User login
AVAHO
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Esophageal cancer: Preoperative chemoradiotherapy benefit in CROSS persists over 10 years
Among patients with locally advanced resectable esophageal or junctional cancer, the overall survival benefit conferred by preoperative chemoradiotherapy persists for at least 10 years, according to long-term results of the Chemoradiotherapy for Esophageal Cancer Followed by Surgery Study (CROSS). As a result of earlier publication of CROSS data, chemoradiotherapy followed by surgery has become one of the standards of care for patients with locally advanced resectable esophageal cancer, stated lead author Ben M. Eyck, MD, of Erasmus University Medical Center, Rotterdam, the Netherlands, and colleagues in the Journal of Clinical Oncology.
In the multicenter, randomized trial, initiated in 2004, 178 patients randomized to chemoradiotherapy with subsequent surgery and 188 patients randomized to surgery alone were followed with overall survival as the primary, and cause-specific survival and risks of locoregional and distant relapse as the secondary endpoints. Chemoradiotherapy consisted of 5 weekly cycles of carboplatin (area under the curve of 2 mg/mL/min) and paclitaxel (50 mg/m2 body surface area on days 1, 8, 15, 22, and 29) with concurrent radiotherapy (41.4 Gy in 23 fractions, 5 days per week. Mean age was 60 years (around 78% male), with squamous cell carcinoma (23%) and adenocarcinoma (75%) as the predominant histologies.
The first analysis showed low short-term toxicity and 2-year survival increased from 50% for patients receiving surgery alone to 67% for neoadjuvant chemoradiotherapy plus surgery. Five-year follow-up data were consistent with initial reporting. Long-term benefits and harms of this regimen remain unclear, according to the researchers. Neoadjuvant chemoradiotherapy’s side effects could lead to long-term death from other causes than esophageal cancer, and may not be preventing but rather merely postponing cancer-related death. The aim of the current analysis was to determine whether the observed benefits persisted beyond 5 years.
As of Dec. 31, 2018, 117/178 patients in the chemoradiotherapy-surgery arm and 144/188 in the surgery arm had died. Median follow-up for surviving patients was 147 months. Patients in the chemoradiotherapy surgery arm had better overall survival than patients in the surgery arm (hazard ratio, 0.70; 95% confidence interval, 0.55-0.89; P = .004), with a 10-year overall survival of 38% (95% CI, 31-45) and 25% (95% CI, 19-32), respectively. No significant subgroup differences were observed for overall survival. Also, there was no evidence of a time-dependent effect of neoadjuvant chemoradiotherapy on overall survival. The major effect of neoadjuvant chemoradiotherapy, landmark analyses showed, was in the first 5 years of follow-up, with the effect on overall survival stabilized thereafter, with a hazard ratio approaching 1.00.
Cause-specific mortality
Eighty-four of 178 patients in the chemoradiotherapy-surgery arm died of esophageal cancer, with 32 dying of other causes. In the surgery arm, 121/188 died of esophageal cancer and 22 of other causes. The hazard ratio for esophageal cancer death in the chemoradiotherapy-surgery arm was 0.60 (95% CI, 0.46 to 0.80), with 10-year absolute risks of 47% (95% CI, 40-54) and 64% (95% CI,57-71), respectively, in the two arms. Death from other causes was comparable, with 10-year absolute risks of 15% (95% CI, 10-21) and 11% (95% CI, 7-16), respectively, for chemoradiotherapy-surgery versus surgery alone.
Locoregional relapse
Locoregional relapse rates were 8% (15/178) and 18% (33/188) in the chemoradiotherapy-surgery and surgery arms, respectively (HR, 0.39; 95% CI, 0.21-0.72). Eighty-seven percent of those developed within 3 years of follow-up in the chemoradiotherapy arm, with the median relapse-free interval at 3.9 months. In the surgery arm, 28 of 33 relapses (85%) developed within 3 years and the median relapse-free interval was 7.1 months. Beyond 6 years, there were no further relapses in either arm.
While synchronous distant plus locoregional relapse developed in 23 of 178 patients (13%) in the chemoradiotherapy-surgery arm and in 42 of 188 patients (22%) in the surgery arm (HR, 0.43; 95% CI, 0.26-0.72), isolated distant relapse developed at similar rates (around 27.5%) in both groups. Risk of distant relapse (with or without locoregional relapse) was lower in the chemoradiotherapy-surgery arm (HR, 0.61; 95%CI, 0.45-0.84). The median relapse-free interval was 15.1 months (interquartile range, 9.3-27.6) in the chemoradiotherapy-surgery arm and 9.0 months (IQR, 5.3-19.7) in the surgery arm.
Safety and health-related quality of life
The combination of paclitaxel and carboplatin with concurrent 41.4 Gy radiotherapy before surgery seems safe in the long term and does not significantly increase the risk of toxicity-related death, the researchers stated. Within the CROSS trial, short-term and long-term health-related quality of life after neoadjuvant chemoradiotherapy plus surgery for surviving patients was comparable to that after surgery alone.
Long-term persistent overall survival benefit
Ten-year CROSS results show that “for locally advanced resectable cancer of the esophagus or esophagogastric junction, preoperative chemoradiotherapy induces a long-term persistent improvement in overall survival.” Also, neoadjuvant chemoradiotherapy does not lead to an increased risk of death from other causes, and the survival benefit of long-term survivors is not compromised, compared with surgery alone. Furthermore, neoadjuvant chemoradiotherapy plus surgery according to CROSS can still be regarded as a standard of care, the researchers added.
Dr. Eyck and colleagues are currently performing the phase II TNT-OES-1 trial. It combines FLOT (fluorouracil, leucovorin, oxaliplatin and docetaxel) chemotherapy followed by CROSS chemoradiotherapy in patients with advanced esophageal and junctional adenocarcinoma. If this regimen appears to be safe in advanced cancer, they plan to perform a phase III trial with this regimen in locally advanced cancer. In addition, they are currently evaluating the implementation of adjuvant nivolumab in clinical practice for patients with pathologically residual disease after CROSS + surgery, based on the recently published CheckMate 577 trial .
“If possible, we prefer adding better systemic therapy to chemoradiotherapy rather than replacing chemoradiotherapy with systemic therapy alone,” Dr. Eyck said in an interview. “The reason for this is that we would like to allow patients with a complete response to neoadjuvant therapy to undergo active surveillance instead of surgery in the near future. … Since the pathologically complete response rate after regimens containing radiotherapy is substantially higher, we still prefer the addition of radiotherapy.”
The study was funded by the Dutch Cancer Foundation (KWF Kankerbestrijding). Dr. Eyck reported no disclosures. Several of the coauthors reported consulting and advisory roles with a variety of pharmaceutical companies.
Among patients with locally advanced resectable esophageal or junctional cancer, the overall survival benefit conferred by preoperative chemoradiotherapy persists for at least 10 years, according to long-term results of the Chemoradiotherapy for Esophageal Cancer Followed by Surgery Study (CROSS). As a result of earlier publication of CROSS data, chemoradiotherapy followed by surgery has become one of the standards of care for patients with locally advanced resectable esophageal cancer, stated lead author Ben M. Eyck, MD, of Erasmus University Medical Center, Rotterdam, the Netherlands, and colleagues in the Journal of Clinical Oncology.
In the multicenter, randomized trial, initiated in 2004, 178 patients randomized to chemoradiotherapy with subsequent surgery and 188 patients randomized to surgery alone were followed with overall survival as the primary, and cause-specific survival and risks of locoregional and distant relapse as the secondary endpoints. Chemoradiotherapy consisted of 5 weekly cycles of carboplatin (area under the curve of 2 mg/mL/min) and paclitaxel (50 mg/m2 body surface area on days 1, 8, 15, 22, and 29) with concurrent radiotherapy (41.4 Gy in 23 fractions, 5 days per week. Mean age was 60 years (around 78% male), with squamous cell carcinoma (23%) and adenocarcinoma (75%) as the predominant histologies.
The first analysis showed low short-term toxicity and 2-year survival increased from 50% for patients receiving surgery alone to 67% for neoadjuvant chemoradiotherapy plus surgery. Five-year follow-up data were consistent with initial reporting. Long-term benefits and harms of this regimen remain unclear, according to the researchers. Neoadjuvant chemoradiotherapy’s side effects could lead to long-term death from other causes than esophageal cancer, and may not be preventing but rather merely postponing cancer-related death. The aim of the current analysis was to determine whether the observed benefits persisted beyond 5 years.
As of Dec. 31, 2018, 117/178 patients in the chemoradiotherapy-surgery arm and 144/188 in the surgery arm had died. Median follow-up for surviving patients was 147 months. Patients in the chemoradiotherapy surgery arm had better overall survival than patients in the surgery arm (hazard ratio, 0.70; 95% confidence interval, 0.55-0.89; P = .004), with a 10-year overall survival of 38% (95% CI, 31-45) and 25% (95% CI, 19-32), respectively. No significant subgroup differences were observed for overall survival. Also, there was no evidence of a time-dependent effect of neoadjuvant chemoradiotherapy on overall survival. The major effect of neoadjuvant chemoradiotherapy, landmark analyses showed, was in the first 5 years of follow-up, with the effect on overall survival stabilized thereafter, with a hazard ratio approaching 1.00.
Cause-specific mortality
Eighty-four of 178 patients in the chemoradiotherapy-surgery arm died of esophageal cancer, with 32 dying of other causes. In the surgery arm, 121/188 died of esophageal cancer and 22 of other causes. The hazard ratio for esophageal cancer death in the chemoradiotherapy-surgery arm was 0.60 (95% CI, 0.46 to 0.80), with 10-year absolute risks of 47% (95% CI, 40-54) and 64% (95% CI,57-71), respectively, in the two arms. Death from other causes was comparable, with 10-year absolute risks of 15% (95% CI, 10-21) and 11% (95% CI, 7-16), respectively, for chemoradiotherapy-surgery versus surgery alone.
Locoregional relapse
Locoregional relapse rates were 8% (15/178) and 18% (33/188) in the chemoradiotherapy-surgery and surgery arms, respectively (HR, 0.39; 95% CI, 0.21-0.72). Eighty-seven percent of those developed within 3 years of follow-up in the chemoradiotherapy arm, with the median relapse-free interval at 3.9 months. In the surgery arm, 28 of 33 relapses (85%) developed within 3 years and the median relapse-free interval was 7.1 months. Beyond 6 years, there were no further relapses in either arm.
While synchronous distant plus locoregional relapse developed in 23 of 178 patients (13%) in the chemoradiotherapy-surgery arm and in 42 of 188 patients (22%) in the surgery arm (HR, 0.43; 95% CI, 0.26-0.72), isolated distant relapse developed at similar rates (around 27.5%) in both groups. Risk of distant relapse (with or without locoregional relapse) was lower in the chemoradiotherapy-surgery arm (HR, 0.61; 95%CI, 0.45-0.84). The median relapse-free interval was 15.1 months (interquartile range, 9.3-27.6) in the chemoradiotherapy-surgery arm and 9.0 months (IQR, 5.3-19.7) in the surgery arm.
Safety and health-related quality of life
The combination of paclitaxel and carboplatin with concurrent 41.4 Gy radiotherapy before surgery seems safe in the long term and does not significantly increase the risk of toxicity-related death, the researchers stated. Within the CROSS trial, short-term and long-term health-related quality of life after neoadjuvant chemoradiotherapy plus surgery for surviving patients was comparable to that after surgery alone.
Long-term persistent overall survival benefit
Ten-year CROSS results show that “for locally advanced resectable cancer of the esophagus or esophagogastric junction, preoperative chemoradiotherapy induces a long-term persistent improvement in overall survival.” Also, neoadjuvant chemoradiotherapy does not lead to an increased risk of death from other causes, and the survival benefit of long-term survivors is not compromised, compared with surgery alone. Furthermore, neoadjuvant chemoradiotherapy plus surgery according to CROSS can still be regarded as a standard of care, the researchers added.
Dr. Eyck and colleagues are currently performing the phase II TNT-OES-1 trial. It combines FLOT (fluorouracil, leucovorin, oxaliplatin and docetaxel) chemotherapy followed by CROSS chemoradiotherapy in patients with advanced esophageal and junctional adenocarcinoma. If this regimen appears to be safe in advanced cancer, they plan to perform a phase III trial with this regimen in locally advanced cancer. In addition, they are currently evaluating the implementation of adjuvant nivolumab in clinical practice for patients with pathologically residual disease after CROSS + surgery, based on the recently published CheckMate 577 trial .
“If possible, we prefer adding better systemic therapy to chemoradiotherapy rather than replacing chemoradiotherapy with systemic therapy alone,” Dr. Eyck said in an interview. “The reason for this is that we would like to allow patients with a complete response to neoadjuvant therapy to undergo active surveillance instead of surgery in the near future. … Since the pathologically complete response rate after regimens containing radiotherapy is substantially higher, we still prefer the addition of radiotherapy.”
The study was funded by the Dutch Cancer Foundation (KWF Kankerbestrijding). Dr. Eyck reported no disclosures. Several of the coauthors reported consulting and advisory roles with a variety of pharmaceutical companies.
Among patients with locally advanced resectable esophageal or junctional cancer, the overall survival benefit conferred by preoperative chemoradiotherapy persists for at least 10 years, according to long-term results of the Chemoradiotherapy for Esophageal Cancer Followed by Surgery Study (CROSS). As a result of earlier publication of CROSS data, chemoradiotherapy followed by surgery has become one of the standards of care for patients with locally advanced resectable esophageal cancer, stated lead author Ben M. Eyck, MD, of Erasmus University Medical Center, Rotterdam, the Netherlands, and colleagues in the Journal of Clinical Oncology.
In the multicenter, randomized trial, initiated in 2004, 178 patients randomized to chemoradiotherapy with subsequent surgery and 188 patients randomized to surgery alone were followed with overall survival as the primary, and cause-specific survival and risks of locoregional and distant relapse as the secondary endpoints. Chemoradiotherapy consisted of 5 weekly cycles of carboplatin (area under the curve of 2 mg/mL/min) and paclitaxel (50 mg/m2 body surface area on days 1, 8, 15, 22, and 29) with concurrent radiotherapy (41.4 Gy in 23 fractions, 5 days per week. Mean age was 60 years (around 78% male), with squamous cell carcinoma (23%) and adenocarcinoma (75%) as the predominant histologies.
The first analysis showed low short-term toxicity and 2-year survival increased from 50% for patients receiving surgery alone to 67% for neoadjuvant chemoradiotherapy plus surgery. Five-year follow-up data were consistent with initial reporting. Long-term benefits and harms of this regimen remain unclear, according to the researchers. Neoadjuvant chemoradiotherapy’s side effects could lead to long-term death from other causes than esophageal cancer, and may not be preventing but rather merely postponing cancer-related death. The aim of the current analysis was to determine whether the observed benefits persisted beyond 5 years.
As of Dec. 31, 2018, 117/178 patients in the chemoradiotherapy-surgery arm and 144/188 in the surgery arm had died. Median follow-up for surviving patients was 147 months. Patients in the chemoradiotherapy surgery arm had better overall survival than patients in the surgery arm (hazard ratio, 0.70; 95% confidence interval, 0.55-0.89; P = .004), with a 10-year overall survival of 38% (95% CI, 31-45) and 25% (95% CI, 19-32), respectively. No significant subgroup differences were observed for overall survival. Also, there was no evidence of a time-dependent effect of neoadjuvant chemoradiotherapy on overall survival. The major effect of neoadjuvant chemoradiotherapy, landmark analyses showed, was in the first 5 years of follow-up, with the effect on overall survival stabilized thereafter, with a hazard ratio approaching 1.00.
Cause-specific mortality
Eighty-four of 178 patients in the chemoradiotherapy-surgery arm died of esophageal cancer, with 32 dying of other causes. In the surgery arm, 121/188 died of esophageal cancer and 22 of other causes. The hazard ratio for esophageal cancer death in the chemoradiotherapy-surgery arm was 0.60 (95% CI, 0.46 to 0.80), with 10-year absolute risks of 47% (95% CI, 40-54) and 64% (95% CI,57-71), respectively, in the two arms. Death from other causes was comparable, with 10-year absolute risks of 15% (95% CI, 10-21) and 11% (95% CI, 7-16), respectively, for chemoradiotherapy-surgery versus surgery alone.
Locoregional relapse
Locoregional relapse rates were 8% (15/178) and 18% (33/188) in the chemoradiotherapy-surgery and surgery arms, respectively (HR, 0.39; 95% CI, 0.21-0.72). Eighty-seven percent of those developed within 3 years of follow-up in the chemoradiotherapy arm, with the median relapse-free interval at 3.9 months. In the surgery arm, 28 of 33 relapses (85%) developed within 3 years and the median relapse-free interval was 7.1 months. Beyond 6 years, there were no further relapses in either arm.
While synchronous distant plus locoregional relapse developed in 23 of 178 patients (13%) in the chemoradiotherapy-surgery arm and in 42 of 188 patients (22%) in the surgery arm (HR, 0.43; 95% CI, 0.26-0.72), isolated distant relapse developed at similar rates (around 27.5%) in both groups. Risk of distant relapse (with or without locoregional relapse) was lower in the chemoradiotherapy-surgery arm (HR, 0.61; 95%CI, 0.45-0.84). The median relapse-free interval was 15.1 months (interquartile range, 9.3-27.6) in the chemoradiotherapy-surgery arm and 9.0 months (IQR, 5.3-19.7) in the surgery arm.
Safety and health-related quality of life
The combination of paclitaxel and carboplatin with concurrent 41.4 Gy radiotherapy before surgery seems safe in the long term and does not significantly increase the risk of toxicity-related death, the researchers stated. Within the CROSS trial, short-term and long-term health-related quality of life after neoadjuvant chemoradiotherapy plus surgery for surviving patients was comparable to that after surgery alone.
Long-term persistent overall survival benefit
Ten-year CROSS results show that “for locally advanced resectable cancer of the esophagus or esophagogastric junction, preoperative chemoradiotherapy induces a long-term persistent improvement in overall survival.” Also, neoadjuvant chemoradiotherapy does not lead to an increased risk of death from other causes, and the survival benefit of long-term survivors is not compromised, compared with surgery alone. Furthermore, neoadjuvant chemoradiotherapy plus surgery according to CROSS can still be regarded as a standard of care, the researchers added.
Dr. Eyck and colleagues are currently performing the phase II TNT-OES-1 trial. It combines FLOT (fluorouracil, leucovorin, oxaliplatin and docetaxel) chemotherapy followed by CROSS chemoradiotherapy in patients with advanced esophageal and junctional adenocarcinoma. If this regimen appears to be safe in advanced cancer, they plan to perform a phase III trial with this regimen in locally advanced cancer. In addition, they are currently evaluating the implementation of adjuvant nivolumab in clinical practice for patients with pathologically residual disease after CROSS + surgery, based on the recently published CheckMate 577 trial .
“If possible, we prefer adding better systemic therapy to chemoradiotherapy rather than replacing chemoradiotherapy with systemic therapy alone,” Dr. Eyck said in an interview. “The reason for this is that we would like to allow patients with a complete response to neoadjuvant therapy to undergo active surveillance instead of surgery in the near future. … Since the pathologically complete response rate after regimens containing radiotherapy is substantially higher, we still prefer the addition of radiotherapy.”
The study was funded by the Dutch Cancer Foundation (KWF Kankerbestrijding). Dr. Eyck reported no disclosures. Several of the coauthors reported consulting and advisory roles with a variety of pharmaceutical companies.
FROM JOURNAL OF CLINICAL ONCOLOGY
Delayed Coronary Vasospasm in a Patient with Metastatic Gastric Cancer Receiving FOLFOX Therapy
A 40-year-old man with stage IV gastric adenocarcinoma was found to have coronary artery vasospasm in the setting of recent 5-fluorouracil administration.
Coronary artery vasospasm is a rare but well-known adverse effect of 5-fluorouracil (5-FU) that can be life threatening if unrecognized. Patients typically present with anginal chest pain and ST elevations on electrocardiogram (ECG) without atherosclerotic disease on coronary angiography. This phenomenon typically occurs during or shortly after infusion and resolves within hours to days after cessation of 5-FU.
In this report, we present an unusual case of coronary artery vasospasm that intermittently recurred for 25 days following 5-FU treatment in a 40-year-old male with stage IV gastric adenocarcinoma. We also review the literature on typical presentation and risk factors for 5-FU-induced coronary vasospasm, findings on coronary angiography, and management options.
5-FU is an IV administered antimetabolite chemotherapy commonly used to treat solid tumors, including gastrointestinal, pancreatic, breast, and head and neck tumors. 5-FU inhibits thymidylate synthase, which reduces levels of thymidine, a key pyrimidine nucleoside required for DNA replication within tumor cells.1 For several decades, 5-FU has remained one of the first-line drugs for colorectal cancer because it may be curative. It is the third most commonly used chemotherapy in the world and is included on the World Health Organization’s list of essential medicines.2
Cardiotoxicity occurs in 1.2 to 18% of patients who receive 5-FU therapy.3 Although there is variability in presentation for acute cardiotoxicity from 5-FU, including sudden death, angina pectoris, myocardial infarction, and ventricular arrhythmias, the mechanism most commonly implicated is coronary artery vasospasm.3 The direct observation of active coronary artery vasospasm during left heart catheterization is rare due its transient nature; however, several case studies have managed to demonstrate this.4,5 The pathophysiology of 5-FU-induced cardiotoxicity is unknown, but adverse effects on cardiac microvasculature, myocyte metabolism, platelet aggregation, and coronary vasoconstriction have all been proposed.3,6In the current case, we present a patient with stage IV gastric adenocarcinoma who complained of chest pain during hospitalization and was found to have coronary artery vasospasm in the setting of recent 5-FU administration. Following coronary angiography that showed a lack of atherosclerotic disease, the patient continued to experience episodes of chest pain with ST elevations on ECG that recurred despite cessation of 5-FU and repeated administration of vasodilatory medications.
Case Presentation
A male aged 40 years was admitted to the hospital for abdominal pain, with initial imaging concerning for partial small bowel obstruction. His history included recently diagnosed stage IV gastric adenocarcinoma complicated by peritoneal carcinomatosis status post initiation of infusional FOLFOX-4 (5-FU, leucovorin, and oxaliplatin) 11 days prior. The patient was treated for small bowel obstruction. However, several days after admission, he developed nonpleuritic, substernal chest pain unrelated to exertion and unrelieved by rest. The patient reported no known risk factors, family history, or personal history of coronary artery disease. Baseline echocardiography and ECG performed several months prior showed normal left ventricular function without ischemic findings.
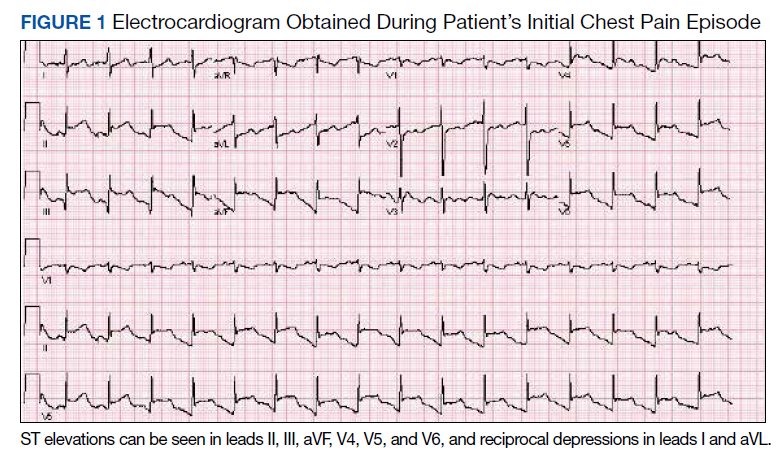
Physical examination at the time of chest pain revealed a heart rate of 140 beats/min. The remainder of his vital signs were within normal range. There were no murmurs, rubs, gallops, or additional heart sounds heard on cardiac auscultation. Chest pain was not reproducible to palpation or positional in nature. An ECG demonstrated dynamic inferolateral ST elevations with reciprocal changes in leads I and aVL (Figure 1). A bedside echocardiogram showed hypokinesis of the septal wall. Troponin-I returned below the detectable level.
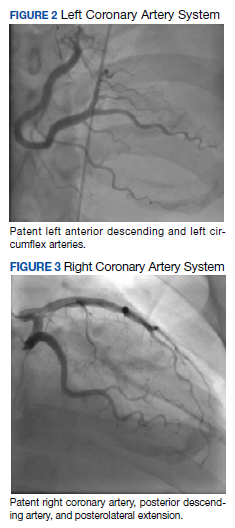
The patient was taken for emergent coronary catheterization, which demonstrated patent epicardial coronary arteries without atherosclerosis, a left ventricular ejection fraction of 60%, and a right dominant heart (Figures 2 and 3). Ventriculogram showed normal wall motion. Repeat troponin-I several hours after catheterization was again below detectable levels.
Given the patient’s acute onset of chest pain and inferolateral ST elevations seen on ECG, the working diagnosis prior to coronary catherization was acute coronary syndrome. The differential diagnosis included other causes of life-threatening chest pain, including pulmonary embolism, pneumonia, aortic dissection, myopericarditis, pericardial effusion, cardiac tamponade, or coronary artery vasospasm. Computed tomography (CT) angiography of the chest was not consistent with pulmonary embolism or other acute cardiopulmonary process. Based on findings from coronary angiography and recent exposure to 5-FU, as well as resolution followed by recurrence of chest pain and ECG changes over weeks, the most likely diagnosis after coronary catheterization was coronary artery vasospasm.
Treatment
Following catheterization, the patient returned to the medical intensive care unit, where he continued to report intermittent episodes of chest pain with ST elevations. In the following days, he was started on isosorbide mononitrate 150 mg daily and amlodipine 10 mg daily. Although these vasodilatory agents reduced the frequency of his chest pain episodes, intermittent chest pain associated with ST elevations on ECG continued even with maximal doses of isosorbide mononitrate and amlodipine. Administration of sublingual nitroglycerin during chest pain episodes effectively relieved his chest pain. Given the severity and frequency of the patient’s chest pain, the oncology consult team recommended foregoing further chemotherapeutic treatment with 5-FU.
Outcome
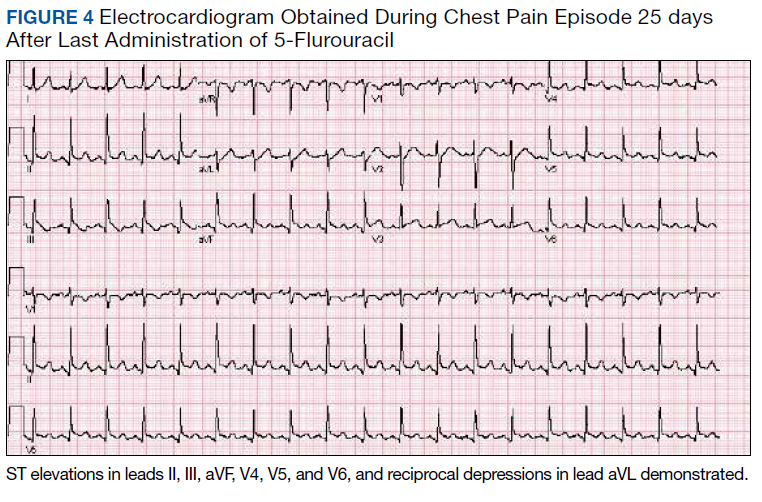
Despite holding 5-FU throughout the patient’s hospitalization and treating the patient with antianginal mediations, frequent chest pain episodes associated with ST elevations continued to recur until 25 days after his last treatment with 5-FU (Figure 4). The patient eventually expired during this hospital stay due to cancer-related complications.
Discussion
Coronary artery vasospasm is a well-known complication of 5-FU that can be life threatening if unrecognized.6-8 As seen in our case, patients typically present with anginal chest pain relieved with nitrates and ST elevations on ECG in the absence of occlusive macrovascular disease on coronary angiography.
A unique aspect of 5-FU is its variability in dose and frequency of administration across chemotherapeutic regimens. Particularly, 5-FU can be administered in daily intravenous bolus doses or as a continuous infusion for a protracted length of time. The spectrum of toxicity from 5-FU differs depending on the dose and frequency of administration. Bolus administration of 5-FU, for example, is thought to be associated with a higher rate of myelosuppression, while infusional administration of 5-FU is thought to be associated with a higher rate of cardiotoxicity and a higher tumor response rate.9
Most cases of coronary vasospasm occur either during infusion of 5-FU or within hours to days after completion. The median time of presentation for 5-FU-induced coronary artery vasospasm is about 12 hours postinfusion, while the most delayed presentation reported in the literature is 72 hours postinfusion.6,8 Delayed presentation of vasospasm may result from the release of potent vasoactive metabolites of 5-FU that accumulate over time; therefore, infusional administration may accentuate this effect.6,9 Remarkably, our patient’s chest pain episodes persisted for 25 days despite treatment with anti-anginal medications, highlighting the extent to which infusional 5-FU can produce a delay in adverse cardiotoxic effects and the importance of ongoing clinical vigilance after 5-FU exposure.
Vasospasm alone does not completely explain the spectrum of cardiac toxicity attributed to 5-FU administration. As in our case, coronary angiography during symptomatic episodes often fails to demonstrate coronary vasospasm.8 Additionally, ergonovine, an alkaloid agent used to assess coronary vasomotor function, failed to induce coronary vasospasm in some patients with suspected 5-FU-induced cardiac toxicity.10 The lack of vasospasm in some patients with 5-FU-induced cardiac toxicity suggests multiple independent effects of 5-FU on cardiac tissue that are poorly understood.
In the absence of obvious macrovascular effects, there also may be a deleterious effect of 5-FU on the coronary microvasculature that may result in coronary artery vasospasm. Though coronary microvasculature cannot be directly visualized, observation of slowed coronary blood velocity indicates a reduction in microvascular flow.8 Thus, the failure to observe epicardial coronary vasospasm in our patient does not preclude a vasospastic pathology.
The heterogeneous presentation of coronary artery vasospasm demands consideration of other disease processes such as atherosclerotic coronary artery disease, pericarditis, myopericarditis, primary arrythmias, and stress-induced cardiomyopathy, all of which have been described in association with 5-FU administration.8 A 12-lead ECG should be performed during a suspected attack. An ECG will typically demonstrate ST elevations corresponding to spasm of the involved vessel. Reciprocal ST depressions in the contralateral leads also may be seen. ECG may be useful in the acute setting to identify regional wall motion abnormalities or to rule out pericardial effusion as a cause. Cardiac biomarkers such as troponin-I, -C, and creatine kinase typically are less useful because they are often normal, even in known coronary artery vasospasm.11
Coronary angiography during an episode may show a localized region of vasospasm in an epicardial artery. Diffuse multivessel vasospasm does occur, and the location of vasospasm may change, but these events are rare. Under normal circumstances, provocative testing involving angiography with administration of acetylcholine, ergot agents, or hyperventilation can be performed. However, this type of investigation should be limited to specialized centers and should not be performed in the acute phase of the disease.12
Treatment of suspected coronary vasospasm in patients receiving 5-FU involves stopping the infusion and administering calcium channel blockers or oral nitrates to relieve anginal symptoms.13 5-FU-induced coronary artery vasospasm has a 90% rate of recurrence with subsequent infusions.8 If possible, alternate chemotherapy regimens should be considered once coronary artery vasospasm has been identified.14,15 If further 5-FU use is required, or if benefits are deemed to outweigh risks, infusions should be given in an inpatient setting with continuous cardiac monitoring.16
Calcium channel blockers and oral nitrates have been found to produce benefit in patients in acute settings; however, there is little evidence to attest to their effectiveness as prophylactic agents in those receiving 5-FU. Some reports demonstrate episodes where both calcium channel blockers and oral nitrates failed to prevent subsequent vasospasms.17 Although this was the case for our patient, short-acting sublingual nitroglycerin seemed to be effective in reducing the frequency of anginal symptoms.
Long-term outcomes have not been well investigated for patients with 5-FU-induced coronary vasospasm. However, many case reports show improvements in left ventricular function between 8 and 15 days after discontinuation of 5-FU.7,10 Although this would be a valuable topic for further research, the rarity of this phenomenon creates limitations.
Conclusions
5-FU is a first-line chemotherapy for gastrointestinal cancers that is generally well tolerated but may be associated with potentially life-threatening cardiotoxic effects, of which coronary artery vasospasm is the most common. Coronary artery vasospasm presents with anginal chest pain and ST elevations on ECG that can be indistinguishable from acute coronary syndrome. Diagnosis requires cardiac catheterization, which will reveal patent coronary arteries. Infusional administration of 5-FU may be more likely to produce late cardiotoxic effects and a longer period of persistent symptoms, necessitating close monitoring for days or even weeks from last administration of 5-FU. Coronary artery vasospasm should be treated with anti-anginal medications, though varying degrees of effectiveness can be seen; clinicians should remain vigilant for recurrent episodes of chest pain despite treatment.
1. Wacker A, Lersch C, Scherpinski U, Reindl L, Seyfarth M. High incidence of angina pectoris in patients treated with 5-fluorouracil. A planned surveillance study with 102 patients. Oncology. 2003;65(2):108-112. doi:10.1159/000072334
2. World Health Organization Model List of Essential Medicines, 21st List, 2019. Accessed April 14, 2021. https://apps.who.int/iris/rest/bitstreams/1237479/retrieve
3. Jensen SA, Sørensen JB. Risk factors and prevention of cardiotoxicity induced by 5-fluorouracil or capecitabine. Cancer Chemother Pharmacol. 2006;58(4):487-493. doi:10.1007/s00280-005-0178-1
4. Shoemaker LK, Arora U, Rocha Lima CM. 5-fluorouracil-induced coronary vasospasm. Cancer Control. 2004;11(1):46-49. doi:10.1177/107327480401100207
5. Luwaert RJ, Descamps O, Majois F, Chaudron JM, Beauduin M. Coronary artery spasm induced by 5-fluorouracil. Eur Heart J. 1991;12(3):468-470. doi:10.1093/oxfordjournals.eurheartj.a059919
6. Saif MW, Shah MM, Shah AR. Fluoropyrimidine-associated cardiotoxicity: revisited. Expert Opin Drug Saf. 2009;8(2):191-202. doi:10.1517/14740330902733961
7. Patel B, Kloner RA, Ensley J, Al-Sarraf M, Kish J, Wynne J. 5-Fluorouracil cardiotoxicity: left ventricular dysfunction and effect of coronary vasodilators. Am J Med Sci. 1987;294(4):238-243. doi:10.1097/00000441-198710000-00004
8. Sara JD, Kaur J, Khodadadi R, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol. 2018;10:1758835918780140. Published 2018 Jun 18. doi:10.1177/1758835918780140
9. Hansen RM, Ryan L, Anderson T, et al. Phase III study of bolus versus infusion fluorouracil with or without cisplatin in advanced colorectal cancer. J Natl Cancer Inst. 1996;88(10):668-674. doi:10.1093/jnci/88.10.668
10. Kim SM, Kwak CH, Lee B, et al. A case of severe coronary spasm associated with 5-fluorouracil chemotherapy. Korean J Intern Med. 2012;27(3):342-345. doi:10.3904/kjim.2012.27.3.342
11. Swarup S, Patibandla S, Grossman SA. Coronary Artery Vasospasm. StatPearls. Treasure Island (FL): StatPearls Publishing LLC.; 2021.
12. Beijk MA, Vlastra WV, Delewi R, et al. Myocardial infarction with non-obstructive coronary arteries: a focus on vasospastic angina. Neth Heart J. 2019;27(5):237-245. doi:10.1007/s12471-019-1232-7
13. Giza DE, Boccalandro F, Lopez-Mattei J, et al. Ischemic heart disease: special considerations in cardio-oncology. Curr Treat Options Cardiovasc Med. 2017;19(5):37. doi:10.1007/s11936-017-0535-5
14. Meydan N, Kundak I, Yavuzsen T, et al. Cardiotoxicity of de Gramont’s regimen: incidence, clinical characteristics and long-term follow-up. Jpn J Clin Oncol. 2005;35(5):265-270. doi:10.1093/jjco/hyi071
15. Senkus E, Jassem J. Cardiovascular effects of systemic cancer treatment. Cancer Treat Rev. 2011;37(4):300-311. doi:10.1016/j.ctrv.2010.11.001
16. Rezkalla S, Kloner RA, Ensley J, et al. Continuous ambulatory ECG monitoring during fluorouracil therapy: a prospective study. J Clin Oncol. 1989;7(4):509-514. doi:10.1200/JCO.1989.7.4.509
17. Akpek G, Hartshorn KL. Failure of oral nitrate and calcium channel blocker therapy to prevent 5-fluorouracil-related myocardial ischemia: a case report. Cancer Chemother Pharmacol. 1999;43(2):157-161. doi:10.1007/s002800050877
A 40-year-old man with stage IV gastric adenocarcinoma was found to have coronary artery vasospasm in the setting of recent 5-fluorouracil administration.
A 40-year-old man with stage IV gastric adenocarcinoma was found to have coronary artery vasospasm in the setting of recent 5-fluorouracil administration.
Coronary artery vasospasm is a rare but well-known adverse effect of 5-fluorouracil (5-FU) that can be life threatening if unrecognized. Patients typically present with anginal chest pain and ST elevations on electrocardiogram (ECG) without atherosclerotic disease on coronary angiography. This phenomenon typically occurs during or shortly after infusion and resolves within hours to days after cessation of 5-FU.
In this report, we present an unusual case of coronary artery vasospasm that intermittently recurred for 25 days following 5-FU treatment in a 40-year-old male with stage IV gastric adenocarcinoma. We also review the literature on typical presentation and risk factors for 5-FU-induced coronary vasospasm, findings on coronary angiography, and management options.
5-FU is an IV administered antimetabolite chemotherapy commonly used to treat solid tumors, including gastrointestinal, pancreatic, breast, and head and neck tumors. 5-FU inhibits thymidylate synthase, which reduces levels of thymidine, a key pyrimidine nucleoside required for DNA replication within tumor cells.1 For several decades, 5-FU has remained one of the first-line drugs for colorectal cancer because it may be curative. It is the third most commonly used chemotherapy in the world and is included on the World Health Organization’s list of essential medicines.2
Cardiotoxicity occurs in 1.2 to 18% of patients who receive 5-FU therapy.3 Although there is variability in presentation for acute cardiotoxicity from 5-FU, including sudden death, angina pectoris, myocardial infarction, and ventricular arrhythmias, the mechanism most commonly implicated is coronary artery vasospasm.3 The direct observation of active coronary artery vasospasm during left heart catheterization is rare due its transient nature; however, several case studies have managed to demonstrate this.4,5 The pathophysiology of 5-FU-induced cardiotoxicity is unknown, but adverse effects on cardiac microvasculature, myocyte metabolism, platelet aggregation, and coronary vasoconstriction have all been proposed.3,6In the current case, we present a patient with stage IV gastric adenocarcinoma who complained of chest pain during hospitalization and was found to have coronary artery vasospasm in the setting of recent 5-FU administration. Following coronary angiography that showed a lack of atherosclerotic disease, the patient continued to experience episodes of chest pain with ST elevations on ECG that recurred despite cessation of 5-FU and repeated administration of vasodilatory medications.
Case Presentation
A male aged 40 years was admitted to the hospital for abdominal pain, with initial imaging concerning for partial small bowel obstruction. His history included recently diagnosed stage IV gastric adenocarcinoma complicated by peritoneal carcinomatosis status post initiation of infusional FOLFOX-4 (5-FU, leucovorin, and oxaliplatin) 11 days prior. The patient was treated for small bowel obstruction. However, several days after admission, he developed nonpleuritic, substernal chest pain unrelated to exertion and unrelieved by rest. The patient reported no known risk factors, family history, or personal history of coronary artery disease. Baseline echocardiography and ECG performed several months prior showed normal left ventricular function without ischemic findings.
Physical examination at the time of chest pain revealed a heart rate of 140 beats/min. The remainder of his vital signs were within normal range. There were no murmurs, rubs, gallops, or additional heart sounds heard on cardiac auscultation. Chest pain was not reproducible to palpation or positional in nature. An ECG demonstrated dynamic inferolateral ST elevations with reciprocal changes in leads I and aVL (Figure 1). A bedside echocardiogram showed hypokinesis of the septal wall. Troponin-I returned below the detectable level.
The patient was taken for emergent coronary catheterization, which demonstrated patent epicardial coronary arteries without atherosclerosis, a left ventricular ejection fraction of 60%, and a right dominant heart (Figures 2 and 3). Ventriculogram showed normal wall motion. Repeat troponin-I several hours after catheterization was again below detectable levels.
Given the patient’s acute onset of chest pain and inferolateral ST elevations seen on ECG, the working diagnosis prior to coronary catherization was acute coronary syndrome. The differential diagnosis included other causes of life-threatening chest pain, including pulmonary embolism, pneumonia, aortic dissection, myopericarditis, pericardial effusion, cardiac tamponade, or coronary artery vasospasm. Computed tomography (CT) angiography of the chest was not consistent with pulmonary embolism or other acute cardiopulmonary process. Based on findings from coronary angiography and recent exposure to 5-FU, as well as resolution followed by recurrence of chest pain and ECG changes over weeks, the most likely diagnosis after coronary catheterization was coronary artery vasospasm.
Treatment
Following catheterization, the patient returned to the medical intensive care unit, where he continued to report intermittent episodes of chest pain with ST elevations. In the following days, he was started on isosorbide mononitrate 150 mg daily and amlodipine 10 mg daily. Although these vasodilatory agents reduced the frequency of his chest pain episodes, intermittent chest pain associated with ST elevations on ECG continued even with maximal doses of isosorbide mononitrate and amlodipine. Administration of sublingual nitroglycerin during chest pain episodes effectively relieved his chest pain. Given the severity and frequency of the patient’s chest pain, the oncology consult team recommended foregoing further chemotherapeutic treatment with 5-FU.
Outcome
Despite holding 5-FU throughout the patient’s hospitalization and treating the patient with antianginal mediations, frequent chest pain episodes associated with ST elevations continued to recur until 25 days after his last treatment with 5-FU (Figure 4). The patient eventually expired during this hospital stay due to cancer-related complications.
Discussion
Coronary artery vasospasm is a well-known complication of 5-FU that can be life threatening if unrecognized.6-8 As seen in our case, patients typically present with anginal chest pain relieved with nitrates and ST elevations on ECG in the absence of occlusive macrovascular disease on coronary angiography.
A unique aspect of 5-FU is its variability in dose and frequency of administration across chemotherapeutic regimens. Particularly, 5-FU can be administered in daily intravenous bolus doses or as a continuous infusion for a protracted length of time. The spectrum of toxicity from 5-FU differs depending on the dose and frequency of administration. Bolus administration of 5-FU, for example, is thought to be associated with a higher rate of myelosuppression, while infusional administration of 5-FU is thought to be associated with a higher rate of cardiotoxicity and a higher tumor response rate.9
Most cases of coronary vasospasm occur either during infusion of 5-FU or within hours to days after completion. The median time of presentation for 5-FU-induced coronary artery vasospasm is about 12 hours postinfusion, while the most delayed presentation reported in the literature is 72 hours postinfusion.6,8 Delayed presentation of vasospasm may result from the release of potent vasoactive metabolites of 5-FU that accumulate over time; therefore, infusional administration may accentuate this effect.6,9 Remarkably, our patient’s chest pain episodes persisted for 25 days despite treatment with anti-anginal medications, highlighting the extent to which infusional 5-FU can produce a delay in adverse cardiotoxic effects and the importance of ongoing clinical vigilance after 5-FU exposure.
Vasospasm alone does not completely explain the spectrum of cardiac toxicity attributed to 5-FU administration. As in our case, coronary angiography during symptomatic episodes often fails to demonstrate coronary vasospasm.8 Additionally, ergonovine, an alkaloid agent used to assess coronary vasomotor function, failed to induce coronary vasospasm in some patients with suspected 5-FU-induced cardiac toxicity.10 The lack of vasospasm in some patients with 5-FU-induced cardiac toxicity suggests multiple independent effects of 5-FU on cardiac tissue that are poorly understood.
In the absence of obvious macrovascular effects, there also may be a deleterious effect of 5-FU on the coronary microvasculature that may result in coronary artery vasospasm. Though coronary microvasculature cannot be directly visualized, observation of slowed coronary blood velocity indicates a reduction in microvascular flow.8 Thus, the failure to observe epicardial coronary vasospasm in our patient does not preclude a vasospastic pathology.
The heterogeneous presentation of coronary artery vasospasm demands consideration of other disease processes such as atherosclerotic coronary artery disease, pericarditis, myopericarditis, primary arrythmias, and stress-induced cardiomyopathy, all of which have been described in association with 5-FU administration.8 A 12-lead ECG should be performed during a suspected attack. An ECG will typically demonstrate ST elevations corresponding to spasm of the involved vessel. Reciprocal ST depressions in the contralateral leads also may be seen. ECG may be useful in the acute setting to identify regional wall motion abnormalities or to rule out pericardial effusion as a cause. Cardiac biomarkers such as troponin-I, -C, and creatine kinase typically are less useful because they are often normal, even in known coronary artery vasospasm.11
Coronary angiography during an episode may show a localized region of vasospasm in an epicardial artery. Diffuse multivessel vasospasm does occur, and the location of vasospasm may change, but these events are rare. Under normal circumstances, provocative testing involving angiography with administration of acetylcholine, ergot agents, or hyperventilation can be performed. However, this type of investigation should be limited to specialized centers and should not be performed in the acute phase of the disease.12
Treatment of suspected coronary vasospasm in patients receiving 5-FU involves stopping the infusion and administering calcium channel blockers or oral nitrates to relieve anginal symptoms.13 5-FU-induced coronary artery vasospasm has a 90% rate of recurrence with subsequent infusions.8 If possible, alternate chemotherapy regimens should be considered once coronary artery vasospasm has been identified.14,15 If further 5-FU use is required, or if benefits are deemed to outweigh risks, infusions should be given in an inpatient setting with continuous cardiac monitoring.16
Calcium channel blockers and oral nitrates have been found to produce benefit in patients in acute settings; however, there is little evidence to attest to their effectiveness as prophylactic agents in those receiving 5-FU. Some reports demonstrate episodes where both calcium channel blockers and oral nitrates failed to prevent subsequent vasospasms.17 Although this was the case for our patient, short-acting sublingual nitroglycerin seemed to be effective in reducing the frequency of anginal symptoms.
Long-term outcomes have not been well investigated for patients with 5-FU-induced coronary vasospasm. However, many case reports show improvements in left ventricular function between 8 and 15 days after discontinuation of 5-FU.7,10 Although this would be a valuable topic for further research, the rarity of this phenomenon creates limitations.
Conclusions
5-FU is a first-line chemotherapy for gastrointestinal cancers that is generally well tolerated but may be associated with potentially life-threatening cardiotoxic effects, of which coronary artery vasospasm is the most common. Coronary artery vasospasm presents with anginal chest pain and ST elevations on ECG that can be indistinguishable from acute coronary syndrome. Diagnosis requires cardiac catheterization, which will reveal patent coronary arteries. Infusional administration of 5-FU may be more likely to produce late cardiotoxic effects and a longer period of persistent symptoms, necessitating close monitoring for days or even weeks from last administration of 5-FU. Coronary artery vasospasm should be treated with anti-anginal medications, though varying degrees of effectiveness can be seen; clinicians should remain vigilant for recurrent episodes of chest pain despite treatment.
Coronary artery vasospasm is a rare but well-known adverse effect of 5-fluorouracil (5-FU) that can be life threatening if unrecognized. Patients typically present with anginal chest pain and ST elevations on electrocardiogram (ECG) without atherosclerotic disease on coronary angiography. This phenomenon typically occurs during or shortly after infusion and resolves within hours to days after cessation of 5-FU.
In this report, we present an unusual case of coronary artery vasospasm that intermittently recurred for 25 days following 5-FU treatment in a 40-year-old male with stage IV gastric adenocarcinoma. We also review the literature on typical presentation and risk factors for 5-FU-induced coronary vasospasm, findings on coronary angiography, and management options.
5-FU is an IV administered antimetabolite chemotherapy commonly used to treat solid tumors, including gastrointestinal, pancreatic, breast, and head and neck tumors. 5-FU inhibits thymidylate synthase, which reduces levels of thymidine, a key pyrimidine nucleoside required for DNA replication within tumor cells.1 For several decades, 5-FU has remained one of the first-line drugs for colorectal cancer because it may be curative. It is the third most commonly used chemotherapy in the world and is included on the World Health Organization’s list of essential medicines.2
Cardiotoxicity occurs in 1.2 to 18% of patients who receive 5-FU therapy.3 Although there is variability in presentation for acute cardiotoxicity from 5-FU, including sudden death, angina pectoris, myocardial infarction, and ventricular arrhythmias, the mechanism most commonly implicated is coronary artery vasospasm.3 The direct observation of active coronary artery vasospasm during left heart catheterization is rare due its transient nature; however, several case studies have managed to demonstrate this.4,5 The pathophysiology of 5-FU-induced cardiotoxicity is unknown, but adverse effects on cardiac microvasculature, myocyte metabolism, platelet aggregation, and coronary vasoconstriction have all been proposed.3,6In the current case, we present a patient with stage IV gastric adenocarcinoma who complained of chest pain during hospitalization and was found to have coronary artery vasospasm in the setting of recent 5-FU administration. Following coronary angiography that showed a lack of atherosclerotic disease, the patient continued to experience episodes of chest pain with ST elevations on ECG that recurred despite cessation of 5-FU and repeated administration of vasodilatory medications.
Case Presentation
A male aged 40 years was admitted to the hospital for abdominal pain, with initial imaging concerning for partial small bowel obstruction. His history included recently diagnosed stage IV gastric adenocarcinoma complicated by peritoneal carcinomatosis status post initiation of infusional FOLFOX-4 (5-FU, leucovorin, and oxaliplatin) 11 days prior. The patient was treated for small bowel obstruction. However, several days after admission, he developed nonpleuritic, substernal chest pain unrelated to exertion and unrelieved by rest. The patient reported no known risk factors, family history, or personal history of coronary artery disease. Baseline echocardiography and ECG performed several months prior showed normal left ventricular function without ischemic findings.
Physical examination at the time of chest pain revealed a heart rate of 140 beats/min. The remainder of his vital signs were within normal range. There were no murmurs, rubs, gallops, or additional heart sounds heard on cardiac auscultation. Chest pain was not reproducible to palpation or positional in nature. An ECG demonstrated dynamic inferolateral ST elevations with reciprocal changes in leads I and aVL (Figure 1). A bedside echocardiogram showed hypokinesis of the septal wall. Troponin-I returned below the detectable level.
The patient was taken for emergent coronary catheterization, which demonstrated patent epicardial coronary arteries without atherosclerosis, a left ventricular ejection fraction of 60%, and a right dominant heart (Figures 2 and 3). Ventriculogram showed normal wall motion. Repeat troponin-I several hours after catheterization was again below detectable levels.
Given the patient’s acute onset of chest pain and inferolateral ST elevations seen on ECG, the working diagnosis prior to coronary catherization was acute coronary syndrome. The differential diagnosis included other causes of life-threatening chest pain, including pulmonary embolism, pneumonia, aortic dissection, myopericarditis, pericardial effusion, cardiac tamponade, or coronary artery vasospasm. Computed tomography (CT) angiography of the chest was not consistent with pulmonary embolism or other acute cardiopulmonary process. Based on findings from coronary angiography and recent exposure to 5-FU, as well as resolution followed by recurrence of chest pain and ECG changes over weeks, the most likely diagnosis after coronary catheterization was coronary artery vasospasm.
Treatment
Following catheterization, the patient returned to the medical intensive care unit, where he continued to report intermittent episodes of chest pain with ST elevations. In the following days, he was started on isosorbide mononitrate 150 mg daily and amlodipine 10 mg daily. Although these vasodilatory agents reduced the frequency of his chest pain episodes, intermittent chest pain associated with ST elevations on ECG continued even with maximal doses of isosorbide mononitrate and amlodipine. Administration of sublingual nitroglycerin during chest pain episodes effectively relieved his chest pain. Given the severity and frequency of the patient’s chest pain, the oncology consult team recommended foregoing further chemotherapeutic treatment with 5-FU.
Outcome
Despite holding 5-FU throughout the patient’s hospitalization and treating the patient with antianginal mediations, frequent chest pain episodes associated with ST elevations continued to recur until 25 days after his last treatment with 5-FU (Figure 4). The patient eventually expired during this hospital stay due to cancer-related complications.
Discussion
Coronary artery vasospasm is a well-known complication of 5-FU that can be life threatening if unrecognized.6-8 As seen in our case, patients typically present with anginal chest pain relieved with nitrates and ST elevations on ECG in the absence of occlusive macrovascular disease on coronary angiography.
A unique aspect of 5-FU is its variability in dose and frequency of administration across chemotherapeutic regimens. Particularly, 5-FU can be administered in daily intravenous bolus doses or as a continuous infusion for a protracted length of time. The spectrum of toxicity from 5-FU differs depending on the dose and frequency of administration. Bolus administration of 5-FU, for example, is thought to be associated with a higher rate of myelosuppression, while infusional administration of 5-FU is thought to be associated with a higher rate of cardiotoxicity and a higher tumor response rate.9
Most cases of coronary vasospasm occur either during infusion of 5-FU or within hours to days after completion. The median time of presentation for 5-FU-induced coronary artery vasospasm is about 12 hours postinfusion, while the most delayed presentation reported in the literature is 72 hours postinfusion.6,8 Delayed presentation of vasospasm may result from the release of potent vasoactive metabolites of 5-FU that accumulate over time; therefore, infusional administration may accentuate this effect.6,9 Remarkably, our patient’s chest pain episodes persisted for 25 days despite treatment with anti-anginal medications, highlighting the extent to which infusional 5-FU can produce a delay in adverse cardiotoxic effects and the importance of ongoing clinical vigilance after 5-FU exposure.
Vasospasm alone does not completely explain the spectrum of cardiac toxicity attributed to 5-FU administration. As in our case, coronary angiography during symptomatic episodes often fails to demonstrate coronary vasospasm.8 Additionally, ergonovine, an alkaloid agent used to assess coronary vasomotor function, failed to induce coronary vasospasm in some patients with suspected 5-FU-induced cardiac toxicity.10 The lack of vasospasm in some patients with 5-FU-induced cardiac toxicity suggests multiple independent effects of 5-FU on cardiac tissue that are poorly understood.
In the absence of obvious macrovascular effects, there also may be a deleterious effect of 5-FU on the coronary microvasculature that may result in coronary artery vasospasm. Though coronary microvasculature cannot be directly visualized, observation of slowed coronary blood velocity indicates a reduction in microvascular flow.8 Thus, the failure to observe epicardial coronary vasospasm in our patient does not preclude a vasospastic pathology.
The heterogeneous presentation of coronary artery vasospasm demands consideration of other disease processes such as atherosclerotic coronary artery disease, pericarditis, myopericarditis, primary arrythmias, and stress-induced cardiomyopathy, all of which have been described in association with 5-FU administration.8 A 12-lead ECG should be performed during a suspected attack. An ECG will typically demonstrate ST elevations corresponding to spasm of the involved vessel. Reciprocal ST depressions in the contralateral leads also may be seen. ECG may be useful in the acute setting to identify regional wall motion abnormalities or to rule out pericardial effusion as a cause. Cardiac biomarkers such as troponin-I, -C, and creatine kinase typically are less useful because they are often normal, even in known coronary artery vasospasm.11
Coronary angiography during an episode may show a localized region of vasospasm in an epicardial artery. Diffuse multivessel vasospasm does occur, and the location of vasospasm may change, but these events are rare. Under normal circumstances, provocative testing involving angiography with administration of acetylcholine, ergot agents, or hyperventilation can be performed. However, this type of investigation should be limited to specialized centers and should not be performed in the acute phase of the disease.12
Treatment of suspected coronary vasospasm in patients receiving 5-FU involves stopping the infusion and administering calcium channel blockers or oral nitrates to relieve anginal symptoms.13 5-FU-induced coronary artery vasospasm has a 90% rate of recurrence with subsequent infusions.8 If possible, alternate chemotherapy regimens should be considered once coronary artery vasospasm has been identified.14,15 If further 5-FU use is required, or if benefits are deemed to outweigh risks, infusions should be given in an inpatient setting with continuous cardiac monitoring.16
Calcium channel blockers and oral nitrates have been found to produce benefit in patients in acute settings; however, there is little evidence to attest to their effectiveness as prophylactic agents in those receiving 5-FU. Some reports demonstrate episodes where both calcium channel blockers and oral nitrates failed to prevent subsequent vasospasms.17 Although this was the case for our patient, short-acting sublingual nitroglycerin seemed to be effective in reducing the frequency of anginal symptoms.
Long-term outcomes have not been well investigated for patients with 5-FU-induced coronary vasospasm. However, many case reports show improvements in left ventricular function between 8 and 15 days after discontinuation of 5-FU.7,10 Although this would be a valuable topic for further research, the rarity of this phenomenon creates limitations.
Conclusions
5-FU is a first-line chemotherapy for gastrointestinal cancers that is generally well tolerated but may be associated with potentially life-threatening cardiotoxic effects, of which coronary artery vasospasm is the most common. Coronary artery vasospasm presents with anginal chest pain and ST elevations on ECG that can be indistinguishable from acute coronary syndrome. Diagnosis requires cardiac catheterization, which will reveal patent coronary arteries. Infusional administration of 5-FU may be more likely to produce late cardiotoxic effects and a longer period of persistent symptoms, necessitating close monitoring for days or even weeks from last administration of 5-FU. Coronary artery vasospasm should be treated with anti-anginal medications, though varying degrees of effectiveness can be seen; clinicians should remain vigilant for recurrent episodes of chest pain despite treatment.
1. Wacker A, Lersch C, Scherpinski U, Reindl L, Seyfarth M. High incidence of angina pectoris in patients treated with 5-fluorouracil. A planned surveillance study with 102 patients. Oncology. 2003;65(2):108-112. doi:10.1159/000072334
2. World Health Organization Model List of Essential Medicines, 21st List, 2019. Accessed April 14, 2021. https://apps.who.int/iris/rest/bitstreams/1237479/retrieve
3. Jensen SA, Sørensen JB. Risk factors and prevention of cardiotoxicity induced by 5-fluorouracil or capecitabine. Cancer Chemother Pharmacol. 2006;58(4):487-493. doi:10.1007/s00280-005-0178-1
4. Shoemaker LK, Arora U, Rocha Lima CM. 5-fluorouracil-induced coronary vasospasm. Cancer Control. 2004;11(1):46-49. doi:10.1177/107327480401100207
5. Luwaert RJ, Descamps O, Majois F, Chaudron JM, Beauduin M. Coronary artery spasm induced by 5-fluorouracil. Eur Heart J. 1991;12(3):468-470. doi:10.1093/oxfordjournals.eurheartj.a059919
6. Saif MW, Shah MM, Shah AR. Fluoropyrimidine-associated cardiotoxicity: revisited. Expert Opin Drug Saf. 2009;8(2):191-202. doi:10.1517/14740330902733961
7. Patel B, Kloner RA, Ensley J, Al-Sarraf M, Kish J, Wynne J. 5-Fluorouracil cardiotoxicity: left ventricular dysfunction and effect of coronary vasodilators. Am J Med Sci. 1987;294(4):238-243. doi:10.1097/00000441-198710000-00004
8. Sara JD, Kaur J, Khodadadi R, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol. 2018;10:1758835918780140. Published 2018 Jun 18. doi:10.1177/1758835918780140
9. Hansen RM, Ryan L, Anderson T, et al. Phase III study of bolus versus infusion fluorouracil with or without cisplatin in advanced colorectal cancer. J Natl Cancer Inst. 1996;88(10):668-674. doi:10.1093/jnci/88.10.668
10. Kim SM, Kwak CH, Lee B, et al. A case of severe coronary spasm associated with 5-fluorouracil chemotherapy. Korean J Intern Med. 2012;27(3):342-345. doi:10.3904/kjim.2012.27.3.342
11. Swarup S, Patibandla S, Grossman SA. Coronary Artery Vasospasm. StatPearls. Treasure Island (FL): StatPearls Publishing LLC.; 2021.
12. Beijk MA, Vlastra WV, Delewi R, et al. Myocardial infarction with non-obstructive coronary arteries: a focus on vasospastic angina. Neth Heart J. 2019;27(5):237-245. doi:10.1007/s12471-019-1232-7
13. Giza DE, Boccalandro F, Lopez-Mattei J, et al. Ischemic heart disease: special considerations in cardio-oncology. Curr Treat Options Cardiovasc Med. 2017;19(5):37. doi:10.1007/s11936-017-0535-5
14. Meydan N, Kundak I, Yavuzsen T, et al. Cardiotoxicity of de Gramont’s regimen: incidence, clinical characteristics and long-term follow-up. Jpn J Clin Oncol. 2005;35(5):265-270. doi:10.1093/jjco/hyi071
15. Senkus E, Jassem J. Cardiovascular effects of systemic cancer treatment. Cancer Treat Rev. 2011;37(4):300-311. doi:10.1016/j.ctrv.2010.11.001
16. Rezkalla S, Kloner RA, Ensley J, et al. Continuous ambulatory ECG monitoring during fluorouracil therapy: a prospective study. J Clin Oncol. 1989;7(4):509-514. doi:10.1200/JCO.1989.7.4.509
17. Akpek G, Hartshorn KL. Failure of oral nitrate and calcium channel blocker therapy to prevent 5-fluorouracil-related myocardial ischemia: a case report. Cancer Chemother Pharmacol. 1999;43(2):157-161. doi:10.1007/s002800050877
1. Wacker A, Lersch C, Scherpinski U, Reindl L, Seyfarth M. High incidence of angina pectoris in patients treated with 5-fluorouracil. A planned surveillance study with 102 patients. Oncology. 2003;65(2):108-112. doi:10.1159/000072334
2. World Health Organization Model List of Essential Medicines, 21st List, 2019. Accessed April 14, 2021. https://apps.who.int/iris/rest/bitstreams/1237479/retrieve
3. Jensen SA, Sørensen JB. Risk factors and prevention of cardiotoxicity induced by 5-fluorouracil or capecitabine. Cancer Chemother Pharmacol. 2006;58(4):487-493. doi:10.1007/s00280-005-0178-1
4. Shoemaker LK, Arora U, Rocha Lima CM. 5-fluorouracil-induced coronary vasospasm. Cancer Control. 2004;11(1):46-49. doi:10.1177/107327480401100207
5. Luwaert RJ, Descamps O, Majois F, Chaudron JM, Beauduin M. Coronary artery spasm induced by 5-fluorouracil. Eur Heart J. 1991;12(3):468-470. doi:10.1093/oxfordjournals.eurheartj.a059919
6. Saif MW, Shah MM, Shah AR. Fluoropyrimidine-associated cardiotoxicity: revisited. Expert Opin Drug Saf. 2009;8(2):191-202. doi:10.1517/14740330902733961
7. Patel B, Kloner RA, Ensley J, Al-Sarraf M, Kish J, Wynne J. 5-Fluorouracil cardiotoxicity: left ventricular dysfunction and effect of coronary vasodilators. Am J Med Sci. 1987;294(4):238-243. doi:10.1097/00000441-198710000-00004
8. Sara JD, Kaur J, Khodadadi R, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol. 2018;10:1758835918780140. Published 2018 Jun 18. doi:10.1177/1758835918780140
9. Hansen RM, Ryan L, Anderson T, et al. Phase III study of bolus versus infusion fluorouracil with or without cisplatin in advanced colorectal cancer. J Natl Cancer Inst. 1996;88(10):668-674. doi:10.1093/jnci/88.10.668
10. Kim SM, Kwak CH, Lee B, et al. A case of severe coronary spasm associated with 5-fluorouracil chemotherapy. Korean J Intern Med. 2012;27(3):342-345. doi:10.3904/kjim.2012.27.3.342
11. Swarup S, Patibandla S, Grossman SA. Coronary Artery Vasospasm. StatPearls. Treasure Island (FL): StatPearls Publishing LLC.; 2021.
12. Beijk MA, Vlastra WV, Delewi R, et al. Myocardial infarction with non-obstructive coronary arteries: a focus on vasospastic angina. Neth Heart J. 2019;27(5):237-245. doi:10.1007/s12471-019-1232-7
13. Giza DE, Boccalandro F, Lopez-Mattei J, et al. Ischemic heart disease: special considerations in cardio-oncology. Curr Treat Options Cardiovasc Med. 2017;19(5):37. doi:10.1007/s11936-017-0535-5
14. Meydan N, Kundak I, Yavuzsen T, et al. Cardiotoxicity of de Gramont’s regimen: incidence, clinical characteristics and long-term follow-up. Jpn J Clin Oncol. 2005;35(5):265-270. doi:10.1093/jjco/hyi071
15. Senkus E, Jassem J. Cardiovascular effects of systemic cancer treatment. Cancer Treat Rev. 2011;37(4):300-311. doi:10.1016/j.ctrv.2010.11.001
16. Rezkalla S, Kloner RA, Ensley J, et al. Continuous ambulatory ECG monitoring during fluorouracil therapy: a prospective study. J Clin Oncol. 1989;7(4):509-514. doi:10.1200/JCO.1989.7.4.509
17. Akpek G, Hartshorn KL. Failure of oral nitrate and calcium channel blocker therapy to prevent 5-fluorouracil-related myocardial ischemia: a case report. Cancer Chemother Pharmacol. 1999;43(2):157-161. doi:10.1007/s002800050877
Beneath the Surface: Massive Retroperitoneal Liposarcoma Masquerading as Meralgia Paresthetica
In patients presenting with focal neurologic findings involving the lower extremities, a thorough abdominal examination should be considered an integral part of the full neurologic work up.
Meralgia paresthetica (MP) is a sensory mononeuropathy of the lateral femoral cutaneous nerve (LFCN), clinically characterized by numbness, pain, and paresthesias involving the anterolateral aspect of the thigh. Estimates of MP incidence are derived largely from observational studies and reported to be about 3.2 to 4.3 cases per 10,000 patient-years.1,2 Although typically arising during midlife and especially in the context of comorbid obesity, diabetes mellitus (DM), and excessive alcohol consumption, MP may occur at any age, and bears a slight predilection for males.2-4
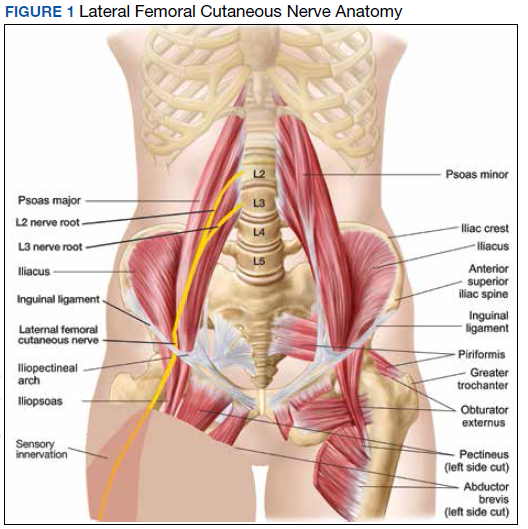
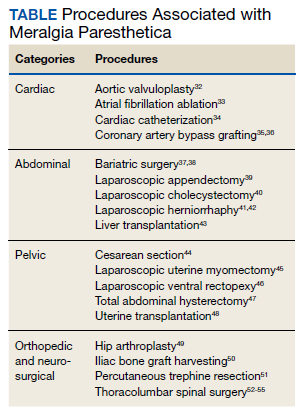
MP may be divided etiologically into iatrogenic and spontaneous subtypes.5 Iatrogenic cases generally are attributable to nerve injury in the setting of direct or indirect trauma (such as with patient malpositioning) arising in the context of multiple forms of procedural or surgical intervention (Table). Spontaneous MP is primarily thought to occur as a result of LFCN compression at the level of the inguinal ligament, wherein internal or external pressures may promote LFCN entrapment and resultant functional disruption (Figure 1).6,7
External forces, such as tight garments, wallets, or even elements of modern body armor, have been reported to provoke MP.8-11 Alternatively, states of increased intraabdominal pressure, such as obesity, ascites, and pregnancy may predispose to LFCN compression.2,12,13 Less commonly, lumbar radiculopathy, pelvic masses, and several forms of retroperitoneal pathology may present with clinical symptomatology indistinguishable from MP.14-17 Importantly, many of these represent must-not-miss diagnoses, and may be suggested via a focused history and physical examination.
Here, we present a case of MP secondary to a massive retroperitoneal sarcoma, ultimately drawing renewed attention to the known association of MP and retroperitoneal pathology, and therein highlighting the utility of a dedicated review of systems to identify red-flag features in patients who present with MP and a thorough abdominal examination in all patients presenting with focal neurologic deficits involving the lower extremities.
Case Presentation
A male Vietnam War veteran aged 69 years presented to a primary care clinic at West Roxbury Veterans Affairs Medical Center (WRVAMC) in Massachusetts with progressive right lower extremity numbness. Three months prior to this visit, he was evaluated in an urgent care clinic at WRVAMC for 6 months of numbness and increasingly painful nocturnal paresthesias involving the same extremity. A targeted physical examination at that visit revealed an obese male wearing tight suspenders, as well as focally diminished sensation to light touch involving the anterolateral aspect of the thigh, extending from just below the right hip to above the knee. Sensation in the medial thigh was spared. Strength and reflexes were normal in the bilateral lower extremities. An abdominal examination was not performed. He received a diagnosis of MP and counseled regarding weight loss, glycemic control, garment optimization, and conservative analgesia with as-needed nonsteroidal anti-inflammatory drugs. He was instructed to follow-up closely with his primary care physician for further monitoring.
During the current visit, the patient reported 2 atraumatic falls the prior 2 months, attributed to escalating right leg weakness. The patient reported that ascending stairs had become difficult, and he was unable to cross his right leg over his left while in a seated position. The territory of numbness expanded to his front and inner thigh. Although previously he was able to hike 4 miles, he now was unable to walk more than half of a mile without developing shortness of breath. He reported frequent urination without hematuria and a recent weight gain of 8 pounds despite early satiety.
His medical history included hypertension, hypercholesterolemia, truncal obesity, noninsulin dependent DM, coronary artery disease, atrial flutter, transient ischemic attack, and benign positional paroxysmal vertigo. He was exposed to Agent Orange during his service in Vietnam. Family history was notable for breast cancer (mother), lung cancer (father), and an unspecified form of lymphoma (brother). He had smoked approximately 2 packs of cigarettes daily for 15 years but quit 38 years prior. He reported consuming on average 3 alcohol-containing drinks per week and no illicit drug use. He was adherent with all medications, including furosemide 40 mg daily, losartan 25 mg daily, metoprolol succinate 50 mg daily, atorvastatin 80 mg daily, metformin 500 mg twice daily, and rivaroxaban 20 mg daily with dinner.
His vital signs included a blood pressure of 123/58 mmHg, a pulse of 74 beats per minute, a respiratory rate of 16 breaths per minute, and an oxygen saturation of 94% on ambient air. His temperature was recorded at 96.7°F, and his weight was 234 pounds with a body mass index (BMI) of 34. He was well groomed and in no acute distress. His cardiopulmonary examination was normal. Carotid, radial, and bilateral dorsalis pedis pulsations were 2+ bilaterally, and no jugular venous distension was observed at 30°. The abdomen was protuberant. Nonshifting dullness to percussion and firmness to palpation was observed throughout right upper and lower quadrants, with hyperactive bowel sounds primarily localized to the left upper and lower quadrants.
Neurologic examination revealed symmetric facies with normal phonation and diction. He was spontaneously moving all extremities, and his gait was normal. Sensation to light touch was severely diminished throughout the anterolateral and medial thigh, extending to the level of the knee, and otherwise reduced in a stocking-type pattern over the bilateral feet and toes. His right hip flexion, adduction, as well as internal and external rotation were focally diminished to 4- out of 5. Right knee extension was 4+ out of 5. Strength was otherwise 5 out of 5. The patient exhibited asymmetric Patellar reflexes—absent on the right and 2+ on the left. Achilles reflexes were absent bilaterally. Straight-leg raise test was negative bilaterally and did not clearly exacerbate his right leg numbness or paresthesias. There were no notable fasciculations. There was 2+ bilateral lower extremity pitting edema appreciated to the level of the midshin (right greater than left), without palpable cords or new skin lesions.
Upon referral to the neurology service, the patient underwent electromyography, which revealed complex repetitive discharges in the right tibialis anterior and pattern of reduced recruitment upon activation of the right vastus medialis, collectively suggestive of an L3-4 plexopathy. The patient was admitted for expedited workup.
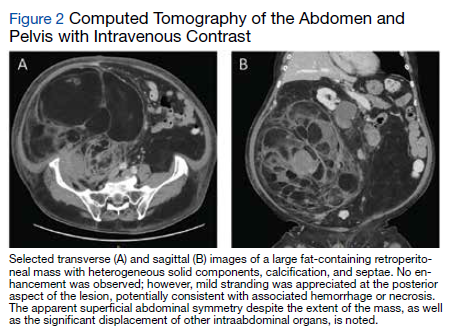
A complete blood count and metabolic panel that were taken in the emergency department were normal, save for a serum bicarbonate of 30 mEq/L. His hemoglobin A1c was 6.6%. Computed tomography (CT) of the abdomen and pelvis with IV contrast was obtained, and notable for a 30 cm fat-containing right-sided retroperitoneal mass with associated solid nodular components and calcification (Figure 2). No enhancement of the lesion was observed. There was significant associated mass effect, with superior displacement of the liver and right hemidiaphragm, as well as superomedial deflection of the right kidney, inferior vena cava, and other intraabdominal organs. Subsequent imaging with a CT of the chest, as well as magnetic resonance imaging of the brain, were without evidence of metastatic disease.
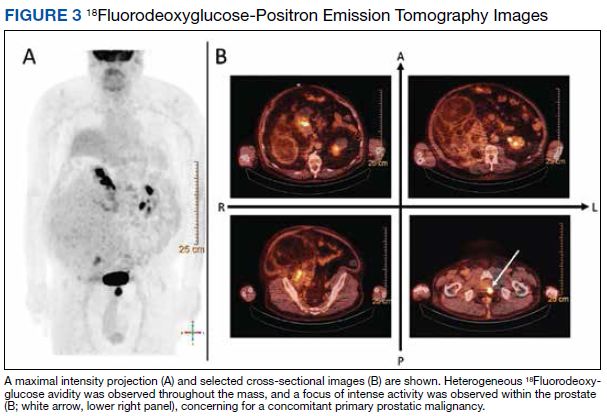
18Fluorodeoxyglucose-positron emission tomography (FDG-PET) was performed and demonstrated heterogeneous FDG avidity throughout the mass (SUVmax 5.9), as well as poor delineation of the boundary of the right psoas major, consistent with muscular invasion (Figure 3). The FDG-PET also revealed intense tracer uptake within the left prostate (SUVmax 26), concerning for a concomitant prostate malignancy.
To facilitate tissue diagnosis, the patient underwent a CT-guided biopsy of the retroperitoneal mass. Subsequent histopathologic analysis revealed a primarily well-differentiated spindle cell lesion with occasional adipocytic atypia, and a superimposed hypercellular element characterized by the presence of pleomorphic high-grade spindled cells. The neoplastic spindle cells were MDM2-positive by both immunohistochemistry and fluorescence in situ hybridization (FISH), and negative for pancytokeratin, smooth muscle myosin, and S100. The findings were collectively consistent with a dedifferentiated liposarcoma (DDLPS).
Given the focus of FDG avidity observed on the PET, the patient underwent a transrectal ultrasound-guided biopsy of the prostate, which yielded diagnosis of a concomitant high-risk (Gleason 4+4) prostate adenocarcinoma. A bone scan did not reveal evidence of osseous metastatic disease.
Outcome
The patient was treated with external beam radiotherapy (EBRT) delivered simultaneously to both the prostate and high-risk retroperitoneal margins of the DDLPS, as well as concurrent androgen deprivation therapy. Five months after completed radiotherapy, resection of the DDLPS was attempted. However, palliative tumor debulking was instead performed due to extensive locoregional invasion with involvement of the posterior peritoneum and ipsilateral quadratus, iliopsoas, and psoas muscles, as well as the adjacent lumbar nerve roots.
At present, the patient is undergoing surveillance imaging every 3 months to reevaluate his underlying disease burden, which has thus far been radiographically stable. Current management at the primary care level is focused on preserving quality of life, particularly maintaining mobility and functional independence.
Discussion
Although generally a benign entrapment neuropathy, MP bears well-established associations with multiple forms of must-not-miss pathology. Here, we present the case of a veteran in whom MP was the index presentation of a massive retroperitoneal liposarcoma, stressing the importance of a thorough history and physical examination in all patients presenting with MP. The case presented herein highlights many of the red-flag signs and symptoms that primary care physicians might encounter in patients with retroperitoneal pathology, including MP and MP-like syndromes (Figure 4).
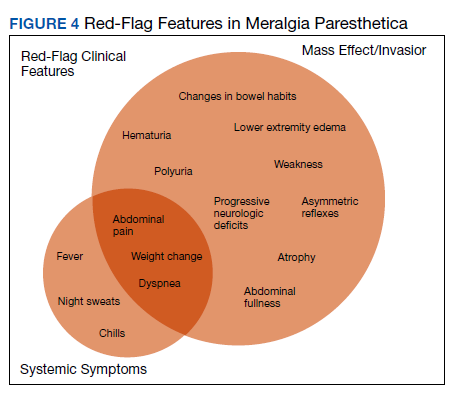
In this case, the pretest probability of a spontaneous and uncomplicated MP was high given the patient’s sex, age, body habitus, and DM; however, there important atypia that emerged as the case evolved, including: (1) the progressive course; (2) proximal right lower extremity weakness; (3) asymmetric patellar reflexes; and (4) numerous clinical stigmata of intraabdominal mass effect. The patient exhibited abnormalities on abdominal examination that suggested the presence of an underlying intraabdominal mass, providing key diagnostic insight into this case. Given the slowly progressive nature of liposarcomas, we feel the abnormalities appreciated on abdominal examination were likely apparent during the initial presentation.18
There are numerous cognitive biases that may explain why an abdominal examination was not prioritized during the initial presentation. Namely, the patient’s numerous risk factors for spontaneous MP, as detailed above, may have contributed to framing bias that limited consideration of alternative diagnoses. In addition, the patient’s physical examination likely contributed to search satisfaction, whereby alternative diagnoses were not further entertained after discovery of findings consistent with spontaneous MP.19 Finally, it remains conceivable that an abdominal examination was not prioritized as it is often perceived as being distinct from, rather than an integral part of, the neurologic examination.20 Given that numerous neurologic disorders may present with abdominal pathology, we feel a thorough abdominal examination should be considered part of the full neurologic examination, especially in cases presenting with focal neurologic findings involving the lower extremities.21
Collectively, this case alludes to the importance of close clinical follow-up, as well as adequate anticipatory patient guidance in cases of suspected MP. In most patients, the clinical course of spontaneous MP is benign and favorable, with up to 85% of patients experiencing resolution within 4 to 6 months of the initial presentation.22 Common conservative measures include weight loss, garment optimization, and nonsteroidal anti-inflammatory drugs as needed for analgesia. In refractory cases, procedural interventions such as with neurolysis or resection of the lateral femoral cutaneous nerve, may be required after the ruling out of alternative diagnoses.23,24
Importantly, in even prolonged and resistant cases of MP, patient discomfort remains localized to the territory of the LFCN. Additional lower motor neuron signs, such as an expanding territory of sensory involvement, muscle weakness, or diminished reflexes, should prompt additional testing for alternative diagnoses. In addition, clinical findings concerning for intraabdominal mass effect, many of which were observed in this case, should lead to further evaluation and expeditious cross-sectional imaging. Although this patient’s early satiety, polyuria, bilateral lower extremity edema, weight gain, and lumbar plexopathy each may be explained by direct compression, invasion, or displacement, his report of progressive exertional dyspnea merits further discussion.
Exertional dyspnea is an uncommon complication of soft tissue sarcoma, reported almost exclusively in cases with cardiac, mediastinal, or other thoracic involvement.25-28 In this case, there was no evidence of thoracic involvement, either through direct extension or metastasis. Instead, the patient’s exertional dyspnea may have been attributable to increased intraabdominal pressure leading to compromised diaphragm excursion and reduced pulmonary reserve. In addition, the radiographic findings also raise the possibility of a potential contribution from preload failure due to IVC compression. Overall, dyspnea is a concerning feature that may suggest advanced disease.
Despite the value of a thorough history and physical examination in patients with MP, major clinical guidelines from neurologic, neurosurgical, and orthopedic organizations do not formally address MP evaluation and management. Further, proposed clinical practice algorithms are inconsistent in their recommendations regarding the identification of red-flag features and ruling out of alternative diagnoses.22,29,30 To supplement the abdominal examination, it would be reasonable to perform a pelvic compression test (PCT) in patients presenting with suspected MP. The PCT is a highly sensitive and specific provocative maneuver shown to enable reliable differentiation between MP and lumbar radiculopathy, and is performed by placing downward force on the anterior superior iliac spine of the affected extremity for 45 seconds with the patient in the lateral recumbent position.31 As this maneuver is intended to force relaxation of the inguinal ligament, thereby relieving pressure on the LFCN, improvement in the patient’s symptoms with the PCT is consistent with MP.
Conclusions
1. van Slobbe AM, Bohnen AM, Bernsen RM, Koes BW, Bierma-Zeinstra SM. Incidence rates and determinants in meralgia paresthetica in general practice. J Neurol. 2004;251(3):294-297. doi:10.1007/s00415-004-0310-x
2. Parisi TJ, Mandrekar J, Dyck PJ, Klein CJ. Meralgia paresthetica: relation to obesity, advanced age, and diabetes mellitus. Neurology. 2011;77(16):1538-1542. doi:10.1212/WNL.0b013e318233b356
3. Ecker AD. Diagnosis of meralgia paresthetica. JAMA. 1985;253(7):976.
4. Massey EW, Pellock JM. Meralgia paraesthetica in a child. J Pediatr. 1978;93(2):325-326. doi:10.1016/s0022-3476(78)80566-6
5. Harney D, Patijn J. Meralgia paresthetica: diagnosis and management strategies. Pain Med. 2007;8(8):669-677. doi:10.1111/j.1526-4637.2006.00227.x
6. Berini SE, Spinner RJ, Jentoft ME, et al. Chronic meralgia paresthetica and neurectomy: a clinical pathologic study. Neurology. 2014;82(17):1551-1555. doi:10.1212/WNL.0000000000000367
7. Payne RA, Harbaugh K, Specht CS, Rizk E. Correlation of histopathology and clinical symptoms in meralgia paresthetica. Cureus. 2017;9(10):e1789. Published 2017 Oct 20. doi:10.7759/cureus.1789
8. Boyce JR. Meralgia paresthetica and tight trousers. JAMA. 1984;251(12):1553.
9. Orton D. Meralgia paresthetica from a wallet. JAMA. 1984;252(24):3368.
10. Fargo MV, Konitzer LN. Meralgia paresthetica due to body armor wear in U.S. soldiers serving in Iraq: a case report and review of the literature. Mil Med. 2007;172(6):663-665. doi:10.7205/milmed.172.6.663
11. Korkmaz N, Ozçakar L. Meralgia paresthetica in a policeman: the belt or the gun. Plast Reconstr Surg. 2004;114(4):1012-1013. doi:10.1097/01.prs.0000138706.86633.01
12. Gooding MS, Evangelista V, Pereira L. Carpal Tunnel Syndrome and Meralgia Paresthetica in Pregnancy. Obstet Gynecol Surv. 2020;75(2):121-126. doi:10.1097/OGX.0000000000000745
13. Pauwels A, Amarenco P, Chazouillères O, Pigot F, Calmus Y, Lévy VG. Une complication rare et méconnue de l’ascite: la méralgie paresthésique [Unusual and unknown complication of ascites: meralgia paresthetica]. Gastroenterol Clin Biol. 1990;14(3):295.
14. Braddom RL. L2 rather than L1 radiculopathy mimics meralgia paresthetica. Muscle Nerve. 2010;42(5):842. doi:10.1002/mus.21826
15. Suber DA, Massey EW. Pelvic mass presenting as meralgia paresthetica. Obstet Gynecol. 1979;53(2):257-258.
16. Flowers RS. Meralgia paresthetica. A clue to retroperitoneal malignant tumor. Am J Surg. 1968;116(1):89-92. doi:10.1016/0002-9610(68)90423-6
17. Yi TI, Yoon TH, Kim JS, Lee GE, Kim BR. Femoral neuropathy and meralgia paresthetica secondary to an iliacus hematoma. Ann Rehabil Med. 2012;36(2):273-277. doi:10.5535/arm.2012.36.2.273
18. Lee ATJ, Thway K, Huang PH, Jones RL. Clinical and molecular spectrum of liposarcoma. J Clin Oncol. 2018;36(2):151-159. doi:10.1200/JCO.2017.74.9598
19. O’Sullivan ED, Schofield SJ. Cognitive bias in clinical medicine. J R Coll Physicians Edinb. 2018;48(3):225-232. doi:10.4997/JRCPE.2018.306
20. Bickley, LS. Bates’ Guide to Physical Examination and History Taking. 12th Edition. Wolters Kluwer Health/Lippincott Williams and Wilkins; 2016.
21. Bhavsar AS, Verma S, Lamba R, Lall CG, Koenigsknecht V, Rajesh A. Abdominal manifestations of neurologic disorders. Radiographics. 2013;33(1):135-153. doi:10.1148/rg.331125097
22. Dureja GP, Gulaya V, Jayalakshmi TS, Mandal P. Management of meralgia paresthetica: a multimodality regimen. Anesth Analg. 1995;80(5):1060-1061. doi:10.1097/00000539-199505000-00043
23. Patijn J, Mekhail N, Hayek S, Lataster A, van Kleef M, Van Zundert J. Meralgia paresthetica. Pain Pract. 2011;11(3):302-308. doi:10.1111/j.1533-2500.2011.00458.x24. Ivins GK. Meralgia paresthetica, the elusive diagnosis: clinical experience with 14 adult patients. Ann Surg. 2000;232(2):281-286. doi:10.1097/00000658-200008000-00019
25. Munin MA, Goerner MS, Raggio I, et al. A rare cause of dyspnea: undifferentiated pleomorphic sarcoma in the left atrium. Cardiol Res. 2017;8(5):241-245. doi:10.14740/cr590w
26. Nguyen A, Awad WI. Cardiac sarcoma arising from malignant transformation of a preexisting atrial myxoma. Ann Thorac Surg. 2016;101(4):1571-1573. doi:10.1016/j.athoracsur.2015.05.129
27. Jiang S, Li J, Zeng Q, Liang J. Pulmonary artery intimal sarcoma misdiagnosed as pulmonary embolism: a case report. Oncol Lett. 2017;13(4):2713-2716. doi:10.3892/ol.2017.5775
28. Cojocaru A, Oliveira PJ, Pellecchia C. A pleural presentation of a rare soft tissue sarcoma. Am J Resp Crit Care Med. 2012;185:A5201. doi:10.1164/ajrccm-conference.2012.185.1_MeetingAbstracts.A5201
29. Grossman MG, Ducey SA, Nadler SS, Levy AS. Meralgia paresthetica: diagnosis and treatment. J Am Acad Orthop Surg. 2001;9(5):336-344. doi:10.5435/00124635-200109000-00007
30. Cheatham SW, Kolber MJ, Salamh PA. Meralgia paresthetica: a review of the literature. Int J Sports Phys Ther. 2013;8(6):883-893.
31. Nouraei SA, Anand B, Spink G, O’Neill KS. A novel approach to the diagnosis and management of meralgia paresthetica. Neurosurgery. 2007;60(4):696-700. doi:10.1227/01.NEU.0000255392.69914.F7
32. Antunes PE, Antunes MJ. Meralgia paresthetica after aortic valve surgery. J Heart Valve Dis. 1997;6(6):589-590.
33. Reddy YM, Singh D, Chikkam V, et al. Postprocedural neuropathy after atrial fibrillation ablation. J Interv Card Electrophysiol. 2013;36(3):279-285. doi:10.1007/s10840-012-9724-z
34. Butler R, Webster MW. Meralgia paresthetica: an unusual complication of cardiac catheterization via the femoral artery. Catheter Cardiovasc Interv. 2002;56(1):69-71. doi:10.1002/ccd.10149
35. Jellish WS, Oftadeh M. Peripheral nerve injury in cardiac surgery. J Cardiothorac Vasc Anesth. 2018;32(1):495-511. doi:10.1053/j.jvca.2017.08.030
36. Parsonnet V, Karasakalides A, Gielchinsky I, Hochberg M, Hussain SM. Meralgia paresthetica after coronary bypass surgery. J Thorac Cardiovasc Surg. 1991;101(2):219-221.
37. Macgregor AM, Thoburn EK. Meralgia paresthetica following bariatric surgery. Obes Surg. 1999;9(4):364-368. doi:10.1381/096089299765552945
38. Grace DM. Meralgia paresthetica after gastroplasty for morbid obesity. Can J Surg. 1987;30(1):64-65.
39. Polidori L, Magarelli M, Tramutoli R. Meralgia paresthetica as a complication of laparoscopic appendectomy. Surg Endosc. 2003;17(5):832. doi:10.1007/s00464-002-4279-1
40. Yamout B, Tayyim A, Farhat W. Meralgia paresthetica as a complication of laparoscopic cholecystectomy. Clin Neurol Neurosurg. 1994;96(2):143-144. doi:10.1016/0303-8467(94)90048-5
41. Broin EO, Horner C, Mealy K, et al. Meralgia paraesthetica following laparoscopic inguinal hernia repair. an anatomical analysis. Surg Endosc. 1995;9(1):76-78. doi:10.1007/BF00187893
42. Eubanks S, Newman L 3rd, Goehring L, et al. Meralgia paresthetica: a complication of laparoscopic herniorrhaphy. Surg Laparosc Endosc. 1993;3(5):381-385.
43. Atamaz F, Hepgüler S, Karasu Z, Kilic M. Meralgia paresthetica after liver transplantation: a case report. Transplant Proc. 2005;37(10):4424-4425. doi:10.1016/j.transproceed.2005.11.047
44. Chung KH, Lee JY, Ko TK, et al. Meralgia paresthetica affecting parturient women who underwent cesarean section -a case report-. Korean J Anesthesiol. 2010;59 Suppl(Suppl):S86-S89. doi:10.4097/kjae.2010.59.S.S86
45. Hutchins FL Jr, Huggins J, Delaney ML. Laparoscopic myomectomy-an unusual cause of meralgia paresthetica. J Am Assoc Gynecol Laparosc. 1998;5(3):309-311. doi:10.1016/s1074-3804(98)80039-x
46. Jones CD, Guiot L, Portelli M, Bullen T, Skaife P. Two interesting cases of meralgia paraesthetica. Pain Physician. 2017;20(6):E987-E989.
47. Peters G, Larner AJ. Meralgia paresthetica following gynecologic and obstetric surgery. Int J Gynaecol Obstet. 2006;95(1):42-43. doi:10.1016/j.ijgo.2006.05.025
48. Kvarnström N, Järvholm S, Johannesson L, Dahm-Kähler P, Olausson M, Brännström M. Live donors of the initial observational study of uterus transplantation-psychological and medical follow-up until 1 year after surgery in the 9 cases. Transplantation. 2017;101(3):664-670. doi:10.1097/TP.0000000000001567
49. Goulding K, Beaulé PE, Kim PR, Fazekas A. Incidence of lateral femoral cutaneous nerve neuropraxia after anterior approach hip arthroplasty. Clin Orthop Relat Res. 2010;468(9):2397-2404. doi:10.1007/s11999-010-1406-5
50. Yamamoto T, Nagira K, Kurosaka M. Meralgia paresthetica occurring 40 years after iliac bone graft harvesting: case report. Neurosurgery. 2001;49(6):1455-1457. doi:10.1097/00006123-200112000-00028
51. Roqueplan F, Porcher R, Hamzé B, et al. Long-term results of percutaneous resection and interstitial laser ablation of osteoid osteomas. Eur Radiol. 2010;20(1):209-217. doi:10.1007/s00330-009-1537-9
52. Gupta A, Muzumdar D, Ramani PS. Meralgia paraesthetica following lumbar spine surgery: a study in 110 consecutive surgically treated cases. Neurol India. 2004;52(1):64-66.
53. Yang SH, Wu CC, Chen PQ. Postoperative meralgia paresthetica after posterior spine surgery: incidence, risk factors, and clinical outcomes. Spine (Phila Pa 1976). 2005;30(18):E547-E550. doi:10.1097/01.brs.0000178821.14102.9d
54. Tejwani SG, Scaduto AA, Bowen RE. Transient meralgia paresthetica after pediatric posterior spine fusion. J Pediatr Orthop. 2006;26(4):530-533. doi:10.1097/01.bpo.0000217721.95480.9e
55. Peker S, Ay B, Sun I, Ozgen S, Pamir M. Meralgia paraesthetica: complications of prone position during lumbar disc surgery. Internet J Anesthesiol. 2003;8(1):24-29.
In patients presenting with focal neurologic findings involving the lower extremities, a thorough abdominal examination should be considered an integral part of the full neurologic work up.
In patients presenting with focal neurologic findings involving the lower extremities, a thorough abdominal examination should be considered an integral part of the full neurologic work up.
Meralgia paresthetica (MP) is a sensory mononeuropathy of the lateral femoral cutaneous nerve (LFCN), clinically characterized by numbness, pain, and paresthesias involving the anterolateral aspect of the thigh. Estimates of MP incidence are derived largely from observational studies and reported to be about 3.2 to 4.3 cases per 10,000 patient-years.1,2 Although typically arising during midlife and especially in the context of comorbid obesity, diabetes mellitus (DM), and excessive alcohol consumption, MP may occur at any age, and bears a slight predilection for males.2-4
MP may be divided etiologically into iatrogenic and spontaneous subtypes.5 Iatrogenic cases generally are attributable to nerve injury in the setting of direct or indirect trauma (such as with patient malpositioning) arising in the context of multiple forms of procedural or surgical intervention (Table). Spontaneous MP is primarily thought to occur as a result of LFCN compression at the level of the inguinal ligament, wherein internal or external pressures may promote LFCN entrapment and resultant functional disruption (Figure 1).6,7
External forces, such as tight garments, wallets, or even elements of modern body armor, have been reported to provoke MP.8-11 Alternatively, states of increased intraabdominal pressure, such as obesity, ascites, and pregnancy may predispose to LFCN compression.2,12,13 Less commonly, lumbar radiculopathy, pelvic masses, and several forms of retroperitoneal pathology may present with clinical symptomatology indistinguishable from MP.14-17 Importantly, many of these represent must-not-miss diagnoses, and may be suggested via a focused history and physical examination.
Here, we present a case of MP secondary to a massive retroperitoneal sarcoma, ultimately drawing renewed attention to the known association of MP and retroperitoneal pathology, and therein highlighting the utility of a dedicated review of systems to identify red-flag features in patients who present with MP and a thorough abdominal examination in all patients presenting with focal neurologic deficits involving the lower extremities.
Case Presentation
A male Vietnam War veteran aged 69 years presented to a primary care clinic at West Roxbury Veterans Affairs Medical Center (WRVAMC) in Massachusetts with progressive right lower extremity numbness. Three months prior to this visit, he was evaluated in an urgent care clinic at WRVAMC for 6 months of numbness and increasingly painful nocturnal paresthesias involving the same extremity. A targeted physical examination at that visit revealed an obese male wearing tight suspenders, as well as focally diminished sensation to light touch involving the anterolateral aspect of the thigh, extending from just below the right hip to above the knee. Sensation in the medial thigh was spared. Strength and reflexes were normal in the bilateral lower extremities. An abdominal examination was not performed. He received a diagnosis of MP and counseled regarding weight loss, glycemic control, garment optimization, and conservative analgesia with as-needed nonsteroidal anti-inflammatory drugs. He was instructed to follow-up closely with his primary care physician for further monitoring.
During the current visit, the patient reported 2 atraumatic falls the prior 2 months, attributed to escalating right leg weakness. The patient reported that ascending stairs had become difficult, and he was unable to cross his right leg over his left while in a seated position. The territory of numbness expanded to his front and inner thigh. Although previously he was able to hike 4 miles, he now was unable to walk more than half of a mile without developing shortness of breath. He reported frequent urination without hematuria and a recent weight gain of 8 pounds despite early satiety.
His medical history included hypertension, hypercholesterolemia, truncal obesity, noninsulin dependent DM, coronary artery disease, atrial flutter, transient ischemic attack, and benign positional paroxysmal vertigo. He was exposed to Agent Orange during his service in Vietnam. Family history was notable for breast cancer (mother), lung cancer (father), and an unspecified form of lymphoma (brother). He had smoked approximately 2 packs of cigarettes daily for 15 years but quit 38 years prior. He reported consuming on average 3 alcohol-containing drinks per week and no illicit drug use. He was adherent with all medications, including furosemide 40 mg daily, losartan 25 mg daily, metoprolol succinate 50 mg daily, atorvastatin 80 mg daily, metformin 500 mg twice daily, and rivaroxaban 20 mg daily with dinner.
His vital signs included a blood pressure of 123/58 mmHg, a pulse of 74 beats per minute, a respiratory rate of 16 breaths per minute, and an oxygen saturation of 94% on ambient air. His temperature was recorded at 96.7°F, and his weight was 234 pounds with a body mass index (BMI) of 34. He was well groomed and in no acute distress. His cardiopulmonary examination was normal. Carotid, radial, and bilateral dorsalis pedis pulsations were 2+ bilaterally, and no jugular venous distension was observed at 30°. The abdomen was protuberant. Nonshifting dullness to percussion and firmness to palpation was observed throughout right upper and lower quadrants, with hyperactive bowel sounds primarily localized to the left upper and lower quadrants.
Neurologic examination revealed symmetric facies with normal phonation and diction. He was spontaneously moving all extremities, and his gait was normal. Sensation to light touch was severely diminished throughout the anterolateral and medial thigh, extending to the level of the knee, and otherwise reduced in a stocking-type pattern over the bilateral feet and toes. His right hip flexion, adduction, as well as internal and external rotation were focally diminished to 4- out of 5. Right knee extension was 4+ out of 5. Strength was otherwise 5 out of 5. The patient exhibited asymmetric Patellar reflexes—absent on the right and 2+ on the left. Achilles reflexes were absent bilaterally. Straight-leg raise test was negative bilaterally and did not clearly exacerbate his right leg numbness or paresthesias. There were no notable fasciculations. There was 2+ bilateral lower extremity pitting edema appreciated to the level of the midshin (right greater than left), without palpable cords or new skin lesions.
Upon referral to the neurology service, the patient underwent electromyography, which revealed complex repetitive discharges in the right tibialis anterior and pattern of reduced recruitment upon activation of the right vastus medialis, collectively suggestive of an L3-4 plexopathy. The patient was admitted for expedited workup.
A complete blood count and metabolic panel that were taken in the emergency department were normal, save for a serum bicarbonate of 30 mEq/L. His hemoglobin A1c was 6.6%. Computed tomography (CT) of the abdomen and pelvis with IV contrast was obtained, and notable for a 30 cm fat-containing right-sided retroperitoneal mass with associated solid nodular components and calcification (Figure 2). No enhancement of the lesion was observed. There was significant associated mass effect, with superior displacement of the liver and right hemidiaphragm, as well as superomedial deflection of the right kidney, inferior vena cava, and other intraabdominal organs. Subsequent imaging with a CT of the chest, as well as magnetic resonance imaging of the brain, were without evidence of metastatic disease.
18Fluorodeoxyglucose-positron emission tomography (FDG-PET) was performed and demonstrated heterogeneous FDG avidity throughout the mass (SUVmax 5.9), as well as poor delineation of the boundary of the right psoas major, consistent with muscular invasion (Figure 3). The FDG-PET also revealed intense tracer uptake within the left prostate (SUVmax 26), concerning for a concomitant prostate malignancy.
To facilitate tissue diagnosis, the patient underwent a CT-guided biopsy of the retroperitoneal mass. Subsequent histopathologic analysis revealed a primarily well-differentiated spindle cell lesion with occasional adipocytic atypia, and a superimposed hypercellular element characterized by the presence of pleomorphic high-grade spindled cells. The neoplastic spindle cells were MDM2-positive by both immunohistochemistry and fluorescence in situ hybridization (FISH), and negative for pancytokeratin, smooth muscle myosin, and S100. The findings were collectively consistent with a dedifferentiated liposarcoma (DDLPS).
Given the focus of FDG avidity observed on the PET, the patient underwent a transrectal ultrasound-guided biopsy of the prostate, which yielded diagnosis of a concomitant high-risk (Gleason 4+4) prostate adenocarcinoma. A bone scan did not reveal evidence of osseous metastatic disease.
Outcome
The patient was treated with external beam radiotherapy (EBRT) delivered simultaneously to both the prostate and high-risk retroperitoneal margins of the DDLPS, as well as concurrent androgen deprivation therapy. Five months after completed radiotherapy, resection of the DDLPS was attempted. However, palliative tumor debulking was instead performed due to extensive locoregional invasion with involvement of the posterior peritoneum and ipsilateral quadratus, iliopsoas, and psoas muscles, as well as the adjacent lumbar nerve roots.
At present, the patient is undergoing surveillance imaging every 3 months to reevaluate his underlying disease burden, which has thus far been radiographically stable. Current management at the primary care level is focused on preserving quality of life, particularly maintaining mobility and functional independence.
Discussion
Although generally a benign entrapment neuropathy, MP bears well-established associations with multiple forms of must-not-miss pathology. Here, we present the case of a veteran in whom MP was the index presentation of a massive retroperitoneal liposarcoma, stressing the importance of a thorough history and physical examination in all patients presenting with MP. The case presented herein highlights many of the red-flag signs and symptoms that primary care physicians might encounter in patients with retroperitoneal pathology, including MP and MP-like syndromes (Figure 4).
In this case, the pretest probability of a spontaneous and uncomplicated MP was high given the patient’s sex, age, body habitus, and DM; however, there important atypia that emerged as the case evolved, including: (1) the progressive course; (2) proximal right lower extremity weakness; (3) asymmetric patellar reflexes; and (4) numerous clinical stigmata of intraabdominal mass effect. The patient exhibited abnormalities on abdominal examination that suggested the presence of an underlying intraabdominal mass, providing key diagnostic insight into this case. Given the slowly progressive nature of liposarcomas, we feel the abnormalities appreciated on abdominal examination were likely apparent during the initial presentation.18
There are numerous cognitive biases that may explain why an abdominal examination was not prioritized during the initial presentation. Namely, the patient’s numerous risk factors for spontaneous MP, as detailed above, may have contributed to framing bias that limited consideration of alternative diagnoses. In addition, the patient’s physical examination likely contributed to search satisfaction, whereby alternative diagnoses were not further entertained after discovery of findings consistent with spontaneous MP.19 Finally, it remains conceivable that an abdominal examination was not prioritized as it is often perceived as being distinct from, rather than an integral part of, the neurologic examination.20 Given that numerous neurologic disorders may present with abdominal pathology, we feel a thorough abdominal examination should be considered part of the full neurologic examination, especially in cases presenting with focal neurologic findings involving the lower extremities.21
Collectively, this case alludes to the importance of close clinical follow-up, as well as adequate anticipatory patient guidance in cases of suspected MP. In most patients, the clinical course of spontaneous MP is benign and favorable, with up to 85% of patients experiencing resolution within 4 to 6 months of the initial presentation.22 Common conservative measures include weight loss, garment optimization, and nonsteroidal anti-inflammatory drugs as needed for analgesia. In refractory cases, procedural interventions such as with neurolysis or resection of the lateral femoral cutaneous nerve, may be required after the ruling out of alternative diagnoses.23,24
Importantly, in even prolonged and resistant cases of MP, patient discomfort remains localized to the territory of the LFCN. Additional lower motor neuron signs, such as an expanding territory of sensory involvement, muscle weakness, or diminished reflexes, should prompt additional testing for alternative diagnoses. In addition, clinical findings concerning for intraabdominal mass effect, many of which were observed in this case, should lead to further evaluation and expeditious cross-sectional imaging. Although this patient’s early satiety, polyuria, bilateral lower extremity edema, weight gain, and lumbar plexopathy each may be explained by direct compression, invasion, or displacement, his report of progressive exertional dyspnea merits further discussion.
Exertional dyspnea is an uncommon complication of soft tissue sarcoma, reported almost exclusively in cases with cardiac, mediastinal, or other thoracic involvement.25-28 In this case, there was no evidence of thoracic involvement, either through direct extension or metastasis. Instead, the patient’s exertional dyspnea may have been attributable to increased intraabdominal pressure leading to compromised diaphragm excursion and reduced pulmonary reserve. In addition, the radiographic findings also raise the possibility of a potential contribution from preload failure due to IVC compression. Overall, dyspnea is a concerning feature that may suggest advanced disease.
Despite the value of a thorough history and physical examination in patients with MP, major clinical guidelines from neurologic, neurosurgical, and orthopedic organizations do not formally address MP evaluation and management. Further, proposed clinical practice algorithms are inconsistent in their recommendations regarding the identification of red-flag features and ruling out of alternative diagnoses.22,29,30 To supplement the abdominal examination, it would be reasonable to perform a pelvic compression test (PCT) in patients presenting with suspected MP. The PCT is a highly sensitive and specific provocative maneuver shown to enable reliable differentiation between MP and lumbar radiculopathy, and is performed by placing downward force on the anterior superior iliac spine of the affected extremity for 45 seconds with the patient in the lateral recumbent position.31 As this maneuver is intended to force relaxation of the inguinal ligament, thereby relieving pressure on the LFCN, improvement in the patient’s symptoms with the PCT is consistent with MP.
Conclusions
Meralgia paresthetica (MP) is a sensory mononeuropathy of the lateral femoral cutaneous nerve (LFCN), clinically characterized by numbness, pain, and paresthesias involving the anterolateral aspect of the thigh. Estimates of MP incidence are derived largely from observational studies and reported to be about 3.2 to 4.3 cases per 10,000 patient-years.1,2 Although typically arising during midlife and especially in the context of comorbid obesity, diabetes mellitus (DM), and excessive alcohol consumption, MP may occur at any age, and bears a slight predilection for males.2-4
MP may be divided etiologically into iatrogenic and spontaneous subtypes.5 Iatrogenic cases generally are attributable to nerve injury in the setting of direct or indirect trauma (such as with patient malpositioning) arising in the context of multiple forms of procedural or surgical intervention (Table). Spontaneous MP is primarily thought to occur as a result of LFCN compression at the level of the inguinal ligament, wherein internal or external pressures may promote LFCN entrapment and resultant functional disruption (Figure 1).6,7
External forces, such as tight garments, wallets, or even elements of modern body armor, have been reported to provoke MP.8-11 Alternatively, states of increased intraabdominal pressure, such as obesity, ascites, and pregnancy may predispose to LFCN compression.2,12,13 Less commonly, lumbar radiculopathy, pelvic masses, and several forms of retroperitoneal pathology may present with clinical symptomatology indistinguishable from MP.14-17 Importantly, many of these represent must-not-miss diagnoses, and may be suggested via a focused history and physical examination.
Here, we present a case of MP secondary to a massive retroperitoneal sarcoma, ultimately drawing renewed attention to the known association of MP and retroperitoneal pathology, and therein highlighting the utility of a dedicated review of systems to identify red-flag features in patients who present with MP and a thorough abdominal examination in all patients presenting with focal neurologic deficits involving the lower extremities.
Case Presentation
A male Vietnam War veteran aged 69 years presented to a primary care clinic at West Roxbury Veterans Affairs Medical Center (WRVAMC) in Massachusetts with progressive right lower extremity numbness. Three months prior to this visit, he was evaluated in an urgent care clinic at WRVAMC for 6 months of numbness and increasingly painful nocturnal paresthesias involving the same extremity. A targeted physical examination at that visit revealed an obese male wearing tight suspenders, as well as focally diminished sensation to light touch involving the anterolateral aspect of the thigh, extending from just below the right hip to above the knee. Sensation in the medial thigh was spared. Strength and reflexes were normal in the bilateral lower extremities. An abdominal examination was not performed. He received a diagnosis of MP and counseled regarding weight loss, glycemic control, garment optimization, and conservative analgesia with as-needed nonsteroidal anti-inflammatory drugs. He was instructed to follow-up closely with his primary care physician for further monitoring.
During the current visit, the patient reported 2 atraumatic falls the prior 2 months, attributed to escalating right leg weakness. The patient reported that ascending stairs had become difficult, and he was unable to cross his right leg over his left while in a seated position. The territory of numbness expanded to his front and inner thigh. Although previously he was able to hike 4 miles, he now was unable to walk more than half of a mile without developing shortness of breath. He reported frequent urination without hematuria and a recent weight gain of 8 pounds despite early satiety.
His medical history included hypertension, hypercholesterolemia, truncal obesity, noninsulin dependent DM, coronary artery disease, atrial flutter, transient ischemic attack, and benign positional paroxysmal vertigo. He was exposed to Agent Orange during his service in Vietnam. Family history was notable for breast cancer (mother), lung cancer (father), and an unspecified form of lymphoma (brother). He had smoked approximately 2 packs of cigarettes daily for 15 years but quit 38 years prior. He reported consuming on average 3 alcohol-containing drinks per week and no illicit drug use. He was adherent with all medications, including furosemide 40 mg daily, losartan 25 mg daily, metoprolol succinate 50 mg daily, atorvastatin 80 mg daily, metformin 500 mg twice daily, and rivaroxaban 20 mg daily with dinner.
His vital signs included a blood pressure of 123/58 mmHg, a pulse of 74 beats per minute, a respiratory rate of 16 breaths per minute, and an oxygen saturation of 94% on ambient air. His temperature was recorded at 96.7°F, and his weight was 234 pounds with a body mass index (BMI) of 34. He was well groomed and in no acute distress. His cardiopulmonary examination was normal. Carotid, radial, and bilateral dorsalis pedis pulsations were 2+ bilaterally, and no jugular venous distension was observed at 30°. The abdomen was protuberant. Nonshifting dullness to percussion and firmness to palpation was observed throughout right upper and lower quadrants, with hyperactive bowel sounds primarily localized to the left upper and lower quadrants.
Neurologic examination revealed symmetric facies with normal phonation and diction. He was spontaneously moving all extremities, and his gait was normal. Sensation to light touch was severely diminished throughout the anterolateral and medial thigh, extending to the level of the knee, and otherwise reduced in a stocking-type pattern over the bilateral feet and toes. His right hip flexion, adduction, as well as internal and external rotation were focally diminished to 4- out of 5. Right knee extension was 4+ out of 5. Strength was otherwise 5 out of 5. The patient exhibited asymmetric Patellar reflexes—absent on the right and 2+ on the left. Achilles reflexes were absent bilaterally. Straight-leg raise test was negative bilaterally and did not clearly exacerbate his right leg numbness or paresthesias. There were no notable fasciculations. There was 2+ bilateral lower extremity pitting edema appreciated to the level of the midshin (right greater than left), without palpable cords or new skin lesions.
Upon referral to the neurology service, the patient underwent electromyography, which revealed complex repetitive discharges in the right tibialis anterior and pattern of reduced recruitment upon activation of the right vastus medialis, collectively suggestive of an L3-4 plexopathy. The patient was admitted for expedited workup.
A complete blood count and metabolic panel that were taken in the emergency department were normal, save for a serum bicarbonate of 30 mEq/L. His hemoglobin A1c was 6.6%. Computed tomography (CT) of the abdomen and pelvis with IV contrast was obtained, and notable for a 30 cm fat-containing right-sided retroperitoneal mass with associated solid nodular components and calcification (Figure 2). No enhancement of the lesion was observed. There was significant associated mass effect, with superior displacement of the liver and right hemidiaphragm, as well as superomedial deflection of the right kidney, inferior vena cava, and other intraabdominal organs. Subsequent imaging with a CT of the chest, as well as magnetic resonance imaging of the brain, were without evidence of metastatic disease.
18Fluorodeoxyglucose-positron emission tomography (FDG-PET) was performed and demonstrated heterogeneous FDG avidity throughout the mass (SUVmax 5.9), as well as poor delineation of the boundary of the right psoas major, consistent with muscular invasion (Figure 3). The FDG-PET also revealed intense tracer uptake within the left prostate (SUVmax 26), concerning for a concomitant prostate malignancy.
To facilitate tissue diagnosis, the patient underwent a CT-guided biopsy of the retroperitoneal mass. Subsequent histopathologic analysis revealed a primarily well-differentiated spindle cell lesion with occasional adipocytic atypia, and a superimposed hypercellular element characterized by the presence of pleomorphic high-grade spindled cells. The neoplastic spindle cells were MDM2-positive by both immunohistochemistry and fluorescence in situ hybridization (FISH), and negative for pancytokeratin, smooth muscle myosin, and S100. The findings were collectively consistent with a dedifferentiated liposarcoma (DDLPS).
Given the focus of FDG avidity observed on the PET, the patient underwent a transrectal ultrasound-guided biopsy of the prostate, which yielded diagnosis of a concomitant high-risk (Gleason 4+4) prostate adenocarcinoma. A bone scan did not reveal evidence of osseous metastatic disease.
Outcome
The patient was treated with external beam radiotherapy (EBRT) delivered simultaneously to both the prostate and high-risk retroperitoneal margins of the DDLPS, as well as concurrent androgen deprivation therapy. Five months after completed radiotherapy, resection of the DDLPS was attempted. However, palliative tumor debulking was instead performed due to extensive locoregional invasion with involvement of the posterior peritoneum and ipsilateral quadratus, iliopsoas, and psoas muscles, as well as the adjacent lumbar nerve roots.
At present, the patient is undergoing surveillance imaging every 3 months to reevaluate his underlying disease burden, which has thus far been radiographically stable. Current management at the primary care level is focused on preserving quality of life, particularly maintaining mobility and functional independence.
Discussion
Although generally a benign entrapment neuropathy, MP bears well-established associations with multiple forms of must-not-miss pathology. Here, we present the case of a veteran in whom MP was the index presentation of a massive retroperitoneal liposarcoma, stressing the importance of a thorough history and physical examination in all patients presenting with MP. The case presented herein highlights many of the red-flag signs and symptoms that primary care physicians might encounter in patients with retroperitoneal pathology, including MP and MP-like syndromes (Figure 4).
In this case, the pretest probability of a spontaneous and uncomplicated MP was high given the patient’s sex, age, body habitus, and DM; however, there important atypia that emerged as the case evolved, including: (1) the progressive course; (2) proximal right lower extremity weakness; (3) asymmetric patellar reflexes; and (4) numerous clinical stigmata of intraabdominal mass effect. The patient exhibited abnormalities on abdominal examination that suggested the presence of an underlying intraabdominal mass, providing key diagnostic insight into this case. Given the slowly progressive nature of liposarcomas, we feel the abnormalities appreciated on abdominal examination were likely apparent during the initial presentation.18
There are numerous cognitive biases that may explain why an abdominal examination was not prioritized during the initial presentation. Namely, the patient’s numerous risk factors for spontaneous MP, as detailed above, may have contributed to framing bias that limited consideration of alternative diagnoses. In addition, the patient’s physical examination likely contributed to search satisfaction, whereby alternative diagnoses were not further entertained after discovery of findings consistent with spontaneous MP.19 Finally, it remains conceivable that an abdominal examination was not prioritized as it is often perceived as being distinct from, rather than an integral part of, the neurologic examination.20 Given that numerous neurologic disorders may present with abdominal pathology, we feel a thorough abdominal examination should be considered part of the full neurologic examination, especially in cases presenting with focal neurologic findings involving the lower extremities.21
Collectively, this case alludes to the importance of close clinical follow-up, as well as adequate anticipatory patient guidance in cases of suspected MP. In most patients, the clinical course of spontaneous MP is benign and favorable, with up to 85% of patients experiencing resolution within 4 to 6 months of the initial presentation.22 Common conservative measures include weight loss, garment optimization, and nonsteroidal anti-inflammatory drugs as needed for analgesia. In refractory cases, procedural interventions such as with neurolysis or resection of the lateral femoral cutaneous nerve, may be required after the ruling out of alternative diagnoses.23,24
Importantly, in even prolonged and resistant cases of MP, patient discomfort remains localized to the territory of the LFCN. Additional lower motor neuron signs, such as an expanding territory of sensory involvement, muscle weakness, or diminished reflexes, should prompt additional testing for alternative diagnoses. In addition, clinical findings concerning for intraabdominal mass effect, many of which were observed in this case, should lead to further evaluation and expeditious cross-sectional imaging. Although this patient’s early satiety, polyuria, bilateral lower extremity edema, weight gain, and lumbar plexopathy each may be explained by direct compression, invasion, or displacement, his report of progressive exertional dyspnea merits further discussion.
Exertional dyspnea is an uncommon complication of soft tissue sarcoma, reported almost exclusively in cases with cardiac, mediastinal, or other thoracic involvement.25-28 In this case, there was no evidence of thoracic involvement, either through direct extension or metastasis. Instead, the patient’s exertional dyspnea may have been attributable to increased intraabdominal pressure leading to compromised diaphragm excursion and reduced pulmonary reserve. In addition, the radiographic findings also raise the possibility of a potential contribution from preload failure due to IVC compression. Overall, dyspnea is a concerning feature that may suggest advanced disease.
Despite the value of a thorough history and physical examination in patients with MP, major clinical guidelines from neurologic, neurosurgical, and orthopedic organizations do not formally address MP evaluation and management. Further, proposed clinical practice algorithms are inconsistent in their recommendations regarding the identification of red-flag features and ruling out of alternative diagnoses.22,29,30 To supplement the abdominal examination, it would be reasonable to perform a pelvic compression test (PCT) in patients presenting with suspected MP. The PCT is a highly sensitive and specific provocative maneuver shown to enable reliable differentiation between MP and lumbar radiculopathy, and is performed by placing downward force on the anterior superior iliac spine of the affected extremity for 45 seconds with the patient in the lateral recumbent position.31 As this maneuver is intended to force relaxation of the inguinal ligament, thereby relieving pressure on the LFCN, improvement in the patient’s symptoms with the PCT is consistent with MP.
Conclusions
1. van Slobbe AM, Bohnen AM, Bernsen RM, Koes BW, Bierma-Zeinstra SM. Incidence rates and determinants in meralgia paresthetica in general practice. J Neurol. 2004;251(3):294-297. doi:10.1007/s00415-004-0310-x
2. Parisi TJ, Mandrekar J, Dyck PJ, Klein CJ. Meralgia paresthetica: relation to obesity, advanced age, and diabetes mellitus. Neurology. 2011;77(16):1538-1542. doi:10.1212/WNL.0b013e318233b356
3. Ecker AD. Diagnosis of meralgia paresthetica. JAMA. 1985;253(7):976.
4. Massey EW, Pellock JM. Meralgia paraesthetica in a child. J Pediatr. 1978;93(2):325-326. doi:10.1016/s0022-3476(78)80566-6
5. Harney D, Patijn J. Meralgia paresthetica: diagnosis and management strategies. Pain Med. 2007;8(8):669-677. doi:10.1111/j.1526-4637.2006.00227.x
6. Berini SE, Spinner RJ, Jentoft ME, et al. Chronic meralgia paresthetica and neurectomy: a clinical pathologic study. Neurology. 2014;82(17):1551-1555. doi:10.1212/WNL.0000000000000367
7. Payne RA, Harbaugh K, Specht CS, Rizk E. Correlation of histopathology and clinical symptoms in meralgia paresthetica. Cureus. 2017;9(10):e1789. Published 2017 Oct 20. doi:10.7759/cureus.1789
8. Boyce JR. Meralgia paresthetica and tight trousers. JAMA. 1984;251(12):1553.
9. Orton D. Meralgia paresthetica from a wallet. JAMA. 1984;252(24):3368.
10. Fargo MV, Konitzer LN. Meralgia paresthetica due to body armor wear in U.S. soldiers serving in Iraq: a case report and review of the literature. Mil Med. 2007;172(6):663-665. doi:10.7205/milmed.172.6.663
11. Korkmaz N, Ozçakar L. Meralgia paresthetica in a policeman: the belt or the gun. Plast Reconstr Surg. 2004;114(4):1012-1013. doi:10.1097/01.prs.0000138706.86633.01
12. Gooding MS, Evangelista V, Pereira L. Carpal Tunnel Syndrome and Meralgia Paresthetica in Pregnancy. Obstet Gynecol Surv. 2020;75(2):121-126. doi:10.1097/OGX.0000000000000745
13. Pauwels A, Amarenco P, Chazouillères O, Pigot F, Calmus Y, Lévy VG. Une complication rare et méconnue de l’ascite: la méralgie paresthésique [Unusual and unknown complication of ascites: meralgia paresthetica]. Gastroenterol Clin Biol. 1990;14(3):295.
14. Braddom RL. L2 rather than L1 radiculopathy mimics meralgia paresthetica. Muscle Nerve. 2010;42(5):842. doi:10.1002/mus.21826
15. Suber DA, Massey EW. Pelvic mass presenting as meralgia paresthetica. Obstet Gynecol. 1979;53(2):257-258.
16. Flowers RS. Meralgia paresthetica. A clue to retroperitoneal malignant tumor. Am J Surg. 1968;116(1):89-92. doi:10.1016/0002-9610(68)90423-6
17. Yi TI, Yoon TH, Kim JS, Lee GE, Kim BR. Femoral neuropathy and meralgia paresthetica secondary to an iliacus hematoma. Ann Rehabil Med. 2012;36(2):273-277. doi:10.5535/arm.2012.36.2.273
18. Lee ATJ, Thway K, Huang PH, Jones RL. Clinical and molecular spectrum of liposarcoma. J Clin Oncol. 2018;36(2):151-159. doi:10.1200/JCO.2017.74.9598
19. O’Sullivan ED, Schofield SJ. Cognitive bias in clinical medicine. J R Coll Physicians Edinb. 2018;48(3):225-232. doi:10.4997/JRCPE.2018.306
20. Bickley, LS. Bates’ Guide to Physical Examination and History Taking. 12th Edition. Wolters Kluwer Health/Lippincott Williams and Wilkins; 2016.
21. Bhavsar AS, Verma S, Lamba R, Lall CG, Koenigsknecht V, Rajesh A. Abdominal manifestations of neurologic disorders. Radiographics. 2013;33(1):135-153. doi:10.1148/rg.331125097
22. Dureja GP, Gulaya V, Jayalakshmi TS, Mandal P. Management of meralgia paresthetica: a multimodality regimen. Anesth Analg. 1995;80(5):1060-1061. doi:10.1097/00000539-199505000-00043
23. Patijn J, Mekhail N, Hayek S, Lataster A, van Kleef M, Van Zundert J. Meralgia paresthetica. Pain Pract. 2011;11(3):302-308. doi:10.1111/j.1533-2500.2011.00458.x24. Ivins GK. Meralgia paresthetica, the elusive diagnosis: clinical experience with 14 adult patients. Ann Surg. 2000;232(2):281-286. doi:10.1097/00000658-200008000-00019
25. Munin MA, Goerner MS, Raggio I, et al. A rare cause of dyspnea: undifferentiated pleomorphic sarcoma in the left atrium. Cardiol Res. 2017;8(5):241-245. doi:10.14740/cr590w
26. Nguyen A, Awad WI. Cardiac sarcoma arising from malignant transformation of a preexisting atrial myxoma. Ann Thorac Surg. 2016;101(4):1571-1573. doi:10.1016/j.athoracsur.2015.05.129
27. Jiang S, Li J, Zeng Q, Liang J. Pulmonary artery intimal sarcoma misdiagnosed as pulmonary embolism: a case report. Oncol Lett. 2017;13(4):2713-2716. doi:10.3892/ol.2017.5775
28. Cojocaru A, Oliveira PJ, Pellecchia C. A pleural presentation of a rare soft tissue sarcoma. Am J Resp Crit Care Med. 2012;185:A5201. doi:10.1164/ajrccm-conference.2012.185.1_MeetingAbstracts.A5201
29. Grossman MG, Ducey SA, Nadler SS, Levy AS. Meralgia paresthetica: diagnosis and treatment. J Am Acad Orthop Surg. 2001;9(5):336-344. doi:10.5435/00124635-200109000-00007
30. Cheatham SW, Kolber MJ, Salamh PA. Meralgia paresthetica: a review of the literature. Int J Sports Phys Ther. 2013;8(6):883-893.
31. Nouraei SA, Anand B, Spink G, O’Neill KS. A novel approach to the diagnosis and management of meralgia paresthetica. Neurosurgery. 2007;60(4):696-700. doi:10.1227/01.NEU.0000255392.69914.F7
32. Antunes PE, Antunes MJ. Meralgia paresthetica after aortic valve surgery. J Heart Valve Dis. 1997;6(6):589-590.
33. Reddy YM, Singh D, Chikkam V, et al. Postprocedural neuropathy after atrial fibrillation ablation. J Interv Card Electrophysiol. 2013;36(3):279-285. doi:10.1007/s10840-012-9724-z
34. Butler R, Webster MW. Meralgia paresthetica: an unusual complication of cardiac catheterization via the femoral artery. Catheter Cardiovasc Interv. 2002;56(1):69-71. doi:10.1002/ccd.10149
35. Jellish WS, Oftadeh M. Peripheral nerve injury in cardiac surgery. J Cardiothorac Vasc Anesth. 2018;32(1):495-511. doi:10.1053/j.jvca.2017.08.030
36. Parsonnet V, Karasakalides A, Gielchinsky I, Hochberg M, Hussain SM. Meralgia paresthetica after coronary bypass surgery. J Thorac Cardiovasc Surg. 1991;101(2):219-221.
37. Macgregor AM, Thoburn EK. Meralgia paresthetica following bariatric surgery. Obes Surg. 1999;9(4):364-368. doi:10.1381/096089299765552945
38. Grace DM. Meralgia paresthetica after gastroplasty for morbid obesity. Can J Surg. 1987;30(1):64-65.
39. Polidori L, Magarelli M, Tramutoli R. Meralgia paresthetica as a complication of laparoscopic appendectomy. Surg Endosc. 2003;17(5):832. doi:10.1007/s00464-002-4279-1
40. Yamout B, Tayyim A, Farhat W. Meralgia paresthetica as a complication of laparoscopic cholecystectomy. Clin Neurol Neurosurg. 1994;96(2):143-144. doi:10.1016/0303-8467(94)90048-5
41. Broin EO, Horner C, Mealy K, et al. Meralgia paraesthetica following laparoscopic inguinal hernia repair. an anatomical analysis. Surg Endosc. 1995;9(1):76-78. doi:10.1007/BF00187893
42. Eubanks S, Newman L 3rd, Goehring L, et al. Meralgia paresthetica: a complication of laparoscopic herniorrhaphy. Surg Laparosc Endosc. 1993;3(5):381-385.
43. Atamaz F, Hepgüler S, Karasu Z, Kilic M. Meralgia paresthetica after liver transplantation: a case report. Transplant Proc. 2005;37(10):4424-4425. doi:10.1016/j.transproceed.2005.11.047
44. Chung KH, Lee JY, Ko TK, et al. Meralgia paresthetica affecting parturient women who underwent cesarean section -a case report-. Korean J Anesthesiol. 2010;59 Suppl(Suppl):S86-S89. doi:10.4097/kjae.2010.59.S.S86
45. Hutchins FL Jr, Huggins J, Delaney ML. Laparoscopic myomectomy-an unusual cause of meralgia paresthetica. J Am Assoc Gynecol Laparosc. 1998;5(3):309-311. doi:10.1016/s1074-3804(98)80039-x
46. Jones CD, Guiot L, Portelli M, Bullen T, Skaife P. Two interesting cases of meralgia paraesthetica. Pain Physician. 2017;20(6):E987-E989.
47. Peters G, Larner AJ. Meralgia paresthetica following gynecologic and obstetric surgery. Int J Gynaecol Obstet. 2006;95(1):42-43. doi:10.1016/j.ijgo.2006.05.025
48. Kvarnström N, Järvholm S, Johannesson L, Dahm-Kähler P, Olausson M, Brännström M. Live donors of the initial observational study of uterus transplantation-psychological and medical follow-up until 1 year after surgery in the 9 cases. Transplantation. 2017;101(3):664-670. doi:10.1097/TP.0000000000001567
49. Goulding K, Beaulé PE, Kim PR, Fazekas A. Incidence of lateral femoral cutaneous nerve neuropraxia after anterior approach hip arthroplasty. Clin Orthop Relat Res. 2010;468(9):2397-2404. doi:10.1007/s11999-010-1406-5
50. Yamamoto T, Nagira K, Kurosaka M. Meralgia paresthetica occurring 40 years after iliac bone graft harvesting: case report. Neurosurgery. 2001;49(6):1455-1457. doi:10.1097/00006123-200112000-00028
51. Roqueplan F, Porcher R, Hamzé B, et al. Long-term results of percutaneous resection and interstitial laser ablation of osteoid osteomas. Eur Radiol. 2010;20(1):209-217. doi:10.1007/s00330-009-1537-9
52. Gupta A, Muzumdar D, Ramani PS. Meralgia paraesthetica following lumbar spine surgery: a study in 110 consecutive surgically treated cases. Neurol India. 2004;52(1):64-66.
53. Yang SH, Wu CC, Chen PQ. Postoperative meralgia paresthetica after posterior spine surgery: incidence, risk factors, and clinical outcomes. Spine (Phila Pa 1976). 2005;30(18):E547-E550. doi:10.1097/01.brs.0000178821.14102.9d
54. Tejwani SG, Scaduto AA, Bowen RE. Transient meralgia paresthetica after pediatric posterior spine fusion. J Pediatr Orthop. 2006;26(4):530-533. doi:10.1097/01.bpo.0000217721.95480.9e
55. Peker S, Ay B, Sun I, Ozgen S, Pamir M. Meralgia paraesthetica: complications of prone position during lumbar disc surgery. Internet J Anesthesiol. 2003;8(1):24-29.
1. van Slobbe AM, Bohnen AM, Bernsen RM, Koes BW, Bierma-Zeinstra SM. Incidence rates and determinants in meralgia paresthetica in general practice. J Neurol. 2004;251(3):294-297. doi:10.1007/s00415-004-0310-x
2. Parisi TJ, Mandrekar J, Dyck PJ, Klein CJ. Meralgia paresthetica: relation to obesity, advanced age, and diabetes mellitus. Neurology. 2011;77(16):1538-1542. doi:10.1212/WNL.0b013e318233b356
3. Ecker AD. Diagnosis of meralgia paresthetica. JAMA. 1985;253(7):976.
4. Massey EW, Pellock JM. Meralgia paraesthetica in a child. J Pediatr. 1978;93(2):325-326. doi:10.1016/s0022-3476(78)80566-6
5. Harney D, Patijn J. Meralgia paresthetica: diagnosis and management strategies. Pain Med. 2007;8(8):669-677. doi:10.1111/j.1526-4637.2006.00227.x
6. Berini SE, Spinner RJ, Jentoft ME, et al. Chronic meralgia paresthetica and neurectomy: a clinical pathologic study. Neurology. 2014;82(17):1551-1555. doi:10.1212/WNL.0000000000000367
7. Payne RA, Harbaugh K, Specht CS, Rizk E. Correlation of histopathology and clinical symptoms in meralgia paresthetica. Cureus. 2017;9(10):e1789. Published 2017 Oct 20. doi:10.7759/cureus.1789
8. Boyce JR. Meralgia paresthetica and tight trousers. JAMA. 1984;251(12):1553.
9. Orton D. Meralgia paresthetica from a wallet. JAMA. 1984;252(24):3368.
10. Fargo MV, Konitzer LN. Meralgia paresthetica due to body armor wear in U.S. soldiers serving in Iraq: a case report and review of the literature. Mil Med. 2007;172(6):663-665. doi:10.7205/milmed.172.6.663
11. Korkmaz N, Ozçakar L. Meralgia paresthetica in a policeman: the belt or the gun. Plast Reconstr Surg. 2004;114(4):1012-1013. doi:10.1097/01.prs.0000138706.86633.01
12. Gooding MS, Evangelista V, Pereira L. Carpal Tunnel Syndrome and Meralgia Paresthetica in Pregnancy. Obstet Gynecol Surv. 2020;75(2):121-126. doi:10.1097/OGX.0000000000000745
13. Pauwels A, Amarenco P, Chazouillères O, Pigot F, Calmus Y, Lévy VG. Une complication rare et méconnue de l’ascite: la méralgie paresthésique [Unusual and unknown complication of ascites: meralgia paresthetica]. Gastroenterol Clin Biol. 1990;14(3):295.
14. Braddom RL. L2 rather than L1 radiculopathy mimics meralgia paresthetica. Muscle Nerve. 2010;42(5):842. doi:10.1002/mus.21826
15. Suber DA, Massey EW. Pelvic mass presenting as meralgia paresthetica. Obstet Gynecol. 1979;53(2):257-258.
16. Flowers RS. Meralgia paresthetica. A clue to retroperitoneal malignant tumor. Am J Surg. 1968;116(1):89-92. doi:10.1016/0002-9610(68)90423-6
17. Yi TI, Yoon TH, Kim JS, Lee GE, Kim BR. Femoral neuropathy and meralgia paresthetica secondary to an iliacus hematoma. Ann Rehabil Med. 2012;36(2):273-277. doi:10.5535/arm.2012.36.2.273
18. Lee ATJ, Thway K, Huang PH, Jones RL. Clinical and molecular spectrum of liposarcoma. J Clin Oncol. 2018;36(2):151-159. doi:10.1200/JCO.2017.74.9598
19. O’Sullivan ED, Schofield SJ. Cognitive bias in clinical medicine. J R Coll Physicians Edinb. 2018;48(3):225-232. doi:10.4997/JRCPE.2018.306
20. Bickley, LS. Bates’ Guide to Physical Examination and History Taking. 12th Edition. Wolters Kluwer Health/Lippincott Williams and Wilkins; 2016.
21. Bhavsar AS, Verma S, Lamba R, Lall CG, Koenigsknecht V, Rajesh A. Abdominal manifestations of neurologic disorders. Radiographics. 2013;33(1):135-153. doi:10.1148/rg.331125097
22. Dureja GP, Gulaya V, Jayalakshmi TS, Mandal P. Management of meralgia paresthetica: a multimodality regimen. Anesth Analg. 1995;80(5):1060-1061. doi:10.1097/00000539-199505000-00043
23. Patijn J, Mekhail N, Hayek S, Lataster A, van Kleef M, Van Zundert J. Meralgia paresthetica. Pain Pract. 2011;11(3):302-308. doi:10.1111/j.1533-2500.2011.00458.x24. Ivins GK. Meralgia paresthetica, the elusive diagnosis: clinical experience with 14 adult patients. Ann Surg. 2000;232(2):281-286. doi:10.1097/00000658-200008000-00019
25. Munin MA, Goerner MS, Raggio I, et al. A rare cause of dyspnea: undifferentiated pleomorphic sarcoma in the left atrium. Cardiol Res. 2017;8(5):241-245. doi:10.14740/cr590w
26. Nguyen A, Awad WI. Cardiac sarcoma arising from malignant transformation of a preexisting atrial myxoma. Ann Thorac Surg. 2016;101(4):1571-1573. doi:10.1016/j.athoracsur.2015.05.129
27. Jiang S, Li J, Zeng Q, Liang J. Pulmonary artery intimal sarcoma misdiagnosed as pulmonary embolism: a case report. Oncol Lett. 2017;13(4):2713-2716. doi:10.3892/ol.2017.5775
28. Cojocaru A, Oliveira PJ, Pellecchia C. A pleural presentation of a rare soft tissue sarcoma. Am J Resp Crit Care Med. 2012;185:A5201. doi:10.1164/ajrccm-conference.2012.185.1_MeetingAbstracts.A5201
29. Grossman MG, Ducey SA, Nadler SS, Levy AS. Meralgia paresthetica: diagnosis and treatment. J Am Acad Orthop Surg. 2001;9(5):336-344. doi:10.5435/00124635-200109000-00007
30. Cheatham SW, Kolber MJ, Salamh PA. Meralgia paresthetica: a review of the literature. Int J Sports Phys Ther. 2013;8(6):883-893.
31. Nouraei SA, Anand B, Spink G, O’Neill KS. A novel approach to the diagnosis and management of meralgia paresthetica. Neurosurgery. 2007;60(4):696-700. doi:10.1227/01.NEU.0000255392.69914.F7
32. Antunes PE, Antunes MJ. Meralgia paresthetica after aortic valve surgery. J Heart Valve Dis. 1997;6(6):589-590.
33. Reddy YM, Singh D, Chikkam V, et al. Postprocedural neuropathy after atrial fibrillation ablation. J Interv Card Electrophysiol. 2013;36(3):279-285. doi:10.1007/s10840-012-9724-z
34. Butler R, Webster MW. Meralgia paresthetica: an unusual complication of cardiac catheterization via the femoral artery. Catheter Cardiovasc Interv. 2002;56(1):69-71. doi:10.1002/ccd.10149
35. Jellish WS, Oftadeh M. Peripheral nerve injury in cardiac surgery. J Cardiothorac Vasc Anesth. 2018;32(1):495-511. doi:10.1053/j.jvca.2017.08.030
36. Parsonnet V, Karasakalides A, Gielchinsky I, Hochberg M, Hussain SM. Meralgia paresthetica after coronary bypass surgery. J Thorac Cardiovasc Surg. 1991;101(2):219-221.
37. Macgregor AM, Thoburn EK. Meralgia paresthetica following bariatric surgery. Obes Surg. 1999;9(4):364-368. doi:10.1381/096089299765552945
38. Grace DM. Meralgia paresthetica after gastroplasty for morbid obesity. Can J Surg. 1987;30(1):64-65.
39. Polidori L, Magarelli M, Tramutoli R. Meralgia paresthetica as a complication of laparoscopic appendectomy. Surg Endosc. 2003;17(5):832. doi:10.1007/s00464-002-4279-1
40. Yamout B, Tayyim A, Farhat W. Meralgia paresthetica as a complication of laparoscopic cholecystectomy. Clin Neurol Neurosurg. 1994;96(2):143-144. doi:10.1016/0303-8467(94)90048-5
41. Broin EO, Horner C, Mealy K, et al. Meralgia paraesthetica following laparoscopic inguinal hernia repair. an anatomical analysis. Surg Endosc. 1995;9(1):76-78. doi:10.1007/BF00187893
42. Eubanks S, Newman L 3rd, Goehring L, et al. Meralgia paresthetica: a complication of laparoscopic herniorrhaphy. Surg Laparosc Endosc. 1993;3(5):381-385.
43. Atamaz F, Hepgüler S, Karasu Z, Kilic M. Meralgia paresthetica after liver transplantation: a case report. Transplant Proc. 2005;37(10):4424-4425. doi:10.1016/j.transproceed.2005.11.047
44. Chung KH, Lee JY, Ko TK, et al. Meralgia paresthetica affecting parturient women who underwent cesarean section -a case report-. Korean J Anesthesiol. 2010;59 Suppl(Suppl):S86-S89. doi:10.4097/kjae.2010.59.S.S86
45. Hutchins FL Jr, Huggins J, Delaney ML. Laparoscopic myomectomy-an unusual cause of meralgia paresthetica. J Am Assoc Gynecol Laparosc. 1998;5(3):309-311. doi:10.1016/s1074-3804(98)80039-x
46. Jones CD, Guiot L, Portelli M, Bullen T, Skaife P. Two interesting cases of meralgia paraesthetica. Pain Physician. 2017;20(6):E987-E989.
47. Peters G, Larner AJ. Meralgia paresthetica following gynecologic and obstetric surgery. Int J Gynaecol Obstet. 2006;95(1):42-43. doi:10.1016/j.ijgo.2006.05.025
48. Kvarnström N, Järvholm S, Johannesson L, Dahm-Kähler P, Olausson M, Brännström M. Live donors of the initial observational study of uterus transplantation-psychological and medical follow-up until 1 year after surgery in the 9 cases. Transplantation. 2017;101(3):664-670. doi:10.1097/TP.0000000000001567
49. Goulding K, Beaulé PE, Kim PR, Fazekas A. Incidence of lateral femoral cutaneous nerve neuropraxia after anterior approach hip arthroplasty. Clin Orthop Relat Res. 2010;468(9):2397-2404. doi:10.1007/s11999-010-1406-5
50. Yamamoto T, Nagira K, Kurosaka M. Meralgia paresthetica occurring 40 years after iliac bone graft harvesting: case report. Neurosurgery. 2001;49(6):1455-1457. doi:10.1097/00006123-200112000-00028
51. Roqueplan F, Porcher R, Hamzé B, et al. Long-term results of percutaneous resection and interstitial laser ablation of osteoid osteomas. Eur Radiol. 2010;20(1):209-217. doi:10.1007/s00330-009-1537-9
52. Gupta A, Muzumdar D, Ramani PS. Meralgia paraesthetica following lumbar spine surgery: a study in 110 consecutive surgically treated cases. Neurol India. 2004;52(1):64-66.
53. Yang SH, Wu CC, Chen PQ. Postoperative meralgia paresthetica after posterior spine surgery: incidence, risk factors, and clinical outcomes. Spine (Phila Pa 1976). 2005;30(18):E547-E550. doi:10.1097/01.brs.0000178821.14102.9d
54. Tejwani SG, Scaduto AA, Bowen RE. Transient meralgia paresthetica after pediatric posterior spine fusion. J Pediatr Orthop. 2006;26(4):530-533. doi:10.1097/01.bpo.0000217721.95480.9e
55. Peker S, Ay B, Sun I, Ozgen S, Pamir M. Meralgia paraesthetica: complications of prone position during lumbar disc surgery. Internet J Anesthesiol. 2003;8(1):24-29.
Standardization of the Discharge Process for Inpatient Hematology and Oncology Using Plan-Do-Study-Act Methodology Improves Follow-Up and Patient Hand-Off
Hematology and oncology patients are a complex patient population that requires timely follow-up to prevent clinical decompensation and delays in treatment. Previous reports have demonstrated that outpatient follow-up within 14 days is associated with decreased 30-day readmissions. The magnitude of this effect is greater for higher-risk patients.1 Therefore, patients being discharged from the hematology and oncology inpatient service should be seen by a hematology and oncology provider within 14 days of discharge. Patients who do not require close oncologic follow-up should be seen by a primary care provider (PCP) within this timeframe.
Background
The Institute of Medicine (IOM) identified the need to focus on quality improvement and patient safety with a 1999 report, To Err Is Human.2 Tremendous strides have been made in the areas of quality improvement and patient safety over the past 2 decades. In a 2013 report, the IOM further identified hematology and oncology care as an area of need due to a combination of growing demand, complexity of cancer and cancer treatment, shrinking workforce, and rising costs. The report concluded that cancer care is not as patient-centered, accessible, coordinated, or evidence based as it could be, with detrimental impacts on patients.3 Patients with cancer have been identified as a high-risk population for hospital readmissions.4,5 Lack of timely follow-up and failed hand-offs have been identified as factors contributing to poor outcomes at time of discharge.6-10
Upon internal review of baseline performance data, we identified areas needing improvement in the discharge process. These included time to hematology and oncology follow-up appointment, percent of patients with PCP appointments scheduled at time of discharge, and electronically alerts for the outpatient hematologist/oncologist to discharge summaries. It was determined that patients discharged from the inpatient service were seen a mean 17 days later by their outpatient hematology and oncology provider and the time to the follow-up appointment varied substantially, with some patients being seen several weeks to months after discharge. Furthermore, only 68% of patients had a primary care appointment scheduled at the time of discharge. These data along with review of data reported in the medical literature supported our initiative for improvement in the transition from inpatient to outpatient care for our hematology and oncology patients.
Plan-Do-Study-Act (PDSA) quality improvement methodology was used to create and implement several interventions to standardize the discharge process for this patient population, with the primary goal of decreasing the mean time to hematology and oncology follow-up from 17 days by 12% to fewer than 14 days. Patients who do not require close oncologic follow-up should be seen by a PCP within this timeframe. Otherwise, PCP follow-up within at least 6 months should be made. Secondary aims included (1) an increase in scheduled PCP visits at time of discharge from 68% to > 90%; and (2) an increase in communication of the discharge summary via electronic alerting of the outpatient hematology and oncology physician from 20% to > 90%. Herein, we report our experience and results of this quality improvement initiative
Methods
The Institutional Review Board at Edward Hines Veteran Affairs Hospital in Hines, Illinois reviewed this single-center study and deemed it to be exempt from oversight. Using PDSA quality improvement methodology, a multidisciplinary team of hematology and oncology staff developed and implemented a standardized discharge process. The multidisciplinary team included a robust representation of inpatient and outpatient staff caring for the hematology and oncology patient population, including attending physicians, fellows, residents, advanced practice nurses, registered nurses, clinical pharmacists, patient care coordinators, clinic schedulers, clinical applications coordinators, quality support staff, and a systems redesign coach. Hospital leadership including chief of staff, chief of medicine, and chief of nursing participated as the management guidance team. Several interviews and group meetings were conducted and a multidisciplinary team collaboratively developed and implemented the interventions and monitored the results.
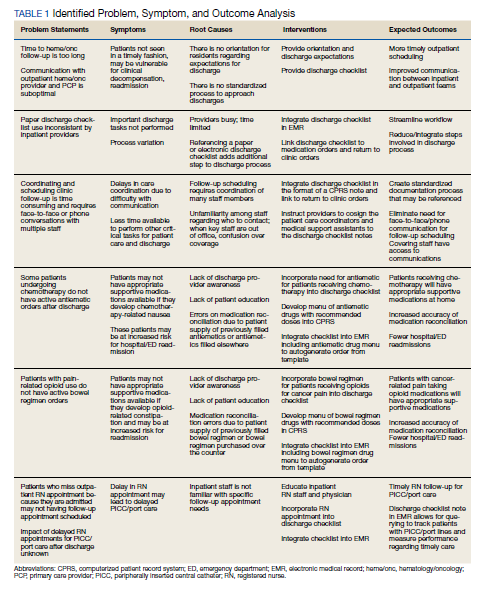
Outcome measures were identified, including time to hematology and oncology clinic visit, primary care follow-up scheduling, and communication of discharge to the outpatient hematology and oncology physician. Baseline data were collected and reviewed. The multidisciplinary team developed a process flow map to understand the steps and resources involved with the transition from inpatient to outpatient care. Gap analysis and root cause analysis were performed. A solutions approach was applied to develop interventions. Table 1 shows a summary of the identified problems, symptoms, associated causes, the interventions aimed to address the problems, and expected outcomes. Rotating resident physicians were trained through online and in-person education. The multidisciplinary team met intermittently to monitor outcomes, provide feedback, further refine interventions, and develop additional interventions.
PDSA Cycle 1
A standardized discharge process was developed in the form of guidelines and expectations. These include an explanation of unique features of the hematology and oncology service and expectations of medication reconciliation with emphasis placed on antiemetics, antimicrobial prophylaxis, and bowel regimen when appropriate, outpatient hematology and oncology follow-up within 14 days, primary care follow-up, communication with the outpatient hematology and oncology physician, written discharge instructions, and bedside teaching when appropriate.
PDSA Cycle 2
Based on team member feedback and further discussions, a discharge checklist was developed. This checklist was available online, reviewed in person, and posted in the team room for rotating residents to use for discharge planning and when discharging patients (Figure 1).

PDSA Cycle 3
Based on ongoing user feedback, group discussions, and data monitoring, the discharge checklist was further refined and updated. An electronic clinical decision support tool was developed and integrated into the electronic medical record (EMR) in the form of a discharge checklist note template directly linked to orders. The tool is a computerized patient record system (CPRS) note template that prompts users to select whether medications or return to clinic orders are needed and offers a menu of frequently used medications. If any of the selections are chosen within the note template, an order is generated automatically in the chart that requires only the user’s signature. Furthermore, the patient care coordinator reviews the prescribed follow-up and works with the medical support assistant to make these appointments. The physician is contacted only when an appointment cannot be made. Therefore, this tool allows many additional actions to be bypassed such as generating medication and return to clinic orders individually and calling schedulers to make follow-up appointments (Figure 2).

Data Analysis
All patients discharged during the 2-month period prior to and discharged after the implementation of the standardized process were reviewed. Patients who followed up with hematology and oncology at another facility, enrolled in hospice, or died during admission were excluded. Follow-up appointment scheduling data and communication between inpatient and outpatient providers were reviewed. Data were analyzed using XmR statistical process control chart and Fisher’s Exact Test using GraphPad. Control limits were calculated for each PDSA cycle as the mean ± the average of the moving range multiplied by 2.66. All data were included in the analysis.
Results
A total of 142 consecutive patients were reviewed from May 1, 2018 to August 31, 2018 and January 1, 2019 to April 30, 2019, including 58 patients prior to the intervention and 84 patients during PDSA cycles. There was a gap in data collection between September 1, 2018 and December 31, 2018 due to limited team member availability. All data were collected by 2 reviewers—a postgraduate year (PGY)-4 chief resident and a PGY-2 internal medicine resident. The median age of patients in the preintervention group was 72 years and 69 years in the postintervention group. All patients were men. Baseline data revealed a mean 17 days to hematology and oncology follow-up. Primary care visits were scheduled for 68% of patients at the time of discharge. The outpatient hematology and oncology physician was alerted electronically to the discharge summary for 20% of the patients (Table 2).
The primary endpoint of time to hematology and oncology follow-up appointment improved to 13 days in PDSA cycles 1 and 2 and 10 days in PDSA cycle 3. The target of mean 14 days to follow-up was achieved. The statistical process control chart shows 5 shifts with clusters of ≥ 7 points below the mean revealing a true signal or change in the data and demonstrating that an improvement was seen (Figure 3). Furthermore, the statistical process control chart demonstrates upper control limit decreased from 58 days at baseline to 21 days in PDSA cycle 3, suggesting a decrease in variation.
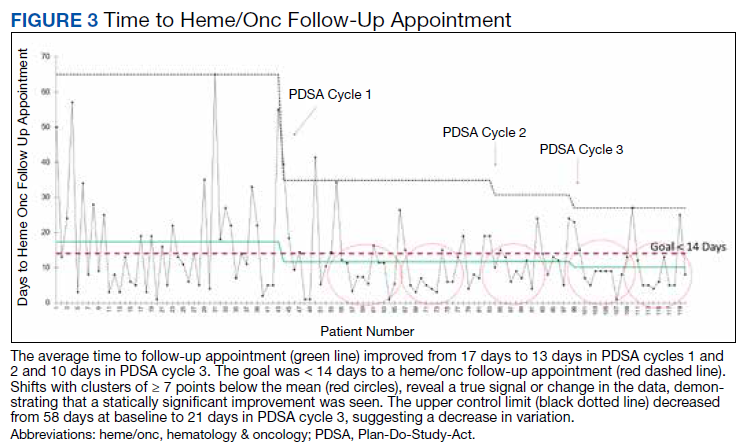
Regarding secondary endpoints, the outpatient hematology and oncology attending physician and/or fellow was alerted electronically to the discharge summary for 62% of patients compared with 20% at baseline (P = .01), and primary care appointments were scheduled for 77% of patients after the intervention compared with 68% at baseline (P = .88) (Table 2).
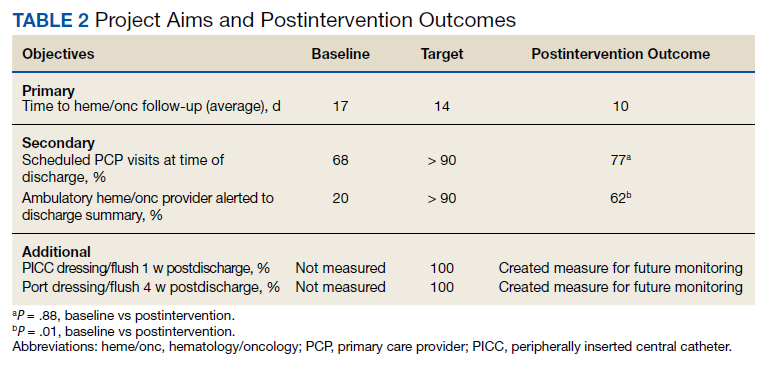
Through ongoing meetings, discussions, and feedback, we identified additional objectives unique to this patient population that had no performance measurement. These included peripherally inserted central catheter (PICC) care nursing visits scheduled 1 week after discharge and port care nursing visits scheduled 4 weeks after discharge. These visits allow nursing staff to dress and flush these catheters for routine maintenance per institutional policy. The implementation of the discharge checklist note creates a mechanism of tracking performance in meeting this goal moving forward, whereas no method was in place to track this metric.
Discussion
The 2013 IOM report Delivering High-Quality Cancer Care: Charting a New Course for a System in Crisis found that that cancer care is not as patient-centered, accessible, coordinated, or evidence-based as it could be, with detrimental impacts on patients.3 The document offered a conceptual framework to improve quality of cancer care that includes the translation of evidence into clinical practice, quality measurement, and performance improvement, as well as using advances in information technology to enhance quality measurement and performance improvement. Our quality initiative uses this framework to work toward the goal as stated by the IOM report: to deliver “comprehensive, patient-centered, evidence-based, high-quality cancer care that is accessible and affordable.”3
Two large studies that evaluated risk factors for 15-day and 30-day hospital readmissions identified cancer diagnosis as a risk factor for increased hospital readmission, highlighting the need to identify strategies to improve the discharge process for these patients.4,5 Timely outpatient follow-up and better patient hand-off may improve clinical outcomes among this high-risk patient population after hospital discharge. Multiple studies have demonstrated that timely follow-up is associated with fewer readmissions.1,8-10 A study by Forster and colleagues that evaluated postdischarge adverse events (AEs) revealed a 23% incidence of AEs with 12% of these identified as preventable. Postdischarge monitoring was deemed inadequate among these patients, with closer follow-up and improved hand-offs between inpatient and outpatient medical teams identified as possible interventions to improve postdischarge patient monitoring and to prevent AEs.7
The present quality initiative to standardize the discharge process for the hematology and oncology service decreased time to hematology and oncology follow-up appointment, improved communication between inpatient and outpatient teams, and decreased process variation. Timelier follow-up for this complex patient population likely will prevent clinical decompensation, delays in treatment, and directly improve patient access to care.
The multidisciplinary nature of this effort was instrumental to successful completion. In a complex health care system, it is challenging to truly understand a problem and identify possible solutions without the perspective of all members of the care team. The involvement of team members with training in quality improvement methodology was important to evaluate and develop interventions in a systematic way. Furthermore, the support and involvement of leadership is important in order to allocate resources appropriately to achieve system changes that improve care. Using quality improvement methodology, the team was able to map our processes and perform gap and root cause analyses. Strategies were identified to improve our performance using a solutions approach. Changes were implemented with continued intermittent meetings for monitoring of progression and discussion of how interventions could be made more efficient, effective, and user friendly. The primary goal was ultimately achieved.
Integration of intervention into the EMR embodies the IOM’s call to use advances in information technology to enhance the quality and delivery of care, quality measurement, and performance improvement.3 This intervention offered the strongest system changes as an electronic clinical decision support tool was developed and embedded into the EMR in the form of a Discharge Checklist Note that is linked to associated orders. This intervention was the most robust, as it provided objective data regarding utilization of the checklist, offered a more efficient way to communicate with team members regarding discharge needs, and streamlined the workflow for the discharging provider. Furthermore, this electronic tool created the ability to measure other important aspects in the care of this patient population that we previously had no mechanism of measuring: timely nursing appointments for routine care of PICC lines and ports.
Limitations
The absence of clinical endpoints was a limitation of this study. The present study was unable to evaluate the effect of the intervention on readmission rates, emergency department visits, hospital length of stay, cost, or mortality. Coordinating this multidisciplinary effort required much time and planning, and additional resources were not available to evaluate these clinical endpoints. Further studies are needed to evaluate whether the increased patient access and closer follow-up would result in improvement in these clinical endpoints. Another consideration for future improvement projects would be to include patients in the multidisciplinary team. The patient perspective would be invaluable in identifying gaps in care delivery and strategies aimed at improving care delivery.
Conclusions
This quality initiative to standardize the discharge process for the hematology and oncology service decreased time to the initial hematology and oncology follow-up appointment, improved communication between inpatient and outpatient teams, and decreased process variation. Timelier follow-up for this complex patient population likely will prevent clinical decompensation, delays in treatment, and directly improve patient access to care.
Acknowledgments
We thank our patients for whom we hope our process improvement efforts will ultimately benefit. We thank all the hematology and oncology staff at Edward Hines Jr. VA Hospital and Loyola University Medical Center residents and fellows who care for our patients and participated in the multidisciplinary team to improve care for our patients. We thank the following professionals for their uncompensated assistance in the coordination and execution of this initiative: Robert Kutter, MS, and Meghan O’Halloran, MD.
1. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-122. doi:10.1370/afm.1753
2. Kohn LT, Corrigan J, Donaldson MS, eds. To Err Is Human: Building a Safer Health System. Washington, DC: National Academy Press; 2000.
3. Levit LA, Balogh E, Nass SJ, Ganz P, Institute of Medicine (U.S.), eds. Delivering High-Quality Cancer Care: Charting a New Course for a System in Crisis. Washington, DC: National Academies Press; 2013.
4. Allaudeen N, Vidyarthi A, Maselli J, Auerbach A. Redefining readmission risk factors for general medicine patients. J Hosp Med. 2011;6(2):54-60. doi:10.1002/jhm.805
5. Dorajoo SR, See V, Chan CT, et al. Identifying potentially avoidable readmissions: a medication-based 15-day readmission risk stratification algorithm. Pharmacotherapy. 2017;37(3):268-277. doi:10.1002/phar.1896
6. Kripalani S, LeFevre F, Phillips CO, Williams MV, Basaviah P, Baker DW. Deficits in communication and information transfer between hospital-based and primary care physicians: implications for patient safety and continuity of care. JAMA. 2007;297(8):831-841. doi:10.1001/jama.297.8.831
7. Forster AJ, Clark HD, Menard A, et al. Adverse events among medical patients after discharge from hospital [published correction appears in CMAJ. 2004 March 2;170(5):771]. CMAJ. 2004;170(3):345-349.
8. Hernandez AF, Greiner MA, Fonarow GC, et al. Relationship between early physician follow-up and 30-day readmission among Medicare beneficiaries hospitalized for heart failure. JAMA. 2010;303(17):1716-1722. doi:10.1001/jama.2010.533
9. Misky GJ, Wald HL, Coleman EA. Post-hospitalization transitions: examining the effects of timing of primary care provider follow-up. J Hosp Med. 2010;5(7):392-397. doi:10.1002/jhm.666
10. Sharma G, Kuo YF, Freeman JL, Zhang DD, Goodwin JS. Outpatient follow-up visit and 30-day emergency department visit and readmission in patients hospitalized for chronic obstructive pulmonary disease. Arch Intern Med. 2010;170(18):1664-1670. doi:10.1001/archinternmed.2010.345
Hematology and oncology patients are a complex patient population that requires timely follow-up to prevent clinical decompensation and delays in treatment. Previous reports have demonstrated that outpatient follow-up within 14 days is associated with decreased 30-day readmissions. The magnitude of this effect is greater for higher-risk patients.1 Therefore, patients being discharged from the hematology and oncology inpatient service should be seen by a hematology and oncology provider within 14 days of discharge. Patients who do not require close oncologic follow-up should be seen by a primary care provider (PCP) within this timeframe.
Background
The Institute of Medicine (IOM) identified the need to focus on quality improvement and patient safety with a 1999 report, To Err Is Human.2 Tremendous strides have been made in the areas of quality improvement and patient safety over the past 2 decades. In a 2013 report, the IOM further identified hematology and oncology care as an area of need due to a combination of growing demand, complexity of cancer and cancer treatment, shrinking workforce, and rising costs. The report concluded that cancer care is not as patient-centered, accessible, coordinated, or evidence based as it could be, with detrimental impacts on patients.3 Patients with cancer have been identified as a high-risk population for hospital readmissions.4,5 Lack of timely follow-up and failed hand-offs have been identified as factors contributing to poor outcomes at time of discharge.6-10
Upon internal review of baseline performance data, we identified areas needing improvement in the discharge process. These included time to hematology and oncology follow-up appointment, percent of patients with PCP appointments scheduled at time of discharge, and electronically alerts for the outpatient hematologist/oncologist to discharge summaries. It was determined that patients discharged from the inpatient service were seen a mean 17 days later by their outpatient hematology and oncology provider and the time to the follow-up appointment varied substantially, with some patients being seen several weeks to months after discharge. Furthermore, only 68% of patients had a primary care appointment scheduled at the time of discharge. These data along with review of data reported in the medical literature supported our initiative for improvement in the transition from inpatient to outpatient care for our hematology and oncology patients.
Plan-Do-Study-Act (PDSA) quality improvement methodology was used to create and implement several interventions to standardize the discharge process for this patient population, with the primary goal of decreasing the mean time to hematology and oncology follow-up from 17 days by 12% to fewer than 14 days. Patients who do not require close oncologic follow-up should be seen by a PCP within this timeframe. Otherwise, PCP follow-up within at least 6 months should be made. Secondary aims included (1) an increase in scheduled PCP visits at time of discharge from 68% to > 90%; and (2) an increase in communication of the discharge summary via electronic alerting of the outpatient hematology and oncology physician from 20% to > 90%. Herein, we report our experience and results of this quality improvement initiative
Methods
The Institutional Review Board at Edward Hines Veteran Affairs Hospital in Hines, Illinois reviewed this single-center study and deemed it to be exempt from oversight. Using PDSA quality improvement methodology, a multidisciplinary team of hematology and oncology staff developed and implemented a standardized discharge process. The multidisciplinary team included a robust representation of inpatient and outpatient staff caring for the hematology and oncology patient population, including attending physicians, fellows, residents, advanced practice nurses, registered nurses, clinical pharmacists, patient care coordinators, clinic schedulers, clinical applications coordinators, quality support staff, and a systems redesign coach. Hospital leadership including chief of staff, chief of medicine, and chief of nursing participated as the management guidance team. Several interviews and group meetings were conducted and a multidisciplinary team collaboratively developed and implemented the interventions and monitored the results.
Outcome measures were identified, including time to hematology and oncology clinic visit, primary care follow-up scheduling, and communication of discharge to the outpatient hematology and oncology physician. Baseline data were collected and reviewed. The multidisciplinary team developed a process flow map to understand the steps and resources involved with the transition from inpatient to outpatient care. Gap analysis and root cause analysis were performed. A solutions approach was applied to develop interventions. Table 1 shows a summary of the identified problems, symptoms, associated causes, the interventions aimed to address the problems, and expected outcomes. Rotating resident physicians were trained through online and in-person education. The multidisciplinary team met intermittently to monitor outcomes, provide feedback, further refine interventions, and develop additional interventions.
PDSA Cycle 1
A standardized discharge process was developed in the form of guidelines and expectations. These include an explanation of unique features of the hematology and oncology service and expectations of medication reconciliation with emphasis placed on antiemetics, antimicrobial prophylaxis, and bowel regimen when appropriate, outpatient hematology and oncology follow-up within 14 days, primary care follow-up, communication with the outpatient hematology and oncology physician, written discharge instructions, and bedside teaching when appropriate.
PDSA Cycle 2
Based on team member feedback and further discussions, a discharge checklist was developed. This checklist was available online, reviewed in person, and posted in the team room for rotating residents to use for discharge planning and when discharging patients (Figure 1).
PDSA Cycle 3
Based on ongoing user feedback, group discussions, and data monitoring, the discharge checklist was further refined and updated. An electronic clinical decision support tool was developed and integrated into the electronic medical record (EMR) in the form of a discharge checklist note template directly linked to orders. The tool is a computerized patient record system (CPRS) note template that prompts users to select whether medications or return to clinic orders are needed and offers a menu of frequently used medications. If any of the selections are chosen within the note template, an order is generated automatically in the chart that requires only the user’s signature. Furthermore, the patient care coordinator reviews the prescribed follow-up and works with the medical support assistant to make these appointments. The physician is contacted only when an appointment cannot be made. Therefore, this tool allows many additional actions to be bypassed such as generating medication and return to clinic orders individually and calling schedulers to make follow-up appointments (Figure 2).
Data Analysis
All patients discharged during the 2-month period prior to and discharged after the implementation of the standardized process were reviewed. Patients who followed up with hematology and oncology at another facility, enrolled in hospice, or died during admission were excluded. Follow-up appointment scheduling data and communication between inpatient and outpatient providers were reviewed. Data were analyzed using XmR statistical process control chart and Fisher’s Exact Test using GraphPad. Control limits were calculated for each PDSA cycle as the mean ± the average of the moving range multiplied by 2.66. All data were included in the analysis.
Results
A total of 142 consecutive patients were reviewed from May 1, 2018 to August 31, 2018 and January 1, 2019 to April 30, 2019, including 58 patients prior to the intervention and 84 patients during PDSA cycles. There was a gap in data collection between September 1, 2018 and December 31, 2018 due to limited team member availability. All data were collected by 2 reviewers—a postgraduate year (PGY)-4 chief resident and a PGY-2 internal medicine resident. The median age of patients in the preintervention group was 72 years and 69 years in the postintervention group. All patients were men. Baseline data revealed a mean 17 days to hematology and oncology follow-up. Primary care visits were scheduled for 68% of patients at the time of discharge. The outpatient hematology and oncology physician was alerted electronically to the discharge summary for 20% of the patients (Table 2).
The primary endpoint of time to hematology and oncology follow-up appointment improved to 13 days in PDSA cycles 1 and 2 and 10 days in PDSA cycle 3. The target of mean 14 days to follow-up was achieved. The statistical process control chart shows 5 shifts with clusters of ≥ 7 points below the mean revealing a true signal or change in the data and demonstrating that an improvement was seen (Figure 3). Furthermore, the statistical process control chart demonstrates upper control limit decreased from 58 days at baseline to 21 days in PDSA cycle 3, suggesting a decrease in variation.
Regarding secondary endpoints, the outpatient hematology and oncology attending physician and/or fellow was alerted electronically to the discharge summary for 62% of patients compared with 20% at baseline (P = .01), and primary care appointments were scheduled for 77% of patients after the intervention compared with 68% at baseline (P = .88) (Table 2).
Through ongoing meetings, discussions, and feedback, we identified additional objectives unique to this patient population that had no performance measurement. These included peripherally inserted central catheter (PICC) care nursing visits scheduled 1 week after discharge and port care nursing visits scheduled 4 weeks after discharge. These visits allow nursing staff to dress and flush these catheters for routine maintenance per institutional policy. The implementation of the discharge checklist note creates a mechanism of tracking performance in meeting this goal moving forward, whereas no method was in place to track this metric.
Discussion
The 2013 IOM report Delivering High-Quality Cancer Care: Charting a New Course for a System in Crisis found that that cancer care is not as patient-centered, accessible, coordinated, or evidence-based as it could be, with detrimental impacts on patients.3 The document offered a conceptual framework to improve quality of cancer care that includes the translation of evidence into clinical practice, quality measurement, and performance improvement, as well as using advances in information technology to enhance quality measurement and performance improvement. Our quality initiative uses this framework to work toward the goal as stated by the IOM report: to deliver “comprehensive, patient-centered, evidence-based, high-quality cancer care that is accessible and affordable.”3
Two large studies that evaluated risk factors for 15-day and 30-day hospital readmissions identified cancer diagnosis as a risk factor for increased hospital readmission, highlighting the need to identify strategies to improve the discharge process for these patients.4,5 Timely outpatient follow-up and better patient hand-off may improve clinical outcomes among this high-risk patient population after hospital discharge. Multiple studies have demonstrated that timely follow-up is associated with fewer readmissions.1,8-10 A study by Forster and colleagues that evaluated postdischarge adverse events (AEs) revealed a 23% incidence of AEs with 12% of these identified as preventable. Postdischarge monitoring was deemed inadequate among these patients, with closer follow-up and improved hand-offs between inpatient and outpatient medical teams identified as possible interventions to improve postdischarge patient monitoring and to prevent AEs.7
The present quality initiative to standardize the discharge process for the hematology and oncology service decreased time to hematology and oncology follow-up appointment, improved communication between inpatient and outpatient teams, and decreased process variation. Timelier follow-up for this complex patient population likely will prevent clinical decompensation, delays in treatment, and directly improve patient access to care.
The multidisciplinary nature of this effort was instrumental to successful completion. In a complex health care system, it is challenging to truly understand a problem and identify possible solutions without the perspective of all members of the care team. The involvement of team members with training in quality improvement methodology was important to evaluate and develop interventions in a systematic way. Furthermore, the support and involvement of leadership is important in order to allocate resources appropriately to achieve system changes that improve care. Using quality improvement methodology, the team was able to map our processes and perform gap and root cause analyses. Strategies were identified to improve our performance using a solutions approach. Changes were implemented with continued intermittent meetings for monitoring of progression and discussion of how interventions could be made more efficient, effective, and user friendly. The primary goal was ultimately achieved.
Integration of intervention into the EMR embodies the IOM’s call to use advances in information technology to enhance the quality and delivery of care, quality measurement, and performance improvement.3 This intervention offered the strongest system changes as an electronic clinical decision support tool was developed and embedded into the EMR in the form of a Discharge Checklist Note that is linked to associated orders. This intervention was the most robust, as it provided objective data regarding utilization of the checklist, offered a more efficient way to communicate with team members regarding discharge needs, and streamlined the workflow for the discharging provider. Furthermore, this electronic tool created the ability to measure other important aspects in the care of this patient population that we previously had no mechanism of measuring: timely nursing appointments for routine care of PICC lines and ports.
Limitations
The absence of clinical endpoints was a limitation of this study. The present study was unable to evaluate the effect of the intervention on readmission rates, emergency department visits, hospital length of stay, cost, or mortality. Coordinating this multidisciplinary effort required much time and planning, and additional resources were not available to evaluate these clinical endpoints. Further studies are needed to evaluate whether the increased patient access and closer follow-up would result in improvement in these clinical endpoints. Another consideration for future improvement projects would be to include patients in the multidisciplinary team. The patient perspective would be invaluable in identifying gaps in care delivery and strategies aimed at improving care delivery.
Conclusions
This quality initiative to standardize the discharge process for the hematology and oncology service decreased time to the initial hematology and oncology follow-up appointment, improved communication between inpatient and outpatient teams, and decreased process variation. Timelier follow-up for this complex patient population likely will prevent clinical decompensation, delays in treatment, and directly improve patient access to care.
Acknowledgments
We thank our patients for whom we hope our process improvement efforts will ultimately benefit. We thank all the hematology and oncology staff at Edward Hines Jr. VA Hospital and Loyola University Medical Center residents and fellows who care for our patients and participated in the multidisciplinary team to improve care for our patients. We thank the following professionals for their uncompensated assistance in the coordination and execution of this initiative: Robert Kutter, MS, and Meghan O’Halloran, MD.
Hematology and oncology patients are a complex patient population that requires timely follow-up to prevent clinical decompensation and delays in treatment. Previous reports have demonstrated that outpatient follow-up within 14 days is associated with decreased 30-day readmissions. The magnitude of this effect is greater for higher-risk patients.1 Therefore, patients being discharged from the hematology and oncology inpatient service should be seen by a hematology and oncology provider within 14 days of discharge. Patients who do not require close oncologic follow-up should be seen by a primary care provider (PCP) within this timeframe.
Background
The Institute of Medicine (IOM) identified the need to focus on quality improvement and patient safety with a 1999 report, To Err Is Human.2 Tremendous strides have been made in the areas of quality improvement and patient safety over the past 2 decades. In a 2013 report, the IOM further identified hematology and oncology care as an area of need due to a combination of growing demand, complexity of cancer and cancer treatment, shrinking workforce, and rising costs. The report concluded that cancer care is not as patient-centered, accessible, coordinated, or evidence based as it could be, with detrimental impacts on patients.3 Patients with cancer have been identified as a high-risk population for hospital readmissions.4,5 Lack of timely follow-up and failed hand-offs have been identified as factors contributing to poor outcomes at time of discharge.6-10
Upon internal review of baseline performance data, we identified areas needing improvement in the discharge process. These included time to hematology and oncology follow-up appointment, percent of patients with PCP appointments scheduled at time of discharge, and electronically alerts for the outpatient hematologist/oncologist to discharge summaries. It was determined that patients discharged from the inpatient service were seen a mean 17 days later by their outpatient hematology and oncology provider and the time to the follow-up appointment varied substantially, with some patients being seen several weeks to months after discharge. Furthermore, only 68% of patients had a primary care appointment scheduled at the time of discharge. These data along with review of data reported in the medical literature supported our initiative for improvement in the transition from inpatient to outpatient care for our hematology and oncology patients.
Plan-Do-Study-Act (PDSA) quality improvement methodology was used to create and implement several interventions to standardize the discharge process for this patient population, with the primary goal of decreasing the mean time to hematology and oncology follow-up from 17 days by 12% to fewer than 14 days. Patients who do not require close oncologic follow-up should be seen by a PCP within this timeframe. Otherwise, PCP follow-up within at least 6 months should be made. Secondary aims included (1) an increase in scheduled PCP visits at time of discharge from 68% to > 90%; and (2) an increase in communication of the discharge summary via electronic alerting of the outpatient hematology and oncology physician from 20% to > 90%. Herein, we report our experience and results of this quality improvement initiative
Methods
The Institutional Review Board at Edward Hines Veteran Affairs Hospital in Hines, Illinois reviewed this single-center study and deemed it to be exempt from oversight. Using PDSA quality improvement methodology, a multidisciplinary team of hematology and oncology staff developed and implemented a standardized discharge process. The multidisciplinary team included a robust representation of inpatient and outpatient staff caring for the hematology and oncology patient population, including attending physicians, fellows, residents, advanced practice nurses, registered nurses, clinical pharmacists, patient care coordinators, clinic schedulers, clinical applications coordinators, quality support staff, and a systems redesign coach. Hospital leadership including chief of staff, chief of medicine, and chief of nursing participated as the management guidance team. Several interviews and group meetings were conducted and a multidisciplinary team collaboratively developed and implemented the interventions and monitored the results.
Outcome measures were identified, including time to hematology and oncology clinic visit, primary care follow-up scheduling, and communication of discharge to the outpatient hematology and oncology physician. Baseline data were collected and reviewed. The multidisciplinary team developed a process flow map to understand the steps and resources involved with the transition from inpatient to outpatient care. Gap analysis and root cause analysis were performed. A solutions approach was applied to develop interventions. Table 1 shows a summary of the identified problems, symptoms, associated causes, the interventions aimed to address the problems, and expected outcomes. Rotating resident physicians were trained through online and in-person education. The multidisciplinary team met intermittently to monitor outcomes, provide feedback, further refine interventions, and develop additional interventions.
PDSA Cycle 1
A standardized discharge process was developed in the form of guidelines and expectations. These include an explanation of unique features of the hematology and oncology service and expectations of medication reconciliation with emphasis placed on antiemetics, antimicrobial prophylaxis, and bowel regimen when appropriate, outpatient hematology and oncology follow-up within 14 days, primary care follow-up, communication with the outpatient hematology and oncology physician, written discharge instructions, and bedside teaching when appropriate.
PDSA Cycle 2
Based on team member feedback and further discussions, a discharge checklist was developed. This checklist was available online, reviewed in person, and posted in the team room for rotating residents to use for discharge planning and when discharging patients (Figure 1).
PDSA Cycle 3
Based on ongoing user feedback, group discussions, and data monitoring, the discharge checklist was further refined and updated. An electronic clinical decision support tool was developed and integrated into the electronic medical record (EMR) in the form of a discharge checklist note template directly linked to orders. The tool is a computerized patient record system (CPRS) note template that prompts users to select whether medications or return to clinic orders are needed and offers a menu of frequently used medications. If any of the selections are chosen within the note template, an order is generated automatically in the chart that requires only the user’s signature. Furthermore, the patient care coordinator reviews the prescribed follow-up and works with the medical support assistant to make these appointments. The physician is contacted only when an appointment cannot be made. Therefore, this tool allows many additional actions to be bypassed such as generating medication and return to clinic orders individually and calling schedulers to make follow-up appointments (Figure 2).
Data Analysis
All patients discharged during the 2-month period prior to and discharged after the implementation of the standardized process were reviewed. Patients who followed up with hematology and oncology at another facility, enrolled in hospice, or died during admission were excluded. Follow-up appointment scheduling data and communication between inpatient and outpatient providers were reviewed. Data were analyzed using XmR statistical process control chart and Fisher’s Exact Test using GraphPad. Control limits were calculated for each PDSA cycle as the mean ± the average of the moving range multiplied by 2.66. All data were included in the analysis.
Results
A total of 142 consecutive patients were reviewed from May 1, 2018 to August 31, 2018 and January 1, 2019 to April 30, 2019, including 58 patients prior to the intervention and 84 patients during PDSA cycles. There was a gap in data collection between September 1, 2018 and December 31, 2018 due to limited team member availability. All data were collected by 2 reviewers—a postgraduate year (PGY)-4 chief resident and a PGY-2 internal medicine resident. The median age of patients in the preintervention group was 72 years and 69 years in the postintervention group. All patients were men. Baseline data revealed a mean 17 days to hematology and oncology follow-up. Primary care visits were scheduled for 68% of patients at the time of discharge. The outpatient hematology and oncology physician was alerted electronically to the discharge summary for 20% of the patients (Table 2).
The primary endpoint of time to hematology and oncology follow-up appointment improved to 13 days in PDSA cycles 1 and 2 and 10 days in PDSA cycle 3. The target of mean 14 days to follow-up was achieved. The statistical process control chart shows 5 shifts with clusters of ≥ 7 points below the mean revealing a true signal or change in the data and demonstrating that an improvement was seen (Figure 3). Furthermore, the statistical process control chart demonstrates upper control limit decreased from 58 days at baseline to 21 days in PDSA cycle 3, suggesting a decrease in variation.
Regarding secondary endpoints, the outpatient hematology and oncology attending physician and/or fellow was alerted electronically to the discharge summary for 62% of patients compared with 20% at baseline (P = .01), and primary care appointments were scheduled for 77% of patients after the intervention compared with 68% at baseline (P = .88) (Table 2).
Through ongoing meetings, discussions, and feedback, we identified additional objectives unique to this patient population that had no performance measurement. These included peripherally inserted central catheter (PICC) care nursing visits scheduled 1 week after discharge and port care nursing visits scheduled 4 weeks after discharge. These visits allow nursing staff to dress and flush these catheters for routine maintenance per institutional policy. The implementation of the discharge checklist note creates a mechanism of tracking performance in meeting this goal moving forward, whereas no method was in place to track this metric.
Discussion
The 2013 IOM report Delivering High-Quality Cancer Care: Charting a New Course for a System in Crisis found that that cancer care is not as patient-centered, accessible, coordinated, or evidence-based as it could be, with detrimental impacts on patients.3 The document offered a conceptual framework to improve quality of cancer care that includes the translation of evidence into clinical practice, quality measurement, and performance improvement, as well as using advances in information technology to enhance quality measurement and performance improvement. Our quality initiative uses this framework to work toward the goal as stated by the IOM report: to deliver “comprehensive, patient-centered, evidence-based, high-quality cancer care that is accessible and affordable.”3
Two large studies that evaluated risk factors for 15-day and 30-day hospital readmissions identified cancer diagnosis as a risk factor for increased hospital readmission, highlighting the need to identify strategies to improve the discharge process for these patients.4,5 Timely outpatient follow-up and better patient hand-off may improve clinical outcomes among this high-risk patient population after hospital discharge. Multiple studies have demonstrated that timely follow-up is associated with fewer readmissions.1,8-10 A study by Forster and colleagues that evaluated postdischarge adverse events (AEs) revealed a 23% incidence of AEs with 12% of these identified as preventable. Postdischarge monitoring was deemed inadequate among these patients, with closer follow-up and improved hand-offs between inpatient and outpatient medical teams identified as possible interventions to improve postdischarge patient monitoring and to prevent AEs.7
The present quality initiative to standardize the discharge process for the hematology and oncology service decreased time to hematology and oncology follow-up appointment, improved communication between inpatient and outpatient teams, and decreased process variation. Timelier follow-up for this complex patient population likely will prevent clinical decompensation, delays in treatment, and directly improve patient access to care.
The multidisciplinary nature of this effort was instrumental to successful completion. In a complex health care system, it is challenging to truly understand a problem and identify possible solutions without the perspective of all members of the care team. The involvement of team members with training in quality improvement methodology was important to evaluate and develop interventions in a systematic way. Furthermore, the support and involvement of leadership is important in order to allocate resources appropriately to achieve system changes that improve care. Using quality improvement methodology, the team was able to map our processes and perform gap and root cause analyses. Strategies were identified to improve our performance using a solutions approach. Changes were implemented with continued intermittent meetings for monitoring of progression and discussion of how interventions could be made more efficient, effective, and user friendly. The primary goal was ultimately achieved.
Integration of intervention into the EMR embodies the IOM’s call to use advances in information technology to enhance the quality and delivery of care, quality measurement, and performance improvement.3 This intervention offered the strongest system changes as an electronic clinical decision support tool was developed and embedded into the EMR in the form of a Discharge Checklist Note that is linked to associated orders. This intervention was the most robust, as it provided objective data regarding utilization of the checklist, offered a more efficient way to communicate with team members regarding discharge needs, and streamlined the workflow for the discharging provider. Furthermore, this electronic tool created the ability to measure other important aspects in the care of this patient population that we previously had no mechanism of measuring: timely nursing appointments for routine care of PICC lines and ports.
Limitations
The absence of clinical endpoints was a limitation of this study. The present study was unable to evaluate the effect of the intervention on readmission rates, emergency department visits, hospital length of stay, cost, or mortality. Coordinating this multidisciplinary effort required much time and planning, and additional resources were not available to evaluate these clinical endpoints. Further studies are needed to evaluate whether the increased patient access and closer follow-up would result in improvement in these clinical endpoints. Another consideration for future improvement projects would be to include patients in the multidisciplinary team. The patient perspective would be invaluable in identifying gaps in care delivery and strategies aimed at improving care delivery.
Conclusions
This quality initiative to standardize the discharge process for the hematology and oncology service decreased time to the initial hematology and oncology follow-up appointment, improved communication between inpatient and outpatient teams, and decreased process variation. Timelier follow-up for this complex patient population likely will prevent clinical decompensation, delays in treatment, and directly improve patient access to care.
Acknowledgments
We thank our patients for whom we hope our process improvement efforts will ultimately benefit. We thank all the hematology and oncology staff at Edward Hines Jr. VA Hospital and Loyola University Medical Center residents and fellows who care for our patients and participated in the multidisciplinary team to improve care for our patients. We thank the following professionals for their uncompensated assistance in the coordination and execution of this initiative: Robert Kutter, MS, and Meghan O’Halloran, MD.
1. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-122. doi:10.1370/afm.1753
2. Kohn LT, Corrigan J, Donaldson MS, eds. To Err Is Human: Building a Safer Health System. Washington, DC: National Academy Press; 2000.
3. Levit LA, Balogh E, Nass SJ, Ganz P, Institute of Medicine (U.S.), eds. Delivering High-Quality Cancer Care: Charting a New Course for a System in Crisis. Washington, DC: National Academies Press; 2013.
4. Allaudeen N, Vidyarthi A, Maselli J, Auerbach A. Redefining readmission risk factors for general medicine patients. J Hosp Med. 2011;6(2):54-60. doi:10.1002/jhm.805
5. Dorajoo SR, See V, Chan CT, et al. Identifying potentially avoidable readmissions: a medication-based 15-day readmission risk stratification algorithm. Pharmacotherapy. 2017;37(3):268-277. doi:10.1002/phar.1896
6. Kripalani S, LeFevre F, Phillips CO, Williams MV, Basaviah P, Baker DW. Deficits in communication and information transfer between hospital-based and primary care physicians: implications for patient safety and continuity of care. JAMA. 2007;297(8):831-841. doi:10.1001/jama.297.8.831
7. Forster AJ, Clark HD, Menard A, et al. Adverse events among medical patients after discharge from hospital [published correction appears in CMAJ. 2004 March 2;170(5):771]. CMAJ. 2004;170(3):345-349.
8. Hernandez AF, Greiner MA, Fonarow GC, et al. Relationship between early physician follow-up and 30-day readmission among Medicare beneficiaries hospitalized for heart failure. JAMA. 2010;303(17):1716-1722. doi:10.1001/jama.2010.533
9. Misky GJ, Wald HL, Coleman EA. Post-hospitalization transitions: examining the effects of timing of primary care provider follow-up. J Hosp Med. 2010;5(7):392-397. doi:10.1002/jhm.666
10. Sharma G, Kuo YF, Freeman JL, Zhang DD, Goodwin JS. Outpatient follow-up visit and 30-day emergency department visit and readmission in patients hospitalized for chronic obstructive pulmonary disease. Arch Intern Med. 2010;170(18):1664-1670. doi:10.1001/archinternmed.2010.345
1. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-122. doi:10.1370/afm.1753
2. Kohn LT, Corrigan J, Donaldson MS, eds. To Err Is Human: Building a Safer Health System. Washington, DC: National Academy Press; 2000.
3. Levit LA, Balogh E, Nass SJ, Ganz P, Institute of Medicine (U.S.), eds. Delivering High-Quality Cancer Care: Charting a New Course for a System in Crisis. Washington, DC: National Academies Press; 2013.
4. Allaudeen N, Vidyarthi A, Maselli J, Auerbach A. Redefining readmission risk factors for general medicine patients. J Hosp Med. 2011;6(2):54-60. doi:10.1002/jhm.805
5. Dorajoo SR, See V, Chan CT, et al. Identifying potentially avoidable readmissions: a medication-based 15-day readmission risk stratification algorithm. Pharmacotherapy. 2017;37(3):268-277. doi:10.1002/phar.1896
6. Kripalani S, LeFevre F, Phillips CO, Williams MV, Basaviah P, Baker DW. Deficits in communication and information transfer between hospital-based and primary care physicians: implications for patient safety and continuity of care. JAMA. 2007;297(8):831-841. doi:10.1001/jama.297.8.831
7. Forster AJ, Clark HD, Menard A, et al. Adverse events among medical patients after discharge from hospital [published correction appears in CMAJ. 2004 March 2;170(5):771]. CMAJ. 2004;170(3):345-349.
8. Hernandez AF, Greiner MA, Fonarow GC, et al. Relationship between early physician follow-up and 30-day readmission among Medicare beneficiaries hospitalized for heart failure. JAMA. 2010;303(17):1716-1722. doi:10.1001/jama.2010.533
9. Misky GJ, Wald HL, Coleman EA. Post-hospitalization transitions: examining the effects of timing of primary care provider follow-up. J Hosp Med. 2010;5(7):392-397. doi:10.1002/jhm.666
10. Sharma G, Kuo YF, Freeman JL, Zhang DD, Goodwin JS. Outpatient follow-up visit and 30-day emergency department visit and readmission in patients hospitalized for chronic obstructive pulmonary disease. Arch Intern Med. 2010;170(18):1664-1670. doi:10.1001/archinternmed.2010.345
Factors Associated with Radiation Toxicity and Survival in Patients with Presumed Early-Stage Non-Small Cell Lung Cancer Receiving Empiric Stereotactic Ablative Radiotherapy
Stereotactic ablative radiotherapy (SABR) has become the standard of care for inoperable early-stage non-small cell lung cancer (NSCLC). Many patients are unable to undergo a biopsy safely because of poor pulmonary function or underlying emphysema and are then empirically treated with radiotherapy if they meet criteria. In these patients, local control can be achieved with SABR with minimal toxicity.1 Considering that median overall survival (OS) among patients with untreated stage I NSCLC has been reported to be as low as 9 months, early treatment with SABR could lead to increased survival of 29 to 60 months.2-4
The RTOG 0236 trial showed a median OS of 48 months and the randomized phase III CHISEL trial showed a median OS of 60 months; however, these survival data were reported in patients who were able to safely undergo a biopsy and had confirmed NSCLC.4,5 For patients without a diagnosis confirmed by biopsy and who are treated with empiric SABR, patient factors that influence radiation toxicity and OS are not well defined.
It is not clear if empiric radiation benefits survival or if treatment causes decline in lung function, considering that underlying chronic lung disease precludes these patients from biopsy. The purpose of this study was to evaluate the factors associated with radiation toxicity with empiric SABR and to evaluate OS in this population without a biopsy-confirmed diagnosis.
Methods
This was a single center retrospective review of patients treated at the radiation oncology department at the Kansas City Veterans Affairs Medical Center from August 2014 to February 2019. Data were collected on 69 patients with pulmonary nodules identified by chest computed tomography (CT) and/or positron emission tomography (PET)-CT that were highly suspicious for primary NSCLC.
These patients were presented at a multidisciplinary meeting that involved pulmonologists, oncologists, radiation oncologists, and thoracic surgeons. Patients were deemed to be poor candidates for biopsy because of severe underlying emphysema, which would put them at high risk for pneumothorax with a percutaneous needle biopsy, or were unable to tolerate general anesthesia for navigational bronchoscopy or surgical biopsy because of poor lung function. These patients were diagnosed with presumed stage I NSCLC using the criteria: minimum of 2 sequential CT scans with enlarging nodule; absence of metastases on PET-CT; the single nodule had to be fluorodeoxyglucose avid with a minimum standardized uptake value of 2.5, and absence of clinical history or physical examination consistent with small cell lung cancer or infection.
After a consensus was reached that patients met these criteria, individuals were referred for empiric SABR. Follow-up visits were at 1 month, 3 months, and every 6 months. Variables analyzed included: patient demographics, pre- and posttreatment pulmonary function tests (PFT) when available, pre-treatment oxygen use, tumor size and location (peripheral, central, or ultra-central), radiation doses, and grade of toxicity as defined by Human and Health Services Common Terminology Criteria for Adverse Events version 5.0 (dyspnea and cough both counted as pulmonary toxicity): acute ≤ 90 days and late > 90 days (Table 1).
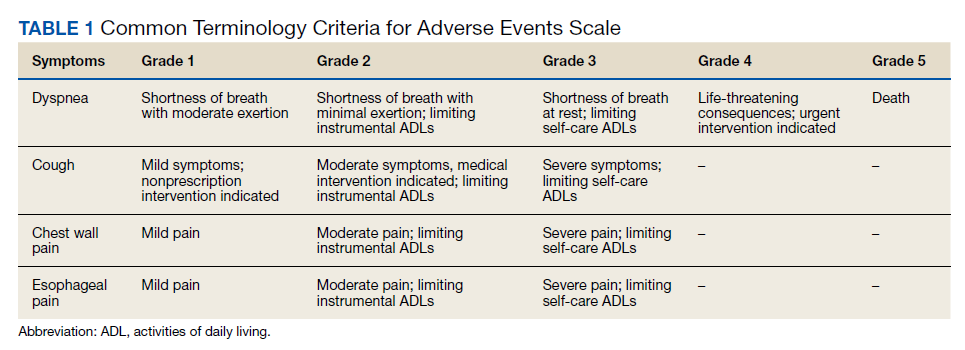
SPSS versions 24 and 26 were used for statistical analysis. Median and range were obtained for continuous variables with a normal distribution. Kaplan-Meier log-rank testing was used to analyze OS. χ2 and Mann-Whitney U tests were used to analyze association between independent variables and OS. Analysis of significant findings were repeated with operable patients excluded for further analysis.
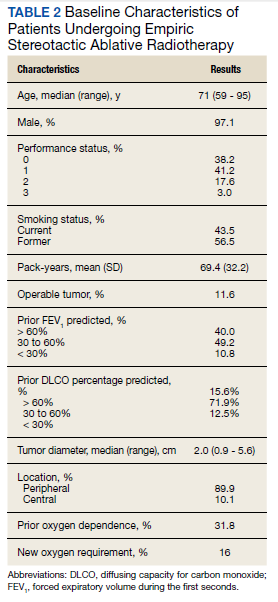
Results
The median follow-up was 18 months (range, 1 to 54). The median age was 71 years (range, 59 to 95) (Table 2). Most patients (97.1%) were male. The majority of patients (79.4%) had a 0 or 1 for the Eastern Cooperative Oncology group performance status, indicating fully active or restricted in physically strenuous activity but ambulatory and able to perform light work. All patients were either current or former smokers with an average pack-year history of 69.4. Only 11.6% of patients had operable disease, but received empiric SABR because they declined surgery. Four patients did not have pretreatment spirometry available and 37 did not have pretreatment diffusing capacity for carbon monoxide (DLCO) data.
Most patients had a pretreatment forced expiratory volume during the first seconds (FEV1) value and DLCO < 60% of predicted (60% and 84% of the patients, respectively). The median tumor diameter was 2 cm. Of the 68.2% of patients who did not have chronic hypoxemic respiratory failure before SABR, 16% developed a new requirement for supplemental oxygen. Sixty-two tumors (89.9%) were peripheral. There were 4 local recurrences (5.7%), 10 regional (different lobe and nodal) failures (14.3%), and 15 distant metastases (21.4%).
Nineteen of 67 patients (26.3%) had acute toxicity of which 9 had acute grade ≥ 2 toxicity; information regarding toxicity was missing on 2 patients. Thirty-two of 65 (49.9%) patients had late toxicity of which 20 (30.8%) had late grade ≥ 2 toxicity. The main factor associated with development of acute toxicity was pretreatment oxygendependence (P = .047). This was not significant when comparing only inoperable patients. Twenty patients (29.9%) developed some type of acute toxicity; pulmonary toxicity was most common (22.4%) (Table 3). All patients with acute toxicity also developed late toxicity except for 1 who died before 3 months. Predominantly, the deaths in our sample were from causes other than the malignancy or treatment, such as sepsis, deconditioning after a fall, cardiovascular complications, etc. Acute toxicity of grade ≥ 2 was significantly associated with late toxicity (P < .001 for both) in both operable and inoperable patients (P < .001).
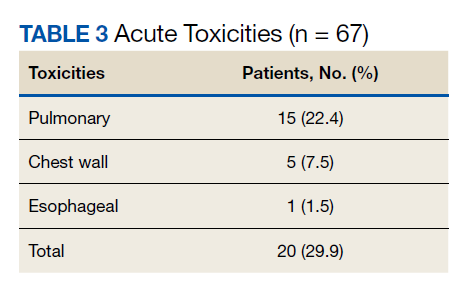
Development of any acute toxicity grade ≥ 2 was significantly associated with oxygendependence at baseline (P = .003), central location (P < .001), and new oxygen requirement (P = .02). Only central tumor location was found to be significant (P = .001) within the inoperable cohort. There were no significant differences in outcome based on pulmonary function testing (FEV1, forced vital capacity, or DLCO) or the analyzed PFT subgroups (FEV1 < 1.0 L, FEV1 < 1.5 L, FEV1 < 30%, and FEV1 < 35%).
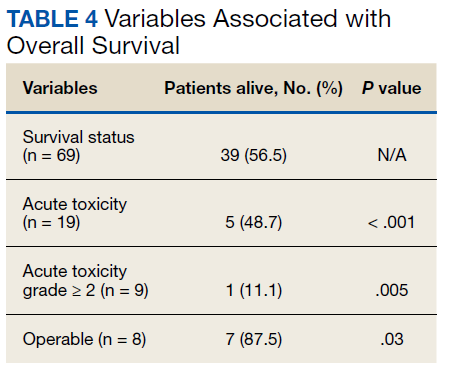
At the time of data collection, 30 patients were deceased (43.5%). There was a statistically significant association between OS and operability (P = .03; Table 4, Figure 1). Decreased OS was significantly associated with acute toxicity (P = .001) and acute toxicity grade ≥ 2 (P = .005; Figures 2 and 3). For the inoperable patients, both acute toxicity (P < .001) and acute toxicity grade ≥ 2 (P = .026) remained significant.
Discussion
SABR is an effective treatment for inoperable early-stage NSCLC, however its therapeutic ratio in a more frail population who cannot withstand biopsy is not well established. Additionally, the prevalence of benign disease in patients with solitary pulmonary nodules can be between 9% and 21%.6 Haidar and colleagues looked at 55 patients who received empiric SABR and found a median OS of 30.2 months with an 8.7% risk of local failure, 13% risk of regional failure with 8.7% acute toxicity, and 13% chronic toxicity.7 Data from Harkenrider and colleagues (n = 34) revealed similar results with a 2-year OS of 85%, local control of 97.1%, and regional control of 80%. The authors noted no grade ≥ 3 acute toxicities and an incidence of grade ≥ 3 late toxicities of 8.8%.1 These findings are concordant with our study results, confirming the safety and efficacy of SABR. Furthermore, a National Cancer Database analysis of observation vs empiric SABR found an OS of 10.1 months and 29 months respectively, with a hazard ratio of 0.64 (P < .001).3 Additionally, Fischer-Valuck and colleagues (n = 88) compared biopsy confirmed vs unbiopsied patients treated with SABR and found no difference in the 3-year local progression-free survival (93.1% vs 94.1%), regional lymph node metastasis and distant metastases free survival (92.5% vs 87.4%), or OS (59.9% vs 58.9%).8 With a median OS of ≤ 1 year for untreated stage I NSCLC,these studies support treating patients with empiric SABR.4
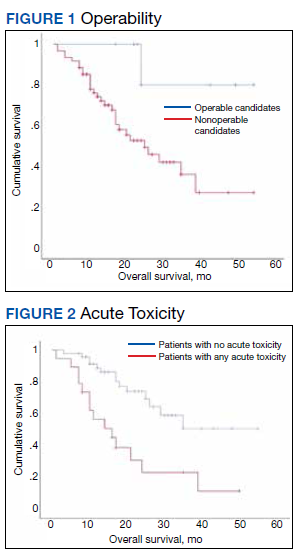
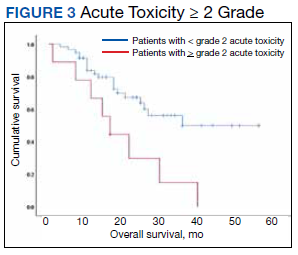
Other researchers have sought parameters to identify patients for whom radiation therapy would be too toxic. Guckenberger and colleagues aimed to establish a lower limit of pretreatment PFT to exclude patients and found only a 7% incidence of grade ≥ 2 adverse effects and toxicity did not increase with lower pulmonary function.9 They concluded that SABR was safe even for patients with poor pulmonary function. Other institutions have confirmed such findings and have been unable to find a cut-off PFT to exclude patients from empiric SABR.10,11 An analysis from the RTOG 0236 trial also noted that poor baseline PFT could not predict pulmonary toxicity or survival. Additionally, the study demonstrated only minimal decreases in patients’ FEV1 (5.8%) and DLCO (6%) at 2 years.12
Our study sought to identify a cut-off on FEV1 or DLCO that could be associated with increased toxicity. We also evaluated the incidence of acute toxicities grade ≥ 2 by stratifying patients according to FEV1 into subgroups: FEV1 < 1.0 L, FEV1 < 1.5 L, FEV1 < 30% of predicted and FEV1 < 35% of predicted. However, similar to other studies, we did not find any value that was significantly associated with increased toxicity that could preclude empiric SABR. One possible reason is that no treatment is offered for patients with extremely poor lung function as deemed by clinical judgement, therefore data on these patients is unavailable. In contradiction to other studies, our study found that oxygen dependence before treatment was significantly associated with development of acute toxicities. The exact mechanism for this association is unknown and could not be elucidated by baseline PFT. One possible explanation is that SABR could lead to oxygen free radical generation. In addition, our study indicated that those who developed acute toxicities had worse OS.
Limitations
Our study is limited by caveats of a retrospective study and its small sample size, but is in line with the reported literature (ranging from 33 to 88 patients).1,7,8 Another limitation is that data on pretreatment DLCO was missing in 37 patients and the lack of statistical robustness in terms of the smaller inoperable cohort, which limits the analyses of these factors in regards to anticipated morbidity from SABR. Also, given this is data collected from the US Department of Veterans Affairs, only 3% of our sample was female.
Conclusions
Empiric SABR for patients with presumed early-stage NSCLC appears to be safe and might positively impact OS. Development of any acute toxicity grade ≥ 2 was significantly associated with dependence on supplemental oxygen before treatment, central tumor location, and development of new oxygen requirement. No association was found in patients with poor pulmonary function before treatment because we could not find a FEV1 or DLCO cutoff that could preclude patients from empiric SABR. Considering the poor survival of untreated early-stage NSCLC, coupled with the efficacy and safety of empiric SABR for those with presumed disease, definitive SABR should be offered selectively within this patient population.
Acknowledgments
Drs. Park, Whiting and Castillo contributed to data collection. Drs. Park, Govindan and Castillo contributed to the statistical analysis and writing the first draft and final manuscript. Drs. Park, Govindan, Huang, and Reddy contributed to the discussion section.
1. Harkenrider MM, Bertke MH, Dunlap NE. Stereotactic body radiation therapy for unbiopsied early-stage lung cancer: a multi-institutional analysis. Am J Clin Oncol. 2014;37(4):337-342. doi:10.1097/COC.0b013e318277d822
2. Raz DJ, Zell JA, Ou SH, Gandara DR, Anton-Culver H, Jablons DM. Natural history of stage I non-small cell lung cancer: implications for early detection. Chest. 2007;132(1):193-199. doi:10.1378/chest.06-3096
3. Nanda RH, Liu Y, Gillespie TW, et al. Stereotactic body radiation therapy versus no treatment for early stage non-small cell lung cancer in medically inoperable elderly patients: a National Cancer Data Base analysis. Cancer. 2015;121(23):4222-4230. doi:10.1002/cncr.29640
4. Ball D, Mai GT, Vinod S, et al. Stereotactic ablative radiotherapy versus standard radiotherapy in stage 1 non-small-cell lung cancer (TROG 09.02 CHISEL): a phase 3, open-label, randomised controlled trial. Lancet Oncol. 2019;20(4):494-503. doi:10.1016/S1470-2045(18)30896-9
5. Timmerman R, Paulus R, Galvin J, et al. Stereotactic body radiation therapy for inoperable early stage lung cancer. JAMA. 2010;303(11):1070-1076. doi:10.1001/jama.2010.261
6. Smith MA, Battafarano RJ, Meyers BF, Zoole JB, Cooper JD, Patterson GA. Prevalence of benign disease in patients undergoing resection for suspected lung cancer. Ann Thorac Surg. 2006;81(5):1824-1828. doi:10.1016/j.athoracsur.2005.11.010
7. Haidar YM, Rahn DA 3rd, Nath S, et al. Comparison of outcomes following stereotactic body radiotherapy for nonsmall cell lung cancer in patients with and without pathological confirmation. Ther Adv Respir Dis. 2014;8(1):3-12. doi:10.1177/1753465813512545
8. Fischer-Valuck BW, Boggs H, Katz S, Durci M, Acharya S, Rosen LR. Comparison of stereotactic body radiation therapy for biopsy-proven versus radiographically diagnosed early-stage non-small lung cancer: a single-institution experience. Tumori. 2015;101(3):287-293. doi:10.5301/tj.5000279
9. Guckenberger M, Kestin LL, Hope AJ, et al. Is there a lower limit of pretreatment pulmonary function for safe and effective stereotactic body radiotherapy for early-stage non-small cell lung cancer? J Thorac Oncol. 2012;7:542-551. doi:10.1097/JTO.0b013e31824165d7
10. Wang J, Cao J, Yuan S, et al. Poor baseline pulmonary function may not increase the risk of radiation-induced lung toxicity. Int J Radiat Oncol Biol Phys. 2013;85(3):798-804. doi:10.1016/j.ijrobp.2012.06.040
11. Henderson M, McGarry R, Yiannoutsos C, et al. Baseline pulmonary function as a predictor for survival and decline in pulmonary function over time in patients undergoing stereotactic body radiotherapy for the treatment of stage I non-small-cell lung cancer. Int J Radiat Oncol Biol Phys. 2008;72(2):404-409. doi:10.1016/j.ijrobp.2007.12.051
12. Stanic S, Paulus R, Timmerman RD, et al. No clinically significant changes in pulmonary function following stereotactic body radiation therapy for early- stage peripheral non-small cell lung cancer: an analysis of RTOG 0236. Int J Radiat Oncol Biol Phys. 2014;88(5):1092-1099. doi:10.1016/j.ijrobp.2013.12.050
Stereotactic ablative radiotherapy (SABR) has become the standard of care for inoperable early-stage non-small cell lung cancer (NSCLC). Many patients are unable to undergo a biopsy safely because of poor pulmonary function or underlying emphysema and are then empirically treated with radiotherapy if they meet criteria. In these patients, local control can be achieved with SABR with minimal toxicity.1 Considering that median overall survival (OS) among patients with untreated stage I NSCLC has been reported to be as low as 9 months, early treatment with SABR could lead to increased survival of 29 to 60 months.2-4
The RTOG 0236 trial showed a median OS of 48 months and the randomized phase III CHISEL trial showed a median OS of 60 months; however, these survival data were reported in patients who were able to safely undergo a biopsy and had confirmed NSCLC.4,5 For patients without a diagnosis confirmed by biopsy and who are treated with empiric SABR, patient factors that influence radiation toxicity and OS are not well defined.
It is not clear if empiric radiation benefits survival or if treatment causes decline in lung function, considering that underlying chronic lung disease precludes these patients from biopsy. The purpose of this study was to evaluate the factors associated with radiation toxicity with empiric SABR and to evaluate OS in this population without a biopsy-confirmed diagnosis.
Methods
This was a single center retrospective review of patients treated at the radiation oncology department at the Kansas City Veterans Affairs Medical Center from August 2014 to February 2019. Data were collected on 69 patients with pulmonary nodules identified by chest computed tomography (CT) and/or positron emission tomography (PET)-CT that were highly suspicious for primary NSCLC.
These patients were presented at a multidisciplinary meeting that involved pulmonologists, oncologists, radiation oncologists, and thoracic surgeons. Patients were deemed to be poor candidates for biopsy because of severe underlying emphysema, which would put them at high risk for pneumothorax with a percutaneous needle biopsy, or were unable to tolerate general anesthesia for navigational bronchoscopy or surgical biopsy because of poor lung function. These patients were diagnosed with presumed stage I NSCLC using the criteria: minimum of 2 sequential CT scans with enlarging nodule; absence of metastases on PET-CT; the single nodule had to be fluorodeoxyglucose avid with a minimum standardized uptake value of 2.5, and absence of clinical history or physical examination consistent with small cell lung cancer or infection.
After a consensus was reached that patients met these criteria, individuals were referred for empiric SABR. Follow-up visits were at 1 month, 3 months, and every 6 months. Variables analyzed included: patient demographics, pre- and posttreatment pulmonary function tests (PFT) when available, pre-treatment oxygen use, tumor size and location (peripheral, central, or ultra-central), radiation doses, and grade of toxicity as defined by Human and Health Services Common Terminology Criteria for Adverse Events version 5.0 (dyspnea and cough both counted as pulmonary toxicity): acute ≤ 90 days and late > 90 days (Table 1).
SPSS versions 24 and 26 were used for statistical analysis. Median and range were obtained for continuous variables with a normal distribution. Kaplan-Meier log-rank testing was used to analyze OS. χ2 and Mann-Whitney U tests were used to analyze association between independent variables and OS. Analysis of significant findings were repeated with operable patients excluded for further analysis.
Results
The median follow-up was 18 months (range, 1 to 54). The median age was 71 years (range, 59 to 95) (Table 2). Most patients (97.1%) were male. The majority of patients (79.4%) had a 0 or 1 for the Eastern Cooperative Oncology group performance status, indicating fully active or restricted in physically strenuous activity but ambulatory and able to perform light work. All patients were either current or former smokers with an average pack-year history of 69.4. Only 11.6% of patients had operable disease, but received empiric SABR because they declined surgery. Four patients did not have pretreatment spirometry available and 37 did not have pretreatment diffusing capacity for carbon monoxide (DLCO) data.
Most patients had a pretreatment forced expiratory volume during the first seconds (FEV1) value and DLCO < 60% of predicted (60% and 84% of the patients, respectively). The median tumor diameter was 2 cm. Of the 68.2% of patients who did not have chronic hypoxemic respiratory failure before SABR, 16% developed a new requirement for supplemental oxygen. Sixty-two tumors (89.9%) were peripheral. There were 4 local recurrences (5.7%), 10 regional (different lobe and nodal) failures (14.3%), and 15 distant metastases (21.4%).
Nineteen of 67 patients (26.3%) had acute toxicity of which 9 had acute grade ≥ 2 toxicity; information regarding toxicity was missing on 2 patients. Thirty-two of 65 (49.9%) patients had late toxicity of which 20 (30.8%) had late grade ≥ 2 toxicity. The main factor associated with development of acute toxicity was pretreatment oxygendependence (P = .047). This was not significant when comparing only inoperable patients. Twenty patients (29.9%) developed some type of acute toxicity; pulmonary toxicity was most common (22.4%) (Table 3). All patients with acute toxicity also developed late toxicity except for 1 who died before 3 months. Predominantly, the deaths in our sample were from causes other than the malignancy or treatment, such as sepsis, deconditioning after a fall, cardiovascular complications, etc. Acute toxicity of grade ≥ 2 was significantly associated with late toxicity (P < .001 for both) in both operable and inoperable patients (P < .001).
Development of any acute toxicity grade ≥ 2 was significantly associated with oxygendependence at baseline (P = .003), central location (P < .001), and new oxygen requirement (P = .02). Only central tumor location was found to be significant (P = .001) within the inoperable cohort. There were no significant differences in outcome based on pulmonary function testing (FEV1, forced vital capacity, or DLCO) or the analyzed PFT subgroups (FEV1 < 1.0 L, FEV1 < 1.5 L, FEV1 < 30%, and FEV1 < 35%).
At the time of data collection, 30 patients were deceased (43.5%). There was a statistically significant association between OS and operability (P = .03; Table 4, Figure 1). Decreased OS was significantly associated with acute toxicity (P = .001) and acute toxicity grade ≥ 2 (P = .005; Figures 2 and 3). For the inoperable patients, both acute toxicity (P < .001) and acute toxicity grade ≥ 2 (P = .026) remained significant.
Discussion
SABR is an effective treatment for inoperable early-stage NSCLC, however its therapeutic ratio in a more frail population who cannot withstand biopsy is not well established. Additionally, the prevalence of benign disease in patients with solitary pulmonary nodules can be between 9% and 21%.6 Haidar and colleagues looked at 55 patients who received empiric SABR and found a median OS of 30.2 months with an 8.7% risk of local failure, 13% risk of regional failure with 8.7% acute toxicity, and 13% chronic toxicity.7 Data from Harkenrider and colleagues (n = 34) revealed similar results with a 2-year OS of 85%, local control of 97.1%, and regional control of 80%. The authors noted no grade ≥ 3 acute toxicities and an incidence of grade ≥ 3 late toxicities of 8.8%.1 These findings are concordant with our study results, confirming the safety and efficacy of SABR. Furthermore, a National Cancer Database analysis of observation vs empiric SABR found an OS of 10.1 months and 29 months respectively, with a hazard ratio of 0.64 (P < .001).3 Additionally, Fischer-Valuck and colleagues (n = 88) compared biopsy confirmed vs unbiopsied patients treated with SABR and found no difference in the 3-year local progression-free survival (93.1% vs 94.1%), regional lymph node metastasis and distant metastases free survival (92.5% vs 87.4%), or OS (59.9% vs 58.9%).8 With a median OS of ≤ 1 year for untreated stage I NSCLC,these studies support treating patients with empiric SABR.4
Other researchers have sought parameters to identify patients for whom radiation therapy would be too toxic. Guckenberger and colleagues aimed to establish a lower limit of pretreatment PFT to exclude patients and found only a 7% incidence of grade ≥ 2 adverse effects and toxicity did not increase with lower pulmonary function.9 They concluded that SABR was safe even for patients with poor pulmonary function. Other institutions have confirmed such findings and have been unable to find a cut-off PFT to exclude patients from empiric SABR.10,11 An analysis from the RTOG 0236 trial also noted that poor baseline PFT could not predict pulmonary toxicity or survival. Additionally, the study demonstrated only minimal decreases in patients’ FEV1 (5.8%) and DLCO (6%) at 2 years.12
Our study sought to identify a cut-off on FEV1 or DLCO that could be associated with increased toxicity. We also evaluated the incidence of acute toxicities grade ≥ 2 by stratifying patients according to FEV1 into subgroups: FEV1 < 1.0 L, FEV1 < 1.5 L, FEV1 < 30% of predicted and FEV1 < 35% of predicted. However, similar to other studies, we did not find any value that was significantly associated with increased toxicity that could preclude empiric SABR. One possible reason is that no treatment is offered for patients with extremely poor lung function as deemed by clinical judgement, therefore data on these patients is unavailable. In contradiction to other studies, our study found that oxygen dependence before treatment was significantly associated with development of acute toxicities. The exact mechanism for this association is unknown and could not be elucidated by baseline PFT. One possible explanation is that SABR could lead to oxygen free radical generation. In addition, our study indicated that those who developed acute toxicities had worse OS.
Limitations
Our study is limited by caveats of a retrospective study and its small sample size, but is in line with the reported literature (ranging from 33 to 88 patients).1,7,8 Another limitation is that data on pretreatment DLCO was missing in 37 patients and the lack of statistical robustness in terms of the smaller inoperable cohort, which limits the analyses of these factors in regards to anticipated morbidity from SABR. Also, given this is data collected from the US Department of Veterans Affairs, only 3% of our sample was female.
Conclusions
Empiric SABR for patients with presumed early-stage NSCLC appears to be safe and might positively impact OS. Development of any acute toxicity grade ≥ 2 was significantly associated with dependence on supplemental oxygen before treatment, central tumor location, and development of new oxygen requirement. No association was found in patients with poor pulmonary function before treatment because we could not find a FEV1 or DLCO cutoff that could preclude patients from empiric SABR. Considering the poor survival of untreated early-stage NSCLC, coupled with the efficacy and safety of empiric SABR for those with presumed disease, definitive SABR should be offered selectively within this patient population.
Acknowledgments
Drs. Park, Whiting and Castillo contributed to data collection. Drs. Park, Govindan and Castillo contributed to the statistical analysis and writing the first draft and final manuscript. Drs. Park, Govindan, Huang, and Reddy contributed to the discussion section.
Stereotactic ablative radiotherapy (SABR) has become the standard of care for inoperable early-stage non-small cell lung cancer (NSCLC). Many patients are unable to undergo a biopsy safely because of poor pulmonary function or underlying emphysema and are then empirically treated with radiotherapy if they meet criteria. In these patients, local control can be achieved with SABR with minimal toxicity.1 Considering that median overall survival (OS) among patients with untreated stage I NSCLC has been reported to be as low as 9 months, early treatment with SABR could lead to increased survival of 29 to 60 months.2-4
The RTOG 0236 trial showed a median OS of 48 months and the randomized phase III CHISEL trial showed a median OS of 60 months; however, these survival data were reported in patients who were able to safely undergo a biopsy and had confirmed NSCLC.4,5 For patients without a diagnosis confirmed by biopsy and who are treated with empiric SABR, patient factors that influence radiation toxicity and OS are not well defined.
It is not clear if empiric radiation benefits survival or if treatment causes decline in lung function, considering that underlying chronic lung disease precludes these patients from biopsy. The purpose of this study was to evaluate the factors associated with radiation toxicity with empiric SABR and to evaluate OS in this population without a biopsy-confirmed diagnosis.
Methods
This was a single center retrospective review of patients treated at the radiation oncology department at the Kansas City Veterans Affairs Medical Center from August 2014 to February 2019. Data were collected on 69 patients with pulmonary nodules identified by chest computed tomography (CT) and/or positron emission tomography (PET)-CT that were highly suspicious for primary NSCLC.
These patients were presented at a multidisciplinary meeting that involved pulmonologists, oncologists, radiation oncologists, and thoracic surgeons. Patients were deemed to be poor candidates for biopsy because of severe underlying emphysema, which would put them at high risk for pneumothorax with a percutaneous needle biopsy, or were unable to tolerate general anesthesia for navigational bronchoscopy or surgical biopsy because of poor lung function. These patients were diagnosed with presumed stage I NSCLC using the criteria: minimum of 2 sequential CT scans with enlarging nodule; absence of metastases on PET-CT; the single nodule had to be fluorodeoxyglucose avid with a minimum standardized uptake value of 2.5, and absence of clinical history or physical examination consistent with small cell lung cancer or infection.
After a consensus was reached that patients met these criteria, individuals were referred for empiric SABR. Follow-up visits were at 1 month, 3 months, and every 6 months. Variables analyzed included: patient demographics, pre- and posttreatment pulmonary function tests (PFT) when available, pre-treatment oxygen use, tumor size and location (peripheral, central, or ultra-central), radiation doses, and grade of toxicity as defined by Human and Health Services Common Terminology Criteria for Adverse Events version 5.0 (dyspnea and cough both counted as pulmonary toxicity): acute ≤ 90 days and late > 90 days (Table 1).
SPSS versions 24 and 26 were used for statistical analysis. Median and range were obtained for continuous variables with a normal distribution. Kaplan-Meier log-rank testing was used to analyze OS. χ2 and Mann-Whitney U tests were used to analyze association between independent variables and OS. Analysis of significant findings were repeated with operable patients excluded for further analysis.
Results
The median follow-up was 18 months (range, 1 to 54). The median age was 71 years (range, 59 to 95) (Table 2). Most patients (97.1%) were male. The majority of patients (79.4%) had a 0 or 1 for the Eastern Cooperative Oncology group performance status, indicating fully active or restricted in physically strenuous activity but ambulatory and able to perform light work. All patients were either current or former smokers with an average pack-year history of 69.4. Only 11.6% of patients had operable disease, but received empiric SABR because they declined surgery. Four patients did not have pretreatment spirometry available and 37 did not have pretreatment diffusing capacity for carbon monoxide (DLCO) data.
Most patients had a pretreatment forced expiratory volume during the first seconds (FEV1) value and DLCO < 60% of predicted (60% and 84% of the patients, respectively). The median tumor diameter was 2 cm. Of the 68.2% of patients who did not have chronic hypoxemic respiratory failure before SABR, 16% developed a new requirement for supplemental oxygen. Sixty-two tumors (89.9%) were peripheral. There were 4 local recurrences (5.7%), 10 regional (different lobe and nodal) failures (14.3%), and 15 distant metastases (21.4%).
Nineteen of 67 patients (26.3%) had acute toxicity of which 9 had acute grade ≥ 2 toxicity; information regarding toxicity was missing on 2 patients. Thirty-two of 65 (49.9%) patients had late toxicity of which 20 (30.8%) had late grade ≥ 2 toxicity. The main factor associated with development of acute toxicity was pretreatment oxygendependence (P = .047). This was not significant when comparing only inoperable patients. Twenty patients (29.9%) developed some type of acute toxicity; pulmonary toxicity was most common (22.4%) (Table 3). All patients with acute toxicity also developed late toxicity except for 1 who died before 3 months. Predominantly, the deaths in our sample were from causes other than the malignancy or treatment, such as sepsis, deconditioning after a fall, cardiovascular complications, etc. Acute toxicity of grade ≥ 2 was significantly associated with late toxicity (P < .001 for both) in both operable and inoperable patients (P < .001).
Development of any acute toxicity grade ≥ 2 was significantly associated with oxygendependence at baseline (P = .003), central location (P < .001), and new oxygen requirement (P = .02). Only central tumor location was found to be significant (P = .001) within the inoperable cohort. There were no significant differences in outcome based on pulmonary function testing (FEV1, forced vital capacity, or DLCO) or the analyzed PFT subgroups (FEV1 < 1.0 L, FEV1 < 1.5 L, FEV1 < 30%, and FEV1 < 35%).
At the time of data collection, 30 patients were deceased (43.5%). There was a statistically significant association between OS and operability (P = .03; Table 4, Figure 1). Decreased OS was significantly associated with acute toxicity (P = .001) and acute toxicity grade ≥ 2 (P = .005; Figures 2 and 3). For the inoperable patients, both acute toxicity (P < .001) and acute toxicity grade ≥ 2 (P = .026) remained significant.
Discussion
SABR is an effective treatment for inoperable early-stage NSCLC, however its therapeutic ratio in a more frail population who cannot withstand biopsy is not well established. Additionally, the prevalence of benign disease in patients with solitary pulmonary nodules can be between 9% and 21%.6 Haidar and colleagues looked at 55 patients who received empiric SABR and found a median OS of 30.2 months with an 8.7% risk of local failure, 13% risk of regional failure with 8.7% acute toxicity, and 13% chronic toxicity.7 Data from Harkenrider and colleagues (n = 34) revealed similar results with a 2-year OS of 85%, local control of 97.1%, and regional control of 80%. The authors noted no grade ≥ 3 acute toxicities and an incidence of grade ≥ 3 late toxicities of 8.8%.1 These findings are concordant with our study results, confirming the safety and efficacy of SABR. Furthermore, a National Cancer Database analysis of observation vs empiric SABR found an OS of 10.1 months and 29 months respectively, with a hazard ratio of 0.64 (P < .001).3 Additionally, Fischer-Valuck and colleagues (n = 88) compared biopsy confirmed vs unbiopsied patients treated with SABR and found no difference in the 3-year local progression-free survival (93.1% vs 94.1%), regional lymph node metastasis and distant metastases free survival (92.5% vs 87.4%), or OS (59.9% vs 58.9%).8 With a median OS of ≤ 1 year for untreated stage I NSCLC,these studies support treating patients with empiric SABR.4
Other researchers have sought parameters to identify patients for whom radiation therapy would be too toxic. Guckenberger and colleagues aimed to establish a lower limit of pretreatment PFT to exclude patients and found only a 7% incidence of grade ≥ 2 adverse effects and toxicity did not increase with lower pulmonary function.9 They concluded that SABR was safe even for patients with poor pulmonary function. Other institutions have confirmed such findings and have been unable to find a cut-off PFT to exclude patients from empiric SABR.10,11 An analysis from the RTOG 0236 trial also noted that poor baseline PFT could not predict pulmonary toxicity or survival. Additionally, the study demonstrated only minimal decreases in patients’ FEV1 (5.8%) and DLCO (6%) at 2 years.12
Our study sought to identify a cut-off on FEV1 or DLCO that could be associated with increased toxicity. We also evaluated the incidence of acute toxicities grade ≥ 2 by stratifying patients according to FEV1 into subgroups: FEV1 < 1.0 L, FEV1 < 1.5 L, FEV1 < 30% of predicted and FEV1 < 35% of predicted. However, similar to other studies, we did not find any value that was significantly associated with increased toxicity that could preclude empiric SABR. One possible reason is that no treatment is offered for patients with extremely poor lung function as deemed by clinical judgement, therefore data on these patients is unavailable. In contradiction to other studies, our study found that oxygen dependence before treatment was significantly associated with development of acute toxicities. The exact mechanism for this association is unknown and could not be elucidated by baseline PFT. One possible explanation is that SABR could lead to oxygen free radical generation. In addition, our study indicated that those who developed acute toxicities had worse OS.
Limitations
Our study is limited by caveats of a retrospective study and its small sample size, but is in line with the reported literature (ranging from 33 to 88 patients).1,7,8 Another limitation is that data on pretreatment DLCO was missing in 37 patients and the lack of statistical robustness in terms of the smaller inoperable cohort, which limits the analyses of these factors in regards to anticipated morbidity from SABR. Also, given this is data collected from the US Department of Veterans Affairs, only 3% of our sample was female.
Conclusions
Empiric SABR for patients with presumed early-stage NSCLC appears to be safe and might positively impact OS. Development of any acute toxicity grade ≥ 2 was significantly associated with dependence on supplemental oxygen before treatment, central tumor location, and development of new oxygen requirement. No association was found in patients with poor pulmonary function before treatment because we could not find a FEV1 or DLCO cutoff that could preclude patients from empiric SABR. Considering the poor survival of untreated early-stage NSCLC, coupled with the efficacy and safety of empiric SABR for those with presumed disease, definitive SABR should be offered selectively within this patient population.
Acknowledgments
Drs. Park, Whiting and Castillo contributed to data collection. Drs. Park, Govindan and Castillo contributed to the statistical analysis and writing the first draft and final manuscript. Drs. Park, Govindan, Huang, and Reddy contributed to the discussion section.
1. Harkenrider MM, Bertke MH, Dunlap NE. Stereotactic body radiation therapy for unbiopsied early-stage lung cancer: a multi-institutional analysis. Am J Clin Oncol. 2014;37(4):337-342. doi:10.1097/COC.0b013e318277d822
2. Raz DJ, Zell JA, Ou SH, Gandara DR, Anton-Culver H, Jablons DM. Natural history of stage I non-small cell lung cancer: implications for early detection. Chest. 2007;132(1):193-199. doi:10.1378/chest.06-3096
3. Nanda RH, Liu Y, Gillespie TW, et al. Stereotactic body radiation therapy versus no treatment for early stage non-small cell lung cancer in medically inoperable elderly patients: a National Cancer Data Base analysis. Cancer. 2015;121(23):4222-4230. doi:10.1002/cncr.29640
4. Ball D, Mai GT, Vinod S, et al. Stereotactic ablative radiotherapy versus standard radiotherapy in stage 1 non-small-cell lung cancer (TROG 09.02 CHISEL): a phase 3, open-label, randomised controlled trial. Lancet Oncol. 2019;20(4):494-503. doi:10.1016/S1470-2045(18)30896-9
5. Timmerman R, Paulus R, Galvin J, et al. Stereotactic body radiation therapy for inoperable early stage lung cancer. JAMA. 2010;303(11):1070-1076. doi:10.1001/jama.2010.261
6. Smith MA, Battafarano RJ, Meyers BF, Zoole JB, Cooper JD, Patterson GA. Prevalence of benign disease in patients undergoing resection for suspected lung cancer. Ann Thorac Surg. 2006;81(5):1824-1828. doi:10.1016/j.athoracsur.2005.11.010
7. Haidar YM, Rahn DA 3rd, Nath S, et al. Comparison of outcomes following stereotactic body radiotherapy for nonsmall cell lung cancer in patients with and without pathological confirmation. Ther Adv Respir Dis. 2014;8(1):3-12. doi:10.1177/1753465813512545
8. Fischer-Valuck BW, Boggs H, Katz S, Durci M, Acharya S, Rosen LR. Comparison of stereotactic body radiation therapy for biopsy-proven versus radiographically diagnosed early-stage non-small lung cancer: a single-institution experience. Tumori. 2015;101(3):287-293. doi:10.5301/tj.5000279
9. Guckenberger M, Kestin LL, Hope AJ, et al. Is there a lower limit of pretreatment pulmonary function for safe and effective stereotactic body radiotherapy for early-stage non-small cell lung cancer? J Thorac Oncol. 2012;7:542-551. doi:10.1097/JTO.0b013e31824165d7
10. Wang J, Cao J, Yuan S, et al. Poor baseline pulmonary function may not increase the risk of radiation-induced lung toxicity. Int J Radiat Oncol Biol Phys. 2013;85(3):798-804. doi:10.1016/j.ijrobp.2012.06.040
11. Henderson M, McGarry R, Yiannoutsos C, et al. Baseline pulmonary function as a predictor for survival and decline in pulmonary function over time in patients undergoing stereotactic body radiotherapy for the treatment of stage I non-small-cell lung cancer. Int J Radiat Oncol Biol Phys. 2008;72(2):404-409. doi:10.1016/j.ijrobp.2007.12.051
12. Stanic S, Paulus R, Timmerman RD, et al. No clinically significant changes in pulmonary function following stereotactic body radiation therapy for early- stage peripheral non-small cell lung cancer: an analysis of RTOG 0236. Int J Radiat Oncol Biol Phys. 2014;88(5):1092-1099. doi:10.1016/j.ijrobp.2013.12.050
1. Harkenrider MM, Bertke MH, Dunlap NE. Stereotactic body radiation therapy for unbiopsied early-stage lung cancer: a multi-institutional analysis. Am J Clin Oncol. 2014;37(4):337-342. doi:10.1097/COC.0b013e318277d822
2. Raz DJ, Zell JA, Ou SH, Gandara DR, Anton-Culver H, Jablons DM. Natural history of stage I non-small cell lung cancer: implications for early detection. Chest. 2007;132(1):193-199. doi:10.1378/chest.06-3096
3. Nanda RH, Liu Y, Gillespie TW, et al. Stereotactic body radiation therapy versus no treatment for early stage non-small cell lung cancer in medically inoperable elderly patients: a National Cancer Data Base analysis. Cancer. 2015;121(23):4222-4230. doi:10.1002/cncr.29640
4. Ball D, Mai GT, Vinod S, et al. Stereotactic ablative radiotherapy versus standard radiotherapy in stage 1 non-small-cell lung cancer (TROG 09.02 CHISEL): a phase 3, open-label, randomised controlled trial. Lancet Oncol. 2019;20(4):494-503. doi:10.1016/S1470-2045(18)30896-9
5. Timmerman R, Paulus R, Galvin J, et al. Stereotactic body radiation therapy for inoperable early stage lung cancer. JAMA. 2010;303(11):1070-1076. doi:10.1001/jama.2010.261
6. Smith MA, Battafarano RJ, Meyers BF, Zoole JB, Cooper JD, Patterson GA. Prevalence of benign disease in patients undergoing resection for suspected lung cancer. Ann Thorac Surg. 2006;81(5):1824-1828. doi:10.1016/j.athoracsur.2005.11.010
7. Haidar YM, Rahn DA 3rd, Nath S, et al. Comparison of outcomes following stereotactic body radiotherapy for nonsmall cell lung cancer in patients with and without pathological confirmation. Ther Adv Respir Dis. 2014;8(1):3-12. doi:10.1177/1753465813512545
8. Fischer-Valuck BW, Boggs H, Katz S, Durci M, Acharya S, Rosen LR. Comparison of stereotactic body radiation therapy for biopsy-proven versus radiographically diagnosed early-stage non-small lung cancer: a single-institution experience. Tumori. 2015;101(3):287-293. doi:10.5301/tj.5000279
9. Guckenberger M, Kestin LL, Hope AJ, et al. Is there a lower limit of pretreatment pulmonary function for safe and effective stereotactic body radiotherapy for early-stage non-small cell lung cancer? J Thorac Oncol. 2012;7:542-551. doi:10.1097/JTO.0b013e31824165d7
10. Wang J, Cao J, Yuan S, et al. Poor baseline pulmonary function may not increase the risk of radiation-induced lung toxicity. Int J Radiat Oncol Biol Phys. 2013;85(3):798-804. doi:10.1016/j.ijrobp.2012.06.040
11. Henderson M, McGarry R, Yiannoutsos C, et al. Baseline pulmonary function as a predictor for survival and decline in pulmonary function over time in patients undergoing stereotactic body radiotherapy for the treatment of stage I non-small-cell lung cancer. Int J Radiat Oncol Biol Phys. 2008;72(2):404-409. doi:10.1016/j.ijrobp.2007.12.051
12. Stanic S, Paulus R, Timmerman RD, et al. No clinically significant changes in pulmonary function following stereotactic body radiation therapy for early- stage peripheral non-small cell lung cancer: an analysis of RTOG 0236. Int J Radiat Oncol Biol Phys. 2014;88(5):1092-1099. doi:10.1016/j.ijrobp.2013.12.050
Impact of an Oral Antineoplastic Renewal Clinic on Medication Possession Ratio and Cost-Savings
Evaluation of oral antineoplastic agent (OAN) adherence patterns have identified correlations between nonadherence or over-adherence and poorer disease-related outcomes. Multiple studies have focused on imatinib use in chronic myeloid leukemia (CML) due to its continuous, long-term use. A study by Ganesan and colleagues found that nonadherence to imatinib showed a significant decrease in 5-year event-free survival between 76.7% of adherent participants compared with 59.8% of nonadherent participants.1 This study found that 44% of patients who were adherent to imatinib achieved complete cytogenetic response vs only 26% of patients who were nonadherent. In another study of imatinib for CML, major molecular response (MMR) was strongly correlated with adherence and no patients with adherence < 80% were able to achieve MMR.2 Similarly, in studies of tamoxifen for breast cancer, < 80% adherence resulted in a 10% decrease in survival when compared to those who were more adherent.3,4
In addition to the clinical implications of nonadherence, there can be a significant cost associated with suboptimal use of these medications. The price of a single dose of OAN medication may cost as much as $440.5
The benefits of multidisciplinary care teams have been identified in many studies.6,7 While studies are limited in oncology, pharmacists provide vital contributions to the oncology multidisciplinary team when managing OANs as these health care professionals have expert knowledge of the medications, potential adverse events (AEs), and necessary monitoring parameters.8 In one study, patients seen by the pharmacist-led oral chemotherapy management program experienced improved clinical outcomes and response to therapy when compared with preintervention patients (early molecular response, 88.9% vs 54.8%, P = .01; major molecular response, 83.3% vs 57.6%, P = .06).9 During the study, 318 AEs were reported, leading to 235 pharmacist interventions to ameliorate AEs and improve adherence.
The primary objective of this study was to measure the impact of a pharmacist-driven OAN renewal clinic on medication adherence. The secondary objective was to estimate cost-savings of this new service.
Methods
Prior to July 2014, several limitations were identified related to OAN prescribing and monitoring at the Richard L. Roudebush Veterans Affairs Medical Center in Indianapolis, Indiana (RLRVAMC). The prescription ordering process relied primarily on the patient to initiate refills, rather than the prescriber OAN prescriptions also lacked consistency for number of refills or quantities dispensed. Furthermore, ordering of antineoplastic products was not limited to hematology/oncology providers. Patients were identified with significant supply on hand at the time of medication discontinuation, creating concerns for medication waste, tolerability, and nonadherence.
As a result, opportunities were identified to improve the prescribing process, recommended monitoring, toxicity and tolerability evaluation, medication reconciliation, and medication adherence. In July of 2014, the RLRVAMC adopted a new chemotherapy order entry system capable of restricting prescriptions to hematology/oncology providers and limiting dispensed quantities and refill amounts. A comprehensive pharmacist driven OAN renewal clinic was implemented on September 1, 2014 with the goal of improving long-term adherence and tolerability, in addition to minimizing medication waste.
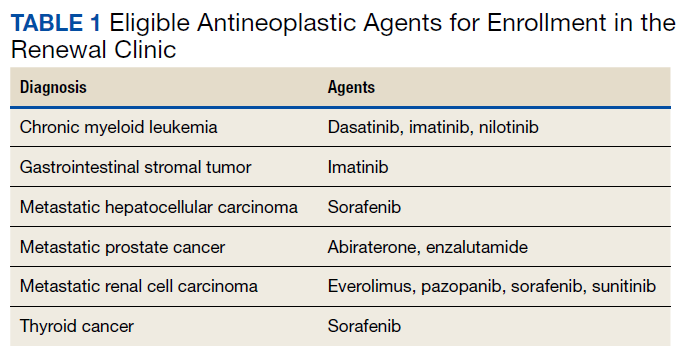
Patients were eligible for enrollment in the clinic if they had a cancer diagnosis and were concomitantly prescribed an OAN outlined in Table 1. All eligible patients were automatically enrolled in the clinic when they were deemed stable on their OAN by a hematology/oncology pharmacy specialist. Stability was defined as ≤ Grade 1 symptoms associated with the toxicities of OAN therapy managed with or without intervention as defined by the Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. Once enrolled in the renewal clinic, patients were called by an oncology pharmacy resident (PGY2) 1 week prior to any OAN refill due date. Patients were asked a series of 5 adherence and tolerability questions (Table 2) to evaluate renewal criteria for approval or need for further evaluation. These questions were developed based on targeted information and published reports on monitoring adherence.10,11 Criteria for renewal included: < 10% self-reported missed doses of the OAN during the previous dispensing period, no hospitalizations or emergency department visits since most recent hematology/oncology provider appointment, no changes to concomitant medication therapies, and no new or worsening medication-related AEs. Patients meeting all criteria were given a 30-day supply of OAN. Prescribing, dispensing, and delivery of OAN were facilitated by the pharmacist. Patient cases that did not meet criteria for renewal were escalated to the hematology/oncology provider or oncology clinical pharmacy specialist for further evaluation.

Study Design and Setting
This was a pre/post retrospective cohort, quality improvement study of patients enrolled in the RLRVAMC OAN pharmacist renewal clinic. The study was deemed exempt from institutional review board (IRB) by the US Department of Veterans Affairs (VA) Research and Development Department.
Study Population
Patients were included in the preimplementation group if they had received at least 2 prescriptions of an eligible OAN. Therapy for the preimplementation group was required to be a monthly duration > 21 days and between the dates of September 1, 2013 and August 31, 2014. Patients were included in the postimplementation group if they had received at least 2 prescriptions of the studied OANs between September 1, 2014 and January 31, 2015. Patients were excluded if they had filled < 2 prescriptions of OAN; were managed by a non-VA oncologist or hematologist; or received an OAN other than those listed in Table 1.
Data Collection
For all patients in both the pre- and postimplementation cohorts, a standardized data collection tool was used to collect the following via electronic health record review by a PGY2 oncology resident: age, race, gender, oral antineoplastic agent, refill dates, days’ supply, estimated unit cost per dose cancer diagnosis, distance from the RLRVAMC, copay status, presence of hospitalizations/ED visits/dosage reductions, discontinuation rates, reasons for discontinuation, and total number of current prescriptions. The presence or absence of dosage reductions were collected to identify concerns for tolerability, but only the original dose for the preimplementation group and dosage at time of clinic enrollment for the postimplementation group was included in the analysis.
Outcomes and Statistical Analyses
The primary outcome was medication adherence defined as the median medication possession ratio (MPR) before and after implementation of the clinic. Secondary outcomes included the proportion of patients who were adherent from before implementation to after implementation and estimated cost-savings of this clinic after implementation. MPR was used to estimate medication adherence by taking the cumulative day supply of medication on hand divided by the number of days on therapy.12 Number of days on therapy was determined by taking the difference on the start date of the new medication regimen and the discontinuation date of the same regimen. Patients were grouped by adherence into one of the following categories: < 0.8, 0.8 to 0.89, 0.9 to 1, and > 1.1. Patients were considered adherent if they reported taking ≥ 90% (MPR ≥ 0.9) of prescribed doses, adopted from the study by Anderson and colleagues.12 A patient with an MPR > 1, likely due to filling prior to the anticipated refill date, was considered 100% adherent (MPR = 1). If a patient switched OAN during the study, both agents were included as separate entities.
A conservative estimate of cost-savings was made by multiplying the RLRVAMC cost per unit of medication at time of initial prescription fill by the number of units taken each day multiplied by the total days’ supply on hand at time of therapy discontinuation. Patients with an MPR < 1 at time of therapy discontinuation were assumed to have zero remaining units on hand and zero cost savings was estimated. Waste, for purposes of cost-savings, was calculated for all MPR values > 1. Additional supply anticipated to be on hand from dose reductions was not included in the estimated cost of unused medication.
Descriptive statistics compared demographic characteristics between the pre- and postimplementation groups. MPR data were not normally distributed, which required the use of nonparametric Mann-Whitney U tests to compare pre- and postMPRs. Pearson χ2 compared the proportion of adherent patients between groups while descriptive statistics were used to estimate cost savings. Significance was determined based on a P value < .05. IBM SPSS Statistics software was used for all statistical analyses. As this was a complete sample of all eligible subjects, no sample size calculation was performed.
Results
In the preimplementation period, 246 patients received an OAN and 61 patients received an OAN in the postimplementation period (Figure 1). Of the 246 patients in the preimplementation period, 98 were eligible and included in the preimplementation group. Similarly, of the 61 patients in the postimplementation period, 35 patients met inclusion criteria for the postimplementation group. The study population was predominantly male with an average age of approximately 70 years in both groups (Table 3). More than 70% of the population in each group was White. No statistically significant differences between groups were identified. The most commonly prescribed OAN in the preimplementation group were abiraterone, imatinib, and enzalutamide (Table 3). In the postimplementation group, the most commonly prescribed agents were abiraterone, imatinib, pazopanib, and dasatinib. No significant differences were observed in prescribing of individual agents between the pre- and postimplementation groups or other characteristics that may affect adherence including patient copay status, number of concomitant medications, and driving distance from the RLRVAMC.
Thirty-six (36.7%) patients in the preimplementation group were considered nonadherent (MPR < 0.9) and 18 (18.4%) had an MPR < 0.8. Fifteen (15.3%) patients in the preimplementation clinic were considered overadherent (MPR > 1.1). Forty-seven (47.9%) patients in the preimplementation group were considered adherent (MPR 0.9 - 1.1) while all 35 (100%) patients in the postimplementation group were considered adherent (MPR 0.9 - 1.1). No non- or overadherent patients were identified in the postimplementation group (Figure 2). The median MPR for all patients in the preimplementation group was 0.94 compared with 1.06 (P < .001) in the postimplementation group.
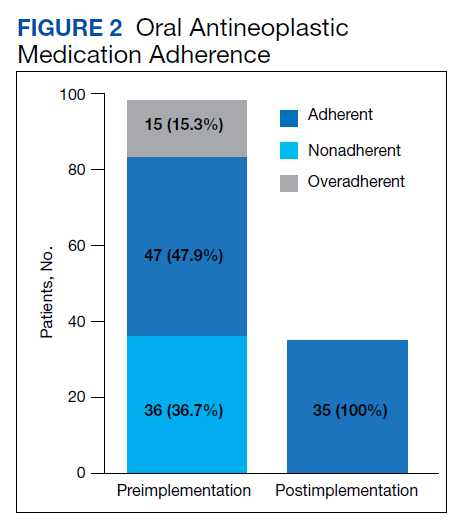

Thirty-five (35.7%) patients had therapy discontinued or held in the preimplementation group compared with 2 (5.7%) patients in the postimplementation group (P < .001). Reasons for discontinuation in the preimplementation group included disease progression (n = 27), death (n = 3), lost to follow up (n = 2), and intolerability of therapy (n = 3). Both patients that discontinued therapy in the postimplementation group did so due to disease progression. Of the 35 patients who had their OAN discontinued or held in the preimplementation group, 14 patients had excess supply on hand at time of discontinuation. The estimated value of the unused medication was $37,890. Nine (25%) of the 35 patients who discontinued therapy had a dosage reduction during the course of therapy and the additional supply was not included in the cost estimate. Similarly, 1 of the 2 patients in the postimplementation group had their OAN discontinued during study. The cost of oversupply of medication at the time of therapy discontinuation was estimated at $1,555. No patients in the postimplementation group had dose reductions. After implementation of the OAN renewal clinic, the total cost savings between pre ($37,890) and postimplementation ($1,555) groups was $36,355.
Discussion
OANs are widely used therapies, with more than 25 million doses administered per year in the United States alone.12 The use of these agents will continue to grow as more targeted agents become available and patients request more convenient treatment options. The role for hematology/oncology clinical pharmacy services must adapt to this increased usage of OANs, including increasing pharmacist involvement in medication education, adherence and tolerability assessments, and proactive drug interaction monitoring.However, additional research is needed to determine optimal management strategies.
Our study aimed to compare OAN adherence among patients at a tertiary care VA hospital before and after implementation of a renewal clinic. The preimplementation population had a median MPR of 0.94 compared with 1.06 in the postimplementation group (P < .001). Although an ideal MPR is 1.0, we aimed for a slightly higher MPR to allow a supply buffer in the event of prescription delivery delays, as more than 90% of prescriptions are mailed to patients from a regional mail-order pharmacy. Importantly, the median MPRs do not adequately convey the impact from this clinic. The proportion of patients who were considered adherent to OANs increased from 47.9% in the preimplementation to 100% in the postimplementation period. These finding suggest that the clinical pharmacist role to assess and encourage adherence through monitoring tolerability of these OANs improved the overall medication taking experience of these patients.
Upon initial evaluation of adherence pre- and postimplementation, median adherence rates in both groups appeared to be above goal at 0.94 and 1.06 respectively. Patients in the postimplementation group intentionally received a 5- to 7-day supply buffer to account for potential prescription delivery delays due to holidays and inclement weather. This would indicate that the patients in the postimplementation group would have 15% oversupply due to the 5-day supply buffer. After correcting for patients with confounding reasons for excess (dose reductions, breaks in treatment, etc.), the median MPR in the prerefill clinic group decreased to 0.9 and the MPR in the postrefill clinic group increased slightly to 1.08. Although the median adherence rate in both the pre- and postimplementation groups were above goal of 0.90, 36% of the patients in the preimplementation group were considered nonadherent (MPR < 0.9) compared with no patients in the postimplementation group. Therefore, our intervention to improve patient adherence appeared to be beneficial at our institution.
In addition to improving adherence, one of the goals of the renewal clinic was to minimize excess supply at the time of therapy discontinuation. This was accomplished by aligning medication fills with medical visits and objective monitoring, as well as limiting supply to no more than 30 days. Of the patients in the postimplementation group, only 1 patient had remaining medication at the time of therapy discontinuation compared with 14 patients in the preimplementation group. The estimated cost savings from excess supply was $36,335. Limiting the amount of unused supply not only saves money for the patient and the institution, but also decreases opportunity for improper hazardous waste disposal and unnecessary exposure of hazardous materials to others.
Our results show the pharmacist intervention in the coordination of renewals improved adherence, minimized medication waste, and saved money. The cost of pharmacist time participating in the refill clinic was not calculated. Each visit was completed in approximately 5 minutes, with subsequent documentation and coordination taking an additional 5 to 10 minutes. During the launch of this service, the oncology pharmacy resident provided all coverage of the clinic. Oversite of the resident was provided by hematology/oncology clinical pharmacy specialists. We have continued to utilize pharmacy resident coverage since that time to meet education needs and keep the estimated cost per visit low. Another option in the case that pharmacy residents are not available would be utilization of a pharmacy technician, intern, or professional student to conduct the adherence and tolerability phone assessments. Our escalation protocol allows intervention by clinical pharmacy specialist and/or other health care providers when necessary. Trainees have only required basic training on how to use the protocol.
Limitations
Due to this study’s retrospective design, an inherent limitation is dependence on prescriber and refill records for documentation of initiation and discontinuation dates. Therefore, only the association of impact of pharmacist intervention on medication adherence can be determined as opposed to causation. We did not take into account discrepancies in day supply secondary to ‘held’ therapies, dose reductions, or doses supplied during an inpatient admission, which may alter estimates of MPR and cost-savings data. Patients in the postimplementation group intentionally received a 5 to 7-day supply buffer to account for potential prescription delivery delays due to holidays and inclement weather. This would indicate that the patients in the postimplementation group would have 15% oversupply due to the 5-day supply buffer, thereby skewing MPR values. This study did not account for cost avoidance resulting from early identification and management of toxicity. Finally, the postimplementation data only spans 4 months and a longer duration of time is needed to more accurately determine sustainability of renewal clinic interventions and provide comprehensive evaluation of cost-avoidance.
Conclusion
Implementation of an OAN renewal clinic was associated with an increase in MPR, improved proportion of patients considered adherent, and an estimated $36,335 cost-savings. However, prospective evaluation and a longer study duration are needed to determine causality of improved adherence and cost-savings associated with a pharmacist-driven OAN renewal clinic.
1. Ganesan P, Sagar TG, Dubashi B, et al. Nonadherence to imatinib adversely affects event free survival in chronic phase chronic myeloid leukemia. Am J Hematol 2011; 86: 471-474. doi:10.1002/ajh.22019
2. Marin D, Bazeos A, Mahon FX, et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J Clin Oncol 2010; 28: 2381-2388. doi:10.1200/JCO.2009.26.3087
3. McCowan C, Shearer J, Donnan PT, et al. Cohort study examining tamoxifen adherence and its relationship to mortality in women with breast cancer. Br J Cancer 2008; 99: 1763-1768. doi:10.1038/sj.bjc.6604758
4. Lexicomp Online. Sunitinib. Hudson, Ohio: Lexi-Comp, Inc; August 20, 2019.
5. Babiker A, El Husseini M, Al Nemri A, et al. Health care professional development: Working as a team to improve patient care. Sudan J Paediatr. 2014;14(2):9-16.
6. Spence MM, Makarem AF, Reyes SL, et al. Evaluation of an outpatient pharmacy clinical services program on adherence and clinical outcomes among patients with diabetes and/or coronary artery disease. J Manag Care Spec Pharm. 2014;20(10):1036-1045. doi:10.18553/jmcp.2014.20.10.1036
7. Holle LM, Puri S, Clement JM. Physician-pharmacist collaboration for oral chemotherapy monitoring: Insights from an academic genitourinary oncology practice. J Oncol Pharm Pract 2015; doi:10.1177/1078155215581524
8. Muluneh B, Schneider M, Faso A, et al. Improved Adherence Rates and Clinical Outcomes of an Integrated, Closed-Loop, Pharmacist-Led Oral Chemotherapy Management Program. Journal of Oncology Practice. 2018;14(6):371-333. doi:10.1200/JOP.17.00039.
9. Font R, Espinas JA, Gil-Gil M, et al. Prescription refill, patient self-report and physician report in assessing adherence to oral endocrine therapy in early breast cancer patients: a retrospective cohort study in Catalonia, Spain. British Journal of Cancer. 2012 ;107(8):1249-1256. doi:10.1038/bjc.2012.389.
10. Anderson KR, Chambers CR, Lam N, et al. Medication adherence among adults prescribed imatinib, dasatinib, or nilotinib for the treatment of chronic myeloid leukemia. J Oncol Pharm Practice. 2015;21(1):19–25. doi:10.1177/1078155213520261
11. Weingart SN, Brown E, Bach PB, et al. NCCN Task Force Report: oral chemotherapy. J Natl Compr Canc Netw. 2008;6(3): S1-S14.
Evaluation of oral antineoplastic agent (OAN) adherence patterns have identified correlations between nonadherence or over-adherence and poorer disease-related outcomes. Multiple studies have focused on imatinib use in chronic myeloid leukemia (CML) due to its continuous, long-term use. A study by Ganesan and colleagues found that nonadherence to imatinib showed a significant decrease in 5-year event-free survival between 76.7% of adherent participants compared with 59.8% of nonadherent participants.1 This study found that 44% of patients who were adherent to imatinib achieved complete cytogenetic response vs only 26% of patients who were nonadherent. In another study of imatinib for CML, major molecular response (MMR) was strongly correlated with adherence and no patients with adherence < 80% were able to achieve MMR.2 Similarly, in studies of tamoxifen for breast cancer, < 80% adherence resulted in a 10% decrease in survival when compared to those who were more adherent.3,4
In addition to the clinical implications of nonadherence, there can be a significant cost associated with suboptimal use of these medications. The price of a single dose of OAN medication may cost as much as $440.5
The benefits of multidisciplinary care teams have been identified in many studies.6,7 While studies are limited in oncology, pharmacists provide vital contributions to the oncology multidisciplinary team when managing OANs as these health care professionals have expert knowledge of the medications, potential adverse events (AEs), and necessary monitoring parameters.8 In one study, patients seen by the pharmacist-led oral chemotherapy management program experienced improved clinical outcomes and response to therapy when compared with preintervention patients (early molecular response, 88.9% vs 54.8%, P = .01; major molecular response, 83.3% vs 57.6%, P = .06).9 During the study, 318 AEs were reported, leading to 235 pharmacist interventions to ameliorate AEs and improve adherence.
The primary objective of this study was to measure the impact of a pharmacist-driven OAN renewal clinic on medication adherence. The secondary objective was to estimate cost-savings of this new service.
Methods
Prior to July 2014, several limitations were identified related to OAN prescribing and monitoring at the Richard L. Roudebush Veterans Affairs Medical Center in Indianapolis, Indiana (RLRVAMC). The prescription ordering process relied primarily on the patient to initiate refills, rather than the prescriber OAN prescriptions also lacked consistency for number of refills or quantities dispensed. Furthermore, ordering of antineoplastic products was not limited to hematology/oncology providers. Patients were identified with significant supply on hand at the time of medication discontinuation, creating concerns for medication waste, tolerability, and nonadherence.
As a result, opportunities were identified to improve the prescribing process, recommended monitoring, toxicity and tolerability evaluation, medication reconciliation, and medication adherence. In July of 2014, the RLRVAMC adopted a new chemotherapy order entry system capable of restricting prescriptions to hematology/oncology providers and limiting dispensed quantities and refill amounts. A comprehensive pharmacist driven OAN renewal clinic was implemented on September 1, 2014 with the goal of improving long-term adherence and tolerability, in addition to minimizing medication waste.
Patients were eligible for enrollment in the clinic if they had a cancer diagnosis and were concomitantly prescribed an OAN outlined in Table 1. All eligible patients were automatically enrolled in the clinic when they were deemed stable on their OAN by a hematology/oncology pharmacy specialist. Stability was defined as ≤ Grade 1 symptoms associated with the toxicities of OAN therapy managed with or without intervention as defined by the Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. Once enrolled in the renewal clinic, patients were called by an oncology pharmacy resident (PGY2) 1 week prior to any OAN refill due date. Patients were asked a series of 5 adherence and tolerability questions (Table 2) to evaluate renewal criteria for approval or need for further evaluation. These questions were developed based on targeted information and published reports on monitoring adherence.10,11 Criteria for renewal included: < 10% self-reported missed doses of the OAN during the previous dispensing period, no hospitalizations or emergency department visits since most recent hematology/oncology provider appointment, no changes to concomitant medication therapies, and no new or worsening medication-related AEs. Patients meeting all criteria were given a 30-day supply of OAN. Prescribing, dispensing, and delivery of OAN were facilitated by the pharmacist. Patient cases that did not meet criteria for renewal were escalated to the hematology/oncology provider or oncology clinical pharmacy specialist for further evaluation.
Study Design and Setting
This was a pre/post retrospective cohort, quality improvement study of patients enrolled in the RLRVAMC OAN pharmacist renewal clinic. The study was deemed exempt from institutional review board (IRB) by the US Department of Veterans Affairs (VA) Research and Development Department.
Study Population
Patients were included in the preimplementation group if they had received at least 2 prescriptions of an eligible OAN. Therapy for the preimplementation group was required to be a monthly duration > 21 days and between the dates of September 1, 2013 and August 31, 2014. Patients were included in the postimplementation group if they had received at least 2 prescriptions of the studied OANs between September 1, 2014 and January 31, 2015. Patients were excluded if they had filled < 2 prescriptions of OAN; were managed by a non-VA oncologist or hematologist; or received an OAN other than those listed in Table 1.
Data Collection
For all patients in both the pre- and postimplementation cohorts, a standardized data collection tool was used to collect the following via electronic health record review by a PGY2 oncology resident: age, race, gender, oral antineoplastic agent, refill dates, days’ supply, estimated unit cost per dose cancer diagnosis, distance from the RLRVAMC, copay status, presence of hospitalizations/ED visits/dosage reductions, discontinuation rates, reasons for discontinuation, and total number of current prescriptions. The presence or absence of dosage reductions were collected to identify concerns for tolerability, but only the original dose for the preimplementation group and dosage at time of clinic enrollment for the postimplementation group was included in the analysis.
Outcomes and Statistical Analyses
The primary outcome was medication adherence defined as the median medication possession ratio (MPR) before and after implementation of the clinic. Secondary outcomes included the proportion of patients who were adherent from before implementation to after implementation and estimated cost-savings of this clinic after implementation. MPR was used to estimate medication adherence by taking the cumulative day supply of medication on hand divided by the number of days on therapy.12 Number of days on therapy was determined by taking the difference on the start date of the new medication regimen and the discontinuation date of the same regimen. Patients were grouped by adherence into one of the following categories: < 0.8, 0.8 to 0.89, 0.9 to 1, and > 1.1. Patients were considered adherent if they reported taking ≥ 90% (MPR ≥ 0.9) of prescribed doses, adopted from the study by Anderson and colleagues.12 A patient with an MPR > 1, likely due to filling prior to the anticipated refill date, was considered 100% adherent (MPR = 1). If a patient switched OAN during the study, both agents were included as separate entities.
A conservative estimate of cost-savings was made by multiplying the RLRVAMC cost per unit of medication at time of initial prescription fill by the number of units taken each day multiplied by the total days’ supply on hand at time of therapy discontinuation. Patients with an MPR < 1 at time of therapy discontinuation were assumed to have zero remaining units on hand and zero cost savings was estimated. Waste, for purposes of cost-savings, was calculated for all MPR values > 1. Additional supply anticipated to be on hand from dose reductions was not included in the estimated cost of unused medication.
Descriptive statistics compared demographic characteristics between the pre- and postimplementation groups. MPR data were not normally distributed, which required the use of nonparametric Mann-Whitney U tests to compare pre- and postMPRs. Pearson χ2 compared the proportion of adherent patients between groups while descriptive statistics were used to estimate cost savings. Significance was determined based on a P value < .05. IBM SPSS Statistics software was used for all statistical analyses. As this was a complete sample of all eligible subjects, no sample size calculation was performed.
Results
In the preimplementation period, 246 patients received an OAN and 61 patients received an OAN in the postimplementation period (Figure 1). Of the 246 patients in the preimplementation period, 98 were eligible and included in the preimplementation group. Similarly, of the 61 patients in the postimplementation period, 35 patients met inclusion criteria for the postimplementation group. The study population was predominantly male with an average age of approximately 70 years in both groups (Table 3). More than 70% of the population in each group was White. No statistically significant differences between groups were identified. The most commonly prescribed OAN in the preimplementation group were abiraterone, imatinib, and enzalutamide (Table 3). In the postimplementation group, the most commonly prescribed agents were abiraterone, imatinib, pazopanib, and dasatinib. No significant differences were observed in prescribing of individual agents between the pre- and postimplementation groups or other characteristics that may affect adherence including patient copay status, number of concomitant medications, and driving distance from the RLRVAMC.
Thirty-six (36.7%) patients in the preimplementation group were considered nonadherent (MPR < 0.9) and 18 (18.4%) had an MPR < 0.8. Fifteen (15.3%) patients in the preimplementation clinic were considered overadherent (MPR > 1.1). Forty-seven (47.9%) patients in the preimplementation group were considered adherent (MPR 0.9 - 1.1) while all 35 (100%) patients in the postimplementation group were considered adherent (MPR 0.9 - 1.1). No non- or overadherent patients were identified in the postimplementation group (Figure 2). The median MPR for all patients in the preimplementation group was 0.94 compared with 1.06 (P < .001) in the postimplementation group.
Thirty-five (35.7%) patients had therapy discontinued or held in the preimplementation group compared with 2 (5.7%) patients in the postimplementation group (P < .001). Reasons for discontinuation in the preimplementation group included disease progression (n = 27), death (n = 3), lost to follow up (n = 2), and intolerability of therapy (n = 3). Both patients that discontinued therapy in the postimplementation group did so due to disease progression. Of the 35 patients who had their OAN discontinued or held in the preimplementation group, 14 patients had excess supply on hand at time of discontinuation. The estimated value of the unused medication was $37,890. Nine (25%) of the 35 patients who discontinued therapy had a dosage reduction during the course of therapy and the additional supply was not included in the cost estimate. Similarly, 1 of the 2 patients in the postimplementation group had their OAN discontinued during study. The cost of oversupply of medication at the time of therapy discontinuation was estimated at $1,555. No patients in the postimplementation group had dose reductions. After implementation of the OAN renewal clinic, the total cost savings between pre ($37,890) and postimplementation ($1,555) groups was $36,355.
Discussion
OANs are widely used therapies, with more than 25 million doses administered per year in the United States alone.12 The use of these agents will continue to grow as more targeted agents become available and patients request more convenient treatment options. The role for hematology/oncology clinical pharmacy services must adapt to this increased usage of OANs, including increasing pharmacist involvement in medication education, adherence and tolerability assessments, and proactive drug interaction monitoring.However, additional research is needed to determine optimal management strategies.
Our study aimed to compare OAN adherence among patients at a tertiary care VA hospital before and after implementation of a renewal clinic. The preimplementation population had a median MPR of 0.94 compared with 1.06 in the postimplementation group (P < .001). Although an ideal MPR is 1.0, we aimed for a slightly higher MPR to allow a supply buffer in the event of prescription delivery delays, as more than 90% of prescriptions are mailed to patients from a regional mail-order pharmacy. Importantly, the median MPRs do not adequately convey the impact from this clinic. The proportion of patients who were considered adherent to OANs increased from 47.9% in the preimplementation to 100% in the postimplementation period. These finding suggest that the clinical pharmacist role to assess and encourage adherence through monitoring tolerability of these OANs improved the overall medication taking experience of these patients.
Upon initial evaluation of adherence pre- and postimplementation, median adherence rates in both groups appeared to be above goal at 0.94 and 1.06 respectively. Patients in the postimplementation group intentionally received a 5- to 7-day supply buffer to account for potential prescription delivery delays due to holidays and inclement weather. This would indicate that the patients in the postimplementation group would have 15% oversupply due to the 5-day supply buffer. After correcting for patients with confounding reasons for excess (dose reductions, breaks in treatment, etc.), the median MPR in the prerefill clinic group decreased to 0.9 and the MPR in the postrefill clinic group increased slightly to 1.08. Although the median adherence rate in both the pre- and postimplementation groups were above goal of 0.90, 36% of the patients in the preimplementation group were considered nonadherent (MPR < 0.9) compared with no patients in the postimplementation group. Therefore, our intervention to improve patient adherence appeared to be beneficial at our institution.
In addition to improving adherence, one of the goals of the renewal clinic was to minimize excess supply at the time of therapy discontinuation. This was accomplished by aligning medication fills with medical visits and objective monitoring, as well as limiting supply to no more than 30 days. Of the patients in the postimplementation group, only 1 patient had remaining medication at the time of therapy discontinuation compared with 14 patients in the preimplementation group. The estimated cost savings from excess supply was $36,335. Limiting the amount of unused supply not only saves money for the patient and the institution, but also decreases opportunity for improper hazardous waste disposal and unnecessary exposure of hazardous materials to others.
Our results show the pharmacist intervention in the coordination of renewals improved adherence, minimized medication waste, and saved money. The cost of pharmacist time participating in the refill clinic was not calculated. Each visit was completed in approximately 5 minutes, with subsequent documentation and coordination taking an additional 5 to 10 minutes. During the launch of this service, the oncology pharmacy resident provided all coverage of the clinic. Oversite of the resident was provided by hematology/oncology clinical pharmacy specialists. We have continued to utilize pharmacy resident coverage since that time to meet education needs and keep the estimated cost per visit low. Another option in the case that pharmacy residents are not available would be utilization of a pharmacy technician, intern, or professional student to conduct the adherence and tolerability phone assessments. Our escalation protocol allows intervention by clinical pharmacy specialist and/or other health care providers when necessary. Trainees have only required basic training on how to use the protocol.
Limitations
Due to this study’s retrospective design, an inherent limitation is dependence on prescriber and refill records for documentation of initiation and discontinuation dates. Therefore, only the association of impact of pharmacist intervention on medication adherence can be determined as opposed to causation. We did not take into account discrepancies in day supply secondary to ‘held’ therapies, dose reductions, or doses supplied during an inpatient admission, which may alter estimates of MPR and cost-savings data. Patients in the postimplementation group intentionally received a 5 to 7-day supply buffer to account for potential prescription delivery delays due to holidays and inclement weather. This would indicate that the patients in the postimplementation group would have 15% oversupply due to the 5-day supply buffer, thereby skewing MPR values. This study did not account for cost avoidance resulting from early identification and management of toxicity. Finally, the postimplementation data only spans 4 months and a longer duration of time is needed to more accurately determine sustainability of renewal clinic interventions and provide comprehensive evaluation of cost-avoidance.
Conclusion
Implementation of an OAN renewal clinic was associated with an increase in MPR, improved proportion of patients considered adherent, and an estimated $36,335 cost-savings. However, prospective evaluation and a longer study duration are needed to determine causality of improved adherence and cost-savings associated with a pharmacist-driven OAN renewal clinic.
Evaluation of oral antineoplastic agent (OAN) adherence patterns have identified correlations between nonadherence or over-adherence and poorer disease-related outcomes. Multiple studies have focused on imatinib use in chronic myeloid leukemia (CML) due to its continuous, long-term use. A study by Ganesan and colleagues found that nonadherence to imatinib showed a significant decrease in 5-year event-free survival between 76.7% of adherent participants compared with 59.8% of nonadherent participants.1 This study found that 44% of patients who were adherent to imatinib achieved complete cytogenetic response vs only 26% of patients who were nonadherent. In another study of imatinib for CML, major molecular response (MMR) was strongly correlated with adherence and no patients with adherence < 80% were able to achieve MMR.2 Similarly, in studies of tamoxifen for breast cancer, < 80% adherence resulted in a 10% decrease in survival when compared to those who were more adherent.3,4
In addition to the clinical implications of nonadherence, there can be a significant cost associated with suboptimal use of these medications. The price of a single dose of OAN medication may cost as much as $440.5
The benefits of multidisciplinary care teams have been identified in many studies.6,7 While studies are limited in oncology, pharmacists provide vital contributions to the oncology multidisciplinary team when managing OANs as these health care professionals have expert knowledge of the medications, potential adverse events (AEs), and necessary monitoring parameters.8 In one study, patients seen by the pharmacist-led oral chemotherapy management program experienced improved clinical outcomes and response to therapy when compared with preintervention patients (early molecular response, 88.9% vs 54.8%, P = .01; major molecular response, 83.3% vs 57.6%, P = .06).9 During the study, 318 AEs were reported, leading to 235 pharmacist interventions to ameliorate AEs and improve adherence.
The primary objective of this study was to measure the impact of a pharmacist-driven OAN renewal clinic on medication adherence. The secondary objective was to estimate cost-savings of this new service.
Methods
Prior to July 2014, several limitations were identified related to OAN prescribing and monitoring at the Richard L. Roudebush Veterans Affairs Medical Center in Indianapolis, Indiana (RLRVAMC). The prescription ordering process relied primarily on the patient to initiate refills, rather than the prescriber OAN prescriptions also lacked consistency for number of refills or quantities dispensed. Furthermore, ordering of antineoplastic products was not limited to hematology/oncology providers. Patients were identified with significant supply on hand at the time of medication discontinuation, creating concerns for medication waste, tolerability, and nonadherence.
As a result, opportunities were identified to improve the prescribing process, recommended monitoring, toxicity and tolerability evaluation, medication reconciliation, and medication adherence. In July of 2014, the RLRVAMC adopted a new chemotherapy order entry system capable of restricting prescriptions to hematology/oncology providers and limiting dispensed quantities and refill amounts. A comprehensive pharmacist driven OAN renewal clinic was implemented on September 1, 2014 with the goal of improving long-term adherence and tolerability, in addition to minimizing medication waste.
Patients were eligible for enrollment in the clinic if they had a cancer diagnosis and were concomitantly prescribed an OAN outlined in Table 1. All eligible patients were automatically enrolled in the clinic when they were deemed stable on their OAN by a hematology/oncology pharmacy specialist. Stability was defined as ≤ Grade 1 symptoms associated with the toxicities of OAN therapy managed with or without intervention as defined by the Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. Once enrolled in the renewal clinic, patients were called by an oncology pharmacy resident (PGY2) 1 week prior to any OAN refill due date. Patients were asked a series of 5 adherence and tolerability questions (Table 2) to evaluate renewal criteria for approval or need for further evaluation. These questions were developed based on targeted information and published reports on monitoring adherence.10,11 Criteria for renewal included: < 10% self-reported missed doses of the OAN during the previous dispensing period, no hospitalizations or emergency department visits since most recent hematology/oncology provider appointment, no changes to concomitant medication therapies, and no new or worsening medication-related AEs. Patients meeting all criteria were given a 30-day supply of OAN. Prescribing, dispensing, and delivery of OAN were facilitated by the pharmacist. Patient cases that did not meet criteria for renewal were escalated to the hematology/oncology provider or oncology clinical pharmacy specialist for further evaluation.
Study Design and Setting
This was a pre/post retrospective cohort, quality improvement study of patients enrolled in the RLRVAMC OAN pharmacist renewal clinic. The study was deemed exempt from institutional review board (IRB) by the US Department of Veterans Affairs (VA) Research and Development Department.
Study Population
Patients were included in the preimplementation group if they had received at least 2 prescriptions of an eligible OAN. Therapy for the preimplementation group was required to be a monthly duration > 21 days and between the dates of September 1, 2013 and August 31, 2014. Patients were included in the postimplementation group if they had received at least 2 prescriptions of the studied OANs between September 1, 2014 and January 31, 2015. Patients were excluded if they had filled < 2 prescriptions of OAN; were managed by a non-VA oncologist or hematologist; or received an OAN other than those listed in Table 1.
Data Collection
For all patients in both the pre- and postimplementation cohorts, a standardized data collection tool was used to collect the following via electronic health record review by a PGY2 oncology resident: age, race, gender, oral antineoplastic agent, refill dates, days’ supply, estimated unit cost per dose cancer diagnosis, distance from the RLRVAMC, copay status, presence of hospitalizations/ED visits/dosage reductions, discontinuation rates, reasons for discontinuation, and total number of current prescriptions. The presence or absence of dosage reductions were collected to identify concerns for tolerability, but only the original dose for the preimplementation group and dosage at time of clinic enrollment for the postimplementation group was included in the analysis.
Outcomes and Statistical Analyses
The primary outcome was medication adherence defined as the median medication possession ratio (MPR) before and after implementation of the clinic. Secondary outcomes included the proportion of patients who were adherent from before implementation to after implementation and estimated cost-savings of this clinic after implementation. MPR was used to estimate medication adherence by taking the cumulative day supply of medication on hand divided by the number of days on therapy.12 Number of days on therapy was determined by taking the difference on the start date of the new medication regimen and the discontinuation date of the same regimen. Patients were grouped by adherence into one of the following categories: < 0.8, 0.8 to 0.89, 0.9 to 1, and > 1.1. Patients were considered adherent if they reported taking ≥ 90% (MPR ≥ 0.9) of prescribed doses, adopted from the study by Anderson and colleagues.12 A patient with an MPR > 1, likely due to filling prior to the anticipated refill date, was considered 100% adherent (MPR = 1). If a patient switched OAN during the study, both agents were included as separate entities.
A conservative estimate of cost-savings was made by multiplying the RLRVAMC cost per unit of medication at time of initial prescription fill by the number of units taken each day multiplied by the total days’ supply on hand at time of therapy discontinuation. Patients with an MPR < 1 at time of therapy discontinuation were assumed to have zero remaining units on hand and zero cost savings was estimated. Waste, for purposes of cost-savings, was calculated for all MPR values > 1. Additional supply anticipated to be on hand from dose reductions was not included in the estimated cost of unused medication.
Descriptive statistics compared demographic characteristics between the pre- and postimplementation groups. MPR data were not normally distributed, which required the use of nonparametric Mann-Whitney U tests to compare pre- and postMPRs. Pearson χ2 compared the proportion of adherent patients between groups while descriptive statistics were used to estimate cost savings. Significance was determined based on a P value < .05. IBM SPSS Statistics software was used for all statistical analyses. As this was a complete sample of all eligible subjects, no sample size calculation was performed.
Results
In the preimplementation period, 246 patients received an OAN and 61 patients received an OAN in the postimplementation period (Figure 1). Of the 246 patients in the preimplementation period, 98 were eligible and included in the preimplementation group. Similarly, of the 61 patients in the postimplementation period, 35 patients met inclusion criteria for the postimplementation group. The study population was predominantly male with an average age of approximately 70 years in both groups (Table 3). More than 70% of the population in each group was White. No statistically significant differences between groups were identified. The most commonly prescribed OAN in the preimplementation group were abiraterone, imatinib, and enzalutamide (Table 3). In the postimplementation group, the most commonly prescribed agents were abiraterone, imatinib, pazopanib, and dasatinib. No significant differences were observed in prescribing of individual agents between the pre- and postimplementation groups or other characteristics that may affect adherence including patient copay status, number of concomitant medications, and driving distance from the RLRVAMC.
Thirty-six (36.7%) patients in the preimplementation group were considered nonadherent (MPR < 0.9) and 18 (18.4%) had an MPR < 0.8. Fifteen (15.3%) patients in the preimplementation clinic were considered overadherent (MPR > 1.1). Forty-seven (47.9%) patients in the preimplementation group were considered adherent (MPR 0.9 - 1.1) while all 35 (100%) patients in the postimplementation group were considered adherent (MPR 0.9 - 1.1). No non- or overadherent patients were identified in the postimplementation group (Figure 2). The median MPR for all patients in the preimplementation group was 0.94 compared with 1.06 (P < .001) in the postimplementation group.
Thirty-five (35.7%) patients had therapy discontinued or held in the preimplementation group compared with 2 (5.7%) patients in the postimplementation group (P < .001). Reasons for discontinuation in the preimplementation group included disease progression (n = 27), death (n = 3), lost to follow up (n = 2), and intolerability of therapy (n = 3). Both patients that discontinued therapy in the postimplementation group did so due to disease progression. Of the 35 patients who had their OAN discontinued or held in the preimplementation group, 14 patients had excess supply on hand at time of discontinuation. The estimated value of the unused medication was $37,890. Nine (25%) of the 35 patients who discontinued therapy had a dosage reduction during the course of therapy and the additional supply was not included in the cost estimate. Similarly, 1 of the 2 patients in the postimplementation group had their OAN discontinued during study. The cost of oversupply of medication at the time of therapy discontinuation was estimated at $1,555. No patients in the postimplementation group had dose reductions. After implementation of the OAN renewal clinic, the total cost savings between pre ($37,890) and postimplementation ($1,555) groups was $36,355.
Discussion
OANs are widely used therapies, with more than 25 million doses administered per year in the United States alone.12 The use of these agents will continue to grow as more targeted agents become available and patients request more convenient treatment options. The role for hematology/oncology clinical pharmacy services must adapt to this increased usage of OANs, including increasing pharmacist involvement in medication education, adherence and tolerability assessments, and proactive drug interaction monitoring.However, additional research is needed to determine optimal management strategies.
Our study aimed to compare OAN adherence among patients at a tertiary care VA hospital before and after implementation of a renewal clinic. The preimplementation population had a median MPR of 0.94 compared with 1.06 in the postimplementation group (P < .001). Although an ideal MPR is 1.0, we aimed for a slightly higher MPR to allow a supply buffer in the event of prescription delivery delays, as more than 90% of prescriptions are mailed to patients from a regional mail-order pharmacy. Importantly, the median MPRs do not adequately convey the impact from this clinic. The proportion of patients who were considered adherent to OANs increased from 47.9% in the preimplementation to 100% in the postimplementation period. These finding suggest that the clinical pharmacist role to assess and encourage adherence through monitoring tolerability of these OANs improved the overall medication taking experience of these patients.
Upon initial evaluation of adherence pre- and postimplementation, median adherence rates in both groups appeared to be above goal at 0.94 and 1.06 respectively. Patients in the postimplementation group intentionally received a 5- to 7-day supply buffer to account for potential prescription delivery delays due to holidays and inclement weather. This would indicate that the patients in the postimplementation group would have 15% oversupply due to the 5-day supply buffer. After correcting for patients with confounding reasons for excess (dose reductions, breaks in treatment, etc.), the median MPR in the prerefill clinic group decreased to 0.9 and the MPR in the postrefill clinic group increased slightly to 1.08. Although the median adherence rate in both the pre- and postimplementation groups were above goal of 0.90, 36% of the patients in the preimplementation group were considered nonadherent (MPR < 0.9) compared with no patients in the postimplementation group. Therefore, our intervention to improve patient adherence appeared to be beneficial at our institution.
In addition to improving adherence, one of the goals of the renewal clinic was to minimize excess supply at the time of therapy discontinuation. This was accomplished by aligning medication fills with medical visits and objective monitoring, as well as limiting supply to no more than 30 days. Of the patients in the postimplementation group, only 1 patient had remaining medication at the time of therapy discontinuation compared with 14 patients in the preimplementation group. The estimated cost savings from excess supply was $36,335. Limiting the amount of unused supply not only saves money for the patient and the institution, but also decreases opportunity for improper hazardous waste disposal and unnecessary exposure of hazardous materials to others.
Our results show the pharmacist intervention in the coordination of renewals improved adherence, minimized medication waste, and saved money. The cost of pharmacist time participating in the refill clinic was not calculated. Each visit was completed in approximately 5 minutes, with subsequent documentation and coordination taking an additional 5 to 10 minutes. During the launch of this service, the oncology pharmacy resident provided all coverage of the clinic. Oversite of the resident was provided by hematology/oncology clinical pharmacy specialists. We have continued to utilize pharmacy resident coverage since that time to meet education needs and keep the estimated cost per visit low. Another option in the case that pharmacy residents are not available would be utilization of a pharmacy technician, intern, or professional student to conduct the adherence and tolerability phone assessments. Our escalation protocol allows intervention by clinical pharmacy specialist and/or other health care providers when necessary. Trainees have only required basic training on how to use the protocol.
Limitations
Due to this study’s retrospective design, an inherent limitation is dependence on prescriber and refill records for documentation of initiation and discontinuation dates. Therefore, only the association of impact of pharmacist intervention on medication adherence can be determined as opposed to causation. We did not take into account discrepancies in day supply secondary to ‘held’ therapies, dose reductions, or doses supplied during an inpatient admission, which may alter estimates of MPR and cost-savings data. Patients in the postimplementation group intentionally received a 5 to 7-day supply buffer to account for potential prescription delivery delays due to holidays and inclement weather. This would indicate that the patients in the postimplementation group would have 15% oversupply due to the 5-day supply buffer, thereby skewing MPR values. This study did not account for cost avoidance resulting from early identification and management of toxicity. Finally, the postimplementation data only spans 4 months and a longer duration of time is needed to more accurately determine sustainability of renewal clinic interventions and provide comprehensive evaluation of cost-avoidance.
Conclusion
Implementation of an OAN renewal clinic was associated with an increase in MPR, improved proportion of patients considered adherent, and an estimated $36,335 cost-savings. However, prospective evaluation and a longer study duration are needed to determine causality of improved adherence and cost-savings associated with a pharmacist-driven OAN renewal clinic.
1. Ganesan P, Sagar TG, Dubashi B, et al. Nonadherence to imatinib adversely affects event free survival in chronic phase chronic myeloid leukemia. Am J Hematol 2011; 86: 471-474. doi:10.1002/ajh.22019
2. Marin D, Bazeos A, Mahon FX, et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J Clin Oncol 2010; 28: 2381-2388. doi:10.1200/JCO.2009.26.3087
3. McCowan C, Shearer J, Donnan PT, et al. Cohort study examining tamoxifen adherence and its relationship to mortality in women with breast cancer. Br J Cancer 2008; 99: 1763-1768. doi:10.1038/sj.bjc.6604758
4. Lexicomp Online. Sunitinib. Hudson, Ohio: Lexi-Comp, Inc; August 20, 2019.
5. Babiker A, El Husseini M, Al Nemri A, et al. Health care professional development: Working as a team to improve patient care. Sudan J Paediatr. 2014;14(2):9-16.
6. Spence MM, Makarem AF, Reyes SL, et al. Evaluation of an outpatient pharmacy clinical services program on adherence and clinical outcomes among patients with diabetes and/or coronary artery disease. J Manag Care Spec Pharm. 2014;20(10):1036-1045. doi:10.18553/jmcp.2014.20.10.1036
7. Holle LM, Puri S, Clement JM. Physician-pharmacist collaboration for oral chemotherapy monitoring: Insights from an academic genitourinary oncology practice. J Oncol Pharm Pract 2015; doi:10.1177/1078155215581524
8. Muluneh B, Schneider M, Faso A, et al. Improved Adherence Rates and Clinical Outcomes of an Integrated, Closed-Loop, Pharmacist-Led Oral Chemotherapy Management Program. Journal of Oncology Practice. 2018;14(6):371-333. doi:10.1200/JOP.17.00039.
9. Font R, Espinas JA, Gil-Gil M, et al. Prescription refill, patient self-report and physician report in assessing adherence to oral endocrine therapy in early breast cancer patients: a retrospective cohort study in Catalonia, Spain. British Journal of Cancer. 2012 ;107(8):1249-1256. doi:10.1038/bjc.2012.389.
10. Anderson KR, Chambers CR, Lam N, et al. Medication adherence among adults prescribed imatinib, dasatinib, or nilotinib for the treatment of chronic myeloid leukemia. J Oncol Pharm Practice. 2015;21(1):19–25. doi:10.1177/1078155213520261
11. Weingart SN, Brown E, Bach PB, et al. NCCN Task Force Report: oral chemotherapy. J Natl Compr Canc Netw. 2008;6(3): S1-S14.
1. Ganesan P, Sagar TG, Dubashi B, et al. Nonadherence to imatinib adversely affects event free survival in chronic phase chronic myeloid leukemia. Am J Hematol 2011; 86: 471-474. doi:10.1002/ajh.22019
2. Marin D, Bazeos A, Mahon FX, et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J Clin Oncol 2010; 28: 2381-2388. doi:10.1200/JCO.2009.26.3087
3. McCowan C, Shearer J, Donnan PT, et al. Cohort study examining tamoxifen adherence and its relationship to mortality in women with breast cancer. Br J Cancer 2008; 99: 1763-1768. doi:10.1038/sj.bjc.6604758
4. Lexicomp Online. Sunitinib. Hudson, Ohio: Lexi-Comp, Inc; August 20, 2019.
5. Babiker A, El Husseini M, Al Nemri A, et al. Health care professional development: Working as a team to improve patient care. Sudan J Paediatr. 2014;14(2):9-16.
6. Spence MM, Makarem AF, Reyes SL, et al. Evaluation of an outpatient pharmacy clinical services program on adherence and clinical outcomes among patients with diabetes and/or coronary artery disease. J Manag Care Spec Pharm. 2014;20(10):1036-1045. doi:10.18553/jmcp.2014.20.10.1036
7. Holle LM, Puri S, Clement JM. Physician-pharmacist collaboration for oral chemotherapy monitoring: Insights from an academic genitourinary oncology practice. J Oncol Pharm Pract 2015; doi:10.1177/1078155215581524
8. Muluneh B, Schneider M, Faso A, et al. Improved Adherence Rates and Clinical Outcomes of an Integrated, Closed-Loop, Pharmacist-Led Oral Chemotherapy Management Program. Journal of Oncology Practice. 2018;14(6):371-333. doi:10.1200/JOP.17.00039.
9. Font R, Espinas JA, Gil-Gil M, et al. Prescription refill, patient self-report and physician report in assessing adherence to oral endocrine therapy in early breast cancer patients: a retrospective cohort study in Catalonia, Spain. British Journal of Cancer. 2012 ;107(8):1249-1256. doi:10.1038/bjc.2012.389.
10. Anderson KR, Chambers CR, Lam N, et al. Medication adherence among adults prescribed imatinib, dasatinib, or nilotinib for the treatment of chronic myeloid leukemia. J Oncol Pharm Practice. 2015;21(1):19–25. doi:10.1177/1078155213520261
11. Weingart SN, Brown E, Bach PB, et al. NCCN Task Force Report: oral chemotherapy. J Natl Compr Canc Netw. 2008;6(3): S1-S14.
Use of Comprehensive Geriatric Assessment in Oncology Patients to Guide Treatment Decisions and Predict Chemotherapy Toxicity
Age is a well recognized risk factor for cancer development. The population of older Americans is growing, and by 2030, 20% of the US population will be aged ≥ 65 years.1 While 25% of all new cancer cases are diagnosed in people aged 65 to 74 years, more than half of cancers occur in individuals aged ≥ 70 years, with even higher rates in those aged ≥ 75 years.2 Although cancer rates have declined slightly overall among people aged ≥ 65 years, this population still has an 11-fold increased incidence of cancer compared with that of younger individuals.3 With a rapidly growing older population, there will be increasing demand for cancer care.
Treatment of cancer in older individuals often is complicated by medical comorbidities, frailty, and poor functional status. Distinguishing patients who can tolerate aggressive therapy from those who require less intensive therapy can be challenging. Age-related physiologic changes predispose older adults to an increased risk of therapy-related toxicities, resulting in suboptimal therapeutic benefit and substantial morbidity. For example, cardiovascular changes can lead to reduction of the cardiac functional reserve, which can increase the risk of congestive heart failure. Similarly, decline in renal function leads to an increased potential for nephrotoxicity.4 Although patients may be of the same chronologic age, their performance, functional, and biologic status may be quite variable; thus, tolerance to aggressive treatment is not easily predicted. The comprehensive geriatric assessment (CGA) may be used as a global assessment tool to risk stratify older patients prior to oncologic treatment decisions.
Health care providers (HCPs), including physician assistants, nurse practitioners, clinical nurse specialists, nurses, and physicians, routinely participate in every aspect of cancer care by ordering and interpreting diagnostic tests, addressing comorbidities, managing symptoms, and discussing cancer treatment recommendations. HCPs in oncology will continue to play a vital role in the coordination and management of older patients with cancer. However, in general, CGA has not been a consistent part of oncology practices, and few HCPs are familiar with the benefits of CGA screening tools.
What Is Geriatric Assessment?
Geriatric assessment is a multidisciplinary, multidimensional process aimed at detecting medical, psychosocial, and functional issues of older adults that are not identified by traditional performance status measures alone. It provides guidance for management of identified problems and improvement in quality of life.6 CGA was developed by geriatricians and multidisciplinary care teams to evaluate the domains of functional, nutritional, cognitive, psychosocial, and economic status; comorbidities; geriatric syndromes; and mood, and it has been tested in both clinics and hospitals.7 Although such assessment requires additional time and resources, its goals are to identify areas of vulnerability, assist in clinical decisions of treatable health problems, and guide therapeutic interventions.6 In oncology practice, the assessment not only addresses these global issues, but also is critical in predicting toxicity and survival outcomes in older oncology patients.
Components of CGA
Advancing age brings many physiologic, psychosocial, and functional challenges, and a cancer diagnosis only adds to these issues. CGA provides a system of assessing older and/or frail patients with cancer through specific domains to identify issues that are not apparent on routine evaluation in a clinic setting before and during chemotherapy treatments. These domains include comorbidity, polypharmacy, functional status, cognition, psychological and social status, and nutrition.8
Comorbidity
The prevalence of multiple medical problems and comorbidities, including cancer, among people aged > 65 years is increasing.9 Studies have shown that two-thirds of patients with cancer had ≥ 2 medical conditions, and nearly one quarter had ≥ 4 medical conditions.10 In older adults, common comorbidities include cardiovascular disease, hypertension, diabetes mellitus, and dementia. These comorbidities can impact treatment decisions, increase the risk of disease, impact treatment-related complications, and affect a patient’s life expectancy.11 Assessing comorbidities is essential to CGA and is done using the Charlson Comorbidity Index and/or the Cumulative Illness Rating Scale.12
The Charlson Comorbidity Index was originally designed to predict 1-year mortality on the basis of a weighted composite score for the following categories: cardiovascular, endocrine, pulmonary, neurologic, renal, hepatic, gastrointestinal, and neoplastic disease.13 It is now the most widely used comorbidity index and has been adapted and verified as applicable and valid for predicting the outcomes and risk of death from many comorbid diseases.14 The Cumulative Illness Rating Scale has been validated as a predictor for readmission for hospitalized older adults, hospitalization within 1 year in a residential setting, and long-term mortality when assessed in inpatient and residential settings.15
Polypharmacy
Polypharmacy (use of ≥ 5 medications) is common in older patients regardless of cancer diagnosis and is often instead defined as “the use of multiple drugs or more than are medically necessary.”16 The use of multiple medications, including those not indicated for existing medical conditions (such as over‐the‐counter, herbal, and complementary/alternative medicines, which patients often fail to declare to their specialist, doctor, or pharmacist) adds to the potential negative aspects of polypharmacy that affect older patients.17
Patients with cancer usually are prescribed an extensive number of medicines, both for the disease and for supportive care, which can increase the chance of drug-drug interactions and adverse reactions.18 While these issues certainly affect quality of life, they also may influence chemotherapy treatment and potentially impact survival. Studies have shown that the presence of polypharmacy has been associated with higher numbers of comorbidities, increased use of inappropriate medications, poor performance status, decline in functional status, and poor survival.18
Functional Status
Although Eastern Cooperative Oncology Group (ECOG) performance status and Karnofsky Performance Status are commonly used by oncologists, these guidelines are limited in focus and do not reliably measure functional status in older patients. Functional status is determined by the ability to perform daily acts of self-care, which includes assessment of activities of daily living (ADLs) and instrumental activities of daily living (IADLs). ADLs refer to such tasks as bathing, dressing, eating, mobility, balance, and toileting.19 IADLs include the ability to perform activities required to live within a community and include shopping, transportation, managing finances, medication management, cooking, and cleaning.11
Physical functionality also can be assessed by measures such as gait speed, grip strength, balance, and lower extremity strength. These are more sensitive and shown to be associated with worse clinical outcomes.20 Grip strength and gait speed, as assessed by the Timed Up and Go test or the Short Physical Performance Battery measure strength and balance.12 Reduction in gait speed and/or grip strength are associated with adverse clinical outcomes and increased risk of mortality.21 Patients with cancer who have difficulty with ADLs are at increased risk for falls, which can limit their functional independence, compromise cancer therapy, and increase the risk of chemotherapy toxicities.11 Impaired hearing and poor vision are added factors that can be barriers to cancer treatment.
Cognition
Cognitive impairment in patients with cancer is becoming more of an issue for oncology HCPs as both cancer and cognitive decline are more common with advancing age. Cognition in cancer patients is important for understanding their diagnosis, prognosis, treatment options, and adherence. Impaired cognition can affect decision making regarding treatment options and administration. Cognition can be assessed through validated screening tools such as the Mini-Mental State Examination and Mini-Cog.11
Psychological and Social Status
A cancer diagnosis has a major impact on the mental and emotional state of patients and family members. Clinically significant anxiety has been reported in approximately 21% of older patients with cancer, and the incidence of depression ranges from 17 to 26%.22 In older patients with, psychologic distress can impact cancer treatment, resulting in less definitive therapy and poorer outcomes.23 All patients with cancer should be screened for psychologic distress using standardized methods, such as the Geriatric Depression Scale or the General Anxiety Disorder-7 scale.24 A positive screen should lead to additional assessments that evaluate the severity of depression and other comorbid psychological problems and medical conditions.
Social isolation and loneliness are factors that can affect both depression and anxiety. Older patients with cancer are at risk for decreased social activities and are already challenged with issues related to home care, comorbidities, functional status, and caregiver support.23 Therefore, it is important to assess the social interactions of an older and/or frail patient with cancer and use social work assistance to address needs for supportive services.
Nutrition
Nutrition is important in any patient with cancer undergoing chemotherapy treatment. However, it is of greater importance in older adults, as malnutrition and weight loss are negative prognostic factors that correlate with poor tolerance to chemotherapy treatment, decline in quality of life, and increased mortality.25 The Mini-Nutritional Assessment is a widely used validated tool to assess nutritional status and risk of malnutrition.11 This tool can help identify those older and/or frail patients with cancer with impaired nutritional status and aid in instituting corrective measures to treat or prevent malnutrition.
Effectiveness of CGA
Multiple randomized controlled clinical trials assessing the effectiveness of CGA have been conducted over the past 3 decades with overall positive outcomes related to its value.26 Benefits of CGA can include overall improved medical care, avoidance of hospitalization or nursing home placement, identification of cognitive impairment, and prevention of geriatric syndrome (a range of conditions representing multiple organ impairment in older adults).27
In oncology, CGA is particularly beneficial, as it can identify issues in nearly 70% of patients that may not be apparent through traditional oncology assessment.28 A systematic review of 36 studies assessing the prognostic value of CGA in elderly patients with cancer receiving chemotherapy concluded that impaired performance and functional status as well as a frail and vulnerable profile are important predictors of severe chemotherapy-related toxicity and are associated with a higher risk of mortality.29 Therefore, CGA should be an integral part of the evaluation of older and/or frail patients with cancer prior to chemotherapy consideration.
Several screening tools have been developed using information from CGA to assess the risk of severe toxicities. The most commonly used tools for predicting toxicity include the Cancer and Aging Research Group (CARG) chemotoxicity calculator and the Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH).30,31 Although these tools are readily available to facilitate CGA, and despite their proven beneficial outcome and recommended usage by national guidelines, implementation of these tools in routine oncology practice has been challenging and slow to spread. Unless these recommended interventions are effectively implemented, the benefits of CGA cannot be realized. With the expected surge in the number of older patients with cancer, hopefully this will change.
Geriatric Assessment Screening Tools
A screening tool recommended for use in older and/or frail patients with cancer allows for a brief assessment to help clinicians identify patients in need of further evaluation by CGA and to provides information on treatment-related toxicities, functional decline, and survival.32 The predictive value and utility of geriatric assessment screening tools have been repeatedly proven to identify older and/or frail adults at risk for treatment-related toxicities.12 The CARG and the CRASH are validated screening tools used in identifying patients at higher risk for chemotherapy toxicity. These screening tools are intended to provide guidance to the clinical oncology practitioner on risk stratification of chemotherapy toxicity in older patients with cancer.33
Both of these screening tools provide similar predictive performance for chemotherapy toxicity in older patients with cancer.34 However, the CARG tool seems to have the advantage of using more data that had already been obtained during regular office visits and is clear and easy to use clinically. The CRASH tool is slightly more involved, as it uses multiple geriatric instruments to determine the predictive risk of both hematologic and nonhematologic toxicities of chemotherapy.
CARG Chemotoxicity Calculator
Hurria and colleagues originally developed the CARG tool from data obtained through a prospective multicenter study involving 500 patients with cancer aged ≥ 65 years.35 They concluded that chemotherapy-related toxicity is common in older adults, with 53% of patients sustaining grade 3 or 4 treatment-related toxicities and 2% treatment-related mortality.12 This predictive model for chemotherapy-related toxicity used 11 variables, both objective (obtained during a regular clinical encounter: age, tumor type, chemotherapy dosing, number of drugs, creatinine, and hemoglobin) and subjective (completed by patient: number of falls, social support, the ability to take medications, hearing impairment, and physical performance), to determine at-risk patients (Table 1).31
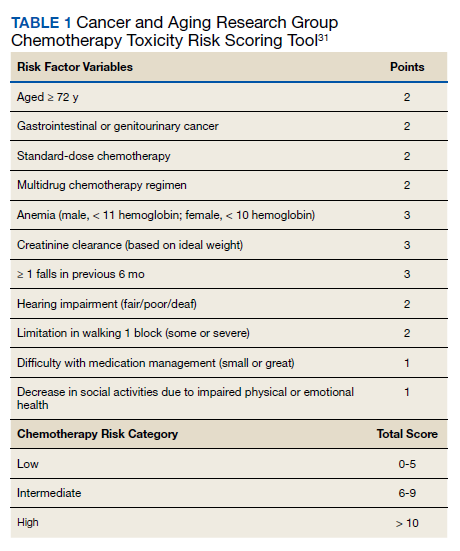
Compared with standard performance status measures in oncology practice, the CARG model was better able to predict chemotherapy-related toxicities. In 2016, Hurria and colleagues published the results of an updated external validation study with a cohort of 250 older patients with cancer receiving chemotherapy that confirmed the prediction of chemotherapy toxicity using the CARG screening tool in this population.31 An appealing feature of this tool is the free online accessibility and the expedited manner in which screening can be conducted.
CRASH Score
The CRASH score was derived from the results of a prospective, multicenter study of 518 patients aged ≥ 70 years who were assessed on 24 parameters prior to starting chemotherapy.30 A total of 64% of patients experienced significant toxicities, including 32% with grade 4 hematologic toxicity and 56% with grade 3 or 4 nonhematologic toxicity. The hematologic and nonhematologic toxicity risks are the 2 categories that comprise the CRASH score. Both baseline patient variables and chemotherapy regimen are incorporated into an 8-item assessment profile that determines the risk categories (Table 2).30
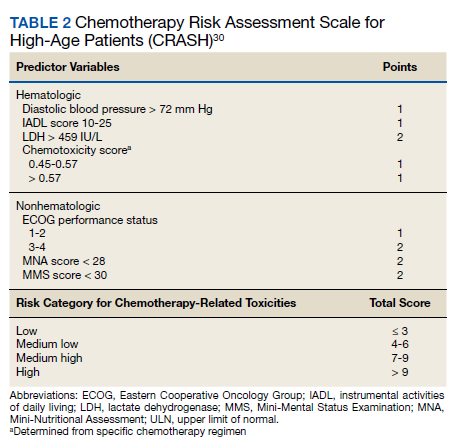
Increased risk of hematologic toxicities was associated with increased diastolic blood pressure, increased lactate dehydrogenase, need for assistance with IADL, and increased toxicity potential of the chemotherapy regimen. Nonhematologic toxicities were associated with ECOG performance score, Mini Mental Status Examination and Mini-Nutritional Assessment, and increased toxicity of the chemotherapy regimen.12 Patient scores are stratified into 4 risk categories: low, medium-low, medium-high, and high.30 Like the CARG tool, the CRASH screening tool also is available as a free online resource and can be used in everyday clinical practice to assess older and/or frail adults with cancer.
Conclusions
In older adults, cancer may significantly impact the natural course of concurrent comorbidities due to physiologic and functional changes. These vulnerabilities predispose older patients with cancer to an increased risk of adverse outcomes, including treatment-related toxicities.36 Given the rapidly aging population, it is critical for oncology clinical teams to be prepared to assess for, prevent, and manage issues for older adults that could impact outcomes, including complications and toxicities from chemotherapy.35 Studies have reported that 78 to 93% of older oncology patients have at least 1 geriatric impairment that could potentially impact oncology treatment plans.37,38 This supports the utility of CGA as a global assessment tool to risk stratify older and/or frail patients prior to deciding on subsequent oncologic treatment approaches.5 In fact, major cooperative groups sponsored by the National Cancer Institute, such as the Alliance for Clinical Trials in Oncology, are including CGA as part of some of their treatment trials. CGA was conducted as part of a multicenter cooperative group study in older patients with acute myeloid leukemia prior to inpatient intensive induction chemotherapy and was determined to be feasible and useful in clinical trials and practice.39
Despite the increasing evidence for benefits of CGA, it has not been a consistent part of oncology practices, and few HCPs are familiar with the benefits of CGA screening tools. Although oncology providers routinely participate in every aspect of cancer care and play a vital role in the coordination and management of older patients with cancer, CGA implementation into routine clinical practice has been slow in part due to lack of knowledge and training regarding the use of GA tools.
Oncology providers can easily incorporate CGA screening tools into the history and physical examination process for older patients with cancer, which will add an important dimension to these patient evaluations. Oncology providers are not only well positioned to administer these screening tools, but also can lead the field in developing innovative ways for effective implementation in busy routine oncology clinics. However, to be successful, oncology providers must be knowledgeable about these tools and understand their utility in guiding treatment decisions and improving quality of care in older patients with cancer.
1. Sharless NE. The challenging landscape of cancer and aging: charting a way forward. Published January 24, 2018. Accessed April 16, 2021. https://www.cancer.gov/news-events/cancer-currents-blog/2018/sharpless-aging-cancer-research
2. National Cancer Institute. Age and cancer risk. Updated March 5, 2021. Accessed April 16, 2021. https://www.cancer.gov/about-cancer/causes-prevention/risk/age
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34. doi:10.3322/caac.21551 4. Sawhney R, Sehl M, Naeim A. Physiologic aspects of aging: impact on cancer management and decision making, part I. Cancer J. 2005;11(6):449-460. doi:10.1097/00130404-200511000-00004
5. Kenis C, Bron D, Libert Y, et al. Relevance of a systematic geriatric screening and assessment in older patients with cancer: results of a prospective multicentric study. Ann Oncol. 2013;24(5):1306-1312. doi:10.1093/annonc/mds619
6. Loh KP, Soto-Perez-de-Celis E, Hsu T, et al. What every oncologist should know about geriatric assessment for older patients with cancer: Young International Society of Geriatric Oncology position paper. J Oncol Pract. 2018;14(2):85-94. doi:10.1200/JOP.2017.026435
7. Cohen HJ. Evolution of geriatric assessment in oncology. J Oncol Pract. 2018;14(2):95-96. doi:10.1200/JOP.18.00017
8. Wildiers H, Heeren P, Puts M, et al. International Society of Geriatric Oncology consensus on geriatric assessment in older patients with cancer. J Clin Oncol. 2014;32(24):2595-2603. doi:10.1200/JCO.2013.54.8347
9. American Cancer Society. Cancer facts & figures 2019. Accessed April 16, 2021. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2019.html
10. Williams GR, Mackenzie A, Magnuson A, et al. Comorbidity in older adults with cancer. J Geriatr Oncol. 2016;7(4):249-257. doi:10.1016/j.jgo.2015.12.002
11. Korc-Grodzicki B, Holmes HM, Shahrokni A. Geriatric assessment for oncologists. Cancer Biol Med. 2015;12(4):261-274. doi:10.7497/j.issn.2095-3941.2015.0082
12. Li D, Soto-Perez-de-Celis E, Hurria A. Geriatric assessment and tools for predicting treatment toxicity in older adults with cancer. Cancer J. 2017;23(4):206-210. doi:10.1097/PPO.0000000000000269
13. Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373-383. doi:10.1016/0021-9681(87)90171-8
14. Huang Y, Gou R, Diao Y, et al. Charlson comorbidity index helps predict the risk of mortality for patients with type 2 diabetic nephropathy. J Zhejiang Univ Sci B. 2014;15(1):58-66. doi:10.1631/jzus.B1300109
15. Osborn KP IV, Nothelle S, Slaven JE, Montz K, Hui S, Torke AM. Cumulative Illness Rating Scale (CIRS) can be used to predict hospital outcomes in older adults. J Geriatric Med Gerontol. 2017;3(2). doi:10.23937/2469-5858/1510030
16. Maher RL, Hanlon J, Hajjar ER. Clinical consequences of polypharmacy in elderly. Expert Opin Drug Saf. 2014;13(1):57-65. doi:10.1517/14740338.2013.827660
17. Shrestha S, Shrestha S, Khanal S. Polypharmacy in elderly cancer patients: challenges and the way clinical pharmacists can contribute in resource-limited settings. Aging Med. 2019;2(1):42-49. doi:10.1002/agm2.12051
18. Sharma M, Loh KP, Nightingale G, Mohile SG, Holmes HM. Polypharmacy and potentially inappropriate medication use in geriatric oncology. J Geriatr Oncol. 2016;7(5):346-353. doi:10.1016/j.jgo.2016.07.010
19. Norburn JE, Bernard SL, Konrad TR, et al. Self-care and assistance from others in coping with functional status limitations among a national sample of older adults. J Gerontol B Psychol Sci Soc Sci. 1995;50(2):S101-S109. doi:10.1093/geronb/50b.2.s101
20. Fragala MS, Alley DE, Shardell MD, et al. Comparison of handgrip and leg extension strength in predicting slow gait speed in older adults. J Am Geriatr Soc. 2016;64(1):144-150. doi:10.1111/jgs.13871
21. Owusu C, Berger NA. Comprehensive geriatric assessment in the older cancer patient: coming of age in clinical cancer care. Clin Pract (Lond). 2014;11(6):749-762. doi:10.2217/cpr.14.72
22. Weiss Wiesel TR, Nelson CJ, Tew WP, et al. The relationship between age, anxiety, and depression in older adults with cancer. Psychooncology. 2015;24(6):712-717. doi:10.1002/pon.3638
23. Soto-Perez-de-Celis E, Li D, Yuan Y, Lau YM, Hurria A. Functional versus chronological age: geriatric assessments to guide decision making in older patients with cancer. Lancet Oncol. 2018;19(6):e305-e316. doi:10.1016/S1470-2045(18)30348-6
24. Andersen BL, DeRubeis RJ, Berman BS, et al. Screening, assessment, and care of anxiety and depressive symptoms in adults with cancer: an American Society of Clinical Oncology guideline adaptation. J Clin Oncol. 2014;32(15):1605-1619. doi:10.1200/JCO.2013.52.4611
25. Muscaritoli M, Lucia S, Farcomeni A, et al. Prevalence of malnutrition in patients at first medical oncology visit: the PreMiO study. Oncotarget. 2017;8(45):79884-79886. doi:10.18632/oncotarget.20168
26. Ekdahl AW, Axmon A, Sandberg M, Steen Carlsson K. Is care based on comprehensive geriatric assessment with mobile teams better than usual care? A study protocol of a randomised controlled trial (the GerMoT study). BMJ Open. 2018;8(10)e23969. doi:10.1136/bmjopen-2018-023969
27. Mohile SG, Dale W, Somerfield MR, et al. Practical assessment and management of vulnerabilities in older patients receiving chemotherapy: ASCO guideline for geriatric oncology. J Clin Oncol. 2018;36(22):2326-2347. doi:10.1200/JCO.2018.78.8687
28. Hernandez Torres C, Hsu T. Comprehensive geriatric assessment in the older adult with cancer: a review. Eur Urol Focus. 2017;3(4-5):330-339. doi:10.1016/j.euf.2017.10.010
29. Janssens K, Specenier P. The prognostic value of the comprehensive geriatric assessment (CGA) in elderly cancer patients (ECP) treated with chemotherapy (CT): a systematic review. Eur J Cancer. 2017;72(1):S164-S165. doi:10.1016/S0959-8049(17)30611-1
30. Extermann M, Boler I, Reich RR, et al. Predicting the risk of chemotherapy toxicity in older patients: The Chemotherapy Risk Assessment Scale for High‐Age Patients (CRASH) score. Cancer. 2012;118(13):3377-3386. doi:10.1002/cncr.26646
31. Hurria A, Mohile S, Gajra A, et al. Validation of a prediction tool for chemotherapy toxicity in older adults with cancer. J Clin Oncol. 2016;34(20):2366-2371. doi:10.1200/JCO.2015.65.4327
32. Decoster L, Van Puyvelde K, Mohile S, et al. Screening tools for multidimensional health problems warranting a geriatric assessment in older cancer patients: an update on SIOG recommendations. Ann Oncol. 2015;26(2):288-300. doi:10.1093/annonc/mdu210
33. Schiefen JK, Madsen LT, Dains JE. Instruments that predict oncology treatment risk in the senior population. J Adv Pract Oncol. 2017;8(5):528-533.
34. Ortland I, Mendel Ott M, Kowar M, et al. Comparing the performance of the CARG and the CRASH score for predicting toxicity in older patients with cancer. J Geriatr Oncol. 2020;11(6):997-1005. doi:10.1016/j.jgo.2019.12.016
35. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol. 2011;29(25):3457-3465. doi:10.1200/JCO.2011.34.7625
36. Mohile SG, Velarde C, Hurria A, et al. Geriatric assessment-guided care processes for older adults: a Delphi consensus of geriatric oncology experts. J Natl Compr Canc Netw. 2015;13(9):1120-1130. doi:10.6004/jnccn.2015.0137
37. Schiphorst AHW, Ten Bokkel Huinink D, Breumelhof R, Burgmans JPJ, Pronk A, Hamaker ME. Geriatric consultation can aid in complex treatment decisions for elderly cancer patients. Eur J Cancer Care (Engl). 2016;25(3):365-370. doi:10.1111/ecc.12349
38. Schulkes KJG, Souwer ETD, Hamaker ME, et al. The effect of a geriatric assessment on treatment decisions for patients with lung cancer. Lung. 2017;195(2):225-231. doi:10.1007/s00408-017-9983-7
39. Klepin HD, Ritchie E, Major-Elechi B, et al. Geriatric assessment among older adults receiving intensive therapy for acute myeloid leukemia: report of CALGB 361006 (Alliance). J Geriatr Oncol. 2020;11(1):107-113. doi:10.1016/j.jgo.2019.10.002
Age is a well recognized risk factor for cancer development. The population of older Americans is growing, and by 2030, 20% of the US population will be aged ≥ 65 years.1 While 25% of all new cancer cases are diagnosed in people aged 65 to 74 years, more than half of cancers occur in individuals aged ≥ 70 years, with even higher rates in those aged ≥ 75 years.2 Although cancer rates have declined slightly overall among people aged ≥ 65 years, this population still has an 11-fold increased incidence of cancer compared with that of younger individuals.3 With a rapidly growing older population, there will be increasing demand for cancer care.
Treatment of cancer in older individuals often is complicated by medical comorbidities, frailty, and poor functional status. Distinguishing patients who can tolerate aggressive therapy from those who require less intensive therapy can be challenging. Age-related physiologic changes predispose older adults to an increased risk of therapy-related toxicities, resulting in suboptimal therapeutic benefit and substantial morbidity. For example, cardiovascular changes can lead to reduction of the cardiac functional reserve, which can increase the risk of congestive heart failure. Similarly, decline in renal function leads to an increased potential for nephrotoxicity.4 Although patients may be of the same chronologic age, their performance, functional, and biologic status may be quite variable; thus, tolerance to aggressive treatment is not easily predicted. The comprehensive geriatric assessment (CGA) may be used as a global assessment tool to risk stratify older patients prior to oncologic treatment decisions.
Health care providers (HCPs), including physician assistants, nurse practitioners, clinical nurse specialists, nurses, and physicians, routinely participate in every aspect of cancer care by ordering and interpreting diagnostic tests, addressing comorbidities, managing symptoms, and discussing cancer treatment recommendations. HCPs in oncology will continue to play a vital role in the coordination and management of older patients with cancer. However, in general, CGA has not been a consistent part of oncology practices, and few HCPs are familiar with the benefits of CGA screening tools.
What Is Geriatric Assessment?
Geriatric assessment is a multidisciplinary, multidimensional process aimed at detecting medical, psychosocial, and functional issues of older adults that are not identified by traditional performance status measures alone. It provides guidance for management of identified problems and improvement in quality of life.6 CGA was developed by geriatricians and multidisciplinary care teams to evaluate the domains of functional, nutritional, cognitive, psychosocial, and economic status; comorbidities; geriatric syndromes; and mood, and it has been tested in both clinics and hospitals.7 Although such assessment requires additional time and resources, its goals are to identify areas of vulnerability, assist in clinical decisions of treatable health problems, and guide therapeutic interventions.6 In oncology practice, the assessment not only addresses these global issues, but also is critical in predicting toxicity and survival outcomes in older oncology patients.
Components of CGA
Advancing age brings many physiologic, psychosocial, and functional challenges, and a cancer diagnosis only adds to these issues. CGA provides a system of assessing older and/or frail patients with cancer through specific domains to identify issues that are not apparent on routine evaluation in a clinic setting before and during chemotherapy treatments. These domains include comorbidity, polypharmacy, functional status, cognition, psychological and social status, and nutrition.8
Comorbidity
The prevalence of multiple medical problems and comorbidities, including cancer, among people aged > 65 years is increasing.9 Studies have shown that two-thirds of patients with cancer had ≥ 2 medical conditions, and nearly one quarter had ≥ 4 medical conditions.10 In older adults, common comorbidities include cardiovascular disease, hypertension, diabetes mellitus, and dementia. These comorbidities can impact treatment decisions, increase the risk of disease, impact treatment-related complications, and affect a patient’s life expectancy.11 Assessing comorbidities is essential to CGA and is done using the Charlson Comorbidity Index and/or the Cumulative Illness Rating Scale.12
The Charlson Comorbidity Index was originally designed to predict 1-year mortality on the basis of a weighted composite score for the following categories: cardiovascular, endocrine, pulmonary, neurologic, renal, hepatic, gastrointestinal, and neoplastic disease.13 It is now the most widely used comorbidity index and has been adapted and verified as applicable and valid for predicting the outcomes and risk of death from many comorbid diseases.14 The Cumulative Illness Rating Scale has been validated as a predictor for readmission for hospitalized older adults, hospitalization within 1 year in a residential setting, and long-term mortality when assessed in inpatient and residential settings.15
Polypharmacy
Polypharmacy (use of ≥ 5 medications) is common in older patients regardless of cancer diagnosis and is often instead defined as “the use of multiple drugs or more than are medically necessary.”16 The use of multiple medications, including those not indicated for existing medical conditions (such as over‐the‐counter, herbal, and complementary/alternative medicines, which patients often fail to declare to their specialist, doctor, or pharmacist) adds to the potential negative aspects of polypharmacy that affect older patients.17
Patients with cancer usually are prescribed an extensive number of medicines, both for the disease and for supportive care, which can increase the chance of drug-drug interactions and adverse reactions.18 While these issues certainly affect quality of life, they also may influence chemotherapy treatment and potentially impact survival. Studies have shown that the presence of polypharmacy has been associated with higher numbers of comorbidities, increased use of inappropriate medications, poor performance status, decline in functional status, and poor survival.18
Functional Status
Although Eastern Cooperative Oncology Group (ECOG) performance status and Karnofsky Performance Status are commonly used by oncologists, these guidelines are limited in focus and do not reliably measure functional status in older patients. Functional status is determined by the ability to perform daily acts of self-care, which includes assessment of activities of daily living (ADLs) and instrumental activities of daily living (IADLs). ADLs refer to such tasks as bathing, dressing, eating, mobility, balance, and toileting.19 IADLs include the ability to perform activities required to live within a community and include shopping, transportation, managing finances, medication management, cooking, and cleaning.11
Physical functionality also can be assessed by measures such as gait speed, grip strength, balance, and lower extremity strength. These are more sensitive and shown to be associated with worse clinical outcomes.20 Grip strength and gait speed, as assessed by the Timed Up and Go test or the Short Physical Performance Battery measure strength and balance.12 Reduction in gait speed and/or grip strength are associated with adverse clinical outcomes and increased risk of mortality.21 Patients with cancer who have difficulty with ADLs are at increased risk for falls, which can limit their functional independence, compromise cancer therapy, and increase the risk of chemotherapy toxicities.11 Impaired hearing and poor vision are added factors that can be barriers to cancer treatment.
Cognition
Cognitive impairment in patients with cancer is becoming more of an issue for oncology HCPs as both cancer and cognitive decline are more common with advancing age. Cognition in cancer patients is important for understanding their diagnosis, prognosis, treatment options, and adherence. Impaired cognition can affect decision making regarding treatment options and administration. Cognition can be assessed through validated screening tools such as the Mini-Mental State Examination and Mini-Cog.11
Psychological and Social Status
A cancer diagnosis has a major impact on the mental and emotional state of patients and family members. Clinically significant anxiety has been reported in approximately 21% of older patients with cancer, and the incidence of depression ranges from 17 to 26%.22 In older patients with, psychologic distress can impact cancer treatment, resulting in less definitive therapy and poorer outcomes.23 All patients with cancer should be screened for psychologic distress using standardized methods, such as the Geriatric Depression Scale or the General Anxiety Disorder-7 scale.24 A positive screen should lead to additional assessments that evaluate the severity of depression and other comorbid psychological problems and medical conditions.
Social isolation and loneliness are factors that can affect both depression and anxiety. Older patients with cancer are at risk for decreased social activities and are already challenged with issues related to home care, comorbidities, functional status, and caregiver support.23 Therefore, it is important to assess the social interactions of an older and/or frail patient with cancer and use social work assistance to address needs for supportive services.
Nutrition
Nutrition is important in any patient with cancer undergoing chemotherapy treatment. However, it is of greater importance in older adults, as malnutrition and weight loss are negative prognostic factors that correlate with poor tolerance to chemotherapy treatment, decline in quality of life, and increased mortality.25 The Mini-Nutritional Assessment is a widely used validated tool to assess nutritional status and risk of malnutrition.11 This tool can help identify those older and/or frail patients with cancer with impaired nutritional status and aid in instituting corrective measures to treat or prevent malnutrition.
Effectiveness of CGA
Multiple randomized controlled clinical trials assessing the effectiveness of CGA have been conducted over the past 3 decades with overall positive outcomes related to its value.26 Benefits of CGA can include overall improved medical care, avoidance of hospitalization or nursing home placement, identification of cognitive impairment, and prevention of geriatric syndrome (a range of conditions representing multiple organ impairment in older adults).27
In oncology, CGA is particularly beneficial, as it can identify issues in nearly 70% of patients that may not be apparent through traditional oncology assessment.28 A systematic review of 36 studies assessing the prognostic value of CGA in elderly patients with cancer receiving chemotherapy concluded that impaired performance and functional status as well as a frail and vulnerable profile are important predictors of severe chemotherapy-related toxicity and are associated with a higher risk of mortality.29 Therefore, CGA should be an integral part of the evaluation of older and/or frail patients with cancer prior to chemotherapy consideration.
Several screening tools have been developed using information from CGA to assess the risk of severe toxicities. The most commonly used tools for predicting toxicity include the Cancer and Aging Research Group (CARG) chemotoxicity calculator and the Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH).30,31 Although these tools are readily available to facilitate CGA, and despite their proven beneficial outcome and recommended usage by national guidelines, implementation of these tools in routine oncology practice has been challenging and slow to spread. Unless these recommended interventions are effectively implemented, the benefits of CGA cannot be realized. With the expected surge in the number of older patients with cancer, hopefully this will change.
Geriatric Assessment Screening Tools
A screening tool recommended for use in older and/or frail patients with cancer allows for a brief assessment to help clinicians identify patients in need of further evaluation by CGA and to provides information on treatment-related toxicities, functional decline, and survival.32 The predictive value and utility of geriatric assessment screening tools have been repeatedly proven to identify older and/or frail adults at risk for treatment-related toxicities.12 The CARG and the CRASH are validated screening tools used in identifying patients at higher risk for chemotherapy toxicity. These screening tools are intended to provide guidance to the clinical oncology practitioner on risk stratification of chemotherapy toxicity in older patients with cancer.33
Both of these screening tools provide similar predictive performance for chemotherapy toxicity in older patients with cancer.34 However, the CARG tool seems to have the advantage of using more data that had already been obtained during regular office visits and is clear and easy to use clinically. The CRASH tool is slightly more involved, as it uses multiple geriatric instruments to determine the predictive risk of both hematologic and nonhematologic toxicities of chemotherapy.
CARG Chemotoxicity Calculator
Hurria and colleagues originally developed the CARG tool from data obtained through a prospective multicenter study involving 500 patients with cancer aged ≥ 65 years.35 They concluded that chemotherapy-related toxicity is common in older adults, with 53% of patients sustaining grade 3 or 4 treatment-related toxicities and 2% treatment-related mortality.12 This predictive model for chemotherapy-related toxicity used 11 variables, both objective (obtained during a regular clinical encounter: age, tumor type, chemotherapy dosing, number of drugs, creatinine, and hemoglobin) and subjective (completed by patient: number of falls, social support, the ability to take medications, hearing impairment, and physical performance), to determine at-risk patients (Table 1).31
Compared with standard performance status measures in oncology practice, the CARG model was better able to predict chemotherapy-related toxicities. In 2016, Hurria and colleagues published the results of an updated external validation study with a cohort of 250 older patients with cancer receiving chemotherapy that confirmed the prediction of chemotherapy toxicity using the CARG screening tool in this population.31 An appealing feature of this tool is the free online accessibility and the expedited manner in which screening can be conducted.
CRASH Score
The CRASH score was derived from the results of a prospective, multicenter study of 518 patients aged ≥ 70 years who were assessed on 24 parameters prior to starting chemotherapy.30 A total of 64% of patients experienced significant toxicities, including 32% with grade 4 hematologic toxicity and 56% with grade 3 or 4 nonhematologic toxicity. The hematologic and nonhematologic toxicity risks are the 2 categories that comprise the CRASH score. Both baseline patient variables and chemotherapy regimen are incorporated into an 8-item assessment profile that determines the risk categories (Table 2).30
Increased risk of hematologic toxicities was associated with increased diastolic blood pressure, increased lactate dehydrogenase, need for assistance with IADL, and increased toxicity potential of the chemotherapy regimen. Nonhematologic toxicities were associated with ECOG performance score, Mini Mental Status Examination and Mini-Nutritional Assessment, and increased toxicity of the chemotherapy regimen.12 Patient scores are stratified into 4 risk categories: low, medium-low, medium-high, and high.30 Like the CARG tool, the CRASH screening tool also is available as a free online resource and can be used in everyday clinical practice to assess older and/or frail adults with cancer.
Conclusions
In older adults, cancer may significantly impact the natural course of concurrent comorbidities due to physiologic and functional changes. These vulnerabilities predispose older patients with cancer to an increased risk of adverse outcomes, including treatment-related toxicities.36 Given the rapidly aging population, it is critical for oncology clinical teams to be prepared to assess for, prevent, and manage issues for older adults that could impact outcomes, including complications and toxicities from chemotherapy.35 Studies have reported that 78 to 93% of older oncology patients have at least 1 geriatric impairment that could potentially impact oncology treatment plans.37,38 This supports the utility of CGA as a global assessment tool to risk stratify older and/or frail patients prior to deciding on subsequent oncologic treatment approaches.5 In fact, major cooperative groups sponsored by the National Cancer Institute, such as the Alliance for Clinical Trials in Oncology, are including CGA as part of some of their treatment trials. CGA was conducted as part of a multicenter cooperative group study in older patients with acute myeloid leukemia prior to inpatient intensive induction chemotherapy and was determined to be feasible and useful in clinical trials and practice.39
Despite the increasing evidence for benefits of CGA, it has not been a consistent part of oncology practices, and few HCPs are familiar with the benefits of CGA screening tools. Although oncology providers routinely participate in every aspect of cancer care and play a vital role in the coordination and management of older patients with cancer, CGA implementation into routine clinical practice has been slow in part due to lack of knowledge and training regarding the use of GA tools.
Oncology providers can easily incorporate CGA screening tools into the history and physical examination process for older patients with cancer, which will add an important dimension to these patient evaluations. Oncology providers are not only well positioned to administer these screening tools, but also can lead the field in developing innovative ways for effective implementation in busy routine oncology clinics. However, to be successful, oncology providers must be knowledgeable about these tools and understand their utility in guiding treatment decisions and improving quality of care in older patients with cancer.
Age is a well recognized risk factor for cancer development. The population of older Americans is growing, and by 2030, 20% of the US population will be aged ≥ 65 years.1 While 25% of all new cancer cases are diagnosed in people aged 65 to 74 years, more than half of cancers occur in individuals aged ≥ 70 years, with even higher rates in those aged ≥ 75 years.2 Although cancer rates have declined slightly overall among people aged ≥ 65 years, this population still has an 11-fold increased incidence of cancer compared with that of younger individuals.3 With a rapidly growing older population, there will be increasing demand for cancer care.
Treatment of cancer in older individuals often is complicated by medical comorbidities, frailty, and poor functional status. Distinguishing patients who can tolerate aggressive therapy from those who require less intensive therapy can be challenging. Age-related physiologic changes predispose older adults to an increased risk of therapy-related toxicities, resulting in suboptimal therapeutic benefit and substantial morbidity. For example, cardiovascular changes can lead to reduction of the cardiac functional reserve, which can increase the risk of congestive heart failure. Similarly, decline in renal function leads to an increased potential for nephrotoxicity.4 Although patients may be of the same chronologic age, their performance, functional, and biologic status may be quite variable; thus, tolerance to aggressive treatment is not easily predicted. The comprehensive geriatric assessment (CGA) may be used as a global assessment tool to risk stratify older patients prior to oncologic treatment decisions.
Health care providers (HCPs), including physician assistants, nurse practitioners, clinical nurse specialists, nurses, and physicians, routinely participate in every aspect of cancer care by ordering and interpreting diagnostic tests, addressing comorbidities, managing symptoms, and discussing cancer treatment recommendations. HCPs in oncology will continue to play a vital role in the coordination and management of older patients with cancer. However, in general, CGA has not been a consistent part of oncology practices, and few HCPs are familiar with the benefits of CGA screening tools.
What Is Geriatric Assessment?
Geriatric assessment is a multidisciplinary, multidimensional process aimed at detecting medical, psychosocial, and functional issues of older adults that are not identified by traditional performance status measures alone. It provides guidance for management of identified problems and improvement in quality of life.6 CGA was developed by geriatricians and multidisciplinary care teams to evaluate the domains of functional, nutritional, cognitive, psychosocial, and economic status; comorbidities; geriatric syndromes; and mood, and it has been tested in both clinics and hospitals.7 Although such assessment requires additional time and resources, its goals are to identify areas of vulnerability, assist in clinical decisions of treatable health problems, and guide therapeutic interventions.6 In oncology practice, the assessment not only addresses these global issues, but also is critical in predicting toxicity and survival outcomes in older oncology patients.
Components of CGA
Advancing age brings many physiologic, psychosocial, and functional challenges, and a cancer diagnosis only adds to these issues. CGA provides a system of assessing older and/or frail patients with cancer through specific domains to identify issues that are not apparent on routine evaluation in a clinic setting before and during chemotherapy treatments. These domains include comorbidity, polypharmacy, functional status, cognition, psychological and social status, and nutrition.8
Comorbidity
The prevalence of multiple medical problems and comorbidities, including cancer, among people aged > 65 years is increasing.9 Studies have shown that two-thirds of patients with cancer had ≥ 2 medical conditions, and nearly one quarter had ≥ 4 medical conditions.10 In older adults, common comorbidities include cardiovascular disease, hypertension, diabetes mellitus, and dementia. These comorbidities can impact treatment decisions, increase the risk of disease, impact treatment-related complications, and affect a patient’s life expectancy.11 Assessing comorbidities is essential to CGA and is done using the Charlson Comorbidity Index and/or the Cumulative Illness Rating Scale.12
The Charlson Comorbidity Index was originally designed to predict 1-year mortality on the basis of a weighted composite score for the following categories: cardiovascular, endocrine, pulmonary, neurologic, renal, hepatic, gastrointestinal, and neoplastic disease.13 It is now the most widely used comorbidity index and has been adapted and verified as applicable and valid for predicting the outcomes and risk of death from many comorbid diseases.14 The Cumulative Illness Rating Scale has been validated as a predictor for readmission for hospitalized older adults, hospitalization within 1 year in a residential setting, and long-term mortality when assessed in inpatient and residential settings.15
Polypharmacy
Polypharmacy (use of ≥ 5 medications) is common in older patients regardless of cancer diagnosis and is often instead defined as “the use of multiple drugs or more than are medically necessary.”16 The use of multiple medications, including those not indicated for existing medical conditions (such as over‐the‐counter, herbal, and complementary/alternative medicines, which patients often fail to declare to their specialist, doctor, or pharmacist) adds to the potential negative aspects of polypharmacy that affect older patients.17
Patients with cancer usually are prescribed an extensive number of medicines, both for the disease and for supportive care, which can increase the chance of drug-drug interactions and adverse reactions.18 While these issues certainly affect quality of life, they also may influence chemotherapy treatment and potentially impact survival. Studies have shown that the presence of polypharmacy has been associated with higher numbers of comorbidities, increased use of inappropriate medications, poor performance status, decline in functional status, and poor survival.18
Functional Status
Although Eastern Cooperative Oncology Group (ECOG) performance status and Karnofsky Performance Status are commonly used by oncologists, these guidelines are limited in focus and do not reliably measure functional status in older patients. Functional status is determined by the ability to perform daily acts of self-care, which includes assessment of activities of daily living (ADLs) and instrumental activities of daily living (IADLs). ADLs refer to such tasks as bathing, dressing, eating, mobility, balance, and toileting.19 IADLs include the ability to perform activities required to live within a community and include shopping, transportation, managing finances, medication management, cooking, and cleaning.11
Physical functionality also can be assessed by measures such as gait speed, grip strength, balance, and lower extremity strength. These are more sensitive and shown to be associated with worse clinical outcomes.20 Grip strength and gait speed, as assessed by the Timed Up and Go test or the Short Physical Performance Battery measure strength and balance.12 Reduction in gait speed and/or grip strength are associated with adverse clinical outcomes and increased risk of mortality.21 Patients with cancer who have difficulty with ADLs are at increased risk for falls, which can limit their functional independence, compromise cancer therapy, and increase the risk of chemotherapy toxicities.11 Impaired hearing and poor vision are added factors that can be barriers to cancer treatment.
Cognition
Cognitive impairment in patients with cancer is becoming more of an issue for oncology HCPs as both cancer and cognitive decline are more common with advancing age. Cognition in cancer patients is important for understanding their diagnosis, prognosis, treatment options, and adherence. Impaired cognition can affect decision making regarding treatment options and administration. Cognition can be assessed through validated screening tools such as the Mini-Mental State Examination and Mini-Cog.11
Psychological and Social Status
A cancer diagnosis has a major impact on the mental and emotional state of patients and family members. Clinically significant anxiety has been reported in approximately 21% of older patients with cancer, and the incidence of depression ranges from 17 to 26%.22 In older patients with, psychologic distress can impact cancer treatment, resulting in less definitive therapy and poorer outcomes.23 All patients with cancer should be screened for psychologic distress using standardized methods, such as the Geriatric Depression Scale or the General Anxiety Disorder-7 scale.24 A positive screen should lead to additional assessments that evaluate the severity of depression and other comorbid psychological problems and medical conditions.
Social isolation and loneliness are factors that can affect both depression and anxiety. Older patients with cancer are at risk for decreased social activities and are already challenged with issues related to home care, comorbidities, functional status, and caregiver support.23 Therefore, it is important to assess the social interactions of an older and/or frail patient with cancer and use social work assistance to address needs for supportive services.
Nutrition
Nutrition is important in any patient with cancer undergoing chemotherapy treatment. However, it is of greater importance in older adults, as malnutrition and weight loss are negative prognostic factors that correlate with poor tolerance to chemotherapy treatment, decline in quality of life, and increased mortality.25 The Mini-Nutritional Assessment is a widely used validated tool to assess nutritional status and risk of malnutrition.11 This tool can help identify those older and/or frail patients with cancer with impaired nutritional status and aid in instituting corrective measures to treat or prevent malnutrition.
Effectiveness of CGA
Multiple randomized controlled clinical trials assessing the effectiveness of CGA have been conducted over the past 3 decades with overall positive outcomes related to its value.26 Benefits of CGA can include overall improved medical care, avoidance of hospitalization or nursing home placement, identification of cognitive impairment, and prevention of geriatric syndrome (a range of conditions representing multiple organ impairment in older adults).27
In oncology, CGA is particularly beneficial, as it can identify issues in nearly 70% of patients that may not be apparent through traditional oncology assessment.28 A systematic review of 36 studies assessing the prognostic value of CGA in elderly patients with cancer receiving chemotherapy concluded that impaired performance and functional status as well as a frail and vulnerable profile are important predictors of severe chemotherapy-related toxicity and are associated with a higher risk of mortality.29 Therefore, CGA should be an integral part of the evaluation of older and/or frail patients with cancer prior to chemotherapy consideration.
Several screening tools have been developed using information from CGA to assess the risk of severe toxicities. The most commonly used tools for predicting toxicity include the Cancer and Aging Research Group (CARG) chemotoxicity calculator and the Chemotherapy Risk Assessment Scale for High-Age Patients (CRASH).30,31 Although these tools are readily available to facilitate CGA, and despite their proven beneficial outcome and recommended usage by national guidelines, implementation of these tools in routine oncology practice has been challenging and slow to spread. Unless these recommended interventions are effectively implemented, the benefits of CGA cannot be realized. With the expected surge in the number of older patients with cancer, hopefully this will change.
Geriatric Assessment Screening Tools
A screening tool recommended for use in older and/or frail patients with cancer allows for a brief assessment to help clinicians identify patients in need of further evaluation by CGA and to provides information on treatment-related toxicities, functional decline, and survival.32 The predictive value and utility of geriatric assessment screening tools have been repeatedly proven to identify older and/or frail adults at risk for treatment-related toxicities.12 The CARG and the CRASH are validated screening tools used in identifying patients at higher risk for chemotherapy toxicity. These screening tools are intended to provide guidance to the clinical oncology practitioner on risk stratification of chemotherapy toxicity in older patients with cancer.33
Both of these screening tools provide similar predictive performance for chemotherapy toxicity in older patients with cancer.34 However, the CARG tool seems to have the advantage of using more data that had already been obtained during regular office visits and is clear and easy to use clinically. The CRASH tool is slightly more involved, as it uses multiple geriatric instruments to determine the predictive risk of both hematologic and nonhematologic toxicities of chemotherapy.
CARG Chemotoxicity Calculator
Hurria and colleagues originally developed the CARG tool from data obtained through a prospective multicenter study involving 500 patients with cancer aged ≥ 65 years.35 They concluded that chemotherapy-related toxicity is common in older adults, with 53% of patients sustaining grade 3 or 4 treatment-related toxicities and 2% treatment-related mortality.12 This predictive model for chemotherapy-related toxicity used 11 variables, both objective (obtained during a regular clinical encounter: age, tumor type, chemotherapy dosing, number of drugs, creatinine, and hemoglobin) and subjective (completed by patient: number of falls, social support, the ability to take medications, hearing impairment, and physical performance), to determine at-risk patients (Table 1).31
Compared with standard performance status measures in oncology practice, the CARG model was better able to predict chemotherapy-related toxicities. In 2016, Hurria and colleagues published the results of an updated external validation study with a cohort of 250 older patients with cancer receiving chemotherapy that confirmed the prediction of chemotherapy toxicity using the CARG screening tool in this population.31 An appealing feature of this tool is the free online accessibility and the expedited manner in which screening can be conducted.
CRASH Score
The CRASH score was derived from the results of a prospective, multicenter study of 518 patients aged ≥ 70 years who were assessed on 24 parameters prior to starting chemotherapy.30 A total of 64% of patients experienced significant toxicities, including 32% with grade 4 hematologic toxicity and 56% with grade 3 or 4 nonhematologic toxicity. The hematologic and nonhematologic toxicity risks are the 2 categories that comprise the CRASH score. Both baseline patient variables and chemotherapy regimen are incorporated into an 8-item assessment profile that determines the risk categories (Table 2).30
Increased risk of hematologic toxicities was associated with increased diastolic blood pressure, increased lactate dehydrogenase, need for assistance with IADL, and increased toxicity potential of the chemotherapy regimen. Nonhematologic toxicities were associated with ECOG performance score, Mini Mental Status Examination and Mini-Nutritional Assessment, and increased toxicity of the chemotherapy regimen.12 Patient scores are stratified into 4 risk categories: low, medium-low, medium-high, and high.30 Like the CARG tool, the CRASH screening tool also is available as a free online resource and can be used in everyday clinical practice to assess older and/or frail adults with cancer.
Conclusions
In older adults, cancer may significantly impact the natural course of concurrent comorbidities due to physiologic and functional changes. These vulnerabilities predispose older patients with cancer to an increased risk of adverse outcomes, including treatment-related toxicities.36 Given the rapidly aging population, it is critical for oncology clinical teams to be prepared to assess for, prevent, and manage issues for older adults that could impact outcomes, including complications and toxicities from chemotherapy.35 Studies have reported that 78 to 93% of older oncology patients have at least 1 geriatric impairment that could potentially impact oncology treatment plans.37,38 This supports the utility of CGA as a global assessment tool to risk stratify older and/or frail patients prior to deciding on subsequent oncologic treatment approaches.5 In fact, major cooperative groups sponsored by the National Cancer Institute, such as the Alliance for Clinical Trials in Oncology, are including CGA as part of some of their treatment trials. CGA was conducted as part of a multicenter cooperative group study in older patients with acute myeloid leukemia prior to inpatient intensive induction chemotherapy and was determined to be feasible and useful in clinical trials and practice.39
Despite the increasing evidence for benefits of CGA, it has not been a consistent part of oncology practices, and few HCPs are familiar with the benefits of CGA screening tools. Although oncology providers routinely participate in every aspect of cancer care and play a vital role in the coordination and management of older patients with cancer, CGA implementation into routine clinical practice has been slow in part due to lack of knowledge and training regarding the use of GA tools.
Oncology providers can easily incorporate CGA screening tools into the history and physical examination process for older patients with cancer, which will add an important dimension to these patient evaluations. Oncology providers are not only well positioned to administer these screening tools, but also can lead the field in developing innovative ways for effective implementation in busy routine oncology clinics. However, to be successful, oncology providers must be knowledgeable about these tools and understand their utility in guiding treatment decisions and improving quality of care in older patients with cancer.
1. Sharless NE. The challenging landscape of cancer and aging: charting a way forward. Published January 24, 2018. Accessed April 16, 2021. https://www.cancer.gov/news-events/cancer-currents-blog/2018/sharpless-aging-cancer-research
2. National Cancer Institute. Age and cancer risk. Updated March 5, 2021. Accessed April 16, 2021. https://www.cancer.gov/about-cancer/causes-prevention/risk/age
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34. doi:10.3322/caac.21551 4. Sawhney R, Sehl M, Naeim A. Physiologic aspects of aging: impact on cancer management and decision making, part I. Cancer J. 2005;11(6):449-460. doi:10.1097/00130404-200511000-00004
5. Kenis C, Bron D, Libert Y, et al. Relevance of a systematic geriatric screening and assessment in older patients with cancer: results of a prospective multicentric study. Ann Oncol. 2013;24(5):1306-1312. doi:10.1093/annonc/mds619
6. Loh KP, Soto-Perez-de-Celis E, Hsu T, et al. What every oncologist should know about geriatric assessment for older patients with cancer: Young International Society of Geriatric Oncology position paper. J Oncol Pract. 2018;14(2):85-94. doi:10.1200/JOP.2017.026435
7. Cohen HJ. Evolution of geriatric assessment in oncology. J Oncol Pract. 2018;14(2):95-96. doi:10.1200/JOP.18.00017
8. Wildiers H, Heeren P, Puts M, et al. International Society of Geriatric Oncology consensus on geriatric assessment in older patients with cancer. J Clin Oncol. 2014;32(24):2595-2603. doi:10.1200/JCO.2013.54.8347
9. American Cancer Society. Cancer facts & figures 2019. Accessed April 16, 2021. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2019.html
10. Williams GR, Mackenzie A, Magnuson A, et al. Comorbidity in older adults with cancer. J Geriatr Oncol. 2016;7(4):249-257. doi:10.1016/j.jgo.2015.12.002
11. Korc-Grodzicki B, Holmes HM, Shahrokni A. Geriatric assessment for oncologists. Cancer Biol Med. 2015;12(4):261-274. doi:10.7497/j.issn.2095-3941.2015.0082
12. Li D, Soto-Perez-de-Celis E, Hurria A. Geriatric assessment and tools for predicting treatment toxicity in older adults with cancer. Cancer J. 2017;23(4):206-210. doi:10.1097/PPO.0000000000000269
13. Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373-383. doi:10.1016/0021-9681(87)90171-8
14. Huang Y, Gou R, Diao Y, et al. Charlson comorbidity index helps predict the risk of mortality for patients with type 2 diabetic nephropathy. J Zhejiang Univ Sci B. 2014;15(1):58-66. doi:10.1631/jzus.B1300109
15. Osborn KP IV, Nothelle S, Slaven JE, Montz K, Hui S, Torke AM. Cumulative Illness Rating Scale (CIRS) can be used to predict hospital outcomes in older adults. J Geriatric Med Gerontol. 2017;3(2). doi:10.23937/2469-5858/1510030
16. Maher RL, Hanlon J, Hajjar ER. Clinical consequences of polypharmacy in elderly. Expert Opin Drug Saf. 2014;13(1):57-65. doi:10.1517/14740338.2013.827660
17. Shrestha S, Shrestha S, Khanal S. Polypharmacy in elderly cancer patients: challenges and the way clinical pharmacists can contribute in resource-limited settings. Aging Med. 2019;2(1):42-49. doi:10.1002/agm2.12051
18. Sharma M, Loh KP, Nightingale G, Mohile SG, Holmes HM. Polypharmacy and potentially inappropriate medication use in geriatric oncology. J Geriatr Oncol. 2016;7(5):346-353. doi:10.1016/j.jgo.2016.07.010
19. Norburn JE, Bernard SL, Konrad TR, et al. Self-care and assistance from others in coping with functional status limitations among a national sample of older adults. J Gerontol B Psychol Sci Soc Sci. 1995;50(2):S101-S109. doi:10.1093/geronb/50b.2.s101
20. Fragala MS, Alley DE, Shardell MD, et al. Comparison of handgrip and leg extension strength in predicting slow gait speed in older adults. J Am Geriatr Soc. 2016;64(1):144-150. doi:10.1111/jgs.13871
21. Owusu C, Berger NA. Comprehensive geriatric assessment in the older cancer patient: coming of age in clinical cancer care. Clin Pract (Lond). 2014;11(6):749-762. doi:10.2217/cpr.14.72
22. Weiss Wiesel TR, Nelson CJ, Tew WP, et al. The relationship between age, anxiety, and depression in older adults with cancer. Psychooncology. 2015;24(6):712-717. doi:10.1002/pon.3638
23. Soto-Perez-de-Celis E, Li D, Yuan Y, Lau YM, Hurria A. Functional versus chronological age: geriatric assessments to guide decision making in older patients with cancer. Lancet Oncol. 2018;19(6):e305-e316. doi:10.1016/S1470-2045(18)30348-6
24. Andersen BL, DeRubeis RJ, Berman BS, et al. Screening, assessment, and care of anxiety and depressive symptoms in adults with cancer: an American Society of Clinical Oncology guideline adaptation. J Clin Oncol. 2014;32(15):1605-1619. doi:10.1200/JCO.2013.52.4611
25. Muscaritoli M, Lucia S, Farcomeni A, et al. Prevalence of malnutrition in patients at first medical oncology visit: the PreMiO study. Oncotarget. 2017;8(45):79884-79886. doi:10.18632/oncotarget.20168
26. Ekdahl AW, Axmon A, Sandberg M, Steen Carlsson K. Is care based on comprehensive geriatric assessment with mobile teams better than usual care? A study protocol of a randomised controlled trial (the GerMoT study). BMJ Open. 2018;8(10)e23969. doi:10.1136/bmjopen-2018-023969
27. Mohile SG, Dale W, Somerfield MR, et al. Practical assessment and management of vulnerabilities in older patients receiving chemotherapy: ASCO guideline for geriatric oncology. J Clin Oncol. 2018;36(22):2326-2347. doi:10.1200/JCO.2018.78.8687
28. Hernandez Torres C, Hsu T. Comprehensive geriatric assessment in the older adult with cancer: a review. Eur Urol Focus. 2017;3(4-5):330-339. doi:10.1016/j.euf.2017.10.010
29. Janssens K, Specenier P. The prognostic value of the comprehensive geriatric assessment (CGA) in elderly cancer patients (ECP) treated with chemotherapy (CT): a systematic review. Eur J Cancer. 2017;72(1):S164-S165. doi:10.1016/S0959-8049(17)30611-1
30. Extermann M, Boler I, Reich RR, et al. Predicting the risk of chemotherapy toxicity in older patients: The Chemotherapy Risk Assessment Scale for High‐Age Patients (CRASH) score. Cancer. 2012;118(13):3377-3386. doi:10.1002/cncr.26646
31. Hurria A, Mohile S, Gajra A, et al. Validation of a prediction tool for chemotherapy toxicity in older adults with cancer. J Clin Oncol. 2016;34(20):2366-2371. doi:10.1200/JCO.2015.65.4327
32. Decoster L, Van Puyvelde K, Mohile S, et al. Screening tools for multidimensional health problems warranting a geriatric assessment in older cancer patients: an update on SIOG recommendations. Ann Oncol. 2015;26(2):288-300. doi:10.1093/annonc/mdu210
33. Schiefen JK, Madsen LT, Dains JE. Instruments that predict oncology treatment risk in the senior population. J Adv Pract Oncol. 2017;8(5):528-533.
34. Ortland I, Mendel Ott M, Kowar M, et al. Comparing the performance of the CARG and the CRASH score for predicting toxicity in older patients with cancer. J Geriatr Oncol. 2020;11(6):997-1005. doi:10.1016/j.jgo.2019.12.016
35. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol. 2011;29(25):3457-3465. doi:10.1200/JCO.2011.34.7625
36. Mohile SG, Velarde C, Hurria A, et al. Geriatric assessment-guided care processes for older adults: a Delphi consensus of geriatric oncology experts. J Natl Compr Canc Netw. 2015;13(9):1120-1130. doi:10.6004/jnccn.2015.0137
37. Schiphorst AHW, Ten Bokkel Huinink D, Breumelhof R, Burgmans JPJ, Pronk A, Hamaker ME. Geriatric consultation can aid in complex treatment decisions for elderly cancer patients. Eur J Cancer Care (Engl). 2016;25(3):365-370. doi:10.1111/ecc.12349
38. Schulkes KJG, Souwer ETD, Hamaker ME, et al. The effect of a geriatric assessment on treatment decisions for patients with lung cancer. Lung. 2017;195(2):225-231. doi:10.1007/s00408-017-9983-7
39. Klepin HD, Ritchie E, Major-Elechi B, et al. Geriatric assessment among older adults receiving intensive therapy for acute myeloid leukemia: report of CALGB 361006 (Alliance). J Geriatr Oncol. 2020;11(1):107-113. doi:10.1016/j.jgo.2019.10.002
1. Sharless NE. The challenging landscape of cancer and aging: charting a way forward. Published January 24, 2018. Accessed April 16, 2021. https://www.cancer.gov/news-events/cancer-currents-blog/2018/sharpless-aging-cancer-research
2. National Cancer Institute. Age and cancer risk. Updated March 5, 2021. Accessed April 16, 2021. https://www.cancer.gov/about-cancer/causes-prevention/risk/age
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34. doi:10.3322/caac.21551 4. Sawhney R, Sehl M, Naeim A. Physiologic aspects of aging: impact on cancer management and decision making, part I. Cancer J. 2005;11(6):449-460. doi:10.1097/00130404-200511000-00004
5. Kenis C, Bron D, Libert Y, et al. Relevance of a systematic geriatric screening and assessment in older patients with cancer: results of a prospective multicentric study. Ann Oncol. 2013;24(5):1306-1312. doi:10.1093/annonc/mds619
6. Loh KP, Soto-Perez-de-Celis E, Hsu T, et al. What every oncologist should know about geriatric assessment for older patients with cancer: Young International Society of Geriatric Oncology position paper. J Oncol Pract. 2018;14(2):85-94. doi:10.1200/JOP.2017.026435
7. Cohen HJ. Evolution of geriatric assessment in oncology. J Oncol Pract. 2018;14(2):95-96. doi:10.1200/JOP.18.00017
8. Wildiers H, Heeren P, Puts M, et al. International Society of Geriatric Oncology consensus on geriatric assessment in older patients with cancer. J Clin Oncol. 2014;32(24):2595-2603. doi:10.1200/JCO.2013.54.8347
9. American Cancer Society. Cancer facts & figures 2019. Accessed April 16, 2021. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2019.html
10. Williams GR, Mackenzie A, Magnuson A, et al. Comorbidity in older adults with cancer. J Geriatr Oncol. 2016;7(4):249-257. doi:10.1016/j.jgo.2015.12.002
11. Korc-Grodzicki B, Holmes HM, Shahrokni A. Geriatric assessment for oncologists. Cancer Biol Med. 2015;12(4):261-274. doi:10.7497/j.issn.2095-3941.2015.0082
12. Li D, Soto-Perez-de-Celis E, Hurria A. Geriatric assessment and tools for predicting treatment toxicity in older adults with cancer. Cancer J. 2017;23(4):206-210. doi:10.1097/PPO.0000000000000269
13. Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis. 1987;40(5):373-383. doi:10.1016/0021-9681(87)90171-8
14. Huang Y, Gou R, Diao Y, et al. Charlson comorbidity index helps predict the risk of mortality for patients with type 2 diabetic nephropathy. J Zhejiang Univ Sci B. 2014;15(1):58-66. doi:10.1631/jzus.B1300109
15. Osborn KP IV, Nothelle S, Slaven JE, Montz K, Hui S, Torke AM. Cumulative Illness Rating Scale (CIRS) can be used to predict hospital outcomes in older adults. J Geriatric Med Gerontol. 2017;3(2). doi:10.23937/2469-5858/1510030
16. Maher RL, Hanlon J, Hajjar ER. Clinical consequences of polypharmacy in elderly. Expert Opin Drug Saf. 2014;13(1):57-65. doi:10.1517/14740338.2013.827660
17. Shrestha S, Shrestha S, Khanal S. Polypharmacy in elderly cancer patients: challenges and the way clinical pharmacists can contribute in resource-limited settings. Aging Med. 2019;2(1):42-49. doi:10.1002/agm2.12051
18. Sharma M, Loh KP, Nightingale G, Mohile SG, Holmes HM. Polypharmacy and potentially inappropriate medication use in geriatric oncology. J Geriatr Oncol. 2016;7(5):346-353. doi:10.1016/j.jgo.2016.07.010
19. Norburn JE, Bernard SL, Konrad TR, et al. Self-care and assistance from others in coping with functional status limitations among a national sample of older adults. J Gerontol B Psychol Sci Soc Sci. 1995;50(2):S101-S109. doi:10.1093/geronb/50b.2.s101
20. Fragala MS, Alley DE, Shardell MD, et al. Comparison of handgrip and leg extension strength in predicting slow gait speed in older adults. J Am Geriatr Soc. 2016;64(1):144-150. doi:10.1111/jgs.13871
21. Owusu C, Berger NA. Comprehensive geriatric assessment in the older cancer patient: coming of age in clinical cancer care. Clin Pract (Lond). 2014;11(6):749-762. doi:10.2217/cpr.14.72
22. Weiss Wiesel TR, Nelson CJ, Tew WP, et al. The relationship between age, anxiety, and depression in older adults with cancer. Psychooncology. 2015;24(6):712-717. doi:10.1002/pon.3638
23. Soto-Perez-de-Celis E, Li D, Yuan Y, Lau YM, Hurria A. Functional versus chronological age: geriatric assessments to guide decision making in older patients with cancer. Lancet Oncol. 2018;19(6):e305-e316. doi:10.1016/S1470-2045(18)30348-6
24. Andersen BL, DeRubeis RJ, Berman BS, et al. Screening, assessment, and care of anxiety and depressive symptoms in adults with cancer: an American Society of Clinical Oncology guideline adaptation. J Clin Oncol. 2014;32(15):1605-1619. doi:10.1200/JCO.2013.52.4611
25. Muscaritoli M, Lucia S, Farcomeni A, et al. Prevalence of malnutrition in patients at first medical oncology visit: the PreMiO study. Oncotarget. 2017;8(45):79884-79886. doi:10.18632/oncotarget.20168
26. Ekdahl AW, Axmon A, Sandberg M, Steen Carlsson K. Is care based on comprehensive geriatric assessment with mobile teams better than usual care? A study protocol of a randomised controlled trial (the GerMoT study). BMJ Open. 2018;8(10)e23969. doi:10.1136/bmjopen-2018-023969
27. Mohile SG, Dale W, Somerfield MR, et al. Practical assessment and management of vulnerabilities in older patients receiving chemotherapy: ASCO guideline for geriatric oncology. J Clin Oncol. 2018;36(22):2326-2347. doi:10.1200/JCO.2018.78.8687
28. Hernandez Torres C, Hsu T. Comprehensive geriatric assessment in the older adult with cancer: a review. Eur Urol Focus. 2017;3(4-5):330-339. doi:10.1016/j.euf.2017.10.010
29. Janssens K, Specenier P. The prognostic value of the comprehensive geriatric assessment (CGA) in elderly cancer patients (ECP) treated with chemotherapy (CT): a systematic review. Eur J Cancer. 2017;72(1):S164-S165. doi:10.1016/S0959-8049(17)30611-1
30. Extermann M, Boler I, Reich RR, et al. Predicting the risk of chemotherapy toxicity in older patients: The Chemotherapy Risk Assessment Scale for High‐Age Patients (CRASH) score. Cancer. 2012;118(13):3377-3386. doi:10.1002/cncr.26646
31. Hurria A, Mohile S, Gajra A, et al. Validation of a prediction tool for chemotherapy toxicity in older adults with cancer. J Clin Oncol. 2016;34(20):2366-2371. doi:10.1200/JCO.2015.65.4327
32. Decoster L, Van Puyvelde K, Mohile S, et al. Screening tools for multidimensional health problems warranting a geriatric assessment in older cancer patients: an update on SIOG recommendations. Ann Oncol. 2015;26(2):288-300. doi:10.1093/annonc/mdu210
33. Schiefen JK, Madsen LT, Dains JE. Instruments that predict oncology treatment risk in the senior population. J Adv Pract Oncol. 2017;8(5):528-533.
34. Ortland I, Mendel Ott M, Kowar M, et al. Comparing the performance of the CARG and the CRASH score for predicting toxicity in older patients with cancer. J Geriatr Oncol. 2020;11(6):997-1005. doi:10.1016/j.jgo.2019.12.016
35. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol. 2011;29(25):3457-3465. doi:10.1200/JCO.2011.34.7625
36. Mohile SG, Velarde C, Hurria A, et al. Geriatric assessment-guided care processes for older adults: a Delphi consensus of geriatric oncology experts. J Natl Compr Canc Netw. 2015;13(9):1120-1130. doi:10.6004/jnccn.2015.0137
37. Schiphorst AHW, Ten Bokkel Huinink D, Breumelhof R, Burgmans JPJ, Pronk A, Hamaker ME. Geriatric consultation can aid in complex treatment decisions for elderly cancer patients. Eur J Cancer Care (Engl). 2016;25(3):365-370. doi:10.1111/ecc.12349
38. Schulkes KJG, Souwer ETD, Hamaker ME, et al. The effect of a geriatric assessment on treatment decisions for patients with lung cancer. Lung. 2017;195(2):225-231. doi:10.1007/s00408-017-9983-7
39. Klepin HD, Ritchie E, Major-Elechi B, et al. Geriatric assessment among older adults receiving intensive therapy for acute myeloid leukemia: report of CALGB 361006 (Alliance). J Geriatr Oncol. 2020;11(1):107-113. doi:10.1016/j.jgo.2019.10.002
Screening High-Risk Women Veterans for Breast Cancer
The number of women seeking care from the Veterans Health Administration (VHA) is increasing.1 In 2015, there were 2 million women veterans in the United States, which is 9.4% of the total veteran population. This group is expected to increase at an average of about 18,000 women per year for the next 10 years.2 The percentage of women veterans who are US Department of Veterans Affairs (VA) users aged 45 to 64 years rose 46% from 2000 to 2015.1,3-4 It is estimated that 15% of veterans who used VA services in 2020 were women.1 Nineteen percent of women veterans are Black.1 The median age of women veterans in 2015 was 50 years.5 Breast cancer is the leading cancer affecting female veterans, and data suggest they have an increased risk of breast cancer based on unique service-related exposures.1,6-9
In the US, about 10 million women are eligible for breast cancer preventive therapy, including, but not limited to, medications, surgery, or lifestyle changes.10 Secondary prevention options include change in surveillance that can reduce their risk or identify cancer at an earlier stage when treatment is more effective. The United States Preventive Services Task Force, the National Comprehensive Cancer Network, the American Society for Clinical Oncology, the National Institute for Health and Care Excellence, and the Oncology Nursing Society recommend screening women aged ≥ 35 years to assess breast cancer risk.11-18 If a woman is at increased risk, she may be a candidate for chemoprevention, prozphylactic surgery, and possibly an enhanced screening regimen.
Urban and minority women are an understudied population. Most veterans (75%) live in urban or suburban settings.19,20 Urban veteran women constitute an important potential study population.
Chemoprevention measures have been underused because of factors involving both women and their health care providers. A large proportion of women are unaware of their higher risk status due to lack of adequate screening and risk assessment.21,22 In addition to patient lack of awareness of their high-risk status, primary care physicians are also reluctant to prescribe chemopreventive agents due to a lack of comfort or familiarity with the risks and benefits.23-26 The STAR2015, BCPT2005, IBIS2014, MAP3 2011, IBIS-I 2014, and IBIS II 2014 studies clearly demonstrate a 49 to 62% reduction in risk for women using chemoprevention such as selective estrogen receptor modulators or aromatase inhibitors, respectively.27-32 Yet only 4 to 9% of high-risk women not enrolled in a clinical trial are using chemoprevention.33-39
The possibility of developing breast cancer also may be increased because of a positive family history or being a member of a family in which there is a known susceptibility gene mutation.40 Based on these risk factors, women may be eligible for tailored follow-up and genetic counseling.41-44
Nationally, 7 to 10% of the civilian US population will experience posttraumatic stress disorder (PTSD).45 The rates are remarkably higher for women veterans, with roughly 20% diagnosed with PTSD.46,47 Anxiety and PTSD have been implicated in poor adherence to medical advice.48,49
In 2014, a national VA multidisciplinary group focused on breast cancer prevention, detection, treatment, and research to address breast health in the growing population of women veterans. High-risk breast cancer screenings are not routinely carried out by the VA in primary care, women’s health, or oncology services. Furthermore, the recording of screening questionnaire results was not synchronized until a standard questionnaire was created and approved as a template by this group in the VA electronic medical record (EMR) in 2015.
Several prediction models can identify which women are at an increased risk of developing breast cancer. The most commonly used risk assessment model, the Gail breast cancer risk assessment tool (BCRAT), has been refined to include women of additional ethnicities (https://www.cancer.gov/bcrisktool).
This pilot project was launched to identify an effective manner to screen women veterans regarding their risk of developing breast cancer and refer them for chemoprevention education or genetic counseling as appropriate.
Methods
A high-risk breast cancer screening questionnaire based on the Gail BCRAT and including lifestyle questions was developed and included as a note template in the VA EMR. The James J. Peters VA Medical Center, Bronx, NY (JJPVAMC) and the Washington DC VA Medical Center (DCVAMC) ran a pilot study between 2015 and 2018 using this breast cancer screening questionnaire to collect data from women veterans. Quality Executive Committee and institutional review board approvals were granted respectively.
Eligibility criteria included women aged ≥ 35 years with no personal history of breast cancer. Most patients were self-referred, but participants also were recruited during VA Breast Cancer Awareness month events, health fairs, or at informational tables in the hospital lobbies. After completing the 20 multiple choice questionnaire with a study team member, either in person or over the phone, a 5-year and lifetime risk of invasive breast cancer was calculated using the Gail BCRAT. A woman is considered high risk and eligible for chemoprevention if her 5-year risk is > 1.66% or her lifetime risk is ≥ 20%. Eligibility for genetic counseling is based on the Breast Cancer Referral Screening Tool, which includes a personal or family history of breast or ovarian cancer and Jewish ancestry.
All patients were notified of their average or high risk status by a clinician. Those who were deemed to be average risk received a follow-up letter in the mail with instructions (eg, to follow-up with a yearly mammogram). Those who were deemed to be high risk for developing breast cancer were asked to come in for an appointment with the study principal investigator (a VA oncologist/breast cancer specialist) to discuss prevention options, further screening, or referrals to genetic counseling. Depending on a patient’s other health factors, a woman at high risk for developing breast cancer also may be a candidate for chemoprevention with tamoxifen, raloxifene, exemestane, anastrozole, or letrozole.
Data on the participant’s lifestyle, including exercise, diet, and smoking, were evaluated to determine whether these factors had an impact on risk status.
Results
The JJP and DC VAMCs screened 103 women veterans between 2015 and 2018. Four patients were excluded for nonveteran (spousal) status, leaving 99 women veterans with a mean age of 54 years. The most common self-reported races were Black (60%), non-Hispanic White (14%), and Hispanic or Latino (13%) (Table 1).

Women veterans in our study were nearly 3-times more likely than the general population were to receive a high-risk Gail Score/BCRAT (35% vs 13%, respectively).50,51 Of this subset, 46% had breast biopsies, and 86% had a positive family history. Thirty-one percent of Black women in our study were high risk, while nationally, 8.2 to 13.3% of Black women aged 50 to 59 years are considered high risk.50,51 Of the Black high-risk group with a high Gail/BCRAT score, 94% had a positive family history, and 33% had a history of breast biopsy (Table 2).
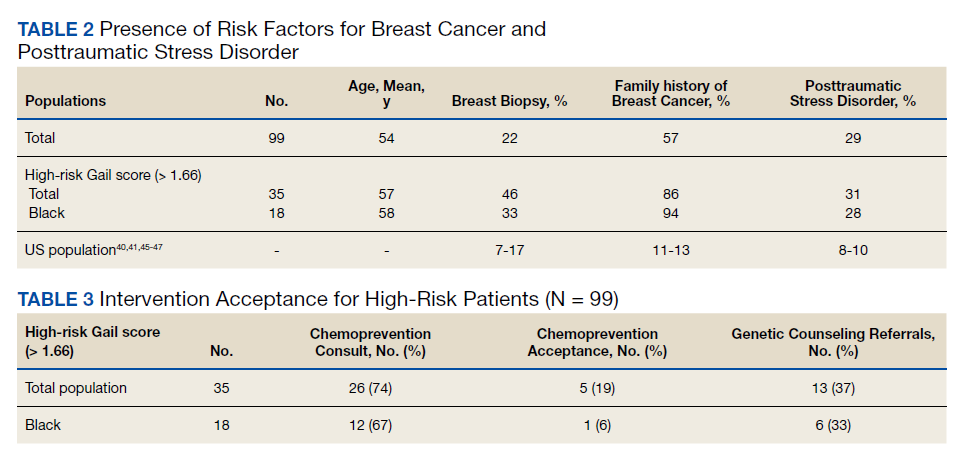
Of the 35 high-risk patients 26 (74%) patients accepted consultations for chemoprevention and 5 (19%) started chemoprevention. Of this high-risk group, 13 (37%) patients were referred for genetic counseling (Table 3).44 The prevalence of PTSD was present in 31% of high-risk women and 29% of the cohort (Figure).The lifestyle questions indicated that, among all participants, 79% had an overweight or obese body mass index; 58% exercised weekly; 51% consumed alcohol; 14% were smokers; and 21% consumed 3 to 4 servings of fruits/vegetables daily.
Discussion
Breast cancer is the most common cancer in women.52 The number of women with breast cancer in the VHA has more than tripled from 1995 to 2012.1 The lifetime risk of developing breast cancer in the general population is about 13%.50 This rate can be affected by risk factors including age, hormone exposure, family history, radiation exposure, and lifestyle factors, such as weight and alcohol use.6,52-56 In the United States, invasive breast cancer affects 1 in 8 women.50,52,57
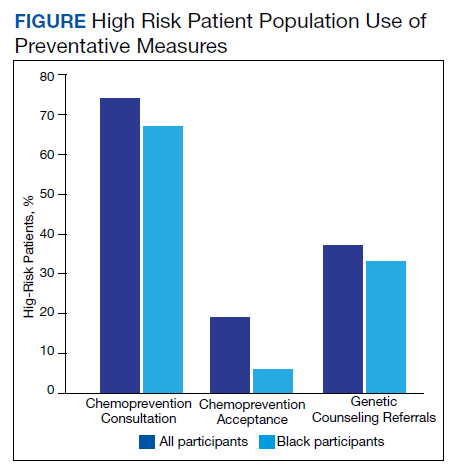
Our screened population showed nearly 3 times as many women veterans were at an increased risk for breast cancer when compared with historical averages in US women. This difference may be based on a high rate of prior breast biopsies or positive family history, although a provocative study using the Surveillance, Epidemiology, and End Results database showed military women to have higher rates of breast cancer as well.9 Historically, Blacks are vastly understudied in clinical research with only 5% representation on a national level.5,58 The urban locations of both pilot sites (Washington, DC and Bronx, NY) allowed for the inclusion of minority patients in our study. We found that the rates of breast cancer in Black women veterans to be higher than seen nationally, possibly prompting further screening initiatives for this understudied population.
Our pilot study’s chemoprevention utilization (19%) was double the < 10% seen in the national population.33-35 The presence of a knowledgeable breast health practitioner to recruit study participants and offer personalized counseling to women veterans is a likely factor in overcoming barriers to chemopreventive acceptance. These participants may have been motivated to seek care for their high-risk status given a strong family history and prior breast biopsies.
Interestingly, a 3-fold higher PTSD rate was seen in this pilot population (29%) when compared with PTSD rates in the general female population (7-10%) and still one-third higher than the general population of women veterans (20%).45-47 Mental health, anxiety, and PTSD have been barriers to patients who sought treatment and have been implicated in poor adherence to medical advice.48,49 Cancer screening can induce anxiety in patients, and it may be amplified in patients with PTSD. It was remarkable that although adherence with screening recommendations is decreased when PTSD is present, our patient population demonstrated a higher rate of screening adherence.
Women who are seen at the VA often use multiple clinical specialties, and their EMR can be accessed across VA medical centers nationwide. Therefore, identifying women veterans who meet screening criteria is easily attainable within the VA.
When comparing high-risk with average risk women, the lifestyle results (BMI, smoking history, exercise and consumption of fruits, vegetables and alcohol) were essentially the same. Lifestyle factors were similar to national population rates and were unlikely to impact risk levels.
Limitations
Study limitations included a high number of self-referrals and the large percentage of patients with a family history of breast cancer, making them more likely to seek screening. The higher-than-average risk of breast cancer may be driven by a high rate of breast biopsies and a strong family history. Lifestyle metrics could not be accurately compared to other national assessments of lifestyle factors due to the difference in data points that we used or the format of our questions.
Conclusions
As the number of women veterans increases and the incidence of breast cancer in women veterans rise, chemoprevention options should follow national guidelines. To our knowledge, this is the only oncology study with 60% Black women veterans. This study had a higher participation rate for Black women veterans than is typically seen in national research studies and shows the VA to be a germane source for further understanding of an understudied population that may benefit from increased screening for breast cancer.
A team-based, multidisciplinary model that meets the unique healthcare needs of women veterans results in a patient-centric delivery of care for assessing breast cancer risk status and prevention options. This model can be replicated nationally by directing primary care physicians and women’s health practitioners to a risk-assessment questionnaire and referring high-risk women for appropriate preventative care. Given that these results show chemoprevention adherence rates doubled those seen nationally, perhaps techniques used within this VA pilot study may be adapted to decrease breast cancer incidence nationally.
Since the rate of PTSD among women veterans is triple the national average, we would expect adherence rates to be lower in our patient cohort. However, the multidisciplinary approach we used in this study (eg, 1:1 consultation with oncologist; genetic counseling referrals; mental health support available), may have improved adherence rates. Perhaps the high rates of PTSD seen in the VA patient population can be a useful way to explore patient adherence rates in those with mental illness and medical conditions.
Future research with a larger cohort may lead to greater insight into the correlation between PTSD and adherence to treatment. Exploring the connection between breast cancer, epigenetics, and specific military service-related exposures could be an area of analysis among this veteran population exhibiting increased breast cancer rates. VAMCs are situated in rural, suburban, and urban locations across the United States and offers a diverse socioeconomic and ethnic patient population for inclusion in clinical investigations. Women veterans make up a small subpopulation of women in the United States, but it is worth considering VA patients as an untapped resource for research collaboration.
Acknowledgements
The authors thank Steven Sanchez and Marissa Vallette, PhD, Breast Health Research Group. This research project was approved by the James J. Peters VA Medical Center Quality Executive Committee and the Washington, DC VA Medical Center Institutional Review Board. This work was supported by the US Department of Veterans Affairs. This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
1. US Department of Veterans Affairs. National Center for Veterans Analysis and Statistics. The past, present and future of women veterans. Published February 2017. Accessed April 28, 2021. https://www.va.gov/vetdata/docs/specialreports/women_veterans_2015_final.pdf.
2. Frayne SM, Carney DV, Bastian L, et al. The VA Women’s Health Practice-Based Research Network: amplifying women veterans’ voices in VA research. J Gen Intern Med. 2013;28 Suppl 2(Suppl 2):S504-S509. doi:10.1007/s11606-013-2476-3
3. US Department of Veterans Affairs, Veterans Health Administration, Women’s Health Evaluation Initiative, Women Veterans Health Strategic Health Care Group. Sourcebook: women veterans in the Veterans Health Administration. Volume 1: Sociodemographic characteristics and use of VHA care. Published December 2010. Accessed April 12, 2021. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=2455
4. Bean-Mayberry B, Yano EM, Bayliss N, Navratil J, Weisman CS, Scholle SH. Federally funded comprehensive women’s health centers: leading innovation in women’s healthcare delivery. J Womens Health (Larchmt). 2007;16(9):1281-1290. doi:10.1089/jwh.2006.0284
5. US Department of Veterans Affairs. National Center for Veterans Analysis and Statistics.VA utilization profile FY 2016. Published November 2017. Accessed April 12, 2021. https://www.va.gov/vetdata/docs/QuickFacts/VA_Utilization_Profile.PDF
6. Ekenga CC, Parks CG, Sandler DP. Chemical exposures in the workplace and breast cancer risk: a prospective cohort study. Int J Cancer. 2015;137(7):1765-1774. doi:10.1002/ijc.29545
7. Rennix CP, Quinn MM, Amoroso PJ, Eisen EA, Wegman DH. Risk of breast cancer among enlisted Army women occupationally exposed to volatile organic compounds. Am J Ind Med. 2005;48(3):157-167. doi:10.1002/ajim.20201
8. Ritz B. Cancer mortality among workers exposed to chemicals during uranium processing. J Occup Environ Med. 1999;41(7):556-566. doi:10.1097/00043764-199907000-00004
9. Zhu K, Devesa SS, Wu H, et al. Cancer incidence in the U.S. military population: comparison with rates from the SEER program. Cancer Epidemiol Biomarkers Prev. 2009;18(6):1740-1745. doi:10.1158/1055-9965.EPI-09-0041
10. Freedman AN, Yu B, Gail MH, et al. Benefit/risk assessment for breast cancer chemoprevention with raloxifene or tamoxifen for women age 50 years or older [published correction appears in J Clin Oncol. 2013 Nov 10;31(32):4167]. J Clin Oncol. 2011;29(17):2327-2333. doi:10.1200/JCO.2010.33.0258
11. Greene, H. Cancer prevention, screening and early detection. In: Gobel BH, Triest-Robertson S, Vogel WH, eds. Advanced Oncology Nursing Certification Review and Resource Manual. 3rd ed. Oncology Nursing Society; 2016:1-34. https://www.ons.org/sites/default/files/publication_pdfs/2%20ADVPrac%20chapter%201.pdf
12. National Comprehensive Cancer Network. NCCN Breast Cancer Risk Reduction. Version 1.2021 NCCN Clinical Practice Guidelines in Oncology. Updated March 24, 2021 Accessed April 12, 2021. https://www.nccn.org/professionals/physician_gls/pdf/breast_risk.pdf
13. US Preventive Services Task Force. Breast cancer: Medications use to reduce risk. Updated September 3, 2019. Accessed April 12, 2021. https://www.uspreventiveservicestaskforce.org/uspstf/recommendation/breast-cancer-medications-for-risk-reduction
14. Moyer VA; U.S. Preventive Services Task Force. Medications to decrease the risk for breast cancer in women: recommendations from the U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159(10):698-708. doi:10.7326/0003-4819-159-10-201311190-00717
15. Boucher JE. Chemoprevention: an overview of pharmacologic agents and nursing considerations. Clin J Oncol Nurs. 2018;22(3):350-353. doi:10.1188/18.CJON.350-353
16. Nichols HB, Stürmer T, Lee VS, et al. Breast cancer chemoprevention in an integrated health care setting. JCO Clin Cancer Inform. 2017;1:1-12. doi:10.1200/CCI.16.00059
17. Bevers TB, Helvie M, Bonaccio E, et al. Breast cancer screening and diagnosis, Version 3.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2018;16(11):1362-1389. doi:10.6004/jnccn.2018.0083
18. Visvanathan K, Hurley P, Bantug E, et al. Use of pharmacologic interventions for breast cancer risk reduction: American Society of Clinical Oncology clinical practice guideline [published correction appears in J Clin Oncol. 2013 Dec 1;31(34):4383]. J Clin Oncol. 2013;31(23):2942-2962. doi:10.1200/JCO.2013.49.3122
19. Sealy-Jefferson S, Roseland ME, Cote ML, et al. rural-urban residence and stage at breast cancer diagnosis among postmenopausal women: The Women’s Health Initiative. J Womens Health (Larchmt). 2019;28(2):276-283. doi:10.1089/jwh.2017.6884
20. Holder KA. Veterans in rural America: 2011-2015. Published January 25, 2017. Accessed April 12, 2021. https://www.census.gov/library/publications/2017/acs/acs-36.html
21. Owens WL, Gallagher TJ, Kincheloe MJ, Ruetten VL. Implementation in a large health system of a program to identify women at high risk for breast cancer. J Oncol Pract. 2011;7(2):85-88. doi:10.1200/JOP.2010.000107
2. Pivot X, Viguier J, Touboul C, et al. Breast cancer screening controversy: too much or not enough?. Eur J Cancer Prev. 2015;24 Suppl:S73-S76. doi:10.1097/CEJ.0000000000000145
23. Bidassie B, Kovach A, Vallette MA, et al. Breast Cancer risk assessment and chemoprevention use among veterans affairs primary care providers: a national online survey. Mil Med. 2020;185(3-4):512-518. doi:10.1093/milmed/usz291
24. Brewster AM, Davidson NE, McCaskill-Stevens W. Chemoprevention for breast cancer: overcoming barriers to treatment. Am Soc Clin Oncol Educ Book. 2012;85-90. doi:10.14694/EdBook_AM.2012.32.152
25. Meyskens FL Jr, Curt GA, Brenner DE, et al. Regulatory approval of cancer risk-reducing (chemopreventive) drugs: moving what we have learned into the clinic. Cancer Prev Res (Phila). 2011;4(3):311-323. doi:10.1158/1940-6207.CAPR-09-0014
26. Tice JA, Kerlikowske K. Screening and prevention of breast cancer in primary care. Prim Care. 2009;36(3):533-558. doi:10.1016/j.pop.2009.04.003
27. Vogel VG. Selective estrogen receptor modulators and aromatase inhibitors for breast cancer chemoprevention. Curr Drug Targets. 2011;12(13):1874-1887. doi:10.2174/138945011798184164
28. Vogel VG, Costantino JP, Wickerham DL, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial [published correction appears in JAMA. 2006 Dec 27;296(24):2926] [published correction appears in JAMA. 2007 Sep 5;298(9):973]. JAMA. 2006;295(23):2727-2741. doi:10.1001/jama.295.23.joc60074
29. Pruthi S, Heisey RE, Bevers TB. Chemoprevention for breast cancer. Ann Surg Oncol. 2015;22(10):3230-3235. doi:10.1245/s10434-015-4715-9
30. Cuzick J, Sestak I, Forbes JF, et al. Anastrozole for prevention of breast cancer in high-risk postmenopausal women (IBIS-II): an international, double-blind, randomised placebo-controlled trial [published correction appears in Lancet. 2014 Mar 22;383(9922):1040] [published correction appears in Lancet. 2017 Mar 11;389(10073):1010]. Lancet. 2014;383(9922):1041-1048. doi:10.1016/S0140-6736(13)62292-8
31. Bozovic-Spasojevic I, Azambuja E, McCaskill-Stevens W, Dinh P, Cardoso F. Chemoprevention for breast cancer. Cancer Treat Rev. 2012;38(5):329-339. doi:10.1016/j.ctrv.2011.07.005
32. Gabriel EM, Jatoi I. Breast cancer chemoprevention. Expert Rev Anticancer Ther. 2012;12(2):223-228. doi:10.1586/era.11.206

33. Crew KD, Albain KS, Hershman DL, Unger JM, Lo SS. How do we increase uptake of tamoxifen and other anti-estrogens for breast cancer prevention?. NPJ Breast Cancer. 2017;3:20. Published 2017 May 19. doi:10.1038/s41523-017-0021-y
34. Ropka ME, Keim J, Philbrick JT. Patient decisions about breast cancer chemoprevention: a systematic review and meta-analysis. J Clin Oncol. 2010;28(18):3090-3095. doi:10.1200/JCO.2009.27.8077
35. Smith SG, Sestak I, Forster A, et al. Factors affecting uptake and adherence to breast cancer chemoprevention: a systematic review and meta-analysis. Ann Oncol. 2016;27(4):575-590. doi:10.1093/annonc/mdv590
36. Grann VR, Patel PR, Jacobson JS, et al. Comparative effectiveness of screening and prevention strategies among BRCA1/2-affected mutation carriers. Breast Cancer Res Treat. 2011 Feb;125(3):837-847. doi:10.1007/s10549-010-1043-4
37. Goss PE, Ingle JN, Alés-Martínez JE, et al. Exemestane for breast-cancer prevention in postmenopausal women [published correction appears in N Engl J Med. 2011 Oct 6;365(14):1361]. N Engl J Med. 2011;364(25):2381-2391. doi:10.1056/NEJMoa1103507
38. Kmietowicz Z. Five in six women reject drugs that could reduce their risk of breast cancer. BMJ. 2015;351:h6650. Published 2015 Dec 8. doi:10.1136/bmj.h6650
39. Nelson HD, Fu R, Griffin JC, Nygren P, Smith ME, Humphrey L. Systematic review: comparative effectiveness of medications to reduce risk for primary breast cancer. Ann Intern Med. 2009;151(10):703-235. doi:10.7326/0003-4819-151-10-200911170-00147
40. Dahabreh IJ, Wieland LS, Adam GP, Halladay C, Lau J, Trikalinos TA. Core needle and open surgery biopsy for diagnosis of breast lesions: an update to the 2009 report. Published September 2014. Accessed April 12, 2021. https://www.ncbi.nlm.nih.gov/books/NBK246878
41. National Cancer Institute. Genetics of breast and ovarian cancer (PDQ)—health profession version. Updated February 12, 2021. Accessed April 12, 2021. http://www.cancer.gov/cancertopics/pdq/genetics/breast-and-ovarian/HealthProfessional
42. US Department of Health and Human Services. National Institutes of Health, National Institute of Environmental Health Sciences The sister study. Accessed April 12, 2021. https://sisterstudy.niehs.nih.gov/english/NIEHS.htm
43. Tutt A, Ashworth A. Can genetic testing guide treatment in breast cancer?. Eur J Cancer. 2008;44(18):2774-2780. doi:10.1016/j.ejca.2008.10.009
44. Katz SJ, Ward KC, Hamilton AS, et al. Gaps in receipt of clinically indicated genetic counseling after diagnosis of breast cancer. J Clin Oncol. 2018;36(12):1218-1224. doi:10.1200/JCO.2017.76.2369
45. US Department of Veterans Affairs. PTSD: National Center for PTSD. How common is PTSD in adults? Updated October 17, 2019. Accessed April 12, 2021. https://www.ptsd.va.gov/understand/common/common_adults.asp
46. US Department of Veterans Affairs. PTSD: National Center for PTSD. How common is PTSD in women? Updated October 16, 2019. Accessed April 12, 2021. https://www.ptsd.va.gov/understand/common/common_women.asp
47. US Department of Veterans Affairs. PTSD: National Center for PTSD. How common is PTSD in veterans? Updated September 24, 2018. Accessed April 12, 2021. https://www.ptsd.va.gov/understand/common/common_veterans.asp
48. Lindberg NM, Wellisch D. Anxiety and compliance among women at high risk for breast cancer. Ann Behav Med. 2001;23(4):298-303. doi:10.1207/S15324796ABM2304_9
49. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107. doi:10.1001/archinte.160.14.2101
50. Centers for Disease Control and Prevention. MMWR appendix: breast cancer rates among black women and white women. Updated October 13, 2016. Accessed April 12, 2021. https://www.cdc.gov/cancer/breast/statistics/trends_invasive.htm
51. Richardson LC, Henley SJ, Miller JW, Massetti G, Thomas CC. Patterns and trends in age-specific black-white differences in breast cancer incidence and mortality - United States, 1999-2014. MMWR Morb Mortal Wkly Rep. 2016;65(40):1093-1098. Published 2016 Oct 14. doi:10.15585/mmwr.mm6540a1
52. Brody JG, Moysich KB, Humblet O, Attfield KR, Beehler GP, Rudel RA. Environmental pollutants and breast cancer: epidemiologic studies. Cancer. 2007;109(12 Suppl):2667-2711. doi:10.1002/cncr.22655
53. Brophy JT, Keith MM, Watterson A, et al. Breast cancer risk in relation to occupations with exposure to carcinogens and endocrine disruptors: a Canadian case-control study. Environ Health. 2012;11:87. Published 2012 Nov 19. doi:10.1186/1476-069X-11-87
54. Labrèche F, Goldberg MS, Valois MF, Nadon L. Postmenopausal breast cancer and occupational exposures. Occup Environ Med. 2010;67(4):263-269. doi:10.1136/oem.2009.049817
55. National Institute of Environmental Health Sciences, Interagency Breast Cancer & Environmental Research Coordinating Committee. Breast cancer and the environment: prioritizing prevention. Updated March 8, 2013. Accessed April 12, 2021. https://www.niehs.nih.gov/about/boards/ibcercc/index.cfm
56. Gail MH, Costantino JP, Pee D, et al. Projecting individualized absolute invasive breast cancer risk in African American women [published correction appears in J Natl Cancer Inst. 2008 Aug 6;100(15):1118] [published correction appears in J Natl Cancer Inst. 2008 Mar 5;100(5):373]. J Natl Cancer Inst. 2007;99(23):1782-1792. doi:10.1093/jnci/djm223
57. Corbie-Smith G, Thomas SB, Williams MV, Moody-Ayers S. Attitudes and beliefs of African Americans toward participation in medical research. J Gen Intern Med. 1999;14(9):537-546. doi:10.1046/j.1525-1497.1999.07048.x
58. Braunstein JB, Sherber NS, Schulman SP, Ding EL, Powe NR. Race, medical researcher distrust, perceived harm, and willingness to participate in cardiovascular prevention trials. Medicine (Baltimore). 2008;87(1):1-9. doi:10.1097/MD.0b013e3181625d78
The number of women seeking care from the Veterans Health Administration (VHA) is increasing.1 In 2015, there were 2 million women veterans in the United States, which is 9.4% of the total veteran population. This group is expected to increase at an average of about 18,000 women per year for the next 10 years.2 The percentage of women veterans who are US Department of Veterans Affairs (VA) users aged 45 to 64 years rose 46% from 2000 to 2015.1,3-4 It is estimated that 15% of veterans who used VA services in 2020 were women.1 Nineteen percent of women veterans are Black.1 The median age of women veterans in 2015 was 50 years.5 Breast cancer is the leading cancer affecting female veterans, and data suggest they have an increased risk of breast cancer based on unique service-related exposures.1,6-9
In the US, about 10 million women are eligible for breast cancer preventive therapy, including, but not limited to, medications, surgery, or lifestyle changes.10 Secondary prevention options include change in surveillance that can reduce their risk or identify cancer at an earlier stage when treatment is more effective. The United States Preventive Services Task Force, the National Comprehensive Cancer Network, the American Society for Clinical Oncology, the National Institute for Health and Care Excellence, and the Oncology Nursing Society recommend screening women aged ≥ 35 years to assess breast cancer risk.11-18 If a woman is at increased risk, she may be a candidate for chemoprevention, prozphylactic surgery, and possibly an enhanced screening regimen.
Urban and minority women are an understudied population. Most veterans (75%) live in urban or suburban settings.19,20 Urban veteran women constitute an important potential study population.
Chemoprevention measures have been underused because of factors involving both women and their health care providers. A large proportion of women are unaware of their higher risk status due to lack of adequate screening and risk assessment.21,22 In addition to patient lack of awareness of their high-risk status, primary care physicians are also reluctant to prescribe chemopreventive agents due to a lack of comfort or familiarity with the risks and benefits.23-26 The STAR2015, BCPT2005, IBIS2014, MAP3 2011, IBIS-I 2014, and IBIS II 2014 studies clearly demonstrate a 49 to 62% reduction in risk for women using chemoprevention such as selective estrogen receptor modulators or aromatase inhibitors, respectively.27-32 Yet only 4 to 9% of high-risk women not enrolled in a clinical trial are using chemoprevention.33-39
The possibility of developing breast cancer also may be increased because of a positive family history or being a member of a family in which there is a known susceptibility gene mutation.40 Based on these risk factors, women may be eligible for tailored follow-up and genetic counseling.41-44
Nationally, 7 to 10% of the civilian US population will experience posttraumatic stress disorder (PTSD).45 The rates are remarkably higher for women veterans, with roughly 20% diagnosed with PTSD.46,47 Anxiety and PTSD have been implicated in poor adherence to medical advice.48,49
In 2014, a national VA multidisciplinary group focused on breast cancer prevention, detection, treatment, and research to address breast health in the growing population of women veterans. High-risk breast cancer screenings are not routinely carried out by the VA in primary care, women’s health, or oncology services. Furthermore, the recording of screening questionnaire results was not synchronized until a standard questionnaire was created and approved as a template by this group in the VA electronic medical record (EMR) in 2015.
Several prediction models can identify which women are at an increased risk of developing breast cancer. The most commonly used risk assessment model, the Gail breast cancer risk assessment tool (BCRAT), has been refined to include women of additional ethnicities (https://www.cancer.gov/bcrisktool).
This pilot project was launched to identify an effective manner to screen women veterans regarding their risk of developing breast cancer and refer them for chemoprevention education or genetic counseling as appropriate.
Methods
A high-risk breast cancer screening questionnaire based on the Gail BCRAT and including lifestyle questions was developed and included as a note template in the VA EMR. The James J. Peters VA Medical Center, Bronx, NY (JJPVAMC) and the Washington DC VA Medical Center (DCVAMC) ran a pilot study between 2015 and 2018 using this breast cancer screening questionnaire to collect data from women veterans. Quality Executive Committee and institutional review board approvals were granted respectively.
Eligibility criteria included women aged ≥ 35 years with no personal history of breast cancer. Most patients were self-referred, but participants also were recruited during VA Breast Cancer Awareness month events, health fairs, or at informational tables in the hospital lobbies. After completing the 20 multiple choice questionnaire with a study team member, either in person or over the phone, a 5-year and lifetime risk of invasive breast cancer was calculated using the Gail BCRAT. A woman is considered high risk and eligible for chemoprevention if her 5-year risk is > 1.66% or her lifetime risk is ≥ 20%. Eligibility for genetic counseling is based on the Breast Cancer Referral Screening Tool, which includes a personal or family history of breast or ovarian cancer and Jewish ancestry.
All patients were notified of their average or high risk status by a clinician. Those who were deemed to be average risk received a follow-up letter in the mail with instructions (eg, to follow-up with a yearly mammogram). Those who were deemed to be high risk for developing breast cancer were asked to come in for an appointment with the study principal investigator (a VA oncologist/breast cancer specialist) to discuss prevention options, further screening, or referrals to genetic counseling. Depending on a patient’s other health factors, a woman at high risk for developing breast cancer also may be a candidate for chemoprevention with tamoxifen, raloxifene, exemestane, anastrozole, or letrozole.
Data on the participant’s lifestyle, including exercise, diet, and smoking, were evaluated to determine whether these factors had an impact on risk status.
Results
The JJP and DC VAMCs screened 103 women veterans between 2015 and 2018. Four patients were excluded for nonveteran (spousal) status, leaving 99 women veterans with a mean age of 54 years. The most common self-reported races were Black (60%), non-Hispanic White (14%), and Hispanic or Latino (13%) (Table 1).
Women veterans in our study were nearly 3-times more likely than the general population were to receive a high-risk Gail Score/BCRAT (35% vs 13%, respectively).50,51 Of this subset, 46% had breast biopsies, and 86% had a positive family history. Thirty-one percent of Black women in our study were high risk, while nationally, 8.2 to 13.3% of Black women aged 50 to 59 years are considered high risk.50,51 Of the Black high-risk group with a high Gail/BCRAT score, 94% had a positive family history, and 33% had a history of breast biopsy (Table 2).
Of the 35 high-risk patients 26 (74%) patients accepted consultations for chemoprevention and 5 (19%) started chemoprevention. Of this high-risk group, 13 (37%) patients were referred for genetic counseling (Table 3).44 The prevalence of PTSD was present in 31% of high-risk women and 29% of the cohort (Figure).The lifestyle questions indicated that, among all participants, 79% had an overweight or obese body mass index; 58% exercised weekly; 51% consumed alcohol; 14% were smokers; and 21% consumed 3 to 4 servings of fruits/vegetables daily.
Discussion
Breast cancer is the most common cancer in women.52 The number of women with breast cancer in the VHA has more than tripled from 1995 to 2012.1 The lifetime risk of developing breast cancer in the general population is about 13%.50 This rate can be affected by risk factors including age, hormone exposure, family history, radiation exposure, and lifestyle factors, such as weight and alcohol use.6,52-56 In the United States, invasive breast cancer affects 1 in 8 women.50,52,57
Our screened population showed nearly 3 times as many women veterans were at an increased risk for breast cancer when compared with historical averages in US women. This difference may be based on a high rate of prior breast biopsies or positive family history, although a provocative study using the Surveillance, Epidemiology, and End Results database showed military women to have higher rates of breast cancer as well.9 Historically, Blacks are vastly understudied in clinical research with only 5% representation on a national level.5,58 The urban locations of both pilot sites (Washington, DC and Bronx, NY) allowed for the inclusion of minority patients in our study. We found that the rates of breast cancer in Black women veterans to be higher than seen nationally, possibly prompting further screening initiatives for this understudied population.
Our pilot study’s chemoprevention utilization (19%) was double the < 10% seen in the national population.33-35 The presence of a knowledgeable breast health practitioner to recruit study participants and offer personalized counseling to women veterans is a likely factor in overcoming barriers to chemopreventive acceptance. These participants may have been motivated to seek care for their high-risk status given a strong family history and prior breast biopsies.
Interestingly, a 3-fold higher PTSD rate was seen in this pilot population (29%) when compared with PTSD rates in the general female population (7-10%) and still one-third higher than the general population of women veterans (20%).45-47 Mental health, anxiety, and PTSD have been barriers to patients who sought treatment and have been implicated in poor adherence to medical advice.48,49 Cancer screening can induce anxiety in patients, and it may be amplified in patients with PTSD. It was remarkable that although adherence with screening recommendations is decreased when PTSD is present, our patient population demonstrated a higher rate of screening adherence.
Women who are seen at the VA often use multiple clinical specialties, and their EMR can be accessed across VA medical centers nationwide. Therefore, identifying women veterans who meet screening criteria is easily attainable within the VA.
When comparing high-risk with average risk women, the lifestyle results (BMI, smoking history, exercise and consumption of fruits, vegetables and alcohol) were essentially the same. Lifestyle factors were similar to national population rates and were unlikely to impact risk levels.
Limitations
Study limitations included a high number of self-referrals and the large percentage of patients with a family history of breast cancer, making them more likely to seek screening. The higher-than-average risk of breast cancer may be driven by a high rate of breast biopsies and a strong family history. Lifestyle metrics could not be accurately compared to other national assessments of lifestyle factors due to the difference in data points that we used or the format of our questions.
Conclusions
As the number of women veterans increases and the incidence of breast cancer in women veterans rise, chemoprevention options should follow national guidelines. To our knowledge, this is the only oncology study with 60% Black women veterans. This study had a higher participation rate for Black women veterans than is typically seen in national research studies and shows the VA to be a germane source for further understanding of an understudied population that may benefit from increased screening for breast cancer.
A team-based, multidisciplinary model that meets the unique healthcare needs of women veterans results in a patient-centric delivery of care for assessing breast cancer risk status and prevention options. This model can be replicated nationally by directing primary care physicians and women’s health practitioners to a risk-assessment questionnaire and referring high-risk women for appropriate preventative care. Given that these results show chemoprevention adherence rates doubled those seen nationally, perhaps techniques used within this VA pilot study may be adapted to decrease breast cancer incidence nationally.
Since the rate of PTSD among women veterans is triple the national average, we would expect adherence rates to be lower in our patient cohort. However, the multidisciplinary approach we used in this study (eg, 1:1 consultation with oncologist; genetic counseling referrals; mental health support available), may have improved adherence rates. Perhaps the high rates of PTSD seen in the VA patient population can be a useful way to explore patient adherence rates in those with mental illness and medical conditions.
Future research with a larger cohort may lead to greater insight into the correlation between PTSD and adherence to treatment. Exploring the connection between breast cancer, epigenetics, and specific military service-related exposures could be an area of analysis among this veteran population exhibiting increased breast cancer rates. VAMCs are situated in rural, suburban, and urban locations across the United States and offers a diverse socioeconomic and ethnic patient population for inclusion in clinical investigations. Women veterans make up a small subpopulation of women in the United States, but it is worth considering VA patients as an untapped resource for research collaboration.
Acknowledgements
The authors thank Steven Sanchez and Marissa Vallette, PhD, Breast Health Research Group. This research project was approved by the James J. Peters VA Medical Center Quality Executive Committee and the Washington, DC VA Medical Center Institutional Review Board. This work was supported by the US Department of Veterans Affairs. This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
The number of women seeking care from the Veterans Health Administration (VHA) is increasing.1 In 2015, there were 2 million women veterans in the United States, which is 9.4% of the total veteran population. This group is expected to increase at an average of about 18,000 women per year for the next 10 years.2 The percentage of women veterans who are US Department of Veterans Affairs (VA) users aged 45 to 64 years rose 46% from 2000 to 2015.1,3-4 It is estimated that 15% of veterans who used VA services in 2020 were women.1 Nineteen percent of women veterans are Black.1 The median age of women veterans in 2015 was 50 years.5 Breast cancer is the leading cancer affecting female veterans, and data suggest they have an increased risk of breast cancer based on unique service-related exposures.1,6-9
In the US, about 10 million women are eligible for breast cancer preventive therapy, including, but not limited to, medications, surgery, or lifestyle changes.10 Secondary prevention options include change in surveillance that can reduce their risk or identify cancer at an earlier stage when treatment is more effective. The United States Preventive Services Task Force, the National Comprehensive Cancer Network, the American Society for Clinical Oncology, the National Institute for Health and Care Excellence, and the Oncology Nursing Society recommend screening women aged ≥ 35 years to assess breast cancer risk.11-18 If a woman is at increased risk, she may be a candidate for chemoprevention, prozphylactic surgery, and possibly an enhanced screening regimen.
Urban and minority women are an understudied population. Most veterans (75%) live in urban or suburban settings.19,20 Urban veteran women constitute an important potential study population.
Chemoprevention measures have been underused because of factors involving both women and their health care providers. A large proportion of women are unaware of their higher risk status due to lack of adequate screening and risk assessment.21,22 In addition to patient lack of awareness of their high-risk status, primary care physicians are also reluctant to prescribe chemopreventive agents due to a lack of comfort or familiarity with the risks and benefits.23-26 The STAR2015, BCPT2005, IBIS2014, MAP3 2011, IBIS-I 2014, and IBIS II 2014 studies clearly demonstrate a 49 to 62% reduction in risk for women using chemoprevention such as selective estrogen receptor modulators or aromatase inhibitors, respectively.27-32 Yet only 4 to 9% of high-risk women not enrolled in a clinical trial are using chemoprevention.33-39
The possibility of developing breast cancer also may be increased because of a positive family history or being a member of a family in which there is a known susceptibility gene mutation.40 Based on these risk factors, women may be eligible for tailored follow-up and genetic counseling.41-44
Nationally, 7 to 10% of the civilian US population will experience posttraumatic stress disorder (PTSD).45 The rates are remarkably higher for women veterans, with roughly 20% diagnosed with PTSD.46,47 Anxiety and PTSD have been implicated in poor adherence to medical advice.48,49
In 2014, a national VA multidisciplinary group focused on breast cancer prevention, detection, treatment, and research to address breast health in the growing population of women veterans. High-risk breast cancer screenings are not routinely carried out by the VA in primary care, women’s health, or oncology services. Furthermore, the recording of screening questionnaire results was not synchronized until a standard questionnaire was created and approved as a template by this group in the VA electronic medical record (EMR) in 2015.
Several prediction models can identify which women are at an increased risk of developing breast cancer. The most commonly used risk assessment model, the Gail breast cancer risk assessment tool (BCRAT), has been refined to include women of additional ethnicities (https://www.cancer.gov/bcrisktool).
This pilot project was launched to identify an effective manner to screen women veterans regarding their risk of developing breast cancer and refer them for chemoprevention education or genetic counseling as appropriate.
Methods
A high-risk breast cancer screening questionnaire based on the Gail BCRAT and including lifestyle questions was developed and included as a note template in the VA EMR. The James J. Peters VA Medical Center, Bronx, NY (JJPVAMC) and the Washington DC VA Medical Center (DCVAMC) ran a pilot study between 2015 and 2018 using this breast cancer screening questionnaire to collect data from women veterans. Quality Executive Committee and institutional review board approvals were granted respectively.
Eligibility criteria included women aged ≥ 35 years with no personal history of breast cancer. Most patients were self-referred, but participants also were recruited during VA Breast Cancer Awareness month events, health fairs, or at informational tables in the hospital lobbies. After completing the 20 multiple choice questionnaire with a study team member, either in person or over the phone, a 5-year and lifetime risk of invasive breast cancer was calculated using the Gail BCRAT. A woman is considered high risk and eligible for chemoprevention if her 5-year risk is > 1.66% or her lifetime risk is ≥ 20%. Eligibility for genetic counseling is based on the Breast Cancer Referral Screening Tool, which includes a personal or family history of breast or ovarian cancer and Jewish ancestry.
All patients were notified of their average or high risk status by a clinician. Those who were deemed to be average risk received a follow-up letter in the mail with instructions (eg, to follow-up with a yearly mammogram). Those who were deemed to be high risk for developing breast cancer were asked to come in for an appointment with the study principal investigator (a VA oncologist/breast cancer specialist) to discuss prevention options, further screening, or referrals to genetic counseling. Depending on a patient’s other health factors, a woman at high risk for developing breast cancer also may be a candidate for chemoprevention with tamoxifen, raloxifene, exemestane, anastrozole, or letrozole.
Data on the participant’s lifestyle, including exercise, diet, and smoking, were evaluated to determine whether these factors had an impact on risk status.
Results
The JJP and DC VAMCs screened 103 women veterans between 2015 and 2018. Four patients were excluded for nonveteran (spousal) status, leaving 99 women veterans with a mean age of 54 years. The most common self-reported races were Black (60%), non-Hispanic White (14%), and Hispanic or Latino (13%) (Table 1).
Women veterans in our study were nearly 3-times more likely than the general population were to receive a high-risk Gail Score/BCRAT (35% vs 13%, respectively).50,51 Of this subset, 46% had breast biopsies, and 86% had a positive family history. Thirty-one percent of Black women in our study were high risk, while nationally, 8.2 to 13.3% of Black women aged 50 to 59 years are considered high risk.50,51 Of the Black high-risk group with a high Gail/BCRAT score, 94% had a positive family history, and 33% had a history of breast biopsy (Table 2).
Of the 35 high-risk patients 26 (74%) patients accepted consultations for chemoprevention and 5 (19%) started chemoprevention. Of this high-risk group, 13 (37%) patients were referred for genetic counseling (Table 3).44 The prevalence of PTSD was present in 31% of high-risk women and 29% of the cohort (Figure).The lifestyle questions indicated that, among all participants, 79% had an overweight or obese body mass index; 58% exercised weekly; 51% consumed alcohol; 14% were smokers; and 21% consumed 3 to 4 servings of fruits/vegetables daily.
Discussion
Breast cancer is the most common cancer in women.52 The number of women with breast cancer in the VHA has more than tripled from 1995 to 2012.1 The lifetime risk of developing breast cancer in the general population is about 13%.50 This rate can be affected by risk factors including age, hormone exposure, family history, radiation exposure, and lifestyle factors, such as weight and alcohol use.6,52-56 In the United States, invasive breast cancer affects 1 in 8 women.50,52,57
Our screened population showed nearly 3 times as many women veterans were at an increased risk for breast cancer when compared with historical averages in US women. This difference may be based on a high rate of prior breast biopsies or positive family history, although a provocative study using the Surveillance, Epidemiology, and End Results database showed military women to have higher rates of breast cancer as well.9 Historically, Blacks are vastly understudied in clinical research with only 5% representation on a national level.5,58 The urban locations of both pilot sites (Washington, DC and Bronx, NY) allowed for the inclusion of minority patients in our study. We found that the rates of breast cancer in Black women veterans to be higher than seen nationally, possibly prompting further screening initiatives for this understudied population.
Our pilot study’s chemoprevention utilization (19%) was double the < 10% seen in the national population.33-35 The presence of a knowledgeable breast health practitioner to recruit study participants and offer personalized counseling to women veterans is a likely factor in overcoming barriers to chemopreventive acceptance. These participants may have been motivated to seek care for their high-risk status given a strong family history and prior breast biopsies.
Interestingly, a 3-fold higher PTSD rate was seen in this pilot population (29%) when compared with PTSD rates in the general female population (7-10%) and still one-third higher than the general population of women veterans (20%).45-47 Mental health, anxiety, and PTSD have been barriers to patients who sought treatment and have been implicated in poor adherence to medical advice.48,49 Cancer screening can induce anxiety in patients, and it may be amplified in patients with PTSD. It was remarkable that although adherence with screening recommendations is decreased when PTSD is present, our patient population demonstrated a higher rate of screening adherence.
Women who are seen at the VA often use multiple clinical specialties, and their EMR can be accessed across VA medical centers nationwide. Therefore, identifying women veterans who meet screening criteria is easily attainable within the VA.
When comparing high-risk with average risk women, the lifestyle results (BMI, smoking history, exercise and consumption of fruits, vegetables and alcohol) were essentially the same. Lifestyle factors were similar to national population rates and were unlikely to impact risk levels.
Limitations
Study limitations included a high number of self-referrals and the large percentage of patients with a family history of breast cancer, making them more likely to seek screening. The higher-than-average risk of breast cancer may be driven by a high rate of breast biopsies and a strong family history. Lifestyle metrics could not be accurately compared to other national assessments of lifestyle factors due to the difference in data points that we used or the format of our questions.
Conclusions
As the number of women veterans increases and the incidence of breast cancer in women veterans rise, chemoprevention options should follow national guidelines. To our knowledge, this is the only oncology study with 60% Black women veterans. This study had a higher participation rate for Black women veterans than is typically seen in national research studies and shows the VA to be a germane source for further understanding of an understudied population that may benefit from increased screening for breast cancer.
A team-based, multidisciplinary model that meets the unique healthcare needs of women veterans results in a patient-centric delivery of care for assessing breast cancer risk status and prevention options. This model can be replicated nationally by directing primary care physicians and women’s health practitioners to a risk-assessment questionnaire and referring high-risk women for appropriate preventative care. Given that these results show chemoprevention adherence rates doubled those seen nationally, perhaps techniques used within this VA pilot study may be adapted to decrease breast cancer incidence nationally.
Since the rate of PTSD among women veterans is triple the national average, we would expect adherence rates to be lower in our patient cohort. However, the multidisciplinary approach we used in this study (eg, 1:1 consultation with oncologist; genetic counseling referrals; mental health support available), may have improved adherence rates. Perhaps the high rates of PTSD seen in the VA patient population can be a useful way to explore patient adherence rates in those with mental illness and medical conditions.
Future research with a larger cohort may lead to greater insight into the correlation between PTSD and adherence to treatment. Exploring the connection between breast cancer, epigenetics, and specific military service-related exposures could be an area of analysis among this veteran population exhibiting increased breast cancer rates. VAMCs are situated in rural, suburban, and urban locations across the United States and offers a diverse socioeconomic and ethnic patient population for inclusion in clinical investigations. Women veterans make up a small subpopulation of women in the United States, but it is worth considering VA patients as an untapped resource for research collaboration.
Acknowledgements
The authors thank Steven Sanchez and Marissa Vallette, PhD, Breast Health Research Group. This research project was approved by the James J. Peters VA Medical Center Quality Executive Committee and the Washington, DC VA Medical Center Institutional Review Board. This work was supported by the US Department of Veterans Affairs. This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
1. US Department of Veterans Affairs. National Center for Veterans Analysis and Statistics. The past, present and future of women veterans. Published February 2017. Accessed April 28, 2021. https://www.va.gov/vetdata/docs/specialreports/women_veterans_2015_final.pdf.
2. Frayne SM, Carney DV, Bastian L, et al. The VA Women’s Health Practice-Based Research Network: amplifying women veterans’ voices in VA research. J Gen Intern Med. 2013;28 Suppl 2(Suppl 2):S504-S509. doi:10.1007/s11606-013-2476-3
3. US Department of Veterans Affairs, Veterans Health Administration, Women’s Health Evaluation Initiative, Women Veterans Health Strategic Health Care Group. Sourcebook: women veterans in the Veterans Health Administration. Volume 1: Sociodemographic characteristics and use of VHA care. Published December 2010. Accessed April 12, 2021. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=2455
4. Bean-Mayberry B, Yano EM, Bayliss N, Navratil J, Weisman CS, Scholle SH. Federally funded comprehensive women’s health centers: leading innovation in women’s healthcare delivery. J Womens Health (Larchmt). 2007;16(9):1281-1290. doi:10.1089/jwh.2006.0284
5. US Department of Veterans Affairs. National Center for Veterans Analysis and Statistics.VA utilization profile FY 2016. Published November 2017. Accessed April 12, 2021. https://www.va.gov/vetdata/docs/QuickFacts/VA_Utilization_Profile.PDF
6. Ekenga CC, Parks CG, Sandler DP. Chemical exposures in the workplace and breast cancer risk: a prospective cohort study. Int J Cancer. 2015;137(7):1765-1774. doi:10.1002/ijc.29545
7. Rennix CP, Quinn MM, Amoroso PJ, Eisen EA, Wegman DH. Risk of breast cancer among enlisted Army women occupationally exposed to volatile organic compounds. Am J Ind Med. 2005;48(3):157-167. doi:10.1002/ajim.20201
8. Ritz B. Cancer mortality among workers exposed to chemicals during uranium processing. J Occup Environ Med. 1999;41(7):556-566. doi:10.1097/00043764-199907000-00004
9. Zhu K, Devesa SS, Wu H, et al. Cancer incidence in the U.S. military population: comparison with rates from the SEER program. Cancer Epidemiol Biomarkers Prev. 2009;18(6):1740-1745. doi:10.1158/1055-9965.EPI-09-0041
10. Freedman AN, Yu B, Gail MH, et al. Benefit/risk assessment for breast cancer chemoprevention with raloxifene or tamoxifen for women age 50 years or older [published correction appears in J Clin Oncol. 2013 Nov 10;31(32):4167]. J Clin Oncol. 2011;29(17):2327-2333. doi:10.1200/JCO.2010.33.0258
11. Greene, H. Cancer prevention, screening and early detection. In: Gobel BH, Triest-Robertson S, Vogel WH, eds. Advanced Oncology Nursing Certification Review and Resource Manual. 3rd ed. Oncology Nursing Society; 2016:1-34. https://www.ons.org/sites/default/files/publication_pdfs/2%20ADVPrac%20chapter%201.pdf
12. National Comprehensive Cancer Network. NCCN Breast Cancer Risk Reduction. Version 1.2021 NCCN Clinical Practice Guidelines in Oncology. Updated March 24, 2021 Accessed April 12, 2021. https://www.nccn.org/professionals/physician_gls/pdf/breast_risk.pdf
13. US Preventive Services Task Force. Breast cancer: Medications use to reduce risk. Updated September 3, 2019. Accessed April 12, 2021. https://www.uspreventiveservicestaskforce.org/uspstf/recommendation/breast-cancer-medications-for-risk-reduction
14. Moyer VA; U.S. Preventive Services Task Force. Medications to decrease the risk for breast cancer in women: recommendations from the U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159(10):698-708. doi:10.7326/0003-4819-159-10-201311190-00717
15. Boucher JE. Chemoprevention: an overview of pharmacologic agents and nursing considerations. Clin J Oncol Nurs. 2018;22(3):350-353. doi:10.1188/18.CJON.350-353
16. Nichols HB, Stürmer T, Lee VS, et al. Breast cancer chemoprevention in an integrated health care setting. JCO Clin Cancer Inform. 2017;1:1-12. doi:10.1200/CCI.16.00059
17. Bevers TB, Helvie M, Bonaccio E, et al. Breast cancer screening and diagnosis, Version 3.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2018;16(11):1362-1389. doi:10.6004/jnccn.2018.0083
18. Visvanathan K, Hurley P, Bantug E, et al. Use of pharmacologic interventions for breast cancer risk reduction: American Society of Clinical Oncology clinical practice guideline [published correction appears in J Clin Oncol. 2013 Dec 1;31(34):4383]. J Clin Oncol. 2013;31(23):2942-2962. doi:10.1200/JCO.2013.49.3122
19. Sealy-Jefferson S, Roseland ME, Cote ML, et al. rural-urban residence and stage at breast cancer diagnosis among postmenopausal women: The Women’s Health Initiative. J Womens Health (Larchmt). 2019;28(2):276-283. doi:10.1089/jwh.2017.6884
20. Holder KA. Veterans in rural America: 2011-2015. Published January 25, 2017. Accessed April 12, 2021. https://www.census.gov/library/publications/2017/acs/acs-36.html
21. Owens WL, Gallagher TJ, Kincheloe MJ, Ruetten VL. Implementation in a large health system of a program to identify women at high risk for breast cancer. J Oncol Pract. 2011;7(2):85-88. doi:10.1200/JOP.2010.000107
2. Pivot X, Viguier J, Touboul C, et al. Breast cancer screening controversy: too much or not enough?. Eur J Cancer Prev. 2015;24 Suppl:S73-S76. doi:10.1097/CEJ.0000000000000145
23. Bidassie B, Kovach A, Vallette MA, et al. Breast Cancer risk assessment and chemoprevention use among veterans affairs primary care providers: a national online survey. Mil Med. 2020;185(3-4):512-518. doi:10.1093/milmed/usz291
24. Brewster AM, Davidson NE, McCaskill-Stevens W. Chemoprevention for breast cancer: overcoming barriers to treatment. Am Soc Clin Oncol Educ Book. 2012;85-90. doi:10.14694/EdBook_AM.2012.32.152
25. Meyskens FL Jr, Curt GA, Brenner DE, et al. Regulatory approval of cancer risk-reducing (chemopreventive) drugs: moving what we have learned into the clinic. Cancer Prev Res (Phila). 2011;4(3):311-323. doi:10.1158/1940-6207.CAPR-09-0014
26. Tice JA, Kerlikowske K. Screening and prevention of breast cancer in primary care. Prim Care. 2009;36(3):533-558. doi:10.1016/j.pop.2009.04.003
27. Vogel VG. Selective estrogen receptor modulators and aromatase inhibitors for breast cancer chemoprevention. Curr Drug Targets. 2011;12(13):1874-1887. doi:10.2174/138945011798184164
28. Vogel VG, Costantino JP, Wickerham DL, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial [published correction appears in JAMA. 2006 Dec 27;296(24):2926] [published correction appears in JAMA. 2007 Sep 5;298(9):973]. JAMA. 2006;295(23):2727-2741. doi:10.1001/jama.295.23.joc60074
29. Pruthi S, Heisey RE, Bevers TB. Chemoprevention for breast cancer. Ann Surg Oncol. 2015;22(10):3230-3235. doi:10.1245/s10434-015-4715-9
30. Cuzick J, Sestak I, Forbes JF, et al. Anastrozole for prevention of breast cancer in high-risk postmenopausal women (IBIS-II): an international, double-blind, randomised placebo-controlled trial [published correction appears in Lancet. 2014 Mar 22;383(9922):1040] [published correction appears in Lancet. 2017 Mar 11;389(10073):1010]. Lancet. 2014;383(9922):1041-1048. doi:10.1016/S0140-6736(13)62292-8
31. Bozovic-Spasojevic I, Azambuja E, McCaskill-Stevens W, Dinh P, Cardoso F. Chemoprevention for breast cancer. Cancer Treat Rev. 2012;38(5):329-339. doi:10.1016/j.ctrv.2011.07.005
32. Gabriel EM, Jatoi I. Breast cancer chemoprevention. Expert Rev Anticancer Ther. 2012;12(2):223-228. doi:10.1586/era.11.206
33. Crew KD, Albain KS, Hershman DL, Unger JM, Lo SS. How do we increase uptake of tamoxifen and other anti-estrogens for breast cancer prevention?. NPJ Breast Cancer. 2017;3:20. Published 2017 May 19. doi:10.1038/s41523-017-0021-y
34. Ropka ME, Keim J, Philbrick JT. Patient decisions about breast cancer chemoprevention: a systematic review and meta-analysis. J Clin Oncol. 2010;28(18):3090-3095. doi:10.1200/JCO.2009.27.8077
35. Smith SG, Sestak I, Forster A, et al. Factors affecting uptake and adherence to breast cancer chemoprevention: a systematic review and meta-analysis. Ann Oncol. 2016;27(4):575-590. doi:10.1093/annonc/mdv590
36. Grann VR, Patel PR, Jacobson JS, et al. Comparative effectiveness of screening and prevention strategies among BRCA1/2-affected mutation carriers. Breast Cancer Res Treat. 2011 Feb;125(3):837-847. doi:10.1007/s10549-010-1043-4
37. Goss PE, Ingle JN, Alés-Martínez JE, et al. Exemestane for breast-cancer prevention in postmenopausal women [published correction appears in N Engl J Med. 2011 Oct 6;365(14):1361]. N Engl J Med. 2011;364(25):2381-2391. doi:10.1056/NEJMoa1103507
38. Kmietowicz Z. Five in six women reject drugs that could reduce their risk of breast cancer. BMJ. 2015;351:h6650. Published 2015 Dec 8. doi:10.1136/bmj.h6650
39. Nelson HD, Fu R, Griffin JC, Nygren P, Smith ME, Humphrey L. Systematic review: comparative effectiveness of medications to reduce risk for primary breast cancer. Ann Intern Med. 2009;151(10):703-235. doi:10.7326/0003-4819-151-10-200911170-00147
40. Dahabreh IJ, Wieland LS, Adam GP, Halladay C, Lau J, Trikalinos TA. Core needle and open surgery biopsy for diagnosis of breast lesions: an update to the 2009 report. Published September 2014. Accessed April 12, 2021. https://www.ncbi.nlm.nih.gov/books/NBK246878
41. National Cancer Institute. Genetics of breast and ovarian cancer (PDQ)—health profession version. Updated February 12, 2021. Accessed April 12, 2021. http://www.cancer.gov/cancertopics/pdq/genetics/breast-and-ovarian/HealthProfessional
42. US Department of Health and Human Services. National Institutes of Health, National Institute of Environmental Health Sciences The sister study. Accessed April 12, 2021. https://sisterstudy.niehs.nih.gov/english/NIEHS.htm
43. Tutt A, Ashworth A. Can genetic testing guide treatment in breast cancer?. Eur J Cancer. 2008;44(18):2774-2780. doi:10.1016/j.ejca.2008.10.009
44. Katz SJ, Ward KC, Hamilton AS, et al. Gaps in receipt of clinically indicated genetic counseling after diagnosis of breast cancer. J Clin Oncol. 2018;36(12):1218-1224. doi:10.1200/JCO.2017.76.2369
45. US Department of Veterans Affairs. PTSD: National Center for PTSD. How common is PTSD in adults? Updated October 17, 2019. Accessed April 12, 2021. https://www.ptsd.va.gov/understand/common/common_adults.asp
46. US Department of Veterans Affairs. PTSD: National Center for PTSD. How common is PTSD in women? Updated October 16, 2019. Accessed April 12, 2021. https://www.ptsd.va.gov/understand/common/common_women.asp
47. US Department of Veterans Affairs. PTSD: National Center for PTSD. How common is PTSD in veterans? Updated September 24, 2018. Accessed April 12, 2021. https://www.ptsd.va.gov/understand/common/common_veterans.asp
48. Lindberg NM, Wellisch D. Anxiety and compliance among women at high risk for breast cancer. Ann Behav Med. 2001;23(4):298-303. doi:10.1207/S15324796ABM2304_9
49. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107. doi:10.1001/archinte.160.14.2101
50. Centers for Disease Control and Prevention. MMWR appendix: breast cancer rates among black women and white women. Updated October 13, 2016. Accessed April 12, 2021. https://www.cdc.gov/cancer/breast/statistics/trends_invasive.htm
51. Richardson LC, Henley SJ, Miller JW, Massetti G, Thomas CC. Patterns and trends in age-specific black-white differences in breast cancer incidence and mortality - United States, 1999-2014. MMWR Morb Mortal Wkly Rep. 2016;65(40):1093-1098. Published 2016 Oct 14. doi:10.15585/mmwr.mm6540a1
52. Brody JG, Moysich KB, Humblet O, Attfield KR, Beehler GP, Rudel RA. Environmental pollutants and breast cancer: epidemiologic studies. Cancer. 2007;109(12 Suppl):2667-2711. doi:10.1002/cncr.22655
53. Brophy JT, Keith MM, Watterson A, et al. Breast cancer risk in relation to occupations with exposure to carcinogens and endocrine disruptors: a Canadian case-control study. Environ Health. 2012;11:87. Published 2012 Nov 19. doi:10.1186/1476-069X-11-87
54. Labrèche F, Goldberg MS, Valois MF, Nadon L. Postmenopausal breast cancer and occupational exposures. Occup Environ Med. 2010;67(4):263-269. doi:10.1136/oem.2009.049817
55. National Institute of Environmental Health Sciences, Interagency Breast Cancer & Environmental Research Coordinating Committee. Breast cancer and the environment: prioritizing prevention. Updated March 8, 2013. Accessed April 12, 2021. https://www.niehs.nih.gov/about/boards/ibcercc/index.cfm
56. Gail MH, Costantino JP, Pee D, et al. Projecting individualized absolute invasive breast cancer risk in African American women [published correction appears in J Natl Cancer Inst. 2008 Aug 6;100(15):1118] [published correction appears in J Natl Cancer Inst. 2008 Mar 5;100(5):373]. J Natl Cancer Inst. 2007;99(23):1782-1792. doi:10.1093/jnci/djm223
57. Corbie-Smith G, Thomas SB, Williams MV, Moody-Ayers S. Attitudes and beliefs of African Americans toward participation in medical research. J Gen Intern Med. 1999;14(9):537-546. doi:10.1046/j.1525-1497.1999.07048.x
58. Braunstein JB, Sherber NS, Schulman SP, Ding EL, Powe NR. Race, medical researcher distrust, perceived harm, and willingness to participate in cardiovascular prevention trials. Medicine (Baltimore). 2008;87(1):1-9. doi:10.1097/MD.0b013e3181625d78
1. US Department of Veterans Affairs. National Center for Veterans Analysis and Statistics. The past, present and future of women veterans. Published February 2017. Accessed April 28, 2021. https://www.va.gov/vetdata/docs/specialreports/women_veterans_2015_final.pdf.
2. Frayne SM, Carney DV, Bastian L, et al. The VA Women’s Health Practice-Based Research Network: amplifying women veterans’ voices in VA research. J Gen Intern Med. 2013;28 Suppl 2(Suppl 2):S504-S509. doi:10.1007/s11606-013-2476-3
3. US Department of Veterans Affairs, Veterans Health Administration, Women’s Health Evaluation Initiative, Women Veterans Health Strategic Health Care Group. Sourcebook: women veterans in the Veterans Health Administration. Volume 1: Sociodemographic characteristics and use of VHA care. Published December 2010. Accessed April 12, 2021. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=2455
4. Bean-Mayberry B, Yano EM, Bayliss N, Navratil J, Weisman CS, Scholle SH. Federally funded comprehensive women’s health centers: leading innovation in women’s healthcare delivery. J Womens Health (Larchmt). 2007;16(9):1281-1290. doi:10.1089/jwh.2006.0284
5. US Department of Veterans Affairs. National Center for Veterans Analysis and Statistics.VA utilization profile FY 2016. Published November 2017. Accessed April 12, 2021. https://www.va.gov/vetdata/docs/QuickFacts/VA_Utilization_Profile.PDF
6. Ekenga CC, Parks CG, Sandler DP. Chemical exposures in the workplace and breast cancer risk: a prospective cohort study. Int J Cancer. 2015;137(7):1765-1774. doi:10.1002/ijc.29545
7. Rennix CP, Quinn MM, Amoroso PJ, Eisen EA, Wegman DH. Risk of breast cancer among enlisted Army women occupationally exposed to volatile organic compounds. Am J Ind Med. 2005;48(3):157-167. doi:10.1002/ajim.20201
8. Ritz B. Cancer mortality among workers exposed to chemicals during uranium processing. J Occup Environ Med. 1999;41(7):556-566. doi:10.1097/00043764-199907000-00004
9. Zhu K, Devesa SS, Wu H, et al. Cancer incidence in the U.S. military population: comparison with rates from the SEER program. Cancer Epidemiol Biomarkers Prev. 2009;18(6):1740-1745. doi:10.1158/1055-9965.EPI-09-0041
10. Freedman AN, Yu B, Gail MH, et al. Benefit/risk assessment for breast cancer chemoprevention with raloxifene or tamoxifen for women age 50 years or older [published correction appears in J Clin Oncol. 2013 Nov 10;31(32):4167]. J Clin Oncol. 2011;29(17):2327-2333. doi:10.1200/JCO.2010.33.0258
11. Greene, H. Cancer prevention, screening and early detection. In: Gobel BH, Triest-Robertson S, Vogel WH, eds. Advanced Oncology Nursing Certification Review and Resource Manual. 3rd ed. Oncology Nursing Society; 2016:1-34. https://www.ons.org/sites/default/files/publication_pdfs/2%20ADVPrac%20chapter%201.pdf
12. National Comprehensive Cancer Network. NCCN Breast Cancer Risk Reduction. Version 1.2021 NCCN Clinical Practice Guidelines in Oncology. Updated March 24, 2021 Accessed April 12, 2021. https://www.nccn.org/professionals/physician_gls/pdf/breast_risk.pdf
13. US Preventive Services Task Force. Breast cancer: Medications use to reduce risk. Updated September 3, 2019. Accessed April 12, 2021. https://www.uspreventiveservicestaskforce.org/uspstf/recommendation/breast-cancer-medications-for-risk-reduction
14. Moyer VA; U.S. Preventive Services Task Force. Medications to decrease the risk for breast cancer in women: recommendations from the U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2013;159(10):698-708. doi:10.7326/0003-4819-159-10-201311190-00717
15. Boucher JE. Chemoprevention: an overview of pharmacologic agents and nursing considerations. Clin J Oncol Nurs. 2018;22(3):350-353. doi:10.1188/18.CJON.350-353
16. Nichols HB, Stürmer T, Lee VS, et al. Breast cancer chemoprevention in an integrated health care setting. JCO Clin Cancer Inform. 2017;1:1-12. doi:10.1200/CCI.16.00059
17. Bevers TB, Helvie M, Bonaccio E, et al. Breast cancer screening and diagnosis, Version 3.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2018;16(11):1362-1389. doi:10.6004/jnccn.2018.0083
18. Visvanathan K, Hurley P, Bantug E, et al. Use of pharmacologic interventions for breast cancer risk reduction: American Society of Clinical Oncology clinical practice guideline [published correction appears in J Clin Oncol. 2013 Dec 1;31(34):4383]. J Clin Oncol. 2013;31(23):2942-2962. doi:10.1200/JCO.2013.49.3122
19. Sealy-Jefferson S, Roseland ME, Cote ML, et al. rural-urban residence and stage at breast cancer diagnosis among postmenopausal women: The Women’s Health Initiative. J Womens Health (Larchmt). 2019;28(2):276-283. doi:10.1089/jwh.2017.6884
20. Holder KA. Veterans in rural America: 2011-2015. Published January 25, 2017. Accessed April 12, 2021. https://www.census.gov/library/publications/2017/acs/acs-36.html
21. Owens WL, Gallagher TJ, Kincheloe MJ, Ruetten VL. Implementation in a large health system of a program to identify women at high risk for breast cancer. J Oncol Pract. 2011;7(2):85-88. doi:10.1200/JOP.2010.000107
2. Pivot X, Viguier J, Touboul C, et al. Breast cancer screening controversy: too much or not enough?. Eur J Cancer Prev. 2015;24 Suppl:S73-S76. doi:10.1097/CEJ.0000000000000145
23. Bidassie B, Kovach A, Vallette MA, et al. Breast Cancer risk assessment and chemoprevention use among veterans affairs primary care providers: a national online survey. Mil Med. 2020;185(3-4):512-518. doi:10.1093/milmed/usz291
24. Brewster AM, Davidson NE, McCaskill-Stevens W. Chemoprevention for breast cancer: overcoming barriers to treatment. Am Soc Clin Oncol Educ Book. 2012;85-90. doi:10.14694/EdBook_AM.2012.32.152
25. Meyskens FL Jr, Curt GA, Brenner DE, et al. Regulatory approval of cancer risk-reducing (chemopreventive) drugs: moving what we have learned into the clinic. Cancer Prev Res (Phila). 2011;4(3):311-323. doi:10.1158/1940-6207.CAPR-09-0014
26. Tice JA, Kerlikowske K. Screening and prevention of breast cancer in primary care. Prim Care. 2009;36(3):533-558. doi:10.1016/j.pop.2009.04.003
27. Vogel VG. Selective estrogen receptor modulators and aromatase inhibitors for breast cancer chemoprevention. Curr Drug Targets. 2011;12(13):1874-1887. doi:10.2174/138945011798184164
28. Vogel VG, Costantino JP, Wickerham DL, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial [published correction appears in JAMA. 2006 Dec 27;296(24):2926] [published correction appears in JAMA. 2007 Sep 5;298(9):973]. JAMA. 2006;295(23):2727-2741. doi:10.1001/jama.295.23.joc60074
29. Pruthi S, Heisey RE, Bevers TB. Chemoprevention for breast cancer. Ann Surg Oncol. 2015;22(10):3230-3235. doi:10.1245/s10434-015-4715-9
30. Cuzick J, Sestak I, Forbes JF, et al. Anastrozole for prevention of breast cancer in high-risk postmenopausal women (IBIS-II): an international, double-blind, randomised placebo-controlled trial [published correction appears in Lancet. 2014 Mar 22;383(9922):1040] [published correction appears in Lancet. 2017 Mar 11;389(10073):1010]. Lancet. 2014;383(9922):1041-1048. doi:10.1016/S0140-6736(13)62292-8
31. Bozovic-Spasojevic I, Azambuja E, McCaskill-Stevens W, Dinh P, Cardoso F. Chemoprevention for breast cancer. Cancer Treat Rev. 2012;38(5):329-339. doi:10.1016/j.ctrv.2011.07.005
32. Gabriel EM, Jatoi I. Breast cancer chemoprevention. Expert Rev Anticancer Ther. 2012;12(2):223-228. doi:10.1586/era.11.206
33. Crew KD, Albain KS, Hershman DL, Unger JM, Lo SS. How do we increase uptake of tamoxifen and other anti-estrogens for breast cancer prevention?. NPJ Breast Cancer. 2017;3:20. Published 2017 May 19. doi:10.1038/s41523-017-0021-y
34. Ropka ME, Keim J, Philbrick JT. Patient decisions about breast cancer chemoprevention: a systematic review and meta-analysis. J Clin Oncol. 2010;28(18):3090-3095. doi:10.1200/JCO.2009.27.8077
35. Smith SG, Sestak I, Forster A, et al. Factors affecting uptake and adherence to breast cancer chemoprevention: a systematic review and meta-analysis. Ann Oncol. 2016;27(4):575-590. doi:10.1093/annonc/mdv590
36. Grann VR, Patel PR, Jacobson JS, et al. Comparative effectiveness of screening and prevention strategies among BRCA1/2-affected mutation carriers. Breast Cancer Res Treat. 2011 Feb;125(3):837-847. doi:10.1007/s10549-010-1043-4
37. Goss PE, Ingle JN, Alés-Martínez JE, et al. Exemestane for breast-cancer prevention in postmenopausal women [published correction appears in N Engl J Med. 2011 Oct 6;365(14):1361]. N Engl J Med. 2011;364(25):2381-2391. doi:10.1056/NEJMoa1103507
38. Kmietowicz Z. Five in six women reject drugs that could reduce their risk of breast cancer. BMJ. 2015;351:h6650. Published 2015 Dec 8. doi:10.1136/bmj.h6650
39. Nelson HD, Fu R, Griffin JC, Nygren P, Smith ME, Humphrey L. Systematic review: comparative effectiveness of medications to reduce risk for primary breast cancer. Ann Intern Med. 2009;151(10):703-235. doi:10.7326/0003-4819-151-10-200911170-00147
40. Dahabreh IJ, Wieland LS, Adam GP, Halladay C, Lau J, Trikalinos TA. Core needle and open surgery biopsy for diagnosis of breast lesions: an update to the 2009 report. Published September 2014. Accessed April 12, 2021. https://www.ncbi.nlm.nih.gov/books/NBK246878
41. National Cancer Institute. Genetics of breast and ovarian cancer (PDQ)—health profession version. Updated February 12, 2021. Accessed April 12, 2021. http://www.cancer.gov/cancertopics/pdq/genetics/breast-and-ovarian/HealthProfessional
42. US Department of Health and Human Services. National Institutes of Health, National Institute of Environmental Health Sciences The sister study. Accessed April 12, 2021. https://sisterstudy.niehs.nih.gov/english/NIEHS.htm
43. Tutt A, Ashworth A. Can genetic testing guide treatment in breast cancer?. Eur J Cancer. 2008;44(18):2774-2780. doi:10.1016/j.ejca.2008.10.009
44. Katz SJ, Ward KC, Hamilton AS, et al. Gaps in receipt of clinically indicated genetic counseling after diagnosis of breast cancer. J Clin Oncol. 2018;36(12):1218-1224. doi:10.1200/JCO.2017.76.2369
45. US Department of Veterans Affairs. PTSD: National Center for PTSD. How common is PTSD in adults? Updated October 17, 2019. Accessed April 12, 2021. https://www.ptsd.va.gov/understand/common/common_adults.asp
46. US Department of Veterans Affairs. PTSD: National Center for PTSD. How common is PTSD in women? Updated October 16, 2019. Accessed April 12, 2021. https://www.ptsd.va.gov/understand/common/common_women.asp
47. US Department of Veterans Affairs. PTSD: National Center for PTSD. How common is PTSD in veterans? Updated September 24, 2018. Accessed April 12, 2021. https://www.ptsd.va.gov/understand/common/common_veterans.asp
48. Lindberg NM, Wellisch D. Anxiety and compliance among women at high risk for breast cancer. Ann Behav Med. 2001;23(4):298-303. doi:10.1207/S15324796ABM2304_9
49. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107. doi:10.1001/archinte.160.14.2101
50. Centers for Disease Control and Prevention. MMWR appendix: breast cancer rates among black women and white women. Updated October 13, 2016. Accessed April 12, 2021. https://www.cdc.gov/cancer/breast/statistics/trends_invasive.htm
51. Richardson LC, Henley SJ, Miller JW, Massetti G, Thomas CC. Patterns and trends in age-specific black-white differences in breast cancer incidence and mortality - United States, 1999-2014. MMWR Morb Mortal Wkly Rep. 2016;65(40):1093-1098. Published 2016 Oct 14. doi:10.15585/mmwr.mm6540a1
52. Brody JG, Moysich KB, Humblet O, Attfield KR, Beehler GP, Rudel RA. Environmental pollutants and breast cancer: epidemiologic studies. Cancer. 2007;109(12 Suppl):2667-2711. doi:10.1002/cncr.22655
53. Brophy JT, Keith MM, Watterson A, et al. Breast cancer risk in relation to occupations with exposure to carcinogens and endocrine disruptors: a Canadian case-control study. Environ Health. 2012;11:87. Published 2012 Nov 19. doi:10.1186/1476-069X-11-87
54. Labrèche F, Goldberg MS, Valois MF, Nadon L. Postmenopausal breast cancer and occupational exposures. Occup Environ Med. 2010;67(4):263-269. doi:10.1136/oem.2009.049817
55. National Institute of Environmental Health Sciences, Interagency Breast Cancer & Environmental Research Coordinating Committee. Breast cancer and the environment: prioritizing prevention. Updated March 8, 2013. Accessed April 12, 2021. https://www.niehs.nih.gov/about/boards/ibcercc/index.cfm
56. Gail MH, Costantino JP, Pee D, et al. Projecting individualized absolute invasive breast cancer risk in African American women [published correction appears in J Natl Cancer Inst. 2008 Aug 6;100(15):1118] [published correction appears in J Natl Cancer Inst. 2008 Mar 5;100(5):373]. J Natl Cancer Inst. 2007;99(23):1782-1792. doi:10.1093/jnci/djm223
57. Corbie-Smith G, Thomas SB, Williams MV, Moody-Ayers S. Attitudes and beliefs of African Americans toward participation in medical research. J Gen Intern Med. 1999;14(9):537-546. doi:10.1046/j.1525-1497.1999.07048.x
58. Braunstein JB, Sherber NS, Schulman SP, Ding EL, Powe NR. Race, medical researcher distrust, perceived harm, and willingness to participate in cardiovascular prevention trials. Medicine (Baltimore). 2008;87(1):1-9. doi:10.1097/MD.0b013e3181625d78
Study supports intensifying chemoradiotherapy for head and neck cancer
Of the 16 treatment options compared and ranked, HFCRT topped the list for overall survival, event-free survival, locoregional control, and cancer-specific death.
The results also suggested that taxane-based induction chemotherapy followed by locoregional therapy, especially with concomitant chemotherapy, “is another good option in selected patients with a good performance status and minor comorbidities,” according to investigator Claire Petit, MD, PhD, of Centre hospitalier de l’Université de Montréal in Canada, and colleagues.
The investigators concluded that further intensifying chemoradiotherapy with these approaches “could improve outcomes over chemoradiotherapy.”
The findings, published in The Lancet Oncology, “could help to guide clinical decision-making in locally advanced head and neck cancer with a high risk of locoregional failure, especially human papillomavirus–negative tumours,” the authors wrote.
However, Jared Weiss, MD, of the University of North Carolina, Chapel Hill, cautioned that this “study is an individual patient data network meta-analysis, not a randomized controlled trial. As the authors note, it can help frame existing data but cannot define standard of care.”
Still, “it does support the efficacy of two commonly considered intensification strategies for high-risk patients – hyperfractionation of the radiation and the addition of preceding induction chemotherapy. Both of these intensifications substantially increase the time commitment from the patient, and many patients find this unacceptable. But, for select patients, hyperfractionation and induction chemotherapy have a role and may be considered for patients at high risk of treatment failure,” Dr. Weiss said.
Study details
The goal of this study was to find the best option among many chemoradiation approaches for head and neck cancer. The investigators pulled together and reanalyzed individual patient data from recently updated meta-analyses.
The current analysis included 115 randomized trials that enrolled patients between Jan. 1, 1980, and April 30, 2012. This encompassed 28,978 patients with 20,579 progression events and 19,253 deaths over a median follow-up of 6.6 years.
Treatments were ranked by P score, with higher scores indicating more effective therapies.
For overall survival, HFCRT had a P score of 97%. The hazard ratio (HR) was 0.63 for the comparison with locoregional therapy alone (surgery, radiotherapy, or both). The absolute benefit at 5 years, compared with locoregional therapy alone, was 16.7% with HFCRT.
The P score for the second most effective treatment option – induction chemotherapy with taxane, cisplatin, and fluorouracil followed by locoregional therapy (ICTaxPF-LRT) – was 89%, with a hazard ratio of 0.69 and an absolute benefit at 5 years of 13.4%, versus locoregional therapy.
The HR of HFCRT versus the accepted standard of care worldwide – locoregional therapy with concomitant platinum-based chemotherapy and radiotherapy (CLRTP) – was 0.82 in favor of HFCRT for overall survival and 0.80 for event-free survival.
For overall survival, the P score for CLRTP was 78%. Three other treatment options had a better P score than CLRTP but not a better HR (0.77). These included ICTaxPF-LRT (P score, 89%; HR, 0.69), accelerated radiotherapy with concomitant chemotherapy (P score, 82%; HR, 0.75), and ICTaxPF-LRT followed by CLRTP (P score, 80%; HR, 0.75).
In the end, the investigators found “superiority of HFCRT over other treatments,” but noted it can be difficult to implement HFCRT in the era of intensity-modulated radiotherapy for head and neck cancer. Even so, HFCRT “could be considered as an option for tertiary centres with a high throughput of patients,” the investigators wrote.
The team noted that one of the limitations of this study is that cancer care has improved substantially since the very earliest trials that were included in the analysis. This introduces potential confounders, including that patients in older trials might have been understaged so that even an experimental local therapy would have been less effective.
Toxicity wasn’t part of this analysis but must be taken into account when making therapeutic decisions, “especially because HFCRT and induction chemotherapy based on taxane, cisplatin, and fluorouracil are known to be toxic,” the investigators wrote.
This research was funded by the French Institut National du Cancer, French Ligue Nationale Contre le Cancer, and Fondation ARC. The authors disclosed relationships with numerous companies, including AbbVie, Lilly, and Merck. Dr. Weiss did not report any relevant conflicts.
Of the 16 treatment options compared and ranked, HFCRT topped the list for overall survival, event-free survival, locoregional control, and cancer-specific death.
The results also suggested that taxane-based induction chemotherapy followed by locoregional therapy, especially with concomitant chemotherapy, “is another good option in selected patients with a good performance status and minor comorbidities,” according to investigator Claire Petit, MD, PhD, of Centre hospitalier de l’Université de Montréal in Canada, and colleagues.
The investigators concluded that further intensifying chemoradiotherapy with these approaches “could improve outcomes over chemoradiotherapy.”
The findings, published in The Lancet Oncology, “could help to guide clinical decision-making in locally advanced head and neck cancer with a high risk of locoregional failure, especially human papillomavirus–negative tumours,” the authors wrote.
However, Jared Weiss, MD, of the University of North Carolina, Chapel Hill, cautioned that this “study is an individual patient data network meta-analysis, not a randomized controlled trial. As the authors note, it can help frame existing data but cannot define standard of care.”
Still, “it does support the efficacy of two commonly considered intensification strategies for high-risk patients – hyperfractionation of the radiation and the addition of preceding induction chemotherapy. Both of these intensifications substantially increase the time commitment from the patient, and many patients find this unacceptable. But, for select patients, hyperfractionation and induction chemotherapy have a role and may be considered for patients at high risk of treatment failure,” Dr. Weiss said.
Study details
The goal of this study was to find the best option among many chemoradiation approaches for head and neck cancer. The investigators pulled together and reanalyzed individual patient data from recently updated meta-analyses.
The current analysis included 115 randomized trials that enrolled patients between Jan. 1, 1980, and April 30, 2012. This encompassed 28,978 patients with 20,579 progression events and 19,253 deaths over a median follow-up of 6.6 years.
Treatments were ranked by P score, with higher scores indicating more effective therapies.
For overall survival, HFCRT had a P score of 97%. The hazard ratio (HR) was 0.63 for the comparison with locoregional therapy alone (surgery, radiotherapy, or both). The absolute benefit at 5 years, compared with locoregional therapy alone, was 16.7% with HFCRT.
The P score for the second most effective treatment option – induction chemotherapy with taxane, cisplatin, and fluorouracil followed by locoregional therapy (ICTaxPF-LRT) – was 89%, with a hazard ratio of 0.69 and an absolute benefit at 5 years of 13.4%, versus locoregional therapy.
The HR of HFCRT versus the accepted standard of care worldwide – locoregional therapy with concomitant platinum-based chemotherapy and radiotherapy (CLRTP) – was 0.82 in favor of HFCRT for overall survival and 0.80 for event-free survival.
For overall survival, the P score for CLRTP was 78%. Three other treatment options had a better P score than CLRTP but not a better HR (0.77). These included ICTaxPF-LRT (P score, 89%; HR, 0.69), accelerated radiotherapy with concomitant chemotherapy (P score, 82%; HR, 0.75), and ICTaxPF-LRT followed by CLRTP (P score, 80%; HR, 0.75).
In the end, the investigators found “superiority of HFCRT over other treatments,” but noted it can be difficult to implement HFCRT in the era of intensity-modulated radiotherapy for head and neck cancer. Even so, HFCRT “could be considered as an option for tertiary centres with a high throughput of patients,” the investigators wrote.
The team noted that one of the limitations of this study is that cancer care has improved substantially since the very earliest trials that were included in the analysis. This introduces potential confounders, including that patients in older trials might have been understaged so that even an experimental local therapy would have been less effective.
Toxicity wasn’t part of this analysis but must be taken into account when making therapeutic decisions, “especially because HFCRT and induction chemotherapy based on taxane, cisplatin, and fluorouracil are known to be toxic,” the investigators wrote.
This research was funded by the French Institut National du Cancer, French Ligue Nationale Contre le Cancer, and Fondation ARC. The authors disclosed relationships with numerous companies, including AbbVie, Lilly, and Merck. Dr. Weiss did not report any relevant conflicts.
Of the 16 treatment options compared and ranked, HFCRT topped the list for overall survival, event-free survival, locoregional control, and cancer-specific death.
The results also suggested that taxane-based induction chemotherapy followed by locoregional therapy, especially with concomitant chemotherapy, “is another good option in selected patients with a good performance status and minor comorbidities,” according to investigator Claire Petit, MD, PhD, of Centre hospitalier de l’Université de Montréal in Canada, and colleagues.
The investigators concluded that further intensifying chemoradiotherapy with these approaches “could improve outcomes over chemoradiotherapy.”
The findings, published in The Lancet Oncology, “could help to guide clinical decision-making in locally advanced head and neck cancer with a high risk of locoregional failure, especially human papillomavirus–negative tumours,” the authors wrote.
However, Jared Weiss, MD, of the University of North Carolina, Chapel Hill, cautioned that this “study is an individual patient data network meta-analysis, not a randomized controlled trial. As the authors note, it can help frame existing data but cannot define standard of care.”
Still, “it does support the efficacy of two commonly considered intensification strategies for high-risk patients – hyperfractionation of the radiation and the addition of preceding induction chemotherapy. Both of these intensifications substantially increase the time commitment from the patient, and many patients find this unacceptable. But, for select patients, hyperfractionation and induction chemotherapy have a role and may be considered for patients at high risk of treatment failure,” Dr. Weiss said.
Study details
The goal of this study was to find the best option among many chemoradiation approaches for head and neck cancer. The investigators pulled together and reanalyzed individual patient data from recently updated meta-analyses.
The current analysis included 115 randomized trials that enrolled patients between Jan. 1, 1980, and April 30, 2012. This encompassed 28,978 patients with 20,579 progression events and 19,253 deaths over a median follow-up of 6.6 years.
Treatments were ranked by P score, with higher scores indicating more effective therapies.
For overall survival, HFCRT had a P score of 97%. The hazard ratio (HR) was 0.63 for the comparison with locoregional therapy alone (surgery, radiotherapy, or both). The absolute benefit at 5 years, compared with locoregional therapy alone, was 16.7% with HFCRT.
The P score for the second most effective treatment option – induction chemotherapy with taxane, cisplatin, and fluorouracil followed by locoregional therapy (ICTaxPF-LRT) – was 89%, with a hazard ratio of 0.69 and an absolute benefit at 5 years of 13.4%, versus locoregional therapy.
The HR of HFCRT versus the accepted standard of care worldwide – locoregional therapy with concomitant platinum-based chemotherapy and radiotherapy (CLRTP) – was 0.82 in favor of HFCRT for overall survival and 0.80 for event-free survival.
For overall survival, the P score for CLRTP was 78%. Three other treatment options had a better P score than CLRTP but not a better HR (0.77). These included ICTaxPF-LRT (P score, 89%; HR, 0.69), accelerated radiotherapy with concomitant chemotherapy (P score, 82%; HR, 0.75), and ICTaxPF-LRT followed by CLRTP (P score, 80%; HR, 0.75).
In the end, the investigators found “superiority of HFCRT over other treatments,” but noted it can be difficult to implement HFCRT in the era of intensity-modulated radiotherapy for head and neck cancer. Even so, HFCRT “could be considered as an option for tertiary centres with a high throughput of patients,” the investigators wrote.
The team noted that one of the limitations of this study is that cancer care has improved substantially since the very earliest trials that were included in the analysis. This introduces potential confounders, including that patients in older trials might have been understaged so that even an experimental local therapy would have been less effective.
Toxicity wasn’t part of this analysis but must be taken into account when making therapeutic decisions, “especially because HFCRT and induction chemotherapy based on taxane, cisplatin, and fluorouracil are known to be toxic,” the investigators wrote.
This research was funded by the French Institut National du Cancer, French Ligue Nationale Contre le Cancer, and Fondation ARC. The authors disclosed relationships with numerous companies, including AbbVie, Lilly, and Merck. Dr. Weiss did not report any relevant conflicts.
FROM THE LANCET ONCOLOGY
Possible obesity effect detected in cancer death rates
“By integrating 20 years of cancer mortality data, we demonstrated that trends in obesity-associated cancer mortality showed signs of recent deceleration, consistent with recent findings for heart disease mortality,” Christy L. Avery, PhD, and associates wrote in JAMA Network Open.
Improvements in mortality related to heart disease slowed after 2011, a phenomenon that has been associated with rising obesity rates. The age-adjusted mortality rate (AAMR) declined at an average of 3.8 deaths per 100,000 persons from 1999 to 2011 but only 0.7 deaths per 100,000 from 2011 to 2018, based on data from the Centers for Disease Control and Prevention’s Wide-Ranging Online Data for Epidemiologic Research (WONDER).
To understand trends in cancer mortality and their possible connection with obesity, data for 1999-2018 from the WONDER database were divided into obesity-associated and non–obesity-associated categories and compared with heart disease mortality, they explained. The database included more than 50 million deaths that matched inclusion criteria.
The analysis showed there was difference between obesity-associated and non–obesity-associated cancers that was obscured when all cancer deaths were considered together. The average annual change in AAMR for obesity-associated cancers slowed from –1.19 deaths per 100,000 in 1999-2011 to –0.83 in 2011-2018, Dr. Avery and associates reported.
For non–obesity-associated cancers, the annual change in AAMR increased from –1.62 per 100,000 for 1999-2011 to –2.29 for 2011-2018, following the trend for all cancers: –1.48 per 100,000 during 1999-2011 and –1.77 in 2011-2018, they said.
“The largest mortality decreases were observed for melanoma of the skin and lung cancer, two cancers not associated with obesity. For obesity-associated cancers, stable or increasing mortality rates have been observed for liver and pancreatic cancer among both men and women as well as for uterine cancer among women,” the investigators wrote.
Demographically, however, the slowing improvement in mortality for obesity-associated cancers did not follow the trend for heart disease. The deceleration for cancer was more pronounced for women and for non-Hispanic Whites and not seen at all in non-Hispanic Asian/Pacific Islander individuals. “For heart disease, evidence of a deceleration was consistent across sex, race, and ethnicity,” they said.
There are “longstanding disparities in obesity” among various populations in the United States, and the recent trend of obesity occurring earlier in life may be having an effect. “Whether the findings of decelerating mortality rates potentially signal a changing profile of cancer and heart disease mortality as the consequences of the obesity epidemic are realized remains to be seen,” they concluded.
The investigators reported receiving grants from the National Institutes of Health during the conduct of the study, but no other disclosures were reported.
“By integrating 20 years of cancer mortality data, we demonstrated that trends in obesity-associated cancer mortality showed signs of recent deceleration, consistent with recent findings for heart disease mortality,” Christy L. Avery, PhD, and associates wrote in JAMA Network Open.
Improvements in mortality related to heart disease slowed after 2011, a phenomenon that has been associated with rising obesity rates. The age-adjusted mortality rate (AAMR) declined at an average of 3.8 deaths per 100,000 persons from 1999 to 2011 but only 0.7 deaths per 100,000 from 2011 to 2018, based on data from the Centers for Disease Control and Prevention’s Wide-Ranging Online Data for Epidemiologic Research (WONDER).
To understand trends in cancer mortality and their possible connection with obesity, data for 1999-2018 from the WONDER database were divided into obesity-associated and non–obesity-associated categories and compared with heart disease mortality, they explained. The database included more than 50 million deaths that matched inclusion criteria.
The analysis showed there was difference between obesity-associated and non–obesity-associated cancers that was obscured when all cancer deaths were considered together. The average annual change in AAMR for obesity-associated cancers slowed from –1.19 deaths per 100,000 in 1999-2011 to –0.83 in 2011-2018, Dr. Avery and associates reported.
For non–obesity-associated cancers, the annual change in AAMR increased from –1.62 per 100,000 for 1999-2011 to –2.29 for 2011-2018, following the trend for all cancers: –1.48 per 100,000 during 1999-2011 and –1.77 in 2011-2018, they said.
“The largest mortality decreases were observed for melanoma of the skin and lung cancer, two cancers not associated with obesity. For obesity-associated cancers, stable or increasing mortality rates have been observed for liver and pancreatic cancer among both men and women as well as for uterine cancer among women,” the investigators wrote.
Demographically, however, the slowing improvement in mortality for obesity-associated cancers did not follow the trend for heart disease. The deceleration for cancer was more pronounced for women and for non-Hispanic Whites and not seen at all in non-Hispanic Asian/Pacific Islander individuals. “For heart disease, evidence of a deceleration was consistent across sex, race, and ethnicity,” they said.
There are “longstanding disparities in obesity” among various populations in the United States, and the recent trend of obesity occurring earlier in life may be having an effect. “Whether the findings of decelerating mortality rates potentially signal a changing profile of cancer and heart disease mortality as the consequences of the obesity epidemic are realized remains to be seen,” they concluded.
The investigators reported receiving grants from the National Institutes of Health during the conduct of the study, but no other disclosures were reported.
“By integrating 20 years of cancer mortality data, we demonstrated that trends in obesity-associated cancer mortality showed signs of recent deceleration, consistent with recent findings for heart disease mortality,” Christy L. Avery, PhD, and associates wrote in JAMA Network Open.
Improvements in mortality related to heart disease slowed after 2011, a phenomenon that has been associated with rising obesity rates. The age-adjusted mortality rate (AAMR) declined at an average of 3.8 deaths per 100,000 persons from 1999 to 2011 but only 0.7 deaths per 100,000 from 2011 to 2018, based on data from the Centers for Disease Control and Prevention’s Wide-Ranging Online Data for Epidemiologic Research (WONDER).
To understand trends in cancer mortality and their possible connection with obesity, data for 1999-2018 from the WONDER database were divided into obesity-associated and non–obesity-associated categories and compared with heart disease mortality, they explained. The database included more than 50 million deaths that matched inclusion criteria.
The analysis showed there was difference between obesity-associated and non–obesity-associated cancers that was obscured when all cancer deaths were considered together. The average annual change in AAMR for obesity-associated cancers slowed from –1.19 deaths per 100,000 in 1999-2011 to –0.83 in 2011-2018, Dr. Avery and associates reported.
For non–obesity-associated cancers, the annual change in AAMR increased from –1.62 per 100,000 for 1999-2011 to –2.29 for 2011-2018, following the trend for all cancers: –1.48 per 100,000 during 1999-2011 and –1.77 in 2011-2018, they said.
“The largest mortality decreases were observed for melanoma of the skin and lung cancer, two cancers not associated with obesity. For obesity-associated cancers, stable or increasing mortality rates have been observed for liver and pancreatic cancer among both men and women as well as for uterine cancer among women,” the investigators wrote.
Demographically, however, the slowing improvement in mortality for obesity-associated cancers did not follow the trend for heart disease. The deceleration for cancer was more pronounced for women and for non-Hispanic Whites and not seen at all in non-Hispanic Asian/Pacific Islander individuals. “For heart disease, evidence of a deceleration was consistent across sex, race, and ethnicity,” they said.
There are “longstanding disparities in obesity” among various populations in the United States, and the recent trend of obesity occurring earlier in life may be having an effect. “Whether the findings of decelerating mortality rates potentially signal a changing profile of cancer and heart disease mortality as the consequences of the obesity epidemic are realized remains to be seen,” they concluded.
The investigators reported receiving grants from the National Institutes of Health during the conduct of the study, but no other disclosures were reported.
FROM JAMA NETWORK OPEN