User login
Drug gets fast track designation for MF

Credit: Peter Anderson
The US Food and Drug Administration (FDA) is expediting its review of pacritinib, a tyrosine kinase inhibitor with activity against JAK2 and FLT3, by granting the drug fast track designation.
Pacritinib is under review as a treatment for patients with intermediate- and high-risk myelofibrosis (MF), including those with disease-related or treatment-induced thrombocytopenia and those who cannot tolerate or do not respond well to other JAK2 therapy.
The FDA’s fast track process is designed to expedite the review of drugs to treat serious conditions and fill an unmet medical need.
The program enables a company—in this case, CTI BioPharma—to submit sections of a new drug application on a rolling basis as data becomes available.
That way, the FDA can review sections of the application as they are received, rather than waiting until every section of the application is completed before the entire application can be reviewed. This often leads to faster approval.
Pacritinib is currently under investigation in two phase 3 clinical trials, known as the PERSIST program, for patients with MF.
One of these trials, known as PERSIST-1, includes a broad set of patients without limitations on platelet counts. The other, PERSIST-2, includes patients with low platelet counts.
PERSIST-1
In July 2014, CTI Biopharma completed enrollment in the PERSIST-1 trial, which was designed to enroll approximately 320 patients.
This randomized trial was designed to compared the efficacy and safety of pacritinib with that of best available therapy, other than JAK inhibitors, in patients with primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF, without exclusion for low platelet counts.
The primary endpoint is the percentage of patients achieving at least a 35% reduction in spleen volume, measured by MRI or CT from baseline to 24 weeks of treatment.
PERSIST-2
In March 2014, CTI announced the initiation of the PERSIST-2 trial, a comparison of pacritinib and best available therapy, including approved JAK2 inhibitors that are dosed according to product label, in patients with MF whose platelet counts are 100,000/uL or lower.
The trial is designed to enroll up to 300 patients in North America, Europe, Australia, and New Zealand. In October 2013, CTI reached agreement with the FDA on a special protocol assessment for the trial, a written agreement between CTI and the FDA regarding the planned design, endpoints, and statistical analysis approach of the trial to be used in support of a potential new drug application.
Under the special protocol assessment, the trial will have two primary endpoints. The first is the percentage of patients achieving a 35% or greater reduction in spleen volume, measured by MRI or CT scan from baseline to 24 weeks of treatment.
The second primary endpoint is the percentage of patients achieving a total symptom score reduction of 50% or greater using 6 key symptoms, as measured by the modified Myeloproliferative Neoplasm Symptom Assessment (MPN-SAF TSS 2.0) diary from baseline to 24 weeks.
More details on the PERSIST-1 and PERSIST-2 trials can be found at www.clinicaltrials.gov. ![]()
Credit: Peter Anderson
The US Food and Drug Administration (FDA) is expediting its review of pacritinib, a tyrosine kinase inhibitor with activity against JAK2 and FLT3, by granting the drug fast track designation.
Pacritinib is under review as a treatment for patients with intermediate- and high-risk myelofibrosis (MF), including those with disease-related or treatment-induced thrombocytopenia and those who cannot tolerate or do not respond well to other JAK2 therapy.
The FDA’s fast track process is designed to expedite the review of drugs to treat serious conditions and fill an unmet medical need.
The program enables a company—in this case, CTI BioPharma—to submit sections of a new drug application on a rolling basis as data becomes available.
That way, the FDA can review sections of the application as they are received, rather than waiting until every section of the application is completed before the entire application can be reviewed. This often leads to faster approval.
Pacritinib is currently under investigation in two phase 3 clinical trials, known as the PERSIST program, for patients with MF.
One of these trials, known as PERSIST-1, includes a broad set of patients without limitations on platelet counts. The other, PERSIST-2, includes patients with low platelet counts.
PERSIST-1
In July 2014, CTI Biopharma completed enrollment in the PERSIST-1 trial, which was designed to enroll approximately 320 patients.
This randomized trial was designed to compared the efficacy and safety of pacritinib with that of best available therapy, other than JAK inhibitors, in patients with primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF, without exclusion for low platelet counts.
The primary endpoint is the percentage of patients achieving at least a 35% reduction in spleen volume, measured by MRI or CT from baseline to 24 weeks of treatment.
PERSIST-2
In March 2014, CTI announced the initiation of the PERSIST-2 trial, a comparison of pacritinib and best available therapy, including approved JAK2 inhibitors that are dosed according to product label, in patients with MF whose platelet counts are 100,000/uL or lower.
The trial is designed to enroll up to 300 patients in North America, Europe, Australia, and New Zealand. In October 2013, CTI reached agreement with the FDA on a special protocol assessment for the trial, a written agreement between CTI and the FDA regarding the planned design, endpoints, and statistical analysis approach of the trial to be used in support of a potential new drug application.
Under the special protocol assessment, the trial will have two primary endpoints. The first is the percentage of patients achieving a 35% or greater reduction in spleen volume, measured by MRI or CT scan from baseline to 24 weeks of treatment.
The second primary endpoint is the percentage of patients achieving a total symptom score reduction of 50% or greater using 6 key symptoms, as measured by the modified Myeloproliferative Neoplasm Symptom Assessment (MPN-SAF TSS 2.0) diary from baseline to 24 weeks.
More details on the PERSIST-1 and PERSIST-2 trials can be found at www.clinicaltrials.gov. ![]()
Credit: Peter Anderson
The US Food and Drug Administration (FDA) is expediting its review of pacritinib, a tyrosine kinase inhibitor with activity against JAK2 and FLT3, by granting the drug fast track designation.
Pacritinib is under review as a treatment for patients with intermediate- and high-risk myelofibrosis (MF), including those with disease-related or treatment-induced thrombocytopenia and those who cannot tolerate or do not respond well to other JAK2 therapy.
The FDA’s fast track process is designed to expedite the review of drugs to treat serious conditions and fill an unmet medical need.
The program enables a company—in this case, CTI BioPharma—to submit sections of a new drug application on a rolling basis as data becomes available.
That way, the FDA can review sections of the application as they are received, rather than waiting until every section of the application is completed before the entire application can be reviewed. This often leads to faster approval.
Pacritinib is currently under investigation in two phase 3 clinical trials, known as the PERSIST program, for patients with MF.
One of these trials, known as PERSIST-1, includes a broad set of patients without limitations on platelet counts. The other, PERSIST-2, includes patients with low platelet counts.
PERSIST-1
In July 2014, CTI Biopharma completed enrollment in the PERSIST-1 trial, which was designed to enroll approximately 320 patients.
This randomized trial was designed to compared the efficacy and safety of pacritinib with that of best available therapy, other than JAK inhibitors, in patients with primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF, without exclusion for low platelet counts.
The primary endpoint is the percentage of patients achieving at least a 35% reduction in spleen volume, measured by MRI or CT from baseline to 24 weeks of treatment.
PERSIST-2
In March 2014, CTI announced the initiation of the PERSIST-2 trial, a comparison of pacritinib and best available therapy, including approved JAK2 inhibitors that are dosed according to product label, in patients with MF whose platelet counts are 100,000/uL or lower.
The trial is designed to enroll up to 300 patients in North America, Europe, Australia, and New Zealand. In October 2013, CTI reached agreement with the FDA on a special protocol assessment for the trial, a written agreement between CTI and the FDA regarding the planned design, endpoints, and statistical analysis approach of the trial to be used in support of a potential new drug application.
Under the special protocol assessment, the trial will have two primary endpoints. The first is the percentage of patients achieving a 35% or greater reduction in spleen volume, measured by MRI or CT scan from baseline to 24 weeks of treatment.
The second primary endpoint is the percentage of patients achieving a total symptom score reduction of 50% or greater using 6 key symptoms, as measured by the modified Myeloproliferative Neoplasm Symptom Assessment (MPN-SAF TSS 2.0) diary from baseline to 24 weeks.
More details on the PERSIST-1 and PERSIST-2 trials can be found at www.clinicaltrials.gov. ![]()
Drugs can increase risk of MDS and AML
A class of immunosuppressive agents appear to increase the risk of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) in patients with inflammatory bowel disease (IBD).
In an observational study of more than 19,000 IBD patients, past exposure to the agents—thiopurines—increased the risk of developing AML or MDS nearly 7-fold, when compared to the general population.
However, the absolute risk to an individual patient was about 1 in 10,000.
The researchers reported these results in Clinical Gastroenterology and Hepatology.
Thiopurines are an established treatment for IBD patients, but the drugs are also used to prevent rejection after a kidney transplant, to treat rheumatoid arthritis, as maintenance therapy for acute lymphocytic leukemia, and to induce remission in patients with AML.
Previous research showed that long-term use of thiopurines can increase a person’s risk of developing lymphoma.
“In order to make appropriate, informed decisions about thiopurines, patients and providers need to be well-educated about the risks and benefits of this treatment,” said study author Laurent Peyrin-Biroulet, MD, PhD, of the University Hospital of Nancy-Brabois in France.
“According to our research, the risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. However, it was increased amongst those taking thiopurines. We hope these findings encourage other researchers to investigate more about the drug and its potentially harmful effects.”
The researchers analyzed 19,486 patients who were enrolled in the Cancers Et Surrisque Associé aux Maladies inflammatoires intestinales En France study from May 2004 through June 2005.
At study entry, 10,810 patients had never received thiopurines, 2810 patients had discontinued such drugs, and 5866 patients were still receiving them.
After 3 years of follow up, 5 patients were diagnosed with incident myeloid disorders—2 with AML and 3 with MDS. Four of these patients had been exposed to thiopurines—1 with ongoing treatment and 3 with past exposure.
The risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. The standardized incidence ratio (SIR) was 1.80.
Similarly, the risk of myeloid disorders was not increased among IBD patients still receiving thiopurine treatment. The SIR was 1.54.
However, patients with prior exposure to thiopurines did have a significantly increased risk of myeloid disorders, with an SIR of 6.98.
The researchers noted that, although these findings provide evidence of a connection between thiopurines and myeloid disorders in IBD patients, the absolute risk to an individual patient was low.
So it seems the link between thiopurines and myeloid disorders remains complex. And physicians must balance the risk against the known benefits of thiopurines in the management of IBD.
The American Gastroenterological Association has developed a guideline-based clinical decision support tool to help providers determine when to use thiopurines in these patients. ![]()
A class of immunosuppressive agents appear to increase the risk of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) in patients with inflammatory bowel disease (IBD).
In an observational study of more than 19,000 IBD patients, past exposure to the agents—thiopurines—increased the risk of developing AML or MDS nearly 7-fold, when compared to the general population.
However, the absolute risk to an individual patient was about 1 in 10,000.
The researchers reported these results in Clinical Gastroenterology and Hepatology.
Thiopurines are an established treatment for IBD patients, but the drugs are also used to prevent rejection after a kidney transplant, to treat rheumatoid arthritis, as maintenance therapy for acute lymphocytic leukemia, and to induce remission in patients with AML.
Previous research showed that long-term use of thiopurines can increase a person’s risk of developing lymphoma.
“In order to make appropriate, informed decisions about thiopurines, patients and providers need to be well-educated about the risks and benefits of this treatment,” said study author Laurent Peyrin-Biroulet, MD, PhD, of the University Hospital of Nancy-Brabois in France.
“According to our research, the risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. However, it was increased amongst those taking thiopurines. We hope these findings encourage other researchers to investigate more about the drug and its potentially harmful effects.”
The researchers analyzed 19,486 patients who were enrolled in the Cancers Et Surrisque Associé aux Maladies inflammatoires intestinales En France study from May 2004 through June 2005.
At study entry, 10,810 patients had never received thiopurines, 2810 patients had discontinued such drugs, and 5866 patients were still receiving them.
After 3 years of follow up, 5 patients were diagnosed with incident myeloid disorders—2 with AML and 3 with MDS. Four of these patients had been exposed to thiopurines—1 with ongoing treatment and 3 with past exposure.
The risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. The standardized incidence ratio (SIR) was 1.80.
Similarly, the risk of myeloid disorders was not increased among IBD patients still receiving thiopurine treatment. The SIR was 1.54.
However, patients with prior exposure to thiopurines did have a significantly increased risk of myeloid disorders, with an SIR of 6.98.
The researchers noted that, although these findings provide evidence of a connection between thiopurines and myeloid disorders in IBD patients, the absolute risk to an individual patient was low.
So it seems the link between thiopurines and myeloid disorders remains complex. And physicians must balance the risk against the known benefits of thiopurines in the management of IBD.
The American Gastroenterological Association has developed a guideline-based clinical decision support tool to help providers determine when to use thiopurines in these patients. ![]()
A class of immunosuppressive agents appear to increase the risk of acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) in patients with inflammatory bowel disease (IBD).
In an observational study of more than 19,000 IBD patients, past exposure to the agents—thiopurines—increased the risk of developing AML or MDS nearly 7-fold, when compared to the general population.
However, the absolute risk to an individual patient was about 1 in 10,000.
The researchers reported these results in Clinical Gastroenterology and Hepatology.
Thiopurines are an established treatment for IBD patients, but the drugs are also used to prevent rejection after a kidney transplant, to treat rheumatoid arthritis, as maintenance therapy for acute lymphocytic leukemia, and to induce remission in patients with AML.
Previous research showed that long-term use of thiopurines can increase a person’s risk of developing lymphoma.
“In order to make appropriate, informed decisions about thiopurines, patients and providers need to be well-educated about the risks and benefits of this treatment,” said study author Laurent Peyrin-Biroulet, MD, PhD, of the University Hospital of Nancy-Brabois in France.
“According to our research, the risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. However, it was increased amongst those taking thiopurines. We hope these findings encourage other researchers to investigate more about the drug and its potentially harmful effects.”
The researchers analyzed 19,486 patients who were enrolled in the Cancers Et Surrisque Associé aux Maladies inflammatoires intestinales En France study from May 2004 through June 2005.
At study entry, 10,810 patients had never received thiopurines, 2810 patients had discontinued such drugs, and 5866 patients were still receiving them.
After 3 years of follow up, 5 patients were diagnosed with incident myeloid disorders—2 with AML and 3 with MDS. Four of these patients had been exposed to thiopurines—1 with ongoing treatment and 3 with past exposure.
The risk of myeloid disorders was not increased among the overall IBD population, compared with the general population. The standardized incidence ratio (SIR) was 1.80.
Similarly, the risk of myeloid disorders was not increased among IBD patients still receiving thiopurine treatment. The SIR was 1.54.
However, patients with prior exposure to thiopurines did have a significantly increased risk of myeloid disorders, with an SIR of 6.98.
The researchers noted that, although these findings provide evidence of a connection between thiopurines and myeloid disorders in IBD patients, the absolute risk to an individual patient was low.
So it seems the link between thiopurines and myeloid disorders remains complex. And physicians must balance the risk against the known benefits of thiopurines in the management of IBD.
The American Gastroenterological Association has developed a guideline-based clinical decision support tool to help providers determine when to use thiopurines in these patients. ![]()
CHMP recommends antifungal agent
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for intravenous (IV) posaconazole (Noxafil), an antifungal agent.
If the European Commission affirms the CHMP opinion, IV posaconazole will be authorized for use in the European Union, Iceland, Liechtenstein, and Norway.
The commission previously granted marketing authorization for posaconazole delayed-release tablets and oral suspension.
Posaconazole is used to prevent invasive fungal infections in severely immunocompromised patients, such as hematopoietic stem cell transplant recipients with graft-vs-host disease or patients with hematologic malignancies and prolonged neutropenia from chemotherapy.
The drug is also used to treat fungal diseases—invasive aspergillosis, fusariosis, chromoblastomycosis, mycetoma, and coccidioidomycosis—when other antifungal agents—amphotericin B, itraconazole, or fluconazole—cannot be tolerated or have failed.
And posaconazole oral suspension is used as a first-line treatment for thrush, a fungal infection of the mouth and throat due to Candida.
Posaconazole injection is administered with a loading dose of 300 mg twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by IV infusion over approximately 90 minutes.
Once combined with a mixture of IV solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2°-8° C (36°-46° F).
The safety and effectiveness of IV posaconazole in patients younger than 18 years has not been established. IV posaconazole should not be used in pediatric patients because of non-clinical safety concerns.
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
Patients with known hypersensitivity to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
Hepatic reactions have been reported as well. This includes mild to moderate elevations in ALT, AST, alkaline phosphatase, total bilirubin, and/or clinical hepatitis. Monitoring or discontinuation may be necessary in patients with hepatic reactions to posaconazole.
IV posaconazole should be avoided in patients with moderate or severe renal impairment (estimated glomerular filtration rate <50 mL/min), unless an assessment of the benefit/risk to the patient justifies the use of posaconazole.
In clinical trials, the adverse events associated with IV posaconazole were generally similar to those in trials of posaconazole oral suspension. The most frequently reported events were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
IV posaconazole is under development by MSD (known as Merck in the US and Canada). ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for intravenous (IV) posaconazole (Noxafil), an antifungal agent.
If the European Commission affirms the CHMP opinion, IV posaconazole will be authorized for use in the European Union, Iceland, Liechtenstein, and Norway.
The commission previously granted marketing authorization for posaconazole delayed-release tablets and oral suspension.
Posaconazole is used to prevent invasive fungal infections in severely immunocompromised patients, such as hematopoietic stem cell transplant recipients with graft-vs-host disease or patients with hematologic malignancies and prolonged neutropenia from chemotherapy.
The drug is also used to treat fungal diseases—invasive aspergillosis, fusariosis, chromoblastomycosis, mycetoma, and coccidioidomycosis—when other antifungal agents—amphotericin B, itraconazole, or fluconazole—cannot be tolerated or have failed.
And posaconazole oral suspension is used as a first-line treatment for thrush, a fungal infection of the mouth and throat due to Candida.
Posaconazole injection is administered with a loading dose of 300 mg twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by IV infusion over approximately 90 minutes.
Once combined with a mixture of IV solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2°-8° C (36°-46° F).
The safety and effectiveness of IV posaconazole in patients younger than 18 years has not been established. IV posaconazole should not be used in pediatric patients because of non-clinical safety concerns.
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
Patients with known hypersensitivity to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
Hepatic reactions have been reported as well. This includes mild to moderate elevations in ALT, AST, alkaline phosphatase, total bilirubin, and/or clinical hepatitis. Monitoring or discontinuation may be necessary in patients with hepatic reactions to posaconazole.
IV posaconazole should be avoided in patients with moderate or severe renal impairment (estimated glomerular filtration rate <50 mL/min), unless an assessment of the benefit/risk to the patient justifies the use of posaconazole.
In clinical trials, the adverse events associated with IV posaconazole were generally similar to those in trials of posaconazole oral suspension. The most frequently reported events were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
IV posaconazole is under development by MSD (known as Merck in the US and Canada). ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for intravenous (IV) posaconazole (Noxafil), an antifungal agent.
If the European Commission affirms the CHMP opinion, IV posaconazole will be authorized for use in the European Union, Iceland, Liechtenstein, and Norway.
The commission previously granted marketing authorization for posaconazole delayed-release tablets and oral suspension.
Posaconazole is used to prevent invasive fungal infections in severely immunocompromised patients, such as hematopoietic stem cell transplant recipients with graft-vs-host disease or patients with hematologic malignancies and prolonged neutropenia from chemotherapy.
The drug is also used to treat fungal diseases—invasive aspergillosis, fusariosis, chromoblastomycosis, mycetoma, and coccidioidomycosis—when other antifungal agents—amphotericin B, itraconazole, or fluconazole—cannot be tolerated or have failed.
And posaconazole oral suspension is used as a first-line treatment for thrush, a fungal infection of the mouth and throat due to Candida.
Posaconazole injection is administered with a loading dose of 300 mg twice a day on the first day of therapy, then 300 mg once a day thereafter. It is given through a central venous line by IV infusion over approximately 90 minutes.
Once combined with a mixture of IV solution (150 mL of 5% dextrose in water or sodium chloride 0.9%), posaconazole should be administered immediately. If not used immediately, the solution can be stored up to 24 hours if refrigerated at 2°-8° C (36°-46° F).
The safety and effectiveness of IV posaconazole in patients younger than 18 years has not been established. IV posaconazole should not be used in pediatric patients because of non-clinical safety concerns.
Co-administration of drugs that can decrease the plasma concentration of posaconazole should be avoided unless the benefit outweighs the risk. If such drugs are necessary, patients should be monitored closely for breakthrough fungal infections.
Patients with known hypersensitivity to posaconazole or other azole antifungal medicines should not receive posaconazole. The drug should not be given with sirolimus, pimozide, quinidine, atorvastatin, lovastatin, simvastatin, or ergot alkaloids.
Drugs such as cyclosporine and tacrolimus require dose adjustments and frequent blood monitoring when administered with posaconazole. Serious side effects, including nephrotoxicity, leukoencephalopathy, and death, have been reported in patients with increased cyclosporine or tacrolimus blood levels.
Healthcare professionals should use caution when administering posaconazole to patients at risk of developing an irregular heart rhythm, as the drug has been shown to prolong the QT interval, and cases of potentially fatal irregular heart rhythm (torsades de pointes) have been reported in patients taking posaconazole.
Hepatic reactions have been reported as well. This includes mild to moderate elevations in ALT, AST, alkaline phosphatase, total bilirubin, and/or clinical hepatitis. Monitoring or discontinuation may be necessary in patients with hepatic reactions to posaconazole.
IV posaconazole should be avoided in patients with moderate or severe renal impairment (estimated glomerular filtration rate <50 mL/min), unless an assessment of the benefit/risk to the patient justifies the use of posaconazole.
In clinical trials, the adverse events associated with IV posaconazole were generally similar to those in trials of posaconazole oral suspension. The most frequently reported events were diarrhea (32%), hypokalemia (22%), fever (21%), and nausea (19%).
IV posaconazole is under development by MSD (known as Merck in the US and Canada). ![]()
Method corrects β-thalassemia mutations

Credit: Salk Institute
Genome editing technology allows for seamless correction of disease-causing mutations in cells from patients with β-thalassemia, investigators have reported in Genome Research.
The team noted that β-thalassemia results from inherited mutations in the hemoglobin beta (HBB) gene, which prompt reduced HBB expression in red blood cells, as well as anemia.
The only established curative treatment is hematopoietic stem cell transplant, but this requires a matched donor.
Gene therapy could eliminate this need.
To correct HBB mutations directly in a patient’s genome, Yuet Wai Kan, MD, of the University of California, San Francisco, and his colleagues first generated induced pluripotent stem cells (iPSCs) from patients’ skin cells.
The team then used CRISPR/Cas9 technology to precisely engineer a double-strand DNA break at the HBB locus in the iPSCs, allowing a donor plasmid with the corrected sites to be efficiently integrated, thus replacing the mutated sites.
The donor plasmid also contained selectable markers to identify cells with corrected copies of the gene. These selectable markers were subsequently removed with transposase and a second round of selection, generating a seamless, corrected version of HBB in the patient’s genome.
The investigators found the corrected iPSCs could differentiate into mature blood cells, and these blood cells showed restored expression of hemoglobin.
However, the team said a lot more work is needed before these cells could be transplanted to treat a patient with β-thalassemia.
“Although we and others are able to differentiate iPSCs into blood cell progenitors as well as mature blood cells, the transplantation of the progenitors into mouse models to test them has, so far, proven very difficult,” Dr Kan said. “I believe it will take quite a few more years before we can apply it in a clinical setting.” ![]()
Credit: Salk Institute
Genome editing technology allows for seamless correction of disease-causing mutations in cells from patients with β-thalassemia, investigators have reported in Genome Research.
The team noted that β-thalassemia results from inherited mutations in the hemoglobin beta (HBB) gene, which prompt reduced HBB expression in red blood cells, as well as anemia.
The only established curative treatment is hematopoietic stem cell transplant, but this requires a matched donor.
Gene therapy could eliminate this need.
To correct HBB mutations directly in a patient’s genome, Yuet Wai Kan, MD, of the University of California, San Francisco, and his colleagues first generated induced pluripotent stem cells (iPSCs) from patients’ skin cells.
The team then used CRISPR/Cas9 technology to precisely engineer a double-strand DNA break at the HBB locus in the iPSCs, allowing a donor plasmid with the corrected sites to be efficiently integrated, thus replacing the mutated sites.
The donor plasmid also contained selectable markers to identify cells with corrected copies of the gene. These selectable markers were subsequently removed with transposase and a second round of selection, generating a seamless, corrected version of HBB in the patient’s genome.
The investigators found the corrected iPSCs could differentiate into mature blood cells, and these blood cells showed restored expression of hemoglobin.
However, the team said a lot more work is needed before these cells could be transplanted to treat a patient with β-thalassemia.
“Although we and others are able to differentiate iPSCs into blood cell progenitors as well as mature blood cells, the transplantation of the progenitors into mouse models to test them has, so far, proven very difficult,” Dr Kan said. “I believe it will take quite a few more years before we can apply it in a clinical setting.” ![]()
Credit: Salk Institute
Genome editing technology allows for seamless correction of disease-causing mutations in cells from patients with β-thalassemia, investigators have reported in Genome Research.
The team noted that β-thalassemia results from inherited mutations in the hemoglobin beta (HBB) gene, which prompt reduced HBB expression in red blood cells, as well as anemia.
The only established curative treatment is hematopoietic stem cell transplant, but this requires a matched donor.
Gene therapy could eliminate this need.
To correct HBB mutations directly in a patient’s genome, Yuet Wai Kan, MD, of the University of California, San Francisco, and his colleagues first generated induced pluripotent stem cells (iPSCs) from patients’ skin cells.
The team then used CRISPR/Cas9 technology to precisely engineer a double-strand DNA break at the HBB locus in the iPSCs, allowing a donor plasmid with the corrected sites to be efficiently integrated, thus replacing the mutated sites.
The donor plasmid also contained selectable markers to identify cells with corrected copies of the gene. These selectable markers were subsequently removed with transposase and a second round of selection, generating a seamless, corrected version of HBB in the patient’s genome.
The investigators found the corrected iPSCs could differentiate into mature blood cells, and these blood cells showed restored expression of hemoglobin.
However, the team said a lot more work is needed before these cells could be transplanted to treat a patient with β-thalassemia.
“Although we and others are able to differentiate iPSCs into blood cell progenitors as well as mature blood cells, the transplantation of the progenitors into mouse models to test them has, so far, proven very difficult,” Dr Kan said. “I believe it will take quite a few more years before we can apply it in a clinical setting.” ![]()
Study reveals why HSCs falter with age

in the bone marrow
A new study helps explain how blood production declines with age and why older individuals are not suitable donors for hematopoietic stem cell (HSC) transplant.
The research also reveals a potential approach for mitigating
the negative effects of aging on the blood, which can lead to anemia,
bone marrow failure, and myeloid malignancies.
The study, conducted in mice, suggests HSCs falter with age because they lose the ability to replicate their DNA accurately and efficiently during cell division.
Emmanuelle Passegué, PhD, of the University of California San Francisco, and her colleagues reported this discovery in Nature.
The researchers analyzed old HSCs in mice and found a scarcity of protein components needed to form the mini-chromosome maintenance helicase. This molecular machine unwinds double-stranded DNA so the cell’s genetic material can be duplicated and allocated to daughter cells later in cell division.
The HSCs were stressed by the loss of this machine’s activity. As a result, they had an increased risk for DNA damage and death when forced to divide.
On the other hand, the cells tended to survive unless they were confronted with a “strong replication challenge” like transplantation.
The researchers also discovered that even after the stress associated with DNA replication, old HSCs retained molecular tags on histones, a feature often associated with DNA damage.
However, these old survivors could repair induced DNA damage as efficiently as young stem cells.
“Old stem cells are not just sitting there with damaged DNA ready to develop cancer, as it has long been postulated,” Dr Passegué said.
Of course, not all was well in the old, surviving HSCs. The molecular tags accumulated on genes needed to make ribosomes.
Dr Passegué said she will further explore the consequences of reduced protein production as part of her ongoing research. She hopes it might be possible to prevent declining stem cell populations by developing a drug to prevent the loss of the helicase components needed to unwind and replicate DNA, thereby avoiding immune system failure. ![]()
in the bone marrow
A new study helps explain how blood production declines with age and why older individuals are not suitable donors for hematopoietic stem cell (HSC) transplant.
The research also reveals a potential approach for mitigating
the negative effects of aging on the blood, which can lead to anemia,
bone marrow failure, and myeloid malignancies.
The study, conducted in mice, suggests HSCs falter with age because they lose the ability to replicate their DNA accurately and efficiently during cell division.
Emmanuelle Passegué, PhD, of the University of California San Francisco, and her colleagues reported this discovery in Nature.
The researchers analyzed old HSCs in mice and found a scarcity of protein components needed to form the mini-chromosome maintenance helicase. This molecular machine unwinds double-stranded DNA so the cell’s genetic material can be duplicated and allocated to daughter cells later in cell division.
The HSCs were stressed by the loss of this machine’s activity. As a result, they had an increased risk for DNA damage and death when forced to divide.
On the other hand, the cells tended to survive unless they were confronted with a “strong replication challenge” like transplantation.
The researchers also discovered that even after the stress associated with DNA replication, old HSCs retained molecular tags on histones, a feature often associated with DNA damage.
However, these old survivors could repair induced DNA damage as efficiently as young stem cells.
“Old stem cells are not just sitting there with damaged DNA ready to develop cancer, as it has long been postulated,” Dr Passegué said.
Of course, not all was well in the old, surviving HSCs. The molecular tags accumulated on genes needed to make ribosomes.
Dr Passegué said she will further explore the consequences of reduced protein production as part of her ongoing research. She hopes it might be possible to prevent declining stem cell populations by developing a drug to prevent the loss of the helicase components needed to unwind and replicate DNA, thereby avoiding immune system failure. ![]()
in the bone marrow
A new study helps explain how blood production declines with age and why older individuals are not suitable donors for hematopoietic stem cell (HSC) transplant.
The research also reveals a potential approach for mitigating
the negative effects of aging on the blood, which can lead to anemia,
bone marrow failure, and myeloid malignancies.
The study, conducted in mice, suggests HSCs falter with age because they lose the ability to replicate their DNA accurately and efficiently during cell division.
Emmanuelle Passegué, PhD, of the University of California San Francisco, and her colleagues reported this discovery in Nature.
The researchers analyzed old HSCs in mice and found a scarcity of protein components needed to form the mini-chromosome maintenance helicase. This molecular machine unwinds double-stranded DNA so the cell’s genetic material can be duplicated and allocated to daughter cells later in cell division.
The HSCs were stressed by the loss of this machine’s activity. As a result, they had an increased risk for DNA damage and death when forced to divide.
On the other hand, the cells tended to survive unless they were confronted with a “strong replication challenge” like transplantation.
The researchers also discovered that even after the stress associated with DNA replication, old HSCs retained molecular tags on histones, a feature often associated with DNA damage.
However, these old survivors could repair induced DNA damage as efficiently as young stem cells.
“Old stem cells are not just sitting there with damaged DNA ready to develop cancer, as it has long been postulated,” Dr Passegué said.
Of course, not all was well in the old, surviving HSCs. The molecular tags accumulated on genes needed to make ribosomes.
Dr Passegué said she will further explore the consequences of reduced protein production as part of her ongoing research. She hopes it might be possible to prevent declining stem cell populations by developing a drug to prevent the loss of the helicase components needed to unwind and replicate DNA, thereby avoiding immune system failure. ![]()
New insight into thalassemia, sickle cell anemia
Credit: Graham Beards
Researchers have found evidence suggesting that beneficial variants of a gene controlling hematopoiesis exist in nearly all human populations.
The team analyzed genomic data from world populations, looking at HMIP-2, a human quantitative trait locus that affects the production of fetal hemoglobin in adults.
The analysis revealed 2 alleles that promote fetal hemoglobin production and can therefore reduce the severity of thalassemia and sickle cell anemia (SCA).
“Patients who have milder versions of [these] blood disorders, thanks to their ability to keep producing fetal hemoglobin, carry genetic clues that are helping us to understand the function of the genes and biological pathways involved in these diseases,” said Stephan Menzel, MD, of King’s College London in the UK.
He and his colleagues conducted this research and reported the results in Annals of Human Genetics.
The researchers noted that HMIP (HBS1L-MYB intergenic polymorphism) on chromosome 6q23.3 was first detected in a large Asian Indian family, where it was shown to be responsible for the persistence of fetal hemoglobin production in adulthood.
And HMIP-2 occupies a 24-kb stretch of DNA that acts as a distal upstream enhancer for MYB, the gene for cMYB, which is essential to hematopoiesis.
While studying 4 groups of SCA patients of diverse African descent, Dr Menzel and his colleagues discovered 2 alleles at HMIP-2 that promote fetal hemoglobin—HMIP-2A and HMIP2-B.
Subsequent analyses revealed the alleles were present, either alone or together, in major human populations and nearly all of the ethnic groups studied.
Both HMIP-2A and HMIP2-B occur in Sub-Saharan Africa, but only at low frequencies. In much of the rest of the world, the alleles have combined, forming HMIP-2A-B, and this combination is relatively common in Europe, South Asia, and China. HMIP-2B alone is common in Far-East Asian peoples and in Amerindians.
The researchers also analyzed genomic data from Neanderthals, Denisovans, and Great Apes, but detected neither HMIP-2A nor HMIP-2B.
The team said these results suggest MYB enhancer variants that modulate the severity of SCA and thalassemia have arisen twice in modern humans, in Africa, and then spread to the rest of the world.
However, this likely occurred long before inherited blood disorders became prevalent, so the environmental factors that favored such variants in these early humans are not clear.
For the next stage of this research, Dr Menzel and his colleagues plan to explore which selection pressures or benefits might have contributed to the present population distribution of the variants.
Selection pressures could include nutritional factors, such as the availability of iron in the diet, or specific demands on red blood cell production, such as adaptation to high altitudes. ![]()
Credit: Graham Beards
Researchers have found evidence suggesting that beneficial variants of a gene controlling hematopoiesis exist in nearly all human populations.
The team analyzed genomic data from world populations, looking at HMIP-2, a human quantitative trait locus that affects the production of fetal hemoglobin in adults.
The analysis revealed 2 alleles that promote fetal hemoglobin production and can therefore reduce the severity of thalassemia and sickle cell anemia (SCA).
“Patients who have milder versions of [these] blood disorders, thanks to their ability to keep producing fetal hemoglobin, carry genetic clues that are helping us to understand the function of the genes and biological pathways involved in these diseases,” said Stephan Menzel, MD, of King’s College London in the UK.
He and his colleagues conducted this research and reported the results in Annals of Human Genetics.
The researchers noted that HMIP (HBS1L-MYB intergenic polymorphism) on chromosome 6q23.3 was first detected in a large Asian Indian family, where it was shown to be responsible for the persistence of fetal hemoglobin production in adulthood.
And HMIP-2 occupies a 24-kb stretch of DNA that acts as a distal upstream enhancer for MYB, the gene for cMYB, which is essential to hematopoiesis.
While studying 4 groups of SCA patients of diverse African descent, Dr Menzel and his colleagues discovered 2 alleles at HMIP-2 that promote fetal hemoglobin—HMIP-2A and HMIP2-B.
Subsequent analyses revealed the alleles were present, either alone or together, in major human populations and nearly all of the ethnic groups studied.
Both HMIP-2A and HMIP2-B occur in Sub-Saharan Africa, but only at low frequencies. In much of the rest of the world, the alleles have combined, forming HMIP-2A-B, and this combination is relatively common in Europe, South Asia, and China. HMIP-2B alone is common in Far-East Asian peoples and in Amerindians.
The researchers also analyzed genomic data from Neanderthals, Denisovans, and Great Apes, but detected neither HMIP-2A nor HMIP-2B.
The team said these results suggest MYB enhancer variants that modulate the severity of SCA and thalassemia have arisen twice in modern humans, in Africa, and then spread to the rest of the world.
However, this likely occurred long before inherited blood disorders became prevalent, so the environmental factors that favored such variants in these early humans are not clear.
For the next stage of this research, Dr Menzel and his colleagues plan to explore which selection pressures or benefits might have contributed to the present population distribution of the variants.
Selection pressures could include nutritional factors, such as the availability of iron in the diet, or specific demands on red blood cell production, such as adaptation to high altitudes. ![]()
Credit: Graham Beards
Researchers have found evidence suggesting that beneficial variants of a gene controlling hematopoiesis exist in nearly all human populations.
The team analyzed genomic data from world populations, looking at HMIP-2, a human quantitative trait locus that affects the production of fetal hemoglobin in adults.
The analysis revealed 2 alleles that promote fetal hemoglobin production and can therefore reduce the severity of thalassemia and sickle cell anemia (SCA).
“Patients who have milder versions of [these] blood disorders, thanks to their ability to keep producing fetal hemoglobin, carry genetic clues that are helping us to understand the function of the genes and biological pathways involved in these diseases,” said Stephan Menzel, MD, of King’s College London in the UK.
He and his colleagues conducted this research and reported the results in Annals of Human Genetics.
The researchers noted that HMIP (HBS1L-MYB intergenic polymorphism) on chromosome 6q23.3 was first detected in a large Asian Indian family, where it was shown to be responsible for the persistence of fetal hemoglobin production in adulthood.
And HMIP-2 occupies a 24-kb stretch of DNA that acts as a distal upstream enhancer for MYB, the gene for cMYB, which is essential to hematopoiesis.
While studying 4 groups of SCA patients of diverse African descent, Dr Menzel and his colleagues discovered 2 alleles at HMIP-2 that promote fetal hemoglobin—HMIP-2A and HMIP2-B.
Subsequent analyses revealed the alleles were present, either alone or together, in major human populations and nearly all of the ethnic groups studied.
Both HMIP-2A and HMIP2-B occur in Sub-Saharan Africa, but only at low frequencies. In much of the rest of the world, the alleles have combined, forming HMIP-2A-B, and this combination is relatively common in Europe, South Asia, and China. HMIP-2B alone is common in Far-East Asian peoples and in Amerindians.
The researchers also analyzed genomic data from Neanderthals, Denisovans, and Great Apes, but detected neither HMIP-2A nor HMIP-2B.
The team said these results suggest MYB enhancer variants that modulate the severity of SCA and thalassemia have arisen twice in modern humans, in Africa, and then spread to the rest of the world.
However, this likely occurred long before inherited blood disorders became prevalent, so the environmental factors that favored such variants in these early humans are not clear.
For the next stage of this research, Dr Menzel and his colleagues plan to explore which selection pressures or benefits might have contributed to the present population distribution of the variants.
Selection pressures could include nutritional factors, such as the availability of iron in the diet, or specific demands on red blood cell production, such as adaptation to high altitudes. ![]()
Drug could prevent thrombocytopenia in MM
in the bone marrow
Researchers say they’ve identified a previously unknown but crucial component of the platelet production process.
And this discovery could help spare multiple myeloma (MM) patients from thrombocytopenia induced by the proteasome inhibitor bortezomib.
The researchers found that proteasome inhibition blocked platelet production in vitro and in vivo.
But fasudil, a Rho kinase inhibitor that is approved for use outside the US, restored platelet counts.
The researchers believe these findings, published in The Journal of Clinical Investigation, could translate to MM patients.
“A low platelet count is a big issue for people who receive bortezomib for this cancer,” said study author Andrew S. Weyrich, PhD, of the University of Utah in Salt Lake City.
“When platelet levels drop too low, it can mean interrupting treatment to allow the platelet count to recover. Fasudil potentially could help keep platelet counts normal while multiple myeloma patients receive bortezomib.”
Dr Weyrich and his colleagues found that bortezomib-induced proteasome inhibition prevented the production of proplatelets in both human and mouse megakaryocytes.
Megakaryocytes isolated from mice lacking PSMC1, an essential subunit of the 26S proteasome, also failed to produce proplatelets.
Further study revealed that the megakaryocytes’ inability to generate platelets was caused by the hyperactivation of RhoA, a protein that helps megakaryocytes maintain the proper shape to produce platelets.
When the researchers inhibited RhoA or its downstream target, Rho-associated protein kinase, in vitro, they were able to restore megakaryocyte proplatelet formation in the setting of proteasome inhibition.
Likewise, the Rho kinase inhibitor fasudil restored platelet counts in adult mice that had thrombocytopenia induced by proteasome inhibition.
Fasudil is approved in Japan and elsewhere to treat cerebral vasospasms, or constricted arteries that arise as a complication of brain aneurysms.
The drug is under investigation in US clinical trials for treating high blood pressure, diabetic macular edema, and other health issues.
There are no trials investigating fasudil’s effects on thrombocytopenia, but Dr Weyrich and his colleagues hope their study might change that. And if clinical trials produce favorable results, fasudil might be made available for MM patients much faster than a new drug.
“If the Food and Drug Administration did approve fasudil for use by multiple myeloma patients, it could, in principle, be moved to the clinic relatively fast in the United States,” Dr Weyrich said. ![]()
in the bone marrow
Researchers say they’ve identified a previously unknown but crucial component of the platelet production process.
And this discovery could help spare multiple myeloma (MM) patients from thrombocytopenia induced by the proteasome inhibitor bortezomib.
The researchers found that proteasome inhibition blocked platelet production in vitro and in vivo.
But fasudil, a Rho kinase inhibitor that is approved for use outside the US, restored platelet counts.
The researchers believe these findings, published in The Journal of Clinical Investigation, could translate to MM patients.
“A low platelet count is a big issue for people who receive bortezomib for this cancer,” said study author Andrew S. Weyrich, PhD, of the University of Utah in Salt Lake City.
“When platelet levels drop too low, it can mean interrupting treatment to allow the platelet count to recover. Fasudil potentially could help keep platelet counts normal while multiple myeloma patients receive bortezomib.”
Dr Weyrich and his colleagues found that bortezomib-induced proteasome inhibition prevented the production of proplatelets in both human and mouse megakaryocytes.
Megakaryocytes isolated from mice lacking PSMC1, an essential subunit of the 26S proteasome, also failed to produce proplatelets.
Further study revealed that the megakaryocytes’ inability to generate platelets was caused by the hyperactivation of RhoA, a protein that helps megakaryocytes maintain the proper shape to produce platelets.
When the researchers inhibited RhoA or its downstream target, Rho-associated protein kinase, in vitro, they were able to restore megakaryocyte proplatelet formation in the setting of proteasome inhibition.
Likewise, the Rho kinase inhibitor fasudil restored platelet counts in adult mice that had thrombocytopenia induced by proteasome inhibition.
Fasudil is approved in Japan and elsewhere to treat cerebral vasospasms, or constricted arteries that arise as a complication of brain aneurysms.
The drug is under investigation in US clinical trials for treating high blood pressure, diabetic macular edema, and other health issues.
There are no trials investigating fasudil’s effects on thrombocytopenia, but Dr Weyrich and his colleagues hope their study might change that. And if clinical trials produce favorable results, fasudil might be made available for MM patients much faster than a new drug.
“If the Food and Drug Administration did approve fasudil for use by multiple myeloma patients, it could, in principle, be moved to the clinic relatively fast in the United States,” Dr Weyrich said. ![]()
in the bone marrow
Researchers say they’ve identified a previously unknown but crucial component of the platelet production process.
And this discovery could help spare multiple myeloma (MM) patients from thrombocytopenia induced by the proteasome inhibitor bortezomib.
The researchers found that proteasome inhibition blocked platelet production in vitro and in vivo.
But fasudil, a Rho kinase inhibitor that is approved for use outside the US, restored platelet counts.
The researchers believe these findings, published in The Journal of Clinical Investigation, could translate to MM patients.
“A low platelet count is a big issue for people who receive bortezomib for this cancer,” said study author Andrew S. Weyrich, PhD, of the University of Utah in Salt Lake City.
“When platelet levels drop too low, it can mean interrupting treatment to allow the platelet count to recover. Fasudil potentially could help keep platelet counts normal while multiple myeloma patients receive bortezomib.”
Dr Weyrich and his colleagues found that bortezomib-induced proteasome inhibition prevented the production of proplatelets in both human and mouse megakaryocytes.
Megakaryocytes isolated from mice lacking PSMC1, an essential subunit of the 26S proteasome, also failed to produce proplatelets.
Further study revealed that the megakaryocytes’ inability to generate platelets was caused by the hyperactivation of RhoA, a protein that helps megakaryocytes maintain the proper shape to produce platelets.
When the researchers inhibited RhoA or its downstream target, Rho-associated protein kinase, in vitro, they were able to restore megakaryocyte proplatelet formation in the setting of proteasome inhibition.
Likewise, the Rho kinase inhibitor fasudil restored platelet counts in adult mice that had thrombocytopenia induced by proteasome inhibition.
Fasudil is approved in Japan and elsewhere to treat cerebral vasospasms, or constricted arteries that arise as a complication of brain aneurysms.
The drug is under investigation in US clinical trials for treating high blood pressure, diabetic macular edema, and other health issues.
There are no trials investigating fasudil’s effects on thrombocytopenia, but Dr Weyrich and his colleagues hope their study might change that. And if clinical trials produce favorable results, fasudil might be made available for MM patients much faster than a new drug.
“If the Food and Drug Administration did approve fasudil for use by multiple myeloma patients, it could, in principle, be moved to the clinic relatively fast in the United States,” Dr Weyrich said.
Biosimilar can treat chemo-induced anemia

Credit: Rhoda Baer
A biosimilar of the erythropoiesis-stimulating agent epoetin alfa can elicit responses in patients with chemotherapy-induced anemia, according to a study published in BMC Cancer.
The agent, epoetin zeta (Retacrit), produced a hemoglobin (Hb) response in more than 80% of patients at 3- and 6-month time points.
Response rates were similar in patients with hematologic malignancies and those with solid tumors.
And the rate of clinically significant adverse events was low. This included thromboembolic events, bleeding, infection, local intolerability, and increased blood pressure.
Mauricette Michallet, MD, PhD, of Centre Hospitalier Lyon in France, and her colleagues conducted this study, known as ORHEO. It was sponsored by Hospira, the makers of epoetin zeta.
The researchers evaluated 2310 adult patients with chemotherapy-induced anemia (Hb<11 g/dL). Patients had solid tumors (n=1838), lymphomas (n=301), or multiple myeloma (n=171).
Patients were taking a number of treatments aside from epoetin zeta and chemotherapy. This included intravenous iron (10%), oral iron (16%), antithrombotic agents (12%), folates (7%), vitamin B (4%), and other vitamins (2%). An additional 17% of patients were reported as being on “other treatments.”
In all, 99.9% of patients received epoetin zeta. The primary endpoint was the rate of response.
Response was defined as an increase in Hb levels to at least 10 g/dL since enrollment, an increase in Hb levels of at least 1 g/dL since enrollment, or reaching target Hb levels set at the start of study, without any blood transfusions in the 3 weeks prior to measurement. In patients with baseline Hb levels of at least 10 g/dL, only those who reached their Hb target or had an increase greater than 1 g/dL were considered responders.
Eighty-two percent of patients achieved a response at 3 months, and 87% had a response at 6 months. The overall mean change in Hb level was 1.52 ± 1.61 at 3 months and 1.72 ± 1.61 g/dL at 6 months. The rate of transfusion was 9% at 3 months and 6% at 6 months.
Between enrollment and month 6, 1202 patients discontinued epoetin zeta. Forty percent stopped because Hb levels were met, 27% stopped because they were stopping or changing chemotherapy, 15% stopped for both of the aforementioned reasons, 11% stopped because epoetin zeta was ineffective, and 2% stopped due to adverse events.
Seventeen percent of patients experienced an adverse event, including thromboembolic events (4%), infection (5%), bleeding (2%), local intolerability (0.2%), increased blood pressure (2%), and “other” events (9%).
Epoetin zeta was approved in Europe in 2007. For epoetin biosimilars to gain approval in the European Union, companies must agree to conduct post-marketing studies.
Credit: Rhoda Baer
A biosimilar of the erythropoiesis-stimulating agent epoetin alfa can elicit responses in patients with chemotherapy-induced anemia, according to a study published in BMC Cancer.
The agent, epoetin zeta (Retacrit), produced a hemoglobin (Hb) response in more than 80% of patients at 3- and 6-month time points.
Response rates were similar in patients with hematologic malignancies and those with solid tumors.
And the rate of clinically significant adverse events was low. This included thromboembolic events, bleeding, infection, local intolerability, and increased blood pressure.
Mauricette Michallet, MD, PhD, of Centre Hospitalier Lyon in France, and her colleagues conducted this study, known as ORHEO. It was sponsored by Hospira, the makers of epoetin zeta.
The researchers evaluated 2310 adult patients with chemotherapy-induced anemia (Hb<11 g/dL). Patients had solid tumors (n=1838), lymphomas (n=301), or multiple myeloma (n=171).
Patients were taking a number of treatments aside from epoetin zeta and chemotherapy. This included intravenous iron (10%), oral iron (16%), antithrombotic agents (12%), folates (7%), vitamin B (4%), and other vitamins (2%). An additional 17% of patients were reported as being on “other treatments.”
In all, 99.9% of patients received epoetin zeta. The primary endpoint was the rate of response.
Response was defined as an increase in Hb levels to at least 10 g/dL since enrollment, an increase in Hb levels of at least 1 g/dL since enrollment, or reaching target Hb levels set at the start of study, without any blood transfusions in the 3 weeks prior to measurement. In patients with baseline Hb levels of at least 10 g/dL, only those who reached their Hb target or had an increase greater than 1 g/dL were considered responders.
Eighty-two percent of patients achieved a response at 3 months, and 87% had a response at 6 months. The overall mean change in Hb level was 1.52 ± 1.61 at 3 months and 1.72 ± 1.61 g/dL at 6 months. The rate of transfusion was 9% at 3 months and 6% at 6 months.
Between enrollment and month 6, 1202 patients discontinued epoetin zeta. Forty percent stopped because Hb levels were met, 27% stopped because they were stopping or changing chemotherapy, 15% stopped for both of the aforementioned reasons, 11% stopped because epoetin zeta was ineffective, and 2% stopped due to adverse events.
Seventeen percent of patients experienced an adverse event, including thromboembolic events (4%), infection (5%), bleeding (2%), local intolerability (0.2%), increased blood pressure (2%), and “other” events (9%).
Epoetin zeta was approved in Europe in 2007. For epoetin biosimilars to gain approval in the European Union, companies must agree to conduct post-marketing studies.
Credit: Rhoda Baer
A biosimilar of the erythropoiesis-stimulating agent epoetin alfa can elicit responses in patients with chemotherapy-induced anemia, according to a study published in BMC Cancer.
The agent, epoetin zeta (Retacrit), produced a hemoglobin (Hb) response in more than 80% of patients at 3- and 6-month time points.
Response rates were similar in patients with hematologic malignancies and those with solid tumors.
And the rate of clinically significant adverse events was low. This included thromboembolic events, bleeding, infection, local intolerability, and increased blood pressure.
Mauricette Michallet, MD, PhD, of Centre Hospitalier Lyon in France, and her colleagues conducted this study, known as ORHEO. It was sponsored by Hospira, the makers of epoetin zeta.
The researchers evaluated 2310 adult patients with chemotherapy-induced anemia (Hb<11 g/dL). Patients had solid tumors (n=1838), lymphomas (n=301), or multiple myeloma (n=171).
Patients were taking a number of treatments aside from epoetin zeta and chemotherapy. This included intravenous iron (10%), oral iron (16%), antithrombotic agents (12%), folates (7%), vitamin B (4%), and other vitamins (2%). An additional 17% of patients were reported as being on “other treatments.”
In all, 99.9% of patients received epoetin zeta. The primary endpoint was the rate of response.
Response was defined as an increase in Hb levels to at least 10 g/dL since enrollment, an increase in Hb levels of at least 1 g/dL since enrollment, or reaching target Hb levels set at the start of study, without any blood transfusions in the 3 weeks prior to measurement. In patients with baseline Hb levels of at least 10 g/dL, only those who reached their Hb target or had an increase greater than 1 g/dL were considered responders.
Eighty-two percent of patients achieved a response at 3 months, and 87% had a response at 6 months. The overall mean change in Hb level was 1.52 ± 1.61 at 3 months and 1.72 ± 1.61 g/dL at 6 months. The rate of transfusion was 9% at 3 months and 6% at 6 months.
Between enrollment and month 6, 1202 patients discontinued epoetin zeta. Forty percent stopped because Hb levels were met, 27% stopped because they were stopping or changing chemotherapy, 15% stopped for both of the aforementioned reasons, 11% stopped because epoetin zeta was ineffective, and 2% stopped due to adverse events.
Seventeen percent of patients experienced an adverse event, including thromboembolic events (4%), infection (5%), bleeding (2%), local intolerability (0.2%), increased blood pressure (2%), and “other” events (9%).
Epoetin zeta was approved in Europe in 2007. For epoetin biosimilars to gain approval in the European Union, companies must agree to conduct post-marketing studies.
FDA approves new product for chronic ITP

Credit: Octapharma USA
The US Food and Drug Administration (FDA) has approved an intravenous immunoglobulin product (octagam 10%) for the treatment of chronic immune thrombocytopenia (ITP).
The product is a solvent/detergent-treated, sterile preparation of highly purified immunoglobulin G derived from large pools of human plasma.
It is intended to raise platelet counts to control or prevent bleeding.
The approval of octagam 10% is based on results of a phase 3 trial (Robak et al, Hematology, Oct. 2010). The trial included 66 patients with chronic ITP and 49 with newly diagnosed ITP.
Among the chronic ITP patients, 81.8% attained the primary efficacy endpoint of clinical response—a platelet count of at least 50×109/L within 7 days of dosing.
Among chronic ITP patients with bleeding at baseline (n=45), 77.7% reported no bleeding at day 7 after treatment.
There were no unexpected tolerability issues, even at the maximum infusion rate of 0.12 mL/kg/minute (720 mg/kg/hour).
The most common treatment-related adverse events in the entire patient cohort were headache (25%), fever (15%), and increased heart rate (11%). The most serious adverse event was headache.
octagam 10% has a black box warning detailing the risk of thrombosis, renal dysfunction, and acute renal failure associated with use of the product. For patients at risk of thrombosis, renal dysfunction, or renal failure, octagam 10% should be given at the minimum infusion rate practicable.
Healthcare providers should ensure adequate hydration in these patients before administering octagam 10%. Providers should also monitor patients for signs and symptoms of thrombosis and assess blood viscosity in patients at risk for hyperviscosity.
octagam 10% is contraindicated in patients who have a history of severe systemic hypersensitivity reactions, such as anaphylaxis, to human immunoglobulin. The product contains trace amounts of IgA (average 106 µg/mL in a 10% solution). It is contraindicated in IgA-deficient patients with antibodies against IgA and a history of hypersensitivity.
For more details, see the full prescribing information.
The makers of octagam 10%, Octapharma USA, said the product should be available in the US in September.
Credit: Octapharma USA
The US Food and Drug Administration (FDA) has approved an intravenous immunoglobulin product (octagam 10%) for the treatment of chronic immune thrombocytopenia (ITP).
The product is a solvent/detergent-treated, sterile preparation of highly purified immunoglobulin G derived from large pools of human plasma.
It is intended to raise platelet counts to control or prevent bleeding.
The approval of octagam 10% is based on results of a phase 3 trial (Robak et al, Hematology, Oct. 2010). The trial included 66 patients with chronic ITP and 49 with newly diagnosed ITP.
Among the chronic ITP patients, 81.8% attained the primary efficacy endpoint of clinical response—a platelet count of at least 50×109/L within 7 days of dosing.
Among chronic ITP patients with bleeding at baseline (n=45), 77.7% reported no bleeding at day 7 after treatment.
There were no unexpected tolerability issues, even at the maximum infusion rate of 0.12 mL/kg/minute (720 mg/kg/hour).
The most common treatment-related adverse events in the entire patient cohort were headache (25%), fever (15%), and increased heart rate (11%). The most serious adverse event was headache.
octagam 10% has a black box warning detailing the risk of thrombosis, renal dysfunction, and acute renal failure associated with use of the product. For patients at risk of thrombosis, renal dysfunction, or renal failure, octagam 10% should be given at the minimum infusion rate practicable.
Healthcare providers should ensure adequate hydration in these patients before administering octagam 10%. Providers should also monitor patients for signs and symptoms of thrombosis and assess blood viscosity in patients at risk for hyperviscosity.
octagam 10% is contraindicated in patients who have a history of severe systemic hypersensitivity reactions, such as anaphylaxis, to human immunoglobulin. The product contains trace amounts of IgA (average 106 µg/mL in a 10% solution). It is contraindicated in IgA-deficient patients with antibodies against IgA and a history of hypersensitivity.
For more details, see the full prescribing information.
The makers of octagam 10%, Octapharma USA, said the product should be available in the US in September.
Credit: Octapharma USA
The US Food and Drug Administration (FDA) has approved an intravenous immunoglobulin product (octagam 10%) for the treatment of chronic immune thrombocytopenia (ITP).
The product is a solvent/detergent-treated, sterile preparation of highly purified immunoglobulin G derived from large pools of human plasma.
It is intended to raise platelet counts to control or prevent bleeding.
The approval of octagam 10% is based on results of a phase 3 trial (Robak et al, Hematology, Oct. 2010). The trial included 66 patients with chronic ITP and 49 with newly diagnosed ITP.
Among the chronic ITP patients, 81.8% attained the primary efficacy endpoint of clinical response—a platelet count of at least 50×109/L within 7 days of dosing.
Among chronic ITP patients with bleeding at baseline (n=45), 77.7% reported no bleeding at day 7 after treatment.
There were no unexpected tolerability issues, even at the maximum infusion rate of 0.12 mL/kg/minute (720 mg/kg/hour).
The most common treatment-related adverse events in the entire patient cohort were headache (25%), fever (15%), and increased heart rate (11%). The most serious adverse event was headache.
octagam 10% has a black box warning detailing the risk of thrombosis, renal dysfunction, and acute renal failure associated with use of the product. For patients at risk of thrombosis, renal dysfunction, or renal failure, octagam 10% should be given at the minimum infusion rate practicable.
Healthcare providers should ensure adequate hydration in these patients before administering octagam 10%. Providers should also monitor patients for signs and symptoms of thrombosis and assess blood viscosity in patients at risk for hyperviscosity.
octagam 10% is contraindicated in patients who have a history of severe systemic hypersensitivity reactions, such as anaphylaxis, to human immunoglobulin. The product contains trace amounts of IgA (average 106 µg/mL in a 10% solution). It is contraindicated in IgA-deficient patients with antibodies against IgA and a history of hypersensitivity.
For more details, see the full prescribing information.
The makers of octagam 10%, Octapharma USA, said the product should be available in the US in September.
Gene editing doesn’t increase mutations in iPSCs
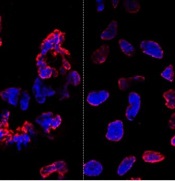
misshapen nuclear envelopes
(red) from iPSCs (DNA in blue).
The right panel shows
gene-edited iPSCs.
Credit: Salk Institute
Results of new research may ease previous concerns that gene-editing techniques could add unwanted mutations to stem cells.
Researchers compared gene editing techniques in lines of induced pluripotent stem cells (iPSCs) derived from a patient with sickle cell disease (SCD).
And they found that neither viral nor nuclease-based gene-editing methods increased the frequency of mutations in the iPSCs.
The team reported these results in Cell Stem Cell.
“The ability to precisely modify the DNA of stem cells has greatly accelerated research on human diseases and cell therapy,” said senior study author Juan Carlos Izpisua Belmonte, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“To successfully translate this technology into the clinic, we first need to scrutinize the safety of these modified stem cells, such as their genome stability and mutational load.”
Previously, Dr Belmonte’s lab pioneered the use of modified viruses, called helper-dependent adenoviral vectors (HDAdVs), to correct the genetic mutation that causes SCD.
He and his colleagues used HDAdVs to replace the mutated gene in a line of iPSCs with a mutant-free version, creating stem cells that could, theoretically, be infused into patients’ bone marrow and help create healthy blood cells.
Before such technologies are applied to humans, though, Dr Belmonte and his colleagues wanted to know whether there were risks related to editing the genes in iPSCs.
“As cells are being reprogrammed into stem cells, they tend to accumulate many mutations,” said Mo Li, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“So people naturally worry that any process you perform with these cells in vitro—including gene editing—might generate even more mutations.”
To find out whether this was the case, the researchers conducted tests in a line of SCD-derived iPSCs.
They edited the genes of some cells using 1 of 2 HDAdV designs. And they edited others using 1 of 2 transcription activator-like effector nuclease (TALEN) proteins.
They kept the rest of the SCD iPSCs in culture without editing them. Then, the team sequenced the entire genome of each cell from the 4 edits and control experiment.
While all of the cells gained a low level of random gene mutations during the experiments, the cells that had undergone gene-editing—whether through HDAdV- or TALEN-based approaches—had no more mutations than the cells kept in culture.
“We were pleasantly surprised by the results,” said Keiichiro Suzuki, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“People have found thousands of mutations introduced during iPSC reprogramming. We found less than a hundred single nucleotide variants in all cases.”
The researchers noted that this finding doesn’t necessarily mean there are no inherent risks to using stem cells with edited genes. However, it does suggest the editing process doesn’t make iPSCs any less safe.
“We concluded that the risk of mutation isn’t inherently connected to gene editing,” Dr Li said. “These cells present the same risks as using any other cells manipulated for cell or gene therapy.”
The Belmonte group is now planning more studies to address whether gene-repair in other cell types, using other approaches, or targeting other genes could be more or less likely to cause unwanted mutations.
For now, they hope their findings encourage those in the field to keep pursuing gene-editing techniques as a potential way to treat genetic diseases in the future. ![]()
misshapen nuclear envelopes
(red) from iPSCs (DNA in blue).
The right panel shows
gene-edited iPSCs.
Credit: Salk Institute
Results of new research may ease previous concerns that gene-editing techniques could add unwanted mutations to stem cells.
Researchers compared gene editing techniques in lines of induced pluripotent stem cells (iPSCs) derived from a patient with sickle cell disease (SCD).
And they found that neither viral nor nuclease-based gene-editing methods increased the frequency of mutations in the iPSCs.
The team reported these results in Cell Stem Cell.
“The ability to precisely modify the DNA of stem cells has greatly accelerated research on human diseases and cell therapy,” said senior study author Juan Carlos Izpisua Belmonte, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“To successfully translate this technology into the clinic, we first need to scrutinize the safety of these modified stem cells, such as their genome stability and mutational load.”
Previously, Dr Belmonte’s lab pioneered the use of modified viruses, called helper-dependent adenoviral vectors (HDAdVs), to correct the genetic mutation that causes SCD.
He and his colleagues used HDAdVs to replace the mutated gene in a line of iPSCs with a mutant-free version, creating stem cells that could, theoretically, be infused into patients’ bone marrow and help create healthy blood cells.
Before such technologies are applied to humans, though, Dr Belmonte and his colleagues wanted to know whether there were risks related to editing the genes in iPSCs.
“As cells are being reprogrammed into stem cells, they tend to accumulate many mutations,” said Mo Li, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“So people naturally worry that any process you perform with these cells in vitro—including gene editing—might generate even more mutations.”
To find out whether this was the case, the researchers conducted tests in a line of SCD-derived iPSCs.
They edited the genes of some cells using 1 of 2 HDAdV designs. And they edited others using 1 of 2 transcription activator-like effector nuclease (TALEN) proteins.
They kept the rest of the SCD iPSCs in culture without editing them. Then, the team sequenced the entire genome of each cell from the 4 edits and control experiment.
While all of the cells gained a low level of random gene mutations during the experiments, the cells that had undergone gene-editing—whether through HDAdV- or TALEN-based approaches—had no more mutations than the cells kept in culture.
“We were pleasantly surprised by the results,” said Keiichiro Suzuki, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“People have found thousands of mutations introduced during iPSC reprogramming. We found less than a hundred single nucleotide variants in all cases.”
The researchers noted that this finding doesn’t necessarily mean there are no inherent risks to using stem cells with edited genes. However, it does suggest the editing process doesn’t make iPSCs any less safe.
“We concluded that the risk of mutation isn’t inherently connected to gene editing,” Dr Li said. “These cells present the same risks as using any other cells manipulated for cell or gene therapy.”
The Belmonte group is now planning more studies to address whether gene-repair in other cell types, using other approaches, or targeting other genes could be more or less likely to cause unwanted mutations.
For now, they hope their findings encourage those in the field to keep pursuing gene-editing techniques as a potential way to treat genetic diseases in the future. ![]()
misshapen nuclear envelopes
(red) from iPSCs (DNA in blue).
The right panel shows
gene-edited iPSCs.
Credit: Salk Institute
Results of new research may ease previous concerns that gene-editing techniques could add unwanted mutations to stem cells.
Researchers compared gene editing techniques in lines of induced pluripotent stem cells (iPSCs) derived from a patient with sickle cell disease (SCD).
And they found that neither viral nor nuclease-based gene-editing methods increased the frequency of mutations in the iPSCs.
The team reported these results in Cell Stem Cell.
“The ability to precisely modify the DNA of stem cells has greatly accelerated research on human diseases and cell therapy,” said senior study author Juan Carlos Izpisua Belmonte, PhD, of the Salk Institute for Biological Studies in La Jolla, California.
“To successfully translate this technology into the clinic, we first need to scrutinize the safety of these modified stem cells, such as their genome stability and mutational load.”
Previously, Dr Belmonte’s lab pioneered the use of modified viruses, called helper-dependent adenoviral vectors (HDAdVs), to correct the genetic mutation that causes SCD.
He and his colleagues used HDAdVs to replace the mutated gene in a line of iPSCs with a mutant-free version, creating stem cells that could, theoretically, be infused into patients’ bone marrow and help create healthy blood cells.
Before such technologies are applied to humans, though, Dr Belmonte and his colleagues wanted to know whether there were risks related to editing the genes in iPSCs.
“As cells are being reprogrammed into stem cells, they tend to accumulate many mutations,” said Mo Li, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“So people naturally worry that any process you perform with these cells in vitro—including gene editing—might generate even more mutations.”
To find out whether this was the case, the researchers conducted tests in a line of SCD-derived iPSCs.
They edited the genes of some cells using 1 of 2 HDAdV designs. And they edited others using 1 of 2 transcription activator-like effector nuclease (TALEN) proteins.
They kept the rest of the SCD iPSCs in culture without editing them. Then, the team sequenced the entire genome of each cell from the 4 edits and control experiment.
While all of the cells gained a low level of random gene mutations during the experiments, the cells that had undergone gene-editing—whether through HDAdV- or TALEN-based approaches—had no more mutations than the cells kept in culture.
“We were pleasantly surprised by the results,” said Keiichiro Suzuki, PhD, a postdoctoral fellow in Dr Belmonte’s lab.
“People have found thousands of mutations introduced during iPSC reprogramming. We found less than a hundred single nucleotide variants in all cases.”
The researchers noted that this finding doesn’t necessarily mean there are no inherent risks to using stem cells with edited genes. However, it does suggest the editing process doesn’t make iPSCs any less safe.
“We concluded that the risk of mutation isn’t inherently connected to gene editing,” Dr Li said. “These cells present the same risks as using any other cells manipulated for cell or gene therapy.”
The Belmonte group is now planning more studies to address whether gene-repair in other cell types, using other approaches, or targeting other genes could be more or less likely to cause unwanted mutations.
For now, they hope their findings encourage those in the field to keep pursuing gene-editing techniques as a potential way to treat genetic diseases in the future. ![]()