User login
Mantle Cell Lymphoma
INTRODUCTION
Mantle cell lymphoma (MCL) is an uncommon, distinct clinical subtype of non-Hodgkin lymphoma (NHL) that comprises approximately 8% of all lymphoma diagnoses in the United States and Europe.1,2 Considered incurable, MCL often presents in advanced stages, particularly with involvement of the lymph nodes, spleen, bone marrow, and gastrointestinal tract in the form of lymphomatous polyps. MCL disproportionately affects males, and incidence rises with age, with a median age at diagnosis of 68 years.2 Historically, the prognosis of patients with MCL has been among the poorest among B-cell lymphoma patients, with a median overall survival (OS) of 3 to 5 years, and time to treatment failure (TTF) of 18 to 24 months, although this is improving in the modern era.3 Less frequently, patients with MCL display isolated bone marrow, peripheral blood, and splenic involvement. These cases tend to behave more indolently with longer survival.4,5 Recent advances in therapy have dramatically impacted treatment alternatives and outcomes for MCL. As such, the therapeutic and prognostic landscape of MCL is evolving rapidly.
PATHOGENESIS
The histologic diagnosis of MCL by morphology alone is often challenging. Accurate diagnosis relies on immunohistochemical staining for the purposes of immunophenotyping.6 MCL typically expresses B-cell markers CD5 and CD20, and lacks both CD10 and CD23. The genetic hallmark of MCL is the t(11;14) (q13;q32) chromosomal translocation leading to upregulation of the cyclin D1 protein, a critical regulator of the G1 phase of the cell cycle. Specifically, the t(11;14) translocation, present in virtually all cases of MCL, juxtaposes the proto-oncogene CCND1 to the immunoglobulin heavy chain gene.7 Consequently, cyclin D1, normally not expressed in B lymphocytes, becomes constitutively overexpressed. This alteration is thought to facilitate the deregulation of the cell cycle at the G1-S phase transition.8
Gene expression profiling studies have underscored the importance of cell cycle deregulation in MCL, and high proliferation is associated with a worse prognosis.9 More than 50% of the genes associated with poor outcomes were derived from the “proliferation signature” that was more highly expressed in dividing cells. In the seminal Rosenwald study, a gene expression–based outcome model was constructed in which the proliferation signature average represents a linear variable that assigns a discrete probability of survival to an individual patient.9 The proliferative index, or proliferative signature, of MCL can be estimated by the percentage of Ki-67–positive cells present in the tumor through immunohistochemistry. This is often used as a marker of poor outcomes, and as a surrogate for the proliferative signature in MCL that can be incorporated into clinical practice (as opposed to gene expression profiling). Statistically significant differences in OS have emerged between groups of MCL patients with Ki-67–positive cells comprising less than 30% of their tumor sample (favorable) and those with Ki-67–positive cells comprising 30% or greater (unfavorable).10
Recent data has also identified the importance of the transcription factor SOX 11 (SRY-related HMG-box), which regulates multiple cellular transcriptional events, including cell proliferation and differentiation, apoptosis, and angiogenesis.11 MCL expressing SOX 11 behaves more aggressively than MCL variants lacking SOX 11 expression, and tends to accumulate more genetic alterations.12 Moreover, lack of SOX 11 expression characterizes a subset of MCL that does not carry the t(11;14) translocation.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 62-year-old man with a history of diabetes mellitus and hypertension presents with cervical lymphadenopathy, fatigue, and early satiety over the past several months. He is otherwise in good health. His Eastern Cooperative Oncology Group (ECOG) performance status is 1. On physical examination, 3-cm lymphadenopathy in the bilateral cervical chain is noted. Bilateral axillary lymph nodes measure 2 to 4 cm. His spleen is enlarged and is palpable at approximately 5 cm below the costal margin. A complete blood count reveals a total white blood cell (WBC) count of 14,000 cells/μL, with 68% lymphocytes and a normal distribution of neutrophils. Hemoglobin is 11 g/dL, and platelet count is 112,000/μL. The lactate dehydrogenase (LDH) level is 322 U/L (upper limit of normal: 225 U/L).
• How is MCL diagnosed?
Diagnosis of MCL requires review by expert hematopathologists.13 Whenever possible, an excisional biopsy should be performed for the adequate characterization of lymph node architecture and evaluation by immunohistochemistry. Aside from the characteristic expression of CD5 and CD20 and absence of CD23, MCL should express cyclin D1, which reflects t(11;14). If cyclin D1 is inconclusive or unavailable, fluorescent in situ hybridization (FISH) for t(11;14) should be performed.8 Patients often have circulating malignant lymphocytes, or leukemic phase MCL. Flow cytometry of the peripheral blood can detect traditional surface markers, and FISH can also be performed on circulating abnormal lymphocytes.
For disease staging, bone marrow biopsy and aspiration are required. Radiographic staging using computed tomography (CT) scans and/or positron emission tomography (PET) scans had traditionally followed the Ann Arbor staging system, but recently the Lugano classification has emerged, which delineates only early or advanced stage.14 Gastrointestinal evaluation of MCL with endoscopy and colonoscopy with blind biopsies has been recommended to evaluate for the presence of lymphomatous polyps, but this is not an absolute requirement.15
RISK STRATIFICATION
At diagnosis, patients should undergo risk stratification in order to understand prognosis and possibly guide treatment. In MCL, the MCL international prognostic index (MIPI) is used. The MIPI is a prognostic tool developed exclusively for patients with MCL using data from 455 patients with advanced-stage MCL treated within 3 European clinical trials.16 The MIPI classified patients into risk groups based on age, ECOG performance status, LDH level, and WBC count. Patients were categorized into low-risk (44% of patients, median OS not reached), intermediate-risk (35%, median OS 51 months), and high-risk groups (21%, median OS 29 months). This is done through a logarithmic calculation, which can be accessed through online calculators (a prototype example can be found at www.qxmd.com/calculate-online/hematology/prognosis-mantle-cell-lymphoma-mipi). Cell proliferation using the Ki-67 index was evaluated in an exploratory analysis (the biologic [“B”] MIPI), and also demonstrated strong prognostic relevance.16 Currently, treatment of MCL patients is not stratified by MIPI outside of a clinical trial, but this useful tool assists in assessing patient prognosis and has been validated for use with both conventional chemoimmunotherapy and in the setting of autologous stem cell transplant (autoSCT).16,17 At this point in time, the MIPI score is not used to stratify treatment, although some clinical trials are incorporating the use of the MIPI score at diagnosis. Nonetheless, given its prognostic importance, the MIPI should be performed for all MCL patients undergoing staging and evaluation for treatment to establish disease risk.
As noted, the proliferative signature, represented by the Ki-67 protein, is also highly prognostic in MCL. Ki-67 is expressed in the late G1, S, G2, and M phases of the cell cycle. The Ki-67 index is defined by the hematopathologist as the percentage of lymphoma cells staining positive for Ki-67 protein, based on the number of cells per high-power field. There is significant interobserver variability in this process, which can be minimized by assessing Ki-67 quantitatively using computer software. The prognostic significance of Ki-67 at diagnosis was established in large studies of MCL patient cohorts, with survival differing by up to 3 years.18,19 Determann et al demonstrated the utility of the proliferative index in patients with MCL treated with standard chemoimmunotherapy.10 In this study, 249 patients with advanced-stage MCL treated within randomized trials conducted by the European MCL Network were analyzed. The Ki-67 index was found to be extremely prognostic of OS, independent of other clinical risk factors, including the MIPI score. As a continuous variable, Ki-67 indices of greater than 10% correlated with poor outcomes. The Ki-67 index has also been confirmed as prognostic in relapsed MCL.20 It is important to note that, as a unique feature, the Ki-67 index has remained an independent prognostic factor, even when incorporated into the “B” MIPI.
TREATMENT
CASE CONTINUED
The patient undergoes an excisional biopsy of a cervical lymph node, which demonstrates an abnormal proliferation of small-medium–sized lymphocytes with slightly irregular nuclear contours. Immunohistochemistry shows that the abnormal lymphocytes are positive for CD20 and CD5, negative for CD10 and CD23, and diffusely positive for cyclin D1, consistent with a diagnosis of MCL. The proliferative index, as measured by the Ki-67 immunostain, is 40%. A bone marrow aspirate and biopsy are then obtained, which show a clonal population of B lymphocytes expressing the same immunophenotype as the lymph node (positive for CD20 and CD5, negative for CD10 and CD23, cyclin D1 positive). A CT scan of the neck, chest, abdomen, and pelvis with contrast is obtained, along with a PET scan. These studies identify extensive hypermetabolic lymphadenopathy in the bilateral cervical chains, supraclavicular areas, mediastinum, and hilum. Mesenteric lymph nodes are also enlarged and hypermetabolic, as are retroperitoneal lymph nodes. The spleen is noted to be enlarged with multiple hypermetabolic lesions. Based on the presence of extensive lymphadenopathy as well as bone marrow involvement, the patient is diagnosed with stage IV MCL. He undergoes risk-stratification with the MIPI. His MIPI score is 6.3, high risk.
• What is the approach to upfront therapy for MCL?
FRONTLINE THERAPY
Role of Watchful Waiting
A small proportion of MCL patients have indolent disease that can be observed. This population is more likely to have leukemic-phase MCL with circulating lymphocytes, splenomegaly, and bone marrow involvement and absent or minimal lymphadenopathy.4,5 A retrospective study of 97 patients established that deferment of initial therapy in MCL is acceptable in some patients.5 In this study, approximately one third of patients with MCL were observed for more than 3 months before initiating systemic therapy, and the median time to treatment for the observation group was 12 months. Most patients undergoing observation had a low-risk MIPI. Patients were not harmed by observation, as no OS differences were observed among groups. This study underscores that deferred treatment can be an acceptable alternative in selected MCL patients for a short period of time. In practice, the type of patient who would be appropriate for this approach is someone who is frail, elderly, and with multiple comorbidities. Additionally, expectant observation could be considered for patients with limited-stage or low-volume MCL, low Ki-67 index, and low-risk MIPI scores.
Approach to Therapy
Treatment of MCL is generally approached by evaluating patient age and fitness for treatment. While there is no accepted standard, for younger patients healthy enough to tolerate aggressive approaches, treatment often involves an intensive cytarabine-containing regimen, which is consolidated with an autoSCT. This approach results in the longest remission duration, with some series suggesting a plateau in survival after 5 years, with no relapses.21 Nonintensive conventional chemotherapy alone is often reserved for the frailer or older patient. Given that remission durations with chemotherapy alone in MCL are short, goals of treatment focus on maximizing benefit and remission duration and minimizing risk of toxicity.
Standard Chemotherapy: Elderly and/or Frail Patients
Conventional chemotherapy alone for the treatment of MCL results in a 70% to 85% overall response rate (ORR) and 7% to 30% complete response (CR) rate.22 Rituximab, a mouse humanized monoclonal IgG1 anti-CD20 antibody, is used as standard of care in combination with chemotherapy, since its addition has been found to increase response rates and extend both progression-free survival (PFS) and OS compared to chemotherapy alone.23,24 However, chemoimmunotherapy approaches do not provide long-term control of MCL and are considered noncurative. Various regimens have been studied and include anthracycline-containing regimens such as R-CHOP (rituximab with cyclophosphamide, doxorubicin, vincristine, prednisone),22 combination chemotherapy with antimetabolites such as R-hyper-CVAD (hyper-fractionated rituximab with cyclophosphamide, vincristine, doxorubicin, dexamethasone, alternating with methotrexate and cytarabine),25 purine analogue–based regimens such as R-FC (rituximab with fludarabine and cyclophosphamide),26 bortezomib-containing regimens,27 and alkylator-based treatment with BR (bendamustine and rituximab) (Table 1).28,29 Among these, the most commonly used are R-CHOP and BR.
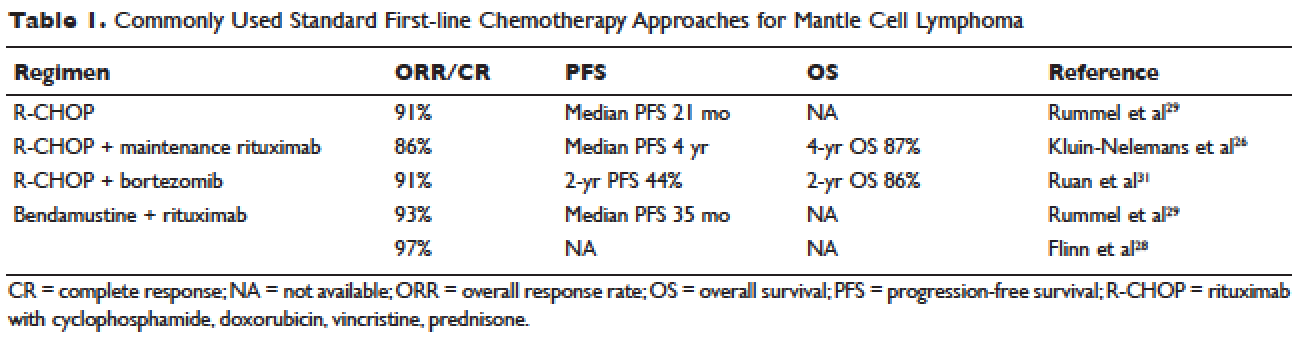
Two large randomized studies compared R-CHOP for 6 cycles to BR for 6 cycles in patients with indolent NHL and MCL. Among MCL patients, BR resulted in superior PFS compared to R-CHOP (69 months versus 26 months) but no benefit in OS.28,29 The ORR to R-CHOP was approximately 90%, with a PFS of 21 months in the Rummel et al study.29 This study included more than 80 centers in Germany and enrolled 549 patients with MCL, follicular lymphoma, small lymphocytic lymphoma, marginal zone lymphoma, and Waldenström macroglobulinemia. Patients were randomized in a 1:1 fashion. Among these, 46 patients received BR and 48 received R-CHOP (18% for both, respectively). It should be noted that patients in the BR group had significantly less toxicity and experienced fewer side effects than did those in the R-CHOP group. Similarly, BR-treated patients had a lower frequency of hematologic side effects and infections of any grade. However, drug-associated skin reactions and allergies were more common with BR compared to R-CHOP. The study by Flinn and colleagues was an international randomized, noninferiority phase 3 study designed to evaluate the efficacy and safety of BR compared with R-CHOP or R-CVP (rituximab plus cyclophosphamide, vincristine, and prednisone) for treatment-naive patients with MCL or other indolent NHL. The primary endpoint was CR. In this study, BR was found to be noninferior to R-CHOP and R-CVP based on CR rate (31% versus 25%, respectively; P = 0.0225). Response rates in general were high: 97% for BR and 91% for R-CHOP/R-CVP (P = 0.0102). Here, BR-treated patients experienced more nausea, emesis, and drug-induced hypersensitivity compared to the R-CHOP and R-CVP groups.
Another approach studied in older patients is the use of R-CHOP with rituximab maintenance. In a large European study, 560 patients 60 years of age or older with advanced-stage MCL were randomly assigned to either R-FC (rituximab, fludarabine, and cyclophosphamide) every 28 days for 6 cycles, or R-CHOP every 21 days for 8 cycles. Patients who had a response then underwent a second randomization, with one group receiving rituximab maintenance therapy. Maintenance was continued until progression of disease. Patients in this study were not eligible for high-dose chemotherapy and autoSCT. The study found that rates of CR were similar with both R-FC and R-CHOP (40% and 34%, respectively; P = 0.10). However, the R-FC arm underperformed in several arenas. Disease progression occurred more frequently with R-FC (14% versus 5% with R-CHOP), and OS was shorter (4-year OS, 47% versus 62%; P = 0.005, respectively). More patients also died in the R-FC group, and there was greater hematologic toxicity compared to R-CHOP. At 4 years, 58% of the patients receiving rituximab remained in remission. Among patients who responded to R-CHOP, rituximab maintenance led to a benefit in OS, reducing the risk of progression or death by 45%.26 At this time, studies are ongoing to establish the benefit of rituximab maintenance after BR.
Bendamustine in combination with other agents has also been studied in the frontline setting. Visco and colleagues evaluated the combination of bendamustine with rituximab and cytarabine (R-BAC) in older patients with MCL (age 65 or older).63 This phase 2, two-stage study enrolled 40 patients and had a dose-finding arm for cytarabine in combination with BR. It permitted relapsed/refractory patients, but 50% had newly diagnosed, previously untreated MCL. The regimen had an impressive ORR of 100%, with CR rates of 95% for previously untreated patients. PFS at 2 years was 95%. R-BAC was well tolerated, with the primary toxicity being reversible myelosuppression.
BR was combined with the proteasome inhibitor bortezomib and dexamethasone in a phase 2 study.64 This Lymphoma Study Association (LYSA) study evaluated 76 patients with newly diagnosed MCL older than age 65 years. BR was administered in standard doses (bendamustine 90 mg/m2 on days 1 and 2 and rituximab 375 mg/m² IV on day 1) and bortezomib was administered subcutaneously on days 1, 4, 8, and 11, with acyclovir for viral prophylaxis. Patients received 6 cycles. The ORR was 87% and the CR was 60%. Patients experienced toxicity, and not all bortezomib doses were administered due to neurotoxic or hematologic side effects.
A randomized phase 3 study compared R-CHOP to the VR-CAP regimen (R-CHOP regimen but bortezomib replaces vincristine on days 1, 4, 8, 11, at 1.3 mg/m2) in 487 newly diagnosed MCL patients.27 Median PFS was superior in the VR-CAP group compared with R-CHOP (14.4 months versus 24.7 months, respectively). Additionally, rates of CR were superior in the VR-CAP group (53% compared to 42% with R-CHOP). However, there was more hematologic toxicity with VR-CAP. On the basis of these findings, the U.S. Food and Drug Administration approved bortezomib for the frontline treatment of MCL.
Other chemoimmunotherapy combinations containing bortezomib have been studied in frontline MCL treatment, with promising results. These include bortezomib in combination with R-CHOP or modified R-hyper-CVAD, as well as bortezomib in combination with CHOP-like treatments and purine analogues.27,30–32 The ongoing ECOG 1411 study is currently evaluating bortezomib added to BR for induction therapy of newly diagnosed MCL in a 4-arm randomized trial. Patients receive BR with or without bortezomib during induction and are then randomly assigned to maintenance with either rituximab alone or rituximab with lenalidomide. Other novel combination agents are actively being studied in frontline MCL treatment, including lenalidomide and rituximab and BR with lenalidomide.
Intensification of Therapy and AutoSCT: Fitter and/or Younger Patients
Short response duration has created the need for post-remission therapy in MCL. One approach to improve remission duration in MCL is to intensify induction through the use of cytarabine-containing regimens and/or consolidation with high-dose chemotherapy, typically using BEAM (carmustine, etoposide, cytarabine, melphalan) and autoSCT (Table 2). The cytarabine-containing R-hyper-CVAD regimen, developed at the MD Anderson Cancer Center, resulted in a 97% ORR and an 87% CR rate, with TTF of nearly 5 years. However, nearly one third of patients were unable to complete treatment due to toxicity, and 5 patients developed secondary myelodysplastic syndrome or acute myeloid leukemia.33 The feasibility of this R-hyper-CVAD regimen was tested in a multicenter cooperative group setting, but similar results were not seen; in this study, nearly 40% of patients were unable to complete the full scheduled course of treatment due to toxicity.34
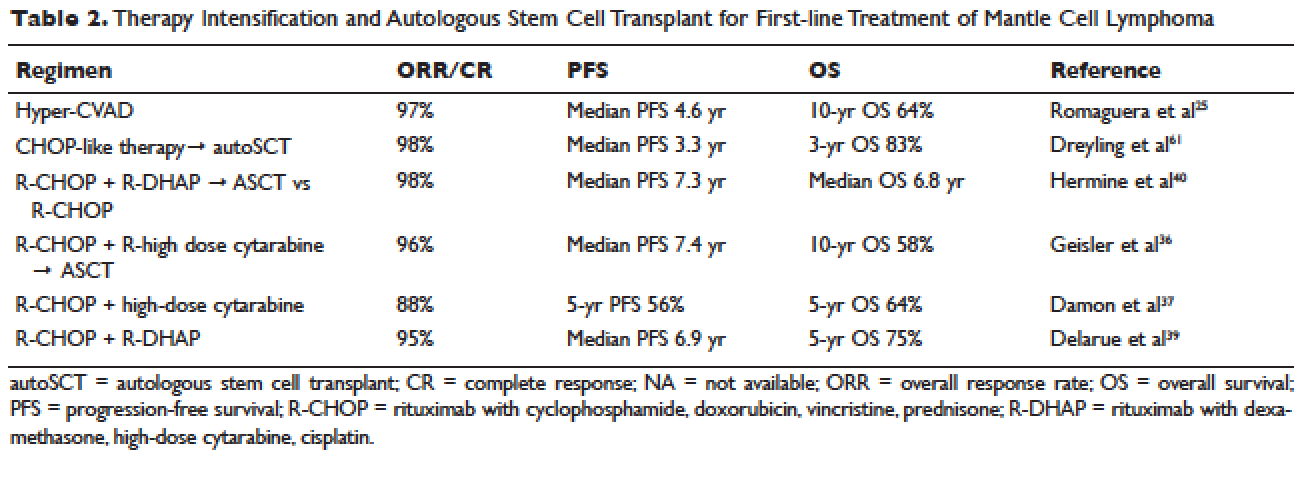
Other ways to intensify therapy in MCL involve adding a second non-cross-resistant cytarabine-containing regimen to R-CHOP after remission, such as DHAP (dexamethasone, high-dose cytarabine, cisplatin), followed by consolidation with an autoSCT. A retrospective registry from the National Comprehensive Cancer Network sought to compare the efficacy of different treatment approaches in the frontline setting. They studied 167 patients with MCL and compared 4 groups: treatment with R-hyper-CVAD, either with or without autoSCT, and treatment with R-CHOP, either with or without autoSCT. This study found that in patients younger than 65, R-CHOP followed by autoSCT or R-hyper-CVAD without autoSCT resulted in similar PF and OS, but was superior to R-CHOP alone for newly diagnosed MCL patients.35 These data support more intensive regimens in younger and fitter patients. Several other prospective and randomized studies have demonstrated clinical benefit for patients with MCL undergoing autoSCT in first remission. Of particular importance is the seminal phase 3 study of the European MCL Network, which established the role of autoSCT in this setting.61 In this prospective randomized trial involving 122 newly diagnosed MCL patients who responded to CHOP-like induction, patients in CR derived a greater benefit from autoSCT.
More recent studies have demonstrated similar benefits using cytarabine-based autoSCT. The Nordic MCL2 study evaluated 160 patients using R-CHOP, alternating with rituximab and high-dose cytarabine, followed by autoSCT. This study used “maxi-CHOP,” an augmented CHOP regimen (cyclophosphamide 1200 mg/m2, doxorubicin 75 mg/m2, but standard doses of vincristine [2 mg] and prednisone [100 mg days 1–5]), alternating with 4 infusions of cytarabine at 2 g/m2 and standard doses of rituximab (375 mg/m2). Patients then received conditioning with BEAM and autoSCT. Patients were evaluated for the presence of minimal residual disease (MRD) and for the t(11;14) or clonal immunoglobulin heavy chain gene rearrangement with polymerase chain reaction (PCR). Patients with MRD were offered therapy with rituximab at 375 mg/m2 weekly for 4 doses. This combination resulted in 10-year OS rates of 58%.36 In a multicenter study involving 78 patients from the Cancer and Leukemia Group B (CALGB), R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT resulted in a 5-year OS of 64%.37 A single-arm phase 2 study from the Netherlands also tested R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT. Nonhematologic toxicities were 22% after high-dose cytarabine, and 55% after BEAM. The ORR was 70%, with a 64% CR rate and 66% OS at 4 years.38 The French GELA group used 3 cycles of R-CHOP and 3 cycles of R-DHAP in a phase 2 study of young (under age 66) MCL patients. Following R-CHOP, the ORR was 93%, and following R-DHAP the ORR was 95%. Five-year OSA was 75%.39 A large randomized phase 3 study by Hermine and colleagues of the EMCLN confirmed the benefit of this approach in 497 patients with newly diagnosed MCL. R-CHOP for 6 cycles followed by autoSCT was compared to R-CHOP for 3 cycles alternating with R-DHAP for 3 cycles and autoSCT with a cytarabine-based conditioning regimen. The addition of cytarabine significantly increased rates of CR, TTF, and OS, without increasing toxicity.40
CASE CONTINUED
The patient is treated with R-CHOP chemotherapy for 3 cycles followed by R-DHAP. His course is complicated by mild tinnitus and acute kidney injury from cisplatin that promptly resolves. Three weeks following treatment, a restaging PET/CT scan shows resolution of all lymphadenopathy, with no hypermetabolic uptake, consistent with a complete remission. A repeat bone marrow biopsy shows no involvement with MCL. He subsequently undergoes an autoSCT, and restaging CT/PET 3 months following autoSCT shows continued remission. He is monitored every 3 to 6 months over the next several years.
He has a 4.5-year disease remission, after which he develops growing palpable lymphadenopathy on exam and progressive anemia and thrombocytopenia. A bone marrow biopsy is repeated, which shows recurrent MCL. Restaging diagnostic imaging with a CT scan reveals lymphadenopathy above and below the diaphragm. An axillary lymph node biopsy also demonstrates recurrent MCL. At this time the patient is otherwise in fairly good health, except for feeling fatigued. His ECOG performance status is 1. He begins therapy with bortezomib at a dose of 1.3 mg/m2 intravenously on days 1, 4, 8, and 11 for 6 cycles. His treatment course is complicated by painful sensory peripheral neuropathy of the bilateral lower extremities. Restaging studies at the completion of therapy demonstrate that he has achieved a partial response, with a 50% reduction in the size of involved lymphadenopathy and some residual areas of hypermetabolic uptake. His peripheral cytopenias improve moderately.
• What are the therapeutic options for relapsed MCL?
TREATMENT OF RELAPSED MCL
Single-Agent and Combination Chemotherapy
Whenever possible, and since there is no standard, patients with relapsed MCL should be offered a clinical trial. Outside of a clinical study, many of the treatment regimens used at diagnosis can also be applied in the relapsed setting. In relapsed MCL, Rummel et al showed that BR for 4 cycles resulted in an ORR of 90%, with a CR of 60%. The median PFS was 24 months.41 Bortezomib, an inhibitor of the proteasome-ubiquitin pathway, leads to apoptosis and cell cycle arrest in MCL.42 Multiple studies have evaluated bortezomib both as a single agent and in combination for patients with relapsed MCL. In 2006, bortezomib became the first agent approved by the FDA in relapsed or refractory MCL, based on the phase 2 PINNACLE study. This prospective multicenter study involving 155 patients demonstrated an ORR of 33%, CR rate of 8%, and median treatment duration of 9 months. The median time to progression was 6 months.43 Subsequently, bortezomib-containing combinations evolved. In a multicenter study of relapsed and refractory indolent NHL and MCL, Friedberg and colleagues evaluated bortezomib in combination with BR.44 In the MCL cohort, the ORR was 71%. These promising results led to the study of this combination in the frontline setting. The ongoing ECOG 1411 study is using BR for the frontline treatment of MCL with or without bortezomib as induction. This study also includes rituximab maintenance, and randomizes patients to undergo maintenance with or without the immunomodulator lenalidomide. Bortezomib has been associated with herpes simplex and herpes zoster reactivation. Neuropathy has also been observed with bortezomib, which can be attenuated by administering it subcutaneously.
Lenalidomide is an immunomodulatory agent derived from thalidomide. It has significant activity and is a mainstay of treatment in multiple myeloma. Lenalidomide acts by enhancing cellular immunity, has antiproliferative effects, and inhibits T-cell function leading to growth inhibitory effects in the tumor microenvironment.45 In MCL, lenalidomide has demonstrated clinical activity both as a single agent and in combination, as well as in preclinical studies establishing its pro-apoptotic effects.46 The pivotal EMERGE study evaluated monotherapy with lenalidomide in heavily pretreated relapsed and refractory MCL. This multicenter international study of 134 patents reported an ORR of 28% with a 7.5% CR rate and median PFS of 4 months. All patients had relapsed or progressed following bortezomib. This led to the approval of lenalidomide by the FDA in 2013 for the treatment of patients with MCL whose disease relapsed or progressed following 2 prior therapies, one of which included bortezomib.47 Lenalidomide has been associated with neutropenia, secondary cancers, and deep venous thrombosis.
In combination with other agents in the relapsed setting, lenalidomide shows broader activity. A phase 1/2 study by Wang and colleagues demonstrated an ORR of 57%; the median response duration was 19 months when lenalidomide was combined with rituximab for relapsed/refractory MCL.48
Novel Therapies
More recently, novel treatment approaches have been tested in MCL based on an increased understanding of aberrant signaling pathways in this disease (Table 3). Constitutive activation of B-cell receptor signaling is critical for the survival and proliferation of lymphomas, and has led to the development of targeted agents inhibiting B-cell receptor–associated protein kinases. Bruton’s tyrosine kinase (BTK) is one essential component of the B-cell receptor.49 In particular, proteins upstream of the BTK pathway have been implicated in growth and proliferation of MCL, suggesting that inhibition of BTK may impede lymphomagenesis.50 Ibrutinib is an oral inhibitor of BTK, and demonstrates activity in multiple lymphoma subtypes. In a phase 1 study of ibrutinib in relapsed and refractory hematologic malignancies, an ORR of 60% was observed in 50 evaluable patients, with 16% CR. Median PFS was 13 months. Among these, 7 of 9 patients with MCL responded, including 3 CRs.51 Given these promising results, a phase 2 multicenter study evaluating ibrutinib in relapsed and refractory MCL was completed.52 At a dose of 560 mg daily, the response rate was 68%, with CR of 21%. The most common observed treatment-related side effects included diarrhea, fatigue, and nausea. Neutropenia and thrombocytopenia were also observed. Of importance, 5% of patients had grade 3 or higher bleeding events, including subdural hematoma, gastrointestinal bleeding, and hematuria. The estimated OS rate was 58% at 18 months. On the basis of this study, the FDA approved ibrutinib for relapsed and refractory MCL in November 2013.
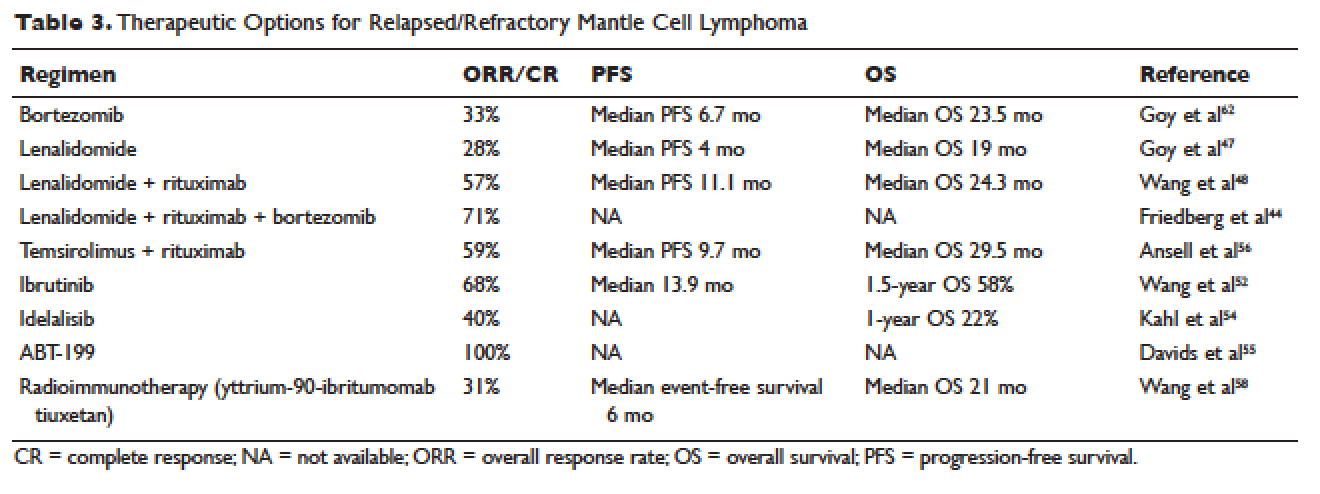
The PI3K pathway is another survival pathway that is dysfunctional in several hematologic disorders, including MCL. Overexpression of PI3K and its downstream targets contributes to MCL pathogenesis.53 Idelalisib is an oral small molecule inhibitor of the delta isoform of PI3K that is dosed daily; it was approved by the FDA for the treatment of relapsed and refractory follicular lymphoma, small lymphocytic lymphoma, and chronic lymphocytic leukemia. It is being further evaluated in MCL. A dose-escalation phase 1 study in heavily pre-treated MCL patients established safety and tolerability.54 Efficacy analysis showed an ORR of 40%, CR of 5%, and 1-year OS of 22%. Further phase 2 studies testing idelalisib as a single agent and in combination for MCL are ongoing. Side effects of idelalisib include elevated liver enzymes, pneumonitis, and diarrhea.
The BCL family of proteins is involved in both pro-and anti-apoptotic functions. BCL2 is an intracellular protein that blocks apoptosis. ABT-199 is an oral BCL2 inhibitor that in early clinical trials has shown very promising activity in MCL. In a phase 1 study of 31 relapsed and refractory NHL patients, all 8 MCL patients (100% ORR) responded to ABT-199 therapy.55 Given these promising initial results, multiple studies evaluating ABT-199 are ongoing in MCL as part of first-line treatment as well as for relapsed disease. ABT-199 has been implicated in tumor lysis syndrome, and in early studies of chronic lymphocytic leukemia, fatal tumor lysis was observed.
The mammalian target of rapamycin (mTOR) inhibitor temsirolimus has been evaluated in relapsed MCL. It is given weekly at 250 mg intravenously. Response rates to single-agent temsirolimus are approximately 20% to 35%, and are higher when combined with rituximab.56,57 The phase 2 study evaluating temsirolimus as a single agent enrolled 35 heavily pre-treated patients. ORR was 38% with only 1 CR. The duration of response was 7 months. Temsirolimus is approved for relapsed MCL in Europe but not in the United States. Similar to the other targeted agents, temsirolimus is actively being studied in combination with other active agents in MCL. Adverse effects noted with temsirolimus include diarrhea, stomatitis, and rash. Thrombocytopenia requiring dose reductions is another frequently observed complication.
Radioimmunotherapy
Radioimmunotherapy (RIT) has been studied extensively in MCL. RIT consists of anti-CD 20 antibodies coupled to radioactive particles that deliver radiation to targeted cells, minimizing toxicity to surrounding tissues. RIT is not used as frequently in the modern era as it had been in the past. At this time, only yttrium-90-ibritumomab tiuxetan is available.
RIT has been evaluated in MCL both at the time of relapse58 and more recently, as part of a conditioning regimen prior to autoSCT, with good tolerability.65–67 Averse events noted with RIT include hematologic toxicity (can be prolonged), hypothyroidism, and in rare cases, myelodysplastic syndrome and acute leukemia. The bone marrow must have less than 25% involvement with disease prior to administration. Wang and colleagues evaluated yttrium-90-ibritumomab tiuxetan in 34 heavily pretreated patients with MCL.58 They observed an ORR of 31%. The median event-free survival (EFS) was 6 months, but in patients achieving either CR or PR, EFS was 28 months. A 21-month OS was noted.
In the upfront setting, RIT has been added as a mechanism of intensification. A recent Nordic group study of RIT with autoSCT did not find benefit with the addition of RIT.59 An ECOG study recently added yttrium-90-ibritumomab tiuxetan after CHOP chemotherapy in the upfront treatment of MCL, with good tolerability.55 However, when added to R-hyper-CVAD, the combination had unexpected high rates of hematologic toxicity, including grade 3/4 cytopenias and an unacceptably high rate of secondary malignancies.68
AutoSCT or Allogeneic Transplant
While many studies noted above have established the beneficial role of autoSCT in MCL in first remission, the role of allogeneic transplant (alloSCT) in MCL remains controversial. A recent large retrospective study conducted by the Center for International Blood and Marrow Transplant Research (CIBMTR) evaluated 519 patients with MCL who underwent both autoSCT and alloSCT.60 Patients were grouped into an early cohort (transplant in first PR or CR, and 2 or fewer treatments) and late cohort (all other patients). The analysis had mature follow up. A multivariate analysis demonstrated that early autoSCT was associated with superior outcomes compared to autoSCT performed later. While it was not possible to demonstrate a survival benefit favoring autoSCT over reduced intensity (RIC) alloSCT, patients transplanted later in their disease course had shorter OS. For patients receiving autoSCT in CR 1 following only 1 prior line of therapy, OS at 5 years was 75% and PFS was 70%. Patients undergoing RIC followed by alloSCT had fewer relapses, but this was negated by higher nonrelapse mortality (25%), resulting in a PFS similar to autoSCT.
CASE CONCLUSION
After treatment with bortezomib the patient is well for 9 months. Subsequently, however, he develops increasing lymphadenopathy and progressive fatigue. He is then started on lenalidomide 25 mg orally daily for 21 out of 28 days. He experiences significant fatigue with lenalidomide and prolonged neutropenia requiring dose delays, despite dose modification to 10 mg orally daily. He requires discontinuation of lenalidomide. Given persistent disease, the patient then begins treatment with ibrutinib. Within a few days of starting ibrutinib therapy, he experiences a marked but transient leukocytosis. Two months later, the patient’s palpable lymphadenopathy has decreased, and his anemia and thrombocytopenia related to MCL are improving. He has tolerated treatment well. His course has been complicated only by a mild, pruritic maculopapular eruption on his chest, back, and arms, that was responsive to topical low-dose steroids. He remains on ibrutinib 1 year later.
CONCLUSION
Advances in our understanding of MCL treatment are revolutionizing the approach to this once deadly disease. Over the next several years, these gains will weave themselves into the current treatment paradigm and likely alter the treatment landscape for MCL as we know it.
- Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
- Zhou Y, Wang H, Fang W, et al. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer 2008;113:791–8.
- Geisler CH. Front-line treatment of mantle cell lymphoma. Haematologica 2010;95:1241–3.
- Fernandez V, Salamero O, Espinet B, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res 2010;70:1408–18.
- Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
- Bertoni F, Ponzoni M. The cellular origin of mantle cell lymphoma. Int J Biochem Cell Biol 2007;39:1747–53.
- de Boer CJ, van Krieken JH, Kluin-Nelemans HC, et al. Cyclin D1 messenger RNA overexpression as a marker for mantle cell lymphoma. Oncogene 1995;10:1833–40.
- Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest 2012;122:3416–23.
- Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003;3:185–97.
- Determann O, Hoster E, Ott G, et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood 2008;111:2385–7.
- Vegliante MC, Palomero J, Perez-Galan P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood 2013;121:2175–85.
- Bea S, Valdes-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A 2013;110:18250–5.
- Zelenetz AD, Abramson JS, Advani RH, et al. Non- Hodgkin’s lymphomas. J Natl Compr Canc Netw 2011;9: 484–560.
- Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
- Zelenetz AD, Abramson JS, Advani RH, et al. NCCN Clinical Practice Guidelines in Oncology: non-Hodgkin’s lymphomas. J Natl Compr Canc Netw 2010;8:288–334.
- Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
- Geisler CH, Kolstad A, Laurell A, et al. The Mantle Cell Lymphoma International Prognostic Index (MIPI) is superior to the International Prognostic Index (IPI) in predicting survival following intensive first-line immunochemotherapy and autologous stem cell transplantation (ASCT). Blood 2010;115:1530–3.
- Tiemann M, Schrader C, Klapper W, et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): a clinicopathological study from the European MCL Network. Br J Haematol 2005;131:29–38.
- Raty R, Franssila K, Joensuu H, et al. Ki-67 expression level, histological subtype, and the International Prognostic Index as outcome predictors in mantle cell lymphoma. Eur J Haematol 2002;69:11–20.
- Vogt N, Klapper W. Variability in morphology and cell proliferation in sequential biopsies of mantle cell lymphoma at diagnosis and relapse: clinical correlation and insights into disease progression. Histopathology 2013;62:334–42.
- Geisler CH, Kolstad A, Laurell A, et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 2008;112:2687–93.
- Howard OM, Gribben JG, Neuberg DS, et al. Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: molecular complete responses are not predictive of progression-free survival. J Clin Oncol 2002;20:1288–94.
- Griffiths R, Mikhael J, Gleeson M, et al. Addition of rituximab to chemotherapy alone as first-line therapy improves overall survival in elderly patients with mantle cell lymphoma. Blood 2011;118:4808–16.
- Lenz G, Dreyling M, Hoster E, et al. immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
- Romaguera JE, Fayad L, Rodriguez MA, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol 2005;23:7013–23.
- Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
- Robak T, Huang H, Jin J, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med 2015;372:944–53.
- Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
- Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
- Houot R, Le Gouill S, Ojeda Uribe M, et al. Combination of rituximab, bortezomib, doxorubicin, dexamethasone and chlorambucil (RiPAD+C) as first-line therapy for elderly mantle cell lymphoma patients: results of a phase II trial from the GOELAMS. Ann Oncol 2012;23:1555–61.
- Ruan J, Martin P, Furman RR, et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol 2011;29:690–7.
- Chang JE, Li H, Smith MR, et al. Phase 2 study of VcR-CVAD with maintenance rituximab for untreated mantle cell lymphoma: an Eastern Cooperative Oncology Group study (E1405). Blood 2014; 123:1665–73.
- Romaguera JE, Fayad LE, Feng L, et al. Ten-year follow-up after intense chemoimmunotherapy with Rituximab-HyperCVAD alternating with Rituximab-high dose methotrexate/cytarabine (R-MA) and without stem cell transplantation in patients with untreated aggressive mantle cell lymphoma. Br J Haematol 2010;150:200–8.
- Bernstein SH, Epner E, Unger JM, et al. A phase II multicenter trial of hyperCVAD MTX/Ara-C and rituximab in patients with previously untreated mantle cell lymphoma; SWOG 0213. Ann Oncol 2013;24:1587–93.
- LaCasce AS, Vandergrift JL, Rodriguez MA, et al. Comparative outcome of initial therapy for younger patients with mantle cell lymphoma: an analysis from the NCCN NHL Database. Blood 2012;119:2093–9.
- Geisler CH, Kolstad A, Laurell A, et al. Nordic MCL2 trial update: six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma followed by BEAM or BEAC + autologous stem-cell support: still very long survival but late relapses do occur. Br J Haematol 2012;158:355–62.
- Damon LE, Johnson JL, Niedzwiecki D, et al. Immunochemotherapy and autologous stem-cell transplantation for untreated patients with mantle-cell lymphoma: CALGB 59909. J Clin Oncol 2009;27:6101–8.
- van ‘t Veer MB, de Jong D, MacKenzie M, et al. High-dose Ara-C and beam with autograft rescue in R-CHOP responsive mantle cell lymphoma patients. Br J Haematol 2009;144:524–30.
- Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
- Hermine O, Hoster E, Walewski J, et al. Alternating courses of 3x CHOP and 3x DHAP plus rituximab followed by a high dose ARA-C containing myeloablative regimen and autologous stem cell transplantation (ASCT) increases overall survival when compared to 6 courses of CHOP plus rituximab followed by myeloablative radiochemotherapy and ASCT in mantle cell lymphoma: final analysis of the MCL Younger Trial of the European Mantle Cell Lymphoma Network (MCL net). In: American Society of Hematology Proceedings. December 8–11, 2012; Atlanta, GA. Abstract 151.
- Rummel MJ, Al-Batran SE, Kim SZ, et al. Bendamustine plus rituximab is effective and has a favorable toxicity profile in the treatment of mantle cell and low-grade non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:3383–9.
- Pham LV, Tamayo AT, Yoshimura LC, et al. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol 2003;171:88–95.
- Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
- Friedberg JW, Vose JM, Kelly JL, et al. The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood 2011;117:2807–12.
- Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 2004;4:314–22.
- Qian Z, Zhang L, Cai Z, et al. Lenalidomide synergizes with dexamethasone to induce growth arrest and apoptosis of mantle cell lymphoma cells in vitro and in vivo. Leuk Res 2011;35:380–6.
- Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
- Wang M, Fayad L, Wagner-Bartak N, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol 2012;13:716–23.
- Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol 2012;31: 119–32.
- Rinaldi A, Kwee I, Taborelli M, et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol 2006;132:303–16.
- Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013; 31:88–94.
- Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
- Rudelius M, Pittaluga S, Nishizuka S, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood 2006;108: 1668–76.
- Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3Kdelta inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014;123:3398–405.
- Davids MS, Seymour JF, Gerecitano JF, et al. Updated results of a phase I first in human study of the BCL-2inhibitor ABT-199 in patients with relapsed/refractory NHL. J Clin Oncol 31, 2013 (suppl; abstr 8520).
- Ansell SM, Tang H, Kurtin PJ, et al. Temsirolimus and rituximab in patients with relapsed or refractory mantle cell lymphoma: a phase 2 study. Lancet Oncol 2011;12:361–8.
- Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 2005;23:5347–56.
- Wang M, Oki Y, Pro B, et al. Phase II study of yttrium-90-ibritumomab tiuxetan in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009;27:5213–8.
- Kolstad A, Laurell A, Jerkeman M, et al. Nordic MCL3 study: 90Y-ibritumomab-tiuxetan added to BEAM/C in non-CR patients before transplant in mantle cell lymphoma. Blood 2014;123:2953–9.
- Fenske TS, Zhang MJ, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
- Dreyling M, Lenz G, Hoster E, et al. Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood 2005;105:2677–84.
- Goy A, Younes A, McLaughlin P, et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:667–75.
- Visco C, Finotto S, Zambello R, et al. Combination of rituximab, bendamustine, and cytarabine for patients with mantle-cell non-Hodgkin lymphoma ineligible for intensive regimens or autologous transplantation. J Clin Oncol 2013;10;31:1442–9.
- Gressin R, Callanan M, Daguindau N, et al. The Ribvd regimen (Rituximab IV, Bendamustine IV, Velcade SC, Dexamethasone IV) offers a high complete response rate In elderly patients with untreated mantle cell lymphoma. Preliminary results of the Lysa trial “Lymphome Du Manteau 2010 SA.” Blood 2013;122:370.
- Krishnan A, Nademanee A, Fung HC, et al. Phase II trial of a transplantation regimen of yttrium-90 ibritumomab tiuxetan and high-dose chemotherapy in patients with non-Hodgkin’s lymphoma. J Clin Oncol 2008;26:90–5.
- Nademanee A, Forman S, Molina A, et al. A phase 1/2 trial of high-dose yttrium-90-ibritumomab tiuxetan in combination with high-dose etoposide and cyclophosphamide followed by autologous stem cell transplantation in patients with poor-risk or relapsed non-Hodgkinlymphoma. Blood 2005;106:2896–902.
- Shimoni A, Avivi I, Rowe JM, et al. A randomized study comparing yttrium-90 ibritumomab tiuxetan (Zevalin) and high-dose BEAM chemotherapy versus BEAM alone as the conditioning regimen before autologous stem cell transplantation in patients with aggressive lymphoma. Cancer 2012;118:4706–14.
- Arranz R, García-Noblejas A, Grande C, et al. First-line treatment with rituximab-hyperCVAD alternating with rituximab-methotrexate-cytarabine and followed by consolidation with 90Y-ibritumomab-tiuxetan in patients with mantle cell lymphoma. Results of a multicenter, phase 2 pilot trial from the GELTAMO group. Haematologica 2013;98:1563-70.
INTRODUCTION
Mantle cell lymphoma (MCL) is an uncommon, distinct clinical subtype of non-Hodgkin lymphoma (NHL) that comprises approximately 8% of all lymphoma diagnoses in the United States and Europe.1,2 Considered incurable, MCL often presents in advanced stages, particularly with involvement of the lymph nodes, spleen, bone marrow, and gastrointestinal tract in the form of lymphomatous polyps. MCL disproportionately affects males, and incidence rises with age, with a median age at diagnosis of 68 years.2 Historically, the prognosis of patients with MCL has been among the poorest among B-cell lymphoma patients, with a median overall survival (OS) of 3 to 5 years, and time to treatment failure (TTF) of 18 to 24 months, although this is improving in the modern era.3 Less frequently, patients with MCL display isolated bone marrow, peripheral blood, and splenic involvement. These cases tend to behave more indolently with longer survival.4,5 Recent advances in therapy have dramatically impacted treatment alternatives and outcomes for MCL. As such, the therapeutic and prognostic landscape of MCL is evolving rapidly.
PATHOGENESIS
The histologic diagnosis of MCL by morphology alone is often challenging. Accurate diagnosis relies on immunohistochemical staining for the purposes of immunophenotyping.6 MCL typically expresses B-cell markers CD5 and CD20, and lacks both CD10 and CD23. The genetic hallmark of MCL is the t(11;14) (q13;q32) chromosomal translocation leading to upregulation of the cyclin D1 protein, a critical regulator of the G1 phase of the cell cycle. Specifically, the t(11;14) translocation, present in virtually all cases of MCL, juxtaposes the proto-oncogene CCND1 to the immunoglobulin heavy chain gene.7 Consequently, cyclin D1, normally not expressed in B lymphocytes, becomes constitutively overexpressed. This alteration is thought to facilitate the deregulation of the cell cycle at the G1-S phase transition.8
Gene expression profiling studies have underscored the importance of cell cycle deregulation in MCL, and high proliferation is associated with a worse prognosis.9 More than 50% of the genes associated with poor outcomes were derived from the “proliferation signature” that was more highly expressed in dividing cells. In the seminal Rosenwald study, a gene expression–based outcome model was constructed in which the proliferation signature average represents a linear variable that assigns a discrete probability of survival to an individual patient.9 The proliferative index, or proliferative signature, of MCL can be estimated by the percentage of Ki-67–positive cells present in the tumor through immunohistochemistry. This is often used as a marker of poor outcomes, and as a surrogate for the proliferative signature in MCL that can be incorporated into clinical practice (as opposed to gene expression profiling). Statistically significant differences in OS have emerged between groups of MCL patients with Ki-67–positive cells comprising less than 30% of their tumor sample (favorable) and those with Ki-67–positive cells comprising 30% or greater (unfavorable).10
Recent data has also identified the importance of the transcription factor SOX 11 (SRY-related HMG-box), which regulates multiple cellular transcriptional events, including cell proliferation and differentiation, apoptosis, and angiogenesis.11 MCL expressing SOX 11 behaves more aggressively than MCL variants lacking SOX 11 expression, and tends to accumulate more genetic alterations.12 Moreover, lack of SOX 11 expression characterizes a subset of MCL that does not carry the t(11;14) translocation.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 62-year-old man with a history of diabetes mellitus and hypertension presents with cervical lymphadenopathy, fatigue, and early satiety over the past several months. He is otherwise in good health. His Eastern Cooperative Oncology Group (ECOG) performance status is 1. On physical examination, 3-cm lymphadenopathy in the bilateral cervical chain is noted. Bilateral axillary lymph nodes measure 2 to 4 cm. His spleen is enlarged and is palpable at approximately 5 cm below the costal margin. A complete blood count reveals a total white blood cell (WBC) count of 14,000 cells/μL, with 68% lymphocytes and a normal distribution of neutrophils. Hemoglobin is 11 g/dL, and platelet count is 112,000/μL. The lactate dehydrogenase (LDH) level is 322 U/L (upper limit of normal: 225 U/L).
• How is MCL diagnosed?
Diagnosis of MCL requires review by expert hematopathologists.13 Whenever possible, an excisional biopsy should be performed for the adequate characterization of lymph node architecture and evaluation by immunohistochemistry. Aside from the characteristic expression of CD5 and CD20 and absence of CD23, MCL should express cyclin D1, which reflects t(11;14). If cyclin D1 is inconclusive or unavailable, fluorescent in situ hybridization (FISH) for t(11;14) should be performed.8 Patients often have circulating malignant lymphocytes, or leukemic phase MCL. Flow cytometry of the peripheral blood can detect traditional surface markers, and FISH can also be performed on circulating abnormal lymphocytes.
For disease staging, bone marrow biopsy and aspiration are required. Radiographic staging using computed tomography (CT) scans and/or positron emission tomography (PET) scans had traditionally followed the Ann Arbor staging system, but recently the Lugano classification has emerged, which delineates only early or advanced stage.14 Gastrointestinal evaluation of MCL with endoscopy and colonoscopy with blind biopsies has been recommended to evaluate for the presence of lymphomatous polyps, but this is not an absolute requirement.15
RISK STRATIFICATION
At diagnosis, patients should undergo risk stratification in order to understand prognosis and possibly guide treatment. In MCL, the MCL international prognostic index (MIPI) is used. The MIPI is a prognostic tool developed exclusively for patients with MCL using data from 455 patients with advanced-stage MCL treated within 3 European clinical trials.16 The MIPI classified patients into risk groups based on age, ECOG performance status, LDH level, and WBC count. Patients were categorized into low-risk (44% of patients, median OS not reached), intermediate-risk (35%, median OS 51 months), and high-risk groups (21%, median OS 29 months). This is done through a logarithmic calculation, which can be accessed through online calculators (a prototype example can be found at www.qxmd.com/calculate-online/hematology/prognosis-mantle-cell-lymphoma-mipi). Cell proliferation using the Ki-67 index was evaluated in an exploratory analysis (the biologic [“B”] MIPI), and also demonstrated strong prognostic relevance.16 Currently, treatment of MCL patients is not stratified by MIPI outside of a clinical trial, but this useful tool assists in assessing patient prognosis and has been validated for use with both conventional chemoimmunotherapy and in the setting of autologous stem cell transplant (autoSCT).16,17 At this point in time, the MIPI score is not used to stratify treatment, although some clinical trials are incorporating the use of the MIPI score at diagnosis. Nonetheless, given its prognostic importance, the MIPI should be performed for all MCL patients undergoing staging and evaluation for treatment to establish disease risk.
As noted, the proliferative signature, represented by the Ki-67 protein, is also highly prognostic in MCL. Ki-67 is expressed in the late G1, S, G2, and M phases of the cell cycle. The Ki-67 index is defined by the hematopathologist as the percentage of lymphoma cells staining positive for Ki-67 protein, based on the number of cells per high-power field. There is significant interobserver variability in this process, which can be minimized by assessing Ki-67 quantitatively using computer software. The prognostic significance of Ki-67 at diagnosis was established in large studies of MCL patient cohorts, with survival differing by up to 3 years.18,19 Determann et al demonstrated the utility of the proliferative index in patients with MCL treated with standard chemoimmunotherapy.10 In this study, 249 patients with advanced-stage MCL treated within randomized trials conducted by the European MCL Network were analyzed. The Ki-67 index was found to be extremely prognostic of OS, independent of other clinical risk factors, including the MIPI score. As a continuous variable, Ki-67 indices of greater than 10% correlated with poor outcomes. The Ki-67 index has also been confirmed as prognostic in relapsed MCL.20 It is important to note that, as a unique feature, the Ki-67 index has remained an independent prognostic factor, even when incorporated into the “B” MIPI.
TREATMENT
CASE CONTINUED
The patient undergoes an excisional biopsy of a cervical lymph node, which demonstrates an abnormal proliferation of small-medium–sized lymphocytes with slightly irregular nuclear contours. Immunohistochemistry shows that the abnormal lymphocytes are positive for CD20 and CD5, negative for CD10 and CD23, and diffusely positive for cyclin D1, consistent with a diagnosis of MCL. The proliferative index, as measured by the Ki-67 immunostain, is 40%. A bone marrow aspirate and biopsy are then obtained, which show a clonal population of B lymphocytes expressing the same immunophenotype as the lymph node (positive for CD20 and CD5, negative for CD10 and CD23, cyclin D1 positive). A CT scan of the neck, chest, abdomen, and pelvis with contrast is obtained, along with a PET scan. These studies identify extensive hypermetabolic lymphadenopathy in the bilateral cervical chains, supraclavicular areas, mediastinum, and hilum. Mesenteric lymph nodes are also enlarged and hypermetabolic, as are retroperitoneal lymph nodes. The spleen is noted to be enlarged with multiple hypermetabolic lesions. Based on the presence of extensive lymphadenopathy as well as bone marrow involvement, the patient is diagnosed with stage IV MCL. He undergoes risk-stratification with the MIPI. His MIPI score is 6.3, high risk.
• What is the approach to upfront therapy for MCL?
FRONTLINE THERAPY
Role of Watchful Waiting
A small proportion of MCL patients have indolent disease that can be observed. This population is more likely to have leukemic-phase MCL with circulating lymphocytes, splenomegaly, and bone marrow involvement and absent or minimal lymphadenopathy.4,5 A retrospective study of 97 patients established that deferment of initial therapy in MCL is acceptable in some patients.5 In this study, approximately one third of patients with MCL were observed for more than 3 months before initiating systemic therapy, and the median time to treatment for the observation group was 12 months. Most patients undergoing observation had a low-risk MIPI. Patients were not harmed by observation, as no OS differences were observed among groups. This study underscores that deferred treatment can be an acceptable alternative in selected MCL patients for a short period of time. In practice, the type of patient who would be appropriate for this approach is someone who is frail, elderly, and with multiple comorbidities. Additionally, expectant observation could be considered for patients with limited-stage or low-volume MCL, low Ki-67 index, and low-risk MIPI scores.
Approach to Therapy
Treatment of MCL is generally approached by evaluating patient age and fitness for treatment. While there is no accepted standard, for younger patients healthy enough to tolerate aggressive approaches, treatment often involves an intensive cytarabine-containing regimen, which is consolidated with an autoSCT. This approach results in the longest remission duration, with some series suggesting a plateau in survival after 5 years, with no relapses.21 Nonintensive conventional chemotherapy alone is often reserved for the frailer or older patient. Given that remission durations with chemotherapy alone in MCL are short, goals of treatment focus on maximizing benefit and remission duration and minimizing risk of toxicity.
Standard Chemotherapy: Elderly and/or Frail Patients
Conventional chemotherapy alone for the treatment of MCL results in a 70% to 85% overall response rate (ORR) and 7% to 30% complete response (CR) rate.22 Rituximab, a mouse humanized monoclonal IgG1 anti-CD20 antibody, is used as standard of care in combination with chemotherapy, since its addition has been found to increase response rates and extend both progression-free survival (PFS) and OS compared to chemotherapy alone.23,24 However, chemoimmunotherapy approaches do not provide long-term control of MCL and are considered noncurative. Various regimens have been studied and include anthracycline-containing regimens such as R-CHOP (rituximab with cyclophosphamide, doxorubicin, vincristine, prednisone),22 combination chemotherapy with antimetabolites such as R-hyper-CVAD (hyper-fractionated rituximab with cyclophosphamide, vincristine, doxorubicin, dexamethasone, alternating with methotrexate and cytarabine),25 purine analogue–based regimens such as R-FC (rituximab with fludarabine and cyclophosphamide),26 bortezomib-containing regimens,27 and alkylator-based treatment with BR (bendamustine and rituximab) (Table 1).28,29 Among these, the most commonly used are R-CHOP and BR.
Two large randomized studies compared R-CHOP for 6 cycles to BR for 6 cycles in patients with indolent NHL and MCL. Among MCL patients, BR resulted in superior PFS compared to R-CHOP (69 months versus 26 months) but no benefit in OS.28,29 The ORR to R-CHOP was approximately 90%, with a PFS of 21 months in the Rummel et al study.29 This study included more than 80 centers in Germany and enrolled 549 patients with MCL, follicular lymphoma, small lymphocytic lymphoma, marginal zone lymphoma, and Waldenström macroglobulinemia. Patients were randomized in a 1:1 fashion. Among these, 46 patients received BR and 48 received R-CHOP (18% for both, respectively). It should be noted that patients in the BR group had significantly less toxicity and experienced fewer side effects than did those in the R-CHOP group. Similarly, BR-treated patients had a lower frequency of hematologic side effects and infections of any grade. However, drug-associated skin reactions and allergies were more common with BR compared to R-CHOP. The study by Flinn and colleagues was an international randomized, noninferiority phase 3 study designed to evaluate the efficacy and safety of BR compared with R-CHOP or R-CVP (rituximab plus cyclophosphamide, vincristine, and prednisone) for treatment-naive patients with MCL or other indolent NHL. The primary endpoint was CR. In this study, BR was found to be noninferior to R-CHOP and R-CVP based on CR rate (31% versus 25%, respectively; P = 0.0225). Response rates in general were high: 97% for BR and 91% for R-CHOP/R-CVP (P = 0.0102). Here, BR-treated patients experienced more nausea, emesis, and drug-induced hypersensitivity compared to the R-CHOP and R-CVP groups.
Another approach studied in older patients is the use of R-CHOP with rituximab maintenance. In a large European study, 560 patients 60 years of age or older with advanced-stage MCL were randomly assigned to either R-FC (rituximab, fludarabine, and cyclophosphamide) every 28 days for 6 cycles, or R-CHOP every 21 days for 8 cycles. Patients who had a response then underwent a second randomization, with one group receiving rituximab maintenance therapy. Maintenance was continued until progression of disease. Patients in this study were not eligible for high-dose chemotherapy and autoSCT. The study found that rates of CR were similar with both R-FC and R-CHOP (40% and 34%, respectively; P = 0.10). However, the R-FC arm underperformed in several arenas. Disease progression occurred more frequently with R-FC (14% versus 5% with R-CHOP), and OS was shorter (4-year OS, 47% versus 62%; P = 0.005, respectively). More patients also died in the R-FC group, and there was greater hematologic toxicity compared to R-CHOP. At 4 years, 58% of the patients receiving rituximab remained in remission. Among patients who responded to R-CHOP, rituximab maintenance led to a benefit in OS, reducing the risk of progression or death by 45%.26 At this time, studies are ongoing to establish the benefit of rituximab maintenance after BR.
Bendamustine in combination with other agents has also been studied in the frontline setting. Visco and colleagues evaluated the combination of bendamustine with rituximab and cytarabine (R-BAC) in older patients with MCL (age 65 or older).63 This phase 2, two-stage study enrolled 40 patients and had a dose-finding arm for cytarabine in combination with BR. It permitted relapsed/refractory patients, but 50% had newly diagnosed, previously untreated MCL. The regimen had an impressive ORR of 100%, with CR rates of 95% for previously untreated patients. PFS at 2 years was 95%. R-BAC was well tolerated, with the primary toxicity being reversible myelosuppression.
BR was combined with the proteasome inhibitor bortezomib and dexamethasone in a phase 2 study.64 This Lymphoma Study Association (LYSA) study evaluated 76 patients with newly diagnosed MCL older than age 65 years. BR was administered in standard doses (bendamustine 90 mg/m2 on days 1 and 2 and rituximab 375 mg/m² IV on day 1) and bortezomib was administered subcutaneously on days 1, 4, 8, and 11, with acyclovir for viral prophylaxis. Patients received 6 cycles. The ORR was 87% and the CR was 60%. Patients experienced toxicity, and not all bortezomib doses were administered due to neurotoxic or hematologic side effects.
A randomized phase 3 study compared R-CHOP to the VR-CAP regimen (R-CHOP regimen but bortezomib replaces vincristine on days 1, 4, 8, 11, at 1.3 mg/m2) in 487 newly diagnosed MCL patients.27 Median PFS was superior in the VR-CAP group compared with R-CHOP (14.4 months versus 24.7 months, respectively). Additionally, rates of CR were superior in the VR-CAP group (53% compared to 42% with R-CHOP). However, there was more hematologic toxicity with VR-CAP. On the basis of these findings, the U.S. Food and Drug Administration approved bortezomib for the frontline treatment of MCL.
Other chemoimmunotherapy combinations containing bortezomib have been studied in frontline MCL treatment, with promising results. These include bortezomib in combination with R-CHOP or modified R-hyper-CVAD, as well as bortezomib in combination with CHOP-like treatments and purine analogues.27,30–32 The ongoing ECOG 1411 study is currently evaluating bortezomib added to BR for induction therapy of newly diagnosed MCL in a 4-arm randomized trial. Patients receive BR with or without bortezomib during induction and are then randomly assigned to maintenance with either rituximab alone or rituximab with lenalidomide. Other novel combination agents are actively being studied in frontline MCL treatment, including lenalidomide and rituximab and BR with lenalidomide.
Intensification of Therapy and AutoSCT: Fitter and/or Younger Patients
Short response duration has created the need for post-remission therapy in MCL. One approach to improve remission duration in MCL is to intensify induction through the use of cytarabine-containing regimens and/or consolidation with high-dose chemotherapy, typically using BEAM (carmustine, etoposide, cytarabine, melphalan) and autoSCT (Table 2). The cytarabine-containing R-hyper-CVAD regimen, developed at the MD Anderson Cancer Center, resulted in a 97% ORR and an 87% CR rate, with TTF of nearly 5 years. However, nearly one third of patients were unable to complete treatment due to toxicity, and 5 patients developed secondary myelodysplastic syndrome or acute myeloid leukemia.33 The feasibility of this R-hyper-CVAD regimen was tested in a multicenter cooperative group setting, but similar results were not seen; in this study, nearly 40% of patients were unable to complete the full scheduled course of treatment due to toxicity.34
Other ways to intensify therapy in MCL involve adding a second non-cross-resistant cytarabine-containing regimen to R-CHOP after remission, such as DHAP (dexamethasone, high-dose cytarabine, cisplatin), followed by consolidation with an autoSCT. A retrospective registry from the National Comprehensive Cancer Network sought to compare the efficacy of different treatment approaches in the frontline setting. They studied 167 patients with MCL and compared 4 groups: treatment with R-hyper-CVAD, either with or without autoSCT, and treatment with R-CHOP, either with or without autoSCT. This study found that in patients younger than 65, R-CHOP followed by autoSCT or R-hyper-CVAD without autoSCT resulted in similar PF and OS, but was superior to R-CHOP alone for newly diagnosed MCL patients.35 These data support more intensive regimens in younger and fitter patients. Several other prospective and randomized studies have demonstrated clinical benefit for patients with MCL undergoing autoSCT in first remission. Of particular importance is the seminal phase 3 study of the European MCL Network, which established the role of autoSCT in this setting.61 In this prospective randomized trial involving 122 newly diagnosed MCL patients who responded to CHOP-like induction, patients in CR derived a greater benefit from autoSCT.
More recent studies have demonstrated similar benefits using cytarabine-based autoSCT. The Nordic MCL2 study evaluated 160 patients using R-CHOP, alternating with rituximab and high-dose cytarabine, followed by autoSCT. This study used “maxi-CHOP,” an augmented CHOP regimen (cyclophosphamide 1200 mg/m2, doxorubicin 75 mg/m2, but standard doses of vincristine [2 mg] and prednisone [100 mg days 1–5]), alternating with 4 infusions of cytarabine at 2 g/m2 and standard doses of rituximab (375 mg/m2). Patients then received conditioning with BEAM and autoSCT. Patients were evaluated for the presence of minimal residual disease (MRD) and for the t(11;14) or clonal immunoglobulin heavy chain gene rearrangement with polymerase chain reaction (PCR). Patients with MRD were offered therapy with rituximab at 375 mg/m2 weekly for 4 doses. This combination resulted in 10-year OS rates of 58%.36 In a multicenter study involving 78 patients from the Cancer and Leukemia Group B (CALGB), R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT resulted in a 5-year OS of 64%.37 A single-arm phase 2 study from the Netherlands also tested R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT. Nonhematologic toxicities were 22% after high-dose cytarabine, and 55% after BEAM. The ORR was 70%, with a 64% CR rate and 66% OS at 4 years.38 The French GELA group used 3 cycles of R-CHOP and 3 cycles of R-DHAP in a phase 2 study of young (under age 66) MCL patients. Following R-CHOP, the ORR was 93%, and following R-DHAP the ORR was 95%. Five-year OSA was 75%.39 A large randomized phase 3 study by Hermine and colleagues of the EMCLN confirmed the benefit of this approach in 497 patients with newly diagnosed MCL. R-CHOP for 6 cycles followed by autoSCT was compared to R-CHOP for 3 cycles alternating with R-DHAP for 3 cycles and autoSCT with a cytarabine-based conditioning regimen. The addition of cytarabine significantly increased rates of CR, TTF, and OS, without increasing toxicity.40
CASE CONTINUED
The patient is treated with R-CHOP chemotherapy for 3 cycles followed by R-DHAP. His course is complicated by mild tinnitus and acute kidney injury from cisplatin that promptly resolves. Three weeks following treatment, a restaging PET/CT scan shows resolution of all lymphadenopathy, with no hypermetabolic uptake, consistent with a complete remission. A repeat bone marrow biopsy shows no involvement with MCL. He subsequently undergoes an autoSCT, and restaging CT/PET 3 months following autoSCT shows continued remission. He is monitored every 3 to 6 months over the next several years.
He has a 4.5-year disease remission, after which he develops growing palpable lymphadenopathy on exam and progressive anemia and thrombocytopenia. A bone marrow biopsy is repeated, which shows recurrent MCL. Restaging diagnostic imaging with a CT scan reveals lymphadenopathy above and below the diaphragm. An axillary lymph node biopsy also demonstrates recurrent MCL. At this time the patient is otherwise in fairly good health, except for feeling fatigued. His ECOG performance status is 1. He begins therapy with bortezomib at a dose of 1.3 mg/m2 intravenously on days 1, 4, 8, and 11 for 6 cycles. His treatment course is complicated by painful sensory peripheral neuropathy of the bilateral lower extremities. Restaging studies at the completion of therapy demonstrate that he has achieved a partial response, with a 50% reduction in the size of involved lymphadenopathy and some residual areas of hypermetabolic uptake. His peripheral cytopenias improve moderately.
• What are the therapeutic options for relapsed MCL?
TREATMENT OF RELAPSED MCL
Single-Agent and Combination Chemotherapy
Whenever possible, and since there is no standard, patients with relapsed MCL should be offered a clinical trial. Outside of a clinical study, many of the treatment regimens used at diagnosis can also be applied in the relapsed setting. In relapsed MCL, Rummel et al showed that BR for 4 cycles resulted in an ORR of 90%, with a CR of 60%. The median PFS was 24 months.41 Bortezomib, an inhibitor of the proteasome-ubiquitin pathway, leads to apoptosis and cell cycle arrest in MCL.42 Multiple studies have evaluated bortezomib both as a single agent and in combination for patients with relapsed MCL. In 2006, bortezomib became the first agent approved by the FDA in relapsed or refractory MCL, based on the phase 2 PINNACLE study. This prospective multicenter study involving 155 patients demonstrated an ORR of 33%, CR rate of 8%, and median treatment duration of 9 months. The median time to progression was 6 months.43 Subsequently, bortezomib-containing combinations evolved. In a multicenter study of relapsed and refractory indolent NHL and MCL, Friedberg and colleagues evaluated bortezomib in combination with BR.44 In the MCL cohort, the ORR was 71%. These promising results led to the study of this combination in the frontline setting. The ongoing ECOG 1411 study is using BR for the frontline treatment of MCL with or without bortezomib as induction. This study also includes rituximab maintenance, and randomizes patients to undergo maintenance with or without the immunomodulator lenalidomide. Bortezomib has been associated with herpes simplex and herpes zoster reactivation. Neuropathy has also been observed with bortezomib, which can be attenuated by administering it subcutaneously.
Lenalidomide is an immunomodulatory agent derived from thalidomide. It has significant activity and is a mainstay of treatment in multiple myeloma. Lenalidomide acts by enhancing cellular immunity, has antiproliferative effects, and inhibits T-cell function leading to growth inhibitory effects in the tumor microenvironment.45 In MCL, lenalidomide has demonstrated clinical activity both as a single agent and in combination, as well as in preclinical studies establishing its pro-apoptotic effects.46 The pivotal EMERGE study evaluated monotherapy with lenalidomide in heavily pretreated relapsed and refractory MCL. This multicenter international study of 134 patents reported an ORR of 28% with a 7.5% CR rate and median PFS of 4 months. All patients had relapsed or progressed following bortezomib. This led to the approval of lenalidomide by the FDA in 2013 for the treatment of patients with MCL whose disease relapsed or progressed following 2 prior therapies, one of which included bortezomib.47 Lenalidomide has been associated with neutropenia, secondary cancers, and deep venous thrombosis.
In combination with other agents in the relapsed setting, lenalidomide shows broader activity. A phase 1/2 study by Wang and colleagues demonstrated an ORR of 57%; the median response duration was 19 months when lenalidomide was combined with rituximab for relapsed/refractory MCL.48
Novel Therapies
More recently, novel treatment approaches have been tested in MCL based on an increased understanding of aberrant signaling pathways in this disease (Table 3). Constitutive activation of B-cell receptor signaling is critical for the survival and proliferation of lymphomas, and has led to the development of targeted agents inhibiting B-cell receptor–associated protein kinases. Bruton’s tyrosine kinase (BTK) is one essential component of the B-cell receptor.49 In particular, proteins upstream of the BTK pathway have been implicated in growth and proliferation of MCL, suggesting that inhibition of BTK may impede lymphomagenesis.50 Ibrutinib is an oral inhibitor of BTK, and demonstrates activity in multiple lymphoma subtypes. In a phase 1 study of ibrutinib in relapsed and refractory hematologic malignancies, an ORR of 60% was observed in 50 evaluable patients, with 16% CR. Median PFS was 13 months. Among these, 7 of 9 patients with MCL responded, including 3 CRs.51 Given these promising results, a phase 2 multicenter study evaluating ibrutinib in relapsed and refractory MCL was completed.52 At a dose of 560 mg daily, the response rate was 68%, with CR of 21%. The most common observed treatment-related side effects included diarrhea, fatigue, and nausea. Neutropenia and thrombocytopenia were also observed. Of importance, 5% of patients had grade 3 or higher bleeding events, including subdural hematoma, gastrointestinal bleeding, and hematuria. The estimated OS rate was 58% at 18 months. On the basis of this study, the FDA approved ibrutinib for relapsed and refractory MCL in November 2013.
The PI3K pathway is another survival pathway that is dysfunctional in several hematologic disorders, including MCL. Overexpression of PI3K and its downstream targets contributes to MCL pathogenesis.53 Idelalisib is an oral small molecule inhibitor of the delta isoform of PI3K that is dosed daily; it was approved by the FDA for the treatment of relapsed and refractory follicular lymphoma, small lymphocytic lymphoma, and chronic lymphocytic leukemia. It is being further evaluated in MCL. A dose-escalation phase 1 study in heavily pre-treated MCL patients established safety and tolerability.54 Efficacy analysis showed an ORR of 40%, CR of 5%, and 1-year OS of 22%. Further phase 2 studies testing idelalisib as a single agent and in combination for MCL are ongoing. Side effects of idelalisib include elevated liver enzymes, pneumonitis, and diarrhea.
The BCL family of proteins is involved in both pro-and anti-apoptotic functions. BCL2 is an intracellular protein that blocks apoptosis. ABT-199 is an oral BCL2 inhibitor that in early clinical trials has shown very promising activity in MCL. In a phase 1 study of 31 relapsed and refractory NHL patients, all 8 MCL patients (100% ORR) responded to ABT-199 therapy.55 Given these promising initial results, multiple studies evaluating ABT-199 are ongoing in MCL as part of first-line treatment as well as for relapsed disease. ABT-199 has been implicated in tumor lysis syndrome, and in early studies of chronic lymphocytic leukemia, fatal tumor lysis was observed.
The mammalian target of rapamycin (mTOR) inhibitor temsirolimus has been evaluated in relapsed MCL. It is given weekly at 250 mg intravenously. Response rates to single-agent temsirolimus are approximately 20% to 35%, and are higher when combined with rituximab.56,57 The phase 2 study evaluating temsirolimus as a single agent enrolled 35 heavily pre-treated patients. ORR was 38% with only 1 CR. The duration of response was 7 months. Temsirolimus is approved for relapsed MCL in Europe but not in the United States. Similar to the other targeted agents, temsirolimus is actively being studied in combination with other active agents in MCL. Adverse effects noted with temsirolimus include diarrhea, stomatitis, and rash. Thrombocytopenia requiring dose reductions is another frequently observed complication.
Radioimmunotherapy
Radioimmunotherapy (RIT) has been studied extensively in MCL. RIT consists of anti-CD 20 antibodies coupled to radioactive particles that deliver radiation to targeted cells, minimizing toxicity to surrounding tissues. RIT is not used as frequently in the modern era as it had been in the past. At this time, only yttrium-90-ibritumomab tiuxetan is available.
RIT has been evaluated in MCL both at the time of relapse58 and more recently, as part of a conditioning regimen prior to autoSCT, with good tolerability.65–67 Averse events noted with RIT include hematologic toxicity (can be prolonged), hypothyroidism, and in rare cases, myelodysplastic syndrome and acute leukemia. The bone marrow must have less than 25% involvement with disease prior to administration. Wang and colleagues evaluated yttrium-90-ibritumomab tiuxetan in 34 heavily pretreated patients with MCL.58 They observed an ORR of 31%. The median event-free survival (EFS) was 6 months, but in patients achieving either CR or PR, EFS was 28 months. A 21-month OS was noted.
In the upfront setting, RIT has been added as a mechanism of intensification. A recent Nordic group study of RIT with autoSCT did not find benefit with the addition of RIT.59 An ECOG study recently added yttrium-90-ibritumomab tiuxetan after CHOP chemotherapy in the upfront treatment of MCL, with good tolerability.55 However, when added to R-hyper-CVAD, the combination had unexpected high rates of hematologic toxicity, including grade 3/4 cytopenias and an unacceptably high rate of secondary malignancies.68
AutoSCT or Allogeneic Transplant
While many studies noted above have established the beneficial role of autoSCT in MCL in first remission, the role of allogeneic transplant (alloSCT) in MCL remains controversial. A recent large retrospective study conducted by the Center for International Blood and Marrow Transplant Research (CIBMTR) evaluated 519 patients with MCL who underwent both autoSCT and alloSCT.60 Patients were grouped into an early cohort (transplant in first PR or CR, and 2 or fewer treatments) and late cohort (all other patients). The analysis had mature follow up. A multivariate analysis demonstrated that early autoSCT was associated with superior outcomes compared to autoSCT performed later. While it was not possible to demonstrate a survival benefit favoring autoSCT over reduced intensity (RIC) alloSCT, patients transplanted later in their disease course had shorter OS. For patients receiving autoSCT in CR 1 following only 1 prior line of therapy, OS at 5 years was 75% and PFS was 70%. Patients undergoing RIC followed by alloSCT had fewer relapses, but this was negated by higher nonrelapse mortality (25%), resulting in a PFS similar to autoSCT.
CASE CONCLUSION
After treatment with bortezomib the patient is well for 9 months. Subsequently, however, he develops increasing lymphadenopathy and progressive fatigue. He is then started on lenalidomide 25 mg orally daily for 21 out of 28 days. He experiences significant fatigue with lenalidomide and prolonged neutropenia requiring dose delays, despite dose modification to 10 mg orally daily. He requires discontinuation of lenalidomide. Given persistent disease, the patient then begins treatment with ibrutinib. Within a few days of starting ibrutinib therapy, he experiences a marked but transient leukocytosis. Two months later, the patient’s palpable lymphadenopathy has decreased, and his anemia and thrombocytopenia related to MCL are improving. He has tolerated treatment well. His course has been complicated only by a mild, pruritic maculopapular eruption on his chest, back, and arms, that was responsive to topical low-dose steroids. He remains on ibrutinib 1 year later.
CONCLUSION
Advances in our understanding of MCL treatment are revolutionizing the approach to this once deadly disease. Over the next several years, these gains will weave themselves into the current treatment paradigm and likely alter the treatment landscape for MCL as we know it.
INTRODUCTION
Mantle cell lymphoma (MCL) is an uncommon, distinct clinical subtype of non-Hodgkin lymphoma (NHL) that comprises approximately 8% of all lymphoma diagnoses in the United States and Europe.1,2 Considered incurable, MCL often presents in advanced stages, particularly with involvement of the lymph nodes, spleen, bone marrow, and gastrointestinal tract in the form of lymphomatous polyps. MCL disproportionately affects males, and incidence rises with age, with a median age at diagnosis of 68 years.2 Historically, the prognosis of patients with MCL has been among the poorest among B-cell lymphoma patients, with a median overall survival (OS) of 3 to 5 years, and time to treatment failure (TTF) of 18 to 24 months, although this is improving in the modern era.3 Less frequently, patients with MCL display isolated bone marrow, peripheral blood, and splenic involvement. These cases tend to behave more indolently with longer survival.4,5 Recent advances in therapy have dramatically impacted treatment alternatives and outcomes for MCL. As such, the therapeutic and prognostic landscape of MCL is evolving rapidly.
PATHOGENESIS
The histologic diagnosis of MCL by morphology alone is often challenging. Accurate diagnosis relies on immunohistochemical staining for the purposes of immunophenotyping.6 MCL typically expresses B-cell markers CD5 and CD20, and lacks both CD10 and CD23. The genetic hallmark of MCL is the t(11;14) (q13;q32) chromosomal translocation leading to upregulation of the cyclin D1 protein, a critical regulator of the G1 phase of the cell cycle. Specifically, the t(11;14) translocation, present in virtually all cases of MCL, juxtaposes the proto-oncogene CCND1 to the immunoglobulin heavy chain gene.7 Consequently, cyclin D1, normally not expressed in B lymphocytes, becomes constitutively overexpressed. This alteration is thought to facilitate the deregulation of the cell cycle at the G1-S phase transition.8
Gene expression profiling studies have underscored the importance of cell cycle deregulation in MCL, and high proliferation is associated with a worse prognosis.9 More than 50% of the genes associated with poor outcomes were derived from the “proliferation signature” that was more highly expressed in dividing cells. In the seminal Rosenwald study, a gene expression–based outcome model was constructed in which the proliferation signature average represents a linear variable that assigns a discrete probability of survival to an individual patient.9 The proliferative index, or proliferative signature, of MCL can be estimated by the percentage of Ki-67–positive cells present in the tumor through immunohistochemistry. This is often used as a marker of poor outcomes, and as a surrogate for the proliferative signature in MCL that can be incorporated into clinical practice (as opposed to gene expression profiling). Statistically significant differences in OS have emerged between groups of MCL patients with Ki-67–positive cells comprising less than 30% of their tumor sample (favorable) and those with Ki-67–positive cells comprising 30% or greater (unfavorable).10
Recent data has also identified the importance of the transcription factor SOX 11 (SRY-related HMG-box), which regulates multiple cellular transcriptional events, including cell proliferation and differentiation, apoptosis, and angiogenesis.11 MCL expressing SOX 11 behaves more aggressively than MCL variants lacking SOX 11 expression, and tends to accumulate more genetic alterations.12 Moreover, lack of SOX 11 expression characterizes a subset of MCL that does not carry the t(11;14) translocation.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 62-year-old man with a history of diabetes mellitus and hypertension presents with cervical lymphadenopathy, fatigue, and early satiety over the past several months. He is otherwise in good health. His Eastern Cooperative Oncology Group (ECOG) performance status is 1. On physical examination, 3-cm lymphadenopathy in the bilateral cervical chain is noted. Bilateral axillary lymph nodes measure 2 to 4 cm. His spleen is enlarged and is palpable at approximately 5 cm below the costal margin. A complete blood count reveals a total white blood cell (WBC) count of 14,000 cells/μL, with 68% lymphocytes and a normal distribution of neutrophils. Hemoglobin is 11 g/dL, and platelet count is 112,000/μL. The lactate dehydrogenase (LDH) level is 322 U/L (upper limit of normal: 225 U/L).
• How is MCL diagnosed?
Diagnosis of MCL requires review by expert hematopathologists.13 Whenever possible, an excisional biopsy should be performed for the adequate characterization of lymph node architecture and evaluation by immunohistochemistry. Aside from the characteristic expression of CD5 and CD20 and absence of CD23, MCL should express cyclin D1, which reflects t(11;14). If cyclin D1 is inconclusive or unavailable, fluorescent in situ hybridization (FISH) for t(11;14) should be performed.8 Patients often have circulating malignant lymphocytes, or leukemic phase MCL. Flow cytometry of the peripheral blood can detect traditional surface markers, and FISH can also be performed on circulating abnormal lymphocytes.
For disease staging, bone marrow biopsy and aspiration are required. Radiographic staging using computed tomography (CT) scans and/or positron emission tomography (PET) scans had traditionally followed the Ann Arbor staging system, but recently the Lugano classification has emerged, which delineates only early or advanced stage.14 Gastrointestinal evaluation of MCL with endoscopy and colonoscopy with blind biopsies has been recommended to evaluate for the presence of lymphomatous polyps, but this is not an absolute requirement.15
RISK STRATIFICATION
At diagnosis, patients should undergo risk stratification in order to understand prognosis and possibly guide treatment. In MCL, the MCL international prognostic index (MIPI) is used. The MIPI is a prognostic tool developed exclusively for patients with MCL using data from 455 patients with advanced-stage MCL treated within 3 European clinical trials.16 The MIPI classified patients into risk groups based on age, ECOG performance status, LDH level, and WBC count. Patients were categorized into low-risk (44% of patients, median OS not reached), intermediate-risk (35%, median OS 51 months), and high-risk groups (21%, median OS 29 months). This is done through a logarithmic calculation, which can be accessed through online calculators (a prototype example can be found at www.qxmd.com/calculate-online/hematology/prognosis-mantle-cell-lymphoma-mipi). Cell proliferation using the Ki-67 index was evaluated in an exploratory analysis (the biologic [“B”] MIPI), and also demonstrated strong prognostic relevance.16 Currently, treatment of MCL patients is not stratified by MIPI outside of a clinical trial, but this useful tool assists in assessing patient prognosis and has been validated for use with both conventional chemoimmunotherapy and in the setting of autologous stem cell transplant (autoSCT).16,17 At this point in time, the MIPI score is not used to stratify treatment, although some clinical trials are incorporating the use of the MIPI score at diagnosis. Nonetheless, given its prognostic importance, the MIPI should be performed for all MCL patients undergoing staging and evaluation for treatment to establish disease risk.
As noted, the proliferative signature, represented by the Ki-67 protein, is also highly prognostic in MCL. Ki-67 is expressed in the late G1, S, G2, and M phases of the cell cycle. The Ki-67 index is defined by the hematopathologist as the percentage of lymphoma cells staining positive for Ki-67 protein, based on the number of cells per high-power field. There is significant interobserver variability in this process, which can be minimized by assessing Ki-67 quantitatively using computer software. The prognostic significance of Ki-67 at diagnosis was established in large studies of MCL patient cohorts, with survival differing by up to 3 years.18,19 Determann et al demonstrated the utility of the proliferative index in patients with MCL treated with standard chemoimmunotherapy.10 In this study, 249 patients with advanced-stage MCL treated within randomized trials conducted by the European MCL Network were analyzed. The Ki-67 index was found to be extremely prognostic of OS, independent of other clinical risk factors, including the MIPI score. As a continuous variable, Ki-67 indices of greater than 10% correlated with poor outcomes. The Ki-67 index has also been confirmed as prognostic in relapsed MCL.20 It is important to note that, as a unique feature, the Ki-67 index has remained an independent prognostic factor, even when incorporated into the “B” MIPI.
TREATMENT
CASE CONTINUED
The patient undergoes an excisional biopsy of a cervical lymph node, which demonstrates an abnormal proliferation of small-medium–sized lymphocytes with slightly irregular nuclear contours. Immunohistochemistry shows that the abnormal lymphocytes are positive for CD20 and CD5, negative for CD10 and CD23, and diffusely positive for cyclin D1, consistent with a diagnosis of MCL. The proliferative index, as measured by the Ki-67 immunostain, is 40%. A bone marrow aspirate and biopsy are then obtained, which show a clonal population of B lymphocytes expressing the same immunophenotype as the lymph node (positive for CD20 and CD5, negative for CD10 and CD23, cyclin D1 positive). A CT scan of the neck, chest, abdomen, and pelvis with contrast is obtained, along with a PET scan. These studies identify extensive hypermetabolic lymphadenopathy in the bilateral cervical chains, supraclavicular areas, mediastinum, and hilum. Mesenteric lymph nodes are also enlarged and hypermetabolic, as are retroperitoneal lymph nodes. The spleen is noted to be enlarged with multiple hypermetabolic lesions. Based on the presence of extensive lymphadenopathy as well as bone marrow involvement, the patient is diagnosed with stage IV MCL. He undergoes risk-stratification with the MIPI. His MIPI score is 6.3, high risk.
• What is the approach to upfront therapy for MCL?
FRONTLINE THERAPY
Role of Watchful Waiting
A small proportion of MCL patients have indolent disease that can be observed. This population is more likely to have leukemic-phase MCL with circulating lymphocytes, splenomegaly, and bone marrow involvement and absent or minimal lymphadenopathy.4,5 A retrospective study of 97 patients established that deferment of initial therapy in MCL is acceptable in some patients.5 In this study, approximately one third of patients with MCL were observed for more than 3 months before initiating systemic therapy, and the median time to treatment for the observation group was 12 months. Most patients undergoing observation had a low-risk MIPI. Patients were not harmed by observation, as no OS differences were observed among groups. This study underscores that deferred treatment can be an acceptable alternative in selected MCL patients for a short period of time. In practice, the type of patient who would be appropriate for this approach is someone who is frail, elderly, and with multiple comorbidities. Additionally, expectant observation could be considered for patients with limited-stage or low-volume MCL, low Ki-67 index, and low-risk MIPI scores.
Approach to Therapy
Treatment of MCL is generally approached by evaluating patient age and fitness for treatment. While there is no accepted standard, for younger patients healthy enough to tolerate aggressive approaches, treatment often involves an intensive cytarabine-containing regimen, which is consolidated with an autoSCT. This approach results in the longest remission duration, with some series suggesting a plateau in survival after 5 years, with no relapses.21 Nonintensive conventional chemotherapy alone is often reserved for the frailer or older patient. Given that remission durations with chemotherapy alone in MCL are short, goals of treatment focus on maximizing benefit and remission duration and minimizing risk of toxicity.
Standard Chemotherapy: Elderly and/or Frail Patients
Conventional chemotherapy alone for the treatment of MCL results in a 70% to 85% overall response rate (ORR) and 7% to 30% complete response (CR) rate.22 Rituximab, a mouse humanized monoclonal IgG1 anti-CD20 antibody, is used as standard of care in combination with chemotherapy, since its addition has been found to increase response rates and extend both progression-free survival (PFS) and OS compared to chemotherapy alone.23,24 However, chemoimmunotherapy approaches do not provide long-term control of MCL and are considered noncurative. Various regimens have been studied and include anthracycline-containing regimens such as R-CHOP (rituximab with cyclophosphamide, doxorubicin, vincristine, prednisone),22 combination chemotherapy with antimetabolites such as R-hyper-CVAD (hyper-fractionated rituximab with cyclophosphamide, vincristine, doxorubicin, dexamethasone, alternating with methotrexate and cytarabine),25 purine analogue–based regimens such as R-FC (rituximab with fludarabine and cyclophosphamide),26 bortezomib-containing regimens,27 and alkylator-based treatment with BR (bendamustine and rituximab) (Table 1).28,29 Among these, the most commonly used are R-CHOP and BR.
Two large randomized studies compared R-CHOP for 6 cycles to BR for 6 cycles in patients with indolent NHL and MCL. Among MCL patients, BR resulted in superior PFS compared to R-CHOP (69 months versus 26 months) but no benefit in OS.28,29 The ORR to R-CHOP was approximately 90%, with a PFS of 21 months in the Rummel et al study.29 This study included more than 80 centers in Germany and enrolled 549 patients with MCL, follicular lymphoma, small lymphocytic lymphoma, marginal zone lymphoma, and Waldenström macroglobulinemia. Patients were randomized in a 1:1 fashion. Among these, 46 patients received BR and 48 received R-CHOP (18% for both, respectively). It should be noted that patients in the BR group had significantly less toxicity and experienced fewer side effects than did those in the R-CHOP group. Similarly, BR-treated patients had a lower frequency of hematologic side effects and infections of any grade. However, drug-associated skin reactions and allergies were more common with BR compared to R-CHOP. The study by Flinn and colleagues was an international randomized, noninferiority phase 3 study designed to evaluate the efficacy and safety of BR compared with R-CHOP or R-CVP (rituximab plus cyclophosphamide, vincristine, and prednisone) for treatment-naive patients with MCL or other indolent NHL. The primary endpoint was CR. In this study, BR was found to be noninferior to R-CHOP and R-CVP based on CR rate (31% versus 25%, respectively; P = 0.0225). Response rates in general were high: 97% for BR and 91% for R-CHOP/R-CVP (P = 0.0102). Here, BR-treated patients experienced more nausea, emesis, and drug-induced hypersensitivity compared to the R-CHOP and R-CVP groups.
Another approach studied in older patients is the use of R-CHOP with rituximab maintenance. In a large European study, 560 patients 60 years of age or older with advanced-stage MCL were randomly assigned to either R-FC (rituximab, fludarabine, and cyclophosphamide) every 28 days for 6 cycles, or R-CHOP every 21 days for 8 cycles. Patients who had a response then underwent a second randomization, with one group receiving rituximab maintenance therapy. Maintenance was continued until progression of disease. Patients in this study were not eligible for high-dose chemotherapy and autoSCT. The study found that rates of CR were similar with both R-FC and R-CHOP (40% and 34%, respectively; P = 0.10). However, the R-FC arm underperformed in several arenas. Disease progression occurred more frequently with R-FC (14% versus 5% with R-CHOP), and OS was shorter (4-year OS, 47% versus 62%; P = 0.005, respectively). More patients also died in the R-FC group, and there was greater hematologic toxicity compared to R-CHOP. At 4 years, 58% of the patients receiving rituximab remained in remission. Among patients who responded to R-CHOP, rituximab maintenance led to a benefit in OS, reducing the risk of progression or death by 45%.26 At this time, studies are ongoing to establish the benefit of rituximab maintenance after BR.
Bendamustine in combination with other agents has also been studied in the frontline setting. Visco and colleagues evaluated the combination of bendamustine with rituximab and cytarabine (R-BAC) in older patients with MCL (age 65 or older).63 This phase 2, two-stage study enrolled 40 patients and had a dose-finding arm for cytarabine in combination with BR. It permitted relapsed/refractory patients, but 50% had newly diagnosed, previously untreated MCL. The regimen had an impressive ORR of 100%, with CR rates of 95% for previously untreated patients. PFS at 2 years was 95%. R-BAC was well tolerated, with the primary toxicity being reversible myelosuppression.
BR was combined with the proteasome inhibitor bortezomib and dexamethasone in a phase 2 study.64 This Lymphoma Study Association (LYSA) study evaluated 76 patients with newly diagnosed MCL older than age 65 years. BR was administered in standard doses (bendamustine 90 mg/m2 on days 1 and 2 and rituximab 375 mg/m² IV on day 1) and bortezomib was administered subcutaneously on days 1, 4, 8, and 11, with acyclovir for viral prophylaxis. Patients received 6 cycles. The ORR was 87% and the CR was 60%. Patients experienced toxicity, and not all bortezomib doses were administered due to neurotoxic or hematologic side effects.
A randomized phase 3 study compared R-CHOP to the VR-CAP regimen (R-CHOP regimen but bortezomib replaces vincristine on days 1, 4, 8, 11, at 1.3 mg/m2) in 487 newly diagnosed MCL patients.27 Median PFS was superior in the VR-CAP group compared with R-CHOP (14.4 months versus 24.7 months, respectively). Additionally, rates of CR were superior in the VR-CAP group (53% compared to 42% with R-CHOP). However, there was more hematologic toxicity with VR-CAP. On the basis of these findings, the U.S. Food and Drug Administration approved bortezomib for the frontline treatment of MCL.
Other chemoimmunotherapy combinations containing bortezomib have been studied in frontline MCL treatment, with promising results. These include bortezomib in combination with R-CHOP or modified R-hyper-CVAD, as well as bortezomib in combination with CHOP-like treatments and purine analogues.27,30–32 The ongoing ECOG 1411 study is currently evaluating bortezomib added to BR for induction therapy of newly diagnosed MCL in a 4-arm randomized trial. Patients receive BR with or without bortezomib during induction and are then randomly assigned to maintenance with either rituximab alone or rituximab with lenalidomide. Other novel combination agents are actively being studied in frontline MCL treatment, including lenalidomide and rituximab and BR with lenalidomide.
Intensification of Therapy and AutoSCT: Fitter and/or Younger Patients
Short response duration has created the need for post-remission therapy in MCL. One approach to improve remission duration in MCL is to intensify induction through the use of cytarabine-containing regimens and/or consolidation with high-dose chemotherapy, typically using BEAM (carmustine, etoposide, cytarabine, melphalan) and autoSCT (Table 2). The cytarabine-containing R-hyper-CVAD regimen, developed at the MD Anderson Cancer Center, resulted in a 97% ORR and an 87% CR rate, with TTF of nearly 5 years. However, nearly one third of patients were unable to complete treatment due to toxicity, and 5 patients developed secondary myelodysplastic syndrome or acute myeloid leukemia.33 The feasibility of this R-hyper-CVAD regimen was tested in a multicenter cooperative group setting, but similar results were not seen; in this study, nearly 40% of patients were unable to complete the full scheduled course of treatment due to toxicity.34
Other ways to intensify therapy in MCL involve adding a second non-cross-resistant cytarabine-containing regimen to R-CHOP after remission, such as DHAP (dexamethasone, high-dose cytarabine, cisplatin), followed by consolidation with an autoSCT. A retrospective registry from the National Comprehensive Cancer Network sought to compare the efficacy of different treatment approaches in the frontline setting. They studied 167 patients with MCL and compared 4 groups: treatment with R-hyper-CVAD, either with or without autoSCT, and treatment with R-CHOP, either with or without autoSCT. This study found that in patients younger than 65, R-CHOP followed by autoSCT or R-hyper-CVAD without autoSCT resulted in similar PF and OS, but was superior to R-CHOP alone for newly diagnosed MCL patients.35 These data support more intensive regimens in younger and fitter patients. Several other prospective and randomized studies have demonstrated clinical benefit for patients with MCL undergoing autoSCT in first remission. Of particular importance is the seminal phase 3 study of the European MCL Network, which established the role of autoSCT in this setting.61 In this prospective randomized trial involving 122 newly diagnosed MCL patients who responded to CHOP-like induction, patients in CR derived a greater benefit from autoSCT.
More recent studies have demonstrated similar benefits using cytarabine-based autoSCT. The Nordic MCL2 study evaluated 160 patients using R-CHOP, alternating with rituximab and high-dose cytarabine, followed by autoSCT. This study used “maxi-CHOP,” an augmented CHOP regimen (cyclophosphamide 1200 mg/m2, doxorubicin 75 mg/m2, but standard doses of vincristine [2 mg] and prednisone [100 mg days 1–5]), alternating with 4 infusions of cytarabine at 2 g/m2 and standard doses of rituximab (375 mg/m2). Patients then received conditioning with BEAM and autoSCT. Patients were evaluated for the presence of minimal residual disease (MRD) and for the t(11;14) or clonal immunoglobulin heavy chain gene rearrangement with polymerase chain reaction (PCR). Patients with MRD were offered therapy with rituximab at 375 mg/m2 weekly for 4 doses. This combination resulted in 10-year OS rates of 58%.36 In a multicenter study involving 78 patients from the Cancer and Leukemia Group B (CALGB), R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT resulted in a 5-year OS of 64%.37 A single-arm phase 2 study from the Netherlands also tested R-CHOP followed by high-dose cytarabine and BEAM-based autoSCT. Nonhematologic toxicities were 22% after high-dose cytarabine, and 55% after BEAM. The ORR was 70%, with a 64% CR rate and 66% OS at 4 years.38 The French GELA group used 3 cycles of R-CHOP and 3 cycles of R-DHAP in a phase 2 study of young (under age 66) MCL patients. Following R-CHOP, the ORR was 93%, and following R-DHAP the ORR was 95%. Five-year OSA was 75%.39 A large randomized phase 3 study by Hermine and colleagues of the EMCLN confirmed the benefit of this approach in 497 patients with newly diagnosed MCL. R-CHOP for 6 cycles followed by autoSCT was compared to R-CHOP for 3 cycles alternating with R-DHAP for 3 cycles and autoSCT with a cytarabine-based conditioning regimen. The addition of cytarabine significantly increased rates of CR, TTF, and OS, without increasing toxicity.40
CASE CONTINUED
The patient is treated with R-CHOP chemotherapy for 3 cycles followed by R-DHAP. His course is complicated by mild tinnitus and acute kidney injury from cisplatin that promptly resolves. Three weeks following treatment, a restaging PET/CT scan shows resolution of all lymphadenopathy, with no hypermetabolic uptake, consistent with a complete remission. A repeat bone marrow biopsy shows no involvement with MCL. He subsequently undergoes an autoSCT, and restaging CT/PET 3 months following autoSCT shows continued remission. He is monitored every 3 to 6 months over the next several years.
He has a 4.5-year disease remission, after which he develops growing palpable lymphadenopathy on exam and progressive anemia and thrombocytopenia. A bone marrow biopsy is repeated, which shows recurrent MCL. Restaging diagnostic imaging with a CT scan reveals lymphadenopathy above and below the diaphragm. An axillary lymph node biopsy also demonstrates recurrent MCL. At this time the patient is otherwise in fairly good health, except for feeling fatigued. His ECOG performance status is 1. He begins therapy with bortezomib at a dose of 1.3 mg/m2 intravenously on days 1, 4, 8, and 11 for 6 cycles. His treatment course is complicated by painful sensory peripheral neuropathy of the bilateral lower extremities. Restaging studies at the completion of therapy demonstrate that he has achieved a partial response, with a 50% reduction in the size of involved lymphadenopathy and some residual areas of hypermetabolic uptake. His peripheral cytopenias improve moderately.
• What are the therapeutic options for relapsed MCL?
TREATMENT OF RELAPSED MCL
Single-Agent and Combination Chemotherapy
Whenever possible, and since there is no standard, patients with relapsed MCL should be offered a clinical trial. Outside of a clinical study, many of the treatment regimens used at diagnosis can also be applied in the relapsed setting. In relapsed MCL, Rummel et al showed that BR for 4 cycles resulted in an ORR of 90%, with a CR of 60%. The median PFS was 24 months.41 Bortezomib, an inhibitor of the proteasome-ubiquitin pathway, leads to apoptosis and cell cycle arrest in MCL.42 Multiple studies have evaluated bortezomib both as a single agent and in combination for patients with relapsed MCL. In 2006, bortezomib became the first agent approved by the FDA in relapsed or refractory MCL, based on the phase 2 PINNACLE study. This prospective multicenter study involving 155 patients demonstrated an ORR of 33%, CR rate of 8%, and median treatment duration of 9 months. The median time to progression was 6 months.43 Subsequently, bortezomib-containing combinations evolved. In a multicenter study of relapsed and refractory indolent NHL and MCL, Friedberg and colleagues evaluated bortezomib in combination with BR.44 In the MCL cohort, the ORR was 71%. These promising results led to the study of this combination in the frontline setting. The ongoing ECOG 1411 study is using BR for the frontline treatment of MCL with or without bortezomib as induction. This study also includes rituximab maintenance, and randomizes patients to undergo maintenance with or without the immunomodulator lenalidomide. Bortezomib has been associated with herpes simplex and herpes zoster reactivation. Neuropathy has also been observed with bortezomib, which can be attenuated by administering it subcutaneously.
Lenalidomide is an immunomodulatory agent derived from thalidomide. It has significant activity and is a mainstay of treatment in multiple myeloma. Lenalidomide acts by enhancing cellular immunity, has antiproliferative effects, and inhibits T-cell function leading to growth inhibitory effects in the tumor microenvironment.45 In MCL, lenalidomide has demonstrated clinical activity both as a single agent and in combination, as well as in preclinical studies establishing its pro-apoptotic effects.46 The pivotal EMERGE study evaluated monotherapy with lenalidomide in heavily pretreated relapsed and refractory MCL. This multicenter international study of 134 patents reported an ORR of 28% with a 7.5% CR rate and median PFS of 4 months. All patients had relapsed or progressed following bortezomib. This led to the approval of lenalidomide by the FDA in 2013 for the treatment of patients with MCL whose disease relapsed or progressed following 2 prior therapies, one of which included bortezomib.47 Lenalidomide has been associated with neutropenia, secondary cancers, and deep venous thrombosis.
In combination with other agents in the relapsed setting, lenalidomide shows broader activity. A phase 1/2 study by Wang and colleagues demonstrated an ORR of 57%; the median response duration was 19 months when lenalidomide was combined with rituximab for relapsed/refractory MCL.48
Novel Therapies
More recently, novel treatment approaches have been tested in MCL based on an increased understanding of aberrant signaling pathways in this disease (Table 3). Constitutive activation of B-cell receptor signaling is critical for the survival and proliferation of lymphomas, and has led to the development of targeted agents inhibiting B-cell receptor–associated protein kinases. Bruton’s tyrosine kinase (BTK) is one essential component of the B-cell receptor.49 In particular, proteins upstream of the BTK pathway have been implicated in growth and proliferation of MCL, suggesting that inhibition of BTK may impede lymphomagenesis.50 Ibrutinib is an oral inhibitor of BTK, and demonstrates activity in multiple lymphoma subtypes. In a phase 1 study of ibrutinib in relapsed and refractory hematologic malignancies, an ORR of 60% was observed in 50 evaluable patients, with 16% CR. Median PFS was 13 months. Among these, 7 of 9 patients with MCL responded, including 3 CRs.51 Given these promising results, a phase 2 multicenter study evaluating ibrutinib in relapsed and refractory MCL was completed.52 At a dose of 560 mg daily, the response rate was 68%, with CR of 21%. The most common observed treatment-related side effects included diarrhea, fatigue, and nausea. Neutropenia and thrombocytopenia were also observed. Of importance, 5% of patients had grade 3 or higher bleeding events, including subdural hematoma, gastrointestinal bleeding, and hematuria. The estimated OS rate was 58% at 18 months. On the basis of this study, the FDA approved ibrutinib for relapsed and refractory MCL in November 2013.
The PI3K pathway is another survival pathway that is dysfunctional in several hematologic disorders, including MCL. Overexpression of PI3K and its downstream targets contributes to MCL pathogenesis.53 Idelalisib is an oral small molecule inhibitor of the delta isoform of PI3K that is dosed daily; it was approved by the FDA for the treatment of relapsed and refractory follicular lymphoma, small lymphocytic lymphoma, and chronic lymphocytic leukemia. It is being further evaluated in MCL. A dose-escalation phase 1 study in heavily pre-treated MCL patients established safety and tolerability.54 Efficacy analysis showed an ORR of 40%, CR of 5%, and 1-year OS of 22%. Further phase 2 studies testing idelalisib as a single agent and in combination for MCL are ongoing. Side effects of idelalisib include elevated liver enzymes, pneumonitis, and diarrhea.
The BCL family of proteins is involved in both pro-and anti-apoptotic functions. BCL2 is an intracellular protein that blocks apoptosis. ABT-199 is an oral BCL2 inhibitor that in early clinical trials has shown very promising activity in MCL. In a phase 1 study of 31 relapsed and refractory NHL patients, all 8 MCL patients (100% ORR) responded to ABT-199 therapy.55 Given these promising initial results, multiple studies evaluating ABT-199 are ongoing in MCL as part of first-line treatment as well as for relapsed disease. ABT-199 has been implicated in tumor lysis syndrome, and in early studies of chronic lymphocytic leukemia, fatal tumor lysis was observed.
The mammalian target of rapamycin (mTOR) inhibitor temsirolimus has been evaluated in relapsed MCL. It is given weekly at 250 mg intravenously. Response rates to single-agent temsirolimus are approximately 20% to 35%, and are higher when combined with rituximab.56,57 The phase 2 study evaluating temsirolimus as a single agent enrolled 35 heavily pre-treated patients. ORR was 38% with only 1 CR. The duration of response was 7 months. Temsirolimus is approved for relapsed MCL in Europe but not in the United States. Similar to the other targeted agents, temsirolimus is actively being studied in combination with other active agents in MCL. Adverse effects noted with temsirolimus include diarrhea, stomatitis, and rash. Thrombocytopenia requiring dose reductions is another frequently observed complication.
Radioimmunotherapy
Radioimmunotherapy (RIT) has been studied extensively in MCL. RIT consists of anti-CD 20 antibodies coupled to radioactive particles that deliver radiation to targeted cells, minimizing toxicity to surrounding tissues. RIT is not used as frequently in the modern era as it had been in the past. At this time, only yttrium-90-ibritumomab tiuxetan is available.
RIT has been evaluated in MCL both at the time of relapse58 and more recently, as part of a conditioning regimen prior to autoSCT, with good tolerability.65–67 Averse events noted with RIT include hematologic toxicity (can be prolonged), hypothyroidism, and in rare cases, myelodysplastic syndrome and acute leukemia. The bone marrow must have less than 25% involvement with disease prior to administration. Wang and colleagues evaluated yttrium-90-ibritumomab tiuxetan in 34 heavily pretreated patients with MCL.58 They observed an ORR of 31%. The median event-free survival (EFS) was 6 months, but in patients achieving either CR or PR, EFS was 28 months. A 21-month OS was noted.
In the upfront setting, RIT has been added as a mechanism of intensification. A recent Nordic group study of RIT with autoSCT did not find benefit with the addition of RIT.59 An ECOG study recently added yttrium-90-ibritumomab tiuxetan after CHOP chemotherapy in the upfront treatment of MCL, with good tolerability.55 However, when added to R-hyper-CVAD, the combination had unexpected high rates of hematologic toxicity, including grade 3/4 cytopenias and an unacceptably high rate of secondary malignancies.68
AutoSCT or Allogeneic Transplant
While many studies noted above have established the beneficial role of autoSCT in MCL in first remission, the role of allogeneic transplant (alloSCT) in MCL remains controversial. A recent large retrospective study conducted by the Center for International Blood and Marrow Transplant Research (CIBMTR) evaluated 519 patients with MCL who underwent both autoSCT and alloSCT.60 Patients were grouped into an early cohort (transplant in first PR or CR, and 2 or fewer treatments) and late cohort (all other patients). The analysis had mature follow up. A multivariate analysis demonstrated that early autoSCT was associated with superior outcomes compared to autoSCT performed later. While it was not possible to demonstrate a survival benefit favoring autoSCT over reduced intensity (RIC) alloSCT, patients transplanted later in their disease course had shorter OS. For patients receiving autoSCT in CR 1 following only 1 prior line of therapy, OS at 5 years was 75% and PFS was 70%. Patients undergoing RIC followed by alloSCT had fewer relapses, but this was negated by higher nonrelapse mortality (25%), resulting in a PFS similar to autoSCT.
CASE CONCLUSION
After treatment with bortezomib the patient is well for 9 months. Subsequently, however, he develops increasing lymphadenopathy and progressive fatigue. He is then started on lenalidomide 25 mg orally daily for 21 out of 28 days. He experiences significant fatigue with lenalidomide and prolonged neutropenia requiring dose delays, despite dose modification to 10 mg orally daily. He requires discontinuation of lenalidomide. Given persistent disease, the patient then begins treatment with ibrutinib. Within a few days of starting ibrutinib therapy, he experiences a marked but transient leukocytosis. Two months later, the patient’s palpable lymphadenopathy has decreased, and his anemia and thrombocytopenia related to MCL are improving. He has tolerated treatment well. His course has been complicated only by a mild, pruritic maculopapular eruption on his chest, back, and arms, that was responsive to topical low-dose steroids. He remains on ibrutinib 1 year later.
CONCLUSION
Advances in our understanding of MCL treatment are revolutionizing the approach to this once deadly disease. Over the next several years, these gains will weave themselves into the current treatment paradigm and likely alter the treatment landscape for MCL as we know it.
- Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
- Zhou Y, Wang H, Fang W, et al. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer 2008;113:791–8.
- Geisler CH. Front-line treatment of mantle cell lymphoma. Haematologica 2010;95:1241–3.
- Fernandez V, Salamero O, Espinet B, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res 2010;70:1408–18.
- Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
- Bertoni F, Ponzoni M. The cellular origin of mantle cell lymphoma. Int J Biochem Cell Biol 2007;39:1747–53.
- de Boer CJ, van Krieken JH, Kluin-Nelemans HC, et al. Cyclin D1 messenger RNA overexpression as a marker for mantle cell lymphoma. Oncogene 1995;10:1833–40.
- Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest 2012;122:3416–23.
- Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003;3:185–97.
- Determann O, Hoster E, Ott G, et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood 2008;111:2385–7.
- Vegliante MC, Palomero J, Perez-Galan P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood 2013;121:2175–85.
- Bea S, Valdes-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A 2013;110:18250–5.
- Zelenetz AD, Abramson JS, Advani RH, et al. Non- Hodgkin’s lymphomas. J Natl Compr Canc Netw 2011;9: 484–560.
- Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
- Zelenetz AD, Abramson JS, Advani RH, et al. NCCN Clinical Practice Guidelines in Oncology: non-Hodgkin’s lymphomas. J Natl Compr Canc Netw 2010;8:288–334.
- Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
- Geisler CH, Kolstad A, Laurell A, et al. The Mantle Cell Lymphoma International Prognostic Index (MIPI) is superior to the International Prognostic Index (IPI) in predicting survival following intensive first-line immunochemotherapy and autologous stem cell transplantation (ASCT). Blood 2010;115:1530–3.
- Tiemann M, Schrader C, Klapper W, et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): a clinicopathological study from the European MCL Network. Br J Haematol 2005;131:29–38.
- Raty R, Franssila K, Joensuu H, et al. Ki-67 expression level, histological subtype, and the International Prognostic Index as outcome predictors in mantle cell lymphoma. Eur J Haematol 2002;69:11–20.
- Vogt N, Klapper W. Variability in morphology and cell proliferation in sequential biopsies of mantle cell lymphoma at diagnosis and relapse: clinical correlation and insights into disease progression. Histopathology 2013;62:334–42.
- Geisler CH, Kolstad A, Laurell A, et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 2008;112:2687–93.
- Howard OM, Gribben JG, Neuberg DS, et al. Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: molecular complete responses are not predictive of progression-free survival. J Clin Oncol 2002;20:1288–94.
- Griffiths R, Mikhael J, Gleeson M, et al. Addition of rituximab to chemotherapy alone as first-line therapy improves overall survival in elderly patients with mantle cell lymphoma. Blood 2011;118:4808–16.
- Lenz G, Dreyling M, Hoster E, et al. immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
- Romaguera JE, Fayad L, Rodriguez MA, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol 2005;23:7013–23.
- Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
- Robak T, Huang H, Jin J, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med 2015;372:944–53.
- Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
- Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
- Houot R, Le Gouill S, Ojeda Uribe M, et al. Combination of rituximab, bortezomib, doxorubicin, dexamethasone and chlorambucil (RiPAD+C) as first-line therapy for elderly mantle cell lymphoma patients: results of a phase II trial from the GOELAMS. Ann Oncol 2012;23:1555–61.
- Ruan J, Martin P, Furman RR, et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol 2011;29:690–7.
- Chang JE, Li H, Smith MR, et al. Phase 2 study of VcR-CVAD with maintenance rituximab for untreated mantle cell lymphoma: an Eastern Cooperative Oncology Group study (E1405). Blood 2014; 123:1665–73.
- Romaguera JE, Fayad LE, Feng L, et al. Ten-year follow-up after intense chemoimmunotherapy with Rituximab-HyperCVAD alternating with Rituximab-high dose methotrexate/cytarabine (R-MA) and without stem cell transplantation in patients with untreated aggressive mantle cell lymphoma. Br J Haematol 2010;150:200–8.
- Bernstein SH, Epner E, Unger JM, et al. A phase II multicenter trial of hyperCVAD MTX/Ara-C and rituximab in patients with previously untreated mantle cell lymphoma; SWOG 0213. Ann Oncol 2013;24:1587–93.
- LaCasce AS, Vandergrift JL, Rodriguez MA, et al. Comparative outcome of initial therapy for younger patients with mantle cell lymphoma: an analysis from the NCCN NHL Database. Blood 2012;119:2093–9.
- Geisler CH, Kolstad A, Laurell A, et al. Nordic MCL2 trial update: six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma followed by BEAM or BEAC + autologous stem-cell support: still very long survival but late relapses do occur. Br J Haematol 2012;158:355–62.
- Damon LE, Johnson JL, Niedzwiecki D, et al. Immunochemotherapy and autologous stem-cell transplantation for untreated patients with mantle-cell lymphoma: CALGB 59909. J Clin Oncol 2009;27:6101–8.
- van ‘t Veer MB, de Jong D, MacKenzie M, et al. High-dose Ara-C and beam with autograft rescue in R-CHOP responsive mantle cell lymphoma patients. Br J Haematol 2009;144:524–30.
- Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
- Hermine O, Hoster E, Walewski J, et al. Alternating courses of 3x CHOP and 3x DHAP plus rituximab followed by a high dose ARA-C containing myeloablative regimen and autologous stem cell transplantation (ASCT) increases overall survival when compared to 6 courses of CHOP plus rituximab followed by myeloablative radiochemotherapy and ASCT in mantle cell lymphoma: final analysis of the MCL Younger Trial of the European Mantle Cell Lymphoma Network (MCL net). In: American Society of Hematology Proceedings. December 8–11, 2012; Atlanta, GA. Abstract 151.
- Rummel MJ, Al-Batran SE, Kim SZ, et al. Bendamustine plus rituximab is effective and has a favorable toxicity profile in the treatment of mantle cell and low-grade non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:3383–9.
- Pham LV, Tamayo AT, Yoshimura LC, et al. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol 2003;171:88–95.
- Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
- Friedberg JW, Vose JM, Kelly JL, et al. The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood 2011;117:2807–12.
- Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 2004;4:314–22.
- Qian Z, Zhang L, Cai Z, et al. Lenalidomide synergizes with dexamethasone to induce growth arrest and apoptosis of mantle cell lymphoma cells in vitro and in vivo. Leuk Res 2011;35:380–6.
- Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
- Wang M, Fayad L, Wagner-Bartak N, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol 2012;13:716–23.
- Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol 2012;31: 119–32.
- Rinaldi A, Kwee I, Taborelli M, et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol 2006;132:303–16.
- Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013; 31:88–94.
- Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
- Rudelius M, Pittaluga S, Nishizuka S, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood 2006;108: 1668–76.
- Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3Kdelta inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014;123:3398–405.
- Davids MS, Seymour JF, Gerecitano JF, et al. Updated results of a phase I first in human study of the BCL-2inhibitor ABT-199 in patients with relapsed/refractory NHL. J Clin Oncol 31, 2013 (suppl; abstr 8520).
- Ansell SM, Tang H, Kurtin PJ, et al. Temsirolimus and rituximab in patients with relapsed or refractory mantle cell lymphoma: a phase 2 study. Lancet Oncol 2011;12:361–8.
- Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 2005;23:5347–56.
- Wang M, Oki Y, Pro B, et al. Phase II study of yttrium-90-ibritumomab tiuxetan in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009;27:5213–8.
- Kolstad A, Laurell A, Jerkeman M, et al. Nordic MCL3 study: 90Y-ibritumomab-tiuxetan added to BEAM/C in non-CR patients before transplant in mantle cell lymphoma. Blood 2014;123:2953–9.
- Fenske TS, Zhang MJ, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
- Dreyling M, Lenz G, Hoster E, et al. Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood 2005;105:2677–84.
- Goy A, Younes A, McLaughlin P, et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:667–75.
- Visco C, Finotto S, Zambello R, et al. Combination of rituximab, bendamustine, and cytarabine for patients with mantle-cell non-Hodgkin lymphoma ineligible for intensive regimens or autologous transplantation. J Clin Oncol 2013;10;31:1442–9.
- Gressin R, Callanan M, Daguindau N, et al. The Ribvd regimen (Rituximab IV, Bendamustine IV, Velcade SC, Dexamethasone IV) offers a high complete response rate In elderly patients with untreated mantle cell lymphoma. Preliminary results of the Lysa trial “Lymphome Du Manteau 2010 SA.” Blood 2013;122:370.
- Krishnan A, Nademanee A, Fung HC, et al. Phase II trial of a transplantation regimen of yttrium-90 ibritumomab tiuxetan and high-dose chemotherapy in patients with non-Hodgkin’s lymphoma. J Clin Oncol 2008;26:90–5.
- Nademanee A, Forman S, Molina A, et al. A phase 1/2 trial of high-dose yttrium-90-ibritumomab tiuxetan in combination with high-dose etoposide and cyclophosphamide followed by autologous stem cell transplantation in patients with poor-risk or relapsed non-Hodgkinlymphoma. Blood 2005;106:2896–902.
- Shimoni A, Avivi I, Rowe JM, et al. A randomized study comparing yttrium-90 ibritumomab tiuxetan (Zevalin) and high-dose BEAM chemotherapy versus BEAM alone as the conditioning regimen before autologous stem cell transplantation in patients with aggressive lymphoma. Cancer 2012;118:4706–14.
- Arranz R, García-Noblejas A, Grande C, et al. First-line treatment with rituximab-hyperCVAD alternating with rituximab-methotrexate-cytarabine and followed by consolidation with 90Y-ibritumomab-tiuxetan in patients with mantle cell lymphoma. Results of a multicenter, phase 2 pilot trial from the GELTAMO group. Haematologica 2013;98:1563-70.
- Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998;16:2780–95.
- Zhou Y, Wang H, Fang W, et al. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer 2008;113:791–8.
- Geisler CH. Front-line treatment of mantle cell lymphoma. Haematologica 2010;95:1241–3.
- Fernandez V, Salamero O, Espinet B, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res 2010;70:1408–18.
- Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol 2009;27:1209–13.
- Bertoni F, Ponzoni M. The cellular origin of mantle cell lymphoma. Int J Biochem Cell Biol 2007;39:1747–53.
- de Boer CJ, van Krieken JH, Kluin-Nelemans HC, et al. Cyclin D1 messenger RNA overexpression as a marker for mantle cell lymphoma. Oncogene 1995;10:1833–40.
- Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest 2012;122:3416–23.
- Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 2003;3:185–97.
- Determann O, Hoster E, Ott G, et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood 2008;111:2385–7.
- Vegliante MC, Palomero J, Perez-Galan P, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood 2013;121:2175–85.
- Bea S, Valdes-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A 2013;110:18250–5.
- Zelenetz AD, Abramson JS, Advani RH, et al. Non- Hodgkin’s lymphomas. J Natl Compr Canc Netw 2011;9: 484–560.
- Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68.
- Zelenetz AD, Abramson JS, Advani RH, et al. NCCN Clinical Practice Guidelines in Oncology: non-Hodgkin’s lymphomas. J Natl Compr Canc Netw 2010;8:288–334.
- Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood 2008;111:558–65.
- Geisler CH, Kolstad A, Laurell A, et al. The Mantle Cell Lymphoma International Prognostic Index (MIPI) is superior to the International Prognostic Index (IPI) in predicting survival following intensive first-line immunochemotherapy and autologous stem cell transplantation (ASCT). Blood 2010;115:1530–3.
- Tiemann M, Schrader C, Klapper W, et al. Histopathology, cell proliferation indices and clinical outcome in 304 patients with mantle cell lymphoma (MCL): a clinicopathological study from the European MCL Network. Br J Haematol 2005;131:29–38.
- Raty R, Franssila K, Joensuu H, et al. Ki-67 expression level, histological subtype, and the International Prognostic Index as outcome predictors in mantle cell lymphoma. Eur J Haematol 2002;69:11–20.
- Vogt N, Klapper W. Variability in morphology and cell proliferation in sequential biopsies of mantle cell lymphoma at diagnosis and relapse: clinical correlation and insights into disease progression. Histopathology 2013;62:334–42.
- Geisler CH, Kolstad A, Laurell A, et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 2008;112:2687–93.
- Howard OM, Gribben JG, Neuberg DS, et al. Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: molecular complete responses are not predictive of progression-free survival. J Clin Oncol 2002;20:1288–94.
- Griffiths R, Mikhael J, Gleeson M, et al. Addition of rituximab to chemotherapy alone as first-line therapy improves overall survival in elderly patients with mantle cell lymphoma. Blood 2011;118:4808–16.
- Lenz G, Dreyling M, Hoster E, et al. immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol 2005;23:1984–92.
- Romaguera JE, Fayad L, Rodriguez MA, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol 2005;23:7013–23.
- Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med 2012;367:520–31.
- Robak T, Huang H, Jin J, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med 2015;372:944–53.
- Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 2014;123:2944–52.
- Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013;381:1203–10.
- Houot R, Le Gouill S, Ojeda Uribe M, et al. Combination of rituximab, bortezomib, doxorubicin, dexamethasone and chlorambucil (RiPAD+C) as first-line therapy for elderly mantle cell lymphoma patients: results of a phase II trial from the GOELAMS. Ann Oncol 2012;23:1555–61.
- Ruan J, Martin P, Furman RR, et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol 2011;29:690–7.
- Chang JE, Li H, Smith MR, et al. Phase 2 study of VcR-CVAD with maintenance rituximab for untreated mantle cell lymphoma: an Eastern Cooperative Oncology Group study (E1405). Blood 2014; 123:1665–73.
- Romaguera JE, Fayad LE, Feng L, et al. Ten-year follow-up after intense chemoimmunotherapy with Rituximab-HyperCVAD alternating with Rituximab-high dose methotrexate/cytarabine (R-MA) and without stem cell transplantation in patients with untreated aggressive mantle cell lymphoma. Br J Haematol 2010;150:200–8.
- Bernstein SH, Epner E, Unger JM, et al. A phase II multicenter trial of hyperCVAD MTX/Ara-C and rituximab in patients with previously untreated mantle cell lymphoma; SWOG 0213. Ann Oncol 2013;24:1587–93.
- LaCasce AS, Vandergrift JL, Rodriguez MA, et al. Comparative outcome of initial therapy for younger patients with mantle cell lymphoma: an analysis from the NCCN NHL Database. Blood 2012;119:2093–9.
- Geisler CH, Kolstad A, Laurell A, et al. Nordic MCL2 trial update: six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma followed by BEAM or BEAC + autologous stem-cell support: still very long survival but late relapses do occur. Br J Haematol 2012;158:355–62.
- Damon LE, Johnson JL, Niedzwiecki D, et al. Immunochemotherapy and autologous stem-cell transplantation for untreated patients with mantle-cell lymphoma: CALGB 59909. J Clin Oncol 2009;27:6101–8.
- van ‘t Veer MB, de Jong D, MacKenzie M, et al. High-dose Ara-C and beam with autograft rescue in R-CHOP responsive mantle cell lymphoma patients. Br J Haematol 2009;144:524–30.
- Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013;121:48–53.
- Hermine O, Hoster E, Walewski J, et al. Alternating courses of 3x CHOP and 3x DHAP plus rituximab followed by a high dose ARA-C containing myeloablative regimen and autologous stem cell transplantation (ASCT) increases overall survival when compared to 6 courses of CHOP plus rituximab followed by myeloablative radiochemotherapy and ASCT in mantle cell lymphoma: final analysis of the MCL Younger Trial of the European Mantle Cell Lymphoma Network (MCL net). In: American Society of Hematology Proceedings. December 8–11, 2012; Atlanta, GA. Abstract 151.
- Rummel MJ, Al-Batran SE, Kim SZ, et al. Bendamustine plus rituximab is effective and has a favorable toxicity profile in the treatment of mantle cell and low-grade non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:3383–9.
- Pham LV, Tamayo AT, Yoshimura LC, et al. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol 2003;171:88–95.
- Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006;24:4867–74.
- Friedberg JW, Vose JM, Kelly JL, et al. The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood 2011;117:2807–12.
- Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 2004;4:314–22.
- Qian Z, Zhang L, Cai Z, et al. Lenalidomide synergizes with dexamethasone to induce growth arrest and apoptosis of mantle cell lymphoma cells in vitro and in vivo. Leuk Res 2011;35:380–6.
- Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013;31:3688–95.
- Wang M, Fayad L, Wagner-Bartak N, et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol 2012;13:716–23.
- Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol 2012;31: 119–32.
- Rinaldi A, Kwee I, Taborelli M, et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol 2006;132:303–16.
- Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013; 31:88–94.
- Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013;369:507–16.
- Rudelius M, Pittaluga S, Nishizuka S, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood 2006;108: 1668–76.
- Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3Kdelta inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014;123:3398–405.
- Davids MS, Seymour JF, Gerecitano JF, et al. Updated results of a phase I first in human study of the BCL-2inhibitor ABT-199 in patients with relapsed/refractory NHL. J Clin Oncol 31, 2013 (suppl; abstr 8520).
- Ansell SM, Tang H, Kurtin PJ, et al. Temsirolimus and rituximab in patients with relapsed or refractory mantle cell lymphoma: a phase 2 study. Lancet Oncol 2011;12:361–8.
- Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 2005;23:5347–56.
- Wang M, Oki Y, Pro B, et al. Phase II study of yttrium-90-ibritumomab tiuxetan in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009;27:5213–8.
- Kolstad A, Laurell A, Jerkeman M, et al. Nordic MCL3 study: 90Y-ibritumomab-tiuxetan added to BEAM/C in non-CR patients before transplant in mantle cell lymphoma. Blood 2014;123:2953–9.
- Fenske TS, Zhang MJ, Carreras J, et al. Autologous or reduced-intensity conditioning allogeneic hematopoietic cell transplantation for chemotherapy-sensitive mantle-cell lymphoma: analysis of transplantation timing and modality. J Clin Oncol 2014;32:273–81.
- Dreyling M, Lenz G, Hoster E, et al. Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood 2005;105:2677–84.
- Goy A, Younes A, McLaughlin P, et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncol 2005;23:667–75.
- Visco C, Finotto S, Zambello R, et al. Combination of rituximab, bendamustine, and cytarabine for patients with mantle-cell non-Hodgkin lymphoma ineligible for intensive regimens or autologous transplantation. J Clin Oncol 2013;10;31:1442–9.
- Gressin R, Callanan M, Daguindau N, et al. The Ribvd regimen (Rituximab IV, Bendamustine IV, Velcade SC, Dexamethasone IV) offers a high complete response rate In elderly patients with untreated mantle cell lymphoma. Preliminary results of the Lysa trial “Lymphome Du Manteau 2010 SA.” Blood 2013;122:370.
- Krishnan A, Nademanee A, Fung HC, et al. Phase II trial of a transplantation regimen of yttrium-90 ibritumomab tiuxetan and high-dose chemotherapy in patients with non-Hodgkin’s lymphoma. J Clin Oncol 2008;26:90–5.
- Nademanee A, Forman S, Molina A, et al. A phase 1/2 trial of high-dose yttrium-90-ibritumomab tiuxetan in combination with high-dose etoposide and cyclophosphamide followed by autologous stem cell transplantation in patients with poor-risk or relapsed non-Hodgkinlymphoma. Blood 2005;106:2896–902.
- Shimoni A, Avivi I, Rowe JM, et al. A randomized study comparing yttrium-90 ibritumomab tiuxetan (Zevalin) and high-dose BEAM chemotherapy versus BEAM alone as the conditioning regimen before autologous stem cell transplantation in patients with aggressive lymphoma. Cancer 2012;118:4706–14.
- Arranz R, García-Noblejas A, Grande C, et al. First-line treatment with rituximab-hyperCVAD alternating with rituximab-methotrexate-cytarabine and followed by consolidation with 90Y-ibritumomab-tiuxetan in patients with mantle cell lymphoma. Results of a multicenter, phase 2 pilot trial from the GELTAMO group. Haematologica 2013;98:1563-70.
Current Therapeutic Approaches to Renal Cell Carcinoma
INTRODUCTION
Renal cell carcinoma (RCC) is the most common malignancy arising in the kidney, comprising 90% of all renal tumors.1 Approximately 55,000 new RCC cases are diagnosed each year.2 Patients with RCC are often asymptomatic, and most cases are discovered as incidental findings on abdominal imaging performed during evaluation of nonrenal complaints. Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20%.2 Advanced RCC is resistant to conventional chemotherapy and radiotherapy, and outcomes for patients with metastatic or unresectable RCC remain poor. However, the recent development of new therapeutic modalities that target tumor molecular pathways has expanded the treatment options for these patients and changed the management of RCC.
EPIDEMIOLOGY AND CLASSIFICATION
Median age at diagnosis in the United States is 64 years. Men have a higher incidence of RCC than women, with the highest incidence seen in American Indian and Alaska Native men (30.1 per 100,000 population). Genetic syndromes account for 2% to 4% of all RCCs.2 Risk factors for RCC include smoking, hypertension, obesity, and acquired cystic kidney disease that is associated with end-stage renal failure.3 Longer duration of tobacco use is associated with a more aggressive course.
The 2004 World Health Organization (WHO) classification of renal tumors summarizes the previous classification systems (including the Heidelberg and Mainz classification systems) to describe different categories of RCC based on histologic and molecular genetics characteristics.2 Using the WHO classification criteria, RCC comprises 90% of all renal tumors, with clear cell being the most common type (80%).2 Other types of renal tumors include papillary, chromophobe, oncocytoma, and collecting-duct or Bellini duct tumors. Approximately 3% to 5% of tumors are unclassified. Oncocytomas are generally considered benign, and chromophobe tumors typically have an indolent course and rarely metastasize. Sarcomatoid differentiation can be seen in any histologic type and is associated with a worse prognosis. While different types of tumors may be seen in the kidney (such as transitional cell or lymphomas), the focus of this review is the primary malignancies of the renal parenchyma.
FAMILIAL SYNDROMES
Several genetic syndromes have been identified by studying families with inherited RCC. Among these, von Hippel-Lindau (VHL) gene mutation is the most commonly found inherited genetic defect. Table 1 summarizes the incidence of gene mutations and the corresponding histologic appearance of the most common sporadic and hereditary RCCs.4
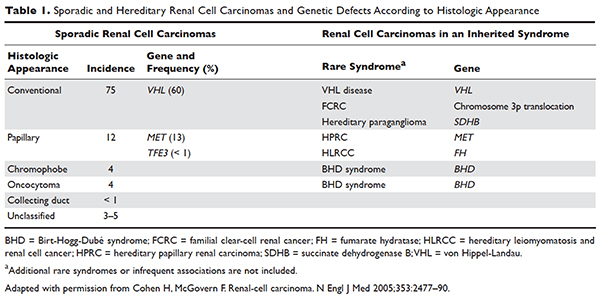
VHL disease is an autosomal dominant familial syndrome. Patients with this mutation are at higher risk for developing RCC (clear cell histology), retinal angiomas, pheochromocytomas, as well as hemangioblastomas of the central nervous system (CNS).4 Of all the genetic mutations seen in RCC, the somatic mutation in the VHL tumor-suppressor gene is by far the most common.5 VHL targets hypoxia–inducible factor-1 alpha (HIF-α) for ubiquitination and subsequent degradation, which has been shown to suppress the growth of clear-cell RCC in mouse models.6–8 HIF expression under hypoxic conditions leads to activation of a number of genes important in blood vessel development, cell proliferation, and glucose metabolism, including vascular endothelial growth factor (VEGF), erythropoietin, platelet-derived growth factor beta (PDGF-β), transforming growth factor alpha (TGF-α), and glucose transporter-1 (GLUT-1). Mutation in the VHL gene prevents degradation of the HIF-α protein, thereby leading to increased expression of these downstream proteins, including MET and Axl. The upregulation of these angiogenic factors is thought to be the underlying process for increased vascularity of CNS hemangioblastomas and clear-cell renal tumors in VHL disease.4–8
Other less common genetic syndromes seen in hereditary RCC include hereditary papillary RCC, hereditary leiomyomatosis, and Birt-Hogg-Dubé (BHD) syndrome.9 In hereditary papillary RCC, the MET gene is mutated. BHD syndrome is a rare, autosomal dominant syndrome characterized by hair follicle hamartomas of the face and neck. About 15% of patients have multiple renal tumors, the majority of which are of the chromophobe or mixed chromophobe-oncocytoma histology. The BHD gene encodes the protein folliculin, which is thought to be a tumor-suppressor gene.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 74-year-old man who works as an airplane mechanic repairman presents to the emergency department with sudden worsening of chronic right upper arm and shoulder pain after lifting a jug of orange juice. He does not have a significant past medical history and initially thought that his pain was due to a work-related injury. Upon initial evaluation in the emergency department he is found to have a fracture of his right humerus. Given that the fracture appears to be pathologic, further work-up is recommended.
• What are common clinical presentations of RCC?
Most patients are asymptomatic until the disease becomes advanced. The classic triad of flank pain, hematuria, and palpable abdominal mass is seen in approximately 10% of patients with RCC, partly because of earlier detection of renal masses by imaging performed for other purposes.10 Less frequently, patients present with signs or symptoms of metastatic disease such as bone pain or fracture (as seen in the case patient), painful adenopathy, and pulmonary symptoms related to mediastinal masses. Fever, weight loss, anemia, and/or varicocele often occur in young patients (≤ 46 years) and may indicate the presence of a hereditary form of the disease. Patients may present with paraneoplastic syndromes seen as abnormalities on routine blood work. These can include polycythemia or elevated liver function tests (LFTs) without the presence of liver metastases (known as Stauffer syndrome), which can be seen in localized renal tumors. Nearly half (45%) of patients present with localized disease, 25% present with locally advanced disease, and 30% present with metastatic disease.11 Bone is the second most common site of distant metastatic spread (following lung) in patients with advanced RCC.
• What is the approach to initial evaluation for a patient with suspected RCC?
Initial evaluation consists of a physical exam, laboratory tests including complete blood count (CBC) and comprehensive metabolic panel (calcium, serum creatinine, LFTs, lactate dehydrogenase [LDH], and urinalysis), and imaging. Imaging studies include computed tomography (CT) scan with contrast of the abdomen and pelvis or magnetic resonance imaging (MRI) of the abdomen and chest imaging. A chest radiograph may be obtained, although a chest CT is more sensitive for the presence of pulmonary metastases. MRI can be used in patients with renal dysfunction to evaluate the renal vein and inferior vena cava (IVC) for thrombus or to determine the presence of local invasion.12 Although bone and brain are common sites for metastases, routine imaging is not indicated unless the patient is symptomatic. The value of positron emission tomography in RCC remains undetermined at this time.
Staging is done according to the American Joint Committee on Cancer (AJCC) staging classification for RCC; the Figure summarizes the staging and 5-year survival data based on this classification scheme.4,13
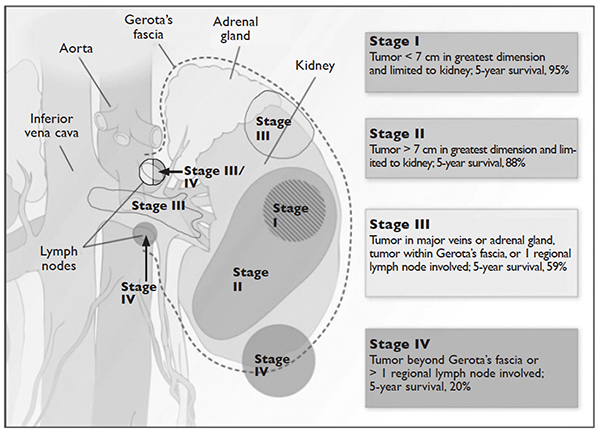
J Med 2005;353:2477–90.)
LIMITED-STAGE DISEASE
• What are the therapeutic options for limited-stage disease?
For patients with nondistant metastases, or limited-stage disease, surgical intervention with curative intent is considered. Convention suggests considering definitive surgery for patients with stage I and II disease, select patients with stage III disease with pathologically enlarged retroperitoneal lymph nodes, patients with IVC and/or cardiac atrium involvement of tumor thrombus, and patients with direct extension of the renal tumor into the ipsilateral adrenal gland if there is no evidence of distant disease. While there may be a role for aggressive surgical intervention in patients with distant metastatic disease, this topic will not be covered in this review.
SURGICAL INTERVENTION
Once patients are determined to be appropriate candidates for surgical removal of a renal tumor, the urologist will perform either a radical nephrectomy or a nephron-sparing nephrectomy, also called a partial nephrectomy. The urologist will evaluate the patient based on his or her body habitus, the location of the tumor, whether multiple tumors in one kidney or bilateral tumors are present, whether the patient has a solitary kidney or otherwise impaired kidney function, and whether the patient has a history of a hereditary syndrome involving kidney cancer as this affects the risk of future kidney tumors.
A radical nephrectomy is surgically preferred in the presence of the following factors: tumor larger than 7 cm in diameter, a more centrally located tumor, suspicion of lymph node involvement, tumor involvement with renal vein or IVC, and/or direct extension of the tumor into the ipsilateral adrenal gland. Nephrectomy involves ligation of the vascular supply (renal artery and vein) followed by removal of the kidney and surrounding Gerota’s fascia. The ipsilateral adrenal gland is removed if there is a high-risk for or presence of invasion of the adrenal gland. Removal of the adrenal gland is not standard since the literature demonstrates there is less than a 10% chance of solitary, ipsilateral adrenal gland involvement of tumor at the time of nephrectomy in the absence of high-risk features, and a recent systematic review suggests that the chance may be as low as 1.8%.14 Preoperative factors that correlated with adrenal involvement included upper pole kidney location, renal vein thrombosis, higher T stage (T3a and greater), multifocal tumors, and evidence for distant metastases or lymph node involvement. Lymphadenectomy previously had been included in radical nephrectomy but now is performed selectively. Radical nephrectomy may be performed as
either an open or laparoscopic procedure, the latter of which may be performed robotically.15 Oncologic outcomes appear to be comparable between the 2 approaches, with equivalent 5-year cancer-specific survival (91% with laparoscopic versus 93% with open approach) and recurrence-free survival (91% with laparoscopic versus 93% with open approach).16 The approach ultimately is selected based on provider- and patient-specific input, though in all cases the goal is to remove the specimen intact.16,17
Conversely, a nephron-sparing approach is preferred for tumors less than 7 cm in diameter, for patients with a solitary kidney or impaired renal function, for patients with multiple small ipsilateral tumors or with bilateral tumors, or for radical nephrectomy candidates with comorbidities for whom a limited intervention is deemed to be a lower-risk procedure. A nephron-sparing procedure may also be performed open or laparoscopically. In nephron-sparing procedures, the tumor is removed along with a small margin of normal parenchyma.15
In summary, the goal of surgical intervention is curative intent with removal of the tumor while maintaining as much residual renal function as possible to limit long-term morbidity of chronic kidney disease and associated cardiovascular events.18 Oncologic outcomes for radical nephrectomy and partial nephrectomy are similar. In one study, overall survival was slightly lower in the partial nephrectomy cohort, but only a small number of the deaths were due to RCC.19
ADJUVANT THERAPY
Adjuvant systemic therapy currently has no role following nephrectomy for RCC because no systemic therapy has been able to reduce the likelihood of relapse. Randomized trials of cytokine therapy (eg, interferon, interleukin 2) or tyrosine kinase inhibitors (TKIs; eg, sorafenib, sunitinib) with observation alone in patients with locally advanced completely resected RCC have shown no delay in time to relapse or improvement of survival with adjuvant therapy.20 Similarly, adjuvant radiation therapy has not shown benefit even in patients with nodal involvement or incomplete resection.21 Therefore, observation remains the standard of care after nephrectomy.
RENAL TUMOR ABLATION
For patients who are deemed not to be surgical candidates due to age, comorbidities, or patient preference and who have tumors less than 4 cm in size (stage I tumors), ablative techniques may be considered. The 2 most well-studied and effective techniques at present are cryoablation and radiofrequency ablation (RFA). Microwave ablation may be an option in some facilities, but the data in RCC are limited. An emerging ablative technique under investigation is irreversible electroporation. At present, the long-term efficacy of all ablative techniques is unknown.
Patient selection is undertaken by urologists and interventional radiologists who evaluate the patient with ultrasound, CT, and/or MRI to determine the location and size of the tumor and the presence or absence of metastatic disease. A pretreatment biopsy is recommended to document the histology of the lesion to confirm a malignancy and to guide future treatment for recurrent or metastatic disease. Contraindications to the procedure include the presence of metastatic disease, a life expectancy of less than 1 year, general medical instability, or uncorrectable coagulopathy due to increased risk of bleeding complications. Tumors in close proximity to the renal hilum or collecting system are a contraindication to the procedure because of the risk for hemorrhage or damage to the collecting system. The location of the tumor in relation to the vasculature is also important to maximize efficacy because the vasculature acts as a “heat sink,” causing dissipation of the thermal energy. Occasionally, stenting of the proximal ureter due to upper tumor location is necessary to prevent thermal injury that could lead to urine leaks.
Selection of the modality to be used primarily depends on operator comfort, which translates to good patient outcomes, such as better cancer control and fewer complications. Cryoablation and RFA have both demonstrated good clinical efficacy and cancer control of 89% and 90%, respectively, with comparable complication rates.22 There have been no studies performed directly comparing the modalities.
Cryoablation
Cryoablation is performed through the insertion of a probe into the tumor, which may be done through a surgical or percutaneous approach. Once the probe is in place, a high- pressure gas (argon, nitrogen) is passed through the probe and upon entering a low pressure region the gas cools. The gas is able to cool to temperatures as low as –185°C. The tissue is then rewarmed through the use of helium, which conversely warms when entering a low pressure area. The process of freezing followed by rewarming subsequently causes cell death/tissue destruction through direct cell injury from cellular dehydration and vascular injury. Clinically, 2 freeze-thaw cycles are used to treat a tumor.23,24
RFA
Radiofrequency ablation, or RFA, targets tumors via an electrode placed within the mass that produces intense frictional heat from medium-frequency alternating current (approximately 500 kHz) produced by a connected generator that is grounded on the patient. The thermal energy created causes coagulative necrosis. Due to the reliance on heat for tumor destruction, central lesions are less amenable to this approach because of the “heat sink” effect from the hilum.24
Microwave Ablation
Microwave ablation, like RFA, relies on the generation of frictional heat to cause cell death by coagulative necrosis. In this case, the friction is created through the activation of water molecules; because of the different thermal kinetics involved with microwave ablation, the “heat sink” effect is minimized when treatment is employed near large vessels, in comparison to RFA.24 The data on this mechanism of ablation are still maturing, with varied outcomes thus far. One study demonstrated outcomes comparable to RFA and cryoablation, with cancer-specific survival of 97.8% at 3 years.25 However, a study by Castle and colleagues26 demonstrated higher recurrence rates. The overarching impediment to widespread adoption of microwave ablation is inconclusive data gleaned from studies with small numbers of patients with limited follow up. The role of this modality will need to be revisited.
Irreversible Electroporation
Irreversible electroporation (IRE) is under investigation. IRE is a non-thermal ablative technique that employs rapid electrical pulses to create pores in cell membranes, leading to cell death. The postulated benefits of IRE include the lack of an effect from “heat sinks” and less collateral damage to the surrounding tissues, when compared with the thermal modalities. In a human phase 1 study of patients undergoing IRE prior to immediate surgical resection, the procedure appeared feasible and safe.27 Significant concerns for this method of ablation possibly inducing cardiac arrhythmias, and the resultant need for sedation with neuromuscular blockade and associated electrocardiography monitoring, may impede its implementation in nonresearch settings.24
ACTIVE SURVEILLANCE
Due to the more frequent use of imaging for various indications, there has been an increase in the discovery of small renal masses (SRM); 85% of RCC that present in an asymptomatic or incidental manner are tumors under 4 cm in diameter.28,29 The role of active surveillance is evolving, but is primarily suggested for patients who are not candidates for more aggressive intervention based on comorbidities. A recent prospective, nonrandomized analysis of data from the Delayed Intervention and Surveillance for Small Renal Masses (DISSRM) registry evaluated outcomes for patients with SRM looking at primary intervention compared with active surveillance.30 The primary intervention selected was at the discretion of the provider; treatments included partial nephrectomy, RFA, and cryoablation, and active surveillance patients were followed with imaging every 6 months. Progression of SRM, with recommendation for delayed intervention, was defined as a growth rate of mass greater than 0.5 cm/year, size greater than 4 cm, or hematuria. Thirty-six of 158 patients on active surveillance met criteria for progression; 21 underwent delayed intervention. Of note, even the patients who progressed but did not undergo delayed intervention did not develop metastatic disease during the follow-up interval. With a median follow-up of 2 years, cancer-specific survival was noted to be 99% and 100% at 5 years for primary intervention and active surveillance, respectively. Overall survival at 2 years for primary intervention was 98% and 96% for active surveillance; at 5 years, the survival rates were 92% and 75% (P = 0.06). Of note, 2 patients in the primary intervention arm died of RCC, while none in the active surveillance arm died. As would be expected, active surveillance patients were older, had a worse performance status, and had more comorbidities. Interestingly, 40% of patients enrolled selected active surveillance as their preferred management for SRM. The DISSRM results were consistent with data from the Renal Cell Consortium of Canada and other retrospective reviews.31–33
• What is the approach to follow-up after treatment of localized RCC?
After a patient undergoes treatment for a localized RCC, the goal is to optimize oncologic outcomes, monitor for treatment sequelae, such as renal failure, and focus on survivorship. At this time, there is no consensus in the literature or across published national and international guidelines with regards to the appropriate schedule for surveillance to achieve these goals. In principle, the greatest risk for recurrence occurs within the first 3 years, so many guidelines focus on this timeframe. Likewise, the route of spread tends to be hematogenous, so patients present with pulmonary, bone, and brain metastases, in addition to local recurrence within the renal bed. Symptomatic recurrences often are seen
with bone and brain metastases, and thus bone scans and brain imaging are not listed as part of routine surveillance protocols in asymptomatic patients. Although there is inconclusive evidence that surveillance protocols improve outcomes in RCC, many professional associations have outlined recommendations based on expert opinion.34 The American Urological Association released guidelines in 2013 and the National Comprehensive Cancer Network (NCCN) released their most recent set of guidelines in 2016.21,35 These guidelines use TNM staging to risk-stratify patients and recommend follow-up.
METASTATIC DISEASE
CASE CONTINUED
CT scan with contrast of the chest, abdomen, and pelvis as well as bone scan are done. CT of the abdomen and pelvis demonstrates a 7.8-cm left renal mass arising from the lower pole of the left kidney. Paraesophageal lymphadenopathy and mesenteric nodules are also noted. CT of the chest demonstrates bilateral pulmonary emboli. Bone scan is significant for increased activity related to the pathological fracture involving the right humerus. The patient undergoes surgery to stabilize the pathologic fracture of his humerus. He is diagnosed with metastatic RCC (clear cell histology) and undergoes palliative debulking nephrectomy.
• How is prognosis defined for metastatic RCC?
PROGNOSTIC MODELS
Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates for stage T1 and T2 disease approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20%.13 Approximately 30% of patients have metastatic disease at diagnosis, and about one-third of patients who have undergone treatment for localized disease experience relapse.36,37 Common sites of metastases include lung, lymph nodes, bone, liver, adrenal gland, and brain.
Prognostic scoring systems have been developed to define risk groups and assist with determining appropriate therapy in the metastatic setting. The most widely used validated prognostic factor model is that from the Memorial Sloan-Kettering Cancer Center (MSKCC), which was developed using a multivariate analysis derived from data of patients enrolled in clinical trials and treated with interferon alfa.38 The factors included in the MSKCC model are Karnofsky performance status less than 80, time from diagnosis to treatment with interferon alfa less than 12 months, hemoglobin level less than lower limit of laboratory’s reference range, LDH level greater than 1.5 times the upper limit of laboratory’s reference range, and corrected serum calcium level greater than 10 mg/dL. Risk groups are categorized as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors).39 Median survival for favorable-, intermediate-, and poor-risk patients was 20, 10, and 4 months, respectively.40
Another prognostic model, the International Metastatic RCC Database Consortium, or Heng, model was developed to evaluate prognosis in patients treated with VEGF-targeted therapy.41 This model was developed from a retrospective study of patients treated with sunitinib, sorafenib, and bevacizumab plus interferon alfa or prior immunotherapy. Prognostic factors in this model include 4 of the 5 MSKCC risk factors (hemoglobin level, corrected serum calcium level, Karnofsky performance status, and time to initial diagnosis). Additionally, this model includes both absolute neutrophil and platelet counts greater than the upper limit of normal. Risk groups are identified as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors). Median survival for favorable-, intermediate-, and poor-risk patients was not reached, 27 months, and 8.8 months, respectively. The University of California, Los Angeles scoring algorithm to predict survival after nephrectomy and immunotherapy (SANI) in patients with metastatic RCC is another prognostic model that can be used. This simplified scoring system incorporates lymph node status, constitutional symptoms, metastases location, histology, and thyroid stimulating hormone (TSH) level.42
The role of debulking or cytoreductive nephrectomy in treatment of metastatic RCC is well established. Large randomized studies have demonstrated a statistically significant median survival benefit for patients undergoing nephrectomy plus interferon alfa therapy compared with patients treated with interferon alfa alone (13.6 months versus 7.8 months, respectively).43 The role of cytoreductive nephrectomy in combination with antiangiogenic agents is less clear. While a retrospective study investigating outcomes of patients with metastatic RCC receiving anti-VEGF agents showed a prolonged survival with nephrectomy, results of large randomized trials are not yet available.44,45 Patients with lung-only metastases, good prognostic features, and a good performance status are historically the most likely to benefit from cytoreductive surgery.
CASE CONTINUED
Based on the MSKCC prognostic factor model, the patient is considered to be in the intermediate-risk group (Karnofsky performance status of 80, calcium 9.5 mg/dL, LDH 204 U/L, hemoglobin 13.6 g/dL). He is started on treatment for his bilateral pulmonary emboli and recovers well from orthopedic surgery as well as palliative debulking nephrectomy.
• What is the appropriate first-line therapy in managing this patient’s metastatic disease?
Several approaches to systemic therapy for advanced RCC have been taken based on the histologic type of the tumor. Clear-cell is by far the predominant histologic type in RCC. Several options are available as first-line treatment for patients with metastatic clear-cell RCC (Table 2).46–54 These include biologic agents such as high-dose interleukin-2 (IL-2) immune therapy, as well as targeted therapies including TKIs and anti-VEGF antibodies. The mammalian target of rapamycin (mTOR) inhibitor temsirolimus is recommended as first-line therapy in patients with poor prognosis only. Second-line therapies for clear-cell RCC following antiangiogenic therapy include TKIs, mTOR inhibitors, nivolumab (PD-1 inhibitor), and the combination of the TKI lenvatinib and mTOR inhibitor everolimus.55 In addition, after initial cytokine therapy, TKIs, temsirolimus, and the anti-VEGF antibody bevacizumab are other treatment options available to patients. Best supportive care should always be provided along with initial and subsequent therapies. Clinical trials are also an appropriate choice as first-line or subsequent therapies. All of these therapies require periodic monitoring to prevent and quickly treat adverse effects. Table 3 lists recommended monitoring parameters for each of these agents.56
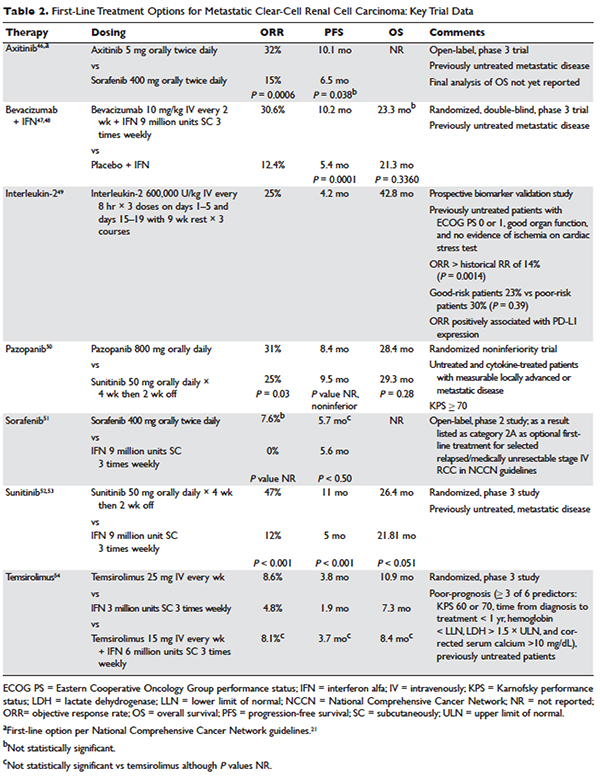
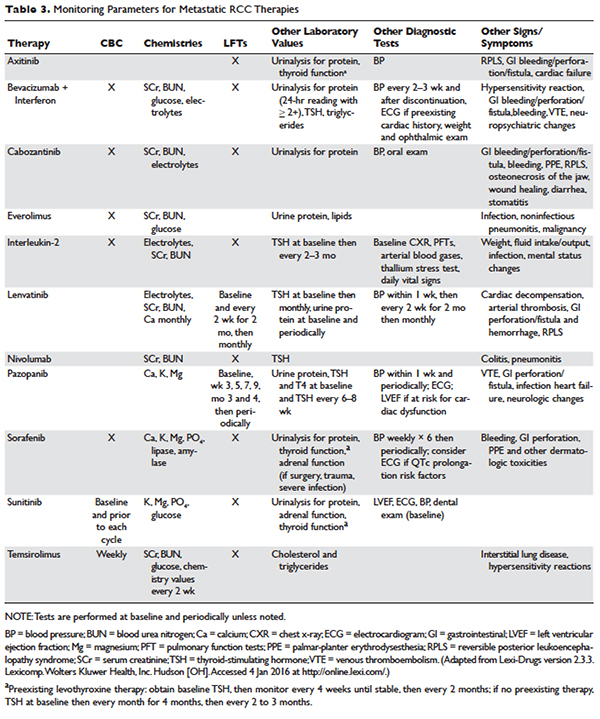
Based on several studies, TKIs seem to be less effective in patients with non–clear-cell type histology.57,58 In these patients, risk factors can guide therapy. In the ASPEN trial, where 108 patients were randomly assigned to everolimus or sunitinib, patients in the good- and intermediate-risk groups had longer overall and median progression-free survival (PFS) on sunitinib (8.3 months versus 5.3 months, respectively). However, those in the poor-risk group had a longer median overall survival with everolimus.59 Given that the role of targeted therapies in non–clear-cell RCCs is less well established, enrollment in clinical trials should be considered as a first-line treatment option.21
Sarcomatoid features can be observed in any of the histologic types of RCC, and RCC with these features has an aggressive course and a poor prognosis. Currently, there is no standard therapy for treatment of patients with metastatic or unresectable RCC with sarcomatoid features.60 Chemotherapeutic regimens used for soft tissue sarcomas, including a trial of ifosfamide and doxorubicin, did not show any objective response.61 A small trial of 10 patients treated with doxorubicin and gemcitabine resulted in complete response in 2 patients and partial response in 1 patient.62
Enrollment in a clinical trial remains a first-line treatment option for these patients. More recently, a phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid (39 patients) and/or poor-risk (33 patients) metastatic RCC showed overall response rates (ORR) of 26% and 24%, respectively. A higher clinical benefit rate (defined as ORR plus stable disease) was seen in patients with tumors containing more than 10% sarcomatoid histology, as compared with patients whose tumors contained less than 10% sarcomatoid histology. Neutropenia (n = 20), anemia (n = 10), and fatigue (n = 7) were the most common grade 3 toxicities seen in all the patients. Although this was a small study, the results showed a trend towards better efficacy of the combination therapy as compared with the single-agent regimen. Currently, another study is underway to further investigate this in a larger group of patients.63
BIOLOGICS
Cytokine therapy, including high-dose IL-2 and interferon alfa, had long been the only first-line treatment option for patients with metastatic or unresectable RCC. Studies of high-dose IL-2 have shown an ORR of 25% and durable response in up to 11% of patients with clear-cell histology.64 Toxicities were similar to those previously observed with high-dose IL-2 treatment; the most commonly observed grade 3 toxicities were hypotension and capillary leak syndrome. IL-2 requires strict monitoring (Table 3). It is important to note that retrospective studies evaluating the safety and efficacy of using IL-2 as second-line treatment in patients previously treated with TKIs demonstrated significant toxicity without achieving partial or complete response in any of the patients.65
Prior to the advent of TKIs in the treatment of RCC, interferon alfa was a first-line treatment option for those who could not receive high-dose IL-2. It has been shown to produce response rates of approximately 20%, with maximum response seen with a higher dose range of 5 to 20 million units daily in 1 study.66,67 However, with the introduction of TKIs, which produce a higher and more durable response, interferon alfa alone is no longer recommended as a treatment option.
VEGF MONOCLONAL ANTIBODIES
Bevacizumab is a recombinant humanized monoclonal antibody that binds and neutralizes VEGF-A. Given overexpression of VEGF in RCC, the role of bevacizumab both as a single agent and in combination with interferon alfa has been investigated. In a randomized phase 2 study involving patients with cytokine-refractory disease, bevacizumab produced a 10% response rate and PFS of 4.8 months compared to patients treated with placebo.68 In the AVOREN trial, the addition of bevacizumab (10 mg/kg intravenously [IV] every 2 weeks) to interferon alfa (9 million units subcutaneously [SC] 3 times weekly) was shown to significantly increase PFS compared with interferon alfa alone (10.2 months versus 5.4 months; P = 0.0001).47,48 Adverse effects of this combination therapy include fatigue and asthenia. Additionally, hypertension, proteinuria, and bleeding occurred.
TYROSINE KINASE INHIBITORS
TKIs have largely replaced IL-2 as first-line therapy for metastatic RCC. Axitinib, pazopanib, sorafenib, and sunitinib and can be used as first-line therapy. All of the TKIs can be used as subsequent therapy.
Sunitinib
Sunitinib is an orally administered TKI that inhibits VEGF receptor (VEGFR) types 1 and 2, PDGF receptors (PDGFR) α and β, stem cell factor receptor (c-Kit), and FLT-3 and RET kinases. Motzer and colleagues52,53 compared sunitinib 50 mg daily orally for 4 weeks with 2 weeks off to the then standard of care, interferon alfa 9 million units SC 3 times weekly. Sunitinib significantly increased the overall objective response rate (47% versus 12%; P < 0.001), PFS (11 versus 5 months; P < 0.001), and overall survival (26.4 versus 21.8 months; hazard ratio [HR], 0.821). The most common side effects are diarrhea, fatigue, nausea/vomiting, anorexia, hypertension, stomatitis, and hand-foot syndrome, occurring in more than 30% of patients. Often patients will require dose reductions or temporary discontinuations to tolerate therapy. Alternative dosing strategies (eg, 50 mg dose orally daily for 2 weeks alternating with 1-week free interval) have been attempted but not prospectively evaluated for efficacy.69–71
Pazopanib
Pazopanib is an oral multi-kinase inhibitor of VEGFR types 1 and 2, PDGFR, and c-KIT. Results of a phase 3 trial comparing pazopanib (800 mg orally daily) to placebo favored the TKI, with a PFS of 9.2 months versus 4.2 months. A subset of treatment-naïve patients had a longer PFS of 11.1 versus 2.8 months and a response rate of 32% versus 4%.72 This led to a noninferiority phase 3 trial comparing pazopanib with sunitinib as first-line therapy.50 In this study, PFS was similar (8.4 versus 9.5 months; HR 1.05), and overall safety and quality-of-life endpoints favored pazopanib. Much less fatigue, stomatitis, hand-foot syndrome, and thrombocytopenia occurred with pazopanib, whereas hair color changes, weight loss, alopecia, and elevations of LFT enzymes occurred more frequently with pazopanib. Hypertension is common with the administration of pazopanib as well.
Sorafenib
Sorafenib is an orally administered inhibitor of Raf, serine/threonine kinase, VEGFR, PDGFR, FLT-3, c-Kit, and RET. The pivotal phase 3 Treatment Approaches in Renal Cancer Global Evaluation Trial (TARGET) compared sorafenib (400 mg orally twice daily) with placebo in patients who had progressed on prior cytokine-based therapy.73 A final analysis, which excluded patients who were allowed to cross over therapies, found improved overall survival times (14.3 versus 1.8 months, P = 0.029).51 Sorafenib is associated with lower rates of diarrhea, rash, fatigue, hand-foot syndrome, alopecia, hypertension, and nausea than sunitinib, although these agents have not been compared to one another.
Axitinib
Axitinib is an oral inhibitor of VEGFRs 1, 2, and 3. Results of the phase 3 AXIS trial comparing axitinib (5 mg orally twice daily) with sorafenib (400 mg orally twice daily) in patients receiving 1 prior systemic therapy showed axitinib was more active than sorafenib in improving ORR (19% versus 9%; P = 0.001) and PFS (6.7 versus 4.7 months; P < 0.001), although no difference in overall survival times was noted.74 In a subsequent phase 3 trial comparing these drugs in the first-line setting, axitinib showed a nonsignificantly higher response rate and PFS. Despite this, the National Comprehensive Cancer Network guidelines consider axitinib an acceptable first-line therapy because activity with acceptable toxicity was demonstrated (Table 2).46 The most common adverse effects of axitinib are diarrhea, hypertension, fatigue, decreased appetite, dysphonia, hypothyroidism, and upper abdominal pain.
CABOZANTINIB
Given that resistance eventually develops in most patients treated with standard treatments, including bevacizumab and TKIs, the need to evaluate the safety and efficacy of novel agents targeting VEGFR and overcoming this resistance is of vital importance. Cabozantinib is an oral small-molecule inhibitor of VEGFR, Met, and Axl, all tyrosine kinases implicated in metastatic RCC. Overexpression of Met and Axl, which occurs as a result of inactivation of the VHL gene, is associated with a poor prognosis in patients with RCC. In a
randomized, open label, phase 3 trial of cabozantinib versus everolimus in advanced RCC, Choueiri and colleagues75 compared the efficacy of cabozantinib with everolimus in patients with metastatic RCC who had progressed on previous VEGFR-targeted therapies. In this study, 658 patients were randomly assigned to receive cabozantinib (60 mg orally daily) or everolimus (10 mg orally daily). Results of the study found that PFS was longer with cabozantinib in patients who had previously been treated with other TKIs (median PFS of 7.4 months versus 3.8 months; HR 0.58), corresponding to a 42% reduction in the rate of disease progression or death. The most common grade 3 and 4 toxicities seen with cabozantinib were similar to its class effect and consisted of hypertension, diarrhea, and fatigue. In the final analysis of the data, the median overall survival was 21.4 months (95% confidence interval [CI] 18.7–not estimable) with cabozantinib and 16.5 months (95% CI 14.7 to 18.8) with everolimus (HR 0.66 [95% CI 0.53 to 0.83]; P = 0.00026). The median follow-up for overall survival and safety was 18.7 months. These results highlight the importance of cabozantinib as a first line option in treatment of previously treated patients with advanced RCC.76
MTOR INHIBITORS
The mTOR inhibitors, temsirolimus and everolimus, are also approved for the treatment of metastatic or advanced RCC. These drugs block mTOR’s phosphorylation and subsequent translation of mRNA to inhibit cell proliferation, cell growth, and angiogenesis.77 Temsirolimus can be used as first-line therapy for patients with a poor prognosis, and everolimus is appropriate as a subsequent therapy.
Temsirolimus is an intravenous prodrug of rapamycin. It was the first of the class to be approved for metastatic RCC for treatment-naïve patients with a poor prognosis (ie, at least 3 of 6 predictors of poor survival based on MSKCC model).54 The pivotal ARCC trial compared temsirolimus (25 mg IV weekly) alone, interferon alfa (3 million units SC 3 times weekly) alone, or the combination (temsirolimus 15 mg IV weekly plus interferon alfa 6 million units SC 3 times weekly). In this trial, temsirolimus monotherapy produced a significantly longer overall survival time than interferon alfa alone (10.9 versus 7.3 months; P = 0.008) and improved PFS time when administered alone or in combination with interferon alfa (3.8 and 3.7 months, respectively, versus 1.9 months). Because no real efficacy advantage of the combination was demonstrated, temsirolimus is administered alone. The most common adverse effects of temsirolimus are asthenia, rash, anemia, nausea, anorexia, pain, and dyspnea. Additionally, hyperglycemia, hyper-cholesterolemia, and hyperlipidemia occur with these agents. Noninfectious pneumonitis is a rare but often fatal complication.
Everolimus is also an orally administered derivative of rapamycin that is approved for use after failure of VEGF-targeted therapies. The results of the landmark trial RECORD-1 demonstrated that everolimus (10 mg orally daily) is effective at prolonging PFS (4 versus 1.9 months; P < 0.001) when compared with best supportive care, a viable treatment option at the time of approval.78 The most common adverse effects of everolimus are stomatitis, rash, fatigue, asthenia, and diarrhea. As with temsirolimus, elevations in glucose, lipids, and triglycerides and noninfectious pneumonitis can occur.
TKI + MTOR INHIBITOR
Lenvatinib is also a small molecule targeting multiple tyrosine kinases, primarily VEGF2. Combined with the mTOR inhibitor everolimus, it has been shown to be an effective regimen in patients with metastatic RCC who have failed other therapies. In a randomized phase 2 study involving patients with advanced or metastatic clear-cell RCC, patients were randomly assigned to receive either lenvatinib (24 mg/day), everolimus (10 mg/day), or lenvatinib plus everolimus (18 mg/day and 5 mg/day, respectively). Patients received the treatment continuously on a 28-day cycle until progression or inability to tolerate toxicity. Patients in the lenvatinib plus everolimus arm had median PFS of 14.6 months (95% CI 5.9 to 20.1) versus 5.5 months (95% CI 3.5 to 7.1) with everlolimus alone (HR 0.40 [95% CI 0.24 to 0.68]; P = 0.0005). PFS with levantinib alone was 7.4 months (95% CI 5.6 to 10.20; HR 0.66 [95% CI 0.30 to 1.10]; P = 0.12). In addition, PFS with levantinib alone was significantly prolonged in comparison with everolimus alone (HR 0.61 [95% CI 0.38 to 0.98]; P = 0.048). Grade 3 or 4 toxicity were less frequent in the everolimus only arm and the most common grade 3 or 4 toxicity in the lenvatinib plus everolimus arm was diarrhea. The results of this study show that the combination of lenvatinib plus everolimus is an acceptable second-line option for treatment of patients with advanced or metastatic RCC.55
CASE CONTINUED
The patient is initially started on pazopanib and tolerates the medication well, with partial response to the treatment. However, on restaging scans he is noted to have small bowel perforation. Pazopanib is discontinued until the patient has a full recovery. He is then started on everolimus. Restaging scans done 3 months after starting everolimus demonstrate disease progression.
• What is the appropriate next step in treatment?
PD1 BLOCKADE
Programmed death 1 (PD-1) protein is a T-cell inhibitory receptor with 2 ligands, PD-L1 and PD-L2. PD-L1 is expressed on many tumors. Blocking the interaction between PD-1 and PD-L1 by anti-PD-1 humanized antibodies potentiates a robust immune response and has been a breakthrough in the field of cancer immunotherapy.79 Previous studies have demonstrated that overexpression of PD-L1 leads to worse outcomes and poor prognosis in patients with RCC.80 Nivolumab, a fully human IgG4 PD-1 immune checkpoint inhibitor, blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2. In a randomized, open-label, phase 3 study comparing nivolumab with everolimus in patients with RCC who had previously undergone treatment with other standard therapies, Motzer and colleagues81 demonstrated a longer overall survival time and fewer adverse effects with nivolumab. In this study, 821 patients with clear-cell RCC were randomly assigned to receive nivolumab (3 mg/kg of body weight IV every 2 weeks) or everolimus (10 mg orally once daily). The median overall survival time with nivolumab was 25 months versus 19.6 months with everolimus (P < 0.0148). Nineteen percent of patients receiving nivolumab experienced grade 3 or 4 toxicities, with fatigue being the most common adverse effect. Grade 3 or 4 toxicities were observed in 37% of patients treated with everolimus, with anemia being the most common. Based on the results of this trial, on November 23, 2015, the U.S. Food and Drug Administration approved nivolumab to treat patients with metastatic RCC who have received a prior antiangiogenic therapy.
CASE CONCLUSION
Both TKI and mTOR inhibitor therapy fail, and the patient is eligible for third-line therapy. Because of his previous GI perforation, other TKIs are not an option. The patient opts for enrollment in hospice due to declining performance status. For other patients in this situation with a good performance status, nivolumab would be a reasonable option.
FUTURE DIRECTIONS
With the approval of nivolumab, multiple treatment options are now available for patients with metastatic or unresectable RCC. Development of other PD-1 inhibitors and immunotherapies as well as multi-targeted TKIs will only serve to expand treatment options for these patients. Given the aggressive course and poor prognosis of non-clear cell renal cell tumors and those with sarcomatoid features, evaluation of systemic and targeted therapies for these subtypes should remain active areas of research and investigation.
- Siegel R, Miller, K, Jemal A. Cancer Statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Eble JN, Sauter G, Epstein JI, Sesterhenn IA. Pathology and genetics. Tumors of the urinary system and male genital organs. Lyon: IARC Press; 2004.
- Chow WH, Gridley G, Fraumeni JF Jr, Jarvholm B. Obesity, hypertension, and the risk of kidney cancer in men. N Engl J Med 2000;343:1305–11.
- Cohen H, McGovern F. Renal-cell carcinoma. N Engl J Med 2005;353:2477–90.
- Yao M, Yoshida M, Kishida T, et al. VHL tumor suppres sor gene alterations associated with good prognosis in sporadic clear-cell renal carcinoma. J Natl Cancer Inst 2002;94:1569–75.
- Iliopoulos O, Kibel A, Gray S, Kaelin WG Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1995;1:822–6
- Chen F, Kishida T, Duh FM, et al. Suppression of growth of renal carcinoma cells by the von Hippel-Lindau tumor suppressor gene. Cancer Res 1995;55:4804–7.
- Iliopoulos O, Levy AP, Jiang C, et al. Negative regulation of hypoxia-inducible genes by the von Hippel Lindau protein. Proc Natl Acad Sci U S A 1996;93:10595–9.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Bir- Hogg-Dube syndrome. Cancer Cell 2002;2:157–64
- Shuch B, Vorganit S, Ricketts CJ, et al. Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 2014;32:431–7.
- Bukowski RM. Immunotherapy in renal cell carcinoma. Oncology 1999;13:801–10.
- Mueller-Lisse UG, Mueller-Lisse UL. Imaging of advanced renal cell carcinoma. World J Urol 2010;28: 253–61.
- Edge SB, Byrd DR, Compton CC, et al, eds. AJCC cancer staging manual, 7th ed. New York: Springer Science and Business Media LLC; 2010.
- O’Malley RL, Godoy G, Kanofsky JA, Taneja SS. The necessity of adrenalectomy at the time of radical nephrectomy: a systematic review. J Urol 2009;181:2009–17.
- McDougal S, Wein AJ, Kavoussi LR, et al. Campbell-Walsh Urology. 10th ed. Philadelphia (PA): Saunders; 2012.
- Colombo JR Jr, Haber GP, Kelovsek JE, et al. Seven years after laparoscopic radical nephrectomy: oncologic and renal functional outcomes. Urology 2008:71:1149–54.
- Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Ca 2013;49: 1374–403.
- Weight CJ, Larson BT, Fergany AF, et al. Nephrectomy induced chronic renal insufficiency is associated with increased risk of cardiovascular death and death from any cause in patients with localized cT1b renal masses. J Urol 2010;183:1317–23.
- Van Poppel H, Da Pozzo L, Albrecht W, et al. A prospective, randomized EORTC intergroup phase 3 study comparing the oncologic outcome of elective nephron-sparing surgery and radical nephrectomy for low-stage renal cell carcinoma. Eur Urol 2011;59:543–52.
- Smaldone MC, Fung C, Uzzo RG, Hass NB. Adjuvant and neoadjuvant therapies in high-risk renal cell carcioma. Hematol Oncol Clin North Am 2011;25:765–91.
- NCCN clinical practice guidelines in oncology. Version 3.2016. www.nccn.org. Accessed July 13, 2016
- El Dib R, Touma NJ, Kapoor A. Cryoablation vs radiofrequency ablation for the treatment of renal cell carcinoma: a meta-amalysis of case series studies. BJU Int 2012;110:510–6.
- Theodorescu D. Cancer cryotherapy: evolution and biology. Rev Urol 2004;6 Suppl 4:S9–S19.
- Khiatani V, Dixon RG. Renal ablation update. Sem Intervent Radiol 2014;31:157–66.
- Yu J, Liang P, Yu XL, et al. US-guided percutaneous microwave ablation of renal cell carcinoma: intermediate-term results. Radiol 2012;263:900–8.
- Castle SM, Salas N, Leveillee RJ. Initial experience using microwave ablation therapy for renal tumor treatment: 18- month follow-up. Urology 2011;77:792–7.
- Pech M, Janitzky A, Wendler JJ, et al. Irreversible electroporation of renal cell carcinoma: a first-in-man phase I clinical study. Cardiovasc Intervent Radiol 2011;34:132–8.
- Chow WH, Devesa SS, Warren JL, Fraumeni JF Jr. Rising incidence of renal cell cancer in the United States. JAMA 1999;281:1628–31.
- Jayson M, Sanders H. Increased incidence of serendipitously discovered renal cell carcinoma. Urology 1998;51:203–5.
- Pierorazio PM, Johnson MH, Ball MW, et al. Five-year analysis of a multi-institutional prospective clinical trial of delayed intervention and surveillance for small renal masses: the DISSRM registry. Eur Urol 2015;68:408–15.
- Jewett MA, Mattar K, Basiuk J, et al. Active surveillance of small renal masses: progression patterns of early stage kidney cancer. Eur Urol 2011;60:39–44.
- Chawla SN, Crispen PL, Hanlon AL, et al. The natural history of observed enhancing renal masses: meta-analysis and review of the world literature. J Urol 2006;175:425–31.
- Smaldone MC, Kutikov A, Egleston BL, et al. Small renal masses progressing to metastases under active surveillance: a systematic review and pooled analysis. Cancer 2012;118:997–1006.
- Williamson TJ, Pearson JR, Ischia J, et al.Guideline of guidelines: follow-up after nephrectomy for renal cell carcinoma. BJU Int 2016;117:555–62.
- Donat S, Diaz M, Bishoff JT, et al. Follow-up for clinically localized renal neoplasms: AUA Guideline. J Urol 2013;190:407–16.
- Janzen NK, Kim HL, Figlin RA, Bell-degrun AS. Surveillance after radical or partial nephrectomy for localized renal cell carcinoma and management of recurrent disease. Urol Clin North Am 2003:30:843–52.
- Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socio-economic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev 2008;34:193–205.
- Mekhail T, Abou-Jawde R, Boumerhi G, et al. Validation and extension of the Memorial Sloan-Kettering Prognostic Factors Model for Survival in patients with previously untreated metastatic renal cell carcinoma. J Clin Oncol 2005;23: 832–41.
- Motzer RJ, Bacik J, Murphy BA, et al. Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J Clin Oncol 2002;20:289–96.
- Motzer RJ, Mazumdar M, Bacik J, et al. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J Clin Oncol 1999;17:2530–40.
- Heng DY, Xie W, Regan MM. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol 2009;27:5794–9.
- Leibovich BC, Han KR, Bui MH, et al. Scoring algorithm to predict survival after nephrectomy and immunotherapy in patients with metastatic renal cell carcinoma: A stratification tool for prospective clinical trials. Cancer 2003;98:2566–77.
- Flanigan RC, Mickisch G, Sylvester R, et al. Cytoreductive nephrectomy in patients with metastatic renal cancer: a combined analysis. J Urol 2004;171:1071–6.
- Choueiri TK, Xie W, Kollmannsberger C, et al. The impact of cytoreductive nephrectomy on survival of patients with metastatic renal cell carcinoma receiving vascular endothelial growth factor targeted therapy. J Urol 2011;185:60–6.
- Chapin BF, Delacroix SE Jr, Culp SH, et al. Safety of presurgical targeted therapy in the setting of metastatic renal cell carcinoma. Eur Urol 2011;60:964–71.
- Hutson TE, Lesovoy V, Al-Shukri S, et al. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal-cell carcinoma: a randomized open-label phase 3 trial. Lancet Oncol 2013;14:1287–94.
- Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metatastic renal cell carcinoma: a randomized, double-blind phase III trial. Lancet 2007;370:2103–11.
- Escudier B, Bellmunt J, Negrier S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol 2010;28:2144–50.
- McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8.
- Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med 2013;369:722–31.
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cell global evaluation trial. J Clin Oncol 2009;27:3312–8.
- Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007;356:115–24.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 2009;27:3584–90.
- Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 2007;356:2271–81.
- Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus and the combination in patients with metastatic renal cell carcinoma: a randomized, phase 2, open label, multicenter trial. Lancet Oncology 2015;16:1473–82.
- Lexi-Comp, Inc. (Lexi-Drugs® ). Lexi-Drugs version 2.3.3. Lexicomp. Wolters Kluwer Health, Inc. Hudson, OH.
- Choueiri TK, Plantade A, Elson P, et al. Efficacy of sunitinib and sorafenib in metastatic papillary and chromophobe renal cell carcinoma. J Clin Oncol 2008;26:127–31.
- Lee JL, Ahn JH, Lim HY, et al. Multicenter phase II study of sunitinib in patients with non-clear cell renal cell carcinoma. Ann Oncol 2012;23:2108–14.
- Armstrong AJ, Broderick S, Eisen T, et al. Final clinical results of a randomized phase II international trial of everolimus vs. sunitinib in patients with metastatic non-clear cell renal cell carcinoma (ASPEN). ASCO Meeting Abstracts 2015;33:4507.
- Chowdhury S, Matrana MR, Tsang C, et al. Systemic therapy for metastatic non-clear-cell renal cell carcinoma: recent progress and future directions. Hematol Oncol Clin North Am 2011;25:853–69.
- Escudier B, Droz JP, Rolland F, et al. Doxorubicin and ifosfamide in patients with metastatic sarcomatoid renal cell carcinoma: a phase II study of the Genitourinary Group of the French Federation of Cancer Centers. J Urol 2002; 168–71
- Nanus DM, Garino A, Milowsky MI, et al. Active chemotherapy for sarcomatoid and rapidly progressing renal cell carcinoma. Cancer 2004;101:1545–51.
- Michaelson MD, McKay RR, Werner L, et al. Phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid and/or poor-risk metastatic renal cell carcinoma. Cancer 2015;121:3435–43.
- McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8
- Cho DC, Puzanov I, Regan MM, et al. Retrospective analysis of the safety and efficacy of interleukin-2 after prior VEGF-targeted therapy in patients with advanced renal cell carcinoma. J Immunother 2009;32:181–5.
- Pyrhönen S, Salminen E, Ruutu M, et al. Prospective randomized trial of interferon alfa-2a plus vinblastine versus vinblastine alone in patients with advanced renal cell cancer. J Clin Oncol 1999;17:2859–67.
- Interferon-alpha and survival in metastatic renal carcinoma: early results of a randomised controlled trial. Medical Research Council Renal Cancer Collaborators. Lancet 1999;353:14–7.
- Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349:427–34.
- Atkinson BJ, Kalra S, Wang X, et al. Clinical outcomes for patients with metastatic renal cell carcinoma treated with alternative sunitinib schedules. J Urol 2014;191:611–8.
- Kollmannsberger C, Bjarnason G, Burnett P, et al. Sunitinib in metastatic renal cell carcinoma: recommendations for management of noncardiovascular toxicities. Oncologist 2011;16:543–53.
- Najjar YG, Mittal K, Elson P, et al. A 2 weeks on and 1 week off schedule of sunitinib is associated with decreased toxicity in metastatic renal cell carcinoma. Eur J Cancer 2014;50:1084–9.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 2010;28:1061–8.
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007;356:125–34
- Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011;378:1931–9.
- Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1814–23.
- Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR) final results from a randomized, open-label, phase 3 trial. Lancet Oncology 2016;17:917–27.
- Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer 2004;4:335–48.
- Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372:449–56.
- Brahmer J, Tykodi S, Chow L, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65.
- Thomson RH, Kuntz SM, Leibovich BC, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow up. Cancer Res 2006;66: 3381–5.
- Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803–13.
INTRODUCTION
Renal cell carcinoma (RCC) is the most common malignancy arising in the kidney, comprising 90% of all renal tumors.1 Approximately 55,000 new RCC cases are diagnosed each year.2 Patients with RCC are often asymptomatic, and most cases are discovered as incidental findings on abdominal imaging performed during evaluation of nonrenal complaints. Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20%.2 Advanced RCC is resistant to conventional chemotherapy and radiotherapy, and outcomes for patients with metastatic or unresectable RCC remain poor. However, the recent development of new therapeutic modalities that target tumor molecular pathways has expanded the treatment options for these patients and changed the management of RCC.
EPIDEMIOLOGY AND CLASSIFICATION
Median age at diagnosis in the United States is 64 years. Men have a higher incidence of RCC than women, with the highest incidence seen in American Indian and Alaska Native men (30.1 per 100,000 population). Genetic syndromes account for 2% to 4% of all RCCs.2 Risk factors for RCC include smoking, hypertension, obesity, and acquired cystic kidney disease that is associated with end-stage renal failure.3 Longer duration of tobacco use is associated with a more aggressive course.
The 2004 World Health Organization (WHO) classification of renal tumors summarizes the previous classification systems (including the Heidelberg and Mainz classification systems) to describe different categories of RCC based on histologic and molecular genetics characteristics.2 Using the WHO classification criteria, RCC comprises 90% of all renal tumors, with clear cell being the most common type (80%).2 Other types of renal tumors include papillary, chromophobe, oncocytoma, and collecting-duct or Bellini duct tumors. Approximately 3% to 5% of tumors are unclassified. Oncocytomas are generally considered benign, and chromophobe tumors typically have an indolent course and rarely metastasize. Sarcomatoid differentiation can be seen in any histologic type and is associated with a worse prognosis. While different types of tumors may be seen in the kidney (such as transitional cell or lymphomas), the focus of this review is the primary malignancies of the renal parenchyma.
FAMILIAL SYNDROMES
Several genetic syndromes have been identified by studying families with inherited RCC. Among these, von Hippel-Lindau (VHL) gene mutation is the most commonly found inherited genetic defect. Table 1 summarizes the incidence of gene mutations and the corresponding histologic appearance of the most common sporadic and hereditary RCCs.4
VHL disease is an autosomal dominant familial syndrome. Patients with this mutation are at higher risk for developing RCC (clear cell histology), retinal angiomas, pheochromocytomas, as well as hemangioblastomas of the central nervous system (CNS).4 Of all the genetic mutations seen in RCC, the somatic mutation in the VHL tumor-suppressor gene is by far the most common.5 VHL targets hypoxia–inducible factor-1 alpha (HIF-α) for ubiquitination and subsequent degradation, which has been shown to suppress the growth of clear-cell RCC in mouse models.6–8 HIF expression under hypoxic conditions leads to activation of a number of genes important in blood vessel development, cell proliferation, and glucose metabolism, including vascular endothelial growth factor (VEGF), erythropoietin, platelet-derived growth factor beta (PDGF-β), transforming growth factor alpha (TGF-α), and glucose transporter-1 (GLUT-1). Mutation in the VHL gene prevents degradation of the HIF-α protein, thereby leading to increased expression of these downstream proteins, including MET and Axl. The upregulation of these angiogenic factors is thought to be the underlying process for increased vascularity of CNS hemangioblastomas and clear-cell renal tumors in VHL disease.4–8
Other less common genetic syndromes seen in hereditary RCC include hereditary papillary RCC, hereditary leiomyomatosis, and Birt-Hogg-Dubé (BHD) syndrome.9 In hereditary papillary RCC, the MET gene is mutated. BHD syndrome is a rare, autosomal dominant syndrome characterized by hair follicle hamartomas of the face and neck. About 15% of patients have multiple renal tumors, the majority of which are of the chromophobe or mixed chromophobe-oncocytoma histology. The BHD gene encodes the protein folliculin, which is thought to be a tumor-suppressor gene.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 74-year-old man who works as an airplane mechanic repairman presents to the emergency department with sudden worsening of chronic right upper arm and shoulder pain after lifting a jug of orange juice. He does not have a significant past medical history and initially thought that his pain was due to a work-related injury. Upon initial evaluation in the emergency department he is found to have a fracture of his right humerus. Given that the fracture appears to be pathologic, further work-up is recommended.
• What are common clinical presentations of RCC?
Most patients are asymptomatic until the disease becomes advanced. The classic triad of flank pain, hematuria, and palpable abdominal mass is seen in approximately 10% of patients with RCC, partly because of earlier detection of renal masses by imaging performed for other purposes.10 Less frequently, patients present with signs or symptoms of metastatic disease such as bone pain or fracture (as seen in the case patient), painful adenopathy, and pulmonary symptoms related to mediastinal masses. Fever, weight loss, anemia, and/or varicocele often occur in young patients (≤ 46 years) and may indicate the presence of a hereditary form of the disease. Patients may present with paraneoplastic syndromes seen as abnormalities on routine blood work. These can include polycythemia or elevated liver function tests (LFTs) without the presence of liver metastases (known as Stauffer syndrome), which can be seen in localized renal tumors. Nearly half (45%) of patients present with localized disease, 25% present with locally advanced disease, and 30% present with metastatic disease.11 Bone is the second most common site of distant metastatic spread (following lung) in patients with advanced RCC.
• What is the approach to initial evaluation for a patient with suspected RCC?
Initial evaluation consists of a physical exam, laboratory tests including complete blood count (CBC) and comprehensive metabolic panel (calcium, serum creatinine, LFTs, lactate dehydrogenase [LDH], and urinalysis), and imaging. Imaging studies include computed tomography (CT) scan with contrast of the abdomen and pelvis or magnetic resonance imaging (MRI) of the abdomen and chest imaging. A chest radiograph may be obtained, although a chest CT is more sensitive for the presence of pulmonary metastases. MRI can be used in patients with renal dysfunction to evaluate the renal vein and inferior vena cava (IVC) for thrombus or to determine the presence of local invasion.12 Although bone and brain are common sites for metastases, routine imaging is not indicated unless the patient is symptomatic. The value of positron emission tomography in RCC remains undetermined at this time.
Staging is done according to the American Joint Committee on Cancer (AJCC) staging classification for RCC; the Figure summarizes the staging and 5-year survival data based on this classification scheme.4,13
J Med 2005;353:2477–90.)
LIMITED-STAGE DISEASE
• What are the therapeutic options for limited-stage disease?
For patients with nondistant metastases, or limited-stage disease, surgical intervention with curative intent is considered. Convention suggests considering definitive surgery for patients with stage I and II disease, select patients with stage III disease with pathologically enlarged retroperitoneal lymph nodes, patients with IVC and/or cardiac atrium involvement of tumor thrombus, and patients with direct extension of the renal tumor into the ipsilateral adrenal gland if there is no evidence of distant disease. While there may be a role for aggressive surgical intervention in patients with distant metastatic disease, this topic will not be covered in this review.
SURGICAL INTERVENTION
Once patients are determined to be appropriate candidates for surgical removal of a renal tumor, the urologist will perform either a radical nephrectomy or a nephron-sparing nephrectomy, also called a partial nephrectomy. The urologist will evaluate the patient based on his or her body habitus, the location of the tumor, whether multiple tumors in one kidney or bilateral tumors are present, whether the patient has a solitary kidney or otherwise impaired kidney function, and whether the patient has a history of a hereditary syndrome involving kidney cancer as this affects the risk of future kidney tumors.
A radical nephrectomy is surgically preferred in the presence of the following factors: tumor larger than 7 cm in diameter, a more centrally located tumor, suspicion of lymph node involvement, tumor involvement with renal vein or IVC, and/or direct extension of the tumor into the ipsilateral adrenal gland. Nephrectomy involves ligation of the vascular supply (renal artery and vein) followed by removal of the kidney and surrounding Gerota’s fascia. The ipsilateral adrenal gland is removed if there is a high-risk for or presence of invasion of the adrenal gland. Removal of the adrenal gland is not standard since the literature demonstrates there is less than a 10% chance of solitary, ipsilateral adrenal gland involvement of tumor at the time of nephrectomy in the absence of high-risk features, and a recent systematic review suggests that the chance may be as low as 1.8%.14 Preoperative factors that correlated with adrenal involvement included upper pole kidney location, renal vein thrombosis, higher T stage (T3a and greater), multifocal tumors, and evidence for distant metastases or lymph node involvement. Lymphadenectomy previously had been included in radical nephrectomy but now is performed selectively. Radical nephrectomy may be performed as
either an open or laparoscopic procedure, the latter of which may be performed robotically.15 Oncologic outcomes appear to be comparable between the 2 approaches, with equivalent 5-year cancer-specific survival (91% with laparoscopic versus 93% with open approach) and recurrence-free survival (91% with laparoscopic versus 93% with open approach).16 The approach ultimately is selected based on provider- and patient-specific input, though in all cases the goal is to remove the specimen intact.16,17
Conversely, a nephron-sparing approach is preferred for tumors less than 7 cm in diameter, for patients with a solitary kidney or impaired renal function, for patients with multiple small ipsilateral tumors or with bilateral tumors, or for radical nephrectomy candidates with comorbidities for whom a limited intervention is deemed to be a lower-risk procedure. A nephron-sparing procedure may also be performed open or laparoscopically. In nephron-sparing procedures, the tumor is removed along with a small margin of normal parenchyma.15
In summary, the goal of surgical intervention is curative intent with removal of the tumor while maintaining as much residual renal function as possible to limit long-term morbidity of chronic kidney disease and associated cardiovascular events.18 Oncologic outcomes for radical nephrectomy and partial nephrectomy are similar. In one study, overall survival was slightly lower in the partial nephrectomy cohort, but only a small number of the deaths were due to RCC.19
ADJUVANT THERAPY
Adjuvant systemic therapy currently has no role following nephrectomy for RCC because no systemic therapy has been able to reduce the likelihood of relapse. Randomized trials of cytokine therapy (eg, interferon, interleukin 2) or tyrosine kinase inhibitors (TKIs; eg, sorafenib, sunitinib) with observation alone in patients with locally advanced completely resected RCC have shown no delay in time to relapse or improvement of survival with adjuvant therapy.20 Similarly, adjuvant radiation therapy has not shown benefit even in patients with nodal involvement or incomplete resection.21 Therefore, observation remains the standard of care after nephrectomy.
RENAL TUMOR ABLATION
For patients who are deemed not to be surgical candidates due to age, comorbidities, or patient preference and who have tumors less than 4 cm in size (stage I tumors), ablative techniques may be considered. The 2 most well-studied and effective techniques at present are cryoablation and radiofrequency ablation (RFA). Microwave ablation may be an option in some facilities, but the data in RCC are limited. An emerging ablative technique under investigation is irreversible electroporation. At present, the long-term efficacy of all ablative techniques is unknown.
Patient selection is undertaken by urologists and interventional radiologists who evaluate the patient with ultrasound, CT, and/or MRI to determine the location and size of the tumor and the presence or absence of metastatic disease. A pretreatment biopsy is recommended to document the histology of the lesion to confirm a malignancy and to guide future treatment for recurrent or metastatic disease. Contraindications to the procedure include the presence of metastatic disease, a life expectancy of less than 1 year, general medical instability, or uncorrectable coagulopathy due to increased risk of bleeding complications. Tumors in close proximity to the renal hilum or collecting system are a contraindication to the procedure because of the risk for hemorrhage or damage to the collecting system. The location of the tumor in relation to the vasculature is also important to maximize efficacy because the vasculature acts as a “heat sink,” causing dissipation of the thermal energy. Occasionally, stenting of the proximal ureter due to upper tumor location is necessary to prevent thermal injury that could lead to urine leaks.
Selection of the modality to be used primarily depends on operator comfort, which translates to good patient outcomes, such as better cancer control and fewer complications. Cryoablation and RFA have both demonstrated good clinical efficacy and cancer control of 89% and 90%, respectively, with comparable complication rates.22 There have been no studies performed directly comparing the modalities.
Cryoablation
Cryoablation is performed through the insertion of a probe into the tumor, which may be done through a surgical or percutaneous approach. Once the probe is in place, a high- pressure gas (argon, nitrogen) is passed through the probe and upon entering a low pressure region the gas cools. The gas is able to cool to temperatures as low as –185°C. The tissue is then rewarmed through the use of helium, which conversely warms when entering a low pressure area. The process of freezing followed by rewarming subsequently causes cell death/tissue destruction through direct cell injury from cellular dehydration and vascular injury. Clinically, 2 freeze-thaw cycles are used to treat a tumor.23,24
RFA
Radiofrequency ablation, or RFA, targets tumors via an electrode placed within the mass that produces intense frictional heat from medium-frequency alternating current (approximately 500 kHz) produced by a connected generator that is grounded on the patient. The thermal energy created causes coagulative necrosis. Due to the reliance on heat for tumor destruction, central lesions are less amenable to this approach because of the “heat sink” effect from the hilum.24
Microwave Ablation
Microwave ablation, like RFA, relies on the generation of frictional heat to cause cell death by coagulative necrosis. In this case, the friction is created through the activation of water molecules; because of the different thermal kinetics involved with microwave ablation, the “heat sink” effect is minimized when treatment is employed near large vessels, in comparison to RFA.24 The data on this mechanism of ablation are still maturing, with varied outcomes thus far. One study demonstrated outcomes comparable to RFA and cryoablation, with cancer-specific survival of 97.8% at 3 years.25 However, a study by Castle and colleagues26 demonstrated higher recurrence rates. The overarching impediment to widespread adoption of microwave ablation is inconclusive data gleaned from studies with small numbers of patients with limited follow up. The role of this modality will need to be revisited.
Irreversible Electroporation
Irreversible electroporation (IRE) is under investigation. IRE is a non-thermal ablative technique that employs rapid electrical pulses to create pores in cell membranes, leading to cell death. The postulated benefits of IRE include the lack of an effect from “heat sinks” and less collateral damage to the surrounding tissues, when compared with the thermal modalities. In a human phase 1 study of patients undergoing IRE prior to immediate surgical resection, the procedure appeared feasible and safe.27 Significant concerns for this method of ablation possibly inducing cardiac arrhythmias, and the resultant need for sedation with neuromuscular blockade and associated electrocardiography monitoring, may impede its implementation in nonresearch settings.24
ACTIVE SURVEILLANCE
Due to the more frequent use of imaging for various indications, there has been an increase in the discovery of small renal masses (SRM); 85% of RCC that present in an asymptomatic or incidental manner are tumors under 4 cm in diameter.28,29 The role of active surveillance is evolving, but is primarily suggested for patients who are not candidates for more aggressive intervention based on comorbidities. A recent prospective, nonrandomized analysis of data from the Delayed Intervention and Surveillance for Small Renal Masses (DISSRM) registry evaluated outcomes for patients with SRM looking at primary intervention compared with active surveillance.30 The primary intervention selected was at the discretion of the provider; treatments included partial nephrectomy, RFA, and cryoablation, and active surveillance patients were followed with imaging every 6 months. Progression of SRM, with recommendation for delayed intervention, was defined as a growth rate of mass greater than 0.5 cm/year, size greater than 4 cm, or hematuria. Thirty-six of 158 patients on active surveillance met criteria for progression; 21 underwent delayed intervention. Of note, even the patients who progressed but did not undergo delayed intervention did not develop metastatic disease during the follow-up interval. With a median follow-up of 2 years, cancer-specific survival was noted to be 99% and 100% at 5 years for primary intervention and active surveillance, respectively. Overall survival at 2 years for primary intervention was 98% and 96% for active surveillance; at 5 years, the survival rates were 92% and 75% (P = 0.06). Of note, 2 patients in the primary intervention arm died of RCC, while none in the active surveillance arm died. As would be expected, active surveillance patients were older, had a worse performance status, and had more comorbidities. Interestingly, 40% of patients enrolled selected active surveillance as their preferred management for SRM. The DISSRM results were consistent with data from the Renal Cell Consortium of Canada and other retrospective reviews.31–33
• What is the approach to follow-up after treatment of localized RCC?
After a patient undergoes treatment for a localized RCC, the goal is to optimize oncologic outcomes, monitor for treatment sequelae, such as renal failure, and focus on survivorship. At this time, there is no consensus in the literature or across published national and international guidelines with regards to the appropriate schedule for surveillance to achieve these goals. In principle, the greatest risk for recurrence occurs within the first 3 years, so many guidelines focus on this timeframe. Likewise, the route of spread tends to be hematogenous, so patients present with pulmonary, bone, and brain metastases, in addition to local recurrence within the renal bed. Symptomatic recurrences often are seen
with bone and brain metastases, and thus bone scans and brain imaging are not listed as part of routine surveillance protocols in asymptomatic patients. Although there is inconclusive evidence that surveillance protocols improve outcomes in RCC, many professional associations have outlined recommendations based on expert opinion.34 The American Urological Association released guidelines in 2013 and the National Comprehensive Cancer Network (NCCN) released their most recent set of guidelines in 2016.21,35 These guidelines use TNM staging to risk-stratify patients and recommend follow-up.
METASTATIC DISEASE
CASE CONTINUED
CT scan with contrast of the chest, abdomen, and pelvis as well as bone scan are done. CT of the abdomen and pelvis demonstrates a 7.8-cm left renal mass arising from the lower pole of the left kidney. Paraesophageal lymphadenopathy and mesenteric nodules are also noted. CT of the chest demonstrates bilateral pulmonary emboli. Bone scan is significant for increased activity related to the pathological fracture involving the right humerus. The patient undergoes surgery to stabilize the pathologic fracture of his humerus. He is diagnosed with metastatic RCC (clear cell histology) and undergoes palliative debulking nephrectomy.
• How is prognosis defined for metastatic RCC?
PROGNOSTIC MODELS
Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates for stage T1 and T2 disease approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20%.13 Approximately 30% of patients have metastatic disease at diagnosis, and about one-third of patients who have undergone treatment for localized disease experience relapse.36,37 Common sites of metastases include lung, lymph nodes, bone, liver, adrenal gland, and brain.
Prognostic scoring systems have been developed to define risk groups and assist with determining appropriate therapy in the metastatic setting. The most widely used validated prognostic factor model is that from the Memorial Sloan-Kettering Cancer Center (MSKCC), which was developed using a multivariate analysis derived from data of patients enrolled in clinical trials and treated with interferon alfa.38 The factors included in the MSKCC model are Karnofsky performance status less than 80, time from diagnosis to treatment with interferon alfa less than 12 months, hemoglobin level less than lower limit of laboratory’s reference range, LDH level greater than 1.5 times the upper limit of laboratory’s reference range, and corrected serum calcium level greater than 10 mg/dL. Risk groups are categorized as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors).39 Median survival for favorable-, intermediate-, and poor-risk patients was 20, 10, and 4 months, respectively.40
Another prognostic model, the International Metastatic RCC Database Consortium, or Heng, model was developed to evaluate prognosis in patients treated with VEGF-targeted therapy.41 This model was developed from a retrospective study of patients treated with sunitinib, sorafenib, and bevacizumab plus interferon alfa or prior immunotherapy. Prognostic factors in this model include 4 of the 5 MSKCC risk factors (hemoglobin level, corrected serum calcium level, Karnofsky performance status, and time to initial diagnosis). Additionally, this model includes both absolute neutrophil and platelet counts greater than the upper limit of normal. Risk groups are identified as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors). Median survival for favorable-, intermediate-, and poor-risk patients was not reached, 27 months, and 8.8 months, respectively. The University of California, Los Angeles scoring algorithm to predict survival after nephrectomy and immunotherapy (SANI) in patients with metastatic RCC is another prognostic model that can be used. This simplified scoring system incorporates lymph node status, constitutional symptoms, metastases location, histology, and thyroid stimulating hormone (TSH) level.42
The role of debulking or cytoreductive nephrectomy in treatment of metastatic RCC is well established. Large randomized studies have demonstrated a statistically significant median survival benefit for patients undergoing nephrectomy plus interferon alfa therapy compared with patients treated with interferon alfa alone (13.6 months versus 7.8 months, respectively).43 The role of cytoreductive nephrectomy in combination with antiangiogenic agents is less clear. While a retrospective study investigating outcomes of patients with metastatic RCC receiving anti-VEGF agents showed a prolonged survival with nephrectomy, results of large randomized trials are not yet available.44,45 Patients with lung-only metastases, good prognostic features, and a good performance status are historically the most likely to benefit from cytoreductive surgery.
CASE CONTINUED
Based on the MSKCC prognostic factor model, the patient is considered to be in the intermediate-risk group (Karnofsky performance status of 80, calcium 9.5 mg/dL, LDH 204 U/L, hemoglobin 13.6 g/dL). He is started on treatment for his bilateral pulmonary emboli and recovers well from orthopedic surgery as well as palliative debulking nephrectomy.
• What is the appropriate first-line therapy in managing this patient’s metastatic disease?
Several approaches to systemic therapy for advanced RCC have been taken based on the histologic type of the tumor. Clear-cell is by far the predominant histologic type in RCC. Several options are available as first-line treatment for patients with metastatic clear-cell RCC (Table 2).46–54 These include biologic agents such as high-dose interleukin-2 (IL-2) immune therapy, as well as targeted therapies including TKIs and anti-VEGF antibodies. The mammalian target of rapamycin (mTOR) inhibitor temsirolimus is recommended as first-line therapy in patients with poor prognosis only. Second-line therapies for clear-cell RCC following antiangiogenic therapy include TKIs, mTOR inhibitors, nivolumab (PD-1 inhibitor), and the combination of the TKI lenvatinib and mTOR inhibitor everolimus.55 In addition, after initial cytokine therapy, TKIs, temsirolimus, and the anti-VEGF antibody bevacizumab are other treatment options available to patients. Best supportive care should always be provided along with initial and subsequent therapies. Clinical trials are also an appropriate choice as first-line or subsequent therapies. All of these therapies require periodic monitoring to prevent and quickly treat adverse effects. Table 3 lists recommended monitoring parameters for each of these agents.56
Based on several studies, TKIs seem to be less effective in patients with non–clear-cell type histology.57,58 In these patients, risk factors can guide therapy. In the ASPEN trial, where 108 patients were randomly assigned to everolimus or sunitinib, patients in the good- and intermediate-risk groups had longer overall and median progression-free survival (PFS) on sunitinib (8.3 months versus 5.3 months, respectively). However, those in the poor-risk group had a longer median overall survival with everolimus.59 Given that the role of targeted therapies in non–clear-cell RCCs is less well established, enrollment in clinical trials should be considered as a first-line treatment option.21
Sarcomatoid features can be observed in any of the histologic types of RCC, and RCC with these features has an aggressive course and a poor prognosis. Currently, there is no standard therapy for treatment of patients with metastatic or unresectable RCC with sarcomatoid features.60 Chemotherapeutic regimens used for soft tissue sarcomas, including a trial of ifosfamide and doxorubicin, did not show any objective response.61 A small trial of 10 patients treated with doxorubicin and gemcitabine resulted in complete response in 2 patients and partial response in 1 patient.62
Enrollment in a clinical trial remains a first-line treatment option for these patients. More recently, a phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid (39 patients) and/or poor-risk (33 patients) metastatic RCC showed overall response rates (ORR) of 26% and 24%, respectively. A higher clinical benefit rate (defined as ORR plus stable disease) was seen in patients with tumors containing more than 10% sarcomatoid histology, as compared with patients whose tumors contained less than 10% sarcomatoid histology. Neutropenia (n = 20), anemia (n = 10), and fatigue (n = 7) were the most common grade 3 toxicities seen in all the patients. Although this was a small study, the results showed a trend towards better efficacy of the combination therapy as compared with the single-agent regimen. Currently, another study is underway to further investigate this in a larger group of patients.63
BIOLOGICS
Cytokine therapy, including high-dose IL-2 and interferon alfa, had long been the only first-line treatment option for patients with metastatic or unresectable RCC. Studies of high-dose IL-2 have shown an ORR of 25% and durable response in up to 11% of patients with clear-cell histology.64 Toxicities were similar to those previously observed with high-dose IL-2 treatment; the most commonly observed grade 3 toxicities were hypotension and capillary leak syndrome. IL-2 requires strict monitoring (Table 3). It is important to note that retrospective studies evaluating the safety and efficacy of using IL-2 as second-line treatment in patients previously treated with TKIs demonstrated significant toxicity without achieving partial or complete response in any of the patients.65
Prior to the advent of TKIs in the treatment of RCC, interferon alfa was a first-line treatment option for those who could not receive high-dose IL-2. It has been shown to produce response rates of approximately 20%, with maximum response seen with a higher dose range of 5 to 20 million units daily in 1 study.66,67 However, with the introduction of TKIs, which produce a higher and more durable response, interferon alfa alone is no longer recommended as a treatment option.
VEGF MONOCLONAL ANTIBODIES
Bevacizumab is a recombinant humanized monoclonal antibody that binds and neutralizes VEGF-A. Given overexpression of VEGF in RCC, the role of bevacizumab both as a single agent and in combination with interferon alfa has been investigated. In a randomized phase 2 study involving patients with cytokine-refractory disease, bevacizumab produced a 10% response rate and PFS of 4.8 months compared to patients treated with placebo.68 In the AVOREN trial, the addition of bevacizumab (10 mg/kg intravenously [IV] every 2 weeks) to interferon alfa (9 million units subcutaneously [SC] 3 times weekly) was shown to significantly increase PFS compared with interferon alfa alone (10.2 months versus 5.4 months; P = 0.0001).47,48 Adverse effects of this combination therapy include fatigue and asthenia. Additionally, hypertension, proteinuria, and bleeding occurred.
TYROSINE KINASE INHIBITORS
TKIs have largely replaced IL-2 as first-line therapy for metastatic RCC. Axitinib, pazopanib, sorafenib, and sunitinib and can be used as first-line therapy. All of the TKIs can be used as subsequent therapy.
Sunitinib
Sunitinib is an orally administered TKI that inhibits VEGF receptor (VEGFR) types 1 and 2, PDGF receptors (PDGFR) α and β, stem cell factor receptor (c-Kit), and FLT-3 and RET kinases. Motzer and colleagues52,53 compared sunitinib 50 mg daily orally for 4 weeks with 2 weeks off to the then standard of care, interferon alfa 9 million units SC 3 times weekly. Sunitinib significantly increased the overall objective response rate (47% versus 12%; P < 0.001), PFS (11 versus 5 months; P < 0.001), and overall survival (26.4 versus 21.8 months; hazard ratio [HR], 0.821). The most common side effects are diarrhea, fatigue, nausea/vomiting, anorexia, hypertension, stomatitis, and hand-foot syndrome, occurring in more than 30% of patients. Often patients will require dose reductions or temporary discontinuations to tolerate therapy. Alternative dosing strategies (eg, 50 mg dose orally daily for 2 weeks alternating with 1-week free interval) have been attempted but not prospectively evaluated for efficacy.69–71
Pazopanib
Pazopanib is an oral multi-kinase inhibitor of VEGFR types 1 and 2, PDGFR, and c-KIT. Results of a phase 3 trial comparing pazopanib (800 mg orally daily) to placebo favored the TKI, with a PFS of 9.2 months versus 4.2 months. A subset of treatment-naïve patients had a longer PFS of 11.1 versus 2.8 months and a response rate of 32% versus 4%.72 This led to a noninferiority phase 3 trial comparing pazopanib with sunitinib as first-line therapy.50 In this study, PFS was similar (8.4 versus 9.5 months; HR 1.05), and overall safety and quality-of-life endpoints favored pazopanib. Much less fatigue, stomatitis, hand-foot syndrome, and thrombocytopenia occurred with pazopanib, whereas hair color changes, weight loss, alopecia, and elevations of LFT enzymes occurred more frequently with pazopanib. Hypertension is common with the administration of pazopanib as well.
Sorafenib
Sorafenib is an orally administered inhibitor of Raf, serine/threonine kinase, VEGFR, PDGFR, FLT-3, c-Kit, and RET. The pivotal phase 3 Treatment Approaches in Renal Cancer Global Evaluation Trial (TARGET) compared sorafenib (400 mg orally twice daily) with placebo in patients who had progressed on prior cytokine-based therapy.73 A final analysis, which excluded patients who were allowed to cross over therapies, found improved overall survival times (14.3 versus 1.8 months, P = 0.029).51 Sorafenib is associated with lower rates of diarrhea, rash, fatigue, hand-foot syndrome, alopecia, hypertension, and nausea than sunitinib, although these agents have not been compared to one another.
Axitinib
Axitinib is an oral inhibitor of VEGFRs 1, 2, and 3. Results of the phase 3 AXIS trial comparing axitinib (5 mg orally twice daily) with sorafenib (400 mg orally twice daily) in patients receiving 1 prior systemic therapy showed axitinib was more active than sorafenib in improving ORR (19% versus 9%; P = 0.001) and PFS (6.7 versus 4.7 months; P < 0.001), although no difference in overall survival times was noted.74 In a subsequent phase 3 trial comparing these drugs in the first-line setting, axitinib showed a nonsignificantly higher response rate and PFS. Despite this, the National Comprehensive Cancer Network guidelines consider axitinib an acceptable first-line therapy because activity with acceptable toxicity was demonstrated (Table 2).46 The most common adverse effects of axitinib are diarrhea, hypertension, fatigue, decreased appetite, dysphonia, hypothyroidism, and upper abdominal pain.
CABOZANTINIB
Given that resistance eventually develops in most patients treated with standard treatments, including bevacizumab and TKIs, the need to evaluate the safety and efficacy of novel agents targeting VEGFR and overcoming this resistance is of vital importance. Cabozantinib is an oral small-molecule inhibitor of VEGFR, Met, and Axl, all tyrosine kinases implicated in metastatic RCC. Overexpression of Met and Axl, which occurs as a result of inactivation of the VHL gene, is associated with a poor prognosis in patients with RCC. In a
randomized, open label, phase 3 trial of cabozantinib versus everolimus in advanced RCC, Choueiri and colleagues75 compared the efficacy of cabozantinib with everolimus in patients with metastatic RCC who had progressed on previous VEGFR-targeted therapies. In this study, 658 patients were randomly assigned to receive cabozantinib (60 mg orally daily) or everolimus (10 mg orally daily). Results of the study found that PFS was longer with cabozantinib in patients who had previously been treated with other TKIs (median PFS of 7.4 months versus 3.8 months; HR 0.58), corresponding to a 42% reduction in the rate of disease progression or death. The most common grade 3 and 4 toxicities seen with cabozantinib were similar to its class effect and consisted of hypertension, diarrhea, and fatigue. In the final analysis of the data, the median overall survival was 21.4 months (95% confidence interval [CI] 18.7–not estimable) with cabozantinib and 16.5 months (95% CI 14.7 to 18.8) with everolimus (HR 0.66 [95% CI 0.53 to 0.83]; P = 0.00026). The median follow-up for overall survival and safety was 18.7 months. These results highlight the importance of cabozantinib as a first line option in treatment of previously treated patients with advanced RCC.76
MTOR INHIBITORS
The mTOR inhibitors, temsirolimus and everolimus, are also approved for the treatment of metastatic or advanced RCC. These drugs block mTOR’s phosphorylation and subsequent translation of mRNA to inhibit cell proliferation, cell growth, and angiogenesis.77 Temsirolimus can be used as first-line therapy for patients with a poor prognosis, and everolimus is appropriate as a subsequent therapy.
Temsirolimus is an intravenous prodrug of rapamycin. It was the first of the class to be approved for metastatic RCC for treatment-naïve patients with a poor prognosis (ie, at least 3 of 6 predictors of poor survival based on MSKCC model).54 The pivotal ARCC trial compared temsirolimus (25 mg IV weekly) alone, interferon alfa (3 million units SC 3 times weekly) alone, or the combination (temsirolimus 15 mg IV weekly plus interferon alfa 6 million units SC 3 times weekly). In this trial, temsirolimus monotherapy produced a significantly longer overall survival time than interferon alfa alone (10.9 versus 7.3 months; P = 0.008) and improved PFS time when administered alone or in combination with interferon alfa (3.8 and 3.7 months, respectively, versus 1.9 months). Because no real efficacy advantage of the combination was demonstrated, temsirolimus is administered alone. The most common adverse effects of temsirolimus are asthenia, rash, anemia, nausea, anorexia, pain, and dyspnea. Additionally, hyperglycemia, hyper-cholesterolemia, and hyperlipidemia occur with these agents. Noninfectious pneumonitis is a rare but often fatal complication.
Everolimus is also an orally administered derivative of rapamycin that is approved for use after failure of VEGF-targeted therapies. The results of the landmark trial RECORD-1 demonstrated that everolimus (10 mg orally daily) is effective at prolonging PFS (4 versus 1.9 months; P < 0.001) when compared with best supportive care, a viable treatment option at the time of approval.78 The most common adverse effects of everolimus are stomatitis, rash, fatigue, asthenia, and diarrhea. As with temsirolimus, elevations in glucose, lipids, and triglycerides and noninfectious pneumonitis can occur.
TKI + MTOR INHIBITOR
Lenvatinib is also a small molecule targeting multiple tyrosine kinases, primarily VEGF2. Combined with the mTOR inhibitor everolimus, it has been shown to be an effective regimen in patients with metastatic RCC who have failed other therapies. In a randomized phase 2 study involving patients with advanced or metastatic clear-cell RCC, patients were randomly assigned to receive either lenvatinib (24 mg/day), everolimus (10 mg/day), or lenvatinib plus everolimus (18 mg/day and 5 mg/day, respectively). Patients received the treatment continuously on a 28-day cycle until progression or inability to tolerate toxicity. Patients in the lenvatinib plus everolimus arm had median PFS of 14.6 months (95% CI 5.9 to 20.1) versus 5.5 months (95% CI 3.5 to 7.1) with everlolimus alone (HR 0.40 [95% CI 0.24 to 0.68]; P = 0.0005). PFS with levantinib alone was 7.4 months (95% CI 5.6 to 10.20; HR 0.66 [95% CI 0.30 to 1.10]; P = 0.12). In addition, PFS with levantinib alone was significantly prolonged in comparison with everolimus alone (HR 0.61 [95% CI 0.38 to 0.98]; P = 0.048). Grade 3 or 4 toxicity were less frequent in the everolimus only arm and the most common grade 3 or 4 toxicity in the lenvatinib plus everolimus arm was diarrhea. The results of this study show that the combination of lenvatinib plus everolimus is an acceptable second-line option for treatment of patients with advanced or metastatic RCC.55
CASE CONTINUED
The patient is initially started on pazopanib and tolerates the medication well, with partial response to the treatment. However, on restaging scans he is noted to have small bowel perforation. Pazopanib is discontinued until the patient has a full recovery. He is then started on everolimus. Restaging scans done 3 months after starting everolimus demonstrate disease progression.
• What is the appropriate next step in treatment?
PD1 BLOCKADE
Programmed death 1 (PD-1) protein is a T-cell inhibitory receptor with 2 ligands, PD-L1 and PD-L2. PD-L1 is expressed on many tumors. Blocking the interaction between PD-1 and PD-L1 by anti-PD-1 humanized antibodies potentiates a robust immune response and has been a breakthrough in the field of cancer immunotherapy.79 Previous studies have demonstrated that overexpression of PD-L1 leads to worse outcomes and poor prognosis in patients with RCC.80 Nivolumab, a fully human IgG4 PD-1 immune checkpoint inhibitor, blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2. In a randomized, open-label, phase 3 study comparing nivolumab with everolimus in patients with RCC who had previously undergone treatment with other standard therapies, Motzer and colleagues81 demonstrated a longer overall survival time and fewer adverse effects with nivolumab. In this study, 821 patients with clear-cell RCC were randomly assigned to receive nivolumab (3 mg/kg of body weight IV every 2 weeks) or everolimus (10 mg orally once daily). The median overall survival time with nivolumab was 25 months versus 19.6 months with everolimus (P < 0.0148). Nineteen percent of patients receiving nivolumab experienced grade 3 or 4 toxicities, with fatigue being the most common adverse effect. Grade 3 or 4 toxicities were observed in 37% of patients treated with everolimus, with anemia being the most common. Based on the results of this trial, on November 23, 2015, the U.S. Food and Drug Administration approved nivolumab to treat patients with metastatic RCC who have received a prior antiangiogenic therapy.
CASE CONCLUSION
Both TKI and mTOR inhibitor therapy fail, and the patient is eligible for third-line therapy. Because of his previous GI perforation, other TKIs are not an option. The patient opts for enrollment in hospice due to declining performance status. For other patients in this situation with a good performance status, nivolumab would be a reasonable option.
FUTURE DIRECTIONS
With the approval of nivolumab, multiple treatment options are now available for patients with metastatic or unresectable RCC. Development of other PD-1 inhibitors and immunotherapies as well as multi-targeted TKIs will only serve to expand treatment options for these patients. Given the aggressive course and poor prognosis of non-clear cell renal cell tumors and those with sarcomatoid features, evaluation of systemic and targeted therapies for these subtypes should remain active areas of research and investigation.
INTRODUCTION
Renal cell carcinoma (RCC) is the most common malignancy arising in the kidney, comprising 90% of all renal tumors.1 Approximately 55,000 new RCC cases are diagnosed each year.2 Patients with RCC are often asymptomatic, and most cases are discovered as incidental findings on abdominal imaging performed during evaluation of nonrenal complaints. Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20%.2 Advanced RCC is resistant to conventional chemotherapy and radiotherapy, and outcomes for patients with metastatic or unresectable RCC remain poor. However, the recent development of new therapeutic modalities that target tumor molecular pathways has expanded the treatment options for these patients and changed the management of RCC.
EPIDEMIOLOGY AND CLASSIFICATION
Median age at diagnosis in the United States is 64 years. Men have a higher incidence of RCC than women, with the highest incidence seen in American Indian and Alaska Native men (30.1 per 100,000 population). Genetic syndromes account for 2% to 4% of all RCCs.2 Risk factors for RCC include smoking, hypertension, obesity, and acquired cystic kidney disease that is associated with end-stage renal failure.3 Longer duration of tobacco use is associated with a more aggressive course.
The 2004 World Health Organization (WHO) classification of renal tumors summarizes the previous classification systems (including the Heidelberg and Mainz classification systems) to describe different categories of RCC based on histologic and molecular genetics characteristics.2 Using the WHO classification criteria, RCC comprises 90% of all renal tumors, with clear cell being the most common type (80%).2 Other types of renal tumors include papillary, chromophobe, oncocytoma, and collecting-duct or Bellini duct tumors. Approximately 3% to 5% of tumors are unclassified. Oncocytomas are generally considered benign, and chromophobe tumors typically have an indolent course and rarely metastasize. Sarcomatoid differentiation can be seen in any histologic type and is associated with a worse prognosis. While different types of tumors may be seen in the kidney (such as transitional cell or lymphomas), the focus of this review is the primary malignancies of the renal parenchyma.
FAMILIAL SYNDROMES
Several genetic syndromes have been identified by studying families with inherited RCC. Among these, von Hippel-Lindau (VHL) gene mutation is the most commonly found inherited genetic defect. Table 1 summarizes the incidence of gene mutations and the corresponding histologic appearance of the most common sporadic and hereditary RCCs.4
VHL disease is an autosomal dominant familial syndrome. Patients with this mutation are at higher risk for developing RCC (clear cell histology), retinal angiomas, pheochromocytomas, as well as hemangioblastomas of the central nervous system (CNS).4 Of all the genetic mutations seen in RCC, the somatic mutation in the VHL tumor-suppressor gene is by far the most common.5 VHL targets hypoxia–inducible factor-1 alpha (HIF-α) for ubiquitination and subsequent degradation, which has been shown to suppress the growth of clear-cell RCC in mouse models.6–8 HIF expression under hypoxic conditions leads to activation of a number of genes important in blood vessel development, cell proliferation, and glucose metabolism, including vascular endothelial growth factor (VEGF), erythropoietin, platelet-derived growth factor beta (PDGF-β), transforming growth factor alpha (TGF-α), and glucose transporter-1 (GLUT-1). Mutation in the VHL gene prevents degradation of the HIF-α protein, thereby leading to increased expression of these downstream proteins, including MET and Axl. The upregulation of these angiogenic factors is thought to be the underlying process for increased vascularity of CNS hemangioblastomas and clear-cell renal tumors in VHL disease.4–8
Other less common genetic syndromes seen in hereditary RCC include hereditary papillary RCC, hereditary leiomyomatosis, and Birt-Hogg-Dubé (BHD) syndrome.9 In hereditary papillary RCC, the MET gene is mutated. BHD syndrome is a rare, autosomal dominant syndrome characterized by hair follicle hamartomas of the face and neck. About 15% of patients have multiple renal tumors, the majority of which are of the chromophobe or mixed chromophobe-oncocytoma histology. The BHD gene encodes the protein folliculin, which is thought to be a tumor-suppressor gene.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 74-year-old man who works as an airplane mechanic repairman presents to the emergency department with sudden worsening of chronic right upper arm and shoulder pain after lifting a jug of orange juice. He does not have a significant past medical history and initially thought that his pain was due to a work-related injury. Upon initial evaluation in the emergency department he is found to have a fracture of his right humerus. Given that the fracture appears to be pathologic, further work-up is recommended.
• What are common clinical presentations of RCC?
Most patients are asymptomatic until the disease becomes advanced. The classic triad of flank pain, hematuria, and palpable abdominal mass is seen in approximately 10% of patients with RCC, partly because of earlier detection of renal masses by imaging performed for other purposes.10 Less frequently, patients present with signs or symptoms of metastatic disease such as bone pain or fracture (as seen in the case patient), painful adenopathy, and pulmonary symptoms related to mediastinal masses. Fever, weight loss, anemia, and/or varicocele often occur in young patients (≤ 46 years) and may indicate the presence of a hereditary form of the disease. Patients may present with paraneoplastic syndromes seen as abnormalities on routine blood work. These can include polycythemia or elevated liver function tests (LFTs) without the presence of liver metastases (known as Stauffer syndrome), which can be seen in localized renal tumors. Nearly half (45%) of patients present with localized disease, 25% present with locally advanced disease, and 30% present with metastatic disease.11 Bone is the second most common site of distant metastatic spread (following lung) in patients with advanced RCC.
• What is the approach to initial evaluation for a patient with suspected RCC?
Initial evaluation consists of a physical exam, laboratory tests including complete blood count (CBC) and comprehensive metabolic panel (calcium, serum creatinine, LFTs, lactate dehydrogenase [LDH], and urinalysis), and imaging. Imaging studies include computed tomography (CT) scan with contrast of the abdomen and pelvis or magnetic resonance imaging (MRI) of the abdomen and chest imaging. A chest radiograph may be obtained, although a chest CT is more sensitive for the presence of pulmonary metastases. MRI can be used in patients with renal dysfunction to evaluate the renal vein and inferior vena cava (IVC) for thrombus or to determine the presence of local invasion.12 Although bone and brain are common sites for metastases, routine imaging is not indicated unless the patient is symptomatic. The value of positron emission tomography in RCC remains undetermined at this time.
Staging is done according to the American Joint Committee on Cancer (AJCC) staging classification for RCC; the Figure summarizes the staging and 5-year survival data based on this classification scheme.4,13
J Med 2005;353:2477–90.)
LIMITED-STAGE DISEASE
• What are the therapeutic options for limited-stage disease?
For patients with nondistant metastases, or limited-stage disease, surgical intervention with curative intent is considered. Convention suggests considering definitive surgery for patients with stage I and II disease, select patients with stage III disease with pathologically enlarged retroperitoneal lymph nodes, patients with IVC and/or cardiac atrium involvement of tumor thrombus, and patients with direct extension of the renal tumor into the ipsilateral adrenal gland if there is no evidence of distant disease. While there may be a role for aggressive surgical intervention in patients with distant metastatic disease, this topic will not be covered in this review.
SURGICAL INTERVENTION
Once patients are determined to be appropriate candidates for surgical removal of a renal tumor, the urologist will perform either a radical nephrectomy or a nephron-sparing nephrectomy, also called a partial nephrectomy. The urologist will evaluate the patient based on his or her body habitus, the location of the tumor, whether multiple tumors in one kidney or bilateral tumors are present, whether the patient has a solitary kidney or otherwise impaired kidney function, and whether the patient has a history of a hereditary syndrome involving kidney cancer as this affects the risk of future kidney tumors.
A radical nephrectomy is surgically preferred in the presence of the following factors: tumor larger than 7 cm in diameter, a more centrally located tumor, suspicion of lymph node involvement, tumor involvement with renal vein or IVC, and/or direct extension of the tumor into the ipsilateral adrenal gland. Nephrectomy involves ligation of the vascular supply (renal artery and vein) followed by removal of the kidney and surrounding Gerota’s fascia. The ipsilateral adrenal gland is removed if there is a high-risk for or presence of invasion of the adrenal gland. Removal of the adrenal gland is not standard since the literature demonstrates there is less than a 10% chance of solitary, ipsilateral adrenal gland involvement of tumor at the time of nephrectomy in the absence of high-risk features, and a recent systematic review suggests that the chance may be as low as 1.8%.14 Preoperative factors that correlated with adrenal involvement included upper pole kidney location, renal vein thrombosis, higher T stage (T3a and greater), multifocal tumors, and evidence for distant metastases or lymph node involvement. Lymphadenectomy previously had been included in radical nephrectomy but now is performed selectively. Radical nephrectomy may be performed as
either an open or laparoscopic procedure, the latter of which may be performed robotically.15 Oncologic outcomes appear to be comparable between the 2 approaches, with equivalent 5-year cancer-specific survival (91% with laparoscopic versus 93% with open approach) and recurrence-free survival (91% with laparoscopic versus 93% with open approach).16 The approach ultimately is selected based on provider- and patient-specific input, though in all cases the goal is to remove the specimen intact.16,17
Conversely, a nephron-sparing approach is preferred for tumors less than 7 cm in diameter, for patients with a solitary kidney or impaired renal function, for patients with multiple small ipsilateral tumors or with bilateral tumors, or for radical nephrectomy candidates with comorbidities for whom a limited intervention is deemed to be a lower-risk procedure. A nephron-sparing procedure may also be performed open or laparoscopically. In nephron-sparing procedures, the tumor is removed along with a small margin of normal parenchyma.15
In summary, the goal of surgical intervention is curative intent with removal of the tumor while maintaining as much residual renal function as possible to limit long-term morbidity of chronic kidney disease and associated cardiovascular events.18 Oncologic outcomes for radical nephrectomy and partial nephrectomy are similar. In one study, overall survival was slightly lower in the partial nephrectomy cohort, but only a small number of the deaths were due to RCC.19
ADJUVANT THERAPY
Adjuvant systemic therapy currently has no role following nephrectomy for RCC because no systemic therapy has been able to reduce the likelihood of relapse. Randomized trials of cytokine therapy (eg, interferon, interleukin 2) or tyrosine kinase inhibitors (TKIs; eg, sorafenib, sunitinib) with observation alone in patients with locally advanced completely resected RCC have shown no delay in time to relapse or improvement of survival with adjuvant therapy.20 Similarly, adjuvant radiation therapy has not shown benefit even in patients with nodal involvement or incomplete resection.21 Therefore, observation remains the standard of care after nephrectomy.
RENAL TUMOR ABLATION
For patients who are deemed not to be surgical candidates due to age, comorbidities, or patient preference and who have tumors less than 4 cm in size (stage I tumors), ablative techniques may be considered. The 2 most well-studied and effective techniques at present are cryoablation and radiofrequency ablation (RFA). Microwave ablation may be an option in some facilities, but the data in RCC are limited. An emerging ablative technique under investigation is irreversible electroporation. At present, the long-term efficacy of all ablative techniques is unknown.
Patient selection is undertaken by urologists and interventional radiologists who evaluate the patient with ultrasound, CT, and/or MRI to determine the location and size of the tumor and the presence or absence of metastatic disease. A pretreatment biopsy is recommended to document the histology of the lesion to confirm a malignancy and to guide future treatment for recurrent or metastatic disease. Contraindications to the procedure include the presence of metastatic disease, a life expectancy of less than 1 year, general medical instability, or uncorrectable coagulopathy due to increased risk of bleeding complications. Tumors in close proximity to the renal hilum or collecting system are a contraindication to the procedure because of the risk for hemorrhage or damage to the collecting system. The location of the tumor in relation to the vasculature is also important to maximize efficacy because the vasculature acts as a “heat sink,” causing dissipation of the thermal energy. Occasionally, stenting of the proximal ureter due to upper tumor location is necessary to prevent thermal injury that could lead to urine leaks.
Selection of the modality to be used primarily depends on operator comfort, which translates to good patient outcomes, such as better cancer control and fewer complications. Cryoablation and RFA have both demonstrated good clinical efficacy and cancer control of 89% and 90%, respectively, with comparable complication rates.22 There have been no studies performed directly comparing the modalities.
Cryoablation
Cryoablation is performed through the insertion of a probe into the tumor, which may be done through a surgical or percutaneous approach. Once the probe is in place, a high- pressure gas (argon, nitrogen) is passed through the probe and upon entering a low pressure region the gas cools. The gas is able to cool to temperatures as low as –185°C. The tissue is then rewarmed through the use of helium, which conversely warms when entering a low pressure area. The process of freezing followed by rewarming subsequently causes cell death/tissue destruction through direct cell injury from cellular dehydration and vascular injury. Clinically, 2 freeze-thaw cycles are used to treat a tumor.23,24
RFA
Radiofrequency ablation, or RFA, targets tumors via an electrode placed within the mass that produces intense frictional heat from medium-frequency alternating current (approximately 500 kHz) produced by a connected generator that is grounded on the patient. The thermal energy created causes coagulative necrosis. Due to the reliance on heat for tumor destruction, central lesions are less amenable to this approach because of the “heat sink” effect from the hilum.24
Microwave Ablation
Microwave ablation, like RFA, relies on the generation of frictional heat to cause cell death by coagulative necrosis. In this case, the friction is created through the activation of water molecules; because of the different thermal kinetics involved with microwave ablation, the “heat sink” effect is minimized when treatment is employed near large vessels, in comparison to RFA.24 The data on this mechanism of ablation are still maturing, with varied outcomes thus far. One study demonstrated outcomes comparable to RFA and cryoablation, with cancer-specific survival of 97.8% at 3 years.25 However, a study by Castle and colleagues26 demonstrated higher recurrence rates. The overarching impediment to widespread adoption of microwave ablation is inconclusive data gleaned from studies with small numbers of patients with limited follow up. The role of this modality will need to be revisited.
Irreversible Electroporation
Irreversible electroporation (IRE) is under investigation. IRE is a non-thermal ablative technique that employs rapid electrical pulses to create pores in cell membranes, leading to cell death. The postulated benefits of IRE include the lack of an effect from “heat sinks” and less collateral damage to the surrounding tissues, when compared with the thermal modalities. In a human phase 1 study of patients undergoing IRE prior to immediate surgical resection, the procedure appeared feasible and safe.27 Significant concerns for this method of ablation possibly inducing cardiac arrhythmias, and the resultant need for sedation with neuromuscular blockade and associated electrocardiography monitoring, may impede its implementation in nonresearch settings.24
ACTIVE SURVEILLANCE
Due to the more frequent use of imaging for various indications, there has been an increase in the discovery of small renal masses (SRM); 85% of RCC that present in an asymptomatic or incidental manner are tumors under 4 cm in diameter.28,29 The role of active surveillance is evolving, but is primarily suggested for patients who are not candidates for more aggressive intervention based on comorbidities. A recent prospective, nonrandomized analysis of data from the Delayed Intervention and Surveillance for Small Renal Masses (DISSRM) registry evaluated outcomes for patients with SRM looking at primary intervention compared with active surveillance.30 The primary intervention selected was at the discretion of the provider; treatments included partial nephrectomy, RFA, and cryoablation, and active surveillance patients were followed with imaging every 6 months. Progression of SRM, with recommendation for delayed intervention, was defined as a growth rate of mass greater than 0.5 cm/year, size greater than 4 cm, or hematuria. Thirty-six of 158 patients on active surveillance met criteria for progression; 21 underwent delayed intervention. Of note, even the patients who progressed but did not undergo delayed intervention did not develop metastatic disease during the follow-up interval. With a median follow-up of 2 years, cancer-specific survival was noted to be 99% and 100% at 5 years for primary intervention and active surveillance, respectively. Overall survival at 2 years for primary intervention was 98% and 96% for active surveillance; at 5 years, the survival rates were 92% and 75% (P = 0.06). Of note, 2 patients in the primary intervention arm died of RCC, while none in the active surveillance arm died. As would be expected, active surveillance patients were older, had a worse performance status, and had more comorbidities. Interestingly, 40% of patients enrolled selected active surveillance as their preferred management for SRM. The DISSRM results were consistent with data from the Renal Cell Consortium of Canada and other retrospective reviews.31–33
• What is the approach to follow-up after treatment of localized RCC?
After a patient undergoes treatment for a localized RCC, the goal is to optimize oncologic outcomes, monitor for treatment sequelae, such as renal failure, and focus on survivorship. At this time, there is no consensus in the literature or across published national and international guidelines with regards to the appropriate schedule for surveillance to achieve these goals. In principle, the greatest risk for recurrence occurs within the first 3 years, so many guidelines focus on this timeframe. Likewise, the route of spread tends to be hematogenous, so patients present with pulmonary, bone, and brain metastases, in addition to local recurrence within the renal bed. Symptomatic recurrences often are seen
with bone and brain metastases, and thus bone scans and brain imaging are not listed as part of routine surveillance protocols in asymptomatic patients. Although there is inconclusive evidence that surveillance protocols improve outcomes in RCC, many professional associations have outlined recommendations based on expert opinion.34 The American Urological Association released guidelines in 2013 and the National Comprehensive Cancer Network (NCCN) released their most recent set of guidelines in 2016.21,35 These guidelines use TNM staging to risk-stratify patients and recommend follow-up.
METASTATIC DISEASE
CASE CONTINUED
CT scan with contrast of the chest, abdomen, and pelvis as well as bone scan are done. CT of the abdomen and pelvis demonstrates a 7.8-cm left renal mass arising from the lower pole of the left kidney. Paraesophageal lymphadenopathy and mesenteric nodules are also noted. CT of the chest demonstrates bilateral pulmonary emboli. Bone scan is significant for increased activity related to the pathological fracture involving the right humerus. The patient undergoes surgery to stabilize the pathologic fracture of his humerus. He is diagnosed with metastatic RCC (clear cell histology) and undergoes palliative debulking nephrectomy.
• How is prognosis defined for metastatic RCC?
PROGNOSTIC MODELS
Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates for stage T1 and T2 disease approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20%.13 Approximately 30% of patients have metastatic disease at diagnosis, and about one-third of patients who have undergone treatment for localized disease experience relapse.36,37 Common sites of metastases include lung, lymph nodes, bone, liver, adrenal gland, and brain.
Prognostic scoring systems have been developed to define risk groups and assist with determining appropriate therapy in the metastatic setting. The most widely used validated prognostic factor model is that from the Memorial Sloan-Kettering Cancer Center (MSKCC), which was developed using a multivariate analysis derived from data of patients enrolled in clinical trials and treated with interferon alfa.38 The factors included in the MSKCC model are Karnofsky performance status less than 80, time from diagnosis to treatment with interferon alfa less than 12 months, hemoglobin level less than lower limit of laboratory’s reference range, LDH level greater than 1.5 times the upper limit of laboratory’s reference range, and corrected serum calcium level greater than 10 mg/dL. Risk groups are categorized as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors).39 Median survival for favorable-, intermediate-, and poor-risk patients was 20, 10, and 4 months, respectively.40
Another prognostic model, the International Metastatic RCC Database Consortium, or Heng, model was developed to evaluate prognosis in patients treated with VEGF-targeted therapy.41 This model was developed from a retrospective study of patients treated with sunitinib, sorafenib, and bevacizumab plus interferon alfa or prior immunotherapy. Prognostic factors in this model include 4 of the 5 MSKCC risk factors (hemoglobin level, corrected serum calcium level, Karnofsky performance status, and time to initial diagnosis). Additionally, this model includes both absolute neutrophil and platelet counts greater than the upper limit of normal. Risk groups are identified as favorable (0 risk factors), intermediate (1 to 2 risk factors), and poor (3 or more risk factors). Median survival for favorable-, intermediate-, and poor-risk patients was not reached, 27 months, and 8.8 months, respectively. The University of California, Los Angeles scoring algorithm to predict survival after nephrectomy and immunotherapy (SANI) in patients with metastatic RCC is another prognostic model that can be used. This simplified scoring system incorporates lymph node status, constitutional symptoms, metastases location, histology, and thyroid stimulating hormone (TSH) level.42
The role of debulking or cytoreductive nephrectomy in treatment of metastatic RCC is well established. Large randomized studies have demonstrated a statistically significant median survival benefit for patients undergoing nephrectomy plus interferon alfa therapy compared with patients treated with interferon alfa alone (13.6 months versus 7.8 months, respectively).43 The role of cytoreductive nephrectomy in combination with antiangiogenic agents is less clear. While a retrospective study investigating outcomes of patients with metastatic RCC receiving anti-VEGF agents showed a prolonged survival with nephrectomy, results of large randomized trials are not yet available.44,45 Patients with lung-only metastases, good prognostic features, and a good performance status are historically the most likely to benefit from cytoreductive surgery.
CASE CONTINUED
Based on the MSKCC prognostic factor model, the patient is considered to be in the intermediate-risk group (Karnofsky performance status of 80, calcium 9.5 mg/dL, LDH 204 U/L, hemoglobin 13.6 g/dL). He is started on treatment for his bilateral pulmonary emboli and recovers well from orthopedic surgery as well as palliative debulking nephrectomy.
• What is the appropriate first-line therapy in managing this patient’s metastatic disease?
Several approaches to systemic therapy for advanced RCC have been taken based on the histologic type of the tumor. Clear-cell is by far the predominant histologic type in RCC. Several options are available as first-line treatment for patients with metastatic clear-cell RCC (Table 2).46–54 These include biologic agents such as high-dose interleukin-2 (IL-2) immune therapy, as well as targeted therapies including TKIs and anti-VEGF antibodies. The mammalian target of rapamycin (mTOR) inhibitor temsirolimus is recommended as first-line therapy in patients with poor prognosis only. Second-line therapies for clear-cell RCC following antiangiogenic therapy include TKIs, mTOR inhibitors, nivolumab (PD-1 inhibitor), and the combination of the TKI lenvatinib and mTOR inhibitor everolimus.55 In addition, after initial cytokine therapy, TKIs, temsirolimus, and the anti-VEGF antibody bevacizumab are other treatment options available to patients. Best supportive care should always be provided along with initial and subsequent therapies. Clinical trials are also an appropriate choice as first-line or subsequent therapies. All of these therapies require periodic monitoring to prevent and quickly treat adverse effects. Table 3 lists recommended monitoring parameters for each of these agents.56
Based on several studies, TKIs seem to be less effective in patients with non–clear-cell type histology.57,58 In these patients, risk factors can guide therapy. In the ASPEN trial, where 108 patients were randomly assigned to everolimus or sunitinib, patients in the good- and intermediate-risk groups had longer overall and median progression-free survival (PFS) on sunitinib (8.3 months versus 5.3 months, respectively). However, those in the poor-risk group had a longer median overall survival with everolimus.59 Given that the role of targeted therapies in non–clear-cell RCCs is less well established, enrollment in clinical trials should be considered as a first-line treatment option.21
Sarcomatoid features can be observed in any of the histologic types of RCC, and RCC with these features has an aggressive course and a poor prognosis. Currently, there is no standard therapy for treatment of patients with metastatic or unresectable RCC with sarcomatoid features.60 Chemotherapeutic regimens used for soft tissue sarcomas, including a trial of ifosfamide and doxorubicin, did not show any objective response.61 A small trial of 10 patients treated with doxorubicin and gemcitabine resulted in complete response in 2 patients and partial response in 1 patient.62
Enrollment in a clinical trial remains a first-line treatment option for these patients. More recently, a phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid (39 patients) and/or poor-risk (33 patients) metastatic RCC showed overall response rates (ORR) of 26% and 24%, respectively. A higher clinical benefit rate (defined as ORR plus stable disease) was seen in patients with tumors containing more than 10% sarcomatoid histology, as compared with patients whose tumors contained less than 10% sarcomatoid histology. Neutropenia (n = 20), anemia (n = 10), and fatigue (n = 7) were the most common grade 3 toxicities seen in all the patients. Although this was a small study, the results showed a trend towards better efficacy of the combination therapy as compared with the single-agent regimen. Currently, another study is underway to further investigate this in a larger group of patients.63
BIOLOGICS
Cytokine therapy, including high-dose IL-2 and interferon alfa, had long been the only first-line treatment option for patients with metastatic or unresectable RCC. Studies of high-dose IL-2 have shown an ORR of 25% and durable response in up to 11% of patients with clear-cell histology.64 Toxicities were similar to those previously observed with high-dose IL-2 treatment; the most commonly observed grade 3 toxicities were hypotension and capillary leak syndrome. IL-2 requires strict monitoring (Table 3). It is important to note that retrospective studies evaluating the safety and efficacy of using IL-2 as second-line treatment in patients previously treated with TKIs demonstrated significant toxicity without achieving partial or complete response in any of the patients.65
Prior to the advent of TKIs in the treatment of RCC, interferon alfa was a first-line treatment option for those who could not receive high-dose IL-2. It has been shown to produce response rates of approximately 20%, with maximum response seen with a higher dose range of 5 to 20 million units daily in 1 study.66,67 However, with the introduction of TKIs, which produce a higher and more durable response, interferon alfa alone is no longer recommended as a treatment option.
VEGF MONOCLONAL ANTIBODIES
Bevacizumab is a recombinant humanized monoclonal antibody that binds and neutralizes VEGF-A. Given overexpression of VEGF in RCC, the role of bevacizumab both as a single agent and in combination with interferon alfa has been investigated. In a randomized phase 2 study involving patients with cytokine-refractory disease, bevacizumab produced a 10% response rate and PFS of 4.8 months compared to patients treated with placebo.68 In the AVOREN trial, the addition of bevacizumab (10 mg/kg intravenously [IV] every 2 weeks) to interferon alfa (9 million units subcutaneously [SC] 3 times weekly) was shown to significantly increase PFS compared with interferon alfa alone (10.2 months versus 5.4 months; P = 0.0001).47,48 Adverse effects of this combination therapy include fatigue and asthenia. Additionally, hypertension, proteinuria, and bleeding occurred.
TYROSINE KINASE INHIBITORS
TKIs have largely replaced IL-2 as first-line therapy for metastatic RCC. Axitinib, pazopanib, sorafenib, and sunitinib and can be used as first-line therapy. All of the TKIs can be used as subsequent therapy.
Sunitinib
Sunitinib is an orally administered TKI that inhibits VEGF receptor (VEGFR) types 1 and 2, PDGF receptors (PDGFR) α and β, stem cell factor receptor (c-Kit), and FLT-3 and RET kinases. Motzer and colleagues52,53 compared sunitinib 50 mg daily orally for 4 weeks with 2 weeks off to the then standard of care, interferon alfa 9 million units SC 3 times weekly. Sunitinib significantly increased the overall objective response rate (47% versus 12%; P < 0.001), PFS (11 versus 5 months; P < 0.001), and overall survival (26.4 versus 21.8 months; hazard ratio [HR], 0.821). The most common side effects are diarrhea, fatigue, nausea/vomiting, anorexia, hypertension, stomatitis, and hand-foot syndrome, occurring in more than 30% of patients. Often patients will require dose reductions or temporary discontinuations to tolerate therapy. Alternative dosing strategies (eg, 50 mg dose orally daily for 2 weeks alternating with 1-week free interval) have been attempted but not prospectively evaluated for efficacy.69–71
Pazopanib
Pazopanib is an oral multi-kinase inhibitor of VEGFR types 1 and 2, PDGFR, and c-KIT. Results of a phase 3 trial comparing pazopanib (800 mg orally daily) to placebo favored the TKI, with a PFS of 9.2 months versus 4.2 months. A subset of treatment-naïve patients had a longer PFS of 11.1 versus 2.8 months and a response rate of 32% versus 4%.72 This led to a noninferiority phase 3 trial comparing pazopanib with sunitinib as first-line therapy.50 In this study, PFS was similar (8.4 versus 9.5 months; HR 1.05), and overall safety and quality-of-life endpoints favored pazopanib. Much less fatigue, stomatitis, hand-foot syndrome, and thrombocytopenia occurred with pazopanib, whereas hair color changes, weight loss, alopecia, and elevations of LFT enzymes occurred more frequently with pazopanib. Hypertension is common with the administration of pazopanib as well.
Sorafenib
Sorafenib is an orally administered inhibitor of Raf, serine/threonine kinase, VEGFR, PDGFR, FLT-3, c-Kit, and RET. The pivotal phase 3 Treatment Approaches in Renal Cancer Global Evaluation Trial (TARGET) compared sorafenib (400 mg orally twice daily) with placebo in patients who had progressed on prior cytokine-based therapy.73 A final analysis, which excluded patients who were allowed to cross over therapies, found improved overall survival times (14.3 versus 1.8 months, P = 0.029).51 Sorafenib is associated with lower rates of diarrhea, rash, fatigue, hand-foot syndrome, alopecia, hypertension, and nausea than sunitinib, although these agents have not been compared to one another.
Axitinib
Axitinib is an oral inhibitor of VEGFRs 1, 2, and 3. Results of the phase 3 AXIS trial comparing axitinib (5 mg orally twice daily) with sorafenib (400 mg orally twice daily) in patients receiving 1 prior systemic therapy showed axitinib was more active than sorafenib in improving ORR (19% versus 9%; P = 0.001) and PFS (6.7 versus 4.7 months; P < 0.001), although no difference in overall survival times was noted.74 In a subsequent phase 3 trial comparing these drugs in the first-line setting, axitinib showed a nonsignificantly higher response rate and PFS. Despite this, the National Comprehensive Cancer Network guidelines consider axitinib an acceptable first-line therapy because activity with acceptable toxicity was demonstrated (Table 2).46 The most common adverse effects of axitinib are diarrhea, hypertension, fatigue, decreased appetite, dysphonia, hypothyroidism, and upper abdominal pain.
CABOZANTINIB
Given that resistance eventually develops in most patients treated with standard treatments, including bevacizumab and TKIs, the need to evaluate the safety and efficacy of novel agents targeting VEGFR and overcoming this resistance is of vital importance. Cabozantinib is an oral small-molecule inhibitor of VEGFR, Met, and Axl, all tyrosine kinases implicated in metastatic RCC. Overexpression of Met and Axl, which occurs as a result of inactivation of the VHL gene, is associated with a poor prognosis in patients with RCC. In a
randomized, open label, phase 3 trial of cabozantinib versus everolimus in advanced RCC, Choueiri and colleagues75 compared the efficacy of cabozantinib with everolimus in patients with metastatic RCC who had progressed on previous VEGFR-targeted therapies. In this study, 658 patients were randomly assigned to receive cabozantinib (60 mg orally daily) or everolimus (10 mg orally daily). Results of the study found that PFS was longer with cabozantinib in patients who had previously been treated with other TKIs (median PFS of 7.4 months versus 3.8 months; HR 0.58), corresponding to a 42% reduction in the rate of disease progression or death. The most common grade 3 and 4 toxicities seen with cabozantinib were similar to its class effect and consisted of hypertension, diarrhea, and fatigue. In the final analysis of the data, the median overall survival was 21.4 months (95% confidence interval [CI] 18.7–not estimable) with cabozantinib and 16.5 months (95% CI 14.7 to 18.8) with everolimus (HR 0.66 [95% CI 0.53 to 0.83]; P = 0.00026). The median follow-up for overall survival and safety was 18.7 months. These results highlight the importance of cabozantinib as a first line option in treatment of previously treated patients with advanced RCC.76
MTOR INHIBITORS
The mTOR inhibitors, temsirolimus and everolimus, are also approved for the treatment of metastatic or advanced RCC. These drugs block mTOR’s phosphorylation and subsequent translation of mRNA to inhibit cell proliferation, cell growth, and angiogenesis.77 Temsirolimus can be used as first-line therapy for patients with a poor prognosis, and everolimus is appropriate as a subsequent therapy.
Temsirolimus is an intravenous prodrug of rapamycin. It was the first of the class to be approved for metastatic RCC for treatment-naïve patients with a poor prognosis (ie, at least 3 of 6 predictors of poor survival based on MSKCC model).54 The pivotal ARCC trial compared temsirolimus (25 mg IV weekly) alone, interferon alfa (3 million units SC 3 times weekly) alone, or the combination (temsirolimus 15 mg IV weekly plus interferon alfa 6 million units SC 3 times weekly). In this trial, temsirolimus monotherapy produced a significantly longer overall survival time than interferon alfa alone (10.9 versus 7.3 months; P = 0.008) and improved PFS time when administered alone or in combination with interferon alfa (3.8 and 3.7 months, respectively, versus 1.9 months). Because no real efficacy advantage of the combination was demonstrated, temsirolimus is administered alone. The most common adverse effects of temsirolimus are asthenia, rash, anemia, nausea, anorexia, pain, and dyspnea. Additionally, hyperglycemia, hyper-cholesterolemia, and hyperlipidemia occur with these agents. Noninfectious pneumonitis is a rare but often fatal complication.
Everolimus is also an orally administered derivative of rapamycin that is approved for use after failure of VEGF-targeted therapies. The results of the landmark trial RECORD-1 demonstrated that everolimus (10 mg orally daily) is effective at prolonging PFS (4 versus 1.9 months; P < 0.001) when compared with best supportive care, a viable treatment option at the time of approval.78 The most common adverse effects of everolimus are stomatitis, rash, fatigue, asthenia, and diarrhea. As with temsirolimus, elevations in glucose, lipids, and triglycerides and noninfectious pneumonitis can occur.
TKI + MTOR INHIBITOR
Lenvatinib is also a small molecule targeting multiple tyrosine kinases, primarily VEGF2. Combined with the mTOR inhibitor everolimus, it has been shown to be an effective regimen in patients with metastatic RCC who have failed other therapies. In a randomized phase 2 study involving patients with advanced or metastatic clear-cell RCC, patients were randomly assigned to receive either lenvatinib (24 mg/day), everolimus (10 mg/day), or lenvatinib plus everolimus (18 mg/day and 5 mg/day, respectively). Patients received the treatment continuously on a 28-day cycle until progression or inability to tolerate toxicity. Patients in the lenvatinib plus everolimus arm had median PFS of 14.6 months (95% CI 5.9 to 20.1) versus 5.5 months (95% CI 3.5 to 7.1) with everlolimus alone (HR 0.40 [95% CI 0.24 to 0.68]; P = 0.0005). PFS with levantinib alone was 7.4 months (95% CI 5.6 to 10.20; HR 0.66 [95% CI 0.30 to 1.10]; P = 0.12). In addition, PFS with levantinib alone was significantly prolonged in comparison with everolimus alone (HR 0.61 [95% CI 0.38 to 0.98]; P = 0.048). Grade 3 or 4 toxicity were less frequent in the everolimus only arm and the most common grade 3 or 4 toxicity in the lenvatinib plus everolimus arm was diarrhea. The results of this study show that the combination of lenvatinib plus everolimus is an acceptable second-line option for treatment of patients with advanced or metastatic RCC.55
CASE CONTINUED
The patient is initially started on pazopanib and tolerates the medication well, with partial response to the treatment. However, on restaging scans he is noted to have small bowel perforation. Pazopanib is discontinued until the patient has a full recovery. He is then started on everolimus. Restaging scans done 3 months after starting everolimus demonstrate disease progression.
• What is the appropriate next step in treatment?
PD1 BLOCKADE
Programmed death 1 (PD-1) protein is a T-cell inhibitory receptor with 2 ligands, PD-L1 and PD-L2. PD-L1 is expressed on many tumors. Blocking the interaction between PD-1 and PD-L1 by anti-PD-1 humanized antibodies potentiates a robust immune response and has been a breakthrough in the field of cancer immunotherapy.79 Previous studies have demonstrated that overexpression of PD-L1 leads to worse outcomes and poor prognosis in patients with RCC.80 Nivolumab, a fully human IgG4 PD-1 immune checkpoint inhibitor, blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2. In a randomized, open-label, phase 3 study comparing nivolumab with everolimus in patients with RCC who had previously undergone treatment with other standard therapies, Motzer and colleagues81 demonstrated a longer overall survival time and fewer adverse effects with nivolumab. In this study, 821 patients with clear-cell RCC were randomly assigned to receive nivolumab (3 mg/kg of body weight IV every 2 weeks) or everolimus (10 mg orally once daily). The median overall survival time with nivolumab was 25 months versus 19.6 months with everolimus (P < 0.0148). Nineteen percent of patients receiving nivolumab experienced grade 3 or 4 toxicities, with fatigue being the most common adverse effect. Grade 3 or 4 toxicities were observed in 37% of patients treated with everolimus, with anemia being the most common. Based on the results of this trial, on November 23, 2015, the U.S. Food and Drug Administration approved nivolumab to treat patients with metastatic RCC who have received a prior antiangiogenic therapy.
CASE CONCLUSION
Both TKI and mTOR inhibitor therapy fail, and the patient is eligible for third-line therapy. Because of his previous GI perforation, other TKIs are not an option. The patient opts for enrollment in hospice due to declining performance status. For other patients in this situation with a good performance status, nivolumab would be a reasonable option.
FUTURE DIRECTIONS
With the approval of nivolumab, multiple treatment options are now available for patients with metastatic or unresectable RCC. Development of other PD-1 inhibitors and immunotherapies as well as multi-targeted TKIs will only serve to expand treatment options for these patients. Given the aggressive course and poor prognosis of non-clear cell renal cell tumors and those with sarcomatoid features, evaluation of systemic and targeted therapies for these subtypes should remain active areas of research and investigation.
- Siegel R, Miller, K, Jemal A. Cancer Statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Eble JN, Sauter G, Epstein JI, Sesterhenn IA. Pathology and genetics. Tumors of the urinary system and male genital organs. Lyon: IARC Press; 2004.
- Chow WH, Gridley G, Fraumeni JF Jr, Jarvholm B. Obesity, hypertension, and the risk of kidney cancer in men. N Engl J Med 2000;343:1305–11.
- Cohen H, McGovern F. Renal-cell carcinoma. N Engl J Med 2005;353:2477–90.
- Yao M, Yoshida M, Kishida T, et al. VHL tumor suppres sor gene alterations associated with good prognosis in sporadic clear-cell renal carcinoma. J Natl Cancer Inst 2002;94:1569–75.
- Iliopoulos O, Kibel A, Gray S, Kaelin WG Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1995;1:822–6
- Chen F, Kishida T, Duh FM, et al. Suppression of growth of renal carcinoma cells by the von Hippel-Lindau tumor suppressor gene. Cancer Res 1995;55:4804–7.
- Iliopoulos O, Levy AP, Jiang C, et al. Negative regulation of hypoxia-inducible genes by the von Hippel Lindau protein. Proc Natl Acad Sci U S A 1996;93:10595–9.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Bir- Hogg-Dube syndrome. Cancer Cell 2002;2:157–64
- Shuch B, Vorganit S, Ricketts CJ, et al. Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 2014;32:431–7.
- Bukowski RM. Immunotherapy in renal cell carcinoma. Oncology 1999;13:801–10.
- Mueller-Lisse UG, Mueller-Lisse UL. Imaging of advanced renal cell carcinoma. World J Urol 2010;28: 253–61.
- Edge SB, Byrd DR, Compton CC, et al, eds. AJCC cancer staging manual, 7th ed. New York: Springer Science and Business Media LLC; 2010.
- O’Malley RL, Godoy G, Kanofsky JA, Taneja SS. The necessity of adrenalectomy at the time of radical nephrectomy: a systematic review. J Urol 2009;181:2009–17.
- McDougal S, Wein AJ, Kavoussi LR, et al. Campbell-Walsh Urology. 10th ed. Philadelphia (PA): Saunders; 2012.
- Colombo JR Jr, Haber GP, Kelovsek JE, et al. Seven years after laparoscopic radical nephrectomy: oncologic and renal functional outcomes. Urology 2008:71:1149–54.
- Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Ca 2013;49: 1374–403.
- Weight CJ, Larson BT, Fergany AF, et al. Nephrectomy induced chronic renal insufficiency is associated with increased risk of cardiovascular death and death from any cause in patients with localized cT1b renal masses. J Urol 2010;183:1317–23.
- Van Poppel H, Da Pozzo L, Albrecht W, et al. A prospective, randomized EORTC intergroup phase 3 study comparing the oncologic outcome of elective nephron-sparing surgery and radical nephrectomy for low-stage renal cell carcinoma. Eur Urol 2011;59:543–52.
- Smaldone MC, Fung C, Uzzo RG, Hass NB. Adjuvant and neoadjuvant therapies in high-risk renal cell carcioma. Hematol Oncol Clin North Am 2011;25:765–91.
- NCCN clinical practice guidelines in oncology. Version 3.2016. www.nccn.org. Accessed July 13, 2016
- El Dib R, Touma NJ, Kapoor A. Cryoablation vs radiofrequency ablation for the treatment of renal cell carcinoma: a meta-amalysis of case series studies. BJU Int 2012;110:510–6.
- Theodorescu D. Cancer cryotherapy: evolution and biology. Rev Urol 2004;6 Suppl 4:S9–S19.
- Khiatani V, Dixon RG. Renal ablation update. Sem Intervent Radiol 2014;31:157–66.
- Yu J, Liang P, Yu XL, et al. US-guided percutaneous microwave ablation of renal cell carcinoma: intermediate-term results. Radiol 2012;263:900–8.
- Castle SM, Salas N, Leveillee RJ. Initial experience using microwave ablation therapy for renal tumor treatment: 18- month follow-up. Urology 2011;77:792–7.
- Pech M, Janitzky A, Wendler JJ, et al. Irreversible electroporation of renal cell carcinoma: a first-in-man phase I clinical study. Cardiovasc Intervent Radiol 2011;34:132–8.
- Chow WH, Devesa SS, Warren JL, Fraumeni JF Jr. Rising incidence of renal cell cancer in the United States. JAMA 1999;281:1628–31.
- Jayson M, Sanders H. Increased incidence of serendipitously discovered renal cell carcinoma. Urology 1998;51:203–5.
- Pierorazio PM, Johnson MH, Ball MW, et al. Five-year analysis of a multi-institutional prospective clinical trial of delayed intervention and surveillance for small renal masses: the DISSRM registry. Eur Urol 2015;68:408–15.
- Jewett MA, Mattar K, Basiuk J, et al. Active surveillance of small renal masses: progression patterns of early stage kidney cancer. Eur Urol 2011;60:39–44.
- Chawla SN, Crispen PL, Hanlon AL, et al. The natural history of observed enhancing renal masses: meta-analysis and review of the world literature. J Urol 2006;175:425–31.
- Smaldone MC, Kutikov A, Egleston BL, et al. Small renal masses progressing to metastases under active surveillance: a systematic review and pooled analysis. Cancer 2012;118:997–1006.
- Williamson TJ, Pearson JR, Ischia J, et al.Guideline of guidelines: follow-up after nephrectomy for renal cell carcinoma. BJU Int 2016;117:555–62.
- Donat S, Diaz M, Bishoff JT, et al. Follow-up for clinically localized renal neoplasms: AUA Guideline. J Urol 2013;190:407–16.
- Janzen NK, Kim HL, Figlin RA, Bell-degrun AS. Surveillance after radical or partial nephrectomy for localized renal cell carcinoma and management of recurrent disease. Urol Clin North Am 2003:30:843–52.
- Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socio-economic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev 2008;34:193–205.
- Mekhail T, Abou-Jawde R, Boumerhi G, et al. Validation and extension of the Memorial Sloan-Kettering Prognostic Factors Model for Survival in patients with previously untreated metastatic renal cell carcinoma. J Clin Oncol 2005;23: 832–41.
- Motzer RJ, Bacik J, Murphy BA, et al. Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J Clin Oncol 2002;20:289–96.
- Motzer RJ, Mazumdar M, Bacik J, et al. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J Clin Oncol 1999;17:2530–40.
- Heng DY, Xie W, Regan MM. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol 2009;27:5794–9.
- Leibovich BC, Han KR, Bui MH, et al. Scoring algorithm to predict survival after nephrectomy and immunotherapy in patients with metastatic renal cell carcinoma: A stratification tool for prospective clinical trials. Cancer 2003;98:2566–77.
- Flanigan RC, Mickisch G, Sylvester R, et al. Cytoreductive nephrectomy in patients with metastatic renal cancer: a combined analysis. J Urol 2004;171:1071–6.
- Choueiri TK, Xie W, Kollmannsberger C, et al. The impact of cytoreductive nephrectomy on survival of patients with metastatic renal cell carcinoma receiving vascular endothelial growth factor targeted therapy. J Urol 2011;185:60–6.
- Chapin BF, Delacroix SE Jr, Culp SH, et al. Safety of presurgical targeted therapy in the setting of metastatic renal cell carcinoma. Eur Urol 2011;60:964–71.
- Hutson TE, Lesovoy V, Al-Shukri S, et al. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal-cell carcinoma: a randomized open-label phase 3 trial. Lancet Oncol 2013;14:1287–94.
- Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metatastic renal cell carcinoma: a randomized, double-blind phase III trial. Lancet 2007;370:2103–11.
- Escudier B, Bellmunt J, Negrier S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol 2010;28:2144–50.
- McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8.
- Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med 2013;369:722–31.
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cell global evaluation trial. J Clin Oncol 2009;27:3312–8.
- Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007;356:115–24.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 2009;27:3584–90.
- Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 2007;356:2271–81.
- Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus and the combination in patients with metastatic renal cell carcinoma: a randomized, phase 2, open label, multicenter trial. Lancet Oncology 2015;16:1473–82.
- Lexi-Comp, Inc. (Lexi-Drugs® ). Lexi-Drugs version 2.3.3. Lexicomp. Wolters Kluwer Health, Inc. Hudson, OH.
- Choueiri TK, Plantade A, Elson P, et al. Efficacy of sunitinib and sorafenib in metastatic papillary and chromophobe renal cell carcinoma. J Clin Oncol 2008;26:127–31.
- Lee JL, Ahn JH, Lim HY, et al. Multicenter phase II study of sunitinib in patients with non-clear cell renal cell carcinoma. Ann Oncol 2012;23:2108–14.
- Armstrong AJ, Broderick S, Eisen T, et al. Final clinical results of a randomized phase II international trial of everolimus vs. sunitinib in patients with metastatic non-clear cell renal cell carcinoma (ASPEN). ASCO Meeting Abstracts 2015;33:4507.
- Chowdhury S, Matrana MR, Tsang C, et al. Systemic therapy for metastatic non-clear-cell renal cell carcinoma: recent progress and future directions. Hematol Oncol Clin North Am 2011;25:853–69.
- Escudier B, Droz JP, Rolland F, et al. Doxorubicin and ifosfamide in patients with metastatic sarcomatoid renal cell carcinoma: a phase II study of the Genitourinary Group of the French Federation of Cancer Centers. J Urol 2002; 168–71
- Nanus DM, Garino A, Milowsky MI, et al. Active chemotherapy for sarcomatoid and rapidly progressing renal cell carcinoma. Cancer 2004;101:1545–51.
- Michaelson MD, McKay RR, Werner L, et al. Phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid and/or poor-risk metastatic renal cell carcinoma. Cancer 2015;121:3435–43.
- McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8
- Cho DC, Puzanov I, Regan MM, et al. Retrospective analysis of the safety and efficacy of interleukin-2 after prior VEGF-targeted therapy in patients with advanced renal cell carcinoma. J Immunother 2009;32:181–5.
- Pyrhönen S, Salminen E, Ruutu M, et al. Prospective randomized trial of interferon alfa-2a plus vinblastine versus vinblastine alone in patients with advanced renal cell cancer. J Clin Oncol 1999;17:2859–67.
- Interferon-alpha and survival in metastatic renal carcinoma: early results of a randomised controlled trial. Medical Research Council Renal Cancer Collaborators. Lancet 1999;353:14–7.
- Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349:427–34.
- Atkinson BJ, Kalra S, Wang X, et al. Clinical outcomes for patients with metastatic renal cell carcinoma treated with alternative sunitinib schedules. J Urol 2014;191:611–8.
- Kollmannsberger C, Bjarnason G, Burnett P, et al. Sunitinib in metastatic renal cell carcinoma: recommendations for management of noncardiovascular toxicities. Oncologist 2011;16:543–53.
- Najjar YG, Mittal K, Elson P, et al. A 2 weeks on and 1 week off schedule of sunitinib is associated with decreased toxicity in metastatic renal cell carcinoma. Eur J Cancer 2014;50:1084–9.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 2010;28:1061–8.
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007;356:125–34
- Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011;378:1931–9.
- Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1814–23.
- Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR) final results from a randomized, open-label, phase 3 trial. Lancet Oncology 2016;17:917–27.
- Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer 2004;4:335–48.
- Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372:449–56.
- Brahmer J, Tykodi S, Chow L, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65.
- Thomson RH, Kuntz SM, Leibovich BC, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow up. Cancer Res 2006;66: 3381–5.
- Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803–13.
- Siegel R, Miller, K, Jemal A. Cancer Statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Eble JN, Sauter G, Epstein JI, Sesterhenn IA. Pathology and genetics. Tumors of the urinary system and male genital organs. Lyon: IARC Press; 2004.
- Chow WH, Gridley G, Fraumeni JF Jr, Jarvholm B. Obesity, hypertension, and the risk of kidney cancer in men. N Engl J Med 2000;343:1305–11.
- Cohen H, McGovern F. Renal-cell carcinoma. N Engl J Med 2005;353:2477–90.
- Yao M, Yoshida M, Kishida T, et al. VHL tumor suppres sor gene alterations associated with good prognosis in sporadic clear-cell renal carcinoma. J Natl Cancer Inst 2002;94:1569–75.
- Iliopoulos O, Kibel A, Gray S, Kaelin WG Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1995;1:822–6
- Chen F, Kishida T, Duh FM, et al. Suppression of growth of renal carcinoma cells by the von Hippel-Lindau tumor suppressor gene. Cancer Res 1995;55:4804–7.
- Iliopoulos O, Levy AP, Jiang C, et al. Negative regulation of hypoxia-inducible genes by the von Hippel Lindau protein. Proc Natl Acad Sci U S A 1996;93:10595–9.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Bir- Hogg-Dube syndrome. Cancer Cell 2002;2:157–64
- Shuch B, Vorganit S, Ricketts CJ, et al. Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 2014;32:431–7.
- Bukowski RM. Immunotherapy in renal cell carcinoma. Oncology 1999;13:801–10.
- Mueller-Lisse UG, Mueller-Lisse UL. Imaging of advanced renal cell carcinoma. World J Urol 2010;28: 253–61.
- Edge SB, Byrd DR, Compton CC, et al, eds. AJCC cancer staging manual, 7th ed. New York: Springer Science and Business Media LLC; 2010.
- O’Malley RL, Godoy G, Kanofsky JA, Taneja SS. The necessity of adrenalectomy at the time of radical nephrectomy: a systematic review. J Urol 2009;181:2009–17.
- McDougal S, Wein AJ, Kavoussi LR, et al. Campbell-Walsh Urology. 10th ed. Philadelphia (PA): Saunders; 2012.
- Colombo JR Jr, Haber GP, Kelovsek JE, et al. Seven years after laparoscopic radical nephrectomy: oncologic and renal functional outcomes. Urology 2008:71:1149–54.
- Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Ca 2013;49: 1374–403.
- Weight CJ, Larson BT, Fergany AF, et al. Nephrectomy induced chronic renal insufficiency is associated with increased risk of cardiovascular death and death from any cause in patients with localized cT1b renal masses. J Urol 2010;183:1317–23.
- Van Poppel H, Da Pozzo L, Albrecht W, et al. A prospective, randomized EORTC intergroup phase 3 study comparing the oncologic outcome of elective nephron-sparing surgery and radical nephrectomy for low-stage renal cell carcinoma. Eur Urol 2011;59:543–52.
- Smaldone MC, Fung C, Uzzo RG, Hass NB. Adjuvant and neoadjuvant therapies in high-risk renal cell carcioma. Hematol Oncol Clin North Am 2011;25:765–91.
- NCCN clinical practice guidelines in oncology. Version 3.2016. www.nccn.org. Accessed July 13, 2016
- El Dib R, Touma NJ, Kapoor A. Cryoablation vs radiofrequency ablation for the treatment of renal cell carcinoma: a meta-amalysis of case series studies. BJU Int 2012;110:510–6.
- Theodorescu D. Cancer cryotherapy: evolution and biology. Rev Urol 2004;6 Suppl 4:S9–S19.
- Khiatani V, Dixon RG. Renal ablation update. Sem Intervent Radiol 2014;31:157–66.
- Yu J, Liang P, Yu XL, et al. US-guided percutaneous microwave ablation of renal cell carcinoma: intermediate-term results. Radiol 2012;263:900–8.
- Castle SM, Salas N, Leveillee RJ. Initial experience using microwave ablation therapy for renal tumor treatment: 18- month follow-up. Urology 2011;77:792–7.
- Pech M, Janitzky A, Wendler JJ, et al. Irreversible electroporation of renal cell carcinoma: a first-in-man phase I clinical study. Cardiovasc Intervent Radiol 2011;34:132–8.
- Chow WH, Devesa SS, Warren JL, Fraumeni JF Jr. Rising incidence of renal cell cancer in the United States. JAMA 1999;281:1628–31.
- Jayson M, Sanders H. Increased incidence of serendipitously discovered renal cell carcinoma. Urology 1998;51:203–5.
- Pierorazio PM, Johnson MH, Ball MW, et al. Five-year analysis of a multi-institutional prospective clinical trial of delayed intervention and surveillance for small renal masses: the DISSRM registry. Eur Urol 2015;68:408–15.
- Jewett MA, Mattar K, Basiuk J, et al. Active surveillance of small renal masses: progression patterns of early stage kidney cancer. Eur Urol 2011;60:39–44.
- Chawla SN, Crispen PL, Hanlon AL, et al. The natural history of observed enhancing renal masses: meta-analysis and review of the world literature. J Urol 2006;175:425–31.
- Smaldone MC, Kutikov A, Egleston BL, et al. Small renal masses progressing to metastases under active surveillance: a systematic review and pooled analysis. Cancer 2012;118:997–1006.
- Williamson TJ, Pearson JR, Ischia J, et al.Guideline of guidelines: follow-up after nephrectomy for renal cell carcinoma. BJU Int 2016;117:555–62.
- Donat S, Diaz M, Bishoff JT, et al. Follow-up for clinically localized renal neoplasms: AUA Guideline. J Urol 2013;190:407–16.
- Janzen NK, Kim HL, Figlin RA, Bell-degrun AS. Surveillance after radical or partial nephrectomy for localized renal cell carcinoma and management of recurrent disease. Urol Clin North Am 2003:30:843–52.
- Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socio-economic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev 2008;34:193–205.
- Mekhail T, Abou-Jawde R, Boumerhi G, et al. Validation and extension of the Memorial Sloan-Kettering Prognostic Factors Model for Survival in patients with previously untreated metastatic renal cell carcinoma. J Clin Oncol 2005;23: 832–41.
- Motzer RJ, Bacik J, Murphy BA, et al. Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J Clin Oncol 2002;20:289–96.
- Motzer RJ, Mazumdar M, Bacik J, et al. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J Clin Oncol 1999;17:2530–40.
- Heng DY, Xie W, Regan MM. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol 2009;27:5794–9.
- Leibovich BC, Han KR, Bui MH, et al. Scoring algorithm to predict survival after nephrectomy and immunotherapy in patients with metastatic renal cell carcinoma: A stratification tool for prospective clinical trials. Cancer 2003;98:2566–77.
- Flanigan RC, Mickisch G, Sylvester R, et al. Cytoreductive nephrectomy in patients with metastatic renal cancer: a combined analysis. J Urol 2004;171:1071–6.
- Choueiri TK, Xie W, Kollmannsberger C, et al. The impact of cytoreductive nephrectomy on survival of patients with metastatic renal cell carcinoma receiving vascular endothelial growth factor targeted therapy. J Urol 2011;185:60–6.
- Chapin BF, Delacroix SE Jr, Culp SH, et al. Safety of presurgical targeted therapy in the setting of metastatic renal cell carcinoma. Eur Urol 2011;60:964–71.
- Hutson TE, Lesovoy V, Al-Shukri S, et al. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal-cell carcinoma: a randomized open-label phase 3 trial. Lancet Oncol 2013;14:1287–94.
- Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metatastic renal cell carcinoma: a randomized, double-blind phase III trial. Lancet 2007;370:2103–11.
- Escudier B, Bellmunt J, Negrier S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol 2010;28:2144–50.
- McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8.
- Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med 2013;369:722–31.
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cell global evaluation trial. J Clin Oncol 2009;27:3312–8.
- Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007;356:115–24.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 2009;27:3584–90.
- Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 2007;356:2271–81.
- Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus and the combination in patients with metastatic renal cell carcinoma: a randomized, phase 2, open label, multicenter trial. Lancet Oncology 2015;16:1473–82.
- Lexi-Comp, Inc. (Lexi-Drugs® ). Lexi-Drugs version 2.3.3. Lexicomp. Wolters Kluwer Health, Inc. Hudson, OH.
- Choueiri TK, Plantade A, Elson P, et al. Efficacy of sunitinib and sorafenib in metastatic papillary and chromophobe renal cell carcinoma. J Clin Oncol 2008;26:127–31.
- Lee JL, Ahn JH, Lim HY, et al. Multicenter phase II study of sunitinib in patients with non-clear cell renal cell carcinoma. Ann Oncol 2012;23:2108–14.
- Armstrong AJ, Broderick S, Eisen T, et al. Final clinical results of a randomized phase II international trial of everolimus vs. sunitinib in patients with metastatic non-clear cell renal cell carcinoma (ASPEN). ASCO Meeting Abstracts 2015;33:4507.
- Chowdhury S, Matrana MR, Tsang C, et al. Systemic therapy for metastatic non-clear-cell renal cell carcinoma: recent progress and future directions. Hematol Oncol Clin North Am 2011;25:853–69.
- Escudier B, Droz JP, Rolland F, et al. Doxorubicin and ifosfamide in patients with metastatic sarcomatoid renal cell carcinoma: a phase II study of the Genitourinary Group of the French Federation of Cancer Centers. J Urol 2002; 168–71
- Nanus DM, Garino A, Milowsky MI, et al. Active chemotherapy for sarcomatoid and rapidly progressing renal cell carcinoma. Cancer 2004;101:1545–51.
- Michaelson MD, McKay RR, Werner L, et al. Phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid and/or poor-risk metastatic renal cell carcinoma. Cancer 2015;121:3435–43.
- McDermott DF, Cheng SC, Signoretti S, et al. The high-dose aldesleukin “select”trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res 2015;21:561–8
- Cho DC, Puzanov I, Regan MM, et al. Retrospective analysis of the safety and efficacy of interleukin-2 after prior VEGF-targeted therapy in patients with advanced renal cell carcinoma. J Immunother 2009;32:181–5.
- Pyrhönen S, Salminen E, Ruutu M, et al. Prospective randomized trial of interferon alfa-2a plus vinblastine versus vinblastine alone in patients with advanced renal cell cancer. J Clin Oncol 1999;17:2859–67.
- Interferon-alpha and survival in metastatic renal carcinoma: early results of a randomised controlled trial. Medical Research Council Renal Cancer Collaborators. Lancet 1999;353:14–7.
- Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349:427–34.
- Atkinson BJ, Kalra S, Wang X, et al. Clinical outcomes for patients with metastatic renal cell carcinoma treated with alternative sunitinib schedules. J Urol 2014;191:611–8.
- Kollmannsberger C, Bjarnason G, Burnett P, et al. Sunitinib in metastatic renal cell carcinoma: recommendations for management of noncardiovascular toxicities. Oncologist 2011;16:543–53.
- Najjar YG, Mittal K, Elson P, et al. A 2 weeks on and 1 week off schedule of sunitinib is associated with decreased toxicity in metastatic renal cell carcinoma. Eur J Cancer 2014;50:1084–9.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 2010;28:1061–8.
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007;356:125–34
- Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011;378:1931–9.
- Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1814–23.
- Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR) final results from a randomized, open-label, phase 3 trial. Lancet Oncology 2016;17:917–27.
- Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer 2004;4:335–48.
- Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372:449–56.
- Brahmer J, Tykodi S, Chow L, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65.
- Thomson RH, Kuntz SM, Leibovich BC, et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow up. Cancer Res 2006;66: 3381–5.
- Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803–13.
Soft Tissue Sarcoma: Diagnosis and Treatment
INTRODUCTION
Soft tissue sarcomas (STSs) are rare adult tumors, with 3.4 new cases per 100,000 persons or 12,310 expected new cases in 2016.1 Sarcomas are a heterogeneous collection of tumors that affect fat, muscle, nerve, nerve sheath, vascular, and connective tissues. There are more than 50 histological subtypes that comprise this diverse category of tumors. Treatment varies by stage, with limb-sparing surgery representing the mainstay of curative-intent treatment. Radiation and chemotherapy may also be considered depending on the size, grade, and location of the tumor. Survival rates have been stagnant until recently, with a disease-specific survival hovering around 65%.1 Given the complexity of these cases, all patients ideally should be evaluated and treated by a multidisciplinary team at an institution with extensive experience treating STS.2
EPIDEMIOLOGY AND CLASSIFICATION
The most common STS subtypes are gastrointestinal stromal tumor (GIST), undifferentiate pleomorphic sarcoma (previously referred to as malignant fibrous histiocytoma), liposarcoma, leiomyosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, and unclassified sarcoma.3 Liposarcoma is one of the most common subtypes, comprising 20% of all STSs; it is subdivided into well-differentiated/dedifferentiated liposarcomas, myxoid/round cell liposarcomas, and pleomorphic liposarcomas. Well-differentiated liposarcomas tend to occur in the retroperitoneum and limbs, while both myxoid and round cell as well as pleomorphic liposarcomas more commonly originate on the limbs. Histology varies based on subtype and ranges from mature-appearing adipocytes and fibroblasts to undifferentiated cells with minimal lipogenic differentiation.4
Leiomyosarcomas are smooth muscle tumors and are usually located in the retroperitoneum, but have also been associated with peripheral soft tissue and vasculature. Typical histology ranges from well-defined areas of spindle-shaped cells to poorly differentiated anaplastic spindle cells.5,6 Synovial sarcomas are a distinct type of STS that can show epithelial differentiation and account for 5% of adult STSs. The extremities are the most common presenting location (90%).7
Rhabdomyosarcomas are skeletal muscle tumors and are further subdivided into embryonal, alveolar, and pleomorphic subtypes. Embryonal histology ranges from primitive mesenchymal-appearing cells to highly differentiated muscle cells. Alveolar rhabdomyosarcoma has the worst prognosis of the subtypes and consists of round cells with high nuclear-to-chromatin ratios that form “glandular-like” or “alveolar” spaces.8 Pleomorphic rhabdomyosarcomas are composed of rhabdomyoblasts that can affect many different locations, but most commonly present on the lower extremities.9
Malignant peripheral nerve sheath tumor (MPNST) comprises 5% to 10% of all STSs. These tumors are associated with neurofibromatosis type 1 (NF-1), with 25% to 50% of tumors occurring in NF-1 patients. Additionally, most patients have a truncating lesion in the NF1 gene on chromosome 17.10 Anghileri et al in their single institution analysis of 205 patients with MPNSTs found the 2 most common presenting sites were the trunk and extremities. Histologically, these tumors have dense fascicles of spindle cells.10
GISTs are the most common STS of the gastrointestinal (GI) tract. Previously, GISTs were classified as smooth muscle tumors and were not accounted for in the literature as a separate entity distinct from leiomyomas, leiomyoblastomas, and leiomyosarcomas.11 GISTs are found throughout the GI tract: the most common sites are the stomach (60%) and small intestine (30%). Less common sites include duodenum (4%–5%), esophagus (1%), rectum (1%–2%), and appendix (< 0.2%).12 GISTs can be spindle cell, epithelioid, or mesenchymal tumors. Immunohistochemically, GISTs are KIT (CD117) positive. Other cell markers that are also commonly positive include CD34 (60%–70%) and smooth muscle actin (SMA) (25%).11 The majority of GISTs (80%) have an activating c-KIT gene mutation. The most common mutation site is exon 11, with less common c-KIT gene mutations also occurring at exon 9 or 13. Not all GISTs have KIT mutations. The second most common mutation is the PDGFRA mutation (5%–10% of GISTs).2 A minority of GISTs are negative for both KIT and PDGFRA mutations. These tumors were previously called wild-type, but as the majority have either a succinate dehydrogenase (SDH) loss of function or loss of SDHB protein expression, they are now referred to as SDH-deficient GISTs.2 GISTs vary in aggressiveness from incidental to aggressive. Typically, small intestine and rectal GISTs are more aggressive than gastric GISTs. Both size and mitotic rate help to predict the metastatic potential of the tumor. Tumors less than 2 cm in size and having a mitotic rate of less than 5 per 50 high-power fields (hpf) have the lowest risk of metastases, while tumors greater than 5 cm and with more than 5 mitoses per 50 hpf have the highest rates of metastases.12
Angiosarcomas are rare tumors comprising 4% of all STSs. Although they can occur in any site, the majority are cutaneous and occur most frequently in the head and neck regions. These tumors are either of vascular or lymphatic origin and are comprised of abnormal, pleomorphic, malignant endothelial cells. The most useful immunohistochemical markers include von Willebrand factor, CD31, and Ulex europaeus agglutinin 1. The majority of these tumors occur sporadically; however, radiation exposure, chronic lymphedema, and certain toxins including vinyl chloride and thorium dioxide are known risk factors.13
Undifferentiated sarcomas have no specific features and typically consist of primitive mesenchymal cells.
CLINICAL EVALUATION
CASE PRESENTATION
Initial Presentation and History
A 55-year-old man presents to his primary care physician with a painless mass in his anterior thigh. The mass has been present for the past 3 months and he believes that it is enlarging. The patient has a history of well-controlled hypertension and hyperlipidemia. His medications include atorvastatin and hydrochlorothiazide. He has no known drug allergies. Family history is notable for diabetes and hypertension. He drinks 4 to 5 alcoholic drinks a week and he is a former smoker. He quit smoking in his 30s and only smoked intermittently prior to quitting. He denies any illicit drug use. He works as a high school principal. Currently, he feels well. His review of systems is otherwise noncontributory.
Physical Examination
On physical exam, he is afebrile with a blood pressure of 132/75 mm Hg, respiratory rate of 10 breaths/min, and oxygen saturation of 99% on room air. He is a well appearing, overweight male. His head and neck exam is unremarkable. Lung exam reveals clear breath sounds, and cardiac exam reveals a regular rate and rhythm. His abdomen is obese, soft, and without hepatosplenomegaly. There is a large, fixed mass on the anterior lateral aspect of his right thigh. He has no appreciable lymphadenopathy. His neurological exam is unremarkable.
• What are risk factors for sarcoma?
There are few known risk factors for sarcoma. Established risks factors include prior radiation therapy, chronic lymphedema, viruses, and genetic cancer syndromes including Li-Fraumeni syndrome, hereditary retinoblastoma, and NF-1. Other environmental exposures include phenoxyacetic acids and chlorophenols.14 The majority of cases are sporadic, with only a minority of patients having one of these known risk factors.15 Up to one third of sarcomas have a specific translocation and are driven by fusion oncogenes (Table 1).

A painless mass is the most typical presenting symptom. Size at presentation varies based on location, with extremity and head and neck locations typically presenting at smaller sizes than retroperitoneal tumors.14 Patients may experience pain and numbness as the mass enlarges and impinges on surrounding structures including nerves and vasculature. The vast majority of patients are without systemic symptoms.
• How is sarcoma staged?
The American Joint Committee on Cancer (AJCC) staging system is the most widely used staging system in the United States. The latest AJCC manual was updated in 2010 to include a 3-tiered grading system where the tumor is classified according to tumor size, lymph node involvement, metastases, and grade at time of diagnosis (Table 2 and Table 3). Additionally, tumor depth in relation to deep fascia is also taken into account, with superficial tumors being assigned a designation of “a” and deep tumors a designation of “b.”

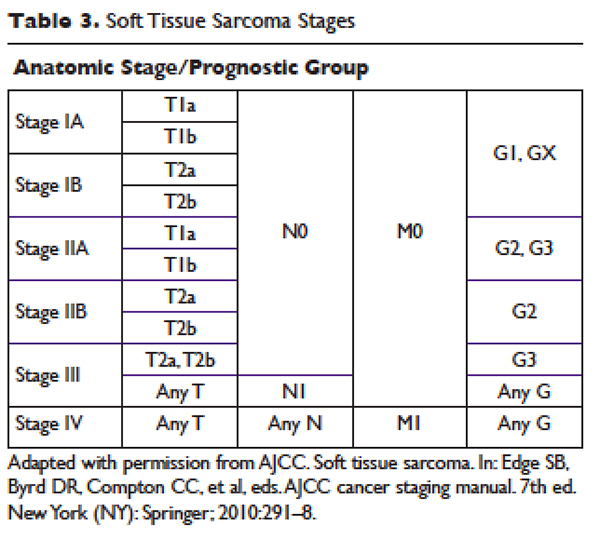
Previously, 2 of the most widely used grading systems were the National Cancer Institute (NCI) and French Federation of Cancer Centers Sarcoma Group (FNCLCC) systems, both 3-tier grading systems. The main components that determine the NCI grade are the tumor’s histologic type and location and the amount of tumor necrosis. The FNCLCC system evaluation focuses on tumor differentiation, mitotic rate, and amount of tumor necrosis. A study that compared the NCI and FNCLCC grading systems found that FNCLCC was a better predictor of mortality and distant metastasis.16 Previously, the AJCC was a 4-tier grading system, but the 2010 version was updated to the 3-tier FNCLCC grading system. Additionally, the AJCC system has reclassified single lymph node disease as stage III as it confers better survival than metastatic disease.17 It is important that pathology be evaluated by a sarcoma specialist as disagreements with regard to histologic subtype and grade are common.18,19
• What are the most important prognostic factors?
Prognostic factors include grade, size, and presence of metastases at presentation. Best survival is associated with low-grade, small tumors with no metastases at time of diagnosis.14
• What imaging should be considered?
Imaging should be undertaken to help differentiate between benign and malignant lesions. Ideally, it should be undertaken before a biopsy is planned as the imaging can be used to plan biopsy as well as provide invaluable prognostic information. There are several imaging modalities that should be considered during the preliminary work-up and staging of STSs. Conventional imaging includes magnetic resonance imaging (MRI) of the original tumor site; computed tomography (CT) to evaluate for pulmonary metastases and, depending on location, liver metastases; and in the case of small, low-grade tumors, chest radiography. MRI is considered the test of choice for soft tissue masses and can help delineate benign masses such as hematomas, lipomas, and hemangiomas from sarcomas.20 It is difficult to compare the accuracy of positron emission tomography (PET)/CT to CT and MRI because most studies have evaluated PET/CT in parallel with CT and MRI.21 Tateishi et al compared the accuracy of conventional imaging, PET/CT, and PET/CT combined with conventional imaging at determining the TNM staging for 117 patients. They found that conventional imaging correctly classified 77% of patients, PET alone correctly classified 70%, PET/CT correctly classified 83%, and PET/CT combined with conventional imaging correctly staged 87%.22
• Which subtypes are most likely to metastasize?
Although the vast majority of sarcomas spread hematogenously, 3 have a propensity to spread lymphogenously: epithelioid sarcoma, rhabdomyosarcoma, and clear-cell sarcoma. Additionally, certain subtypes are more likely to metastasize: leiomyosarcomas, synovial sarcomas, neurogenic sarcomas, rhabdomyosarcomas, and epithelioid sarcomas.23 Sarcomas metastasize to the lungs more frequently than to the liver. The metastatic pattern is defined primarily by sarcoma subtype and site of primary tumor. Sarcomas rarely metastasize to the brain (~1%).
MANAGEMENT
CASE CONTINUED
The patient undergoes an ultrasound to better visualize the mass. Given the heterogeneous character of the mass, he is referred for an MRI to evaluate the mass and a CT scan of the chest, abdomen, and pelvis to evaluate for distant metastases. MRI reveals a 5.1 cm × 4.6 cm heterogeneous mass invading the superficial fascia of the rectus femoris muscle. No suspicious lymph nodes or other masses are identified on imaging. The patient next undergoes an image-guided core needle biopsy. Pathology from that procedure is consistent with a stage III, T2bNxMx, grade 3, dedifferentiated liposarcoma.
• What is the best management approach for this patient?
SURGERY
Surgery is the mainstay of treatment for STS. Patients with the best prognosis are those who undergo complete resection with negative surgical margins.24,25 Goal tumor-free margin is 1 to 3 cm.26 Complete resection confers the best long-term survival. Both local and metastatic recurrence is higher in patients with incomplete resection and positive margins.24,25 In a study that analyzed 2084 localized primary STSs, patients with negative margins had a local recurrence rate of 15% versus a rate of 28% in patients with positive margins. This translated into higher 5-year local recurrence-free survival for patients with negative surgical margins (82%) compared to patients with positive margins (65%).27 Another study similarly found that patients with negative margins at referral to their institution who underwent postoperative radiation had high local control rates of 93% (95% confidence interval [CI] 87% to 97%) at 5, 10, and 15 years.26 Although radiation improves local control, neither preoperative or postoperative radiation has been shown to improve progression-free or overall survival.28 Other factors that are associated with risk of recurrence are tumor location, history of previous recurrence, age of patient, histopathology, tumor grade, and tumor size. Approximately 40% to 50% of patients with high-grade tumors (defined as size > 5 cm, deep location, and high grade) will develop distant metastases.29
Zagars et al found that positive or uncertain resection margin had a relative risk of local recurrence of 2.0 (95% CI 1.3 to 3.1; P = 0.002), and presentation with locally recurrent disease (vs new tumor) had a relative risk of local recurrence of 2.0 (95% CI 1.2 to 3.4; P = 0.013).26 Patients with STS of head and neck and deep trunk have higher recurrence rates than those with superficial trunk and extremity STS. A single-institution retrospective review demonstrated that patients with completely resectable retroperitoneal sarcomas have longer median survival (103 months) compared to patients with incompletely resected abdominal sarcomas (18 months).25
Rosenberg and colleagues compared amputation to limb-sparing surgery and radiation.24 Their prospective analysis of 65 patients found no difference in disease-free and overall survival between the 2 treatment groups. The limb-sparing treatment group had higher rates of local recurrence, which was highly correlated with positive surgical margins on pathology.24 Evidence from this and similar studies has resulted in radical amputations being replaced by conservative limb-sparing procedures and radiation therapy. In those found to have positive margins, re-resection is an option for some. Patients who undergo re-resection have higher local control rates than patients with positive margins who do not undergo re-resection. The 5-year control rate for patients who undergo re-resection is 85% (95% CI 80% to 89%) compared to 78% (95% CI 71% to 83%) for those who do not undergo re-resection. Similarly, patients who undergo re-resection have lower rates of metastases at 5, 10, and 15 years as well as higher 5-, 10-, and 15-year disease-free survival rates.26
CASE CONTINUED
The patient is referred for limb-sparing surgery after presentation at a multidisciplinary tumor board. Prior to undergoing resection of the tumor, he is also referred to radiation-oncology to discuss the risks and benefits of combination radiotherapy and surgery as opposed to surgical resection alone.
• What is the evidence for radiation therapy?
RADIATION THERAPY
Radiation therapy is used in the preoperative, intraoperative, and postoperative settings to reduce the risk of local recurrence. There are several options for radiation, including external beam radiation therapy (EBRT), intraoperative radiation, and brachytherapy. A newer strategy, intensity-modulated radiation therapy (IMRT), utilizes 3-dimensional modeling to reduce radiation dosages. Overall there are no differences in overall survival or local recurrence rates between preoperative and postoperative radiation in STS.28
The rationale behind preoperative radiation is that it reduces seeding of tumor cells, especially at the time of surgery.30 Additionally, for EBRT, preoperative radiation has smaller field sizes and lower radiation doses. It can also help to reduce the size of the tumor prior to resection. Intraoperative radiation is often paired with preoperative radiation as a boost dose given only to the area of residual tumor.
Suit et al reviewed patients treated at a single institution with limb-sparing surgery and different radiation strategies. Local control rates between preoperative and postoperative radiation groups were not statistically significant. Local recurrence was linked to grade and size of the tumor in both groups. The authors did note, however, that the preoperative radiation group tended to have larger tumor sizes at baseline compared to the patients who received postoperative radiation.30 A study that compared 190 patients who received preoperative and postoperative EBRT or brachytherapy (primary end point was wound complications, and local control was a secondary end point) showed a trend towards greater local control with preoperative radiation; however, the preoperative radiation group had significantly more wound complications compared to the postoperative radiation group.31
Yang et al found that postoperative EBRT decreases rates of local recurrence compared to surgery alone in high-grade extremity sarcomas.32 However, there were no differences in rates of distant metastases and overall survival between the 2 treatment groups. Similarly, in patients with low-grade sarcoma, there were fewer local recurrences in those who received EBRT and surgery as compared to surgery alone.32 Another study that evaluated 164 patients who received either adjuvant brachytherapy or no further therapy after complete resection found that brachytherapy reduced local recurrence in high-grade sarcomas. No difference in local recurrence rates was found in patients with low-grade sarcomas, nor was a significant difference found in the rates of distant metastases and overall survival between the 2 treatment groups.33 With regards to IMRT, a single institution cohort experience with 41 patients who received IMRT following limb-sparing surgery had similar local control rates when compared to historical controls.34
CASE CONTINUED
After discussion of the risks and benefits of radiation therapy, the patient opts for preoperative radiation prior to resection of his liposarcoma. He receives 50 Gy of EBRT prior to undergoing resection. Resection results in R1 margin consistent with microscopic disease. He receives 16 Gy of EBRT as a boost after recovery from his resection.2
• What is the evidence for neoadjuvant and adjuvant chemotherapy for stage I tumors?
CHEMOTHERAPY
Localized Sarcoma
For localized sarcoma, limb-sparing resection with or without radiation forms the backbone of treatment. Studies have evaluated chemotherapy in both the neoadjuvant and adjuvant settings, with the vast majority of studies evaluating doxorubicin-based chemotherapy regimens in the adjuvant settings. Due to the rare nature of sarcomas, most studies are not sufficiently powered to detect significant benefit from chemotherapy. Several trials evaluating chemotherapy regimens in the neoadjuvant and adjuvant settings needed to be terminated prematurely due to inadequate enrollment into the study. 35,36
For stage IA (T1a-Tb, N0, M0, low grade) tumors, no additional therapy is recommended after limb-sparing surgery with appropriate surgical margins. For stage IB (T2a-2b, N0, M0, low grade) tumors with insufficient margins, re-resection and radiation therapy should be considered, while for stage IIA (T1a-1b, N0, M0, G2-3) tumors preoperative or postoperative radiation therapy is recommended.2 Studies have not found benefit of adjuvant chemotherapy in these low-grade, stage I tumors in terms of progression-free survival and overall survival.37
• At what stage should chemotherapy be considered?
For stage IIb and stage III tumors, surgery and radiation therapy again form the backbone of therapy; however, neoadjuvant and adjuvant chemotherapy are also recommended as considerations. Anthracycline-based chemotherapy with either single-agent doxorubicin or doxorubicin and ifosfamide in combination are considered first-line chemotherapy agents in locally advanced STS.2,29,37
Evidence regarding the efficacy of both neoadjuvant and adjuvant chemotherapy regimens in the setting of locally advanced high-grade STS has been mixed. The Sarcoma Meta-analysis Collaboration evaluated 14 trials of doxorubicin-based adjuvant chemotherapy and found a trend towards overall survival in the treatment groups that received chemotherapy.37 All trials included in the meta-analysis compared patients with localized resectable soft-tissue sarcomas who were randomized to either adjuvant chemotherapy or no adjuvant chemotherapy after limb-sparing surgery with or without radiation therapy. None of the individual trials showed a significant benefit, and all trials had large confidence intervals; however, the meta-analysis showed significant benefit in the chemotherapy treatment groups with regard to local recurrence, distant recurrence, and progression-free survival. No significant difference in overall survival was found.37 Pervais et al updated the Sarcoma Meta-analysis Collaboration’s 1997 meta-analysis with the inclusion of 4 new trials that evaluated doxorubicin combined with ifosfamide and found that both patients who received doxorubicin-based regimens or doxorubicin with ifosfamide had significant decreases in distant and overall recurrences. Only the trials that utilized doxorubicin and ifosfamide had an improved overall survival that was statistically significant (hazard ratio 0.56 [95% CI 0.36 to 0.85]; P = 0.01).29 Although no significant heterogeneity was found among the trials included in either meta-analysis, a variety of sarcomas were included in each clinical trial evaluated. Given the extremely small number of each sarcoma subtype present in each trial, subgroup analysis is difficult and prone to inaccuracies. As a result, it is not known if certain histological subtypes are more or less responsive to chemotherapy.37–39
One randomized controlled trial evaluated neoadjuvant chemotherapy in high-risk sarcomas defined as tumors greater than 8 cm or grade II/III tumors. This study evaluated doxorubicin and ifosfamide and found no significant difference in disease-free and overall survival in the neoadjuvant therapy group compared to the control group.35 There remains controversy in the literature with regards to adjuvant chemotherapy. Many oncologists offer adjuvant chemotherapy to patients with certain stage III subtypes. Examples of subtypes that may be offered adjuvant therapy include myxoid liposarcomas, synovial sarcomas, and leiomyosarcomas.2 With regards to how many cycles of chemotherapy should be considered, a noninferiority study compared 3 cycles of epirubicin and ifosfamide to 5 cycles of epirubicin and ifosfamide in patients with high-risk locally advanced adult STSs. Three cycles of preoperative epirubicin and ifosfamide was found to be noninferior to 5 cycles with regards to overall survival.38
• What is this patient’s risk for recurrence?
The patient is at intermediate risk for recurrence. Numerous studies have demonstrated that tumor size, grade, and location are the most important factors to determine risk of recurrence, with larger size, higher grades, and deeper locations being associated with higher risk of recurrence. In an analysis of 1041 patients with STS of the extremities, high grade was the most important risk factor for distant metastases.39 The highest risk of recurrence is within the first 2 years. Given that the patient’s initial tumor was located in the extremity, he is more likely to have a distant metastasis as his site of recurrence; individuals with retroperitoneal tumors and visceral tumors are more likely to recur locally.40 For STSs of the extremity, distant metastases determine overall survival, whereas patients with retroperitoneal sarcomas can die from complications of local metastases.41 Once a patient develops distant metastases, the most important prognostic factor is the size of the tumor, with tumors larger than 10 cm having a relative risk of 1.5 (95% CI 1.0 to 2.0).39
• What are the recommendations for surveillance?
Surveillance recommendations are based on the stage of the sarcoma. Stage I tumors are the least likely to recur either locally or distally. As a result, it is recommended that stage I tumors be followed with history and physical exam every 3 to 6 months for the first 2 to 3 years, and then annually after the first 2 to 3 years. Chest x-rays should be considered every 6 to 12 months.2 For stage II–IV tumors, history and physical exam is recommended every 3 to 6 months for the first 2 to 3 years. Chest and distant metastases imaging should also be performed every 3 to 6 months during this time frame. For the next 2 years, history and physical exam and imaging are recommended every 6 months. After the first 4 to 5 years, annual follow-up is recommended.2
A study that followed 141 patients with primary extremity STSs for a median interval of 49 months found that high-grade tumors were most likely to recur during the first 2 years, with 20% of their patients recurring locally and 40% recurring distally. Chest x-rays performed during surveillance follow-up found distant lung metastases in 36 asymptomatic patients and had a positive predictive value of 92%, a negative predictive value of 97%, and a quality-adjusted life-year of $30,000.40,41 No laboratory testing was found to aid in detection of recurrence.
CASE CONTINUED
The patient does well for 1 year. With physical therapy, he regains most of the strength and coordination of the lower extremity. He is followed every 3 months with chest x-rays and a MRI of the thigh for the first year. On his fourth follow-up clinic visit, he describes increased dyspnea on exertion over the previous few weeks and is found to have multiple lung metastases in both lungs on chest x-ray. He undergoes further evaluation for metastases and is not found to have any other metastatic lesions. Bronchoscopy and biopsy of 1 of the lung nodules confirms recurrent dedifferentiated liposarcoma.
• Should this patient undergo metastectomy?
An analysis of 3149 patients with STS treated at Memorial Sloan-Kettering who developed lung metastases found that patients with pulmonary metastases have survival rates of 25%. The most important prognostic factor for survival was complete resection of all metastases.42 For stage IV disease, surgery is used only in certain instances. In instances where tumor is more localized or limited, removal of metastases or metastectomy can play a role in management.2
CASE CONTINUED
Because the patient’s metastases are limited to the lungs, he is referred for metastectomy. He undergoes wedge resection for definitive diagnosis but it is not possible to completely resect all of the metastases. He is thus referred to a medical oncologist to discuss his treatment options.
• What are treatment options for unresectable or metastatic disease?
Metastatic Disease
Unlike local and locally advanced disease, chemotherapy forms the backbone of treatment in stage IV disease. Doxorubicin and olaratumab or doxorubicin and ifosfamide in combination are considered first line in metastatic disease. Response rates for single-agent doxorubicin range from 16% to 27%, while phase 2 and phase 3 studies of doxorubicin and ifosfamide have found response rates ranging from 18% to 36%.43 In addition, the effectiveness of doxorubicin and ifosfamide phase 2 and 3 trials varied. Edmonson et al found a tumor regression rate of 34% for doxorubicin and ifosfamide as compared to 20% for doxorubicin alone.44 In comparison, Santoro et al found a response rate of 21.3% for doxorubicin alone and 25.2% for doxorubicin and ifosfamide.45 Neither study found increased survival benefit for doxorubicin and ifosfamide when compared to doxorubicin alone. In a Cochrane review evaluating randomized trials that compared doxorubicin and combination chemotherapy regimens, response rates varied from 14% for doxorubicin in combination with streptomycin to 34% for doxorubicin and ifosfamide. Most trials did not show a significant benefit for combination therapies when compared to doxorubicin alone.43 Mean survival with doxorubicin or doxorubicin and ifosfamide is 12 months. High rates of recurrence highlight the need for additional chemotherapy regimens.
The newest approved agent is olaratumab, a monoclonal antibody that binds platelet-derived growth factor receptor alpha and prevents receptor activation. A phase 1-b and phase 2 trial evaluated patients with locally advanced and metastatic STS and randomly assigned them to either olaratumab and doxorubicin or doxorubicin alone.46 Progression-free survival for olaratumab/doxorubicin was 6.6 months (95% CI 4.1 to 8.3) compared to 4.1 months (95% CI 2.8 to 5.4) for doxorubicin alone. The objective response rate was 18.2% (95% CI 9.8 to 29.6) for olaratumab/doxorubicin compared to 7.5% (95% CI 2.5 to 6.6) for doxorubicin alone. Furthermore, the median overall survival for olaratumab plus doxorubicin was 26.5 months (95% CI 20.9 to 31.7) compared to 14.7 months for doxorubicin alone (95% CI 5.5 to 26.0). Impressively, this improved response was notable across histological types. Furthermore, patients who had previously been treated with more than 1 regimen and those who were treatment naïve had similar response rates.46
• What are second-line treatment options?
Doxorubicin has been used in combination with several other agents including dacarbazine (DTIC) as well as DTIC and ifosfamide (MAID). Borden et al evaluated patients with metastatic STS and randomly assigned the patients to either doxorubicin or doxorubicin and DTIC. Combination therapy demonstrated better tumor response than doxorubicin alone: 30% complete or partial response for combination therapy and 18% for doxorubicin alone.47 However, Omura et al
found similar rates of efficacy between doxorubicin and combination doxorubicin and DTIC in women with recurrent or nonresectable uterine sarcomas.48 MAID has never been directly compared in a randomized trial to doxorubicin alone. In a study that compared MAID to doxorubicin and DTIC (AD) in patients with unresectable or metastatic sarcomas, MAID had superior response rates (32% versus 17%), but there was no difference with regards to overall survival (mean survival of 12.5 months).49
Several additional regimens have undergone evaluation in metastatic and recurrent STSs. Gemcitabine has been used both as a single agent and as part of combination therapy in many studies. Studies with gemcitabine in combination with either docetaxel or DTIC have been the most efficacious. In a phase 2 trial, patients with metastatic STS were randomly assigned to either gemcitabine alone or gemcitabine and docetaxel. Combination therapy had a higher response rate (16% versus 8%) and longer overall survival (17.9 months versus 11.5 months) than gemcitabine alone.50 Furthermore, a phase 2 trial of gemcitabine and docetaxel in patients with unresectable leiomyosarcoma showed an overall response rate of 56%, with 3 complete and 15 partial responses among the 34 patients enrolled in the study.51
A phase 2 trial randomly assigned patients with unresectable or metastatic STS to either DTIC or combination gemcitabine and DTIC.52 Gemcitabine-DTIC had a superior progression-free survival at 3 months (56% [95% CI 43% to 69%]) as compared to DTIC alone (37% [95% CI 23.5% to 50%]). Furthermore, mean progression-free survival and overall survival were improved in the gemcitabine-DTIC group (4.2 months and 16.8 months) as compared to the DTIC group (2.0 months and 8.2 months).52 DTIC has a single-agent response rate of 16%, but has been shown to be particularly effective in the setting of leiomyosarcomas.49
• Does response to treatment regimens differ by histologic subtype?
The majority of STS trials include many different histologic subtypes. Given the rarity of sarcomas as a whole, many trials have had difficulty recruiting adequate numbers of patients to have sufficient power to definitely determine if the treatment under investigation has clinical benefit. Furthermore, the patients recruited have been heterogeneous with regard to subtype. Many older studies hypothesized that the efficacy of chemotherapeutic agents vary based on histologic subtype; however, for most subtypes the number of individuals included in those trials was too low to evaluate efficacy based on subtype.
Some exceptions exist, however. For example, both gemcitabine-DTIC and gemcitabine-docetaxel have been found to be particularly effective in the treatment of leiomyosarcomas.50,52 Additionally, a retrospective study found a 51% overall response rate for patients with myxoid liposarcomas treated with trabectedin.53 Studies of patients with angiosarcoma treated with paclitaxel have demonstrated response rates of 43% and 53%.54,55
• What are the newest approved and investigational agents?
A recently approved agent is trabectedin, a tris tetrahydroisoquinoline alkaloid isolated from ascidians that binds to the minor groove of DNA and causes disruptions in the cell cycle. Samuels et al reported data from a single-arm, open-label expanded access trial that evaluated patients with advanced metastatic sarcomas.56 In this study, patients with liposarcomas and leiomyosarcomas had an objective response rate of 6.9% (95% CI 4.8 to 9.6) as compared to a rate of 5.9% (95% CI 4.4 to 7.8) for all assessable patients. Median survival was 11.9 months for all patients, with improved median survivals for liposarcoma and leiomyosarcomas of 16.2 months (95% CI 14.1 to 19.5) compared to 8.4 months (95% CI 7.1 to 10.7 months) for other subtypes.56
Schöffski et al evaluated eribulin, a chemotherapeutic agent that affects microtubule dynamics, in a phase 2 trial of patients with progressive or high-grade STS with progression on previous chemotherapy. They found a median progression-free survival of 2.6 months (95% CI 1.7 to 6.2) for adipocytic sarcoma, 2.9 months (95% CI 2.4 to 4.6) for leiomyosarcoma, 2.6 months (95% CI 2.3 to 4.3) for synovial sarcoma, and 2.1 months (95% CI 1.4 to 2.9) for other sarcomas.57
Van der Graaf and colleagues randomly assigned patients with metastatic nonadipocytic STS to pazopanib or placebo in a phase 3 trial. Pazopanib is a small-molecule endothelial growth factor inhibitor with activity against vascular endothelial growth factors 1, 2, and 3 as well as platelet-derived growth factors. Median progression-free survival was 4.6 months (95% CI 3.7 to 4.8) with pazopanib compared to 1.6 months (95% CI 0.9 to 1.8) with placebo.58 Adipocytic sarcomas (liposarcomas) were excluded from the trial because phase 2 trials had found a lower rate of progression-free survival (26%) for them compared to other subtypes.
• What are the most common toxicities associated with the approved and investigational chemotherapeutic agents?
Toxicities were seen with each of the regimens studied and were common in the randomized trials, with higher rates of toxicities in the combination chemotherapy regimens. The most common toxicities are myelosuppression, nausea, and vomiting. In the doxorubicin trials, the most common toxicities were myelosuppression, nausea, and vomiting.44
Ifosfamide both as an individual agent and in combination with doxorubicin has higher rates and higher grades of toxicity than doxorubicin alone. Myelosuppression is the most common toxicity associated with ifosfamide, and the most commonly affected cell line is leukocytes.44 Combination doxorubicin and ifosfamide also had high rates of nausea and vomiting (95%) and alopecia (100%).35
Neutropenia is the most common toxicity associated with gemcitabine and dacarbazine, while their most common nonhematologic toxicities are fatigue and nausea.52,59 Trabectedin’s most common toxicities are nausea (29%), neutropenia (24%), and fatigue (23%). It has also been shown to cause increased alkaline phosphatase (20%) and alanine aminotransferase (19%) levels.56 In a phase 2 study of eribulin, 50% of patients had neutropenia, and other toxicities included fatigue, alopecia, nausea, sensory neuropathy, and thrombocytopenia.57 Pazopanib is generally well tolerated; the most common toxicities are fatigue (65%), diarrhea (58%), nausea (54%), and hypertension (41%).58 Higher rates of neutropenia, mucositis, nausea, vomiting, diarrhea, and transfusion reactions were seen with olaratumab and doxorubicin compared to doxorubicin alone in phase 1b and 2 studies.46
CASE CONCLUSION
Given his poor prognosis with unresectable metastatic undifferentiated liposarcoma, the patient considers a clinical trial prior to undergoing combined therapy with doxorubicin and ifosfamide. He tolerates therapy well with stable disease at 6 months.
CONCLUSION
STSs are a heterogeneous collection of rare tumors. Low-grade, localized tumors have the best prognosis, and patients who undergo complete resection have the best long-term survival. Due to the rarity of STSs, trials often have limited enrollment, and little progress has been made with regards to treatment and survival rates for metastatic and unresectable disease. All patients should be evaluated and treated at specialized sarcoma centers. This case highlights the need for continued research and clinical trials to improve overall survival of patients with sarcoma.
- American Cancer Society. Cancer facts and figures 2016. American Cancer Society Web site. www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf. Accessed December 20, 2016.
- National Comprehensive Cancer Network. NCCN clinical guidelines in oncology: soft tissue sarcoma. 2016
- Coindre J, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001;91:1914–26.
- Dei Tos A. Liposarcoma: new entities and evolving concepts. Ann Diagn Pathol 2000;4:252–66.
- Wile AG, Evans HL, Romsdahl MM. Leiomyosarcoma of soft tissue: a clinicopathologic study. Cancer 1981;48:1022–32.
- Hashimoto H, Daimaru Y, Tsuneyoshi M, Enjoji M. Leiomyosarcoma of the external soft tissues. A clinicopathologic, immunohistochemical, and electron microscopic study. Cancer 1986;57:2077–88
- Fisher C. Synovial sarcoma. Ann Diagn Pathol 1998;2:401–21.
- Newton WA Jr, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 1995;76:1073–85.
- Furlong MA. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14:595–603.
- Anghileri M, Miceli R, Fiore M. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006;107:1065–74.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Archive 2001;438:1–12.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006;23:70–83.
- Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol 2010;11:983–91.
- Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin 2004;54:94–109.
- Penel N, Grosjean J, Robin YM, et al. Frequency of certain established risk factors in soft tissue sarcomas in adults: a prospective descriptive study of 658 cases. Sarcoma 2008;2008:459386.
- Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997;15:350–62.
- Maki RG, Moraco N, Antonescu CR, et al. Toward better soft tissue sarcoma staging: building on American joint committee on cancer staging systems versions 6 and 7. Ann Surg Oncol 2013;20:3377–83.
- Shiraki M, Enterline HT, Brooks JJ, et al. Pathologic analysis of advanced adult soft tissue sarcomas, bone sarcomas, and mesotheliomas. The Eastern Cooperative Oncology Group (ECOG) experience. Cancer 1989;64:484–90.
- Presant CA, Russell WO, Alexander RW, Fu YS. Soft-tissue and bone sarcoma histopathology peer review: The frequency of disagreement in diagnosis and the need for second pathology opinions. The Southeastern Cancer Study Group experience. J Clin Oncol 1986; 4:1658–61.
- Sundaram M, McLeod RA. MR imaging of tumor and tumorlike lesions of bone and soft tissue. AJR Am J Roentgenol 1990;155:817–24.
- Ioannidis JP, Lau J. 18F-FDG PET for the diagnosis and grading of soft-tissue sarcoma: a meta-analysis. J Nucl Med 2003;44:717–24.
- Tateishi U, Yamaguchi U, Seki K, et al. Bone and soft-tissue sarcoma: preoperative staging with fluorine 18 fluorodeoxyglucose PET/CT and conventional imaging. Radiology 2007;245:839–47.
- Zagars GK, Ballo MT, Pisters PW, et al. Prognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer 2003;97:2530–43
- Rosenberg S, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 1982;196:305–14.
- Lewis J, Leung D, Woodruff J, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 1998;288:355–65.
- Zagars GK, Ballo MT, Pisters PW, et al. Surgical margins and reresection in the management of patients with soft tissue sarcoma using conservative surgery and radiation therapy. Cancer 2003;97:2544–53.
- Stojadinovic A, Leung DH, Hoos A. Analysis of the prognostic significance of microscopic margins in 2,084 localized primary adult soft tisusse sarcomas. Ann Surg 2002;235:424–34.
- O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
- Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008;113:573–81.
- Suit HD, Mankin HJ, Wood WC, Proppe KH. Preoperative, intraoperative, and postoperative radiation in the treatment of primary soft tissue sarcoma. Cancer 1985;55:2659–67
- O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
- Yang J, Chang A, Baker A, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 1998;16:197–203.
- Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 1996;14:859–68.
- Alektiar KM, Brennan MF, Healey JH, Singer S. Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 2008;26:3440–5.
- Gortzak E, Azzarelli A, Buesa J, et al. A randomized phase II study on neo-adjuvant chemotherapy for ‘high-risk’ adult soft-tissue sarcoma. Eur J Cancer 2001;37:1096–1103.
- Fakhari N, Ebm C, Kostler WJ, et al. Intensified adjuvant IFADIC chemotherapy in combination with radiotherapy versus radiotherapy alone for soft tissue sarcoma: long-term follow-up of a prospective randomized feasibility trial. Wein Klin Wochenschr 2010;122:614–9.
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997;350:1647–54.
- Gronchi A, Frustaci S, Mercuri M, et al. Short, full-dose adjuvant chemotherapy in high-risk adult soft tissue sarcomas: a randomized clinical trial from the Italian Sarcoma Group and the Spanish Sarcoma Group. J Clin Oncol 2012;30:850–56.
- Pisters PW, Leung DH, Woodruff J. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679–89.
- Whooley B, Gibbs J, Mooney M. Primary Extremity Sarcoma: What is the Appropriate Follow-up? Annals of Surg Oncol 2000; 7: 9-14.
- Whooley BP, Mooney MN, Gibbs JF, Graybill WG. Effective follow-up strategies in soft tissue sarcoma. Sem Surg Oncol 1999;17:83–87.
- Billingsley KG, Burt ME, Jara E, et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg 1999;229:602–10.
- Bramwell VH, Anderson D, Charette ML; Sarcoma Disease Site Group. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev 2003;(3):CD003293.
- Edmonson J, Ryan L, Blum R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993;11:1269–75.
- Santoro A, Tursz T, Mouridsen H. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1995;13:1537–45.
- Tap WD, Jones RL, Van Tine B, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 2016;388:488–97.
- Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol 1987;5:840–50.
- Omura GA, Major FJ, Blessing JA, et al. A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 1983;52:626–32.
- Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276–85.
- Maki R, Wathen K, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 2007; 25: 2755–63.
- Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 2002;12:2824–31.
- Garcia-del-Muro X, Lopez-Pousa A, Maurel J, et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. J Clin Oncol 2011;29:2528–33.
- Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 2007;7:595–602.
- Italiano A, Cioffi A, Penel N, et al. Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas. Cancer 2012;118:3330–6.
- Penel N, Italiano A, Ray-Coquard I, et al. Metastatic angiosarcomas: doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve outcome. Ann Oncol 2012;23:517–23.
- Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 2013;24:1703–9.
- Schöffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histolical subtypes. Lancet 2011;11:1045–52.
- Van der Graaf W, Blay JY, Chawla S, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86.
- Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer 2007;109:1863–9.
INTRODUCTION
Soft tissue sarcomas (STSs) are rare adult tumors, with 3.4 new cases per 100,000 persons or 12,310 expected new cases in 2016.1 Sarcomas are a heterogeneous collection of tumors that affect fat, muscle, nerve, nerve sheath, vascular, and connective tissues. There are more than 50 histological subtypes that comprise this diverse category of tumors. Treatment varies by stage, with limb-sparing surgery representing the mainstay of curative-intent treatment. Radiation and chemotherapy may also be considered depending on the size, grade, and location of the tumor. Survival rates have been stagnant until recently, with a disease-specific survival hovering around 65%.1 Given the complexity of these cases, all patients ideally should be evaluated and treated by a multidisciplinary team at an institution with extensive experience treating STS.2
EPIDEMIOLOGY AND CLASSIFICATION
The most common STS subtypes are gastrointestinal stromal tumor (GIST), undifferentiate pleomorphic sarcoma (previously referred to as malignant fibrous histiocytoma), liposarcoma, leiomyosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, and unclassified sarcoma.3 Liposarcoma is one of the most common subtypes, comprising 20% of all STSs; it is subdivided into well-differentiated/dedifferentiated liposarcomas, myxoid/round cell liposarcomas, and pleomorphic liposarcomas. Well-differentiated liposarcomas tend to occur in the retroperitoneum and limbs, while both myxoid and round cell as well as pleomorphic liposarcomas more commonly originate on the limbs. Histology varies based on subtype and ranges from mature-appearing adipocytes and fibroblasts to undifferentiated cells with minimal lipogenic differentiation.4
Leiomyosarcomas are smooth muscle tumors and are usually located in the retroperitoneum, but have also been associated with peripheral soft tissue and vasculature. Typical histology ranges from well-defined areas of spindle-shaped cells to poorly differentiated anaplastic spindle cells.5,6 Synovial sarcomas are a distinct type of STS that can show epithelial differentiation and account for 5% of adult STSs. The extremities are the most common presenting location (90%).7
Rhabdomyosarcomas are skeletal muscle tumors and are further subdivided into embryonal, alveolar, and pleomorphic subtypes. Embryonal histology ranges from primitive mesenchymal-appearing cells to highly differentiated muscle cells. Alveolar rhabdomyosarcoma has the worst prognosis of the subtypes and consists of round cells with high nuclear-to-chromatin ratios that form “glandular-like” or “alveolar” spaces.8 Pleomorphic rhabdomyosarcomas are composed of rhabdomyoblasts that can affect many different locations, but most commonly present on the lower extremities.9
Malignant peripheral nerve sheath tumor (MPNST) comprises 5% to 10% of all STSs. These tumors are associated with neurofibromatosis type 1 (NF-1), with 25% to 50% of tumors occurring in NF-1 patients. Additionally, most patients have a truncating lesion in the NF1 gene on chromosome 17.10 Anghileri et al in their single institution analysis of 205 patients with MPNSTs found the 2 most common presenting sites were the trunk and extremities. Histologically, these tumors have dense fascicles of spindle cells.10
GISTs are the most common STS of the gastrointestinal (GI) tract. Previously, GISTs were classified as smooth muscle tumors and were not accounted for in the literature as a separate entity distinct from leiomyomas, leiomyoblastomas, and leiomyosarcomas.11 GISTs are found throughout the GI tract: the most common sites are the stomach (60%) and small intestine (30%). Less common sites include duodenum (4%–5%), esophagus (1%), rectum (1%–2%), and appendix (< 0.2%).12 GISTs can be spindle cell, epithelioid, or mesenchymal tumors. Immunohistochemically, GISTs are KIT (CD117) positive. Other cell markers that are also commonly positive include CD34 (60%–70%) and smooth muscle actin (SMA) (25%).11 The majority of GISTs (80%) have an activating c-KIT gene mutation. The most common mutation site is exon 11, with less common c-KIT gene mutations also occurring at exon 9 or 13. Not all GISTs have KIT mutations. The second most common mutation is the PDGFRA mutation (5%–10% of GISTs).2 A minority of GISTs are negative for both KIT and PDGFRA mutations. These tumors were previously called wild-type, but as the majority have either a succinate dehydrogenase (SDH) loss of function or loss of SDHB protein expression, they are now referred to as SDH-deficient GISTs.2 GISTs vary in aggressiveness from incidental to aggressive. Typically, small intestine and rectal GISTs are more aggressive than gastric GISTs. Both size and mitotic rate help to predict the metastatic potential of the tumor. Tumors less than 2 cm in size and having a mitotic rate of less than 5 per 50 high-power fields (hpf) have the lowest risk of metastases, while tumors greater than 5 cm and with more than 5 mitoses per 50 hpf have the highest rates of metastases.12
Angiosarcomas are rare tumors comprising 4% of all STSs. Although they can occur in any site, the majority are cutaneous and occur most frequently in the head and neck regions. These tumors are either of vascular or lymphatic origin and are comprised of abnormal, pleomorphic, malignant endothelial cells. The most useful immunohistochemical markers include von Willebrand factor, CD31, and Ulex europaeus agglutinin 1. The majority of these tumors occur sporadically; however, radiation exposure, chronic lymphedema, and certain toxins including vinyl chloride and thorium dioxide are known risk factors.13
Undifferentiated sarcomas have no specific features and typically consist of primitive mesenchymal cells.
CLINICAL EVALUATION
CASE PRESENTATION
Initial Presentation and History
A 55-year-old man presents to his primary care physician with a painless mass in his anterior thigh. The mass has been present for the past 3 months and he believes that it is enlarging. The patient has a history of well-controlled hypertension and hyperlipidemia. His medications include atorvastatin and hydrochlorothiazide. He has no known drug allergies. Family history is notable for diabetes and hypertension. He drinks 4 to 5 alcoholic drinks a week and he is a former smoker. He quit smoking in his 30s and only smoked intermittently prior to quitting. He denies any illicit drug use. He works as a high school principal. Currently, he feels well. His review of systems is otherwise noncontributory.
Physical Examination
On physical exam, he is afebrile with a blood pressure of 132/75 mm Hg, respiratory rate of 10 breaths/min, and oxygen saturation of 99% on room air. He is a well appearing, overweight male. His head and neck exam is unremarkable. Lung exam reveals clear breath sounds, and cardiac exam reveals a regular rate and rhythm. His abdomen is obese, soft, and without hepatosplenomegaly. There is a large, fixed mass on the anterior lateral aspect of his right thigh. He has no appreciable lymphadenopathy. His neurological exam is unremarkable.
• What are risk factors for sarcoma?
There are few known risk factors for sarcoma. Established risks factors include prior radiation therapy, chronic lymphedema, viruses, and genetic cancer syndromes including Li-Fraumeni syndrome, hereditary retinoblastoma, and NF-1. Other environmental exposures include phenoxyacetic acids and chlorophenols.14 The majority of cases are sporadic, with only a minority of patients having one of these known risk factors.15 Up to one third of sarcomas have a specific translocation and are driven by fusion oncogenes (Table 1).
A painless mass is the most typical presenting symptom. Size at presentation varies based on location, with extremity and head and neck locations typically presenting at smaller sizes than retroperitoneal tumors.14 Patients may experience pain and numbness as the mass enlarges and impinges on surrounding structures including nerves and vasculature. The vast majority of patients are without systemic symptoms.
• How is sarcoma staged?
The American Joint Committee on Cancer (AJCC) staging system is the most widely used staging system in the United States. The latest AJCC manual was updated in 2010 to include a 3-tiered grading system where the tumor is classified according to tumor size, lymph node involvement, metastases, and grade at time of diagnosis (Table 2 and Table 3). Additionally, tumor depth in relation to deep fascia is also taken into account, with superficial tumors being assigned a designation of “a” and deep tumors a designation of “b.”
Previously, 2 of the most widely used grading systems were the National Cancer Institute (NCI) and French Federation of Cancer Centers Sarcoma Group (FNCLCC) systems, both 3-tier grading systems. The main components that determine the NCI grade are the tumor’s histologic type and location and the amount of tumor necrosis. The FNCLCC system evaluation focuses on tumor differentiation, mitotic rate, and amount of tumor necrosis. A study that compared the NCI and FNCLCC grading systems found that FNCLCC was a better predictor of mortality and distant metastasis.16 Previously, the AJCC was a 4-tier grading system, but the 2010 version was updated to the 3-tier FNCLCC grading system. Additionally, the AJCC system has reclassified single lymph node disease as stage III as it confers better survival than metastatic disease.17 It is important that pathology be evaluated by a sarcoma specialist as disagreements with regard to histologic subtype and grade are common.18,19
• What are the most important prognostic factors?
Prognostic factors include grade, size, and presence of metastases at presentation. Best survival is associated with low-grade, small tumors with no metastases at time of diagnosis.14
• What imaging should be considered?
Imaging should be undertaken to help differentiate between benign and malignant lesions. Ideally, it should be undertaken before a biopsy is planned as the imaging can be used to plan biopsy as well as provide invaluable prognostic information. There are several imaging modalities that should be considered during the preliminary work-up and staging of STSs. Conventional imaging includes magnetic resonance imaging (MRI) of the original tumor site; computed tomography (CT) to evaluate for pulmonary metastases and, depending on location, liver metastases; and in the case of small, low-grade tumors, chest radiography. MRI is considered the test of choice for soft tissue masses and can help delineate benign masses such as hematomas, lipomas, and hemangiomas from sarcomas.20 It is difficult to compare the accuracy of positron emission tomography (PET)/CT to CT and MRI because most studies have evaluated PET/CT in parallel with CT and MRI.21 Tateishi et al compared the accuracy of conventional imaging, PET/CT, and PET/CT combined with conventional imaging at determining the TNM staging for 117 patients. They found that conventional imaging correctly classified 77% of patients, PET alone correctly classified 70%, PET/CT correctly classified 83%, and PET/CT combined with conventional imaging correctly staged 87%.22
• Which subtypes are most likely to metastasize?
Although the vast majority of sarcomas spread hematogenously, 3 have a propensity to spread lymphogenously: epithelioid sarcoma, rhabdomyosarcoma, and clear-cell sarcoma. Additionally, certain subtypes are more likely to metastasize: leiomyosarcomas, synovial sarcomas, neurogenic sarcomas, rhabdomyosarcomas, and epithelioid sarcomas.23 Sarcomas metastasize to the lungs more frequently than to the liver. The metastatic pattern is defined primarily by sarcoma subtype and site of primary tumor. Sarcomas rarely metastasize to the brain (~1%).
MANAGEMENT
CASE CONTINUED
The patient undergoes an ultrasound to better visualize the mass. Given the heterogeneous character of the mass, he is referred for an MRI to evaluate the mass and a CT scan of the chest, abdomen, and pelvis to evaluate for distant metastases. MRI reveals a 5.1 cm × 4.6 cm heterogeneous mass invading the superficial fascia of the rectus femoris muscle. No suspicious lymph nodes or other masses are identified on imaging. The patient next undergoes an image-guided core needle biopsy. Pathology from that procedure is consistent with a stage III, T2bNxMx, grade 3, dedifferentiated liposarcoma.
• What is the best management approach for this patient?
SURGERY
Surgery is the mainstay of treatment for STS. Patients with the best prognosis are those who undergo complete resection with negative surgical margins.24,25 Goal tumor-free margin is 1 to 3 cm.26 Complete resection confers the best long-term survival. Both local and metastatic recurrence is higher in patients with incomplete resection and positive margins.24,25 In a study that analyzed 2084 localized primary STSs, patients with negative margins had a local recurrence rate of 15% versus a rate of 28% in patients with positive margins. This translated into higher 5-year local recurrence-free survival for patients with negative surgical margins (82%) compared to patients with positive margins (65%).27 Another study similarly found that patients with negative margins at referral to their institution who underwent postoperative radiation had high local control rates of 93% (95% confidence interval [CI] 87% to 97%) at 5, 10, and 15 years.26 Although radiation improves local control, neither preoperative or postoperative radiation has been shown to improve progression-free or overall survival.28 Other factors that are associated with risk of recurrence are tumor location, history of previous recurrence, age of patient, histopathology, tumor grade, and tumor size. Approximately 40% to 50% of patients with high-grade tumors (defined as size > 5 cm, deep location, and high grade) will develop distant metastases.29
Zagars et al found that positive or uncertain resection margin had a relative risk of local recurrence of 2.0 (95% CI 1.3 to 3.1; P = 0.002), and presentation with locally recurrent disease (vs new tumor) had a relative risk of local recurrence of 2.0 (95% CI 1.2 to 3.4; P = 0.013).26 Patients with STS of head and neck and deep trunk have higher recurrence rates than those with superficial trunk and extremity STS. A single-institution retrospective review demonstrated that patients with completely resectable retroperitoneal sarcomas have longer median survival (103 months) compared to patients with incompletely resected abdominal sarcomas (18 months).25
Rosenberg and colleagues compared amputation to limb-sparing surgery and radiation.24 Their prospective analysis of 65 patients found no difference in disease-free and overall survival between the 2 treatment groups. The limb-sparing treatment group had higher rates of local recurrence, which was highly correlated with positive surgical margins on pathology.24 Evidence from this and similar studies has resulted in radical amputations being replaced by conservative limb-sparing procedures and radiation therapy. In those found to have positive margins, re-resection is an option for some. Patients who undergo re-resection have higher local control rates than patients with positive margins who do not undergo re-resection. The 5-year control rate for patients who undergo re-resection is 85% (95% CI 80% to 89%) compared to 78% (95% CI 71% to 83%) for those who do not undergo re-resection. Similarly, patients who undergo re-resection have lower rates of metastases at 5, 10, and 15 years as well as higher 5-, 10-, and 15-year disease-free survival rates.26
CASE CONTINUED
The patient is referred for limb-sparing surgery after presentation at a multidisciplinary tumor board. Prior to undergoing resection of the tumor, he is also referred to radiation-oncology to discuss the risks and benefits of combination radiotherapy and surgery as opposed to surgical resection alone.
• What is the evidence for radiation therapy?
RADIATION THERAPY
Radiation therapy is used in the preoperative, intraoperative, and postoperative settings to reduce the risk of local recurrence. There are several options for radiation, including external beam radiation therapy (EBRT), intraoperative radiation, and brachytherapy. A newer strategy, intensity-modulated radiation therapy (IMRT), utilizes 3-dimensional modeling to reduce radiation dosages. Overall there are no differences in overall survival or local recurrence rates between preoperative and postoperative radiation in STS.28
The rationale behind preoperative radiation is that it reduces seeding of tumor cells, especially at the time of surgery.30 Additionally, for EBRT, preoperative radiation has smaller field sizes and lower radiation doses. It can also help to reduce the size of the tumor prior to resection. Intraoperative radiation is often paired with preoperative radiation as a boost dose given only to the area of residual tumor.
Suit et al reviewed patients treated at a single institution with limb-sparing surgery and different radiation strategies. Local control rates between preoperative and postoperative radiation groups were not statistically significant. Local recurrence was linked to grade and size of the tumor in both groups. The authors did note, however, that the preoperative radiation group tended to have larger tumor sizes at baseline compared to the patients who received postoperative radiation.30 A study that compared 190 patients who received preoperative and postoperative EBRT or brachytherapy (primary end point was wound complications, and local control was a secondary end point) showed a trend towards greater local control with preoperative radiation; however, the preoperative radiation group had significantly more wound complications compared to the postoperative radiation group.31
Yang et al found that postoperative EBRT decreases rates of local recurrence compared to surgery alone in high-grade extremity sarcomas.32 However, there were no differences in rates of distant metastases and overall survival between the 2 treatment groups. Similarly, in patients with low-grade sarcoma, there were fewer local recurrences in those who received EBRT and surgery as compared to surgery alone.32 Another study that evaluated 164 patients who received either adjuvant brachytherapy or no further therapy after complete resection found that brachytherapy reduced local recurrence in high-grade sarcomas. No difference in local recurrence rates was found in patients with low-grade sarcomas, nor was a significant difference found in the rates of distant metastases and overall survival between the 2 treatment groups.33 With regards to IMRT, a single institution cohort experience with 41 patients who received IMRT following limb-sparing surgery had similar local control rates when compared to historical controls.34
CASE CONTINUED
After discussion of the risks and benefits of radiation therapy, the patient opts for preoperative radiation prior to resection of his liposarcoma. He receives 50 Gy of EBRT prior to undergoing resection. Resection results in R1 margin consistent with microscopic disease. He receives 16 Gy of EBRT as a boost after recovery from his resection.2
• What is the evidence for neoadjuvant and adjuvant chemotherapy for stage I tumors?
CHEMOTHERAPY
Localized Sarcoma
For localized sarcoma, limb-sparing resection with or without radiation forms the backbone of treatment. Studies have evaluated chemotherapy in both the neoadjuvant and adjuvant settings, with the vast majority of studies evaluating doxorubicin-based chemotherapy regimens in the adjuvant settings. Due to the rare nature of sarcomas, most studies are not sufficiently powered to detect significant benefit from chemotherapy. Several trials evaluating chemotherapy regimens in the neoadjuvant and adjuvant settings needed to be terminated prematurely due to inadequate enrollment into the study. 35,36
For stage IA (T1a-Tb, N0, M0, low grade) tumors, no additional therapy is recommended after limb-sparing surgery with appropriate surgical margins. For stage IB (T2a-2b, N0, M0, low grade) tumors with insufficient margins, re-resection and radiation therapy should be considered, while for stage IIA (T1a-1b, N0, M0, G2-3) tumors preoperative or postoperative radiation therapy is recommended.2 Studies have not found benefit of adjuvant chemotherapy in these low-grade, stage I tumors in terms of progression-free survival and overall survival.37
• At what stage should chemotherapy be considered?
For stage IIb and stage III tumors, surgery and radiation therapy again form the backbone of therapy; however, neoadjuvant and adjuvant chemotherapy are also recommended as considerations. Anthracycline-based chemotherapy with either single-agent doxorubicin or doxorubicin and ifosfamide in combination are considered first-line chemotherapy agents in locally advanced STS.2,29,37
Evidence regarding the efficacy of both neoadjuvant and adjuvant chemotherapy regimens in the setting of locally advanced high-grade STS has been mixed. The Sarcoma Meta-analysis Collaboration evaluated 14 trials of doxorubicin-based adjuvant chemotherapy and found a trend towards overall survival in the treatment groups that received chemotherapy.37 All trials included in the meta-analysis compared patients with localized resectable soft-tissue sarcomas who were randomized to either adjuvant chemotherapy or no adjuvant chemotherapy after limb-sparing surgery with or without radiation therapy. None of the individual trials showed a significant benefit, and all trials had large confidence intervals; however, the meta-analysis showed significant benefit in the chemotherapy treatment groups with regard to local recurrence, distant recurrence, and progression-free survival. No significant difference in overall survival was found.37 Pervais et al updated the Sarcoma Meta-analysis Collaboration’s 1997 meta-analysis with the inclusion of 4 new trials that evaluated doxorubicin combined with ifosfamide and found that both patients who received doxorubicin-based regimens or doxorubicin with ifosfamide had significant decreases in distant and overall recurrences. Only the trials that utilized doxorubicin and ifosfamide had an improved overall survival that was statistically significant (hazard ratio 0.56 [95% CI 0.36 to 0.85]; P = 0.01).29 Although no significant heterogeneity was found among the trials included in either meta-analysis, a variety of sarcomas were included in each clinical trial evaluated. Given the extremely small number of each sarcoma subtype present in each trial, subgroup analysis is difficult and prone to inaccuracies. As a result, it is not known if certain histological subtypes are more or less responsive to chemotherapy.37–39
One randomized controlled trial evaluated neoadjuvant chemotherapy in high-risk sarcomas defined as tumors greater than 8 cm or grade II/III tumors. This study evaluated doxorubicin and ifosfamide and found no significant difference in disease-free and overall survival in the neoadjuvant therapy group compared to the control group.35 There remains controversy in the literature with regards to adjuvant chemotherapy. Many oncologists offer adjuvant chemotherapy to patients with certain stage III subtypes. Examples of subtypes that may be offered adjuvant therapy include myxoid liposarcomas, synovial sarcomas, and leiomyosarcomas.2 With regards to how many cycles of chemotherapy should be considered, a noninferiority study compared 3 cycles of epirubicin and ifosfamide to 5 cycles of epirubicin and ifosfamide in patients with high-risk locally advanced adult STSs. Three cycles of preoperative epirubicin and ifosfamide was found to be noninferior to 5 cycles with regards to overall survival.38
• What is this patient’s risk for recurrence?
The patient is at intermediate risk for recurrence. Numerous studies have demonstrated that tumor size, grade, and location are the most important factors to determine risk of recurrence, with larger size, higher grades, and deeper locations being associated with higher risk of recurrence. In an analysis of 1041 patients with STS of the extremities, high grade was the most important risk factor for distant metastases.39 The highest risk of recurrence is within the first 2 years. Given that the patient’s initial tumor was located in the extremity, he is more likely to have a distant metastasis as his site of recurrence; individuals with retroperitoneal tumors and visceral tumors are more likely to recur locally.40 For STSs of the extremity, distant metastases determine overall survival, whereas patients with retroperitoneal sarcomas can die from complications of local metastases.41 Once a patient develops distant metastases, the most important prognostic factor is the size of the tumor, with tumors larger than 10 cm having a relative risk of 1.5 (95% CI 1.0 to 2.0).39
• What are the recommendations for surveillance?
Surveillance recommendations are based on the stage of the sarcoma. Stage I tumors are the least likely to recur either locally or distally. As a result, it is recommended that stage I tumors be followed with history and physical exam every 3 to 6 months for the first 2 to 3 years, and then annually after the first 2 to 3 years. Chest x-rays should be considered every 6 to 12 months.2 For stage II–IV tumors, history and physical exam is recommended every 3 to 6 months for the first 2 to 3 years. Chest and distant metastases imaging should also be performed every 3 to 6 months during this time frame. For the next 2 years, history and physical exam and imaging are recommended every 6 months. After the first 4 to 5 years, annual follow-up is recommended.2
A study that followed 141 patients with primary extremity STSs for a median interval of 49 months found that high-grade tumors were most likely to recur during the first 2 years, with 20% of their patients recurring locally and 40% recurring distally. Chest x-rays performed during surveillance follow-up found distant lung metastases in 36 asymptomatic patients and had a positive predictive value of 92%, a negative predictive value of 97%, and a quality-adjusted life-year of $30,000.40,41 No laboratory testing was found to aid in detection of recurrence.
CASE CONTINUED
The patient does well for 1 year. With physical therapy, he regains most of the strength and coordination of the lower extremity. He is followed every 3 months with chest x-rays and a MRI of the thigh for the first year. On his fourth follow-up clinic visit, he describes increased dyspnea on exertion over the previous few weeks and is found to have multiple lung metastases in both lungs on chest x-ray. He undergoes further evaluation for metastases and is not found to have any other metastatic lesions. Bronchoscopy and biopsy of 1 of the lung nodules confirms recurrent dedifferentiated liposarcoma.
• Should this patient undergo metastectomy?
An analysis of 3149 patients with STS treated at Memorial Sloan-Kettering who developed lung metastases found that patients with pulmonary metastases have survival rates of 25%. The most important prognostic factor for survival was complete resection of all metastases.42 For stage IV disease, surgery is used only in certain instances. In instances where tumor is more localized or limited, removal of metastases or metastectomy can play a role in management.2
CASE CONTINUED
Because the patient’s metastases are limited to the lungs, he is referred for metastectomy. He undergoes wedge resection for definitive diagnosis but it is not possible to completely resect all of the metastases. He is thus referred to a medical oncologist to discuss his treatment options.
• What are treatment options for unresectable or metastatic disease?
Metastatic Disease
Unlike local and locally advanced disease, chemotherapy forms the backbone of treatment in stage IV disease. Doxorubicin and olaratumab or doxorubicin and ifosfamide in combination are considered first line in metastatic disease. Response rates for single-agent doxorubicin range from 16% to 27%, while phase 2 and phase 3 studies of doxorubicin and ifosfamide have found response rates ranging from 18% to 36%.43 In addition, the effectiveness of doxorubicin and ifosfamide phase 2 and 3 trials varied. Edmonson et al found a tumor regression rate of 34% for doxorubicin and ifosfamide as compared to 20% for doxorubicin alone.44 In comparison, Santoro et al found a response rate of 21.3% for doxorubicin alone and 25.2% for doxorubicin and ifosfamide.45 Neither study found increased survival benefit for doxorubicin and ifosfamide when compared to doxorubicin alone. In a Cochrane review evaluating randomized trials that compared doxorubicin and combination chemotherapy regimens, response rates varied from 14% for doxorubicin in combination with streptomycin to 34% for doxorubicin and ifosfamide. Most trials did not show a significant benefit for combination therapies when compared to doxorubicin alone.43 Mean survival with doxorubicin or doxorubicin and ifosfamide is 12 months. High rates of recurrence highlight the need for additional chemotherapy regimens.
The newest approved agent is olaratumab, a monoclonal antibody that binds platelet-derived growth factor receptor alpha and prevents receptor activation. A phase 1-b and phase 2 trial evaluated patients with locally advanced and metastatic STS and randomly assigned them to either olaratumab and doxorubicin or doxorubicin alone.46 Progression-free survival for olaratumab/doxorubicin was 6.6 months (95% CI 4.1 to 8.3) compared to 4.1 months (95% CI 2.8 to 5.4) for doxorubicin alone. The objective response rate was 18.2% (95% CI 9.8 to 29.6) for olaratumab/doxorubicin compared to 7.5% (95% CI 2.5 to 6.6) for doxorubicin alone. Furthermore, the median overall survival for olaratumab plus doxorubicin was 26.5 months (95% CI 20.9 to 31.7) compared to 14.7 months for doxorubicin alone (95% CI 5.5 to 26.0). Impressively, this improved response was notable across histological types. Furthermore, patients who had previously been treated with more than 1 regimen and those who were treatment naïve had similar response rates.46
• What are second-line treatment options?
Doxorubicin has been used in combination with several other agents including dacarbazine (DTIC) as well as DTIC and ifosfamide (MAID). Borden et al evaluated patients with metastatic STS and randomly assigned the patients to either doxorubicin or doxorubicin and DTIC. Combination therapy demonstrated better tumor response than doxorubicin alone: 30% complete or partial response for combination therapy and 18% for doxorubicin alone.47 However, Omura et al
found similar rates of efficacy between doxorubicin and combination doxorubicin and DTIC in women with recurrent or nonresectable uterine sarcomas.48 MAID has never been directly compared in a randomized trial to doxorubicin alone. In a study that compared MAID to doxorubicin and DTIC (AD) in patients with unresectable or metastatic sarcomas, MAID had superior response rates (32% versus 17%), but there was no difference with regards to overall survival (mean survival of 12.5 months).49
Several additional regimens have undergone evaluation in metastatic and recurrent STSs. Gemcitabine has been used both as a single agent and as part of combination therapy in many studies. Studies with gemcitabine in combination with either docetaxel or DTIC have been the most efficacious. In a phase 2 trial, patients with metastatic STS were randomly assigned to either gemcitabine alone or gemcitabine and docetaxel. Combination therapy had a higher response rate (16% versus 8%) and longer overall survival (17.9 months versus 11.5 months) than gemcitabine alone.50 Furthermore, a phase 2 trial of gemcitabine and docetaxel in patients with unresectable leiomyosarcoma showed an overall response rate of 56%, with 3 complete and 15 partial responses among the 34 patients enrolled in the study.51
A phase 2 trial randomly assigned patients with unresectable or metastatic STS to either DTIC or combination gemcitabine and DTIC.52 Gemcitabine-DTIC had a superior progression-free survival at 3 months (56% [95% CI 43% to 69%]) as compared to DTIC alone (37% [95% CI 23.5% to 50%]). Furthermore, mean progression-free survival and overall survival were improved in the gemcitabine-DTIC group (4.2 months and 16.8 months) as compared to the DTIC group (2.0 months and 8.2 months).52 DTIC has a single-agent response rate of 16%, but has been shown to be particularly effective in the setting of leiomyosarcomas.49
• Does response to treatment regimens differ by histologic subtype?
The majority of STS trials include many different histologic subtypes. Given the rarity of sarcomas as a whole, many trials have had difficulty recruiting adequate numbers of patients to have sufficient power to definitely determine if the treatment under investigation has clinical benefit. Furthermore, the patients recruited have been heterogeneous with regard to subtype. Many older studies hypothesized that the efficacy of chemotherapeutic agents vary based on histologic subtype; however, for most subtypes the number of individuals included in those trials was too low to evaluate efficacy based on subtype.
Some exceptions exist, however. For example, both gemcitabine-DTIC and gemcitabine-docetaxel have been found to be particularly effective in the treatment of leiomyosarcomas.50,52 Additionally, a retrospective study found a 51% overall response rate for patients with myxoid liposarcomas treated with trabectedin.53 Studies of patients with angiosarcoma treated with paclitaxel have demonstrated response rates of 43% and 53%.54,55
• What are the newest approved and investigational agents?
A recently approved agent is trabectedin, a tris tetrahydroisoquinoline alkaloid isolated from ascidians that binds to the minor groove of DNA and causes disruptions in the cell cycle. Samuels et al reported data from a single-arm, open-label expanded access trial that evaluated patients with advanced metastatic sarcomas.56 In this study, patients with liposarcomas and leiomyosarcomas had an objective response rate of 6.9% (95% CI 4.8 to 9.6) as compared to a rate of 5.9% (95% CI 4.4 to 7.8) for all assessable patients. Median survival was 11.9 months for all patients, with improved median survivals for liposarcoma and leiomyosarcomas of 16.2 months (95% CI 14.1 to 19.5) compared to 8.4 months (95% CI 7.1 to 10.7 months) for other subtypes.56
Schöffski et al evaluated eribulin, a chemotherapeutic agent that affects microtubule dynamics, in a phase 2 trial of patients with progressive or high-grade STS with progression on previous chemotherapy. They found a median progression-free survival of 2.6 months (95% CI 1.7 to 6.2) for adipocytic sarcoma, 2.9 months (95% CI 2.4 to 4.6) for leiomyosarcoma, 2.6 months (95% CI 2.3 to 4.3) for synovial sarcoma, and 2.1 months (95% CI 1.4 to 2.9) for other sarcomas.57
Van der Graaf and colleagues randomly assigned patients with metastatic nonadipocytic STS to pazopanib or placebo in a phase 3 trial. Pazopanib is a small-molecule endothelial growth factor inhibitor with activity against vascular endothelial growth factors 1, 2, and 3 as well as platelet-derived growth factors. Median progression-free survival was 4.6 months (95% CI 3.7 to 4.8) with pazopanib compared to 1.6 months (95% CI 0.9 to 1.8) with placebo.58 Adipocytic sarcomas (liposarcomas) were excluded from the trial because phase 2 trials had found a lower rate of progression-free survival (26%) for them compared to other subtypes.
• What are the most common toxicities associated with the approved and investigational chemotherapeutic agents?
Toxicities were seen with each of the regimens studied and were common in the randomized trials, with higher rates of toxicities in the combination chemotherapy regimens. The most common toxicities are myelosuppression, nausea, and vomiting. In the doxorubicin trials, the most common toxicities were myelosuppression, nausea, and vomiting.44
Ifosfamide both as an individual agent and in combination with doxorubicin has higher rates and higher grades of toxicity than doxorubicin alone. Myelosuppression is the most common toxicity associated with ifosfamide, and the most commonly affected cell line is leukocytes.44 Combination doxorubicin and ifosfamide also had high rates of nausea and vomiting (95%) and alopecia (100%).35
Neutropenia is the most common toxicity associated with gemcitabine and dacarbazine, while their most common nonhematologic toxicities are fatigue and nausea.52,59 Trabectedin’s most common toxicities are nausea (29%), neutropenia (24%), and fatigue (23%). It has also been shown to cause increased alkaline phosphatase (20%) and alanine aminotransferase (19%) levels.56 In a phase 2 study of eribulin, 50% of patients had neutropenia, and other toxicities included fatigue, alopecia, nausea, sensory neuropathy, and thrombocytopenia.57 Pazopanib is generally well tolerated; the most common toxicities are fatigue (65%), diarrhea (58%), nausea (54%), and hypertension (41%).58 Higher rates of neutropenia, mucositis, nausea, vomiting, diarrhea, and transfusion reactions were seen with olaratumab and doxorubicin compared to doxorubicin alone in phase 1b and 2 studies.46
CASE CONCLUSION
Given his poor prognosis with unresectable metastatic undifferentiated liposarcoma, the patient considers a clinical trial prior to undergoing combined therapy with doxorubicin and ifosfamide. He tolerates therapy well with stable disease at 6 months.
CONCLUSION
STSs are a heterogeneous collection of rare tumors. Low-grade, localized tumors have the best prognosis, and patients who undergo complete resection have the best long-term survival. Due to the rarity of STSs, trials often have limited enrollment, and little progress has been made with regards to treatment and survival rates for metastatic and unresectable disease. All patients should be evaluated and treated at specialized sarcoma centers. This case highlights the need for continued research and clinical trials to improve overall survival of patients with sarcoma.
INTRODUCTION
Soft tissue sarcomas (STSs) are rare adult tumors, with 3.4 new cases per 100,000 persons or 12,310 expected new cases in 2016.1 Sarcomas are a heterogeneous collection of tumors that affect fat, muscle, nerve, nerve sheath, vascular, and connective tissues. There are more than 50 histological subtypes that comprise this diverse category of tumors. Treatment varies by stage, with limb-sparing surgery representing the mainstay of curative-intent treatment. Radiation and chemotherapy may also be considered depending on the size, grade, and location of the tumor. Survival rates have been stagnant until recently, with a disease-specific survival hovering around 65%.1 Given the complexity of these cases, all patients ideally should be evaluated and treated by a multidisciplinary team at an institution with extensive experience treating STS.2
EPIDEMIOLOGY AND CLASSIFICATION
The most common STS subtypes are gastrointestinal stromal tumor (GIST), undifferentiate pleomorphic sarcoma (previously referred to as malignant fibrous histiocytoma), liposarcoma, leiomyosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, and unclassified sarcoma.3 Liposarcoma is one of the most common subtypes, comprising 20% of all STSs; it is subdivided into well-differentiated/dedifferentiated liposarcomas, myxoid/round cell liposarcomas, and pleomorphic liposarcomas. Well-differentiated liposarcomas tend to occur in the retroperitoneum and limbs, while both myxoid and round cell as well as pleomorphic liposarcomas more commonly originate on the limbs. Histology varies based on subtype and ranges from mature-appearing adipocytes and fibroblasts to undifferentiated cells with minimal lipogenic differentiation.4
Leiomyosarcomas are smooth muscle tumors and are usually located in the retroperitoneum, but have also been associated with peripheral soft tissue and vasculature. Typical histology ranges from well-defined areas of spindle-shaped cells to poorly differentiated anaplastic spindle cells.5,6 Synovial sarcomas are a distinct type of STS that can show epithelial differentiation and account for 5% of adult STSs. The extremities are the most common presenting location (90%).7
Rhabdomyosarcomas are skeletal muscle tumors and are further subdivided into embryonal, alveolar, and pleomorphic subtypes. Embryonal histology ranges from primitive mesenchymal-appearing cells to highly differentiated muscle cells. Alveolar rhabdomyosarcoma has the worst prognosis of the subtypes and consists of round cells with high nuclear-to-chromatin ratios that form “glandular-like” or “alveolar” spaces.8 Pleomorphic rhabdomyosarcomas are composed of rhabdomyoblasts that can affect many different locations, but most commonly present on the lower extremities.9
Malignant peripheral nerve sheath tumor (MPNST) comprises 5% to 10% of all STSs. These tumors are associated with neurofibromatosis type 1 (NF-1), with 25% to 50% of tumors occurring in NF-1 patients. Additionally, most patients have a truncating lesion in the NF1 gene on chromosome 17.10 Anghileri et al in their single institution analysis of 205 patients with MPNSTs found the 2 most common presenting sites were the trunk and extremities. Histologically, these tumors have dense fascicles of spindle cells.10
GISTs are the most common STS of the gastrointestinal (GI) tract. Previously, GISTs were classified as smooth muscle tumors and were not accounted for in the literature as a separate entity distinct from leiomyomas, leiomyoblastomas, and leiomyosarcomas.11 GISTs are found throughout the GI tract: the most common sites are the stomach (60%) and small intestine (30%). Less common sites include duodenum (4%–5%), esophagus (1%), rectum (1%–2%), and appendix (< 0.2%).12 GISTs can be spindle cell, epithelioid, or mesenchymal tumors. Immunohistochemically, GISTs are KIT (CD117) positive. Other cell markers that are also commonly positive include CD34 (60%–70%) and smooth muscle actin (SMA) (25%).11 The majority of GISTs (80%) have an activating c-KIT gene mutation. The most common mutation site is exon 11, with less common c-KIT gene mutations also occurring at exon 9 or 13. Not all GISTs have KIT mutations. The second most common mutation is the PDGFRA mutation (5%–10% of GISTs).2 A minority of GISTs are negative for both KIT and PDGFRA mutations. These tumors were previously called wild-type, but as the majority have either a succinate dehydrogenase (SDH) loss of function or loss of SDHB protein expression, they are now referred to as SDH-deficient GISTs.2 GISTs vary in aggressiveness from incidental to aggressive. Typically, small intestine and rectal GISTs are more aggressive than gastric GISTs. Both size and mitotic rate help to predict the metastatic potential of the tumor. Tumors less than 2 cm in size and having a mitotic rate of less than 5 per 50 high-power fields (hpf) have the lowest risk of metastases, while tumors greater than 5 cm and with more than 5 mitoses per 50 hpf have the highest rates of metastases.12
Angiosarcomas are rare tumors comprising 4% of all STSs. Although they can occur in any site, the majority are cutaneous and occur most frequently in the head and neck regions. These tumors are either of vascular or lymphatic origin and are comprised of abnormal, pleomorphic, malignant endothelial cells. The most useful immunohistochemical markers include von Willebrand factor, CD31, and Ulex europaeus agglutinin 1. The majority of these tumors occur sporadically; however, radiation exposure, chronic lymphedema, and certain toxins including vinyl chloride and thorium dioxide are known risk factors.13
Undifferentiated sarcomas have no specific features and typically consist of primitive mesenchymal cells.
CLINICAL EVALUATION
CASE PRESENTATION
Initial Presentation and History
A 55-year-old man presents to his primary care physician with a painless mass in his anterior thigh. The mass has been present for the past 3 months and he believes that it is enlarging. The patient has a history of well-controlled hypertension and hyperlipidemia. His medications include atorvastatin and hydrochlorothiazide. He has no known drug allergies. Family history is notable for diabetes and hypertension. He drinks 4 to 5 alcoholic drinks a week and he is a former smoker. He quit smoking in his 30s and only smoked intermittently prior to quitting. He denies any illicit drug use. He works as a high school principal. Currently, he feels well. His review of systems is otherwise noncontributory.
Physical Examination
On physical exam, he is afebrile with a blood pressure of 132/75 mm Hg, respiratory rate of 10 breaths/min, and oxygen saturation of 99% on room air. He is a well appearing, overweight male. His head and neck exam is unremarkable. Lung exam reveals clear breath sounds, and cardiac exam reveals a regular rate and rhythm. His abdomen is obese, soft, and without hepatosplenomegaly. There is a large, fixed mass on the anterior lateral aspect of his right thigh. He has no appreciable lymphadenopathy. His neurological exam is unremarkable.
• What are risk factors for sarcoma?
There are few known risk factors for sarcoma. Established risks factors include prior radiation therapy, chronic lymphedema, viruses, and genetic cancer syndromes including Li-Fraumeni syndrome, hereditary retinoblastoma, and NF-1. Other environmental exposures include phenoxyacetic acids and chlorophenols.14 The majority of cases are sporadic, with only a minority of patients having one of these known risk factors.15 Up to one third of sarcomas have a specific translocation and are driven by fusion oncogenes (Table 1).
A painless mass is the most typical presenting symptom. Size at presentation varies based on location, with extremity and head and neck locations typically presenting at smaller sizes than retroperitoneal tumors.14 Patients may experience pain and numbness as the mass enlarges and impinges on surrounding structures including nerves and vasculature. The vast majority of patients are without systemic symptoms.
• How is sarcoma staged?
The American Joint Committee on Cancer (AJCC) staging system is the most widely used staging system in the United States. The latest AJCC manual was updated in 2010 to include a 3-tiered grading system where the tumor is classified according to tumor size, lymph node involvement, metastases, and grade at time of diagnosis (Table 2 and Table 3). Additionally, tumor depth in relation to deep fascia is also taken into account, with superficial tumors being assigned a designation of “a” and deep tumors a designation of “b.”
Previously, 2 of the most widely used grading systems were the National Cancer Institute (NCI) and French Federation of Cancer Centers Sarcoma Group (FNCLCC) systems, both 3-tier grading systems. The main components that determine the NCI grade are the tumor’s histologic type and location and the amount of tumor necrosis. The FNCLCC system evaluation focuses on tumor differentiation, mitotic rate, and amount of tumor necrosis. A study that compared the NCI and FNCLCC grading systems found that FNCLCC was a better predictor of mortality and distant metastasis.16 Previously, the AJCC was a 4-tier grading system, but the 2010 version was updated to the 3-tier FNCLCC grading system. Additionally, the AJCC system has reclassified single lymph node disease as stage III as it confers better survival than metastatic disease.17 It is important that pathology be evaluated by a sarcoma specialist as disagreements with regard to histologic subtype and grade are common.18,19
• What are the most important prognostic factors?
Prognostic factors include grade, size, and presence of metastases at presentation. Best survival is associated with low-grade, small tumors with no metastases at time of diagnosis.14
• What imaging should be considered?
Imaging should be undertaken to help differentiate between benign and malignant lesions. Ideally, it should be undertaken before a biopsy is planned as the imaging can be used to plan biopsy as well as provide invaluable prognostic information. There are several imaging modalities that should be considered during the preliminary work-up and staging of STSs. Conventional imaging includes magnetic resonance imaging (MRI) of the original tumor site; computed tomography (CT) to evaluate for pulmonary metastases and, depending on location, liver metastases; and in the case of small, low-grade tumors, chest radiography. MRI is considered the test of choice for soft tissue masses and can help delineate benign masses such as hematomas, lipomas, and hemangiomas from sarcomas.20 It is difficult to compare the accuracy of positron emission tomography (PET)/CT to CT and MRI because most studies have evaluated PET/CT in parallel with CT and MRI.21 Tateishi et al compared the accuracy of conventional imaging, PET/CT, and PET/CT combined with conventional imaging at determining the TNM staging for 117 patients. They found that conventional imaging correctly classified 77% of patients, PET alone correctly classified 70%, PET/CT correctly classified 83%, and PET/CT combined with conventional imaging correctly staged 87%.22
• Which subtypes are most likely to metastasize?
Although the vast majority of sarcomas spread hematogenously, 3 have a propensity to spread lymphogenously: epithelioid sarcoma, rhabdomyosarcoma, and clear-cell sarcoma. Additionally, certain subtypes are more likely to metastasize: leiomyosarcomas, synovial sarcomas, neurogenic sarcomas, rhabdomyosarcomas, and epithelioid sarcomas.23 Sarcomas metastasize to the lungs more frequently than to the liver. The metastatic pattern is defined primarily by sarcoma subtype and site of primary tumor. Sarcomas rarely metastasize to the brain (~1%).
MANAGEMENT
CASE CONTINUED
The patient undergoes an ultrasound to better visualize the mass. Given the heterogeneous character of the mass, he is referred for an MRI to evaluate the mass and a CT scan of the chest, abdomen, and pelvis to evaluate for distant metastases. MRI reveals a 5.1 cm × 4.6 cm heterogeneous mass invading the superficial fascia of the rectus femoris muscle. No suspicious lymph nodes or other masses are identified on imaging. The patient next undergoes an image-guided core needle biopsy. Pathology from that procedure is consistent with a stage III, T2bNxMx, grade 3, dedifferentiated liposarcoma.
• What is the best management approach for this patient?
SURGERY
Surgery is the mainstay of treatment for STS. Patients with the best prognosis are those who undergo complete resection with negative surgical margins.24,25 Goal tumor-free margin is 1 to 3 cm.26 Complete resection confers the best long-term survival. Both local and metastatic recurrence is higher in patients with incomplete resection and positive margins.24,25 In a study that analyzed 2084 localized primary STSs, patients with negative margins had a local recurrence rate of 15% versus a rate of 28% in patients with positive margins. This translated into higher 5-year local recurrence-free survival for patients with negative surgical margins (82%) compared to patients with positive margins (65%).27 Another study similarly found that patients with negative margins at referral to their institution who underwent postoperative radiation had high local control rates of 93% (95% confidence interval [CI] 87% to 97%) at 5, 10, and 15 years.26 Although radiation improves local control, neither preoperative or postoperative radiation has been shown to improve progression-free or overall survival.28 Other factors that are associated with risk of recurrence are tumor location, history of previous recurrence, age of patient, histopathology, tumor grade, and tumor size. Approximately 40% to 50% of patients with high-grade tumors (defined as size > 5 cm, deep location, and high grade) will develop distant metastases.29
Zagars et al found that positive or uncertain resection margin had a relative risk of local recurrence of 2.0 (95% CI 1.3 to 3.1; P = 0.002), and presentation with locally recurrent disease (vs new tumor) had a relative risk of local recurrence of 2.0 (95% CI 1.2 to 3.4; P = 0.013).26 Patients with STS of head and neck and deep trunk have higher recurrence rates than those with superficial trunk and extremity STS. A single-institution retrospective review demonstrated that patients with completely resectable retroperitoneal sarcomas have longer median survival (103 months) compared to patients with incompletely resected abdominal sarcomas (18 months).25
Rosenberg and colleagues compared amputation to limb-sparing surgery and radiation.24 Their prospective analysis of 65 patients found no difference in disease-free and overall survival between the 2 treatment groups. The limb-sparing treatment group had higher rates of local recurrence, which was highly correlated with positive surgical margins on pathology.24 Evidence from this and similar studies has resulted in radical amputations being replaced by conservative limb-sparing procedures and radiation therapy. In those found to have positive margins, re-resection is an option for some. Patients who undergo re-resection have higher local control rates than patients with positive margins who do not undergo re-resection. The 5-year control rate for patients who undergo re-resection is 85% (95% CI 80% to 89%) compared to 78% (95% CI 71% to 83%) for those who do not undergo re-resection. Similarly, patients who undergo re-resection have lower rates of metastases at 5, 10, and 15 years as well as higher 5-, 10-, and 15-year disease-free survival rates.26
CASE CONTINUED
The patient is referred for limb-sparing surgery after presentation at a multidisciplinary tumor board. Prior to undergoing resection of the tumor, he is also referred to radiation-oncology to discuss the risks and benefits of combination radiotherapy and surgery as opposed to surgical resection alone.
• What is the evidence for radiation therapy?
RADIATION THERAPY
Radiation therapy is used in the preoperative, intraoperative, and postoperative settings to reduce the risk of local recurrence. There are several options for radiation, including external beam radiation therapy (EBRT), intraoperative radiation, and brachytherapy. A newer strategy, intensity-modulated radiation therapy (IMRT), utilizes 3-dimensional modeling to reduce radiation dosages. Overall there are no differences in overall survival or local recurrence rates between preoperative and postoperative radiation in STS.28
The rationale behind preoperative radiation is that it reduces seeding of tumor cells, especially at the time of surgery.30 Additionally, for EBRT, preoperative radiation has smaller field sizes and lower radiation doses. It can also help to reduce the size of the tumor prior to resection. Intraoperative radiation is often paired with preoperative radiation as a boost dose given only to the area of residual tumor.
Suit et al reviewed patients treated at a single institution with limb-sparing surgery and different radiation strategies. Local control rates between preoperative and postoperative radiation groups were not statistically significant. Local recurrence was linked to grade and size of the tumor in both groups. The authors did note, however, that the preoperative radiation group tended to have larger tumor sizes at baseline compared to the patients who received postoperative radiation.30 A study that compared 190 patients who received preoperative and postoperative EBRT or brachytherapy (primary end point was wound complications, and local control was a secondary end point) showed a trend towards greater local control with preoperative radiation; however, the preoperative radiation group had significantly more wound complications compared to the postoperative radiation group.31
Yang et al found that postoperative EBRT decreases rates of local recurrence compared to surgery alone in high-grade extremity sarcomas.32 However, there were no differences in rates of distant metastases and overall survival between the 2 treatment groups. Similarly, in patients with low-grade sarcoma, there were fewer local recurrences in those who received EBRT and surgery as compared to surgery alone.32 Another study that evaluated 164 patients who received either adjuvant brachytherapy or no further therapy after complete resection found that brachytherapy reduced local recurrence in high-grade sarcomas. No difference in local recurrence rates was found in patients with low-grade sarcomas, nor was a significant difference found in the rates of distant metastases and overall survival between the 2 treatment groups.33 With regards to IMRT, a single institution cohort experience with 41 patients who received IMRT following limb-sparing surgery had similar local control rates when compared to historical controls.34
CASE CONTINUED
After discussion of the risks and benefits of radiation therapy, the patient opts for preoperative radiation prior to resection of his liposarcoma. He receives 50 Gy of EBRT prior to undergoing resection. Resection results in R1 margin consistent with microscopic disease. He receives 16 Gy of EBRT as a boost after recovery from his resection.2
• What is the evidence for neoadjuvant and adjuvant chemotherapy for stage I tumors?
CHEMOTHERAPY
Localized Sarcoma
For localized sarcoma, limb-sparing resection with or without radiation forms the backbone of treatment. Studies have evaluated chemotherapy in both the neoadjuvant and adjuvant settings, with the vast majority of studies evaluating doxorubicin-based chemotherapy regimens in the adjuvant settings. Due to the rare nature of sarcomas, most studies are not sufficiently powered to detect significant benefit from chemotherapy. Several trials evaluating chemotherapy regimens in the neoadjuvant and adjuvant settings needed to be terminated prematurely due to inadequate enrollment into the study. 35,36
For stage IA (T1a-Tb, N0, M0, low grade) tumors, no additional therapy is recommended after limb-sparing surgery with appropriate surgical margins. For stage IB (T2a-2b, N0, M0, low grade) tumors with insufficient margins, re-resection and radiation therapy should be considered, while for stage IIA (T1a-1b, N0, M0, G2-3) tumors preoperative or postoperative radiation therapy is recommended.2 Studies have not found benefit of adjuvant chemotherapy in these low-grade, stage I tumors in terms of progression-free survival and overall survival.37
• At what stage should chemotherapy be considered?
For stage IIb and stage III tumors, surgery and radiation therapy again form the backbone of therapy; however, neoadjuvant and adjuvant chemotherapy are also recommended as considerations. Anthracycline-based chemotherapy with either single-agent doxorubicin or doxorubicin and ifosfamide in combination are considered first-line chemotherapy agents in locally advanced STS.2,29,37
Evidence regarding the efficacy of both neoadjuvant and adjuvant chemotherapy regimens in the setting of locally advanced high-grade STS has been mixed. The Sarcoma Meta-analysis Collaboration evaluated 14 trials of doxorubicin-based adjuvant chemotherapy and found a trend towards overall survival in the treatment groups that received chemotherapy.37 All trials included in the meta-analysis compared patients with localized resectable soft-tissue sarcomas who were randomized to either adjuvant chemotherapy or no adjuvant chemotherapy after limb-sparing surgery with or without radiation therapy. None of the individual trials showed a significant benefit, and all trials had large confidence intervals; however, the meta-analysis showed significant benefit in the chemotherapy treatment groups with regard to local recurrence, distant recurrence, and progression-free survival. No significant difference in overall survival was found.37 Pervais et al updated the Sarcoma Meta-analysis Collaboration’s 1997 meta-analysis with the inclusion of 4 new trials that evaluated doxorubicin combined with ifosfamide and found that both patients who received doxorubicin-based regimens or doxorubicin with ifosfamide had significant decreases in distant and overall recurrences. Only the trials that utilized doxorubicin and ifosfamide had an improved overall survival that was statistically significant (hazard ratio 0.56 [95% CI 0.36 to 0.85]; P = 0.01).29 Although no significant heterogeneity was found among the trials included in either meta-analysis, a variety of sarcomas were included in each clinical trial evaluated. Given the extremely small number of each sarcoma subtype present in each trial, subgroup analysis is difficult and prone to inaccuracies. As a result, it is not known if certain histological subtypes are more or less responsive to chemotherapy.37–39
One randomized controlled trial evaluated neoadjuvant chemotherapy in high-risk sarcomas defined as tumors greater than 8 cm or grade II/III tumors. This study evaluated doxorubicin and ifosfamide and found no significant difference in disease-free and overall survival in the neoadjuvant therapy group compared to the control group.35 There remains controversy in the literature with regards to adjuvant chemotherapy. Many oncologists offer adjuvant chemotherapy to patients with certain stage III subtypes. Examples of subtypes that may be offered adjuvant therapy include myxoid liposarcomas, synovial sarcomas, and leiomyosarcomas.2 With regards to how many cycles of chemotherapy should be considered, a noninferiority study compared 3 cycles of epirubicin and ifosfamide to 5 cycles of epirubicin and ifosfamide in patients with high-risk locally advanced adult STSs. Three cycles of preoperative epirubicin and ifosfamide was found to be noninferior to 5 cycles with regards to overall survival.38
• What is this patient’s risk for recurrence?
The patient is at intermediate risk for recurrence. Numerous studies have demonstrated that tumor size, grade, and location are the most important factors to determine risk of recurrence, with larger size, higher grades, and deeper locations being associated with higher risk of recurrence. In an analysis of 1041 patients with STS of the extremities, high grade was the most important risk factor for distant metastases.39 The highest risk of recurrence is within the first 2 years. Given that the patient’s initial tumor was located in the extremity, he is more likely to have a distant metastasis as his site of recurrence; individuals with retroperitoneal tumors and visceral tumors are more likely to recur locally.40 For STSs of the extremity, distant metastases determine overall survival, whereas patients with retroperitoneal sarcomas can die from complications of local metastases.41 Once a patient develops distant metastases, the most important prognostic factor is the size of the tumor, with tumors larger than 10 cm having a relative risk of 1.5 (95% CI 1.0 to 2.0).39
• What are the recommendations for surveillance?
Surveillance recommendations are based on the stage of the sarcoma. Stage I tumors are the least likely to recur either locally or distally. As a result, it is recommended that stage I tumors be followed with history and physical exam every 3 to 6 months for the first 2 to 3 years, and then annually after the first 2 to 3 years. Chest x-rays should be considered every 6 to 12 months.2 For stage II–IV tumors, history and physical exam is recommended every 3 to 6 months for the first 2 to 3 years. Chest and distant metastases imaging should also be performed every 3 to 6 months during this time frame. For the next 2 years, history and physical exam and imaging are recommended every 6 months. After the first 4 to 5 years, annual follow-up is recommended.2
A study that followed 141 patients with primary extremity STSs for a median interval of 49 months found that high-grade tumors were most likely to recur during the first 2 years, with 20% of their patients recurring locally and 40% recurring distally. Chest x-rays performed during surveillance follow-up found distant lung metastases in 36 asymptomatic patients and had a positive predictive value of 92%, a negative predictive value of 97%, and a quality-adjusted life-year of $30,000.40,41 No laboratory testing was found to aid in detection of recurrence.
CASE CONTINUED
The patient does well for 1 year. With physical therapy, he regains most of the strength and coordination of the lower extremity. He is followed every 3 months with chest x-rays and a MRI of the thigh for the first year. On his fourth follow-up clinic visit, he describes increased dyspnea on exertion over the previous few weeks and is found to have multiple lung metastases in both lungs on chest x-ray. He undergoes further evaluation for metastases and is not found to have any other metastatic lesions. Bronchoscopy and biopsy of 1 of the lung nodules confirms recurrent dedifferentiated liposarcoma.
• Should this patient undergo metastectomy?
An analysis of 3149 patients with STS treated at Memorial Sloan-Kettering who developed lung metastases found that patients with pulmonary metastases have survival rates of 25%. The most important prognostic factor for survival was complete resection of all metastases.42 For stage IV disease, surgery is used only in certain instances. In instances where tumor is more localized or limited, removal of metastases or metastectomy can play a role in management.2
CASE CONTINUED
Because the patient’s metastases are limited to the lungs, he is referred for metastectomy. He undergoes wedge resection for definitive diagnosis but it is not possible to completely resect all of the metastases. He is thus referred to a medical oncologist to discuss his treatment options.
• What are treatment options for unresectable or metastatic disease?
Metastatic Disease
Unlike local and locally advanced disease, chemotherapy forms the backbone of treatment in stage IV disease. Doxorubicin and olaratumab or doxorubicin and ifosfamide in combination are considered first line in metastatic disease. Response rates for single-agent doxorubicin range from 16% to 27%, while phase 2 and phase 3 studies of doxorubicin and ifosfamide have found response rates ranging from 18% to 36%.43 In addition, the effectiveness of doxorubicin and ifosfamide phase 2 and 3 trials varied. Edmonson et al found a tumor regression rate of 34% for doxorubicin and ifosfamide as compared to 20% for doxorubicin alone.44 In comparison, Santoro et al found a response rate of 21.3% for doxorubicin alone and 25.2% for doxorubicin and ifosfamide.45 Neither study found increased survival benefit for doxorubicin and ifosfamide when compared to doxorubicin alone. In a Cochrane review evaluating randomized trials that compared doxorubicin and combination chemotherapy regimens, response rates varied from 14% for doxorubicin in combination with streptomycin to 34% for doxorubicin and ifosfamide. Most trials did not show a significant benefit for combination therapies when compared to doxorubicin alone.43 Mean survival with doxorubicin or doxorubicin and ifosfamide is 12 months. High rates of recurrence highlight the need for additional chemotherapy regimens.
The newest approved agent is olaratumab, a monoclonal antibody that binds platelet-derived growth factor receptor alpha and prevents receptor activation. A phase 1-b and phase 2 trial evaluated patients with locally advanced and metastatic STS and randomly assigned them to either olaratumab and doxorubicin or doxorubicin alone.46 Progression-free survival for olaratumab/doxorubicin was 6.6 months (95% CI 4.1 to 8.3) compared to 4.1 months (95% CI 2.8 to 5.4) for doxorubicin alone. The objective response rate was 18.2% (95% CI 9.8 to 29.6) for olaratumab/doxorubicin compared to 7.5% (95% CI 2.5 to 6.6) for doxorubicin alone. Furthermore, the median overall survival for olaratumab plus doxorubicin was 26.5 months (95% CI 20.9 to 31.7) compared to 14.7 months for doxorubicin alone (95% CI 5.5 to 26.0). Impressively, this improved response was notable across histological types. Furthermore, patients who had previously been treated with more than 1 regimen and those who were treatment naïve had similar response rates.46
• What are second-line treatment options?
Doxorubicin has been used in combination with several other agents including dacarbazine (DTIC) as well as DTIC and ifosfamide (MAID). Borden et al evaluated patients with metastatic STS and randomly assigned the patients to either doxorubicin or doxorubicin and DTIC. Combination therapy demonstrated better tumor response than doxorubicin alone: 30% complete or partial response for combination therapy and 18% for doxorubicin alone.47 However, Omura et al
found similar rates of efficacy between doxorubicin and combination doxorubicin and DTIC in women with recurrent or nonresectable uterine sarcomas.48 MAID has never been directly compared in a randomized trial to doxorubicin alone. In a study that compared MAID to doxorubicin and DTIC (AD) in patients with unresectable or metastatic sarcomas, MAID had superior response rates (32% versus 17%), but there was no difference with regards to overall survival (mean survival of 12.5 months).49
Several additional regimens have undergone evaluation in metastatic and recurrent STSs. Gemcitabine has been used both as a single agent and as part of combination therapy in many studies. Studies with gemcitabine in combination with either docetaxel or DTIC have been the most efficacious. In a phase 2 trial, patients with metastatic STS were randomly assigned to either gemcitabine alone or gemcitabine and docetaxel. Combination therapy had a higher response rate (16% versus 8%) and longer overall survival (17.9 months versus 11.5 months) than gemcitabine alone.50 Furthermore, a phase 2 trial of gemcitabine and docetaxel in patients with unresectable leiomyosarcoma showed an overall response rate of 56%, with 3 complete and 15 partial responses among the 34 patients enrolled in the study.51
A phase 2 trial randomly assigned patients with unresectable or metastatic STS to either DTIC or combination gemcitabine and DTIC.52 Gemcitabine-DTIC had a superior progression-free survival at 3 months (56% [95% CI 43% to 69%]) as compared to DTIC alone (37% [95% CI 23.5% to 50%]). Furthermore, mean progression-free survival and overall survival were improved in the gemcitabine-DTIC group (4.2 months and 16.8 months) as compared to the DTIC group (2.0 months and 8.2 months).52 DTIC has a single-agent response rate of 16%, but has been shown to be particularly effective in the setting of leiomyosarcomas.49
• Does response to treatment regimens differ by histologic subtype?
The majority of STS trials include many different histologic subtypes. Given the rarity of sarcomas as a whole, many trials have had difficulty recruiting adequate numbers of patients to have sufficient power to definitely determine if the treatment under investigation has clinical benefit. Furthermore, the patients recruited have been heterogeneous with regard to subtype. Many older studies hypothesized that the efficacy of chemotherapeutic agents vary based on histologic subtype; however, for most subtypes the number of individuals included in those trials was too low to evaluate efficacy based on subtype.
Some exceptions exist, however. For example, both gemcitabine-DTIC and gemcitabine-docetaxel have been found to be particularly effective in the treatment of leiomyosarcomas.50,52 Additionally, a retrospective study found a 51% overall response rate for patients with myxoid liposarcomas treated with trabectedin.53 Studies of patients with angiosarcoma treated with paclitaxel have demonstrated response rates of 43% and 53%.54,55
• What are the newest approved and investigational agents?
A recently approved agent is trabectedin, a tris tetrahydroisoquinoline alkaloid isolated from ascidians that binds to the minor groove of DNA and causes disruptions in the cell cycle. Samuels et al reported data from a single-arm, open-label expanded access trial that evaluated patients with advanced metastatic sarcomas.56 In this study, patients with liposarcomas and leiomyosarcomas had an objective response rate of 6.9% (95% CI 4.8 to 9.6) as compared to a rate of 5.9% (95% CI 4.4 to 7.8) for all assessable patients. Median survival was 11.9 months for all patients, with improved median survivals for liposarcoma and leiomyosarcomas of 16.2 months (95% CI 14.1 to 19.5) compared to 8.4 months (95% CI 7.1 to 10.7 months) for other subtypes.56
Schöffski et al evaluated eribulin, a chemotherapeutic agent that affects microtubule dynamics, in a phase 2 trial of patients with progressive or high-grade STS with progression on previous chemotherapy. They found a median progression-free survival of 2.6 months (95% CI 1.7 to 6.2) for adipocytic sarcoma, 2.9 months (95% CI 2.4 to 4.6) for leiomyosarcoma, 2.6 months (95% CI 2.3 to 4.3) for synovial sarcoma, and 2.1 months (95% CI 1.4 to 2.9) for other sarcomas.57
Van der Graaf and colleagues randomly assigned patients with metastatic nonadipocytic STS to pazopanib or placebo in a phase 3 trial. Pazopanib is a small-molecule endothelial growth factor inhibitor with activity against vascular endothelial growth factors 1, 2, and 3 as well as platelet-derived growth factors. Median progression-free survival was 4.6 months (95% CI 3.7 to 4.8) with pazopanib compared to 1.6 months (95% CI 0.9 to 1.8) with placebo.58 Adipocytic sarcomas (liposarcomas) were excluded from the trial because phase 2 trials had found a lower rate of progression-free survival (26%) for them compared to other subtypes.
• What are the most common toxicities associated with the approved and investigational chemotherapeutic agents?
Toxicities were seen with each of the regimens studied and were common in the randomized trials, with higher rates of toxicities in the combination chemotherapy regimens. The most common toxicities are myelosuppression, nausea, and vomiting. In the doxorubicin trials, the most common toxicities were myelosuppression, nausea, and vomiting.44
Ifosfamide both as an individual agent and in combination with doxorubicin has higher rates and higher grades of toxicity than doxorubicin alone. Myelosuppression is the most common toxicity associated with ifosfamide, and the most commonly affected cell line is leukocytes.44 Combination doxorubicin and ifosfamide also had high rates of nausea and vomiting (95%) and alopecia (100%).35
Neutropenia is the most common toxicity associated with gemcitabine and dacarbazine, while their most common nonhematologic toxicities are fatigue and nausea.52,59 Trabectedin’s most common toxicities are nausea (29%), neutropenia (24%), and fatigue (23%). It has also been shown to cause increased alkaline phosphatase (20%) and alanine aminotransferase (19%) levels.56 In a phase 2 study of eribulin, 50% of patients had neutropenia, and other toxicities included fatigue, alopecia, nausea, sensory neuropathy, and thrombocytopenia.57 Pazopanib is generally well tolerated; the most common toxicities are fatigue (65%), diarrhea (58%), nausea (54%), and hypertension (41%).58 Higher rates of neutropenia, mucositis, nausea, vomiting, diarrhea, and transfusion reactions were seen with olaratumab and doxorubicin compared to doxorubicin alone in phase 1b and 2 studies.46
CASE CONCLUSION
Given his poor prognosis with unresectable metastatic undifferentiated liposarcoma, the patient considers a clinical trial prior to undergoing combined therapy with doxorubicin and ifosfamide. He tolerates therapy well with stable disease at 6 months.
CONCLUSION
STSs are a heterogeneous collection of rare tumors. Low-grade, localized tumors have the best prognosis, and patients who undergo complete resection have the best long-term survival. Due to the rarity of STSs, trials often have limited enrollment, and little progress has been made with regards to treatment and survival rates for metastatic and unresectable disease. All patients should be evaluated and treated at specialized sarcoma centers. This case highlights the need for continued research and clinical trials to improve overall survival of patients with sarcoma.
- American Cancer Society. Cancer facts and figures 2016. American Cancer Society Web site. www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf. Accessed December 20, 2016.
- National Comprehensive Cancer Network. NCCN clinical guidelines in oncology: soft tissue sarcoma. 2016
- Coindre J, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001;91:1914–26.
- Dei Tos A. Liposarcoma: new entities and evolving concepts. Ann Diagn Pathol 2000;4:252–66.
- Wile AG, Evans HL, Romsdahl MM. Leiomyosarcoma of soft tissue: a clinicopathologic study. Cancer 1981;48:1022–32.
- Hashimoto H, Daimaru Y, Tsuneyoshi M, Enjoji M. Leiomyosarcoma of the external soft tissues. A clinicopathologic, immunohistochemical, and electron microscopic study. Cancer 1986;57:2077–88
- Fisher C. Synovial sarcoma. Ann Diagn Pathol 1998;2:401–21.
- Newton WA Jr, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 1995;76:1073–85.
- Furlong MA. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14:595–603.
- Anghileri M, Miceli R, Fiore M. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006;107:1065–74.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Archive 2001;438:1–12.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006;23:70–83.
- Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol 2010;11:983–91.
- Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin 2004;54:94–109.
- Penel N, Grosjean J, Robin YM, et al. Frequency of certain established risk factors in soft tissue sarcomas in adults: a prospective descriptive study of 658 cases. Sarcoma 2008;2008:459386.
- Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997;15:350–62.
- Maki RG, Moraco N, Antonescu CR, et al. Toward better soft tissue sarcoma staging: building on American joint committee on cancer staging systems versions 6 and 7. Ann Surg Oncol 2013;20:3377–83.
- Shiraki M, Enterline HT, Brooks JJ, et al. Pathologic analysis of advanced adult soft tissue sarcomas, bone sarcomas, and mesotheliomas. The Eastern Cooperative Oncology Group (ECOG) experience. Cancer 1989;64:484–90.
- Presant CA, Russell WO, Alexander RW, Fu YS. Soft-tissue and bone sarcoma histopathology peer review: The frequency of disagreement in diagnosis and the need for second pathology opinions. The Southeastern Cancer Study Group experience. J Clin Oncol 1986; 4:1658–61.
- Sundaram M, McLeod RA. MR imaging of tumor and tumorlike lesions of bone and soft tissue. AJR Am J Roentgenol 1990;155:817–24.
- Ioannidis JP, Lau J. 18F-FDG PET for the diagnosis and grading of soft-tissue sarcoma: a meta-analysis. J Nucl Med 2003;44:717–24.
- Tateishi U, Yamaguchi U, Seki K, et al. Bone and soft-tissue sarcoma: preoperative staging with fluorine 18 fluorodeoxyglucose PET/CT and conventional imaging. Radiology 2007;245:839–47.
- Zagars GK, Ballo MT, Pisters PW, et al. Prognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer 2003;97:2530–43
- Rosenberg S, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 1982;196:305–14.
- Lewis J, Leung D, Woodruff J, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 1998;288:355–65.
- Zagars GK, Ballo MT, Pisters PW, et al. Surgical margins and reresection in the management of patients with soft tissue sarcoma using conservative surgery and radiation therapy. Cancer 2003;97:2544–53.
- Stojadinovic A, Leung DH, Hoos A. Analysis of the prognostic significance of microscopic margins in 2,084 localized primary adult soft tisusse sarcomas. Ann Surg 2002;235:424–34.
- O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
- Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008;113:573–81.
- Suit HD, Mankin HJ, Wood WC, Proppe KH. Preoperative, intraoperative, and postoperative radiation in the treatment of primary soft tissue sarcoma. Cancer 1985;55:2659–67
- O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
- Yang J, Chang A, Baker A, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 1998;16:197–203.
- Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 1996;14:859–68.
- Alektiar KM, Brennan MF, Healey JH, Singer S. Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 2008;26:3440–5.
- Gortzak E, Azzarelli A, Buesa J, et al. A randomized phase II study on neo-adjuvant chemotherapy for ‘high-risk’ adult soft-tissue sarcoma. Eur J Cancer 2001;37:1096–1103.
- Fakhari N, Ebm C, Kostler WJ, et al. Intensified adjuvant IFADIC chemotherapy in combination with radiotherapy versus radiotherapy alone for soft tissue sarcoma: long-term follow-up of a prospective randomized feasibility trial. Wein Klin Wochenschr 2010;122:614–9.
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997;350:1647–54.
- Gronchi A, Frustaci S, Mercuri M, et al. Short, full-dose adjuvant chemotherapy in high-risk adult soft tissue sarcomas: a randomized clinical trial from the Italian Sarcoma Group and the Spanish Sarcoma Group. J Clin Oncol 2012;30:850–56.
- Pisters PW, Leung DH, Woodruff J. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679–89.
- Whooley B, Gibbs J, Mooney M. Primary Extremity Sarcoma: What is the Appropriate Follow-up? Annals of Surg Oncol 2000; 7: 9-14.
- Whooley BP, Mooney MN, Gibbs JF, Graybill WG. Effective follow-up strategies in soft tissue sarcoma. Sem Surg Oncol 1999;17:83–87.
- Billingsley KG, Burt ME, Jara E, et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg 1999;229:602–10.
- Bramwell VH, Anderson D, Charette ML; Sarcoma Disease Site Group. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev 2003;(3):CD003293.
- Edmonson J, Ryan L, Blum R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993;11:1269–75.
- Santoro A, Tursz T, Mouridsen H. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1995;13:1537–45.
- Tap WD, Jones RL, Van Tine B, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 2016;388:488–97.
- Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol 1987;5:840–50.
- Omura GA, Major FJ, Blessing JA, et al. A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 1983;52:626–32.
- Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276–85.
- Maki R, Wathen K, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 2007; 25: 2755–63.
- Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 2002;12:2824–31.
- Garcia-del-Muro X, Lopez-Pousa A, Maurel J, et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. J Clin Oncol 2011;29:2528–33.
- Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 2007;7:595–602.
- Italiano A, Cioffi A, Penel N, et al. Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas. Cancer 2012;118:3330–6.
- Penel N, Italiano A, Ray-Coquard I, et al. Metastatic angiosarcomas: doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve outcome. Ann Oncol 2012;23:517–23.
- Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 2013;24:1703–9.
- Schöffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histolical subtypes. Lancet 2011;11:1045–52.
- Van der Graaf W, Blay JY, Chawla S, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86.
- Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer 2007;109:1863–9.
- American Cancer Society. Cancer facts and figures 2016. American Cancer Society Web site. www.cancer.org/acs/groups/content/@research/documents/document/acspc-047079.pdf. Accessed December 20, 2016.
- National Comprehensive Cancer Network. NCCN clinical guidelines in oncology: soft tissue sarcoma. 2016
- Coindre J, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001;91:1914–26.
- Dei Tos A. Liposarcoma: new entities and evolving concepts. Ann Diagn Pathol 2000;4:252–66.
- Wile AG, Evans HL, Romsdahl MM. Leiomyosarcoma of soft tissue: a clinicopathologic study. Cancer 1981;48:1022–32.
- Hashimoto H, Daimaru Y, Tsuneyoshi M, Enjoji M. Leiomyosarcoma of the external soft tissues. A clinicopathologic, immunohistochemical, and electron microscopic study. Cancer 1986;57:2077–88
- Fisher C. Synovial sarcoma. Ann Diagn Pathol 1998;2:401–21.
- Newton WA Jr, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study. Cancer 1995;76:1073–85.
- Furlong MA. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14:595–603.
- Anghileri M, Miceli R, Fiore M. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006;107:1065–74.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors–definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Archive 2001;438:1–12.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006;23:70–83.
- Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol 2010;11:983–91.
- Cormier JN, Pollock RE. Soft tissue sarcomas. CA Cancer J Clin 2004;54:94–109.
- Penel N, Grosjean J, Robin YM, et al. Frequency of certain established risk factors in soft tissue sarcomas in adults: a prospective descriptive study of 658 cases. Sarcoma 2008;2008:459386.
- Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997;15:350–62.
- Maki RG, Moraco N, Antonescu CR, et al. Toward better soft tissue sarcoma staging: building on American joint committee on cancer staging systems versions 6 and 7. Ann Surg Oncol 2013;20:3377–83.
- Shiraki M, Enterline HT, Brooks JJ, et al. Pathologic analysis of advanced adult soft tissue sarcomas, bone sarcomas, and mesotheliomas. The Eastern Cooperative Oncology Group (ECOG) experience. Cancer 1989;64:484–90.
- Presant CA, Russell WO, Alexander RW, Fu YS. Soft-tissue and bone sarcoma histopathology peer review: The frequency of disagreement in diagnosis and the need for second pathology opinions. The Southeastern Cancer Study Group experience. J Clin Oncol 1986; 4:1658–61.
- Sundaram M, McLeod RA. MR imaging of tumor and tumorlike lesions of bone and soft tissue. AJR Am J Roentgenol 1990;155:817–24.
- Ioannidis JP, Lau J. 18F-FDG PET for the diagnosis and grading of soft-tissue sarcoma: a meta-analysis. J Nucl Med 2003;44:717–24.
- Tateishi U, Yamaguchi U, Seki K, et al. Bone and soft-tissue sarcoma: preoperative staging with fluorine 18 fluorodeoxyglucose PET/CT and conventional imaging. Radiology 2007;245:839–47.
- Zagars GK, Ballo MT, Pisters PW, et al. Prognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 1225 patients. Cancer 2003;97:2530–43
- Rosenberg S, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 1982;196:305–14.
- Lewis J, Leung D, Woodruff J, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 1998;288:355–65.
- Zagars GK, Ballo MT, Pisters PW, et al. Surgical margins and reresection in the management of patients with soft tissue sarcoma using conservative surgery and radiation therapy. Cancer 2003;97:2544–53.
- Stojadinovic A, Leung DH, Hoos A. Analysis of the prognostic significance of microscopic margins in 2,084 localized primary adult soft tisusse sarcomas. Ann Surg 2002;235:424–34.
- O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
- Pervaiz N, Colterjohn N, Farrokhyar F, et al. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008;113:573–81.
- Suit HD, Mankin HJ, Wood WC, Proppe KH. Preoperative, intraoperative, and postoperative radiation in the treatment of primary soft tissue sarcoma. Cancer 1985;55:2659–67
- O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomized trial. Lancet 2002;359:2235–41.
- Yang J, Chang A, Baker A, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 1998;16:197–203.
- Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 1996;14:859–68.
- Alektiar KM, Brennan MF, Healey JH, Singer S. Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 2008;26:3440–5.
- Gortzak E, Azzarelli A, Buesa J, et al. A randomized phase II study on neo-adjuvant chemotherapy for ‘high-risk’ adult soft-tissue sarcoma. Eur J Cancer 2001;37:1096–1103.
- Fakhari N, Ebm C, Kostler WJ, et al. Intensified adjuvant IFADIC chemotherapy in combination with radiotherapy versus radiotherapy alone for soft tissue sarcoma: long-term follow-up of a prospective randomized feasibility trial. Wein Klin Wochenschr 2010;122:614–9.
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997;350:1647–54.
- Gronchi A, Frustaci S, Mercuri M, et al. Short, full-dose adjuvant chemotherapy in high-risk adult soft tissue sarcomas: a randomized clinical trial from the Italian Sarcoma Group and the Spanish Sarcoma Group. J Clin Oncol 2012;30:850–56.
- Pisters PW, Leung DH, Woodruff J. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679–89.
- Whooley B, Gibbs J, Mooney M. Primary Extremity Sarcoma: What is the Appropriate Follow-up? Annals of Surg Oncol 2000; 7: 9-14.
- Whooley BP, Mooney MN, Gibbs JF, Graybill WG. Effective follow-up strategies in soft tissue sarcoma. Sem Surg Oncol 1999;17:83–87.
- Billingsley KG, Burt ME, Jara E, et al. Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg 1999;229:602–10.
- Bramwell VH, Anderson D, Charette ML; Sarcoma Disease Site Group. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev 2003;(3):CD003293.
- Edmonson J, Ryan L, Blum R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993;11:1269–75.
- Santoro A, Tursz T, Mouridsen H. Doxorubicin versus CYVADIC versus doxorubicin plus ifosfamide in first-line treatment of advanced soft tissue sarcomas: a randomized study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 1995;13:1537–45.
- Tap WD, Jones RL, Van Tine B, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet 2016;388:488–97.
- Borden EC, Amato DA, Rosenbaum C, et al. Randomized comparison of three adriamycin regimens for metastatic soft tissue sarcomas. J Clin Oncol 1987;5:840–50.
- Omura GA, Major FJ, Blessing JA, et al. A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 1983;52:626–32.
- Antman K, Crowley J, Balcerzak SP, et al. An intergroup phase III randomized study of doxorubicin and dacarbazine with or without ifosfamide and mesna in advanced soft tissue and bone sarcomas. J Clin Oncol 1993;11:1276–85.
- Maki R, Wathen K, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 2007; 25: 2755–63.
- Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 2002;12:2824–31.
- Garcia-del-Muro X, Lopez-Pousa A, Maurel J, et al. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. J Clin Oncol 2011;29:2528–33.
- Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 2007;7:595–602.
- Italiano A, Cioffi A, Penel N, et al. Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas. Cancer 2012;118:3330–6.
- Penel N, Italiano A, Ray-Coquard I, et al. Metastatic angiosarcomas: doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve outcome. Ann Oncol 2012;23:517–23.
- Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 2013;24:1703–9.
- Schöffski P, Ray-Coquard IL, Cioffi A, et al. Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histolical subtypes. Lancet 2011;11:1045–52.
- Van der Graaf W, Blay JY, Chawla S, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86.
- Dileo P, Morgan JA, Zahrieh D, et al. Gemcitabine and vinorelbine combination chemotherapy for patients with advanced soft tissue sarcomas: results of a phase II trial. Cancer 2007;109:1863–9.
Sickle Cell Disease
INTRODUCTION
Sickle cell disease is the most common inherited blood disorder in the world. It affects more than 100,000 individuals in the United States, and millions more worldwide.1 Sickle cell disease is most commonly found in individuals of African heritage, but the disease also occurs in Hispanics and people of Middle Eastern and subcontinent Indian heritage.2 The distribution of the sickle hemoglobin (hemoglobin S [HbS]) allele overlaps with the distribution of malaria; HbS carriers, or individuals with sickle cell trait, have protection against malaria,3 and are not considered to have sickle cell disease.
Sickle cell disease is a severe monogenic disorder marked by significant morbidity and mortality, affecting every organ in the body.4 The term sickle cell disease refers to all genotypes that cause sickling; the most common are the homozygous hemoglobin SS (HbSS) and compound heterozygotes hemoglobin SC (HbSC), hemoglobin S–β0-thalassemia (HbSβ0), and hemoglobin S–β+-thalassemia (HbSβ+), although HbS and several rarer hemoglobin variants such as HbSO(Arab) and HbSD(Punjab) can also cause sickle cell disease. The term sickle cell anemia refers exclusively to the most severe genotypes, HbSS and HbSβ0.5 Common sickling genotypes along with their relative clinical severity are shown in Table 1.6–11
| Table 1. Genotypes of Sickling Syndromes and Their Relative Severities | ||
Genotype | Severity | Characteristics |
HbSS | Severe | Most common form |
HbSβ0 | Severe | Clinically indistinguishable from HbSS6 |
HbSO-Arab | Severe | Relatively rare6 |
HbSD-Punjab | Severe | Mostly in northern India6 |
HbSC-Harlem | Severe | Migrates like HbSC, but rare double β-globin mutation7 |
HbCS-Antilles | Severe | Rare double β-globin mutation8 |
HbSC | Moderate | 25% of SCD9 |
HbSβ+, Mediterranean | Moderate | 5%–16% HbA6 |
HbAS-Oman | Moderate | Dominant rare double β-globin mutation10 |
HbSβ+, African | Mild | 16%–30% HbA6 |
HbSE | Mild | HbE found mostly in Southeast Asia11 |
HbS-HPFH | Very mild | Large deletions in β-globin gene complex; > 30% HbF6 |
| HbA = hemoglobin A; HbE = hemoglobin E; HbF = fetal hemoglobin; HbS-HPFH = HbS and gene deletion HPFH; HbSC = heterozygous hemoglobin SC; HbSS = homozygous hemoglobin SS; HbSβ0 = hemoglobin S-β thalassemia0; HbSβ+ = hemoglobin S-β thalassemia+; SCD = sickle cell disease. | ||
This article reviews the pathophysiology of sickle cell disease, common clinical complications, and available therapies. A complex case which illustrates diagnostic and management challenges is presented as well.
PATHOPHYSIOLOGY
HbS is the result of a substitution of valine for glutamic acid in the sixth amino acid of the β-globin chain.12 The change from a hydrophilic to a hydrophobic amino acid causes the hemoglobin molecules to stack, or polymerize, when deoxygenated. This rigid rod of hemoglobin distorts the cell, producing the characteristic crescent or sickle shape that gives the disease its name.13 Polymerization of hemoglobin within the cell is promoted by dehydration, which increases the concentration of HbS.13,14 Polymerization occurs when hemoglobin is in the deoxygenated state.13
The sickle red blood cell is abnormal; it is rigid and dense, and lacks the deformability needed to navigate the microvasculature.15 Blockages of blood flow result in painful vaso-occlusion that is the hallmark of the disease, and that also can cause damage to the spleen, kidneys, and liver.16 The sickle red cell is also fragile, with a lifespan of only 20 days compared to the 120-day lifespan of a normal red blood cell.13 Frequent hemolysis results in anemia and the release of free hemoglobin, which both scavenges nitric oxide and impairs the production of more nitric oxide, which is essential for vasodilatation.17 This contributes to vascular dysfunction and an increased risk for stroke.18 If untreated, the natural course of sickle cell anemia is mortality in early childhood in most cases.19 Common chronic and acute sickle cell disease–related complications and recommended therapies, based on 2014 National Institutes of Health guidelines, are shown in Table 2 and Table 3.20
| Table 2. Common Adult Sickle Cell Disease Chronic Complications and Recommended Therapies | ||
Chronic Complication | Recommended Therapy | Strength of Recommendation |
Chronic pain | Opioids | Consensus |
Avascular necrosis | Analgesics and physical therapy | Consensus |
Proliferative sickle retinopathy | Laser photocoagulation | Strong |
Leg ulcers | Standard wound care | Moderate |
Recurrent priapism | Consult urology | Moderate |
| Data from Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48. | ||
| Table 3. Common Adult Sickle Cell Disease Acute Complications and Recommended Therapies | ||
Acute Complication | Recommended Therapy | Strength of Recommendation |
Vaso-occlusive crisis | NSAIDs, opioids for severe pain | Moderate-consensus |
ACS | Antibiotics, oxygen | Strong |
Simple transfusiona | Weak | |
Urgent exchange transfusionb | Strong | |
Acute stroke | Exchange transfusion | Strong |
Priapism ≥ 4 hr | Aggressive hydration, pain control, and urology consult | Strong-consensus |
Gallstones, symptomatic | Cholecystectomy, laparoscopic | Strong |
Splenic sequestration | Intravenous fluids, transfuse cautiously, discuss surgical splenectomy | Strong-moderate |
Acute renal failure | Consult nephrologyc | Consensus |
ACS = acute chest syndrome; NSAIDs = nonsteroidal anti-inflammatory drugs. a For symptomatic ACS with hemoglobin > 1 g/dL below baseline but > 9.0 g/dL. b When there is progression of ACS (SpO2 < 90% despite supplemental oxygen, increasing respiratory distress, progressive pulmonary infiltrates despite simple transfusion). c For acute rise in creatinine ≥ 0.3 mg/dL; do not give transfusions unless there are other indications. Data from Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48. | ||
One of the most challenging aspects of sickle cell disease is its clinical variability. While in general, HbSS and HbSβ0 are the most severe genotypes, there are patients with HbSC and HbSb+ who have significant sickle-cell–related complications, and may have a more severe clinical course than a HbSS patient.21 A great deal of this clinical variability cannot be explained, but some can be attributed to endogenous fetal hemoglobin (HbF) levels.22–24 The importance of HbF levels in sickle cell disease was first noted by a pediatrician in the 1940s.25 She observed that sickle cell disease complications in children under the age of 1 were rare, and attributed it to the presence of HbF.25 HbF levels decline more slowly in individuals with hemoglobinopathies, reaching their nadir after the age of 5 rather than within 6 months of birth in individuals without hemoglobinopathies.26 HbF levels remain elevated lifelong in most sickle cell disease patients, especially those with the HbSS and HbSβ0 genotypes. Levels of HbF vary widely between individuals, from zero to 20% to 30%, with a median of 10%.26–28 Individuals who produce more HbF have a milder course, in general.24 An association between the 4 β-globin haplotypes and HbF levels has been reported in the past,27,29 but more sophisticated next-generation sequencing has revealed causal variants in BCL11A and HBS1L-MYB that contribute approximately 50% of the observed variability in HbF levels.30–33
Co-inheritance of α-thalassemia also modifies disease course; less available α-globin chains results in a lower hemoglobin concentration within the cell. Paradoxically, this results in a higher overall hemoglobin level, as there is a reduction in polymerization, and therefore sickling due to lower HbS concentrations in the cell. Patients therefore are less anemic, reducing the risk of stroke in childhood,34,35 but blood viscosity may be higher, resulting in more frequent pain crises and increased risk36 of avascular necrosis.34,35,37 It is often helpful to think of sickle cell patients as falling into 1 of 2 groups: high hemolysis/low hemoglobin and high viscosity/high hemoglobin. Individuals with high rates of hemolysis are at greater risk for stroke, pulmonary hypertension, and acute chest syndrome (ACS). Higher rates of hemolysis result in higher levels of free hemoglobin, which scavenges nitric oxide. This leads to the vascular damage and dysfunction that contributes to the associated clinical complications. This phenotype is most commonly seen in HbSS and HbSβ0.38 High hemoglobin/high viscosity phenotypes are most often found in HbSC patients and in sickle cell anemia with α-thalassemia coinheritance.39–42
TREATMENT OPTIONS
In high-resource countries with newborn screening, the initiation of penicillin prophylaxis has dramatically altered the natural history of the disease, allowing the majority of patients to reach adulthood.43 Penicillin prophylaxis is usually discontinued at age 5 years; however, individuals who have undergone surgical splenectomy or have had pneumococcal sepsis on penicillin prophylaxis may remain on penicillin to age 18 or beyond.20
Another advance in sickle cell care is screening for stroke risk through transcranial Doppler ultrasound (TCD).44–47 This screening tool has reduced the incidence of childhood stroke from 10% by age 11 to 1%. TCDs typically cannot be performed after the age of 16 due to changes in the skull. Individuals found to have abnormal (elevated) TCD velocities are placed on chronic transfusion therapy for primary stroke prevention. They may remain on monthly chronic transfusions, with the goal of suppressing the percentage of HbS to 30% to 50% indefinitely. A clinical trial (STOPII) designed to determine if pediatric sickle cell disease patients on chronic transfusion therapy for primary stroke prevention could be safely taken off transfusion therapy was discontinued early due to an excess of strokes and conversion to abnormal TCD velocities in the untransfused arm.44 Individuals who have experienced an ischemic stroke have a 70% risk of another stroke, and must remain on chronic transfusion therapy indefinitely. Chronic transfusion reduces their stroke risk to 13%.
The only widely used pharmacologic therapy for sickle cell disease is hydroxyurea.12,48–50 A significant portion of the benefit of hydroxyurea stems from its induction of HbF.51 HbF does not sickle, and it interrupts the polymerization of HbS in the cell, if present in high enough concentrations.50 The level of HbF needed to achieve clinical improvement is not known, but in vitro assays suggest 20% HbF is needed to prevent sickling.52,53 However, endogenous and hydroxyurea-induced HbF is not distributed evenly through the red cells, so sickling is possible regardless of the level of HbF induced.54,55 Hydroxyurea likely has other disease-modifying effects as well, including reduction of white blood cell count and reticulocyte count and reduction of red cell adhesion to the endothelium.56–58 Clinical criteria for initiation of hydroxyurea in adult sickle cell disease patients are shown in Table 4.20 Hydroxyurea is given daily and is dosed to maximum tolerated dose for the individual by following the absolute neutrophil count (ANC). The goal ANC is between 2000 and 4000/µL. At times, absolute reticulocyte count (ARC) can be dose-limiting; goal ARC is greater than 70,000/µL.59 Platelet counts may be reduced as well, especially in HbSC patients.60,61
| Table 4. Indications for Hydroxyurea in Adult Patients with Sickle Cell Disease | |
Indication | Strength of Recommendation |
SCA with ≥ 3 pain crises per year | Strong |
SCA with pain that interferes with ADL and QoL | Strong |
History of severe or recurrent ACS | Strong |
Chronic kidney disease on epoetin | Weak |
HbSβ+ and HbSC with pain that interferes with ADL and QoL; consult sickle cell disease expert | Moderate |
| ACS = acute chest syndrome; ADL = activities of daily living; QoL = quality of life; SCA = sickle cell anemia. | |
The only curative therapy for sickle cell disease is hematopoietic stem cell transplant.62 Transplant use is limited by availability of matched sibling donors,62 and even at experienced centers transplant carries a small risk for mortality, graft rejection, and graft-versus-host disease. Furthermore, consensus on disease complications for which transplant is recommended is also lacking.63–65 Clinical trials of gene therapy for sickle cell disease and thalassemia are ongoing.66
COMPLICATIONS AND DISEASE-SPECIFIC THERAPIES
CASE PRESENTATION
A 26-year-old African-American man who works as a school bus driver presents to an academic center’s emergency department complaining of pain in his left leg, similar to prior pain events. He is described as having sickle cell trait, although no hemoglobin profile is available in his chart. He describes the pain as dull and aching, 10/10 in intensity. A complete blood count (CBC) is obtained; it reveals a hemoglobin of 14.5 g/dL, white blood cell (WBC) count of 5600/µL, and platelet count of 344,000/µL. His CBC is also notable for a mean corpuscular volume (MCV) of 72 fL, a mean corpuscular hemoglobin concentration (MCHC) of 37 g/dL, and a red blood cell distribution width (RDW) of 12. Slide review of a peripheral blood smear shows 2+ target cells (Figure).

The patient is given 6 mg of morphine, which provides some relief of his pain, and is discharged with a prescription for hydrocodone bitartrate/acetaminophen 5/325 mg. The diagnosis given is musculoskeletal pain, and he is instructed to follow-up with a primary care physician. His past medical history is significant for 4 or 5 visits to the emergency department per year in the past 4 years. Prior to 4 years ago, he rarely required medical attention.
• What laboratory and clinical features might lead you to question the diagnosis of sickle cell trait in this patient?
The patient’s hemoglobin is within normal range, which is consistent with sickle cell trait; however, he is microcytic, with a normal RDW. It is possible to be mildly microcytic in the early stages of iron deficiency, prior to the development of anemia, but the RDW would typically be elevated, demonstrating the presence of newer, smaller cells produced under conditions of iron deficiency.67 It is also possible that his microcytosis with a normal RDW could represent sickle cell trait with co-inheritance of β-thalassemia. Up to 30% of African Americans have β-thalassemia,2 and 1 in 10 have sickle cell trait.68 However, a high MCHC, indicating the presence of dense cells, and target cells noted on slide review are most consistent with HbSC.9 HbSC patients, especially males, can have hemoglobin levels in the normal range.4 The biggest inconsistency with the diagnosis of sickle cell trait is his history of frequent pain events. Individuals with sickle cell trait rarely present with pain crises, except under extreme conditions of dehydration or high altitude.68 Sickle cell trait is generally regarded as a benign condition, although a study of U.S. military recruits found a 30-fold higher risk of sudden death during basic training in persons with sickle cell trait.69 Additional sickle cell trait–related complications include hematuria, risk of splenic sequestration or infarct under extreme conditions and high altitude, and a rare and usually fatal renal malignancy, renal medullary carcinoma, which is vanishingly rare in individuals without sickle cell trait.70,71 Although the patient reported having sickle cell trait, this diagnosis should have been verified with a hemoglobin panel, given his atypical presentation.20
• What is the approach to managing pain episodes in sickle cell disease?
In sickle cell disease, vaso-occlusive pain events can be common, often beginning in early childhood.17 This disease complication accounts for 95% of all adult sickle cell disease hospitalizations.72 There is a great deal of variability in pain symptoms between individuals, and within individuals at various times in their lives:73 30% have no pain events, 50% have occasional events, and 20% have monthly or more frequent events that require hospitalization.74 The frequency and severity of pain events are modulated by HbF levels, β-thalassemia status, genotypes, therapies like hydroxyurea, or in rare cases, chronic transfusion therapy.23 Personal factors, such as psychosocial stressors, also contribute to the frequency of pain events.75 Pain event triggers include exposure to cold water, windy or cold weather, temperature changes, and extreme temperatures.76–79 Patient age also contributes to pain event frequency. Many patients see an increase in pain event frequency in their late 20s, and a marked decrease in their 40s.23,73 More than 3 pain events per year is associated with reduced life expectancy.23
Acute management of pain episodes involves nonsteroidal anti-inflammatory drugs, oral opioids, and when hospitalization is required, intravenous opioids, often delivered via patient-controlled analgesia (PCA) pumps.79 As sickle cell disease patients become teenagers and young adults, some experience an increased frequency of pain episodes, with fewer pain-free days, or a failure to return to baseline before the next pain crisis occurs.80,81 This is characteristic of emerging chronic pain.82 Chronic pain is a significant problem in adult patients with sickle cell disease, with up to 85% reporting pain on most days.72,80 The development of chronic pain may be reduced by early and aggressive treatment of acute pain events, as well as use of hydroxyurea to reduce the number of pain events. Many adult sickle cell patients with chronic pain are treated with daily opioids.20 Given the significant side effects of chronic opioid use—sedation, respiratory depression, itching, nausea, and impairment of function and quality of life—non-opioid therapies are under investigation.83 Many chronic pain patients have symptoms of neuropathic pain, and may benefit from neuropathic agents like gabapentin, both to reduce opioid use and to more effectively treat chronic neuropathic pain, which is known to respond poorly to opioids.84–86
• Is the patient’s peripheral blood smear consistent with a diagnosis of sickle cell trait?
Several target cells are visible, which is not typical of sickle cell trait, but may be seen in HbSC or thalassemia. The finding of an intracellular crystal is pathognomonic for HbSC or HbCC. HbC polymerizes in high oxygen conditions, opposite of HbS, which polymerizes in low oxygen conditions.9
CASE CONTINUED
The patient’s family history is significant for a sister who died at age 3 from sickle cell–related complications, and a sister with sickle cell trait who had a cholecystectomy for gallstones at age 22. His father died at age 38 due to unknown causes. The sickle cell trait status of his parents is unknown. His mother is alive, and has hypertension.
• Is the medical history of this patient’s family members consistent with sickle cell trait?
It is unlikely that sickle cell trait would result in early death in childhood, or in gallstones at age 22. Gallstones in early adulthood is a common presentation for HbSC patients not diagnosed by newborn screening.87 Any hemolytic condition can lead to the formation of hemoglobin-containing pigmented gallstones, biliary sludge, and obstruction of the gallbladder. In the presence of right-sided abdominal pain, a serum bilirubin level of more than 4 mg/dL should lead to measurement of direct bilirubin; if greater than 10% of total, imaging of the gallbladder should be obtained. In sickle cell disease, 30% of patients will have gallstones by 18 years of age. The low hemolysis/high viscosity phenotype patients are typically older at diagnosis. Co-inheritance of Gilbert syndrome and sickle cell disease is not uncommon, and can result in formation of gallstones at a young age; Gilbert syndrome alone typically results in gallstones in mid-life.88
CASE CONTINUED
Two months later, the patient presents again to the emergency department with the same complaint of leg pain, as well as abdominal pain. His hemoglobin is 12.5 g/dL, and his platelet count is 134,000/µL. His pain is not improved with 3 doses of morphine 6 mg intravenously, and he is admitted to the medicine service. A hemoglobin profile is obtained, revealing 52% HbS, 45% HbC, and 1.5% HbF, consistent with HbSC. In sickle cell trait, the hemoglobin profile is 60% HbA and 40% HbS (available α-globin prefers to pair with a normal β-globin, so the ratio of HbA to HbS is 60:40, not 50:50).
On the second hospital day, the patient’s hemoglobin drops to 7.2 g/dL and his platelet count decreases to 44,000/µL. His abdomen is distended and diffusely tender. The internist transfuses him with 2 units of packed red blood cells (PRBC), after which his hemoglobin increases to 11 g/dL, while his platelet count increases to 112,000/µL. Following the transfusion, his abdominal pain resolves, as does his anemia and thrombocytopenia.
• What caused this patient’s anemia and thrombocytopenia?
High on the differential diagnosis is a splenic sequestration. Acute splenic sequestration occurs when red cells are trapped in the splenic sinuses. Massive splenic enlargement may occur over several hours.89,90 Unrecognized splenic sequestration has a high mortality rate from severe anemia and splenic rupture.90 Splenic sequestration must be ruled out in a sickle cell patient with abdominal pain accompanied by dropping platelet and red cell counts, especially in milder subtypes that often have splenic function preserved into adolescence and adulthood. Sickle cell anemia patients usually become functionally asplenic in early childhood.89,91,92 The rise in hemoglobin, more than would be expected from 2 units of PRBC, plus the improvement in platelet count without a platelet transfusion observed in the case patient strongly supports the diagnosis of splenic sequestration.
Splenic sequestration can occur in any sickle cell patient whose spleen has not fibrosed. Splenic sequestration in adulthood is not uncommon in HbSC patients, who often have preserved splenic function into adulthood.93–95
Clinical signs of splenic sequestration include a rapid drop in hemoglobin, rise in reticulocyte count, a tender, enlarged spleen, and, in severe cases, hypovolemia.89,93 It is treated with prompt blood transfusion, but care must be taken not to overtransfuse the patient, as the spleen can trap several grams of hemoglobin, which may be released upon transfusion, potentially causing life-threatening hyperviscosity.89 Hemoglobin levels must be checked following transfusion in suspected splenic sequestration, and “mini transfusions” of 5 mL/kg are recommended in sickle cell disease patients who are hemodynamically stable.20
Hepatic sequestration may also occur, but it is much less common than splenic sequestration.96 Other conditions on the differential diagnosis include thrombotic thrombocytopenic purpura, which would be unlikely to respond to a transfusion. ACS can cause a drop in hemoglobin, and is treated with simple or exchange transfusions.97 ACS is less likely without respiratory symptoms or oxygen requirement, and usually is not associated with thrombocytopenia. Sepsis may also cause anemia and thrombocytopenia, but again would not likely respond to a simple transfusion. The patient’s response to transfusion is consistent with a sequestering event, not a destructive event as in the case of sepsis.
CASE CONTINUED
Imaging reveals a grossly enlarged spleen, which is having a mass effect on the left kidney. The patient is started on hydroxyurea therapy at 500 mg 3 times daily. Discharge instructions include following up with his primary care physician, continuing hydroxyurea therapy, and receiving yearly dilated eye exams to evaluate for proliferative sickle retinopathy.
• Are these discharge instructions complete?
Splenic sequestration has a 50% recurrence rate.98 In very young children, watchful waiting or chronic transfusion may be implemented to preserve the immunologic function of the spleen and reduce the risk of sepsis.89 Splenectomy after a single episode of sequestration in adults is a matter of debate, with experts advising both watchful waiting99 and splenectomy after recovery from the first sequestering event.100 The patient should have been informed of the risk for recurrence, and the signs and symptoms of splenic sequestration as well as the need for emergency medical attention should have been discussed. Splenic sequestration may be milder in adults than in children, but fatal sequestrations have been reported.95,101–103
Proliferative sickle cell retinopathy is a high viscosity/high hemoglobin complication that may occur more frequently in HbSC than HbSS, with an incidence of 33% in HbSC.42,104 Spontaneous regression of retinopathy occurs in approximately 32% of eyes, and laser or scatter photocoagulation is an effective intervention.105
• Would the patient need to be transfused prior to splenectomy?
Preoperative transfusion therapy is standard of care for HbSS patients undergoing general anesthesia. The TRAP study found that simple “top off” transfusion to a hemoglobin of 10 g/dL was as effective at preventing postoperative sickle cell–related complications as exchange transfusion to HbS of 30% or less, and had fewer transfusion-related complications like alloimmunization.106 There is little data regarding preoperative transfusions in HbSC disease. A retrospective study suggests that HbSC patients undergoing abdominal surgeries should be transfused.107 The higher hemoglobin level of the typical HbSC patient necessitates exchange transfusion to avoid hyperviscosity.
• Is hydroxyurea therapy indicated in this patient?
• Has it been dosed appropriately?
If the patient had the HbSS subtype, hydroxyurea would be clearly indicated, given his frequent pain events.20 HbSC patients may be placed on hydroxyurea on a case-by-case basis, but evidence for its efficacy in this sickle cell subtype is lacking.108 Large clinical trials like the Multi-Center Study of Hydroxyurea (MSH) that established the safety and efficacy of hydroxyurea in sickle cell anemia excluded HbSC and HbSβ+ patients.109 These mild to moderate subtypes produce less HbF at baseline, and typically have a minimal to modest rise in HbF on hydroxyurea.110 In sickle cell anemia, hydroxyurea is titrated to maximum tolerated dose, defined as an ANC of 2000 to 4000/µL and an ARC of 70,000/µL or higher.53 Because of their lower levels of chronic inflammation and lower reticulocyte counts due to higher hemoglobin levels, many HbSC and HbSβ+ patients have values in that range before initiating hydroxyurea therapy.9 Cytopenias, particularly of platelets in HbSC, occur at low doses of hydroxyurea.111
Of note, although the half-life of hydroxyurea would suggest that 3 times daily dosing is indicated, daily dosing has been found to have equal response and is preferred. Another concern is the monitoring of this myelosuppressive medication. This patient has repeatedly failed to obtain a primary care physician or a hematologist, and hydroxyurea requires laboratory monitoring at least every 2 months, especially in a HbSC patient with a very large spleen who is at significant risk for thrombocytopenia and neutropenia.9
CASE CONTINUED
A week after discharge from his admission for abdominal pain diagnosed as splenic sequestration, the patient presents again to the emergency department with abdominal pain which he reports is his typical sickle cell pain. Hemoglobin is 13.8 g/dL, platelet count is 388,000/µL, and alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels are both 10 times their prior value. Creatinine is 1.2 mg/dL (0.75 mg/dL on his prior admission), and total bilirubin is 3 mg/dL, with 0.3 mg/dL direct bilirubin. He undergoes an ultrasound exam of his gallbladder, which reveals sludge and a possible gallstone. There is no evidence of cholecystitis. General surgery performs a laparoscopic cholecystectomy.
• Was this cholecystectomy necessary?
In patients with sickle cell disease, symptomatic gallstones and gallbladder sludge should be observed; recurrent abdominal pain without a significant change in bilirubin may not be due to gallstones or sludge, and therefore may not be relieved by cholecystectomy.112,113 In sickle cell disease, 40% of patients with gallbladder sludge do not develop gallstones.87 The patient’s bilirubin level was at baseline, and there was no increase in the direct (conjugated) fraction. Watchful waiting would have been appropriate, with cholecystectomy being performed if he experienced recurrent symptoms associated with fatty foods accompanied by an elevation in direct bilirubin.
More concerning and deserving of investigation was his elevated liver enzymes. Patients with sickle cell disease may experience recurrent ischemia and reperfusion injuries in the liver, which is called right upper quadrant syndrome. On autopsy of 70 sickle cell patients, 91% had hepatomegaly and 34% had focal necrosis.114 AST is often elevated in sickle cell disease, as it is affected by hemolysis. In this patient, both AST and ALT are elevated, consistent with a hepatocellular disorder. His abdominal pain and ALT rise may be a sign of a hepatic crisis.115 Rapid resolution of ALT elevation in a matter of days suggests a vaso-occlusive, inflammatory event that is self- limiting. Prolonged AST elevation requires further investigation, with consideration of autoimmune hepatitis, viral hepatitis, or iron overload. Iron overload is unlikely in this patient given his lifetime history of only 1 transfusion. Hepatic iron overload typically occurs in sickle cell disease after a minimum of 10 transfusions.115
CASE CONTINUED
The patient is discharged on the day after the procedure, with instructions to continue his hydroxyurea.
• Should the patient resume hydroxyurea therapy?
Hydroxyurea is hepatically cleared and thus it should be held until his liver function tests normalize.106
CASE CONTINUED
Two months later, the patient presents to the emergency department with abdominal pain that moves to his left leg. A CBC is obtained, showing a hemoglobin of 11.8 g/dL and a platelet count of 144,000/µL. He is given 2 doses of morphine 6 mg intravenously, and reports that his leg pain is now a 4/10. He is discharged home with a prescription for hydrocodone/acetaminophen.
• Is the emergency department evaluation sufficient?
This patient remains at high risk for splenic sequestration,93 with a hemoglobin 2 g lower than it was 2 months ago and platelets less than half. This decline could be consistent with early splenic sequestration.20 Additionally, he had elevated liver function tests on a recent admission, as well as rising creatinine, without evidence of resolution. It is not appropriate to discharge him without checking a chemistry and liver panel, and abdominal imaging should be considered. The best plan would be to admit him for observation, given his risk for splenic sequestration, and consult surgery for an elective splenectomy if he has a second episode of splenic sequestration 2 months after the first.100 His abdominal pain that migrates to his left leg could be due to his massive splenomegaly compressing his left kidney, as noted on imaging during his recent admission for splenic sequestration
CASE CONTINUED
An hour after discharge from the emergency department, EMS is called to his home for intractable pain. He is found lying on the floor, and reports excruciating left leg pain. He is brought to the closest hospital, a community hospital that he has not visited previously. There, he is admitted for hydration and pain control and placed on hydromorphone 2 mg every 4 hours as needed for pain. His hemoglobin is 10.8 g/dL, and platelets are 121,000/µL. A chemistry panel is remarkable for a creatinine level of 1.5 mg/dL and a potassium level of 3.2 mEq/L. Liver function tests are not obtained. After 3 doses of hydromorphone, he falls asleep. He is not in a monitored bed, and intravenous fluids, while ordered, are not started. At 6:30 AM the day after admission, he cannot be aroused on a routine vital sign check; he has an SpO2 of 60%, a blood pressure of 80/60 mm Hg, and heart rate of 148 beats/min. A rapid response is called, and naloxone is administered along with oxygen by face mask and several fluid boluses. His systolic blood pressure increases to 100 mm Hg from a low of 70 mm Hg. His SpO2 increases to 92%, and he is arousable and alert, although he reports 10/10 leg pain. His abdomen is noted to be distended and tender.
• What may have contributed to his clinical condition?
The patient is opioid tolerant and has received equivalent doses of opioids in the past without excess sedation. He may have liver dysfunction making him unable to metabolize opioids effectively. His hemoglobin and platelets continue to decline, raising concern for splenic sequestration versus sepsis. Failure to place him on a monitor allowed his hypoxia to continue for an unknown amount of time, placing him at high risk for developing ACS. Lack of intravenous hydration while he was too sedated to drink likely exacerbated his sickling.
CASE CONTINUED
At 9:20 AM, a CBC is obtained and reveals a hemoglobin of 4.8 g/dL and a platelet count of 44,000/µL. Two units of stat O negative blood are administered, and preparations are made to administer an exchange transfusion. A liver panel is obtained 3 hours later, which reveals an AST level of 1200 U/L and an ALT level of 1050 U/L. His bilirubin is 10 mg/dL, and his lactate dehydrogenase level is 1800 U/L. His urine is dark and is positive for bilirubin and ketones. He is transferred to the intensive care unit. A chest X-ray shows pulmonary congestion. Hematology/oncology is consulted.
He receives a 7-unit red blood cell exchange, which reduces his HbS to 11%. He continues to be hypotensive, and requires norepinephrine to support his blood pressure. Antibiotic therapy is started. His creatinine concentration rises to 2.3 mg/dL, potassium is 7.8 mEq/L, and bicarbonate is 12 mEq/L. He is placed on hemodialysis.
Computed tomography of the chest and abdomen reveals lower posterior lung infiltrates and a grossly enlarged spleen. He requires intubation. He is given a diagnosis of ACS in addition to kidney failure, liver failure, and “sickle crisis.” He continues to require daily to twice daily transfusions to maintain a hemoglobin of 7 to 9 g/dL, and his abdominal distension increases. As his condition worsens, surgery is consulted to discuss a liver transplant. He is deemed to not be a surgical candidate, and he passes away 6 days after entering the hospital. The immediate cause of death is listed as vaso-occlusive crisis, with ACS and sickle crisis listed as contributors.
• Are the causes of death accurate and complete?
If vaso-occlusive crisis is used to indicate a pain event, it is not an accurate cause of death. Pain is one of the most distressing complications of sickle cell disease, and frequent pain events are associated with early mortality,4,80 but they are not in themselves fatal. ACS is the number one cause of death in sickle cell disease,4 and it likely contributed to this patient’s death. Sickle crisis is a vague term that should not be used in this context. Causes of death should include splenic sequestration and multisystem organ failure. Multisystem organ failure in sickle cell disease often responds to aggressive transfusion therapy, which this patient received.116–118
CONCLUSION
Sickle cell disease is a complex chronic disease that impacts almost every organ system in the body. Clinicians may be inclined to attribute most pain in a patient with sickle cell disease to a simple vaso-occlusive crisis, treat them for this, and not investigate further. As the case presented here demonstrates, failure to identify the actual life-threatening process occurring in a patient with sickle cell disease presenting with pain can result in preventable early mortality. Clinicians must approach a sickle cell patient reporting pain in a thoughtful manner, and consider a complete differential diagnosis, including both sickle cell disease complications and those unrelated to sickle cell disease. Knowledge of the disease courses of the different sickle cell genotypes is essential, and must go beyond a superficial hierarchy of severity, but rather include an understanding of the complications each genotype is most prone to, and at what ages. Complete laboratory assessment, including a comprehensive metabolic panel, should be performed on all admitted patients, not just a complete blood count. Treating pain with high-dose opioids, while appropriate in an uncomplicated pain crisis, can lead to ACS or even respiratory failure in a patient with uninvestigated liver and kidney dysfunction. The most important lesson to remember is that even the sickle cell disease patient who has been given the unfortunate and pejorative label of “frequent flyer” by some providers has the potential for rapid deterioration into multisystem organ failure and death.
- Weatherall D, Hofman K, Rodgers G, Ruffin J, Hrynkow S. A case for developing North-South partnerships for research in sickle cell disease. Blood 2005;105:921–3.
- Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol 1998;11:1–51.
- Allison AC. The distribution of the sickle-cell trait in East Africa and elsewhere, and its apparent relationship to the incidence of subtertian malaria. Trans R Soc Trop Med Hyg 1954;48:312–8.
- Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639–44.
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010;376:2018–31.
- Serjeant GR, Serjeant BE. Sickle cell disease. 3rd ed. New York: Oxford University Press; 2001.
- Moo-Penn W, Bechtel K, Jue D, et al. The presence of hemoglobin S and C Harlem in an individual in the United States. Blood 1975;46:363–7.
- Monplaisir N, Merault G, Poyart C, et al. Hemoglobin S Antilles: a variant with lower solubility than hemoglobin S and producing sickle cell disease in heterozygotes. Proc Natl Acad Sci U S A 1986;83:9363–7.
- Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood Rev 2003;17:167–78.
- Nagel RL, Daar S, Romero JR, et al. HbS-oman heterozygote: a new dominant sickle syndrome. Blood 1998;92:4375–82.
- Masiello D, Heeney MM, Adewoye AH, et al. Hemoglobin SE disease: a concise review. Am J Hematol 2007;82:643–9.
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med 1997;337:762–9.
- Brittenham GM, Schechter AN, Noguchi CT. Hemoglobin S polymerization: primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood 1985;65:183–9.
- .Nagel RL, Bookchin RM, Johnson J, et al. Structural bases of the inhibitory effects of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S. Proc Natl Acad Sci U S A 1979;76:670–2.
- .Ballas SK, Dover GJ, Charache S. Effect of hydroxyurea on the rheological properties of sickle erythrocytes in vivo. Am J Hematol 1989;32:104–11.
- Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol 2002;9:101–6.
- Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nature Med 2002;8:1383–9.
- Nouraie M, Lee JS, Zhang Y, et al. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica 2013;98:464–72.
- Serjeant GR. The natural history of sickle cell disease. Cold Spring Harb Perspect Med 2013;3:a011783.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48.
- Serjeant GR. Natural history and determinants of clinical severity of sickle cell disease. Curr Opin Hematol 1995;2:103–8.
- Odenheimer DJ, Sarnaik SA, Whitten CF, et al. The relationship between fetal hemoglobin and disease severity in children with sickle cell anemia. Am J Med Genet 1987;27:525–35.
- Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med 1991;325:11–6.
- Falusi AG, Olatunji PO. Effects of alpha thalassaemia and haemoglobin F (HbF) level on the clinical severity of sickle-cell anaemia. Eur J Haematol 1994;52:13–5.
- Watson J. The significance of the paucity of sickle cells in newborn Negro infants. Am J Med Sci 1948;215:419–23.
- Marcus SJ, Ware RE. Physiologic decline in fetal hemoglobin parameters in infants with sickle cell disease: implications for pharmacological intervention. J Pediatr Hematol Oncol 1999;21:407–11.
- Schroeder WA, Powars DR, Kay LM, et al. Beta-cluster haplotypes, alpha-gene status, and hematological data from SS, SC, and S-beta-thalassemia patients in southern California. Hemoglobin 1989;13:325–53.
- Steinberg MH, Voskaridou E, Kutlar A, et al. Concordant fetal hemoglobin response to hydroxyurea in siblings with sickle cell disease. Am J Hematol 2003;72:121–6.
- Ngo D, Bae H, Steinberg MH, et al. Fetal hemoglobin in sickle cell anemia: genetic studies of the Arab-Indian haplotype. Blood Cells Mol Dis 2013;51:22–6.
- Galarneau G, Palmer CD, Sankaran VG, et al. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet 2010;42:1049–51.
- Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U S A 2008;105:11869–74.
- Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 2008;322:1839–42.
- Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A 2008;105:1620–5.
- Adams RJ, Kutlar A, McKie V, et al. Alpha thalassemia and stroke risk in sickle cell anemia. Am J Hematol 1994;45:279–82.
- Ballas SK. Effect of alpha-globin genotype on the pathophysiology of sickle cell disease. Pediatr Pathol Mol Med 2001;20:107–21.
- Gaston MH, Verter JI, Woods G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med 1986;314:1593–9.
- Ballas SK, Talacki CA, Rao VM, Steiner RM. The prevalence of avascular necrosis in sickle cell anemia: correlation with alpha-thalassemia. Hemoglobin 1989;13:649–55.
- .Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev 2007;21:37–47.
- Murphy JR, Wengard M, Brereton W. Rheological studies of Hb SS blood: influence of hematocrit, hypertonicity, separation of cells, deoxygenation, and mixture with normal cells. J Lab Clin Med 1976;87:475–86.
- Fabry ME, Kaul DK, Raventos-Suarez C, Chang H, Nagel RL. SC erythrocytes have an abnormally high intracellular hemoglobin concentration. Pathophysiological consequences. J Clin Invest 1982;70:1315–9.
- Stuart J, Johnson CS. Rheology of the sickle cell disorders. Baillieres Clin Haematol 1987;1:747–75.
- Lionnet F, Hammoudi N, Stojanovic KS, et al. Hemoglobin sickle cell disease complications: a clinical study of 179 cases. Haematologica 2012;97:1136-41.
- Adamkiewicz TV, Sarnaik S, Buchanan GR, et al. Invasive pneumococcal infections in children with sickle cell disease in the era of penicillin prophylaxis, antibiotic resistance, and 23-valent pneumococcal polysaccharide vaccination. J Pediatr 2003;143:438–44.
- Abboud MR, Yim E, Musallam KM, Adams RJ, Investigators SIS. Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from STOP II. Blood 2011;118:894–8.
- Adams RJ. Lessons from the Stroke Prevention Trial in Sickle Cell Anemia (STOP) study. J Child Neurol 2000;15:344–9.
- Adams RJ, Brambilla DJ, Granger S, et al. Stroke and conversion to high risk in children screened with transcranial Doppler ultrasound during the STOP study. Blood 2004;103:3689–94.
- Lee MT, Piomelli S, Granger S, et al. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow-up and final results. Blood 2006;108:847–52.
- Charache S, Dover GJ, Moore RD, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood 1992;79:2555–65.
- Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995;332:1317–22.
- Charache S. Mechanism of action of hydroxyurea in the management of sickle cell anemia in adults. Semin Hematol 1997;34:15–21.
- Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol 2010;85:403–8.
- Noguchi CT, Rodgers GP, Serjeant G, Schechter AN. Levels of fetal hemoglobin necessary for treatment of sickle cell disease. N Engl J Med 1988;318:96–9.
- Powars DR, Weiss JN, Chan LS, Schroeder WA. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood 1984;63:921–6.
- Maier-Redelsperger M, de Montalembert M, Flahault A, et al. Fetal hemoglobin and F-cell responses to long-term hydroxyurea treatment in young sickle cell patients. The French Study Group on Sickle Cell Disease. Blood 1998;91:4472–9.
- Steinberg MH, Chui DH, Dover GJ, et al. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood 2014;123:481–5.
- Adragna NC, Fonseca P, Lauf PK. Hydroxyurea affects cell morphology, cation transport, and red blood cell adhesion in cultured vascular endothelial cells. Blood 1994;83:553–60.
- Bridges KR, Barabino GD, Brugnara C, et al. A multiparameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood 1996;88:4701–10.
- Jiang J, Jordan SJ, Barr DP, et al. In vivo production of nitric oxide in rats after administration of hydroxyurea. Mol Pharmacol 1997;52:1081–6.
- Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 2010;115:5300–11.
- Yates AM, Dedeken L, Smeltzer MP, et al. Hydroxyurea treatment of children with hemoglobin SC disease. Pediatr Blood Cancer 2013;60:323–5.
- Barbosa CG, Aleluia AC, Pacheco AP, et al. Genetic modulation of HbF in Brazilians with HbSC disease and sickle cell anemia. Am J Hematol 2013;88:923–4.
- Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med 2009;361:2309–17.
- King A, Shenoy S. Evidence-based focused review of the status of hematopoietic stem cell transplantation as treatment of sickle cell disease and thalassemia. Blood 2014;123:3089–94.
- Oringanje C, Nemecek E, Oniyangi O. Hematopoietic stem cell transplantation for people with sickle cell disease. Cochrane Database Syst Rev 2013;5:CD007001.
- Freed J, Talano J, Small T, et al. Allogeneic cellular and autologous stem cell therapy for sickle cell disease: ‘whom, when and how’. Bone Marrow Transplant 2012;47:1489–98.
- Urbinati F, Hargrove PW, Geiger S, et al. Potentially therapeutic levels of anti-sickling globin gene expression following lentivirus-mediated gene transfer in sickle cell disease bone marrow CD34 cells. Exp Hematol 2015;43:346–51.
- Brugnara C, Mohandas N. Red cell indices in classification and treatment of anemias: from M.M. Wintrobes’s original 1934 classification to the third millennium. Curr Opin Hematol 2013;20:222–30.
- Key NS, Derebail VK. Sickle-cell trait: novel clinical significance. Hematology Am Soc Hematol Educ Program 2010;2010:418–22.
- Kark JA, Posey DM, Schumacher HR, Ruehle CJ. Sickle-cell trait as a risk factor for sudden death in physical training. N Engl J Med 1987;317:781–7.
- Goldsmith JC, Bonham VL, Joiner CH, et al. Framing the research agenda for sickle cell trait: building on the current understanding of clinical events and their potential implications. Am J Hematol 2012;87:340–6.
- Grant AM, Parker CS, Jordan LB, et al. Public health implications of sickle cell trait: a report of the CDC meeting. Am J Prev Med 2011;41:S435–9.
- Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance. Am J Hematol 2005;79:17–25.
- Serjeant GR, Ceulaer CD, Lethbridge R, et al. The painful crisis of homozygous sickle cell disease: clinical features. Br J Haematol 1994;87:586–91.
- Vichinsky EP, Johnson R, Lubin BH. Multidisciplinary approach to pain management in sickle cell disease. Am J Pediatr Hematol Oncol 1982;4:328–33.
- Gil KM, Carson JW, Porter LS, et al. Daily mood and stress predict pain, health care use, and work activity in African American adults with sickle-cell disease. Health Psychol 2004;23:267–74.
- Amjad H, Bannerman RM, Judisch JM. Letter: Sickling pain and season. Br Med J 1974;2:54.
- Ibrahim AS. Relationship between meteorological changes and occurrence of painful sickle cell crises in Kuwait. Trans R Soc Trop Med Hyg 1980;74:159–61.
- Jones S, Duncan ER, Thomas N, et al. Windy weather and low humidity are associated with an increased number of hospital admissions for acute pain and sickle cell disease in an urban environment with a maritime temperate climate. Br J Haematol 2005;131:530–3.
- Resar LM, Oski FA. Cold water exposure and vaso-occlusive crises in sickle cell anemia. J Pediatr 1991;118:407–9.
- Darbari DS, Ballas SK, Clauw DJ. Thinking beyond sickling to better understand pain in sickle cell disease. Eur J Haematol 2014;93:89–95.
- Darbari DS, Onyekwere O, Nouraie M, et al. Markers of severe vaso-occlusive painful episode frequency in children and adolescents with sickle cell anemia. J Pediatr 2011;160:286–90.
- Hollins M, Stonerock GL, Kisaalita NR, et al. Detecting the emergence of chronic pain in sickle cell disease. J Pain Symptom Manage 2012;43:1082–93.
- Ballas SK, Darbari DS. Neuropathy, neuropathic pain, and sickle cell disease. Am J Hematol 2013;88:927–9.
- Brandow AM, Farley RA, Panepinto JA. Early insights into the neurobiology of pain in sickle cell disease: A systematic review of the literature. Pediatr Blood Cancer 2015 May 13. doi: 10.1002/pbc.25574. [Epub ahead of print].
- Brandow AM, Farley RA, Panepinto JA. Neuropathic pain in patients with sickle cell disease. Pediatr Blood Cancer 2014;61:512–7.
- Brandow AM, Farley RA, Dasgupta M, et al. The use of neuropathic pain drugs in children with sickle cell disease is associated with older age, female sex, and longer length of hospital stay. J Pediatr Hematol Oncol 2015;37:10–5.
- Walker TM, Hambleton IR, Serjeant GR. Gallstones in sickle cell disease: observations from The Jamaican Cohort study. J Pediatr 2000;136:80–5.
- Penner E, Mayr WR, Djawan S, et al. [The genetics of Gilbert syndrome]. Schweiz Med Wochenschr 1976;106:860–2. [German]
- Powell RW, Levine GL, Yang YM, Mankad VN. Acute splenic sequestration crisis in sickle cell disease: early detection and treatment. J Pediatr Surg 1992;27:215–8.
- Al Salem AH, Qaisaruddin S, Nasserullah Z, et al. Splenectomy and acute splenic sequestration crises in sickle cell disease. Pediatr Surg Int 1996;11:26–8.
- Pearson HA, Spencer RP, Cornelius EA. Functional asplenia in sickle-cell anemia. N Engl J Med 1969;281:923–6.
- Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011;377:1663–72.
- Brousse V, Buffet P, Rees D. The spleen and sickle cell disease: the sick(led) spleen. Br J Haematol 2014;166:165–76.
- Orringer EP, Fowler VG Jr, Owens CM, et al. Case report: splenic infarction and acute splenic sequestration in adults with hemoglobin SC disease. Am J Med Sci 1991;302:374–9.
- Michel JB, Hernandez JA, Buchanan GR. A fatal case of acute splenic sequestration in a 53–year-old woman with sickle-hemoglobin C disease. Am J Med 1992;92:97–100.
- Hatton CS, Bunch C, Weatherall DJ. Hepatic sequestration in sickle cell anaemia. Br Med J (Clin Res Ed) 1985;290:744–5.
- Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood 1994;84:643–9.
- Gill FM, Sleeper LA, Weiner SJ, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood 1995;86:776–83.
- Owusu-Ofori S, Hirst C. Splenectomy versus conservative management for acute sequestration crises in people with sickle cell disease. Cochrane Database Syst Rev 2013;5:CD003425.
- 00.Al-Salem AH. Splenic complications of sickle cell anemia and the role of splenectomy. ISRN Hematol 2011;2011:864257.
- Sabarense AP, Lima GO, Silva LM, Viana MB. Characterization of mortality in children with sickle cell disease diagnosed through the Newborn Screening Program. J Pediatr (Rio J) 2015;91:242–7.
- Aslam AF, Aslam AK, Dipillo F. Fatal splenic sequestration crisis with multiorgan failure in an adult woman with sickle cell-beta+ thalassemia. Am J Med Sci 2005;329:141–3.
- Berry RA, Odumakinde EA, Lewis JP. Massive splenic infarction in doubly abnormal heterozygous sickling disorders. A new complication of acute splenic sequestration syndrome. The West J Med 1991;155:531–2.
- Bonanomi MT, Lavezzo MM. Sickle cell retinopathy: diagnosis and treatment. Arq Bras de Oftalmol 2013;76:320–7.
- Chen RW, Flynn HW Jr, Lee WH, et al. Vitreoretinal management and surgical outcomes in proliferative sickle retinopathy: a case series. Am J Ophthalmol 2014;157:870–5 e1.
- Howard J, Malfroy M, Llewelyn C, et al. The Transfusion Alternatives Preoperatively in Sickle Cell Disease (TAPS) study: a randomised, controlled, multicentre clinical trial. Lancet 2013;381:930–8.
- Neumayr L, Koshy M, Haberkern C, et al. Surgery in patients with hemoglobin SC disease. Preoperative Transfusion in Sickle Cell Disease Study Group. Am J Hematol 1998;57:101–8.
- 08. Savage WJ, Buchanan GR, Yawn BP, et al. Evidence gaps in the management of sickle cell disease: A summary of needed research. Am J Hematol 2015;90:273–5.
- Charache S, Barton FB, Moore RD, et al. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive “switching” agent. The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine (Baltimore) 1996;75:300–26.
- Steinberg MH, Nagel RL, Brugnara C. Cellular effects of hydroxyurea in Hb SC disease. Br J Haematol 1997;98:838–44.
- Wang W, Brugnara C, Snyder C, et al. The effects of hydroxycarbamide and magnesium on haemoglobin SC disease: results of the multi-centre CHAMPS trial. Br J Haematol 2011;152:771–6.
- Jungst C, Kullak-Ublick GA, Jungst D. Gallstone disease: Microlithiasis and sludge. Best Pract Res Clin Gastroenterol 2006;20:1053–62.
- Walker TM, Serjeant GR. Biliary sludge in sickle cell disease. J Pediatr 1996;129:443–5.
- Bauer TW, Moore GW, Hutchins GM. The liver in sickle cell disease. A clinicopathologic study of 70 patients. Am J Med 1980;69:833–7.
- Ebert EC, Nagar M, Hagspiel KD. Gastrointestinal and hepatic complications of sickle cell disease. Clin Gastroenterol Hepatol 2010;8:483–9.
- Hiran S. Multiorgan dysfunction syndrome in sickle cell disease. J Assoc Physicians India 2005;53:19–22.
- Shao SH, Orringer EP. Case report: splenic sequestration and multiorgan failure as the presenting manifestation of hemoglobin SC disease. Am J Med Sci 1996;311:139–41.
- Hassell KL, Eckman JR, Lane PA. Acute multiorgan failure syndrome: a potentially catastrophic complication of severe sickle cell pain episodes. Am J Med 1994;96:155–62.
INTRODUCTION
Sickle cell disease is the most common inherited blood disorder in the world. It affects more than 100,000 individuals in the United States, and millions more worldwide.1 Sickle cell disease is most commonly found in individuals of African heritage, but the disease also occurs in Hispanics and people of Middle Eastern and subcontinent Indian heritage.2 The distribution of the sickle hemoglobin (hemoglobin S [HbS]) allele overlaps with the distribution of malaria; HbS carriers, or individuals with sickle cell trait, have protection against malaria,3 and are not considered to have sickle cell disease.
Sickle cell disease is a severe monogenic disorder marked by significant morbidity and mortality, affecting every organ in the body.4 The term sickle cell disease refers to all genotypes that cause sickling; the most common are the homozygous hemoglobin SS (HbSS) and compound heterozygotes hemoglobin SC (HbSC), hemoglobin S–β0-thalassemia (HbSβ0), and hemoglobin S–β+-thalassemia (HbSβ+), although HbS and several rarer hemoglobin variants such as HbSO(Arab) and HbSD(Punjab) can also cause sickle cell disease. The term sickle cell anemia refers exclusively to the most severe genotypes, HbSS and HbSβ0.5 Common sickling genotypes along with their relative clinical severity are shown in Table 1.6–11
| Table 1. Genotypes of Sickling Syndromes and Their Relative Severities | ||
Genotype | Severity | Characteristics |
HbSS | Severe | Most common form |
HbSβ0 | Severe | Clinically indistinguishable from HbSS6 |
HbSO-Arab | Severe | Relatively rare6 |
HbSD-Punjab | Severe | Mostly in northern India6 |
HbSC-Harlem | Severe | Migrates like HbSC, but rare double β-globin mutation7 |
HbCS-Antilles | Severe | Rare double β-globin mutation8 |
HbSC | Moderate | 25% of SCD9 |
HbSβ+, Mediterranean | Moderate | 5%–16% HbA6 |
HbAS-Oman | Moderate | Dominant rare double β-globin mutation10 |
HbSβ+, African | Mild | 16%–30% HbA6 |
HbSE | Mild | HbE found mostly in Southeast Asia11 |
HbS-HPFH | Very mild | Large deletions in β-globin gene complex; > 30% HbF6 |
| HbA = hemoglobin A; HbE = hemoglobin E; HbF = fetal hemoglobin; HbS-HPFH = HbS and gene deletion HPFH; HbSC = heterozygous hemoglobin SC; HbSS = homozygous hemoglobin SS; HbSβ0 = hemoglobin S-β thalassemia0; HbSβ+ = hemoglobin S-β thalassemia+; SCD = sickle cell disease. | ||
This article reviews the pathophysiology of sickle cell disease, common clinical complications, and available therapies. A complex case which illustrates diagnostic and management challenges is presented as well.
PATHOPHYSIOLOGY
HbS is the result of a substitution of valine for glutamic acid in the sixth amino acid of the β-globin chain.12 The change from a hydrophilic to a hydrophobic amino acid causes the hemoglobin molecules to stack, or polymerize, when deoxygenated. This rigid rod of hemoglobin distorts the cell, producing the characteristic crescent or sickle shape that gives the disease its name.13 Polymerization of hemoglobin within the cell is promoted by dehydration, which increases the concentration of HbS.13,14 Polymerization occurs when hemoglobin is in the deoxygenated state.13
The sickle red blood cell is abnormal; it is rigid and dense, and lacks the deformability needed to navigate the microvasculature.15 Blockages of blood flow result in painful vaso-occlusion that is the hallmark of the disease, and that also can cause damage to the spleen, kidneys, and liver.16 The sickle red cell is also fragile, with a lifespan of only 20 days compared to the 120-day lifespan of a normal red blood cell.13 Frequent hemolysis results in anemia and the release of free hemoglobin, which both scavenges nitric oxide and impairs the production of more nitric oxide, which is essential for vasodilatation.17 This contributes to vascular dysfunction and an increased risk for stroke.18 If untreated, the natural course of sickle cell anemia is mortality in early childhood in most cases.19 Common chronic and acute sickle cell disease–related complications and recommended therapies, based on 2014 National Institutes of Health guidelines, are shown in Table 2 and Table 3.20
| Table 2. Common Adult Sickle Cell Disease Chronic Complications and Recommended Therapies | ||
Chronic Complication | Recommended Therapy | Strength of Recommendation |
Chronic pain | Opioids | Consensus |
Avascular necrosis | Analgesics and physical therapy | Consensus |
Proliferative sickle retinopathy | Laser photocoagulation | Strong |
Leg ulcers | Standard wound care | Moderate |
Recurrent priapism | Consult urology | Moderate |
| Data from Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48. | ||
| Table 3. Common Adult Sickle Cell Disease Acute Complications and Recommended Therapies | ||
Acute Complication | Recommended Therapy | Strength of Recommendation |
Vaso-occlusive crisis | NSAIDs, opioids for severe pain | Moderate-consensus |
ACS | Antibiotics, oxygen | Strong |
Simple transfusiona | Weak | |
Urgent exchange transfusionb | Strong | |
Acute stroke | Exchange transfusion | Strong |
Priapism ≥ 4 hr | Aggressive hydration, pain control, and urology consult | Strong-consensus |
Gallstones, symptomatic | Cholecystectomy, laparoscopic | Strong |
Splenic sequestration | Intravenous fluids, transfuse cautiously, discuss surgical splenectomy | Strong-moderate |
Acute renal failure | Consult nephrologyc | Consensus |
ACS = acute chest syndrome; NSAIDs = nonsteroidal anti-inflammatory drugs. a For symptomatic ACS with hemoglobin > 1 g/dL below baseline but > 9.0 g/dL. b When there is progression of ACS (SpO2 < 90% despite supplemental oxygen, increasing respiratory distress, progressive pulmonary infiltrates despite simple transfusion). c For acute rise in creatinine ≥ 0.3 mg/dL; do not give transfusions unless there are other indications. Data from Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48. | ||
One of the most challenging aspects of sickle cell disease is its clinical variability. While in general, HbSS and HbSβ0 are the most severe genotypes, there are patients with HbSC and HbSb+ who have significant sickle-cell–related complications, and may have a more severe clinical course than a HbSS patient.21 A great deal of this clinical variability cannot be explained, but some can be attributed to endogenous fetal hemoglobin (HbF) levels.22–24 The importance of HbF levels in sickle cell disease was first noted by a pediatrician in the 1940s.25 She observed that sickle cell disease complications in children under the age of 1 were rare, and attributed it to the presence of HbF.25 HbF levels decline more slowly in individuals with hemoglobinopathies, reaching their nadir after the age of 5 rather than within 6 months of birth in individuals without hemoglobinopathies.26 HbF levels remain elevated lifelong in most sickle cell disease patients, especially those with the HbSS and HbSβ0 genotypes. Levels of HbF vary widely between individuals, from zero to 20% to 30%, with a median of 10%.26–28 Individuals who produce more HbF have a milder course, in general.24 An association between the 4 β-globin haplotypes and HbF levels has been reported in the past,27,29 but more sophisticated next-generation sequencing has revealed causal variants in BCL11A and HBS1L-MYB that contribute approximately 50% of the observed variability in HbF levels.30–33
Co-inheritance of α-thalassemia also modifies disease course; less available α-globin chains results in a lower hemoglobin concentration within the cell. Paradoxically, this results in a higher overall hemoglobin level, as there is a reduction in polymerization, and therefore sickling due to lower HbS concentrations in the cell. Patients therefore are less anemic, reducing the risk of stroke in childhood,34,35 but blood viscosity may be higher, resulting in more frequent pain crises and increased risk36 of avascular necrosis.34,35,37 It is often helpful to think of sickle cell patients as falling into 1 of 2 groups: high hemolysis/low hemoglobin and high viscosity/high hemoglobin. Individuals with high rates of hemolysis are at greater risk for stroke, pulmonary hypertension, and acute chest syndrome (ACS). Higher rates of hemolysis result in higher levels of free hemoglobin, which scavenges nitric oxide. This leads to the vascular damage and dysfunction that contributes to the associated clinical complications. This phenotype is most commonly seen in HbSS and HbSβ0.38 High hemoglobin/high viscosity phenotypes are most often found in HbSC patients and in sickle cell anemia with α-thalassemia coinheritance.39–42
TREATMENT OPTIONS
In high-resource countries with newborn screening, the initiation of penicillin prophylaxis has dramatically altered the natural history of the disease, allowing the majority of patients to reach adulthood.43 Penicillin prophylaxis is usually discontinued at age 5 years; however, individuals who have undergone surgical splenectomy or have had pneumococcal sepsis on penicillin prophylaxis may remain on penicillin to age 18 or beyond.20
Another advance in sickle cell care is screening for stroke risk through transcranial Doppler ultrasound (TCD).44–47 This screening tool has reduced the incidence of childhood stroke from 10% by age 11 to 1%. TCDs typically cannot be performed after the age of 16 due to changes in the skull. Individuals found to have abnormal (elevated) TCD velocities are placed on chronic transfusion therapy for primary stroke prevention. They may remain on monthly chronic transfusions, with the goal of suppressing the percentage of HbS to 30% to 50% indefinitely. A clinical trial (STOPII) designed to determine if pediatric sickle cell disease patients on chronic transfusion therapy for primary stroke prevention could be safely taken off transfusion therapy was discontinued early due to an excess of strokes and conversion to abnormal TCD velocities in the untransfused arm.44 Individuals who have experienced an ischemic stroke have a 70% risk of another stroke, and must remain on chronic transfusion therapy indefinitely. Chronic transfusion reduces their stroke risk to 13%.
The only widely used pharmacologic therapy for sickle cell disease is hydroxyurea.12,48–50 A significant portion of the benefit of hydroxyurea stems from its induction of HbF.51 HbF does not sickle, and it interrupts the polymerization of HbS in the cell, if present in high enough concentrations.50 The level of HbF needed to achieve clinical improvement is not known, but in vitro assays suggest 20% HbF is needed to prevent sickling.52,53 However, endogenous and hydroxyurea-induced HbF is not distributed evenly through the red cells, so sickling is possible regardless of the level of HbF induced.54,55 Hydroxyurea likely has other disease-modifying effects as well, including reduction of white blood cell count and reticulocyte count and reduction of red cell adhesion to the endothelium.56–58 Clinical criteria for initiation of hydroxyurea in adult sickle cell disease patients are shown in Table 4.20 Hydroxyurea is given daily and is dosed to maximum tolerated dose for the individual by following the absolute neutrophil count (ANC). The goal ANC is between 2000 and 4000/µL. At times, absolute reticulocyte count (ARC) can be dose-limiting; goal ARC is greater than 70,000/µL.59 Platelet counts may be reduced as well, especially in HbSC patients.60,61
| Table 4. Indications for Hydroxyurea in Adult Patients with Sickle Cell Disease | |
Indication | Strength of Recommendation |
SCA with ≥ 3 pain crises per year | Strong |
SCA with pain that interferes with ADL and QoL | Strong |
History of severe or recurrent ACS | Strong |
Chronic kidney disease on epoetin | Weak |
HbSβ+ and HbSC with pain that interferes with ADL and QoL; consult sickle cell disease expert | Moderate |
| ACS = acute chest syndrome; ADL = activities of daily living; QoL = quality of life; SCA = sickle cell anemia. | |
The only curative therapy for sickle cell disease is hematopoietic stem cell transplant.62 Transplant use is limited by availability of matched sibling donors,62 and even at experienced centers transplant carries a small risk for mortality, graft rejection, and graft-versus-host disease. Furthermore, consensus on disease complications for which transplant is recommended is also lacking.63–65 Clinical trials of gene therapy for sickle cell disease and thalassemia are ongoing.66
COMPLICATIONS AND DISEASE-SPECIFIC THERAPIES
CASE PRESENTATION
A 26-year-old African-American man who works as a school bus driver presents to an academic center’s emergency department complaining of pain in his left leg, similar to prior pain events. He is described as having sickle cell trait, although no hemoglobin profile is available in his chart. He describes the pain as dull and aching, 10/10 in intensity. A complete blood count (CBC) is obtained; it reveals a hemoglobin of 14.5 g/dL, white blood cell (WBC) count of 5600/µL, and platelet count of 344,000/µL. His CBC is also notable for a mean corpuscular volume (MCV) of 72 fL, a mean corpuscular hemoglobin concentration (MCHC) of 37 g/dL, and a red blood cell distribution width (RDW) of 12. Slide review of a peripheral blood smear shows 2+ target cells (Figure).
The patient is given 6 mg of morphine, which provides some relief of his pain, and is discharged with a prescription for hydrocodone bitartrate/acetaminophen 5/325 mg. The diagnosis given is musculoskeletal pain, and he is instructed to follow-up with a primary care physician. His past medical history is significant for 4 or 5 visits to the emergency department per year in the past 4 years. Prior to 4 years ago, he rarely required medical attention.
• What laboratory and clinical features might lead you to question the diagnosis of sickle cell trait in this patient?
The patient’s hemoglobin is within normal range, which is consistent with sickle cell trait; however, he is microcytic, with a normal RDW. It is possible to be mildly microcytic in the early stages of iron deficiency, prior to the development of anemia, but the RDW would typically be elevated, demonstrating the presence of newer, smaller cells produced under conditions of iron deficiency.67 It is also possible that his microcytosis with a normal RDW could represent sickle cell trait with co-inheritance of β-thalassemia. Up to 30% of African Americans have β-thalassemia,2 and 1 in 10 have sickle cell trait.68 However, a high MCHC, indicating the presence of dense cells, and target cells noted on slide review are most consistent with HbSC.9 HbSC patients, especially males, can have hemoglobin levels in the normal range.4 The biggest inconsistency with the diagnosis of sickle cell trait is his history of frequent pain events. Individuals with sickle cell trait rarely present with pain crises, except under extreme conditions of dehydration or high altitude.68 Sickle cell trait is generally regarded as a benign condition, although a study of U.S. military recruits found a 30-fold higher risk of sudden death during basic training in persons with sickle cell trait.69 Additional sickle cell trait–related complications include hematuria, risk of splenic sequestration or infarct under extreme conditions and high altitude, and a rare and usually fatal renal malignancy, renal medullary carcinoma, which is vanishingly rare in individuals without sickle cell trait.70,71 Although the patient reported having sickle cell trait, this diagnosis should have been verified with a hemoglobin panel, given his atypical presentation.20
• What is the approach to managing pain episodes in sickle cell disease?
In sickle cell disease, vaso-occlusive pain events can be common, often beginning in early childhood.17 This disease complication accounts for 95% of all adult sickle cell disease hospitalizations.72 There is a great deal of variability in pain symptoms between individuals, and within individuals at various times in their lives:73 30% have no pain events, 50% have occasional events, and 20% have monthly or more frequent events that require hospitalization.74 The frequency and severity of pain events are modulated by HbF levels, β-thalassemia status, genotypes, therapies like hydroxyurea, or in rare cases, chronic transfusion therapy.23 Personal factors, such as psychosocial stressors, also contribute to the frequency of pain events.75 Pain event triggers include exposure to cold water, windy or cold weather, temperature changes, and extreme temperatures.76–79 Patient age also contributes to pain event frequency. Many patients see an increase in pain event frequency in their late 20s, and a marked decrease in their 40s.23,73 More than 3 pain events per year is associated with reduced life expectancy.23
Acute management of pain episodes involves nonsteroidal anti-inflammatory drugs, oral opioids, and when hospitalization is required, intravenous opioids, often delivered via patient-controlled analgesia (PCA) pumps.79 As sickle cell disease patients become teenagers and young adults, some experience an increased frequency of pain episodes, with fewer pain-free days, or a failure to return to baseline before the next pain crisis occurs.80,81 This is characteristic of emerging chronic pain.82 Chronic pain is a significant problem in adult patients with sickle cell disease, with up to 85% reporting pain on most days.72,80 The development of chronic pain may be reduced by early and aggressive treatment of acute pain events, as well as use of hydroxyurea to reduce the number of pain events. Many adult sickle cell patients with chronic pain are treated with daily opioids.20 Given the significant side effects of chronic opioid use—sedation, respiratory depression, itching, nausea, and impairment of function and quality of life—non-opioid therapies are under investigation.83 Many chronic pain patients have symptoms of neuropathic pain, and may benefit from neuropathic agents like gabapentin, both to reduce opioid use and to more effectively treat chronic neuropathic pain, which is known to respond poorly to opioids.84–86
• Is the patient’s peripheral blood smear consistent with a diagnosis of sickle cell trait?
Several target cells are visible, which is not typical of sickle cell trait, but may be seen in HbSC or thalassemia. The finding of an intracellular crystal is pathognomonic for HbSC or HbCC. HbC polymerizes in high oxygen conditions, opposite of HbS, which polymerizes in low oxygen conditions.9
CASE CONTINUED
The patient’s family history is significant for a sister who died at age 3 from sickle cell–related complications, and a sister with sickle cell trait who had a cholecystectomy for gallstones at age 22. His father died at age 38 due to unknown causes. The sickle cell trait status of his parents is unknown. His mother is alive, and has hypertension.
• Is the medical history of this patient’s family members consistent with sickle cell trait?
It is unlikely that sickle cell trait would result in early death in childhood, or in gallstones at age 22. Gallstones in early adulthood is a common presentation for HbSC patients not diagnosed by newborn screening.87 Any hemolytic condition can lead to the formation of hemoglobin-containing pigmented gallstones, biliary sludge, and obstruction of the gallbladder. In the presence of right-sided abdominal pain, a serum bilirubin level of more than 4 mg/dL should lead to measurement of direct bilirubin; if greater than 10% of total, imaging of the gallbladder should be obtained. In sickle cell disease, 30% of patients will have gallstones by 18 years of age. The low hemolysis/high viscosity phenotype patients are typically older at diagnosis. Co-inheritance of Gilbert syndrome and sickle cell disease is not uncommon, and can result in formation of gallstones at a young age; Gilbert syndrome alone typically results in gallstones in mid-life.88
CASE CONTINUED
Two months later, the patient presents again to the emergency department with the same complaint of leg pain, as well as abdominal pain. His hemoglobin is 12.5 g/dL, and his platelet count is 134,000/µL. His pain is not improved with 3 doses of morphine 6 mg intravenously, and he is admitted to the medicine service. A hemoglobin profile is obtained, revealing 52% HbS, 45% HbC, and 1.5% HbF, consistent with HbSC. In sickle cell trait, the hemoglobin profile is 60% HbA and 40% HbS (available α-globin prefers to pair with a normal β-globin, so the ratio of HbA to HbS is 60:40, not 50:50).
On the second hospital day, the patient’s hemoglobin drops to 7.2 g/dL and his platelet count decreases to 44,000/µL. His abdomen is distended and diffusely tender. The internist transfuses him with 2 units of packed red blood cells (PRBC), after which his hemoglobin increases to 11 g/dL, while his platelet count increases to 112,000/µL. Following the transfusion, his abdominal pain resolves, as does his anemia and thrombocytopenia.
• What caused this patient’s anemia and thrombocytopenia?
High on the differential diagnosis is a splenic sequestration. Acute splenic sequestration occurs when red cells are trapped in the splenic sinuses. Massive splenic enlargement may occur over several hours.89,90 Unrecognized splenic sequestration has a high mortality rate from severe anemia and splenic rupture.90 Splenic sequestration must be ruled out in a sickle cell patient with abdominal pain accompanied by dropping platelet and red cell counts, especially in milder subtypes that often have splenic function preserved into adolescence and adulthood. Sickle cell anemia patients usually become functionally asplenic in early childhood.89,91,92 The rise in hemoglobin, more than would be expected from 2 units of PRBC, plus the improvement in platelet count without a platelet transfusion observed in the case patient strongly supports the diagnosis of splenic sequestration.
Splenic sequestration can occur in any sickle cell patient whose spleen has not fibrosed. Splenic sequestration in adulthood is not uncommon in HbSC patients, who often have preserved splenic function into adulthood.93–95
Clinical signs of splenic sequestration include a rapid drop in hemoglobin, rise in reticulocyte count, a tender, enlarged spleen, and, in severe cases, hypovolemia.89,93 It is treated with prompt blood transfusion, but care must be taken not to overtransfuse the patient, as the spleen can trap several grams of hemoglobin, which may be released upon transfusion, potentially causing life-threatening hyperviscosity.89 Hemoglobin levels must be checked following transfusion in suspected splenic sequestration, and “mini transfusions” of 5 mL/kg are recommended in sickle cell disease patients who are hemodynamically stable.20
Hepatic sequestration may also occur, but it is much less common than splenic sequestration.96 Other conditions on the differential diagnosis include thrombotic thrombocytopenic purpura, which would be unlikely to respond to a transfusion. ACS can cause a drop in hemoglobin, and is treated with simple or exchange transfusions.97 ACS is less likely without respiratory symptoms or oxygen requirement, and usually is not associated with thrombocytopenia. Sepsis may also cause anemia and thrombocytopenia, but again would not likely respond to a simple transfusion. The patient’s response to transfusion is consistent with a sequestering event, not a destructive event as in the case of sepsis.
CASE CONTINUED
Imaging reveals a grossly enlarged spleen, which is having a mass effect on the left kidney. The patient is started on hydroxyurea therapy at 500 mg 3 times daily. Discharge instructions include following up with his primary care physician, continuing hydroxyurea therapy, and receiving yearly dilated eye exams to evaluate for proliferative sickle retinopathy.
• Are these discharge instructions complete?
Splenic sequestration has a 50% recurrence rate.98 In very young children, watchful waiting or chronic transfusion may be implemented to preserve the immunologic function of the spleen and reduce the risk of sepsis.89 Splenectomy after a single episode of sequestration in adults is a matter of debate, with experts advising both watchful waiting99 and splenectomy after recovery from the first sequestering event.100 The patient should have been informed of the risk for recurrence, and the signs and symptoms of splenic sequestration as well as the need for emergency medical attention should have been discussed. Splenic sequestration may be milder in adults than in children, but fatal sequestrations have been reported.95,101–103
Proliferative sickle cell retinopathy is a high viscosity/high hemoglobin complication that may occur more frequently in HbSC than HbSS, with an incidence of 33% in HbSC.42,104 Spontaneous regression of retinopathy occurs in approximately 32% of eyes, and laser or scatter photocoagulation is an effective intervention.105
• Would the patient need to be transfused prior to splenectomy?
Preoperative transfusion therapy is standard of care for HbSS patients undergoing general anesthesia. The TRAP study found that simple “top off” transfusion to a hemoglobin of 10 g/dL was as effective at preventing postoperative sickle cell–related complications as exchange transfusion to HbS of 30% or less, and had fewer transfusion-related complications like alloimmunization.106 There is little data regarding preoperative transfusions in HbSC disease. A retrospective study suggests that HbSC patients undergoing abdominal surgeries should be transfused.107 The higher hemoglobin level of the typical HbSC patient necessitates exchange transfusion to avoid hyperviscosity.
• Is hydroxyurea therapy indicated in this patient?
• Has it been dosed appropriately?
If the patient had the HbSS subtype, hydroxyurea would be clearly indicated, given his frequent pain events.20 HbSC patients may be placed on hydroxyurea on a case-by-case basis, but evidence for its efficacy in this sickle cell subtype is lacking.108 Large clinical trials like the Multi-Center Study of Hydroxyurea (MSH) that established the safety and efficacy of hydroxyurea in sickle cell anemia excluded HbSC and HbSβ+ patients.109 These mild to moderate subtypes produce less HbF at baseline, and typically have a minimal to modest rise in HbF on hydroxyurea.110 In sickle cell anemia, hydroxyurea is titrated to maximum tolerated dose, defined as an ANC of 2000 to 4000/µL and an ARC of 70,000/µL or higher.53 Because of their lower levels of chronic inflammation and lower reticulocyte counts due to higher hemoglobin levels, many HbSC and HbSβ+ patients have values in that range before initiating hydroxyurea therapy.9 Cytopenias, particularly of platelets in HbSC, occur at low doses of hydroxyurea.111
Of note, although the half-life of hydroxyurea would suggest that 3 times daily dosing is indicated, daily dosing has been found to have equal response and is preferred. Another concern is the monitoring of this myelosuppressive medication. This patient has repeatedly failed to obtain a primary care physician or a hematologist, and hydroxyurea requires laboratory monitoring at least every 2 months, especially in a HbSC patient with a very large spleen who is at significant risk for thrombocytopenia and neutropenia.9
CASE CONTINUED
A week after discharge from his admission for abdominal pain diagnosed as splenic sequestration, the patient presents again to the emergency department with abdominal pain which he reports is his typical sickle cell pain. Hemoglobin is 13.8 g/dL, platelet count is 388,000/µL, and alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels are both 10 times their prior value. Creatinine is 1.2 mg/dL (0.75 mg/dL on his prior admission), and total bilirubin is 3 mg/dL, with 0.3 mg/dL direct bilirubin. He undergoes an ultrasound exam of his gallbladder, which reveals sludge and a possible gallstone. There is no evidence of cholecystitis. General surgery performs a laparoscopic cholecystectomy.
• Was this cholecystectomy necessary?
In patients with sickle cell disease, symptomatic gallstones and gallbladder sludge should be observed; recurrent abdominal pain without a significant change in bilirubin may not be due to gallstones or sludge, and therefore may not be relieved by cholecystectomy.112,113 In sickle cell disease, 40% of patients with gallbladder sludge do not develop gallstones.87 The patient’s bilirubin level was at baseline, and there was no increase in the direct (conjugated) fraction. Watchful waiting would have been appropriate, with cholecystectomy being performed if he experienced recurrent symptoms associated with fatty foods accompanied by an elevation in direct bilirubin.
More concerning and deserving of investigation was his elevated liver enzymes. Patients with sickle cell disease may experience recurrent ischemia and reperfusion injuries in the liver, which is called right upper quadrant syndrome. On autopsy of 70 sickle cell patients, 91% had hepatomegaly and 34% had focal necrosis.114 AST is often elevated in sickle cell disease, as it is affected by hemolysis. In this patient, both AST and ALT are elevated, consistent with a hepatocellular disorder. His abdominal pain and ALT rise may be a sign of a hepatic crisis.115 Rapid resolution of ALT elevation in a matter of days suggests a vaso-occlusive, inflammatory event that is self- limiting. Prolonged AST elevation requires further investigation, with consideration of autoimmune hepatitis, viral hepatitis, or iron overload. Iron overload is unlikely in this patient given his lifetime history of only 1 transfusion. Hepatic iron overload typically occurs in sickle cell disease after a minimum of 10 transfusions.115
CASE CONTINUED
The patient is discharged on the day after the procedure, with instructions to continue his hydroxyurea.
• Should the patient resume hydroxyurea therapy?
Hydroxyurea is hepatically cleared and thus it should be held until his liver function tests normalize.106
CASE CONTINUED
Two months later, the patient presents to the emergency department with abdominal pain that moves to his left leg. A CBC is obtained, showing a hemoglobin of 11.8 g/dL and a platelet count of 144,000/µL. He is given 2 doses of morphine 6 mg intravenously, and reports that his leg pain is now a 4/10. He is discharged home with a prescription for hydrocodone/acetaminophen.
• Is the emergency department evaluation sufficient?
This patient remains at high risk for splenic sequestration,93 with a hemoglobin 2 g lower than it was 2 months ago and platelets less than half. This decline could be consistent with early splenic sequestration.20 Additionally, he had elevated liver function tests on a recent admission, as well as rising creatinine, without evidence of resolution. It is not appropriate to discharge him without checking a chemistry and liver panel, and abdominal imaging should be considered. The best plan would be to admit him for observation, given his risk for splenic sequestration, and consult surgery for an elective splenectomy if he has a second episode of splenic sequestration 2 months after the first.100 His abdominal pain that migrates to his left leg could be due to his massive splenomegaly compressing his left kidney, as noted on imaging during his recent admission for splenic sequestration
CASE CONTINUED
An hour after discharge from the emergency department, EMS is called to his home for intractable pain. He is found lying on the floor, and reports excruciating left leg pain. He is brought to the closest hospital, a community hospital that he has not visited previously. There, he is admitted for hydration and pain control and placed on hydromorphone 2 mg every 4 hours as needed for pain. His hemoglobin is 10.8 g/dL, and platelets are 121,000/µL. A chemistry panel is remarkable for a creatinine level of 1.5 mg/dL and a potassium level of 3.2 mEq/L. Liver function tests are not obtained. After 3 doses of hydromorphone, he falls asleep. He is not in a monitored bed, and intravenous fluids, while ordered, are not started. At 6:30 AM the day after admission, he cannot be aroused on a routine vital sign check; he has an SpO2 of 60%, a blood pressure of 80/60 mm Hg, and heart rate of 148 beats/min. A rapid response is called, and naloxone is administered along with oxygen by face mask and several fluid boluses. His systolic blood pressure increases to 100 mm Hg from a low of 70 mm Hg. His SpO2 increases to 92%, and he is arousable and alert, although he reports 10/10 leg pain. His abdomen is noted to be distended and tender.
• What may have contributed to his clinical condition?
The patient is opioid tolerant and has received equivalent doses of opioids in the past without excess sedation. He may have liver dysfunction making him unable to metabolize opioids effectively. His hemoglobin and platelets continue to decline, raising concern for splenic sequestration versus sepsis. Failure to place him on a monitor allowed his hypoxia to continue for an unknown amount of time, placing him at high risk for developing ACS. Lack of intravenous hydration while he was too sedated to drink likely exacerbated his sickling.
CASE CONTINUED
At 9:20 AM, a CBC is obtained and reveals a hemoglobin of 4.8 g/dL and a platelet count of 44,000/µL. Two units of stat O negative blood are administered, and preparations are made to administer an exchange transfusion. A liver panel is obtained 3 hours later, which reveals an AST level of 1200 U/L and an ALT level of 1050 U/L. His bilirubin is 10 mg/dL, and his lactate dehydrogenase level is 1800 U/L. His urine is dark and is positive for bilirubin and ketones. He is transferred to the intensive care unit. A chest X-ray shows pulmonary congestion. Hematology/oncology is consulted.
He receives a 7-unit red blood cell exchange, which reduces his HbS to 11%. He continues to be hypotensive, and requires norepinephrine to support his blood pressure. Antibiotic therapy is started. His creatinine concentration rises to 2.3 mg/dL, potassium is 7.8 mEq/L, and bicarbonate is 12 mEq/L. He is placed on hemodialysis.
Computed tomography of the chest and abdomen reveals lower posterior lung infiltrates and a grossly enlarged spleen. He requires intubation. He is given a diagnosis of ACS in addition to kidney failure, liver failure, and “sickle crisis.” He continues to require daily to twice daily transfusions to maintain a hemoglobin of 7 to 9 g/dL, and his abdominal distension increases. As his condition worsens, surgery is consulted to discuss a liver transplant. He is deemed to not be a surgical candidate, and he passes away 6 days after entering the hospital. The immediate cause of death is listed as vaso-occlusive crisis, with ACS and sickle crisis listed as contributors.
• Are the causes of death accurate and complete?
If vaso-occlusive crisis is used to indicate a pain event, it is not an accurate cause of death. Pain is one of the most distressing complications of sickle cell disease, and frequent pain events are associated with early mortality,4,80 but they are not in themselves fatal. ACS is the number one cause of death in sickle cell disease,4 and it likely contributed to this patient’s death. Sickle crisis is a vague term that should not be used in this context. Causes of death should include splenic sequestration and multisystem organ failure. Multisystem organ failure in sickle cell disease often responds to aggressive transfusion therapy, which this patient received.116–118
CONCLUSION
Sickle cell disease is a complex chronic disease that impacts almost every organ system in the body. Clinicians may be inclined to attribute most pain in a patient with sickle cell disease to a simple vaso-occlusive crisis, treat them for this, and not investigate further. As the case presented here demonstrates, failure to identify the actual life-threatening process occurring in a patient with sickle cell disease presenting with pain can result in preventable early mortality. Clinicians must approach a sickle cell patient reporting pain in a thoughtful manner, and consider a complete differential diagnosis, including both sickle cell disease complications and those unrelated to sickle cell disease. Knowledge of the disease courses of the different sickle cell genotypes is essential, and must go beyond a superficial hierarchy of severity, but rather include an understanding of the complications each genotype is most prone to, and at what ages. Complete laboratory assessment, including a comprehensive metabolic panel, should be performed on all admitted patients, not just a complete blood count. Treating pain with high-dose opioids, while appropriate in an uncomplicated pain crisis, can lead to ACS or even respiratory failure in a patient with uninvestigated liver and kidney dysfunction. The most important lesson to remember is that even the sickle cell disease patient who has been given the unfortunate and pejorative label of “frequent flyer” by some providers has the potential for rapid deterioration into multisystem organ failure and death.
INTRODUCTION
Sickle cell disease is the most common inherited blood disorder in the world. It affects more than 100,000 individuals in the United States, and millions more worldwide.1 Sickle cell disease is most commonly found in individuals of African heritage, but the disease also occurs in Hispanics and people of Middle Eastern and subcontinent Indian heritage.2 The distribution of the sickle hemoglobin (hemoglobin S [HbS]) allele overlaps with the distribution of malaria; HbS carriers, or individuals with sickle cell trait, have protection against malaria,3 and are not considered to have sickle cell disease.
Sickle cell disease is a severe monogenic disorder marked by significant morbidity and mortality, affecting every organ in the body.4 The term sickle cell disease refers to all genotypes that cause sickling; the most common are the homozygous hemoglobin SS (HbSS) and compound heterozygotes hemoglobin SC (HbSC), hemoglobin S–β0-thalassemia (HbSβ0), and hemoglobin S–β+-thalassemia (HbSβ+), although HbS and several rarer hemoglobin variants such as HbSO(Arab) and HbSD(Punjab) can also cause sickle cell disease. The term sickle cell anemia refers exclusively to the most severe genotypes, HbSS and HbSβ0.5 Common sickling genotypes along with their relative clinical severity are shown in Table 1.6–11
| Table 1. Genotypes of Sickling Syndromes and Their Relative Severities | ||
Genotype | Severity | Characteristics |
HbSS | Severe | Most common form |
HbSβ0 | Severe | Clinically indistinguishable from HbSS6 |
HbSO-Arab | Severe | Relatively rare6 |
HbSD-Punjab | Severe | Mostly in northern India6 |
HbSC-Harlem | Severe | Migrates like HbSC, but rare double β-globin mutation7 |
HbCS-Antilles | Severe | Rare double β-globin mutation8 |
HbSC | Moderate | 25% of SCD9 |
HbSβ+, Mediterranean | Moderate | 5%–16% HbA6 |
HbAS-Oman | Moderate | Dominant rare double β-globin mutation10 |
HbSβ+, African | Mild | 16%–30% HbA6 |
HbSE | Mild | HbE found mostly in Southeast Asia11 |
HbS-HPFH | Very mild | Large deletions in β-globin gene complex; > 30% HbF6 |
| HbA = hemoglobin A; HbE = hemoglobin E; HbF = fetal hemoglobin; HbS-HPFH = HbS and gene deletion HPFH; HbSC = heterozygous hemoglobin SC; HbSS = homozygous hemoglobin SS; HbSβ0 = hemoglobin S-β thalassemia0; HbSβ+ = hemoglobin S-β thalassemia+; SCD = sickle cell disease. | ||
This article reviews the pathophysiology of sickle cell disease, common clinical complications, and available therapies. A complex case which illustrates diagnostic and management challenges is presented as well.
PATHOPHYSIOLOGY
HbS is the result of a substitution of valine for glutamic acid in the sixth amino acid of the β-globin chain.12 The change from a hydrophilic to a hydrophobic amino acid causes the hemoglobin molecules to stack, or polymerize, when deoxygenated. This rigid rod of hemoglobin distorts the cell, producing the characteristic crescent or sickle shape that gives the disease its name.13 Polymerization of hemoglobin within the cell is promoted by dehydration, which increases the concentration of HbS.13,14 Polymerization occurs when hemoglobin is in the deoxygenated state.13
The sickle red blood cell is abnormal; it is rigid and dense, and lacks the deformability needed to navigate the microvasculature.15 Blockages of blood flow result in painful vaso-occlusion that is the hallmark of the disease, and that also can cause damage to the spleen, kidneys, and liver.16 The sickle red cell is also fragile, with a lifespan of only 20 days compared to the 120-day lifespan of a normal red blood cell.13 Frequent hemolysis results in anemia and the release of free hemoglobin, which both scavenges nitric oxide and impairs the production of more nitric oxide, which is essential for vasodilatation.17 This contributes to vascular dysfunction and an increased risk for stroke.18 If untreated, the natural course of sickle cell anemia is mortality in early childhood in most cases.19 Common chronic and acute sickle cell disease–related complications and recommended therapies, based on 2014 National Institutes of Health guidelines, are shown in Table 2 and Table 3.20
| Table 2. Common Adult Sickle Cell Disease Chronic Complications and Recommended Therapies | ||
Chronic Complication | Recommended Therapy | Strength of Recommendation |
Chronic pain | Opioids | Consensus |
Avascular necrosis | Analgesics and physical therapy | Consensus |
Proliferative sickle retinopathy | Laser photocoagulation | Strong |
Leg ulcers | Standard wound care | Moderate |
Recurrent priapism | Consult urology | Moderate |
| Data from Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48. | ||
| Table 3. Common Adult Sickle Cell Disease Acute Complications and Recommended Therapies | ||
Acute Complication | Recommended Therapy | Strength of Recommendation |
Vaso-occlusive crisis | NSAIDs, opioids for severe pain | Moderate-consensus |
ACS | Antibiotics, oxygen | Strong |
Simple transfusiona | Weak | |
Urgent exchange transfusionb | Strong | |
Acute stroke | Exchange transfusion | Strong |
Priapism ≥ 4 hr | Aggressive hydration, pain control, and urology consult | Strong-consensus |
Gallstones, symptomatic | Cholecystectomy, laparoscopic | Strong |
Splenic sequestration | Intravenous fluids, transfuse cautiously, discuss surgical splenectomy | Strong-moderate |
Acute renal failure | Consult nephrologyc | Consensus |
ACS = acute chest syndrome; NSAIDs = nonsteroidal anti-inflammatory drugs. a For symptomatic ACS with hemoglobin > 1 g/dL below baseline but > 9.0 g/dL. b When there is progression of ACS (SpO2 < 90% despite supplemental oxygen, increasing respiratory distress, progressive pulmonary infiltrates despite simple transfusion). c For acute rise in creatinine ≥ 0.3 mg/dL; do not give transfusions unless there are other indications. Data from Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48. | ||
One of the most challenging aspects of sickle cell disease is its clinical variability. While in general, HbSS and HbSβ0 are the most severe genotypes, there are patients with HbSC and HbSb+ who have significant sickle-cell–related complications, and may have a more severe clinical course than a HbSS patient.21 A great deal of this clinical variability cannot be explained, but some can be attributed to endogenous fetal hemoglobin (HbF) levels.22–24 The importance of HbF levels in sickle cell disease was first noted by a pediatrician in the 1940s.25 She observed that sickle cell disease complications in children under the age of 1 were rare, and attributed it to the presence of HbF.25 HbF levels decline more slowly in individuals with hemoglobinopathies, reaching their nadir after the age of 5 rather than within 6 months of birth in individuals without hemoglobinopathies.26 HbF levels remain elevated lifelong in most sickle cell disease patients, especially those with the HbSS and HbSβ0 genotypes. Levels of HbF vary widely between individuals, from zero to 20% to 30%, with a median of 10%.26–28 Individuals who produce more HbF have a milder course, in general.24 An association between the 4 β-globin haplotypes and HbF levels has been reported in the past,27,29 but more sophisticated next-generation sequencing has revealed causal variants in BCL11A and HBS1L-MYB that contribute approximately 50% of the observed variability in HbF levels.30–33
Co-inheritance of α-thalassemia also modifies disease course; less available α-globin chains results in a lower hemoglobin concentration within the cell. Paradoxically, this results in a higher overall hemoglobin level, as there is a reduction in polymerization, and therefore sickling due to lower HbS concentrations in the cell. Patients therefore are less anemic, reducing the risk of stroke in childhood,34,35 but blood viscosity may be higher, resulting in more frequent pain crises and increased risk36 of avascular necrosis.34,35,37 It is often helpful to think of sickle cell patients as falling into 1 of 2 groups: high hemolysis/low hemoglobin and high viscosity/high hemoglobin. Individuals with high rates of hemolysis are at greater risk for stroke, pulmonary hypertension, and acute chest syndrome (ACS). Higher rates of hemolysis result in higher levels of free hemoglobin, which scavenges nitric oxide. This leads to the vascular damage and dysfunction that contributes to the associated clinical complications. This phenotype is most commonly seen in HbSS and HbSβ0.38 High hemoglobin/high viscosity phenotypes are most often found in HbSC patients and in sickle cell anemia with α-thalassemia coinheritance.39–42
TREATMENT OPTIONS
In high-resource countries with newborn screening, the initiation of penicillin prophylaxis has dramatically altered the natural history of the disease, allowing the majority of patients to reach adulthood.43 Penicillin prophylaxis is usually discontinued at age 5 years; however, individuals who have undergone surgical splenectomy or have had pneumococcal sepsis on penicillin prophylaxis may remain on penicillin to age 18 or beyond.20
Another advance in sickle cell care is screening for stroke risk through transcranial Doppler ultrasound (TCD).44–47 This screening tool has reduced the incidence of childhood stroke from 10% by age 11 to 1%. TCDs typically cannot be performed after the age of 16 due to changes in the skull. Individuals found to have abnormal (elevated) TCD velocities are placed on chronic transfusion therapy for primary stroke prevention. They may remain on monthly chronic transfusions, with the goal of suppressing the percentage of HbS to 30% to 50% indefinitely. A clinical trial (STOPII) designed to determine if pediatric sickle cell disease patients on chronic transfusion therapy for primary stroke prevention could be safely taken off transfusion therapy was discontinued early due to an excess of strokes and conversion to abnormal TCD velocities in the untransfused arm.44 Individuals who have experienced an ischemic stroke have a 70% risk of another stroke, and must remain on chronic transfusion therapy indefinitely. Chronic transfusion reduces their stroke risk to 13%.
The only widely used pharmacologic therapy for sickle cell disease is hydroxyurea.12,48–50 A significant portion of the benefit of hydroxyurea stems from its induction of HbF.51 HbF does not sickle, and it interrupts the polymerization of HbS in the cell, if present in high enough concentrations.50 The level of HbF needed to achieve clinical improvement is not known, but in vitro assays suggest 20% HbF is needed to prevent sickling.52,53 However, endogenous and hydroxyurea-induced HbF is not distributed evenly through the red cells, so sickling is possible regardless of the level of HbF induced.54,55 Hydroxyurea likely has other disease-modifying effects as well, including reduction of white blood cell count and reticulocyte count and reduction of red cell adhesion to the endothelium.56–58 Clinical criteria for initiation of hydroxyurea in adult sickle cell disease patients are shown in Table 4.20 Hydroxyurea is given daily and is dosed to maximum tolerated dose for the individual by following the absolute neutrophil count (ANC). The goal ANC is between 2000 and 4000/µL. At times, absolute reticulocyte count (ARC) can be dose-limiting; goal ARC is greater than 70,000/µL.59 Platelet counts may be reduced as well, especially in HbSC patients.60,61
| Table 4. Indications for Hydroxyurea in Adult Patients with Sickle Cell Disease | |
Indication | Strength of Recommendation |
SCA with ≥ 3 pain crises per year | Strong |
SCA with pain that interferes with ADL and QoL | Strong |
History of severe or recurrent ACS | Strong |
Chronic kidney disease on epoetin | Weak |
HbSβ+ and HbSC with pain that interferes with ADL and QoL; consult sickle cell disease expert | Moderate |
| ACS = acute chest syndrome; ADL = activities of daily living; QoL = quality of life; SCA = sickle cell anemia. | |
The only curative therapy for sickle cell disease is hematopoietic stem cell transplant.62 Transplant use is limited by availability of matched sibling donors,62 and even at experienced centers transplant carries a small risk for mortality, graft rejection, and graft-versus-host disease. Furthermore, consensus on disease complications for which transplant is recommended is also lacking.63–65 Clinical trials of gene therapy for sickle cell disease and thalassemia are ongoing.66
COMPLICATIONS AND DISEASE-SPECIFIC THERAPIES
CASE PRESENTATION
A 26-year-old African-American man who works as a school bus driver presents to an academic center’s emergency department complaining of pain in his left leg, similar to prior pain events. He is described as having sickle cell trait, although no hemoglobin profile is available in his chart. He describes the pain as dull and aching, 10/10 in intensity. A complete blood count (CBC) is obtained; it reveals a hemoglobin of 14.5 g/dL, white blood cell (WBC) count of 5600/µL, and platelet count of 344,000/µL. His CBC is also notable for a mean corpuscular volume (MCV) of 72 fL, a mean corpuscular hemoglobin concentration (MCHC) of 37 g/dL, and a red blood cell distribution width (RDW) of 12. Slide review of a peripheral blood smear shows 2+ target cells (Figure).
The patient is given 6 mg of morphine, which provides some relief of his pain, and is discharged with a prescription for hydrocodone bitartrate/acetaminophen 5/325 mg. The diagnosis given is musculoskeletal pain, and he is instructed to follow-up with a primary care physician. His past medical history is significant for 4 or 5 visits to the emergency department per year in the past 4 years. Prior to 4 years ago, he rarely required medical attention.
• What laboratory and clinical features might lead you to question the diagnosis of sickle cell trait in this patient?
The patient’s hemoglobin is within normal range, which is consistent with sickle cell trait; however, he is microcytic, with a normal RDW. It is possible to be mildly microcytic in the early stages of iron deficiency, prior to the development of anemia, but the RDW would typically be elevated, demonstrating the presence of newer, smaller cells produced under conditions of iron deficiency.67 It is also possible that his microcytosis with a normal RDW could represent sickle cell trait with co-inheritance of β-thalassemia. Up to 30% of African Americans have β-thalassemia,2 and 1 in 10 have sickle cell trait.68 However, a high MCHC, indicating the presence of dense cells, and target cells noted on slide review are most consistent with HbSC.9 HbSC patients, especially males, can have hemoglobin levels in the normal range.4 The biggest inconsistency with the diagnosis of sickle cell trait is his history of frequent pain events. Individuals with sickle cell trait rarely present with pain crises, except under extreme conditions of dehydration or high altitude.68 Sickle cell trait is generally regarded as a benign condition, although a study of U.S. military recruits found a 30-fold higher risk of sudden death during basic training in persons with sickle cell trait.69 Additional sickle cell trait–related complications include hematuria, risk of splenic sequestration or infarct under extreme conditions and high altitude, and a rare and usually fatal renal malignancy, renal medullary carcinoma, which is vanishingly rare in individuals without sickle cell trait.70,71 Although the patient reported having sickle cell trait, this diagnosis should have been verified with a hemoglobin panel, given his atypical presentation.20
• What is the approach to managing pain episodes in sickle cell disease?
In sickle cell disease, vaso-occlusive pain events can be common, often beginning in early childhood.17 This disease complication accounts for 95% of all adult sickle cell disease hospitalizations.72 There is a great deal of variability in pain symptoms between individuals, and within individuals at various times in their lives:73 30% have no pain events, 50% have occasional events, and 20% have monthly or more frequent events that require hospitalization.74 The frequency and severity of pain events are modulated by HbF levels, β-thalassemia status, genotypes, therapies like hydroxyurea, or in rare cases, chronic transfusion therapy.23 Personal factors, such as psychosocial stressors, also contribute to the frequency of pain events.75 Pain event triggers include exposure to cold water, windy or cold weather, temperature changes, and extreme temperatures.76–79 Patient age also contributes to pain event frequency. Many patients see an increase in pain event frequency in their late 20s, and a marked decrease in their 40s.23,73 More than 3 pain events per year is associated with reduced life expectancy.23
Acute management of pain episodes involves nonsteroidal anti-inflammatory drugs, oral opioids, and when hospitalization is required, intravenous opioids, often delivered via patient-controlled analgesia (PCA) pumps.79 As sickle cell disease patients become teenagers and young adults, some experience an increased frequency of pain episodes, with fewer pain-free days, or a failure to return to baseline before the next pain crisis occurs.80,81 This is characteristic of emerging chronic pain.82 Chronic pain is a significant problem in adult patients with sickle cell disease, with up to 85% reporting pain on most days.72,80 The development of chronic pain may be reduced by early and aggressive treatment of acute pain events, as well as use of hydroxyurea to reduce the number of pain events. Many adult sickle cell patients with chronic pain are treated with daily opioids.20 Given the significant side effects of chronic opioid use—sedation, respiratory depression, itching, nausea, and impairment of function and quality of life—non-opioid therapies are under investigation.83 Many chronic pain patients have symptoms of neuropathic pain, and may benefit from neuropathic agents like gabapentin, both to reduce opioid use and to more effectively treat chronic neuropathic pain, which is known to respond poorly to opioids.84–86
• Is the patient’s peripheral blood smear consistent with a diagnosis of sickle cell trait?
Several target cells are visible, which is not typical of sickle cell trait, but may be seen in HbSC or thalassemia. The finding of an intracellular crystal is pathognomonic for HbSC or HbCC. HbC polymerizes in high oxygen conditions, opposite of HbS, which polymerizes in low oxygen conditions.9
CASE CONTINUED
The patient’s family history is significant for a sister who died at age 3 from sickle cell–related complications, and a sister with sickle cell trait who had a cholecystectomy for gallstones at age 22. His father died at age 38 due to unknown causes. The sickle cell trait status of his parents is unknown. His mother is alive, and has hypertension.
• Is the medical history of this patient’s family members consistent with sickle cell trait?
It is unlikely that sickle cell trait would result in early death in childhood, or in gallstones at age 22. Gallstones in early adulthood is a common presentation for HbSC patients not diagnosed by newborn screening.87 Any hemolytic condition can lead to the formation of hemoglobin-containing pigmented gallstones, biliary sludge, and obstruction of the gallbladder. In the presence of right-sided abdominal pain, a serum bilirubin level of more than 4 mg/dL should lead to measurement of direct bilirubin; if greater than 10% of total, imaging of the gallbladder should be obtained. In sickle cell disease, 30% of patients will have gallstones by 18 years of age. The low hemolysis/high viscosity phenotype patients are typically older at diagnosis. Co-inheritance of Gilbert syndrome and sickle cell disease is not uncommon, and can result in formation of gallstones at a young age; Gilbert syndrome alone typically results in gallstones in mid-life.88
CASE CONTINUED
Two months later, the patient presents again to the emergency department with the same complaint of leg pain, as well as abdominal pain. His hemoglobin is 12.5 g/dL, and his platelet count is 134,000/µL. His pain is not improved with 3 doses of morphine 6 mg intravenously, and he is admitted to the medicine service. A hemoglobin profile is obtained, revealing 52% HbS, 45% HbC, and 1.5% HbF, consistent with HbSC. In sickle cell trait, the hemoglobin profile is 60% HbA and 40% HbS (available α-globin prefers to pair with a normal β-globin, so the ratio of HbA to HbS is 60:40, not 50:50).
On the second hospital day, the patient’s hemoglobin drops to 7.2 g/dL and his platelet count decreases to 44,000/µL. His abdomen is distended and diffusely tender. The internist transfuses him with 2 units of packed red blood cells (PRBC), after which his hemoglobin increases to 11 g/dL, while his platelet count increases to 112,000/µL. Following the transfusion, his abdominal pain resolves, as does his anemia and thrombocytopenia.
• What caused this patient’s anemia and thrombocytopenia?
High on the differential diagnosis is a splenic sequestration. Acute splenic sequestration occurs when red cells are trapped in the splenic sinuses. Massive splenic enlargement may occur over several hours.89,90 Unrecognized splenic sequestration has a high mortality rate from severe anemia and splenic rupture.90 Splenic sequestration must be ruled out in a sickle cell patient with abdominal pain accompanied by dropping platelet and red cell counts, especially in milder subtypes that often have splenic function preserved into adolescence and adulthood. Sickle cell anemia patients usually become functionally asplenic in early childhood.89,91,92 The rise in hemoglobin, more than would be expected from 2 units of PRBC, plus the improvement in platelet count without a platelet transfusion observed in the case patient strongly supports the diagnosis of splenic sequestration.
Splenic sequestration can occur in any sickle cell patient whose spleen has not fibrosed. Splenic sequestration in adulthood is not uncommon in HbSC patients, who often have preserved splenic function into adulthood.93–95
Clinical signs of splenic sequestration include a rapid drop in hemoglobin, rise in reticulocyte count, a tender, enlarged spleen, and, in severe cases, hypovolemia.89,93 It is treated with prompt blood transfusion, but care must be taken not to overtransfuse the patient, as the spleen can trap several grams of hemoglobin, which may be released upon transfusion, potentially causing life-threatening hyperviscosity.89 Hemoglobin levels must be checked following transfusion in suspected splenic sequestration, and “mini transfusions” of 5 mL/kg are recommended in sickle cell disease patients who are hemodynamically stable.20
Hepatic sequestration may also occur, but it is much less common than splenic sequestration.96 Other conditions on the differential diagnosis include thrombotic thrombocytopenic purpura, which would be unlikely to respond to a transfusion. ACS can cause a drop in hemoglobin, and is treated with simple or exchange transfusions.97 ACS is less likely without respiratory symptoms or oxygen requirement, and usually is not associated with thrombocytopenia. Sepsis may also cause anemia and thrombocytopenia, but again would not likely respond to a simple transfusion. The patient’s response to transfusion is consistent with a sequestering event, not a destructive event as in the case of sepsis.
CASE CONTINUED
Imaging reveals a grossly enlarged spleen, which is having a mass effect on the left kidney. The patient is started on hydroxyurea therapy at 500 mg 3 times daily. Discharge instructions include following up with his primary care physician, continuing hydroxyurea therapy, and receiving yearly dilated eye exams to evaluate for proliferative sickle retinopathy.
• Are these discharge instructions complete?
Splenic sequestration has a 50% recurrence rate.98 In very young children, watchful waiting or chronic transfusion may be implemented to preserve the immunologic function of the spleen and reduce the risk of sepsis.89 Splenectomy after a single episode of sequestration in adults is a matter of debate, with experts advising both watchful waiting99 and splenectomy after recovery from the first sequestering event.100 The patient should have been informed of the risk for recurrence, and the signs and symptoms of splenic sequestration as well as the need for emergency medical attention should have been discussed. Splenic sequestration may be milder in adults than in children, but fatal sequestrations have been reported.95,101–103
Proliferative sickle cell retinopathy is a high viscosity/high hemoglobin complication that may occur more frequently in HbSC than HbSS, with an incidence of 33% in HbSC.42,104 Spontaneous regression of retinopathy occurs in approximately 32% of eyes, and laser or scatter photocoagulation is an effective intervention.105
• Would the patient need to be transfused prior to splenectomy?
Preoperative transfusion therapy is standard of care for HbSS patients undergoing general anesthesia. The TRAP study found that simple “top off” transfusion to a hemoglobin of 10 g/dL was as effective at preventing postoperative sickle cell–related complications as exchange transfusion to HbS of 30% or less, and had fewer transfusion-related complications like alloimmunization.106 There is little data regarding preoperative transfusions in HbSC disease. A retrospective study suggests that HbSC patients undergoing abdominal surgeries should be transfused.107 The higher hemoglobin level of the typical HbSC patient necessitates exchange transfusion to avoid hyperviscosity.
• Is hydroxyurea therapy indicated in this patient?
• Has it been dosed appropriately?
If the patient had the HbSS subtype, hydroxyurea would be clearly indicated, given his frequent pain events.20 HbSC patients may be placed on hydroxyurea on a case-by-case basis, but evidence for its efficacy in this sickle cell subtype is lacking.108 Large clinical trials like the Multi-Center Study of Hydroxyurea (MSH) that established the safety and efficacy of hydroxyurea in sickle cell anemia excluded HbSC and HbSβ+ patients.109 These mild to moderate subtypes produce less HbF at baseline, and typically have a minimal to modest rise in HbF on hydroxyurea.110 In sickle cell anemia, hydroxyurea is titrated to maximum tolerated dose, defined as an ANC of 2000 to 4000/µL and an ARC of 70,000/µL or higher.53 Because of their lower levels of chronic inflammation and lower reticulocyte counts due to higher hemoglobin levels, many HbSC and HbSβ+ patients have values in that range before initiating hydroxyurea therapy.9 Cytopenias, particularly of platelets in HbSC, occur at low doses of hydroxyurea.111
Of note, although the half-life of hydroxyurea would suggest that 3 times daily dosing is indicated, daily dosing has been found to have equal response and is preferred. Another concern is the monitoring of this myelosuppressive medication. This patient has repeatedly failed to obtain a primary care physician or a hematologist, and hydroxyurea requires laboratory monitoring at least every 2 months, especially in a HbSC patient with a very large spleen who is at significant risk for thrombocytopenia and neutropenia.9
CASE CONTINUED
A week after discharge from his admission for abdominal pain diagnosed as splenic sequestration, the patient presents again to the emergency department with abdominal pain which he reports is his typical sickle cell pain. Hemoglobin is 13.8 g/dL, platelet count is 388,000/µL, and alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels are both 10 times their prior value. Creatinine is 1.2 mg/dL (0.75 mg/dL on his prior admission), and total bilirubin is 3 mg/dL, with 0.3 mg/dL direct bilirubin. He undergoes an ultrasound exam of his gallbladder, which reveals sludge and a possible gallstone. There is no evidence of cholecystitis. General surgery performs a laparoscopic cholecystectomy.
• Was this cholecystectomy necessary?
In patients with sickle cell disease, symptomatic gallstones and gallbladder sludge should be observed; recurrent abdominal pain without a significant change in bilirubin may not be due to gallstones or sludge, and therefore may not be relieved by cholecystectomy.112,113 In sickle cell disease, 40% of patients with gallbladder sludge do not develop gallstones.87 The patient’s bilirubin level was at baseline, and there was no increase in the direct (conjugated) fraction. Watchful waiting would have been appropriate, with cholecystectomy being performed if he experienced recurrent symptoms associated with fatty foods accompanied by an elevation in direct bilirubin.
More concerning and deserving of investigation was his elevated liver enzymes. Patients with sickle cell disease may experience recurrent ischemia and reperfusion injuries in the liver, which is called right upper quadrant syndrome. On autopsy of 70 sickle cell patients, 91% had hepatomegaly and 34% had focal necrosis.114 AST is often elevated in sickle cell disease, as it is affected by hemolysis. In this patient, both AST and ALT are elevated, consistent with a hepatocellular disorder. His abdominal pain and ALT rise may be a sign of a hepatic crisis.115 Rapid resolution of ALT elevation in a matter of days suggests a vaso-occlusive, inflammatory event that is self- limiting. Prolonged AST elevation requires further investigation, with consideration of autoimmune hepatitis, viral hepatitis, or iron overload. Iron overload is unlikely in this patient given his lifetime history of only 1 transfusion. Hepatic iron overload typically occurs in sickle cell disease after a minimum of 10 transfusions.115
CASE CONTINUED
The patient is discharged on the day after the procedure, with instructions to continue his hydroxyurea.
• Should the patient resume hydroxyurea therapy?
Hydroxyurea is hepatically cleared and thus it should be held until his liver function tests normalize.106
CASE CONTINUED
Two months later, the patient presents to the emergency department with abdominal pain that moves to his left leg. A CBC is obtained, showing a hemoglobin of 11.8 g/dL and a platelet count of 144,000/µL. He is given 2 doses of morphine 6 mg intravenously, and reports that his leg pain is now a 4/10. He is discharged home with a prescription for hydrocodone/acetaminophen.
• Is the emergency department evaluation sufficient?
This patient remains at high risk for splenic sequestration,93 with a hemoglobin 2 g lower than it was 2 months ago and platelets less than half. This decline could be consistent with early splenic sequestration.20 Additionally, he had elevated liver function tests on a recent admission, as well as rising creatinine, without evidence of resolution. It is not appropriate to discharge him without checking a chemistry and liver panel, and abdominal imaging should be considered. The best plan would be to admit him for observation, given his risk for splenic sequestration, and consult surgery for an elective splenectomy if he has a second episode of splenic sequestration 2 months after the first.100 His abdominal pain that migrates to his left leg could be due to his massive splenomegaly compressing his left kidney, as noted on imaging during his recent admission for splenic sequestration
CASE CONTINUED
An hour after discharge from the emergency department, EMS is called to his home for intractable pain. He is found lying on the floor, and reports excruciating left leg pain. He is brought to the closest hospital, a community hospital that he has not visited previously. There, he is admitted for hydration and pain control and placed on hydromorphone 2 mg every 4 hours as needed for pain. His hemoglobin is 10.8 g/dL, and platelets are 121,000/µL. A chemistry panel is remarkable for a creatinine level of 1.5 mg/dL and a potassium level of 3.2 mEq/L. Liver function tests are not obtained. After 3 doses of hydromorphone, he falls asleep. He is not in a monitored bed, and intravenous fluids, while ordered, are not started. At 6:30 AM the day after admission, he cannot be aroused on a routine vital sign check; he has an SpO2 of 60%, a blood pressure of 80/60 mm Hg, and heart rate of 148 beats/min. A rapid response is called, and naloxone is administered along with oxygen by face mask and several fluid boluses. His systolic blood pressure increases to 100 mm Hg from a low of 70 mm Hg. His SpO2 increases to 92%, and he is arousable and alert, although he reports 10/10 leg pain. His abdomen is noted to be distended and tender.
• What may have contributed to his clinical condition?
The patient is opioid tolerant and has received equivalent doses of opioids in the past without excess sedation. He may have liver dysfunction making him unable to metabolize opioids effectively. His hemoglobin and platelets continue to decline, raising concern for splenic sequestration versus sepsis. Failure to place him on a monitor allowed his hypoxia to continue for an unknown amount of time, placing him at high risk for developing ACS. Lack of intravenous hydration while he was too sedated to drink likely exacerbated his sickling.
CASE CONTINUED
At 9:20 AM, a CBC is obtained and reveals a hemoglobin of 4.8 g/dL and a platelet count of 44,000/µL. Two units of stat O negative blood are administered, and preparations are made to administer an exchange transfusion. A liver panel is obtained 3 hours later, which reveals an AST level of 1200 U/L and an ALT level of 1050 U/L. His bilirubin is 10 mg/dL, and his lactate dehydrogenase level is 1800 U/L. His urine is dark and is positive for bilirubin and ketones. He is transferred to the intensive care unit. A chest X-ray shows pulmonary congestion. Hematology/oncology is consulted.
He receives a 7-unit red blood cell exchange, which reduces his HbS to 11%. He continues to be hypotensive, and requires norepinephrine to support his blood pressure. Antibiotic therapy is started. His creatinine concentration rises to 2.3 mg/dL, potassium is 7.8 mEq/L, and bicarbonate is 12 mEq/L. He is placed on hemodialysis.
Computed tomography of the chest and abdomen reveals lower posterior lung infiltrates and a grossly enlarged spleen. He requires intubation. He is given a diagnosis of ACS in addition to kidney failure, liver failure, and “sickle crisis.” He continues to require daily to twice daily transfusions to maintain a hemoglobin of 7 to 9 g/dL, and his abdominal distension increases. As his condition worsens, surgery is consulted to discuss a liver transplant. He is deemed to not be a surgical candidate, and he passes away 6 days after entering the hospital. The immediate cause of death is listed as vaso-occlusive crisis, with ACS and sickle crisis listed as contributors.
• Are the causes of death accurate and complete?
If vaso-occlusive crisis is used to indicate a pain event, it is not an accurate cause of death. Pain is one of the most distressing complications of sickle cell disease, and frequent pain events are associated with early mortality,4,80 but they are not in themselves fatal. ACS is the number one cause of death in sickle cell disease,4 and it likely contributed to this patient’s death. Sickle crisis is a vague term that should not be used in this context. Causes of death should include splenic sequestration and multisystem organ failure. Multisystem organ failure in sickle cell disease often responds to aggressive transfusion therapy, which this patient received.116–118
CONCLUSION
Sickle cell disease is a complex chronic disease that impacts almost every organ system in the body. Clinicians may be inclined to attribute most pain in a patient with sickle cell disease to a simple vaso-occlusive crisis, treat them for this, and not investigate further. As the case presented here demonstrates, failure to identify the actual life-threatening process occurring in a patient with sickle cell disease presenting with pain can result in preventable early mortality. Clinicians must approach a sickle cell patient reporting pain in a thoughtful manner, and consider a complete differential diagnosis, including both sickle cell disease complications and those unrelated to sickle cell disease. Knowledge of the disease courses of the different sickle cell genotypes is essential, and must go beyond a superficial hierarchy of severity, but rather include an understanding of the complications each genotype is most prone to, and at what ages. Complete laboratory assessment, including a comprehensive metabolic panel, should be performed on all admitted patients, not just a complete blood count. Treating pain with high-dose opioids, while appropriate in an uncomplicated pain crisis, can lead to ACS or even respiratory failure in a patient with uninvestigated liver and kidney dysfunction. The most important lesson to remember is that even the sickle cell disease patient who has been given the unfortunate and pejorative label of “frequent flyer” by some providers has the potential for rapid deterioration into multisystem organ failure and death.
- Weatherall D, Hofman K, Rodgers G, Ruffin J, Hrynkow S. A case for developing North-South partnerships for research in sickle cell disease. Blood 2005;105:921–3.
- Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol 1998;11:1–51.
- Allison AC. The distribution of the sickle-cell trait in East Africa and elsewhere, and its apparent relationship to the incidence of subtertian malaria. Trans R Soc Trop Med Hyg 1954;48:312–8.
- Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639–44.
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010;376:2018–31.
- Serjeant GR, Serjeant BE. Sickle cell disease. 3rd ed. New York: Oxford University Press; 2001.
- Moo-Penn W, Bechtel K, Jue D, et al. The presence of hemoglobin S and C Harlem in an individual in the United States. Blood 1975;46:363–7.
- Monplaisir N, Merault G, Poyart C, et al. Hemoglobin S Antilles: a variant with lower solubility than hemoglobin S and producing sickle cell disease in heterozygotes. Proc Natl Acad Sci U S A 1986;83:9363–7.
- Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood Rev 2003;17:167–78.
- Nagel RL, Daar S, Romero JR, et al. HbS-oman heterozygote: a new dominant sickle syndrome. Blood 1998;92:4375–82.
- Masiello D, Heeney MM, Adewoye AH, et al. Hemoglobin SE disease: a concise review. Am J Hematol 2007;82:643–9.
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med 1997;337:762–9.
- Brittenham GM, Schechter AN, Noguchi CT. Hemoglobin S polymerization: primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood 1985;65:183–9.
- .Nagel RL, Bookchin RM, Johnson J, et al. Structural bases of the inhibitory effects of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S. Proc Natl Acad Sci U S A 1979;76:670–2.
- .Ballas SK, Dover GJ, Charache S. Effect of hydroxyurea on the rheological properties of sickle erythrocytes in vivo. Am J Hematol 1989;32:104–11.
- Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol 2002;9:101–6.
- Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nature Med 2002;8:1383–9.
- Nouraie M, Lee JS, Zhang Y, et al. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica 2013;98:464–72.
- Serjeant GR. The natural history of sickle cell disease. Cold Spring Harb Perspect Med 2013;3:a011783.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48.
- Serjeant GR. Natural history and determinants of clinical severity of sickle cell disease. Curr Opin Hematol 1995;2:103–8.
- Odenheimer DJ, Sarnaik SA, Whitten CF, et al. The relationship between fetal hemoglobin and disease severity in children with sickle cell anemia. Am J Med Genet 1987;27:525–35.
- Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med 1991;325:11–6.
- Falusi AG, Olatunji PO. Effects of alpha thalassaemia and haemoglobin F (HbF) level on the clinical severity of sickle-cell anaemia. Eur J Haematol 1994;52:13–5.
- Watson J. The significance of the paucity of sickle cells in newborn Negro infants. Am J Med Sci 1948;215:419–23.
- Marcus SJ, Ware RE. Physiologic decline in fetal hemoglobin parameters in infants with sickle cell disease: implications for pharmacological intervention. J Pediatr Hematol Oncol 1999;21:407–11.
- Schroeder WA, Powars DR, Kay LM, et al. Beta-cluster haplotypes, alpha-gene status, and hematological data from SS, SC, and S-beta-thalassemia patients in southern California. Hemoglobin 1989;13:325–53.
- Steinberg MH, Voskaridou E, Kutlar A, et al. Concordant fetal hemoglobin response to hydroxyurea in siblings with sickle cell disease. Am J Hematol 2003;72:121–6.
- Ngo D, Bae H, Steinberg MH, et al. Fetal hemoglobin in sickle cell anemia: genetic studies of the Arab-Indian haplotype. Blood Cells Mol Dis 2013;51:22–6.
- Galarneau G, Palmer CD, Sankaran VG, et al. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet 2010;42:1049–51.
- Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U S A 2008;105:11869–74.
- Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 2008;322:1839–42.
- Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A 2008;105:1620–5.
- Adams RJ, Kutlar A, McKie V, et al. Alpha thalassemia and stroke risk in sickle cell anemia. Am J Hematol 1994;45:279–82.
- Ballas SK. Effect of alpha-globin genotype on the pathophysiology of sickle cell disease. Pediatr Pathol Mol Med 2001;20:107–21.
- Gaston MH, Verter JI, Woods G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med 1986;314:1593–9.
- Ballas SK, Talacki CA, Rao VM, Steiner RM. The prevalence of avascular necrosis in sickle cell anemia: correlation with alpha-thalassemia. Hemoglobin 1989;13:649–55.
- .Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev 2007;21:37–47.
- Murphy JR, Wengard M, Brereton W. Rheological studies of Hb SS blood: influence of hematocrit, hypertonicity, separation of cells, deoxygenation, and mixture with normal cells. J Lab Clin Med 1976;87:475–86.
- Fabry ME, Kaul DK, Raventos-Suarez C, Chang H, Nagel RL. SC erythrocytes have an abnormally high intracellular hemoglobin concentration. Pathophysiological consequences. J Clin Invest 1982;70:1315–9.
- Stuart J, Johnson CS. Rheology of the sickle cell disorders. Baillieres Clin Haematol 1987;1:747–75.
- Lionnet F, Hammoudi N, Stojanovic KS, et al. Hemoglobin sickle cell disease complications: a clinical study of 179 cases. Haematologica 2012;97:1136-41.
- Adamkiewicz TV, Sarnaik S, Buchanan GR, et al. Invasive pneumococcal infections in children with sickle cell disease in the era of penicillin prophylaxis, antibiotic resistance, and 23-valent pneumococcal polysaccharide vaccination. J Pediatr 2003;143:438–44.
- Abboud MR, Yim E, Musallam KM, Adams RJ, Investigators SIS. Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from STOP II. Blood 2011;118:894–8.
- Adams RJ. Lessons from the Stroke Prevention Trial in Sickle Cell Anemia (STOP) study. J Child Neurol 2000;15:344–9.
- Adams RJ, Brambilla DJ, Granger S, et al. Stroke and conversion to high risk in children screened with transcranial Doppler ultrasound during the STOP study. Blood 2004;103:3689–94.
- Lee MT, Piomelli S, Granger S, et al. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow-up and final results. Blood 2006;108:847–52.
- Charache S, Dover GJ, Moore RD, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood 1992;79:2555–65.
- Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995;332:1317–22.
- Charache S. Mechanism of action of hydroxyurea in the management of sickle cell anemia in adults. Semin Hematol 1997;34:15–21.
- Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol 2010;85:403–8.
- Noguchi CT, Rodgers GP, Serjeant G, Schechter AN. Levels of fetal hemoglobin necessary for treatment of sickle cell disease. N Engl J Med 1988;318:96–9.
- Powars DR, Weiss JN, Chan LS, Schroeder WA. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood 1984;63:921–6.
- Maier-Redelsperger M, de Montalembert M, Flahault A, et al. Fetal hemoglobin and F-cell responses to long-term hydroxyurea treatment in young sickle cell patients. The French Study Group on Sickle Cell Disease. Blood 1998;91:4472–9.
- Steinberg MH, Chui DH, Dover GJ, et al. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood 2014;123:481–5.
- Adragna NC, Fonseca P, Lauf PK. Hydroxyurea affects cell morphology, cation transport, and red blood cell adhesion in cultured vascular endothelial cells. Blood 1994;83:553–60.
- Bridges KR, Barabino GD, Brugnara C, et al. A multiparameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood 1996;88:4701–10.
- Jiang J, Jordan SJ, Barr DP, et al. In vivo production of nitric oxide in rats after administration of hydroxyurea. Mol Pharmacol 1997;52:1081–6.
- Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 2010;115:5300–11.
- Yates AM, Dedeken L, Smeltzer MP, et al. Hydroxyurea treatment of children with hemoglobin SC disease. Pediatr Blood Cancer 2013;60:323–5.
- Barbosa CG, Aleluia AC, Pacheco AP, et al. Genetic modulation of HbF in Brazilians with HbSC disease and sickle cell anemia. Am J Hematol 2013;88:923–4.
- Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med 2009;361:2309–17.
- King A, Shenoy S. Evidence-based focused review of the status of hematopoietic stem cell transplantation as treatment of sickle cell disease and thalassemia. Blood 2014;123:3089–94.
- Oringanje C, Nemecek E, Oniyangi O. Hematopoietic stem cell transplantation for people with sickle cell disease. Cochrane Database Syst Rev 2013;5:CD007001.
- Freed J, Talano J, Small T, et al. Allogeneic cellular and autologous stem cell therapy for sickle cell disease: ‘whom, when and how’. Bone Marrow Transplant 2012;47:1489–98.
- Urbinati F, Hargrove PW, Geiger S, et al. Potentially therapeutic levels of anti-sickling globin gene expression following lentivirus-mediated gene transfer in sickle cell disease bone marrow CD34 cells. Exp Hematol 2015;43:346–51.
- Brugnara C, Mohandas N. Red cell indices in classification and treatment of anemias: from M.M. Wintrobes’s original 1934 classification to the third millennium. Curr Opin Hematol 2013;20:222–30.
- Key NS, Derebail VK. Sickle-cell trait: novel clinical significance. Hematology Am Soc Hematol Educ Program 2010;2010:418–22.
- Kark JA, Posey DM, Schumacher HR, Ruehle CJ. Sickle-cell trait as a risk factor for sudden death in physical training. N Engl J Med 1987;317:781–7.
- Goldsmith JC, Bonham VL, Joiner CH, et al. Framing the research agenda for sickle cell trait: building on the current understanding of clinical events and their potential implications. Am J Hematol 2012;87:340–6.
- Grant AM, Parker CS, Jordan LB, et al. Public health implications of sickle cell trait: a report of the CDC meeting. Am J Prev Med 2011;41:S435–9.
- Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance. Am J Hematol 2005;79:17–25.
- Serjeant GR, Ceulaer CD, Lethbridge R, et al. The painful crisis of homozygous sickle cell disease: clinical features. Br J Haematol 1994;87:586–91.
- Vichinsky EP, Johnson R, Lubin BH. Multidisciplinary approach to pain management in sickle cell disease. Am J Pediatr Hematol Oncol 1982;4:328–33.
- Gil KM, Carson JW, Porter LS, et al. Daily mood and stress predict pain, health care use, and work activity in African American adults with sickle-cell disease. Health Psychol 2004;23:267–74.
- Amjad H, Bannerman RM, Judisch JM. Letter: Sickling pain and season. Br Med J 1974;2:54.
- Ibrahim AS. Relationship between meteorological changes and occurrence of painful sickle cell crises in Kuwait. Trans R Soc Trop Med Hyg 1980;74:159–61.
- Jones S, Duncan ER, Thomas N, et al. Windy weather and low humidity are associated with an increased number of hospital admissions for acute pain and sickle cell disease in an urban environment with a maritime temperate climate. Br J Haematol 2005;131:530–3.
- Resar LM, Oski FA. Cold water exposure and vaso-occlusive crises in sickle cell anemia. J Pediatr 1991;118:407–9.
- Darbari DS, Ballas SK, Clauw DJ. Thinking beyond sickling to better understand pain in sickle cell disease. Eur J Haematol 2014;93:89–95.
- Darbari DS, Onyekwere O, Nouraie M, et al. Markers of severe vaso-occlusive painful episode frequency in children and adolescents with sickle cell anemia. J Pediatr 2011;160:286–90.
- Hollins M, Stonerock GL, Kisaalita NR, et al. Detecting the emergence of chronic pain in sickle cell disease. J Pain Symptom Manage 2012;43:1082–93.
- Ballas SK, Darbari DS. Neuropathy, neuropathic pain, and sickle cell disease. Am J Hematol 2013;88:927–9.
- Brandow AM, Farley RA, Panepinto JA. Early insights into the neurobiology of pain in sickle cell disease: A systematic review of the literature. Pediatr Blood Cancer 2015 May 13. doi: 10.1002/pbc.25574. [Epub ahead of print].
- Brandow AM, Farley RA, Panepinto JA. Neuropathic pain in patients with sickle cell disease. Pediatr Blood Cancer 2014;61:512–7.
- Brandow AM, Farley RA, Dasgupta M, et al. The use of neuropathic pain drugs in children with sickle cell disease is associated with older age, female sex, and longer length of hospital stay. J Pediatr Hematol Oncol 2015;37:10–5.
- Walker TM, Hambleton IR, Serjeant GR. Gallstones in sickle cell disease: observations from The Jamaican Cohort study. J Pediatr 2000;136:80–5.
- Penner E, Mayr WR, Djawan S, et al. [The genetics of Gilbert syndrome]. Schweiz Med Wochenschr 1976;106:860–2. [German]
- Powell RW, Levine GL, Yang YM, Mankad VN. Acute splenic sequestration crisis in sickle cell disease: early detection and treatment. J Pediatr Surg 1992;27:215–8.
- Al Salem AH, Qaisaruddin S, Nasserullah Z, et al. Splenectomy and acute splenic sequestration crises in sickle cell disease. Pediatr Surg Int 1996;11:26–8.
- Pearson HA, Spencer RP, Cornelius EA. Functional asplenia in sickle-cell anemia. N Engl J Med 1969;281:923–6.
- Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011;377:1663–72.
- Brousse V, Buffet P, Rees D. The spleen and sickle cell disease: the sick(led) spleen. Br J Haematol 2014;166:165–76.
- Orringer EP, Fowler VG Jr, Owens CM, et al. Case report: splenic infarction and acute splenic sequestration in adults with hemoglobin SC disease. Am J Med Sci 1991;302:374–9.
- Michel JB, Hernandez JA, Buchanan GR. A fatal case of acute splenic sequestration in a 53–year-old woman with sickle-hemoglobin C disease. Am J Med 1992;92:97–100.
- Hatton CS, Bunch C, Weatherall DJ. Hepatic sequestration in sickle cell anaemia. Br Med J (Clin Res Ed) 1985;290:744–5.
- Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood 1994;84:643–9.
- Gill FM, Sleeper LA, Weiner SJ, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood 1995;86:776–83.
- Owusu-Ofori S, Hirst C. Splenectomy versus conservative management for acute sequestration crises in people with sickle cell disease. Cochrane Database Syst Rev 2013;5:CD003425.
- 00.Al-Salem AH. Splenic complications of sickle cell anemia and the role of splenectomy. ISRN Hematol 2011;2011:864257.
- Sabarense AP, Lima GO, Silva LM, Viana MB. Characterization of mortality in children with sickle cell disease diagnosed through the Newborn Screening Program. J Pediatr (Rio J) 2015;91:242–7.
- Aslam AF, Aslam AK, Dipillo F. Fatal splenic sequestration crisis with multiorgan failure in an adult woman with sickle cell-beta+ thalassemia. Am J Med Sci 2005;329:141–3.
- Berry RA, Odumakinde EA, Lewis JP. Massive splenic infarction in doubly abnormal heterozygous sickling disorders. A new complication of acute splenic sequestration syndrome. The West J Med 1991;155:531–2.
- Bonanomi MT, Lavezzo MM. Sickle cell retinopathy: diagnosis and treatment. Arq Bras de Oftalmol 2013;76:320–7.
- Chen RW, Flynn HW Jr, Lee WH, et al. Vitreoretinal management and surgical outcomes in proliferative sickle retinopathy: a case series. Am J Ophthalmol 2014;157:870–5 e1.
- Howard J, Malfroy M, Llewelyn C, et al. The Transfusion Alternatives Preoperatively in Sickle Cell Disease (TAPS) study: a randomised, controlled, multicentre clinical trial. Lancet 2013;381:930–8.
- Neumayr L, Koshy M, Haberkern C, et al. Surgery in patients with hemoglobin SC disease. Preoperative Transfusion in Sickle Cell Disease Study Group. Am J Hematol 1998;57:101–8.
- 08. Savage WJ, Buchanan GR, Yawn BP, et al. Evidence gaps in the management of sickle cell disease: A summary of needed research. Am J Hematol 2015;90:273–5.
- Charache S, Barton FB, Moore RD, et al. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive “switching” agent. The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine (Baltimore) 1996;75:300–26.
- Steinberg MH, Nagel RL, Brugnara C. Cellular effects of hydroxyurea in Hb SC disease. Br J Haematol 1997;98:838–44.
- Wang W, Brugnara C, Snyder C, et al. The effects of hydroxycarbamide and magnesium on haemoglobin SC disease: results of the multi-centre CHAMPS trial. Br J Haematol 2011;152:771–6.
- Jungst C, Kullak-Ublick GA, Jungst D. Gallstone disease: Microlithiasis and sludge. Best Pract Res Clin Gastroenterol 2006;20:1053–62.
- Walker TM, Serjeant GR. Biliary sludge in sickle cell disease. J Pediatr 1996;129:443–5.
- Bauer TW, Moore GW, Hutchins GM. The liver in sickle cell disease. A clinicopathologic study of 70 patients. Am J Med 1980;69:833–7.
- Ebert EC, Nagar M, Hagspiel KD. Gastrointestinal and hepatic complications of sickle cell disease. Clin Gastroenterol Hepatol 2010;8:483–9.
- Hiran S. Multiorgan dysfunction syndrome in sickle cell disease. J Assoc Physicians India 2005;53:19–22.
- Shao SH, Orringer EP. Case report: splenic sequestration and multiorgan failure as the presenting manifestation of hemoglobin SC disease. Am J Med Sci 1996;311:139–41.
- Hassell KL, Eckman JR, Lane PA. Acute multiorgan failure syndrome: a potentially catastrophic complication of severe sickle cell pain episodes. Am J Med 1994;96:155–62.
- Weatherall D, Hofman K, Rodgers G, Ruffin J, Hrynkow S. A case for developing North-South partnerships for research in sickle cell disease. Blood 2005;105:921–3.
- Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol 1998;11:1–51.
- Allison AC. The distribution of the sickle-cell trait in East Africa and elsewhere, and its apparent relationship to the incidence of subtertian malaria. Trans R Soc Trop Med Hyg 1954;48:312–8.
- Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994;330:1639–44.
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010;376:2018–31.
- Serjeant GR, Serjeant BE. Sickle cell disease. 3rd ed. New York: Oxford University Press; 2001.
- Moo-Penn W, Bechtel K, Jue D, et al. The presence of hemoglobin S and C Harlem in an individual in the United States. Blood 1975;46:363–7.
- Monplaisir N, Merault G, Poyart C, et al. Hemoglobin S Antilles: a variant with lower solubility than hemoglobin S and producing sickle cell disease in heterozygotes. Proc Natl Acad Sci U S A 1986;83:9363–7.
- Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood Rev 2003;17:167–78.
- Nagel RL, Daar S, Romero JR, et al. HbS-oman heterozygote: a new dominant sickle syndrome. Blood 1998;92:4375–82.
- Masiello D, Heeney MM, Adewoye AH, et al. Hemoglobin SE disease: a concise review. Am J Hematol 2007;82:643–9.
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med 1997;337:762–9.
- Brittenham GM, Schechter AN, Noguchi CT. Hemoglobin S polymerization: primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood 1985;65:183–9.
- .Nagel RL, Bookchin RM, Johnson J, et al. Structural bases of the inhibitory effects of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S. Proc Natl Acad Sci U S A 1979;76:670–2.
- .Ballas SK, Dover GJ, Charache S. Effect of hydroxyurea on the rheological properties of sickle erythrocytes in vivo. Am J Hematol 1989;32:104–11.
- Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol 2002;9:101–6.
- Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nature Med 2002;8:1383–9.
- Nouraie M, Lee JS, Zhang Y, et al. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica 2013;98:464–72.
- Serjeant GR. The natural history of sickle cell disease. Cold Spring Harb Perspect Med 2013;3:a011783.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033–48.
- Serjeant GR. Natural history and determinants of clinical severity of sickle cell disease. Curr Opin Hematol 1995;2:103–8.
- Odenheimer DJ, Sarnaik SA, Whitten CF, et al. The relationship between fetal hemoglobin and disease severity in children with sickle cell anemia. Am J Med Genet 1987;27:525–35.
- Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med 1991;325:11–6.
- Falusi AG, Olatunji PO. Effects of alpha thalassaemia and haemoglobin F (HbF) level on the clinical severity of sickle-cell anaemia. Eur J Haematol 1994;52:13–5.
- Watson J. The significance of the paucity of sickle cells in newborn Negro infants. Am J Med Sci 1948;215:419–23.
- Marcus SJ, Ware RE. Physiologic decline in fetal hemoglobin parameters in infants with sickle cell disease: implications for pharmacological intervention. J Pediatr Hematol Oncol 1999;21:407–11.
- Schroeder WA, Powars DR, Kay LM, et al. Beta-cluster haplotypes, alpha-gene status, and hematological data from SS, SC, and S-beta-thalassemia patients in southern California. Hemoglobin 1989;13:325–53.
- Steinberg MH, Voskaridou E, Kutlar A, et al. Concordant fetal hemoglobin response to hydroxyurea in siblings with sickle cell disease. Am J Hematol 2003;72:121–6.
- Ngo D, Bae H, Steinberg MH, et al. Fetal hemoglobin in sickle cell anemia: genetic studies of the Arab-Indian haplotype. Blood Cells Mol Dis 2013;51:22–6.
- Galarneau G, Palmer CD, Sankaran VG, et al. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet 2010;42:1049–51.
- Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U S A 2008;105:11869–74.
- Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 2008;322:1839–42.
- Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A 2008;105:1620–5.
- Adams RJ, Kutlar A, McKie V, et al. Alpha thalassemia and stroke risk in sickle cell anemia. Am J Hematol 1994;45:279–82.
- Ballas SK. Effect of alpha-globin genotype on the pathophysiology of sickle cell disease. Pediatr Pathol Mol Med 2001;20:107–21.
- Gaston MH, Verter JI, Woods G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med 1986;314:1593–9.
- Ballas SK, Talacki CA, Rao VM, Steiner RM. The prevalence of avascular necrosis in sickle cell anemia: correlation with alpha-thalassemia. Hemoglobin 1989;13:649–55.
- .Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev 2007;21:37–47.
- Murphy JR, Wengard M, Brereton W. Rheological studies of Hb SS blood: influence of hematocrit, hypertonicity, separation of cells, deoxygenation, and mixture with normal cells. J Lab Clin Med 1976;87:475–86.
- Fabry ME, Kaul DK, Raventos-Suarez C, Chang H, Nagel RL. SC erythrocytes have an abnormally high intracellular hemoglobin concentration. Pathophysiological consequences. J Clin Invest 1982;70:1315–9.
- Stuart J, Johnson CS. Rheology of the sickle cell disorders. Baillieres Clin Haematol 1987;1:747–75.
- Lionnet F, Hammoudi N, Stojanovic KS, et al. Hemoglobin sickle cell disease complications: a clinical study of 179 cases. Haematologica 2012;97:1136-41.
- Adamkiewicz TV, Sarnaik S, Buchanan GR, et al. Invasive pneumococcal infections in children with sickle cell disease in the era of penicillin prophylaxis, antibiotic resistance, and 23-valent pneumococcal polysaccharide vaccination. J Pediatr 2003;143:438–44.
- Abboud MR, Yim E, Musallam KM, Adams RJ, Investigators SIS. Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from STOP II. Blood 2011;118:894–8.
- Adams RJ. Lessons from the Stroke Prevention Trial in Sickle Cell Anemia (STOP) study. J Child Neurol 2000;15:344–9.
- Adams RJ, Brambilla DJ, Granger S, et al. Stroke and conversion to high risk in children screened with transcranial Doppler ultrasound during the STOP study. Blood 2004;103:3689–94.
- Lee MT, Piomelli S, Granger S, et al. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow-up and final results. Blood 2006;108:847–52.
- Charache S, Dover GJ, Moore RD, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood 1992;79:2555–65.
- Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995;332:1317–22.
- Charache S. Mechanism of action of hydroxyurea in the management of sickle cell anemia in adults. Semin Hematol 1997;34:15–21.
- Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol 2010;85:403–8.
- Noguchi CT, Rodgers GP, Serjeant G, Schechter AN. Levels of fetal hemoglobin necessary for treatment of sickle cell disease. N Engl J Med 1988;318:96–9.
- Powars DR, Weiss JN, Chan LS, Schroeder WA. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood 1984;63:921–6.
- Maier-Redelsperger M, de Montalembert M, Flahault A, et al. Fetal hemoglobin and F-cell responses to long-term hydroxyurea treatment in young sickle cell patients. The French Study Group on Sickle Cell Disease. Blood 1998;91:4472–9.
- Steinberg MH, Chui DH, Dover GJ, et al. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood 2014;123:481–5.
- Adragna NC, Fonseca P, Lauf PK. Hydroxyurea affects cell morphology, cation transport, and red blood cell adhesion in cultured vascular endothelial cells. Blood 1994;83:553–60.
- Bridges KR, Barabino GD, Brugnara C, et al. A multiparameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood 1996;88:4701–10.
- Jiang J, Jordan SJ, Barr DP, et al. In vivo production of nitric oxide in rats after administration of hydroxyurea. Mol Pharmacol 1997;52:1081–6.
- Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 2010;115:5300–11.
- Yates AM, Dedeken L, Smeltzer MP, et al. Hydroxyurea treatment of children with hemoglobin SC disease. Pediatr Blood Cancer 2013;60:323–5.
- Barbosa CG, Aleluia AC, Pacheco AP, et al. Genetic modulation of HbF in Brazilians with HbSC disease and sickle cell anemia. Am J Hematol 2013;88:923–4.
- Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med 2009;361:2309–17.
- King A, Shenoy S. Evidence-based focused review of the status of hematopoietic stem cell transplantation as treatment of sickle cell disease and thalassemia. Blood 2014;123:3089–94.
- Oringanje C, Nemecek E, Oniyangi O. Hematopoietic stem cell transplantation for people with sickle cell disease. Cochrane Database Syst Rev 2013;5:CD007001.
- Freed J, Talano J, Small T, et al. Allogeneic cellular and autologous stem cell therapy for sickle cell disease: ‘whom, when and how’. Bone Marrow Transplant 2012;47:1489–98.
- Urbinati F, Hargrove PW, Geiger S, et al. Potentially therapeutic levels of anti-sickling globin gene expression following lentivirus-mediated gene transfer in sickle cell disease bone marrow CD34 cells. Exp Hematol 2015;43:346–51.
- Brugnara C, Mohandas N. Red cell indices in classification and treatment of anemias: from M.M. Wintrobes’s original 1934 classification to the third millennium. Curr Opin Hematol 2013;20:222–30.
- Key NS, Derebail VK. Sickle-cell trait: novel clinical significance. Hematology Am Soc Hematol Educ Program 2010;2010:418–22.
- Kark JA, Posey DM, Schumacher HR, Ruehle CJ. Sickle-cell trait as a risk factor for sudden death in physical training. N Engl J Med 1987;317:781–7.
- Goldsmith JC, Bonham VL, Joiner CH, et al. Framing the research agenda for sickle cell trait: building on the current understanding of clinical events and their potential implications. Am J Hematol 2012;87:340–6.
- Grant AM, Parker CS, Jordan LB, et al. Public health implications of sickle cell trait: a report of the CDC meeting. Am J Prev Med 2011;41:S435–9.
- Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance. Am J Hematol 2005;79:17–25.
- Serjeant GR, Ceulaer CD, Lethbridge R, et al. The painful crisis of homozygous sickle cell disease: clinical features. Br J Haematol 1994;87:586–91.
- Vichinsky EP, Johnson R, Lubin BH. Multidisciplinary approach to pain management in sickle cell disease. Am J Pediatr Hematol Oncol 1982;4:328–33.
- Gil KM, Carson JW, Porter LS, et al. Daily mood and stress predict pain, health care use, and work activity in African American adults with sickle-cell disease. Health Psychol 2004;23:267–74.
- Amjad H, Bannerman RM, Judisch JM. Letter: Sickling pain and season. Br Med J 1974;2:54.
- Ibrahim AS. Relationship between meteorological changes and occurrence of painful sickle cell crises in Kuwait. Trans R Soc Trop Med Hyg 1980;74:159–61.
- Jones S, Duncan ER, Thomas N, et al. Windy weather and low humidity are associated with an increased number of hospital admissions for acute pain and sickle cell disease in an urban environment with a maritime temperate climate. Br J Haematol 2005;131:530–3.
- Resar LM, Oski FA. Cold water exposure and vaso-occlusive crises in sickle cell anemia. J Pediatr 1991;118:407–9.
- Darbari DS, Ballas SK, Clauw DJ. Thinking beyond sickling to better understand pain in sickle cell disease. Eur J Haematol 2014;93:89–95.
- Darbari DS, Onyekwere O, Nouraie M, et al. Markers of severe vaso-occlusive painful episode frequency in children and adolescents with sickle cell anemia. J Pediatr 2011;160:286–90.
- Hollins M, Stonerock GL, Kisaalita NR, et al. Detecting the emergence of chronic pain in sickle cell disease. J Pain Symptom Manage 2012;43:1082–93.
- Ballas SK, Darbari DS. Neuropathy, neuropathic pain, and sickle cell disease. Am J Hematol 2013;88:927–9.
- Brandow AM, Farley RA, Panepinto JA. Early insights into the neurobiology of pain in sickle cell disease: A systematic review of the literature. Pediatr Blood Cancer 2015 May 13. doi: 10.1002/pbc.25574. [Epub ahead of print].
- Brandow AM, Farley RA, Panepinto JA. Neuropathic pain in patients with sickle cell disease. Pediatr Blood Cancer 2014;61:512–7.
- Brandow AM, Farley RA, Dasgupta M, et al. The use of neuropathic pain drugs in children with sickle cell disease is associated with older age, female sex, and longer length of hospital stay. J Pediatr Hematol Oncol 2015;37:10–5.
- Walker TM, Hambleton IR, Serjeant GR. Gallstones in sickle cell disease: observations from The Jamaican Cohort study. J Pediatr 2000;136:80–5.
- Penner E, Mayr WR, Djawan S, et al. [The genetics of Gilbert syndrome]. Schweiz Med Wochenschr 1976;106:860–2. [German]
- Powell RW, Levine GL, Yang YM, Mankad VN. Acute splenic sequestration crisis in sickle cell disease: early detection and treatment. J Pediatr Surg 1992;27:215–8.
- Al Salem AH, Qaisaruddin S, Nasserullah Z, et al. Splenectomy and acute splenic sequestration crises in sickle cell disease. Pediatr Surg Int 1996;11:26–8.
- Pearson HA, Spencer RP, Cornelius EA. Functional asplenia in sickle-cell anemia. N Engl J Med 1969;281:923–6.
- Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet 2011;377:1663–72.
- Brousse V, Buffet P, Rees D. The spleen and sickle cell disease: the sick(led) spleen. Br J Haematol 2014;166:165–76.
- Orringer EP, Fowler VG Jr, Owens CM, et al. Case report: splenic infarction and acute splenic sequestration in adults with hemoglobin SC disease. Am J Med Sci 1991;302:374–9.
- Michel JB, Hernandez JA, Buchanan GR. A fatal case of acute splenic sequestration in a 53–year-old woman with sickle-hemoglobin C disease. Am J Med 1992;92:97–100.
- Hatton CS, Bunch C, Weatherall DJ. Hepatic sequestration in sickle cell anaemia. Br Med J (Clin Res Ed) 1985;290:744–5.
- Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood 1994;84:643–9.
- Gill FM, Sleeper LA, Weiner SJ, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood 1995;86:776–83.
- Owusu-Ofori S, Hirst C. Splenectomy versus conservative management for acute sequestration crises in people with sickle cell disease. Cochrane Database Syst Rev 2013;5:CD003425.
- 00.Al-Salem AH. Splenic complications of sickle cell anemia and the role of splenectomy. ISRN Hematol 2011;2011:864257.
- Sabarense AP, Lima GO, Silva LM, Viana MB. Characterization of mortality in children with sickle cell disease diagnosed through the Newborn Screening Program. J Pediatr (Rio J) 2015;91:242–7.
- Aslam AF, Aslam AK, Dipillo F. Fatal splenic sequestration crisis with multiorgan failure in an adult woman with sickle cell-beta+ thalassemia. Am J Med Sci 2005;329:141–3.
- Berry RA, Odumakinde EA, Lewis JP. Massive splenic infarction in doubly abnormal heterozygous sickling disorders. A new complication of acute splenic sequestration syndrome. The West J Med 1991;155:531–2.
- Bonanomi MT, Lavezzo MM. Sickle cell retinopathy: diagnosis and treatment. Arq Bras de Oftalmol 2013;76:320–7.
- Chen RW, Flynn HW Jr, Lee WH, et al. Vitreoretinal management and surgical outcomes in proliferative sickle retinopathy: a case series. Am J Ophthalmol 2014;157:870–5 e1.
- Howard J, Malfroy M, Llewelyn C, et al. The Transfusion Alternatives Preoperatively in Sickle Cell Disease (TAPS) study: a randomised, controlled, multicentre clinical trial. Lancet 2013;381:930–8.
- Neumayr L, Koshy M, Haberkern C, et al. Surgery in patients with hemoglobin SC disease. Preoperative Transfusion in Sickle Cell Disease Study Group. Am J Hematol 1998;57:101–8.
- 08. Savage WJ, Buchanan GR, Yawn BP, et al. Evidence gaps in the management of sickle cell disease: A summary of needed research. Am J Hematol 2015;90:273–5.
- Charache S, Barton FB, Moore RD, et al. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive “switching” agent. The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine (Baltimore) 1996;75:300–26.
- Steinberg MH, Nagel RL, Brugnara C. Cellular effects of hydroxyurea in Hb SC disease. Br J Haematol 1997;98:838–44.
- Wang W, Brugnara C, Snyder C, et al. The effects of hydroxycarbamide and magnesium on haemoglobin SC disease: results of the multi-centre CHAMPS trial. Br J Haematol 2011;152:771–6.
- Jungst C, Kullak-Ublick GA, Jungst D. Gallstone disease: Microlithiasis and sludge. Best Pract Res Clin Gastroenterol 2006;20:1053–62.
- Walker TM, Serjeant GR. Biliary sludge in sickle cell disease. J Pediatr 1996;129:443–5.
- Bauer TW, Moore GW, Hutchins GM. The liver in sickle cell disease. A clinicopathologic study of 70 patients. Am J Med 1980;69:833–7.
- Ebert EC, Nagar M, Hagspiel KD. Gastrointestinal and hepatic complications of sickle cell disease. Clin Gastroenterol Hepatol 2010;8:483–9.
- Hiran S. Multiorgan dysfunction syndrome in sickle cell disease. J Assoc Physicians India 2005;53:19–22.
- Shao SH, Orringer EP. Case report: splenic sequestration and multiorgan failure as the presenting manifestation of hemoglobin SC disease. Am J Med Sci 1996;311:139–41.
- Hassell KL, Eckman JR, Lane PA. Acute multiorgan failure syndrome: a potentially catastrophic complication of severe sickle cell pain episodes. Am J Med 1994;96:155–62.
Adjuvant Systemic Therapy for Early-Stage Breast Cancer
Over the past 20 years, substantial progress has been achieved in our understanding of breast cancer and in breast cancer treatment, with mortality from breast cancer declining by more than 25% over this time. This progress has been characterized by a greater understanding of the molecular biology of breast cancer, rational drug design, development of agents with specific cellular targets and pathways, development of better prognostic and predictive multigene assays, and marked improvements in supportive care.
To read the full article in PDF:
Over the past 20 years, substantial progress has been achieved in our understanding of breast cancer and in breast cancer treatment, with mortality from breast cancer declining by more than 25% over this time. This progress has been characterized by a greater understanding of the molecular biology of breast cancer, rational drug design, development of agents with specific cellular targets and pathways, development of better prognostic and predictive multigene assays, and marked improvements in supportive care.
To read the full article in PDF:
Over the past 20 years, substantial progress has been achieved in our understanding of breast cancer and in breast cancer treatment, with mortality from breast cancer declining by more than 25% over this time. This progress has been characterized by a greater understanding of the molecular biology of breast cancer, rational drug design, development of agents with specific cellular targets and pathways, development of better prognostic and predictive multigene assays, and marked improvements in supportive care.
To read the full article in PDF:
Management of Locally Advanced Rectal Adenocarcinoma
Colorectal cancers are among the most common cancers worldwide, and there is a high mortality rate for advanced-stage disease. Approximately 132,000 new cases of colorectal cancer will be diagnosed in the United States in 2015, and approximately 40,000 of these cases will be primary rectal cancers. The incidence and mortality rates have been steadily declining over the past two decades, largely through advances in screening and improvements in treatment. However, rectal cancer remains a significant cause of morbidity and mortality in the United States and worldwide.
To read the full article in PDF:
Colorectal cancers are among the most common cancers worldwide, and there is a high mortality rate for advanced-stage disease. Approximately 132,000 new cases of colorectal cancer will be diagnosed in the United States in 2015, and approximately 40,000 of these cases will be primary rectal cancers. The incidence and mortality rates have been steadily declining over the past two decades, largely through advances in screening and improvements in treatment. However, rectal cancer remains a significant cause of morbidity and mortality in the United States and worldwide.
To read the full article in PDF:
Colorectal cancers are among the most common cancers worldwide, and there is a high mortality rate for advanced-stage disease. Approximately 132,000 new cases of colorectal cancer will be diagnosed in the United States in 2015, and approximately 40,000 of these cases will be primary rectal cancers. The incidence and mortality rates have been steadily declining over the past two decades, largely through advances in screening and improvements in treatment. However, rectal cancer remains a significant cause of morbidity and mortality in the United States and worldwide.
To read the full article in PDF:
Chronic Myeloid Leukemia
The first connection between cancer and a patient’s genome was documented by Peter Nowell and David Hungerford when they identified a unique chromosome in the metaphase spread of 7 patients diagnosed with chronic myeloid leukemia (CML). In 1973, renowned cytopathologist Janet Rowley determined that this chromosome is part of a chromosomal translocation between chromosome 9 and chromosome 22. Further delineation of this translocation showed that the gene ABL1, normally located on chromosome 9, is translocated to the Philadelphia (Ph+) chromosome in patients with CML. ABL1 was found to be located downstream of a specific genetic region in each patient, and this region became known as the BCR, or “breakpoint cluster region.” The BCR-ABL1 translocation found in patients with CML creates a constitutively active tyrosine kinase necessary for cellular transformation.
To read the full article in PDF:
The first connection between cancer and a patient’s genome was documented by Peter Nowell and David Hungerford when they identified a unique chromosome in the metaphase spread of 7 patients diagnosed with chronic myeloid leukemia (CML). In 1973, renowned cytopathologist Janet Rowley determined that this chromosome is part of a chromosomal translocation between chromosome 9 and chromosome 22. Further delineation of this translocation showed that the gene ABL1, normally located on chromosome 9, is translocated to the Philadelphia (Ph+) chromosome in patients with CML. ABL1 was found to be located downstream of a specific genetic region in each patient, and this region became known as the BCR, or “breakpoint cluster region.” The BCR-ABL1 translocation found in patients with CML creates a constitutively active tyrosine kinase necessary for cellular transformation.
To read the full article in PDF:
The first connection between cancer and a patient’s genome was documented by Peter Nowell and David Hungerford when they identified a unique chromosome in the metaphase spread of 7 patients diagnosed with chronic myeloid leukemia (CML). In 1973, renowned cytopathologist Janet Rowley determined that this chromosome is part of a chromosomal translocation between chromosome 9 and chromosome 22. Further delineation of this translocation showed that the gene ABL1, normally located on chromosome 9, is translocated to the Philadelphia (Ph+) chromosome in patients with CML. ABL1 was found to be located downstream of a specific genetic region in each patient, and this region became known as the BCR, or “breakpoint cluster region.” The BCR-ABL1 translocation found in patients with CML creates a constitutively active tyrosine kinase necessary for cellular transformation.
To read the full article in PDF:
Epithelial Ovarian Cancer: Management of Advanced Disease
Edited by: Arthur T. Skarin, MD, FACP, FCCP
Epithelial ovarian cancer is the fifth leading cause of cancer death among women in the United States. Most women with ovarian cancer present at an advanced stage (International Federation of Gynecology and Obstetrics stage III), for which the standard treatment remains cytoreductive surgery followed by platinum- and taxane-based combination chemotherapy. Although this treatment frequently is curative for patients with early-stage disease, more than 60% of women with advanced disease will develop recurrent disease with progressively shorter disease-free intervals. However, there are many clinical trials in progress that are aimed at refining current therapy and evaluating different approaches to postoperative therapy, with the goal of improving prognosis and quality of life.
To read the full article in PDF:
Edited by: Arthur T. Skarin, MD, FACP, FCCP
Epithelial ovarian cancer is the fifth leading cause of cancer death among women in the United States. Most women with ovarian cancer present at an advanced stage (International Federation of Gynecology and Obstetrics stage III), for which the standard treatment remains cytoreductive surgery followed by platinum- and taxane-based combination chemotherapy. Although this treatment frequently is curative for patients with early-stage disease, more than 60% of women with advanced disease will develop recurrent disease with progressively shorter disease-free intervals. However, there are many clinical trials in progress that are aimed at refining current therapy and evaluating different approaches to postoperative therapy, with the goal of improving prognosis and quality of life.
To read the full article in PDF:
Edited by: Arthur T. Skarin, MD, FACP, FCCP
Epithelial ovarian cancer is the fifth leading cause of cancer death among women in the United States. Most women with ovarian cancer present at an advanced stage (International Federation of Gynecology and Obstetrics stage III), for which the standard treatment remains cytoreductive surgery followed by platinum- and taxane-based combination chemotherapy. Although this treatment frequently is curative for patients with early-stage disease, more than 60% of women with advanced disease will develop recurrent disease with progressively shorter disease-free intervals. However, there are many clinical trials in progress that are aimed at refining current therapy and evaluating different approaches to postoperative therapy, with the goal of improving prognosis and quality of life.
To read the full article in PDF:
Epithelial Ovarian Cancer: Evaluation, Staging, Surgery, and Stage I and II Disease Management
Edited by: Arthur T. Skarin, MD, FACP, FCCP
Ovarian cancer is the second most common gynecologic cancer among women in the United States. It is also the fifth leading cause of cancer mortality in women and the leading cause of death among women with gynecologic malignancies. The American Cancer Society statistics released in 2015 estimate that 21,290 new cases of ovarian cancer will occur during the year, with approximately 14,180 deaths. Globally, there were 238,719 new cases of ovarian cancer diagnosed in 2012, representing 3.6% of all cancers in women, and nearly 151,905 deaths. The highest incidence of ovarian cancer occurs in northern, central, and eastern Europe, followed by western Europe and North America, with the lowest incidence in parts of Africa and Asia. The majority of women presenting with ovarian cancer will present at an advanced stage, and the 5-year survival in this group is less than 30%.
To read the full article in PDF:
Edited by: Arthur T. Skarin, MD, FACP, FCCP
Ovarian cancer is the second most common gynecologic cancer among women in the United States. It is also the fifth leading cause of cancer mortality in women and the leading cause of death among women with gynecologic malignancies. The American Cancer Society statistics released in 2015 estimate that 21,290 new cases of ovarian cancer will occur during the year, with approximately 14,180 deaths. Globally, there were 238,719 new cases of ovarian cancer diagnosed in 2012, representing 3.6% of all cancers in women, and nearly 151,905 deaths. The highest incidence of ovarian cancer occurs in northern, central, and eastern Europe, followed by western Europe and North America, with the lowest incidence in parts of Africa and Asia. The majority of women presenting with ovarian cancer will present at an advanced stage, and the 5-year survival in this group is less than 30%.
To read the full article in PDF:
Edited by: Arthur T. Skarin, MD, FACP, FCCP
Ovarian cancer is the second most common gynecologic cancer among women in the United States. It is also the fifth leading cause of cancer mortality in women and the leading cause of death among women with gynecologic malignancies. The American Cancer Society statistics released in 2015 estimate that 21,290 new cases of ovarian cancer will occur during the year, with approximately 14,180 deaths. Globally, there were 238,719 new cases of ovarian cancer diagnosed in 2012, representing 3.6% of all cancers in women, and nearly 151,905 deaths. The highest incidence of ovarian cancer occurs in northern, central, and eastern Europe, followed by western Europe and North America, with the lowest incidence in parts of Africa and Asia. The majority of women presenting with ovarian cancer will present at an advanced stage, and the 5-year survival in this group is less than 30%.
To read the full article in PDF: