User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Sleep improved with urinary incontinence treatment
Treating urgency urinary incontinence in women may have the added benefit of improving their quality of sleep, according to a paper published online Jan. 9 in Obstetrics & Gynecology.
Researchers analyzed data from a multicenter, double-blind, randomized, controlled trial of daily antimuscarinic therapy (4-8 mg fesoterodine) or placebo in 645 women with urgency-predominant incontinence, which also evaluated sleep quality and daytime sleepiness.
The antimuscarinic treatment was also associated with a significant 0.48 point improvement in Pittsburgh Sleep Quality Index score (P = .02), compared with the placebo group, as well as significant improvements in sleep duration and sleep efficiency subscales. However, there were no significant differences between the two groups in Epworth Sleepiness Scale scores.
“Both fewer voids at night and decreased urge incontinence reduce the number of awakenings during the night, which may be reflected in higher sleep efficiency and longer sleep duration,” wrote Qurratul A. Warsi, MBBS, of the University of California, San Francisco, and her coauthors.
Antimuscarinic medications such as fesoterodine may have a sedating effect, the authors noted, which could also improve the quality of sleep. The study also did not control for sleep disorders such as obstructive sleep apnea and restless leg syndrome.
“This analysis provides new data that indicate initiating pharmacologic treatment for UUI in ambulatory women is associated with improvement in important domains of sleep,” they wrote. “Among community-dwelling women with UUI, flexible-dose antimuscarinic therapy not only resulted in improvement in incontinence measures, but was also associated with significant improvements in overall quality of sleep, sleep duration, and sleep efficiency.”
Pfizer funded the study and provided the study medication. Four authors declared research grants from the pharmaceutical sector, including three who had received grants or consultancies from Pfizer. One author declared royalties and stipends from the publishing industry. No other conflicts of interest were declared.
SOURCE: Warsi Q et al. Obstet Gynecol. 2018 Feb;131(2):204-11.
Treating urgency urinary incontinence in women may have the added benefit of improving their quality of sleep, according to a paper published online Jan. 9 in Obstetrics & Gynecology.
Researchers analyzed data from a multicenter, double-blind, randomized, controlled trial of daily antimuscarinic therapy (4-8 mg fesoterodine) or placebo in 645 women with urgency-predominant incontinence, which also evaluated sleep quality and daytime sleepiness.
The antimuscarinic treatment was also associated with a significant 0.48 point improvement in Pittsburgh Sleep Quality Index score (P = .02), compared with the placebo group, as well as significant improvements in sleep duration and sleep efficiency subscales. However, there were no significant differences between the two groups in Epworth Sleepiness Scale scores.
“Both fewer voids at night and decreased urge incontinence reduce the number of awakenings during the night, which may be reflected in higher sleep efficiency and longer sleep duration,” wrote Qurratul A. Warsi, MBBS, of the University of California, San Francisco, and her coauthors.
Antimuscarinic medications such as fesoterodine may have a sedating effect, the authors noted, which could also improve the quality of sleep. The study also did not control for sleep disorders such as obstructive sleep apnea and restless leg syndrome.
“This analysis provides new data that indicate initiating pharmacologic treatment for UUI in ambulatory women is associated with improvement in important domains of sleep,” they wrote. “Among community-dwelling women with UUI, flexible-dose antimuscarinic therapy not only resulted in improvement in incontinence measures, but was also associated with significant improvements in overall quality of sleep, sleep duration, and sleep efficiency.”
Pfizer funded the study and provided the study medication. Four authors declared research grants from the pharmaceutical sector, including three who had received grants or consultancies from Pfizer. One author declared royalties and stipends from the publishing industry. No other conflicts of interest were declared.
SOURCE: Warsi Q et al. Obstet Gynecol. 2018 Feb;131(2):204-11.
Treating urgency urinary incontinence in women may have the added benefit of improving their quality of sleep, according to a paper published online Jan. 9 in Obstetrics & Gynecology.
Researchers analyzed data from a multicenter, double-blind, randomized, controlled trial of daily antimuscarinic therapy (4-8 mg fesoterodine) or placebo in 645 women with urgency-predominant incontinence, which also evaluated sleep quality and daytime sleepiness.
The antimuscarinic treatment was also associated with a significant 0.48 point improvement in Pittsburgh Sleep Quality Index score (P = .02), compared with the placebo group, as well as significant improvements in sleep duration and sleep efficiency subscales. However, there were no significant differences between the two groups in Epworth Sleepiness Scale scores.
“Both fewer voids at night and decreased urge incontinence reduce the number of awakenings during the night, which may be reflected in higher sleep efficiency and longer sleep duration,” wrote Qurratul A. Warsi, MBBS, of the University of California, San Francisco, and her coauthors.
Antimuscarinic medications such as fesoterodine may have a sedating effect, the authors noted, which could also improve the quality of sleep. The study also did not control for sleep disorders such as obstructive sleep apnea and restless leg syndrome.
“This analysis provides new data that indicate initiating pharmacologic treatment for UUI in ambulatory women is associated with improvement in important domains of sleep,” they wrote. “Among community-dwelling women with UUI, flexible-dose antimuscarinic therapy not only resulted in improvement in incontinence measures, but was also associated with significant improvements in overall quality of sleep, sleep duration, and sleep efficiency.”
Pfizer funded the study and provided the study medication. Four authors declared research grants from the pharmaceutical sector, including three who had received grants or consultancies from Pfizer. One author declared royalties and stipends from the publishing industry. No other conflicts of interest were declared.
SOURCE: Warsi Q et al. Obstet Gynecol. 2018 Feb;131(2):204-11.
FROM OBSTETRICS & GYNECOLOGY
Key clinical point: Treating urinary incontinence may also result in improved sleep quality and duration.
Major finding: Women treated with antimuscarinic therapy had significantly improved Pittsburgh Sleep Quality Index scores, compared with those given placebo.
Data source: Analysis of data from a randomized, placebo-controlled trial in 645 women with urgency-predominant incontinence.
Disclosures: The study was funded by Pfizer, which also provided the study medication. Four authors declared research grants from the pharmaceutical sector, including three who had received grants or consultancies from Pfizer. One author declared royalties and stipends from the publishing industry. No other conflicts of interest were declared.
Source: Warsi Q et al. Obstet Gynecol. 2018 Feb;131(2):204-11.
Women filling more ADHD prescriptions
, according to the Centers for Disease Control and Prevention.
In 2003, 0.9% of women aged 15-44 years with private employer–sponsored insurance filled a prescription for an ADHD medication. By 2015, that figure had gone up to 4.0% for an increase of 344% that was unevenly split by medication class: prescriptions for stimulants were up by 388%, but nonstimulants had no change, wrote Kayla N. Anderson, PhD, and her associates in the Morbidity and Mortality Weekly Report.
“The substantial increase in the percentage of reproductive-aged women filling ADHD medication prescriptions from 2003 to 2015 ... is of public health concern given the high percentage of unintended pregnancies and uncertainty concerning the safety of ADHD medication exposure before and during pregnancy,” they wrote.
This analysis was restricted to women with at least 11 months of enrollment in a private health insurance plan that included prescription drug coverage during the year of interest. The sample included a median of 4.6 million women each year.
SOURCE: Anderson K et al. MMWR. 2018 Jan 19;76(2):66-70.
, according to the Centers for Disease Control and Prevention.
In 2003, 0.9% of women aged 15-44 years with private employer–sponsored insurance filled a prescription for an ADHD medication. By 2015, that figure had gone up to 4.0% for an increase of 344% that was unevenly split by medication class: prescriptions for stimulants were up by 388%, but nonstimulants had no change, wrote Kayla N. Anderson, PhD, and her associates in the Morbidity and Mortality Weekly Report.
“The substantial increase in the percentage of reproductive-aged women filling ADHD medication prescriptions from 2003 to 2015 ... is of public health concern given the high percentage of unintended pregnancies and uncertainty concerning the safety of ADHD medication exposure before and during pregnancy,” they wrote.
This analysis was restricted to women with at least 11 months of enrollment in a private health insurance plan that included prescription drug coverage during the year of interest. The sample included a median of 4.6 million women each year.
SOURCE: Anderson K et al. MMWR. 2018 Jan 19;76(2):66-70.
, according to the Centers for Disease Control and Prevention.
In 2003, 0.9% of women aged 15-44 years with private employer–sponsored insurance filled a prescription for an ADHD medication. By 2015, that figure had gone up to 4.0% for an increase of 344% that was unevenly split by medication class: prescriptions for stimulants were up by 388%, but nonstimulants had no change, wrote Kayla N. Anderson, PhD, and her associates in the Morbidity and Mortality Weekly Report.
“The substantial increase in the percentage of reproductive-aged women filling ADHD medication prescriptions from 2003 to 2015 ... is of public health concern given the high percentage of unintended pregnancies and uncertainty concerning the safety of ADHD medication exposure before and during pregnancy,” they wrote.
This analysis was restricted to women with at least 11 months of enrollment in a private health insurance plan that included prescription drug coverage during the year of interest. The sample included a median of 4.6 million women each year.
SOURCE: Anderson K et al. MMWR. 2018 Jan 19;76(2):66-70.
FROM MMWR
Who fares best after successful ECT?
PARIS – , Pascal Sienaert, MD, PhD, reported at the annual congress of the European College of Neuropsychopharmacology.
This conclusion is based on the results of two prospective studies by ResPECT – the Research in Psychiatry and ECT by the Flemish-Dutch Research Consortium – which, in turn, confirm the findings of an earlier metaanalysis of 32 studies including 702 patients conducted by investigators at Trinity College Dublin, noted Dr. Sienaert, a psychiatrist at the Catholic University of Leuven (Belgium) Academic Center for ECT and Neuromodulation.

Dr. Sienaert noted that the relapse rate in the ECT metaanalysis is nearly identical to that reported in the landmark Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial in real-world patients with major depression who achieved remission in response to second-step or later antidepressant medication.
“It’s a common misconception that relapse is higher after ECT than medication,” the psychiatrist said.
In the ECT metaanalysis, continuation ECT after induction of remission did not substantially affect the relapse risk. But that’s because the prevailing maintenance ECT strategy in the studies included in the 2013 metaanalysis relied upon a fixed-dose treatment schedule, according to Dr. Sienaert.
“In most studies, fixed-schedule maintenance ECT is used and with rather high relapse rates. Most clinicians have the experience that flexible, clinically driven on an as-needed-basis maintenance ECT has lower relapse rates,” he said. “Still, relapse remains the most pressing issue in the field, and it is very difficult for us as clinicians to predict which patients will relapse and which will not.”
That’s where the two ResPECT studies come into play.
In one of the studies, 116 patients with major depression at three tertiary psychiatric hospitals were randomized double blind to twice-weekly high-dose ultrabrief pulse (0.3-0.4 milliseconds) right unilateral or high-dose brief pulse (1.0 millisecond) right unilateral ECT. The dosing was at eight times the seizure threshold until remission as defined by a Montgomery-Åsberg Depression Rating Scale (MADRS) score below 10 or for a maximum of 6 weeks. Among the 87 completers, the remission rate was 68% in the brief pulse group, significantly higher than the 49% rate with ultrabrief ECT. Cognitive effects on semantic and lexical memory, and retrograde amnesia were the same in the two groups (J Clin Psychiatry. 2013 Nov;74[11]:e1029-36).
Dr. Sienaert and his coinvestigators then prospectively followed the 50 remitters for 6 months, during which all but one patient remained on antidepressant medication. The relapse rate, defined as rehospitalization for depression, restart of ECT, suicide, or a MADRS score above 15, was 25% at 3 months and about 40% at 6 months. The investigators found several predictors of a lower relapse rate. The strongest was early complete remission as defined by a Clinical Global Impressions Scale score of 1 out of a maximum of a possible 7 points within the first four ECT sessions: The 6-month relapse rate was 10% among those early complete remitters versus 63% in the other remitters (J Affect Disord. 2015 Sep 15;184:137-44).
“These are very small numbers in these groups, but the signal that emerges is the same as we have seen in the Irish metaanalysis: Early complete remitters were older, had shorter current episodes of depression, and showed more baseline psychotic features,” Dr. Sienaert said.
In a more recent ResPECT consortium study, the Mood Disorders in Elderly Treated With ECT (MODECT) study, 110 patients aged 55 and older with unipolar depression treated by ECT were followed with serial brain imaging studies prior to and for 6 months post treatment in an effort to gain insight into the mechanism of the particularly strong benefit of ECT in late-life depression. The response rate to ECT was significantly higher in those with onset of depression at age 55 or older than in those with disease onset before age 55, by a margin of 87% vs. 67%. The presence of baseline psychotic symptoms also was associated with a higher response rate.
In contrast, treatment response proved unrelated to changes in hippocampal volume, white matter hypersensitivities, amyloid load, or serum brain-derived neurotrophic factor, which is believed to be an important mediator of neuroplasticity. Thus, ECT’s mechanism of action in late-life depression remains elusive, the authors reported (Am J Geriatr Psychiatry. 2017 Feb;25[2]:178-89).
In a separate study, Dr. Sienaert and his colleagues found that ECT’s superior efficacy, compared with antidepressant medication in patients with late-life depression, was independent of their vascular disease burden. The study population was comprised of 81 patients in an antidepressant drug trial and 43 in an ECT trial, all of whom were inpatients with unipolar major depression. Their mean age was in the mid-70s.
The investigators gauged vascular burden by adding up each patient’s number of vascular risk factors, namely, diabetes, hypertension, smoking, hypercholesterolemia, known cardiovascular disease, and cerebrovascular disease. The depression remission rate was 80% in the ECT patients with no vascular risk factors, dropping to 58% in those with one or more. In the antidepressant drug trial participants, the remission rate was 38% in those with no vascular risk factors, compared with 32% in patients with one or more. Using different cutoffs for the number of vascular risk factors did not significantly alter the results (Int J Geriatr Psychiatry. 2017 Jun 28. doi: 10.1002/gps.4754).
At present, once a patient has achieved remission in response to ECT, most psychiatrists stop the therapy altogether. That’s often a mistake, according to session cochair Eduard Vieta, MD, PhD.
“. In many cases you need to continue ECT. Especially in patients who are refractory or treatment resistant, I don’t see a reason why maintenance ECT shouldn’t be the first choice. Yet in the guidelines, ECT is always the third- or fourth-line therapy,” said Dr. Vieta, professor of psychiatry at the University of Barcelona and scientific director of the Spanish Research Network on Mental Diseases.
Dr. Sienaert concurred, adding that he has patients who are on weekly maintenance ECT for as long as 16 years, with continued good results.
He reported having received honoraria from Mecta, a manufacturer of ECT equipment.
[email protected]
PARIS – , Pascal Sienaert, MD, PhD, reported at the annual congress of the European College of Neuropsychopharmacology.
This conclusion is based on the results of two prospective studies by ResPECT – the Research in Psychiatry and ECT by the Flemish-Dutch Research Consortium – which, in turn, confirm the findings of an earlier metaanalysis of 32 studies including 702 patients conducted by investigators at Trinity College Dublin, noted Dr. Sienaert, a psychiatrist at the Catholic University of Leuven (Belgium) Academic Center for ECT and Neuromodulation.
Dr. Sienaert noted that the relapse rate in the ECT metaanalysis is nearly identical to that reported in the landmark Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial in real-world patients with major depression who achieved remission in response to second-step or later antidepressant medication.
“It’s a common misconception that relapse is higher after ECT than medication,” the psychiatrist said.
In the ECT metaanalysis, continuation ECT after induction of remission did not substantially affect the relapse risk. But that’s because the prevailing maintenance ECT strategy in the studies included in the 2013 metaanalysis relied upon a fixed-dose treatment schedule, according to Dr. Sienaert.
“In most studies, fixed-schedule maintenance ECT is used and with rather high relapse rates. Most clinicians have the experience that flexible, clinically driven on an as-needed-basis maintenance ECT has lower relapse rates,” he said. “Still, relapse remains the most pressing issue in the field, and it is very difficult for us as clinicians to predict which patients will relapse and which will not.”
That’s where the two ResPECT studies come into play.
In one of the studies, 116 patients with major depression at three tertiary psychiatric hospitals were randomized double blind to twice-weekly high-dose ultrabrief pulse (0.3-0.4 milliseconds) right unilateral or high-dose brief pulse (1.0 millisecond) right unilateral ECT. The dosing was at eight times the seizure threshold until remission as defined by a Montgomery-Åsberg Depression Rating Scale (MADRS) score below 10 or for a maximum of 6 weeks. Among the 87 completers, the remission rate was 68% in the brief pulse group, significantly higher than the 49% rate with ultrabrief ECT. Cognitive effects on semantic and lexical memory, and retrograde amnesia were the same in the two groups (J Clin Psychiatry. 2013 Nov;74[11]:e1029-36).
Dr. Sienaert and his coinvestigators then prospectively followed the 50 remitters for 6 months, during which all but one patient remained on antidepressant medication. The relapse rate, defined as rehospitalization for depression, restart of ECT, suicide, or a MADRS score above 15, was 25% at 3 months and about 40% at 6 months. The investigators found several predictors of a lower relapse rate. The strongest was early complete remission as defined by a Clinical Global Impressions Scale score of 1 out of a maximum of a possible 7 points within the first four ECT sessions: The 6-month relapse rate was 10% among those early complete remitters versus 63% in the other remitters (J Affect Disord. 2015 Sep 15;184:137-44).
“These are very small numbers in these groups, but the signal that emerges is the same as we have seen in the Irish metaanalysis: Early complete remitters were older, had shorter current episodes of depression, and showed more baseline psychotic features,” Dr. Sienaert said.
In a more recent ResPECT consortium study, the Mood Disorders in Elderly Treated With ECT (MODECT) study, 110 patients aged 55 and older with unipolar depression treated by ECT were followed with serial brain imaging studies prior to and for 6 months post treatment in an effort to gain insight into the mechanism of the particularly strong benefit of ECT in late-life depression. The response rate to ECT was significantly higher in those with onset of depression at age 55 or older than in those with disease onset before age 55, by a margin of 87% vs. 67%. The presence of baseline psychotic symptoms also was associated with a higher response rate.
In contrast, treatment response proved unrelated to changes in hippocampal volume, white matter hypersensitivities, amyloid load, or serum brain-derived neurotrophic factor, which is believed to be an important mediator of neuroplasticity. Thus, ECT’s mechanism of action in late-life depression remains elusive, the authors reported (Am J Geriatr Psychiatry. 2017 Feb;25[2]:178-89).
In a separate study, Dr. Sienaert and his colleagues found that ECT’s superior efficacy, compared with antidepressant medication in patients with late-life depression, was independent of their vascular disease burden. The study population was comprised of 81 patients in an antidepressant drug trial and 43 in an ECT trial, all of whom were inpatients with unipolar major depression. Their mean age was in the mid-70s.
The investigators gauged vascular burden by adding up each patient’s number of vascular risk factors, namely, diabetes, hypertension, smoking, hypercholesterolemia, known cardiovascular disease, and cerebrovascular disease. The depression remission rate was 80% in the ECT patients with no vascular risk factors, dropping to 58% in those with one or more. In the antidepressant drug trial participants, the remission rate was 38% in those with no vascular risk factors, compared with 32% in patients with one or more. Using different cutoffs for the number of vascular risk factors did not significantly alter the results (Int J Geriatr Psychiatry. 2017 Jun 28. doi: 10.1002/gps.4754).
At present, once a patient has achieved remission in response to ECT, most psychiatrists stop the therapy altogether. That’s often a mistake, according to session cochair Eduard Vieta, MD, PhD.
“. In many cases you need to continue ECT. Especially in patients who are refractory or treatment resistant, I don’t see a reason why maintenance ECT shouldn’t be the first choice. Yet in the guidelines, ECT is always the third- or fourth-line therapy,” said Dr. Vieta, professor of psychiatry at the University of Barcelona and scientific director of the Spanish Research Network on Mental Diseases.
Dr. Sienaert concurred, adding that he has patients who are on weekly maintenance ECT for as long as 16 years, with continued good results.
He reported having received honoraria from Mecta, a manufacturer of ECT equipment.
[email protected]
PARIS – , Pascal Sienaert, MD, PhD, reported at the annual congress of the European College of Neuropsychopharmacology.
This conclusion is based on the results of two prospective studies by ResPECT – the Research in Psychiatry and ECT by the Flemish-Dutch Research Consortium – which, in turn, confirm the findings of an earlier metaanalysis of 32 studies including 702 patients conducted by investigators at Trinity College Dublin, noted Dr. Sienaert, a psychiatrist at the Catholic University of Leuven (Belgium) Academic Center for ECT and Neuromodulation.
Dr. Sienaert noted that the relapse rate in the ECT metaanalysis is nearly identical to that reported in the landmark Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial in real-world patients with major depression who achieved remission in response to second-step or later antidepressant medication.
“It’s a common misconception that relapse is higher after ECT than medication,” the psychiatrist said.
In the ECT metaanalysis, continuation ECT after induction of remission did not substantially affect the relapse risk. But that’s because the prevailing maintenance ECT strategy in the studies included in the 2013 metaanalysis relied upon a fixed-dose treatment schedule, according to Dr. Sienaert.
“In most studies, fixed-schedule maintenance ECT is used and with rather high relapse rates. Most clinicians have the experience that flexible, clinically driven on an as-needed-basis maintenance ECT has lower relapse rates,” he said. “Still, relapse remains the most pressing issue in the field, and it is very difficult for us as clinicians to predict which patients will relapse and which will not.”
That’s where the two ResPECT studies come into play.
In one of the studies, 116 patients with major depression at three tertiary psychiatric hospitals were randomized double blind to twice-weekly high-dose ultrabrief pulse (0.3-0.4 milliseconds) right unilateral or high-dose brief pulse (1.0 millisecond) right unilateral ECT. The dosing was at eight times the seizure threshold until remission as defined by a Montgomery-Åsberg Depression Rating Scale (MADRS) score below 10 or for a maximum of 6 weeks. Among the 87 completers, the remission rate was 68% in the brief pulse group, significantly higher than the 49% rate with ultrabrief ECT. Cognitive effects on semantic and lexical memory, and retrograde amnesia were the same in the two groups (J Clin Psychiatry. 2013 Nov;74[11]:e1029-36).
Dr. Sienaert and his coinvestigators then prospectively followed the 50 remitters for 6 months, during which all but one patient remained on antidepressant medication. The relapse rate, defined as rehospitalization for depression, restart of ECT, suicide, or a MADRS score above 15, was 25% at 3 months and about 40% at 6 months. The investigators found several predictors of a lower relapse rate. The strongest was early complete remission as defined by a Clinical Global Impressions Scale score of 1 out of a maximum of a possible 7 points within the first four ECT sessions: The 6-month relapse rate was 10% among those early complete remitters versus 63% in the other remitters (J Affect Disord. 2015 Sep 15;184:137-44).
“These are very small numbers in these groups, but the signal that emerges is the same as we have seen in the Irish metaanalysis: Early complete remitters were older, had shorter current episodes of depression, and showed more baseline psychotic features,” Dr. Sienaert said.
In a more recent ResPECT consortium study, the Mood Disorders in Elderly Treated With ECT (MODECT) study, 110 patients aged 55 and older with unipolar depression treated by ECT were followed with serial brain imaging studies prior to and for 6 months post treatment in an effort to gain insight into the mechanism of the particularly strong benefit of ECT in late-life depression. The response rate to ECT was significantly higher in those with onset of depression at age 55 or older than in those with disease onset before age 55, by a margin of 87% vs. 67%. The presence of baseline psychotic symptoms also was associated with a higher response rate.
In contrast, treatment response proved unrelated to changes in hippocampal volume, white matter hypersensitivities, amyloid load, or serum brain-derived neurotrophic factor, which is believed to be an important mediator of neuroplasticity. Thus, ECT’s mechanism of action in late-life depression remains elusive, the authors reported (Am J Geriatr Psychiatry. 2017 Feb;25[2]:178-89).
In a separate study, Dr. Sienaert and his colleagues found that ECT’s superior efficacy, compared with antidepressant medication in patients with late-life depression, was independent of their vascular disease burden. The study population was comprised of 81 patients in an antidepressant drug trial and 43 in an ECT trial, all of whom were inpatients with unipolar major depression. Their mean age was in the mid-70s.
The investigators gauged vascular burden by adding up each patient’s number of vascular risk factors, namely, diabetes, hypertension, smoking, hypercholesterolemia, known cardiovascular disease, and cerebrovascular disease. The depression remission rate was 80% in the ECT patients with no vascular risk factors, dropping to 58% in those with one or more. In the antidepressant drug trial participants, the remission rate was 38% in those with no vascular risk factors, compared with 32% in patients with one or more. Using different cutoffs for the number of vascular risk factors did not significantly alter the results (Int J Geriatr Psychiatry. 2017 Jun 28. doi: 10.1002/gps.4754).
At present, once a patient has achieved remission in response to ECT, most psychiatrists stop the therapy altogether. That’s often a mistake, according to session cochair Eduard Vieta, MD, PhD.
“. In many cases you need to continue ECT. Especially in patients who are refractory or treatment resistant, I don’t see a reason why maintenance ECT shouldn’t be the first choice. Yet in the guidelines, ECT is always the third- or fourth-line therapy,” said Dr. Vieta, professor of psychiatry at the University of Barcelona and scientific director of the Spanish Research Network on Mental Diseases.
Dr. Sienaert concurred, adding that he has patients who are on weekly maintenance ECT for as long as 16 years, with continued good results.
He reported having received honoraria from Mecta, a manufacturer of ECT equipment.
[email protected]
REPORTING FROM THE ECNP CONGRESS
Disorders of diminished motivation: What they are, and how to treat them
Disorders of diminished motivation (DDM)—including apathy, abulia, and akinetic mutism—are characterized by impairment in goal-directed behavior, thought, and emotion.1 These disorders can be observed clinically as a gross underproduction of speech, movement, and emotional response.
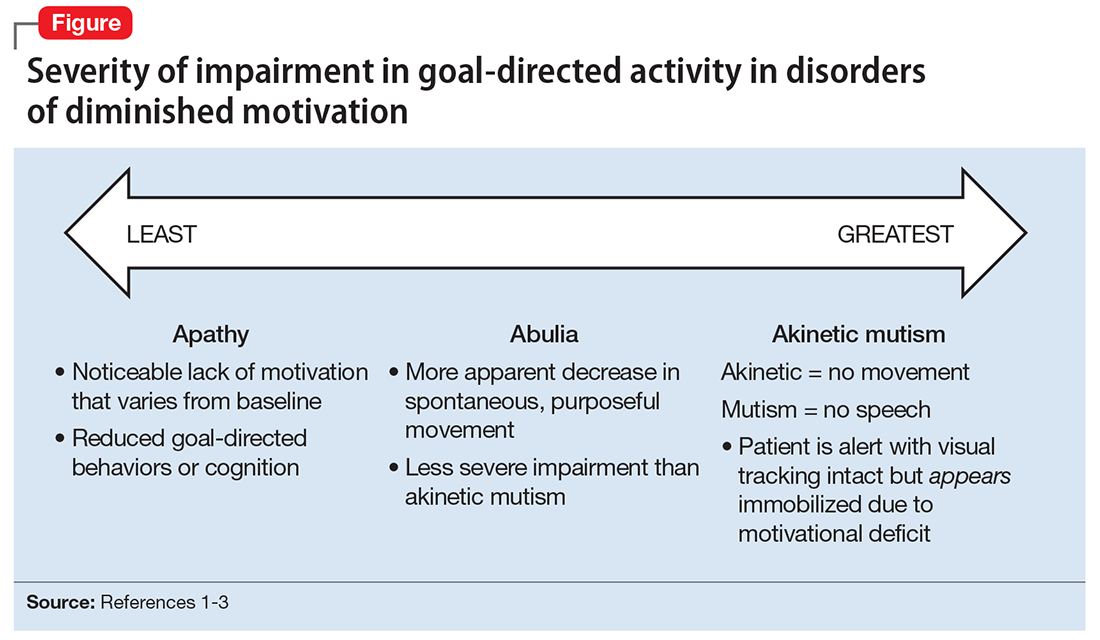
DDM are not classified as disorders within DSM-5, and it remains unclear if they are distinct disorders or symptoms that overlap in other conditions. Some sources support distinct diagnoses, while the traditional position is that DDM are variations along a spectrum, with apathy as the mildest form and akinetic mutism as the most severe form (Figure).1-3 DDM can result from various neurologic, medical, psychiatric, socioeconomic, and drug-induced pathologies, and may represent differing severity of the same underlying pathology.1,4 It is postulated that DDM arise from disruptions in the dopaminergic frontal-subcortical-mesolimbic networks.1,4
We present 2 cases of patients who developed distinct phenotypes within DDM. Despite differences in presentation and symptom severity, both patients showed clinical improvement on methylphenidate (not the only treatment option) as assessed by the Neuropsychiatric Inventory (NPI),5 a scale used to measure dementia-related behavioral symptoms that includes an Apathy/Indifference (A/I) subscale.
CASE 1
Apathy secondary to glioblastoma multiforme
Ms. E, age 59, presents with wound drainage 3 weeks after a repeat right craniotomy for recurrent glioblastoma multiforme (GBM) of the temporal lobe. Her medical history is not believed to have contributed to her current presentation.
On hospital day 2, Ms. E undergoes debridement and reclosure at the craniotomy site. Prior to the procedure, the patient was noted to have anhedonia and flat affect. Her family reports that she seems to get little enjoyment from life and “only slept and ate.” Psychiatry is consulted on hospital day 3 for evaluation and management of a perceived depressed mood.
On initial psychiatric evaluation, Ms. E continues to have a constricted affect with delayed psychomotor processing speed. However, she denies dysphoria or anhedonia. Richmond Agitation-Sedation Scale6 score is 0 (alert and calm) and test of sustained attention (‘Vigilant A’) is intact (ie, based on the Confusion Assessment Method for the Intensive Care Unit [CAM-ICU],7 Ms. E does not have delirium). The NPI A/I frequency score is 15, with a severity score of 3, for a total score of 45, indicating moderate behavioral disturbance on the NPI A/I subsection. A diagnosis of neuropsychiatric apathy due to recurrent GBM or craniotomy is made, although substance-induced mood disorder due to concurrent dexamethasone and opiate use is considered. Methylphenidate, 2.5 mg/d, is started, and Ms. E’s blood pressure remains stable with the initial dose.
Methylphenidate is titrated to 5 mg, twice daily, over a 1-week period. Ms. E’s NPI A/I subscale score improves to 3 (mild behavioral problem), with 3 points for frequency and a multiplier of 1 for mild severity, reflecting an improvement in neuropsychiatric apathy, and she is transferred to a long-term care rehabilitation center.
CASE 2
Akinetic mutism secondary to subarachnoid hemorrhage
Ms. G, age 47, is brought to an outside hospital with syncope and a severe headache radiating to her neck. Upon arrival, she is unconscious and requires intubation. A non-contrast head CT scan shows diffuse subarachnoid hemorrhage, 6 mm right midline shift, and a small left frontal subdural hematoma. A CT angiography of her head and neck reveals a 0.7 cm anterior paraclinoid left internal carotid artery aneurysm with ophthalmic involvement. Evidence of underlying left and right carotid fibromuscular dysplasia is also seen. Ms. G is transferred to our facility for neurosurgical intervention.
Neurosurgery proceeds with aneurysm coiling, followed by left craniotomy with subdural evacuation and ventriculostomy placement. Her postoperative course is complicated by prolonged nasogastric hyperalimentation, mild hypernatremia and hyperglycemia, tracheostomy, and recurrent central fever. She also develops persistent vasospasm, which requires balloon angioplasty of the left middle cerebral artery.
The psychiatry team is consulted on postoperative day 29 to assess for delirium. The CAM-ICU is positive for delirium, with nocturnal accentuation of agitation. Ms. G demonstrates paucity of speech and minimal verbal comprehension. She starts oral ziprasidone, 5 mg/d at bedtime. In addition to her original CNS insult, scopolamine patch, 1.5 mg, to decrease respiratory secretions, and IV metronidazole, 500 mg every 8 hours, for skin-site infection, may have been contributing to her delirium.
Ms. G’s delirium quickly resolves; however, on day 32 she continues to demonstrate behavioral and cognitive slowing; The NPI A/I frequency score is 28, with a severity score of 3, for a total score of 84, indicating severe behavioral disturbance on the NPI A/I subsection. Methylphenidate, 2.5 mg/d, is started and the next day is increased to 5 mg twice a day to treat severe akinetic mutism. Ms. G also is switched from ziprasidone to olanzapine, 2.5 mg/d at night.
By day 37, the tracheostomy is decannulated, and Ms. G demonstrates a full level of alertness, awareness, and attention. Her affect is full range and appropriate; however, she demonstrates residual language deficits, including dysnomia. On day 38, Ms. G is discharged with an NPI A/I subscale score of 5, indicating a mild behavioral problem.
What these cases demonstrate about DDM
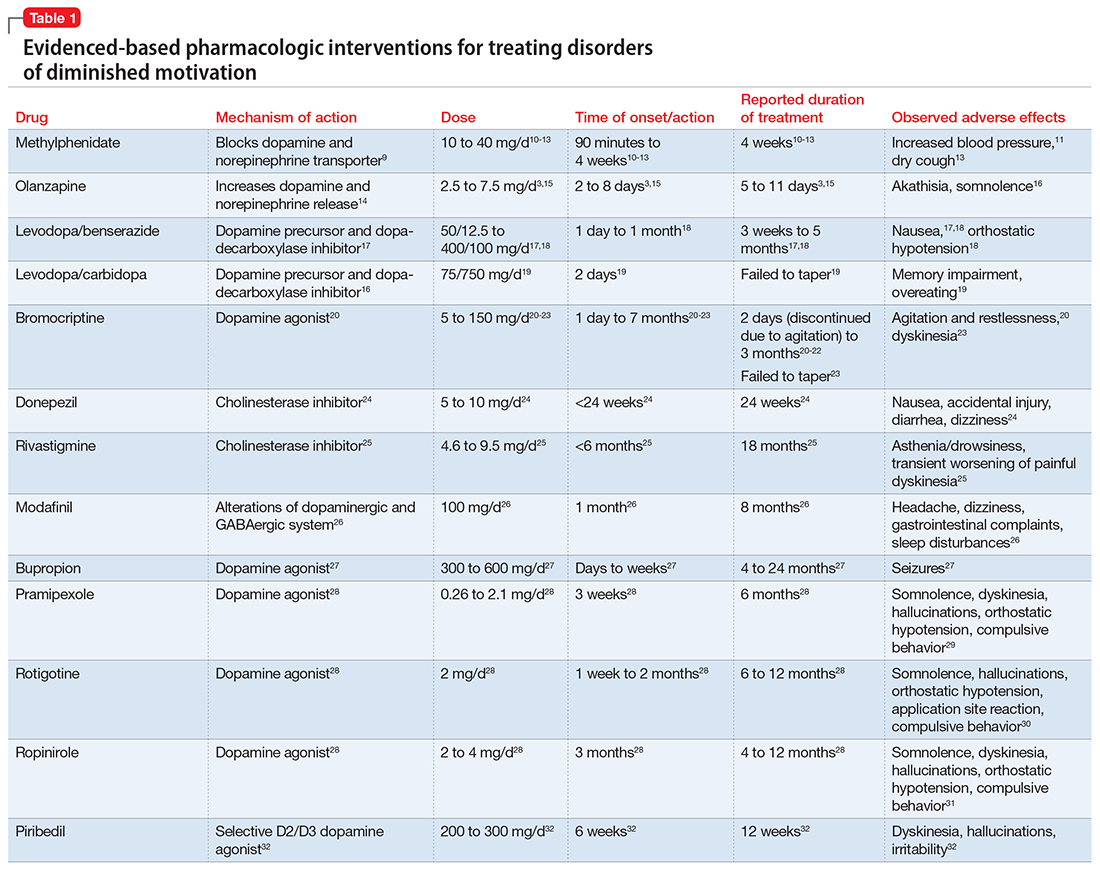
These 2 cases are part of a larger, emerging conversation about the role of dopamine in DDM. Although not fully elucidated, the pathophysiology of abulia, apathy, and akinetic mutism is thought to be related to multiple neurotransmitters—especially dopamine—involved in the cortico-striatal-pallidal-thalamic network.1,8 This position has been supported by reports of clinical improvement in patients with DDM who are given dopaminergic agonists (Table 1).3,9-32
The clinical improvement seen in both of our patients after initiating methylphenidate is consistent with previous reports.10-13 Methylphenidate was selected because of its favorable adverse effect profile and potentially rapid onset of action in DDM.10-13 In cases where oral medication cannot be administered, such as in patients with akinetic mutism, short-term adjunctive IM olanzapine may be helpful, although it is not a first-line treatment.3,15
Interestingly, both of our patients showed improvement with low doses of methylphenidate. Ms. E showed rapid improvement at 2.5 mg/d, but eventually was increased to 10 mg/d. For Ms. G, who demonstrated severe akinetic mutism, rapid improvement was noted after the initial 2.5 mg/d dose; however, because of reports of efficacy of olanzapine in treating akinetic mutism, it is possible that these medications worked synergistically. The proposed mechanism of action of olanzapine in akinetic mutism is through increased dopamine transmission in the medial prefrontal cortex.3,15 Ms. G’s methylphenidate dose was increased to 5 mg/d, which was still “subtherapeutic,” because most reports have used dosages ranging from 10 to 40 mg/d.10-13 Although there were favorable acute results in both patients, their long-term requirements are unknown because of a lack of follow-up. Our findings are also limited by the fact that both patients were recovering from neurosurgical procedures, which could lead to natural improvement in symptoms over time.
Prevalence of DDM in psychiatric disorders
The successful treatment of DDM with dopaminergic drugs is meaningful because of the coexistence of DDM in various neuropsychiatric conditions. In Alzheimer’s disease (AD), disturbances in the dopaminergic system may explain the high comorbidity of apathy, which ranges from 47% in mild AD to 80% in moderate AD.33 In the dopamine-reduced states of cocaine and amphetamine withdrawal, 67% of patients report apathy and lack of motivation.8,34 Additionally, the prevalence of apathy is reported at 27% in Parkinson’s disease, 43% in mild cognitive impairment, 70% in mixed dementia, 94% in a major depressive episode, and 53% in schizophrenia.35 In schizophrenia with predominately negative symptoms, in vivo and postmortem studies have found reduced dopamine receptors.8 Meanwhile, the high rate of akinetic mutism in Creutzfeldt-Jakob disease allows for its use as a reliable diagnostic criteria in this disorder.36
However, the prevalence of DDM is best documented as it relates to stroke and traumatic brain injury (TBI). For instance, after experiencing a stroke, 20% to 25% of patients suffer from apathy.37 Many case reports describe abulia and akinetic mutism after cerebral infarction or hemorrhage, although the incidence of these disorders is unknown.2,38-40 Apathy following TBI is common, especially in younger patients who have sustained a severe injury.41 One study evaluated the prevalence of apathy after TBI among 83 consecutive patients in a neuropsychiatric clinic. Of the 83 patients, 10.84% had apathy without depression, and an equal number were depressed without apathy; another 60% of patients exhibited both apathy and depression. Younger patients (mean age, 29 years) were more likely to be apathetic than older patients, who were more likely to be depressed or depressed and apathetic (mean age, 42 and 38 years, respectively).41 Interestingly, DDM often are associated with cerebral lesions in distinct and distant anatomical locations that are not clearly connected to the neural circuits of motivational pathways. This phenomenon may be explained by the concept of diaschisis, which states that injury to one part of an interconnected neural network can affect other, separate parts of that network.2 If this concept is accurate, it may broaden the impact of DDM, especially as it relates to stroke and TBI.
The differential diagnosis of DDM includes depression and hypokinetic delirium (Table 21,3,42-50). A potential overlapping but confounding condition is stuporous catatonia, with symptoms that include psychomotor slowing such as immobility, staring, and stupor.47 It is important to differentiate these disorders because the treatment for each differs. As previously discussed, there is a clear role for dopamine receptor agonists in the treatment of DDM.

Although major depressive disorder can be treated with medications that increase dopaminergic transmission, selective serotonin reuptake inhibitors (SSRIs) are more commonly used as first-line agents.44 However, an SSRI would theoretically be contraindicated in DDM, because increased serotonin transmission decreases dopamine release from the midbrain, and therefore an SSRI may not only result in a lack of improvement but may worsen DDM.48 Finally, although delirium is treated with atypical or conventional antipsychotics vis-a-vis dopamine type 2 receptor antagonism,45 stuporous catatonia is preferentially treated with gamma-aminobutyric acid-A receptor agonists such as lorazepam.50
What to do when your patient’s presentation suggests DDM
Assessment of DDM should be structured, with input from the patient and the caregiver, and should incorporate the physician’s perspective. A history should be obtained applying recent criteria of apathy. The 3 core domains of apathy—behavior, cognition, and emotion—need to be evaluated. The revised criteria are based on the premise that change in motivation can be measured by examining a patient’s responsiveness to internal or external stimuli. Therefore, each of the 3 domains includes 2 symptoms: (1) self-initiated or “internal” behaviors, cognitions, and emotions (initiation symptom), and (2) the patient’s responsiveness to “external” stimuli (responsiveness symptom).51
One of the main diagnostic dilemmas is how to separate DDM from depression. The differentiation is difficult because of substantial overlap in the manifestation of key symptoms, such as a lack of interest, anergia, psychomotor slowing, and fatigue. Caregivers often mistakenly describe DDM as a depressive state, even though a lack of sadness, desperation, crying, and a depressive mood distinguish DDM from depression. Usually, DDM patients lack negative thoughts, emotional distress, sadness, vegetative symptoms, and somatic concerns, which are frequently observed in mood disorders.51
Several instruments have been developed for assessing neuropsychiatric symptoms. Some were specifically designed to measure apathy, whereas others were designed to provide a broader neuropsychiatric assessment. The NPI is the most widely used multidimensional instrument for assessing neuropsychiatric functioning in patients with neurocognitive disorders (NCDs). It is a valid, reliable instrument that consists of an interview of the patient’s caregiver. It is designed to assess the presence and severity of 10 symptoms, including apathy. The NPI includes both apathy and depression items, which can help clinicians distinguish the 2 conditions. Although beyond the scope of this article, more recent standardized instruments that can assess DDM include the Apathy Inventory, the Dementia Apathy Interview and Rating, and the Structured Clinical Interview for Apathy.52
As previously mentioned, researchers have proposed that DDM are simply a continuum of severity of reduced behavior, and akinetic mutism may be the extreme form. The dilemma is how to formally diagnose states of abulia and akinetic mutism, given the lack of diagnostic criteria and paucity of standardized instruments. Thus, distinguishing between abulia and akinetic mutism (and apathy) is more of a quantitative than qualitative exercise. One could hypothesize that higher scores on a standardized scale to measure apathy (ie, NPI) could imply abulia or akinetic mutism, although to the best of our knowledge, no formal “cut-off scores” exist.53
Treatment of apathy. The duration of pharmacotherapy to treat apathy is unknown and their usage is off-label. Further studies, including double-blind, randomized controlled trials (RCTs), are needed. Nonetheless, the 2 classes of medications that have the most evidence for treating apathy/DDM are psychostimulants and acetylcholinesterase inhibitors (AChEIs).
AChEIs are primarily used for treating cognitive symptoms in NCDs, but recent findings indicate that they have beneficial effects on noncognitive symptoms such as apathy. Of all medications used to treat apathy in NCDs, AChEIs have been used to treat the largest number of patients. Of 26 studies, 24 demonstrated improvement in apathy, with 21 demonstrating statistical significance. These studies ranged in duration from 8 weeks to 1 year, and most were open-label.54
Five studies (3 RCTs and 2 open-label studies) assessed the efficacy of methylphenidate for treating apathy due to AD. All the studies demonstrated at least some benefit in apathy scores after treatment with methylphenidate. These studies ranged from 5 to 12 weeks in duration. Notably, some patients reported adverse effects, including delusions and irritability.54
Although available evidence suggests AChEIs may be the most effective medications for treating apathy in NCDs, methylphenidate has been demonstrated to work faster.55 Thus, in cases where apathy can significantly affect activities of daily living or instrumental activities of daily living, a quicker response may dictate treatment with methylphenidate. It is imperative to note that safety studies and more large-scale double-blind RCTs are needed to further demonstrate the effectiveness and safety of methylphenidate.
Published in 2007, the American Psychiatric Association (APA) guidelines56 state that psychostimulants are a possible treatment option for patients with severe apathy. At the same time, clinicians are reminded that these agents—especially at higher doses—can produce various problematic adverse effects, including tachycardia, hypertension, restlessness, dyskinesia, agitation, sleep disturbances, psychosis, confusion, and decreased appetite. The APA guidelines recommend using low initial doses, with slow and careful titration. For example, methylphenidate should be started at 2.5 to 5 mg once in the morning, with daily doses not to exceed 30 to 40 mg. In our clinical experience, doses >20 mg/d have not been necessary.57
Treatment of akinetic mutism and abulia. In patients with akinetic mutism and possible abulia, for whom oral medication administration is either impossible or contraindicated (ie, due to the potential risk of aspiration pneumonia), atypical antipsychotics, such as IM olanazapine, have produced a therapeutic response in apathetic patients with NCD. However, extensive use of antipsychotics in NCD is not recommended because this class of medications has been associated with serious adverse effects, including an increased risk of death.55
Bottom Line
Apathy, abulia, and akinetic mutism have been categorized as disorders of diminished motivation (DDM). They commonly present after a stroke or traumatic brain injury, and should be differentiated from depression, hypokinetic delirium, and stuporous catatonia. DDM can be successfully treated with dopamine agonists.
Related Resources
- Barnhart WJ, Makela EH, Latocha MJ. SSRI-induced apathy syndrome: a clinical review. J Psychiatr Pract. 2004;10(3):196-199.
- Dell’Osso B, Benatti B, Altamura AC, et al. Prevalence of selective serotonin reuptake inhibitor-related apathy in patients with obsessive compulsive disorder. J Clin Psychopharmacol. 2016;36(6):725-726.
- D’Souza G, Kakoullis A, Hegde N, et al. Recognition and management of abulia in the elderly. Prog Neurol Psychiatry. 2010;14(6):24-28.
Drug Brand Names
Bromocriptine • Parlodel
Bupropion • Wellbutrin XL, Zyban
Carbidopa • Lodosyn
Dexamethasone • DexPak, Ozurde
Donepezil • Aricept
Levodopa/benserazide • Prolopa
Levodopa/carbidopa • Pacopa Rytary Sinemet
Lorazepam • Ativan
Methylphenidate • Concerta, Methylin
Metronidazole • Flagyl, Metrogel
Modafinil • Provigil
Olanzapine • Zyprexa
Pramipexole • Mirapex
Rivastigmine • Exelon
Ropinirole • Requip
Rotigotine • Neurpro
Scopolamine • Transderm Scop
Ziprasidone • Geodon
1. Marin RS, Wilkosz PA. Disorders of diminished motivation. J Head Trauma Rehabil. 2005;20(4):377-388.
2. Ghoshal S, Gokhale S, Rebovich G, et al. The neurology of decreased activity: abulia. Rev Neurol Dis. 2011;8(3-4):e55-e67.
3. Spiegel DR, Chatterjee A. A case of abulia, status/post right middle cerebral artery territory infarct, treated successfully with olanzapine. Clin Neuropharmacol. 2014;37(6):186-189.
4. Marin RS. Differential diagnosis and classification of apathy. Am J Psychiatry. 1990;147(1):22-30.
5. Cummings JL, Mega M, Gray K, et al. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44(12):2308-2314.
6. Sessler CN, Gosnell MS, Grap MJ, et al. The Richmond Agitation-Sedation Scale: validity and reliability in adult intensive care unit patients. Am J Respir Crit Care Med. 2002;166(10):1338-1344.
7. Ely EW, Margolin R, Francis J, et al. Evaluation of delirium in critically ill patients: validation of the Confusion Assessment Method for the intensive care unit (CAM-ICU). Crit Care Med. 2001;29(7):1370-1379.
8. Al-Adawi S, Dawe GS, Al-Hussaini AA. Aboulia: neurobehavioural dysfunction of dopaminergic system? Med Hypotheses. 2000;54(4):523-530.
9. Volkow ND, Fowler JS, Wang G, et al. Mechanism of action of methylphenidate: insights from PET imaging studies. J Atten Disord. 2002;6(suppl 1):S31-S43.
10. Chatterjee A, Fahn S. Methylphenidate treats apathy in Parkinson’s disease. J Neuropsychiatry Clin Neurosci. 2002;14(4):461-462.
11. Keenan S, Mavaddat N, Iddon J, et al. Effects of methylphenidate on cognition and apathy in normal pressure hydrocephalus: a case study and review. Br J Neurosurg. 2005;19(1):46-50.
12. Padala PR, Petty F, Bhatia SC. Methylphenidate may treat apathy independent of depression. Ann Pharmacother. 2005;39(11):1947-1949.
13. Padala PR, Burke WJ, Bhatia SC, et al. Treatment of apathy with methylphenidate. J Neuropsychiatry Clin Neurosci. 2007;19(1):81-83.
14. Li XM, Perry KW, Wong DT, et al. Olanzapine increases in vivo dopamine and norepinephrine release in rat prefrontal cortex, nucleus accumbens and striatum. Psychopharmacology (Berl). 1998;136(2):153-161.
15. Spiegel DR, Casella DP, Callender DM, et al. Treatment of akinetic mutism with intramuscular olanzapine: a case series. J Neuropsychiatry Clin Neurosci. 2008;20(1):93-95.
16. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
17. Bakheit AM, Fletcher K, Brennan A. Successful treatment of severe abulia with co-beneldopa. NeuroRehabilitation. 2011;29(4):347-351.
18. Debette S, Kozlowski O, Steinling M, et al. Levodopa and bromocriptine in hypoxic brain injury. J Neurol. 2002;249(12):1678-1682.
19. Combarros O, Infante J, Berciano J. Akinetic mutism from frontal lobe damage responding to levodopa. J Neurol. 2000;247(7):568-569.
20. Echiverri HC, Tatum WO, Merens TA, et al. Akinetic mutism: pharmacologic probe of the dopaminergic mesencephalofrontal activating system. Pediatr Neurol. 1988;4(4):228-230.
21. Psarros T, Zouros A, Coimbra C. Bromocriptine-responsive akinetic mutism following endoscopy for ventricular neurocysticercosis. Case report and review of the literature. J Neurosurg. 2003;99(2):397-401.
22. Naik VD. Abulia following an episode of cardiac arrest [published online July 1, 2015]. BMJ Case Rep. doi: 10.1136/bcr-2015-209357.
23. Kim MS, Rhee JJ, Lee SJ, et al. Akinetic mutism responsive to bromocriptine following subdural hematoma evacuation in a patient with hydrocephalus. Neurol Med Chir (Tokyo). 2007;47(9):419-423.
24. Rockwood K, Black S, Bedard MA; TOPS Study Investigators. Specific symptomatic changes following donepezil treatment of Alzheimer’s disease: a multi-centre, primary care, open-label study. Int J Geriatr Psychiatry. 2007;22(4):312-319.
25. Devos D, Moreau C, Maltête D, et al. Rivastigmine in apathetic but dementia and depression-free patients with Parkinson’s disease: a double-blind, placebo-controlled, randomised clinical trial. J Neurol Neurosurg Psychiatry. 2014;85(6):668-674.
26. Camargos EF, Quintas JL. Apathy syndrome treated successfully with modafinil [published online November 15, 2011]. BMJ Case Rep. doi: 10.1136/bcr.08.2011.4652.
27. Corcoran C, Wong ML, O’Keane V. Bupropion in the management of apathy. J Psychopharmacol. 2004;18(1):133-135.
28. Blundo C, Gerace C. Dopamine agonists can improve pure apathy associated with lesions of the prefrontal-basal ganglia functional system. Neurol Sci. 2015;36(7):1197-1201.
29. Mirapex [package insert]. Ridgefield, CT: Boehringer Ingelheim International GmbH; 2016.
30. Neupro [package insert]. Smyrna, GA: UBC, Inc.; 2012.
31. Requip [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2017.
32. Thobois S, Lhommée E, Klinger H, et al. Parkinsonian apathy responds to dopaminergic stimulation of D2/D3 receptors with piribedil. Brain. 2013;136(pt 5):1568-1577.
33. Mitchell RA, Herrmann N, Lanctôt KL. The role of dopamine in symptoms and treatment of apathy in Alzheimer’s disease. CNS Neurosci Ther. 2011;17(5):411-427.
34. Brower KJ, Maddahian E, Blow FC, et al. A comparison of self-reported symptoms and DSM-III-R criteria for cocaine withdrawal. Am J Drug Alcohol Abuse. 1988;14(3):347-356.
35. Mulin E, Leone E, Dujardin K, et al. Diagnostic criteria for apathy in clinical practice. Int J Geriatr Psychiatry. 2011;26(2):158-165.
36. Otto A, Zerr I, Lantsch M, et al. Akinetic mutism as a classification criterion for the diagnosis of Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 1998;64(4):524-528.
37. Jorge RE, Starkstein SE, Robinson RG. Apathy following stroke. Can J Psychiatry. 2010;55(6):350-354.
38. Hastak SM, Gorawara PS, Mishra NK. Abulia: no will, no way. J Assoc Physicians India. 2005;53:814-818.
39. Nagaratnam N, Nagaratnam K, Ng K, et al. Akinetic mutism following stroke. J Clin Neurosci. 2004;11(1):25-30.
40. Freemon FR. Akinetic mutism and bilateral anterior cerebral artery occlusion. J Neurol Neurosurg Psychiatry. 1971;34(6):693-698.
41. Schwarzbold M, Diaz A, Martins ET, et al. Psychiatric disorders and traumatic brain injury. Neuropsychiatr Dis Treat. 2008;4(4):797-816.
42. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
43. Levy ML, Cummings JL, Fairbanks LA, et al. Apathy is not depression. J Neuropsychiatry Clin Neurosci. 1998;10(3):314-319.
44. Snow V, Lascher S, Mottur-Pilson C. Pharmacologic treatment of acute major depression and dysthymia. American College of Physicians-American Society of Internal Medicine. Ann Intern Med. 2000;132(9):738-742.
45. Schwartz AC, Fisher TJ, Greenspan HN, et al. Pharmacologic and nonpharmacologic approaches to the prevention and management of delirium. Int J Psychiatry Med. 2016;51(2):160-170.
46. Kang H, Zhao F, You L, et al. Pseudo-dementia: a neuropsychological review. Ann Indian Acad Neurol. 2014;17(2):147-154.
47. Fricchione GL, Beach SR, Huffman J, et al. Life-threatening conditions in psychiatry: catatonia, neuroleptic malignant syndrome, and serotonin syndrome. In: Stern TA, Fava M, Wilens TE, eds. Massachusetts General Hospital comprehensive clinical psychiatry. London, United Kingdom: Elsevier; 2016:608-617.
48. Rogers RD. The roles of dopamine and serotonin in decision making: evidence from pharmacological experiments in humans. Neuropsychopharmacology. 2011;36(1):114-132.
49. Stransky M, Schmidt C, Ganslmeier P, et al. Hypoactive delirium after cardiac surgery as an independent risk factor for prolonged mechanical ventilation. J Cardiothorac Vasc Anesth. 2011;25(6):968-974.
50. Wilcox JA, Reid Duffy P. The syndrome of catatonia. Behav Sci (Basel). 2015;5(4):576-588.
51. Robert PH, Mulin E, Malléa P, et al. REVIEW: apathy diagnosis, assessment, and treatment in Alzheimer’s disease. CNS Neurosci Ther. 2010;16(5):263-271.
52. Cipriani G, Lucetti C, Danti S, et al. Apathy and dementia. Nosology, assessment and management. J Nerv Ment Dis. 2014;202(10):718-724.
53. Starkstein SE, Leentjens AF. The nosological position of apathy in clinical practice. J Neurol Neurosurg Psychiatry. 2008;79(10):1088-1092.54. Berman K, Brodaty H, Withall A, et al. Pharmacologic treatment of apathy in dementia. Am J Geriatr Psychiatry. 2012;20(2):104-122.
55. Theleritis C, Siarkos K, Katirtzoglou E, et al. Pharmacological and nonpharmacological treatment for apathy in Alzheimer disease: a systematic review across modalities. J Geriatr Psychiatry Neurol. 2017;30(1):26-49.
56. APA Work Group on Alzheimer’s Disease and other Dementias; Rabins PV, Blacker D, Rovner BW, et al. American Psychiatric Association practice guideline for the treatment of patients with Alzheimer’s disease and other dementias. Second edition. Am J Psychiatry. 2007;164(suppl 12):5-56.
57. Dolder CR, Davis LN, McKinsey J. Use of psychostimulants in patients with dementia. Ann Pharmacother. 2010;44(10):1624-1632.
Disorders of diminished motivation (DDM)—including apathy, abulia, and akinetic mutism—are characterized by impairment in goal-directed behavior, thought, and emotion.1 These disorders can be observed clinically as a gross underproduction of speech, movement, and emotional response.
DDM are not classified as disorders within DSM-5, and it remains unclear if they are distinct disorders or symptoms that overlap in other conditions. Some sources support distinct diagnoses, while the traditional position is that DDM are variations along a spectrum, with apathy as the mildest form and akinetic mutism as the most severe form (Figure).1-3 DDM can result from various neurologic, medical, psychiatric, socioeconomic, and drug-induced pathologies, and may represent differing severity of the same underlying pathology.1,4 It is postulated that DDM arise from disruptions in the dopaminergic frontal-subcortical-mesolimbic networks.1,4
We present 2 cases of patients who developed distinct phenotypes within DDM. Despite differences in presentation and symptom severity, both patients showed clinical improvement on methylphenidate (not the only treatment option) as assessed by the Neuropsychiatric Inventory (NPI),5 a scale used to measure dementia-related behavioral symptoms that includes an Apathy/Indifference (A/I) subscale.
CASE 1
Apathy secondary to glioblastoma multiforme
Ms. E, age 59, presents with wound drainage 3 weeks after a repeat right craniotomy for recurrent glioblastoma multiforme (GBM) of the temporal lobe. Her medical history is not believed to have contributed to her current presentation.
On hospital day 2, Ms. E undergoes debridement and reclosure at the craniotomy site. Prior to the procedure, the patient was noted to have anhedonia and flat affect. Her family reports that she seems to get little enjoyment from life and “only slept and ate.” Psychiatry is consulted on hospital day 3 for evaluation and management of a perceived depressed mood.
On initial psychiatric evaluation, Ms. E continues to have a constricted affect with delayed psychomotor processing speed. However, she denies dysphoria or anhedonia. Richmond Agitation-Sedation Scale6 score is 0 (alert and calm) and test of sustained attention (‘Vigilant A’) is intact (ie, based on the Confusion Assessment Method for the Intensive Care Unit [CAM-ICU],7 Ms. E does not have delirium). The NPI A/I frequency score is 15, with a severity score of 3, for a total score of 45, indicating moderate behavioral disturbance on the NPI A/I subsection. A diagnosis of neuropsychiatric apathy due to recurrent GBM or craniotomy is made, although substance-induced mood disorder due to concurrent dexamethasone and opiate use is considered. Methylphenidate, 2.5 mg/d, is started, and Ms. E’s blood pressure remains stable with the initial dose.
Methylphenidate is titrated to 5 mg, twice daily, over a 1-week period. Ms. E’s NPI A/I subscale score improves to 3 (mild behavioral problem), with 3 points for frequency and a multiplier of 1 for mild severity, reflecting an improvement in neuropsychiatric apathy, and she is transferred to a long-term care rehabilitation center.
CASE 2
Akinetic mutism secondary to subarachnoid hemorrhage
Ms. G, age 47, is brought to an outside hospital with syncope and a severe headache radiating to her neck. Upon arrival, she is unconscious and requires intubation. A non-contrast head CT scan shows diffuse subarachnoid hemorrhage, 6 mm right midline shift, and a small left frontal subdural hematoma. A CT angiography of her head and neck reveals a 0.7 cm anterior paraclinoid left internal carotid artery aneurysm with ophthalmic involvement. Evidence of underlying left and right carotid fibromuscular dysplasia is also seen. Ms. G is transferred to our facility for neurosurgical intervention.
Neurosurgery proceeds with aneurysm coiling, followed by left craniotomy with subdural evacuation and ventriculostomy placement. Her postoperative course is complicated by prolonged nasogastric hyperalimentation, mild hypernatremia and hyperglycemia, tracheostomy, and recurrent central fever. She also develops persistent vasospasm, which requires balloon angioplasty of the left middle cerebral artery.
The psychiatry team is consulted on postoperative day 29 to assess for delirium. The CAM-ICU is positive for delirium, with nocturnal accentuation of agitation. Ms. G demonstrates paucity of speech and minimal verbal comprehension. She starts oral ziprasidone, 5 mg/d at bedtime. In addition to her original CNS insult, scopolamine patch, 1.5 mg, to decrease respiratory secretions, and IV metronidazole, 500 mg every 8 hours, for skin-site infection, may have been contributing to her delirium.
Ms. G’s delirium quickly resolves; however, on day 32 she continues to demonstrate behavioral and cognitive slowing; The NPI A/I frequency score is 28, with a severity score of 3, for a total score of 84, indicating severe behavioral disturbance on the NPI A/I subsection. Methylphenidate, 2.5 mg/d, is started and the next day is increased to 5 mg twice a day to treat severe akinetic mutism. Ms. G also is switched from ziprasidone to olanzapine, 2.5 mg/d at night.
By day 37, the tracheostomy is decannulated, and Ms. G demonstrates a full level of alertness, awareness, and attention. Her affect is full range and appropriate; however, she demonstrates residual language deficits, including dysnomia. On day 38, Ms. G is discharged with an NPI A/I subscale score of 5, indicating a mild behavioral problem.
What these cases demonstrate about DDM
These 2 cases are part of a larger, emerging conversation about the role of dopamine in DDM. Although not fully elucidated, the pathophysiology of abulia, apathy, and akinetic mutism is thought to be related to multiple neurotransmitters—especially dopamine—involved in the cortico-striatal-pallidal-thalamic network.1,8 This position has been supported by reports of clinical improvement in patients with DDM who are given dopaminergic agonists (Table 1).3,9-32
The clinical improvement seen in both of our patients after initiating methylphenidate is consistent with previous reports.10-13 Methylphenidate was selected because of its favorable adverse effect profile and potentially rapid onset of action in DDM.10-13 In cases where oral medication cannot be administered, such as in patients with akinetic mutism, short-term adjunctive IM olanzapine may be helpful, although it is not a first-line treatment.3,15
Interestingly, both of our patients showed improvement with low doses of methylphenidate. Ms. E showed rapid improvement at 2.5 mg/d, but eventually was increased to 10 mg/d. For Ms. G, who demonstrated severe akinetic mutism, rapid improvement was noted after the initial 2.5 mg/d dose; however, because of reports of efficacy of olanzapine in treating akinetic mutism, it is possible that these medications worked synergistically. The proposed mechanism of action of olanzapine in akinetic mutism is through increased dopamine transmission in the medial prefrontal cortex.3,15 Ms. G’s methylphenidate dose was increased to 5 mg/d, which was still “subtherapeutic,” because most reports have used dosages ranging from 10 to 40 mg/d.10-13 Although there were favorable acute results in both patients, their long-term requirements are unknown because of a lack of follow-up. Our findings are also limited by the fact that both patients were recovering from neurosurgical procedures, which could lead to natural improvement in symptoms over time.
Prevalence of DDM in psychiatric disorders
The successful treatment of DDM with dopaminergic drugs is meaningful because of the coexistence of DDM in various neuropsychiatric conditions. In Alzheimer’s disease (AD), disturbances in the dopaminergic system may explain the high comorbidity of apathy, which ranges from 47% in mild AD to 80% in moderate AD.33 In the dopamine-reduced states of cocaine and amphetamine withdrawal, 67% of patients report apathy and lack of motivation.8,34 Additionally, the prevalence of apathy is reported at 27% in Parkinson’s disease, 43% in mild cognitive impairment, 70% in mixed dementia, 94% in a major depressive episode, and 53% in schizophrenia.35 In schizophrenia with predominately negative symptoms, in vivo and postmortem studies have found reduced dopamine receptors.8 Meanwhile, the high rate of akinetic mutism in Creutzfeldt-Jakob disease allows for its use as a reliable diagnostic criteria in this disorder.36
However, the prevalence of DDM is best documented as it relates to stroke and traumatic brain injury (TBI). For instance, after experiencing a stroke, 20% to 25% of patients suffer from apathy.37 Many case reports describe abulia and akinetic mutism after cerebral infarction or hemorrhage, although the incidence of these disorders is unknown.2,38-40 Apathy following TBI is common, especially in younger patients who have sustained a severe injury.41 One study evaluated the prevalence of apathy after TBI among 83 consecutive patients in a neuropsychiatric clinic. Of the 83 patients, 10.84% had apathy without depression, and an equal number were depressed without apathy; another 60% of patients exhibited both apathy and depression. Younger patients (mean age, 29 years) were more likely to be apathetic than older patients, who were more likely to be depressed or depressed and apathetic (mean age, 42 and 38 years, respectively).41 Interestingly, DDM often are associated with cerebral lesions in distinct and distant anatomical locations that are not clearly connected to the neural circuits of motivational pathways. This phenomenon may be explained by the concept of diaschisis, which states that injury to one part of an interconnected neural network can affect other, separate parts of that network.2 If this concept is accurate, it may broaden the impact of DDM, especially as it relates to stroke and TBI.
The differential diagnosis of DDM includes depression and hypokinetic delirium (Table 21,3,42-50). A potential overlapping but confounding condition is stuporous catatonia, with symptoms that include psychomotor slowing such as immobility, staring, and stupor.47 It is important to differentiate these disorders because the treatment for each differs. As previously discussed, there is a clear role for dopamine receptor agonists in the treatment of DDM.
Although major depressive disorder can be treated with medications that increase dopaminergic transmission, selective serotonin reuptake inhibitors (SSRIs) are more commonly used as first-line agents.44 However, an SSRI would theoretically be contraindicated in DDM, because increased serotonin transmission decreases dopamine release from the midbrain, and therefore an SSRI may not only result in a lack of improvement but may worsen DDM.48 Finally, although delirium is treated with atypical or conventional antipsychotics vis-a-vis dopamine type 2 receptor antagonism,45 stuporous catatonia is preferentially treated with gamma-aminobutyric acid-A receptor agonists such as lorazepam.50
What to do when your patient’s presentation suggests DDM
Assessment of DDM should be structured, with input from the patient and the caregiver, and should incorporate the physician’s perspective. A history should be obtained applying recent criteria of apathy. The 3 core domains of apathy—behavior, cognition, and emotion—need to be evaluated. The revised criteria are based on the premise that change in motivation can be measured by examining a patient’s responsiveness to internal or external stimuli. Therefore, each of the 3 domains includes 2 symptoms: (1) self-initiated or “internal” behaviors, cognitions, and emotions (initiation symptom), and (2) the patient’s responsiveness to “external” stimuli (responsiveness symptom).51
One of the main diagnostic dilemmas is how to separate DDM from depression. The differentiation is difficult because of substantial overlap in the manifestation of key symptoms, such as a lack of interest, anergia, psychomotor slowing, and fatigue. Caregivers often mistakenly describe DDM as a depressive state, even though a lack of sadness, desperation, crying, and a depressive mood distinguish DDM from depression. Usually, DDM patients lack negative thoughts, emotional distress, sadness, vegetative symptoms, and somatic concerns, which are frequently observed in mood disorders.51
Several instruments have been developed for assessing neuropsychiatric symptoms. Some were specifically designed to measure apathy, whereas others were designed to provide a broader neuropsychiatric assessment. The NPI is the most widely used multidimensional instrument for assessing neuropsychiatric functioning in patients with neurocognitive disorders (NCDs). It is a valid, reliable instrument that consists of an interview of the patient’s caregiver. It is designed to assess the presence and severity of 10 symptoms, including apathy. The NPI includes both apathy and depression items, which can help clinicians distinguish the 2 conditions. Although beyond the scope of this article, more recent standardized instruments that can assess DDM include the Apathy Inventory, the Dementia Apathy Interview and Rating, and the Structured Clinical Interview for Apathy.52
As previously mentioned, researchers have proposed that DDM are simply a continuum of severity of reduced behavior, and akinetic mutism may be the extreme form. The dilemma is how to formally diagnose states of abulia and akinetic mutism, given the lack of diagnostic criteria and paucity of standardized instruments. Thus, distinguishing between abulia and akinetic mutism (and apathy) is more of a quantitative than qualitative exercise. One could hypothesize that higher scores on a standardized scale to measure apathy (ie, NPI) could imply abulia or akinetic mutism, although to the best of our knowledge, no formal “cut-off scores” exist.53
Treatment of apathy. The duration of pharmacotherapy to treat apathy is unknown and their usage is off-label. Further studies, including double-blind, randomized controlled trials (RCTs), are needed. Nonetheless, the 2 classes of medications that have the most evidence for treating apathy/DDM are psychostimulants and acetylcholinesterase inhibitors (AChEIs).
AChEIs are primarily used for treating cognitive symptoms in NCDs, but recent findings indicate that they have beneficial effects on noncognitive symptoms such as apathy. Of all medications used to treat apathy in NCDs, AChEIs have been used to treat the largest number of patients. Of 26 studies, 24 demonstrated improvement in apathy, with 21 demonstrating statistical significance. These studies ranged in duration from 8 weeks to 1 year, and most were open-label.54
Five studies (3 RCTs and 2 open-label studies) assessed the efficacy of methylphenidate for treating apathy due to AD. All the studies demonstrated at least some benefit in apathy scores after treatment with methylphenidate. These studies ranged from 5 to 12 weeks in duration. Notably, some patients reported adverse effects, including delusions and irritability.54
Although available evidence suggests AChEIs may be the most effective medications for treating apathy in NCDs, methylphenidate has been demonstrated to work faster.55 Thus, in cases where apathy can significantly affect activities of daily living or instrumental activities of daily living, a quicker response may dictate treatment with methylphenidate. It is imperative to note that safety studies and more large-scale double-blind RCTs are needed to further demonstrate the effectiveness and safety of methylphenidate.
Published in 2007, the American Psychiatric Association (APA) guidelines56 state that psychostimulants are a possible treatment option for patients with severe apathy. At the same time, clinicians are reminded that these agents—especially at higher doses—can produce various problematic adverse effects, including tachycardia, hypertension, restlessness, dyskinesia, agitation, sleep disturbances, psychosis, confusion, and decreased appetite. The APA guidelines recommend using low initial doses, with slow and careful titration. For example, methylphenidate should be started at 2.5 to 5 mg once in the morning, with daily doses not to exceed 30 to 40 mg. In our clinical experience, doses >20 mg/d have not been necessary.57
Treatment of akinetic mutism and abulia. In patients with akinetic mutism and possible abulia, for whom oral medication administration is either impossible or contraindicated (ie, due to the potential risk of aspiration pneumonia), atypical antipsychotics, such as IM olanazapine, have produced a therapeutic response in apathetic patients with NCD. However, extensive use of antipsychotics in NCD is not recommended because this class of medications has been associated with serious adverse effects, including an increased risk of death.55
Bottom Line
Apathy, abulia, and akinetic mutism have been categorized as disorders of diminished motivation (DDM). They commonly present after a stroke or traumatic brain injury, and should be differentiated from depression, hypokinetic delirium, and stuporous catatonia. DDM can be successfully treated with dopamine agonists.
Related Resources
- Barnhart WJ, Makela EH, Latocha MJ. SSRI-induced apathy syndrome: a clinical review. J Psychiatr Pract. 2004;10(3):196-199.
- Dell’Osso B, Benatti B, Altamura AC, et al. Prevalence of selective serotonin reuptake inhibitor-related apathy in patients with obsessive compulsive disorder. J Clin Psychopharmacol. 2016;36(6):725-726.
- D’Souza G, Kakoullis A, Hegde N, et al. Recognition and management of abulia in the elderly. Prog Neurol Psychiatry. 2010;14(6):24-28.
Drug Brand Names
Bromocriptine • Parlodel
Bupropion • Wellbutrin XL, Zyban
Carbidopa • Lodosyn
Dexamethasone • DexPak, Ozurde
Donepezil • Aricept
Levodopa/benserazide • Prolopa
Levodopa/carbidopa • Pacopa Rytary Sinemet
Lorazepam • Ativan
Methylphenidate • Concerta, Methylin
Metronidazole • Flagyl, Metrogel
Modafinil • Provigil
Olanzapine • Zyprexa
Pramipexole • Mirapex
Rivastigmine • Exelon
Ropinirole • Requip
Rotigotine • Neurpro
Scopolamine • Transderm Scop
Ziprasidone • Geodon
Disorders of diminished motivation (DDM)—including apathy, abulia, and akinetic mutism—are characterized by impairment in goal-directed behavior, thought, and emotion.1 These disorders can be observed clinically as a gross underproduction of speech, movement, and emotional response.
DDM are not classified as disorders within DSM-5, and it remains unclear if they are distinct disorders or symptoms that overlap in other conditions. Some sources support distinct diagnoses, while the traditional position is that DDM are variations along a spectrum, with apathy as the mildest form and akinetic mutism as the most severe form (Figure).1-3 DDM can result from various neurologic, medical, psychiatric, socioeconomic, and drug-induced pathologies, and may represent differing severity of the same underlying pathology.1,4 It is postulated that DDM arise from disruptions in the dopaminergic frontal-subcortical-mesolimbic networks.1,4
We present 2 cases of patients who developed distinct phenotypes within DDM. Despite differences in presentation and symptom severity, both patients showed clinical improvement on methylphenidate (not the only treatment option) as assessed by the Neuropsychiatric Inventory (NPI),5 a scale used to measure dementia-related behavioral symptoms that includes an Apathy/Indifference (A/I) subscale.
CASE 1
Apathy secondary to glioblastoma multiforme
Ms. E, age 59, presents with wound drainage 3 weeks after a repeat right craniotomy for recurrent glioblastoma multiforme (GBM) of the temporal lobe. Her medical history is not believed to have contributed to her current presentation.
On hospital day 2, Ms. E undergoes debridement and reclosure at the craniotomy site. Prior to the procedure, the patient was noted to have anhedonia and flat affect. Her family reports that she seems to get little enjoyment from life and “only slept and ate.” Psychiatry is consulted on hospital day 3 for evaluation and management of a perceived depressed mood.
On initial psychiatric evaluation, Ms. E continues to have a constricted affect with delayed psychomotor processing speed. However, she denies dysphoria or anhedonia. Richmond Agitation-Sedation Scale6 score is 0 (alert and calm) and test of sustained attention (‘Vigilant A’) is intact (ie, based on the Confusion Assessment Method for the Intensive Care Unit [CAM-ICU],7 Ms. E does not have delirium). The NPI A/I frequency score is 15, with a severity score of 3, for a total score of 45, indicating moderate behavioral disturbance on the NPI A/I subsection. A diagnosis of neuropsychiatric apathy due to recurrent GBM or craniotomy is made, although substance-induced mood disorder due to concurrent dexamethasone and opiate use is considered. Methylphenidate, 2.5 mg/d, is started, and Ms. E’s blood pressure remains stable with the initial dose.
Methylphenidate is titrated to 5 mg, twice daily, over a 1-week period. Ms. E’s NPI A/I subscale score improves to 3 (mild behavioral problem), with 3 points for frequency and a multiplier of 1 for mild severity, reflecting an improvement in neuropsychiatric apathy, and she is transferred to a long-term care rehabilitation center.
CASE 2
Akinetic mutism secondary to subarachnoid hemorrhage
Ms. G, age 47, is brought to an outside hospital with syncope and a severe headache radiating to her neck. Upon arrival, she is unconscious and requires intubation. A non-contrast head CT scan shows diffuse subarachnoid hemorrhage, 6 mm right midline shift, and a small left frontal subdural hematoma. A CT angiography of her head and neck reveals a 0.7 cm anterior paraclinoid left internal carotid artery aneurysm with ophthalmic involvement. Evidence of underlying left and right carotid fibromuscular dysplasia is also seen. Ms. G is transferred to our facility for neurosurgical intervention.
Neurosurgery proceeds with aneurysm coiling, followed by left craniotomy with subdural evacuation and ventriculostomy placement. Her postoperative course is complicated by prolonged nasogastric hyperalimentation, mild hypernatremia and hyperglycemia, tracheostomy, and recurrent central fever. She also develops persistent vasospasm, which requires balloon angioplasty of the left middle cerebral artery.
The psychiatry team is consulted on postoperative day 29 to assess for delirium. The CAM-ICU is positive for delirium, with nocturnal accentuation of agitation. Ms. G demonstrates paucity of speech and minimal verbal comprehension. She starts oral ziprasidone, 5 mg/d at bedtime. In addition to her original CNS insult, scopolamine patch, 1.5 mg, to decrease respiratory secretions, and IV metronidazole, 500 mg every 8 hours, for skin-site infection, may have been contributing to her delirium.
Ms. G’s delirium quickly resolves; however, on day 32 she continues to demonstrate behavioral and cognitive slowing; The NPI A/I frequency score is 28, with a severity score of 3, for a total score of 84, indicating severe behavioral disturbance on the NPI A/I subsection. Methylphenidate, 2.5 mg/d, is started and the next day is increased to 5 mg twice a day to treat severe akinetic mutism. Ms. G also is switched from ziprasidone to olanzapine, 2.5 mg/d at night.
By day 37, the tracheostomy is decannulated, and Ms. G demonstrates a full level of alertness, awareness, and attention. Her affect is full range and appropriate; however, she demonstrates residual language deficits, including dysnomia. On day 38, Ms. G is discharged with an NPI A/I subscale score of 5, indicating a mild behavioral problem.
What these cases demonstrate about DDM
These 2 cases are part of a larger, emerging conversation about the role of dopamine in DDM. Although not fully elucidated, the pathophysiology of abulia, apathy, and akinetic mutism is thought to be related to multiple neurotransmitters—especially dopamine—involved in the cortico-striatal-pallidal-thalamic network.1,8 This position has been supported by reports of clinical improvement in patients with DDM who are given dopaminergic agonists (Table 1).3,9-32
The clinical improvement seen in both of our patients after initiating methylphenidate is consistent with previous reports.10-13 Methylphenidate was selected because of its favorable adverse effect profile and potentially rapid onset of action in DDM.10-13 In cases where oral medication cannot be administered, such as in patients with akinetic mutism, short-term adjunctive IM olanzapine may be helpful, although it is not a first-line treatment.3,15
Interestingly, both of our patients showed improvement with low doses of methylphenidate. Ms. E showed rapid improvement at 2.5 mg/d, but eventually was increased to 10 mg/d. For Ms. G, who demonstrated severe akinetic mutism, rapid improvement was noted after the initial 2.5 mg/d dose; however, because of reports of efficacy of olanzapine in treating akinetic mutism, it is possible that these medications worked synergistically. The proposed mechanism of action of olanzapine in akinetic mutism is through increased dopamine transmission in the medial prefrontal cortex.3,15 Ms. G’s methylphenidate dose was increased to 5 mg/d, which was still “subtherapeutic,” because most reports have used dosages ranging from 10 to 40 mg/d.10-13 Although there were favorable acute results in both patients, their long-term requirements are unknown because of a lack of follow-up. Our findings are also limited by the fact that both patients were recovering from neurosurgical procedures, which could lead to natural improvement in symptoms over time.
Prevalence of DDM in psychiatric disorders
The successful treatment of DDM with dopaminergic drugs is meaningful because of the coexistence of DDM in various neuropsychiatric conditions. In Alzheimer’s disease (AD), disturbances in the dopaminergic system may explain the high comorbidity of apathy, which ranges from 47% in mild AD to 80% in moderate AD.33 In the dopamine-reduced states of cocaine and amphetamine withdrawal, 67% of patients report apathy and lack of motivation.8,34 Additionally, the prevalence of apathy is reported at 27% in Parkinson’s disease, 43% in mild cognitive impairment, 70% in mixed dementia, 94% in a major depressive episode, and 53% in schizophrenia.35 In schizophrenia with predominately negative symptoms, in vivo and postmortem studies have found reduced dopamine receptors.8 Meanwhile, the high rate of akinetic mutism in Creutzfeldt-Jakob disease allows for its use as a reliable diagnostic criteria in this disorder.36
However, the prevalence of DDM is best documented as it relates to stroke and traumatic brain injury (TBI). For instance, after experiencing a stroke, 20% to 25% of patients suffer from apathy.37 Many case reports describe abulia and akinetic mutism after cerebral infarction or hemorrhage, although the incidence of these disorders is unknown.2,38-40 Apathy following TBI is common, especially in younger patients who have sustained a severe injury.41 One study evaluated the prevalence of apathy after TBI among 83 consecutive patients in a neuropsychiatric clinic. Of the 83 patients, 10.84% had apathy without depression, and an equal number were depressed without apathy; another 60% of patients exhibited both apathy and depression. Younger patients (mean age, 29 years) were more likely to be apathetic than older patients, who were more likely to be depressed or depressed and apathetic (mean age, 42 and 38 years, respectively).41 Interestingly, DDM often are associated with cerebral lesions in distinct and distant anatomical locations that are not clearly connected to the neural circuits of motivational pathways. This phenomenon may be explained by the concept of diaschisis, which states that injury to one part of an interconnected neural network can affect other, separate parts of that network.2 If this concept is accurate, it may broaden the impact of DDM, especially as it relates to stroke and TBI.
The differential diagnosis of DDM includes depression and hypokinetic delirium (Table 21,3,42-50). A potential overlapping but confounding condition is stuporous catatonia, with symptoms that include psychomotor slowing such as immobility, staring, and stupor.47 It is important to differentiate these disorders because the treatment for each differs. As previously discussed, there is a clear role for dopamine receptor agonists in the treatment of DDM.
Although major depressive disorder can be treated with medications that increase dopaminergic transmission, selective serotonin reuptake inhibitors (SSRIs) are more commonly used as first-line agents.44 However, an SSRI would theoretically be contraindicated in DDM, because increased serotonin transmission decreases dopamine release from the midbrain, and therefore an SSRI may not only result in a lack of improvement but may worsen DDM.48 Finally, although delirium is treated with atypical or conventional antipsychotics vis-a-vis dopamine type 2 receptor antagonism,45 stuporous catatonia is preferentially treated with gamma-aminobutyric acid-A receptor agonists such as lorazepam.50
What to do when your patient’s presentation suggests DDM
Assessment of DDM should be structured, with input from the patient and the caregiver, and should incorporate the physician’s perspective. A history should be obtained applying recent criteria of apathy. The 3 core domains of apathy—behavior, cognition, and emotion—need to be evaluated. The revised criteria are based on the premise that change in motivation can be measured by examining a patient’s responsiveness to internal or external stimuli. Therefore, each of the 3 domains includes 2 symptoms: (1) self-initiated or “internal” behaviors, cognitions, and emotions (initiation symptom), and (2) the patient’s responsiveness to “external” stimuli (responsiveness symptom).51
One of the main diagnostic dilemmas is how to separate DDM from depression. The differentiation is difficult because of substantial overlap in the manifestation of key symptoms, such as a lack of interest, anergia, psychomotor slowing, and fatigue. Caregivers often mistakenly describe DDM as a depressive state, even though a lack of sadness, desperation, crying, and a depressive mood distinguish DDM from depression. Usually, DDM patients lack negative thoughts, emotional distress, sadness, vegetative symptoms, and somatic concerns, which are frequently observed in mood disorders.51
Several instruments have been developed for assessing neuropsychiatric symptoms. Some were specifically designed to measure apathy, whereas others were designed to provide a broader neuropsychiatric assessment. The NPI is the most widely used multidimensional instrument for assessing neuropsychiatric functioning in patients with neurocognitive disorders (NCDs). It is a valid, reliable instrument that consists of an interview of the patient’s caregiver. It is designed to assess the presence and severity of 10 symptoms, including apathy. The NPI includes both apathy and depression items, which can help clinicians distinguish the 2 conditions. Although beyond the scope of this article, more recent standardized instruments that can assess DDM include the Apathy Inventory, the Dementia Apathy Interview and Rating, and the Structured Clinical Interview for Apathy.52
As previously mentioned, researchers have proposed that DDM are simply a continuum of severity of reduced behavior, and akinetic mutism may be the extreme form. The dilemma is how to formally diagnose states of abulia and akinetic mutism, given the lack of diagnostic criteria and paucity of standardized instruments. Thus, distinguishing between abulia and akinetic mutism (and apathy) is more of a quantitative than qualitative exercise. One could hypothesize that higher scores on a standardized scale to measure apathy (ie, NPI) could imply abulia or akinetic mutism, although to the best of our knowledge, no formal “cut-off scores” exist.53
Treatment of apathy. The duration of pharmacotherapy to treat apathy is unknown and their usage is off-label. Further studies, including double-blind, randomized controlled trials (RCTs), are needed. Nonetheless, the 2 classes of medications that have the most evidence for treating apathy/DDM are psychostimulants and acetylcholinesterase inhibitors (AChEIs).
AChEIs are primarily used for treating cognitive symptoms in NCDs, but recent findings indicate that they have beneficial effects on noncognitive symptoms such as apathy. Of all medications used to treat apathy in NCDs, AChEIs have been used to treat the largest number of patients. Of 26 studies, 24 demonstrated improvement in apathy, with 21 demonstrating statistical significance. These studies ranged in duration from 8 weeks to 1 year, and most were open-label.54
Five studies (3 RCTs and 2 open-label studies) assessed the efficacy of methylphenidate for treating apathy due to AD. All the studies demonstrated at least some benefit in apathy scores after treatment with methylphenidate. These studies ranged from 5 to 12 weeks in duration. Notably, some patients reported adverse effects, including delusions and irritability.54
Although available evidence suggests AChEIs may be the most effective medications for treating apathy in NCDs, methylphenidate has been demonstrated to work faster.55 Thus, in cases where apathy can significantly affect activities of daily living or instrumental activities of daily living, a quicker response may dictate treatment with methylphenidate. It is imperative to note that safety studies and more large-scale double-blind RCTs are needed to further demonstrate the effectiveness and safety of methylphenidate.
Published in 2007, the American Psychiatric Association (APA) guidelines56 state that psychostimulants are a possible treatment option for patients with severe apathy. At the same time, clinicians are reminded that these agents—especially at higher doses—can produce various problematic adverse effects, including tachycardia, hypertension, restlessness, dyskinesia, agitation, sleep disturbances, psychosis, confusion, and decreased appetite. The APA guidelines recommend using low initial doses, with slow and careful titration. For example, methylphenidate should be started at 2.5 to 5 mg once in the morning, with daily doses not to exceed 30 to 40 mg. In our clinical experience, doses >20 mg/d have not been necessary.57
Treatment of akinetic mutism and abulia. In patients with akinetic mutism and possible abulia, for whom oral medication administration is either impossible or contraindicated (ie, due to the potential risk of aspiration pneumonia), atypical antipsychotics, such as IM olanazapine, have produced a therapeutic response in apathetic patients with NCD. However, extensive use of antipsychotics in NCD is not recommended because this class of medications has been associated with serious adverse effects, including an increased risk of death.55
Bottom Line
Apathy, abulia, and akinetic mutism have been categorized as disorders of diminished motivation (DDM). They commonly present after a stroke or traumatic brain injury, and should be differentiated from depression, hypokinetic delirium, and stuporous catatonia. DDM can be successfully treated with dopamine agonists.
Related Resources
- Barnhart WJ, Makela EH, Latocha MJ. SSRI-induced apathy syndrome: a clinical review. J Psychiatr Pract. 2004;10(3):196-199.
- Dell’Osso B, Benatti B, Altamura AC, et al. Prevalence of selective serotonin reuptake inhibitor-related apathy in patients with obsessive compulsive disorder. J Clin Psychopharmacol. 2016;36(6):725-726.
- D’Souza G, Kakoullis A, Hegde N, et al. Recognition and management of abulia in the elderly. Prog Neurol Psychiatry. 2010;14(6):24-28.
Drug Brand Names
Bromocriptine • Parlodel
Bupropion • Wellbutrin XL, Zyban
Carbidopa • Lodosyn
Dexamethasone • DexPak, Ozurde
Donepezil • Aricept
Levodopa/benserazide • Prolopa
Levodopa/carbidopa • Pacopa Rytary Sinemet
Lorazepam • Ativan
Methylphenidate • Concerta, Methylin
Metronidazole • Flagyl, Metrogel
Modafinil • Provigil
Olanzapine • Zyprexa
Pramipexole • Mirapex
Rivastigmine • Exelon
Ropinirole • Requip
Rotigotine • Neurpro
Scopolamine • Transderm Scop
Ziprasidone • Geodon
1. Marin RS, Wilkosz PA. Disorders of diminished motivation. J Head Trauma Rehabil. 2005;20(4):377-388.
2. Ghoshal S, Gokhale S, Rebovich G, et al. The neurology of decreased activity: abulia. Rev Neurol Dis. 2011;8(3-4):e55-e67.
3. Spiegel DR, Chatterjee A. A case of abulia, status/post right middle cerebral artery territory infarct, treated successfully with olanzapine. Clin Neuropharmacol. 2014;37(6):186-189.
4. Marin RS. Differential diagnosis and classification of apathy. Am J Psychiatry. 1990;147(1):22-30.
5. Cummings JL, Mega M, Gray K, et al. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44(12):2308-2314.
6. Sessler CN, Gosnell MS, Grap MJ, et al. The Richmond Agitation-Sedation Scale: validity and reliability in adult intensive care unit patients. Am J Respir Crit Care Med. 2002;166(10):1338-1344.
7. Ely EW, Margolin R, Francis J, et al. Evaluation of delirium in critically ill patients: validation of the Confusion Assessment Method for the intensive care unit (CAM-ICU). Crit Care Med. 2001;29(7):1370-1379.
8. Al-Adawi S, Dawe GS, Al-Hussaini AA. Aboulia: neurobehavioural dysfunction of dopaminergic system? Med Hypotheses. 2000;54(4):523-530.
9. Volkow ND, Fowler JS, Wang G, et al. Mechanism of action of methylphenidate: insights from PET imaging studies. J Atten Disord. 2002;6(suppl 1):S31-S43.
10. Chatterjee A, Fahn S. Methylphenidate treats apathy in Parkinson’s disease. J Neuropsychiatry Clin Neurosci. 2002;14(4):461-462.
11. Keenan S, Mavaddat N, Iddon J, et al. Effects of methylphenidate on cognition and apathy in normal pressure hydrocephalus: a case study and review. Br J Neurosurg. 2005;19(1):46-50.
12. Padala PR, Petty F, Bhatia SC. Methylphenidate may treat apathy independent of depression. Ann Pharmacother. 2005;39(11):1947-1949.
13. Padala PR, Burke WJ, Bhatia SC, et al. Treatment of apathy with methylphenidate. J Neuropsychiatry Clin Neurosci. 2007;19(1):81-83.
14. Li XM, Perry KW, Wong DT, et al. Olanzapine increases in vivo dopamine and norepinephrine release in rat prefrontal cortex, nucleus accumbens and striatum. Psychopharmacology (Berl). 1998;136(2):153-161.
15. Spiegel DR, Casella DP, Callender DM, et al. Treatment of akinetic mutism with intramuscular olanzapine: a case series. J Neuropsychiatry Clin Neurosci. 2008;20(1):93-95.
16. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
17. Bakheit AM, Fletcher K, Brennan A. Successful treatment of severe abulia with co-beneldopa. NeuroRehabilitation. 2011;29(4):347-351.
18. Debette S, Kozlowski O, Steinling M, et al. Levodopa and bromocriptine in hypoxic brain injury. J Neurol. 2002;249(12):1678-1682.
19. Combarros O, Infante J, Berciano J. Akinetic mutism from frontal lobe damage responding to levodopa. J Neurol. 2000;247(7):568-569.
20. Echiverri HC, Tatum WO, Merens TA, et al. Akinetic mutism: pharmacologic probe of the dopaminergic mesencephalofrontal activating system. Pediatr Neurol. 1988;4(4):228-230.
21. Psarros T, Zouros A, Coimbra C. Bromocriptine-responsive akinetic mutism following endoscopy for ventricular neurocysticercosis. Case report and review of the literature. J Neurosurg. 2003;99(2):397-401.
22. Naik VD. Abulia following an episode of cardiac arrest [published online July 1, 2015]. BMJ Case Rep. doi: 10.1136/bcr-2015-209357.
23. Kim MS, Rhee JJ, Lee SJ, et al. Akinetic mutism responsive to bromocriptine following subdural hematoma evacuation in a patient with hydrocephalus. Neurol Med Chir (Tokyo). 2007;47(9):419-423.
24. Rockwood K, Black S, Bedard MA; TOPS Study Investigators. Specific symptomatic changes following donepezil treatment of Alzheimer’s disease: a multi-centre, primary care, open-label study. Int J Geriatr Psychiatry. 2007;22(4):312-319.
25. Devos D, Moreau C, Maltête D, et al. Rivastigmine in apathetic but dementia and depression-free patients with Parkinson’s disease: a double-blind, placebo-controlled, randomised clinical trial. J Neurol Neurosurg Psychiatry. 2014;85(6):668-674.
26. Camargos EF, Quintas JL. Apathy syndrome treated successfully with modafinil [published online November 15, 2011]. BMJ Case Rep. doi: 10.1136/bcr.08.2011.4652.
27. Corcoran C, Wong ML, O’Keane V. Bupropion in the management of apathy. J Psychopharmacol. 2004;18(1):133-135.
28. Blundo C, Gerace C. Dopamine agonists can improve pure apathy associated with lesions of the prefrontal-basal ganglia functional system. Neurol Sci. 2015;36(7):1197-1201.
29. Mirapex [package insert]. Ridgefield, CT: Boehringer Ingelheim International GmbH; 2016.
30. Neupro [package insert]. Smyrna, GA: UBC, Inc.; 2012.
31. Requip [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2017.
32. Thobois S, Lhommée E, Klinger H, et al. Parkinsonian apathy responds to dopaminergic stimulation of D2/D3 receptors with piribedil. Brain. 2013;136(pt 5):1568-1577.
33. Mitchell RA, Herrmann N, Lanctôt KL. The role of dopamine in symptoms and treatment of apathy in Alzheimer’s disease. CNS Neurosci Ther. 2011;17(5):411-427.
34. Brower KJ, Maddahian E, Blow FC, et al. A comparison of self-reported symptoms and DSM-III-R criteria for cocaine withdrawal. Am J Drug Alcohol Abuse. 1988;14(3):347-356.
35. Mulin E, Leone E, Dujardin K, et al. Diagnostic criteria for apathy in clinical practice. Int J Geriatr Psychiatry. 2011;26(2):158-165.
36. Otto A, Zerr I, Lantsch M, et al. Akinetic mutism as a classification criterion for the diagnosis of Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 1998;64(4):524-528.
37. Jorge RE, Starkstein SE, Robinson RG. Apathy following stroke. Can J Psychiatry. 2010;55(6):350-354.
38. Hastak SM, Gorawara PS, Mishra NK. Abulia: no will, no way. J Assoc Physicians India. 2005;53:814-818.
39. Nagaratnam N, Nagaratnam K, Ng K, et al. Akinetic mutism following stroke. J Clin Neurosci. 2004;11(1):25-30.
40. Freemon FR. Akinetic mutism and bilateral anterior cerebral artery occlusion. J Neurol Neurosurg Psychiatry. 1971;34(6):693-698.
41. Schwarzbold M, Diaz A, Martins ET, et al. Psychiatric disorders and traumatic brain injury. Neuropsychiatr Dis Treat. 2008;4(4):797-816.
42. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
43. Levy ML, Cummings JL, Fairbanks LA, et al. Apathy is not depression. J Neuropsychiatry Clin Neurosci. 1998;10(3):314-319.
44. Snow V, Lascher S, Mottur-Pilson C. Pharmacologic treatment of acute major depression and dysthymia. American College of Physicians-American Society of Internal Medicine. Ann Intern Med. 2000;132(9):738-742.
45. Schwartz AC, Fisher TJ, Greenspan HN, et al. Pharmacologic and nonpharmacologic approaches to the prevention and management of delirium. Int J Psychiatry Med. 2016;51(2):160-170.
46. Kang H, Zhao F, You L, et al. Pseudo-dementia: a neuropsychological review. Ann Indian Acad Neurol. 2014;17(2):147-154.
47. Fricchione GL, Beach SR, Huffman J, et al. Life-threatening conditions in psychiatry: catatonia, neuroleptic malignant syndrome, and serotonin syndrome. In: Stern TA, Fava M, Wilens TE, eds. Massachusetts General Hospital comprehensive clinical psychiatry. London, United Kingdom: Elsevier; 2016:608-617.
48. Rogers RD. The roles of dopamine and serotonin in decision making: evidence from pharmacological experiments in humans. Neuropsychopharmacology. 2011;36(1):114-132.
49. Stransky M, Schmidt C, Ganslmeier P, et al. Hypoactive delirium after cardiac surgery as an independent risk factor for prolonged mechanical ventilation. J Cardiothorac Vasc Anesth. 2011;25(6):968-974.
50. Wilcox JA, Reid Duffy P. The syndrome of catatonia. Behav Sci (Basel). 2015;5(4):576-588.
51. Robert PH, Mulin E, Malléa P, et al. REVIEW: apathy diagnosis, assessment, and treatment in Alzheimer’s disease. CNS Neurosci Ther. 2010;16(5):263-271.
52. Cipriani G, Lucetti C, Danti S, et al. Apathy and dementia. Nosology, assessment and management. J Nerv Ment Dis. 2014;202(10):718-724.
53. Starkstein SE, Leentjens AF. The nosological position of apathy in clinical practice. J Neurol Neurosurg Psychiatry. 2008;79(10):1088-1092.54. Berman K, Brodaty H, Withall A, et al. Pharmacologic treatment of apathy in dementia. Am J Geriatr Psychiatry. 2012;20(2):104-122.
55. Theleritis C, Siarkos K, Katirtzoglou E, et al. Pharmacological and nonpharmacological treatment for apathy in Alzheimer disease: a systematic review across modalities. J Geriatr Psychiatry Neurol. 2017;30(1):26-49.
56. APA Work Group on Alzheimer’s Disease and other Dementias; Rabins PV, Blacker D, Rovner BW, et al. American Psychiatric Association practice guideline for the treatment of patients with Alzheimer’s disease and other dementias. Second edition. Am J Psychiatry. 2007;164(suppl 12):5-56.
57. Dolder CR, Davis LN, McKinsey J. Use of psychostimulants in patients with dementia. Ann Pharmacother. 2010;44(10):1624-1632.
1. Marin RS, Wilkosz PA. Disorders of diminished motivation. J Head Trauma Rehabil. 2005;20(4):377-388.
2. Ghoshal S, Gokhale S, Rebovich G, et al. The neurology of decreased activity: abulia. Rev Neurol Dis. 2011;8(3-4):e55-e67.
3. Spiegel DR, Chatterjee A. A case of abulia, status/post right middle cerebral artery territory infarct, treated successfully with olanzapine. Clin Neuropharmacol. 2014;37(6):186-189.
4. Marin RS. Differential diagnosis and classification of apathy. Am J Psychiatry. 1990;147(1):22-30.
5. Cummings JL, Mega M, Gray K, et al. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44(12):2308-2314.
6. Sessler CN, Gosnell MS, Grap MJ, et al. The Richmond Agitation-Sedation Scale: validity and reliability in adult intensive care unit patients. Am J Respir Crit Care Med. 2002;166(10):1338-1344.
7. Ely EW, Margolin R, Francis J, et al. Evaluation of delirium in critically ill patients: validation of the Confusion Assessment Method for the intensive care unit (CAM-ICU). Crit Care Med. 2001;29(7):1370-1379.
8. Al-Adawi S, Dawe GS, Al-Hussaini AA. Aboulia: neurobehavioural dysfunction of dopaminergic system? Med Hypotheses. 2000;54(4):523-530.
9. Volkow ND, Fowler JS, Wang G, et al. Mechanism of action of methylphenidate: insights from PET imaging studies. J Atten Disord. 2002;6(suppl 1):S31-S43.
10. Chatterjee A, Fahn S. Methylphenidate treats apathy in Parkinson’s disease. J Neuropsychiatry Clin Neurosci. 2002;14(4):461-462.
11. Keenan S, Mavaddat N, Iddon J, et al. Effects of methylphenidate on cognition and apathy in normal pressure hydrocephalus: a case study and review. Br J Neurosurg. 2005;19(1):46-50.
12. Padala PR, Petty F, Bhatia SC. Methylphenidate may treat apathy independent of depression. Ann Pharmacother. 2005;39(11):1947-1949.
13. Padala PR, Burke WJ, Bhatia SC, et al. Treatment of apathy with methylphenidate. J Neuropsychiatry Clin Neurosci. 2007;19(1):81-83.
14. Li XM, Perry KW, Wong DT, et al. Olanzapine increases in vivo dopamine and norepinephrine release in rat prefrontal cortex, nucleus accumbens and striatum. Psychopharmacology (Berl). 1998;136(2):153-161.
15. Spiegel DR, Casella DP, Callender DM, et al. Treatment of akinetic mutism with intramuscular olanzapine: a case series. J Neuropsychiatry Clin Neurosci. 2008;20(1):93-95.
16. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
17. Bakheit AM, Fletcher K, Brennan A. Successful treatment of severe abulia with co-beneldopa. NeuroRehabilitation. 2011;29(4):347-351.
18. Debette S, Kozlowski O, Steinling M, et al. Levodopa and bromocriptine in hypoxic brain injury. J Neurol. 2002;249(12):1678-1682.
19. Combarros O, Infante J, Berciano J. Akinetic mutism from frontal lobe damage responding to levodopa. J Neurol. 2000;247(7):568-569.
20. Echiverri HC, Tatum WO, Merens TA, et al. Akinetic mutism: pharmacologic probe of the dopaminergic mesencephalofrontal activating system. Pediatr Neurol. 1988;4(4):228-230.
21. Psarros T, Zouros A, Coimbra C. Bromocriptine-responsive akinetic mutism following endoscopy for ventricular neurocysticercosis. Case report and review of the literature. J Neurosurg. 2003;99(2):397-401.
22. Naik VD. Abulia following an episode of cardiac arrest [published online July 1, 2015]. BMJ Case Rep. doi: 10.1136/bcr-2015-209357.
23. Kim MS, Rhee JJ, Lee SJ, et al. Akinetic mutism responsive to bromocriptine following subdural hematoma evacuation in a patient with hydrocephalus. Neurol Med Chir (Tokyo). 2007;47(9):419-423.
24. Rockwood K, Black S, Bedard MA; TOPS Study Investigators. Specific symptomatic changes following donepezil treatment of Alzheimer’s disease: a multi-centre, primary care, open-label study. Int J Geriatr Psychiatry. 2007;22(4):312-319.
25. Devos D, Moreau C, Maltête D, et al. Rivastigmine in apathetic but dementia and depression-free patients with Parkinson’s disease: a double-blind, placebo-controlled, randomised clinical trial. J Neurol Neurosurg Psychiatry. 2014;85(6):668-674.
26. Camargos EF, Quintas JL. Apathy syndrome treated successfully with modafinil [published online November 15, 2011]. BMJ Case Rep. doi: 10.1136/bcr.08.2011.4652.
27. Corcoran C, Wong ML, O’Keane V. Bupropion in the management of apathy. J Psychopharmacol. 2004;18(1):133-135.
28. Blundo C, Gerace C. Dopamine agonists can improve pure apathy associated with lesions of the prefrontal-basal ganglia functional system. Neurol Sci. 2015;36(7):1197-1201.
29. Mirapex [package insert]. Ridgefield, CT: Boehringer Ingelheim International GmbH; 2016.
30. Neupro [package insert]. Smyrna, GA: UBC, Inc.; 2012.
31. Requip [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2017.
32. Thobois S, Lhommée E, Klinger H, et al. Parkinsonian apathy responds to dopaminergic stimulation of D2/D3 receptors with piribedil. Brain. 2013;136(pt 5):1568-1577.
33. Mitchell RA, Herrmann N, Lanctôt KL. The role of dopamine in symptoms and treatment of apathy in Alzheimer’s disease. CNS Neurosci Ther. 2011;17(5):411-427.
34. Brower KJ, Maddahian E, Blow FC, et al. A comparison of self-reported symptoms and DSM-III-R criteria for cocaine withdrawal. Am J Drug Alcohol Abuse. 1988;14(3):347-356.
35. Mulin E, Leone E, Dujardin K, et al. Diagnostic criteria for apathy in clinical practice. Int J Geriatr Psychiatry. 2011;26(2):158-165.
36. Otto A, Zerr I, Lantsch M, et al. Akinetic mutism as a classification criterion for the diagnosis of Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 1998;64(4):524-528.
37. Jorge RE, Starkstein SE, Robinson RG. Apathy following stroke. Can J Psychiatry. 2010;55(6):350-354.
38. Hastak SM, Gorawara PS, Mishra NK. Abulia: no will, no way. J Assoc Physicians India. 2005;53:814-818.
39. Nagaratnam N, Nagaratnam K, Ng K, et al. Akinetic mutism following stroke. J Clin Neurosci. 2004;11(1):25-30.
40. Freemon FR. Akinetic mutism and bilateral anterior cerebral artery occlusion. J Neurol Neurosurg Psychiatry. 1971;34(6):693-698.
41. Schwarzbold M, Diaz A, Martins ET, et al. Psychiatric disorders and traumatic brain injury. Neuropsychiatr Dis Treat. 2008;4(4):797-816.
42. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
43. Levy ML, Cummings JL, Fairbanks LA, et al. Apathy is not depression. J Neuropsychiatry Clin Neurosci. 1998;10(3):314-319.
44. Snow V, Lascher S, Mottur-Pilson C. Pharmacologic treatment of acute major depression and dysthymia. American College of Physicians-American Society of Internal Medicine. Ann Intern Med. 2000;132(9):738-742.
45. Schwartz AC, Fisher TJ, Greenspan HN, et al. Pharmacologic and nonpharmacologic approaches to the prevention and management of delirium. Int J Psychiatry Med. 2016;51(2):160-170.
46. Kang H, Zhao F, You L, et al. Pseudo-dementia: a neuropsychological review. Ann Indian Acad Neurol. 2014;17(2):147-154.
47. Fricchione GL, Beach SR, Huffman J, et al. Life-threatening conditions in psychiatry: catatonia, neuroleptic malignant syndrome, and serotonin syndrome. In: Stern TA, Fava M, Wilens TE, eds. Massachusetts General Hospital comprehensive clinical psychiatry. London, United Kingdom: Elsevier; 2016:608-617.
48. Rogers RD. The roles of dopamine and serotonin in decision making: evidence from pharmacological experiments in humans. Neuropsychopharmacology. 2011;36(1):114-132.
49. Stransky M, Schmidt C, Ganslmeier P, et al. Hypoactive delirium after cardiac surgery as an independent risk factor for prolonged mechanical ventilation. J Cardiothorac Vasc Anesth. 2011;25(6):968-974.
50. Wilcox JA, Reid Duffy P. The syndrome of catatonia. Behav Sci (Basel). 2015;5(4):576-588.
51. Robert PH, Mulin E, Malléa P, et al. REVIEW: apathy diagnosis, assessment, and treatment in Alzheimer’s disease. CNS Neurosci Ther. 2010;16(5):263-271.
52. Cipriani G, Lucetti C, Danti S, et al. Apathy and dementia. Nosology, assessment and management. J Nerv Ment Dis. 2014;202(10):718-724.
53. Starkstein SE, Leentjens AF. The nosological position of apathy in clinical practice. J Neurol Neurosurg Psychiatry. 2008;79(10):1088-1092.54. Berman K, Brodaty H, Withall A, et al. Pharmacologic treatment of apathy in dementia. Am J Geriatr Psychiatry. 2012;20(2):104-122.
55. Theleritis C, Siarkos K, Katirtzoglou E, et al. Pharmacological and nonpharmacological treatment for apathy in Alzheimer disease: a systematic review across modalities. J Geriatr Psychiatry Neurol. 2017;30(1):26-49.
56. APA Work Group on Alzheimer’s Disease and other Dementias; Rabins PV, Blacker D, Rovner BW, et al. American Psychiatric Association practice guideline for the treatment of patients with Alzheimer’s disease and other dementias. Second edition. Am J Psychiatry. 2007;164(suppl 12):5-56.
57. Dolder CR, Davis LN, McKinsey J. Use of psychostimulants in patients with dementia. Ann Pharmacother. 2010;44(10):1624-1632.

Etiologies of DDM

Medical marijuana: Do the benefits outweigh the risks?
There is a need for additional treatment options to improve symptoms, enhance the quality of life (QOL), and reduce suffering among patients who have chronic medical illness. Medical marijuana (MM) has the potential to help patients who have certain medical conditions in states where it is legal for prescription by a licensed medical provider.
Cannabis has a long history of medicinal use (Box 11-12). Two derivatives of the Cannabis plant—cannabinoid delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD)—are responsible for most of its effects. Some of these effects, including analgesia, decreased muscle spasticity, and reduced eye pressure, have been harnessed for their potential therapeutic effects (Box 213-19). As of November 2017, 29 states had legalized Cannabis for medical use, and several had legalized its recreational use.12
With the increasing availability of MM, psychiatrists are likely to encounter patients who are using it or who will ask them about it. This article reviews evidence related to using MM to treat patients with neuropathic pain; chemotherapyinduced nausea and vomiting (CINV); epilepsy; multiple sclerosis (MS); glaucoma; Crohn’s disease; Parkinson’s disease; amyotrophic lateral sclerosis; dementia-related behavioral disturbances; posttraumatic stress disorder (PTSD); and anxiety.
Box 1
Cannabis: A history of medicinal use
Cannabis has been cultivated since ancient times, beginning in China and India. The earliest reference of its use for healing purposes may have been in the Chinese Pharmacopeia, circa 1500 BC.1 In 1839, Dr. William Brooke O’Shaughnessy introduced Cannabis Indica, or “Indian hemp,” to the western world after a professorship in Calcutta, India.2 In the early 1840s, an English physician, Dr. John Clendinning, prescribed Cannabis for migraine headache.3 In the 19th and early 20th centuries, several prominent physicians advocated using Cannabis for migraines; Sir William Osler did so in his textbook, The principles and practice of medicine.4 It was listed in the U.S. Pharmacopeia in 1850 but removed in 1942.5,6
Until 1937, Cannabis was used in the United States for medicinal purposes, such as for treating inflamed skin, incontinence, and sexually transmitted diseases.7 In 1937, the Marihuana Tax Act, which prohibited the production, importation, possession, use, and dispersal of Cannabis, was passed.8 Cannabis became a Schedule I drug under the Controlled Substance Act of 1970.9
In 1999, based on available evidence, the Institute of Medicine (IOM) concluded Cannabis had less likelihood of dependence than benzodiazepines, opiates, cocaine, or nicotine. The IOM also concluded that the symptoms of withdrawal were mild in comparison with benzodiazepines or opiates. Finally, the IOM stated that Cannabis was not a “gateway” drug.10
In 1996, California was the first state to reimplement medicinal use of Cannabis under the Compassionate Use Act, also known as Proposition 215.11 This act allowed individuals to retain or produce Cannabis for personal consumption with a physician’s approval. Many states eventually followed California’s lead. As of November 2017, 29 states, the District of Columbia, Guam, and Puerto Rico had regulated Cannabis use for medical purposes,12 and recreational use had been approved in 7 states and the District of Columbia.
Medical illnesses
Neuropathic pain. Chronic neuropathic pain affects an estimated 7% to 8% of adults.20 Patients with neuropathic pain are often treated with anticonvulsants, antidepressants, opioids, and local anesthetics21; however, these medications may not provide substantial relief. Research has revealed that THC and CBD can improve central and peripheral neuropathic pain, as well as pain associated with rheumatoid arthritis and fibromyalgia.22
Wilsey et al23 evaluated the analgesic effects of smoked MM for neuropathic pain in a small (N = 38) double-blind, randomized controlled trial (RCT). Patients in this study had a preexisting diagnosis of complex regional pain syndrome, spinal cord injury, peripheral neuropathy, or nerve injury. To prevent any unforeseen adverse outcomes related to Cannabis use, participants were required to have previous exposure to Cannabis. Patients were excluded if they had major mental illness, substance abuse, or other major medical ailments.
Participants smoked high-dose Cannabis cigarettes (7% THC), low-dose Cannabis cigarettes (3.5% THC), or placebo cigarettes. Pain was measured on a visual analog scale (VAS) that ranged from 0 (no pain) to 100 (worst possible pain). Compared with the placebo group, significant analgesia was achieved in both Cannabis groups (P = .016). The high-dose group had greater neurocognitive impairment.
Ware et al24 conducted a crossover RCT (N = 23) to determine the efficacy of smoked MM for neuropathic pain. Participants had neuropathic pain for at least 3 months that was caused by trauma or surgery, with an average weekly pain intensity score >4 on scale of 0 to 10. Patients with pain due to cancer, nociceptive causes, unstable medical conditions, current substance abuse, history of a psychotic disorder, or suicidal ideation were excluded. Participants were assigned to a 9.4% THC group or a 0% THC group. Pain intensity was evaluated daily via telephone. Participants in the 9.4% THC group had statistically lower pain intensity compared with the 0% THC group (P = .023). Common adverse effects reported by those in the 9.4% group included headache, dry eyes, burning sensation, dizziness, numbness, and cough.
Box 2
The effects of Cannabis
Marijuana is harvested from the plant Cannabis sativa and composed of 400 lipophilic chemical compounds, including phytocannabinoids, terpenoids, and flavonoids.13 The plant contains compounds termed “cannabinoids.” Two of these derivatives in particular are responsible for most of the effects of marijuana: cannabinoid delta-9- tetrahydrocannabinol (THC) and cannabidiol (CBD). THC has a comparable structure and binding mechanism to anandamide, a naturally occurring fatty acid neurotransmitter present within the human brain.14-16 The endogenous endocannabinoid system and its receptors are found throughout the entire body (brain, organs, glands, immune cells, and connective tissues).
THC binds to cannabinoid receptors CB1 and CB2. CB1 is found predominantly in the CNS. CB2 is found predominantly outside the CNS and is associated with the immune system.14-16 The effects of THC include euphoria, relaxation, appetite stimulation, improvement of nausea and vomiting, analgesia, decreased muscle spasticity, and reduced eye pressure.14,15 CBD may have anxiolytic, antipsychotic, anticonvulsive, and analgesic effects.
The rate of absorption of THC and CBD depends both on the potency of the cannabinoid as well as the mechanism of consumption. Cannabis can be administered by multiple routes, including via smoking, oral ingestion, or IV.16 When Cannabis is smoked (the route for the most rapid delivery), THC is transported from the lungs to the bloodstream and reaches peak concentrations in 3 to 10 minutes. Oral ingestion (capsules, tinctures, sprays, and edibles) has a more flexible onset of action, usually occurring in 30 to 120 minutes, with effects lasting 5 to 6 hours. IV administration has rapid effects; the onset can occur within seconds to minutes, and effects can last 2 to 3 hours. The IV form allows 90% of THC to be distributed in plasma and can rapidly penetrate highly vascularized tissues, such as the liver, heart, fat, lungs, and muscles.
Pharmaceutical manufacturers have used cannabinoid derivatives to produce Cannabis-based medications for treating medical conditions. Nabilone, a potent agonist of the CB1 receptor, became available as a Schedule II medication in 1981 and was approved for patients with chemotherapy-induced nausea and vomiting (CINV).17 In 1985, dronabinol was introduced as an antiemetic for CINV as well as an appetite stimulant for patients with conditions associated with excessive weight loss.18 Another option, nabiximols, is an oral mucosal spray that consists of THC and CBD in a 1:1 ratio.19 Nabiximols is approved in Canada for pain relief in end-stage cancer patients and pain associated with multiple sclerosis.19
In an RCT of vaporized Cannabis, 39 patients with a diagnosis of complex regional pain syndrome, thalamic pain, spinal cord injury, peripheral neuropathy, radiculopathy, or nerve injury were assigned to a medium-dose (3.53% THC), low-dose (1.29% THC), or placebo group.25 Serious mental illness, substance abuse, and medical conditions were cause for exclusion. Participants received vaporized marijuana (average 8 to 12 puffs per visit) over 3 sessions. A 30% pain reduction was achieved by 26% of those in the placebo group, 57% of those in the low-dose group, and 61% of individuals in the high-dose group; the difference between placebo and each Cannabis group was statistically significant.
Chemotherapy-induced nausea and vomiting. Up to 80% of patients who receive chemotherapy experience CINV, which occurs from 24 hours to 7 days after receiving such therapy.26 CINV negatively influences a patient’s QOL and may impact the decision to continue with chemotherapy. Use of MM can help to diminish vomiting by binding to central CB1 receptors and averting the proemetic effects of dopamine and serotonin.27 Two synthetically derived cannabinoids, dronabinol and nabilone, are FDA-approved for treating CINV.
In a small (N = 64) parallel-group RCT, Meiri et al27 compared dronabinol with the commonly used antiemetic ondansetron and with a combination of dronabinol and ondansetron for treating CINV in adults. The primary outcome was prevention of delayed-onset CINV. Patients were eligible for this study if they had a malignancy that did not involve bone marrow, were receiving treatment with a moderately to highly emetogenic regimen, were not pregnant, and had an estimated life expectancy of at least 6 weeks after chemotherapy. The patients were randomized to 1 of 4 treatment groups: dronabinol alone, ondansetron alone, dronabinol plus ondansetron, or placebo. Overall, 47% to 58% of the active treatment groups improved, compared with 20% of the placebo group. Combination therapy did not provide any benefit beyond any single agent alone. All active treatments reduced nausea compared with placebo; there was no difference between active treatment groups. This study was limited by low enrollment.
Tramèr et al28 conducted a systematic review of 30 randomized comparisons of MM with placebo or antiemetics. The reviewed studies were completed between 1975 to 1997 and analyzed a total of 1,366 patients. Nabilone was evaluated in 16 trials; dronabinol was utilized in 13 trials; and IM levonantradol, a synthetic cannabinoid analog of dronabinol, was used in 1 trial. These agents were found to be more effective as an antiemetic compared with prochlorperazine, metoclopramide, chlorpromazine, thiethylperazine, haloperidol, domperidone, or alizapride. In addition, 38% to 90% of patients in these studies preferred MM over the traditional antiemetics.
A Cochrane review29 suggested that MM may be a viable option for treatment-resistant CINV; however, further studies are needed because current studies have methodological limitations.
Epilepsy. Maa and Figi30 reported a case of a 5-year-old girl who had Dravet syndrome, which resulted in 50 generalized tonic-clonic seizures daily; multiple anticonvulsants did not alleviate these seizures. Because of her recurring seizures, the patient had multiple cognitive and motor delays and needed a feeding tube. In addition to her existing antiepileptic drug regimen, she was started on adjunctive therapy with a sublingual Cannabis extract containing a high concentration of CBD. Her seizures decreased from 50 per day to 2 to 3 nocturnal convulsions per month. The treatment enabled her to stop using a feeding tube, resume walking and talking, and sleep soundly.
dos Santos et al31 reviewed studies of MM for treating epilepsy. One was a double-blind, placebo-controlled trial that included 15 patients ages 14 to 49 who had secondary generalized epilepsy with a temporal lobe focus. Eight patients received 200 to 300 mg/d of oral CBD for 8 to 18 weeks, and 7 received placebo. Seven patients had fewer seizures and 4 had no seizures. Only 1 patient in the placebo group demonstrated any improvement. Another study in this review included 19 children with treatment-resistant epilepsy: Dravet syndrome (n = 13), Doose syndrome (n = 4), Lennox-Gastaut syndrome (n = 1), or idiopathic epilepsy (n = 1). These patients experienced various types of seizures with a frequency ranging from 2 per week to 250 per day. Overall, 84% of children treated with CBD had fewer seizures: 11% were seizure-free, 42% had a >80% reduction in seizures, and 32% had a 25% to 60% reduction in seizures. Parents also noted additional benefits, including increased attention, improved mood, and improved sleep. CBD was well tolerated in most patients in both studies.
Despite these results, a Cochrane review32 found that no reliable conclusions can be drawn regarding the efficacy of MM for treating epilepsy.
Multiple sclerosis. According to American Academy of Neurology guidelines, physicians may provide MM as an alternative treatment for patients with MS-related spasticity.33 Multiple studies have tested MM and MM-related extracts for treating spasticity related to MS.34,35 In a placebo-controlled crossover study, Corey-Bloom et al34 reported a significant reduction in spasticity, measured using the modified Ashworth scale, in MS patients receiving Cannabis cigarettes vs placebo cigarettes (P < .0001). However, compared with the placebo group, patients who received MM had significant adverse effects, primarily cognitive impairment (P = .003).
In a multicenter RCT (N = 572 patients with refractory MS spasticity), Novotna et al36 evaluated nabiximols, an oral mucosal spray of a formulated extract of Cannabis that contains THC and CBD in a 1:1 ratio. They assessed spasticity using the Numerical Spasticity Rating Scale (NRS). Results were confirmed by measuring the number of daily spasms, self-report of sleep quality, and activities of daily living. After 4 weeks of single-blind treatment, patients who responded to nabiximols (≥20% improvement in spasticity) were randomized to a placebo group or nabiximols group for 12 additional weeks. After 12 weeks, compared with those who received placebo, those in the nabiximols group experienced a statistically significant reduction in spasticity based on NRS score (P = .0002).
For a summary of evidence on MM for treating glaucoma, Crohn’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, see Box 3.37-43
Box 3
Cannabis for treating glaucoma, Crohn’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis
Glaucoma. In a placebo-controlled study, oromucosal administration of medical marijuana (MM) reduced intraocular pressure from 28 mm Hg to 22 mm Hg, with a duration of action of 3.5 hours.37However, the American Academy of Ophthalmologists does not recommend treating glaucoma with MM because the effect is short-lasting, and MM causes significant cognitive impairment compared with other standardized treatments.38 MM also leads to decreased blood pressure, which lowers blood flow to the optic nerve, thus increasing the risk of blindness.
Crohn’s disease. A randomized controlled trial (RCT) of MM for Crohn’s disease was conducted using the Crohn’s Disease Activity Index (CDAI) to assess for remission. In this 8-week study,21 individuals with Crohn’s disease were administered smoked MM (115 mg of delta-9-tetrahydrocannabinol [THC]) or placebo.39 Eligible patients were at least 20 years old, had active Crohn’s disease (CDAI >200), and had not responded to medical treatment for the illness. Compared with those who received placebo, patients who received MM experienced a statistically significant reduction in CDAI scores (P < .05). However, at follow-up 2 weeks after the study, when MM was no longer administered, there was no difference in mean CDAI scores between the 2 groups. Five of the 11 patients in the MM group achieved clinical remission, compared with 1 of 10 in the placebo group, but this difference was not statistically significant.
Parkinson’s disease (PD). According to the American Academy of Neurology, oral Cannabis extracts are “probably ineffective” for levodopa-induced dyskinesia in patients with PD.40 Reported benefits have come mainly from self-report studies. A 2014 survey (22 patients) found a significant reduction in PD symptoms—mainly relief from drug-induced tremor and pain—when measured using the Unified Parkinson’s Disease Rating Scale (UPDRS). Patients also reported better sleep and reduced pain (measured with a visual analog scale [VAS]). An exploratory double-blind placebo trial (N = 119) found no difference in mean UPDRS and no difference in any neuroprotective measures.41 However, the experimental group had a significantly higher quality of life (QOL; P = .05). A similar double-blind crossover study that included 19 patients found no significant difference in dyskinesia, as measured with the UPDRS, in the group receiving oral Cannabis extract compared with the placebo group.42
Amyotrophic lateral sclerosis (ALS). A randomized double-blind crossover trial of 27 ALS patients found that an oral THC extract (dronabinol, 5 mg, twice daily) had no significant effects on spasticity, as measured with the VAS.43 There was also no significant difference between the experimental and placebo groups on number of spasms (also measured with a VAS), quality of sleep (measured with the Sleep Disorders Questionnaire), or QOL (measured with the Amyotrophic Lateral Sclerosis Assessment questionnaire).
Psychiatric illnesses
Dementia-related behavioral disturbances. A few clinical trials with small sample sizes have found evidence supporting the use of MM compounds for alleviating neuropsychiatric symptoms of patients with dementia. An open-label pilot study of 6 individuals with late-stage dementia who received dronabinol, 2.5 mg/d, for 2 weeks, found a significant reduction (compared with baseline) in nighttime motor activity as measured with an actometer (P < .0028).44 The secondary Neuropsychiatric Inventory (NPI) assessment found reductions in aberrant motor behavior (P = .042), agitation (P = .042), and nighttime behaviors (P = .42).
A 2014 retrospective analysis of 40 inpatients with dementia-related agitation and appetite loss who were treated with dronabinol (mean dosage: 7.03 mg/d) found reductions in all aspects of agitation, including aberrant vocalization, motor agitation, aggressiveness, and treatment resistance, as measured with the Pittsburgh Agitation Scale (P < .0001).45 The study found no significant improvements in appetite, Global Assessment of Functioning mean score, or number of times patients awoke during the night. Adverse effects included sedation and delirium.
A RCT of 50 dementia patients with clinically relevant neuropsychiatric symptoms found no significant difference in mean NPI scores between patients given placebo and those who received nabiximols, 1.5 mg, 3 times daily.46 There were no significant differences found in agitation, QOL, life activities, or caregiver-scored Caregiver Global Impression of Change scale.
In a small RCT, THC was safe and well tolerated in 10 older patients with dementia.47 A 2009 Cochrane review48 concluded that there was no evidence for the efficacy of MM in treating the neuropsychiatric symptoms related to dementia.
PTSD. Preclinical evidence shows that the endocannabinoid system is involved in regulating emotional memory. Evidence also suggests that cannabinoids may facilitate the extinction of aversive memories.49,50
In 2009, New Mexico became the first state to authorize the use of MM for patients with PTSD. In a study of patients applying for the New Mexico Medical Cannabis Program, researchers used the Clinician Administered Posttraumatic Scale (CAPS) to assess PTSD symptoms.51 A retrospective chart review of the first 80 patients evaluated found significant (P < .0001) reductions of several PTSD symptoms, including intrusive memories, distressing dreams, flashbacks, numbing and avoidance, and hyperarousal, in the group using MM vs those not using MM. There also was a significant difference in CAPS total score (P < .0001). Patients reported a 75% reduction in PTSD symptoms while using MM. This study has several limitations: It was a retrospective review, not an RCT, and patients were prescreened and knew before the study began that MM helped their PTSD symptoms.
In another retrospective study, researchers evaluated treatment with nabilone, 0.5 to 6 mg/d, in 104 incarcerated men with various major mental illnesses; most (91%) met criteria for Cannabis dependence.52 They found significant improvements in sleep and PTSD symptoms.
A double-blind RCT evaluated MM in 10 Canadian male soldiers with PTSD who experienced nightmares despite standard medication treatment. Adjunctive nabilone (maximum dose: 3 mg/d) resulted in a reduction in nightmares as measured by the CAPS recurrent distressing dream of the event item score.53
Currently, there are no adequately powered RCTs of MM in a diverse group of PTSD patients. Most studies are open-label, enriched design, and included white male veterans. No well-conducted trials have evaluated patients with noncombat-related PTSD. Most of the relevant literature consists of case reports of Cannabis use by patients with PTSD.
Anxiety disorders.Patients frequently indicate that smoking Cannabis helps relieve their anxiety, although there is no replicated evidence based on double-blind RCTs to support this. However, in rat models CBD has been shown to facilitate extinction of conditioned fear via the endocannabinoid system.54-56 The mechanism of action is not completely understood. CBD has been shown to have antagonistic action at CB1 and CB2 receptors. It may have similar effects on memory extinction and may be an adjunct to exposure therapies for anxiety disorders.
Das et al57 studied the effects of CBD (32 mg) on extinction and consolidation of memory related to contextual fear in 48 individuals. They found that CBD can enhance extinction learning, and suggested it may have potential as an adjunct to extinction-based therapies for anxiety disorders.
Caveats: Adverse effects, lack of RCTs
Cannabis use causes impairment of learning, memory, attention, and working memory. Adolescents are particularly vulnerable to the effects of Cannabis on brain development at a time when synaptic pruning and increased myelination occur. Normal brain development could be disrupted. Some studies have linked Cannabis use to abnormalities in the amygdala, hippocampus, frontal lobe, and cerebellum. From 1995 to 2014, the potency of Cannabis (THC concentration) increased from 4% to 12%.58 This has substantial implications for increased abuse among adolescents and the deleterious effects of Cannabis on the brain.
Heavy Cannabis use impairs motivation and could precipitate psychosis in vulnerable individuals. Cannabis use may be linked to the development of schizophrenia.59
There are no well-conducted RCTs on the efficacy of MM, and adequate safety data are lacking. There is also lack of consensus among qualified experts. There is soft evidence that MM may be helpful in some medical conditions, including but not limited to CINV, neuropathic pain, epilepsy, and MS-related spasticity. Currently, the benefits of using MM do not appear to outweigh the risks.
Bottom Line
Limited evidence suggests medical marijuana (MM) may be beneficial for treating a few medical conditions, including neuropathic pain and chemotherapy-induced nausea and vomiting. There is no clear and convincing evidence MM is beneficial for psychiatric disorders, and Cannabis can impair cognition and attention and may precipitate psychosis. The risk of deleterious effects are greater in adolescents.
Related Resources
- Nguyen DH, Thant TM. Caring for medical marijuana patients who request controlled prescriptions. Current Psychiatry. 2017;16(8):50-51.
- National Institute on Drug Abuse. Marijuana as medicine. https://www.drugabuse.gov/publications/drugfacts/ marijuana-medicine.
Drug Brand Names
Alizapride • Litican, Superan
Chlorpromazine • Thorazine
Domperidone • Motilium
Dronabinol • Marinol, Syndros
Haloperidol • Haldol
Metoclopramide • Reglan
Nabilone • Cesamet
Nabiximols • Sativex
Ondansetron • Zofran, Zuplenz
Prochlorperazine • Compazine
Thiethylperazine • Torecan
1. National Institute on Drug Abuse (NIDA). Marijuana Research Findings 1976. NIDA research monograph 14. https://archives.drugabuse.gov/sites/default/files/monograph14.pdf. Published July 1977. Accessed November 15, 2017.
2. O’Shaughnessy WB. On the preparations of the Indian hemp, or gunjah- cannabis indica their effects on the animal system in health, and their utility in the treatment of tetanus and other convulsive diseases. Prov Med J Retrosp Med Sci. 1843;5(123):363-369.
3. Clendinning J. Observations on the medical properties of the Cannabis Sativa of India. Med Chir Trans. 1843;26:188-210.
4. Osler W, McCrae T. The principles and practice of medicine. 9th ed. New York, NY: D. Appleton and Company; 1921.
5. The pharmacopoeia of the United States of America. 3rd ed. Philadelphia, PA: Lippincott; 1851.
6. The pharmacopoeia of the United States of America. 12th ed. Easton, PA: Mack Printing Company; 1942.
7. Philipsen N, Butler RD, Simon C, et al. Medical marijuana: a primer on ethics, evidence, and politics. Journal Nurse Pract. 2014;10(9):633-640.
8. Marihuana Tax Act of 1937, Pub L No. 75-238, 75th Cong, 50 Stat 551 (1937).
9. Controlled Substances Act, 21 USC §812.
10. Watson SJ, Benson JA, Joy JE, eds. Marijuana and medicine: assessing the science base. Washington, DC: National Academy Press; 1999.
11. California Proposition 215, the medical marijuana initiative (1996). https://ballotpedia.org/California_Proposition_215,_the_Medical_Marijuana_Initiative_(1996). Accessed November 16, 2017.
12. National Conference of State Legislatures. State medical marijuana laws. http://www.ncsl.org/research/health/state-medical-marijuana-laws.aspx. Updated September 14, 2017. Accessed November 16, 2017.
13. Russo EB. Taming THC: potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br J Pharmacol. 2011;163(7):1344-1364.
14. Alger BE. Getting high on the endocannabinoid system. Cerebrum. 2013:14. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3997295. Accessed December 5, 2017.
15. Galal AM, Slade D, Gul W, et al. Naturally occurring and related synthetic cannabinoids and their potential therapeutic applications. Recent Pat CNS Drug Discov. 2009;4(2):112-136.
16. Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. 2007;4(8):1770-1804.
17. Cesamet [package insert]. Somerset, NJ: Meda Pharmaceuticals; 2013.
18. Marinol [package insert]. Chicago, IL: AbbVie Inc.; 2017.
19. Sativex [package insert]. Mississauga, Ontario: Bayer Inc.; 2015.
20. Torrance N, Ferguson JA, Afolabi E, et al. Neuropathic pain in the community: more under-treated than refractory? Pain. 2013;154(5):690-699.
21. Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14(2):162-173.
22. Whiting PF, Wolff RF, Deshpande S, et al. Cannabinoids for medical use: a systematic review and meta-analysis. JAMA. 2015;313(24):2456-2473.
23. Wilsey B, Marcotte, T, Tsodikov A, et al. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. J Pain. 2008;9(6):506-521.
24. Ware MA, Wang T, Shapiro S, et al. Smoked cannabis for chronic neuropathic pain: a randomized controlled trial. CMAJ. 2010;182(14):E694-E701.
25. Wilsey B, Marcotte T, Deutsch R, et al. Low-dose vaporized cannabis significantly improves neuropathic pain. J Pain. 2013;14(2):136-148.
26. National Cancer Institute. Treatment-related nausea and vomiting (PDQ®)-health professional version. https://www.cancer.gov/about-cancer/treatment/side-effects/nausea/nausea-hp-pdq. Updated May 10, 2017. Accessed November 7, 2017.
27. Meiri E, Jhangiani H, Vrendenburgh JJ, et al. Efficacy of dronabinol alone and in combination with ondansetron versus ondansetron alone for delayed chemotherapy-induced nausea and vomiting. Curr Med Res Opin. 2007;23(3):533-543.
28. Tramèr MR, Carroll D, Campbell FA, et al. Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review. BMJ. 2001;323(7303):16-21.
29. Smith LA, Azariah F, Lavender VT, et al. Cannabinoids for nausea and vomiting in adults with cancer receiving chemotherapy. Cochrane Database Syst Rev. 2015;(11):CD009464.
30. Maa E, Figi P. The case for medical marijuana in epilepsy. Epilepsia. 2014;55(6):783-786.
31. dos Santos RG, Hallak JE, Leite JP, et al. Phytocannabinoids and epilepsy. J Clin Pharm Ther. 2015;40(2):135-143.
32. Gloss D, Vickrey B. Cannabinoids for epilepsy. Cochrane Database Syst Rev. 2014;(3):CD009270.
33. Yadav V, Bever C Jr, Bowen J, et al. Summary of evidence-based guideline: complementary and alternative medicine in multiple sclerosis: report of the guideline development subcommittee of the American Academy of Neurology. Neurology. 2014;82(12):1083-1092.
34. Corey-Bloom J, Wolfson T, Gamst A, et al. Smoked cannabis for spasticity in multiple sclerosis: a randomized, placebo-controlled trial. CMAJ. 2012;184(10):1143-1150.
35. Zajicek J, Ball S, Wright D, et al; CUPID investigator group. Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): a randomised, placebo-controlled trial. Lancet Neurol. 2013;12(9):857-865.
36. Novotna A, Mares J, Ratcliffe S, et al; Sativex Spasticity Study Group. A randomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols* (Sativex(®)), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. Eur J Neurol. 2011;18(9):1122-1131.
37. Merritt JC, Crawford WJ, Alexander PC, et al. Effect of marihuana on intraocular and blood pressure in glaucoma. Ophthalmology. 1980;87(3):222-228.
38. American Academy of Ophthalmology. American Academy of Ophthalmology reiterates position that marijuana is not a proven treatment for glaucoma. https://www.aao.org/newsroom/news-releases/detail/american-academy-of-ophthalmology-reiterates-posit. Published June 27, 2014. Accessed May 29, 2017.
39. Naftali T, Bar-Lev Schleider L, Dotan I, et al. Cannabis induces a clinical response in patients with Crohn’s disease: a prospective placebo-controlled study. Clin Gastroenterol Hepatol. 2013;11(10):1276.e1-1280.e1.
40. Koppel BS Brust JC, Fife T, et al. Systematic review: efficacy and safety of medical marijuana in certain neurological disorders. Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2014;82(17):1556-1563.
41. Chagas MH, Zuardi AW, Tumas V, et al. Effects of cannabidiol in the treatment of patients with Parkinson’s disease: an exploratory double-blind trial. J Psychopharmacol. 2014;28(11):1088-1098.
42. Carroll CB, Bain PG, Teare L, et al. Cannabis for dyskinesia in Parkinson disease: a randomized double-blind crossover study. Neurology. 2004;63(7):1245-1250.
43. Weber M, Goldman B, Truniger S. Tetrahydrocannabinol (THC) for cramps in amyotrophic lateral sclerosis: a randomised, double-blind crossover trial. J Neurol Neurosurg Psychiatry. 2010;81(10):1135-1140.
44. Walther S, Mahlberg R, Eichmann U, et al. Delta-9-tetrahydrocannabinol for nighttime agitation in severe dementia. Psychopharmacology (Berl). 2006;185(4):524-528.
45. Woodward MR, Harper DG, Stolyar A, et al. Dronabinol for the treatment of agitation and aggressive behavior in acutely hospitalized severely demented patients with noncognitive behavioral symptoms. Am J Geriatr Psychiatry. 2014;22(4):415-419.
46. van den Elsen GA, Ahmed A, Verkes RJ, et al. Tetrahydrocannabinol for neuropsychiatric symptoms in dementia: a randomized controlled trial. Neurology. 2015;84(23):2338-2346.
47. Ahmed AI, van den Elsen GA, Colbers A, et al. Safety, pharmacodynamics, and pharmacokinetics of multiple oral doses of delta-9-tetrahydrocannabinol in older persons with dementia. Psychopharmacology (Berl). 2015;232(14):25872595.
48. Krishnan S, Cairns R, Howard R. Cannabinoids for the treatment of dementia. Cochrane Database Syst Rev. 2009;(2):CD007204.
49. de Bitencourt RM, Pamplona FA, Takahashi RN. A current overview of cannabinoids and glucocorticoids in facilitating extinction of aversive memories: potential extinction enhancers. Neuropharmacology. 2013;64:389-395.
50. Fraser GA. The use of a synthetic cannabinoid in the management of treatment-resistant nightmares in posttraumatic stress disorder (PTSD). CNS Neurosci Ther. 2009;15(1):84-88.
51. Greer GR, Grob CS, Halberstadt AL. PTSD symptom reports of patients evaluated for the New Mexico Medical Cannabis Program. J Psychoactive Drugs. 2014;46(1):73-77.
52. Cameron C, Watson D, Robinson J. Use of a synthetic cannabinoid in a correctional population for posttraumatic stress disorder-related insomnia and nightmares, chronic pain, harm reduction, and other indications: a retrospective evaluation. J Clin Psychopharmacol. 2014;34(5):559-564.
53. Jetly R, Heber A, Fraser G, et al. The efficacy of nabilone, a synthetic cannabinoid, in the treatment of PTSD-associated nightmares: a preliminary randomized, double-blind, placebo-controlled cross-over design study. Psychoneuroendocrinology. 2015;51:585-588.
54. Bitencourt RM, Pamplona FA, Takahashi RN. Facilitation of contextual fear, memory extinction, and anti-anxiogenic effects of AM404 and cannabidiol in conditioned rats. Eur Neuropsychopharmacol. 2008;18(12):849-859.
55. Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153(2):199-215.
56. Thomas A, Baillie GL, Phillips AM, et al. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol. 2007;150(5):613-623.
57. Das RK, Kamboj SK, Ramadas M, et al. Cannabidiol enhances consolidation of explicit fear extinction in humans. Psychopharmacology (Berl). 2013;226(4):781-792.
58. ElSohly MA, Mehmedic Z, Foster S, et al. Changes in cannabis potency over the last 2 decades (1995-2014): analysis of current data in the United States. Biol Psychiatry. 2016;79(7):613-619.
59. Volkow ND, Swanson JM, Evins AE, et al. Effects of cannabis use on human behavior, including cognition, motivation, and psychosis: a review. JAMA Psychiatry. 2016;73(3):292297.
There is a need for additional treatment options to improve symptoms, enhance the quality of life (QOL), and reduce suffering among patients who have chronic medical illness. Medical marijuana (MM) has the potential to help patients who have certain medical conditions in states where it is legal for prescription by a licensed medical provider.
Cannabis has a long history of medicinal use (Box 11-12). Two derivatives of the Cannabis plant—cannabinoid delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD)—are responsible for most of its effects. Some of these effects, including analgesia, decreased muscle spasticity, and reduced eye pressure, have been harnessed for their potential therapeutic effects (Box 213-19). As of November 2017, 29 states had legalized Cannabis for medical use, and several had legalized its recreational use.12
With the increasing availability of MM, psychiatrists are likely to encounter patients who are using it or who will ask them about it. This article reviews evidence related to using MM to treat patients with neuropathic pain; chemotherapyinduced nausea and vomiting (CINV); epilepsy; multiple sclerosis (MS); glaucoma; Crohn’s disease; Parkinson’s disease; amyotrophic lateral sclerosis; dementia-related behavioral disturbances; posttraumatic stress disorder (PTSD); and anxiety.
Box 1
Cannabis: A history of medicinal use
Cannabis has been cultivated since ancient times, beginning in China and India. The earliest reference of its use for healing purposes may have been in the Chinese Pharmacopeia, circa 1500 BC.1 In 1839, Dr. William Brooke O’Shaughnessy introduced Cannabis Indica, or “Indian hemp,” to the western world after a professorship in Calcutta, India.2 In the early 1840s, an English physician, Dr. John Clendinning, prescribed Cannabis for migraine headache.3 In the 19th and early 20th centuries, several prominent physicians advocated using Cannabis for migraines; Sir William Osler did so in his textbook, The principles and practice of medicine.4 It was listed in the U.S. Pharmacopeia in 1850 but removed in 1942.5,6
Until 1937, Cannabis was used in the United States for medicinal purposes, such as for treating inflamed skin, incontinence, and sexually transmitted diseases.7 In 1937, the Marihuana Tax Act, which prohibited the production, importation, possession, use, and dispersal of Cannabis, was passed.8 Cannabis became a Schedule I drug under the Controlled Substance Act of 1970.9
In 1999, based on available evidence, the Institute of Medicine (IOM) concluded Cannabis had less likelihood of dependence than benzodiazepines, opiates, cocaine, or nicotine. The IOM also concluded that the symptoms of withdrawal were mild in comparison with benzodiazepines or opiates. Finally, the IOM stated that Cannabis was not a “gateway” drug.10
In 1996, California was the first state to reimplement medicinal use of Cannabis under the Compassionate Use Act, also known as Proposition 215.11 This act allowed individuals to retain or produce Cannabis for personal consumption with a physician’s approval. Many states eventually followed California’s lead. As of November 2017, 29 states, the District of Columbia, Guam, and Puerto Rico had regulated Cannabis use for medical purposes,12 and recreational use had been approved in 7 states and the District of Columbia.
Medical illnesses
Neuropathic pain. Chronic neuropathic pain affects an estimated 7% to 8% of adults.20 Patients with neuropathic pain are often treated with anticonvulsants, antidepressants, opioids, and local anesthetics21; however, these medications may not provide substantial relief. Research has revealed that THC and CBD can improve central and peripheral neuropathic pain, as well as pain associated with rheumatoid arthritis and fibromyalgia.22
Wilsey et al23 evaluated the analgesic effects of smoked MM for neuropathic pain in a small (N = 38) double-blind, randomized controlled trial (RCT). Patients in this study had a preexisting diagnosis of complex regional pain syndrome, spinal cord injury, peripheral neuropathy, or nerve injury. To prevent any unforeseen adverse outcomes related to Cannabis use, participants were required to have previous exposure to Cannabis. Patients were excluded if they had major mental illness, substance abuse, or other major medical ailments.
Participants smoked high-dose Cannabis cigarettes (7% THC), low-dose Cannabis cigarettes (3.5% THC), or placebo cigarettes. Pain was measured on a visual analog scale (VAS) that ranged from 0 (no pain) to 100 (worst possible pain). Compared with the placebo group, significant analgesia was achieved in both Cannabis groups (P = .016). The high-dose group had greater neurocognitive impairment.
Ware et al24 conducted a crossover RCT (N = 23) to determine the efficacy of smoked MM for neuropathic pain. Participants had neuropathic pain for at least 3 months that was caused by trauma or surgery, with an average weekly pain intensity score >4 on scale of 0 to 10. Patients with pain due to cancer, nociceptive causes, unstable medical conditions, current substance abuse, history of a psychotic disorder, or suicidal ideation were excluded. Participants were assigned to a 9.4% THC group or a 0% THC group. Pain intensity was evaluated daily via telephone. Participants in the 9.4% THC group had statistically lower pain intensity compared with the 0% THC group (P = .023). Common adverse effects reported by those in the 9.4% group included headache, dry eyes, burning sensation, dizziness, numbness, and cough.
Box 2
The effects of Cannabis
Marijuana is harvested from the plant Cannabis sativa and composed of 400 lipophilic chemical compounds, including phytocannabinoids, terpenoids, and flavonoids.13 The plant contains compounds termed “cannabinoids.” Two of these derivatives in particular are responsible for most of the effects of marijuana: cannabinoid delta-9- tetrahydrocannabinol (THC) and cannabidiol (CBD). THC has a comparable structure and binding mechanism to anandamide, a naturally occurring fatty acid neurotransmitter present within the human brain.14-16 The endogenous endocannabinoid system and its receptors are found throughout the entire body (brain, organs, glands, immune cells, and connective tissues).
THC binds to cannabinoid receptors CB1 and CB2. CB1 is found predominantly in the CNS. CB2 is found predominantly outside the CNS and is associated with the immune system.14-16 The effects of THC include euphoria, relaxation, appetite stimulation, improvement of nausea and vomiting, analgesia, decreased muscle spasticity, and reduced eye pressure.14,15 CBD may have anxiolytic, antipsychotic, anticonvulsive, and analgesic effects.
The rate of absorption of THC and CBD depends both on the potency of the cannabinoid as well as the mechanism of consumption. Cannabis can be administered by multiple routes, including via smoking, oral ingestion, or IV.16 When Cannabis is smoked (the route for the most rapid delivery), THC is transported from the lungs to the bloodstream and reaches peak concentrations in 3 to 10 minutes. Oral ingestion (capsules, tinctures, sprays, and edibles) has a more flexible onset of action, usually occurring in 30 to 120 minutes, with effects lasting 5 to 6 hours. IV administration has rapid effects; the onset can occur within seconds to minutes, and effects can last 2 to 3 hours. The IV form allows 90% of THC to be distributed in plasma and can rapidly penetrate highly vascularized tissues, such as the liver, heart, fat, lungs, and muscles.
Pharmaceutical manufacturers have used cannabinoid derivatives to produce Cannabis-based medications for treating medical conditions. Nabilone, a potent agonist of the CB1 receptor, became available as a Schedule II medication in 1981 and was approved for patients with chemotherapy-induced nausea and vomiting (CINV).17 In 1985, dronabinol was introduced as an antiemetic for CINV as well as an appetite stimulant for patients with conditions associated with excessive weight loss.18 Another option, nabiximols, is an oral mucosal spray that consists of THC and CBD in a 1:1 ratio.19 Nabiximols is approved in Canada for pain relief in end-stage cancer patients and pain associated with multiple sclerosis.19
In an RCT of vaporized Cannabis, 39 patients with a diagnosis of complex regional pain syndrome, thalamic pain, spinal cord injury, peripheral neuropathy, radiculopathy, or nerve injury were assigned to a medium-dose (3.53% THC), low-dose (1.29% THC), or placebo group.25 Serious mental illness, substance abuse, and medical conditions were cause for exclusion. Participants received vaporized marijuana (average 8 to 12 puffs per visit) over 3 sessions. A 30% pain reduction was achieved by 26% of those in the placebo group, 57% of those in the low-dose group, and 61% of individuals in the high-dose group; the difference between placebo and each Cannabis group was statistically significant.
Chemotherapy-induced nausea and vomiting. Up to 80% of patients who receive chemotherapy experience CINV, which occurs from 24 hours to 7 days after receiving such therapy.26 CINV negatively influences a patient’s QOL and may impact the decision to continue with chemotherapy. Use of MM can help to diminish vomiting by binding to central CB1 receptors and averting the proemetic effects of dopamine and serotonin.27 Two synthetically derived cannabinoids, dronabinol and nabilone, are FDA-approved for treating CINV.
In a small (N = 64) parallel-group RCT, Meiri et al27 compared dronabinol with the commonly used antiemetic ondansetron and with a combination of dronabinol and ondansetron for treating CINV in adults. The primary outcome was prevention of delayed-onset CINV. Patients were eligible for this study if they had a malignancy that did not involve bone marrow, were receiving treatment with a moderately to highly emetogenic regimen, were not pregnant, and had an estimated life expectancy of at least 6 weeks after chemotherapy. The patients were randomized to 1 of 4 treatment groups: dronabinol alone, ondansetron alone, dronabinol plus ondansetron, or placebo. Overall, 47% to 58% of the active treatment groups improved, compared with 20% of the placebo group. Combination therapy did not provide any benefit beyond any single agent alone. All active treatments reduced nausea compared with placebo; there was no difference between active treatment groups. This study was limited by low enrollment.
Tramèr et al28 conducted a systematic review of 30 randomized comparisons of MM with placebo or antiemetics. The reviewed studies were completed between 1975 to 1997 and analyzed a total of 1,366 patients. Nabilone was evaluated in 16 trials; dronabinol was utilized in 13 trials; and IM levonantradol, a synthetic cannabinoid analog of dronabinol, was used in 1 trial. These agents were found to be more effective as an antiemetic compared with prochlorperazine, metoclopramide, chlorpromazine, thiethylperazine, haloperidol, domperidone, or alizapride. In addition, 38% to 90% of patients in these studies preferred MM over the traditional antiemetics.
A Cochrane review29 suggested that MM may be a viable option for treatment-resistant CINV; however, further studies are needed because current studies have methodological limitations.
Epilepsy. Maa and Figi30 reported a case of a 5-year-old girl who had Dravet syndrome, which resulted in 50 generalized tonic-clonic seizures daily; multiple anticonvulsants did not alleviate these seizures. Because of her recurring seizures, the patient had multiple cognitive and motor delays and needed a feeding tube. In addition to her existing antiepileptic drug regimen, she was started on adjunctive therapy with a sublingual Cannabis extract containing a high concentration of CBD. Her seizures decreased from 50 per day to 2 to 3 nocturnal convulsions per month. The treatment enabled her to stop using a feeding tube, resume walking and talking, and sleep soundly.
dos Santos et al31 reviewed studies of MM for treating epilepsy. One was a double-blind, placebo-controlled trial that included 15 patients ages 14 to 49 who had secondary generalized epilepsy with a temporal lobe focus. Eight patients received 200 to 300 mg/d of oral CBD for 8 to 18 weeks, and 7 received placebo. Seven patients had fewer seizures and 4 had no seizures. Only 1 patient in the placebo group demonstrated any improvement. Another study in this review included 19 children with treatment-resistant epilepsy: Dravet syndrome (n = 13), Doose syndrome (n = 4), Lennox-Gastaut syndrome (n = 1), or idiopathic epilepsy (n = 1). These patients experienced various types of seizures with a frequency ranging from 2 per week to 250 per day. Overall, 84% of children treated with CBD had fewer seizures: 11% were seizure-free, 42% had a >80% reduction in seizures, and 32% had a 25% to 60% reduction in seizures. Parents also noted additional benefits, including increased attention, improved mood, and improved sleep. CBD was well tolerated in most patients in both studies.
Despite these results, a Cochrane review32 found that no reliable conclusions can be drawn regarding the efficacy of MM for treating epilepsy.
Multiple sclerosis. According to American Academy of Neurology guidelines, physicians may provide MM as an alternative treatment for patients with MS-related spasticity.33 Multiple studies have tested MM and MM-related extracts for treating spasticity related to MS.34,35 In a placebo-controlled crossover study, Corey-Bloom et al34 reported a significant reduction in spasticity, measured using the modified Ashworth scale, in MS patients receiving Cannabis cigarettes vs placebo cigarettes (P < .0001). However, compared with the placebo group, patients who received MM had significant adverse effects, primarily cognitive impairment (P = .003).
In a multicenter RCT (N = 572 patients with refractory MS spasticity), Novotna et al36 evaluated nabiximols, an oral mucosal spray of a formulated extract of Cannabis that contains THC and CBD in a 1:1 ratio. They assessed spasticity using the Numerical Spasticity Rating Scale (NRS). Results were confirmed by measuring the number of daily spasms, self-report of sleep quality, and activities of daily living. After 4 weeks of single-blind treatment, patients who responded to nabiximols (≥20% improvement in spasticity) were randomized to a placebo group or nabiximols group for 12 additional weeks. After 12 weeks, compared with those who received placebo, those in the nabiximols group experienced a statistically significant reduction in spasticity based on NRS score (P = .0002).
For a summary of evidence on MM for treating glaucoma, Crohn’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, see Box 3.37-43
Box 3
Cannabis for treating glaucoma, Crohn’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis
Glaucoma. In a placebo-controlled study, oromucosal administration of medical marijuana (MM) reduced intraocular pressure from 28 mm Hg to 22 mm Hg, with a duration of action of 3.5 hours.37However, the American Academy of Ophthalmologists does not recommend treating glaucoma with MM because the effect is short-lasting, and MM causes significant cognitive impairment compared with other standardized treatments.38 MM also leads to decreased blood pressure, which lowers blood flow to the optic nerve, thus increasing the risk of blindness.
Crohn’s disease. A randomized controlled trial (RCT) of MM for Crohn’s disease was conducted using the Crohn’s Disease Activity Index (CDAI) to assess for remission. In this 8-week study,21 individuals with Crohn’s disease were administered smoked MM (115 mg of delta-9-tetrahydrocannabinol [THC]) or placebo.39 Eligible patients were at least 20 years old, had active Crohn’s disease (CDAI >200), and had not responded to medical treatment for the illness. Compared with those who received placebo, patients who received MM experienced a statistically significant reduction in CDAI scores (P < .05). However, at follow-up 2 weeks after the study, when MM was no longer administered, there was no difference in mean CDAI scores between the 2 groups. Five of the 11 patients in the MM group achieved clinical remission, compared with 1 of 10 in the placebo group, but this difference was not statistically significant.
Parkinson’s disease (PD). According to the American Academy of Neurology, oral Cannabis extracts are “probably ineffective” for levodopa-induced dyskinesia in patients with PD.40 Reported benefits have come mainly from self-report studies. A 2014 survey (22 patients) found a significant reduction in PD symptoms—mainly relief from drug-induced tremor and pain—when measured using the Unified Parkinson’s Disease Rating Scale (UPDRS). Patients also reported better sleep and reduced pain (measured with a visual analog scale [VAS]). An exploratory double-blind placebo trial (N = 119) found no difference in mean UPDRS and no difference in any neuroprotective measures.41 However, the experimental group had a significantly higher quality of life (QOL; P = .05). A similar double-blind crossover study that included 19 patients found no significant difference in dyskinesia, as measured with the UPDRS, in the group receiving oral Cannabis extract compared with the placebo group.42
Amyotrophic lateral sclerosis (ALS). A randomized double-blind crossover trial of 27 ALS patients found that an oral THC extract (dronabinol, 5 mg, twice daily) had no significant effects on spasticity, as measured with the VAS.43 There was also no significant difference between the experimental and placebo groups on number of spasms (also measured with a VAS), quality of sleep (measured with the Sleep Disorders Questionnaire), or QOL (measured with the Amyotrophic Lateral Sclerosis Assessment questionnaire).
Psychiatric illnesses
Dementia-related behavioral disturbances. A few clinical trials with small sample sizes have found evidence supporting the use of MM compounds for alleviating neuropsychiatric symptoms of patients with dementia. An open-label pilot study of 6 individuals with late-stage dementia who received dronabinol, 2.5 mg/d, for 2 weeks, found a significant reduction (compared with baseline) in nighttime motor activity as measured with an actometer (P < .0028).44 The secondary Neuropsychiatric Inventory (NPI) assessment found reductions in aberrant motor behavior (P = .042), agitation (P = .042), and nighttime behaviors (P = .42).
A 2014 retrospective analysis of 40 inpatients with dementia-related agitation and appetite loss who were treated with dronabinol (mean dosage: 7.03 mg/d) found reductions in all aspects of agitation, including aberrant vocalization, motor agitation, aggressiveness, and treatment resistance, as measured with the Pittsburgh Agitation Scale (P < .0001).45 The study found no significant improvements in appetite, Global Assessment of Functioning mean score, or number of times patients awoke during the night. Adverse effects included sedation and delirium.
A RCT of 50 dementia patients with clinically relevant neuropsychiatric symptoms found no significant difference in mean NPI scores between patients given placebo and those who received nabiximols, 1.5 mg, 3 times daily.46 There were no significant differences found in agitation, QOL, life activities, or caregiver-scored Caregiver Global Impression of Change scale.
In a small RCT, THC was safe and well tolerated in 10 older patients with dementia.47 A 2009 Cochrane review48 concluded that there was no evidence for the efficacy of MM in treating the neuropsychiatric symptoms related to dementia.
PTSD. Preclinical evidence shows that the endocannabinoid system is involved in regulating emotional memory. Evidence also suggests that cannabinoids may facilitate the extinction of aversive memories.49,50
In 2009, New Mexico became the first state to authorize the use of MM for patients with PTSD. In a study of patients applying for the New Mexico Medical Cannabis Program, researchers used the Clinician Administered Posttraumatic Scale (CAPS) to assess PTSD symptoms.51 A retrospective chart review of the first 80 patients evaluated found significant (P < .0001) reductions of several PTSD symptoms, including intrusive memories, distressing dreams, flashbacks, numbing and avoidance, and hyperarousal, in the group using MM vs those not using MM. There also was a significant difference in CAPS total score (P < .0001). Patients reported a 75% reduction in PTSD symptoms while using MM. This study has several limitations: It was a retrospective review, not an RCT, and patients were prescreened and knew before the study began that MM helped their PTSD symptoms.
In another retrospective study, researchers evaluated treatment with nabilone, 0.5 to 6 mg/d, in 104 incarcerated men with various major mental illnesses; most (91%) met criteria for Cannabis dependence.52 They found significant improvements in sleep and PTSD symptoms.
A double-blind RCT evaluated MM in 10 Canadian male soldiers with PTSD who experienced nightmares despite standard medication treatment. Adjunctive nabilone (maximum dose: 3 mg/d) resulted in a reduction in nightmares as measured by the CAPS recurrent distressing dream of the event item score.53
Currently, there are no adequately powered RCTs of MM in a diverse group of PTSD patients. Most studies are open-label, enriched design, and included white male veterans. No well-conducted trials have evaluated patients with noncombat-related PTSD. Most of the relevant literature consists of case reports of Cannabis use by patients with PTSD.
Anxiety disorders.Patients frequently indicate that smoking Cannabis helps relieve their anxiety, although there is no replicated evidence based on double-blind RCTs to support this. However, in rat models CBD has been shown to facilitate extinction of conditioned fear via the endocannabinoid system.54-56 The mechanism of action is not completely understood. CBD has been shown to have antagonistic action at CB1 and CB2 receptors. It may have similar effects on memory extinction and may be an adjunct to exposure therapies for anxiety disorders.
Das et al57 studied the effects of CBD (32 mg) on extinction and consolidation of memory related to contextual fear in 48 individuals. They found that CBD can enhance extinction learning, and suggested it may have potential as an adjunct to extinction-based therapies for anxiety disorders.
Caveats: Adverse effects, lack of RCTs
Cannabis use causes impairment of learning, memory, attention, and working memory. Adolescents are particularly vulnerable to the effects of Cannabis on brain development at a time when synaptic pruning and increased myelination occur. Normal brain development could be disrupted. Some studies have linked Cannabis use to abnormalities in the amygdala, hippocampus, frontal lobe, and cerebellum. From 1995 to 2014, the potency of Cannabis (THC concentration) increased from 4% to 12%.58 This has substantial implications for increased abuse among adolescents and the deleterious effects of Cannabis on the brain.
Heavy Cannabis use impairs motivation and could precipitate psychosis in vulnerable individuals. Cannabis use may be linked to the development of schizophrenia.59
There are no well-conducted RCTs on the efficacy of MM, and adequate safety data are lacking. There is also lack of consensus among qualified experts. There is soft evidence that MM may be helpful in some medical conditions, including but not limited to CINV, neuropathic pain, epilepsy, and MS-related spasticity. Currently, the benefits of using MM do not appear to outweigh the risks.
Bottom Line
Limited evidence suggests medical marijuana (MM) may be beneficial for treating a few medical conditions, including neuropathic pain and chemotherapy-induced nausea and vomiting. There is no clear and convincing evidence MM is beneficial for psychiatric disorders, and Cannabis can impair cognition and attention and may precipitate psychosis. The risk of deleterious effects are greater in adolescents.
Related Resources
- Nguyen DH, Thant TM. Caring for medical marijuana patients who request controlled prescriptions. Current Psychiatry. 2017;16(8):50-51.
- National Institute on Drug Abuse. Marijuana as medicine. https://www.drugabuse.gov/publications/drugfacts/ marijuana-medicine.
Drug Brand Names
Alizapride • Litican, Superan
Chlorpromazine • Thorazine
Domperidone • Motilium
Dronabinol • Marinol, Syndros
Haloperidol • Haldol
Metoclopramide • Reglan
Nabilone • Cesamet
Nabiximols • Sativex
Ondansetron • Zofran, Zuplenz
Prochlorperazine • Compazine
Thiethylperazine • Torecan
There is a need for additional treatment options to improve symptoms, enhance the quality of life (QOL), and reduce suffering among patients who have chronic medical illness. Medical marijuana (MM) has the potential to help patients who have certain medical conditions in states where it is legal for prescription by a licensed medical provider.
Cannabis has a long history of medicinal use (Box 11-12). Two derivatives of the Cannabis plant—cannabinoid delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD)—are responsible for most of its effects. Some of these effects, including analgesia, decreased muscle spasticity, and reduced eye pressure, have been harnessed for their potential therapeutic effects (Box 213-19). As of November 2017, 29 states had legalized Cannabis for medical use, and several had legalized its recreational use.12
With the increasing availability of MM, psychiatrists are likely to encounter patients who are using it or who will ask them about it. This article reviews evidence related to using MM to treat patients with neuropathic pain; chemotherapyinduced nausea and vomiting (CINV); epilepsy; multiple sclerosis (MS); glaucoma; Crohn’s disease; Parkinson’s disease; amyotrophic lateral sclerosis; dementia-related behavioral disturbances; posttraumatic stress disorder (PTSD); and anxiety.
Box 1
Cannabis: A history of medicinal use
Cannabis has been cultivated since ancient times, beginning in China and India. The earliest reference of its use for healing purposes may have been in the Chinese Pharmacopeia, circa 1500 BC.1 In 1839, Dr. William Brooke O’Shaughnessy introduced Cannabis Indica, or “Indian hemp,” to the western world after a professorship in Calcutta, India.2 In the early 1840s, an English physician, Dr. John Clendinning, prescribed Cannabis for migraine headache.3 In the 19th and early 20th centuries, several prominent physicians advocated using Cannabis for migraines; Sir William Osler did so in his textbook, The principles and practice of medicine.4 It was listed in the U.S. Pharmacopeia in 1850 but removed in 1942.5,6
Until 1937, Cannabis was used in the United States for medicinal purposes, such as for treating inflamed skin, incontinence, and sexually transmitted diseases.7 In 1937, the Marihuana Tax Act, which prohibited the production, importation, possession, use, and dispersal of Cannabis, was passed.8 Cannabis became a Schedule I drug under the Controlled Substance Act of 1970.9
In 1999, based on available evidence, the Institute of Medicine (IOM) concluded Cannabis had less likelihood of dependence than benzodiazepines, opiates, cocaine, or nicotine. The IOM also concluded that the symptoms of withdrawal were mild in comparison with benzodiazepines or opiates. Finally, the IOM stated that Cannabis was not a “gateway” drug.10
In 1996, California was the first state to reimplement medicinal use of Cannabis under the Compassionate Use Act, also known as Proposition 215.11 This act allowed individuals to retain or produce Cannabis for personal consumption with a physician’s approval. Many states eventually followed California’s lead. As of November 2017, 29 states, the District of Columbia, Guam, and Puerto Rico had regulated Cannabis use for medical purposes,12 and recreational use had been approved in 7 states and the District of Columbia.
Medical illnesses
Neuropathic pain. Chronic neuropathic pain affects an estimated 7% to 8% of adults.20 Patients with neuropathic pain are often treated with anticonvulsants, antidepressants, opioids, and local anesthetics21; however, these medications may not provide substantial relief. Research has revealed that THC and CBD can improve central and peripheral neuropathic pain, as well as pain associated with rheumatoid arthritis and fibromyalgia.22
Wilsey et al23 evaluated the analgesic effects of smoked MM for neuropathic pain in a small (N = 38) double-blind, randomized controlled trial (RCT). Patients in this study had a preexisting diagnosis of complex regional pain syndrome, spinal cord injury, peripheral neuropathy, or nerve injury. To prevent any unforeseen adverse outcomes related to Cannabis use, participants were required to have previous exposure to Cannabis. Patients were excluded if they had major mental illness, substance abuse, or other major medical ailments.
Participants smoked high-dose Cannabis cigarettes (7% THC), low-dose Cannabis cigarettes (3.5% THC), or placebo cigarettes. Pain was measured on a visual analog scale (VAS) that ranged from 0 (no pain) to 100 (worst possible pain). Compared with the placebo group, significant analgesia was achieved in both Cannabis groups (P = .016). The high-dose group had greater neurocognitive impairment.
Ware et al24 conducted a crossover RCT (N = 23) to determine the efficacy of smoked MM for neuropathic pain. Participants had neuropathic pain for at least 3 months that was caused by trauma or surgery, with an average weekly pain intensity score >4 on scale of 0 to 10. Patients with pain due to cancer, nociceptive causes, unstable medical conditions, current substance abuse, history of a psychotic disorder, or suicidal ideation were excluded. Participants were assigned to a 9.4% THC group or a 0% THC group. Pain intensity was evaluated daily via telephone. Participants in the 9.4% THC group had statistically lower pain intensity compared with the 0% THC group (P = .023). Common adverse effects reported by those in the 9.4% group included headache, dry eyes, burning sensation, dizziness, numbness, and cough.
Box 2
The effects of Cannabis
Marijuana is harvested from the plant Cannabis sativa and composed of 400 lipophilic chemical compounds, including phytocannabinoids, terpenoids, and flavonoids.13 The plant contains compounds termed “cannabinoids.” Two of these derivatives in particular are responsible for most of the effects of marijuana: cannabinoid delta-9- tetrahydrocannabinol (THC) and cannabidiol (CBD). THC has a comparable structure and binding mechanism to anandamide, a naturally occurring fatty acid neurotransmitter present within the human brain.14-16 The endogenous endocannabinoid system and its receptors are found throughout the entire body (brain, organs, glands, immune cells, and connective tissues).
THC binds to cannabinoid receptors CB1 and CB2. CB1 is found predominantly in the CNS. CB2 is found predominantly outside the CNS and is associated with the immune system.14-16 The effects of THC include euphoria, relaxation, appetite stimulation, improvement of nausea and vomiting, analgesia, decreased muscle spasticity, and reduced eye pressure.14,15 CBD may have anxiolytic, antipsychotic, anticonvulsive, and analgesic effects.
The rate of absorption of THC and CBD depends both on the potency of the cannabinoid as well as the mechanism of consumption. Cannabis can be administered by multiple routes, including via smoking, oral ingestion, or IV.16 When Cannabis is smoked (the route for the most rapid delivery), THC is transported from the lungs to the bloodstream and reaches peak concentrations in 3 to 10 minutes. Oral ingestion (capsules, tinctures, sprays, and edibles) has a more flexible onset of action, usually occurring in 30 to 120 minutes, with effects lasting 5 to 6 hours. IV administration has rapid effects; the onset can occur within seconds to minutes, and effects can last 2 to 3 hours. The IV form allows 90% of THC to be distributed in plasma and can rapidly penetrate highly vascularized tissues, such as the liver, heart, fat, lungs, and muscles.
Pharmaceutical manufacturers have used cannabinoid derivatives to produce Cannabis-based medications for treating medical conditions. Nabilone, a potent agonist of the CB1 receptor, became available as a Schedule II medication in 1981 and was approved for patients with chemotherapy-induced nausea and vomiting (CINV).17 In 1985, dronabinol was introduced as an antiemetic for CINV as well as an appetite stimulant for patients with conditions associated with excessive weight loss.18 Another option, nabiximols, is an oral mucosal spray that consists of THC and CBD in a 1:1 ratio.19 Nabiximols is approved in Canada for pain relief in end-stage cancer patients and pain associated with multiple sclerosis.19
In an RCT of vaporized Cannabis, 39 patients with a diagnosis of complex regional pain syndrome, thalamic pain, spinal cord injury, peripheral neuropathy, radiculopathy, or nerve injury were assigned to a medium-dose (3.53% THC), low-dose (1.29% THC), or placebo group.25 Serious mental illness, substance abuse, and medical conditions were cause for exclusion. Participants received vaporized marijuana (average 8 to 12 puffs per visit) over 3 sessions. A 30% pain reduction was achieved by 26% of those in the placebo group, 57% of those in the low-dose group, and 61% of individuals in the high-dose group; the difference between placebo and each Cannabis group was statistically significant.
Chemotherapy-induced nausea and vomiting. Up to 80% of patients who receive chemotherapy experience CINV, which occurs from 24 hours to 7 days after receiving such therapy.26 CINV negatively influences a patient’s QOL and may impact the decision to continue with chemotherapy. Use of MM can help to diminish vomiting by binding to central CB1 receptors and averting the proemetic effects of dopamine and serotonin.27 Two synthetically derived cannabinoids, dronabinol and nabilone, are FDA-approved for treating CINV.
In a small (N = 64) parallel-group RCT, Meiri et al27 compared dronabinol with the commonly used antiemetic ondansetron and with a combination of dronabinol and ondansetron for treating CINV in adults. The primary outcome was prevention of delayed-onset CINV. Patients were eligible for this study if they had a malignancy that did not involve bone marrow, were receiving treatment with a moderately to highly emetogenic regimen, were not pregnant, and had an estimated life expectancy of at least 6 weeks after chemotherapy. The patients were randomized to 1 of 4 treatment groups: dronabinol alone, ondansetron alone, dronabinol plus ondansetron, or placebo. Overall, 47% to 58% of the active treatment groups improved, compared with 20% of the placebo group. Combination therapy did not provide any benefit beyond any single agent alone. All active treatments reduced nausea compared with placebo; there was no difference between active treatment groups. This study was limited by low enrollment.
Tramèr et al28 conducted a systematic review of 30 randomized comparisons of MM with placebo or antiemetics. The reviewed studies were completed between 1975 to 1997 and analyzed a total of 1,366 patients. Nabilone was evaluated in 16 trials; dronabinol was utilized in 13 trials; and IM levonantradol, a synthetic cannabinoid analog of dronabinol, was used in 1 trial. These agents were found to be more effective as an antiemetic compared with prochlorperazine, metoclopramide, chlorpromazine, thiethylperazine, haloperidol, domperidone, or alizapride. In addition, 38% to 90% of patients in these studies preferred MM over the traditional antiemetics.
A Cochrane review29 suggested that MM may be a viable option for treatment-resistant CINV; however, further studies are needed because current studies have methodological limitations.
Epilepsy. Maa and Figi30 reported a case of a 5-year-old girl who had Dravet syndrome, which resulted in 50 generalized tonic-clonic seizures daily; multiple anticonvulsants did not alleviate these seizures. Because of her recurring seizures, the patient had multiple cognitive and motor delays and needed a feeding tube. In addition to her existing antiepileptic drug regimen, she was started on adjunctive therapy with a sublingual Cannabis extract containing a high concentration of CBD. Her seizures decreased from 50 per day to 2 to 3 nocturnal convulsions per month. The treatment enabled her to stop using a feeding tube, resume walking and talking, and sleep soundly.
dos Santos et al31 reviewed studies of MM for treating epilepsy. One was a double-blind, placebo-controlled trial that included 15 patients ages 14 to 49 who had secondary generalized epilepsy with a temporal lobe focus. Eight patients received 200 to 300 mg/d of oral CBD for 8 to 18 weeks, and 7 received placebo. Seven patients had fewer seizures and 4 had no seizures. Only 1 patient in the placebo group demonstrated any improvement. Another study in this review included 19 children with treatment-resistant epilepsy: Dravet syndrome (n = 13), Doose syndrome (n = 4), Lennox-Gastaut syndrome (n = 1), or idiopathic epilepsy (n = 1). These patients experienced various types of seizures with a frequency ranging from 2 per week to 250 per day. Overall, 84% of children treated with CBD had fewer seizures: 11% were seizure-free, 42% had a >80% reduction in seizures, and 32% had a 25% to 60% reduction in seizures. Parents also noted additional benefits, including increased attention, improved mood, and improved sleep. CBD was well tolerated in most patients in both studies.
Despite these results, a Cochrane review32 found that no reliable conclusions can be drawn regarding the efficacy of MM for treating epilepsy.
Multiple sclerosis. According to American Academy of Neurology guidelines, physicians may provide MM as an alternative treatment for patients with MS-related spasticity.33 Multiple studies have tested MM and MM-related extracts for treating spasticity related to MS.34,35 In a placebo-controlled crossover study, Corey-Bloom et al34 reported a significant reduction in spasticity, measured using the modified Ashworth scale, in MS patients receiving Cannabis cigarettes vs placebo cigarettes (P < .0001). However, compared with the placebo group, patients who received MM had significant adverse effects, primarily cognitive impairment (P = .003).
In a multicenter RCT (N = 572 patients with refractory MS spasticity), Novotna et al36 evaluated nabiximols, an oral mucosal spray of a formulated extract of Cannabis that contains THC and CBD in a 1:1 ratio. They assessed spasticity using the Numerical Spasticity Rating Scale (NRS). Results were confirmed by measuring the number of daily spasms, self-report of sleep quality, and activities of daily living. After 4 weeks of single-blind treatment, patients who responded to nabiximols (≥20% improvement in spasticity) were randomized to a placebo group or nabiximols group for 12 additional weeks. After 12 weeks, compared with those who received placebo, those in the nabiximols group experienced a statistically significant reduction in spasticity based on NRS score (P = .0002).
For a summary of evidence on MM for treating glaucoma, Crohn’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, see Box 3.37-43
Box 3
Cannabis for treating glaucoma, Crohn’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis
Glaucoma. In a placebo-controlled study, oromucosal administration of medical marijuana (MM) reduced intraocular pressure from 28 mm Hg to 22 mm Hg, with a duration of action of 3.5 hours.37However, the American Academy of Ophthalmologists does not recommend treating glaucoma with MM because the effect is short-lasting, and MM causes significant cognitive impairment compared with other standardized treatments.38 MM also leads to decreased blood pressure, which lowers blood flow to the optic nerve, thus increasing the risk of blindness.
Crohn’s disease. A randomized controlled trial (RCT) of MM for Crohn’s disease was conducted using the Crohn’s Disease Activity Index (CDAI) to assess for remission. In this 8-week study,21 individuals with Crohn’s disease were administered smoked MM (115 mg of delta-9-tetrahydrocannabinol [THC]) or placebo.39 Eligible patients were at least 20 years old, had active Crohn’s disease (CDAI >200), and had not responded to medical treatment for the illness. Compared with those who received placebo, patients who received MM experienced a statistically significant reduction in CDAI scores (P < .05). However, at follow-up 2 weeks after the study, when MM was no longer administered, there was no difference in mean CDAI scores between the 2 groups. Five of the 11 patients in the MM group achieved clinical remission, compared with 1 of 10 in the placebo group, but this difference was not statistically significant.
Parkinson’s disease (PD). According to the American Academy of Neurology, oral Cannabis extracts are “probably ineffective” for levodopa-induced dyskinesia in patients with PD.40 Reported benefits have come mainly from self-report studies. A 2014 survey (22 patients) found a significant reduction in PD symptoms—mainly relief from drug-induced tremor and pain—when measured using the Unified Parkinson’s Disease Rating Scale (UPDRS). Patients also reported better sleep and reduced pain (measured with a visual analog scale [VAS]). An exploratory double-blind placebo trial (N = 119) found no difference in mean UPDRS and no difference in any neuroprotective measures.41 However, the experimental group had a significantly higher quality of life (QOL; P = .05). A similar double-blind crossover study that included 19 patients found no significant difference in dyskinesia, as measured with the UPDRS, in the group receiving oral Cannabis extract compared with the placebo group.42
Amyotrophic lateral sclerosis (ALS). A randomized double-blind crossover trial of 27 ALS patients found that an oral THC extract (dronabinol, 5 mg, twice daily) had no significant effects on spasticity, as measured with the VAS.43 There was also no significant difference between the experimental and placebo groups on number of spasms (also measured with a VAS), quality of sleep (measured with the Sleep Disorders Questionnaire), or QOL (measured with the Amyotrophic Lateral Sclerosis Assessment questionnaire).
Psychiatric illnesses
Dementia-related behavioral disturbances. A few clinical trials with small sample sizes have found evidence supporting the use of MM compounds for alleviating neuropsychiatric symptoms of patients with dementia. An open-label pilot study of 6 individuals with late-stage dementia who received dronabinol, 2.5 mg/d, for 2 weeks, found a significant reduction (compared with baseline) in nighttime motor activity as measured with an actometer (P < .0028).44 The secondary Neuropsychiatric Inventory (NPI) assessment found reductions in aberrant motor behavior (P = .042), agitation (P = .042), and nighttime behaviors (P = .42).
A 2014 retrospective analysis of 40 inpatients with dementia-related agitation and appetite loss who were treated with dronabinol (mean dosage: 7.03 mg/d) found reductions in all aspects of agitation, including aberrant vocalization, motor agitation, aggressiveness, and treatment resistance, as measured with the Pittsburgh Agitation Scale (P < .0001).45 The study found no significant improvements in appetite, Global Assessment of Functioning mean score, or number of times patients awoke during the night. Adverse effects included sedation and delirium.
A RCT of 50 dementia patients with clinically relevant neuropsychiatric symptoms found no significant difference in mean NPI scores between patients given placebo and those who received nabiximols, 1.5 mg, 3 times daily.46 There were no significant differences found in agitation, QOL, life activities, or caregiver-scored Caregiver Global Impression of Change scale.
In a small RCT, THC was safe and well tolerated in 10 older patients with dementia.47 A 2009 Cochrane review48 concluded that there was no evidence for the efficacy of MM in treating the neuropsychiatric symptoms related to dementia.
PTSD. Preclinical evidence shows that the endocannabinoid system is involved in regulating emotional memory. Evidence also suggests that cannabinoids may facilitate the extinction of aversive memories.49,50
In 2009, New Mexico became the first state to authorize the use of MM for patients with PTSD. In a study of patients applying for the New Mexico Medical Cannabis Program, researchers used the Clinician Administered Posttraumatic Scale (CAPS) to assess PTSD symptoms.51 A retrospective chart review of the first 80 patients evaluated found significant (P < .0001) reductions of several PTSD symptoms, including intrusive memories, distressing dreams, flashbacks, numbing and avoidance, and hyperarousal, in the group using MM vs those not using MM. There also was a significant difference in CAPS total score (P < .0001). Patients reported a 75% reduction in PTSD symptoms while using MM. This study has several limitations: It was a retrospective review, not an RCT, and patients were prescreened and knew before the study began that MM helped their PTSD symptoms.
In another retrospective study, researchers evaluated treatment with nabilone, 0.5 to 6 mg/d, in 104 incarcerated men with various major mental illnesses; most (91%) met criteria for Cannabis dependence.52 They found significant improvements in sleep and PTSD symptoms.
A double-blind RCT evaluated MM in 10 Canadian male soldiers with PTSD who experienced nightmares despite standard medication treatment. Adjunctive nabilone (maximum dose: 3 mg/d) resulted in a reduction in nightmares as measured by the CAPS recurrent distressing dream of the event item score.53
Currently, there are no adequately powered RCTs of MM in a diverse group of PTSD patients. Most studies are open-label, enriched design, and included white male veterans. No well-conducted trials have evaluated patients with noncombat-related PTSD. Most of the relevant literature consists of case reports of Cannabis use by patients with PTSD.
Anxiety disorders.Patients frequently indicate that smoking Cannabis helps relieve their anxiety, although there is no replicated evidence based on double-blind RCTs to support this. However, in rat models CBD has been shown to facilitate extinction of conditioned fear via the endocannabinoid system.54-56 The mechanism of action is not completely understood. CBD has been shown to have antagonistic action at CB1 and CB2 receptors. It may have similar effects on memory extinction and may be an adjunct to exposure therapies for anxiety disorders.
Das et al57 studied the effects of CBD (32 mg) on extinction and consolidation of memory related to contextual fear in 48 individuals. They found that CBD can enhance extinction learning, and suggested it may have potential as an adjunct to extinction-based therapies for anxiety disorders.
Caveats: Adverse effects, lack of RCTs
Cannabis use causes impairment of learning, memory, attention, and working memory. Adolescents are particularly vulnerable to the effects of Cannabis on brain development at a time when synaptic pruning and increased myelination occur. Normal brain development could be disrupted. Some studies have linked Cannabis use to abnormalities in the amygdala, hippocampus, frontal lobe, and cerebellum. From 1995 to 2014, the potency of Cannabis (THC concentration) increased from 4% to 12%.58 This has substantial implications for increased abuse among adolescents and the deleterious effects of Cannabis on the brain.
Heavy Cannabis use impairs motivation and could precipitate psychosis in vulnerable individuals. Cannabis use may be linked to the development of schizophrenia.59
There are no well-conducted RCTs on the efficacy of MM, and adequate safety data are lacking. There is also lack of consensus among qualified experts. There is soft evidence that MM may be helpful in some medical conditions, including but not limited to CINV, neuropathic pain, epilepsy, and MS-related spasticity. Currently, the benefits of using MM do not appear to outweigh the risks.
Bottom Line
Limited evidence suggests medical marijuana (MM) may be beneficial for treating a few medical conditions, including neuropathic pain and chemotherapy-induced nausea and vomiting. There is no clear and convincing evidence MM is beneficial for psychiatric disorders, and Cannabis can impair cognition and attention and may precipitate psychosis. The risk of deleterious effects are greater in adolescents.
Related Resources
- Nguyen DH, Thant TM. Caring for medical marijuana patients who request controlled prescriptions. Current Psychiatry. 2017;16(8):50-51.
- National Institute on Drug Abuse. Marijuana as medicine. https://www.drugabuse.gov/publications/drugfacts/ marijuana-medicine.
Drug Brand Names
Alizapride • Litican, Superan
Chlorpromazine • Thorazine
Domperidone • Motilium
Dronabinol • Marinol, Syndros
Haloperidol • Haldol
Metoclopramide • Reglan
Nabilone • Cesamet
Nabiximols • Sativex
Ondansetron • Zofran, Zuplenz
Prochlorperazine • Compazine
Thiethylperazine • Torecan
1. National Institute on Drug Abuse (NIDA). Marijuana Research Findings 1976. NIDA research monograph 14. https://archives.drugabuse.gov/sites/default/files/monograph14.pdf. Published July 1977. Accessed November 15, 2017.
2. O’Shaughnessy WB. On the preparations of the Indian hemp, or gunjah- cannabis indica their effects on the animal system in health, and their utility in the treatment of tetanus and other convulsive diseases. Prov Med J Retrosp Med Sci. 1843;5(123):363-369.
3. Clendinning J. Observations on the medical properties of the Cannabis Sativa of India. Med Chir Trans. 1843;26:188-210.
4. Osler W, McCrae T. The principles and practice of medicine. 9th ed. New York, NY: D. Appleton and Company; 1921.
5. The pharmacopoeia of the United States of America. 3rd ed. Philadelphia, PA: Lippincott; 1851.
6. The pharmacopoeia of the United States of America. 12th ed. Easton, PA: Mack Printing Company; 1942.
7. Philipsen N, Butler RD, Simon C, et al. Medical marijuana: a primer on ethics, evidence, and politics. Journal Nurse Pract. 2014;10(9):633-640.
8. Marihuana Tax Act of 1937, Pub L No. 75-238, 75th Cong, 50 Stat 551 (1937).
9. Controlled Substances Act, 21 USC §812.
10. Watson SJ, Benson JA, Joy JE, eds. Marijuana and medicine: assessing the science base. Washington, DC: National Academy Press; 1999.
11. California Proposition 215, the medical marijuana initiative (1996). https://ballotpedia.org/California_Proposition_215,_the_Medical_Marijuana_Initiative_(1996). Accessed November 16, 2017.
12. National Conference of State Legislatures. State medical marijuana laws. http://www.ncsl.org/research/health/state-medical-marijuana-laws.aspx. Updated September 14, 2017. Accessed November 16, 2017.
13. Russo EB. Taming THC: potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br J Pharmacol. 2011;163(7):1344-1364.
14. Alger BE. Getting high on the endocannabinoid system. Cerebrum. 2013:14. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3997295. Accessed December 5, 2017.
15. Galal AM, Slade D, Gul W, et al. Naturally occurring and related synthetic cannabinoids and their potential therapeutic applications. Recent Pat CNS Drug Discov. 2009;4(2):112-136.
16. Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. 2007;4(8):1770-1804.
17. Cesamet [package insert]. Somerset, NJ: Meda Pharmaceuticals; 2013.
18. Marinol [package insert]. Chicago, IL: AbbVie Inc.; 2017.
19. Sativex [package insert]. Mississauga, Ontario: Bayer Inc.; 2015.
20. Torrance N, Ferguson JA, Afolabi E, et al. Neuropathic pain in the community: more under-treated than refractory? Pain. 2013;154(5):690-699.
21. Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14(2):162-173.
22. Whiting PF, Wolff RF, Deshpande S, et al. Cannabinoids for medical use: a systematic review and meta-analysis. JAMA. 2015;313(24):2456-2473.
23. Wilsey B, Marcotte, T, Tsodikov A, et al. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. J Pain. 2008;9(6):506-521.
24. Ware MA, Wang T, Shapiro S, et al. Smoked cannabis for chronic neuropathic pain: a randomized controlled trial. CMAJ. 2010;182(14):E694-E701.
25. Wilsey B, Marcotte T, Deutsch R, et al. Low-dose vaporized cannabis significantly improves neuropathic pain. J Pain. 2013;14(2):136-148.
26. National Cancer Institute. Treatment-related nausea and vomiting (PDQ®)-health professional version. https://www.cancer.gov/about-cancer/treatment/side-effects/nausea/nausea-hp-pdq. Updated May 10, 2017. Accessed November 7, 2017.
27. Meiri E, Jhangiani H, Vrendenburgh JJ, et al. Efficacy of dronabinol alone and in combination with ondansetron versus ondansetron alone for delayed chemotherapy-induced nausea and vomiting. Curr Med Res Opin. 2007;23(3):533-543.
28. Tramèr MR, Carroll D, Campbell FA, et al. Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review. BMJ. 2001;323(7303):16-21.
29. Smith LA, Azariah F, Lavender VT, et al. Cannabinoids for nausea and vomiting in adults with cancer receiving chemotherapy. Cochrane Database Syst Rev. 2015;(11):CD009464.
30. Maa E, Figi P. The case for medical marijuana in epilepsy. Epilepsia. 2014;55(6):783-786.
31. dos Santos RG, Hallak JE, Leite JP, et al. Phytocannabinoids and epilepsy. J Clin Pharm Ther. 2015;40(2):135-143.
32. Gloss D, Vickrey B. Cannabinoids for epilepsy. Cochrane Database Syst Rev. 2014;(3):CD009270.
33. Yadav V, Bever C Jr, Bowen J, et al. Summary of evidence-based guideline: complementary and alternative medicine in multiple sclerosis: report of the guideline development subcommittee of the American Academy of Neurology. Neurology. 2014;82(12):1083-1092.
34. Corey-Bloom J, Wolfson T, Gamst A, et al. Smoked cannabis for spasticity in multiple sclerosis: a randomized, placebo-controlled trial. CMAJ. 2012;184(10):1143-1150.
35. Zajicek J, Ball S, Wright D, et al; CUPID investigator group. Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): a randomised, placebo-controlled trial. Lancet Neurol. 2013;12(9):857-865.
36. Novotna A, Mares J, Ratcliffe S, et al; Sativex Spasticity Study Group. A randomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols* (Sativex(®)), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. Eur J Neurol. 2011;18(9):1122-1131.
37. Merritt JC, Crawford WJ, Alexander PC, et al. Effect of marihuana on intraocular and blood pressure in glaucoma. Ophthalmology. 1980;87(3):222-228.
38. American Academy of Ophthalmology. American Academy of Ophthalmology reiterates position that marijuana is not a proven treatment for glaucoma. https://www.aao.org/newsroom/news-releases/detail/american-academy-of-ophthalmology-reiterates-posit. Published June 27, 2014. Accessed May 29, 2017.
39. Naftali T, Bar-Lev Schleider L, Dotan I, et al. Cannabis induces a clinical response in patients with Crohn’s disease: a prospective placebo-controlled study. Clin Gastroenterol Hepatol. 2013;11(10):1276.e1-1280.e1.
40. Koppel BS Brust JC, Fife T, et al. Systematic review: efficacy and safety of medical marijuana in certain neurological disorders. Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2014;82(17):1556-1563.
41. Chagas MH, Zuardi AW, Tumas V, et al. Effects of cannabidiol in the treatment of patients with Parkinson’s disease: an exploratory double-blind trial. J Psychopharmacol. 2014;28(11):1088-1098.
42. Carroll CB, Bain PG, Teare L, et al. Cannabis for dyskinesia in Parkinson disease: a randomized double-blind crossover study. Neurology. 2004;63(7):1245-1250.
43. Weber M, Goldman B, Truniger S. Tetrahydrocannabinol (THC) for cramps in amyotrophic lateral sclerosis: a randomised, double-blind crossover trial. J Neurol Neurosurg Psychiatry. 2010;81(10):1135-1140.
44. Walther S, Mahlberg R, Eichmann U, et al. Delta-9-tetrahydrocannabinol for nighttime agitation in severe dementia. Psychopharmacology (Berl). 2006;185(4):524-528.
45. Woodward MR, Harper DG, Stolyar A, et al. Dronabinol for the treatment of agitation and aggressive behavior in acutely hospitalized severely demented patients with noncognitive behavioral symptoms. Am J Geriatr Psychiatry. 2014;22(4):415-419.
46. van den Elsen GA, Ahmed A, Verkes RJ, et al. Tetrahydrocannabinol for neuropsychiatric symptoms in dementia: a randomized controlled trial. Neurology. 2015;84(23):2338-2346.
47. Ahmed AI, van den Elsen GA, Colbers A, et al. Safety, pharmacodynamics, and pharmacokinetics of multiple oral doses of delta-9-tetrahydrocannabinol in older persons with dementia. Psychopharmacology (Berl). 2015;232(14):25872595.
48. Krishnan S, Cairns R, Howard R. Cannabinoids for the treatment of dementia. Cochrane Database Syst Rev. 2009;(2):CD007204.
49. de Bitencourt RM, Pamplona FA, Takahashi RN. A current overview of cannabinoids and glucocorticoids in facilitating extinction of aversive memories: potential extinction enhancers. Neuropharmacology. 2013;64:389-395.
50. Fraser GA. The use of a synthetic cannabinoid in the management of treatment-resistant nightmares in posttraumatic stress disorder (PTSD). CNS Neurosci Ther. 2009;15(1):84-88.
51. Greer GR, Grob CS, Halberstadt AL. PTSD symptom reports of patients evaluated for the New Mexico Medical Cannabis Program. J Psychoactive Drugs. 2014;46(1):73-77.
52. Cameron C, Watson D, Robinson J. Use of a synthetic cannabinoid in a correctional population for posttraumatic stress disorder-related insomnia and nightmares, chronic pain, harm reduction, and other indications: a retrospective evaluation. J Clin Psychopharmacol. 2014;34(5):559-564.
53. Jetly R, Heber A, Fraser G, et al. The efficacy of nabilone, a synthetic cannabinoid, in the treatment of PTSD-associated nightmares: a preliminary randomized, double-blind, placebo-controlled cross-over design study. Psychoneuroendocrinology. 2015;51:585-588.
54. Bitencourt RM, Pamplona FA, Takahashi RN. Facilitation of contextual fear, memory extinction, and anti-anxiogenic effects of AM404 and cannabidiol in conditioned rats. Eur Neuropsychopharmacol. 2008;18(12):849-859.
55. Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153(2):199-215.
56. Thomas A, Baillie GL, Phillips AM, et al. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol. 2007;150(5):613-623.
57. Das RK, Kamboj SK, Ramadas M, et al. Cannabidiol enhances consolidation of explicit fear extinction in humans. Psychopharmacology (Berl). 2013;226(4):781-792.
58. ElSohly MA, Mehmedic Z, Foster S, et al. Changes in cannabis potency over the last 2 decades (1995-2014): analysis of current data in the United States. Biol Psychiatry. 2016;79(7):613-619.
59. Volkow ND, Swanson JM, Evins AE, et al. Effects of cannabis use on human behavior, including cognition, motivation, and psychosis: a review. JAMA Psychiatry. 2016;73(3):292297.
1. National Institute on Drug Abuse (NIDA). Marijuana Research Findings 1976. NIDA research monograph 14. https://archives.drugabuse.gov/sites/default/files/monograph14.pdf. Published July 1977. Accessed November 15, 2017.
2. O’Shaughnessy WB. On the preparations of the Indian hemp, or gunjah- cannabis indica their effects on the animal system in health, and their utility in the treatment of tetanus and other convulsive diseases. Prov Med J Retrosp Med Sci. 1843;5(123):363-369.
3. Clendinning J. Observations on the medical properties of the Cannabis Sativa of India. Med Chir Trans. 1843;26:188-210.
4. Osler W, McCrae T. The principles and practice of medicine. 9th ed. New York, NY: D. Appleton and Company; 1921.
5. The pharmacopoeia of the United States of America. 3rd ed. Philadelphia, PA: Lippincott; 1851.
6. The pharmacopoeia of the United States of America. 12th ed. Easton, PA: Mack Printing Company; 1942.
7. Philipsen N, Butler RD, Simon C, et al. Medical marijuana: a primer on ethics, evidence, and politics. Journal Nurse Pract. 2014;10(9):633-640.
8. Marihuana Tax Act of 1937, Pub L No. 75-238, 75th Cong, 50 Stat 551 (1937).
9. Controlled Substances Act, 21 USC §812.
10. Watson SJ, Benson JA, Joy JE, eds. Marijuana and medicine: assessing the science base. Washington, DC: National Academy Press; 1999.
11. California Proposition 215, the medical marijuana initiative (1996). https://ballotpedia.org/California_Proposition_215,_the_Medical_Marijuana_Initiative_(1996). Accessed November 16, 2017.
12. National Conference of State Legislatures. State medical marijuana laws. http://www.ncsl.org/research/health/state-medical-marijuana-laws.aspx. Updated September 14, 2017. Accessed November 16, 2017.
13. Russo EB. Taming THC: potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br J Pharmacol. 2011;163(7):1344-1364.
14. Alger BE. Getting high on the endocannabinoid system. Cerebrum. 2013:14. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3997295. Accessed December 5, 2017.
15. Galal AM, Slade D, Gul W, et al. Naturally occurring and related synthetic cannabinoids and their potential therapeutic applications. Recent Pat CNS Drug Discov. 2009;4(2):112-136.
16. Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. 2007;4(8):1770-1804.
17. Cesamet [package insert]. Somerset, NJ: Meda Pharmaceuticals; 2013.
18. Marinol [package insert]. Chicago, IL: AbbVie Inc.; 2017.
19. Sativex [package insert]. Mississauga, Ontario: Bayer Inc.; 2015.
20. Torrance N, Ferguson JA, Afolabi E, et al. Neuropathic pain in the community: more under-treated than refractory? Pain. 2013;154(5):690-699.
21. Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14(2):162-173.
22. Whiting PF, Wolff RF, Deshpande S, et al. Cannabinoids for medical use: a systematic review and meta-analysis. JAMA. 2015;313(24):2456-2473.
23. Wilsey B, Marcotte, T, Tsodikov A, et al. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. J Pain. 2008;9(6):506-521.
24. Ware MA, Wang T, Shapiro S, et al. Smoked cannabis for chronic neuropathic pain: a randomized controlled trial. CMAJ. 2010;182(14):E694-E701.
25. Wilsey B, Marcotte T, Deutsch R, et al. Low-dose vaporized cannabis significantly improves neuropathic pain. J Pain. 2013;14(2):136-148.
26. National Cancer Institute. Treatment-related nausea and vomiting (PDQ®)-health professional version. https://www.cancer.gov/about-cancer/treatment/side-effects/nausea/nausea-hp-pdq. Updated May 10, 2017. Accessed November 7, 2017.
27. Meiri E, Jhangiani H, Vrendenburgh JJ, et al. Efficacy of dronabinol alone and in combination with ondansetron versus ondansetron alone for delayed chemotherapy-induced nausea and vomiting. Curr Med Res Opin. 2007;23(3):533-543.
28. Tramèr MR, Carroll D, Campbell FA, et al. Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review. BMJ. 2001;323(7303):16-21.
29. Smith LA, Azariah F, Lavender VT, et al. Cannabinoids for nausea and vomiting in adults with cancer receiving chemotherapy. Cochrane Database Syst Rev. 2015;(11):CD009464.
30. Maa E, Figi P. The case for medical marijuana in epilepsy. Epilepsia. 2014;55(6):783-786.
31. dos Santos RG, Hallak JE, Leite JP, et al. Phytocannabinoids and epilepsy. J Clin Pharm Ther. 2015;40(2):135-143.
32. Gloss D, Vickrey B. Cannabinoids for epilepsy. Cochrane Database Syst Rev. 2014;(3):CD009270.
33. Yadav V, Bever C Jr, Bowen J, et al. Summary of evidence-based guideline: complementary and alternative medicine in multiple sclerosis: report of the guideline development subcommittee of the American Academy of Neurology. Neurology. 2014;82(12):1083-1092.
34. Corey-Bloom J, Wolfson T, Gamst A, et al. Smoked cannabis for spasticity in multiple sclerosis: a randomized, placebo-controlled trial. CMAJ. 2012;184(10):1143-1150.
35. Zajicek J, Ball S, Wright D, et al; CUPID investigator group. Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): a randomised, placebo-controlled trial. Lancet Neurol. 2013;12(9):857-865.
36. Novotna A, Mares J, Ratcliffe S, et al; Sativex Spasticity Study Group. A randomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols* (Sativex(®)), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. Eur J Neurol. 2011;18(9):1122-1131.
37. Merritt JC, Crawford WJ, Alexander PC, et al. Effect of marihuana on intraocular and blood pressure in glaucoma. Ophthalmology. 1980;87(3):222-228.
38. American Academy of Ophthalmology. American Academy of Ophthalmology reiterates position that marijuana is not a proven treatment for glaucoma. https://www.aao.org/newsroom/news-releases/detail/american-academy-of-ophthalmology-reiterates-posit. Published June 27, 2014. Accessed May 29, 2017.
39. Naftali T, Bar-Lev Schleider L, Dotan I, et al. Cannabis induces a clinical response in patients with Crohn’s disease: a prospective placebo-controlled study. Clin Gastroenterol Hepatol. 2013;11(10):1276.e1-1280.e1.
40. Koppel BS Brust JC, Fife T, et al. Systematic review: efficacy and safety of medical marijuana in certain neurological disorders. Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2014;82(17):1556-1563.
41. Chagas MH, Zuardi AW, Tumas V, et al. Effects of cannabidiol in the treatment of patients with Parkinson’s disease: an exploratory double-blind trial. J Psychopharmacol. 2014;28(11):1088-1098.
42. Carroll CB, Bain PG, Teare L, et al. Cannabis for dyskinesia in Parkinson disease: a randomized double-blind crossover study. Neurology. 2004;63(7):1245-1250.
43. Weber M, Goldman B, Truniger S. Tetrahydrocannabinol (THC) for cramps in amyotrophic lateral sclerosis: a randomised, double-blind crossover trial. J Neurol Neurosurg Psychiatry. 2010;81(10):1135-1140.
44. Walther S, Mahlberg R, Eichmann U, et al. Delta-9-tetrahydrocannabinol for nighttime agitation in severe dementia. Psychopharmacology (Berl). 2006;185(4):524-528.
45. Woodward MR, Harper DG, Stolyar A, et al. Dronabinol for the treatment of agitation and aggressive behavior in acutely hospitalized severely demented patients with noncognitive behavioral symptoms. Am J Geriatr Psychiatry. 2014;22(4):415-419.
46. van den Elsen GA, Ahmed A, Verkes RJ, et al. Tetrahydrocannabinol for neuropsychiatric symptoms in dementia: a randomized controlled trial. Neurology. 2015;84(23):2338-2346.
47. Ahmed AI, van den Elsen GA, Colbers A, et al. Safety, pharmacodynamics, and pharmacokinetics of multiple oral doses of delta-9-tetrahydrocannabinol in older persons with dementia. Psychopharmacology (Berl). 2015;232(14):25872595.
48. Krishnan S, Cairns R, Howard R. Cannabinoids for the treatment of dementia. Cochrane Database Syst Rev. 2009;(2):CD007204.
49. de Bitencourt RM, Pamplona FA, Takahashi RN. A current overview of cannabinoids and glucocorticoids in facilitating extinction of aversive memories: potential extinction enhancers. Neuropharmacology. 2013;64:389-395.
50. Fraser GA. The use of a synthetic cannabinoid in the management of treatment-resistant nightmares in posttraumatic stress disorder (PTSD). CNS Neurosci Ther. 2009;15(1):84-88.
51. Greer GR, Grob CS, Halberstadt AL. PTSD symptom reports of patients evaluated for the New Mexico Medical Cannabis Program. J Psychoactive Drugs. 2014;46(1):73-77.
52. Cameron C, Watson D, Robinson J. Use of a synthetic cannabinoid in a correctional population for posttraumatic stress disorder-related insomnia and nightmares, chronic pain, harm reduction, and other indications: a retrospective evaluation. J Clin Psychopharmacol. 2014;34(5):559-564.
53. Jetly R, Heber A, Fraser G, et al. The efficacy of nabilone, a synthetic cannabinoid, in the treatment of PTSD-associated nightmares: a preliminary randomized, double-blind, placebo-controlled cross-over design study. Psychoneuroendocrinology. 2015;51:585-588.
54. Bitencourt RM, Pamplona FA, Takahashi RN. Facilitation of contextual fear, memory extinction, and anti-anxiogenic effects of AM404 and cannabidiol in conditioned rats. Eur Neuropsychopharmacol. 2008;18(12):849-859.
55. Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153(2):199-215.
56. Thomas A, Baillie GL, Phillips AM, et al. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol. 2007;150(5):613-623.
57. Das RK, Kamboj SK, Ramadas M, et al. Cannabidiol enhances consolidation of explicit fear extinction in humans. Psychopharmacology (Berl). 2013;226(4):781-792.
58. ElSohly MA, Mehmedic Z, Foster S, et al. Changes in cannabis potency over the last 2 decades (1995-2014): analysis of current data in the United States. Biol Psychiatry. 2016;79(7):613-619.
59. Volkow ND, Swanson JM, Evins AE, et al. Effects of cannabis use on human behavior, including cognition, motivation, and psychosis: a review. JAMA Psychiatry. 2016;73(3):292297.

The evidence on medical marijuana

A concise guide to monoamine oxidase inhibitors: How to avoid drug interactions
Monoamine oxidase inhibitors (MAOIs) have well-established efficacy for treating depression, panic disorder, and social phobia. However, a lack of familiarity with these agents and misconceptions about the risks associated with their use have led to MAOIs being substantially underutilized. The goal of this 2-part guide to MAOIs is to educate clinicians about this often-overlooked class of medications. Part 1 (“A concise guide to monoamine inhibitors,”
MAOIs and potential drug interactions
One source of concern in patients receiving irreversible nonselective MAOIs is the development of excessive serotonergic neurotransmission resulting in SS. In the 1960s, researchers noted that administering large doses of
- mild symptoms: tremor, akathisia, inducible clonus
- moderate symptoms: spontaneous or sustained clonus, muscular hypertonicity
- severe symptoms: hyperthermia, diaphoresis.2
Although SS can be induced by significant exposure to individual agents that promote excess synaptic serotonin (eg, overdose of selective serotonin reuptake inhibitors [SSRIs]), the majority of fatal cases have occurred among patients taking MAOIs who were coadministered an agent that inhibited serotonin reuptake (Table 13). Animal studies have determined that excessive stimulation of the 5HT2A receptor is primarily responsible for SS,4 and that 5HT2A antagonists, such as mirtazapine, can block the development of SS in a mouse coadministered

Risk for SS. Most medications that promote serotonergic activity are well known for their use as antidepressants, but other agents that have 5HT reuptake properties (eg, the antihistamine chlorpheniramine) must be avoided. Although older literature suggests that the use of lower doses of certain tricyclic antidepressants concurrently with MAOIs may not be as dangerous as once believed,6 there are sufficient reports of serious outcomes that tricyclics should be avoided in patients taking MAOIs because of the risk of SS, and also because, in general, tricyclics are poorly tolerated.7
Desipramine, a potent norepinephrine transporter (NET) inhibitor, blocks the entry of tyramine into cells by NET, thereby preventing hypertensive events in animal models of tyramine overexposure. However, in some assays, the affinity for the serotonin transporter is not insignificant, so at higher doses desipramine may pose the same theoretical risk for SS as seen with other tricyclics.3
Lastly
Astute clinicians will recognize that antidepressants that lack 5HT reuptake (eg, bupropion, mirtazapine) are not on this list of agents that may increase SS risk when taken with an MAOI. Older papers often list mirtazapine, but as a 5HT2A antagonist, it does not possess a plausible mechanism by which it can induce 5HT toxicity.9,10 Most atypical antipsychotics have significant 5HT2A antagonism and can be combined with MAOIs, but ziprasidone is an exception: as a moderate SNRI, it has been associated with SS when administered with an MAOI.11
Pressor reactions. The only theoretical sources of concern for pressor effects are medications that act as norepinephrine releasers through interactions at the trace amine-associated receptor 1 (TAAR1) (for more information on TAAR1, see
Starting a patient on an MAOI
Contraindicated medications need to be tapered before beginning MAOI treatment. The duration of the washout period depends on the half-life of the medication and any active metabolites. Antidepressants with half-lives of approximately ≤24 hours should be tapered over 7 to 14 days (depending on the dose) to minimize the risk of withdrawal syndromes, while those with long half-lives (eg, fluoxetine,
Initiation of an MAOI is always based on whether the patient can reliably follow the basic dietary advice (see “A concise guide to monoamine inhibitors,”
The orthostasis management strategy is similar to that employed for
Augmentation options for patients taking MAOIs
For depressed patients who do not achieve remission of symptoms from MAOI therapy, augmentation options should be sought, as patients who respond but fail to remit are at increased risk of relapse.26 Lithium augmentation is one of the more common strategies, with abundant data supporting its use.27,28 Case reports dating back >12 years describe the concurrent use o
1. Krishnamoorthy S, Ma Z, Zhang G, et al. Involvement of 5-HT2A receptors in the serotonin (5-HT) syndrome caused by excessive 5-HT efflux in rat brain. Basic Clin Pharmacol Toxicol. 2010;107(4):830-841.
2. Sternbach H. The serotonin syndrome. Am J Psychiatry 1991;148(6):705-713.
3. Gillman PK. Monoamine oxidase inhibitors: a review concerning dietary tyramine and drug interactions. PsychoTropical Commentaries. 2016;16(6):1-90.
4. Haberzettl R, Fink H, Bert B. Role of 5-HT(1A)- and 5-HT(2A) receptors for the murine model of the serotonin syndrome. J Pharmacol Toxicol Methods. 2014;70(2):129-133.
5. Shioda K, Nisijima K, Yoshino T, et al. Mirtazapine abolishes hyperthermia in an animal model of serotonin syndrome. Neurosci Lett. 2010;482(3):216-219.
6. White K, Simpson G. Combined MAOI-tricyclic antidepressant treatment: a reevaluation. J Clin Psychopharmacol. 1981;1(5):264-282.
7. Otte W, Birkenhager TK, van den Broek WW. Fatal interaction between tranylcypromine and imipramine. Eur Psychiatry. 2003;18(5):264-265.
8. Panisset M, Chen JJ, Rhyee SH, et al. Serotonin toxicity association with concomitant antidepressants and rasagiline treatment: retrospective study (STACCATO). Pharmacotherapy. 2014;34(12):1250-1258.
9. Gillman PK. Mirtazapine: unable to induce serotonin toxicity? Clin Neuropharmacol. 2003;26(6):288-289; author reply 289-290.
10. Gillman PK. A systematic review of the serotonergic effects of mirtazapine in humans: implications for its dual action status. Hum Psychopharmacol. 2006;21(2):117-125.
11. Meyer JM, Cummings MA, Proctor G. Augmentation of phenelzine with aripiprazole and quetiapine in a treatment resistant patient with psychotic unipolar depression: case report and literature review. CNS Spectr. 2017;22(5):391-396.
12. Feinberg SS. Combining stimulants with monoamine oxidase inhibitors: a review of uses and one possible additional indication. J Clin Psychiatry. 2004;65(11):1520-1524.
13. Israel JA. Combining stimulants and monoamine oxidase inhibitors: a reexamination of the literature and a report of a new treatment combination. Prim Care Companion CNS Disord. 2015;17(6). doi: 10.4088/PCC.15br01836.
14. Simmler LD, Buchy D, Chaboz S, et al. In vitro characterization of psychoactive substances at rat, mouse, and human trace amine-associated receptor 1. J Pharmacol Exp Ther. 2016;357(1):134-144.
15. Froimowitz M, Gu Y, Dakin LA, et al. Slow-onset, long-duration, alkyl analogues of methylphenidate with enhanced selectivity for the dopamine transporter. J Med Chem. 2007;50(2):219-232.
16. Stahl SM, Felker A. Monoamine oxidase inhibitors: a modern guide to an unrequited class of antidepressants. CNS Spectr. 2008;13(10):855-780.
17. Hiemke C, Härtter S. Pharmacokinetics of selective serotonin reuptake inhibitors. Pharmacol Ther. 2000;85(1):11-28.
18. Pristiq [package insert]. New York, NY: Pfizer Inc; 2016.
19. Savella [package insert]. Irvine, CA: Allergan USA Inc; 2016.
20. Viibryd [package insert]. Irvine, CA: Allergan USA Inc; 2016.
21. Trintellix [package insert]. Deerfield, IL: Takeda Pharmaceuticals America Inc; 2016.
22. Fetzima [package insert]. Irvine, CA: Allergan USA Inc; 2017.
23. Nardil [package insert]. New York, NY: Pfizer Inc; 2009.
24. Testani M Jr. Clozapine-induced orthostatic hypotension treated with fludrocortisone. J Clin Psychiatry. 1994;55(11):497-498.
25. Emsam [package insert]. Morgantown, WV: Somerset Pharmaceuticals Inc; 2015.
26. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905-1917.
27. Tariot PN, Murphy DL, Sunderland T, et al. Rapid antidepressant effect of addition of lithium to tranylcypromine. J Clin Psychopharmacol. 1986;6(3):165-167.
28. Kok RM, Vink D, Heeren TJ, et al. Lithium augmentation compared with phenelzine in treatment-resistant depression in the elderly: an open, randomized, controlled trial. J Clin Psychiatry. 2007;68(8):1177-1185.
29. Quante A, Zeugmann S. Tranylcypromine and bupropion combination therapy in treatment-resistant major depression: a report of 2 cases. J Clin Psychopharmacol. 2012;32(4):572-574.
30. Joffe RT. Triiodothyronine potentiation of the antidepressant effect of phenelzine. J Clin Psychiatry. 1988;49(10):409-410.
31. Hullett FJ, Bidder TG. Phenelzine plus triiodothyronine combination in a case of refractory depression. J Nerv Ment Dis. 1983;171(5):318-320.
Monoamine oxidase inhibitors (MAOIs) have well-established efficacy for treating depression, panic disorder, and social phobia. However, a lack of familiarity with these agents and misconceptions about the risks associated with their use have led to MAOIs being substantially underutilized. The goal of this 2-part guide to MAOIs is to educate clinicians about this often-overlooked class of medications. Part 1 (“A concise guide to monoamine inhibitors,”
MAOIs and potential drug interactions
One source of concern in patients receiving irreversible nonselective MAOIs is the development of excessive serotonergic neurotransmission resulting in SS. In the 1960s, researchers noted that administering large doses of
- mild symptoms: tremor, akathisia, inducible clonus
- moderate symptoms: spontaneous or sustained clonus, muscular hypertonicity
- severe symptoms: hyperthermia, diaphoresis.2
Although SS can be induced by significant exposure to individual agents that promote excess synaptic serotonin (eg, overdose of selective serotonin reuptake inhibitors [SSRIs]), the majority of fatal cases have occurred among patients taking MAOIs who were coadministered an agent that inhibited serotonin reuptake (Table 13). Animal studies have determined that excessive stimulation of the 5HT2A receptor is primarily responsible for SS,4 and that 5HT2A antagonists, such as mirtazapine, can block the development of SS in a mouse coadministered
Risk for SS. Most medications that promote serotonergic activity are well known for their use as antidepressants, but other agents that have 5HT reuptake properties (eg, the antihistamine chlorpheniramine) must be avoided. Although older literature suggests that the use of lower doses of certain tricyclic antidepressants concurrently with MAOIs may not be as dangerous as once believed,6 there are sufficient reports of serious outcomes that tricyclics should be avoided in patients taking MAOIs because of the risk of SS, and also because, in general, tricyclics are poorly tolerated.7
Desipramine, a potent norepinephrine transporter (NET) inhibitor, blocks the entry of tyramine into cells by NET, thereby preventing hypertensive events in animal models of tyramine overexposure. However, in some assays, the affinity for the serotonin transporter is not insignificant, so at higher doses desipramine may pose the same theoretical risk for SS as seen with other tricyclics.3
Lastly
Astute clinicians will recognize that antidepressants that lack 5HT reuptake (eg, bupropion, mirtazapine) are not on this list of agents that may increase SS risk when taken with an MAOI. Older papers often list mirtazapine, but as a 5HT2A antagonist, it does not possess a plausible mechanism by which it can induce 5HT toxicity.9,10 Most atypical antipsychotics have significant 5HT2A antagonism and can be combined with MAOIs, but ziprasidone is an exception: as a moderate SNRI, it has been associated with SS when administered with an MAOI.11
Pressor reactions. The only theoretical sources of concern for pressor effects are medications that act as norepinephrine releasers through interactions at the trace amine-associated receptor 1 (TAAR1) (for more information on TAAR1, see
Starting a patient on an MAOI
Contraindicated medications need to be tapered before beginning MAOI treatment. The duration of the washout period depends on the half-life of the medication and any active metabolites. Antidepressants with half-lives of approximately ≤24 hours should be tapered over 7 to 14 days (depending on the dose) to minimize the risk of withdrawal syndromes, while those with long half-lives (eg, fluoxetine,
Initiation of an MAOI is always based on whether the patient can reliably follow the basic dietary advice (see “A concise guide to monoamine inhibitors,”
The orthostasis management strategy is similar to that employed for
Augmentation options for patients taking MAOIs
For depressed patients who do not achieve remission of symptoms from MAOI therapy, augmentation options should be sought, as patients who respond but fail to remit are at increased risk of relapse.26 Lithium augmentation is one of the more common strategies, with abundant data supporting its use.27,28 Case reports dating back >12 years describe the concurrent use o
Monoamine oxidase inhibitors (MAOIs) have well-established efficacy for treating depression, panic disorder, and social phobia. However, a lack of familiarity with these agents and misconceptions about the risks associated with their use have led to MAOIs being substantially underutilized. The goal of this 2-part guide to MAOIs is to educate clinicians about this often-overlooked class of medications. Part 1 (“A concise guide to monoamine inhibitors,”
MAOIs and potential drug interactions
One source of concern in patients receiving irreversible nonselective MAOIs is the development of excessive serotonergic neurotransmission resulting in SS. In the 1960s, researchers noted that administering large doses of
- mild symptoms: tremor, akathisia, inducible clonus
- moderate symptoms: spontaneous or sustained clonus, muscular hypertonicity
- severe symptoms: hyperthermia, diaphoresis.2
Although SS can be induced by significant exposure to individual agents that promote excess synaptic serotonin (eg, overdose of selective serotonin reuptake inhibitors [SSRIs]), the majority of fatal cases have occurred among patients taking MAOIs who were coadministered an agent that inhibited serotonin reuptake (Table 13). Animal studies have determined that excessive stimulation of the 5HT2A receptor is primarily responsible for SS,4 and that 5HT2A antagonists, such as mirtazapine, can block the development of SS in a mouse coadministered
Risk for SS. Most medications that promote serotonergic activity are well known for their use as antidepressants, but other agents that have 5HT reuptake properties (eg, the antihistamine chlorpheniramine) must be avoided. Although older literature suggests that the use of lower doses of certain tricyclic antidepressants concurrently with MAOIs may not be as dangerous as once believed,6 there are sufficient reports of serious outcomes that tricyclics should be avoided in patients taking MAOIs because of the risk of SS, and also because, in general, tricyclics are poorly tolerated.7
Desipramine, a potent norepinephrine transporter (NET) inhibitor, blocks the entry of tyramine into cells by NET, thereby preventing hypertensive events in animal models of tyramine overexposure. However, in some assays, the affinity for the serotonin transporter is not insignificant, so at higher doses desipramine may pose the same theoretical risk for SS as seen with other tricyclics.3
Lastly
Astute clinicians will recognize that antidepressants that lack 5HT reuptake (eg, bupropion, mirtazapine) are not on this list of agents that may increase SS risk when taken with an MAOI. Older papers often list mirtazapine, but as a 5HT2A antagonist, it does not possess a plausible mechanism by which it can induce 5HT toxicity.9,10 Most atypical antipsychotics have significant 5HT2A antagonism and can be combined with MAOIs, but ziprasidone is an exception: as a moderate SNRI, it has been associated with SS when administered with an MAOI.11
Pressor reactions. The only theoretical sources of concern for pressor effects are medications that act as norepinephrine releasers through interactions at the trace amine-associated receptor 1 (TAAR1) (for more information on TAAR1, see
Starting a patient on an MAOI
Contraindicated medications need to be tapered before beginning MAOI treatment. The duration of the washout period depends on the half-life of the medication and any active metabolites. Antidepressants with half-lives of approximately ≤24 hours should be tapered over 7 to 14 days (depending on the dose) to minimize the risk of withdrawal syndromes, while those with long half-lives (eg, fluoxetine,
Initiation of an MAOI is always based on whether the patient can reliably follow the basic dietary advice (see “A concise guide to monoamine inhibitors,”
The orthostasis management strategy is similar to that employed for
Augmentation options for patients taking MAOIs
For depressed patients who do not achieve remission of symptoms from MAOI therapy, augmentation options should be sought, as patients who respond but fail to remit are at increased risk of relapse.26 Lithium augmentation is one of the more common strategies, with abundant data supporting its use.27,28 Case reports dating back >12 years describe the concurrent use o
1. Krishnamoorthy S, Ma Z, Zhang G, et al. Involvement of 5-HT2A receptors in the serotonin (5-HT) syndrome caused by excessive 5-HT efflux in rat brain. Basic Clin Pharmacol Toxicol. 2010;107(4):830-841.
2. Sternbach H. The serotonin syndrome. Am J Psychiatry 1991;148(6):705-713.
3. Gillman PK. Monoamine oxidase inhibitors: a review concerning dietary tyramine and drug interactions. PsychoTropical Commentaries. 2016;16(6):1-90.
4. Haberzettl R, Fink H, Bert B. Role of 5-HT(1A)- and 5-HT(2A) receptors for the murine model of the serotonin syndrome. J Pharmacol Toxicol Methods. 2014;70(2):129-133.
5. Shioda K, Nisijima K, Yoshino T, et al. Mirtazapine abolishes hyperthermia in an animal model of serotonin syndrome. Neurosci Lett. 2010;482(3):216-219.
6. White K, Simpson G. Combined MAOI-tricyclic antidepressant treatment: a reevaluation. J Clin Psychopharmacol. 1981;1(5):264-282.
7. Otte W, Birkenhager TK, van den Broek WW. Fatal interaction between tranylcypromine and imipramine. Eur Psychiatry. 2003;18(5):264-265.
8. Panisset M, Chen JJ, Rhyee SH, et al. Serotonin toxicity association with concomitant antidepressants and rasagiline treatment: retrospective study (STACCATO). Pharmacotherapy. 2014;34(12):1250-1258.
9. Gillman PK. Mirtazapine: unable to induce serotonin toxicity? Clin Neuropharmacol. 2003;26(6):288-289; author reply 289-290.
10. Gillman PK. A systematic review of the serotonergic effects of mirtazapine in humans: implications for its dual action status. Hum Psychopharmacol. 2006;21(2):117-125.
11. Meyer JM, Cummings MA, Proctor G. Augmentation of phenelzine with aripiprazole and quetiapine in a treatment resistant patient with psychotic unipolar depression: case report and literature review. CNS Spectr. 2017;22(5):391-396.
12. Feinberg SS. Combining stimulants with monoamine oxidase inhibitors: a review of uses and one possible additional indication. J Clin Psychiatry. 2004;65(11):1520-1524.
13. Israel JA. Combining stimulants and monoamine oxidase inhibitors: a reexamination of the literature and a report of a new treatment combination. Prim Care Companion CNS Disord. 2015;17(6). doi: 10.4088/PCC.15br01836.
14. Simmler LD, Buchy D, Chaboz S, et al. In vitro characterization of psychoactive substances at rat, mouse, and human trace amine-associated receptor 1. J Pharmacol Exp Ther. 2016;357(1):134-144.
15. Froimowitz M, Gu Y, Dakin LA, et al. Slow-onset, long-duration, alkyl analogues of methylphenidate with enhanced selectivity for the dopamine transporter. J Med Chem. 2007;50(2):219-232.
16. Stahl SM, Felker A. Monoamine oxidase inhibitors: a modern guide to an unrequited class of antidepressants. CNS Spectr. 2008;13(10):855-780.
17. Hiemke C, Härtter S. Pharmacokinetics of selective serotonin reuptake inhibitors. Pharmacol Ther. 2000;85(1):11-28.
18. Pristiq [package insert]. New York, NY: Pfizer Inc; 2016.
19. Savella [package insert]. Irvine, CA: Allergan USA Inc; 2016.
20. Viibryd [package insert]. Irvine, CA: Allergan USA Inc; 2016.
21. Trintellix [package insert]. Deerfield, IL: Takeda Pharmaceuticals America Inc; 2016.
22. Fetzima [package insert]. Irvine, CA: Allergan USA Inc; 2017.
23. Nardil [package insert]. New York, NY: Pfizer Inc; 2009.
24. Testani M Jr. Clozapine-induced orthostatic hypotension treated with fludrocortisone. J Clin Psychiatry. 1994;55(11):497-498.
25. Emsam [package insert]. Morgantown, WV: Somerset Pharmaceuticals Inc; 2015.
26. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905-1917.
27. Tariot PN, Murphy DL, Sunderland T, et al. Rapid antidepressant effect of addition of lithium to tranylcypromine. J Clin Psychopharmacol. 1986;6(3):165-167.
28. Kok RM, Vink D, Heeren TJ, et al. Lithium augmentation compared with phenelzine in treatment-resistant depression in the elderly: an open, randomized, controlled trial. J Clin Psychiatry. 2007;68(8):1177-1185.
29. Quante A, Zeugmann S. Tranylcypromine and bupropion combination therapy in treatment-resistant major depression: a report of 2 cases. J Clin Psychopharmacol. 2012;32(4):572-574.
30. Joffe RT. Triiodothyronine potentiation of the antidepressant effect of phenelzine. J Clin Psychiatry. 1988;49(10):409-410.
31. Hullett FJ, Bidder TG. Phenelzine plus triiodothyronine combination in a case of refractory depression. J Nerv Ment Dis. 1983;171(5):318-320.
1. Krishnamoorthy S, Ma Z, Zhang G, et al. Involvement of 5-HT2A receptors in the serotonin (5-HT) syndrome caused by excessive 5-HT efflux in rat brain. Basic Clin Pharmacol Toxicol. 2010;107(4):830-841.
2. Sternbach H. The serotonin syndrome. Am J Psychiatry 1991;148(6):705-713.
3. Gillman PK. Monoamine oxidase inhibitors: a review concerning dietary tyramine and drug interactions. PsychoTropical Commentaries. 2016;16(6):1-90.
4. Haberzettl R, Fink H, Bert B. Role of 5-HT(1A)- and 5-HT(2A) receptors for the murine model of the serotonin syndrome. J Pharmacol Toxicol Methods. 2014;70(2):129-133.
5. Shioda K, Nisijima K, Yoshino T, et al. Mirtazapine abolishes hyperthermia in an animal model of serotonin syndrome. Neurosci Lett. 2010;482(3):216-219.
6. White K, Simpson G. Combined MAOI-tricyclic antidepressant treatment: a reevaluation. J Clin Psychopharmacol. 1981;1(5):264-282.
7. Otte W, Birkenhager TK, van den Broek WW. Fatal interaction between tranylcypromine and imipramine. Eur Psychiatry. 2003;18(5):264-265.
8. Panisset M, Chen JJ, Rhyee SH, et al. Serotonin toxicity association with concomitant antidepressants and rasagiline treatment: retrospective study (STACCATO). Pharmacotherapy. 2014;34(12):1250-1258.
9. Gillman PK. Mirtazapine: unable to induce serotonin toxicity? Clin Neuropharmacol. 2003;26(6):288-289; author reply 289-290.
10. Gillman PK. A systematic review of the serotonergic effects of mirtazapine in humans: implications for its dual action status. Hum Psychopharmacol. 2006;21(2):117-125.
11. Meyer JM, Cummings MA, Proctor G. Augmentation of phenelzine with aripiprazole and quetiapine in a treatment resistant patient with psychotic unipolar depression: case report and literature review. CNS Spectr. 2017;22(5):391-396.
12. Feinberg SS. Combining stimulants with monoamine oxidase inhibitors: a review of uses and one possible additional indication. J Clin Psychiatry. 2004;65(11):1520-1524.
13. Israel JA. Combining stimulants and monoamine oxidase inhibitors: a reexamination of the literature and a report of a new treatment combination. Prim Care Companion CNS Disord. 2015;17(6). doi: 10.4088/PCC.15br01836.
14. Simmler LD, Buchy D, Chaboz S, et al. In vitro characterization of psychoactive substances at rat, mouse, and human trace amine-associated receptor 1. J Pharmacol Exp Ther. 2016;357(1):134-144.
15. Froimowitz M, Gu Y, Dakin LA, et al. Slow-onset, long-duration, alkyl analogues of methylphenidate with enhanced selectivity for the dopamine transporter. J Med Chem. 2007;50(2):219-232.
16. Stahl SM, Felker A. Monoamine oxidase inhibitors: a modern guide to an unrequited class of antidepressants. CNS Spectr. 2008;13(10):855-780.
17. Hiemke C, Härtter S. Pharmacokinetics of selective serotonin reuptake inhibitors. Pharmacol Ther. 2000;85(1):11-28.
18. Pristiq [package insert]. New York, NY: Pfizer Inc; 2016.
19. Savella [package insert]. Irvine, CA: Allergan USA Inc; 2016.
20. Viibryd [package insert]. Irvine, CA: Allergan USA Inc; 2016.
21. Trintellix [package insert]. Deerfield, IL: Takeda Pharmaceuticals America Inc; 2016.
22. Fetzima [package insert]. Irvine, CA: Allergan USA Inc; 2017.
23. Nardil [package insert]. New York, NY: Pfizer Inc; 2009.
24. Testani M Jr. Clozapine-induced orthostatic hypotension treated with fludrocortisone. J Clin Psychiatry. 1994;55(11):497-498.
25. Emsam [package insert]. Morgantown, WV: Somerset Pharmaceuticals Inc; 2015.
26. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905-1917.
27. Tariot PN, Murphy DL, Sunderland T, et al. Rapid antidepressant effect of addition of lithium to tranylcypromine. J Clin Psychopharmacol. 1986;6(3):165-167.
28. Kok RM, Vink D, Heeren TJ, et al. Lithium augmentation compared with phenelzine in treatment-resistant depression in the elderly: an open, randomized, controlled trial. J Clin Psychiatry. 2007;68(8):1177-1185.
29. Quante A, Zeugmann S. Tranylcypromine and bupropion combination therapy in treatment-resistant major depression: a report of 2 cases. J Clin Psychopharmacol. 2012;32(4):572-574.
30. Joffe RT. Triiodothyronine potentiation of the antidepressant effect of phenelzine. J Clin Psychiatry. 1988;49(10):409-410.
31. Hullett FJ, Bidder TG. Phenelzine plus triiodothyronine combination in a case of refractory depression. J Nerv Ment Dis. 1983;171(5):318-320.

The puzzling relationship between cholesterol and psychopathology
Cholesterol generally is regarded as a cardiovascular risk factor when elevated. However, numerous studies suggest that cholesterol levels—both high and low—may be associated with various psychiatric brai
The relationship between cholesterol and mental illness is fascinating, complex, and perplexing. Whether elevated or reduced, cholesterol’s effects can be deleterious or salutary, but the literature is riddled with conflicting reports. Physicians should measure their patients’ serum cholesterol levels not only to assess cardiovascular risk, but because cholesterol can be associated with certain neuropsychiatric disorders or may predict the lack of response to psychopharmacotherapy.2
The fact that lowering total cholesterol levels in people with hypercholesterolemia reduces the risk of coronary heart disease is indisputable. Large-scale cardiology clinical trials have shown a significant reduction in mortality from heart disease or stroke with cholesterol-lowering drugs (statins). However, the same trials found an uptick in “unnatural deaths,” mostly suicide or homicide.3 Those findings triggered numerous intriguing reports of the association between cholesterol levels and psychopathology.
Consider the following:
- Low cholesterol levels have been associated with depression, antisocial personality disorder, borderline personality disorder, and dissociative disorder.4
- High cholesterol levels have been associated with schizophrenia, obsessive-compulsive disorder, panic disorder, generalized anxiety disorder, and posttraumatic stress disorder.4
- Some studies suggest that high cholesterol levels are associated with better mental health, mental processing speed, social skills, responsibility, self-control, and self-awareness.5
- In the Clinical Antipsychotic Trials of Intervention Effectiveness schizophrenia study, better cognitive scores were found in patients with higher fasting cholesterol and triglyceride levels (H.A.N., unpublished data, 2017).
The brain is only 2% of body weight, but it contains 25% of the body’s cholesterol.6 Cholesterol is important for brain function and neurotransmission because neuroactive steroids (NASs) are synthesized from cholesterol and they modulate brain processes and interact with γ-aminobutyric acid, N-methyl-
Interestingly, both extremes in cholesterol levels represent a high risk for premature mortality.10 Hypercholesterolemia leads to early death from coronary artery disease. Studies that evaluated statins to lower cholesterol found increased mortality from suicide, accidents, and violence.11 Even without statin treatment, among persons with naturally low cholesterol, there is a significant increase in mortality from non-medical causes.12 However, some studies did not find an association between hypocholesterolemia and suicide.13,14
There also is some evidence that elevated cholesterol may play a role in dementia.15 Reducing cholesterol with statins decreases beta-amyloid in mice, while the opposite occurs with elevated cholesterol.2 Another possible mechanism by which high cholesterol worsens dementia is that neurodegeneration in Alzheimer’s disease (AD) breaks down neuronal cell membranes, which releases the neurotoxic metabolite of cholesterol (24-hydroxycholesterol), which leads to further neurodegeneration.16 Statins may decrease the production of 24-hydroxycholesterol in AD patients and slow down neurodegeneration.16
A large study of 4,444 consecutive patients in Taiwan found that those with low total cholesterol (<160 mg/dL) had higher scores of anxiety, phobia, psychoticism, and aggressive hostility.17 In the same study, women with low high-density lipoprotein cholesterol (<35 mg/dL) had significantly higher scores for depression, phobia, anxiety, interpersonal sensitivity, somatization, and aggressive hostility.17
Not surprisingly, low cholesterol has been proposed as a biomarker for mood dysregulation, depression, and suicidality,18 as well as a predictor of the depression severity and increased suicide risk.19 Clinical recovery in depression may be accompanied by a significant increase of total cholesterol20 but, interestingly, a decrease in cholesterol levels after treatment of mania. High cholesterol was reported to predict poorer response to selective serotonin reuptake inhibitors, and total cholesterol levels >200 mg/dL were associated with lack of response to fluoxetine and nortriptyline.2 Interestingly, clozapine, which elevates lipids, exerts a strong anti-suicide effect in schizophrenia and schizoaffective disorder, but that may not be the main reason for its efficacy in preventing suicide in patients with psychosis.
Cholesterol is an important lipid for brain function. At lower levels, it appears to be associated with depression, suicide, violence, anxiety, schizophrenia, and severe personality disorders (including antisocial personality disorder and borderline personality disorder). However, at high levels, it may improve cognition in schizophrenia and ameliorate the pace of AD and neurodegeneration. Psychiatrists should monitor patients for hypercholesterolemia and hypocholesterolemia, both of which are common among psychiatric patients. High levels may be genetic or the result of weight gain, hypercortisolemia, diabetes, or immune or inflammatory processes. Similarly, low levels may be genetic or secondary to statin therapy.
The bottom line: As psychiatric physicians, we should protect both the hearts and brains of our patients.
1. Hallahan B, Garland MR. Essential fatty acids and mental health. British J Psychiatry. 2005;186(4):275-277.
2. Papakostas GI, Ongür D, Iosifescu DV, et al. Cholesterol in mood and anxiety disorders: review of the literature and new hypotheses. Eur Neuropsychopharmacol. 2004;14(2):135-142.
3. Muldoon MF, Manuck SB, Matthews KA, et al. Lowering cholesterol concentrations and mortality: a quantitative review of primary prevention trials. BMJ. 1990;301(647):309-314.
4. Jakovljevic
5. Rogers PJ. A healthy body, a healthy mind: long-term impact of diet on mood and cognitive function. Pro Nutr Soc. 2001;60(1):135-143.
6. Björkhem I. Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J Intern Med. 2006;260(6):493-508.
7. Tuem KB, Atey TM. Neuroactive steroids: receptor interactions and responses. Front Neurol. 2017;8:442.
8. Borroni MV, Vallés AS, Barrantes FJ. The lipid habitats of neurotransmitter receptors in the brain. Biochim Biophys Acta. 2016;1858(1):2662-2670.
9. Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci. 2003;60(6):1158-1171.
10. Graham I, Atar D, Borch-Johnsen K, et al; European Society of Cardiology (ESC); European Association for Cardiovascular Prevention and Rehabilitation (EACPR); Council on Cardiovascular Nursing; European Association for Study of Diabetes (EASD); International Diabetes Federation Europe (IDF-Europe); European Stroke Initiative (EUSI); Society of Behavioural Medicine (ISBM); European Society of Hypertension (ESH); WONCA Europe (European Society of General Practice/Family Medicine); European Heart Network (EHN); European Atherosclerosis Society (EAS). European guidelines on cardiovascular disease prevention in clinical practice: full text. Fourth Joint Task Force of the European Society of Cardiology and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of none societies and by invited experts). Eur J Cardiovasc Prev Rehabil. 2007;14(suppl 2):S1-S113.
11. Almeida-Montes LG, Valles-Sanchez V, Moreno-Aguilar J, et al. Relation of serum cholesterol, lipid, serotonin and tryptophan levels to severity of depression and to suicide attempts. J Psychiatry Neurosci. 2000;25(4):371-377.
12. Ryman A. Cholesterol, violent death, and mental disorder. BMJ. 1994;309(69525):421-422.
13. Wardle J. Cholesterol and psychological well-being. J Psychosom Res. 1995;39(5):549-562.
14. Irribarren C, Reed DM, Chen R, et al. Low serum cholesterol and mortality. Which is the cause and which is the effect? Circulation. 1995;92(9):2396-2403.
15. Stampfer MJ. Cardiovascular disease and Alzheimer’s disease: common links. J Intern Med. 2006;260(3):211-223.
16. Raffai RL, Weisgraber KH. Cholesterol: from heart attacks to Alzheimer’s disease. J Lipid Res. 2003;44(8):1423-1430.
17. Chen CC, Lu FH, Wu JS, et al. Correlation between serum lipid concentrations and psychological distress. Psychiatry Res. 2003;102(2):153-162.
18. Mössmer R, Mikova O, Koutsilieri E, et al. Consensus paper of the WFSBP Task Force on Biological Markers: biological markers in depression. World J Biol Psychiatry. 2007;8(3):141-174.
19. Papakostas GI, Petersen T, Sonawalla SB, et al. Serum cholesterol in treatment-resistant depression. Neuropsychobiology. 2003;47(3):146-151.
20. Gabriel A. Changes in plasma cholesterol in mood disorder patients: does treatment make a difference? J Affect Disord. 2007;99(1-3):273-278.
Cholesterol generally is regarded as a cardiovascular risk factor when elevated. However, numerous studies suggest that cholesterol levels—both high and low—may be associated with various psychiatric brai
The relationship between cholesterol and mental illness is fascinating, complex, and perplexing. Whether elevated or reduced, cholesterol’s effects can be deleterious or salutary, but the literature is riddled with conflicting reports. Physicians should measure their patients’ serum cholesterol levels not only to assess cardiovascular risk, but because cholesterol can be associated with certain neuropsychiatric disorders or may predict the lack of response to psychopharmacotherapy.2
The fact that lowering total cholesterol levels in people with hypercholesterolemia reduces the risk of coronary heart disease is indisputable. Large-scale cardiology clinical trials have shown a significant reduction in mortality from heart disease or stroke with cholesterol-lowering drugs (statins). However, the same trials found an uptick in “unnatural deaths,” mostly suicide or homicide.3 Those findings triggered numerous intriguing reports of the association between cholesterol levels and psychopathology.
Consider the following:
- Low cholesterol levels have been associated with depression, antisocial personality disorder, borderline personality disorder, and dissociative disorder.4
- High cholesterol levels have been associated with schizophrenia, obsessive-compulsive disorder, panic disorder, generalized anxiety disorder, and posttraumatic stress disorder.4
- Some studies suggest that high cholesterol levels are associated with better mental health, mental processing speed, social skills, responsibility, self-control, and self-awareness.5
- In the Clinical Antipsychotic Trials of Intervention Effectiveness schizophrenia study, better cognitive scores were found in patients with higher fasting cholesterol and triglyceride levels (H.A.N., unpublished data, 2017).
The brain is only 2% of body weight, but it contains 25% of the body’s cholesterol.6 Cholesterol is important for brain function and neurotransmission because neuroactive steroids (NASs) are synthesized from cholesterol and they modulate brain processes and interact with γ-aminobutyric acid, N-methyl-
Interestingly, both extremes in cholesterol levels represent a high risk for premature mortality.10 Hypercholesterolemia leads to early death from coronary artery disease. Studies that evaluated statins to lower cholesterol found increased mortality from suicide, accidents, and violence.11 Even without statin treatment, among persons with naturally low cholesterol, there is a significant increase in mortality from non-medical causes.12 However, some studies did not find an association between hypocholesterolemia and suicide.13,14
There also is some evidence that elevated cholesterol may play a role in dementia.15 Reducing cholesterol with statins decreases beta-amyloid in mice, while the opposite occurs with elevated cholesterol.2 Another possible mechanism by which high cholesterol worsens dementia is that neurodegeneration in Alzheimer’s disease (AD) breaks down neuronal cell membranes, which releases the neurotoxic metabolite of cholesterol (24-hydroxycholesterol), which leads to further neurodegeneration.16 Statins may decrease the production of 24-hydroxycholesterol in AD patients and slow down neurodegeneration.16
A large study of 4,444 consecutive patients in Taiwan found that those with low total cholesterol (<160 mg/dL) had higher scores of anxiety, phobia, psychoticism, and aggressive hostility.17 In the same study, women with low high-density lipoprotein cholesterol (<35 mg/dL) had significantly higher scores for depression, phobia, anxiety, interpersonal sensitivity, somatization, and aggressive hostility.17
Not surprisingly, low cholesterol has been proposed as a biomarker for mood dysregulation, depression, and suicidality,18 as well as a predictor of the depression severity and increased suicide risk.19 Clinical recovery in depression may be accompanied by a significant increase of total cholesterol20 but, interestingly, a decrease in cholesterol levels after treatment of mania. High cholesterol was reported to predict poorer response to selective serotonin reuptake inhibitors, and total cholesterol levels >200 mg/dL were associated with lack of response to fluoxetine and nortriptyline.2 Interestingly, clozapine, which elevates lipids, exerts a strong anti-suicide effect in schizophrenia and schizoaffective disorder, but that may not be the main reason for its efficacy in preventing suicide in patients with psychosis.
Cholesterol is an important lipid for brain function. At lower levels, it appears to be associated with depression, suicide, violence, anxiety, schizophrenia, and severe personality disorders (including antisocial personality disorder and borderline personality disorder). However, at high levels, it may improve cognition in schizophrenia and ameliorate the pace of AD and neurodegeneration. Psychiatrists should monitor patients for hypercholesterolemia and hypocholesterolemia, both of which are common among psychiatric patients. High levels may be genetic or the result of weight gain, hypercortisolemia, diabetes, or immune or inflammatory processes. Similarly, low levels may be genetic or secondary to statin therapy.
The bottom line: As psychiatric physicians, we should protect both the hearts and brains of our patients.
Cholesterol generally is regarded as a cardiovascular risk factor when elevated. However, numerous studies suggest that cholesterol levels—both high and low—may be associated with various psychiatric brai
The relationship between cholesterol and mental illness is fascinating, complex, and perplexing. Whether elevated or reduced, cholesterol’s effects can be deleterious or salutary, but the literature is riddled with conflicting reports. Physicians should measure their patients’ serum cholesterol levels not only to assess cardiovascular risk, but because cholesterol can be associated with certain neuropsychiatric disorders or may predict the lack of response to psychopharmacotherapy.2
The fact that lowering total cholesterol levels in people with hypercholesterolemia reduces the risk of coronary heart disease is indisputable. Large-scale cardiology clinical trials have shown a significant reduction in mortality from heart disease or stroke with cholesterol-lowering drugs (statins). However, the same trials found an uptick in “unnatural deaths,” mostly suicide or homicide.3 Those findings triggered numerous intriguing reports of the association between cholesterol levels and psychopathology.
Consider the following:
- Low cholesterol levels have been associated with depression, antisocial personality disorder, borderline personality disorder, and dissociative disorder.4
- High cholesterol levels have been associated with schizophrenia, obsessive-compulsive disorder, panic disorder, generalized anxiety disorder, and posttraumatic stress disorder.4
- Some studies suggest that high cholesterol levels are associated with better mental health, mental processing speed, social skills, responsibility, self-control, and self-awareness.5
- In the Clinical Antipsychotic Trials of Intervention Effectiveness schizophrenia study, better cognitive scores were found in patients with higher fasting cholesterol and triglyceride levels (H.A.N., unpublished data, 2017).
The brain is only 2% of body weight, but it contains 25% of the body’s cholesterol.6 Cholesterol is important for brain function and neurotransmission because neuroactive steroids (NASs) are synthesized from cholesterol and they modulate brain processes and interact with γ-aminobutyric acid, N-methyl-
Interestingly, both extremes in cholesterol levels represent a high risk for premature mortality.10 Hypercholesterolemia leads to early death from coronary artery disease. Studies that evaluated statins to lower cholesterol found increased mortality from suicide, accidents, and violence.11 Even without statin treatment, among persons with naturally low cholesterol, there is a significant increase in mortality from non-medical causes.12 However, some studies did not find an association between hypocholesterolemia and suicide.13,14
There also is some evidence that elevated cholesterol may play a role in dementia.15 Reducing cholesterol with statins decreases beta-amyloid in mice, while the opposite occurs with elevated cholesterol.2 Another possible mechanism by which high cholesterol worsens dementia is that neurodegeneration in Alzheimer’s disease (AD) breaks down neuronal cell membranes, which releases the neurotoxic metabolite of cholesterol (24-hydroxycholesterol), which leads to further neurodegeneration.16 Statins may decrease the production of 24-hydroxycholesterol in AD patients and slow down neurodegeneration.16
A large study of 4,444 consecutive patients in Taiwan found that those with low total cholesterol (<160 mg/dL) had higher scores of anxiety, phobia, psychoticism, and aggressive hostility.17 In the same study, women with low high-density lipoprotein cholesterol (<35 mg/dL) had significantly higher scores for depression, phobia, anxiety, interpersonal sensitivity, somatization, and aggressive hostility.17
Not surprisingly, low cholesterol has been proposed as a biomarker for mood dysregulation, depression, and suicidality,18 as well as a predictor of the depression severity and increased suicide risk.19 Clinical recovery in depression may be accompanied by a significant increase of total cholesterol20 but, interestingly, a decrease in cholesterol levels after treatment of mania. High cholesterol was reported to predict poorer response to selective serotonin reuptake inhibitors, and total cholesterol levels >200 mg/dL were associated with lack of response to fluoxetine and nortriptyline.2 Interestingly, clozapine, which elevates lipids, exerts a strong anti-suicide effect in schizophrenia and schizoaffective disorder, but that may not be the main reason for its efficacy in preventing suicide in patients with psychosis.
Cholesterol is an important lipid for brain function. At lower levels, it appears to be associated with depression, suicide, violence, anxiety, schizophrenia, and severe personality disorders (including antisocial personality disorder and borderline personality disorder). However, at high levels, it may improve cognition in schizophrenia and ameliorate the pace of AD and neurodegeneration. Psychiatrists should monitor patients for hypercholesterolemia and hypocholesterolemia, both of which are common among psychiatric patients. High levels may be genetic or the result of weight gain, hypercortisolemia, diabetes, or immune or inflammatory processes. Similarly, low levels may be genetic or secondary to statin therapy.
The bottom line: As psychiatric physicians, we should protect both the hearts and brains of our patients.
1. Hallahan B, Garland MR. Essential fatty acids and mental health. British J Psychiatry. 2005;186(4):275-277.
2. Papakostas GI, Ongür D, Iosifescu DV, et al. Cholesterol in mood and anxiety disorders: review of the literature and new hypotheses. Eur Neuropsychopharmacol. 2004;14(2):135-142.
3. Muldoon MF, Manuck SB, Matthews KA, et al. Lowering cholesterol concentrations and mortality: a quantitative review of primary prevention trials. BMJ. 1990;301(647):309-314.
4. Jakovljevic
5. Rogers PJ. A healthy body, a healthy mind: long-term impact of diet on mood and cognitive function. Pro Nutr Soc. 2001;60(1):135-143.
6. Björkhem I. Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J Intern Med. 2006;260(6):493-508.
7. Tuem KB, Atey TM. Neuroactive steroids: receptor interactions and responses. Front Neurol. 2017;8:442.
8. Borroni MV, Vallés AS, Barrantes FJ. The lipid habitats of neurotransmitter receptors in the brain. Biochim Biophys Acta. 2016;1858(1):2662-2670.
9. Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci. 2003;60(6):1158-1171.
10. Graham I, Atar D, Borch-Johnsen K, et al; European Society of Cardiology (ESC); European Association for Cardiovascular Prevention and Rehabilitation (EACPR); Council on Cardiovascular Nursing; European Association for Study of Diabetes (EASD); International Diabetes Federation Europe (IDF-Europe); European Stroke Initiative (EUSI); Society of Behavioural Medicine (ISBM); European Society of Hypertension (ESH); WONCA Europe (European Society of General Practice/Family Medicine); European Heart Network (EHN); European Atherosclerosis Society (EAS). European guidelines on cardiovascular disease prevention in clinical practice: full text. Fourth Joint Task Force of the European Society of Cardiology and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of none societies and by invited experts). Eur J Cardiovasc Prev Rehabil. 2007;14(suppl 2):S1-S113.
11. Almeida-Montes LG, Valles-Sanchez V, Moreno-Aguilar J, et al. Relation of serum cholesterol, lipid, serotonin and tryptophan levels to severity of depression and to suicide attempts. J Psychiatry Neurosci. 2000;25(4):371-377.
12. Ryman A. Cholesterol, violent death, and mental disorder. BMJ. 1994;309(69525):421-422.
13. Wardle J. Cholesterol and psychological well-being. J Psychosom Res. 1995;39(5):549-562.
14. Irribarren C, Reed DM, Chen R, et al. Low serum cholesterol and mortality. Which is the cause and which is the effect? Circulation. 1995;92(9):2396-2403.
15. Stampfer MJ. Cardiovascular disease and Alzheimer’s disease: common links. J Intern Med. 2006;260(3):211-223.
16. Raffai RL, Weisgraber KH. Cholesterol: from heart attacks to Alzheimer’s disease. J Lipid Res. 2003;44(8):1423-1430.
17. Chen CC, Lu FH, Wu JS, et al. Correlation between serum lipid concentrations and psychological distress. Psychiatry Res. 2003;102(2):153-162.
18. Mössmer R, Mikova O, Koutsilieri E, et al. Consensus paper of the WFSBP Task Force on Biological Markers: biological markers in depression. World J Biol Psychiatry. 2007;8(3):141-174.
19. Papakostas GI, Petersen T, Sonawalla SB, et al. Serum cholesterol in treatment-resistant depression. Neuropsychobiology. 2003;47(3):146-151.
20. Gabriel A. Changes in plasma cholesterol in mood disorder patients: does treatment make a difference? J Affect Disord. 2007;99(1-3):273-278.
1. Hallahan B, Garland MR. Essential fatty acids and mental health. British J Psychiatry. 2005;186(4):275-277.
2. Papakostas GI, Ongür D, Iosifescu DV, et al. Cholesterol in mood and anxiety disorders: review of the literature and new hypotheses. Eur Neuropsychopharmacol. 2004;14(2):135-142.
3. Muldoon MF, Manuck SB, Matthews KA, et al. Lowering cholesterol concentrations and mortality: a quantitative review of primary prevention trials. BMJ. 1990;301(647):309-314.
4. Jakovljevic
5. Rogers PJ. A healthy body, a healthy mind: long-term impact of diet on mood and cognitive function. Pro Nutr Soc. 2001;60(1):135-143.
6. Björkhem I. Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J Intern Med. 2006;260(6):493-508.
7. Tuem KB, Atey TM. Neuroactive steroids: receptor interactions and responses. Front Neurol. 2017;8:442.
8. Borroni MV, Vallés AS, Barrantes FJ. The lipid habitats of neurotransmitter receptors in the brain. Biochim Biophys Acta. 2016;1858(1):2662-2670.
9. Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci. 2003;60(6):1158-1171.
10. Graham I, Atar D, Borch-Johnsen K, et al; European Society of Cardiology (ESC); European Association for Cardiovascular Prevention and Rehabilitation (EACPR); Council on Cardiovascular Nursing; European Association for Study of Diabetes (EASD); International Diabetes Federation Europe (IDF-Europe); European Stroke Initiative (EUSI); Society of Behavioural Medicine (ISBM); European Society of Hypertension (ESH); WONCA Europe (European Society of General Practice/Family Medicine); European Heart Network (EHN); European Atherosclerosis Society (EAS). European guidelines on cardiovascular disease prevention in clinical practice: full text. Fourth Joint Task Force of the European Society of Cardiology and other societies on cardiovascular disease prevention in clinical practice (constituted by representatives of none societies and by invited experts). Eur J Cardiovasc Prev Rehabil. 2007;14(suppl 2):S1-S113.
11. Almeida-Montes LG, Valles-Sanchez V, Moreno-Aguilar J, et al. Relation of serum cholesterol, lipid, serotonin and tryptophan levels to severity of depression and to suicide attempts. J Psychiatry Neurosci. 2000;25(4):371-377.
12. Ryman A. Cholesterol, violent death, and mental disorder. BMJ. 1994;309(69525):421-422.
13. Wardle J. Cholesterol and psychological well-being. J Psychosom Res. 1995;39(5):549-562.
14. Irribarren C, Reed DM, Chen R, et al. Low serum cholesterol and mortality. Which is the cause and which is the effect? Circulation. 1995;92(9):2396-2403.
15. Stampfer MJ. Cardiovascular disease and Alzheimer’s disease: common links. J Intern Med. 2006;260(3):211-223.
16. Raffai RL, Weisgraber KH. Cholesterol: from heart attacks to Alzheimer’s disease. J Lipid Res. 2003;44(8):1423-1430.
17. Chen CC, Lu FH, Wu JS, et al. Correlation between serum lipid concentrations and psychological distress. Psychiatry Res. 2003;102(2):153-162.
18. Mössmer R, Mikova O, Koutsilieri E, et al. Consensus paper of the WFSBP Task Force on Biological Markers: biological markers in depression. World J Biol Psychiatry. 2007;8(3):141-174.
19. Papakostas GI, Petersen T, Sonawalla SB, et al. Serum cholesterol in treatment-resistant depression. Neuropsychobiology. 2003;47(3):146-151.
20. Gabriel A. Changes in plasma cholesterol in mood disorder patients: does treatment make a difference? J Affect Disord. 2007;99(1-3):273-278.
