User login
Molecular Markers and Targeted Therapies in the Management of Non-Small Cell Lung Cancer
INTRODUCTION
Lung cancer is the second most common type of cancer in the United States, with 222,500 estimated new cases in 2017, according to the American Cancer Society.1 However, it is by far the number one cause of death due to cancer, with an estimated 155,870 lung cancer–related deaths occurring in 2017, which is higher than the number of deaths due to breast cancer, prostate cancer, and colorectal cancer combined.1,2 Despite slightly decreasing incidence and mortality over the past decade, largely due to smoking cessation, the 5-year survival rate of lung cancer remains dismal at approximately 18%.2–4
Non-small cell lung cancer (NSCLC) accounts for 80% to 85% of all lung cancer cases.4 Traditionally, it is further divided based on histology: adenocarcinoma, squamous cell carcinoma, large cell carcinoma, and not otherwise specified.5 Chemotherapy had been the cornerstone of treatment for stage IV NSCLC. It is not target-specific and is most effective against rapidly growing cells. Common adverse effects include alopecia, nausea/vomiting, myelosuppression, cardiotoxicity, neuropathy, and nephrotoxicity. However, this paradigm has shifted following the discovery of mutations of the epidermal growth factor receptor (EGFR) gene as an oncogenic driver that confers sensitivity to small molecule tyrosine kinase inhibitors (TKIs) targeting EGFR.6 The EGFR inhibitors are given orally and have a spectrum of toxicities (eg, such as rash, diarrhea, and elevated transaminases) different from that of systemic chemotherapy, which is often administered intravenously. Following the discovery of EGFR mutations, rearrangements of the anaplastic lymphoma kinase (ALK) gene7 and ROS1 gene8 were identified as targetable driver mutations in NSCLC. The frequency of both rearrangements is lower than that of EGFR mutations. Additionally, BRAF V600E mutation has been identified in NSCLC.9–12 This activation mutation is commonly seen in melanoma. Agents that have already been approved for the treatment of melanoma with the BRAF V600E mutation are being tested in NSCLC patients with this mutation.13–16
Given the effectiveness and tolerability of targeted therapy, identifying this distinct molecular subset of NSCLC patients is critical in treatment. Currently, molecular testing is mandatory in all stage IV patients with non-squamous cell carcinoma, as a preponderance of patients with driver mutations have this histology subtype.5,17–19 For patients with squamous cell carcinoma, molecular testing should be considered if the biopsy specimen is small, there is mixed histology, or the patient is a nonsmoker.5,20 Several techniques are commonly utilized in detecting these genetic alterations. EGFR mutation can be detected by polymerase chain reaction (PCR), ALK or ROS1 rearrangement can be detected by fluorescence in-situ hybridization (FISH), and immunohistochemistry (IHC) can also be used to detect ALK rearrangement. The current guideline is to use comprehensive genomic profiling to capture all the potential molecular targets simultaneously instead of running stepwise tests just for EGFR, ALK, and ROS1.5 BRAF V600E mutation,13–16 MET exon 14 skipping mutation,21–24 RET rearrangements,25–27 and HER2 mutations28–30 are among the emergent genetic alterations with various responses to targeted therapy.31 Some of these targeted agents have been approved for other types of malignancy, and others are still in the development phase.
Several initiatives worldwide have reported better outcomes of patients with driver mutations treated with targeted therapy. For instance, the Lung Cancer Mutation Consortium in the United States demonstrated that the median survival of patients without driver mutations, with drivers mutations but not treated with targeted therapy, and with driver mutations and treated with targeted therapy was 2.08 years, 2.38 years, and 3.49 years, respectively.32 The French Cooperative Thoracic Intergroup-French National Cancer Institute demonstrated that the median survival for patients with driver mutations versus those without driver mutations was 16.5 months versus 11.8 months.33 The Spanish Lung Cancer Group demonstrated that the overall survival (OS) for patients with EGFR mutations treated with erlotinib was 27 months.34 The mutations in lung cancer, their frequencies, and the downstream signaling pathways are depicted in the Figure.35
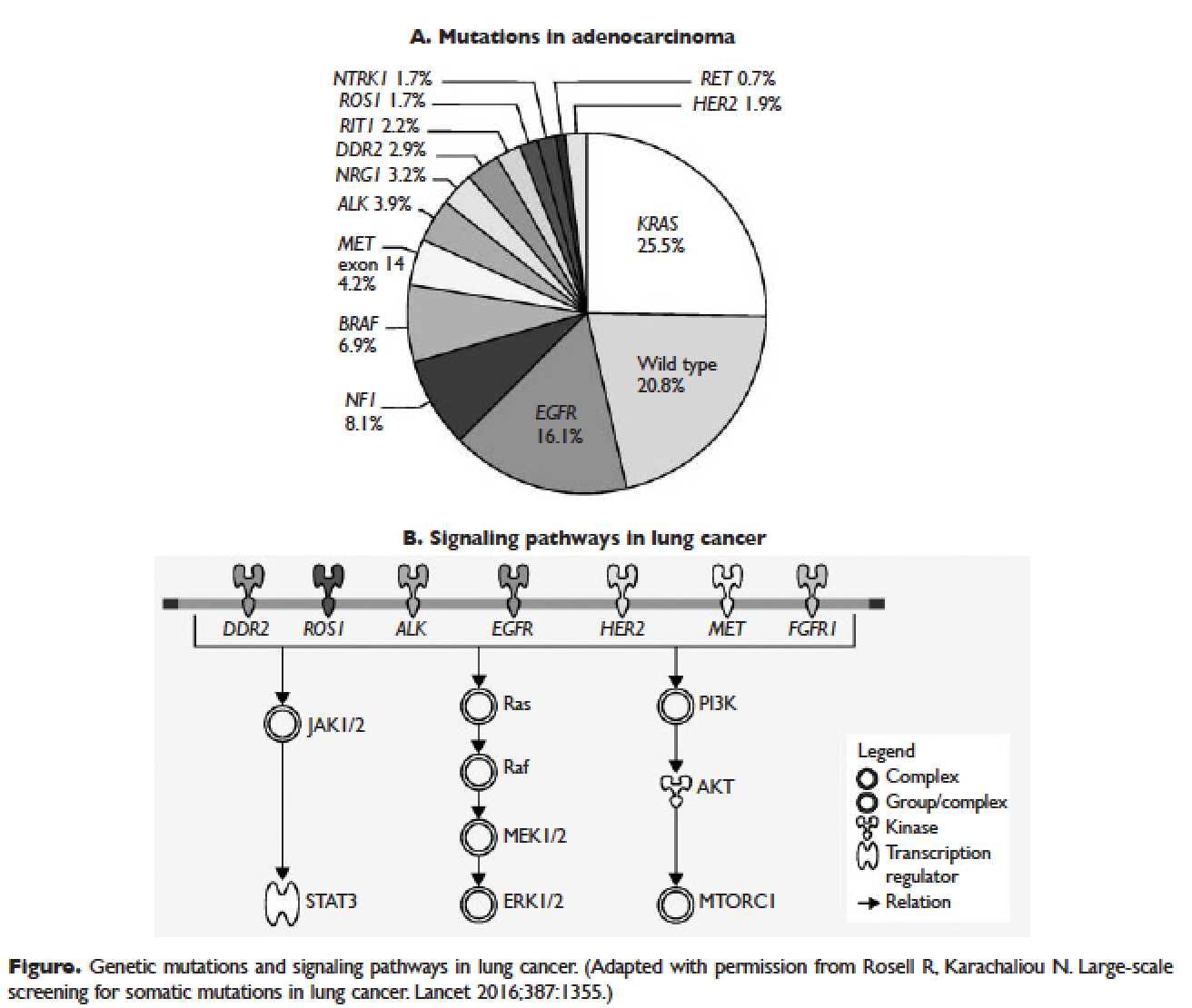
In this article, we discuss targeted therapy for patients with EGFR mutations, ALK rearrangements, ROS1 rearrangements, and BRAF V600E mutation. We also discuss the management of patients with EGFR mutations who develop a secondary mutation after TKI therapy. Almost all of the targeted agents discussed herein have been approved by the US Food and Drug Administration (FDA), so they are considered standard of care. All available phase 3 trials pertinent to these targeted therapies are included in the discussion.
EGFR MUTATIONS
CASE PRESENTATION 1
A 54-year-old Caucasian man who is a former smoker with a 10 pack-year history and past medical history of hypertension and dyslipidemia presents with progressive dyspnea for several weeks. A chest x-ray shows moderate pleural effusion on the left side with possible mass-like opacity on the left upper lung field. An ultrasound-guided thoracentesis is performed and cytology is positive for adenocarcinoma of likely pulmonary origin. Staging workup including positron emission tomography (PET)/computed tomography (CT) and magnetic resonance imaging of the brain with and without contrast is done. PET/CT shows a 5.5-cm mass in the left upper lobe of the lung with high fluorodeoxyglucose (FDG) uptake, several 1- to 2-cm mediastinal lymph nodes with moderate FDG uptake, and small pleural effusion on both sides with moderate FDG uptake. MRI-brain is negative for malignancy. The patient subsequently undergoes a CT-guided biopsy of the lung mass, which shows moderately differentiated adenocarcinoma. Comprehensive molecular profiling reveals EGFR L858R mutation only. The patient now presents for the initial consultation. Of note, his Eastern Cooperative Oncology Group performance status is 1.
What is the next step in the management of this patient?
FIRST-LINE TKI FOR SENSITIZING EGFR MUTATIONS
The 2 most common EGFR mutations are deletions in exon 19 and substitution of arginine for leucine in exon 21 (L858R), found in approximately 45% and 40% of patients with EGFR mutations, respectively.36 Both mutations are sensitive to EGFR TKIs. The benefit may be greater in patients with exon 19 deletions as compared to exon 21 L858R substitution,37,38 but this has not been demonstrated consistently in clinical trials.39-43 In the United States, EGFR mutations are found in approximately 10% of patients with NSCLC, while the incidence can be as high as 50% in Asia.44 Even though the cobas EGFR mutation test is the companion diagnostic approved by the US FDA, a positive test result from any laboratory with the Clinical Laboratory Improvement Amendments (CLIA) certificate should prompt the use of an EGFR TKI as the initial treatment.
Three EGFR TKIs that have been approved as first-line therapy in the United States are available: erlotinib, afatinib, and gefitinib.5 Both erlotinib and gefitinib are considered first-generation TKIs. They have higher binding affinity for the 2 common EGFR mutations than wild-type EGFR. In addition, they reversibly bind to the intracellular tyrosine kinase domain, resulting in inhibition of autophosphorylation of the tyrosine residues. Afatinib, a second-generation and irreversible TKI, targets EGFR (HER1) as well as HER2 and HER4.45
The superior efficacy of the EGFR TKIs over platinum doublet chemotherapy in treatment-naïve patients with EGFR mutations has been demonstrated in 7 randomized trials to date (Table).46 Erlotinib was the TKI arm for the OPTIMAL,41 EURTAC,42 and ENSURE trials;38 afatinib was the TKI arm for LUX-LUNG 337 and 6;43 gefitinib was the TKI arm for NEJ00239,47 and WJTOG3405.40 A meta-analysis of these 7 trials by Lee et al showed that progression-free survival (PFS) was significantly prolonged by EGFR TKIs (hazard ratio [HR] 0.37 [95% confidence interval {CI} 0.32 to 0.42]).46 For instance, in the EURTAC trial, median PFS was 9.7 months for patients treated with erlotinib as compared to 5.2 months for patients treated with platinum/gemcitabine or platinum/docetaxel.42 In this meta-analysis, prespecified subgroups included age, sex, ethnicity, smoking status, performance status, tumor histology, and EGFR mutation subtype. The superior outcome with TKIs was observed in all subgroups. Furthermore, patients with exon 19 deletions, nonsmokers, and women had even better outcomes.46
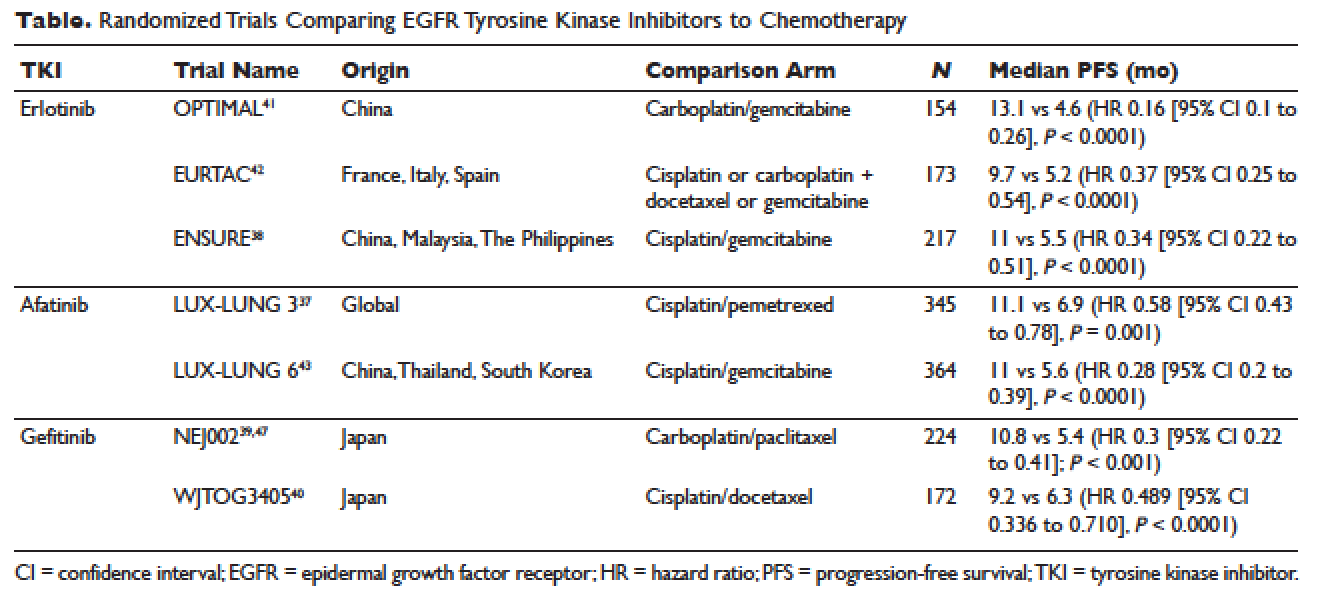
Erlotinib is the most commonly used TKI in the United States largely because gefitinib was off the market for some time until it was re-approved by the FDA in 2015. Interestingly, this “re-approval” was not based on either 1 of the 2 prospective trials (NEJ00239,47 and WJTOG340540), but rather was based on an exploratory analysis of the IPASS trial48,49 as well as a prospective phase 4, single-arm trial in Europe (IFUM).50 The superior efficacy of gefitinib over carboplatin/paclitaxel among patients with EGFR mutations in the IPASS trial was confirmed by blind independent central review, with longer PFS (HR 0.54 [95% CI 0.38 to 0.79] P = 0.0012) and higher objective response rate (ORR; odds ratio 3 [95% CI 1.63 to 5.54], P = 0.0004).49
CASE 1 CONTINUED
Based on the EGFR L858R mutation status, the patient is started on erlotinib. He is quite happy that he does not need intravenous chemotherapy but wants to know what toxicities he might potentially have with erlotinib.
What are the common adverse effects (AEs) of EGFR TKIs? How are AEs of TKIs managed?
Safety Profile
The important toxicities associated with EGFR TKIs are rash, gastrointestinal toxicity, hepatic toxicity, and pulmonary toxicity. Rash is an AE specific to all agents blocking the EGFR pathway, including small molecules and monoclonal antibodies such as cetuximab. The epidermis has a high level of expression of EGFR, which contributes to this toxicity.51 Rash usually presents as dry skin or acneiform eruption. Prophylactic treatment with oral tetracyclines and topical corticosteroids is generally recommended upon initiation of TKI therapy. Diarrhea is the most prevalent gastrointestinal toxicity. All patients starting treatment should be given prescriptions to manage diarrhea such as loperamide and be advised to call when it occurs. Hepatic toxicity is often manifested as elevated transaminases or bilirubin. Interstitial lung disease (ILD) is a rare but potentially fatal pulmonary toxicity.
Rash of any grade was reported in 49.2% of patients treated with erlotinib in clinical trials, while grade 3 rash occurred in 6% of patients and no grade 4 was reported. Diarrhea of any grade was reported in 20.3% of patients, grade 3 diarrhea occurred in 1.8%, and no grade 4 was reported. Grade 2 and 3 alanine aminotransferase (ALT) elevations were seen in 2% and 1% of patients, respectively. Grade 2 and 3 bilirubin elevations were seen in 4% and less than 1% of patients, respectively. The incidence of serious ILD-like events was less than 1%.52
Afatinib is associated with higher incidences of rash and diarrhea. Specifically, diarrhea and rash of all grades were reported in 96% and 90% of patients treated with afatinib, respectively. Paronychia of all grades occurred in 58% of patients. Elevated ALT of all grades was seen in 11% of patients. Approximately 1.5% of patients treated with afatinib across clinical trials had ILD or ILD-like AEs.53
Gefitinib, the most commonly used TKI outside United States, has a toxicity profile similar to erlotinib, except for hepatic toxicity. For instance, rash of all grades occurred in 47% of patients, diarrhea of all grades occurred in 29% of patients, and ILD or ILD-like AEs occurred in 1.3% of patients across clinical trials. In comparison, elevated ALT and aspartate aminotransferase (AST) of all grades was seen in 38% and 40% of patients, respectively.54 Therefore, close monitoring of liver function is clinically warranted. In particular, patients need to be advised to avoid concomitant use of herbal supplements, a common practice in Asian countries.
CASE 1 CONTINUED
The patient does well while on erlotinib at 150 mg orally once daily for about 8 months, until he develops increasing abdominal pain. A CT scan of the abdomen and pelvis with contrast shows a new 8-cm right adrenal mass. Additionally, a repeat CT scan of the chest with contrast shows a stable lung mass but enlarging mediastinal lymphadenopathy.
How would you manage the patient at this point?
MANAGEMENT OF T790M MUTATION AFTER PROGRESSION ON FIRST-LINE EGFR TKIS
As mentioned above, the median PFS of patients with EGFR mutations treated with 1 of the 3 TKIs is around 9 to 13 months.46 Of the various resistance mechanisms that have been described, the T790M mutation is found in approximately 60% of patients who progress after treatment with first-line TKIs.55,56 Other mechanisms, such as HER2 amplification, MET amplification, or rarely small cell transformation, have been reported.56 The first- and second-generation EGFR TKIs function by binding to the ATP-binding domain of mutated EGFR, leading to inhibition of the downstream signaling pathways (Figure, part B) and ultimately cell death.35 The T790M mutation hinders the interaction between the ATP-binding domain of EGFR kinase and TKIs, resulting in treatment resistance and disease progression.57,58
Osimertinib is a third-generation irreversible EGFR TKI with activity against both sensitizing EGFR and resistant T790M mutations. It has low affinity for wide-type EGFR as well as insulin receptor and insulin-like growth factor receptor.59 Osimertinib has been fully approved for NSCLC patients with EGFR mutations who have progressed on first-line EGFR TKIs with the development of T790M mutation. An international phase 3 trial (AURA3) randomly assigned 419 patients in a 2:1 ratio to either osimertinib or platinum/pemetrexed. Eligible patients all had the documented EGFR mutations and disease progression after first-line EGFR TKIs. Central confirmation of the T790M mutation was required. Median PFS by investigator assessment, the trial’s primary end point, was 10.1 months for osimertinib versus 4.4 months for chemotherapy (HR 0.3 [95% CI 0.23 to 0.41]; P < 0.001). ORR was 71% for osimertinib versus 31% for chemotherapy (HR 5.39 [95% CI 3.47 to 8.48], P < 0.001). A total of 144 patients with stable and asymptomatic brain metastases were also eligible. Median PFS for this subset of patients treated with osimertinib and chemotherapy was 8.5 months and 4.2 months, respectively (HR 0.32 [95% CI 0.21 to 0.49]). In the AURA3 trial, osimertinib was better tolerated than chemotherapy, with 23% of patients treated with osimertinib experiencing grade 3 or 4 AEs as compared to 47% of chemotherapy-treated patients. The most common AEs of any grade were diarrhea (41%), rash (34%), dry skin (23%), and paronychia (22%).60
For the case patient, a reasonable approach would be to obtain a tissue biopsy of the adrenal mass and more importantly to check for the T790M mutation. Similar to the companion diagnostic for EGFR mutations, the cobas EGFR mutation test v2 is the FDA-approved test for T790M. However, if this resistance mutation is detected by any CLIA-certified laboratories, osimertinib should be the recommended treatment option. If tissue biopsy is not feasible, plasma-based testing should be considered. A blood-based companion diagnostic also is FDA approved.
ALK REARRANGEMENTS
CASE 2 PRESENTATION
A 42-year-old Korean woman who is a non-smoker with no significant past medical history presents with fatigue, unintentional weight loss of 20 lb in the past 4 months, and vague abdominal pain. A CT can of the abdomen and pelvis without contrast shows multiple foci in the liver and an indeterminate nodule in the right lung base. She subsequently undergoes PET/CT, which confirms multiple liver nodules/masses ranging from 1 to 3 cm with moderate FDG uptake. In addition, there is a 3.5-cm pleura-based lung mass on the right side with moderate FDG uptake. MRI-brain with and without contrast is negative for malignancy. A CT-guided biopsy of 1 of the liver masses is ordered and pathology returns positive for poorly differentiated adenocarcinoma consistent with lung primary. Molecular analysis reveals an echinoderm microtubule-associated protein-like 4 (EML4)-ALK rearrangement. She is placed on crizotinib by an outside oncologist and after about 3 weeks of therapy is doing well. She is now in your clinic for a second opinion. She says that some of her friends told her about another medication called ceritinib and was wondering if she would need to switch her cancer treatment.
How would you respond to this patient’s inquiry?
FIRST-LINE TKIS FOR ALK REARRANGEMENTS
ALK rearrangements are found in 2% to 7% of NSCLC, with EML4-ALK being the most prevalent fusion variant.61 The inversion of chromosome 2p leads to the fusion of the EML4 gene and the ALK gene, which causes the constitutive activation of the fusion protein and ultimately increased transformation and tumorigenicity.7,61 Patients harboring ALK rearrangements tend to be non-smokers. Adenocarcinoma, especially signet ring cell subtype, is the predominant histology. Compared to EGFR mutations, patients with ALK mutations are significantly younger and more likely to be men.62 ALK rearrangements can be detected by either FISH or IHC, and most next-generation sequencing (NGS) panels have the ability to identify this driver mutation.
Crizotinib is the first approved ALK inhibitor for the treatment of NSCLC in this molecular subset of patients.63 PROFILE 1014 is a phase 3 randomized trial that compared crizotinib with chemotherapy containing platinum/pemetrexed for up to 6 cycles. Crossover to crizotinib was allowed for patients with disease progression on chemotherapy. The primary end point was PFS by independent radiologic review. The crizotinib arm demonstrated superior PFS (10.9 months versus 7 months; HR 0.45 [95% CI 0.35 to 0.6], P < 0.001) and ORR (74% versus 45%, P < 0.001). Median survival was not reached in either arm (HR 0.82 [95% CI 0.54 to 1.26], P = 0.36).64 Based on this international trial, crizotinib is considered standard of care in the United States for treatment-naïve patients with advanced NSCLC harboring ALK rearrangements. The current recommended dose is 250 mg orally twice daily. Common treatment-related AEs of all grades include vision disorder (62%), nausea (53%), diarrhea (43%), vomiting (40%), edema (28%), and constipation (27%).65 PROFILE 1007 compared crizotinib with pemetrexed or docetaxel in ALK-rearranged NSCLC patients with prior exposure to 1 platinum-based chemotherapy. The median PFS was 7.7 months for crizotinib as compared to 3 months for chemotherapy (HR 0.49 [95% CI 0.37 to 0.64], P < 0.001). The response rates were 65% and 20% for crizotinib and chemotherapy, respectively (P < 0.001).66 In other countries, crizotinib following 1 prior platinum-based regimen may be considered standard of care based on this trial.
Ceritinib is an oral second-generation ALK inhibitor that is 20 times more potent than crizotinib based on enzymatic assays.67 It also targets ROS1 and insulin-like growth factor 1 receptor but not c-MET. It was first approved by the FDA in April 2014 for metastatic ALK-rearranged NSCLC following crizotinib.68 In May 2017, the FDA granted approval of ceritinib for treatment-naïve patients. This decision was based on the results of the ASCEND-4 trial, a randomized phase 3 trial assessing the efficacy and safety of ceritinib over chemotherapy in the first-line setting. The trial assigned 376 patients to either ceritinib at 750 mg once daily or platinum/pemetrexed for 4 cycles followed by maintenance pemetrexed. Median PFS was 16.6 months for ceritinib versus 8.1 months for chemotherapy (HR 0.55 [95% CI 0.42 to 0.73]; P < 0.00001).69 Toxicities of ceritinib are not negligible, with gastrointestinal toxicity being the most prevalent. For instance, diarrhea, nausea, vomiting, abdominal pain, and constipation of all grades were seen in 86%, 80%, 60%, 54%, and 29% of patients, respectively. Furthermore, fatigue and decreased appetite occurred in 52% and 34% of patients, respectively. In terms of laboratory abnormalities, 84% of patients experienced decreased hemoglobin of all grades; 80% increased ALT; 75% increased AST; 58% increased creatinine; 49% increased glucose; 36% decreased phosphate; and 28% increased lipase. Due to these AEs, the incidence of dose reduction was about 58% and the median onset was around 7 weeks.70
Alectinib is another oral second-generation ALK inhibitor that was approved by the FDA in December 2015 for the treatment of NSCLC patients with ALK rearrangements who have progressed on or are intolerant to crizotinib.71 Its indication will soon be broadened to the first-line setting based on the ALEX trial.72 Alectinib is a potent and highly selective TKI of ALK73 with activity against known resistant mutations to crizotinib.74,75 It also inhibits RET but not ROS1 or c-MET.76 ALEX, a randomized phase 3 study, compared alectinib with crizotinib in treatment-naïve patients with NSCLC harboring ALK rearrangements. The trial enrolled 303 patients and the median follow-up was approximately 18 months. The alectinib arm (600 mg twice daily) demonstrated significantly higher PFS by investigator-assessment, the trial’s primary end point. The 12-month event-free survival was 68.4% (95% CI 61% to 75.9%) versus 48.7% (95% CI 40.4% to 56.9%) for alectinib and crizotinib, respectively (HR 0.47 [95% CI 0.34 to 0.65], P < 0.001). The median PFS was not reached in the alectinib arm (95% CI 17.7 months to not estimable) as compared to 11.1 months in the crizotinib arm (95% CI 9.1 to 13.1 months).72 Alectinib is generally well tolerated. Common AEs of all grades include fatigue (41%), constipation (34%), edema (30%), and myalgia (29%). As alectinib can cause anemia, lymphopenia, hepatic toxicity, increased creatine phosphokinase, hyperglycemia, electrolyte abnormalities, and increased creatinine, periodic monitoring of these laboratory values is important, although most of these abnormalities are grade 1 or 2.77
Brigatinib, another oral second-generation ALK inhibitor, was granted accelerated approval by the FDA in April 2017 for ALK-rearranged and crizotinib-resistant NSCLC based on the ALTA trial. This randomized phase 2 study of brigatinib showed an ORR by investigator assessment of 54% (97.5% CI 43% to 65%) in the 180 mg once daily arm with lead-in of 90 mg once daily for 7 days. Median PFS was 12.9 months (95% CI 11.1 months to not reached [NR]).78 Currently, a phase 3 study of brigatinib versus crizotinib in ALK inhibitor–naïve patients is recruiting participants (ALTA-1L). It will be interesting to see if brigatinib can achieve a front-line indication.
Starting the case patient on crizotinib is well within the treatment guidelines. One may consider ceritinib or alectinib in the first-line setting, but both TKIs can be reserved upon disease progression. We would recommend a repeat biopsy at that point to look for resistant mechanisms, as certain secondary ALK mutations may be rescued by certain next-generation ALK inhibitors. For instance, the F1174V mutation has been reported to confer resistance to ceritinib but sensitivity to alectinib, while the opposite is true for I1171T. The G1202R mutation is resistant to ceritinib, alectinib, and brigatinib, but lorlatinib, a third-generation ALK inhibitor, has shown activity against this mutation.79 Furthermore, brain metastasis represents a treatment challenge for patients with ALK rearrangements. It is also an efficacy measure of next-generation ALK inhibitors, all of which have demonstrated better central nervous system activity than crizotinib.69,78,80 If the case patient were found to have brain metastasis at the initial diagnosis, either ceritinib or alectinib would be a reasonable choice since crizotinib has limited penetration of blood-brain barrier.81
ROS1 REARRANGEMENTS
CASE PRESENTATION 3
A 66-year-old Chinese woman who is a non-smoker with a past medical history of hypertension and hypothyroidism presents to the emergency department for worsening lower back pain. Initial workup includes x-ray of the lumbar spine followed by MRI with contrast, which shows a soft tissue mass at L3-4 without cord compression. CT of the chest, abdomen, and pelvis with contrast shows a 7-cm right hilar mass, bilateral small lung nodules, mediastinal lymphadenopathy, and multiple lytic lesions in ribs, lumbar spine, and pelvis. MRI-brain with and without contrast is negative for malignancy. She undergoes endo-bronchial ultrasound and biopsy of the right hilar mass, which shows poorly differentiated adenocarcinoma. While waiting for the result of the molecular analysis, the patient undergoes palliative radiation therapy to L2-5 with good pain relief. She is discharged from the hospital and presents to your clinic for follow up. Molecular analysis now reveals ROS1 rearrangement with CD74-ROS1 fusion.
What treatment plan should be put in place for this patient?
FIRST-LINE THERAPY FOR ROS1 REARRANGEMENTS
Approximately 2.4% of lung adenocarcinomas harbor ROS1 rearrangements.82 This distinct genetic alteration occurs more frequently in NSCLC patients who are younger, female, and never-smokers, and who have adenocarcinomas.8 It has been shown that ROS1 rearrangements rarely overlap with other genetic alterations including KRAS mutations, EGFR mutations, and ALK rearrangements.83 As a receptor tyrosine kinase, ROS1 is similar to ALK and insulin receptor family members.84 Crizotinib, which targets ALK, ROS1, and c-MET, was approved by the FDA on March 11, 2016, for the treatment of metastatic ROS1-rearranged NSCLC.85 The approval was based on a phase 2 expansion cohort of the original phase 1 study. Among 50 US patients enrolled in this expansion cohort, 3 had complete responses and 33 had partial responses with ORR of 72% (95% CI 58% to 84%). Median PFS was 19.2 months (95% CI 14.4 months to NR) and median duration of response (DOR) was 17.6 months (95% CI 14.5 months to NR).86 During longer follow-up, independent radiology review confirmed high ORR of 66% and median DOR of 18.3 months.85
Interestingly, no companion diagnostic assay has been approved for the detection of ROS1 rearrangements with the approval of crizotinib. In the United States, break apart FISH is the most common detection method. In fact, in the above mentioned phase 2 study, ROS1 rearrangements were detected in 49 out of 50 patients by this method.86 FISH can be technically challenging when dealing with high volume and multiple targets. Reverse transcriptase-PCR is another detection method, but it requires knowledge of the fusion partners. To date, at least 14 ROS1 fusion partners have been reported, with CD74 being the most common.87 NGS with appropriate design and validation can also be used to detect ROS1 rearrangements.
For the case patient, the recommendation would be to start her on crizotinib at 250 mg twice daily. Monitoring for vision disturbance, gastrointestinal complaints, and edema is warranted. Because the estimated onset of response is around 7.9 weeks,86 plans should be made to repeat her scans in approximately 2 months.
BRAF V600E MUTATIONS
CASE PRESENTATION 4
A 71-year-old Caucasian man with a past medical history of hypertension, dyslipidemia, and ischemic cerebrovascular accident without residual deficits was diagnosed with stage IV adenocarcinoma of the lung about 8 months ago. He has a 40 pack-year smoking history and quit smoking when he was diagnosed with lung cancer. His disease burden involved a large mediastinal mass, scattered pleural nodules, multiple lymphadenopathy, and several soft tissue masses. His outside oncologist started him on chemotherapy containing carboplatin and pemetrexed for 6 cycles followed by maintenance pemetrexed. The most recent restaging scans show disease progression with enlarging soft tissue masses and several new lytic bone lesions. MRI-brain with and without contrast shows 2 subcentimeter enhancing lesions. He transferred care to you approximately 4 weeks ago. You ordered a repeat biopsy of 1 of the enlarging soft tissue masses. Molecular analysis revealed BRAF V600E mutation. In the interim, he underwent stereotactic radiosurgery for the 2 brain lesions without any complications. The patient is now in your clinic for follow up.
What would be your recommended systemic treatment?
TARGETED THERAPIES FOR BRAF V600E MUTATION
BRAF mutations were first recognized as activating mutations in advanced melanomas, with BRAF V600E, resulting from the substitution of glutamic acid for valine at amino acid 600, being the most common. BRAF plays an important role in the mitogen-activated protein kinase (MAPK) signaling pathway. Briefly, the activation of MAPK pathway occurs upon ligand binding of receptor tyrosine kinases, which then involves RAS/BRAF/MEK/ERK in a stepwise manner, ultimately leading to cell survival. BRAF mutations have been increasingly recognized also as driver mutations in NSCLC.9–12 They can be detected by PCR or NGS method. The characteristics of NSCLC patients harboring BRAF mutations have been described by various groups.9–12 For instance, 1 case series showed that the incidence was 2.2% among patients with advanced lung adenocarcinoma; 50% of mutations were V600E, while G469A and D594G accounted for the remaining 39% and 11% of patients, respectively. All patients were either current or former smokers. The median OS of patients with BRAF mutations in this case series was NR, while it was 37 months for patients with EGFR mutations (P = 0.73) and NR for patients with ALK rearrangements (P = 0.64).9
For patients with BRAF V600E–mutant NSCLC who have progressed on platinum-based chemotherapy, the combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) may represent a new treatment paradigm. This was illustrated in a phase 2, nonrandomized, open-label study. A total of 57 patients were enrolled and 36 patients (63.2% [95% CI 49.3% to 75.6%]) achieved an overall response by investigator assessment, the trial’s primary end point. Disease control rate was 78.9% (95% CI 66.1% to 88.6%), with 4% complete response, 60% partial response, and 16% stable disease. PFS was 9.7 months (95% CI [6.9 to 19.6 months]). The safety profile was comparable to what had been observed in patients with melanoma treated with this regimen. More specifically, 56% of patients on this trial reported serious AEs, including pyrexia (16%), anemia (5%), confusional state (4%), decreased appetite (4%), hemoptysis (4%), hypercalcemia (4%), nausea (4%), and cutaneous squamous cell carcinoma (4%). In addition, neutropenia (9%) and hyponatremia (7%) were the most common grade 3-4 AEs.16
The case patient has experienced disease progression after 1 line of platinum-based chemotherapy, so the combination of dabrafenib and trametinib would be a robust systemic treatment option. dabrafenib as a single agent has also been studied in BRAF V600E–mutant NSCLC in a phase 2 trial. The overall response by investigator assessment among 84 patients was 33% (95% CI 23% to 45%).14 Vemurafenib, another oral BRAF TKI, has demonstrated efficacy for NSCLC patients harboring BRAF V600E mutation. In the cohort of 20 patients with NSCLC, the response rate was 42% (95% CI 20% to 67%) and median PFS was 7.3 months (95% CI 3.5 to 10.8 months).13 Patients with non-V600E mutations have shown variable responses to targeted therapies. MEK TKIs may be considered in this setting; however, the details of this discussion are beyond the scope of this review.
CONCLUSION
The management of advanced NSCLC with driver mutations has seen revolutionary changes over the past decade. Tremendous research has been done in order to first understand the molecular pathogenesis of NSCLC and then discover driver mutations that would lead to development of targeted therapies with clinically significant efficacy as well as tolerability. More recently, increasing efforts have focused on how to conquer acquired resistance in patients with disease progression after first-line TKIs. The field of EGFR-mutant NSCLC has set a successful example, but the work is nowhere near finished. The goals are to search for more driver mutations and to design agents that could potentially block cell survival signals once and for all.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
- Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol 2016;893:1–19.
- Alberg AJ, Brock MV, Ford JG, et al. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013;143:e1S–29S.
- Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-3013, based on November 2015 SEER data submission, posted to the SEER website, April 2016. Bethesda (MD): National Cancer Institute; 2016.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Non-Small Cell Lung Cancer: 1–190.
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–39.
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6.
- Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancer. J Clin Oncol 2012;30:863–70.
- Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–51.
- Kinno T, Tsuta K, Shiraishi K, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 2014;25:138–42.
- Litvak AM, Paik PK, Woo KM, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 2014;9:1669–74.
- Villaruz LC, Socinski MA, Abberbock S, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015;121:448–56.
- Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–36.
- Planchard D, Kim TM, Mazieres J, et al. DaBRAFenib in patients with BRAF V600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:642–50.
- Gautschi O, Milia J, Cabarrou B, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol 2015;10:1451–7.
- Planchard D, Besse B, Groen HJ, et al. DaBRAFenib plus trametinib in patients with previously treated BRAF V600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984–93.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. J Thorac Oncol 2013;8:823–59.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. Arch Pathol Lab Med 2013;137:828–60.
- Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology guideline. J Clin Oncol 2014;32:3673–9.
- Paik PK, Varghese AM, Sima CS, et al. Response to erlotinib in patients with EGFR mutant advanced non-small cell lung cancers with a squamous or squamous-like component. Mol Cancer Ther 2012;11:2535–40.
- Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5:842–9.
- Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age, and stage-dependent MET genomic amplification, and c-MET overexpression. J Clin Oncol 2016;34:721–30.
- Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol 2016;11:1493–502.
- Reungwetwattana T, Liang Y, Zhu V, et al. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017;103:27–37.
- Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 2013;3:630–5.
- Lin JJ, Kennedy E, Sequist LV, et al. Clinical activity of alectinib in advanced RET-rearranged non-small cell lung cancer. J Thorac Oncol 2016;11:2027–32.
- Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 2016;17:1653–60.
- Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med 2006;354:2619–21.
- Mazieres J. Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targted drugs: results from the European EUHER2 cohort. Annal Oncol 2016;27:281–6.
- Ou SH, Schrock AB, Bocharov EV, et al. HER2 transmembrane (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 2017;12:446–57.
- Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emergent therapies. Cancer Discov 2017;7:596–609.
- Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006.
- Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016;387:1415–26.
- Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958–67.
- Rosell R, Karachaliou N, et al. Large-scale screening for somatic mutations in lung cancer. Lancet 2016;387:1354–6.
- Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005;97:339–46.
- Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327–34.
- Wu YL, Chou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol 2015;26:1883–9.
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Eng J Med 2010;362:2380–8.
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121–8.
- Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735–42.
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239–46.
- Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:213–22.
- Hirsch FR, Bunn PA Jr. EGFR testing in lung cancer is ready for prime time. Lancet Oncol 2009;10:432–3.
- Nelson V, Ziehr J, Aqulnik M, et al. Afatinib: emerging next-generation tyrosine kinase inhibitor for NSCLC. Onco Targets Ther 2013;5:135–43.
- Lee CK, Wu YL, Ding PN, et al. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: a meta-analysis. J Clin Oncol 2015;33:1958–65.
- Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol 2013;24:54–9.
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57.
- Wu YL, Saijo N, Thongprasert S, et al. Efficacy according to blind independent central review: post-hoc analyses from the phase III, randomized, multicenter, IPASS study of first-line gefitinib versus carboplatin/paclitaxel in Asian patients with EFGR mutation-positive advanced NSCLC. Lung Cancer 2017;104:119–25.
- Douillard JY, Ostoros G, Cobo M, et al. First-line gefitinib in Caucasian EGFR-mutation positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer 2014;110:55–62.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol 2007;56:317–26.
- Tarceva [package insert]. South San Francisco (CA): Genentech, Inc; 2010. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021743s14s16lbl.pdf. Accessed April 23, 2017.
- Gilotrif [package insert.] Ridgefield (CT): Boehringer Ingelheim, Inc; 2013. www.accessdata.fda.gov/drugsatfda_docs/label/2013/201292s000lbl.pdf. Accessed April 23, 2017.
- Iressa [package insert]. Wilmington (DE): AstraZeneca, Inc; 2015. Error! Hyperlink reference not valid. Accessed April 23, 2017.
- Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616–22.
- Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR mutant lung cancers. Clin Cancer Res 2013;19:2240–7.
- Yun CH, Mengwasser KE, Tom AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5.
- Sos ML, Rode HB, Heynck S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res 2010;70:868–74.
- Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T190M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046–61.
- Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629–40.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N Engl J Med 2010;363:1693–703.
- Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247–53.
- Kazandjian D, Blumenthal GM, Chen HY, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014;19:e5–11.
- Solomon BJ, Mok T, Kim DW, et al. First-ling crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167–77.
- Xalkori [package insert]. New York: Pfizer, Inc; 2011. www.accessdata.fda.gov/drugsatfda_docs/label/2012/202570s002lbl.pdf. Accessed April 23, 2017.
- Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385–94.
- Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013;56:5675–90.
- Khozin S, Blumenthal GM, Zhang L, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res 2015;21:2436–9.
- Soria JC, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 2017;389:917–29.
- Zykadia [package insert]. East Hanover (NJ): Novartis Pharmaceuticals Corporation, Inc; 2016. www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/zykadia.pdf. Accessed April 23, 2017.
- Larkins E, Blumenthal GM, Chen H, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res 2016;22:5171–6.
- Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. New Engl J Med 2017 June 6 [Epub ahead of print].
- Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem 2012;20:1271–80.
- Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679–90.
- Kodama T, Tsukaguchi T, Yoshida M, et al. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett 2014;351:215–21.
- Kodama T, Tsukaguchi T, Satoh T, et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther 2014;13:2910–8.
- Alecensa [package insert]. South San Francisco (CA): Genentech, Inc; 2015. www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf. Accessed April 23, 2017.
- Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol 2017 May 5 [Epub ahead of print].
- Zhu V, Ou SH. Safety of alectinib for the treatment of metastatic ALK-rearranged non-small cell lung cancer. Expert Opin Drug Saf 2017;16:509–14.
- Gadgeel SM, Shaw AT, Govindan R, et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small cell lung cancer. J Clin Oncol 2016;34:4079–85.
- Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29:e443–5.
- Zhu Q, Zhan P, Zhang X, et al. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res 2015;4:300–9.
- Lin JJ, Ritterhouse LL, Ali SM, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol 2017;12:872–7.
- Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta 2009;1795:37–52.
- Kazandjian D, Blumenthal G, Luo L, et al. Benefit-Risk summary of crizotinib for the treatment of patients with ROS1 alteration-positive metastatic NSCLC. Oncologist 2016;21:974–80.
- Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014;371:1963–71.
- Zhu VW, Upadhyay D, Schrock AB, et al. TPD52L1-ROS1, a new ROS1 fusion variant in lung adenosquamous cell carcinoma identified by comprehensive genomic profiling. Lung Cancer 2016;97:48–50.
INTRODUCTION
Lung cancer is the second most common type of cancer in the United States, with 222,500 estimated new cases in 2017, according to the American Cancer Society.1 However, it is by far the number one cause of death due to cancer, with an estimated 155,870 lung cancer–related deaths occurring in 2017, which is higher than the number of deaths due to breast cancer, prostate cancer, and colorectal cancer combined.1,2 Despite slightly decreasing incidence and mortality over the past decade, largely due to smoking cessation, the 5-year survival rate of lung cancer remains dismal at approximately 18%.2–4
Non-small cell lung cancer (NSCLC) accounts for 80% to 85% of all lung cancer cases.4 Traditionally, it is further divided based on histology: adenocarcinoma, squamous cell carcinoma, large cell carcinoma, and not otherwise specified.5 Chemotherapy had been the cornerstone of treatment for stage IV NSCLC. It is not target-specific and is most effective against rapidly growing cells. Common adverse effects include alopecia, nausea/vomiting, myelosuppression, cardiotoxicity, neuropathy, and nephrotoxicity. However, this paradigm has shifted following the discovery of mutations of the epidermal growth factor receptor (EGFR) gene as an oncogenic driver that confers sensitivity to small molecule tyrosine kinase inhibitors (TKIs) targeting EGFR.6 The EGFR inhibitors are given orally and have a spectrum of toxicities (eg, such as rash, diarrhea, and elevated transaminases) different from that of systemic chemotherapy, which is often administered intravenously. Following the discovery of EGFR mutations, rearrangements of the anaplastic lymphoma kinase (ALK) gene7 and ROS1 gene8 were identified as targetable driver mutations in NSCLC. The frequency of both rearrangements is lower than that of EGFR mutations. Additionally, BRAF V600E mutation has been identified in NSCLC.9–12 This activation mutation is commonly seen in melanoma. Agents that have already been approved for the treatment of melanoma with the BRAF V600E mutation are being tested in NSCLC patients with this mutation.13–16
Given the effectiveness and tolerability of targeted therapy, identifying this distinct molecular subset of NSCLC patients is critical in treatment. Currently, molecular testing is mandatory in all stage IV patients with non-squamous cell carcinoma, as a preponderance of patients with driver mutations have this histology subtype.5,17–19 For patients with squamous cell carcinoma, molecular testing should be considered if the biopsy specimen is small, there is mixed histology, or the patient is a nonsmoker.5,20 Several techniques are commonly utilized in detecting these genetic alterations. EGFR mutation can be detected by polymerase chain reaction (PCR), ALK or ROS1 rearrangement can be detected by fluorescence in-situ hybridization (FISH), and immunohistochemistry (IHC) can also be used to detect ALK rearrangement. The current guideline is to use comprehensive genomic profiling to capture all the potential molecular targets simultaneously instead of running stepwise tests just for EGFR, ALK, and ROS1.5 BRAF V600E mutation,13–16 MET exon 14 skipping mutation,21–24 RET rearrangements,25–27 and HER2 mutations28–30 are among the emergent genetic alterations with various responses to targeted therapy.31 Some of these targeted agents have been approved for other types of malignancy, and others are still in the development phase.
Several initiatives worldwide have reported better outcomes of patients with driver mutations treated with targeted therapy. For instance, the Lung Cancer Mutation Consortium in the United States demonstrated that the median survival of patients without driver mutations, with drivers mutations but not treated with targeted therapy, and with driver mutations and treated with targeted therapy was 2.08 years, 2.38 years, and 3.49 years, respectively.32 The French Cooperative Thoracic Intergroup-French National Cancer Institute demonstrated that the median survival for patients with driver mutations versus those without driver mutations was 16.5 months versus 11.8 months.33 The Spanish Lung Cancer Group demonstrated that the overall survival (OS) for patients with EGFR mutations treated with erlotinib was 27 months.34 The mutations in lung cancer, their frequencies, and the downstream signaling pathways are depicted in the Figure.35
In this article, we discuss targeted therapy for patients with EGFR mutations, ALK rearrangements, ROS1 rearrangements, and BRAF V600E mutation. We also discuss the management of patients with EGFR mutations who develop a secondary mutation after TKI therapy. Almost all of the targeted agents discussed herein have been approved by the US Food and Drug Administration (FDA), so they are considered standard of care. All available phase 3 trials pertinent to these targeted therapies are included in the discussion.
EGFR MUTATIONS
CASE PRESENTATION 1
A 54-year-old Caucasian man who is a former smoker with a 10 pack-year history and past medical history of hypertension and dyslipidemia presents with progressive dyspnea for several weeks. A chest x-ray shows moderate pleural effusion on the left side with possible mass-like opacity on the left upper lung field. An ultrasound-guided thoracentesis is performed and cytology is positive for adenocarcinoma of likely pulmonary origin. Staging workup including positron emission tomography (PET)/computed tomography (CT) and magnetic resonance imaging of the brain with and without contrast is done. PET/CT shows a 5.5-cm mass in the left upper lobe of the lung with high fluorodeoxyglucose (FDG) uptake, several 1- to 2-cm mediastinal lymph nodes with moderate FDG uptake, and small pleural effusion on both sides with moderate FDG uptake. MRI-brain is negative for malignancy. The patient subsequently undergoes a CT-guided biopsy of the lung mass, which shows moderately differentiated adenocarcinoma. Comprehensive molecular profiling reveals EGFR L858R mutation only. The patient now presents for the initial consultation. Of note, his Eastern Cooperative Oncology Group performance status is 1.
What is the next step in the management of this patient?
FIRST-LINE TKI FOR SENSITIZING EGFR MUTATIONS
The 2 most common EGFR mutations are deletions in exon 19 and substitution of arginine for leucine in exon 21 (L858R), found in approximately 45% and 40% of patients with EGFR mutations, respectively.36 Both mutations are sensitive to EGFR TKIs. The benefit may be greater in patients with exon 19 deletions as compared to exon 21 L858R substitution,37,38 but this has not been demonstrated consistently in clinical trials.39-43 In the United States, EGFR mutations are found in approximately 10% of patients with NSCLC, while the incidence can be as high as 50% in Asia.44 Even though the cobas EGFR mutation test is the companion diagnostic approved by the US FDA, a positive test result from any laboratory with the Clinical Laboratory Improvement Amendments (CLIA) certificate should prompt the use of an EGFR TKI as the initial treatment.
Three EGFR TKIs that have been approved as first-line therapy in the United States are available: erlotinib, afatinib, and gefitinib.5 Both erlotinib and gefitinib are considered first-generation TKIs. They have higher binding affinity for the 2 common EGFR mutations than wild-type EGFR. In addition, they reversibly bind to the intracellular tyrosine kinase domain, resulting in inhibition of autophosphorylation of the tyrosine residues. Afatinib, a second-generation and irreversible TKI, targets EGFR (HER1) as well as HER2 and HER4.45
The superior efficacy of the EGFR TKIs over platinum doublet chemotherapy in treatment-naïve patients with EGFR mutations has been demonstrated in 7 randomized trials to date (Table).46 Erlotinib was the TKI arm for the OPTIMAL,41 EURTAC,42 and ENSURE trials;38 afatinib was the TKI arm for LUX-LUNG 337 and 6;43 gefitinib was the TKI arm for NEJ00239,47 and WJTOG3405.40 A meta-analysis of these 7 trials by Lee et al showed that progression-free survival (PFS) was significantly prolonged by EGFR TKIs (hazard ratio [HR] 0.37 [95% confidence interval {CI} 0.32 to 0.42]).46 For instance, in the EURTAC trial, median PFS was 9.7 months for patients treated with erlotinib as compared to 5.2 months for patients treated with platinum/gemcitabine or platinum/docetaxel.42 In this meta-analysis, prespecified subgroups included age, sex, ethnicity, smoking status, performance status, tumor histology, and EGFR mutation subtype. The superior outcome with TKIs was observed in all subgroups. Furthermore, patients with exon 19 deletions, nonsmokers, and women had even better outcomes.46
Erlotinib is the most commonly used TKI in the United States largely because gefitinib was off the market for some time until it was re-approved by the FDA in 2015. Interestingly, this “re-approval” was not based on either 1 of the 2 prospective trials (NEJ00239,47 and WJTOG340540), but rather was based on an exploratory analysis of the IPASS trial48,49 as well as a prospective phase 4, single-arm trial in Europe (IFUM).50 The superior efficacy of gefitinib over carboplatin/paclitaxel among patients with EGFR mutations in the IPASS trial was confirmed by blind independent central review, with longer PFS (HR 0.54 [95% CI 0.38 to 0.79] P = 0.0012) and higher objective response rate (ORR; odds ratio 3 [95% CI 1.63 to 5.54], P = 0.0004).49
CASE 1 CONTINUED
Based on the EGFR L858R mutation status, the patient is started on erlotinib. He is quite happy that he does not need intravenous chemotherapy but wants to know what toxicities he might potentially have with erlotinib.
What are the common adverse effects (AEs) of EGFR TKIs? How are AEs of TKIs managed?
Safety Profile
The important toxicities associated with EGFR TKIs are rash, gastrointestinal toxicity, hepatic toxicity, and pulmonary toxicity. Rash is an AE specific to all agents blocking the EGFR pathway, including small molecules and monoclonal antibodies such as cetuximab. The epidermis has a high level of expression of EGFR, which contributes to this toxicity.51 Rash usually presents as dry skin or acneiform eruption. Prophylactic treatment with oral tetracyclines and topical corticosteroids is generally recommended upon initiation of TKI therapy. Diarrhea is the most prevalent gastrointestinal toxicity. All patients starting treatment should be given prescriptions to manage diarrhea such as loperamide and be advised to call when it occurs. Hepatic toxicity is often manifested as elevated transaminases or bilirubin. Interstitial lung disease (ILD) is a rare but potentially fatal pulmonary toxicity.
Rash of any grade was reported in 49.2% of patients treated with erlotinib in clinical trials, while grade 3 rash occurred in 6% of patients and no grade 4 was reported. Diarrhea of any grade was reported in 20.3% of patients, grade 3 diarrhea occurred in 1.8%, and no grade 4 was reported. Grade 2 and 3 alanine aminotransferase (ALT) elevations were seen in 2% and 1% of patients, respectively. Grade 2 and 3 bilirubin elevations were seen in 4% and less than 1% of patients, respectively. The incidence of serious ILD-like events was less than 1%.52
Afatinib is associated with higher incidences of rash and diarrhea. Specifically, diarrhea and rash of all grades were reported in 96% and 90% of patients treated with afatinib, respectively. Paronychia of all grades occurred in 58% of patients. Elevated ALT of all grades was seen in 11% of patients. Approximately 1.5% of patients treated with afatinib across clinical trials had ILD or ILD-like AEs.53
Gefitinib, the most commonly used TKI outside United States, has a toxicity profile similar to erlotinib, except for hepatic toxicity. For instance, rash of all grades occurred in 47% of patients, diarrhea of all grades occurred in 29% of patients, and ILD or ILD-like AEs occurred in 1.3% of patients across clinical trials. In comparison, elevated ALT and aspartate aminotransferase (AST) of all grades was seen in 38% and 40% of patients, respectively.54 Therefore, close monitoring of liver function is clinically warranted. In particular, patients need to be advised to avoid concomitant use of herbal supplements, a common practice in Asian countries.
CASE 1 CONTINUED
The patient does well while on erlotinib at 150 mg orally once daily for about 8 months, until he develops increasing abdominal pain. A CT scan of the abdomen and pelvis with contrast shows a new 8-cm right adrenal mass. Additionally, a repeat CT scan of the chest with contrast shows a stable lung mass but enlarging mediastinal lymphadenopathy.
How would you manage the patient at this point?
MANAGEMENT OF T790M MUTATION AFTER PROGRESSION ON FIRST-LINE EGFR TKIS
As mentioned above, the median PFS of patients with EGFR mutations treated with 1 of the 3 TKIs is around 9 to 13 months.46 Of the various resistance mechanisms that have been described, the T790M mutation is found in approximately 60% of patients who progress after treatment with first-line TKIs.55,56 Other mechanisms, such as HER2 amplification, MET amplification, or rarely small cell transformation, have been reported.56 The first- and second-generation EGFR TKIs function by binding to the ATP-binding domain of mutated EGFR, leading to inhibition of the downstream signaling pathways (Figure, part B) and ultimately cell death.35 The T790M mutation hinders the interaction between the ATP-binding domain of EGFR kinase and TKIs, resulting in treatment resistance and disease progression.57,58
Osimertinib is a third-generation irreversible EGFR TKI with activity against both sensitizing EGFR and resistant T790M mutations. It has low affinity for wide-type EGFR as well as insulin receptor and insulin-like growth factor receptor.59 Osimertinib has been fully approved for NSCLC patients with EGFR mutations who have progressed on first-line EGFR TKIs with the development of T790M mutation. An international phase 3 trial (AURA3) randomly assigned 419 patients in a 2:1 ratio to either osimertinib or platinum/pemetrexed. Eligible patients all had the documented EGFR mutations and disease progression after first-line EGFR TKIs. Central confirmation of the T790M mutation was required. Median PFS by investigator assessment, the trial’s primary end point, was 10.1 months for osimertinib versus 4.4 months for chemotherapy (HR 0.3 [95% CI 0.23 to 0.41]; P < 0.001). ORR was 71% for osimertinib versus 31% for chemotherapy (HR 5.39 [95% CI 3.47 to 8.48], P < 0.001). A total of 144 patients with stable and asymptomatic brain metastases were also eligible. Median PFS for this subset of patients treated with osimertinib and chemotherapy was 8.5 months and 4.2 months, respectively (HR 0.32 [95% CI 0.21 to 0.49]). In the AURA3 trial, osimertinib was better tolerated than chemotherapy, with 23% of patients treated with osimertinib experiencing grade 3 or 4 AEs as compared to 47% of chemotherapy-treated patients. The most common AEs of any grade were diarrhea (41%), rash (34%), dry skin (23%), and paronychia (22%).60
For the case patient, a reasonable approach would be to obtain a tissue biopsy of the adrenal mass and more importantly to check for the T790M mutation. Similar to the companion diagnostic for EGFR mutations, the cobas EGFR mutation test v2 is the FDA-approved test for T790M. However, if this resistance mutation is detected by any CLIA-certified laboratories, osimertinib should be the recommended treatment option. If tissue biopsy is not feasible, plasma-based testing should be considered. A blood-based companion diagnostic also is FDA approved.
ALK REARRANGEMENTS
CASE 2 PRESENTATION
A 42-year-old Korean woman who is a non-smoker with no significant past medical history presents with fatigue, unintentional weight loss of 20 lb in the past 4 months, and vague abdominal pain. A CT can of the abdomen and pelvis without contrast shows multiple foci in the liver and an indeterminate nodule in the right lung base. She subsequently undergoes PET/CT, which confirms multiple liver nodules/masses ranging from 1 to 3 cm with moderate FDG uptake. In addition, there is a 3.5-cm pleura-based lung mass on the right side with moderate FDG uptake. MRI-brain with and without contrast is negative for malignancy. A CT-guided biopsy of 1 of the liver masses is ordered and pathology returns positive for poorly differentiated adenocarcinoma consistent with lung primary. Molecular analysis reveals an echinoderm microtubule-associated protein-like 4 (EML4)-ALK rearrangement. She is placed on crizotinib by an outside oncologist and after about 3 weeks of therapy is doing well. She is now in your clinic for a second opinion. She says that some of her friends told her about another medication called ceritinib and was wondering if she would need to switch her cancer treatment.
How would you respond to this patient’s inquiry?
FIRST-LINE TKIS FOR ALK REARRANGEMENTS
ALK rearrangements are found in 2% to 7% of NSCLC, with EML4-ALK being the most prevalent fusion variant.61 The inversion of chromosome 2p leads to the fusion of the EML4 gene and the ALK gene, which causes the constitutive activation of the fusion protein and ultimately increased transformation and tumorigenicity.7,61 Patients harboring ALK rearrangements tend to be non-smokers. Adenocarcinoma, especially signet ring cell subtype, is the predominant histology. Compared to EGFR mutations, patients with ALK mutations are significantly younger and more likely to be men.62 ALK rearrangements can be detected by either FISH or IHC, and most next-generation sequencing (NGS) panels have the ability to identify this driver mutation.
Crizotinib is the first approved ALK inhibitor for the treatment of NSCLC in this molecular subset of patients.63 PROFILE 1014 is a phase 3 randomized trial that compared crizotinib with chemotherapy containing platinum/pemetrexed for up to 6 cycles. Crossover to crizotinib was allowed for patients with disease progression on chemotherapy. The primary end point was PFS by independent radiologic review. The crizotinib arm demonstrated superior PFS (10.9 months versus 7 months; HR 0.45 [95% CI 0.35 to 0.6], P < 0.001) and ORR (74% versus 45%, P < 0.001). Median survival was not reached in either arm (HR 0.82 [95% CI 0.54 to 1.26], P = 0.36).64 Based on this international trial, crizotinib is considered standard of care in the United States for treatment-naïve patients with advanced NSCLC harboring ALK rearrangements. The current recommended dose is 250 mg orally twice daily. Common treatment-related AEs of all grades include vision disorder (62%), nausea (53%), diarrhea (43%), vomiting (40%), edema (28%), and constipation (27%).65 PROFILE 1007 compared crizotinib with pemetrexed or docetaxel in ALK-rearranged NSCLC patients with prior exposure to 1 platinum-based chemotherapy. The median PFS was 7.7 months for crizotinib as compared to 3 months for chemotherapy (HR 0.49 [95% CI 0.37 to 0.64], P < 0.001). The response rates were 65% and 20% for crizotinib and chemotherapy, respectively (P < 0.001).66 In other countries, crizotinib following 1 prior platinum-based regimen may be considered standard of care based on this trial.
Ceritinib is an oral second-generation ALK inhibitor that is 20 times more potent than crizotinib based on enzymatic assays.67 It also targets ROS1 and insulin-like growth factor 1 receptor but not c-MET. It was first approved by the FDA in April 2014 for metastatic ALK-rearranged NSCLC following crizotinib.68 In May 2017, the FDA granted approval of ceritinib for treatment-naïve patients. This decision was based on the results of the ASCEND-4 trial, a randomized phase 3 trial assessing the efficacy and safety of ceritinib over chemotherapy in the first-line setting. The trial assigned 376 patients to either ceritinib at 750 mg once daily or platinum/pemetrexed for 4 cycles followed by maintenance pemetrexed. Median PFS was 16.6 months for ceritinib versus 8.1 months for chemotherapy (HR 0.55 [95% CI 0.42 to 0.73]; P < 0.00001).69 Toxicities of ceritinib are not negligible, with gastrointestinal toxicity being the most prevalent. For instance, diarrhea, nausea, vomiting, abdominal pain, and constipation of all grades were seen in 86%, 80%, 60%, 54%, and 29% of patients, respectively. Furthermore, fatigue and decreased appetite occurred in 52% and 34% of patients, respectively. In terms of laboratory abnormalities, 84% of patients experienced decreased hemoglobin of all grades; 80% increased ALT; 75% increased AST; 58% increased creatinine; 49% increased glucose; 36% decreased phosphate; and 28% increased lipase. Due to these AEs, the incidence of dose reduction was about 58% and the median onset was around 7 weeks.70
Alectinib is another oral second-generation ALK inhibitor that was approved by the FDA in December 2015 for the treatment of NSCLC patients with ALK rearrangements who have progressed on or are intolerant to crizotinib.71 Its indication will soon be broadened to the first-line setting based on the ALEX trial.72 Alectinib is a potent and highly selective TKI of ALK73 with activity against known resistant mutations to crizotinib.74,75 It also inhibits RET but not ROS1 or c-MET.76 ALEX, a randomized phase 3 study, compared alectinib with crizotinib in treatment-naïve patients with NSCLC harboring ALK rearrangements. The trial enrolled 303 patients and the median follow-up was approximately 18 months. The alectinib arm (600 mg twice daily) demonstrated significantly higher PFS by investigator-assessment, the trial’s primary end point. The 12-month event-free survival was 68.4% (95% CI 61% to 75.9%) versus 48.7% (95% CI 40.4% to 56.9%) for alectinib and crizotinib, respectively (HR 0.47 [95% CI 0.34 to 0.65], P < 0.001). The median PFS was not reached in the alectinib arm (95% CI 17.7 months to not estimable) as compared to 11.1 months in the crizotinib arm (95% CI 9.1 to 13.1 months).72 Alectinib is generally well tolerated. Common AEs of all grades include fatigue (41%), constipation (34%), edema (30%), and myalgia (29%). As alectinib can cause anemia, lymphopenia, hepatic toxicity, increased creatine phosphokinase, hyperglycemia, electrolyte abnormalities, and increased creatinine, periodic monitoring of these laboratory values is important, although most of these abnormalities are grade 1 or 2.77
Brigatinib, another oral second-generation ALK inhibitor, was granted accelerated approval by the FDA in April 2017 for ALK-rearranged and crizotinib-resistant NSCLC based on the ALTA trial. This randomized phase 2 study of brigatinib showed an ORR by investigator assessment of 54% (97.5% CI 43% to 65%) in the 180 mg once daily arm with lead-in of 90 mg once daily for 7 days. Median PFS was 12.9 months (95% CI 11.1 months to not reached [NR]).78 Currently, a phase 3 study of brigatinib versus crizotinib in ALK inhibitor–naïve patients is recruiting participants (ALTA-1L). It will be interesting to see if brigatinib can achieve a front-line indication.
Starting the case patient on crizotinib is well within the treatment guidelines. One may consider ceritinib or alectinib in the first-line setting, but both TKIs can be reserved upon disease progression. We would recommend a repeat biopsy at that point to look for resistant mechanisms, as certain secondary ALK mutations may be rescued by certain next-generation ALK inhibitors. For instance, the F1174V mutation has been reported to confer resistance to ceritinib but sensitivity to alectinib, while the opposite is true for I1171T. The G1202R mutation is resistant to ceritinib, alectinib, and brigatinib, but lorlatinib, a third-generation ALK inhibitor, has shown activity against this mutation.79 Furthermore, brain metastasis represents a treatment challenge for patients with ALK rearrangements. It is also an efficacy measure of next-generation ALK inhibitors, all of which have demonstrated better central nervous system activity than crizotinib.69,78,80 If the case patient were found to have brain metastasis at the initial diagnosis, either ceritinib or alectinib would be a reasonable choice since crizotinib has limited penetration of blood-brain barrier.81
ROS1 REARRANGEMENTS
CASE PRESENTATION 3
A 66-year-old Chinese woman who is a non-smoker with a past medical history of hypertension and hypothyroidism presents to the emergency department for worsening lower back pain. Initial workup includes x-ray of the lumbar spine followed by MRI with contrast, which shows a soft tissue mass at L3-4 without cord compression. CT of the chest, abdomen, and pelvis with contrast shows a 7-cm right hilar mass, bilateral small lung nodules, mediastinal lymphadenopathy, and multiple lytic lesions in ribs, lumbar spine, and pelvis. MRI-brain with and without contrast is negative for malignancy. She undergoes endo-bronchial ultrasound and biopsy of the right hilar mass, which shows poorly differentiated adenocarcinoma. While waiting for the result of the molecular analysis, the patient undergoes palliative radiation therapy to L2-5 with good pain relief. She is discharged from the hospital and presents to your clinic for follow up. Molecular analysis now reveals ROS1 rearrangement with CD74-ROS1 fusion.
What treatment plan should be put in place for this patient?
FIRST-LINE THERAPY FOR ROS1 REARRANGEMENTS
Approximately 2.4% of lung adenocarcinomas harbor ROS1 rearrangements.82 This distinct genetic alteration occurs more frequently in NSCLC patients who are younger, female, and never-smokers, and who have adenocarcinomas.8 It has been shown that ROS1 rearrangements rarely overlap with other genetic alterations including KRAS mutations, EGFR mutations, and ALK rearrangements.83 As a receptor tyrosine kinase, ROS1 is similar to ALK and insulin receptor family members.84 Crizotinib, which targets ALK, ROS1, and c-MET, was approved by the FDA on March 11, 2016, for the treatment of metastatic ROS1-rearranged NSCLC.85 The approval was based on a phase 2 expansion cohort of the original phase 1 study. Among 50 US patients enrolled in this expansion cohort, 3 had complete responses and 33 had partial responses with ORR of 72% (95% CI 58% to 84%). Median PFS was 19.2 months (95% CI 14.4 months to NR) and median duration of response (DOR) was 17.6 months (95% CI 14.5 months to NR).86 During longer follow-up, independent radiology review confirmed high ORR of 66% and median DOR of 18.3 months.85
Interestingly, no companion diagnostic assay has been approved for the detection of ROS1 rearrangements with the approval of crizotinib. In the United States, break apart FISH is the most common detection method. In fact, in the above mentioned phase 2 study, ROS1 rearrangements were detected in 49 out of 50 patients by this method.86 FISH can be technically challenging when dealing with high volume and multiple targets. Reverse transcriptase-PCR is another detection method, but it requires knowledge of the fusion partners. To date, at least 14 ROS1 fusion partners have been reported, with CD74 being the most common.87 NGS with appropriate design and validation can also be used to detect ROS1 rearrangements.
For the case patient, the recommendation would be to start her on crizotinib at 250 mg twice daily. Monitoring for vision disturbance, gastrointestinal complaints, and edema is warranted. Because the estimated onset of response is around 7.9 weeks,86 plans should be made to repeat her scans in approximately 2 months.
BRAF V600E MUTATIONS
CASE PRESENTATION 4
A 71-year-old Caucasian man with a past medical history of hypertension, dyslipidemia, and ischemic cerebrovascular accident without residual deficits was diagnosed with stage IV adenocarcinoma of the lung about 8 months ago. He has a 40 pack-year smoking history and quit smoking when he was diagnosed with lung cancer. His disease burden involved a large mediastinal mass, scattered pleural nodules, multiple lymphadenopathy, and several soft tissue masses. His outside oncologist started him on chemotherapy containing carboplatin and pemetrexed for 6 cycles followed by maintenance pemetrexed. The most recent restaging scans show disease progression with enlarging soft tissue masses and several new lytic bone lesions. MRI-brain with and without contrast shows 2 subcentimeter enhancing lesions. He transferred care to you approximately 4 weeks ago. You ordered a repeat biopsy of 1 of the enlarging soft tissue masses. Molecular analysis revealed BRAF V600E mutation. In the interim, he underwent stereotactic radiosurgery for the 2 brain lesions without any complications. The patient is now in your clinic for follow up.
What would be your recommended systemic treatment?
TARGETED THERAPIES FOR BRAF V600E MUTATION
BRAF mutations were first recognized as activating mutations in advanced melanomas, with BRAF V600E, resulting from the substitution of glutamic acid for valine at amino acid 600, being the most common. BRAF plays an important role in the mitogen-activated protein kinase (MAPK) signaling pathway. Briefly, the activation of MAPK pathway occurs upon ligand binding of receptor tyrosine kinases, which then involves RAS/BRAF/MEK/ERK in a stepwise manner, ultimately leading to cell survival. BRAF mutations have been increasingly recognized also as driver mutations in NSCLC.9–12 They can be detected by PCR or NGS method. The characteristics of NSCLC patients harboring BRAF mutations have been described by various groups.9–12 For instance, 1 case series showed that the incidence was 2.2% among patients with advanced lung adenocarcinoma; 50% of mutations were V600E, while G469A and D594G accounted for the remaining 39% and 11% of patients, respectively. All patients were either current or former smokers. The median OS of patients with BRAF mutations in this case series was NR, while it was 37 months for patients with EGFR mutations (P = 0.73) and NR for patients with ALK rearrangements (P = 0.64).9
For patients with BRAF V600E–mutant NSCLC who have progressed on platinum-based chemotherapy, the combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) may represent a new treatment paradigm. This was illustrated in a phase 2, nonrandomized, open-label study. A total of 57 patients were enrolled and 36 patients (63.2% [95% CI 49.3% to 75.6%]) achieved an overall response by investigator assessment, the trial’s primary end point. Disease control rate was 78.9% (95% CI 66.1% to 88.6%), with 4% complete response, 60% partial response, and 16% stable disease. PFS was 9.7 months (95% CI [6.9 to 19.6 months]). The safety profile was comparable to what had been observed in patients with melanoma treated with this regimen. More specifically, 56% of patients on this trial reported serious AEs, including pyrexia (16%), anemia (5%), confusional state (4%), decreased appetite (4%), hemoptysis (4%), hypercalcemia (4%), nausea (4%), and cutaneous squamous cell carcinoma (4%). In addition, neutropenia (9%) and hyponatremia (7%) were the most common grade 3-4 AEs.16
The case patient has experienced disease progression after 1 line of platinum-based chemotherapy, so the combination of dabrafenib and trametinib would be a robust systemic treatment option. dabrafenib as a single agent has also been studied in BRAF V600E–mutant NSCLC in a phase 2 trial. The overall response by investigator assessment among 84 patients was 33% (95% CI 23% to 45%).14 Vemurafenib, another oral BRAF TKI, has demonstrated efficacy for NSCLC patients harboring BRAF V600E mutation. In the cohort of 20 patients with NSCLC, the response rate was 42% (95% CI 20% to 67%) and median PFS was 7.3 months (95% CI 3.5 to 10.8 months).13 Patients with non-V600E mutations have shown variable responses to targeted therapies. MEK TKIs may be considered in this setting; however, the details of this discussion are beyond the scope of this review.
CONCLUSION
The management of advanced NSCLC with driver mutations has seen revolutionary changes over the past decade. Tremendous research has been done in order to first understand the molecular pathogenesis of NSCLC and then discover driver mutations that would lead to development of targeted therapies with clinically significant efficacy as well as tolerability. More recently, increasing efforts have focused on how to conquer acquired resistance in patients with disease progression after first-line TKIs. The field of EGFR-mutant NSCLC has set a successful example, but the work is nowhere near finished. The goals are to search for more driver mutations and to design agents that could potentially block cell survival signals once and for all.
INTRODUCTION
Lung cancer is the second most common type of cancer in the United States, with 222,500 estimated new cases in 2017, according to the American Cancer Society.1 However, it is by far the number one cause of death due to cancer, with an estimated 155,870 lung cancer–related deaths occurring in 2017, which is higher than the number of deaths due to breast cancer, prostate cancer, and colorectal cancer combined.1,2 Despite slightly decreasing incidence and mortality over the past decade, largely due to smoking cessation, the 5-year survival rate of lung cancer remains dismal at approximately 18%.2–4
Non-small cell lung cancer (NSCLC) accounts for 80% to 85% of all lung cancer cases.4 Traditionally, it is further divided based on histology: adenocarcinoma, squamous cell carcinoma, large cell carcinoma, and not otherwise specified.5 Chemotherapy had been the cornerstone of treatment for stage IV NSCLC. It is not target-specific and is most effective against rapidly growing cells. Common adverse effects include alopecia, nausea/vomiting, myelosuppression, cardiotoxicity, neuropathy, and nephrotoxicity. However, this paradigm has shifted following the discovery of mutations of the epidermal growth factor receptor (EGFR) gene as an oncogenic driver that confers sensitivity to small molecule tyrosine kinase inhibitors (TKIs) targeting EGFR.6 The EGFR inhibitors are given orally and have a spectrum of toxicities (eg, such as rash, diarrhea, and elevated transaminases) different from that of systemic chemotherapy, which is often administered intravenously. Following the discovery of EGFR mutations, rearrangements of the anaplastic lymphoma kinase (ALK) gene7 and ROS1 gene8 were identified as targetable driver mutations in NSCLC. The frequency of both rearrangements is lower than that of EGFR mutations. Additionally, BRAF V600E mutation has been identified in NSCLC.9–12 This activation mutation is commonly seen in melanoma. Agents that have already been approved for the treatment of melanoma with the BRAF V600E mutation are being tested in NSCLC patients with this mutation.13–16
Given the effectiveness and tolerability of targeted therapy, identifying this distinct molecular subset of NSCLC patients is critical in treatment. Currently, molecular testing is mandatory in all stage IV patients with non-squamous cell carcinoma, as a preponderance of patients with driver mutations have this histology subtype.5,17–19 For patients with squamous cell carcinoma, molecular testing should be considered if the biopsy specimen is small, there is mixed histology, or the patient is a nonsmoker.5,20 Several techniques are commonly utilized in detecting these genetic alterations. EGFR mutation can be detected by polymerase chain reaction (PCR), ALK or ROS1 rearrangement can be detected by fluorescence in-situ hybridization (FISH), and immunohistochemistry (IHC) can also be used to detect ALK rearrangement. The current guideline is to use comprehensive genomic profiling to capture all the potential molecular targets simultaneously instead of running stepwise tests just for EGFR, ALK, and ROS1.5 BRAF V600E mutation,13–16 MET exon 14 skipping mutation,21–24 RET rearrangements,25–27 and HER2 mutations28–30 are among the emergent genetic alterations with various responses to targeted therapy.31 Some of these targeted agents have been approved for other types of malignancy, and others are still in the development phase.
Several initiatives worldwide have reported better outcomes of patients with driver mutations treated with targeted therapy. For instance, the Lung Cancer Mutation Consortium in the United States demonstrated that the median survival of patients without driver mutations, with drivers mutations but not treated with targeted therapy, and with driver mutations and treated with targeted therapy was 2.08 years, 2.38 years, and 3.49 years, respectively.32 The French Cooperative Thoracic Intergroup-French National Cancer Institute demonstrated that the median survival for patients with driver mutations versus those without driver mutations was 16.5 months versus 11.8 months.33 The Spanish Lung Cancer Group demonstrated that the overall survival (OS) for patients with EGFR mutations treated with erlotinib was 27 months.34 The mutations in lung cancer, their frequencies, and the downstream signaling pathways are depicted in the Figure.35
In this article, we discuss targeted therapy for patients with EGFR mutations, ALK rearrangements, ROS1 rearrangements, and BRAF V600E mutation. We also discuss the management of patients with EGFR mutations who develop a secondary mutation after TKI therapy. Almost all of the targeted agents discussed herein have been approved by the US Food and Drug Administration (FDA), so they are considered standard of care. All available phase 3 trials pertinent to these targeted therapies are included in the discussion.
EGFR MUTATIONS
CASE PRESENTATION 1
A 54-year-old Caucasian man who is a former smoker with a 10 pack-year history and past medical history of hypertension and dyslipidemia presents with progressive dyspnea for several weeks. A chest x-ray shows moderate pleural effusion on the left side with possible mass-like opacity on the left upper lung field. An ultrasound-guided thoracentesis is performed and cytology is positive for adenocarcinoma of likely pulmonary origin. Staging workup including positron emission tomography (PET)/computed tomography (CT) and magnetic resonance imaging of the brain with and without contrast is done. PET/CT shows a 5.5-cm mass in the left upper lobe of the lung with high fluorodeoxyglucose (FDG) uptake, several 1- to 2-cm mediastinal lymph nodes with moderate FDG uptake, and small pleural effusion on both sides with moderate FDG uptake. MRI-brain is negative for malignancy. The patient subsequently undergoes a CT-guided biopsy of the lung mass, which shows moderately differentiated adenocarcinoma. Comprehensive molecular profiling reveals EGFR L858R mutation only. The patient now presents for the initial consultation. Of note, his Eastern Cooperative Oncology Group performance status is 1.
What is the next step in the management of this patient?
FIRST-LINE TKI FOR SENSITIZING EGFR MUTATIONS
The 2 most common EGFR mutations are deletions in exon 19 and substitution of arginine for leucine in exon 21 (L858R), found in approximately 45% and 40% of patients with EGFR mutations, respectively.36 Both mutations are sensitive to EGFR TKIs. The benefit may be greater in patients with exon 19 deletions as compared to exon 21 L858R substitution,37,38 but this has not been demonstrated consistently in clinical trials.39-43 In the United States, EGFR mutations are found in approximately 10% of patients with NSCLC, while the incidence can be as high as 50% in Asia.44 Even though the cobas EGFR mutation test is the companion diagnostic approved by the US FDA, a positive test result from any laboratory with the Clinical Laboratory Improvement Amendments (CLIA) certificate should prompt the use of an EGFR TKI as the initial treatment.
Three EGFR TKIs that have been approved as first-line therapy in the United States are available: erlotinib, afatinib, and gefitinib.5 Both erlotinib and gefitinib are considered first-generation TKIs. They have higher binding affinity for the 2 common EGFR mutations than wild-type EGFR. In addition, they reversibly bind to the intracellular tyrosine kinase domain, resulting in inhibition of autophosphorylation of the tyrosine residues. Afatinib, a second-generation and irreversible TKI, targets EGFR (HER1) as well as HER2 and HER4.45
The superior efficacy of the EGFR TKIs over platinum doublet chemotherapy in treatment-naïve patients with EGFR mutations has been demonstrated in 7 randomized trials to date (Table).46 Erlotinib was the TKI arm for the OPTIMAL,41 EURTAC,42 and ENSURE trials;38 afatinib was the TKI arm for LUX-LUNG 337 and 6;43 gefitinib was the TKI arm for NEJ00239,47 and WJTOG3405.40 A meta-analysis of these 7 trials by Lee et al showed that progression-free survival (PFS) was significantly prolonged by EGFR TKIs (hazard ratio [HR] 0.37 [95% confidence interval {CI} 0.32 to 0.42]).46 For instance, in the EURTAC trial, median PFS was 9.7 months for patients treated with erlotinib as compared to 5.2 months for patients treated with platinum/gemcitabine or platinum/docetaxel.42 In this meta-analysis, prespecified subgroups included age, sex, ethnicity, smoking status, performance status, tumor histology, and EGFR mutation subtype. The superior outcome with TKIs was observed in all subgroups. Furthermore, patients with exon 19 deletions, nonsmokers, and women had even better outcomes.46
Erlotinib is the most commonly used TKI in the United States largely because gefitinib was off the market for some time until it was re-approved by the FDA in 2015. Interestingly, this “re-approval” was not based on either 1 of the 2 prospective trials (NEJ00239,47 and WJTOG340540), but rather was based on an exploratory analysis of the IPASS trial48,49 as well as a prospective phase 4, single-arm trial in Europe (IFUM).50 The superior efficacy of gefitinib over carboplatin/paclitaxel among patients with EGFR mutations in the IPASS trial was confirmed by blind independent central review, with longer PFS (HR 0.54 [95% CI 0.38 to 0.79] P = 0.0012) and higher objective response rate (ORR; odds ratio 3 [95% CI 1.63 to 5.54], P = 0.0004).49
CASE 1 CONTINUED
Based on the EGFR L858R mutation status, the patient is started on erlotinib. He is quite happy that he does not need intravenous chemotherapy but wants to know what toxicities he might potentially have with erlotinib.
What are the common adverse effects (AEs) of EGFR TKIs? How are AEs of TKIs managed?
Safety Profile
The important toxicities associated with EGFR TKIs are rash, gastrointestinal toxicity, hepatic toxicity, and pulmonary toxicity. Rash is an AE specific to all agents blocking the EGFR pathway, including small molecules and monoclonal antibodies such as cetuximab. The epidermis has a high level of expression of EGFR, which contributes to this toxicity.51 Rash usually presents as dry skin or acneiform eruption. Prophylactic treatment with oral tetracyclines and topical corticosteroids is generally recommended upon initiation of TKI therapy. Diarrhea is the most prevalent gastrointestinal toxicity. All patients starting treatment should be given prescriptions to manage diarrhea such as loperamide and be advised to call when it occurs. Hepatic toxicity is often manifested as elevated transaminases or bilirubin. Interstitial lung disease (ILD) is a rare but potentially fatal pulmonary toxicity.
Rash of any grade was reported in 49.2% of patients treated with erlotinib in clinical trials, while grade 3 rash occurred in 6% of patients and no grade 4 was reported. Diarrhea of any grade was reported in 20.3% of patients, grade 3 diarrhea occurred in 1.8%, and no grade 4 was reported. Grade 2 and 3 alanine aminotransferase (ALT) elevations were seen in 2% and 1% of patients, respectively. Grade 2 and 3 bilirubin elevations were seen in 4% and less than 1% of patients, respectively. The incidence of serious ILD-like events was less than 1%.52
Afatinib is associated with higher incidences of rash and diarrhea. Specifically, diarrhea and rash of all grades were reported in 96% and 90% of patients treated with afatinib, respectively. Paronychia of all grades occurred in 58% of patients. Elevated ALT of all grades was seen in 11% of patients. Approximately 1.5% of patients treated with afatinib across clinical trials had ILD or ILD-like AEs.53
Gefitinib, the most commonly used TKI outside United States, has a toxicity profile similar to erlotinib, except for hepatic toxicity. For instance, rash of all grades occurred in 47% of patients, diarrhea of all grades occurred in 29% of patients, and ILD or ILD-like AEs occurred in 1.3% of patients across clinical trials. In comparison, elevated ALT and aspartate aminotransferase (AST) of all grades was seen in 38% and 40% of patients, respectively.54 Therefore, close monitoring of liver function is clinically warranted. In particular, patients need to be advised to avoid concomitant use of herbal supplements, a common practice in Asian countries.
CASE 1 CONTINUED
The patient does well while on erlotinib at 150 mg orally once daily for about 8 months, until he develops increasing abdominal pain. A CT scan of the abdomen and pelvis with contrast shows a new 8-cm right adrenal mass. Additionally, a repeat CT scan of the chest with contrast shows a stable lung mass but enlarging mediastinal lymphadenopathy.
How would you manage the patient at this point?
MANAGEMENT OF T790M MUTATION AFTER PROGRESSION ON FIRST-LINE EGFR TKIS
As mentioned above, the median PFS of patients with EGFR mutations treated with 1 of the 3 TKIs is around 9 to 13 months.46 Of the various resistance mechanisms that have been described, the T790M mutation is found in approximately 60% of patients who progress after treatment with first-line TKIs.55,56 Other mechanisms, such as HER2 amplification, MET amplification, or rarely small cell transformation, have been reported.56 The first- and second-generation EGFR TKIs function by binding to the ATP-binding domain of mutated EGFR, leading to inhibition of the downstream signaling pathways (Figure, part B) and ultimately cell death.35 The T790M mutation hinders the interaction between the ATP-binding domain of EGFR kinase and TKIs, resulting in treatment resistance and disease progression.57,58
Osimertinib is a third-generation irreversible EGFR TKI with activity against both sensitizing EGFR and resistant T790M mutations. It has low affinity for wide-type EGFR as well as insulin receptor and insulin-like growth factor receptor.59 Osimertinib has been fully approved for NSCLC patients with EGFR mutations who have progressed on first-line EGFR TKIs with the development of T790M mutation. An international phase 3 trial (AURA3) randomly assigned 419 patients in a 2:1 ratio to either osimertinib or platinum/pemetrexed. Eligible patients all had the documented EGFR mutations and disease progression after first-line EGFR TKIs. Central confirmation of the T790M mutation was required. Median PFS by investigator assessment, the trial’s primary end point, was 10.1 months for osimertinib versus 4.4 months for chemotherapy (HR 0.3 [95% CI 0.23 to 0.41]; P < 0.001). ORR was 71% for osimertinib versus 31% for chemotherapy (HR 5.39 [95% CI 3.47 to 8.48], P < 0.001). A total of 144 patients with stable and asymptomatic brain metastases were also eligible. Median PFS for this subset of patients treated with osimertinib and chemotherapy was 8.5 months and 4.2 months, respectively (HR 0.32 [95% CI 0.21 to 0.49]). In the AURA3 trial, osimertinib was better tolerated than chemotherapy, with 23% of patients treated with osimertinib experiencing grade 3 or 4 AEs as compared to 47% of chemotherapy-treated patients. The most common AEs of any grade were diarrhea (41%), rash (34%), dry skin (23%), and paronychia (22%).60
For the case patient, a reasonable approach would be to obtain a tissue biopsy of the adrenal mass and more importantly to check for the T790M mutation. Similar to the companion diagnostic for EGFR mutations, the cobas EGFR mutation test v2 is the FDA-approved test for T790M. However, if this resistance mutation is detected by any CLIA-certified laboratories, osimertinib should be the recommended treatment option. If tissue biopsy is not feasible, plasma-based testing should be considered. A blood-based companion diagnostic also is FDA approved.
ALK REARRANGEMENTS
CASE 2 PRESENTATION
A 42-year-old Korean woman who is a non-smoker with no significant past medical history presents with fatigue, unintentional weight loss of 20 lb in the past 4 months, and vague abdominal pain. A CT can of the abdomen and pelvis without contrast shows multiple foci in the liver and an indeterminate nodule in the right lung base. She subsequently undergoes PET/CT, which confirms multiple liver nodules/masses ranging from 1 to 3 cm with moderate FDG uptake. In addition, there is a 3.5-cm pleura-based lung mass on the right side with moderate FDG uptake. MRI-brain with and without contrast is negative for malignancy. A CT-guided biopsy of 1 of the liver masses is ordered and pathology returns positive for poorly differentiated adenocarcinoma consistent with lung primary. Molecular analysis reveals an echinoderm microtubule-associated protein-like 4 (EML4)-ALK rearrangement. She is placed on crizotinib by an outside oncologist and after about 3 weeks of therapy is doing well. She is now in your clinic for a second opinion. She says that some of her friends told her about another medication called ceritinib and was wondering if she would need to switch her cancer treatment.
How would you respond to this patient’s inquiry?
FIRST-LINE TKIS FOR ALK REARRANGEMENTS
ALK rearrangements are found in 2% to 7% of NSCLC, with EML4-ALK being the most prevalent fusion variant.61 The inversion of chromosome 2p leads to the fusion of the EML4 gene and the ALK gene, which causes the constitutive activation of the fusion protein and ultimately increased transformation and tumorigenicity.7,61 Patients harboring ALK rearrangements tend to be non-smokers. Adenocarcinoma, especially signet ring cell subtype, is the predominant histology. Compared to EGFR mutations, patients with ALK mutations are significantly younger and more likely to be men.62 ALK rearrangements can be detected by either FISH or IHC, and most next-generation sequencing (NGS) panels have the ability to identify this driver mutation.
Crizotinib is the first approved ALK inhibitor for the treatment of NSCLC in this molecular subset of patients.63 PROFILE 1014 is a phase 3 randomized trial that compared crizotinib with chemotherapy containing platinum/pemetrexed for up to 6 cycles. Crossover to crizotinib was allowed for patients with disease progression on chemotherapy. The primary end point was PFS by independent radiologic review. The crizotinib arm demonstrated superior PFS (10.9 months versus 7 months; HR 0.45 [95% CI 0.35 to 0.6], P < 0.001) and ORR (74% versus 45%, P < 0.001). Median survival was not reached in either arm (HR 0.82 [95% CI 0.54 to 1.26], P = 0.36).64 Based on this international trial, crizotinib is considered standard of care in the United States for treatment-naïve patients with advanced NSCLC harboring ALK rearrangements. The current recommended dose is 250 mg orally twice daily. Common treatment-related AEs of all grades include vision disorder (62%), nausea (53%), diarrhea (43%), vomiting (40%), edema (28%), and constipation (27%).65 PROFILE 1007 compared crizotinib with pemetrexed or docetaxel in ALK-rearranged NSCLC patients with prior exposure to 1 platinum-based chemotherapy. The median PFS was 7.7 months for crizotinib as compared to 3 months for chemotherapy (HR 0.49 [95% CI 0.37 to 0.64], P < 0.001). The response rates were 65% and 20% for crizotinib and chemotherapy, respectively (P < 0.001).66 In other countries, crizotinib following 1 prior platinum-based regimen may be considered standard of care based on this trial.
Ceritinib is an oral second-generation ALK inhibitor that is 20 times more potent than crizotinib based on enzymatic assays.67 It also targets ROS1 and insulin-like growth factor 1 receptor but not c-MET. It was first approved by the FDA in April 2014 for metastatic ALK-rearranged NSCLC following crizotinib.68 In May 2017, the FDA granted approval of ceritinib for treatment-naïve patients. This decision was based on the results of the ASCEND-4 trial, a randomized phase 3 trial assessing the efficacy and safety of ceritinib over chemotherapy in the first-line setting. The trial assigned 376 patients to either ceritinib at 750 mg once daily or platinum/pemetrexed for 4 cycles followed by maintenance pemetrexed. Median PFS was 16.6 months for ceritinib versus 8.1 months for chemotherapy (HR 0.55 [95% CI 0.42 to 0.73]; P < 0.00001).69 Toxicities of ceritinib are not negligible, with gastrointestinal toxicity being the most prevalent. For instance, diarrhea, nausea, vomiting, abdominal pain, and constipation of all grades were seen in 86%, 80%, 60%, 54%, and 29% of patients, respectively. Furthermore, fatigue and decreased appetite occurred in 52% and 34% of patients, respectively. In terms of laboratory abnormalities, 84% of patients experienced decreased hemoglobin of all grades; 80% increased ALT; 75% increased AST; 58% increased creatinine; 49% increased glucose; 36% decreased phosphate; and 28% increased lipase. Due to these AEs, the incidence of dose reduction was about 58% and the median onset was around 7 weeks.70
Alectinib is another oral second-generation ALK inhibitor that was approved by the FDA in December 2015 for the treatment of NSCLC patients with ALK rearrangements who have progressed on or are intolerant to crizotinib.71 Its indication will soon be broadened to the first-line setting based on the ALEX trial.72 Alectinib is a potent and highly selective TKI of ALK73 with activity against known resistant mutations to crizotinib.74,75 It also inhibits RET but not ROS1 or c-MET.76 ALEX, a randomized phase 3 study, compared alectinib with crizotinib in treatment-naïve patients with NSCLC harboring ALK rearrangements. The trial enrolled 303 patients and the median follow-up was approximately 18 months. The alectinib arm (600 mg twice daily) demonstrated significantly higher PFS by investigator-assessment, the trial’s primary end point. The 12-month event-free survival was 68.4% (95% CI 61% to 75.9%) versus 48.7% (95% CI 40.4% to 56.9%) for alectinib and crizotinib, respectively (HR 0.47 [95% CI 0.34 to 0.65], P < 0.001). The median PFS was not reached in the alectinib arm (95% CI 17.7 months to not estimable) as compared to 11.1 months in the crizotinib arm (95% CI 9.1 to 13.1 months).72 Alectinib is generally well tolerated. Common AEs of all grades include fatigue (41%), constipation (34%), edema (30%), and myalgia (29%). As alectinib can cause anemia, lymphopenia, hepatic toxicity, increased creatine phosphokinase, hyperglycemia, electrolyte abnormalities, and increased creatinine, periodic monitoring of these laboratory values is important, although most of these abnormalities are grade 1 or 2.77
Brigatinib, another oral second-generation ALK inhibitor, was granted accelerated approval by the FDA in April 2017 for ALK-rearranged and crizotinib-resistant NSCLC based on the ALTA trial. This randomized phase 2 study of brigatinib showed an ORR by investigator assessment of 54% (97.5% CI 43% to 65%) in the 180 mg once daily arm with lead-in of 90 mg once daily for 7 days. Median PFS was 12.9 months (95% CI 11.1 months to not reached [NR]).78 Currently, a phase 3 study of brigatinib versus crizotinib in ALK inhibitor–naïve patients is recruiting participants (ALTA-1L). It will be interesting to see if brigatinib can achieve a front-line indication.
Starting the case patient on crizotinib is well within the treatment guidelines. One may consider ceritinib or alectinib in the first-line setting, but both TKIs can be reserved upon disease progression. We would recommend a repeat biopsy at that point to look for resistant mechanisms, as certain secondary ALK mutations may be rescued by certain next-generation ALK inhibitors. For instance, the F1174V mutation has been reported to confer resistance to ceritinib but sensitivity to alectinib, while the opposite is true for I1171T. The G1202R mutation is resistant to ceritinib, alectinib, and brigatinib, but lorlatinib, a third-generation ALK inhibitor, has shown activity against this mutation.79 Furthermore, brain metastasis represents a treatment challenge for patients with ALK rearrangements. It is also an efficacy measure of next-generation ALK inhibitors, all of which have demonstrated better central nervous system activity than crizotinib.69,78,80 If the case patient were found to have brain metastasis at the initial diagnosis, either ceritinib or alectinib would be a reasonable choice since crizotinib has limited penetration of blood-brain barrier.81
ROS1 REARRANGEMENTS
CASE PRESENTATION 3
A 66-year-old Chinese woman who is a non-smoker with a past medical history of hypertension and hypothyroidism presents to the emergency department for worsening lower back pain. Initial workup includes x-ray of the lumbar spine followed by MRI with contrast, which shows a soft tissue mass at L3-4 without cord compression. CT of the chest, abdomen, and pelvis with contrast shows a 7-cm right hilar mass, bilateral small lung nodules, mediastinal lymphadenopathy, and multiple lytic lesions in ribs, lumbar spine, and pelvis. MRI-brain with and without contrast is negative for malignancy. She undergoes endo-bronchial ultrasound and biopsy of the right hilar mass, which shows poorly differentiated adenocarcinoma. While waiting for the result of the molecular analysis, the patient undergoes palliative radiation therapy to L2-5 with good pain relief. She is discharged from the hospital and presents to your clinic for follow up. Molecular analysis now reveals ROS1 rearrangement with CD74-ROS1 fusion.
What treatment plan should be put in place for this patient?
FIRST-LINE THERAPY FOR ROS1 REARRANGEMENTS
Approximately 2.4% of lung adenocarcinomas harbor ROS1 rearrangements.82 This distinct genetic alteration occurs more frequently in NSCLC patients who are younger, female, and never-smokers, and who have adenocarcinomas.8 It has been shown that ROS1 rearrangements rarely overlap with other genetic alterations including KRAS mutations, EGFR mutations, and ALK rearrangements.83 As a receptor tyrosine kinase, ROS1 is similar to ALK and insulin receptor family members.84 Crizotinib, which targets ALK, ROS1, and c-MET, was approved by the FDA on March 11, 2016, for the treatment of metastatic ROS1-rearranged NSCLC.85 The approval was based on a phase 2 expansion cohort of the original phase 1 study. Among 50 US patients enrolled in this expansion cohort, 3 had complete responses and 33 had partial responses with ORR of 72% (95% CI 58% to 84%). Median PFS was 19.2 months (95% CI 14.4 months to NR) and median duration of response (DOR) was 17.6 months (95% CI 14.5 months to NR).86 During longer follow-up, independent radiology review confirmed high ORR of 66% and median DOR of 18.3 months.85
Interestingly, no companion diagnostic assay has been approved for the detection of ROS1 rearrangements with the approval of crizotinib. In the United States, break apart FISH is the most common detection method. In fact, in the above mentioned phase 2 study, ROS1 rearrangements were detected in 49 out of 50 patients by this method.86 FISH can be technically challenging when dealing with high volume and multiple targets. Reverse transcriptase-PCR is another detection method, but it requires knowledge of the fusion partners. To date, at least 14 ROS1 fusion partners have been reported, with CD74 being the most common.87 NGS with appropriate design and validation can also be used to detect ROS1 rearrangements.
For the case patient, the recommendation would be to start her on crizotinib at 250 mg twice daily. Monitoring for vision disturbance, gastrointestinal complaints, and edema is warranted. Because the estimated onset of response is around 7.9 weeks,86 plans should be made to repeat her scans in approximately 2 months.
BRAF V600E MUTATIONS
CASE PRESENTATION 4
A 71-year-old Caucasian man with a past medical history of hypertension, dyslipidemia, and ischemic cerebrovascular accident without residual deficits was diagnosed with stage IV adenocarcinoma of the lung about 8 months ago. He has a 40 pack-year smoking history and quit smoking when he was diagnosed with lung cancer. His disease burden involved a large mediastinal mass, scattered pleural nodules, multiple lymphadenopathy, and several soft tissue masses. His outside oncologist started him on chemotherapy containing carboplatin and pemetrexed for 6 cycles followed by maintenance pemetrexed. The most recent restaging scans show disease progression with enlarging soft tissue masses and several new lytic bone lesions. MRI-brain with and without contrast shows 2 subcentimeter enhancing lesions. He transferred care to you approximately 4 weeks ago. You ordered a repeat biopsy of 1 of the enlarging soft tissue masses. Molecular analysis revealed BRAF V600E mutation. In the interim, he underwent stereotactic radiosurgery for the 2 brain lesions without any complications. The patient is now in your clinic for follow up.
What would be your recommended systemic treatment?
TARGETED THERAPIES FOR BRAF V600E MUTATION
BRAF mutations were first recognized as activating mutations in advanced melanomas, with BRAF V600E, resulting from the substitution of glutamic acid for valine at amino acid 600, being the most common. BRAF plays an important role in the mitogen-activated protein kinase (MAPK) signaling pathway. Briefly, the activation of MAPK pathway occurs upon ligand binding of receptor tyrosine kinases, which then involves RAS/BRAF/MEK/ERK in a stepwise manner, ultimately leading to cell survival. BRAF mutations have been increasingly recognized also as driver mutations in NSCLC.9–12 They can be detected by PCR or NGS method. The characteristics of NSCLC patients harboring BRAF mutations have been described by various groups.9–12 For instance, 1 case series showed that the incidence was 2.2% among patients with advanced lung adenocarcinoma; 50% of mutations were V600E, while G469A and D594G accounted for the remaining 39% and 11% of patients, respectively. All patients were either current or former smokers. The median OS of patients with BRAF mutations in this case series was NR, while it was 37 months for patients with EGFR mutations (P = 0.73) and NR for patients with ALK rearrangements (P = 0.64).9
For patients with BRAF V600E–mutant NSCLC who have progressed on platinum-based chemotherapy, the combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) may represent a new treatment paradigm. This was illustrated in a phase 2, nonrandomized, open-label study. A total of 57 patients were enrolled and 36 patients (63.2% [95% CI 49.3% to 75.6%]) achieved an overall response by investigator assessment, the trial’s primary end point. Disease control rate was 78.9% (95% CI 66.1% to 88.6%), with 4% complete response, 60% partial response, and 16% stable disease. PFS was 9.7 months (95% CI [6.9 to 19.6 months]). The safety profile was comparable to what had been observed in patients with melanoma treated with this regimen. More specifically, 56% of patients on this trial reported serious AEs, including pyrexia (16%), anemia (5%), confusional state (4%), decreased appetite (4%), hemoptysis (4%), hypercalcemia (4%), nausea (4%), and cutaneous squamous cell carcinoma (4%). In addition, neutropenia (9%) and hyponatremia (7%) were the most common grade 3-4 AEs.16
The case patient has experienced disease progression after 1 line of platinum-based chemotherapy, so the combination of dabrafenib and trametinib would be a robust systemic treatment option. dabrafenib as a single agent has also been studied in BRAF V600E–mutant NSCLC in a phase 2 trial. The overall response by investigator assessment among 84 patients was 33% (95% CI 23% to 45%).14 Vemurafenib, another oral BRAF TKI, has demonstrated efficacy for NSCLC patients harboring BRAF V600E mutation. In the cohort of 20 patients with NSCLC, the response rate was 42% (95% CI 20% to 67%) and median PFS was 7.3 months (95% CI 3.5 to 10.8 months).13 Patients with non-V600E mutations have shown variable responses to targeted therapies. MEK TKIs may be considered in this setting; however, the details of this discussion are beyond the scope of this review.
CONCLUSION
The management of advanced NSCLC with driver mutations has seen revolutionary changes over the past decade. Tremendous research has been done in order to first understand the molecular pathogenesis of NSCLC and then discover driver mutations that would lead to development of targeted therapies with clinically significant efficacy as well as tolerability. More recently, increasing efforts have focused on how to conquer acquired resistance in patients with disease progression after first-line TKIs. The field of EGFR-mutant NSCLC has set a successful example, but the work is nowhere near finished. The goals are to search for more driver mutations and to design agents that could potentially block cell survival signals once and for all.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
- Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol 2016;893:1–19.
- Alberg AJ, Brock MV, Ford JG, et al. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013;143:e1S–29S.
- Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-3013, based on November 2015 SEER data submission, posted to the SEER website, April 2016. Bethesda (MD): National Cancer Institute; 2016.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Non-Small Cell Lung Cancer: 1–190.
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–39.
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6.
- Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancer. J Clin Oncol 2012;30:863–70.
- Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–51.
- Kinno T, Tsuta K, Shiraishi K, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 2014;25:138–42.
- Litvak AM, Paik PK, Woo KM, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 2014;9:1669–74.
- Villaruz LC, Socinski MA, Abberbock S, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015;121:448–56.
- Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–36.
- Planchard D, Kim TM, Mazieres J, et al. DaBRAFenib in patients with BRAF V600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:642–50.
- Gautschi O, Milia J, Cabarrou B, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol 2015;10:1451–7.
- Planchard D, Besse B, Groen HJ, et al. DaBRAFenib plus trametinib in patients with previously treated BRAF V600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984–93.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. J Thorac Oncol 2013;8:823–59.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. Arch Pathol Lab Med 2013;137:828–60.
- Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology guideline. J Clin Oncol 2014;32:3673–9.
- Paik PK, Varghese AM, Sima CS, et al. Response to erlotinib in patients with EGFR mutant advanced non-small cell lung cancers with a squamous or squamous-like component. Mol Cancer Ther 2012;11:2535–40.
- Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5:842–9.
- Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age, and stage-dependent MET genomic amplification, and c-MET overexpression. J Clin Oncol 2016;34:721–30.
- Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol 2016;11:1493–502.
- Reungwetwattana T, Liang Y, Zhu V, et al. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017;103:27–37.
- Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 2013;3:630–5.
- Lin JJ, Kennedy E, Sequist LV, et al. Clinical activity of alectinib in advanced RET-rearranged non-small cell lung cancer. J Thorac Oncol 2016;11:2027–32.
- Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 2016;17:1653–60.
- Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med 2006;354:2619–21.
- Mazieres J. Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targted drugs: results from the European EUHER2 cohort. Annal Oncol 2016;27:281–6.
- Ou SH, Schrock AB, Bocharov EV, et al. HER2 transmembrane (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 2017;12:446–57.
- Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emergent therapies. Cancer Discov 2017;7:596–609.
- Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006.
- Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016;387:1415–26.
- Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958–67.
- Rosell R, Karachaliou N, et al. Large-scale screening for somatic mutations in lung cancer. Lancet 2016;387:1354–6.
- Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005;97:339–46.
- Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327–34.
- Wu YL, Chou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol 2015;26:1883–9.
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Eng J Med 2010;362:2380–8.
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121–8.
- Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735–42.
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239–46.
- Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:213–22.
- Hirsch FR, Bunn PA Jr. EGFR testing in lung cancer is ready for prime time. Lancet Oncol 2009;10:432–3.
- Nelson V, Ziehr J, Aqulnik M, et al. Afatinib: emerging next-generation tyrosine kinase inhibitor for NSCLC. Onco Targets Ther 2013;5:135–43.
- Lee CK, Wu YL, Ding PN, et al. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: a meta-analysis. J Clin Oncol 2015;33:1958–65.
- Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol 2013;24:54–9.
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57.
- Wu YL, Saijo N, Thongprasert S, et al. Efficacy according to blind independent central review: post-hoc analyses from the phase III, randomized, multicenter, IPASS study of first-line gefitinib versus carboplatin/paclitaxel in Asian patients with EFGR mutation-positive advanced NSCLC. Lung Cancer 2017;104:119–25.
- Douillard JY, Ostoros G, Cobo M, et al. First-line gefitinib in Caucasian EGFR-mutation positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer 2014;110:55–62.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol 2007;56:317–26.
- Tarceva [package insert]. South San Francisco (CA): Genentech, Inc; 2010. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021743s14s16lbl.pdf. Accessed April 23, 2017.
- Gilotrif [package insert.] Ridgefield (CT): Boehringer Ingelheim, Inc; 2013. www.accessdata.fda.gov/drugsatfda_docs/label/2013/201292s000lbl.pdf. Accessed April 23, 2017.
- Iressa [package insert]. Wilmington (DE): AstraZeneca, Inc; 2015. Error! Hyperlink reference not valid. Accessed April 23, 2017.
- Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616–22.
- Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR mutant lung cancers. Clin Cancer Res 2013;19:2240–7.
- Yun CH, Mengwasser KE, Tom AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5.
- Sos ML, Rode HB, Heynck S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res 2010;70:868–74.
- Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T190M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046–61.
- Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629–40.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N Engl J Med 2010;363:1693–703.
- Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247–53.
- Kazandjian D, Blumenthal GM, Chen HY, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014;19:e5–11.
- Solomon BJ, Mok T, Kim DW, et al. First-ling crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167–77.
- Xalkori [package insert]. New York: Pfizer, Inc; 2011. www.accessdata.fda.gov/drugsatfda_docs/label/2012/202570s002lbl.pdf. Accessed April 23, 2017.
- Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385–94.
- Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013;56:5675–90.
- Khozin S, Blumenthal GM, Zhang L, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res 2015;21:2436–9.
- Soria JC, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 2017;389:917–29.
- Zykadia [package insert]. East Hanover (NJ): Novartis Pharmaceuticals Corporation, Inc; 2016. www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/zykadia.pdf. Accessed April 23, 2017.
- Larkins E, Blumenthal GM, Chen H, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res 2016;22:5171–6.
- Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. New Engl J Med 2017 June 6 [Epub ahead of print].
- Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem 2012;20:1271–80.
- Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679–90.
- Kodama T, Tsukaguchi T, Yoshida M, et al. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett 2014;351:215–21.
- Kodama T, Tsukaguchi T, Satoh T, et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther 2014;13:2910–8.
- Alecensa [package insert]. South San Francisco (CA): Genentech, Inc; 2015. www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf. Accessed April 23, 2017.
- Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol 2017 May 5 [Epub ahead of print].
- Zhu V, Ou SH. Safety of alectinib for the treatment of metastatic ALK-rearranged non-small cell lung cancer. Expert Opin Drug Saf 2017;16:509–14.
- Gadgeel SM, Shaw AT, Govindan R, et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small cell lung cancer. J Clin Oncol 2016;34:4079–85.
- Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29:e443–5.
- Zhu Q, Zhan P, Zhang X, et al. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res 2015;4:300–9.
- Lin JJ, Ritterhouse LL, Ali SM, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol 2017;12:872–7.
- Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta 2009;1795:37–52.
- Kazandjian D, Blumenthal G, Luo L, et al. Benefit-Risk summary of crizotinib for the treatment of patients with ROS1 alteration-positive metastatic NSCLC. Oncologist 2016;21:974–80.
- Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014;371:1963–71.
- Zhu VW, Upadhyay D, Schrock AB, et al. TPD52L1-ROS1, a new ROS1 fusion variant in lung adenosquamous cell carcinoma identified by comprehensive genomic profiling. Lung Cancer 2016;97:48–50.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
- Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol 2016;893:1–19.
- Alberg AJ, Brock MV, Ford JG, et al. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013;143:e1S–29S.
- Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-3013, based on November 2015 SEER data submission, posted to the SEER website, April 2016. Bethesda (MD): National Cancer Institute; 2016.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Non-Small Cell Lung Cancer: 1–190.
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–39.
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6.
- Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancer. J Clin Oncol 2012;30:863–70.
- Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–51.
- Kinno T, Tsuta K, Shiraishi K, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 2014;25:138–42.
- Litvak AM, Paik PK, Woo KM, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 2014;9:1669–74.
- Villaruz LC, Socinski MA, Abberbock S, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015;121:448–56.
- Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–36.
- Planchard D, Kim TM, Mazieres J, et al. DaBRAFenib in patients with BRAF V600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:642–50.
- Gautschi O, Milia J, Cabarrou B, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol 2015;10:1451–7.
- Planchard D, Besse B, Groen HJ, et al. DaBRAFenib plus trametinib in patients with previously treated BRAF V600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984–93.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. J Thorac Oncol 2013;8:823–59.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. Arch Pathol Lab Med 2013;137:828–60.
- Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology guideline. J Clin Oncol 2014;32:3673–9.
- Paik PK, Varghese AM, Sima CS, et al. Response to erlotinib in patients with EGFR mutant advanced non-small cell lung cancers with a squamous or squamous-like component. Mol Cancer Ther 2012;11:2535–40.
- Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5:842–9.
- Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age, and stage-dependent MET genomic amplification, and c-MET overexpression. J Clin Oncol 2016;34:721–30.
- Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol 2016;11:1493–502.
- Reungwetwattana T, Liang Y, Zhu V, et al. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017;103:27–37.
- Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 2013;3:630–5.
- Lin JJ, Kennedy E, Sequist LV, et al. Clinical activity of alectinib in advanced RET-rearranged non-small cell lung cancer. J Thorac Oncol 2016;11:2027–32.
- Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 2016;17:1653–60.
- Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med 2006;354:2619–21.
- Mazieres J. Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targted drugs: results from the European EUHER2 cohort. Annal Oncol 2016;27:281–6.
- Ou SH, Schrock AB, Bocharov EV, et al. HER2 transmembrane (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 2017;12:446–57.
- Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emergent therapies. Cancer Discov 2017;7:596–609.
- Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006.
- Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016;387:1415–26.
- Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958–67.
- Rosell R, Karachaliou N, et al. Large-scale screening for somatic mutations in lung cancer. Lancet 2016;387:1354–6.
- Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005;97:339–46.
- Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327–34.
- Wu YL, Chou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol 2015;26:1883–9.
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Eng J Med 2010;362:2380–8.
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121–8.
- Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735–42.
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239–46.
- Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:213–22.
- Hirsch FR, Bunn PA Jr. EGFR testing in lung cancer is ready for prime time. Lancet Oncol 2009;10:432–3.
- Nelson V, Ziehr J, Aqulnik M, et al. Afatinib: emerging next-generation tyrosine kinase inhibitor for NSCLC. Onco Targets Ther 2013;5:135–43.
- Lee CK, Wu YL, Ding PN, et al. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: a meta-analysis. J Clin Oncol 2015;33:1958–65.
- Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol 2013;24:54–9.
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57.
- Wu YL, Saijo N, Thongprasert S, et al. Efficacy according to blind independent central review: post-hoc analyses from the phase III, randomized, multicenter, IPASS study of first-line gefitinib versus carboplatin/paclitaxel in Asian patients with EFGR mutation-positive advanced NSCLC. Lung Cancer 2017;104:119–25.
- Douillard JY, Ostoros G, Cobo M, et al. First-line gefitinib in Caucasian EGFR-mutation positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer 2014;110:55–62.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol 2007;56:317–26.
- Tarceva [package insert]. South San Francisco (CA): Genentech, Inc; 2010. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021743s14s16lbl.pdf. Accessed April 23, 2017.
- Gilotrif [package insert.] Ridgefield (CT): Boehringer Ingelheim, Inc; 2013. www.accessdata.fda.gov/drugsatfda_docs/label/2013/201292s000lbl.pdf. Accessed April 23, 2017.
- Iressa [package insert]. Wilmington (DE): AstraZeneca, Inc; 2015. Error! Hyperlink reference not valid. Accessed April 23, 2017.
- Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616–22.
- Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR mutant lung cancers. Clin Cancer Res 2013;19:2240–7.
- Yun CH, Mengwasser KE, Tom AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5.
- Sos ML, Rode HB, Heynck S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res 2010;70:868–74.
- Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T190M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046–61.
- Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629–40.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N Engl J Med 2010;363:1693–703.
- Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247–53.
- Kazandjian D, Blumenthal GM, Chen HY, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014;19:e5–11.
- Solomon BJ, Mok T, Kim DW, et al. First-ling crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167–77.
- Xalkori [package insert]. New York: Pfizer, Inc; 2011. www.accessdata.fda.gov/drugsatfda_docs/label/2012/202570s002lbl.pdf. Accessed April 23, 2017.
- Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385–94.
- Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013;56:5675–90.
- Khozin S, Blumenthal GM, Zhang L, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res 2015;21:2436–9.
- Soria JC, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 2017;389:917–29.
- Zykadia [package insert]. East Hanover (NJ): Novartis Pharmaceuticals Corporation, Inc; 2016. www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/zykadia.pdf. Accessed April 23, 2017.
- Larkins E, Blumenthal GM, Chen H, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res 2016;22:5171–6.
- Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. New Engl J Med 2017 June 6 [Epub ahead of print].
- Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem 2012;20:1271–80.
- Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679–90.
- Kodama T, Tsukaguchi T, Yoshida M, et al. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett 2014;351:215–21.
- Kodama T, Tsukaguchi T, Satoh T, et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther 2014;13:2910–8.
- Alecensa [package insert]. South San Francisco (CA): Genentech, Inc; 2015. www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf. Accessed April 23, 2017.
- Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol 2017 May 5 [Epub ahead of print].
- Zhu V, Ou SH. Safety of alectinib for the treatment of metastatic ALK-rearranged non-small cell lung cancer. Expert Opin Drug Saf 2017;16:509–14.
- Gadgeel SM, Shaw AT, Govindan R, et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small cell lung cancer. J Clin Oncol 2016;34:4079–85.
- Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29:e443–5.
- Zhu Q, Zhan P, Zhang X, et al. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res 2015;4:300–9.
- Lin JJ, Ritterhouse LL, Ali SM, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol 2017;12:872–7.
- Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta 2009;1795:37–52.
- Kazandjian D, Blumenthal G, Luo L, et al. Benefit-Risk summary of crizotinib for the treatment of patients with ROS1 alteration-positive metastatic NSCLC. Oncologist 2016;21:974–80.
- Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014;371:1963–71.
- Zhu VW, Upadhyay D, Schrock AB, et al. TPD52L1-ROS1, a new ROS1 fusion variant in lung adenosquamous cell carcinoma identified by comprehensive genomic profiling. Lung Cancer 2016;97:48–50.
Targeted Therapy and Immunotherapy in the Treatment of Metastatic Cutaneous Melanoma
INTRODUCTION
The incidence of cutaneous melanoma has increased over the past 2 decades, with SEER estimates indicating that the number of new cases of melanoma diagnosed annually rose from 38,300 in 1996 to 76,000 in 2016.1 Among persons younger than 50 years, the incidence is higher in females, and younger women (aged 15–39 years) are especially vulnerable.2 Among persons older than 50, melanoma incidence in men is nearly twice that of women, in whom melanomas are often thicker and often associated with worse outcomes.1,2 Approximately 85% of melanomas are diagnosed at early stages when surgery is curative, but the lifetime probability of developing invasive disease is 3% in men and 2% in women.
Prior to the advent of effective immunotherapies and targeted therapies, melanoma was often managed with chemotherapy, which had dismal response rates and commensurately poor outcomes. Advances in the understanding of the molecular etiopathogenesis and immune escape responses of cutaneous metastatic melanoma have transformed therapeutic approaches. Specifically, improved understanding of the genetic mutations driving melanoma tumorigenesis coupled with insights into mechanisms of tumor-mediated immune evasion resulted in development of inhibitors of mitogen-activated protein kinases (MAPK; BRAF and MEK) along with inhibitors of negative regulatory immune checkpoints (cytotoxic T lymphocyte–associated antigen 4 [CTLA-4] and programmed cell death-1 [PD-1]). In this review, we discuss the role of immune therapy, targeted therapy, and combinations of these in the treatment of metastatic cutaneous melanoma. We limit the immuno-therapy discussion to approved CTLA-4/PD-1 inhibitors and the targeted therapy discussion to approved BRAF/NRAS/MEK inhibitors and do not discuss non-checkpoint immunotherapies including cytokines (HD IL-2), vaccines, or adoptive T-cell approaches. Interested readers are directed to other excellent works covering these important topics.26–29
DEVELOPMENT OF TARGETED AND NOVEL IMMUNE THERAPIES
For many years the degree of ultraviolet (UV) light exposure was considered the sole major risk factor for melanoma oncogenesis, even though its mechanism was largely unknown.3 However, clinical observations regarding the occurrence of melanoma on less exposed areas (trunk and limbs) in individuals with intermittent sun exposure led to the proposition that melanomas that arose in younger patients with intermittent sun exposure were distinct from melanomas that arose in older patients in association with markers of chronic sun exposure—the “divergent pathway” hypothesis.3 Critical to this understanding were whole-exome sequencing data from multiple groups, including The Cancer Genome Atlas, that identified patterns of mutations in oncogenic drivers that were distinct in patients with and without chronically sun-damaged (CSD) skin.4–7 It is now clear that based on its association with CSD skin, melanoma can be subclassified into CSD or non-CSD melanoma. CSD and non-CSD melanoma have distinct clinico-pathological characteristics and are associated with different driver mutations. CSD melanomas typically arise in older patients on sun-exposed areas (head/neck, dorsal surfaces of distal extremities) and are associated with particular driver mutations (BRAF non-V600E, NRAS, NF1, or KIT) and genetic signatures of UV-induced DNA damage (G > T [UVA] or C > T [UVB]) transitions. Conversely, non-CSD melanomas typically arise in younger (< 55 years) patients on intermittently sun-exposed areas (trunk, proximal extremities) and are associated with BRAF V600E/K driver mutations and often lack genetic signatures of UV mutagenesis.
Identification of driver mutations in components of the MAPK pathway, including BRAF and NRAS, facilitated the development of targeted inhibitors. The BRAF inhibitors vemurafenib and dabrafenib have been shown in pivotal phase 3 studies to significantly improve overall and progression-free survival in patients with metastatic melanoma compared with chemotherapy and garnered regulatory approval (vemurafenib, BRIM-3;8,9 dabrafenib, BREAK-310). Concomitant MEK and BRAF inhibition extends the duration of benefit by preventing downstream kinase activation in the MAPK pathway. Notably, concomitant MEK inhibition alters the side-effect profile of BRAF inhibitors, with reduced incidence of keratoacanthomas and cutaneous squamous cell carcinomas that are attributable to on-target, off-tumor effects of BRAF inhibitors. Combined BRAF and MEK inhibition (vemurafenib/cobimetinib and dabrafenib/trametinib) further improved overall and progression-free survival compared to single-agent BRAF inhibition in phase 3 studies (COMBI-d,11 COMBI-v,12 and coBRIM13). Although often deep, the responses seen with the use of targeted kinase inhibitors are not often durable, with the vast majority of patients progressing after 12 to 15 months of therapy.In parallel, work primarily done in murine models of chronic viral infection uncovered the role played by co-inhibitory or co-excitatory immune checkpoints in mediating T-cell immune responses. These efforts clarified that tumor-mediated immune suppression primarily occurs through enhancement of inhibitory signals via the negative T-cell immune checkpoints CTLA-4 or PD-1.14,15 Blockade of negative T-cell immune checkpoints resulted in activation of the adaptive immune system, resulting in durable anti-tumor responses as demonstrated in studies of the CTLA-4 inhibitor ipilimumab (CA184-02016 and CA184-02417) and the PD-1 inhibitors nivolumab (CA209-003,18 CheckMate 037,19 and CheckMate 06620) and pembrolizumab (KEYNOTE-00121 and KEYNOTE-00622). Compared to the deep but short-lived responses seen with targeted kinase inhibitors, patients treated with CTLA-4 or PD-1 immune checkpoint blockade often developed durable responses that persisted even after completion of therapy. Combined CTLA-4 and PD-1 blockade results in greater magnitude of response with proportionately increased toxicity.23–25
IMMUNOTHERAPY
CTLA-4 AND PD-1 IMMUNE CHECKPOINT INHIBITORS
The novel success of immunotherapy in recent decades is largely attributable to improved understanding of adaptive immune physiology, specifically T-cell activation and regulation. T-cell activation requires 2 independent signaling events: it is initiated upon recognition of the antigen-MHC class II-receptor complex on antigen-presenting cells (APC), and requires a secondary co-stimulatory interaction of CD80/CD86 (B7.1/B7.2) on APCs and CD28 molecule on T-cells; without this second event, T-cells enter an anergic state.30–32 Upon successful signaling and co-stimulation, newly activated T-cells upregulate CTLA-4, which can bind to B7 molecules with a nearly 100-fold greater affinity than CD28.33,34 Unlike CD28, CTLA-4 engagement negatively regulates T-cell activation. The opposing signals produced by CD28 and CTLA-4 are integrated by the T-cell to determine eventual response to activation, and provide a means by which T-cell activation is homeostatically regulated to prevent exaggerated physiologic immune responses.35 It was hypothesized that CTLA-4 blockade would permit T-cell activation, which is thwarted in the tumor microenvironment by tumor-mediated CTLA-4 engagement, thereby unleashing an anti-tumor immune response.36
PD-1 is a member of the CD28 and CTLA-4 immunoglobulin super family and, similar to CTLA-4, binds activated T-cells. PD-1 has 2 ligands on activated T-cells: PD-L1 and PD-L2.37 PD-L1 is constitutively expressed by a variety of immune and non-immune cells, particularly in inflammatory environments including tumor microenvironments, in response to the release of inflammatory cytokines such as interferon (IFN)-γ.37,38 Conversely, PD-L2 is only minimally expressed constitutively, although its expression on immune and non-immune cells can be induced by similar cues from inflammatory microenvironments. PD-L1 and PD-L2 cross-compete for binding to PD-1, with PD-L2 exhibiting 2- to 6-fold greater relative affinity than PD-L1.39 PD-L1/PD-1 binding results in phosphorylation of 2 tyrosinases in the intracellular portion of PD-1, which contains immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM). PD-1 ITSM subsequently recruits either of 2 SH2-domain–containing protein tyrosine phosphatases: SHP-1 and SHP-2. SHP-2 signaling suppresses PI3K/Akt activation, down-regulates Bcl-xL, and suppresses expression of multiple transcription factors that mediate T-cell effector function including GATA-3, Eomes, and T-bet.40–42 The net effect of PD-L1/PD-1 engagement is to suppress T-cell proliferation, cytokine production, cytolytic function, and survival. Unlike CTLA-4, which primarily affects the priming phase of naive T-cell activation, PD-1 chiefly regulates the effector phase of T-cell function. Furthermore, because PD-L1/PD-L2 expression is limited to inflammatory microenvironments, the effects of PD-1 are less generalized than those of CTLA-4.
SINGLE AGENT ACTIVITY OF CTLA-4 AND PD-1 INHIBITORS
Ipilimumab (MDX-010) is a human IgG1 monoclonal antibody shown to inhibit CTLA-4.43 Early studies tested different formulations (transfectoma-derived and hybridoma-derived), doses, and schedules of ipilimumab primarily in patients with advanced refractory melanoma.44–46 Although responses were infrequent, responding patients experienced durable remissions at 1- and 2-year time points. Notably, in a foreshadowing of changes to response criteria used to evaluate these agents, several treated patients who initially had radiographically stable disease upon completion of therapy subsequently experienced a gradual decline in tumor burden.
Ipilimumab was subsequently evaluated in 2 phase 3 trials. The first study (MDX010-020/CA184-020), which involved 676 HLA-A*0201–positive patients with advanced melanoma, compared ipilimumab 3 mg/kg every 3 weeks for 4 doses either singly or in combination with gp100 vaccine with a gp100-only control arm.16 Ipilimumab administration resulted in objective responses in 11% of patients and improved progression-free and overall survival compared to gp100 alone. Of note, ipilimumab monotherapy was superior to ipilimumab/gp100 combination, possibly related to timing of vaccine in relation to ipilimumab. A confirmatory study (CA184-024) compared a higher dose of ipilimumab (10 mg/kg) in combination with dacarbazine to dacarbazine monotherapy in previously untreated melanoma and was positive.17 Given the lack of augmented efficacy with the higher (10 mg/kg) dose, ipilimumab received regulatory approval in 2011 for the treatment of melanoma at the lower dose: 3 mg/kg administered every 3 weeks for 4 doses (Table 1). Survival data was strikingly similar to patterns observed in prior phase 2 studies, with survival curves plateauing after 2 years at 23.5% to 28.5% of treated patients. Pooled survival data from prospective and retrospective studies of ipilimumab corroborate the plateau of 22% (26% treated; 20% untreated) reached at year 3 regardless of prior therapy or ipilimumab dose, underscoring the durability of long-term survival in ipilimumab-treated patients.47 Ipilimumab administration resulted in an unusual spectrum of toxicities including diarrhea, rash, hepatitis, and hypophysitis (termed immune-related adverse events, or irAEs) in up to a third of patients.
Pembrolizumab and nivolumab are humanized IgG4 monoclonal antibodies that target the PD-1 receptor found on activated T cells, B cells, and myeloid cells. Pembrolizumab and nivolumab are engineered similarly: by immunizing transgenic mice with recombinant human PD-1-Fc protein and subsequently screening murine splenic cells fused with myeloma cells for hybridomas producing antibodies reactive to PD-1-Fc.48,49 Unlike IgG1, the IgG4 moiety neither engages Fc receptors nor activates complement, avoiding cytotoxic effects of the antibody upon binding to the T cells that it is intended to activate. Both pembrolizumab and nivolumab bind PD-1 with high affinity and specificity, effectively inhibiting the interaction between PD-1 and ligands PD-L1 and PD-L2.
Nivolumab was first studied in a phase 1 study (CA209-003) of 296 patients with advanced cancers who received 1, 3, or 10 mg/kg administered every 2 weeks.18 Histologies tested included melanoma, non–small-cell lung cancer (NSCLC), renal-cell cancer (RCC), castration-resistant prostate cancer (CRPC), and colorectal cancer (CRC). Responses were seen in melanoma and RCC and unusually in NSCLC, including in both squamous and non-squamous tumors. Objective responses were noted in 41% of the 107 melanoma patients treated at 3 mg/kg. Survival was improved, with 1- and 2-year survival rates of 62% and 43% at extended follow up.50
Subsequently, nivolumab was compared to chemotherapy in a pair of phase 3 studies involving both previously untreated (Checkmate 066) and ipilimumab/BRAF inhibitor–refractory (CheckMate 037) patients.19,20 In both studies, nivolumab produced durable responses in 32% to 34% of patients and improved survival over chemotherapy. Compared to ipilimumab, the incidence of irAEs was much lower with nivolumab. The depth and magnitude of responses observed led to regulatory approval for nivolumab in both indications (untreated and ipilimumab/BRAF inhibitor–treated melanoma) in 2014. Data from both studies are summarized in Table 1.
Pembrolizumab was first evaluated in a phase 1 study of 30 patients with a variety of solid organ malignancies in which no dose-limiting toxicities were observed and no defined maximal tolerated dose was reached.51 Per protocol, maximal administered dose was 10 mg/kg every 2 weeks. Following startling responses including 2 complete responses of long duration, pembrolizumab was evaluated in a large phase 1 study (KEYNOTE-001) of 1260 patients that evaluated 3 doses (10 mg/kg every 2 weeks, 10 mg/kg every 3 weeks, and 2 mg/kg every 3 weeks) in separate melanoma and NSCLC substudies.21 Both ipilimumab-naïve and ipilimumab-treated patients were enrolled in the melanoma substudy. Objective responses were seen in 38% ofpatients across all 3 dosing schedules and were similar in both ipilimumab-naïve and ipilimumab-treated patients. Similar to nivolumab, most responders experienced durable remissions.
Pembrolizumab was subsequently compared to ipilimumab in untreated patients (KEYNOTE-006) in which patients were randomly assigned to receive either ipilimumab or pembrolizumab at 1 of 2 doses: 10 mg/kg every 2 weeks and pembrolizumab 10 mg/kg every 3 weeks.22 Response rates were greater with pembrolizumab than ipilimumab, with commensurately greater 1-year survival rates. Rates of treatment-related adverse events requiring discontinuation of study drug were much lower with pembrolizumab than ipilimumab. This trial was instrumental in proving the superior profile of pembrolizumab over ipilimumab. The US Food and Drug Administration (FDA) granted pembrolizumab accelerated approval for second-line treatment of melanoma in 2014, and updated this to include a first-line indication in 2015 (Table 1).
EFFICACY OF COMBINED CTLA-4 AND PD-1 INHIBITION
Preclinical studies demonstrated that PD-1 blockade was more effective than CTLA-4 blockade and combination PD-1/CTLA-4 blockade was synergistic, with complete rejection of tumors in approximately half of the treated animals.14 This hypothesis was evaluated in a phase 1 study that explored both concurrent and sequential combinations of ipilimumab and nivolumab along with increasing doses of both agents in PD-1/CTLA-4–naïve advanced melanoma.23 Responses were greater in the concurrent arm (40%) than in the sequential arm (20%) across dose-levels with a small fraction of patients treated in the concurrent arm experiencing a profound reduction (80%) in tumor burden.
The superiority of ipilimumab/nivolumab combination to ipilimumab monotherapy was demonstrated in a randomized blinded phase 2 study (CheckMate 069).24 Of the 4 different ipilimumab/nivolumab doses explored in the phase 1 study (3 mg/kg and 0.3 mg/kg, 3 mg/kg and 1 mg/kg, 1 mg/kg and 3 mg/kg, 3 mg/kg and 3 mg/kg), ipilimumab 3 mg/kg and nivolumab 1 mg/kg (followed by nivolumab 3 mg/kg) was compared to ipilimumab and nivolumab-matched placebo. Responses were significantly greater with dual PD-1/CTLA-4 blockade compared to CTLA-4 blockade alone (59% versus 11%). Concurrently, a 3-arm randomized phase 3 study compared the same dose of ipilimumab/nivolumab to ipilimumab and nivolumab in previously untreated advanced melanoma (CheckMate 067).25 Similar to CheckMate 069, CheckMate 067 demonstrated that ipilimumab/nivolumab combination resulted in more profound responses (58%) than either ipilimumab (19%) or nivolumab (44%) alone. Toxicity, primarily diarrhea, fatigue, pruritus, and rash, was considerable in the combination arm (55% grade 3/4 adverse events) and resulted in treatment discontinuation in 30% of patients. The profound and durable responses observed led to accelerated approval of ipilimumab/nivolumab combination in 2015 (Table 1).
Efforts to improve the toxicity/benefit ratio of ipilimumab/nivolumab combination have centered around studying lower doses and/or extended dosing schedules of ipilimumab, including ipilimumab 1 mg/kg every 6 or 12 weeks with nivolumab dosed at 3 mg/kg every 2 weeks or 480 mg every 4 weeks. Promising data from a first-line study in NSCLC (CheckMate 012) support the evaluation of nivolumab in combination with lower-dosed ipilimumab (1 mg/kg every 6 or 12 weeks).52 This approach is being tested against platinum doublet chemotherapy in a confirmatory phase 3 study in NSCLC (CheckMate 227).
TARGETED THERAPY
MAPK KINASE PATHWAY IN MELANOMA TUMORIGENESIS
The MAPK pathway mediates cellular responses to growth signals. RAF kinases are central mediators in the MAPK pathway and exert their effect primarily through MEK phosphorylation and activation following dimerization (hetero- or homo-) of RAF molecules. As a result, RAF is integral to multiple cellular processes, including transcriptional regulation, cellular differentiation, and cell proliferation. MAPK pathway activation is a common event in many cancers, primarily due to activating mutations in BRAF or RAS. Alternatively, MAPK pathway activation can occur in the absence of activating mutations in BRAF or NRAS through down-regulation of MAPK pathway inhibitory proteins (RAF-1 inhibitory protein or SPRY-2), C-MET overexpression, or activating mutations in non-BRAF/NRAS kinases including CRAF, HRAS, and NRAS.53,54
Somatic point mutations in BRAF are frequently observed (37%–50%) in malignant melanomas and at lower frequency in a range of human cancers including NSCLC, colorectal cancer, papillary thyroid cancer, ovarian cancer, glioma, and gastrointestinal stromal tumor.6,55,56 BRAF mutations in melanoma typically occur within the activation segment of the kinase domain (exon 15). Between 80% and 90% of activating mutations result in an amino acid substitution of glutamate (E) for valine (V) at position 600: V600E.57,58 V600E mutations are true oncogenic drivers, resulting in increased kinase activity with demonstrable transformational capacity in vitro. BRAF mutations are usually mutually exclusive, with tumors typically containing no other driver mutations in NRAS, KIT, NF1, or other genes.
NRAS mutations are less common than BRAF mutations, having a reported frequency of 13% to 25% in melanoma.4 NRAS mutations generally occur within the P-loop region of the G domain (exon 2), or less commonly in the switch II region of the G domain (exon 3). Most NRAS exon 2 mutations comprise amino acid substitutions at position 61 from glutamine (Q) to arginine (R; 35%), lysine (K; 34%) and less often to glutamate (E), leucine (L), or proline (P). Preclinical data suggest that NRAS mutations paradoxically stimulate the MAPK pathway and thus enhance tumor growth in vitro.59,60 Several important phenotypic differences distinguish NRAS- from BRAF-mutated melanoma. NRAS-mutated tumors are typically associated with increasing age and CSD skin, while BRAF-mutated tumors arise in younger patients in non-CSD skin. A large population-based study suggested that NRAS-mutated melanomas were associated with mitoses and lower tumor infiltrating lymphocytes (TIL) grade, and arose in anatomic sites other than the head/neck, while BRAF-mutated tumors were associated with mitoses and superficial spreading histology.61 Although the lower TIL grade seen with NRAS-mutated melanomas suggests a more immunosuppressed microenvironment and argues for poorer responses to immune therapies, clinical studies comparing responses to immunotherapies in various categories of driver mutations provide conflicting results for the prognostic role of NRAS mutations in relation to immune checkpoint blockade and other immune therapies.62–64
NF1 represents the third known driver in cutaneous melanoma, with mutations reported in 12% of cases.6,7 NF1 encodes neurofibromin, which has GTPase activity and regulates RAS proteins; NF1 loss results in increased RAS.65 Unlike BRAF or NRAS, which are usually mutually exclusive, NF1 mutations in melanoma can occur singly or in combination with either BRAF or NRAS mutations. In these settings, NF1 mutations are associated with RAS activation, MEK-dependence, and resistance to RAF inhibition.66
MAPK PATHWAY INHIBITION SINGLY AND IN COMBINATION
Although multiple MEK 1/2 inhibitors (AS703026, AZD8330/ARRY-704, AZD6244, CH5126766, CI-1040, GSK1120212, PD0325901, RDEA119, and XL518) and RAF inhibitors (ARQ 680, GDC-0879, GSK2118436, PLX4032, RAF265, sorafenib, XL281/BMS-908662) were developed, the initial evaluation of MAPK pathway inhibitors in advanced human cancers began with CI-1040. Preclinical data suggested that CI-1040 potently and selectively inhibited both MEK1 and MEK2, but phase 1 and 2 human trial results were disappointing, likely because these trials were not selectively enriched for NRAS/BRAF–mutated tumors or cancers in which these oncogenic mutations were most commonly detected, such as melanoma.67,68 The subsequent evaluation of selumetinib (AZD6244/ARRY-142886) in a phase 2 study was also negative. Although investigators enrolled a presumably enriched population (cutaneous melanoma), the incidence of NRAS/BRAF–mutated tumors was not ascertained to determine this, but rather assumed, which led to a discrepancy between the assumed (prestudy) and observed (on-study) proportions of BRAF/NRAS mutations that was not accounted for in power calculations.69,70 Lessons learned from these earlier misadventures informed the current paradigm of targeted therapy development: (1) identification of a highly specific and potent inhibitor through high-throughput screening; (2) establishment of maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) in unselected patients; (3) confirmation of RP2D in selected tumor types enriched for target of interest; and (4) confirmatory study against standard comparator to seek regulatory approval.
Vemurafenib and dabrafenib were evaluated in this tiered fashion in phase 1 dose-finding studies comprising unselected patients, followed by phase 2 studies in advanced BRAF V600E–mutated melanoma. Both were subsequently evaluated in randomized phase 3 trials (vemurafenib, BRIM-38; dabrafenib, BREAK-310) that compared them with dacarbazine (1000 mg/m2 intravenously every 3 weeks) in the treatment of advanced BRAF V600E–mutated melanoma. Response kinetics for both agents were remarkably similar: single-agent BRAF inhibitors resulted in rapid (time to response 2–3 months), profound (approximately 50% objective responses) reductions in tumor burden that lasted 6 to 7 months. Adverse events common to both agents included rash, fatigue, and arthralgia, although clinically significant photosensitivity was more common with vemurafenib and clinically significant pyrexia was more common with dabrafenib. Class-specific adverse events included the development of cutaneous squamous-cell carcinomas and keratoacanthomas secondary to paradoxical activation of MAPK pathway signaling either through activating mutations in HRAS or mutations or amplifications in receptor tyrosine kinases upstream of BRAF, resulting in elevated levels of RAS–guanosine triphosphate complexes.71 Results of these studies resulted in regulatory approval of single-agent BRAF inhibitors for the treatment of BRAF V600E (and later V600K)–mutated melanoma (vemurafenib in 2011; dabrafenib in 2013). Details regarding trial populations, study interventions, efficacy, and adverse events are summarized in Table 2.
Responses to BRAF inhibitors are typically profound but temporary. Mechanisms of acquired resistance are diverse and include reactivation of MAPK pathway–dependent signaling (RAS activation or increased RAF expression), and development of MAPK pathway–independent signaling (COT overexpression; increased PI3K or AKT signaling) that permits bypass of inhibited BRAF signaling within the MAPK pathway.72–76 These findings suggested that upfront inhibition of both MEK and mutant BRAF may produce more durable responses than BRAF inhibition alone. Three pivotal phase 3 studies established the superiority of combination BRAF and MEK inhibition over BRAF inhibition alone: COMBI-d11 (dabrafenib/trametinib versus dabrafenib/placebo), COMBI-v12 (dabrafenib/trametinib versus vemurafenib), and coBRIM13 (vemurafenib/cobimetinib versus vemurafenib/placebo). As expected, compared to BRAF inhibitor monotherapy, combination BRAF and MEK inhibition produced greater responses and improved progression-free and overall survival (Table 2). Interestingly, the rate of cutaneous squamous-cell carcinomas was much lower with combination therapy, reflecting the more profound degree of MAPK pathway inhibition achieved with combination BRAF and MEK inhibition. Based on these results, FDA approval was granted for both dabrafenib/trametinib and vemurafenib/cobimetinib combinations in 2015. Although the dabrafenib/trametinib combination was only approved in 2015, trametinib had independently gained FDA approval in 2013 for the treatment of BRAF V600E/K–mutated melanoma on the basis of the phase 3 METRIC study.77
Encorafenib (LGX818) and binimetinib (MEK162, ARRY-162, ARRY-438162) are new BRAF and MEK inhibitors currently being evaluated in clinical trials. Encorafenib/binimetinib combination was first evaluated in a phase 3 study (COLUMBUS) that compared it with vemurafenib monotherapy in BRAF-mutant melanoma.78 Unsurprisingly, encorafenib/binimetinib combination produced greater and more durable responses compared to vemurafenib monotherapy. The median progression-free survival of the encorafenib/binimetinib combination (14.9 months) was greater than vemurafenib monotherapy (7.3 months) in this study, and intriguingly greater than that seen with vemurafenib/cobimetinib (coBRIM 9.9 months) and dabrafenib/trametinib (COMBI-d 9.3 months; COMBI-v 11.4 months). Of note, although encorafenib has an IC50 midway between dabrafenib and vemurafenib in cell-free assays (0.8 nM dabrafenib, 4 nM encorafenib, and 31 nM vemurafenib), it has an extremely slower off-rate from BRAF V600E, which results in significantly greater target inhibition in cells following drug wash-out.79 This may account for the significantly greater clinical benefit seen with encorafenib/binimetinib in clinical trials. Final study data are eagerly awaited. Regulatory approval has been sought, and is pending at this time.
Binimetinib has been compared to dacarbazine in a phase 3 study (NEMO) of patients with NRAS-mutant melanoma, most of whom had been previously treated with immunotherapy.80 Response rates were low in both arms, although slightly greater with binimetinib than dacarbazine (15% versus 9%), commensurate with a modest improvement in progression-free survival. FDA approval has been sought and remains pending at this time.
KIT INHIBITION SINGLY AND IN COMBINATION
The KIT receptor protein tyrosine kinase is a transmembrane protein consisting of extracellular and intracellular domains. Activating KIT mutations occur in 2% to 8% of all melanoma patients and may be found in all melanoma subtypes but are commonest in acral melanomas (10%–20%) and mucosal melanomas (15%–20%). Activating KIT mutations primarily occur in exons 11 and 13, which code for the juxtamembrane and kinase domains, respectively.5,81–83
Imatinib mesylate is a tyrosine kinase inhibitor of the 2-phenyl amino pyrimidine class that occupies the tyrosine kinase active site with resultant blocking of tyrosine kinase activity. Imatinib mesylate is known to block KIT and has been extensively studied in patients with gastrointestinal stromal tumors (GIST), 80% of whom harbor KIT mutations, in both the adjuvant and the metastatic settings. In melanoma, imatinib mesylate was studied in a Chinese open-label, phase 2 study of imatinib mesylate monotherapy in metastatic melanoma harboring KIT mutation or amplification; 25% of the study patients had mucosal disease and the rest had cutaneous disease, with acral involvement in 50% of all patients.84 Overall response rate was 23%, while 51% of patients remained alive at 1 year with no differences in response rate and/or survival being noted between patients with either KIT mutations or amplifications. In a separate study of imatinib mesylate at 400 mg daily or 400 mg twice daily in Caucasian patients with KIT-mutated/amplified melanoma, similar response and survival rates were reported, although patients with KIT mutations did nonsignificantly better than those with KIT amplifications.85
Other novel studies evaluating KIT inhibitors include KIT inhibition in combination with the VEGF inhibitor bevacizumab and a study of selective BCR-ABL kinase inhibitor nilotinib in imatinib-resistant melanoma. In the former phase 1/2 study, Flaherty and colleagues studied imatinib 800 mg daily and bevacizumab at 10 mg/kg every 2 weeks in 63 patients with advanced tumors, including 23 with metastatic melanoma. Although the combination was relatively nontoxic, no significant efficacy signal was seen and further accrual to the phase 2 portion was halted after the first stage was completed.86 Nilotinib is a BCR-ABL1 tyrosine kinase inhibitor intelligently designed based on the structure of the ABL-imatinib complex that is 10 to 30 times more potent than imatinib in inhibiting BCR-ABL1 tyrosine kinase activity. Nilotinib is approved for the treatment of imatinib-resistant chronic myelogenous leukemia (CML), with reported efficacy in patients with central nervous system (CNS) involvement.87,88 Nilotinib has been studied in a single study of KIT-mutated/amplified melanoma that included patients with imatinib-resistance and those with treated CNS disease. Nilotinib appeared to be active in imatinib-resistant melanoma, although no responses were seen in the CNS disease cohort.89 Overall, the response rates observed with KIT inhibition in melanoma are much lower than those observed in CML and GIST.
CONCLUSION AND FUTURE DIRECTIONS
Prior to 2011, the only approved agents for the treatment of advanced melanoma were dacarbazine and high-dose interleukin-2. Since 2011, drug approvals in melanoma have proceeded at a frenetic pace unmatched in any other disease. The primary events underlying this are advances in our understanding of the gene mutation landscape driving melanoma tumorigenesis, accompanied by insights into the means by which tumors circumvent the induction of effective anti-tumor T-cell responses. These insights have resulted in the development of inhibitors targeting MAPK pathway kinases BRAF, MEK, and NRAS), KIT, and regulatory immune checkpoints (CTLA-4 and PD-1). Although BRAF/MEK inhibition results in profound reductions and even occasional complete responses in patients, these responses are typically short lived, rarely lasting more than 9 to 11 months; the encorafenib/binimetinib combination may improve that duration marginally. However, the signature therapeutic advance in melanoma of the past decade is immunotherapy, particularly the development of inhibitors of CTLA-4 and PD-1 immune checkpoints. With these agents, significant proportions of treated patients remain free of progression off-therapy (ipilimumab 23%; nivolumab 34%; pembrolizumab 35%; ipilimumab/nivolumab 64%), and some patients can be successfully re-induced after delayed progression. Separately, the high response rates observed with the use of KIT inhibitors in CML and GIST have not been observed in KIT mutated/amplified melanoma and development of agents in this space has been limited. The challenges ahead center around identifying predictive biomarkers and circumventing primary or acquired resistance, with the eventual goal of producing durable remissions in the majority of treated patients.
Our improved understanding of the mechanisms of acquired resistance to BRAF/MEK inhibitors suggests that anti-tumor activity may be achieved by targeting multiple pathways, possibly with combination regimens comprising other inhibitors and/or immunotherapy. Preclinical data supports the use of combination strategies targeting both ERK and PI3K/mTOR to circumvent acquired resistance.90 Ongoing studies are evaluating combinations with biguanides (metformin: NCT02143050 and NCT01638676; phenformin: NCT03026517), HSP90 inhibitors (XL888: NCT02721459; AT13387: NCT02097225), and decitabine (NCT01876641).
One complexity affecting management of resistance in the targeted therapy landscape remains tumor heterogeneity, particularly intra- and intertumoral heterogeneity, which may explain the apparent contradiction between continued efficacy of BRAF inhibitors in BRAF-resistant tumors and preclinical data predicting slower progression of resistant tumors on cessation of BRAF inhibitors.91–94 These data provide a rationale to investigate intermittent dosing regimens with BRAF/MEK inhibitors; several studies exploring this approach are ongoing (NCT01894672 and NCT02583516).
Given the specificity, adaptability, and memory response associated with immunotherapy, it is likely that these agents will be used to treat the majority of patients regardless of mutational status. Hence, identifying predictive biomarkers of response to immune checkpoint inhibitors is vital. The presence of CD8+ T-cell infiltrate and IFN-γ gene signature, which indicate an “inflamed” tumor microenvironment, are highly predictive of clinical benefit from PD-1 inhibitors.95,96 However, not all PD-1 responders have “inflamed” tumor microenvironments, and not all patients with an “inflamed” tumor microenvironment respond to immune checkpoint inhibitors. The complexity of the immune system is reflected in the multiple non-redundant immunologic pathways, both positive and negative, with checkpoints and ligands that emerge dynamically in response to treatment. Given the dynamic nature of the immune response, it is unlikely that any single immunologic biomarker identified pre-treatment will be completely predictive. Rather, the complexity of the biomarker approach must match the complexity of the immune response elicited, and will likely incorporate multifarious elements including CD8+ T-cell infiltrate, IFN-γ gene signature, and additional elements including microbiome, genetic polymorphisms, and tumor mutation load. The goal is to use multiple markers to guide development of combinations and then, depending on initial response, to examine tumors for alterations to guide decisions about additional treatment(s) to improve responses, with the eventual goal being durable clinical responses for all patients.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Guy GP, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982-2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6.
- Anderson WF, Pfeiffer RM, Tucker MA, Rosenberg PS. Divergent cancer pathways for early-onset and late-onset cutaneous malignant melanoma. Cancer 2009;115:4176–85.
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135–47.
- Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340–6.
- Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63.
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681–96.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 2014;15:323–32.
- Hauschild A, Grob J-J, Demidov LV, et al. DaBRAFenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined daBRAFenib and trametinib. N Engl J Med 2015;372:30–9.
- Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76.
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107:4275–80.
- Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577–81.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54.
- Weber JS, Angelo SP D’, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134–44.
- Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33.
- Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372:2006–17.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
- Eggermont AMM. Therapeutic vaccines in solid tumours: can they be harmful? Eur J Cancer 2009;45:2087–90.
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012;4:127ps8.
- Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol 2014;192:5451–8.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62–8.
- Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992;356:607–9.
- Greenfield EA, Nguyen KA, Kuchroo VK. CD28/B7 costimulation: a review. Crit Rev Immunol 1998;18:389–418.
- Sharpe AH, Abbas AK. T-cell costimulation--biology, therapeutic potential, and challenges. N Engl J Med 2006;355:973–5.
- Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001;19:565–94.
- Collins AV, Brodie DW, Gilbert RJC, et al. The interaction properties of costimulatory molecules revisited. Immunity 2002;17:201–10.
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459–65.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734–6.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704.
- Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 2002;169:5538–45.
- Youngnak P, Kozono Y, Kozono H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun 2003;307:672–7.
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945–54.
- Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005;25:9543–53.
- Riley JL. PD-1 signaling in primary T cells. Immunol Rev 2009;229:114–25.
- Wolchok JD, Hodi FS, Weber JS, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013;1291:1–13.
- Weber JS, O’Day S, Urba W, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol 2008;26:5950–6.
- Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A 2008;105:3005–10.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 2010;11:155–64.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–94.
- Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2014;2:846–56.
- Poole RM. Pembrolizumab: first global approval. Drugs 2014;74:1973–81.
- Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30.
- Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21:4286–93.
- Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18:31–41.
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am 2009;23:529–45, ix.
- Satyamoorthy K, Li G, Gerrero MR, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res 2003;63:756–9.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54.
- Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14.
- Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010;8:67.
- Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS ONE 2012;7:e35309.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431–5.
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30.
- Thomas NE, Edmiston SN, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncology 2015;1:359–68.
- Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother 2012;35:66–72.
- Johnson DB, Lovly CM, Flavin M, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015;3:288–95.
- Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;4:959–67.
- Kiuru M, Busam KJ. The NF1 gene in tumor syndromes and melanoma. Lab Invest 2017;97:146–57.
- Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 2014;74:2340–50.
- Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol 2005;23:5281–93.
- Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004;22:4456–62.
- Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res 2012;18:555–67.
- Davar D, Kirkwood JM. CCR 20th anniversary commentary: MAPK/ERK pathway inhibition in melanoma-kinase inhibition redux. Clin Cancer Res 2015;21:5412–4.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207–15.
- Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010;468:968–72.
- Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7.
- Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov 2014;4:69–79.
- Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80–93.
- Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4:94–109.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14.
- Dummer R, Ascierto PA, Gogas HJ, et al. Results of COLUMBUS Part 1: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in BRAF-mutant melanoma. Presented at Society for Melanoma Research 2016 Congress. November 6-9, 2016. Boston (MA).
- Adelmann CH, Ching G, Du L, et al. Comparative profiles of BRAF inhibitors: the paradox index as a predictor of clinical toxicity. Oncotarget 2016;7:30453–60.
- Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45.
- Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol 2005;36:486–93.
- Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 2008;14:6821–8.
- Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res 2010;23:210–5.
- Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 2011;29:2904–9.
- Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 2013;31:3182–90.
- Flaherty KT, Hamilton BK, Rosen MA, et al. Phase I/II trial of imatinib and bevacizumab in patients with advanced melanoma and other advanced cancers. Oncologist 2015;20:952–9.
- Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia 2013;27:107–12.
- Reinwald M, Schleyer E, Kiewe P, et al. Efficacy and pharmacologic data of second-generation tyrosine kinase inhibitor nilotinib in BCR-ABL-positive leukemia patients with central nervous system relapse after allogeneic stem cell transplantation. Biomed Res Int 2014;2014:637059.
- Carvajal RD, Lawrence DP, Weber JS, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res 2015;21:2289–96.
- Carlino MS, Todd JR, Gowrishankar K, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol 2014;8:544–54.
- Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther 2013;12:1332–42.
- Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer 2014;120:3142–53.
- Thakur M Das, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–5.
- Thakur M Das, Stuart DD. Molecular pathways: response and resistance to BRAF and MEK inhibitors in BRAF(V600E) tumors. Clin Cancer Res 2014;20:1074–80.
- Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71.
- Ayers M, Lunceford J, Nebozhyn M, et al. Relationship between immune gene signatures and clinical response to PD-1 blockade with pembrolizumab (MK-3475) in patients with advanced solid tumors. J Immunotherapy Cancer 2015;3(Suppl 2):P80.
INTRODUCTION
The incidence of cutaneous melanoma has increased over the past 2 decades, with SEER estimates indicating that the number of new cases of melanoma diagnosed annually rose from 38,300 in 1996 to 76,000 in 2016.1 Among persons younger than 50 years, the incidence is higher in females, and younger women (aged 15–39 years) are especially vulnerable.2 Among persons older than 50, melanoma incidence in men is nearly twice that of women, in whom melanomas are often thicker and often associated with worse outcomes.1,2 Approximately 85% of melanomas are diagnosed at early stages when surgery is curative, but the lifetime probability of developing invasive disease is 3% in men and 2% in women.
Prior to the advent of effective immunotherapies and targeted therapies, melanoma was often managed with chemotherapy, which had dismal response rates and commensurately poor outcomes. Advances in the understanding of the molecular etiopathogenesis and immune escape responses of cutaneous metastatic melanoma have transformed therapeutic approaches. Specifically, improved understanding of the genetic mutations driving melanoma tumorigenesis coupled with insights into mechanisms of tumor-mediated immune evasion resulted in development of inhibitors of mitogen-activated protein kinases (MAPK; BRAF and MEK) along with inhibitors of negative regulatory immune checkpoints (cytotoxic T lymphocyte–associated antigen 4 [CTLA-4] and programmed cell death-1 [PD-1]). In this review, we discuss the role of immune therapy, targeted therapy, and combinations of these in the treatment of metastatic cutaneous melanoma. We limit the immuno-therapy discussion to approved CTLA-4/PD-1 inhibitors and the targeted therapy discussion to approved BRAF/NRAS/MEK inhibitors and do not discuss non-checkpoint immunotherapies including cytokines (HD IL-2), vaccines, or adoptive T-cell approaches. Interested readers are directed to other excellent works covering these important topics.26–29
DEVELOPMENT OF TARGETED AND NOVEL IMMUNE THERAPIES
For many years the degree of ultraviolet (UV) light exposure was considered the sole major risk factor for melanoma oncogenesis, even though its mechanism was largely unknown.3 However, clinical observations regarding the occurrence of melanoma on less exposed areas (trunk and limbs) in individuals with intermittent sun exposure led to the proposition that melanomas that arose in younger patients with intermittent sun exposure were distinct from melanomas that arose in older patients in association with markers of chronic sun exposure—the “divergent pathway” hypothesis.3 Critical to this understanding were whole-exome sequencing data from multiple groups, including The Cancer Genome Atlas, that identified patterns of mutations in oncogenic drivers that were distinct in patients with and without chronically sun-damaged (CSD) skin.4–7 It is now clear that based on its association with CSD skin, melanoma can be subclassified into CSD or non-CSD melanoma. CSD and non-CSD melanoma have distinct clinico-pathological characteristics and are associated with different driver mutations. CSD melanomas typically arise in older patients on sun-exposed areas (head/neck, dorsal surfaces of distal extremities) and are associated with particular driver mutations (BRAF non-V600E, NRAS, NF1, or KIT) and genetic signatures of UV-induced DNA damage (G > T [UVA] or C > T [UVB]) transitions. Conversely, non-CSD melanomas typically arise in younger (< 55 years) patients on intermittently sun-exposed areas (trunk, proximal extremities) and are associated with BRAF V600E/K driver mutations and often lack genetic signatures of UV mutagenesis.
Identification of driver mutations in components of the MAPK pathway, including BRAF and NRAS, facilitated the development of targeted inhibitors. The BRAF inhibitors vemurafenib and dabrafenib have been shown in pivotal phase 3 studies to significantly improve overall and progression-free survival in patients with metastatic melanoma compared with chemotherapy and garnered regulatory approval (vemurafenib, BRIM-3;8,9 dabrafenib, BREAK-310). Concomitant MEK and BRAF inhibition extends the duration of benefit by preventing downstream kinase activation in the MAPK pathway. Notably, concomitant MEK inhibition alters the side-effect profile of BRAF inhibitors, with reduced incidence of keratoacanthomas and cutaneous squamous cell carcinomas that are attributable to on-target, off-tumor effects of BRAF inhibitors. Combined BRAF and MEK inhibition (vemurafenib/cobimetinib and dabrafenib/trametinib) further improved overall and progression-free survival compared to single-agent BRAF inhibition in phase 3 studies (COMBI-d,11 COMBI-v,12 and coBRIM13). Although often deep, the responses seen with the use of targeted kinase inhibitors are not often durable, with the vast majority of patients progressing after 12 to 15 months of therapy.In parallel, work primarily done in murine models of chronic viral infection uncovered the role played by co-inhibitory or co-excitatory immune checkpoints in mediating T-cell immune responses. These efforts clarified that tumor-mediated immune suppression primarily occurs through enhancement of inhibitory signals via the negative T-cell immune checkpoints CTLA-4 or PD-1.14,15 Blockade of negative T-cell immune checkpoints resulted in activation of the adaptive immune system, resulting in durable anti-tumor responses as demonstrated in studies of the CTLA-4 inhibitor ipilimumab (CA184-02016 and CA184-02417) and the PD-1 inhibitors nivolumab (CA209-003,18 CheckMate 037,19 and CheckMate 06620) and pembrolizumab (KEYNOTE-00121 and KEYNOTE-00622). Compared to the deep but short-lived responses seen with targeted kinase inhibitors, patients treated with CTLA-4 or PD-1 immune checkpoint blockade often developed durable responses that persisted even after completion of therapy. Combined CTLA-4 and PD-1 blockade results in greater magnitude of response with proportionately increased toxicity.23–25
IMMUNOTHERAPY
CTLA-4 AND PD-1 IMMUNE CHECKPOINT INHIBITORS
The novel success of immunotherapy in recent decades is largely attributable to improved understanding of adaptive immune physiology, specifically T-cell activation and regulation. T-cell activation requires 2 independent signaling events: it is initiated upon recognition of the antigen-MHC class II-receptor complex on antigen-presenting cells (APC), and requires a secondary co-stimulatory interaction of CD80/CD86 (B7.1/B7.2) on APCs and CD28 molecule on T-cells; without this second event, T-cells enter an anergic state.30–32 Upon successful signaling and co-stimulation, newly activated T-cells upregulate CTLA-4, which can bind to B7 molecules with a nearly 100-fold greater affinity than CD28.33,34 Unlike CD28, CTLA-4 engagement negatively regulates T-cell activation. The opposing signals produced by CD28 and CTLA-4 are integrated by the T-cell to determine eventual response to activation, and provide a means by which T-cell activation is homeostatically regulated to prevent exaggerated physiologic immune responses.35 It was hypothesized that CTLA-4 blockade would permit T-cell activation, which is thwarted in the tumor microenvironment by tumor-mediated CTLA-4 engagement, thereby unleashing an anti-tumor immune response.36
PD-1 is a member of the CD28 and CTLA-4 immunoglobulin super family and, similar to CTLA-4, binds activated T-cells. PD-1 has 2 ligands on activated T-cells: PD-L1 and PD-L2.37 PD-L1 is constitutively expressed by a variety of immune and non-immune cells, particularly in inflammatory environments including tumor microenvironments, in response to the release of inflammatory cytokines such as interferon (IFN)-γ.37,38 Conversely, PD-L2 is only minimally expressed constitutively, although its expression on immune and non-immune cells can be induced by similar cues from inflammatory microenvironments. PD-L1 and PD-L2 cross-compete for binding to PD-1, with PD-L2 exhibiting 2- to 6-fold greater relative affinity than PD-L1.39 PD-L1/PD-1 binding results in phosphorylation of 2 tyrosinases in the intracellular portion of PD-1, which contains immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM). PD-1 ITSM subsequently recruits either of 2 SH2-domain–containing protein tyrosine phosphatases: SHP-1 and SHP-2. SHP-2 signaling suppresses PI3K/Akt activation, down-regulates Bcl-xL, and suppresses expression of multiple transcription factors that mediate T-cell effector function including GATA-3, Eomes, and T-bet.40–42 The net effect of PD-L1/PD-1 engagement is to suppress T-cell proliferation, cytokine production, cytolytic function, and survival. Unlike CTLA-4, which primarily affects the priming phase of naive T-cell activation, PD-1 chiefly regulates the effector phase of T-cell function. Furthermore, because PD-L1/PD-L2 expression is limited to inflammatory microenvironments, the effects of PD-1 are less generalized than those of CTLA-4.
SINGLE AGENT ACTIVITY OF CTLA-4 AND PD-1 INHIBITORS
Ipilimumab (MDX-010) is a human IgG1 monoclonal antibody shown to inhibit CTLA-4.43 Early studies tested different formulations (transfectoma-derived and hybridoma-derived), doses, and schedules of ipilimumab primarily in patients with advanced refractory melanoma.44–46 Although responses were infrequent, responding patients experienced durable remissions at 1- and 2-year time points. Notably, in a foreshadowing of changes to response criteria used to evaluate these agents, several treated patients who initially had radiographically stable disease upon completion of therapy subsequently experienced a gradual decline in tumor burden.
Ipilimumab was subsequently evaluated in 2 phase 3 trials. The first study (MDX010-020/CA184-020), which involved 676 HLA-A*0201–positive patients with advanced melanoma, compared ipilimumab 3 mg/kg every 3 weeks for 4 doses either singly or in combination with gp100 vaccine with a gp100-only control arm.16 Ipilimumab administration resulted in objective responses in 11% of patients and improved progression-free and overall survival compared to gp100 alone. Of note, ipilimumab monotherapy was superior to ipilimumab/gp100 combination, possibly related to timing of vaccine in relation to ipilimumab. A confirmatory study (CA184-024) compared a higher dose of ipilimumab (10 mg/kg) in combination with dacarbazine to dacarbazine monotherapy in previously untreated melanoma and was positive.17 Given the lack of augmented efficacy with the higher (10 mg/kg) dose, ipilimumab received regulatory approval in 2011 for the treatment of melanoma at the lower dose: 3 mg/kg administered every 3 weeks for 4 doses (Table 1). Survival data was strikingly similar to patterns observed in prior phase 2 studies, with survival curves plateauing after 2 years at 23.5% to 28.5% of treated patients. Pooled survival data from prospective and retrospective studies of ipilimumab corroborate the plateau of 22% (26% treated; 20% untreated) reached at year 3 regardless of prior therapy or ipilimumab dose, underscoring the durability of long-term survival in ipilimumab-treated patients.47 Ipilimumab administration resulted in an unusual spectrum of toxicities including diarrhea, rash, hepatitis, and hypophysitis (termed immune-related adverse events, or irAEs) in up to a third of patients.
Pembrolizumab and nivolumab are humanized IgG4 monoclonal antibodies that target the PD-1 receptor found on activated T cells, B cells, and myeloid cells. Pembrolizumab and nivolumab are engineered similarly: by immunizing transgenic mice with recombinant human PD-1-Fc protein and subsequently screening murine splenic cells fused with myeloma cells for hybridomas producing antibodies reactive to PD-1-Fc.48,49 Unlike IgG1, the IgG4 moiety neither engages Fc receptors nor activates complement, avoiding cytotoxic effects of the antibody upon binding to the T cells that it is intended to activate. Both pembrolizumab and nivolumab bind PD-1 with high affinity and specificity, effectively inhibiting the interaction between PD-1 and ligands PD-L1 and PD-L2.
Nivolumab was first studied in a phase 1 study (CA209-003) of 296 patients with advanced cancers who received 1, 3, or 10 mg/kg administered every 2 weeks.18 Histologies tested included melanoma, non–small-cell lung cancer (NSCLC), renal-cell cancer (RCC), castration-resistant prostate cancer (CRPC), and colorectal cancer (CRC). Responses were seen in melanoma and RCC and unusually in NSCLC, including in both squamous and non-squamous tumors. Objective responses were noted in 41% of the 107 melanoma patients treated at 3 mg/kg. Survival was improved, with 1- and 2-year survival rates of 62% and 43% at extended follow up.50
Subsequently, nivolumab was compared to chemotherapy in a pair of phase 3 studies involving both previously untreated (Checkmate 066) and ipilimumab/BRAF inhibitor–refractory (CheckMate 037) patients.19,20 In both studies, nivolumab produced durable responses in 32% to 34% of patients and improved survival over chemotherapy. Compared to ipilimumab, the incidence of irAEs was much lower with nivolumab. The depth and magnitude of responses observed led to regulatory approval for nivolumab in both indications (untreated and ipilimumab/BRAF inhibitor–treated melanoma) in 2014. Data from both studies are summarized in Table 1.
Pembrolizumab was first evaluated in a phase 1 study of 30 patients with a variety of solid organ malignancies in which no dose-limiting toxicities were observed and no defined maximal tolerated dose was reached.51 Per protocol, maximal administered dose was 10 mg/kg every 2 weeks. Following startling responses including 2 complete responses of long duration, pembrolizumab was evaluated in a large phase 1 study (KEYNOTE-001) of 1260 patients that evaluated 3 doses (10 mg/kg every 2 weeks, 10 mg/kg every 3 weeks, and 2 mg/kg every 3 weeks) in separate melanoma and NSCLC substudies.21 Both ipilimumab-naïve and ipilimumab-treated patients were enrolled in the melanoma substudy. Objective responses were seen in 38% ofpatients across all 3 dosing schedules and were similar in both ipilimumab-naïve and ipilimumab-treated patients. Similar to nivolumab, most responders experienced durable remissions.
Pembrolizumab was subsequently compared to ipilimumab in untreated patients (KEYNOTE-006) in which patients were randomly assigned to receive either ipilimumab or pembrolizumab at 1 of 2 doses: 10 mg/kg every 2 weeks and pembrolizumab 10 mg/kg every 3 weeks.22 Response rates were greater with pembrolizumab than ipilimumab, with commensurately greater 1-year survival rates. Rates of treatment-related adverse events requiring discontinuation of study drug were much lower with pembrolizumab than ipilimumab. This trial was instrumental in proving the superior profile of pembrolizumab over ipilimumab. The US Food and Drug Administration (FDA) granted pembrolizumab accelerated approval for second-line treatment of melanoma in 2014, and updated this to include a first-line indication in 2015 (Table 1).
EFFICACY OF COMBINED CTLA-4 AND PD-1 INHIBITION
Preclinical studies demonstrated that PD-1 blockade was more effective than CTLA-4 blockade and combination PD-1/CTLA-4 blockade was synergistic, with complete rejection of tumors in approximately half of the treated animals.14 This hypothesis was evaluated in a phase 1 study that explored both concurrent and sequential combinations of ipilimumab and nivolumab along with increasing doses of both agents in PD-1/CTLA-4–naïve advanced melanoma.23 Responses were greater in the concurrent arm (40%) than in the sequential arm (20%) across dose-levels with a small fraction of patients treated in the concurrent arm experiencing a profound reduction (80%) in tumor burden.
The superiority of ipilimumab/nivolumab combination to ipilimumab monotherapy was demonstrated in a randomized blinded phase 2 study (CheckMate 069).24 Of the 4 different ipilimumab/nivolumab doses explored in the phase 1 study (3 mg/kg and 0.3 mg/kg, 3 mg/kg and 1 mg/kg, 1 mg/kg and 3 mg/kg, 3 mg/kg and 3 mg/kg), ipilimumab 3 mg/kg and nivolumab 1 mg/kg (followed by nivolumab 3 mg/kg) was compared to ipilimumab and nivolumab-matched placebo. Responses were significantly greater with dual PD-1/CTLA-4 blockade compared to CTLA-4 blockade alone (59% versus 11%). Concurrently, a 3-arm randomized phase 3 study compared the same dose of ipilimumab/nivolumab to ipilimumab and nivolumab in previously untreated advanced melanoma (CheckMate 067).25 Similar to CheckMate 069, CheckMate 067 demonstrated that ipilimumab/nivolumab combination resulted in more profound responses (58%) than either ipilimumab (19%) or nivolumab (44%) alone. Toxicity, primarily diarrhea, fatigue, pruritus, and rash, was considerable in the combination arm (55% grade 3/4 adverse events) and resulted in treatment discontinuation in 30% of patients. The profound and durable responses observed led to accelerated approval of ipilimumab/nivolumab combination in 2015 (Table 1).
Efforts to improve the toxicity/benefit ratio of ipilimumab/nivolumab combination have centered around studying lower doses and/or extended dosing schedules of ipilimumab, including ipilimumab 1 mg/kg every 6 or 12 weeks with nivolumab dosed at 3 mg/kg every 2 weeks or 480 mg every 4 weeks. Promising data from a first-line study in NSCLC (CheckMate 012) support the evaluation of nivolumab in combination with lower-dosed ipilimumab (1 mg/kg every 6 or 12 weeks).52 This approach is being tested against platinum doublet chemotherapy in a confirmatory phase 3 study in NSCLC (CheckMate 227).
TARGETED THERAPY
MAPK KINASE PATHWAY IN MELANOMA TUMORIGENESIS
The MAPK pathway mediates cellular responses to growth signals. RAF kinases are central mediators in the MAPK pathway and exert their effect primarily through MEK phosphorylation and activation following dimerization (hetero- or homo-) of RAF molecules. As a result, RAF is integral to multiple cellular processes, including transcriptional regulation, cellular differentiation, and cell proliferation. MAPK pathway activation is a common event in many cancers, primarily due to activating mutations in BRAF or RAS. Alternatively, MAPK pathway activation can occur in the absence of activating mutations in BRAF or NRAS through down-regulation of MAPK pathway inhibitory proteins (RAF-1 inhibitory protein or SPRY-2), C-MET overexpression, or activating mutations in non-BRAF/NRAS kinases including CRAF, HRAS, and NRAS.53,54
Somatic point mutations in BRAF are frequently observed (37%–50%) in malignant melanomas and at lower frequency in a range of human cancers including NSCLC, colorectal cancer, papillary thyroid cancer, ovarian cancer, glioma, and gastrointestinal stromal tumor.6,55,56 BRAF mutations in melanoma typically occur within the activation segment of the kinase domain (exon 15). Between 80% and 90% of activating mutations result in an amino acid substitution of glutamate (E) for valine (V) at position 600: V600E.57,58 V600E mutations are true oncogenic drivers, resulting in increased kinase activity with demonstrable transformational capacity in vitro. BRAF mutations are usually mutually exclusive, with tumors typically containing no other driver mutations in NRAS, KIT, NF1, or other genes.
NRAS mutations are less common than BRAF mutations, having a reported frequency of 13% to 25% in melanoma.4 NRAS mutations generally occur within the P-loop region of the G domain (exon 2), or less commonly in the switch II region of the G domain (exon 3). Most NRAS exon 2 mutations comprise amino acid substitutions at position 61 from glutamine (Q) to arginine (R; 35%), lysine (K; 34%) and less often to glutamate (E), leucine (L), or proline (P). Preclinical data suggest that NRAS mutations paradoxically stimulate the MAPK pathway and thus enhance tumor growth in vitro.59,60 Several important phenotypic differences distinguish NRAS- from BRAF-mutated melanoma. NRAS-mutated tumors are typically associated with increasing age and CSD skin, while BRAF-mutated tumors arise in younger patients in non-CSD skin. A large population-based study suggested that NRAS-mutated melanomas were associated with mitoses and lower tumor infiltrating lymphocytes (TIL) grade, and arose in anatomic sites other than the head/neck, while BRAF-mutated tumors were associated with mitoses and superficial spreading histology.61 Although the lower TIL grade seen with NRAS-mutated melanomas suggests a more immunosuppressed microenvironment and argues for poorer responses to immune therapies, clinical studies comparing responses to immunotherapies in various categories of driver mutations provide conflicting results for the prognostic role of NRAS mutations in relation to immune checkpoint blockade and other immune therapies.62–64
NF1 represents the third known driver in cutaneous melanoma, with mutations reported in 12% of cases.6,7 NF1 encodes neurofibromin, which has GTPase activity and regulates RAS proteins; NF1 loss results in increased RAS.65 Unlike BRAF or NRAS, which are usually mutually exclusive, NF1 mutations in melanoma can occur singly or in combination with either BRAF or NRAS mutations. In these settings, NF1 mutations are associated with RAS activation, MEK-dependence, and resistance to RAF inhibition.66
MAPK PATHWAY INHIBITION SINGLY AND IN COMBINATION
Although multiple MEK 1/2 inhibitors (AS703026, AZD8330/ARRY-704, AZD6244, CH5126766, CI-1040, GSK1120212, PD0325901, RDEA119, and XL518) and RAF inhibitors (ARQ 680, GDC-0879, GSK2118436, PLX4032, RAF265, sorafenib, XL281/BMS-908662) were developed, the initial evaluation of MAPK pathway inhibitors in advanced human cancers began with CI-1040. Preclinical data suggested that CI-1040 potently and selectively inhibited both MEK1 and MEK2, but phase 1 and 2 human trial results were disappointing, likely because these trials were not selectively enriched for NRAS/BRAF–mutated tumors or cancers in which these oncogenic mutations were most commonly detected, such as melanoma.67,68 The subsequent evaluation of selumetinib (AZD6244/ARRY-142886) in a phase 2 study was also negative. Although investigators enrolled a presumably enriched population (cutaneous melanoma), the incidence of NRAS/BRAF–mutated tumors was not ascertained to determine this, but rather assumed, which led to a discrepancy between the assumed (prestudy) and observed (on-study) proportions of BRAF/NRAS mutations that was not accounted for in power calculations.69,70 Lessons learned from these earlier misadventures informed the current paradigm of targeted therapy development: (1) identification of a highly specific and potent inhibitor through high-throughput screening; (2) establishment of maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) in unselected patients; (3) confirmation of RP2D in selected tumor types enriched for target of interest; and (4) confirmatory study against standard comparator to seek regulatory approval.
Vemurafenib and dabrafenib were evaluated in this tiered fashion in phase 1 dose-finding studies comprising unselected patients, followed by phase 2 studies in advanced BRAF V600E–mutated melanoma. Both were subsequently evaluated in randomized phase 3 trials (vemurafenib, BRIM-38; dabrafenib, BREAK-310) that compared them with dacarbazine (1000 mg/m2 intravenously every 3 weeks) in the treatment of advanced BRAF V600E–mutated melanoma. Response kinetics for both agents were remarkably similar: single-agent BRAF inhibitors resulted in rapid (time to response 2–3 months), profound (approximately 50% objective responses) reductions in tumor burden that lasted 6 to 7 months. Adverse events common to both agents included rash, fatigue, and arthralgia, although clinically significant photosensitivity was more common with vemurafenib and clinically significant pyrexia was more common with dabrafenib. Class-specific adverse events included the development of cutaneous squamous-cell carcinomas and keratoacanthomas secondary to paradoxical activation of MAPK pathway signaling either through activating mutations in HRAS or mutations or amplifications in receptor tyrosine kinases upstream of BRAF, resulting in elevated levels of RAS–guanosine triphosphate complexes.71 Results of these studies resulted in regulatory approval of single-agent BRAF inhibitors for the treatment of BRAF V600E (and later V600K)–mutated melanoma (vemurafenib in 2011; dabrafenib in 2013). Details regarding trial populations, study interventions, efficacy, and adverse events are summarized in Table 2.
Responses to BRAF inhibitors are typically profound but temporary. Mechanisms of acquired resistance are diverse and include reactivation of MAPK pathway–dependent signaling (RAS activation or increased RAF expression), and development of MAPK pathway–independent signaling (COT overexpression; increased PI3K or AKT signaling) that permits bypass of inhibited BRAF signaling within the MAPK pathway.72–76 These findings suggested that upfront inhibition of both MEK and mutant BRAF may produce more durable responses than BRAF inhibition alone. Three pivotal phase 3 studies established the superiority of combination BRAF and MEK inhibition over BRAF inhibition alone: COMBI-d11 (dabrafenib/trametinib versus dabrafenib/placebo), COMBI-v12 (dabrafenib/trametinib versus vemurafenib), and coBRIM13 (vemurafenib/cobimetinib versus vemurafenib/placebo). As expected, compared to BRAF inhibitor monotherapy, combination BRAF and MEK inhibition produced greater responses and improved progression-free and overall survival (Table 2). Interestingly, the rate of cutaneous squamous-cell carcinomas was much lower with combination therapy, reflecting the more profound degree of MAPK pathway inhibition achieved with combination BRAF and MEK inhibition. Based on these results, FDA approval was granted for both dabrafenib/trametinib and vemurafenib/cobimetinib combinations in 2015. Although the dabrafenib/trametinib combination was only approved in 2015, trametinib had independently gained FDA approval in 2013 for the treatment of BRAF V600E/K–mutated melanoma on the basis of the phase 3 METRIC study.77
Encorafenib (LGX818) and binimetinib (MEK162, ARRY-162, ARRY-438162) are new BRAF and MEK inhibitors currently being evaluated in clinical trials. Encorafenib/binimetinib combination was first evaluated in a phase 3 study (COLUMBUS) that compared it with vemurafenib monotherapy in BRAF-mutant melanoma.78 Unsurprisingly, encorafenib/binimetinib combination produced greater and more durable responses compared to vemurafenib monotherapy. The median progression-free survival of the encorafenib/binimetinib combination (14.9 months) was greater than vemurafenib monotherapy (7.3 months) in this study, and intriguingly greater than that seen with vemurafenib/cobimetinib (coBRIM 9.9 months) and dabrafenib/trametinib (COMBI-d 9.3 months; COMBI-v 11.4 months). Of note, although encorafenib has an IC50 midway between dabrafenib and vemurafenib in cell-free assays (0.8 nM dabrafenib, 4 nM encorafenib, and 31 nM vemurafenib), it has an extremely slower off-rate from BRAF V600E, which results in significantly greater target inhibition in cells following drug wash-out.79 This may account for the significantly greater clinical benefit seen with encorafenib/binimetinib in clinical trials. Final study data are eagerly awaited. Regulatory approval has been sought, and is pending at this time.
Binimetinib has been compared to dacarbazine in a phase 3 study (NEMO) of patients with NRAS-mutant melanoma, most of whom had been previously treated with immunotherapy.80 Response rates were low in both arms, although slightly greater with binimetinib than dacarbazine (15% versus 9%), commensurate with a modest improvement in progression-free survival. FDA approval has been sought and remains pending at this time.
KIT INHIBITION SINGLY AND IN COMBINATION
The KIT receptor protein tyrosine kinase is a transmembrane protein consisting of extracellular and intracellular domains. Activating KIT mutations occur in 2% to 8% of all melanoma patients and may be found in all melanoma subtypes but are commonest in acral melanomas (10%–20%) and mucosal melanomas (15%–20%). Activating KIT mutations primarily occur in exons 11 and 13, which code for the juxtamembrane and kinase domains, respectively.5,81–83
Imatinib mesylate is a tyrosine kinase inhibitor of the 2-phenyl amino pyrimidine class that occupies the tyrosine kinase active site with resultant blocking of tyrosine kinase activity. Imatinib mesylate is known to block KIT and has been extensively studied in patients with gastrointestinal stromal tumors (GIST), 80% of whom harbor KIT mutations, in both the adjuvant and the metastatic settings. In melanoma, imatinib mesylate was studied in a Chinese open-label, phase 2 study of imatinib mesylate monotherapy in metastatic melanoma harboring KIT mutation or amplification; 25% of the study patients had mucosal disease and the rest had cutaneous disease, with acral involvement in 50% of all patients.84 Overall response rate was 23%, while 51% of patients remained alive at 1 year with no differences in response rate and/or survival being noted between patients with either KIT mutations or amplifications. In a separate study of imatinib mesylate at 400 mg daily or 400 mg twice daily in Caucasian patients with KIT-mutated/amplified melanoma, similar response and survival rates were reported, although patients with KIT mutations did nonsignificantly better than those with KIT amplifications.85
Other novel studies evaluating KIT inhibitors include KIT inhibition in combination with the VEGF inhibitor bevacizumab and a study of selective BCR-ABL kinase inhibitor nilotinib in imatinib-resistant melanoma. In the former phase 1/2 study, Flaherty and colleagues studied imatinib 800 mg daily and bevacizumab at 10 mg/kg every 2 weeks in 63 patients with advanced tumors, including 23 with metastatic melanoma. Although the combination was relatively nontoxic, no significant efficacy signal was seen and further accrual to the phase 2 portion was halted after the first stage was completed.86 Nilotinib is a BCR-ABL1 tyrosine kinase inhibitor intelligently designed based on the structure of the ABL-imatinib complex that is 10 to 30 times more potent than imatinib in inhibiting BCR-ABL1 tyrosine kinase activity. Nilotinib is approved for the treatment of imatinib-resistant chronic myelogenous leukemia (CML), with reported efficacy in patients with central nervous system (CNS) involvement.87,88 Nilotinib has been studied in a single study of KIT-mutated/amplified melanoma that included patients with imatinib-resistance and those with treated CNS disease. Nilotinib appeared to be active in imatinib-resistant melanoma, although no responses were seen in the CNS disease cohort.89 Overall, the response rates observed with KIT inhibition in melanoma are much lower than those observed in CML and GIST.
CONCLUSION AND FUTURE DIRECTIONS
Prior to 2011, the only approved agents for the treatment of advanced melanoma were dacarbazine and high-dose interleukin-2. Since 2011, drug approvals in melanoma have proceeded at a frenetic pace unmatched in any other disease. The primary events underlying this are advances in our understanding of the gene mutation landscape driving melanoma tumorigenesis, accompanied by insights into the means by which tumors circumvent the induction of effective anti-tumor T-cell responses. These insights have resulted in the development of inhibitors targeting MAPK pathway kinases BRAF, MEK, and NRAS), KIT, and regulatory immune checkpoints (CTLA-4 and PD-1). Although BRAF/MEK inhibition results in profound reductions and even occasional complete responses in patients, these responses are typically short lived, rarely lasting more than 9 to 11 months; the encorafenib/binimetinib combination may improve that duration marginally. However, the signature therapeutic advance in melanoma of the past decade is immunotherapy, particularly the development of inhibitors of CTLA-4 and PD-1 immune checkpoints. With these agents, significant proportions of treated patients remain free of progression off-therapy (ipilimumab 23%; nivolumab 34%; pembrolizumab 35%; ipilimumab/nivolumab 64%), and some patients can be successfully re-induced after delayed progression. Separately, the high response rates observed with the use of KIT inhibitors in CML and GIST have not been observed in KIT mutated/amplified melanoma and development of agents in this space has been limited. The challenges ahead center around identifying predictive biomarkers and circumventing primary or acquired resistance, with the eventual goal of producing durable remissions in the majority of treated patients.
Our improved understanding of the mechanisms of acquired resistance to BRAF/MEK inhibitors suggests that anti-tumor activity may be achieved by targeting multiple pathways, possibly with combination regimens comprising other inhibitors and/or immunotherapy. Preclinical data supports the use of combination strategies targeting both ERK and PI3K/mTOR to circumvent acquired resistance.90 Ongoing studies are evaluating combinations with biguanides (metformin: NCT02143050 and NCT01638676; phenformin: NCT03026517), HSP90 inhibitors (XL888: NCT02721459; AT13387: NCT02097225), and decitabine (NCT01876641).
One complexity affecting management of resistance in the targeted therapy landscape remains tumor heterogeneity, particularly intra- and intertumoral heterogeneity, which may explain the apparent contradiction between continued efficacy of BRAF inhibitors in BRAF-resistant tumors and preclinical data predicting slower progression of resistant tumors on cessation of BRAF inhibitors.91–94 These data provide a rationale to investigate intermittent dosing regimens with BRAF/MEK inhibitors; several studies exploring this approach are ongoing (NCT01894672 and NCT02583516).
Given the specificity, adaptability, and memory response associated with immunotherapy, it is likely that these agents will be used to treat the majority of patients regardless of mutational status. Hence, identifying predictive biomarkers of response to immune checkpoint inhibitors is vital. The presence of CD8+ T-cell infiltrate and IFN-γ gene signature, which indicate an “inflamed” tumor microenvironment, are highly predictive of clinical benefit from PD-1 inhibitors.95,96 However, not all PD-1 responders have “inflamed” tumor microenvironments, and not all patients with an “inflamed” tumor microenvironment respond to immune checkpoint inhibitors. The complexity of the immune system is reflected in the multiple non-redundant immunologic pathways, both positive and negative, with checkpoints and ligands that emerge dynamically in response to treatment. Given the dynamic nature of the immune response, it is unlikely that any single immunologic biomarker identified pre-treatment will be completely predictive. Rather, the complexity of the biomarker approach must match the complexity of the immune response elicited, and will likely incorporate multifarious elements including CD8+ T-cell infiltrate, IFN-γ gene signature, and additional elements including microbiome, genetic polymorphisms, and tumor mutation load. The goal is to use multiple markers to guide development of combinations and then, depending on initial response, to examine tumors for alterations to guide decisions about additional treatment(s) to improve responses, with the eventual goal being durable clinical responses for all patients.
INTRODUCTION
The incidence of cutaneous melanoma has increased over the past 2 decades, with SEER estimates indicating that the number of new cases of melanoma diagnosed annually rose from 38,300 in 1996 to 76,000 in 2016.1 Among persons younger than 50 years, the incidence is higher in females, and younger women (aged 15–39 years) are especially vulnerable.2 Among persons older than 50, melanoma incidence in men is nearly twice that of women, in whom melanomas are often thicker and often associated with worse outcomes.1,2 Approximately 85% of melanomas are diagnosed at early stages when surgery is curative, but the lifetime probability of developing invasive disease is 3% in men and 2% in women.
Prior to the advent of effective immunotherapies and targeted therapies, melanoma was often managed with chemotherapy, which had dismal response rates and commensurately poor outcomes. Advances in the understanding of the molecular etiopathogenesis and immune escape responses of cutaneous metastatic melanoma have transformed therapeutic approaches. Specifically, improved understanding of the genetic mutations driving melanoma tumorigenesis coupled with insights into mechanisms of tumor-mediated immune evasion resulted in development of inhibitors of mitogen-activated protein kinases (MAPK; BRAF and MEK) along with inhibitors of negative regulatory immune checkpoints (cytotoxic T lymphocyte–associated antigen 4 [CTLA-4] and programmed cell death-1 [PD-1]). In this review, we discuss the role of immune therapy, targeted therapy, and combinations of these in the treatment of metastatic cutaneous melanoma. We limit the immuno-therapy discussion to approved CTLA-4/PD-1 inhibitors and the targeted therapy discussion to approved BRAF/NRAS/MEK inhibitors and do not discuss non-checkpoint immunotherapies including cytokines (HD IL-2), vaccines, or adoptive T-cell approaches. Interested readers are directed to other excellent works covering these important topics.26–29
DEVELOPMENT OF TARGETED AND NOVEL IMMUNE THERAPIES
For many years the degree of ultraviolet (UV) light exposure was considered the sole major risk factor for melanoma oncogenesis, even though its mechanism was largely unknown.3 However, clinical observations regarding the occurrence of melanoma on less exposed areas (trunk and limbs) in individuals with intermittent sun exposure led to the proposition that melanomas that arose in younger patients with intermittent sun exposure were distinct from melanomas that arose in older patients in association with markers of chronic sun exposure—the “divergent pathway” hypothesis.3 Critical to this understanding were whole-exome sequencing data from multiple groups, including The Cancer Genome Atlas, that identified patterns of mutations in oncogenic drivers that were distinct in patients with and without chronically sun-damaged (CSD) skin.4–7 It is now clear that based on its association with CSD skin, melanoma can be subclassified into CSD or non-CSD melanoma. CSD and non-CSD melanoma have distinct clinico-pathological characteristics and are associated with different driver mutations. CSD melanomas typically arise in older patients on sun-exposed areas (head/neck, dorsal surfaces of distal extremities) and are associated with particular driver mutations (BRAF non-V600E, NRAS, NF1, or KIT) and genetic signatures of UV-induced DNA damage (G > T [UVA] or C > T [UVB]) transitions. Conversely, non-CSD melanomas typically arise in younger (< 55 years) patients on intermittently sun-exposed areas (trunk, proximal extremities) and are associated with BRAF V600E/K driver mutations and often lack genetic signatures of UV mutagenesis.
Identification of driver mutations in components of the MAPK pathway, including BRAF and NRAS, facilitated the development of targeted inhibitors. The BRAF inhibitors vemurafenib and dabrafenib have been shown in pivotal phase 3 studies to significantly improve overall and progression-free survival in patients with metastatic melanoma compared with chemotherapy and garnered regulatory approval (vemurafenib, BRIM-3;8,9 dabrafenib, BREAK-310). Concomitant MEK and BRAF inhibition extends the duration of benefit by preventing downstream kinase activation in the MAPK pathway. Notably, concomitant MEK inhibition alters the side-effect profile of BRAF inhibitors, with reduced incidence of keratoacanthomas and cutaneous squamous cell carcinomas that are attributable to on-target, off-tumor effects of BRAF inhibitors. Combined BRAF and MEK inhibition (vemurafenib/cobimetinib and dabrafenib/trametinib) further improved overall and progression-free survival compared to single-agent BRAF inhibition in phase 3 studies (COMBI-d,11 COMBI-v,12 and coBRIM13). Although often deep, the responses seen with the use of targeted kinase inhibitors are not often durable, with the vast majority of patients progressing after 12 to 15 months of therapy.In parallel, work primarily done in murine models of chronic viral infection uncovered the role played by co-inhibitory or co-excitatory immune checkpoints in mediating T-cell immune responses. These efforts clarified that tumor-mediated immune suppression primarily occurs through enhancement of inhibitory signals via the negative T-cell immune checkpoints CTLA-4 or PD-1.14,15 Blockade of negative T-cell immune checkpoints resulted in activation of the adaptive immune system, resulting in durable anti-tumor responses as demonstrated in studies of the CTLA-4 inhibitor ipilimumab (CA184-02016 and CA184-02417) and the PD-1 inhibitors nivolumab (CA209-003,18 CheckMate 037,19 and CheckMate 06620) and pembrolizumab (KEYNOTE-00121 and KEYNOTE-00622). Compared to the deep but short-lived responses seen with targeted kinase inhibitors, patients treated with CTLA-4 or PD-1 immune checkpoint blockade often developed durable responses that persisted even after completion of therapy. Combined CTLA-4 and PD-1 blockade results in greater magnitude of response with proportionately increased toxicity.23–25
IMMUNOTHERAPY
CTLA-4 AND PD-1 IMMUNE CHECKPOINT INHIBITORS
The novel success of immunotherapy in recent decades is largely attributable to improved understanding of adaptive immune physiology, specifically T-cell activation and regulation. T-cell activation requires 2 independent signaling events: it is initiated upon recognition of the antigen-MHC class II-receptor complex on antigen-presenting cells (APC), and requires a secondary co-stimulatory interaction of CD80/CD86 (B7.1/B7.2) on APCs and CD28 molecule on T-cells; without this second event, T-cells enter an anergic state.30–32 Upon successful signaling and co-stimulation, newly activated T-cells upregulate CTLA-4, which can bind to B7 molecules with a nearly 100-fold greater affinity than CD28.33,34 Unlike CD28, CTLA-4 engagement negatively regulates T-cell activation. The opposing signals produced by CD28 and CTLA-4 are integrated by the T-cell to determine eventual response to activation, and provide a means by which T-cell activation is homeostatically regulated to prevent exaggerated physiologic immune responses.35 It was hypothesized that CTLA-4 blockade would permit T-cell activation, which is thwarted in the tumor microenvironment by tumor-mediated CTLA-4 engagement, thereby unleashing an anti-tumor immune response.36
PD-1 is a member of the CD28 and CTLA-4 immunoglobulin super family and, similar to CTLA-4, binds activated T-cells. PD-1 has 2 ligands on activated T-cells: PD-L1 and PD-L2.37 PD-L1 is constitutively expressed by a variety of immune and non-immune cells, particularly in inflammatory environments including tumor microenvironments, in response to the release of inflammatory cytokines such as interferon (IFN)-γ.37,38 Conversely, PD-L2 is only minimally expressed constitutively, although its expression on immune and non-immune cells can be induced by similar cues from inflammatory microenvironments. PD-L1 and PD-L2 cross-compete for binding to PD-1, with PD-L2 exhibiting 2- to 6-fold greater relative affinity than PD-L1.39 PD-L1/PD-1 binding results in phosphorylation of 2 tyrosinases in the intracellular portion of PD-1, which contains immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM). PD-1 ITSM subsequently recruits either of 2 SH2-domain–containing protein tyrosine phosphatases: SHP-1 and SHP-2. SHP-2 signaling suppresses PI3K/Akt activation, down-regulates Bcl-xL, and suppresses expression of multiple transcription factors that mediate T-cell effector function including GATA-3, Eomes, and T-bet.40–42 The net effect of PD-L1/PD-1 engagement is to suppress T-cell proliferation, cytokine production, cytolytic function, and survival. Unlike CTLA-4, which primarily affects the priming phase of naive T-cell activation, PD-1 chiefly regulates the effector phase of T-cell function. Furthermore, because PD-L1/PD-L2 expression is limited to inflammatory microenvironments, the effects of PD-1 are less generalized than those of CTLA-4.
SINGLE AGENT ACTIVITY OF CTLA-4 AND PD-1 INHIBITORS
Ipilimumab (MDX-010) is a human IgG1 monoclonal antibody shown to inhibit CTLA-4.43 Early studies tested different formulations (transfectoma-derived and hybridoma-derived), doses, and schedules of ipilimumab primarily in patients with advanced refractory melanoma.44–46 Although responses were infrequent, responding patients experienced durable remissions at 1- and 2-year time points. Notably, in a foreshadowing of changes to response criteria used to evaluate these agents, several treated patients who initially had radiographically stable disease upon completion of therapy subsequently experienced a gradual decline in tumor burden.
Ipilimumab was subsequently evaluated in 2 phase 3 trials. The first study (MDX010-020/CA184-020), which involved 676 HLA-A*0201–positive patients with advanced melanoma, compared ipilimumab 3 mg/kg every 3 weeks for 4 doses either singly or in combination with gp100 vaccine with a gp100-only control arm.16 Ipilimumab administration resulted in objective responses in 11% of patients and improved progression-free and overall survival compared to gp100 alone. Of note, ipilimumab monotherapy was superior to ipilimumab/gp100 combination, possibly related to timing of vaccine in relation to ipilimumab. A confirmatory study (CA184-024) compared a higher dose of ipilimumab (10 mg/kg) in combination with dacarbazine to dacarbazine monotherapy in previously untreated melanoma and was positive.17 Given the lack of augmented efficacy with the higher (10 mg/kg) dose, ipilimumab received regulatory approval in 2011 for the treatment of melanoma at the lower dose: 3 mg/kg administered every 3 weeks for 4 doses (Table 1). Survival data was strikingly similar to patterns observed in prior phase 2 studies, with survival curves plateauing after 2 years at 23.5% to 28.5% of treated patients. Pooled survival data from prospective and retrospective studies of ipilimumab corroborate the plateau of 22% (26% treated; 20% untreated) reached at year 3 regardless of prior therapy or ipilimumab dose, underscoring the durability of long-term survival in ipilimumab-treated patients.47 Ipilimumab administration resulted in an unusual spectrum of toxicities including diarrhea, rash, hepatitis, and hypophysitis (termed immune-related adverse events, or irAEs) in up to a third of patients.
Pembrolizumab and nivolumab are humanized IgG4 monoclonal antibodies that target the PD-1 receptor found on activated T cells, B cells, and myeloid cells. Pembrolizumab and nivolumab are engineered similarly: by immunizing transgenic mice with recombinant human PD-1-Fc protein and subsequently screening murine splenic cells fused with myeloma cells for hybridomas producing antibodies reactive to PD-1-Fc.48,49 Unlike IgG1, the IgG4 moiety neither engages Fc receptors nor activates complement, avoiding cytotoxic effects of the antibody upon binding to the T cells that it is intended to activate. Both pembrolizumab and nivolumab bind PD-1 with high affinity and specificity, effectively inhibiting the interaction between PD-1 and ligands PD-L1 and PD-L2.
Nivolumab was first studied in a phase 1 study (CA209-003) of 296 patients with advanced cancers who received 1, 3, or 10 mg/kg administered every 2 weeks.18 Histologies tested included melanoma, non–small-cell lung cancer (NSCLC), renal-cell cancer (RCC), castration-resistant prostate cancer (CRPC), and colorectal cancer (CRC). Responses were seen in melanoma and RCC and unusually in NSCLC, including in both squamous and non-squamous tumors. Objective responses were noted in 41% of the 107 melanoma patients treated at 3 mg/kg. Survival was improved, with 1- and 2-year survival rates of 62% and 43% at extended follow up.50
Subsequently, nivolumab was compared to chemotherapy in a pair of phase 3 studies involving both previously untreated (Checkmate 066) and ipilimumab/BRAF inhibitor–refractory (CheckMate 037) patients.19,20 In both studies, nivolumab produced durable responses in 32% to 34% of patients and improved survival over chemotherapy. Compared to ipilimumab, the incidence of irAEs was much lower with nivolumab. The depth and magnitude of responses observed led to regulatory approval for nivolumab in both indications (untreated and ipilimumab/BRAF inhibitor–treated melanoma) in 2014. Data from both studies are summarized in Table 1.
Pembrolizumab was first evaluated in a phase 1 study of 30 patients with a variety of solid organ malignancies in which no dose-limiting toxicities were observed and no defined maximal tolerated dose was reached.51 Per protocol, maximal administered dose was 10 mg/kg every 2 weeks. Following startling responses including 2 complete responses of long duration, pembrolizumab was evaluated in a large phase 1 study (KEYNOTE-001) of 1260 patients that evaluated 3 doses (10 mg/kg every 2 weeks, 10 mg/kg every 3 weeks, and 2 mg/kg every 3 weeks) in separate melanoma and NSCLC substudies.21 Both ipilimumab-naïve and ipilimumab-treated patients were enrolled in the melanoma substudy. Objective responses were seen in 38% ofpatients across all 3 dosing schedules and were similar in both ipilimumab-naïve and ipilimumab-treated patients. Similar to nivolumab, most responders experienced durable remissions.
Pembrolizumab was subsequently compared to ipilimumab in untreated patients (KEYNOTE-006) in which patients were randomly assigned to receive either ipilimumab or pembrolizumab at 1 of 2 doses: 10 mg/kg every 2 weeks and pembrolizumab 10 mg/kg every 3 weeks.22 Response rates were greater with pembrolizumab than ipilimumab, with commensurately greater 1-year survival rates. Rates of treatment-related adverse events requiring discontinuation of study drug were much lower with pembrolizumab than ipilimumab. This trial was instrumental in proving the superior profile of pembrolizumab over ipilimumab. The US Food and Drug Administration (FDA) granted pembrolizumab accelerated approval for second-line treatment of melanoma in 2014, and updated this to include a first-line indication in 2015 (Table 1).
EFFICACY OF COMBINED CTLA-4 AND PD-1 INHIBITION
Preclinical studies demonstrated that PD-1 blockade was more effective than CTLA-4 blockade and combination PD-1/CTLA-4 blockade was synergistic, with complete rejection of tumors in approximately half of the treated animals.14 This hypothesis was evaluated in a phase 1 study that explored both concurrent and sequential combinations of ipilimumab and nivolumab along with increasing doses of both agents in PD-1/CTLA-4–naïve advanced melanoma.23 Responses were greater in the concurrent arm (40%) than in the sequential arm (20%) across dose-levels with a small fraction of patients treated in the concurrent arm experiencing a profound reduction (80%) in tumor burden.
The superiority of ipilimumab/nivolumab combination to ipilimumab monotherapy was demonstrated in a randomized blinded phase 2 study (CheckMate 069).24 Of the 4 different ipilimumab/nivolumab doses explored in the phase 1 study (3 mg/kg and 0.3 mg/kg, 3 mg/kg and 1 mg/kg, 1 mg/kg and 3 mg/kg, 3 mg/kg and 3 mg/kg), ipilimumab 3 mg/kg and nivolumab 1 mg/kg (followed by nivolumab 3 mg/kg) was compared to ipilimumab and nivolumab-matched placebo. Responses were significantly greater with dual PD-1/CTLA-4 blockade compared to CTLA-4 blockade alone (59% versus 11%). Concurrently, a 3-arm randomized phase 3 study compared the same dose of ipilimumab/nivolumab to ipilimumab and nivolumab in previously untreated advanced melanoma (CheckMate 067).25 Similar to CheckMate 069, CheckMate 067 demonstrated that ipilimumab/nivolumab combination resulted in more profound responses (58%) than either ipilimumab (19%) or nivolumab (44%) alone. Toxicity, primarily diarrhea, fatigue, pruritus, and rash, was considerable in the combination arm (55% grade 3/4 adverse events) and resulted in treatment discontinuation in 30% of patients. The profound and durable responses observed led to accelerated approval of ipilimumab/nivolumab combination in 2015 (Table 1).
Efforts to improve the toxicity/benefit ratio of ipilimumab/nivolumab combination have centered around studying lower doses and/or extended dosing schedules of ipilimumab, including ipilimumab 1 mg/kg every 6 or 12 weeks with nivolumab dosed at 3 mg/kg every 2 weeks or 480 mg every 4 weeks. Promising data from a first-line study in NSCLC (CheckMate 012) support the evaluation of nivolumab in combination with lower-dosed ipilimumab (1 mg/kg every 6 or 12 weeks).52 This approach is being tested against platinum doublet chemotherapy in a confirmatory phase 3 study in NSCLC (CheckMate 227).
TARGETED THERAPY
MAPK KINASE PATHWAY IN MELANOMA TUMORIGENESIS
The MAPK pathway mediates cellular responses to growth signals. RAF kinases are central mediators in the MAPK pathway and exert their effect primarily through MEK phosphorylation and activation following dimerization (hetero- or homo-) of RAF molecules. As a result, RAF is integral to multiple cellular processes, including transcriptional regulation, cellular differentiation, and cell proliferation. MAPK pathway activation is a common event in many cancers, primarily due to activating mutations in BRAF or RAS. Alternatively, MAPK pathway activation can occur in the absence of activating mutations in BRAF or NRAS through down-regulation of MAPK pathway inhibitory proteins (RAF-1 inhibitory protein or SPRY-2), C-MET overexpression, or activating mutations in non-BRAF/NRAS kinases including CRAF, HRAS, and NRAS.53,54
Somatic point mutations in BRAF are frequently observed (37%–50%) in malignant melanomas and at lower frequency in a range of human cancers including NSCLC, colorectal cancer, papillary thyroid cancer, ovarian cancer, glioma, and gastrointestinal stromal tumor.6,55,56 BRAF mutations in melanoma typically occur within the activation segment of the kinase domain (exon 15). Between 80% and 90% of activating mutations result in an amino acid substitution of glutamate (E) for valine (V) at position 600: V600E.57,58 V600E mutations are true oncogenic drivers, resulting in increased kinase activity with demonstrable transformational capacity in vitro. BRAF mutations are usually mutually exclusive, with tumors typically containing no other driver mutations in NRAS, KIT, NF1, or other genes.
NRAS mutations are less common than BRAF mutations, having a reported frequency of 13% to 25% in melanoma.4 NRAS mutations generally occur within the P-loop region of the G domain (exon 2), or less commonly in the switch II region of the G domain (exon 3). Most NRAS exon 2 mutations comprise amino acid substitutions at position 61 from glutamine (Q) to arginine (R; 35%), lysine (K; 34%) and less often to glutamate (E), leucine (L), or proline (P). Preclinical data suggest that NRAS mutations paradoxically stimulate the MAPK pathway and thus enhance tumor growth in vitro.59,60 Several important phenotypic differences distinguish NRAS- from BRAF-mutated melanoma. NRAS-mutated tumors are typically associated with increasing age and CSD skin, while BRAF-mutated tumors arise in younger patients in non-CSD skin. A large population-based study suggested that NRAS-mutated melanomas were associated with mitoses and lower tumor infiltrating lymphocytes (TIL) grade, and arose in anatomic sites other than the head/neck, while BRAF-mutated tumors were associated with mitoses and superficial spreading histology.61 Although the lower TIL grade seen with NRAS-mutated melanomas suggests a more immunosuppressed microenvironment and argues for poorer responses to immune therapies, clinical studies comparing responses to immunotherapies in various categories of driver mutations provide conflicting results for the prognostic role of NRAS mutations in relation to immune checkpoint blockade and other immune therapies.62–64
NF1 represents the third known driver in cutaneous melanoma, with mutations reported in 12% of cases.6,7 NF1 encodes neurofibromin, which has GTPase activity and regulates RAS proteins; NF1 loss results in increased RAS.65 Unlike BRAF or NRAS, which are usually mutually exclusive, NF1 mutations in melanoma can occur singly or in combination with either BRAF or NRAS mutations. In these settings, NF1 mutations are associated with RAS activation, MEK-dependence, and resistance to RAF inhibition.66
MAPK PATHWAY INHIBITION SINGLY AND IN COMBINATION
Although multiple MEK 1/2 inhibitors (AS703026, AZD8330/ARRY-704, AZD6244, CH5126766, CI-1040, GSK1120212, PD0325901, RDEA119, and XL518) and RAF inhibitors (ARQ 680, GDC-0879, GSK2118436, PLX4032, RAF265, sorafenib, XL281/BMS-908662) were developed, the initial evaluation of MAPK pathway inhibitors in advanced human cancers began with CI-1040. Preclinical data suggested that CI-1040 potently and selectively inhibited both MEK1 and MEK2, but phase 1 and 2 human trial results were disappointing, likely because these trials were not selectively enriched for NRAS/BRAF–mutated tumors or cancers in which these oncogenic mutations were most commonly detected, such as melanoma.67,68 The subsequent evaluation of selumetinib (AZD6244/ARRY-142886) in a phase 2 study was also negative. Although investigators enrolled a presumably enriched population (cutaneous melanoma), the incidence of NRAS/BRAF–mutated tumors was not ascertained to determine this, but rather assumed, which led to a discrepancy between the assumed (prestudy) and observed (on-study) proportions of BRAF/NRAS mutations that was not accounted for in power calculations.69,70 Lessons learned from these earlier misadventures informed the current paradigm of targeted therapy development: (1) identification of a highly specific and potent inhibitor through high-throughput screening; (2) establishment of maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) in unselected patients; (3) confirmation of RP2D in selected tumor types enriched for target of interest; and (4) confirmatory study against standard comparator to seek regulatory approval.
Vemurafenib and dabrafenib were evaluated in this tiered fashion in phase 1 dose-finding studies comprising unselected patients, followed by phase 2 studies in advanced BRAF V600E–mutated melanoma. Both were subsequently evaluated in randomized phase 3 trials (vemurafenib, BRIM-38; dabrafenib, BREAK-310) that compared them with dacarbazine (1000 mg/m2 intravenously every 3 weeks) in the treatment of advanced BRAF V600E–mutated melanoma. Response kinetics for both agents were remarkably similar: single-agent BRAF inhibitors resulted in rapid (time to response 2–3 months), profound (approximately 50% objective responses) reductions in tumor burden that lasted 6 to 7 months. Adverse events common to both agents included rash, fatigue, and arthralgia, although clinically significant photosensitivity was more common with vemurafenib and clinically significant pyrexia was more common with dabrafenib. Class-specific adverse events included the development of cutaneous squamous-cell carcinomas and keratoacanthomas secondary to paradoxical activation of MAPK pathway signaling either through activating mutations in HRAS or mutations or amplifications in receptor tyrosine kinases upstream of BRAF, resulting in elevated levels of RAS–guanosine triphosphate complexes.71 Results of these studies resulted in regulatory approval of single-agent BRAF inhibitors for the treatment of BRAF V600E (and later V600K)–mutated melanoma (vemurafenib in 2011; dabrafenib in 2013). Details regarding trial populations, study interventions, efficacy, and adverse events are summarized in Table 2.
Responses to BRAF inhibitors are typically profound but temporary. Mechanisms of acquired resistance are diverse and include reactivation of MAPK pathway–dependent signaling (RAS activation or increased RAF expression), and development of MAPK pathway–independent signaling (COT overexpression; increased PI3K or AKT signaling) that permits bypass of inhibited BRAF signaling within the MAPK pathway.72–76 These findings suggested that upfront inhibition of both MEK and mutant BRAF may produce more durable responses than BRAF inhibition alone. Three pivotal phase 3 studies established the superiority of combination BRAF and MEK inhibition over BRAF inhibition alone: COMBI-d11 (dabrafenib/trametinib versus dabrafenib/placebo), COMBI-v12 (dabrafenib/trametinib versus vemurafenib), and coBRIM13 (vemurafenib/cobimetinib versus vemurafenib/placebo). As expected, compared to BRAF inhibitor monotherapy, combination BRAF and MEK inhibition produced greater responses and improved progression-free and overall survival (Table 2). Interestingly, the rate of cutaneous squamous-cell carcinomas was much lower with combination therapy, reflecting the more profound degree of MAPK pathway inhibition achieved with combination BRAF and MEK inhibition. Based on these results, FDA approval was granted for both dabrafenib/trametinib and vemurafenib/cobimetinib combinations in 2015. Although the dabrafenib/trametinib combination was only approved in 2015, trametinib had independently gained FDA approval in 2013 for the treatment of BRAF V600E/K–mutated melanoma on the basis of the phase 3 METRIC study.77
Encorafenib (LGX818) and binimetinib (MEK162, ARRY-162, ARRY-438162) are new BRAF and MEK inhibitors currently being evaluated in clinical trials. Encorafenib/binimetinib combination was first evaluated in a phase 3 study (COLUMBUS) that compared it with vemurafenib monotherapy in BRAF-mutant melanoma.78 Unsurprisingly, encorafenib/binimetinib combination produced greater and more durable responses compared to vemurafenib monotherapy. The median progression-free survival of the encorafenib/binimetinib combination (14.9 months) was greater than vemurafenib monotherapy (7.3 months) in this study, and intriguingly greater than that seen with vemurafenib/cobimetinib (coBRIM 9.9 months) and dabrafenib/trametinib (COMBI-d 9.3 months; COMBI-v 11.4 months). Of note, although encorafenib has an IC50 midway between dabrafenib and vemurafenib in cell-free assays (0.8 nM dabrafenib, 4 nM encorafenib, and 31 nM vemurafenib), it has an extremely slower off-rate from BRAF V600E, which results in significantly greater target inhibition in cells following drug wash-out.79 This may account for the significantly greater clinical benefit seen with encorafenib/binimetinib in clinical trials. Final study data are eagerly awaited. Regulatory approval has been sought, and is pending at this time.
Binimetinib has been compared to dacarbazine in a phase 3 study (NEMO) of patients with NRAS-mutant melanoma, most of whom had been previously treated with immunotherapy.80 Response rates were low in both arms, although slightly greater with binimetinib than dacarbazine (15% versus 9%), commensurate with a modest improvement in progression-free survival. FDA approval has been sought and remains pending at this time.
KIT INHIBITION SINGLY AND IN COMBINATION
The KIT receptor protein tyrosine kinase is a transmembrane protein consisting of extracellular and intracellular domains. Activating KIT mutations occur in 2% to 8% of all melanoma patients and may be found in all melanoma subtypes but are commonest in acral melanomas (10%–20%) and mucosal melanomas (15%–20%). Activating KIT mutations primarily occur in exons 11 and 13, which code for the juxtamembrane and kinase domains, respectively.5,81–83
Imatinib mesylate is a tyrosine kinase inhibitor of the 2-phenyl amino pyrimidine class that occupies the tyrosine kinase active site with resultant blocking of tyrosine kinase activity. Imatinib mesylate is known to block KIT and has been extensively studied in patients with gastrointestinal stromal tumors (GIST), 80% of whom harbor KIT mutations, in both the adjuvant and the metastatic settings. In melanoma, imatinib mesylate was studied in a Chinese open-label, phase 2 study of imatinib mesylate monotherapy in metastatic melanoma harboring KIT mutation or amplification; 25% of the study patients had mucosal disease and the rest had cutaneous disease, with acral involvement in 50% of all patients.84 Overall response rate was 23%, while 51% of patients remained alive at 1 year with no differences in response rate and/or survival being noted between patients with either KIT mutations or amplifications. In a separate study of imatinib mesylate at 400 mg daily or 400 mg twice daily in Caucasian patients with KIT-mutated/amplified melanoma, similar response and survival rates were reported, although patients with KIT mutations did nonsignificantly better than those with KIT amplifications.85
Other novel studies evaluating KIT inhibitors include KIT inhibition in combination with the VEGF inhibitor bevacizumab and a study of selective BCR-ABL kinase inhibitor nilotinib in imatinib-resistant melanoma. In the former phase 1/2 study, Flaherty and colleagues studied imatinib 800 mg daily and bevacizumab at 10 mg/kg every 2 weeks in 63 patients with advanced tumors, including 23 with metastatic melanoma. Although the combination was relatively nontoxic, no significant efficacy signal was seen and further accrual to the phase 2 portion was halted after the first stage was completed.86 Nilotinib is a BCR-ABL1 tyrosine kinase inhibitor intelligently designed based on the structure of the ABL-imatinib complex that is 10 to 30 times more potent than imatinib in inhibiting BCR-ABL1 tyrosine kinase activity. Nilotinib is approved for the treatment of imatinib-resistant chronic myelogenous leukemia (CML), with reported efficacy in patients with central nervous system (CNS) involvement.87,88 Nilotinib has been studied in a single study of KIT-mutated/amplified melanoma that included patients with imatinib-resistance and those with treated CNS disease. Nilotinib appeared to be active in imatinib-resistant melanoma, although no responses were seen in the CNS disease cohort.89 Overall, the response rates observed with KIT inhibition in melanoma are much lower than those observed in CML and GIST.
CONCLUSION AND FUTURE DIRECTIONS
Prior to 2011, the only approved agents for the treatment of advanced melanoma were dacarbazine and high-dose interleukin-2. Since 2011, drug approvals in melanoma have proceeded at a frenetic pace unmatched in any other disease. The primary events underlying this are advances in our understanding of the gene mutation landscape driving melanoma tumorigenesis, accompanied by insights into the means by which tumors circumvent the induction of effective anti-tumor T-cell responses. These insights have resulted in the development of inhibitors targeting MAPK pathway kinases BRAF, MEK, and NRAS), KIT, and regulatory immune checkpoints (CTLA-4 and PD-1). Although BRAF/MEK inhibition results in profound reductions and even occasional complete responses in patients, these responses are typically short lived, rarely lasting more than 9 to 11 months; the encorafenib/binimetinib combination may improve that duration marginally. However, the signature therapeutic advance in melanoma of the past decade is immunotherapy, particularly the development of inhibitors of CTLA-4 and PD-1 immune checkpoints. With these agents, significant proportions of treated patients remain free of progression off-therapy (ipilimumab 23%; nivolumab 34%; pembrolizumab 35%; ipilimumab/nivolumab 64%), and some patients can be successfully re-induced after delayed progression. Separately, the high response rates observed with the use of KIT inhibitors in CML and GIST have not been observed in KIT mutated/amplified melanoma and development of agents in this space has been limited. The challenges ahead center around identifying predictive biomarkers and circumventing primary or acquired resistance, with the eventual goal of producing durable remissions in the majority of treated patients.
Our improved understanding of the mechanisms of acquired resistance to BRAF/MEK inhibitors suggests that anti-tumor activity may be achieved by targeting multiple pathways, possibly with combination regimens comprising other inhibitors and/or immunotherapy. Preclinical data supports the use of combination strategies targeting both ERK and PI3K/mTOR to circumvent acquired resistance.90 Ongoing studies are evaluating combinations with biguanides (metformin: NCT02143050 and NCT01638676; phenformin: NCT03026517), HSP90 inhibitors (XL888: NCT02721459; AT13387: NCT02097225), and decitabine (NCT01876641).
One complexity affecting management of resistance in the targeted therapy landscape remains tumor heterogeneity, particularly intra- and intertumoral heterogeneity, which may explain the apparent contradiction between continued efficacy of BRAF inhibitors in BRAF-resistant tumors and preclinical data predicting slower progression of resistant tumors on cessation of BRAF inhibitors.91–94 These data provide a rationale to investigate intermittent dosing regimens with BRAF/MEK inhibitors; several studies exploring this approach are ongoing (NCT01894672 and NCT02583516).
Given the specificity, adaptability, and memory response associated with immunotherapy, it is likely that these agents will be used to treat the majority of patients regardless of mutational status. Hence, identifying predictive biomarkers of response to immune checkpoint inhibitors is vital. The presence of CD8+ T-cell infiltrate and IFN-γ gene signature, which indicate an “inflamed” tumor microenvironment, are highly predictive of clinical benefit from PD-1 inhibitors.95,96 However, not all PD-1 responders have “inflamed” tumor microenvironments, and not all patients with an “inflamed” tumor microenvironment respond to immune checkpoint inhibitors. The complexity of the immune system is reflected in the multiple non-redundant immunologic pathways, both positive and negative, with checkpoints and ligands that emerge dynamically in response to treatment. Given the dynamic nature of the immune response, it is unlikely that any single immunologic biomarker identified pre-treatment will be completely predictive. Rather, the complexity of the biomarker approach must match the complexity of the immune response elicited, and will likely incorporate multifarious elements including CD8+ T-cell infiltrate, IFN-γ gene signature, and additional elements including microbiome, genetic polymorphisms, and tumor mutation load. The goal is to use multiple markers to guide development of combinations and then, depending on initial response, to examine tumors for alterations to guide decisions about additional treatment(s) to improve responses, with the eventual goal being durable clinical responses for all patients.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Guy GP, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982-2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6.
- Anderson WF, Pfeiffer RM, Tucker MA, Rosenberg PS. Divergent cancer pathways for early-onset and late-onset cutaneous malignant melanoma. Cancer 2009;115:4176–85.
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135–47.
- Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340–6.
- Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63.
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681–96.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 2014;15:323–32.
- Hauschild A, Grob J-J, Demidov LV, et al. DaBRAFenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined daBRAFenib and trametinib. N Engl J Med 2015;372:30–9.
- Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76.
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107:4275–80.
- Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577–81.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54.
- Weber JS, Angelo SP D’, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134–44.
- Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33.
- Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372:2006–17.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
- Eggermont AMM. Therapeutic vaccines in solid tumours: can they be harmful? Eur J Cancer 2009;45:2087–90.
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012;4:127ps8.
- Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol 2014;192:5451–8.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62–8.
- Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992;356:607–9.
- Greenfield EA, Nguyen KA, Kuchroo VK. CD28/B7 costimulation: a review. Crit Rev Immunol 1998;18:389–418.
- Sharpe AH, Abbas AK. T-cell costimulation--biology, therapeutic potential, and challenges. N Engl J Med 2006;355:973–5.
- Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001;19:565–94.
- Collins AV, Brodie DW, Gilbert RJC, et al. The interaction properties of costimulatory molecules revisited. Immunity 2002;17:201–10.
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459–65.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734–6.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704.
- Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 2002;169:5538–45.
- Youngnak P, Kozono Y, Kozono H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun 2003;307:672–7.
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945–54.
- Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005;25:9543–53.
- Riley JL. PD-1 signaling in primary T cells. Immunol Rev 2009;229:114–25.
- Wolchok JD, Hodi FS, Weber JS, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013;1291:1–13.
- Weber JS, O’Day S, Urba W, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol 2008;26:5950–6.
- Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A 2008;105:3005–10.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 2010;11:155–64.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–94.
- Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2014;2:846–56.
- Poole RM. Pembrolizumab: first global approval. Drugs 2014;74:1973–81.
- Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30.
- Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21:4286–93.
- Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18:31–41.
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am 2009;23:529–45, ix.
- Satyamoorthy K, Li G, Gerrero MR, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res 2003;63:756–9.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54.
- Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14.
- Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010;8:67.
- Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS ONE 2012;7:e35309.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431–5.
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30.
- Thomas NE, Edmiston SN, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncology 2015;1:359–68.
- Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother 2012;35:66–72.
- Johnson DB, Lovly CM, Flavin M, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015;3:288–95.
- Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;4:959–67.
- Kiuru M, Busam KJ. The NF1 gene in tumor syndromes and melanoma. Lab Invest 2017;97:146–57.
- Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 2014;74:2340–50.
- Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol 2005;23:5281–93.
- Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004;22:4456–62.
- Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res 2012;18:555–67.
- Davar D, Kirkwood JM. CCR 20th anniversary commentary: MAPK/ERK pathway inhibition in melanoma-kinase inhibition redux. Clin Cancer Res 2015;21:5412–4.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207–15.
- Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010;468:968–72.
- Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7.
- Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov 2014;4:69–79.
- Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80–93.
- Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4:94–109.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14.
- Dummer R, Ascierto PA, Gogas HJ, et al. Results of COLUMBUS Part 1: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in BRAF-mutant melanoma. Presented at Society for Melanoma Research 2016 Congress. November 6-9, 2016. Boston (MA).
- Adelmann CH, Ching G, Du L, et al. Comparative profiles of BRAF inhibitors: the paradox index as a predictor of clinical toxicity. Oncotarget 2016;7:30453–60.
- Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45.
- Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol 2005;36:486–93.
- Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 2008;14:6821–8.
- Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res 2010;23:210–5.
- Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 2011;29:2904–9.
- Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 2013;31:3182–90.
- Flaherty KT, Hamilton BK, Rosen MA, et al. Phase I/II trial of imatinib and bevacizumab in patients with advanced melanoma and other advanced cancers. Oncologist 2015;20:952–9.
- Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia 2013;27:107–12.
- Reinwald M, Schleyer E, Kiewe P, et al. Efficacy and pharmacologic data of second-generation tyrosine kinase inhibitor nilotinib in BCR-ABL-positive leukemia patients with central nervous system relapse after allogeneic stem cell transplantation. Biomed Res Int 2014;2014:637059.
- Carvajal RD, Lawrence DP, Weber JS, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res 2015;21:2289–96.
- Carlino MS, Todd JR, Gowrishankar K, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol 2014;8:544–54.
- Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther 2013;12:1332–42.
- Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer 2014;120:3142–53.
- Thakur M Das, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–5.
- Thakur M Das, Stuart DD. Molecular pathways: response and resistance to BRAF and MEK inhibitors in BRAF(V600E) tumors. Clin Cancer Res 2014;20:1074–80.
- Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71.
- Ayers M, Lunceford J, Nebozhyn M, et al. Relationship between immune gene signatures and clinical response to PD-1 blockade with pembrolizumab (MK-3475) in patients with advanced solid tumors. J Immunotherapy Cancer 2015;3(Suppl 2):P80.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Guy GP, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982-2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6.
- Anderson WF, Pfeiffer RM, Tucker MA, Rosenberg PS. Divergent cancer pathways for early-onset and late-onset cutaneous malignant melanoma. Cancer 2009;115:4176–85.
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135–47.
- Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340–6.
- Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63.
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681–96.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 2014;15:323–32.
- Hauschild A, Grob J-J, Demidov LV, et al. DaBRAFenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined daBRAFenib and trametinib. N Engl J Med 2015;372:30–9.
- Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76.
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107:4275–80.
- Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577–81.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54.
- Weber JS, Angelo SP D’, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134–44.
- Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33.
- Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372:2006–17.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
- Eggermont AMM. Therapeutic vaccines in solid tumours: can they be harmful? Eur J Cancer 2009;45:2087–90.
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012;4:127ps8.
- Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol 2014;192:5451–8.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62–8.
- Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992;356:607–9.
- Greenfield EA, Nguyen KA, Kuchroo VK. CD28/B7 costimulation: a review. Crit Rev Immunol 1998;18:389–418.
- Sharpe AH, Abbas AK. T-cell costimulation--biology, therapeutic potential, and challenges. N Engl J Med 2006;355:973–5.
- Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001;19:565–94.
- Collins AV, Brodie DW, Gilbert RJC, et al. The interaction properties of costimulatory molecules revisited. Immunity 2002;17:201–10.
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459–65.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734–6.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704.
- Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 2002;169:5538–45.
- Youngnak P, Kozono Y, Kozono H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun 2003;307:672–7.
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945–54.
- Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005;25:9543–53.
- Riley JL. PD-1 signaling in primary T cells. Immunol Rev 2009;229:114–25.
- Wolchok JD, Hodi FS, Weber JS, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013;1291:1–13.
- Weber JS, O’Day S, Urba W, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol 2008;26:5950–6.
- Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A 2008;105:3005–10.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 2010;11:155–64.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–94.
- Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2014;2:846–56.
- Poole RM. Pembrolizumab: first global approval. Drugs 2014;74:1973–81.
- Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30.
- Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21:4286–93.
- Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18:31–41.
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am 2009;23:529–45, ix.
- Satyamoorthy K, Li G, Gerrero MR, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res 2003;63:756–9.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54.
- Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14.
- Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010;8:67.
- Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS ONE 2012;7:e35309.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431–5.
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30.
- Thomas NE, Edmiston SN, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncology 2015;1:359–68.
- Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother 2012;35:66–72.
- Johnson DB, Lovly CM, Flavin M, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015;3:288–95.
- Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;4:959–67.
- Kiuru M, Busam KJ. The NF1 gene in tumor syndromes and melanoma. Lab Invest 2017;97:146–57.
- Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 2014;74:2340–50.
- Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol 2005;23:5281–93.
- Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004;22:4456–62.
- Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res 2012;18:555–67.
- Davar D, Kirkwood JM. CCR 20th anniversary commentary: MAPK/ERK pathway inhibition in melanoma-kinase inhibition redux. Clin Cancer Res 2015;21:5412–4.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207–15.
- Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010;468:968–72.
- Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7.
- Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov 2014;4:69–79.
- Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80–93.
- Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4:94–109.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14.
- Dummer R, Ascierto PA, Gogas HJ, et al. Results of COLUMBUS Part 1: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in BRAF-mutant melanoma. Presented at Society for Melanoma Research 2016 Congress. November 6-9, 2016. Boston (MA).
- Adelmann CH, Ching G, Du L, et al. Comparative profiles of BRAF inhibitors: the paradox index as a predictor of clinical toxicity. Oncotarget 2016;7:30453–60.
- Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45.
- Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol 2005;36:486–93.
- Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 2008;14:6821–8.
- Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res 2010;23:210–5.
- Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 2011;29:2904–9.
- Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 2013;31:3182–90.
- Flaherty KT, Hamilton BK, Rosen MA, et al. Phase I/II trial of imatinib and bevacizumab in patients with advanced melanoma and other advanced cancers. Oncologist 2015;20:952–9.
- Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia 2013;27:107–12.
- Reinwald M, Schleyer E, Kiewe P, et al. Efficacy and pharmacologic data of second-generation tyrosine kinase inhibitor nilotinib in BCR-ABL-positive leukemia patients with central nervous system relapse after allogeneic stem cell transplantation. Biomed Res Int 2014;2014:637059.
- Carvajal RD, Lawrence DP, Weber JS, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res 2015;21:2289–96.
- Carlino MS, Todd JR, Gowrishankar K, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol 2014;8:544–54.
- Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther 2013;12:1332–42.
- Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer 2014;120:3142–53.
- Thakur M Das, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–5.
- Thakur M Das, Stuart DD. Molecular pathways: response and resistance to BRAF and MEK inhibitors in BRAF(V600E) tumors. Clin Cancer Res 2014;20:1074–80.
- Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71.
- Ayers M, Lunceford J, Nebozhyn M, et al. Relationship between immune gene signatures and clinical response to PD-1 blockade with pembrolizumab (MK-3475) in patients with advanced solid tumors. J Immunotherapy Cancer 2015;3(Suppl 2):P80.
Adjuvant Chemotherapy in the Treatment of Colon Cancer
INTRODUCTION
Colorectal cancer (CRC) is one of the most prevalent malignancies and is the fourth most common cancer in the United States, with an estimated 133,490 new cases diagnosed in 2016. Of these, approximately 95,520 are located in the colon and 39,970 are in the rectum.1 CRC is the third leading cause of cancer death in women and the second leading cause of cancer death in men, with an estimated 49,190 total deaths in 2016.2 The incidence appears to be increasing,3 especially in patients younger than 55 years of age;4 the reason for this increase remains uncertain.
A number of risk factors for the development of CRC have been identified. Numerous hered-itary CRC syndromes have been described, including familial adenomatous polyposis,5 hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome,6 and MUTYH-associated polyposis.7,8 A family history of CRC doubles the risk of developing CRC,9 and current guidelines support lowering the age of screening in individuals with a family history of CRC to 10 years younger than the age of diagnosis of the family member or 40 years of age, whichever is lower.10 Patients with a personal history of adenomatous polyps are at increased risk for developing CRC, as are patients with a personal history of CRC, with a relative risk ranging from 3 to 6.11 Ulcerative colitis and Crohn’s disease are associated with the development of CRC and also influence screening, though evidence suggests good control of these diseases may mitigate risk.12 Finally, modifiable risk factors for the development of CRC include high red meat consumption,13 diets low in fiber,14 obesity,13 smoking, alcohol use,15 and physical inactivity16; lifestyle modification targeting these factors has been shown to decrease rates of CRC.17 The majority of colon cancers present with clinical symptoms, often with rectal bleeding, abdominal pain, change in bowel habits, or obstructive symptoms. More rarely, these tumors are detected during screening colonoscopy, in which case they tend to be at an early stage.
SURGICAL MANAGEMENT
A critical goal in the resection of early-stage colon cancer is attaining R0 resection. Patients who achieve R0 resection as compared to R1 (microscopic residual tumor) and R2 (macroscopic residual tumor)18 have significantly improved long-term overall survival.19 Traditionally, open resection of the involved colonic segment was employed, with end-end anastomosis of the uninvolved free margins. Laparoscopic resection for early-stage disease has been utilized in attempts to decrease morbidity of open procedures, with similar outcomes and node sampling.20 Laparoscopic resection appears to provide similar outcomes even in locally advanced disease.21 Right-sided lesions are treated with right colectomy and primary ileocolic anastomosis.22 For patients presenting with obstructing masses, the Hartmann procedure is the most commonly performed operation. This involves creation of an ostomy with subtotal colectomy and subsequent ostomy reversal in a 2- or 3-stage protocol.23 Patients with locally advanced disease and invasion into surrounding structures require multivisceral resection, which involves resection en bloc with secondarily involved organs.24 Intestinal perforation presents a unique challenge and is associated with surgical complications, infection, and lower overall survival (OS) and 5-year disease-free survival (DFS). Complete mesocolic excision is a newer technique that has been performed with reports of better oncologic outcome at some centers; however, this approach is not currently considered standard of care.25
STAGING
According to a report by the National Cancer Institute, the estimated 5-year relative survival rates for localized colon cancer (lymph node negative), regional (lymph node positive) disease, and distant (metastatic) disease are 89.9%, 71.3%, and 13.9%, respectively.1 However, efforts have been made to further classify patients into distinct categories to allow fine-tuning of prognostication. In the current system, staging of colon cancer utilizes the American Joint Committee on Cancer tumor/node/metastasis (TNM) system.20 Clinical and pathologic features include depth of invasion, local invasion of other organs, nodal involvement, and presence of distant metastasis (Table 1). Studies completed prior to the adoption of the TNM system used the Dukes criteria, which divided colon cancer into A, B, and C, corresponding to TNM stage I, stage IIA–IIC, and stage IIIA-IIIC. This classification is rarely used in more contemporary studies.
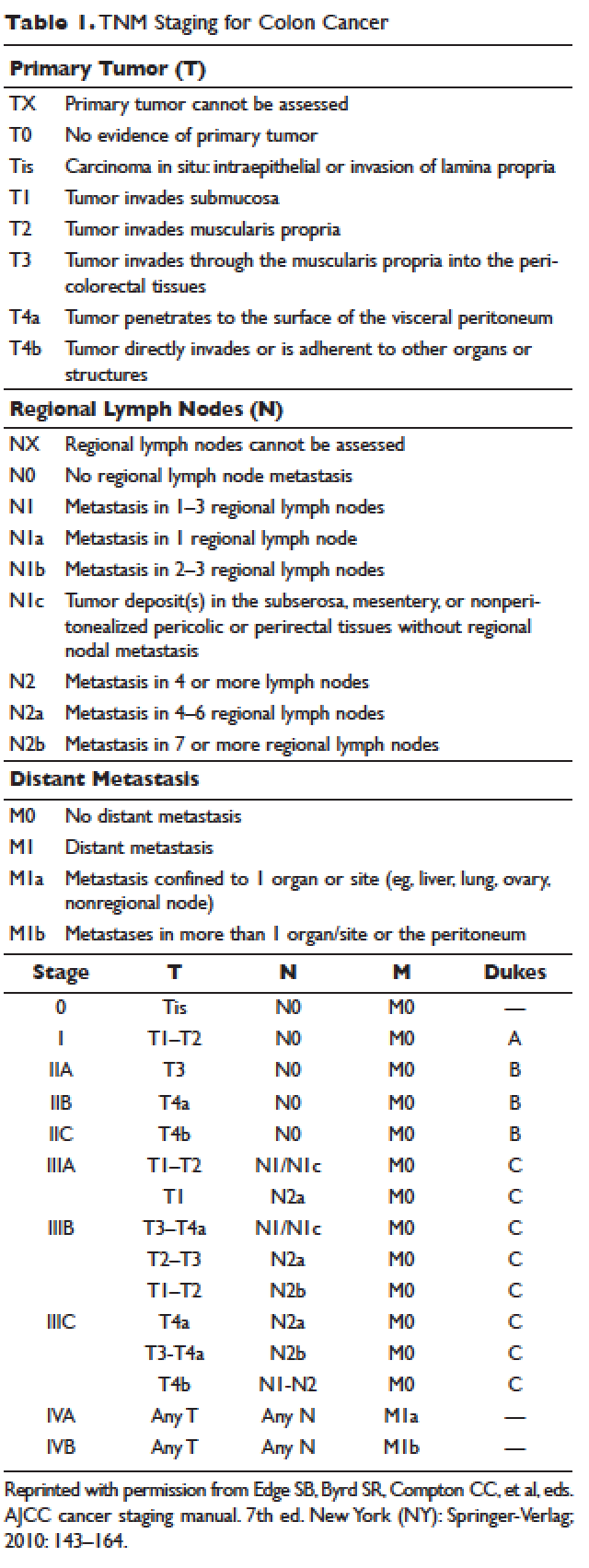
APPROACH TO ADJUVANT CHEMOTHERAPY
Adjuvant chemotherapy seeks to eliminate micrometastatic disease present following curative surgical resection. When stage 0 cancer is discovered incidentally during colonoscopy, endoscopic resection alone is the management of choice, as presence of micrometastatic disease is exceedingly unlikely.26 Stage I–III CRCs are treated with surgical resection withcurative intent. The 5-year survival rate for stage I and early-stage II CRC is estimated at 97% with surgery alone.27,28 The survival rate drops to about 60% for high-risk stage II tumors (T4aN0), and down to 50% or less for stage II-T4N0 or stage III cancers. Adjuvant chemotherapy is generally recommended to further decrease the rates of distant recurrence in certain cases of stage II and in all stage III tumors.
DETERMINATION OF BENEFIT FROM CHEMOTHERAPY: PROGNOSTIC MARKERS
Prior to administration of adjuvant chemotherapy, a clinical evaluation by the medical oncologist to determine appropriateness and safety of treatment is paramount. Poor performance status and comorbid conditions may indicate risk for excessive toxicity and minimal benefit from chemotherapy. CRC commonly presents in older individuals, with the median age at diagnosis of 69 years for men and 73 years for women.29 In this patient population, comorbidities such as cardiovascular disease, diabetes, and renal dysfunction are more prevalent.30 Decisions regarding adjuvant chemotherapy in this patient population have to take into consideration the fact that older patients may experience higher rates of toxicity with chemotherapy, including gastrointestinal toxicities and marrow suppression.31 Though some reports indicate patients older than 70 years derive similar benefit from adjuvant chemotherapy,32,33 a large pooled analysis of the ACCENT database, which included 7 adjuvant therapy trials and 14,528 patients, suggested limited benefit from the addition of oxaliplatin to fluorouracil in elderly patients.32 Other factors that weigh on the decision include stage, pathology, and presence of high-risk features. A common concern in the postoperative setting is delaying initiation of chemotherapy to allow adequate wound healing; however, evidence suggests that delays longer than 8 weeks leads to worse overall survival, with hazard ratios (HR) ranging from 1.4 to 1.7.34,35 Thus, the start of adjuvant therapy should ideally be within this time frame.
HIGH-RISK FEATURES
Multiple factors have been found to predict worse outcome and are classified as high-risk features (Table 2). Histologically, high-grade or poorly differentiated tumors are associated with higher recurrence rate and worse outcome.36 Certain histological subtypes, including mucinous and signet-ring, both appear to have more aggressive biology.37 Presence of microscopic invasion into surrounding blood vessels (vascular invasion) and nerves (perineural invasion) is associated with lower survival.38 Penetration of the cancer through the visceral peritoneum (T4a) or into surrounding structures (T4b) is associated with lower survival.36 During surgical resection, multiple lymph nodes are removed along with the primary tumor to evaluate for metastasis to the regional nodes. Multiple analyses have demonstrated that removal and pathologic assessment of fewer than 12 lymph nodes is associated with high risk of missing a positive node, and is thus equated with high risk.39–41 In addition, extension of tumor beyond the capsules of any single lymph node, termed extracapsular extension, is associated with an increased risk of all-cause mortality.42 Tumor deposits, or focal aggregates of adenocarcinoma in the pericolic fat that are not contiguous with the primary tumor and are not associated with lymph nodes, are currently classified as lymph nodes as N1c in the current TNM staging system. Presence of these deposits has been found to predict poor outcome stage for stage.43 Obstruction and/or perforation secondary to the tumor are also considered high-risk features that predict poor outcome.

SIDEDNESS
As reported at the 2016 American Society of Clinical Oncology annual meeting, tumor location predicts outcome in the metastatic setting. A report by Venook and colleagues based on a post-hoc analysis found that in the metastatic setting, location of the tumor primary in the left side is associated with longer OS (33.3 months) when compared to the right side of the colon (19.4 months).44 A retrospective analysis of multiple databases presented by Schrag and colleagues similarly reported inferior outcomes in patients with stage III and IV disease who had right-sided primary tumors.45 However, the prognostic implications for stage II disease remain uncertain.
BIOMARKERS
Given the controversy regarding adjuvant therapy of patients with stage II colon cancer, multiple biomarkers have been evaluated as possible predictive markers that can assist in this decision. The mismatch repair (MMR) system is a complex cellular enzymatic mechanism that identifies and corrects DNA errors during cell division and prevents mutagenesis.46 The familial cancer syndrome HNPCC is linked to alteration in a variety of MMR genes, leading to deficient mismatch repair (dMMR), also termed microsatellite instability-high (MSI-high).47,48 Epigenetic modification can also lead to silencing of the same implicated genes and accounts for 15% to 20% of sporadic colorectal cancer.49 These epigenetic modifications lead to hypermethylation of the promotor region of MLH1 in 70% of cases.50 The 4 MMR genes most commonly tested are MLH-1, MSH2, MSH6, and PMS2. Testing can be performed by immunohistochemistry or polymerase chain reaction.51 Across tumor histology and stage, MSI status is prognostic. Patients with MSI-high tumors have been shown to have improved prognosis and longer OS both in stage II and III disease52–54 and in the metastatic setting.55 However, despite this survival benefit, there is conflicting data as to whether patients with stage II, MSI-high colon cancer may benefit less from adjuvant chemotherapy. One early retrospective study compared outcomes of 70 patients with stage II and III disease and dMMR to those of 387 patients with stage II and III disease and proficient mismatch repair (pMMR). Adjuvant fluorouracil with leucovorin improved DFS for patients with pMMR (HR 0.67) but not for those with dMMR (HR 1.10). In addition, for patients with stage II disease and dMMR, the HR for OS was inferior at 2.95.56 Data collected from randomized clinical trials using fluorouracil-based adjuvant chemotherapy were analyzed in an attempt to predict benefit based on MSI status. Benefit was only seen in pMMR patients, with a HR of 0.72; this was not seen in the dMMR patients.57 Subsequent studies have had different findings and did not demonstrate a detrimental effect of fluorouracil in dMMR.58,59 For stage III patients, MSI status does not appear to affect benefit from chemotherapy, as analysis of data from the NSABP C-07 trial (Table 3) demonstrated benefit of FOLFOX (leucovorin, fluorouracil, oxaliplatin) in patients with dMMR status and stage III disease.59
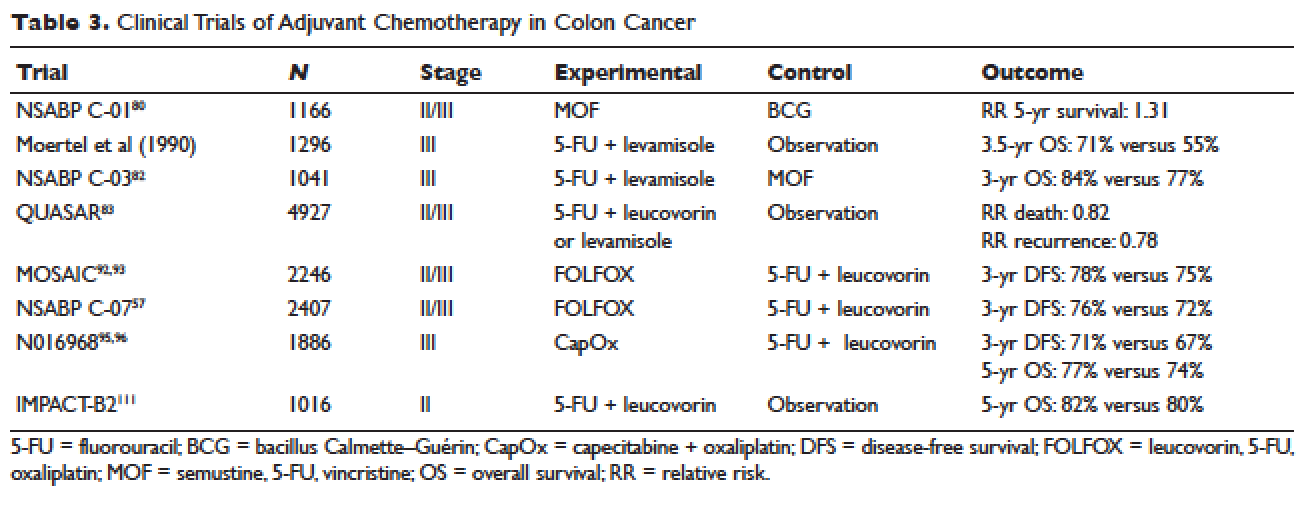
Another genetic abnormality identified in colon cancers is chromosome 18q loss of heterozygosity (LOH). The presence of 18q LOH appears to be inversely associated with MSI-high status. Some reports have linked presence of 18q with worse outcome,60 but others question this, arguing the finding may simply be related to MSI status.61,62 This biomarker has not been established as a clear prognostic marker that can aid clinical decisions.
Most recently, expression of caudal-type homeobox transcription factor 2 (CDX2) has been reported as a novel prognostic and predictive tool. A 2015 report linked lack of expression of CDX2 to worse outcome; in this study, 5-year DFS was 41% in patients with CDX2-negative tumors versus 74% in the CDX2-positive tumors, with a HR of disease recurrence of 2.73 for CDX2-negative tumors.63 Similar numbers were observed in patients with stage II disease, with 5-year OS of 40% in patients with CDX2-negative tumors versus 70% in those with CDX2-positive tumors. Treatment of CDX2-negative patients with adjuvant chemotherapy improved outcomes: 5-year DFS in the stage II subgroup was 91% with chemotherapy versus 56% without, and in the stage III subgroup, 74% with chemotherapy versus 37% without. The authors concluded that patients with stage II and III colon cancer that is CDX2-negative may benefit from adjuvant chemotherapy. Importantly, CDX2-negativity is a rare event, occurring in only 6.9% of evaluable tumors.
RISK ASSESSMENT TOOLS
Several risk assessment tools have been developed in an attempt to aid clinical decision making regarding adjuvant chemotherapy for patients with stage II colon cancer. The Oncotype DX Colon Assay analyses a 12-gene signature in the pathologic sample and was developed with the goal to improve prognostication and aid in treatment decision making. The test utilizes reverse transcription-PCR on RNA extracted from the tumor.64 After evaluating 12 genes, a recurrence score is generated that predicts the risk of disease recurrence. This score was validated using data from 3 large clinical trials.65–67 Unlike the Oncotype Dx score used in breast cancer, the test in colon cancer has not been found to predict the benefit from chemotherapy and has not been incorporated widely into clinical practice.
Adjuvant! Online (available at www.adjuvantonline.com) is a web-based tool that combines clinical and histological features to estimate outcome. Calculations are based on US SEER tumor registry-reported outcomes.68 A second web-based tool, Numeracy (available at www.mayoclinic.com/calcs), was developed by the Mayo Clinic using pooled data from 7 randomized clinical trials including 3341 patients.68 Both tools seek to predict absolute benefit for patients treated with fluorouracil, though data suggests Adjuvant! Online may be more reliable in its predictive ability.69 Adjuvant! Online has also been validated in an Asian population70 and patients older than 70 years.71
MUTATIONAL ANALYSIS
Multiple mutations in proto-oncogenes have been found in colon cancer cells. One such proto-oncogene is BRAF, which encodes a serine-threonine kinase in the rapidly accelerated fibrosarcoma (RAF). Mutations in BRAF have been found in 5% to 10% of colon cancers and are associated with right-sided tumors.72 As a prognostic marker, some studies have associated BRAF mutations with worse prognosis, including shorter time to relapse and shorter OS.73,74 Two other proto-oncogenes are Kristen rat sarcoma viral oncogene homolog (KRAS) and neuroblastoma rat sarcoma viral oncogene homolog (NRAS), both of which encode proteins downstream of epidermal growth factor receptor (EGFR). KRAS and NRAS mutations have been shown to be predictive in the metastatic setting where they predict resistance to the EGFR inhibitors cetuximab and panitumumab.75,76 The effect of KRAS and NRAS mutations on outcome in stage II and III colon cancer is uncertain. Some studies suggest worse outcome in KRAS-mutated cancers,77 while others failed to demonstrate this finding.73
CASE PRESENTATION 1
A 53-year-old man with no past medical history presents to the emergency department with early satiety and generalized abdominal pain. Laboratory evaluation shows a microcytic anemia with normal white blood cell count, platelet count, renal function, and liver function tests. Computed tomography (CT) scan of the abdomen and pelvis show a 4-cm mass in the transverse colon without obstruction and without abnormality in the liver. CT scan of the chest does not demonstrate pathologic lymphadenopathy or other findings. He undergoes robotic laparoscopic transverse colon resection and appendectomy. Pathology confirms a 3.5-cm focus of adenocarcinoma of the colon with invasion through the muscularis propria and 5 of 27 regional lymph nodes positive for adenocarcinoma and uninvolved proximal, distal, and radial margins. He is given a stage of IIIB pT3 pN2a M0 and referred to medical oncology for further management, where 6 months of adjuvant FOLFOX chemotherapy is recommended.
ADJUVANT CHEMOTHERAPY IN STAGE III COLON CANCER
Postoperative adjuvant chemotherapy is the standard of care for patients with stage III disease. In the 1960s, infusional fluorouracil was first used to treat inoperable colon cancer.78,79 After encouraging results, the agent was used both intraluminally and intravenously as an adjuvant therapy for patients undergoing resection with curative intent; however, only modest benefits were described.80,81 The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-01 trial (Table 3) was the first study to demonstrate a benefit from adjuvant chemotherapy in colon cancer. This study randomly assigned patients with stage II and III colon cancer to surgery alone, postoperative chemotherapy with fluorouracil, semustine, and vincristine (MOF), or postoperative bacillus Calmette-Guérin (BCG). DFS and OS were significantly improved with MOF chemotherapy.82 In 1990, a landmark study reported on outcomes after treatment of 1296 patients with stage III colon cancer with adjuvant fluorouracil and levamisole for 12 months. The combination was associated with a 41% reduction in risk of cancer recurrence and a 33% reduction in risk of death.83 The NSABP C-03 trial (Table 3) compared MOF to the combination of fluorouracil and leucovorin and demonstrated improved 3-year DFS (69% versus 73%) and 3-year OS (77% versus 84%) in patients with stage III disease.84 Building on these outcomes, the QUASAR study (Table 3) compared fluorouracil in combination with one of levamisole, low-dose leucovorin, or high-dose leucovorin. The study enrolled 4927 patients and found worse outcomes with fluorouracil plus levamisole and no difference in low-doseversus high-dose leucovorin.85 Levamisole fell out of use after associations with development of multifocal leukoencephalopathy,86 and was later shown to have inferior outcomes versus leucovorin when combined with fluorouracil.87,88 Intravenous fluorouracil has shown similar benefit when administered by bolus or infusion,89 although continuous infusion has been associated with lower incidence of severe toxicity.90 The efficacy of the oral fluoropyrimidine capecitabine has been shown to be equivalent to that of fluorouracil.91
Fluorouracil-based treatment remained the standard of care until the introduction of oxaliplatin in the mid-1990s. After encouraging results in the metastatic setting,92,93 the agent was moved to the adjuvant setting. The MOSAIC trial (Table 3) randomly assigned patients with stage II and III colon cancer to fluorouracil with leucovorin (FULV) versus FOLFOX given once every 2 weeks for 12 cycles. Analysis with respect to stage III patients showed a clear survival benefit, with a 10-year OS of 67.1% with FOLFOX chemotherapy versus 59% with fluorouracil and leucovorin.94,95 The NSABP C-07 (Table 3) trial used a similar trial design but employed bolus fluorouracil. More than 2400 patients with stage II and III colon cancer were randomly assigned to bolus FULV or bolus fluorouracil, leucovorin, and oxaliplatin (FLOX). The addition of oxaliplatin significantly improved outcomes, with 4-year DFS of 67% versus 71.8% for FULV and FLOX, respectively, and a HR of death of 0.80 with FLOX.59,96 The multicenter N016968 trial (Table 3) randomly assigned 1886 patients with stage III colon cancer to adjuvant capecitabine plus oxaliplatin (XELOX) or bolus fluorouracil plus leucovorin (FU/FA). The 3-year DFS was 70.9% versus 66.5% with XELOX and FU/FA, respectively, and 5-year OS was 77.6% versus 74.2%, respectively.97,98
In the metastatic setting, additional agents have shown efficacy, including irinotecan,99,100 bevacizumab,101,102 cetuximab,103,104 and regorafenib.105 This observation led to testing of these agents in earlier stage disease. The CALGB 89803 trial compared fluorouracil, leucovorin, and irinotecan to fluorouracil with leucovorin alone. No benefit in 5-year DFS or OS was seen.106 Similarly, infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) was not found to improve 5-year DFS as compared to fluorouracil with leucovorin alone in the PETACC-3 trial.107 The NSABP C-08 trial considered the addition of bevacizumab to FOLFOX. When compared to FOLFOX alone, the combination of bevacizumab to FOLFOX had similar 3-year DFS (77.9% versus 75.1%) and 5-year OS (82.5% versus 80.7%).108 This finding was confirmed in the Avant trial.109 The addition of cetuximab to FOLFOX was equally disappointing, as shown in the N0147 trial110 and PETACC-8 trial.111 Data on regorafenib in the adjuvant setting for stage III colon cancer is lacking; however, 2 ongoing clinical trials, NCT02425683 and NCT02664077, are each studying the use of regorafenib following completion of FOLFOX for patients with stage III disease.
Thus, after multiple trials comparing various regimens and despite attempts to improve outcomes by the addition of a third agent, the standard of care per National Comprehensive Cancer Network (NCCN) guidelines for management of stage III colon cancer remains 12 cycles of FOLFOX chemotherapy. Therapy should be initiated within 8 weeks of surgery. Data are emerging to support a short duration of therapy for patients with low-risk stage III tumors, as shown in an abstract presented at the 2017 American Society of Clinical Oncology annual meeting. The IDEA trial was a pooled analysis of 6 randomized clinical trials across multiple countries, all of which evaluated 3 versus 6 months of FOLFOX or capecitabine and oxaliplatin in the treatment of stage III colon cancer. The analysis was designed to test non-inferiority of 3 months of therapy as compared to 6 months. The analysis included 6088 patients across 244 centers in 6 countries. The overall analysis failed to establish noninferiority. The 3-year DFS rate was 74.6% for 3 months and 75.5% for 6 months, with a DFS HR of 1.07 and a confidence interval that did not meet the prespecified endpoint. Subgroup analysis suggested noninferiority for lower stage disease (T1–3 or N1) but not for higher stage disease (T4 or N2). Given the high rates of neuropathy with 6 months of oxaliplatin, these results suggest that 3 months of adjuvant therapy can be considered for patients with T1–3 or N1 disease in an attempt to limit toxicity.112
CASE PRESENTATION 2
A 57-year-old woman presents to the emergency department with fever and abdominal pain. CT of the abdomen and pelvis demonstrates a left-sided colonic mass with surrounding fat stranding and pelvic abscess. She is taken emergently for left hemicolectomy, cholecystectomy, and evacuation of pelvic abscess. Pathology reveals a 5-cm adenocarcinoma with invasion through the visceral peritoneum; 0/22 lymph nodes are involved. She is given a diagnosis of stage IIC and referred to medical oncology for further management. Due to her young age and presence of high-risk features, she is recommended adjuvant therapy with FOLFOX for 6 months.
ADJUVANT CHEMOTHERAPY IN STAGE II COLON CANCER
Because of excellent outcomes with surgical resection alone for stage II cancers, the use of adjuvant chemotherapy for patients with stage II disease is controversial. Limited prospective data is available to guide adjuvant treatment decisions for stage II patients. The QUASAR trial, which compared observation to adjuvant fluorouracil and leucovorin in patients with early-stage colon cancer, included 2963 patients with stage II disease and found a relative risk (RR) of death or recurrence of 0.82 and 0.78, respectively. Importantly, the absolute benefit of therapy was less than 5%.113 The IMPACT-B2 trial (Table 3) combined data from 5 separate trials and analyzed 1016 patients with stage II colon cancer who received fluorouracil with leucovorin or observation. Event-free survival was 0.86 versus 0.83 and 5-year OS was 82% versus 80%, suggesting no benefit.114 The benefit of addition of oxaliplatin to fluorouracil in stage II disease appears to be less than the benefit of adding this agent in the treatment of stage III CRC. As noted above, the MOSAIC trial randomly assigned patients with stage II and III colon cancer to receive adjuvant fluorouracil and leucovorin with or without oxaliplatin for 12 cycles. After a median follow-up of 9.5 years, 10-year OS rates for patients with stage II disease were 78.4% versus 79.5%. For patients with high-risk stage II disease (defined as T4, bowel perforation, or fewer than 10 lymph nodes examined), 10-year OS was 71.7% and 75.4% respectively, but these differences were not statistically significant.94
Because of conflicting data as to the benefit of adding oxaliplatin in stage II disease, oxaliplatin is not recommended for standard-risk stage II patients. The use of oxaliplatin in high-risk stage II tumors should be weighed carefully given the toxicity risk. Oxaliplatin is recognized to cause sensory neuropathy in many patients, which can become painful and debilitating.115 Two types of neuropathy are associated with oxaliplatin: acute and chronic. Acute neuropathy manifests most often as cold-induced paresthesias in the fingers and toes and is quite common, affecting up to 90% of patients. These symptoms are self-limited and resolve usually within 1 week of each treatment.116 Some patients, with reports ranging from 10% to 79%, develop chronic neuropathy that persists for 1 year or more and causes significant decrements in quality of life.117 Patients older than age 70 may be at greater risk for oxaliplatin-induced neuropathy, which would increase risk of falls in this population.118 In addition to neuropathy, oxaliplatin is associated with hypersensitivity reactions that can be severe and even fatal.119 In a single institution series, the incidence of severe reactions was 2%.120 Desensitization following hypersensitivity reactions is possible but requires a time-intensive protocol.121
Based on the inconclusive efficacy findings and due to concerns over toxicity, each decision must be individualized to fit patient characteristics and preferences. In general, for patients with stage II disease without high-risk features, an individualized discussion should be held as to the risks and benefits of single-agent fluorouracil, and this treatment should be offered in cases where the patient or provider would like to be aggressive. Patients with stage II cancer who have 1 or more high-risk features are often recommended adjuvant chemotherapy. Whether treatment with fluorouracil plus leucovorin or FOLFOX is preferred remains uncertain, and thus the risks and the potential gains of oxaliplatin must be discussed with the individual patient. MMR status can also influence the treatment recommendation for patients with stage II disease. In general, patients with standard-risk stage II tumors that are pMMR are offered MMR with leucovorin or oral capecitabine for 12 cycles. FOLFOX is considered for patients with MSI-high disease and those with multiple high-risk features.
MONITORING AFTER THERAPY
After completion of adjuvant chemotherapy, patients enter a period of survivorship. Patients are seen in clinic for symptom and laboratory monitoring of the complete blood count, liver function tests, and carcinoembryonic antigen (CEA). NCCN guidelines support history and physical examination with CEA testing every 3 to 6 months for the first 2 years, then every 6 months for the next 3 years, after which many patients continue to be seen annually. CT imaging of the chest, abdomen, and pelvis for monitoring of disease recurrence is recommended every 6 to 12 months for a total of 5 years. New elevations in CEA or liver function tests should prompt early imaging. Colonoscopy should be performed 1 year after completion of therapy; however, if no preoperative colonoscopy was performed, this should be done 3 to 6 months after completion. Colonoscopy is then repeated in 3 years and then every 5 years unless advanced adenomas are present.122
SUMMARY
The addition of chemotherapy to surgical management of colon cancer has lowered the rate of disease recurrence and improved long-term survival. Adjuvant FOLFOX for 12 cycles is the standard of care for patients with stage III colon cancer and for patients with stage II disease with certain high-risk features. Use of adjuvant chemotherapy in stage II disease without high-risk features is controversial, and treatment decisions should be individualized. Biologic markers such as MSI and CDX2 status as well as patient-related factors including age, overall health, and personal preferences can inform treatment decisions. If chemotherapy is recommended in this setting, it would be with single-agent fluorouracil in an infusional or oral formulation, unless the tumor has the MSI-high feature. Following completion of adjuvant therapy, patients should be followed with clinical evaluation, laboratory testing, and imaging for a total of 5 years as per recommended guidelines.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67(1):7–30.
- United States Cancer Statistics. 1999–2013 incidence and mortality web-based report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute, 2016. www.cdc.gov/uscs. Accessed June 12, 2017.
- Ahnen DJ, Wade SW, Jones WF, et al. The increasing incidence of young-onset colorectal cancer: a call to action. Mayo Clin Proc 2014;89:216–24.
- Jemal A, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States, 1974–2013. J Natl Cancer Inst 2017;109(8).
- Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer 2013;49:3680–5.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60: 950–7.
- van Puijenbroek M, Nielsen M, Tops CM, et al. Identification of patients with (atypical) MUTYH-associated polyposis by KRAS2 c.34G > T prescreening followed by MUTYH hotspot analysis in formalin-fixed paraffin-embedded tissue. Clin Cancer Res 2008;14:139–42.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006;119:807–14.
- Tuohy TM, Rowe KG, Mineau GP, et al. Risk of colorectal cancer and adenomas in the families of patients with adenomas: a population-based study in Utah. Cancer 2014;120:35–42.
- Choi Y, Sateia HF, Peairs KS, Stewart RW. Screening for colorectal cancer. Semin Oncol 2017; 44:34–44.
- Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med 1992;326:658–62.
- Rutter MD. Surveillance programmes for neoplasia in colitis. J Gastroenterol 2011;46 Suppl 1:1–5.
- Giovannucci E. Modifiable risk factors for colon cancer. Gastroenterol Clin North Am 2002;31:925–43.
- Michels KB, Fuchs GS, Giovannucci E, et al. Fiber intake and incidence of colorectal cancer among 76,947 women and 47,279 men. Cancer Epidemiol Biomarkers Prev 2005;14:842–9.
- Omata F, Brown WR, Tokuda Y, et al. Modifiable risk factors for colorectal neoplasms and hyperplastic polyps. Intern Med 2009;48:123–8.
- Friedenreich CM, Neilson HK, Lynch BM. State of the epidemiological evidence on physical activity and cancer prevention. Eur J Cancer 2010;46:2593–604.
- Aleksandrova K, Pischon T, Jenab M, et al. Combined impact of healthy lifestyle factors on colorectal cancer: a large European cohort study. BMC Med 2014;12:168.
- Hermanek P, Wittekind C. The pathologist and the residual tumor (R) classification. Pathol Res Pract 1994;190:115–23.
- Lehnert T, Methner M, Pollok A, et al. Multivisceral resection for locally advanced primary colon and rectal cancer: an analysis of prognostic factors in 201 patients. Ann Surg 2002;235:217–25.
- Feinberg AE, et al. Oncologic outcomes following laparoscopic versus open resection of pT4 colon cancer: a systematic review and meta-analysis. Dis Colon Rectum 2017;60:116–125.
- Vignali A, et al. Laparoscopic treatment of advanced colonic cancer: a case-matched control with open surgery. Colorectal Dis 2013;15:944–8.
- Gainant A. Emergency management of acute colonic cancer obstruction. J Visc Surg 2012;149: e3–e10.
- Rosenman LD. Hartmann’s operation. Am J Surg 1994;168:283–4.
- Lee-Kong S, Lisle D. Surgical management of complicated colon cancer. Clin Colon Rectal Surg 2015;28:228–33.
- Bertelsen CA. Complete mesocolic excision an assessment of feasibility and outcome. Dan Med J 2017;64(2).
- Wolff WI SH. Definitive treatment of “malignant” polyps of the colon. Ann Surg 1975;182:516–25.
- Clinical Outcomes of Surgical Therapy Study Group, Nelson H, Sargent DJ, Wieand HS, et al. A comparison of laparoscopically assisted and open colectomy for colon cancer. N Engl J Med 2004;350:2050–9.
- Gunderson LL, Jessup JM, Sarjent DJ, et al. Revised tumor and node categorization for rectal cancer based on surveillance, epidemiology, and end results and rectal pooled analysis outcomes. J Clin Oncol 2010;28:256–63.
- Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
- Alves A, Panis Y, Mathieu P, et al. Postoperative mortality and morbidity in French patients undergoing colorectal surgery: results of a prospective multicenter study. Arch Surg 2005;140:278–83.
- Popescu RA, Norman A, Ross PJ, et al, Adjuvant or palliative chemotherapy for colorectal cancer in patients 70 years or older. J Clin Oncol 1999;17:2412–8.
- McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
- Tominaga T, Nonaka T, Sumida Y, et al. Effectiveness of adjuvant chemotherapy for elderly patients with lymph node-positive colorectal cancer. World J Surg Oncol 2016;14:197.
- Bos AC, van Erning FN, van Gestel YR, et al. Timing of adjuvant chemotherapy and its relation to survival among patients with stage III colon cancer. Eur J Cancer 2015;51:2553–61.
- Peixoto RD, Kumar A, Speers C, et al. Effect of delay in adjuvant oxaliplatin-based chemotherapy for stage III colon cancer. Clin Colorectal Cancer 2015;14:25–30.
- Compton CC, Fielding LP, Burgart LJ, et al. Prognostic factors in colorectal cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med 2000;124:979–94.
- Lieu CH, Lambert LA, Wolff RA, et al. Systemic chemotherapy and surgical cytoreduction for poorly differentiated and signet ring cell adenocarcinomas of the appendix. Ann Oncol 2012;23:652–8.
- Krasna MJ, Flancbaum L, Cody RP, et al. Vascular and neural invasion in colorectal carcinoma. Incidence and prognostic significance. Cancer 1988;61:1018–23.
- Cianchi F, Palomba A, Boddi V, et al. Lymph node recovery from colorectal tumor specimens: recommendation for a minimum number of lymph nodes to be examined. World J Surg 2002;26:384–9.
- Yoshimatsu K, et al. How many lymph nodes should be examined in Dukes’ B colorectal cancer? Determination on the basis of cumulative survival rate. Hepatogastroenterology 2005;52:1703–6.
- Caplin S, Cerottini JP, Bosman FT, et al. For patients with Dukes’ B (TNM Stage II) colorectal carcinoma, examination of six or fewer lymph nodes is related to poor prognosis. Cancer 1998;83:666–72.
- Veronese N, Nottegar A, Pea A, et al. Prognostic impact and implications of extracapsular lymph node involvement in colorectal cancer: a systematic review with meta-analysis. Ann Oncol 2016;27:42–8.
- Li J, Yang S, Hu J, et al. Tumor deposits counted as positive lymph nodes in TNM staging for advanced colorectal cancer: a retrospective multicenter study. Oncotarget 2016;7:18269–79.
- Venook A, Niedzwiecki D, Innocenti Fet al. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016;34 no. 15 suppl. Abstract 3504.
- Schrag D, Brooks G, Meyerhardt JA ,et al. The relationship between primary tumor sidedness and prognosis in colorectal cancer. J Clin Oncol 2016;34 no. 15 suppl. Abstract 3505.
- Larrea AA, Lujan SA, Kunkel TA. SnapShot: DNA mismatch repair. Cell 2010;141:730 e1.
- Jass JR. Pathology of hereditary nonpolyposis colorectal cancer. Ann N Y Acad Sci 2000;910:62–73.
- Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer 1996;78:1149–67.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812–6.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol 2017;12:24.
- Bupathi M, Wu C. Biomarkers for immune therapy in colorectal cancer: mismatch-repair deficiency and others. J Gastrointest Oncol 2016;7:713–20.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
- Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
- Ogino S, Kuchiba A, Qian ZR, et al. Prognostic significance and molecular associations of 18q loss of heterozygosity: a cohort study of microsatellite stable colorectal cancers. J Clin Oncol 2009; 27:4591–8.
- Kim ST, Lee J, Park SH, et al. The effect of DNA mismatch repair (MMR) status on oxaliplatin-based first-line chemotherapy as in recurrent or metastatic colon cancer. Med Oncol 2010;27:1277–85.
- Sargent DJ, Monges G, Thibodeau SN, et al. Therapy in colon cancer. J Clin Oncol 2010;28:4664.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
- Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 2011;29:1261–270.
- Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses J Clin Oncol 2011;29:3768–74.
- Chang SC, Lin JK, Lin TC, Liang WY. Loss of heterozygosity: an independent prognostic factor of colorectal cancer. World J Gastroenterol 2005;11:778–84.
- Bertagnolli MM, Niedzwiecki D, Compton CC, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol 2009;27:1814–21.
- Bertagnolli MM, Redston M, Compton CC, et al. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: prospective evaluation of biomarkers for stages II and III colon cancer--a study of CALGB 9581 and 89803. J Clin Oncol 2011;29:3153–62.
- Dalerba P, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016;374: 211–22.
- Clark-Langone KM, Wu JY, Sangli C, et al. Biomarker discovery for colon cancer using a 761 gene RT-PCR assay. BMC Genomics 2007;8:279.
- Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011;29:4611–9.
- Niedzwiecki D, Bertagnolli MM, Warren RS, et al. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: results from CALGB 9581. J Clin Oncol 2011;29:3146–52.
- Yothers G, O’Connell MJ, Lee M, et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol 2013;31:4512–9.
- Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
- Gill S, Loprinzi C, Kennecke H, et al. Prognostic web-based models for stage II and III colon cancer: A population and clinical trials-based validation of numeracy and adjuvant! online. Cancer 2011;117:4155–65.
- Jung M, Kim GW, Jung I, et al. Application of the Western-based adjuvant online model to Korean colon cancer patients; a single institution experience. BMC Cancer 2012;12:471.
- Papamichael D, Renfro LA, Matthaiou C, et al. Validity of Adjuvant! Online in older patients with stage III colon cancer based on 2967 patients from the ACCENT database. J Geriatr Oncol 2016;7:422–9.
- Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011;117:4623–32.
- Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466–74.
- Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–8.
- Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol 2014;53:852–64.
- Taieb J, Le Malicot K, Shi Q, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J Natl Cancer Inst 2017;109(5).
- Palumbo LT, Sharpe WS, Henry JS. Cancer of the colon and rectum; analysis of 300 cases. Am J Surg 1965;109:439–44.
- Sharp GS, Benefiel WW. 5-Fluorouracil in the treatment of inoperable carcinoma of the colon and rectum. Cancer Chemother Rep 1962;20:97–101.
- Lawrence W Jr, Terz JJ, Horsley JS 3rd, et al. Chemotherapy as an adjuvant to surgery for colorectal cancer. Ann Surg 1975;181:616–23.
- Grage TD, et al. Adjuvant chemotherapy with 5-fluorouracil after surgical resection of colorectal carcinoma (COG protocol 7041). A preliminary report. Am J Surg 1977;133:59–66.
- Wolmark N, Fisher B, Rockette H, et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: results from NSABP protocol C-01. J Natl Cancer Inst 1988;80:30–6.
- Moertel CG, Fleming TR, Macdonald JS, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N Engl J Med 1990;322:352–8.
- Wolmark N, Rockette H, Fisher B, et al. The benefit of leucovorin-modulated fluorouracil as postoperative adjuvant therapy for primary colon cancer: results from National Surgical Adjuvant Breast and Bowel Project protocol C-03. J Clin Oncol 1993;11:1879–87.
- Comparison of fluorouracil with additional levamisole, higher-dose folinic acid, or both, as adjuvant chemotherapy for colorectal cancer: a randomised trial. QUASAR Collaborative Group. Lancet 2000;355(9215):1588–96.
- Chen TC, Hinton DR, Leichman L, et al. Multifocal inflammatory leukoencephalopathy associated with levamisole and 5-fluorouracil: case report. Neurosurgery 1994;35:1138-42.
- Porschen R, Bermann A, Löffler T, et al. Fluorouracil plus leucovorin as effective adjuvant chemotherapy in curatively resected stage III colon cancer: results of the trial adjCCA-01. J Clin Oncol 2001;19:1787–94.
- Arkenau HT, Bermann A, Rettig K, et al. 5-Fluorouracil plus leucovorin is an effective adjuvant chemotherapy in curatively resected stage III colon cancer: long-term follow-up results of the adjCCA-01 trial. Ann Oncol 2003;14:395–9.
- Weinerman B, Shah A, Fields A, et al. Systemic infusion versus bolus chemotherapy with 5-fluorouracil in measurable metastatic colorectal cancer. Am J Clin Oncol 1992;15:518–23.
- Poplin EA, Benedetti JK, Estes NC, et al. Phase III Southwest Oncology Group 9415/Intergroup 0153 randomized trial of fluorouracil, leucovorin, and levamisole versus fluorouracil continuous infusion and levamisole for adjuvant treatment of stage III and high-risk stage II colon cancer. J Clin Oncol 2005;23:1819–25.
- Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
- de Gramont A, Vignoud J, Tournigand C, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur J Cancer 1997;33:214–9.
- Diaz-Rubio E, Sastre J, Zaniboni A, et al. Oxaliplatin as single agent in previously untreated colorectal carcinoma patients: a phase II multicentric study. Ann Oncol 1998;9:105–8.
- André T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF mutation and mismatch repair status of the MOSAIC Study. J Clin Oncol 2015;33:4176–87.
- Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004;350:2343–51.
- Kuebler JP, Wieand HS, O’Connell MJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2007;25:2198–204.
- Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
- Schmoll HJ, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
- Colucci G, Gebbia V, Paoletti G, et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J Clin Oncol 2005;23:4866–75.
- Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 2004;22:229–37.
- Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42.
- Saltz LB, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008;26:2013–9.
- Cremolini C, Loupakis F, Ruzzo A, et al. Predictors of benefit in colorectal cancer treated with cetuximab: are we getting “Lost in TranslationAL”? J Clin Oncol 2010;28:e173–4.
- Sorich MJ, Wiese MD, Rowland D, et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015;26:13–21.
- Grothey A, van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):303–12.
- Saltz LB, Niedzwiecki D, Hollis D, et al. Irinotecan fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: results of CALGB 89803. J Clin Oncol 2007;25:3456–61.
- Van Cutsem E, et al. Randomized phase III trial comparing biweekly infusional fluorouracil/leucovorin alone or with irinotecan in the adjuvant treatment of stage III colon cancer: PETACC-3. J Clin Oncol 2009;27:3117–25.
- Allegra CJ, et al. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J Clin Oncol 2013;31:359–64.
- de Gramont A, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol 2012;13:1225–33.
- Alberts SR, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA 2012;307:1383–93.
- Taieb J, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:862–73.
- Shi Q, Sobrero AF, Shields AF, et al. Prospective pooled analysis of six phase III trials investigating duration of adjuvant (adjuvant) oxaliplatin-based therapy (3 vs 6 months) for patients (pts) with stage III colon cancer (CC): The IDEA (International Duration Evaluation of Adjuvant chemotherapy) collaboration. In: Proceedings from the American Society of Clinical Oncology; June 1–5, 2017; Chicago. Abstract LBA1.
- Quasar Collaborative Group; Gray R, Barnwell J, McConkey C, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370(9604):2020–9.
- Efficacy of adjuvant fluorouracil and folinic acid in B2 colon cancer. International Multicentre Pooled Analysis of B2 Colon Cancer Trials (IMPACT B2) Investigators. J Clin Oncol 1999;17:1356–63.
- Kidwell KM, et al. Long-term neurotoxicity effects of oxaliplatin added to fluorouracil and leucovorin as adjuvant therapy for colon cancer: results from National Surgical Adjuvant Breast and Bowel Project trials C-07 and LTS-01. Cancer 2012;118:5614–22.
- Beijers AJ, Mols F, Vreugdenhil G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer 2014;22:1999–2007.
- Mols F, Beijers T, Lemmens V, et al. Chemotherapy-induced neuropathy and its association with quality of life among 2- to 11-year colorectal cancer survivors: results from the population-based PROFILES registry. J Clin Oncol 2013;31:2699–707.
- Raphael MJ, Fischer HD, Fung K, et al. Neurotoxicity outcomes in a population-based cohort of elderly patients treated with adjuvant oxaliplatin for colorectal cancer. Clin Colorectal Cancer 2017 March 24.
- Toki MI, Saif MW, Syrigos KN. Hypersensitivity reactions associated with oxaliplatin and their clinical management. Expert Opin Drug Saf 2014;13:1545–54.
- Siu SW, Chan RT, Au GK. Hypersensitivity reactions to oxaliplatin: experience in a single institute. Ann Oncol 2006;17:259–61.
- Wong JT, Ling M, Patil S, et al. Oxaliplatin hypersensitivity: evaluation, implications of skin testing, and desensitization. J Allergy Clin Immunol Pract 2014;2:40–5.
- Benson AB 3rd, Venook AP, Cederquist L, et al. NCCN Guidelines Colon Cancer Version 2.2017. www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed May 8, 2017.
- Wolmark N, Rockette H, Mamounas E, et al. Clinical trial to assess the relative efficacy of fluorouracil and leucovorin, fluorouracil and levamisole, and fluorouracil, leucovorin, and levamisole in patients with Dukes’ B and C carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project C-04. J Clin Oncol 1999;17:3553–9.
INTRODUCTION
Colorectal cancer (CRC) is one of the most prevalent malignancies and is the fourth most common cancer in the United States, with an estimated 133,490 new cases diagnosed in 2016. Of these, approximately 95,520 are located in the colon and 39,970 are in the rectum.1 CRC is the third leading cause of cancer death in women and the second leading cause of cancer death in men, with an estimated 49,190 total deaths in 2016.2 The incidence appears to be increasing,3 especially in patients younger than 55 years of age;4 the reason for this increase remains uncertain.
A number of risk factors for the development of CRC have been identified. Numerous hered-itary CRC syndromes have been described, including familial adenomatous polyposis,5 hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome,6 and MUTYH-associated polyposis.7,8 A family history of CRC doubles the risk of developing CRC,9 and current guidelines support lowering the age of screening in individuals with a family history of CRC to 10 years younger than the age of diagnosis of the family member or 40 years of age, whichever is lower.10 Patients with a personal history of adenomatous polyps are at increased risk for developing CRC, as are patients with a personal history of CRC, with a relative risk ranging from 3 to 6.11 Ulcerative colitis and Crohn’s disease are associated with the development of CRC and also influence screening, though evidence suggests good control of these diseases may mitigate risk.12 Finally, modifiable risk factors for the development of CRC include high red meat consumption,13 diets low in fiber,14 obesity,13 smoking, alcohol use,15 and physical inactivity16; lifestyle modification targeting these factors has been shown to decrease rates of CRC.17 The majority of colon cancers present with clinical symptoms, often with rectal bleeding, abdominal pain, change in bowel habits, or obstructive symptoms. More rarely, these tumors are detected during screening colonoscopy, in which case they tend to be at an early stage.
SURGICAL MANAGEMENT
A critical goal in the resection of early-stage colon cancer is attaining R0 resection. Patients who achieve R0 resection as compared to R1 (microscopic residual tumor) and R2 (macroscopic residual tumor)18 have significantly improved long-term overall survival.19 Traditionally, open resection of the involved colonic segment was employed, with end-end anastomosis of the uninvolved free margins. Laparoscopic resection for early-stage disease has been utilized in attempts to decrease morbidity of open procedures, with similar outcomes and node sampling.20 Laparoscopic resection appears to provide similar outcomes even in locally advanced disease.21 Right-sided lesions are treated with right colectomy and primary ileocolic anastomosis.22 For patients presenting with obstructing masses, the Hartmann procedure is the most commonly performed operation. This involves creation of an ostomy with subtotal colectomy and subsequent ostomy reversal in a 2- or 3-stage protocol.23 Patients with locally advanced disease and invasion into surrounding structures require multivisceral resection, which involves resection en bloc with secondarily involved organs.24 Intestinal perforation presents a unique challenge and is associated with surgical complications, infection, and lower overall survival (OS) and 5-year disease-free survival (DFS). Complete mesocolic excision is a newer technique that has been performed with reports of better oncologic outcome at some centers; however, this approach is not currently considered standard of care.25
STAGING
According to a report by the National Cancer Institute, the estimated 5-year relative survival rates for localized colon cancer (lymph node negative), regional (lymph node positive) disease, and distant (metastatic) disease are 89.9%, 71.3%, and 13.9%, respectively.1 However, efforts have been made to further classify patients into distinct categories to allow fine-tuning of prognostication. In the current system, staging of colon cancer utilizes the American Joint Committee on Cancer tumor/node/metastasis (TNM) system.20 Clinical and pathologic features include depth of invasion, local invasion of other organs, nodal involvement, and presence of distant metastasis (Table 1). Studies completed prior to the adoption of the TNM system used the Dukes criteria, which divided colon cancer into A, B, and C, corresponding to TNM stage I, stage IIA–IIC, and stage IIIA-IIIC. This classification is rarely used in more contemporary studies.
APPROACH TO ADJUVANT CHEMOTHERAPY
Adjuvant chemotherapy seeks to eliminate micrometastatic disease present following curative surgical resection. When stage 0 cancer is discovered incidentally during colonoscopy, endoscopic resection alone is the management of choice, as presence of micrometastatic disease is exceedingly unlikely.26 Stage I–III CRCs are treated with surgical resection withcurative intent. The 5-year survival rate for stage I and early-stage II CRC is estimated at 97% with surgery alone.27,28 The survival rate drops to about 60% for high-risk stage II tumors (T4aN0), and down to 50% or less for stage II-T4N0 or stage III cancers. Adjuvant chemotherapy is generally recommended to further decrease the rates of distant recurrence in certain cases of stage II and in all stage III tumors.
DETERMINATION OF BENEFIT FROM CHEMOTHERAPY: PROGNOSTIC MARKERS
Prior to administration of adjuvant chemotherapy, a clinical evaluation by the medical oncologist to determine appropriateness and safety of treatment is paramount. Poor performance status and comorbid conditions may indicate risk for excessive toxicity and minimal benefit from chemotherapy. CRC commonly presents in older individuals, with the median age at diagnosis of 69 years for men and 73 years for women.29 In this patient population, comorbidities such as cardiovascular disease, diabetes, and renal dysfunction are more prevalent.30 Decisions regarding adjuvant chemotherapy in this patient population have to take into consideration the fact that older patients may experience higher rates of toxicity with chemotherapy, including gastrointestinal toxicities and marrow suppression.31 Though some reports indicate patients older than 70 years derive similar benefit from adjuvant chemotherapy,32,33 a large pooled analysis of the ACCENT database, which included 7 adjuvant therapy trials and 14,528 patients, suggested limited benefit from the addition of oxaliplatin to fluorouracil in elderly patients.32 Other factors that weigh on the decision include stage, pathology, and presence of high-risk features. A common concern in the postoperative setting is delaying initiation of chemotherapy to allow adequate wound healing; however, evidence suggests that delays longer than 8 weeks leads to worse overall survival, with hazard ratios (HR) ranging from 1.4 to 1.7.34,35 Thus, the start of adjuvant therapy should ideally be within this time frame.
HIGH-RISK FEATURES
Multiple factors have been found to predict worse outcome and are classified as high-risk features (Table 2). Histologically, high-grade or poorly differentiated tumors are associated with higher recurrence rate and worse outcome.36 Certain histological subtypes, including mucinous and signet-ring, both appear to have more aggressive biology.37 Presence of microscopic invasion into surrounding blood vessels (vascular invasion) and nerves (perineural invasion) is associated with lower survival.38 Penetration of the cancer through the visceral peritoneum (T4a) or into surrounding structures (T4b) is associated with lower survival.36 During surgical resection, multiple lymph nodes are removed along with the primary tumor to evaluate for metastasis to the regional nodes. Multiple analyses have demonstrated that removal and pathologic assessment of fewer than 12 lymph nodes is associated with high risk of missing a positive node, and is thus equated with high risk.39–41 In addition, extension of tumor beyond the capsules of any single lymph node, termed extracapsular extension, is associated with an increased risk of all-cause mortality.42 Tumor deposits, or focal aggregates of adenocarcinoma in the pericolic fat that are not contiguous with the primary tumor and are not associated with lymph nodes, are currently classified as lymph nodes as N1c in the current TNM staging system. Presence of these deposits has been found to predict poor outcome stage for stage.43 Obstruction and/or perforation secondary to the tumor are also considered high-risk features that predict poor outcome.
SIDEDNESS
As reported at the 2016 American Society of Clinical Oncology annual meeting, tumor location predicts outcome in the metastatic setting. A report by Venook and colleagues based on a post-hoc analysis found that in the metastatic setting, location of the tumor primary in the left side is associated with longer OS (33.3 months) when compared to the right side of the colon (19.4 months).44 A retrospective analysis of multiple databases presented by Schrag and colleagues similarly reported inferior outcomes in patients with stage III and IV disease who had right-sided primary tumors.45 However, the prognostic implications for stage II disease remain uncertain.
BIOMARKERS
Given the controversy regarding adjuvant therapy of patients with stage II colon cancer, multiple biomarkers have been evaluated as possible predictive markers that can assist in this decision. The mismatch repair (MMR) system is a complex cellular enzymatic mechanism that identifies and corrects DNA errors during cell division and prevents mutagenesis.46 The familial cancer syndrome HNPCC is linked to alteration in a variety of MMR genes, leading to deficient mismatch repair (dMMR), also termed microsatellite instability-high (MSI-high).47,48 Epigenetic modification can also lead to silencing of the same implicated genes and accounts for 15% to 20% of sporadic colorectal cancer.49 These epigenetic modifications lead to hypermethylation of the promotor region of MLH1 in 70% of cases.50 The 4 MMR genes most commonly tested are MLH-1, MSH2, MSH6, and PMS2. Testing can be performed by immunohistochemistry or polymerase chain reaction.51 Across tumor histology and stage, MSI status is prognostic. Patients with MSI-high tumors have been shown to have improved prognosis and longer OS both in stage II and III disease52–54 and in the metastatic setting.55 However, despite this survival benefit, there is conflicting data as to whether patients with stage II, MSI-high colon cancer may benefit less from adjuvant chemotherapy. One early retrospective study compared outcomes of 70 patients with stage II and III disease and dMMR to those of 387 patients with stage II and III disease and proficient mismatch repair (pMMR). Adjuvant fluorouracil with leucovorin improved DFS for patients with pMMR (HR 0.67) but not for those with dMMR (HR 1.10). In addition, for patients with stage II disease and dMMR, the HR for OS was inferior at 2.95.56 Data collected from randomized clinical trials using fluorouracil-based adjuvant chemotherapy were analyzed in an attempt to predict benefit based on MSI status. Benefit was only seen in pMMR patients, with a HR of 0.72; this was not seen in the dMMR patients.57 Subsequent studies have had different findings and did not demonstrate a detrimental effect of fluorouracil in dMMR.58,59 For stage III patients, MSI status does not appear to affect benefit from chemotherapy, as analysis of data from the NSABP C-07 trial (Table 3) demonstrated benefit of FOLFOX (leucovorin, fluorouracil, oxaliplatin) in patients with dMMR status and stage III disease.59
Another genetic abnormality identified in colon cancers is chromosome 18q loss of heterozygosity (LOH). The presence of 18q LOH appears to be inversely associated with MSI-high status. Some reports have linked presence of 18q with worse outcome,60 but others question this, arguing the finding may simply be related to MSI status.61,62 This biomarker has not been established as a clear prognostic marker that can aid clinical decisions.
Most recently, expression of caudal-type homeobox transcription factor 2 (CDX2) has been reported as a novel prognostic and predictive tool. A 2015 report linked lack of expression of CDX2 to worse outcome; in this study, 5-year DFS was 41% in patients with CDX2-negative tumors versus 74% in the CDX2-positive tumors, with a HR of disease recurrence of 2.73 for CDX2-negative tumors.63 Similar numbers were observed in patients with stage II disease, with 5-year OS of 40% in patients with CDX2-negative tumors versus 70% in those with CDX2-positive tumors. Treatment of CDX2-negative patients with adjuvant chemotherapy improved outcomes: 5-year DFS in the stage II subgroup was 91% with chemotherapy versus 56% without, and in the stage III subgroup, 74% with chemotherapy versus 37% without. The authors concluded that patients with stage II and III colon cancer that is CDX2-negative may benefit from adjuvant chemotherapy. Importantly, CDX2-negativity is a rare event, occurring in only 6.9% of evaluable tumors.
RISK ASSESSMENT TOOLS
Several risk assessment tools have been developed in an attempt to aid clinical decision making regarding adjuvant chemotherapy for patients with stage II colon cancer. The Oncotype DX Colon Assay analyses a 12-gene signature in the pathologic sample and was developed with the goal to improve prognostication and aid in treatment decision making. The test utilizes reverse transcription-PCR on RNA extracted from the tumor.64 After evaluating 12 genes, a recurrence score is generated that predicts the risk of disease recurrence. This score was validated using data from 3 large clinical trials.65–67 Unlike the Oncotype Dx score used in breast cancer, the test in colon cancer has not been found to predict the benefit from chemotherapy and has not been incorporated widely into clinical practice.
Adjuvant! Online (available at www.adjuvantonline.com) is a web-based tool that combines clinical and histological features to estimate outcome. Calculations are based on US SEER tumor registry-reported outcomes.68 A second web-based tool, Numeracy (available at www.mayoclinic.com/calcs), was developed by the Mayo Clinic using pooled data from 7 randomized clinical trials including 3341 patients.68 Both tools seek to predict absolute benefit for patients treated with fluorouracil, though data suggests Adjuvant! Online may be more reliable in its predictive ability.69 Adjuvant! Online has also been validated in an Asian population70 and patients older than 70 years.71
MUTATIONAL ANALYSIS
Multiple mutations in proto-oncogenes have been found in colon cancer cells. One such proto-oncogene is BRAF, which encodes a serine-threonine kinase in the rapidly accelerated fibrosarcoma (RAF). Mutations in BRAF have been found in 5% to 10% of colon cancers and are associated with right-sided tumors.72 As a prognostic marker, some studies have associated BRAF mutations with worse prognosis, including shorter time to relapse and shorter OS.73,74 Two other proto-oncogenes are Kristen rat sarcoma viral oncogene homolog (KRAS) and neuroblastoma rat sarcoma viral oncogene homolog (NRAS), both of which encode proteins downstream of epidermal growth factor receptor (EGFR). KRAS and NRAS mutations have been shown to be predictive in the metastatic setting where they predict resistance to the EGFR inhibitors cetuximab and panitumumab.75,76 The effect of KRAS and NRAS mutations on outcome in stage II and III colon cancer is uncertain. Some studies suggest worse outcome in KRAS-mutated cancers,77 while others failed to demonstrate this finding.73
CASE PRESENTATION 1
A 53-year-old man with no past medical history presents to the emergency department with early satiety and generalized abdominal pain. Laboratory evaluation shows a microcytic anemia with normal white blood cell count, platelet count, renal function, and liver function tests. Computed tomography (CT) scan of the abdomen and pelvis show a 4-cm mass in the transverse colon without obstruction and without abnormality in the liver. CT scan of the chest does not demonstrate pathologic lymphadenopathy or other findings. He undergoes robotic laparoscopic transverse colon resection and appendectomy. Pathology confirms a 3.5-cm focus of adenocarcinoma of the colon with invasion through the muscularis propria and 5 of 27 regional lymph nodes positive for adenocarcinoma and uninvolved proximal, distal, and radial margins. He is given a stage of IIIB pT3 pN2a M0 and referred to medical oncology for further management, where 6 months of adjuvant FOLFOX chemotherapy is recommended.
ADJUVANT CHEMOTHERAPY IN STAGE III COLON CANCER
Postoperative adjuvant chemotherapy is the standard of care for patients with stage III disease. In the 1960s, infusional fluorouracil was first used to treat inoperable colon cancer.78,79 After encouraging results, the agent was used both intraluminally and intravenously as an adjuvant therapy for patients undergoing resection with curative intent; however, only modest benefits were described.80,81 The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-01 trial (Table 3) was the first study to demonstrate a benefit from adjuvant chemotherapy in colon cancer. This study randomly assigned patients with stage II and III colon cancer to surgery alone, postoperative chemotherapy with fluorouracil, semustine, and vincristine (MOF), or postoperative bacillus Calmette-Guérin (BCG). DFS and OS were significantly improved with MOF chemotherapy.82 In 1990, a landmark study reported on outcomes after treatment of 1296 patients with stage III colon cancer with adjuvant fluorouracil and levamisole for 12 months. The combination was associated with a 41% reduction in risk of cancer recurrence and a 33% reduction in risk of death.83 The NSABP C-03 trial (Table 3) compared MOF to the combination of fluorouracil and leucovorin and demonstrated improved 3-year DFS (69% versus 73%) and 3-year OS (77% versus 84%) in patients with stage III disease.84 Building on these outcomes, the QUASAR study (Table 3) compared fluorouracil in combination with one of levamisole, low-dose leucovorin, or high-dose leucovorin. The study enrolled 4927 patients and found worse outcomes with fluorouracil plus levamisole and no difference in low-doseversus high-dose leucovorin.85 Levamisole fell out of use after associations with development of multifocal leukoencephalopathy,86 and was later shown to have inferior outcomes versus leucovorin when combined with fluorouracil.87,88 Intravenous fluorouracil has shown similar benefit when administered by bolus or infusion,89 although continuous infusion has been associated with lower incidence of severe toxicity.90 The efficacy of the oral fluoropyrimidine capecitabine has been shown to be equivalent to that of fluorouracil.91
Fluorouracil-based treatment remained the standard of care until the introduction of oxaliplatin in the mid-1990s. After encouraging results in the metastatic setting,92,93 the agent was moved to the adjuvant setting. The MOSAIC trial (Table 3) randomly assigned patients with stage II and III colon cancer to fluorouracil with leucovorin (FULV) versus FOLFOX given once every 2 weeks for 12 cycles. Analysis with respect to stage III patients showed a clear survival benefit, with a 10-year OS of 67.1% with FOLFOX chemotherapy versus 59% with fluorouracil and leucovorin.94,95 The NSABP C-07 (Table 3) trial used a similar trial design but employed bolus fluorouracil. More than 2400 patients with stage II and III colon cancer were randomly assigned to bolus FULV or bolus fluorouracil, leucovorin, and oxaliplatin (FLOX). The addition of oxaliplatin significantly improved outcomes, with 4-year DFS of 67% versus 71.8% for FULV and FLOX, respectively, and a HR of death of 0.80 with FLOX.59,96 The multicenter N016968 trial (Table 3) randomly assigned 1886 patients with stage III colon cancer to adjuvant capecitabine plus oxaliplatin (XELOX) or bolus fluorouracil plus leucovorin (FU/FA). The 3-year DFS was 70.9% versus 66.5% with XELOX and FU/FA, respectively, and 5-year OS was 77.6% versus 74.2%, respectively.97,98
In the metastatic setting, additional agents have shown efficacy, including irinotecan,99,100 bevacizumab,101,102 cetuximab,103,104 and regorafenib.105 This observation led to testing of these agents in earlier stage disease. The CALGB 89803 trial compared fluorouracil, leucovorin, and irinotecan to fluorouracil with leucovorin alone. No benefit in 5-year DFS or OS was seen.106 Similarly, infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) was not found to improve 5-year DFS as compared to fluorouracil with leucovorin alone in the PETACC-3 trial.107 The NSABP C-08 trial considered the addition of bevacizumab to FOLFOX. When compared to FOLFOX alone, the combination of bevacizumab to FOLFOX had similar 3-year DFS (77.9% versus 75.1%) and 5-year OS (82.5% versus 80.7%).108 This finding was confirmed in the Avant trial.109 The addition of cetuximab to FOLFOX was equally disappointing, as shown in the N0147 trial110 and PETACC-8 trial.111 Data on regorafenib in the adjuvant setting for stage III colon cancer is lacking; however, 2 ongoing clinical trials, NCT02425683 and NCT02664077, are each studying the use of regorafenib following completion of FOLFOX for patients with stage III disease.
Thus, after multiple trials comparing various regimens and despite attempts to improve outcomes by the addition of a third agent, the standard of care per National Comprehensive Cancer Network (NCCN) guidelines for management of stage III colon cancer remains 12 cycles of FOLFOX chemotherapy. Therapy should be initiated within 8 weeks of surgery. Data are emerging to support a short duration of therapy for patients with low-risk stage III tumors, as shown in an abstract presented at the 2017 American Society of Clinical Oncology annual meeting. The IDEA trial was a pooled analysis of 6 randomized clinical trials across multiple countries, all of which evaluated 3 versus 6 months of FOLFOX or capecitabine and oxaliplatin in the treatment of stage III colon cancer. The analysis was designed to test non-inferiority of 3 months of therapy as compared to 6 months. The analysis included 6088 patients across 244 centers in 6 countries. The overall analysis failed to establish noninferiority. The 3-year DFS rate was 74.6% for 3 months and 75.5% for 6 months, with a DFS HR of 1.07 and a confidence interval that did not meet the prespecified endpoint. Subgroup analysis suggested noninferiority for lower stage disease (T1–3 or N1) but not for higher stage disease (T4 or N2). Given the high rates of neuropathy with 6 months of oxaliplatin, these results suggest that 3 months of adjuvant therapy can be considered for patients with T1–3 or N1 disease in an attempt to limit toxicity.112
CASE PRESENTATION 2
A 57-year-old woman presents to the emergency department with fever and abdominal pain. CT of the abdomen and pelvis demonstrates a left-sided colonic mass with surrounding fat stranding and pelvic abscess. She is taken emergently for left hemicolectomy, cholecystectomy, and evacuation of pelvic abscess. Pathology reveals a 5-cm adenocarcinoma with invasion through the visceral peritoneum; 0/22 lymph nodes are involved. She is given a diagnosis of stage IIC and referred to medical oncology for further management. Due to her young age and presence of high-risk features, she is recommended adjuvant therapy with FOLFOX for 6 months.
ADJUVANT CHEMOTHERAPY IN STAGE II COLON CANCER
Because of excellent outcomes with surgical resection alone for stage II cancers, the use of adjuvant chemotherapy for patients with stage II disease is controversial. Limited prospective data is available to guide adjuvant treatment decisions for stage II patients. The QUASAR trial, which compared observation to adjuvant fluorouracil and leucovorin in patients with early-stage colon cancer, included 2963 patients with stage II disease and found a relative risk (RR) of death or recurrence of 0.82 and 0.78, respectively. Importantly, the absolute benefit of therapy was less than 5%.113 The IMPACT-B2 trial (Table 3) combined data from 5 separate trials and analyzed 1016 patients with stage II colon cancer who received fluorouracil with leucovorin or observation. Event-free survival was 0.86 versus 0.83 and 5-year OS was 82% versus 80%, suggesting no benefit.114 The benefit of addition of oxaliplatin to fluorouracil in stage II disease appears to be less than the benefit of adding this agent in the treatment of stage III CRC. As noted above, the MOSAIC trial randomly assigned patients with stage II and III colon cancer to receive adjuvant fluorouracil and leucovorin with or without oxaliplatin for 12 cycles. After a median follow-up of 9.5 years, 10-year OS rates for patients with stage II disease were 78.4% versus 79.5%. For patients with high-risk stage II disease (defined as T4, bowel perforation, or fewer than 10 lymph nodes examined), 10-year OS was 71.7% and 75.4% respectively, but these differences were not statistically significant.94
Because of conflicting data as to the benefit of adding oxaliplatin in stage II disease, oxaliplatin is not recommended for standard-risk stage II patients. The use of oxaliplatin in high-risk stage II tumors should be weighed carefully given the toxicity risk. Oxaliplatin is recognized to cause sensory neuropathy in many patients, which can become painful and debilitating.115 Two types of neuropathy are associated with oxaliplatin: acute and chronic. Acute neuropathy manifests most often as cold-induced paresthesias in the fingers and toes and is quite common, affecting up to 90% of patients. These symptoms are self-limited and resolve usually within 1 week of each treatment.116 Some patients, with reports ranging from 10% to 79%, develop chronic neuropathy that persists for 1 year or more and causes significant decrements in quality of life.117 Patients older than age 70 may be at greater risk for oxaliplatin-induced neuropathy, which would increase risk of falls in this population.118 In addition to neuropathy, oxaliplatin is associated with hypersensitivity reactions that can be severe and even fatal.119 In a single institution series, the incidence of severe reactions was 2%.120 Desensitization following hypersensitivity reactions is possible but requires a time-intensive protocol.121
Based on the inconclusive efficacy findings and due to concerns over toxicity, each decision must be individualized to fit patient characteristics and preferences. In general, for patients with stage II disease without high-risk features, an individualized discussion should be held as to the risks and benefits of single-agent fluorouracil, and this treatment should be offered in cases where the patient or provider would like to be aggressive. Patients with stage II cancer who have 1 or more high-risk features are often recommended adjuvant chemotherapy. Whether treatment with fluorouracil plus leucovorin or FOLFOX is preferred remains uncertain, and thus the risks and the potential gains of oxaliplatin must be discussed with the individual patient. MMR status can also influence the treatment recommendation for patients with stage II disease. In general, patients with standard-risk stage II tumors that are pMMR are offered MMR with leucovorin or oral capecitabine for 12 cycles. FOLFOX is considered for patients with MSI-high disease and those with multiple high-risk features.
MONITORING AFTER THERAPY
After completion of adjuvant chemotherapy, patients enter a period of survivorship. Patients are seen in clinic for symptom and laboratory monitoring of the complete blood count, liver function tests, and carcinoembryonic antigen (CEA). NCCN guidelines support history and physical examination with CEA testing every 3 to 6 months for the first 2 years, then every 6 months for the next 3 years, after which many patients continue to be seen annually. CT imaging of the chest, abdomen, and pelvis for monitoring of disease recurrence is recommended every 6 to 12 months for a total of 5 years. New elevations in CEA or liver function tests should prompt early imaging. Colonoscopy should be performed 1 year after completion of therapy; however, if no preoperative colonoscopy was performed, this should be done 3 to 6 months after completion. Colonoscopy is then repeated in 3 years and then every 5 years unless advanced adenomas are present.122
SUMMARY
The addition of chemotherapy to surgical management of colon cancer has lowered the rate of disease recurrence and improved long-term survival. Adjuvant FOLFOX for 12 cycles is the standard of care for patients with stage III colon cancer and for patients with stage II disease with certain high-risk features. Use of adjuvant chemotherapy in stage II disease without high-risk features is controversial, and treatment decisions should be individualized. Biologic markers such as MSI and CDX2 status as well as patient-related factors including age, overall health, and personal preferences can inform treatment decisions. If chemotherapy is recommended in this setting, it would be with single-agent fluorouracil in an infusional or oral formulation, unless the tumor has the MSI-high feature. Following completion of adjuvant therapy, patients should be followed with clinical evaluation, laboratory testing, and imaging for a total of 5 years as per recommended guidelines.
INTRODUCTION
Colorectal cancer (CRC) is one of the most prevalent malignancies and is the fourth most common cancer in the United States, with an estimated 133,490 new cases diagnosed in 2016. Of these, approximately 95,520 are located in the colon and 39,970 are in the rectum.1 CRC is the third leading cause of cancer death in women and the second leading cause of cancer death in men, with an estimated 49,190 total deaths in 2016.2 The incidence appears to be increasing,3 especially in patients younger than 55 years of age;4 the reason for this increase remains uncertain.
A number of risk factors for the development of CRC have been identified. Numerous hered-itary CRC syndromes have been described, including familial adenomatous polyposis,5 hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome,6 and MUTYH-associated polyposis.7,8 A family history of CRC doubles the risk of developing CRC,9 and current guidelines support lowering the age of screening in individuals with a family history of CRC to 10 years younger than the age of diagnosis of the family member or 40 years of age, whichever is lower.10 Patients with a personal history of adenomatous polyps are at increased risk for developing CRC, as are patients with a personal history of CRC, with a relative risk ranging from 3 to 6.11 Ulcerative colitis and Crohn’s disease are associated with the development of CRC and also influence screening, though evidence suggests good control of these diseases may mitigate risk.12 Finally, modifiable risk factors for the development of CRC include high red meat consumption,13 diets low in fiber,14 obesity,13 smoking, alcohol use,15 and physical inactivity16; lifestyle modification targeting these factors has been shown to decrease rates of CRC.17 The majority of colon cancers present with clinical symptoms, often with rectal bleeding, abdominal pain, change in bowel habits, or obstructive symptoms. More rarely, these tumors are detected during screening colonoscopy, in which case they tend to be at an early stage.
SURGICAL MANAGEMENT
A critical goal in the resection of early-stage colon cancer is attaining R0 resection. Patients who achieve R0 resection as compared to R1 (microscopic residual tumor) and R2 (macroscopic residual tumor)18 have significantly improved long-term overall survival.19 Traditionally, open resection of the involved colonic segment was employed, with end-end anastomosis of the uninvolved free margins. Laparoscopic resection for early-stage disease has been utilized in attempts to decrease morbidity of open procedures, with similar outcomes and node sampling.20 Laparoscopic resection appears to provide similar outcomes even in locally advanced disease.21 Right-sided lesions are treated with right colectomy and primary ileocolic anastomosis.22 For patients presenting with obstructing masses, the Hartmann procedure is the most commonly performed operation. This involves creation of an ostomy with subtotal colectomy and subsequent ostomy reversal in a 2- or 3-stage protocol.23 Patients with locally advanced disease and invasion into surrounding structures require multivisceral resection, which involves resection en bloc with secondarily involved organs.24 Intestinal perforation presents a unique challenge and is associated with surgical complications, infection, and lower overall survival (OS) and 5-year disease-free survival (DFS). Complete mesocolic excision is a newer technique that has been performed with reports of better oncologic outcome at some centers; however, this approach is not currently considered standard of care.25
STAGING
According to a report by the National Cancer Institute, the estimated 5-year relative survival rates for localized colon cancer (lymph node negative), regional (lymph node positive) disease, and distant (metastatic) disease are 89.9%, 71.3%, and 13.9%, respectively.1 However, efforts have been made to further classify patients into distinct categories to allow fine-tuning of prognostication. In the current system, staging of colon cancer utilizes the American Joint Committee on Cancer tumor/node/metastasis (TNM) system.20 Clinical and pathologic features include depth of invasion, local invasion of other organs, nodal involvement, and presence of distant metastasis (Table 1). Studies completed prior to the adoption of the TNM system used the Dukes criteria, which divided colon cancer into A, B, and C, corresponding to TNM stage I, stage IIA–IIC, and stage IIIA-IIIC. This classification is rarely used in more contemporary studies.
APPROACH TO ADJUVANT CHEMOTHERAPY
Adjuvant chemotherapy seeks to eliminate micrometastatic disease present following curative surgical resection. When stage 0 cancer is discovered incidentally during colonoscopy, endoscopic resection alone is the management of choice, as presence of micrometastatic disease is exceedingly unlikely.26 Stage I–III CRCs are treated with surgical resection withcurative intent. The 5-year survival rate for stage I and early-stage II CRC is estimated at 97% with surgery alone.27,28 The survival rate drops to about 60% for high-risk stage II tumors (T4aN0), and down to 50% or less for stage II-T4N0 or stage III cancers. Adjuvant chemotherapy is generally recommended to further decrease the rates of distant recurrence in certain cases of stage II and in all stage III tumors.
DETERMINATION OF BENEFIT FROM CHEMOTHERAPY: PROGNOSTIC MARKERS
Prior to administration of adjuvant chemotherapy, a clinical evaluation by the medical oncologist to determine appropriateness and safety of treatment is paramount. Poor performance status and comorbid conditions may indicate risk for excessive toxicity and minimal benefit from chemotherapy. CRC commonly presents in older individuals, with the median age at diagnosis of 69 years for men and 73 years for women.29 In this patient population, comorbidities such as cardiovascular disease, diabetes, and renal dysfunction are more prevalent.30 Decisions regarding adjuvant chemotherapy in this patient population have to take into consideration the fact that older patients may experience higher rates of toxicity with chemotherapy, including gastrointestinal toxicities and marrow suppression.31 Though some reports indicate patients older than 70 years derive similar benefit from adjuvant chemotherapy,32,33 a large pooled analysis of the ACCENT database, which included 7 adjuvant therapy trials and 14,528 patients, suggested limited benefit from the addition of oxaliplatin to fluorouracil in elderly patients.32 Other factors that weigh on the decision include stage, pathology, and presence of high-risk features. A common concern in the postoperative setting is delaying initiation of chemotherapy to allow adequate wound healing; however, evidence suggests that delays longer than 8 weeks leads to worse overall survival, with hazard ratios (HR) ranging from 1.4 to 1.7.34,35 Thus, the start of adjuvant therapy should ideally be within this time frame.
HIGH-RISK FEATURES
Multiple factors have been found to predict worse outcome and are classified as high-risk features (Table 2). Histologically, high-grade or poorly differentiated tumors are associated with higher recurrence rate and worse outcome.36 Certain histological subtypes, including mucinous and signet-ring, both appear to have more aggressive biology.37 Presence of microscopic invasion into surrounding blood vessels (vascular invasion) and nerves (perineural invasion) is associated with lower survival.38 Penetration of the cancer through the visceral peritoneum (T4a) or into surrounding structures (T4b) is associated with lower survival.36 During surgical resection, multiple lymph nodes are removed along with the primary tumor to evaluate for metastasis to the regional nodes. Multiple analyses have demonstrated that removal and pathologic assessment of fewer than 12 lymph nodes is associated with high risk of missing a positive node, and is thus equated with high risk.39–41 In addition, extension of tumor beyond the capsules of any single lymph node, termed extracapsular extension, is associated with an increased risk of all-cause mortality.42 Tumor deposits, or focal aggregates of adenocarcinoma in the pericolic fat that are not contiguous with the primary tumor and are not associated with lymph nodes, are currently classified as lymph nodes as N1c in the current TNM staging system. Presence of these deposits has been found to predict poor outcome stage for stage.43 Obstruction and/or perforation secondary to the tumor are also considered high-risk features that predict poor outcome.
SIDEDNESS
As reported at the 2016 American Society of Clinical Oncology annual meeting, tumor location predicts outcome in the metastatic setting. A report by Venook and colleagues based on a post-hoc analysis found that in the metastatic setting, location of the tumor primary in the left side is associated with longer OS (33.3 months) when compared to the right side of the colon (19.4 months).44 A retrospective analysis of multiple databases presented by Schrag and colleagues similarly reported inferior outcomes in patients with stage III and IV disease who had right-sided primary tumors.45 However, the prognostic implications for stage II disease remain uncertain.
BIOMARKERS
Given the controversy regarding adjuvant therapy of patients with stage II colon cancer, multiple biomarkers have been evaluated as possible predictive markers that can assist in this decision. The mismatch repair (MMR) system is a complex cellular enzymatic mechanism that identifies and corrects DNA errors during cell division and prevents mutagenesis.46 The familial cancer syndrome HNPCC is linked to alteration in a variety of MMR genes, leading to deficient mismatch repair (dMMR), also termed microsatellite instability-high (MSI-high).47,48 Epigenetic modification can also lead to silencing of the same implicated genes and accounts for 15% to 20% of sporadic colorectal cancer.49 These epigenetic modifications lead to hypermethylation of the promotor region of MLH1 in 70% of cases.50 The 4 MMR genes most commonly tested are MLH-1, MSH2, MSH6, and PMS2. Testing can be performed by immunohistochemistry or polymerase chain reaction.51 Across tumor histology and stage, MSI status is prognostic. Patients with MSI-high tumors have been shown to have improved prognosis and longer OS both in stage II and III disease52–54 and in the metastatic setting.55 However, despite this survival benefit, there is conflicting data as to whether patients with stage II, MSI-high colon cancer may benefit less from adjuvant chemotherapy. One early retrospective study compared outcomes of 70 patients with stage II and III disease and dMMR to those of 387 patients with stage II and III disease and proficient mismatch repair (pMMR). Adjuvant fluorouracil with leucovorin improved DFS for patients with pMMR (HR 0.67) but not for those with dMMR (HR 1.10). In addition, for patients with stage II disease and dMMR, the HR for OS was inferior at 2.95.56 Data collected from randomized clinical trials using fluorouracil-based adjuvant chemotherapy were analyzed in an attempt to predict benefit based on MSI status. Benefit was only seen in pMMR patients, with a HR of 0.72; this was not seen in the dMMR patients.57 Subsequent studies have had different findings and did not demonstrate a detrimental effect of fluorouracil in dMMR.58,59 For stage III patients, MSI status does not appear to affect benefit from chemotherapy, as analysis of data from the NSABP C-07 trial (Table 3) demonstrated benefit of FOLFOX (leucovorin, fluorouracil, oxaliplatin) in patients with dMMR status and stage III disease.59
Another genetic abnormality identified in colon cancers is chromosome 18q loss of heterozygosity (LOH). The presence of 18q LOH appears to be inversely associated with MSI-high status. Some reports have linked presence of 18q with worse outcome,60 but others question this, arguing the finding may simply be related to MSI status.61,62 This biomarker has not been established as a clear prognostic marker that can aid clinical decisions.
Most recently, expression of caudal-type homeobox transcription factor 2 (CDX2) has been reported as a novel prognostic and predictive tool. A 2015 report linked lack of expression of CDX2 to worse outcome; in this study, 5-year DFS was 41% in patients with CDX2-negative tumors versus 74% in the CDX2-positive tumors, with a HR of disease recurrence of 2.73 for CDX2-negative tumors.63 Similar numbers were observed in patients with stage II disease, with 5-year OS of 40% in patients with CDX2-negative tumors versus 70% in those with CDX2-positive tumors. Treatment of CDX2-negative patients with adjuvant chemotherapy improved outcomes: 5-year DFS in the stage II subgroup was 91% with chemotherapy versus 56% without, and in the stage III subgroup, 74% with chemotherapy versus 37% without. The authors concluded that patients with stage II and III colon cancer that is CDX2-negative may benefit from adjuvant chemotherapy. Importantly, CDX2-negativity is a rare event, occurring in only 6.9% of evaluable tumors.
RISK ASSESSMENT TOOLS
Several risk assessment tools have been developed in an attempt to aid clinical decision making regarding adjuvant chemotherapy for patients with stage II colon cancer. The Oncotype DX Colon Assay analyses a 12-gene signature in the pathologic sample and was developed with the goal to improve prognostication and aid in treatment decision making. The test utilizes reverse transcription-PCR on RNA extracted from the tumor.64 After evaluating 12 genes, a recurrence score is generated that predicts the risk of disease recurrence. This score was validated using data from 3 large clinical trials.65–67 Unlike the Oncotype Dx score used in breast cancer, the test in colon cancer has not been found to predict the benefit from chemotherapy and has not been incorporated widely into clinical practice.
Adjuvant! Online (available at www.adjuvantonline.com) is a web-based tool that combines clinical and histological features to estimate outcome. Calculations are based on US SEER tumor registry-reported outcomes.68 A second web-based tool, Numeracy (available at www.mayoclinic.com/calcs), was developed by the Mayo Clinic using pooled data from 7 randomized clinical trials including 3341 patients.68 Both tools seek to predict absolute benefit for patients treated with fluorouracil, though data suggests Adjuvant! Online may be more reliable in its predictive ability.69 Adjuvant! Online has also been validated in an Asian population70 and patients older than 70 years.71
MUTATIONAL ANALYSIS
Multiple mutations in proto-oncogenes have been found in colon cancer cells. One such proto-oncogene is BRAF, which encodes a serine-threonine kinase in the rapidly accelerated fibrosarcoma (RAF). Mutations in BRAF have been found in 5% to 10% of colon cancers and are associated with right-sided tumors.72 As a prognostic marker, some studies have associated BRAF mutations with worse prognosis, including shorter time to relapse and shorter OS.73,74 Two other proto-oncogenes are Kristen rat sarcoma viral oncogene homolog (KRAS) and neuroblastoma rat sarcoma viral oncogene homolog (NRAS), both of which encode proteins downstream of epidermal growth factor receptor (EGFR). KRAS and NRAS mutations have been shown to be predictive in the metastatic setting where they predict resistance to the EGFR inhibitors cetuximab and panitumumab.75,76 The effect of KRAS and NRAS mutations on outcome in stage II and III colon cancer is uncertain. Some studies suggest worse outcome in KRAS-mutated cancers,77 while others failed to demonstrate this finding.73
CASE PRESENTATION 1
A 53-year-old man with no past medical history presents to the emergency department with early satiety and generalized abdominal pain. Laboratory evaluation shows a microcytic anemia with normal white blood cell count, platelet count, renal function, and liver function tests. Computed tomography (CT) scan of the abdomen and pelvis show a 4-cm mass in the transverse colon without obstruction and without abnormality in the liver. CT scan of the chest does not demonstrate pathologic lymphadenopathy or other findings. He undergoes robotic laparoscopic transverse colon resection and appendectomy. Pathology confirms a 3.5-cm focus of adenocarcinoma of the colon with invasion through the muscularis propria and 5 of 27 regional lymph nodes positive for adenocarcinoma and uninvolved proximal, distal, and radial margins. He is given a stage of IIIB pT3 pN2a M0 and referred to medical oncology for further management, where 6 months of adjuvant FOLFOX chemotherapy is recommended.
ADJUVANT CHEMOTHERAPY IN STAGE III COLON CANCER
Postoperative adjuvant chemotherapy is the standard of care for patients with stage III disease. In the 1960s, infusional fluorouracil was first used to treat inoperable colon cancer.78,79 After encouraging results, the agent was used both intraluminally and intravenously as an adjuvant therapy for patients undergoing resection with curative intent; however, only modest benefits were described.80,81 The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-01 trial (Table 3) was the first study to demonstrate a benefit from adjuvant chemotherapy in colon cancer. This study randomly assigned patients with stage II and III colon cancer to surgery alone, postoperative chemotherapy with fluorouracil, semustine, and vincristine (MOF), or postoperative bacillus Calmette-Guérin (BCG). DFS and OS were significantly improved with MOF chemotherapy.82 In 1990, a landmark study reported on outcomes after treatment of 1296 patients with stage III colon cancer with adjuvant fluorouracil and levamisole for 12 months. The combination was associated with a 41% reduction in risk of cancer recurrence and a 33% reduction in risk of death.83 The NSABP C-03 trial (Table 3) compared MOF to the combination of fluorouracil and leucovorin and demonstrated improved 3-year DFS (69% versus 73%) and 3-year OS (77% versus 84%) in patients with stage III disease.84 Building on these outcomes, the QUASAR study (Table 3) compared fluorouracil in combination with one of levamisole, low-dose leucovorin, or high-dose leucovorin. The study enrolled 4927 patients and found worse outcomes with fluorouracil plus levamisole and no difference in low-doseversus high-dose leucovorin.85 Levamisole fell out of use after associations with development of multifocal leukoencephalopathy,86 and was later shown to have inferior outcomes versus leucovorin when combined with fluorouracil.87,88 Intravenous fluorouracil has shown similar benefit when administered by bolus or infusion,89 although continuous infusion has been associated with lower incidence of severe toxicity.90 The efficacy of the oral fluoropyrimidine capecitabine has been shown to be equivalent to that of fluorouracil.91
Fluorouracil-based treatment remained the standard of care until the introduction of oxaliplatin in the mid-1990s. After encouraging results in the metastatic setting,92,93 the agent was moved to the adjuvant setting. The MOSAIC trial (Table 3) randomly assigned patients with stage II and III colon cancer to fluorouracil with leucovorin (FULV) versus FOLFOX given once every 2 weeks for 12 cycles. Analysis with respect to stage III patients showed a clear survival benefit, with a 10-year OS of 67.1% with FOLFOX chemotherapy versus 59% with fluorouracil and leucovorin.94,95 The NSABP C-07 (Table 3) trial used a similar trial design but employed bolus fluorouracil. More than 2400 patients with stage II and III colon cancer were randomly assigned to bolus FULV or bolus fluorouracil, leucovorin, and oxaliplatin (FLOX). The addition of oxaliplatin significantly improved outcomes, with 4-year DFS of 67% versus 71.8% for FULV and FLOX, respectively, and a HR of death of 0.80 with FLOX.59,96 The multicenter N016968 trial (Table 3) randomly assigned 1886 patients with stage III colon cancer to adjuvant capecitabine plus oxaliplatin (XELOX) or bolus fluorouracil plus leucovorin (FU/FA). The 3-year DFS was 70.9% versus 66.5% with XELOX and FU/FA, respectively, and 5-year OS was 77.6% versus 74.2%, respectively.97,98
In the metastatic setting, additional agents have shown efficacy, including irinotecan,99,100 bevacizumab,101,102 cetuximab,103,104 and regorafenib.105 This observation led to testing of these agents in earlier stage disease. The CALGB 89803 trial compared fluorouracil, leucovorin, and irinotecan to fluorouracil with leucovorin alone. No benefit in 5-year DFS or OS was seen.106 Similarly, infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) was not found to improve 5-year DFS as compared to fluorouracil with leucovorin alone in the PETACC-3 trial.107 The NSABP C-08 trial considered the addition of bevacizumab to FOLFOX. When compared to FOLFOX alone, the combination of bevacizumab to FOLFOX had similar 3-year DFS (77.9% versus 75.1%) and 5-year OS (82.5% versus 80.7%).108 This finding was confirmed in the Avant trial.109 The addition of cetuximab to FOLFOX was equally disappointing, as shown in the N0147 trial110 and PETACC-8 trial.111 Data on regorafenib in the adjuvant setting for stage III colon cancer is lacking; however, 2 ongoing clinical trials, NCT02425683 and NCT02664077, are each studying the use of regorafenib following completion of FOLFOX for patients with stage III disease.
Thus, after multiple trials comparing various regimens and despite attempts to improve outcomes by the addition of a third agent, the standard of care per National Comprehensive Cancer Network (NCCN) guidelines for management of stage III colon cancer remains 12 cycles of FOLFOX chemotherapy. Therapy should be initiated within 8 weeks of surgery. Data are emerging to support a short duration of therapy for patients with low-risk stage III tumors, as shown in an abstract presented at the 2017 American Society of Clinical Oncology annual meeting. The IDEA trial was a pooled analysis of 6 randomized clinical trials across multiple countries, all of which evaluated 3 versus 6 months of FOLFOX or capecitabine and oxaliplatin in the treatment of stage III colon cancer. The analysis was designed to test non-inferiority of 3 months of therapy as compared to 6 months. The analysis included 6088 patients across 244 centers in 6 countries. The overall analysis failed to establish noninferiority. The 3-year DFS rate was 74.6% for 3 months and 75.5% for 6 months, with a DFS HR of 1.07 and a confidence interval that did not meet the prespecified endpoint. Subgroup analysis suggested noninferiority for lower stage disease (T1–3 or N1) but not for higher stage disease (T4 or N2). Given the high rates of neuropathy with 6 months of oxaliplatin, these results suggest that 3 months of adjuvant therapy can be considered for patients with T1–3 or N1 disease in an attempt to limit toxicity.112
CASE PRESENTATION 2
A 57-year-old woman presents to the emergency department with fever and abdominal pain. CT of the abdomen and pelvis demonstrates a left-sided colonic mass with surrounding fat stranding and pelvic abscess. She is taken emergently for left hemicolectomy, cholecystectomy, and evacuation of pelvic abscess. Pathology reveals a 5-cm adenocarcinoma with invasion through the visceral peritoneum; 0/22 lymph nodes are involved. She is given a diagnosis of stage IIC and referred to medical oncology for further management. Due to her young age and presence of high-risk features, she is recommended adjuvant therapy with FOLFOX for 6 months.
ADJUVANT CHEMOTHERAPY IN STAGE II COLON CANCER
Because of excellent outcomes with surgical resection alone for stage II cancers, the use of adjuvant chemotherapy for patients with stage II disease is controversial. Limited prospective data is available to guide adjuvant treatment decisions for stage II patients. The QUASAR trial, which compared observation to adjuvant fluorouracil and leucovorin in patients with early-stage colon cancer, included 2963 patients with stage II disease and found a relative risk (RR) of death or recurrence of 0.82 and 0.78, respectively. Importantly, the absolute benefit of therapy was less than 5%.113 The IMPACT-B2 trial (Table 3) combined data from 5 separate trials and analyzed 1016 patients with stage II colon cancer who received fluorouracil with leucovorin or observation. Event-free survival was 0.86 versus 0.83 and 5-year OS was 82% versus 80%, suggesting no benefit.114 The benefit of addition of oxaliplatin to fluorouracil in stage II disease appears to be less than the benefit of adding this agent in the treatment of stage III CRC. As noted above, the MOSAIC trial randomly assigned patients with stage II and III colon cancer to receive adjuvant fluorouracil and leucovorin with or without oxaliplatin for 12 cycles. After a median follow-up of 9.5 years, 10-year OS rates for patients with stage II disease were 78.4% versus 79.5%. For patients with high-risk stage II disease (defined as T4, bowel perforation, or fewer than 10 lymph nodes examined), 10-year OS was 71.7% and 75.4% respectively, but these differences were not statistically significant.94
Because of conflicting data as to the benefit of adding oxaliplatin in stage II disease, oxaliplatin is not recommended for standard-risk stage II patients. The use of oxaliplatin in high-risk stage II tumors should be weighed carefully given the toxicity risk. Oxaliplatin is recognized to cause sensory neuropathy in many patients, which can become painful and debilitating.115 Two types of neuropathy are associated with oxaliplatin: acute and chronic. Acute neuropathy manifests most often as cold-induced paresthesias in the fingers and toes and is quite common, affecting up to 90% of patients. These symptoms are self-limited and resolve usually within 1 week of each treatment.116 Some patients, with reports ranging from 10% to 79%, develop chronic neuropathy that persists for 1 year or more and causes significant decrements in quality of life.117 Patients older than age 70 may be at greater risk for oxaliplatin-induced neuropathy, which would increase risk of falls in this population.118 In addition to neuropathy, oxaliplatin is associated with hypersensitivity reactions that can be severe and even fatal.119 In a single institution series, the incidence of severe reactions was 2%.120 Desensitization following hypersensitivity reactions is possible but requires a time-intensive protocol.121
Based on the inconclusive efficacy findings and due to concerns over toxicity, each decision must be individualized to fit patient characteristics and preferences. In general, for patients with stage II disease without high-risk features, an individualized discussion should be held as to the risks and benefits of single-agent fluorouracil, and this treatment should be offered in cases where the patient or provider would like to be aggressive. Patients with stage II cancer who have 1 or more high-risk features are often recommended adjuvant chemotherapy. Whether treatment with fluorouracil plus leucovorin or FOLFOX is preferred remains uncertain, and thus the risks and the potential gains of oxaliplatin must be discussed with the individual patient. MMR status can also influence the treatment recommendation for patients with stage II disease. In general, patients with standard-risk stage II tumors that are pMMR are offered MMR with leucovorin or oral capecitabine for 12 cycles. FOLFOX is considered for patients with MSI-high disease and those with multiple high-risk features.
MONITORING AFTER THERAPY
After completion of adjuvant chemotherapy, patients enter a period of survivorship. Patients are seen in clinic for symptom and laboratory monitoring of the complete blood count, liver function tests, and carcinoembryonic antigen (CEA). NCCN guidelines support history and physical examination with CEA testing every 3 to 6 months for the first 2 years, then every 6 months for the next 3 years, after which many patients continue to be seen annually. CT imaging of the chest, abdomen, and pelvis for monitoring of disease recurrence is recommended every 6 to 12 months for a total of 5 years. New elevations in CEA or liver function tests should prompt early imaging. Colonoscopy should be performed 1 year after completion of therapy; however, if no preoperative colonoscopy was performed, this should be done 3 to 6 months after completion. Colonoscopy is then repeated in 3 years and then every 5 years unless advanced adenomas are present.122
SUMMARY
The addition of chemotherapy to surgical management of colon cancer has lowered the rate of disease recurrence and improved long-term survival. Adjuvant FOLFOX for 12 cycles is the standard of care for patients with stage III colon cancer and for patients with stage II disease with certain high-risk features. Use of adjuvant chemotherapy in stage II disease without high-risk features is controversial, and treatment decisions should be individualized. Biologic markers such as MSI and CDX2 status as well as patient-related factors including age, overall health, and personal preferences can inform treatment decisions. If chemotherapy is recommended in this setting, it would be with single-agent fluorouracil in an infusional or oral formulation, unless the tumor has the MSI-high feature. Following completion of adjuvant therapy, patients should be followed with clinical evaluation, laboratory testing, and imaging for a total of 5 years as per recommended guidelines.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67(1):7–30.
- United States Cancer Statistics. 1999–2013 incidence and mortality web-based report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute, 2016. www.cdc.gov/uscs. Accessed June 12, 2017.
- Ahnen DJ, Wade SW, Jones WF, et al. The increasing incidence of young-onset colorectal cancer: a call to action. Mayo Clin Proc 2014;89:216–24.
- Jemal A, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States, 1974–2013. J Natl Cancer Inst 2017;109(8).
- Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer 2013;49:3680–5.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60: 950–7.
- van Puijenbroek M, Nielsen M, Tops CM, et al. Identification of patients with (atypical) MUTYH-associated polyposis by KRAS2 c.34G > T prescreening followed by MUTYH hotspot analysis in formalin-fixed paraffin-embedded tissue. Clin Cancer Res 2008;14:139–42.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006;119:807–14.
- Tuohy TM, Rowe KG, Mineau GP, et al. Risk of colorectal cancer and adenomas in the families of patients with adenomas: a population-based study in Utah. Cancer 2014;120:35–42.
- Choi Y, Sateia HF, Peairs KS, Stewart RW. Screening for colorectal cancer. Semin Oncol 2017; 44:34–44.
- Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med 1992;326:658–62.
- Rutter MD. Surveillance programmes for neoplasia in colitis. J Gastroenterol 2011;46 Suppl 1:1–5.
- Giovannucci E. Modifiable risk factors for colon cancer. Gastroenterol Clin North Am 2002;31:925–43.
- Michels KB, Fuchs GS, Giovannucci E, et al. Fiber intake and incidence of colorectal cancer among 76,947 women and 47,279 men. Cancer Epidemiol Biomarkers Prev 2005;14:842–9.
- Omata F, Brown WR, Tokuda Y, et al. Modifiable risk factors for colorectal neoplasms and hyperplastic polyps. Intern Med 2009;48:123–8.
- Friedenreich CM, Neilson HK, Lynch BM. State of the epidemiological evidence on physical activity and cancer prevention. Eur J Cancer 2010;46:2593–604.
- Aleksandrova K, Pischon T, Jenab M, et al. Combined impact of healthy lifestyle factors on colorectal cancer: a large European cohort study. BMC Med 2014;12:168.
- Hermanek P, Wittekind C. The pathologist and the residual tumor (R) classification. Pathol Res Pract 1994;190:115–23.
- Lehnert T, Methner M, Pollok A, et al. Multivisceral resection for locally advanced primary colon and rectal cancer: an analysis of prognostic factors in 201 patients. Ann Surg 2002;235:217–25.
- Feinberg AE, et al. Oncologic outcomes following laparoscopic versus open resection of pT4 colon cancer: a systematic review and meta-analysis. Dis Colon Rectum 2017;60:116–125.
- Vignali A, et al. Laparoscopic treatment of advanced colonic cancer: a case-matched control with open surgery. Colorectal Dis 2013;15:944–8.
- Gainant A. Emergency management of acute colonic cancer obstruction. J Visc Surg 2012;149: e3–e10.
- Rosenman LD. Hartmann’s operation. Am J Surg 1994;168:283–4.
- Lee-Kong S, Lisle D. Surgical management of complicated colon cancer. Clin Colon Rectal Surg 2015;28:228–33.
- Bertelsen CA. Complete mesocolic excision an assessment of feasibility and outcome. Dan Med J 2017;64(2).
- Wolff WI SH. Definitive treatment of “malignant” polyps of the colon. Ann Surg 1975;182:516–25.
- Clinical Outcomes of Surgical Therapy Study Group, Nelson H, Sargent DJ, Wieand HS, et al. A comparison of laparoscopically assisted and open colectomy for colon cancer. N Engl J Med 2004;350:2050–9.
- Gunderson LL, Jessup JM, Sarjent DJ, et al. Revised tumor and node categorization for rectal cancer based on surveillance, epidemiology, and end results and rectal pooled analysis outcomes. J Clin Oncol 2010;28:256–63.
- Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
- Alves A, Panis Y, Mathieu P, et al. Postoperative mortality and morbidity in French patients undergoing colorectal surgery: results of a prospective multicenter study. Arch Surg 2005;140:278–83.
- Popescu RA, Norman A, Ross PJ, et al, Adjuvant or palliative chemotherapy for colorectal cancer in patients 70 years or older. J Clin Oncol 1999;17:2412–8.
- McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
- Tominaga T, Nonaka T, Sumida Y, et al. Effectiveness of adjuvant chemotherapy for elderly patients with lymph node-positive colorectal cancer. World J Surg Oncol 2016;14:197.
- Bos AC, van Erning FN, van Gestel YR, et al. Timing of adjuvant chemotherapy and its relation to survival among patients with stage III colon cancer. Eur J Cancer 2015;51:2553–61.
- Peixoto RD, Kumar A, Speers C, et al. Effect of delay in adjuvant oxaliplatin-based chemotherapy for stage III colon cancer. Clin Colorectal Cancer 2015;14:25–30.
- Compton CC, Fielding LP, Burgart LJ, et al. Prognostic factors in colorectal cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med 2000;124:979–94.
- Lieu CH, Lambert LA, Wolff RA, et al. Systemic chemotherapy and surgical cytoreduction for poorly differentiated and signet ring cell adenocarcinomas of the appendix. Ann Oncol 2012;23:652–8.
- Krasna MJ, Flancbaum L, Cody RP, et al. Vascular and neural invasion in colorectal carcinoma. Incidence and prognostic significance. Cancer 1988;61:1018–23.
- Cianchi F, Palomba A, Boddi V, et al. Lymph node recovery from colorectal tumor specimens: recommendation for a minimum number of lymph nodes to be examined. World J Surg 2002;26:384–9.
- Yoshimatsu K, et al. How many lymph nodes should be examined in Dukes’ B colorectal cancer? Determination on the basis of cumulative survival rate. Hepatogastroenterology 2005;52:1703–6.
- Caplin S, Cerottini JP, Bosman FT, et al. For patients with Dukes’ B (TNM Stage II) colorectal carcinoma, examination of six or fewer lymph nodes is related to poor prognosis. Cancer 1998;83:666–72.
- Veronese N, Nottegar A, Pea A, et al. Prognostic impact and implications of extracapsular lymph node involvement in colorectal cancer: a systematic review with meta-analysis. Ann Oncol 2016;27:42–8.
- Li J, Yang S, Hu J, et al. Tumor deposits counted as positive lymph nodes in TNM staging for advanced colorectal cancer: a retrospective multicenter study. Oncotarget 2016;7:18269–79.
- Venook A, Niedzwiecki D, Innocenti Fet al. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016;34 no. 15 suppl. Abstract 3504.
- Schrag D, Brooks G, Meyerhardt JA ,et al. The relationship between primary tumor sidedness and prognosis in colorectal cancer. J Clin Oncol 2016;34 no. 15 suppl. Abstract 3505.
- Larrea AA, Lujan SA, Kunkel TA. SnapShot: DNA mismatch repair. Cell 2010;141:730 e1.
- Jass JR. Pathology of hereditary nonpolyposis colorectal cancer. Ann N Y Acad Sci 2000;910:62–73.
- Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer 1996;78:1149–67.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812–6.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol 2017;12:24.
- Bupathi M, Wu C. Biomarkers for immune therapy in colorectal cancer: mismatch-repair deficiency and others. J Gastrointest Oncol 2016;7:713–20.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
- Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
- Ogino S, Kuchiba A, Qian ZR, et al. Prognostic significance and molecular associations of 18q loss of heterozygosity: a cohort study of microsatellite stable colorectal cancers. J Clin Oncol 2009; 27:4591–8.
- Kim ST, Lee J, Park SH, et al. The effect of DNA mismatch repair (MMR) status on oxaliplatin-based first-line chemotherapy as in recurrent or metastatic colon cancer. Med Oncol 2010;27:1277–85.
- Sargent DJ, Monges G, Thibodeau SN, et al. Therapy in colon cancer. J Clin Oncol 2010;28:4664.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
- Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 2011;29:1261–270.
- Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses J Clin Oncol 2011;29:3768–74.
- Chang SC, Lin JK, Lin TC, Liang WY. Loss of heterozygosity: an independent prognostic factor of colorectal cancer. World J Gastroenterol 2005;11:778–84.
- Bertagnolli MM, Niedzwiecki D, Compton CC, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol 2009;27:1814–21.
- Bertagnolli MM, Redston M, Compton CC, et al. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: prospective evaluation of biomarkers for stages II and III colon cancer--a study of CALGB 9581 and 89803. J Clin Oncol 2011;29:3153–62.
- Dalerba P, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016;374: 211–22.
- Clark-Langone KM, Wu JY, Sangli C, et al. Biomarker discovery for colon cancer using a 761 gene RT-PCR assay. BMC Genomics 2007;8:279.
- Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011;29:4611–9.
- Niedzwiecki D, Bertagnolli MM, Warren RS, et al. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: results from CALGB 9581. J Clin Oncol 2011;29:3146–52.
- Yothers G, O’Connell MJ, Lee M, et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol 2013;31:4512–9.
- Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
- Gill S, Loprinzi C, Kennecke H, et al. Prognostic web-based models for stage II and III colon cancer: A population and clinical trials-based validation of numeracy and adjuvant! online. Cancer 2011;117:4155–65.
- Jung M, Kim GW, Jung I, et al. Application of the Western-based adjuvant online model to Korean colon cancer patients; a single institution experience. BMC Cancer 2012;12:471.
- Papamichael D, Renfro LA, Matthaiou C, et al. Validity of Adjuvant! Online in older patients with stage III colon cancer based on 2967 patients from the ACCENT database. J Geriatr Oncol 2016;7:422–9.
- Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011;117:4623–32.
- Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466–74.
- Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–8.
- Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol 2014;53:852–64.
- Taieb J, Le Malicot K, Shi Q, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J Natl Cancer Inst 2017;109(5).
- Palumbo LT, Sharpe WS, Henry JS. Cancer of the colon and rectum; analysis of 300 cases. Am J Surg 1965;109:439–44.
- Sharp GS, Benefiel WW. 5-Fluorouracil in the treatment of inoperable carcinoma of the colon and rectum. Cancer Chemother Rep 1962;20:97–101.
- Lawrence W Jr, Terz JJ, Horsley JS 3rd, et al. Chemotherapy as an adjuvant to surgery for colorectal cancer. Ann Surg 1975;181:616–23.
- Grage TD, et al. Adjuvant chemotherapy with 5-fluorouracil after surgical resection of colorectal carcinoma (COG protocol 7041). A preliminary report. Am J Surg 1977;133:59–66.
- Wolmark N, Fisher B, Rockette H, et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: results from NSABP protocol C-01. J Natl Cancer Inst 1988;80:30–6.
- Moertel CG, Fleming TR, Macdonald JS, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N Engl J Med 1990;322:352–8.
- Wolmark N, Rockette H, Fisher B, et al. The benefit of leucovorin-modulated fluorouracil as postoperative adjuvant therapy for primary colon cancer: results from National Surgical Adjuvant Breast and Bowel Project protocol C-03. J Clin Oncol 1993;11:1879–87.
- Comparison of fluorouracil with additional levamisole, higher-dose folinic acid, or both, as adjuvant chemotherapy for colorectal cancer: a randomised trial. QUASAR Collaborative Group. Lancet 2000;355(9215):1588–96.
- Chen TC, Hinton DR, Leichman L, et al. Multifocal inflammatory leukoencephalopathy associated with levamisole and 5-fluorouracil: case report. Neurosurgery 1994;35:1138-42.
- Porschen R, Bermann A, Löffler T, et al. Fluorouracil plus leucovorin as effective adjuvant chemotherapy in curatively resected stage III colon cancer: results of the trial adjCCA-01. J Clin Oncol 2001;19:1787–94.
- Arkenau HT, Bermann A, Rettig K, et al. 5-Fluorouracil plus leucovorin is an effective adjuvant chemotherapy in curatively resected stage III colon cancer: long-term follow-up results of the adjCCA-01 trial. Ann Oncol 2003;14:395–9.
- Weinerman B, Shah A, Fields A, et al. Systemic infusion versus bolus chemotherapy with 5-fluorouracil in measurable metastatic colorectal cancer. Am J Clin Oncol 1992;15:518–23.
- Poplin EA, Benedetti JK, Estes NC, et al. Phase III Southwest Oncology Group 9415/Intergroup 0153 randomized trial of fluorouracil, leucovorin, and levamisole versus fluorouracil continuous infusion and levamisole for adjuvant treatment of stage III and high-risk stage II colon cancer. J Clin Oncol 2005;23:1819–25.
- Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
- de Gramont A, Vignoud J, Tournigand C, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur J Cancer 1997;33:214–9.
- Diaz-Rubio E, Sastre J, Zaniboni A, et al. Oxaliplatin as single agent in previously untreated colorectal carcinoma patients: a phase II multicentric study. Ann Oncol 1998;9:105–8.
- André T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF mutation and mismatch repair status of the MOSAIC Study. J Clin Oncol 2015;33:4176–87.
- Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004;350:2343–51.
- Kuebler JP, Wieand HS, O’Connell MJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2007;25:2198–204.
- Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
- Schmoll HJ, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
- Colucci G, Gebbia V, Paoletti G, et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J Clin Oncol 2005;23:4866–75.
- Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 2004;22:229–37.
- Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42.
- Saltz LB, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008;26:2013–9.
- Cremolini C, Loupakis F, Ruzzo A, et al. Predictors of benefit in colorectal cancer treated with cetuximab: are we getting “Lost in TranslationAL”? J Clin Oncol 2010;28:e173–4.
- Sorich MJ, Wiese MD, Rowland D, et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015;26:13–21.
- Grothey A, van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):303–12.
- Saltz LB, Niedzwiecki D, Hollis D, et al. Irinotecan fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: results of CALGB 89803. J Clin Oncol 2007;25:3456–61.
- Van Cutsem E, et al. Randomized phase III trial comparing biweekly infusional fluorouracil/leucovorin alone or with irinotecan in the adjuvant treatment of stage III colon cancer: PETACC-3. J Clin Oncol 2009;27:3117–25.
- Allegra CJ, et al. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J Clin Oncol 2013;31:359–64.
- de Gramont A, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol 2012;13:1225–33.
- Alberts SR, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA 2012;307:1383–93.
- Taieb J, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:862–73.
- Shi Q, Sobrero AF, Shields AF, et al. Prospective pooled analysis of six phase III trials investigating duration of adjuvant (adjuvant) oxaliplatin-based therapy (3 vs 6 months) for patients (pts) with stage III colon cancer (CC): The IDEA (International Duration Evaluation of Adjuvant chemotherapy) collaboration. In: Proceedings from the American Society of Clinical Oncology; June 1–5, 2017; Chicago. Abstract LBA1.
- Quasar Collaborative Group; Gray R, Barnwell J, McConkey C, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370(9604):2020–9.
- Efficacy of adjuvant fluorouracil and folinic acid in B2 colon cancer. International Multicentre Pooled Analysis of B2 Colon Cancer Trials (IMPACT B2) Investigators. J Clin Oncol 1999;17:1356–63.
- Kidwell KM, et al. Long-term neurotoxicity effects of oxaliplatin added to fluorouracil and leucovorin as adjuvant therapy for colon cancer: results from National Surgical Adjuvant Breast and Bowel Project trials C-07 and LTS-01. Cancer 2012;118:5614–22.
- Beijers AJ, Mols F, Vreugdenhil G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer 2014;22:1999–2007.
- Mols F, Beijers T, Lemmens V, et al. Chemotherapy-induced neuropathy and its association with quality of life among 2- to 11-year colorectal cancer survivors: results from the population-based PROFILES registry. J Clin Oncol 2013;31:2699–707.
- Raphael MJ, Fischer HD, Fung K, et al. Neurotoxicity outcomes in a population-based cohort of elderly patients treated with adjuvant oxaliplatin for colorectal cancer. Clin Colorectal Cancer 2017 March 24.
- Toki MI, Saif MW, Syrigos KN. Hypersensitivity reactions associated with oxaliplatin and their clinical management. Expert Opin Drug Saf 2014;13:1545–54.
- Siu SW, Chan RT, Au GK. Hypersensitivity reactions to oxaliplatin: experience in a single institute. Ann Oncol 2006;17:259–61.
- Wong JT, Ling M, Patil S, et al. Oxaliplatin hypersensitivity: evaluation, implications of skin testing, and desensitization. J Allergy Clin Immunol Pract 2014;2:40–5.
- Benson AB 3rd, Venook AP, Cederquist L, et al. NCCN Guidelines Colon Cancer Version 2.2017. www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed May 8, 2017.
- Wolmark N, Rockette H, Mamounas E, et al. Clinical trial to assess the relative efficacy of fluorouracil and leucovorin, fluorouracil and levamisole, and fluorouracil, leucovorin, and levamisole in patients with Dukes’ B and C carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project C-04. J Clin Oncol 1999;17:3553–9.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67(1):7–30.
- United States Cancer Statistics. 1999–2013 incidence and mortality web-based report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute, 2016. www.cdc.gov/uscs. Accessed June 12, 2017.
- Ahnen DJ, Wade SW, Jones WF, et al. The increasing incidence of young-onset colorectal cancer: a call to action. Mayo Clin Proc 2014;89:216–24.
- Jemal A, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States, 1974–2013. J Natl Cancer Inst 2017;109(8).
- Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer 2013;49:3680–5.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60: 950–7.
- van Puijenbroek M, Nielsen M, Tops CM, et al. Identification of patients with (atypical) MUTYH-associated polyposis by KRAS2 c.34G > T prescreening followed by MUTYH hotspot analysis in formalin-fixed paraffin-embedded tissue. Clin Cancer Res 2008;14:139–42.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006;119:807–14.
- Tuohy TM, Rowe KG, Mineau GP, et al. Risk of colorectal cancer and adenomas in the families of patients with adenomas: a population-based study in Utah. Cancer 2014;120:35–42.
- Choi Y, Sateia HF, Peairs KS, Stewart RW. Screening for colorectal cancer. Semin Oncol 2017; 44:34–44.
- Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med 1992;326:658–62.
- Rutter MD. Surveillance programmes for neoplasia in colitis. J Gastroenterol 2011;46 Suppl 1:1–5.
- Giovannucci E. Modifiable risk factors for colon cancer. Gastroenterol Clin North Am 2002;31:925–43.
- Michels KB, Fuchs GS, Giovannucci E, et al. Fiber intake and incidence of colorectal cancer among 76,947 women and 47,279 men. Cancer Epidemiol Biomarkers Prev 2005;14:842–9.
- Omata F, Brown WR, Tokuda Y, et al. Modifiable risk factors for colorectal neoplasms and hyperplastic polyps. Intern Med 2009;48:123–8.
- Friedenreich CM, Neilson HK, Lynch BM. State of the epidemiological evidence on physical activity and cancer prevention. Eur J Cancer 2010;46:2593–604.
- Aleksandrova K, Pischon T, Jenab M, et al. Combined impact of healthy lifestyle factors on colorectal cancer: a large European cohort study. BMC Med 2014;12:168.
- Hermanek P, Wittekind C. The pathologist and the residual tumor (R) classification. Pathol Res Pract 1994;190:115–23.
- Lehnert T, Methner M, Pollok A, et al. Multivisceral resection for locally advanced primary colon and rectal cancer: an analysis of prognostic factors in 201 patients. Ann Surg 2002;235:217–25.
- Feinberg AE, et al. Oncologic outcomes following laparoscopic versus open resection of pT4 colon cancer: a systematic review and meta-analysis. Dis Colon Rectum 2017;60:116–125.
- Vignali A, et al. Laparoscopic treatment of advanced colonic cancer: a case-matched control with open surgery. Colorectal Dis 2013;15:944–8.
- Gainant A. Emergency management of acute colonic cancer obstruction. J Visc Surg 2012;149: e3–e10.
- Rosenman LD. Hartmann’s operation. Am J Surg 1994;168:283–4.
- Lee-Kong S, Lisle D. Surgical management of complicated colon cancer. Clin Colon Rectal Surg 2015;28:228–33.
- Bertelsen CA. Complete mesocolic excision an assessment of feasibility and outcome. Dan Med J 2017;64(2).
- Wolff WI SH. Definitive treatment of “malignant” polyps of the colon. Ann Surg 1975;182:516–25.
- Clinical Outcomes of Surgical Therapy Study Group, Nelson H, Sargent DJ, Wieand HS, et al. A comparison of laparoscopically assisted and open colectomy for colon cancer. N Engl J Med 2004;350:2050–9.
- Gunderson LL, Jessup JM, Sarjent DJ, et al. Revised tumor and node categorization for rectal cancer based on surveillance, epidemiology, and end results and rectal pooled analysis outcomes. J Clin Oncol 2010;28:256–63.
- Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
- Alves A, Panis Y, Mathieu P, et al. Postoperative mortality and morbidity in French patients undergoing colorectal surgery: results of a prospective multicenter study. Arch Surg 2005;140:278–83.
- Popescu RA, Norman A, Ross PJ, et al, Adjuvant or palliative chemotherapy for colorectal cancer in patients 70 years or older. J Clin Oncol 1999;17:2412–8.
- McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
- Tominaga T, Nonaka T, Sumida Y, et al. Effectiveness of adjuvant chemotherapy for elderly patients with lymph node-positive colorectal cancer. World J Surg Oncol 2016;14:197.
- Bos AC, van Erning FN, van Gestel YR, et al. Timing of adjuvant chemotherapy and its relation to survival among patients with stage III colon cancer. Eur J Cancer 2015;51:2553–61.
- Peixoto RD, Kumar A, Speers C, et al. Effect of delay in adjuvant oxaliplatin-based chemotherapy for stage III colon cancer. Clin Colorectal Cancer 2015;14:25–30.
- Compton CC, Fielding LP, Burgart LJ, et al. Prognostic factors in colorectal cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med 2000;124:979–94.
- Lieu CH, Lambert LA, Wolff RA, et al. Systemic chemotherapy and surgical cytoreduction for poorly differentiated and signet ring cell adenocarcinomas of the appendix. Ann Oncol 2012;23:652–8.
- Krasna MJ, Flancbaum L, Cody RP, et al. Vascular and neural invasion in colorectal carcinoma. Incidence and prognostic significance. Cancer 1988;61:1018–23.
- Cianchi F, Palomba A, Boddi V, et al. Lymph node recovery from colorectal tumor specimens: recommendation for a minimum number of lymph nodes to be examined. World J Surg 2002;26:384–9.
- Yoshimatsu K, et al. How many lymph nodes should be examined in Dukes’ B colorectal cancer? Determination on the basis of cumulative survival rate. Hepatogastroenterology 2005;52:1703–6.
- Caplin S, Cerottini JP, Bosman FT, et al. For patients with Dukes’ B (TNM Stage II) colorectal carcinoma, examination of six or fewer lymph nodes is related to poor prognosis. Cancer 1998;83:666–72.
- Veronese N, Nottegar A, Pea A, et al. Prognostic impact and implications of extracapsular lymph node involvement in colorectal cancer: a systematic review with meta-analysis. Ann Oncol 2016;27:42–8.
- Li J, Yang S, Hu J, et al. Tumor deposits counted as positive lymph nodes in TNM staging for advanced colorectal cancer: a retrospective multicenter study. Oncotarget 2016;7:18269–79.
- Venook A, Niedzwiecki D, Innocenti Fet al. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016;34 no. 15 suppl. Abstract 3504.
- Schrag D, Brooks G, Meyerhardt JA ,et al. The relationship between primary tumor sidedness and prognosis in colorectal cancer. J Clin Oncol 2016;34 no. 15 suppl. Abstract 3505.
- Larrea AA, Lujan SA, Kunkel TA. SnapShot: DNA mismatch repair. Cell 2010;141:730 e1.
- Jass JR. Pathology of hereditary nonpolyposis colorectal cancer. Ann N Y Acad Sci 2000;910:62–73.
- Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer 1996;78:1149–67.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812–6.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol 2017;12:24.
- Bupathi M, Wu C. Biomarkers for immune therapy in colorectal cancer: mismatch-repair deficiency and others. J Gastrointest Oncol 2016;7:713–20.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
- Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
- Ogino S, Kuchiba A, Qian ZR, et al. Prognostic significance and molecular associations of 18q loss of heterozygosity: a cohort study of microsatellite stable colorectal cancers. J Clin Oncol 2009; 27:4591–8.
- Kim ST, Lee J, Park SH, et al. The effect of DNA mismatch repair (MMR) status on oxaliplatin-based first-line chemotherapy as in recurrent or metastatic colon cancer. Med Oncol 2010;27:1277–85.
- Sargent DJ, Monges G, Thibodeau SN, et al. Therapy in colon cancer. J Clin Oncol 2010;28:4664.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
- Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 2011;29:1261–270.
- Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses J Clin Oncol 2011;29:3768–74.
- Chang SC, Lin JK, Lin TC, Liang WY. Loss of heterozygosity: an independent prognostic factor of colorectal cancer. World J Gastroenterol 2005;11:778–84.
- Bertagnolli MM, Niedzwiecki D, Compton CC, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol 2009;27:1814–21.
- Bertagnolli MM, Redston M, Compton CC, et al. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: prospective evaluation of biomarkers for stages II and III colon cancer--a study of CALGB 9581 and 89803. J Clin Oncol 2011;29:3153–62.
- Dalerba P, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016;374: 211–22.
- Clark-Langone KM, Wu JY, Sangli C, et al. Biomarker discovery for colon cancer using a 761 gene RT-PCR assay. BMC Genomics 2007;8:279.
- Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011;29:4611–9.
- Niedzwiecki D, Bertagnolli MM, Warren RS, et al. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: results from CALGB 9581. J Clin Oncol 2011;29:3146–52.
- Yothers G, O’Connell MJ, Lee M, et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol 2013;31:4512–9.
- Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
- Gill S, Loprinzi C, Kennecke H, et al. Prognostic web-based models for stage II and III colon cancer: A population and clinical trials-based validation of numeracy and adjuvant! online. Cancer 2011;117:4155–65.
- Jung M, Kim GW, Jung I, et al. Application of the Western-based adjuvant online model to Korean colon cancer patients; a single institution experience. BMC Cancer 2012;12:471.
- Papamichael D, Renfro LA, Matthaiou C, et al. Validity of Adjuvant! Online in older patients with stage III colon cancer based on 2967 patients from the ACCENT database. J Geriatr Oncol 2016;7:422–9.
- Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011;117:4623–32.
- Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466–74.
- Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–8.
- Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol 2014;53:852–64.
- Taieb J, Le Malicot K, Shi Q, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J Natl Cancer Inst 2017;109(5).
- Palumbo LT, Sharpe WS, Henry JS. Cancer of the colon and rectum; analysis of 300 cases. Am J Surg 1965;109:439–44.
- Sharp GS, Benefiel WW. 5-Fluorouracil in the treatment of inoperable carcinoma of the colon and rectum. Cancer Chemother Rep 1962;20:97–101.
- Lawrence W Jr, Terz JJ, Horsley JS 3rd, et al. Chemotherapy as an adjuvant to surgery for colorectal cancer. Ann Surg 1975;181:616–23.
- Grage TD, et al. Adjuvant chemotherapy with 5-fluorouracil after surgical resection of colorectal carcinoma (COG protocol 7041). A preliminary report. Am J Surg 1977;133:59–66.
- Wolmark N, Fisher B, Rockette H, et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: results from NSABP protocol C-01. J Natl Cancer Inst 1988;80:30–6.
- Moertel CG, Fleming TR, Macdonald JS, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N Engl J Med 1990;322:352–8.
- Wolmark N, Rockette H, Fisher B, et al. The benefit of leucovorin-modulated fluorouracil as postoperative adjuvant therapy for primary colon cancer: results from National Surgical Adjuvant Breast and Bowel Project protocol C-03. J Clin Oncol 1993;11:1879–87.
- Comparison of fluorouracil with additional levamisole, higher-dose folinic acid, or both, as adjuvant chemotherapy for colorectal cancer: a randomised trial. QUASAR Collaborative Group. Lancet 2000;355(9215):1588–96.
- Chen TC, Hinton DR, Leichman L, et al. Multifocal inflammatory leukoencephalopathy associated with levamisole and 5-fluorouracil: case report. Neurosurgery 1994;35:1138-42.
- Porschen R, Bermann A, Löffler T, et al. Fluorouracil plus leucovorin as effective adjuvant chemotherapy in curatively resected stage III colon cancer: results of the trial adjCCA-01. J Clin Oncol 2001;19:1787–94.
- Arkenau HT, Bermann A, Rettig K, et al. 5-Fluorouracil plus leucovorin is an effective adjuvant chemotherapy in curatively resected stage III colon cancer: long-term follow-up results of the adjCCA-01 trial. Ann Oncol 2003;14:395–9.
- Weinerman B, Shah A, Fields A, et al. Systemic infusion versus bolus chemotherapy with 5-fluorouracil in measurable metastatic colorectal cancer. Am J Clin Oncol 1992;15:518–23.
- Poplin EA, Benedetti JK, Estes NC, et al. Phase III Southwest Oncology Group 9415/Intergroup 0153 randomized trial of fluorouracil, leucovorin, and levamisole versus fluorouracil continuous infusion and levamisole for adjuvant treatment of stage III and high-risk stage II colon cancer. J Clin Oncol 2005;23:1819–25.
- Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
- de Gramont A, Vignoud J, Tournigand C, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur J Cancer 1997;33:214–9.
- Diaz-Rubio E, Sastre J, Zaniboni A, et al. Oxaliplatin as single agent in previously untreated colorectal carcinoma patients: a phase II multicentric study. Ann Oncol 1998;9:105–8.
- André T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF mutation and mismatch repair status of the MOSAIC Study. J Clin Oncol 2015;33:4176–87.
- Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004;350:2343–51.
- Kuebler JP, Wieand HS, O’Connell MJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2007;25:2198–204.
- Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
- Schmoll HJ, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
- Colucci G, Gebbia V, Paoletti G, et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J Clin Oncol 2005;23:4866–75.
- Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 2004;22:229–37.
- Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42.
- Saltz LB, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008;26:2013–9.
- Cremolini C, Loupakis F, Ruzzo A, et al. Predictors of benefit in colorectal cancer treated with cetuximab: are we getting “Lost in TranslationAL”? J Clin Oncol 2010;28:e173–4.
- Sorich MJ, Wiese MD, Rowland D, et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015;26:13–21.
- Grothey A, van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):303–12.
- Saltz LB, Niedzwiecki D, Hollis D, et al. Irinotecan fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: results of CALGB 89803. J Clin Oncol 2007;25:3456–61.
- Van Cutsem E, et al. Randomized phase III trial comparing biweekly infusional fluorouracil/leucovorin alone or with irinotecan in the adjuvant treatment of stage III colon cancer: PETACC-3. J Clin Oncol 2009;27:3117–25.
- Allegra CJ, et al. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J Clin Oncol 2013;31:359–64.
- de Gramont A, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol 2012;13:1225–33.
- Alberts SR, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA 2012;307:1383–93.
- Taieb J, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:862–73.
- Shi Q, Sobrero AF, Shields AF, et al. Prospective pooled analysis of six phase III trials investigating duration of adjuvant (adjuvant) oxaliplatin-based therapy (3 vs 6 months) for patients (pts) with stage III colon cancer (CC): The IDEA (International Duration Evaluation of Adjuvant chemotherapy) collaboration. In: Proceedings from the American Society of Clinical Oncology; June 1–5, 2017; Chicago. Abstract LBA1.
- Quasar Collaborative Group; Gray R, Barnwell J, McConkey C, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370(9604):2020–9.
- Efficacy of adjuvant fluorouracil and folinic acid in B2 colon cancer. International Multicentre Pooled Analysis of B2 Colon Cancer Trials (IMPACT B2) Investigators. J Clin Oncol 1999;17:1356–63.
- Kidwell KM, et al. Long-term neurotoxicity effects of oxaliplatin added to fluorouracil and leucovorin as adjuvant therapy for colon cancer: results from National Surgical Adjuvant Breast and Bowel Project trials C-07 and LTS-01. Cancer 2012;118:5614–22.
- Beijers AJ, Mols F, Vreugdenhil G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer 2014;22:1999–2007.
- Mols F, Beijers T, Lemmens V, et al. Chemotherapy-induced neuropathy and its association with quality of life among 2- to 11-year colorectal cancer survivors: results from the population-based PROFILES registry. J Clin Oncol 2013;31:2699–707.
- Raphael MJ, Fischer HD, Fung K, et al. Neurotoxicity outcomes in a population-based cohort of elderly patients treated with adjuvant oxaliplatin for colorectal cancer. Clin Colorectal Cancer 2017 March 24.
- Toki MI, Saif MW, Syrigos KN. Hypersensitivity reactions associated with oxaliplatin and their clinical management. Expert Opin Drug Saf 2014;13:1545–54.
- Siu SW, Chan RT, Au GK. Hypersensitivity reactions to oxaliplatin: experience in a single institute. Ann Oncol 2006;17:259–61.
- Wong JT, Ling M, Patil S, et al. Oxaliplatin hypersensitivity: evaluation, implications of skin testing, and desensitization. J Allergy Clin Immunol Pract 2014;2:40–5.
- Benson AB 3rd, Venook AP, Cederquist L, et al. NCCN Guidelines Colon Cancer Version 2.2017. www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed May 8, 2017.
- Wolmark N, Rockette H, Mamounas E, et al. Clinical trial to assess the relative efficacy of fluorouracil and leucovorin, fluorouracil and levamisole, and fluorouracil, leucovorin, and levamisole in patients with Dukes’ B and C carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project C-04. J Clin Oncol 1999;17:3553–9.
Thrombosis in Pregnancy
INTRODUCTION
Venous thromboembolism (VTE), comprising deep vein thrombosis (DVT) and pulmonary embolism (PE), is a leading nonobstetric cause of maternal death in the United States and in developed countries.1,2 During pregnancy, the risk for VTE increases four- to six-fold, and although the risk is present throughout pregnancy, the mother is at highest risk immediately postpartum.3–5
VTE risk is increased due to physiologic and anatomic changes that occur in pregnancy. These changes include hypercoagulability, progesterone-induced venous stasis, decreased venous outflow, compression of the inferior vena cava and pelvic veins by the expanding uterus, and decreased mobility. The hypercoagulability of pregnancy is due to increased levels of coagulation factors I (fibrinogen), VII, VIII, and X, and von Willebrand factor; decreased free protein S, a natural anticoagulant; acquired resistance to activated protein C; and decreased fibrinolysis due to increased levels of plasminogen activator inhibitor-1 and -2.6,7 These changes confer increased hemostasis to the mother for delivery but also place her at higher risk for thrombosis.
A review of the literature found that more than 70% of pregnancy-associated DVTs are located in the ileofemoral region, as compared with approximately 9% in non-pregnant patients.8 The proximal location is associated with a higher risk for post-thrombotic syndrome and embolization as compared with calf DVTs.9 Proximal postnatal thrombosis, smoking, and older age are independent predictors of the development of post-thrombotic syndrome.10
RISK FACTORS
Clinical risk factors that increase the risk for VTE during pregnancy include a prior history of estrogen-related or unprovoked VTE, being a carrier of severe inherited thrombophilia (homozygotes for factor V Leiden or factor II G20210A variants, double heterozygotes, or persons with antithrombin, protein C, or protein S deficiencies), and the presence of antiphospholipid (aPL) antibodies.11 Women with systemic lupus erythematosus, diabetes, sickle cell disease, and heart disease also have a high risk for VTE during pregnancy.12 Other risk factors predisposing to thrombosis include black ethnicity, smoking, operative procedures, conception after assisted reproductive techniques, high body mass index, antepartum immobilization, severe preeclampsia, advanced age and parity, and a family history of VTE.13 A prospective cohort study of 1,297,037 pregnancies and related puerperium identified the following risk factors for thrombosis: hospitalization, infection, hyperemesis, multiple pregnancies, preeclampsia, obesity, cesarean section, major postpartum hemorrhage, intrauterine growth restriction, and fetal death.14 Risk factors identified in an Agency for Healthcare Research and Quality study include: age 35 or older, black ethnicity, lupus, sickle cell disease, heart disease, postpartum infection, and transfusion.15 The combination of more than one risk factor increases the risk for VTE. All these factors have to be considered when deciding on prophylactic or therapeutic anticoagulation therapy in pregnancy. In addition, the risks of anticoagulation, including bruising, bleeding, and other side effects (eg, reduced bone mineral density with therapeutic-dose unfractionated heparin), allergic reactions, and rarely thrombocytopenia, must be considered.
EVALUATION AND DIAGNOSIS
CASE PRESENTATION I
A 31-year-old woman G1P0 at 10 weeks’ gestation with no personal or family history of thrombosis presents with acute onset of shortness of breath and left-sided chest pain that awoke her the morning of presentation. Her vital signs are significant for a heart rate of 106 beats/min, respiration rate of 22 breaths/min, blood pressure of 105/76 mm Hg, and pulse oximetry of 98% on room air. The patient denies previous exposure to oral contraceptives. She does not smoke. She reports that she had noticed left calf pain and swelling, which worsened with walking after a 4-hour drive 2 days prior.
What is the approach to diagnosis of thromboembolism in pregnant patients?
DEEP VEIN THROMBOSIS
Although a clinical diagnosis of DVT in pregnancy is unreliable, a history and physical examination are necessary to exclude other diagnoses and to assess the likelihood of thrombosis. Unfortunately, studies of the accuracy of history and physical examination for detecting DVT and PE have not included pregnant patients. In most pregnant patients with clinically suspected DVT, the diagnosis is not confirmed. Other causes of leg pain and swelling are not uncommon during pregnancy and include cellulitis, ruptured Baker’s cyst, or muscular pain.
A cross-sectional study described the derivation of the LEFt clinical decision rule, which relies on 3 variables in pregnant women with suspected DVT: left leg presentation (L), ≥ 2 cm calf circumference difference (E for edema), and first trimester presentation (Ft). If none of these variables is present, the negative predictive value is 100%.16 A validation study suggested that a negative LEFt rule accurately identifies pregnant women in whom the risk for confirmed DVT appears to be very low. The rule should not be used as an individual test for excluding DVT during pregnancy, but could be applied in a diagnostic approach in association with D-dimer measurement and compression ultrasonography (CUS); however, it has not been prospectively validated for safety and efficacy.17 In a study of 149 consecutive pregnant women with suspected DVT, a whole-blood agglutination D-dimer had a sensitivity of 100% and specificity of 60%.18 A 2006 systematic review found only 4 diagnostic studies of VTE in pregnancy in the literature. One of these studies showed that a combination of a negative CUS and normal D-dimer can accurately exclude DVT.19
Serial CUS is necessary for pregnant women with a high clinical suspicion of DVT but a negative initial investigation. In a study of 221 pregnant women in whom DVT was clinically suspected, 16 women (7.2%) were diagnosed with DVT by initial CUS, and none were diagnosed with DVT onserial testing.20 During follow-up (≥ 3 months), 6 of the 205 women with normal serial CUS results presented with symptoms of DVT, PE, or both, and 1 of them was diagnosed with DVT and PE. The sensitivity of serial CUS with Doppler imaging was 94.1% (95% confidence interval [CI] 69.2% to 99.7%), and the negative predictive value was 99.5% (95% CI 96.9% to 100%).20 All ultrasounds undertaken for investigation of pregnancy-associated DVT should include imaging of the iliac veins if there is a high index of suspicion and the CUS is negative for femoral DVT. Serial CUS with Doppler imaging of the iliac vein performed over a 7-day period excludes DVT in symptomatic pregnant women.20 Repeat CUS may be done 2 to 4 days and 6 to 8 days after the initial scan.
Ileofemoral vein thrombosis accounts for approximately 90% of proximal thromboses in pregnancy, occurring most often in the left lower extremity.20 The incidence of isolated iliac vein thrombosis in pregnancy is low, but when it does occur, delay in diagnosis can lead to significant morbidity. Therefore, for women with suspected isolated iliac vein thrombosis in whom CUS is negative or nondiagnostic, magnetic resonance direct thrombus imaging (MRDTI) should be performed.21 Patients with iliac vein thrombosis may present with unexplained inguinal, pelvic, or abdominal pain, which may be accompanied by back pain, and they usually present with swelling of the entire leg. MRDTI does not require gadolinium contrast and its accuracy appears to be similar to that of venography for iliac vein thrombi in the nonpregnant population.21 Exposure to gadolinium during pregnancy is associated with an increased risk for rheumatologic, inflammatory, or infiltrative skin conditions and stillbirth or neonatal death.22
Ovarian vein thrombosis is a rare but serious diagnosis. It occurs mostly in the postpartum period, mainly after cesarean delivery, and usually affects the right ovarian vein. The diagnosis is confirmed by ultrasound, computed tomography (CT), or magnetic resonance imaging.23
PULMONARY EMBOLISM
PE is more difficult to diagnose than DVT, particularly because clinical signs of PE are unreliable in the pregnant patient. The mortality rate of untreated PE is high, ranging from 18% to 38%, and approximately one-third of patients with untreated thromboembolic disease develop recurrent embolism.24 Studies have reported a PE prevalence between 1.4% and 4.2% in pregnant women with suspected clinical diagnosis of PE.25
The clinical presentation of PE and associated laboratory testing results may be subtler in pregnant than in nonpregnant patients. Arterial blood gases (ABG) may show hypoxemia or hypocapnia. The ABG in pregnancy has a sensitivity of 76.9%, specificity of 20.2%, and negative and positive predictive values of 80% and 11.5% for PE, respectively.26 The alveolar-arterial oxygen gradient is a poor screening test for PE during pregnancy and postpartum. A retrospective chart review of 17 pregnant women with documented PE showed that 58% had normal alveolar-arterial gradients.27 Therefore, in a pregnant woman with a history suspicious for PE, objective imaging studies should be performed even if the patient has normal ABG.
The 2011 guidelines from the American Thoracic Society (ATS) and the Society of Thoracic Radiology (STR) recommend against using D-dimer to diagnose PE in pregnancy.28 In addition, lower extremity CUS should only be performed as the first diagnostic imaging procedure if the patient has signs or symptoms of DVT. Instead, the ATS/STR guidelines recommend a plain radiograph of the chest as the first imaging test. If the chest radiograph is normal, a ventilation/perfusion scan (V/Q) scan is preferred over CT pulmonary angiography (CTPA). Diagnostic accuracy of the V/Q scan may be superior to CTPA in pregnancy, and it is preferable because of the lower prevalence of indeterminate V/Q scan in pregnant women.29 Moreover, there is lower radiation exposure to the maternal breast and lung tissue with a V/Q scan than with CTPA. CTPA confers lower fetal radiation doses than V/Q scans (0.03–0.66 mGy versus 0.32–0.74 mGy, respectively) but higher total body maternal radiation (4–16 mSv versus 1–2.5 mSv).30 A quantitative approach to lung scan interpretation, based on the distribution histogram of V/Q ratios, may be helpful in categorizing patients with suspected PE.28 A study of 121 suspected episodes of PE in 120 pregnant women showed that 104 women with normal or nondiagnostic scans did not develop subsequent episodes of VTE during a mean follow-up period of 20 months.31
If the baseline chest radiograph is abnormal in a pregnant woman with clinical suspicion of PE, a CTPA should be performed. As noted, fetal radiation doses for CTPA examinations in which the fetus is not directly imaged are minimal. If CTPA is recommended for the diagnosis of PE, the patient should be informed that radiation to the breast may increase her baseline risk for breast cancer. The ATS guidelines state that “given the lack of evidence documenting clear superiority of any one diagnostic test, the values and preferences of a patient and her physician likely will and should determine the final choice and sequence of tests performed.”28
CASE I CONTINUED
Upon presentation to the emergency department, the circumference of the patient’s left leg is not significantly greater than that of her right leg, and her leg pain has resolved. Bilateral CUS is negative for proximal or distal DVT. Chest radiograph shows an opacification of her left lower lobe. CTPA shows bilateral segmental and subsegmental lower lobe pulmonary emboli.
How does risk for VTE change throughout pregnancy?
Women are at increased risk for VTE throughout the entire pregnancy, starting from conception, but mainly during the postpartum period. A Danish historical controlled cohort study of 819,751 pregnant women (ages 15–49 years) over a 10-year period identified 727 women with VTE. The absolute risk for VTE per 10,000 pregnancy-years increased from 4.1 (95% CI 3.2 to 5.2) during weeks 1 to 11 to 59.0 (95% CI 46.1 to 76.4) in week 40 and decreased in the postpartum period from 60 (95% CI 47.2 to 76.4) during the first week after birth to 2.1 during weeks 9–12 after birth (95% CI 1.1 to 4.2).32 This study showed that the risk of VTE increases throughout pregnancy and reaches its maximum during the peripartum period and is not significantly increased after 6 weeks post-delivery. In a retrospective cross-over cohort study of 1,687,930 women in California who delivered their first newborn, an elevated risk of VTE persisted until at least 12 weeks after delivery. However, the absolute increase in risk after 6 weeks postpartum was low.33
CASE 1 CONCLUSION
The patient is started on anticoagulation therapy and carefully monitored during the remainder of the pregnancy and postpartum period. Anticoagulation is discontinued 6 weeks after delivery.
TREATMENT
ANTICOAGULATION THERAPY
The treatment of VTE can be lifesaving. In a study comparing 35 patients with PE randomly assigned to treatment with anticoagulants versus no treatment, 5 of 19 patients in the untreated group died from PE and an additional 5 had nonfatal recurrences, as compared with none in the treated group.24 Unfractionated heparin (UFH) and low-molecular-weight heparin (LMWH) are both safe and effective anticoagulants during pregnancy as neither crosses the placenta. In a review of 186 reports of fetal and infant outcomes following anticoagulant therapy during pregnancy in 1325 pregnancies, outcomes in UFH-treated patients were similar to those in the normal population after excluding pregnancies with comorbid conditions independently associated with adverse outcomes.34 A 2005 systematic review of LMWH for prophylaxis and treatment of VTE during pregnancy included 64 studies of 277 pregnancies. There were no maternal deaths, live births resulted from 94.7% of the pregnancies, VTE or arterial thrombosis occurred in 0.86%, and significant bleeding occurred in 1.98%.35
The standard UFH regimen is an initial bolus of 5000 units subcutaneously and 17,500 units every 12 hours, with dose adjustment made based on a mid-interval activated partial thromboplastin time (aPTT).36 Although still controversial, it has been suggested that the anti-Xa assay with a mid-dosing interval target of 0.3 to 0.7 U/mL is a more reliable measure of therapeutic UFH activity than the aPTT, as the aPTT response is suppressed due to a pregnancy-related increase in factor VIII. LMWH is dosed based on weight; regimens are enoxaparin 1 mg/kg subcutaneously twice daily or 1.5 mg/kg subcutaneously once daily, and dalteparin 100 units/kg every 12 hours or 150 units/kg daily.
A 2017 Cochrane review of the effect of LMWH compared with UFH for the treatment of VTE in the nonpregnant setting included 23 studies with 9587 patients. Thrombotic complications (odds ratio [OR] 0.70 [CI 0.57 to 0.85]) and major hemorrhage (OR 0.58 [CI 0.40 to 0.83]) were lower in patients receiving LMWH, with a trend toward lower mortality.37 In addition, the incidence of bleeding complications in patients treated with subcutaneous LMWH versus intravenous heparin was compared in a 2012 systematic review of 27 randomized controlled trials with a total of 28,637 patients. In patients treated with LMWH, there was a nonstatistically significant lower incidence of major bleeding events (OR 0.79 [95% CI 0.60 to 1.04]) and a statistically significant reduction in bleeding risk (OR 0.68 [95% CI 0.47 to 1.00]) compared to patients treated with UFH.38 Additionally, a trial comparing the use of standard UFH versus LMWH found a significantly lower incidence of thrombocytopenia in patients treated with LMWH.39,40 Overall, LMWH is more effective at decreasing both thrombotic and bleeding complications, and the risk for osteoporosis is lower with LMWH. Based on these results, the American College of Chest Physicians (ACCP) recommends LMWH as the first-line treatment for VTE in pregnancy.41
In specific clinical situations, such as patients with renal dysfunction with creatinine clearance (CrCl) less than 30 mL/min, UFH is indicated. In a study of 103 pregnancies in 93 women given anti-coagulation during pregnancy, 89.3% received UFH. There were no maternal deaths, and fetal demise occurred in 8 pregnancies (7.8%) at a median of 14 weeks’ gestation. There were 2 episodes of PE (1.9%) and 2 major bleeding events requiring transfusion (1.9%).42 UFH costs much less than LMWH, and therefore UFH remains an important, inexpensive, and efficacious anticoagulant option for pregnant women who require anticoagulation and cannot afford LMWH.43
Due to the physiologic changes associated with pregnancy, LMWH and UFH dosages may need to be adjusted. An observational study of 20 pregnant women with acute VTE found no recurrent VTE or major bleeding after treatment with dalteparin. Dalteparin doses approximately 10% to 20% higher than those recommended in nonpregnant women were required to reach therapeutic target anti-Xa activity.44
Caution Regarding Oral Anticoagulants
Due to its teratogenicity, warfarin is not a first-line anticoagulation option. It is strictly contraindicated during the first trimester during organogenesis, and its use during pregnancy is restricted to women with mechanical heart valves. Warfarin crosses the placenta and has been associated with nasal hypoplasia, stippled epiphyses, and growth restriction, particularly between 6 to 9 weeks’ gestation. Every effort should be made to substitute UFH or LMWH for warfarin between 6 and 12 weeks of gestation. The bridging process should begin as early in the gestational age as possible due to the long half-life of warfarin.45 When used later in gestation, warfarin has been associated with fetal hemorrhage and central nervous system abnormalities. Other complications from use during the second and third trimesters include microcephaly, blindness, deafness, and fetal growth restriction.46,47 Its use also increases the risk for abortion and fetal death in utero.48–50
The direct oral anticoagulants (DOACs) are not approved for use in pregnancy. Although there are limited anecdotal reports of DOAC use in pregnancy,51 there is preclinical evidence of placental transfer with the DOACs rivaroxaban and apixaban (direct Xa inhibitors) and the oral thrombin inhibitor dabigatran, thus increasing the risk to the fetus.52–54 Edoxaban, another direct Xa inhibitor, should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. It should be discontinued in nursing mothers.55
THROMBOLYSIS
Fetal as well as maternal survival is dependent on adequate maternal perfusion and oxygenation. The risk of death from PE is significant, with a cross-sectional study of 58 patients with acute, massive PE showing a 55% mortality rate.56 Thus, pregnancy is not an absolute contraindication to mechanical or systemic (recombinant tissue plasminogen activator or streptokinase) thrombolysis in an unstable patient at high risk for death.57–59 There are no major studies of this approach, although a small review of 13 cases using systemic thrombolysis showed no increased risk of maternal mortality.58 Thrombolysis should be considered for appropriate indications in pregnant patients as it would be in nonpregnant patients. However, caution is required when drawing conclusions regarding maternal and fetal safety, given the lack of controlled clinical trials including pregnant women.
SURGICAL PULMONARY EMBOLECTOMY
Surgical pulmonary embolectomy is an important therapeutic and potentially life-saving option in women presenting with massive PE in the immediate postpartum period. Because of the risk of massive uterine bleeding immediately postpartum, thrombolytic therapy should not be used.60
INFERIOR VENA CAVA FILTER
Placement of an inferior vena cava (IVC) filter is indicated in patients who have an acute VTE with absolute contraindications for anticoagulation. In addition, it can be considered in patients with extensive ileofemoral venous thrombosis within 2 weeks prior to expected delivery.61 In a systematic review of 44 studies of IVC filters placed in pregnant patients, the IVC filter complication rate was 8.87% and the failure-to-retrieve rate was 11.25%.62 The complication rate is similar to that found in the nonpregnant population. Thus, IVC filters may be used when appropriately indicated and should be removed as soon as clinically feasible.
RECURRENT THROMBOSIS AND THROMBOPHILIAS
CASE PRESENTATION 2
A 34-year-old pregnant woman G1P0 at 38 weeks’ gestation presents with a painful, swollen left calf that is associated with difficulty on walking; the circumference of the left calf is 2 cm greater than that of the right. She has no shortness of breath or chest pain. She has a prior history of distal right lower extremity DVT while on combined oral contraceptives. Her mother also has a history of DVT while bedbound during a prolonged hospitalization at an older age. CUS is negative, and the patient is discharged home. However, 24 hours later she returns to the hospital with worsening swelling and pain in her left leg. Magnetic resonance venography demonstrates a large left external iliac and common iliac DVT. She is admitted and is started on UFH, and a retrievable IVC filter is placed in anticipation of delivery.
What is the risk for VTE recurrence during pregnancy?
A personal and family history of VTE should be obtained when evaluating pregnant patients. A retrospective study of 109 women with prior history of VTE showed recurrence rates per patient-year of 10.9% during pregnancy and 3.7% in the nonpregnant period; the relative risk of recurrent VTE during pregnancy was 3.5 (95% CI 1.6 to 7.8).63 Two large European retrospective cohort studies of VTE in pregnancy showed that the recurrence rate of VTE in women with a history of thrombosis is around 6% during pregnancy, equally distributed among trimesters. The highest incidence of recurrence was in the postpartum period, ranging from 8.3% to 10%.64 The recurrence risk during pregnancy in women with a history of a single episode of VTE was 2.4% antepartum (95% CI 0.2% to 6.9%).65 These risks may be lower in women without thrombophilia or with a temporary risk factor associated with their previous thromboembolic event.65 Recurrence risk is higher if the previous VTE was estrogen-related, either due to pregnancy or through hormonal contraception (10%), than if the previous VTE was non-estrogen-related (2.7%).64,66
The timing of the case patient’s presentation is consistent with reports of increased risk of VTE during the peripartum period. Her prior history of estrogen-related DVT is concerning for a risk of recurrence, particularly during pregnancy. A retrospective cohort study of 1104 women with previous VTE, 88 of whom became pregnant without receiving thromboprophylaxis, showed that the overall rate of VTE recurrence was 5.8% (95% CI 3.0% to 10.6%) and 8.3% (95% CI 4.5% to 14.6%) during pregnancy and postpartum, respectively. The risk of VTE recurrence was absent if the first VTE was related to a transient risk factor other than pregnancy, postpartum period, or hormonal contraception.67 However, the recurrence rate of VTE in women with prior unprovoked VTE and/or thrombophilia has been reported as 5.9% (95% CI 1.2% to 16.2%).65 The presence of an underlying hypercoagulable state can increase the recurrence risk by 25% to 50%, depending on the disorder.68 A retrospective cohort study of 270 pregnancies in 105 carriers of factor V Leiden, identified because of a symptomatic relative with the factor V Leiden mutation, found a VTE risk (mostly in the postpartum period) of 6.4% for heterozygous women, 16.7% for homozygous women, 20% for double heterozygous women, and 1.2% for noncarriers.69
Should the patient be screened for a thrombophilia disorder?
Half of all index thromboses in patients with thrombophilia occur in association with an additional risk factor. In women of child-bearing age, pregnancy, the postpartum period, and the use of combined hormonal contraception are all risk factors for VTE. A 2010 guideline from the British hematology community recommended testing for thrombophilia in women with prior VTE secondary to a minor provoking factor before or during pregnancy, but not testing women with unprovoked VTE (who would receive prophylaxis regardless) or those with VTE secondary to a major provoking factor (who would not require prophylaxis).70 Indications to screen for aPL antibodies include: women with (1) 3 unexplained recurrent first-trimester pregnancy losses or 1 second or third trimester fetal loss of morphologically normal fetuses; (2) severe preeclampsia; (3) intrauterine growth restriction; or (4) premature labor (< 34 weeks’ gestation).71,72
CASE 2 CONCLUSION
The patient is subsequently screened for inherited thrombophilia disorders and is found to be heterozygous for factor V Leiden.
CASE PRESENTATION 3
A 25-year-old woman is diagnosed with antiphospholipid syndrome (APS) during her second pregnancy when she experiences fetal loss during her second trimester. Pathologic examination of the placenta reveals infarcts. Laboratory evaluation reveals positive high-titer anticardiolipin and anti-beta-2 glycoprotein 1 antibodies (IgG isotype) and lupus anticoagulant on 2 separate occasions 12 weeks apart. In a subsequent pregnancy, she is started on prophylactic LMWH and daily low-dose aspirin (81 mg). At 36 weeks’ gestation, she presents with a blood pressure of 210/104 mm Hg and a platelet count of 94,000 cells/µL. She is diagnosed with preeclampsia and hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome and is induced for early delivery. About 2 weeks after vaginal delivery, she notices shortness of breath and chest pain. A CTPA demonstrates a right lower lobe lobar defect consistent with a PE. Her anticoagulation is increased to therapeutic dosage LMWH.
To what extent does thrombophilia increase the risk for VTE in pregnancy?
Approximately 50% of pregnancy-related VTEs are associated with inherited thrombophilia. A systematic review of 79 studies, in which 9 studies (n = 2526 patients) assessed the risk of VTE associated with inherited thrombophilia in pregnancy, revealed that the odds ratio for individuals with thrombophilia to develop VTE ranged from 0.74 to 34.40.73 Although women with thrombophilia have an increased relative risk of developing VTE in pregnancy, the absolute risk of VTE remains low (Table 1).41,73,74
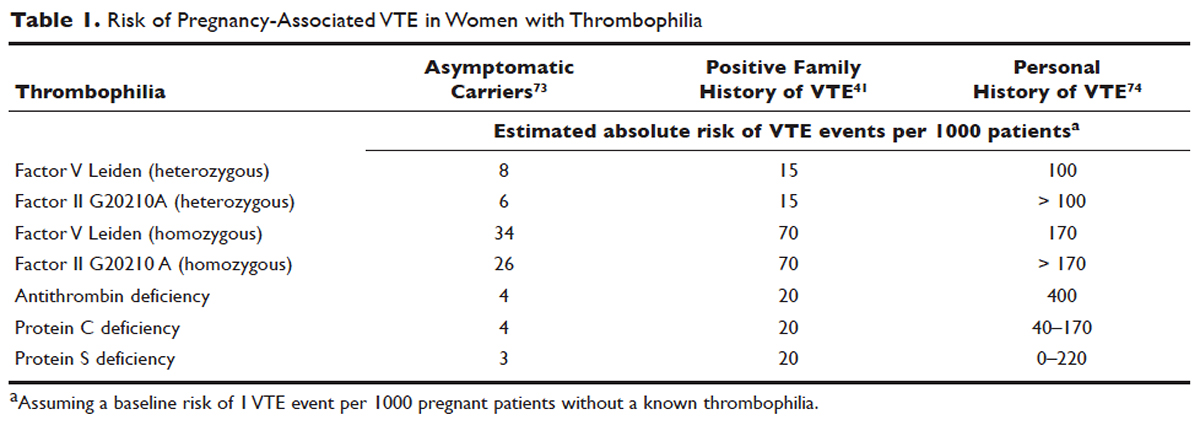
How is APS managed in pregnant patients?
Women with history of recurrent early pregnancy loss (< 10 weeks’ gestation) related to the presence of aPL antibodies are managed with low-dose aspirin and prophylactic-dose UFH or LMWH. This treatment increases the rate of subsequent successful pregnancy outcomes and reduces the risk for thrombosis. A 2010 systematic review and meta-analysis of UFH plus low-dose aspirin compared with low-dose aspirin alone in patients with APS and recurrent pregnancy loss included 5 trials and 334 patients. Patients receiving dual therapy had higher rates of live births (74.3%; relative risk [RR] 1.30 [CI 1.04 to 1.63]) compared to the aspirin-only group (55.8%).75 A 2009 randomized controlled trial compared low-dose aspirin to low-dose aspirin plus LMWH in women with recurrent pregnancy loss and either aPL antibodies, antinuclear antibody, or inherited thrombophilia. The study was stopped early after 4 years and found no difference in rates of live births between the groups (77.8% versus 79.1%).76 However, a randomized case-control trial of women with aPL antibodies and recurrent miscarriage found a 72% live birth rate in 47 women randomly assigned to low-dose aspirin and LMWH.77 A 2012 guideline from the American College of Chest Physicians (ACCP) recommends that women with aPL antibodies with a history of 3 or more pregnancy losses receive low-dose aspirin plus prophylactic-dose LMWH or UFH.78 A 2014 systematic review and meta-analysis showed that the combination of low-dose aspirin and UFH resulted in a higher live-birth rate than aspirin alone in 803 women with APS (RR 1.54 [95% CI 1.25 to 1.89]).79 Further large randomized controlled trials are needed to confirm optimal management of recurrent miscarriage and aPL antibodies.
The addition of prednisone to aspirin, heparin, or both has shown no benefits in pregnant women with aPL antibodies. Indeed, prolonged use of steroids may cause serious pregnancy complications, such as prematurity and hypertension.80–83 Intravenous infusions of immunoglobulin (IVIG) have not been shown to be superior to heparin and aspirin. This finding was confirmed in a multicenter clinical trial that tested the effects of IVIG compared with LMWH plus low-dose aspirin for the treatment of women with aPL antibodies and recurrent miscarriage. The rate of live-birth was 72.5% in the group treated with heparin plus low-dose aspirin compared with 39.5% in the IVIG group.84
Preeclampsia and HELLP syndrome complicated the case patient’s pregnancy even though she was being treated with prophylactic-dose LMWH and low-dose aspirin, the current standard of care for pregnant women with APS (UFH can be used as well). It is important to note that complications may still occur despite standard treatment. Indeed, PE is more common in the postpartum than in the antepartum period. Prompt diagnosis is paramount to initiate the appropriate treatment; in this case the dose of LMWH was increased from prophylactic to therapeutic dose. However, additional therapeutic modalities are necessary to improve outcomes. A randomized controlled trial comparing standard of care with or without hydroxychloroquine is under way to address this issue.
PROPHYLAXIS
CASE PRESENTATION 4
A 34-year-old woman G1P0 at 6 weeks’ gestation with a past medical history of a proximal lower extremity DVT while on oral contraception is treated with warfarin anticoagulation for 6 months. Her obstetrician consults the hematologist to advise regarding antithrombotic management during this pregnancy.
What is the approach to prophylaxis in women at high risk for pregnancy-associated VTE?
All women at high risk for pregnancy-associated VTE should be counseled about the signs and symptoms of DVT or PE during preconception and pregnancy and have a plan developed should these symptoms arise. The ACCP guidelines on antithrombotic therapy outline recommendations ranging from clinical vigilance to prophylactic and intermediate-dose anticoagulation, depending on the risk for VTE recurrence, based on the personal and family history of VTE and type of thrombophilia (Table 2).78 These recommendations range from grade 2B to 2C.

For women with a history of estrogen-related VTE, single unprovoked VTE, or recurrent unprovoked VTE not on chronic anticoagulation, antepartum and postpartum pharmacologic thromboprophylaxis with either prophylactic or intermediate-dose LMWH is recommended (grade 2C). In patients with prior history of provoked VTE (non-estrogen related), antepartum clinical vigilance and postpartum pharmacologic thromboprophylaxis is recommended (grade 2C, 2B).
In asymptomatic pregnant women who are homozygote carriers for factor V Leiden or prothrombin G20210A variants and have a positive family history of thrombosis, antepartum and postpartum pharmacologic thromboprophylaxis is recommended (grade 2B). In asymptomatic homozygote carriers of factor V Leiden or prothrombin G20210A variants with no family history of thrombosis and women with all other thrombophilias with a positive family history of thrombosis, postpartum pharmacologic thromboprophylaxis is indicated (grade 2B and 2C, respectively). For women with confirmed APS and clinical criteria of obstetric APS with recurrent pregnancy loss, antepartum thromboprophylaxis with LMWH and low-dose aspirin is recommended (grade 1B). For pregnant women with all other thrombophilias with no personal or family history of thrombosis, clinical vigilance is suggested (grade 2 C).78
As an alternative to LMWH, vitamin K antagonists (VKA) such as warfarin can be used for postpartum thromboprophylaxis; in patients with protein C or S deficiency, due to the risk of warfarin-induced skin necrosis, a rapid-onset anticoagulant must be concomitantly administered. Warfarin and LMWH are safe anticoagulants during lactation, but there are no clinical data on the effects of the DOACs on infants during lactation. Data from animal studies indicate that DOACs are secreted into breast milk.85
What risks are associated with anticoagulant therapy in pregnancy?
VKAs cross the placenta and can cause teratogenicity, pregnancy loss, fetal bleeding, and neurodevelopmental deficits. Therefore, discontinuation of VKAs prior to the sixth week of gestation is necessary to avoid warfarin embryopathy. DOACs have been shown to readily cross the placenta but with unknown human reproductive risks. Fondaparinux, a synthetic pentasaccharide, crosses the placenta in small quantities. Though there are reports of the successful use of fondaparinux in pregnancy, there is limited reported experience of its use in the first trimester.86
The risk for bleeding with anticoagulation is notably acceptable. In a case-control study of 88 pregnant women receiving therapeutic-dose anticoagulation, the risk of postpartum hemorrhage (PPH) after vaginal delivery was 30% in those who received LMWH anticoagulation versus 18% in those who did not (OR 1.9 [95% CI 1.1 to 3.5]).87 However, the risk for severe PPH (≥ 500 mL) was similar (5.6% versus 5.0%; OR 1.1 [95% CI 0.4 to 3.6]). The risk for PPH after cesarean section was 12% in LMWH users versus 4% in LMWH non-users (OR 2.9 [95% CI 0.5 to 19.4]). The risk for PPH associated with delivery within 24 hours after the last dose of LMWH was 1.2 times higher (95% CI 0.4 to 3.6) compared to a longer interval. Therefore, therapeutic LMWH increases the risk for blood loss after vaginal delivery, but not the risk for severe PPH. The risk for PPH is influenced by the interval between the last dose of LMWH and delivery. Of note in this study, per the institution’s protocol, the anticoagulation was stopped with signs of labor or determination of need for delivery. The risk for blood loss may be mitigated in more planned delivery scenarios.87
CASE 4 CONTINUED
The patient is placed on prophylactic-dose LMWH with good tolerance and delivers at 39 weeks' gestation via caesarian section due to nonprogression of labor. Postpartum she is restarted on prophylactic-dose anticoagulation with LMWH. Two weeks after discharge from the hospital, she presents with right calf pain and mild shortness of breath. On physical exam, her leg circumferences are equal. A D-dimer assay is 3375 ng/mL (normal 0–229). CUS of the right leg shows a complete occlusive DVT of the mid-distal superficial femoral and popliteal veins and partially occlusive acute DVT of the right posterior tibial and peroneal veins. CTPA reveals a right lower lobe PE. Because she had developed VTE despite prophylactic LMWH, her anticoagulation is changed to therapeutic dose. She is treated with anticoagulation with LMWH for a total of 3 months, after which a repeat CUS shows no residual thrombosis.
What is the recommended dosing of heparin and LMWH during pregnancy?
A prospective study of 14 pregnant women receiving UFH prophylaxis found that a prophylactic dose of 5000 units twice a day was inadequate to achieve prophylactic heparin levels in any patient in the second or third trimester.88 Similar to treatment dosage, there is no consensus evidence for prophylactic dosing, and dosage recommendations are based on expert opinion. In a retrospective study of 25 pregnant women on intermediate-dose UFH, the mean UFH dose required to achieve a target anti-factor Xa level of 0.1 to 0.3 units/mL was 236.9 units/kg/day.89 However, the use of anti-factor Xa levels for monitoring is controversial as there is no data to support a difference in outcomes with its use in prophylactic or therapeutic dosing.
The timing of the previous VTE history is important when deciding on the anticoagulant dose in pregnancy. In pregnant women with a VTE that occurred within the previous 4 to 6 weeks, full-dose anticoagulation with LMWH should be considered; an intermediate dose (three-fourths of a therapeutic dose) may be used if the thrombotic episode occurred more than 6 weeks earlier but still within a year. Prophylactic dosing may be sufficient if the episode occurred more than a year earlier.90 A clinical trial (High-Low) is under way to explore the optimal dose of LMWH in pregnant women with prior history of VTE who are not on chronic anticoagulation therapy.91
How is anticoagulation therapy managed in the peripartum period?
Neuraxial anesthesia during active labor while on anticoagulation increases the risk for central nervous system bleeding. Therefore, if spontaneous labor occurs in women on therapeutic dose anticoagulation, neuraxial anesthesia cannot be used. However, in the event of elective induction of labor or caesarean section, neuroaxial anesthesia may be performed 12 hours after the administration of the last prophylactic dose of LMWH or 24 hours after the last therapeutic dose of LMWH. Intravenous UFH should be stopped for 6 hours before induction of labor with a confirmed normal aPTT before placement of neuraxial anesthesia. There is no contraindication for using neuraxial anesthesia during subcutaneous standard UFH at total doses of 10,000 units daily. The risk of spinal hematoma with larger daily subcutaneous doses is unclear; therefore, a documented normal aPTT must be obtained before placement of neuroaxial anesthesia.
Postpartum, reinitiation of prophylactic-dose LMWH should be delayed for at least 12 hours after the removal of an epidural catheter. Therapeutic-dose LMWH should be administered no earlier than 24 hours after neuraxial anesthesia, providing that proper hemostasis is achieved. In the absence of persistent bleeding, if no regional anesthesia was used, LMWH may be resumed 12 hours after delivery.92 Anticoagulation with either LMWH or warfarin is recommended for at least 6 to 12 weeks postpartum.33
COUNSELING
Patients should be advised to manage controllable risk factors, including avoiding prolonged immobilization, avoiding excessive weight gain in pregnancy, and stopping smoking. Periods of immobilization tend to cause reduced blood flow (stasis), which predisposes to thrombosis. In a systematic review of records of all patients with confirmed PE after arrival at Charles de Gaulle airport in Paris during a 13-year period, women had a higher risk of PE after a long-distance flight than men, with an estimated incidence of 0.61 per million passengers versus 0.20, respectively; the incidence reached 7.24 and 2.35 cases, respectively, in passengers traveling more than 10,000 kilometers.93,94
The risk of air travel-related thrombosis in pregnant women is estimated to be between 0.03% and 0.1%. Physicians must decide on an individual basis how to prevent travel-related thrombosis in their pregnant patients. In most passengers, prevention can be limited to encouraging exercise, avoidance of long sleeping periods, and not using a window seat. Women at high risk for VTE, such as women with a prior history of VTE who are not on anticoagulation or women with known asymptomatic thrombophilia or other risk factors for thrombosis such as obesity, may benefit from a short period (1–3 days) of LMWH starting 2 hours before a long-distance flight.95
Activation of the coagulation system has been demonstrated in cigarette smokers.96 Heavy smoking was found to be a significant risk factor for VTE in a cross-sectional analysis of 2404 men and women.97 An increased risk for thrombosis during pregnancy is seen in cigarette smokers15,98 and is enhanced with the concomitant use of illicit drugs.99 Other obstetric complications associated with smoking and illicit drug use during pregnancy include preterm labor, spontaneous abortion, perinatal death, low birth weight, and abruption placenta. The efficacy of nicotine replacement therapy in pregnancy is uncertain.100 Recommendations are to advise patients to stop smoking, obtain psychosocial counseling, and utilize adjunctive therapies, which have been shown to have some effect on abstinence rates.101
CONCLUSION
Women are at increased risk for VTE during pregnancy and the postpartum period. Awareness of risk factors and the signs and symptoms of VTE is paramount. Prompt diagnosis and treatment is mandatory to decrease complications of VTE. LMWH is the mainstay treatment of VTE in pregnancy, as it does not cross the placenta. Both LMWH and warfarin are safe during lactation. Close communication among the patient, obstetrician, hematologist, anesthesiologist, and neonatologist is crucial to optimize the care of these patients.
- Knight M, Nour M, Tuffnell D, et al, eds. on behalf of MBRRACE-UK. Saving lives, improving mothers’ care—surveillance of maternal deaths in the UK 2012-14 and lessons learned to inform maternity care from the UK and Ireland confidential enquiries into maternal deaths and morbidity 2009-14. Oxford: National Perinatal Epidemiology Unit, University of Oxford; 2016: 69–75.
- Berg CJ, Callaghan WM, Syverson C, Henderson Z. Pregnancy-related mortality in the United States, 1998 to 2005. Obstet Gynecol 2010;116:1302–9.
- Salonen Ros H, Lichtenstein P, Bellocco R, et al. Increased risks of circulatory diseases in late pregnancy and puerperium. Epidemiology 2001;12:456–60.
- Heit JA, Kobbervig CE, James AH, et al. Trends in the incidence of venous thromboembolism during pregnancy or postpartum: a 30-year population-based study. Ann Intern Med 2005;143:697–706.
- Greer IA. Thrombosis in pregnancy: updates in diagnosis and management. Hematology Am Soc Hematol Educ Program 2012;2012:203–7.
- Hellgren M. Hemostasis during normal pregnancy and puerperium. Semin Thromb Hemost 2003;29:125–30.
- Brenner B. Haemostatic changes in pregnancy. Thromb Res 2004;114:409–14.
- Chan W-S, Spencer FA, Ginsberg JS. Anatomic distribution of deep vein thrombosis in pregnancy. CMAJ 2010;182:657–60.
- Greer IA. Prevention and management of venous thromboembolism in pregnancy. Clin Chest Med 2003;24:123–37.
- Wik HS, Jacobsen AF, Sandvik L, Sandset PM. Prevalence and predictors for post-thrombotic syndrome 3 to 16 years after pregnancy-related venous thrombosis: a population-based, cross-sectional, case-control study. J Thromb Haemost 2012;10:840–7.
- Pomp ER, Lenselink AM, Rosendaal FR, Doggen CJM. Pregnancy, the postpartum period and prothrombotic defects: risk of venous thrombosis in the MEGA study. J Thromb Haemost 2008;6:632–7.
- Marik PE, Plante LA. Venous thromboembolic disease and pregnancy. N Engl J Med 2008;359:2025–33.
- Jacobsen AF, Skjeldestad FE, Sandset PM. Ante- and postnatal risk factors of venous thrombosis: a hospital-based case-control study. J Thromb Haemost 2008;6:905–12.
- Virkus RA, Løkkegaard E, Lidegaard Ø, et al. Risk factors for venous thromboembolism in 1.3 million pregnancies: a nationwide prospective cohort. PloS One 2014;9:e96495.
- James AH, Jamison MG, Brancazio LR, Myers ER. Venous thromboembolism during pregnancy and the postpartum period: incidence, risk factors, and mortality. Am J Obstet Gynecol 2006;194:1311–5.
- Chan W-S, Lee A, Spencer FA, et al. Predicting deep venous thrombosis in pregnancy: out in “LEFt” field? Ann Intern Med 2009;151:85–92.
- Righini M, Jobic C, Boehlen F, et al. Predicting deep venous thrombosis in pregnancy: external validation of the LEFT clinical prediction rule. Haematologica 2013;98:545–8.
- Chan W-S, Chunilal S, Lee A, et al. A red blood cell agglutination D-dimer test to exclude deep venous thrombosis in pregnancy. Ann Intern Med 2007;147:165–70.
- Nijkeuter M, Ginsberg JS, Huisman MV. Diagnosis of deep vein thrombosis and pulmonary embolism in pregnancy: a systematic review. J Thromb Haemost 2006;4:496–500.
- Chan W-S, Spencer FA, Lee AY, et al. Safety of withholding anticoagulation in pregnant women with suspected deep vein thrombosis following negative serial compression ultrasound and iliac vein imaging. CMAJ 2013;185:E194–200.
- Dronkers CE, Srámek A, Huisman MV, Klok FA. Accurate diagnosis of iliac vein thrombosis in pregnancy with magnetic resonance direct thrombus imaging (MRDTI). BMJ Case Rep 2016;2016. pii: bcr2016218091.
- Ray JG, Vermeulen MJ, Bharatha A, et al. Association between MRI exposure during pregnancy and fetal and childhood outcomes. JAMA 2016;316:952–61.
- Royal College of Obstretricians and Gynaecologists. Thromboembolic disease in pregnancy and the puerperium: acute management. Green-top Guideline No. 37b. London: RCOG; 2015.
- Barritt DW, Jordan SC. Anticoagulant drugs in the treatment of pulmonary embolism. A controlled trial. Lancet 1960;1(7138):1309–12.
- Bourjeily G, Khalil H, Raker C, et al. Outcomes of negative multidetector computed tomography with pulmonary angiography in pregnant women suspected of pulmonary embolism. Lung 2012;190:105–11.
- Deutsch AB, Twitty P, Downes K, Parsons MT. Assessment of the alveolar-arterial oxygen gradient as a screening test for pulmonary embolism in pregnancy. Am J Obstet Gynecol 2010;203:373.e1–4.
- Powrie RO, Larson L, Rosene-Montella K, et al. Alveolar-arterial oxygen gradient in acute pulmonary embolism in pregnancy. Am J Obstet Gynecol 1998;178:394–6.
- Leung AN, Bull TM, Jaeschke R, et al. American Thoracic Society documents: an official American Thoracic Society/Society of Thoracic Radiology clinical practice guideline--evaluation of suspected pulmonary embolism in pregnancy. Radiology 2012;262:635–46.
- Cahill AG, Stout MJ, Macones GA, Bhalla S. Diagnosing pulmonary embolism in pregnancy using computed-tomographic angiography or ventilation-perfusion. Obstet Gynecol 2009;114:124–9.
- Bourjeily G, Paidas M, Khalil H, et al. Pulmonary embolism in pregnancy. Lancet 2010;375(9713):500–12.
- Chan WS, Ray JG, Murray S, et al. Suspected pulmonary embolism in pregnancy: clinical presentation, results of lung scanning, and subsequent maternal and pediatric outcomes. Arch Intern Med 2002;162:1170–5.
- Virkus RA, Løkkegaard ECL, Bergholt T, et al. Venous thromboembolism in pregnant and puerperal women in Denmark 1995-2005. A national cohort study. Thromb Haemost 2011;106:304–9.
- Kamel H, Navi BB, Sriram N, et al. Risk of a thrombotic event after the 6-week postpartum period. N Engl J Med 2014;370:1307–15.
- Ginsberg JS, Hirsh J, Turner DC, et al. Risks to the fetus of anticoagulant therapy during pregnancy. Thromb Haemost 1989;61:197–203.
- Greer IA, Nelson-Piercy C. Low-molecular-weight heparins for thromboprophylaxis and treatment of venous thromboembolism in pregnancy: a systematic review of safety and efficacy. Blood 2005;106:401–7.
- Prandoni P, Carnovali M, Marchiori A, Galilei Investigators. Subcutaneous adjusted-dose unfractionated heparin vs fixed-dose low-molecular-weight heparin in the initial treatment of venous thromboembolism. Arch Intern Med 2004;164:1077–83.
- Robertson L, Jones LE. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev 2017;(9):eb9;2:CD001100.
- Costantino G, Ceriani E, Rusconi AM, et al. Bleeding risk during treatment of acute thrombotic events with subcutaneous LMWH compared to intravenous unfractionated heparin; a systematic review. PloS One 2012;7:e44553.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med 1995;332:1330–5.
- Junqueira DRG, Perini E, Penholati RRM, Carvalho MG. Unfractionated heparin versus low molecular weight heparin for avoiding heparin-induced thrombocytopenia in postoperative patients. Cochrane Database Syst Rev 2012;(9):CD007557.
- Bates SM, Greer IA, Middeldorp S, et al. VTE, thrombophilia, antithrombotic therapy, and pregnancy. Chest 2012;141:e691S–e736S.
- Clark NP, Delate T, Witt DM, et al. A descriptive evaluation of unfractionated heparin use during pregnancy. J Thromb Thrombolysis 2009;27:267–73.
- Clark NP, Delate T, Cleary SJ, Witt DM. Analysis of unfractionated heparin dose requirements to target therapeutic anti-Xa intensity during pregnancy. Thromb Res 2010;125:402–5.
- Jacobsen AF, Qvigstad E, Sandset PM. Low molecular weight heparin (dalteparin) for the treatment of venous thromboembolism in pregnancy. BJOG 2003;110:139–44.
- Walfisch A, Koren G. The “warfarin window” in pregnancy: the importance of half-life. J Obstet Gynaecol Can 2010;32:988–9.
- Bates SM, Ginsberg JS. Anticoagulants in pregnancy: fetal effects. Baillières Clin Obstet Gynaecol 1997;11:479–88.
- Stevenson RE, Burton OM, Ferlauto GJ, Taylor HA. Hazards of oral anticoagulants during pregnancy. JAMA 1980;243:1549–51.
- Ginsberg JS, Hirsh J. Anticoagulants during pregnancy. Annu Rev Med 1989;40:79–86.
- Wong V, Cheng CH, Chan KC. Fetal and neonatal outcome of exposure to anticoagulants during pregnancy. Am J Med Genet 1993;45:17–21.
- Blickstein D, Blickstein I. The risk of fetal loss associated with Warfarin anticoagulation. Int J Gynaecol Obstet 2002;78:221–5.
- Burnett AE, Mahan CE, Vazquez SR, et al. Guidance for the practical management of the direct oral anticoagulants (DOACs) in VTE treatment. J Thromb Thrombolysis 2016;41:206–32.
- Bapat P, Pinto LSR, Lubetsky A, et al. Examining the transplacental passage of apixaban using the dually perfused human placenta. J Thromb Haemost 2016;14:1436–41.
- Bapat P, Pinto LSR, Lubetsky A, et al. Rivaroxaban transfer across the dually perfused isolated human placental cotyledon. Am J Obstet Gynecol 2015;213:710.e1–6.
- Bapat P, Kedar R, Lubetsky A, et al. Transfer of dabigatran and dabigatran etexilate mesylate across the dually perfused human placenta. Obstet Gynecol 2014;123:1256–61.
- Savaysa [package insert]. Parsippany (NJ): Daiichi Sankyo, Inc; 2015.
- Filipecki S, Tomkowski W, Hajduk B, et al. [Outcome of patients with clinically acute massive pulmonary embolism]. Pneumonol Alergol Pol 1994;62:132–7.
- Holden EL, Ranu H, Sheth A, et al. Thrombolysis for massive pulmonary embolism in pregnancy--a report of three cases and follow up over a two year period. Thromb Res 2011;127:58–9.
- te Raa GD, Ribbert LS, Snijder RJ, Biesma DH. Treatment options in massive pulmonary embolism during pregnancy; a case-report and review of literature. Thromb Res 2009;124:1–5.
- Leonhardt G, Gaul C, Nietsch HH, et al. Thrombolytic therapy in pregnancy. J Thromb Thrombolysis 2006;21:271–6.
- Colombier S, Niclauss L. Successful surgical pulmonary embolectomy for massive perinatal embolism after emergency cesarean section. Ann Vasc Surg 2015;29:1452.e1–4.
- British Committee for Standards in Haematology Writing Group, Baglin TP, Brush J, Streiff M. Guidelines on use of vena cava filters. Br J Haematol 2006;134:590–5.
- Harris SA, Velineni R, Davies AH. Inferior vena cava filters in pregnancy: a systematic review. J Vasc Interv Radiol 2016;27:354–360.
- Pabinger I, Grafenhofer H, Kyrle PA, et al. Temporary increase in the risk for recurrence during pregnancy in women with a history of venous thromboembolism. Blood 2002;100:1060–2.
- Pabinger I, Grafenhofer H, Kaider A, et al. Risk of pregnancy-associated recurrent venous thromboembolism in women with a history of venous thrombosis. J Thromb Haemost 2005;3:949–54.
- Brill-Edwards P, Ginsberg JS, Gent M, et al. Safety of withholding heparin in pregnant women with a history of venous thromboembolism. Recurrence of Clot in This Pregnancy Study Group. N Engl J Med 2000;343:1439–44.
- De Stefano V, Martinelli I, Rossi E, et al. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006;135:386–91.
- De Stefano V, Martinelli I, Rossi E, et al. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006;135:386–91.
- Lim W, Eikelboom JW, Ginsberg JS. Inherited thrombophilia and pregnancy associated venous thromboembolism. BMJ 2007;334:1318–21.
- Tormene D, Simioni P, Prandoni P, et al. Factor V Leiden mutation and the risk of venous thromboembolism in pregnant women. Haematologica 2001;86:1305–9.
- Baglin T, Gray E, Greaves M, et al. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010;149:209–20.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4:295–306.
- Pengo V, Tripodi A, Reber G, et al. Update of the guidelines for lupus anticoagulant detection. J Thromb Haemost 2009;7:1737–40.
- Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol 2006;132:171–96.
- American College of Obstetricians and Gynecologists Women’s Health Care Physicians. ACOG Practice Bulletin No. 138: Inherited thrombophilias in pregnancy. Obstet Gynecol 2013;122:706–17.
- Mak A, Cheung MW, Cheak AA, Ho RC. Combination of heparin and aspirin is superior to aspirin alone in enhancing live births in patients with recurrent pregnancy loss and positive anti-phospholipid antibodies: a meta-analysis of randomized controlled trials and meta-regression. Rheumatology (Oxf) 2010;49:281–8.
- Laskin CA, Spitzer KA, Clark CA, et al. Low molecular weight heparin and aspirin for recurrent pregnancy loss: results from the randomized, controlled HepASA Trial. J Rheumatol 2009;36:279–87.
- Farquharson RG, Quenby S, Greaves M. Antiphospholipid syndrome in pregnancy: a randomized, controlled trial of treatment. Obstet Gynecol 2002;100:408–13.
- Bates SM, Jaeschke R, Stevens SM, et al. Diagnosis of DVT: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012;141(2 Suppl):e351S–418S.
- Lubbe WF, Butler WS, Palmer SJ, Liggins GC. Fetal survival after prednisone suppression of maternal lupus-anticoagulant. Lancet 1983;1(8338):1361–3.
- Lockshin MD, Druzin ML, Qamar T. Prednisone does not prevent recurrent fetal death in women with antiphospholipid antibody. Am J Obstet Gynecol 1989;160:439–43.
- Silver RK, MacGregor SN, Sholl JS, et al. Comparative trial of prednisone plus aspirin versus aspirin alone in the treatment of anticardiolipin antibody-positive obstetric patients. Am J Obstet Gynecol 1993;169:1411–7.
- Cowchock FS, Reece EA, Balaban D, et al. Repeated fetal losses associated with antiphospholipid antibodies: a collaborative randomized trial comparing prednisone with low-dose heparin treatment. Am J Obstet Gynecol 1992;166:1318–23.
- Laskin CA, Bombardier C, Hannah ME, et al. Prednisone and aspirin in women with autoantibodies and unexplained recurrent fetal loss. N Engl J Med 1997;337:148–53.
- Dendrinos S, Sakkas E, Makrakis E. Low-molecular-weight heparin versus intravenous immunoglobulin for recurrent abortion associated with antiphospholipid antibody syndrome. Int J Gynaecol Obstet 2009;104:223–5.
- Cohen H, Arachchillage DRJ, Beyer-Westendorf J, et al. Direct oral anticoagulants and women. Semin Thromb Hemost 2016;42:789–97.
- Bates SM, Middeldorp S, Rodger M, et al. Guidance for the treatment and prevention of obstetric-associated venous thromboembolism. J Thromb Thrombolysis 2016;41:92–128.
- Knol HM, Schultinge L, Veeger NJ, et al. The risk of postpartum hemorrhage in women using high dose of low-molecular-weight heparins during pregnancy. Thromb Res 2012;130:334–8.
- Barbour LA, Smith JM, Marlar RA. Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol 1995;173:1869–73.
- Bergqvist A, Bergqvist D, Lindhagen A, Mätzsch T. Late symptoms after pregnancy-related deep vein thrombosis. Br J Obstet Gynaecol 1990;97:338–41.
- Rodger M. Evidence base for the management of venous thromboembolism in pregnancy. Hematology Am Soc Hematol Educ Program. 2010;2010:173–80.
- Bleker SM, Buchmüller A, Chauleur C, et al. Low-molecular-weight heparin to prevent recurrent venous thromboembolism in pregnancy: Rationale and design of the Highlow study, a randomised trial of two doses. Thromb Res 2016;144:62–8.
- Horlocker TT, Wedel DJ, Rowlingson JC, et al. Regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy: American Society of Regional Anesthesia and Pain Medicine evidence-based guidelines (third edition). Reg Anesth Pain Med 2010;35:64–101.
- Lapostolle F, Surget V, Borron SW, et al. Severe pulmonary embolism associated with air travel. N Engl J Med 2001;345:779–83.
- Lapostolle F, Le Toumelin P, Chassery C, et al. Gender as a risk factor for pulmonary embolism after air travel. Thromb Haemost 2009;102:1165–8.
- Cannegieter SC, Rosendaal FR. Pregnancy and travel-related thromboembolism. Thromb Res 2013;131 Suppl 1:S55–58.
- Miller GJ, Bauer KA, Cooper JA, Rosenberg RD. Activation of the coagulant pathway in cigarette smokers. Thromb Haemost 1998;79:549–53.
- Golomb BA, Chan VT, Denenberg JO, et al. Risk marker associations with venous thrombotic events: a cross-sectional analysis. BMJ Open 2014;4:e003208.
- Lindqvist P, Dahlbäck B, Marŝál K. Thrombotic risk during pregnancy: a population study. Obstet Gynecol 1999;94:595–9.
- Black M, Bhattacharya S, Fairley T, et al. Outcomes of pregnancy in women using illegal drugs and in women who smoke cigarettes. Acta Obstet Gynecol Scand 2013;92:47–52.
- Mendelsohn C, Gould GS, Oncken C. Management of smoking in pregnant women. Aust Fam Physician 2014;43:46–51.
- Chamberlain C, O’Mara-Eves A, Oliver S, et al. Psychosocial interventions for supporting women to stop smoking in pregnancy. Cochrane Database Syst Rev 2013;10:CD001055.
INTRODUCTION
Venous thromboembolism (VTE), comprising deep vein thrombosis (DVT) and pulmonary embolism (PE), is a leading nonobstetric cause of maternal death in the United States and in developed countries.1,2 During pregnancy, the risk for VTE increases four- to six-fold, and although the risk is present throughout pregnancy, the mother is at highest risk immediately postpartum.3–5
VTE risk is increased due to physiologic and anatomic changes that occur in pregnancy. These changes include hypercoagulability, progesterone-induced venous stasis, decreased venous outflow, compression of the inferior vena cava and pelvic veins by the expanding uterus, and decreased mobility. The hypercoagulability of pregnancy is due to increased levels of coagulation factors I (fibrinogen), VII, VIII, and X, and von Willebrand factor; decreased free protein S, a natural anticoagulant; acquired resistance to activated protein C; and decreased fibrinolysis due to increased levels of plasminogen activator inhibitor-1 and -2.6,7 These changes confer increased hemostasis to the mother for delivery but also place her at higher risk for thrombosis.
A review of the literature found that more than 70% of pregnancy-associated DVTs are located in the ileofemoral region, as compared with approximately 9% in non-pregnant patients.8 The proximal location is associated with a higher risk for post-thrombotic syndrome and embolization as compared with calf DVTs.9 Proximal postnatal thrombosis, smoking, and older age are independent predictors of the development of post-thrombotic syndrome.10
RISK FACTORS
Clinical risk factors that increase the risk for VTE during pregnancy include a prior history of estrogen-related or unprovoked VTE, being a carrier of severe inherited thrombophilia (homozygotes for factor V Leiden or factor II G20210A variants, double heterozygotes, or persons with antithrombin, protein C, or protein S deficiencies), and the presence of antiphospholipid (aPL) antibodies.11 Women with systemic lupus erythematosus, diabetes, sickle cell disease, and heart disease also have a high risk for VTE during pregnancy.12 Other risk factors predisposing to thrombosis include black ethnicity, smoking, operative procedures, conception after assisted reproductive techniques, high body mass index, antepartum immobilization, severe preeclampsia, advanced age and parity, and a family history of VTE.13 A prospective cohort study of 1,297,037 pregnancies and related puerperium identified the following risk factors for thrombosis: hospitalization, infection, hyperemesis, multiple pregnancies, preeclampsia, obesity, cesarean section, major postpartum hemorrhage, intrauterine growth restriction, and fetal death.14 Risk factors identified in an Agency for Healthcare Research and Quality study include: age 35 or older, black ethnicity, lupus, sickle cell disease, heart disease, postpartum infection, and transfusion.15 The combination of more than one risk factor increases the risk for VTE. All these factors have to be considered when deciding on prophylactic or therapeutic anticoagulation therapy in pregnancy. In addition, the risks of anticoagulation, including bruising, bleeding, and other side effects (eg, reduced bone mineral density with therapeutic-dose unfractionated heparin), allergic reactions, and rarely thrombocytopenia, must be considered.
EVALUATION AND DIAGNOSIS
CASE PRESENTATION I
A 31-year-old woman G1P0 at 10 weeks’ gestation with no personal or family history of thrombosis presents with acute onset of shortness of breath and left-sided chest pain that awoke her the morning of presentation. Her vital signs are significant for a heart rate of 106 beats/min, respiration rate of 22 breaths/min, blood pressure of 105/76 mm Hg, and pulse oximetry of 98% on room air. The patient denies previous exposure to oral contraceptives. She does not smoke. She reports that she had noticed left calf pain and swelling, which worsened with walking after a 4-hour drive 2 days prior.
What is the approach to diagnosis of thromboembolism in pregnant patients?
DEEP VEIN THROMBOSIS
Although a clinical diagnosis of DVT in pregnancy is unreliable, a history and physical examination are necessary to exclude other diagnoses and to assess the likelihood of thrombosis. Unfortunately, studies of the accuracy of history and physical examination for detecting DVT and PE have not included pregnant patients. In most pregnant patients with clinically suspected DVT, the diagnosis is not confirmed. Other causes of leg pain and swelling are not uncommon during pregnancy and include cellulitis, ruptured Baker’s cyst, or muscular pain.
A cross-sectional study described the derivation of the LEFt clinical decision rule, which relies on 3 variables in pregnant women with suspected DVT: left leg presentation (L), ≥ 2 cm calf circumference difference (E for edema), and first trimester presentation (Ft). If none of these variables is present, the negative predictive value is 100%.16 A validation study suggested that a negative LEFt rule accurately identifies pregnant women in whom the risk for confirmed DVT appears to be very low. The rule should not be used as an individual test for excluding DVT during pregnancy, but could be applied in a diagnostic approach in association with D-dimer measurement and compression ultrasonography (CUS); however, it has not been prospectively validated for safety and efficacy.17 In a study of 149 consecutive pregnant women with suspected DVT, a whole-blood agglutination D-dimer had a sensitivity of 100% and specificity of 60%.18 A 2006 systematic review found only 4 diagnostic studies of VTE in pregnancy in the literature. One of these studies showed that a combination of a negative CUS and normal D-dimer can accurately exclude DVT.19
Serial CUS is necessary for pregnant women with a high clinical suspicion of DVT but a negative initial investigation. In a study of 221 pregnant women in whom DVT was clinically suspected, 16 women (7.2%) were diagnosed with DVT by initial CUS, and none were diagnosed with DVT onserial testing.20 During follow-up (≥ 3 months), 6 of the 205 women with normal serial CUS results presented with symptoms of DVT, PE, or both, and 1 of them was diagnosed with DVT and PE. The sensitivity of serial CUS with Doppler imaging was 94.1% (95% confidence interval [CI] 69.2% to 99.7%), and the negative predictive value was 99.5% (95% CI 96.9% to 100%).20 All ultrasounds undertaken for investigation of pregnancy-associated DVT should include imaging of the iliac veins if there is a high index of suspicion and the CUS is negative for femoral DVT. Serial CUS with Doppler imaging of the iliac vein performed over a 7-day period excludes DVT in symptomatic pregnant women.20 Repeat CUS may be done 2 to 4 days and 6 to 8 days after the initial scan.
Ileofemoral vein thrombosis accounts for approximately 90% of proximal thromboses in pregnancy, occurring most often in the left lower extremity.20 The incidence of isolated iliac vein thrombosis in pregnancy is low, but when it does occur, delay in diagnosis can lead to significant morbidity. Therefore, for women with suspected isolated iliac vein thrombosis in whom CUS is negative or nondiagnostic, magnetic resonance direct thrombus imaging (MRDTI) should be performed.21 Patients with iliac vein thrombosis may present with unexplained inguinal, pelvic, or abdominal pain, which may be accompanied by back pain, and they usually present with swelling of the entire leg. MRDTI does not require gadolinium contrast and its accuracy appears to be similar to that of venography for iliac vein thrombi in the nonpregnant population.21 Exposure to gadolinium during pregnancy is associated with an increased risk for rheumatologic, inflammatory, or infiltrative skin conditions and stillbirth or neonatal death.22
Ovarian vein thrombosis is a rare but serious diagnosis. It occurs mostly in the postpartum period, mainly after cesarean delivery, and usually affects the right ovarian vein. The diagnosis is confirmed by ultrasound, computed tomography (CT), or magnetic resonance imaging.23
PULMONARY EMBOLISM
PE is more difficult to diagnose than DVT, particularly because clinical signs of PE are unreliable in the pregnant patient. The mortality rate of untreated PE is high, ranging from 18% to 38%, and approximately one-third of patients with untreated thromboembolic disease develop recurrent embolism.24 Studies have reported a PE prevalence between 1.4% and 4.2% in pregnant women with suspected clinical diagnosis of PE.25
The clinical presentation of PE and associated laboratory testing results may be subtler in pregnant than in nonpregnant patients. Arterial blood gases (ABG) may show hypoxemia or hypocapnia. The ABG in pregnancy has a sensitivity of 76.9%, specificity of 20.2%, and negative and positive predictive values of 80% and 11.5% for PE, respectively.26 The alveolar-arterial oxygen gradient is a poor screening test for PE during pregnancy and postpartum. A retrospective chart review of 17 pregnant women with documented PE showed that 58% had normal alveolar-arterial gradients.27 Therefore, in a pregnant woman with a history suspicious for PE, objective imaging studies should be performed even if the patient has normal ABG.
The 2011 guidelines from the American Thoracic Society (ATS) and the Society of Thoracic Radiology (STR) recommend against using D-dimer to diagnose PE in pregnancy.28 In addition, lower extremity CUS should only be performed as the first diagnostic imaging procedure if the patient has signs or symptoms of DVT. Instead, the ATS/STR guidelines recommend a plain radiograph of the chest as the first imaging test. If the chest radiograph is normal, a ventilation/perfusion scan (V/Q) scan is preferred over CT pulmonary angiography (CTPA). Diagnostic accuracy of the V/Q scan may be superior to CTPA in pregnancy, and it is preferable because of the lower prevalence of indeterminate V/Q scan in pregnant women.29 Moreover, there is lower radiation exposure to the maternal breast and lung tissue with a V/Q scan than with CTPA. CTPA confers lower fetal radiation doses than V/Q scans (0.03–0.66 mGy versus 0.32–0.74 mGy, respectively) but higher total body maternal radiation (4–16 mSv versus 1–2.5 mSv).30 A quantitative approach to lung scan interpretation, based on the distribution histogram of V/Q ratios, may be helpful in categorizing patients with suspected PE.28 A study of 121 suspected episodes of PE in 120 pregnant women showed that 104 women with normal or nondiagnostic scans did not develop subsequent episodes of VTE during a mean follow-up period of 20 months.31
If the baseline chest radiograph is abnormal in a pregnant woman with clinical suspicion of PE, a CTPA should be performed. As noted, fetal radiation doses for CTPA examinations in which the fetus is not directly imaged are minimal. If CTPA is recommended for the diagnosis of PE, the patient should be informed that radiation to the breast may increase her baseline risk for breast cancer. The ATS guidelines state that “given the lack of evidence documenting clear superiority of any one diagnostic test, the values and preferences of a patient and her physician likely will and should determine the final choice and sequence of tests performed.”28
CASE I CONTINUED
Upon presentation to the emergency department, the circumference of the patient’s left leg is not significantly greater than that of her right leg, and her leg pain has resolved. Bilateral CUS is negative for proximal or distal DVT. Chest radiograph shows an opacification of her left lower lobe. CTPA shows bilateral segmental and subsegmental lower lobe pulmonary emboli.
How does risk for VTE change throughout pregnancy?
Women are at increased risk for VTE throughout the entire pregnancy, starting from conception, but mainly during the postpartum period. A Danish historical controlled cohort study of 819,751 pregnant women (ages 15–49 years) over a 10-year period identified 727 women with VTE. The absolute risk for VTE per 10,000 pregnancy-years increased from 4.1 (95% CI 3.2 to 5.2) during weeks 1 to 11 to 59.0 (95% CI 46.1 to 76.4) in week 40 and decreased in the postpartum period from 60 (95% CI 47.2 to 76.4) during the first week after birth to 2.1 during weeks 9–12 after birth (95% CI 1.1 to 4.2).32 This study showed that the risk of VTE increases throughout pregnancy and reaches its maximum during the peripartum period and is not significantly increased after 6 weeks post-delivery. In a retrospective cross-over cohort study of 1,687,930 women in California who delivered their first newborn, an elevated risk of VTE persisted until at least 12 weeks after delivery. However, the absolute increase in risk after 6 weeks postpartum was low.33
CASE 1 CONCLUSION
The patient is started on anticoagulation therapy and carefully monitored during the remainder of the pregnancy and postpartum period. Anticoagulation is discontinued 6 weeks after delivery.
TREATMENT
ANTICOAGULATION THERAPY
The treatment of VTE can be lifesaving. In a study comparing 35 patients with PE randomly assigned to treatment with anticoagulants versus no treatment, 5 of 19 patients in the untreated group died from PE and an additional 5 had nonfatal recurrences, as compared with none in the treated group.24 Unfractionated heparin (UFH) and low-molecular-weight heparin (LMWH) are both safe and effective anticoagulants during pregnancy as neither crosses the placenta. In a review of 186 reports of fetal and infant outcomes following anticoagulant therapy during pregnancy in 1325 pregnancies, outcomes in UFH-treated patients were similar to those in the normal population after excluding pregnancies with comorbid conditions independently associated with adverse outcomes.34 A 2005 systematic review of LMWH for prophylaxis and treatment of VTE during pregnancy included 64 studies of 277 pregnancies. There were no maternal deaths, live births resulted from 94.7% of the pregnancies, VTE or arterial thrombosis occurred in 0.86%, and significant bleeding occurred in 1.98%.35
The standard UFH regimen is an initial bolus of 5000 units subcutaneously and 17,500 units every 12 hours, with dose adjustment made based on a mid-interval activated partial thromboplastin time (aPTT).36 Although still controversial, it has been suggested that the anti-Xa assay with a mid-dosing interval target of 0.3 to 0.7 U/mL is a more reliable measure of therapeutic UFH activity than the aPTT, as the aPTT response is suppressed due to a pregnancy-related increase in factor VIII. LMWH is dosed based on weight; regimens are enoxaparin 1 mg/kg subcutaneously twice daily or 1.5 mg/kg subcutaneously once daily, and dalteparin 100 units/kg every 12 hours or 150 units/kg daily.
A 2017 Cochrane review of the effect of LMWH compared with UFH for the treatment of VTE in the nonpregnant setting included 23 studies with 9587 patients. Thrombotic complications (odds ratio [OR] 0.70 [CI 0.57 to 0.85]) and major hemorrhage (OR 0.58 [CI 0.40 to 0.83]) were lower in patients receiving LMWH, with a trend toward lower mortality.37 In addition, the incidence of bleeding complications in patients treated with subcutaneous LMWH versus intravenous heparin was compared in a 2012 systematic review of 27 randomized controlled trials with a total of 28,637 patients. In patients treated with LMWH, there was a nonstatistically significant lower incidence of major bleeding events (OR 0.79 [95% CI 0.60 to 1.04]) and a statistically significant reduction in bleeding risk (OR 0.68 [95% CI 0.47 to 1.00]) compared to patients treated with UFH.38 Additionally, a trial comparing the use of standard UFH versus LMWH found a significantly lower incidence of thrombocytopenia in patients treated with LMWH.39,40 Overall, LMWH is more effective at decreasing both thrombotic and bleeding complications, and the risk for osteoporosis is lower with LMWH. Based on these results, the American College of Chest Physicians (ACCP) recommends LMWH as the first-line treatment for VTE in pregnancy.41
In specific clinical situations, such as patients with renal dysfunction with creatinine clearance (CrCl) less than 30 mL/min, UFH is indicated. In a study of 103 pregnancies in 93 women given anti-coagulation during pregnancy, 89.3% received UFH. There were no maternal deaths, and fetal demise occurred in 8 pregnancies (7.8%) at a median of 14 weeks’ gestation. There were 2 episodes of PE (1.9%) and 2 major bleeding events requiring transfusion (1.9%).42 UFH costs much less than LMWH, and therefore UFH remains an important, inexpensive, and efficacious anticoagulant option for pregnant women who require anticoagulation and cannot afford LMWH.43
Due to the physiologic changes associated with pregnancy, LMWH and UFH dosages may need to be adjusted. An observational study of 20 pregnant women with acute VTE found no recurrent VTE or major bleeding after treatment with dalteparin. Dalteparin doses approximately 10% to 20% higher than those recommended in nonpregnant women were required to reach therapeutic target anti-Xa activity.44
Caution Regarding Oral Anticoagulants
Due to its teratogenicity, warfarin is not a first-line anticoagulation option. It is strictly contraindicated during the first trimester during organogenesis, and its use during pregnancy is restricted to women with mechanical heart valves. Warfarin crosses the placenta and has been associated with nasal hypoplasia, stippled epiphyses, and growth restriction, particularly between 6 to 9 weeks’ gestation. Every effort should be made to substitute UFH or LMWH for warfarin between 6 and 12 weeks of gestation. The bridging process should begin as early in the gestational age as possible due to the long half-life of warfarin.45 When used later in gestation, warfarin has been associated with fetal hemorrhage and central nervous system abnormalities. Other complications from use during the second and third trimesters include microcephaly, blindness, deafness, and fetal growth restriction.46,47 Its use also increases the risk for abortion and fetal death in utero.48–50
The direct oral anticoagulants (DOACs) are not approved for use in pregnancy. Although there are limited anecdotal reports of DOAC use in pregnancy,51 there is preclinical evidence of placental transfer with the DOACs rivaroxaban and apixaban (direct Xa inhibitors) and the oral thrombin inhibitor dabigatran, thus increasing the risk to the fetus.52–54 Edoxaban, another direct Xa inhibitor, should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. It should be discontinued in nursing mothers.55
THROMBOLYSIS
Fetal as well as maternal survival is dependent on adequate maternal perfusion and oxygenation. The risk of death from PE is significant, with a cross-sectional study of 58 patients with acute, massive PE showing a 55% mortality rate.56 Thus, pregnancy is not an absolute contraindication to mechanical or systemic (recombinant tissue plasminogen activator or streptokinase) thrombolysis in an unstable patient at high risk for death.57–59 There are no major studies of this approach, although a small review of 13 cases using systemic thrombolysis showed no increased risk of maternal mortality.58 Thrombolysis should be considered for appropriate indications in pregnant patients as it would be in nonpregnant patients. However, caution is required when drawing conclusions regarding maternal and fetal safety, given the lack of controlled clinical trials including pregnant women.
SURGICAL PULMONARY EMBOLECTOMY
Surgical pulmonary embolectomy is an important therapeutic and potentially life-saving option in women presenting with massive PE in the immediate postpartum period. Because of the risk of massive uterine bleeding immediately postpartum, thrombolytic therapy should not be used.60
INFERIOR VENA CAVA FILTER
Placement of an inferior vena cava (IVC) filter is indicated in patients who have an acute VTE with absolute contraindications for anticoagulation. In addition, it can be considered in patients with extensive ileofemoral venous thrombosis within 2 weeks prior to expected delivery.61 In a systematic review of 44 studies of IVC filters placed in pregnant patients, the IVC filter complication rate was 8.87% and the failure-to-retrieve rate was 11.25%.62 The complication rate is similar to that found in the nonpregnant population. Thus, IVC filters may be used when appropriately indicated and should be removed as soon as clinically feasible.
RECURRENT THROMBOSIS AND THROMBOPHILIAS
CASE PRESENTATION 2
A 34-year-old pregnant woman G1P0 at 38 weeks’ gestation presents with a painful, swollen left calf that is associated with difficulty on walking; the circumference of the left calf is 2 cm greater than that of the right. She has no shortness of breath or chest pain. She has a prior history of distal right lower extremity DVT while on combined oral contraceptives. Her mother also has a history of DVT while bedbound during a prolonged hospitalization at an older age. CUS is negative, and the patient is discharged home. However, 24 hours later she returns to the hospital with worsening swelling and pain in her left leg. Magnetic resonance venography demonstrates a large left external iliac and common iliac DVT. She is admitted and is started on UFH, and a retrievable IVC filter is placed in anticipation of delivery.
What is the risk for VTE recurrence during pregnancy?
A personal and family history of VTE should be obtained when evaluating pregnant patients. A retrospective study of 109 women with prior history of VTE showed recurrence rates per patient-year of 10.9% during pregnancy and 3.7% in the nonpregnant period; the relative risk of recurrent VTE during pregnancy was 3.5 (95% CI 1.6 to 7.8).63 Two large European retrospective cohort studies of VTE in pregnancy showed that the recurrence rate of VTE in women with a history of thrombosis is around 6% during pregnancy, equally distributed among trimesters. The highest incidence of recurrence was in the postpartum period, ranging from 8.3% to 10%.64 The recurrence risk during pregnancy in women with a history of a single episode of VTE was 2.4% antepartum (95% CI 0.2% to 6.9%).65 These risks may be lower in women without thrombophilia or with a temporary risk factor associated with their previous thromboembolic event.65 Recurrence risk is higher if the previous VTE was estrogen-related, either due to pregnancy or through hormonal contraception (10%), than if the previous VTE was non-estrogen-related (2.7%).64,66
The timing of the case patient’s presentation is consistent with reports of increased risk of VTE during the peripartum period. Her prior history of estrogen-related DVT is concerning for a risk of recurrence, particularly during pregnancy. A retrospective cohort study of 1104 women with previous VTE, 88 of whom became pregnant without receiving thromboprophylaxis, showed that the overall rate of VTE recurrence was 5.8% (95% CI 3.0% to 10.6%) and 8.3% (95% CI 4.5% to 14.6%) during pregnancy and postpartum, respectively. The risk of VTE recurrence was absent if the first VTE was related to a transient risk factor other than pregnancy, postpartum period, or hormonal contraception.67 However, the recurrence rate of VTE in women with prior unprovoked VTE and/or thrombophilia has been reported as 5.9% (95% CI 1.2% to 16.2%).65 The presence of an underlying hypercoagulable state can increase the recurrence risk by 25% to 50%, depending on the disorder.68 A retrospective cohort study of 270 pregnancies in 105 carriers of factor V Leiden, identified because of a symptomatic relative with the factor V Leiden mutation, found a VTE risk (mostly in the postpartum period) of 6.4% for heterozygous women, 16.7% for homozygous women, 20% for double heterozygous women, and 1.2% for noncarriers.69
Should the patient be screened for a thrombophilia disorder?
Half of all index thromboses in patients with thrombophilia occur in association with an additional risk factor. In women of child-bearing age, pregnancy, the postpartum period, and the use of combined hormonal contraception are all risk factors for VTE. A 2010 guideline from the British hematology community recommended testing for thrombophilia in women with prior VTE secondary to a minor provoking factor before or during pregnancy, but not testing women with unprovoked VTE (who would receive prophylaxis regardless) or those with VTE secondary to a major provoking factor (who would not require prophylaxis).70 Indications to screen for aPL antibodies include: women with (1) 3 unexplained recurrent first-trimester pregnancy losses or 1 second or third trimester fetal loss of morphologically normal fetuses; (2) severe preeclampsia; (3) intrauterine growth restriction; or (4) premature labor (< 34 weeks’ gestation).71,72
CASE 2 CONCLUSION
The patient is subsequently screened for inherited thrombophilia disorders and is found to be heterozygous for factor V Leiden.
CASE PRESENTATION 3
A 25-year-old woman is diagnosed with antiphospholipid syndrome (APS) during her second pregnancy when she experiences fetal loss during her second trimester. Pathologic examination of the placenta reveals infarcts. Laboratory evaluation reveals positive high-titer anticardiolipin and anti-beta-2 glycoprotein 1 antibodies (IgG isotype) and lupus anticoagulant on 2 separate occasions 12 weeks apart. In a subsequent pregnancy, she is started on prophylactic LMWH and daily low-dose aspirin (81 mg). At 36 weeks’ gestation, she presents with a blood pressure of 210/104 mm Hg and a platelet count of 94,000 cells/µL. She is diagnosed with preeclampsia and hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome and is induced for early delivery. About 2 weeks after vaginal delivery, she notices shortness of breath and chest pain. A CTPA demonstrates a right lower lobe lobar defect consistent with a PE. Her anticoagulation is increased to therapeutic dosage LMWH.
To what extent does thrombophilia increase the risk for VTE in pregnancy?
Approximately 50% of pregnancy-related VTEs are associated with inherited thrombophilia. A systematic review of 79 studies, in which 9 studies (n = 2526 patients) assessed the risk of VTE associated with inherited thrombophilia in pregnancy, revealed that the odds ratio for individuals with thrombophilia to develop VTE ranged from 0.74 to 34.40.73 Although women with thrombophilia have an increased relative risk of developing VTE in pregnancy, the absolute risk of VTE remains low (Table 1).41,73,74
How is APS managed in pregnant patients?
Women with history of recurrent early pregnancy loss (< 10 weeks’ gestation) related to the presence of aPL antibodies are managed with low-dose aspirin and prophylactic-dose UFH or LMWH. This treatment increases the rate of subsequent successful pregnancy outcomes and reduces the risk for thrombosis. A 2010 systematic review and meta-analysis of UFH plus low-dose aspirin compared with low-dose aspirin alone in patients with APS and recurrent pregnancy loss included 5 trials and 334 patients. Patients receiving dual therapy had higher rates of live births (74.3%; relative risk [RR] 1.30 [CI 1.04 to 1.63]) compared to the aspirin-only group (55.8%).75 A 2009 randomized controlled trial compared low-dose aspirin to low-dose aspirin plus LMWH in women with recurrent pregnancy loss and either aPL antibodies, antinuclear antibody, or inherited thrombophilia. The study was stopped early after 4 years and found no difference in rates of live births between the groups (77.8% versus 79.1%).76 However, a randomized case-control trial of women with aPL antibodies and recurrent miscarriage found a 72% live birth rate in 47 women randomly assigned to low-dose aspirin and LMWH.77 A 2012 guideline from the American College of Chest Physicians (ACCP) recommends that women with aPL antibodies with a history of 3 or more pregnancy losses receive low-dose aspirin plus prophylactic-dose LMWH or UFH.78 A 2014 systematic review and meta-analysis showed that the combination of low-dose aspirin and UFH resulted in a higher live-birth rate than aspirin alone in 803 women with APS (RR 1.54 [95% CI 1.25 to 1.89]).79 Further large randomized controlled trials are needed to confirm optimal management of recurrent miscarriage and aPL antibodies.
The addition of prednisone to aspirin, heparin, or both has shown no benefits in pregnant women with aPL antibodies. Indeed, prolonged use of steroids may cause serious pregnancy complications, such as prematurity and hypertension.80–83 Intravenous infusions of immunoglobulin (IVIG) have not been shown to be superior to heparin and aspirin. This finding was confirmed in a multicenter clinical trial that tested the effects of IVIG compared with LMWH plus low-dose aspirin for the treatment of women with aPL antibodies and recurrent miscarriage. The rate of live-birth was 72.5% in the group treated with heparin plus low-dose aspirin compared with 39.5% in the IVIG group.84
Preeclampsia and HELLP syndrome complicated the case patient’s pregnancy even though she was being treated with prophylactic-dose LMWH and low-dose aspirin, the current standard of care for pregnant women with APS (UFH can be used as well). It is important to note that complications may still occur despite standard treatment. Indeed, PE is more common in the postpartum than in the antepartum period. Prompt diagnosis is paramount to initiate the appropriate treatment; in this case the dose of LMWH was increased from prophylactic to therapeutic dose. However, additional therapeutic modalities are necessary to improve outcomes. A randomized controlled trial comparing standard of care with or without hydroxychloroquine is under way to address this issue.
PROPHYLAXIS
CASE PRESENTATION 4
A 34-year-old woman G1P0 at 6 weeks’ gestation with a past medical history of a proximal lower extremity DVT while on oral contraception is treated with warfarin anticoagulation for 6 months. Her obstetrician consults the hematologist to advise regarding antithrombotic management during this pregnancy.
What is the approach to prophylaxis in women at high risk for pregnancy-associated VTE?
All women at high risk for pregnancy-associated VTE should be counseled about the signs and symptoms of DVT or PE during preconception and pregnancy and have a plan developed should these symptoms arise. The ACCP guidelines on antithrombotic therapy outline recommendations ranging from clinical vigilance to prophylactic and intermediate-dose anticoagulation, depending on the risk for VTE recurrence, based on the personal and family history of VTE and type of thrombophilia (Table 2).78 These recommendations range from grade 2B to 2C.
For women with a history of estrogen-related VTE, single unprovoked VTE, or recurrent unprovoked VTE not on chronic anticoagulation, antepartum and postpartum pharmacologic thromboprophylaxis with either prophylactic or intermediate-dose LMWH is recommended (grade 2C). In patients with prior history of provoked VTE (non-estrogen related), antepartum clinical vigilance and postpartum pharmacologic thromboprophylaxis is recommended (grade 2C, 2B).
In asymptomatic pregnant women who are homozygote carriers for factor V Leiden or prothrombin G20210A variants and have a positive family history of thrombosis, antepartum and postpartum pharmacologic thromboprophylaxis is recommended (grade 2B). In asymptomatic homozygote carriers of factor V Leiden or prothrombin G20210A variants with no family history of thrombosis and women with all other thrombophilias with a positive family history of thrombosis, postpartum pharmacologic thromboprophylaxis is indicated (grade 2B and 2C, respectively). For women with confirmed APS and clinical criteria of obstetric APS with recurrent pregnancy loss, antepartum thromboprophylaxis with LMWH and low-dose aspirin is recommended (grade 1B). For pregnant women with all other thrombophilias with no personal or family history of thrombosis, clinical vigilance is suggested (grade 2 C).78
As an alternative to LMWH, vitamin K antagonists (VKA) such as warfarin can be used for postpartum thromboprophylaxis; in patients with protein C or S deficiency, due to the risk of warfarin-induced skin necrosis, a rapid-onset anticoagulant must be concomitantly administered. Warfarin and LMWH are safe anticoagulants during lactation, but there are no clinical data on the effects of the DOACs on infants during lactation. Data from animal studies indicate that DOACs are secreted into breast milk.85
What risks are associated with anticoagulant therapy in pregnancy?
VKAs cross the placenta and can cause teratogenicity, pregnancy loss, fetal bleeding, and neurodevelopmental deficits. Therefore, discontinuation of VKAs prior to the sixth week of gestation is necessary to avoid warfarin embryopathy. DOACs have been shown to readily cross the placenta but with unknown human reproductive risks. Fondaparinux, a synthetic pentasaccharide, crosses the placenta in small quantities. Though there are reports of the successful use of fondaparinux in pregnancy, there is limited reported experience of its use in the first trimester.86
The risk for bleeding with anticoagulation is notably acceptable. In a case-control study of 88 pregnant women receiving therapeutic-dose anticoagulation, the risk of postpartum hemorrhage (PPH) after vaginal delivery was 30% in those who received LMWH anticoagulation versus 18% in those who did not (OR 1.9 [95% CI 1.1 to 3.5]).87 However, the risk for severe PPH (≥ 500 mL) was similar (5.6% versus 5.0%; OR 1.1 [95% CI 0.4 to 3.6]). The risk for PPH after cesarean section was 12% in LMWH users versus 4% in LMWH non-users (OR 2.9 [95% CI 0.5 to 19.4]). The risk for PPH associated with delivery within 24 hours after the last dose of LMWH was 1.2 times higher (95% CI 0.4 to 3.6) compared to a longer interval. Therefore, therapeutic LMWH increases the risk for blood loss after vaginal delivery, but not the risk for severe PPH. The risk for PPH is influenced by the interval between the last dose of LMWH and delivery. Of note in this study, per the institution’s protocol, the anticoagulation was stopped with signs of labor or determination of need for delivery. The risk for blood loss may be mitigated in more planned delivery scenarios.87
CASE 4 CONTINUED
The patient is placed on prophylactic-dose LMWH with good tolerance and delivers at 39 weeks' gestation via caesarian section due to nonprogression of labor. Postpartum she is restarted on prophylactic-dose anticoagulation with LMWH. Two weeks after discharge from the hospital, she presents with right calf pain and mild shortness of breath. On physical exam, her leg circumferences are equal. A D-dimer assay is 3375 ng/mL (normal 0–229). CUS of the right leg shows a complete occlusive DVT of the mid-distal superficial femoral and popliteal veins and partially occlusive acute DVT of the right posterior tibial and peroneal veins. CTPA reveals a right lower lobe PE. Because she had developed VTE despite prophylactic LMWH, her anticoagulation is changed to therapeutic dose. She is treated with anticoagulation with LMWH for a total of 3 months, after which a repeat CUS shows no residual thrombosis.
What is the recommended dosing of heparin and LMWH during pregnancy?
A prospective study of 14 pregnant women receiving UFH prophylaxis found that a prophylactic dose of 5000 units twice a day was inadequate to achieve prophylactic heparin levels in any patient in the second or third trimester.88 Similar to treatment dosage, there is no consensus evidence for prophylactic dosing, and dosage recommendations are based on expert opinion. In a retrospective study of 25 pregnant women on intermediate-dose UFH, the mean UFH dose required to achieve a target anti-factor Xa level of 0.1 to 0.3 units/mL was 236.9 units/kg/day.89 However, the use of anti-factor Xa levels for monitoring is controversial as there is no data to support a difference in outcomes with its use in prophylactic or therapeutic dosing.
The timing of the previous VTE history is important when deciding on the anticoagulant dose in pregnancy. In pregnant women with a VTE that occurred within the previous 4 to 6 weeks, full-dose anticoagulation with LMWH should be considered; an intermediate dose (three-fourths of a therapeutic dose) may be used if the thrombotic episode occurred more than 6 weeks earlier but still within a year. Prophylactic dosing may be sufficient if the episode occurred more than a year earlier.90 A clinical trial (High-Low) is under way to explore the optimal dose of LMWH in pregnant women with prior history of VTE who are not on chronic anticoagulation therapy.91
How is anticoagulation therapy managed in the peripartum period?
Neuraxial anesthesia during active labor while on anticoagulation increases the risk for central nervous system bleeding. Therefore, if spontaneous labor occurs in women on therapeutic dose anticoagulation, neuraxial anesthesia cannot be used. However, in the event of elective induction of labor or caesarean section, neuroaxial anesthesia may be performed 12 hours after the administration of the last prophylactic dose of LMWH or 24 hours after the last therapeutic dose of LMWH. Intravenous UFH should be stopped for 6 hours before induction of labor with a confirmed normal aPTT before placement of neuraxial anesthesia. There is no contraindication for using neuraxial anesthesia during subcutaneous standard UFH at total doses of 10,000 units daily. The risk of spinal hematoma with larger daily subcutaneous doses is unclear; therefore, a documented normal aPTT must be obtained before placement of neuroaxial anesthesia.
Postpartum, reinitiation of prophylactic-dose LMWH should be delayed for at least 12 hours after the removal of an epidural catheter. Therapeutic-dose LMWH should be administered no earlier than 24 hours after neuraxial anesthesia, providing that proper hemostasis is achieved. In the absence of persistent bleeding, if no regional anesthesia was used, LMWH may be resumed 12 hours after delivery.92 Anticoagulation with either LMWH or warfarin is recommended for at least 6 to 12 weeks postpartum.33
COUNSELING
Patients should be advised to manage controllable risk factors, including avoiding prolonged immobilization, avoiding excessive weight gain in pregnancy, and stopping smoking. Periods of immobilization tend to cause reduced blood flow (stasis), which predisposes to thrombosis. In a systematic review of records of all patients with confirmed PE after arrival at Charles de Gaulle airport in Paris during a 13-year period, women had a higher risk of PE after a long-distance flight than men, with an estimated incidence of 0.61 per million passengers versus 0.20, respectively; the incidence reached 7.24 and 2.35 cases, respectively, in passengers traveling more than 10,000 kilometers.93,94
The risk of air travel-related thrombosis in pregnant women is estimated to be between 0.03% and 0.1%. Physicians must decide on an individual basis how to prevent travel-related thrombosis in their pregnant patients. In most passengers, prevention can be limited to encouraging exercise, avoidance of long sleeping periods, and not using a window seat. Women at high risk for VTE, such as women with a prior history of VTE who are not on anticoagulation or women with known asymptomatic thrombophilia or other risk factors for thrombosis such as obesity, may benefit from a short period (1–3 days) of LMWH starting 2 hours before a long-distance flight.95
Activation of the coagulation system has been demonstrated in cigarette smokers.96 Heavy smoking was found to be a significant risk factor for VTE in a cross-sectional analysis of 2404 men and women.97 An increased risk for thrombosis during pregnancy is seen in cigarette smokers15,98 and is enhanced with the concomitant use of illicit drugs.99 Other obstetric complications associated with smoking and illicit drug use during pregnancy include preterm labor, spontaneous abortion, perinatal death, low birth weight, and abruption placenta. The efficacy of nicotine replacement therapy in pregnancy is uncertain.100 Recommendations are to advise patients to stop smoking, obtain psychosocial counseling, and utilize adjunctive therapies, which have been shown to have some effect on abstinence rates.101
CONCLUSION
Women are at increased risk for VTE during pregnancy and the postpartum period. Awareness of risk factors and the signs and symptoms of VTE is paramount. Prompt diagnosis and treatment is mandatory to decrease complications of VTE. LMWH is the mainstay treatment of VTE in pregnancy, as it does not cross the placenta. Both LMWH and warfarin are safe during lactation. Close communication among the patient, obstetrician, hematologist, anesthesiologist, and neonatologist is crucial to optimize the care of these patients.
INTRODUCTION
Venous thromboembolism (VTE), comprising deep vein thrombosis (DVT) and pulmonary embolism (PE), is a leading nonobstetric cause of maternal death in the United States and in developed countries.1,2 During pregnancy, the risk for VTE increases four- to six-fold, and although the risk is present throughout pregnancy, the mother is at highest risk immediately postpartum.3–5
VTE risk is increased due to physiologic and anatomic changes that occur in pregnancy. These changes include hypercoagulability, progesterone-induced venous stasis, decreased venous outflow, compression of the inferior vena cava and pelvic veins by the expanding uterus, and decreased mobility. The hypercoagulability of pregnancy is due to increased levels of coagulation factors I (fibrinogen), VII, VIII, and X, and von Willebrand factor; decreased free protein S, a natural anticoagulant; acquired resistance to activated protein C; and decreased fibrinolysis due to increased levels of plasminogen activator inhibitor-1 and -2.6,7 These changes confer increased hemostasis to the mother for delivery but also place her at higher risk for thrombosis.
A review of the literature found that more than 70% of pregnancy-associated DVTs are located in the ileofemoral region, as compared with approximately 9% in non-pregnant patients.8 The proximal location is associated with a higher risk for post-thrombotic syndrome and embolization as compared with calf DVTs.9 Proximal postnatal thrombosis, smoking, and older age are independent predictors of the development of post-thrombotic syndrome.10
RISK FACTORS
Clinical risk factors that increase the risk for VTE during pregnancy include a prior history of estrogen-related or unprovoked VTE, being a carrier of severe inherited thrombophilia (homozygotes for factor V Leiden or factor II G20210A variants, double heterozygotes, or persons with antithrombin, protein C, or protein S deficiencies), and the presence of antiphospholipid (aPL) antibodies.11 Women with systemic lupus erythematosus, diabetes, sickle cell disease, and heart disease also have a high risk for VTE during pregnancy.12 Other risk factors predisposing to thrombosis include black ethnicity, smoking, operative procedures, conception after assisted reproductive techniques, high body mass index, antepartum immobilization, severe preeclampsia, advanced age and parity, and a family history of VTE.13 A prospective cohort study of 1,297,037 pregnancies and related puerperium identified the following risk factors for thrombosis: hospitalization, infection, hyperemesis, multiple pregnancies, preeclampsia, obesity, cesarean section, major postpartum hemorrhage, intrauterine growth restriction, and fetal death.14 Risk factors identified in an Agency for Healthcare Research and Quality study include: age 35 or older, black ethnicity, lupus, sickle cell disease, heart disease, postpartum infection, and transfusion.15 The combination of more than one risk factor increases the risk for VTE. All these factors have to be considered when deciding on prophylactic or therapeutic anticoagulation therapy in pregnancy. In addition, the risks of anticoagulation, including bruising, bleeding, and other side effects (eg, reduced bone mineral density with therapeutic-dose unfractionated heparin), allergic reactions, and rarely thrombocytopenia, must be considered.
EVALUATION AND DIAGNOSIS
CASE PRESENTATION I
A 31-year-old woman G1P0 at 10 weeks’ gestation with no personal or family history of thrombosis presents with acute onset of shortness of breath and left-sided chest pain that awoke her the morning of presentation. Her vital signs are significant for a heart rate of 106 beats/min, respiration rate of 22 breaths/min, blood pressure of 105/76 mm Hg, and pulse oximetry of 98% on room air. The patient denies previous exposure to oral contraceptives. She does not smoke. She reports that she had noticed left calf pain and swelling, which worsened with walking after a 4-hour drive 2 days prior.
What is the approach to diagnosis of thromboembolism in pregnant patients?
DEEP VEIN THROMBOSIS
Although a clinical diagnosis of DVT in pregnancy is unreliable, a history and physical examination are necessary to exclude other diagnoses and to assess the likelihood of thrombosis. Unfortunately, studies of the accuracy of history and physical examination for detecting DVT and PE have not included pregnant patients. In most pregnant patients with clinically suspected DVT, the diagnosis is not confirmed. Other causes of leg pain and swelling are not uncommon during pregnancy and include cellulitis, ruptured Baker’s cyst, or muscular pain.
A cross-sectional study described the derivation of the LEFt clinical decision rule, which relies on 3 variables in pregnant women with suspected DVT: left leg presentation (L), ≥ 2 cm calf circumference difference (E for edema), and first trimester presentation (Ft). If none of these variables is present, the negative predictive value is 100%.16 A validation study suggested that a negative LEFt rule accurately identifies pregnant women in whom the risk for confirmed DVT appears to be very low. The rule should not be used as an individual test for excluding DVT during pregnancy, but could be applied in a diagnostic approach in association with D-dimer measurement and compression ultrasonography (CUS); however, it has not been prospectively validated for safety and efficacy.17 In a study of 149 consecutive pregnant women with suspected DVT, a whole-blood agglutination D-dimer had a sensitivity of 100% and specificity of 60%.18 A 2006 systematic review found only 4 diagnostic studies of VTE in pregnancy in the literature. One of these studies showed that a combination of a negative CUS and normal D-dimer can accurately exclude DVT.19
Serial CUS is necessary for pregnant women with a high clinical suspicion of DVT but a negative initial investigation. In a study of 221 pregnant women in whom DVT was clinically suspected, 16 women (7.2%) were diagnosed with DVT by initial CUS, and none were diagnosed with DVT onserial testing.20 During follow-up (≥ 3 months), 6 of the 205 women with normal serial CUS results presented with symptoms of DVT, PE, or both, and 1 of them was diagnosed with DVT and PE. The sensitivity of serial CUS with Doppler imaging was 94.1% (95% confidence interval [CI] 69.2% to 99.7%), and the negative predictive value was 99.5% (95% CI 96.9% to 100%).20 All ultrasounds undertaken for investigation of pregnancy-associated DVT should include imaging of the iliac veins if there is a high index of suspicion and the CUS is negative for femoral DVT. Serial CUS with Doppler imaging of the iliac vein performed over a 7-day period excludes DVT in symptomatic pregnant women.20 Repeat CUS may be done 2 to 4 days and 6 to 8 days after the initial scan.
Ileofemoral vein thrombosis accounts for approximately 90% of proximal thromboses in pregnancy, occurring most often in the left lower extremity.20 The incidence of isolated iliac vein thrombosis in pregnancy is low, but when it does occur, delay in diagnosis can lead to significant morbidity. Therefore, for women with suspected isolated iliac vein thrombosis in whom CUS is negative or nondiagnostic, magnetic resonance direct thrombus imaging (MRDTI) should be performed.21 Patients with iliac vein thrombosis may present with unexplained inguinal, pelvic, or abdominal pain, which may be accompanied by back pain, and they usually present with swelling of the entire leg. MRDTI does not require gadolinium contrast and its accuracy appears to be similar to that of venography for iliac vein thrombi in the nonpregnant population.21 Exposure to gadolinium during pregnancy is associated with an increased risk for rheumatologic, inflammatory, or infiltrative skin conditions and stillbirth or neonatal death.22
Ovarian vein thrombosis is a rare but serious diagnosis. It occurs mostly in the postpartum period, mainly after cesarean delivery, and usually affects the right ovarian vein. The diagnosis is confirmed by ultrasound, computed tomography (CT), or magnetic resonance imaging.23
PULMONARY EMBOLISM
PE is more difficult to diagnose than DVT, particularly because clinical signs of PE are unreliable in the pregnant patient. The mortality rate of untreated PE is high, ranging from 18% to 38%, and approximately one-third of patients with untreated thromboembolic disease develop recurrent embolism.24 Studies have reported a PE prevalence between 1.4% and 4.2% in pregnant women with suspected clinical diagnosis of PE.25
The clinical presentation of PE and associated laboratory testing results may be subtler in pregnant than in nonpregnant patients. Arterial blood gases (ABG) may show hypoxemia or hypocapnia. The ABG in pregnancy has a sensitivity of 76.9%, specificity of 20.2%, and negative and positive predictive values of 80% and 11.5% for PE, respectively.26 The alveolar-arterial oxygen gradient is a poor screening test for PE during pregnancy and postpartum. A retrospective chart review of 17 pregnant women with documented PE showed that 58% had normal alveolar-arterial gradients.27 Therefore, in a pregnant woman with a history suspicious for PE, objective imaging studies should be performed even if the patient has normal ABG.
The 2011 guidelines from the American Thoracic Society (ATS) and the Society of Thoracic Radiology (STR) recommend against using D-dimer to diagnose PE in pregnancy.28 In addition, lower extremity CUS should only be performed as the first diagnostic imaging procedure if the patient has signs or symptoms of DVT. Instead, the ATS/STR guidelines recommend a plain radiograph of the chest as the first imaging test. If the chest radiograph is normal, a ventilation/perfusion scan (V/Q) scan is preferred over CT pulmonary angiography (CTPA). Diagnostic accuracy of the V/Q scan may be superior to CTPA in pregnancy, and it is preferable because of the lower prevalence of indeterminate V/Q scan in pregnant women.29 Moreover, there is lower radiation exposure to the maternal breast and lung tissue with a V/Q scan than with CTPA. CTPA confers lower fetal radiation doses than V/Q scans (0.03–0.66 mGy versus 0.32–0.74 mGy, respectively) but higher total body maternal radiation (4–16 mSv versus 1–2.5 mSv).30 A quantitative approach to lung scan interpretation, based on the distribution histogram of V/Q ratios, may be helpful in categorizing patients with suspected PE.28 A study of 121 suspected episodes of PE in 120 pregnant women showed that 104 women with normal or nondiagnostic scans did not develop subsequent episodes of VTE during a mean follow-up period of 20 months.31
If the baseline chest radiograph is abnormal in a pregnant woman with clinical suspicion of PE, a CTPA should be performed. As noted, fetal radiation doses for CTPA examinations in which the fetus is not directly imaged are minimal. If CTPA is recommended for the diagnosis of PE, the patient should be informed that radiation to the breast may increase her baseline risk for breast cancer. The ATS guidelines state that “given the lack of evidence documenting clear superiority of any one diagnostic test, the values and preferences of a patient and her physician likely will and should determine the final choice and sequence of tests performed.”28
CASE I CONTINUED
Upon presentation to the emergency department, the circumference of the patient’s left leg is not significantly greater than that of her right leg, and her leg pain has resolved. Bilateral CUS is negative for proximal or distal DVT. Chest radiograph shows an opacification of her left lower lobe. CTPA shows bilateral segmental and subsegmental lower lobe pulmonary emboli.
How does risk for VTE change throughout pregnancy?
Women are at increased risk for VTE throughout the entire pregnancy, starting from conception, but mainly during the postpartum period. A Danish historical controlled cohort study of 819,751 pregnant women (ages 15–49 years) over a 10-year period identified 727 women with VTE. The absolute risk for VTE per 10,000 pregnancy-years increased from 4.1 (95% CI 3.2 to 5.2) during weeks 1 to 11 to 59.0 (95% CI 46.1 to 76.4) in week 40 and decreased in the postpartum period from 60 (95% CI 47.2 to 76.4) during the first week after birth to 2.1 during weeks 9–12 after birth (95% CI 1.1 to 4.2).32 This study showed that the risk of VTE increases throughout pregnancy and reaches its maximum during the peripartum period and is not significantly increased after 6 weeks post-delivery. In a retrospective cross-over cohort study of 1,687,930 women in California who delivered their first newborn, an elevated risk of VTE persisted until at least 12 weeks after delivery. However, the absolute increase in risk after 6 weeks postpartum was low.33
CASE 1 CONCLUSION
The patient is started on anticoagulation therapy and carefully monitored during the remainder of the pregnancy and postpartum period. Anticoagulation is discontinued 6 weeks after delivery.
TREATMENT
ANTICOAGULATION THERAPY
The treatment of VTE can be lifesaving. In a study comparing 35 patients with PE randomly assigned to treatment with anticoagulants versus no treatment, 5 of 19 patients in the untreated group died from PE and an additional 5 had nonfatal recurrences, as compared with none in the treated group.24 Unfractionated heparin (UFH) and low-molecular-weight heparin (LMWH) are both safe and effective anticoagulants during pregnancy as neither crosses the placenta. In a review of 186 reports of fetal and infant outcomes following anticoagulant therapy during pregnancy in 1325 pregnancies, outcomes in UFH-treated patients were similar to those in the normal population after excluding pregnancies with comorbid conditions independently associated with adverse outcomes.34 A 2005 systematic review of LMWH for prophylaxis and treatment of VTE during pregnancy included 64 studies of 277 pregnancies. There were no maternal deaths, live births resulted from 94.7% of the pregnancies, VTE or arterial thrombosis occurred in 0.86%, and significant bleeding occurred in 1.98%.35
The standard UFH regimen is an initial bolus of 5000 units subcutaneously and 17,500 units every 12 hours, with dose adjustment made based on a mid-interval activated partial thromboplastin time (aPTT).36 Although still controversial, it has been suggested that the anti-Xa assay with a mid-dosing interval target of 0.3 to 0.7 U/mL is a more reliable measure of therapeutic UFH activity than the aPTT, as the aPTT response is suppressed due to a pregnancy-related increase in factor VIII. LMWH is dosed based on weight; regimens are enoxaparin 1 mg/kg subcutaneously twice daily or 1.5 mg/kg subcutaneously once daily, and dalteparin 100 units/kg every 12 hours or 150 units/kg daily.
A 2017 Cochrane review of the effect of LMWH compared with UFH for the treatment of VTE in the nonpregnant setting included 23 studies with 9587 patients. Thrombotic complications (odds ratio [OR] 0.70 [CI 0.57 to 0.85]) and major hemorrhage (OR 0.58 [CI 0.40 to 0.83]) were lower in patients receiving LMWH, with a trend toward lower mortality.37 In addition, the incidence of bleeding complications in patients treated with subcutaneous LMWH versus intravenous heparin was compared in a 2012 systematic review of 27 randomized controlled trials with a total of 28,637 patients. In patients treated with LMWH, there was a nonstatistically significant lower incidence of major bleeding events (OR 0.79 [95% CI 0.60 to 1.04]) and a statistically significant reduction in bleeding risk (OR 0.68 [95% CI 0.47 to 1.00]) compared to patients treated with UFH.38 Additionally, a trial comparing the use of standard UFH versus LMWH found a significantly lower incidence of thrombocytopenia in patients treated with LMWH.39,40 Overall, LMWH is more effective at decreasing both thrombotic and bleeding complications, and the risk for osteoporosis is lower with LMWH. Based on these results, the American College of Chest Physicians (ACCP) recommends LMWH as the first-line treatment for VTE in pregnancy.41
In specific clinical situations, such as patients with renal dysfunction with creatinine clearance (CrCl) less than 30 mL/min, UFH is indicated. In a study of 103 pregnancies in 93 women given anti-coagulation during pregnancy, 89.3% received UFH. There were no maternal deaths, and fetal demise occurred in 8 pregnancies (7.8%) at a median of 14 weeks’ gestation. There were 2 episodes of PE (1.9%) and 2 major bleeding events requiring transfusion (1.9%).42 UFH costs much less than LMWH, and therefore UFH remains an important, inexpensive, and efficacious anticoagulant option for pregnant women who require anticoagulation and cannot afford LMWH.43
Due to the physiologic changes associated with pregnancy, LMWH and UFH dosages may need to be adjusted. An observational study of 20 pregnant women with acute VTE found no recurrent VTE or major bleeding after treatment with dalteparin. Dalteparin doses approximately 10% to 20% higher than those recommended in nonpregnant women were required to reach therapeutic target anti-Xa activity.44
Caution Regarding Oral Anticoagulants
Due to its teratogenicity, warfarin is not a first-line anticoagulation option. It is strictly contraindicated during the first trimester during organogenesis, and its use during pregnancy is restricted to women with mechanical heart valves. Warfarin crosses the placenta and has been associated with nasal hypoplasia, stippled epiphyses, and growth restriction, particularly between 6 to 9 weeks’ gestation. Every effort should be made to substitute UFH or LMWH for warfarin between 6 and 12 weeks of gestation. The bridging process should begin as early in the gestational age as possible due to the long half-life of warfarin.45 When used later in gestation, warfarin has been associated with fetal hemorrhage and central nervous system abnormalities. Other complications from use during the second and third trimesters include microcephaly, blindness, deafness, and fetal growth restriction.46,47 Its use also increases the risk for abortion and fetal death in utero.48–50
The direct oral anticoagulants (DOACs) are not approved for use in pregnancy. Although there are limited anecdotal reports of DOAC use in pregnancy,51 there is preclinical evidence of placental transfer with the DOACs rivaroxaban and apixaban (direct Xa inhibitors) and the oral thrombin inhibitor dabigatran, thus increasing the risk to the fetus.52–54 Edoxaban, another direct Xa inhibitor, should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. It should be discontinued in nursing mothers.55
THROMBOLYSIS
Fetal as well as maternal survival is dependent on adequate maternal perfusion and oxygenation. The risk of death from PE is significant, with a cross-sectional study of 58 patients with acute, massive PE showing a 55% mortality rate.56 Thus, pregnancy is not an absolute contraindication to mechanical or systemic (recombinant tissue plasminogen activator or streptokinase) thrombolysis in an unstable patient at high risk for death.57–59 There are no major studies of this approach, although a small review of 13 cases using systemic thrombolysis showed no increased risk of maternal mortality.58 Thrombolysis should be considered for appropriate indications in pregnant patients as it would be in nonpregnant patients. However, caution is required when drawing conclusions regarding maternal and fetal safety, given the lack of controlled clinical trials including pregnant women.
SURGICAL PULMONARY EMBOLECTOMY
Surgical pulmonary embolectomy is an important therapeutic and potentially life-saving option in women presenting with massive PE in the immediate postpartum period. Because of the risk of massive uterine bleeding immediately postpartum, thrombolytic therapy should not be used.60
INFERIOR VENA CAVA FILTER
Placement of an inferior vena cava (IVC) filter is indicated in patients who have an acute VTE with absolute contraindications for anticoagulation. In addition, it can be considered in patients with extensive ileofemoral venous thrombosis within 2 weeks prior to expected delivery.61 In a systematic review of 44 studies of IVC filters placed in pregnant patients, the IVC filter complication rate was 8.87% and the failure-to-retrieve rate was 11.25%.62 The complication rate is similar to that found in the nonpregnant population. Thus, IVC filters may be used when appropriately indicated and should be removed as soon as clinically feasible.
RECURRENT THROMBOSIS AND THROMBOPHILIAS
CASE PRESENTATION 2
A 34-year-old pregnant woman G1P0 at 38 weeks’ gestation presents with a painful, swollen left calf that is associated with difficulty on walking; the circumference of the left calf is 2 cm greater than that of the right. She has no shortness of breath or chest pain. She has a prior history of distal right lower extremity DVT while on combined oral contraceptives. Her mother also has a history of DVT while bedbound during a prolonged hospitalization at an older age. CUS is negative, and the patient is discharged home. However, 24 hours later she returns to the hospital with worsening swelling and pain in her left leg. Magnetic resonance venography demonstrates a large left external iliac and common iliac DVT. She is admitted and is started on UFH, and a retrievable IVC filter is placed in anticipation of delivery.
What is the risk for VTE recurrence during pregnancy?
A personal and family history of VTE should be obtained when evaluating pregnant patients. A retrospective study of 109 women with prior history of VTE showed recurrence rates per patient-year of 10.9% during pregnancy and 3.7% in the nonpregnant period; the relative risk of recurrent VTE during pregnancy was 3.5 (95% CI 1.6 to 7.8).63 Two large European retrospective cohort studies of VTE in pregnancy showed that the recurrence rate of VTE in women with a history of thrombosis is around 6% during pregnancy, equally distributed among trimesters. The highest incidence of recurrence was in the postpartum period, ranging from 8.3% to 10%.64 The recurrence risk during pregnancy in women with a history of a single episode of VTE was 2.4% antepartum (95% CI 0.2% to 6.9%).65 These risks may be lower in women without thrombophilia or with a temporary risk factor associated with their previous thromboembolic event.65 Recurrence risk is higher if the previous VTE was estrogen-related, either due to pregnancy or through hormonal contraception (10%), than if the previous VTE was non-estrogen-related (2.7%).64,66
The timing of the case patient’s presentation is consistent with reports of increased risk of VTE during the peripartum period. Her prior history of estrogen-related DVT is concerning for a risk of recurrence, particularly during pregnancy. A retrospective cohort study of 1104 women with previous VTE, 88 of whom became pregnant without receiving thromboprophylaxis, showed that the overall rate of VTE recurrence was 5.8% (95% CI 3.0% to 10.6%) and 8.3% (95% CI 4.5% to 14.6%) during pregnancy and postpartum, respectively. The risk of VTE recurrence was absent if the first VTE was related to a transient risk factor other than pregnancy, postpartum period, or hormonal contraception.67 However, the recurrence rate of VTE in women with prior unprovoked VTE and/or thrombophilia has been reported as 5.9% (95% CI 1.2% to 16.2%).65 The presence of an underlying hypercoagulable state can increase the recurrence risk by 25% to 50%, depending on the disorder.68 A retrospective cohort study of 270 pregnancies in 105 carriers of factor V Leiden, identified because of a symptomatic relative with the factor V Leiden mutation, found a VTE risk (mostly in the postpartum period) of 6.4% for heterozygous women, 16.7% for homozygous women, 20% for double heterozygous women, and 1.2% for noncarriers.69
Should the patient be screened for a thrombophilia disorder?
Half of all index thromboses in patients with thrombophilia occur in association with an additional risk factor. In women of child-bearing age, pregnancy, the postpartum period, and the use of combined hormonal contraception are all risk factors for VTE. A 2010 guideline from the British hematology community recommended testing for thrombophilia in women with prior VTE secondary to a minor provoking factor before or during pregnancy, but not testing women with unprovoked VTE (who would receive prophylaxis regardless) or those with VTE secondary to a major provoking factor (who would not require prophylaxis).70 Indications to screen for aPL antibodies include: women with (1) 3 unexplained recurrent first-trimester pregnancy losses or 1 second or third trimester fetal loss of morphologically normal fetuses; (2) severe preeclampsia; (3) intrauterine growth restriction; or (4) premature labor (< 34 weeks’ gestation).71,72
CASE 2 CONCLUSION
The patient is subsequently screened for inherited thrombophilia disorders and is found to be heterozygous for factor V Leiden.
CASE PRESENTATION 3
A 25-year-old woman is diagnosed with antiphospholipid syndrome (APS) during her second pregnancy when she experiences fetal loss during her second trimester. Pathologic examination of the placenta reveals infarcts. Laboratory evaluation reveals positive high-titer anticardiolipin and anti-beta-2 glycoprotein 1 antibodies (IgG isotype) and lupus anticoagulant on 2 separate occasions 12 weeks apart. In a subsequent pregnancy, she is started on prophylactic LMWH and daily low-dose aspirin (81 mg). At 36 weeks’ gestation, she presents with a blood pressure of 210/104 mm Hg and a platelet count of 94,000 cells/µL. She is diagnosed with preeclampsia and hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome and is induced for early delivery. About 2 weeks after vaginal delivery, she notices shortness of breath and chest pain. A CTPA demonstrates a right lower lobe lobar defect consistent with a PE. Her anticoagulation is increased to therapeutic dosage LMWH.
To what extent does thrombophilia increase the risk for VTE in pregnancy?
Approximately 50% of pregnancy-related VTEs are associated with inherited thrombophilia. A systematic review of 79 studies, in which 9 studies (n = 2526 patients) assessed the risk of VTE associated with inherited thrombophilia in pregnancy, revealed that the odds ratio for individuals with thrombophilia to develop VTE ranged from 0.74 to 34.40.73 Although women with thrombophilia have an increased relative risk of developing VTE in pregnancy, the absolute risk of VTE remains low (Table 1).41,73,74
How is APS managed in pregnant patients?
Women with history of recurrent early pregnancy loss (< 10 weeks’ gestation) related to the presence of aPL antibodies are managed with low-dose aspirin and prophylactic-dose UFH or LMWH. This treatment increases the rate of subsequent successful pregnancy outcomes and reduces the risk for thrombosis. A 2010 systematic review and meta-analysis of UFH plus low-dose aspirin compared with low-dose aspirin alone in patients with APS and recurrent pregnancy loss included 5 trials and 334 patients. Patients receiving dual therapy had higher rates of live births (74.3%; relative risk [RR] 1.30 [CI 1.04 to 1.63]) compared to the aspirin-only group (55.8%).75 A 2009 randomized controlled trial compared low-dose aspirin to low-dose aspirin plus LMWH in women with recurrent pregnancy loss and either aPL antibodies, antinuclear antibody, or inherited thrombophilia. The study was stopped early after 4 years and found no difference in rates of live births between the groups (77.8% versus 79.1%).76 However, a randomized case-control trial of women with aPL antibodies and recurrent miscarriage found a 72% live birth rate in 47 women randomly assigned to low-dose aspirin and LMWH.77 A 2012 guideline from the American College of Chest Physicians (ACCP) recommends that women with aPL antibodies with a history of 3 or more pregnancy losses receive low-dose aspirin plus prophylactic-dose LMWH or UFH.78 A 2014 systematic review and meta-analysis showed that the combination of low-dose aspirin and UFH resulted in a higher live-birth rate than aspirin alone in 803 women with APS (RR 1.54 [95% CI 1.25 to 1.89]).79 Further large randomized controlled trials are needed to confirm optimal management of recurrent miscarriage and aPL antibodies.
The addition of prednisone to aspirin, heparin, or both has shown no benefits in pregnant women with aPL antibodies. Indeed, prolonged use of steroids may cause serious pregnancy complications, such as prematurity and hypertension.80–83 Intravenous infusions of immunoglobulin (IVIG) have not been shown to be superior to heparin and aspirin. This finding was confirmed in a multicenter clinical trial that tested the effects of IVIG compared with LMWH plus low-dose aspirin for the treatment of women with aPL antibodies and recurrent miscarriage. The rate of live-birth was 72.5% in the group treated with heparin plus low-dose aspirin compared with 39.5% in the IVIG group.84
Preeclampsia and HELLP syndrome complicated the case patient’s pregnancy even though she was being treated with prophylactic-dose LMWH and low-dose aspirin, the current standard of care for pregnant women with APS (UFH can be used as well). It is important to note that complications may still occur despite standard treatment. Indeed, PE is more common in the postpartum than in the antepartum period. Prompt diagnosis is paramount to initiate the appropriate treatment; in this case the dose of LMWH was increased from prophylactic to therapeutic dose. However, additional therapeutic modalities are necessary to improve outcomes. A randomized controlled trial comparing standard of care with or without hydroxychloroquine is under way to address this issue.
PROPHYLAXIS
CASE PRESENTATION 4
A 34-year-old woman G1P0 at 6 weeks’ gestation with a past medical history of a proximal lower extremity DVT while on oral contraception is treated with warfarin anticoagulation for 6 months. Her obstetrician consults the hematologist to advise regarding antithrombotic management during this pregnancy.
What is the approach to prophylaxis in women at high risk for pregnancy-associated VTE?
All women at high risk for pregnancy-associated VTE should be counseled about the signs and symptoms of DVT or PE during preconception and pregnancy and have a plan developed should these symptoms arise. The ACCP guidelines on antithrombotic therapy outline recommendations ranging from clinical vigilance to prophylactic and intermediate-dose anticoagulation, depending on the risk for VTE recurrence, based on the personal and family history of VTE and type of thrombophilia (Table 2).78 These recommendations range from grade 2B to 2C.
For women with a history of estrogen-related VTE, single unprovoked VTE, or recurrent unprovoked VTE not on chronic anticoagulation, antepartum and postpartum pharmacologic thromboprophylaxis with either prophylactic or intermediate-dose LMWH is recommended (grade 2C). In patients with prior history of provoked VTE (non-estrogen related), antepartum clinical vigilance and postpartum pharmacologic thromboprophylaxis is recommended (grade 2C, 2B).
In asymptomatic pregnant women who are homozygote carriers for factor V Leiden or prothrombin G20210A variants and have a positive family history of thrombosis, antepartum and postpartum pharmacologic thromboprophylaxis is recommended (grade 2B). In asymptomatic homozygote carriers of factor V Leiden or prothrombin G20210A variants with no family history of thrombosis and women with all other thrombophilias with a positive family history of thrombosis, postpartum pharmacologic thromboprophylaxis is indicated (grade 2B and 2C, respectively). For women with confirmed APS and clinical criteria of obstetric APS with recurrent pregnancy loss, antepartum thromboprophylaxis with LMWH and low-dose aspirin is recommended (grade 1B). For pregnant women with all other thrombophilias with no personal or family history of thrombosis, clinical vigilance is suggested (grade 2 C).78
As an alternative to LMWH, vitamin K antagonists (VKA) such as warfarin can be used for postpartum thromboprophylaxis; in patients with protein C or S deficiency, due to the risk of warfarin-induced skin necrosis, a rapid-onset anticoagulant must be concomitantly administered. Warfarin and LMWH are safe anticoagulants during lactation, but there are no clinical data on the effects of the DOACs on infants during lactation. Data from animal studies indicate that DOACs are secreted into breast milk.85
What risks are associated with anticoagulant therapy in pregnancy?
VKAs cross the placenta and can cause teratogenicity, pregnancy loss, fetal bleeding, and neurodevelopmental deficits. Therefore, discontinuation of VKAs prior to the sixth week of gestation is necessary to avoid warfarin embryopathy. DOACs have been shown to readily cross the placenta but with unknown human reproductive risks. Fondaparinux, a synthetic pentasaccharide, crosses the placenta in small quantities. Though there are reports of the successful use of fondaparinux in pregnancy, there is limited reported experience of its use in the first trimester.86
The risk for bleeding with anticoagulation is notably acceptable. In a case-control study of 88 pregnant women receiving therapeutic-dose anticoagulation, the risk of postpartum hemorrhage (PPH) after vaginal delivery was 30% in those who received LMWH anticoagulation versus 18% in those who did not (OR 1.9 [95% CI 1.1 to 3.5]).87 However, the risk for severe PPH (≥ 500 mL) was similar (5.6% versus 5.0%; OR 1.1 [95% CI 0.4 to 3.6]). The risk for PPH after cesarean section was 12% in LMWH users versus 4% in LMWH non-users (OR 2.9 [95% CI 0.5 to 19.4]). The risk for PPH associated with delivery within 24 hours after the last dose of LMWH was 1.2 times higher (95% CI 0.4 to 3.6) compared to a longer interval. Therefore, therapeutic LMWH increases the risk for blood loss after vaginal delivery, but not the risk for severe PPH. The risk for PPH is influenced by the interval between the last dose of LMWH and delivery. Of note in this study, per the institution’s protocol, the anticoagulation was stopped with signs of labor or determination of need for delivery. The risk for blood loss may be mitigated in more planned delivery scenarios.87
CASE 4 CONTINUED
The patient is placed on prophylactic-dose LMWH with good tolerance and delivers at 39 weeks' gestation via caesarian section due to nonprogression of labor. Postpartum she is restarted on prophylactic-dose anticoagulation with LMWH. Two weeks after discharge from the hospital, she presents with right calf pain and mild shortness of breath. On physical exam, her leg circumferences are equal. A D-dimer assay is 3375 ng/mL (normal 0–229). CUS of the right leg shows a complete occlusive DVT of the mid-distal superficial femoral and popliteal veins and partially occlusive acute DVT of the right posterior tibial and peroneal veins. CTPA reveals a right lower lobe PE. Because she had developed VTE despite prophylactic LMWH, her anticoagulation is changed to therapeutic dose. She is treated with anticoagulation with LMWH for a total of 3 months, after which a repeat CUS shows no residual thrombosis.
What is the recommended dosing of heparin and LMWH during pregnancy?
A prospective study of 14 pregnant women receiving UFH prophylaxis found that a prophylactic dose of 5000 units twice a day was inadequate to achieve prophylactic heparin levels in any patient in the second or third trimester.88 Similar to treatment dosage, there is no consensus evidence for prophylactic dosing, and dosage recommendations are based on expert opinion. In a retrospective study of 25 pregnant women on intermediate-dose UFH, the mean UFH dose required to achieve a target anti-factor Xa level of 0.1 to 0.3 units/mL was 236.9 units/kg/day.89 However, the use of anti-factor Xa levels for monitoring is controversial as there is no data to support a difference in outcomes with its use in prophylactic or therapeutic dosing.
The timing of the previous VTE history is important when deciding on the anticoagulant dose in pregnancy. In pregnant women with a VTE that occurred within the previous 4 to 6 weeks, full-dose anticoagulation with LMWH should be considered; an intermediate dose (three-fourths of a therapeutic dose) may be used if the thrombotic episode occurred more than 6 weeks earlier but still within a year. Prophylactic dosing may be sufficient if the episode occurred more than a year earlier.90 A clinical trial (High-Low) is under way to explore the optimal dose of LMWH in pregnant women with prior history of VTE who are not on chronic anticoagulation therapy.91
How is anticoagulation therapy managed in the peripartum period?
Neuraxial anesthesia during active labor while on anticoagulation increases the risk for central nervous system bleeding. Therefore, if spontaneous labor occurs in women on therapeutic dose anticoagulation, neuraxial anesthesia cannot be used. However, in the event of elective induction of labor or caesarean section, neuroaxial anesthesia may be performed 12 hours after the administration of the last prophylactic dose of LMWH or 24 hours after the last therapeutic dose of LMWH. Intravenous UFH should be stopped for 6 hours before induction of labor with a confirmed normal aPTT before placement of neuraxial anesthesia. There is no contraindication for using neuraxial anesthesia during subcutaneous standard UFH at total doses of 10,000 units daily. The risk of spinal hematoma with larger daily subcutaneous doses is unclear; therefore, a documented normal aPTT must be obtained before placement of neuroaxial anesthesia.
Postpartum, reinitiation of prophylactic-dose LMWH should be delayed for at least 12 hours after the removal of an epidural catheter. Therapeutic-dose LMWH should be administered no earlier than 24 hours after neuraxial anesthesia, providing that proper hemostasis is achieved. In the absence of persistent bleeding, if no regional anesthesia was used, LMWH may be resumed 12 hours after delivery.92 Anticoagulation with either LMWH or warfarin is recommended for at least 6 to 12 weeks postpartum.33
COUNSELING
Patients should be advised to manage controllable risk factors, including avoiding prolonged immobilization, avoiding excessive weight gain in pregnancy, and stopping smoking. Periods of immobilization tend to cause reduced blood flow (stasis), which predisposes to thrombosis. In a systematic review of records of all patients with confirmed PE after arrival at Charles de Gaulle airport in Paris during a 13-year period, women had a higher risk of PE after a long-distance flight than men, with an estimated incidence of 0.61 per million passengers versus 0.20, respectively; the incidence reached 7.24 and 2.35 cases, respectively, in passengers traveling more than 10,000 kilometers.93,94
The risk of air travel-related thrombosis in pregnant women is estimated to be between 0.03% and 0.1%. Physicians must decide on an individual basis how to prevent travel-related thrombosis in their pregnant patients. In most passengers, prevention can be limited to encouraging exercise, avoidance of long sleeping periods, and not using a window seat. Women at high risk for VTE, such as women with a prior history of VTE who are not on anticoagulation or women with known asymptomatic thrombophilia or other risk factors for thrombosis such as obesity, may benefit from a short period (1–3 days) of LMWH starting 2 hours before a long-distance flight.95
Activation of the coagulation system has been demonstrated in cigarette smokers.96 Heavy smoking was found to be a significant risk factor for VTE in a cross-sectional analysis of 2404 men and women.97 An increased risk for thrombosis during pregnancy is seen in cigarette smokers15,98 and is enhanced with the concomitant use of illicit drugs.99 Other obstetric complications associated with smoking and illicit drug use during pregnancy include preterm labor, spontaneous abortion, perinatal death, low birth weight, and abruption placenta. The efficacy of nicotine replacement therapy in pregnancy is uncertain.100 Recommendations are to advise patients to stop smoking, obtain psychosocial counseling, and utilize adjunctive therapies, which have been shown to have some effect on abstinence rates.101
CONCLUSION
Women are at increased risk for VTE during pregnancy and the postpartum period. Awareness of risk factors and the signs and symptoms of VTE is paramount. Prompt diagnosis and treatment is mandatory to decrease complications of VTE. LMWH is the mainstay treatment of VTE in pregnancy, as it does not cross the placenta. Both LMWH and warfarin are safe during lactation. Close communication among the patient, obstetrician, hematologist, anesthesiologist, and neonatologist is crucial to optimize the care of these patients.
- Knight M, Nour M, Tuffnell D, et al, eds. on behalf of MBRRACE-UK. Saving lives, improving mothers’ care—surveillance of maternal deaths in the UK 2012-14 and lessons learned to inform maternity care from the UK and Ireland confidential enquiries into maternal deaths and morbidity 2009-14. Oxford: National Perinatal Epidemiology Unit, University of Oxford; 2016: 69–75.
- Berg CJ, Callaghan WM, Syverson C, Henderson Z. Pregnancy-related mortality in the United States, 1998 to 2005. Obstet Gynecol 2010;116:1302–9.
- Salonen Ros H, Lichtenstein P, Bellocco R, et al. Increased risks of circulatory diseases in late pregnancy and puerperium. Epidemiology 2001;12:456–60.
- Heit JA, Kobbervig CE, James AH, et al. Trends in the incidence of venous thromboembolism during pregnancy or postpartum: a 30-year population-based study. Ann Intern Med 2005;143:697–706.
- Greer IA. Thrombosis in pregnancy: updates in diagnosis and management. Hematology Am Soc Hematol Educ Program 2012;2012:203–7.
- Hellgren M. Hemostasis during normal pregnancy and puerperium. Semin Thromb Hemost 2003;29:125–30.
- Brenner B. Haemostatic changes in pregnancy. Thromb Res 2004;114:409–14.
- Chan W-S, Spencer FA, Ginsberg JS. Anatomic distribution of deep vein thrombosis in pregnancy. CMAJ 2010;182:657–60.
- Greer IA. Prevention and management of venous thromboembolism in pregnancy. Clin Chest Med 2003;24:123–37.
- Wik HS, Jacobsen AF, Sandvik L, Sandset PM. Prevalence and predictors for post-thrombotic syndrome 3 to 16 years after pregnancy-related venous thrombosis: a population-based, cross-sectional, case-control study. J Thromb Haemost 2012;10:840–7.
- Pomp ER, Lenselink AM, Rosendaal FR, Doggen CJM. Pregnancy, the postpartum period and prothrombotic defects: risk of venous thrombosis in the MEGA study. J Thromb Haemost 2008;6:632–7.
- Marik PE, Plante LA. Venous thromboembolic disease and pregnancy. N Engl J Med 2008;359:2025–33.
- Jacobsen AF, Skjeldestad FE, Sandset PM. Ante- and postnatal risk factors of venous thrombosis: a hospital-based case-control study. J Thromb Haemost 2008;6:905–12.
- Virkus RA, Løkkegaard E, Lidegaard Ø, et al. Risk factors for venous thromboembolism in 1.3 million pregnancies: a nationwide prospective cohort. PloS One 2014;9:e96495.
- James AH, Jamison MG, Brancazio LR, Myers ER. Venous thromboembolism during pregnancy and the postpartum period: incidence, risk factors, and mortality. Am J Obstet Gynecol 2006;194:1311–5.
- Chan W-S, Lee A, Spencer FA, et al. Predicting deep venous thrombosis in pregnancy: out in “LEFt” field? Ann Intern Med 2009;151:85–92.
- Righini M, Jobic C, Boehlen F, et al. Predicting deep venous thrombosis in pregnancy: external validation of the LEFT clinical prediction rule. Haematologica 2013;98:545–8.
- Chan W-S, Chunilal S, Lee A, et al. A red blood cell agglutination D-dimer test to exclude deep venous thrombosis in pregnancy. Ann Intern Med 2007;147:165–70.
- Nijkeuter M, Ginsberg JS, Huisman MV. Diagnosis of deep vein thrombosis and pulmonary embolism in pregnancy: a systematic review. J Thromb Haemost 2006;4:496–500.
- Chan W-S, Spencer FA, Lee AY, et al. Safety of withholding anticoagulation in pregnant women with suspected deep vein thrombosis following negative serial compression ultrasound and iliac vein imaging. CMAJ 2013;185:E194–200.
- Dronkers CE, Srámek A, Huisman MV, Klok FA. Accurate diagnosis of iliac vein thrombosis in pregnancy with magnetic resonance direct thrombus imaging (MRDTI). BMJ Case Rep 2016;2016. pii: bcr2016218091.
- Ray JG, Vermeulen MJ, Bharatha A, et al. Association between MRI exposure during pregnancy and fetal and childhood outcomes. JAMA 2016;316:952–61.
- Royal College of Obstretricians and Gynaecologists. Thromboembolic disease in pregnancy and the puerperium: acute management. Green-top Guideline No. 37b. London: RCOG; 2015.
- Barritt DW, Jordan SC. Anticoagulant drugs in the treatment of pulmonary embolism. A controlled trial. Lancet 1960;1(7138):1309–12.
- Bourjeily G, Khalil H, Raker C, et al. Outcomes of negative multidetector computed tomography with pulmonary angiography in pregnant women suspected of pulmonary embolism. Lung 2012;190:105–11.
- Deutsch AB, Twitty P, Downes K, Parsons MT. Assessment of the alveolar-arterial oxygen gradient as a screening test for pulmonary embolism in pregnancy. Am J Obstet Gynecol 2010;203:373.e1–4.
- Powrie RO, Larson L, Rosene-Montella K, et al. Alveolar-arterial oxygen gradient in acute pulmonary embolism in pregnancy. Am J Obstet Gynecol 1998;178:394–6.
- Leung AN, Bull TM, Jaeschke R, et al. American Thoracic Society documents: an official American Thoracic Society/Society of Thoracic Radiology clinical practice guideline--evaluation of suspected pulmonary embolism in pregnancy. Radiology 2012;262:635–46.
- Cahill AG, Stout MJ, Macones GA, Bhalla S. Diagnosing pulmonary embolism in pregnancy using computed-tomographic angiography or ventilation-perfusion. Obstet Gynecol 2009;114:124–9.
- Bourjeily G, Paidas M, Khalil H, et al. Pulmonary embolism in pregnancy. Lancet 2010;375(9713):500–12.
- Chan WS, Ray JG, Murray S, et al. Suspected pulmonary embolism in pregnancy: clinical presentation, results of lung scanning, and subsequent maternal and pediatric outcomes. Arch Intern Med 2002;162:1170–5.
- Virkus RA, Løkkegaard ECL, Bergholt T, et al. Venous thromboembolism in pregnant and puerperal women in Denmark 1995-2005. A national cohort study. Thromb Haemost 2011;106:304–9.
- Kamel H, Navi BB, Sriram N, et al. Risk of a thrombotic event after the 6-week postpartum period. N Engl J Med 2014;370:1307–15.
- Ginsberg JS, Hirsh J, Turner DC, et al. Risks to the fetus of anticoagulant therapy during pregnancy. Thromb Haemost 1989;61:197–203.
- Greer IA, Nelson-Piercy C. Low-molecular-weight heparins for thromboprophylaxis and treatment of venous thromboembolism in pregnancy: a systematic review of safety and efficacy. Blood 2005;106:401–7.
- Prandoni P, Carnovali M, Marchiori A, Galilei Investigators. Subcutaneous adjusted-dose unfractionated heparin vs fixed-dose low-molecular-weight heparin in the initial treatment of venous thromboembolism. Arch Intern Med 2004;164:1077–83.
- Robertson L, Jones LE. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev 2017;(9):eb9;2:CD001100.
- Costantino G, Ceriani E, Rusconi AM, et al. Bleeding risk during treatment of acute thrombotic events with subcutaneous LMWH compared to intravenous unfractionated heparin; a systematic review. PloS One 2012;7:e44553.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med 1995;332:1330–5.
- Junqueira DRG, Perini E, Penholati RRM, Carvalho MG. Unfractionated heparin versus low molecular weight heparin for avoiding heparin-induced thrombocytopenia in postoperative patients. Cochrane Database Syst Rev 2012;(9):CD007557.
- Bates SM, Greer IA, Middeldorp S, et al. VTE, thrombophilia, antithrombotic therapy, and pregnancy. Chest 2012;141:e691S–e736S.
- Clark NP, Delate T, Witt DM, et al. A descriptive evaluation of unfractionated heparin use during pregnancy. J Thromb Thrombolysis 2009;27:267–73.
- Clark NP, Delate T, Cleary SJ, Witt DM. Analysis of unfractionated heparin dose requirements to target therapeutic anti-Xa intensity during pregnancy. Thromb Res 2010;125:402–5.
- Jacobsen AF, Qvigstad E, Sandset PM. Low molecular weight heparin (dalteparin) for the treatment of venous thromboembolism in pregnancy. BJOG 2003;110:139–44.
- Walfisch A, Koren G. The “warfarin window” in pregnancy: the importance of half-life. J Obstet Gynaecol Can 2010;32:988–9.
- Bates SM, Ginsberg JS. Anticoagulants in pregnancy: fetal effects. Baillières Clin Obstet Gynaecol 1997;11:479–88.
- Stevenson RE, Burton OM, Ferlauto GJ, Taylor HA. Hazards of oral anticoagulants during pregnancy. JAMA 1980;243:1549–51.
- Ginsberg JS, Hirsh J. Anticoagulants during pregnancy. Annu Rev Med 1989;40:79–86.
- Wong V, Cheng CH, Chan KC. Fetal and neonatal outcome of exposure to anticoagulants during pregnancy. Am J Med Genet 1993;45:17–21.
- Blickstein D, Blickstein I. The risk of fetal loss associated with Warfarin anticoagulation. Int J Gynaecol Obstet 2002;78:221–5.
- Burnett AE, Mahan CE, Vazquez SR, et al. Guidance for the practical management of the direct oral anticoagulants (DOACs) in VTE treatment. J Thromb Thrombolysis 2016;41:206–32.
- Bapat P, Pinto LSR, Lubetsky A, et al. Examining the transplacental passage of apixaban using the dually perfused human placenta. J Thromb Haemost 2016;14:1436–41.
- Bapat P, Pinto LSR, Lubetsky A, et al. Rivaroxaban transfer across the dually perfused isolated human placental cotyledon. Am J Obstet Gynecol 2015;213:710.e1–6.
- Bapat P, Kedar R, Lubetsky A, et al. Transfer of dabigatran and dabigatran etexilate mesylate across the dually perfused human placenta. Obstet Gynecol 2014;123:1256–61.
- Savaysa [package insert]. Parsippany (NJ): Daiichi Sankyo, Inc; 2015.
- Filipecki S, Tomkowski W, Hajduk B, et al. [Outcome of patients with clinically acute massive pulmonary embolism]. Pneumonol Alergol Pol 1994;62:132–7.
- Holden EL, Ranu H, Sheth A, et al. Thrombolysis for massive pulmonary embolism in pregnancy--a report of three cases and follow up over a two year period. Thromb Res 2011;127:58–9.
- te Raa GD, Ribbert LS, Snijder RJ, Biesma DH. Treatment options in massive pulmonary embolism during pregnancy; a case-report and review of literature. Thromb Res 2009;124:1–5.
- Leonhardt G, Gaul C, Nietsch HH, et al. Thrombolytic therapy in pregnancy. J Thromb Thrombolysis 2006;21:271–6.
- Colombier S, Niclauss L. Successful surgical pulmonary embolectomy for massive perinatal embolism after emergency cesarean section. Ann Vasc Surg 2015;29:1452.e1–4.
- British Committee for Standards in Haematology Writing Group, Baglin TP, Brush J, Streiff M. Guidelines on use of vena cava filters. Br J Haematol 2006;134:590–5.
- Harris SA, Velineni R, Davies AH. Inferior vena cava filters in pregnancy: a systematic review. J Vasc Interv Radiol 2016;27:354–360.
- Pabinger I, Grafenhofer H, Kyrle PA, et al. Temporary increase in the risk for recurrence during pregnancy in women with a history of venous thromboembolism. Blood 2002;100:1060–2.
- Pabinger I, Grafenhofer H, Kaider A, et al. Risk of pregnancy-associated recurrent venous thromboembolism in women with a history of venous thrombosis. J Thromb Haemost 2005;3:949–54.
- Brill-Edwards P, Ginsberg JS, Gent M, et al. Safety of withholding heparin in pregnant women with a history of venous thromboembolism. Recurrence of Clot in This Pregnancy Study Group. N Engl J Med 2000;343:1439–44.
- De Stefano V, Martinelli I, Rossi E, et al. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006;135:386–91.
- De Stefano V, Martinelli I, Rossi E, et al. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006;135:386–91.
- Lim W, Eikelboom JW, Ginsberg JS. Inherited thrombophilia and pregnancy associated venous thromboembolism. BMJ 2007;334:1318–21.
- Tormene D, Simioni P, Prandoni P, et al. Factor V Leiden mutation and the risk of venous thromboembolism in pregnant women. Haematologica 2001;86:1305–9.
- Baglin T, Gray E, Greaves M, et al. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010;149:209–20.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4:295–306.
- Pengo V, Tripodi A, Reber G, et al. Update of the guidelines for lupus anticoagulant detection. J Thromb Haemost 2009;7:1737–40.
- Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol 2006;132:171–96.
- American College of Obstetricians and Gynecologists Women’s Health Care Physicians. ACOG Practice Bulletin No. 138: Inherited thrombophilias in pregnancy. Obstet Gynecol 2013;122:706–17.
- Mak A, Cheung MW, Cheak AA, Ho RC. Combination of heparin and aspirin is superior to aspirin alone in enhancing live births in patients with recurrent pregnancy loss and positive anti-phospholipid antibodies: a meta-analysis of randomized controlled trials and meta-regression. Rheumatology (Oxf) 2010;49:281–8.
- Laskin CA, Spitzer KA, Clark CA, et al. Low molecular weight heparin and aspirin for recurrent pregnancy loss: results from the randomized, controlled HepASA Trial. J Rheumatol 2009;36:279–87.
- Farquharson RG, Quenby S, Greaves M. Antiphospholipid syndrome in pregnancy: a randomized, controlled trial of treatment. Obstet Gynecol 2002;100:408–13.
- Bates SM, Jaeschke R, Stevens SM, et al. Diagnosis of DVT: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012;141(2 Suppl):e351S–418S.
- Lubbe WF, Butler WS, Palmer SJ, Liggins GC. Fetal survival after prednisone suppression of maternal lupus-anticoagulant. Lancet 1983;1(8338):1361–3.
- Lockshin MD, Druzin ML, Qamar T. Prednisone does not prevent recurrent fetal death in women with antiphospholipid antibody. Am J Obstet Gynecol 1989;160:439–43.
- Silver RK, MacGregor SN, Sholl JS, et al. Comparative trial of prednisone plus aspirin versus aspirin alone in the treatment of anticardiolipin antibody-positive obstetric patients. Am J Obstet Gynecol 1993;169:1411–7.
- Cowchock FS, Reece EA, Balaban D, et al. Repeated fetal losses associated with antiphospholipid antibodies: a collaborative randomized trial comparing prednisone with low-dose heparin treatment. Am J Obstet Gynecol 1992;166:1318–23.
- Laskin CA, Bombardier C, Hannah ME, et al. Prednisone and aspirin in women with autoantibodies and unexplained recurrent fetal loss. N Engl J Med 1997;337:148–53.
- Dendrinos S, Sakkas E, Makrakis E. Low-molecular-weight heparin versus intravenous immunoglobulin for recurrent abortion associated with antiphospholipid antibody syndrome. Int J Gynaecol Obstet 2009;104:223–5.
- Cohen H, Arachchillage DRJ, Beyer-Westendorf J, et al. Direct oral anticoagulants and women. Semin Thromb Hemost 2016;42:789–97.
- Bates SM, Middeldorp S, Rodger M, et al. Guidance for the treatment and prevention of obstetric-associated venous thromboembolism. J Thromb Thrombolysis 2016;41:92–128.
- Knol HM, Schultinge L, Veeger NJ, et al. The risk of postpartum hemorrhage in women using high dose of low-molecular-weight heparins during pregnancy. Thromb Res 2012;130:334–8.
- Barbour LA, Smith JM, Marlar RA. Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol 1995;173:1869–73.
- Bergqvist A, Bergqvist D, Lindhagen A, Mätzsch T. Late symptoms after pregnancy-related deep vein thrombosis. Br J Obstet Gynaecol 1990;97:338–41.
- Rodger M. Evidence base for the management of venous thromboembolism in pregnancy. Hematology Am Soc Hematol Educ Program. 2010;2010:173–80.
- Bleker SM, Buchmüller A, Chauleur C, et al. Low-molecular-weight heparin to prevent recurrent venous thromboembolism in pregnancy: Rationale and design of the Highlow study, a randomised trial of two doses. Thromb Res 2016;144:62–8.
- Horlocker TT, Wedel DJ, Rowlingson JC, et al. Regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy: American Society of Regional Anesthesia and Pain Medicine evidence-based guidelines (third edition). Reg Anesth Pain Med 2010;35:64–101.
- Lapostolle F, Surget V, Borron SW, et al. Severe pulmonary embolism associated with air travel. N Engl J Med 2001;345:779–83.
- Lapostolle F, Le Toumelin P, Chassery C, et al. Gender as a risk factor for pulmonary embolism after air travel. Thromb Haemost 2009;102:1165–8.
- Cannegieter SC, Rosendaal FR. Pregnancy and travel-related thromboembolism. Thromb Res 2013;131 Suppl 1:S55–58.
- Miller GJ, Bauer KA, Cooper JA, Rosenberg RD. Activation of the coagulant pathway in cigarette smokers. Thromb Haemost 1998;79:549–53.
- Golomb BA, Chan VT, Denenberg JO, et al. Risk marker associations with venous thrombotic events: a cross-sectional analysis. BMJ Open 2014;4:e003208.
- Lindqvist P, Dahlbäck B, Marŝál K. Thrombotic risk during pregnancy: a population study. Obstet Gynecol 1999;94:595–9.
- Black M, Bhattacharya S, Fairley T, et al. Outcomes of pregnancy in women using illegal drugs and in women who smoke cigarettes. Acta Obstet Gynecol Scand 2013;92:47–52.
- Mendelsohn C, Gould GS, Oncken C. Management of smoking in pregnant women. Aust Fam Physician 2014;43:46–51.
- Chamberlain C, O’Mara-Eves A, Oliver S, et al. Psychosocial interventions for supporting women to stop smoking in pregnancy. Cochrane Database Syst Rev 2013;10:CD001055.
- Knight M, Nour M, Tuffnell D, et al, eds. on behalf of MBRRACE-UK. Saving lives, improving mothers’ care—surveillance of maternal deaths in the UK 2012-14 and lessons learned to inform maternity care from the UK and Ireland confidential enquiries into maternal deaths and morbidity 2009-14. Oxford: National Perinatal Epidemiology Unit, University of Oxford; 2016: 69–75.
- Berg CJ, Callaghan WM, Syverson C, Henderson Z. Pregnancy-related mortality in the United States, 1998 to 2005. Obstet Gynecol 2010;116:1302–9.
- Salonen Ros H, Lichtenstein P, Bellocco R, et al. Increased risks of circulatory diseases in late pregnancy and puerperium. Epidemiology 2001;12:456–60.
- Heit JA, Kobbervig CE, James AH, et al. Trends in the incidence of venous thromboembolism during pregnancy or postpartum: a 30-year population-based study. Ann Intern Med 2005;143:697–706.
- Greer IA. Thrombosis in pregnancy: updates in diagnosis and management. Hematology Am Soc Hematol Educ Program 2012;2012:203–7.
- Hellgren M. Hemostasis during normal pregnancy and puerperium. Semin Thromb Hemost 2003;29:125–30.
- Brenner B. Haemostatic changes in pregnancy. Thromb Res 2004;114:409–14.
- Chan W-S, Spencer FA, Ginsberg JS. Anatomic distribution of deep vein thrombosis in pregnancy. CMAJ 2010;182:657–60.
- Greer IA. Prevention and management of venous thromboembolism in pregnancy. Clin Chest Med 2003;24:123–37.
- Wik HS, Jacobsen AF, Sandvik L, Sandset PM. Prevalence and predictors for post-thrombotic syndrome 3 to 16 years after pregnancy-related venous thrombosis: a population-based, cross-sectional, case-control study. J Thromb Haemost 2012;10:840–7.
- Pomp ER, Lenselink AM, Rosendaal FR, Doggen CJM. Pregnancy, the postpartum period and prothrombotic defects: risk of venous thrombosis in the MEGA study. J Thromb Haemost 2008;6:632–7.
- Marik PE, Plante LA. Venous thromboembolic disease and pregnancy. N Engl J Med 2008;359:2025–33.
- Jacobsen AF, Skjeldestad FE, Sandset PM. Ante- and postnatal risk factors of venous thrombosis: a hospital-based case-control study. J Thromb Haemost 2008;6:905–12.
- Virkus RA, Løkkegaard E, Lidegaard Ø, et al. Risk factors for venous thromboembolism in 1.3 million pregnancies: a nationwide prospective cohort. PloS One 2014;9:e96495.
- James AH, Jamison MG, Brancazio LR, Myers ER. Venous thromboembolism during pregnancy and the postpartum period: incidence, risk factors, and mortality. Am J Obstet Gynecol 2006;194:1311–5.
- Chan W-S, Lee A, Spencer FA, et al. Predicting deep venous thrombosis in pregnancy: out in “LEFt” field? Ann Intern Med 2009;151:85–92.
- Righini M, Jobic C, Boehlen F, et al. Predicting deep venous thrombosis in pregnancy: external validation of the LEFT clinical prediction rule. Haematologica 2013;98:545–8.
- Chan W-S, Chunilal S, Lee A, et al. A red blood cell agglutination D-dimer test to exclude deep venous thrombosis in pregnancy. Ann Intern Med 2007;147:165–70.
- Nijkeuter M, Ginsberg JS, Huisman MV. Diagnosis of deep vein thrombosis and pulmonary embolism in pregnancy: a systematic review. J Thromb Haemost 2006;4:496–500.
- Chan W-S, Spencer FA, Lee AY, et al. Safety of withholding anticoagulation in pregnant women with suspected deep vein thrombosis following negative serial compression ultrasound and iliac vein imaging. CMAJ 2013;185:E194–200.
- Dronkers CE, Srámek A, Huisman MV, Klok FA. Accurate diagnosis of iliac vein thrombosis in pregnancy with magnetic resonance direct thrombus imaging (MRDTI). BMJ Case Rep 2016;2016. pii: bcr2016218091.
- Ray JG, Vermeulen MJ, Bharatha A, et al. Association between MRI exposure during pregnancy and fetal and childhood outcomes. JAMA 2016;316:952–61.
- Royal College of Obstretricians and Gynaecologists. Thromboembolic disease in pregnancy and the puerperium: acute management. Green-top Guideline No. 37b. London: RCOG; 2015.
- Barritt DW, Jordan SC. Anticoagulant drugs in the treatment of pulmonary embolism. A controlled trial. Lancet 1960;1(7138):1309–12.
- Bourjeily G, Khalil H, Raker C, et al. Outcomes of negative multidetector computed tomography with pulmonary angiography in pregnant women suspected of pulmonary embolism. Lung 2012;190:105–11.
- Deutsch AB, Twitty P, Downes K, Parsons MT. Assessment of the alveolar-arterial oxygen gradient as a screening test for pulmonary embolism in pregnancy. Am J Obstet Gynecol 2010;203:373.e1–4.
- Powrie RO, Larson L, Rosene-Montella K, et al. Alveolar-arterial oxygen gradient in acute pulmonary embolism in pregnancy. Am J Obstet Gynecol 1998;178:394–6.
- Leung AN, Bull TM, Jaeschke R, et al. American Thoracic Society documents: an official American Thoracic Society/Society of Thoracic Radiology clinical practice guideline--evaluation of suspected pulmonary embolism in pregnancy. Radiology 2012;262:635–46.
- Cahill AG, Stout MJ, Macones GA, Bhalla S. Diagnosing pulmonary embolism in pregnancy using computed-tomographic angiography or ventilation-perfusion. Obstet Gynecol 2009;114:124–9.
- Bourjeily G, Paidas M, Khalil H, et al. Pulmonary embolism in pregnancy. Lancet 2010;375(9713):500–12.
- Chan WS, Ray JG, Murray S, et al. Suspected pulmonary embolism in pregnancy: clinical presentation, results of lung scanning, and subsequent maternal and pediatric outcomes. Arch Intern Med 2002;162:1170–5.
- Virkus RA, Løkkegaard ECL, Bergholt T, et al. Venous thromboembolism in pregnant and puerperal women in Denmark 1995-2005. A national cohort study. Thromb Haemost 2011;106:304–9.
- Kamel H, Navi BB, Sriram N, et al. Risk of a thrombotic event after the 6-week postpartum period. N Engl J Med 2014;370:1307–15.
- Ginsberg JS, Hirsh J, Turner DC, et al. Risks to the fetus of anticoagulant therapy during pregnancy. Thromb Haemost 1989;61:197–203.
- Greer IA, Nelson-Piercy C. Low-molecular-weight heparins for thromboprophylaxis and treatment of venous thromboembolism in pregnancy: a systematic review of safety and efficacy. Blood 2005;106:401–7.
- Prandoni P, Carnovali M, Marchiori A, Galilei Investigators. Subcutaneous adjusted-dose unfractionated heparin vs fixed-dose low-molecular-weight heparin in the initial treatment of venous thromboembolism. Arch Intern Med 2004;164:1077–83.
- Robertson L, Jones LE. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev 2017;(9):eb9;2:CD001100.
- Costantino G, Ceriani E, Rusconi AM, et al. Bleeding risk during treatment of acute thrombotic events with subcutaneous LMWH compared to intravenous unfractionated heparin; a systematic review. PloS One 2012;7:e44553.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med 1995;332:1330–5.
- Junqueira DRG, Perini E, Penholati RRM, Carvalho MG. Unfractionated heparin versus low molecular weight heparin for avoiding heparin-induced thrombocytopenia in postoperative patients. Cochrane Database Syst Rev 2012;(9):CD007557.
- Bates SM, Greer IA, Middeldorp S, et al. VTE, thrombophilia, antithrombotic therapy, and pregnancy. Chest 2012;141:e691S–e736S.
- Clark NP, Delate T, Witt DM, et al. A descriptive evaluation of unfractionated heparin use during pregnancy. J Thromb Thrombolysis 2009;27:267–73.
- Clark NP, Delate T, Cleary SJ, Witt DM. Analysis of unfractionated heparin dose requirements to target therapeutic anti-Xa intensity during pregnancy. Thromb Res 2010;125:402–5.
- Jacobsen AF, Qvigstad E, Sandset PM. Low molecular weight heparin (dalteparin) for the treatment of venous thromboembolism in pregnancy. BJOG 2003;110:139–44.
- Walfisch A, Koren G. The “warfarin window” in pregnancy: the importance of half-life. J Obstet Gynaecol Can 2010;32:988–9.
- Bates SM, Ginsberg JS. Anticoagulants in pregnancy: fetal effects. Baillières Clin Obstet Gynaecol 1997;11:479–88.
- Stevenson RE, Burton OM, Ferlauto GJ, Taylor HA. Hazards of oral anticoagulants during pregnancy. JAMA 1980;243:1549–51.
- Ginsberg JS, Hirsh J. Anticoagulants during pregnancy. Annu Rev Med 1989;40:79–86.
- Wong V, Cheng CH, Chan KC. Fetal and neonatal outcome of exposure to anticoagulants during pregnancy. Am J Med Genet 1993;45:17–21.
- Blickstein D, Blickstein I. The risk of fetal loss associated with Warfarin anticoagulation. Int J Gynaecol Obstet 2002;78:221–5.
- Burnett AE, Mahan CE, Vazquez SR, et al. Guidance for the practical management of the direct oral anticoagulants (DOACs) in VTE treatment. J Thromb Thrombolysis 2016;41:206–32.
- Bapat P, Pinto LSR, Lubetsky A, et al. Examining the transplacental passage of apixaban using the dually perfused human placenta. J Thromb Haemost 2016;14:1436–41.
- Bapat P, Pinto LSR, Lubetsky A, et al. Rivaroxaban transfer across the dually perfused isolated human placental cotyledon. Am J Obstet Gynecol 2015;213:710.e1–6.
- Bapat P, Kedar R, Lubetsky A, et al. Transfer of dabigatran and dabigatran etexilate mesylate across the dually perfused human placenta. Obstet Gynecol 2014;123:1256–61.
- Savaysa [package insert]. Parsippany (NJ): Daiichi Sankyo, Inc; 2015.
- Filipecki S, Tomkowski W, Hajduk B, et al. [Outcome of patients with clinically acute massive pulmonary embolism]. Pneumonol Alergol Pol 1994;62:132–7.
- Holden EL, Ranu H, Sheth A, et al. Thrombolysis for massive pulmonary embolism in pregnancy--a report of three cases and follow up over a two year period. Thromb Res 2011;127:58–9.
- te Raa GD, Ribbert LS, Snijder RJ, Biesma DH. Treatment options in massive pulmonary embolism during pregnancy; a case-report and review of literature. Thromb Res 2009;124:1–5.
- Leonhardt G, Gaul C, Nietsch HH, et al. Thrombolytic therapy in pregnancy. J Thromb Thrombolysis 2006;21:271–6.
- Colombier S, Niclauss L. Successful surgical pulmonary embolectomy for massive perinatal embolism after emergency cesarean section. Ann Vasc Surg 2015;29:1452.e1–4.
- British Committee for Standards in Haematology Writing Group, Baglin TP, Brush J, Streiff M. Guidelines on use of vena cava filters. Br J Haematol 2006;134:590–5.
- Harris SA, Velineni R, Davies AH. Inferior vena cava filters in pregnancy: a systematic review. J Vasc Interv Radiol 2016;27:354–360.
- Pabinger I, Grafenhofer H, Kyrle PA, et al. Temporary increase in the risk for recurrence during pregnancy in women with a history of venous thromboembolism. Blood 2002;100:1060–2.
- Pabinger I, Grafenhofer H, Kaider A, et al. Risk of pregnancy-associated recurrent venous thromboembolism in women with a history of venous thrombosis. J Thromb Haemost 2005;3:949–54.
- Brill-Edwards P, Ginsberg JS, Gent M, et al. Safety of withholding heparin in pregnant women with a history of venous thromboembolism. Recurrence of Clot in This Pregnancy Study Group. N Engl J Med 2000;343:1439–44.
- De Stefano V, Martinelli I, Rossi E, et al. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006;135:386–91.
- De Stefano V, Martinelli I, Rossi E, et al. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006;135:386–91.
- Lim W, Eikelboom JW, Ginsberg JS. Inherited thrombophilia and pregnancy associated venous thromboembolism. BMJ 2007;334:1318–21.
- Tormene D, Simioni P, Prandoni P, et al. Factor V Leiden mutation and the risk of venous thromboembolism in pregnant women. Haematologica 2001;86:1305–9.
- Baglin T, Gray E, Greaves M, et al. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010;149:209–20.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4:295–306.
- Pengo V, Tripodi A, Reber G, et al. Update of the guidelines for lupus anticoagulant detection. J Thromb Haemost 2009;7:1737–40.
- Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol 2006;132:171–96.
- American College of Obstetricians and Gynecologists Women’s Health Care Physicians. ACOG Practice Bulletin No. 138: Inherited thrombophilias in pregnancy. Obstet Gynecol 2013;122:706–17.
- Mak A, Cheung MW, Cheak AA, Ho RC. Combination of heparin and aspirin is superior to aspirin alone in enhancing live births in patients with recurrent pregnancy loss and positive anti-phospholipid antibodies: a meta-analysis of randomized controlled trials and meta-regression. Rheumatology (Oxf) 2010;49:281–8.
- Laskin CA, Spitzer KA, Clark CA, et al. Low molecular weight heparin and aspirin for recurrent pregnancy loss: results from the randomized, controlled HepASA Trial. J Rheumatol 2009;36:279–87.
- Farquharson RG, Quenby S, Greaves M. Antiphospholipid syndrome in pregnancy: a randomized, controlled trial of treatment. Obstet Gynecol 2002;100:408–13.
- Bates SM, Jaeschke R, Stevens SM, et al. Diagnosis of DVT: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012;141(2 Suppl):e351S–418S.
- Lubbe WF, Butler WS, Palmer SJ, Liggins GC. Fetal survival after prednisone suppression of maternal lupus-anticoagulant. Lancet 1983;1(8338):1361–3.
- Lockshin MD, Druzin ML, Qamar T. Prednisone does not prevent recurrent fetal death in women with antiphospholipid antibody. Am J Obstet Gynecol 1989;160:439–43.
- Silver RK, MacGregor SN, Sholl JS, et al. Comparative trial of prednisone plus aspirin versus aspirin alone in the treatment of anticardiolipin antibody-positive obstetric patients. Am J Obstet Gynecol 1993;169:1411–7.
- Cowchock FS, Reece EA, Balaban D, et al. Repeated fetal losses associated with antiphospholipid antibodies: a collaborative randomized trial comparing prednisone with low-dose heparin treatment. Am J Obstet Gynecol 1992;166:1318–23.
- Laskin CA, Bombardier C, Hannah ME, et al. Prednisone and aspirin in women with autoantibodies and unexplained recurrent fetal loss. N Engl J Med 1997;337:148–53.
- Dendrinos S, Sakkas E, Makrakis E. Low-molecular-weight heparin versus intravenous immunoglobulin for recurrent abortion associated with antiphospholipid antibody syndrome. Int J Gynaecol Obstet 2009;104:223–5.
- Cohen H, Arachchillage DRJ, Beyer-Westendorf J, et al. Direct oral anticoagulants and women. Semin Thromb Hemost 2016;42:789–97.
- Bates SM, Middeldorp S, Rodger M, et al. Guidance for the treatment and prevention of obstetric-associated venous thromboembolism. J Thromb Thrombolysis 2016;41:92–128.
- Knol HM, Schultinge L, Veeger NJ, et al. The risk of postpartum hemorrhage in women using high dose of low-molecular-weight heparins during pregnancy. Thromb Res 2012;130:334–8.
- Barbour LA, Smith JM, Marlar RA. Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol 1995;173:1869–73.
- Bergqvist A, Bergqvist D, Lindhagen A, Mätzsch T. Late symptoms after pregnancy-related deep vein thrombosis. Br J Obstet Gynaecol 1990;97:338–41.
- Rodger M. Evidence base for the management of venous thromboembolism in pregnancy. Hematology Am Soc Hematol Educ Program. 2010;2010:173–80.
- Bleker SM, Buchmüller A, Chauleur C, et al. Low-molecular-weight heparin to prevent recurrent venous thromboembolism in pregnancy: Rationale and design of the Highlow study, a randomised trial of two doses. Thromb Res 2016;144:62–8.
- Horlocker TT, Wedel DJ, Rowlingson JC, et al. Regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy: American Society of Regional Anesthesia and Pain Medicine evidence-based guidelines (third edition). Reg Anesth Pain Med 2010;35:64–101.
- Lapostolle F, Surget V, Borron SW, et al. Severe pulmonary embolism associated with air travel. N Engl J Med 2001;345:779–83.
- Lapostolle F, Le Toumelin P, Chassery C, et al. Gender as a risk factor for pulmonary embolism after air travel. Thromb Haemost 2009;102:1165–8.
- Cannegieter SC, Rosendaal FR. Pregnancy and travel-related thromboembolism. Thromb Res 2013;131 Suppl 1:S55–58.
- Miller GJ, Bauer KA, Cooper JA, Rosenberg RD. Activation of the coagulant pathway in cigarette smokers. Thromb Haemost 1998;79:549–53.
- Golomb BA, Chan VT, Denenberg JO, et al. Risk marker associations with venous thrombotic events: a cross-sectional analysis. BMJ Open 2014;4:e003208.
- Lindqvist P, Dahlbäck B, Marŝál K. Thrombotic risk during pregnancy: a population study. Obstet Gynecol 1999;94:595–9.
- Black M, Bhattacharya S, Fairley T, et al. Outcomes of pregnancy in women using illegal drugs and in women who smoke cigarettes. Acta Obstet Gynecol Scand 2013;92:47–52.
- Mendelsohn C, Gould GS, Oncken C. Management of smoking in pregnant women. Aust Fam Physician 2014;43:46–51.
- Chamberlain C, O’Mara-Eves A, Oliver S, et al. Psychosocial interventions for supporting women to stop smoking in pregnancy. Cochrane Database Syst Rev 2013;10:CD001055.
Locally Advanced Pancreatic Cancer
INTRODUCTION
Pancreatic cancer is one of the most rapidly rising causes of mortality in the United States. In 2016, the number of deaths from pancreatic cancer exceeded those from breast cancer, making it the third leading cause of cancer-related death in the United States.1 It is projected that by 2020 pancreatic cancer will overtake colorectal malignancies to become the second most common cause of cancer death in this country.1,2 The term pancreatic cancer encompasses both exocrine and endocrine tumors. However, since 80% of pancreatic cancers are classified as pancreatic ductal adenocarcinoma (PDA), when speaking about pancreatic cancer most clinicians and scientists are referring to PDA.
Even with advances in chemotherapy and radiotherapy over the past decade, the only curative option for PDA is surgical resection. Unfortunately, only 20% of patients are appropriate surgical candidates at the time of diagnosis.3 Considering the lack of screening options and the ambiguity of symptomatology, roughly 4 out 5 patients with PDA are diagnosed as having locally advanced or metastatic disease that is initially not amenable to surgery.
Locally advanced pancreatic adenocarcinoma presents unique challenges in management and treatment. Treatment options include multi-agent chemotherapy, chemoradiation, or radiotherapy. Some patients can be successfully down-staged with these therapies and be deemed surgical candidates. Other challenges include selecting the appropriate sequence of therapies and stratifying therapies based on comorbidities. In this article, we review the epidemiology, biology, and diagnostic approach to PDA and focus on current treatment strategies for locally advanced pancreatic cancer (LAPC).
EPIDEMIOLOGY
In 2012, GLOBOCAN estimated that PDA caused 331,000 deaths per year, accounting for 4% of all worldwide mortality.4,5 Despite high incidence rates internationally, PDA is a disease of Western and industrialized nations. In the Unites States, PDA is a malignancy of middle to late adulthood, with a sharp upsurge in incidence after age 50 years.6 More than one third of new cases are diagnosed in patients older than 70 years, and more than half of patients diagnosed are older than 60 years of age.2 The incidence of pancreatic cancer is fairly equal among men and women, with a slightly higher rate for the male sex. It has an incidence preference for African-Americans by 4.8 cases per 100,000 persons nationally.7
Risk factors for the development of exocrine pancreatic cancer include hereditary disposition, underlying medical conditions, and environmental factors. One of the most significant environmental risk factors for the development of PDA is smoking,8 which is associated with up to 25% of all cases.9 Smoking cessation leads to a rapid reduction in risk for pancreatic cancer, with the risk among former smokers approaching that for never smokers less than 10 years after quitting.9 Other environmental factors that contribute to the development of pancreatic cancer include increased body mass index, a high-salt and high-saturated fat diet, heavy alcohol intake, and increased utilization of nonsteroidal inflammatory drugs.10–13
There is a strong association between new-onset diabetes and increased risk for developing PDA.14,15 Data also suggest that diabetes may be a risk factor and/or a consequence of tissue destruction that arises during the development or progression of PDA.16,17 Interestingly, ABO blood grouping is another underlying medical disposition that confers an altered risk profile. Studies have shown that patients with blood group O were less likely than those with type A, B, or AB to develop pancreatic cancer.18
Genetic predisposition syndromes can elevate an individual patient’s risk for developing PDA. Genetic syndromes and gene alterations that increase the risk for PDA include BRCA1/2, Peutz-Jeghers syndrome, and Lynch syndrome risk.19–21 Up to 10% to 15% of PDA cases may be due to an inherited familial cancer.22 Having a first-degree relative with PDA increases the odds of developing PDA 1.76-fold compared to those without a family history.23 The exact biologic and molecular mechanisms of familial pancreatic cancer are unclear. It is estimated that about 10% of patients with familial pancreatic cancer (FPC) carry BRCA2 mutations.24 Individuals at risk for FPC should undergo genetic screening for the presence of the most frequently inherited pancreatic cancer susceptibility genetic defects: BRCA2, PALB2, and ATM germline mutations.25 Carriers of BRCA2, who are also at increased risk for developing breast, ovarian, and prostate cancer, should be monitored closely. Of all hereditary conditions, hereditary pancreatitis confers the highest risk for developing PDA, with an approximate risk elevation of 40% to 50%.26,27 Although several genetic predisposition syndromes have been identified, most cases of pancreatic adenocarcinoma are thought to be sporadic.
CANCER BIOLOGY AND PATHOLOGY
The pathologic predecessor of PDA is pancreatic intraepithelial neoplasia (PIN). With further dysplastic changes that result from increasing genetic alterations, these precancerous lesions progress from low- to high-grade and finally to adenocarcinoma. More than 90% of all PINs across all grades have oncogenic KRAS mutations.28 Additionally, inactivating mutations in the tumor suppressor genes SMAD4, p53, and CDKN2A are found with increasing frequency in higher grade PINs. The frequency and presence of mutations in both oncogenes and tumor suppressor genes in precursor neoplasias mirror the genetic mutations noted in advanced PDA.29 Among all mutations, KRAS is the most common and most functionally important for pancreatic cancer cell survival. KRAS mutations not only have profound effects on downstream mediators of tumor growth and metastasis, but they are implicated in reprograming of cellular metabolism.30,31
Pancreatic adenocarcinoma has a unique microenvironment that makes it a difficult target for current therapeutic modalities. First, it is one of the most stroma-rich malignancies. The dense stroma surrounding pancreatic tumor cells leads to increased tumor pressures and alterations in tumor vascular perfusion.32 It also serves as a barrier that prevents chemotherapeutic drugs from reaching the tumor cells. Thus, clinical trials are under way to investigate agents such has hyaluronidase, which may degrade components of the extracellular matrix that supports thestromal environment. Additionally, there is data to suggest that the microenvironment of PDA downregulates immune monitoring, leading to further tumor growth.27,33 The molecular, cellular, and immunologic complexity of PDA may contribute to its resistance to traditional therapeutics.
EVALUATION AND DIAGNOSIS
CASE PRESENTATION
A 61-year-old man with a history of type 2 diabetes mellitus and chronic tobacco use presents to the emergency department (ED) with a 4-month history of progressively worsening abdominal discomfort and fatigue. He has also noticed darkening of his urine and slight yellow discoloration of his eyes. His weight measured 5 months ago in his primary care physician’s office was 91 kg (200 lb, BMI 29.5) and in the ED is 75 kg (165 lb, BMI 24.4). He has noticed bulky, malodorous, oily stools for about 2 months. Preliminary laboratory studies reveal elevated levels of total bilirubin (2.7 mg/dL) and alkaline phosphatase (204 IU/L). Transabdominal ultrasound (US) is obtained and reveals a 3-cm pancreatic mass with biliary tract dilation.
Does this patient have pancreatic cancer?
CLINICAL SIGNS AND SYMPTOMS
Establishing the diagnosis of pancreatic cancer in a patient who presents with a high index of suspicion is critical. Patients with pancreatic cancer usually present after a period of nonspecific and vague symptoms, which typically are experienced as abdominal discomfort, weight loss, and weakness. It is estimated that approximately 25% of patients may complain of vague abdominal pain up to 6 months prior to diagnosis. Up to 15% of patients may seek medical attention more than 6 months prior to establishing a diagnosis of PDA.34 The most common symptoms associated with pancreatic cancer in order of decreasing reported frequency are weight loss, anorexia, abdominal/epigastric pain, dark-colored urine, jaundice, nausea, back pain, and diarrhea with associated steatorrhea.35 Upwards of 15% of patients present with painless jaundice, a term that is often associated with pancreatic cancer.36 On exam these patients may have scleral icterus, sublingual jaundice, epigastric pain on palpation, weight loss, hepatomegaly, lymphadenopathy and a nontender, distended, palpable gallbladder (also known as Courvoisier sign).34 Abdominal signs and symptoms arise from tumor growth into surrounding vessels, tissues, and ducts within the abdominal cavity. Compression of the common bile duct accounts for the development of jaundice. Tumor growth around the stomach and duodenum can lead to delayed gastric emptying and subsequently nausea and vomiting. Constriction of the pancreatic duct leads to pancreatic insufficiency, precipitation of weight loss, and steatorrhea. Pancreatic insufficiency can worsen abdominal pain, and lead to increased weight loss and flatulence.
Less common symptoms include pain, erythema, and edema involving the lower extremities, which may be reflective of migratory thrombophlebitis (commonly known as Trousseau syndrome). Thromboembolic disease, including pulmonary embolism, portal vein, and deep vein thromboses are frequently encountered complications of pancreatic cancer. The incidence of thromboembolic events in patients with PDA has been reported to be as high as 54%.37 Of all signs encountered, weight loss is the most common and most profound. Patients with advanced PDA have severe degrees of cachexia. Some patients present with as much as a 5 kg/m2 decrease in their BMI from pre-illness baseline BMI, and lose another 3 to 4 kg/m2 through disease progression.38 At the time of diagnosis, many patients have already undergone significant weight loss, which can have substantial implications on treatment planning and clinical outcomes.
What other studies can be done to assist in making the diagnosis?
LABORATORY ABNORMALITIES AND TUMOR MARKERS
Elevations in alkaline phosphatase, γ-glutamyltransferase (GGT), serum aspartate aminotransferase (AST), serum alanine aminotransferase (ALT), and direct fractions of bilirubin are common in patients with PDA. Patients will usually have an obstructive pattern on their liver panel, with predominant elevations in direct bilirubin, alkaline phosphatase, and GGT, as compared with AST and ALT. Other baseline laboratory studies, including a complete blood count and basic metabolic panel, should be obtained because patients commonly have thrombocytosis, anemia, and electrolyte abnormalities due to the tumor itself and pancreatic insufficiency (Table 1).

Measurement of glycated hemoglobin (HBA1C) is an emerging and important diagnostic test in the diagnosis of pancreatic cancer. Recently, data has emerged to suggest that new-onset diabetes is present in about 50% of patients diagnosed with pancreatic cancer.39 The temporal relationship of pancreatic cancer and diabetes is supported by evidence showing that patients who undergo resection commonly have resolution of their diabetes.17 This study suggested that hyperglycemia, elevated HBA1C, and symptoms of diabetes in patients older than 50 years may identify patients who have early pancreatic cancer. The entity of pancreatic cancer–associated diabetes needs to be better defined and the algorithmic approach to evaluation and diagnosis, utilizing signs, symptoms, and laboratory values associated with diabetes, needs to be clearly established.
The only serum marker for PDA is carbohydrate antigen 19-9 (CA 19-9), also known as sialylated Lewis antigen or cancer-associated antigen. It was first identified in pancreatic cancer patients in 1981.40,41 The sensitivity and specificity of CA 19-9 ranges from 70% to approximately 90%.42,43 Hereditary predispositions and comorbid disease cross-reactivity contribute to the diminished sensitivity and specificity of CA 19-9. In about 5% to 10% of the population, CA 19-9 is not expressed (Lewis antigen A and B negative). Additionally, since CA 19-9 is expressed in the cells that line the biliary tree, diseases that lead to pancreatic or liver inflammation may falsely elevate CA 19-9.44 As a result, CA 19-9 is not an ideal screening test. However, data has shown that CA 19-9 may have prognostic value postoperatively and serve as a marker for therapeutic response.45,46
Is biopsy needed for this patient and if so, what is the most appropriate technique?
ENDOSCOPIC ULTRASOUND
Generally, diagnosis with tissue is not necessary for patients who clearly have resectable disease and will proceed directly to surgery for management. Nevertheless, it is still commonly obtained in this group of patients. However, in patients with LAPC or with features suggestive of LAPC, such as tumor approximation to critical vessels such as the superior mesenteric artery (SMA) or celiac axis, biopsy is necessary. These patients will receive neoadjuvant therapy, and biopsy is important in establishing a diagnosis. The ideal way to obtain a biopsy is through fine-needle aspiration (FNA) or biopsy (FNB) utilizing endoscopic ultrasound (EUS). Percutaneous and computed tomography (CT)–guided FNB can also be used to obtain a biopsy for diagnosis. In comparison to percutaneous and CT-guided FNB, EUS-FNA/FNB has low rates of complications, a decreased rate of peritoneal seeding, and is cost effective.47,48
CASE CONTINUED
Abdominal CT obtained following abdominal ultrasound reveals a 3.5-cm mass in the head of the pancreas in close approximation to the SMA and celiac axis.
Does the patient have borderline resectable or unresectable disease?
IMAGING
Abdominal ultrasound is a reasonable, inexpensive, and safe alternative to abdominal CT as it does not utilize ionizing radiation. It is particularly useful in patients who present with jaundice or have concern for biliary obstruction based on laboratory evaluation. It is particularly sensitive for detecting tumors greater than 3 cm in size.49,50 In patients whose abdominal ultrasound is unrevealing and whose index of suspicion remains high for PDA, abdominal CT should be the next imaging modality.
Abdominal CT obtained utilizing a pancreatic protocol is ideal for detection and staging of pancreatic tumors. By implementing a triple-phase protocol with arterial, late arterial, and venous phases, tumors, which have a density different from that of the pancreatic parenchyma, are accentuated. Abdominal CT is also able to provide critical information about tumor resectability.51 By revealing the degree of tumor encasement around the aorta, level of destruction of the superior mesenteric vein, or degree of involvement of the SMA or celiac vessels, abdominal CT determines if a patient should be deemed resectable, borderline resectable, or unresectable (Table 2).52,53 Resectability is based on thorough imaging evaluation, expert opinion of a multidisciplinary team, and guidelines proposed by American Hepatopancreaticobiliary Association, Society of Surgical Oncology, Society for Surgery of the Alimentary Tract, and the NCCN.54
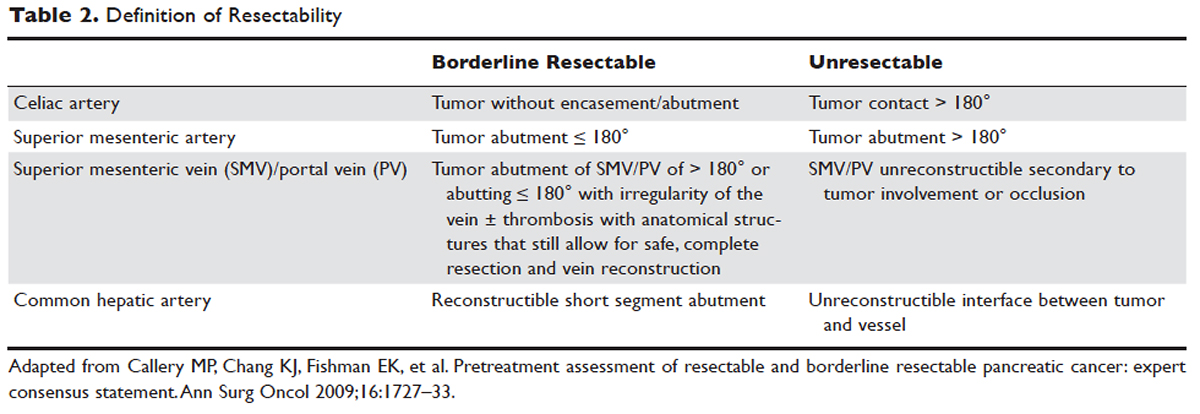
Other imaging modalities have a less clearly established role in the diagnostic approach to PDA. In patients who have contraindications to obtaining CT, magnetic resonance imaging can be utilized as a secondary imaging modality.55 The role of positron emission tomography 18F-fluorodeoxyglucose (PET-FDG) is not clearly defined among clinicians, nor reflected in consensus guidelines by the National Comprehensive Cancer Network (NCCN). In clinical practice, it is still often combined with CT to detect metastatic disease, particularly in high-risk patients such has those with LAPC. The role of PET-CT in staging and its impact on clinical outcomes has not been fully established.
Endoscopic retrograde cholangiopancreatography (ERCP) and magnetic resonance cholangiopancreatography (MRCP) can also assist in the diagnosis and management of PDA. In patients with obstructive jaundice, both MRCP and ERCP visualize obstructions and dilations within the biliary tree, with the latter having the ability to intervene. ERCP allows for the collection of tissue to aid in diagnosis, and has the ability to relieve biliary obstruction via stenting.56
TREATMENT
CASE CONTINUED
After an abdominal CT is obtained, the patient is referred to an outpatient oncologist because of concern for pancreatic adenocarcinoma. After consultation, the patient is advised to obtain EUS with biopsy and to return immediately afterwards for further treatment planning. The pathology report following EUS confirms that the mass is a poorly differentiated PDA. The patient’s case is discussed at a multidisciplinary meeting with radiation, surgical, and medical oncology. The abdominal CT and PET-CT scan are thoroughly reviewed. After imaging review, the multidisciplinary team concludes that the tumor is in contact with the SMA at 120° and with the common hepatic artery without extension in the celiac axis and without evidence of metastasis.
What is the appropriate management of borderline resectable pancreatic cancer?
BORDERLINE RESECTABLE CANCER
Patients who have nonmetastatic disease and are deemed resectable and without contraindications to surgery or high-risk features, as defined by NCCN guidelines, should proceed directly to surgery. A large body of evidence suggests that complete surgical resection with negative margins is a significant predictor of survival and currently provides the only option for cure.57–59 Despite the curative intent of surgery, the rate of recurrence remains high in patients who undergo surgical resection. Even in patients with negative resection margins (R0 resection), the 5-year survival is 20% to 30%, with a median survival ranging from 12 to 25 months, suggesting the presence of regional and distant occult disease at the time of diagnosis.60–62
Additionally, in half the patients who undergo surgical resection with resultant positive microscopic (R1 resection) or gross (R2 resection) margins, the median survival is no greater than 12 months. In this subset of patients, clinical outcomes are similar to outcomes in patients with locally advanced and metastatic pancreatic cancer, suggesting that upfront surgery and adjuvant therapy may not be the ideal therapeutic option. This raises 2 important points: first, resectability should be assessed carefully in all patients with LAPC, and second, for those patients who are deemed borderline resectable, neoadjuvant therapy should be considered.63 Borderline resectability is defined as tumor abutment ≤ 180° of the celiac artery, and tumor abutment of the superior mesenteric vein /portal vein of > 180° or abutting ≤ 180° with irregularity of the vein with or without thrombosis with anatomical structures that still allows for safe and complete resection and vein reconstruction (Table 2).
Neoadjuvant Therapy
The goal of neoadjuvant therapy is to minimize the negative impact of upfront surgery in patients who have a high likelihood of having microscopic or grossly positive margins. Research has suggested that neoadjuvant therapy may improve resectability, decrease the rate of recurrence, and improve overall survival.64–66
There is no clear consensus on the ideal management of patients with borderline resectable disease. However, expert guidelines are in agreement that upfront surgery in patients with LAPC is not appropriate, as most patients will not be able to achieve an R0 resection.67 As staging and management of patients with LAPC is difficult, expertise of a multidisciplinary team can be helpful.68
Several studies and the NCCN guidelines support the use of neoadjuvant therapy in patients deemed borderline resectable.69,70 Treatment of borderline resectable disease is similar to unresectable LAPC and generally involves 2 chemotherapy treatment backbones: FOLFIRINOX (folinic acid [leucovorin], fluorouracil [5-FU], irinotecan, and oxaliplatin) or gemcitabine-based therapy.
Phase 1 to 2/3 clinical trials conducted by Conroy et al from 2005 to 2011, including the landmark ACCORD-11 trial, established the safety and role of FOLFIRINOX in metastatic pancreatic cancer and also demonstrated an improved overall survival with the use of this therapy in these patients.71,72 These findings led to interest in FOLFIRINOX as a neoadjuvant therapy for patients with LAPC. Since then, multiple prospective and retrospective studies have shown that 54% to 100% of patients with borderline resectable LAPC who were treated with FOLFIRINOX were down-staged significantly enough to undergo resection. Of those patients, more than 90% had a R0 resection following surgery (Table 3).73–79
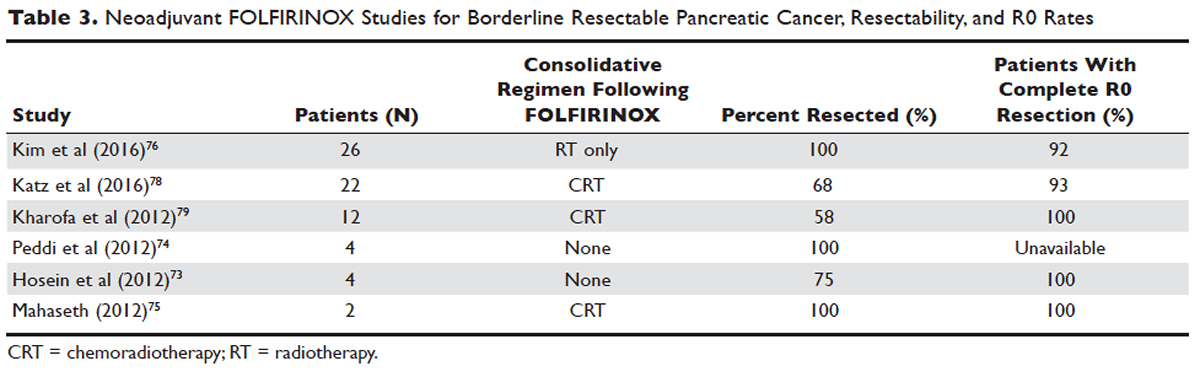
Data over the past 7 years suggests that neoadjuvant FOLFIRINOX improves overall survival and resectability in patients with borderline disease. However, treatment with FOLFIRINOX is not without limitations. FOLFIRINOX is associated with higher rates of febrile neutropenia, thrombocytopenia, diarrhea, and sensory neuropathy as compared with gemcitabine-based therapy.72 Other less commonly observed toxicities associated with FOLFIRINOX include mucositis, hand-foot syndrome, pulmonary toxicity, and alopecia. Dose-attenuated FOLFIRINOX-based regimens, including those that exclude the bolus fluorouracil dose and augment upfront filgrastim, have demonstrated improved safety and comparable efficacy as compared to standard FOLFIRINOX.80
Gemcitabine has been the fundamental treatment backbone for PDA since the results of the phase 3 CONKO-001 trial were published.81 Gemcitabine is a pyrimidine antimetabolite and potent inhibitor of DNA polymerase and ribonucleotide reductase.82 In recent years, multiple combination therapies with gemcitabine have been investigated, including regimens with nab-paclitaxel, oxaliplatin, or docetaxel. Resection rates and negative margin outcomes have been shown to be comparable to patients who received FOLFIRINOX in the neoadjuvant setting with borderline locally advanced disease.83–85 In addition to having a more tolerable side effect profile in comparison to fluorouracil-based regimens, gemcitabine is considered to be a potent radiosensitizer.86 For this reason, studies have also investigated the role of radiotherapy in conjunction with gemcitabine, revealing negative margin resection rates above 80% in patients with borderline resectable disease.87,88
Because very few studies directly comparing FOLFIRINOX with gemcitabine-based combination regimens have been completed, there is no clear consensus on the preferred treatment regimen, in both borderline and unresectable LAPC. Decisions to treat are influenced predominantly by comorbidities, adverse effect profiles, and performance status of patients, as FOLFIRINOX is the more toxic of the 2 treatment backbones. Therefore, FOLFIRINOX has mostly been utilized in patients with relatively good functional status (Eastern Cooperative Oncology Group [ECOG] performance status 0 to 1).89 In elderly patients and those with poor functional status, ECOG 2 to 4, gemcitabine as a single agent is a reasonable alternative in the neoadjuvant setting of borderline resectable disease.
The exact role of radiation therapy in addition to induction chemotherapy in borderline resectable pancreatic cancer has not been clearly established because of the lack of prospective studies in this area. Multiple large retrospective series have identified high rates of conversion to margin-negative resection with neoadjuvant chemoradiation alone.90 Based on available data, it is reasonable for patients with borderline resectable disease to proceed with any of the following treatment options: chemotherapy, chemoradiation, or induction chemotherapy followed by chemoradiation (Figure). Chemotherapy and chemoradiation are generally more appropriate with patients with high CA 19-9 levels or those at an elevated risk of having positive margins or occult metastatic disease.91 Obtaining negative margin resections is the predominant goal of neoadjuvant radiotherapy.89 Many studies have identified margin status to be one of the most significant prognostic factors in PDA.57,59,92,93 Additionally, several studies have highlighted that radiotherapy in the neoadjuvant setting could improve negative margin resection rates, local control, and clinical outcomes in patients with borderline resectable locally advanced disease.94–97 A common multimodal regimen utilized in the neoadjuvant setting combines capecitabine, an oral prodrug that is converted to fluorouracil, with radiation therapy. This combination has also been shown to improve resectability rates and long-term clinical outcomes in patients with borderline resectable disease.98 Additionally, neoadjuvant radiation therapy can potentially downstage patients with unresectable disease at the time of diagnosis to become surgical candidates.99 Despite the paucity of data, interval scans utilizing CT following neoadjuvant therapy should be obtained 2 to 4 months after completion of therapy to determine therapeutic response, evaluate for disease progression, and, most important, reassess surgical stage/resectability. It is clinically acceptable to proceed to resection with radiographically stable disease post-neoadjuvant therapy.

Many patients classified as borderline resectable are able to proceed with surgery following neoadjuvant therapy. Unfortunately, specific data on adjuvant therapy following neoadjuvant chemotherapy or chemoradiotherapy and surgical resection in borderline resectable patients is scarce. Clinical practice guidelines are extrapolated from studies where upfront resection in clearly resectable patients was followed by adjuvant therapy. Based on these data, approximately 6 months of perioperative chemotherapy with or without chemoradiotherapy is a reasonable consideration. Nevertheless, about 80% of patients at the time of diagnosis are deemed to be unresectable, and a smaller number do not proceed to surgery despite an initial classification as borderline resectable. Of the 80% of patients with advanced disease, about half are metastatic at presentation and the remaining 30% to 40% are defined as having unresectable LAPC.100
CASE CONTINUED
The patient is deemed borderline resectable. He receives neoadjuvant therapy with gemcitabine and nab-paclitaxel. Two months after therapy, interval imaging with abdominal CT does not show improvement in tumor size and there is now evidence that the tumor has invaded the celiac axis and is abutting more than 180° of the SMA. The patient presents to the oncologist to discuss further management. He has lost about 15 lb since his last evaluation, is capable of self-care, but is unable to carry on with any work activities.
What is the appropriate management of unresectable nonmetastatic LAPC?
UNRESECTABLE LOCALLY ADVANCED CANCER
As in the case of borderline resectable disease, there are many treatment options for patients with unresectable LAPC. Timing, optimal chemotherapy regimen, and the addition of regularly and hypofractionated radiotherapy are issues currently under investigation. However, there are some general considerations and principles that are followed as reflected in the NCCN guidelines and recent studies. The primary therapeutic aims in patients with unresectable locally advanced disease are to increase survival and improve palliation.
The elderly comprise a large percentage of the patients diagnosed with unresectable locally advanced disease. Pharmacokinetics and toxicity profiles are altered in the elderly population.101,102 Therefore, it is important to assess functional status and comorbidities as these are critical factors in determining treatment regimens, similar to patients with borderline resectable disease. Currently, the most common first-line therapies in advanced pancreatic cancer are gemcitabine alone, gemcitabine and nab-paclitaxel, FOLFIRINOX, gemcitabine/capecitabine, and gemcitabine/oxaliplatin.103 The overall treatment approach to unresectable locally advanced pancreatic adenocarcinoma closely mirrors that of patients with borderline resectable disease and metastatic disease. Much of the data supporting treatment regimens in unresectable LAPC is extrapolated from clinical trials looking at advanced or metastatic pancreatic cancer.
Consensus opinions domestically and from Europe recommend that patients with locally advanced unresectable disease undergo upfront chemotherapy (Figure).104 This is based on the premise that initial chemotherapy may destroy occult metastatic cells and increase the efficacy of consolidative chemotherapy, particularly with radiation in the future. Upfront chemoradiotherapy has only been investigated in a small series of trials in which no clear survival benefit was observed and has the added consequence of treatment-related toxicity.105 However, data is limited in this regard, with variations in treatment protocols and cohort compositions contributing to the inconclusive findings.
Despite advances in immunotherapy, targeted therapies, and gene sequencing, initial chemotherapy for unresectable disease is still either gemcitabine-based combination therapy or FOLFIRINOX. Across numerous studies, patients with unresectable LAPC receiving FOLFIRINOX have a median progression-free survival of 3 to 20 months and a median overall survival of 10 to 32.7 months.106 As with borderline resectable patients, FOLFIRINOX (Table 4) is generally reserved for unresectable patients with good functional status (ECOG 0–1 or Karnofsky Performance Status 90–100) and those at low risk for developing grade 3 or 4 systemic toxicities.103 For these reasons it has generally not been frequently combined with other chemotherapeutic agents. However, FOLFIRINOX has been combined with radiation therapy in the consolidative neoadjuvant setting after induction chemotherapy. There have also been studies where traditional FOLFIRONIX was modified to improve tolerability, as evidenced by Gunturu et al’s study, which dose-reduced both fluorouracil and irinotecan by 25%, without compromising efficacy and simultaneously increasing tolerability.107 Additionally, FOLFIRINOX requires infusional administration of the fluorouracil component, which may not be practical in certain patients. In that subset, capecitabine can be substituted. Among radiosensitizers during neoadjuvant therapy for unresectable LAPC, capecitabine has been shown to be as efficacious and less toxic than even gemcitabine.108
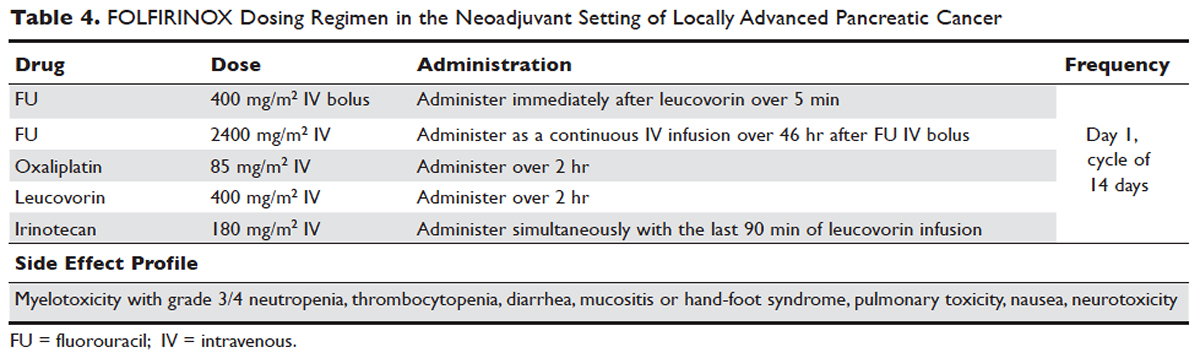

No head-to-head studies investigating FOLFIRINOX versus nab-paclitaxel and gemcitabine in patients with locally advanced disease have been published, but clinical trials are under way. Other combination therapies have been looked at through small retrospective or prospective studies, but no robust, large-scale clinical trials have been completed. For this reason, NCCN guidelines recommend enrollment of patients with LAPC into active clinical trials.
What is the role of radiation therapy in unresectable LAPC?
Despite the reported advantages of neoadjuvant radiation in patients with potentially resectable disease, there is significant debate regarding the timing and role of neoadjuvant radiation in patients with unresectable disease. Numerous comprehensive analyses and studiest indicate that chemoradiotherapy leads to significantly better overall survival compared to no therapy or radiation therapy alone in LAPC.68,110,111 However, conflicting data support the use of upfront chemoradiotherapy in unresectable LAPC when compared to chemotherapy alone. Unfortunately, most prospective studies investigating the role of radiotherapy were performed following administration of single-agent gemcitabine, which is no longer considered standard of care for patients with LAPC. In spite of this, ECOG 4201 identified a statistically significant improvement in median overall survival following the addition of gemcitabine-based radiotherapy. Huguet et al in his review pointed out that upfront chemoradiotherapy was not superior to chemotherapy only and was associated with increased treatment toxicity.105 Additionally, a recent phase 3 study looking at chemoradiotherapy versus chemotherapy alone in patients treated with gemcitabine found no difference in overall survival.112 This can potentially be attributed to the fact that about 30% of patients with LAPC develop metastatic disease in the early phases of treatment due to poor control of local and systemically occult disease.113 Given the propensity for high rates of occult metastatic disease in LAPC, treatment paradigms and consensus guidelines recommend multi-agent systemic chemotherapy followed by chemoradiotherapy in select patients.
Based on current studies and until further clinical investigations are completed, consensus opinion indicates that the most appropriate approach in unresectable LAPC is to begin with induction chemotherapy (with either gemcitabine plus nab-paclitaxel, FOLFIRINOX, capecitabine, or other treatment combinations), followed by chemoradiation in the absence of disease progression when the first repeat imaging evaluation is completed (Figure). One important caveat regarding reimaging with CT in the neoadjuvant setting is that radiologic response may not correlate with pathologic response.114 PET-CT may have a role in predicting response to neoadjuvant therapy. Overall, induction chemotherapy followed by consolidative chemoradiation may confer numerous benefits: it removes the unnecessary burden and toxicity associated with radiotherapy in the nearly one third of patients who have pervasive disease progression during initial treatment; it allows testing and increases the chances of tolerating full-dose systemic chemotherapy; and it raises the likelihood of converting patients who do not progress to metastasis during the initial phase of treatment from unresectable to resectable status.103,115 Despite the lack of strong conclusive data, the general agreement is that neoadjuvant chemoradiotherapy converts about one third of borderline and unresectable LAPC to an R0 resection.95,103 There are very specific rationales for the addition of radiotherapy in LAPC, and these functions need to be better defined through further clinical trials.
PALLIATIVE CARE
CASE CONTINUED
The patient is unable to tolerate his first round of second-line therapy with modified FOLFIRINOX. His overall treatment plan is transitioned to palliation. He continues to have pain, despite increasing doses of narcotics.
What is the next step for patients in whom second-line therapy fails and who have intractable pain while on high-dose narcotics?
A subset of patients with unresectable LAPC may not be amenable to chemotherapy with or without radiation due to significant comorbidities or because they present with or progress to ECOG scores 3 or 4. The goal in these patients should be palliation. Pain is one of the most predominant and difficult to manage symptoms in progressive LAPC. Opioid-based medications are the primary treatment for pain in LAPC. However, patients sometimes become refractory to opioid medications. In this group of patients, it is reasonable to consider palliative radiation as an alternative method for pain control.116
An alternative to palliative radiation in the setting of progressive pain in PDA is celiac plexus block or neurolysis. By injecting an anesthetic or alcohol into the celiac plexus, neural signaling pathways involved in the propagation of pain are inhibited without leading to significant nerve destruction. Additionally, chemical splanchnicectomy allows for reduced opioid medication use and associated side effects.117
In general patients with LAPC have profound weight loss prior to and during treatment. This has significant implications prognostically and on treatment options. The underlying etiology is multifactorial, but one of the primary driving factors is pancreatic insufficiency. An estimated 65% of pancreatic cancer patients have fat malabsorption, and 50% have protein malabsorption, leading to steatorrhea and weight loss.118 Patients diagnosed with pancreatic cancer should be given enzyme replacement with formulations that include lipase, amylase, and protease. A minimum dose of enzyme replacement should include 40,000 to 50,000 U of lipase during meals and 25,000 U during snack intake. If maldigestion, symptoms, or nutritional endpoints (BMI, albumin, prealbumin, cholesterol) do not improve, the pancreatic enzyme dose should be escalated and a proton-pump inhibitor (PPI) added. In patients with pancreatic insufficiency, PPIs have been shown to improve fat absorption.119 Enzyme replacement therapy has been shown to prevent weight loss in patients with unresectable pancreatic cancer.120
As most patients with LAPC go on to develop progressive disease, palliative care becomes an integral aspect of the therapeutic paradigm. Palliation in LAPC has a significant role in determining quality of life and ensuring patient’s goals of care have been meet. Studies have suggested that pancreatic cancer is second only to lung cancer in terms of the number of emergency department visits in the later stages of disease.120 Additionally, aggressive care in the setting of incurable diseases such as LAPC has been associated with poor quality of life.121 More recently it has been shown that involvement of palliative care in patients with advanced pancreatic is associated with less aggressive care near death.122 Therefore, the incorporation of palliative or supportive care teams in the treatment of patients with progressive LAPC can improve quality of life and alleviate suffering associated with increasing symptom burden.
CONCLUSION
LAPC is a difficult disease for both provider and patient. There is a paucity of robust clinical trials in the neoadjuvant setting for LAPC. Current research is complicated by varying consensus definitions of resectability and the varying treatment configurations across studies. The optimal type, timing, and sequence of treatment and whether to add radiation therapy in LAPC have not been clearly defined. However, based on the available studies and consensus guidelines, patients who are deemed to have LAPC should have neoadjuvant therapy. FOLFIRINOX or gemcitabine with nab-paclitaxel should be considered first-line treatments. Patients with LAPC who respond to chemotherapy or are ineligible for multi-drug chemotherapy may benefit from chemoradiotherapy. In patients with unresectable disease, chemoradiotherapy has been shown to enhance survival as compared to best supportive care or radiation alone. For borderline resectable disease, it is reasonable to treat patients with either chemoradiotherapy, chemotherapy alone, or chemotherapy followed by chemoradiotherapy.
Considering the invasive nature of LAPC and the controversy around neoadjuvant treatment protocols, enrollment of patients with LAPC into clinical trials is important and will help determine the optimal treatment regimen for future patients.
- Network PCA. Pancreatic cancer facts 2016. 2016. https://www.pancan.org/wp-content/uploads/2016/02/2016-GAA-PC-Facts.pdf. Accessed April 24, 2017.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Konstantinidis IT, Warshaw AL, Allen JN, et al. Pancreatic ductal adenocarcinoma: is there a survival difference for R1 resections versus locally advanced unresectable tumors? What is a “true” R0 resection? Ann Surg 2013;257:731–6.
- Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol 2016;22:9694–705.
- Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69–90.
- Zhang J, Dhakal I, Ning B, Kesteloot H. Patterns and trends of pancreatic cancer mortality rates in Arkansas, 1969-2002: a comparison with the US population. Eur J Cancer Prev 2008;17:18–27.
- National Cancer Institute. SEER cancer statistics review, 1975-2013. http://seer.cancer.gov/csr/1975_2013/. Accessed April 24, 2017.
- Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol 2006;20:197–209.
- Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med 1996;156:2255–60.
- Lucenteforte E, La Vecchia C, Silverman D, et al. Alcohol consumption and pancreatic cancer: a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann Oncol 2012;23:374–82.
- Schernhammer ES, Kang JH, Chan AT, et al. A prospective study of aspirin use and the risk of pancreatic cancer in women. J Natl Cancer Inst 2004;96:22–28.
- Michaud DS, Giovannucci E, Willett WC, et al. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001;286:921–9.
- Nothlings U, Wilkens LR, Murphy SP, et al. Meat and fat intake as risk factors for pancreatic cancer: the multiethnic cohort study. J Natl Cancer Inst 2005;97:1458–65.
- Chari ST, Leibson CL, Rabe KG, et al. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology 2005;129:504–11.
- Batabyal P, Vander Hoorn S, Christophi C, Nikfarjam M. Association of diabetes mellitus and pancreatic adenocarcinoma: a meta-analysis of 88 studies. Ann Surg Oncol 2014;21:2453–62.
- Chari ST, Leibson CL, Rabe KG, et al. Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology 2008;134:95–101.
- Pannala R, Basu A, Petersen GM, Chari ST. New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol 2009;10:88–95.
- Wolpin BM, Chan AT, Hartge P, et al. ABO blood group and the risk of pancreatic cancer. J Natl Cancer Inst 2009;101:424–31.
- Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
- Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000;119:1447–53.
- Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA 2009;302:1790–5.
- Brand RE, Lynch HT. Hereditary pancreatic adenocarcinoma. A clinical perspective. Med Clin North Am 2000;84:665–75.
- Jacobs EJ, Chanock SJ, Fuchs CS, et al. Family history of cancer and risk of pancreatic cancer: a pooled analysis from the Pancreatic Cancer Cohort Consortium (PanScan). Int J Cancer 2010;127:1421–8.
- Rustgi AK. Familial pancreatic cancer: genetic advances. Genes Dev 2014;28:1–7.
- Reznik R, Hendifar AE, Tuli R. Genetic determinants and potential therapeutic targets for pancreatic adenocarcinoma. Front Physiol 2014;5:87.
- Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst 1997;89:442–6.
- Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014;371:2140–1.
- Kanda M, Matthaei H, Wu J, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012;142:730–3.
- Feldmann G, Maitra A. Molecular genetics of pancreatic ductal adenocarcinomas and recent implications for translational efforts. J Mol Diagn 2008;10:111–22.
- Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 2014;111:817–22.
- Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic KRAS maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656–70.
- Provenzano PP, Cuevas C, Chang AE, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:418–29.
- Vonderheide RH, Bayne LJ. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr Opin Immunol 2013;25:200–5.
- DiMagno EP. Pancreatic cancer: Clinical presentation, pitfalls and early clues. Ann Oncol 1999;10(suppl 4):S140–S142.
- Porta M, Fabregat X, Malats N, et al. Exocrine pancreatic cancer: symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol 2005;7:189–97.
- Gullo L, Tomassetti P, Migliori M, et al. Do early symptoms of pancreatic cancer exist that can allow an earlier diagnosis? Pancreas 2001;22:210–3.
- Khorana AA, Fine RL. Pancreatic cancer and thromboembolic disease. Lancet Oncol 2004;5(11):655-663.
- Wigmore SJ, Plester CE, Richardson RA, Fearon KC. Changes in nutritional status associated with unresectable pancreatic cancer. Br J Cancer 1997;75:106–9.
- Aggarwal G, Rabe KG, Petersen GM, Chari ST. New-onset diabetes in pancreatic cancer: a study in the primary care setting. Pancreatology 2012;12:156–61.
- Koprowski H, Herlyn M, Steplewski Z, Sears HF. Specific antigen in serum of patients with colon carcinoma. Science 1981;212:53–5.
- Bond-Smith G, Banga N, Hammond TM, Imber CJ. Pancreatic adenocarcinoma. BMJ 2012;344:e2476.
- Cwik G, Wallner G, Skoczylas T, et al. Cancer antigens 19-9 and 125 in the differential diagnosis of pancreatic mass lesions. Arch Surg 2006;141:968–73.
- van den Bosch RP, van Eijck CH, Mulder PG, Jeekel J. Serum CA19-9 determination in the management of pancreatic cancer. Hepatogastroenterology 1996;43:710–3.
- Lamerz R. Role of tumour markers, cytogenetics. Ann Oncol 1999;10 Suppl 4:145–9.
- Hess V, Glimelius B, Grawe P, et al. CA 19-9 tumour-marker response to chemotherapy in patients with advanced pancreatic cancer enrolled in a randomised controlled trial. Lancet Oncol 2008;9:132–8.
- Montgomery RC, Hoffman JP, Riley LB, et al. Prediction of recurrence and survival by post-resection CA 19-9 values in patients with adenocarcinoma of the pancreas. Ann Surg Oncol 1997;4:551–6.
- Zamboni GA, D’Onofrio M, Idili A, et al. Ultrasound-guided percutaneous fine-needle aspiration of 545 focal pancreatic lesions. AJR Am J Roentgenol 2009;193:1691–5.
- Micames C, Jowell PS, White R, et al. Lower frequency of peritoneal carcinomatosis in patients with pancreatic cancer diagnosed by EUS-guided FNA vs. percutaneous FNA. Gastrointest Endosc 2003;58:690–5.
- Brambs HJ, Claussen CD. Pancreatic and ampullary carcinoma. Ultrasound, computed tomography, magnetic resonance imaging and angiography. Endoscopy 1993;25:58–68.
- Karlson BM, Ekbom A, Lindgren PG, et al. Abdominal US for diagnosis of pancreatic tumor: prospective cohort analysis. Radiology 1999;213:107–11.
- Imbriaco M, Megibow AJ, Camera L, et al. Dual-phase versus single-phase helical CT to detect and assess resectability of pancreatic carcinoma. AJR Am J Roentgenol 2002;178:1473–9.
- Schrag D. Optimizing treatment for locally advanced pancreas cancer: progress but no precision. JAMA 2016;315:1837–8.
- House MG, Yeo CJ, Cameron JL, et al. Predicting resectability of periampullary cancer with three-dimensional computed tomography. J Gastrointest Surg 2004;8:280–8.
- Callery MP, Chang KJ, Fishman EK, et al. Pretreatment assessment of resectable and borderline resectable pancreatic cancer: expert consensus statement. Ann Surg Oncol 2009;16:1727–33.
- Horton KM, Fishman EK. Adenocarcinoma of the pancreas: CT imaging. Radiol Clin North Am 2002;40:1263–72.
- Ross WA, Wasan SM, Evans DB, et al. Combined EUS with FNA and ERCP for the evaluation of patients with obstructive jaundice from presumed pancreatic malignancy. Gastrointest Endosc 2008;68:461–6.
- Hartwig W, Hackert T, Hinz U, et al. Pancreatic cancer surgery in the new millennium: better prediction of outcome. Ann Surg 2011;254:311–9.
- Jamieson NB, Chan NI, Foulis AK, et al. The prognostic influence of resection margin clearance following pancreaticoduodenectomy for pancreatic ductal adenocarcinoma. J Gastrointest Surg 2013;17:511–21.
- Ethun CG, Kooby DA. The importance of surgical margins in pancreatic cancer. J Surg Oncol 2016;113:283–8.
- Fischer R, Breidert M, Keck T, et al. Early recurrence of pancreatic cancer after resection and during adjuvant chemotherapy. Saudi J Gastroenterol 2012;18:118–21.
- Neoptolemos JP, Stocken DD, Friess H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med 2004;350:1200–10.
- Neoptolemos JP, Palmer DH, Ghaneh P, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389(10073):1011–24.
- Cardenes HR, Chiorean EG, Dewitt J, et al. Locally advanced pancreatic cancer: current therapeutic approach. Oncologist 2006;11:612–23.
- Yeung RS, Weese JL, Hoffman JP, et al. Neoadjuvant chemoradiation in pancreatic and duodenal carcinoma. A Phase II Study. Cancer 1993;72:2124–33.
- Spitz FR, Abbruzzese JL, Lee JE, et al. Preoperative and postoperative chemoradiation strategies in patients treated with pancreaticoduodenectomy for adenocarcinoma of the pancreas. J Clin Oncol 1997;15:928–37.
- McClaine RJ, Lowy AM, Sussman JJ, et al. Neoadjuvant therapy may lead to successful surgical resection and improved survival in patients with borderline resectable pancreatic cancer. HPB (Oxford) 2010;12:73–9.
- Balaban EP, Mangu PB, Khorana AA, et al. Locally advanced, unresectable pancreatic cCancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2016;34:2654–68.
- Shaib WL, Ip A, Cardona K, et al. Contemporary management of borderline resectable and locally advanced unresectable pancreatic cancer. Oncologist 2016;21:178–87.
- Chun YS, Milestone BN, Watson JC, et al. Defining venous involvement in borderline resectable pancreatic cancer. Ann Surg Oncol 2010;17:2832–8.
- Evans DB, Erickson BA, Ritch P. Borderline resectable pancreatic cancer: definitions and the importance of multimodality therapy. Ann Surg Oncol 2010;17:2803–5.
- Conroy T, Paillot B, Francois E, et al. Irinotecan plus oxaliplatin and leucovorin-modulated fluorouracil in advanced pancreatic cancer--a Groupe Tumeurs Digestives of the Federation Nationale des Centres de Lutte Contre le Cancer study. J Clin Oncol 2005;23:1228–36.
- Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
- Hosein PJ, Macintyre J, Kawamura C, et al. A retrospective study of neoadjuvant FOLFIRINOX in unresectable or borderline-resectable locally advanced pancreatic adenocarcinoma. BMC Cancer 2012;12:199.
- Peddi PF, Lubner S, McWilliams R, et al. Multi-institutional experience with FOLFIRINOX in pancreatic adenocarcinoma. JOP 2012;13:497–501.
- Mahaseth H, Kauh JS, Brutcher E, et al. Safety and efficacy of modified FOLFIRINOX in pancreatic cancer: A retrospective experience. J Clin Oncol 2012;30 (suppl; abstr e14614).
- Kim SS, Nakakura EK, Wang ZJ, et al. Preoperative FOLFIRINOX for borderline resectable pancreatic cancer: Is radiation necessary in the modern era of chemotherapy? J Surg Oncol 2016;114:587–96.
- Conroy T, Gavoille C, Samalin E, et al. The role of the FOLFIRINOX regimen for advanced pancreatic cancer. Curr Oncol Rep 2013;15:182–9.
- Katz MH, Shi Q, Ahmad SA, et al. Preoperative modified FOLFIRINOX treatment followed by capecitabine-based chemoradiation for borderline resectable pancreatic cancer: Alliance for Clinical Trials in Oncology Trial A021101. JAMA Surg 2016;151:e161137.
- Kharofa J, Kelly TR, Ritch PS, et al. 5-FU/leucovorin, irinotecan, oxaliplatin (FOLFIRINOX) induction followed by chemoXRT in borderline resectable pancreatic cancer. J Clin Oncol 2012;30 (suppl; abstr e14613).
- Blazer M, Wu C, Goldberg RM, et al. Neoadjuvant modified (m) FOLFIRINOX for locally advanced unresectable (LAPC) and borderline resectable (BRPC) adenocarcinoma of the pancreas. Ann Surg Oncol 2015;22:1153–9.
- Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
- Plunkett W, Huang P, Xu YZ, et al. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol 1995;22(4 Suppl 11):3–10.
- Sahora K, Kuehrer I, Eisenhut A, et al. NeoGemOx: Gemcitabine and oxaliplatin as neoadjuvant treatment for locally advanced, nonmetastasized pancreatic cancer. Surgery 2011;149(3):311–20.
- Lee JL, Kim SC, Kim JH, et al. Prospective efficacy and safety study of neoadjuvant gemcitabine with capecitabine combination chemotherapy for borderline-resectable or unresectable locally advanced pancreatic adenocarcinoma. Surgery 2012;152:851–62.
- Leone F, Gatti M, Massucco P, et al. Induction gemcitabine and oxaliplatin therapy followed by a twice-weekly infusion of gemcitabine and concurrent external-beam radiation for neoadjuvant treatment of locally advanced pancreatic cancer: a single institutional experience. Cancer 2013;119:277–84.
- Lawrence TS, Eisbruch A, Shewach DS. Gemcitabine-mediated radiosensitization. Semin Oncol 1997;24(2 Suppl 7):S7–24-S27–28.
- Kang CM, Chung YE, Park JY, et al. Potential contribution of preoperative neoadjuvant concurrent chemoradiation therapy on margin-negative resection in borderline resectable pancreatic cancer. J Gastrointest Surg 2012;16:509–17.
- Chuong MD, Hayman TJ, Patel MR, et al. Comparison of 1-, 2-, and 3-dimensional tumor response assessment after neoadjuvant GTX-RT in borderline-resectable pancreatic cancer. Gastrointest Cancer Res 2011;4:128–34.
- Loehrer AP, Kinnier CV, Ferrone CR. Treatment of locally advanced pancreatic ductal adenocarcinoma. Adv Surg 2016;50:115–28.
- Katz MH, Wang H, Balachandran A, et al. Effect of neoadjuvant chemoradiation and surgical technique on recurrence of localized pancreatic cancer. J Gastrointest Surg 2012;16:68–78.
- Franke AJ, Rosati LM, Pawlik TM, et al. The role of radiation therapy in pancreatic ductal adenocarcinoma in the neoadjuvant and adjuvant settings. Semin Oncol 2015;42:144–62.
- Butturini G, Stocken DD, Wente MN, et al. Influence of resection margins and treatment on survival in patients with pancreatic cancer: meta-analysis of randomized controlled trials. Arch Surg 2008;143:75–83.
- Paniccia A, Hosokawa P, Henderson W, et al. Characteristics of 10-year survivors of pancreatic ductal adenocarcinoma. JAMA Surg 2015;150:701–10.
- Massucco P, Capussotti L, Magnino A, et al. Pancreatic resections after chemoradiotherapy for locally advanced ductal adenocarcinoma: analysis of perioperative outcome and survival. Ann Surg Oncol 2006;13:1201–8.
- Patel M, Hoffe S, Malafa M, et al. Neoadjuvant GTX chemotherapy and IMRT-based chemoradiation for borderline resectable pancreatic cancer. J Surg Oncol 2011;104:155–161.
- Landry J, Catalano PJ, Staley C, et al. Randomized phase II study of gemcitabine plus radiotherapy versus gemcitabine, 5-fluorouracil, and cisplatin followed by radiotherapy and 5-fluorouracil for patients with locally advanced, potentially resectable pancreatic adenocarcinoma. J Surg Oncol 2010;101:587–92.
- Lamb R, Ozsvari B, Lisanti CL, et al. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget 2015;6:4569–84.
- Stokes JB, Nolan NJ, Stelow EB, et al. Preoperative capecitabine and concurrent radiation for borderline resectable pancreatic cancer. Ann Surg Oncol 2011;18:619–27.
- White R, Lee C, Anscher M, et al. Preoperative chemoradiation for patients with locally advanced adenocarcinoma of the pancreas. Ann Surg Oncol 1999;6:38–45.
- Martin RC 2nd. Management of locally advanced pancreatic cancer. Surg Clin North Am 2016;96:1371–89.
- Higuera O, Ghanem I, Nasimi R, et al. Management of pancreatic cancer in the elderly. World J Gastroenterol 2016;22:764–75.
- Hurria A, Lichtman SM. Clinical pharmacology of cancer therapies in older adults. Br J Cancer 2008;98:517–22.
- Spadi R, Brusa F, Ponzetti A, et al. Current therapeutic strategies for advanced pancreatic cancer: A review for clinicians. World J Clin Oncol 2016;7:27–43.
- Seufferlein T, Bachet JB, Van Cutsem E, Rougier P, Group EGW. Pancreatic adenocarcinoma: ESMO-ESDO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012;23 Suppl 7:vii33–40.
- Huguet F, Girard N, Guerche CS, et al. Chemoradiotherapy in the management of locally advanced pancreatic carcinoma: a qualitative systematic review. J Clin Oncol 2009;27:2269–77.
- Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol 2016;17:801–10.
- Gunturu KS, Yao X, Cong X, et al. FOLFIRINOX for locally advanced and metastatic pancreatic cancer: single institution retrospective review of efficacy and toxicity. Med Oncol 2013;30:361.
- Mukherjee S, Hurt CN, Bridgewater J, et al. Gemcitabine-based or capecitabine-based chemoradiotherapy for locally advanced pancreatic cancer (SCALOP): a multicentre, randomised, phase 2 trial. Lancet Oncol 2013;14:317–26.
- Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
- Krzyzanowska MK, Weeks JC, Earle CC. Treatment of locally advanced pancreatic cancer in the real world: population-based practices and effectiveness. J Clin Oncol 2003;21:3409–14.
- Sultana A, Tudur Smith C, Cunningham D, et al. Meta-analyses of chemotherapy for locally advanced and metastatic pancreatic cancer: results of secondary end points analyses. Br J Cancer 2008;99:6–13.
- Hammel P, Huguet F, van Laethem JL, et al. Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: the LAP07 randomized clinical trial. JAMA 2016;315:1844–53.
- Huguet F, Andre T, Hammel P, et al. Impact of chemoradiotherapy after disease control with chemotherapy in locally advanced pancreatic adenocarcinoma in GERCOR phase II and III studies. J Clin Oncol 2007;25:326–31.
- Dholakia AS, Hacker-Prietz A, Wild AT, et al. Resection of borderline resectable pancreatic cancer after neoadjuvant chemoradiation does not depend on improved radiographic appearance of tumor-vessel relationships. J Radiat Oncol 2013;2:413–25.
- Heinemann V, Haas M, Boeck S. Neoadjuvant treatment of borderline resectable and non-resectable pancreatic cancer. Ann Oncol 2013;24:2484–92.
- Morganti AG, Trodella L, Valentini V, et al. Pain relief with short-term irradiation in locally advanced carcinoma of the pancreas. J Palliat Care 2003;19:258–62.
- Arcidiacono PG, Calori G, Carrara S, et al. Celiac plexus block for pancreatic cancer pain in adults. Cochrane Database Syst Rev 2011(3):CD007519.
- Pezzilli R, Andriulli A, Bassi C, et al. Exocrine pancreatic insufficiency in adults: a shared position statement of the Italian Association for the Study of the Pancreas. World J Gastroenterol 2013;19:7930–46.
- Dominguez-Munoz JE. Pancreatic exocrine insufficiency: diagnosis and treatment. J Gastroenterol Hepatol 2011;26 Suppl 2:12–16.
- Bruno MJ, Haverkort EB, Tijssen GP, et al. Placebo controlled trial of enteric coated pancreatin microsphere treatment in patients with unresectable cancer of the pancreatic head region. Gut 1998;42:92–6.
- Wright AA, Keating NL, Balboni TA, et al. Place of death: correlations with quality of life of patients with cancer and predictors of bereaved caregivers’ mental health. J Clin Oncol 2010;28:4457–64.
- Jang RW, Krzyzanowska MK, Zimmermann C, et al. Palliative care and the aggressiveness of end-of-life care in patients with advanced pancreatic cancer. J Natl Cancer Inst 2015;107(3). pii: dju424.
INTRODUCTION
Pancreatic cancer is one of the most rapidly rising causes of mortality in the United States. In 2016, the number of deaths from pancreatic cancer exceeded those from breast cancer, making it the third leading cause of cancer-related death in the United States.1 It is projected that by 2020 pancreatic cancer will overtake colorectal malignancies to become the second most common cause of cancer death in this country.1,2 The term pancreatic cancer encompasses both exocrine and endocrine tumors. However, since 80% of pancreatic cancers are classified as pancreatic ductal adenocarcinoma (PDA), when speaking about pancreatic cancer most clinicians and scientists are referring to PDA.
Even with advances in chemotherapy and radiotherapy over the past decade, the only curative option for PDA is surgical resection. Unfortunately, only 20% of patients are appropriate surgical candidates at the time of diagnosis.3 Considering the lack of screening options and the ambiguity of symptomatology, roughly 4 out 5 patients with PDA are diagnosed as having locally advanced or metastatic disease that is initially not amenable to surgery.
Locally advanced pancreatic adenocarcinoma presents unique challenges in management and treatment. Treatment options include multi-agent chemotherapy, chemoradiation, or radiotherapy. Some patients can be successfully down-staged with these therapies and be deemed surgical candidates. Other challenges include selecting the appropriate sequence of therapies and stratifying therapies based on comorbidities. In this article, we review the epidemiology, biology, and diagnostic approach to PDA and focus on current treatment strategies for locally advanced pancreatic cancer (LAPC).
EPIDEMIOLOGY
In 2012, GLOBOCAN estimated that PDA caused 331,000 deaths per year, accounting for 4% of all worldwide mortality.4,5 Despite high incidence rates internationally, PDA is a disease of Western and industrialized nations. In the Unites States, PDA is a malignancy of middle to late adulthood, with a sharp upsurge in incidence after age 50 years.6 More than one third of new cases are diagnosed in patients older than 70 years, and more than half of patients diagnosed are older than 60 years of age.2 The incidence of pancreatic cancer is fairly equal among men and women, with a slightly higher rate for the male sex. It has an incidence preference for African-Americans by 4.8 cases per 100,000 persons nationally.7
Risk factors for the development of exocrine pancreatic cancer include hereditary disposition, underlying medical conditions, and environmental factors. One of the most significant environmental risk factors for the development of PDA is smoking,8 which is associated with up to 25% of all cases.9 Smoking cessation leads to a rapid reduction in risk for pancreatic cancer, with the risk among former smokers approaching that for never smokers less than 10 years after quitting.9 Other environmental factors that contribute to the development of pancreatic cancer include increased body mass index, a high-salt and high-saturated fat diet, heavy alcohol intake, and increased utilization of nonsteroidal inflammatory drugs.10–13
There is a strong association between new-onset diabetes and increased risk for developing PDA.14,15 Data also suggest that diabetes may be a risk factor and/or a consequence of tissue destruction that arises during the development or progression of PDA.16,17 Interestingly, ABO blood grouping is another underlying medical disposition that confers an altered risk profile. Studies have shown that patients with blood group O were less likely than those with type A, B, or AB to develop pancreatic cancer.18
Genetic predisposition syndromes can elevate an individual patient’s risk for developing PDA. Genetic syndromes and gene alterations that increase the risk for PDA include BRCA1/2, Peutz-Jeghers syndrome, and Lynch syndrome risk.19–21 Up to 10% to 15% of PDA cases may be due to an inherited familial cancer.22 Having a first-degree relative with PDA increases the odds of developing PDA 1.76-fold compared to those without a family history.23 The exact biologic and molecular mechanisms of familial pancreatic cancer are unclear. It is estimated that about 10% of patients with familial pancreatic cancer (FPC) carry BRCA2 mutations.24 Individuals at risk for FPC should undergo genetic screening for the presence of the most frequently inherited pancreatic cancer susceptibility genetic defects: BRCA2, PALB2, and ATM germline mutations.25 Carriers of BRCA2, who are also at increased risk for developing breast, ovarian, and prostate cancer, should be monitored closely. Of all hereditary conditions, hereditary pancreatitis confers the highest risk for developing PDA, with an approximate risk elevation of 40% to 50%.26,27 Although several genetic predisposition syndromes have been identified, most cases of pancreatic adenocarcinoma are thought to be sporadic.
CANCER BIOLOGY AND PATHOLOGY
The pathologic predecessor of PDA is pancreatic intraepithelial neoplasia (PIN). With further dysplastic changes that result from increasing genetic alterations, these precancerous lesions progress from low- to high-grade and finally to adenocarcinoma. More than 90% of all PINs across all grades have oncogenic KRAS mutations.28 Additionally, inactivating mutations in the tumor suppressor genes SMAD4, p53, and CDKN2A are found with increasing frequency in higher grade PINs. The frequency and presence of mutations in both oncogenes and tumor suppressor genes in precursor neoplasias mirror the genetic mutations noted in advanced PDA.29 Among all mutations, KRAS is the most common and most functionally important for pancreatic cancer cell survival. KRAS mutations not only have profound effects on downstream mediators of tumor growth and metastasis, but they are implicated in reprograming of cellular metabolism.30,31
Pancreatic adenocarcinoma has a unique microenvironment that makes it a difficult target for current therapeutic modalities. First, it is one of the most stroma-rich malignancies. The dense stroma surrounding pancreatic tumor cells leads to increased tumor pressures and alterations in tumor vascular perfusion.32 It also serves as a barrier that prevents chemotherapeutic drugs from reaching the tumor cells. Thus, clinical trials are under way to investigate agents such has hyaluronidase, which may degrade components of the extracellular matrix that supports thestromal environment. Additionally, there is data to suggest that the microenvironment of PDA downregulates immune monitoring, leading to further tumor growth.27,33 The molecular, cellular, and immunologic complexity of PDA may contribute to its resistance to traditional therapeutics.
EVALUATION AND DIAGNOSIS
CASE PRESENTATION
A 61-year-old man with a history of type 2 diabetes mellitus and chronic tobacco use presents to the emergency department (ED) with a 4-month history of progressively worsening abdominal discomfort and fatigue. He has also noticed darkening of his urine and slight yellow discoloration of his eyes. His weight measured 5 months ago in his primary care physician’s office was 91 kg (200 lb, BMI 29.5) and in the ED is 75 kg (165 lb, BMI 24.4). He has noticed bulky, malodorous, oily stools for about 2 months. Preliminary laboratory studies reveal elevated levels of total bilirubin (2.7 mg/dL) and alkaline phosphatase (204 IU/L). Transabdominal ultrasound (US) is obtained and reveals a 3-cm pancreatic mass with biliary tract dilation.
Does this patient have pancreatic cancer?
CLINICAL SIGNS AND SYMPTOMS
Establishing the diagnosis of pancreatic cancer in a patient who presents with a high index of suspicion is critical. Patients with pancreatic cancer usually present after a period of nonspecific and vague symptoms, which typically are experienced as abdominal discomfort, weight loss, and weakness. It is estimated that approximately 25% of patients may complain of vague abdominal pain up to 6 months prior to diagnosis. Up to 15% of patients may seek medical attention more than 6 months prior to establishing a diagnosis of PDA.34 The most common symptoms associated with pancreatic cancer in order of decreasing reported frequency are weight loss, anorexia, abdominal/epigastric pain, dark-colored urine, jaundice, nausea, back pain, and diarrhea with associated steatorrhea.35 Upwards of 15% of patients present with painless jaundice, a term that is often associated with pancreatic cancer.36 On exam these patients may have scleral icterus, sublingual jaundice, epigastric pain on palpation, weight loss, hepatomegaly, lymphadenopathy and a nontender, distended, palpable gallbladder (also known as Courvoisier sign).34 Abdominal signs and symptoms arise from tumor growth into surrounding vessels, tissues, and ducts within the abdominal cavity. Compression of the common bile duct accounts for the development of jaundice. Tumor growth around the stomach and duodenum can lead to delayed gastric emptying and subsequently nausea and vomiting. Constriction of the pancreatic duct leads to pancreatic insufficiency, precipitation of weight loss, and steatorrhea. Pancreatic insufficiency can worsen abdominal pain, and lead to increased weight loss and flatulence.
Less common symptoms include pain, erythema, and edema involving the lower extremities, which may be reflective of migratory thrombophlebitis (commonly known as Trousseau syndrome). Thromboembolic disease, including pulmonary embolism, portal vein, and deep vein thromboses are frequently encountered complications of pancreatic cancer. The incidence of thromboembolic events in patients with PDA has been reported to be as high as 54%.37 Of all signs encountered, weight loss is the most common and most profound. Patients with advanced PDA have severe degrees of cachexia. Some patients present with as much as a 5 kg/m2 decrease in their BMI from pre-illness baseline BMI, and lose another 3 to 4 kg/m2 through disease progression.38 At the time of diagnosis, many patients have already undergone significant weight loss, which can have substantial implications on treatment planning and clinical outcomes.
What other studies can be done to assist in making the diagnosis?
LABORATORY ABNORMALITIES AND TUMOR MARKERS
Elevations in alkaline phosphatase, γ-glutamyltransferase (GGT), serum aspartate aminotransferase (AST), serum alanine aminotransferase (ALT), and direct fractions of bilirubin are common in patients with PDA. Patients will usually have an obstructive pattern on their liver panel, with predominant elevations in direct bilirubin, alkaline phosphatase, and GGT, as compared with AST and ALT. Other baseline laboratory studies, including a complete blood count and basic metabolic panel, should be obtained because patients commonly have thrombocytosis, anemia, and electrolyte abnormalities due to the tumor itself and pancreatic insufficiency (Table 1).
Measurement of glycated hemoglobin (HBA1C) is an emerging and important diagnostic test in the diagnosis of pancreatic cancer. Recently, data has emerged to suggest that new-onset diabetes is present in about 50% of patients diagnosed with pancreatic cancer.39 The temporal relationship of pancreatic cancer and diabetes is supported by evidence showing that patients who undergo resection commonly have resolution of their diabetes.17 This study suggested that hyperglycemia, elevated HBA1C, and symptoms of diabetes in patients older than 50 years may identify patients who have early pancreatic cancer. The entity of pancreatic cancer–associated diabetes needs to be better defined and the algorithmic approach to evaluation and diagnosis, utilizing signs, symptoms, and laboratory values associated with diabetes, needs to be clearly established.
The only serum marker for PDA is carbohydrate antigen 19-9 (CA 19-9), also known as sialylated Lewis antigen or cancer-associated antigen. It was first identified in pancreatic cancer patients in 1981.40,41 The sensitivity and specificity of CA 19-9 ranges from 70% to approximately 90%.42,43 Hereditary predispositions and comorbid disease cross-reactivity contribute to the diminished sensitivity and specificity of CA 19-9. In about 5% to 10% of the population, CA 19-9 is not expressed (Lewis antigen A and B negative). Additionally, since CA 19-9 is expressed in the cells that line the biliary tree, diseases that lead to pancreatic or liver inflammation may falsely elevate CA 19-9.44 As a result, CA 19-9 is not an ideal screening test. However, data has shown that CA 19-9 may have prognostic value postoperatively and serve as a marker for therapeutic response.45,46
Is biopsy needed for this patient and if so, what is the most appropriate technique?
ENDOSCOPIC ULTRASOUND
Generally, diagnosis with tissue is not necessary for patients who clearly have resectable disease and will proceed directly to surgery for management. Nevertheless, it is still commonly obtained in this group of patients. However, in patients with LAPC or with features suggestive of LAPC, such as tumor approximation to critical vessels such as the superior mesenteric artery (SMA) or celiac axis, biopsy is necessary. These patients will receive neoadjuvant therapy, and biopsy is important in establishing a diagnosis. The ideal way to obtain a biopsy is through fine-needle aspiration (FNA) or biopsy (FNB) utilizing endoscopic ultrasound (EUS). Percutaneous and computed tomography (CT)–guided FNB can also be used to obtain a biopsy for diagnosis. In comparison to percutaneous and CT-guided FNB, EUS-FNA/FNB has low rates of complications, a decreased rate of peritoneal seeding, and is cost effective.47,48
CASE CONTINUED
Abdominal CT obtained following abdominal ultrasound reveals a 3.5-cm mass in the head of the pancreas in close approximation to the SMA and celiac axis.
Does the patient have borderline resectable or unresectable disease?
IMAGING
Abdominal ultrasound is a reasonable, inexpensive, and safe alternative to abdominal CT as it does not utilize ionizing radiation. It is particularly useful in patients who present with jaundice or have concern for biliary obstruction based on laboratory evaluation. It is particularly sensitive for detecting tumors greater than 3 cm in size.49,50 In patients whose abdominal ultrasound is unrevealing and whose index of suspicion remains high for PDA, abdominal CT should be the next imaging modality.
Abdominal CT obtained utilizing a pancreatic protocol is ideal for detection and staging of pancreatic tumors. By implementing a triple-phase protocol with arterial, late arterial, and venous phases, tumors, which have a density different from that of the pancreatic parenchyma, are accentuated. Abdominal CT is also able to provide critical information about tumor resectability.51 By revealing the degree of tumor encasement around the aorta, level of destruction of the superior mesenteric vein, or degree of involvement of the SMA or celiac vessels, abdominal CT determines if a patient should be deemed resectable, borderline resectable, or unresectable (Table 2).52,53 Resectability is based on thorough imaging evaluation, expert opinion of a multidisciplinary team, and guidelines proposed by American Hepatopancreaticobiliary Association, Society of Surgical Oncology, Society for Surgery of the Alimentary Tract, and the NCCN.54
Other imaging modalities have a less clearly established role in the diagnostic approach to PDA. In patients who have contraindications to obtaining CT, magnetic resonance imaging can be utilized as a secondary imaging modality.55 The role of positron emission tomography 18F-fluorodeoxyglucose (PET-FDG) is not clearly defined among clinicians, nor reflected in consensus guidelines by the National Comprehensive Cancer Network (NCCN). In clinical practice, it is still often combined with CT to detect metastatic disease, particularly in high-risk patients such has those with LAPC. The role of PET-CT in staging and its impact on clinical outcomes has not been fully established.
Endoscopic retrograde cholangiopancreatography (ERCP) and magnetic resonance cholangiopancreatography (MRCP) can also assist in the diagnosis and management of PDA. In patients with obstructive jaundice, both MRCP and ERCP visualize obstructions and dilations within the biliary tree, with the latter having the ability to intervene. ERCP allows for the collection of tissue to aid in diagnosis, and has the ability to relieve biliary obstruction via stenting.56
TREATMENT
CASE CONTINUED
After an abdominal CT is obtained, the patient is referred to an outpatient oncologist because of concern for pancreatic adenocarcinoma. After consultation, the patient is advised to obtain EUS with biopsy and to return immediately afterwards for further treatment planning. The pathology report following EUS confirms that the mass is a poorly differentiated PDA. The patient’s case is discussed at a multidisciplinary meeting with radiation, surgical, and medical oncology. The abdominal CT and PET-CT scan are thoroughly reviewed. After imaging review, the multidisciplinary team concludes that the tumor is in contact with the SMA at 120° and with the common hepatic artery without extension in the celiac axis and without evidence of metastasis.
What is the appropriate management of borderline resectable pancreatic cancer?
BORDERLINE RESECTABLE CANCER
Patients who have nonmetastatic disease and are deemed resectable and without contraindications to surgery or high-risk features, as defined by NCCN guidelines, should proceed directly to surgery. A large body of evidence suggests that complete surgical resection with negative margins is a significant predictor of survival and currently provides the only option for cure.57–59 Despite the curative intent of surgery, the rate of recurrence remains high in patients who undergo surgical resection. Even in patients with negative resection margins (R0 resection), the 5-year survival is 20% to 30%, with a median survival ranging from 12 to 25 months, suggesting the presence of regional and distant occult disease at the time of diagnosis.60–62
Additionally, in half the patients who undergo surgical resection with resultant positive microscopic (R1 resection) or gross (R2 resection) margins, the median survival is no greater than 12 months. In this subset of patients, clinical outcomes are similar to outcomes in patients with locally advanced and metastatic pancreatic cancer, suggesting that upfront surgery and adjuvant therapy may not be the ideal therapeutic option. This raises 2 important points: first, resectability should be assessed carefully in all patients with LAPC, and second, for those patients who are deemed borderline resectable, neoadjuvant therapy should be considered.63 Borderline resectability is defined as tumor abutment ≤ 180° of the celiac artery, and tumor abutment of the superior mesenteric vein /portal vein of > 180° or abutting ≤ 180° with irregularity of the vein with or without thrombosis with anatomical structures that still allows for safe and complete resection and vein reconstruction (Table 2).
Neoadjuvant Therapy
The goal of neoadjuvant therapy is to minimize the negative impact of upfront surgery in patients who have a high likelihood of having microscopic or grossly positive margins. Research has suggested that neoadjuvant therapy may improve resectability, decrease the rate of recurrence, and improve overall survival.64–66
There is no clear consensus on the ideal management of patients with borderline resectable disease. However, expert guidelines are in agreement that upfront surgery in patients with LAPC is not appropriate, as most patients will not be able to achieve an R0 resection.67 As staging and management of patients with LAPC is difficult, expertise of a multidisciplinary team can be helpful.68
Several studies and the NCCN guidelines support the use of neoadjuvant therapy in patients deemed borderline resectable.69,70 Treatment of borderline resectable disease is similar to unresectable LAPC and generally involves 2 chemotherapy treatment backbones: FOLFIRINOX (folinic acid [leucovorin], fluorouracil [5-FU], irinotecan, and oxaliplatin) or gemcitabine-based therapy.
Phase 1 to 2/3 clinical trials conducted by Conroy et al from 2005 to 2011, including the landmark ACCORD-11 trial, established the safety and role of FOLFIRINOX in metastatic pancreatic cancer and also demonstrated an improved overall survival with the use of this therapy in these patients.71,72 These findings led to interest in FOLFIRINOX as a neoadjuvant therapy for patients with LAPC. Since then, multiple prospective and retrospective studies have shown that 54% to 100% of patients with borderline resectable LAPC who were treated with FOLFIRINOX were down-staged significantly enough to undergo resection. Of those patients, more than 90% had a R0 resection following surgery (Table 3).73–79
Data over the past 7 years suggests that neoadjuvant FOLFIRINOX improves overall survival and resectability in patients with borderline disease. However, treatment with FOLFIRINOX is not without limitations. FOLFIRINOX is associated with higher rates of febrile neutropenia, thrombocytopenia, diarrhea, and sensory neuropathy as compared with gemcitabine-based therapy.72 Other less commonly observed toxicities associated with FOLFIRINOX include mucositis, hand-foot syndrome, pulmonary toxicity, and alopecia. Dose-attenuated FOLFIRINOX-based regimens, including those that exclude the bolus fluorouracil dose and augment upfront filgrastim, have demonstrated improved safety and comparable efficacy as compared to standard FOLFIRINOX.80
Gemcitabine has been the fundamental treatment backbone for PDA since the results of the phase 3 CONKO-001 trial were published.81 Gemcitabine is a pyrimidine antimetabolite and potent inhibitor of DNA polymerase and ribonucleotide reductase.82 In recent years, multiple combination therapies with gemcitabine have been investigated, including regimens with nab-paclitaxel, oxaliplatin, or docetaxel. Resection rates and negative margin outcomes have been shown to be comparable to patients who received FOLFIRINOX in the neoadjuvant setting with borderline locally advanced disease.83–85 In addition to having a more tolerable side effect profile in comparison to fluorouracil-based regimens, gemcitabine is considered to be a potent radiosensitizer.86 For this reason, studies have also investigated the role of radiotherapy in conjunction with gemcitabine, revealing negative margin resection rates above 80% in patients with borderline resectable disease.87,88
Because very few studies directly comparing FOLFIRINOX with gemcitabine-based combination regimens have been completed, there is no clear consensus on the preferred treatment regimen, in both borderline and unresectable LAPC. Decisions to treat are influenced predominantly by comorbidities, adverse effect profiles, and performance status of patients, as FOLFIRINOX is the more toxic of the 2 treatment backbones. Therefore, FOLFIRINOX has mostly been utilized in patients with relatively good functional status (Eastern Cooperative Oncology Group [ECOG] performance status 0 to 1).89 In elderly patients and those with poor functional status, ECOG 2 to 4, gemcitabine as a single agent is a reasonable alternative in the neoadjuvant setting of borderline resectable disease.
The exact role of radiation therapy in addition to induction chemotherapy in borderline resectable pancreatic cancer has not been clearly established because of the lack of prospective studies in this area. Multiple large retrospective series have identified high rates of conversion to margin-negative resection with neoadjuvant chemoradiation alone.90 Based on available data, it is reasonable for patients with borderline resectable disease to proceed with any of the following treatment options: chemotherapy, chemoradiation, or induction chemotherapy followed by chemoradiation (Figure). Chemotherapy and chemoradiation are generally more appropriate with patients with high CA 19-9 levels or those at an elevated risk of having positive margins or occult metastatic disease.91 Obtaining negative margin resections is the predominant goal of neoadjuvant radiotherapy.89 Many studies have identified margin status to be one of the most significant prognostic factors in PDA.57,59,92,93 Additionally, several studies have highlighted that radiotherapy in the neoadjuvant setting could improve negative margin resection rates, local control, and clinical outcomes in patients with borderline resectable locally advanced disease.94–97 A common multimodal regimen utilized in the neoadjuvant setting combines capecitabine, an oral prodrug that is converted to fluorouracil, with radiation therapy. This combination has also been shown to improve resectability rates and long-term clinical outcomes in patients with borderline resectable disease.98 Additionally, neoadjuvant radiation therapy can potentially downstage patients with unresectable disease at the time of diagnosis to become surgical candidates.99 Despite the paucity of data, interval scans utilizing CT following neoadjuvant therapy should be obtained 2 to 4 months after completion of therapy to determine therapeutic response, evaluate for disease progression, and, most important, reassess surgical stage/resectability. It is clinically acceptable to proceed to resection with radiographically stable disease post-neoadjuvant therapy.
Many patients classified as borderline resectable are able to proceed with surgery following neoadjuvant therapy. Unfortunately, specific data on adjuvant therapy following neoadjuvant chemotherapy or chemoradiotherapy and surgical resection in borderline resectable patients is scarce. Clinical practice guidelines are extrapolated from studies where upfront resection in clearly resectable patients was followed by adjuvant therapy. Based on these data, approximately 6 months of perioperative chemotherapy with or without chemoradiotherapy is a reasonable consideration. Nevertheless, about 80% of patients at the time of diagnosis are deemed to be unresectable, and a smaller number do not proceed to surgery despite an initial classification as borderline resectable. Of the 80% of patients with advanced disease, about half are metastatic at presentation and the remaining 30% to 40% are defined as having unresectable LAPC.100
CASE CONTINUED
The patient is deemed borderline resectable. He receives neoadjuvant therapy with gemcitabine and nab-paclitaxel. Two months after therapy, interval imaging with abdominal CT does not show improvement in tumor size and there is now evidence that the tumor has invaded the celiac axis and is abutting more than 180° of the SMA. The patient presents to the oncologist to discuss further management. He has lost about 15 lb since his last evaluation, is capable of self-care, but is unable to carry on with any work activities.
What is the appropriate management of unresectable nonmetastatic LAPC?
UNRESECTABLE LOCALLY ADVANCED CANCER
As in the case of borderline resectable disease, there are many treatment options for patients with unresectable LAPC. Timing, optimal chemotherapy regimen, and the addition of regularly and hypofractionated radiotherapy are issues currently under investigation. However, there are some general considerations and principles that are followed as reflected in the NCCN guidelines and recent studies. The primary therapeutic aims in patients with unresectable locally advanced disease are to increase survival and improve palliation.
The elderly comprise a large percentage of the patients diagnosed with unresectable locally advanced disease. Pharmacokinetics and toxicity profiles are altered in the elderly population.101,102 Therefore, it is important to assess functional status and comorbidities as these are critical factors in determining treatment regimens, similar to patients with borderline resectable disease. Currently, the most common first-line therapies in advanced pancreatic cancer are gemcitabine alone, gemcitabine and nab-paclitaxel, FOLFIRINOX, gemcitabine/capecitabine, and gemcitabine/oxaliplatin.103 The overall treatment approach to unresectable locally advanced pancreatic adenocarcinoma closely mirrors that of patients with borderline resectable disease and metastatic disease. Much of the data supporting treatment regimens in unresectable LAPC is extrapolated from clinical trials looking at advanced or metastatic pancreatic cancer.
Consensus opinions domestically and from Europe recommend that patients with locally advanced unresectable disease undergo upfront chemotherapy (Figure).104 This is based on the premise that initial chemotherapy may destroy occult metastatic cells and increase the efficacy of consolidative chemotherapy, particularly with radiation in the future. Upfront chemoradiotherapy has only been investigated in a small series of trials in which no clear survival benefit was observed and has the added consequence of treatment-related toxicity.105 However, data is limited in this regard, with variations in treatment protocols and cohort compositions contributing to the inconclusive findings.
Despite advances in immunotherapy, targeted therapies, and gene sequencing, initial chemotherapy for unresectable disease is still either gemcitabine-based combination therapy or FOLFIRINOX. Across numerous studies, patients with unresectable LAPC receiving FOLFIRINOX have a median progression-free survival of 3 to 20 months and a median overall survival of 10 to 32.7 months.106 As with borderline resectable patients, FOLFIRINOX (Table 4) is generally reserved for unresectable patients with good functional status (ECOG 0–1 or Karnofsky Performance Status 90–100) and those at low risk for developing grade 3 or 4 systemic toxicities.103 For these reasons it has generally not been frequently combined with other chemotherapeutic agents. However, FOLFIRINOX has been combined with radiation therapy in the consolidative neoadjuvant setting after induction chemotherapy. There have also been studies where traditional FOLFIRONIX was modified to improve tolerability, as evidenced by Gunturu et al’s study, which dose-reduced both fluorouracil and irinotecan by 25%, without compromising efficacy and simultaneously increasing tolerability.107 Additionally, FOLFIRINOX requires infusional administration of the fluorouracil component, which may not be practical in certain patients. In that subset, capecitabine can be substituted. Among radiosensitizers during neoadjuvant therapy for unresectable LAPC, capecitabine has been shown to be as efficacious and less toxic than even gemcitabine.108
No head-to-head studies investigating FOLFIRINOX versus nab-paclitaxel and gemcitabine in patients with locally advanced disease have been published, but clinical trials are under way. Other combination therapies have been looked at through small retrospective or prospective studies, but no robust, large-scale clinical trials have been completed. For this reason, NCCN guidelines recommend enrollment of patients with LAPC into active clinical trials.
What is the role of radiation therapy in unresectable LAPC?
Despite the reported advantages of neoadjuvant radiation in patients with potentially resectable disease, there is significant debate regarding the timing and role of neoadjuvant radiation in patients with unresectable disease. Numerous comprehensive analyses and studiest indicate that chemoradiotherapy leads to significantly better overall survival compared to no therapy or radiation therapy alone in LAPC.68,110,111 However, conflicting data support the use of upfront chemoradiotherapy in unresectable LAPC when compared to chemotherapy alone. Unfortunately, most prospective studies investigating the role of radiotherapy were performed following administration of single-agent gemcitabine, which is no longer considered standard of care for patients with LAPC. In spite of this, ECOG 4201 identified a statistically significant improvement in median overall survival following the addition of gemcitabine-based radiotherapy. Huguet et al in his review pointed out that upfront chemoradiotherapy was not superior to chemotherapy only and was associated with increased treatment toxicity.105 Additionally, a recent phase 3 study looking at chemoradiotherapy versus chemotherapy alone in patients treated with gemcitabine found no difference in overall survival.112 This can potentially be attributed to the fact that about 30% of patients with LAPC develop metastatic disease in the early phases of treatment due to poor control of local and systemically occult disease.113 Given the propensity for high rates of occult metastatic disease in LAPC, treatment paradigms and consensus guidelines recommend multi-agent systemic chemotherapy followed by chemoradiotherapy in select patients.
Based on current studies and until further clinical investigations are completed, consensus opinion indicates that the most appropriate approach in unresectable LAPC is to begin with induction chemotherapy (with either gemcitabine plus nab-paclitaxel, FOLFIRINOX, capecitabine, or other treatment combinations), followed by chemoradiation in the absence of disease progression when the first repeat imaging evaluation is completed (Figure). One important caveat regarding reimaging with CT in the neoadjuvant setting is that radiologic response may not correlate with pathologic response.114 PET-CT may have a role in predicting response to neoadjuvant therapy. Overall, induction chemotherapy followed by consolidative chemoradiation may confer numerous benefits: it removes the unnecessary burden and toxicity associated with radiotherapy in the nearly one third of patients who have pervasive disease progression during initial treatment; it allows testing and increases the chances of tolerating full-dose systemic chemotherapy; and it raises the likelihood of converting patients who do not progress to metastasis during the initial phase of treatment from unresectable to resectable status.103,115 Despite the lack of strong conclusive data, the general agreement is that neoadjuvant chemoradiotherapy converts about one third of borderline and unresectable LAPC to an R0 resection.95,103 There are very specific rationales for the addition of radiotherapy in LAPC, and these functions need to be better defined through further clinical trials.
PALLIATIVE CARE
CASE CONTINUED
The patient is unable to tolerate his first round of second-line therapy with modified FOLFIRINOX. His overall treatment plan is transitioned to palliation. He continues to have pain, despite increasing doses of narcotics.
What is the next step for patients in whom second-line therapy fails and who have intractable pain while on high-dose narcotics?
A subset of patients with unresectable LAPC may not be amenable to chemotherapy with or without radiation due to significant comorbidities or because they present with or progress to ECOG scores 3 or 4. The goal in these patients should be palliation. Pain is one of the most predominant and difficult to manage symptoms in progressive LAPC. Opioid-based medications are the primary treatment for pain in LAPC. However, patients sometimes become refractory to opioid medications. In this group of patients, it is reasonable to consider palliative radiation as an alternative method for pain control.116
An alternative to palliative radiation in the setting of progressive pain in PDA is celiac plexus block or neurolysis. By injecting an anesthetic or alcohol into the celiac plexus, neural signaling pathways involved in the propagation of pain are inhibited without leading to significant nerve destruction. Additionally, chemical splanchnicectomy allows for reduced opioid medication use and associated side effects.117
In general patients with LAPC have profound weight loss prior to and during treatment. This has significant implications prognostically and on treatment options. The underlying etiology is multifactorial, but one of the primary driving factors is pancreatic insufficiency. An estimated 65% of pancreatic cancer patients have fat malabsorption, and 50% have protein malabsorption, leading to steatorrhea and weight loss.118 Patients diagnosed with pancreatic cancer should be given enzyme replacement with formulations that include lipase, amylase, and protease. A minimum dose of enzyme replacement should include 40,000 to 50,000 U of lipase during meals and 25,000 U during snack intake. If maldigestion, symptoms, or nutritional endpoints (BMI, albumin, prealbumin, cholesterol) do not improve, the pancreatic enzyme dose should be escalated and a proton-pump inhibitor (PPI) added. In patients with pancreatic insufficiency, PPIs have been shown to improve fat absorption.119 Enzyme replacement therapy has been shown to prevent weight loss in patients with unresectable pancreatic cancer.120
As most patients with LAPC go on to develop progressive disease, palliative care becomes an integral aspect of the therapeutic paradigm. Palliation in LAPC has a significant role in determining quality of life and ensuring patient’s goals of care have been meet. Studies have suggested that pancreatic cancer is second only to lung cancer in terms of the number of emergency department visits in the later stages of disease.120 Additionally, aggressive care in the setting of incurable diseases such as LAPC has been associated with poor quality of life.121 More recently it has been shown that involvement of palliative care in patients with advanced pancreatic is associated with less aggressive care near death.122 Therefore, the incorporation of palliative or supportive care teams in the treatment of patients with progressive LAPC can improve quality of life and alleviate suffering associated with increasing symptom burden.
CONCLUSION
LAPC is a difficult disease for both provider and patient. There is a paucity of robust clinical trials in the neoadjuvant setting for LAPC. Current research is complicated by varying consensus definitions of resectability and the varying treatment configurations across studies. The optimal type, timing, and sequence of treatment and whether to add radiation therapy in LAPC have not been clearly defined. However, based on the available studies and consensus guidelines, patients who are deemed to have LAPC should have neoadjuvant therapy. FOLFIRINOX or gemcitabine with nab-paclitaxel should be considered first-line treatments. Patients with LAPC who respond to chemotherapy or are ineligible for multi-drug chemotherapy may benefit from chemoradiotherapy. In patients with unresectable disease, chemoradiotherapy has been shown to enhance survival as compared to best supportive care or radiation alone. For borderline resectable disease, it is reasonable to treat patients with either chemoradiotherapy, chemotherapy alone, or chemotherapy followed by chemoradiotherapy.
Considering the invasive nature of LAPC and the controversy around neoadjuvant treatment protocols, enrollment of patients with LAPC into clinical trials is important and will help determine the optimal treatment regimen for future patients.
INTRODUCTION
Pancreatic cancer is one of the most rapidly rising causes of mortality in the United States. In 2016, the number of deaths from pancreatic cancer exceeded those from breast cancer, making it the third leading cause of cancer-related death in the United States.1 It is projected that by 2020 pancreatic cancer will overtake colorectal malignancies to become the second most common cause of cancer death in this country.1,2 The term pancreatic cancer encompasses both exocrine and endocrine tumors. However, since 80% of pancreatic cancers are classified as pancreatic ductal adenocarcinoma (PDA), when speaking about pancreatic cancer most clinicians and scientists are referring to PDA.
Even with advances in chemotherapy and radiotherapy over the past decade, the only curative option for PDA is surgical resection. Unfortunately, only 20% of patients are appropriate surgical candidates at the time of diagnosis.3 Considering the lack of screening options and the ambiguity of symptomatology, roughly 4 out 5 patients with PDA are diagnosed as having locally advanced or metastatic disease that is initially not amenable to surgery.
Locally advanced pancreatic adenocarcinoma presents unique challenges in management and treatment. Treatment options include multi-agent chemotherapy, chemoradiation, or radiotherapy. Some patients can be successfully down-staged with these therapies and be deemed surgical candidates. Other challenges include selecting the appropriate sequence of therapies and stratifying therapies based on comorbidities. In this article, we review the epidemiology, biology, and diagnostic approach to PDA and focus on current treatment strategies for locally advanced pancreatic cancer (LAPC).
EPIDEMIOLOGY
In 2012, GLOBOCAN estimated that PDA caused 331,000 deaths per year, accounting for 4% of all worldwide mortality.4,5 Despite high incidence rates internationally, PDA is a disease of Western and industrialized nations. In the Unites States, PDA is a malignancy of middle to late adulthood, with a sharp upsurge in incidence after age 50 years.6 More than one third of new cases are diagnosed in patients older than 70 years, and more than half of patients diagnosed are older than 60 years of age.2 The incidence of pancreatic cancer is fairly equal among men and women, with a slightly higher rate for the male sex. It has an incidence preference for African-Americans by 4.8 cases per 100,000 persons nationally.7
Risk factors for the development of exocrine pancreatic cancer include hereditary disposition, underlying medical conditions, and environmental factors. One of the most significant environmental risk factors for the development of PDA is smoking,8 which is associated with up to 25% of all cases.9 Smoking cessation leads to a rapid reduction in risk for pancreatic cancer, with the risk among former smokers approaching that for never smokers less than 10 years after quitting.9 Other environmental factors that contribute to the development of pancreatic cancer include increased body mass index, a high-salt and high-saturated fat diet, heavy alcohol intake, and increased utilization of nonsteroidal inflammatory drugs.10–13
There is a strong association between new-onset diabetes and increased risk for developing PDA.14,15 Data also suggest that diabetes may be a risk factor and/or a consequence of tissue destruction that arises during the development or progression of PDA.16,17 Interestingly, ABO blood grouping is another underlying medical disposition that confers an altered risk profile. Studies have shown that patients with blood group O were less likely than those with type A, B, or AB to develop pancreatic cancer.18
Genetic predisposition syndromes can elevate an individual patient’s risk for developing PDA. Genetic syndromes and gene alterations that increase the risk for PDA include BRCA1/2, Peutz-Jeghers syndrome, and Lynch syndrome risk.19–21 Up to 10% to 15% of PDA cases may be due to an inherited familial cancer.22 Having a first-degree relative with PDA increases the odds of developing PDA 1.76-fold compared to those without a family history.23 The exact biologic and molecular mechanisms of familial pancreatic cancer are unclear. It is estimated that about 10% of patients with familial pancreatic cancer (FPC) carry BRCA2 mutations.24 Individuals at risk for FPC should undergo genetic screening for the presence of the most frequently inherited pancreatic cancer susceptibility genetic defects: BRCA2, PALB2, and ATM germline mutations.25 Carriers of BRCA2, who are also at increased risk for developing breast, ovarian, and prostate cancer, should be monitored closely. Of all hereditary conditions, hereditary pancreatitis confers the highest risk for developing PDA, with an approximate risk elevation of 40% to 50%.26,27 Although several genetic predisposition syndromes have been identified, most cases of pancreatic adenocarcinoma are thought to be sporadic.
CANCER BIOLOGY AND PATHOLOGY
The pathologic predecessor of PDA is pancreatic intraepithelial neoplasia (PIN). With further dysplastic changes that result from increasing genetic alterations, these precancerous lesions progress from low- to high-grade and finally to adenocarcinoma. More than 90% of all PINs across all grades have oncogenic KRAS mutations.28 Additionally, inactivating mutations in the tumor suppressor genes SMAD4, p53, and CDKN2A are found with increasing frequency in higher grade PINs. The frequency and presence of mutations in both oncogenes and tumor suppressor genes in precursor neoplasias mirror the genetic mutations noted in advanced PDA.29 Among all mutations, KRAS is the most common and most functionally important for pancreatic cancer cell survival. KRAS mutations not only have profound effects on downstream mediators of tumor growth and metastasis, but they are implicated in reprograming of cellular metabolism.30,31
Pancreatic adenocarcinoma has a unique microenvironment that makes it a difficult target for current therapeutic modalities. First, it is one of the most stroma-rich malignancies. The dense stroma surrounding pancreatic tumor cells leads to increased tumor pressures and alterations in tumor vascular perfusion.32 It also serves as a barrier that prevents chemotherapeutic drugs from reaching the tumor cells. Thus, clinical trials are under way to investigate agents such has hyaluronidase, which may degrade components of the extracellular matrix that supports thestromal environment. Additionally, there is data to suggest that the microenvironment of PDA downregulates immune monitoring, leading to further tumor growth.27,33 The molecular, cellular, and immunologic complexity of PDA may contribute to its resistance to traditional therapeutics.
EVALUATION AND DIAGNOSIS
CASE PRESENTATION
A 61-year-old man with a history of type 2 diabetes mellitus and chronic tobacco use presents to the emergency department (ED) with a 4-month history of progressively worsening abdominal discomfort and fatigue. He has also noticed darkening of his urine and slight yellow discoloration of his eyes. His weight measured 5 months ago in his primary care physician’s office was 91 kg (200 lb, BMI 29.5) and in the ED is 75 kg (165 lb, BMI 24.4). He has noticed bulky, malodorous, oily stools for about 2 months. Preliminary laboratory studies reveal elevated levels of total bilirubin (2.7 mg/dL) and alkaline phosphatase (204 IU/L). Transabdominal ultrasound (US) is obtained and reveals a 3-cm pancreatic mass with biliary tract dilation.
Does this patient have pancreatic cancer?
CLINICAL SIGNS AND SYMPTOMS
Establishing the diagnosis of pancreatic cancer in a patient who presents with a high index of suspicion is critical. Patients with pancreatic cancer usually present after a period of nonspecific and vague symptoms, which typically are experienced as abdominal discomfort, weight loss, and weakness. It is estimated that approximately 25% of patients may complain of vague abdominal pain up to 6 months prior to diagnosis. Up to 15% of patients may seek medical attention more than 6 months prior to establishing a diagnosis of PDA.34 The most common symptoms associated with pancreatic cancer in order of decreasing reported frequency are weight loss, anorexia, abdominal/epigastric pain, dark-colored urine, jaundice, nausea, back pain, and diarrhea with associated steatorrhea.35 Upwards of 15% of patients present with painless jaundice, a term that is often associated with pancreatic cancer.36 On exam these patients may have scleral icterus, sublingual jaundice, epigastric pain on palpation, weight loss, hepatomegaly, lymphadenopathy and a nontender, distended, palpable gallbladder (also known as Courvoisier sign).34 Abdominal signs and symptoms arise from tumor growth into surrounding vessels, tissues, and ducts within the abdominal cavity. Compression of the common bile duct accounts for the development of jaundice. Tumor growth around the stomach and duodenum can lead to delayed gastric emptying and subsequently nausea and vomiting. Constriction of the pancreatic duct leads to pancreatic insufficiency, precipitation of weight loss, and steatorrhea. Pancreatic insufficiency can worsen abdominal pain, and lead to increased weight loss and flatulence.
Less common symptoms include pain, erythema, and edema involving the lower extremities, which may be reflective of migratory thrombophlebitis (commonly known as Trousseau syndrome). Thromboembolic disease, including pulmonary embolism, portal vein, and deep vein thromboses are frequently encountered complications of pancreatic cancer. The incidence of thromboembolic events in patients with PDA has been reported to be as high as 54%.37 Of all signs encountered, weight loss is the most common and most profound. Patients with advanced PDA have severe degrees of cachexia. Some patients present with as much as a 5 kg/m2 decrease in their BMI from pre-illness baseline BMI, and lose another 3 to 4 kg/m2 through disease progression.38 At the time of diagnosis, many patients have already undergone significant weight loss, which can have substantial implications on treatment planning and clinical outcomes.
What other studies can be done to assist in making the diagnosis?
LABORATORY ABNORMALITIES AND TUMOR MARKERS
Elevations in alkaline phosphatase, γ-glutamyltransferase (GGT), serum aspartate aminotransferase (AST), serum alanine aminotransferase (ALT), and direct fractions of bilirubin are common in patients with PDA. Patients will usually have an obstructive pattern on their liver panel, with predominant elevations in direct bilirubin, alkaline phosphatase, and GGT, as compared with AST and ALT. Other baseline laboratory studies, including a complete blood count and basic metabolic panel, should be obtained because patients commonly have thrombocytosis, anemia, and electrolyte abnormalities due to the tumor itself and pancreatic insufficiency (Table 1).
Measurement of glycated hemoglobin (HBA1C) is an emerging and important diagnostic test in the diagnosis of pancreatic cancer. Recently, data has emerged to suggest that new-onset diabetes is present in about 50% of patients diagnosed with pancreatic cancer.39 The temporal relationship of pancreatic cancer and diabetes is supported by evidence showing that patients who undergo resection commonly have resolution of their diabetes.17 This study suggested that hyperglycemia, elevated HBA1C, and symptoms of diabetes in patients older than 50 years may identify patients who have early pancreatic cancer. The entity of pancreatic cancer–associated diabetes needs to be better defined and the algorithmic approach to evaluation and diagnosis, utilizing signs, symptoms, and laboratory values associated with diabetes, needs to be clearly established.
The only serum marker for PDA is carbohydrate antigen 19-9 (CA 19-9), also known as sialylated Lewis antigen or cancer-associated antigen. It was first identified in pancreatic cancer patients in 1981.40,41 The sensitivity and specificity of CA 19-9 ranges from 70% to approximately 90%.42,43 Hereditary predispositions and comorbid disease cross-reactivity contribute to the diminished sensitivity and specificity of CA 19-9. In about 5% to 10% of the population, CA 19-9 is not expressed (Lewis antigen A and B negative). Additionally, since CA 19-9 is expressed in the cells that line the biliary tree, diseases that lead to pancreatic or liver inflammation may falsely elevate CA 19-9.44 As a result, CA 19-9 is not an ideal screening test. However, data has shown that CA 19-9 may have prognostic value postoperatively and serve as a marker for therapeutic response.45,46
Is biopsy needed for this patient and if so, what is the most appropriate technique?
ENDOSCOPIC ULTRASOUND
Generally, diagnosis with tissue is not necessary for patients who clearly have resectable disease and will proceed directly to surgery for management. Nevertheless, it is still commonly obtained in this group of patients. However, in patients with LAPC or with features suggestive of LAPC, such as tumor approximation to critical vessels such as the superior mesenteric artery (SMA) or celiac axis, biopsy is necessary. These patients will receive neoadjuvant therapy, and biopsy is important in establishing a diagnosis. The ideal way to obtain a biopsy is through fine-needle aspiration (FNA) or biopsy (FNB) utilizing endoscopic ultrasound (EUS). Percutaneous and computed tomography (CT)–guided FNB can also be used to obtain a biopsy for diagnosis. In comparison to percutaneous and CT-guided FNB, EUS-FNA/FNB has low rates of complications, a decreased rate of peritoneal seeding, and is cost effective.47,48
CASE CONTINUED
Abdominal CT obtained following abdominal ultrasound reveals a 3.5-cm mass in the head of the pancreas in close approximation to the SMA and celiac axis.
Does the patient have borderline resectable or unresectable disease?
IMAGING
Abdominal ultrasound is a reasonable, inexpensive, and safe alternative to abdominal CT as it does not utilize ionizing radiation. It is particularly useful in patients who present with jaundice or have concern for biliary obstruction based on laboratory evaluation. It is particularly sensitive for detecting tumors greater than 3 cm in size.49,50 In patients whose abdominal ultrasound is unrevealing and whose index of suspicion remains high for PDA, abdominal CT should be the next imaging modality.
Abdominal CT obtained utilizing a pancreatic protocol is ideal for detection and staging of pancreatic tumors. By implementing a triple-phase protocol with arterial, late arterial, and venous phases, tumors, which have a density different from that of the pancreatic parenchyma, are accentuated. Abdominal CT is also able to provide critical information about tumor resectability.51 By revealing the degree of tumor encasement around the aorta, level of destruction of the superior mesenteric vein, or degree of involvement of the SMA or celiac vessels, abdominal CT determines if a patient should be deemed resectable, borderline resectable, or unresectable (Table 2).52,53 Resectability is based on thorough imaging evaluation, expert opinion of a multidisciplinary team, and guidelines proposed by American Hepatopancreaticobiliary Association, Society of Surgical Oncology, Society for Surgery of the Alimentary Tract, and the NCCN.54
Other imaging modalities have a less clearly established role in the diagnostic approach to PDA. In patients who have contraindications to obtaining CT, magnetic resonance imaging can be utilized as a secondary imaging modality.55 The role of positron emission tomography 18F-fluorodeoxyglucose (PET-FDG) is not clearly defined among clinicians, nor reflected in consensus guidelines by the National Comprehensive Cancer Network (NCCN). In clinical practice, it is still often combined with CT to detect metastatic disease, particularly in high-risk patients such has those with LAPC. The role of PET-CT in staging and its impact on clinical outcomes has not been fully established.
Endoscopic retrograde cholangiopancreatography (ERCP) and magnetic resonance cholangiopancreatography (MRCP) can also assist in the diagnosis and management of PDA. In patients with obstructive jaundice, both MRCP and ERCP visualize obstructions and dilations within the biliary tree, with the latter having the ability to intervene. ERCP allows for the collection of tissue to aid in diagnosis, and has the ability to relieve biliary obstruction via stenting.56
TREATMENT
CASE CONTINUED
After an abdominal CT is obtained, the patient is referred to an outpatient oncologist because of concern for pancreatic adenocarcinoma. After consultation, the patient is advised to obtain EUS with biopsy and to return immediately afterwards for further treatment planning. The pathology report following EUS confirms that the mass is a poorly differentiated PDA. The patient’s case is discussed at a multidisciplinary meeting with radiation, surgical, and medical oncology. The abdominal CT and PET-CT scan are thoroughly reviewed. After imaging review, the multidisciplinary team concludes that the tumor is in contact with the SMA at 120° and with the common hepatic artery without extension in the celiac axis and without evidence of metastasis.
What is the appropriate management of borderline resectable pancreatic cancer?
BORDERLINE RESECTABLE CANCER
Patients who have nonmetastatic disease and are deemed resectable and without contraindications to surgery or high-risk features, as defined by NCCN guidelines, should proceed directly to surgery. A large body of evidence suggests that complete surgical resection with negative margins is a significant predictor of survival and currently provides the only option for cure.57–59 Despite the curative intent of surgery, the rate of recurrence remains high in patients who undergo surgical resection. Even in patients with negative resection margins (R0 resection), the 5-year survival is 20% to 30%, with a median survival ranging from 12 to 25 months, suggesting the presence of regional and distant occult disease at the time of diagnosis.60–62
Additionally, in half the patients who undergo surgical resection with resultant positive microscopic (R1 resection) or gross (R2 resection) margins, the median survival is no greater than 12 months. In this subset of patients, clinical outcomes are similar to outcomes in patients with locally advanced and metastatic pancreatic cancer, suggesting that upfront surgery and adjuvant therapy may not be the ideal therapeutic option. This raises 2 important points: first, resectability should be assessed carefully in all patients with LAPC, and second, for those patients who are deemed borderline resectable, neoadjuvant therapy should be considered.63 Borderline resectability is defined as tumor abutment ≤ 180° of the celiac artery, and tumor abutment of the superior mesenteric vein /portal vein of > 180° or abutting ≤ 180° with irregularity of the vein with or without thrombosis with anatomical structures that still allows for safe and complete resection and vein reconstruction (Table 2).
Neoadjuvant Therapy
The goal of neoadjuvant therapy is to minimize the negative impact of upfront surgery in patients who have a high likelihood of having microscopic or grossly positive margins. Research has suggested that neoadjuvant therapy may improve resectability, decrease the rate of recurrence, and improve overall survival.64–66
There is no clear consensus on the ideal management of patients with borderline resectable disease. However, expert guidelines are in agreement that upfront surgery in patients with LAPC is not appropriate, as most patients will not be able to achieve an R0 resection.67 As staging and management of patients with LAPC is difficult, expertise of a multidisciplinary team can be helpful.68
Several studies and the NCCN guidelines support the use of neoadjuvant therapy in patients deemed borderline resectable.69,70 Treatment of borderline resectable disease is similar to unresectable LAPC and generally involves 2 chemotherapy treatment backbones: FOLFIRINOX (folinic acid [leucovorin], fluorouracil [5-FU], irinotecan, and oxaliplatin) or gemcitabine-based therapy.
Phase 1 to 2/3 clinical trials conducted by Conroy et al from 2005 to 2011, including the landmark ACCORD-11 trial, established the safety and role of FOLFIRINOX in metastatic pancreatic cancer and also demonstrated an improved overall survival with the use of this therapy in these patients.71,72 These findings led to interest in FOLFIRINOX as a neoadjuvant therapy for patients with LAPC. Since then, multiple prospective and retrospective studies have shown that 54% to 100% of patients with borderline resectable LAPC who were treated with FOLFIRINOX were down-staged significantly enough to undergo resection. Of those patients, more than 90% had a R0 resection following surgery (Table 3).73–79
Data over the past 7 years suggests that neoadjuvant FOLFIRINOX improves overall survival and resectability in patients with borderline disease. However, treatment with FOLFIRINOX is not without limitations. FOLFIRINOX is associated with higher rates of febrile neutropenia, thrombocytopenia, diarrhea, and sensory neuropathy as compared with gemcitabine-based therapy.72 Other less commonly observed toxicities associated with FOLFIRINOX include mucositis, hand-foot syndrome, pulmonary toxicity, and alopecia. Dose-attenuated FOLFIRINOX-based regimens, including those that exclude the bolus fluorouracil dose and augment upfront filgrastim, have demonstrated improved safety and comparable efficacy as compared to standard FOLFIRINOX.80
Gemcitabine has been the fundamental treatment backbone for PDA since the results of the phase 3 CONKO-001 trial were published.81 Gemcitabine is a pyrimidine antimetabolite and potent inhibitor of DNA polymerase and ribonucleotide reductase.82 In recent years, multiple combination therapies with gemcitabine have been investigated, including regimens with nab-paclitaxel, oxaliplatin, or docetaxel. Resection rates and negative margin outcomes have been shown to be comparable to patients who received FOLFIRINOX in the neoadjuvant setting with borderline locally advanced disease.83–85 In addition to having a more tolerable side effect profile in comparison to fluorouracil-based regimens, gemcitabine is considered to be a potent radiosensitizer.86 For this reason, studies have also investigated the role of radiotherapy in conjunction with gemcitabine, revealing negative margin resection rates above 80% in patients with borderline resectable disease.87,88
Because very few studies directly comparing FOLFIRINOX with gemcitabine-based combination regimens have been completed, there is no clear consensus on the preferred treatment regimen, in both borderline and unresectable LAPC. Decisions to treat are influenced predominantly by comorbidities, adverse effect profiles, and performance status of patients, as FOLFIRINOX is the more toxic of the 2 treatment backbones. Therefore, FOLFIRINOX has mostly been utilized in patients with relatively good functional status (Eastern Cooperative Oncology Group [ECOG] performance status 0 to 1).89 In elderly patients and those with poor functional status, ECOG 2 to 4, gemcitabine as a single agent is a reasonable alternative in the neoadjuvant setting of borderline resectable disease.
The exact role of radiation therapy in addition to induction chemotherapy in borderline resectable pancreatic cancer has not been clearly established because of the lack of prospective studies in this area. Multiple large retrospective series have identified high rates of conversion to margin-negative resection with neoadjuvant chemoradiation alone.90 Based on available data, it is reasonable for patients with borderline resectable disease to proceed with any of the following treatment options: chemotherapy, chemoradiation, or induction chemotherapy followed by chemoradiation (Figure). Chemotherapy and chemoradiation are generally more appropriate with patients with high CA 19-9 levels or those at an elevated risk of having positive margins or occult metastatic disease.91 Obtaining negative margin resections is the predominant goal of neoadjuvant radiotherapy.89 Many studies have identified margin status to be one of the most significant prognostic factors in PDA.57,59,92,93 Additionally, several studies have highlighted that radiotherapy in the neoadjuvant setting could improve negative margin resection rates, local control, and clinical outcomes in patients with borderline resectable locally advanced disease.94–97 A common multimodal regimen utilized in the neoadjuvant setting combines capecitabine, an oral prodrug that is converted to fluorouracil, with radiation therapy. This combination has also been shown to improve resectability rates and long-term clinical outcomes in patients with borderline resectable disease.98 Additionally, neoadjuvant radiation therapy can potentially downstage patients with unresectable disease at the time of diagnosis to become surgical candidates.99 Despite the paucity of data, interval scans utilizing CT following neoadjuvant therapy should be obtained 2 to 4 months after completion of therapy to determine therapeutic response, evaluate for disease progression, and, most important, reassess surgical stage/resectability. It is clinically acceptable to proceed to resection with radiographically stable disease post-neoadjuvant therapy.
Many patients classified as borderline resectable are able to proceed with surgery following neoadjuvant therapy. Unfortunately, specific data on adjuvant therapy following neoadjuvant chemotherapy or chemoradiotherapy and surgical resection in borderline resectable patients is scarce. Clinical practice guidelines are extrapolated from studies where upfront resection in clearly resectable patients was followed by adjuvant therapy. Based on these data, approximately 6 months of perioperative chemotherapy with or without chemoradiotherapy is a reasonable consideration. Nevertheless, about 80% of patients at the time of diagnosis are deemed to be unresectable, and a smaller number do not proceed to surgery despite an initial classification as borderline resectable. Of the 80% of patients with advanced disease, about half are metastatic at presentation and the remaining 30% to 40% are defined as having unresectable LAPC.100
CASE CONTINUED
The patient is deemed borderline resectable. He receives neoadjuvant therapy with gemcitabine and nab-paclitaxel. Two months after therapy, interval imaging with abdominal CT does not show improvement in tumor size and there is now evidence that the tumor has invaded the celiac axis and is abutting more than 180° of the SMA. The patient presents to the oncologist to discuss further management. He has lost about 15 lb since his last evaluation, is capable of self-care, but is unable to carry on with any work activities.
What is the appropriate management of unresectable nonmetastatic LAPC?
UNRESECTABLE LOCALLY ADVANCED CANCER
As in the case of borderline resectable disease, there are many treatment options for patients with unresectable LAPC. Timing, optimal chemotherapy regimen, and the addition of regularly and hypofractionated radiotherapy are issues currently under investigation. However, there are some general considerations and principles that are followed as reflected in the NCCN guidelines and recent studies. The primary therapeutic aims in patients with unresectable locally advanced disease are to increase survival and improve palliation.
The elderly comprise a large percentage of the patients diagnosed with unresectable locally advanced disease. Pharmacokinetics and toxicity profiles are altered in the elderly population.101,102 Therefore, it is important to assess functional status and comorbidities as these are critical factors in determining treatment regimens, similar to patients with borderline resectable disease. Currently, the most common first-line therapies in advanced pancreatic cancer are gemcitabine alone, gemcitabine and nab-paclitaxel, FOLFIRINOX, gemcitabine/capecitabine, and gemcitabine/oxaliplatin.103 The overall treatment approach to unresectable locally advanced pancreatic adenocarcinoma closely mirrors that of patients with borderline resectable disease and metastatic disease. Much of the data supporting treatment regimens in unresectable LAPC is extrapolated from clinical trials looking at advanced or metastatic pancreatic cancer.
Consensus opinions domestically and from Europe recommend that patients with locally advanced unresectable disease undergo upfront chemotherapy (Figure).104 This is based on the premise that initial chemotherapy may destroy occult metastatic cells and increase the efficacy of consolidative chemotherapy, particularly with radiation in the future. Upfront chemoradiotherapy has only been investigated in a small series of trials in which no clear survival benefit was observed and has the added consequence of treatment-related toxicity.105 However, data is limited in this regard, with variations in treatment protocols and cohort compositions contributing to the inconclusive findings.
Despite advances in immunotherapy, targeted therapies, and gene sequencing, initial chemotherapy for unresectable disease is still either gemcitabine-based combination therapy or FOLFIRINOX. Across numerous studies, patients with unresectable LAPC receiving FOLFIRINOX have a median progression-free survival of 3 to 20 months and a median overall survival of 10 to 32.7 months.106 As with borderline resectable patients, FOLFIRINOX (Table 4) is generally reserved for unresectable patients with good functional status (ECOG 0–1 or Karnofsky Performance Status 90–100) and those at low risk for developing grade 3 or 4 systemic toxicities.103 For these reasons it has generally not been frequently combined with other chemotherapeutic agents. However, FOLFIRINOX has been combined with radiation therapy in the consolidative neoadjuvant setting after induction chemotherapy. There have also been studies where traditional FOLFIRONIX was modified to improve tolerability, as evidenced by Gunturu et al’s study, which dose-reduced both fluorouracil and irinotecan by 25%, without compromising efficacy and simultaneously increasing tolerability.107 Additionally, FOLFIRINOX requires infusional administration of the fluorouracil component, which may not be practical in certain patients. In that subset, capecitabine can be substituted. Among radiosensitizers during neoadjuvant therapy for unresectable LAPC, capecitabine has been shown to be as efficacious and less toxic than even gemcitabine.108
No head-to-head studies investigating FOLFIRINOX versus nab-paclitaxel and gemcitabine in patients with locally advanced disease have been published, but clinical trials are under way. Other combination therapies have been looked at through small retrospective or prospective studies, but no robust, large-scale clinical trials have been completed. For this reason, NCCN guidelines recommend enrollment of patients with LAPC into active clinical trials.
What is the role of radiation therapy in unresectable LAPC?
Despite the reported advantages of neoadjuvant radiation in patients with potentially resectable disease, there is significant debate regarding the timing and role of neoadjuvant radiation in patients with unresectable disease. Numerous comprehensive analyses and studiest indicate that chemoradiotherapy leads to significantly better overall survival compared to no therapy or radiation therapy alone in LAPC.68,110,111 However, conflicting data support the use of upfront chemoradiotherapy in unresectable LAPC when compared to chemotherapy alone. Unfortunately, most prospective studies investigating the role of radiotherapy were performed following administration of single-agent gemcitabine, which is no longer considered standard of care for patients with LAPC. In spite of this, ECOG 4201 identified a statistically significant improvement in median overall survival following the addition of gemcitabine-based radiotherapy. Huguet et al in his review pointed out that upfront chemoradiotherapy was not superior to chemotherapy only and was associated with increased treatment toxicity.105 Additionally, a recent phase 3 study looking at chemoradiotherapy versus chemotherapy alone in patients treated with gemcitabine found no difference in overall survival.112 This can potentially be attributed to the fact that about 30% of patients with LAPC develop metastatic disease in the early phases of treatment due to poor control of local and systemically occult disease.113 Given the propensity for high rates of occult metastatic disease in LAPC, treatment paradigms and consensus guidelines recommend multi-agent systemic chemotherapy followed by chemoradiotherapy in select patients.
Based on current studies and until further clinical investigations are completed, consensus opinion indicates that the most appropriate approach in unresectable LAPC is to begin with induction chemotherapy (with either gemcitabine plus nab-paclitaxel, FOLFIRINOX, capecitabine, or other treatment combinations), followed by chemoradiation in the absence of disease progression when the first repeat imaging evaluation is completed (Figure). One important caveat regarding reimaging with CT in the neoadjuvant setting is that radiologic response may not correlate with pathologic response.114 PET-CT may have a role in predicting response to neoadjuvant therapy. Overall, induction chemotherapy followed by consolidative chemoradiation may confer numerous benefits: it removes the unnecessary burden and toxicity associated with radiotherapy in the nearly one third of patients who have pervasive disease progression during initial treatment; it allows testing and increases the chances of tolerating full-dose systemic chemotherapy; and it raises the likelihood of converting patients who do not progress to metastasis during the initial phase of treatment from unresectable to resectable status.103,115 Despite the lack of strong conclusive data, the general agreement is that neoadjuvant chemoradiotherapy converts about one third of borderline and unresectable LAPC to an R0 resection.95,103 There are very specific rationales for the addition of radiotherapy in LAPC, and these functions need to be better defined through further clinical trials.
PALLIATIVE CARE
CASE CONTINUED
The patient is unable to tolerate his first round of second-line therapy with modified FOLFIRINOX. His overall treatment plan is transitioned to palliation. He continues to have pain, despite increasing doses of narcotics.
What is the next step for patients in whom second-line therapy fails and who have intractable pain while on high-dose narcotics?
A subset of patients with unresectable LAPC may not be amenable to chemotherapy with or without radiation due to significant comorbidities or because they present with or progress to ECOG scores 3 or 4. The goal in these patients should be palliation. Pain is one of the most predominant and difficult to manage symptoms in progressive LAPC. Opioid-based medications are the primary treatment for pain in LAPC. However, patients sometimes become refractory to opioid medications. In this group of patients, it is reasonable to consider palliative radiation as an alternative method for pain control.116
An alternative to palliative radiation in the setting of progressive pain in PDA is celiac plexus block or neurolysis. By injecting an anesthetic or alcohol into the celiac plexus, neural signaling pathways involved in the propagation of pain are inhibited without leading to significant nerve destruction. Additionally, chemical splanchnicectomy allows for reduced opioid medication use and associated side effects.117
In general patients with LAPC have profound weight loss prior to and during treatment. This has significant implications prognostically and on treatment options. The underlying etiology is multifactorial, but one of the primary driving factors is pancreatic insufficiency. An estimated 65% of pancreatic cancer patients have fat malabsorption, and 50% have protein malabsorption, leading to steatorrhea and weight loss.118 Patients diagnosed with pancreatic cancer should be given enzyme replacement with formulations that include lipase, amylase, and protease. A minimum dose of enzyme replacement should include 40,000 to 50,000 U of lipase during meals and 25,000 U during snack intake. If maldigestion, symptoms, or nutritional endpoints (BMI, albumin, prealbumin, cholesterol) do not improve, the pancreatic enzyme dose should be escalated and a proton-pump inhibitor (PPI) added. In patients with pancreatic insufficiency, PPIs have been shown to improve fat absorption.119 Enzyme replacement therapy has been shown to prevent weight loss in patients with unresectable pancreatic cancer.120
As most patients with LAPC go on to develop progressive disease, palliative care becomes an integral aspect of the therapeutic paradigm. Palliation in LAPC has a significant role in determining quality of life and ensuring patient’s goals of care have been meet. Studies have suggested that pancreatic cancer is second only to lung cancer in terms of the number of emergency department visits in the later stages of disease.120 Additionally, aggressive care in the setting of incurable diseases such as LAPC has been associated with poor quality of life.121 More recently it has been shown that involvement of palliative care in patients with advanced pancreatic is associated with less aggressive care near death.122 Therefore, the incorporation of palliative or supportive care teams in the treatment of patients with progressive LAPC can improve quality of life and alleviate suffering associated with increasing symptom burden.
CONCLUSION
LAPC is a difficult disease for both provider and patient. There is a paucity of robust clinical trials in the neoadjuvant setting for LAPC. Current research is complicated by varying consensus definitions of resectability and the varying treatment configurations across studies. The optimal type, timing, and sequence of treatment and whether to add radiation therapy in LAPC have not been clearly defined. However, based on the available studies and consensus guidelines, patients who are deemed to have LAPC should have neoadjuvant therapy. FOLFIRINOX or gemcitabine with nab-paclitaxel should be considered first-line treatments. Patients with LAPC who respond to chemotherapy or are ineligible for multi-drug chemotherapy may benefit from chemoradiotherapy. In patients with unresectable disease, chemoradiotherapy has been shown to enhance survival as compared to best supportive care or radiation alone. For borderline resectable disease, it is reasonable to treat patients with either chemoradiotherapy, chemotherapy alone, or chemotherapy followed by chemoradiotherapy.
Considering the invasive nature of LAPC and the controversy around neoadjuvant treatment protocols, enrollment of patients with LAPC into clinical trials is important and will help determine the optimal treatment regimen for future patients.
- Network PCA. Pancreatic cancer facts 2016. 2016. https://www.pancan.org/wp-content/uploads/2016/02/2016-GAA-PC-Facts.pdf. Accessed April 24, 2017.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Konstantinidis IT, Warshaw AL, Allen JN, et al. Pancreatic ductal adenocarcinoma: is there a survival difference for R1 resections versus locally advanced unresectable tumors? What is a “true” R0 resection? Ann Surg 2013;257:731–6.
- Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol 2016;22:9694–705.
- Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69–90.
- Zhang J, Dhakal I, Ning B, Kesteloot H. Patterns and trends of pancreatic cancer mortality rates in Arkansas, 1969-2002: a comparison with the US population. Eur J Cancer Prev 2008;17:18–27.
- National Cancer Institute. SEER cancer statistics review, 1975-2013. http://seer.cancer.gov/csr/1975_2013/. Accessed April 24, 2017.
- Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol 2006;20:197–209.
- Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med 1996;156:2255–60.
- Lucenteforte E, La Vecchia C, Silverman D, et al. Alcohol consumption and pancreatic cancer: a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann Oncol 2012;23:374–82.
- Schernhammer ES, Kang JH, Chan AT, et al. A prospective study of aspirin use and the risk of pancreatic cancer in women. J Natl Cancer Inst 2004;96:22–28.
- Michaud DS, Giovannucci E, Willett WC, et al. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001;286:921–9.
- Nothlings U, Wilkens LR, Murphy SP, et al. Meat and fat intake as risk factors for pancreatic cancer: the multiethnic cohort study. J Natl Cancer Inst 2005;97:1458–65.
- Chari ST, Leibson CL, Rabe KG, et al. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology 2005;129:504–11.
- Batabyal P, Vander Hoorn S, Christophi C, Nikfarjam M. Association of diabetes mellitus and pancreatic adenocarcinoma: a meta-analysis of 88 studies. Ann Surg Oncol 2014;21:2453–62.
- Chari ST, Leibson CL, Rabe KG, et al. Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology 2008;134:95–101.
- Pannala R, Basu A, Petersen GM, Chari ST. New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol 2009;10:88–95.
- Wolpin BM, Chan AT, Hartge P, et al. ABO blood group and the risk of pancreatic cancer. J Natl Cancer Inst 2009;101:424–31.
- Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
- Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000;119:1447–53.
- Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA 2009;302:1790–5.
- Brand RE, Lynch HT. Hereditary pancreatic adenocarcinoma. A clinical perspective. Med Clin North Am 2000;84:665–75.
- Jacobs EJ, Chanock SJ, Fuchs CS, et al. Family history of cancer and risk of pancreatic cancer: a pooled analysis from the Pancreatic Cancer Cohort Consortium (PanScan). Int J Cancer 2010;127:1421–8.
- Rustgi AK. Familial pancreatic cancer: genetic advances. Genes Dev 2014;28:1–7.
- Reznik R, Hendifar AE, Tuli R. Genetic determinants and potential therapeutic targets for pancreatic adenocarcinoma. Front Physiol 2014;5:87.
- Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst 1997;89:442–6.
- Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014;371:2140–1.
- Kanda M, Matthaei H, Wu J, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012;142:730–3.
- Feldmann G, Maitra A. Molecular genetics of pancreatic ductal adenocarcinomas and recent implications for translational efforts. J Mol Diagn 2008;10:111–22.
- Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 2014;111:817–22.
- Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic KRAS maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656–70.
- Provenzano PP, Cuevas C, Chang AE, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:418–29.
- Vonderheide RH, Bayne LJ. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr Opin Immunol 2013;25:200–5.
- DiMagno EP. Pancreatic cancer: Clinical presentation, pitfalls and early clues. Ann Oncol 1999;10(suppl 4):S140–S142.
- Porta M, Fabregat X, Malats N, et al. Exocrine pancreatic cancer: symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol 2005;7:189–97.
- Gullo L, Tomassetti P, Migliori M, et al. Do early symptoms of pancreatic cancer exist that can allow an earlier diagnosis? Pancreas 2001;22:210–3.
- Khorana AA, Fine RL. Pancreatic cancer and thromboembolic disease. Lancet Oncol 2004;5(11):655-663.
- Wigmore SJ, Plester CE, Richardson RA, Fearon KC. Changes in nutritional status associated with unresectable pancreatic cancer. Br J Cancer 1997;75:106–9.
- Aggarwal G, Rabe KG, Petersen GM, Chari ST. New-onset diabetes in pancreatic cancer: a study in the primary care setting. Pancreatology 2012;12:156–61.
- Koprowski H, Herlyn M, Steplewski Z, Sears HF. Specific antigen in serum of patients with colon carcinoma. Science 1981;212:53–5.
- Bond-Smith G, Banga N, Hammond TM, Imber CJ. Pancreatic adenocarcinoma. BMJ 2012;344:e2476.
- Cwik G, Wallner G, Skoczylas T, et al. Cancer antigens 19-9 and 125 in the differential diagnosis of pancreatic mass lesions. Arch Surg 2006;141:968–73.
- van den Bosch RP, van Eijck CH, Mulder PG, Jeekel J. Serum CA19-9 determination in the management of pancreatic cancer. Hepatogastroenterology 1996;43:710–3.
- Lamerz R. Role of tumour markers, cytogenetics. Ann Oncol 1999;10 Suppl 4:145–9.
- Hess V, Glimelius B, Grawe P, et al. CA 19-9 tumour-marker response to chemotherapy in patients with advanced pancreatic cancer enrolled in a randomised controlled trial. Lancet Oncol 2008;9:132–8.
- Montgomery RC, Hoffman JP, Riley LB, et al. Prediction of recurrence and survival by post-resection CA 19-9 values in patients with adenocarcinoma of the pancreas. Ann Surg Oncol 1997;4:551–6.
- Zamboni GA, D’Onofrio M, Idili A, et al. Ultrasound-guided percutaneous fine-needle aspiration of 545 focal pancreatic lesions. AJR Am J Roentgenol 2009;193:1691–5.
- Micames C, Jowell PS, White R, et al. Lower frequency of peritoneal carcinomatosis in patients with pancreatic cancer diagnosed by EUS-guided FNA vs. percutaneous FNA. Gastrointest Endosc 2003;58:690–5.
- Brambs HJ, Claussen CD. Pancreatic and ampullary carcinoma. Ultrasound, computed tomography, magnetic resonance imaging and angiography. Endoscopy 1993;25:58–68.
- Karlson BM, Ekbom A, Lindgren PG, et al. Abdominal US for diagnosis of pancreatic tumor: prospective cohort analysis. Radiology 1999;213:107–11.
- Imbriaco M, Megibow AJ, Camera L, et al. Dual-phase versus single-phase helical CT to detect and assess resectability of pancreatic carcinoma. AJR Am J Roentgenol 2002;178:1473–9.
- Schrag D. Optimizing treatment for locally advanced pancreas cancer: progress but no precision. JAMA 2016;315:1837–8.
- House MG, Yeo CJ, Cameron JL, et al. Predicting resectability of periampullary cancer with three-dimensional computed tomography. J Gastrointest Surg 2004;8:280–8.
- Callery MP, Chang KJ, Fishman EK, et al. Pretreatment assessment of resectable and borderline resectable pancreatic cancer: expert consensus statement. Ann Surg Oncol 2009;16:1727–33.
- Horton KM, Fishman EK. Adenocarcinoma of the pancreas: CT imaging. Radiol Clin North Am 2002;40:1263–72.
- Ross WA, Wasan SM, Evans DB, et al. Combined EUS with FNA and ERCP for the evaluation of patients with obstructive jaundice from presumed pancreatic malignancy. Gastrointest Endosc 2008;68:461–6.
- Hartwig W, Hackert T, Hinz U, et al. Pancreatic cancer surgery in the new millennium: better prediction of outcome. Ann Surg 2011;254:311–9.
- Jamieson NB, Chan NI, Foulis AK, et al. The prognostic influence of resection margin clearance following pancreaticoduodenectomy for pancreatic ductal adenocarcinoma. J Gastrointest Surg 2013;17:511–21.
- Ethun CG, Kooby DA. The importance of surgical margins in pancreatic cancer. J Surg Oncol 2016;113:283–8.
- Fischer R, Breidert M, Keck T, et al. Early recurrence of pancreatic cancer after resection and during adjuvant chemotherapy. Saudi J Gastroenterol 2012;18:118–21.
- Neoptolemos JP, Stocken DD, Friess H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med 2004;350:1200–10.
- Neoptolemos JP, Palmer DH, Ghaneh P, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389(10073):1011–24.
- Cardenes HR, Chiorean EG, Dewitt J, et al. Locally advanced pancreatic cancer: current therapeutic approach. Oncologist 2006;11:612–23.
- Yeung RS, Weese JL, Hoffman JP, et al. Neoadjuvant chemoradiation in pancreatic and duodenal carcinoma. A Phase II Study. Cancer 1993;72:2124–33.
- Spitz FR, Abbruzzese JL, Lee JE, et al. Preoperative and postoperative chemoradiation strategies in patients treated with pancreaticoduodenectomy for adenocarcinoma of the pancreas. J Clin Oncol 1997;15:928–37.
- McClaine RJ, Lowy AM, Sussman JJ, et al. Neoadjuvant therapy may lead to successful surgical resection and improved survival in patients with borderline resectable pancreatic cancer. HPB (Oxford) 2010;12:73–9.
- Balaban EP, Mangu PB, Khorana AA, et al. Locally advanced, unresectable pancreatic cCancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2016;34:2654–68.
- Shaib WL, Ip A, Cardona K, et al. Contemporary management of borderline resectable and locally advanced unresectable pancreatic cancer. Oncologist 2016;21:178–87.
- Chun YS, Milestone BN, Watson JC, et al. Defining venous involvement in borderline resectable pancreatic cancer. Ann Surg Oncol 2010;17:2832–8.
- Evans DB, Erickson BA, Ritch P. Borderline resectable pancreatic cancer: definitions and the importance of multimodality therapy. Ann Surg Oncol 2010;17:2803–5.
- Conroy T, Paillot B, Francois E, et al. Irinotecan plus oxaliplatin and leucovorin-modulated fluorouracil in advanced pancreatic cancer--a Groupe Tumeurs Digestives of the Federation Nationale des Centres de Lutte Contre le Cancer study. J Clin Oncol 2005;23:1228–36.
- Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
- Hosein PJ, Macintyre J, Kawamura C, et al. A retrospective study of neoadjuvant FOLFIRINOX in unresectable or borderline-resectable locally advanced pancreatic adenocarcinoma. BMC Cancer 2012;12:199.
- Peddi PF, Lubner S, McWilliams R, et al. Multi-institutional experience with FOLFIRINOX in pancreatic adenocarcinoma. JOP 2012;13:497–501.
- Mahaseth H, Kauh JS, Brutcher E, et al. Safety and efficacy of modified FOLFIRINOX in pancreatic cancer: A retrospective experience. J Clin Oncol 2012;30 (suppl; abstr e14614).
- Kim SS, Nakakura EK, Wang ZJ, et al. Preoperative FOLFIRINOX for borderline resectable pancreatic cancer: Is radiation necessary in the modern era of chemotherapy? J Surg Oncol 2016;114:587–96.
- Conroy T, Gavoille C, Samalin E, et al. The role of the FOLFIRINOX regimen for advanced pancreatic cancer. Curr Oncol Rep 2013;15:182–9.
- Katz MH, Shi Q, Ahmad SA, et al. Preoperative modified FOLFIRINOX treatment followed by capecitabine-based chemoradiation for borderline resectable pancreatic cancer: Alliance for Clinical Trials in Oncology Trial A021101. JAMA Surg 2016;151:e161137.
- Kharofa J, Kelly TR, Ritch PS, et al. 5-FU/leucovorin, irinotecan, oxaliplatin (FOLFIRINOX) induction followed by chemoXRT in borderline resectable pancreatic cancer. J Clin Oncol 2012;30 (suppl; abstr e14613).
- Blazer M, Wu C, Goldberg RM, et al. Neoadjuvant modified (m) FOLFIRINOX for locally advanced unresectable (LAPC) and borderline resectable (BRPC) adenocarcinoma of the pancreas. Ann Surg Oncol 2015;22:1153–9.
- Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
- Plunkett W, Huang P, Xu YZ, et al. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol 1995;22(4 Suppl 11):3–10.
- Sahora K, Kuehrer I, Eisenhut A, et al. NeoGemOx: Gemcitabine and oxaliplatin as neoadjuvant treatment for locally advanced, nonmetastasized pancreatic cancer. Surgery 2011;149(3):311–20.
- Lee JL, Kim SC, Kim JH, et al. Prospective efficacy and safety study of neoadjuvant gemcitabine with capecitabine combination chemotherapy for borderline-resectable or unresectable locally advanced pancreatic adenocarcinoma. Surgery 2012;152:851–62.
- Leone F, Gatti M, Massucco P, et al. Induction gemcitabine and oxaliplatin therapy followed by a twice-weekly infusion of gemcitabine and concurrent external-beam radiation for neoadjuvant treatment of locally advanced pancreatic cancer: a single institutional experience. Cancer 2013;119:277–84.
- Lawrence TS, Eisbruch A, Shewach DS. Gemcitabine-mediated radiosensitization. Semin Oncol 1997;24(2 Suppl 7):S7–24-S27–28.
- Kang CM, Chung YE, Park JY, et al. Potential contribution of preoperative neoadjuvant concurrent chemoradiation therapy on margin-negative resection in borderline resectable pancreatic cancer. J Gastrointest Surg 2012;16:509–17.
- Chuong MD, Hayman TJ, Patel MR, et al. Comparison of 1-, 2-, and 3-dimensional tumor response assessment after neoadjuvant GTX-RT in borderline-resectable pancreatic cancer. Gastrointest Cancer Res 2011;4:128–34.
- Loehrer AP, Kinnier CV, Ferrone CR. Treatment of locally advanced pancreatic ductal adenocarcinoma. Adv Surg 2016;50:115–28.
- Katz MH, Wang H, Balachandran A, et al. Effect of neoadjuvant chemoradiation and surgical technique on recurrence of localized pancreatic cancer. J Gastrointest Surg 2012;16:68–78.
- Franke AJ, Rosati LM, Pawlik TM, et al. The role of radiation therapy in pancreatic ductal adenocarcinoma in the neoadjuvant and adjuvant settings. Semin Oncol 2015;42:144–62.
- Butturini G, Stocken DD, Wente MN, et al. Influence of resection margins and treatment on survival in patients with pancreatic cancer: meta-analysis of randomized controlled trials. Arch Surg 2008;143:75–83.
- Paniccia A, Hosokawa P, Henderson W, et al. Characteristics of 10-year survivors of pancreatic ductal adenocarcinoma. JAMA Surg 2015;150:701–10.
- Massucco P, Capussotti L, Magnino A, et al. Pancreatic resections after chemoradiotherapy for locally advanced ductal adenocarcinoma: analysis of perioperative outcome and survival. Ann Surg Oncol 2006;13:1201–8.
- Patel M, Hoffe S, Malafa M, et al. Neoadjuvant GTX chemotherapy and IMRT-based chemoradiation for borderline resectable pancreatic cancer. J Surg Oncol 2011;104:155–161.
- Landry J, Catalano PJ, Staley C, et al. Randomized phase II study of gemcitabine plus radiotherapy versus gemcitabine, 5-fluorouracil, and cisplatin followed by radiotherapy and 5-fluorouracil for patients with locally advanced, potentially resectable pancreatic adenocarcinoma. J Surg Oncol 2010;101:587–92.
- Lamb R, Ozsvari B, Lisanti CL, et al. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget 2015;6:4569–84.
- Stokes JB, Nolan NJ, Stelow EB, et al. Preoperative capecitabine and concurrent radiation for borderline resectable pancreatic cancer. Ann Surg Oncol 2011;18:619–27.
- White R, Lee C, Anscher M, et al. Preoperative chemoradiation for patients with locally advanced adenocarcinoma of the pancreas. Ann Surg Oncol 1999;6:38–45.
- Martin RC 2nd. Management of locally advanced pancreatic cancer. Surg Clin North Am 2016;96:1371–89.
- Higuera O, Ghanem I, Nasimi R, et al. Management of pancreatic cancer in the elderly. World J Gastroenterol 2016;22:764–75.
- Hurria A, Lichtman SM. Clinical pharmacology of cancer therapies in older adults. Br J Cancer 2008;98:517–22.
- Spadi R, Brusa F, Ponzetti A, et al. Current therapeutic strategies for advanced pancreatic cancer: A review for clinicians. World J Clin Oncol 2016;7:27–43.
- Seufferlein T, Bachet JB, Van Cutsem E, Rougier P, Group EGW. Pancreatic adenocarcinoma: ESMO-ESDO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012;23 Suppl 7:vii33–40.
- Huguet F, Girard N, Guerche CS, et al. Chemoradiotherapy in the management of locally advanced pancreatic carcinoma: a qualitative systematic review. J Clin Oncol 2009;27:2269–77.
- Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol 2016;17:801–10.
- Gunturu KS, Yao X, Cong X, et al. FOLFIRINOX for locally advanced and metastatic pancreatic cancer: single institution retrospective review of efficacy and toxicity. Med Oncol 2013;30:361.
- Mukherjee S, Hurt CN, Bridgewater J, et al. Gemcitabine-based or capecitabine-based chemoradiotherapy for locally advanced pancreatic cancer (SCALOP): a multicentre, randomised, phase 2 trial. Lancet Oncol 2013;14:317–26.
- Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
- Krzyzanowska MK, Weeks JC, Earle CC. Treatment of locally advanced pancreatic cancer in the real world: population-based practices and effectiveness. J Clin Oncol 2003;21:3409–14.
- Sultana A, Tudur Smith C, Cunningham D, et al. Meta-analyses of chemotherapy for locally advanced and metastatic pancreatic cancer: results of secondary end points analyses. Br J Cancer 2008;99:6–13.
- Hammel P, Huguet F, van Laethem JL, et al. Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: the LAP07 randomized clinical trial. JAMA 2016;315:1844–53.
- Huguet F, Andre T, Hammel P, et al. Impact of chemoradiotherapy after disease control with chemotherapy in locally advanced pancreatic adenocarcinoma in GERCOR phase II and III studies. J Clin Oncol 2007;25:326–31.
- Dholakia AS, Hacker-Prietz A, Wild AT, et al. Resection of borderline resectable pancreatic cancer after neoadjuvant chemoradiation does not depend on improved radiographic appearance of tumor-vessel relationships. J Radiat Oncol 2013;2:413–25.
- Heinemann V, Haas M, Boeck S. Neoadjuvant treatment of borderline resectable and non-resectable pancreatic cancer. Ann Oncol 2013;24:2484–92.
- Morganti AG, Trodella L, Valentini V, et al. Pain relief with short-term irradiation in locally advanced carcinoma of the pancreas. J Palliat Care 2003;19:258–62.
- Arcidiacono PG, Calori G, Carrara S, et al. Celiac plexus block for pancreatic cancer pain in adults. Cochrane Database Syst Rev 2011(3):CD007519.
- Pezzilli R, Andriulli A, Bassi C, et al. Exocrine pancreatic insufficiency in adults: a shared position statement of the Italian Association for the Study of the Pancreas. World J Gastroenterol 2013;19:7930–46.
- Dominguez-Munoz JE. Pancreatic exocrine insufficiency: diagnosis and treatment. J Gastroenterol Hepatol 2011;26 Suppl 2:12–16.
- Bruno MJ, Haverkort EB, Tijssen GP, et al. Placebo controlled trial of enteric coated pancreatin microsphere treatment in patients with unresectable cancer of the pancreatic head region. Gut 1998;42:92–6.
- Wright AA, Keating NL, Balboni TA, et al. Place of death: correlations with quality of life of patients with cancer and predictors of bereaved caregivers’ mental health. J Clin Oncol 2010;28:4457–64.
- Jang RW, Krzyzanowska MK, Zimmermann C, et al. Palliative care and the aggressiveness of end-of-life care in patients with advanced pancreatic cancer. J Natl Cancer Inst 2015;107(3). pii: dju424.
- Network PCA. Pancreatic cancer facts 2016. 2016. https://www.pancan.org/wp-content/uploads/2016/02/2016-GAA-PC-Facts.pdf. Accessed April 24, 2017.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Konstantinidis IT, Warshaw AL, Allen JN, et al. Pancreatic ductal adenocarcinoma: is there a survival difference for R1 resections versus locally advanced unresectable tumors? What is a “true” R0 resection? Ann Surg 2013;257:731–6.
- Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol 2016;22:9694–705.
- Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69–90.
- Zhang J, Dhakal I, Ning B, Kesteloot H. Patterns and trends of pancreatic cancer mortality rates in Arkansas, 1969-2002: a comparison with the US population. Eur J Cancer Prev 2008;17:18–27.
- National Cancer Institute. SEER cancer statistics review, 1975-2013. http://seer.cancer.gov/csr/1975_2013/. Accessed April 24, 2017.
- Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol 2006;20:197–209.
- Fuchs CS, Colditz GA, Stampfer MJ, et al. A prospective study of cigarette smoking and the risk of pancreatic cancer. Arch Intern Med 1996;156:2255–60.
- Lucenteforte E, La Vecchia C, Silverman D, et al. Alcohol consumption and pancreatic cancer: a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann Oncol 2012;23:374–82.
- Schernhammer ES, Kang JH, Chan AT, et al. A prospective study of aspirin use and the risk of pancreatic cancer in women. J Natl Cancer Inst 2004;96:22–28.
- Michaud DS, Giovannucci E, Willett WC, et al. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001;286:921–9.
- Nothlings U, Wilkens LR, Murphy SP, et al. Meat and fat intake as risk factors for pancreatic cancer: the multiethnic cohort study. J Natl Cancer Inst 2005;97:1458–65.
- Chari ST, Leibson CL, Rabe KG, et al. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology 2005;129:504–11.
- Batabyal P, Vander Hoorn S, Christophi C, Nikfarjam M. Association of diabetes mellitus and pancreatic adenocarcinoma: a meta-analysis of 88 studies. Ann Surg Oncol 2014;21:2453–62.
- Chari ST, Leibson CL, Rabe KG, et al. Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology 2008;134:95–101.
- Pannala R, Basu A, Petersen GM, Chari ST. New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol 2009;10:88–95.
- Wolpin BM, Chan AT, Hartge P, et al. ABO blood group and the risk of pancreatic cancer. J Natl Cancer Inst 2009;101:424–31.
- Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer 2012;107:2005–9.
- Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000;119:1447–53.
- Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA 2009;302:1790–5.
- Brand RE, Lynch HT. Hereditary pancreatic adenocarcinoma. A clinical perspective. Med Clin North Am 2000;84:665–75.
- Jacobs EJ, Chanock SJ, Fuchs CS, et al. Family history of cancer and risk of pancreatic cancer: a pooled analysis from the Pancreatic Cancer Cohort Consortium (PanScan). Int J Cancer 2010;127:1421–8.
- Rustgi AK. Familial pancreatic cancer: genetic advances. Genes Dev 2014;28:1–7.
- Reznik R, Hendifar AE, Tuli R. Genetic determinants and potential therapeutic targets for pancreatic adenocarcinoma. Front Physiol 2014;5:87.
- Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst 1997;89:442–6.
- Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014;371:2140–1.
- Kanda M, Matthaei H, Wu J, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012;142:730–3.
- Feldmann G, Maitra A. Molecular genetics of pancreatic ductal adenocarcinomas and recent implications for translational efforts. J Mol Diagn 2008;10:111–22.
- Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 2014;111:817–22.
- Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic KRAS maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656–70.
- Provenzano PP, Cuevas C, Chang AE, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:418–29.
- Vonderheide RH, Bayne LJ. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr Opin Immunol 2013;25:200–5.
- DiMagno EP. Pancreatic cancer: Clinical presentation, pitfalls and early clues. Ann Oncol 1999;10(suppl 4):S140–S142.
- Porta M, Fabregat X, Malats N, et al. Exocrine pancreatic cancer: symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol 2005;7:189–97.
- Gullo L, Tomassetti P, Migliori M, et al. Do early symptoms of pancreatic cancer exist that can allow an earlier diagnosis? Pancreas 2001;22:210–3.
- Khorana AA, Fine RL. Pancreatic cancer and thromboembolic disease. Lancet Oncol 2004;5(11):655-663.
- Wigmore SJ, Plester CE, Richardson RA, Fearon KC. Changes in nutritional status associated with unresectable pancreatic cancer. Br J Cancer 1997;75:106–9.
- Aggarwal G, Rabe KG, Petersen GM, Chari ST. New-onset diabetes in pancreatic cancer: a study in the primary care setting. Pancreatology 2012;12:156–61.
- Koprowski H, Herlyn M, Steplewski Z, Sears HF. Specific antigen in serum of patients with colon carcinoma. Science 1981;212:53–5.
- Bond-Smith G, Banga N, Hammond TM, Imber CJ. Pancreatic adenocarcinoma. BMJ 2012;344:e2476.
- Cwik G, Wallner G, Skoczylas T, et al. Cancer antigens 19-9 and 125 in the differential diagnosis of pancreatic mass lesions. Arch Surg 2006;141:968–73.
- van den Bosch RP, van Eijck CH, Mulder PG, Jeekel J. Serum CA19-9 determination in the management of pancreatic cancer. Hepatogastroenterology 1996;43:710–3.
- Lamerz R. Role of tumour markers, cytogenetics. Ann Oncol 1999;10 Suppl 4:145–9.
- Hess V, Glimelius B, Grawe P, et al. CA 19-9 tumour-marker response to chemotherapy in patients with advanced pancreatic cancer enrolled in a randomised controlled trial. Lancet Oncol 2008;9:132–8.
- Montgomery RC, Hoffman JP, Riley LB, et al. Prediction of recurrence and survival by post-resection CA 19-9 values in patients with adenocarcinoma of the pancreas. Ann Surg Oncol 1997;4:551–6.
- Zamboni GA, D’Onofrio M, Idili A, et al. Ultrasound-guided percutaneous fine-needle aspiration of 545 focal pancreatic lesions. AJR Am J Roentgenol 2009;193:1691–5.
- Micames C, Jowell PS, White R, et al. Lower frequency of peritoneal carcinomatosis in patients with pancreatic cancer diagnosed by EUS-guided FNA vs. percutaneous FNA. Gastrointest Endosc 2003;58:690–5.
- Brambs HJ, Claussen CD. Pancreatic and ampullary carcinoma. Ultrasound, computed tomography, magnetic resonance imaging and angiography. Endoscopy 1993;25:58–68.
- Karlson BM, Ekbom A, Lindgren PG, et al. Abdominal US for diagnosis of pancreatic tumor: prospective cohort analysis. Radiology 1999;213:107–11.
- Imbriaco M, Megibow AJ, Camera L, et al. Dual-phase versus single-phase helical CT to detect and assess resectability of pancreatic carcinoma. AJR Am J Roentgenol 2002;178:1473–9.
- Schrag D. Optimizing treatment for locally advanced pancreas cancer: progress but no precision. JAMA 2016;315:1837–8.
- House MG, Yeo CJ, Cameron JL, et al. Predicting resectability of periampullary cancer with three-dimensional computed tomography. J Gastrointest Surg 2004;8:280–8.
- Callery MP, Chang KJ, Fishman EK, et al. Pretreatment assessment of resectable and borderline resectable pancreatic cancer: expert consensus statement. Ann Surg Oncol 2009;16:1727–33.
- Horton KM, Fishman EK. Adenocarcinoma of the pancreas: CT imaging. Radiol Clin North Am 2002;40:1263–72.
- Ross WA, Wasan SM, Evans DB, et al. Combined EUS with FNA and ERCP for the evaluation of patients with obstructive jaundice from presumed pancreatic malignancy. Gastrointest Endosc 2008;68:461–6.
- Hartwig W, Hackert T, Hinz U, et al. Pancreatic cancer surgery in the new millennium: better prediction of outcome. Ann Surg 2011;254:311–9.
- Jamieson NB, Chan NI, Foulis AK, et al. The prognostic influence of resection margin clearance following pancreaticoduodenectomy for pancreatic ductal adenocarcinoma. J Gastrointest Surg 2013;17:511–21.
- Ethun CG, Kooby DA. The importance of surgical margins in pancreatic cancer. J Surg Oncol 2016;113:283–8.
- Fischer R, Breidert M, Keck T, et al. Early recurrence of pancreatic cancer after resection and during adjuvant chemotherapy. Saudi J Gastroenterol 2012;18:118–21.
- Neoptolemos JP, Stocken DD, Friess H, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med 2004;350:1200–10.
- Neoptolemos JP, Palmer DH, Ghaneh P, et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 2017;389(10073):1011–24.
- Cardenes HR, Chiorean EG, Dewitt J, et al. Locally advanced pancreatic cancer: current therapeutic approach. Oncologist 2006;11:612–23.
- Yeung RS, Weese JL, Hoffman JP, et al. Neoadjuvant chemoradiation in pancreatic and duodenal carcinoma. A Phase II Study. Cancer 1993;72:2124–33.
- Spitz FR, Abbruzzese JL, Lee JE, et al. Preoperative and postoperative chemoradiation strategies in patients treated with pancreaticoduodenectomy for adenocarcinoma of the pancreas. J Clin Oncol 1997;15:928–37.
- McClaine RJ, Lowy AM, Sussman JJ, et al. Neoadjuvant therapy may lead to successful surgical resection and improved survival in patients with borderline resectable pancreatic cancer. HPB (Oxford) 2010;12:73–9.
- Balaban EP, Mangu PB, Khorana AA, et al. Locally advanced, unresectable pancreatic cCancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2016;34:2654–68.
- Shaib WL, Ip A, Cardona K, et al. Contemporary management of borderline resectable and locally advanced unresectable pancreatic cancer. Oncologist 2016;21:178–87.
- Chun YS, Milestone BN, Watson JC, et al. Defining venous involvement in borderline resectable pancreatic cancer. Ann Surg Oncol 2010;17:2832–8.
- Evans DB, Erickson BA, Ritch P. Borderline resectable pancreatic cancer: definitions and the importance of multimodality therapy. Ann Surg Oncol 2010;17:2803–5.
- Conroy T, Paillot B, Francois E, et al. Irinotecan plus oxaliplatin and leucovorin-modulated fluorouracil in advanced pancreatic cancer--a Groupe Tumeurs Digestives of the Federation Nationale des Centres de Lutte Contre le Cancer study. J Clin Oncol 2005;23:1228–36.
- Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25.
- Hosein PJ, Macintyre J, Kawamura C, et al. A retrospective study of neoadjuvant FOLFIRINOX in unresectable or borderline-resectable locally advanced pancreatic adenocarcinoma. BMC Cancer 2012;12:199.
- Peddi PF, Lubner S, McWilliams R, et al. Multi-institutional experience with FOLFIRINOX in pancreatic adenocarcinoma. JOP 2012;13:497–501.
- Mahaseth H, Kauh JS, Brutcher E, et al. Safety and efficacy of modified FOLFIRINOX in pancreatic cancer: A retrospective experience. J Clin Oncol 2012;30 (suppl; abstr e14614).
- Kim SS, Nakakura EK, Wang ZJ, et al. Preoperative FOLFIRINOX for borderline resectable pancreatic cancer: Is radiation necessary in the modern era of chemotherapy? J Surg Oncol 2016;114:587–96.
- Conroy T, Gavoille C, Samalin E, et al. The role of the FOLFIRINOX regimen for advanced pancreatic cancer. Curr Oncol Rep 2013;15:182–9.
- Katz MH, Shi Q, Ahmad SA, et al. Preoperative modified FOLFIRINOX treatment followed by capecitabine-based chemoradiation for borderline resectable pancreatic cancer: Alliance for Clinical Trials in Oncology Trial A021101. JAMA Surg 2016;151:e161137.
- Kharofa J, Kelly TR, Ritch PS, et al. 5-FU/leucovorin, irinotecan, oxaliplatin (FOLFIRINOX) induction followed by chemoXRT in borderline resectable pancreatic cancer. J Clin Oncol 2012;30 (suppl; abstr e14613).
- Blazer M, Wu C, Goldberg RM, et al. Neoadjuvant modified (m) FOLFIRINOX for locally advanced unresectable (LAPC) and borderline resectable (BRPC) adenocarcinoma of the pancreas. Ann Surg Oncol 2015;22:1153–9.
- Oettle H, Post S, Neuhaus P, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA 2007;297:267–77.
- Plunkett W, Huang P, Xu YZ, et al. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol 1995;22(4 Suppl 11):3–10.
- Sahora K, Kuehrer I, Eisenhut A, et al. NeoGemOx: Gemcitabine and oxaliplatin as neoadjuvant treatment for locally advanced, nonmetastasized pancreatic cancer. Surgery 2011;149(3):311–20.
- Lee JL, Kim SC, Kim JH, et al. Prospective efficacy and safety study of neoadjuvant gemcitabine with capecitabine combination chemotherapy for borderline-resectable or unresectable locally advanced pancreatic adenocarcinoma. Surgery 2012;152:851–62.
- Leone F, Gatti M, Massucco P, et al. Induction gemcitabine and oxaliplatin therapy followed by a twice-weekly infusion of gemcitabine and concurrent external-beam radiation for neoadjuvant treatment of locally advanced pancreatic cancer: a single institutional experience. Cancer 2013;119:277–84.
- Lawrence TS, Eisbruch A, Shewach DS. Gemcitabine-mediated radiosensitization. Semin Oncol 1997;24(2 Suppl 7):S7–24-S27–28.
- Kang CM, Chung YE, Park JY, et al. Potential contribution of preoperative neoadjuvant concurrent chemoradiation therapy on margin-negative resection in borderline resectable pancreatic cancer. J Gastrointest Surg 2012;16:509–17.
- Chuong MD, Hayman TJ, Patel MR, et al. Comparison of 1-, 2-, and 3-dimensional tumor response assessment after neoadjuvant GTX-RT in borderline-resectable pancreatic cancer. Gastrointest Cancer Res 2011;4:128–34.
- Loehrer AP, Kinnier CV, Ferrone CR. Treatment of locally advanced pancreatic ductal adenocarcinoma. Adv Surg 2016;50:115–28.
- Katz MH, Wang H, Balachandran A, et al. Effect of neoadjuvant chemoradiation and surgical technique on recurrence of localized pancreatic cancer. J Gastrointest Surg 2012;16:68–78.
- Franke AJ, Rosati LM, Pawlik TM, et al. The role of radiation therapy in pancreatic ductal adenocarcinoma in the neoadjuvant and adjuvant settings. Semin Oncol 2015;42:144–62.
- Butturini G, Stocken DD, Wente MN, et al. Influence of resection margins and treatment on survival in patients with pancreatic cancer: meta-analysis of randomized controlled trials. Arch Surg 2008;143:75–83.
- Paniccia A, Hosokawa P, Henderson W, et al. Characteristics of 10-year survivors of pancreatic ductal adenocarcinoma. JAMA Surg 2015;150:701–10.
- Massucco P, Capussotti L, Magnino A, et al. Pancreatic resections after chemoradiotherapy for locally advanced ductal adenocarcinoma: analysis of perioperative outcome and survival. Ann Surg Oncol 2006;13:1201–8.
- Patel M, Hoffe S, Malafa M, et al. Neoadjuvant GTX chemotherapy and IMRT-based chemoradiation for borderline resectable pancreatic cancer. J Surg Oncol 2011;104:155–161.
- Landry J, Catalano PJ, Staley C, et al. Randomized phase II study of gemcitabine plus radiotherapy versus gemcitabine, 5-fluorouracil, and cisplatin followed by radiotherapy and 5-fluorouracil for patients with locally advanced, potentially resectable pancreatic adenocarcinoma. J Surg Oncol 2010;101:587–92.
- Lamb R, Ozsvari B, Lisanti CL, et al. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget 2015;6:4569–84.
- Stokes JB, Nolan NJ, Stelow EB, et al. Preoperative capecitabine and concurrent radiation for borderline resectable pancreatic cancer. Ann Surg Oncol 2011;18:619–27.
- White R, Lee C, Anscher M, et al. Preoperative chemoradiation for patients with locally advanced adenocarcinoma of the pancreas. Ann Surg Oncol 1999;6:38–45.
- Martin RC 2nd. Management of locally advanced pancreatic cancer. Surg Clin North Am 2016;96:1371–89.
- Higuera O, Ghanem I, Nasimi R, et al. Management of pancreatic cancer in the elderly. World J Gastroenterol 2016;22:764–75.
- Hurria A, Lichtman SM. Clinical pharmacology of cancer therapies in older adults. Br J Cancer 2008;98:517–22.
- Spadi R, Brusa F, Ponzetti A, et al. Current therapeutic strategies for advanced pancreatic cancer: A review for clinicians. World J Clin Oncol 2016;7:27–43.
- Seufferlein T, Bachet JB, Van Cutsem E, Rougier P, Group EGW. Pancreatic adenocarcinoma: ESMO-ESDO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012;23 Suppl 7:vii33–40.
- Huguet F, Girard N, Guerche CS, et al. Chemoradiotherapy in the management of locally advanced pancreatic carcinoma: a qualitative systematic review. J Clin Oncol 2009;27:2269–77.
- Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol 2016;17:801–10.
- Gunturu KS, Yao X, Cong X, et al. FOLFIRINOX for locally advanced and metastatic pancreatic cancer: single institution retrospective review of efficacy and toxicity. Med Oncol 2013;30:361.
- Mukherjee S, Hurt CN, Bridgewater J, et al. Gemcitabine-based or capecitabine-based chemoradiotherapy for locally advanced pancreatic cancer (SCALOP): a multicentre, randomised, phase 2 trial. Lancet Oncol 2013;14:317–26.
- Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
- Krzyzanowska MK, Weeks JC, Earle CC. Treatment of locally advanced pancreatic cancer in the real world: population-based practices and effectiveness. J Clin Oncol 2003;21:3409–14.
- Sultana A, Tudur Smith C, Cunningham D, et al. Meta-analyses of chemotherapy for locally advanced and metastatic pancreatic cancer: results of secondary end points analyses. Br J Cancer 2008;99:6–13.
- Hammel P, Huguet F, van Laethem JL, et al. Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: the LAP07 randomized clinical trial. JAMA 2016;315:1844–53.
- Huguet F, Andre T, Hammel P, et al. Impact of chemoradiotherapy after disease control with chemotherapy in locally advanced pancreatic adenocarcinoma in GERCOR phase II and III studies. J Clin Oncol 2007;25:326–31.
- Dholakia AS, Hacker-Prietz A, Wild AT, et al. Resection of borderline resectable pancreatic cancer after neoadjuvant chemoradiation does not depend on improved radiographic appearance of tumor-vessel relationships. J Radiat Oncol 2013;2:413–25.
- Heinemann V, Haas M, Boeck S. Neoadjuvant treatment of borderline resectable and non-resectable pancreatic cancer. Ann Oncol 2013;24:2484–92.
- Morganti AG, Trodella L, Valentini V, et al. Pain relief with short-term irradiation in locally advanced carcinoma of the pancreas. J Palliat Care 2003;19:258–62.
- Arcidiacono PG, Calori G, Carrara S, et al. Celiac plexus block for pancreatic cancer pain in adults. Cochrane Database Syst Rev 2011(3):CD007519.
- Pezzilli R, Andriulli A, Bassi C, et al. Exocrine pancreatic insufficiency in adults: a shared position statement of the Italian Association for the Study of the Pancreas. World J Gastroenterol 2013;19:7930–46.
- Dominguez-Munoz JE. Pancreatic exocrine insufficiency: diagnosis and treatment. J Gastroenterol Hepatol 2011;26 Suppl 2:12–16.
- Bruno MJ, Haverkort EB, Tijssen GP, et al. Placebo controlled trial of enteric coated pancreatin microsphere treatment in patients with unresectable cancer of the pancreatic head region. Gut 1998;42:92–6.
- Wright AA, Keating NL, Balboni TA, et al. Place of death: correlations with quality of life of patients with cancer and predictors of bereaved caregivers’ mental health. J Clin Oncol 2010;28:4457–64.
- Jang RW, Krzyzanowska MK, Zimmermann C, et al. Palliative care and the aggressiveness of end-of-life care in patients with advanced pancreatic cancer. J Natl Cancer Inst 2015;107(3). pii: dju424.
Early-Stage Hodgkin Lymphoma
INTRODUCTION
Hodgkin lymphoma, previously known as Hodgkin’s disease, is a B-cell malignancy with unique pathological and epidemiological features for which highly effective therapies exist. The disease is characterized by the presence of mononuclear and multinucleate giant cells called Hodgkin and Reed-Sternberg (HRS) cells.1
Hodgkin lymphoma is unique compared to other B-cell lymphomas because of the scarcity of the malignant cells in the tumor tissue. The HRS cells usually account for only 0.1% to 10% of the cells in the affected tissues, and the HRS cells induce accumulation of nonmalignant lymphocytes, macrophages, granulocytes, eosinophils, plasma cells, and histiocytes, which constitute more than 90% of tumor cellularity.2 Although the disease was first described by Sir Thomas Hodgkin in 1832, in part because of this unique histopathology, not until 1991 was it conclusively demonstrated that HRS cells are in fact monoclonal germinal center–derived B-cells. This article reviews management and frontline treatment options for limited-stage classical Hodgkin lymphoma and nodular lymphocyte predominant Hodgkin lymphoma. Treatment of advanced stage and relapsed/refractory Hodgkin lymphoma will be discussed in a separate article.
EPIDEMIOLOGY
Hodgkin lymphoma accounts for 0.5% of all malignancies and 11.7% of all lymphomas among adults in the United States.3 The incidence of Hodgkin lymphoma has been steadily increasing over the past 4 decades and was estimated to be 8260 cases in the United States in 2017, with a slight male predominance. Hodgkin lymphoma is expected to cause 1070 deaths in 2017, accounting for 0.2% of all cancer deaths.3 First-degree relatives of patients with Hodgkin lymphoma have a 3- to 9-fold increased risk of having the disease compared to the general population,4 and monozygotic twin siblings of Hodgkin lymphoma patients have a greatly increased risk for developing the disease—up to 100-fold—compared to normal cohorts.5 The incidence is highest among Caucasians, African Americans, and Hispanics, and lower in Asians and American Indians.3 Hodgkin lymphoma incidence shows a bimodal peak distribution, with 1 peak between the ages of 15 and 44 years, and another peak after age 65 years.6
ETIOLOGY/PATHOGENESIS
The cause of Hodgkin lymphoma is unknown. Epstein-Barr virus (EBV) infection is present in up to 40% of Hodgkin lymphoma cases, suggesting a role of this virus in the pathogenesis of some cases. The risk of EBV-positive Hodgkin lymphoma was found to be higher following an episode of infectious mononucleosis, while the risk of EBV-negative Hodgkin lymphoma remained unchanged.7 The incidence of Hodgkin lymphoma is 5 to 14 times higher in HIV-infected patients than in noninfected patients.8 It is not considered an AIDS-defining illness, but has become more frequent with the growth and aging of the HIV-positive population.9,10 Hodgkin lymphoma patients with HIV typically have CD4 lymphocyte counts greater than 200 cells/μL,11 with the incidence of Hodgkin lymphoma actually declining with lower CD4 lymphocyte counts.12 HIV-related Hodgkin lymphoma tends to have an aggressive course, with high rates of EBV positivity.13 The incidence of Hodgkin lymphoma is 1.8 times higher among smokers, and the risk appears to increase with duration of smoking.14,15
The cell of origin of Hodgkin lymphoma, while long suspected to be the HRS cell, remained unproven until the 1990s when micro-dissection and single-cell polymerase chain reaction techniques allowed for confirmation that the HRS cell was in fact a monoclonal germinal center derived B cell.16,17 These HRS cells lack immunoglobulin due to defective transcription regulation and not due to crippling mutations.18,19 The cellular infiltrate in Hodgkin lymphoma appears to play a decisive role in allowing the HRS cells to survive by providing an environment that suppresses cytotoxic immune responses as well as by providing cellular interactions and cytokines that support their growth and survival. The extensive inflammatory infiltrate in classical Hodgkin lymphoma is comprised of T helper 2 (Th2) and regulatory T cells and lacks T helper 1 (Th1) cells, CD8 cytotoxic T cells, and natural killer cells.20 The HRS cells escape apoptosis by several mechanisms which include latent EBV infection and constitutive nuclear factor (NF)-kB pathways, as well as other deregulated signaling pathways that promote survival, such as EBV nuclear antigen 1 (EBNA1) protein, EBV latent infection membrane protein 1 (LMP1), and LMP2.21,22
Genetic alterations in the 9p24 locus which encodes PD-L1/PD-L2 are nearly universally present in classical Hodgkin lymphoma and are now considered a disease-defining feature.23
PATHOLOGIC CLASSIFICATION
According to the 2008 World Health Organization (WHO) classification, Hodgkin lymphoma has 2 clearly defined entities: classical Hodgkin lymphoma (cHL), which accounts for approximately 95% cases, and nodular lymphocyte predominant Hodgkin lymphoma (NLPHL), which accounts for the remaining cases.24 These 2 entities differ in their clinical, pathological, and biological features, which in turn affect prognosis and treatment options. Classical Hodgkin lymphoma is characterized by a paucity of HRS cells surrounded by a background of mixed inflammatory infiltrate comprised of histiocytes, small lymphocytes, eosinophils, neutrophils, plasma cells, fibroblasts, and collagen. Depending on the particular combinations of these elements and the specific features of the neoplastic cells, cases can be subclassified into several cHL subtypes: the nodular sclerosis, mixed cellularity, lymphocyte-rich, and lymphocyte-depleted types.25
The diagnosis of cHL is made based on a combination of morphology of HRS cells and the other cells infiltrating the tissue, combined with immunohistochemical staining. Because of the rare nature of the malignant (clonal) cell in Hodgkin lymphoma specimens, flow cytometry is generally of little value. The HRS cells in cHL are CD30-positive and CD45 negative in virtually all cases, and CD15-positive in 85% of cases.26 B-cell antigens are typically negative except for CD20, which is positive in about 20% cases.27
Nodular sclerosis Hodgkin lymphoma (NSHL) is the most common subtype of cHL, accounting for 65% to 75% of cases. It is common among young adults and tends to involve the mediastinal, supraclavicular, and cervical lymph nodes. NSHL is characterized by the presence of collagen bands that divide the lymphoid tissue into circumscribed nodules. This subtype usually presents as stage I or II disease, typically with neck and/or mediastinal disease, and evidence of EBV infection is present in approximately 10% to 40% of North American cases.7 Patients diagnosed with NSHL generally have a very good prognosis.
Mixed cellularity Hodgkin lymphoma (MCHL) constitutes about 20% to 25% of cHL cases. It affects a somewhat older population, with a median age at diagnosis of 38 years. The typical bimodal age distribution is not seen with MCHL. MCHL has a male predominance (70%), and is more frequent in HIV-infected patients (70% of whom also have EBV infection). Lymphoid tissues have classic HRS cells and significant inflammatory infiltrates. Approximately 50% of patients with MCHL present as stage III or IV with abdominal lymphadenopathy or splenic involvement, and B symptoms are frequent.24
Lymphocyte-rich Hodgkin lymphoma (LRHL) is uncommon, accounting for only 3% to 5% of cases of cHL.28 The disease usually presents at an older age and has a 2:1 male predominance. HRS cells are commonly seen and a large number of reactive lymphocytes are also present. Although on the basis of morphology and immunohistochemistry LRHL belongs to the cHL group, clinically it more closely resembles LPHL. Patients usually present at early stage and rarely have B symptoms. LRHL carries an excellent prognosis, with a greater than 90% PFS after 5 years.23,29
Lymphocyte-depleted Hodgkin lymphoma (LDHL) is the least common form of cHL, accounting for less than 5% of cases. Many cases previously placed in this category are now recognized as diffuse large B-cell lymphoma (DLBCL), anaplastic large-cell lymphoma (ALCL), or NSHL with lymphocyte depletion.30 HRS cells are frequently seen, but reactive inflammatory cells are relatively sparse. EBV infection is seen in up to 90% of cases, commonly associated with HIV-infected individuals. Advanced-stage and symptomatic disease are more common. Prognosis is slightly worse compared to other categories.
NLPHL accounts for approximately 5% of cases of Hodgkin lymphoma. It has a unimodal age distribution, with the peak incidence in the fourth decade, and male predilection of 3:1.28 NLPHL is characterized by large primary lymphoid follicles, with polytypic small B lymphocytes and extensive meshworks of follicular dendritic cells. The lymphocytic/histiocytic (L and H), or “popcorn,” cells scattered within the nodules differ from classic HRS cells, both in their morphology and in their biochemical profile, being frequently negative for CD15, CD30 and for the EBV genome, and usually positive for B-cell antigens such as CD20, suggesting that L and H cells may be immunoglobulin-synthesizing monoclonal B cells. CD45 is also typically positive in NLPHL, in distinction from cHL. NLPHL has an indolent course compared to cHL, and long-term survival is common.19,31 NLPHL commonly presents with limited-stage disease. NLPHL may eventually transform into a more aggressive lymphoma, such as diffuse large B-cell lymphoma (including centroblastic, immunoblastic, or T-cell/histiocyte–rich subtypes), at a rate of 4% to 12%. This can occur even 15 to 20 years after the initial diagnosis of NLPHL.32,33 In a recent large retrospective study of 222 patients with NLPHL, the rate of transformation to DLBCL was 7.6%, with a median time to transformation of 35 months. Overall survival was not adversely affected in patients undergoing transformation compared to those without transformation.34
PRESENTATION
Classical Hodgkin lymphoma usually presents with asymptomatic mediastinal or cervical lymphadenopathy. Half of patients present with stage I or stage II disease.35 A mediastinal mass is seen in most patients with NSHL, at times with bulky disease, with “bulky” defined as a mediastinal mass measuring one-third or more of the maximal thoracic diameter on chest x-ray, or 10 cm on computed tomography (CT) scan. Systemic symptoms, or "B" symptoms—fevers (> 38°C), drenching night sweats, and unexplained weight loss (> 10% of baseline body weight over the preceding 6 months or less)—are detected in approximately 25% of patients. Between 10% and 15% will have extranodal disease, most commonly involving lung, bone, and liver. NLPHL usually presents with limited-stage disease without B symptoms; it typically has a more indolent presentation and clinical course than cHL.
INITIAL EVALUATION AND STAGING
The initial workup includes a complete blood count (CBC), erythrocyte sedimentation rate (ESR), lactate dehydrogenase (LDH), and chemistry studies to evaluate renal function and liver function. Fine-needle aspiration will usually fail to identify the infrequent HRS cells, and instead only reveal the reactive background of inflammatory cells. Generous (large gauge) core needle biopsies may provide diagnosis effectively in some cases, but in general, an excisional lymph node biopsy is preferred to ensure an accurate diagnosis and avoid the need for repeated biopsy procedures. In cases where an excisional biopsy would be difficult or risky, a core needle biopsy procedure is a reasonable first step, with the understanding that a subsequent surgical procedure may still be necessary.
Baseline imaging includes CT scans of the neck, chest, abdomen, and pelvis. Use of positron emission tomography (PET) scanning is now standard in the initial evaluation and assessment of treatment response in Hodgkin lymphoma.36 Due to the increased sensitivity of PET or PET/CT scan, additional lesions may be identified that were not seen on conventional CT scans. This will alter the staging, and potentially the treatment plan, in up to 25% to 30% of patients.37,38 PET/CT scan performed during initial evaluation also facilitates optimal interpretation of post-therapy PET/CT scans and is therefore strongly encouraged as a part of the initial staging evaluation.39
Recent studies have shown that bone marrow biopsy is not routinely needed in the initial staging of cHL. A study of 454 patients concluded that bone marrow biopsy would not have altered the stage in any stage I or II patients. It was further concluded that overall treatment strategy would not have been altered for any of the patients.40 Based on this study and others, it is now clear that FDG-PET has a high sensitivity, and when PET scan is negative (in the bone marrow and skeleton), a bone marrow biopsy provides little additional value. For patients with significant cytopenias, a bone marrow biopsy is reasonable. Such patients may benefit from a bilateral biopsy, which increases the probability of demonstrating bone marrow involvement by 16% to 33%.41,42 Techniques such as staging laparotomy and lymphangiography are now considered obsolete.
Hodgkin lymphoma is staged according to the Ann Arbor staging system (Table 1). The original Ann Arbor staging was published in 1971,43 and in 1989 the “Cotswold modifications” extended the definitions of stage IV disease and the suffix “X” was added to denote bulky disease.44 Both systems were developed for the staging of Hodgkin lymphoma, but are now used for staging non-Hodgkin lymphoma as well.

PROGNOSTIC FACTORS
For the purposes of prognosis and selection of treatment, Hodgkin lymphoma is commonly classified into early-stage favorable, early-stage unfavorable, and advanced stage. Early-stage Hodgkin lymphoma refers to patients with Ann Arbor stage I or stage II disease. With early-stage Hodgkin lymphoma, the prognosis varies significantly based on factors such as the presence of B symptoms, elevated erythrocyte sedimentation rate ([ESR] > 50 mm/hr), number of nodal sites involved, older age, and a large mediastinal mass. For this reason, most clinical trials to evaluate treatment strategies for early-stage Hodgkin lymphoma are based on various combinations of these risk factors. The definition of early-stage unfavorable Hodgkin lymphoma varies across different clinical trial study groups, and it is important to understand the definition in interpreting the results of these trials (Table 2).45,46

In the German Hodgkin Study Group (GHSG) trials, early-stage Hodgkin lymphoma is stratified into a high risk (“unfavorable”) group defined by any of the following: a large mediastinal mass (one third of maximum thoracic diameter), extra-nodal disease, 3 or more nodal areas, and an ESR of > 50 mm/hr in asymptomatic patients or > 30 mm/hr in patients with B symptoms. Low-risk (“favorable”) patients lack all of these factors.47 The European Organization for Research and Treatment of Cancer (EORTC) defines the unfavorable prognostic group as older than 50 years of age, large mediastinal adenopathy (maximum width on a chest radiograph of at least one third of the internal transverse diameter of the thorax at the level of T5 through T6 or any mass of ≥ 10 cm in the largest dimension), an ESR of 50 mm/hr and no B symptoms, or with an ESR of 30 mm/hr in those who have B symptoms, and/or 4 or more regions of involvement.48 The National Cancer Institute of Canada (NCIC) Clinical Trials Group and the Eastern Cooperative Oncology Group (ECOG) define high-risk groups as presence of B symptoms, bulky disease with a mediastinal mass width of at least one third of the maximum chest wall diameter, or any mass greater than 10 cm, and patients with intra-abdominal disease.49,50
Gene-expression profiling in Hodgkin lymphoma has identified a gene signature of tumor-associated macrophages that was able to identify patients with a higher risk for primary treatment failure. In an independent cohort of patients, an increased number of CD68-positive macrophages was correlated with inferior outcomes.51,52 Studies such as these underscore the importance of the tumor “microenvironment” (ie, the nonmalignant cells within a tumor) in determining the overall clinical behavior of a malignancy. While quantification of CD68-positive macrophages has potential to be applied to routine clinical practice, prospective data using CD68 as a tool for risk-adapted therapy is lacking.
Genetic alterations and amplifications in the 9p24.1 locus have recently been found to be a defining genetic feature of cHL. Amplification of 9p24.1 has been associated with unfavorable outcomes. Amplification of 9p24.1 (which includes the loci encoding the PD-L1 and PD-L2 genes) is more common in patients with advanced stage disease and is associated with shorter PFS.23
A recent study attempted to integrate several different prognostic factors in cHL patients who were treated with ABVD (adriamycin [doxorubicin], bleomycin, vinblastine, and dacarbazine) and underwent an interim PET (iPET) scan after 2 cycles of ABVD. Focusing on those with a negative iPET scan, it was found that expression of CD68 and PD-1 in microenvironment cells, and STAT1 negativity in HRS cells identified a subset of PET-2 negative patients with a 3-year PFS significantly lower than that of the remaining PET-2 negative population (64% versus 95%). The algorithm correctly predicted the response to treatment in more than half of the patients who had relapse or disease progression despite a negative PET-2 scan. It therefore appears feasible, using tissue biomarkers at diagnosis, to identify patients at increased risk for poor outcome, even if the iPET scan is negative.53
ROLE OF PET/CT IN ASSESSMENT OF RESPONSE TO THERAPY
PET/CT has been increasingly used for response assessment at various stages in lymphoma in recent years. Almost all types of lymphomas are fluorodeoxyglucose (FDG) avid; however, Hodgkin lymphoma is FDG avid in 97% to 100% of cases. In 2009, a 5-point scale was developed to score PET images with regard to treatment response, either partway through treatment (iPET) or at the end of therapy.54 It was recommended as the standard reporting tool at the First International Workshop on PET in Lymphoma in Deauville, France, in 2009, and is thus now referred to as the Deauville score. A score of 1 is given if there is no uptake, 2 if the uptake ≤ mediastinum, 3 if > mediastinum but ≤ liver, 4 if uptake moderately higher than liver, 5 if uptake is markedly higher than liver and/or new lesions. X designates new areas of uptake unlikely to be related to lymphoma. In most trials, a score of 1 or 2 is considered a complete response and a score of 4 or 5 is considered a treatment failure. A score of 3 is sometimes considered a complete response, depending on the study. The Deauville criteria have been widely used in newer clinical trials utilizing response-adapted treatment as defined by PET response. PET/CT is recommended for staging and restaging at the end of therapy, in clinical practice, and clinical trials. Interim PET/CT scan, while commonly performed in clinical practice, is only recommended if the results will alter therapy (eg, if that information will result in the clinician omitting radiation therapy [RT] or altering the chemotherapy plan).
Early studies of iPET showed that achieving PET negativity early in the course of treatment was strongly associated with PFS and overall survival.55 Subsequent studies confirmed the importance of achieving a negative iPET. As a result, considerable efforts have been put into designing response-adapted treatment approaches using iPET (see Treatment section), with some of these approaches now being listed in the National Comprehensive Cancer Network (NCCN) guidelines and being used in standard practice.
TREATMENT
EVOLUTION OF TREATMENT
The treatment of Hodgkin lymphoma has evolved over the past century, starting with the discovery of RT as effective treatment in the early 20th century. Long-term survival of patients with Hodgkin lymphoma treated with involved-field radiation therapy (IFRT) was first reported in the 1960s.56,57 Outcomes improved further with the introduction of combined modality treatment (CMT) using chemotherapy and RT, with the overall 5-year relative survival for patients with Hodgkin lymphoma (all stages) treated in 2006–2012 being 85.4% to 87.3%.3 Since the majority of patients are now cured with modern therapy, treatment-related complications have become an important competing cause of mortality. Recent studies have therefore focused on maintaining efficacy while reducing toxicities, and refining the process of selecting patients who might benefit from more aggressive therapy. While RT was the first treatment modality shown to be curative for Hodgkin lymphoma,56,57 multiple subsequent studies showed that CMT is superior to RT alone in terms of relapse-free survival.58–63 In the GHSG H8-F trial, the estimated 5-year event-free survival and overall survival rates were significantly higher after 3 cycles of MOPP-ABV (mechlorethamine, vincristine, procarbazine, and prednisone combined with doxorubicin, bleomycin, and vinblastine) plus IFRT than after subtotal nodal radiotherapy alone. The 10-year overall survival estimates were 97% and 92%, respectively (P = 0.001).64 As a result, CMT replaced RT alone as the standard of care for limited-stage Hodgkin lymphoma. However, for elderly or infirm patients, or those with other comorbidities making them poor chemotherapy candidates, RT alone may be a very reasonable option.65 More recently, an increasing body of evidence has accumulated to support the use of chemotherapy alone in early stage cHL. This literature has consistently shown that omission of RT is associated with a modest increase in relapse, without a clear compromise in long-term overall survival. For some patients, the trade-off in terms of avoiding radiation (and the associated late effects) may be worth the small increase in relapse risk, since long-term survival does not appear to be substantially worse with chemo-therapy alone. Table 3 and Table 4 provide a summary of recent key studies which have defined treatment options for early-stage cHL.48,66–71
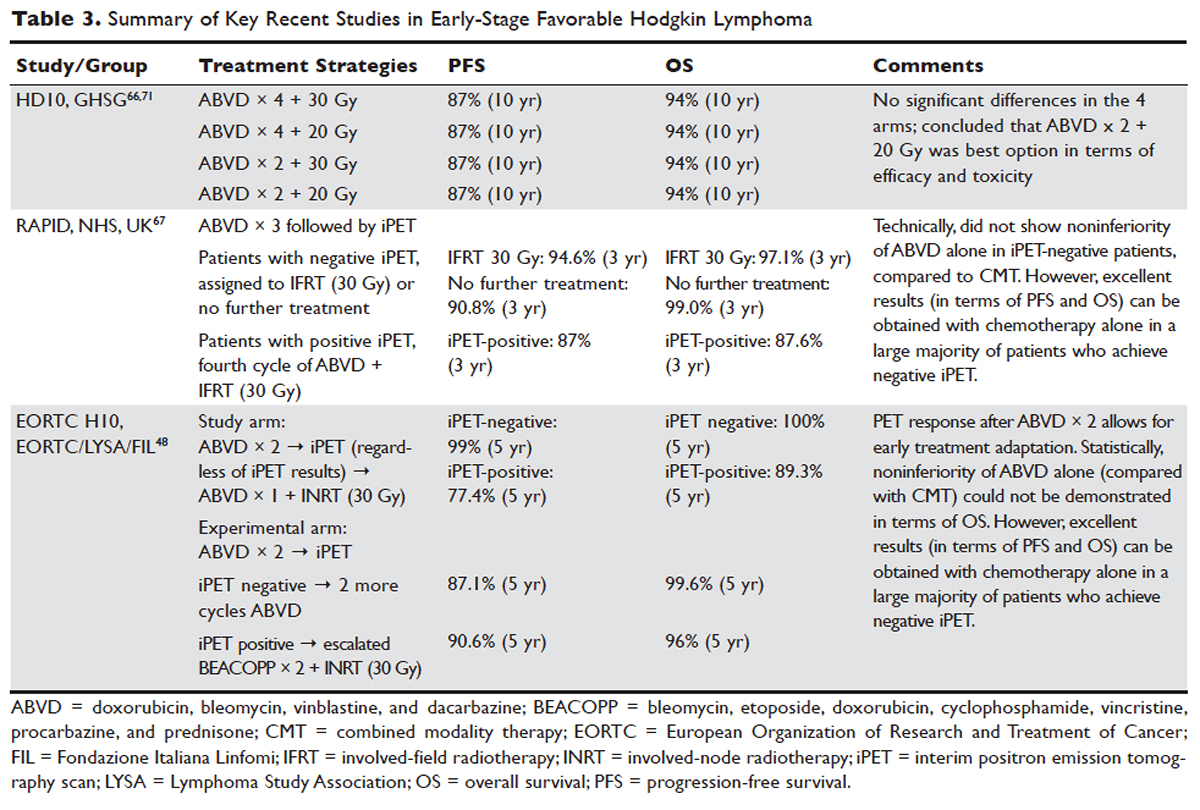

EARLY-STAGE NLPHL
NLPHL usually presents with limited-stage disease without B symptoms and has an indolent course with a slightly better prognosis compared to cHL.72 Due to the rarity of the disease, treatment guidelines are mostly based on retrospective analyses from single or multi-institution studies or subgroup analyses, often with relatively short follow-up. These studies must be interpreted with caution because of the possibility of inaccuracies in the pathologic diagnosis, small sample sizes, and selection bias. Treatment options for limited-stage NLPHL include observation, single-agent rituximab, IFRT (or involved-site radiation therapy [ISRT]) alone, or CMT.46
Historically, patients with limited-stage NLPHL have been treated with RT alone, with 80% to 85% PFS and 85% to 95% overall survival rates.73–75 Patients who relapse or progress after RT in general can successfully undergo salvage therapy.74 In one study, rates of PFS and overall survival were similar among patients who had limited-field, regional-field, or extended-field RT,75 indicating that IFRT is preferred. Studies comparing RT alone and CMT are limited. The GHSG HD7 trial included a subset of NLPHL patients, with a trend towards improved freedom from treatment failure (96% versus 83%) favoring CMT. This, however, did not translate into improved overall survival.47 A retrospective analysis of the British Columbia Cancer Agency database compared patients with limited-stage NLPHL treated with RT alone to patients who received 2 cycles of ABVD followed by RT. A significant improvement in PFS (91% versus 65%) and overall survival (93% versus 84%) was seen, favoring CMT.76
Chemotherapy alone is not recommended for limited-stage NLPHL since studies evaluating chemotherapy alone are quite limited and indicate relatively high rates of treatment failure. Given that the malignant cells in NLPHL are CD20-positive, single-agent rituximab has also been studied in this disease, including a study as frontline therapy in limited-stage patients. In this phase 2 trial in newly diagnosed patients with stage IA disease, an overall response rate (ORR) of 100% was seen, with an 85% complete response (CR) rate.77 At 3 years, overall survival was 100% and PFS was 81%, indicating that the responses with single-agent rituximab are less durable than those with RT.
Advani et al evaluated rituximab followed by observation versus rituximab (R) followed by maintenance rituximab (MR) for 2 years in 39 new or previously treated patients. At 4 weeks the ORR was 100% (with CR in 67%, and partial response in 33%). At a median follow up of 9.8 years for R alone, and 5 years for R+MR, median PFS was 3 and 5.6 years, respectively (P = 0.26). Estimated 5-yr PFS and overall survival in patients treated with R versus R+MR were 39.1% and 95.7% versus 58.9% and 85.7%, with Pvalues of 0.26 (PFS) and 0.38 (overall), respectively. Maintenance rituximab therefore appears to prolong remission, although the results did not quite reach statistical significance.78 Even though rituximab does not appear to be curative in NLPHL, it is a reasonable option for patients with early-stage NLPHL who are not good candidates for definitive RT. Whether combining rituximab with RT or CMT might further improve outcomes in early-stage NLPHL has not yet been determined.
In children, surgery alone may lead to long-term remission or possibly cure of limited-stage NLPHL. In a European multicenter retrospective study, 58 patients underwent surgery for limited-stage NLPHL. Among the 51 patients who achieved complete remission following surgery, 67% remained progression-free and 100% were alive at a median follow up of 43 months.79 In adults, there is no data to support surgical treatment alone for NLPHL. Finally, observation may be a reasonable option in elderly or infirm patients for whom NLPHL is unlikely to affect life expectancy. For younger patients, given the excellent outcome with modern therapy and the long-term risk of transformation of NLPHL into an aggressive non-Hodgkin lymphoma, observation is generally not recommended.
The NCCN recommends RT (ISRT or IFRT, 30–36 Gy) as the preferred treatment for stage IA and IIA non-bulky NLPHL. In patients with stage IA disease with complete excision of solitary nodule, observation may be appropriate. A course of chemotherapy with ISRT with or without rituximab is recommended for patients with stage IB or IIB disease, or patients with stage IA or IIA bulky disease.
FIRST-LINE TREATMENT OF LIMITED-STAGE CHL
Early-Stage Favorable cHL
There is lack of consensus regarding the ideal treatment approach for patients with early-stage favorable cHL. However, there are several excellent options available, with overall survival rates exceeding 90%. Most of these regimens involve CMT, although some chemotherapy-alone approaches have been evaluated as well. Concurrent with the demonstration of excellent long-term remission rates with CMT, it became apparent that the long-term survival and quality of life of these patients is determined in large part by the risk of serious (and potentially fatal) treatment-related toxicities. Such toxicities consist primarily of secondary malignancies and cardiovascular events, and can continue to cause significant morbidity and mortality even 2 to 3 decades after treatment.80–82 As a result, treatment decisions for these patients are complicated and require balancing efficacy against risk of late complications.
In the United States, until recently, CMT was generally considered the standard of care, with robust long-term data regarding efficacy. The most commonly used regimen has been ABVD for 2 to 4 cycles followed by IFRT. In some German studies, escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone) has been used, but this is not a general standard of care in the United States for early-stage patients.
More recent data suggests that the rate of serious late complications in Hodgkin lymphoma patients is decreasing, likely due to less extensive radiation fields, lower radiation doses, and a movement away from the MOPP regimen to ABVD.83,84 For patients who meet the “favorable” criteria set forth in the GHSG HD10 trial (see Table 2), 2 cycles of ABVD followed by 20 Gy of IFRT is an attractive option, with efficacy preserved and a low anticipated rate of late effects.66 With this approach, and with long-term (10 years) follow up, all 4 arms had similar PFS (87%) and overall survival (94%), whether 2 or 4 cycles of ABVD were given. When the effects of 20-Gy and 30-Gy doses of RT were compared, there were also no significant differences in freedom from treatment failure or overall survival. Adverse events and acute toxic effects of treatment were most common in the patients who received 4 cycles of ABVD and 30 Gy of RT.66,71
In recent years, in an attempt to reduce late effects further, regimens consisting of chemotherapy alone have been investigated. In a study by Meyer et al, at 12 years the rate of overall survival was 94% among those receiving ABVD alone, as compared with 87% among those receiving subtotal nodal RT; the rates of freedom from disease progression were 87% and 92% in the 2 groups; and the rates of event-free survival were 85% and 80%, respectively.50 However, it is important to note that this study did not include a CMT arm for the early favorable patients, and did not utilize modern RT techniques. Nevertheless, this early study and others60 suggested that chemotherapy alone may be a reasonable option for some early-stage cHL patients, particularly for patients who are felt to be at increased risk for late toxicities from RT. As a result, additional studies have been conducted evaluating CMT versus chemotherapy alone for early-stage cHL. Many of these studies have incorporated interim PET/CT scan to develop a response-adapted approach to decide which patients are least likely to benefit from RT.
The HD-13 study was a follow-up study for HD-10, looking at deletion of bleomycin, dacarbazine, or both from the ABVD backbone. The ABD arm was closed early, because of an excess rate of treatment failure. Among the 1243 patents assigned to either the ABVD or AVD arm at 5 years of follow-up, there was 4.3% difference in PFS. This study was not able to demonstrate that 2 cycles of AVD was noninferior to 2 cycles of ABVD, each followed by 30 Gy IFRT, even though there was no difference in all 4 groups. It confirmed 2 cycles of ABVD as the preferred regimen in early favorable Hodgkin lymphoma, when CMT is the plan of care. However, for patients over age 60 to 65 years, or those with underlying cardiac or pulmonary comorbidities, bleomycin has significant risk of toxicity. In that setting, AVD is a safer option, with only a very modest decrease in 5-year PFS.
Based on the observation that iPET scan is highly predictive of outcome in Hodgkin lymphoma,55,85 several trials have employed the use of an iPET scan to guide therapy. It is hoped that such studies will lead to new PET-directed treatment algorithms in which patients who require more aggressive therapy (eg, with CMT, or escalated BEACOPP) can be identified, and the remaining patients can be safely treated less aggressively (eg, with chemotherapy alone).
In the EORTC H10 trial, performed to evaluate treatment adaptation on the basis of iPET scan results in stage I and II Hodgkin lymphoma, a control arm received standard combined modality treatment (3 or 4 cycles of ABVD with INRT) irrespective of PET scan results. In the experimental arm, patients with a negative PET scan after 2 cycles of ABVD continued with 1 or 2 cycles of ABVD and did not receive RT. The iPET-positive patients received either standard treatment with ABVD plus INRT or escalated BEACOPP plus INRT. The iPET-negative patients received either ABVD only or ABVD plus INRT. The final results of this study, published recently, showed that in the iPET-positive patients the 5-year PFS was improved from 77.4% with standard ABVD plus INRT to 90.6% with escalated BEACOPP plus INRT (P = 0.002). In iPET-negative patients, 5-year PFS in the favorable group was 99% versus 87.1% in favor of ABVD plus INRT. The H10 study suggested that PET results after 2 cycles of ABVD can be integrated into treatment planning, In iPET-negative patients, the study was technically not able to demonstrate the noninferiority of the ABVD only regimen, owing to a higher risk of relapse if INRT is omitted, particularly in the favorable group.48 However, this study does show that excellent outcomes can be obtained with omission of RT in patients with a negative iPET scan. This study provides a cautionary lesson though, in that the increase in relapse rate associated with omission of RT was more substantial (12%) for favorable versus unfavorable early-stage patients (2.5%), and this difference was only apparent after longer (5 years) follow-up. Despite this, chemotherapy alone is considered a reasonable treatment option, especially for patients felt to be at increased risk for late toxicities of RT or for patients who wish to avoid the risks of RT, with over 99% of patients alive at 5 years.
Similar results were shown in the RAPID trial, in which patients with limited-stage cHL underwent 3 cycles of ABVD followed by PET assessment.67 Patients with a negative PET (n = 426) were then randomized to RT (n = 209) versus no further therapy (n = 211). At a median of 60 months of follow-up, 3-year PFS was 94.6% in the RT group and 90.8% in the chemotherapy alone group. Similar to the H10 trial, it was concluded that chemotherapy alone was statistically inferior to CMT in terms of PFS. However, also similar to the H10 trial, the RAPID trial demonstrated that excellent results can be obtained in early-stage cHL patients with omission of RT, if iPET scan is negative after 3 cycles of ABVD, as there was no survival difference. These findings indicate that, when relapses occur as a result of omission of RT, such patients can be effectively treated later.
In the ongoing GHSG HD16 trial, patients with early-stage favorable cHL will be randomly assigned to a standard approach (ABVD × 2 cycles followed by 20-Gy IFRT) versus an experimental approach in which they receive ABVD for 2 cycles and then undergo PET scan. If the PET remains positive, they will receive 20-Gy IFRT. If the PET is negative, they will receive no further therapy. This trial could ultimately define ABVD for 2 cycles as a treatment option.
It is clear from these studies that omission of RT results in a somewhat higher rate of relapse but can be considered in selected patients. However, taking a less aggressive frontline approach may also be justified by the fact that, for those who do relapse, successful salvage therapies are available. Aggressive salvage therapy with autologous stem cell transplantation historically can cure approximately 50% of relapsed patients. With new and emerging therapies for relapsed disease, such as brentuximab vedotin and the PD-1 inhibitors (eg, nivolumab and pembrolizumab), the ability to cure relapsed patients may improve even more, further calling into question the practice of applying CMT uniformly for early-stage patients undergoing first-line therapy. Unfortunately, there is insufficient data from large randomized studies with long-term follow-up to fully address this issue currently, and there remains some controversy around this issue. NCCN recommends restaging PET/CT after 3 cycles of ABVD if a chemotherapy alone treatment modality is chosen. If the Deauville score is 1 or 2, either observation or 1 additional cycle of ABVD is recommended.46
Early-Stage Unfavorable cHL
In the United States, historically early-stage unfavorable Hodgkin lymphoma has been treated with CMT, most commonly 4 to 6 cycles of ABVD followed by consolidative RT. With this approach one can expect a 5-year PFS of approximately 80% to 85%.58,64,86 The GHSG HD8 trial showed that RT volume size reduction from extended-field to involved-field after COPP + ABVD chemotherapy for 2 cycles produced similar results and less toxicity in patients with early-stage unfavorable cHL.86 The GHSG trial HD11 established ABVD for 4 cycles plus 30-Gy IFRT as a standard for early unfavorable Hodgkin lymphoma. The freedom from treatment failure at 5 years was 85.0%, and overall survival was 94.5%.68
In the HD14 study by the GHSG, patients with early unfavorable cHL were treated with 2 cycles of escalated BEACOPP followed by 2 cycles of ABVD, versus 4 cycles of ABVD. All patients then received 30 Gy of consolidative IFRT. A 5-year PFS of 95% was seen in the experimental arm, compared with 89% in the standard (ABVD) arm. As expected, this regimen was associated with more acute hematologic toxicity, and there was no difference between the 2 regimens with respect to overall survival or fertility.69 Given the lack of improved survival and increased toxicity, ABVD has remained the standard chemotherapy regimen for early unfavorable cHL in the United States. NCCN recommends a restaging PET scan after 2 cycles of ABVD and to continue with 2 to 4 cycles of ABVD or escalated BEACOPP with or without ISRT based on Deauville scores.
Another viable treatment option is the Stanford V regimen, a condensed, 12-week regimen that includes mechlorethamine, doxorubicin, vinblastine, prednisone, vincristine, etoposide, and bleomycin, followed by IFRT.87 In a randomized phase 3 trial conducted by ECOG (E2496), patients with stage I/II Hodgkin lymphoma with bulky mediastinal disease or advanced-stage disease were randomized to ABVD × 6 to 8 cycles versus Stanford V. RT was given (36 Gy) for those with bulky mediastinal disease or to sites of disease greater than 5 cm in the Stanford V arm. In a subset analysis focusing only on those with stage I/II bulky mediastinal disease, the 5-year failure free survival was 85% versus 79% and the 5-year overall survival was 96% versus 92% for the ABVD versus Stanford V arms, respectively. These differences were not statistically significant.70 While the Stanford V regimen has the advantages of a 12-week treatment duration and a lower cumulative amount of bleomycin and doxorubicin, the Stanford V arm had higher rates of grade 3 lymphopenia and grade 3 to 4 peripheral neuropathies. In addition, Stanford V requires that most patients undergo RT (to original sites of disease measuring 5 cm or more plus contiguous areas). As a result, the investigators concluded that ABVD × 4 cycles plus IFRT remains the standard of care for patients with early unfavorable Hodgkin lymphoma with bulky mediastinal disease.
Other regimens have been studied in hopes of reducing toxicity, including the EVE regimen (epirubicin, vinblastine, and etoposide). This regimen was compared to ABVD in early unfavorable Hodgkin lymphoma patients, with all patients undergoing the same RT program. No differences were observed between the ABVD and EVE arms in terms of complete remission rate and overall survival. However, patients who received EVE had a significantly worse outcome than those who received ABVD in terms of relapse-free survival and failure-free survival.88 EBVP (epirubicin, bleomycin, vinblastine, and prednisone) followed by IFRT was less efficacious compared with MOPP/ABV–type therapy.58
An area of active investigation is whether RT can be safely omitted in patients with early- stage unfavorable cHL. The EORTC H10 study showed that, for patients with a negative iPET scan (after 2 cycles), the 5-year PFS rates were 92.1% versus 89.6% for ABVD plus INRT versus ABVD alone, respectively. While this technically did not meet criteria for noninferiority of ABVD alone, this study demonstrated that, for those with negative iPET, ABVD × 6 cycles (without radiation) can result in long-term remission in a high proportion (89%) of patients. For iPET-positive patients, 2 cycles of escalated BEACOPP were given followed by 30 Gy of IFRT on the experimental arm. This resulted in a 5-year PFS of 90.6% versus 77.4%, suggesting this may be a preferred approach for early-stage unfavorable patients with a positive iPET.48 Even though the noninferiority of ABVD alone could not be established based on the statistical design of the study, the current NCCN guidelines recommend restaging after 2 cycles of ABVD for stage I or II unfavorable cHL and using that iPET as a guide, based on Deauville scores. For scores 1–3, ABVD × 2 cycles (total 4 cycles) plus ISRT or AVD × 4 (total 6) with or without ISRT is recommended. For a Deauville score of 4, escalated BEACOPP × 2 cycles or ABVD × 2 cycles (total 4) followed by ISRT is recommended. If the Deauville score is 5, further treatment decisions should be made based on repeat biopsy results. A follow up PET/CT is recommended for Deauville scores of 4 and 5 to confirm complete response.46
LATE EFFECTS AND THE EVOLUTION OF RADIATION THERAPY
The RT given in Hodgkin lymphoma has evolved considerably over the years, from extended field or subtotal nodal fields developed in the 1960s, to the more focused involved-field or even involved-site radiation commonly given now. This approach reduces radiation volumes, and it already is becoming evident that the relative risk of breast cancer among young females receiving mediastinal RT for Hodgkin lymphoma is declining.89 Cardiac dose is reduced significantly with IFRT compared to older radiation techniques as well. The extent of radiation may be reduced even further with involved-nodal/involved site or intensity-modulated approaches.90
With new RT techniques allowing for more focused therapy and lower doses of radiation, models predict that the rate of long-term complications will decline further.91,92 Furthermore, response-adapted (ie, PET-directed) approaches, as discussed in detail earlier in the article, are expected to increasingly allow for identification of patients who can safely avoid radiation entirely, which will hopefully lead to an even lower rate of late complications of therapy.
MONITORING FOR RELAPSE
A number of recent studies have shown that, for patients who achieve complete remission with first-line therapy, performing repeated scheduled surveillance imaging does not improve outcomes. In fact, most relapses are detected by the patient (due to symptoms or recurrence of lymph node enlargement). It is rare that a relapse would be detected by surveillance imaging alone. Furthermore, surveillance that includes routine imaging has not been associated with improved survival.93 As a result, it is now recommended that patients undergo regular follow-up with symptom review, physical exam, and basic laboratory studies. Imaging studies should be obtained as needed for patients who develop signs, symptoms, exam findings, or laboratory values concerning for relapse.
More important than scheduled surveillance imaging for relapse is monitoring for late effects of therapy. These fall into several broad categories such as cardiovascular disease (coronary disease, congestive heart failure, valvular disease, carotid artery disease), pulmonary disease, hypothyroidism, and secondary malignancies. Aggressive surveillance for breast cancer is especially warranted in female patients who underwent chest radiation.46
CONCLUSION
Hodgkin lymphoma is characterized pathologically by the presence of HRS cells accompanied by a polymorphous cellular infiltrate. It is a disease with a bimodal age distribution, several pathologic subtypes, and numerous treatment options. Overall, the prognosis for patients with early-stage disease is excellent, and although a majority of patients can now be cured, further studies are needed to optimize treatment such that short- and long-term treatment-related toxicities are minimized, without compromising disease control and cure.
- Küppers R, Rajewsky K, Zhao M, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A 1994;91:10962–6.
- Küppers R. The biology of Hodgkin›s lymphoma. Nat Rev Cancer 2009;9:15–27.
- National Cancer Institute. SEER cancer statistics review, 1975–2014. 2017. http://seer.cancer.gov/csr/1975_2013/. Accessed April 27, 2017.
- Haim N, Cohen Y, Robinson E. Malignant lymphoma in first-degree blood relatives. Cancer 1982;49:2197–200.
- Mack TM, Cozen W, Shibata DK, et al. Concordance for Hodgkin’s disease in identical twins suggesting genetic susceptibility to the young-adult form of the disease. N Engl J Med 1995;332:413–8.
- Sant M, Allemani C, Tereanu C, et al. Incidence of hematologic malignancies in Europe by morphologic subtype: results of the HAEMACARE project. Blood 2010;116:3724–34.
- Hjalgrim H, Askling J, Rostgaard K, et al. Characteristics of Hodgkin’s lymphoma after infectious mononucleosis. N Engl J Med 2003;349:1324–32.
- Hessol NA, Katz MH, Liu JY, et al. Increased incidence of Hodgkin disease in homosexual men with HIV infection. Ann Intern Med 1992;117:309–11.
- Shiels MS, Pfeiffer RM, Gail MH, et al. Cancer burden in the HIV-infected population in the United States. J Natl Cancer Inst 2011;103:753–62.
- Powles T, Robinson D, Stebbing J, et al. Highly active antiretroviral therapy and the incidence of non-AIDS-defining cancers in people with HIV infection. J Clin Oncol 2009;27:884–90.
- Bedimo RJ, McGinnis KA, Dunlap M, et al. Incidence of non-AIDS-defining malignancies in HIV-infected versus noninfected patients in the HAART era: impact of immunosuppression. J Acquir Immune Defic Syndr 2009;52:203–8.
- Biggar RJ, Jaffe ES, Goedert JJ, et al. Hodgkin lymphoma and immunodeficiency in persons with HIV/AIDS. Blood 2006;108:3786–91.
- Thompson LD, Fisher SI, Chu WS, et al. HIV-associated Hodgkin lymphoma: a clinicopathologic and immunophenotypic study of 45 cases. Am J Clin Pathol 2004;121:727–38.
- Briggs NC, Hall HI, Brann EA, et al. Cigarette smoking and risk of Hodgkin’s disease: a population-based case-control study. Am J Epidemiol 2002;156:1011–20.
- Castillo JJ, Dalia S, Shum H. Meta-analysis of the association between cigarette smoking and incidence of Hodgkin’s Lymphoma. J Clin Oncol 2011;29:3900–6.
- Kanzler H, Kuppers R, Hansmann ML, Rajewsky K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med 1996;184:1495–505.
- Stein H, Hummel M. Cellular origin and clonality of classic Hodgkin’s lymphoma: immunophenotypic and molecular studies. Semin Hematol 1999;36:233-41.
- Marafioti T, Hummel M, Foss HD, et al. Hodgkin and reed-sternberg cells represent an expansion of a single clone originating from a germinal center B-cell with functional immunoglobulin gene rearrangements but defective immunoglobulin transcription. Blood 2000;95:1443–50.
- Marafioti T, Hummel M, Anagnostopoulos I, et al. Origin of nodular lymphocyte-predominant Hodgkin’s disease from a clonal expansion of highly mutated germinal-center B cells. N Engl J Med 1997;337:453–8.
- van den Berg A, Visser L, Poppema S. High expression of the CC chemokine TARC in Reed-Sternberg cells. A possible explanation for the characteristic T-cell infiltratein Hodgkin’s lymphoma. Am J Pathol 1999;154:1685–91.
- Bargou RC, Emmerich F, Krappmann D, et al. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J Clin Invest 1997;100:2961–9.
- Luftig M, Yasui T, Soni V, et al. Epstein-Barr virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKK alpha-dependent noncanonical NF-kappaB activation. Proc Natl Acad Sci U S A 2004;101:141–6.
- Roemer MGM, Advani RH, Ligon AH, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol 2016;34:2690–7.
- Swerdlow SH CE, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008.
- Campo E, Swerdlow SH, Harris NL, et al. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 2011;117:5019–32.
- von Wasielewski R, Mengel M, Fischer R, et al. Classical Hodgkin’s disease. Clinical impact of the immunophenotype. Am J Pathol 1997;151:1123–30.
- Tzankov A, Krugmann J, Fend F, et al. Prognostic significance of CD20 expression in classical Hodgkin lymphoma: a clinicopathological study of 119 cases. Clin Cancer Res 2003;9:1381–6.
- Diehl V, Sextro M, Franklin J, et al. Clinical presentation, course, and prognostic factors in lymphocyte-predominant Hodgkin’s disease and lymphocyte-rich classical Hodgkin’s disease: report from the European Task Force on Lymphoma Project on Lymphocyte-Predominant Hodgkin’s Disease. J Clin Oncol 1999;17:776–83.
- Shimabukuro-Vornhagen A, Haverkamp H, Engert A, et al. Lymphocyte-rich classical Hodgkin’s lymphoma: clinical presentation and treatment outcome in 100 patients treated within German Hodgkin’s Study Group trials. J Clin Oncol 2005;23:5739–45.
- Slack GW, Ferry JA, Hasserjian RP, et al. Lymphocyte depleted Hodgkin lymphoma: an evaluation with immunophenotyping and genetic analysis. Leuk Lymphoma 2009;50:937–43.
- Mason DY, Banks PM, Chan J, et al. Nodular lymphocyte predominance Hodgkin’s disease. A distinct clinicopathological entity. Am J Surg Pathol 1994;18:526–30.
- Rudiger T, Gascoyne RD, Jaffe ES, et al. Workshop on the relationship between nodular lymphocyte predominant Hodgkin’s lymphoma and T cell/histiocyte-rich B cell lymphoma. Ann Oncol 2002;13 Suppl 1:44–51.
- Sundeen JT, Cossman J, Jaffe ES. Lymphocyte predominant Hodgkin’s disease nodular subtype with coexistent “large cell lymphoma”. Histological progression or composite malignancy? Am J Surg Pathol 1988;12:599–606.
- Kenderian SS, Habermann TM, Macon WR, et al. Large B-cell transformation in nodular lymphocyte-predominant Hodgkin lymphoma: 40-year experience from a single institution. Blood. 2016;127:1960–6.
- Mauch PM, Kalish LA, Kadin M, et al. Patterns of presentation of Hodgkin disease. Implications for etiology and pathogenesis. Cancer 1993;71:2062–71.
- Juweid ME, Cheson BD. Positron-emission tomography and assessment of cancer therapy. N J Engl Med 2006;354:496–507.
- Hutchings M, Loft A, Hansen M, et al. Position emission tomography with or without computed tomography in the primary staging of Hodgkin’s lymphoma. Haematologica 2006;91:482–9.
- Naumann R, Beuthien-Baumann B, Reiss A, et al. Substantial impact of FDG PET imaging on the therapy decision in patients with early-stage Hodgkin’s lymphoma. Br J Cancer 2004;90:620–5.
- Juweid ME, Stroobants S, Hoekstra OS, et al. Use of positron emission tomography for response assessment of lymphoma: consensus of the Imaging Subcommittee of International Harmonization Project in Lymphoma. J Clin Oncol 2007;25:571–8.
- El-Galaly TC, d’Amore F, Mylam KJ, et al. Routine bone marrow biopsy has little or no therapeutic consequence for positron emission tomography/computed tomography-staged treatment-naive patients with Hodgkin lymphoma. J Clin Oncol 2012;30:4508–14.
- Wang J, Weiss LM, Chang KL, et al. Diagnostic utility of bilateral bone marrow examination: significance of morphologic and ancillary technique study in malignancy. Cancer 2002;94:1522–31.
- Menon NC, Buchanan JG. Bilateral trephine bone marrow biopsies in Hodgkin’s and non-Hodgkin’s lymphoma. Pathology 1979;11:53–7.
- Carbone PP, Kaplan HS, Musshoff K, et al. Report of the Committee on Hodgkin’s Disease Staging Classification. Cancer Res 1971;31:1860–1.
- Lister TA, Crowther D, Sutcliffe SB, et al. Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin’s disease: Cotswolds meeting. J Clin Oncol 1989;7:1630–6.
- Armitage JO. Early-stage Hodgkin’s lymphoma. N Engl J Med 2010;363:653–62.
- National Comprehensive Cancer Network I. NCCN Guidelines Version 3.2016 Hodgkin lymphoma. 2017.
- Engert A, Franklin J, Eich HT, et al. Two cycles of doxorubicin, bleomycin, vinblastine, and dacarbazine plus extended-field radiotherapy is superior to radiotherapy alone in early favorable Hodgkin's lymphoma: final results of the GHSG HD7 trial. J Clin Oncol 2007;25:3495–502.
- Andre MP, Girinsky T, Federico M, et al. Early positron emission tomography response-adapted treatment in stage I and II Hodgkin lymphoma: final results of the randomized EORTC/LYSA/FIL H10 trial. J Clin Oncol 2017:Jco2016686394.
- Meyer RM, Gospodarowicz MK, Connors JM, et al. Randomized comparison of ABVD chemotherapy with a strategy that includes radiation therapy in patients with limited-stage Hodgkin’s lymphoma: National Cancer Institute of Canada Clinical Trials Group and the Eastern Cooperative Oncology Group. J Clin Oncol 2005;23:4634–42.
- Meyer RM, Gospodarowicz MK, Connors JM, Pearcey RG, Wells WA, Winter JN, et al. ABVD alone versus radiation-based therapy in limited-stage Hodgkin’s lymphoma. N Engl J Med 2012;366:399–408.
- Steidl C, Lee T, Shah SP, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med 2010;362:875–85.
- Kamper P, Bendix K, Hamilton-Dutoit S, et al. Tumor-infiltrating macrophages correlate with adverse prognosis and Epstein-Barr virus status in classical Hodgkin’s lymphoma. Haematologica 2011;96:269–76.
- Agostinelli C, Gallamini A, Stracqualursi L, et al. The combined role of biomarkers and interim PET scan in prediction of treatment outcome in classical Hodgkin’s lymphoma: a retrospective, European, multicentre cohort study. Lancet Haematol 2016;3:e467–e79.
- Meignan M, Gallamini A, Meignan M, et al. Report on the First International Workshop on Interim-PET-Scan in Lymphoma. Leuk Lymph 2009;50:1257–60.
- Gallamini A, Hutchings M, Rigacci L, et al. Early interim 2-[18F]fluoro-2-deoxy-D-glucose positron emission tomography is prognostically superior to international prognostic score in advanced-stage Hodgkin’s lymphoma: a report from a joint Italian-Danish study. J Clin Oncol 2007;25:3746–52.
- Easson EC, Russell MH. Cure of Hodgkin’s Disease. Br Med J 1963;1(5347):1704–7.
- Kaplan HS. The radical radiotherapy of regionally localized Hodgkin’s disease. Radiology 1962;78:553–61.
- Noordijk EM, Carde P, Dupouy N, et al. Combined-modality therapy for clinical stage I or II Hodgkin’s lymphoma: long-term results of the European Organisation for Research and Treatment of Cancer H7 randomized controlled trials. J Clin Oncol 2006;24:3128–35.
- Eghbali H, Raemaekers J, Carde P. The EORTC strategy in the treatment of Hodgkin’s lymphoma. Eur J Haematol Suppl 2005:135–40.
- Bloomfield CD PT, Glicksman AS, et al. Chemotherapy and combined modality therapy for Hodgkin’s disease: A progress report on cancer and leukemia group B studies. Cancer Treat Rep 1982;66:835–46.
- Pavlovsky S, Maschio M, Santarelli MT, et al. Randomized trial of chemotherapy versus chemotherapy plus radiotherapy for stage I-II Hodgkin’s disease. J Natl Cancer Inst 1988;80:1466–73.
- Aviles A, Delgado S. A prospective clinical trial comparing chemotherapy, radiotherapy and combined therapy in the treatment of early stage Hodgkin’s disease with bulky disease. Clin Lab Haematol 1998;20:95–9.
- Herbst C, Rehan FA, Brillant C, et al. Combined modality treatment improves tumor control and overall survival in patients with early stage Hodgkin’s lymphoma: a systematic review. Haematologica 2010;95:494–500.
- Ferme C, Eghbali H, Meerwaldt JH, et al. Chemotherapy plus involved-field radiation in early-stage Hodgkin’s disease. N Engl J Med 2007;357:1916–27.
- Landgren O, Axdorph U, Fears TR, et al. A population-based cohort study on early-stage Hodgkin lymphoma treated with radiotherapy alone: with special reference to older patients. Ann Oncol 2006;17:1290–5.
- Engert A, Plutschow A, Eich HT, et al. Reduced treatment intensity in patients with early-stage Hodgkin's lymphoma. N Engl J Med 2010;363:640–52.
- Radford J, Illidge T, Counsell N, et al. Results of a trial of PET-directed therapy for early-stage Hodgkin's lymphoma. N Eng J Med 2015;372:1598–607.
- Eich HT, Diehl V, Gorgen H, et al. Intensified chemotherapy and dose-reduced involved-field radiotherapy in patients with early unfavorable Hodgkin’s lymphoma: final analysis of the German Hodgkin Study Group HD11 trial. J Clin Oncol 2010;28:4199–206.
- von Tresckow B, Plutschow A, Fuchs M, et al. Dose-intensification in early unfavorable Hodgkin’s lymphoma: final analysis of the German Hodgkin Study Group HD14 trial. J Clin Oncol 2012;30:907–13.
- Advani RH, Hong F, Fisher RI, et al. Randomized phase III trial comparing ABVD plus radiotherapy with the Stanford V regimen in patients with stages I or II locally extensive, bulky mediastinal Hodgkin lymphoma: a subset analysis of the North American Intergroup E2496 Trial. J Clin Oncol 2015;33:1936–42.
- Sasse S, Brockelmann PJ, Georgen H, et al. Long-term follow-up of contemporary treatment in early-stage Hodgkin lymphoma: Updated analyses of the German Hodgkin Study Group HD7, HD8, HD10 and HD11 trials. J Clin Oncol 2017 Apr 18:JCO2016709410. doi: 10.1200/JCO.2016.70.9410. [Epub ahead of print]
- Nogova L, Reineke T, Brillant C, et al. Lymphocyte-predominant and classical Hodgkin’s lymphoma: a comprehensive analysis from the German Hodgkin Study Group. J Clin Oncol 2008;26:434–9.
- Wirth A, Yuen K, Barton M, et al. Long-term outcome after radiotherapy alone for lymphocyte-predominant Hodgkin lymphoma: a retrospective multicenter study of the Australasian Radiation Oncology Lymphoma Group. Cancer 2005;104:1221–9.
- Chera BS, Olivier K, Morris CG, et al. Clinical presentation and outcomes of lymphocyte-predominant Hodgkin disease at the University of Florida. Am J Clin Oncol 2007;30:601–6.
- Chen RC, Chin MS, Ng AK, et al. Early-stage, lymphocyte-predominant Hodgkin’s lymphoma: patient outcomes from a large, single-institution series with long follow-up. J Clin Oncol 2010;28:136–41.
- Savage KJ, Skinnider B, Al-Mansour M, et al. Treating limited-stage nodular lymphocyte predominant Hodgkin lymphoma similarly to classical Hodgkin lymphoma with ABVD may improve outcome. Blood 2011;118:4585–90.
- Eichenauer DA FM, Pluetschow A, et al. Phase 2 study of rituximab in newly diagnosed stage IA nodular lymphocytepredominant Hodgkin lymphoma: a report from the German Hodgkin Study Group. Blood 2011;118:4363–5.
- Advani RH, Horning SJ, Hoppe RT, et al. Mature results of a phase II study of rituximab therapy for nodular lymphocyte-predominant Hodgkin lymphoma. J Clin Oncol 2014;32:912–8.
- Mauz-Korholz C, Gorde-Grosjean S, Hasenclever D, et al. Resection alone in 58 children with limited stage, lymphocyte-predominant Hodgkin lymphoma-experience from the European network group on pediatric Hodgkin lymphoma. Cancer 2007;110:179–85.
- Ng AK. Review of the cardiac long-term effects of therapy for Hodgkin lymphoma. Br J Haematol 2011;154:23–31.
- Ng AK, LaCasce A, Travis LB. Long-term complications of lymphoma and its treatment. J Clin Oncol 2011;29:1885–92.
- Aleman BM, van den Belt-Dusebout AW, Klokman WJ, et al. Long-term cause-specific mortality of patients treated for Hodgkin’s disease. J Clin Oncol 2003;21:3431–9.
- Girinsky T, van der Maazen R, Specht L, et al. Involved-node radiotherapy (INRT) in patients with early Hodgkin lymphoma: concepts and guidelines. Radiother Oncol 2006;79:270–7.
- Campbell BA, Voss N, Pickles T, et al. Involved-nodal radiation therapy as a component of combination therapy for limited-stage Hodgkin’s lymphoma: a question of field size. J Clin Oncol 2008;26:5170–4.
- Advani R, Maeda L, Lavori P, et al. Impact of positive positron emission tomography on prediction of freedom from progression after Stanford V chemotherapy in Hodgkin’s disease. J Clin Oncol 2007;25:3902–7.
- Engert A, Schiller P, Josting A, et al. Involved-field radiotherapy is equally effective and less toxic compared with extended-field radiotherapy after four cycles of chemotherapy in patients with early-stage unfavorable Hodgkin’s lymphoma: results of the HD8 trial of the German Hodgkin’s Lymphoma Study Group. J Clin Oncol 2003;21:3601–8.
- Horning SJ, Hoppe RT, Breslin S, et al. Stanford V and radiotherapy for locally extensive and advanced Hodgkin’s disease: mature results of a prospective clinical trial. J Clin Oncol 2002;20:630–7.
- Pavone V, Ricardi U, Luminari S, et al. ABVD plus radiotherapy versus EVE plus radiotherapy in unfavorable stage IA and IIA Hodgkin’s lymphoma: results from an Intergruppo Italiano Linfomi randomized study. Ann Oncol 2008;19:763–8.
- De Bruin ML, Sparidans J, van’t Veer MB, et al. Breast cancer risk in female survivors of Hodgkin’s lymphoma: lower risk after smaller radiation volumes. J Clin Oncol 2009;27:4239–46.
- Hodgson DC. Late effects in the era of modern therapy for Hodgkin lymphoma. Hematology Am Soc Hematol Educ Program 2011;2011:323–9.
- Maraldo MV, Brodin NP, Vogelius IR, et al. Risk of developing cardiovascular disease after involved node radiotherapy versus mantle field for Hodgkin lymphoma. Int J Radiat Oncol Biol Phys 2012;83:1232–7.
- Campbell BA, Hornby C, Cunninghame J, et al. Minimising critical organ irradiation in limited stage Hodgkin lymphoma: a dosimetric study of the benefit of involved node radiotherapy. Ann Oncol 2012;23:1259–66.
- Pingali SR, Jewell SE, Havlat L, et al. Limited utility of routine surveillance imaging for classical Hodgkin lymphoma patients in first complete remission. Cancer 2014;120:2122–9.
INTRODUCTION
Hodgkin lymphoma, previously known as Hodgkin’s disease, is a B-cell malignancy with unique pathological and epidemiological features for which highly effective therapies exist. The disease is characterized by the presence of mononuclear and multinucleate giant cells called Hodgkin and Reed-Sternberg (HRS) cells.1
Hodgkin lymphoma is unique compared to other B-cell lymphomas because of the scarcity of the malignant cells in the tumor tissue. The HRS cells usually account for only 0.1% to 10% of the cells in the affected tissues, and the HRS cells induce accumulation of nonmalignant lymphocytes, macrophages, granulocytes, eosinophils, plasma cells, and histiocytes, which constitute more than 90% of tumor cellularity.2 Although the disease was first described by Sir Thomas Hodgkin in 1832, in part because of this unique histopathology, not until 1991 was it conclusively demonstrated that HRS cells are in fact monoclonal germinal center–derived B-cells. This article reviews management and frontline treatment options for limited-stage classical Hodgkin lymphoma and nodular lymphocyte predominant Hodgkin lymphoma. Treatment of advanced stage and relapsed/refractory Hodgkin lymphoma will be discussed in a separate article.
EPIDEMIOLOGY
Hodgkin lymphoma accounts for 0.5% of all malignancies and 11.7% of all lymphomas among adults in the United States.3 The incidence of Hodgkin lymphoma has been steadily increasing over the past 4 decades and was estimated to be 8260 cases in the United States in 2017, with a slight male predominance. Hodgkin lymphoma is expected to cause 1070 deaths in 2017, accounting for 0.2% of all cancer deaths.3 First-degree relatives of patients with Hodgkin lymphoma have a 3- to 9-fold increased risk of having the disease compared to the general population,4 and monozygotic twin siblings of Hodgkin lymphoma patients have a greatly increased risk for developing the disease—up to 100-fold—compared to normal cohorts.5 The incidence is highest among Caucasians, African Americans, and Hispanics, and lower in Asians and American Indians.3 Hodgkin lymphoma incidence shows a bimodal peak distribution, with 1 peak between the ages of 15 and 44 years, and another peak after age 65 years.6
ETIOLOGY/PATHOGENESIS
The cause of Hodgkin lymphoma is unknown. Epstein-Barr virus (EBV) infection is present in up to 40% of Hodgkin lymphoma cases, suggesting a role of this virus in the pathogenesis of some cases. The risk of EBV-positive Hodgkin lymphoma was found to be higher following an episode of infectious mononucleosis, while the risk of EBV-negative Hodgkin lymphoma remained unchanged.7 The incidence of Hodgkin lymphoma is 5 to 14 times higher in HIV-infected patients than in noninfected patients.8 It is not considered an AIDS-defining illness, but has become more frequent with the growth and aging of the HIV-positive population.9,10 Hodgkin lymphoma patients with HIV typically have CD4 lymphocyte counts greater than 200 cells/μL,11 with the incidence of Hodgkin lymphoma actually declining with lower CD4 lymphocyte counts.12 HIV-related Hodgkin lymphoma tends to have an aggressive course, with high rates of EBV positivity.13 The incidence of Hodgkin lymphoma is 1.8 times higher among smokers, and the risk appears to increase with duration of smoking.14,15
The cell of origin of Hodgkin lymphoma, while long suspected to be the HRS cell, remained unproven until the 1990s when micro-dissection and single-cell polymerase chain reaction techniques allowed for confirmation that the HRS cell was in fact a monoclonal germinal center derived B cell.16,17 These HRS cells lack immunoglobulin due to defective transcription regulation and not due to crippling mutations.18,19 The cellular infiltrate in Hodgkin lymphoma appears to play a decisive role in allowing the HRS cells to survive by providing an environment that suppresses cytotoxic immune responses as well as by providing cellular interactions and cytokines that support their growth and survival. The extensive inflammatory infiltrate in classical Hodgkin lymphoma is comprised of T helper 2 (Th2) and regulatory T cells and lacks T helper 1 (Th1) cells, CD8 cytotoxic T cells, and natural killer cells.20 The HRS cells escape apoptosis by several mechanisms which include latent EBV infection and constitutive nuclear factor (NF)-kB pathways, as well as other deregulated signaling pathways that promote survival, such as EBV nuclear antigen 1 (EBNA1) protein, EBV latent infection membrane protein 1 (LMP1), and LMP2.21,22
Genetic alterations in the 9p24 locus which encodes PD-L1/PD-L2 are nearly universally present in classical Hodgkin lymphoma and are now considered a disease-defining feature.23
PATHOLOGIC CLASSIFICATION
According to the 2008 World Health Organization (WHO) classification, Hodgkin lymphoma has 2 clearly defined entities: classical Hodgkin lymphoma (cHL), which accounts for approximately 95% cases, and nodular lymphocyte predominant Hodgkin lymphoma (NLPHL), which accounts for the remaining cases.24 These 2 entities differ in their clinical, pathological, and biological features, which in turn affect prognosis and treatment options. Classical Hodgkin lymphoma is characterized by a paucity of HRS cells surrounded by a background of mixed inflammatory infiltrate comprised of histiocytes, small lymphocytes, eosinophils, neutrophils, plasma cells, fibroblasts, and collagen. Depending on the particular combinations of these elements and the specific features of the neoplastic cells, cases can be subclassified into several cHL subtypes: the nodular sclerosis, mixed cellularity, lymphocyte-rich, and lymphocyte-depleted types.25
The diagnosis of cHL is made based on a combination of morphology of HRS cells and the other cells infiltrating the tissue, combined with immunohistochemical staining. Because of the rare nature of the malignant (clonal) cell in Hodgkin lymphoma specimens, flow cytometry is generally of little value. The HRS cells in cHL are CD30-positive and CD45 negative in virtually all cases, and CD15-positive in 85% of cases.26 B-cell antigens are typically negative except for CD20, which is positive in about 20% cases.27
Nodular sclerosis Hodgkin lymphoma (NSHL) is the most common subtype of cHL, accounting for 65% to 75% of cases. It is common among young adults and tends to involve the mediastinal, supraclavicular, and cervical lymph nodes. NSHL is characterized by the presence of collagen bands that divide the lymphoid tissue into circumscribed nodules. This subtype usually presents as stage I or II disease, typically with neck and/or mediastinal disease, and evidence of EBV infection is present in approximately 10% to 40% of North American cases.7 Patients diagnosed with NSHL generally have a very good prognosis.
Mixed cellularity Hodgkin lymphoma (MCHL) constitutes about 20% to 25% of cHL cases. It affects a somewhat older population, with a median age at diagnosis of 38 years. The typical bimodal age distribution is not seen with MCHL. MCHL has a male predominance (70%), and is more frequent in HIV-infected patients (70% of whom also have EBV infection). Lymphoid tissues have classic HRS cells and significant inflammatory infiltrates. Approximately 50% of patients with MCHL present as stage III or IV with abdominal lymphadenopathy or splenic involvement, and B symptoms are frequent.24
Lymphocyte-rich Hodgkin lymphoma (LRHL) is uncommon, accounting for only 3% to 5% of cases of cHL.28 The disease usually presents at an older age and has a 2:1 male predominance. HRS cells are commonly seen and a large number of reactive lymphocytes are also present. Although on the basis of morphology and immunohistochemistry LRHL belongs to the cHL group, clinically it more closely resembles LPHL. Patients usually present at early stage and rarely have B symptoms. LRHL carries an excellent prognosis, with a greater than 90% PFS after 5 years.23,29
Lymphocyte-depleted Hodgkin lymphoma (LDHL) is the least common form of cHL, accounting for less than 5% of cases. Many cases previously placed in this category are now recognized as diffuse large B-cell lymphoma (DLBCL), anaplastic large-cell lymphoma (ALCL), or NSHL with lymphocyte depletion.30 HRS cells are frequently seen, but reactive inflammatory cells are relatively sparse. EBV infection is seen in up to 90% of cases, commonly associated with HIV-infected individuals. Advanced-stage and symptomatic disease are more common. Prognosis is slightly worse compared to other categories.
NLPHL accounts for approximately 5% of cases of Hodgkin lymphoma. It has a unimodal age distribution, with the peak incidence in the fourth decade, and male predilection of 3:1.28 NLPHL is characterized by large primary lymphoid follicles, with polytypic small B lymphocytes and extensive meshworks of follicular dendritic cells. The lymphocytic/histiocytic (L and H), or “popcorn,” cells scattered within the nodules differ from classic HRS cells, both in their morphology and in their biochemical profile, being frequently negative for CD15, CD30 and for the EBV genome, and usually positive for B-cell antigens such as CD20, suggesting that L and H cells may be immunoglobulin-synthesizing monoclonal B cells. CD45 is also typically positive in NLPHL, in distinction from cHL. NLPHL has an indolent course compared to cHL, and long-term survival is common.19,31 NLPHL commonly presents with limited-stage disease. NLPHL may eventually transform into a more aggressive lymphoma, such as diffuse large B-cell lymphoma (including centroblastic, immunoblastic, or T-cell/histiocyte–rich subtypes), at a rate of 4% to 12%. This can occur even 15 to 20 years after the initial diagnosis of NLPHL.32,33 In a recent large retrospective study of 222 patients with NLPHL, the rate of transformation to DLBCL was 7.6%, with a median time to transformation of 35 months. Overall survival was not adversely affected in patients undergoing transformation compared to those without transformation.34
PRESENTATION
Classical Hodgkin lymphoma usually presents with asymptomatic mediastinal or cervical lymphadenopathy. Half of patients present with stage I or stage II disease.35 A mediastinal mass is seen in most patients with NSHL, at times with bulky disease, with “bulky” defined as a mediastinal mass measuring one-third or more of the maximal thoracic diameter on chest x-ray, or 10 cm on computed tomography (CT) scan. Systemic symptoms, or "B" symptoms—fevers (> 38°C), drenching night sweats, and unexplained weight loss (> 10% of baseline body weight over the preceding 6 months or less)—are detected in approximately 25% of patients. Between 10% and 15% will have extranodal disease, most commonly involving lung, bone, and liver. NLPHL usually presents with limited-stage disease without B symptoms; it typically has a more indolent presentation and clinical course than cHL.
INITIAL EVALUATION AND STAGING
The initial workup includes a complete blood count (CBC), erythrocyte sedimentation rate (ESR), lactate dehydrogenase (LDH), and chemistry studies to evaluate renal function and liver function. Fine-needle aspiration will usually fail to identify the infrequent HRS cells, and instead only reveal the reactive background of inflammatory cells. Generous (large gauge) core needle biopsies may provide diagnosis effectively in some cases, but in general, an excisional lymph node biopsy is preferred to ensure an accurate diagnosis and avoid the need for repeated biopsy procedures. In cases where an excisional biopsy would be difficult or risky, a core needle biopsy procedure is a reasonable first step, with the understanding that a subsequent surgical procedure may still be necessary.
Baseline imaging includes CT scans of the neck, chest, abdomen, and pelvis. Use of positron emission tomography (PET) scanning is now standard in the initial evaluation and assessment of treatment response in Hodgkin lymphoma.36 Due to the increased sensitivity of PET or PET/CT scan, additional lesions may be identified that were not seen on conventional CT scans. This will alter the staging, and potentially the treatment plan, in up to 25% to 30% of patients.37,38 PET/CT scan performed during initial evaluation also facilitates optimal interpretation of post-therapy PET/CT scans and is therefore strongly encouraged as a part of the initial staging evaluation.39
Recent studies have shown that bone marrow biopsy is not routinely needed in the initial staging of cHL. A study of 454 patients concluded that bone marrow biopsy would not have altered the stage in any stage I or II patients. It was further concluded that overall treatment strategy would not have been altered for any of the patients.40 Based on this study and others, it is now clear that FDG-PET has a high sensitivity, and when PET scan is negative (in the bone marrow and skeleton), a bone marrow biopsy provides little additional value. For patients with significant cytopenias, a bone marrow biopsy is reasonable. Such patients may benefit from a bilateral biopsy, which increases the probability of demonstrating bone marrow involvement by 16% to 33%.41,42 Techniques such as staging laparotomy and lymphangiography are now considered obsolete.
Hodgkin lymphoma is staged according to the Ann Arbor staging system (Table 1). The original Ann Arbor staging was published in 1971,43 and in 1989 the “Cotswold modifications” extended the definitions of stage IV disease and the suffix “X” was added to denote bulky disease.44 Both systems were developed for the staging of Hodgkin lymphoma, but are now used for staging non-Hodgkin lymphoma as well.
PROGNOSTIC FACTORS
For the purposes of prognosis and selection of treatment, Hodgkin lymphoma is commonly classified into early-stage favorable, early-stage unfavorable, and advanced stage. Early-stage Hodgkin lymphoma refers to patients with Ann Arbor stage I or stage II disease. With early-stage Hodgkin lymphoma, the prognosis varies significantly based on factors such as the presence of B symptoms, elevated erythrocyte sedimentation rate ([ESR] > 50 mm/hr), number of nodal sites involved, older age, and a large mediastinal mass. For this reason, most clinical trials to evaluate treatment strategies for early-stage Hodgkin lymphoma are based on various combinations of these risk factors. The definition of early-stage unfavorable Hodgkin lymphoma varies across different clinical trial study groups, and it is important to understand the definition in interpreting the results of these trials (Table 2).45,46
In the German Hodgkin Study Group (GHSG) trials, early-stage Hodgkin lymphoma is stratified into a high risk (“unfavorable”) group defined by any of the following: a large mediastinal mass (one third of maximum thoracic diameter), extra-nodal disease, 3 or more nodal areas, and an ESR of > 50 mm/hr in asymptomatic patients or > 30 mm/hr in patients with B symptoms. Low-risk (“favorable”) patients lack all of these factors.47 The European Organization for Research and Treatment of Cancer (EORTC) defines the unfavorable prognostic group as older than 50 years of age, large mediastinal adenopathy (maximum width on a chest radiograph of at least one third of the internal transverse diameter of the thorax at the level of T5 through T6 or any mass of ≥ 10 cm in the largest dimension), an ESR of 50 mm/hr and no B symptoms, or with an ESR of 30 mm/hr in those who have B symptoms, and/or 4 or more regions of involvement.48 The National Cancer Institute of Canada (NCIC) Clinical Trials Group and the Eastern Cooperative Oncology Group (ECOG) define high-risk groups as presence of B symptoms, bulky disease with a mediastinal mass width of at least one third of the maximum chest wall diameter, or any mass greater than 10 cm, and patients with intra-abdominal disease.49,50
Gene-expression profiling in Hodgkin lymphoma has identified a gene signature of tumor-associated macrophages that was able to identify patients with a higher risk for primary treatment failure. In an independent cohort of patients, an increased number of CD68-positive macrophages was correlated with inferior outcomes.51,52 Studies such as these underscore the importance of the tumor “microenvironment” (ie, the nonmalignant cells within a tumor) in determining the overall clinical behavior of a malignancy. While quantification of CD68-positive macrophages has potential to be applied to routine clinical practice, prospective data using CD68 as a tool for risk-adapted therapy is lacking.
Genetic alterations and amplifications in the 9p24.1 locus have recently been found to be a defining genetic feature of cHL. Amplification of 9p24.1 has been associated with unfavorable outcomes. Amplification of 9p24.1 (which includes the loci encoding the PD-L1 and PD-L2 genes) is more common in patients with advanced stage disease and is associated with shorter PFS.23
A recent study attempted to integrate several different prognostic factors in cHL patients who were treated with ABVD (adriamycin [doxorubicin], bleomycin, vinblastine, and dacarbazine) and underwent an interim PET (iPET) scan after 2 cycles of ABVD. Focusing on those with a negative iPET scan, it was found that expression of CD68 and PD-1 in microenvironment cells, and STAT1 negativity in HRS cells identified a subset of PET-2 negative patients with a 3-year PFS significantly lower than that of the remaining PET-2 negative population (64% versus 95%). The algorithm correctly predicted the response to treatment in more than half of the patients who had relapse or disease progression despite a negative PET-2 scan. It therefore appears feasible, using tissue biomarkers at diagnosis, to identify patients at increased risk for poor outcome, even if the iPET scan is negative.53
ROLE OF PET/CT IN ASSESSMENT OF RESPONSE TO THERAPY
PET/CT has been increasingly used for response assessment at various stages in lymphoma in recent years. Almost all types of lymphomas are fluorodeoxyglucose (FDG) avid; however, Hodgkin lymphoma is FDG avid in 97% to 100% of cases. In 2009, a 5-point scale was developed to score PET images with regard to treatment response, either partway through treatment (iPET) or at the end of therapy.54 It was recommended as the standard reporting tool at the First International Workshop on PET in Lymphoma in Deauville, France, in 2009, and is thus now referred to as the Deauville score. A score of 1 is given if there is no uptake, 2 if the uptake ≤ mediastinum, 3 if > mediastinum but ≤ liver, 4 if uptake moderately higher than liver, 5 if uptake is markedly higher than liver and/or new lesions. X designates new areas of uptake unlikely to be related to lymphoma. In most trials, a score of 1 or 2 is considered a complete response and a score of 4 or 5 is considered a treatment failure. A score of 3 is sometimes considered a complete response, depending on the study. The Deauville criteria have been widely used in newer clinical trials utilizing response-adapted treatment as defined by PET response. PET/CT is recommended for staging and restaging at the end of therapy, in clinical practice, and clinical trials. Interim PET/CT scan, while commonly performed in clinical practice, is only recommended if the results will alter therapy (eg, if that information will result in the clinician omitting radiation therapy [RT] or altering the chemotherapy plan).
Early studies of iPET showed that achieving PET negativity early in the course of treatment was strongly associated with PFS and overall survival.55 Subsequent studies confirmed the importance of achieving a negative iPET. As a result, considerable efforts have been put into designing response-adapted treatment approaches using iPET (see Treatment section), with some of these approaches now being listed in the National Comprehensive Cancer Network (NCCN) guidelines and being used in standard practice.
TREATMENT
EVOLUTION OF TREATMENT
The treatment of Hodgkin lymphoma has evolved over the past century, starting with the discovery of RT as effective treatment in the early 20th century. Long-term survival of patients with Hodgkin lymphoma treated with involved-field radiation therapy (IFRT) was first reported in the 1960s.56,57 Outcomes improved further with the introduction of combined modality treatment (CMT) using chemotherapy and RT, with the overall 5-year relative survival for patients with Hodgkin lymphoma (all stages) treated in 2006–2012 being 85.4% to 87.3%.3 Since the majority of patients are now cured with modern therapy, treatment-related complications have become an important competing cause of mortality. Recent studies have therefore focused on maintaining efficacy while reducing toxicities, and refining the process of selecting patients who might benefit from more aggressive therapy. While RT was the first treatment modality shown to be curative for Hodgkin lymphoma,56,57 multiple subsequent studies showed that CMT is superior to RT alone in terms of relapse-free survival.58–63 In the GHSG H8-F trial, the estimated 5-year event-free survival and overall survival rates were significantly higher after 3 cycles of MOPP-ABV (mechlorethamine, vincristine, procarbazine, and prednisone combined with doxorubicin, bleomycin, and vinblastine) plus IFRT than after subtotal nodal radiotherapy alone. The 10-year overall survival estimates were 97% and 92%, respectively (P = 0.001).64 As a result, CMT replaced RT alone as the standard of care for limited-stage Hodgkin lymphoma. However, for elderly or infirm patients, or those with other comorbidities making them poor chemotherapy candidates, RT alone may be a very reasonable option.65 More recently, an increasing body of evidence has accumulated to support the use of chemotherapy alone in early stage cHL. This literature has consistently shown that omission of RT is associated with a modest increase in relapse, without a clear compromise in long-term overall survival. For some patients, the trade-off in terms of avoiding radiation (and the associated late effects) may be worth the small increase in relapse risk, since long-term survival does not appear to be substantially worse with chemo-therapy alone. Table 3 and Table 4 provide a summary of recent key studies which have defined treatment options for early-stage cHL.48,66–71
EARLY-STAGE NLPHL
NLPHL usually presents with limited-stage disease without B symptoms and has an indolent course with a slightly better prognosis compared to cHL.72 Due to the rarity of the disease, treatment guidelines are mostly based on retrospective analyses from single or multi-institution studies or subgroup analyses, often with relatively short follow-up. These studies must be interpreted with caution because of the possibility of inaccuracies in the pathologic diagnosis, small sample sizes, and selection bias. Treatment options for limited-stage NLPHL include observation, single-agent rituximab, IFRT (or involved-site radiation therapy [ISRT]) alone, or CMT.46
Historically, patients with limited-stage NLPHL have been treated with RT alone, with 80% to 85% PFS and 85% to 95% overall survival rates.73–75 Patients who relapse or progress after RT in general can successfully undergo salvage therapy.74 In one study, rates of PFS and overall survival were similar among patients who had limited-field, regional-field, or extended-field RT,75 indicating that IFRT is preferred. Studies comparing RT alone and CMT are limited. The GHSG HD7 trial included a subset of NLPHL patients, with a trend towards improved freedom from treatment failure (96% versus 83%) favoring CMT. This, however, did not translate into improved overall survival.47 A retrospective analysis of the British Columbia Cancer Agency database compared patients with limited-stage NLPHL treated with RT alone to patients who received 2 cycles of ABVD followed by RT. A significant improvement in PFS (91% versus 65%) and overall survival (93% versus 84%) was seen, favoring CMT.76
Chemotherapy alone is not recommended for limited-stage NLPHL since studies evaluating chemotherapy alone are quite limited and indicate relatively high rates of treatment failure. Given that the malignant cells in NLPHL are CD20-positive, single-agent rituximab has also been studied in this disease, including a study as frontline therapy in limited-stage patients. In this phase 2 trial in newly diagnosed patients with stage IA disease, an overall response rate (ORR) of 100% was seen, with an 85% complete response (CR) rate.77 At 3 years, overall survival was 100% and PFS was 81%, indicating that the responses with single-agent rituximab are less durable than those with RT.
Advani et al evaluated rituximab followed by observation versus rituximab (R) followed by maintenance rituximab (MR) for 2 years in 39 new or previously treated patients. At 4 weeks the ORR was 100% (with CR in 67%, and partial response in 33%). At a median follow up of 9.8 years for R alone, and 5 years for R+MR, median PFS was 3 and 5.6 years, respectively (P = 0.26). Estimated 5-yr PFS and overall survival in patients treated with R versus R+MR were 39.1% and 95.7% versus 58.9% and 85.7%, with Pvalues of 0.26 (PFS) and 0.38 (overall), respectively. Maintenance rituximab therefore appears to prolong remission, although the results did not quite reach statistical significance.78 Even though rituximab does not appear to be curative in NLPHL, it is a reasonable option for patients with early-stage NLPHL who are not good candidates for definitive RT. Whether combining rituximab with RT or CMT might further improve outcomes in early-stage NLPHL has not yet been determined.
In children, surgery alone may lead to long-term remission or possibly cure of limited-stage NLPHL. In a European multicenter retrospective study, 58 patients underwent surgery for limited-stage NLPHL. Among the 51 patients who achieved complete remission following surgery, 67% remained progression-free and 100% were alive at a median follow up of 43 months.79 In adults, there is no data to support surgical treatment alone for NLPHL. Finally, observation may be a reasonable option in elderly or infirm patients for whom NLPHL is unlikely to affect life expectancy. For younger patients, given the excellent outcome with modern therapy and the long-term risk of transformation of NLPHL into an aggressive non-Hodgkin lymphoma, observation is generally not recommended.
The NCCN recommends RT (ISRT or IFRT, 30–36 Gy) as the preferred treatment for stage IA and IIA non-bulky NLPHL. In patients with stage IA disease with complete excision of solitary nodule, observation may be appropriate. A course of chemotherapy with ISRT with or without rituximab is recommended for patients with stage IB or IIB disease, or patients with stage IA or IIA bulky disease.
FIRST-LINE TREATMENT OF LIMITED-STAGE CHL
Early-Stage Favorable cHL
There is lack of consensus regarding the ideal treatment approach for patients with early-stage favorable cHL. However, there are several excellent options available, with overall survival rates exceeding 90%. Most of these regimens involve CMT, although some chemotherapy-alone approaches have been evaluated as well. Concurrent with the demonstration of excellent long-term remission rates with CMT, it became apparent that the long-term survival and quality of life of these patients is determined in large part by the risk of serious (and potentially fatal) treatment-related toxicities. Such toxicities consist primarily of secondary malignancies and cardiovascular events, and can continue to cause significant morbidity and mortality even 2 to 3 decades after treatment.80–82 As a result, treatment decisions for these patients are complicated and require balancing efficacy against risk of late complications.
In the United States, until recently, CMT was generally considered the standard of care, with robust long-term data regarding efficacy. The most commonly used regimen has been ABVD for 2 to 4 cycles followed by IFRT. In some German studies, escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone) has been used, but this is not a general standard of care in the United States for early-stage patients.
More recent data suggests that the rate of serious late complications in Hodgkin lymphoma patients is decreasing, likely due to less extensive radiation fields, lower radiation doses, and a movement away from the MOPP regimen to ABVD.83,84 For patients who meet the “favorable” criteria set forth in the GHSG HD10 trial (see Table 2), 2 cycles of ABVD followed by 20 Gy of IFRT is an attractive option, with efficacy preserved and a low anticipated rate of late effects.66 With this approach, and with long-term (10 years) follow up, all 4 arms had similar PFS (87%) and overall survival (94%), whether 2 or 4 cycles of ABVD were given. When the effects of 20-Gy and 30-Gy doses of RT were compared, there were also no significant differences in freedom from treatment failure or overall survival. Adverse events and acute toxic effects of treatment were most common in the patients who received 4 cycles of ABVD and 30 Gy of RT.66,71
In recent years, in an attempt to reduce late effects further, regimens consisting of chemotherapy alone have been investigated. In a study by Meyer et al, at 12 years the rate of overall survival was 94% among those receiving ABVD alone, as compared with 87% among those receiving subtotal nodal RT; the rates of freedom from disease progression were 87% and 92% in the 2 groups; and the rates of event-free survival were 85% and 80%, respectively.50 However, it is important to note that this study did not include a CMT arm for the early favorable patients, and did not utilize modern RT techniques. Nevertheless, this early study and others60 suggested that chemotherapy alone may be a reasonable option for some early-stage cHL patients, particularly for patients who are felt to be at increased risk for late toxicities from RT. As a result, additional studies have been conducted evaluating CMT versus chemotherapy alone for early-stage cHL. Many of these studies have incorporated interim PET/CT scan to develop a response-adapted approach to decide which patients are least likely to benefit from RT.
The HD-13 study was a follow-up study for HD-10, looking at deletion of bleomycin, dacarbazine, or both from the ABVD backbone. The ABD arm was closed early, because of an excess rate of treatment failure. Among the 1243 patents assigned to either the ABVD or AVD arm at 5 years of follow-up, there was 4.3% difference in PFS. This study was not able to demonstrate that 2 cycles of AVD was noninferior to 2 cycles of ABVD, each followed by 30 Gy IFRT, even though there was no difference in all 4 groups. It confirmed 2 cycles of ABVD as the preferred regimen in early favorable Hodgkin lymphoma, when CMT is the plan of care. However, for patients over age 60 to 65 years, or those with underlying cardiac or pulmonary comorbidities, bleomycin has significant risk of toxicity. In that setting, AVD is a safer option, with only a very modest decrease in 5-year PFS.
Based on the observation that iPET scan is highly predictive of outcome in Hodgkin lymphoma,55,85 several trials have employed the use of an iPET scan to guide therapy. It is hoped that such studies will lead to new PET-directed treatment algorithms in which patients who require more aggressive therapy (eg, with CMT, or escalated BEACOPP) can be identified, and the remaining patients can be safely treated less aggressively (eg, with chemotherapy alone).
In the EORTC H10 trial, performed to evaluate treatment adaptation on the basis of iPET scan results in stage I and II Hodgkin lymphoma, a control arm received standard combined modality treatment (3 or 4 cycles of ABVD with INRT) irrespective of PET scan results. In the experimental arm, patients with a negative PET scan after 2 cycles of ABVD continued with 1 or 2 cycles of ABVD and did not receive RT. The iPET-positive patients received either standard treatment with ABVD plus INRT or escalated BEACOPP plus INRT. The iPET-negative patients received either ABVD only or ABVD plus INRT. The final results of this study, published recently, showed that in the iPET-positive patients the 5-year PFS was improved from 77.4% with standard ABVD plus INRT to 90.6% with escalated BEACOPP plus INRT (P = 0.002). In iPET-negative patients, 5-year PFS in the favorable group was 99% versus 87.1% in favor of ABVD plus INRT. The H10 study suggested that PET results after 2 cycles of ABVD can be integrated into treatment planning, In iPET-negative patients, the study was technically not able to demonstrate the noninferiority of the ABVD only regimen, owing to a higher risk of relapse if INRT is omitted, particularly in the favorable group.48 However, this study does show that excellent outcomes can be obtained with omission of RT in patients with a negative iPET scan. This study provides a cautionary lesson though, in that the increase in relapse rate associated with omission of RT was more substantial (12%) for favorable versus unfavorable early-stage patients (2.5%), and this difference was only apparent after longer (5 years) follow-up. Despite this, chemotherapy alone is considered a reasonable treatment option, especially for patients felt to be at increased risk for late toxicities of RT or for patients who wish to avoid the risks of RT, with over 99% of patients alive at 5 years.
Similar results were shown in the RAPID trial, in which patients with limited-stage cHL underwent 3 cycles of ABVD followed by PET assessment.67 Patients with a negative PET (n = 426) were then randomized to RT (n = 209) versus no further therapy (n = 211). At a median of 60 months of follow-up, 3-year PFS was 94.6% in the RT group and 90.8% in the chemotherapy alone group. Similar to the H10 trial, it was concluded that chemotherapy alone was statistically inferior to CMT in terms of PFS. However, also similar to the H10 trial, the RAPID trial demonstrated that excellent results can be obtained in early-stage cHL patients with omission of RT, if iPET scan is negative after 3 cycles of ABVD, as there was no survival difference. These findings indicate that, when relapses occur as a result of omission of RT, such patients can be effectively treated later.
In the ongoing GHSG HD16 trial, patients with early-stage favorable cHL will be randomly assigned to a standard approach (ABVD × 2 cycles followed by 20-Gy IFRT) versus an experimental approach in which they receive ABVD for 2 cycles and then undergo PET scan. If the PET remains positive, they will receive 20-Gy IFRT. If the PET is negative, they will receive no further therapy. This trial could ultimately define ABVD for 2 cycles as a treatment option.
It is clear from these studies that omission of RT results in a somewhat higher rate of relapse but can be considered in selected patients. However, taking a less aggressive frontline approach may also be justified by the fact that, for those who do relapse, successful salvage therapies are available. Aggressive salvage therapy with autologous stem cell transplantation historically can cure approximately 50% of relapsed patients. With new and emerging therapies for relapsed disease, such as brentuximab vedotin and the PD-1 inhibitors (eg, nivolumab and pembrolizumab), the ability to cure relapsed patients may improve even more, further calling into question the practice of applying CMT uniformly for early-stage patients undergoing first-line therapy. Unfortunately, there is insufficient data from large randomized studies with long-term follow-up to fully address this issue currently, and there remains some controversy around this issue. NCCN recommends restaging PET/CT after 3 cycles of ABVD if a chemotherapy alone treatment modality is chosen. If the Deauville score is 1 or 2, either observation or 1 additional cycle of ABVD is recommended.46
Early-Stage Unfavorable cHL
In the United States, historically early-stage unfavorable Hodgkin lymphoma has been treated with CMT, most commonly 4 to 6 cycles of ABVD followed by consolidative RT. With this approach one can expect a 5-year PFS of approximately 80% to 85%.58,64,86 The GHSG HD8 trial showed that RT volume size reduction from extended-field to involved-field after COPP + ABVD chemotherapy for 2 cycles produced similar results and less toxicity in patients with early-stage unfavorable cHL.86 The GHSG trial HD11 established ABVD for 4 cycles plus 30-Gy IFRT as a standard for early unfavorable Hodgkin lymphoma. The freedom from treatment failure at 5 years was 85.0%, and overall survival was 94.5%.68
In the HD14 study by the GHSG, patients with early unfavorable cHL were treated with 2 cycles of escalated BEACOPP followed by 2 cycles of ABVD, versus 4 cycles of ABVD. All patients then received 30 Gy of consolidative IFRT. A 5-year PFS of 95% was seen in the experimental arm, compared with 89% in the standard (ABVD) arm. As expected, this regimen was associated with more acute hematologic toxicity, and there was no difference between the 2 regimens with respect to overall survival or fertility.69 Given the lack of improved survival and increased toxicity, ABVD has remained the standard chemotherapy regimen for early unfavorable cHL in the United States. NCCN recommends a restaging PET scan after 2 cycles of ABVD and to continue with 2 to 4 cycles of ABVD or escalated BEACOPP with or without ISRT based on Deauville scores.
Another viable treatment option is the Stanford V regimen, a condensed, 12-week regimen that includes mechlorethamine, doxorubicin, vinblastine, prednisone, vincristine, etoposide, and bleomycin, followed by IFRT.87 In a randomized phase 3 trial conducted by ECOG (E2496), patients with stage I/II Hodgkin lymphoma with bulky mediastinal disease or advanced-stage disease were randomized to ABVD × 6 to 8 cycles versus Stanford V. RT was given (36 Gy) for those with bulky mediastinal disease or to sites of disease greater than 5 cm in the Stanford V arm. In a subset analysis focusing only on those with stage I/II bulky mediastinal disease, the 5-year failure free survival was 85% versus 79% and the 5-year overall survival was 96% versus 92% for the ABVD versus Stanford V arms, respectively. These differences were not statistically significant.70 While the Stanford V regimen has the advantages of a 12-week treatment duration and a lower cumulative amount of bleomycin and doxorubicin, the Stanford V arm had higher rates of grade 3 lymphopenia and grade 3 to 4 peripheral neuropathies. In addition, Stanford V requires that most patients undergo RT (to original sites of disease measuring 5 cm or more plus contiguous areas). As a result, the investigators concluded that ABVD × 4 cycles plus IFRT remains the standard of care for patients with early unfavorable Hodgkin lymphoma with bulky mediastinal disease.
Other regimens have been studied in hopes of reducing toxicity, including the EVE regimen (epirubicin, vinblastine, and etoposide). This regimen was compared to ABVD in early unfavorable Hodgkin lymphoma patients, with all patients undergoing the same RT program. No differences were observed between the ABVD and EVE arms in terms of complete remission rate and overall survival. However, patients who received EVE had a significantly worse outcome than those who received ABVD in terms of relapse-free survival and failure-free survival.88 EBVP (epirubicin, bleomycin, vinblastine, and prednisone) followed by IFRT was less efficacious compared with MOPP/ABV–type therapy.58
An area of active investigation is whether RT can be safely omitted in patients with early- stage unfavorable cHL. The EORTC H10 study showed that, for patients with a negative iPET scan (after 2 cycles), the 5-year PFS rates were 92.1% versus 89.6% for ABVD plus INRT versus ABVD alone, respectively. While this technically did not meet criteria for noninferiority of ABVD alone, this study demonstrated that, for those with negative iPET, ABVD × 6 cycles (without radiation) can result in long-term remission in a high proportion (89%) of patients. For iPET-positive patients, 2 cycles of escalated BEACOPP were given followed by 30 Gy of IFRT on the experimental arm. This resulted in a 5-year PFS of 90.6% versus 77.4%, suggesting this may be a preferred approach for early-stage unfavorable patients with a positive iPET.48 Even though the noninferiority of ABVD alone could not be established based on the statistical design of the study, the current NCCN guidelines recommend restaging after 2 cycles of ABVD for stage I or II unfavorable cHL and using that iPET as a guide, based on Deauville scores. For scores 1–3, ABVD × 2 cycles (total 4 cycles) plus ISRT or AVD × 4 (total 6) with or without ISRT is recommended. For a Deauville score of 4, escalated BEACOPP × 2 cycles or ABVD × 2 cycles (total 4) followed by ISRT is recommended. If the Deauville score is 5, further treatment decisions should be made based on repeat biopsy results. A follow up PET/CT is recommended for Deauville scores of 4 and 5 to confirm complete response.46
LATE EFFECTS AND THE EVOLUTION OF RADIATION THERAPY
The RT given in Hodgkin lymphoma has evolved considerably over the years, from extended field or subtotal nodal fields developed in the 1960s, to the more focused involved-field or even involved-site radiation commonly given now. This approach reduces radiation volumes, and it already is becoming evident that the relative risk of breast cancer among young females receiving mediastinal RT for Hodgkin lymphoma is declining.89 Cardiac dose is reduced significantly with IFRT compared to older radiation techniques as well. The extent of radiation may be reduced even further with involved-nodal/involved site or intensity-modulated approaches.90
With new RT techniques allowing for more focused therapy and lower doses of radiation, models predict that the rate of long-term complications will decline further.91,92 Furthermore, response-adapted (ie, PET-directed) approaches, as discussed in detail earlier in the article, are expected to increasingly allow for identification of patients who can safely avoid radiation entirely, which will hopefully lead to an even lower rate of late complications of therapy.
MONITORING FOR RELAPSE
A number of recent studies have shown that, for patients who achieve complete remission with first-line therapy, performing repeated scheduled surveillance imaging does not improve outcomes. In fact, most relapses are detected by the patient (due to symptoms or recurrence of lymph node enlargement). It is rare that a relapse would be detected by surveillance imaging alone. Furthermore, surveillance that includes routine imaging has not been associated with improved survival.93 As a result, it is now recommended that patients undergo regular follow-up with symptom review, physical exam, and basic laboratory studies. Imaging studies should be obtained as needed for patients who develop signs, symptoms, exam findings, or laboratory values concerning for relapse.
More important than scheduled surveillance imaging for relapse is monitoring for late effects of therapy. These fall into several broad categories such as cardiovascular disease (coronary disease, congestive heart failure, valvular disease, carotid artery disease), pulmonary disease, hypothyroidism, and secondary malignancies. Aggressive surveillance for breast cancer is especially warranted in female patients who underwent chest radiation.46
CONCLUSION
Hodgkin lymphoma is characterized pathologically by the presence of HRS cells accompanied by a polymorphous cellular infiltrate. It is a disease with a bimodal age distribution, several pathologic subtypes, and numerous treatment options. Overall, the prognosis for patients with early-stage disease is excellent, and although a majority of patients can now be cured, further studies are needed to optimize treatment such that short- and long-term treatment-related toxicities are minimized, without compromising disease control and cure.
INTRODUCTION
Hodgkin lymphoma, previously known as Hodgkin’s disease, is a B-cell malignancy with unique pathological and epidemiological features for which highly effective therapies exist. The disease is characterized by the presence of mononuclear and multinucleate giant cells called Hodgkin and Reed-Sternberg (HRS) cells.1
Hodgkin lymphoma is unique compared to other B-cell lymphomas because of the scarcity of the malignant cells in the tumor tissue. The HRS cells usually account for only 0.1% to 10% of the cells in the affected tissues, and the HRS cells induce accumulation of nonmalignant lymphocytes, macrophages, granulocytes, eosinophils, plasma cells, and histiocytes, which constitute more than 90% of tumor cellularity.2 Although the disease was first described by Sir Thomas Hodgkin in 1832, in part because of this unique histopathology, not until 1991 was it conclusively demonstrated that HRS cells are in fact monoclonal germinal center–derived B-cells. This article reviews management and frontline treatment options for limited-stage classical Hodgkin lymphoma and nodular lymphocyte predominant Hodgkin lymphoma. Treatment of advanced stage and relapsed/refractory Hodgkin lymphoma will be discussed in a separate article.
EPIDEMIOLOGY
Hodgkin lymphoma accounts for 0.5% of all malignancies and 11.7% of all lymphomas among adults in the United States.3 The incidence of Hodgkin lymphoma has been steadily increasing over the past 4 decades and was estimated to be 8260 cases in the United States in 2017, with a slight male predominance. Hodgkin lymphoma is expected to cause 1070 deaths in 2017, accounting for 0.2% of all cancer deaths.3 First-degree relatives of patients with Hodgkin lymphoma have a 3- to 9-fold increased risk of having the disease compared to the general population,4 and monozygotic twin siblings of Hodgkin lymphoma patients have a greatly increased risk for developing the disease—up to 100-fold—compared to normal cohorts.5 The incidence is highest among Caucasians, African Americans, and Hispanics, and lower in Asians and American Indians.3 Hodgkin lymphoma incidence shows a bimodal peak distribution, with 1 peak between the ages of 15 and 44 years, and another peak after age 65 years.6
ETIOLOGY/PATHOGENESIS
The cause of Hodgkin lymphoma is unknown. Epstein-Barr virus (EBV) infection is present in up to 40% of Hodgkin lymphoma cases, suggesting a role of this virus in the pathogenesis of some cases. The risk of EBV-positive Hodgkin lymphoma was found to be higher following an episode of infectious mononucleosis, while the risk of EBV-negative Hodgkin lymphoma remained unchanged.7 The incidence of Hodgkin lymphoma is 5 to 14 times higher in HIV-infected patients than in noninfected patients.8 It is not considered an AIDS-defining illness, but has become more frequent with the growth and aging of the HIV-positive population.9,10 Hodgkin lymphoma patients with HIV typically have CD4 lymphocyte counts greater than 200 cells/μL,11 with the incidence of Hodgkin lymphoma actually declining with lower CD4 lymphocyte counts.12 HIV-related Hodgkin lymphoma tends to have an aggressive course, with high rates of EBV positivity.13 The incidence of Hodgkin lymphoma is 1.8 times higher among smokers, and the risk appears to increase with duration of smoking.14,15
The cell of origin of Hodgkin lymphoma, while long suspected to be the HRS cell, remained unproven until the 1990s when micro-dissection and single-cell polymerase chain reaction techniques allowed for confirmation that the HRS cell was in fact a monoclonal germinal center derived B cell.16,17 These HRS cells lack immunoglobulin due to defective transcription regulation and not due to crippling mutations.18,19 The cellular infiltrate in Hodgkin lymphoma appears to play a decisive role in allowing the HRS cells to survive by providing an environment that suppresses cytotoxic immune responses as well as by providing cellular interactions and cytokines that support their growth and survival. The extensive inflammatory infiltrate in classical Hodgkin lymphoma is comprised of T helper 2 (Th2) and regulatory T cells and lacks T helper 1 (Th1) cells, CD8 cytotoxic T cells, and natural killer cells.20 The HRS cells escape apoptosis by several mechanisms which include latent EBV infection and constitutive nuclear factor (NF)-kB pathways, as well as other deregulated signaling pathways that promote survival, such as EBV nuclear antigen 1 (EBNA1) protein, EBV latent infection membrane protein 1 (LMP1), and LMP2.21,22
Genetic alterations in the 9p24 locus which encodes PD-L1/PD-L2 are nearly universally present in classical Hodgkin lymphoma and are now considered a disease-defining feature.23
PATHOLOGIC CLASSIFICATION
According to the 2008 World Health Organization (WHO) classification, Hodgkin lymphoma has 2 clearly defined entities: classical Hodgkin lymphoma (cHL), which accounts for approximately 95% cases, and nodular lymphocyte predominant Hodgkin lymphoma (NLPHL), which accounts for the remaining cases.24 These 2 entities differ in their clinical, pathological, and biological features, which in turn affect prognosis and treatment options. Classical Hodgkin lymphoma is characterized by a paucity of HRS cells surrounded by a background of mixed inflammatory infiltrate comprised of histiocytes, small lymphocytes, eosinophils, neutrophils, plasma cells, fibroblasts, and collagen. Depending on the particular combinations of these elements and the specific features of the neoplastic cells, cases can be subclassified into several cHL subtypes: the nodular sclerosis, mixed cellularity, lymphocyte-rich, and lymphocyte-depleted types.25
The diagnosis of cHL is made based on a combination of morphology of HRS cells and the other cells infiltrating the tissue, combined with immunohistochemical staining. Because of the rare nature of the malignant (clonal) cell in Hodgkin lymphoma specimens, flow cytometry is generally of little value. The HRS cells in cHL are CD30-positive and CD45 negative in virtually all cases, and CD15-positive in 85% of cases.26 B-cell antigens are typically negative except for CD20, which is positive in about 20% cases.27
Nodular sclerosis Hodgkin lymphoma (NSHL) is the most common subtype of cHL, accounting for 65% to 75% of cases. It is common among young adults and tends to involve the mediastinal, supraclavicular, and cervical lymph nodes. NSHL is characterized by the presence of collagen bands that divide the lymphoid tissue into circumscribed nodules. This subtype usually presents as stage I or II disease, typically with neck and/or mediastinal disease, and evidence of EBV infection is present in approximately 10% to 40% of North American cases.7 Patients diagnosed with NSHL generally have a very good prognosis.
Mixed cellularity Hodgkin lymphoma (MCHL) constitutes about 20% to 25% of cHL cases. It affects a somewhat older population, with a median age at diagnosis of 38 years. The typical bimodal age distribution is not seen with MCHL. MCHL has a male predominance (70%), and is more frequent in HIV-infected patients (70% of whom also have EBV infection). Lymphoid tissues have classic HRS cells and significant inflammatory infiltrates. Approximately 50% of patients with MCHL present as stage III or IV with abdominal lymphadenopathy or splenic involvement, and B symptoms are frequent.24
Lymphocyte-rich Hodgkin lymphoma (LRHL) is uncommon, accounting for only 3% to 5% of cases of cHL.28 The disease usually presents at an older age and has a 2:1 male predominance. HRS cells are commonly seen and a large number of reactive lymphocytes are also present. Although on the basis of morphology and immunohistochemistry LRHL belongs to the cHL group, clinically it more closely resembles LPHL. Patients usually present at early stage and rarely have B symptoms. LRHL carries an excellent prognosis, with a greater than 90% PFS after 5 years.23,29
Lymphocyte-depleted Hodgkin lymphoma (LDHL) is the least common form of cHL, accounting for less than 5% of cases. Many cases previously placed in this category are now recognized as diffuse large B-cell lymphoma (DLBCL), anaplastic large-cell lymphoma (ALCL), or NSHL with lymphocyte depletion.30 HRS cells are frequently seen, but reactive inflammatory cells are relatively sparse. EBV infection is seen in up to 90% of cases, commonly associated with HIV-infected individuals. Advanced-stage and symptomatic disease are more common. Prognosis is slightly worse compared to other categories.
NLPHL accounts for approximately 5% of cases of Hodgkin lymphoma. It has a unimodal age distribution, with the peak incidence in the fourth decade, and male predilection of 3:1.28 NLPHL is characterized by large primary lymphoid follicles, with polytypic small B lymphocytes and extensive meshworks of follicular dendritic cells. The lymphocytic/histiocytic (L and H), or “popcorn,” cells scattered within the nodules differ from classic HRS cells, both in their morphology and in their biochemical profile, being frequently negative for CD15, CD30 and for the EBV genome, and usually positive for B-cell antigens such as CD20, suggesting that L and H cells may be immunoglobulin-synthesizing monoclonal B cells. CD45 is also typically positive in NLPHL, in distinction from cHL. NLPHL has an indolent course compared to cHL, and long-term survival is common.19,31 NLPHL commonly presents with limited-stage disease. NLPHL may eventually transform into a more aggressive lymphoma, such as diffuse large B-cell lymphoma (including centroblastic, immunoblastic, or T-cell/histiocyte–rich subtypes), at a rate of 4% to 12%. This can occur even 15 to 20 years after the initial diagnosis of NLPHL.32,33 In a recent large retrospective study of 222 patients with NLPHL, the rate of transformation to DLBCL was 7.6%, with a median time to transformation of 35 months. Overall survival was not adversely affected in patients undergoing transformation compared to those without transformation.34
PRESENTATION
Classical Hodgkin lymphoma usually presents with asymptomatic mediastinal or cervical lymphadenopathy. Half of patients present with stage I or stage II disease.35 A mediastinal mass is seen in most patients with NSHL, at times with bulky disease, with “bulky” defined as a mediastinal mass measuring one-third or more of the maximal thoracic diameter on chest x-ray, or 10 cm on computed tomography (CT) scan. Systemic symptoms, or "B" symptoms—fevers (> 38°C), drenching night sweats, and unexplained weight loss (> 10% of baseline body weight over the preceding 6 months or less)—are detected in approximately 25% of patients. Between 10% and 15% will have extranodal disease, most commonly involving lung, bone, and liver. NLPHL usually presents with limited-stage disease without B symptoms; it typically has a more indolent presentation and clinical course than cHL.
INITIAL EVALUATION AND STAGING
The initial workup includes a complete blood count (CBC), erythrocyte sedimentation rate (ESR), lactate dehydrogenase (LDH), and chemistry studies to evaluate renal function and liver function. Fine-needle aspiration will usually fail to identify the infrequent HRS cells, and instead only reveal the reactive background of inflammatory cells. Generous (large gauge) core needle biopsies may provide diagnosis effectively in some cases, but in general, an excisional lymph node biopsy is preferred to ensure an accurate diagnosis and avoid the need for repeated biopsy procedures. In cases where an excisional biopsy would be difficult or risky, a core needle biopsy procedure is a reasonable first step, with the understanding that a subsequent surgical procedure may still be necessary.
Baseline imaging includes CT scans of the neck, chest, abdomen, and pelvis. Use of positron emission tomography (PET) scanning is now standard in the initial evaluation and assessment of treatment response in Hodgkin lymphoma.36 Due to the increased sensitivity of PET or PET/CT scan, additional lesions may be identified that were not seen on conventional CT scans. This will alter the staging, and potentially the treatment plan, in up to 25% to 30% of patients.37,38 PET/CT scan performed during initial evaluation also facilitates optimal interpretation of post-therapy PET/CT scans and is therefore strongly encouraged as a part of the initial staging evaluation.39
Recent studies have shown that bone marrow biopsy is not routinely needed in the initial staging of cHL. A study of 454 patients concluded that bone marrow biopsy would not have altered the stage in any stage I or II patients. It was further concluded that overall treatment strategy would not have been altered for any of the patients.40 Based on this study and others, it is now clear that FDG-PET has a high sensitivity, and when PET scan is negative (in the bone marrow and skeleton), a bone marrow biopsy provides little additional value. For patients with significant cytopenias, a bone marrow biopsy is reasonable. Such patients may benefit from a bilateral biopsy, which increases the probability of demonstrating bone marrow involvement by 16% to 33%.41,42 Techniques such as staging laparotomy and lymphangiography are now considered obsolete.
Hodgkin lymphoma is staged according to the Ann Arbor staging system (Table 1). The original Ann Arbor staging was published in 1971,43 and in 1989 the “Cotswold modifications” extended the definitions of stage IV disease and the suffix “X” was added to denote bulky disease.44 Both systems were developed for the staging of Hodgkin lymphoma, but are now used for staging non-Hodgkin lymphoma as well.
PROGNOSTIC FACTORS
For the purposes of prognosis and selection of treatment, Hodgkin lymphoma is commonly classified into early-stage favorable, early-stage unfavorable, and advanced stage. Early-stage Hodgkin lymphoma refers to patients with Ann Arbor stage I or stage II disease. With early-stage Hodgkin lymphoma, the prognosis varies significantly based on factors such as the presence of B symptoms, elevated erythrocyte sedimentation rate ([ESR] > 50 mm/hr), number of nodal sites involved, older age, and a large mediastinal mass. For this reason, most clinical trials to evaluate treatment strategies for early-stage Hodgkin lymphoma are based on various combinations of these risk factors. The definition of early-stage unfavorable Hodgkin lymphoma varies across different clinical trial study groups, and it is important to understand the definition in interpreting the results of these trials (Table 2).45,46
In the German Hodgkin Study Group (GHSG) trials, early-stage Hodgkin lymphoma is stratified into a high risk (“unfavorable”) group defined by any of the following: a large mediastinal mass (one third of maximum thoracic diameter), extra-nodal disease, 3 or more nodal areas, and an ESR of > 50 mm/hr in asymptomatic patients or > 30 mm/hr in patients with B symptoms. Low-risk (“favorable”) patients lack all of these factors.47 The European Organization for Research and Treatment of Cancer (EORTC) defines the unfavorable prognostic group as older than 50 years of age, large mediastinal adenopathy (maximum width on a chest radiograph of at least one third of the internal transverse diameter of the thorax at the level of T5 through T6 or any mass of ≥ 10 cm in the largest dimension), an ESR of 50 mm/hr and no B symptoms, or with an ESR of 30 mm/hr in those who have B symptoms, and/or 4 or more regions of involvement.48 The National Cancer Institute of Canada (NCIC) Clinical Trials Group and the Eastern Cooperative Oncology Group (ECOG) define high-risk groups as presence of B symptoms, bulky disease with a mediastinal mass width of at least one third of the maximum chest wall diameter, or any mass greater than 10 cm, and patients with intra-abdominal disease.49,50
Gene-expression profiling in Hodgkin lymphoma has identified a gene signature of tumor-associated macrophages that was able to identify patients with a higher risk for primary treatment failure. In an independent cohort of patients, an increased number of CD68-positive macrophages was correlated with inferior outcomes.51,52 Studies such as these underscore the importance of the tumor “microenvironment” (ie, the nonmalignant cells within a tumor) in determining the overall clinical behavior of a malignancy. While quantification of CD68-positive macrophages has potential to be applied to routine clinical practice, prospective data using CD68 as a tool for risk-adapted therapy is lacking.
Genetic alterations and amplifications in the 9p24.1 locus have recently been found to be a defining genetic feature of cHL. Amplification of 9p24.1 has been associated with unfavorable outcomes. Amplification of 9p24.1 (which includes the loci encoding the PD-L1 and PD-L2 genes) is more common in patients with advanced stage disease and is associated with shorter PFS.23
A recent study attempted to integrate several different prognostic factors in cHL patients who were treated with ABVD (adriamycin [doxorubicin], bleomycin, vinblastine, and dacarbazine) and underwent an interim PET (iPET) scan after 2 cycles of ABVD. Focusing on those with a negative iPET scan, it was found that expression of CD68 and PD-1 in microenvironment cells, and STAT1 negativity in HRS cells identified a subset of PET-2 negative patients with a 3-year PFS significantly lower than that of the remaining PET-2 negative population (64% versus 95%). The algorithm correctly predicted the response to treatment in more than half of the patients who had relapse or disease progression despite a negative PET-2 scan. It therefore appears feasible, using tissue biomarkers at diagnosis, to identify patients at increased risk for poor outcome, even if the iPET scan is negative.53
ROLE OF PET/CT IN ASSESSMENT OF RESPONSE TO THERAPY
PET/CT has been increasingly used for response assessment at various stages in lymphoma in recent years. Almost all types of lymphomas are fluorodeoxyglucose (FDG) avid; however, Hodgkin lymphoma is FDG avid in 97% to 100% of cases. In 2009, a 5-point scale was developed to score PET images with regard to treatment response, either partway through treatment (iPET) or at the end of therapy.54 It was recommended as the standard reporting tool at the First International Workshop on PET in Lymphoma in Deauville, France, in 2009, and is thus now referred to as the Deauville score. A score of 1 is given if there is no uptake, 2 if the uptake ≤ mediastinum, 3 if > mediastinum but ≤ liver, 4 if uptake moderately higher than liver, 5 if uptake is markedly higher than liver and/or new lesions. X designates new areas of uptake unlikely to be related to lymphoma. In most trials, a score of 1 or 2 is considered a complete response and a score of 4 or 5 is considered a treatment failure. A score of 3 is sometimes considered a complete response, depending on the study. The Deauville criteria have been widely used in newer clinical trials utilizing response-adapted treatment as defined by PET response. PET/CT is recommended for staging and restaging at the end of therapy, in clinical practice, and clinical trials. Interim PET/CT scan, while commonly performed in clinical practice, is only recommended if the results will alter therapy (eg, if that information will result in the clinician omitting radiation therapy [RT] or altering the chemotherapy plan).
Early studies of iPET showed that achieving PET negativity early in the course of treatment was strongly associated with PFS and overall survival.55 Subsequent studies confirmed the importance of achieving a negative iPET. As a result, considerable efforts have been put into designing response-adapted treatment approaches using iPET (see Treatment section), with some of these approaches now being listed in the National Comprehensive Cancer Network (NCCN) guidelines and being used in standard practice.
TREATMENT
EVOLUTION OF TREATMENT
The treatment of Hodgkin lymphoma has evolved over the past century, starting with the discovery of RT as effective treatment in the early 20th century. Long-term survival of patients with Hodgkin lymphoma treated with involved-field radiation therapy (IFRT) was first reported in the 1960s.56,57 Outcomes improved further with the introduction of combined modality treatment (CMT) using chemotherapy and RT, with the overall 5-year relative survival for patients with Hodgkin lymphoma (all stages) treated in 2006–2012 being 85.4% to 87.3%.3 Since the majority of patients are now cured with modern therapy, treatment-related complications have become an important competing cause of mortality. Recent studies have therefore focused on maintaining efficacy while reducing toxicities, and refining the process of selecting patients who might benefit from more aggressive therapy. While RT was the first treatment modality shown to be curative for Hodgkin lymphoma,56,57 multiple subsequent studies showed that CMT is superior to RT alone in terms of relapse-free survival.58–63 In the GHSG H8-F trial, the estimated 5-year event-free survival and overall survival rates were significantly higher after 3 cycles of MOPP-ABV (mechlorethamine, vincristine, procarbazine, and prednisone combined with doxorubicin, bleomycin, and vinblastine) plus IFRT than after subtotal nodal radiotherapy alone. The 10-year overall survival estimates were 97% and 92%, respectively (P = 0.001).64 As a result, CMT replaced RT alone as the standard of care for limited-stage Hodgkin lymphoma. However, for elderly or infirm patients, or those with other comorbidities making them poor chemotherapy candidates, RT alone may be a very reasonable option.65 More recently, an increasing body of evidence has accumulated to support the use of chemotherapy alone in early stage cHL. This literature has consistently shown that omission of RT is associated with a modest increase in relapse, without a clear compromise in long-term overall survival. For some patients, the trade-off in terms of avoiding radiation (and the associated late effects) may be worth the small increase in relapse risk, since long-term survival does not appear to be substantially worse with chemo-therapy alone. Table 3 and Table 4 provide a summary of recent key studies which have defined treatment options for early-stage cHL.48,66–71
EARLY-STAGE NLPHL
NLPHL usually presents with limited-stage disease without B symptoms and has an indolent course with a slightly better prognosis compared to cHL.72 Due to the rarity of the disease, treatment guidelines are mostly based on retrospective analyses from single or multi-institution studies or subgroup analyses, often with relatively short follow-up. These studies must be interpreted with caution because of the possibility of inaccuracies in the pathologic diagnosis, small sample sizes, and selection bias. Treatment options for limited-stage NLPHL include observation, single-agent rituximab, IFRT (or involved-site radiation therapy [ISRT]) alone, or CMT.46
Historically, patients with limited-stage NLPHL have been treated with RT alone, with 80% to 85% PFS and 85% to 95% overall survival rates.73–75 Patients who relapse or progress after RT in general can successfully undergo salvage therapy.74 In one study, rates of PFS and overall survival were similar among patients who had limited-field, regional-field, or extended-field RT,75 indicating that IFRT is preferred. Studies comparing RT alone and CMT are limited. The GHSG HD7 trial included a subset of NLPHL patients, with a trend towards improved freedom from treatment failure (96% versus 83%) favoring CMT. This, however, did not translate into improved overall survival.47 A retrospective analysis of the British Columbia Cancer Agency database compared patients with limited-stage NLPHL treated with RT alone to patients who received 2 cycles of ABVD followed by RT. A significant improvement in PFS (91% versus 65%) and overall survival (93% versus 84%) was seen, favoring CMT.76
Chemotherapy alone is not recommended for limited-stage NLPHL since studies evaluating chemotherapy alone are quite limited and indicate relatively high rates of treatment failure. Given that the malignant cells in NLPHL are CD20-positive, single-agent rituximab has also been studied in this disease, including a study as frontline therapy in limited-stage patients. In this phase 2 trial in newly diagnosed patients with stage IA disease, an overall response rate (ORR) of 100% was seen, with an 85% complete response (CR) rate.77 At 3 years, overall survival was 100% and PFS was 81%, indicating that the responses with single-agent rituximab are less durable than those with RT.
Advani et al evaluated rituximab followed by observation versus rituximab (R) followed by maintenance rituximab (MR) for 2 years in 39 new or previously treated patients. At 4 weeks the ORR was 100% (with CR in 67%, and partial response in 33%). At a median follow up of 9.8 years for R alone, and 5 years for R+MR, median PFS was 3 and 5.6 years, respectively (P = 0.26). Estimated 5-yr PFS and overall survival in patients treated with R versus R+MR were 39.1% and 95.7% versus 58.9% and 85.7%, with Pvalues of 0.26 (PFS) and 0.38 (overall), respectively. Maintenance rituximab therefore appears to prolong remission, although the results did not quite reach statistical significance.78 Even though rituximab does not appear to be curative in NLPHL, it is a reasonable option for patients with early-stage NLPHL who are not good candidates for definitive RT. Whether combining rituximab with RT or CMT might further improve outcomes in early-stage NLPHL has not yet been determined.
In children, surgery alone may lead to long-term remission or possibly cure of limited-stage NLPHL. In a European multicenter retrospective study, 58 patients underwent surgery for limited-stage NLPHL. Among the 51 patients who achieved complete remission following surgery, 67% remained progression-free and 100% were alive at a median follow up of 43 months.79 In adults, there is no data to support surgical treatment alone for NLPHL. Finally, observation may be a reasonable option in elderly or infirm patients for whom NLPHL is unlikely to affect life expectancy. For younger patients, given the excellent outcome with modern therapy and the long-term risk of transformation of NLPHL into an aggressive non-Hodgkin lymphoma, observation is generally not recommended.
The NCCN recommends RT (ISRT or IFRT, 30–36 Gy) as the preferred treatment for stage IA and IIA non-bulky NLPHL. In patients with stage IA disease with complete excision of solitary nodule, observation may be appropriate. A course of chemotherapy with ISRT with or without rituximab is recommended for patients with stage IB or IIB disease, or patients with stage IA or IIA bulky disease.
FIRST-LINE TREATMENT OF LIMITED-STAGE CHL
Early-Stage Favorable cHL
There is lack of consensus regarding the ideal treatment approach for patients with early-stage favorable cHL. However, there are several excellent options available, with overall survival rates exceeding 90%. Most of these regimens involve CMT, although some chemotherapy-alone approaches have been evaluated as well. Concurrent with the demonstration of excellent long-term remission rates with CMT, it became apparent that the long-term survival and quality of life of these patients is determined in large part by the risk of serious (and potentially fatal) treatment-related toxicities. Such toxicities consist primarily of secondary malignancies and cardiovascular events, and can continue to cause significant morbidity and mortality even 2 to 3 decades after treatment.80–82 As a result, treatment decisions for these patients are complicated and require balancing efficacy against risk of late complications.
In the United States, until recently, CMT was generally considered the standard of care, with robust long-term data regarding efficacy. The most commonly used regimen has been ABVD for 2 to 4 cycles followed by IFRT. In some German studies, escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone) has been used, but this is not a general standard of care in the United States for early-stage patients.
More recent data suggests that the rate of serious late complications in Hodgkin lymphoma patients is decreasing, likely due to less extensive radiation fields, lower radiation doses, and a movement away from the MOPP regimen to ABVD.83,84 For patients who meet the “favorable” criteria set forth in the GHSG HD10 trial (see Table 2), 2 cycles of ABVD followed by 20 Gy of IFRT is an attractive option, with efficacy preserved and a low anticipated rate of late effects.66 With this approach, and with long-term (10 years) follow up, all 4 arms had similar PFS (87%) and overall survival (94%), whether 2 or 4 cycles of ABVD were given. When the effects of 20-Gy and 30-Gy doses of RT were compared, there were also no significant differences in freedom from treatment failure or overall survival. Adverse events and acute toxic effects of treatment were most common in the patients who received 4 cycles of ABVD and 30 Gy of RT.66,71
In recent years, in an attempt to reduce late effects further, regimens consisting of chemotherapy alone have been investigated. In a study by Meyer et al, at 12 years the rate of overall survival was 94% among those receiving ABVD alone, as compared with 87% among those receiving subtotal nodal RT; the rates of freedom from disease progression were 87% and 92% in the 2 groups; and the rates of event-free survival were 85% and 80%, respectively.50 However, it is important to note that this study did not include a CMT arm for the early favorable patients, and did not utilize modern RT techniques. Nevertheless, this early study and others60 suggested that chemotherapy alone may be a reasonable option for some early-stage cHL patients, particularly for patients who are felt to be at increased risk for late toxicities from RT. As a result, additional studies have been conducted evaluating CMT versus chemotherapy alone for early-stage cHL. Many of these studies have incorporated interim PET/CT scan to develop a response-adapted approach to decide which patients are least likely to benefit from RT.
The HD-13 study was a follow-up study for HD-10, looking at deletion of bleomycin, dacarbazine, or both from the ABVD backbone. The ABD arm was closed early, because of an excess rate of treatment failure. Among the 1243 patents assigned to either the ABVD or AVD arm at 5 years of follow-up, there was 4.3% difference in PFS. This study was not able to demonstrate that 2 cycles of AVD was noninferior to 2 cycles of ABVD, each followed by 30 Gy IFRT, even though there was no difference in all 4 groups. It confirmed 2 cycles of ABVD as the preferred regimen in early favorable Hodgkin lymphoma, when CMT is the plan of care. However, for patients over age 60 to 65 years, or those with underlying cardiac or pulmonary comorbidities, bleomycin has significant risk of toxicity. In that setting, AVD is a safer option, with only a very modest decrease in 5-year PFS.
Based on the observation that iPET scan is highly predictive of outcome in Hodgkin lymphoma,55,85 several trials have employed the use of an iPET scan to guide therapy. It is hoped that such studies will lead to new PET-directed treatment algorithms in which patients who require more aggressive therapy (eg, with CMT, or escalated BEACOPP) can be identified, and the remaining patients can be safely treated less aggressively (eg, with chemotherapy alone).
In the EORTC H10 trial, performed to evaluate treatment adaptation on the basis of iPET scan results in stage I and II Hodgkin lymphoma, a control arm received standard combined modality treatment (3 or 4 cycles of ABVD with INRT) irrespective of PET scan results. In the experimental arm, patients with a negative PET scan after 2 cycles of ABVD continued with 1 or 2 cycles of ABVD and did not receive RT. The iPET-positive patients received either standard treatment with ABVD plus INRT or escalated BEACOPP plus INRT. The iPET-negative patients received either ABVD only or ABVD plus INRT. The final results of this study, published recently, showed that in the iPET-positive patients the 5-year PFS was improved from 77.4% with standard ABVD plus INRT to 90.6% with escalated BEACOPP plus INRT (P = 0.002). In iPET-negative patients, 5-year PFS in the favorable group was 99% versus 87.1% in favor of ABVD plus INRT. The H10 study suggested that PET results after 2 cycles of ABVD can be integrated into treatment planning, In iPET-negative patients, the study was technically not able to demonstrate the noninferiority of the ABVD only regimen, owing to a higher risk of relapse if INRT is omitted, particularly in the favorable group.48 However, this study does show that excellent outcomes can be obtained with omission of RT in patients with a negative iPET scan. This study provides a cautionary lesson though, in that the increase in relapse rate associated with omission of RT was more substantial (12%) for favorable versus unfavorable early-stage patients (2.5%), and this difference was only apparent after longer (5 years) follow-up. Despite this, chemotherapy alone is considered a reasonable treatment option, especially for patients felt to be at increased risk for late toxicities of RT or for patients who wish to avoid the risks of RT, with over 99% of patients alive at 5 years.
Similar results were shown in the RAPID trial, in which patients with limited-stage cHL underwent 3 cycles of ABVD followed by PET assessment.67 Patients with a negative PET (n = 426) were then randomized to RT (n = 209) versus no further therapy (n = 211). At a median of 60 months of follow-up, 3-year PFS was 94.6% in the RT group and 90.8% in the chemotherapy alone group. Similar to the H10 trial, it was concluded that chemotherapy alone was statistically inferior to CMT in terms of PFS. However, also similar to the H10 trial, the RAPID trial demonstrated that excellent results can be obtained in early-stage cHL patients with omission of RT, if iPET scan is negative after 3 cycles of ABVD, as there was no survival difference. These findings indicate that, when relapses occur as a result of omission of RT, such patients can be effectively treated later.
In the ongoing GHSG HD16 trial, patients with early-stage favorable cHL will be randomly assigned to a standard approach (ABVD × 2 cycles followed by 20-Gy IFRT) versus an experimental approach in which they receive ABVD for 2 cycles and then undergo PET scan. If the PET remains positive, they will receive 20-Gy IFRT. If the PET is negative, they will receive no further therapy. This trial could ultimately define ABVD for 2 cycles as a treatment option.
It is clear from these studies that omission of RT results in a somewhat higher rate of relapse but can be considered in selected patients. However, taking a less aggressive frontline approach may also be justified by the fact that, for those who do relapse, successful salvage therapies are available. Aggressive salvage therapy with autologous stem cell transplantation historically can cure approximately 50% of relapsed patients. With new and emerging therapies for relapsed disease, such as brentuximab vedotin and the PD-1 inhibitors (eg, nivolumab and pembrolizumab), the ability to cure relapsed patients may improve even more, further calling into question the practice of applying CMT uniformly for early-stage patients undergoing first-line therapy. Unfortunately, there is insufficient data from large randomized studies with long-term follow-up to fully address this issue currently, and there remains some controversy around this issue. NCCN recommends restaging PET/CT after 3 cycles of ABVD if a chemotherapy alone treatment modality is chosen. If the Deauville score is 1 or 2, either observation or 1 additional cycle of ABVD is recommended.46
Early-Stage Unfavorable cHL
In the United States, historically early-stage unfavorable Hodgkin lymphoma has been treated with CMT, most commonly 4 to 6 cycles of ABVD followed by consolidative RT. With this approach one can expect a 5-year PFS of approximately 80% to 85%.58,64,86 The GHSG HD8 trial showed that RT volume size reduction from extended-field to involved-field after COPP + ABVD chemotherapy for 2 cycles produced similar results and less toxicity in patients with early-stage unfavorable cHL.86 The GHSG trial HD11 established ABVD for 4 cycles plus 30-Gy IFRT as a standard for early unfavorable Hodgkin lymphoma. The freedom from treatment failure at 5 years was 85.0%, and overall survival was 94.5%.68
In the HD14 study by the GHSG, patients with early unfavorable cHL were treated with 2 cycles of escalated BEACOPP followed by 2 cycles of ABVD, versus 4 cycles of ABVD. All patients then received 30 Gy of consolidative IFRT. A 5-year PFS of 95% was seen in the experimental arm, compared with 89% in the standard (ABVD) arm. As expected, this regimen was associated with more acute hematologic toxicity, and there was no difference between the 2 regimens with respect to overall survival or fertility.69 Given the lack of improved survival and increased toxicity, ABVD has remained the standard chemotherapy regimen for early unfavorable cHL in the United States. NCCN recommends a restaging PET scan after 2 cycles of ABVD and to continue with 2 to 4 cycles of ABVD or escalated BEACOPP with or without ISRT based on Deauville scores.
Another viable treatment option is the Stanford V regimen, a condensed, 12-week regimen that includes mechlorethamine, doxorubicin, vinblastine, prednisone, vincristine, etoposide, and bleomycin, followed by IFRT.87 In a randomized phase 3 trial conducted by ECOG (E2496), patients with stage I/II Hodgkin lymphoma with bulky mediastinal disease or advanced-stage disease were randomized to ABVD × 6 to 8 cycles versus Stanford V. RT was given (36 Gy) for those with bulky mediastinal disease or to sites of disease greater than 5 cm in the Stanford V arm. In a subset analysis focusing only on those with stage I/II bulky mediastinal disease, the 5-year failure free survival was 85% versus 79% and the 5-year overall survival was 96% versus 92% for the ABVD versus Stanford V arms, respectively. These differences were not statistically significant.70 While the Stanford V regimen has the advantages of a 12-week treatment duration and a lower cumulative amount of bleomycin and doxorubicin, the Stanford V arm had higher rates of grade 3 lymphopenia and grade 3 to 4 peripheral neuropathies. In addition, Stanford V requires that most patients undergo RT (to original sites of disease measuring 5 cm or more plus contiguous areas). As a result, the investigators concluded that ABVD × 4 cycles plus IFRT remains the standard of care for patients with early unfavorable Hodgkin lymphoma with bulky mediastinal disease.
Other regimens have been studied in hopes of reducing toxicity, including the EVE regimen (epirubicin, vinblastine, and etoposide). This regimen was compared to ABVD in early unfavorable Hodgkin lymphoma patients, with all patients undergoing the same RT program. No differences were observed between the ABVD and EVE arms in terms of complete remission rate and overall survival. However, patients who received EVE had a significantly worse outcome than those who received ABVD in terms of relapse-free survival and failure-free survival.88 EBVP (epirubicin, bleomycin, vinblastine, and prednisone) followed by IFRT was less efficacious compared with MOPP/ABV–type therapy.58
An area of active investigation is whether RT can be safely omitted in patients with early- stage unfavorable cHL. The EORTC H10 study showed that, for patients with a negative iPET scan (after 2 cycles), the 5-year PFS rates were 92.1% versus 89.6% for ABVD plus INRT versus ABVD alone, respectively. While this technically did not meet criteria for noninferiority of ABVD alone, this study demonstrated that, for those with negative iPET, ABVD × 6 cycles (without radiation) can result in long-term remission in a high proportion (89%) of patients. For iPET-positive patients, 2 cycles of escalated BEACOPP were given followed by 30 Gy of IFRT on the experimental arm. This resulted in a 5-year PFS of 90.6% versus 77.4%, suggesting this may be a preferred approach for early-stage unfavorable patients with a positive iPET.48 Even though the noninferiority of ABVD alone could not be established based on the statistical design of the study, the current NCCN guidelines recommend restaging after 2 cycles of ABVD for stage I or II unfavorable cHL and using that iPET as a guide, based on Deauville scores. For scores 1–3, ABVD × 2 cycles (total 4 cycles) plus ISRT or AVD × 4 (total 6) with or without ISRT is recommended. For a Deauville score of 4, escalated BEACOPP × 2 cycles or ABVD × 2 cycles (total 4) followed by ISRT is recommended. If the Deauville score is 5, further treatment decisions should be made based on repeat biopsy results. A follow up PET/CT is recommended for Deauville scores of 4 and 5 to confirm complete response.46
LATE EFFECTS AND THE EVOLUTION OF RADIATION THERAPY
The RT given in Hodgkin lymphoma has evolved considerably over the years, from extended field or subtotal nodal fields developed in the 1960s, to the more focused involved-field or even involved-site radiation commonly given now. This approach reduces radiation volumes, and it already is becoming evident that the relative risk of breast cancer among young females receiving mediastinal RT for Hodgkin lymphoma is declining.89 Cardiac dose is reduced significantly with IFRT compared to older radiation techniques as well. The extent of radiation may be reduced even further with involved-nodal/involved site or intensity-modulated approaches.90
With new RT techniques allowing for more focused therapy and lower doses of radiation, models predict that the rate of long-term complications will decline further.91,92 Furthermore, response-adapted (ie, PET-directed) approaches, as discussed in detail earlier in the article, are expected to increasingly allow for identification of patients who can safely avoid radiation entirely, which will hopefully lead to an even lower rate of late complications of therapy.
MONITORING FOR RELAPSE
A number of recent studies have shown that, for patients who achieve complete remission with first-line therapy, performing repeated scheduled surveillance imaging does not improve outcomes. In fact, most relapses are detected by the patient (due to symptoms or recurrence of lymph node enlargement). It is rare that a relapse would be detected by surveillance imaging alone. Furthermore, surveillance that includes routine imaging has not been associated with improved survival.93 As a result, it is now recommended that patients undergo regular follow-up with symptom review, physical exam, and basic laboratory studies. Imaging studies should be obtained as needed for patients who develop signs, symptoms, exam findings, or laboratory values concerning for relapse.
More important than scheduled surveillance imaging for relapse is monitoring for late effects of therapy. These fall into several broad categories such as cardiovascular disease (coronary disease, congestive heart failure, valvular disease, carotid artery disease), pulmonary disease, hypothyroidism, and secondary malignancies. Aggressive surveillance for breast cancer is especially warranted in female patients who underwent chest radiation.46
CONCLUSION
Hodgkin lymphoma is characterized pathologically by the presence of HRS cells accompanied by a polymorphous cellular infiltrate. It is a disease with a bimodal age distribution, several pathologic subtypes, and numerous treatment options. Overall, the prognosis for patients with early-stage disease is excellent, and although a majority of patients can now be cured, further studies are needed to optimize treatment such that short- and long-term treatment-related toxicities are minimized, without compromising disease control and cure.
- Küppers R, Rajewsky K, Zhao M, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A 1994;91:10962–6.
- Küppers R. The biology of Hodgkin›s lymphoma. Nat Rev Cancer 2009;9:15–27.
- National Cancer Institute. SEER cancer statistics review, 1975–2014. 2017. http://seer.cancer.gov/csr/1975_2013/. Accessed April 27, 2017.
- Haim N, Cohen Y, Robinson E. Malignant lymphoma in first-degree blood relatives. Cancer 1982;49:2197–200.
- Mack TM, Cozen W, Shibata DK, et al. Concordance for Hodgkin’s disease in identical twins suggesting genetic susceptibility to the young-adult form of the disease. N Engl J Med 1995;332:413–8.
- Sant M, Allemani C, Tereanu C, et al. Incidence of hematologic malignancies in Europe by morphologic subtype: results of the HAEMACARE project. Blood 2010;116:3724–34.
- Hjalgrim H, Askling J, Rostgaard K, et al. Characteristics of Hodgkin’s lymphoma after infectious mononucleosis. N Engl J Med 2003;349:1324–32.
- Hessol NA, Katz MH, Liu JY, et al. Increased incidence of Hodgkin disease in homosexual men with HIV infection. Ann Intern Med 1992;117:309–11.
- Shiels MS, Pfeiffer RM, Gail MH, et al. Cancer burden in the HIV-infected population in the United States. J Natl Cancer Inst 2011;103:753–62.
- Powles T, Robinson D, Stebbing J, et al. Highly active antiretroviral therapy and the incidence of non-AIDS-defining cancers in people with HIV infection. J Clin Oncol 2009;27:884–90.
- Bedimo RJ, McGinnis KA, Dunlap M, et al. Incidence of non-AIDS-defining malignancies in HIV-infected versus noninfected patients in the HAART era: impact of immunosuppression. J Acquir Immune Defic Syndr 2009;52:203–8.
- Biggar RJ, Jaffe ES, Goedert JJ, et al. Hodgkin lymphoma and immunodeficiency in persons with HIV/AIDS. Blood 2006;108:3786–91.
- Thompson LD, Fisher SI, Chu WS, et al. HIV-associated Hodgkin lymphoma: a clinicopathologic and immunophenotypic study of 45 cases. Am J Clin Pathol 2004;121:727–38.
- Briggs NC, Hall HI, Brann EA, et al. Cigarette smoking and risk of Hodgkin’s disease: a population-based case-control study. Am J Epidemiol 2002;156:1011–20.
- Castillo JJ, Dalia S, Shum H. Meta-analysis of the association between cigarette smoking and incidence of Hodgkin’s Lymphoma. J Clin Oncol 2011;29:3900–6.
- Kanzler H, Kuppers R, Hansmann ML, Rajewsky K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med 1996;184:1495–505.
- Stein H, Hummel M. Cellular origin and clonality of classic Hodgkin’s lymphoma: immunophenotypic and molecular studies. Semin Hematol 1999;36:233-41.
- Marafioti T, Hummel M, Foss HD, et al. Hodgkin and reed-sternberg cells represent an expansion of a single clone originating from a germinal center B-cell with functional immunoglobulin gene rearrangements but defective immunoglobulin transcription. Blood 2000;95:1443–50.
- Marafioti T, Hummel M, Anagnostopoulos I, et al. Origin of nodular lymphocyte-predominant Hodgkin’s disease from a clonal expansion of highly mutated germinal-center B cells. N Engl J Med 1997;337:453–8.
- van den Berg A, Visser L, Poppema S. High expression of the CC chemokine TARC in Reed-Sternberg cells. A possible explanation for the characteristic T-cell infiltratein Hodgkin’s lymphoma. Am J Pathol 1999;154:1685–91.
- Bargou RC, Emmerich F, Krappmann D, et al. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J Clin Invest 1997;100:2961–9.
- Luftig M, Yasui T, Soni V, et al. Epstein-Barr virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKK alpha-dependent noncanonical NF-kappaB activation. Proc Natl Acad Sci U S A 2004;101:141–6.
- Roemer MGM, Advani RH, Ligon AH, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol 2016;34:2690–7.
- Swerdlow SH CE, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008.
- Campo E, Swerdlow SH, Harris NL, et al. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 2011;117:5019–32.
- von Wasielewski R, Mengel M, Fischer R, et al. Classical Hodgkin’s disease. Clinical impact of the immunophenotype. Am J Pathol 1997;151:1123–30.
- Tzankov A, Krugmann J, Fend F, et al. Prognostic significance of CD20 expression in classical Hodgkin lymphoma: a clinicopathological study of 119 cases. Clin Cancer Res 2003;9:1381–6.
- Diehl V, Sextro M, Franklin J, et al. Clinical presentation, course, and prognostic factors in lymphocyte-predominant Hodgkin’s disease and lymphocyte-rich classical Hodgkin’s disease: report from the European Task Force on Lymphoma Project on Lymphocyte-Predominant Hodgkin’s Disease. J Clin Oncol 1999;17:776–83.
- Shimabukuro-Vornhagen A, Haverkamp H, Engert A, et al. Lymphocyte-rich classical Hodgkin’s lymphoma: clinical presentation and treatment outcome in 100 patients treated within German Hodgkin’s Study Group trials. J Clin Oncol 2005;23:5739–45.
- Slack GW, Ferry JA, Hasserjian RP, et al. Lymphocyte depleted Hodgkin lymphoma: an evaluation with immunophenotyping and genetic analysis. Leuk Lymphoma 2009;50:937–43.
- Mason DY, Banks PM, Chan J, et al. Nodular lymphocyte predominance Hodgkin’s disease. A distinct clinicopathological entity. Am J Surg Pathol 1994;18:526–30.
- Rudiger T, Gascoyne RD, Jaffe ES, et al. Workshop on the relationship between nodular lymphocyte predominant Hodgkin’s lymphoma and T cell/histiocyte-rich B cell lymphoma. Ann Oncol 2002;13 Suppl 1:44–51.
- Sundeen JT, Cossman J, Jaffe ES. Lymphocyte predominant Hodgkin’s disease nodular subtype with coexistent “large cell lymphoma”. Histological progression or composite malignancy? Am J Surg Pathol 1988;12:599–606.
- Kenderian SS, Habermann TM, Macon WR, et al. Large B-cell transformation in nodular lymphocyte-predominant Hodgkin lymphoma: 40-year experience from a single institution. Blood. 2016;127:1960–6.
- Mauch PM, Kalish LA, Kadin M, et al. Patterns of presentation of Hodgkin disease. Implications for etiology and pathogenesis. Cancer 1993;71:2062–71.
- Juweid ME, Cheson BD. Positron-emission tomography and assessment of cancer therapy. N J Engl Med 2006;354:496–507.
- Hutchings M, Loft A, Hansen M, et al. Position emission tomography with or without computed tomography in the primary staging of Hodgkin’s lymphoma. Haematologica 2006;91:482–9.
- Naumann R, Beuthien-Baumann B, Reiss A, et al. Substantial impact of FDG PET imaging on the therapy decision in patients with early-stage Hodgkin’s lymphoma. Br J Cancer 2004;90:620–5.
- Juweid ME, Stroobants S, Hoekstra OS, et al. Use of positron emission tomography for response assessment of lymphoma: consensus of the Imaging Subcommittee of International Harmonization Project in Lymphoma. J Clin Oncol 2007;25:571–8.
- El-Galaly TC, d’Amore F, Mylam KJ, et al. Routine bone marrow biopsy has little or no therapeutic consequence for positron emission tomography/computed tomography-staged treatment-naive patients with Hodgkin lymphoma. J Clin Oncol 2012;30:4508–14.
- Wang J, Weiss LM, Chang KL, et al. Diagnostic utility of bilateral bone marrow examination: significance of morphologic and ancillary technique study in malignancy. Cancer 2002;94:1522–31.
- Menon NC, Buchanan JG. Bilateral trephine bone marrow biopsies in Hodgkin’s and non-Hodgkin’s lymphoma. Pathology 1979;11:53–7.
- Carbone PP, Kaplan HS, Musshoff K, et al. Report of the Committee on Hodgkin’s Disease Staging Classification. Cancer Res 1971;31:1860–1.
- Lister TA, Crowther D, Sutcliffe SB, et al. Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin’s disease: Cotswolds meeting. J Clin Oncol 1989;7:1630–6.
- Armitage JO. Early-stage Hodgkin’s lymphoma. N Engl J Med 2010;363:653–62.
- National Comprehensive Cancer Network I. NCCN Guidelines Version 3.2016 Hodgkin lymphoma. 2017.
- Engert A, Franklin J, Eich HT, et al. Two cycles of doxorubicin, bleomycin, vinblastine, and dacarbazine plus extended-field radiotherapy is superior to radiotherapy alone in early favorable Hodgkin's lymphoma: final results of the GHSG HD7 trial. J Clin Oncol 2007;25:3495–502.
- Andre MP, Girinsky T, Federico M, et al. Early positron emission tomography response-adapted treatment in stage I and II Hodgkin lymphoma: final results of the randomized EORTC/LYSA/FIL H10 trial. J Clin Oncol 2017:Jco2016686394.
- Meyer RM, Gospodarowicz MK, Connors JM, et al. Randomized comparison of ABVD chemotherapy with a strategy that includes radiation therapy in patients with limited-stage Hodgkin’s lymphoma: National Cancer Institute of Canada Clinical Trials Group and the Eastern Cooperative Oncology Group. J Clin Oncol 2005;23:4634–42.
- Meyer RM, Gospodarowicz MK, Connors JM, Pearcey RG, Wells WA, Winter JN, et al. ABVD alone versus radiation-based therapy in limited-stage Hodgkin’s lymphoma. N Engl J Med 2012;366:399–408.
- Steidl C, Lee T, Shah SP, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med 2010;362:875–85.
- Kamper P, Bendix K, Hamilton-Dutoit S, et al. Tumor-infiltrating macrophages correlate with adverse prognosis and Epstein-Barr virus status in classical Hodgkin’s lymphoma. Haematologica 2011;96:269–76.
- Agostinelli C, Gallamini A, Stracqualursi L, et al. The combined role of biomarkers and interim PET scan in prediction of treatment outcome in classical Hodgkin’s lymphoma: a retrospective, European, multicentre cohort study. Lancet Haematol 2016;3:e467–e79.
- Meignan M, Gallamini A, Meignan M, et al. Report on the First International Workshop on Interim-PET-Scan in Lymphoma. Leuk Lymph 2009;50:1257–60.
- Gallamini A, Hutchings M, Rigacci L, et al. Early interim 2-[18F]fluoro-2-deoxy-D-glucose positron emission tomography is prognostically superior to international prognostic score in advanced-stage Hodgkin’s lymphoma: a report from a joint Italian-Danish study. J Clin Oncol 2007;25:3746–52.
- Easson EC, Russell MH. Cure of Hodgkin’s Disease. Br Med J 1963;1(5347):1704–7.
- Kaplan HS. The radical radiotherapy of regionally localized Hodgkin’s disease. Radiology 1962;78:553–61.
- Noordijk EM, Carde P, Dupouy N, et al. Combined-modality therapy for clinical stage I or II Hodgkin’s lymphoma: long-term results of the European Organisation for Research and Treatment of Cancer H7 randomized controlled trials. J Clin Oncol 2006;24:3128–35.
- Eghbali H, Raemaekers J, Carde P. The EORTC strategy in the treatment of Hodgkin’s lymphoma. Eur J Haematol Suppl 2005:135–40.
- Bloomfield CD PT, Glicksman AS, et al. Chemotherapy and combined modality therapy for Hodgkin’s disease: A progress report on cancer and leukemia group B studies. Cancer Treat Rep 1982;66:835–46.
- Pavlovsky S, Maschio M, Santarelli MT, et al. Randomized trial of chemotherapy versus chemotherapy plus radiotherapy for stage I-II Hodgkin’s disease. J Natl Cancer Inst 1988;80:1466–73.
- Aviles A, Delgado S. A prospective clinical trial comparing chemotherapy, radiotherapy and combined therapy in the treatment of early stage Hodgkin’s disease with bulky disease. Clin Lab Haematol 1998;20:95–9.
- Herbst C, Rehan FA, Brillant C, et al. Combined modality treatment improves tumor control and overall survival in patients with early stage Hodgkin’s lymphoma: a systematic review. Haematologica 2010;95:494–500.
- Ferme C, Eghbali H, Meerwaldt JH, et al. Chemotherapy plus involved-field radiation in early-stage Hodgkin’s disease. N Engl J Med 2007;357:1916–27.
- Landgren O, Axdorph U, Fears TR, et al. A population-based cohort study on early-stage Hodgkin lymphoma treated with radiotherapy alone: with special reference to older patients. Ann Oncol 2006;17:1290–5.
- Engert A, Plutschow A, Eich HT, et al. Reduced treatment intensity in patients with early-stage Hodgkin's lymphoma. N Engl J Med 2010;363:640–52.
- Radford J, Illidge T, Counsell N, et al. Results of a trial of PET-directed therapy for early-stage Hodgkin's lymphoma. N Eng J Med 2015;372:1598–607.
- Eich HT, Diehl V, Gorgen H, et al. Intensified chemotherapy and dose-reduced involved-field radiotherapy in patients with early unfavorable Hodgkin’s lymphoma: final analysis of the German Hodgkin Study Group HD11 trial. J Clin Oncol 2010;28:4199–206.
- von Tresckow B, Plutschow A, Fuchs M, et al. Dose-intensification in early unfavorable Hodgkin’s lymphoma: final analysis of the German Hodgkin Study Group HD14 trial. J Clin Oncol 2012;30:907–13.
- Advani RH, Hong F, Fisher RI, et al. Randomized phase III trial comparing ABVD plus radiotherapy with the Stanford V regimen in patients with stages I or II locally extensive, bulky mediastinal Hodgkin lymphoma: a subset analysis of the North American Intergroup E2496 Trial. J Clin Oncol 2015;33:1936–42.
- Sasse S, Brockelmann PJ, Georgen H, et al. Long-term follow-up of contemporary treatment in early-stage Hodgkin lymphoma: Updated analyses of the German Hodgkin Study Group HD7, HD8, HD10 and HD11 trials. J Clin Oncol 2017 Apr 18:JCO2016709410. doi: 10.1200/JCO.2016.70.9410. [Epub ahead of print]
- Nogova L, Reineke T, Brillant C, et al. Lymphocyte-predominant and classical Hodgkin’s lymphoma: a comprehensive analysis from the German Hodgkin Study Group. J Clin Oncol 2008;26:434–9.
- Wirth A, Yuen K, Barton M, et al. Long-term outcome after radiotherapy alone for lymphocyte-predominant Hodgkin lymphoma: a retrospective multicenter study of the Australasian Radiation Oncology Lymphoma Group. Cancer 2005;104:1221–9.
- Chera BS, Olivier K, Morris CG, et al. Clinical presentation and outcomes of lymphocyte-predominant Hodgkin disease at the University of Florida. Am J Clin Oncol 2007;30:601–6.
- Chen RC, Chin MS, Ng AK, et al. Early-stage, lymphocyte-predominant Hodgkin’s lymphoma: patient outcomes from a large, single-institution series with long follow-up. J Clin Oncol 2010;28:136–41.
- Savage KJ, Skinnider B, Al-Mansour M, et al. Treating limited-stage nodular lymphocyte predominant Hodgkin lymphoma similarly to classical Hodgkin lymphoma with ABVD may improve outcome. Blood 2011;118:4585–90.
- Eichenauer DA FM, Pluetschow A, et al. Phase 2 study of rituximab in newly diagnosed stage IA nodular lymphocytepredominant Hodgkin lymphoma: a report from the German Hodgkin Study Group. Blood 2011;118:4363–5.
- Advani RH, Horning SJ, Hoppe RT, et al. Mature results of a phase II study of rituximab therapy for nodular lymphocyte-predominant Hodgkin lymphoma. J Clin Oncol 2014;32:912–8.
- Mauz-Korholz C, Gorde-Grosjean S, Hasenclever D, et al. Resection alone in 58 children with limited stage, lymphocyte-predominant Hodgkin lymphoma-experience from the European network group on pediatric Hodgkin lymphoma. Cancer 2007;110:179–85.
- Ng AK. Review of the cardiac long-term effects of therapy for Hodgkin lymphoma. Br J Haematol 2011;154:23–31.
- Ng AK, LaCasce A, Travis LB. Long-term complications of lymphoma and its treatment. J Clin Oncol 2011;29:1885–92.
- Aleman BM, van den Belt-Dusebout AW, Klokman WJ, et al. Long-term cause-specific mortality of patients treated for Hodgkin’s disease. J Clin Oncol 2003;21:3431–9.
- Girinsky T, van der Maazen R, Specht L, et al. Involved-node radiotherapy (INRT) in patients with early Hodgkin lymphoma: concepts and guidelines. Radiother Oncol 2006;79:270–7.
- Campbell BA, Voss N, Pickles T, et al. Involved-nodal radiation therapy as a component of combination therapy for limited-stage Hodgkin’s lymphoma: a question of field size. J Clin Oncol 2008;26:5170–4.
- Advani R, Maeda L, Lavori P, et al. Impact of positive positron emission tomography on prediction of freedom from progression after Stanford V chemotherapy in Hodgkin’s disease. J Clin Oncol 2007;25:3902–7.
- Engert A, Schiller P, Josting A, et al. Involved-field radiotherapy is equally effective and less toxic compared with extended-field radiotherapy after four cycles of chemotherapy in patients with early-stage unfavorable Hodgkin’s lymphoma: results of the HD8 trial of the German Hodgkin’s Lymphoma Study Group. J Clin Oncol 2003;21:3601–8.
- Horning SJ, Hoppe RT, Breslin S, et al. Stanford V and radiotherapy for locally extensive and advanced Hodgkin’s disease: mature results of a prospective clinical trial. J Clin Oncol 2002;20:630–7.
- Pavone V, Ricardi U, Luminari S, et al. ABVD plus radiotherapy versus EVE plus radiotherapy in unfavorable stage IA and IIA Hodgkin’s lymphoma: results from an Intergruppo Italiano Linfomi randomized study. Ann Oncol 2008;19:763–8.
- De Bruin ML, Sparidans J, van’t Veer MB, et al. Breast cancer risk in female survivors of Hodgkin’s lymphoma: lower risk after smaller radiation volumes. J Clin Oncol 2009;27:4239–46.
- Hodgson DC. Late effects in the era of modern therapy for Hodgkin lymphoma. Hematology Am Soc Hematol Educ Program 2011;2011:323–9.
- Maraldo MV, Brodin NP, Vogelius IR, et al. Risk of developing cardiovascular disease after involved node radiotherapy versus mantle field for Hodgkin lymphoma. Int J Radiat Oncol Biol Phys 2012;83:1232–7.
- Campbell BA, Hornby C, Cunninghame J, et al. Minimising critical organ irradiation in limited stage Hodgkin lymphoma: a dosimetric study of the benefit of involved node radiotherapy. Ann Oncol 2012;23:1259–66.
- Pingali SR, Jewell SE, Havlat L, et al. Limited utility of routine surveillance imaging for classical Hodgkin lymphoma patients in first complete remission. Cancer 2014;120:2122–9.
- Küppers R, Rajewsky K, Zhao M, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A 1994;91:10962–6.
- Küppers R. The biology of Hodgkin›s lymphoma. Nat Rev Cancer 2009;9:15–27.
- National Cancer Institute. SEER cancer statistics review, 1975–2014. 2017. http://seer.cancer.gov/csr/1975_2013/. Accessed April 27, 2017.
- Haim N, Cohen Y, Robinson E. Malignant lymphoma in first-degree blood relatives. Cancer 1982;49:2197–200.
- Mack TM, Cozen W, Shibata DK, et al. Concordance for Hodgkin’s disease in identical twins suggesting genetic susceptibility to the young-adult form of the disease. N Engl J Med 1995;332:413–8.
- Sant M, Allemani C, Tereanu C, et al. Incidence of hematologic malignancies in Europe by morphologic subtype: results of the HAEMACARE project. Blood 2010;116:3724–34.
- Hjalgrim H, Askling J, Rostgaard K, et al. Characteristics of Hodgkin’s lymphoma after infectious mononucleosis. N Engl J Med 2003;349:1324–32.
- Hessol NA, Katz MH, Liu JY, et al. Increased incidence of Hodgkin disease in homosexual men with HIV infection. Ann Intern Med 1992;117:309–11.
- Shiels MS, Pfeiffer RM, Gail MH, et al. Cancer burden in the HIV-infected population in the United States. J Natl Cancer Inst 2011;103:753–62.
- Powles T, Robinson D, Stebbing J, et al. Highly active antiretroviral therapy and the incidence of non-AIDS-defining cancers in people with HIV infection. J Clin Oncol 2009;27:884–90.
- Bedimo RJ, McGinnis KA, Dunlap M, et al. Incidence of non-AIDS-defining malignancies in HIV-infected versus noninfected patients in the HAART era: impact of immunosuppression. J Acquir Immune Defic Syndr 2009;52:203–8.
- Biggar RJ, Jaffe ES, Goedert JJ, et al. Hodgkin lymphoma and immunodeficiency in persons with HIV/AIDS. Blood 2006;108:3786–91.
- Thompson LD, Fisher SI, Chu WS, et al. HIV-associated Hodgkin lymphoma: a clinicopathologic and immunophenotypic study of 45 cases. Am J Clin Pathol 2004;121:727–38.
- Briggs NC, Hall HI, Brann EA, et al. Cigarette smoking and risk of Hodgkin’s disease: a population-based case-control study. Am J Epidemiol 2002;156:1011–20.
- Castillo JJ, Dalia S, Shum H. Meta-analysis of the association between cigarette smoking and incidence of Hodgkin’s Lymphoma. J Clin Oncol 2011;29:3900–6.
- Kanzler H, Kuppers R, Hansmann ML, Rajewsky K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med 1996;184:1495–505.
- Stein H, Hummel M. Cellular origin and clonality of classic Hodgkin’s lymphoma: immunophenotypic and molecular studies. Semin Hematol 1999;36:233-41.
- Marafioti T, Hummel M, Foss HD, et al. Hodgkin and reed-sternberg cells represent an expansion of a single clone originating from a germinal center B-cell with functional immunoglobulin gene rearrangements but defective immunoglobulin transcription. Blood 2000;95:1443–50.
- Marafioti T, Hummel M, Anagnostopoulos I, et al. Origin of nodular lymphocyte-predominant Hodgkin’s disease from a clonal expansion of highly mutated germinal-center B cells. N Engl J Med 1997;337:453–8.
- van den Berg A, Visser L, Poppema S. High expression of the CC chemokine TARC in Reed-Sternberg cells. A possible explanation for the characteristic T-cell infiltratein Hodgkin’s lymphoma. Am J Pathol 1999;154:1685–91.
- Bargou RC, Emmerich F, Krappmann D, et al. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J Clin Invest 1997;100:2961–9.
- Luftig M, Yasui T, Soni V, et al. Epstein-Barr virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKK alpha-dependent noncanonical NF-kappaB activation. Proc Natl Acad Sci U S A 2004;101:141–6.
- Roemer MGM, Advani RH, Ligon AH, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol 2016;34:2690–7.
- Swerdlow SH CE, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008.
- Campo E, Swerdlow SH, Harris NL, et al. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood 2011;117:5019–32.
- von Wasielewski R, Mengel M, Fischer R, et al. Classical Hodgkin’s disease. Clinical impact of the immunophenotype. Am J Pathol 1997;151:1123–30.
- Tzankov A, Krugmann J, Fend F, et al. Prognostic significance of CD20 expression in classical Hodgkin lymphoma: a clinicopathological study of 119 cases. Clin Cancer Res 2003;9:1381–6.
- Diehl V, Sextro M, Franklin J, et al. Clinical presentation, course, and prognostic factors in lymphocyte-predominant Hodgkin’s disease and lymphocyte-rich classical Hodgkin’s disease: report from the European Task Force on Lymphoma Project on Lymphocyte-Predominant Hodgkin’s Disease. J Clin Oncol 1999;17:776–83.
- Shimabukuro-Vornhagen A, Haverkamp H, Engert A, et al. Lymphocyte-rich classical Hodgkin’s lymphoma: clinical presentation and treatment outcome in 100 patients treated within German Hodgkin’s Study Group trials. J Clin Oncol 2005;23:5739–45.
- Slack GW, Ferry JA, Hasserjian RP, et al. Lymphocyte depleted Hodgkin lymphoma: an evaluation with immunophenotyping and genetic analysis. Leuk Lymphoma 2009;50:937–43.
- Mason DY, Banks PM, Chan J, et al. Nodular lymphocyte predominance Hodgkin’s disease. A distinct clinicopathological entity. Am J Surg Pathol 1994;18:526–30.
- Rudiger T, Gascoyne RD, Jaffe ES, et al. Workshop on the relationship between nodular lymphocyte predominant Hodgkin’s lymphoma and T cell/histiocyte-rich B cell lymphoma. Ann Oncol 2002;13 Suppl 1:44–51.
- Sundeen JT, Cossman J, Jaffe ES. Lymphocyte predominant Hodgkin’s disease nodular subtype with coexistent “large cell lymphoma”. Histological progression or composite malignancy? Am J Surg Pathol 1988;12:599–606.
- Kenderian SS, Habermann TM, Macon WR, et al. Large B-cell transformation in nodular lymphocyte-predominant Hodgkin lymphoma: 40-year experience from a single institution. Blood. 2016;127:1960–6.
- Mauch PM, Kalish LA, Kadin M, et al. Patterns of presentation of Hodgkin disease. Implications for etiology and pathogenesis. Cancer 1993;71:2062–71.
- Juweid ME, Cheson BD. Positron-emission tomography and assessment of cancer therapy. N J Engl Med 2006;354:496–507.
- Hutchings M, Loft A, Hansen M, et al. Position emission tomography with or without computed tomography in the primary staging of Hodgkin’s lymphoma. Haematologica 2006;91:482–9.
- Naumann R, Beuthien-Baumann B, Reiss A, et al. Substantial impact of FDG PET imaging on the therapy decision in patients with early-stage Hodgkin’s lymphoma. Br J Cancer 2004;90:620–5.
- Juweid ME, Stroobants S, Hoekstra OS, et al. Use of positron emission tomography for response assessment of lymphoma: consensus of the Imaging Subcommittee of International Harmonization Project in Lymphoma. J Clin Oncol 2007;25:571–8.
- El-Galaly TC, d’Amore F, Mylam KJ, et al. Routine bone marrow biopsy has little or no therapeutic consequence for positron emission tomography/computed tomography-staged treatment-naive patients with Hodgkin lymphoma. J Clin Oncol 2012;30:4508–14.
- Wang J, Weiss LM, Chang KL, et al. Diagnostic utility of bilateral bone marrow examination: significance of morphologic and ancillary technique study in malignancy. Cancer 2002;94:1522–31.
- Menon NC, Buchanan JG. Bilateral trephine bone marrow biopsies in Hodgkin’s and non-Hodgkin’s lymphoma. Pathology 1979;11:53–7.
- Carbone PP, Kaplan HS, Musshoff K, et al. Report of the Committee on Hodgkin’s Disease Staging Classification. Cancer Res 1971;31:1860–1.
- Lister TA, Crowther D, Sutcliffe SB, et al. Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin’s disease: Cotswolds meeting. J Clin Oncol 1989;7:1630–6.
- Armitage JO. Early-stage Hodgkin’s lymphoma. N Engl J Med 2010;363:653–62.
- National Comprehensive Cancer Network I. NCCN Guidelines Version 3.2016 Hodgkin lymphoma. 2017.
- Engert A, Franklin J, Eich HT, et al. Two cycles of doxorubicin, bleomycin, vinblastine, and dacarbazine plus extended-field radiotherapy is superior to radiotherapy alone in early favorable Hodgkin's lymphoma: final results of the GHSG HD7 trial. J Clin Oncol 2007;25:3495–502.
- Andre MP, Girinsky T, Federico M, et al. Early positron emission tomography response-adapted treatment in stage I and II Hodgkin lymphoma: final results of the randomized EORTC/LYSA/FIL H10 trial. J Clin Oncol 2017:Jco2016686394.
- Meyer RM, Gospodarowicz MK, Connors JM, et al. Randomized comparison of ABVD chemotherapy with a strategy that includes radiation therapy in patients with limited-stage Hodgkin’s lymphoma: National Cancer Institute of Canada Clinical Trials Group and the Eastern Cooperative Oncology Group. J Clin Oncol 2005;23:4634–42.
- Meyer RM, Gospodarowicz MK, Connors JM, Pearcey RG, Wells WA, Winter JN, et al. ABVD alone versus radiation-based therapy in limited-stage Hodgkin’s lymphoma. N Engl J Med 2012;366:399–408.
- Steidl C, Lee T, Shah SP, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med 2010;362:875–85.
- Kamper P, Bendix K, Hamilton-Dutoit S, et al. Tumor-infiltrating macrophages correlate with adverse prognosis and Epstein-Barr virus status in classical Hodgkin’s lymphoma. Haematologica 2011;96:269–76.
- Agostinelli C, Gallamini A, Stracqualursi L, et al. The combined role of biomarkers and interim PET scan in prediction of treatment outcome in classical Hodgkin’s lymphoma: a retrospective, European, multicentre cohort study. Lancet Haematol 2016;3:e467–e79.
- Meignan M, Gallamini A, Meignan M, et al. Report on the First International Workshop on Interim-PET-Scan in Lymphoma. Leuk Lymph 2009;50:1257–60.
- Gallamini A, Hutchings M, Rigacci L, et al. Early interim 2-[18F]fluoro-2-deoxy-D-glucose positron emission tomography is prognostically superior to international prognostic score in advanced-stage Hodgkin’s lymphoma: a report from a joint Italian-Danish study. J Clin Oncol 2007;25:3746–52.
- Easson EC, Russell MH. Cure of Hodgkin’s Disease. Br Med J 1963;1(5347):1704–7.
- Kaplan HS. The radical radiotherapy of regionally localized Hodgkin’s disease. Radiology 1962;78:553–61.
- Noordijk EM, Carde P, Dupouy N, et al. Combined-modality therapy for clinical stage I or II Hodgkin’s lymphoma: long-term results of the European Organisation for Research and Treatment of Cancer H7 randomized controlled trials. J Clin Oncol 2006;24:3128–35.
- Eghbali H, Raemaekers J, Carde P. The EORTC strategy in the treatment of Hodgkin’s lymphoma. Eur J Haematol Suppl 2005:135–40.
- Bloomfield CD PT, Glicksman AS, et al. Chemotherapy and combined modality therapy for Hodgkin’s disease: A progress report on cancer and leukemia group B studies. Cancer Treat Rep 1982;66:835–46.
- Pavlovsky S, Maschio M, Santarelli MT, et al. Randomized trial of chemotherapy versus chemotherapy plus radiotherapy for stage I-II Hodgkin’s disease. J Natl Cancer Inst 1988;80:1466–73.
- Aviles A, Delgado S. A prospective clinical trial comparing chemotherapy, radiotherapy and combined therapy in the treatment of early stage Hodgkin’s disease with bulky disease. Clin Lab Haematol 1998;20:95–9.
- Herbst C, Rehan FA, Brillant C, et al. Combined modality treatment improves tumor control and overall survival in patients with early stage Hodgkin’s lymphoma: a systematic review. Haematologica 2010;95:494–500.
- Ferme C, Eghbali H, Meerwaldt JH, et al. Chemotherapy plus involved-field radiation in early-stage Hodgkin’s disease. N Engl J Med 2007;357:1916–27.
- Landgren O, Axdorph U, Fears TR, et al. A population-based cohort study on early-stage Hodgkin lymphoma treated with radiotherapy alone: with special reference to older patients. Ann Oncol 2006;17:1290–5.
- Engert A, Plutschow A, Eich HT, et al. Reduced treatment intensity in patients with early-stage Hodgkin's lymphoma. N Engl J Med 2010;363:640–52.
- Radford J, Illidge T, Counsell N, et al. Results of a trial of PET-directed therapy for early-stage Hodgkin's lymphoma. N Eng J Med 2015;372:1598–607.
- Eich HT, Diehl V, Gorgen H, et al. Intensified chemotherapy and dose-reduced involved-field radiotherapy in patients with early unfavorable Hodgkin’s lymphoma: final analysis of the German Hodgkin Study Group HD11 trial. J Clin Oncol 2010;28:4199–206.
- von Tresckow B, Plutschow A, Fuchs M, et al. Dose-intensification in early unfavorable Hodgkin’s lymphoma: final analysis of the German Hodgkin Study Group HD14 trial. J Clin Oncol 2012;30:907–13.
- Advani RH, Hong F, Fisher RI, et al. Randomized phase III trial comparing ABVD plus radiotherapy with the Stanford V regimen in patients with stages I or II locally extensive, bulky mediastinal Hodgkin lymphoma: a subset analysis of the North American Intergroup E2496 Trial. J Clin Oncol 2015;33:1936–42.
- Sasse S, Brockelmann PJ, Georgen H, et al. Long-term follow-up of contemporary treatment in early-stage Hodgkin lymphoma: Updated analyses of the German Hodgkin Study Group HD7, HD8, HD10 and HD11 trials. J Clin Oncol 2017 Apr 18:JCO2016709410. doi: 10.1200/JCO.2016.70.9410. [Epub ahead of print]
- Nogova L, Reineke T, Brillant C, et al. Lymphocyte-predominant and classical Hodgkin’s lymphoma: a comprehensive analysis from the German Hodgkin Study Group. J Clin Oncol 2008;26:434–9.
- Wirth A, Yuen K, Barton M, et al. Long-term outcome after radiotherapy alone for lymphocyte-predominant Hodgkin lymphoma: a retrospective multicenter study of the Australasian Radiation Oncology Lymphoma Group. Cancer 2005;104:1221–9.
- Chera BS, Olivier K, Morris CG, et al. Clinical presentation and outcomes of lymphocyte-predominant Hodgkin disease at the University of Florida. Am J Clin Oncol 2007;30:601–6.
- Chen RC, Chin MS, Ng AK, et al. Early-stage, lymphocyte-predominant Hodgkin’s lymphoma: patient outcomes from a large, single-institution series with long follow-up. J Clin Oncol 2010;28:136–41.
- Savage KJ, Skinnider B, Al-Mansour M, et al. Treating limited-stage nodular lymphocyte predominant Hodgkin lymphoma similarly to classical Hodgkin lymphoma with ABVD may improve outcome. Blood 2011;118:4585–90.
- Eichenauer DA FM, Pluetschow A, et al. Phase 2 study of rituximab in newly diagnosed stage IA nodular lymphocytepredominant Hodgkin lymphoma: a report from the German Hodgkin Study Group. Blood 2011;118:4363–5.
- Advani RH, Horning SJ, Hoppe RT, et al. Mature results of a phase II study of rituximab therapy for nodular lymphocyte-predominant Hodgkin lymphoma. J Clin Oncol 2014;32:912–8.
- Mauz-Korholz C, Gorde-Grosjean S, Hasenclever D, et al. Resection alone in 58 children with limited stage, lymphocyte-predominant Hodgkin lymphoma-experience from the European network group on pediatric Hodgkin lymphoma. Cancer 2007;110:179–85.
- Ng AK. Review of the cardiac long-term effects of therapy for Hodgkin lymphoma. Br J Haematol 2011;154:23–31.
- Ng AK, LaCasce A, Travis LB. Long-term complications of lymphoma and its treatment. J Clin Oncol 2011;29:1885–92.
- Aleman BM, van den Belt-Dusebout AW, Klokman WJ, et al. Long-term cause-specific mortality of patients treated for Hodgkin’s disease. J Clin Oncol 2003;21:3431–9.
- Girinsky T, van der Maazen R, Specht L, et al. Involved-node radiotherapy (INRT) in patients with early Hodgkin lymphoma: concepts and guidelines. Radiother Oncol 2006;79:270–7.
- Campbell BA, Voss N, Pickles T, et al. Involved-nodal radiation therapy as a component of combination therapy for limited-stage Hodgkin’s lymphoma: a question of field size. J Clin Oncol 2008;26:5170–4.
- Advani R, Maeda L, Lavori P, et al. Impact of positive positron emission tomography on prediction of freedom from progression after Stanford V chemotherapy in Hodgkin’s disease. J Clin Oncol 2007;25:3902–7.
- Engert A, Schiller P, Josting A, et al. Involved-field radiotherapy is equally effective and less toxic compared with extended-field radiotherapy after four cycles of chemotherapy in patients with early-stage unfavorable Hodgkin’s lymphoma: results of the HD8 trial of the German Hodgkin’s Lymphoma Study Group. J Clin Oncol 2003;21:3601–8.
- Horning SJ, Hoppe RT, Breslin S, et al. Stanford V and radiotherapy for locally extensive and advanced Hodgkin’s disease: mature results of a prospective clinical trial. J Clin Oncol 2002;20:630–7.
- Pavone V, Ricardi U, Luminari S, et al. ABVD plus radiotherapy versus EVE plus radiotherapy in unfavorable stage IA and IIA Hodgkin’s lymphoma: results from an Intergruppo Italiano Linfomi randomized study. Ann Oncol 2008;19:763–8.
- De Bruin ML, Sparidans J, van’t Veer MB, et al. Breast cancer risk in female survivors of Hodgkin’s lymphoma: lower risk after smaller radiation volumes. J Clin Oncol 2009;27:4239–46.
- Hodgson DC. Late effects in the era of modern therapy for Hodgkin lymphoma. Hematology Am Soc Hematol Educ Program 2011;2011:323–9.
- Maraldo MV, Brodin NP, Vogelius IR, et al. Risk of developing cardiovascular disease after involved node radiotherapy versus mantle field for Hodgkin lymphoma. Int J Radiat Oncol Biol Phys 2012;83:1232–7.
- Campbell BA, Hornby C, Cunninghame J, et al. Minimising critical organ irradiation in limited stage Hodgkin lymphoma: a dosimetric study of the benefit of involved node radiotherapy. Ann Oncol 2012;23:1259–66.
- Pingali SR, Jewell SE, Havlat L, et al. Limited utility of routine surveillance imaging for classical Hodgkin lymphoma patients in first complete remission. Cancer 2014;120:2122–9.
Neoadjuvant and Adjuvant Therapy for Gastric Cancer
INTRODUCTION
Gastric cancer is the fifth most common cancer worldwide and the third leading cause of cancer death in both females and males.1 More than 70% of gastric cancer cases occur in the developing world, with approximately 50% occurring in East Asia.2 Gastric cancer is less common in the United States, with an incidence of 12.3 cases in males and 6.0 cases in females per 100,000 per year and a disproportionately higher incidence in Asians.3 According to the Surveillance, Epidemiology, and End Results Program, approximately 26,370 new cases of stomach cancer were diagnosed in the United States in 2016, and an estimated 10,730 people died of this disease.4 Since the 1970s, the 5-year relative survival rate for gastric cancer in the United States has improved from 15% in 1975 to 29% in 2009.5 In contrast, in Japan and Korea, where screening programs have been implemented, the 5-year survival rate approaches 70%.6
RISK FACTORS AND CLASSIFICATION
A variety of risk factors have been linked to gastric cancer. Diets high in salt, salt-preserved foods, and/or processed meats have been associated with an increased risk for developing gastric cancer.7,8 Obesity and smoking have also been implicated in gastric cancer.9,10 Several studies have demonstrated a strong association between Helicobacter pylori and the development of gastric cancer.11–13 It is believed that H. pylori infection leads to chronic active gastritis, atrophic gastritis, and intestinal metaplasia. Interestingly, mass eradication of H. pylori has not been shown to reduce the risk for gastric cancer.14 Therefore, treatment of H. pylori should only be considered in patients with active peptic ulcer disease.15 Other risk factors include Epstein-Barr virus (EBV), prior gastric surgery, and radiation exposure.16–18 Family history of gastric cancer, hereditary nonpolyposis colon cancer, Li-Fraumeni syndrome, and hereditary diffuse gastric cancer caused by mutations in the E-cadherin gene increase the risk.17
The anatomic distinction between gastric cancer and cancer of the gastroesophageal junction (GEJ) has been a topic of debate. The Siewert classification is the most widely used system and divides GEJ adenocarcinoma into 3 categories:20 type I tumor: adenocarcinoma of distal esophagus, located 1 cm to 5 cm above the GEJ; type II tumor: true carcinoma of gastric cardia, located within 1 cm above and 2 cm below the GEJ; type III tumor: subcardial gastric carcinoma, located 2 cm to 5 cm below the GEJ, and infiltrates esophagus from below.
The American Joint Committee on Cancer (AJCC) has updated the latest (7th) edition of TMN staging for stomach cancer to include tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ.21
In the following sections, neoadjuvant and adjuvant therapy in gastric cancer are discussed using a case presentation to illustrate important concepts.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 43-year old male with no significant past medical history presents with epigastric abdominal pain and heart burn for the past few weeks. He denies nausea, vomiting, melena, or hematochezia. His primary care physician (PCP) diagnoses him with gastroesophageal reflux disease (GERD) and initiates a trial of pantoprazole. Over the next 2 to 3 months, his symptoms do not improve and he has an associated 40-lb weight loss. Both social history and family history are noncontributory. Physical exam reveals epigastric tenderness without rebound or guarding. Laboratory evaluation reveals a hemoglobin of 12.6 g/dL with a mean corpuscular volume of 72 fL. A comprehensive chemistry profile is within normal limits. Given the constellation of presenting symptoms, especially the unintentional weight loss and the presence of microcytic anemia, his PCP suspects a malignant process and refers the patient to a gastroenterologist.
• What are the next appropriate steps for diagnosis?
The most common presenting symptoms of gastric cancer are weight loss and abdominal pain.22 Less commonly, patients exhibit nausea, anorexia, and dysphagia with proximal tumors. Melena is seen in only about 20% of patients. In Japan, where gastric cancer is more prevalent, mass screening programs allow for detection at an earlier stage, which partially accounts for the better survival rates seen in Asia as compared to the United States. Diagnostic work-up includes esophagogastroduodenoscopy (EGD) to assess Siewert category and to obtain a tissue sample for diagnosis. Full staging requires a complete blood count (CBC) with differential; comprehensive chemistry profile; computed tomography (CT) of chest/abdomen/pelvis with oral and intravenous contrast; endoscopic ultrasound (EUS) if no M1 disease is identified; positron emission tomography (PET)-CT if there is no evidence of M1 disease and if clinically indicated; and laparoscopy with cytology for clinical stage T1b or higher.23 Patients should be staged according to the TMN staging system (Table 1).
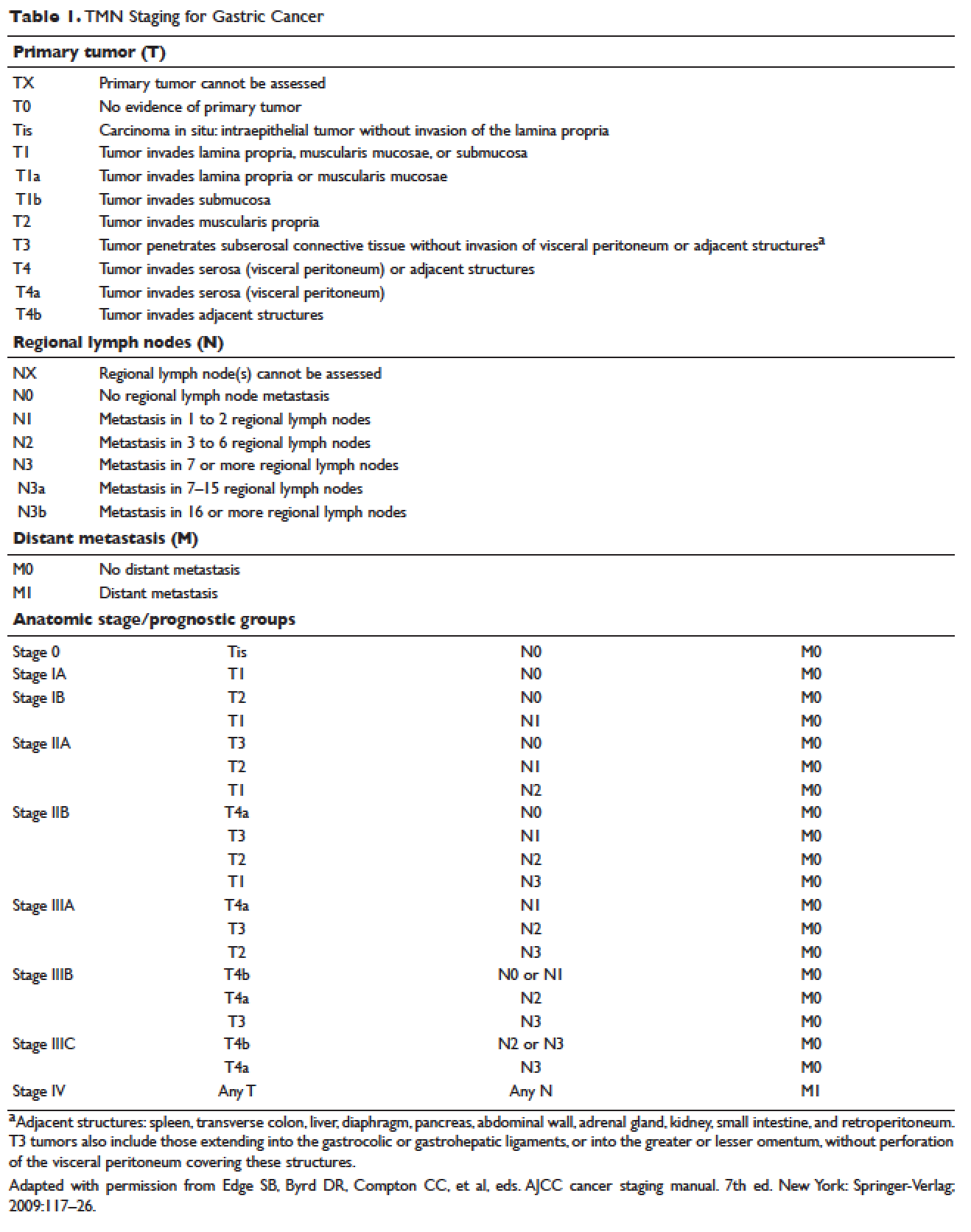
MANAGEMENT OF NONMETASTATIC DISEASE
CASE CONTINUED
The patient undergoes EGD, which reveals a large ulcerated, partially circumferential mass measuring approximately 4 cm. The mass extends from the gastric body to the cardia. Biopsy of the mass reveals poorly differentiated adenocarcinoma as well as H. pylori–associated gastritis. He is given antibiotic therapy and undergoes complete work-up of his newly diagnosed gastric adenocarcinoma. CT of the chest/abdomen/pelvis demonstrates a large gastric mass with gastrohepatic and distal perigastric adenopathy, compatible with locally advance primary gastric cancer. There is no evidence of distant metastasis. PET scan shows a large hypermetabolic mass in the stomach body and increased FDG activity in 3 small nodes along the lesser gastric curvature and in 1 node in the gastrohepatic region. EUS reveals a malignant gastric tumor in the body of the stomach, which is staged as T3, and a few malignant-appearing lymph nodes in the perigastric region. Fine-needle aspiration of the perigastric lymph node is performed and the sample obtained is positive for malignant cells. Diagnostic laparoscopy with peritoneal washings is performed and cytology is negative for malignant cells. The patient is staged as clinical stage IIB (T3N1M0).
• How should this patient with newly diagnosed, locally advanced, resectable gastric cancer be managed?
SURGERY
Surgical resection for localized gastric cancer is the mainstay of treatment with curative intent. Only very early stage (Tis or T1a) tumors can be considered for endoscopic mucosal resection. Regarding surgical resection, distal gastric cancers are typically treated with subtotal gastrectomy because there is no survival difference between subtotal and total gastrectomy.24,25 Moreover, subtotal gastrectomy is associated with better nutritional status and quality of life. For proximal tumors, total gastrectomy is preferred as subtotal gastrectomy has been associated with a higher incidence of reflux esophagitis and anastomotic stenosis.26 In terms of surgical approach, multiple studies have shown that a laparoscopic approach has a lower complication rate and similar outcomes in terms of cancer recurrence and long-term survival when compared to open gastrectomy.27–29 Thus, a laparoscopic approach is often used in academic centers with highly experienced surgeons.
The extent of lymph node dissection remains a topic of debate. A D1 dissection involves the removal of perigastric lymph nodes. A D2 dissection is a D1 dissection plus the removal of lymph nodes along the left gastric artery, common hepatic artery, celiac artery, splenic hilum, and splenic artery. D2 lymphadenectomy has become the standard of care in Eastern countries where gastric cancer is more prevalent, such as Japan and Korea.30 In Western countries, including the United States, less extensive lymphadenectomies are performed. Both randomized clinical trials and meta-analyses have failed to demonstrate an overall survival advantage of D2 dissection over D1 dissection.31,32 A Dutch trial by Bonenkamp et al involving 711 patients, one of the largest randomized trials of D1 and D2 lymphadenectomy, showed that D2 patients had a higher operative mortality rate than D1 patients (10% versus 4%, P = 0.004) and experienced more complications (43% versus 25%, P < 0.001).33 In a 15-year follow-up of this study, patients who had a D2 resection had lower locoregional recurrence and gastric-cancer–related death rates compared to those who had a D1 resection; however, D2 resection was associated with a significantly higher operative mortality and complication rate compared to D1.34 In addition, a 2015 Cochrane meta-analysis has demonstrated improved disease-specific survival (DSS) with D2 dissection (hazard ratio [HR] 0.81 [95% confidence interval {CI} 0.71 to 0.92]).35 Currently, the National Comprehensive Cancer Network (NCCN) recommends a D1 or a modified D2 gastrectomy with at least 15 lymph nodes removed for examination, with D2 lymphadenectomies only to be performed at experienced centers.23
SYSTEMIC CHEMOTHERAPY
Locally advanced gastric cancer (T3-T4 or node positive) requires systemic chemotherapy in addition to surgery, as this intervention improves the 5-year overall survival by 10% to 15%.36 Systemic therapy should also be considered in patients with T2N0 disease with high-risk features: poorly differentiated or high-grade cancer; lymphovascular invasion; neural invasion; age younger than 50 years; and patients who did not undergo D2 dissection.23 Currently, there is no global consensus on the best treatment approach. In the United States, where a less aggressive lymph-node dissection is performed, adjuvant chemoradiotherapy after surgery is more commonly seen. In Europe, perioperative (preoperative and postoperative) chemotherapy is the standard treatment. In Japan, adjuvant chemotherapy after D2 lymphadenectomy is the standard of care.37 These regional preferences are largely due to randomized clinical trials that have shown benefit for each approach. The landmark trials are discussed in the following sections and are summarized in Table 2.
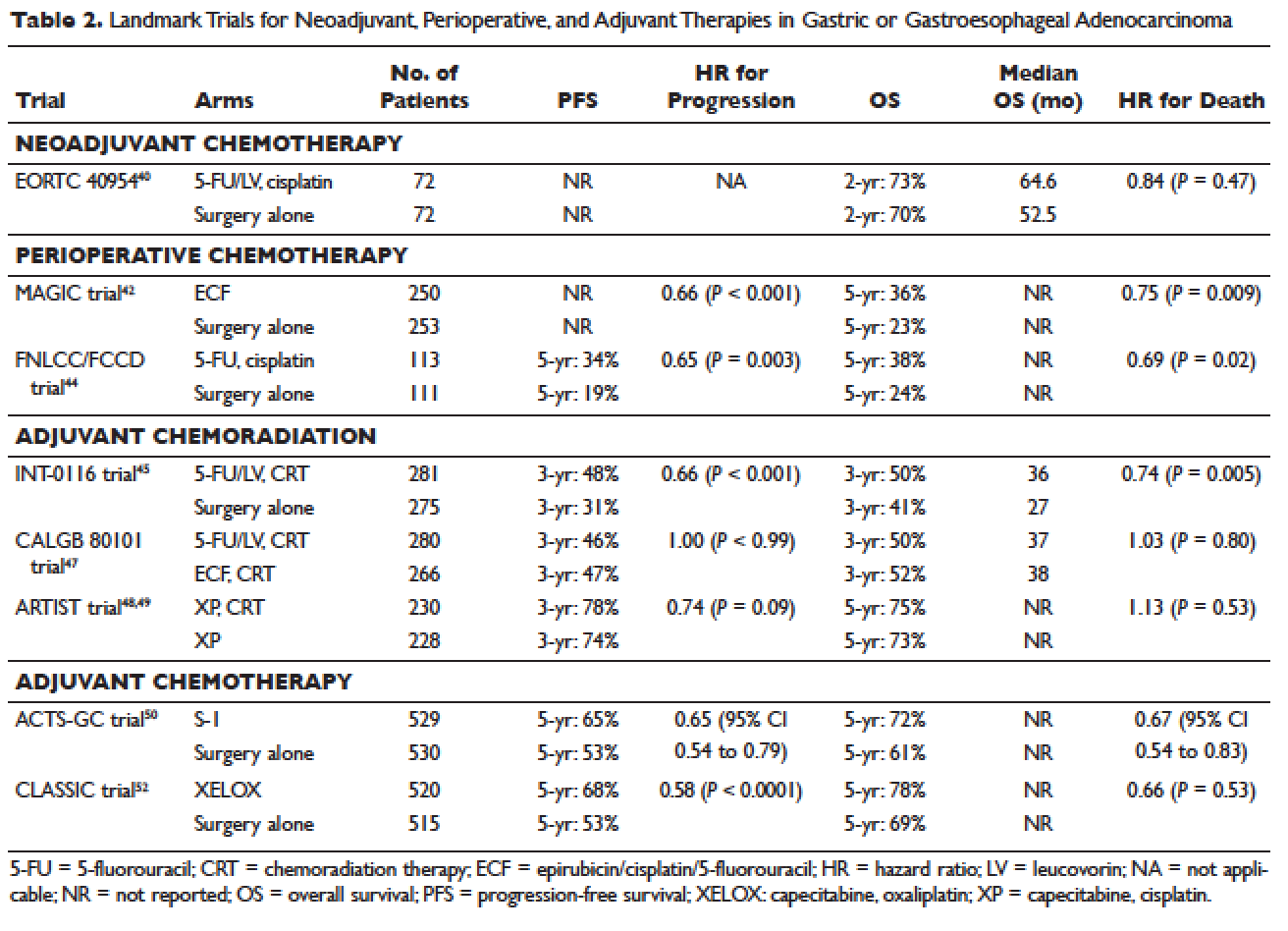
Neoadjuvant Chemotherapy
Neoadjuvant chemotherapy has the benefit of “downstaging” locally advanced tumors to allow for curative resection. Phase 2 clinical trials have also demonstrated good pathologic response rates and high R0 resection rates following neoadjuvant chemotherapy.38,39 However, phase 3 trials to support this treatment approach are lacking. In the European Organisation for Research and Treatment of Cancer (EORTC) 40954 trial, patients with stage III or IV gastric or GEJ cancer were randomly assigned to surgery with or without preoperative cisplatin, leucovorin, and infusional fluorouracil (5-FU).40 The trial was stopped early due to poor accrual after 144 patients were randomized. The neoadjuvant chemotherapy arm had a higher R0 resection rate compared to the surgery alone arm (82% versus 67%, respectively, P = 0.036) but a higher postoperative complication rate (27% versus 16%, respectively, P = 0.09). More important, after a median follow-up of 4.4 years, a survival benefit could not be shown, with 2-year survival rates of 72.7% and 69.9% in the neoadjuvant and surgery-only arms, respectively (HR 0.84 [95% CI 0.52 to 1.35], P = 0.466). Due to the lack of large trials, a meta-analysis assessing the effectiveness of neoadjuvant chemotherapy combined with surgery versus surgery alone in advanced gastric and gastroesophageal cancer was performed.41 The analysis included 12 randomized controlled trials with a total of 1820 patients. Neoadjuvant chemotherapy was shown to slightly improve the survival rate (odds ratio [OR] 1.32 [95% CI 1.07 to 1.64], P = 0.01). It significantly improved the 3-year progression-free survival (PFS; OR 1.85 [95% CI 1.39 to 2.46], P < 0.0001), tumor down-staging rate (OR 1.71 [95% CI 1.26 to 2.33], P = 0.0006), and R0 resection rate (OR 1.38 [95% CI 1.08 to 1.78], P = 0.01). There were no differences between the 2 arms in terms of relapse rates, operative complications, perioperative mortality, and grade 3/4 adverse effects. While these results are encouraging, further randomized clinical trials are needed to clarify the role of neoadjuvant chemotherapy and its impact on overall survival.
Perioperative Chemotherapy
The results of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial published in 2006 established perioperative chemotherapy as standard of care in patients with resectable gastric and gastroesophageal adenocarcinoma.42 A total of 503 patients with potentially resectable gastric and lower esophageal adenocarcinoma were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. Perioperative chemotherapy consisted of 3 preoperative and postoperative cycles of epirubicin, cisplatin, and infusional 5-FU (ECF). At a median follow-up of 4 years, the perioperative-chemotherapy group had a significantly better PFS (HR 0.66 [95% CI 0.53 to 0.81], P < 0.001) as well as overall survival (HR 0.75 [95% CI 0.60 to 0.93], P = 0.009). The 5-year overall survival rate was 36.3% in the perioperative chemotherapy group and 23% in the surgery group. Of note, there was a greater proportion of stage T1/T2 tumors (52% versus 37%, P = 0.002) and N0/N1 disease (84% versus 71%) in the perioperative-chemotherapy group compared to the surgery alone group. In addition, only 42% of patients in the perioperative chemotherapy group completed all 6 cycles of chemotherapy.
The administration of ECF is often difficult since the 5-FU component requires a central venous access device and an ambulatory infusion pump and the cisplatin component is associated with nephrotoxicity and ototoxicity. The REAL-2 trial was a randomized phase 3 clinical trial that assessed whether 5-FU could be replaced by capecitabine and cisplatin by oxaliplatin in the ECF regimen.43 Between June 2000 and May 2005, a total of 1002 patients with locally advanced esophageal/GEJ/gastric cancer were enrolled. Patients were randomly assigned to 1 of 4 triplet therapies: epirubicin and cisplatin plus either 5-FU (ECF) or capecitabine (ECX) or epirubicin and oxaliplatin plus either 5-FU (EOF) or capecitabine (EOX). After a median follow-up of approximately 18 months, the overall survival in the capecitabine groups did not differ significantly from that in the 5-FU groups (HR 0.88 [95% CI 0.77 to 1.00], P = 0.06), nor did overall survival in the oxaliplatin groups differ significantly from that in the cisplatin groups (HR 0.91 [95% CI 0.79 to 1.04], P = 0.16). Interestingly, the 1-year survival rate was longer in the EOX group than in the ECF group (46.8% versus 37.7%, respectively; HR 0.80 [95% CI 0.66 to 0.97], P = 0.02). This translated into an overall survival of 11.2 months for the EOX group and 9.9 months for the ECF group. Therefore, EOX is preferred over ECF in clinical practice.
The French FNLCC/FFCD trial published in 2011 provided further support for perioperative chemotherapy.44 A total of 224 patients with adenocarcinoma of the lower esophagus, GEJ, or stomach were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. The perioperative-chemotherapy group received 2 to 3 cycles of preoperative chemotherapy and 3 to 4 cycles of postoperative chemotherapy, consisting of infusional 5-FU (800 mg/m2 daily for days 1 to 5) and cisplatin (100 mg/m2 on day 1). In patients receiving preoperative chemotherapy, 38% experienced at least grade 3 to 4 toxicity. Among the 109 patients who received at least 1 cycle of preoperative chemotherapy, only 54 patients (50%) received postoperative chemotherapy. Despite this, the perioperative-chemotherapy group had a statistically significant higher R0 resection rate (84% versus 74%, P = 0.04) compared to the surgery alone group. At a median follow-up of 5.7 years, the perioperative chemotherapy group had an improved overall survival (HR for death 0.69 [95% CI 0.50 to 0.95], P = 0.02) and disease-free survival (DFS; HR for recurrence or death 0.65 [95% CI 0.48 to 0.89], P = 0.003). This translated into 5-year overall survival rates of 38% versus 24% and 5-year DFS rates of 34% versus 19%. One caveat to this study is that the majority of patients (64%) had GEJ cancer and only 25% had gastric cancer. In the multivariate analysis, the 2 significant prognostic factors for overall survival were the administration of preoperative chemotherapy (P = 0.01) and tumor site at the GEJ (P < 0.01).
Adjuvant Chemoradiotherapy
The INT-0116 (Intergroup 0116) study published in 2001 established adjuvant chemoradiotherapy as the standard approach for resectable gastric cancer in the United States. In this study, a total of 556 patients with resected gastric or GEJ cancer were randomly assigned to surgery alone or surgery followed by adjuvant 5-FU/leucovorin bolus chemotherapy, sandwiched with 5-FU–based chemoradiation (45 Gy).45 In the chemoradiotherapy group, 64% of patients completed treatment and grade 3 and 4 toxicity occurred in 41% and 32%, respectively. However, only 3 patients (1%) died from treatment-related toxicity. At a median follow-up of 5 years, the median overall survival was 36 months in the chemoradiation group and 27 months in the surgery group. Overall survival rate was 50% in the combined modality group and 41% in the surgery-alone group, with a HR of 1.35 (95% CI 1.09 to 1.66, P = 0.005). The 3-year DFS was 48% in the chemoradiotherapy group and 31% in the surgery-alone group, corresponding to a DFS of 30 months and 19 months, respectively. Even after a median follow-up of 10 years, these positive results were maintained, with a HR for survival of 1.32 (95% CI 1.10 to 1.60, P = 0.0046) and HR for DFS of 1.51 (95% CI 1.25 to 1.83, P < 0.001).46 A criticism of the INT-0116 study is that 54% of patients had less than a D1 lymph node dissection, suggesting that adjuvant chemoradiation may have compensated for suboptimal surgery.
CALGB 80101, a United States Intergroup study, compared the INT-0116 protocol regimen (bolus 5-FU/leucovorin with 5-FU plus concurrent radiotherapy) to postoperative ECF sandwiched with 5-FU plus concurrent radiotherapy.47 The study included patients with resected gastric or GEJ adenocarcinoma that extended beyond the muscularis propria or was node positive. The percentage of patients with gastric versus GEJ cancer was not reported. A total of 546 patients were randomized. Preliminary results were presented at the 2011 American Society of Clinical Oncology meeting. The ECF arm had lower rates of grade 3/4 toxicities, including neutropenia, diarrhea, and mucositis. However, there was no difference in overall survival (3-year overall survival of 52% versus 50% for ECF and 5-FU/leucovorin, respectively) or DFS (3-year DFS of 47% versus 46% for ECF and 5-FU/leucovorin, respectively). The trial was not adequately powered to assess noninferiority. The location of the primary tumor (GEJ versus proximal versus distal stomach) did not have any effect on treatment outcome.
The Adjuvant Chemoradiation Therapy in Stomach Cancer (ARTIST) trial was the first study to compare adjuvant chemoradiotherapy with adjuvant chemotherapy in patients with D2-resected gastric cancer.48 A total of 458 patients were randomly assigned to 6 cycles of XP chemotherapy (capecitabine 2000 mg/m2 per day on days 1–14 and cisplatin 60 mg/m2 on day 1, every 3 weeks) or XP/radiotherapy/XP (2 cycles of XP followed by 45 Gy radiotherapy with concurrent daily capecitabine [825 mg/m2 twice daily] and 2 cycles of XP). After a median follow-up of 84 months, there was no difference in DFS or overall survival between treatment arms (HR for progression 0.74 [95% CI 0.52 to 1.05], P = 0.09; HR for death 1.13 [95% CI 0.78 to 1.65], P = 0.53).49 However, subgroup analysis showed that chemoradiotherapy significantly improved DFS in patients with node-positive disease (3-year DFS 76% versus 72%, P = 0.004).
Adjuvant Chemotherapy
Data supporting the use of adjuvant chemotherapy alone is largely derived from trials done in Asia, typically after a D2 lymph node dissection, and thus adjuvant chemotherapy has become the standard of care in that region. In the Japanese Adjuvant Chemotherapy Trial of S-1 for Gastric Cancer (ACTS-GC), a total of 1059 patients with stage II or III gastric cancer who had undergone surgery with a D2 lymphadenectomy were randomly assigned to 1 year of S-1 (an oral fluoropyrimidine) or surgery alone.50 The 5-year overall survival rate was 72% in the S-1 group and 61% in the surgery-only group (HR 0.669 [95% CI 0.54 to 0.83]).51 The 5-year relapse-free survival was 65% in the S-1 group and 53% in the surgery-only group (HR 0.65 [95% CI 0.537 to 0.793]). Of note, both arms of the ACTS-GC trial had significantly higher 5-year overall survival rates compared to the INT-0116 and MAGIC trials: 43% versus 28% and 36% versus 23% for the treatment and control groups, respectively.42,45 Consequently, it is unclear if the benefit of adjuvant chemotherapy can be translated to Western countries.
The Korean Capecitabine and Oxaliplatin Adjuvant Study in Stomach Cancer (CLASSIC) trial published in 2012 also established the role of adjuvant chemotherapy after D2 gastrectomy.52 A total of 1035 patients with stage II-IIIB gastric cancer who had curative D2 gastrectomy were randomly assigned to 8 cycles of adjuvant XELOX (capecitabine 1000 mg/m2 twice daily on days 1–14 plus oxaliplatin 130 mg/m2 on day 1, 21-day cycle) or surgery alone. Median follow-up was 34 months in both arms and 67% of patients in the chemotherapy arm completed all 8 cycles of planned chemotherapy. The 3-year DFS was 74% in the chemotherapy group and 59% in the surgery-only group (HR 0.56 [95% CI 0.44 to 0.72], P < 0.0001). There was a trend toward improvement in overall survival (83% versus 78%, HR 0.72 [95% CI 0.52 to 1.00]). After 5 years of follow-up, the improvement in overall survival became statistically significant (78% versus 69%, HR 0.66 [95% CI 0.51 to 0.85]).53
The benefit of adjuvant chemotherapy was reinforced by a 2010 meta-analysis comparing adjuvant chemotherapy to surgery alone in patients with resected gastric cancer.54 A total of 17 randomized controlled trials were included. Adjuvant fluorouracil-based chemotherapy was associated with a statistically significant improved overall survival (HR 0.82 [95% CI 0.76 to 0.90], P < 0.001) and DFS (HR 0.82 [95% CI 0.75 to 0.90], P < 0.001). Five-year overall survival increased from 49.6% to 55.3% with chemotherapy.
SELECTION OF TREATMENT APPROACH
Since data exists for all 3 approaches (perioperative chemotherapy, adjuvant chemoradiotherapy, and adjuvant chemotherapy), various meta-analyses have been done to clarify which approach is the best. In a recent meta-analysis of 6 randomized controlled trials reported between 2010 and 2012, which involved 1171 patients with resected gastric cancer, adjuvant chemotherapy was compared to adjuvant chemoradiotherapy.55 Five of the studies were from East Asia, while one was from a Western country. Adjuvant chemoradiation was associated with a lower local-regional recurrence rate (OR 0.46 [95% CI 0.32 to 0.67]) and better 5-year DFS rate (OR 1.56 [95% CI 1.09 to 2.24]). However, there was no statistical difference in 5-year overall survival rate (OR 1.32 [95% CI 0.92 to 1.88]). Similar results were reported by Zhou et al in 2016.56 This meta-analysis included 4 randomized controlled trials reported between 2010 and 2015, with a total of 960 patients who had undergone a D2 resection for gastric cancer. Compared to adjuvant chemotherapy, adjuvant chemoradiotherapy significantly reduced the loco-regional recurrence rate (LRRR; relative risk [RR] 0.50 [95% CI 0.34 to 0.74], P = 0.0005) and improved DFS (HR 0.73 [95% CI 0.60 to 0.89], P = 0.002). Again, no difference in overall survival was seen (HR 0.91 [95% CI 0.74 to 1.11], P = 0.34).
Adjuvant chemotherapy and perioperative chemotherapy have also been compared. In a recent meta-analysis of 14 randomized controlled trials (8 Asian, 6 European) involving 2093 patients with resected gastric or GEJ cancer, perioperative chemotherapy was associated with improved overall survival when compared to adjuvant chemotherapy (HR 0.48 [95% CI 0.35 to 0.67], P < 0.001).57 The benefit of perioperative chemotherapy over adjuvant chemotherapy has also been reported in a 2016 meta-analysis by Zhao et al.58 A total of 1240 patients were included from 5 randomized controlled trials and 6 clinical controlled trials, all from Asian countries. The 5-year overall survival rate was significantly better in the perioperative chemotherapy group compared to the adjuvant chemotherapy group (RR 0.77 [95% CI 0.64 to 0.92], P < 0.01). Furthermore, the 2 groups showed no significant differences in the postoperative complication rates (RR 0.98 [95% CI 0.63 to 1.51], P = 0.91) or adverse effects of chemotherapy (P > 0.05 for all adverse effects).
While these meta-analyses may offer some insight on the best treatment approach, they should be interpreted with caution. Most studies included in these meta-analyses were from Asian countries, and their findings may not be applicable to Western countries. Furthermore, the heterogeneity of trials and inclusion of nonrandomized trials make it difficult to draw conclusions. There are several ongoing trials that will help to define the optimal treatment approach.
CASE CONTINUED
The patient is presented at tumor board and the consensus is to proceed with the perioperative chemotherapy approach. The patient undergoes echocardiography, which reveals a normal ejection fraction. He receives 3 cycles of neoadjuvant EOX (epirubicin, oxaliplatin, and capecitabine). After 3 cycles of neoadjuvant EOX, the patient has a repeat CT that shows marked interval reduction in the size of the primary gastric neoplasm and interval decrease in the size of the small perigastric lymph nodes. He subsequently undergoes a total gastrectomy with J-tube placement. Pathology shows ypT3N0 disease with 0 out of 47 lymph nodes involved and negative margins. He then receives 3 cycles of adjuvant EOX.
• What are the recommendations for surveillance?
According to the current NCCN guidelines, a history and physical exam should be performed every 3 to 6 months for 1 to 2 years, then every 6 to 12 months for 3 to 5 years, and then annually.23 Labs, CT chest/abdomen, and EGD should be done as clinically indicated. Patients who have undergone surgical resection should be monitored for nutritional deficiencies (vitamin B12 and iron).
GASTROESOPHAGEAL JUNCTION TUMORS
Tumors arising in the GEJ or gastric cardia within 5 cm of the GEJ that extend into the GEJ or distal esophagus are staged and treated as esophageal cancers.21 The primary treatment for T1/T2N0 tumors is surgical resection. In patients with T3 or higher or node-positive adenocarcinoma of the GEJ, a combined modality approach is preferred, with preoperative chemoradiotherapy followed by surgical resection.59 The CROSS trial demonstrated a significant survival benefit with preoperative chemoradiation using carboplatin/paclitaxel compared to surgery alone in patients with esophageal or GEJ cancer (49 months versus 24 months, HR 0.66, P = 0.003).60
ONGOING TRIALS
As mentioned previously, several randomized clinical trials are in progress to clarify the optimal treatment approach. The MAGIC-B/MRC-ST03 is a randomized phase 2/3 trial looking at perioperative epirubicin, cisplatin, and capecitabine (ECX) with or without bevacizumab in patients with resectable lower esophageal, GEJ, or gastric cancer.61 The TOPGEAR trial, a randomized phase 2/3 study being conducted in Canada and Europe, is comparing perioperative ECF chemotherapy with preoperative chemoradiation plus perioperative ECF chemotherapy.62 In Asia, the PRODIGY trial is a phase 3, open-label, randomized study comparing neoadjuvant docetaxel, oxaliplatin, and S-1 followed by surgery and adjuvant S-1 versus surgery plus adjuvant S-1 in patients with locally advanced gastric cancer (T2-T4 or node positive).63 Primary endpoint is PFS and secondary endpoints are overall survival, R0 resection rate, and safety.
Trials comparing adjuvant chemotherapy to adjuvant chemoradiotherapy are also being conducted. In the Dutch CRITICS study, a randomized phase 3 trial, patients with stage Ib-Iva resectable gastric cancer were given 3 cycles of epirubicin, cisplatin/oxaliplatin, and capecitabine (ECC/EOC), followed by D2 resection and either 3 cycles of ECC/EOC or chemoradiation with weekly cisplatin and daily capecitabine.64 Between January 2007 and April 2015, a total of 788 patients were enrolled. In a preliminary report presented at ASCO in 2016, the 5-year survival rate was similar between the 2 arms (41.3% for chemotherapy arm and 40.9% for chemoradiotherapy arm, P = 0.99). The Korean ARTIST II trial is comparing adjuvant S-1 and S-1/oxaliplatin with or without radiotherapy in patients with D2-resected gastric cancer.65 Similarly, the NCT01711242 trial is comparing adjuvant XELOX alone versus XELOX with concurrent capecitabine/radiotherapy in patients with resected D2 gastric cancer.66
The ToGA trial established a survival benefit of trastuzumab in combination with chemotherapy in HER2-positive metastatic gastric cancer.67 Consequently, there are ongoing clinical trials to assess the role of trastuzumab in nonmetastatic gastric cancer. The TOXAG study is a phase 2 trial looking at the safety and tolerability of adjuvant oxaliplatin, capecitabine, and trastuzumab with radiation in patients with resected HER2-positive gastric or GEJ adenocarcinoma.68 The NCT01130337 clinical trial is evaluating perioperative XELOX/trastuzumab in patients with resectable gastric or GEJ adeno-carcinoma.69
SUMMARY
Gastric cancer is the fifth most common cancer worldwide, with the greatest incidence in East Asia. Survival outcomes are better in Asian countries when compared to the United States. This difference in survival may be related to the presence of mass screening programs in Asia, which allows for detection at an earlier stage and the use of a more extensive surgical approach (ie, D2 resection). Risk factors for developing gastric cancer include: diets high in salt/salt-preserved foods or processed meats, obesity, smoking, H. pylori infection, EBV, prior gastric surgery, radiation exposure, and positive family history.
According to the latest edition of TMN staging, gastric cancer includes tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ. Diagnostic work-up includes: EGD with biopsy; basic labs; CT chest/abdomen/pelvis with oral and intravenous contrast; EUS if no M1 disease is identified; PET-CT if there is no M1 disease and if clinically indicated; and diagnostic laparoscopy with cytology for clinical stage T1b or higher.
The mainstay of treatment is surgical resection. Laparoscopic approach is preferred over open gastrectomy due to lower complication rates and similar survival outcomes. Current NCCN guidelines recommend a D1 or a modified D2 lymph node dissection with at least 15 lymph nodes removed for examination. Systemic chemotherapy is required in locally advanced gastric cancer (T3-T4 or node positive) and should be considered in T2N0 disease with high-risk features. Currently, there is no global consensus on the optimal treatment approach. Data from various trials have shown benefit for each approach. Regional preferences are: perioperative chemotherapy in Europe; adjuvant chemoradiotherapy in the United States; and adjuvant chemotherapy in Asia. In an effort to better define the optimal treatment approach, several randomized clinical trials are being conducted. According to the current NCCN guidelines, the following treatment approaches are acceptable and are supported by data in the trial listed in parentheses:
• Perioperative chemotherapy
° 5-FU/cisplatin (French FNLCC/FCCD trial)44 or
° ECF (MAGIC trial)42 or
° ECF modifications: EOX, EOF, ECX (REAL-2 trial)43
• Adjuvant chemoradiotherapy
° 5-FU/leucovorin sandwiched with 5-FU-based chemoradiation (INT-0116 trial)45
• Adjuvant chemotherapy (after D2 resection)
° Capecitabine/oxaliplatin (CLASSIC trial)52 or
° Capecitabine/cisplatin (ARTIST trial)48,49
- World Health Organization. GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence Worldwide in 2012. France, Lyon: IARC; 2012.
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127:2893–917.
- Lui FH, Tuan B, Swenson SL, et al. Ethnic disparities in gastric cancer incidence and survival in the USA: an updated analysis of 1992-2009 SEER data. Dig Dis Sci 2014;59:3027–34.
- Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2013. National Cancer Institute. http://seer.cancer.gov/csr/1975_2013/. Based on November 2015 SEER data submission, posted to the SEER web site April 2016.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
- Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer 2007;10:75.
- Bouvard V, Loomis D, Guyton KZ, et al. Carcinogenicity of consumption of red and processed meat. Lancet Oncol 2015;16:1599–600.
- Yang P, Zhou Y, Chen B, et al. Overweight, obesity and gastric cancer risk: results from a meta-analysis of cohort studies. Eur J Cancer 2009;45:2867–73.
- González CA, Pera G, Agudo A, et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int J Cancer 2003;107:629–34.
- Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998;114:1169–79.
- Eslick GD, Lim LL, Byles JE, et al. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol 1999;94:2373–9.
- An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993;341:1359–62.
- Parsonnet J, Forman D. Helicobacter pylori infection and gastric cancer—for want of more outcomes. JAMA 2004;291:244–5.
- Malfertheiner P, Megraud F, O'Morain CA, et al. Management of Helicobacter pylori infection—the Maastricht IV/ Florence Consensus Report. Gut 2012;61:646–64.
- Fukayama M. Epstein-Barr virus and gastric carcinoma. Pathol Int 2010;60:337–50.
- Takeno S, Hashimoto T, Maki K, et al. Gastric cancer arising from the remnant stomach after distal gastrectomy: a review. World J Gastroenterol 2014;20:13734–40.
- Morton LM, Dores GM, Curtis RE, et al. Stomach cancer risk after treatment for Hodgkin lymphoma. J Clin Oncol 2013;31:3369–77.
- van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361–74.
- Siewert J, Stein H. Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg 1998;85:1457–9.
- Edge S, Byrd DR, Compton CC, et al. AJCC cancer staging manual. 7th ed. New York: Springer New York; 2009.
- Wanebo HJ, Kennedy BJ, Chmiel J, et al. Cancer of the stomach. A patient care study by the American College of Surgeons. Ann Surg 1993;218:583–92.
- National Comprehensive Cancer Network. Gastric cancer (version 3.2016www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed December 14, 2016.
- Bozzetti F, Marubini E, Bonfanti G, et al. Subtotal versus total gastrectomy for gastric cancer: five-year survival rates in a multicenter randomized Italian trial. Italian Gastrointestinal Tumor Study Group. Ann Surg 1999;230:170–8.
- Gouzi JL, Huguier M, Fagniez PL, et al. Total versus subtotal gastrectomy for adenocarcinoma of the gastric antrum. A French prospective controlled study. Ann Surg 1989;209:162–6.
- Pu YW, Gong W, Wu YY, et al. Proximal gastrectomy versus total gastrectomy for proximal gastric carcinoma. A meta-analysis on postoperative complications, 5-year survival, and recurrence rate. Saudi Med J 2013;34:1223–8.
- Chen K, Xu XW, Mou YP, et al. Systematic review and meta-analysis of laparoscopic and open gastrectomy for advanced gastric cancer. World J Surg Oncol 2013;11:182.
- Fang C, Hua J, Li J, et al. Comparison of long-term results between laparoscopy-assisted gastrectomy and open gastrectomy with D2 lymphadenectomy for advanced gastric cancer. Am J Surg 2014;208:391–6.
- Wang W, Li Z, Tang J, et al. Laparoscopic versus open total gastrectomy with D2 dissection for gastric cancer: a meta-analysis. J Cancer Res Clin Oncol 2013;139:1721–34.
- Schmidt B, Yoon SS. D1 versus D2 lymphadenectomy for gastric cancer. J Surg Oncol 2013;107:259–64.
- Jiang L, Yang KH, Guan QL, et al. Survival and recurrence free benefits with different lymphadenectomy for resectable gastric cancer: a meta-analysis. J Surg Oncol 2013;107:807–14.
- Degiuli M, Sasako M, Ponti A, et al. Randomized clinical trial comparing survival after D1 or D2 gastrectomy for gastric cancer. Br J Surg 2014;101:23–31.
- Bonenkamp JJ, Songun I, Hermans J, et al. Randomized comparison of morbidity after D1 and D2 dissection for gastric cancer in 996 Dutch patients. Lancet 1995;345:745–8.
- Songun I, Putter H, Kranenbarg EM, et al. Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. Lancet Oncol 2010;11:439–49.
- Mocellin S, McCulloch P, Kazi H, et al. Extent of lymph node dissection for adenocarcinoma of the stomach. Cochrane Database Syst Rev 2015;8:CD001964.
- Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
- Quéro L, Guillerm S, Hennequin C. Neoadjuvant or adjuvant therapy for gastric cancer. World J Gastrointest Oncol 2015;7:102–10.
- Okabe H, Hata H, Ueda S, et al. A phase II study of neoadjuvant chemotherapy with S-1 and cisplatin for stage III gastric cancer: KUGC03. J Surg Oncol 2016 Jan;113:36–41.
- Wang X, Zhao L, Liu H et al. A phase II study of a modified FOLFOX6 regimen as neoadjuvant chemotherapy for locally advanced gastric cancer. Br J Cancer 2016;114:1326-33.
- Schuhmacher C, Gretschel S, Lordick F, et al. Neoadjuvant chemotherapy compared with surgery alone for locally advanced cancer of the stomach and cardia: European Organisation for Research and Treatment of Cancer randomized trial 40954. J Clin Oncol 2010;28:5210–18.
- Xiong BH, Cheng Y, Ma L, Zhang CQ. An updated meta-analysis of randomized controlled trial assessing the effect of neoadjuvant chemotherapy in advanced gastric cancer. Cancer Invest 2014;32:272–84.
- Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006;355:11–20.
- Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008;358:36–46.
- Ychou M, Boige V, Pignon JP, et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol 2011;29:1715–21.
- Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med 2001;345:725–30.
- Smalley SR, Benedetti JK, Haller DG, et al. Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. J Clin Oncol 2012;30:2327–33.
- Fuchs CS, Tepper JE, Niedzwiecki D, et al. Postoperative adjuvant chemoradiation for gastric or gastroesophageal junction (GEJ) adenocarcinoma using epirubicin, cisplatin, and infusional (CI) 5-FU (ECF) before and after CI 5-FU and radiotherapy (CRT) compared with bolus 5-FU/LV before and after CRT: Intergroup trial CALGB 80101. J Clin Oncol 2011;29:256s. Abstract 4003.
- Lee J, Lim do H, Kim S, et al. Phase III trial comparing capecitabine plus cisplatin versus capecitabine plus cisplatin with concurrent capecitabine radiotherapy in completely resected gastric cancer with D2 lymph node dissection: the ARTIST trial. J Clin Oncol 2012;30:268–73
- Park SH, Sohn TS, Lee J, et al. Phase III trial to compare adjuvant chemotherapy with capecitabine and cisplatin versus concurrent chemoradiotherapy in gastric cancer: final report of the adjuvant chemoradiotherapy in stomach tumors trial, including survival and subset analyses. J Clin Oncol 2015;33:3130–6.
- Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 2007;357:1810–20.
- Sasako M, Sakuramoto S, Katai H, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol 2011;29:4387–93.
- Bang YJ, Kim YW, Yang HK, et al. Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet 2012;379:315–21.
- Noh SH, Park SR, Yang HK, et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:1389–96.
- Paoletti X, Oba K, Burzykowski T, et al. Benefit of adjuvant chemotherapy for resectable gastric cancer: a meta-analysis. JAMA 2010; 303:1729–37.
- Dai Q, Jiang L, Lin RJ, et al. Adjuvant chemoradiotherapy versus chemotherapy for gastric cancer: a meta-analysis of randomized controlled trials. J Surg Oncol 2015;111:277–84.
- Zhou M, Kang M, Li G, et al. Postoperative chemoradiotherapy versus chemotherapy for R0 resected gastric cancer with D2 lymph node dissection: an up-to-date meta-analysis. World J Surg Oncol 2016;14:209.
- Yang Y, Yin X, Sheng L, et al. Perioperative chemotherapy more of a benefit for overall survival than adjuvant chemotherapy for operable gastric cancer: an updated meta-analysis. Sci Rep 2015;5:12850.
- Zhao JH, Gao P, Song YX, et al. Which is better for gastric cancer patients, perioperative or adjuvant chemotherapy: a meta-analysis. BMC Cancer 2016;16:631.
- Narsule CK, Montgomery MM, and Fernando HC. Evidence-based review of the management of cancers of the gastroesophageal junction. Thorac Surg Clin 2012;22:109–21.
- van Hagen P, Hulshof MCCM, van Lanschot JJB, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Eng J Med 2012;266:2074–84.
- Cunningham D. Chemotherapy with or without bevacizumab or lapatinib to treat operable oesophagogastric cancer (ST03). ClinicalTrials.gov. https://clinicaltrials.gov/show/NCT00450203. NLM Identifier: NCT00450203. Accessed December 14, 2016.
- Leong T, Smithers BM, Michael M, et al. TOPGEAR: a randomised phase III trial of perioperative ECF chemotherapy versus preoperative chemoradiation plus perioperative ECF chemotherapy for resectable gastric cancer (an international, intergroup trial of the AGITG/TROG/EORTC/NCIC CTG). BMC Cancer 2015;15:532.
- Docetaxel+oxaliplatin+S-1 (DOS) regimen as neoadjuvant chemotherapy in advanced gastric cancer (PRODIGY). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01515748 NLM. Identifier: NCT01515748. Accessed December 14, 2016.
- Verheij M, Jansen EP, Cats A, et al. A multicenter randomized phase III trial of neo-adjuvant chemotherapy followed by surgery and chemotherapy or by surgery and chemoradiotherapy in resectable gastric cancer: First results from the CRITICS study. J Clin Oncol 2016;34 (suppl). Abstract 4000.
- Kang WK. Phase III randomized trial of adjuvant chemotherapy with S-1 vs S-1/oxaliplatin ± radiotherapy for completely resected gastric adenocarcinoma : The ARTIST II Trial (ARTIST-II). ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01761461. NLM Identifier: NCT01761461. Accessed December 14, 2016.
- Trial of adjuvant XELOX chemotherapy and concurrent capecitabine and radiotherapy for resected gastric carcinoma. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01711242. NLM Identifier: NCT01711242. Accessed December 14, 2016.
- Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
- Roche HL. A Study of the combination of oxaliplatin, capecitabine and herceptin (trastuzumab) and chemoradiotherapy in the adjuvant setting in operated patients with HER2+ gastric or gastro-esophageal junction cancer (TOXAG Study). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01748773. NLM Identifer: NCT01748773. Accessed December 14, 2016.
- A study of capecitabine [Xeloda] in combination with trastuzumab [herceptin] and oxaliplatine in patients with resectable gastric cancer. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01130337. NLM Identifier: NCT01130337. Accessed December 14, 2016.
INTRODUCTION
Gastric cancer is the fifth most common cancer worldwide and the third leading cause of cancer death in both females and males.1 More than 70% of gastric cancer cases occur in the developing world, with approximately 50% occurring in East Asia.2 Gastric cancer is less common in the United States, with an incidence of 12.3 cases in males and 6.0 cases in females per 100,000 per year and a disproportionately higher incidence in Asians.3 According to the Surveillance, Epidemiology, and End Results Program, approximately 26,370 new cases of stomach cancer were diagnosed in the United States in 2016, and an estimated 10,730 people died of this disease.4 Since the 1970s, the 5-year relative survival rate for gastric cancer in the United States has improved from 15% in 1975 to 29% in 2009.5 In contrast, in Japan and Korea, where screening programs have been implemented, the 5-year survival rate approaches 70%.6
RISK FACTORS AND CLASSIFICATION
A variety of risk factors have been linked to gastric cancer. Diets high in salt, salt-preserved foods, and/or processed meats have been associated with an increased risk for developing gastric cancer.7,8 Obesity and smoking have also been implicated in gastric cancer.9,10 Several studies have demonstrated a strong association between Helicobacter pylori and the development of gastric cancer.11–13 It is believed that H. pylori infection leads to chronic active gastritis, atrophic gastritis, and intestinal metaplasia. Interestingly, mass eradication of H. pylori has not been shown to reduce the risk for gastric cancer.14 Therefore, treatment of H. pylori should only be considered in patients with active peptic ulcer disease.15 Other risk factors include Epstein-Barr virus (EBV), prior gastric surgery, and radiation exposure.16–18 Family history of gastric cancer, hereditary nonpolyposis colon cancer, Li-Fraumeni syndrome, and hereditary diffuse gastric cancer caused by mutations in the E-cadherin gene increase the risk.17
The anatomic distinction between gastric cancer and cancer of the gastroesophageal junction (GEJ) has been a topic of debate. The Siewert classification is the most widely used system and divides GEJ adenocarcinoma into 3 categories:20 type I tumor: adenocarcinoma of distal esophagus, located 1 cm to 5 cm above the GEJ; type II tumor: true carcinoma of gastric cardia, located within 1 cm above and 2 cm below the GEJ; type III tumor: subcardial gastric carcinoma, located 2 cm to 5 cm below the GEJ, and infiltrates esophagus from below.
The American Joint Committee on Cancer (AJCC) has updated the latest (7th) edition of TMN staging for stomach cancer to include tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ.21
In the following sections, neoadjuvant and adjuvant therapy in gastric cancer are discussed using a case presentation to illustrate important concepts.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 43-year old male with no significant past medical history presents with epigastric abdominal pain and heart burn for the past few weeks. He denies nausea, vomiting, melena, or hematochezia. His primary care physician (PCP) diagnoses him with gastroesophageal reflux disease (GERD) and initiates a trial of pantoprazole. Over the next 2 to 3 months, his symptoms do not improve and he has an associated 40-lb weight loss. Both social history and family history are noncontributory. Physical exam reveals epigastric tenderness without rebound or guarding. Laboratory evaluation reveals a hemoglobin of 12.6 g/dL with a mean corpuscular volume of 72 fL. A comprehensive chemistry profile is within normal limits. Given the constellation of presenting symptoms, especially the unintentional weight loss and the presence of microcytic anemia, his PCP suspects a malignant process and refers the patient to a gastroenterologist.
• What are the next appropriate steps for diagnosis?
The most common presenting symptoms of gastric cancer are weight loss and abdominal pain.22 Less commonly, patients exhibit nausea, anorexia, and dysphagia with proximal tumors. Melena is seen in only about 20% of patients. In Japan, where gastric cancer is more prevalent, mass screening programs allow for detection at an earlier stage, which partially accounts for the better survival rates seen in Asia as compared to the United States. Diagnostic work-up includes esophagogastroduodenoscopy (EGD) to assess Siewert category and to obtain a tissue sample for diagnosis. Full staging requires a complete blood count (CBC) with differential; comprehensive chemistry profile; computed tomography (CT) of chest/abdomen/pelvis with oral and intravenous contrast; endoscopic ultrasound (EUS) if no M1 disease is identified; positron emission tomography (PET)-CT if there is no evidence of M1 disease and if clinically indicated; and laparoscopy with cytology for clinical stage T1b or higher.23 Patients should be staged according to the TMN staging system (Table 1).
MANAGEMENT OF NONMETASTATIC DISEASE
CASE CONTINUED
The patient undergoes EGD, which reveals a large ulcerated, partially circumferential mass measuring approximately 4 cm. The mass extends from the gastric body to the cardia. Biopsy of the mass reveals poorly differentiated adenocarcinoma as well as H. pylori–associated gastritis. He is given antibiotic therapy and undergoes complete work-up of his newly diagnosed gastric adenocarcinoma. CT of the chest/abdomen/pelvis demonstrates a large gastric mass with gastrohepatic and distal perigastric adenopathy, compatible with locally advance primary gastric cancer. There is no evidence of distant metastasis. PET scan shows a large hypermetabolic mass in the stomach body and increased FDG activity in 3 small nodes along the lesser gastric curvature and in 1 node in the gastrohepatic region. EUS reveals a malignant gastric tumor in the body of the stomach, which is staged as T3, and a few malignant-appearing lymph nodes in the perigastric region. Fine-needle aspiration of the perigastric lymph node is performed and the sample obtained is positive for malignant cells. Diagnostic laparoscopy with peritoneal washings is performed and cytology is negative for malignant cells. The patient is staged as clinical stage IIB (T3N1M0).
• How should this patient with newly diagnosed, locally advanced, resectable gastric cancer be managed?
SURGERY
Surgical resection for localized gastric cancer is the mainstay of treatment with curative intent. Only very early stage (Tis or T1a) tumors can be considered for endoscopic mucosal resection. Regarding surgical resection, distal gastric cancers are typically treated with subtotal gastrectomy because there is no survival difference between subtotal and total gastrectomy.24,25 Moreover, subtotal gastrectomy is associated with better nutritional status and quality of life. For proximal tumors, total gastrectomy is preferred as subtotal gastrectomy has been associated with a higher incidence of reflux esophagitis and anastomotic stenosis.26 In terms of surgical approach, multiple studies have shown that a laparoscopic approach has a lower complication rate and similar outcomes in terms of cancer recurrence and long-term survival when compared to open gastrectomy.27–29 Thus, a laparoscopic approach is often used in academic centers with highly experienced surgeons.
The extent of lymph node dissection remains a topic of debate. A D1 dissection involves the removal of perigastric lymph nodes. A D2 dissection is a D1 dissection plus the removal of lymph nodes along the left gastric artery, common hepatic artery, celiac artery, splenic hilum, and splenic artery. D2 lymphadenectomy has become the standard of care in Eastern countries where gastric cancer is more prevalent, such as Japan and Korea.30 In Western countries, including the United States, less extensive lymphadenectomies are performed. Both randomized clinical trials and meta-analyses have failed to demonstrate an overall survival advantage of D2 dissection over D1 dissection.31,32 A Dutch trial by Bonenkamp et al involving 711 patients, one of the largest randomized trials of D1 and D2 lymphadenectomy, showed that D2 patients had a higher operative mortality rate than D1 patients (10% versus 4%, P = 0.004) and experienced more complications (43% versus 25%, P < 0.001).33 In a 15-year follow-up of this study, patients who had a D2 resection had lower locoregional recurrence and gastric-cancer–related death rates compared to those who had a D1 resection; however, D2 resection was associated with a significantly higher operative mortality and complication rate compared to D1.34 In addition, a 2015 Cochrane meta-analysis has demonstrated improved disease-specific survival (DSS) with D2 dissection (hazard ratio [HR] 0.81 [95% confidence interval {CI} 0.71 to 0.92]).35 Currently, the National Comprehensive Cancer Network (NCCN) recommends a D1 or a modified D2 gastrectomy with at least 15 lymph nodes removed for examination, with D2 lymphadenectomies only to be performed at experienced centers.23
SYSTEMIC CHEMOTHERAPY
Locally advanced gastric cancer (T3-T4 or node positive) requires systemic chemotherapy in addition to surgery, as this intervention improves the 5-year overall survival by 10% to 15%.36 Systemic therapy should also be considered in patients with T2N0 disease with high-risk features: poorly differentiated or high-grade cancer; lymphovascular invasion; neural invasion; age younger than 50 years; and patients who did not undergo D2 dissection.23 Currently, there is no global consensus on the best treatment approach. In the United States, where a less aggressive lymph-node dissection is performed, adjuvant chemoradiotherapy after surgery is more commonly seen. In Europe, perioperative (preoperative and postoperative) chemotherapy is the standard treatment. In Japan, adjuvant chemotherapy after D2 lymphadenectomy is the standard of care.37 These regional preferences are largely due to randomized clinical trials that have shown benefit for each approach. The landmark trials are discussed in the following sections and are summarized in Table 2.
Neoadjuvant Chemotherapy
Neoadjuvant chemotherapy has the benefit of “downstaging” locally advanced tumors to allow for curative resection. Phase 2 clinical trials have also demonstrated good pathologic response rates and high R0 resection rates following neoadjuvant chemotherapy.38,39 However, phase 3 trials to support this treatment approach are lacking. In the European Organisation for Research and Treatment of Cancer (EORTC) 40954 trial, patients with stage III or IV gastric or GEJ cancer were randomly assigned to surgery with or without preoperative cisplatin, leucovorin, and infusional fluorouracil (5-FU).40 The trial was stopped early due to poor accrual after 144 patients were randomized. The neoadjuvant chemotherapy arm had a higher R0 resection rate compared to the surgery alone arm (82% versus 67%, respectively, P = 0.036) but a higher postoperative complication rate (27% versus 16%, respectively, P = 0.09). More important, after a median follow-up of 4.4 years, a survival benefit could not be shown, with 2-year survival rates of 72.7% and 69.9% in the neoadjuvant and surgery-only arms, respectively (HR 0.84 [95% CI 0.52 to 1.35], P = 0.466). Due to the lack of large trials, a meta-analysis assessing the effectiveness of neoadjuvant chemotherapy combined with surgery versus surgery alone in advanced gastric and gastroesophageal cancer was performed.41 The analysis included 12 randomized controlled trials with a total of 1820 patients. Neoadjuvant chemotherapy was shown to slightly improve the survival rate (odds ratio [OR] 1.32 [95% CI 1.07 to 1.64], P = 0.01). It significantly improved the 3-year progression-free survival (PFS; OR 1.85 [95% CI 1.39 to 2.46], P < 0.0001), tumor down-staging rate (OR 1.71 [95% CI 1.26 to 2.33], P = 0.0006), and R0 resection rate (OR 1.38 [95% CI 1.08 to 1.78], P = 0.01). There were no differences between the 2 arms in terms of relapse rates, operative complications, perioperative mortality, and grade 3/4 adverse effects. While these results are encouraging, further randomized clinical trials are needed to clarify the role of neoadjuvant chemotherapy and its impact on overall survival.
Perioperative Chemotherapy
The results of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial published in 2006 established perioperative chemotherapy as standard of care in patients with resectable gastric and gastroesophageal adenocarcinoma.42 A total of 503 patients with potentially resectable gastric and lower esophageal adenocarcinoma were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. Perioperative chemotherapy consisted of 3 preoperative and postoperative cycles of epirubicin, cisplatin, and infusional 5-FU (ECF). At a median follow-up of 4 years, the perioperative-chemotherapy group had a significantly better PFS (HR 0.66 [95% CI 0.53 to 0.81], P < 0.001) as well as overall survival (HR 0.75 [95% CI 0.60 to 0.93], P = 0.009). The 5-year overall survival rate was 36.3% in the perioperative chemotherapy group and 23% in the surgery group. Of note, there was a greater proportion of stage T1/T2 tumors (52% versus 37%, P = 0.002) and N0/N1 disease (84% versus 71%) in the perioperative-chemotherapy group compared to the surgery alone group. In addition, only 42% of patients in the perioperative chemotherapy group completed all 6 cycles of chemotherapy.
The administration of ECF is often difficult since the 5-FU component requires a central venous access device and an ambulatory infusion pump and the cisplatin component is associated with nephrotoxicity and ototoxicity. The REAL-2 trial was a randomized phase 3 clinical trial that assessed whether 5-FU could be replaced by capecitabine and cisplatin by oxaliplatin in the ECF regimen.43 Between June 2000 and May 2005, a total of 1002 patients with locally advanced esophageal/GEJ/gastric cancer were enrolled. Patients were randomly assigned to 1 of 4 triplet therapies: epirubicin and cisplatin plus either 5-FU (ECF) or capecitabine (ECX) or epirubicin and oxaliplatin plus either 5-FU (EOF) or capecitabine (EOX). After a median follow-up of approximately 18 months, the overall survival in the capecitabine groups did not differ significantly from that in the 5-FU groups (HR 0.88 [95% CI 0.77 to 1.00], P = 0.06), nor did overall survival in the oxaliplatin groups differ significantly from that in the cisplatin groups (HR 0.91 [95% CI 0.79 to 1.04], P = 0.16). Interestingly, the 1-year survival rate was longer in the EOX group than in the ECF group (46.8% versus 37.7%, respectively; HR 0.80 [95% CI 0.66 to 0.97], P = 0.02). This translated into an overall survival of 11.2 months for the EOX group and 9.9 months for the ECF group. Therefore, EOX is preferred over ECF in clinical practice.
The French FNLCC/FFCD trial published in 2011 provided further support for perioperative chemotherapy.44 A total of 224 patients with adenocarcinoma of the lower esophagus, GEJ, or stomach were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. The perioperative-chemotherapy group received 2 to 3 cycles of preoperative chemotherapy and 3 to 4 cycles of postoperative chemotherapy, consisting of infusional 5-FU (800 mg/m2 daily for days 1 to 5) and cisplatin (100 mg/m2 on day 1). In patients receiving preoperative chemotherapy, 38% experienced at least grade 3 to 4 toxicity. Among the 109 patients who received at least 1 cycle of preoperative chemotherapy, only 54 patients (50%) received postoperative chemotherapy. Despite this, the perioperative-chemotherapy group had a statistically significant higher R0 resection rate (84% versus 74%, P = 0.04) compared to the surgery alone group. At a median follow-up of 5.7 years, the perioperative chemotherapy group had an improved overall survival (HR for death 0.69 [95% CI 0.50 to 0.95], P = 0.02) and disease-free survival (DFS; HR for recurrence or death 0.65 [95% CI 0.48 to 0.89], P = 0.003). This translated into 5-year overall survival rates of 38% versus 24% and 5-year DFS rates of 34% versus 19%. One caveat to this study is that the majority of patients (64%) had GEJ cancer and only 25% had gastric cancer. In the multivariate analysis, the 2 significant prognostic factors for overall survival were the administration of preoperative chemotherapy (P = 0.01) and tumor site at the GEJ (P < 0.01).
Adjuvant Chemoradiotherapy
The INT-0116 (Intergroup 0116) study published in 2001 established adjuvant chemoradiotherapy as the standard approach for resectable gastric cancer in the United States. In this study, a total of 556 patients with resected gastric or GEJ cancer were randomly assigned to surgery alone or surgery followed by adjuvant 5-FU/leucovorin bolus chemotherapy, sandwiched with 5-FU–based chemoradiation (45 Gy).45 In the chemoradiotherapy group, 64% of patients completed treatment and grade 3 and 4 toxicity occurred in 41% and 32%, respectively. However, only 3 patients (1%) died from treatment-related toxicity. At a median follow-up of 5 years, the median overall survival was 36 months in the chemoradiation group and 27 months in the surgery group. Overall survival rate was 50% in the combined modality group and 41% in the surgery-alone group, with a HR of 1.35 (95% CI 1.09 to 1.66, P = 0.005). The 3-year DFS was 48% in the chemoradiotherapy group and 31% in the surgery-alone group, corresponding to a DFS of 30 months and 19 months, respectively. Even after a median follow-up of 10 years, these positive results were maintained, with a HR for survival of 1.32 (95% CI 1.10 to 1.60, P = 0.0046) and HR for DFS of 1.51 (95% CI 1.25 to 1.83, P < 0.001).46 A criticism of the INT-0116 study is that 54% of patients had less than a D1 lymph node dissection, suggesting that adjuvant chemoradiation may have compensated for suboptimal surgery.
CALGB 80101, a United States Intergroup study, compared the INT-0116 protocol regimen (bolus 5-FU/leucovorin with 5-FU plus concurrent radiotherapy) to postoperative ECF sandwiched with 5-FU plus concurrent radiotherapy.47 The study included patients with resected gastric or GEJ adenocarcinoma that extended beyond the muscularis propria or was node positive. The percentage of patients with gastric versus GEJ cancer was not reported. A total of 546 patients were randomized. Preliminary results were presented at the 2011 American Society of Clinical Oncology meeting. The ECF arm had lower rates of grade 3/4 toxicities, including neutropenia, diarrhea, and mucositis. However, there was no difference in overall survival (3-year overall survival of 52% versus 50% for ECF and 5-FU/leucovorin, respectively) or DFS (3-year DFS of 47% versus 46% for ECF and 5-FU/leucovorin, respectively). The trial was not adequately powered to assess noninferiority. The location of the primary tumor (GEJ versus proximal versus distal stomach) did not have any effect on treatment outcome.
The Adjuvant Chemoradiation Therapy in Stomach Cancer (ARTIST) trial was the first study to compare adjuvant chemoradiotherapy with adjuvant chemotherapy in patients with D2-resected gastric cancer.48 A total of 458 patients were randomly assigned to 6 cycles of XP chemotherapy (capecitabine 2000 mg/m2 per day on days 1–14 and cisplatin 60 mg/m2 on day 1, every 3 weeks) or XP/radiotherapy/XP (2 cycles of XP followed by 45 Gy radiotherapy with concurrent daily capecitabine [825 mg/m2 twice daily] and 2 cycles of XP). After a median follow-up of 84 months, there was no difference in DFS or overall survival between treatment arms (HR for progression 0.74 [95% CI 0.52 to 1.05], P = 0.09; HR for death 1.13 [95% CI 0.78 to 1.65], P = 0.53).49 However, subgroup analysis showed that chemoradiotherapy significantly improved DFS in patients with node-positive disease (3-year DFS 76% versus 72%, P = 0.004).
Adjuvant Chemotherapy
Data supporting the use of adjuvant chemotherapy alone is largely derived from trials done in Asia, typically after a D2 lymph node dissection, and thus adjuvant chemotherapy has become the standard of care in that region. In the Japanese Adjuvant Chemotherapy Trial of S-1 for Gastric Cancer (ACTS-GC), a total of 1059 patients with stage II or III gastric cancer who had undergone surgery with a D2 lymphadenectomy were randomly assigned to 1 year of S-1 (an oral fluoropyrimidine) or surgery alone.50 The 5-year overall survival rate was 72% in the S-1 group and 61% in the surgery-only group (HR 0.669 [95% CI 0.54 to 0.83]).51 The 5-year relapse-free survival was 65% in the S-1 group and 53% in the surgery-only group (HR 0.65 [95% CI 0.537 to 0.793]). Of note, both arms of the ACTS-GC trial had significantly higher 5-year overall survival rates compared to the INT-0116 and MAGIC trials: 43% versus 28% and 36% versus 23% for the treatment and control groups, respectively.42,45 Consequently, it is unclear if the benefit of adjuvant chemotherapy can be translated to Western countries.
The Korean Capecitabine and Oxaliplatin Adjuvant Study in Stomach Cancer (CLASSIC) trial published in 2012 also established the role of adjuvant chemotherapy after D2 gastrectomy.52 A total of 1035 patients with stage II-IIIB gastric cancer who had curative D2 gastrectomy were randomly assigned to 8 cycles of adjuvant XELOX (capecitabine 1000 mg/m2 twice daily on days 1–14 plus oxaliplatin 130 mg/m2 on day 1, 21-day cycle) or surgery alone. Median follow-up was 34 months in both arms and 67% of patients in the chemotherapy arm completed all 8 cycles of planned chemotherapy. The 3-year DFS was 74% in the chemotherapy group and 59% in the surgery-only group (HR 0.56 [95% CI 0.44 to 0.72], P < 0.0001). There was a trend toward improvement in overall survival (83% versus 78%, HR 0.72 [95% CI 0.52 to 1.00]). After 5 years of follow-up, the improvement in overall survival became statistically significant (78% versus 69%, HR 0.66 [95% CI 0.51 to 0.85]).53
The benefit of adjuvant chemotherapy was reinforced by a 2010 meta-analysis comparing adjuvant chemotherapy to surgery alone in patients with resected gastric cancer.54 A total of 17 randomized controlled trials were included. Adjuvant fluorouracil-based chemotherapy was associated with a statistically significant improved overall survival (HR 0.82 [95% CI 0.76 to 0.90], P < 0.001) and DFS (HR 0.82 [95% CI 0.75 to 0.90], P < 0.001). Five-year overall survival increased from 49.6% to 55.3% with chemotherapy.
SELECTION OF TREATMENT APPROACH
Since data exists for all 3 approaches (perioperative chemotherapy, adjuvant chemoradiotherapy, and adjuvant chemotherapy), various meta-analyses have been done to clarify which approach is the best. In a recent meta-analysis of 6 randomized controlled trials reported between 2010 and 2012, which involved 1171 patients with resected gastric cancer, adjuvant chemotherapy was compared to adjuvant chemoradiotherapy.55 Five of the studies were from East Asia, while one was from a Western country. Adjuvant chemoradiation was associated with a lower local-regional recurrence rate (OR 0.46 [95% CI 0.32 to 0.67]) and better 5-year DFS rate (OR 1.56 [95% CI 1.09 to 2.24]). However, there was no statistical difference in 5-year overall survival rate (OR 1.32 [95% CI 0.92 to 1.88]). Similar results were reported by Zhou et al in 2016.56 This meta-analysis included 4 randomized controlled trials reported between 2010 and 2015, with a total of 960 patients who had undergone a D2 resection for gastric cancer. Compared to adjuvant chemotherapy, adjuvant chemoradiotherapy significantly reduced the loco-regional recurrence rate (LRRR; relative risk [RR] 0.50 [95% CI 0.34 to 0.74], P = 0.0005) and improved DFS (HR 0.73 [95% CI 0.60 to 0.89], P = 0.002). Again, no difference in overall survival was seen (HR 0.91 [95% CI 0.74 to 1.11], P = 0.34).
Adjuvant chemotherapy and perioperative chemotherapy have also been compared. In a recent meta-analysis of 14 randomized controlled trials (8 Asian, 6 European) involving 2093 patients with resected gastric or GEJ cancer, perioperative chemotherapy was associated with improved overall survival when compared to adjuvant chemotherapy (HR 0.48 [95% CI 0.35 to 0.67], P < 0.001).57 The benefit of perioperative chemotherapy over adjuvant chemotherapy has also been reported in a 2016 meta-analysis by Zhao et al.58 A total of 1240 patients were included from 5 randomized controlled trials and 6 clinical controlled trials, all from Asian countries. The 5-year overall survival rate was significantly better in the perioperative chemotherapy group compared to the adjuvant chemotherapy group (RR 0.77 [95% CI 0.64 to 0.92], P < 0.01). Furthermore, the 2 groups showed no significant differences in the postoperative complication rates (RR 0.98 [95% CI 0.63 to 1.51], P = 0.91) or adverse effects of chemotherapy (P > 0.05 for all adverse effects).
While these meta-analyses may offer some insight on the best treatment approach, they should be interpreted with caution. Most studies included in these meta-analyses were from Asian countries, and their findings may not be applicable to Western countries. Furthermore, the heterogeneity of trials and inclusion of nonrandomized trials make it difficult to draw conclusions. There are several ongoing trials that will help to define the optimal treatment approach.
CASE CONTINUED
The patient is presented at tumor board and the consensus is to proceed with the perioperative chemotherapy approach. The patient undergoes echocardiography, which reveals a normal ejection fraction. He receives 3 cycles of neoadjuvant EOX (epirubicin, oxaliplatin, and capecitabine). After 3 cycles of neoadjuvant EOX, the patient has a repeat CT that shows marked interval reduction in the size of the primary gastric neoplasm and interval decrease in the size of the small perigastric lymph nodes. He subsequently undergoes a total gastrectomy with J-tube placement. Pathology shows ypT3N0 disease with 0 out of 47 lymph nodes involved and negative margins. He then receives 3 cycles of adjuvant EOX.
• What are the recommendations for surveillance?
According to the current NCCN guidelines, a history and physical exam should be performed every 3 to 6 months for 1 to 2 years, then every 6 to 12 months for 3 to 5 years, and then annually.23 Labs, CT chest/abdomen, and EGD should be done as clinically indicated. Patients who have undergone surgical resection should be monitored for nutritional deficiencies (vitamin B12 and iron).
GASTROESOPHAGEAL JUNCTION TUMORS
Tumors arising in the GEJ or gastric cardia within 5 cm of the GEJ that extend into the GEJ or distal esophagus are staged and treated as esophageal cancers.21 The primary treatment for T1/T2N0 tumors is surgical resection. In patients with T3 or higher or node-positive adenocarcinoma of the GEJ, a combined modality approach is preferred, with preoperative chemoradiotherapy followed by surgical resection.59 The CROSS trial demonstrated a significant survival benefit with preoperative chemoradiation using carboplatin/paclitaxel compared to surgery alone in patients with esophageal or GEJ cancer (49 months versus 24 months, HR 0.66, P = 0.003).60
ONGOING TRIALS
As mentioned previously, several randomized clinical trials are in progress to clarify the optimal treatment approach. The MAGIC-B/MRC-ST03 is a randomized phase 2/3 trial looking at perioperative epirubicin, cisplatin, and capecitabine (ECX) with or without bevacizumab in patients with resectable lower esophageal, GEJ, or gastric cancer.61 The TOPGEAR trial, a randomized phase 2/3 study being conducted in Canada and Europe, is comparing perioperative ECF chemotherapy with preoperative chemoradiation plus perioperative ECF chemotherapy.62 In Asia, the PRODIGY trial is a phase 3, open-label, randomized study comparing neoadjuvant docetaxel, oxaliplatin, and S-1 followed by surgery and adjuvant S-1 versus surgery plus adjuvant S-1 in patients with locally advanced gastric cancer (T2-T4 or node positive).63 Primary endpoint is PFS and secondary endpoints are overall survival, R0 resection rate, and safety.
Trials comparing adjuvant chemotherapy to adjuvant chemoradiotherapy are also being conducted. In the Dutch CRITICS study, a randomized phase 3 trial, patients with stage Ib-Iva resectable gastric cancer were given 3 cycles of epirubicin, cisplatin/oxaliplatin, and capecitabine (ECC/EOC), followed by D2 resection and either 3 cycles of ECC/EOC or chemoradiation with weekly cisplatin and daily capecitabine.64 Between January 2007 and April 2015, a total of 788 patients were enrolled. In a preliminary report presented at ASCO in 2016, the 5-year survival rate was similar between the 2 arms (41.3% for chemotherapy arm and 40.9% for chemoradiotherapy arm, P = 0.99). The Korean ARTIST II trial is comparing adjuvant S-1 and S-1/oxaliplatin with or without radiotherapy in patients with D2-resected gastric cancer.65 Similarly, the NCT01711242 trial is comparing adjuvant XELOX alone versus XELOX with concurrent capecitabine/radiotherapy in patients with resected D2 gastric cancer.66
The ToGA trial established a survival benefit of trastuzumab in combination with chemotherapy in HER2-positive metastatic gastric cancer.67 Consequently, there are ongoing clinical trials to assess the role of trastuzumab in nonmetastatic gastric cancer. The TOXAG study is a phase 2 trial looking at the safety and tolerability of adjuvant oxaliplatin, capecitabine, and trastuzumab with radiation in patients with resected HER2-positive gastric or GEJ adenocarcinoma.68 The NCT01130337 clinical trial is evaluating perioperative XELOX/trastuzumab in patients with resectable gastric or GEJ adeno-carcinoma.69
SUMMARY
Gastric cancer is the fifth most common cancer worldwide, with the greatest incidence in East Asia. Survival outcomes are better in Asian countries when compared to the United States. This difference in survival may be related to the presence of mass screening programs in Asia, which allows for detection at an earlier stage and the use of a more extensive surgical approach (ie, D2 resection). Risk factors for developing gastric cancer include: diets high in salt/salt-preserved foods or processed meats, obesity, smoking, H. pylori infection, EBV, prior gastric surgery, radiation exposure, and positive family history.
According to the latest edition of TMN staging, gastric cancer includes tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ. Diagnostic work-up includes: EGD with biopsy; basic labs; CT chest/abdomen/pelvis with oral and intravenous contrast; EUS if no M1 disease is identified; PET-CT if there is no M1 disease and if clinically indicated; and diagnostic laparoscopy with cytology for clinical stage T1b or higher.
The mainstay of treatment is surgical resection. Laparoscopic approach is preferred over open gastrectomy due to lower complication rates and similar survival outcomes. Current NCCN guidelines recommend a D1 or a modified D2 lymph node dissection with at least 15 lymph nodes removed for examination. Systemic chemotherapy is required in locally advanced gastric cancer (T3-T4 or node positive) and should be considered in T2N0 disease with high-risk features. Currently, there is no global consensus on the optimal treatment approach. Data from various trials have shown benefit for each approach. Regional preferences are: perioperative chemotherapy in Europe; adjuvant chemoradiotherapy in the United States; and adjuvant chemotherapy in Asia. In an effort to better define the optimal treatment approach, several randomized clinical trials are being conducted. According to the current NCCN guidelines, the following treatment approaches are acceptable and are supported by data in the trial listed in parentheses:
• Perioperative chemotherapy
° 5-FU/cisplatin (French FNLCC/FCCD trial)44 or
° ECF (MAGIC trial)42 or
° ECF modifications: EOX, EOF, ECX (REAL-2 trial)43
• Adjuvant chemoradiotherapy
° 5-FU/leucovorin sandwiched with 5-FU-based chemoradiation (INT-0116 trial)45
• Adjuvant chemotherapy (after D2 resection)
° Capecitabine/oxaliplatin (CLASSIC trial)52 or
° Capecitabine/cisplatin (ARTIST trial)48,49
INTRODUCTION
Gastric cancer is the fifth most common cancer worldwide and the third leading cause of cancer death in both females and males.1 More than 70% of gastric cancer cases occur in the developing world, with approximately 50% occurring in East Asia.2 Gastric cancer is less common in the United States, with an incidence of 12.3 cases in males and 6.0 cases in females per 100,000 per year and a disproportionately higher incidence in Asians.3 According to the Surveillance, Epidemiology, and End Results Program, approximately 26,370 new cases of stomach cancer were diagnosed in the United States in 2016, and an estimated 10,730 people died of this disease.4 Since the 1970s, the 5-year relative survival rate for gastric cancer in the United States has improved from 15% in 1975 to 29% in 2009.5 In contrast, in Japan and Korea, where screening programs have been implemented, the 5-year survival rate approaches 70%.6
RISK FACTORS AND CLASSIFICATION
A variety of risk factors have been linked to gastric cancer. Diets high in salt, salt-preserved foods, and/or processed meats have been associated with an increased risk for developing gastric cancer.7,8 Obesity and smoking have also been implicated in gastric cancer.9,10 Several studies have demonstrated a strong association between Helicobacter pylori and the development of gastric cancer.11–13 It is believed that H. pylori infection leads to chronic active gastritis, atrophic gastritis, and intestinal metaplasia. Interestingly, mass eradication of H. pylori has not been shown to reduce the risk for gastric cancer.14 Therefore, treatment of H. pylori should only be considered in patients with active peptic ulcer disease.15 Other risk factors include Epstein-Barr virus (EBV), prior gastric surgery, and radiation exposure.16–18 Family history of gastric cancer, hereditary nonpolyposis colon cancer, Li-Fraumeni syndrome, and hereditary diffuse gastric cancer caused by mutations in the E-cadherin gene increase the risk.17
The anatomic distinction between gastric cancer and cancer of the gastroesophageal junction (GEJ) has been a topic of debate. The Siewert classification is the most widely used system and divides GEJ adenocarcinoma into 3 categories:20 type I tumor: adenocarcinoma of distal esophagus, located 1 cm to 5 cm above the GEJ; type II tumor: true carcinoma of gastric cardia, located within 1 cm above and 2 cm below the GEJ; type III tumor: subcardial gastric carcinoma, located 2 cm to 5 cm below the GEJ, and infiltrates esophagus from below.
The American Joint Committee on Cancer (AJCC) has updated the latest (7th) edition of TMN staging for stomach cancer to include tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ.21
In the following sections, neoadjuvant and adjuvant therapy in gastric cancer are discussed using a case presentation to illustrate important concepts.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 43-year old male with no significant past medical history presents with epigastric abdominal pain and heart burn for the past few weeks. He denies nausea, vomiting, melena, or hematochezia. His primary care physician (PCP) diagnoses him with gastroesophageal reflux disease (GERD) and initiates a trial of pantoprazole. Over the next 2 to 3 months, his symptoms do not improve and he has an associated 40-lb weight loss. Both social history and family history are noncontributory. Physical exam reveals epigastric tenderness without rebound or guarding. Laboratory evaluation reveals a hemoglobin of 12.6 g/dL with a mean corpuscular volume of 72 fL. A comprehensive chemistry profile is within normal limits. Given the constellation of presenting symptoms, especially the unintentional weight loss and the presence of microcytic anemia, his PCP suspects a malignant process and refers the patient to a gastroenterologist.
• What are the next appropriate steps for diagnosis?
The most common presenting symptoms of gastric cancer are weight loss and abdominal pain.22 Less commonly, patients exhibit nausea, anorexia, and dysphagia with proximal tumors. Melena is seen in only about 20% of patients. In Japan, where gastric cancer is more prevalent, mass screening programs allow for detection at an earlier stage, which partially accounts for the better survival rates seen in Asia as compared to the United States. Diagnostic work-up includes esophagogastroduodenoscopy (EGD) to assess Siewert category and to obtain a tissue sample for diagnosis. Full staging requires a complete blood count (CBC) with differential; comprehensive chemistry profile; computed tomography (CT) of chest/abdomen/pelvis with oral and intravenous contrast; endoscopic ultrasound (EUS) if no M1 disease is identified; positron emission tomography (PET)-CT if there is no evidence of M1 disease and if clinically indicated; and laparoscopy with cytology for clinical stage T1b or higher.23 Patients should be staged according to the TMN staging system (Table 1).
MANAGEMENT OF NONMETASTATIC DISEASE
CASE CONTINUED
The patient undergoes EGD, which reveals a large ulcerated, partially circumferential mass measuring approximately 4 cm. The mass extends from the gastric body to the cardia. Biopsy of the mass reveals poorly differentiated adenocarcinoma as well as H. pylori–associated gastritis. He is given antibiotic therapy and undergoes complete work-up of his newly diagnosed gastric adenocarcinoma. CT of the chest/abdomen/pelvis demonstrates a large gastric mass with gastrohepatic and distal perigastric adenopathy, compatible with locally advance primary gastric cancer. There is no evidence of distant metastasis. PET scan shows a large hypermetabolic mass in the stomach body and increased FDG activity in 3 small nodes along the lesser gastric curvature and in 1 node in the gastrohepatic region. EUS reveals a malignant gastric tumor in the body of the stomach, which is staged as T3, and a few malignant-appearing lymph nodes in the perigastric region. Fine-needle aspiration of the perigastric lymph node is performed and the sample obtained is positive for malignant cells. Diagnostic laparoscopy with peritoneal washings is performed and cytology is negative for malignant cells. The patient is staged as clinical stage IIB (T3N1M0).
• How should this patient with newly diagnosed, locally advanced, resectable gastric cancer be managed?
SURGERY
Surgical resection for localized gastric cancer is the mainstay of treatment with curative intent. Only very early stage (Tis or T1a) tumors can be considered for endoscopic mucosal resection. Regarding surgical resection, distal gastric cancers are typically treated with subtotal gastrectomy because there is no survival difference between subtotal and total gastrectomy.24,25 Moreover, subtotal gastrectomy is associated with better nutritional status and quality of life. For proximal tumors, total gastrectomy is preferred as subtotal gastrectomy has been associated with a higher incidence of reflux esophagitis and anastomotic stenosis.26 In terms of surgical approach, multiple studies have shown that a laparoscopic approach has a lower complication rate and similar outcomes in terms of cancer recurrence and long-term survival when compared to open gastrectomy.27–29 Thus, a laparoscopic approach is often used in academic centers with highly experienced surgeons.
The extent of lymph node dissection remains a topic of debate. A D1 dissection involves the removal of perigastric lymph nodes. A D2 dissection is a D1 dissection plus the removal of lymph nodes along the left gastric artery, common hepatic artery, celiac artery, splenic hilum, and splenic artery. D2 lymphadenectomy has become the standard of care in Eastern countries where gastric cancer is more prevalent, such as Japan and Korea.30 In Western countries, including the United States, less extensive lymphadenectomies are performed. Both randomized clinical trials and meta-analyses have failed to demonstrate an overall survival advantage of D2 dissection over D1 dissection.31,32 A Dutch trial by Bonenkamp et al involving 711 patients, one of the largest randomized trials of D1 and D2 lymphadenectomy, showed that D2 patients had a higher operative mortality rate than D1 patients (10% versus 4%, P = 0.004) and experienced more complications (43% versus 25%, P < 0.001).33 In a 15-year follow-up of this study, patients who had a D2 resection had lower locoregional recurrence and gastric-cancer–related death rates compared to those who had a D1 resection; however, D2 resection was associated with a significantly higher operative mortality and complication rate compared to D1.34 In addition, a 2015 Cochrane meta-analysis has demonstrated improved disease-specific survival (DSS) with D2 dissection (hazard ratio [HR] 0.81 [95% confidence interval {CI} 0.71 to 0.92]).35 Currently, the National Comprehensive Cancer Network (NCCN) recommends a D1 or a modified D2 gastrectomy with at least 15 lymph nodes removed for examination, with D2 lymphadenectomies only to be performed at experienced centers.23
SYSTEMIC CHEMOTHERAPY
Locally advanced gastric cancer (T3-T4 or node positive) requires systemic chemotherapy in addition to surgery, as this intervention improves the 5-year overall survival by 10% to 15%.36 Systemic therapy should also be considered in patients with T2N0 disease with high-risk features: poorly differentiated or high-grade cancer; lymphovascular invasion; neural invasion; age younger than 50 years; and patients who did not undergo D2 dissection.23 Currently, there is no global consensus on the best treatment approach. In the United States, where a less aggressive lymph-node dissection is performed, adjuvant chemoradiotherapy after surgery is more commonly seen. In Europe, perioperative (preoperative and postoperative) chemotherapy is the standard treatment. In Japan, adjuvant chemotherapy after D2 lymphadenectomy is the standard of care.37 These regional preferences are largely due to randomized clinical trials that have shown benefit for each approach. The landmark trials are discussed in the following sections and are summarized in Table 2.
Neoadjuvant Chemotherapy
Neoadjuvant chemotherapy has the benefit of “downstaging” locally advanced tumors to allow for curative resection. Phase 2 clinical trials have also demonstrated good pathologic response rates and high R0 resection rates following neoadjuvant chemotherapy.38,39 However, phase 3 trials to support this treatment approach are lacking. In the European Organisation for Research and Treatment of Cancer (EORTC) 40954 trial, patients with stage III or IV gastric or GEJ cancer were randomly assigned to surgery with or without preoperative cisplatin, leucovorin, and infusional fluorouracil (5-FU).40 The trial was stopped early due to poor accrual after 144 patients were randomized. The neoadjuvant chemotherapy arm had a higher R0 resection rate compared to the surgery alone arm (82% versus 67%, respectively, P = 0.036) but a higher postoperative complication rate (27% versus 16%, respectively, P = 0.09). More important, after a median follow-up of 4.4 years, a survival benefit could not be shown, with 2-year survival rates of 72.7% and 69.9% in the neoadjuvant and surgery-only arms, respectively (HR 0.84 [95% CI 0.52 to 1.35], P = 0.466). Due to the lack of large trials, a meta-analysis assessing the effectiveness of neoadjuvant chemotherapy combined with surgery versus surgery alone in advanced gastric and gastroesophageal cancer was performed.41 The analysis included 12 randomized controlled trials with a total of 1820 patients. Neoadjuvant chemotherapy was shown to slightly improve the survival rate (odds ratio [OR] 1.32 [95% CI 1.07 to 1.64], P = 0.01). It significantly improved the 3-year progression-free survival (PFS; OR 1.85 [95% CI 1.39 to 2.46], P < 0.0001), tumor down-staging rate (OR 1.71 [95% CI 1.26 to 2.33], P = 0.0006), and R0 resection rate (OR 1.38 [95% CI 1.08 to 1.78], P = 0.01). There were no differences between the 2 arms in terms of relapse rates, operative complications, perioperative mortality, and grade 3/4 adverse effects. While these results are encouraging, further randomized clinical trials are needed to clarify the role of neoadjuvant chemotherapy and its impact on overall survival.
Perioperative Chemotherapy
The results of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial published in 2006 established perioperative chemotherapy as standard of care in patients with resectable gastric and gastroesophageal adenocarcinoma.42 A total of 503 patients with potentially resectable gastric and lower esophageal adenocarcinoma were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. Perioperative chemotherapy consisted of 3 preoperative and postoperative cycles of epirubicin, cisplatin, and infusional 5-FU (ECF). At a median follow-up of 4 years, the perioperative-chemotherapy group had a significantly better PFS (HR 0.66 [95% CI 0.53 to 0.81], P < 0.001) as well as overall survival (HR 0.75 [95% CI 0.60 to 0.93], P = 0.009). The 5-year overall survival rate was 36.3% in the perioperative chemotherapy group and 23% in the surgery group. Of note, there was a greater proportion of stage T1/T2 tumors (52% versus 37%, P = 0.002) and N0/N1 disease (84% versus 71%) in the perioperative-chemotherapy group compared to the surgery alone group. In addition, only 42% of patients in the perioperative chemotherapy group completed all 6 cycles of chemotherapy.
The administration of ECF is often difficult since the 5-FU component requires a central venous access device and an ambulatory infusion pump and the cisplatin component is associated with nephrotoxicity and ototoxicity. The REAL-2 trial was a randomized phase 3 clinical trial that assessed whether 5-FU could be replaced by capecitabine and cisplatin by oxaliplatin in the ECF regimen.43 Between June 2000 and May 2005, a total of 1002 patients with locally advanced esophageal/GEJ/gastric cancer were enrolled. Patients were randomly assigned to 1 of 4 triplet therapies: epirubicin and cisplatin plus either 5-FU (ECF) or capecitabine (ECX) or epirubicin and oxaliplatin plus either 5-FU (EOF) or capecitabine (EOX). After a median follow-up of approximately 18 months, the overall survival in the capecitabine groups did not differ significantly from that in the 5-FU groups (HR 0.88 [95% CI 0.77 to 1.00], P = 0.06), nor did overall survival in the oxaliplatin groups differ significantly from that in the cisplatin groups (HR 0.91 [95% CI 0.79 to 1.04], P = 0.16). Interestingly, the 1-year survival rate was longer in the EOX group than in the ECF group (46.8% versus 37.7%, respectively; HR 0.80 [95% CI 0.66 to 0.97], P = 0.02). This translated into an overall survival of 11.2 months for the EOX group and 9.9 months for the ECF group. Therefore, EOX is preferred over ECF in clinical practice.
The French FNLCC/FFCD trial published in 2011 provided further support for perioperative chemotherapy.44 A total of 224 patients with adenocarcinoma of the lower esophagus, GEJ, or stomach were randomly assigned to perioperative chemotherapy plus surgery or surgery alone. The perioperative-chemotherapy group received 2 to 3 cycles of preoperative chemotherapy and 3 to 4 cycles of postoperative chemotherapy, consisting of infusional 5-FU (800 mg/m2 daily for days 1 to 5) and cisplatin (100 mg/m2 on day 1). In patients receiving preoperative chemotherapy, 38% experienced at least grade 3 to 4 toxicity. Among the 109 patients who received at least 1 cycle of preoperative chemotherapy, only 54 patients (50%) received postoperative chemotherapy. Despite this, the perioperative-chemotherapy group had a statistically significant higher R0 resection rate (84% versus 74%, P = 0.04) compared to the surgery alone group. At a median follow-up of 5.7 years, the perioperative chemotherapy group had an improved overall survival (HR for death 0.69 [95% CI 0.50 to 0.95], P = 0.02) and disease-free survival (DFS; HR for recurrence or death 0.65 [95% CI 0.48 to 0.89], P = 0.003). This translated into 5-year overall survival rates of 38% versus 24% and 5-year DFS rates of 34% versus 19%. One caveat to this study is that the majority of patients (64%) had GEJ cancer and only 25% had gastric cancer. In the multivariate analysis, the 2 significant prognostic factors for overall survival were the administration of preoperative chemotherapy (P = 0.01) and tumor site at the GEJ (P < 0.01).
Adjuvant Chemoradiotherapy
The INT-0116 (Intergroup 0116) study published in 2001 established adjuvant chemoradiotherapy as the standard approach for resectable gastric cancer in the United States. In this study, a total of 556 patients with resected gastric or GEJ cancer were randomly assigned to surgery alone or surgery followed by adjuvant 5-FU/leucovorin bolus chemotherapy, sandwiched with 5-FU–based chemoradiation (45 Gy).45 In the chemoradiotherapy group, 64% of patients completed treatment and grade 3 and 4 toxicity occurred in 41% and 32%, respectively. However, only 3 patients (1%) died from treatment-related toxicity. At a median follow-up of 5 years, the median overall survival was 36 months in the chemoradiation group and 27 months in the surgery group. Overall survival rate was 50% in the combined modality group and 41% in the surgery-alone group, with a HR of 1.35 (95% CI 1.09 to 1.66, P = 0.005). The 3-year DFS was 48% in the chemoradiotherapy group and 31% in the surgery-alone group, corresponding to a DFS of 30 months and 19 months, respectively. Even after a median follow-up of 10 years, these positive results were maintained, with a HR for survival of 1.32 (95% CI 1.10 to 1.60, P = 0.0046) and HR for DFS of 1.51 (95% CI 1.25 to 1.83, P < 0.001).46 A criticism of the INT-0116 study is that 54% of patients had less than a D1 lymph node dissection, suggesting that adjuvant chemoradiation may have compensated for suboptimal surgery.
CALGB 80101, a United States Intergroup study, compared the INT-0116 protocol regimen (bolus 5-FU/leucovorin with 5-FU plus concurrent radiotherapy) to postoperative ECF sandwiched with 5-FU plus concurrent radiotherapy.47 The study included patients with resected gastric or GEJ adenocarcinoma that extended beyond the muscularis propria or was node positive. The percentage of patients with gastric versus GEJ cancer was not reported. A total of 546 patients were randomized. Preliminary results were presented at the 2011 American Society of Clinical Oncology meeting. The ECF arm had lower rates of grade 3/4 toxicities, including neutropenia, diarrhea, and mucositis. However, there was no difference in overall survival (3-year overall survival of 52% versus 50% for ECF and 5-FU/leucovorin, respectively) or DFS (3-year DFS of 47% versus 46% for ECF and 5-FU/leucovorin, respectively). The trial was not adequately powered to assess noninferiority. The location of the primary tumor (GEJ versus proximal versus distal stomach) did not have any effect on treatment outcome.
The Adjuvant Chemoradiation Therapy in Stomach Cancer (ARTIST) trial was the first study to compare adjuvant chemoradiotherapy with adjuvant chemotherapy in patients with D2-resected gastric cancer.48 A total of 458 patients were randomly assigned to 6 cycles of XP chemotherapy (capecitabine 2000 mg/m2 per day on days 1–14 and cisplatin 60 mg/m2 on day 1, every 3 weeks) or XP/radiotherapy/XP (2 cycles of XP followed by 45 Gy radiotherapy with concurrent daily capecitabine [825 mg/m2 twice daily] and 2 cycles of XP). After a median follow-up of 84 months, there was no difference in DFS or overall survival between treatment arms (HR for progression 0.74 [95% CI 0.52 to 1.05], P = 0.09; HR for death 1.13 [95% CI 0.78 to 1.65], P = 0.53).49 However, subgroup analysis showed that chemoradiotherapy significantly improved DFS in patients with node-positive disease (3-year DFS 76% versus 72%, P = 0.004).
Adjuvant Chemotherapy
Data supporting the use of adjuvant chemotherapy alone is largely derived from trials done in Asia, typically after a D2 lymph node dissection, and thus adjuvant chemotherapy has become the standard of care in that region. In the Japanese Adjuvant Chemotherapy Trial of S-1 for Gastric Cancer (ACTS-GC), a total of 1059 patients with stage II or III gastric cancer who had undergone surgery with a D2 lymphadenectomy were randomly assigned to 1 year of S-1 (an oral fluoropyrimidine) or surgery alone.50 The 5-year overall survival rate was 72% in the S-1 group and 61% in the surgery-only group (HR 0.669 [95% CI 0.54 to 0.83]).51 The 5-year relapse-free survival was 65% in the S-1 group and 53% in the surgery-only group (HR 0.65 [95% CI 0.537 to 0.793]). Of note, both arms of the ACTS-GC trial had significantly higher 5-year overall survival rates compared to the INT-0116 and MAGIC trials: 43% versus 28% and 36% versus 23% for the treatment and control groups, respectively.42,45 Consequently, it is unclear if the benefit of adjuvant chemotherapy can be translated to Western countries.
The Korean Capecitabine and Oxaliplatin Adjuvant Study in Stomach Cancer (CLASSIC) trial published in 2012 also established the role of adjuvant chemotherapy after D2 gastrectomy.52 A total of 1035 patients with stage II-IIIB gastric cancer who had curative D2 gastrectomy were randomly assigned to 8 cycles of adjuvant XELOX (capecitabine 1000 mg/m2 twice daily on days 1–14 plus oxaliplatin 130 mg/m2 on day 1, 21-day cycle) or surgery alone. Median follow-up was 34 months in both arms and 67% of patients in the chemotherapy arm completed all 8 cycles of planned chemotherapy. The 3-year DFS was 74% in the chemotherapy group and 59% in the surgery-only group (HR 0.56 [95% CI 0.44 to 0.72], P < 0.0001). There was a trend toward improvement in overall survival (83% versus 78%, HR 0.72 [95% CI 0.52 to 1.00]). After 5 years of follow-up, the improvement in overall survival became statistically significant (78% versus 69%, HR 0.66 [95% CI 0.51 to 0.85]).53
The benefit of adjuvant chemotherapy was reinforced by a 2010 meta-analysis comparing adjuvant chemotherapy to surgery alone in patients with resected gastric cancer.54 A total of 17 randomized controlled trials were included. Adjuvant fluorouracil-based chemotherapy was associated with a statistically significant improved overall survival (HR 0.82 [95% CI 0.76 to 0.90], P < 0.001) and DFS (HR 0.82 [95% CI 0.75 to 0.90], P < 0.001). Five-year overall survival increased from 49.6% to 55.3% with chemotherapy.
SELECTION OF TREATMENT APPROACH
Since data exists for all 3 approaches (perioperative chemotherapy, adjuvant chemoradiotherapy, and adjuvant chemotherapy), various meta-analyses have been done to clarify which approach is the best. In a recent meta-analysis of 6 randomized controlled trials reported between 2010 and 2012, which involved 1171 patients with resected gastric cancer, adjuvant chemotherapy was compared to adjuvant chemoradiotherapy.55 Five of the studies were from East Asia, while one was from a Western country. Adjuvant chemoradiation was associated with a lower local-regional recurrence rate (OR 0.46 [95% CI 0.32 to 0.67]) and better 5-year DFS rate (OR 1.56 [95% CI 1.09 to 2.24]). However, there was no statistical difference in 5-year overall survival rate (OR 1.32 [95% CI 0.92 to 1.88]). Similar results were reported by Zhou et al in 2016.56 This meta-analysis included 4 randomized controlled trials reported between 2010 and 2015, with a total of 960 patients who had undergone a D2 resection for gastric cancer. Compared to adjuvant chemotherapy, adjuvant chemoradiotherapy significantly reduced the loco-regional recurrence rate (LRRR; relative risk [RR] 0.50 [95% CI 0.34 to 0.74], P = 0.0005) and improved DFS (HR 0.73 [95% CI 0.60 to 0.89], P = 0.002). Again, no difference in overall survival was seen (HR 0.91 [95% CI 0.74 to 1.11], P = 0.34).
Adjuvant chemotherapy and perioperative chemotherapy have also been compared. In a recent meta-analysis of 14 randomized controlled trials (8 Asian, 6 European) involving 2093 patients with resected gastric or GEJ cancer, perioperative chemotherapy was associated with improved overall survival when compared to adjuvant chemotherapy (HR 0.48 [95% CI 0.35 to 0.67], P < 0.001).57 The benefit of perioperative chemotherapy over adjuvant chemotherapy has also been reported in a 2016 meta-analysis by Zhao et al.58 A total of 1240 patients were included from 5 randomized controlled trials and 6 clinical controlled trials, all from Asian countries. The 5-year overall survival rate was significantly better in the perioperative chemotherapy group compared to the adjuvant chemotherapy group (RR 0.77 [95% CI 0.64 to 0.92], P < 0.01). Furthermore, the 2 groups showed no significant differences in the postoperative complication rates (RR 0.98 [95% CI 0.63 to 1.51], P = 0.91) or adverse effects of chemotherapy (P > 0.05 for all adverse effects).
While these meta-analyses may offer some insight on the best treatment approach, they should be interpreted with caution. Most studies included in these meta-analyses were from Asian countries, and their findings may not be applicable to Western countries. Furthermore, the heterogeneity of trials and inclusion of nonrandomized trials make it difficult to draw conclusions. There are several ongoing trials that will help to define the optimal treatment approach.
CASE CONTINUED
The patient is presented at tumor board and the consensus is to proceed with the perioperative chemotherapy approach. The patient undergoes echocardiography, which reveals a normal ejection fraction. He receives 3 cycles of neoadjuvant EOX (epirubicin, oxaliplatin, and capecitabine). After 3 cycles of neoadjuvant EOX, the patient has a repeat CT that shows marked interval reduction in the size of the primary gastric neoplasm and interval decrease in the size of the small perigastric lymph nodes. He subsequently undergoes a total gastrectomy with J-tube placement. Pathology shows ypT3N0 disease with 0 out of 47 lymph nodes involved and negative margins. He then receives 3 cycles of adjuvant EOX.
• What are the recommendations for surveillance?
According to the current NCCN guidelines, a history and physical exam should be performed every 3 to 6 months for 1 to 2 years, then every 6 to 12 months for 3 to 5 years, and then annually.23 Labs, CT chest/abdomen, and EGD should be done as clinically indicated. Patients who have undergone surgical resection should be monitored for nutritional deficiencies (vitamin B12 and iron).
GASTROESOPHAGEAL JUNCTION TUMORS
Tumors arising in the GEJ or gastric cardia within 5 cm of the GEJ that extend into the GEJ or distal esophagus are staged and treated as esophageal cancers.21 The primary treatment for T1/T2N0 tumors is surgical resection. In patients with T3 or higher or node-positive adenocarcinoma of the GEJ, a combined modality approach is preferred, with preoperative chemoradiotherapy followed by surgical resection.59 The CROSS trial demonstrated a significant survival benefit with preoperative chemoradiation using carboplatin/paclitaxel compared to surgery alone in patients with esophageal or GEJ cancer (49 months versus 24 months, HR 0.66, P = 0.003).60
ONGOING TRIALS
As mentioned previously, several randomized clinical trials are in progress to clarify the optimal treatment approach. The MAGIC-B/MRC-ST03 is a randomized phase 2/3 trial looking at perioperative epirubicin, cisplatin, and capecitabine (ECX) with or without bevacizumab in patients with resectable lower esophageal, GEJ, or gastric cancer.61 The TOPGEAR trial, a randomized phase 2/3 study being conducted in Canada and Europe, is comparing perioperative ECF chemotherapy with preoperative chemoradiation plus perioperative ECF chemotherapy.62 In Asia, the PRODIGY trial is a phase 3, open-label, randomized study comparing neoadjuvant docetaxel, oxaliplatin, and S-1 followed by surgery and adjuvant S-1 versus surgery plus adjuvant S-1 in patients with locally advanced gastric cancer (T2-T4 or node positive).63 Primary endpoint is PFS and secondary endpoints are overall survival, R0 resection rate, and safety.
Trials comparing adjuvant chemotherapy to adjuvant chemoradiotherapy are also being conducted. In the Dutch CRITICS study, a randomized phase 3 trial, patients with stage Ib-Iva resectable gastric cancer were given 3 cycles of epirubicin, cisplatin/oxaliplatin, and capecitabine (ECC/EOC), followed by D2 resection and either 3 cycles of ECC/EOC or chemoradiation with weekly cisplatin and daily capecitabine.64 Between January 2007 and April 2015, a total of 788 patients were enrolled. In a preliminary report presented at ASCO in 2016, the 5-year survival rate was similar between the 2 arms (41.3% for chemotherapy arm and 40.9% for chemoradiotherapy arm, P = 0.99). The Korean ARTIST II trial is comparing adjuvant S-1 and S-1/oxaliplatin with or without radiotherapy in patients with D2-resected gastric cancer.65 Similarly, the NCT01711242 trial is comparing adjuvant XELOX alone versus XELOX with concurrent capecitabine/radiotherapy in patients with resected D2 gastric cancer.66
The ToGA trial established a survival benefit of trastuzumab in combination with chemotherapy in HER2-positive metastatic gastric cancer.67 Consequently, there are ongoing clinical trials to assess the role of trastuzumab in nonmetastatic gastric cancer. The TOXAG study is a phase 2 trial looking at the safety and tolerability of adjuvant oxaliplatin, capecitabine, and trastuzumab with radiation in patients with resected HER2-positive gastric or GEJ adenocarcinoma.68 The NCT01130337 clinical trial is evaluating perioperative XELOX/trastuzumab in patients with resectable gastric or GEJ adeno-carcinoma.69
SUMMARY
Gastric cancer is the fifth most common cancer worldwide, with the greatest incidence in East Asia. Survival outcomes are better in Asian countries when compared to the United States. This difference in survival may be related to the presence of mass screening programs in Asia, which allows for detection at an earlier stage and the use of a more extensive surgical approach (ie, D2 resection). Risk factors for developing gastric cancer include: diets high in salt/salt-preserved foods or processed meats, obesity, smoking, H. pylori infection, EBV, prior gastric surgery, radiation exposure, and positive family history.
According to the latest edition of TMN staging, gastric cancer includes tumors arising more than 5 cm distally of the GEJ or within 5 cm of the GEJ but without extension to the esophagus or GEJ. Diagnostic work-up includes: EGD with biopsy; basic labs; CT chest/abdomen/pelvis with oral and intravenous contrast; EUS if no M1 disease is identified; PET-CT if there is no M1 disease and if clinically indicated; and diagnostic laparoscopy with cytology for clinical stage T1b or higher.
The mainstay of treatment is surgical resection. Laparoscopic approach is preferred over open gastrectomy due to lower complication rates and similar survival outcomes. Current NCCN guidelines recommend a D1 or a modified D2 lymph node dissection with at least 15 lymph nodes removed for examination. Systemic chemotherapy is required in locally advanced gastric cancer (T3-T4 or node positive) and should be considered in T2N0 disease with high-risk features. Currently, there is no global consensus on the optimal treatment approach. Data from various trials have shown benefit for each approach. Regional preferences are: perioperative chemotherapy in Europe; adjuvant chemoradiotherapy in the United States; and adjuvant chemotherapy in Asia. In an effort to better define the optimal treatment approach, several randomized clinical trials are being conducted. According to the current NCCN guidelines, the following treatment approaches are acceptable and are supported by data in the trial listed in parentheses:
• Perioperative chemotherapy
° 5-FU/cisplatin (French FNLCC/FCCD trial)44 or
° ECF (MAGIC trial)42 or
° ECF modifications: EOX, EOF, ECX (REAL-2 trial)43
• Adjuvant chemoradiotherapy
° 5-FU/leucovorin sandwiched with 5-FU-based chemoradiation (INT-0116 trial)45
• Adjuvant chemotherapy (after D2 resection)
° Capecitabine/oxaliplatin (CLASSIC trial)52 or
° Capecitabine/cisplatin (ARTIST trial)48,49
- World Health Organization. GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence Worldwide in 2012. France, Lyon: IARC; 2012.
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127:2893–917.
- Lui FH, Tuan B, Swenson SL, et al. Ethnic disparities in gastric cancer incidence and survival in the USA: an updated analysis of 1992-2009 SEER data. Dig Dis Sci 2014;59:3027–34.
- Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2013. National Cancer Institute. http://seer.cancer.gov/csr/1975_2013/. Based on November 2015 SEER data submission, posted to the SEER web site April 2016.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
- Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer 2007;10:75.
- Bouvard V, Loomis D, Guyton KZ, et al. Carcinogenicity of consumption of red and processed meat. Lancet Oncol 2015;16:1599–600.
- Yang P, Zhou Y, Chen B, et al. Overweight, obesity and gastric cancer risk: results from a meta-analysis of cohort studies. Eur J Cancer 2009;45:2867–73.
- González CA, Pera G, Agudo A, et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int J Cancer 2003;107:629–34.
- Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998;114:1169–79.
- Eslick GD, Lim LL, Byles JE, et al. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol 1999;94:2373–9.
- An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993;341:1359–62.
- Parsonnet J, Forman D. Helicobacter pylori infection and gastric cancer—for want of more outcomes. JAMA 2004;291:244–5.
- Malfertheiner P, Megraud F, O'Morain CA, et al. Management of Helicobacter pylori infection—the Maastricht IV/ Florence Consensus Report. Gut 2012;61:646–64.
- Fukayama M. Epstein-Barr virus and gastric carcinoma. Pathol Int 2010;60:337–50.
- Takeno S, Hashimoto T, Maki K, et al. Gastric cancer arising from the remnant stomach after distal gastrectomy: a review. World J Gastroenterol 2014;20:13734–40.
- Morton LM, Dores GM, Curtis RE, et al. Stomach cancer risk after treatment for Hodgkin lymphoma. J Clin Oncol 2013;31:3369–77.
- van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361–74.
- Siewert J, Stein H. Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg 1998;85:1457–9.
- Edge S, Byrd DR, Compton CC, et al. AJCC cancer staging manual. 7th ed. New York: Springer New York; 2009.
- Wanebo HJ, Kennedy BJ, Chmiel J, et al. Cancer of the stomach. A patient care study by the American College of Surgeons. Ann Surg 1993;218:583–92.
- National Comprehensive Cancer Network. Gastric cancer (version 3.2016www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed December 14, 2016.
- Bozzetti F, Marubini E, Bonfanti G, et al. Subtotal versus total gastrectomy for gastric cancer: five-year survival rates in a multicenter randomized Italian trial. Italian Gastrointestinal Tumor Study Group. Ann Surg 1999;230:170–8.
- Gouzi JL, Huguier M, Fagniez PL, et al. Total versus subtotal gastrectomy for adenocarcinoma of the gastric antrum. A French prospective controlled study. Ann Surg 1989;209:162–6.
- Pu YW, Gong W, Wu YY, et al. Proximal gastrectomy versus total gastrectomy for proximal gastric carcinoma. A meta-analysis on postoperative complications, 5-year survival, and recurrence rate. Saudi Med J 2013;34:1223–8.
- Chen K, Xu XW, Mou YP, et al. Systematic review and meta-analysis of laparoscopic and open gastrectomy for advanced gastric cancer. World J Surg Oncol 2013;11:182.
- Fang C, Hua J, Li J, et al. Comparison of long-term results between laparoscopy-assisted gastrectomy and open gastrectomy with D2 lymphadenectomy for advanced gastric cancer. Am J Surg 2014;208:391–6.
- Wang W, Li Z, Tang J, et al. Laparoscopic versus open total gastrectomy with D2 dissection for gastric cancer: a meta-analysis. J Cancer Res Clin Oncol 2013;139:1721–34.
- Schmidt B, Yoon SS. D1 versus D2 lymphadenectomy for gastric cancer. J Surg Oncol 2013;107:259–64.
- Jiang L, Yang KH, Guan QL, et al. Survival and recurrence free benefits with different lymphadenectomy for resectable gastric cancer: a meta-analysis. J Surg Oncol 2013;107:807–14.
- Degiuli M, Sasako M, Ponti A, et al. Randomized clinical trial comparing survival after D1 or D2 gastrectomy for gastric cancer. Br J Surg 2014;101:23–31.
- Bonenkamp JJ, Songun I, Hermans J, et al. Randomized comparison of morbidity after D1 and D2 dissection for gastric cancer in 996 Dutch patients. Lancet 1995;345:745–8.
- Songun I, Putter H, Kranenbarg EM, et al. Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. Lancet Oncol 2010;11:439–49.
- Mocellin S, McCulloch P, Kazi H, et al. Extent of lymph node dissection for adenocarcinoma of the stomach. Cochrane Database Syst Rev 2015;8:CD001964.
- Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
- Quéro L, Guillerm S, Hennequin C. Neoadjuvant or adjuvant therapy for gastric cancer. World J Gastrointest Oncol 2015;7:102–10.
- Okabe H, Hata H, Ueda S, et al. A phase II study of neoadjuvant chemotherapy with S-1 and cisplatin for stage III gastric cancer: KUGC03. J Surg Oncol 2016 Jan;113:36–41.
- Wang X, Zhao L, Liu H et al. A phase II study of a modified FOLFOX6 regimen as neoadjuvant chemotherapy for locally advanced gastric cancer. Br J Cancer 2016;114:1326-33.
- Schuhmacher C, Gretschel S, Lordick F, et al. Neoadjuvant chemotherapy compared with surgery alone for locally advanced cancer of the stomach and cardia: European Organisation for Research and Treatment of Cancer randomized trial 40954. J Clin Oncol 2010;28:5210–18.
- Xiong BH, Cheng Y, Ma L, Zhang CQ. An updated meta-analysis of randomized controlled trial assessing the effect of neoadjuvant chemotherapy in advanced gastric cancer. Cancer Invest 2014;32:272–84.
- Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006;355:11–20.
- Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008;358:36–46.
- Ychou M, Boige V, Pignon JP, et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol 2011;29:1715–21.
- Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med 2001;345:725–30.
- Smalley SR, Benedetti JK, Haller DG, et al. Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. J Clin Oncol 2012;30:2327–33.
- Fuchs CS, Tepper JE, Niedzwiecki D, et al. Postoperative adjuvant chemoradiation for gastric or gastroesophageal junction (GEJ) adenocarcinoma using epirubicin, cisplatin, and infusional (CI) 5-FU (ECF) before and after CI 5-FU and radiotherapy (CRT) compared with bolus 5-FU/LV before and after CRT: Intergroup trial CALGB 80101. J Clin Oncol 2011;29:256s. Abstract 4003.
- Lee J, Lim do H, Kim S, et al. Phase III trial comparing capecitabine plus cisplatin versus capecitabine plus cisplatin with concurrent capecitabine radiotherapy in completely resected gastric cancer with D2 lymph node dissection: the ARTIST trial. J Clin Oncol 2012;30:268–73
- Park SH, Sohn TS, Lee J, et al. Phase III trial to compare adjuvant chemotherapy with capecitabine and cisplatin versus concurrent chemoradiotherapy in gastric cancer: final report of the adjuvant chemoradiotherapy in stomach tumors trial, including survival and subset analyses. J Clin Oncol 2015;33:3130–6.
- Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 2007;357:1810–20.
- Sasako M, Sakuramoto S, Katai H, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol 2011;29:4387–93.
- Bang YJ, Kim YW, Yang HK, et al. Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet 2012;379:315–21.
- Noh SH, Park SR, Yang HK, et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:1389–96.
- Paoletti X, Oba K, Burzykowski T, et al. Benefit of adjuvant chemotherapy for resectable gastric cancer: a meta-analysis. JAMA 2010; 303:1729–37.
- Dai Q, Jiang L, Lin RJ, et al. Adjuvant chemoradiotherapy versus chemotherapy for gastric cancer: a meta-analysis of randomized controlled trials. J Surg Oncol 2015;111:277–84.
- Zhou M, Kang M, Li G, et al. Postoperative chemoradiotherapy versus chemotherapy for R0 resected gastric cancer with D2 lymph node dissection: an up-to-date meta-analysis. World J Surg Oncol 2016;14:209.
- Yang Y, Yin X, Sheng L, et al. Perioperative chemotherapy more of a benefit for overall survival than adjuvant chemotherapy for operable gastric cancer: an updated meta-analysis. Sci Rep 2015;5:12850.
- Zhao JH, Gao P, Song YX, et al. Which is better for gastric cancer patients, perioperative or adjuvant chemotherapy: a meta-analysis. BMC Cancer 2016;16:631.
- Narsule CK, Montgomery MM, and Fernando HC. Evidence-based review of the management of cancers of the gastroesophageal junction. Thorac Surg Clin 2012;22:109–21.
- van Hagen P, Hulshof MCCM, van Lanschot JJB, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Eng J Med 2012;266:2074–84.
- Cunningham D. Chemotherapy with or without bevacizumab or lapatinib to treat operable oesophagogastric cancer (ST03). ClinicalTrials.gov. https://clinicaltrials.gov/show/NCT00450203. NLM Identifier: NCT00450203. Accessed December 14, 2016.
- Leong T, Smithers BM, Michael M, et al. TOPGEAR: a randomised phase III trial of perioperative ECF chemotherapy versus preoperative chemoradiation plus perioperative ECF chemotherapy for resectable gastric cancer (an international, intergroup trial of the AGITG/TROG/EORTC/NCIC CTG). BMC Cancer 2015;15:532.
- Docetaxel+oxaliplatin+S-1 (DOS) regimen as neoadjuvant chemotherapy in advanced gastric cancer (PRODIGY). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01515748 NLM. Identifier: NCT01515748. Accessed December 14, 2016.
- Verheij M, Jansen EP, Cats A, et al. A multicenter randomized phase III trial of neo-adjuvant chemotherapy followed by surgery and chemotherapy or by surgery and chemoradiotherapy in resectable gastric cancer: First results from the CRITICS study. J Clin Oncol 2016;34 (suppl). Abstract 4000.
- Kang WK. Phase III randomized trial of adjuvant chemotherapy with S-1 vs S-1/oxaliplatin ± radiotherapy for completely resected gastric adenocarcinoma : The ARTIST II Trial (ARTIST-II). ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01761461. NLM Identifier: NCT01761461. Accessed December 14, 2016.
- Trial of adjuvant XELOX chemotherapy and concurrent capecitabine and radiotherapy for resected gastric carcinoma. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01711242. NLM Identifier: NCT01711242. Accessed December 14, 2016.
- Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
- Roche HL. A Study of the combination of oxaliplatin, capecitabine and herceptin (trastuzumab) and chemoradiotherapy in the adjuvant setting in operated patients with HER2+ gastric or gastro-esophageal junction cancer (TOXAG Study). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01748773. NLM Identifer: NCT01748773. Accessed December 14, 2016.
- A study of capecitabine [Xeloda] in combination with trastuzumab [herceptin] and oxaliplatine in patients with resectable gastric cancer. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01130337. NLM Identifier: NCT01130337. Accessed December 14, 2016.
- World Health Organization. GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence Worldwide in 2012. France, Lyon: IARC; 2012.
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010;127:2893–917.
- Lui FH, Tuan B, Swenson SL, et al. Ethnic disparities in gastric cancer incidence and survival in the USA: an updated analysis of 1992-2009 SEER data. Dig Dis Sci 2014;59:3027–34.
- Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2013. National Cancer Institute. http://seer.cancer.gov/csr/1975_2013/. Based on November 2015 SEER data submission, posted to the SEER web site April 2016.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29.
- Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer 2011;14:301–16.
- Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer 2007;10:75.
- Bouvard V, Loomis D, Guyton KZ, et al. Carcinogenicity of consumption of red and processed meat. Lancet Oncol 2015;16:1599–600.
- Yang P, Zhou Y, Chen B, et al. Overweight, obesity and gastric cancer risk: results from a meta-analysis of cohort studies. Eur J Cancer 2009;45:2867–73.
- González CA, Pera G, Agudo A, et al. Smoking and the risk of gastric cancer in the European Prospective Investigation Into Cancer and Nutrition (EPIC). Int J Cancer 2003;107:629–34.
- Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998;114:1169–79.
- Eslick GD, Lim LL, Byles JE, et al. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol 1999;94:2373–9.
- An international association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993;341:1359–62.
- Parsonnet J, Forman D. Helicobacter pylori infection and gastric cancer—for want of more outcomes. JAMA 2004;291:244–5.
- Malfertheiner P, Megraud F, O'Morain CA, et al. Management of Helicobacter pylori infection—the Maastricht IV/ Florence Consensus Report. Gut 2012;61:646–64.
- Fukayama M. Epstein-Barr virus and gastric carcinoma. Pathol Int 2010;60:337–50.
- Takeno S, Hashimoto T, Maki K, et al. Gastric cancer arising from the remnant stomach after distal gastrectomy: a review. World J Gastroenterol 2014;20:13734–40.
- Morton LM, Dores GM, Curtis RE, et al. Stomach cancer risk after treatment for Hodgkin lymphoma. J Clin Oncol 2013;31:3369–77.
- van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet 2015;52:361–74.
- Siewert J, Stein H. Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg 1998;85:1457–9.
- Edge S, Byrd DR, Compton CC, et al. AJCC cancer staging manual. 7th ed. New York: Springer New York; 2009.
- Wanebo HJ, Kennedy BJ, Chmiel J, et al. Cancer of the stomach. A patient care study by the American College of Surgeons. Ann Surg 1993;218:583–92.
- National Comprehensive Cancer Network. Gastric cancer (version 3.2016www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed December 14, 2016.
- Bozzetti F, Marubini E, Bonfanti G, et al. Subtotal versus total gastrectomy for gastric cancer: five-year survival rates in a multicenter randomized Italian trial. Italian Gastrointestinal Tumor Study Group. Ann Surg 1999;230:170–8.
- Gouzi JL, Huguier M, Fagniez PL, et al. Total versus subtotal gastrectomy for adenocarcinoma of the gastric antrum. A French prospective controlled study. Ann Surg 1989;209:162–6.
- Pu YW, Gong W, Wu YY, et al. Proximal gastrectomy versus total gastrectomy for proximal gastric carcinoma. A meta-analysis on postoperative complications, 5-year survival, and recurrence rate. Saudi Med J 2013;34:1223–8.
- Chen K, Xu XW, Mou YP, et al. Systematic review and meta-analysis of laparoscopic and open gastrectomy for advanced gastric cancer. World J Surg Oncol 2013;11:182.
- Fang C, Hua J, Li J, et al. Comparison of long-term results between laparoscopy-assisted gastrectomy and open gastrectomy with D2 lymphadenectomy for advanced gastric cancer. Am J Surg 2014;208:391–6.
- Wang W, Li Z, Tang J, et al. Laparoscopic versus open total gastrectomy with D2 dissection for gastric cancer: a meta-analysis. J Cancer Res Clin Oncol 2013;139:1721–34.
- Schmidt B, Yoon SS. D1 versus D2 lymphadenectomy for gastric cancer. J Surg Oncol 2013;107:259–64.
- Jiang L, Yang KH, Guan QL, et al. Survival and recurrence free benefits with different lymphadenectomy for resectable gastric cancer: a meta-analysis. J Surg Oncol 2013;107:807–14.
- Degiuli M, Sasako M, Ponti A, et al. Randomized clinical trial comparing survival after D1 or D2 gastrectomy for gastric cancer. Br J Surg 2014;101:23–31.
- Bonenkamp JJ, Songun I, Hermans J, et al. Randomized comparison of morbidity after D1 and D2 dissection for gastric cancer in 996 Dutch patients. Lancet 1995;345:745–8.
- Songun I, Putter H, Kranenbarg EM, et al. Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. Lancet Oncol 2010;11:439–49.
- Mocellin S, McCulloch P, Kazi H, et al. Extent of lymph node dissection for adenocarcinoma of the stomach. Cochrane Database Syst Rev 2015;8:CD001964.
- Van Cutsem E, Sagaert X, Topal B, et al. Gastric cancer. Lancet 2016;388:2654–64.
- Quéro L, Guillerm S, Hennequin C. Neoadjuvant or adjuvant therapy for gastric cancer. World J Gastrointest Oncol 2015;7:102–10.
- Okabe H, Hata H, Ueda S, et al. A phase II study of neoadjuvant chemotherapy with S-1 and cisplatin for stage III gastric cancer: KUGC03. J Surg Oncol 2016 Jan;113:36–41.
- Wang X, Zhao L, Liu H et al. A phase II study of a modified FOLFOX6 regimen as neoadjuvant chemotherapy for locally advanced gastric cancer. Br J Cancer 2016;114:1326-33.
- Schuhmacher C, Gretschel S, Lordick F, et al. Neoadjuvant chemotherapy compared with surgery alone for locally advanced cancer of the stomach and cardia: European Organisation for Research and Treatment of Cancer randomized trial 40954. J Clin Oncol 2010;28:5210–18.
- Xiong BH, Cheng Y, Ma L, Zhang CQ. An updated meta-analysis of randomized controlled trial assessing the effect of neoadjuvant chemotherapy in advanced gastric cancer. Cancer Invest 2014;32:272–84.
- Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006;355:11–20.
- Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008;358:36–46.
- Ychou M, Boige V, Pignon JP, et al. Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol 2011;29:1715–21.
- Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med 2001;345:725–30.
- Smalley SR, Benedetti JK, Haller DG, et al. Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. J Clin Oncol 2012;30:2327–33.
- Fuchs CS, Tepper JE, Niedzwiecki D, et al. Postoperative adjuvant chemoradiation for gastric or gastroesophageal junction (GEJ) adenocarcinoma using epirubicin, cisplatin, and infusional (CI) 5-FU (ECF) before and after CI 5-FU and radiotherapy (CRT) compared with bolus 5-FU/LV before and after CRT: Intergroup trial CALGB 80101. J Clin Oncol 2011;29:256s. Abstract 4003.
- Lee J, Lim do H, Kim S, et al. Phase III trial comparing capecitabine plus cisplatin versus capecitabine plus cisplatin with concurrent capecitabine radiotherapy in completely resected gastric cancer with D2 lymph node dissection: the ARTIST trial. J Clin Oncol 2012;30:268–73
- Park SH, Sohn TS, Lee J, et al. Phase III trial to compare adjuvant chemotherapy with capecitabine and cisplatin versus concurrent chemoradiotherapy in gastric cancer: final report of the adjuvant chemoradiotherapy in stomach tumors trial, including survival and subset analyses. J Clin Oncol 2015;33:3130–6.
- Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med 2007;357:1810–20.
- Sasako M, Sakuramoto S, Katai H, et al. Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol 2011;29:4387–93.
- Bang YJ, Kim YW, Yang HK, et al. Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet 2012;379:315–21.
- Noh SH, Park SR, Yang HK, et al. Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:1389–96.
- Paoletti X, Oba K, Burzykowski T, et al. Benefit of adjuvant chemotherapy for resectable gastric cancer: a meta-analysis. JAMA 2010; 303:1729–37.
- Dai Q, Jiang L, Lin RJ, et al. Adjuvant chemoradiotherapy versus chemotherapy for gastric cancer: a meta-analysis of randomized controlled trials. J Surg Oncol 2015;111:277–84.
- Zhou M, Kang M, Li G, et al. Postoperative chemoradiotherapy versus chemotherapy for R0 resected gastric cancer with D2 lymph node dissection: an up-to-date meta-analysis. World J Surg Oncol 2016;14:209.
- Yang Y, Yin X, Sheng L, et al. Perioperative chemotherapy more of a benefit for overall survival than adjuvant chemotherapy for operable gastric cancer: an updated meta-analysis. Sci Rep 2015;5:12850.
- Zhao JH, Gao P, Song YX, et al. Which is better for gastric cancer patients, perioperative or adjuvant chemotherapy: a meta-analysis. BMC Cancer 2016;16:631.
- Narsule CK, Montgomery MM, and Fernando HC. Evidence-based review of the management of cancers of the gastroesophageal junction. Thorac Surg Clin 2012;22:109–21.
- van Hagen P, Hulshof MCCM, van Lanschot JJB, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Eng J Med 2012;266:2074–84.
- Cunningham D. Chemotherapy with or without bevacizumab or lapatinib to treat operable oesophagogastric cancer (ST03). ClinicalTrials.gov. https://clinicaltrials.gov/show/NCT00450203. NLM Identifier: NCT00450203. Accessed December 14, 2016.
- Leong T, Smithers BM, Michael M, et al. TOPGEAR: a randomised phase III trial of perioperative ECF chemotherapy versus preoperative chemoradiation plus perioperative ECF chemotherapy for resectable gastric cancer (an international, intergroup trial of the AGITG/TROG/EORTC/NCIC CTG). BMC Cancer 2015;15:532.
- Docetaxel+oxaliplatin+S-1 (DOS) regimen as neoadjuvant chemotherapy in advanced gastric cancer (PRODIGY). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01515748 NLM. Identifier: NCT01515748. Accessed December 14, 2016.
- Verheij M, Jansen EP, Cats A, et al. A multicenter randomized phase III trial of neo-adjuvant chemotherapy followed by surgery and chemotherapy or by surgery and chemoradiotherapy in resectable gastric cancer: First results from the CRITICS study. J Clin Oncol 2016;34 (suppl). Abstract 4000.
- Kang WK. Phase III randomized trial of adjuvant chemotherapy with S-1 vs S-1/oxaliplatin ± radiotherapy for completely resected gastric adenocarcinoma : The ARTIST II Trial (ARTIST-II). ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01761461. NLM Identifier: NCT01761461. Accessed December 14, 2016.
- Trial of adjuvant XELOX chemotherapy and concurrent capecitabine and radiotherapy for resected gastric carcinoma. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01711242. NLM Identifier: NCT01711242. Accessed December 14, 2016.
- Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97.
- Roche HL. A Study of the combination of oxaliplatin, capecitabine and herceptin (trastuzumab) and chemoradiotherapy in the adjuvant setting in operated patients with HER2+ gastric or gastro-esophageal junction cancer (TOXAG Study). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01748773. NLM Identifer: NCT01748773. Accessed December 14, 2016.
- A study of capecitabine [Xeloda] in combination with trastuzumab [herceptin] and oxaliplatine in patients with resectable gastric cancer. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01130337. NLM Identifier: NCT01130337. Accessed December 14, 2016.
Hemophilia A and B: An Overview
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).

MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
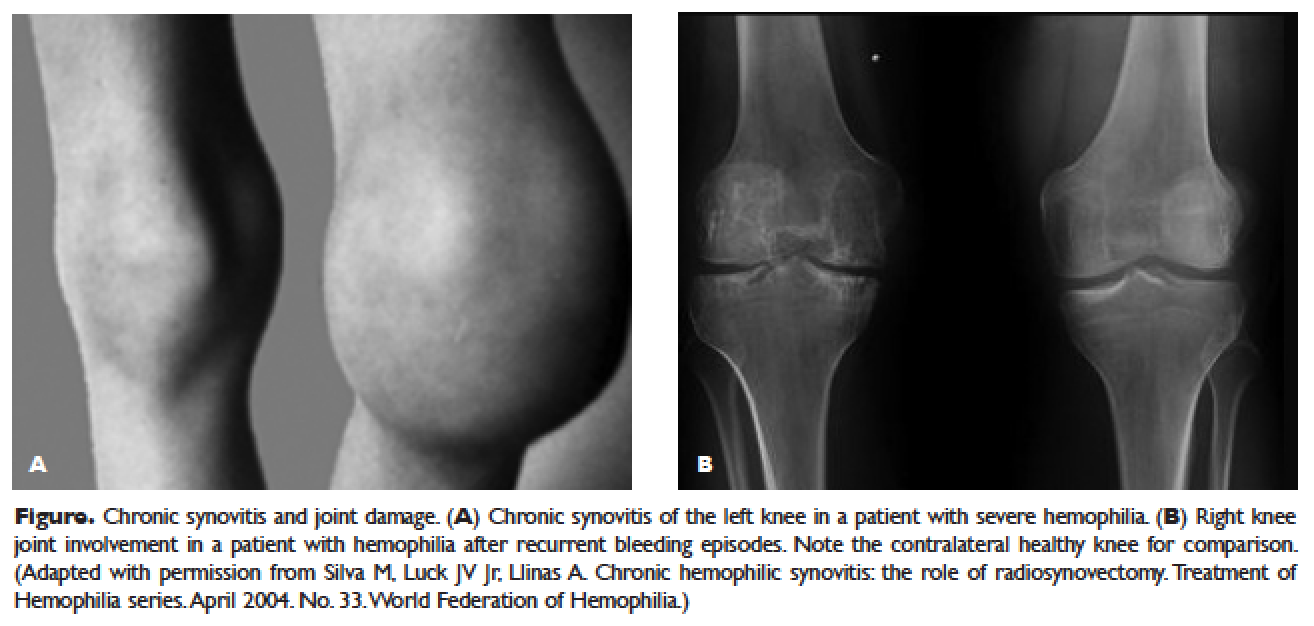
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
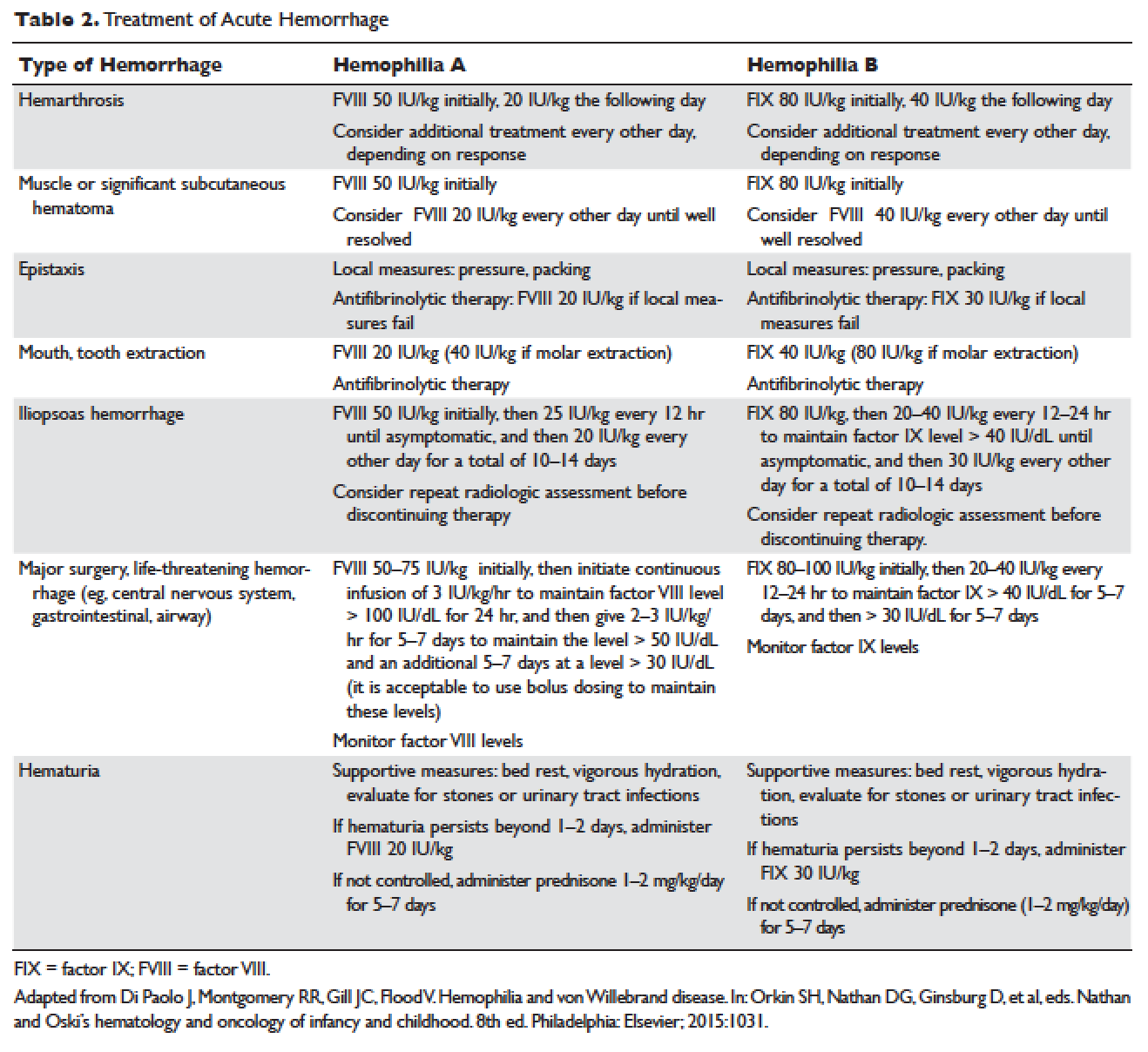
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
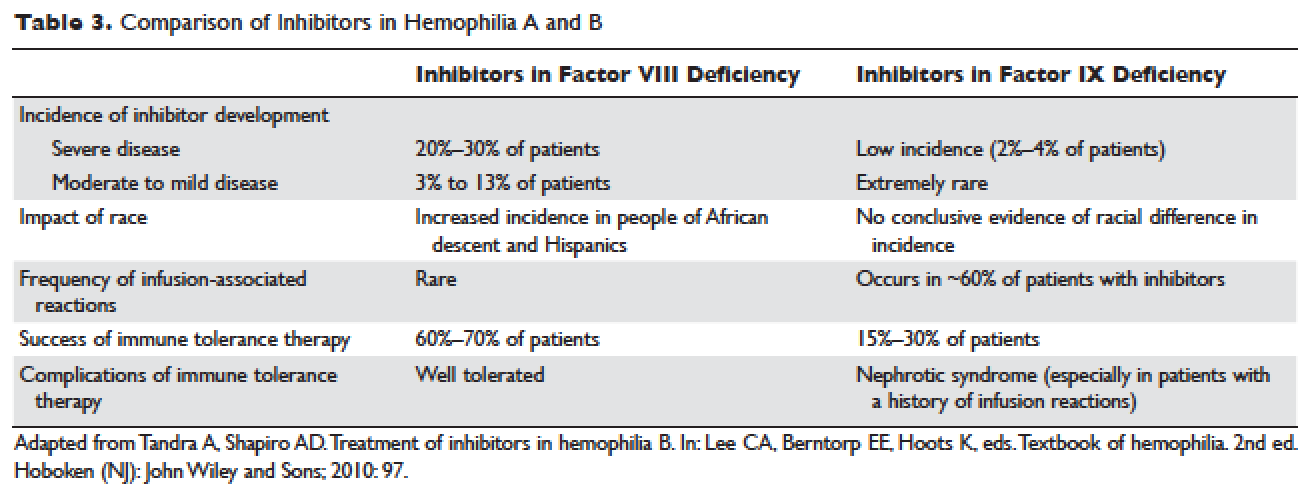
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.

Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82

ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).
MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.
Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82
ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).
MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.
Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82
ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.