User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Strategies for working with patients with personality disorders
Patients with personality disorders can disrupt the treatment relationship, and may leave us feeling angry, ineffective, inadequate, and defeated. Although their behaviors may appear volitional and purposeful, they often are the result of a dysfunctional personality structure.1 These patients’ unbending patterns of viewing themselves, interacting with others, and navigating the world can be problematic in an inpatient or outpatient setting, causing distress for both the staff and patient. Because no 2 personalities are identical, there is no algorithm for managing patients with personality disorders. However, there are strategies that we can apply to provide effective clinical care.1,2
Discuss the responses the patient evokes. Patients with personality disorders can elicit strong responses from the treatment team. Each clinician can have a different response to the same patient, ranging from feeling the need to protect the patient to strongly disliking him or her. Because cohesion among staff is essential for effective patient care, we need to discuss these responses in an open forum with our team members so we can effectively manage our responses and provide the patient with consistent interactions. Limiting the delivery of inconsistent or conflicting messages will decrease staff splitting and increase team unity.
Reinforce appropriate behaviors. Patients with personality disorders usually have negative interpersonal interactions, such as acting out, misinterpreting neutral social cues, and seeking constant attention. However, when they are not engaging in detrimental behaviors, we should provide positive reinforcement for appropriate behaviors, such as remaining composed, that help maintain the treatment relationship. When a patient displays disruptive behaviors, take a neutral approach by stating, “You appear upset. I will come back later when you are feeling better.”1
Set limits. These patients are likely to have difficulty conforming to appropriate social boundaries. Our reflex reaction may be to set concrete rules that fit our preferences. This could lead to a power struggle between us and our patients, which is not helpful. Rather than a “one-size-fits-all” approach to rules, it may be prudent to tailor boundaries according to each patient’s unique personality. Also, allowing the patient to help set these limits could increase the chances that he or she will follow your treatment plan and reinforce the more positive aspects of his or her personality structure.
Offer empathy. Empathy can be conceptualized as a step further than sympathy; in addition to expressing concern and compassion, empathy involves recognizing and sharing the patient’s emotions. Seek to comprehend the reasons behind a patient’s negative reactions by
1. Riddle M, Meeks T, Alvarez C, et al. When personality is the problem: managing patients with difficult personalitie s on the acute care unit. J Hosp Med. 2016;11(12):873-878.
2. Strous RD, Ulman AM, Kotler M. The hateful patient revisited: relevance for 21st century medicine. Eur J Intern Med. 2006;17(6):387-393.
Patients with personality disorders can disrupt the treatment relationship, and may leave us feeling angry, ineffective, inadequate, and defeated. Although their behaviors may appear volitional and purposeful, they often are the result of a dysfunctional personality structure.1 These patients’ unbending patterns of viewing themselves, interacting with others, and navigating the world can be problematic in an inpatient or outpatient setting, causing distress for both the staff and patient. Because no 2 personalities are identical, there is no algorithm for managing patients with personality disorders. However, there are strategies that we can apply to provide effective clinical care.1,2
Discuss the responses the patient evokes. Patients with personality disorders can elicit strong responses from the treatment team. Each clinician can have a different response to the same patient, ranging from feeling the need to protect the patient to strongly disliking him or her. Because cohesion among staff is essential for effective patient care, we need to discuss these responses in an open forum with our team members so we can effectively manage our responses and provide the patient with consistent interactions. Limiting the delivery of inconsistent or conflicting messages will decrease staff splitting and increase team unity.
Reinforce appropriate behaviors. Patients with personality disorders usually have negative interpersonal interactions, such as acting out, misinterpreting neutral social cues, and seeking constant attention. However, when they are not engaging in detrimental behaviors, we should provide positive reinforcement for appropriate behaviors, such as remaining composed, that help maintain the treatment relationship. When a patient displays disruptive behaviors, take a neutral approach by stating, “You appear upset. I will come back later when you are feeling better.”1
Set limits. These patients are likely to have difficulty conforming to appropriate social boundaries. Our reflex reaction may be to set concrete rules that fit our preferences. This could lead to a power struggle between us and our patients, which is not helpful. Rather than a “one-size-fits-all” approach to rules, it may be prudent to tailor boundaries according to each patient’s unique personality. Also, allowing the patient to help set these limits could increase the chances that he or she will follow your treatment plan and reinforce the more positive aspects of his or her personality structure.
Offer empathy. Empathy can be conceptualized as a step further than sympathy; in addition to expressing concern and compassion, empathy involves recognizing and sharing the patient’s emotions. Seek to comprehend the reasons behind a patient’s negative reactions by
Patients with personality disorders can disrupt the treatment relationship, and may leave us feeling angry, ineffective, inadequate, and defeated. Although their behaviors may appear volitional and purposeful, they often are the result of a dysfunctional personality structure.1 These patients’ unbending patterns of viewing themselves, interacting with others, and navigating the world can be problematic in an inpatient or outpatient setting, causing distress for both the staff and patient. Because no 2 personalities are identical, there is no algorithm for managing patients with personality disorders. However, there are strategies that we can apply to provide effective clinical care.1,2
Discuss the responses the patient evokes. Patients with personality disorders can elicit strong responses from the treatment team. Each clinician can have a different response to the same patient, ranging from feeling the need to protect the patient to strongly disliking him or her. Because cohesion among staff is essential for effective patient care, we need to discuss these responses in an open forum with our team members so we can effectively manage our responses and provide the patient with consistent interactions. Limiting the delivery of inconsistent or conflicting messages will decrease staff splitting and increase team unity.
Reinforce appropriate behaviors. Patients with personality disorders usually have negative interpersonal interactions, such as acting out, misinterpreting neutral social cues, and seeking constant attention. However, when they are not engaging in detrimental behaviors, we should provide positive reinforcement for appropriate behaviors, such as remaining composed, that help maintain the treatment relationship. When a patient displays disruptive behaviors, take a neutral approach by stating, “You appear upset. I will come back later when you are feeling better.”1
Set limits. These patients are likely to have difficulty conforming to appropriate social boundaries. Our reflex reaction may be to set concrete rules that fit our preferences. This could lead to a power struggle between us and our patients, which is not helpful. Rather than a “one-size-fits-all” approach to rules, it may be prudent to tailor boundaries according to each patient’s unique personality. Also, allowing the patient to help set these limits could increase the chances that he or she will follow your treatment plan and reinforce the more positive aspects of his or her personality structure.
Offer empathy. Empathy can be conceptualized as a step further than sympathy; in addition to expressing concern and compassion, empathy involves recognizing and sharing the patient’s emotions. Seek to comprehend the reasons behind a patient’s negative reactions by
1. Riddle M, Meeks T, Alvarez C, et al. When personality is the problem: managing patients with difficult personalitie s on the acute care unit. J Hosp Med. 2016;11(12):873-878.
2. Strous RD, Ulman AM, Kotler M. The hateful patient revisited: relevance for 21st century medicine. Eur J Intern Med. 2006;17(6):387-393.
1. Riddle M, Meeks T, Alvarez C, et al. When personality is the problem: managing patients with difficult personalitie s on the acute care unit. J Hosp Med. 2016;11(12):873-878.
2. Strous RD, Ulman AM, Kotler M. The hateful patient revisited: relevance for 21st century medicine. Eur J Intern Med. 2006;17(6):387-393.
‘Nocebo’ effects: Address these 4 psychosocial factors
Sorting out the causes of unexplained adverse effects from psychotropic medications can be challenging. Treatment may be further complicated by ‘nocebo’ effects, which are adverse effects based on the patient’s conscious and unconscious expectations of harm. Having strategies for managing nocebo effects can help clinicians better understand and treat patients who have complex medication complaints. When your patient experiences nocebo effects, consider the following 4 psychosocial factors.1
Pills. The impact of a medication is not solely based on its chemical makeup. For example, the appearance of a medication can affect treatment outcomes. Substituting generic medications for branded ones has been shown to negatively impact patient adherence and increase reports of adverse effects that have no physiologic cause.2 Educating patients about medication manufacturing and distribution practices may decrease such consequences.
Patient. A sense of powerlessness is fertile ground for nocebo effects. Patients with an external locus of control may unconsciously employ nocebo effects to express themselves when other outlets are limited. Having a psychosocial formulation of your patient can help you anticipate pitfalls, offer pertinent insights, and mobilize the patient’s adaptive coping mechanisms. Also, clinicians can bolster their patients’ self-agency by encouraging them to participate in healthy activities.
Provider. Irrational factors in the clinician, such as countertransference, may also affect medication outcomes. Unprocessed countertransference can contribute to clinician burnout and impact the therapeutic relationship negatively. Nocebo effects may indicate that the clinician is not “tuned in” to the patient or is acting out harmful unconscious thoughts. Additionally, countertransference can lead to unnecessary prescribing and polypharmacy that confounds nocebo effects. Therefore self-care, consultation, and supervision may be vital in promoting therapeutic outcomes.
Partnership. The doctor–patient relationship can contribute to nocebo effects. A 2016 Gallup Poll found that Americans had low confidence in the honesty and ethics of psychiatrists compared with other healthcare professionals.3 It is important to have conversations with your patients about their reservations and perceived stigma of mental health. Such conversations can bring a patient’s ambivalence into treatment so that it can be further explored and addressed. Psychoeducation about treatment limitations, motivational interviewing techniques, and involving patients in decision-making can be useful tools for fostering a therapeutic alliance and positive outcomes.
Take an active approach
Evidence demonstrates that psychosocial factors significantly impact treatment outcomes.1 Incorporating this evidence into practice and attending to the 4 factors discussed here can enhance a clinician’s ability to flexibly respond to their patients’ complaints, especially in relation to nocebo effects.
1. Mallo CJ, Mintz DL. Teaching all the evidence bases: reintegrating psychodynamic aspects of prescribing into psychopharmacology training. Psychodyn Psychiatry. 2013;41(1):13-37.
2. Weissenfeld J, Stock S, Lüngen M, et al. The nocebo effect: a reason for patients’ non-adherence to generic substitution? Pharmazie. 2010;65(7):451-456.
3. Norman J. Americans rate healthcare providers high on honesty, ethics. Gallup. http://news.gallup.com/poll/200057/americans-rate-healthcare-providers-high-honesty-ethics.aspx. Published December 19, 2016. Accessed October 22, 2017.
Sorting out the causes of unexplained adverse effects from psychotropic medications can be challenging. Treatment may be further complicated by ‘nocebo’ effects, which are adverse effects based on the patient’s conscious and unconscious expectations of harm. Having strategies for managing nocebo effects can help clinicians better understand and treat patients who have complex medication complaints. When your patient experiences nocebo effects, consider the following 4 psychosocial factors.1
Pills. The impact of a medication is not solely based on its chemical makeup. For example, the appearance of a medication can affect treatment outcomes. Substituting generic medications for branded ones has been shown to negatively impact patient adherence and increase reports of adverse effects that have no physiologic cause.2 Educating patients about medication manufacturing and distribution practices may decrease such consequences.
Patient. A sense of powerlessness is fertile ground for nocebo effects. Patients with an external locus of control may unconsciously employ nocebo effects to express themselves when other outlets are limited. Having a psychosocial formulation of your patient can help you anticipate pitfalls, offer pertinent insights, and mobilize the patient’s adaptive coping mechanisms. Also, clinicians can bolster their patients’ self-agency by encouraging them to participate in healthy activities.
Provider. Irrational factors in the clinician, such as countertransference, may also affect medication outcomes. Unprocessed countertransference can contribute to clinician burnout and impact the therapeutic relationship negatively. Nocebo effects may indicate that the clinician is not “tuned in” to the patient or is acting out harmful unconscious thoughts. Additionally, countertransference can lead to unnecessary prescribing and polypharmacy that confounds nocebo effects. Therefore self-care, consultation, and supervision may be vital in promoting therapeutic outcomes.
Partnership. The doctor–patient relationship can contribute to nocebo effects. A 2016 Gallup Poll found that Americans had low confidence in the honesty and ethics of psychiatrists compared with other healthcare professionals.3 It is important to have conversations with your patients about their reservations and perceived stigma of mental health. Such conversations can bring a patient’s ambivalence into treatment so that it can be further explored and addressed. Psychoeducation about treatment limitations, motivational interviewing techniques, and involving patients in decision-making can be useful tools for fostering a therapeutic alliance and positive outcomes.
Take an active approach
Evidence demonstrates that psychosocial factors significantly impact treatment outcomes.1 Incorporating this evidence into practice and attending to the 4 factors discussed here can enhance a clinician’s ability to flexibly respond to their patients’ complaints, especially in relation to nocebo effects.
Sorting out the causes of unexplained adverse effects from psychotropic medications can be challenging. Treatment may be further complicated by ‘nocebo’ effects, which are adverse effects based on the patient’s conscious and unconscious expectations of harm. Having strategies for managing nocebo effects can help clinicians better understand and treat patients who have complex medication complaints. When your patient experiences nocebo effects, consider the following 4 psychosocial factors.1
Pills. The impact of a medication is not solely based on its chemical makeup. For example, the appearance of a medication can affect treatment outcomes. Substituting generic medications for branded ones has been shown to negatively impact patient adherence and increase reports of adverse effects that have no physiologic cause.2 Educating patients about medication manufacturing and distribution practices may decrease such consequences.
Patient. A sense of powerlessness is fertile ground for nocebo effects. Patients with an external locus of control may unconsciously employ nocebo effects to express themselves when other outlets are limited. Having a psychosocial formulation of your patient can help you anticipate pitfalls, offer pertinent insights, and mobilize the patient’s adaptive coping mechanisms. Also, clinicians can bolster their patients’ self-agency by encouraging them to participate in healthy activities.
Provider. Irrational factors in the clinician, such as countertransference, may also affect medication outcomes. Unprocessed countertransference can contribute to clinician burnout and impact the therapeutic relationship negatively. Nocebo effects may indicate that the clinician is not “tuned in” to the patient or is acting out harmful unconscious thoughts. Additionally, countertransference can lead to unnecessary prescribing and polypharmacy that confounds nocebo effects. Therefore self-care, consultation, and supervision may be vital in promoting therapeutic outcomes.
Partnership. The doctor–patient relationship can contribute to nocebo effects. A 2016 Gallup Poll found that Americans had low confidence in the honesty and ethics of psychiatrists compared with other healthcare professionals.3 It is important to have conversations with your patients about their reservations and perceived stigma of mental health. Such conversations can bring a patient’s ambivalence into treatment so that it can be further explored and addressed. Psychoeducation about treatment limitations, motivational interviewing techniques, and involving patients in decision-making can be useful tools for fostering a therapeutic alliance and positive outcomes.
Take an active approach
Evidence demonstrates that psychosocial factors significantly impact treatment outcomes.1 Incorporating this evidence into practice and attending to the 4 factors discussed here can enhance a clinician’s ability to flexibly respond to their patients’ complaints, especially in relation to nocebo effects.
1. Mallo CJ, Mintz DL. Teaching all the evidence bases: reintegrating psychodynamic aspects of prescribing into psychopharmacology training. Psychodyn Psychiatry. 2013;41(1):13-37.
2. Weissenfeld J, Stock S, Lüngen M, et al. The nocebo effect: a reason for patients’ non-adherence to generic substitution? Pharmazie. 2010;65(7):451-456.
3. Norman J. Americans rate healthcare providers high on honesty, ethics. Gallup. http://news.gallup.com/poll/200057/americans-rate-healthcare-providers-high-honesty-ethics.aspx. Published December 19, 2016. Accessed October 22, 2017.
1. Mallo CJ, Mintz DL. Teaching all the evidence bases: reintegrating psychodynamic aspects of prescribing into psychopharmacology training. Psychodyn Psychiatry. 2013;41(1):13-37.
2. Weissenfeld J, Stock S, Lüngen M, et al. The nocebo effect: a reason for patients’ non-adherence to generic substitution? Pharmazie. 2010;65(7):451-456.
3. Norman J. Americans rate healthcare providers high on honesty, ethics. Gallup. http://news.gallup.com/poll/200057/americans-rate-healthcare-providers-high-honesty-ethics.aspx. Published December 19, 2016. Accessed October 22, 2017.
Aripiprazole, brexpiprazole, and cariprazine: Not all the same
Aripiprazole, brexpiprazole, and cariprazine are dopamine receptor partial agonists, and on the surface, they appear similar. However, there are key differences in terms of available indications, formulations, pharmacodynamics, pharmacokinetics, dosing, drug interactions, tolerability, and other factors related to successful use.1 This review will cover the main points that the knowledgeable clinician will need to be mindful of when prescribing these agents.
Aripiprazole
Aripiprazole was launched in the United States in 20022 as the first dopamine receptor partial agonist approved for the treatment of schizophrenia; it later received additional indications for adults with manic or mixed episodes associated with bipolar I disorder and the maintenance treatment of bipolar I disorder, as well as for the adjunctive treatment of major depressive disorder (MDD). Pediatric indications include schizophrenia, acute treatment of manic or mixed episodes associated with bipolar I disorder, irritability associated with autistic disorder, and Tourette’s disorder.
Several formulations also became available, including a short-acting injection indicated for agitation associated with schizophrenia or bipolar mania, and oral disintegrating tablets and an oral solution that could substitute for the regular tablet. Presently the medication has gone “generic,” and not all formulations are being manufactured. The long-acting formulations of aripiprazole (aripiprazole monohydrate and aripiprazole lauroxil) are considered different products, each with its own product insert, with indications that are more limited in scope than for the oral forms.3,4
Although dopamine D2 receptor partial agonism is a relevant mechanism of action, partial agonist activity at serotonin 5-HT1A receptors and antagonist activity at 5-HT2A receptors also play a role.2 Actions at receptors other than dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A may explain some of the other clinical effects of aripiprazole. In terms of binding, aripiprazole has very high binding affinities (Ki) to dopamine D2 (0.34 nM), dopamine D3 (0.8 nM), and serotonin 5-HT2B (0.36 nM) receptors, and high binding affinities to serotonin 5-HT1A (1.7 nM) and serotonin 5-HT2A (3.4 nM) receptors.
Dosage recommendations for adults with schizophrenia suggest a starting and maintenance dose of 10 to 15 mg/d.2 Although the maximum dose is 30 mg/d, there is no evidence that doses >15 mg/d are superior to lower doses.5 In adolescents with schizophrenia, the product label recommends a starting dose of 2 mg/d, a maintenance dose of 10 mg/d, and a maximum dose of 30 mg/d. Recommendations for dosing in bipolar mania are similar. Dosing for the other indications is lower.
Efficacy in schizophrenia can be quantified using number needed to treat (NNT) for response vs placebo. The NNT answers the question “How many patients need to be randomized to aripiprazole vs placebo before expecting to encounter one additional responder?”6 From the 4 positive pivotal short-term acute schizophrenia trials for aripiprazole in adults,7-10 using the definition of response as a ≥30% decrease in the Positive and Negative Syndrome Scale (PANSS) total score or a Clinical Global Impressions–Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved), and pooling the data for aripiprazole doses 10 to 30 mg/d, response rates were 38% for aripiprazole vs 24% for placebo, resulting in a NNT of 8 (95% confidence interval [CI] 6 to 13).
From the 4 positive pivotal short-term acute bipolar mania trials for aripiprazole monotherapy in adults11-14 using the definition of response as a ≥50% decrease in the Young Mania Rating Scale (YMRS) total score, and pooling the data for aripiprazole doses 15 to 30 mg/d, response rates were 47% for aripiprazole vs 31% for placebo, resulting in a NNT of 7 (95% CI 5 to 11).1 Similar results were observed in the adjunctive aripiprazole acute bipolar mania trial15 where the NNT for response was also 7.1
Continue to: From the 2 positive pivotal short-term...
From the 2 positive pivotal short-term acute MDD trials for aripiprazole,16,17 using the definition of response as a ≥50% decrease in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score, and pooling the data (aripiprazole flexibly dosed 2 to 20 mg/d, with a median dose of 10 mg/d), response rates were 33% for aripiprazole vs 20% for placebo, resulting in a NNT of 8 (95% CI 6 to 17). After including a third trial not described in product labeling,18 the NNT became a more robust 7 (95% CI 5 to 11).1
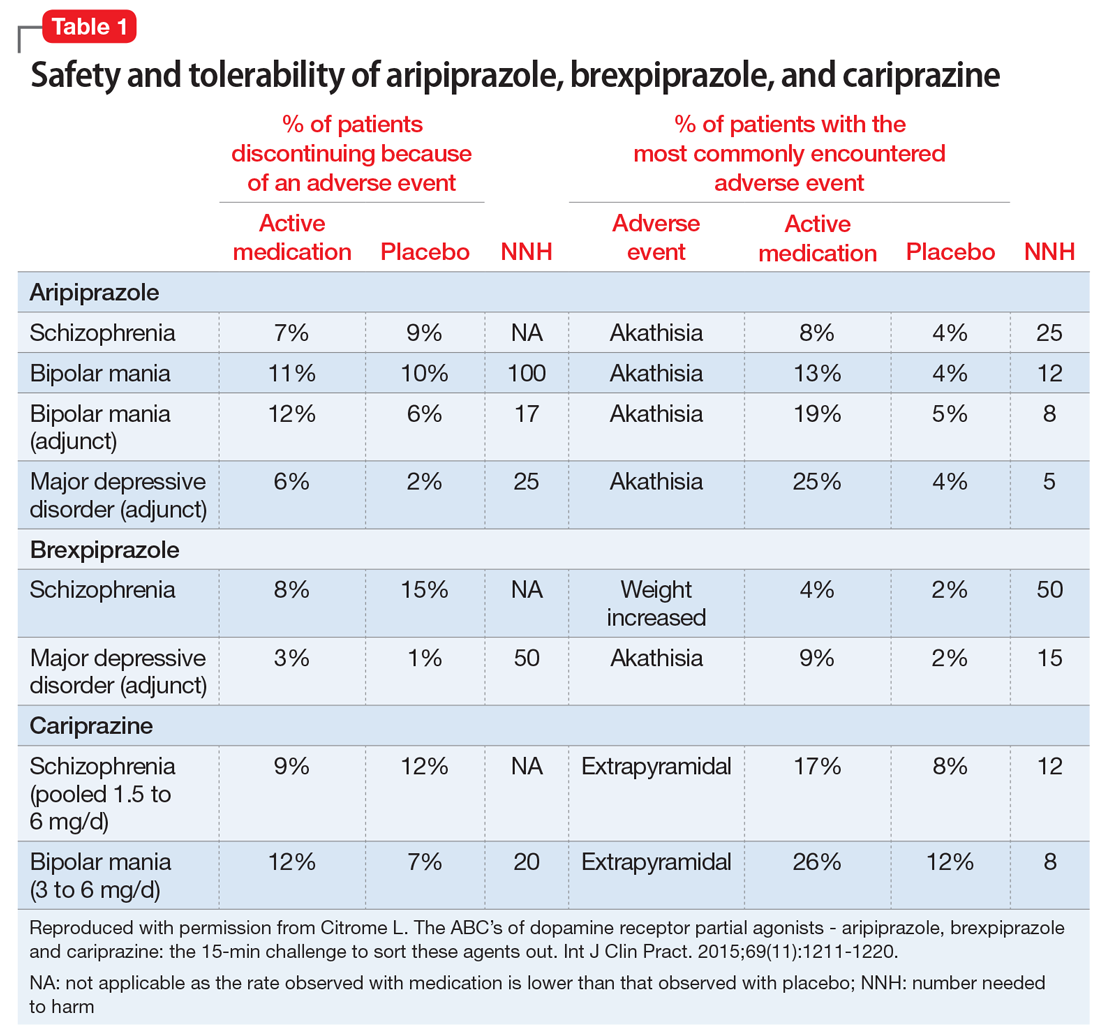
The most commonly encountered adverse events (incidence ≥5% and at least twice the rate of placebo) in the pivotal trials were akathisia (schizophrenia); akathisia, sedation, restlessness, tremor, and extrapyramidal disorder (bipolar mania, monotherapy); akathisia, insomnia, and extrapyramidal disorder (bipolar mania, adjunctive therapy); akathisia, restlessness, insomnia, constipation, fatigue, and blurred vision (MDD); and nausea (short-acting IM formulation). Table 11 summarizes the tolerability information regarding rate of discontinuation due to adverse events (an overall indicator of tolerability), and the incidence of the most common adverse event, together with the calculated number needed to harm (NNH). Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability; for the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability for these indications.
Brexpiprazole
Brexpiprazole was launched in the United States in 2015 for 2 indications: schizophrenia and the adjunctive treatment of MDD, both in adults.19 In terms of binding, brexpiprazole has very high binding affinities to serotonin 5-HT1A (0.12 nM), adrenergic α1B (0.17 nM), dopamine D2 (0.30 nM), serotonin 5-HT2A (0.47 nM), and adrenergic α2C (0.59 nM) receptors, and high binding affinities to dopamine D3 (1.1 nM), serotonin 5-HT2B (1.9 nM), adrenergic α1D (2.6 nM), serotonin 5-HT7 (3.7 nM), and adrenergic α1A (3.8 nM) receptors.
The 1-mg/d starting dose for brexpiprazole is lower than the recommended dose range of 2 to 4 mg/d for schizophrenia or the recommended dose of 2 mg/d for MDD.19 Thus brexpiprazole requires titration. The recommended rate of titration depends on the disease state being treated. For schizophrenia, the recommended titration schedule is to increase the dose to 2 mg/d on Day 5 through Day 7, then to 4 mg/d (the maximum recommended dose) on Day 8 based on the patient’s clinical response and tolerability. For MDD, there is the option of starting at 0.5 mg/d and the titration process is slower, with dosage increases occurring at weekly intervals, and with a maximum dose of 3 mg/d.
Using the identical definition of response in persons with schizophrenia as for the aripiprazole data described above, pooling together all the available data for the recommended target dose of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 studies listed in the product label,20,21 the percentage of responders was 46%, compared with 31% for the pooled placebo groups, yielding a NNT of 7 (95% CI 5 to 12).22
Continue to: For MDD...
For MDD, using the definition of response as a ≥50% decrease in MADRS total score, and pooling the results for brexpiprazole 1, 2, and 3 mg/d from the 2 pivotal trials,23,24 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI 8 to 26).22 Including the 1.5-mg/d dose arm and the placebo arm from the phase II study for which results are also available but not included in product labelling, the NNT becomes a slightly more robust 11 (95% CI 8 to 20).22 Although the magnitude of the NNT effect size is stronger for aripiprazole than for brexpiprazole, the 95% CIs do overlap.
The most commonly encountered adverse event in the short-term trials in schizophrenia (incidence ≥4% and at least twice the rate of placebo) was increased weight. The most commonly encountered adverse events in the short-term trials in MDD (incidence ≥5% and at least twice the rate of placebo) were increased weight and akathisia. Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability, and for MDD the NNH vs placebo on discontinuation because of an adverse event was 50, representing reasonable overall tolerability for this indication as well (Table 11).
Cariprazine
Cariprazine was launched in the United States in 2015 for 2 indications: schizophrenia, and the acute treatment of manic or mixed episodes associated with bipolar I disorder, both in adults.25 In terms of binding, cariprazine has very high binding affinities to dopamine D3 (0.085 nM), dopamine D2L (0.49 nM), serotonin 5-HT2B (0.58 nM), and dopamine D2S (0.69 nM) receptors, and high binding affinity to serotonin 5-HT1A (2.6 nM) receptors. Cariprazine forms 2 major metabolites, desmethyl cariprazine and didesmethyl cariprazine, that have in vitro receptor binding profiles similar to the parent drug. This latter metabolite, didesmethyl cariprazine, has a half-life of 1 to 3 weeks, and is the active moiety responsible for the majority of cariprazine’s effect when in steady state. Thus, following discontinuation of cariprazine, the decline in plasma concentrations of active drug will be slow.
The starting dose for cariprazine for schizophrenia, 1.5 mg/d, can be therapeutic. The dosage can be increased to 3 mg/d on Day 2. Depending upon clinical response and tolerability, further dose adjustments can be made in 1.5-mg or 3-mg increments to a maximum dose of 6 mg/d. For the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d; this can be done on Day 2. Cariprazine has been tested in clinical trials at higher doses; however, doses that exceed 6 mg/d did not confer significant additional benefit.25
A more conservative definition of response was used in the reporting of the cariprazine acute schizophrenia studies. This was simply a ≥30% decrease in the PANSS total score, and did not include the option of including patients who scored a 1 or 2 on the CGI-I. For pooled doses of cariprazine 1.5 to 6 mg/d,26-28 the percentage of responders was 31%, compared with 21% for the pooled placebo groups, yielding a NNT of 10 (95% CI 7 to 18).1 Although the magnitude of the NNT effect size is weaker for cariprazine than the other dopamine receptor partial agonists, the 95% CI overlaps with that of aripiprazole and brexpiprazole. An appropriately designed head-to-head trial would be necessary to directly test noninferiority.
Continue to: Pooling the data...
Pooling the data from the 3 pivotal short-term acute bipolar mania trials for cariprazine monotherapy in adults29-31 and using the definition of response as a ≥50% decrease in the YMRS total score for the recommended target dose of 3 to 6 mg/d, the percentage of responders was 57%, compared with 36% for the pooled placebo groups, yielding a NNT of 5 (95% CI 4 to 8).1 The magnitude of the NNT effect size is stronger for cariprazine than for aripiprazole, but the 95% CIs overlap.
The most commonly encountered adverse events in the short-term trials (incidence ≥5% and at least twice the rate of placebo) were extrapyramidal symptoms and akathisia (schizophrenia); and extrapyramidal symptoms, akathisia, dyspepsia, vomiting, somnolence, and restlessness (bipolar mania). In the schizophrenia studies, rates of discontinuation because of an adverse event were not higher for active medication vs placebo, suggesting excellent overall tolerability, and for bipolar disorder the NNH vs placebo on discontinuation because of an adverse event was 20, representing reasonable overall tolerability for this indication as well (Table 1).
Differences to consider
Indications. Although all 3 medications are approved for the treatment of schizophrenia, both aripiprazole and brexpiprazole are also approved for adjunctive treatment of MDD, and both aripiprazole and cariprazine are also approved for acute treatment of manic or mixed episodes associated with bipolar I disorder. In addition, aripiprazole is approved for a number of different disease states in pediatric patients. Aripiprazole has also been approved in a number of different formulations (oral and IM), but brexpiprazole and cariprazine are presently available only as oral pills (tablets for brexpiprazole, capsules for cariprazine).
Contraindications. All 3 agents are contraindicated in patients with a known hypersensitivity reaction to the product. All 3 also have a “black-box” warning for increased mortality in geriatric patients with dementia-related psychosis, a warning that is found in all antipsychotic medication labels. Additional black-box warnings are included regarding suicidality in the product labels of aripiprazole and brexpiprazole by virtue of their approval for the treatment of MDD.
Pharmacodynamics. All 3 agents describe a similar mechanism of action in their respective product labels: “efficacy … could be mediated through a combination of partial agonist activity at central dopamine D2 and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors.”2,19,25
Continue to: However, binding affinities differ...
However, binding affinities differ substantially among the agents (for example, cariprazine has only moderate binding affinity at serotonin 5-HT2A receptors [18.8 nM]), and differences also exist in terms of intrinsic activity at the receptors where partial agonism is operative. Compared with aripiprazole, brexpiprazole has lower intrinsic activity at the dopamine D2 receptor (and thus is expected to cause less akathisia), and has an approximately 10-fold higher affinity for serotonin 5-HT1A and 5-HT2A receptors, also potentially enhancing tolerability and perhaps anxiolytic activity.32,33 When cariprazine was compared with aripiprazole in functional assays for dopamine D2 and D3 receptors, similar D2 and higher D3 antagonist-partial agonist affinity and a 3- to 10-fold greater D3 vs D2 selectivity was observed for cariprazine.34 Whether specifically targeting the dopamine D3 receptor over the dopamine D2 receptor is clinically advantageous remains unknown, but in preclinical studies, dopamine D3–preferring agents may exert pro-cognitive effects.35-37 All 3 agents have only moderate binding affinities to histamine H1 receptors, thus sedation should not be prominent for any of them. None of the 3 agents have appreciable binding at muscarinic receptors, thus adverse effects related to antimuscarinic activity should not be present as well.
Schizophrenia is a heterogenous disorder. We know from clinical practice that patients respond differently to specific antipsychotics. Having different pharmacodynamic “fingerprints” to choose from allows for flexibility in treatment. Moreover, dopamine receptor partial agonists provide an alternative to the array of dopamine receptor antagonists, such as the other second-generation antipsychotics and all first-generation antipsychotics.
Dosing. Although all 3 agents are dosed once daily, only for aripiprazole is the recommended starting dose the same as the recommended maintenance dose in adults with schizophrenia or bipolar mania. Although the starting dose for cariprazine for schizophrenia can be therapeutic (1.5 mg/d), for the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d.
Half-life. Aripiprazole and brexpiprazole share a similar elimination half-life: approximately 75 hours and 94 hours for aripiprazole and its active metabolite dehydro-aripiprazole, respectively, and 91 hours and 86 hours for brexpiprazole and its major metabolite, DM-3411 (inactive), respectively. Cariprazine is strikingly different, with an elimination half-life of 2 to 4 days, and approximately 1 to 3 weeks for its active metabolite didesmethyl cariprazine.
Drug interactions. Both aripiprazole and brexpiprazole are metabolized via cytochrome P450 (CYP) 2D6 and CYP3A4, and thus the dose may need to be adjusted in the presence of CYP2D6 inhibitors or CYP3A4 inhibitors/inducers; with inhibitors, the dose is decreased by half or more, and with inducers, the dose is doubled. In contrast, cariprazine is primarily metabolized by CYP3A4 and thus potential drug–drug interactions are primarily focused on CYP3A4 inhibitors (decrease cariprazine dose by half) and inducers (co-prescribing of cariprazine with a CYP3A4 inducer is not recommended).
Continue to: Tolerability
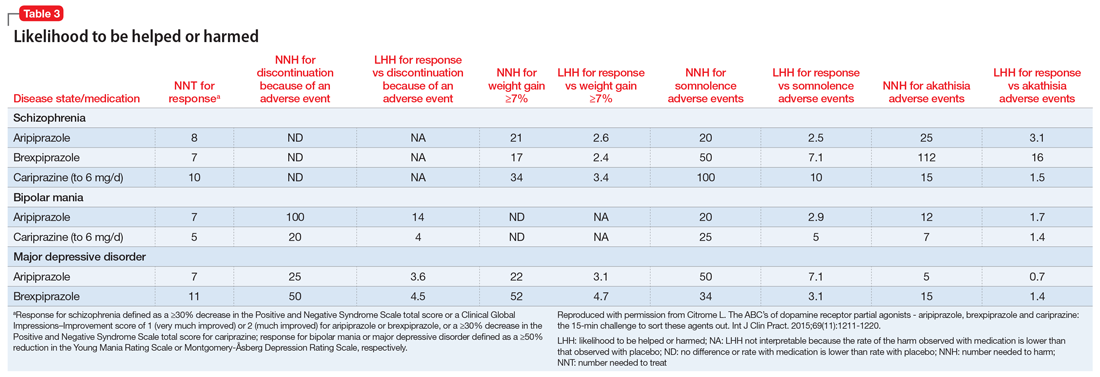
Tolerability. For all 3 agents, rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability.2,19,25 For the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability. For the most commonly encountered adverse event for each medication, the NNH values ranged from 5 (akathisia for aripiprazole for adjunctive use in MDD) to 50 (increased weight for brexpiprazole for schizophrenia). Of special interest are the adverse events of weight gain ≥7% from baseline, somnolence adverse events, and akathisia adverse events; the NNH values vs placebo for these are listed in Table 21. Pragmatically, NNH values <10 are likely to be more clinically relevant. For aripiprazole, brexpiprazole, and cariprazine for the treatment of schizophrenia, none of the NNH values for weight gain, somnolence, or akathisia were <10; however, this was not the case for the mood disorders, where in general, akathisia was more frequently observed for each of the agents. For the indication of schizophrenia, the rank order for propensity for weight gain appears to be brexpiprazole > aripiprazole > cariprazine, the propensity for somnolence aripiprazole > brexpiprazole > cariprazine, and the propensity for akathisia cariprazine > aripiprazole > brexpiprazole; however, this is by indirect comparison, and appropriately designed head-to-head clinical trials will be necessary in order to accurately assess these potential differences.

Because of the partial agonist activity at the dopamine D2 receptor, aripiprazole, brexpiprazole, and cariprazine are less likely to cause hyperprolactinemia than other first-line first- or second-generation antipsychotics. Other differentiating features of the dopamine receptor partial agonists compared with other choices include a relative lack of effect on the QT interval.38 In general, as predicted by their relatively lower binding affinities to histamine H1 receptors, the dopamine receptor partial agonists are not especially sedating.39
Likelihood to be helped or harmed
The concept of likelihood to be helped or harmed (LHH) can be useful to assess benefit vs risk, provided you select a relevant harm to contrast with the expected benefit.40 Table 31 provides the NNT for response, NNH for discontinuation because of an adverse event (where applicable), the NNHs for weight gain ≥7%, somnolence adverse events, and akathisia adverse events, together with the calculated LHH (where applicable). With the exception of aripiprazole for the treatment of MDD when comparing response vs akathisia, all LHH values are >1.0, and thus the benefit (response) would be encountered more often than the harm. When LHH values are ≥10, this can be interpreted that one would encounter a response at least 10 times more often than the adverse event of interest. This was observed for brexpiprazole for the treatment of schizophrenia when comparing response vs akathisia, for cariprazine for schizophrenia when comparing response vs somnolence, for aripiprazole for bipolar mania when comparing response vs discontinuation because of an adverse event, and for cariprazine for bipolar mania when comparing response vs somnolence.
Beyond acute studies
When treating patients with schizophrenia, delaying time to relapse is a main goal. In placebo-controlled randomized withdrawal studies of oral aripiprazole, brexpiprazole, and cariprazine in patients with schizophrenia, observed relapse rates vs placebo were reported, allowing the calculation of NNT vs placebo for the avoidance of relapse.41-44 These NNT values were similar and ranged from 4 to 5. For aripiprazole, relapse rates vs placebo in the 26-week study were 34% vs 57%, resulting in a NNT of 5 (95% CI 3 to 9); brexpiprazole, 52-week study, 13.5% vs 38.5%, NNT of 4 (95% CI 3 to 8); and cariprazine, 72-week study, 25% vs 47.5%, NNT of 5 (95% CI 3 to 11). In addition, cariprazine, 4.5 mg/d, has been directly compared with risperidone, 4 mg/d, in a 26-week double-blind study in non-geriatric adult patients with schizophrenia and predominant negative symptoms for at least 6 months.45 Cariprazine was superior to risperidone on the PANSS–Negative Factor Score, and response to treatment (decrease ≥20% in PANSS–Negative Factor Score) was achieved by more patients treated with cariprazine by 26 weeks than those treated with risperidone (69% vs 58%, NNT 9 [95% CI 5 to 44]).
Caveats
The harms discussed in this article are primarily from acute studies and do not reflect effects that can take time to develop, such as tardive dyskinesia, the long-term accumulation of body weight, and the development of insulin resistance/type 2 diabetes mellitus.40 The data presented are from carefully conducted registration trials that enrolled subjects who fulfilled restrictive inclusion/exclusion criteria. Such patients may differ from those encountered in routine clinical practice. Keep in mind that adverse events may differ in terms of impact and may not be clinically relevant if the adverse event is mild, time-limited, or easily managed. Moreover, different patients carry different propensities to experience different adverse events or to achieve a therapeutic response.
Continue to: Bottom Line
Bottom Line
Although aripiprazole, brexpiprazole, and cariprazine are all dopamine receptor partial agonists with demonstrated efficacy in psychiatric disorders, they differ in terms of available formulations, indications, pharmacodynamics, pharmacokinetics, titration requirements, and tolerability. Careful consideration of these factors can increase the likelihood of successful treatment.
Related Resources
- Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
- Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411.
- U.S. Food & Drug Administration. Drugs@FDA: FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/daf.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole lauroxil • Aristada
Aripiprazole monohydrate • Abilify Maintena
Brexpiprazole • Rexulti
Cariprazine • Vraylar
1. C
2. Otsuka. Abilify (aripiprazole) tablets, ABILIFY DISCMELT (aripiprazole) orally disintegrating tablets, ABILIFY (aripiprazole) oral solution, Abilify (aripiprazole) injection for intramuscular use only. Prescribing information. http://www.otsuka-us.com/Documents/Abilify.PI.pdf. Revised February 2018. Accessed March 14, 2018.
3. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
4. Citrome L. Long-acting injectable antipsychotics update: lengthening the dosing interval and expanding the diagnostic indications. Expert Rev Neurother. 2017;17(10):1029-1043.
5. Mace S, Taylor D. Aripiprazole: dose-response relationship in schizophrenia and schizoaffective disorder. CNS Drugs. 2009;23(9):773-780.
6. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
7. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry. 2002;63(9):763-771.
8. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2003;60(7):681-690.
9. McEvoy JP, Daniel DG, Carson WH Jr, et al. A randomized, double-blind, placebo-controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. J Psychiatr Res. 2007;41(11):895-905.
10. Cutler AJ, Marcus RN, Hardy SA, et al. The efficacy and safety of lower doses of aripiprazole for the treatment of patients with acute exacerbation of schizophrenia. CNS Spectr. 2006;11(9):691-702.
11. Sachs G, Sanchez R, Marcus R, et al; Aripiprazole Study Group. Aripiprazole in the treatment of acute manic or mixed episodes in patients with bipolar I disorder: a 3-week placebo-controlled study. J Psychopharmacol. 2006;20(4):536-546.
12. Keck PE Jr, Marcus R, Tourkodimitris S, et al; Aripiprazole Study Group. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-1658.
13. Keck PE, Orsulak PJ, Cutler AJ, et al; CN138-135 Study Group. Aripiprazole monotherapy in the treatment of acute bipolar I mania: a randomized, double-blind, placebo- and lithium-controlled study. J Affect Disord. 2009;112(1-3):36-49.
14. Young AH, Oren DA, Lowy A, et al. Aripiprazole monotherapy in acute mania: 12-week randomised placebo- and haloperidol-controlled study. Br J Psychiatry. 2009;194(1):40-48.
15. Vieta E, T’joen C, McQuade RD, et al. Efficacy of adjunctive aripiprazole to either valproate or lithium in bipolar mania patients partially nonresponsive to valproate/lithium monotherapy: a placebo-controlled study. Am J Psychiatry. 2008;165(10):1316-1325.
16. Marcus RN, McQuade RD, Carson WH, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a second multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol. 2008;28(2):156-165.
17. Berman RM, Marcus RN, Swanink R, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(6):843-853.
18. Berman RM, Fava M, Thase ME, et al. Aripiprazole augmentation in major depressive disorder: a double-blind, placebo-controlled study in patients with inadequate response to antidepressants. CNS Spectr. 2009;14(4):197-206.
19. Otsuka. Rexulti (brexpiprazole) tablets, for oral use. Prescribing information. http://www.otsuka-us.com/Products/Documents/Rexulti.PI.pdf. Revised February 2018. Accessed March 14, 2018.
20. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
21. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
22. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
23. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
24. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9):1232-1240.
25. Allergan. Vraylar (cariprazine) capsules, for oral use. Prescribing information. https://www.allergan.com/assets/pdf/vraylar_pi. Revised November 2017. Accessed March 14, 2018.
26. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
27. Durgam S, Cutler AJ, Lu K, et al. Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase 3, randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry. 2015;76(12):e1574-e1582.
28. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
29. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
30. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
31. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
32. Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. 2015;15(10):1219-1229.
33. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
34. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
35. Zimnisky R, Chang G, Gyertyán I, et al. Cariprazine, a dopamine D3-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology (Berl). 2013; 226(1):91-100.
36. Neill JC, Grayson B, Kiss B, et al. Effects of cariprazine, a novel antipsychotic, on cognitive deficit and negative symptoms in a rodent model of schizophrenia symptomatology. Eur Neuropsychopharmacol. 2016;26(1):3-14.
37. Gyertyán I, Kiss B, Sághy K, et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int. 2011;59(6):925-935.
38. Leucht S, Leucht C, Huhn M, et al. Sixty years of placebo-controlled antipsychotic drug trials in acute schizophrenia: systematic review, Bayesian meta-analysis, and meta-regression of efficacy predictors. Am J Psychiatry. 2017;174(10):927-942.
39. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
40. Citrome L, Kantrowitz J. Antipsychotics for the treatment of schizophrenia: likelihood to be helped or harmed, understanding proximal and distal benefits and risks. Expert Rev Neurother. 2008;8(7):1079-1091.
41. Pigott TA, Carson WH, Saha AR, et al; Aripiprazole Study Group. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry. 2003;64(9):1048-1056.
42. Fleischhacker WW, Hobart M, Ouyang J, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol. 2016;20(1):11-21.
43. Durgam S, Earley W, Li R, et al. Long-term cariprazine treatment for the prevention of relapse in patients with schizophrenia: a randomized, double-blind, placebo-controlled trial. Schizophr Res. 2016;176(2-3):264-271.
44. Citrome L. Schizophrenia relapse, patient considerations, and potential role of lurasidone. Patient Prefer Adherence. 2016;10:1529-1537.
45. Németh G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.
Aripiprazole, brexpiprazole, and cariprazine are dopamine receptor partial agonists, and on the surface, they appear similar. However, there are key differences in terms of available indications, formulations, pharmacodynamics, pharmacokinetics, dosing, drug interactions, tolerability, and other factors related to successful use.1 This review will cover the main points that the knowledgeable clinician will need to be mindful of when prescribing these agents.
Aripiprazole
Aripiprazole was launched in the United States in 20022 as the first dopamine receptor partial agonist approved for the treatment of schizophrenia; it later received additional indications for adults with manic or mixed episodes associated with bipolar I disorder and the maintenance treatment of bipolar I disorder, as well as for the adjunctive treatment of major depressive disorder (MDD). Pediatric indications include schizophrenia, acute treatment of manic or mixed episodes associated with bipolar I disorder, irritability associated with autistic disorder, and Tourette’s disorder.
Several formulations also became available, including a short-acting injection indicated for agitation associated with schizophrenia or bipolar mania, and oral disintegrating tablets and an oral solution that could substitute for the regular tablet. Presently the medication has gone “generic,” and not all formulations are being manufactured. The long-acting formulations of aripiprazole (aripiprazole monohydrate and aripiprazole lauroxil) are considered different products, each with its own product insert, with indications that are more limited in scope than for the oral forms.3,4
Although dopamine D2 receptor partial agonism is a relevant mechanism of action, partial agonist activity at serotonin 5-HT1A receptors and antagonist activity at 5-HT2A receptors also play a role.2 Actions at receptors other than dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A may explain some of the other clinical effects of aripiprazole. In terms of binding, aripiprazole has very high binding affinities (Ki) to dopamine D2 (0.34 nM), dopamine D3 (0.8 nM), and serotonin 5-HT2B (0.36 nM) receptors, and high binding affinities to serotonin 5-HT1A (1.7 nM) and serotonin 5-HT2A (3.4 nM) receptors.
Dosage recommendations for adults with schizophrenia suggest a starting and maintenance dose of 10 to 15 mg/d.2 Although the maximum dose is 30 mg/d, there is no evidence that doses >15 mg/d are superior to lower doses.5 In adolescents with schizophrenia, the product label recommends a starting dose of 2 mg/d, a maintenance dose of 10 mg/d, and a maximum dose of 30 mg/d. Recommendations for dosing in bipolar mania are similar. Dosing for the other indications is lower.
Efficacy in schizophrenia can be quantified using number needed to treat (NNT) for response vs placebo. The NNT answers the question “How many patients need to be randomized to aripiprazole vs placebo before expecting to encounter one additional responder?”6 From the 4 positive pivotal short-term acute schizophrenia trials for aripiprazole in adults,7-10 using the definition of response as a ≥30% decrease in the Positive and Negative Syndrome Scale (PANSS) total score or a Clinical Global Impressions–Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved), and pooling the data for aripiprazole doses 10 to 30 mg/d, response rates were 38% for aripiprazole vs 24% for placebo, resulting in a NNT of 8 (95% confidence interval [CI] 6 to 13).
From the 4 positive pivotal short-term acute bipolar mania trials for aripiprazole monotherapy in adults11-14 using the definition of response as a ≥50% decrease in the Young Mania Rating Scale (YMRS) total score, and pooling the data for aripiprazole doses 15 to 30 mg/d, response rates were 47% for aripiprazole vs 31% for placebo, resulting in a NNT of 7 (95% CI 5 to 11).1 Similar results were observed in the adjunctive aripiprazole acute bipolar mania trial15 where the NNT for response was also 7.1
Continue to: From the 2 positive pivotal short-term...
From the 2 positive pivotal short-term acute MDD trials for aripiprazole,16,17 using the definition of response as a ≥50% decrease in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score, and pooling the data (aripiprazole flexibly dosed 2 to 20 mg/d, with a median dose of 10 mg/d), response rates were 33% for aripiprazole vs 20% for placebo, resulting in a NNT of 8 (95% CI 6 to 17). After including a third trial not described in product labeling,18 the NNT became a more robust 7 (95% CI 5 to 11).1
The most commonly encountered adverse events (incidence ≥5% and at least twice the rate of placebo) in the pivotal trials were akathisia (schizophrenia); akathisia, sedation, restlessness, tremor, and extrapyramidal disorder (bipolar mania, monotherapy); akathisia, insomnia, and extrapyramidal disorder (bipolar mania, adjunctive therapy); akathisia, restlessness, insomnia, constipation, fatigue, and blurred vision (MDD); and nausea (short-acting IM formulation). Table 11 summarizes the tolerability information regarding rate of discontinuation due to adverse events (an overall indicator of tolerability), and the incidence of the most common adverse event, together with the calculated number needed to harm (NNH). Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability; for the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability for these indications.
Brexpiprazole
Brexpiprazole was launched in the United States in 2015 for 2 indications: schizophrenia and the adjunctive treatment of MDD, both in adults.19 In terms of binding, brexpiprazole has very high binding affinities to serotonin 5-HT1A (0.12 nM), adrenergic α1B (0.17 nM), dopamine D2 (0.30 nM), serotonin 5-HT2A (0.47 nM), and adrenergic α2C (0.59 nM) receptors, and high binding affinities to dopamine D3 (1.1 nM), serotonin 5-HT2B (1.9 nM), adrenergic α1D (2.6 nM), serotonin 5-HT7 (3.7 nM), and adrenergic α1A (3.8 nM) receptors.
The 1-mg/d starting dose for brexpiprazole is lower than the recommended dose range of 2 to 4 mg/d for schizophrenia or the recommended dose of 2 mg/d for MDD.19 Thus brexpiprazole requires titration. The recommended rate of titration depends on the disease state being treated. For schizophrenia, the recommended titration schedule is to increase the dose to 2 mg/d on Day 5 through Day 7, then to 4 mg/d (the maximum recommended dose) on Day 8 based on the patient’s clinical response and tolerability. For MDD, there is the option of starting at 0.5 mg/d and the titration process is slower, with dosage increases occurring at weekly intervals, and with a maximum dose of 3 mg/d.
Using the identical definition of response in persons with schizophrenia as for the aripiprazole data described above, pooling together all the available data for the recommended target dose of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 studies listed in the product label,20,21 the percentage of responders was 46%, compared with 31% for the pooled placebo groups, yielding a NNT of 7 (95% CI 5 to 12).22
Continue to: For MDD...
For MDD, using the definition of response as a ≥50% decrease in MADRS total score, and pooling the results for brexpiprazole 1, 2, and 3 mg/d from the 2 pivotal trials,23,24 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI 8 to 26).22 Including the 1.5-mg/d dose arm and the placebo arm from the phase II study for which results are also available but not included in product labelling, the NNT becomes a slightly more robust 11 (95% CI 8 to 20).22 Although the magnitude of the NNT effect size is stronger for aripiprazole than for brexpiprazole, the 95% CIs do overlap.
The most commonly encountered adverse event in the short-term trials in schizophrenia (incidence ≥4% and at least twice the rate of placebo) was increased weight. The most commonly encountered adverse events in the short-term trials in MDD (incidence ≥5% and at least twice the rate of placebo) were increased weight and akathisia. Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability, and for MDD the NNH vs placebo on discontinuation because of an adverse event was 50, representing reasonable overall tolerability for this indication as well (Table 11).
Cariprazine
Cariprazine was launched in the United States in 2015 for 2 indications: schizophrenia, and the acute treatment of manic or mixed episodes associated with bipolar I disorder, both in adults.25 In terms of binding, cariprazine has very high binding affinities to dopamine D3 (0.085 nM), dopamine D2L (0.49 nM), serotonin 5-HT2B (0.58 nM), and dopamine D2S (0.69 nM) receptors, and high binding affinity to serotonin 5-HT1A (2.6 nM) receptors. Cariprazine forms 2 major metabolites, desmethyl cariprazine and didesmethyl cariprazine, that have in vitro receptor binding profiles similar to the parent drug. This latter metabolite, didesmethyl cariprazine, has a half-life of 1 to 3 weeks, and is the active moiety responsible for the majority of cariprazine’s effect when in steady state. Thus, following discontinuation of cariprazine, the decline in plasma concentrations of active drug will be slow.
The starting dose for cariprazine for schizophrenia, 1.5 mg/d, can be therapeutic. The dosage can be increased to 3 mg/d on Day 2. Depending upon clinical response and tolerability, further dose adjustments can be made in 1.5-mg or 3-mg increments to a maximum dose of 6 mg/d. For the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d; this can be done on Day 2. Cariprazine has been tested in clinical trials at higher doses; however, doses that exceed 6 mg/d did not confer significant additional benefit.25
A more conservative definition of response was used in the reporting of the cariprazine acute schizophrenia studies. This was simply a ≥30% decrease in the PANSS total score, and did not include the option of including patients who scored a 1 or 2 on the CGI-I. For pooled doses of cariprazine 1.5 to 6 mg/d,26-28 the percentage of responders was 31%, compared with 21% for the pooled placebo groups, yielding a NNT of 10 (95% CI 7 to 18).1 Although the magnitude of the NNT effect size is weaker for cariprazine than the other dopamine receptor partial agonists, the 95% CI overlaps with that of aripiprazole and brexpiprazole. An appropriately designed head-to-head trial would be necessary to directly test noninferiority.
Continue to: Pooling the data...
Pooling the data from the 3 pivotal short-term acute bipolar mania trials for cariprazine monotherapy in adults29-31 and using the definition of response as a ≥50% decrease in the YMRS total score for the recommended target dose of 3 to 6 mg/d, the percentage of responders was 57%, compared with 36% for the pooled placebo groups, yielding a NNT of 5 (95% CI 4 to 8).1 The magnitude of the NNT effect size is stronger for cariprazine than for aripiprazole, but the 95% CIs overlap.
The most commonly encountered adverse events in the short-term trials (incidence ≥5% and at least twice the rate of placebo) were extrapyramidal symptoms and akathisia (schizophrenia); and extrapyramidal symptoms, akathisia, dyspepsia, vomiting, somnolence, and restlessness (bipolar mania). In the schizophrenia studies, rates of discontinuation because of an adverse event were not higher for active medication vs placebo, suggesting excellent overall tolerability, and for bipolar disorder the NNH vs placebo on discontinuation because of an adverse event was 20, representing reasonable overall tolerability for this indication as well (Table 1).
Differences to consider
Indications. Although all 3 medications are approved for the treatment of schizophrenia, both aripiprazole and brexpiprazole are also approved for adjunctive treatment of MDD, and both aripiprazole and cariprazine are also approved for acute treatment of manic or mixed episodes associated with bipolar I disorder. In addition, aripiprazole is approved for a number of different disease states in pediatric patients. Aripiprazole has also been approved in a number of different formulations (oral and IM), but brexpiprazole and cariprazine are presently available only as oral pills (tablets for brexpiprazole, capsules for cariprazine).
Contraindications. All 3 agents are contraindicated in patients with a known hypersensitivity reaction to the product. All 3 also have a “black-box” warning for increased mortality in geriatric patients with dementia-related psychosis, a warning that is found in all antipsychotic medication labels. Additional black-box warnings are included regarding suicidality in the product labels of aripiprazole and brexpiprazole by virtue of their approval for the treatment of MDD.
Pharmacodynamics. All 3 agents describe a similar mechanism of action in their respective product labels: “efficacy … could be mediated through a combination of partial agonist activity at central dopamine D2 and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors.”2,19,25
Continue to: However, binding affinities differ...
However, binding affinities differ substantially among the agents (for example, cariprazine has only moderate binding affinity at serotonin 5-HT2A receptors [18.8 nM]), and differences also exist in terms of intrinsic activity at the receptors where partial agonism is operative. Compared with aripiprazole, brexpiprazole has lower intrinsic activity at the dopamine D2 receptor (and thus is expected to cause less akathisia), and has an approximately 10-fold higher affinity for serotonin 5-HT1A and 5-HT2A receptors, also potentially enhancing tolerability and perhaps anxiolytic activity.32,33 When cariprazine was compared with aripiprazole in functional assays for dopamine D2 and D3 receptors, similar D2 and higher D3 antagonist-partial agonist affinity and a 3- to 10-fold greater D3 vs D2 selectivity was observed for cariprazine.34 Whether specifically targeting the dopamine D3 receptor over the dopamine D2 receptor is clinically advantageous remains unknown, but in preclinical studies, dopamine D3–preferring agents may exert pro-cognitive effects.35-37 All 3 agents have only moderate binding affinities to histamine H1 receptors, thus sedation should not be prominent for any of them. None of the 3 agents have appreciable binding at muscarinic receptors, thus adverse effects related to antimuscarinic activity should not be present as well.
Schizophrenia is a heterogenous disorder. We know from clinical practice that patients respond differently to specific antipsychotics. Having different pharmacodynamic “fingerprints” to choose from allows for flexibility in treatment. Moreover, dopamine receptor partial agonists provide an alternative to the array of dopamine receptor antagonists, such as the other second-generation antipsychotics and all first-generation antipsychotics.
Dosing. Although all 3 agents are dosed once daily, only for aripiprazole is the recommended starting dose the same as the recommended maintenance dose in adults with schizophrenia or bipolar mania. Although the starting dose for cariprazine for schizophrenia can be therapeutic (1.5 mg/d), for the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d.
Half-life. Aripiprazole and brexpiprazole share a similar elimination half-life: approximately 75 hours and 94 hours for aripiprazole and its active metabolite dehydro-aripiprazole, respectively, and 91 hours and 86 hours for brexpiprazole and its major metabolite, DM-3411 (inactive), respectively. Cariprazine is strikingly different, with an elimination half-life of 2 to 4 days, and approximately 1 to 3 weeks for its active metabolite didesmethyl cariprazine.
Drug interactions. Both aripiprazole and brexpiprazole are metabolized via cytochrome P450 (CYP) 2D6 and CYP3A4, and thus the dose may need to be adjusted in the presence of CYP2D6 inhibitors or CYP3A4 inhibitors/inducers; with inhibitors, the dose is decreased by half or more, and with inducers, the dose is doubled. In contrast, cariprazine is primarily metabolized by CYP3A4 and thus potential drug–drug interactions are primarily focused on CYP3A4 inhibitors (decrease cariprazine dose by half) and inducers (co-prescribing of cariprazine with a CYP3A4 inducer is not recommended).
Continue to: Tolerability
Tolerability. For all 3 agents, rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability.2,19,25 For the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability. For the most commonly encountered adverse event for each medication, the NNH values ranged from 5 (akathisia for aripiprazole for adjunctive use in MDD) to 50 (increased weight for brexpiprazole for schizophrenia). Of special interest are the adverse events of weight gain ≥7% from baseline, somnolence adverse events, and akathisia adverse events; the NNH values vs placebo for these are listed in Table 21. Pragmatically, NNH values <10 are likely to be more clinically relevant. For aripiprazole, brexpiprazole, and cariprazine for the treatment of schizophrenia, none of the NNH values for weight gain, somnolence, or akathisia were <10; however, this was not the case for the mood disorders, where in general, akathisia was more frequently observed for each of the agents. For the indication of schizophrenia, the rank order for propensity for weight gain appears to be brexpiprazole > aripiprazole > cariprazine, the propensity for somnolence aripiprazole > brexpiprazole > cariprazine, and the propensity for akathisia cariprazine > aripiprazole > brexpiprazole; however, this is by indirect comparison, and appropriately designed head-to-head clinical trials will be necessary in order to accurately assess these potential differences.
Because of the partial agonist activity at the dopamine D2 receptor, aripiprazole, brexpiprazole, and cariprazine are less likely to cause hyperprolactinemia than other first-line first- or second-generation antipsychotics. Other differentiating features of the dopamine receptor partial agonists compared with other choices include a relative lack of effect on the QT interval.38 In general, as predicted by their relatively lower binding affinities to histamine H1 receptors, the dopamine receptor partial agonists are not especially sedating.39
Likelihood to be helped or harmed
The concept of likelihood to be helped or harmed (LHH) can be useful to assess benefit vs risk, provided you select a relevant harm to contrast with the expected benefit.40 Table 31 provides the NNT for response, NNH for discontinuation because of an adverse event (where applicable), the NNHs for weight gain ≥7%, somnolence adverse events, and akathisia adverse events, together with the calculated LHH (where applicable). With the exception of aripiprazole for the treatment of MDD when comparing response vs akathisia, all LHH values are >1.0, and thus the benefit (response) would be encountered more often than the harm. When LHH values are ≥10, this can be interpreted that one would encounter a response at least 10 times more often than the adverse event of interest. This was observed for brexpiprazole for the treatment of schizophrenia when comparing response vs akathisia, for cariprazine for schizophrenia when comparing response vs somnolence, for aripiprazole for bipolar mania when comparing response vs discontinuation because of an adverse event, and for cariprazine for bipolar mania when comparing response vs somnolence.
Beyond acute studies
When treating patients with schizophrenia, delaying time to relapse is a main goal. In placebo-controlled randomized withdrawal studies of oral aripiprazole, brexpiprazole, and cariprazine in patients with schizophrenia, observed relapse rates vs placebo were reported, allowing the calculation of NNT vs placebo for the avoidance of relapse.41-44 These NNT values were similar and ranged from 4 to 5. For aripiprazole, relapse rates vs placebo in the 26-week study were 34% vs 57%, resulting in a NNT of 5 (95% CI 3 to 9); brexpiprazole, 52-week study, 13.5% vs 38.5%, NNT of 4 (95% CI 3 to 8); and cariprazine, 72-week study, 25% vs 47.5%, NNT of 5 (95% CI 3 to 11). In addition, cariprazine, 4.5 mg/d, has been directly compared with risperidone, 4 mg/d, in a 26-week double-blind study in non-geriatric adult patients with schizophrenia and predominant negative symptoms for at least 6 months.45 Cariprazine was superior to risperidone on the PANSS–Negative Factor Score, and response to treatment (decrease ≥20% in PANSS–Negative Factor Score) was achieved by more patients treated with cariprazine by 26 weeks than those treated with risperidone (69% vs 58%, NNT 9 [95% CI 5 to 44]).
Caveats
The harms discussed in this article are primarily from acute studies and do not reflect effects that can take time to develop, such as tardive dyskinesia, the long-term accumulation of body weight, and the development of insulin resistance/type 2 diabetes mellitus.40 The data presented are from carefully conducted registration trials that enrolled subjects who fulfilled restrictive inclusion/exclusion criteria. Such patients may differ from those encountered in routine clinical practice. Keep in mind that adverse events may differ in terms of impact and may not be clinically relevant if the adverse event is mild, time-limited, or easily managed. Moreover, different patients carry different propensities to experience different adverse events or to achieve a therapeutic response.
Continue to: Bottom Line
Bottom Line
Although aripiprazole, brexpiprazole, and cariprazine are all dopamine receptor partial agonists with demonstrated efficacy in psychiatric disorders, they differ in terms of available formulations, indications, pharmacodynamics, pharmacokinetics, titration requirements, and tolerability. Careful consideration of these factors can increase the likelihood of successful treatment.
Related Resources
- Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
- Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411.
- U.S. Food & Drug Administration. Drugs@FDA: FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/daf.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole lauroxil • Aristada
Aripiprazole monohydrate • Abilify Maintena
Brexpiprazole • Rexulti
Cariprazine • Vraylar
Aripiprazole, brexpiprazole, and cariprazine are dopamine receptor partial agonists, and on the surface, they appear similar. However, there are key differences in terms of available indications, formulations, pharmacodynamics, pharmacokinetics, dosing, drug interactions, tolerability, and other factors related to successful use.1 This review will cover the main points that the knowledgeable clinician will need to be mindful of when prescribing these agents.
Aripiprazole
Aripiprazole was launched in the United States in 20022 as the first dopamine receptor partial agonist approved for the treatment of schizophrenia; it later received additional indications for adults with manic or mixed episodes associated with bipolar I disorder and the maintenance treatment of bipolar I disorder, as well as for the adjunctive treatment of major depressive disorder (MDD). Pediatric indications include schizophrenia, acute treatment of manic or mixed episodes associated with bipolar I disorder, irritability associated with autistic disorder, and Tourette’s disorder.
Several formulations also became available, including a short-acting injection indicated for agitation associated with schizophrenia or bipolar mania, and oral disintegrating tablets and an oral solution that could substitute for the regular tablet. Presently the medication has gone “generic,” and not all formulations are being manufactured. The long-acting formulations of aripiprazole (aripiprazole monohydrate and aripiprazole lauroxil) are considered different products, each with its own product insert, with indications that are more limited in scope than for the oral forms.3,4
Although dopamine D2 receptor partial agonism is a relevant mechanism of action, partial agonist activity at serotonin 5-HT1A receptors and antagonist activity at 5-HT2A receptors also play a role.2 Actions at receptors other than dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A may explain some of the other clinical effects of aripiprazole. In terms of binding, aripiprazole has very high binding affinities (Ki) to dopamine D2 (0.34 nM), dopamine D3 (0.8 nM), and serotonin 5-HT2B (0.36 nM) receptors, and high binding affinities to serotonin 5-HT1A (1.7 nM) and serotonin 5-HT2A (3.4 nM) receptors.
Dosage recommendations for adults with schizophrenia suggest a starting and maintenance dose of 10 to 15 mg/d.2 Although the maximum dose is 30 mg/d, there is no evidence that doses >15 mg/d are superior to lower doses.5 In adolescents with schizophrenia, the product label recommends a starting dose of 2 mg/d, a maintenance dose of 10 mg/d, and a maximum dose of 30 mg/d. Recommendations for dosing in bipolar mania are similar. Dosing for the other indications is lower.
Efficacy in schizophrenia can be quantified using number needed to treat (NNT) for response vs placebo. The NNT answers the question “How many patients need to be randomized to aripiprazole vs placebo before expecting to encounter one additional responder?”6 From the 4 positive pivotal short-term acute schizophrenia trials for aripiprazole in adults,7-10 using the definition of response as a ≥30% decrease in the Positive and Negative Syndrome Scale (PANSS) total score or a Clinical Global Impressions–Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved), and pooling the data for aripiprazole doses 10 to 30 mg/d, response rates were 38% for aripiprazole vs 24% for placebo, resulting in a NNT of 8 (95% confidence interval [CI] 6 to 13).
From the 4 positive pivotal short-term acute bipolar mania trials for aripiprazole monotherapy in adults11-14 using the definition of response as a ≥50% decrease in the Young Mania Rating Scale (YMRS) total score, and pooling the data for aripiprazole doses 15 to 30 mg/d, response rates were 47% for aripiprazole vs 31% for placebo, resulting in a NNT of 7 (95% CI 5 to 11).1 Similar results were observed in the adjunctive aripiprazole acute bipolar mania trial15 where the NNT for response was also 7.1
Continue to: From the 2 positive pivotal short-term...
From the 2 positive pivotal short-term acute MDD trials for aripiprazole,16,17 using the definition of response as a ≥50% decrease in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score, and pooling the data (aripiprazole flexibly dosed 2 to 20 mg/d, with a median dose of 10 mg/d), response rates were 33% for aripiprazole vs 20% for placebo, resulting in a NNT of 8 (95% CI 6 to 17). After including a third trial not described in product labeling,18 the NNT became a more robust 7 (95% CI 5 to 11).1
The most commonly encountered adverse events (incidence ≥5% and at least twice the rate of placebo) in the pivotal trials were akathisia (schizophrenia); akathisia, sedation, restlessness, tremor, and extrapyramidal disorder (bipolar mania, monotherapy); akathisia, insomnia, and extrapyramidal disorder (bipolar mania, adjunctive therapy); akathisia, restlessness, insomnia, constipation, fatigue, and blurred vision (MDD); and nausea (short-acting IM formulation). Table 11 summarizes the tolerability information regarding rate of discontinuation due to adverse events (an overall indicator of tolerability), and the incidence of the most common adverse event, together with the calculated number needed to harm (NNH). Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability; for the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability for these indications.
Brexpiprazole
Brexpiprazole was launched in the United States in 2015 for 2 indications: schizophrenia and the adjunctive treatment of MDD, both in adults.19 In terms of binding, brexpiprazole has very high binding affinities to serotonin 5-HT1A (0.12 nM), adrenergic α1B (0.17 nM), dopamine D2 (0.30 nM), serotonin 5-HT2A (0.47 nM), and adrenergic α2C (0.59 nM) receptors, and high binding affinities to dopamine D3 (1.1 nM), serotonin 5-HT2B (1.9 nM), adrenergic α1D (2.6 nM), serotonin 5-HT7 (3.7 nM), and adrenergic α1A (3.8 nM) receptors.
The 1-mg/d starting dose for brexpiprazole is lower than the recommended dose range of 2 to 4 mg/d for schizophrenia or the recommended dose of 2 mg/d for MDD.19 Thus brexpiprazole requires titration. The recommended rate of titration depends on the disease state being treated. For schizophrenia, the recommended titration schedule is to increase the dose to 2 mg/d on Day 5 through Day 7, then to 4 mg/d (the maximum recommended dose) on Day 8 based on the patient’s clinical response and tolerability. For MDD, there is the option of starting at 0.5 mg/d and the titration process is slower, with dosage increases occurring at weekly intervals, and with a maximum dose of 3 mg/d.
Using the identical definition of response in persons with schizophrenia as for the aripiprazole data described above, pooling together all the available data for the recommended target dose of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 studies listed in the product label,20,21 the percentage of responders was 46%, compared with 31% for the pooled placebo groups, yielding a NNT of 7 (95% CI 5 to 12).22
Continue to: For MDD...
For MDD, using the definition of response as a ≥50% decrease in MADRS total score, and pooling the results for brexpiprazole 1, 2, and 3 mg/d from the 2 pivotal trials,23,24 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI 8 to 26).22 Including the 1.5-mg/d dose arm and the placebo arm from the phase II study for which results are also available but not included in product labelling, the NNT becomes a slightly more robust 11 (95% CI 8 to 20).22 Although the magnitude of the NNT effect size is stronger for aripiprazole than for brexpiprazole, the 95% CIs do overlap.
The most commonly encountered adverse event in the short-term trials in schizophrenia (incidence ≥4% and at least twice the rate of placebo) was increased weight. The most commonly encountered adverse events in the short-term trials in MDD (incidence ≥5% and at least twice the rate of placebo) were increased weight and akathisia. Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability, and for MDD the NNH vs placebo on discontinuation because of an adverse event was 50, representing reasonable overall tolerability for this indication as well (Table 11).
Cariprazine
Cariprazine was launched in the United States in 2015 for 2 indications: schizophrenia, and the acute treatment of manic or mixed episodes associated with bipolar I disorder, both in adults.25 In terms of binding, cariprazine has very high binding affinities to dopamine D3 (0.085 nM), dopamine D2L (0.49 nM), serotonin 5-HT2B (0.58 nM), and dopamine D2S (0.69 nM) receptors, and high binding affinity to serotonin 5-HT1A (2.6 nM) receptors. Cariprazine forms 2 major metabolites, desmethyl cariprazine and didesmethyl cariprazine, that have in vitro receptor binding profiles similar to the parent drug. This latter metabolite, didesmethyl cariprazine, has a half-life of 1 to 3 weeks, and is the active moiety responsible for the majority of cariprazine’s effect when in steady state. Thus, following discontinuation of cariprazine, the decline in plasma concentrations of active drug will be slow.
The starting dose for cariprazine for schizophrenia, 1.5 mg/d, can be therapeutic. The dosage can be increased to 3 mg/d on Day 2. Depending upon clinical response and tolerability, further dose adjustments can be made in 1.5-mg or 3-mg increments to a maximum dose of 6 mg/d. For the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d; this can be done on Day 2. Cariprazine has been tested in clinical trials at higher doses; however, doses that exceed 6 mg/d did not confer significant additional benefit.25
A more conservative definition of response was used in the reporting of the cariprazine acute schizophrenia studies. This was simply a ≥30% decrease in the PANSS total score, and did not include the option of including patients who scored a 1 or 2 on the CGI-I. For pooled doses of cariprazine 1.5 to 6 mg/d,26-28 the percentage of responders was 31%, compared with 21% for the pooled placebo groups, yielding a NNT of 10 (95% CI 7 to 18).1 Although the magnitude of the NNT effect size is weaker for cariprazine than the other dopamine receptor partial agonists, the 95% CI overlaps with that of aripiprazole and brexpiprazole. An appropriately designed head-to-head trial would be necessary to directly test noninferiority.
Continue to: Pooling the data...
Pooling the data from the 3 pivotal short-term acute bipolar mania trials for cariprazine monotherapy in adults29-31 and using the definition of response as a ≥50% decrease in the YMRS total score for the recommended target dose of 3 to 6 mg/d, the percentage of responders was 57%, compared with 36% for the pooled placebo groups, yielding a NNT of 5 (95% CI 4 to 8).1 The magnitude of the NNT effect size is stronger for cariprazine than for aripiprazole, but the 95% CIs overlap.
The most commonly encountered adverse events in the short-term trials (incidence ≥5% and at least twice the rate of placebo) were extrapyramidal symptoms and akathisia (schizophrenia); and extrapyramidal symptoms, akathisia, dyspepsia, vomiting, somnolence, and restlessness (bipolar mania). In the schizophrenia studies, rates of discontinuation because of an adverse event were not higher for active medication vs placebo, suggesting excellent overall tolerability, and for bipolar disorder the NNH vs placebo on discontinuation because of an adverse event was 20, representing reasonable overall tolerability for this indication as well (Table 1).
Differences to consider
Indications. Although all 3 medications are approved for the treatment of schizophrenia, both aripiprazole and brexpiprazole are also approved for adjunctive treatment of MDD, and both aripiprazole and cariprazine are also approved for acute treatment of manic or mixed episodes associated with bipolar I disorder. In addition, aripiprazole is approved for a number of different disease states in pediatric patients. Aripiprazole has also been approved in a number of different formulations (oral and IM), but brexpiprazole and cariprazine are presently available only as oral pills (tablets for brexpiprazole, capsules for cariprazine).
Contraindications. All 3 agents are contraindicated in patients with a known hypersensitivity reaction to the product. All 3 also have a “black-box” warning for increased mortality in geriatric patients with dementia-related psychosis, a warning that is found in all antipsychotic medication labels. Additional black-box warnings are included regarding suicidality in the product labels of aripiprazole and brexpiprazole by virtue of their approval for the treatment of MDD.
Pharmacodynamics. All 3 agents describe a similar mechanism of action in their respective product labels: “efficacy … could be mediated through a combination of partial agonist activity at central dopamine D2 and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors.”2,19,25
Continue to: However, binding affinities differ...
However, binding affinities differ substantially among the agents (for example, cariprazine has only moderate binding affinity at serotonin 5-HT2A receptors [18.8 nM]), and differences also exist in terms of intrinsic activity at the receptors where partial agonism is operative. Compared with aripiprazole, brexpiprazole has lower intrinsic activity at the dopamine D2 receptor (and thus is expected to cause less akathisia), and has an approximately 10-fold higher affinity for serotonin 5-HT1A and 5-HT2A receptors, also potentially enhancing tolerability and perhaps anxiolytic activity.32,33 When cariprazine was compared with aripiprazole in functional assays for dopamine D2 and D3 receptors, similar D2 and higher D3 antagonist-partial agonist affinity and a 3- to 10-fold greater D3 vs D2 selectivity was observed for cariprazine.34 Whether specifically targeting the dopamine D3 receptor over the dopamine D2 receptor is clinically advantageous remains unknown, but in preclinical studies, dopamine D3–preferring agents may exert pro-cognitive effects.35-37 All 3 agents have only moderate binding affinities to histamine H1 receptors, thus sedation should not be prominent for any of them. None of the 3 agents have appreciable binding at muscarinic receptors, thus adverse effects related to antimuscarinic activity should not be present as well.
Schizophrenia is a heterogenous disorder. We know from clinical practice that patients respond differently to specific antipsychotics. Having different pharmacodynamic “fingerprints” to choose from allows for flexibility in treatment. Moreover, dopamine receptor partial agonists provide an alternative to the array of dopamine receptor antagonists, such as the other second-generation antipsychotics and all first-generation antipsychotics.
Dosing. Although all 3 agents are dosed once daily, only for aripiprazole is the recommended starting dose the same as the recommended maintenance dose in adults with schizophrenia or bipolar mania. Although the starting dose for cariprazine for schizophrenia can be therapeutic (1.5 mg/d), for the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d.
Half-life. Aripiprazole and brexpiprazole share a similar elimination half-life: approximately 75 hours and 94 hours for aripiprazole and its active metabolite dehydro-aripiprazole, respectively, and 91 hours and 86 hours for brexpiprazole and its major metabolite, DM-3411 (inactive), respectively. Cariprazine is strikingly different, with an elimination half-life of 2 to 4 days, and approximately 1 to 3 weeks for its active metabolite didesmethyl cariprazine.
Drug interactions. Both aripiprazole and brexpiprazole are metabolized via cytochrome P450 (CYP) 2D6 and CYP3A4, and thus the dose may need to be adjusted in the presence of CYP2D6 inhibitors or CYP3A4 inhibitors/inducers; with inhibitors, the dose is decreased by half or more, and with inducers, the dose is doubled. In contrast, cariprazine is primarily metabolized by CYP3A4 and thus potential drug–drug interactions are primarily focused on CYP3A4 inhibitors (decrease cariprazine dose by half) and inducers (co-prescribing of cariprazine with a CYP3A4 inducer is not recommended).
Continue to: Tolerability
Tolerability. For all 3 agents, rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability.2,19,25 For the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability. For the most commonly encountered adverse event for each medication, the NNH values ranged from 5 (akathisia for aripiprazole for adjunctive use in MDD) to 50 (increased weight for brexpiprazole for schizophrenia). Of special interest are the adverse events of weight gain ≥7% from baseline, somnolence adverse events, and akathisia adverse events; the NNH values vs placebo for these are listed in Table 21. Pragmatically, NNH values <10 are likely to be more clinically relevant. For aripiprazole, brexpiprazole, and cariprazine for the treatment of schizophrenia, none of the NNH values for weight gain, somnolence, or akathisia were <10; however, this was not the case for the mood disorders, where in general, akathisia was more frequently observed for each of the agents. For the indication of schizophrenia, the rank order for propensity for weight gain appears to be brexpiprazole > aripiprazole > cariprazine, the propensity for somnolence aripiprazole > brexpiprazole > cariprazine, and the propensity for akathisia cariprazine > aripiprazole > brexpiprazole; however, this is by indirect comparison, and appropriately designed head-to-head clinical trials will be necessary in order to accurately assess these potential differences.
Because of the partial agonist activity at the dopamine D2 receptor, aripiprazole, brexpiprazole, and cariprazine are less likely to cause hyperprolactinemia than other first-line first- or second-generation antipsychotics. Other differentiating features of the dopamine receptor partial agonists compared with other choices include a relative lack of effect on the QT interval.38 In general, as predicted by their relatively lower binding affinities to histamine H1 receptors, the dopamine receptor partial agonists are not especially sedating.39
Likelihood to be helped or harmed
The concept of likelihood to be helped or harmed (LHH) can be useful to assess benefit vs risk, provided you select a relevant harm to contrast with the expected benefit.40 Table 31 provides the NNT for response, NNH for discontinuation because of an adverse event (where applicable), the NNHs for weight gain ≥7%, somnolence adverse events, and akathisia adverse events, together with the calculated LHH (where applicable). With the exception of aripiprazole for the treatment of MDD when comparing response vs akathisia, all LHH values are >1.0, and thus the benefit (response) would be encountered more often than the harm. When LHH values are ≥10, this can be interpreted that one would encounter a response at least 10 times more often than the adverse event of interest. This was observed for brexpiprazole for the treatment of schizophrenia when comparing response vs akathisia, for cariprazine for schizophrenia when comparing response vs somnolence, for aripiprazole for bipolar mania when comparing response vs discontinuation because of an adverse event, and for cariprazine for bipolar mania when comparing response vs somnolence.
Beyond acute studies
When treating patients with schizophrenia, delaying time to relapse is a main goal. In placebo-controlled randomized withdrawal studies of oral aripiprazole, brexpiprazole, and cariprazine in patients with schizophrenia, observed relapse rates vs placebo were reported, allowing the calculation of NNT vs placebo for the avoidance of relapse.41-44 These NNT values were similar and ranged from 4 to 5. For aripiprazole, relapse rates vs placebo in the 26-week study were 34% vs 57%, resulting in a NNT of 5 (95% CI 3 to 9); brexpiprazole, 52-week study, 13.5% vs 38.5%, NNT of 4 (95% CI 3 to 8); and cariprazine, 72-week study, 25% vs 47.5%, NNT of 5 (95% CI 3 to 11). In addition, cariprazine, 4.5 mg/d, has been directly compared with risperidone, 4 mg/d, in a 26-week double-blind study in non-geriatric adult patients with schizophrenia and predominant negative symptoms for at least 6 months.45 Cariprazine was superior to risperidone on the PANSS–Negative Factor Score, and response to treatment (decrease ≥20% in PANSS–Negative Factor Score) was achieved by more patients treated with cariprazine by 26 weeks than those treated with risperidone (69% vs 58%, NNT 9 [95% CI 5 to 44]).
Caveats
The harms discussed in this article are primarily from acute studies and do not reflect effects that can take time to develop, such as tardive dyskinesia, the long-term accumulation of body weight, and the development of insulin resistance/type 2 diabetes mellitus.40 The data presented are from carefully conducted registration trials that enrolled subjects who fulfilled restrictive inclusion/exclusion criteria. Such patients may differ from those encountered in routine clinical practice. Keep in mind that adverse events may differ in terms of impact and may not be clinically relevant if the adverse event is mild, time-limited, or easily managed. Moreover, different patients carry different propensities to experience different adverse events or to achieve a therapeutic response.
Continue to: Bottom Line
Bottom Line
Although aripiprazole, brexpiprazole, and cariprazine are all dopamine receptor partial agonists with demonstrated efficacy in psychiatric disorders, they differ in terms of available formulations, indications, pharmacodynamics, pharmacokinetics, titration requirements, and tolerability. Careful consideration of these factors can increase the likelihood of successful treatment.
Related Resources
- Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
- Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411.
- U.S. Food & Drug Administration. Drugs@FDA: FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/daf.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole lauroxil • Aristada
Aripiprazole monohydrate • Abilify Maintena
Brexpiprazole • Rexulti
Cariprazine • Vraylar
1. C
2. Otsuka. Abilify (aripiprazole) tablets, ABILIFY DISCMELT (aripiprazole) orally disintegrating tablets, ABILIFY (aripiprazole) oral solution, Abilify (aripiprazole) injection for intramuscular use only. Prescribing information. http://www.otsuka-us.com/Documents/Abilify.PI.pdf. Revised February 2018. Accessed March 14, 2018.
3. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
4. Citrome L. Long-acting injectable antipsychotics update: lengthening the dosing interval and expanding the diagnostic indications. Expert Rev Neurother. 2017;17(10):1029-1043.
5. Mace S, Taylor D. Aripiprazole: dose-response relationship in schizophrenia and schizoaffective disorder. CNS Drugs. 2009;23(9):773-780.
6. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
7. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry. 2002;63(9):763-771.
8. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2003;60(7):681-690.
9. McEvoy JP, Daniel DG, Carson WH Jr, et al. A randomized, double-blind, placebo-controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. J Psychiatr Res. 2007;41(11):895-905.
10. Cutler AJ, Marcus RN, Hardy SA, et al. The efficacy and safety of lower doses of aripiprazole for the treatment of patients with acute exacerbation of schizophrenia. CNS Spectr. 2006;11(9):691-702.
11. Sachs G, Sanchez R, Marcus R, et al; Aripiprazole Study Group. Aripiprazole in the treatment of acute manic or mixed episodes in patients with bipolar I disorder: a 3-week placebo-controlled study. J Psychopharmacol. 2006;20(4):536-546.
12. Keck PE Jr, Marcus R, Tourkodimitris S, et al; Aripiprazole Study Group. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-1658.
13. Keck PE, Orsulak PJ, Cutler AJ, et al; CN138-135 Study Group. Aripiprazole monotherapy in the treatment of acute bipolar I mania: a randomized, double-blind, placebo- and lithium-controlled study. J Affect Disord. 2009;112(1-3):36-49.
14. Young AH, Oren DA, Lowy A, et al. Aripiprazole monotherapy in acute mania: 12-week randomised placebo- and haloperidol-controlled study. Br J Psychiatry. 2009;194(1):40-48.
15. Vieta E, T’joen C, McQuade RD, et al. Efficacy of adjunctive aripiprazole to either valproate or lithium in bipolar mania patients partially nonresponsive to valproate/lithium monotherapy: a placebo-controlled study. Am J Psychiatry. 2008;165(10):1316-1325.
16. Marcus RN, McQuade RD, Carson WH, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a second multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol. 2008;28(2):156-165.
17. Berman RM, Marcus RN, Swanink R, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(6):843-853.
18. Berman RM, Fava M, Thase ME, et al. Aripiprazole augmentation in major depressive disorder: a double-blind, placebo-controlled study in patients with inadequate response to antidepressants. CNS Spectr. 2009;14(4):197-206.
19. Otsuka. Rexulti (brexpiprazole) tablets, for oral use. Prescribing information. http://www.otsuka-us.com/Products/Documents/Rexulti.PI.pdf. Revised February 2018. Accessed March 14, 2018.
20. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
21. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
22. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
23. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
24. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9):1232-1240.
25. Allergan. Vraylar (cariprazine) capsules, for oral use. Prescribing information. https://www.allergan.com/assets/pdf/vraylar_pi. Revised November 2017. Accessed March 14, 2018.
26. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
27. Durgam S, Cutler AJ, Lu K, et al. Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase 3, randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry. 2015;76(12):e1574-e1582.
28. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
29. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
30. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
31. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
32. Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. 2015;15(10):1219-1229.
33. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
34. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
35. Zimnisky R, Chang G, Gyertyán I, et al. Cariprazine, a dopamine D3-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology (Berl). 2013; 226(1):91-100.
36. Neill JC, Grayson B, Kiss B, et al. Effects of cariprazine, a novel antipsychotic, on cognitive deficit and negative symptoms in a rodent model of schizophrenia symptomatology. Eur Neuropsychopharmacol. 2016;26(1):3-14.
37. Gyertyán I, Kiss B, Sághy K, et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int. 2011;59(6):925-935.
38. Leucht S, Leucht C, Huhn M, et al. Sixty years of placebo-controlled antipsychotic drug trials in acute schizophrenia: systematic review, Bayesian meta-analysis, and meta-regression of efficacy predictors. Am J Psychiatry. 2017;174(10):927-942.
39. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
40. Citrome L, Kantrowitz J. Antipsychotics for the treatment of schizophrenia: likelihood to be helped or harmed, understanding proximal and distal benefits and risks. Expert Rev Neurother. 2008;8(7):1079-1091.
41. Pigott TA, Carson WH, Saha AR, et al; Aripiprazole Study Group. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry. 2003;64(9):1048-1056.
42. Fleischhacker WW, Hobart M, Ouyang J, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol. 2016;20(1):11-21.
43. Durgam S, Earley W, Li R, et al. Long-term cariprazine treatment for the prevention of relapse in patients with schizophrenia: a randomized, double-blind, placebo-controlled trial. Schizophr Res. 2016;176(2-3):264-271.
44. Citrome L. Schizophrenia relapse, patient considerations, and potential role of lurasidone. Patient Prefer Adherence. 2016;10:1529-1537.
45. Németh G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.
1. C
2. Otsuka. Abilify (aripiprazole) tablets, ABILIFY DISCMELT (aripiprazole) orally disintegrating tablets, ABILIFY (aripiprazole) oral solution, Abilify (aripiprazole) injection for intramuscular use only. Prescribing information. http://www.otsuka-us.com/Documents/Abilify.PI.pdf. Revised February 2018. Accessed March 14, 2018.
3. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
4. Citrome L. Long-acting injectable antipsychotics update: lengthening the dosing interval and expanding the diagnostic indications. Expert Rev Neurother. 2017;17(10):1029-1043.
5. Mace S, Taylor D. Aripiprazole: dose-response relationship in schizophrenia and schizoaffective disorder. CNS Drugs. 2009;23(9):773-780.
6. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
7. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry. 2002;63(9):763-771.
8. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2003;60(7):681-690.
9. McEvoy JP, Daniel DG, Carson WH Jr, et al. A randomized, double-blind, placebo-controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. J Psychiatr Res. 2007;41(11):895-905.
10. Cutler AJ, Marcus RN, Hardy SA, et al. The efficacy and safety of lower doses of aripiprazole for the treatment of patients with acute exacerbation of schizophrenia. CNS Spectr. 2006;11(9):691-702.
11. Sachs G, Sanchez R, Marcus R, et al; Aripiprazole Study Group. Aripiprazole in the treatment of acute manic or mixed episodes in patients with bipolar I disorder: a 3-week placebo-controlled study. J Psychopharmacol. 2006;20(4):536-546.
12. Keck PE Jr, Marcus R, Tourkodimitris S, et al; Aripiprazole Study Group. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-1658.
13. Keck PE, Orsulak PJ, Cutler AJ, et al; CN138-135 Study Group. Aripiprazole monotherapy in the treatment of acute bipolar I mania: a randomized, double-blind, placebo- and lithium-controlled study. J Affect Disord. 2009;112(1-3):36-49.
14. Young AH, Oren DA, Lowy A, et al. Aripiprazole monotherapy in acute mania: 12-week randomised placebo- and haloperidol-controlled study. Br J Psychiatry. 2009;194(1):40-48.
15. Vieta E, T’joen C, McQuade RD, et al. Efficacy of adjunctive aripiprazole to either valproate or lithium in bipolar mania patients partially nonresponsive to valproate/lithium monotherapy: a placebo-controlled study. Am J Psychiatry. 2008;165(10):1316-1325.
16. Marcus RN, McQuade RD, Carson WH, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a second multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol. 2008;28(2):156-165.
17. Berman RM, Marcus RN, Swanink R, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(6):843-853.
18. Berman RM, Fava M, Thase ME, et al. Aripiprazole augmentation in major depressive disorder: a double-blind, placebo-controlled study in patients with inadequate response to antidepressants. CNS Spectr. 2009;14(4):197-206.
19. Otsuka. Rexulti (brexpiprazole) tablets, for oral use. Prescribing information. http://www.otsuka-us.com/Products/Documents/Rexulti.PI.pdf. Revised February 2018. Accessed March 14, 2018.
20. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
21. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
22. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
23. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
24. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9):1232-1240.
25. Allergan. Vraylar (cariprazine) capsules, for oral use. Prescribing information. https://www.allergan.com/assets/pdf/vraylar_pi. Revised November 2017. Accessed March 14, 2018.
26. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
27. Durgam S, Cutler AJ, Lu K, et al. Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase 3, randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry. 2015;76(12):e1574-e1582.
28. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
29. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
30. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
31. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
32. Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. 2015;15(10):1219-1229.
33. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
34. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
35. Zimnisky R, Chang G, Gyertyán I, et al. Cariprazine, a dopamine D3-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology (Berl). 2013; 226(1):91-100.
36. Neill JC, Grayson B, Kiss B, et al. Effects of cariprazine, a novel antipsychotic, on cognitive deficit and negative symptoms in a rodent model of schizophrenia symptomatology. Eur Neuropsychopharmacol. 2016;26(1):3-14.
37. Gyertyán I, Kiss B, Sághy K, et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int. 2011;59(6):925-935.
38. Leucht S, Leucht C, Huhn M, et al. Sixty years of placebo-controlled antipsychotic drug trials in acute schizophrenia: systematic review, Bayesian meta-analysis, and meta-regression of efficacy predictors. Am J Psychiatry. 2017;174(10):927-942.
39. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
40. Citrome L, Kantrowitz J. Antipsychotics for the treatment of schizophrenia: likelihood to be helped or harmed, understanding proximal and distal benefits and risks. Expert Rev Neurother. 2008;8(7):1079-1091.
41. Pigott TA, Carson WH, Saha AR, et al; Aripiprazole Study Group. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry. 2003;64(9):1048-1056.
42. Fleischhacker WW, Hobart M, Ouyang J, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol. 2016;20(1):11-21.
43. Durgam S, Earley W, Li R, et al. Long-term cariprazine treatment for the prevention of relapse in patients with schizophrenia: a randomized, double-blind, placebo-controlled trial. Schizophr Res. 2016;176(2-3):264-271.
44. Citrome L. Schizophrenia relapse, patient considerations, and potential role of lurasidone. Patient Prefer Adherence. 2016;10:1529-1537.
45. Németh G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.

Anxiety and joint hypermobility: An unexpected association
Joint hypermobility syndrome (JHS)—also known as Ehlers-Danlos type 3–hypermobile type (hEDS)1—is a poorly recognized connective tissue disorder characterized by increased joint laxity that may affect 10% to 25% of the general population.2 Researchers are increasingly recognizing an association between JHS/hEDS and psychiatric symptoms and disorders, specifically anxiety. In this review, we describe the clinical presentation of JHS/hEDS, propose a new “Neuroconnective phenotype” based on the link between anxiety and JHS/hEDS, and discuss factors to consider when treating anxiety in a patient who has JHS/hEDS.
JHS/hEDS: A complex disorder
Although JHS/hEDS is a heritable condition, several factors are known to influence its prevalence and visibility, including age, sex, and ethnicity; the prevalence is higher among younger patients, females, and African Americans.2 Its known basis is the type and distribution pattern of collagen, and one of the key features used to identify this syndrome is greater joint laxity, meaning increased distensibility of the joints in passive movements as well as a hypermobility in active movements.
Although first described by two dermatologists (Edvard Ehlers and Henri-Alexandre Danlos) at the beginning of the 20th century, JHS/hEDS is now considered a multi-systemic condition. Thus, JHS/hEDS includes a wide range of musculoskeletal features, and over the recent years, extra-articular symptoms, such as easy bruising or hypertrophic scarring, have gained recognition.3 Moreover, individuals with JHS/hEDS frequently present with stress-sensitive illnesses, such as fibromyalgia, or chronic fatigue syndrome.4 The Table2,5,6 provides a description of musculoskeletal and extra-articular features of JHS/hEDS.

The link between JHS/hEDS and anxiety
Psychiatric symptoms are being increasingly recognized as a key feature of JHS/hEDS. Our group published the first case control study on the association between JHS/hEDS and anxiety in 1988.7 Additional studies have consistently replicated and confirmed these findings in clinical and nonclinical populations, and in adult and geriatric patients.8-12 Specifically, JHS/hEDS has been associated with a higher frequency and greater intensity of fears, greater anxiety severity and somatic concerns, and higher frequency of the so-called endogenous anxiety disorders.6,13 There also is limited but growing evidence that JHS/hEDS is associated with depressive disorders, eating disorders, and neurodevelopmental disorders as well as alcohol and tobacco misuse.6,8,11,14,15
Moving toward a new phenotype. Whereas there is increasing evidence of somatic comorbidity in several major psychiatric disorders, present psychiatric nosology does not include specific psychiatric illnesses associated with medical conditions other than organic dementias and secondary psychiatric conditions. However, the overwhelming data on clinical comorbidity (both somatic and psychiatric) require new nosologic approaches. Following the accumulated evidence on this topic over the past 30 years, our group described the “Neuroconnective phenotype” (Figure 1) on the basis of the collected genetic, neurophysiological, neuroimaging, and clinical data.6 The core of the phenotype includes the “anxiety-joint laxity” association and has 5 dimensions that allow for minor overlap (somatic symptoms, somatic illnesses, psychopathology, behavioral dimensions, and somatosensory symptoms). Each of the 5 dimensions includes features that may be present at different degrees with individual variations.
Continue to: Biologic hypotheses...
Biologic hypotheses that have been proposed to explain the link between anxiety and JHS/hEDS are described in the Box6,16-28.
Box
What underlying mechanisms link anxiety and joint hypermobility?
Interestingly, both anxiety and joint hypermobility syndrome/Ehlers-Danlos type 3-hypermobile type (JHS/hEDS) are often underdiagnosed and undertreated, and have similar prevalence in the general population. While it is possible that some psychiatric symptoms can be a consequence of adaptation and difficulties in dealing with chronic illnesses, biologic hypotheses have been considered to explain the association between JHS/hEDS and anxiety. The most accepted biologic hypotheses include:
- genetic risks
- interoceptive sensitivity
- somatosensory amplification
- emotion processing variances
- autonomic nervous system dysfunction.
A duplication of chromosome 15 (DUP-25) was found in patients with both JHS/hEDS and an anxiety disorder,16 but to date, this finding has not been replicated.17,18 The fact that both conditions are highly heritable suggests high likelihood of a genetic linkage. Other theories about the neural connections between mind and body have been proposed. Brain and body are intrinsically and dynamically coupled; perceptions, emotions, and cognitions respond to and change the state of the body.19 In this sense, body perception and dysautonomia have gained recognition.
Patients with JHS/hEDS have higher interoception,20 meaning greater signaling and perception of internal bodily sensations. This is in line with Critchley's hypothesis, in which he describes the influence of visceral inputs over thoughts, feelings, and behavior.21 Consistent with Critchley's views, Porges described the Polyvagal Theory,22 which is phylogenetic approach relating the autonomic nervous system to behavior. Atypical body awareness is a feature of multiple disorders, including anxiety, depression, and JHS/hEDS.19,23-25 Interestingly, a recent neuroimaging study found that interception sensitivity mediated the relationship between anxiety and hypermobility.20
JHS/hEDS patients have greater exteroception (perception of environment), nocioception (pain perception), and somatosensory amplification.6,26 At the same time, they also have decreased proprioception,27 which could explain the coordination difficulties they experience. Neuroimaging studies have confirmed that individuals with JHS/hEDS have structural differences in key emotion processing regions, notably affecting the amygdala bilaterally.28
Together, these findings increase our understanding about the mechanisms through which vulnerability to anxiety disorders and somatic symptoms arises in certain patients.
Continue to: How JHS/hEDS is diagnosed
How JHS/hEDS is diagnosed
The Beighton criteria are the most common set of criteria used to diagnose JHS/hEDS.29 In 2000, Grahame et al30 developed the Brighton criteria, which include some extra-articular features. The “Hospital del Mar” criteria31 (also known as the “Bulbena criteria”) were obtained after a multivariate analysis of margins from the Beighton criteria and the original set of criteria described by Rotés. They showed consistent indicators of reliability, internal consistency, and better predictive validity.31
Recently, several self-assessment questionnaires have been developed. Specifically, based on the Hakim and Grahame questionnaire,32 our group developed a novel self-assessment questionnaire that includes pictures to facilitate the diagnosis.33
However, despite multiple ways of assessing JHS/hEDS, it remains mostly undiagnosed and untreated. Because of this, a new clinician-administered checklist has been developed,34 although this checklist does not include the psychiatric aspects of the disorder, so clinicians who use this checklist should ensure that the patient receives additional psychiatric assessment.
Transforming the clinical value into specific interventions
Anxiety disorders are chronic, disabling, and represent the 6th leading cause of disability worldwide.35 They have a significant impact due to the high cost of frequent medical evaluations and treatment of the physical components of the disorder.36 As a clinical marker for a homogeneous type of anxiety, JHS/hEDS can provide valuable information about a patient’s complete clinical picture, especially about the somatic aspects of the disorder.
No randomized controlled trials have been conducted to evaluate pharmacotherapy as treatment for JHS/hEDS. In a cohort study, the overall use of psychotropi
Continue to: Current nosology of anxiety disorders...
Current nosology of anxiety disorders neglects the somatic aspects and physical manifestations of anxiety, and in general, therapeutic interventions focus only cognitive/psychological aspects of anxiety. Cognitive-behavioral therapy (CBT) may be effective in treating the cognitive distortions associated with the chronicity of the illness and negative emotions. Baeza-Velasco et al38 found that patients with JHS/hEDS have a tendency toward dysfunctional coping strategies, and CBT may be useful to address those symptoms. Moreover, these individuals often suffer from kinesiophobia and hyperalgesia. Some pilot CBT strategies have been developed, and research suggests that along with exercise, CBT can be a valuable pain management tool in patients with JHS/hEDS.39
Nonetheless, these patients often suffer from several somatic complaints and bodily manifestations (eg, somatosensory amplification, dysautonomia) that require treatment. Thus, interventions that address mind and body connections should be implemented. Some research found meditative therapies for anxiety disorders can be effective,40,41 although further randomized controlled trials are needed.
Based on our proposed “Neuroconnective phenotype,” we suggest a new therapeutic approach to address the 5 dimensions of this phenotype.
Somatic symptoms, such as blue sclera, dislocations, scars, easy bruising, and leptosomatic somatotype, do not require specific intervention, but they provide information about the physical phenotype of JHS/hEDS and can facilitate the diagnosis.
Somatic illnesses. Treatment must address often-found comorbid medical conditions, such as irritable bowel syndrome, other gastrointestinal conditions, temporomandibular dysfunction, fatigue, fibromyalgia, and dysautonomia. Obviously specific attention must be paid to JHS/hEDS, which responds relatively well to physical treatments, including aerobic exercise, and particularly well to expert physiotherapy. Relaxation and meditation techniques also are effective.
Continue to: Psychopathology
Psychopathology. Ensure proper assessment and treatment not only of the anxiety disorder and its dimensions (ie, anticipatory anxiety, high loss sensitivity, depersonalization, impulse phobias, or avoidance behavior), but also of the other related conditions, such as mood disorders, substance use disorders, or eating disorders.
Behavioral dimensions. Defense mechanisms often take individuals with JHS/hEDS to the extremes of a circumflex behavioral model in which the most typical axes include the following: me/others, loss/excess of control, avoidance/invasion, fight/flight, and dependency/isolation. A rich psychotherapeutic approach that focuses on these defense mechanisms and behavioral axes is required to balance these mechanisms.
Somatosensory symptoms. Be aware of, validate, and provide understanding of the patient’s increased sensitivities, including greater pain, body perception, meteorosensitivity, and higher sensitivity to medications and adverse effects.
Additional research is needed
Future directions for exploring the link between anxiety and JHS/hEDS should include the development of new nosologic approaches, the expansion of the therapeutic dimension, and unmasking the common biologic mechanisms using evolutionary models.
1. Tinkle BT, Bird HA, Grahame R, et al. The lack of clinical distinction between the hypermobility type of Ehlers-Danlos syndrome and the joint hypermobility syndrome (a.k.a. hypermobility syndrome). Am J Med Genet A. 2009;149A(11):2368-2370.
2. Hakim A, Grahame R. Joint hypermobility. Best Pract Res Clin Rheumatol. 2003;17(6):989-1004.
3. Hakim AJ, Grahame R. Non-musculoskeletal symptoms in joint hypermobility syndrome. Indirect evidence for autonomic dysfunction? Rheumatology (Oxford). 2004;43(9):1194-1195.
4. Grahame R, Hakim AJ. Hypermobility. Curr Opin Rheumatol. 2008;20(1):106-110.
5. Castori M. Ehlers-Danlos syndrome, hypermobility type: an underdiagnosed hereditary connective tissue disorder with mucocutaneous, articular, and systemic manifestations. ISRN Dermatol. 2012;2012:751768. doi: 10.5402/2012/751768.
6. Bulbena A, Baeza-Velasco C, Bulbena-Cabré A, et al. Psychiatric and psychological aspects in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):237-245.
7. Bulbena A, Duro JC, Mateo A, et al. Joint hypermobility syndrome and anxiety disorders. Lancet. 1988;332(8612):694.
8. Bulbena-Cabré A, Pailhez G, Cabrera A, et al. Body perception in a sample of nonclinical youngsters with joint hypermobility. Ansiedad y Estrés. 2017;23(2-3):99-103.
9. Martín-Santos R, Bulbena A, Porta M, et al. Association between joint hypermobility syndrome and panic disorder. Am J Psychiatry. 1998;155(11):1578-1583.
10. Bulbena A, Agulló A, Pailhez G, et al. Is joint hypermobility related to anxiety in a nonclinical population also? Psychosomatics. 2004;45(5):432-437.
11. Bulbena-Cabré A, Baeza-Velasco C, Pailhez G, et al. Psicopatología de la hiperlaxitud articular [in Spanish]. Cuadernos de Neuropsicología/Panamerican Journal of Neuropsychology 2016;10(3):61-70.
12. Bulbena‐Cabré A, Rojo C, Pailhez G, et al. Joint hypermobility is also associated with anxiety disorders in the elderly population. Int J Geriatr Psychiatry. 2018;33(1):e113-e119.
13. Bulbena A, Pailhez G, Bulbena-Cabré A, et al. Joint hypermobility, anxiety and psychosomatics: two and a half decades of progress toward a new phenotype. Adv Psychosom Med. 2015;34:143-157.
14. Smith TO, Easton V, Bacon H, et al. The relationship between benign joint hypermobility syndrome and psychological distress: a systematic review and meta-analysis. Rheumatology (Oxford). 2014;53(1):114-122.
15. Cederlöf M, Larsson H, Lichtenstein P, et al. Nationwide population-based cohort study of psychiatric disorders in individuals with Ehlers-Danlos syndrome or hypermobility syndrome and their siblings. BMC Psychiatry. 2016;16(1):207.
16. Gratacòs M, Nadal M, Martín-Santos R, et al. A polymorphic genomic duplication on human chromosome 15 is a susceptibility factor for panic and phobic disorders. Cell. 2001;106(3):367-379.
17. Tabiner M, Youings S, Dennis N, A et al. Failure to find DUP25 in patients with anxiety disorders, in control individuals, or in previously reported positive control cell lines. Am J Hum Genet. 2003;72(3):535-538.
18. Henrichsen CN, Delorme R, Boucherie M, et al. No association between DUP25 and anxiety disorders. Am J Med Genet B Neuropsychiatr Genet. 2004;128B(1):80-83.
19. Eccles JA, Owens AP, Mathias CJ, et al. Neurovisceral phenotypes in the expression of psychiatric symptoms. Front Neurosci. 2015;9:4. doi: 10.3389/fnins.2015.00004.
20. Mallorqui-Bagué N, Garfinkel SN, Engels M, et al. Neuroimaging and psychophysiological investigation of the link between anxiety, enhanced affective reactivity and interoception in people with joint hypermobility. Front Psychol. 2014;5:1162. doi: 10.3389/fpsyg.2014.01162.
21. Critchley HD, Harrison NA. Visceral influences on brain and behavior. Neuron. 2013;77(4):624-638.
22. Porges SW. The polyvagal theory: phylogenetic substrates of a social nervous system. Int J Psychophysiol. 2001;42(2):123-146.
23. Cameron OG. Interoception: the inside story—a model for psychosomatic processes. Psychosom Med. 2001;63(5):697-710.
24. Domschke K, Stevens S, Pfleiderer B, et al. Interoceptive sensitivity in anxiety and anxiety disorders: an overview and integration of neurobiological findings. Clin Psychol Rev. 2010;30(1):1-11.
25. Wiebking C, Bauer A, de Greck M, et al. Abnormal body perception and neural activity in the insula in depression: an fMRI study of the depressed “material me.” World J Biol Psychiatry. 2010;11(3):538-549.
26. Baeza-Velasco C, Gely-Nargeot MC, Vilarrasa AB, et al. Association between psychopathological factors and joint hypermobility syndrome in a group of undergraduates from a French university. Int J Psychiatry Med. 2011;41(2):187-201.
27. Smith TO, Jerman E, Easton V, et al. Do people with benign joint hypermobility syndrome (BJHS) have reduced joint proprioception? A systematic review and meta-analysis. Rheumatol Int. 2013;33(11):2709-2716.
28. Eccles JA, Beacher FD, Gray MA, et al. Brain structure and joint hypermobility: relevance to the expression of psychiatric symptoms. Br J Psychiatry. 2012;200(6):508-509.
29. Beighton P, Horan F. Orthopaedic aspects of the Ehlers-Danlos syndrome. J Bone Joint Surg Br. 1969;51(3):444-453.
30. Grahame R, Bird HA, Child A. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS). J Rheumatol. 2000;27(7):1777-1779.
31. Bulbena A, Duró JC, Porta M, et al. Clinical assessment of hypermobility of joints: assembling criteria. J Rheumatol. 1992;19(1):115-122.
32. Hakim AJ, Grahame R. A simple questionnaire to detect hypermobility: an adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract. 2003;57(3):163-166.
33. Bulbena A, Mallorquí-Bagué N, Pailhez G, et al. Self-reported screening questionnaire for the assessment of Joint Hypermobility Syndrome (SQ-CH), a collagen condition, in Spanish population. Eur J Psychiat. 2014;28(1):17-26.
34. Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8-26.
35. Baxter AJ, Vos T, Scott KM, et al. The global burden of anxiety disorders in 2010. Psychol Med. 2014;44(11):2363-2374.
36. Bystritsky A. Treatment-resistant anxiety disorders. Mol Psychiatry. 2006;11(9):805-814.
37. Bulbena A, Gago J, Pailhez G, et al. Joint hypermobility syndrome is a risk factor trait for anxiety disorders: a 15-year follow-up cohort study. Gen Hosp Psychiatry. 2011;33(4):363-370.
38. Baeza-Velasco C, Gély-Nargeot MC, Bulbena Vilarrasa A, et al. Joint hypermobility syndrome: problems that require psychological intervention. Rheumatol Int. 2011;31(9):1131-1136.
39. Bathen T, Hangmann AB, Hoff M, et al. Multidisciplinary treatment of disability in ehlers-danlos syndrome hypermobility type/hypermobility syndrome: A pilot study using a combination of physical and cognitive-behavioral therapy on 12 women. Am J Med Genet A. 2013;161A(12): 3005-3011.
40. Chen KW, Berger CC, Manheimer E, et al. Meditative therapies for reducing anxiety: a systematic review and meta-analysis of randomized controlled trials. Depress Anxiety. 2012;29(7):545-562.
41. Krisanaprakornkit T, Sriraj W, Piyavhatkul N, et al. Meditation therapy for anxiety disorders. Cochrane Database Syst Rev. 2006;(1):CD004998.
Joint hypermobility syndrome (JHS)—also known as Ehlers-Danlos type 3–hypermobile type (hEDS)1—is a poorly recognized connective tissue disorder characterized by increased joint laxity that may affect 10% to 25% of the general population.2 Researchers are increasingly recognizing an association between JHS/hEDS and psychiatric symptoms and disorders, specifically anxiety. In this review, we describe the clinical presentation of JHS/hEDS, propose a new “Neuroconnective phenotype” based on the link between anxiety and JHS/hEDS, and discuss factors to consider when treating anxiety in a patient who has JHS/hEDS.
JHS/hEDS: A complex disorder
Although JHS/hEDS is a heritable condition, several factors are known to influence its prevalence and visibility, including age, sex, and ethnicity; the prevalence is higher among younger patients, females, and African Americans.2 Its known basis is the type and distribution pattern of collagen, and one of the key features used to identify this syndrome is greater joint laxity, meaning increased distensibility of the joints in passive movements as well as a hypermobility in active movements.
Although first described by two dermatologists (Edvard Ehlers and Henri-Alexandre Danlos) at the beginning of the 20th century, JHS/hEDS is now considered a multi-systemic condition. Thus, JHS/hEDS includes a wide range of musculoskeletal features, and over the recent years, extra-articular symptoms, such as easy bruising or hypertrophic scarring, have gained recognition.3 Moreover, individuals with JHS/hEDS frequently present with stress-sensitive illnesses, such as fibromyalgia, or chronic fatigue syndrome.4 The Table2,5,6 provides a description of musculoskeletal and extra-articular features of JHS/hEDS.
The link between JHS/hEDS and anxiety
Psychiatric symptoms are being increasingly recognized as a key feature of JHS/hEDS. Our group published the first case control study on the association between JHS/hEDS and anxiety in 1988.7 Additional studies have consistently replicated and confirmed these findings in clinical and nonclinical populations, and in adult and geriatric patients.8-12 Specifically, JHS/hEDS has been associated with a higher frequency and greater intensity of fears, greater anxiety severity and somatic concerns, and higher frequency of the so-called endogenous anxiety disorders.6,13 There also is limited but growing evidence that JHS/hEDS is associated with depressive disorders, eating disorders, and neurodevelopmental disorders as well as alcohol and tobacco misuse.6,8,11,14,15
Moving toward a new phenotype. Whereas there is increasing evidence of somatic comorbidity in several major psychiatric disorders, present psychiatric nosology does not include specific psychiatric illnesses associated with medical conditions other than organic dementias and secondary psychiatric conditions. However, the overwhelming data on clinical comorbidity (both somatic and psychiatric) require new nosologic approaches. Following the accumulated evidence on this topic over the past 30 years, our group described the “Neuroconnective phenotype” (Figure 1) on the basis of the collected genetic, neurophysiological, neuroimaging, and clinical data.6 The core of the phenotype includes the “anxiety-joint laxity” association and has 5 dimensions that allow for minor overlap (somatic symptoms, somatic illnesses, psychopathology, behavioral dimensions, and somatosensory symptoms). Each of the 5 dimensions includes features that may be present at different degrees with individual variations.
Continue to: Biologic hypotheses...
Biologic hypotheses that have been proposed to explain the link between anxiety and JHS/hEDS are described in the Box6,16-28.
Box
What underlying mechanisms link anxiety and joint hypermobility?
Interestingly, both anxiety and joint hypermobility syndrome/Ehlers-Danlos type 3-hypermobile type (JHS/hEDS) are often underdiagnosed and undertreated, and have similar prevalence in the general population. While it is possible that some psychiatric symptoms can be a consequence of adaptation and difficulties in dealing with chronic illnesses, biologic hypotheses have been considered to explain the association between JHS/hEDS and anxiety. The most accepted biologic hypotheses include:
- genetic risks
- interoceptive sensitivity
- somatosensory amplification
- emotion processing variances
- autonomic nervous system dysfunction.
A duplication of chromosome 15 (DUP-25) was found in patients with both JHS/hEDS and an anxiety disorder,16 but to date, this finding has not been replicated.17,18 The fact that both conditions are highly heritable suggests high likelihood of a genetic linkage. Other theories about the neural connections between mind and body have been proposed. Brain and body are intrinsically and dynamically coupled; perceptions, emotions, and cognitions respond to and change the state of the body.19 In this sense, body perception and dysautonomia have gained recognition.
Patients with JHS/hEDS have higher interoception,20 meaning greater signaling and perception of internal bodily sensations. This is in line with Critchley's hypothesis, in which he describes the influence of visceral inputs over thoughts, feelings, and behavior.21 Consistent with Critchley's views, Porges described the Polyvagal Theory,22 which is phylogenetic approach relating the autonomic nervous system to behavior. Atypical body awareness is a feature of multiple disorders, including anxiety, depression, and JHS/hEDS.19,23-25 Interestingly, a recent neuroimaging study found that interception sensitivity mediated the relationship between anxiety and hypermobility.20
JHS/hEDS patients have greater exteroception (perception of environment), nocioception (pain perception), and somatosensory amplification.6,26 At the same time, they also have decreased proprioception,27 which could explain the coordination difficulties they experience. Neuroimaging studies have confirmed that individuals with JHS/hEDS have structural differences in key emotion processing regions, notably affecting the amygdala bilaterally.28
Together, these findings increase our understanding about the mechanisms through which vulnerability to anxiety disorders and somatic symptoms arises in certain patients.
Continue to: How JHS/hEDS is diagnosed
How JHS/hEDS is diagnosed
The Beighton criteria are the most common set of criteria used to diagnose JHS/hEDS.29 In 2000, Grahame et al30 developed the Brighton criteria, which include some extra-articular features. The “Hospital del Mar” criteria31 (also known as the “Bulbena criteria”) were obtained after a multivariate analysis of margins from the Beighton criteria and the original set of criteria described by Rotés. They showed consistent indicators of reliability, internal consistency, and better predictive validity.31
Recently, several self-assessment questionnaires have been developed. Specifically, based on the Hakim and Grahame questionnaire,32 our group developed a novel self-assessment questionnaire that includes pictures to facilitate the diagnosis.33
However, despite multiple ways of assessing JHS/hEDS, it remains mostly undiagnosed and untreated. Because of this, a new clinician-administered checklist has been developed,34 although this checklist does not include the psychiatric aspects of the disorder, so clinicians who use this checklist should ensure that the patient receives additional psychiatric assessment.
Transforming the clinical value into specific interventions
Anxiety disorders are chronic, disabling, and represent the 6th leading cause of disability worldwide.35 They have a significant impact due to the high cost of frequent medical evaluations and treatment of the physical components of the disorder.36 As a clinical marker for a homogeneous type of anxiety, JHS/hEDS can provide valuable information about a patient’s complete clinical picture, especially about the somatic aspects of the disorder.
No randomized controlled trials have been conducted to evaluate pharmacotherapy as treatment for JHS/hEDS. In a cohort study, the overall use of psychotropi
Continue to: Current nosology of anxiety disorders...
Current nosology of anxiety disorders neglects the somatic aspects and physical manifestations of anxiety, and in general, therapeutic interventions focus only cognitive/psychological aspects of anxiety. Cognitive-behavioral therapy (CBT) may be effective in treating the cognitive distortions associated with the chronicity of the illness and negative emotions. Baeza-Velasco et al38 found that patients with JHS/hEDS have a tendency toward dysfunctional coping strategies, and CBT may be useful to address those symptoms. Moreover, these individuals often suffer from kinesiophobia and hyperalgesia. Some pilot CBT strategies have been developed, and research suggests that along with exercise, CBT can be a valuable pain management tool in patients with JHS/hEDS.39
Nonetheless, these patients often suffer from several somatic complaints and bodily manifestations (eg, somatosensory amplification, dysautonomia) that require treatment. Thus, interventions that address mind and body connections should be implemented. Some research found meditative therapies for anxiety disorders can be effective,40,41 although further randomized controlled trials are needed.
Based on our proposed “Neuroconnective phenotype,” we suggest a new therapeutic approach to address the 5 dimensions of this phenotype.
Somatic symptoms, such as blue sclera, dislocations, scars, easy bruising, and leptosomatic somatotype, do not require specific intervention, but they provide information about the physical phenotype of JHS/hEDS and can facilitate the diagnosis.
Somatic illnesses. Treatment must address often-found comorbid medical conditions, such as irritable bowel syndrome, other gastrointestinal conditions, temporomandibular dysfunction, fatigue, fibromyalgia, and dysautonomia. Obviously specific attention must be paid to JHS/hEDS, which responds relatively well to physical treatments, including aerobic exercise, and particularly well to expert physiotherapy. Relaxation and meditation techniques also are effective.
Continue to: Psychopathology
Psychopathology. Ensure proper assessment and treatment not only of the anxiety disorder and its dimensions (ie, anticipatory anxiety, high loss sensitivity, depersonalization, impulse phobias, or avoidance behavior), but also of the other related conditions, such as mood disorders, substance use disorders, or eating disorders.
Behavioral dimensions. Defense mechanisms often take individuals with JHS/hEDS to the extremes of a circumflex behavioral model in which the most typical axes include the following: me/others, loss/excess of control, avoidance/invasion, fight/flight, and dependency/isolation. A rich psychotherapeutic approach that focuses on these defense mechanisms and behavioral axes is required to balance these mechanisms.
Somatosensory symptoms. Be aware of, validate, and provide understanding of the patient’s increased sensitivities, including greater pain, body perception, meteorosensitivity, and higher sensitivity to medications and adverse effects.
Additional research is needed
Future directions for exploring the link between anxiety and JHS/hEDS should include the development of new nosologic approaches, the expansion of the therapeutic dimension, and unmasking the common biologic mechanisms using evolutionary models.
Joint hypermobility syndrome (JHS)—also known as Ehlers-Danlos type 3–hypermobile type (hEDS)1—is a poorly recognized connective tissue disorder characterized by increased joint laxity that may affect 10% to 25% of the general population.2 Researchers are increasingly recognizing an association between JHS/hEDS and psychiatric symptoms and disorders, specifically anxiety. In this review, we describe the clinical presentation of JHS/hEDS, propose a new “Neuroconnective phenotype” based on the link between anxiety and JHS/hEDS, and discuss factors to consider when treating anxiety in a patient who has JHS/hEDS.
JHS/hEDS: A complex disorder
Although JHS/hEDS is a heritable condition, several factors are known to influence its prevalence and visibility, including age, sex, and ethnicity; the prevalence is higher among younger patients, females, and African Americans.2 Its known basis is the type and distribution pattern of collagen, and one of the key features used to identify this syndrome is greater joint laxity, meaning increased distensibility of the joints in passive movements as well as a hypermobility in active movements.
Although first described by two dermatologists (Edvard Ehlers and Henri-Alexandre Danlos) at the beginning of the 20th century, JHS/hEDS is now considered a multi-systemic condition. Thus, JHS/hEDS includes a wide range of musculoskeletal features, and over the recent years, extra-articular symptoms, such as easy bruising or hypertrophic scarring, have gained recognition.3 Moreover, individuals with JHS/hEDS frequently present with stress-sensitive illnesses, such as fibromyalgia, or chronic fatigue syndrome.4 The Table2,5,6 provides a description of musculoskeletal and extra-articular features of JHS/hEDS.
The link between JHS/hEDS and anxiety
Psychiatric symptoms are being increasingly recognized as a key feature of JHS/hEDS. Our group published the first case control study on the association between JHS/hEDS and anxiety in 1988.7 Additional studies have consistently replicated and confirmed these findings in clinical and nonclinical populations, and in adult and geriatric patients.8-12 Specifically, JHS/hEDS has been associated with a higher frequency and greater intensity of fears, greater anxiety severity and somatic concerns, and higher frequency of the so-called endogenous anxiety disorders.6,13 There also is limited but growing evidence that JHS/hEDS is associated with depressive disorders, eating disorders, and neurodevelopmental disorders as well as alcohol and tobacco misuse.6,8,11,14,15
Moving toward a new phenotype. Whereas there is increasing evidence of somatic comorbidity in several major psychiatric disorders, present psychiatric nosology does not include specific psychiatric illnesses associated with medical conditions other than organic dementias and secondary psychiatric conditions. However, the overwhelming data on clinical comorbidity (both somatic and psychiatric) require new nosologic approaches. Following the accumulated evidence on this topic over the past 30 years, our group described the “Neuroconnective phenotype” (Figure 1) on the basis of the collected genetic, neurophysiological, neuroimaging, and clinical data.6 The core of the phenotype includes the “anxiety-joint laxity” association and has 5 dimensions that allow for minor overlap (somatic symptoms, somatic illnesses, psychopathology, behavioral dimensions, and somatosensory symptoms). Each of the 5 dimensions includes features that may be present at different degrees with individual variations.
Continue to: Biologic hypotheses...
Biologic hypotheses that have been proposed to explain the link between anxiety and JHS/hEDS are described in the Box6,16-28.
Box
What underlying mechanisms link anxiety and joint hypermobility?
Interestingly, both anxiety and joint hypermobility syndrome/Ehlers-Danlos type 3-hypermobile type (JHS/hEDS) are often underdiagnosed and undertreated, and have similar prevalence in the general population. While it is possible that some psychiatric symptoms can be a consequence of adaptation and difficulties in dealing with chronic illnesses, biologic hypotheses have been considered to explain the association between JHS/hEDS and anxiety. The most accepted biologic hypotheses include:
- genetic risks
- interoceptive sensitivity
- somatosensory amplification
- emotion processing variances
- autonomic nervous system dysfunction.
A duplication of chromosome 15 (DUP-25) was found in patients with both JHS/hEDS and an anxiety disorder,16 but to date, this finding has not been replicated.17,18 The fact that both conditions are highly heritable suggests high likelihood of a genetic linkage. Other theories about the neural connections between mind and body have been proposed. Brain and body are intrinsically and dynamically coupled; perceptions, emotions, and cognitions respond to and change the state of the body.19 In this sense, body perception and dysautonomia have gained recognition.
Patients with JHS/hEDS have higher interoception,20 meaning greater signaling and perception of internal bodily sensations. This is in line with Critchley's hypothesis, in which he describes the influence of visceral inputs over thoughts, feelings, and behavior.21 Consistent with Critchley's views, Porges described the Polyvagal Theory,22 which is phylogenetic approach relating the autonomic nervous system to behavior. Atypical body awareness is a feature of multiple disorders, including anxiety, depression, and JHS/hEDS.19,23-25 Interestingly, a recent neuroimaging study found that interception sensitivity mediated the relationship between anxiety and hypermobility.20
JHS/hEDS patients have greater exteroception (perception of environment), nocioception (pain perception), and somatosensory amplification.6,26 At the same time, they also have decreased proprioception,27 which could explain the coordination difficulties they experience. Neuroimaging studies have confirmed that individuals with JHS/hEDS have structural differences in key emotion processing regions, notably affecting the amygdala bilaterally.28
Together, these findings increase our understanding about the mechanisms through which vulnerability to anxiety disorders and somatic symptoms arises in certain patients.
Continue to: How JHS/hEDS is diagnosed
How JHS/hEDS is diagnosed
The Beighton criteria are the most common set of criteria used to diagnose JHS/hEDS.29 In 2000, Grahame et al30 developed the Brighton criteria, which include some extra-articular features. The “Hospital del Mar” criteria31 (also known as the “Bulbena criteria”) were obtained after a multivariate analysis of margins from the Beighton criteria and the original set of criteria described by Rotés. They showed consistent indicators of reliability, internal consistency, and better predictive validity.31
Recently, several self-assessment questionnaires have been developed. Specifically, based on the Hakim and Grahame questionnaire,32 our group developed a novel self-assessment questionnaire that includes pictures to facilitate the diagnosis.33
However, despite multiple ways of assessing JHS/hEDS, it remains mostly undiagnosed and untreated. Because of this, a new clinician-administered checklist has been developed,34 although this checklist does not include the psychiatric aspects of the disorder, so clinicians who use this checklist should ensure that the patient receives additional psychiatric assessment.
Transforming the clinical value into specific interventions
Anxiety disorders are chronic, disabling, and represent the 6th leading cause of disability worldwide.35 They have a significant impact due to the high cost of frequent medical evaluations and treatment of the physical components of the disorder.36 As a clinical marker for a homogeneous type of anxiety, JHS/hEDS can provide valuable information about a patient’s complete clinical picture, especially about the somatic aspects of the disorder.
No randomized controlled trials have been conducted to evaluate pharmacotherapy as treatment for JHS/hEDS. In a cohort study, the overall use of psychotropi
Continue to: Current nosology of anxiety disorders...
Current nosology of anxiety disorders neglects the somatic aspects and physical manifestations of anxiety, and in general, therapeutic interventions focus only cognitive/psychological aspects of anxiety. Cognitive-behavioral therapy (CBT) may be effective in treating the cognitive distortions associated with the chronicity of the illness and negative emotions. Baeza-Velasco et al38 found that patients with JHS/hEDS have a tendency toward dysfunctional coping strategies, and CBT may be useful to address those symptoms. Moreover, these individuals often suffer from kinesiophobia and hyperalgesia. Some pilot CBT strategies have been developed, and research suggests that along with exercise, CBT can be a valuable pain management tool in patients with JHS/hEDS.39
Nonetheless, these patients often suffer from several somatic complaints and bodily manifestations (eg, somatosensory amplification, dysautonomia) that require treatment. Thus, interventions that address mind and body connections should be implemented. Some research found meditative therapies for anxiety disorders can be effective,40,41 although further randomized controlled trials are needed.
Based on our proposed “Neuroconnective phenotype,” we suggest a new therapeutic approach to address the 5 dimensions of this phenotype.
Somatic symptoms, such as blue sclera, dislocations, scars, easy bruising, and leptosomatic somatotype, do not require specific intervention, but they provide information about the physical phenotype of JHS/hEDS and can facilitate the diagnosis.
Somatic illnesses. Treatment must address often-found comorbid medical conditions, such as irritable bowel syndrome, other gastrointestinal conditions, temporomandibular dysfunction, fatigue, fibromyalgia, and dysautonomia. Obviously specific attention must be paid to JHS/hEDS, which responds relatively well to physical treatments, including aerobic exercise, and particularly well to expert physiotherapy. Relaxation and meditation techniques also are effective.
Continue to: Psychopathology
Psychopathology. Ensure proper assessment and treatment not only of the anxiety disorder and its dimensions (ie, anticipatory anxiety, high loss sensitivity, depersonalization, impulse phobias, or avoidance behavior), but also of the other related conditions, such as mood disorders, substance use disorders, or eating disorders.
Behavioral dimensions. Defense mechanisms often take individuals with JHS/hEDS to the extremes of a circumflex behavioral model in which the most typical axes include the following: me/others, loss/excess of control, avoidance/invasion, fight/flight, and dependency/isolation. A rich psychotherapeutic approach that focuses on these defense mechanisms and behavioral axes is required to balance these mechanisms.
Somatosensory symptoms. Be aware of, validate, and provide understanding of the patient’s increased sensitivities, including greater pain, body perception, meteorosensitivity, and higher sensitivity to medications and adverse effects.
Additional research is needed
Future directions for exploring the link between anxiety and JHS/hEDS should include the development of new nosologic approaches, the expansion of the therapeutic dimension, and unmasking the common biologic mechanisms using evolutionary models.
1. Tinkle BT, Bird HA, Grahame R, et al. The lack of clinical distinction between the hypermobility type of Ehlers-Danlos syndrome and the joint hypermobility syndrome (a.k.a. hypermobility syndrome). Am J Med Genet A. 2009;149A(11):2368-2370.
2. Hakim A, Grahame R. Joint hypermobility. Best Pract Res Clin Rheumatol. 2003;17(6):989-1004.
3. Hakim AJ, Grahame R. Non-musculoskeletal symptoms in joint hypermobility syndrome. Indirect evidence for autonomic dysfunction? Rheumatology (Oxford). 2004;43(9):1194-1195.
4. Grahame R, Hakim AJ. Hypermobility. Curr Opin Rheumatol. 2008;20(1):106-110.
5. Castori M. Ehlers-Danlos syndrome, hypermobility type: an underdiagnosed hereditary connective tissue disorder with mucocutaneous, articular, and systemic manifestations. ISRN Dermatol. 2012;2012:751768. doi: 10.5402/2012/751768.
6. Bulbena A, Baeza-Velasco C, Bulbena-Cabré A, et al. Psychiatric and psychological aspects in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):237-245.
7. Bulbena A, Duro JC, Mateo A, et al. Joint hypermobility syndrome and anxiety disorders. Lancet. 1988;332(8612):694.
8. Bulbena-Cabré A, Pailhez G, Cabrera A, et al. Body perception in a sample of nonclinical youngsters with joint hypermobility. Ansiedad y Estrés. 2017;23(2-3):99-103.
9. Martín-Santos R, Bulbena A, Porta M, et al. Association between joint hypermobility syndrome and panic disorder. Am J Psychiatry. 1998;155(11):1578-1583.
10. Bulbena A, Agulló A, Pailhez G, et al. Is joint hypermobility related to anxiety in a nonclinical population also? Psychosomatics. 2004;45(5):432-437.
11. Bulbena-Cabré A, Baeza-Velasco C, Pailhez G, et al. Psicopatología de la hiperlaxitud articular [in Spanish]. Cuadernos de Neuropsicología/Panamerican Journal of Neuropsychology 2016;10(3):61-70.
12. Bulbena‐Cabré A, Rojo C, Pailhez G, et al. Joint hypermobility is also associated with anxiety disorders in the elderly population. Int J Geriatr Psychiatry. 2018;33(1):e113-e119.
13. Bulbena A, Pailhez G, Bulbena-Cabré A, et al. Joint hypermobility, anxiety and psychosomatics: two and a half decades of progress toward a new phenotype. Adv Psychosom Med. 2015;34:143-157.
14. Smith TO, Easton V, Bacon H, et al. The relationship between benign joint hypermobility syndrome and psychological distress: a systematic review and meta-analysis. Rheumatology (Oxford). 2014;53(1):114-122.
15. Cederlöf M, Larsson H, Lichtenstein P, et al. Nationwide population-based cohort study of psychiatric disorders in individuals with Ehlers-Danlos syndrome or hypermobility syndrome and their siblings. BMC Psychiatry. 2016;16(1):207.
16. Gratacòs M, Nadal M, Martín-Santos R, et al. A polymorphic genomic duplication on human chromosome 15 is a susceptibility factor for panic and phobic disorders. Cell. 2001;106(3):367-379.
17. Tabiner M, Youings S, Dennis N, A et al. Failure to find DUP25 in patients with anxiety disorders, in control individuals, or in previously reported positive control cell lines. Am J Hum Genet. 2003;72(3):535-538.
18. Henrichsen CN, Delorme R, Boucherie M, et al. No association between DUP25 and anxiety disorders. Am J Med Genet B Neuropsychiatr Genet. 2004;128B(1):80-83.
19. Eccles JA, Owens AP, Mathias CJ, et al. Neurovisceral phenotypes in the expression of psychiatric symptoms. Front Neurosci. 2015;9:4. doi: 10.3389/fnins.2015.00004.
20. Mallorqui-Bagué N, Garfinkel SN, Engels M, et al. Neuroimaging and psychophysiological investigation of the link between anxiety, enhanced affective reactivity and interoception in people with joint hypermobility. Front Psychol. 2014;5:1162. doi: 10.3389/fpsyg.2014.01162.
21. Critchley HD, Harrison NA. Visceral influences on brain and behavior. Neuron. 2013;77(4):624-638.
22. Porges SW. The polyvagal theory: phylogenetic substrates of a social nervous system. Int J Psychophysiol. 2001;42(2):123-146.
23. Cameron OG. Interoception: the inside story—a model for psychosomatic processes. Psychosom Med. 2001;63(5):697-710.
24. Domschke K, Stevens S, Pfleiderer B, et al. Interoceptive sensitivity in anxiety and anxiety disorders: an overview and integration of neurobiological findings. Clin Psychol Rev. 2010;30(1):1-11.
25. Wiebking C, Bauer A, de Greck M, et al. Abnormal body perception and neural activity in the insula in depression: an fMRI study of the depressed “material me.” World J Biol Psychiatry. 2010;11(3):538-549.
26. Baeza-Velasco C, Gely-Nargeot MC, Vilarrasa AB, et al. Association between psychopathological factors and joint hypermobility syndrome in a group of undergraduates from a French university. Int J Psychiatry Med. 2011;41(2):187-201.
27. Smith TO, Jerman E, Easton V, et al. Do people with benign joint hypermobility syndrome (BJHS) have reduced joint proprioception? A systematic review and meta-analysis. Rheumatol Int. 2013;33(11):2709-2716.
28. Eccles JA, Beacher FD, Gray MA, et al. Brain structure and joint hypermobility: relevance to the expression of psychiatric symptoms. Br J Psychiatry. 2012;200(6):508-509.
29. Beighton P, Horan F. Orthopaedic aspects of the Ehlers-Danlos syndrome. J Bone Joint Surg Br. 1969;51(3):444-453.
30. Grahame R, Bird HA, Child A. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS). J Rheumatol. 2000;27(7):1777-1779.
31. Bulbena A, Duró JC, Porta M, et al. Clinical assessment of hypermobility of joints: assembling criteria. J Rheumatol. 1992;19(1):115-122.
32. Hakim AJ, Grahame R. A simple questionnaire to detect hypermobility: an adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract. 2003;57(3):163-166.
33. Bulbena A, Mallorquí-Bagué N, Pailhez G, et al. Self-reported screening questionnaire for the assessment of Joint Hypermobility Syndrome (SQ-CH), a collagen condition, in Spanish population. Eur J Psychiat. 2014;28(1):17-26.
34. Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8-26.
35. Baxter AJ, Vos T, Scott KM, et al. The global burden of anxiety disorders in 2010. Psychol Med. 2014;44(11):2363-2374.
36. Bystritsky A. Treatment-resistant anxiety disorders. Mol Psychiatry. 2006;11(9):805-814.
37. Bulbena A, Gago J, Pailhez G, et al. Joint hypermobility syndrome is a risk factor trait for anxiety disorders: a 15-year follow-up cohort study. Gen Hosp Psychiatry. 2011;33(4):363-370.
38. Baeza-Velasco C, Gély-Nargeot MC, Bulbena Vilarrasa A, et al. Joint hypermobility syndrome: problems that require psychological intervention. Rheumatol Int. 2011;31(9):1131-1136.
39. Bathen T, Hangmann AB, Hoff M, et al. Multidisciplinary treatment of disability in ehlers-danlos syndrome hypermobility type/hypermobility syndrome: A pilot study using a combination of physical and cognitive-behavioral therapy on 12 women. Am J Med Genet A. 2013;161A(12): 3005-3011.
40. Chen KW, Berger CC, Manheimer E, et al. Meditative therapies for reducing anxiety: a systematic review and meta-analysis of randomized controlled trials. Depress Anxiety. 2012;29(7):545-562.
41. Krisanaprakornkit T, Sriraj W, Piyavhatkul N, et al. Meditation therapy for anxiety disorders. Cochrane Database Syst Rev. 2006;(1):CD004998.
1. Tinkle BT, Bird HA, Grahame R, et al. The lack of clinical distinction between the hypermobility type of Ehlers-Danlos syndrome and the joint hypermobility syndrome (a.k.a. hypermobility syndrome). Am J Med Genet A. 2009;149A(11):2368-2370.
2. Hakim A, Grahame R. Joint hypermobility. Best Pract Res Clin Rheumatol. 2003;17(6):989-1004.
3. Hakim AJ, Grahame R. Non-musculoskeletal symptoms in joint hypermobility syndrome. Indirect evidence for autonomic dysfunction? Rheumatology (Oxford). 2004;43(9):1194-1195.
4. Grahame R, Hakim AJ. Hypermobility. Curr Opin Rheumatol. 2008;20(1):106-110.
5. Castori M. Ehlers-Danlos syndrome, hypermobility type: an underdiagnosed hereditary connective tissue disorder with mucocutaneous, articular, and systemic manifestations. ISRN Dermatol. 2012;2012:751768. doi: 10.5402/2012/751768.
6. Bulbena A, Baeza-Velasco C, Bulbena-Cabré A, et al. Psychiatric and psychological aspects in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):237-245.
7. Bulbena A, Duro JC, Mateo A, et al. Joint hypermobility syndrome and anxiety disorders. Lancet. 1988;332(8612):694.
8. Bulbena-Cabré A, Pailhez G, Cabrera A, et al. Body perception in a sample of nonclinical youngsters with joint hypermobility. Ansiedad y Estrés. 2017;23(2-3):99-103.
9. Martín-Santos R, Bulbena A, Porta M, et al. Association between joint hypermobility syndrome and panic disorder. Am J Psychiatry. 1998;155(11):1578-1583.
10. Bulbena A, Agulló A, Pailhez G, et al. Is joint hypermobility related to anxiety in a nonclinical population also? Psychosomatics. 2004;45(5):432-437.
11. Bulbena-Cabré A, Baeza-Velasco C, Pailhez G, et al. Psicopatología de la hiperlaxitud articular [in Spanish]. Cuadernos de Neuropsicología/Panamerican Journal of Neuropsychology 2016;10(3):61-70.
12. Bulbena‐Cabré A, Rojo C, Pailhez G, et al. Joint hypermobility is also associated with anxiety disorders in the elderly population. Int J Geriatr Psychiatry. 2018;33(1):e113-e119.
13. Bulbena A, Pailhez G, Bulbena-Cabré A, et al. Joint hypermobility, anxiety and psychosomatics: two and a half decades of progress toward a new phenotype. Adv Psychosom Med. 2015;34:143-157.
14. Smith TO, Easton V, Bacon H, et al. The relationship between benign joint hypermobility syndrome and psychological distress: a systematic review and meta-analysis. Rheumatology (Oxford). 2014;53(1):114-122.
15. Cederlöf M, Larsson H, Lichtenstein P, et al. Nationwide population-based cohort study of psychiatric disorders in individuals with Ehlers-Danlos syndrome or hypermobility syndrome and their siblings. BMC Psychiatry. 2016;16(1):207.
16. Gratacòs M, Nadal M, Martín-Santos R, et al. A polymorphic genomic duplication on human chromosome 15 is a susceptibility factor for panic and phobic disorders. Cell. 2001;106(3):367-379.
17. Tabiner M, Youings S, Dennis N, A et al. Failure to find DUP25 in patients with anxiety disorders, in control individuals, or in previously reported positive control cell lines. Am J Hum Genet. 2003;72(3):535-538.
18. Henrichsen CN, Delorme R, Boucherie M, et al. No association between DUP25 and anxiety disorders. Am J Med Genet B Neuropsychiatr Genet. 2004;128B(1):80-83.
19. Eccles JA, Owens AP, Mathias CJ, et al. Neurovisceral phenotypes in the expression of psychiatric symptoms. Front Neurosci. 2015;9:4. doi: 10.3389/fnins.2015.00004.
20. Mallorqui-Bagué N, Garfinkel SN, Engels M, et al. Neuroimaging and psychophysiological investigation of the link between anxiety, enhanced affective reactivity and interoception in people with joint hypermobility. Front Psychol. 2014;5:1162. doi: 10.3389/fpsyg.2014.01162.
21. Critchley HD, Harrison NA. Visceral influences on brain and behavior. Neuron. 2013;77(4):624-638.
22. Porges SW. The polyvagal theory: phylogenetic substrates of a social nervous system. Int J Psychophysiol. 2001;42(2):123-146.
23. Cameron OG. Interoception: the inside story—a model for psychosomatic processes. Psychosom Med. 2001;63(5):697-710.
24. Domschke K, Stevens S, Pfleiderer B, et al. Interoceptive sensitivity in anxiety and anxiety disorders: an overview and integration of neurobiological findings. Clin Psychol Rev. 2010;30(1):1-11.
25. Wiebking C, Bauer A, de Greck M, et al. Abnormal body perception and neural activity in the insula in depression: an fMRI study of the depressed “material me.” World J Biol Psychiatry. 2010;11(3):538-549.
26. Baeza-Velasco C, Gely-Nargeot MC, Vilarrasa AB, et al. Association between psychopathological factors and joint hypermobility syndrome in a group of undergraduates from a French university. Int J Psychiatry Med. 2011;41(2):187-201.
27. Smith TO, Jerman E, Easton V, et al. Do people with benign joint hypermobility syndrome (BJHS) have reduced joint proprioception? A systematic review and meta-analysis. Rheumatol Int. 2013;33(11):2709-2716.
28. Eccles JA, Beacher FD, Gray MA, et al. Brain structure and joint hypermobility: relevance to the expression of psychiatric symptoms. Br J Psychiatry. 2012;200(6):508-509.
29. Beighton P, Horan F. Orthopaedic aspects of the Ehlers-Danlos syndrome. J Bone Joint Surg Br. 1969;51(3):444-453.
30. Grahame R, Bird HA, Child A. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS). J Rheumatol. 2000;27(7):1777-1779.
31. Bulbena A, Duró JC, Porta M, et al. Clinical assessment of hypermobility of joints: assembling criteria. J Rheumatol. 1992;19(1):115-122.
32. Hakim AJ, Grahame R. A simple questionnaire to detect hypermobility: an adjunct to the assessment of patients with diffuse musculoskeletal pain. Int J Clin Pract. 2003;57(3):163-166.
33. Bulbena A, Mallorquí-Bagué N, Pailhez G, et al. Self-reported screening questionnaire for the assessment of Joint Hypermobility Syndrome (SQ-CH), a collagen condition, in Spanish population. Eur J Psychiat. 2014;28(1):17-26.
34. Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8-26.
35. Baxter AJ, Vos T, Scott KM, et al. The global burden of anxiety disorders in 2010. Psychol Med. 2014;44(11):2363-2374.
36. Bystritsky A. Treatment-resistant anxiety disorders. Mol Psychiatry. 2006;11(9):805-814.
37. Bulbena A, Gago J, Pailhez G, et al. Joint hypermobility syndrome is a risk factor trait for anxiety disorders: a 15-year follow-up cohort study. Gen Hosp Psychiatry. 2011;33(4):363-370.
38. Baeza-Velasco C, Gély-Nargeot MC, Bulbena Vilarrasa A, et al. Joint hypermobility syndrome: problems that require psychological intervention. Rheumatol Int. 2011;31(9):1131-1136.
39. Bathen T, Hangmann AB, Hoff M, et al. Multidisciplinary treatment of disability in ehlers-danlos syndrome hypermobility type/hypermobility syndrome: A pilot study using a combination of physical and cognitive-behavioral therapy on 12 women. Am J Med Genet A. 2013;161A(12): 3005-3011.
40. Chen KW, Berger CC, Manheimer E, et al. Meditative therapies for reducing anxiety: a systematic review and meta-analysis of randomized controlled trials. Depress Anxiety. 2012;29(7):545-562.
41. Krisanaprakornkit T, Sriraj W, Piyavhatkul N, et al. Meditation therapy for anxiety disorders. Cochrane Database Syst Rev. 2006;(1):CD004998.
PTSD: A systematic approach to diagnosis and treatment
Posttraumatic stress disorder (PTSD) has increasingly become a part of American culture since its introduction in the American Psychiatric Association’s third edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-III) in 1980.1 Since then, a proliferation of material about this disorder—both academic and popular—has been generated, yet much confusion persists surrounding the definition of the disorder, its prevalence, and its management. This review addresses the essential elements for diagnosis and treatment of PTSD.
Diagnosis: A closer look at the criteria
Criteria for the diagnosis of PTSD have evolved since 1980, with changes in the definition of trauma and the addition of symptoms and symptom groups.2 Table 13 summarizes the current DSM-5 criteria for PTSD.

Trauma exposure. An essential first step in the diagnosis of PTSD is to determine whether the individual has experienced exposure to trauma. This concept is defined in Criterion A (trauma exposure).3 PTSD is nonconformist among the psychiatric diagnoses in that it requires a specific external event as part of its definition. Misapplication of the trauma exposure criterion by many clinicians and researchers has led to misdiagnosis and erroneously high prevalence estimates of PTSD.4,5
A traumatic event is one that represents a threat to life or limb, specifically defined as “actual or threatened death, serious injury, or sexual violence.”3 DSM-5 does not allow for just any stressful event to be considered trauma. For example, no matter how distressing, failing an important test at school or being served with divorce proceedings do not represent a requisite trauma6 because these examples do not entail a threat to life or limb.
DSM-5 PTSD Criterion A also requires a qualifying exposure to the traumatic event. There are 4 types of qualifying exposures:
- direct experience of immediate serious physical danger
- eyewitness of trauma to others
- indirect exposure via violent or accidental trauma experienced by a close family member or close friend
- repeated or extreme exposure to aversive details of trauma, such as first responders collecting human remains or law enforcement officers being repeatedly exposed to horrific details of child abuse.3
Witnessed trauma must be in person; thus, viewing trauma in media reports would not constitute a qualifying exposure. Indirect trauma exposure can occur through learning of the experience of a qualifying trauma exposure by a close family member or personal friend.
It is critical to differentiate exposure to trauma (an objective construct) from the subjective distress that may be associated with it. If trauma has not occurred or a qualifying exposure is not established, no amount of distress associated with it can establish the experience as meeting Criterion A for PTSD. This does not mean that nonqualifying experiences of stressful events are not distressing; in fact, such experiences can result in substantial psychological angst. Conversely, exposure to trauma is not tantamount to a diagnosis of PTSD, as most trauma exposures do not result in PTSD.7,8
Continue to: Symptom groups
Symptom groups. DSM-5 symptom criteria for PTSD include 4 symptom groups, Criteria B to E, respectively:
- intrusion
- avoidance
- negative cognitions and mood (numbing)
- hyperarousal/reactivity.
A specific number of symptoms must be present in all 4 of the symptom groups to fulfill diagnostic criteria. Importantly, these symptoms must be linked temporally and conceptually to the traumatic exposure to qualify as PTSD symptoms. Specifically, the symptoms must be new or substantially worsened after the event. For example, continuing sleep disturbance in someone who has had lifetime difficulty sleeping would not count as a trauma-related symptom. Most symptom checklists do not properly assess diagnostic criteria for PTSD because they do not anchor the symptoms in an exposure to a traumatic event; diagnosis requires an interview to fully assess all the diagnostic criteria. Finally, the symptoms must have been present for >1 month for the diagnosis, and the symptoms must have resulted in clinically significant distress or functional impairment to qualify.
The Algorithm provides a practical way to systematically assess all DSM-5 criteria for PTSD to arrive at a diagnosis. The clinician begins by determining whether a traumatic event has occurred and whether the individual had a qualifying exposure to it. If not, PTSD cannot be diagnosed. Alternative diagnoses to consider for new disorders that arise in the context of trauma among patients who are not exposed to trauma include major depressive disorder, adjustment disorder, and bereavement, as well as acute stress disorder (which is not validated but has potential utility as a billable diagnosis).
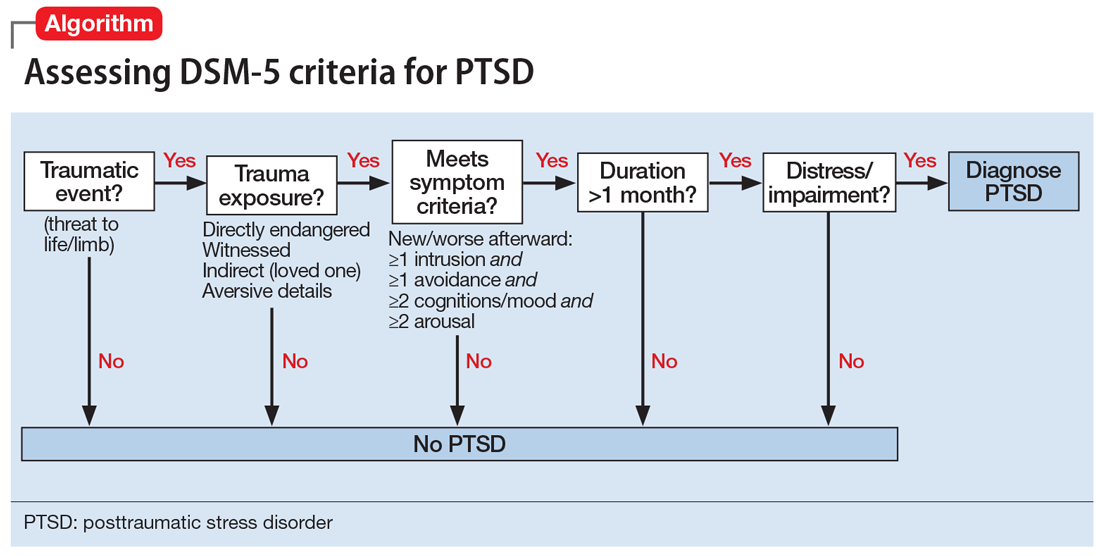
Avoidance and numbing symptoms (present in Criteria C and D) have been shown to represent markers of illness and can be useful in predicting PTSD.8-10 Unlike symptoms of intrusion and hyperarousal (Criteria B and E, respectively), which are very common and by themselves are nonpathological, avoidance/numbing symptoms occur much less commonly, are associated with functional impairment and other indicators of illness, and are strongly associated with PTSD.6 Prominent avoidance/numbing profiles have been demonstrated to predict PTSD in the first 1 to 2 weeks after trauma exposure, before PTSD can be formally diagnosed.11 Posttraumatic stress symptoms are nearly universal after trauma exposure, even in people who do not develop PTSD.5 Intrusion and hyperarousal symptoms constitute most of such symptoms,7 and these symptoms in the absence of prominent avoidance/numbing can be considered normative distress responses to trauma exposure.12
Some PTSD symptoms may seem quite similar to symptoms of depressive disorders and anxiety disorders. PTSD can be differentiated from these other disorders by linking the symptoms temporally and contextually to a qualifying exposure to a traumatic event. More often than not, PTSD presents with comorbid psychiatric disorders, especially depressive disorders, anxiety disorders, and/or substance use disorders.
Continue to: Treatment: Medication, psychotherapy, or both
Treatment: Medication, psychotherapy, or both
Both pharmacotherapy and psychotherapy—as monotherapy or in combination—are beneficial for treatment of PTSD. Research has not conclusively shown either treatment modality to be superior, because adequate head-to-head trials have not been conducted.4 Therefore, the choice of initial treatment is based on individual circumstances, such as patient preference for medication and/or psychotherapy, or the availability of therapists trained in evidence-based PTSD psychotherapy. Pharmacotherapeutic approaches are considered especially beneficial for depressive- and anxiety-like symptoms of PTSD, and trauma-focused psychotherapies are presumed to address the neuropathology of conditioned fear and anxiety responses involved in PTSD.14 Table 214-25 provides a list of published treatment guidelines and reviews to help clinicians seeking further detail beyond that provided in this article.
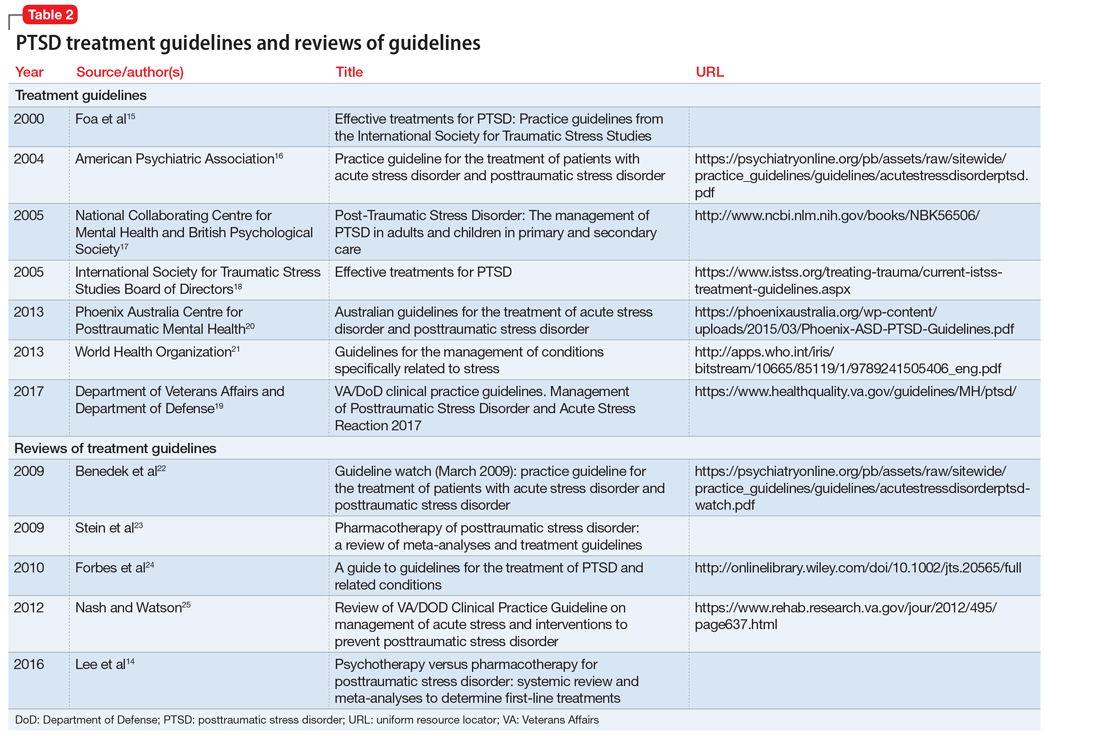
Antidepressants are the mainstay of pharmacotherapy for PTSD. These medications are effective for treating major depressive disorder, and have beneficial properties for PTSD independent of their antidepressant effects. The serotonin selective reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for the treatment of PTSD.6 Other recommended medications include the serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine, and nefazodone, an atypical serotoninergic agent.13 Other antidepressants with less published evidence of effectiveness are used as second-line pharmacotherapies for PTSD, including fluoxetine (SSRI), and mirtazapine, a noradrenergic and specific serotonergic antidepressant (NaSSA).4 Older medications, such as the tricyclic antidepressant amitriptyline and the monoamine oxidase inhibitor phenelzine, have also been used successfully as second-line treatments, but evidence of their benefit is less convincing than that supporting the first-line SSRIs/SNRIs. Additionally, their less favorable adverse effect and safety profiles make them less attractive treatment choices.13 Table 314-25 provides a list of first- and second-line medications for PTSD with recommended dosages and adverse effect profiles.
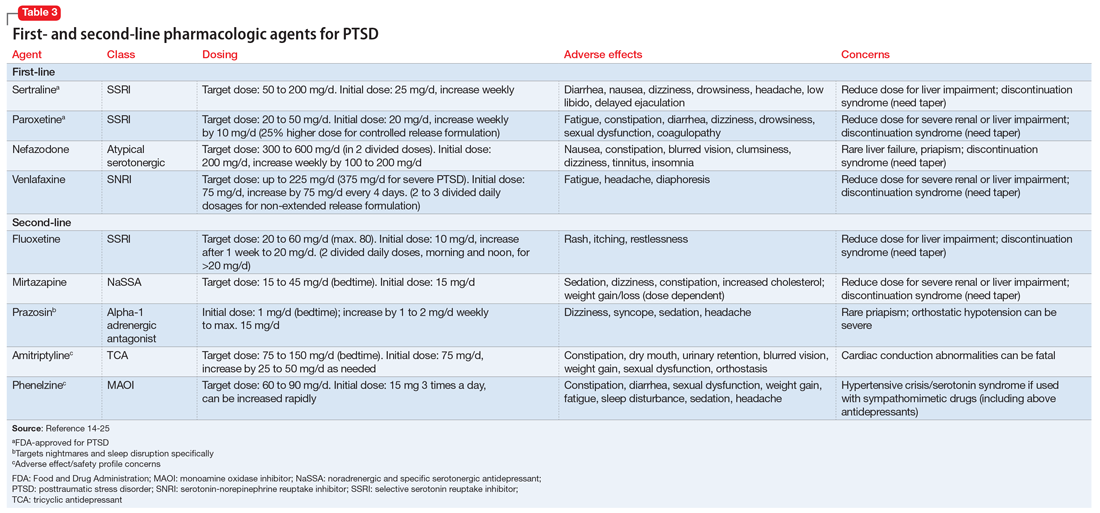
Other medications. Antiepileptics, antipsychotics, and benzodiazepines have not been demonstrated to have efficacy for primary treatment of PTSD, and none of the medications are considered first-line treatments, although sometimes they are used adjunctively in attempts to enhance the effectiveness of antidepressants. Benzodiazepines are sometimes used to target symptoms, such as sleep disturbance or hyperarousal, but only for very short periods. Several authoritative reviews strongly recommend against practices of polypharmacy that commonly involves use of these agents.4,14 Prazosin, an alpha-1 adrenergic antagonist, has been demonstrated to be an effective treatment for nightmares and sleep disturbances, and has grown increasingly popular for treating these symptoms in PTSD, especially in military veterans.13
A well-established barrier to effective pharmacotherapy of PTSD is medication nonadherence.13 Two common underlying sources of nonadherence are inconsistency with the patient’s treatment preference and intolerable adverse effects. Because SSRIs/SNRIs require 8 to 12 weeks of adequate dosing for symptom relief,13 medication adherence is vital. Explaining to patients that it takes many weeks of consistent dosing for clinical effects and reassuring them that the antidepressant agents used to treat PTSD are not habit-forming may help improve adherence.4
Psychotherapy. Prolonged exposure therapy and cognitive processing therapy—both trauma-focused therapies—have the best empirical evidence for efficacy for PTSD.4,14,26 Some patients are too anxious or avoidant to participate in trauma-focused psychotherapy and may benefit from a course of antidepressant treatment before initiating psychotherapy to reduce hyperarousal and avoidance symptoms enough to allow them to tolerate therapy that incorporates trauma memories.6 However, current PTSD treatment guidelines no longer recommend stabilization with medication or preparatory therapy as a routine prerequisite to trauma-focused psychotherapy.4
Continue to: Eye movement desensitization and reprocessing (EMDR) therapy...
Eye movement desensitization and reprocessing (EMDR) therapy has emerged as a popular trauma-focused therapy with documented effectiveness. During EMDR, the patient attends to emotionally disturbing material in brief sequential doses (which varies with individual patients) while simultaneously focusing on an external stimulus, typically therapist-directed lateral eye movements. Critics of EMDR point out that the theoretical concepts and therapeutic maneuvers (eg, finger movements to guide eye gaze) in EMDR are not consistent with current understanding of the neurobiological processes involved in PTSD. Further, studies testing separate components of the therapy have not established independent effectiveness of the therapeutic maneuvers beyond the therapeutic effects of the psychotherapy components of the procedure.4
Other psychotherapies might also be beneficial, but not enough research has been conducted to provide evidence for their effectiveness.4 Non-trauma–focused psychotherapies used for PTSD include supportive therapy, motivational interviewing, relaxation, and mindfulness. Because these therapies have less evidence of effectiveness, they are now widely considered second-line options. Psychological first aid is not a treatment for PTSD, but rather a nontreatment intervention for distress that is widely used by first responders and crisis counselors to provide compassion, support, and stabilization for people exposed to trauma, whether or not they have developed PTSD. Psychological first aid is supported by expert consensus, but it has not been studied enough to demonstrate how helpful it is as a treatment.6
Comorbidities require careful consideration
PTSD in the presence of other psychiatric disorders may require a unique and specialized approach to pharmacotherapy and psychotherapy. For instance, for a patient who has a comorbid substance use disorder, acute substance withdrawal can exacerbate PTSD symptoms. Sertraline is considered a medication of choice for these patients,13 and having a substance abuse specialist on the treatment team is desirable.4,13 A patient with comorbid traumatic brain injury (TBI) may have reduced tolerance to medications, and may require an individually-tailored and elongated titration strategy. Additionally, stimulants sometimes used to improve cognition for patients with comorbid TBI can exacerbate symptoms of hyperarousal, and these patients may need stabilization before beginning PTSD treatment. Antidepressant treatment for PTSD among patients with comorbid bipolar disorder has the potential to induce mania. Psychiatrists must consider these issues when formulating treatment plans for patients with PTSD and specific psychiatric comorbidities.4,6
PTSD symptoms can be chronic, sometimes lasting many years or even decades.27 In a longitudinal study of 716 survivors of 10 different disasters, 62% of those diagnosed with PTSD were still symptomatic 1 to 3 years after the disaster, demonstrating the enduring nature of PTSD symptoms.12 Similarly, a follow-up study of survivors of the Oklahoma City bombing found 58% of those with PTSD and 39% of those without PTSD were still reporting posttraumatic stress symptoms 7 years after the incident.28 Remarkably, these same individuals reported substantially improved functioning at work, with family and personal activities, and social interactions,28 and long-term employment disability specifically related to PTSD is highly unusual.29 Even individuals who continued to report active posttraumatic stress symptoms experienced a return of functioning equivalent to levels in individuals with no PTSD.28 These data suggest that treating psychiatrists and other mental health clinicians can be optimistic that functioning can improve remarkably over the long term, even if posttraumatic stress symptoms persist.
Bottom Line
A thorough understanding of the criteria for posttraumatic stress disorder (PTSD) is necessary for accurate diagnosis and treatment. Evidence-based treatment options for adults with PTSD include certain antidepressants and trauma-focused psychotherapies.
Related Resources
- Bernadino M, Nelson KJ. FIGHT to remember PTSD. Current Psychiatry. 2017;16(8):17.
- Koola MM. Prazosin and doxazosin for PTSD are underutilized and underdosed. Current Psychiatry. 2017;16(3):19-20,47,e1.
Drug Brand Names
Amitriptyline • Elavil, Endep
Fluoxetine • Prozac, Sarafem
Mirtazapine • Remeron
Nefazodone • Serzone
Paroxetine • Paxil
Phenelzine • Nardil
Prazosin • Minipress
Sertraline • Zoloft
Venlafaxine • Effexor
1. Diagnostic and Statistical Manual of Mental Disorders, 3rd ed. Washington, DC: American Psychiatric Association; 1980.
2. North CS, Surís AM, Smith RP, et al. The evolution of PTSD criteria across editions of the DSM. Ann Clin Psychiatry. 2016;28(3):197-208.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013
4. Downs DL, North CS. Trauma-related disorders. Overview of posttraumatic stress disorder. https://www.deckerip.com/products/scientific-american-psychiatry/table-of-contents/. Published July 2017. Accessed February 27, 2018.
5. North CS. Disaster mental health epidemiology: methodological review and interpretation of research findings. Psychiatry. 2016; 79(2):130-146.
6. North CS, Yutzy SH. Goodwin and Guze’s Psychiatric Diagnosis, 6th ed. New York, NY: Oxford University Press; 2010.
7. North CS, Nixon SJ, Shariat S, et al. Psychiatric disorders among survivors of the Oklahoma City bombing. JAMA. 1999;282(8):755-762.
8. North CS, Pfefferbaum B. Mental Health Response to Community Disasters: A Systematic Review. JAMA. 2013;310(5):507-518.
9. North CS, Pollio DE, Smith, RP, et al. Trauma exposure and posttraumatic stress disorder among employees of New York City companies affected by the September 11, 2001 attacks on the World Trade Center. Disaster Med Public Health Prep. 2011;5(suppl 2):S205-S213.
10. North CS, Oliver J, Pandya A. Examining a comprehensive model of disaster-related posttraumatic stress disorder in systematically studied survivors of 10 disasters. Am J Public Health. 2012;102(10):e40-e48.
11. Whitman JB, North CS, Downs DL, et al. A prospective study of the onset of PTSD symptoms in the first month after trauma exposure. Ann Clin Psychiatry. 2013;25(3):163-172.
12. North CS, Oliver J. Analysis of the longitudinal course of PTSD in 716 survivors of 10 disasters. Soc Psychiatry Psychiatr Epidemiol. 2013;48(8):1189-1197.
13. Jeffreys M, Capehart B, Friedman MJ. Pharmacotherapy for posttraumatic stress disorder: review with clinical applications. J Rehabil Res Dev. 2012;49(5):703-715.
14. Lee DJ, Schnitzlein CW, Wolf JP, et al. Psychotherapy versus pharmacotherapy for posttraumatic stress disorder: systemic review and meta-analyses to determine first-line treatments. Depress Anxiety. 2016;33(9):792-806.
15. Foa EB, Keane T, Friedman MJ. Effective treatments for PTSD: practice guidelines from the International Society for traumatic stress studies. New York, NY: The Guilford Press; 2000.
16. Ursano RJ, Bell C, Eth S, et al; Work Group on ASD and PTSD. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Arlington, VA: American Psychiatric Association Publishing; 2004.
17. National Collaborating Centre for Mental Health. Post-traumatic stress disorder: the management of PTSD in adults and children in primary and secondary care. London, UK: Gaskell and the British Psychological Society; 2005.
18. Foa EB, Keane TM, Friedman MJ, eds; The Board of Directors of the International Society for Traumatic Stress Studies. Effective treatments for PTSD. 2nd ed. Oakbrook Terrace, IL: The Guilford Press; 2005.
19. Department of Veterans Affairs and Department of Defense. VA/DoD clinical practice guidelines. Management of Posttraumatic Stress Disorder and Acute Stress Reaction 2017. https://www.healthquality.va.gov/guidelines/MH/ptsd/. Published June 2017. Accessed February 26, 2018.
20. Phoenix Australia -Centre for Posttraumatic Mental Health. Australian guidelines for the treatment of acute stress disorder and posttraumatic stress disorder. Melbourne, Australia: Phoenix Australia Centre for Posttraumatic Mental Health; 2013.
21. World Health Organization. Guidelines for the management of conditions specifically related to stress. Geneva, Switzerland: World Health Organization Press; 2013.
22. Benedek DM, Friedman MJ, Zatzick D, et al. Guideline watch (March 2009): practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Focus. 2009;7(2):201-213.
23. Stein DJ, Ipser J, McAnda N. Pharmacotherapy of posttraumatic stress disorder: a review of meta-analyses and treatment guidelines. CNS Spectr. 2009;14(suppl 1):25-31.
24. Forbes D, Creamer M, Bisson JI, et al. A guide to guidelines for the treatment of PTSD and related conditions. J Trauma Stress. 2010;23(5):537-552.
25. Nash WP, Watson PJ. Review of VA/DOD clinical practice guideline on management of acute stress and interventions to prevent posttraumatic stress disorder. J Rehabil Res Dev. 2012;49(5):637-648.
26. Birur B, Moore NC, Davis LL. An evidence-based review of early intervention and prevention of posttraumatic stress disorder. Community Ment Health J. 2017;53(2):183-201.
27. Breslau N, Davis GC. Posttraumatic stress disorder in an urban population of young adults: Risk factors for chronicity. Am J Psychiatry. 1992;149(5):671-675.
28. North CS, Pfefferbaum B, Kawasaki A, et al. Psychosocial adjustment of directly exposed survivors seven years after the Oklahoma City bombing. Compr Psychiatry. 2011;52(1):1-8
29. Rasco SS, North CS. An empirical study of employment and disability over three years among survivors of major disasters. J Am Acad Psychiatry Law. 2010;38(1):80-86.
Posttraumatic stress disorder (PTSD) has increasingly become a part of American culture since its introduction in the American Psychiatric Association’s third edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-III) in 1980.1 Since then, a proliferation of material about this disorder—both academic and popular—has been generated, yet much confusion persists surrounding the definition of the disorder, its prevalence, and its management. This review addresses the essential elements for diagnosis and treatment of PTSD.
Diagnosis: A closer look at the criteria
Criteria for the diagnosis of PTSD have evolved since 1980, with changes in the definition of trauma and the addition of symptoms and symptom groups.2 Table 13 summarizes the current DSM-5 criteria for PTSD.
Trauma exposure. An essential first step in the diagnosis of PTSD is to determine whether the individual has experienced exposure to trauma. This concept is defined in Criterion A (trauma exposure).3 PTSD is nonconformist among the psychiatric diagnoses in that it requires a specific external event as part of its definition. Misapplication of the trauma exposure criterion by many clinicians and researchers has led to misdiagnosis and erroneously high prevalence estimates of PTSD.4,5
A traumatic event is one that represents a threat to life or limb, specifically defined as “actual or threatened death, serious injury, or sexual violence.”3 DSM-5 does not allow for just any stressful event to be considered trauma. For example, no matter how distressing, failing an important test at school or being served with divorce proceedings do not represent a requisite trauma6 because these examples do not entail a threat to life or limb.
DSM-5 PTSD Criterion A also requires a qualifying exposure to the traumatic event. There are 4 types of qualifying exposures:
- direct experience of immediate serious physical danger
- eyewitness of trauma to others
- indirect exposure via violent or accidental trauma experienced by a close family member or close friend
- repeated or extreme exposure to aversive details of trauma, such as first responders collecting human remains or law enforcement officers being repeatedly exposed to horrific details of child abuse.3
Witnessed trauma must be in person; thus, viewing trauma in media reports would not constitute a qualifying exposure. Indirect trauma exposure can occur through learning of the experience of a qualifying trauma exposure by a close family member or personal friend.
It is critical to differentiate exposure to trauma (an objective construct) from the subjective distress that may be associated with it. If trauma has not occurred or a qualifying exposure is not established, no amount of distress associated with it can establish the experience as meeting Criterion A for PTSD. This does not mean that nonqualifying experiences of stressful events are not distressing; in fact, such experiences can result in substantial psychological angst. Conversely, exposure to trauma is not tantamount to a diagnosis of PTSD, as most trauma exposures do not result in PTSD.7,8
Continue to: Symptom groups
Symptom groups. DSM-5 symptom criteria for PTSD include 4 symptom groups, Criteria B to E, respectively:
- intrusion
- avoidance
- negative cognitions and mood (numbing)
- hyperarousal/reactivity.
A specific number of symptoms must be present in all 4 of the symptom groups to fulfill diagnostic criteria. Importantly, these symptoms must be linked temporally and conceptually to the traumatic exposure to qualify as PTSD symptoms. Specifically, the symptoms must be new or substantially worsened after the event. For example, continuing sleep disturbance in someone who has had lifetime difficulty sleeping would not count as a trauma-related symptom. Most symptom checklists do not properly assess diagnostic criteria for PTSD because they do not anchor the symptoms in an exposure to a traumatic event; diagnosis requires an interview to fully assess all the diagnostic criteria. Finally, the symptoms must have been present for >1 month for the diagnosis, and the symptoms must have resulted in clinically significant distress or functional impairment to qualify.
The Algorithm provides a practical way to systematically assess all DSM-5 criteria for PTSD to arrive at a diagnosis. The clinician begins by determining whether a traumatic event has occurred and whether the individual had a qualifying exposure to it. If not, PTSD cannot be diagnosed. Alternative diagnoses to consider for new disorders that arise in the context of trauma among patients who are not exposed to trauma include major depressive disorder, adjustment disorder, and bereavement, as well as acute stress disorder (which is not validated but has potential utility as a billable diagnosis).
Avoidance and numbing symptoms (present in Criteria C and D) have been shown to represent markers of illness and can be useful in predicting PTSD.8-10 Unlike symptoms of intrusion and hyperarousal (Criteria B and E, respectively), which are very common and by themselves are nonpathological, avoidance/numbing symptoms occur much less commonly, are associated with functional impairment and other indicators of illness, and are strongly associated with PTSD.6 Prominent avoidance/numbing profiles have been demonstrated to predict PTSD in the first 1 to 2 weeks after trauma exposure, before PTSD can be formally diagnosed.11 Posttraumatic stress symptoms are nearly universal after trauma exposure, even in people who do not develop PTSD.5 Intrusion and hyperarousal symptoms constitute most of such symptoms,7 and these symptoms in the absence of prominent avoidance/numbing can be considered normative distress responses to trauma exposure.12
Some PTSD symptoms may seem quite similar to symptoms of depressive disorders and anxiety disorders. PTSD can be differentiated from these other disorders by linking the symptoms temporally and contextually to a qualifying exposure to a traumatic event. More often than not, PTSD presents with comorbid psychiatric disorders, especially depressive disorders, anxiety disorders, and/or substance use disorders.
Continue to: Treatment: Medication, psychotherapy, or both
Treatment: Medication, psychotherapy, or both
Both pharmacotherapy and psychotherapy—as monotherapy or in combination—are beneficial for treatment of PTSD. Research has not conclusively shown either treatment modality to be superior, because adequate head-to-head trials have not been conducted.4 Therefore, the choice of initial treatment is based on individual circumstances, such as patient preference for medication and/or psychotherapy, or the availability of therapists trained in evidence-based PTSD psychotherapy. Pharmacotherapeutic approaches are considered especially beneficial for depressive- and anxiety-like symptoms of PTSD, and trauma-focused psychotherapies are presumed to address the neuropathology of conditioned fear and anxiety responses involved in PTSD.14 Table 214-25 provides a list of published treatment guidelines and reviews to help clinicians seeking further detail beyond that provided in this article.
Antidepressants are the mainstay of pharmacotherapy for PTSD. These medications are effective for treating major depressive disorder, and have beneficial properties for PTSD independent of their antidepressant effects. The serotonin selective reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for the treatment of PTSD.6 Other recommended medications include the serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine, and nefazodone, an atypical serotoninergic agent.13 Other antidepressants with less published evidence of effectiveness are used as second-line pharmacotherapies for PTSD, including fluoxetine (SSRI), and mirtazapine, a noradrenergic and specific serotonergic antidepressant (NaSSA).4 Older medications, such as the tricyclic antidepressant amitriptyline and the monoamine oxidase inhibitor phenelzine, have also been used successfully as second-line treatments, but evidence of their benefit is less convincing than that supporting the first-line SSRIs/SNRIs. Additionally, their less favorable adverse effect and safety profiles make them less attractive treatment choices.13 Table 314-25 provides a list of first- and second-line medications for PTSD with recommended dosages and adverse effect profiles.
Other medications. Antiepileptics, antipsychotics, and benzodiazepines have not been demonstrated to have efficacy for primary treatment of PTSD, and none of the medications are considered first-line treatments, although sometimes they are used adjunctively in attempts to enhance the effectiveness of antidepressants. Benzodiazepines are sometimes used to target symptoms, such as sleep disturbance or hyperarousal, but only for very short periods. Several authoritative reviews strongly recommend against practices of polypharmacy that commonly involves use of these agents.4,14 Prazosin, an alpha-1 adrenergic antagonist, has been demonstrated to be an effective treatment for nightmares and sleep disturbances, and has grown increasingly popular for treating these symptoms in PTSD, especially in military veterans.13
A well-established barrier to effective pharmacotherapy of PTSD is medication nonadherence.13 Two common underlying sources of nonadherence are inconsistency with the patient’s treatment preference and intolerable adverse effects. Because SSRIs/SNRIs require 8 to 12 weeks of adequate dosing for symptom relief,13 medication adherence is vital. Explaining to patients that it takes many weeks of consistent dosing for clinical effects and reassuring them that the antidepressant agents used to treat PTSD are not habit-forming may help improve adherence.4
Psychotherapy. Prolonged exposure therapy and cognitive processing therapy—both trauma-focused therapies—have the best empirical evidence for efficacy for PTSD.4,14,26 Some patients are too anxious or avoidant to participate in trauma-focused psychotherapy and may benefit from a course of antidepressant treatment before initiating psychotherapy to reduce hyperarousal and avoidance symptoms enough to allow them to tolerate therapy that incorporates trauma memories.6 However, current PTSD treatment guidelines no longer recommend stabilization with medication or preparatory therapy as a routine prerequisite to trauma-focused psychotherapy.4
Continue to: Eye movement desensitization and reprocessing (EMDR) therapy...
Eye movement desensitization and reprocessing (EMDR) therapy has emerged as a popular trauma-focused therapy with documented effectiveness. During EMDR, the patient attends to emotionally disturbing material in brief sequential doses (which varies with individual patients) while simultaneously focusing on an external stimulus, typically therapist-directed lateral eye movements. Critics of EMDR point out that the theoretical concepts and therapeutic maneuvers (eg, finger movements to guide eye gaze) in EMDR are not consistent with current understanding of the neurobiological processes involved in PTSD. Further, studies testing separate components of the therapy have not established independent effectiveness of the therapeutic maneuvers beyond the therapeutic effects of the psychotherapy components of the procedure.4
Other psychotherapies might also be beneficial, but not enough research has been conducted to provide evidence for their effectiveness.4 Non-trauma–focused psychotherapies used for PTSD include supportive therapy, motivational interviewing, relaxation, and mindfulness. Because these therapies have less evidence of effectiveness, they are now widely considered second-line options. Psychological first aid is not a treatment for PTSD, but rather a nontreatment intervention for distress that is widely used by first responders and crisis counselors to provide compassion, support, and stabilization for people exposed to trauma, whether or not they have developed PTSD. Psychological first aid is supported by expert consensus, but it has not been studied enough to demonstrate how helpful it is as a treatment.6
Comorbidities require careful consideration
PTSD in the presence of other psychiatric disorders may require a unique and specialized approach to pharmacotherapy and psychotherapy. For instance, for a patient who has a comorbid substance use disorder, acute substance withdrawal can exacerbate PTSD symptoms. Sertraline is considered a medication of choice for these patients,13 and having a substance abuse specialist on the treatment team is desirable.4,13 A patient with comorbid traumatic brain injury (TBI) may have reduced tolerance to medications, and may require an individually-tailored and elongated titration strategy. Additionally, stimulants sometimes used to improve cognition for patients with comorbid TBI can exacerbate symptoms of hyperarousal, and these patients may need stabilization before beginning PTSD treatment. Antidepressant treatment for PTSD among patients with comorbid bipolar disorder has the potential to induce mania. Psychiatrists must consider these issues when formulating treatment plans for patients with PTSD and specific psychiatric comorbidities.4,6
PTSD symptoms can be chronic, sometimes lasting many years or even decades.27 In a longitudinal study of 716 survivors of 10 different disasters, 62% of those diagnosed with PTSD were still symptomatic 1 to 3 years after the disaster, demonstrating the enduring nature of PTSD symptoms.12 Similarly, a follow-up study of survivors of the Oklahoma City bombing found 58% of those with PTSD and 39% of those without PTSD were still reporting posttraumatic stress symptoms 7 years after the incident.28 Remarkably, these same individuals reported substantially improved functioning at work, with family and personal activities, and social interactions,28 and long-term employment disability specifically related to PTSD is highly unusual.29 Even individuals who continued to report active posttraumatic stress symptoms experienced a return of functioning equivalent to levels in individuals with no PTSD.28 These data suggest that treating psychiatrists and other mental health clinicians can be optimistic that functioning can improve remarkably over the long term, even if posttraumatic stress symptoms persist.
Bottom Line
A thorough understanding of the criteria for posttraumatic stress disorder (PTSD) is necessary for accurate diagnosis and treatment. Evidence-based treatment options for adults with PTSD include certain antidepressants and trauma-focused psychotherapies.
Related Resources
- Bernadino M, Nelson KJ. FIGHT to remember PTSD. Current Psychiatry. 2017;16(8):17.
- Koola MM. Prazosin and doxazosin for PTSD are underutilized and underdosed. Current Psychiatry. 2017;16(3):19-20,47,e1.
Drug Brand Names
Amitriptyline • Elavil, Endep
Fluoxetine • Prozac, Sarafem
Mirtazapine • Remeron
Nefazodone • Serzone
Paroxetine • Paxil
Phenelzine • Nardil
Prazosin • Minipress
Sertraline • Zoloft
Venlafaxine • Effexor
Posttraumatic stress disorder (PTSD) has increasingly become a part of American culture since its introduction in the American Psychiatric Association’s third edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-III) in 1980.1 Since then, a proliferation of material about this disorder—both academic and popular—has been generated, yet much confusion persists surrounding the definition of the disorder, its prevalence, and its management. This review addresses the essential elements for diagnosis and treatment of PTSD.
Diagnosis: A closer look at the criteria
Criteria for the diagnosis of PTSD have evolved since 1980, with changes in the definition of trauma and the addition of symptoms and symptom groups.2 Table 13 summarizes the current DSM-5 criteria for PTSD.
Trauma exposure. An essential first step in the diagnosis of PTSD is to determine whether the individual has experienced exposure to trauma. This concept is defined in Criterion A (trauma exposure).3 PTSD is nonconformist among the psychiatric diagnoses in that it requires a specific external event as part of its definition. Misapplication of the trauma exposure criterion by many clinicians and researchers has led to misdiagnosis and erroneously high prevalence estimates of PTSD.4,5
A traumatic event is one that represents a threat to life or limb, specifically defined as “actual or threatened death, serious injury, or sexual violence.”3 DSM-5 does not allow for just any stressful event to be considered trauma. For example, no matter how distressing, failing an important test at school or being served with divorce proceedings do not represent a requisite trauma6 because these examples do not entail a threat to life or limb.
DSM-5 PTSD Criterion A also requires a qualifying exposure to the traumatic event. There are 4 types of qualifying exposures:
- direct experience of immediate serious physical danger
- eyewitness of trauma to others
- indirect exposure via violent or accidental trauma experienced by a close family member or close friend
- repeated or extreme exposure to aversive details of trauma, such as first responders collecting human remains or law enforcement officers being repeatedly exposed to horrific details of child abuse.3
Witnessed trauma must be in person; thus, viewing trauma in media reports would not constitute a qualifying exposure. Indirect trauma exposure can occur through learning of the experience of a qualifying trauma exposure by a close family member or personal friend.
It is critical to differentiate exposure to trauma (an objective construct) from the subjective distress that may be associated with it. If trauma has not occurred or a qualifying exposure is not established, no amount of distress associated with it can establish the experience as meeting Criterion A for PTSD. This does not mean that nonqualifying experiences of stressful events are not distressing; in fact, such experiences can result in substantial psychological angst. Conversely, exposure to trauma is not tantamount to a diagnosis of PTSD, as most trauma exposures do not result in PTSD.7,8
Continue to: Symptom groups
Symptom groups. DSM-5 symptom criteria for PTSD include 4 symptom groups, Criteria B to E, respectively:
- intrusion
- avoidance
- negative cognitions and mood (numbing)
- hyperarousal/reactivity.
A specific number of symptoms must be present in all 4 of the symptom groups to fulfill diagnostic criteria. Importantly, these symptoms must be linked temporally and conceptually to the traumatic exposure to qualify as PTSD symptoms. Specifically, the symptoms must be new or substantially worsened after the event. For example, continuing sleep disturbance in someone who has had lifetime difficulty sleeping would not count as a trauma-related symptom. Most symptom checklists do not properly assess diagnostic criteria for PTSD because they do not anchor the symptoms in an exposure to a traumatic event; diagnosis requires an interview to fully assess all the diagnostic criteria. Finally, the symptoms must have been present for >1 month for the diagnosis, and the symptoms must have resulted in clinically significant distress or functional impairment to qualify.
The Algorithm provides a practical way to systematically assess all DSM-5 criteria for PTSD to arrive at a diagnosis. The clinician begins by determining whether a traumatic event has occurred and whether the individual had a qualifying exposure to it. If not, PTSD cannot be diagnosed. Alternative diagnoses to consider for new disorders that arise in the context of trauma among patients who are not exposed to trauma include major depressive disorder, adjustment disorder, and bereavement, as well as acute stress disorder (which is not validated but has potential utility as a billable diagnosis).
Avoidance and numbing symptoms (present in Criteria C and D) have been shown to represent markers of illness and can be useful in predicting PTSD.8-10 Unlike symptoms of intrusion and hyperarousal (Criteria B and E, respectively), which are very common and by themselves are nonpathological, avoidance/numbing symptoms occur much less commonly, are associated with functional impairment and other indicators of illness, and are strongly associated with PTSD.6 Prominent avoidance/numbing profiles have been demonstrated to predict PTSD in the first 1 to 2 weeks after trauma exposure, before PTSD can be formally diagnosed.11 Posttraumatic stress symptoms are nearly universal after trauma exposure, even in people who do not develop PTSD.5 Intrusion and hyperarousal symptoms constitute most of such symptoms,7 and these symptoms in the absence of prominent avoidance/numbing can be considered normative distress responses to trauma exposure.12
Some PTSD symptoms may seem quite similar to symptoms of depressive disorders and anxiety disorders. PTSD can be differentiated from these other disorders by linking the symptoms temporally and contextually to a qualifying exposure to a traumatic event. More often than not, PTSD presents with comorbid psychiatric disorders, especially depressive disorders, anxiety disorders, and/or substance use disorders.
Continue to: Treatment: Medication, psychotherapy, or both
Treatment: Medication, psychotherapy, or both
Both pharmacotherapy and psychotherapy—as monotherapy or in combination—are beneficial for treatment of PTSD. Research has not conclusively shown either treatment modality to be superior, because adequate head-to-head trials have not been conducted.4 Therefore, the choice of initial treatment is based on individual circumstances, such as patient preference for medication and/or psychotherapy, or the availability of therapists trained in evidence-based PTSD psychotherapy. Pharmacotherapeutic approaches are considered especially beneficial for depressive- and anxiety-like symptoms of PTSD, and trauma-focused psychotherapies are presumed to address the neuropathology of conditioned fear and anxiety responses involved in PTSD.14 Table 214-25 provides a list of published treatment guidelines and reviews to help clinicians seeking further detail beyond that provided in this article.
Antidepressants are the mainstay of pharmacotherapy for PTSD. These medications are effective for treating major depressive disorder, and have beneficial properties for PTSD independent of their antidepressant effects. The serotonin selective reuptake inhibitors (SSRIs) sertraline and paroxetine are FDA-approved for the treatment of PTSD.6 Other recommended medications include the serotonin-norepinephrine reuptake inhibitor (SNRI) venlafaxine, and nefazodone, an atypical serotoninergic agent.13 Other antidepressants with less published evidence of effectiveness are used as second-line pharmacotherapies for PTSD, including fluoxetine (SSRI), and mirtazapine, a noradrenergic and specific serotonergic antidepressant (NaSSA).4 Older medications, such as the tricyclic antidepressant amitriptyline and the monoamine oxidase inhibitor phenelzine, have also been used successfully as second-line treatments, but evidence of their benefit is less convincing than that supporting the first-line SSRIs/SNRIs. Additionally, their less favorable adverse effect and safety profiles make them less attractive treatment choices.13 Table 314-25 provides a list of first- and second-line medications for PTSD with recommended dosages and adverse effect profiles.
Other medications. Antiepileptics, antipsychotics, and benzodiazepines have not been demonstrated to have efficacy for primary treatment of PTSD, and none of the medications are considered first-line treatments, although sometimes they are used adjunctively in attempts to enhance the effectiveness of antidepressants. Benzodiazepines are sometimes used to target symptoms, such as sleep disturbance or hyperarousal, but only for very short periods. Several authoritative reviews strongly recommend against practices of polypharmacy that commonly involves use of these agents.4,14 Prazosin, an alpha-1 adrenergic antagonist, has been demonstrated to be an effective treatment for nightmares and sleep disturbances, and has grown increasingly popular for treating these symptoms in PTSD, especially in military veterans.13
A well-established barrier to effective pharmacotherapy of PTSD is medication nonadherence.13 Two common underlying sources of nonadherence are inconsistency with the patient’s treatment preference and intolerable adverse effects. Because SSRIs/SNRIs require 8 to 12 weeks of adequate dosing for symptom relief,13 medication adherence is vital. Explaining to patients that it takes many weeks of consistent dosing for clinical effects and reassuring them that the antidepressant agents used to treat PTSD are not habit-forming may help improve adherence.4
Psychotherapy. Prolonged exposure therapy and cognitive processing therapy—both trauma-focused therapies—have the best empirical evidence for efficacy for PTSD.4,14,26 Some patients are too anxious or avoidant to participate in trauma-focused psychotherapy and may benefit from a course of antidepressant treatment before initiating psychotherapy to reduce hyperarousal and avoidance symptoms enough to allow them to tolerate therapy that incorporates trauma memories.6 However, current PTSD treatment guidelines no longer recommend stabilization with medication or preparatory therapy as a routine prerequisite to trauma-focused psychotherapy.4
Continue to: Eye movement desensitization and reprocessing (EMDR) therapy...
Eye movement desensitization and reprocessing (EMDR) therapy has emerged as a popular trauma-focused therapy with documented effectiveness. During EMDR, the patient attends to emotionally disturbing material in brief sequential doses (which varies with individual patients) while simultaneously focusing on an external stimulus, typically therapist-directed lateral eye movements. Critics of EMDR point out that the theoretical concepts and therapeutic maneuvers (eg, finger movements to guide eye gaze) in EMDR are not consistent with current understanding of the neurobiological processes involved in PTSD. Further, studies testing separate components of the therapy have not established independent effectiveness of the therapeutic maneuvers beyond the therapeutic effects of the psychotherapy components of the procedure.4
Other psychotherapies might also be beneficial, but not enough research has been conducted to provide evidence for their effectiveness.4 Non-trauma–focused psychotherapies used for PTSD include supportive therapy, motivational interviewing, relaxation, and mindfulness. Because these therapies have less evidence of effectiveness, they are now widely considered second-line options. Psychological first aid is not a treatment for PTSD, but rather a nontreatment intervention for distress that is widely used by first responders and crisis counselors to provide compassion, support, and stabilization for people exposed to trauma, whether or not they have developed PTSD. Psychological first aid is supported by expert consensus, but it has not been studied enough to demonstrate how helpful it is as a treatment.6
Comorbidities require careful consideration
PTSD in the presence of other psychiatric disorders may require a unique and specialized approach to pharmacotherapy and psychotherapy. For instance, for a patient who has a comorbid substance use disorder, acute substance withdrawal can exacerbate PTSD symptoms. Sertraline is considered a medication of choice for these patients,13 and having a substance abuse specialist on the treatment team is desirable.4,13 A patient with comorbid traumatic brain injury (TBI) may have reduced tolerance to medications, and may require an individually-tailored and elongated titration strategy. Additionally, stimulants sometimes used to improve cognition for patients with comorbid TBI can exacerbate symptoms of hyperarousal, and these patients may need stabilization before beginning PTSD treatment. Antidepressant treatment for PTSD among patients with comorbid bipolar disorder has the potential to induce mania. Psychiatrists must consider these issues when formulating treatment plans for patients with PTSD and specific psychiatric comorbidities.4,6
PTSD symptoms can be chronic, sometimes lasting many years or even decades.27 In a longitudinal study of 716 survivors of 10 different disasters, 62% of those diagnosed with PTSD were still symptomatic 1 to 3 years after the disaster, demonstrating the enduring nature of PTSD symptoms.12 Similarly, a follow-up study of survivors of the Oklahoma City bombing found 58% of those with PTSD and 39% of those without PTSD were still reporting posttraumatic stress symptoms 7 years after the incident.28 Remarkably, these same individuals reported substantially improved functioning at work, with family and personal activities, and social interactions,28 and long-term employment disability specifically related to PTSD is highly unusual.29 Even individuals who continued to report active posttraumatic stress symptoms experienced a return of functioning equivalent to levels in individuals with no PTSD.28 These data suggest that treating psychiatrists and other mental health clinicians can be optimistic that functioning can improve remarkably over the long term, even if posttraumatic stress symptoms persist.
Bottom Line
A thorough understanding of the criteria for posttraumatic stress disorder (PTSD) is necessary for accurate diagnosis and treatment. Evidence-based treatment options for adults with PTSD include certain antidepressants and trauma-focused psychotherapies.
Related Resources
- Bernadino M, Nelson KJ. FIGHT to remember PTSD. Current Psychiatry. 2017;16(8):17.
- Koola MM. Prazosin and doxazosin for PTSD are underutilized and underdosed. Current Psychiatry. 2017;16(3):19-20,47,e1.
Drug Brand Names
Amitriptyline • Elavil, Endep
Fluoxetine • Prozac, Sarafem
Mirtazapine • Remeron
Nefazodone • Serzone
Paroxetine • Paxil
Phenelzine • Nardil
Prazosin • Minipress
Sertraline • Zoloft
Venlafaxine • Effexor
1. Diagnostic and Statistical Manual of Mental Disorders, 3rd ed. Washington, DC: American Psychiatric Association; 1980.
2. North CS, Surís AM, Smith RP, et al. The evolution of PTSD criteria across editions of the DSM. Ann Clin Psychiatry. 2016;28(3):197-208.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013
4. Downs DL, North CS. Trauma-related disorders. Overview of posttraumatic stress disorder. https://www.deckerip.com/products/scientific-american-psychiatry/table-of-contents/. Published July 2017. Accessed February 27, 2018.
5. North CS. Disaster mental health epidemiology: methodological review and interpretation of research findings. Psychiatry. 2016; 79(2):130-146.
6. North CS, Yutzy SH. Goodwin and Guze’s Psychiatric Diagnosis, 6th ed. New York, NY: Oxford University Press; 2010.
7. North CS, Nixon SJ, Shariat S, et al. Psychiatric disorders among survivors of the Oklahoma City bombing. JAMA. 1999;282(8):755-762.
8. North CS, Pfefferbaum B. Mental Health Response to Community Disasters: A Systematic Review. JAMA. 2013;310(5):507-518.
9. North CS, Pollio DE, Smith, RP, et al. Trauma exposure and posttraumatic stress disorder among employees of New York City companies affected by the September 11, 2001 attacks on the World Trade Center. Disaster Med Public Health Prep. 2011;5(suppl 2):S205-S213.
10. North CS, Oliver J, Pandya A. Examining a comprehensive model of disaster-related posttraumatic stress disorder in systematically studied survivors of 10 disasters. Am J Public Health. 2012;102(10):e40-e48.
11. Whitman JB, North CS, Downs DL, et al. A prospective study of the onset of PTSD symptoms in the first month after trauma exposure. Ann Clin Psychiatry. 2013;25(3):163-172.
12. North CS, Oliver J. Analysis of the longitudinal course of PTSD in 716 survivors of 10 disasters. Soc Psychiatry Psychiatr Epidemiol. 2013;48(8):1189-1197.
13. Jeffreys M, Capehart B, Friedman MJ. Pharmacotherapy for posttraumatic stress disorder: review with clinical applications. J Rehabil Res Dev. 2012;49(5):703-715.
14. Lee DJ, Schnitzlein CW, Wolf JP, et al. Psychotherapy versus pharmacotherapy for posttraumatic stress disorder: systemic review and meta-analyses to determine first-line treatments. Depress Anxiety. 2016;33(9):792-806.
15. Foa EB, Keane T, Friedman MJ. Effective treatments for PTSD: practice guidelines from the International Society for traumatic stress studies. New York, NY: The Guilford Press; 2000.
16. Ursano RJ, Bell C, Eth S, et al; Work Group on ASD and PTSD. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Arlington, VA: American Psychiatric Association Publishing; 2004.
17. National Collaborating Centre for Mental Health. Post-traumatic stress disorder: the management of PTSD in adults and children in primary and secondary care. London, UK: Gaskell and the British Psychological Society; 2005.
18. Foa EB, Keane TM, Friedman MJ, eds; The Board of Directors of the International Society for Traumatic Stress Studies. Effective treatments for PTSD. 2nd ed. Oakbrook Terrace, IL: The Guilford Press; 2005.
19. Department of Veterans Affairs and Department of Defense. VA/DoD clinical practice guidelines. Management of Posttraumatic Stress Disorder and Acute Stress Reaction 2017. https://www.healthquality.va.gov/guidelines/MH/ptsd/. Published June 2017. Accessed February 26, 2018.
20. Phoenix Australia -Centre for Posttraumatic Mental Health. Australian guidelines for the treatment of acute stress disorder and posttraumatic stress disorder. Melbourne, Australia: Phoenix Australia Centre for Posttraumatic Mental Health; 2013.
21. World Health Organization. Guidelines for the management of conditions specifically related to stress. Geneva, Switzerland: World Health Organization Press; 2013.
22. Benedek DM, Friedman MJ, Zatzick D, et al. Guideline watch (March 2009): practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Focus. 2009;7(2):201-213.
23. Stein DJ, Ipser J, McAnda N. Pharmacotherapy of posttraumatic stress disorder: a review of meta-analyses and treatment guidelines. CNS Spectr. 2009;14(suppl 1):25-31.
24. Forbes D, Creamer M, Bisson JI, et al. A guide to guidelines for the treatment of PTSD and related conditions. J Trauma Stress. 2010;23(5):537-552.
25. Nash WP, Watson PJ. Review of VA/DOD clinical practice guideline on management of acute stress and interventions to prevent posttraumatic stress disorder. J Rehabil Res Dev. 2012;49(5):637-648.
26. Birur B, Moore NC, Davis LL. An evidence-based review of early intervention and prevention of posttraumatic stress disorder. Community Ment Health J. 2017;53(2):183-201.
27. Breslau N, Davis GC. Posttraumatic stress disorder in an urban population of young adults: Risk factors for chronicity. Am J Psychiatry. 1992;149(5):671-675.
28. North CS, Pfefferbaum B, Kawasaki A, et al. Psychosocial adjustment of directly exposed survivors seven years after the Oklahoma City bombing. Compr Psychiatry. 2011;52(1):1-8
29. Rasco SS, North CS. An empirical study of employment and disability over three years among survivors of major disasters. J Am Acad Psychiatry Law. 2010;38(1):80-86.
1. Diagnostic and Statistical Manual of Mental Disorders, 3rd ed. Washington, DC: American Psychiatric Association; 1980.
2. North CS, Surís AM, Smith RP, et al. The evolution of PTSD criteria across editions of the DSM. Ann Clin Psychiatry. 2016;28(3):197-208.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013
4. Downs DL, North CS. Trauma-related disorders. Overview of posttraumatic stress disorder. https://www.deckerip.com/products/scientific-american-psychiatry/table-of-contents/. Published July 2017. Accessed February 27, 2018.
5. North CS. Disaster mental health epidemiology: methodological review and interpretation of research findings. Psychiatry. 2016; 79(2):130-146.
6. North CS, Yutzy SH. Goodwin and Guze’s Psychiatric Diagnosis, 6th ed. New York, NY: Oxford University Press; 2010.
7. North CS, Nixon SJ, Shariat S, et al. Psychiatric disorders among survivors of the Oklahoma City bombing. JAMA. 1999;282(8):755-762.
8. North CS, Pfefferbaum B. Mental Health Response to Community Disasters: A Systematic Review. JAMA. 2013;310(5):507-518.
9. North CS, Pollio DE, Smith, RP, et al. Trauma exposure and posttraumatic stress disorder among employees of New York City companies affected by the September 11, 2001 attacks on the World Trade Center. Disaster Med Public Health Prep. 2011;5(suppl 2):S205-S213.
10. North CS, Oliver J, Pandya A. Examining a comprehensive model of disaster-related posttraumatic stress disorder in systematically studied survivors of 10 disasters. Am J Public Health. 2012;102(10):e40-e48.
11. Whitman JB, North CS, Downs DL, et al. A prospective study of the onset of PTSD symptoms in the first month after trauma exposure. Ann Clin Psychiatry. 2013;25(3):163-172.
12. North CS, Oliver J. Analysis of the longitudinal course of PTSD in 716 survivors of 10 disasters. Soc Psychiatry Psychiatr Epidemiol. 2013;48(8):1189-1197.
13. Jeffreys M, Capehart B, Friedman MJ. Pharmacotherapy for posttraumatic stress disorder: review with clinical applications. J Rehabil Res Dev. 2012;49(5):703-715.
14. Lee DJ, Schnitzlein CW, Wolf JP, et al. Psychotherapy versus pharmacotherapy for posttraumatic stress disorder: systemic review and meta-analyses to determine first-line treatments. Depress Anxiety. 2016;33(9):792-806.
15. Foa EB, Keane T, Friedman MJ. Effective treatments for PTSD: practice guidelines from the International Society for traumatic stress studies. New York, NY: The Guilford Press; 2000.
16. Ursano RJ, Bell C, Eth S, et al; Work Group on ASD and PTSD. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Arlington, VA: American Psychiatric Association Publishing; 2004.
17. National Collaborating Centre for Mental Health. Post-traumatic stress disorder: the management of PTSD in adults and children in primary and secondary care. London, UK: Gaskell and the British Psychological Society; 2005.
18. Foa EB, Keane TM, Friedman MJ, eds; The Board of Directors of the International Society for Traumatic Stress Studies. Effective treatments for PTSD. 2nd ed. Oakbrook Terrace, IL: The Guilford Press; 2005.
19. Department of Veterans Affairs and Department of Defense. VA/DoD clinical practice guidelines. Management of Posttraumatic Stress Disorder and Acute Stress Reaction 2017. https://www.healthquality.va.gov/guidelines/MH/ptsd/. Published June 2017. Accessed February 26, 2018.
20. Phoenix Australia -Centre for Posttraumatic Mental Health. Australian guidelines for the treatment of acute stress disorder and posttraumatic stress disorder. Melbourne, Australia: Phoenix Australia Centre for Posttraumatic Mental Health; 2013.
21. World Health Organization. Guidelines for the management of conditions specifically related to stress. Geneva, Switzerland: World Health Organization Press; 2013.
22. Benedek DM, Friedman MJ, Zatzick D, et al. Guideline watch (March 2009): practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Focus. 2009;7(2):201-213.
23. Stein DJ, Ipser J, McAnda N. Pharmacotherapy of posttraumatic stress disorder: a review of meta-analyses and treatment guidelines. CNS Spectr. 2009;14(suppl 1):25-31.
24. Forbes D, Creamer M, Bisson JI, et al. A guide to guidelines for the treatment of PTSD and related conditions. J Trauma Stress. 2010;23(5):537-552.
25. Nash WP, Watson PJ. Review of VA/DOD clinical practice guideline on management of acute stress and interventions to prevent posttraumatic stress disorder. J Rehabil Res Dev. 2012;49(5):637-648.
26. Birur B, Moore NC, Davis LL. An evidence-based review of early intervention and prevention of posttraumatic stress disorder. Community Ment Health J. 2017;53(2):183-201.
27. Breslau N, Davis GC. Posttraumatic stress disorder in an urban population of young adults: Risk factors for chronicity. Am J Psychiatry. 1992;149(5):671-675.
28. North CS, Pfefferbaum B, Kawasaki A, et al. Psychosocial adjustment of directly exposed survivors seven years after the Oklahoma City bombing. Compr Psychiatry. 2011;52(1):1-8
29. Rasco SS, North CS. An empirical study of employment and disability over three years among survivors of major disasters. J Am Acad Psychiatry Law. 2010;38(1):80-86.

Visual hallucinations and severe anxiety in the ICU after surgery
CASE Anxiety in the ICU
Mr. B, age 42, an African American man, is admitted to the inpatient medical unit for surgical treatment of peritoneal carcinomatosis with pelvic exenteration. He has a history of metastatic rectal cancer, chronic pain, and hypertension, but no psychiatric history. Mr. B’s postsurgical hospital stay is complicated by treatment-resistant tachycardia and hypertension, and he requires a lengthy stay in the ICU. In the ICU, Mr. B reports having visual hallucinations where he sees an individual placing a drug in his IV line. Additionally, he reports severe anxiety related to this experience. His anxiety and visual hallucinations are treated with coadministration of IV lorazepam, diphenhydramine, and haloperidol. These medications resolve the hallucinations, but his anxiety worsens and he becomes restless. He receives additional doses of IV haloperidol administered in 5 mg increments and reaching a cumulative 12-hour dose of 50 mg. Mr. B continues to report anxiety, so the psychiatry consultation-liaison (C-L) service is called.
[polldaddy:9970907]
The authors’ observations
Determining the cause of Mr. B’s anxiety is challenging because of his prolonged medical course, com
From a medical perspective, in a post-surgical patient treated in the ICU, the consulting practitioner must pay particular attention to delirium. ICU delirium is common—one report indicated that it occurs in 32.3% of ICU patients1—and frequently confused with psychiatric morbidity.2 Identifying delirium as the cause of impairment is important because delirium has potentially modifiable underlying etiologies. Symptomatically, delirium presents as impairment and fluctuation in attention, awareness, and at least one other cognitive domain, with a clear indication that the impairment occurred over a short period of time and represents a departure from baseline.3 In Mr. B’s case, these symptoms have not been excluded and should be considered by the C-L psychiatrists.
In addition to delirium, the C-L team must consider psychiatric comorbidity. Mr. B has no psychiatric history and a sudden first occurrence of hallucinations; therefore, it is unlikely that he has developed a primary psychotic disorder. Because he reported his symptoms had been present only for several days, he would not meet criteria for schizophrenia, which according to DSM-5 criteria require at least 1 month of ≥2 symptoms (including delusions, hallucinations, disorganized speech, disorganized behavior, or negative symptoms) and 6 months of declining function.3 However, although it is improbable, the C-L team must consider a primary psychotic illness, particularly given the potential devastating consequence of being misdiagnosed and mismanaged for an alternative illness. Unlike psychotic disorders, anxiety disorders are significantly more prevalent in the U.S. general population than primary psychotic disorders.4 Furthermore, the prevalence of anxiety disorders increases in the ICU setting; one study found that up to 61% of ICU patients setting experience “anxiety features.”5 Therefore, anxiety disorders and stress disorders should be considered in ICU patients who exhibit psychiatric symptoms.
Clinicians also should consider medication-induced adverse effects. In the ICU, patients are frequently managed on multiple medications, which increase their risk of developing adverse effects and adverse reactions.6 One potential consequence of polypharmacy is delirium, which remains a relevant potential diagnosis for Mr. B.7 Alternative consequences vary by medication and their respective pharmacodynamics. We take into consideration Mr. B’s exposure to high doses of the high-potency antipsychotic agent, haloperidol. Exposure to haloperidol can result in extrapyramidal symptoms, including akathisia,8,9,10 and the rare, but potentially fatal, NMS.11 These reactions can often be distinguished by taking a thorough history and a physical evaluation. In the case of akathisia, the clinician should look for medication exposure, titration, or taper. Most commonly, akathisia occurs secondary to antipsychotic exposure,12 followed by the onset of a combination of subjective symptoms, such as restlessness, anxiety, and irritability, and an objective symptom of increased motor activity.3 NMS, on the other hand, is distinguished by symptoms that include hyperthermia (>38ºC), diaphoresis, severe rigidity, urinary incontinence, vital instability, alterations in mental health status, and elevations in creatine kinase greater than 4-fold the upper limit, usually in the setting of treatment with antipsychotics.3 Nearly all cases of NMS occur within the first 30 days of antipsychotic exposure.3 While, overtly, NMS may appear to be less subtle than akathisia, clinicians should still be weary to rule out this admittedly rare, though potentially lethal diagnosis, especially in an ICU patient, where the diagnosis can be muddied by medical comorbidities that may mask the syndrome.
Continue to: EVALUATION Focus on akathisia
EVALUATION Focus on akathisia
On interview by the C-L team, Mr. B is visibly restless, moving all 4 extremities. He reports increased anxiety and irritability over the past 2 to 3 days. Mr. B states that he is aware of his increased motor movements and can briefly suppress them. However, after several seconds, he again begins spontaneously fidgeting, moving all 4 extremities and shifting from side to side in bed, saying, “I just feel anxious.” He denies having visual hallucinations, and says that the previous hallucinations had spontaneously presented and remitted after surgery. He denies the use of psychotropics for mental illness, prior similar symptoms to this presentation, a family history of mental illness, recent illicit substance use, or excessive alcohol use prior to presentation. This history is corroborated by collateral information from his brother, who was present in the ICU. On physical examination, Mr. B is afebrile and his vital signs are within normal limits. He does not have muscular rigidity or neck dystonia. His laboratory values, including complete blood count, electrolytes, liver function tests, and creatine phosphokinase, are within normal limits.
His medication administration record includes 46 standing agents, 16 “as-needed” agents, and 8 infusions. Several of the standing agents had psychotropic properties; however, the most salient were several opioids, ketamine, midazolam, lorazepam, dexamethasone, haloperidol, and olanzapine.
[polldaddy:9970908]
The authors’ observations
We determined that the most likely diagnosis for Mr. B’s symptoms was medication-induced akathisia secondary to haloperidol. Akathisia, coined by Haskovec in 1901,12,13 is from Greek, meaning an “inability to sit.”12 DSM-5 describes 2 forms of akathisia: medication-induced acute akathisia, and tardive akathisia.3 In the literature, others have described additional classifications, including chronic akathisia, withdrawal akathisia, and pseudoakathisia (Table 13,14-17). In Mr. B’s case, given his sudden development of symptoms and their direct chronologic relationship to antipsychotic treatment, and his combined subjective and objective symptoms, we believed that Mr. B’s symptoms were consistent with medication-induced acute akathisia (MIA). The identification and treatment of this clinical entity is important for several reasons, including reducing patient morbidity and maximizing patient comfort. Additionally, because akathisia has been associated with poor medication adherence, increased agitation/aggression, increased suicidality, and the eventual development of tardive dyskinesia,18 it is a relevant prognostic consideration when deciding to treat a patient with antipsychotics.

Pathophysiologically, we have yet to fully shed light on the exact underpinnings of akathisia. Much of our present knowledge stems from patient response to pharmacologic agents. While dopamine blockade has been linked to akathisia, the exact mechanism is not completely understood. Previous theories linking nigrostriatal pathways have been expanded to include mesocortical and mesolimbic considerations.12,17,18 Similarly surmised from medication effects, the transmitters y-aminobutyric acid, serotonin/5-hydroxytryptamine (5-HT), norepinephrine, and acetylcholine also have been linked to this syndrome, though as of yet, exact gross pathophysiologic mechanisms have not been fully elucidated.12 More recently, Stahl and Loonen19 described a novel mechanism by which they link the shell of the nucleus accumbens to akathisia. In their report, they indicate that the potential reduction in dopaminergic activity, secondary to antipsychotic administration, can result in compensatory noradrenergic activation of the locus coeruleus.19 The increased noradrenergic activity results in the downstream activation of the shell of the nucleus accumbens.19 The activation of the nucleus accumbens shell, which has been linked to unconditioned feeding and fear behavior, can then result in a cascade of effects that would phenotypically present as the syndrome we recognize to be akathisia.19
Numerous etiologies have been linked to MIA. Of these, high-potency antipsychotics are believed to remain the greatest risk factor for akathisia,18 although atypical antipsychotics, selective serotonin reuptake inhibitors, and serotonin-norepinephrine reuptake inhibitors, have been linked to the disorder.18,19
Continue to: Regarding antipsychotics...
Regarding antipsychotics, risk factors for akathisia include drug potency, dose, and rapidity of titration.20 All of these factors were relevant in our patient’s case. Risk across antipsychotic classes is not well understood; few head-to-head studies have comparing antipsychotics. However, general estimates suggest a 15% to 40% prevalence in patients exposed to typical antipsychotics, as compared with 0% to 12% exposed to atypical antipsychotics.8 The literature-reported difference in risk, as well as our patient’s comparative difference in exposure to large doses of haloperidol (50 mg) as compared with 1 dose of olanzapine (5 mg), led us to believe his akathisia developed primarily due to his exposure to haloperidol. Conclusively linking his symptoms to haloperidol alone, however, is not possible, and we did consider that olanzapine may in fact have had some effect in worsening Mr. B’s akathisia.
[polldaddy:9970909]
The authors’ observations
While there are reports on the efficacy of various agents in the treatment of akathisia, the most commonly evaluated agents are propranolol, anticholinergics, and benzodiazepines.17, 21
Propranolol is a nonselective beta-adrenergic blocker with numerous indications.17 Despite a 2004 Cochrane Review indicating that there is no evidence in support of central acting beta-blockers for treating akathisia,22 propranolol is not yet recognized as an appropriate treatment.17 The reason for this discrepancy is likely due to the Cochrane Review’s restrictive inclusion criteria, which prevented the analysis of much of the literature.22 In fact, several reports cite evidence for the treatment efficacy of propranolol17 and, to date, some reports continue to advocate for its use as a first-line agent in the treatment of akathisia. Admittedly, besides the Cochrane Review,22 other reports have found propranolol to be ineffective for treating akathisia,23 although these tend to be limited by their population size and generalizability.
As with propranolol, a 2006 Cochrane Review found “no reliable evidence to support or refute” using anticholinergic agents in the treatment of akathisia.24 We suspect that the review’s findings were likely secondary to its strict inclusion criteria.24 In fact, several reports support using anticholinergic agents for treating akathisia.25 Here we focus on benztropine and diphenhydramine.
Two reviews—Blaisdell26 (1994) and Poyurovsky27 (2010)—suggest modest benefits from benztropine, primarily in patients with comorbid Parkinson’s disease. Despite these benefits, head-to-head trials seem to either point to the superiority of propranolol or to no difference between these agents for treating akathisia.28,29 In a review, we only found 1 trial demonstrating benztropine’s superiority over propranolol,23 but this trial was constrained by its small population (6 patients). Therefore, the data suggest that, when indicated, clinicians should lean towards using propranolol for treating akathisia.
Continue to: Diphenhydramine, a first-generation antihistamine...
Diphenhydramine, a first-generation antihistamine with antimuscarinic properties, has been studied for its efficacy in treating metoclopramide-induced akathisia in the emergency setting.30 There are several reports on the efficacy of this agent, including a large randomized study involving 281 patients that found it effective for preventing metoclopramide-induced akathisia.30 Another head-to-head trial reported the benefit of the diphenhydramine vs midazolam.31 Both agents were efficacious for treating akathisia; however, midazolam had a more rapid onset. Despite these positive reports, double-blind trials have found diphenhydramine to be ineffective,17 which suggests propranolol should be the first-line agent, assuming it is not contraindicated.
Benzodiazepines have also been found to be efficacious for treating akathisia. A 1999 Cochrane Review included 2 randomized controlled trials that assessed the efficacy of clonazepam vs placebo for treating akathisia.32 It found evidence of benefit for clonazepam, but questioned the generalizability of these studies.32 This review did not include several other reports that suggest benefits of other benzodiazepines for treating akathisia. Other than clonazepam, reports suggest benefit for diazepam, lorazepam, and midazolam for treating akathisia.17 Despite this evidence and the findings from this Cochrane Review, the literature does not appear to point to clear dominance of these agents over propranolol. Given the safety concerns when prescribing benzodiazepines, it would be prudent to utilize propranolol as a first-line agent for treating akathisia.
Finally, other reports have cited treatment efficacy linked to serotonin 2A receptor (5-HT2A) antagonists (mianserin, mirtazapine, and trazodone), clonidine, gabapentin, amantadine, and other agents.17 If treatment with propranolol is ineffective or contraindicated, clinicians should utilize their clinical judgement in deciding on the use of one agent over another.
OUTCOME Complete resolution
Haloperidol is discontinued and diphenhydramine, 50 mg IV, is administered. (Diphenhydramine was used instead of propranolol due to immediacy of availability.) Most of Mr. B’s signs and symptoms resolve on a repeat interview 3 hours later. He receives another dose of diphenhydramine, 25 mg IV, for persistent mild irritability. By Day 2 of follow-up, his symptoms completely resolve as measured on the Barnes Akathisia Scale33 (Table 2).

Continue to: Bottom Line
Bottom Line
Akathisia is an elusive adverse effect of antipsychotics and can be misdiagnosed as anxiety. Close consideration should be given to potential medical, psychiatric, and drug-related etiologies in patients who have a prolonged medical course, comorbidities, and exposure to multiple pharmacologic agents.
Related Resources
- Factor SA, Leffler JB, Murray CF. Drug-induced movement disorders: a clinical review. Medscape. http://www.medscape.org/viewarticle/586881.
- Marder S, Stroup TS. Pharmacotherapy for schizophrenia: side effect management. UpToDate. https://www.uptodate.com/contents/pharmacotherapy-for-schizophrenia-side-effect-management.
Drug Brand Names
Amantadine • Symmetrel
Benztropine • Cogentin
Clonazepam • Klonopin
Clonidine • Catapres
Dexamethasone • Decadron
Diazepam • Valium
Diphenhydramine • Benadryl
Gabapentin • Neurontin
Haloperidol • Haldol
Ketamine • Ketalar
Lorazepam • Ativan
Metoclopramide • Reglan
Mianserin • Tolvon
Midazolam • Versed
Mirtazapine • Remeron
Olanzapine • Zyprexa
Propranolol • Inderal
Rivastigmine • Exelon
Trazodone • Oleptro
1. Cavallazzi R, Saad M, Marik PE. Delirium in the ICU: an overview. Ann Intensive Care. 2012;2:49.
2. Farrell KR, Ganzini L. Misdiagnosing delirium as depression in medically ill elderly patients. Arch Intern Med. 1995;155(22):2459-2464.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. National Alliance on Mental Illness. Mental health by the numbers. https://www.nami.org/learn-more/mental-health-by-the-numbers. Accessed March 4, 2018.
5. Jacka MJ, Mitchell N, Perez-Parada J. Incidence and prevalence of anxiety, depression, and post-traumatic stress disorder among critical care patients, families, and practitioners. J Anest & Inten Care Med. 2016;1(1):55555. doi: 10.19080/JAICM.2016.01.555555.
6. Reis AM, Cassiani SH. Adverse drug events in an intensive care unit of a university hospital. Eur J Clin Pharmacol. 2011;67(6):625-632.
7. Garpestad E, Devlin JW. Polypharmacy and delirium in critically ill older adults: recognition and prevention. Clin Geriatr Med. 2017;33(2):189-203.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Van Putten T, Marder SR. Toward a more reliable diagnosis of akathisia. Arch Gen Psychiatry. 1986;43(10):1015-1016.
10. Penders TM, Agarwal S, Rohaidy R. Persistent akathisia masquerading as agitated depression after use of ziprasidone in the treatment of bipolar depression. Neuropsychiatr Dis Treat. 2013;9:463-465.
11. Naganuma H, Fujii I. Incidence and risk factors in neuroleptic malignant syndrome. Acta Psychiatr Scand. 1994;90(6):424-426.
12. Forcen FE, Matsoukas K, Alici Y. Antipsychotic-induced akathisia in delirium: a systematic review. Palliat Support Care. 2016;14(1):77-84.
13. Brune M, Sachdev, PS. Ladislav Haskovec and 100 years of akathisia. American Journal of Psychiatry. 2002;159(5):727-727.
14. Havaki-Kontaxaki BJ, Kontaxakis VP, Christodoulou GN. Prevalence and characteristics of patients with pseudoakathisia. Eur Neuropsychopharmacol. 2000;10(5):333-336.
15. Lang AE. Withdrawal akathisia: case reports and a proposed classification of chronic akathisia. Mov Disord. 1994;9(2):188-192.
16. Sachdev P. The epidemiology of drug-induced akathisia: Part II. Chronic, tardive, and withdrawal akathisias. Schizophr Bull. 1995;21(3):451-461.
17. Kern DS, Lang AE. Acute akathisia. In: Friedman JH, ed. Medication-induced movement disorders. Cambridge, United Kingdom: Cambridge University Press; 2015:12-24.
18. Adler LA, Angrist B, Reiter S, et al. Neuroleptic-induced akathisia: a review. Psychopharmacology (Berl). 1989;97(1):1-11.
19. Stahl SM, Loonen AJM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
20. Sachdev P, Kruk J. Clinical characteristics and predisposing factors in acute drug-induced akathisia. Arch Gen Psychiatry. 1994;51(12):963-974.
21. Laoutidis ZG, Luckhaus C. 5-HT2A receptor antagonists for the treatment of neuroleptic-induced akathisia: a systematic review and meta-analysis. Int J Neuropsychopharmacol. 2014;17(5):823-832.
22. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
23. Sachdev P, Loneragan C. Intravenous benztropine and propranolol challenges in acute neuroleptic-induced akathisia. Clin Neuropharmacol. 1993;16(4):324-331.
24. Lima AR, Weiser KV, Bacaltchuk J, et al. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(1):CD003727.
25. Fleischhacker WW, Roth SD, Kane JM. The pharmacologic treatment of neuroleptic-induced akathisia. J Clin Psychopharmacol. 1990;10(1):12-21.
26. Blaisdell GD. Akathisia: a comprehensive review and treatment summary. Pharmacopsychiatry. 1994;27(4):139-146.
27. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
28. Adler LA, Reiter S, Corwin J, et al. Neuroleptic-induced akathisia: propranolol versus benztropine. Biol Psychiatry. 1988;23(2):211-213.
29. Adler LA, Peselow E, Rosenthal M, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Bender B, Friedman B, Davitt M, et al. 118: metoclopramide in the emergency department: a randomized factorial design study to determine the influence of dose and diphenhydramine on akathisia. Ann of Emerg Med. 2008;52(4):S78.
31. Parlak I, Erdur B, Parlak M, et al. Midazolam vs. diphenhydramine for the treatment of metoclopramide-induced akathisia: a randomized controlled trial. Acad Emerg Med. 2007;14(8):715-721.
32. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 1999;(4):CD001950.
33. Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154(5):672-676..
CASE Anxiety in the ICU
Mr. B, age 42, an African American man, is admitted to the inpatient medical unit for surgical treatment of peritoneal carcinomatosis with pelvic exenteration. He has a history of metastatic rectal cancer, chronic pain, and hypertension, but no psychiatric history. Mr. B’s postsurgical hospital stay is complicated by treatment-resistant tachycardia and hypertension, and he requires a lengthy stay in the ICU. In the ICU, Mr. B reports having visual hallucinations where he sees an individual placing a drug in his IV line. Additionally, he reports severe anxiety related to this experience. His anxiety and visual hallucinations are treated with coadministration of IV lorazepam, diphenhydramine, and haloperidol. These medications resolve the hallucinations, but his anxiety worsens and he becomes restless. He receives additional doses of IV haloperidol administered in 5 mg increments and reaching a cumulative 12-hour dose of 50 mg. Mr. B continues to report anxiety, so the psychiatry consultation-liaison (C-L) service is called.
[polldaddy:9970907]
The authors’ observations
Determining the cause of Mr. B’s anxiety is challenging because of his prolonged medical course, com
From a medical perspective, in a post-surgical patient treated in the ICU, the consulting practitioner must pay particular attention to delirium. ICU delirium is common—one report indicated that it occurs in 32.3% of ICU patients1—and frequently confused with psychiatric morbidity.2 Identifying delirium as the cause of impairment is important because delirium has potentially modifiable underlying etiologies. Symptomatically, delirium presents as impairment and fluctuation in attention, awareness, and at least one other cognitive domain, with a clear indication that the impairment occurred over a short period of time and represents a departure from baseline.3 In Mr. B’s case, these symptoms have not been excluded and should be considered by the C-L psychiatrists.
In addition to delirium, the C-L team must consider psychiatric comorbidity. Mr. B has no psychiatric history and a sudden first occurrence of hallucinations; therefore, it is unlikely that he has developed a primary psychotic disorder. Because he reported his symptoms had been present only for several days, he would not meet criteria for schizophrenia, which according to DSM-5 criteria require at least 1 month of ≥2 symptoms (including delusions, hallucinations, disorganized speech, disorganized behavior, or negative symptoms) and 6 months of declining function.3 However, although it is improbable, the C-L team must consider a primary psychotic illness, particularly given the potential devastating consequence of being misdiagnosed and mismanaged for an alternative illness. Unlike psychotic disorders, anxiety disorders are significantly more prevalent in the U.S. general population than primary psychotic disorders.4 Furthermore, the prevalence of anxiety disorders increases in the ICU setting; one study found that up to 61% of ICU patients setting experience “anxiety features.”5 Therefore, anxiety disorders and stress disorders should be considered in ICU patients who exhibit psychiatric symptoms.
Clinicians also should consider medication-induced adverse effects. In the ICU, patients are frequently managed on multiple medications, which increase their risk of developing adverse effects and adverse reactions.6 One potential consequence of polypharmacy is delirium, which remains a relevant potential diagnosis for Mr. B.7 Alternative consequences vary by medication and their respective pharmacodynamics. We take into consideration Mr. B’s exposure to high doses of the high-potency antipsychotic agent, haloperidol. Exposure to haloperidol can result in extrapyramidal symptoms, including akathisia,8,9,10 and the rare, but potentially fatal, NMS.11 These reactions can often be distinguished by taking a thorough history and a physical evaluation. In the case of akathisia, the clinician should look for medication exposure, titration, or taper. Most commonly, akathisia occurs secondary to antipsychotic exposure,12 followed by the onset of a combination of subjective symptoms, such as restlessness, anxiety, and irritability, and an objective symptom of increased motor activity.3 NMS, on the other hand, is distinguished by symptoms that include hyperthermia (>38ºC), diaphoresis, severe rigidity, urinary incontinence, vital instability, alterations in mental health status, and elevations in creatine kinase greater than 4-fold the upper limit, usually in the setting of treatment with antipsychotics.3 Nearly all cases of NMS occur within the first 30 days of antipsychotic exposure.3 While, overtly, NMS may appear to be less subtle than akathisia, clinicians should still be weary to rule out this admittedly rare, though potentially lethal diagnosis, especially in an ICU patient, where the diagnosis can be muddied by medical comorbidities that may mask the syndrome.
Continue to: EVALUATION Focus on akathisia
EVALUATION Focus on akathisia
On interview by the C-L team, Mr. B is visibly restless, moving all 4 extremities. He reports increased anxiety and irritability over the past 2 to 3 days. Mr. B states that he is aware of his increased motor movements and can briefly suppress them. However, after several seconds, he again begins spontaneously fidgeting, moving all 4 extremities and shifting from side to side in bed, saying, “I just feel anxious.” He denies having visual hallucinations, and says that the previous hallucinations had spontaneously presented and remitted after surgery. He denies the use of psychotropics for mental illness, prior similar symptoms to this presentation, a family history of mental illness, recent illicit substance use, or excessive alcohol use prior to presentation. This history is corroborated by collateral information from his brother, who was present in the ICU. On physical examination, Mr. B is afebrile and his vital signs are within normal limits. He does not have muscular rigidity or neck dystonia. His laboratory values, including complete blood count, electrolytes, liver function tests, and creatine phosphokinase, are within normal limits.
His medication administration record includes 46 standing agents, 16 “as-needed” agents, and 8 infusions. Several of the standing agents had psychotropic properties; however, the most salient were several opioids, ketamine, midazolam, lorazepam, dexamethasone, haloperidol, and olanzapine.
[polldaddy:9970908]
The authors’ observations
We determined that the most likely diagnosis for Mr. B’s symptoms was medication-induced akathisia secondary to haloperidol. Akathisia, coined by Haskovec in 1901,12,13 is from Greek, meaning an “inability to sit.”12 DSM-5 describes 2 forms of akathisia: medication-induced acute akathisia, and tardive akathisia.3 In the literature, others have described additional classifications, including chronic akathisia, withdrawal akathisia, and pseudoakathisia (Table 13,14-17). In Mr. B’s case, given his sudden development of symptoms and their direct chronologic relationship to antipsychotic treatment, and his combined subjective and objective symptoms, we believed that Mr. B’s symptoms were consistent with medication-induced acute akathisia (MIA). The identification and treatment of this clinical entity is important for several reasons, including reducing patient morbidity and maximizing patient comfort. Additionally, because akathisia has been associated with poor medication adherence, increased agitation/aggression, increased suicidality, and the eventual development of tardive dyskinesia,18 it is a relevant prognostic consideration when deciding to treat a patient with antipsychotics.
Pathophysiologically, we have yet to fully shed light on the exact underpinnings of akathisia. Much of our present knowledge stems from patient response to pharmacologic agents. While dopamine blockade has been linked to akathisia, the exact mechanism is not completely understood. Previous theories linking nigrostriatal pathways have been expanded to include mesocortical and mesolimbic considerations.12,17,18 Similarly surmised from medication effects, the transmitters y-aminobutyric acid, serotonin/5-hydroxytryptamine (5-HT), norepinephrine, and acetylcholine also have been linked to this syndrome, though as of yet, exact gross pathophysiologic mechanisms have not been fully elucidated.12 More recently, Stahl and Loonen19 described a novel mechanism by which they link the shell of the nucleus accumbens to akathisia. In their report, they indicate that the potential reduction in dopaminergic activity, secondary to antipsychotic administration, can result in compensatory noradrenergic activation of the locus coeruleus.19 The increased noradrenergic activity results in the downstream activation of the shell of the nucleus accumbens.19 The activation of the nucleus accumbens shell, which has been linked to unconditioned feeding and fear behavior, can then result in a cascade of effects that would phenotypically present as the syndrome we recognize to be akathisia.19
Numerous etiologies have been linked to MIA. Of these, high-potency antipsychotics are believed to remain the greatest risk factor for akathisia,18 although atypical antipsychotics, selective serotonin reuptake inhibitors, and serotonin-norepinephrine reuptake inhibitors, have been linked to the disorder.18,19
Continue to: Regarding antipsychotics...
Regarding antipsychotics, risk factors for akathisia include drug potency, dose, and rapidity of titration.20 All of these factors were relevant in our patient’s case. Risk across antipsychotic classes is not well understood; few head-to-head studies have comparing antipsychotics. However, general estimates suggest a 15% to 40% prevalence in patients exposed to typical antipsychotics, as compared with 0% to 12% exposed to atypical antipsychotics.8 The literature-reported difference in risk, as well as our patient’s comparative difference in exposure to large doses of haloperidol (50 mg) as compared with 1 dose of olanzapine (5 mg), led us to believe his akathisia developed primarily due to his exposure to haloperidol. Conclusively linking his symptoms to haloperidol alone, however, is not possible, and we did consider that olanzapine may in fact have had some effect in worsening Mr. B’s akathisia.
[polldaddy:9970909]
The authors’ observations
While there are reports on the efficacy of various agents in the treatment of akathisia, the most commonly evaluated agents are propranolol, anticholinergics, and benzodiazepines.17, 21
Propranolol is a nonselective beta-adrenergic blocker with numerous indications.17 Despite a 2004 Cochrane Review indicating that there is no evidence in support of central acting beta-blockers for treating akathisia,22 propranolol is not yet recognized as an appropriate treatment.17 The reason for this discrepancy is likely due to the Cochrane Review’s restrictive inclusion criteria, which prevented the analysis of much of the literature.22 In fact, several reports cite evidence for the treatment efficacy of propranolol17 and, to date, some reports continue to advocate for its use as a first-line agent in the treatment of akathisia. Admittedly, besides the Cochrane Review,22 other reports have found propranolol to be ineffective for treating akathisia,23 although these tend to be limited by their population size and generalizability.
As with propranolol, a 2006 Cochrane Review found “no reliable evidence to support or refute” using anticholinergic agents in the treatment of akathisia.24 We suspect that the review’s findings were likely secondary to its strict inclusion criteria.24 In fact, several reports support using anticholinergic agents for treating akathisia.25 Here we focus on benztropine and diphenhydramine.
Two reviews—Blaisdell26 (1994) and Poyurovsky27 (2010)—suggest modest benefits from benztropine, primarily in patients with comorbid Parkinson’s disease. Despite these benefits, head-to-head trials seem to either point to the superiority of propranolol or to no difference between these agents for treating akathisia.28,29 In a review, we only found 1 trial demonstrating benztropine’s superiority over propranolol,23 but this trial was constrained by its small population (6 patients). Therefore, the data suggest that, when indicated, clinicians should lean towards using propranolol for treating akathisia.
Continue to: Diphenhydramine, a first-generation antihistamine...
Diphenhydramine, a first-generation antihistamine with antimuscarinic properties, has been studied for its efficacy in treating metoclopramide-induced akathisia in the emergency setting.30 There are several reports on the efficacy of this agent, including a large randomized study involving 281 patients that found it effective for preventing metoclopramide-induced akathisia.30 Another head-to-head trial reported the benefit of the diphenhydramine vs midazolam.31 Both agents were efficacious for treating akathisia; however, midazolam had a more rapid onset. Despite these positive reports, double-blind trials have found diphenhydramine to be ineffective,17 which suggests propranolol should be the first-line agent, assuming it is not contraindicated.
Benzodiazepines have also been found to be efficacious for treating akathisia. A 1999 Cochrane Review included 2 randomized controlled trials that assessed the efficacy of clonazepam vs placebo for treating akathisia.32 It found evidence of benefit for clonazepam, but questioned the generalizability of these studies.32 This review did not include several other reports that suggest benefits of other benzodiazepines for treating akathisia. Other than clonazepam, reports suggest benefit for diazepam, lorazepam, and midazolam for treating akathisia.17 Despite this evidence and the findings from this Cochrane Review, the literature does not appear to point to clear dominance of these agents over propranolol. Given the safety concerns when prescribing benzodiazepines, it would be prudent to utilize propranolol as a first-line agent for treating akathisia.
Finally, other reports have cited treatment efficacy linked to serotonin 2A receptor (5-HT2A) antagonists (mianserin, mirtazapine, and trazodone), clonidine, gabapentin, amantadine, and other agents.17 If treatment with propranolol is ineffective or contraindicated, clinicians should utilize their clinical judgement in deciding on the use of one agent over another.
OUTCOME Complete resolution
Haloperidol is discontinued and diphenhydramine, 50 mg IV, is administered. (Diphenhydramine was used instead of propranolol due to immediacy of availability.) Most of Mr. B’s signs and symptoms resolve on a repeat interview 3 hours later. He receives another dose of diphenhydramine, 25 mg IV, for persistent mild irritability. By Day 2 of follow-up, his symptoms completely resolve as measured on the Barnes Akathisia Scale33 (Table 2).
Continue to: Bottom Line
Bottom Line
Akathisia is an elusive adverse effect of antipsychotics and can be misdiagnosed as anxiety. Close consideration should be given to potential medical, psychiatric, and drug-related etiologies in patients who have a prolonged medical course, comorbidities, and exposure to multiple pharmacologic agents.
Related Resources
- Factor SA, Leffler JB, Murray CF. Drug-induced movement disorders: a clinical review. Medscape. http://www.medscape.org/viewarticle/586881.
- Marder S, Stroup TS. Pharmacotherapy for schizophrenia: side effect management. UpToDate. https://www.uptodate.com/contents/pharmacotherapy-for-schizophrenia-side-effect-management.
Drug Brand Names
Amantadine • Symmetrel
Benztropine • Cogentin
Clonazepam • Klonopin
Clonidine • Catapres
Dexamethasone • Decadron
Diazepam • Valium
Diphenhydramine • Benadryl
Gabapentin • Neurontin
Haloperidol • Haldol
Ketamine • Ketalar
Lorazepam • Ativan
Metoclopramide • Reglan
Mianserin • Tolvon
Midazolam • Versed
Mirtazapine • Remeron
Olanzapine • Zyprexa
Propranolol • Inderal
Rivastigmine • Exelon
Trazodone • Oleptro
CASE Anxiety in the ICU
Mr. B, age 42, an African American man, is admitted to the inpatient medical unit for surgical treatment of peritoneal carcinomatosis with pelvic exenteration. He has a history of metastatic rectal cancer, chronic pain, and hypertension, but no psychiatric history. Mr. B’s postsurgical hospital stay is complicated by treatment-resistant tachycardia and hypertension, and he requires a lengthy stay in the ICU. In the ICU, Mr. B reports having visual hallucinations where he sees an individual placing a drug in his IV line. Additionally, he reports severe anxiety related to this experience. His anxiety and visual hallucinations are treated with coadministration of IV lorazepam, diphenhydramine, and haloperidol. These medications resolve the hallucinations, but his anxiety worsens and he becomes restless. He receives additional doses of IV haloperidol administered in 5 mg increments and reaching a cumulative 12-hour dose of 50 mg. Mr. B continues to report anxiety, so the psychiatry consultation-liaison (C-L) service is called.
[polldaddy:9970907]
The authors’ observations
Determining the cause of Mr. B’s anxiety is challenging because of his prolonged medical course, com
From a medical perspective, in a post-surgical patient treated in the ICU, the consulting practitioner must pay particular attention to delirium. ICU delirium is common—one report indicated that it occurs in 32.3% of ICU patients1—and frequently confused with psychiatric morbidity.2 Identifying delirium as the cause of impairment is important because delirium has potentially modifiable underlying etiologies. Symptomatically, delirium presents as impairment and fluctuation in attention, awareness, and at least one other cognitive domain, with a clear indication that the impairment occurred over a short period of time and represents a departure from baseline.3 In Mr. B’s case, these symptoms have not been excluded and should be considered by the C-L psychiatrists.
In addition to delirium, the C-L team must consider psychiatric comorbidity. Mr. B has no psychiatric history and a sudden first occurrence of hallucinations; therefore, it is unlikely that he has developed a primary psychotic disorder. Because he reported his symptoms had been present only for several days, he would not meet criteria for schizophrenia, which according to DSM-5 criteria require at least 1 month of ≥2 symptoms (including delusions, hallucinations, disorganized speech, disorganized behavior, or negative symptoms) and 6 months of declining function.3 However, although it is improbable, the C-L team must consider a primary psychotic illness, particularly given the potential devastating consequence of being misdiagnosed and mismanaged for an alternative illness. Unlike psychotic disorders, anxiety disorders are significantly more prevalent in the U.S. general population than primary psychotic disorders.4 Furthermore, the prevalence of anxiety disorders increases in the ICU setting; one study found that up to 61% of ICU patients setting experience “anxiety features.”5 Therefore, anxiety disorders and stress disorders should be considered in ICU patients who exhibit psychiatric symptoms.
Clinicians also should consider medication-induced adverse effects. In the ICU, patients are frequently managed on multiple medications, which increase their risk of developing adverse effects and adverse reactions.6 One potential consequence of polypharmacy is delirium, which remains a relevant potential diagnosis for Mr. B.7 Alternative consequences vary by medication and their respective pharmacodynamics. We take into consideration Mr. B’s exposure to high doses of the high-potency antipsychotic agent, haloperidol. Exposure to haloperidol can result in extrapyramidal symptoms, including akathisia,8,9,10 and the rare, but potentially fatal, NMS.11 These reactions can often be distinguished by taking a thorough history and a physical evaluation. In the case of akathisia, the clinician should look for medication exposure, titration, or taper. Most commonly, akathisia occurs secondary to antipsychotic exposure,12 followed by the onset of a combination of subjective symptoms, such as restlessness, anxiety, and irritability, and an objective symptom of increased motor activity.3 NMS, on the other hand, is distinguished by symptoms that include hyperthermia (>38ºC), diaphoresis, severe rigidity, urinary incontinence, vital instability, alterations in mental health status, and elevations in creatine kinase greater than 4-fold the upper limit, usually in the setting of treatment with antipsychotics.3 Nearly all cases of NMS occur within the first 30 days of antipsychotic exposure.3 While, overtly, NMS may appear to be less subtle than akathisia, clinicians should still be weary to rule out this admittedly rare, though potentially lethal diagnosis, especially in an ICU patient, where the diagnosis can be muddied by medical comorbidities that may mask the syndrome.
Continue to: EVALUATION Focus on akathisia
EVALUATION Focus on akathisia
On interview by the C-L team, Mr. B is visibly restless, moving all 4 extremities. He reports increased anxiety and irritability over the past 2 to 3 days. Mr. B states that he is aware of his increased motor movements and can briefly suppress them. However, after several seconds, he again begins spontaneously fidgeting, moving all 4 extremities and shifting from side to side in bed, saying, “I just feel anxious.” He denies having visual hallucinations, and says that the previous hallucinations had spontaneously presented and remitted after surgery. He denies the use of psychotropics for mental illness, prior similar symptoms to this presentation, a family history of mental illness, recent illicit substance use, or excessive alcohol use prior to presentation. This history is corroborated by collateral information from his brother, who was present in the ICU. On physical examination, Mr. B is afebrile and his vital signs are within normal limits. He does not have muscular rigidity or neck dystonia. His laboratory values, including complete blood count, electrolytes, liver function tests, and creatine phosphokinase, are within normal limits.
His medication administration record includes 46 standing agents, 16 “as-needed” agents, and 8 infusions. Several of the standing agents had psychotropic properties; however, the most salient were several opioids, ketamine, midazolam, lorazepam, dexamethasone, haloperidol, and olanzapine.
[polldaddy:9970908]
The authors’ observations
We determined that the most likely diagnosis for Mr. B’s symptoms was medication-induced akathisia secondary to haloperidol. Akathisia, coined by Haskovec in 1901,12,13 is from Greek, meaning an “inability to sit.”12 DSM-5 describes 2 forms of akathisia: medication-induced acute akathisia, and tardive akathisia.3 In the literature, others have described additional classifications, including chronic akathisia, withdrawal akathisia, and pseudoakathisia (Table 13,14-17). In Mr. B’s case, given his sudden development of symptoms and their direct chronologic relationship to antipsychotic treatment, and his combined subjective and objective symptoms, we believed that Mr. B’s symptoms were consistent with medication-induced acute akathisia (MIA). The identification and treatment of this clinical entity is important for several reasons, including reducing patient morbidity and maximizing patient comfort. Additionally, because akathisia has been associated with poor medication adherence, increased agitation/aggression, increased suicidality, and the eventual development of tardive dyskinesia,18 it is a relevant prognostic consideration when deciding to treat a patient with antipsychotics.
Pathophysiologically, we have yet to fully shed light on the exact underpinnings of akathisia. Much of our present knowledge stems from patient response to pharmacologic agents. While dopamine blockade has been linked to akathisia, the exact mechanism is not completely understood. Previous theories linking nigrostriatal pathways have been expanded to include mesocortical and mesolimbic considerations.12,17,18 Similarly surmised from medication effects, the transmitters y-aminobutyric acid, serotonin/5-hydroxytryptamine (5-HT), norepinephrine, and acetylcholine also have been linked to this syndrome, though as of yet, exact gross pathophysiologic mechanisms have not been fully elucidated.12 More recently, Stahl and Loonen19 described a novel mechanism by which they link the shell of the nucleus accumbens to akathisia. In their report, they indicate that the potential reduction in dopaminergic activity, secondary to antipsychotic administration, can result in compensatory noradrenergic activation of the locus coeruleus.19 The increased noradrenergic activity results in the downstream activation of the shell of the nucleus accumbens.19 The activation of the nucleus accumbens shell, which has been linked to unconditioned feeding and fear behavior, can then result in a cascade of effects that would phenotypically present as the syndrome we recognize to be akathisia.19
Numerous etiologies have been linked to MIA. Of these, high-potency antipsychotics are believed to remain the greatest risk factor for akathisia,18 although atypical antipsychotics, selective serotonin reuptake inhibitors, and serotonin-norepinephrine reuptake inhibitors, have been linked to the disorder.18,19
Continue to: Regarding antipsychotics...
Regarding antipsychotics, risk factors for akathisia include drug potency, dose, and rapidity of titration.20 All of these factors were relevant in our patient’s case. Risk across antipsychotic classes is not well understood; few head-to-head studies have comparing antipsychotics. However, general estimates suggest a 15% to 40% prevalence in patients exposed to typical antipsychotics, as compared with 0% to 12% exposed to atypical antipsychotics.8 The literature-reported difference in risk, as well as our patient’s comparative difference in exposure to large doses of haloperidol (50 mg) as compared with 1 dose of olanzapine (5 mg), led us to believe his akathisia developed primarily due to his exposure to haloperidol. Conclusively linking his symptoms to haloperidol alone, however, is not possible, and we did consider that olanzapine may in fact have had some effect in worsening Mr. B’s akathisia.
[polldaddy:9970909]
The authors’ observations
While there are reports on the efficacy of various agents in the treatment of akathisia, the most commonly evaluated agents are propranolol, anticholinergics, and benzodiazepines.17, 21
Propranolol is a nonselective beta-adrenergic blocker with numerous indications.17 Despite a 2004 Cochrane Review indicating that there is no evidence in support of central acting beta-blockers for treating akathisia,22 propranolol is not yet recognized as an appropriate treatment.17 The reason for this discrepancy is likely due to the Cochrane Review’s restrictive inclusion criteria, which prevented the analysis of much of the literature.22 In fact, several reports cite evidence for the treatment efficacy of propranolol17 and, to date, some reports continue to advocate for its use as a first-line agent in the treatment of akathisia. Admittedly, besides the Cochrane Review,22 other reports have found propranolol to be ineffective for treating akathisia,23 although these tend to be limited by their population size and generalizability.
As with propranolol, a 2006 Cochrane Review found “no reliable evidence to support or refute” using anticholinergic agents in the treatment of akathisia.24 We suspect that the review’s findings were likely secondary to its strict inclusion criteria.24 In fact, several reports support using anticholinergic agents for treating akathisia.25 Here we focus on benztropine and diphenhydramine.
Two reviews—Blaisdell26 (1994) and Poyurovsky27 (2010)—suggest modest benefits from benztropine, primarily in patients with comorbid Parkinson’s disease. Despite these benefits, head-to-head trials seem to either point to the superiority of propranolol or to no difference between these agents for treating akathisia.28,29 In a review, we only found 1 trial demonstrating benztropine’s superiority over propranolol,23 but this trial was constrained by its small population (6 patients). Therefore, the data suggest that, when indicated, clinicians should lean towards using propranolol for treating akathisia.
Continue to: Diphenhydramine, a first-generation antihistamine...
Diphenhydramine, a first-generation antihistamine with antimuscarinic properties, has been studied for its efficacy in treating metoclopramide-induced akathisia in the emergency setting.30 There are several reports on the efficacy of this agent, including a large randomized study involving 281 patients that found it effective for preventing metoclopramide-induced akathisia.30 Another head-to-head trial reported the benefit of the diphenhydramine vs midazolam.31 Both agents were efficacious for treating akathisia; however, midazolam had a more rapid onset. Despite these positive reports, double-blind trials have found diphenhydramine to be ineffective,17 which suggests propranolol should be the first-line agent, assuming it is not contraindicated.
Benzodiazepines have also been found to be efficacious for treating akathisia. A 1999 Cochrane Review included 2 randomized controlled trials that assessed the efficacy of clonazepam vs placebo for treating akathisia.32 It found evidence of benefit for clonazepam, but questioned the generalizability of these studies.32 This review did not include several other reports that suggest benefits of other benzodiazepines for treating akathisia. Other than clonazepam, reports suggest benefit for diazepam, lorazepam, and midazolam for treating akathisia.17 Despite this evidence and the findings from this Cochrane Review, the literature does not appear to point to clear dominance of these agents over propranolol. Given the safety concerns when prescribing benzodiazepines, it would be prudent to utilize propranolol as a first-line agent for treating akathisia.
Finally, other reports have cited treatment efficacy linked to serotonin 2A receptor (5-HT2A) antagonists (mianserin, mirtazapine, and trazodone), clonidine, gabapentin, amantadine, and other agents.17 If treatment with propranolol is ineffective or contraindicated, clinicians should utilize their clinical judgement in deciding on the use of one agent over another.
OUTCOME Complete resolution
Haloperidol is discontinued and diphenhydramine, 50 mg IV, is administered. (Diphenhydramine was used instead of propranolol due to immediacy of availability.) Most of Mr. B’s signs and symptoms resolve on a repeat interview 3 hours later. He receives another dose of diphenhydramine, 25 mg IV, for persistent mild irritability. By Day 2 of follow-up, his symptoms completely resolve as measured on the Barnes Akathisia Scale33 (Table 2).
Continue to: Bottom Line
Bottom Line
Akathisia is an elusive adverse effect of antipsychotics and can be misdiagnosed as anxiety. Close consideration should be given to potential medical, psychiatric, and drug-related etiologies in patients who have a prolonged medical course, comorbidities, and exposure to multiple pharmacologic agents.
Related Resources
- Factor SA, Leffler JB, Murray CF. Drug-induced movement disorders: a clinical review. Medscape. http://www.medscape.org/viewarticle/586881.
- Marder S, Stroup TS. Pharmacotherapy for schizophrenia: side effect management. UpToDate. https://www.uptodate.com/contents/pharmacotherapy-for-schizophrenia-side-effect-management.
Drug Brand Names
Amantadine • Symmetrel
Benztropine • Cogentin
Clonazepam • Klonopin
Clonidine • Catapres
Dexamethasone • Decadron
Diazepam • Valium
Diphenhydramine • Benadryl
Gabapentin • Neurontin
Haloperidol • Haldol
Ketamine • Ketalar
Lorazepam • Ativan
Metoclopramide • Reglan
Mianserin • Tolvon
Midazolam • Versed
Mirtazapine • Remeron
Olanzapine • Zyprexa
Propranolol • Inderal
Rivastigmine • Exelon
Trazodone • Oleptro
1. Cavallazzi R, Saad M, Marik PE. Delirium in the ICU: an overview. Ann Intensive Care. 2012;2:49.
2. Farrell KR, Ganzini L. Misdiagnosing delirium as depression in medically ill elderly patients. Arch Intern Med. 1995;155(22):2459-2464.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. National Alliance on Mental Illness. Mental health by the numbers. https://www.nami.org/learn-more/mental-health-by-the-numbers. Accessed March 4, 2018.
5. Jacka MJ, Mitchell N, Perez-Parada J. Incidence and prevalence of anxiety, depression, and post-traumatic stress disorder among critical care patients, families, and practitioners. J Anest & Inten Care Med. 2016;1(1):55555. doi: 10.19080/JAICM.2016.01.555555.
6. Reis AM, Cassiani SH. Adverse drug events in an intensive care unit of a university hospital. Eur J Clin Pharmacol. 2011;67(6):625-632.
7. Garpestad E, Devlin JW. Polypharmacy and delirium in critically ill older adults: recognition and prevention. Clin Geriatr Med. 2017;33(2):189-203.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Van Putten T, Marder SR. Toward a more reliable diagnosis of akathisia. Arch Gen Psychiatry. 1986;43(10):1015-1016.
10. Penders TM, Agarwal S, Rohaidy R. Persistent akathisia masquerading as agitated depression after use of ziprasidone in the treatment of bipolar depression. Neuropsychiatr Dis Treat. 2013;9:463-465.
11. Naganuma H, Fujii I. Incidence and risk factors in neuroleptic malignant syndrome. Acta Psychiatr Scand. 1994;90(6):424-426.
12. Forcen FE, Matsoukas K, Alici Y. Antipsychotic-induced akathisia in delirium: a systematic review. Palliat Support Care. 2016;14(1):77-84.
13. Brune M, Sachdev, PS. Ladislav Haskovec and 100 years of akathisia. American Journal of Psychiatry. 2002;159(5):727-727.
14. Havaki-Kontaxaki BJ, Kontaxakis VP, Christodoulou GN. Prevalence and characteristics of patients with pseudoakathisia. Eur Neuropsychopharmacol. 2000;10(5):333-336.
15. Lang AE. Withdrawal akathisia: case reports and a proposed classification of chronic akathisia. Mov Disord. 1994;9(2):188-192.
16. Sachdev P. The epidemiology of drug-induced akathisia: Part II. Chronic, tardive, and withdrawal akathisias. Schizophr Bull. 1995;21(3):451-461.
17. Kern DS, Lang AE. Acute akathisia. In: Friedman JH, ed. Medication-induced movement disorders. Cambridge, United Kingdom: Cambridge University Press; 2015:12-24.
18. Adler LA, Angrist B, Reiter S, et al. Neuroleptic-induced akathisia: a review. Psychopharmacology (Berl). 1989;97(1):1-11.
19. Stahl SM, Loonen AJM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
20. Sachdev P, Kruk J. Clinical characteristics and predisposing factors in acute drug-induced akathisia. Arch Gen Psychiatry. 1994;51(12):963-974.
21. Laoutidis ZG, Luckhaus C. 5-HT2A receptor antagonists for the treatment of neuroleptic-induced akathisia: a systematic review and meta-analysis. Int J Neuropsychopharmacol. 2014;17(5):823-832.
22. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
23. Sachdev P, Loneragan C. Intravenous benztropine and propranolol challenges in acute neuroleptic-induced akathisia. Clin Neuropharmacol. 1993;16(4):324-331.
24. Lima AR, Weiser KV, Bacaltchuk J, et al. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(1):CD003727.
25. Fleischhacker WW, Roth SD, Kane JM. The pharmacologic treatment of neuroleptic-induced akathisia. J Clin Psychopharmacol. 1990;10(1):12-21.
26. Blaisdell GD. Akathisia: a comprehensive review and treatment summary. Pharmacopsychiatry. 1994;27(4):139-146.
27. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
28. Adler LA, Reiter S, Corwin J, et al. Neuroleptic-induced akathisia: propranolol versus benztropine. Biol Psychiatry. 1988;23(2):211-213.
29. Adler LA, Peselow E, Rosenthal M, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Bender B, Friedman B, Davitt M, et al. 118: metoclopramide in the emergency department: a randomized factorial design study to determine the influence of dose and diphenhydramine on akathisia. Ann of Emerg Med. 2008;52(4):S78.
31. Parlak I, Erdur B, Parlak M, et al. Midazolam vs. diphenhydramine for the treatment of metoclopramide-induced akathisia: a randomized controlled trial. Acad Emerg Med. 2007;14(8):715-721.
32. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 1999;(4):CD001950.
33. Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154(5):672-676..
1. Cavallazzi R, Saad M, Marik PE. Delirium in the ICU: an overview. Ann Intensive Care. 2012;2:49.
2. Farrell KR, Ganzini L. Misdiagnosing delirium as depression in medically ill elderly patients. Arch Intern Med. 1995;155(22):2459-2464.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. National Alliance on Mental Illness. Mental health by the numbers. https://www.nami.org/learn-more/mental-health-by-the-numbers. Accessed March 4, 2018.
5. Jacka MJ, Mitchell N, Perez-Parada J. Incidence and prevalence of anxiety, depression, and post-traumatic stress disorder among critical care patients, families, and practitioners. J Anest & Inten Care Med. 2016;1(1):55555. doi: 10.19080/JAICM.2016.01.555555.
6. Reis AM, Cassiani SH. Adverse drug events in an intensive care unit of a university hospital. Eur J Clin Pharmacol. 2011;67(6):625-632.
7. Garpestad E, Devlin JW. Polypharmacy and delirium in critically ill older adults: recognition and prevention. Clin Geriatr Med. 2017;33(2):189-203.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Van Putten T, Marder SR. Toward a more reliable diagnosis of akathisia. Arch Gen Psychiatry. 1986;43(10):1015-1016.
10. Penders TM, Agarwal S, Rohaidy R. Persistent akathisia masquerading as agitated depression after use of ziprasidone in the treatment of bipolar depression. Neuropsychiatr Dis Treat. 2013;9:463-465.
11. Naganuma H, Fujii I. Incidence and risk factors in neuroleptic malignant syndrome. Acta Psychiatr Scand. 1994;90(6):424-426.
12. Forcen FE, Matsoukas K, Alici Y. Antipsychotic-induced akathisia in delirium: a systematic review. Palliat Support Care. 2016;14(1):77-84.
13. Brune M, Sachdev, PS. Ladislav Haskovec and 100 years of akathisia. American Journal of Psychiatry. 2002;159(5):727-727.
14. Havaki-Kontaxaki BJ, Kontaxakis VP, Christodoulou GN. Prevalence and characteristics of patients with pseudoakathisia. Eur Neuropsychopharmacol. 2000;10(5):333-336.
15. Lang AE. Withdrawal akathisia: case reports and a proposed classification of chronic akathisia. Mov Disord. 1994;9(2):188-192.
16. Sachdev P. The epidemiology of drug-induced akathisia: Part II. Chronic, tardive, and withdrawal akathisias. Schizophr Bull. 1995;21(3):451-461.
17. Kern DS, Lang AE. Acute akathisia. In: Friedman JH, ed. Medication-induced movement disorders. Cambridge, United Kingdom: Cambridge University Press; 2015:12-24.
18. Adler LA, Angrist B, Reiter S, et al. Neuroleptic-induced akathisia: a review. Psychopharmacology (Berl). 1989;97(1):1-11.
19. Stahl SM, Loonen AJM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
20. Sachdev P, Kruk J. Clinical characteristics and predisposing factors in acute drug-induced akathisia. Arch Gen Psychiatry. 1994;51(12):963-974.
21. Laoutidis ZG, Luckhaus C. 5-HT2A receptor antagonists for the treatment of neuroleptic-induced akathisia: a systematic review and meta-analysis. Int J Neuropsychopharmacol. 2014;17(5):823-832.
22. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
23. Sachdev P, Loneragan C. Intravenous benztropine and propranolol challenges in acute neuroleptic-induced akathisia. Clin Neuropharmacol. 1993;16(4):324-331.
24. Lima AR, Weiser KV, Bacaltchuk J, et al. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(1):CD003727.
25. Fleischhacker WW, Roth SD, Kane JM. The pharmacologic treatment of neuroleptic-induced akathisia. J Clin Psychopharmacol. 1990;10(1):12-21.
26. Blaisdell GD. Akathisia: a comprehensive review and treatment summary. Pharmacopsychiatry. 1994;27(4):139-146.
27. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
28. Adler LA, Reiter S, Corwin J, et al. Neuroleptic-induced akathisia: propranolol versus benztropine. Biol Psychiatry. 1988;23(2):211-213.
29. Adler LA, Peselow E, Rosenthal M, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Bender B, Friedman B, Davitt M, et al. 118: metoclopramide in the emergency department: a randomized factorial design study to determine the influence of dose and diphenhydramine on akathisia. Ann of Emerg Med. 2008;52(4):S78.
31. Parlak I, Erdur B, Parlak M, et al. Midazolam vs. diphenhydramine for the treatment of metoclopramide-induced akathisia: a randomized controlled trial. Acad Emerg Med. 2007;14(8):715-721.
32. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 1999;(4):CD001950.
33. Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154(5):672-676..
Depression and substance abuse

The crisis of poor physical health and early mortality of psychiatric patients
It is well established that general medical conditions can be associated with various psychiatric disorders. But the reverse is less recognized: That serious mental illness is associated with many physical maladies, often leading to early mortality. Thus, it is a bidirectional medical reality.
The multisystem adverse effects of psychotropic medications, such as metabolic dysregulation, often are blamed for the serious medical problems afflicting psychiatrically ill patients. However, evidence is mounting that while iatrogenic effects play a role, the larger effect appears to be due to a genetic link between psychiatric disorders and cardiovascular risk.1 Unhealthy lifestyles, including sedentary living, poor dietary habits, smoking, and alcohol/substance use, also play a role in the rapid deterioration of physical health and early mortality of individuals afflicted by mood disorders, psychotic disorders, and anxiety disorders. The mantra of “healthy body, healthy mind” is well known, but “unhealthy mind, unhealthy body” should be equally emphasized as a reason for high morbidity and premature mortality in patients with serious mental disorders.
Consider the following alarming findings:
- A recent study revealed that even before the onset of the first psychotic episode, young patients with schizophrenia already suffer from a wide variety of medical conditions.2 In a large sample of 954,351 Danish persons followed from birth to adulthood, of whom 4,371 developed schizophrenia, 95.6% of patients with schizophrenia had a history of hospitalization for somatic problems, including gastrointestinal, endocrine, genitourinary, metabolic, and circulatory system diseases; cancer; and epilepsy. Those findings suggest genetic, physiological, immunological, or developmental overlap between schizophrenia and medical conditions.
- A survey of 67,609 individuals with mood, anxiety, eating, impulse control, or substance use disorders followed for 10 years found that persons with those psychiatric disorders had a significantly higher risk of chronic medical conditions, including heart disease, stroke, hypertension, diabetes, asthma, arthritis, lung disease, peptic ulcer, and cancer.3
- A 7-year follow-up study of 1,138,853 individuals with schizophrenia in the United States found a 350% increase in mortality among this group of patients, who ranged in age from 20 to 64 years, compared with the general population, matched for age, sex, race, ethnicity, and geographic regions.4 An editorial accompanying this study urged psychiatrists to urgently address the “deadly consequences” of major psychiatric disorders.5
- A study of 18,380 individuals with schizophrenia, schizoaffective disorder, or bipolar disorder in London found that these patients were frequently hospitalized for general medical conditions, most commonly urinary, digestive, respiratory, endocrine/metabolic, hematologic, neurologic, dermatologic, and infectious disorders, neoplasm, and poisoning.6 The authors attributed those nonpsychiatric hospitalizations to self-neglect, self-harm, and poor health care access, as well as to “medically unexplained” causes.
- An extremely elevated mortality rate (24-fold higher than the general population) was reported in a 12-month study of young individuals (age 16 to 30 years) diagnosed with psychosis.7 The investigators also found that 61% of the cohort did not fill their antipsychotic prescriptions during that year, and 62% had ≥1 hospitalizations and/or emergency room visits during that year. The relationship between high mortality and lack of treatment with antipsychotics in schizophrenia was confirmed by another recent study,8 a 7-year follow-up of 29,823 persons with schizophrenia in Sweden that measured all-cause mortality. These researchers found the highest mortality among patients not receiving any antipsychotics, while the lowest mortality was among those receiving a long-acting injectable second-generation antipsychotic.
- A recent systematic review of 16 studies that examined glucose homeostasis in first-episode psychosis9 revealed that even at the onset of schizophrenia, glucose homeostasis was already altered, suggesting that predisposition to type 2 diabetes mellitus is a medical condition associated with schizophrenia, and not simply an iatrogenic effect of antipsychotic pharmacotherapy. This adds fodder to the possibility of a genetic overlap between schizophrenia and somatic disorders, including diabetes.10
- In a meta-analysis of 47 studies of young people at “ultra-high risk” for schizophrenia, cardiovascular risk was found to be high, mostly as a result of lifestyle factors such as low levels of physical activity and high rates of smoking and alcohol use, even before the onset of psychosis.11
- The risk of stroke was found to be higher in 80,569 patients with schizophrenia compared with 241,707 age- and sex-matched control subjects.12
- A meta-analysis of the risk of stroke in 6 cohorts with schizophrenia found that there is a higher risk for stroke in schizophrenia, and that this may be related to natural history of the illness itself, not just due to comorbid metabolic risk factors.13
- The high rate of cardiovascular disease in depression has been attributed to neuroinflammation14 or possibly to increased platelet reactivity.15
Continue to: As psychiatric physicians...
As psychiatric physicians, we always screen our patients for past and current medical conditions that are comorbid with their psychiatric disorders. We are aware of the lifestyle factors that increase these patients’ physical morbidity and mortality, above and beyond their suicide-related mortality. Our patients with schizophrenia and mood disorders have triple the smoking rates of the general population, and they tend to be sedentary with poor eating habits that lead to obesity, obstructive sleep apnea, diabetes, hypertension, and dyslipidemia, which increases their risk for heart attack, stroke, and cancer. Self-neglect during acute episodes of depression or psychosis increases the risk of infection, malnutrition, and tooth decay. We also see skin damage in obsessive-compulsive disorder patients who are compelled to wash their hands numerous times a day, the life-threatening effects of anorexia nervosa, and various types of medical ailments caused by incomplete suicidal attempts. Poverty and substance use among chronically mentally ill patients also increase the odds of physical ailments.
So we need to act diligently to reduce the alarming medical morbidity and mortality of the psychiatric population. Collaborative care with a primary care provider is a must, not an option, for every patient, because studies indicate that without collaborative care, patients receive inadequate primary care.16 Providing rapid access to standard medical care is the single most critical step for the prevention or amelioration of physical disorders in our psychiatric patients, concurrently with stabilizing their ailing brains and minds. If we focus only on treating psychopathology, then we will win the battle against mental illness, but lose the war of life and death.
1. Azad MC, Shoesmith WD, Al Mamun M, et al. Cardiovascular diseases among patients with schizophrenia. Asian J Psychiatr. 2016;19:28-36.
2. Sørensen HJ, Nielsen PR, Benros ME, et al. Somatic diseases and conditions before the first diagnosis of schizophrenia: a nationwide population-based cohort study in more than 900 000 individuals. Schizophr Bull. 2015;41(2):513-521.
3. Scott KM, Lim C, Al-Hamzawi A, et al. Association of mental disorders with subsequent chronic physical conditions: world mental health surveys from 17 countries. JAMA Psychiatry. 2016;73(2):150-158.
4. Olfson M, Gerhard T, Huang C, et al. Premature mortality among adults with schizophrenia in the United States. JAMA Psychiatry. 2015;72(12):1172-1181.
5. Suetani S, Whiteford HA, McGrath JJ. An urgent call to address the deadly consequences of serious mental disorders. JAMA Psychiatry. 2015;72(12):1166-1167.
6. Jayatilleke N, Hayes RD, Chang CK, et al. Acute general hospital admissions in people with serious mental illness [published online February 28, 2018]. Psychol Med. 2018;1-8.
7. Schoenbaum M, Sutherland JM, Chappel A, et al. Twelve-month health care use and mortality in commercially insured young people with incident psychosis in the United States. Schizophr Bull. 2017;43(6):1262-1272.
8. Taipale H, Mittendorfer-Rutz E, Alexanderson K, et al. Antipsychotics and mortality in a nationwide cohort of 29,823 patients with schizophrenia [published online December 20, 2017]. Schizophr Res. pii: S0920-9964(17)30762-4. doi: 10.1016/j.schres.2017.12.010.
9. Pillinger T, Beck K, Gobjila C, et al. Impaired glucose homeostasis in first-episode schizophrenia: a systematic review and meta-analysis. JAMA Psychiatry. 2017;74(3):261-269.
10. Dieset I, Andreassen OA, Haukvik UK. Somatic comorbidity in schizophrenia: some possible biological mechanisms across the life span. Schizophr Bull. 2016;42(6):1316-1319.
11. Carney R, Cotter J, Bradshaw T, et al. Cardiometabolic risk factors in young people at ultra-high risk for psychosis: a systematic review and meta-analysis. Schizophr Res. 2016;170(2-3):290-300.
12. Tsai KY, Lee CC, Chou YM, et al. The incidence and relative risk of stroke in patients with schizophrenia: a five-year follow-up study. Schizophr Res. 2012;138(1):41-47.
13. Li M, Fan YL, Tang ZY, et al. Schizophrenia and risk of stroke: a meta-analysis of cohort studies. Int J Cardiol. 2014;173(3):588-590.
14. Halaris A. Inflammation-associated co-morbidity between depression and cardiovascular disease. Curr Top Behav Neurosci. 2017;31:45-70.
15. Nemeroff CB, Musselman DL. Are platelets the link between depression and ischemic heart disease? Am Heart J. 2000;140(suppl 4):57-62.
16. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
It is well established that general medical conditions can be associated with various psychiatric disorders. But the reverse is less recognized: That serious mental illness is associated with many physical maladies, often leading to early mortality. Thus, it is a bidirectional medical reality.
The multisystem adverse effects of psychotropic medications, such as metabolic dysregulation, often are blamed for the serious medical problems afflicting psychiatrically ill patients. However, evidence is mounting that while iatrogenic effects play a role, the larger effect appears to be due to a genetic link between psychiatric disorders and cardiovascular risk.1 Unhealthy lifestyles, including sedentary living, poor dietary habits, smoking, and alcohol/substance use, also play a role in the rapid deterioration of physical health and early mortality of individuals afflicted by mood disorders, psychotic disorders, and anxiety disorders. The mantra of “healthy body, healthy mind” is well known, but “unhealthy mind, unhealthy body” should be equally emphasized as a reason for high morbidity and premature mortality in patients with serious mental disorders.
Consider the following alarming findings:
- A recent study revealed that even before the onset of the first psychotic episode, young patients with schizophrenia already suffer from a wide variety of medical conditions.2 In a large sample of 954,351 Danish persons followed from birth to adulthood, of whom 4,371 developed schizophrenia, 95.6% of patients with schizophrenia had a history of hospitalization for somatic problems, including gastrointestinal, endocrine, genitourinary, metabolic, and circulatory system diseases; cancer; and epilepsy. Those findings suggest genetic, physiological, immunological, or developmental overlap between schizophrenia and medical conditions.
- A survey of 67,609 individuals with mood, anxiety, eating, impulse control, or substance use disorders followed for 10 years found that persons with those psychiatric disorders had a significantly higher risk of chronic medical conditions, including heart disease, stroke, hypertension, diabetes, asthma, arthritis, lung disease, peptic ulcer, and cancer.3
- A 7-year follow-up study of 1,138,853 individuals with schizophrenia in the United States found a 350% increase in mortality among this group of patients, who ranged in age from 20 to 64 years, compared with the general population, matched for age, sex, race, ethnicity, and geographic regions.4 An editorial accompanying this study urged psychiatrists to urgently address the “deadly consequences” of major psychiatric disorders.5
- A study of 18,380 individuals with schizophrenia, schizoaffective disorder, or bipolar disorder in London found that these patients were frequently hospitalized for general medical conditions, most commonly urinary, digestive, respiratory, endocrine/metabolic, hematologic, neurologic, dermatologic, and infectious disorders, neoplasm, and poisoning.6 The authors attributed those nonpsychiatric hospitalizations to self-neglect, self-harm, and poor health care access, as well as to “medically unexplained” causes.
- An extremely elevated mortality rate (24-fold higher than the general population) was reported in a 12-month study of young individuals (age 16 to 30 years) diagnosed with psychosis.7 The investigators also found that 61% of the cohort did not fill their antipsychotic prescriptions during that year, and 62% had ≥1 hospitalizations and/or emergency room visits during that year. The relationship between high mortality and lack of treatment with antipsychotics in schizophrenia was confirmed by another recent study,8 a 7-year follow-up of 29,823 persons with schizophrenia in Sweden that measured all-cause mortality. These researchers found the highest mortality among patients not receiving any antipsychotics, while the lowest mortality was among those receiving a long-acting injectable second-generation antipsychotic.
- A recent systematic review of 16 studies that examined glucose homeostasis in first-episode psychosis9 revealed that even at the onset of schizophrenia, glucose homeostasis was already altered, suggesting that predisposition to type 2 diabetes mellitus is a medical condition associated with schizophrenia, and not simply an iatrogenic effect of antipsychotic pharmacotherapy. This adds fodder to the possibility of a genetic overlap between schizophrenia and somatic disorders, including diabetes.10
- In a meta-analysis of 47 studies of young people at “ultra-high risk” for schizophrenia, cardiovascular risk was found to be high, mostly as a result of lifestyle factors such as low levels of physical activity and high rates of smoking and alcohol use, even before the onset of psychosis.11
- The risk of stroke was found to be higher in 80,569 patients with schizophrenia compared with 241,707 age- and sex-matched control subjects.12
- A meta-analysis of the risk of stroke in 6 cohorts with schizophrenia found that there is a higher risk for stroke in schizophrenia, and that this may be related to natural history of the illness itself, not just due to comorbid metabolic risk factors.13
- The high rate of cardiovascular disease in depression has been attributed to neuroinflammation14 or possibly to increased platelet reactivity.15
Continue to: As psychiatric physicians...
As psychiatric physicians, we always screen our patients for past and current medical conditions that are comorbid with their psychiatric disorders. We are aware of the lifestyle factors that increase these patients’ physical morbidity and mortality, above and beyond their suicide-related mortality. Our patients with schizophrenia and mood disorders have triple the smoking rates of the general population, and they tend to be sedentary with poor eating habits that lead to obesity, obstructive sleep apnea, diabetes, hypertension, and dyslipidemia, which increases their risk for heart attack, stroke, and cancer. Self-neglect during acute episodes of depression or psychosis increases the risk of infection, malnutrition, and tooth decay. We also see skin damage in obsessive-compulsive disorder patients who are compelled to wash their hands numerous times a day, the life-threatening effects of anorexia nervosa, and various types of medical ailments caused by incomplete suicidal attempts. Poverty and substance use among chronically mentally ill patients also increase the odds of physical ailments.
So we need to act diligently to reduce the alarming medical morbidity and mortality of the psychiatric population. Collaborative care with a primary care provider is a must, not an option, for every patient, because studies indicate that without collaborative care, patients receive inadequate primary care.16 Providing rapid access to standard medical care is the single most critical step for the prevention or amelioration of physical disorders in our psychiatric patients, concurrently with stabilizing their ailing brains and minds. If we focus only on treating psychopathology, then we will win the battle against mental illness, but lose the war of life and death.
It is well established that general medical conditions can be associated with various psychiatric disorders. But the reverse is less recognized: That serious mental illness is associated with many physical maladies, often leading to early mortality. Thus, it is a bidirectional medical reality.
The multisystem adverse effects of psychotropic medications, such as metabolic dysregulation, often are blamed for the serious medical problems afflicting psychiatrically ill patients. However, evidence is mounting that while iatrogenic effects play a role, the larger effect appears to be due to a genetic link between psychiatric disorders and cardiovascular risk.1 Unhealthy lifestyles, including sedentary living, poor dietary habits, smoking, and alcohol/substance use, also play a role in the rapid deterioration of physical health and early mortality of individuals afflicted by mood disorders, psychotic disorders, and anxiety disorders. The mantra of “healthy body, healthy mind” is well known, but “unhealthy mind, unhealthy body” should be equally emphasized as a reason for high morbidity and premature mortality in patients with serious mental disorders.
Consider the following alarming findings:
- A recent study revealed that even before the onset of the first psychotic episode, young patients with schizophrenia already suffer from a wide variety of medical conditions.2 In a large sample of 954,351 Danish persons followed from birth to adulthood, of whom 4,371 developed schizophrenia, 95.6% of patients with schizophrenia had a history of hospitalization for somatic problems, including gastrointestinal, endocrine, genitourinary, metabolic, and circulatory system diseases; cancer; and epilepsy. Those findings suggest genetic, physiological, immunological, or developmental overlap between schizophrenia and medical conditions.
- A survey of 67,609 individuals with mood, anxiety, eating, impulse control, or substance use disorders followed for 10 years found that persons with those psychiatric disorders had a significantly higher risk of chronic medical conditions, including heart disease, stroke, hypertension, diabetes, asthma, arthritis, lung disease, peptic ulcer, and cancer.3
- A 7-year follow-up study of 1,138,853 individuals with schizophrenia in the United States found a 350% increase in mortality among this group of patients, who ranged in age from 20 to 64 years, compared with the general population, matched for age, sex, race, ethnicity, and geographic regions.4 An editorial accompanying this study urged psychiatrists to urgently address the “deadly consequences” of major psychiatric disorders.5
- A study of 18,380 individuals with schizophrenia, schizoaffective disorder, or bipolar disorder in London found that these patients were frequently hospitalized for general medical conditions, most commonly urinary, digestive, respiratory, endocrine/metabolic, hematologic, neurologic, dermatologic, and infectious disorders, neoplasm, and poisoning.6 The authors attributed those nonpsychiatric hospitalizations to self-neglect, self-harm, and poor health care access, as well as to “medically unexplained” causes.
- An extremely elevated mortality rate (24-fold higher than the general population) was reported in a 12-month study of young individuals (age 16 to 30 years) diagnosed with psychosis.7 The investigators also found that 61% of the cohort did not fill their antipsychotic prescriptions during that year, and 62% had ≥1 hospitalizations and/or emergency room visits during that year. The relationship between high mortality and lack of treatment with antipsychotics in schizophrenia was confirmed by another recent study,8 a 7-year follow-up of 29,823 persons with schizophrenia in Sweden that measured all-cause mortality. These researchers found the highest mortality among patients not receiving any antipsychotics, while the lowest mortality was among those receiving a long-acting injectable second-generation antipsychotic.
- A recent systematic review of 16 studies that examined glucose homeostasis in first-episode psychosis9 revealed that even at the onset of schizophrenia, glucose homeostasis was already altered, suggesting that predisposition to type 2 diabetes mellitus is a medical condition associated with schizophrenia, and not simply an iatrogenic effect of antipsychotic pharmacotherapy. This adds fodder to the possibility of a genetic overlap between schizophrenia and somatic disorders, including diabetes.10
- In a meta-analysis of 47 studies of young people at “ultra-high risk” for schizophrenia, cardiovascular risk was found to be high, mostly as a result of lifestyle factors such as low levels of physical activity and high rates of smoking and alcohol use, even before the onset of psychosis.11
- The risk of stroke was found to be higher in 80,569 patients with schizophrenia compared with 241,707 age- and sex-matched control subjects.12
- A meta-analysis of the risk of stroke in 6 cohorts with schizophrenia found that there is a higher risk for stroke in schizophrenia, and that this may be related to natural history of the illness itself, not just due to comorbid metabolic risk factors.13
- The high rate of cardiovascular disease in depression has been attributed to neuroinflammation14 or possibly to increased platelet reactivity.15
Continue to: As psychiatric physicians...
As psychiatric physicians, we always screen our patients for past and current medical conditions that are comorbid with their psychiatric disorders. We are aware of the lifestyle factors that increase these patients’ physical morbidity and mortality, above and beyond their suicide-related mortality. Our patients with schizophrenia and mood disorders have triple the smoking rates of the general population, and they tend to be sedentary with poor eating habits that lead to obesity, obstructive sleep apnea, diabetes, hypertension, and dyslipidemia, which increases their risk for heart attack, stroke, and cancer. Self-neglect during acute episodes of depression or psychosis increases the risk of infection, malnutrition, and tooth decay. We also see skin damage in obsessive-compulsive disorder patients who are compelled to wash their hands numerous times a day, the life-threatening effects of anorexia nervosa, and various types of medical ailments caused by incomplete suicidal attempts. Poverty and substance use among chronically mentally ill patients also increase the odds of physical ailments.
So we need to act diligently to reduce the alarming medical morbidity and mortality of the psychiatric population. Collaborative care with a primary care provider is a must, not an option, for every patient, because studies indicate that without collaborative care, patients receive inadequate primary care.16 Providing rapid access to standard medical care is the single most critical step for the prevention or amelioration of physical disorders in our psychiatric patients, concurrently with stabilizing their ailing brains and minds. If we focus only on treating psychopathology, then we will win the battle against mental illness, but lose the war of life and death.
1. Azad MC, Shoesmith WD, Al Mamun M, et al. Cardiovascular diseases among patients with schizophrenia. Asian J Psychiatr. 2016;19:28-36.
2. Sørensen HJ, Nielsen PR, Benros ME, et al. Somatic diseases and conditions before the first diagnosis of schizophrenia: a nationwide population-based cohort study in more than 900 000 individuals. Schizophr Bull. 2015;41(2):513-521.
3. Scott KM, Lim C, Al-Hamzawi A, et al. Association of mental disorders with subsequent chronic physical conditions: world mental health surveys from 17 countries. JAMA Psychiatry. 2016;73(2):150-158.
4. Olfson M, Gerhard T, Huang C, et al. Premature mortality among adults with schizophrenia in the United States. JAMA Psychiatry. 2015;72(12):1172-1181.
5. Suetani S, Whiteford HA, McGrath JJ. An urgent call to address the deadly consequences of serious mental disorders. JAMA Psychiatry. 2015;72(12):1166-1167.
6. Jayatilleke N, Hayes RD, Chang CK, et al. Acute general hospital admissions in people with serious mental illness [published online February 28, 2018]. Psychol Med. 2018;1-8.
7. Schoenbaum M, Sutherland JM, Chappel A, et al. Twelve-month health care use and mortality in commercially insured young people with incident psychosis in the United States. Schizophr Bull. 2017;43(6):1262-1272.
8. Taipale H, Mittendorfer-Rutz E, Alexanderson K, et al. Antipsychotics and mortality in a nationwide cohort of 29,823 patients with schizophrenia [published online December 20, 2017]. Schizophr Res. pii: S0920-9964(17)30762-4. doi: 10.1016/j.schres.2017.12.010.
9. Pillinger T, Beck K, Gobjila C, et al. Impaired glucose homeostasis in first-episode schizophrenia: a systematic review and meta-analysis. JAMA Psychiatry. 2017;74(3):261-269.
10. Dieset I, Andreassen OA, Haukvik UK. Somatic comorbidity in schizophrenia: some possible biological mechanisms across the life span. Schizophr Bull. 2016;42(6):1316-1319.
11. Carney R, Cotter J, Bradshaw T, et al. Cardiometabolic risk factors in young people at ultra-high risk for psychosis: a systematic review and meta-analysis. Schizophr Res. 2016;170(2-3):290-300.
12. Tsai KY, Lee CC, Chou YM, et al. The incidence and relative risk of stroke in patients with schizophrenia: a five-year follow-up study. Schizophr Res. 2012;138(1):41-47.
13. Li M, Fan YL, Tang ZY, et al. Schizophrenia and risk of stroke: a meta-analysis of cohort studies. Int J Cardiol. 2014;173(3):588-590.
14. Halaris A. Inflammation-associated co-morbidity between depression and cardiovascular disease. Curr Top Behav Neurosci. 2017;31:45-70.
15. Nemeroff CB, Musselman DL. Are platelets the link between depression and ischemic heart disease? Am Heart J. 2000;140(suppl 4):57-62.
16. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
1. Azad MC, Shoesmith WD, Al Mamun M, et al. Cardiovascular diseases among patients with schizophrenia. Asian J Psychiatr. 2016;19:28-36.
2. Sørensen HJ, Nielsen PR, Benros ME, et al. Somatic diseases and conditions before the first diagnosis of schizophrenia: a nationwide population-based cohort study in more than 900 000 individuals. Schizophr Bull. 2015;41(2):513-521.
3. Scott KM, Lim C, Al-Hamzawi A, et al. Association of mental disorders with subsequent chronic physical conditions: world mental health surveys from 17 countries. JAMA Psychiatry. 2016;73(2):150-158.
4. Olfson M, Gerhard T, Huang C, et al. Premature mortality among adults with schizophrenia in the United States. JAMA Psychiatry. 2015;72(12):1172-1181.
5. Suetani S, Whiteford HA, McGrath JJ. An urgent call to address the deadly consequences of serious mental disorders. JAMA Psychiatry. 2015;72(12):1166-1167.
6. Jayatilleke N, Hayes RD, Chang CK, et al. Acute general hospital admissions in people with serious mental illness [published online February 28, 2018]. Psychol Med. 2018;1-8.
7. Schoenbaum M, Sutherland JM, Chappel A, et al. Twelve-month health care use and mortality in commercially insured young people with incident psychosis in the United States. Schizophr Bull. 2017;43(6):1262-1272.
8. Taipale H, Mittendorfer-Rutz E, Alexanderson K, et al. Antipsychotics and mortality in a nationwide cohort of 29,823 patients with schizophrenia [published online December 20, 2017]. Schizophr Res. pii: S0920-9964(17)30762-4. doi: 10.1016/j.schres.2017.12.010.
9. Pillinger T, Beck K, Gobjila C, et al. Impaired glucose homeostasis in first-episode schizophrenia: a systematic review and meta-analysis. JAMA Psychiatry. 2017;74(3):261-269.
10. Dieset I, Andreassen OA, Haukvik UK. Somatic comorbidity in schizophrenia: some possible biological mechanisms across the life span. Schizophr Bull. 2016;42(6):1316-1319.
11. Carney R, Cotter J, Bradshaw T, et al. Cardiometabolic risk factors in young people at ultra-high risk for psychosis: a systematic review and meta-analysis. Schizophr Res. 2016;170(2-3):290-300.
12. Tsai KY, Lee CC, Chou YM, et al. The incidence and relative risk of stroke in patients with schizophrenia: a five-year follow-up study. Schizophr Res. 2012;138(1):41-47.
13. Li M, Fan YL, Tang ZY, et al. Schizophrenia and risk of stroke: a meta-analysis of cohort studies. Int J Cardiol. 2014;173(3):588-590.
14. Halaris A. Inflammation-associated co-morbidity between depression and cardiovascular disease. Curr Top Behav Neurosci. 2017;31:45-70.
15. Nemeroff CB, Musselman DL. Are platelets the link between depression and ischemic heart disease? Am Heart J. 2000;140(suppl 4):57-62.
16. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.

How precision psychiatry helped my patient; Ketamine: The next ‘opioid crisis’?
How precision psychiatry helped my patient
I applaud Dr. Nasrallah’s editorial “The dawn of precision psychiatry” (From the Editor,
Ms. G, age 14, presented with periodic emotional “meltdowns,” which would occur in any setting, and I determined that they were precipitated by a high glycemic intake. By carefully controlling her glycemic intake and starting her on caprylic acid (a medium-chain triglyceride, which was used to maintain a ketotic state), 1 tablespoon 3 times daily, we were able to reduce the frequency of her episodes by 80% to 90%. Using data from commercially available DNA testing, I determined that she had single nucleotide polymorphisms (SNPs) in an alpha-ketoglutarate dehydrogenase gene, which is primarily located in the prefrontal cortex (PFC), and whose function is supported by thiamine and impaired by high glycemic intake.1 After adding oral thiamine hydrochloride, 100 mg twice a day, and correcting other abnormalities (eg, she was hypothyroid), her episodes are now rare. She is functioning well, has been getting high grades, and recently wrote a 40-page short story.
Once she improved, she was able to describe having a partial seizure, with a rising sensation, which often improves with ketosis. Clearly, disruption of her PFC energetics due to the SNPs described above contributed to the disinhibition of the temporal lobe structures. Furthermore, she has an APOE3/4 status, which puts her at risk for Alzheimer’s disease. Her mother was educated about the importance of good health habits, which is personalized and preventative medicine.
Robert Hedaya, MD, DLFAPA
Clinical Professor
MedStar Georgetown University Hospital
Washington, DC
Faculty
Institute for Functional Medicine
Gig Harbor, Washington, DC
Founder
National Center for Whole Psychiatry
Rockville, Maryland
Reference
1. Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci. 2005;360(1464):2335-2345.
Dr. Nasrallah responds
My thanks to Dr. Hedaya for his letter and for providing an excellent example of precision psychiatry. His brief case vignette brings it to life! I commend him for practicing on the cutting-edge of psychiatry’s scientific frontier.
Continue to: Ketamine: The next 'opioid crisis'?
Ketamine: The next ‘opioid crisis’?
The chief of the FDA, Scott Gottlieb, MD, recently discussed the
There are many similarities between the use of opioids to treat pain and the potential use of ketamine to treat suicidality. Physical and mental pain are subjective, qualitative, and difficult to quantify, which makes it difficult to develop accurate measurements of symptom severity. Chronic physical pain and suicidality are not illnesses, but symptoms of myriad types of pathologies with differing etiologies and treatment options.5 Due to the ambiguous and subjective experience of physical and mental pain, we tend to lump them together as 1 pathological category without understanding pathophysiologic differences. The most commonly reported types of pain include low back pain, migraine/headache, neck pain, and facial pain.6 However, each of these pain types would likely have a different pathophysiology and treatment. Likewise, suicide can be associated with various psychiatric conditions,7 and suicidality resulting from these conditions may require a different etiology and treatment.
We already know that both opioids and ketamine are addictive. For example, there is a report of a nurse stealing a hospital’s supply of ketamine and self-treating for depression, which led to an inpatient detox admission after she developed toxicity and addiction.8 Some ketamine research supports its safe use, but it may be biased due to conflicts of interest. For example, several authors of a recent study proclaiming the effectiveness of a single dose of ketamine in treating suicidal ideation
Warnings stating how both opioid and ketamine should be used were published years ago but have since been ignored. For example, a 1977 article advocated that opioids should only be used for a “short duration and limited to patients with acute diseases or inoperable or metastatic cancer who require long-term relief.”10 The rationale for this distinction was foretelling of the current opioid epidemic: “Continued and prolonged use of narcotics in patients with chronic benign pain is not recommended because of serious behavioral consequences, the development of tolerance, and addiction liability. Long-term use of analgesic drugs in chronic pain usually produces negative behavioral complications that are more difficult to manage than the pain it was desired to eliminate.”10 We knew better then.
The earliest report of ketamine dependency I could find was published in 1987, which predates its classification as a controlled substance.11 More recently, ketamine dependency has been associated with adverse effects that are similar to “not only cocaine and amphetamine but also with opiates, alcohol and cannabis, as well as the psychological attractions of its distinctive psychedelic properties.”12 We should consider ourselves warned.
Michael Shapiro, MD
Assistant Professor
Department of Psychiatry
University of Florida
Gainesville, Florida
References
1. Jayne O’Donnell. FDA chief supports opioid prescription limits, regrets agency’s prior inaction. USA TODAY. https://www.usatoday.com/story/news/politics/2017/10/23/fda-chief-supports-opioid-prescription-limits-regrets-agencys-prior-inaction/774007001. Published October 23, 2017. Accessed January 25, 2018.
2. Bill Whitaker. Ex-DEA agent: opioid crisis fueled by drug industry and Congress. CBS News. https://www.cbsnews.com/news/ex-dea-agent-opioid-crisis-fueled-by-drug-industry-and-congress. Published October 15, 2017. Accessed January 25, 2018.
3. Drug Enforcement Administration. Diversion of Control Division. Ketamine. https://www.deadiversion.usdoj.gov/drug_chem_info/ketamine.pdf. Published August 2013. Accessed January 25, 2018.
4. Bell RF. Ketamine for chronic noncancer pain: concerns regarding toxicity. Curr Opin Support Palliat Care. 2012;6(2):183-187.
5. Barzilay S, Apter A. Psychological models of suicide. Arch Suicide Res. 2014;18(4):295-312.
6. American Academy of Pain Medicine. AAPM facts and figures on pain. http://www.painmed.org/patientcenter/facts_on_pain.aspx.
7. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
8. Bonnet U. Long-term ketamine self-injections in major depressive disorder: focus on tolerance in ketamine’s antidepressant response and the development of ketamine addiction. J Psychoactive Drugs. 2015;47(4):276-85.
9. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry 2017. https://doi.org/10.1176/appi.ajp.2017.17040472
10. Halpern LM. Analgesic drugs in the management of pain. Arch Surg. 1977;112(7):861-869.
11. Kamaya H, Krishna PR. Anesthesiology. 1987;67(5):861-862.
12. Jansen KL, Darracot-Cankovic R. The nonmedical use of ketamine, part two: a review of problem use and dependence. J Psychoactive Drugs. 2001;33(2):151-158.
How precision psychiatry helped my patient
I applaud Dr. Nasrallah’s editorial “The dawn of precision psychiatry” (From the Editor,
Ms. G, age 14, presented with periodic emotional “meltdowns,” which would occur in any setting, and I determined that they were precipitated by a high glycemic intake. By carefully controlling her glycemic intake and starting her on caprylic acid (a medium-chain triglyceride, which was used to maintain a ketotic state), 1 tablespoon 3 times daily, we were able to reduce the frequency of her episodes by 80% to 90%. Using data from commercially available DNA testing, I determined that she had single nucleotide polymorphisms (SNPs) in an alpha-ketoglutarate dehydrogenase gene, which is primarily located in the prefrontal cortex (PFC), and whose function is supported by thiamine and impaired by high glycemic intake.1 After adding oral thiamine hydrochloride, 100 mg twice a day, and correcting other abnormalities (eg, she was hypothyroid), her episodes are now rare. She is functioning well, has been getting high grades, and recently wrote a 40-page short story.
Once she improved, she was able to describe having a partial seizure, with a rising sensation, which often improves with ketosis. Clearly, disruption of her PFC energetics due to the SNPs described above contributed to the disinhibition of the temporal lobe structures. Furthermore, she has an APOE3/4 status, which puts her at risk for Alzheimer’s disease. Her mother was educated about the importance of good health habits, which is personalized and preventative medicine.
Robert Hedaya, MD, DLFAPA
Clinical Professor
MedStar Georgetown University Hospital
Washington, DC
Faculty
Institute for Functional Medicine
Gig Harbor, Washington, DC
Founder
National Center for Whole Psychiatry
Rockville, Maryland
Reference
1. Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci. 2005;360(1464):2335-2345.
Dr. Nasrallah responds
My thanks to Dr. Hedaya for his letter and for providing an excellent example of precision psychiatry. His brief case vignette brings it to life! I commend him for practicing on the cutting-edge of psychiatry’s scientific frontier.
Continue to: Ketamine: The next 'opioid crisis'?
Ketamine: The next ‘opioid crisis’?
The chief of the FDA, Scott Gottlieb, MD, recently discussed the
There are many similarities between the use of opioids to treat pain and the potential use of ketamine to treat suicidality. Physical and mental pain are subjective, qualitative, and difficult to quantify, which makes it difficult to develop accurate measurements of symptom severity. Chronic physical pain and suicidality are not illnesses, but symptoms of myriad types of pathologies with differing etiologies and treatment options.5 Due to the ambiguous and subjective experience of physical and mental pain, we tend to lump them together as 1 pathological category without understanding pathophysiologic differences. The most commonly reported types of pain include low back pain, migraine/headache, neck pain, and facial pain.6 However, each of these pain types would likely have a different pathophysiology and treatment. Likewise, suicide can be associated with various psychiatric conditions,7 and suicidality resulting from these conditions may require a different etiology and treatment.
We already know that both opioids and ketamine are addictive. For example, there is a report of a nurse stealing a hospital’s supply of ketamine and self-treating for depression, which led to an inpatient detox admission after she developed toxicity and addiction.8 Some ketamine research supports its safe use, but it may be biased due to conflicts of interest. For example, several authors of a recent study proclaiming the effectiveness of a single dose of ketamine in treating suicidal ideation
Warnings stating how both opioid and ketamine should be used were published years ago but have since been ignored. For example, a 1977 article advocated that opioids should only be used for a “short duration and limited to patients with acute diseases or inoperable or metastatic cancer who require long-term relief.”10 The rationale for this distinction was foretelling of the current opioid epidemic: “Continued and prolonged use of narcotics in patients with chronic benign pain is not recommended because of serious behavioral consequences, the development of tolerance, and addiction liability. Long-term use of analgesic drugs in chronic pain usually produces negative behavioral complications that are more difficult to manage than the pain it was desired to eliminate.”10 We knew better then.
The earliest report of ketamine dependency I could find was published in 1987, which predates its classification as a controlled substance.11 More recently, ketamine dependency has been associated with adverse effects that are similar to “not only cocaine and amphetamine but also with opiates, alcohol and cannabis, as well as the psychological attractions of its distinctive psychedelic properties.”12 We should consider ourselves warned.
Michael Shapiro, MD
Assistant Professor
Department of Psychiatry
University of Florida
Gainesville, Florida
References
1. Jayne O’Donnell. FDA chief supports opioid prescription limits, regrets agency’s prior inaction. USA TODAY. https://www.usatoday.com/story/news/politics/2017/10/23/fda-chief-supports-opioid-prescription-limits-regrets-agencys-prior-inaction/774007001. Published October 23, 2017. Accessed January 25, 2018.
2. Bill Whitaker. Ex-DEA agent: opioid crisis fueled by drug industry and Congress. CBS News. https://www.cbsnews.com/news/ex-dea-agent-opioid-crisis-fueled-by-drug-industry-and-congress. Published October 15, 2017. Accessed January 25, 2018.
3. Drug Enforcement Administration. Diversion of Control Division. Ketamine. https://www.deadiversion.usdoj.gov/drug_chem_info/ketamine.pdf. Published August 2013. Accessed January 25, 2018.
4. Bell RF. Ketamine for chronic noncancer pain: concerns regarding toxicity. Curr Opin Support Palliat Care. 2012;6(2):183-187.
5. Barzilay S, Apter A. Psychological models of suicide. Arch Suicide Res. 2014;18(4):295-312.
6. American Academy of Pain Medicine. AAPM facts and figures on pain. http://www.painmed.org/patientcenter/facts_on_pain.aspx.
7. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
8. Bonnet U. Long-term ketamine self-injections in major depressive disorder: focus on tolerance in ketamine’s antidepressant response and the development of ketamine addiction. J Psychoactive Drugs. 2015;47(4):276-85.
9. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry 2017. https://doi.org/10.1176/appi.ajp.2017.17040472
10. Halpern LM. Analgesic drugs in the management of pain. Arch Surg. 1977;112(7):861-869.
11. Kamaya H, Krishna PR. Anesthesiology. 1987;67(5):861-862.
12. Jansen KL, Darracot-Cankovic R. The nonmedical use of ketamine, part two: a review of problem use and dependence. J Psychoactive Drugs. 2001;33(2):151-158.
How precision psychiatry helped my patient
I applaud Dr. Nasrallah’s editorial “The dawn of precision psychiatry” (From the Editor,
Ms. G, age 14, presented with periodic emotional “meltdowns,” which would occur in any setting, and I determined that they were precipitated by a high glycemic intake. By carefully controlling her glycemic intake and starting her on caprylic acid (a medium-chain triglyceride, which was used to maintain a ketotic state), 1 tablespoon 3 times daily, we were able to reduce the frequency of her episodes by 80% to 90%. Using data from commercially available DNA testing, I determined that she had single nucleotide polymorphisms (SNPs) in an alpha-ketoglutarate dehydrogenase gene, which is primarily located in the prefrontal cortex (PFC), and whose function is supported by thiamine and impaired by high glycemic intake.1 After adding oral thiamine hydrochloride, 100 mg twice a day, and correcting other abnormalities (eg, she was hypothyroid), her episodes are now rare. She is functioning well, has been getting high grades, and recently wrote a 40-page short story.
Once she improved, she was able to describe having a partial seizure, with a rising sensation, which often improves with ketosis. Clearly, disruption of her PFC energetics due to the SNPs described above contributed to the disinhibition of the temporal lobe structures. Furthermore, she has an APOE3/4 status, which puts her at risk for Alzheimer’s disease. Her mother was educated about the importance of good health habits, which is personalized and preventative medicine.
Robert Hedaya, MD, DLFAPA
Clinical Professor
MedStar Georgetown University Hospital
Washington, DC
Faculty
Institute for Functional Medicine
Gig Harbor, Washington, DC
Founder
National Center for Whole Psychiatry
Rockville, Maryland
Reference
1. Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci. 2005;360(1464):2335-2345.
Dr. Nasrallah responds
My thanks to Dr. Hedaya for his letter and for providing an excellent example of precision psychiatry. His brief case vignette brings it to life! I commend him for practicing on the cutting-edge of psychiatry’s scientific frontier.
Continue to: Ketamine: The next 'opioid crisis'?
Ketamine: The next ‘opioid crisis’?
The chief of the FDA, Scott Gottlieb, MD, recently discussed the
There are many similarities between the use of opioids to treat pain and the potential use of ketamine to treat suicidality. Physical and mental pain are subjective, qualitative, and difficult to quantify, which makes it difficult to develop accurate measurements of symptom severity. Chronic physical pain and suicidality are not illnesses, but symptoms of myriad types of pathologies with differing etiologies and treatment options.5 Due to the ambiguous and subjective experience of physical and mental pain, we tend to lump them together as 1 pathological category without understanding pathophysiologic differences. The most commonly reported types of pain include low back pain, migraine/headache, neck pain, and facial pain.6 However, each of these pain types would likely have a different pathophysiology and treatment. Likewise, suicide can be associated with various psychiatric conditions,7 and suicidality resulting from these conditions may require a different etiology and treatment.
We already know that both opioids and ketamine are addictive. For example, there is a report of a nurse stealing a hospital’s supply of ketamine and self-treating for depression, which led to an inpatient detox admission after she developed toxicity and addiction.8 Some ketamine research supports its safe use, but it may be biased due to conflicts of interest. For example, several authors of a recent study proclaiming the effectiveness of a single dose of ketamine in treating suicidal ideation
Warnings stating how both opioid and ketamine should be used were published years ago but have since been ignored. For example, a 1977 article advocated that opioids should only be used for a “short duration and limited to patients with acute diseases or inoperable or metastatic cancer who require long-term relief.”10 The rationale for this distinction was foretelling of the current opioid epidemic: “Continued and prolonged use of narcotics in patients with chronic benign pain is not recommended because of serious behavioral consequences, the development of tolerance, and addiction liability. Long-term use of analgesic drugs in chronic pain usually produces negative behavioral complications that are more difficult to manage than the pain it was desired to eliminate.”10 We knew better then.
The earliest report of ketamine dependency I could find was published in 1987, which predates its classification as a controlled substance.11 More recently, ketamine dependency has been associated with adverse effects that are similar to “not only cocaine and amphetamine but also with opiates, alcohol and cannabis, as well as the psychological attractions of its distinctive psychedelic properties.”12 We should consider ourselves warned.
Michael Shapiro, MD
Assistant Professor
Department of Psychiatry
University of Florida
Gainesville, Florida
References
1. Jayne O’Donnell. FDA chief supports opioid prescription limits, regrets agency’s prior inaction. USA TODAY. https://www.usatoday.com/story/news/politics/2017/10/23/fda-chief-supports-opioid-prescription-limits-regrets-agencys-prior-inaction/774007001. Published October 23, 2017. Accessed January 25, 2018.
2. Bill Whitaker. Ex-DEA agent: opioid crisis fueled by drug industry and Congress. CBS News. https://www.cbsnews.com/news/ex-dea-agent-opioid-crisis-fueled-by-drug-industry-and-congress. Published October 15, 2017. Accessed January 25, 2018.
3. Drug Enforcement Administration. Diversion of Control Division. Ketamine. https://www.deadiversion.usdoj.gov/drug_chem_info/ketamine.pdf. Published August 2013. Accessed January 25, 2018.
4. Bell RF. Ketamine for chronic noncancer pain: concerns regarding toxicity. Curr Opin Support Palliat Care. 2012;6(2):183-187.
5. Barzilay S, Apter A. Psychological models of suicide. Arch Suicide Res. 2014;18(4):295-312.
6. American Academy of Pain Medicine. AAPM facts and figures on pain. http://www.painmed.org/patientcenter/facts_on_pain.aspx.
7. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
8. Bonnet U. Long-term ketamine self-injections in major depressive disorder: focus on tolerance in ketamine’s antidepressant response and the development of ketamine addiction. J Psychoactive Drugs. 2015;47(4):276-85.
9. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry 2017. https://doi.org/10.1176/appi.ajp.2017.17040472
10. Halpern LM. Analgesic drugs in the management of pain. Arch Surg. 1977;112(7):861-869.
11. Kamaya H, Krishna PR. Anesthesiology. 1987;67(5):861-862.
12. Jansen KL, Darracot-Cankovic R. The nonmedical use of ketamine, part two: a review of problem use and dependence. J Psychoactive Drugs. 2001;33(2):151-158.
