User login
CHMP recommends expanding use of drug in CLL

Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
NCCN issues challenge to ‘bag’ vincristine
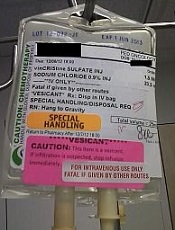
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()
Health Canada approves drug for patients with VTE, NVAF

Image by Andre E.X. Brown
Health Canada has approved edoxaban (Lixiana®), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF in whom anticoagulation is appropriate.
Edoxaban was discovered and developed by Daiichi Sankyo Co., Ltd., but Servier Canada will market the drug in Canada.
Edoxaban has been approved in the US, European Union, Switzerland, Japan, South Korea, Taiwan, and Hong Kong. The drug is marketed as Savaysa® in the US and as Lixiana® elsewhere.
Health Canada’s approval of edoxaban is based on data from a pair of phase 3 trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001). ![]()
Image by Andre E.X. Brown
Health Canada has approved edoxaban (Lixiana®), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF in whom anticoagulation is appropriate.
Edoxaban was discovered and developed by Daiichi Sankyo Co., Ltd., but Servier Canada will market the drug in Canada.
Edoxaban has been approved in the US, European Union, Switzerland, Japan, South Korea, Taiwan, and Hong Kong. The drug is marketed as Savaysa® in the US and as Lixiana® elsewhere.
Health Canada’s approval of edoxaban is based on data from a pair of phase 3 trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001). ![]()
Image by Andre E.X. Brown
Health Canada has approved edoxaban (Lixiana®), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF in whom anticoagulation is appropriate.
Edoxaban was discovered and developed by Daiichi Sankyo Co., Ltd., but Servier Canada will market the drug in Canada.
Edoxaban has been approved in the US, European Union, Switzerland, Japan, South Korea, Taiwan, and Hong Kong. The drug is marketed as Savaysa® in the US and as Lixiana® elsewhere.
Health Canada’s approval of edoxaban is based on data from a pair of phase 3 trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001). ![]()
Treating RBCs with NO may make them safer

Sheep Experiment Station
Research conducted in sheep indicates that pretreating red blood cells (RBCs) with nitric oxide (NO) may make it safer to transfuse blood nearing its expiration date.
Past studies have suggested that RBCs stored for more than 30 days are less likely than “fresher” RBCs to survive after transfusion, and receiving a transfusion of RBCs nearing their expiration date of 42 days may increase the risk of pulmonary hypertension.
However, a new study published in Anesthesiology suggests that pretreating older RBCs with NO may increase their likelihood of survival after transfusion and reduce the risk of pulmonary hypertension in the recipient.
“Extended storage of RBCs makes them rigid and decreases their ability to change shape, which is necessary as they travel through small blood vessels,” said study author Warren M. Zapol, MD, of Massachusetts General Hospital in Boston.
“We found that pretreatment with nitric oxide actually rejuvenates RBCs, making them more flexible so they can more easily travel through blood vessels. This can further reduce the risk of pulmonary hypertension.”
Dr Zapol and his colleagues performed their experiments on RBCs derived from lambs. The team treated RBCs with NO gas, a short-lived NO donor, or gas without NO (control).
The RBCs were then stored for either 2 days (hereafter referred to as “fresh” RBCs) or 40 days (referred to as “stored” RBCs) and transfused back into the original lambs.
RBC survival
The researchers found that treatment with NO gas improved the early post-transfusion survival of stored RBCs.
At 1 hour after transfusion, 75.3 ± 5.8% of the control-treated stored RBCs remained in the circulation, compared to 86.8 ± 8.1% of the NO-treated stored RBCs and 94.2 ± 4.6% of the fresh RBCs.
At 24 hours after transfusion, 73.4 ± 3.8% of the control-treated stored RBCs remained in the circulation, compared to 78.3 ± 6.3% of the NO-treated stored RBCs, 90.8 ± 4.1% of control-treated fresh RBCs, and 91.4 ± 1.4% of NO-treated fresh RBCs.
The differences between stored RBCs that were treated with NO gas and stored control RBCs was statistically significant at both 1 hour and 24 hours, with P values of 0.002 and 0.046, respectively.
Seven days after transfusion, there was no significant difference in the percentage of NO-treated and control-treated RBCs in the circulation.
Pulmonary hypertension
The researchers found that pretreating RBCs with NO prevented transfusion-associated pulmonary hypertension in the lambs.
Lambs that received control-treated stored RBCs had an increase in pulmonary arterial pressure (PAP) during and after transfusion—from 13.4 ± 0.8 mmHg at baseline to a maximum of 22.7 ± 2.2 mmHg.
However, lambs that received stored RBCs treated with NO gas did not have an increase in PAP when compared to lambs that received fresh RBCs.
At 20 minutes, PAP was 14.5 ± 1.4 mmHg for NO-treated stored RBCs, 13.9 ± 0.6 mmHg for control-treated fresh RBCs, and 14 ± 1.2 mmHg for NO-treated fresh RBCs.
The researchers also found that transfusion of stored RBCs caused a transient increase in the pulmonary vascular resistance index (PVRI) from 10 minutes to 30 minutes after transfusion, but pretreatment with NO gas prevented this increase.
At 20 minutes, the PVRI was 211.1 ± 44.4 dyn·sec·cm−5·m−2 for control-treated stored RBCs and 114.6 ± 18.9 dyn·sec·cm−5·m−2 for NO-treated stored RBCs (P<0.0001).
Transfusion of fresh RBCs, with or without prior NO exposure, did not alter the PVRI.
Finally, the researchers found that treating stored RBCs with the NO donor compound MAHMA NONOate prevented transfusion-associated pulmonary hypertension and pulmonary vasoconstriction in awake lambs.
The team said studies with human RBCs are required to confirm the beneficial effects of NO exposure observed in this study. ![]()
Sheep Experiment Station
Research conducted in sheep indicates that pretreating red blood cells (RBCs) with nitric oxide (NO) may make it safer to transfuse blood nearing its expiration date.
Past studies have suggested that RBCs stored for more than 30 days are less likely than “fresher” RBCs to survive after transfusion, and receiving a transfusion of RBCs nearing their expiration date of 42 days may increase the risk of pulmonary hypertension.
However, a new study published in Anesthesiology suggests that pretreating older RBCs with NO may increase their likelihood of survival after transfusion and reduce the risk of pulmonary hypertension in the recipient.
“Extended storage of RBCs makes them rigid and decreases their ability to change shape, which is necessary as they travel through small blood vessels,” said study author Warren M. Zapol, MD, of Massachusetts General Hospital in Boston.
“We found that pretreatment with nitric oxide actually rejuvenates RBCs, making them more flexible so they can more easily travel through blood vessels. This can further reduce the risk of pulmonary hypertension.”
Dr Zapol and his colleagues performed their experiments on RBCs derived from lambs. The team treated RBCs with NO gas, a short-lived NO donor, or gas without NO (control).
The RBCs were then stored for either 2 days (hereafter referred to as “fresh” RBCs) or 40 days (referred to as “stored” RBCs) and transfused back into the original lambs.
RBC survival
The researchers found that treatment with NO gas improved the early post-transfusion survival of stored RBCs.
At 1 hour after transfusion, 75.3 ± 5.8% of the control-treated stored RBCs remained in the circulation, compared to 86.8 ± 8.1% of the NO-treated stored RBCs and 94.2 ± 4.6% of the fresh RBCs.
At 24 hours after transfusion, 73.4 ± 3.8% of the control-treated stored RBCs remained in the circulation, compared to 78.3 ± 6.3% of the NO-treated stored RBCs, 90.8 ± 4.1% of control-treated fresh RBCs, and 91.4 ± 1.4% of NO-treated fresh RBCs.
The differences between stored RBCs that were treated with NO gas and stored control RBCs was statistically significant at both 1 hour and 24 hours, with P values of 0.002 and 0.046, respectively.
Seven days after transfusion, there was no significant difference in the percentage of NO-treated and control-treated RBCs in the circulation.
Pulmonary hypertension
The researchers found that pretreating RBCs with NO prevented transfusion-associated pulmonary hypertension in the lambs.
Lambs that received control-treated stored RBCs had an increase in pulmonary arterial pressure (PAP) during and after transfusion—from 13.4 ± 0.8 mmHg at baseline to a maximum of 22.7 ± 2.2 mmHg.
However, lambs that received stored RBCs treated with NO gas did not have an increase in PAP when compared to lambs that received fresh RBCs.
At 20 minutes, PAP was 14.5 ± 1.4 mmHg for NO-treated stored RBCs, 13.9 ± 0.6 mmHg for control-treated fresh RBCs, and 14 ± 1.2 mmHg for NO-treated fresh RBCs.
The researchers also found that transfusion of stored RBCs caused a transient increase in the pulmonary vascular resistance index (PVRI) from 10 minutes to 30 minutes after transfusion, but pretreatment with NO gas prevented this increase.
At 20 minutes, the PVRI was 211.1 ± 44.4 dyn·sec·cm−5·m−2 for control-treated stored RBCs and 114.6 ± 18.9 dyn·sec·cm−5·m−2 for NO-treated stored RBCs (P<0.0001).
Transfusion of fresh RBCs, with or without prior NO exposure, did not alter the PVRI.
Finally, the researchers found that treating stored RBCs with the NO donor compound MAHMA NONOate prevented transfusion-associated pulmonary hypertension and pulmonary vasoconstriction in awake lambs.
The team said studies with human RBCs are required to confirm the beneficial effects of NO exposure observed in this study. ![]()
Sheep Experiment Station
Research conducted in sheep indicates that pretreating red blood cells (RBCs) with nitric oxide (NO) may make it safer to transfuse blood nearing its expiration date.
Past studies have suggested that RBCs stored for more than 30 days are less likely than “fresher” RBCs to survive after transfusion, and receiving a transfusion of RBCs nearing their expiration date of 42 days may increase the risk of pulmonary hypertension.
However, a new study published in Anesthesiology suggests that pretreating older RBCs with NO may increase their likelihood of survival after transfusion and reduce the risk of pulmonary hypertension in the recipient.
“Extended storage of RBCs makes them rigid and decreases their ability to change shape, which is necessary as they travel through small blood vessels,” said study author Warren M. Zapol, MD, of Massachusetts General Hospital in Boston.
“We found that pretreatment with nitric oxide actually rejuvenates RBCs, making them more flexible so they can more easily travel through blood vessels. This can further reduce the risk of pulmonary hypertension.”
Dr Zapol and his colleagues performed their experiments on RBCs derived from lambs. The team treated RBCs with NO gas, a short-lived NO donor, or gas without NO (control).
The RBCs were then stored for either 2 days (hereafter referred to as “fresh” RBCs) or 40 days (referred to as “stored” RBCs) and transfused back into the original lambs.
RBC survival
The researchers found that treatment with NO gas improved the early post-transfusion survival of stored RBCs.
At 1 hour after transfusion, 75.3 ± 5.8% of the control-treated stored RBCs remained in the circulation, compared to 86.8 ± 8.1% of the NO-treated stored RBCs and 94.2 ± 4.6% of the fresh RBCs.
At 24 hours after transfusion, 73.4 ± 3.8% of the control-treated stored RBCs remained in the circulation, compared to 78.3 ± 6.3% of the NO-treated stored RBCs, 90.8 ± 4.1% of control-treated fresh RBCs, and 91.4 ± 1.4% of NO-treated fresh RBCs.
The differences between stored RBCs that were treated with NO gas and stored control RBCs was statistically significant at both 1 hour and 24 hours, with P values of 0.002 and 0.046, respectively.
Seven days after transfusion, there was no significant difference in the percentage of NO-treated and control-treated RBCs in the circulation.
Pulmonary hypertension
The researchers found that pretreating RBCs with NO prevented transfusion-associated pulmonary hypertension in the lambs.
Lambs that received control-treated stored RBCs had an increase in pulmonary arterial pressure (PAP) during and after transfusion—from 13.4 ± 0.8 mmHg at baseline to a maximum of 22.7 ± 2.2 mmHg.
However, lambs that received stored RBCs treated with NO gas did not have an increase in PAP when compared to lambs that received fresh RBCs.
At 20 minutes, PAP was 14.5 ± 1.4 mmHg for NO-treated stored RBCs, 13.9 ± 0.6 mmHg for control-treated fresh RBCs, and 14 ± 1.2 mmHg for NO-treated fresh RBCs.
The researchers also found that transfusion of stored RBCs caused a transient increase in the pulmonary vascular resistance index (PVRI) from 10 minutes to 30 minutes after transfusion, but pretreatment with NO gas prevented this increase.
At 20 minutes, the PVRI was 211.1 ± 44.4 dyn·sec·cm−5·m−2 for control-treated stored RBCs and 114.6 ± 18.9 dyn·sec·cm−5·m−2 for NO-treated stored RBCs (P<0.0001).
Transfusion of fresh RBCs, with or without prior NO exposure, did not alter the PVRI.
Finally, the researchers found that treating stored RBCs with the NO donor compound MAHMA NONOate prevented transfusion-associated pulmonary hypertension and pulmonary vasoconstriction in awake lambs.
The team said studies with human RBCs are required to confirm the beneficial effects of NO exposure observed in this study. ![]()
Tool provides info for cancer patients, survivors

receiving treatment
Photo by Rhoda Baer
The American Cancer Society and National Cancer Institute have launched an online tool for cancer patients and survivors.
The tool, Springboard Beyond Cancer, was designed to help these individuals address medical, psychosocial, and wellness needs during and after treatment.
Springboard Beyond Cancer provides information to help cancer patients and survivors manage ongoing cancer-related symptoms, deal with stress, ensure healthy behavior, communicate better with healthcare teams, and seek support from friends and family.
“With Springboard Beyond Cancer, we want to empower cancer survivors by giving them the information they need to help identify issues, set goals, and create a plan to more smoothly navigate the cancer journey and take control of their health,” said Corinne Leach, PhD, a behavioral scientist and strategic director in the Behavioral Research Center at the American Cancer Society.
“We hope that Springboard Beyond Cancer, along with the close collaboration of their medical team, can help cancer survivors reduce their disease burden and improve their overall wellbeing,” added Erik Augustson, PhD, program director at the National Cancer Institute. ![]()
receiving treatment
Photo by Rhoda Baer
The American Cancer Society and National Cancer Institute have launched an online tool for cancer patients and survivors.
The tool, Springboard Beyond Cancer, was designed to help these individuals address medical, psychosocial, and wellness needs during and after treatment.
Springboard Beyond Cancer provides information to help cancer patients and survivors manage ongoing cancer-related symptoms, deal with stress, ensure healthy behavior, communicate better with healthcare teams, and seek support from friends and family.
“With Springboard Beyond Cancer, we want to empower cancer survivors by giving them the information they need to help identify issues, set goals, and create a plan to more smoothly navigate the cancer journey and take control of their health,” said Corinne Leach, PhD, a behavioral scientist and strategic director in the Behavioral Research Center at the American Cancer Society.
“We hope that Springboard Beyond Cancer, along with the close collaboration of their medical team, can help cancer survivors reduce their disease burden and improve their overall wellbeing,” added Erik Augustson, PhD, program director at the National Cancer Institute. ![]()
receiving treatment
Photo by Rhoda Baer
The American Cancer Society and National Cancer Institute have launched an online tool for cancer patients and survivors.
The tool, Springboard Beyond Cancer, was designed to help these individuals address medical, psychosocial, and wellness needs during and after treatment.
Springboard Beyond Cancer provides information to help cancer patients and survivors manage ongoing cancer-related symptoms, deal with stress, ensure healthy behavior, communicate better with healthcare teams, and seek support from friends and family.
“With Springboard Beyond Cancer, we want to empower cancer survivors by giving them the information they need to help identify issues, set goals, and create a plan to more smoothly navigate the cancer journey and take control of their health,” said Corinne Leach, PhD, a behavioral scientist and strategic director in the Behavioral Research Center at the American Cancer Society.
“We hope that Springboard Beyond Cancer, along with the close collaboration of their medical team, can help cancer survivors reduce their disease burden and improve their overall wellbeing,” added Erik Augustson, PhD, program director at the National Cancer Institute. ![]()
Blood test can predict outcomes in DLBCL, team says

Photo by Juan D. Alfonso
A blood test can reveal genetic features linked to outcomes in patients with diffuse large B-cell lymphoma (DLBCL), according to research published in Science Translational Medicine.
Investigators used targeted sequencing to analyze circulating tumor DNA (ctDNA) in blood samples from DLBCL patients.
This allowed the team to identify the cell of origin, detect minimal residual disease (MRD), and predict progression-free survival (PFS) in these patients.
Florian Scherer, MD, of Stanford University in California, and his colleagues conducted this research.
They used cancer personalized profiling by deep sequencing (CAPP-Seq) to analyze tumor biopsies and cell-free DNA samples from 92 patients with DLBCL and 24 healthy controls.
The investigators found that CAPP-Seq could effectively detect somatic mutations in DLBCL plasma samples as well as tumor biopsies. They said their results suggest ctDNA is a “robust surrogate for direct assessment of primary tumor genotypes” in most DLBCL patients.
In addition, ctDNA profiling with CAPP-Seq revealed mutations associated with resistance to the BTK inhibitor ibrutinib.
The investigators also said their results suggest ctDNA profiling can be used to classify DLBCL subtypes. The overall concordance in cell of origin predictions between tumor tissue and plasma genotyping was 88%.
Another key finding of this study is that the amount of ctDNA at DLBCL diagnosis was predictive of PFS. The investigators said higher ctDNA levels at diagnosis were “continuously and independently” correlated with inferior PFS.
Dr Scherer and his colleagues also discovered that ctDNA profiling could detect MRD with greater accuracy than immunoglobulin sequencing and radiographic imaging. And patients with ctDNA in their plasma had significantly worse PFS than patients with undetectable ctDNA.
Finally, the investigators found evidence to suggest that ctDNA profiling could provide early detection of disease transformation. They identified “distinct patterns of clonal evolution” by which they could distinguish indolent follicular lymphomas from follicular lymphomas that transformed into DLBCL. ![]()
Photo by Juan D. Alfonso
A blood test can reveal genetic features linked to outcomes in patients with diffuse large B-cell lymphoma (DLBCL), according to research published in Science Translational Medicine.
Investigators used targeted sequencing to analyze circulating tumor DNA (ctDNA) in blood samples from DLBCL patients.
This allowed the team to identify the cell of origin, detect minimal residual disease (MRD), and predict progression-free survival (PFS) in these patients.
Florian Scherer, MD, of Stanford University in California, and his colleagues conducted this research.
They used cancer personalized profiling by deep sequencing (CAPP-Seq) to analyze tumor biopsies and cell-free DNA samples from 92 patients with DLBCL and 24 healthy controls.
The investigators found that CAPP-Seq could effectively detect somatic mutations in DLBCL plasma samples as well as tumor biopsies. They said their results suggest ctDNA is a “robust surrogate for direct assessment of primary tumor genotypes” in most DLBCL patients.
In addition, ctDNA profiling with CAPP-Seq revealed mutations associated with resistance to the BTK inhibitor ibrutinib.
The investigators also said their results suggest ctDNA profiling can be used to classify DLBCL subtypes. The overall concordance in cell of origin predictions between tumor tissue and plasma genotyping was 88%.
Another key finding of this study is that the amount of ctDNA at DLBCL diagnosis was predictive of PFS. The investigators said higher ctDNA levels at diagnosis were “continuously and independently” correlated with inferior PFS.
Dr Scherer and his colleagues also discovered that ctDNA profiling could detect MRD with greater accuracy than immunoglobulin sequencing and radiographic imaging. And patients with ctDNA in their plasma had significantly worse PFS than patients with undetectable ctDNA.
Finally, the investigators found evidence to suggest that ctDNA profiling could provide early detection of disease transformation. They identified “distinct patterns of clonal evolution” by which they could distinguish indolent follicular lymphomas from follicular lymphomas that transformed into DLBCL. ![]()
Photo by Juan D. Alfonso
A blood test can reveal genetic features linked to outcomes in patients with diffuse large B-cell lymphoma (DLBCL), according to research published in Science Translational Medicine.
Investigators used targeted sequencing to analyze circulating tumor DNA (ctDNA) in blood samples from DLBCL patients.
This allowed the team to identify the cell of origin, detect minimal residual disease (MRD), and predict progression-free survival (PFS) in these patients.
Florian Scherer, MD, of Stanford University in California, and his colleagues conducted this research.
They used cancer personalized profiling by deep sequencing (CAPP-Seq) to analyze tumor biopsies and cell-free DNA samples from 92 patients with DLBCL and 24 healthy controls.
The investigators found that CAPP-Seq could effectively detect somatic mutations in DLBCL plasma samples as well as tumor biopsies. They said their results suggest ctDNA is a “robust surrogate for direct assessment of primary tumor genotypes” in most DLBCL patients.
In addition, ctDNA profiling with CAPP-Seq revealed mutations associated with resistance to the BTK inhibitor ibrutinib.
The investigators also said their results suggest ctDNA profiling can be used to classify DLBCL subtypes. The overall concordance in cell of origin predictions between tumor tissue and plasma genotyping was 88%.
Another key finding of this study is that the amount of ctDNA at DLBCL diagnosis was predictive of PFS. The investigators said higher ctDNA levels at diagnosis were “continuously and independently” correlated with inferior PFS.
Dr Scherer and his colleagues also discovered that ctDNA profiling could detect MRD with greater accuracy than immunoglobulin sequencing and radiographic imaging. And patients with ctDNA in their plasma had significantly worse PFS than patients with undetectable ctDNA.
Finally, the investigators found evidence to suggest that ctDNA profiling could provide early detection of disease transformation. They identified “distinct patterns of clonal evolution” by which they could distinguish indolent follicular lymphomas from follicular lymphomas that transformed into DLBCL. ![]()
Link between hemolysis and infection explained

plate showing a positive
streptococcus infection
Photo by Bill Branson
Patients who suffer from hemolysis have an increased risk of developing bacterial infections, and new research provides an explanation for this phenomenon.
The study refutes the idea that excess circulating iron is to blame.
Instead, it suggests that heme prevents macrophages from engulfing bacteria. And targeting this activity might reduce the risk of bacterial infection in patients with hemolytic disorders.
Sylvia Knapp, MD, PhD, of the Medical University of Vienna in Austria, and her colleagues reported these findings in Nature Immunology.
For decades, iron has been considered the prime suspect responsible for the high rate of bacterial infections in patients with hemolysis. Iron has long been established as an essential nutrient for bacteria.
Since hemolysis leads to the release of iron-containing heme, the threat of serious bacterial infections in these patients was attributed to the excess availability of circulating iron (heme).
However, Dr Knapp and her colleagues found that heme does not act as a willing nutrient to bacteria.
“Using in vitro and preclinical models, we could clearly demonstrate that heme-derived iron is not at all vital for bacterial growth,” said Rui Martins, a PhD student at the Medical University of Vienna.
“In contrast, we found that heme acts on macrophages, the most significant immune cells that are required for mounting an antibacterial response, and it furthermore prevented these cells from eliminating bacteria.”
Heme interfered with the cytoskeleton of macrophages, thereby immobilizing them.
“Heme causes cells to form numerous spikes—like hair standing on end—and then ‘stuns’ the cells within minutes,” Martins explained. “It is reminiscent of a cartoon character sticking his finger in an electrical outlet.”
The cytoskeleton is crucial for the basic functions of macrophages. It consists of long, branching filaments that act as the cell’s internal, highly flexible, and mobile framework.
Through targeted build-up and breakdown of these filaments, phagocytes can move in any direction and engulf invading bacteria. However, this requires a finely tuned signaling program in which the protein DOCK8 plays a central role.
“Through chemical proteomics and biochemical experiments, we discovered that heme interacted with DOCK8, which led to the permanent activation of its downstream target, Cdc42, with deleterious effects,” Dr Knapp said.
In the presence of heme, the cytoskeletal resilience was lost, as filaments grew rampant in all directions, resulting in macrophage paralysis. The cells lost their ability to shape-shift and could no longer chase down and engulf the invading bacteria, allowing the bacteria to multiply virtually unrestricted.
However, Dr Knapp and her colleagues found that an antimalarial drug can restore the functionality of these paralyzed macrophages.
“Quinine, which is clinically used to treat malaria and is suspected to bind heme, blocks the interaction of heme with DOCK8 and thereby improves the outcome from sepsis,” Dr Knapp said.
“This is very promising. We conclusively demonstrate that it is indeed feasible to therapeutically ‘protect’ immune cells and to restore the body’s immune defense against bacteria in hemolytic conditions.” ![]()
plate showing a positive
streptococcus infection
Photo by Bill Branson
Patients who suffer from hemolysis have an increased risk of developing bacterial infections, and new research provides an explanation for this phenomenon.
The study refutes the idea that excess circulating iron is to blame.
Instead, it suggests that heme prevents macrophages from engulfing bacteria. And targeting this activity might reduce the risk of bacterial infection in patients with hemolytic disorders.
Sylvia Knapp, MD, PhD, of the Medical University of Vienna in Austria, and her colleagues reported these findings in Nature Immunology.
For decades, iron has been considered the prime suspect responsible for the high rate of bacterial infections in patients with hemolysis. Iron has long been established as an essential nutrient for bacteria.
Since hemolysis leads to the release of iron-containing heme, the threat of serious bacterial infections in these patients was attributed to the excess availability of circulating iron (heme).
However, Dr Knapp and her colleagues found that heme does not act as a willing nutrient to bacteria.
“Using in vitro and preclinical models, we could clearly demonstrate that heme-derived iron is not at all vital for bacterial growth,” said Rui Martins, a PhD student at the Medical University of Vienna.
“In contrast, we found that heme acts on macrophages, the most significant immune cells that are required for mounting an antibacterial response, and it furthermore prevented these cells from eliminating bacteria.”
Heme interfered with the cytoskeleton of macrophages, thereby immobilizing them.
“Heme causes cells to form numerous spikes—like hair standing on end—and then ‘stuns’ the cells within minutes,” Martins explained. “It is reminiscent of a cartoon character sticking his finger in an electrical outlet.”
The cytoskeleton is crucial for the basic functions of macrophages. It consists of long, branching filaments that act as the cell’s internal, highly flexible, and mobile framework.
Through targeted build-up and breakdown of these filaments, phagocytes can move in any direction and engulf invading bacteria. However, this requires a finely tuned signaling program in which the protein DOCK8 plays a central role.
“Through chemical proteomics and biochemical experiments, we discovered that heme interacted with DOCK8, which led to the permanent activation of its downstream target, Cdc42, with deleterious effects,” Dr Knapp said.
In the presence of heme, the cytoskeletal resilience was lost, as filaments grew rampant in all directions, resulting in macrophage paralysis. The cells lost their ability to shape-shift and could no longer chase down and engulf the invading bacteria, allowing the bacteria to multiply virtually unrestricted.
However, Dr Knapp and her colleagues found that an antimalarial drug can restore the functionality of these paralyzed macrophages.
“Quinine, which is clinically used to treat malaria and is suspected to bind heme, blocks the interaction of heme with DOCK8 and thereby improves the outcome from sepsis,” Dr Knapp said.
“This is very promising. We conclusively demonstrate that it is indeed feasible to therapeutically ‘protect’ immune cells and to restore the body’s immune defense against bacteria in hemolytic conditions.” ![]()
plate showing a positive
streptococcus infection
Photo by Bill Branson
Patients who suffer from hemolysis have an increased risk of developing bacterial infections, and new research provides an explanation for this phenomenon.
The study refutes the idea that excess circulating iron is to blame.
Instead, it suggests that heme prevents macrophages from engulfing bacteria. And targeting this activity might reduce the risk of bacterial infection in patients with hemolytic disorders.
Sylvia Knapp, MD, PhD, of the Medical University of Vienna in Austria, and her colleagues reported these findings in Nature Immunology.
For decades, iron has been considered the prime suspect responsible for the high rate of bacterial infections in patients with hemolysis. Iron has long been established as an essential nutrient for bacteria.
Since hemolysis leads to the release of iron-containing heme, the threat of serious bacterial infections in these patients was attributed to the excess availability of circulating iron (heme).
However, Dr Knapp and her colleagues found that heme does not act as a willing nutrient to bacteria.
“Using in vitro and preclinical models, we could clearly demonstrate that heme-derived iron is not at all vital for bacterial growth,” said Rui Martins, a PhD student at the Medical University of Vienna.
“In contrast, we found that heme acts on macrophages, the most significant immune cells that are required for mounting an antibacterial response, and it furthermore prevented these cells from eliminating bacteria.”
Heme interfered with the cytoskeleton of macrophages, thereby immobilizing them.
“Heme causes cells to form numerous spikes—like hair standing on end—and then ‘stuns’ the cells within minutes,” Martins explained. “It is reminiscent of a cartoon character sticking his finger in an electrical outlet.”
The cytoskeleton is crucial for the basic functions of macrophages. It consists of long, branching filaments that act as the cell’s internal, highly flexible, and mobile framework.
Through targeted build-up and breakdown of these filaments, phagocytes can move in any direction and engulf invading bacteria. However, this requires a finely tuned signaling program in which the protein DOCK8 plays a central role.
“Through chemical proteomics and biochemical experiments, we discovered that heme interacted with DOCK8, which led to the permanent activation of its downstream target, Cdc42, with deleterious effects,” Dr Knapp said.
In the presence of heme, the cytoskeletal resilience was lost, as filaments grew rampant in all directions, resulting in macrophage paralysis. The cells lost their ability to shape-shift and could no longer chase down and engulf the invading bacteria, allowing the bacteria to multiply virtually unrestricted.
However, Dr Knapp and her colleagues found that an antimalarial drug can restore the functionality of these paralyzed macrophages.
“Quinine, which is clinically used to treat malaria and is suspected to bind heme, blocks the interaction of heme with DOCK8 and thereby improves the outcome from sepsis,” Dr Knapp said.
“This is very promising. We conclusively demonstrate that it is indeed feasible to therapeutically ‘protect’ immune cells and to restore the body’s immune defense against bacteria in hemolytic conditions.”
Generic bivalirudin available in US
Photo from Business Wire
Fresenius Kabi’s Bivalirudin for Injection, a generic alternative to The Medicines Company’s Angiomax, is now available in the US.
Bivalirudin is a direct thrombin inhibitor indicated for use as an anticoagulant.
Fresenius Kabi’s Bivalirudin for Injection was approved by the US Food and Drug Administration in October.
Bivalirudin for Injection is now available in single-dose vials, each containing 250 mg of bivalirudin.
Bivalirudin for Injection was developed and is manufactured in the US.
Bivalirudin for Injection is indicated for use in:
- Patients with unstable angina who are undergoing percutaneous transluminal coronary angioplasty (PTCA)
- Patients undergoing percutaneous coronary intervention (PCI) with provisional use of glycoprotein IIb/IIIa inhibitors, as in the REPLACE-2 study
- Patients with, or at risk of, heparin-induced thrombocytopenia or heparin-induced thrombocytopenia and thrombosis syndrome who are undergoing PCI.
Bivalirudin for Injection is intended for use with aspirin.
The safety and effectiveness of Bivalirudin for Injection has not been established in patients with acute coronary syndromes who are not undergoing PTCA or PCI.
Photo from Business Wire
Fresenius Kabi’s Bivalirudin for Injection, a generic alternative to The Medicines Company’s Angiomax, is now available in the US.
Bivalirudin is a direct thrombin inhibitor indicated for use as an anticoagulant.
Fresenius Kabi’s Bivalirudin for Injection was approved by the US Food and Drug Administration in October.
Bivalirudin for Injection is now available in single-dose vials, each containing 250 mg of bivalirudin.
Bivalirudin for Injection was developed and is manufactured in the US.
Bivalirudin for Injection is indicated for use in:
- Patients with unstable angina who are undergoing percutaneous transluminal coronary angioplasty (PTCA)
- Patients undergoing percutaneous coronary intervention (PCI) with provisional use of glycoprotein IIb/IIIa inhibitors, as in the REPLACE-2 study
- Patients with, or at risk of, heparin-induced thrombocytopenia or heparin-induced thrombocytopenia and thrombosis syndrome who are undergoing PCI.
Bivalirudin for Injection is intended for use with aspirin.
The safety and effectiveness of Bivalirudin for Injection has not been established in patients with acute coronary syndromes who are not undergoing PTCA or PCI.
Photo from Business Wire
Fresenius Kabi’s Bivalirudin for Injection, a generic alternative to The Medicines Company’s Angiomax, is now available in the US.
Bivalirudin is a direct thrombin inhibitor indicated for use as an anticoagulant.
Fresenius Kabi’s Bivalirudin for Injection was approved by the US Food and Drug Administration in October.
Bivalirudin for Injection is now available in single-dose vials, each containing 250 mg of bivalirudin.
Bivalirudin for Injection was developed and is manufactured in the US.
Bivalirudin for Injection is indicated for use in:
- Patients with unstable angina who are undergoing percutaneous transluminal coronary angioplasty (PTCA)
- Patients undergoing percutaneous coronary intervention (PCI) with provisional use of glycoprotein IIb/IIIa inhibitors, as in the REPLACE-2 study
- Patients with, or at risk of, heparin-induced thrombocytopenia or heparin-induced thrombocytopenia and thrombosis syndrome who are undergoing PCI.
Bivalirudin for Injection is intended for use with aspirin.
The safety and effectiveness of Bivalirudin for Injection has not been established in patients with acute coronary syndromes who are not undergoing PTCA or PCI.
Phase 2 trial of MM drug placed on clinical hold

A phase 2 study of the antibody BI-505 in patients with multiple myeloma (MM) has been placed on full clinical hold.
BioInvent International, the company developing BI-505, said it has received verbal notice of the clinical hold from the US Food and Drug Administration (FDA).
The clinical hold means no new subjects can be enrolled on the trial, and there can be no further dosing of subjects who are already enrolled.
BioInvent International said it has yet to receive written notice of the clinical hold from the FDA. However, based on verbal communications, the clinical hold is due to an adverse cardiopulmonary event.
The study (NCT02756728) is being conducted by BioInvent International in collaboration with investigators at the University of Pennsylvania.
The goal of the study is to determine if BI-505 can deepen therapeutic response and thereby prevent or delay relapse in MM patients undergoing autologous stem cell transplant with high-dose melphalan.
BioInvent International said it will analyze the possibility to obtain release of the clinical hold and will provide updates when there is further information to report.
About BI-505
BI-505 is a human antibody targeting ICAM-1, a protein that is elevated in MM cells. BI-505 has been shown to attack MM in 2 ways—by inducing apoptosis in MM cells and by engaging macrophages to attack and kill MM cells.
According to BioInvent International, the development strategy for BI-505 is focused on eliminating residual disease by combining the antibody with modern standard-of-care drugs used to treat MM.
The company said BI-505 proved safe in a phase 1 trial of patients with relapsed/refractory MM, as well as demonstrating “signs of a positive effect against the disease.” This study was published in Clinical Cancer Research in June 2015.
BI-505 has received orphan drug designation as a treatment for MM from both the FDA and the European Medicines Agency.
A phase 2 study of the antibody BI-505 in patients with multiple myeloma (MM) has been placed on full clinical hold.
BioInvent International, the company developing BI-505, said it has received verbal notice of the clinical hold from the US Food and Drug Administration (FDA).
The clinical hold means no new subjects can be enrolled on the trial, and there can be no further dosing of subjects who are already enrolled.
BioInvent International said it has yet to receive written notice of the clinical hold from the FDA. However, based on verbal communications, the clinical hold is due to an adverse cardiopulmonary event.
The study (NCT02756728) is being conducted by BioInvent International in collaboration with investigators at the University of Pennsylvania.
The goal of the study is to determine if BI-505 can deepen therapeutic response and thereby prevent or delay relapse in MM patients undergoing autologous stem cell transplant with high-dose melphalan.
BioInvent International said it will analyze the possibility to obtain release of the clinical hold and will provide updates when there is further information to report.
About BI-505
BI-505 is a human antibody targeting ICAM-1, a protein that is elevated in MM cells. BI-505 has been shown to attack MM in 2 ways—by inducing apoptosis in MM cells and by engaging macrophages to attack and kill MM cells.
According to BioInvent International, the development strategy for BI-505 is focused on eliminating residual disease by combining the antibody with modern standard-of-care drugs used to treat MM.
The company said BI-505 proved safe in a phase 1 trial of patients with relapsed/refractory MM, as well as demonstrating “signs of a positive effect against the disease.” This study was published in Clinical Cancer Research in June 2015.
BI-505 has received orphan drug designation as a treatment for MM from both the FDA and the European Medicines Agency.
A phase 2 study of the antibody BI-505 in patients with multiple myeloma (MM) has been placed on full clinical hold.
BioInvent International, the company developing BI-505, said it has received verbal notice of the clinical hold from the US Food and Drug Administration (FDA).
The clinical hold means no new subjects can be enrolled on the trial, and there can be no further dosing of subjects who are already enrolled.
BioInvent International said it has yet to receive written notice of the clinical hold from the FDA. However, based on verbal communications, the clinical hold is due to an adverse cardiopulmonary event.
The study (NCT02756728) is being conducted by BioInvent International in collaboration with investigators at the University of Pennsylvania.
The goal of the study is to determine if BI-505 can deepen therapeutic response and thereby prevent or delay relapse in MM patients undergoing autologous stem cell transplant with high-dose melphalan.
BioInvent International said it will analyze the possibility to obtain release of the clinical hold and will provide updates when there is further information to report.
About BI-505
BI-505 is a human antibody targeting ICAM-1, a protein that is elevated in MM cells. BI-505 has been shown to attack MM in 2 ways—by inducing apoptosis in MM cells and by engaging macrophages to attack and kill MM cells.
According to BioInvent International, the development strategy for BI-505 is focused on eliminating residual disease by combining the antibody with modern standard-of-care drugs used to treat MM.
The company said BI-505 proved safe in a phase 1 trial of patients with relapsed/refractory MM, as well as demonstrating “signs of a positive effect against the disease.” This study was published in Clinical Cancer Research in June 2015.
BI-505 has received orphan drug designation as a treatment for MM from both the FDA and the European Medicines Agency.
Panobinostat might treat high-risk ALL subtype
Photo courtesy of Novartis
Researchers say they have identified a high-risk subtype of acute lymphoblastic leukemia (ALL) that may respond to treatment with the histone deacetylase (HDAC) inhibitor panobinostat.
The ALL subtype is characterized by chromosomal rearrangements that involve the MEF2D gene and 1 of 6 partner genes, most often BCL9.
The researchers described this subtype, known as MEF2D-rearranged ALL, in Nature Communications.
“MEF2D is a transcription factor that switches on expression of other genes during normal development,” said study author Charles Mullighan, MD, MBBS, of the St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We found that MEF2D chromosomal rearrangements disrupt expression of those genes and create a vulnerability to at least one targeted therapy, the drug panobinostat.”
Dr Mullighan and his colleagues performed genomic analyses on samples from a total of 1724 children, adolescents, and adults with ALL. This revealed 52 patients with MEF2D rearrangements.
MEF2D-rearranged ALL
The researchers calculated that MEF2D-rearranged ALL accounted for 5.3% of the ALL cases whose genetic basis was unknown.
The team also noted that MEF2D-rearranged ALL occurred most frequently in adolescents. Although, overall, ALL occurs most often in children between 3 and 5 years old, the average patient with MEF2D-rearranged ALL was 14.
In addition, MEF2D-rearranged ALL was associated with reduced survival when compared to some other ALL subtypes. The 5-year cancer-free survival for MEF2D-rearranged ALL patients was 71.6%.
The researchers also found that a fusion protein resulting from the MEF2D rearrangement led to sustained growth of mouse cells when compared to wild-type MEF2D or other proteins.
“That indicates the MEF2D fusion is a key step in transforming a normal white blood cell with a finite lifespan into a leukemic cell that is immortal,” Dr Mullighan said.
Role for panobinostat
MEF2D-rearranged leukemic cells produced high levels of HDAC9, which is targeted by panobinostat.
The researchers tested panobinostat in the lab and found the drug stopped proliferation of human MEF2D-rearranged leukemic cells.
Dr Mullighan said MEF2D-rearranged leukemic cells were exquisitely sensitive to panobinostat, which suggested the drug might function in a more targeted manner against cells with the rearrangement.
“If further testing of panobinostat, either alone or in combination therapy, confirms the anti-proliferative activity, that would lay the foundation for a clinical trial in patients, particularly patients with high-risk disease or those who have relapsed,” he said.
Photo courtesy of Novartis
Researchers say they have identified a high-risk subtype of acute lymphoblastic leukemia (ALL) that may respond to treatment with the histone deacetylase (HDAC) inhibitor panobinostat.
The ALL subtype is characterized by chromosomal rearrangements that involve the MEF2D gene and 1 of 6 partner genes, most often BCL9.
The researchers described this subtype, known as MEF2D-rearranged ALL, in Nature Communications.
“MEF2D is a transcription factor that switches on expression of other genes during normal development,” said study author Charles Mullighan, MD, MBBS, of the St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We found that MEF2D chromosomal rearrangements disrupt expression of those genes and create a vulnerability to at least one targeted therapy, the drug panobinostat.”
Dr Mullighan and his colleagues performed genomic analyses on samples from a total of 1724 children, adolescents, and adults with ALL. This revealed 52 patients with MEF2D rearrangements.
MEF2D-rearranged ALL
The researchers calculated that MEF2D-rearranged ALL accounted for 5.3% of the ALL cases whose genetic basis was unknown.
The team also noted that MEF2D-rearranged ALL occurred most frequently in adolescents. Although, overall, ALL occurs most often in children between 3 and 5 years old, the average patient with MEF2D-rearranged ALL was 14.
In addition, MEF2D-rearranged ALL was associated with reduced survival when compared to some other ALL subtypes. The 5-year cancer-free survival for MEF2D-rearranged ALL patients was 71.6%.
The researchers also found that a fusion protein resulting from the MEF2D rearrangement led to sustained growth of mouse cells when compared to wild-type MEF2D or other proteins.
“That indicates the MEF2D fusion is a key step in transforming a normal white blood cell with a finite lifespan into a leukemic cell that is immortal,” Dr Mullighan said.
Role for panobinostat
MEF2D-rearranged leukemic cells produced high levels of HDAC9, which is targeted by panobinostat.
The researchers tested panobinostat in the lab and found the drug stopped proliferation of human MEF2D-rearranged leukemic cells.
Dr Mullighan said MEF2D-rearranged leukemic cells were exquisitely sensitive to panobinostat, which suggested the drug might function in a more targeted manner against cells with the rearrangement.
“If further testing of panobinostat, either alone or in combination therapy, confirms the anti-proliferative activity, that would lay the foundation for a clinical trial in patients, particularly patients with high-risk disease or those who have relapsed,” he said.
Photo courtesy of Novartis
Researchers say they have identified a high-risk subtype of acute lymphoblastic leukemia (ALL) that may respond to treatment with the histone deacetylase (HDAC) inhibitor panobinostat.
The ALL subtype is characterized by chromosomal rearrangements that involve the MEF2D gene and 1 of 6 partner genes, most often BCL9.
The researchers described this subtype, known as MEF2D-rearranged ALL, in Nature Communications.
“MEF2D is a transcription factor that switches on expression of other genes during normal development,” said study author Charles Mullighan, MD, MBBS, of the St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We found that MEF2D chromosomal rearrangements disrupt expression of those genes and create a vulnerability to at least one targeted therapy, the drug panobinostat.”
Dr Mullighan and his colleagues performed genomic analyses on samples from a total of 1724 children, adolescents, and adults with ALL. This revealed 52 patients with MEF2D rearrangements.
MEF2D-rearranged ALL
The researchers calculated that MEF2D-rearranged ALL accounted for 5.3% of the ALL cases whose genetic basis was unknown.
The team also noted that MEF2D-rearranged ALL occurred most frequently in adolescents. Although, overall, ALL occurs most often in children between 3 and 5 years old, the average patient with MEF2D-rearranged ALL was 14.
In addition, MEF2D-rearranged ALL was associated with reduced survival when compared to some other ALL subtypes. The 5-year cancer-free survival for MEF2D-rearranged ALL patients was 71.6%.
The researchers also found that a fusion protein resulting from the MEF2D rearrangement led to sustained growth of mouse cells when compared to wild-type MEF2D or other proteins.
“That indicates the MEF2D fusion is a key step in transforming a normal white blood cell with a finite lifespan into a leukemic cell that is immortal,” Dr Mullighan said.
Role for panobinostat
MEF2D-rearranged leukemic cells produced high levels of HDAC9, which is targeted by panobinostat.
The researchers tested panobinostat in the lab and found the drug stopped proliferation of human MEF2D-rearranged leukemic cells.
Dr Mullighan said MEF2D-rearranged leukemic cells were exquisitely sensitive to panobinostat, which suggested the drug might function in a more targeted manner against cells with the rearrangement.
“If further testing of panobinostat, either alone or in combination therapy, confirms the anti-proliferative activity, that would lay the foundation for a clinical trial in patients, particularly patients with high-risk disease or those who have relapsed,” he said.