User login
AVAHO
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Denosumab now dominant therapy for osteoporosis linked to cancer
Amid a substantial expansion of therapies in several drug classes for the treatment of osteoporosis, there has been a notable increase in the prescription of denosumab for patients with a cancer-related indication.
In an analysis of claims data from January 2009 to March 2020, the bisphosphonate alendronate represented more than 50% of all prescriptions for bone-directed therapies, but growth in the use of the monoclonal antibody denosumab overall and in cancer-related indications particularly was steady throughout the study period.
“In the malignancy cohort, alendronate and zoledronic acid were each used in approximately 30% of individuals at the onset of the study, but use of both then declined,” Sara Cromer, MD, reported at the annual meeting of the Endocrine Society.
For malignancy-based prescriptions, denosumab surpassed either bisphosphonate by 2013 and then continued to rise.
Denosumab use “reached approximately 50% of all bone-directed medication use in the malignancy cohort” by the end of the study period, said Dr. Cromer, a clinical research fellow in endocrinology at Massachusetts General Hospital, Boston.
The claims data for this analysis was drawn from the Clinformatics Data Mart. The analysis was restricted to individuals aged older than 50 years who received a prescription for a bone-directed therapy. The 15.48 million prescriptions evaluated were drawn from 1.46 million unique individuals. The mean age was 69 years, and 89% of those prescribed a drug were women.
Oncologic indications one of two tracked cohorts
In the context of a large expansion of treatment options in several drug classes for osteoporosis, the objective of this claims analysis was to document trends in treatment choice, according to Dr. Cromer. She and her coinvestigators looked at prescriptions overall as well as in two cohorts defined by ICD codes. One included patients prescribed a prescription by an oncologist. The other included everyone else.
When all prescriptions for bone-directed therapy were evaluated over the study period, alendronate was the most commonly prescribed therapy, and its use increased over time. Prescriptions of zoledronic acid also rose, doubling over the study period, but use was very low in the beginning and it never climbed above 5%.
The proportion of prescriptions written for bisphosphonates other than alendronate and zoledronic acid “declined steadily” over the study period, Dr. Cromer reported.
Denosumab, a monoclonal antibody that targets a step in the process important to maturation of osteoclasts, was approved in 2010. It accounted for 10% of all prescriptions for osteoporosis by 2015 and 15% by 2018. It was still rising through the end of the study period.
In contrast, prescriptions of raloxifene, a selective estrogen receptor modulator, began to decline after 2013. In general, the rates of prescriptions for other agents, including some of the more recently approved drugs, such as teriparatide, abaloparatide, and romosozumab, changed very little over the study period. None of these therapies ever represented more than 2% of prescriptions.
When looking at the cohort of patients who received a bone-directed reason for a noncancer indication, the trends “paralleled those in the all-user analysis,” Dr. Cromer reported.
Denosumab use greater in privately insured
In the malignancy cohort, the decline in the use of bisphosphonates and the rise in the use of denosumab were most pronounced in patients who were privately insured. The increased use of denosumab over the study period “outpaced gains in use of other agents despite guidelines,” said Dr. Cromer, referring to the those issued by the Endocrine Society in 2019 .
In those guidelines, written for management of postmenopausal women at high risk of fractures, bisphosphonates are recommended for initial treatment while denosumab is recommended as an alternative. However, those guidelines do not provide specific recommendations for therapies directed at osteoporosis associated with cancer.
Guidelines for this population exist, including one published by the American Society of Clinical Oncology in 2019.
In the ASCO guidelines, oral bisphosphonates, intravenous bisphosphonates, and subcutaneous denosumab were all identified as “efficacious options,” according to Charles L. Shapiro, MD, director of breast cancer translational research, Mount Sinai Health System, New York.
Specifically, “all three of them work to reduce fractures and improve bone density in women with breast cancer in whom you are trying to prevent or treat osteoporosis,” Dr. Shapiro said in an interview.
There might be relative advantages for one therapy over another in specific subgroups defined by type of cancer or stage of cancer, but trials are not definitive for such outcomes as overall survival. Citing one comparative study associating denosumab with an 18% delay to first skeletal event in women with metastatic breast cancer, Dr. Shapiro observed, “I personally don’t consider an 18% delay [for this outcome] to be that clinically meaningful.”
Although major guidelines from ASCO have not so far favored denosumab over any bisphosphonate in routine care, Dr. Shapiro did not rule out the possibility that future studies will show differences.
Dr. Comer and Dr. Shapiro reported no relevant conflicts of interest.
Amid a substantial expansion of therapies in several drug classes for the treatment of osteoporosis, there has been a notable increase in the prescription of denosumab for patients with a cancer-related indication.
In an analysis of claims data from January 2009 to March 2020, the bisphosphonate alendronate represented more than 50% of all prescriptions for bone-directed therapies, but growth in the use of the monoclonal antibody denosumab overall and in cancer-related indications particularly was steady throughout the study period.
“In the malignancy cohort, alendronate and zoledronic acid were each used in approximately 30% of individuals at the onset of the study, but use of both then declined,” Sara Cromer, MD, reported at the annual meeting of the Endocrine Society.
For malignancy-based prescriptions, denosumab surpassed either bisphosphonate by 2013 and then continued to rise.
Denosumab use “reached approximately 50% of all bone-directed medication use in the malignancy cohort” by the end of the study period, said Dr. Cromer, a clinical research fellow in endocrinology at Massachusetts General Hospital, Boston.
The claims data for this analysis was drawn from the Clinformatics Data Mart. The analysis was restricted to individuals aged older than 50 years who received a prescription for a bone-directed therapy. The 15.48 million prescriptions evaluated were drawn from 1.46 million unique individuals. The mean age was 69 years, and 89% of those prescribed a drug were women.
Oncologic indications one of two tracked cohorts
In the context of a large expansion of treatment options in several drug classes for osteoporosis, the objective of this claims analysis was to document trends in treatment choice, according to Dr. Cromer. She and her coinvestigators looked at prescriptions overall as well as in two cohorts defined by ICD codes. One included patients prescribed a prescription by an oncologist. The other included everyone else.
When all prescriptions for bone-directed therapy were evaluated over the study period, alendronate was the most commonly prescribed therapy, and its use increased over time. Prescriptions of zoledronic acid also rose, doubling over the study period, but use was very low in the beginning and it never climbed above 5%.
The proportion of prescriptions written for bisphosphonates other than alendronate and zoledronic acid “declined steadily” over the study period, Dr. Cromer reported.
Denosumab, a monoclonal antibody that targets a step in the process important to maturation of osteoclasts, was approved in 2010. It accounted for 10% of all prescriptions for osteoporosis by 2015 and 15% by 2018. It was still rising through the end of the study period.
In contrast, prescriptions of raloxifene, a selective estrogen receptor modulator, began to decline after 2013. In general, the rates of prescriptions for other agents, including some of the more recently approved drugs, such as teriparatide, abaloparatide, and romosozumab, changed very little over the study period. None of these therapies ever represented more than 2% of prescriptions.
When looking at the cohort of patients who received a bone-directed reason for a noncancer indication, the trends “paralleled those in the all-user analysis,” Dr. Cromer reported.
Denosumab use greater in privately insured
In the malignancy cohort, the decline in the use of bisphosphonates and the rise in the use of denosumab were most pronounced in patients who were privately insured. The increased use of denosumab over the study period “outpaced gains in use of other agents despite guidelines,” said Dr. Cromer, referring to the those issued by the Endocrine Society in 2019 .
In those guidelines, written for management of postmenopausal women at high risk of fractures, bisphosphonates are recommended for initial treatment while denosumab is recommended as an alternative. However, those guidelines do not provide specific recommendations for therapies directed at osteoporosis associated with cancer.
Guidelines for this population exist, including one published by the American Society of Clinical Oncology in 2019.
In the ASCO guidelines, oral bisphosphonates, intravenous bisphosphonates, and subcutaneous denosumab were all identified as “efficacious options,” according to Charles L. Shapiro, MD, director of breast cancer translational research, Mount Sinai Health System, New York.
Specifically, “all three of them work to reduce fractures and improve bone density in women with breast cancer in whom you are trying to prevent or treat osteoporosis,” Dr. Shapiro said in an interview.
There might be relative advantages for one therapy over another in specific subgroups defined by type of cancer or stage of cancer, but trials are not definitive for such outcomes as overall survival. Citing one comparative study associating denosumab with an 18% delay to first skeletal event in women with metastatic breast cancer, Dr. Shapiro observed, “I personally don’t consider an 18% delay [for this outcome] to be that clinically meaningful.”
Although major guidelines from ASCO have not so far favored denosumab over any bisphosphonate in routine care, Dr. Shapiro did not rule out the possibility that future studies will show differences.
Dr. Comer and Dr. Shapiro reported no relevant conflicts of interest.
Amid a substantial expansion of therapies in several drug classes for the treatment of osteoporosis, there has been a notable increase in the prescription of denosumab for patients with a cancer-related indication.
In an analysis of claims data from January 2009 to March 2020, the bisphosphonate alendronate represented more than 50% of all prescriptions for bone-directed therapies, but growth in the use of the monoclonal antibody denosumab overall and in cancer-related indications particularly was steady throughout the study period.
“In the malignancy cohort, alendronate and zoledronic acid were each used in approximately 30% of individuals at the onset of the study, but use of both then declined,” Sara Cromer, MD, reported at the annual meeting of the Endocrine Society.
For malignancy-based prescriptions, denosumab surpassed either bisphosphonate by 2013 and then continued to rise.
Denosumab use “reached approximately 50% of all bone-directed medication use in the malignancy cohort” by the end of the study period, said Dr. Cromer, a clinical research fellow in endocrinology at Massachusetts General Hospital, Boston.
The claims data for this analysis was drawn from the Clinformatics Data Mart. The analysis was restricted to individuals aged older than 50 years who received a prescription for a bone-directed therapy. The 15.48 million prescriptions evaluated were drawn from 1.46 million unique individuals. The mean age was 69 years, and 89% of those prescribed a drug were women.
Oncologic indications one of two tracked cohorts
In the context of a large expansion of treatment options in several drug classes for osteoporosis, the objective of this claims analysis was to document trends in treatment choice, according to Dr. Cromer. She and her coinvestigators looked at prescriptions overall as well as in two cohorts defined by ICD codes. One included patients prescribed a prescription by an oncologist. The other included everyone else.
When all prescriptions for bone-directed therapy were evaluated over the study period, alendronate was the most commonly prescribed therapy, and its use increased over time. Prescriptions of zoledronic acid also rose, doubling over the study period, but use was very low in the beginning and it never climbed above 5%.
The proportion of prescriptions written for bisphosphonates other than alendronate and zoledronic acid “declined steadily” over the study period, Dr. Cromer reported.
Denosumab, a monoclonal antibody that targets a step in the process important to maturation of osteoclasts, was approved in 2010. It accounted for 10% of all prescriptions for osteoporosis by 2015 and 15% by 2018. It was still rising through the end of the study period.
In contrast, prescriptions of raloxifene, a selective estrogen receptor modulator, began to decline after 2013. In general, the rates of prescriptions for other agents, including some of the more recently approved drugs, such as teriparatide, abaloparatide, and romosozumab, changed very little over the study period. None of these therapies ever represented more than 2% of prescriptions.
When looking at the cohort of patients who received a bone-directed reason for a noncancer indication, the trends “paralleled those in the all-user analysis,” Dr. Cromer reported.
Denosumab use greater in privately insured
In the malignancy cohort, the decline in the use of bisphosphonates and the rise in the use of denosumab were most pronounced in patients who were privately insured. The increased use of denosumab over the study period “outpaced gains in use of other agents despite guidelines,” said Dr. Cromer, referring to the those issued by the Endocrine Society in 2019 .
In those guidelines, written for management of postmenopausal women at high risk of fractures, bisphosphonates are recommended for initial treatment while denosumab is recommended as an alternative. However, those guidelines do not provide specific recommendations for therapies directed at osteoporosis associated with cancer.
Guidelines for this population exist, including one published by the American Society of Clinical Oncology in 2019.
In the ASCO guidelines, oral bisphosphonates, intravenous bisphosphonates, and subcutaneous denosumab were all identified as “efficacious options,” according to Charles L. Shapiro, MD, director of breast cancer translational research, Mount Sinai Health System, New York.
Specifically, “all three of them work to reduce fractures and improve bone density in women with breast cancer in whom you are trying to prevent or treat osteoporosis,” Dr. Shapiro said in an interview.
There might be relative advantages for one therapy over another in specific subgroups defined by type of cancer or stage of cancer, but trials are not definitive for such outcomes as overall survival. Citing one comparative study associating denosumab with an 18% delay to first skeletal event in women with metastatic breast cancer, Dr. Shapiro observed, “I personally don’t consider an 18% delay [for this outcome] to be that clinically meaningful.”
Although major guidelines from ASCO have not so far favored denosumab over any bisphosphonate in routine care, Dr. Shapiro did not rule out the possibility that future studies will show differences.
Dr. Comer and Dr. Shapiro reported no relevant conflicts of interest.
FROM ENDO 2021
Paving the way for diversity in clinical trials
“I’m the first person in my circle of family and friends to participate in a clinical trial.”
Five years ago, Rhonda Long was diagnosed with cholangiocarcinoma, a rare bile duct cancer that’s seen in only about 8,000 Americans each year.
At the time, Mrs. Long, who is Black, said her doctor in Dayton, Ohio, told her she was not a candidate for surgery and suggested palliative care. After seeking a second opinion at Duke University Medical Center, Durham, N.C., where her sister worked, the 51-year-old wife and mother of two had surgery, radiation, and chemotherapy there in North Carolina. When the chemo stopped working after 3 months, her oncologist at Duke referred her to a colleague at Massachusetts General Hospital in Boston, where she was accepted into a clinical trial.
“In 2019, I traveled to Boston from Dayton, Ohio, every 3 weeks for labs and scans, to make sure that the drug wasn’t doing more harm than good, making sure that the drug as developed was maintaining, shrinking, or even eliminating the disease. Physically and financially, it takes a toll on you and loved ones.”
Her medical insurance did not cover the direct expenses from the clinical trial, and she was spending $1,000-$1,500 each trip. Sometimes they drove the 15 hours to Boston, and sometimes they flew on the cheapest flight they could find.
It’s not an unfamiliar story: people traveling, often long distances, to take part in clinical trials they hope will save their lives.
The Lazarex Cancer Foundation of Danville, Calif., helped Mrs. Long do just that.
Marya Shegog, PhD, health equity and diversity coordinator at Lazarex, said that a patient travels an average of 500 miles to participate in a trial.
The financial hurdles often prevent patients from taking part in clinical trials, Dr. Shegog said. “When you are sick, and you have a disease that may be terminal, you start thinking about setting your things in order.”
Many patients have to make a decision.
“Do I bankrupt my family on trying and hoping that this drug works and helps me live longer, or do I start setting things in order so that when I’m gone, they’re okay or at least better than if I wouldn’t have spent all the money traveling back and forth.”
Dr. Shegog, a 17-year cancer survivor, says when she was battling cervical cancer, a clinical trial was never offered or explored.
Lazarex has been helping cancer patients who have run out of options for 15 years. It identifies clinical trial opportunities and reimburses patients for all travel costs. Last year, Lazarex reimbursed more than 1,000 cancer patients. And it has supported more than 6,000 people since opening its doors.
“Lazarex exists to help remove the barriers of people not being able to participate in trials,” Dr. Shegog said. “It’s systemic that the medical system does not treat patients the same and oftentimes does not offer or make aware the opportunities for African Americans to participate.”
But now, thanks in part to COVID-19, new possibilities are taking shape. The pandemic has changed the landscape for trials, forcing many of them to go virtual, which allows patients to schedule telehealth visits and get some services like bloodwork and CT or MRI scans closer to home. Mrs. Long’s trial eventually went virtual.
“It was absolutely fantastic,” she said. “Having the trial locally, it saves us money, it saves wear and tear on my body. Being in the car, being in an airport or in a plane and in a hotel, all of that wears on you physically.”
The move to virtual studies may have lasting effects on research and treatment.
“The current pandemic has forced us to reexamine all of the traditional burdens we place on patients as it relates to receiving cancer treatment,” said Hala Borno, MD, an assistant professor of medicine at the University of California, San Francisco. “Whether they’re coming to our health care facility to see a clinician, for diagnostics such as blood draws and scans, or to receive therapy, this pandemic has challenged us to explore other possibilities that minimize the risk of exposure to SARS-CoV-2. What I find striking is that it has helped us operationalize use of telemedicine and the delivery of care closer to home.”
This is especially encouraging news for minority patients whose participation in trials has for years lagged well behind that of Whites.
But travel is not the only reason. Racial disparities in clinical trials have long been an issue that’s just another part of the implicit bias in health care.
Compared with White people, Black people are largely at higher risk for heart disease, cancer, stroke, diabetes, asthma, and even mental health problems.
And it’s not just African Americans. Asians, Hispanics, Native Americans, and Alaska Natives are all underrepresented in trials at a time when there is growing evidence that drugs may have different effects on different populations.
Dr. Borno is an oncologist who specializes in prostate cancer, a disease that she says shows a “significant disparity,” where Black men are two times more likely to die from advanced prostate cancer, compared with white men. Yet Black men make up just 3% of advanced therapeutic trials.
“A lack of diversity and inclusion in clinical trials is unacceptable,” she said. “If we continue to underrecruit racial/ethnic minorities and older adults to therapeutic clinical trials, we will not be powered to make valid conclusions regarding safety and efficacy in those patient populations. As a result, we can do harm.”
Dr. Borno said that telehealth and telemedicine are not cure-alls, and digital health solutions don’t work for all patients. Approaches, she says, must be tailored to the individual, or disparities could worsen.
In 2020, the Food and Drug Administration approved 53 new drugs. Overall, 32,000 patients took part in these trials. On average, 75% were White, 8% were Black, 6% were Asian, and 11% were Hispanic.
Here’s one stark example of the issue. In 2015, the FDA approved ixazomib (Ninlaro), a promising new drug for multiple myeloma, a blood cancer that affects Black people at disproportionately higher rates than White people. In the United States, one in five people diagnosed with multiple myeloma are Black people. They are more than twice as likely to get the disease as White people. Yet during the clinical trial of 722 participants, only 13 patients, or 1.8%, were Black.
The American Cancer Society estimates that more than 600,000 Americans will die from cancer this year. Historically, Black Americans have the highest death rate and the shortest survival of any racial or ethnic group, stemming largely, it concluded, from centuries of structural racism.
According to Jamie Freedman, MD, head of U.S. medical affairs at Genentech, a global pharmaceutical company, the lack of diversity is often tied to where studies are run.
“Companies tend to choose major academic medical centers where there is a high volume of clinical trial work. When you go to the same tried and true hospitals repeatedly, the pool of patients becomes very homogeneous and tends to be primarily white,” he said. “It’s critical to bring more trials into the community setting by including new sites that can reach underrepresented groups, and Genentech is making significant progress in that area.”
Dr. Freedman believes that, while access is a big hurdle, it doesn’t end there.
“Many patients have a lack of trust in the health care system,” he said. “There are also issues around underserved communities being able to afford quality care, so it’s important to keep time and financial burdens in mind when designing trials to help mitigate barriers such as travel, parking, time off work, and child care.”
Genentech started its diversity and inclusion effort several years ago. Dr. Freeman said that, until more trials become diverse, Black Americans will continue to pay the price. “I think they’re losing their lives in part due to lack of access to these trials. And that is why Genentech and all of us in the health care industry need to change how we design and enroll these studies. We have a long way to go, but I think the steps we’re taking are leading us in the right direction.”
Jennifer Jones-McMeans, PhD, director of global clinical affairs at Abbott Pharmaceuticals, is a clinical research scientist who has designed and led many clinical trials.
She said that Abbott is actively working on solutions.
“We have designed our trials to reduce the barriers to participation and expand access,” she said. “This can be as simple as providing transportation services or home visits for those who are housebound. We’re taking it a step further and providing home health services where someone comes to the home and provides follow-up visits there.”
They also provide interpretation services to address any language barriers.
“We are reaching out to a new set of talented investigators who work closely with underrepresented communities. They are very much wedded and supportive of the communities they treat. By working with doctors within these communities, it expands access to new therapies.”
Spokesperson Keanna Ghazvini said that Pfizer Pharmaceuticals is also committed to increasing minority participation in trials.
“We know that if historically underserved populations are left out of clinical trials, they risk not benefiting from medical breakthroughs down the line,” she said.
The National Institutes of Health’s National Library of Medicine maintains the clinicaltrials.gov database.
There, you can find information on nearly 372,000 publicly and privately supported clinical trials happening in all 50 states and 219 countries. Many are funded by the NIH, but not all of these studies have been evaluated by the U.S. government.
Andrea Denicoff, a nurse consultant at the National Cancer Institute and head of clinical trials operations for the NCI’s National Clinical Trials Network, has been involved in clinical research at the NIH for 35 years.
“It’s really important that our publicly funded trials represent the people of the country,” she said. “There are some cancers that we’re doing a good job in enrolling minorities, and other cancers we need to do a much better job in having a diverse representation in our trials.”
Ms. Denicoff believed opening trials in places where people live is key, but having a diverse clinical trials team is as important.
“We need to reinforce that cancer centers across the country have open doors, and anyone with cancer feels comfortable getting care at that center, and that also includes discussing the option to participate in clinical trials when one might be available. We know from research that when people are invited and asked about trial participation and educated about them, they’ll be much more interested in joining them.”
Ms. Denicoff said that, during the pandemic, the NCI quickly came up with guidance to allow trial sites to send patients their oral study drugs and set up virtual visits. She believes it may help increase future access.
‘Lola Fashoyin-Aje, MD, associate director for science and policy to address disparities in the Oncology Center of Excellence at the FDA, says the agency firmly believes clinical trials should represent the patients who will ultimately get the drug if it’s approved.
But the FDA’s power to require diversity in trials is limited.
“It is important to point out that there are legal constraints which limit’s FDA’s authority to require specific proportional representation in clinical trials by demographic factors,” Dr. Fashoyin-Aje said.
Still, some researchers feel the FDA should play a bigger role. The question is: Should diversity be mandated?
Rhonda Long is now back in Boston to start a new trial, with a new drug that targets her specific mutation. She will be there for 2 months. Once again, Lazarex will help cover some of the cost.
She wants people of color to understand that they are missing out on the promise of new cancer drugs and extended life.
“I feel like there’s not enough emphasis on clinical trials, I don’t believe there’s enough emphasis on second opinions, I don’t think there’s enough emphasis that medicine happens outside our borders, outside of our communities. Clinical trials that don’t have a broad range of participants, how do we know how effective they are if Black and brown people, Asian or Latin American people aren’t represented in the trial?”
And with more trials adopting virtual elements, she said it’s time for minorities to get on board.
Dr. Freedman believed the groundwork is being laid for that to happen. “I don’t think we’ll ever return back to the way we used to do things, where everything has to be done at the clinical trial site. I just don’t think we’re ever going back.”
A version of this article first appeared on WebMD.com.
“I’m the first person in my circle of family and friends to participate in a clinical trial.”
Five years ago, Rhonda Long was diagnosed with cholangiocarcinoma, a rare bile duct cancer that’s seen in only about 8,000 Americans each year.
At the time, Mrs. Long, who is Black, said her doctor in Dayton, Ohio, told her she was not a candidate for surgery and suggested palliative care. After seeking a second opinion at Duke University Medical Center, Durham, N.C., where her sister worked, the 51-year-old wife and mother of two had surgery, radiation, and chemotherapy there in North Carolina. When the chemo stopped working after 3 months, her oncologist at Duke referred her to a colleague at Massachusetts General Hospital in Boston, where she was accepted into a clinical trial.
“In 2019, I traveled to Boston from Dayton, Ohio, every 3 weeks for labs and scans, to make sure that the drug wasn’t doing more harm than good, making sure that the drug as developed was maintaining, shrinking, or even eliminating the disease. Physically and financially, it takes a toll on you and loved ones.”
Her medical insurance did not cover the direct expenses from the clinical trial, and she was spending $1,000-$1,500 each trip. Sometimes they drove the 15 hours to Boston, and sometimes they flew on the cheapest flight they could find.
It’s not an unfamiliar story: people traveling, often long distances, to take part in clinical trials they hope will save their lives.
The Lazarex Cancer Foundation of Danville, Calif., helped Mrs. Long do just that.
Marya Shegog, PhD, health equity and diversity coordinator at Lazarex, said that a patient travels an average of 500 miles to participate in a trial.
The financial hurdles often prevent patients from taking part in clinical trials, Dr. Shegog said. “When you are sick, and you have a disease that may be terminal, you start thinking about setting your things in order.”
Many patients have to make a decision.
“Do I bankrupt my family on trying and hoping that this drug works and helps me live longer, or do I start setting things in order so that when I’m gone, they’re okay or at least better than if I wouldn’t have spent all the money traveling back and forth.”
Dr. Shegog, a 17-year cancer survivor, says when she was battling cervical cancer, a clinical trial was never offered or explored.
Lazarex has been helping cancer patients who have run out of options for 15 years. It identifies clinical trial opportunities and reimburses patients for all travel costs. Last year, Lazarex reimbursed more than 1,000 cancer patients. And it has supported more than 6,000 people since opening its doors.
“Lazarex exists to help remove the barriers of people not being able to participate in trials,” Dr. Shegog said. “It’s systemic that the medical system does not treat patients the same and oftentimes does not offer or make aware the opportunities for African Americans to participate.”
But now, thanks in part to COVID-19, new possibilities are taking shape. The pandemic has changed the landscape for trials, forcing many of them to go virtual, which allows patients to schedule telehealth visits and get some services like bloodwork and CT or MRI scans closer to home. Mrs. Long’s trial eventually went virtual.
“It was absolutely fantastic,” she said. “Having the trial locally, it saves us money, it saves wear and tear on my body. Being in the car, being in an airport or in a plane and in a hotel, all of that wears on you physically.”
The move to virtual studies may have lasting effects on research and treatment.
“The current pandemic has forced us to reexamine all of the traditional burdens we place on patients as it relates to receiving cancer treatment,” said Hala Borno, MD, an assistant professor of medicine at the University of California, San Francisco. “Whether they’re coming to our health care facility to see a clinician, for diagnostics such as blood draws and scans, or to receive therapy, this pandemic has challenged us to explore other possibilities that minimize the risk of exposure to SARS-CoV-2. What I find striking is that it has helped us operationalize use of telemedicine and the delivery of care closer to home.”
This is especially encouraging news for minority patients whose participation in trials has for years lagged well behind that of Whites.
But travel is not the only reason. Racial disparities in clinical trials have long been an issue that’s just another part of the implicit bias in health care.
Compared with White people, Black people are largely at higher risk for heart disease, cancer, stroke, diabetes, asthma, and even mental health problems.
And it’s not just African Americans. Asians, Hispanics, Native Americans, and Alaska Natives are all underrepresented in trials at a time when there is growing evidence that drugs may have different effects on different populations.
Dr. Borno is an oncologist who specializes in prostate cancer, a disease that she says shows a “significant disparity,” where Black men are two times more likely to die from advanced prostate cancer, compared with white men. Yet Black men make up just 3% of advanced therapeutic trials.
“A lack of diversity and inclusion in clinical trials is unacceptable,” she said. “If we continue to underrecruit racial/ethnic minorities and older adults to therapeutic clinical trials, we will not be powered to make valid conclusions regarding safety and efficacy in those patient populations. As a result, we can do harm.”
Dr. Borno said that telehealth and telemedicine are not cure-alls, and digital health solutions don’t work for all patients. Approaches, she says, must be tailored to the individual, or disparities could worsen.
In 2020, the Food and Drug Administration approved 53 new drugs. Overall, 32,000 patients took part in these trials. On average, 75% were White, 8% were Black, 6% were Asian, and 11% were Hispanic.
Here’s one stark example of the issue. In 2015, the FDA approved ixazomib (Ninlaro), a promising new drug for multiple myeloma, a blood cancer that affects Black people at disproportionately higher rates than White people. In the United States, one in five people diagnosed with multiple myeloma are Black people. They are more than twice as likely to get the disease as White people. Yet during the clinical trial of 722 participants, only 13 patients, or 1.8%, were Black.
The American Cancer Society estimates that more than 600,000 Americans will die from cancer this year. Historically, Black Americans have the highest death rate and the shortest survival of any racial or ethnic group, stemming largely, it concluded, from centuries of structural racism.
According to Jamie Freedman, MD, head of U.S. medical affairs at Genentech, a global pharmaceutical company, the lack of diversity is often tied to where studies are run.
“Companies tend to choose major academic medical centers where there is a high volume of clinical trial work. When you go to the same tried and true hospitals repeatedly, the pool of patients becomes very homogeneous and tends to be primarily white,” he said. “It’s critical to bring more trials into the community setting by including new sites that can reach underrepresented groups, and Genentech is making significant progress in that area.”
Dr. Freedman believes that, while access is a big hurdle, it doesn’t end there.
“Many patients have a lack of trust in the health care system,” he said. “There are also issues around underserved communities being able to afford quality care, so it’s important to keep time and financial burdens in mind when designing trials to help mitigate barriers such as travel, parking, time off work, and child care.”
Genentech started its diversity and inclusion effort several years ago. Dr. Freeman said that, until more trials become diverse, Black Americans will continue to pay the price. “I think they’re losing their lives in part due to lack of access to these trials. And that is why Genentech and all of us in the health care industry need to change how we design and enroll these studies. We have a long way to go, but I think the steps we’re taking are leading us in the right direction.”
Jennifer Jones-McMeans, PhD, director of global clinical affairs at Abbott Pharmaceuticals, is a clinical research scientist who has designed and led many clinical trials.
She said that Abbott is actively working on solutions.
“We have designed our trials to reduce the barriers to participation and expand access,” she said. “This can be as simple as providing transportation services or home visits for those who are housebound. We’re taking it a step further and providing home health services where someone comes to the home and provides follow-up visits there.”
They also provide interpretation services to address any language barriers.
“We are reaching out to a new set of talented investigators who work closely with underrepresented communities. They are very much wedded and supportive of the communities they treat. By working with doctors within these communities, it expands access to new therapies.”
Spokesperson Keanna Ghazvini said that Pfizer Pharmaceuticals is also committed to increasing minority participation in trials.
“We know that if historically underserved populations are left out of clinical trials, they risk not benefiting from medical breakthroughs down the line,” she said.
The National Institutes of Health’s National Library of Medicine maintains the clinicaltrials.gov database.
There, you can find information on nearly 372,000 publicly and privately supported clinical trials happening in all 50 states and 219 countries. Many are funded by the NIH, but not all of these studies have been evaluated by the U.S. government.
Andrea Denicoff, a nurse consultant at the National Cancer Institute and head of clinical trials operations for the NCI’s National Clinical Trials Network, has been involved in clinical research at the NIH for 35 years.
“It’s really important that our publicly funded trials represent the people of the country,” she said. “There are some cancers that we’re doing a good job in enrolling minorities, and other cancers we need to do a much better job in having a diverse representation in our trials.”
Ms. Denicoff believed opening trials in places where people live is key, but having a diverse clinical trials team is as important.
“We need to reinforce that cancer centers across the country have open doors, and anyone with cancer feels comfortable getting care at that center, and that also includes discussing the option to participate in clinical trials when one might be available. We know from research that when people are invited and asked about trial participation and educated about them, they’ll be much more interested in joining them.”
Ms. Denicoff said that, during the pandemic, the NCI quickly came up with guidance to allow trial sites to send patients their oral study drugs and set up virtual visits. She believes it may help increase future access.
‘Lola Fashoyin-Aje, MD, associate director for science and policy to address disparities in the Oncology Center of Excellence at the FDA, says the agency firmly believes clinical trials should represent the patients who will ultimately get the drug if it’s approved.
But the FDA’s power to require diversity in trials is limited.
“It is important to point out that there are legal constraints which limit’s FDA’s authority to require specific proportional representation in clinical trials by demographic factors,” Dr. Fashoyin-Aje said.
Still, some researchers feel the FDA should play a bigger role. The question is: Should diversity be mandated?
Rhonda Long is now back in Boston to start a new trial, with a new drug that targets her specific mutation. She will be there for 2 months. Once again, Lazarex will help cover some of the cost.
She wants people of color to understand that they are missing out on the promise of new cancer drugs and extended life.
“I feel like there’s not enough emphasis on clinical trials, I don’t believe there’s enough emphasis on second opinions, I don’t think there’s enough emphasis that medicine happens outside our borders, outside of our communities. Clinical trials that don’t have a broad range of participants, how do we know how effective they are if Black and brown people, Asian or Latin American people aren’t represented in the trial?”
And with more trials adopting virtual elements, she said it’s time for minorities to get on board.
Dr. Freedman believed the groundwork is being laid for that to happen. “I don’t think we’ll ever return back to the way we used to do things, where everything has to be done at the clinical trial site. I just don’t think we’re ever going back.”
A version of this article first appeared on WebMD.com.
“I’m the first person in my circle of family and friends to participate in a clinical trial.”
Five years ago, Rhonda Long was diagnosed with cholangiocarcinoma, a rare bile duct cancer that’s seen in only about 8,000 Americans each year.
At the time, Mrs. Long, who is Black, said her doctor in Dayton, Ohio, told her she was not a candidate for surgery and suggested palliative care. After seeking a second opinion at Duke University Medical Center, Durham, N.C., where her sister worked, the 51-year-old wife and mother of two had surgery, radiation, and chemotherapy there in North Carolina. When the chemo stopped working after 3 months, her oncologist at Duke referred her to a colleague at Massachusetts General Hospital in Boston, where she was accepted into a clinical trial.
“In 2019, I traveled to Boston from Dayton, Ohio, every 3 weeks for labs and scans, to make sure that the drug wasn’t doing more harm than good, making sure that the drug as developed was maintaining, shrinking, or even eliminating the disease. Physically and financially, it takes a toll on you and loved ones.”
Her medical insurance did not cover the direct expenses from the clinical trial, and she was spending $1,000-$1,500 each trip. Sometimes they drove the 15 hours to Boston, and sometimes they flew on the cheapest flight they could find.
It’s not an unfamiliar story: people traveling, often long distances, to take part in clinical trials they hope will save their lives.
The Lazarex Cancer Foundation of Danville, Calif., helped Mrs. Long do just that.
Marya Shegog, PhD, health equity and diversity coordinator at Lazarex, said that a patient travels an average of 500 miles to participate in a trial.
The financial hurdles often prevent patients from taking part in clinical trials, Dr. Shegog said. “When you are sick, and you have a disease that may be terminal, you start thinking about setting your things in order.”
Many patients have to make a decision.
“Do I bankrupt my family on trying and hoping that this drug works and helps me live longer, or do I start setting things in order so that when I’m gone, they’re okay or at least better than if I wouldn’t have spent all the money traveling back and forth.”
Dr. Shegog, a 17-year cancer survivor, says when she was battling cervical cancer, a clinical trial was never offered or explored.
Lazarex has been helping cancer patients who have run out of options for 15 years. It identifies clinical trial opportunities and reimburses patients for all travel costs. Last year, Lazarex reimbursed more than 1,000 cancer patients. And it has supported more than 6,000 people since opening its doors.
“Lazarex exists to help remove the barriers of people not being able to participate in trials,” Dr. Shegog said. “It’s systemic that the medical system does not treat patients the same and oftentimes does not offer or make aware the opportunities for African Americans to participate.”
But now, thanks in part to COVID-19, new possibilities are taking shape. The pandemic has changed the landscape for trials, forcing many of them to go virtual, which allows patients to schedule telehealth visits and get some services like bloodwork and CT or MRI scans closer to home. Mrs. Long’s trial eventually went virtual.
“It was absolutely fantastic,” she said. “Having the trial locally, it saves us money, it saves wear and tear on my body. Being in the car, being in an airport or in a plane and in a hotel, all of that wears on you physically.”
The move to virtual studies may have lasting effects on research and treatment.
“The current pandemic has forced us to reexamine all of the traditional burdens we place on patients as it relates to receiving cancer treatment,” said Hala Borno, MD, an assistant professor of medicine at the University of California, San Francisco. “Whether they’re coming to our health care facility to see a clinician, for diagnostics such as blood draws and scans, or to receive therapy, this pandemic has challenged us to explore other possibilities that minimize the risk of exposure to SARS-CoV-2. What I find striking is that it has helped us operationalize use of telemedicine and the delivery of care closer to home.”
This is especially encouraging news for minority patients whose participation in trials has for years lagged well behind that of Whites.
But travel is not the only reason. Racial disparities in clinical trials have long been an issue that’s just another part of the implicit bias in health care.
Compared with White people, Black people are largely at higher risk for heart disease, cancer, stroke, diabetes, asthma, and even mental health problems.
And it’s not just African Americans. Asians, Hispanics, Native Americans, and Alaska Natives are all underrepresented in trials at a time when there is growing evidence that drugs may have different effects on different populations.
Dr. Borno is an oncologist who specializes in prostate cancer, a disease that she says shows a “significant disparity,” where Black men are two times more likely to die from advanced prostate cancer, compared with white men. Yet Black men make up just 3% of advanced therapeutic trials.
“A lack of diversity and inclusion in clinical trials is unacceptable,” she said. “If we continue to underrecruit racial/ethnic minorities and older adults to therapeutic clinical trials, we will not be powered to make valid conclusions regarding safety and efficacy in those patient populations. As a result, we can do harm.”
Dr. Borno said that telehealth and telemedicine are not cure-alls, and digital health solutions don’t work for all patients. Approaches, she says, must be tailored to the individual, or disparities could worsen.
In 2020, the Food and Drug Administration approved 53 new drugs. Overall, 32,000 patients took part in these trials. On average, 75% were White, 8% were Black, 6% were Asian, and 11% were Hispanic.
Here’s one stark example of the issue. In 2015, the FDA approved ixazomib (Ninlaro), a promising new drug for multiple myeloma, a blood cancer that affects Black people at disproportionately higher rates than White people. In the United States, one in five people diagnosed with multiple myeloma are Black people. They are more than twice as likely to get the disease as White people. Yet during the clinical trial of 722 participants, only 13 patients, or 1.8%, were Black.
The American Cancer Society estimates that more than 600,000 Americans will die from cancer this year. Historically, Black Americans have the highest death rate and the shortest survival of any racial or ethnic group, stemming largely, it concluded, from centuries of structural racism.
According to Jamie Freedman, MD, head of U.S. medical affairs at Genentech, a global pharmaceutical company, the lack of diversity is often tied to where studies are run.
“Companies tend to choose major academic medical centers where there is a high volume of clinical trial work. When you go to the same tried and true hospitals repeatedly, the pool of patients becomes very homogeneous and tends to be primarily white,” he said. “It’s critical to bring more trials into the community setting by including new sites that can reach underrepresented groups, and Genentech is making significant progress in that area.”
Dr. Freedman believes that, while access is a big hurdle, it doesn’t end there.
“Many patients have a lack of trust in the health care system,” he said. “There are also issues around underserved communities being able to afford quality care, so it’s important to keep time and financial burdens in mind when designing trials to help mitigate barriers such as travel, parking, time off work, and child care.”
Genentech started its diversity and inclusion effort several years ago. Dr. Freeman said that, until more trials become diverse, Black Americans will continue to pay the price. “I think they’re losing their lives in part due to lack of access to these trials. And that is why Genentech and all of us in the health care industry need to change how we design and enroll these studies. We have a long way to go, but I think the steps we’re taking are leading us in the right direction.”
Jennifer Jones-McMeans, PhD, director of global clinical affairs at Abbott Pharmaceuticals, is a clinical research scientist who has designed and led many clinical trials.
She said that Abbott is actively working on solutions.
“We have designed our trials to reduce the barriers to participation and expand access,” she said. “This can be as simple as providing transportation services or home visits for those who are housebound. We’re taking it a step further and providing home health services where someone comes to the home and provides follow-up visits there.”
They also provide interpretation services to address any language barriers.
“We are reaching out to a new set of talented investigators who work closely with underrepresented communities. They are very much wedded and supportive of the communities they treat. By working with doctors within these communities, it expands access to new therapies.”
Spokesperson Keanna Ghazvini said that Pfizer Pharmaceuticals is also committed to increasing minority participation in trials.
“We know that if historically underserved populations are left out of clinical trials, they risk not benefiting from medical breakthroughs down the line,” she said.
The National Institutes of Health’s National Library of Medicine maintains the clinicaltrials.gov database.
There, you can find information on nearly 372,000 publicly and privately supported clinical trials happening in all 50 states and 219 countries. Many are funded by the NIH, but not all of these studies have been evaluated by the U.S. government.
Andrea Denicoff, a nurse consultant at the National Cancer Institute and head of clinical trials operations for the NCI’s National Clinical Trials Network, has been involved in clinical research at the NIH for 35 years.
“It’s really important that our publicly funded trials represent the people of the country,” she said. “There are some cancers that we’re doing a good job in enrolling minorities, and other cancers we need to do a much better job in having a diverse representation in our trials.”
Ms. Denicoff believed opening trials in places where people live is key, but having a diverse clinical trials team is as important.
“We need to reinforce that cancer centers across the country have open doors, and anyone with cancer feels comfortable getting care at that center, and that also includes discussing the option to participate in clinical trials when one might be available. We know from research that when people are invited and asked about trial participation and educated about them, they’ll be much more interested in joining them.”
Ms. Denicoff said that, during the pandemic, the NCI quickly came up with guidance to allow trial sites to send patients their oral study drugs and set up virtual visits. She believes it may help increase future access.
‘Lola Fashoyin-Aje, MD, associate director for science and policy to address disparities in the Oncology Center of Excellence at the FDA, says the agency firmly believes clinical trials should represent the patients who will ultimately get the drug if it’s approved.
But the FDA’s power to require diversity in trials is limited.
“It is important to point out that there are legal constraints which limit’s FDA’s authority to require specific proportional representation in clinical trials by demographic factors,” Dr. Fashoyin-Aje said.
Still, some researchers feel the FDA should play a bigger role. The question is: Should diversity be mandated?
Rhonda Long is now back in Boston to start a new trial, with a new drug that targets her specific mutation. She will be there for 2 months. Once again, Lazarex will help cover some of the cost.
She wants people of color to understand that they are missing out on the promise of new cancer drugs and extended life.
“I feel like there’s not enough emphasis on clinical trials, I don’t believe there’s enough emphasis on second opinions, I don’t think there’s enough emphasis that medicine happens outside our borders, outside of our communities. Clinical trials that don’t have a broad range of participants, how do we know how effective they are if Black and brown people, Asian or Latin American people aren’t represented in the trial?”
And with more trials adopting virtual elements, she said it’s time for minorities to get on board.
Dr. Freedman believed the groundwork is being laid for that to happen. “I don’t think we’ll ever return back to the way we used to do things, where everything has to be done at the clinical trial site. I just don’t think we’re ever going back.”
A version of this article first appeared on WebMD.com.
Gynecologic cancer patients at risk of insurance loss, ‘catastrophic’ costs
A retrospective study of respondents to the Medical Expenditure Panel Survey showed that more than one in five gynecologic cancer patients reported losing health insurance for at least 1 month every year, and more than one in four reported having catastrophic health expenses annually.
Benjamin Albright, MD, of Duke University Medical Center in Durham, N.C., presented these results at the Society of Gynecologic Oncology’s Virtual Annual Meeting on Women’s Cancer (Abstract 10303).
“We found gynecologic cancer patients to have high rates of insurance churn and catastrophic health expenditures, particularly among the poor,” Dr. Albright said. “Traditional static measurements clearly underestimate the impact of uninsurance, with over 20% of patients reporting some period of uninsurance annually.”
There was no evidence of improvement in any outcome after the implementation of the ACA, compared with the pre-ACA period, “though our assessment was limited in estimate precision by small sample size,” Dr. Albright acknowledged.
Dynamic, not static
Oncology researchers who study access to care and financial toxicities often consider insurance status as a static characteristic, but in the U.S. health care system, the reality is quite different, with insurance status fluctuating by employment or ability to pay, sometimes on a month-to-month basis, according to Dr. Albright.
Citing the Commonwealth Fund’s definition of catastrophic health expenditures as “spending over 10% of income on health care,” Dr. Albright noted that the prevalence of catastrophic out-of-pocket costs “is also relatively poorly described among cancer patients, particularly in accounting for family spending and income dynamics.
“The Affordable Care Act contained measures to address both of these concerns, including coverage protections and expansions, and spending regulations,” he said.
Dr. Albright and colleagues at Duke and Memorial Sloan Kettering Cancer Center in New York assessed insurance churn and catastrophic health expenditures among gynecologic cancer patients, attempting to determine whether the ACA had helped to limit insurance churn and keep costs manageable.
Representative sample
The investigators conducted a retrospective study of data from Medical Expenditure Panel Survey respondents from 2006 through 2017, a period that spanned the implementation of the ACA in 2010.
The sample included 684 women younger than 65 years reporting care in the given year related to a gynecologic cancer diagnosis. The civilian, noninstitutionalized sample was weighted to represent an estimated average annual population of 533,000 persons. The population was majority White (87%) and non-Hispanic (85.5%).
The investigators found that, compared with the overall U.S. population of people under 65, gynecologic cancer patients were more likely to have incomes of 250% or less of the federal poverty line (45.1% vs. 32.2%, P < .001).
The cancer patients were more likely than was the general population to have less than full-time employment, with 15.2% and 10.5%, respectively, reporting a job change or job loss; 55.3% and 44.1%, respectively, being employed only part of a given year; and 38.6% and 32.4%, respectively, being unemployed for a full year (P < .05 for each comparison).
Gynecologic cancer patients continued to experience insurance troubles and financial hardships after the ACA went into effect, with 8.8% reporting loss of insurance, 18.7% reporting a change in insurance, 21.7% being uninsured for at least 1 month, and 8.4% being uninsured for an entire year.
In addition, 12.8% of gynecologic cancer patients reported catastrophic health expenditures in out-of-pocket costs alone, and 28.0% spent more than 10% of their income on health care when the cost of premiums was factored in.
The numbers were even worse for non-White and Hispanic patients, with 25.9% reporting an insurance change (vs. 16.3% for non-Hispanic Whites) and 30.2% reporting a period of not being insured (vs. 18.7% for non-Hispanic Whites). There were no differences in catastrophic health expenditures by race/ethnicity, however.
Not surprisingly, patients from low-income families had significantly higher probability of having catastrophic expenditures, at 22.7% vs. 3.0% for higher-income families for out-of-pocket expenses alone (P < .001), and 35.3% vs. 20.8%, respectively, when the cost of premiums was included (P = .01).
On the other hand, patients with full-year Medicaid coverage were less likely to suffer from catastrophic costs than were privately-insured patients, at 15.3% vs. 31.3% in the overall sample (P = .02), and 11.5% vs. 62.1% of low-income vs. higher-income patients (P < .001).
There was a trend toward lower catastrophic health expenditures among low-income patients after full implementation of the ACA – 2014-2017 – compared with 2006-2009, but this difference was not statistically significant.
How to change it
In a panel discussion following the presentation, comoderator Eloise Chapman-Davis, MD, of Weill Cornell Medicine in New York, said to Dr. Albright, “As we look to improve equity within our subspecialty, I would like to ask you to comment on how you believe your abstract will inform our gyn-oncology culture and speak to what changes that you believe are needed to better advocate for our patients.”
“I think that our abstract really shows the prevalence of the problems of financial toxicity and of instability in the insurance market in the U.S.,” he replied. “I think it points out that we need to be more proactive about identifying patients and seeking out patients who may be having issues related to financial toxicity, to try to refer people to resources sooner and upfront.”
The investigators did not list a funding source for the study. Dr. Albright and Dr. Chapman-Davis reported having no conflicts of interest.
A retrospective study of respondents to the Medical Expenditure Panel Survey showed that more than one in five gynecologic cancer patients reported losing health insurance for at least 1 month every year, and more than one in four reported having catastrophic health expenses annually.
Benjamin Albright, MD, of Duke University Medical Center in Durham, N.C., presented these results at the Society of Gynecologic Oncology’s Virtual Annual Meeting on Women’s Cancer (Abstract 10303).
“We found gynecologic cancer patients to have high rates of insurance churn and catastrophic health expenditures, particularly among the poor,” Dr. Albright said. “Traditional static measurements clearly underestimate the impact of uninsurance, with over 20% of patients reporting some period of uninsurance annually.”
There was no evidence of improvement in any outcome after the implementation of the ACA, compared with the pre-ACA period, “though our assessment was limited in estimate precision by small sample size,” Dr. Albright acknowledged.
Dynamic, not static
Oncology researchers who study access to care and financial toxicities often consider insurance status as a static characteristic, but in the U.S. health care system, the reality is quite different, with insurance status fluctuating by employment or ability to pay, sometimes on a month-to-month basis, according to Dr. Albright.
Citing the Commonwealth Fund’s definition of catastrophic health expenditures as “spending over 10% of income on health care,” Dr. Albright noted that the prevalence of catastrophic out-of-pocket costs “is also relatively poorly described among cancer patients, particularly in accounting for family spending and income dynamics.
“The Affordable Care Act contained measures to address both of these concerns, including coverage protections and expansions, and spending regulations,” he said.
Dr. Albright and colleagues at Duke and Memorial Sloan Kettering Cancer Center in New York assessed insurance churn and catastrophic health expenditures among gynecologic cancer patients, attempting to determine whether the ACA had helped to limit insurance churn and keep costs manageable.
Representative sample
The investigators conducted a retrospective study of data from Medical Expenditure Panel Survey respondents from 2006 through 2017, a period that spanned the implementation of the ACA in 2010.
The sample included 684 women younger than 65 years reporting care in the given year related to a gynecologic cancer diagnosis. The civilian, noninstitutionalized sample was weighted to represent an estimated average annual population of 533,000 persons. The population was majority White (87%) and non-Hispanic (85.5%).
The investigators found that, compared with the overall U.S. population of people under 65, gynecologic cancer patients were more likely to have incomes of 250% or less of the federal poverty line (45.1% vs. 32.2%, P < .001).
The cancer patients were more likely than was the general population to have less than full-time employment, with 15.2% and 10.5%, respectively, reporting a job change or job loss; 55.3% and 44.1%, respectively, being employed only part of a given year; and 38.6% and 32.4%, respectively, being unemployed for a full year (P < .05 for each comparison).
Gynecologic cancer patients continued to experience insurance troubles and financial hardships after the ACA went into effect, with 8.8% reporting loss of insurance, 18.7% reporting a change in insurance, 21.7% being uninsured for at least 1 month, and 8.4% being uninsured for an entire year.
In addition, 12.8% of gynecologic cancer patients reported catastrophic health expenditures in out-of-pocket costs alone, and 28.0% spent more than 10% of their income on health care when the cost of premiums was factored in.
The numbers were even worse for non-White and Hispanic patients, with 25.9% reporting an insurance change (vs. 16.3% for non-Hispanic Whites) and 30.2% reporting a period of not being insured (vs. 18.7% for non-Hispanic Whites). There were no differences in catastrophic health expenditures by race/ethnicity, however.
Not surprisingly, patients from low-income families had significantly higher probability of having catastrophic expenditures, at 22.7% vs. 3.0% for higher-income families for out-of-pocket expenses alone (P < .001), and 35.3% vs. 20.8%, respectively, when the cost of premiums was included (P = .01).
On the other hand, patients with full-year Medicaid coverage were less likely to suffer from catastrophic costs than were privately-insured patients, at 15.3% vs. 31.3% in the overall sample (P = .02), and 11.5% vs. 62.1% of low-income vs. higher-income patients (P < .001).
There was a trend toward lower catastrophic health expenditures among low-income patients after full implementation of the ACA – 2014-2017 – compared with 2006-2009, but this difference was not statistically significant.
How to change it
In a panel discussion following the presentation, comoderator Eloise Chapman-Davis, MD, of Weill Cornell Medicine in New York, said to Dr. Albright, “As we look to improve equity within our subspecialty, I would like to ask you to comment on how you believe your abstract will inform our gyn-oncology culture and speak to what changes that you believe are needed to better advocate for our patients.”
“I think that our abstract really shows the prevalence of the problems of financial toxicity and of instability in the insurance market in the U.S.,” he replied. “I think it points out that we need to be more proactive about identifying patients and seeking out patients who may be having issues related to financial toxicity, to try to refer people to resources sooner and upfront.”
The investigators did not list a funding source for the study. Dr. Albright and Dr. Chapman-Davis reported having no conflicts of interest.
A retrospective study of respondents to the Medical Expenditure Panel Survey showed that more than one in five gynecologic cancer patients reported losing health insurance for at least 1 month every year, and more than one in four reported having catastrophic health expenses annually.
Benjamin Albright, MD, of Duke University Medical Center in Durham, N.C., presented these results at the Society of Gynecologic Oncology’s Virtual Annual Meeting on Women’s Cancer (Abstract 10303).
“We found gynecologic cancer patients to have high rates of insurance churn and catastrophic health expenditures, particularly among the poor,” Dr. Albright said. “Traditional static measurements clearly underestimate the impact of uninsurance, with over 20% of patients reporting some period of uninsurance annually.”
There was no evidence of improvement in any outcome after the implementation of the ACA, compared with the pre-ACA period, “though our assessment was limited in estimate precision by small sample size,” Dr. Albright acknowledged.
Dynamic, not static
Oncology researchers who study access to care and financial toxicities often consider insurance status as a static characteristic, but in the U.S. health care system, the reality is quite different, with insurance status fluctuating by employment or ability to pay, sometimes on a month-to-month basis, according to Dr. Albright.
Citing the Commonwealth Fund’s definition of catastrophic health expenditures as “spending over 10% of income on health care,” Dr. Albright noted that the prevalence of catastrophic out-of-pocket costs “is also relatively poorly described among cancer patients, particularly in accounting for family spending and income dynamics.
“The Affordable Care Act contained measures to address both of these concerns, including coverage protections and expansions, and spending regulations,” he said.
Dr. Albright and colleagues at Duke and Memorial Sloan Kettering Cancer Center in New York assessed insurance churn and catastrophic health expenditures among gynecologic cancer patients, attempting to determine whether the ACA had helped to limit insurance churn and keep costs manageable.
Representative sample
The investigators conducted a retrospective study of data from Medical Expenditure Panel Survey respondents from 2006 through 2017, a period that spanned the implementation of the ACA in 2010.
The sample included 684 women younger than 65 years reporting care in the given year related to a gynecologic cancer diagnosis. The civilian, noninstitutionalized sample was weighted to represent an estimated average annual population of 533,000 persons. The population was majority White (87%) and non-Hispanic (85.5%).
The investigators found that, compared with the overall U.S. population of people under 65, gynecologic cancer patients were more likely to have incomes of 250% or less of the federal poverty line (45.1% vs. 32.2%, P < .001).
The cancer patients were more likely than was the general population to have less than full-time employment, with 15.2% and 10.5%, respectively, reporting a job change or job loss; 55.3% and 44.1%, respectively, being employed only part of a given year; and 38.6% and 32.4%, respectively, being unemployed for a full year (P < .05 for each comparison).
Gynecologic cancer patients continued to experience insurance troubles and financial hardships after the ACA went into effect, with 8.8% reporting loss of insurance, 18.7% reporting a change in insurance, 21.7% being uninsured for at least 1 month, and 8.4% being uninsured for an entire year.
In addition, 12.8% of gynecologic cancer patients reported catastrophic health expenditures in out-of-pocket costs alone, and 28.0% spent more than 10% of their income on health care when the cost of premiums was factored in.
The numbers were even worse for non-White and Hispanic patients, with 25.9% reporting an insurance change (vs. 16.3% for non-Hispanic Whites) and 30.2% reporting a period of not being insured (vs. 18.7% for non-Hispanic Whites). There were no differences in catastrophic health expenditures by race/ethnicity, however.
Not surprisingly, patients from low-income families had significantly higher probability of having catastrophic expenditures, at 22.7% vs. 3.0% for higher-income families for out-of-pocket expenses alone (P < .001), and 35.3% vs. 20.8%, respectively, when the cost of premiums was included (P = .01).
On the other hand, patients with full-year Medicaid coverage were less likely to suffer from catastrophic costs than were privately-insured patients, at 15.3% vs. 31.3% in the overall sample (P = .02), and 11.5% vs. 62.1% of low-income vs. higher-income patients (P < .001).
There was a trend toward lower catastrophic health expenditures among low-income patients after full implementation of the ACA – 2014-2017 – compared with 2006-2009, but this difference was not statistically significant.
How to change it
In a panel discussion following the presentation, comoderator Eloise Chapman-Davis, MD, of Weill Cornell Medicine in New York, said to Dr. Albright, “As we look to improve equity within our subspecialty, I would like to ask you to comment on how you believe your abstract will inform our gyn-oncology culture and speak to what changes that you believe are needed to better advocate for our patients.”
“I think that our abstract really shows the prevalence of the problems of financial toxicity and of instability in the insurance market in the U.S.,” he replied. “I think it points out that we need to be more proactive about identifying patients and seeking out patients who may be having issues related to financial toxicity, to try to refer people to resources sooner and upfront.”
The investigators did not list a funding source for the study. Dr. Albright and Dr. Chapman-Davis reported having no conflicts of interest.
FROM SGO 2021
Melatonin not recommended for early-stage NSCLC
There was a hint of benefit with melatonin among patients with stage III/IV NSCLC. These patients had a hazard reduction of 25% in 5-year DFS. However, the median DFS for patients with advanced disease was the same whether they received melatonin or placebo – 18 months.
In the overall study population, melatonin had no beneficial effects on quality of life, sleep, anxiety, depression, pain, or fatigue, and it did not reduce adverse events from chemotherapy or radiation.
These results were reported in EClinicalMedicine.
“In light of the results, we do not recommend the inclusion of adjuvant melatonin for patients with early-stage NSCLC. Evidence suggests there may be a benefit for those with late-stage disease,” the authors wrote. “However, because of the mixed findings observed, we recommend a follow-up randomized, controlled trial involving a larger population focusing on later-stage resected lung cancer to clarify these results.”
“I would very much like to pursue another controlled study of melatonin specifically in a group of late-stage lung cancer and possibly in other more advanced cancer types,” said lead author Dugald Seely, ND, of the Canadian College of Naturopathic Medicine in Toronto.
Study rationale and design
Melatonin has shown promise for treating patients with lung cancer, Dr. Seely and colleagues noted. Melatonin is often recommended by naturopathic doctors following lung cancer surgery, but until now there was no high-level evidence regarding the practice.
For their study, Dr. Seely and colleagues evaluated 709 patients who had undergone NSCLC resection. The patients were randomized to receive placebo (n = 353) or melatonin (n = 356) 1 hour before bedtime for 1 year. A 20-mg melatonin dose was used, which is common in clinical practice and research.
The study arms were well matched, with no “clinically meaningful” differences in demographics, surgery type, cancer type, stage of cancer, or preoperative comorbidities, according to the researchers.
The mean age in both treatment arms was 67 years. Overall, 134 participants received adjuvant chemotherapy (66 melatonin, 68 placebo), and 43 had adjuvant radiation (22 melatonin, 21 placebo).
Results
For 2-year DFS, melatonin showed an adjusted relative risk of 1.01 (95% confidence interval, 0.83-1.22; P = .94) versus placebo. The adjusted relative risk in the per-protocol analysis was 1.12 (95% CI, 0.96-1.32; P = .14.)
At 5 years, the median DFS was not reached in either treatment arm. Melatonin showed a hazard ratio of 0.97 (95% CI, 0.86-1.09; P = .84) for 5-year DFS.
Among patients with stage I-II NSCLC, the median DFS was not reached at 5 years in either treatment arm. Among patients with stage III-IV NSCLC, the median DFS was 18 months in both arms.
Melatonin showed a hazard ratio of 0.97 (95% CI, 0.85-1.11; P = .66) in patients with early-stage NSCLC and a hazard reduction of 25% (HR, 0.75; 95% CI, 0.61-0.92; P = .005) in patients with late-stage NSCLC.
For the entire cohort, there were no significant differences between treatment arms in the number, severity, or seriousness of adverse events. Likewise, there were no significant differences between the treatment arms with regard to fatigue, quality of life, or sleep at 1 or 2 years.
Dr. Seely said the most surprising thing about this study was that melatonin didn’t help with sleep.
“Since initiation of the trial, my thinking on the right dose of melatonin to support sleep has changed. Clinically, I see extended-release and, indeed, lower doses to be more effective than 20 mg nightly,” he noted.
Dr. Seely and colleagues also assessed proposed mechanisms for melatonin’s possible benefit in NSCLC but found no effect on natural killer cell cytotoxicity or phenotype and no effect on blood levels of inflammatory cytokines in a substudy of 92 patients.
This research was funded by the Lotte and John Hecht Memorial Foundation and the Gateway for Cancer Research Foundation. The researchers had no relevant disclosures.
There was a hint of benefit with melatonin among patients with stage III/IV NSCLC. These patients had a hazard reduction of 25% in 5-year DFS. However, the median DFS for patients with advanced disease was the same whether they received melatonin or placebo – 18 months.
In the overall study population, melatonin had no beneficial effects on quality of life, sleep, anxiety, depression, pain, or fatigue, and it did not reduce adverse events from chemotherapy or radiation.
These results were reported in EClinicalMedicine.
“In light of the results, we do not recommend the inclusion of adjuvant melatonin for patients with early-stage NSCLC. Evidence suggests there may be a benefit for those with late-stage disease,” the authors wrote. “However, because of the mixed findings observed, we recommend a follow-up randomized, controlled trial involving a larger population focusing on later-stage resected lung cancer to clarify these results.”
“I would very much like to pursue another controlled study of melatonin specifically in a group of late-stage lung cancer and possibly in other more advanced cancer types,” said lead author Dugald Seely, ND, of the Canadian College of Naturopathic Medicine in Toronto.
Study rationale and design
Melatonin has shown promise for treating patients with lung cancer, Dr. Seely and colleagues noted. Melatonin is often recommended by naturopathic doctors following lung cancer surgery, but until now there was no high-level evidence regarding the practice.
For their study, Dr. Seely and colleagues evaluated 709 patients who had undergone NSCLC resection. The patients were randomized to receive placebo (n = 353) or melatonin (n = 356) 1 hour before bedtime for 1 year. A 20-mg melatonin dose was used, which is common in clinical practice and research.
The study arms were well matched, with no “clinically meaningful” differences in demographics, surgery type, cancer type, stage of cancer, or preoperative comorbidities, according to the researchers.
The mean age in both treatment arms was 67 years. Overall, 134 participants received adjuvant chemotherapy (66 melatonin, 68 placebo), and 43 had adjuvant radiation (22 melatonin, 21 placebo).
Results
For 2-year DFS, melatonin showed an adjusted relative risk of 1.01 (95% confidence interval, 0.83-1.22; P = .94) versus placebo. The adjusted relative risk in the per-protocol analysis was 1.12 (95% CI, 0.96-1.32; P = .14.)
At 5 years, the median DFS was not reached in either treatment arm. Melatonin showed a hazard ratio of 0.97 (95% CI, 0.86-1.09; P = .84) for 5-year DFS.
Among patients with stage I-II NSCLC, the median DFS was not reached at 5 years in either treatment arm. Among patients with stage III-IV NSCLC, the median DFS was 18 months in both arms.
Melatonin showed a hazard ratio of 0.97 (95% CI, 0.85-1.11; P = .66) in patients with early-stage NSCLC and a hazard reduction of 25% (HR, 0.75; 95% CI, 0.61-0.92; P = .005) in patients with late-stage NSCLC.
For the entire cohort, there were no significant differences between treatment arms in the number, severity, or seriousness of adverse events. Likewise, there were no significant differences between the treatment arms with regard to fatigue, quality of life, or sleep at 1 or 2 years.
Dr. Seely said the most surprising thing about this study was that melatonin didn’t help with sleep.
“Since initiation of the trial, my thinking on the right dose of melatonin to support sleep has changed. Clinically, I see extended-release and, indeed, lower doses to be more effective than 20 mg nightly,” he noted.
Dr. Seely and colleagues also assessed proposed mechanisms for melatonin’s possible benefit in NSCLC but found no effect on natural killer cell cytotoxicity or phenotype and no effect on blood levels of inflammatory cytokines in a substudy of 92 patients.
This research was funded by the Lotte and John Hecht Memorial Foundation and the Gateway for Cancer Research Foundation. The researchers had no relevant disclosures.
There was a hint of benefit with melatonin among patients with stage III/IV NSCLC. These patients had a hazard reduction of 25% in 5-year DFS. However, the median DFS for patients with advanced disease was the same whether they received melatonin or placebo – 18 months.
In the overall study population, melatonin had no beneficial effects on quality of life, sleep, anxiety, depression, pain, or fatigue, and it did not reduce adverse events from chemotherapy or radiation.
These results were reported in EClinicalMedicine.
“In light of the results, we do not recommend the inclusion of adjuvant melatonin for patients with early-stage NSCLC. Evidence suggests there may be a benefit for those with late-stage disease,” the authors wrote. “However, because of the mixed findings observed, we recommend a follow-up randomized, controlled trial involving a larger population focusing on later-stage resected lung cancer to clarify these results.”
“I would very much like to pursue another controlled study of melatonin specifically in a group of late-stage lung cancer and possibly in other more advanced cancer types,” said lead author Dugald Seely, ND, of the Canadian College of Naturopathic Medicine in Toronto.
Study rationale and design
Melatonin has shown promise for treating patients with lung cancer, Dr. Seely and colleagues noted. Melatonin is often recommended by naturopathic doctors following lung cancer surgery, but until now there was no high-level evidence regarding the practice.
For their study, Dr. Seely and colleagues evaluated 709 patients who had undergone NSCLC resection. The patients were randomized to receive placebo (n = 353) or melatonin (n = 356) 1 hour before bedtime for 1 year. A 20-mg melatonin dose was used, which is common in clinical practice and research.
The study arms were well matched, with no “clinically meaningful” differences in demographics, surgery type, cancer type, stage of cancer, or preoperative comorbidities, according to the researchers.
The mean age in both treatment arms was 67 years. Overall, 134 participants received adjuvant chemotherapy (66 melatonin, 68 placebo), and 43 had adjuvant radiation (22 melatonin, 21 placebo).
Results
For 2-year DFS, melatonin showed an adjusted relative risk of 1.01 (95% confidence interval, 0.83-1.22; P = .94) versus placebo. The adjusted relative risk in the per-protocol analysis was 1.12 (95% CI, 0.96-1.32; P = .14.)
At 5 years, the median DFS was not reached in either treatment arm. Melatonin showed a hazard ratio of 0.97 (95% CI, 0.86-1.09; P = .84) for 5-year DFS.
Among patients with stage I-II NSCLC, the median DFS was not reached at 5 years in either treatment arm. Among patients with stage III-IV NSCLC, the median DFS was 18 months in both arms.
Melatonin showed a hazard ratio of 0.97 (95% CI, 0.85-1.11; P = .66) in patients with early-stage NSCLC and a hazard reduction of 25% (HR, 0.75; 95% CI, 0.61-0.92; P = .005) in patients with late-stage NSCLC.
For the entire cohort, there were no significant differences between treatment arms in the number, severity, or seriousness of adverse events. Likewise, there were no significant differences between the treatment arms with regard to fatigue, quality of life, or sleep at 1 or 2 years.
Dr. Seely said the most surprising thing about this study was that melatonin didn’t help with sleep.
“Since initiation of the trial, my thinking on the right dose of melatonin to support sleep has changed. Clinically, I see extended-release and, indeed, lower doses to be more effective than 20 mg nightly,” he noted.
Dr. Seely and colleagues also assessed proposed mechanisms for melatonin’s possible benefit in NSCLC but found no effect on natural killer cell cytotoxicity or phenotype and no effect on blood levels of inflammatory cytokines in a substudy of 92 patients.
This research was funded by the Lotte and John Hecht Memorial Foundation and the Gateway for Cancer Research Foundation. The researchers had no relevant disclosures.
FROM ECLINICALMEDICINE
FDA scrutinizes cancer therapies granted accelerated approval
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
High-dose chemo no better than standard dose for B-cell lymphoma
After 10 years of follow-up, event-free survival and overall survival were similar between conventional chemotherapy treated patients with aggressive B-cell lymphoma and those receiving high-dose chemotherapy followed by autologous hematopoietic stem-cell transplantation (HSCT), according to a report published online in the Lancet Hematology.
The open-label, randomized, phase 3 trial (NCT00129090) was conducted across 61 centers in Germany on patients aged 18-60 years who had newly diagnosed, high-risk, aggressive B-cell lymphoma, according to Fabian Frontzek, MD, of the University Hospital Münster (Germany) and colleagues.
Between March 2003 and April 2009, patients were randomly assigned to eight cycles of conventional chemotherapy (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisolone) plus rituximab (R-CHOEP-14) or four cycles of high-dose chemotherapy plus rituximab followed by autologous HSCT (R-MegaCHOEP). The intention-to-treat population comprised 130 patients in the R-CHOEP-14 group and 132 patients in the R-MegaCHOEP group. The median follow-up was 9.3 years.
Similar outcomes
The 10-year event-free survival was 51% in the R-MegaCHOEP group and 57% in the R-CHOEP-14 group, a nonsignificant difference (P = .23). Similarly, the 10-year progression-free survival was 59% in the
R-MegaCHOEP group and 60% (P = .64). The 10-year overall survival was 66% in the R-MegaCHOEP group and 72% in the R-CHOEP-14 group (P = .26). Among the 190 patients who had complete remission or unconfirmed complete remission, relapse occurred in 30 (16%); 17 (17%) of 100 patients in the R-CHOEP-14 group and 13 (14%) of 90 patients in the R-MegaCHOEP group.
In terms of secondary malignancies, 22 were reported in the intention-to-treat population; comprising 12 (9%) of 127 patients in the R-CHOEP-14 group and 10 (8%) of 126 patients in the R-MegaCHOEP group.
Patients who relapsed with aggressive histology and with CNS involvement in particular had worse outcomes and “represent a group with an unmet medical need, for which new molecular and cellular therapies should be studied,” the authors stated.
“This study shows that, in the rituximab era, high-dose therapy and autologous HSCT in first-line treatment does not improve long-term survival of younger high-risk patients with aggressive B-cell lymphoma. The R-CHOEP-14 regimen led to favorable outcomes, supporting its continued use in such patients,” the researchers concluded.
In an accompanying commentary, Gita Thanarajasingam, MD, of the Mayo Clinic, Rochester, Minn., and colleagues added that the issue of long-term outcomes is critical to evaluating these new regimens.
They applauded the inclusion of secondary malignancies in the long-term follow-up, but regretted the lack of the, admittedly resource-intensive, information on long-term nonneoplastic adverse events. They added that “the burden of late adverse events such as cardiotoxicity, cumulative neuropathy, delayed infections, or lasting cognitive effects, among others that might drive substantial morbidity, does matter to lymphoma survivors.”
They also commented on the importance of considering effects on fertility in these patients, noting that R-MegaCHOEP patients would be unable to conceive naturally, but that the effect of R-CHOEP-14 was less clear.
“We encourage ongoing emphasis on this type of longitudinal follow-up of secondary malignancies and other nonneoplastic late toxicities in phase 3 studies as well as in the real world in hematological malignancies, so that after prioritizing cure in the front-line setting, we do not neglect the life we have helped survivors achieve for years and decades to come,” they concluded.
The study was sponsored by the German High-Grade Non-Hodgkin’s Lymphoma Study Group. The authors reported grants, personal fees, and non-financial support from multiple pharmaceutical and biotechnology companies. Dr. Thanarajasingam and her colleagues reported that they had no competing interests.
After 10 years of follow-up, event-free survival and overall survival were similar between conventional chemotherapy treated patients with aggressive B-cell lymphoma and those receiving high-dose chemotherapy followed by autologous hematopoietic stem-cell transplantation (HSCT), according to a report published online in the Lancet Hematology.
The open-label, randomized, phase 3 trial (NCT00129090) was conducted across 61 centers in Germany on patients aged 18-60 years who had newly diagnosed, high-risk, aggressive B-cell lymphoma, according to Fabian Frontzek, MD, of the University Hospital Münster (Germany) and colleagues.
Between March 2003 and April 2009, patients were randomly assigned to eight cycles of conventional chemotherapy (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisolone) plus rituximab (R-CHOEP-14) or four cycles of high-dose chemotherapy plus rituximab followed by autologous HSCT (R-MegaCHOEP). The intention-to-treat population comprised 130 patients in the R-CHOEP-14 group and 132 patients in the R-MegaCHOEP group. The median follow-up was 9.3 years.
Similar outcomes
The 10-year event-free survival was 51% in the R-MegaCHOEP group and 57% in the R-CHOEP-14 group, a nonsignificant difference (P = .23). Similarly, the 10-year progression-free survival was 59% in the
R-MegaCHOEP group and 60% (P = .64). The 10-year overall survival was 66% in the R-MegaCHOEP group and 72% in the R-CHOEP-14 group (P = .26). Among the 190 patients who had complete remission or unconfirmed complete remission, relapse occurred in 30 (16%); 17 (17%) of 100 patients in the R-CHOEP-14 group and 13 (14%) of 90 patients in the R-MegaCHOEP group.
In terms of secondary malignancies, 22 were reported in the intention-to-treat population; comprising 12 (9%) of 127 patients in the R-CHOEP-14 group and 10 (8%) of 126 patients in the R-MegaCHOEP group.
Patients who relapsed with aggressive histology and with CNS involvement in particular had worse outcomes and “represent a group with an unmet medical need, for which new molecular and cellular therapies should be studied,” the authors stated.
“This study shows that, in the rituximab era, high-dose therapy and autologous HSCT in first-line treatment does not improve long-term survival of younger high-risk patients with aggressive B-cell lymphoma. The R-CHOEP-14 regimen led to favorable outcomes, supporting its continued use in such patients,” the researchers concluded.
In an accompanying commentary, Gita Thanarajasingam, MD, of the Mayo Clinic, Rochester, Minn., and colleagues added that the issue of long-term outcomes is critical to evaluating these new regimens.
They applauded the inclusion of secondary malignancies in the long-term follow-up, but regretted the lack of the, admittedly resource-intensive, information on long-term nonneoplastic adverse events. They added that “the burden of late adverse events such as cardiotoxicity, cumulative neuropathy, delayed infections, or lasting cognitive effects, among others that might drive substantial morbidity, does matter to lymphoma survivors.”
They also commented on the importance of considering effects on fertility in these patients, noting that R-MegaCHOEP patients would be unable to conceive naturally, but that the effect of R-CHOEP-14 was less clear.
“We encourage ongoing emphasis on this type of longitudinal follow-up of secondary malignancies and other nonneoplastic late toxicities in phase 3 studies as well as in the real world in hematological malignancies, so that after prioritizing cure in the front-line setting, we do not neglect the life we have helped survivors achieve for years and decades to come,” they concluded.
The study was sponsored by the German High-Grade Non-Hodgkin’s Lymphoma Study Group. The authors reported grants, personal fees, and non-financial support from multiple pharmaceutical and biotechnology companies. Dr. Thanarajasingam and her colleagues reported that they had no competing interests.
After 10 years of follow-up, event-free survival and overall survival were similar between conventional chemotherapy treated patients with aggressive B-cell lymphoma and those receiving high-dose chemotherapy followed by autologous hematopoietic stem-cell transplantation (HSCT), according to a report published online in the Lancet Hematology.
The open-label, randomized, phase 3 trial (NCT00129090) was conducted across 61 centers in Germany on patients aged 18-60 years who had newly diagnosed, high-risk, aggressive B-cell lymphoma, according to Fabian Frontzek, MD, of the University Hospital Münster (Germany) and colleagues.
Between March 2003 and April 2009, patients were randomly assigned to eight cycles of conventional chemotherapy (cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisolone) plus rituximab (R-CHOEP-14) or four cycles of high-dose chemotherapy plus rituximab followed by autologous HSCT (R-MegaCHOEP). The intention-to-treat population comprised 130 patients in the R-CHOEP-14 group and 132 patients in the R-MegaCHOEP group. The median follow-up was 9.3 years.
Similar outcomes
The 10-year event-free survival was 51% in the R-MegaCHOEP group and 57% in the R-CHOEP-14 group, a nonsignificant difference (P = .23). Similarly, the 10-year progression-free survival was 59% in the
R-MegaCHOEP group and 60% (P = .64). The 10-year overall survival was 66% in the R-MegaCHOEP group and 72% in the R-CHOEP-14 group (P = .26). Among the 190 patients who had complete remission or unconfirmed complete remission, relapse occurred in 30 (16%); 17 (17%) of 100 patients in the R-CHOEP-14 group and 13 (14%) of 90 patients in the R-MegaCHOEP group.
In terms of secondary malignancies, 22 were reported in the intention-to-treat population; comprising 12 (9%) of 127 patients in the R-CHOEP-14 group and 10 (8%) of 126 patients in the R-MegaCHOEP group.
Patients who relapsed with aggressive histology and with CNS involvement in particular had worse outcomes and “represent a group with an unmet medical need, for which new molecular and cellular therapies should be studied,” the authors stated.
“This study shows that, in the rituximab era, high-dose therapy and autologous HSCT in first-line treatment does not improve long-term survival of younger high-risk patients with aggressive B-cell lymphoma. The R-CHOEP-14 regimen led to favorable outcomes, supporting its continued use in such patients,” the researchers concluded.
In an accompanying commentary, Gita Thanarajasingam, MD, of the Mayo Clinic, Rochester, Minn., and colleagues added that the issue of long-term outcomes is critical to evaluating these new regimens.
They applauded the inclusion of secondary malignancies in the long-term follow-up, but regretted the lack of the, admittedly resource-intensive, information on long-term nonneoplastic adverse events. They added that “the burden of late adverse events such as cardiotoxicity, cumulative neuropathy, delayed infections, or lasting cognitive effects, among others that might drive substantial morbidity, does matter to lymphoma survivors.”
They also commented on the importance of considering effects on fertility in these patients, noting that R-MegaCHOEP patients would be unable to conceive naturally, but that the effect of R-CHOEP-14 was less clear.
“We encourage ongoing emphasis on this type of longitudinal follow-up of secondary malignancies and other nonneoplastic late toxicities in phase 3 studies as well as in the real world in hematological malignancies, so that after prioritizing cure in the front-line setting, we do not neglect the life we have helped survivors achieve for years and decades to come,” they concluded.
The study was sponsored by the German High-Grade Non-Hodgkin’s Lymphoma Study Group. The authors reported grants, personal fees, and non-financial support from multiple pharmaceutical and biotechnology companies. Dr. Thanarajasingam and her colleagues reported that they had no competing interests.
FROM THE LANCET HEMATOLOGY
Don’t delay: Cancer patients need both doses of COVID vaccine
The new findings, which are soon to be published as a preprint, cast doubt on the current U.K. policy of delaying the second dose of the vaccine.
Delaying the second dose can leave most patients with cancer wholly or partially unprotected, according to the researchers. Moreover, such a delay has implications for transmission of SARS-CoV-2 in the cancer patient’s environs as well as for the evolution of virus variants that could be of concern, the researchers concluded.
The data come from a British study that included 151 patients with cancer and 54 healthy control persons. All participants received the COVID-19 mRNA BNT162b2 vaccine (Pfizer-BioNTech).
This vaccine requires two doses. The first few participants in this study were given the second dose 21 days after they had received the first dose, but then national guidelines changed, and the remaining participants had to wait 12 weeks to receive their second dose.
The researchers reported that, among health controls, the immune efficacy of the first dose was very high (97% efficacious). By contrast, among patients with solid tumors, the immune efficacy of a single dose was strikingly low (39%), and it was even lower in patients with hematologic malignancies (13%).
The second dose of vaccine greatly and rapidly increased the immune efficacy in patients with solid tumors (95% within 2 weeks of receiving the second dose), the researchers added.
Too few patients with hematologic cancers had received the second dose before the study ended for clear conclusions to be drawn. Nevertheless, the available data suggest that 50% of patients with hematologic cancers who had received the booster at day 21 were seropositive at 5 weeks vs. only 8% of those who had not received the booster.
“Our data provide the first real-world evidence of immune efficacy following one dose of the Pfizer vaccine in immunocompromised patient populations [and] clearly show that the poor one-dose efficacy in cancer patients can be rescued with an early booster at day 21,” commented senior author Sheeba Irshad, MD, senior clinical lecturer, King’s College London.
“Based on our findings, we would recommend an urgent review of the vaccine strategy for clinically extremely vulnerable groups. Until then, it is important that cancer patients continue to observe all public health measures in place, such as social distancing and shielding when attending hospitals, even after vaccination,” Dr. Irshad added.
The paper, with first author Leticia Monin-Aldama, PhD, is scheduled to appear on the preprint server medRxiv. It has not undergone peer review. The paper was distributed to journalists, with comments from experts not involved in the study, by the UK Science Media Centre.
These data are “of immediate importance” to patients with cancer, commented Shoba Amarnath, PhD, Newcastle University research fellow, Laboratory of T-cell Regulation, Newcastle University Center for Cancer, Newcastle upon Tyne, England.
“These findings are consistent with our understanding. … We know that the immune system within cancer patients is compromised as compared to healthy controls,” Dr. Amarnath said. “The data in the study support the notion that, in solid cancer patients, a considerable delay in second dose will extend the period when cancer patients are at risk of SARS-CoV-2 infection.”
Although more data are required, “this study does raise the issue of whether patients with cancer, other diseases, or those undergoing therapies that affect the body’s immune response should be fast-tracked for their second vaccine dose,” commented Lawrence Young, PhD, professor of molecular oncology and director of the Warwick Cancer Research Center, University of Warwick, Coventry, England.
Stephen Evans, MSc, professor of pharmacoepidemiology, London School of Hygiene and Tropical Medicine, underlined that the study is “essentially” observational and “inevitable limitations must be taken into account.
“Nevertheless, these results do suggest that the vaccines may well not protect those patients with cancer as well as those without cancer,” Mr. Evans said. He added that it is “important that this population continues to observe all COVID-19–associated measures, such as social distancing and shielding when attending hospitals, even after vaccination.”
Study details
Previous studies have shown that some patients with cancer have prolonged responses to SARS-CoV-2 infection, with ongoing immune dysregulation, inefficient seroconversion, and prolonged viral shedding.
There are few data, however, on how these patients respond to COVID-19 vaccination. The authors point out that, among the 18,860 individuals who received the Pfizer vaccine during its development trials, “none with an active oncological diagnosis was included.”
To investigate this issue, they launched the SARS-CoV-2 for Cancer Patients (SOAP-02) study.
The 151 patients with cancer who participated in this study were mostly elderly, the authors noted (75% were older than 65 years; the median age was 73 years). The majority (63%) had solid-tumor malignancies. Of those, 8% had late-stage disease and had been living with their cancer for more than 24 months.
The healthy control persons were vaccine-eligible primary health care workers who were not age matched to the cancer patients.
All participants received the first dose of vaccine; 31 (of 151) patients with cancer and 16 (of 54) healthy control persons received the second dose on day 21.
The remaining participants were scheduled to receive their second dose 12 weeks later (after the study ended), in line with the changes in the national guidelines.
The team reported that, approximately 21 days after receiving the first vaccine dose, the immune efficacy of the vaccine was estimated to be 97% among healthy control persons vs. 39% for patients with solid tumors and only 13% for those with hematologic malignancies (P < .0001 for both).
T-cell responses, as assessed via interferon-gamma and/or interleukin-2 production, were observed in 82% of healthy control persons, 71% of patients with solid tumors, and 50% of those with hematologic cancers.
Vaccine boosting at day 21 resulted in immune efficacy of 100% for healthy control persons and 95% for patients with solid tumors. In contrast, only 43% of those who did not receive the second dose were seropositive 2 weeks later.
Further analysis suggested that participants who did not have a serologic response were “spread evenly” across different cancer types, but the reduced responses were more frequent among patients who had received the vaccine within 15 days of cancer treatment, especially chemotherapy, and had undergone intensive treatments.
The SOAP study is sponsored by King’s College London and Guy’s and St. Thomas Trust Foundation NHS Trust. It is funded from grants from the KCL Charity, Cancer Research UK, and program grants from Breast Cancer Now. The investigators have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The new findings, which are soon to be published as a preprint, cast doubt on the current U.K. policy of delaying the second dose of the vaccine.
Delaying the second dose can leave most patients with cancer wholly or partially unprotected, according to the researchers. Moreover, such a delay has implications for transmission of SARS-CoV-2 in the cancer patient’s environs as well as for the evolution of virus variants that could be of concern, the researchers concluded.
The data come from a British study that included 151 patients with cancer and 54 healthy control persons. All participants received the COVID-19 mRNA BNT162b2 vaccine (Pfizer-BioNTech).
This vaccine requires two doses. The first few participants in this study were given the second dose 21 days after they had received the first dose, but then national guidelines changed, and the remaining participants had to wait 12 weeks to receive their second dose.
The researchers reported that, among health controls, the immune efficacy of the first dose was very high (97% efficacious). By contrast, among patients with solid tumors, the immune efficacy of a single dose was strikingly low (39%), and it was even lower in patients with hematologic malignancies (13%).
The second dose of vaccine greatly and rapidly increased the immune efficacy in patients with solid tumors (95% within 2 weeks of receiving the second dose), the researchers added.
Too few patients with hematologic cancers had received the second dose before the study ended for clear conclusions to be drawn. Nevertheless, the available data suggest that 50% of patients with hematologic cancers who had received the booster at day 21 were seropositive at 5 weeks vs. only 8% of those who had not received the booster.
“Our data provide the first real-world evidence of immune efficacy following one dose of the Pfizer vaccine in immunocompromised patient populations [and] clearly show that the poor one-dose efficacy in cancer patients can be rescued with an early booster at day 21,” commented senior author Sheeba Irshad, MD, senior clinical lecturer, King’s College London.
“Based on our findings, we would recommend an urgent review of the vaccine strategy for clinically extremely vulnerable groups. Until then, it is important that cancer patients continue to observe all public health measures in place, such as social distancing and shielding when attending hospitals, even after vaccination,” Dr. Irshad added.
The paper, with first author Leticia Monin-Aldama, PhD, is scheduled to appear on the preprint server medRxiv. It has not undergone peer review. The paper was distributed to journalists, with comments from experts not involved in the study, by the UK Science Media Centre.
These data are “of immediate importance” to patients with cancer, commented Shoba Amarnath, PhD, Newcastle University research fellow, Laboratory of T-cell Regulation, Newcastle University Center for Cancer, Newcastle upon Tyne, England.
“These findings are consistent with our understanding. … We know that the immune system within cancer patients is compromised as compared to healthy controls,” Dr. Amarnath said. “The data in the study support the notion that, in solid cancer patients, a considerable delay in second dose will extend the period when cancer patients are at risk of SARS-CoV-2 infection.”
Although more data are required, “this study does raise the issue of whether patients with cancer, other diseases, or those undergoing therapies that affect the body’s immune response should be fast-tracked for their second vaccine dose,” commented Lawrence Young, PhD, professor of molecular oncology and director of the Warwick Cancer Research Center, University of Warwick, Coventry, England.
Stephen Evans, MSc, professor of pharmacoepidemiology, London School of Hygiene and Tropical Medicine, underlined that the study is “essentially” observational and “inevitable limitations must be taken into account.
“Nevertheless, these results do suggest that the vaccines may well not protect those patients with cancer as well as those without cancer,” Mr. Evans said. He added that it is “important that this population continues to observe all COVID-19–associated measures, such as social distancing and shielding when attending hospitals, even after vaccination.”
Study details
Previous studies have shown that some patients with cancer have prolonged responses to SARS-CoV-2 infection, with ongoing immune dysregulation, inefficient seroconversion, and prolonged viral shedding.
There are few data, however, on how these patients respond to COVID-19 vaccination. The authors point out that, among the 18,860 individuals who received the Pfizer vaccine during its development trials, “none with an active oncological diagnosis was included.”
To investigate this issue, they launched the SARS-CoV-2 for Cancer Patients (SOAP-02) study.
The 151 patients with cancer who participated in this study were mostly elderly, the authors noted (75% were older than 65 years; the median age was 73 years). The majority (63%) had solid-tumor malignancies. Of those, 8% had late-stage disease and had been living with their cancer for more than 24 months.
The healthy control persons were vaccine-eligible primary health care workers who were not age matched to the cancer patients.
All participants received the first dose of vaccine; 31 (of 151) patients with cancer and 16 (of 54) healthy control persons received the second dose on day 21.
The remaining participants were scheduled to receive their second dose 12 weeks later (after the study ended), in line with the changes in the national guidelines.
The team reported that, approximately 21 days after receiving the first vaccine dose, the immune efficacy of the vaccine was estimated to be 97% among healthy control persons vs. 39% for patients with solid tumors and only 13% for those with hematologic malignancies (P < .0001 for both).
T-cell responses, as assessed via interferon-gamma and/or interleukin-2 production, were observed in 82% of healthy control persons, 71% of patients with solid tumors, and 50% of those with hematologic cancers.
Vaccine boosting at day 21 resulted in immune efficacy of 100% for healthy control persons and 95% for patients with solid tumors. In contrast, only 43% of those who did not receive the second dose were seropositive 2 weeks later.
Further analysis suggested that participants who did not have a serologic response were “spread evenly” across different cancer types, but the reduced responses were more frequent among patients who had received the vaccine within 15 days of cancer treatment, especially chemotherapy, and had undergone intensive treatments.
The SOAP study is sponsored by King’s College London and Guy’s and St. Thomas Trust Foundation NHS Trust. It is funded from grants from the KCL Charity, Cancer Research UK, and program grants from Breast Cancer Now. The investigators have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The new findings, which are soon to be published as a preprint, cast doubt on the current U.K. policy of delaying the second dose of the vaccine.
Delaying the second dose can leave most patients with cancer wholly or partially unprotected, according to the researchers. Moreover, such a delay has implications for transmission of SARS-CoV-2 in the cancer patient’s environs as well as for the evolution of virus variants that could be of concern, the researchers concluded.
The data come from a British study that included 151 patients with cancer and 54 healthy control persons. All participants received the COVID-19 mRNA BNT162b2 vaccine (Pfizer-BioNTech).
This vaccine requires two doses. The first few participants in this study were given the second dose 21 days after they had received the first dose, but then national guidelines changed, and the remaining participants had to wait 12 weeks to receive their second dose.
The researchers reported that, among health controls, the immune efficacy of the first dose was very high (97% efficacious). By contrast, among patients with solid tumors, the immune efficacy of a single dose was strikingly low (39%), and it was even lower in patients with hematologic malignancies (13%).
The second dose of vaccine greatly and rapidly increased the immune efficacy in patients with solid tumors (95% within 2 weeks of receiving the second dose), the researchers added.
Too few patients with hematologic cancers had received the second dose before the study ended for clear conclusions to be drawn. Nevertheless, the available data suggest that 50% of patients with hematologic cancers who had received the booster at day 21 were seropositive at 5 weeks vs. only 8% of those who had not received the booster.
“Our data provide the first real-world evidence of immune efficacy following one dose of the Pfizer vaccine in immunocompromised patient populations [and] clearly show that the poor one-dose efficacy in cancer patients can be rescued with an early booster at day 21,” commented senior author Sheeba Irshad, MD, senior clinical lecturer, King’s College London.
“Based on our findings, we would recommend an urgent review of the vaccine strategy for clinically extremely vulnerable groups. Until then, it is important that cancer patients continue to observe all public health measures in place, such as social distancing and shielding when attending hospitals, even after vaccination,” Dr. Irshad added.
The paper, with first author Leticia Monin-Aldama, PhD, is scheduled to appear on the preprint server medRxiv. It has not undergone peer review. The paper was distributed to journalists, with comments from experts not involved in the study, by the UK Science Media Centre.
These data are “of immediate importance” to patients with cancer, commented Shoba Amarnath, PhD, Newcastle University research fellow, Laboratory of T-cell Regulation, Newcastle University Center for Cancer, Newcastle upon Tyne, England.
“These findings are consistent with our understanding. … We know that the immune system within cancer patients is compromised as compared to healthy controls,” Dr. Amarnath said. “The data in the study support the notion that, in solid cancer patients, a considerable delay in second dose will extend the period when cancer patients are at risk of SARS-CoV-2 infection.”
Although more data are required, “this study does raise the issue of whether patients with cancer, other diseases, or those undergoing therapies that affect the body’s immune response should be fast-tracked for their second vaccine dose,” commented Lawrence Young, PhD, professor of molecular oncology and director of the Warwick Cancer Research Center, University of Warwick, Coventry, England.
Stephen Evans, MSc, professor of pharmacoepidemiology, London School of Hygiene and Tropical Medicine, underlined that the study is “essentially” observational and “inevitable limitations must be taken into account.
“Nevertheless, these results do suggest that the vaccines may well not protect those patients with cancer as well as those without cancer,” Mr. Evans said. He added that it is “important that this population continues to observe all COVID-19–associated measures, such as social distancing and shielding when attending hospitals, even after vaccination.”
Study details
Previous studies have shown that some patients with cancer have prolonged responses to SARS-CoV-2 infection, with ongoing immune dysregulation, inefficient seroconversion, and prolonged viral shedding.
There are few data, however, on how these patients respond to COVID-19 vaccination. The authors point out that, among the 18,860 individuals who received the Pfizer vaccine during its development trials, “none with an active oncological diagnosis was included.”
To investigate this issue, they launched the SARS-CoV-2 for Cancer Patients (SOAP-02) study.
The 151 patients with cancer who participated in this study were mostly elderly, the authors noted (75% were older than 65 years; the median age was 73 years). The majority (63%) had solid-tumor malignancies. Of those, 8% had late-stage disease and had been living with their cancer for more than 24 months.
The healthy control persons were vaccine-eligible primary health care workers who were not age matched to the cancer patients.
All participants received the first dose of vaccine; 31 (of 151) patients with cancer and 16 (of 54) healthy control persons received the second dose on day 21.
The remaining participants were scheduled to receive their second dose 12 weeks later (after the study ended), in line with the changes in the national guidelines.
The team reported that, approximately 21 days after receiving the first vaccine dose, the immune efficacy of the vaccine was estimated to be 97% among healthy control persons vs. 39% for patients with solid tumors and only 13% for those with hematologic malignancies (P < .0001 for both).
T-cell responses, as assessed via interferon-gamma and/or interleukin-2 production, were observed in 82% of healthy control persons, 71% of patients with solid tumors, and 50% of those with hematologic cancers.
Vaccine boosting at day 21 resulted in immune efficacy of 100% for healthy control persons and 95% for patients with solid tumors. In contrast, only 43% of those who did not receive the second dose were seropositive 2 weeks later.
Further analysis suggested that participants who did not have a serologic response were “spread evenly” across different cancer types, but the reduced responses were more frequent among patients who had received the vaccine within 15 days of cancer treatment, especially chemotherapy, and had undergone intensive treatments.
The SOAP study is sponsored by King’s College London and Guy’s and St. Thomas Trust Foundation NHS Trust. It is funded from grants from the KCL Charity, Cancer Research UK, and program grants from Breast Cancer Now. The investigators have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
HBV viremia linked to HCC risk in HIV/HBV coinfection
Any level of hepatitis B virus (HBV) viremia was associated with increased hepatocellular carcinoma (HCC) risk in adults with HIV/HBV coinfection, according to new research presented at the Conference on Retroviruses and Opportunistic Infections (Abstract 136).
“Chronic HBV coinfection is common among people with HIV, but the determinants of HBV-associated HCC are not well characterized,” said presenter H. Nina Kim MD, MSc, of the University of Washington, Seattle. “We sought to identify factors that contribute to HCC development in persons with HIV/HBV coinfection to guide early detection and prevention measures.”
The researchers conducted a longitudinal cohort study within the North American AIDS Cohort Collaboration on Research and Design (NA-ACCORD), a collaboration of single-site and multisite cohorts throughout the United States and Canada; 22 cohorts from NA-ACCORD were included in the analysis.
Potential HIV and HBV risk factors were examined, including viremia and CD4 percentage, as well as HBV DNA levels. Traditional risk factors for liver disease progression, including age, sex, and heavy alcohol use, were also assessed.
Eligible patients were 18 years of age or older who were followed for at least 6 months, had evidence of chronic HBV, and had HIV RNA or CD4+ cell measurement during this period. Persons with prevalent HCC at baseline were excluded.
The primary outcome was first occurrence of HCC, which was adjudicated by medical chart review and/or cancer registry. Multivariable Cox regression was used to determine adjusted hazard ratios of risk factors.
Results
Among 9,383 HIV/HBV-coinfected individuals identified, 8,354 (89%) were included in the analysis. The median age of participants was 43 years and 93.1% were male. Heavy alcohol use (35.3%) and chronic hepatitis C virus (HCV) coinfection (21.6%) were common among participants.
Among 8,354 eligible participants, 115 developed HCC over a median 6.9 years of follow-up (incidence rate, 1.8 events per 1,000 person-years; 95% confidence interval [CI], 1.5-2.1).
Independent risk factors for HCC were chronic HCV coinfection (adjusted hazard ratio [aHR], 1.60 [95% confidence interval, 1.07-2.39]), age 40 years and older (aHR, 2.14 [1.36-3.37]), and heavy alcohol use (aHR, 1.51 [1.03-2.21]); however, time-updated CD4+ percentage less than 14% (aHR, 1.03 [0.56-1.90]) and time-updated HIV RNA level over 500 copies/mL (aHR, 0.88 [0.55-1.41]) were not associated with HCC risk.
In a second model, among 3,054 patients who had HBV DNA measured, the risk of HCC was higher with HBV DNA levels greater than 200 IU/mL (aHR, 2.70 [1.23-5.93]), and the risk was particularly elevated at levels greater than 200,000 IU/mL (aHR, 4.34 [1.72-10.94]).
The researchers also found that the risk of HCC was significantly lower in patients with HBV DNA suppression less than 200 IU/mL receiving HBV-active ART for 1 year or more (aHR, 0.42 [0.24-0.73]). In addition, a dose-response relationship was observed between the duration of suppression and this protective effect.
Dr. Nina Kim acknowledged that a key limitation of the study was inconsistent monitoring of HBV DNA level while patients were on treatment. Furthermore, given the demographics of the cohort, these results may not be generalizable outside of North America.
“Our study was the first to show that any level of HBV viremia using 200 as a threshold of detection was associated with HCC risk in a large regionally diverse cohort of adults outside of Asia,” Dr. Kim said. “To gain maximal protective benefit from antiviral therapy for HCC prevention, sustained and ideally uninterrupted suppression of HBV may be necessary over years.”
“HIV/HBV coinfected patients can take much longer than a year to achieve less than 200 copies on HBV DNA due to their baseline levels, but we still don’t know if HBV therapy intensification could hasten this process,” said moderator Robert T. Schooley, MD, of the University of California, San Diego.
Dr. Kim disclosed no conflicts of interest. The study was supported by multiple sources, including the National Institutes of Health, the Centers for Disease Control and Prevention, and the National Cancer Institute.
Any level of hepatitis B virus (HBV) viremia was associated with increased hepatocellular carcinoma (HCC) risk in adults with HIV/HBV coinfection, according to new research presented at the Conference on Retroviruses and Opportunistic Infections (Abstract 136).
“Chronic HBV coinfection is common among people with HIV, but the determinants of HBV-associated HCC are not well characterized,” said presenter H. Nina Kim MD, MSc, of the University of Washington, Seattle. “We sought to identify factors that contribute to HCC development in persons with HIV/HBV coinfection to guide early detection and prevention measures.”
The researchers conducted a longitudinal cohort study within the North American AIDS Cohort Collaboration on Research and Design (NA-ACCORD), a collaboration of single-site and multisite cohorts throughout the United States and Canada; 22 cohorts from NA-ACCORD were included in the analysis.
Potential HIV and HBV risk factors were examined, including viremia and CD4 percentage, as well as HBV DNA levels. Traditional risk factors for liver disease progression, including age, sex, and heavy alcohol use, were also assessed.
Eligible patients were 18 years of age or older who were followed for at least 6 months, had evidence of chronic HBV, and had HIV RNA or CD4+ cell measurement during this period. Persons with prevalent HCC at baseline were excluded.
The primary outcome was first occurrence of HCC, which was adjudicated by medical chart review and/or cancer registry. Multivariable Cox regression was used to determine adjusted hazard ratios of risk factors.
Results
Among 9,383 HIV/HBV-coinfected individuals identified, 8,354 (89%) were included in the analysis. The median age of participants was 43 years and 93.1% were male. Heavy alcohol use (35.3%) and chronic hepatitis C virus (HCV) coinfection (21.6%) were common among participants.
Among 8,354 eligible participants, 115 developed HCC over a median 6.9 years of follow-up (incidence rate, 1.8 events per 1,000 person-years; 95% confidence interval [CI], 1.5-2.1).
Independent risk factors for HCC were chronic HCV coinfection (adjusted hazard ratio [aHR], 1.60 [95% confidence interval, 1.07-2.39]), age 40 years and older (aHR, 2.14 [1.36-3.37]), and heavy alcohol use (aHR, 1.51 [1.03-2.21]); however, time-updated CD4+ percentage less than 14% (aHR, 1.03 [0.56-1.90]) and time-updated HIV RNA level over 500 copies/mL (aHR, 0.88 [0.55-1.41]) were not associated with HCC risk.
In a second model, among 3,054 patients who had HBV DNA measured, the risk of HCC was higher with HBV DNA levels greater than 200 IU/mL (aHR, 2.70 [1.23-5.93]), and the risk was particularly elevated at levels greater than 200,000 IU/mL (aHR, 4.34 [1.72-10.94]).
The researchers also found that the risk of HCC was significantly lower in patients with HBV DNA suppression less than 200 IU/mL receiving HBV-active ART for 1 year or more (aHR, 0.42 [0.24-0.73]). In addition, a dose-response relationship was observed between the duration of suppression and this protective effect.
Dr. Nina Kim acknowledged that a key limitation of the study was inconsistent monitoring of HBV DNA level while patients were on treatment. Furthermore, given the demographics of the cohort, these results may not be generalizable outside of North America.
“Our study was the first to show that any level of HBV viremia using 200 as a threshold of detection was associated with HCC risk in a large regionally diverse cohort of adults outside of Asia,” Dr. Kim said. “To gain maximal protective benefit from antiviral therapy for HCC prevention, sustained and ideally uninterrupted suppression of HBV may be necessary over years.”
“HIV/HBV coinfected patients can take much longer than a year to achieve less than 200 copies on HBV DNA due to their baseline levels, but we still don’t know if HBV therapy intensification could hasten this process,” said moderator Robert T. Schooley, MD, of the University of California, San Diego.
Dr. Kim disclosed no conflicts of interest. The study was supported by multiple sources, including the National Institutes of Health, the Centers for Disease Control and Prevention, and the National Cancer Institute.
Any level of hepatitis B virus (HBV) viremia was associated with increased hepatocellular carcinoma (HCC) risk in adults with HIV/HBV coinfection, according to new research presented at the Conference on Retroviruses and Opportunistic Infections (Abstract 136).
“Chronic HBV coinfection is common among people with HIV, but the determinants of HBV-associated HCC are not well characterized,” said presenter H. Nina Kim MD, MSc, of the University of Washington, Seattle. “We sought to identify factors that contribute to HCC development in persons with HIV/HBV coinfection to guide early detection and prevention measures.”
The researchers conducted a longitudinal cohort study within the North American AIDS Cohort Collaboration on Research and Design (NA-ACCORD), a collaboration of single-site and multisite cohorts throughout the United States and Canada; 22 cohorts from NA-ACCORD were included in the analysis.
Potential HIV and HBV risk factors were examined, including viremia and CD4 percentage, as well as HBV DNA levels. Traditional risk factors for liver disease progression, including age, sex, and heavy alcohol use, were also assessed.
Eligible patients were 18 years of age or older who were followed for at least 6 months, had evidence of chronic HBV, and had HIV RNA or CD4+ cell measurement during this period. Persons with prevalent HCC at baseline were excluded.
The primary outcome was first occurrence of HCC, which was adjudicated by medical chart review and/or cancer registry. Multivariable Cox regression was used to determine adjusted hazard ratios of risk factors.
Results
Among 9,383 HIV/HBV-coinfected individuals identified, 8,354 (89%) were included in the analysis. The median age of participants was 43 years and 93.1% were male. Heavy alcohol use (35.3%) and chronic hepatitis C virus (HCV) coinfection (21.6%) were common among participants.
Among 8,354 eligible participants, 115 developed HCC over a median 6.9 years of follow-up (incidence rate, 1.8 events per 1,000 person-years; 95% confidence interval [CI], 1.5-2.1).
Independent risk factors for HCC were chronic HCV coinfection (adjusted hazard ratio [aHR], 1.60 [95% confidence interval, 1.07-2.39]), age 40 years and older (aHR, 2.14 [1.36-3.37]), and heavy alcohol use (aHR, 1.51 [1.03-2.21]); however, time-updated CD4+ percentage less than 14% (aHR, 1.03 [0.56-1.90]) and time-updated HIV RNA level over 500 copies/mL (aHR, 0.88 [0.55-1.41]) were not associated with HCC risk.
In a second model, among 3,054 patients who had HBV DNA measured, the risk of HCC was higher with HBV DNA levels greater than 200 IU/mL (aHR, 2.70 [1.23-5.93]), and the risk was particularly elevated at levels greater than 200,000 IU/mL (aHR, 4.34 [1.72-10.94]).
The researchers also found that the risk of HCC was significantly lower in patients with HBV DNA suppression less than 200 IU/mL receiving HBV-active ART for 1 year or more (aHR, 0.42 [0.24-0.73]). In addition, a dose-response relationship was observed between the duration of suppression and this protective effect.
Dr. Nina Kim acknowledged that a key limitation of the study was inconsistent monitoring of HBV DNA level while patients were on treatment. Furthermore, given the demographics of the cohort, these results may not be generalizable outside of North America.
“Our study was the first to show that any level of HBV viremia using 200 as a threshold of detection was associated with HCC risk in a large regionally diverse cohort of adults outside of Asia,” Dr. Kim said. “To gain maximal protective benefit from antiviral therapy for HCC prevention, sustained and ideally uninterrupted suppression of HBV may be necessary over years.”
“HIV/HBV coinfected patients can take much longer than a year to achieve less than 200 copies on HBV DNA due to their baseline levels, but we still don’t know if HBV therapy intensification could hasten this process,” said moderator Robert T. Schooley, MD, of the University of California, San Diego.
Dr. Kim disclosed no conflicts of interest. The study was supported by multiple sources, including the National Institutes of Health, the Centers for Disease Control and Prevention, and the National Cancer Institute.
FROM CROI 2021
USPSTF expands criteria for lung cancer screening
“This is great news because it means that nearly twice as many people are eligible to be screened, which we hope will allow clinicians to save more lives and help people remain healthy longer,” commented John Wong, MD, chief science officer, vice chair for clinical affairs, and chief of the Division of Clinical Decision Making at USPSTF.
The updated final recommendations were published online on March 9 in JAMA.
The USPSTF recommends annual screening with low-dose CT for adults aged 50-80 years who have a 20 pack-year smoking history and currently smoke or have quit within the past 15 years.
This updates guidance issued in 2013, which recommended annual screening for lung cancer for adults aged 55-80 years who had a 30 pack-year smoking history and who were either current smokers or had quit within the past 15 years.
The move will nearly double the number of people are now eligible for screening, up to 14.5 million individuals – an increase of 81% (6.4 million adults) from the 2013 recommendations.
The expanded criteria may help increase screening among Black individuals and women. Data show that both groups tend to smoke fewer cigarettes than White men and that Black persons are at higher risk for lung cancer than White persons. In addition, research has shown that about one-third of Black patients with lung cancer were diagnosed before the age of 55 years, which means they would not have been recommended for screening under the previous guidelines.
Uptake has been limited
To date, uptake of lung cancer screening has been very limited, from 6% to 18% of individuals who meet the eligibility criteria.
The new recommendations will open up screening to many more people, but challenges to implementation remain.
“The science is clear that lung cancer screening has the potential to save lives,” Dr. Wong told this news organization. “We recognize that there are existing barriers to screening everyone who is eligible, but clinicians and patients both deserve to know that screening can detect lung cancer early, when treatment has the best chance of being beneficial.”
He added that the hope is that these recommendations will encourage clinicians to examine the barriers to effective lung cancer screening in their communities and to do what they can to improve implementation. “We also hope to encourage patients to have conversations with their clinicians about whether they are eligible for screening and to discuss smoking cessation treatments if they are still smoking,” Dr. Wong added.
In an accompanying editorial, Louise M. Henderson, PhD, M. Patricia Rivera, MD, FCCP, and Ethan Basch, MD, all from the University of North Carolina at Chapel Hill, address some of the current challenges in implementation.
They note that reimbursement for lung cancer screening by Medicare requires submission of data to a Centers for Medicare & Medicaid Services–approved registry, and this can present problems for facilities serving less affluent communities or that have limited resources.
Medicaid coverage is also uneven. As of September 2020, lung cancer screening was covered by 38 Medicaid programs, but not by 9. For three programs, data on coverage were not available.
“With the new recommendations lowering the screening-eligible age to 50 years, many eligible individuals who are uninsured or who are receiving Medicaid and living in states that do not cover screening will have financial barriers to undergo screening,” they write.
In addition, many individuals in at-risk populations lack adequate geographic access to comprehensive lung cancer screening programs.
Expanding eligibility criteria is important, the editorialists point out, but barriers to screening, which include lack of insurance coverage and limited physical access to high-quality screening programs, highlight the complex problems with implementation that need to be addressed.
“A concerted effort to increase the reach of lung cancer screening is needed,” they write. “The 2021 USPSTF recommendation statement represents a leap forward in evidence and offers promise to prevent more cancer deaths and address screening disparities. But the greatest work lies ahead to ensure this promise is actualized.”
Advocacy needed
When approached for comment, Jianjun Zhang, MD, PhD, from the department of thoracic/head and neck medical oncology, University of Texas MD Anderson Cancer Center, Houston, said he supports the new guidelines, and they will lower mortality. “The data are pretty strong overall,” he said in an interview.
Although the uptake of screening is currently very low, he pointed out that, even if uptake remains the same, more lives will be saved because eligibility has been expanded. “More people will be getting screened, so it’s a start,” he said.
Aside from factors such as insurance and access, another problem involves primary care. “Time is very limited in primary care,” he said. “You have about 15 minutes, and it can be really hard to fit everything into a visit. Screening may get left out or may only get a brief mention.”
Advocacy is needed, Dr. Zhang pointed out. “Breast cancer has strong voices and advocacy, and people are more aware of mammography,” he said. “The information is disseminated out into the community. We need the same for lung cancer.”
Dr. Zhang emphasized that, even with the expanded criteria, many individuals will still be missed. “There are other risk factors besides smoking,” he said. “About 10% of lung cancers occur in never-smokers.”
Other risk factors include a family history of lung cancer, exposure to certain materials and chemicals, working in the mining industry, and genetics.
“We will move on to more personalized screening at some point,” he said. “But right now, we can’t make it too complicated for patients and doctors. We need to concentrate on increasing screening rates within these current criteria.”
The updated guidelines have been given a B recommendation, meaning the USPSTF recommends that clinicians provide the service to eligible patients, there is at least fair evidence that this service improves important health outcomes, and benefits outweigh harms.
The USPSTF is an independent, voluntary body. The U.S. Congress mandates that the Agency for Healthcare Research and Quality support the operations of the USPSTF. All members of the USPSTF receive travel reimbursement and an honorarium for participating in USPSTF meetings. The original article lists relevant financial relationships of task force members. Dr. Zhang has received grants from Johnson & Johnson and Merck, and adversary/consulting/honoraria fees from AstraZeneca, Bristol-Myers Squibb, GenePlus, Innovent, OrigMed, and Roche.
A version of this article first appeared on Medscape.com.
“This is great news because it means that nearly twice as many people are eligible to be screened, which we hope will allow clinicians to save more lives and help people remain healthy longer,” commented John Wong, MD, chief science officer, vice chair for clinical affairs, and chief of the Division of Clinical Decision Making at USPSTF.
The updated final recommendations were published online on March 9 in JAMA.
The USPSTF recommends annual screening with low-dose CT for adults aged 50-80 years who have a 20 pack-year smoking history and currently smoke or have quit within the past 15 years.
This updates guidance issued in 2013, which recommended annual screening for lung cancer for adults aged 55-80 years who had a 30 pack-year smoking history and who were either current smokers or had quit within the past 15 years.
The move will nearly double the number of people are now eligible for screening, up to 14.5 million individuals – an increase of 81% (6.4 million adults) from the 2013 recommendations.
The expanded criteria may help increase screening among Black individuals and women. Data show that both groups tend to smoke fewer cigarettes than White men and that Black persons are at higher risk for lung cancer than White persons. In addition, research has shown that about one-third of Black patients with lung cancer were diagnosed before the age of 55 years, which means they would not have been recommended for screening under the previous guidelines.
Uptake has been limited
To date, uptake of lung cancer screening has been very limited, from 6% to 18% of individuals who meet the eligibility criteria.
The new recommendations will open up screening to many more people, but challenges to implementation remain.
“The science is clear that lung cancer screening has the potential to save lives,” Dr. Wong told this news organization. “We recognize that there are existing barriers to screening everyone who is eligible, but clinicians and patients both deserve to know that screening can detect lung cancer early, when treatment has the best chance of being beneficial.”
He added that the hope is that these recommendations will encourage clinicians to examine the barriers to effective lung cancer screening in their communities and to do what they can to improve implementation. “We also hope to encourage patients to have conversations with their clinicians about whether they are eligible for screening and to discuss smoking cessation treatments if they are still smoking,” Dr. Wong added.
In an accompanying editorial, Louise M. Henderson, PhD, M. Patricia Rivera, MD, FCCP, and Ethan Basch, MD, all from the University of North Carolina at Chapel Hill, address some of the current challenges in implementation.
They note that reimbursement for lung cancer screening by Medicare requires submission of data to a Centers for Medicare & Medicaid Services–approved registry, and this can present problems for facilities serving less affluent communities or that have limited resources.
Medicaid coverage is also uneven. As of September 2020, lung cancer screening was covered by 38 Medicaid programs, but not by 9. For three programs, data on coverage were not available.
“With the new recommendations lowering the screening-eligible age to 50 years, many eligible individuals who are uninsured or who are receiving Medicaid and living in states that do not cover screening will have financial barriers to undergo screening,” they write.
In addition, many individuals in at-risk populations lack adequate geographic access to comprehensive lung cancer screening programs.
Expanding eligibility criteria is important, the editorialists point out, but barriers to screening, which include lack of insurance coverage and limited physical access to high-quality screening programs, highlight the complex problems with implementation that need to be addressed.
“A concerted effort to increase the reach of lung cancer screening is needed,” they write. “The 2021 USPSTF recommendation statement represents a leap forward in evidence and offers promise to prevent more cancer deaths and address screening disparities. But the greatest work lies ahead to ensure this promise is actualized.”
Advocacy needed
When approached for comment, Jianjun Zhang, MD, PhD, from the department of thoracic/head and neck medical oncology, University of Texas MD Anderson Cancer Center, Houston, said he supports the new guidelines, and they will lower mortality. “The data are pretty strong overall,” he said in an interview.
Although the uptake of screening is currently very low, he pointed out that, even if uptake remains the same, more lives will be saved because eligibility has been expanded. “More people will be getting screened, so it’s a start,” he said.
Aside from factors such as insurance and access, another problem involves primary care. “Time is very limited in primary care,” he said. “You have about 15 minutes, and it can be really hard to fit everything into a visit. Screening may get left out or may only get a brief mention.”
Advocacy is needed, Dr. Zhang pointed out. “Breast cancer has strong voices and advocacy, and people are more aware of mammography,” he said. “The information is disseminated out into the community. We need the same for lung cancer.”
Dr. Zhang emphasized that, even with the expanded criteria, many individuals will still be missed. “There are other risk factors besides smoking,” he said. “About 10% of lung cancers occur in never-smokers.”
Other risk factors include a family history of lung cancer, exposure to certain materials and chemicals, working in the mining industry, and genetics.
“We will move on to more personalized screening at some point,” he said. “But right now, we can’t make it too complicated for patients and doctors. We need to concentrate on increasing screening rates within these current criteria.”
The updated guidelines have been given a B recommendation, meaning the USPSTF recommends that clinicians provide the service to eligible patients, there is at least fair evidence that this service improves important health outcomes, and benefits outweigh harms.
The USPSTF is an independent, voluntary body. The U.S. Congress mandates that the Agency for Healthcare Research and Quality support the operations of the USPSTF. All members of the USPSTF receive travel reimbursement and an honorarium for participating in USPSTF meetings. The original article lists relevant financial relationships of task force members. Dr. Zhang has received grants from Johnson & Johnson and Merck, and adversary/consulting/honoraria fees from AstraZeneca, Bristol-Myers Squibb, GenePlus, Innovent, OrigMed, and Roche.
A version of this article first appeared on Medscape.com.
“This is great news because it means that nearly twice as many people are eligible to be screened, which we hope will allow clinicians to save more lives and help people remain healthy longer,” commented John Wong, MD, chief science officer, vice chair for clinical affairs, and chief of the Division of Clinical Decision Making at USPSTF.
The updated final recommendations were published online on March 9 in JAMA.
The USPSTF recommends annual screening with low-dose CT for adults aged 50-80 years who have a 20 pack-year smoking history and currently smoke or have quit within the past 15 years.
This updates guidance issued in 2013, which recommended annual screening for lung cancer for adults aged 55-80 years who had a 30 pack-year smoking history and who were either current smokers or had quit within the past 15 years.
The move will nearly double the number of people are now eligible for screening, up to 14.5 million individuals – an increase of 81% (6.4 million adults) from the 2013 recommendations.
The expanded criteria may help increase screening among Black individuals and women. Data show that both groups tend to smoke fewer cigarettes than White men and that Black persons are at higher risk for lung cancer than White persons. In addition, research has shown that about one-third of Black patients with lung cancer were diagnosed before the age of 55 years, which means they would not have been recommended for screening under the previous guidelines.
Uptake has been limited
To date, uptake of lung cancer screening has been very limited, from 6% to 18% of individuals who meet the eligibility criteria.
The new recommendations will open up screening to many more people, but challenges to implementation remain.
“The science is clear that lung cancer screening has the potential to save lives,” Dr. Wong told this news organization. “We recognize that there are existing barriers to screening everyone who is eligible, but clinicians and patients both deserve to know that screening can detect lung cancer early, when treatment has the best chance of being beneficial.”
He added that the hope is that these recommendations will encourage clinicians to examine the barriers to effective lung cancer screening in their communities and to do what they can to improve implementation. “We also hope to encourage patients to have conversations with their clinicians about whether they are eligible for screening and to discuss smoking cessation treatments if they are still smoking,” Dr. Wong added.
In an accompanying editorial, Louise M. Henderson, PhD, M. Patricia Rivera, MD, FCCP, and Ethan Basch, MD, all from the University of North Carolina at Chapel Hill, address some of the current challenges in implementation.
They note that reimbursement for lung cancer screening by Medicare requires submission of data to a Centers for Medicare & Medicaid Services–approved registry, and this can present problems for facilities serving less affluent communities or that have limited resources.
Medicaid coverage is also uneven. As of September 2020, lung cancer screening was covered by 38 Medicaid programs, but not by 9. For three programs, data on coverage were not available.
“With the new recommendations lowering the screening-eligible age to 50 years, many eligible individuals who are uninsured or who are receiving Medicaid and living in states that do not cover screening will have financial barriers to undergo screening,” they write.
In addition, many individuals in at-risk populations lack adequate geographic access to comprehensive lung cancer screening programs.
Expanding eligibility criteria is important, the editorialists point out, but barriers to screening, which include lack of insurance coverage and limited physical access to high-quality screening programs, highlight the complex problems with implementation that need to be addressed.
“A concerted effort to increase the reach of lung cancer screening is needed,” they write. “The 2021 USPSTF recommendation statement represents a leap forward in evidence and offers promise to prevent more cancer deaths and address screening disparities. But the greatest work lies ahead to ensure this promise is actualized.”
Advocacy needed
When approached for comment, Jianjun Zhang, MD, PhD, from the department of thoracic/head and neck medical oncology, University of Texas MD Anderson Cancer Center, Houston, said he supports the new guidelines, and they will lower mortality. “The data are pretty strong overall,” he said in an interview.
Although the uptake of screening is currently very low, he pointed out that, even if uptake remains the same, more lives will be saved because eligibility has been expanded. “More people will be getting screened, so it’s a start,” he said.
Aside from factors such as insurance and access, another problem involves primary care. “Time is very limited in primary care,” he said. “You have about 15 minutes, and it can be really hard to fit everything into a visit. Screening may get left out or may only get a brief mention.”
Advocacy is needed, Dr. Zhang pointed out. “Breast cancer has strong voices and advocacy, and people are more aware of mammography,” he said. “The information is disseminated out into the community. We need the same for lung cancer.”
Dr. Zhang emphasized that, even with the expanded criteria, many individuals will still be missed. “There are other risk factors besides smoking,” he said. “About 10% of lung cancers occur in never-smokers.”
Other risk factors include a family history of lung cancer, exposure to certain materials and chemicals, working in the mining industry, and genetics.
“We will move on to more personalized screening at some point,” he said. “But right now, we can’t make it too complicated for patients and doctors. We need to concentrate on increasing screening rates within these current criteria.”
The updated guidelines have been given a B recommendation, meaning the USPSTF recommends that clinicians provide the service to eligible patients, there is at least fair evidence that this service improves important health outcomes, and benefits outweigh harms.
The USPSTF is an independent, voluntary body. The U.S. Congress mandates that the Agency for Healthcare Research and Quality support the operations of the USPSTF. All members of the USPSTF receive travel reimbursement and an honorarium for participating in USPSTF meetings. The original article lists relevant financial relationships of task force members. Dr. Zhang has received grants from Johnson & Johnson and Merck, and adversary/consulting/honoraria fees from AstraZeneca, Bristol-Myers Squibb, GenePlus, Innovent, OrigMed, and Roche.
A version of this article first appeared on Medscape.com.
Impact of an Oral Antineoplastic Renewal Clinic on Medication Possession Ratio and Cost-Savings
Evaluation of oral antineoplastic agent (OAN) adherence patterns have identified correlations between nonadherence or over-adherence and poorer disease-related outcomes. Multiple studies have focused on imatinib use in chronic myeloid leukemia (CML) due to its continuous, long-term use. A study by Ganesan and colleagues found that nonadherence to imatinib showed a significant decrease in 5-year event-free survival between 76.7% of adherent participants compared with 59.8% of nonadherent participants.1 This study found that 44% of patients who were adherent to imatinib achieved complete cytogenetic response vs only 26% of patients who were nonadherent. In another study of imatinib for CML, major molecular response (MMR) was strongly correlated with adherence and no patients with adherence < 80% were able to achieve MMR.2 Similarly, in studies of tamoxifen for breast cancer, < 80% adherence resulted in a 10% decrease in survival when compared to those who were more adherent.3,4
In addition to the clinical implications of nonadherence, there can be a significant cost associated with suboptimal use of these medications. The price of a single dose of OAN medication may cost as much as $440.5
The benefits of multidisciplinary care teams have been identified in many studies.6,7 While studies are limited in oncology, pharmacists provide vital contributions to the oncology multidisciplinary team when managing OANs as these health care professionals have expert knowledge of the medications, potential adverse events (AEs), and necessary monitoring parameters.8 In one study, patients seen by the pharmacist-led oral chemotherapy management program experienced improved clinical outcomes and response to therapy when compared with preintervention patients (early molecular response, 88.9% vs 54.8%, P = .01; major molecular response, 83.3% vs 57.6%, P = .06).9 During the study, 318 AEs were reported, leading to 235 pharmacist interventions to ameliorate AEs and improve adherence.
The primary objective of this study was to measure the impact of a pharmacist-driven OAN renewal clinic on medication adherence. The secondary objective was to estimate cost-savings of this new service.
Methods
Prior to July 2014, several limitations were identified related to OAN prescribing and monitoring at the Richard L. Roudebush Veterans Affairs Medical Center in Indianapolis, Indiana (RLRVAMC). The prescription ordering process relied primarily on the patient to initiate refills, rather than the prescriber OAN prescriptions also lacked consistency for number of refills or quantities dispensed. Furthermore, ordering of antineoplastic products was not limited to hematology/oncology providers. Patients were identified with significant supply on hand at the time of medication discontinuation, creating concerns for medication waste, tolerability, and nonadherence.
As a result, opportunities were identified to improve the prescribing process, recommended monitoring, toxicity and tolerability evaluation, medication reconciliation, and medication adherence. In July of 2014, the RLRVAMC adopted a new chemotherapy order entry system capable of restricting prescriptions to hematology/oncology providers and limiting dispensed quantities and refill amounts. A comprehensive pharmacist driven OAN renewal clinic was implemented on September 1, 2014 with the goal of improving long-term adherence and tolerability, in addition to minimizing medication waste.

Patients were eligible for enrollment in the clinic if they had a cancer diagnosis and were concomitantly prescribed an OAN outlined in Table 1. All eligible patients were automatically enrolled in the clinic when they were deemed stable on their OAN by a hematology/oncology pharmacy specialist. Stability was defined as ≤ Grade 1 symptoms associated with the toxicities of OAN therapy managed with or without intervention as defined by the Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. Once enrolled in the renewal clinic, patients were called by an oncology pharmacy resident (PGY2) 1 week prior to any OAN refill due date. Patients were asked a series of 5 adherence and tolerability questions (Table 2) to evaluate renewal criteria for approval or need for further evaluation. These questions were developed based on targeted information and published reports on monitoring adherence.10,11 Criteria for renewal included: < 10% self-reported missed doses of the OAN during the previous dispensing period, no hospitalizations or emergency department visits since most recent hematology/oncology provider appointment, no changes to concomitant medication therapies, and no new or worsening medication-related AEs. Patients meeting all criteria were given a 30-day supply of OAN. Prescribing, dispensing, and delivery of OAN were facilitated by the pharmacist. Patient cases that did not meet criteria for renewal were escalated to the hematology/oncology provider or oncology clinical pharmacy specialist for further evaluation.
Study Design and Setting
This was a pre/post retrospective cohort, quality improvement study of patients enrolled in the RLRVAMC OAN pharmacist renewal clinic. The study was deemed exempt from institutional review board (IRB) by the US Department of Veterans Affairs (VA) Research and Development Department.
Study Population
Patients were included in the preimplementation group if they had received at least 2 prescriptions of an eligible OAN. Therapy for the preimplementation group was required to be a monthly duration > 21 days and between the dates of September 1, 2013 and August 31, 2014. Patients were included in the postimplementation group if they had received at least 2 prescriptions of the studied OANs between September 1, 2014 and January 31, 2015. Patients were excluded if they had filled < 2 prescriptions of OAN; were managed by a non-VA oncologist or hematologist; or received an OAN other than those listed in Table 1.
Data Collection
For all patients in both the pre- and postimplementation cohorts, a standardized data collection tool was used to collect the following via electronic health record review by a PGY2 oncology resident: age, race, gender, oral antineoplastic agent, refill dates, days’ supply, estimated unit cost per dose cancer diagnosis, distance from the RLRVAMC, copay status, presence of hospitalizations/ED visits/dosage reductions, discontinuation rates, reasons for discontinuation, and total number of current prescriptions. The presence or absence of dosage reductions were collected to identify concerns for tolerability, but only the original dose for the preimplementation group and dosage at time of clinic enrollment for the postimplementation group was included in the analysis.
Outcomes and Statistical Analyses
The primary outcome was medication adherence defined as the median medication possession ratio (MPR) before and after implementation of the clinic. Secondary outcomes included the proportion of patients who were adherent from before implementation to after and estimated cost-savings of this clinic after implementation. MPR was used to estimate medication adherence by taking the cumulative day supply of medication on hand divided by the number of days on therapy.12 Number of days on therapy was determined by taking the difference on the start date of the new medication regimen and the discontinuation date of the same regimen. Patients were grouped by adherence into one of the following categories: < 0.8, 0.8 to 0.89, 0.9 to 1, and > 1.1. Patients were considered adherent if they reported taking ≥ 90% (MPR ≥ 0.9) of prescribed doses, adopted from the study by Anderson and colleagues.12 A patient with an MPR > 1, likely due to filling prior to the anticipated refill date, was considered 100% adherent (MPR = 1). If a patient switched OAN during the study, both agents were included as separate entities.
A conservative estimate of cost-savings was made by multiplying the RLRVAMC cost per unit of medication at time of initial prescription fill by the number of units taken each day multiplied by the total days’ supply on hand at time of therapy discontinuation. Patients with an MPR < 1 at time of therapy discontinuation were assumed to have zero remaining units on hand and zero cost savings was estimated. Waste, for purposes of cost-savings, was calculated for all MPR values > 1. Additional supply anticipated to be on hand from dose reductions was not included in the estimated cost of unused medication.
Descriptive statistics compared demographic characteristics between the pre- and postimplementation groups. MPR data were not normally distributed, which required the use of nonparametric Mann-Whitney U tests to compare pre- and postMPRs. Pearson χ2 compared the proportion of adherent patients between groups while descriptive statistics were used to estimate cost savings. Significance was determined based on a P value < .05. IBM SPSS Statistics software was used for all statistical analyses. As this was a complete sample of all eligible subjects, no sample size calculation was performed.
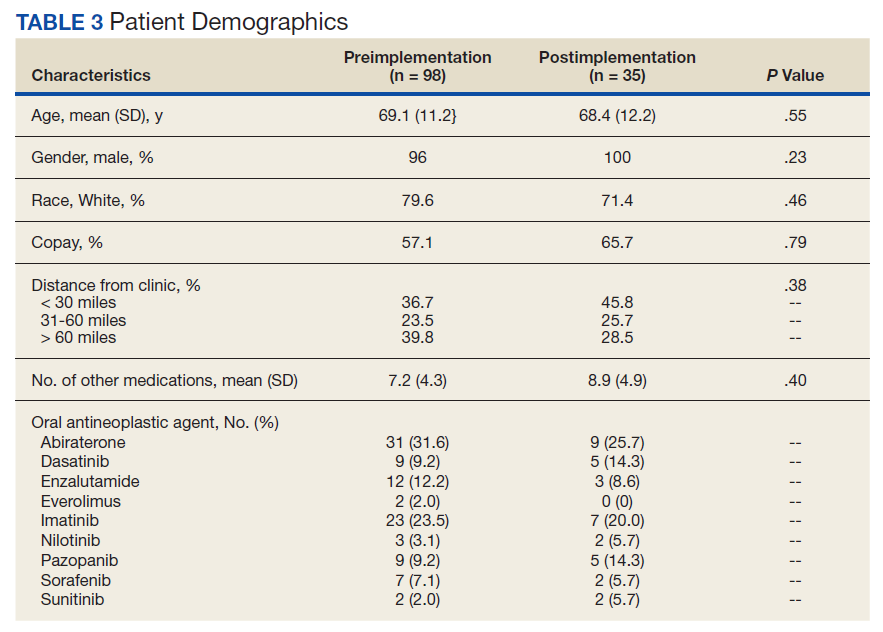
Results
In the preimplementation period, 246 patients received an OAN and 61 patients received an OAN in the postimplementation period (Figure 1). Of the 246 patients in the preimplementation period, 98 were eligible and included in the preimplementation group. Similarly, of the 61 patients in the postimplementation period, 35 patients met inclusion criteria for the postimplementation group. The study population was predominantly male with an average age of approximately 70 years in both groups (Table 3). More than 70% of the population in each group was White. No statistically significant differences between groups were identified. The most commonly prescribed OAN in the preimplementation group were abiraterone, imatinib, and enzalutamide (Table 3). In the postimplementation group, the most commonly prescribed agents were abiraterone, imatinib, pazopanib, and dasatinib. No significant differences were observed in prescribing of individual agents between the pre- and postimplementation groups or other characteristics that may affect adherence including patient copay status, number of concomitant medications, and driving distance from the RLRVAMC.
Thirty-six (36.7%) patients in the preimplementation group were considered nonadherent (MPR < 0.9) and 18 (18.4%) had an MPR < 0.8. Fifteen (15.3%) patients in the preimplementation clinic were considered overadherent (MPR > 1.1). Forty-seven (47.9%) patients in the preimplementation group were considered adherent (MPR 0.9 - 1.1) while all 35 (100%) patients in the postimplementation group were considered adherent (MPR 0.9 - 1.1). No non- or overadherent patients were identified in the postimplementation group (Figure 2). The median MPR for all patients in the preimplementation group was 0.94 compared with 1.06 (P < .001) in the postimplementation group.

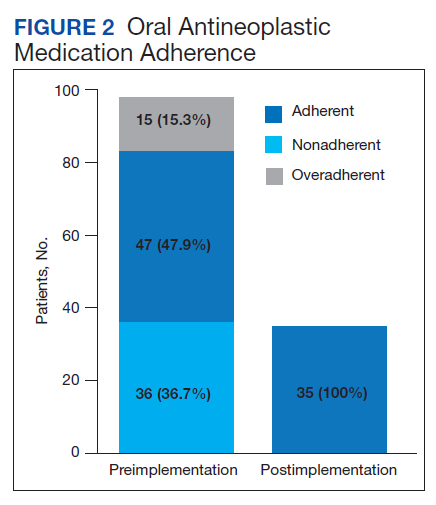
Thirty-five (35.7%) patients had therapy discontinued or held in the preimplementation group compared with 2 (5.7%) patients in the postimplementation group (P < .001). Reasons for discontinuation in the preimplementation group included disease progression (n = 27), death (n = 3), lost to follow up (n = 2), and intolerability of therapy (n = 3). Both patients that discontinued therapy in the postimplementation group did so due to disease progression. Of the 35 patients who had their OAN discontinued or held in the preimplementation group, 14 patients had excess supply on hand at time of discontinuation. The estimated value of the unused medication was $37,890. Nine (25%) of the 35 patients who discontinued therapy had a dosage reduction during the course of therapy and the additional supply was not included in the cost estimate. Similarly, 1 of the 2 patients in the postimplementation group had their OAN discontinued during study. The cost of oversupply of medication at the time of therapy discontinuation was estimated at $1,555. No patients in the postimplementation group had dose reductions. After implementation of the OAN renewal clinic, the total cost savings between pre ($37,890) and postimplementation ($1,555) groups was $36,355.
Discussion
OANs are widely used therapies, with more than 25 million doses administered per year in the United States alone.12 The use of these agents will continue to grow as more targeted agents become available and patients request more convenient treatment options. The role for hematology/oncology clinical pharmacy services must adapt to this increased usage of OANs, including increasing pharmacist involvement in medication education, adherence and tolerability assessments, and proactive drug interaction monitoring.However, additional research is needed to determine optimal management strategies.
Our study aimed to compare OAN adherence among patients at a tertiary care VA hospital before and after implementation of a renewal clinic. The preimplementation population had a median MPR of 0.94 compared with 1.06 in the postimplementation group (P < .001). Although an ideal MPR is 1.0, we aimed for a slightly higher MPR to allow a supply buffer in the event of prescription delivery delays, as more than 90% of prescriptions are mailed to patients from a regional mail-order pharmacy. Importantly, the median MPRs do not adequately convey the impact from this clinic. The proportion of patients who were considered adherent to OANs increased from 47.9% in the preimplementation to 100% in the postimplementation period. These finding suggest that the clinical pharmacist role to assess and encourage adherence through monitoring tolerability of these OANs improved the overall medication taking experience of these patients.
Upon initial evaluation of adherence pre- and postimplementation, median adherence rates in both groups appeared to be above goal at 0.94 and 1.06 respectively. Patients in the postimplementation group intentionally received a 5- to 7-day supply buffer to account for potential prescription delivery delays due to holidays and inclement weather. This would indicate that the patients in the postimplementation group would have 15% oversupply due to the 5-day supply buffer. After correcting for patients with confounding reasons for excess (dose reductions, breaks in treatment, etc.), the median MPR in the prerefill clinic group decreased to 0.9 and the MPR in the postrefill clinic group increased slightly to 1.08. Although the median adherence rate in both the pre- and postimplementation groups were above goal of 0.90, 36% of the patients in the preimplementation group were considered nonadherent (MPR < 0.9) compared with no patients in the postimplementation group. Therefore, our intervention to improve patient adherence appeared to be beneficial at our institution.
In addition to improving adherence, one of the goals of the renewal clinic was to minimize excess supply at the time of therapy discontinuation. This was accomplished by aligning medication fills with medical visits and objective monitoring, as well as limiting supply to no more than 30 days. Of the patients in the postimplementation group, only 1 patient had remaining medication at the time of therapy discontinuation compared with 14 patients in the preimplementation group. The estimated cost savings from excess supply was $36,335. Limiting the amount of unused supply not only saves money for the patient and the institution, but also decreases opportunity for improper hazardous waste disposal and unnecessary exposure of hazardous materials to others.
Our results show the pharmacist intervention in the coordination of renewals improved adherence, minimized medication waste, and saved money. The cost of pharmacist time participating in the refill clinic was not calculated. Each visit was completed in approximately 5 minutes, with subsequent documentation and coordination taking an additional 5 to 10 minutes. During the launch of this service, the oncology pharmacy resident provided all coverage of the clinic. Oversite of the resident was provided by hematology/oncology clinical pharmacy specialists. We have continued to utilize pharmacy resident coverage since that time to meet education needs and keep the estimated cost per visit low. Another option in the case that pharmacy residents are not available would be utilization of a pharmacy technician, intern, or professional student to conduct the adherence and tolerability phone assessments. Our escalation protocol allows intervention by clinical pharmacy specialist and/or other health care providers when necessary. Trainees have only required basic training on how to use the protocol.
Limitations
Due to this study’s retrospective design, an inherent limitation is dependence on prescriber and refill records for documentation of initiation and discontinuation dates. Therefore, only the association of impact of pharmacist intervention on medication adherence can be determined as opposed to causation. We did not take into account discrepancies in day supply secondary to ‘held’ therapies, dose reductions, or doses supplied during an inpatient admission, which may alter estimates of MPR and cost-savings data. Patients in the postimplementation group intentionally received a 5 to 7-day supply buffer to account for potential prescription delivery delays due to holidays and inclement weather. This would indicate that the patients in the postimplementation group would have 15% oversupply due to the 5-day supply buffer, thereby skewing MPR values. This study did not account for cost avoidance resulting from early identification and management of toxicity. Finally, the postimplementation data only spans 4 months and a longer duration of time is needed to more accurately determine sustainability of renewal clinic interventions and provide comprehensive evaluation of cost-avoidance.
Conclusion
Implementation of an OAN renewal clinic was associated with an increase in MPR, improved proportion of patients considered adherent, and an estimated $36,335 cost-savings. However, prospective evaluation and a longer study duration are needed to determine causality of improved adherence and cost-savings associated with a pharmacist-driven OAN renewal clinic.
1. Ganesan P, Sagar TG, Dubashi B, et al. Nonadherence to imatinib adversely affects event free survival in chronic phase chronic myeloid leukemia. Am J Hematol 2011; 86: 471-474. doi:10.1002/ajh.22019
2. Marin D, Bazeos A, Mahon FX, et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J Clin Oncol 2010; 28: 2381-2388. doi:10.1200/JCO.2009.26.3087
3. McCowan C, Shearer J, Donnan PT, et al. Cohort study examining tamoxifen adherence and its relationship to mortality in women with breast cancer. Br J Cancer 2008; 99: 1763-1768. doi:10.1038/sj.bjc.6604758
4. Lexicomp Online. Sunitinib. Hudson, Ohio: Lexi-Comp, Inc; August 20, 2019.
5. Babiker A, El Husseini M, Al Nemri A, et al. Health care professional development: Working as a team to improve patient care. Sudan J Paediatr. 2014;14(2):9-16.
6. Spence MM, Makarem AF, Reyes SL, et al. Evaluation of an outpatient pharmacy clinical services program on adherence and clinical outcomes among patients with diabetes and/or coronary artery disease. J Manag Care Spec Pharm. 2014;20(10):1036-1045. doi:10.18553/jmcp.2014.20.10.1036
7. Holle LM, Puri S, Clement JM. Physician-pharmacist collaboration for oral chemotherapy monitoring: Insights from an academic genitourinary oncology practice. J Oncol Pharm Pract 2015; doi:10.1177/1078155215581524
8. Muluneh B, Schneider M, Faso A, et al. Improved Adherence Rates and Clinical Outcomes of an Integrated, Closed-Loop, Pharmacist-Led Oral Chemotherapy Management Program. Journal of Oncology Practice. 2018;14(6):371-333. doi:10.1200/JOP.17.00039.
9. Font R, Espinas JA, Gil-Gil M, et al. Prescription refill, patient self-report and physician report in assessing adherence to oral endocrine therapy in early breast cancer patients: a retrospective cohort study in Catalonia, Spain. British Journal of Cancer. 2012 ;107(8):1249-1256. doi:10.1038/bjc.2012.389.
10. Anderson KR, Chambers CR, Lam N, et al. Medication adherence among adults prescribed imatinib, dasatinib, or nilotinib for the treatment of chronic myeloid leukemia. J Oncol Pharm Practice. 2015;21(1):19–25. doi:10.1177/1078155213520261
11. Weingart SN, Brown E, Bach PB, et al. NCCN Task Force Report: oral chemotherapy. J Natl Compr Canc Netw. 2008;6(3): S1-S14.
Evaluation of oral antineoplastic agent (OAN) adherence patterns have identified correlations between nonadherence or over-adherence and poorer disease-related outcomes. Multiple studies have focused on imatinib use in chronic myeloid leukemia (CML) due to its continuous, long-term use. A study by Ganesan and colleagues found that nonadherence to imatinib showed a significant decrease in 5-year event-free survival between 76.7% of adherent participants compared with 59.8% of nonadherent participants.1 This study found that 44% of patients who were adherent to imatinib achieved complete cytogenetic response vs only 26% of patients who were nonadherent. In another study of imatinib for CML, major molecular response (MMR) was strongly correlated with adherence and no patients with adherence < 80% were able to achieve MMR.2 Similarly, in studies of tamoxifen for breast cancer, < 80% adherence resulted in a 10% decrease in survival when compared to those who were more adherent.3,4
In addition to the clinical implications of nonadherence, there can be a significant cost associated with suboptimal use of these medications. The price of a single dose of OAN medication may cost as much as $440.5
The benefits of multidisciplinary care teams have been identified in many studies.6,7 While studies are limited in oncology, pharmacists provide vital contributions to the oncology multidisciplinary team when managing OANs as these health care professionals have expert knowledge of the medications, potential adverse events (AEs), and necessary monitoring parameters.8 In one study, patients seen by the pharmacist-led oral chemotherapy management program experienced improved clinical outcomes and response to therapy when compared with preintervention patients (early molecular response, 88.9% vs 54.8%, P = .01; major molecular response, 83.3% vs 57.6%, P = .06).9 During the study, 318 AEs were reported, leading to 235 pharmacist interventions to ameliorate AEs and improve adherence.
The primary objective of this study was to measure the impact of a pharmacist-driven OAN renewal clinic on medication adherence. The secondary objective was to estimate cost-savings of this new service.
Methods
Prior to July 2014, several limitations were identified related to OAN prescribing and monitoring at the Richard L. Roudebush Veterans Affairs Medical Center in Indianapolis, Indiana (RLRVAMC). The prescription ordering process relied primarily on the patient to initiate refills, rather than the prescriber OAN prescriptions also lacked consistency for number of refills or quantities dispensed. Furthermore, ordering of antineoplastic products was not limited to hematology/oncology providers. Patients were identified with significant supply on hand at the time of medication discontinuation, creating concerns for medication waste, tolerability, and nonadherence.
As a result, opportunities were identified to improve the prescribing process, recommended monitoring, toxicity and tolerability evaluation, medication reconciliation, and medication adherence. In July of 2014, the RLRVAMC adopted a new chemotherapy order entry system capable of restricting prescriptions to hematology/oncology providers and limiting dispensed quantities and refill amounts. A comprehensive pharmacist driven OAN renewal clinic was implemented on September 1, 2014 with the goal of improving long-term adherence and tolerability, in addition to minimizing medication waste.
Patients were eligible for enrollment in the clinic if they had a cancer diagnosis and were concomitantly prescribed an OAN outlined in Table 1. All eligible patients were automatically enrolled in the clinic when they were deemed stable on their OAN by a hematology/oncology pharmacy specialist. Stability was defined as ≤ Grade 1 symptoms associated with the toxicities of OAN therapy managed with or without intervention as defined by the Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. Once enrolled in the renewal clinic, patients were called by an oncology pharmacy resident (PGY2) 1 week prior to any OAN refill due date. Patients were asked a series of 5 adherence and tolerability questions (Table 2) to evaluate renewal criteria for approval or need for further evaluation. These questions were developed based on targeted information and published reports on monitoring adherence.10,11 Criteria for renewal included: < 10% self-reported missed doses of the OAN during the previous dispensing period, no hospitalizations or emergency department visits since most recent hematology/oncology provider appointment, no changes to concomitant medication therapies, and no new or worsening medication-related AEs. Patients meeting all criteria were given a 30-day supply of OAN. Prescribing, dispensing, and delivery of OAN were facilitated by the pharmacist. Patient cases that did not meet criteria for renewal were escalated to the hematology/oncology provider or oncology clinical pharmacy specialist for further evaluation.
Study Design and Setting
This was a pre/post retrospective cohort, quality improvement study of patients enrolled in the RLRVAMC OAN pharmacist renewal clinic. The study was deemed exempt from institutional review board (IRB) by the US Department of Veterans Affairs (VA) Research and Development Department.
Study Population
Patients were included in the preimplementation group if they had received at least 2 prescriptions of an eligible OAN. Therapy for the preimplementation group was required to be a monthly duration > 21 days and between the dates of September 1, 2013 and August 31, 2014. Patients were included in the postimplementation group if they had received at least 2 prescriptions of the studied OANs between September 1, 2014 and January 31, 2015. Patients were excluded if they had filled < 2 prescriptions of OAN; were managed by a non-VA oncologist or hematologist; or received an OAN other than those listed in Table 1.
Data Collection
For all patients in both the pre- and postimplementation cohorts, a standardized data collection tool was used to collect the following via electronic health record review by a PGY2 oncology resident: age, race, gender, oral antineoplastic agent, refill dates, days’ supply, estimated unit cost per dose cancer diagnosis, distance from the RLRVAMC, copay status, presence of hospitalizations/ED visits/dosage reductions, discontinuation rates, reasons for discontinuation, and total number of current prescriptions. The presence or absence of dosage reductions were collected to identify concerns for tolerability, but only the original dose for the preimplementation group and dosage at time of clinic enrollment for the postimplementation group was included in the analysis.
Outcomes and Statistical Analyses
The primary outcome was medication adherence defined as the median medication possession ratio (MPR) before and after implementation of the clinic. Secondary outcomes included the proportion of patients who were adherent from before implementation to after and estimated cost-savings of this clinic after implementation. MPR was used to estimate medication adherence by taking the cumulative day supply of medication on hand divided by the number of days on therapy.12 Number of days on therapy was determined by taking the difference on the start date of the new medication regimen and the discontinuation date of the same regimen. Patients were grouped by adherence into one of the following categories: < 0.8, 0.8 to 0.89, 0.9 to 1, and > 1.1. Patients were considered adherent if they reported taking ≥ 90% (MPR ≥ 0.9) of prescribed doses, adopted from the study by Anderson and colleagues.12 A patient with an MPR > 1, likely due to filling prior to the anticipated refill date, was considered 100% adherent (MPR = 1). If a patient switched OAN during the study, both agents were included as separate entities.
A conservative estimate of cost-savings was made by multiplying the RLRVAMC cost per unit of medication at time of initial prescription fill by the number of units taken each day multiplied by the total days’ supply on hand at time of therapy discontinuation. Patients with an MPR < 1 at time of therapy discontinuation were assumed to have zero remaining units on hand and zero cost savings was estimated. Waste, for purposes of cost-savings, was calculated for all MPR values > 1. Additional supply anticipated to be on hand from dose reductions was not included in the estimated cost of unused medication.
Descriptive statistics compared demographic characteristics between the pre- and postimplementation groups. MPR data were not normally distributed, which required the use of nonparametric Mann-Whitney U tests to compare pre- and postMPRs. Pearson χ2 compared the proportion of adherent patients between groups while descriptive statistics were used to estimate cost savings. Significance was determined based on a P value < .05. IBM SPSS Statistics software was used for all statistical analyses. As this was a complete sample of all eligible subjects, no sample size calculation was performed.
Results
In the preimplementation period, 246 patients received an OAN and 61 patients received an OAN in the postimplementation period (Figure 1). Of the 246 patients in the preimplementation period, 98 were eligible and included in the preimplementation group. Similarly, of the 61 patients in the postimplementation period, 35 patients met inclusion criteria for the postimplementation group. The study population was predominantly male with an average age of approximately 70 years in both groups (Table 3). More than 70% of the population in each group was White. No statistically significant differences between groups were identified. The most commonly prescribed OAN in the preimplementation group were abiraterone, imatinib, and enzalutamide (Table 3). In the postimplementation group, the most commonly prescribed agents were abiraterone, imatinib, pazopanib, and dasatinib. No significant differences were observed in prescribing of individual agents between the pre- and postimplementation groups or other characteristics that may affect adherence including patient copay status, number of concomitant medications, and driving distance from the RLRVAMC.
Thirty-six (36.7%) patients in the preimplementation group were considered nonadherent (MPR < 0.9) and 18 (18.4%) had an MPR < 0.8. Fifteen (15.3%) patients in the preimplementation clinic were considered overadherent (MPR > 1.1). Forty-seven (47.9%) patients in the preimplementation group were considered adherent (MPR 0.9 - 1.1) while all 35 (100%) patients in the postimplementation group were considered adherent (MPR 0.9 - 1.1). No non- or overadherent patients were identified in the postimplementation group (Figure 2). The median MPR for all patients in the preimplementation group was 0.94 compared with 1.06 (P < .001) in the postimplementation group.
Thirty-five (35.7%) patients had therapy discontinued or held in the preimplementation group compared with 2 (5.7%) patients in the postimplementation group (P < .001). Reasons for discontinuation in the preimplementation group included disease progression (n = 27), death (n = 3), lost to follow up (n = 2), and intolerability of therapy (n = 3). Both patients that discontinued therapy in the postimplementation group did so due to disease progression. Of the 35 patients who had their OAN discontinued or held in the preimplementation group, 14 patients had excess supply on hand at time of discontinuation. The estimated value of the unused medication was $37,890. Nine (25%) of the 35 patients who discontinued therapy had a dosage reduction during the course of therapy and the additional supply was not included in the cost estimate. Similarly, 1 of the 2 patients in the postimplementation group had their OAN discontinued during study. The cost of oversupply of medication at the time of therapy discontinuation was estimated at $1,555. No patients in the postimplementation group had dose reductions. After implementation of the OAN renewal clinic, the total cost savings between pre ($37,890) and postimplementation ($1,555) groups was $36,355.
Discussion
OANs are widely used therapies, with more than 25 million doses administered per year in the United States alone.12 The use of these agents will continue to grow as more targeted agents become available and patients request more convenient treatment options. The role for hematology/oncology clinical pharmacy services must adapt to this increased usage of OANs, including increasing pharmacist involvement in medication education, adherence and tolerability assessments, and proactive drug interaction monitoring.However, additional research is needed to determine optimal management strategies.
Our study aimed to compare OAN adherence among patients at a tertiary care VA hospital before and after implementation of a renewal clinic. The preimplementation population had a median MPR of 0.94 compared with 1.06 in the postimplementation group (P < .001). Although an ideal MPR is 1.0, we aimed for a slightly higher MPR to allow a supply buffer in the event of prescription delivery delays, as more than 90% of prescriptions are mailed to patients from a regional mail-order pharmacy. Importantly, the median MPRs do not adequately convey the impact from this clinic. The proportion of patients who were considered adherent to OANs increased from 47.9% in the preimplementation to 100% in the postimplementation period. These finding suggest that the clinical pharmacist role to assess and encourage adherence through monitoring tolerability of these OANs improved the overall medication taking experience of these patients.
Upon initial evaluation of adherence pre- and postimplementation, median adherence rates in both groups appeared to be above goal at 0.94 and 1.06 respectively. Patients in the postimplementation group intentionally received a 5- to 7-day supply buffer to account for potential prescription delivery delays due to holidays and inclement weather. This would indicate that the patients in the postimplementation group would have 15% oversupply due to the 5-day supply buffer. After correcting for patients with confounding reasons for excess (dose reductions, breaks in treatment, etc.), the median MPR in the prerefill clinic group decreased to 0.9 and the MPR in the postrefill clinic group increased slightly to 1.08. Although the median adherence rate in both the pre- and postimplementation groups were above goal of 0.90, 36% of the patients in the preimplementation group were considered nonadherent (MPR < 0.9) compared with no patients in the postimplementation group. Therefore, our intervention to improve patient adherence appeared to be beneficial at our institution.
In addition to improving adherence, one of the goals of the renewal clinic was to minimize excess supply at the time of therapy discontinuation. This was accomplished by aligning medication fills with medical visits and objective monitoring, as well as limiting supply to no more than 30 days. Of the patients in the postimplementation group, only 1 patient had remaining medication at the time of therapy discontinuation compared with 14 patients in the preimplementation group. The estimated cost savings from excess supply was $36,335. Limiting the amount of unused supply not only saves money for the patient and the institution, but also decreases opportunity for improper hazardous waste disposal and unnecessary exposure of hazardous materials to others.
Our results show the pharmacist intervention in the coordination of renewals improved adherence, minimized medication waste, and saved money. The cost of pharmacist time participating in the refill clinic was not calculated. Each visit was completed in approximately 5 minutes, with subsequent documentation and coordination taking an additional 5 to 10 minutes. During the launch of this service, the oncology pharmacy resident provided all coverage of the clinic. Oversite of the resident was provided by hematology/oncology clinical pharmacy specialists. We have continued to utilize pharmacy resident coverage since that time to meet education needs and keep the estimated cost per visit low. Another option in the case that pharmacy residents are not available would be utilization of a pharmacy technician, intern, or professional student to conduct the adherence and tolerability phone assessments. Our escalation protocol allows intervention by clinical pharmacy specialist and/or other health care providers when necessary. Trainees have only required basic training on how to use the protocol.
Limitations
Due to this study’s retrospective design, an inherent limitation is dependence on prescriber and refill records for documentation of initiation and discontinuation dates. Therefore, only the association of impact of pharmacist intervention on medication adherence can be determined as opposed to causation. We did not take into account discrepancies in day supply secondary to ‘held’ therapies, dose reductions, or doses supplied during an inpatient admission, which may alter estimates of MPR and cost-savings data. Patients in the postimplementation group intentionally received a 5 to 7-day supply buffer to account for potential prescription delivery delays due to holidays and inclement weather. This would indicate that the patients in the postimplementation group would have 15% oversupply due to the 5-day supply buffer, thereby skewing MPR values. This study did not account for cost avoidance resulting from early identification and management of toxicity. Finally, the postimplementation data only spans 4 months and a longer duration of time is needed to more accurately determine sustainability of renewal clinic interventions and provide comprehensive evaluation of cost-avoidance.
Conclusion
Implementation of an OAN renewal clinic was associated with an increase in MPR, improved proportion of patients considered adherent, and an estimated $36,335 cost-savings. However, prospective evaluation and a longer study duration are needed to determine causality of improved adherence and cost-savings associated with a pharmacist-driven OAN renewal clinic.
Evaluation of oral antineoplastic agent (OAN) adherence patterns have identified correlations between nonadherence or over-adherence and poorer disease-related outcomes. Multiple studies have focused on imatinib use in chronic myeloid leukemia (CML) due to its continuous, long-term use. A study by Ganesan and colleagues found that nonadherence to imatinib showed a significant decrease in 5-year event-free survival between 76.7% of adherent participants compared with 59.8% of nonadherent participants.1 This study found that 44% of patients who were adherent to imatinib achieved complete cytogenetic response vs only 26% of patients who were nonadherent. In another study of imatinib for CML, major molecular response (MMR) was strongly correlated with adherence and no patients with adherence < 80% were able to achieve MMR.2 Similarly, in studies of tamoxifen for breast cancer, < 80% adherence resulted in a 10% decrease in survival when compared to those who were more adherent.3,4
In addition to the clinical implications of nonadherence, there can be a significant cost associated with suboptimal use of these medications. The price of a single dose of OAN medication may cost as much as $440.5
The benefits of multidisciplinary care teams have been identified in many studies.6,7 While studies are limited in oncology, pharmacists provide vital contributions to the oncology multidisciplinary team when managing OANs as these health care professionals have expert knowledge of the medications, potential adverse events (AEs), and necessary monitoring parameters.8 In one study, patients seen by the pharmacist-led oral chemotherapy management program experienced improved clinical outcomes and response to therapy when compared with preintervention patients (early molecular response, 88.9% vs 54.8%, P = .01; major molecular response, 83.3% vs 57.6%, P = .06).9 During the study, 318 AEs were reported, leading to 235 pharmacist interventions to ameliorate AEs and improve adherence.
The primary objective of this study was to measure the impact of a pharmacist-driven OAN renewal clinic on medication adherence. The secondary objective was to estimate cost-savings of this new service.
Methods
Prior to July 2014, several limitations were identified related to OAN prescribing and monitoring at the Richard L. Roudebush Veterans Affairs Medical Center in Indianapolis, Indiana (RLRVAMC). The prescription ordering process relied primarily on the patient to initiate refills, rather than the prescriber OAN prescriptions also lacked consistency for number of refills or quantities dispensed. Furthermore, ordering of antineoplastic products was not limited to hematology/oncology providers. Patients were identified with significant supply on hand at the time of medication discontinuation, creating concerns for medication waste, tolerability, and nonadherence.
As a result, opportunities were identified to improve the prescribing process, recommended monitoring, toxicity and tolerability evaluation, medication reconciliation, and medication adherence. In July of 2014, the RLRVAMC adopted a new chemotherapy order entry system capable of restricting prescriptions to hematology/oncology providers and limiting dispensed quantities and refill amounts. A comprehensive pharmacist driven OAN renewal clinic was implemented on September 1, 2014 with the goal of improving long-term adherence and tolerability, in addition to minimizing medication waste.
Patients were eligible for enrollment in the clinic if they had a cancer diagnosis and were concomitantly prescribed an OAN outlined in Table 1. All eligible patients were automatically enrolled in the clinic when they were deemed stable on their OAN by a hematology/oncology pharmacy specialist. Stability was defined as ≤ Grade 1 symptoms associated with the toxicities of OAN therapy managed with or without intervention as defined by the Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. Once enrolled in the renewal clinic, patients were called by an oncology pharmacy resident (PGY2) 1 week prior to any OAN refill due date. Patients were asked a series of 5 adherence and tolerability questions (Table 2) to evaluate renewal criteria for approval or need for further evaluation. These questions were developed based on targeted information and published reports on monitoring adherence.10,11 Criteria for renewal included: < 10% self-reported missed doses of the OAN during the previous dispensing period, no hospitalizations or emergency department visits since most recent hematology/oncology provider appointment, no changes to concomitant medication therapies, and no new or worsening medication-related AEs. Patients meeting all criteria were given a 30-day supply of OAN. Prescribing, dispensing, and delivery of OAN were facilitated by the pharmacist. Patient cases that did not meet criteria for renewal were escalated to the hematology/oncology provider or oncology clinical pharmacy specialist for further evaluation.
Study Design and Setting
This was a pre/post retrospective cohort, quality improvement study of patients enrolled in the RLRVAMC OAN pharmacist renewal clinic. The study was deemed exempt from institutional review board (IRB) by the US Department of Veterans Affairs (VA) Research and Development Department.
Study Population
Patients were included in the preimplementation group if they had received at least 2 prescriptions of an eligible OAN. Therapy for the preimplementation group was required to be a monthly duration > 21 days and between the dates of September 1, 2013 and August 31, 2014. Patients were included in the postimplementation group if they had received at least 2 prescriptions of the studied OANs between September 1, 2014 and January 31, 2015. Patients were excluded if they had filled < 2 prescriptions of OAN; were managed by a non-VA oncologist or hematologist; or received an OAN other than those listed in Table 1.
Data Collection
For all patients in both the pre- and postimplementation cohorts, a standardized data collection tool was used to collect the following via electronic health record review by a PGY2 oncology resident: age, race, gender, oral antineoplastic agent, refill dates, days’ supply, estimated unit cost per dose cancer diagnosis, distance from the RLRVAMC, copay status, presence of hospitalizations/ED visits/dosage reductions, discontinuation rates, reasons for discontinuation, and total number of current prescriptions. The presence or absence of dosage reductions were collected to identify concerns for tolerability, but only the original dose for the preimplementation group and dosage at time of clinic enrollment for the postimplementation group was included in the analysis.
Outcomes and Statistical Analyses
The primary outcome was medication adherence defined as the median medication possession ratio (MPR) before and after implementation of the clinic. Secondary outcomes included the proportion of patients who were adherent from before implementation to after and estimated cost-savings of this clinic after implementation. MPR was used to estimate medication adherence by taking the cumulative day supply of medication on hand divided by the number of days on therapy.12 Number of days on therapy was determined by taking the difference on the start date of the new medication regimen and the discontinuation date of the same regimen. Patients were grouped by adherence into one of the following categories: < 0.8, 0.8 to 0.89, 0.9 to 1, and > 1.1. Patients were considered adherent if they reported taking ≥ 90% (MPR ≥ 0.9) of prescribed doses, adopted from the study by Anderson and colleagues.12 A patient with an MPR > 1, likely due to filling prior to the anticipated refill date, was considered 100% adherent (MPR = 1). If a patient switched OAN during the study, both agents were included as separate entities.
A conservative estimate of cost-savings was made by multiplying the RLRVAMC cost per unit of medication at time of initial prescription fill by the number of units taken each day multiplied by the total days’ supply on hand at time of therapy discontinuation. Patients with an MPR < 1 at time of therapy discontinuation were assumed to have zero remaining units on hand and zero cost savings was estimated. Waste, for purposes of cost-savings, was calculated for all MPR values > 1. Additional supply anticipated to be on hand from dose reductions was not included in the estimated cost of unused medication.
Descriptive statistics compared demographic characteristics between the pre- and postimplementation groups. MPR data were not normally distributed, which required the use of nonparametric Mann-Whitney U tests to compare pre- and postMPRs. Pearson χ2 compared the proportion of adherent patients between groups while descriptive statistics were used to estimate cost savings. Significance was determined based on a P value < .05. IBM SPSS Statistics software was used for all statistical analyses. As this was a complete sample of all eligible subjects, no sample size calculation was performed.
Results
In the preimplementation period, 246 patients received an OAN and 61 patients received an OAN in the postimplementation period (Figure 1). Of the 246 patients in the preimplementation period, 98 were eligible and included in the preimplementation group. Similarly, of the 61 patients in the postimplementation period, 35 patients met inclusion criteria for the postimplementation group. The study population was predominantly male with an average age of approximately 70 years in both groups (Table 3). More than 70% of the population in each group was White. No statistically significant differences between groups were identified. The most commonly prescribed OAN in the preimplementation group were abiraterone, imatinib, and enzalutamide (Table 3). In the postimplementation group, the most commonly prescribed agents were abiraterone, imatinib, pazopanib, and dasatinib. No significant differences were observed in prescribing of individual agents between the pre- and postimplementation groups or other characteristics that may affect adherence including patient copay status, number of concomitant medications, and driving distance from the RLRVAMC.
Thirty-six (36.7%) patients in the preimplementation group were considered nonadherent (MPR < 0.9) and 18 (18.4%) had an MPR < 0.8. Fifteen (15.3%) patients in the preimplementation clinic were considered overadherent (MPR > 1.1). Forty-seven (47.9%) patients in the preimplementation group were considered adherent (MPR 0.9 - 1.1) while all 35 (100%) patients in the postimplementation group were considered adherent (MPR 0.9 - 1.1). No non- or overadherent patients were identified in the postimplementation group (Figure 2). The median MPR for all patients in the preimplementation group was 0.94 compared with 1.06 (P < .001) in the postimplementation group.
Thirty-five (35.7%) patients had therapy discontinued or held in the preimplementation group compared with 2 (5.7%) patients in the postimplementation group (P < .001). Reasons for discontinuation in the preimplementation group included disease progression (n = 27), death (n = 3), lost to follow up (n = 2), and intolerability of therapy (n = 3). Both patients that discontinued therapy in the postimplementation group did so due to disease progression. Of the 35 patients who had their OAN discontinued or held in the preimplementation group, 14 patients had excess supply on hand at time of discontinuation. The estimated value of the unused medication was $37,890. Nine (25%) of the 35 patients who discontinued therapy had a dosage reduction during the course of therapy and the additional supply was not included in the cost estimate. Similarly, 1 of the 2 patients in the postimplementation group had their OAN discontinued during study. The cost of oversupply of medication at the time of therapy discontinuation was estimated at $1,555. No patients in the postimplementation group had dose reductions. After implementation of the OAN renewal clinic, the total cost savings between pre ($37,890) and postimplementation ($1,555) groups was $36,355.
Discussion
OANs are widely used therapies, with more than 25 million doses administered per year in the United States alone.12 The use of these agents will continue to grow as more targeted agents become available and patients request more convenient treatment options. The role for hematology/oncology clinical pharmacy services must adapt to this increased usage of OANs, including increasing pharmacist involvement in medication education, adherence and tolerability assessments, and proactive drug interaction monitoring.However, additional research is needed to determine optimal management strategies.
Our study aimed to compare OAN adherence among patients at a tertiary care VA hospital before and after implementation of a renewal clinic. The preimplementation population had a median MPR of 0.94 compared with 1.06 in the postimplementation group (P < .001). Although an ideal MPR is 1.0, we aimed for a slightly higher MPR to allow a supply buffer in the event of prescription delivery delays, as more than 90% of prescriptions are mailed to patients from a regional mail-order pharmacy. Importantly, the median MPRs do not adequately convey the impact from this clinic. The proportion of patients who were considered adherent to OANs increased from 47.9% in the preimplementation to 100% in the postimplementation period. These finding suggest that the clinical pharmacist role to assess and encourage adherence through monitoring tolerability of these OANs improved the overall medication taking experience of these patients.
Upon initial evaluation of adherence pre- and postimplementation, median adherence rates in both groups appeared to be above goal at 0.94 and 1.06 respectively. Patients in the postimplementation group intentionally received a 5- to 7-day supply buffer to account for potential prescription delivery delays due to holidays and inclement weather. This would indicate that the patients in the postimplementation group would have 15% oversupply due to the 5-day supply buffer. After correcting for patients with confounding reasons for excess (dose reductions, breaks in treatment, etc.), the median MPR in the prerefill clinic group decreased to 0.9 and the MPR in the postrefill clinic group increased slightly to 1.08. Although the median adherence rate in both the pre- and postimplementation groups were above goal of 0.90, 36% of the patients in the preimplementation group were considered nonadherent (MPR < 0.9) compared with no patients in the postimplementation group. Therefore, our intervention to improve patient adherence appeared to be beneficial at our institution.
In addition to improving adherence, one of the goals of the renewal clinic was to minimize excess supply at the time of therapy discontinuation. This was accomplished by aligning medication fills with medical visits and objective monitoring, as well as limiting supply to no more than 30 days. Of the patients in the postimplementation group, only 1 patient had remaining medication at the time of therapy discontinuation compared with 14 patients in the preimplementation group. The estimated cost savings from excess supply was $36,335. Limiting the amount of unused supply not only saves money for the patient and the institution, but also decreases opportunity for improper hazardous waste disposal and unnecessary exposure of hazardous materials to others.
Our results show the pharmacist intervention in the coordination of renewals improved adherence, minimized medication waste, and saved money. The cost of pharmacist time participating in the refill clinic was not calculated. Each visit was completed in approximately 5 minutes, with subsequent documentation and coordination taking an additional 5 to 10 minutes. During the launch of this service, the oncology pharmacy resident provided all coverage of the clinic. Oversite of the resident was provided by hematology/oncology clinical pharmacy specialists. We have continued to utilize pharmacy resident coverage since that time to meet education needs and keep the estimated cost per visit low. Another option in the case that pharmacy residents are not available would be utilization of a pharmacy technician, intern, or professional student to conduct the adherence and tolerability phone assessments. Our escalation protocol allows intervention by clinical pharmacy specialist and/or other health care providers when necessary. Trainees have only required basic training on how to use the protocol.
Limitations
Due to this study’s retrospective design, an inherent limitation is dependence on prescriber and refill records for documentation of initiation and discontinuation dates. Therefore, only the association of impact of pharmacist intervention on medication adherence can be determined as opposed to causation. We did not take into account discrepancies in day supply secondary to ‘held’ therapies, dose reductions, or doses supplied during an inpatient admission, which may alter estimates of MPR and cost-savings data. Patients in the postimplementation group intentionally received a 5 to 7-day supply buffer to account for potential prescription delivery delays due to holidays and inclement weather. This would indicate that the patients in the postimplementation group would have 15% oversupply due to the 5-day supply buffer, thereby skewing MPR values. This study did not account for cost avoidance resulting from early identification and management of toxicity. Finally, the postimplementation data only spans 4 months and a longer duration of time is needed to more accurately determine sustainability of renewal clinic interventions and provide comprehensive evaluation of cost-avoidance.
Conclusion
Implementation of an OAN renewal clinic was associated with an increase in MPR, improved proportion of patients considered adherent, and an estimated $36,335 cost-savings. However, prospective evaluation and a longer study duration are needed to determine causality of improved adherence and cost-savings associated with a pharmacist-driven OAN renewal clinic.
1. Ganesan P, Sagar TG, Dubashi B, et al. Nonadherence to imatinib adversely affects event free survival in chronic phase chronic myeloid leukemia. Am J Hematol 2011; 86: 471-474. doi:10.1002/ajh.22019
2. Marin D, Bazeos A, Mahon FX, et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J Clin Oncol 2010; 28: 2381-2388. doi:10.1200/JCO.2009.26.3087
3. McCowan C, Shearer J, Donnan PT, et al. Cohort study examining tamoxifen adherence and its relationship to mortality in women with breast cancer. Br J Cancer 2008; 99: 1763-1768. doi:10.1038/sj.bjc.6604758
4. Lexicomp Online. Sunitinib. Hudson, Ohio: Lexi-Comp, Inc; August 20, 2019.
5. Babiker A, El Husseini M, Al Nemri A, et al. Health care professional development: Working as a team to improve patient care. Sudan J Paediatr. 2014;14(2):9-16.
6. Spence MM, Makarem AF, Reyes SL, et al. Evaluation of an outpatient pharmacy clinical services program on adherence and clinical outcomes among patients with diabetes and/or coronary artery disease. J Manag Care Spec Pharm. 2014;20(10):1036-1045. doi:10.18553/jmcp.2014.20.10.1036
7. Holle LM, Puri S, Clement JM. Physician-pharmacist collaboration for oral chemotherapy monitoring: Insights from an academic genitourinary oncology practice. J Oncol Pharm Pract 2015; doi:10.1177/1078155215581524
8. Muluneh B, Schneider M, Faso A, et al. Improved Adherence Rates and Clinical Outcomes of an Integrated, Closed-Loop, Pharmacist-Led Oral Chemotherapy Management Program. Journal of Oncology Practice. 2018;14(6):371-333. doi:10.1200/JOP.17.00039.
9. Font R, Espinas JA, Gil-Gil M, et al. Prescription refill, patient self-report and physician report in assessing adherence to oral endocrine therapy in early breast cancer patients: a retrospective cohort study in Catalonia, Spain. British Journal of Cancer. 2012 ;107(8):1249-1256. doi:10.1038/bjc.2012.389.
10. Anderson KR, Chambers CR, Lam N, et al. Medication adherence among adults prescribed imatinib, dasatinib, or nilotinib for the treatment of chronic myeloid leukemia. J Oncol Pharm Practice. 2015;21(1):19–25. doi:10.1177/1078155213520261
11. Weingart SN, Brown E, Bach PB, et al. NCCN Task Force Report: oral chemotherapy. J Natl Compr Canc Netw. 2008;6(3): S1-S14.
1. Ganesan P, Sagar TG, Dubashi B, et al. Nonadherence to imatinib adversely affects event free survival in chronic phase chronic myeloid leukemia. Am J Hematol 2011; 86: 471-474. doi:10.1002/ajh.22019
2. Marin D, Bazeos A, Mahon FX, et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J Clin Oncol 2010; 28: 2381-2388. doi:10.1200/JCO.2009.26.3087
3. McCowan C, Shearer J, Donnan PT, et al. Cohort study examining tamoxifen adherence and its relationship to mortality in women with breast cancer. Br J Cancer 2008; 99: 1763-1768. doi:10.1038/sj.bjc.6604758
4. Lexicomp Online. Sunitinib. Hudson, Ohio: Lexi-Comp, Inc; August 20, 2019.
5. Babiker A, El Husseini M, Al Nemri A, et al. Health care professional development: Working as a team to improve patient care. Sudan J Paediatr. 2014;14(2):9-16.
6. Spence MM, Makarem AF, Reyes SL, et al. Evaluation of an outpatient pharmacy clinical services program on adherence and clinical outcomes among patients with diabetes and/or coronary artery disease. J Manag Care Spec Pharm. 2014;20(10):1036-1045. doi:10.18553/jmcp.2014.20.10.1036
7. Holle LM, Puri S, Clement JM. Physician-pharmacist collaboration for oral chemotherapy monitoring: Insights from an academic genitourinary oncology practice. J Oncol Pharm Pract 2015; doi:10.1177/1078155215581524
8. Muluneh B, Schneider M, Faso A, et al. Improved Adherence Rates and Clinical Outcomes of an Integrated, Closed-Loop, Pharmacist-Led Oral Chemotherapy Management Program. Journal of Oncology Practice. 2018;14(6):371-333. doi:10.1200/JOP.17.00039.
9. Font R, Espinas JA, Gil-Gil M, et al. Prescription refill, patient self-report and physician report in assessing adherence to oral endocrine therapy in early breast cancer patients: a retrospective cohort study in Catalonia, Spain. British Journal of Cancer. 2012 ;107(8):1249-1256. doi:10.1038/bjc.2012.389.
10. Anderson KR, Chambers CR, Lam N, et al. Medication adherence among adults prescribed imatinib, dasatinib, or nilotinib for the treatment of chronic myeloid leukemia. J Oncol Pharm Practice. 2015;21(1):19–25. doi:10.1177/1078155213520261
11. Weingart SN, Brown E, Bach PB, et al. NCCN Task Force Report: oral chemotherapy. J Natl Compr Canc Netw. 2008;6(3): S1-S14.