices for minimally invasive gynecologic surgery. OBG Manag. 2013;25(11):40-41,44,46-48,72.
- Hasson HM. A modified instrument and method for laparoscopy. Am J Obstet Gynecol. 1971;110(6):886-887.
- Ott J, Jaeger-Lansky A, Poschalko G, et al. Entry techniques in gynecologic laparoscopy—a review. Gynecol Surg. 2012;9(2):139-146.
- Ahmad G, Gent D, Henderson D, et al. Laparoscopic entry techniques. Cochrane Database Syst Rev. 2015;8:CD006583.
- Hasson HM, Rotman C, Rana N, et al. Open laparoscopy: 29-year experience. Obstet Gynecol. 2000;96(5 Pt 1):763-766.
- Schäfer M, Lauper M, Krähenbühl L. Trocar and Veress needle injuries during laparoscopy. Surg Endosc. 2001;15(3):275- 280.
- Immediately in effect guidance document: product labeling for laparoscopic power morcellators. Rockville, MD: US Department of Health and Human Services, Food and Drug Administration Center for Devices and Radiological Health; November 25, 2014. www.fda.gov/downloads /MedicalDevices/DeviceRegulationandGuidance /GuidanceDocuments/UCM424123.pdf. Accessed November 3, 2018.
- Tissue Extraction Task Force Members. Morcellation during uterine tissue extraction: an update. J Minim Invasive Gynecol. 2018;25(4):543-550.
- Chang WC, Li TC, Lin CC. The effect of physician experience on costs and clinical outcomes of laparoscopic-assisted vaginal hysterectomy: a multivariate analysis. J Am Assoc Gynecol Laparosc. 2003;10(3):356-359.

Epistaxis: A guide to assessment and management
Epistaxis is a common presenting complaint in family medicine. Successful treatment requires knowledge of nasal anatomy, possible causes, and a step-wise approach.
Epistaxis predominantly affects children between the ages of 2 and 10 years and older adults between the ages of 45 and 65.1-4 Many presentations are spontaneous and self-limiting; often all that is required is proper first aid. It is important, however, to recognize the signs and symptoms that are suggestive of more worrisome conditions.
Management of epistaxis requires good preparation, appropriate equipment, and adequate assistance. If any of these are lacking, prompt nasal packing followed by referral to an emergency department or ear, nose, and throat (ENT) service is recommended.
Anatomy of the nasal cavity
The nasal cavity has a rich and highly varied blood supply arising from the internal and external carotid arteries with multiple anastomoses and a crossover between the left and right arterial systems.2,4,5 The internal maxillary artery (IMAX) supplies 80% of the nasal vault.2 The sphenopalatine artery (SPA) supplies most of the nasal septum and the turbinates, while the greater palatine artery (GPA) supplies the floor of the nasal septum.3,5 The ethmoidal arteries course through the cribriform plate to supply the roof of the nasal cavity. The ethmoidal arteries communicates with branches of the SPA posteriorly and several branches anteriorly (FIGURE 1).

Kiesselbach’s plexus is a highly vascularized region of cartilaginous nasal septum anteroinferiorly that is also known as Little’s area. It is supplied by the SPA, GPA, superior labial artery, and ethmoidal arteries.5 Woodruff’s plexus is the richly vascularized posterior aspect of the nasal cavity primarily supplied by the SPA.3,5
Is the bleed anterior or posterior; primary or secondary?
Epistaxis is classified as anterior or posterior based on the arterial supply and the location of the bleed in relation to the piriform aperture.2,3 Anterior epistaxis occurs in >90% of patients and arises in Little’s area.6 Posterior epistaxis arises from Woodruff’s plexus in the posterior nasal septum or lateral nasal wall. It occurs in 5% to 10% of patients, is usually arterial in origin, and leads to a greater risk of airway compromise, aspiration, and difficulty in controlling the hemorrhage.2,6
Epistaxis can be classified further as primary or secondary hemorrhage. Primary epistaxis is idiopathic, spontaneous bleeds without any precipitants.2 Blood vessels within the nasal mucosa run superficially and are relatively unprotected. Damage to this mucosa and to vessel walls can result in bleeding.4 Spontaneous rupture of vessels may occur occasionally, during, say the Valsalva maneuver or when straining to lift heavy objects.4 Secondary epistaxis occurs when there is a clear and definite cause (eg trauma, anticoagulant use, or surgery).
Continue to: Numerous causes...
Numerous causes: From trauma to medications
Epistaxis can be caused by local, systemic, or environmental factors; medications; or be idiopathic in nature (TABLE 12). It commonly arises due to self-inflicted trauma from nose picking, particularly in children; trauma to nasal bones or septum; and mucosal irritation from topical nasal drugs, such as corticosteroids and antihistamines. Other local factors include septal abnormalities, such as septal perforation, inflammatory diseases, rhinosinusitis, illicit drug use (eg cocaine), iatrogenic causes, and neoplasia.

Red flags for neoplasia include unilateral or asymmetric symptoms, such as nasal blockage, facial pain, rhinorrhea, headaches, facial swelling or deformity, and cranial neuropathies (ie, facial numbness or double vision). Other red flags include Southeast Asian origin (nasopharyngeal carcinoma), loose maxillary teeth, and deep otalgia (TABLE 22). In adolescent males, it is important to consider juvenile nasopharyngeal angiofibroma, a benign tumor that can bleed extensively.

Systemic factors include age, hypertension, alcohol use, acquired coagulopathies due to liver or renal disease, hematologic abnormalities, circadian rhythms, and genetic disorders such as hereditary hemorrhagic telangiectasia (HHT), hemophilia, and von Willebrand’s disease.2
Medications that contribute to epistaxis include antiplatelet agents, such as aspirin and clopidogrel; nonsteroidal anti-inflammatory drugs (NSAIDs); warfarin and novel oral anticoagulants (NOACs); and complementary and alternative medicines, such garlic, gingko, and ginseng. Environmental factors include temperature and humidity.2
Ask about trauma, but also about upper GI hemorrhage
Resuscitation and control of bleeding (which we’ll discuss in a moment) should always take priority. A thorough history and examination are also essential. It’s important to elicit details of the acute episode and any previous episodes, including the duration, severity, frequency, laterality of bleed, and contributing or inciting factors.1,2 Posterior epistaxis often occurs from both nostrils and feels as though blood is dripping down the throat rather than the nose.
Continue to: Hematemesis and melena from upper gastrointestinal hemorrhage...
Hematemesis and melena from upper gastrointestinal hemorrhage can often be overlooked. Elicit history of local trauma, including nose picking, possible foreign body (particularly batteries in children), and recurrent upper respiratory tract infections.
Treatments, including methods previously used to control episodes, can be instructive. Pinching over the nasal bones—rather than the soft cartilaginous part of the nose—unfortunately remains relatively common. Ask about any past medical history that can give clues to the cause of bleeding, such as hypertension, hepatic impairment, easy bruising, family history of coagulation disorders, and social history including alcohol intake, smoking, and recreational drug use—particularly cocaine use. A detailed medication history, as discussed earlier, is vital.
Initial management: Digital pressure
Epistaxis is potentially a life-threatening event. All patients who are actively bleeding require full assessment, resuscitation, and control of the bleeding.4 To protect the airway sit the patient upright and lean them forward to prevent aspiration of blood posteriorly into the pharynx. To control bleeding, get the patient to apply digital pressure at the cartilaginous part of the nose for a minimum of 10 minutes. This provides tamponade of the anterior septal vessels. Applying ice packs around the neck and having the patient suck on ice significantly reduces nasal mucosa blood flow and can slow down the bleeding.7
If there is significant bleeding
Monitor the patient’s vital signs, in particular, the pulse and respiratory rate. Assess the patient’s hemodynamic stability and look for signs of shock, such as sweating and pallor. Insert 2 large-bore (16 G) intravenous cannula and draw blood for type and crossmatch for potential transfusion if significant bleeding has occurred, in high risk patients (eg patients who are elderly or anticoagulated or have a suspected bleeding diathesis), or if further bleeding is likely to occur.2
Consider fluid resuscitation with intravenous saline initially and blood transfusions based on hemoglobin level, symptoms, and history of ischemic heart disease.3,6 Routine clotting studies need to be performed if there is a suspected bleeding diathesis or the patient is anticoagulated. Test for hepatic or renal dysfunction in patients with systemic conditions that could lead to coagulopathy. The clinical state of an elderly patient may deteriorate rapidly, so aggressive resuscitation is vital.4
Continue to: Getting a better look requires the proper equipment
Getting a better look requires the proper equipment
Universal precautions including facemask, eye protection, and gloves should be worn. Have equipment easily accessible, including sufficient lighting and suction. A headlight enables the use of both hands to assess and treat the patient. The nasal cavity often is obscured by clots, so ask the patient to blow and clear their nose. Although this may lead to a recurrence of bleeding, it could assist in identifying the bleeding point.2
Local anesthetic with a vasoconstrictor should be applied to the nasal mucosa over Little’s area either via a solution applied on cotton-tipped applicator or as a nasal spray. Once adequate local anesthesia is achieved, the nasal cavity can be examined and treatment instigated to stem the hemorrhage. Perform anterior rhinoscopy with a Thudicum’s speculum with one hand (FIGURE 2) while suctioning simultaneously with the other. Assess the nasal cavity systematically, paying particular attention to the septum and Little’s area for an anterior bleed. Look for scabbed and excoriated areas.

Anterior bleeds can be managed safely in primary care, provided that appropriate equipment is available. Consider transfer to an emergency department or referral to an ENT specialist if bleeding continues or if a posterior bleed is suspected.2 Examination of the entire nasal cavity via nasendoscopy may be required to identify the source of bleeding—especially with posterior bleeds.
Nonsurgical management
Topical agents
Topical vasoconstrictor and local anaesthetic agents are widely available, and their limited adverse effect profiles make them a convenient first-line therapy.6,8 These agents reduce hemorrhage to allow for better visualization and analgesia for possible cautery or nasal packing.2 Common preparations include cophenylcaine (topical 5% lidocaine solution with 0.5% phenylephrine) and lidocaine injection (0.5%, 1%, or 2%) with epinephrine 1 in 200,000 and cocaine topical solutions (2% or 5%). Topical tranexamic acid has shown significant benefits in acute epistaxis in a systematic review.9
Cautery
If direct pressure and medical therapy fail to stop the bleeding, cautery or nasal packing can be performed.2,8 Chemical cautery entails application of 75% silver nitrate sticks to the bleeding point with firm pressure for 5 to 10 seconds to produce a local chemical burn.4 Only one side of the septum should be cauterized, as there is a small risk of septal perforation resulting from decreased vascularization to the septal cartilage.2,4,8 This can be performed at 4 to 6 week intervals. Electric bipolar cautery with a metal loop is performed by otolaryngologists under local anesthesia.4 Compared with electric cautery, silver nitrate cautery is cheap, readily available, easy to perform, equal in effectiveness, and has fewer complications.10
Continue to: Nasal packing...
Nasal packing
Nasal packing can be performed if cautery is unsuccessful in controlling the bleed or if no bleeding point is seen on examination.2 It provides direct mechanical compression and acts as a platelet aggregator, thereby facilitating coagulation.
Anterior packing. Packs should be directed posteriorly along the floor of the nasal cavity, rather than superiorly.2 After packing, examine the patient for ongoing bleeding from the contralateral nares or posteriorly in the oropharynx using a tongue depressor. If bleeding is seen, consider packing the other side before removal of the already inserted pack to increase the tamponade pressure over the septum.4
Anterior packs are effective, easy to use, widely available, and inexpensive.8 Types of packs include traditional packing, nasal tampons, and absorbable packing materials.
The Rapid Rhino is also an option. It’s an inflatable balloon pack coated with a lubricating compound. It remains in contact with the mucosa when deflated and can be left in situ for up to 4 days (FIGURE 3). It has the same rate of control of epistaxis when compared with polyvinyl alcohol. Both patients and physicians found insertion and removal of the Rapid Rhino easier with less patient discomfort.11-13

Absorbable packs do not require formal removal and are useful for patients with or without coagulopathies. They can be applied topically with a syringe that conforms to the 3-dimensional structure of the nasal cavity.1 The decision regarding which product to use is based on availability, cost, and physician preference.
Continue to: Posterior packing...
Posterior packing may be required if epistaxis continues despite anterior packing and may take the form of a balloon or a formal pack. A Foley catheter inflated with 3 to 4 mL of water or air is inserted through the anterior nares, along the floor of the nasal cavity into the posterior pharynx and pulled forward until the balloon engages the posterior choana (FIGURE 4). This provides local tamponade and tamponade at the sphenopalatine foramen.2,4 The balloon is held firmly in place with an umbilical clamp at the anterior nares. To prevent pressure necrosis, the columella can be protected with a soft dressing that is regularly checked by the nursing staff. The nasal cavity is then packed anteriorly with ribbon gauze or a nasal sponge to stem any potential anterior bleeds.

Potential complications include posterior displacement of the balloon with potential airway compromise, deflation in situ, and rupture of the balloon—which could result in aspiration.4 It is important to note the Foley catheter is, in fact, not licensed for nasal use.4 Insertion only should be performed by a clinician who has been trained in this skill.
Traditional nasopharyngeal packs are rolled gauze attached to tapes or sutured to a catheter. Compared with balloons, they were found to be more effective in controlling epistaxis and produce less short- and long-term complications.2 However, they are rather uncomfortable and hence normally performed under general anesthesia.4 Posterior packing has many disadvantages. They have a 50% failure rate, which increases to 70% in patients with bleeding disorders.8
Complications vary from mild and self-limiting such as infection, hemorrhage, and pressure effects to severe such as toxic shock syndrome, myocardial infarction, and death (TABLE 32-4,6,8). There is little evidence supporting the use of prophylactic oral antibiotics after packing. Prophylactic antibiotics are reserved for those with posterior packs or if packs remain in situ for more than 24 hours.6

Warm water irrigation
Warm water irrigation via Foley catheter has a reported 82% success rate.8 It results in earlier discharge, less pain, less trauma to the nose, and reduced hospital length of stay.13 The balloon catheter is used to close off the posterior choana and water irrigation is applied at 45° C to 50° C for about 3 minutes with the help of a caloric stimulator.4,8 It helps clear blood clots from the nose and reduces local blood flow by causing mucosal edema, which compresses the bleeding vessels.
Continue to: Surgical management
Surgical management
Any bleeding that fails to stop, despite an escalation of management, requires surgical intervention. This includes cases in which the bleeding continues after pack removal.4 Options include4:
- Diathermy, with bipolar or radiofrequency laser, can be used to localize the bleeding site.
- Septoplasty allows for better access to the nasal cavity, reduction of blood flow to the nasal mucosa by raising a mucoperichondrial flap, correction of a deviated septum, and removal of a septal spur that may be responsible for the epistaxis.
- Arterial ligation involves identification of the bleeding vessel that is clipped or coagulated with bipolar diathermy.
- Endoscopic SPA ligation is an excellent, well-tolerated, and cost-effective method of treating recurrent epistaxis.6,14 It controls 98% of posterior epistaxis, and is superior to posterior nasal packing and embolization.2,3,10 It results in a shorter hospital stay, reduction in repeated hemorrhage and painful packing procedures, and a cost saving of >$7,000 per patient if performed early.7 Concomitant ligation of the anterior ethmoidal artery may be performed in traumatic epistaxis or when severe bleeding is from the ethmoidal region.4,6
- Ligation of the IMAX and external carotid arteries is performed rarely due to potential complications and high failure rates.
Arterial embolization
When arterial ligation fails, or is not possible due to anesthetic concerns, selective embolization of the maxillary or facial arteries by specialist radiologists can be considered.6 Access to the vascular system through a femoral punch leads to identification of the bleeding point. A catheter is then placed in the artery and the bleeding vessel is embolized. Possible candidates include patients with HHT, bleeding tumors, poor surgical candidates, or patient preference.3
Other management considerations
Once bleeding is controlled, factors that contributed to the epistaxis should be addressed.3 Hypertension needs to be managed. Antiplatelet or anticoagulant therapy may need to be temporarily halted in consultation with specialist physicians. Local treatments such as cautery are unlikely to be effective in patients who are anticoagulated. Nasal packing with a ‘procoagulant’ dressing, such as Kaltostat or Rapid Rhino, is often required.
Patient education and follow-up
Patients should be started on saline sprays or irrigation to maintain nasal hygiene after acute epistaxis. It’s a good idea to teach patients about proper first aid for recurrence (eg, sitting upright with digital pressure applied to the cartilaginous part of the nose, ice packs around the neck and ice to suck) and to encourage them to refrain from activities that may stimulate bleeding (blowing or picking the nose, heavy lifting, strenuous exercise). Also advise patients to abstain from alcohol and hot drinks that cause vasodilatation of nasal vessels as much as possible.4 Advise patients that topical gels, lotions, and ointments such as kenacomb, nasalate, or paraffin can be used to moisturise the mucosa and promote healing.1
All patients with a history of severe or recurrent epistaxis require formal examination of the nasal cavity to rule out a neoplastic lesion.
CORRESPONDENCE
Amy Wong, BMBS, Department ENT/Head and Neck Surgery, Monash ENT Building, PO Box 72, Rear 867 Centre Road, Bentleigh East 3165 Australia, [email protected].
1. Schlosser RJ. Clinical practice. Epistaxis. N Engl J Med. 2009;360:784-789.
2. Yau S. An update on epistaxis. Aust Fam Physician. 2015;44:653-656.
3. McClurg SW, Carrau R. Endoscopic management of posterior epistaxis: a review. Acta Otorhinolaryngol Ital. 2014;34:1-8.
4. Pope LE, Hobbs CG. Epistaxis: an update on current management. Postgrad Med J. 2005;81:309-314.
5. Dubel GJ, Ahn SH, Soares GM. Transcatheter embolization in the management of epistaxis. Semin Intervent Radiol. 2013;30:249-262.
6. Spielmann PM, Barnes ML, White PS. Controversies in the specialist management of adult epistaxis: an evidence-based review. Clin Otolaryngol. 2012;37:382-389.
7. Porter M, Marais J, Tolley N. The effect of ice packs upon nasal mucosal blood flow. Acta Otolaryngol. 1991;111:1122-1125.
8. Traboulsi H, Alam E, Hadi U. Changing Trends in the Management of Epistaxis. Int J Otolaryngol. 2015;2015:263987.
9. Kamhieh Y, Fox H. Tranexamic acid in epistaxis: a systematic review. Clin Otolaryngol. 2016;41:771-776.
10. Stangerup SE, Dommerby H, Siim C, et al. New modification of hot-water irrigation in the treatment of posterior epistaxis. Arch Otolaryngol Head Neck Surg. 1999;125:686-690.
11. Douglas R, Wormald PJ. Update on epistaxis. Curr Opin Otolaryngol Head Neck Surg. 2007;15:180-183.
12. Badran K, Malik TH, Belloso A, et al. Randomized controlled trial comparing Merocel and RapidRhino packing in the management of anterior epistaxis. Clin Otolaryngol. 2005;30:333-337.
13. Moumoulidis I, Draper MR, Patel H, et al. A prospective randomised controlled trial comparing Merocel and Rapid Rhino nasal tampons in the treatment of epistaxis. Eur Arch Otorhinolaryngol. 2006;263:719-722.
14. Moshaver A, Harris JR, Liu R, et al. Early operative intervention versus conventional treatment in epistaxis: randomized prospective trial. J Otolaryngol. 2004;33:185-188.
Epistaxis is a common presenting complaint in family medicine. Successful treatment requires knowledge of nasal anatomy, possible causes, and a step-wise approach.
Epistaxis predominantly affects children between the ages of 2 and 10 years and older adults between the ages of 45 and 65.1-4 Many presentations are spontaneous and self-limiting; often all that is required is proper first aid. It is important, however, to recognize the signs and symptoms that are suggestive of more worrisome conditions.
Management of epistaxis requires good preparation, appropriate equipment, and adequate assistance. If any of these are lacking, prompt nasal packing followed by referral to an emergency department or ear, nose, and throat (ENT) service is recommended.
Anatomy of the nasal cavity
The nasal cavity has a rich and highly varied blood supply arising from the internal and external carotid arteries with multiple anastomoses and a crossover between the left and right arterial systems.2,4,5 The internal maxillary artery (IMAX) supplies 80% of the nasal vault.2 The sphenopalatine artery (SPA) supplies most of the nasal septum and the turbinates, while the greater palatine artery (GPA) supplies the floor of the nasal septum.3,5 The ethmoidal arteries course through the cribriform plate to supply the roof of the nasal cavity. The ethmoidal arteries communicates with branches of the SPA posteriorly and several branches anteriorly (FIGURE 1).
Kiesselbach’s plexus is a highly vascularized region of cartilaginous nasal septum anteroinferiorly that is also known as Little’s area. It is supplied by the SPA, GPA, superior labial artery, and ethmoidal arteries.5 Woodruff’s plexus is the richly vascularized posterior aspect of the nasal cavity primarily supplied by the SPA.3,5
Is the bleed anterior or posterior; primary or secondary?
Epistaxis is classified as anterior or posterior based on the arterial supply and the location of the bleed in relation to the piriform aperture.2,3 Anterior epistaxis occurs in >90% of patients and arises in Little’s area.6 Posterior epistaxis arises from Woodruff’s plexus in the posterior nasal septum or lateral nasal wall. It occurs in 5% to 10% of patients, is usually arterial in origin, and leads to a greater risk of airway compromise, aspiration, and difficulty in controlling the hemorrhage.2,6
Epistaxis can be classified further as primary or secondary hemorrhage. Primary epistaxis is idiopathic, spontaneous bleeds without any precipitants.2 Blood vessels within the nasal mucosa run superficially and are relatively unprotected. Damage to this mucosa and to vessel walls can result in bleeding.4 Spontaneous rupture of vessels may occur occasionally, during, say the Valsalva maneuver or when straining to lift heavy objects.4 Secondary epistaxis occurs when there is a clear and definite cause (eg trauma, anticoagulant use, or surgery).
Continue to: Numerous causes...
Numerous causes: From trauma to medications
Epistaxis can be caused by local, systemic, or environmental factors; medications; or be idiopathic in nature (TABLE 12). It commonly arises due to self-inflicted trauma from nose picking, particularly in children; trauma to nasal bones or septum; and mucosal irritation from topical nasal drugs, such as corticosteroids and antihistamines. Other local factors include septal abnormalities, such as septal perforation, inflammatory diseases, rhinosinusitis, illicit drug use (eg cocaine), iatrogenic causes, and neoplasia.
Red flags for neoplasia include unilateral or asymmetric symptoms, such as nasal blockage, facial pain, rhinorrhea, headaches, facial swelling or deformity, and cranial neuropathies (ie, facial numbness or double vision). Other red flags include Southeast Asian origin (nasopharyngeal carcinoma), loose maxillary teeth, and deep otalgia (TABLE 22). In adolescent males, it is important to consider juvenile nasopharyngeal angiofibroma, a benign tumor that can bleed extensively.
Systemic factors include age, hypertension, alcohol use, acquired coagulopathies due to liver or renal disease, hematologic abnormalities, circadian rhythms, and genetic disorders such as hereditary hemorrhagic telangiectasia (HHT), hemophilia, and von Willebrand’s disease.2
Medications that contribute to epistaxis include antiplatelet agents, such as aspirin and clopidogrel; nonsteroidal anti-inflammatory drugs (NSAIDs); warfarin and novel oral anticoagulants (NOACs); and complementary and alternative medicines, such garlic, gingko, and ginseng. Environmental factors include temperature and humidity.2
Ask about trauma, but also about upper GI hemorrhage
Resuscitation and control of bleeding (which we’ll discuss in a moment) should always take priority. A thorough history and examination are also essential. It’s important to elicit details of the acute episode and any previous episodes, including the duration, severity, frequency, laterality of bleed, and contributing or inciting factors.1,2 Posterior epistaxis often occurs from both nostrils and feels as though blood is dripping down the throat rather than the nose.
Continue to: Hematemesis and melena from upper gastrointestinal hemorrhage...
Hematemesis and melena from upper gastrointestinal hemorrhage can often be overlooked. Elicit history of local trauma, including nose picking, possible foreign body (particularly batteries in children), and recurrent upper respiratory tract infections.
Treatments, including methods previously used to control episodes, can be instructive. Pinching over the nasal bones—rather than the soft cartilaginous part of the nose—unfortunately remains relatively common. Ask about any past medical history that can give clues to the cause of bleeding, such as hypertension, hepatic impairment, easy bruising, family history of coagulation disorders, and social history including alcohol intake, smoking, and recreational drug use—particularly cocaine use. A detailed medication history, as discussed earlier, is vital.
Initial management: Digital pressure
Epistaxis is potentially a life-threatening event. All patients who are actively bleeding require full assessment, resuscitation, and control of the bleeding.4 To protect the airway sit the patient upright and lean them forward to prevent aspiration of blood posteriorly into the pharynx. To control bleeding, get the patient to apply digital pressure at the cartilaginous part of the nose for a minimum of 10 minutes. This provides tamponade of the anterior septal vessels. Applying ice packs around the neck and having the patient suck on ice significantly reduces nasal mucosa blood flow and can slow down the bleeding.7
If there is significant bleeding
Monitor the patient’s vital signs, in particular, the pulse and respiratory rate. Assess the patient’s hemodynamic stability and look for signs of shock, such as sweating and pallor. Insert 2 large-bore (16 G) intravenous cannula and draw blood for type and crossmatch for potential transfusion if significant bleeding has occurred, in high risk patients (eg patients who are elderly or anticoagulated or have a suspected bleeding diathesis), or if further bleeding is likely to occur.2
Consider fluid resuscitation with intravenous saline initially and blood transfusions based on hemoglobin level, symptoms, and history of ischemic heart disease.3,6 Routine clotting studies need to be performed if there is a suspected bleeding diathesis or the patient is anticoagulated. Test for hepatic or renal dysfunction in patients with systemic conditions that could lead to coagulopathy. The clinical state of an elderly patient may deteriorate rapidly, so aggressive resuscitation is vital.4
Continue to: Getting a better look requires the proper equipment
Getting a better look requires the proper equipment
Universal precautions including facemask, eye protection, and gloves should be worn. Have equipment easily accessible, including sufficient lighting and suction. A headlight enables the use of both hands to assess and treat the patient. The nasal cavity often is obscured by clots, so ask the patient to blow and clear their nose. Although this may lead to a recurrence of bleeding, it could assist in identifying the bleeding point.2
Local anesthetic with a vasoconstrictor should be applied to the nasal mucosa over Little’s area either via a solution applied on cotton-tipped applicator or as a nasal spray. Once adequate local anesthesia is achieved, the nasal cavity can be examined and treatment instigated to stem the hemorrhage. Perform anterior rhinoscopy with a Thudicum’s speculum with one hand (FIGURE 2) while suctioning simultaneously with the other. Assess the nasal cavity systematically, paying particular attention to the septum and Little’s area for an anterior bleed. Look for scabbed and excoriated areas.
Anterior bleeds can be managed safely in primary care, provided that appropriate equipment is available. Consider transfer to an emergency department or referral to an ENT specialist if bleeding continues or if a posterior bleed is suspected.2 Examination of the entire nasal cavity via nasendoscopy may be required to identify the source of bleeding—especially with posterior bleeds.
Nonsurgical management
Topical agents
Topical vasoconstrictor and local anaesthetic agents are widely available, and their limited adverse effect profiles make them a convenient first-line therapy.6,8 These agents reduce hemorrhage to allow for better visualization and analgesia for possible cautery or nasal packing.2 Common preparations include cophenylcaine (topical 5% lidocaine solution with 0.5% phenylephrine) and lidocaine injection (0.5%, 1%, or 2%) with epinephrine 1 in 200,000 and cocaine topical solutions (2% or 5%). Topical tranexamic acid has shown significant benefits in acute epistaxis in a systematic review.9
Cautery
If direct pressure and medical therapy fail to stop the bleeding, cautery or nasal packing can be performed.2,8 Chemical cautery entails application of 75% silver nitrate sticks to the bleeding point with firm pressure for 5 to 10 seconds to produce a local chemical burn.4 Only one side of the septum should be cauterized, as there is a small risk of septal perforation resulting from decreased vascularization to the septal cartilage.2,4,8 This can be performed at 4 to 6 week intervals. Electric bipolar cautery with a metal loop is performed by otolaryngologists under local anesthesia.4 Compared with electric cautery, silver nitrate cautery is cheap, readily available, easy to perform, equal in effectiveness, and has fewer complications.10
Continue to: Nasal packing...
Nasal packing
Nasal packing can be performed if cautery is unsuccessful in controlling the bleed or if no bleeding point is seen on examination.2 It provides direct mechanical compression and acts as a platelet aggregator, thereby facilitating coagulation.
Anterior packing. Packs should be directed posteriorly along the floor of the nasal cavity, rather than superiorly.2 After packing, examine the patient for ongoing bleeding from the contralateral nares or posteriorly in the oropharynx using a tongue depressor. If bleeding is seen, consider packing the other side before removal of the already inserted pack to increase the tamponade pressure over the septum.4
Anterior packs are effective, easy to use, widely available, and inexpensive.8 Types of packs include traditional packing, nasal tampons, and absorbable packing materials.
The Rapid Rhino is also an option. It’s an inflatable balloon pack coated with a lubricating compound. It remains in contact with the mucosa when deflated and can be left in situ for up to 4 days (FIGURE 3). It has the same rate of control of epistaxis when compared with polyvinyl alcohol. Both patients and physicians found insertion and removal of the Rapid Rhino easier with less patient discomfort.11-13
Absorbable packs do not require formal removal and are useful for patients with or without coagulopathies. They can be applied topically with a syringe that conforms to the 3-dimensional structure of the nasal cavity.1 The decision regarding which product to use is based on availability, cost, and physician preference.
Continue to: Posterior packing...
Posterior packing may be required if epistaxis continues despite anterior packing and may take the form of a balloon or a formal pack. A Foley catheter inflated with 3 to 4 mL of water or air is inserted through the anterior nares, along the floor of the nasal cavity into the posterior pharynx and pulled forward until the balloon engages the posterior choana (FIGURE 4). This provides local tamponade and tamponade at the sphenopalatine foramen.2,4 The balloon is held firmly in place with an umbilical clamp at the anterior nares. To prevent pressure necrosis, the columella can be protected with a soft dressing that is regularly checked by the nursing staff. The nasal cavity is then packed anteriorly with ribbon gauze or a nasal sponge to stem any potential anterior bleeds.
Potential complications include posterior displacement of the balloon with potential airway compromise, deflation in situ, and rupture of the balloon—which could result in aspiration.4 It is important to note the Foley catheter is, in fact, not licensed for nasal use.4 Insertion only should be performed by a clinician who has been trained in this skill.
Traditional nasopharyngeal packs are rolled gauze attached to tapes or sutured to a catheter. Compared with balloons, they were found to be more effective in controlling epistaxis and produce less short- and long-term complications.2 However, they are rather uncomfortable and hence normally performed under general anesthesia.4 Posterior packing has many disadvantages. They have a 50% failure rate, which increases to 70% in patients with bleeding disorders.8
Complications vary from mild and self-limiting such as infection, hemorrhage, and pressure effects to severe such as toxic shock syndrome, myocardial infarction, and death (TABLE 32-4,6,8). There is little evidence supporting the use of prophylactic oral antibiotics after packing. Prophylactic antibiotics are reserved for those with posterior packs or if packs remain in situ for more than 24 hours.6
Warm water irrigation
Warm water irrigation via Foley catheter has a reported 82% success rate.8 It results in earlier discharge, less pain, less trauma to the nose, and reduced hospital length of stay.13 The balloon catheter is used to close off the posterior choana and water irrigation is applied at 45° C to 50° C for about 3 minutes with the help of a caloric stimulator.4,8 It helps clear blood clots from the nose and reduces local blood flow by causing mucosal edema, which compresses the bleeding vessels.
Continue to: Surgical management
Surgical management
Any bleeding that fails to stop, despite an escalation of management, requires surgical intervention. This includes cases in which the bleeding continues after pack removal.4 Options include4:
- Diathermy, with bipolar or radiofrequency laser, can be used to localize the bleeding site.
- Septoplasty allows for better access to the nasal cavity, reduction of blood flow to the nasal mucosa by raising a mucoperichondrial flap, correction of a deviated septum, and removal of a septal spur that may be responsible for the epistaxis.
- Arterial ligation involves identification of the bleeding vessel that is clipped or coagulated with bipolar diathermy.
- Endoscopic SPA ligation is an excellent, well-tolerated, and cost-effective method of treating recurrent epistaxis.6,14 It controls 98% of posterior epistaxis, and is superior to posterior nasal packing and embolization.2,3,10 It results in a shorter hospital stay, reduction in repeated hemorrhage and painful packing procedures, and a cost saving of >$7,000 per patient if performed early.7 Concomitant ligation of the anterior ethmoidal artery may be performed in traumatic epistaxis or when severe bleeding is from the ethmoidal region.4,6
- Ligation of the IMAX and external carotid arteries is performed rarely due to potential complications and high failure rates.
Arterial embolization
When arterial ligation fails, or is not possible due to anesthetic concerns, selective embolization of the maxillary or facial arteries by specialist radiologists can be considered.6 Access to the vascular system through a femoral punch leads to identification of the bleeding point. A catheter is then placed in the artery and the bleeding vessel is embolized. Possible candidates include patients with HHT, bleeding tumors, poor surgical candidates, or patient preference.3
Other management considerations
Once bleeding is controlled, factors that contributed to the epistaxis should be addressed.3 Hypertension needs to be managed. Antiplatelet or anticoagulant therapy may need to be temporarily halted in consultation with specialist physicians. Local treatments such as cautery are unlikely to be effective in patients who are anticoagulated. Nasal packing with a ‘procoagulant’ dressing, such as Kaltostat or Rapid Rhino, is often required.
Patient education and follow-up
Patients should be started on saline sprays or irrigation to maintain nasal hygiene after acute epistaxis. It’s a good idea to teach patients about proper first aid for recurrence (eg, sitting upright with digital pressure applied to the cartilaginous part of the nose, ice packs around the neck and ice to suck) and to encourage them to refrain from activities that may stimulate bleeding (blowing or picking the nose, heavy lifting, strenuous exercise). Also advise patients to abstain from alcohol and hot drinks that cause vasodilatation of nasal vessels as much as possible.4 Advise patients that topical gels, lotions, and ointments such as kenacomb, nasalate, or paraffin can be used to moisturise the mucosa and promote healing.1
All patients with a history of severe or recurrent epistaxis require formal examination of the nasal cavity to rule out a neoplastic lesion.
CORRESPONDENCE
Amy Wong, BMBS, Department ENT/Head and Neck Surgery, Monash ENT Building, PO Box 72, Rear 867 Centre Road, Bentleigh East 3165 Australia, [email protected].
Epistaxis is a common presenting complaint in family medicine. Successful treatment requires knowledge of nasal anatomy, possible causes, and a step-wise approach.
Epistaxis predominantly affects children between the ages of 2 and 10 years and older adults between the ages of 45 and 65.1-4 Many presentations are spontaneous and self-limiting; often all that is required is proper first aid. It is important, however, to recognize the signs and symptoms that are suggestive of more worrisome conditions.
Management of epistaxis requires good preparation, appropriate equipment, and adequate assistance. If any of these are lacking, prompt nasal packing followed by referral to an emergency department or ear, nose, and throat (ENT) service is recommended.
Anatomy of the nasal cavity
The nasal cavity has a rich and highly varied blood supply arising from the internal and external carotid arteries with multiple anastomoses and a crossover between the left and right arterial systems.2,4,5 The internal maxillary artery (IMAX) supplies 80% of the nasal vault.2 The sphenopalatine artery (SPA) supplies most of the nasal septum and the turbinates, while the greater palatine artery (GPA) supplies the floor of the nasal septum.3,5 The ethmoidal arteries course through the cribriform plate to supply the roof of the nasal cavity. The ethmoidal arteries communicates with branches of the SPA posteriorly and several branches anteriorly (FIGURE 1).
Kiesselbach’s plexus is a highly vascularized region of cartilaginous nasal septum anteroinferiorly that is also known as Little’s area. It is supplied by the SPA, GPA, superior labial artery, and ethmoidal arteries.5 Woodruff’s plexus is the richly vascularized posterior aspect of the nasal cavity primarily supplied by the SPA.3,5
Is the bleed anterior or posterior; primary or secondary?
Epistaxis is classified as anterior or posterior based on the arterial supply and the location of the bleed in relation to the piriform aperture.2,3 Anterior epistaxis occurs in >90% of patients and arises in Little’s area.6 Posterior epistaxis arises from Woodruff’s plexus in the posterior nasal septum or lateral nasal wall. It occurs in 5% to 10% of patients, is usually arterial in origin, and leads to a greater risk of airway compromise, aspiration, and difficulty in controlling the hemorrhage.2,6
Epistaxis can be classified further as primary or secondary hemorrhage. Primary epistaxis is idiopathic, spontaneous bleeds without any precipitants.2 Blood vessels within the nasal mucosa run superficially and are relatively unprotected. Damage to this mucosa and to vessel walls can result in bleeding.4 Spontaneous rupture of vessels may occur occasionally, during, say the Valsalva maneuver or when straining to lift heavy objects.4 Secondary epistaxis occurs when there is a clear and definite cause (eg trauma, anticoagulant use, or surgery).
Continue to: Numerous causes...
Numerous causes: From trauma to medications
Epistaxis can be caused by local, systemic, or environmental factors; medications; or be idiopathic in nature (TABLE 12). It commonly arises due to self-inflicted trauma from nose picking, particularly in children; trauma to nasal bones or septum; and mucosal irritation from topical nasal drugs, such as corticosteroids and antihistamines. Other local factors include septal abnormalities, such as septal perforation, inflammatory diseases, rhinosinusitis, illicit drug use (eg cocaine), iatrogenic causes, and neoplasia.
Red flags for neoplasia include unilateral or asymmetric symptoms, such as nasal blockage, facial pain, rhinorrhea, headaches, facial swelling or deformity, and cranial neuropathies (ie, facial numbness or double vision). Other red flags include Southeast Asian origin (nasopharyngeal carcinoma), loose maxillary teeth, and deep otalgia (TABLE 22). In adolescent males, it is important to consider juvenile nasopharyngeal angiofibroma, a benign tumor that can bleed extensively.
Systemic factors include age, hypertension, alcohol use, acquired coagulopathies due to liver or renal disease, hematologic abnormalities, circadian rhythms, and genetic disorders such as hereditary hemorrhagic telangiectasia (HHT), hemophilia, and von Willebrand’s disease.2
Medications that contribute to epistaxis include antiplatelet agents, such as aspirin and clopidogrel; nonsteroidal anti-inflammatory drugs (NSAIDs); warfarin and novel oral anticoagulants (NOACs); and complementary and alternative medicines, such garlic, gingko, and ginseng. Environmental factors include temperature and humidity.2
Ask about trauma, but also about upper GI hemorrhage
Resuscitation and control of bleeding (which we’ll discuss in a moment) should always take priority. A thorough history and examination are also essential. It’s important to elicit details of the acute episode and any previous episodes, including the duration, severity, frequency, laterality of bleed, and contributing or inciting factors.1,2 Posterior epistaxis often occurs from both nostrils and feels as though blood is dripping down the throat rather than the nose.
Continue to: Hematemesis and melena from upper gastrointestinal hemorrhage...
Hematemesis and melena from upper gastrointestinal hemorrhage can often be overlooked. Elicit history of local trauma, including nose picking, possible foreign body (particularly batteries in children), and recurrent upper respiratory tract infections.
Treatments, including methods previously used to control episodes, can be instructive. Pinching over the nasal bones—rather than the soft cartilaginous part of the nose—unfortunately remains relatively common. Ask about any past medical history that can give clues to the cause of bleeding, such as hypertension, hepatic impairment, easy bruising, family history of coagulation disorders, and social history including alcohol intake, smoking, and recreational drug use—particularly cocaine use. A detailed medication history, as discussed earlier, is vital.
Initial management: Digital pressure
Epistaxis is potentially a life-threatening event. All patients who are actively bleeding require full assessment, resuscitation, and control of the bleeding.4 To protect the airway sit the patient upright and lean them forward to prevent aspiration of blood posteriorly into the pharynx. To control bleeding, get the patient to apply digital pressure at the cartilaginous part of the nose for a minimum of 10 minutes. This provides tamponade of the anterior septal vessels. Applying ice packs around the neck and having the patient suck on ice significantly reduces nasal mucosa blood flow and can slow down the bleeding.7
If there is significant bleeding
Monitor the patient’s vital signs, in particular, the pulse and respiratory rate. Assess the patient’s hemodynamic stability and look for signs of shock, such as sweating and pallor. Insert 2 large-bore (16 G) intravenous cannula and draw blood for type and crossmatch for potential transfusion if significant bleeding has occurred, in high risk patients (eg patients who are elderly or anticoagulated or have a suspected bleeding diathesis), or if further bleeding is likely to occur.2
Consider fluid resuscitation with intravenous saline initially and blood transfusions based on hemoglobin level, symptoms, and history of ischemic heart disease.3,6 Routine clotting studies need to be performed if there is a suspected bleeding diathesis or the patient is anticoagulated. Test for hepatic or renal dysfunction in patients with systemic conditions that could lead to coagulopathy. The clinical state of an elderly patient may deteriorate rapidly, so aggressive resuscitation is vital.4
Continue to: Getting a better look requires the proper equipment
Getting a better look requires the proper equipment
Universal precautions including facemask, eye protection, and gloves should be worn. Have equipment easily accessible, including sufficient lighting and suction. A headlight enables the use of both hands to assess and treat the patient. The nasal cavity often is obscured by clots, so ask the patient to blow and clear their nose. Although this may lead to a recurrence of bleeding, it could assist in identifying the bleeding point.2
Local anesthetic with a vasoconstrictor should be applied to the nasal mucosa over Little’s area either via a solution applied on cotton-tipped applicator or as a nasal spray. Once adequate local anesthesia is achieved, the nasal cavity can be examined and treatment instigated to stem the hemorrhage. Perform anterior rhinoscopy with a Thudicum’s speculum with one hand (FIGURE 2) while suctioning simultaneously with the other. Assess the nasal cavity systematically, paying particular attention to the septum and Little’s area for an anterior bleed. Look for scabbed and excoriated areas.
Anterior bleeds can be managed safely in primary care, provided that appropriate equipment is available. Consider transfer to an emergency department or referral to an ENT specialist if bleeding continues or if a posterior bleed is suspected.2 Examination of the entire nasal cavity via nasendoscopy may be required to identify the source of bleeding—especially with posterior bleeds.
Nonsurgical management
Topical agents
Topical vasoconstrictor and local anaesthetic agents are widely available, and their limited adverse effect profiles make them a convenient first-line therapy.6,8 These agents reduce hemorrhage to allow for better visualization and analgesia for possible cautery or nasal packing.2 Common preparations include cophenylcaine (topical 5% lidocaine solution with 0.5% phenylephrine) and lidocaine injection (0.5%, 1%, or 2%) with epinephrine 1 in 200,000 and cocaine topical solutions (2% or 5%). Topical tranexamic acid has shown significant benefits in acute epistaxis in a systematic review.9
Cautery
If direct pressure and medical therapy fail to stop the bleeding, cautery or nasal packing can be performed.2,8 Chemical cautery entails application of 75% silver nitrate sticks to the bleeding point with firm pressure for 5 to 10 seconds to produce a local chemical burn.4 Only one side of the septum should be cauterized, as there is a small risk of septal perforation resulting from decreased vascularization to the septal cartilage.2,4,8 This can be performed at 4 to 6 week intervals. Electric bipolar cautery with a metal loop is performed by otolaryngologists under local anesthesia.4 Compared with electric cautery, silver nitrate cautery is cheap, readily available, easy to perform, equal in effectiveness, and has fewer complications.10
Continue to: Nasal packing...
Nasal packing
Nasal packing can be performed if cautery is unsuccessful in controlling the bleed or if no bleeding point is seen on examination.2 It provides direct mechanical compression and acts as a platelet aggregator, thereby facilitating coagulation.
Anterior packing. Packs should be directed posteriorly along the floor of the nasal cavity, rather than superiorly.2 After packing, examine the patient for ongoing bleeding from the contralateral nares or posteriorly in the oropharynx using a tongue depressor. If bleeding is seen, consider packing the other side before removal of the already inserted pack to increase the tamponade pressure over the septum.4
Anterior packs are effective, easy to use, widely available, and inexpensive.8 Types of packs include traditional packing, nasal tampons, and absorbable packing materials.
The Rapid Rhino is also an option. It’s an inflatable balloon pack coated with a lubricating compound. It remains in contact with the mucosa when deflated and can be left in situ for up to 4 days (FIGURE 3). It has the same rate of control of epistaxis when compared with polyvinyl alcohol. Both patients and physicians found insertion and removal of the Rapid Rhino easier with less patient discomfort.11-13
Absorbable packs do not require formal removal and are useful for patients with or without coagulopathies. They can be applied topically with a syringe that conforms to the 3-dimensional structure of the nasal cavity.1 The decision regarding which product to use is based on availability, cost, and physician preference.
Continue to: Posterior packing...
Posterior packing may be required if epistaxis continues despite anterior packing and may take the form of a balloon or a formal pack. A Foley catheter inflated with 3 to 4 mL of water or air is inserted through the anterior nares, along the floor of the nasal cavity into the posterior pharynx and pulled forward until the balloon engages the posterior choana (FIGURE 4). This provides local tamponade and tamponade at the sphenopalatine foramen.2,4 The balloon is held firmly in place with an umbilical clamp at the anterior nares. To prevent pressure necrosis, the columella can be protected with a soft dressing that is regularly checked by the nursing staff. The nasal cavity is then packed anteriorly with ribbon gauze or a nasal sponge to stem any potential anterior bleeds.
Potential complications include posterior displacement of the balloon with potential airway compromise, deflation in situ, and rupture of the balloon—which could result in aspiration.4 It is important to note the Foley catheter is, in fact, not licensed for nasal use.4 Insertion only should be performed by a clinician who has been trained in this skill.
Traditional nasopharyngeal packs are rolled gauze attached to tapes or sutured to a catheter. Compared with balloons, they were found to be more effective in controlling epistaxis and produce less short- and long-term complications.2 However, they are rather uncomfortable and hence normally performed under general anesthesia.4 Posterior packing has many disadvantages. They have a 50% failure rate, which increases to 70% in patients with bleeding disorders.8
Complications vary from mild and self-limiting such as infection, hemorrhage, and pressure effects to severe such as toxic shock syndrome, myocardial infarction, and death (TABLE 32-4,6,8). There is little evidence supporting the use of prophylactic oral antibiotics after packing. Prophylactic antibiotics are reserved for those with posterior packs or if packs remain in situ for more than 24 hours.6
Warm water irrigation
Warm water irrigation via Foley catheter has a reported 82% success rate.8 It results in earlier discharge, less pain, less trauma to the nose, and reduced hospital length of stay.13 The balloon catheter is used to close off the posterior choana and water irrigation is applied at 45° C to 50° C for about 3 minutes with the help of a caloric stimulator.4,8 It helps clear blood clots from the nose and reduces local blood flow by causing mucosal edema, which compresses the bleeding vessels.
Continue to: Surgical management
Surgical management
Any bleeding that fails to stop, despite an escalation of management, requires surgical intervention. This includes cases in which the bleeding continues after pack removal.4 Options include4:
- Diathermy, with bipolar or radiofrequency laser, can be used to localize the bleeding site.
- Septoplasty allows for better access to the nasal cavity, reduction of blood flow to the nasal mucosa by raising a mucoperichondrial flap, correction of a deviated septum, and removal of a septal spur that may be responsible for the epistaxis.
- Arterial ligation involves identification of the bleeding vessel that is clipped or coagulated with bipolar diathermy.
- Endoscopic SPA ligation is an excellent, well-tolerated, and cost-effective method of treating recurrent epistaxis.6,14 It controls 98% of posterior epistaxis, and is superior to posterior nasal packing and embolization.2,3,10 It results in a shorter hospital stay, reduction in repeated hemorrhage and painful packing procedures, and a cost saving of >$7,000 per patient if performed early.7 Concomitant ligation of the anterior ethmoidal artery may be performed in traumatic epistaxis or when severe bleeding is from the ethmoidal region.4,6
- Ligation of the IMAX and external carotid arteries is performed rarely due to potential complications and high failure rates.
Arterial embolization
When arterial ligation fails, or is not possible due to anesthetic concerns, selective embolization of the maxillary or facial arteries by specialist radiologists can be considered.6 Access to the vascular system through a femoral punch leads to identification of the bleeding point. A catheter is then placed in the artery and the bleeding vessel is embolized. Possible candidates include patients with HHT, bleeding tumors, poor surgical candidates, or patient preference.3
Other management considerations
Once bleeding is controlled, factors that contributed to the epistaxis should be addressed.3 Hypertension needs to be managed. Antiplatelet or anticoagulant therapy may need to be temporarily halted in consultation with specialist physicians. Local treatments such as cautery are unlikely to be effective in patients who are anticoagulated. Nasal packing with a ‘procoagulant’ dressing, such as Kaltostat or Rapid Rhino, is often required.
Patient education and follow-up
Patients should be started on saline sprays or irrigation to maintain nasal hygiene after acute epistaxis. It’s a good idea to teach patients about proper first aid for recurrence (eg, sitting upright with digital pressure applied to the cartilaginous part of the nose, ice packs around the neck and ice to suck) and to encourage them to refrain from activities that may stimulate bleeding (blowing or picking the nose, heavy lifting, strenuous exercise). Also advise patients to abstain from alcohol and hot drinks that cause vasodilatation of nasal vessels as much as possible.4 Advise patients that topical gels, lotions, and ointments such as kenacomb, nasalate, or paraffin can be used to moisturise the mucosa and promote healing.1
All patients with a history of severe or recurrent epistaxis require formal examination of the nasal cavity to rule out a neoplastic lesion.
CORRESPONDENCE
Amy Wong, BMBS, Department ENT/Head and Neck Surgery, Monash ENT Building, PO Box 72, Rear 867 Centre Road, Bentleigh East 3165 Australia, [email protected].
1. Schlosser RJ. Clinical practice. Epistaxis. N Engl J Med. 2009;360:784-789.
2. Yau S. An update on epistaxis. Aust Fam Physician. 2015;44:653-656.
3. McClurg SW, Carrau R. Endoscopic management of posterior epistaxis: a review. Acta Otorhinolaryngol Ital. 2014;34:1-8.
4. Pope LE, Hobbs CG. Epistaxis: an update on current management. Postgrad Med J. 2005;81:309-314.
5. Dubel GJ, Ahn SH, Soares GM. Transcatheter embolization in the management of epistaxis. Semin Intervent Radiol. 2013;30:249-262.
6. Spielmann PM, Barnes ML, White PS. Controversies in the specialist management of adult epistaxis: an evidence-based review. Clin Otolaryngol. 2012;37:382-389.
7. Porter M, Marais J, Tolley N. The effect of ice packs upon nasal mucosal blood flow. Acta Otolaryngol. 1991;111:1122-1125.
8. Traboulsi H, Alam E, Hadi U. Changing Trends in the Management of Epistaxis. Int J Otolaryngol. 2015;2015:263987.
9. Kamhieh Y, Fox H. Tranexamic acid in epistaxis: a systematic review. Clin Otolaryngol. 2016;41:771-776.
10. Stangerup SE, Dommerby H, Siim C, et al. New modification of hot-water irrigation in the treatment of posterior epistaxis. Arch Otolaryngol Head Neck Surg. 1999;125:686-690.
11. Douglas R, Wormald PJ. Update on epistaxis. Curr Opin Otolaryngol Head Neck Surg. 2007;15:180-183.
12. Badran K, Malik TH, Belloso A, et al. Randomized controlled trial comparing Merocel and RapidRhino packing in the management of anterior epistaxis. Clin Otolaryngol. 2005;30:333-337.
13. Moumoulidis I, Draper MR, Patel H, et al. A prospective randomised controlled trial comparing Merocel and Rapid Rhino nasal tampons in the treatment of epistaxis. Eur Arch Otorhinolaryngol. 2006;263:719-722.
14. Moshaver A, Harris JR, Liu R, et al. Early operative intervention versus conventional treatment in epistaxis: randomized prospective trial. J Otolaryngol. 2004;33:185-188.
1. Schlosser RJ. Clinical practice. Epistaxis. N Engl J Med. 2009;360:784-789.
2. Yau S. An update on epistaxis. Aust Fam Physician. 2015;44:653-656.
3. McClurg SW, Carrau R. Endoscopic management of posterior epistaxis: a review. Acta Otorhinolaryngol Ital. 2014;34:1-8.
4. Pope LE, Hobbs CG. Epistaxis: an update on current management. Postgrad Med J. 2005;81:309-314.
5. Dubel GJ, Ahn SH, Soares GM. Transcatheter embolization in the management of epistaxis. Semin Intervent Radiol. 2013;30:249-262.
6. Spielmann PM, Barnes ML, White PS. Controversies in the specialist management of adult epistaxis: an evidence-based review. Clin Otolaryngol. 2012;37:382-389.
7. Porter M, Marais J, Tolley N. The effect of ice packs upon nasal mucosal blood flow. Acta Otolaryngol. 1991;111:1122-1125.
8. Traboulsi H, Alam E, Hadi U. Changing Trends in the Management of Epistaxis. Int J Otolaryngol. 2015;2015:263987.
9. Kamhieh Y, Fox H. Tranexamic acid in epistaxis: a systematic review. Clin Otolaryngol. 2016;41:771-776.
10. Stangerup SE, Dommerby H, Siim C, et al. New modification of hot-water irrigation in the treatment of posterior epistaxis. Arch Otolaryngol Head Neck Surg. 1999;125:686-690.
11. Douglas R, Wormald PJ. Update on epistaxis. Curr Opin Otolaryngol Head Neck Surg. 2007;15:180-183.
12. Badran K, Malik TH, Belloso A, et al. Randomized controlled trial comparing Merocel and RapidRhino packing in the management of anterior epistaxis. Clin Otolaryngol. 2005;30:333-337.
13. Moumoulidis I, Draper MR, Patel H, et al. A prospective randomised controlled trial comparing Merocel and Rapid Rhino nasal tampons in the treatment of epistaxis. Eur Arch Otorhinolaryngol. 2006;263:719-722.
14. Moshaver A, Harris JR, Liu R, et al. Early operative intervention versus conventional treatment in epistaxis: randomized prospective trial. J Otolaryngol. 2004;33:185-188.
From The Journal of Family Practice | 2018;67(12):E13-E20.
PRACTICE RECOMMENDATIONS
› Use topical vasoconstrictor and local anesthetic agents as a first line therapy for epistaxis. Consider the additional use of topical tranexamic acid. A
› Perform chemical cautery with silver nitrate in cases of anterior epistaxis. This approach is cheap, easy to perform, and silver nitrate is readily available. A
› Consider endoscopic sphenopalatine artery ligation in the acute management of posterior epistaxis. It is superior to posterior nasal packing and embolization when it comes to pain, cost-effectiveness, risk, and overall control of bleeding. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

Unit-based assignments: Pros and cons
Geographic cohorting shows ‘varying success’
A relatively recent practice catching on in many different hospitalist groups is geographic cohorting, or unit-based assignments. Traditionally, most hospitalists have had patients assigned on multiple different units. Unit-based assignments have been touted as a way of improving interdisciplinary communication and provider and patient satisfaction.1

How frequently are hospital medicine groups using unit-based assignments? SHM sought to quantify this trend in the recently published 2018 State of Hospital Medicine Report. Overall, among hospital medicine groups serving adults only, a little over one-third (36.4%) of groups reported utilizing unit-based assignments. However, there was significant variation, particularly dependent on group size. Geographic cohorting was used only in 7.6% of groups with 4 or fewer full-time equivalents, and in 68.8% of groups with 30 or more FTE. These data seem logical, as the potential gains from cohorting likely increase with group/hospital size, where physicians would otherwise round on an increasingly large number of units.
As has been shared in the hospital medicine literature, groups have experienced variable success with geographic cohorting. Improvements have been achieved in interprofessional collaboration, efficiency, nursing satisfaction,2 and, in some instances, length of stay. Unit-based assignments have allowed some groups to pilot other interventions, such as interdisciplinary rounds.
But geographic cohorting comes with its implementation challenges, too. For example, in many hospitals, some units have differing telemetry or nursing capabilities. And, in other institutions, there are units providing specialized care, such as care for neurology or oncology patients. The workload for hospitalists caring for particular types of patients may vary, and with specialty units, it may be more difficult to keep a similar census assigned to each hospitalist.
While some groups have noted increased professional satisfaction, others have noted decreases in satisfaction. One reason is that, while the frequency of paging may decrease, this is replaced by an increase in face-to-face interruptions. Also, unit-based assignments in some groups have resulted in hospitalists perceiving they are working in silos because of a decrease in interactions and camaraderie among providers in the same hospital medicine group.
At my home institution, University of California, San Diego, geographic cohorting has largely been a successful and positively perceived change. Our efforts have been particularly successful at one of our two campuses where most units have telemetry capabilities and where we have a dedicated daytime admitter (there are data on this in the Report as well, and a dedicated daytime admitter is the topic of a future Survey Insights column). Unit-based assignments have allowed the implementation of what we’ve termed focused interdisciplinary rounds.
Our unit-based assignments are not perfect – we re-cohort each week when new hospitalists come on service, and some hospitalists are assigned a small number of patients off their home unit. Our internal data have shown a significant increase in patient satisfaction scores, but we have not realized a decrease in length of stay. Despite an overall positive perception, hospitalists have sometimes noted an imbalanced workload – we have a particularly challenging oncology/palliative unit and a daytime admitter that is at times very busy. Our system also requires the use of physician time to assign patients each morning and each week.
In contrast, while we’ve aimed to achieve the same success with unit-based assignments at our other campus, we’ve faced more challenges there. Our other facility is older, and fewer units have telemetry capabilities. A more traditional teaching structure also means that teams take turns with on-call admitting days, as opposed to a daytime admitter structure, and there may not be beds available in the unit assigned to the admitting team of the day.
Overall, geographic cohorting is likely to be considered or implemented in many hospital medicine groups, and efforts have met with varying success. There are certainly pros and cons to every model, and if your group is looking at redesigning services to include unit-based assignments, it’s worth examining the intended outcomes. While unit-based assignments are not for every group, there’s no doubt that this trend has been driven by our specialty’s commitment to outcome-driven process improvement.
Addendum added Feb. 15, 2019: The impact of UC San Diego's efforts discussed in this article are the author's own opinions through limited participation in focused interdisciplinary rounds, and have not been validated with formal data analysis. More study is in progress on the impact of focused interdiscplinary rounds on communication, utilization, and quality metrics. Sarah Horman, MD ([email protected]), Daniel Bouland, MD ([email protected]), and William Frederick, MD ([email protected]), have led efforts at UC San Diego to develop and implement focused interdisciplinary rounds, and may be contacted for further information.
Dr. Huang is physician advisor for care management and associate clinical professor in the division of hospital medicine at the University of California, San Diego. He is a member of SHM’s practice analysis subcommittee.
References
1. O’Leary KJ et al. Interdisciplinary teamwork in hospitals: A review and practical recommendations for improvement. J Hosp Med. 2012 Jan;7(1):48-54.
2. Kara A et al. Hospital-based clinicians’ perceptions of geographic cohorting: Identifying opportunities for improvement. Am J Med Qual. 2018 May/Jun;33(3):303-12.
Geographic cohorting shows ‘varying success’
Geographic cohorting shows ‘varying success’
A relatively recent practice catching on in many different hospitalist groups is geographic cohorting, or unit-based assignments. Traditionally, most hospitalists have had patients assigned on multiple different units. Unit-based assignments have been touted as a way of improving interdisciplinary communication and provider and patient satisfaction.1
How frequently are hospital medicine groups using unit-based assignments? SHM sought to quantify this trend in the recently published 2018 State of Hospital Medicine Report. Overall, among hospital medicine groups serving adults only, a little over one-third (36.4%) of groups reported utilizing unit-based assignments. However, there was significant variation, particularly dependent on group size. Geographic cohorting was used only in 7.6% of groups with 4 or fewer full-time equivalents, and in 68.8% of groups with 30 or more FTE. These data seem logical, as the potential gains from cohorting likely increase with group/hospital size, where physicians would otherwise round on an increasingly large number of units.
As has been shared in the hospital medicine literature, groups have experienced variable success with geographic cohorting. Improvements have been achieved in interprofessional collaboration, efficiency, nursing satisfaction,2 and, in some instances, length of stay. Unit-based assignments have allowed some groups to pilot other interventions, such as interdisciplinary rounds.
But geographic cohorting comes with its implementation challenges, too. For example, in many hospitals, some units have differing telemetry or nursing capabilities. And, in other institutions, there are units providing specialized care, such as care for neurology or oncology patients. The workload for hospitalists caring for particular types of patients may vary, and with specialty units, it may be more difficult to keep a similar census assigned to each hospitalist.
While some groups have noted increased professional satisfaction, others have noted decreases in satisfaction. One reason is that, while the frequency of paging may decrease, this is replaced by an increase in face-to-face interruptions. Also, unit-based assignments in some groups have resulted in hospitalists perceiving they are working in silos because of a decrease in interactions and camaraderie among providers in the same hospital medicine group.
At my home institution, University of California, San Diego, geographic cohorting has largely been a successful and positively perceived change. Our efforts have been particularly successful at one of our two campuses where most units have telemetry capabilities and where we have a dedicated daytime admitter (there are data on this in the Report as well, and a dedicated daytime admitter is the topic of a future Survey Insights column). Unit-based assignments have allowed the implementation of what we’ve termed focused interdisciplinary rounds.
Our unit-based assignments are not perfect – we re-cohort each week when new hospitalists come on service, and some hospitalists are assigned a small number of patients off their home unit. Our internal data have shown a significant increase in patient satisfaction scores, but we have not realized a decrease in length of stay. Despite an overall positive perception, hospitalists have sometimes noted an imbalanced workload – we have a particularly challenging oncology/palliative unit and a daytime admitter that is at times very busy. Our system also requires the use of physician time to assign patients each morning and each week.
In contrast, while we’ve aimed to achieve the same success with unit-based assignments at our other campus, we’ve faced more challenges there. Our other facility is older, and fewer units have telemetry capabilities. A more traditional teaching structure also means that teams take turns with on-call admitting days, as opposed to a daytime admitter structure, and there may not be beds available in the unit assigned to the admitting team of the day.
Overall, geographic cohorting is likely to be considered or implemented in many hospital medicine groups, and efforts have met with varying success. There are certainly pros and cons to every model, and if your group is looking at redesigning services to include unit-based assignments, it’s worth examining the intended outcomes. While unit-based assignments are not for every group, there’s no doubt that this trend has been driven by our specialty’s commitment to outcome-driven process improvement.
Addendum added Feb. 15, 2019: The impact of UC San Diego's efforts discussed in this article are the author's own opinions through limited participation in focused interdisciplinary rounds, and have not been validated with formal data analysis. More study is in progress on the impact of focused interdiscplinary rounds on communication, utilization, and quality metrics. Sarah Horman, MD ([email protected]), Daniel Bouland, MD ([email protected]), and William Frederick, MD ([email protected]), have led efforts at UC San Diego to develop and implement focused interdisciplinary rounds, and may be contacted for further information.
Dr. Huang is physician advisor for care management and associate clinical professor in the division of hospital medicine at the University of California, San Diego. He is a member of SHM’s practice analysis subcommittee.
References
1. O’Leary KJ et al. Interdisciplinary teamwork in hospitals: A review and practical recommendations for improvement. J Hosp Med. 2012 Jan;7(1):48-54.
2. Kara A et al. Hospital-based clinicians’ perceptions of geographic cohorting: Identifying opportunities for improvement. Am J Med Qual. 2018 May/Jun;33(3):303-12.
A relatively recent practice catching on in many different hospitalist groups is geographic cohorting, or unit-based assignments. Traditionally, most hospitalists have had patients assigned on multiple different units. Unit-based assignments have been touted as a way of improving interdisciplinary communication and provider and patient satisfaction.1
How frequently are hospital medicine groups using unit-based assignments? SHM sought to quantify this trend in the recently published 2018 State of Hospital Medicine Report. Overall, among hospital medicine groups serving adults only, a little over one-third (36.4%) of groups reported utilizing unit-based assignments. However, there was significant variation, particularly dependent on group size. Geographic cohorting was used only in 7.6% of groups with 4 or fewer full-time equivalents, and in 68.8% of groups with 30 or more FTE. These data seem logical, as the potential gains from cohorting likely increase with group/hospital size, where physicians would otherwise round on an increasingly large number of units.
As has been shared in the hospital medicine literature, groups have experienced variable success with geographic cohorting. Improvements have been achieved in interprofessional collaboration, efficiency, nursing satisfaction,2 and, in some instances, length of stay. Unit-based assignments have allowed some groups to pilot other interventions, such as interdisciplinary rounds.
But geographic cohorting comes with its implementation challenges, too. For example, in many hospitals, some units have differing telemetry or nursing capabilities. And, in other institutions, there are units providing specialized care, such as care for neurology or oncology patients. The workload for hospitalists caring for particular types of patients may vary, and with specialty units, it may be more difficult to keep a similar census assigned to each hospitalist.
While some groups have noted increased professional satisfaction, others have noted decreases in satisfaction. One reason is that, while the frequency of paging may decrease, this is replaced by an increase in face-to-face interruptions. Also, unit-based assignments in some groups have resulted in hospitalists perceiving they are working in silos because of a decrease in interactions and camaraderie among providers in the same hospital medicine group.
At my home institution, University of California, San Diego, geographic cohorting has largely been a successful and positively perceived change. Our efforts have been particularly successful at one of our two campuses where most units have telemetry capabilities and where we have a dedicated daytime admitter (there are data on this in the Report as well, and a dedicated daytime admitter is the topic of a future Survey Insights column). Unit-based assignments have allowed the implementation of what we’ve termed focused interdisciplinary rounds.
Our unit-based assignments are not perfect – we re-cohort each week when new hospitalists come on service, and some hospitalists are assigned a small number of patients off their home unit. Our internal data have shown a significant increase in patient satisfaction scores, but we have not realized a decrease in length of stay. Despite an overall positive perception, hospitalists have sometimes noted an imbalanced workload – we have a particularly challenging oncology/palliative unit and a daytime admitter that is at times very busy. Our system also requires the use of physician time to assign patients each morning and each week.
In contrast, while we’ve aimed to achieve the same success with unit-based assignments at our other campus, we’ve faced more challenges there. Our other facility is older, and fewer units have telemetry capabilities. A more traditional teaching structure also means that teams take turns with on-call admitting days, as opposed to a daytime admitter structure, and there may not be beds available in the unit assigned to the admitting team of the day.
Overall, geographic cohorting is likely to be considered or implemented in many hospital medicine groups, and efforts have met with varying success. There are certainly pros and cons to every model, and if your group is looking at redesigning services to include unit-based assignments, it’s worth examining the intended outcomes. While unit-based assignments are not for every group, there’s no doubt that this trend has been driven by our specialty’s commitment to outcome-driven process improvement.
Addendum added Feb. 15, 2019: The impact of UC San Diego's efforts discussed in this article are the author's own opinions through limited participation in focused interdisciplinary rounds, and have not been validated with formal data analysis. More study is in progress on the impact of focused interdiscplinary rounds on communication, utilization, and quality metrics. Sarah Horman, MD ([email protected]), Daniel Bouland, MD ([email protected]), and William Frederick, MD ([email protected]), have led efforts at UC San Diego to develop and implement focused interdisciplinary rounds, and may be contacted for further information.
Dr. Huang is physician advisor for care management and associate clinical professor in the division of hospital medicine at the University of California, San Diego. He is a member of SHM’s practice analysis subcommittee.
References
1. O’Leary KJ et al. Interdisciplinary teamwork in hospitals: A review and practical recommendations for improvement. J Hosp Med. 2012 Jan;7(1):48-54.
2. Kara A et al. Hospital-based clinicians’ perceptions of geographic cohorting: Identifying opportunities for improvement. Am J Med Qual. 2018 May/Jun;33(3):303-12.