User login
Multiple Firm Papules on the Wrists and Forearms
THE DIAGNOSIS: Acral Persistent Papular Mucinosis
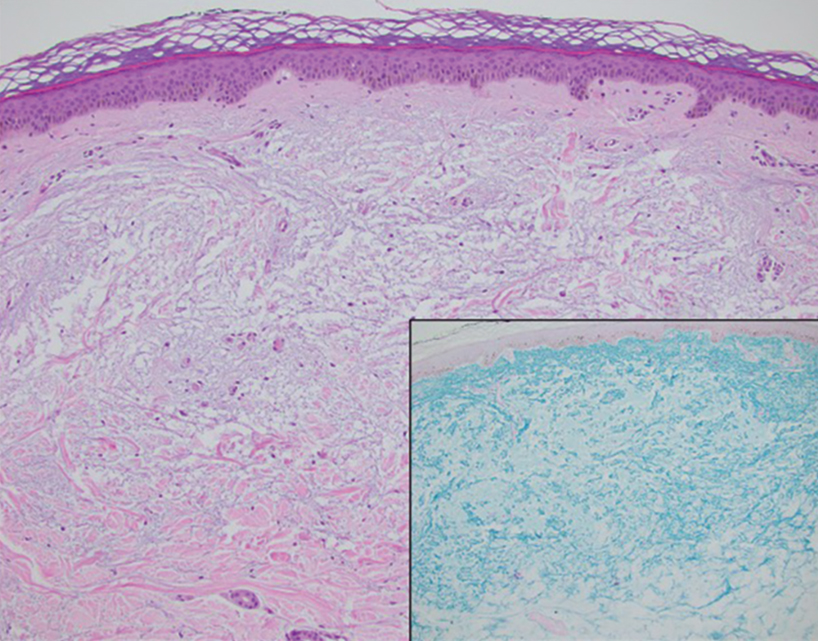
Histopathologic analysis revealed conspicuous interstitial mucin deposition throughout the upper to mid reticular dermis in the absence of a cellular infiltrate or fibroplasia. Colloidal iron staining confirmed the presence of mucin. In correlation with the clinical presentation, a diagnosis of acral persistent papular mucinosis (APPM) was made. The patient was counseled on the benign disease course and lack of associated comorbidities, and additional treatment was not pursued.
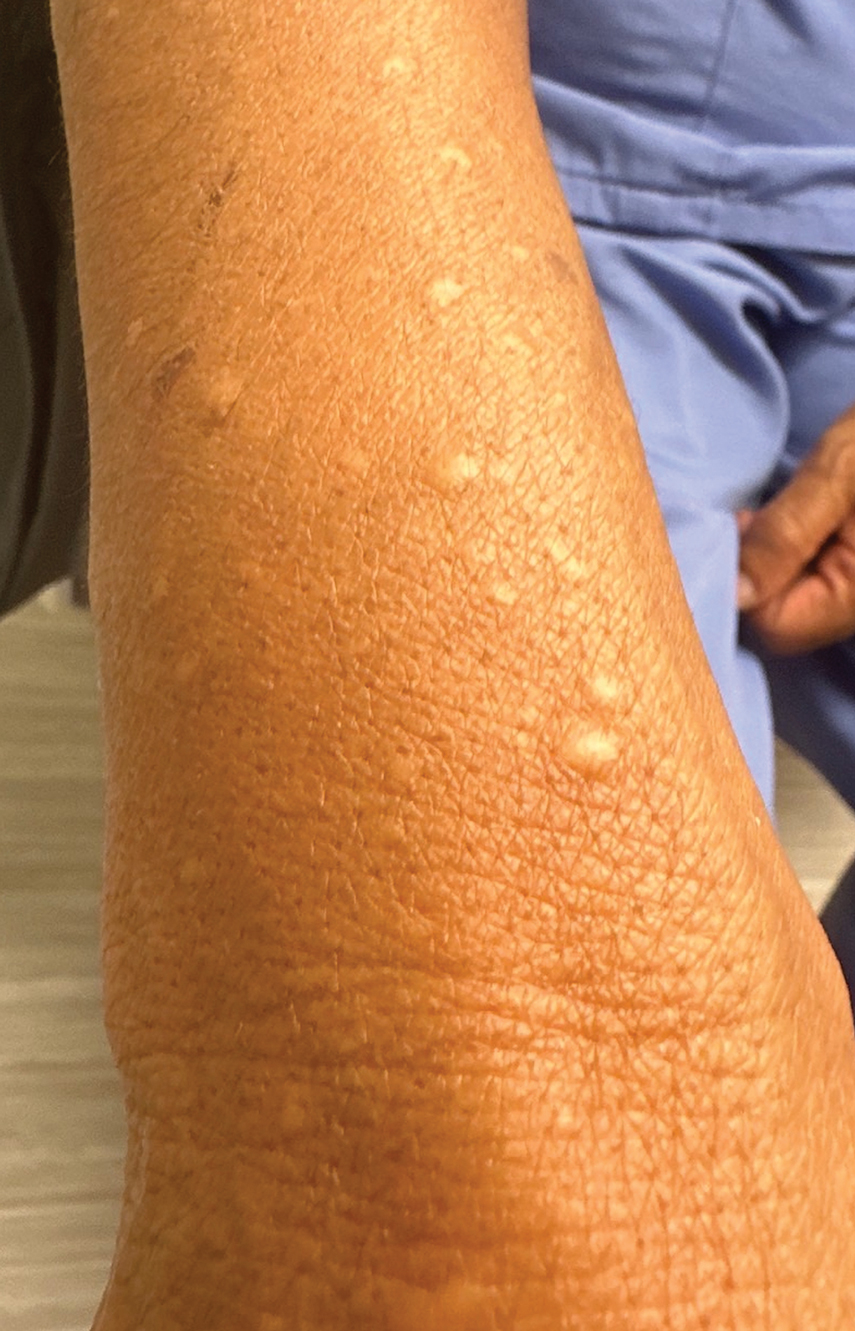
Acral persistent papular mucinosis is a rare distinct subtype of cutaneous mucinosis that initially was described by Rongioletti et al1 in 1986. As a localized form of lichen myxedematosus, APPM is characterized by mucin deposition in the dermis with no systemic involvement. The precise pathogenesis remains unclear, although some investigators have suggested that cytokine-mediated stimulation of glycosaminoglycan production may contribute to increased mucin accumulation in the dermis.2 Acral persistent papular mucinosis predominantly affects middle-aged women with a 5:1 female-to-male predominance.3 Clinically, patients present with discrete, nonfollicular, waxy papules that typically measure 2 to 5 mm and are distributed symmetrically on the extensor surfaces of the wrists and forearms. While the lesions generally are asymptomatic, some patients may report mild pruritus. The condition is chronic, with lesions seldom resolving and often increasing in number over time.3
Histologically, APPM is characterized by focal deposits of mucin in the upper reticular dermis with no evidence of increased fibroblast proliferation or fibrosis.4 This feature is pivotal in differentiating APPM from other subtypes of localized lichen myxedematosus and similar dermatoses. Diagnosis of APPM requires exclusion of systemic involvement, including thyroid abnormalities and monoclonal gammopathy, aligning with its classification as a purely cutaneous condition.5 Management of APPM is unclear due to its rarity. Reassurance for patients of its benign nature as well as clinical observation are recommended, though some reports cite benefits of treatment with topical corticosteroids or calcineurin inhibitors.6,7 The long-term prognosis for patients with APPM is favorable, although the persistence of and potential increase in lesions over time can be a cosmetic concern.
The differential diagnoses for APPM include scleromyxedema, scleredema, and other cutaneous eruptions that manifest as smooth flesh-colored papules, such as granuloma annulare and lichen nitidus.3 Scleromyxedema is a systemic cutaneous mucinosis that is part of the same disease spectrum as lichen myxedematosus. The papular eruption of scleromyxedema is much more widespread, and coalescing of the lesions may lead to characteristic skin thickening, creating leonine facies and deep furrowing over the trunk.8 Extracutaneous manifestations are frequent in scleromyxedema, and up to 90% of patients exhibit evidence of an underlying plasma cell dyscrasia.2 Histopathologically, scleromyxedema shows extensive fibroblast proliferation and fibrosis, in contrast to the findings of APPM (Figure 1).
The histopathology of APPM is most similar to scleredema, a rare fibromucinous disorder of the skin associated with diabetes, infection (especially poststreptococcal), or monoclonal gammopathy.9 Biopsy evaluation of scleredema reveals a normal epidermis with mucin deposition between collagen bundles predominantly in the deep reticular dermis as well as absent fibroblast proliferation (Figure 2). Unlike APPM, scleredema manifests with diffuse woody induration with erythema and hyperpigmentation on the posterior neck and upper back.9 On physical examination, the distinct clinical features of scleredema distinguish this condition from APPM and scleromyxedema.
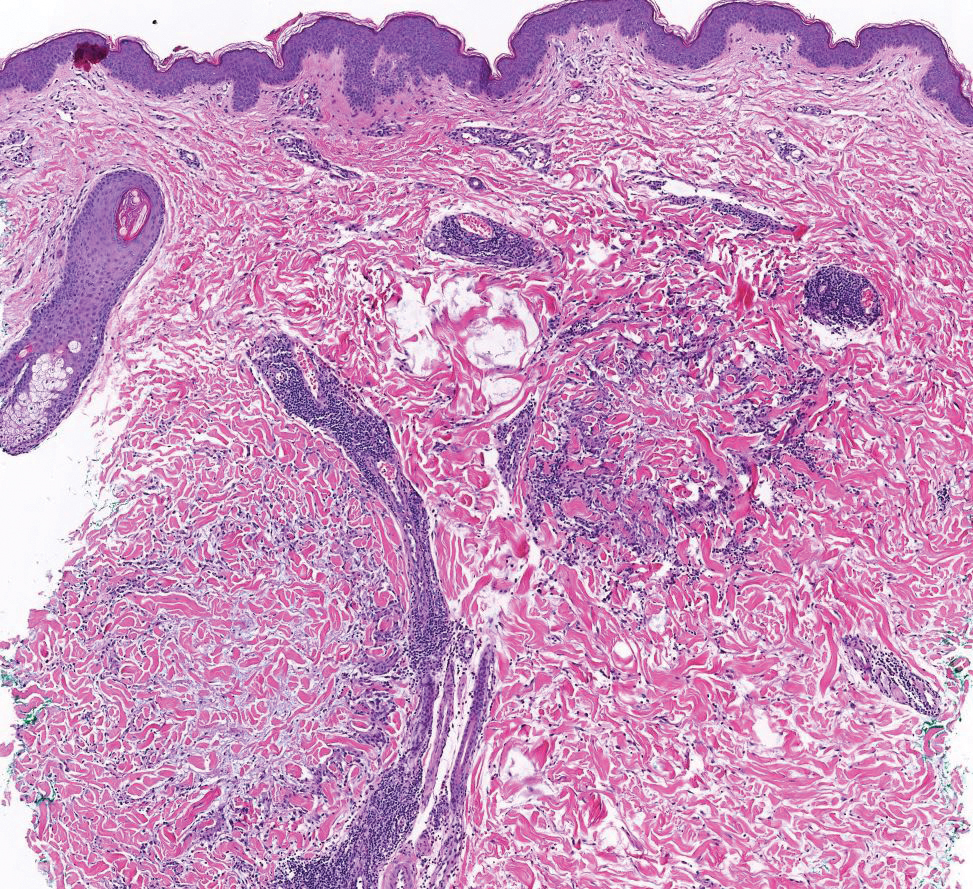
Papular granuloma annulare also was considered in our patient due to the presence of small flesh-colored papules. Histologically, granuloma annulare is characterized by palisading granulomas and mucin deposition in the dermis.10 However, the pattern of mucin deposition differs from that seen in APPM. In granuloma annulare, mucin is observed around foci of degenerated collagen (Figure 3), which was not observed in our patient.10 Additionally, the absence of an inflammatory infiltrate in our patient further ruled out this diagnosis.
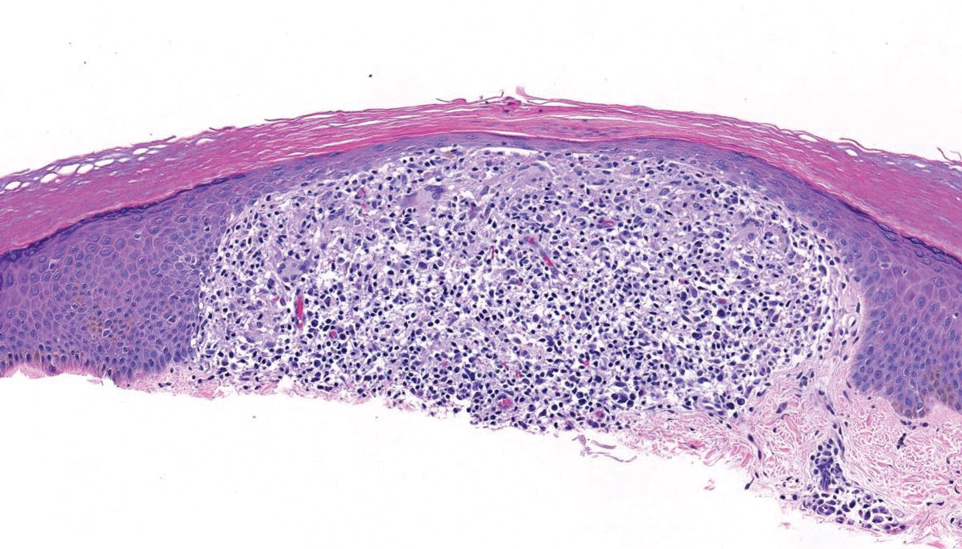
Lichen nitidus also could be considered in the differential diagnosis for ACCM. It typically manifests with minute, clustered, monomorphous papules with a predilection for the chest, abdomen, flexural forearms, and genitalia. The histology of lichen nitidus is distinct, showing a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis bordered by epidermal ridges, resembling a ball and clutch appearance (Figure 4).11
Although the clinical differential diagnosis in our patient was broad, histopathologic evaluation played a crucial role in confirming the diagnosis of APPM. This benign condition could be overlooked by patients and physicians; thorough clinical evaluation is necessary to rule out systemic mucinoses, which are associated with higher risks of morbidity and mortality.
- Rongioletti F, Rebora A. Acral persistent papular mucinosis: a new entity. Arch Dermatol. 1986;122:1237-1239. doi:10.1001 /archderm.1986.01660230027002
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosus (LM)(discrete papular type). Dermatol Online J. 2017;23:13030/qt3xp109qd.
- Rongioletti F, Ferreli C, Atzori L. Acral persistent papular mucinosis. Clin Dermatol. 2021;39:211-214. doi:10.1016/j.clindermatol.2020.10.001
- Rongioletti F, Rebora A. Cutaneous mucinoses: microscopic criteria for diagnosis. Am J Dermatopathol. 2001;23:257-267. doi:10.1097/00000372- 200106000-00022
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104. doi:10.1016/j.sder.2006.04.001
- Jun JY, Oh SH, Shim JH, et al. Acral persistent papular mucinosis with partial response to tacrolimus ointment. Ann Dermatol. 2016;28:517-519. doi:10.5021/ad.2016.28.4.517
- Rongioletti F, Zaccaria E, Cozzani E, et al. Treatment of localized lichen myxedematosus of discrete type with tacrolimus ointment. J Am Acad Dermatol. 2008;58:530-532. doi:10.1016/j.jaad.2006.10.021
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72. doi:10.1016 /j.jaad.2013.01.007
- Rongioletti F, Kaiser F, Cinotti E, et al. Scleredema. a multicentre study of characteristics, comorbidities, course and therapy in 44 patients. J Eur Acad Dermatol Venereol. 2015;29:2399-2404. doi:10.1111/jdv.13272
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465. doi:10.1016/j.jaad.2015.03.054
- Al-Mutairi N, Hassanein A, Nour-Eldin O, et al. Generalized lichen nitidus. Pediatr Dermatol. 2005;22:158-160. doi:10.1111 /j.1525-1470.2005.22215.x
THE DIAGNOSIS: Acral Persistent Papular Mucinosis
Histopathologic analysis revealed conspicuous interstitial mucin deposition throughout the upper to mid reticular dermis in the absence of a cellular infiltrate or fibroplasia. Colloidal iron staining confirmed the presence of mucin. In correlation with the clinical presentation, a diagnosis of acral persistent papular mucinosis (APPM) was made. The patient was counseled on the benign disease course and lack of associated comorbidities, and additional treatment was not pursued.
Acral persistent papular mucinosis is a rare distinct subtype of cutaneous mucinosis that initially was described by Rongioletti et al1 in 1986. As a localized form of lichen myxedematosus, APPM is characterized by mucin deposition in the dermis with no systemic involvement. The precise pathogenesis remains unclear, although some investigators have suggested that cytokine-mediated stimulation of glycosaminoglycan production may contribute to increased mucin accumulation in the dermis.2 Acral persistent papular mucinosis predominantly affects middle-aged women with a 5:1 female-to-male predominance.3 Clinically, patients present with discrete, nonfollicular, waxy papules that typically measure 2 to 5 mm and are distributed symmetrically on the extensor surfaces of the wrists and forearms. While the lesions generally are asymptomatic, some patients may report mild pruritus. The condition is chronic, with lesions seldom resolving and often increasing in number over time.3
Histologically, APPM is characterized by focal deposits of mucin in the upper reticular dermis with no evidence of increased fibroblast proliferation or fibrosis.4 This feature is pivotal in differentiating APPM from other subtypes of localized lichen myxedematosus and similar dermatoses. Diagnosis of APPM requires exclusion of systemic involvement, including thyroid abnormalities and monoclonal gammopathy, aligning with its classification as a purely cutaneous condition.5 Management of APPM is unclear due to its rarity. Reassurance for patients of its benign nature as well as clinical observation are recommended, though some reports cite benefits of treatment with topical corticosteroids or calcineurin inhibitors.6,7 The long-term prognosis for patients with APPM is favorable, although the persistence of and potential increase in lesions over time can be a cosmetic concern.
The differential diagnoses for APPM include scleromyxedema, scleredema, and other cutaneous eruptions that manifest as smooth flesh-colored papules, such as granuloma annulare and lichen nitidus.3 Scleromyxedema is a systemic cutaneous mucinosis that is part of the same disease spectrum as lichen myxedematosus. The papular eruption of scleromyxedema is much more widespread, and coalescing of the lesions may lead to characteristic skin thickening, creating leonine facies and deep furrowing over the trunk.8 Extracutaneous manifestations are frequent in scleromyxedema, and up to 90% of patients exhibit evidence of an underlying plasma cell dyscrasia.2 Histopathologically, scleromyxedema shows extensive fibroblast proliferation and fibrosis, in contrast to the findings of APPM (Figure 1).
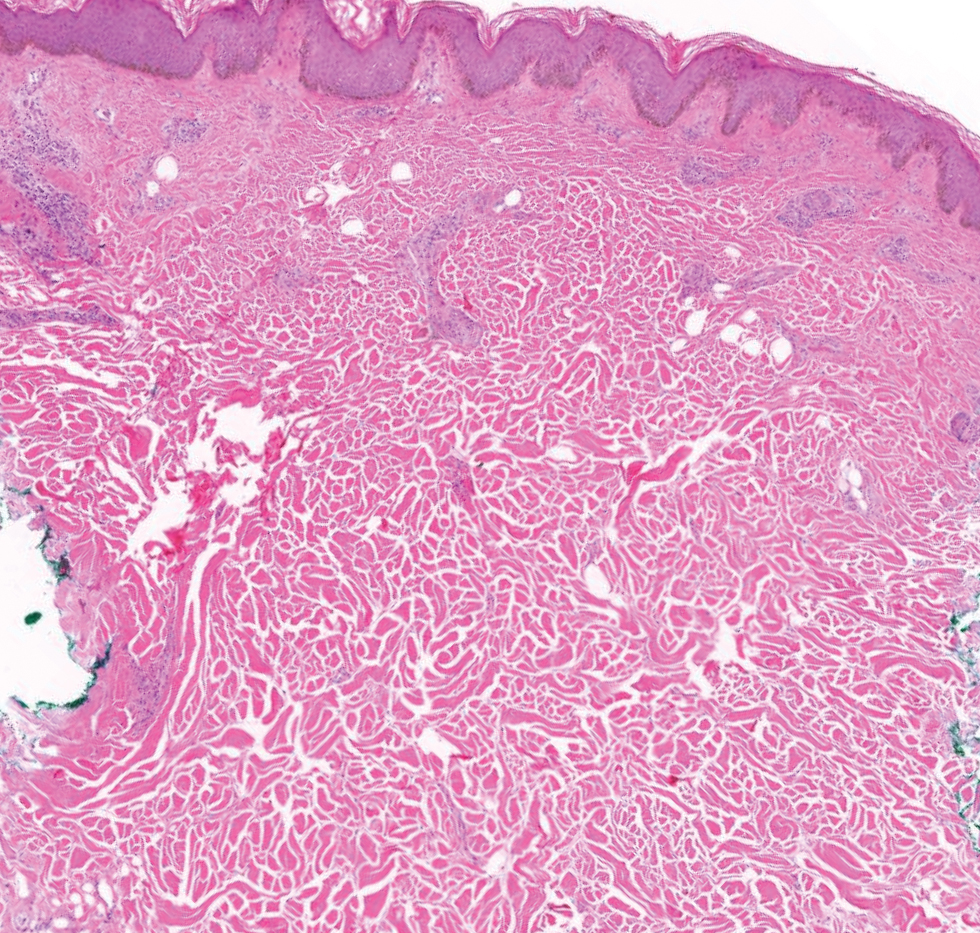
The histopathology of APPM is most similar to scleredema, a rare fibromucinous disorder of the skin associated with diabetes, infection (especially poststreptococcal), or monoclonal gammopathy.9 Biopsy evaluation of scleredema reveals a normal epidermis with mucin deposition between collagen bundles predominantly in the deep reticular dermis as well as absent fibroblast proliferation (Figure 2). Unlike APPM, scleredema manifests with diffuse woody induration with erythema and hyperpigmentation on the posterior neck and upper back.9 On physical examination, the distinct clinical features of scleredema distinguish this condition from APPM and scleromyxedema.
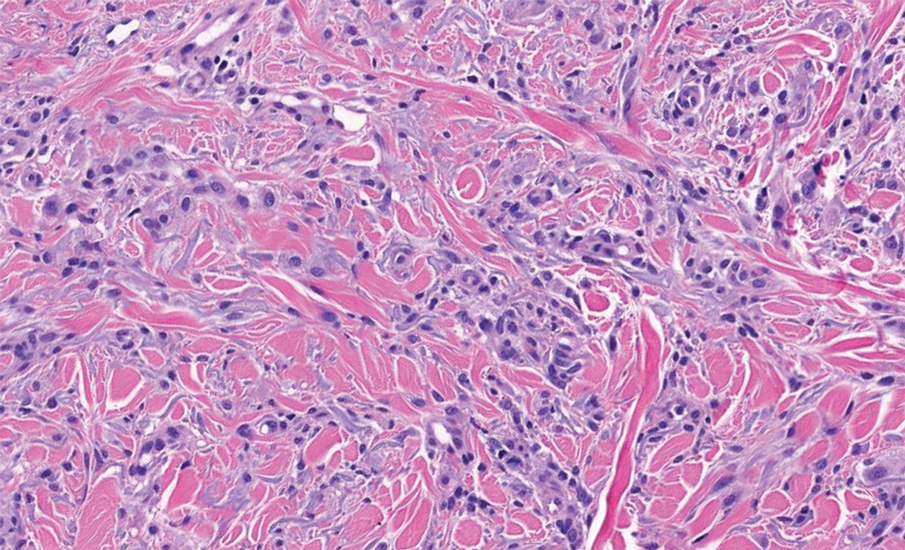
Papular granuloma annulare also was considered in our patient due to the presence of small flesh-colored papules. Histologically, granuloma annulare is characterized by palisading granulomas and mucin deposition in the dermis.10 However, the pattern of mucin deposition differs from that seen in APPM. In granuloma annulare, mucin is observed around foci of degenerated collagen (Figure 3), which was not observed in our patient.10 Additionally, the absence of an inflammatory infiltrate in our patient further ruled out this diagnosis.
Lichen nitidus also could be considered in the differential diagnosis for ACCM. It typically manifests with minute, clustered, monomorphous papules with a predilection for the chest, abdomen, flexural forearms, and genitalia. The histology of lichen nitidus is distinct, showing a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis bordered by epidermal ridges, resembling a ball and clutch appearance (Figure 4).11
Although the clinical differential diagnosis in our patient was broad, histopathologic evaluation played a crucial role in confirming the diagnosis of APPM. This benign condition could be overlooked by patients and physicians; thorough clinical evaluation is necessary to rule out systemic mucinoses, which are associated with higher risks of morbidity and mortality.
THE DIAGNOSIS: Acral Persistent Papular Mucinosis
Histopathologic analysis revealed conspicuous interstitial mucin deposition throughout the upper to mid reticular dermis in the absence of a cellular infiltrate or fibroplasia. Colloidal iron staining confirmed the presence of mucin. In correlation with the clinical presentation, a diagnosis of acral persistent papular mucinosis (APPM) was made. The patient was counseled on the benign disease course and lack of associated comorbidities, and additional treatment was not pursued.
Acral persistent papular mucinosis is a rare distinct subtype of cutaneous mucinosis that initially was described by Rongioletti et al1 in 1986. As a localized form of lichen myxedematosus, APPM is characterized by mucin deposition in the dermis with no systemic involvement. The precise pathogenesis remains unclear, although some investigators have suggested that cytokine-mediated stimulation of glycosaminoglycan production may contribute to increased mucin accumulation in the dermis.2 Acral persistent papular mucinosis predominantly affects middle-aged women with a 5:1 female-to-male predominance.3 Clinically, patients present with discrete, nonfollicular, waxy papules that typically measure 2 to 5 mm and are distributed symmetrically on the extensor surfaces of the wrists and forearms. While the lesions generally are asymptomatic, some patients may report mild pruritus. The condition is chronic, with lesions seldom resolving and often increasing in number over time.3
Histologically, APPM is characterized by focal deposits of mucin in the upper reticular dermis with no evidence of increased fibroblast proliferation or fibrosis.4 This feature is pivotal in differentiating APPM from other subtypes of localized lichen myxedematosus and similar dermatoses. Diagnosis of APPM requires exclusion of systemic involvement, including thyroid abnormalities and monoclonal gammopathy, aligning with its classification as a purely cutaneous condition.5 Management of APPM is unclear due to its rarity. Reassurance for patients of its benign nature as well as clinical observation are recommended, though some reports cite benefits of treatment with topical corticosteroids or calcineurin inhibitors.6,7 The long-term prognosis for patients with APPM is favorable, although the persistence of and potential increase in lesions over time can be a cosmetic concern.
The differential diagnoses for APPM include scleromyxedema, scleredema, and other cutaneous eruptions that manifest as smooth flesh-colored papules, such as granuloma annulare and lichen nitidus.3 Scleromyxedema is a systemic cutaneous mucinosis that is part of the same disease spectrum as lichen myxedematosus. The papular eruption of scleromyxedema is much more widespread, and coalescing of the lesions may lead to characteristic skin thickening, creating leonine facies and deep furrowing over the trunk.8 Extracutaneous manifestations are frequent in scleromyxedema, and up to 90% of patients exhibit evidence of an underlying plasma cell dyscrasia.2 Histopathologically, scleromyxedema shows extensive fibroblast proliferation and fibrosis, in contrast to the findings of APPM (Figure 1).
The histopathology of APPM is most similar to scleredema, a rare fibromucinous disorder of the skin associated with diabetes, infection (especially poststreptococcal), or monoclonal gammopathy.9 Biopsy evaluation of scleredema reveals a normal epidermis with mucin deposition between collagen bundles predominantly in the deep reticular dermis as well as absent fibroblast proliferation (Figure 2). Unlike APPM, scleredema manifests with diffuse woody induration with erythema and hyperpigmentation on the posterior neck and upper back.9 On physical examination, the distinct clinical features of scleredema distinguish this condition from APPM and scleromyxedema.
Papular granuloma annulare also was considered in our patient due to the presence of small flesh-colored papules. Histologically, granuloma annulare is characterized by palisading granulomas and mucin deposition in the dermis.10 However, the pattern of mucin deposition differs from that seen in APPM. In granuloma annulare, mucin is observed around foci of degenerated collagen (Figure 3), which was not observed in our patient.10 Additionally, the absence of an inflammatory infiltrate in our patient further ruled out this diagnosis.
Lichen nitidus also could be considered in the differential diagnosis for ACCM. It typically manifests with minute, clustered, monomorphous papules with a predilection for the chest, abdomen, flexural forearms, and genitalia. The histology of lichen nitidus is distinct, showing a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis bordered by epidermal ridges, resembling a ball and clutch appearance (Figure 4).11
Although the clinical differential diagnosis in our patient was broad, histopathologic evaluation played a crucial role in confirming the diagnosis of APPM. This benign condition could be overlooked by patients and physicians; thorough clinical evaluation is necessary to rule out systemic mucinoses, which are associated with higher risks of morbidity and mortality.
- Rongioletti F, Rebora A. Acral persistent papular mucinosis: a new entity. Arch Dermatol. 1986;122:1237-1239. doi:10.1001 /archderm.1986.01660230027002
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosus (LM)(discrete papular type). Dermatol Online J. 2017;23:13030/qt3xp109qd.
- Rongioletti F, Ferreli C, Atzori L. Acral persistent papular mucinosis. Clin Dermatol. 2021;39:211-214. doi:10.1016/j.clindermatol.2020.10.001
- Rongioletti F, Rebora A. Cutaneous mucinoses: microscopic criteria for diagnosis. Am J Dermatopathol. 2001;23:257-267. doi:10.1097/00000372- 200106000-00022
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104. doi:10.1016/j.sder.2006.04.001
- Jun JY, Oh SH, Shim JH, et al. Acral persistent papular mucinosis with partial response to tacrolimus ointment. Ann Dermatol. 2016;28:517-519. doi:10.5021/ad.2016.28.4.517
- Rongioletti F, Zaccaria E, Cozzani E, et al. Treatment of localized lichen myxedematosus of discrete type with tacrolimus ointment. J Am Acad Dermatol. 2008;58:530-532. doi:10.1016/j.jaad.2006.10.021
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72. doi:10.1016 /j.jaad.2013.01.007
- Rongioletti F, Kaiser F, Cinotti E, et al. Scleredema. a multicentre study of characteristics, comorbidities, course and therapy in 44 patients. J Eur Acad Dermatol Venereol. 2015;29:2399-2404. doi:10.1111/jdv.13272
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465. doi:10.1016/j.jaad.2015.03.054
- Al-Mutairi N, Hassanein A, Nour-Eldin O, et al. Generalized lichen nitidus. Pediatr Dermatol. 2005;22:158-160. doi:10.1111 /j.1525-1470.2005.22215.x
- Rongioletti F, Rebora A. Acral persistent papular mucinosis: a new entity. Arch Dermatol. 1986;122:1237-1239. doi:10.1001 /archderm.1986.01660230027002
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosus (LM)(discrete papular type). Dermatol Online J. 2017;23:13030/qt3xp109qd.
- Rongioletti F, Ferreli C, Atzori L. Acral persistent papular mucinosis. Clin Dermatol. 2021;39:211-214. doi:10.1016/j.clindermatol.2020.10.001
- Rongioletti F, Rebora A. Cutaneous mucinoses: microscopic criteria for diagnosis. Am J Dermatopathol. 2001;23:257-267. doi:10.1097/00000372- 200106000-00022
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104. doi:10.1016/j.sder.2006.04.001
- Jun JY, Oh SH, Shim JH, et al. Acral persistent papular mucinosis with partial response to tacrolimus ointment. Ann Dermatol. 2016;28:517-519. doi:10.5021/ad.2016.28.4.517
- Rongioletti F, Zaccaria E, Cozzani E, et al. Treatment of localized lichen myxedematosus of discrete type with tacrolimus ointment. J Am Acad Dermatol. 2008;58:530-532. doi:10.1016/j.jaad.2006.10.021
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72. doi:10.1016 /j.jaad.2013.01.007
- Rongioletti F, Kaiser F, Cinotti E, et al. Scleredema. a multicentre study of characteristics, comorbidities, course and therapy in 44 patients. J Eur Acad Dermatol Venereol. 2015;29:2399-2404. doi:10.1111/jdv.13272
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465. doi:10.1016/j.jaad.2015.03.054
- Al-Mutairi N, Hassanein A, Nour-Eldin O, et al. Generalized lichen nitidus. Pediatr Dermatol. 2005;22:158-160. doi:10.1111 /j.1525-1470.2005.22215.x
Multiple Firm Papules on the Wrists and Forearms
Multiple Firm Papules on the Wrists and Forearms
A 69-year-old woman presented to the dermatology department with persistent asymptomatic skin lesions on the wrists and forearms of several months’ duration. The lesions had slowly grown in number over the past few months with no identifiable triggers. The patient reported no known history of injury or trauma to the affected sites and was not taking any prescription medications other than daily vitamins. She denied any family history of similar lesions and was otherwise healthy. Physical examination revealed multiple waxy, firm, hypopigmented, 3- to 5-mm papules located exclusively on the dorsal wrists and forearms. No extracutaneous involvement was observed. A 4-mm punch biopsy from the forearm was obtained.