User login
CCJM delivers practical clinical articles relevant to internists, cardiologists, endocrinologists, and other specialists, all written by known experts.
Copyright © 2019 Cleveland Clinic. All rights reserved. The information provided is for educational purposes only. Use of this website is subject to the disclaimer and privacy policy.
gambling
compulsive behaviors
ammunition
assault rifle
black jack
Boko Haram
bondage
child abuse
cocaine
Daech
drug paraphernalia
explosion
gun
human trafficking
ISIL
ISIS
Islamic caliphate
Islamic state
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
pedophile
pedophilia
poker
porn
pornography
psychedelic drug
recreational drug
sex slave rings
slot machine
terrorism
terrorist
Texas hold 'em
UFC
substance abuse
abuseed
abuseer
abusees
abuseing
abusely
abuses
aeolus
aeolused
aeoluser
aeoluses
aeolusing
aeolusly
aeoluss
ahole
aholeed
aholeer
aholees
aholeing
aholely
aholes
alcohol
alcoholed
alcoholer
alcoholes
alcoholing
alcoholly
alcohols
allman
allmaned
allmaner
allmanes
allmaning
allmanly
allmans
alted
altes
alting
altly
alts
analed
analer
anales
analing
anally
analprobe
analprobeed
analprobeer
analprobees
analprobeing
analprobely
analprobes
anals
anilingus
anilingused
anilinguser
anilinguses
anilingusing
anilingusly
anilinguss
anus
anused
anuser
anuses
anusing
anusly
anuss
areola
areolaed
areolaer
areolaes
areolaing
areolaly
areolas
areole
areoleed
areoleer
areolees
areoleing
areolely
areoles
arian
arianed
arianer
arianes
arianing
arianly
arians
aryan
aryaned
aryaner
aryanes
aryaning
aryanly
aryans
asiaed
asiaer
asiaes
asiaing
asialy
asias
ass
ass hole
ass lick
ass licked
ass licker
ass lickes
ass licking
ass lickly
ass licks
assbang
assbanged
assbangeded
assbangeder
assbangedes
assbangeding
assbangedly
assbangeds
assbanger
assbanges
assbanging
assbangly
assbangs
assbangsed
assbangser
assbangses
assbangsing
assbangsly
assbangss
assed
asser
asses
assesed
asseser
asseses
assesing
assesly
assess
assfuck
assfucked
assfucker
assfuckered
assfuckerer
assfuckeres
assfuckering
assfuckerly
assfuckers
assfuckes
assfucking
assfuckly
assfucks
asshat
asshated
asshater
asshates
asshating
asshatly
asshats
assholeed
assholeer
assholees
assholeing
assholely
assholes
assholesed
assholeser
assholeses
assholesing
assholesly
assholess
assing
assly
assmaster
assmastered
assmasterer
assmasteres
assmastering
assmasterly
assmasters
assmunch
assmunched
assmuncher
assmunches
assmunching
assmunchly
assmunchs
asss
asswipe
asswipeed
asswipeer
asswipees
asswipeing
asswipely
asswipes
asswipesed
asswipeser
asswipeses
asswipesing
asswipesly
asswipess
azz
azzed
azzer
azzes
azzing
azzly
azzs
babeed
babeer
babees
babeing
babely
babes
babesed
babeser
babeses
babesing
babesly
babess
ballsac
ballsaced
ballsacer
ballsaces
ballsacing
ballsack
ballsacked
ballsacker
ballsackes
ballsacking
ballsackly
ballsacks
ballsacly
ballsacs
ballsed
ballser
ballses
ballsing
ballsly
ballss
barf
barfed
barfer
barfes
barfing
barfly
barfs
bastard
bastarded
bastarder
bastardes
bastarding
bastardly
bastards
bastardsed
bastardser
bastardses
bastardsing
bastardsly
bastardss
bawdy
bawdyed
bawdyer
bawdyes
bawdying
bawdyly
bawdys
beaner
beanered
beanerer
beaneres
beanering
beanerly
beaners
beardedclam
beardedclamed
beardedclamer
beardedclames
beardedclaming
beardedclamly
beardedclams
beastiality
beastialityed
beastialityer
beastialityes
beastialitying
beastialityly
beastialitys
beatch
beatched
beatcher
beatches
beatching
beatchly
beatchs
beater
beatered
beaterer
beateres
beatering
beaterly
beaters
beered
beerer
beeres
beering
beerly
beeyotch
beeyotched
beeyotcher
beeyotches
beeyotching
beeyotchly
beeyotchs
beotch
beotched
beotcher
beotches
beotching
beotchly
beotchs
biatch
biatched
biatcher
biatches
biatching
biatchly
biatchs
big tits
big titsed
big titser
big titses
big titsing
big titsly
big titss
bigtits
bigtitsed
bigtitser
bigtitses
bigtitsing
bigtitsly
bigtitss
bimbo
bimboed
bimboer
bimboes
bimboing
bimboly
bimbos
bisexualed
bisexualer
bisexuales
bisexualing
bisexually
bisexuals
bitch
bitched
bitcheded
bitcheder
bitchedes
bitcheding
bitchedly
bitcheds
bitcher
bitches
bitchesed
bitcheser
bitcheses
bitchesing
bitchesly
bitchess
bitching
bitchly
bitchs
bitchy
bitchyed
bitchyer
bitchyes
bitchying
bitchyly
bitchys
bleached
bleacher
bleaches
bleaching
bleachly
bleachs
blow job
blow jobed
blow jober
blow jobes
blow jobing
blow jobly
blow jobs
blowed
blower
blowes
blowing
blowjob
blowjobed
blowjober
blowjobes
blowjobing
blowjobly
blowjobs
blowjobsed
blowjobser
blowjobses
blowjobsing
blowjobsly
blowjobss
blowly
blows
boink
boinked
boinker
boinkes
boinking
boinkly
boinks
bollock
bollocked
bollocker
bollockes
bollocking
bollockly
bollocks
bollocksed
bollockser
bollockses
bollocksing
bollocksly
bollockss
bollok
bolloked
bolloker
bollokes
bolloking
bollokly
bolloks
boner
bonered
bonerer
boneres
bonering
bonerly
boners
bonersed
bonerser
bonerses
bonersing
bonersly
bonerss
bong
bonged
bonger
bonges
bonging
bongly
bongs
boob
boobed
boober
boobes
boobies
boobiesed
boobieser
boobieses
boobiesing
boobiesly
boobiess
boobing
boobly
boobs
boobsed
boobser
boobses
boobsing
boobsly
boobss
booby
boobyed
boobyer
boobyes
boobying
boobyly
boobys
booger
boogered
boogerer
boogeres
boogering
boogerly
boogers
bookie
bookieed
bookieer
bookiees
bookieing
bookiely
bookies
bootee
booteeed
booteeer
booteees
booteeing
booteely
bootees
bootie
bootieed
bootieer
bootiees
bootieing
bootiely
booties
booty
bootyed
bootyer
bootyes
bootying
bootyly
bootys
boozeed
boozeer
boozees
boozeing
boozely
boozer
boozered
boozerer
boozeres
boozering
boozerly
boozers
boozes
boozy
boozyed
boozyer
boozyes
boozying
boozyly
boozys
bosomed
bosomer
bosomes
bosoming
bosomly
bosoms
bosomy
bosomyed
bosomyer
bosomyes
bosomying
bosomyly
bosomys
bugger
buggered
buggerer
buggeres
buggering
buggerly
buggers
bukkake
bukkakeed
bukkakeer
bukkakees
bukkakeing
bukkakely
bukkakes
bull shit
bull shited
bull shiter
bull shites
bull shiting
bull shitly
bull shits
bullshit
bullshited
bullshiter
bullshites
bullshiting
bullshitly
bullshits
bullshitsed
bullshitser
bullshitses
bullshitsing
bullshitsly
bullshitss
bullshitted
bullshitteded
bullshitteder
bullshittedes
bullshitteding
bullshittedly
bullshitteds
bullturds
bullturdsed
bullturdser
bullturdses
bullturdsing
bullturdsly
bullturdss
bung
bunged
bunger
bunges
bunging
bungly
bungs
busty
bustyed
bustyer
bustyes
bustying
bustyly
bustys
butt
butt fuck
butt fucked
butt fucker
butt fuckes
butt fucking
butt fuckly
butt fucks
butted
buttes
buttfuck
buttfucked
buttfucker
buttfuckered
buttfuckerer
buttfuckeres
buttfuckering
buttfuckerly
buttfuckers
buttfuckes
buttfucking
buttfuckly
buttfucks
butting
buttly
buttplug
buttpluged
buttpluger
buttpluges
buttpluging
buttplugly
buttplugs
butts
caca
cacaed
cacaer
cacaes
cacaing
cacaly
cacas
cahone
cahoneed
cahoneer
cahonees
cahoneing
cahonely
cahones
cameltoe
cameltoeed
cameltoeer
cameltoees
cameltoeing
cameltoely
cameltoes
carpetmuncher
carpetmunchered
carpetmuncherer
carpetmuncheres
carpetmunchering
carpetmuncherly
carpetmunchers
cawk
cawked
cawker
cawkes
cawking
cawkly
cawks
chinc
chinced
chincer
chinces
chincing
chincly
chincs
chincsed
chincser
chincses
chincsing
chincsly
chincss
chink
chinked
chinker
chinkes
chinking
chinkly
chinks
chode
chodeed
chodeer
chodees
chodeing
chodely
chodes
chodesed
chodeser
chodeses
chodesing
chodesly
chodess
clit
clited
cliter
clites
cliting
clitly
clitoris
clitorised
clitoriser
clitorises
clitorising
clitorisly
clitoriss
clitorus
clitorused
clitoruser
clitoruses
clitorusing
clitorusly
clitoruss
clits
clitsed
clitser
clitses
clitsing
clitsly
clitss
clitty
clittyed
clittyer
clittyes
clittying
clittyly
clittys
cocain
cocaine
cocained
cocaineed
cocaineer
cocainees
cocaineing
cocainely
cocainer
cocaines
cocaining
cocainly
cocains
cock
cock sucker
cock suckered
cock suckerer
cock suckeres
cock suckering
cock suckerly
cock suckers
cockblock
cockblocked
cockblocker
cockblockes
cockblocking
cockblockly
cockblocks
cocked
cocker
cockes
cockholster
cockholstered
cockholsterer
cockholsteres
cockholstering
cockholsterly
cockholsters
cocking
cockknocker
cockknockered
cockknockerer
cockknockeres
cockknockering
cockknockerly
cockknockers
cockly
cocks
cocksed
cockser
cockses
cocksing
cocksly
cocksmoker
cocksmokered
cocksmokerer
cocksmokeres
cocksmokering
cocksmokerly
cocksmokers
cockss
cocksucker
cocksuckered
cocksuckerer
cocksuckeres
cocksuckering
cocksuckerly
cocksuckers
coital
coitaled
coitaler
coitales
coitaling
coitally
coitals
commie
commieed
commieer
commiees
commieing
commiely
commies
condomed
condomer
condomes
condoming
condomly
condoms
coon
cooned
cooner
coones
cooning
coonly
coons
coonsed
coonser
coonses
coonsing
coonsly
coonss
corksucker
corksuckered
corksuckerer
corksuckeres
corksuckering
corksuckerly
corksuckers
cracked
crackwhore
crackwhoreed
crackwhoreer
crackwhorees
crackwhoreing
crackwhorely
crackwhores
crap
craped
craper
crapes
craping
craply
crappy
crappyed
crappyer
crappyes
crappying
crappyly
crappys
cum
cumed
cumer
cumes
cuming
cumly
cummin
cummined
cumminer
cummines
cumming
cumminged
cumminger
cumminges
cumminging
cummingly
cummings
cummining
cumminly
cummins
cums
cumshot
cumshoted
cumshoter
cumshotes
cumshoting
cumshotly
cumshots
cumshotsed
cumshotser
cumshotses
cumshotsing
cumshotsly
cumshotss
cumslut
cumsluted
cumsluter
cumslutes
cumsluting
cumslutly
cumsluts
cumstain
cumstained
cumstainer
cumstaines
cumstaining
cumstainly
cumstains
cunilingus
cunilingused
cunilinguser
cunilinguses
cunilingusing
cunilingusly
cunilinguss
cunnilingus
cunnilingused
cunnilinguser
cunnilinguses
cunnilingusing
cunnilingusly
cunnilinguss
cunny
cunnyed
cunnyer
cunnyes
cunnying
cunnyly
cunnys
cunt
cunted
cunter
cuntes
cuntface
cuntfaceed
cuntfaceer
cuntfacees
cuntfaceing
cuntfacely
cuntfaces
cunthunter
cunthuntered
cunthunterer
cunthunteres
cunthuntering
cunthunterly
cunthunters
cunting
cuntlick
cuntlicked
cuntlicker
cuntlickered
cuntlickerer
cuntlickeres
cuntlickering
cuntlickerly
cuntlickers
cuntlickes
cuntlicking
cuntlickly
cuntlicks
cuntly
cunts
cuntsed
cuntser
cuntses
cuntsing
cuntsly
cuntss
dago
dagoed
dagoer
dagoes
dagoing
dagoly
dagos
dagosed
dagoser
dagoses
dagosing
dagosly
dagoss
dammit
dammited
dammiter
dammites
dammiting
dammitly
dammits
damn
damned
damneded
damneder
damnedes
damneding
damnedly
damneds
damner
damnes
damning
damnit
damnited
damniter
damnites
damniting
damnitly
damnits
damnly
damns
dick
dickbag
dickbaged
dickbager
dickbages
dickbaging
dickbagly
dickbags
dickdipper
dickdippered
dickdipperer
dickdipperes
dickdippering
dickdipperly
dickdippers
dicked
dicker
dickes
dickface
dickfaceed
dickfaceer
dickfacees
dickfaceing
dickfacely
dickfaces
dickflipper
dickflippered
dickflipperer
dickflipperes
dickflippering
dickflipperly
dickflippers
dickhead
dickheaded
dickheader
dickheades
dickheading
dickheadly
dickheads
dickheadsed
dickheadser
dickheadses
dickheadsing
dickheadsly
dickheadss
dicking
dickish
dickished
dickisher
dickishes
dickishing
dickishly
dickishs
dickly
dickripper
dickrippered
dickripperer
dickripperes
dickrippering
dickripperly
dickrippers
dicks
dicksipper
dicksippered
dicksipperer
dicksipperes
dicksippering
dicksipperly
dicksippers
dickweed
dickweeded
dickweeder
dickweedes
dickweeding
dickweedly
dickweeds
dickwhipper
dickwhippered
dickwhipperer
dickwhipperes
dickwhippering
dickwhipperly
dickwhippers
dickzipper
dickzippered
dickzipperer
dickzipperes
dickzippering
dickzipperly
dickzippers
diddle
diddleed
diddleer
diddlees
diddleing
diddlely
diddles
dike
dikeed
dikeer
dikees
dikeing
dikely
dikes
dildo
dildoed
dildoer
dildoes
dildoing
dildoly
dildos
dildosed
dildoser
dildoses
dildosing
dildosly
dildoss
diligaf
diligafed
diligafer
diligafes
diligafing
diligafly
diligafs
dillweed
dillweeded
dillweeder
dillweedes
dillweeding
dillweedly
dillweeds
dimwit
dimwited
dimwiter
dimwites
dimwiting
dimwitly
dimwits
dingle
dingleed
dingleer
dinglees
dingleing
dinglely
dingles
dipship
dipshiped
dipshiper
dipshipes
dipshiping
dipshiply
dipships
dizzyed
dizzyer
dizzyes
dizzying
dizzyly
dizzys
doggiestyleed
doggiestyleer
doggiestylees
doggiestyleing
doggiestylely
doggiestyles
doggystyleed
doggystyleer
doggystylees
doggystyleing
doggystylely
doggystyles
dong
donged
donger
donges
donging
dongly
dongs
doofus
doofused
doofuser
doofuses
doofusing
doofusly
doofuss
doosh
dooshed
doosher
dooshes
dooshing
dooshly
dooshs
dopeyed
dopeyer
dopeyes
dopeying
dopeyly
dopeys
douchebag
douchebaged
douchebager
douchebages
douchebaging
douchebagly
douchebags
douchebagsed
douchebagser
douchebagses
douchebagsing
douchebagsly
douchebagss
doucheed
doucheer
douchees
doucheing
douchely
douches
douchey
doucheyed
doucheyer
doucheyes
doucheying
doucheyly
doucheys
drunk
drunked
drunker
drunkes
drunking
drunkly
drunks
dumass
dumassed
dumasser
dumasses
dumassing
dumassly
dumasss
dumbass
dumbassed
dumbasser
dumbasses
dumbassesed
dumbasseser
dumbasseses
dumbassesing
dumbassesly
dumbassess
dumbassing
dumbassly
dumbasss
dummy
dummyed
dummyer
dummyes
dummying
dummyly
dummys
dyke
dykeed
dykeer
dykees
dykeing
dykely
dykes
dykesed
dykeser
dykeses
dykesing
dykesly
dykess
erotic
eroticed
eroticer
erotices
eroticing
eroticly
erotics
extacy
extacyed
extacyer
extacyes
extacying
extacyly
extacys
extasy
extasyed
extasyer
extasyes
extasying
extasyly
extasys
fack
facked
facker
fackes
facking
fackly
facks
fag
faged
fager
fages
fagg
fagged
faggeded
faggeder
faggedes
faggeding
faggedly
faggeds
fagger
fagges
fagging
faggit
faggited
faggiter
faggites
faggiting
faggitly
faggits
faggly
faggot
faggoted
faggoter
faggotes
faggoting
faggotly
faggots
faggs
faging
fagly
fagot
fagoted
fagoter
fagotes
fagoting
fagotly
fagots
fags
fagsed
fagser
fagses
fagsing
fagsly
fagss
faig
faiged
faiger
faiges
faiging
faigly
faigs
faigt
faigted
faigter
faigtes
faigting
faigtly
faigts
fannybandit
fannybandited
fannybanditer
fannybandites
fannybanditing
fannybanditly
fannybandits
farted
farter
fartes
farting
fartknocker
fartknockered
fartknockerer
fartknockeres
fartknockering
fartknockerly
fartknockers
fartly
farts
felch
felched
felcher
felchered
felcherer
felcheres
felchering
felcherly
felchers
felches
felching
felchinged
felchinger
felchinges
felchinging
felchingly
felchings
felchly
felchs
fellate
fellateed
fellateer
fellatees
fellateing
fellately
fellates
fellatio
fellatioed
fellatioer
fellatioes
fellatioing
fellatioly
fellatios
feltch
feltched
feltcher
feltchered
feltcherer
feltcheres
feltchering
feltcherly
feltchers
feltches
feltching
feltchly
feltchs
feom
feomed
feomer
feomes
feoming
feomly
feoms
fisted
fisteded
fisteder
fistedes
fisteding
fistedly
fisteds
fisting
fistinged
fistinger
fistinges
fistinging
fistingly
fistings
fisty
fistyed
fistyer
fistyes
fistying
fistyly
fistys
floozy
floozyed
floozyer
floozyes
floozying
floozyly
floozys
foad
foaded
foader
foades
foading
foadly
foads
fondleed
fondleer
fondlees
fondleing
fondlely
fondles
foobar
foobared
foobarer
foobares
foobaring
foobarly
foobars
freex
freexed
freexer
freexes
freexing
freexly
freexs
frigg
frigga
friggaed
friggaer
friggaes
friggaing
friggaly
friggas
frigged
frigger
frigges
frigging
friggly
friggs
fubar
fubared
fubarer
fubares
fubaring
fubarly
fubars
fuck
fuckass
fuckassed
fuckasser
fuckasses
fuckassing
fuckassly
fuckasss
fucked
fuckeded
fuckeder
fuckedes
fuckeding
fuckedly
fuckeds
fucker
fuckered
fuckerer
fuckeres
fuckering
fuckerly
fuckers
fuckes
fuckface
fuckfaceed
fuckfaceer
fuckfacees
fuckfaceing
fuckfacely
fuckfaces
fuckin
fuckined
fuckiner
fuckines
fucking
fuckinged
fuckinger
fuckinges
fuckinging
fuckingly
fuckings
fuckining
fuckinly
fuckins
fuckly
fucknugget
fucknuggeted
fucknuggeter
fucknuggetes
fucknuggeting
fucknuggetly
fucknuggets
fucknut
fucknuted
fucknuter
fucknutes
fucknuting
fucknutly
fucknuts
fuckoff
fuckoffed
fuckoffer
fuckoffes
fuckoffing
fuckoffly
fuckoffs
fucks
fucksed
fuckser
fuckses
fucksing
fucksly
fuckss
fucktard
fucktarded
fucktarder
fucktardes
fucktarding
fucktardly
fucktards
fuckup
fuckuped
fuckuper
fuckupes
fuckuping
fuckuply
fuckups
fuckwad
fuckwaded
fuckwader
fuckwades
fuckwading
fuckwadly
fuckwads
fuckwit
fuckwited
fuckwiter
fuckwites
fuckwiting
fuckwitly
fuckwits
fudgepacker
fudgepackered
fudgepackerer
fudgepackeres
fudgepackering
fudgepackerly
fudgepackers
fuk
fuked
fuker
fukes
fuking
fukly
fuks
fvck
fvcked
fvcker
fvckes
fvcking
fvckly
fvcks
fxck
fxcked
fxcker
fxckes
fxcking
fxckly
fxcks
gae
gaeed
gaeer
gaees
gaeing
gaely
gaes
gai
gaied
gaier
gaies
gaiing
gaily
gais
ganja
ganjaed
ganjaer
ganjaes
ganjaing
ganjaly
ganjas
gayed
gayer
gayes
gaying
gayly
gays
gaysed
gayser
gayses
gaysing
gaysly
gayss
gey
geyed
geyer
geyes
geying
geyly
geys
gfc
gfced
gfcer
gfces
gfcing
gfcly
gfcs
gfy
gfyed
gfyer
gfyes
gfying
gfyly
gfys
ghay
ghayed
ghayer
ghayes
ghaying
ghayly
ghays
ghey
gheyed
gheyer
gheyes
gheying
gheyly
gheys
gigolo
gigoloed
gigoloer
gigoloes
gigoloing
gigololy
gigolos
goatse
goatseed
goatseer
goatsees
goatseing
goatsely
goatses
godamn
godamned
godamner
godamnes
godamning
godamnit
godamnited
godamniter
godamnites
godamniting
godamnitly
godamnits
godamnly
godamns
goddam
goddamed
goddamer
goddames
goddaming
goddamly
goddammit
goddammited
goddammiter
goddammites
goddammiting
goddammitly
goddammits
goddamn
goddamned
goddamner
goddamnes
goddamning
goddamnly
goddamns
goddams
goldenshower
goldenshowered
goldenshowerer
goldenshoweres
goldenshowering
goldenshowerly
goldenshowers
gonad
gonaded
gonader
gonades
gonading
gonadly
gonads
gonadsed
gonadser
gonadses
gonadsing
gonadsly
gonadss
gook
gooked
gooker
gookes
gooking
gookly
gooks
gooksed
gookser
gookses
gooksing
gooksly
gookss
gringo
gringoed
gringoer
gringoes
gringoing
gringoly
gringos
gspot
gspoted
gspoter
gspotes
gspoting
gspotly
gspots
gtfo
gtfoed
gtfoer
gtfoes
gtfoing
gtfoly
gtfos
guido
guidoed
guidoer
guidoes
guidoing
guidoly
guidos
handjob
handjobed
handjober
handjobes
handjobing
handjobly
handjobs
hard on
hard oned
hard oner
hard ones
hard oning
hard only
hard ons
hardknight
hardknighted
hardknighter
hardknightes
hardknighting
hardknightly
hardknights
hebe
hebeed
hebeer
hebees
hebeing
hebely
hebes
heeb
heebed
heeber
heebes
heebing
heebly
heebs
hell
helled
heller
helles
helling
hellly
hells
hemp
hemped
hemper
hempes
hemping
hemply
hemps
heroined
heroiner
heroines
heroining
heroinly
heroins
herp
herped
herper
herpes
herpesed
herpeser
herpeses
herpesing
herpesly
herpess
herping
herply
herps
herpy
herpyed
herpyer
herpyes
herpying
herpyly
herpys
hitler
hitlered
hitlerer
hitleres
hitlering
hitlerly
hitlers
hived
hiver
hives
hiving
hivly
hivs
hobag
hobaged
hobager
hobages
hobaging
hobagly
hobags
homey
homeyed
homeyer
homeyes
homeying
homeyly
homeys
homo
homoed
homoer
homoes
homoey
homoeyed
homoeyer
homoeyes
homoeying
homoeyly
homoeys
homoing
homoly
homos
honky
honkyed
honkyer
honkyes
honkying
honkyly
honkys
hooch
hooched
hoocher
hooches
hooching
hoochly
hoochs
hookah
hookahed
hookaher
hookahes
hookahing
hookahly
hookahs
hooker
hookered
hookerer
hookeres
hookering
hookerly
hookers
hoor
hoored
hoorer
hoores
hooring
hoorly
hoors
hootch
hootched
hootcher
hootches
hootching
hootchly
hootchs
hooter
hootered
hooterer
hooteres
hootering
hooterly
hooters
hootersed
hooterser
hooterses
hootersing
hootersly
hooterss
horny
hornyed
hornyer
hornyes
hornying
hornyly
hornys
houstoned
houstoner
houstones
houstoning
houstonly
houstons
hump
humped
humpeded
humpeder
humpedes
humpeding
humpedly
humpeds
humper
humpes
humping
humpinged
humpinger
humpinges
humpinging
humpingly
humpings
humply
humps
husbanded
husbander
husbandes
husbanding
husbandly
husbands
hussy
hussyed
hussyer
hussyes
hussying
hussyly
hussys
hymened
hymener
hymenes
hymening
hymenly
hymens
inbred
inbreded
inbreder
inbredes
inbreding
inbredly
inbreds
incest
incested
incester
incestes
incesting
incestly
incests
injun
injuned
injuner
injunes
injuning
injunly
injuns
jackass
jackassed
jackasser
jackasses
jackassing
jackassly
jackasss
jackhole
jackholeed
jackholeer
jackholees
jackholeing
jackholely
jackholes
jackoff
jackoffed
jackoffer
jackoffes
jackoffing
jackoffly
jackoffs
jap
japed
japer
japes
japing
japly
japs
japsed
japser
japses
japsing
japsly
japss
jerkoff
jerkoffed
jerkoffer
jerkoffes
jerkoffing
jerkoffly
jerkoffs
jerks
jism
jismed
jismer
jismes
jisming
jismly
jisms
jiz
jized
jizer
jizes
jizing
jizly
jizm
jizmed
jizmer
jizmes
jizming
jizmly
jizms
jizs
jizz
jizzed
jizzeded
jizzeder
jizzedes
jizzeding
jizzedly
jizzeds
jizzer
jizzes
jizzing
jizzly
jizzs
junkie
junkieed
junkieer
junkiees
junkieing
junkiely
junkies
junky
junkyed
junkyer
junkyes
junkying
junkyly
junkys
kike
kikeed
kikeer
kikees
kikeing
kikely
kikes
kikesed
kikeser
kikeses
kikesing
kikesly
kikess
killed
killer
killes
killing
killly
kills
kinky
kinkyed
kinkyer
kinkyes
kinkying
kinkyly
kinkys
kkk
kkked
kkker
kkkes
kkking
kkkly
kkks
klan
klaned
klaner
klanes
klaning
klanly
klans
knobend
knobended
knobender
knobendes
knobending
knobendly
knobends
kooch
kooched
koocher
kooches
koochesed
koocheser
koocheses
koochesing
koochesly
koochess
kooching
koochly
koochs
kootch
kootched
kootcher
kootches
kootching
kootchly
kootchs
kraut
krauted
krauter
krautes
krauting
krautly
krauts
kyke
kykeed
kykeer
kykees
kykeing
kykely
kykes
lech
leched
lecher
leches
leching
lechly
lechs
leper
lepered
leperer
leperes
lepering
leperly
lepers
lesbiansed
lesbianser
lesbianses
lesbiansing
lesbiansly
lesbianss
lesbo
lesboed
lesboer
lesboes
lesboing
lesboly
lesbos
lesbosed
lesboser
lesboses
lesbosing
lesbosly
lesboss
lez
lezbianed
lezbianer
lezbianes
lezbianing
lezbianly
lezbians
lezbiansed
lezbianser
lezbianses
lezbiansing
lezbiansly
lezbianss
lezbo
lezboed
lezboer
lezboes
lezboing
lezboly
lezbos
lezbosed
lezboser
lezboses
lezbosing
lezbosly
lezboss
lezed
lezer
lezes
lezing
lezly
lezs
lezzie
lezzieed
lezzieer
lezziees
lezzieing
lezziely
lezzies
lezziesed
lezzieser
lezzieses
lezziesing
lezziesly
lezziess
lezzy
lezzyed
lezzyer
lezzyes
lezzying
lezzyly
lezzys
lmaoed
lmaoer
lmaoes
lmaoing
lmaoly
lmaos
lmfao
lmfaoed
lmfaoer
lmfaoes
lmfaoing
lmfaoly
lmfaos
loined
loiner
loines
loining
loinly
loins
loinsed
loinser
loinses
loinsing
loinsly
loinss
lubeed
lubeer
lubees
lubeing
lubely
lubes
lusty
lustyed
lustyer
lustyes
lustying
lustyly
lustys
massa
massaed
massaer
massaes
massaing
massaly
massas
masterbate
masterbateed
masterbateer
masterbatees
masterbateing
masterbately
masterbates
masterbating
masterbatinged
masterbatinger
masterbatinges
masterbatinging
masterbatingly
masterbatings
masterbation
masterbationed
masterbationer
masterbationes
masterbationing
masterbationly
masterbations
masturbate
masturbateed
masturbateer
masturbatees
masturbateing
masturbately
masturbates
masturbating
masturbatinged
masturbatinger
masturbatinges
masturbatinging
masturbatingly
masturbatings
masturbation
masturbationed
masturbationer
masturbationes
masturbationing
masturbationly
masturbations
methed
mether
methes
mething
methly
meths
militaryed
militaryer
militaryes
militarying
militaryly
militarys
mofo
mofoed
mofoer
mofoes
mofoing
mofoly
mofos
molest
molested
molester
molestes
molesting
molestly
molests
moolie
moolieed
moolieer
mooliees
moolieing
mooliely
moolies
moron
moroned
moroner
morones
moroning
moronly
morons
motherfucka
motherfuckaed
motherfuckaer
motherfuckaes
motherfuckaing
motherfuckaly
motherfuckas
motherfucker
motherfuckered
motherfuckerer
motherfuckeres
motherfuckering
motherfuckerly
motherfuckers
motherfucking
motherfuckinged
motherfuckinger
motherfuckinges
motherfuckinging
motherfuckingly
motherfuckings
mtherfucker
mtherfuckered
mtherfuckerer
mtherfuckeres
mtherfuckering
mtherfuckerly
mtherfuckers
mthrfucker
mthrfuckered
mthrfuckerer
mthrfuckeres
mthrfuckering
mthrfuckerly
mthrfuckers
mthrfucking
mthrfuckinged
mthrfuckinger
mthrfuckinges
mthrfuckinging
mthrfuckingly
mthrfuckings
muff
muffdiver
muffdivered
muffdiverer
muffdiveres
muffdivering
muffdiverly
muffdivers
muffed
muffer
muffes
muffing
muffly
muffs
murdered
murderer
murderes
murdering
murderly
murders
muthafuckaz
muthafuckazed
muthafuckazer
muthafuckazes
muthafuckazing
muthafuckazly
muthafuckazs
muthafucker
muthafuckered
muthafuckerer
muthafuckeres
muthafuckering
muthafuckerly
muthafuckers
mutherfucker
mutherfuckered
mutherfuckerer
mutherfuckeres
mutherfuckering
mutherfuckerly
mutherfuckers
mutherfucking
mutherfuckinged
mutherfuckinger
mutherfuckinges
mutherfuckinging
mutherfuckingly
mutherfuckings
muthrfucking
muthrfuckinged
muthrfuckinger
muthrfuckinges
muthrfuckinging
muthrfuckingly
muthrfuckings
nad
naded
nader
nades
nading
nadly
nads
nadsed
nadser
nadses
nadsing
nadsly
nadss
nakeded
nakeder
nakedes
nakeding
nakedly
nakeds
napalm
napalmed
napalmer
napalmes
napalming
napalmly
napalms
nappy
nappyed
nappyer
nappyes
nappying
nappyly
nappys
nazi
nazied
nazier
nazies
naziing
nazily
nazis
nazism
nazismed
nazismer
nazismes
nazisming
nazismly
nazisms
negro
negroed
negroer
negroes
negroing
negroly
negros
nigga
niggaed
niggaer
niggaes
niggah
niggahed
niggaher
niggahes
niggahing
niggahly
niggahs
niggaing
niggaly
niggas
niggased
niggaser
niggases
niggasing
niggasly
niggass
niggaz
niggazed
niggazer
niggazes
niggazing
niggazly
niggazs
nigger
niggered
niggerer
niggeres
niggering
niggerly
niggers
niggersed
niggerser
niggerses
niggersing
niggersly
niggerss
niggle
niggleed
niggleer
nigglees
niggleing
nigglely
niggles
niglet
nigleted
nigleter
nigletes
nigleting
nigletly
niglets
nimrod
nimroded
nimroder
nimrodes
nimroding
nimrodly
nimrods
ninny
ninnyed
ninnyer
ninnyes
ninnying
ninnyly
ninnys
nooky
nookyed
nookyer
nookyes
nookying
nookyly
nookys
nuccitelli
nuccitellied
nuccitellier
nuccitellies
nuccitelliing
nuccitellily
nuccitellis
nympho
nymphoed
nymphoer
nymphoes
nymphoing
nympholy
nymphos
opium
opiumed
opiumer
opiumes
opiuming
opiumly
opiums
orgies
orgiesed
orgieser
orgieses
orgiesing
orgiesly
orgiess
orgy
orgyed
orgyer
orgyes
orgying
orgyly
orgys
paddy
paddyed
paddyer
paddyes
paddying
paddyly
paddys
paki
pakied
pakier
pakies
pakiing
pakily
pakis
pantie
pantieed
pantieer
pantiees
pantieing
pantiely
panties
pantiesed
pantieser
pantieses
pantiesing
pantiesly
pantiess
panty
pantyed
pantyer
pantyes
pantying
pantyly
pantys
pastie
pastieed
pastieer
pastiees
pastieing
pastiely
pasties
pasty
pastyed
pastyer
pastyes
pastying
pastyly
pastys
pecker
peckered
peckerer
peckeres
peckering
peckerly
peckers
pedo
pedoed
pedoer
pedoes
pedoing
pedoly
pedophile
pedophileed
pedophileer
pedophilees
pedophileing
pedophilely
pedophiles
pedophilia
pedophiliac
pedophiliaced
pedophiliacer
pedophiliaces
pedophiliacing
pedophiliacly
pedophiliacs
pedophiliaed
pedophiliaer
pedophiliaes
pedophiliaing
pedophilialy
pedophilias
pedos
penial
penialed
penialer
peniales
penialing
penially
penials
penile
penileed
penileer
penilees
penileing
penilely
peniles
penis
penised
peniser
penises
penising
penisly
peniss
perversion
perversioned
perversioner
perversiones
perversioning
perversionly
perversions
peyote
peyoteed
peyoteer
peyotees
peyoteing
peyotely
peyotes
phuck
phucked
phucker
phuckes
phucking
phuckly
phucks
pillowbiter
pillowbitered
pillowbiterer
pillowbiteres
pillowbitering
pillowbiterly
pillowbiters
pimp
pimped
pimper
pimpes
pimping
pimply
pimps
pinko
pinkoed
pinkoer
pinkoes
pinkoing
pinkoly
pinkos
pissed
pisseded
pisseder
pissedes
pisseding
pissedly
pisseds
pisser
pisses
pissing
pissly
pissoff
pissoffed
pissoffer
pissoffes
pissoffing
pissoffly
pissoffs
pisss
polack
polacked
polacker
polackes
polacking
polackly
polacks
pollock
pollocked
pollocker
pollockes
pollocking
pollockly
pollocks
poon
pooned
pooner
poones
pooning
poonly
poons
poontang
poontanged
poontanger
poontanges
poontanging
poontangly
poontangs
porn
porned
porner
pornes
porning
pornly
porno
pornoed
pornoer
pornoes
pornography
pornographyed
pornographyer
pornographyes
pornographying
pornographyly
pornographys
pornoing
pornoly
pornos
porns
prick
pricked
pricker
prickes
pricking
prickly
pricks
prig
priged
priger
priges
priging
prigly
prigs
prostitute
prostituteed
prostituteer
prostitutees
prostituteing
prostitutely
prostitutes
prude
prudeed
prudeer
prudees
prudeing
prudely
prudes
punkass
punkassed
punkasser
punkasses
punkassing
punkassly
punkasss
punky
punkyed
punkyer
punkyes
punkying
punkyly
punkys
puss
pussed
pusser
pusses
pussies
pussiesed
pussieser
pussieses
pussiesing
pussiesly
pussiess
pussing
pussly
pusss
pussy
pussyed
pussyer
pussyes
pussying
pussyly
pussypounder
pussypoundered
pussypounderer
pussypounderes
pussypoundering
pussypounderly
pussypounders
pussys
puto
putoed
putoer
putoes
putoing
putoly
putos
queaf
queafed
queafer
queafes
queafing
queafly
queafs
queef
queefed
queefer
queefes
queefing
queefly
queefs
queer
queered
queerer
queeres
queering
queerly
queero
queeroed
queeroer
queeroes
queeroing
queeroly
queeros
queers
queersed
queerser
queerses
queersing
queersly
queerss
quicky
quickyed
quickyer
quickyes
quickying
quickyly
quickys
quim
quimed
quimer
quimes
quiming
quimly
quims
racy
racyed
racyer
racyes
racying
racyly
racys
rape
raped
rapeded
rapeder
rapedes
rapeding
rapedly
rapeds
rapeed
rapeer
rapees
rapeing
rapely
raper
rapered
raperer
raperes
rapering
raperly
rapers
rapes
rapist
rapisted
rapister
rapistes
rapisting
rapistly
rapists
raunch
raunched
rauncher
raunches
raunching
raunchly
raunchs
rectus
rectused
rectuser
rectuses
rectusing
rectusly
rectuss
reefer
reefered
reeferer
reeferes
reefering
reeferly
reefers
reetard
reetarded
reetarder
reetardes
reetarding
reetardly
reetards
reich
reiched
reicher
reiches
reiching
reichly
reichs
retard
retarded
retardeded
retardeder
retardedes
retardeding
retardedly
retardeds
retarder
retardes
retarding
retardly
retards
rimjob
rimjobed
rimjober
rimjobes
rimjobing
rimjobly
rimjobs
ritard
ritarded
ritarder
ritardes
ritarding
ritardly
ritards
rtard
rtarded
rtarder
rtardes
rtarding
rtardly
rtards
rum
rumed
rumer
rumes
ruming
rumly
rump
rumped
rumper
rumpes
rumping
rumply
rumprammer
rumprammered
rumprammerer
rumprammeres
rumprammering
rumprammerly
rumprammers
rumps
rums
ruski
ruskied
ruskier
ruskies
ruskiing
ruskily
ruskis
sadism
sadismed
sadismer
sadismes
sadisming
sadismly
sadisms
sadist
sadisted
sadister
sadistes
sadisting
sadistly
sadists
scag
scaged
scager
scages
scaging
scagly
scags
scantily
scantilyed
scantilyer
scantilyes
scantilying
scantilyly
scantilys
schlong
schlonged
schlonger
schlonges
schlonging
schlongly
schlongs
scrog
scroged
scroger
scroges
scroging
scrogly
scrogs
scrot
scrote
scroted
scroteed
scroteer
scrotees
scroteing
scrotely
scroter
scrotes
scroting
scrotly
scrots
scrotum
scrotumed
scrotumer
scrotumes
scrotuming
scrotumly
scrotums
scrud
scruded
scruder
scrudes
scruding
scrudly
scruds
scum
scumed
scumer
scumes
scuming
scumly
scums
seaman
seamaned
seamaner
seamanes
seamaning
seamanly
seamans
seamen
seamened
seamener
seamenes
seamening
seamenly
seamens
seduceed
seduceer
seducees
seduceing
seducely
seduces
semen
semened
semener
semenes
semening
semenly
semens
shamedame
shamedameed
shamedameer
shamedamees
shamedameing
shamedamely
shamedames
shit
shite
shiteater
shiteatered
shiteaterer
shiteateres
shiteatering
shiteaterly
shiteaters
shited
shiteed
shiteer
shitees
shiteing
shitely
shiter
shites
shitface
shitfaceed
shitfaceer
shitfacees
shitfaceing
shitfacely
shitfaces
shithead
shitheaded
shitheader
shitheades
shitheading
shitheadly
shitheads
shithole
shitholeed
shitholeer
shitholees
shitholeing
shitholely
shitholes
shithouse
shithouseed
shithouseer
shithousees
shithouseing
shithousely
shithouses
shiting
shitly
shits
shitsed
shitser
shitses
shitsing
shitsly
shitss
shitt
shitted
shitteded
shitteder
shittedes
shitteding
shittedly
shitteds
shitter
shittered
shitterer
shitteres
shittering
shitterly
shitters
shittes
shitting
shittly
shitts
shitty
shittyed
shittyer
shittyes
shittying
shittyly
shittys
shiz
shized
shizer
shizes
shizing
shizly
shizs
shooted
shooter
shootes
shooting
shootly
shoots
sissy
sissyed
sissyer
sissyes
sissying
sissyly
sissys
skag
skaged
skager
skages
skaging
skagly
skags
skank
skanked
skanker
skankes
skanking
skankly
skanks
slave
slaveed
slaveer
slavees
slaveing
slavely
slaves
sleaze
sleazeed
sleazeer
sleazees
sleazeing
sleazely
sleazes
sleazy
sleazyed
sleazyer
sleazyes
sleazying
sleazyly
sleazys
slut
slutdumper
slutdumpered
slutdumperer
slutdumperes
slutdumpering
slutdumperly
slutdumpers
sluted
sluter
slutes
sluting
slutkiss
slutkissed
slutkisser
slutkisses
slutkissing
slutkissly
slutkisss
slutly
sluts
slutsed
slutser
slutses
slutsing
slutsly
slutss
smegma
smegmaed
smegmaer
smegmaes
smegmaing
smegmaly
smegmas
smut
smuted
smuter
smutes
smuting
smutly
smuts
smutty
smuttyed
smuttyer
smuttyes
smuttying
smuttyly
smuttys
snatch
snatched
snatcher
snatches
snatching
snatchly
snatchs
sniper
snipered
sniperer
sniperes
snipering
sniperly
snipers
snort
snorted
snorter
snortes
snorting
snortly
snorts
snuff
snuffed
snuffer
snuffes
snuffing
snuffly
snuffs
sodom
sodomed
sodomer
sodomes
sodoming
sodomly
sodoms
spic
spiced
spicer
spices
spicing
spick
spicked
spicker
spickes
spicking
spickly
spicks
spicly
spics
spik
spoof
spoofed
spoofer
spoofes
spoofing
spoofly
spoofs
spooge
spoogeed
spoogeer
spoogees
spoogeing
spoogely
spooges
spunk
spunked
spunker
spunkes
spunking
spunkly
spunks
steamyed
steamyer
steamyes
steamying
steamyly
steamys
stfu
stfued
stfuer
stfues
stfuing
stfuly
stfus
stiffy
stiffyed
stiffyer
stiffyes
stiffying
stiffyly
stiffys
stoneded
stoneder
stonedes
stoneding
stonedly
stoneds
stupided
stupider
stupides
stupiding
stupidly
stupids
suckeded
suckeder
suckedes
suckeding
suckedly
suckeds
sucker
suckes
sucking
suckinged
suckinger
suckinges
suckinging
suckingly
suckings
suckly
sucks
sumofabiatch
sumofabiatched
sumofabiatcher
sumofabiatches
sumofabiatching
sumofabiatchly
sumofabiatchs
tard
tarded
tarder
tardes
tarding
tardly
tards
tawdry
tawdryed
tawdryer
tawdryes
tawdrying
tawdryly
tawdrys
teabagging
teabagginged
teabagginger
teabagginges
teabagginging
teabaggingly
teabaggings
terd
terded
terder
terdes
terding
terdly
terds
teste
testee
testeed
testeeed
testeeer
testeees
testeeing
testeely
testeer
testees
testeing
testely
testes
testesed
testeser
testeses
testesing
testesly
testess
testicle
testicleed
testicleer
testiclees
testicleing
testiclely
testicles
testis
testised
testiser
testises
testising
testisly
testiss
thrusted
thruster
thrustes
thrusting
thrustly
thrusts
thug
thuged
thuger
thuges
thuging
thugly
thugs
tinkle
tinkleed
tinkleer
tinklees
tinkleing
tinklely
tinkles
tit
tited
titer
tites
titfuck
titfucked
titfucker
titfuckes
titfucking
titfuckly
titfucks
titi
titied
titier
tities
titiing
titily
titing
titis
titly
tits
titsed
titser
titses
titsing
titsly
titss
tittiefucker
tittiefuckered
tittiefuckerer
tittiefuckeres
tittiefuckering
tittiefuckerly
tittiefuckers
titties
tittiesed
tittieser
tittieses
tittiesing
tittiesly
tittiess
titty
tittyed
tittyer
tittyes
tittyfuck
tittyfucked
tittyfucker
tittyfuckered
tittyfuckerer
tittyfuckeres
tittyfuckering
tittyfuckerly
tittyfuckers
tittyfuckes
tittyfucking
tittyfuckly
tittyfucks
tittying
tittyly
tittys
toke
tokeed
tokeer
tokees
tokeing
tokely
tokes
toots
tootsed
tootser
tootses
tootsing
tootsly
tootss
tramp
tramped
tramper
trampes
tramping
tramply
tramps
transsexualed
transsexualer
transsexuales
transsexualing
transsexually
transsexuals
trashy
trashyed
trashyer
trashyes
trashying
trashyly
trashys
tubgirl
tubgirled
tubgirler
tubgirles
tubgirling
tubgirlly
tubgirls
turd
turded
turder
turdes
turding
turdly
turds
tush
tushed
tusher
tushes
tushing
tushly
tushs
twat
twated
twater
twates
twating
twatly
twats
twatsed
twatser
twatses
twatsing
twatsly
twatss
undies
undiesed
undieser
undieses
undiesing
undiesly
undiess
unweded
unweder
unwedes
unweding
unwedly
unweds
uzi
uzied
uzier
uzies
uziing
uzily
uzis
vag
vaged
vager
vages
vaging
vagly
vags
valium
valiumed
valiumer
valiumes
valiuming
valiumly
valiums
venous
virgined
virginer
virgines
virgining
virginly
virgins
vixen
vixened
vixener
vixenes
vixening
vixenly
vixens
vodkaed
vodkaer
vodkaes
vodkaing
vodkaly
vodkas
voyeur
voyeured
voyeurer
voyeures
voyeuring
voyeurly
voyeurs
vulgar
vulgared
vulgarer
vulgares
vulgaring
vulgarly
vulgars
wang
wanged
wanger
wanges
wanging
wangly
wangs
wank
wanked
wanker
wankered
wankerer
wankeres
wankering
wankerly
wankers
wankes
wanking
wankly
wanks
wazoo
wazooed
wazooer
wazooes
wazooing
wazooly
wazoos
wedgie
wedgieed
wedgieer
wedgiees
wedgieing
wedgiely
wedgies
weeded
weeder
weedes
weeding
weedly
weeds
weenie
weenieed
weenieer
weeniees
weenieing
weeniely
weenies
weewee
weeweeed
weeweeer
weeweees
weeweeing
weeweely
weewees
weiner
weinered
weinerer
weineres
weinering
weinerly
weiners
weirdo
weirdoed
weirdoer
weirdoes
weirdoing
weirdoly
weirdos
wench
wenched
wencher
wenches
wenching
wenchly
wenchs
wetback
wetbacked
wetbacker
wetbackes
wetbacking
wetbackly
wetbacks
whitey
whiteyed
whiteyer
whiteyes
whiteying
whiteyly
whiteys
whiz
whized
whizer
whizes
whizing
whizly
whizs
whoralicious
whoralicioused
whoraliciouser
whoraliciouses
whoraliciousing
whoraliciously
whoraliciouss
whore
whorealicious
whorealicioused
whorealiciouser
whorealiciouses
whorealiciousing
whorealiciously
whorealiciouss
whored
whoreded
whoreder
whoredes
whoreding
whoredly
whoreds
whoreed
whoreer
whorees
whoreface
whorefaceed
whorefaceer
whorefacees
whorefaceing
whorefacely
whorefaces
whorehopper
whorehoppered
whorehopperer
whorehopperes
whorehoppering
whorehopperly
whorehoppers
whorehouse
whorehouseed
whorehouseer
whorehousees
whorehouseing
whorehousely
whorehouses
whoreing
whorely
whores
whoresed
whoreser
whoreses
whoresing
whoresly
whoress
whoring
whoringed
whoringer
whoringes
whoringing
whoringly
whorings
wigger
wiggered
wiggerer
wiggeres
wiggering
wiggerly
wiggers
woody
woodyed
woodyer
woodyes
woodying
woodyly
woodys
wop
woped
woper
wopes
woping
woply
wops
wtf
wtfed
wtfer
wtfes
wtfing
wtfly
wtfs
xxx
xxxed
xxxer
xxxes
xxxing
xxxly
xxxs
yeasty
yeastyed
yeastyer
yeastyes
yeastying
yeastyly
yeastys
yobbo
yobboed
yobboer
yobboes
yobboing
yobboly
yobbos
zoophile
zoophileed
zoophileer
zoophilees
zoophileing
zoophilely
zoophiles
anal
ass
ass lick
balls
ballsac
bisexual
bleach
causas
cheap
cost of miracles
cunt
display network stats
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gfc
humira AND expensive
illegal
madvocate
masturbation
nuccitelli
overdose
porn
shit
snort
texarkana
direct\-acting antivirals
assistance
ombitasvir
support path
harvoni
abbvie
direct-acting antivirals
paritaprevir
advocacy
ledipasvir
vpak
ritonavir with dasabuvir
program
gilead
greedy
financial
needy
fake-ovir
viekira pak
v pak
sofosbuvir
support
oasis
discount
dasabuvir
protest
ritonavir
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-cleveland-clinic')]
div[contains(@class, 'pane-pub-home-cleveland-clinic')]
div[contains(@class, 'pane-pub-topic-cleveland-clinic')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Hypertrophic cardiomyopathy: A complex disease
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
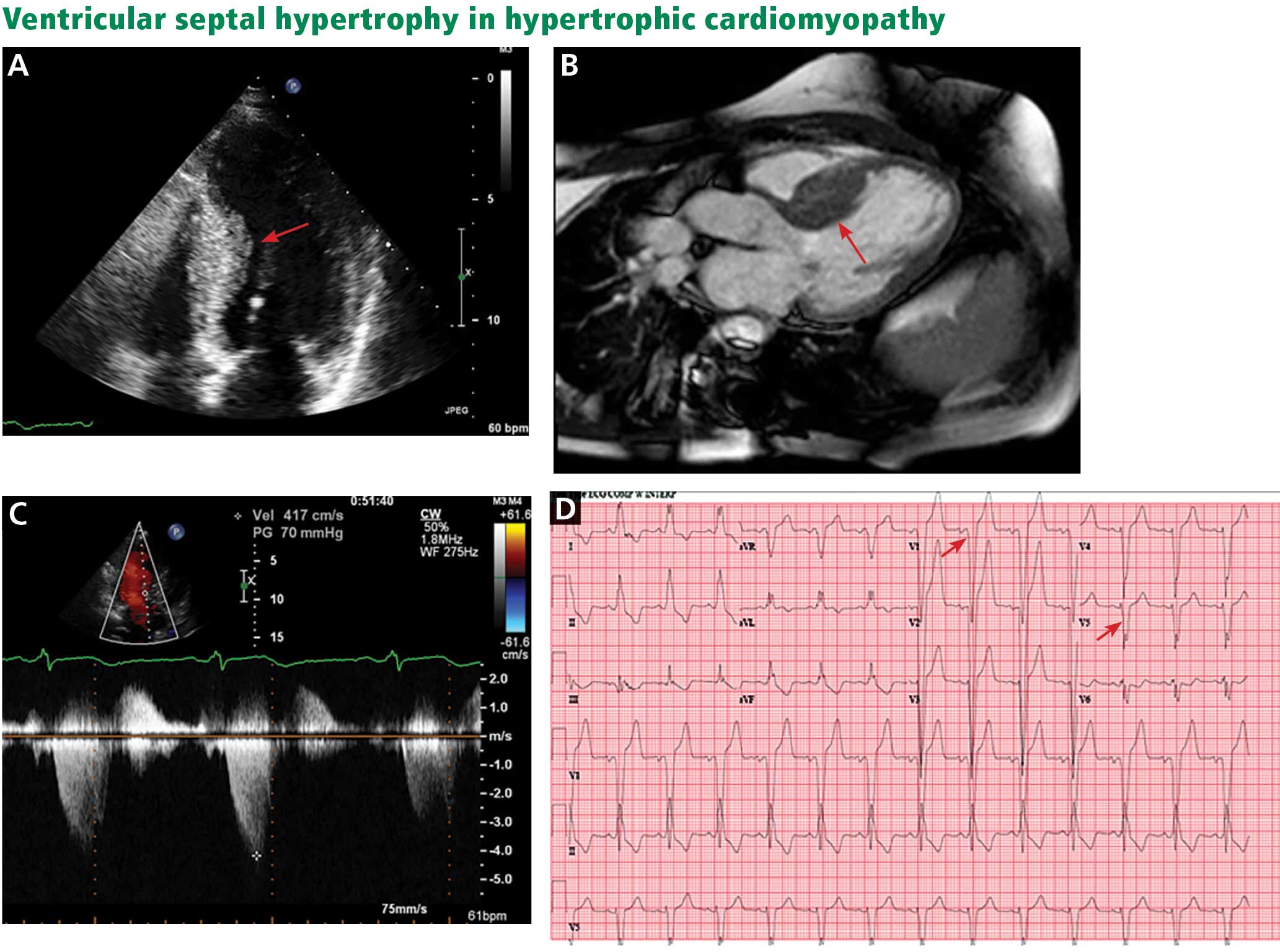
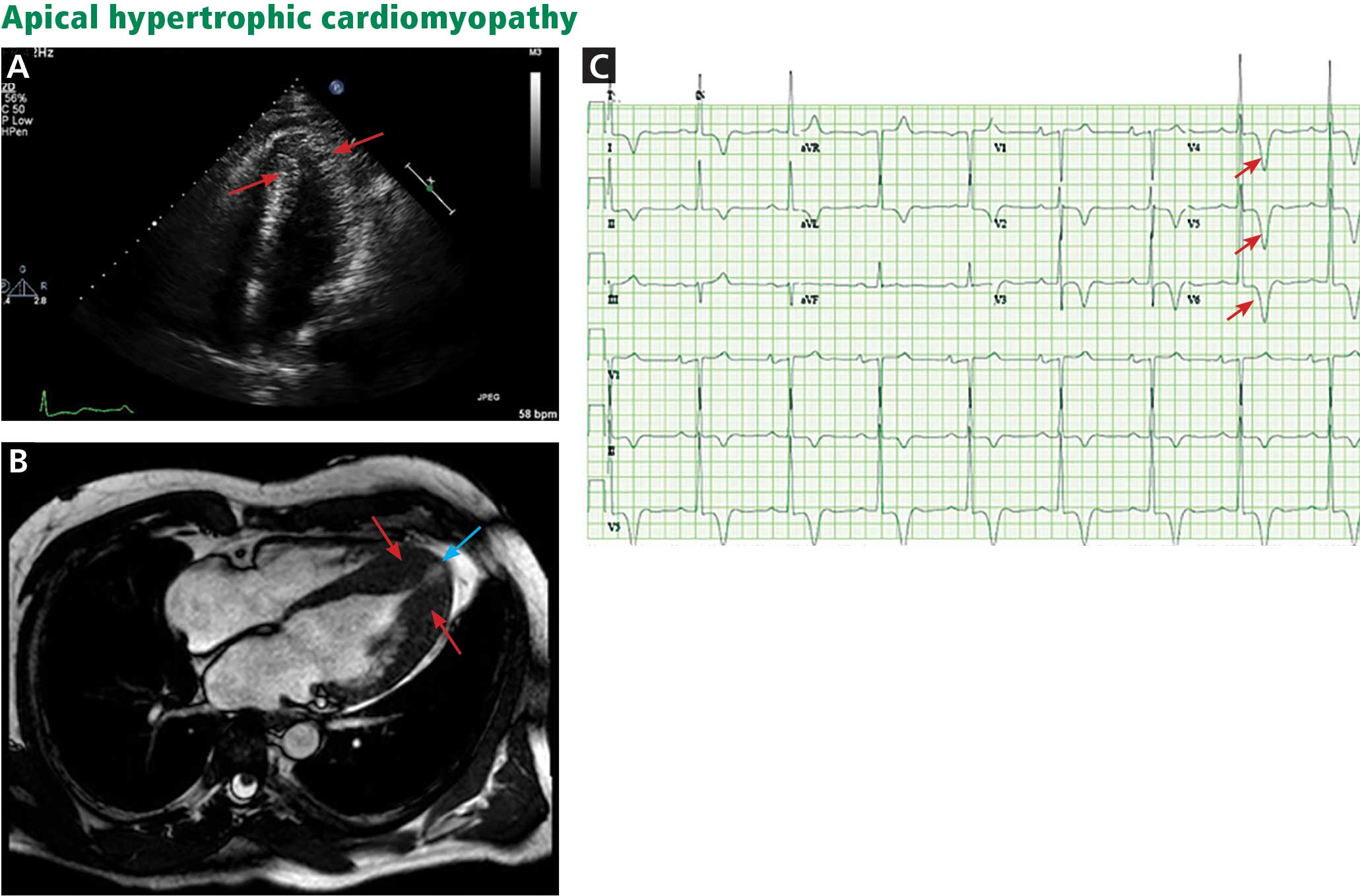
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
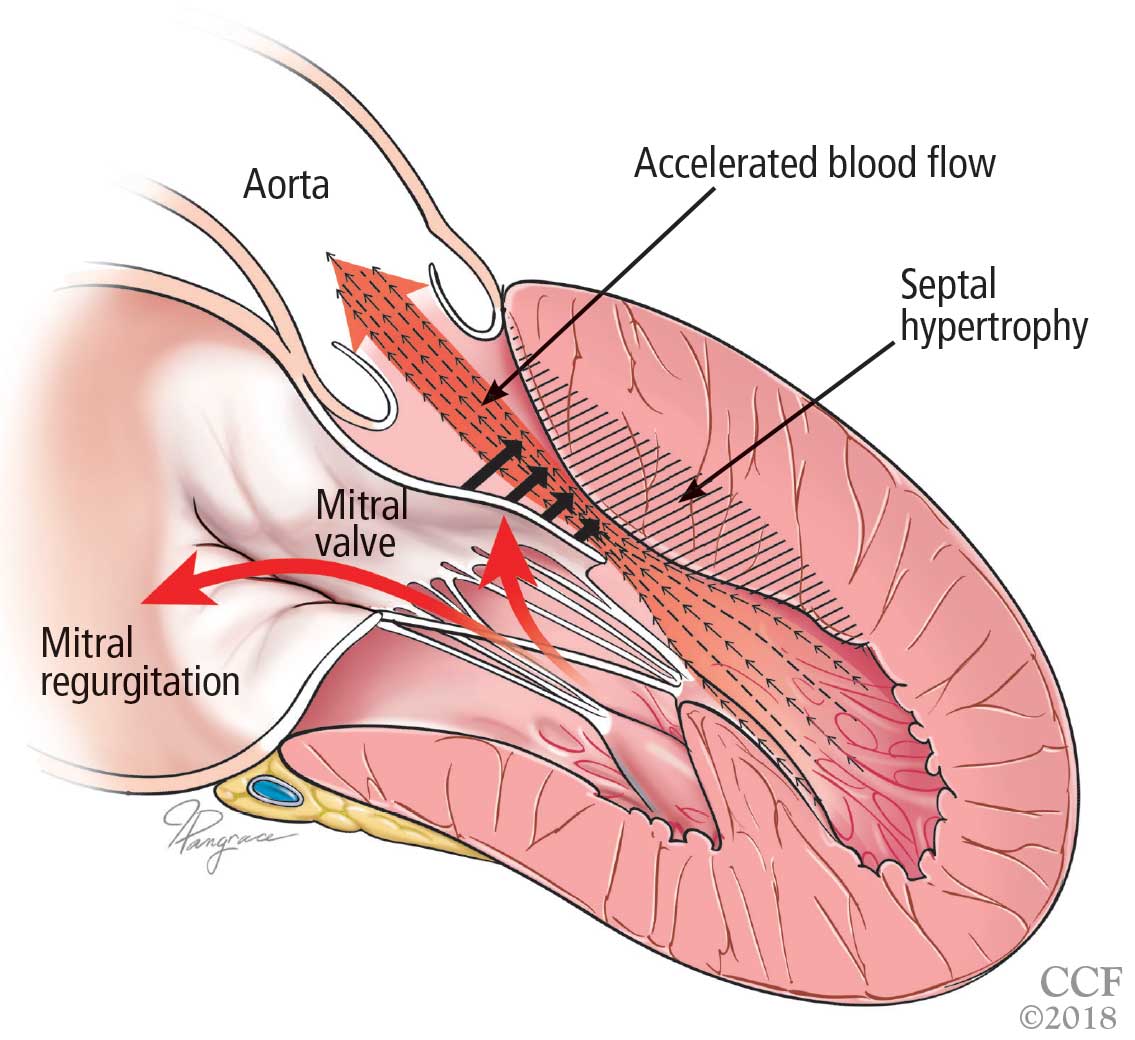
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
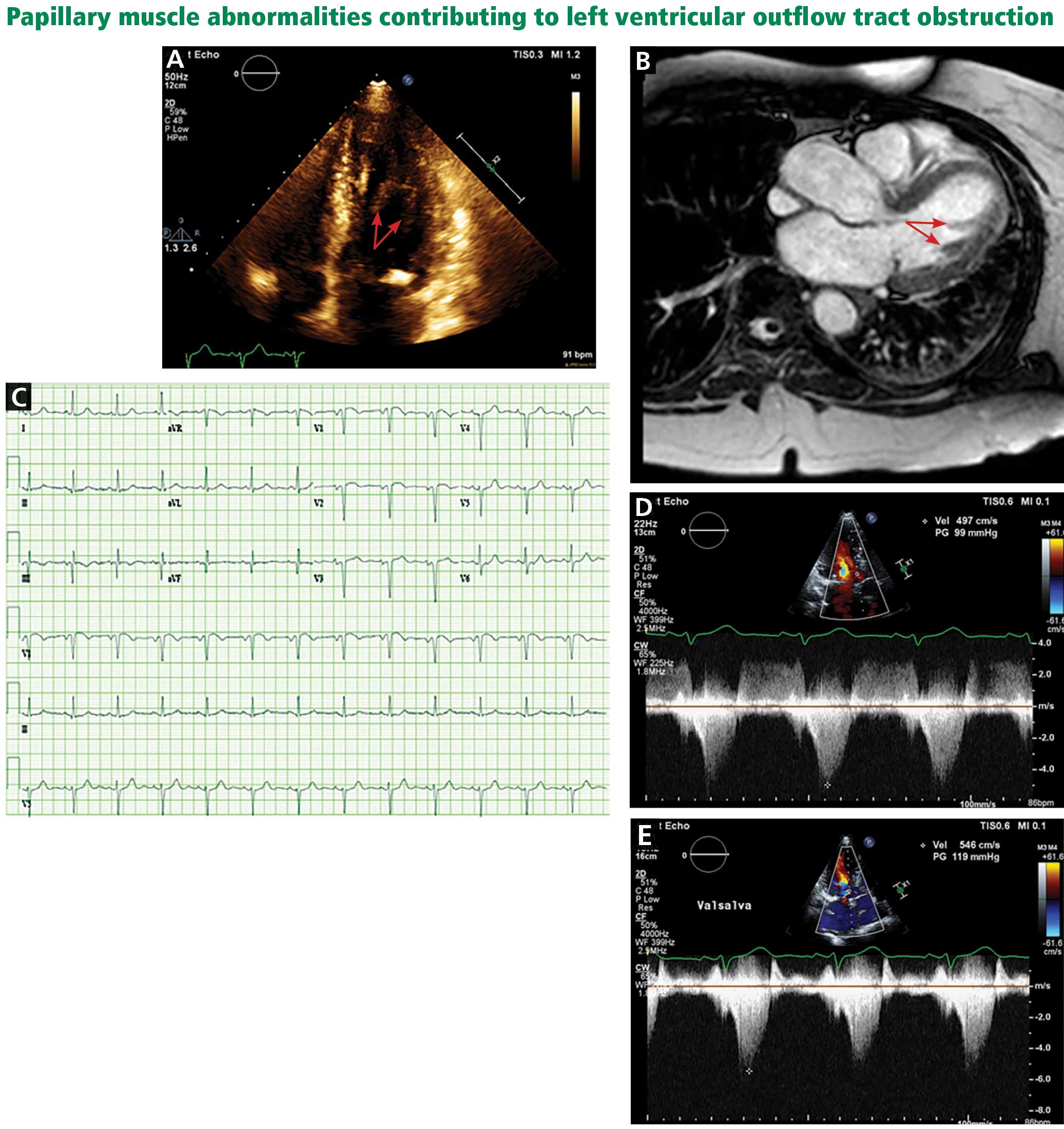
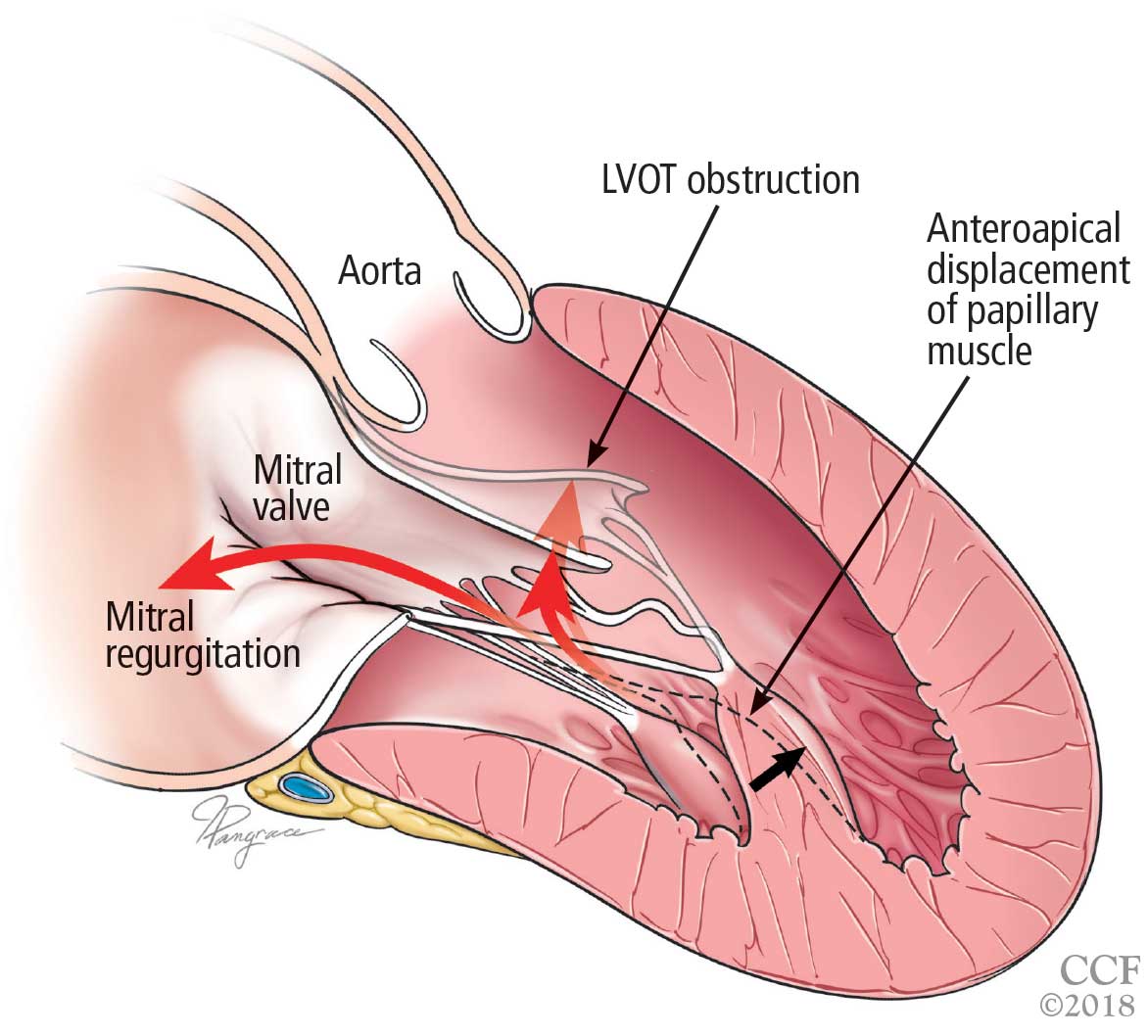
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
Physical findings are nonspecific

It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM

A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models

The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.

Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
Physical findings are nonspecific
It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM
A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models
The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.
Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
Physical findings are nonspecific
It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM
A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models
The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.
Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
KEY POINTS
- Obstruction of the left ventricular outflow tract is a key pathophysiologic mechanism in HCM.
- Because most of the genetic variants that contribute to HCM are autosomal dominant, genetic counseling and testing are suggested for patients and their first-degree relatives.
- Transthoracic echocardiography is the first-line imaging test, followed by magnetic resonance imaging.
- Beta-blockers are the first-line drugs for treating symptoms of HCM.
- An implantable cardioverter-defibrillator can be considered for patients at risk of sudden cardiac death.
- When medical therapy fails or is not tolerated in patients with severe symptoms of obstructive HCM, surgery to reduce the size of the ventricular septum can be considered. Alcohol septal ablation is an alternative.

Idiopathic pulmonary fibrosis: What primary care physicians need to know
Idiopathic pulmonary fibrosis (IPF) is a devastating and fatal lung disease that generally affects older adults. It is characterized by a radiographic and histopathologic pattern of usual interstitial pneumonia (UIP) that has no other known etiology.
Accurate diagnosis of IPF is crucial. We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis, start appropriate therapy, advise the patient on prognosis and enrollment in disease registries and clinical trials, and determine candidacy for lung transplant.
Primary care physicians are uniquely positioned to encounter patients with IPF, whether because of a patient complaint or as an incidental finding on computed tomography. The goal of this article is to delineate the features of IPF so that it may be recognized early and thus expedite referral to a center with expertise in interstitial lung disease for a thorough evaluation and appropriate management.
WHAT IS IDIOPATHIC PULMONARY FIBROSIS?

MORE COMMON THAN ONCE THOUGHT
The true incidence and prevalence of IPF are difficult to assess. IPF is generally considered a rare disease, but it is more common than once thought. In 2011, Raghu et al3 estimated the prevalence in Medicare beneficiaries to be 495 cases per 100,000. Based on this estimate and the current US population, up to 160,000 Americans could have IPF.4 Raghu et al also showed that IPF more often affects adults over age 65, which suggests that as the US population ages, the incidence of IPF may rise. Studies have also reported an increased incidence of IPF worldwide.5
Further, with the rising use of low-dose computed tomography to screen for lung cancer, more incidental cases of IPF will likely be found.6–8
Older data showed a lag from symptom onset to accurate diagnosis of 1 to 2 years.9 A more recent study found a lag in referral of patients with IPF to tertiary care centers, and this delay was associated with a higher rate of death independent of disease severity.10
TYPICALLY PROGRESSIVE, OFTEN FATAL
IPF is typically progressive and limited to the lungs, and it portends a poor prognosis. The median survival is commonly cited as 2 to 5 years from diagnosis, although this is based on older observations that may not reflect current best practice and newer therapies. More recent studies suggest higher survival rates if patients have preserved lung function.11
As the name indicates, the etiology of IPF is unknown, but studies have indicated genetic underpinnings in a notable proportion of cases.12 Regardless of the cause, the pathogenesis and progression of IPF are thought to be the result of an abnormal and persistent wound-repair response. The progressive deposition of scar tissue disrupts normal lung architecture and function, eventually causing clinical disease.13
SYMPTOMS AND KEY FEATURES
Patients with IPF typically present with the insidious onset of dyspnea on exertion, with or without chronic cough. Risk factors include male sex, increasing age, and a history of smoking. Patients with undiagnosed IPF who present with dyspnea and a history of smoking are often treated empirically for chronic obstructive pulmonary disease (COPD).
Rales are a common finding on auscultation in IPF, and this can lead to an exhaustive cardiac evaluation and empiric treatment for heart failure. Digital clubbing is also relatively common.14 Hypoxemia with exertion is another common feature that also often correlates with disease severity and prognosis. Resting hypoxemia is more common in advanced disease.
On spirometry, patients with IPF typically demonstrate restrictive physiology, suggested by a normal or elevated ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1/FVC) (> 70% predicted or above the lower limit of normal) combined with a lower than normal FVC. Restrictive physiology is definitively demonstrated by a decreased total lung capacity (< 80% predicted or below the lower limit of normal) on plethysmography. Impaired gas exchange, manifested by a decreased diffusing capacity of the lungs for carbon monoxide (DLCO) on pulmonary function testing, is also common. Because pulmonary perfusion is higher in the lung bases, where IPF is also predominant, the DLCO is often reduced to a greater extent than the FVC.
PROGNOSTIC INDICATORS

Clinicians typically view IPF as a relentless and progressive process, but its course is variable and can be uncertain in an individual patient (Figure 1).15,16 Nevertheless, over time, most patients have a decline in lung function leading to respiratory failure. Respiratory failure, often preceded by a subacute deterioration (over weeks to months) or an acute deterioration (< 4 weeks), is the most common cause of death, but comorbid diseases such as lung cancer, infection, and heart failure are also common causes of death in these patients.17,18
Predictors of mortality include worsening FVC, DLCO, symptoms, and physiologic impairment, manifested by a decline in the 6-minute walking test or worsening exertional hypoxemia.19–22 Other common comorbidities linked with impaired quality of life and poor prognosis include obstructive sleep apnea, gastroesophageal reflux disease, and depression.16,23 Retrospective studies suggest that most IPF patients die 2 to 5 years after symptom onset. With the lag from symptom onset to final diagnosis, the average life expectancy is as little as 2 years from the time of diagnosis.9,18,24,25
Two staging systems have been developed to predict short-term and long-term mortality risk based on sex, age, and physiologic parameters.23,24 The GAP (gender, age, physiology) index provides an estimate of the risk of death for a cohort of patients: a score of 0 to 8 is calculated, and the score is then categorized as stage I, II, or III. Each stage is associated with 1-, 2-, and 3-year mortality rates, with stage III having the highest rates. The GAP calculator (www.acponline.org/journals/annals/extras/gap) provides an estimate of the risk of death for an individual patient. The application of these tools for the management of IPF is evolving; however, they may be helpful for counseling patients about disease prognosis.
CLUES TO DIAGNOSIS
Histologic patterns
")
UIP on histologic study is also seen in fibrotic lung diseases other than IPF, including connective tissue disease-associated interstitial lung disease, inhalational or occupational interstitial lung disease, and chronic hypersensitivity pneumonitis.26–29 Consequently, the diagnosis of IPF requires exclusion of other known causes of UIP.
According to the 2011 guidelines,16 the histology of interstitial lung disease can be categorized as definite UIP, probable UIP, or possible UIP, or as an atypical pattern suggesting another diagnosis. If no definite cause of the interstitial lung abnormality is found, the level of certainty of the histopathologic pattern of UIP helps formulate the clinical diagnosis and management plan.
Clues on computed tomography
The UIP nomenclature also describes patterns on high-resolution computed tomography (HRCT). HRCT is done without contrast and produces thin-sliced images (usually < 1.5 mm) in inspiratory, expiratory, and prone views; this allows detection of air trapping, which may indicate an airway-centric alternative diagnosis.
On HRCT, UIP appears as reticular opacities, often with traction bronchiectasis or bronchiolectasis, usually with a basilar and peripheral predominance. Honeycombing is a key feature and appears as clustered cystic spaces with well-defined walls in the periphery of the lung parenchyma. Ground-glass opacities are not a prominent feature of UIP, and although they do not exclude a UIP pattern, they should spur consideration of other diagnoses.16 Reactive mediastinal and hilar lymphadenopathy is another common feature of UIP.

When evaluating results of HRCT for UIP, the radiologist categorizes the pattern as definite UIP, possible UIP, or inconsistent. The definite pattern meets all the above features and has none of the features suggesting an alternative diagnosis (Figure 3). The possible pattern includes all the above features with the exception of honeycombing. If the predominant features on HRCT include any atypical finding listed above, then the study is considered inconsistent with UIP. If the pattern on HRCT is considered definite, evaluation of pathology is not necessary. If the pattern is categorized as possible or is inconsistent, then surgical lung biopsy-confirmed UIP is necessary for the definitive diagnosis of IPF.
However, evidence is emerging that in the correct clinical scenario, possible UIP behaves similarly to definite UIP and may be sufficient to make the clinical diagnosis of IPF even without surgical biopsy confirmation.30
A DIAGNOSTIC ALGORITHM FOR IPF
Given the multitude of interstitial lung diseases, their complexities, and the lack of a gold standard definitive diagnostic test, the diagnosis of IPF can be difficult, requiring the integration of clinical, radiologic, and, if necessary, pathologic findings.
")
Demographic features
The patient’s demographic features and risk factors dictate the initial clinical suspicion of IPF compared with other interstitial lung diseases. The incidence of IPF increases with age, and IPF is more common in men. A history of smoking is another risk factor.31 A 45-year-old never-smoker is much less likely to have IPF than a 70-year-old former smoker, and a 70-year-old man is more likely to have IPF than a woman of the same age. Thus, the finding of interstitial lung disease in a patient with a demographic profile that is not typical (ie, a younger woman who never smoked) should prompt an exhaustive investigation for another diagnosis such as hypersensitivity pneumonitis or connective tissue disease.
Key elements of the history
After considering the demographic profile and risk factors, the next step in the evaluation is a thorough and accurate medical history. This should include assessment of the severity of dyspnea and cough, signs and symptoms of connective tissue disease (eg, arthralgias, sicca symptoms, Raynaud phenomenon, difficulty swallowing), and gastroesophageal reflux disease, which can be associated with connective tissue disease and, independently, with IPF.
It is also important to identify any environmental exposures that suggest pneumoconiosis or chronic hypersensitivity pneumonitis. The most common risk factors for hypersensitivity pneumonitis are birds and bird feathers, molds, fungi, hot tub use, and some industrial chemicals.32
A medication history is important. Many medications are associated with interstitial lung disease, but amiodarone, bleomycin, methotrexate, and nitrofurantoin are among the common offenders.33
A thorough family history is necessary, as there are familial forms of IPF.
Focus of the physical examination
The physical examination must include careful auscultation for rales. While rales are not specific for IPF, they are the most common pulmonary abnormality. Detailed skin, musculoskeletal, and cardiovascular examinations are also important to evaluate for rheumatologic signs, clubbing, or evidence of heart failure or pulmonary hypertension.
Laboratory tests
Laboratory testing should include a serologic autoantibody panel to evaluate for connective tissue diseases that can manifest as interstitial lung disease, including rheumatoid arthritis, dermatopolymyositis, scleroderma, Sjögren syndrome, and undifferentiated or mixed connective tissue disease. Typical preliminary laboratory tests are antinuclear antibody, rheumatoid factor, erythrocyte sedimentation rate, and C-reactive protein. Others may include anticyclic citrullinated peptide (anti-CCP), anti-Scl-70, anti-RNP, anti-SS-A, anti-SS-B, and anti-Jo-1.16 The breadth of the panel should depend on patient demographics and findings in the history or physical examination that increase or decrease the likelihood of a connective tissue disease.
Lung function testing
Assessing the patient’s pulmonary physiology should include spirometry, DLCO, and body plethysmography (lung volumes). In most cases, IPF manifests with restrictive physiology. Once restrictive physiology is confirmed with a low total lung capacity, FVC testing can be used as a longitudinal surrogate, as it is less expensive and easier for the patient to perform. In general, a lower total lung capacity or FVC indicates more severe impairment.
The DLCO serves as another marker of severity but is less reliable due to baseline variability and difficulties performing the maneuver.
A 6-minute walk test is another crucial physiologic assessment tool that can quantify exertional hypoxemia and functional status (ie, distance walked), and can assist in prognosis.
Imaging
Most patients undergo chest radiography in the workup for undiagnosed dyspnea. However, chest radiography is not adequate to formulate an accurate diagnosis in suspected interstitial lung disease, and a normal radiograph cannot exclude changes that might reflect early phases of the disease. As the disease progresses, the plain radiograph can show reticulonodular opacities and honeycombing in the peripheral and lower lung zones (Figure 3).34
The decision whether to order HRCT in the workup for a patient who has dyspnea and a normal chest radiograph is challenging. We recommend cross-sectional imaging when physiologic testing shows restriction or low DLCO, or when there is a high index of suspicion for underlying lung disease as the cause of symptoms.
Expert consultation can aid with this decision, especially when the underlying cause of dyspnea remains unclear after initial studies have been completed. Otherwise, HRCT is an essential test in the evaluation of interstitial lung disease.
Bronchoscopy’s role controversial
If the pattern on HRCT is nondiagnostic, then surgical biopsy is necessary, and the diagnosis of IPF requires a histologic pattern of UIP as described above.16,35
Although bronchoscopy can be valuable if an alternative diagnosis such as sarcoidosis or chronic hypersensitivity pneumonitis is suspected, the role of bronchoscopic biopsy in the workup of IPF is controversial. The patchy nature of UIP does not lend itself to the relatively small biopsy samples obtained through bronchoscopy.36,37
Surgical biopsy options
The favored biopsy approach is surgical, using either an open or a video-assisted thoracoscopic technique. The latter is preferred as it is less invasive, requires a shorter length of hospital stay, and allows a faster recovery.38 Bronchoscopic cryobiopsy, currently under investigation, is a potentially valuable tool whose role in diagnosing IPF is evolving.
Frequently, neither HRCT nor surgical lung biopsy demonstrates UIP, making the definitive diagnosis of IPF difficult. Moreover, some patients with nondiagnostic HRCT results are unable to tolerate surgical lung biopsy because of severely impaired lung function or other comorbidities.
The role of multidisciplinary discussions
When surgical lung biopsy is not possible, current practice at leading centers uses a multidisciplinary approach to allow for a confident diagnosis.30,39 Discussions take place between pulmonologists, pathologists, radiologists, and other specialists to collectively consider all aspects of a case before rendering a consensus opinion on the diagnosis and the management plan. If the discussion leads to a consensus diagnosis of IPF, then the patient’s clinician can move forward with treatment options. If not, the group can recommend further workup or alternative diagnoses and treatment regimens. The multidisciplinary group is also well positioned to consider the relative risks and benefits of moving forward with surgical lung biopsy for individual patients.
This approach illustrates the importance of referring these patients to centers of excellence in diagnosing and managing complex cases of interstitial lung disease, including IPF.40
TREATMENT OF IPF
Antifibrotic therapy
Antifibrotic therapy is a choice between pirfenidone and nintedanib.
Pirfenidone, which has an undefined molecular target, was approved based on the results of 3 trials.41,42 Pooled analyses from these trials showed a reduction in the decline from baseline in FVC percent predicted and improved progression-free survival.43 Pooled and meta-analyses of pirfenidone clinical trials have shown a mortality benefit, although no individual study has shown such an effect on mortality rates.44
The major adverse effects of pirfenidone are gastrointestinal distress and photosensitivity rash.
Nintedanib is a triple tyrosine kinase inhibitor that broadly targets fibroblast growth factor, vascular endothelial growth factor, and platelet-derived growth factor receptors. Combined analysis of 2 concurrent trials45 showed that nintedanib reduced the decline in FVC, similarly to pirfenidone. The major adverse event associated with nintedanib was diarrhea. Since it inhibits vascular endothelial growth factor, there is a risk of hematologic complications such as bleeding or clotting events.
Because pirfenidone and nintedanib can increase aminotransferase levels, regular monitoring is recommended.
To date, no trial has compared pirfenidone and nintedanib in terms of their efficacy and tolerability. Therefore, the choice of agent is based on the patient’s preference after a discussion of potential risks and expected benefits, a review of each drug’s side effects, and consideration of comorbid conditions and physician experience.
Patients need to understand that these drugs slow the rate of decline in FVC but have not been shown to improve symptoms or functional status.
Corticosteroids are not routine
Corticosteroids should not be used routinely in the treatment of IPF. Although steroids, alone or in combination with other immunosuppressive medications, were commonly used for IPF in the past, such use was not based on results of randomized controlled trials.46 Retrospective controlled studies have failed to show that corticosteroids improve mortality rates in IPF; indeed, they have shown that corticosteroids confer substantial morbidity.47,48 In addition, a randomized controlled trial combining corticosteroids with N-acetylcysteine and azathioprine was stopped early due to an increased risk of death and hospitalization.49 Collectively, these data suggest that corticosteroids confer no benefit and are potentially harmful. Their use in IPF is discouraged, and the joint international guidelines recommend against immunosuppression to treat IPF.16
Other treatments
The guidelines offer additional suggestions for the management of IPF.
Preliminary evidence suggests that microaspiration associated with abnormal gastroesophageal acid reflux is a risk factor for IPF. As such, there is a weak recommendation for aggressive treatment of reflux disease.50 However, because evidence suggests that proton-pump inhibitor therapy may be associated with adverse renal or central nervous system effects, this recommendation bears caution. It is hoped that ongoing studies will provide further insight into the role of acid-suppression in the management of IPF.51,52
Further treatment recommendations include best supportive management such as supplemental oxygen, pulmonary rehabilitation, and vaccinations.

Prompt referral for lung transplant is imperative. IPF is now the most common indication for lung transplant, and given the poor overall prognosis of advanced IPF, transplant confers a survival benefit in appropriately selected patients.53,54 Table 2 provides an evidence-based checklist for the workup and management of IPF.
ACUTE EXACERBATIONS OF IPF
The unpredictable nature of IPF can manifest in the form of acute exacerbations without an identifiable cause. The loosely defined diagnostic criteria for the diagnosis of acute exacerbations are a previous or new diagnosis of IPF, worsening or development of dyspnea in the last 30 days, and new bilateral ground-glass or consolidative changes with a background of UIP on HRCT.16
A new definition has been proposed55 to facilitate research in the characterization and treatment of acute exacerbations of IPF. The new definition includes all causes of respiratory deterioration except for heart failure and volume overload. It is less strict about the 30-day time frame. This newer definition is based on the lack of evidence differentiating outcomes when an acute deterioration is associated with known or unknown etiologies.55
The incidence of acute exacerbations is variable, with a 1- and 3-year incidence ranging between 8.6% and 23.9% depending on the criteria used.56 In general, acute exacerbations carry a grim prognosis, with a median life expectancy of 2.2 months.57
There is no approved therapy for exacerbations of IPF. Rather, treatment is mainly supportive with supplemental oxygen and mechanical ventilation. Current guidelines have a weak recommendation for the use of corticosteroids, but there are no recommendations regarding dose, route, or duration of therapy. Other treatments, primarily immunomodulatory agents, have been suggested but lack evidence of benefit.
Acknowledgments: Pathology images were provided by Carol Farver, MD, Pathology Institute, Cleveland Clinic. Radiology images were provided by Ruchi Yadav, MD, Imaging Institute, Cleveland Clinic.
- Brown KK, Raghu G. Medical treatment for pulmonary fibrosis: current trends, concepts, and prospects. Clin Chest Med 2004; 25(4):759–772, vii. doi:10.1016/j.ccm.2004.08.003
- Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 2013; 19(5):453–459. doi:10.1097/MCP.0b013e328363f48d
- Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014; 2(7):566–572. doi:10.1016/S2213-2600(14)70101-8
- Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; 21(126):355–361. doi:10.1183/09059180.00002512
- Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46(3):795–806. doi:10.1183/09031936.00185114
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268(2):563–571. doi:10.1148/radiol.13120816
- Southern BD, Scheraga RG, Yadav R. Managing interstitial lung disease detected on CT during lung cancer screening. Cleve Clin J Med 2016; 83(1):55–65. doi:10.3949/ccjm.83a.14157
- King TE Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001; 164(5):1025–1032. doi:10.1164/ajrccm.164.6.2001056
- Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184(7):842–847. doi:10.1164/rccm.201104-0668OC
- Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med 2018; 18(1):19. doi:10.1186/s12890-018-0575-y
- Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res 2015; 165(1):48–60. doi:10.1016/j.trsl.2014.03.011
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378(9807):1949–1961. doi:10.1016/S0140-6736(11)60052-4
- Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis 2008; 3:8. doi:10.1186/1750-1172-3-8
- Raghu G. Idiopathic pulmonary fibrosis. A rational clinical approach. Chest 1987; 92(1):148–154. doi:10.1378/chest.92.1.148
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Panos RJ, Mortenson RL, Niccoli SA, King TE Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med 1990; 88(4):396–404. doi:10.1016/0002-9343(90)90495-Y
- Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183(4):431–440. doi:10.1164/rccm.201006-0894CI
- Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003; 168(5):538–542. doi:10.1164/rccm.200211-1311OC
- Flaherty KR, Andrei AC, Murray S, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174(7):803–809. doi:10.1164/rccm.200604-488OC
- Jegal Y, Kim DS, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171(6):639–644. doi:10.1164/rccm.200403-331OC
- Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003; 168(5):531–537. doi:10.1164/rccm.200210-1245OC
- King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med 2017; 5(1):72–84. doi:10.1016/S2213-2600(16)30222-3
- Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156(1):684–691. doi:10.7326/0003-4819-156-10-201205150-00004
- Rudd RM, Prescott RJ, Chalmers JC, Johnston ID; Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. British Thoracic Society study on cryptogenic fibrosing alveolitis: response to treatment and survival. Thorax 2007; 62(1):62–66. doi:10.1136/thx.2005.045591
- Gutsche M, Rosen GD, Swigris JJ. Connective tissue disease-associated interstitial lung disease: a review. Curr Respir Care Rep 2012; 1:224–232. doi:10.1007/s13665-012-0028-7
- Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007; 175(7):705–711. doi:10.1164/rccm.200607-912OC
- Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006; 3(4):293–298. doi:10.1513/pats.200512-131TK
- Vourlekis JS, Schwarz MI, Cherniack RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med 2004; 116(10):662–668. doi:10.1016/j.amjmed.2003.12.030
- Brownell R, Moua T, Henry TS, et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax 2017; 72(5):424–429. doi:10.1136/thoraxjnl-2016-209671
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997; 155(1):242–248. doi:10.1164/ajrccm.155.1.9001319
- Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012; 186(4):314–324. doi:10.1164/rccm.201203-0513CI
- Schwaiblmair M, Behr W, Haeckel T, Markl B, Foerg W, Berghaus T. Drug induced interstitial lung disease. Open Respir Med J 2012; 6:63–74. doi:10.2174/1874306401206010063
- Grenier P, Valeyre D, Cluzel P, Brauner MW, Lenoir S, Chastang C. Chronic diffuse interstitial lung disease: diagnostic value of chest radiography and high-resolution CT. Radiology 1991; 179(1):123–132. doi:10.1148/radiology.179.1.2006262
- Lynch JP 3rd, Huynh RH, Fishbein MC, Saggar R, Belperio JA, Weigt SS. Idiopathic pulmonary fibrosis: epidemiology, clinical features, prognosis, and management. Semin Respir Crit Care Med 2016; 37(3):331–357. doi:10.1055/s-0036-1582011
- Berbescu EA, Katzenstein AL, Snow JL, Zisman DA. Transbronchial biopsy in usual interstitial pneumonia. Chest 2006; 129(5):1126–1131. doi:10.1378/chest.129.5.1126
- Ohshimo S, Bonella F, Cui A, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179(11):1043–1047. doi:10.1164/rccm.200808-1313OC
- Oparka J, Yan TD, Ryan E, Dunning J. Does video-assisted thoracic surgery provide a safe alternative to conventional techniques in patients with limited pulmonary function who are otherwise suitable for lung resection? Interact Cardiovasc Thorac Surg 2013; 17(1):159–162. doi:10.1093/icvts/ivt097
- Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170(8):904–910. doi:10.1164/rccm.200402-147OC
- Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med 2016; 4(7):557–565. doi:10.1016/S2213-2600(16)30033-9
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779):1760–1769. doi:10.1016/S0140-6736(11)60405-4
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016; 47(1):243–253. doi:10.1183/13993003.00026-2015
- Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5(1):33–41. doi:10.1016/S2213-2600(16)30326-5
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Richeldi L, Davies HR, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2003: 3:CD002880. doi:10.1002/14651858.CD002880
- Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med 2000; 161(4 pt 1):1172–1178. doi:10.1164/ajrccm.161.4.9907002
- Gay SE, Kazerooni EA, Toews GB, et al. Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Crit Care Med 1998; 157(4 pt 1):1063–1072. doi:10.1164/ajrccm.157.4.9703022
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J 2006; 27(1):136–142. doi:10.1183/09031936.06.00037005
- Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia: a pharmacoepidemiological claims data analysis. JAMA Neurol 2016; 73(4):410–416. doi:10.1001/jamaneurol.2015.4791
- Xie Y, Bowe B, Li T, Xian H, Balasubramanian S, Al-Aly Z. Proton pump inhibitors and risk of incident CKD and progression to ESRD. J Am Soc Nephrol 2016; 27(10):3153–3163. doi:10.1681/ASN.2015121377
- Thabut G, Mal H, Castier Y, et al. Survival benefit of lung transplantation for patients with idiopathic pulmonary fibrosis. J Thorac Cardiovasc Surg 2003; 126(2):469–475. doi:10.1016/S0022-5223(03)00600-7
- Valapour M, Skeans MA, Smith JM, et al. Lung. Am J Transplant 2016; 16(suppl 2):141–168. doi:10.1111/ajt.13671
- Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med 2016; 194(3):265–275. doi:10.1164/rccm.201604-0801CI
- Kondoh Y, Taniguchi H, Katsuta T, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27(2):103–110. doi:10.1016/j.resinv.2015.04.005
- Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37(2):356–363. doi:10.1183/09031936.00159709
Idiopathic pulmonary fibrosis (IPF) is a devastating and fatal lung disease that generally affects older adults. It is characterized by a radiographic and histopathologic pattern of usual interstitial pneumonia (UIP) that has no other known etiology.
Accurate diagnosis of IPF is crucial. We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis, start appropriate therapy, advise the patient on prognosis and enrollment in disease registries and clinical trials, and determine candidacy for lung transplant.
Primary care physicians are uniquely positioned to encounter patients with IPF, whether because of a patient complaint or as an incidental finding on computed tomography. The goal of this article is to delineate the features of IPF so that it may be recognized early and thus expedite referral to a center with expertise in interstitial lung disease for a thorough evaluation and appropriate management.
WHAT IS IDIOPATHIC PULMONARY FIBROSIS?
MORE COMMON THAN ONCE THOUGHT
The true incidence and prevalence of IPF are difficult to assess. IPF is generally considered a rare disease, but it is more common than once thought. In 2011, Raghu et al3 estimated the prevalence in Medicare beneficiaries to be 495 cases per 100,000. Based on this estimate and the current US population, up to 160,000 Americans could have IPF.4 Raghu et al also showed that IPF more often affects adults over age 65, which suggests that as the US population ages, the incidence of IPF may rise. Studies have also reported an increased incidence of IPF worldwide.5
Further, with the rising use of low-dose computed tomography to screen for lung cancer, more incidental cases of IPF will likely be found.6–8
Older data showed a lag from symptom onset to accurate diagnosis of 1 to 2 years.9 A more recent study found a lag in referral of patients with IPF to tertiary care centers, and this delay was associated with a higher rate of death independent of disease severity.10
TYPICALLY PROGRESSIVE, OFTEN FATAL
IPF is typically progressive and limited to the lungs, and it portends a poor prognosis. The median survival is commonly cited as 2 to 5 years from diagnosis, although this is based on older observations that may not reflect current best practice and newer therapies. More recent studies suggest higher survival rates if patients have preserved lung function.11
As the name indicates, the etiology of IPF is unknown, but studies have indicated genetic underpinnings in a notable proportion of cases.12 Regardless of the cause, the pathogenesis and progression of IPF are thought to be the result of an abnormal and persistent wound-repair response. The progressive deposition of scar tissue disrupts normal lung architecture and function, eventually causing clinical disease.13
SYMPTOMS AND KEY FEATURES
Patients with IPF typically present with the insidious onset of dyspnea on exertion, with or without chronic cough. Risk factors include male sex, increasing age, and a history of smoking. Patients with undiagnosed IPF who present with dyspnea and a history of smoking are often treated empirically for chronic obstructive pulmonary disease (COPD).
Rales are a common finding on auscultation in IPF, and this can lead to an exhaustive cardiac evaluation and empiric treatment for heart failure. Digital clubbing is also relatively common.14 Hypoxemia with exertion is another common feature that also often correlates with disease severity and prognosis. Resting hypoxemia is more common in advanced disease.
On spirometry, patients with IPF typically demonstrate restrictive physiology, suggested by a normal or elevated ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1/FVC) (> 70% predicted or above the lower limit of normal) combined with a lower than normal FVC. Restrictive physiology is definitively demonstrated by a decreased total lung capacity (< 80% predicted or below the lower limit of normal) on plethysmography. Impaired gas exchange, manifested by a decreased diffusing capacity of the lungs for carbon monoxide (DLCO) on pulmonary function testing, is also common. Because pulmonary perfusion is higher in the lung bases, where IPF is also predominant, the DLCO is often reduced to a greater extent than the FVC.
PROGNOSTIC INDICATORS
Clinicians typically view IPF as a relentless and progressive process, but its course is variable and can be uncertain in an individual patient (Figure 1).15,16 Nevertheless, over time, most patients have a decline in lung function leading to respiratory failure. Respiratory failure, often preceded by a subacute deterioration (over weeks to months) or an acute deterioration (< 4 weeks), is the most common cause of death, but comorbid diseases such as lung cancer, infection, and heart failure are also common causes of death in these patients.17,18
Predictors of mortality include worsening FVC, DLCO, symptoms, and physiologic impairment, manifested by a decline in the 6-minute walking test or worsening exertional hypoxemia.19–22 Other common comorbidities linked with impaired quality of life and poor prognosis include obstructive sleep apnea, gastroesophageal reflux disease, and depression.16,23 Retrospective studies suggest that most IPF patients die 2 to 5 years after symptom onset. With the lag from symptom onset to final diagnosis, the average life expectancy is as little as 2 years from the time of diagnosis.9,18,24,25
Two staging systems have been developed to predict short-term and long-term mortality risk based on sex, age, and physiologic parameters.23,24 The GAP (gender, age, physiology) index provides an estimate of the risk of death for a cohort of patients: a score of 0 to 8 is calculated, and the score is then categorized as stage I, II, or III. Each stage is associated with 1-, 2-, and 3-year mortality rates, with stage III having the highest rates. The GAP calculator (www.acponline.org/journals/annals/extras/gap) provides an estimate of the risk of death for an individual patient. The application of these tools for the management of IPF is evolving; however, they may be helpful for counseling patients about disease prognosis.
CLUES TO DIAGNOSIS
Histologic patterns
UIP on histologic study is also seen in fibrotic lung diseases other than IPF, including connective tissue disease-associated interstitial lung disease, inhalational or occupational interstitial lung disease, and chronic hypersensitivity pneumonitis.26–29 Consequently, the diagnosis of IPF requires exclusion of other known causes of UIP.
According to the 2011 guidelines,16 the histology of interstitial lung disease can be categorized as definite UIP, probable UIP, or possible UIP, or as an atypical pattern suggesting another diagnosis. If no definite cause of the interstitial lung abnormality is found, the level of certainty of the histopathologic pattern of UIP helps formulate the clinical diagnosis and management plan.
Clues on computed tomography
The UIP nomenclature also describes patterns on high-resolution computed tomography (HRCT). HRCT is done without contrast and produces thin-sliced images (usually < 1.5 mm) in inspiratory, expiratory, and prone views; this allows detection of air trapping, which may indicate an airway-centric alternative diagnosis.
On HRCT, UIP appears as reticular opacities, often with traction bronchiectasis or bronchiolectasis, usually with a basilar and peripheral predominance. Honeycombing is a key feature and appears as clustered cystic spaces with well-defined walls in the periphery of the lung parenchyma. Ground-glass opacities are not a prominent feature of UIP, and although they do not exclude a UIP pattern, they should spur consideration of other diagnoses.16 Reactive mediastinal and hilar lymphadenopathy is another common feature of UIP.
When evaluating results of HRCT for UIP, the radiologist categorizes the pattern as definite UIP, possible UIP, or inconsistent. The definite pattern meets all the above features and has none of the features suggesting an alternative diagnosis (Figure 3). The possible pattern includes all the above features with the exception of honeycombing. If the predominant features on HRCT include any atypical finding listed above, then the study is considered inconsistent with UIP. If the pattern on HRCT is considered definite, evaluation of pathology is not necessary. If the pattern is categorized as possible or is inconsistent, then surgical lung biopsy-confirmed UIP is necessary for the definitive diagnosis of IPF.
However, evidence is emerging that in the correct clinical scenario, possible UIP behaves similarly to definite UIP and may be sufficient to make the clinical diagnosis of IPF even without surgical biopsy confirmation.30
A DIAGNOSTIC ALGORITHM FOR IPF
Given the multitude of interstitial lung diseases, their complexities, and the lack of a gold standard definitive diagnostic test, the diagnosis of IPF can be difficult, requiring the integration of clinical, radiologic, and, if necessary, pathologic findings.
Demographic features
The patient’s demographic features and risk factors dictate the initial clinical suspicion of IPF compared with other interstitial lung diseases. The incidence of IPF increases with age, and IPF is more common in men. A history of smoking is another risk factor.31 A 45-year-old never-smoker is much less likely to have IPF than a 70-year-old former smoker, and a 70-year-old man is more likely to have IPF than a woman of the same age. Thus, the finding of interstitial lung disease in a patient with a demographic profile that is not typical (ie, a younger woman who never smoked) should prompt an exhaustive investigation for another diagnosis such as hypersensitivity pneumonitis or connective tissue disease.
Key elements of the history
After considering the demographic profile and risk factors, the next step in the evaluation is a thorough and accurate medical history. This should include assessment of the severity of dyspnea and cough, signs and symptoms of connective tissue disease (eg, arthralgias, sicca symptoms, Raynaud phenomenon, difficulty swallowing), and gastroesophageal reflux disease, which can be associated with connective tissue disease and, independently, with IPF.
It is also important to identify any environmental exposures that suggest pneumoconiosis or chronic hypersensitivity pneumonitis. The most common risk factors for hypersensitivity pneumonitis are birds and bird feathers, molds, fungi, hot tub use, and some industrial chemicals.32
A medication history is important. Many medications are associated with interstitial lung disease, but amiodarone, bleomycin, methotrexate, and nitrofurantoin are among the common offenders.33
A thorough family history is necessary, as there are familial forms of IPF.
Focus of the physical examination
The physical examination must include careful auscultation for rales. While rales are not specific for IPF, they are the most common pulmonary abnormality. Detailed skin, musculoskeletal, and cardiovascular examinations are also important to evaluate for rheumatologic signs, clubbing, or evidence of heart failure or pulmonary hypertension.
Laboratory tests
Laboratory testing should include a serologic autoantibody panel to evaluate for connective tissue diseases that can manifest as interstitial lung disease, including rheumatoid arthritis, dermatopolymyositis, scleroderma, Sjögren syndrome, and undifferentiated or mixed connective tissue disease. Typical preliminary laboratory tests are antinuclear antibody, rheumatoid factor, erythrocyte sedimentation rate, and C-reactive protein. Others may include anticyclic citrullinated peptide (anti-CCP), anti-Scl-70, anti-RNP, anti-SS-A, anti-SS-B, and anti-Jo-1.16 The breadth of the panel should depend on patient demographics and findings in the history or physical examination that increase or decrease the likelihood of a connective tissue disease.
Lung function testing
Assessing the patient’s pulmonary physiology should include spirometry, DLCO, and body plethysmography (lung volumes). In most cases, IPF manifests with restrictive physiology. Once restrictive physiology is confirmed with a low total lung capacity, FVC testing can be used as a longitudinal surrogate, as it is less expensive and easier for the patient to perform. In general, a lower total lung capacity or FVC indicates more severe impairment.
The DLCO serves as another marker of severity but is less reliable due to baseline variability and difficulties performing the maneuver.
A 6-minute walk test is another crucial physiologic assessment tool that can quantify exertional hypoxemia and functional status (ie, distance walked), and can assist in prognosis.
Imaging
Most patients undergo chest radiography in the workup for undiagnosed dyspnea. However, chest radiography is not adequate to formulate an accurate diagnosis in suspected interstitial lung disease, and a normal radiograph cannot exclude changes that might reflect early phases of the disease. As the disease progresses, the plain radiograph can show reticulonodular opacities and honeycombing in the peripheral and lower lung zones (Figure 3).34
The decision whether to order HRCT in the workup for a patient who has dyspnea and a normal chest radiograph is challenging. We recommend cross-sectional imaging when physiologic testing shows restriction or low DLCO, or when there is a high index of suspicion for underlying lung disease as the cause of symptoms.
Expert consultation can aid with this decision, especially when the underlying cause of dyspnea remains unclear after initial studies have been completed. Otherwise, HRCT is an essential test in the evaluation of interstitial lung disease.
Bronchoscopy’s role controversial
If the pattern on HRCT is nondiagnostic, then surgical biopsy is necessary, and the diagnosis of IPF requires a histologic pattern of UIP as described above.16,35
Although bronchoscopy can be valuable if an alternative diagnosis such as sarcoidosis or chronic hypersensitivity pneumonitis is suspected, the role of bronchoscopic biopsy in the workup of IPF is controversial. The patchy nature of UIP does not lend itself to the relatively small biopsy samples obtained through bronchoscopy.36,37
Surgical biopsy options
The favored biopsy approach is surgical, using either an open or a video-assisted thoracoscopic technique. The latter is preferred as it is less invasive, requires a shorter length of hospital stay, and allows a faster recovery.38 Bronchoscopic cryobiopsy, currently under investigation, is a potentially valuable tool whose role in diagnosing IPF is evolving.
Frequently, neither HRCT nor surgical lung biopsy demonstrates UIP, making the definitive diagnosis of IPF difficult. Moreover, some patients with nondiagnostic HRCT results are unable to tolerate surgical lung biopsy because of severely impaired lung function or other comorbidities.
The role of multidisciplinary discussions
When surgical lung biopsy is not possible, current practice at leading centers uses a multidisciplinary approach to allow for a confident diagnosis.30,39 Discussions take place between pulmonologists, pathologists, radiologists, and other specialists to collectively consider all aspects of a case before rendering a consensus opinion on the diagnosis and the management plan. If the discussion leads to a consensus diagnosis of IPF, then the patient’s clinician can move forward with treatment options. If not, the group can recommend further workup or alternative diagnoses and treatment regimens. The multidisciplinary group is also well positioned to consider the relative risks and benefits of moving forward with surgical lung biopsy for individual patients.
This approach illustrates the importance of referring these patients to centers of excellence in diagnosing and managing complex cases of interstitial lung disease, including IPF.40
TREATMENT OF IPF
Antifibrotic therapy
Antifibrotic therapy is a choice between pirfenidone and nintedanib.
Pirfenidone, which has an undefined molecular target, was approved based on the results of 3 trials.41,42 Pooled analyses from these trials showed a reduction in the decline from baseline in FVC percent predicted and improved progression-free survival.43 Pooled and meta-analyses of pirfenidone clinical trials have shown a mortality benefit, although no individual study has shown such an effect on mortality rates.44
The major adverse effects of pirfenidone are gastrointestinal distress and photosensitivity rash.
Nintedanib is a triple tyrosine kinase inhibitor that broadly targets fibroblast growth factor, vascular endothelial growth factor, and platelet-derived growth factor receptors. Combined analysis of 2 concurrent trials45 showed that nintedanib reduced the decline in FVC, similarly to pirfenidone. The major adverse event associated with nintedanib was diarrhea. Since it inhibits vascular endothelial growth factor, there is a risk of hematologic complications such as bleeding or clotting events.
Because pirfenidone and nintedanib can increase aminotransferase levels, regular monitoring is recommended.
To date, no trial has compared pirfenidone and nintedanib in terms of their efficacy and tolerability. Therefore, the choice of agent is based on the patient’s preference after a discussion of potential risks and expected benefits, a review of each drug’s side effects, and consideration of comorbid conditions and physician experience.
Patients need to understand that these drugs slow the rate of decline in FVC but have not been shown to improve symptoms or functional status.
Corticosteroids are not routine
Corticosteroids should not be used routinely in the treatment of IPF. Although steroids, alone or in combination with other immunosuppressive medications, were commonly used for IPF in the past, such use was not based on results of randomized controlled trials.46 Retrospective controlled studies have failed to show that corticosteroids improve mortality rates in IPF; indeed, they have shown that corticosteroids confer substantial morbidity.47,48 In addition, a randomized controlled trial combining corticosteroids with N-acetylcysteine and azathioprine was stopped early due to an increased risk of death and hospitalization.49 Collectively, these data suggest that corticosteroids confer no benefit and are potentially harmful. Their use in IPF is discouraged, and the joint international guidelines recommend against immunosuppression to treat IPF.16
Other treatments
The guidelines offer additional suggestions for the management of IPF.
Preliminary evidence suggests that microaspiration associated with abnormal gastroesophageal acid reflux is a risk factor for IPF. As such, there is a weak recommendation for aggressive treatment of reflux disease.50 However, because evidence suggests that proton-pump inhibitor therapy may be associated with adverse renal or central nervous system effects, this recommendation bears caution. It is hoped that ongoing studies will provide further insight into the role of acid-suppression in the management of IPF.51,52
Further treatment recommendations include best supportive management such as supplemental oxygen, pulmonary rehabilitation, and vaccinations.
Prompt referral for lung transplant is imperative. IPF is now the most common indication for lung transplant, and given the poor overall prognosis of advanced IPF, transplant confers a survival benefit in appropriately selected patients.53,54 Table 2 provides an evidence-based checklist for the workup and management of IPF.
ACUTE EXACERBATIONS OF IPF
The unpredictable nature of IPF can manifest in the form of acute exacerbations without an identifiable cause. The loosely defined diagnostic criteria for the diagnosis of acute exacerbations are a previous or new diagnosis of IPF, worsening or development of dyspnea in the last 30 days, and new bilateral ground-glass or consolidative changes with a background of UIP on HRCT.16
A new definition has been proposed55 to facilitate research in the characterization and treatment of acute exacerbations of IPF. The new definition includes all causes of respiratory deterioration except for heart failure and volume overload. It is less strict about the 30-day time frame. This newer definition is based on the lack of evidence differentiating outcomes when an acute deterioration is associated with known or unknown etiologies.55
The incidence of acute exacerbations is variable, with a 1- and 3-year incidence ranging between 8.6% and 23.9% depending on the criteria used.56 In general, acute exacerbations carry a grim prognosis, with a median life expectancy of 2.2 months.57
There is no approved therapy for exacerbations of IPF. Rather, treatment is mainly supportive with supplemental oxygen and mechanical ventilation. Current guidelines have a weak recommendation for the use of corticosteroids, but there are no recommendations regarding dose, route, or duration of therapy. Other treatments, primarily immunomodulatory agents, have been suggested but lack evidence of benefit.
Acknowledgments: Pathology images were provided by Carol Farver, MD, Pathology Institute, Cleveland Clinic. Radiology images were provided by Ruchi Yadav, MD, Imaging Institute, Cleveland Clinic.
Idiopathic pulmonary fibrosis (IPF) is a devastating and fatal lung disease that generally affects older adults. It is characterized by a radiographic and histopathologic pattern of usual interstitial pneumonia (UIP) that has no other known etiology.
Accurate diagnosis of IPF is crucial. We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis, start appropriate therapy, advise the patient on prognosis and enrollment in disease registries and clinical trials, and determine candidacy for lung transplant.
Primary care physicians are uniquely positioned to encounter patients with IPF, whether because of a patient complaint or as an incidental finding on computed tomography. The goal of this article is to delineate the features of IPF so that it may be recognized early and thus expedite referral to a center with expertise in interstitial lung disease for a thorough evaluation and appropriate management.
WHAT IS IDIOPATHIC PULMONARY FIBROSIS?
MORE COMMON THAN ONCE THOUGHT
The true incidence and prevalence of IPF are difficult to assess. IPF is generally considered a rare disease, but it is more common than once thought. In 2011, Raghu et al3 estimated the prevalence in Medicare beneficiaries to be 495 cases per 100,000. Based on this estimate and the current US population, up to 160,000 Americans could have IPF.4 Raghu et al also showed that IPF more often affects adults over age 65, which suggests that as the US population ages, the incidence of IPF may rise. Studies have also reported an increased incidence of IPF worldwide.5
Further, with the rising use of low-dose computed tomography to screen for lung cancer, more incidental cases of IPF will likely be found.6–8
Older data showed a lag from symptom onset to accurate diagnosis of 1 to 2 years.9 A more recent study found a lag in referral of patients with IPF to tertiary care centers, and this delay was associated with a higher rate of death independent of disease severity.10
TYPICALLY PROGRESSIVE, OFTEN FATAL
IPF is typically progressive and limited to the lungs, and it portends a poor prognosis. The median survival is commonly cited as 2 to 5 years from diagnosis, although this is based on older observations that may not reflect current best practice and newer therapies. More recent studies suggest higher survival rates if patients have preserved lung function.11
As the name indicates, the etiology of IPF is unknown, but studies have indicated genetic underpinnings in a notable proportion of cases.12 Regardless of the cause, the pathogenesis and progression of IPF are thought to be the result of an abnormal and persistent wound-repair response. The progressive deposition of scar tissue disrupts normal lung architecture and function, eventually causing clinical disease.13
SYMPTOMS AND KEY FEATURES
Patients with IPF typically present with the insidious onset of dyspnea on exertion, with or without chronic cough. Risk factors include male sex, increasing age, and a history of smoking. Patients with undiagnosed IPF who present with dyspnea and a history of smoking are often treated empirically for chronic obstructive pulmonary disease (COPD).
Rales are a common finding on auscultation in IPF, and this can lead to an exhaustive cardiac evaluation and empiric treatment for heart failure. Digital clubbing is also relatively common.14 Hypoxemia with exertion is another common feature that also often correlates with disease severity and prognosis. Resting hypoxemia is more common in advanced disease.
On spirometry, patients with IPF typically demonstrate restrictive physiology, suggested by a normal or elevated ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1/FVC) (> 70% predicted or above the lower limit of normal) combined with a lower than normal FVC. Restrictive physiology is definitively demonstrated by a decreased total lung capacity (< 80% predicted or below the lower limit of normal) on plethysmography. Impaired gas exchange, manifested by a decreased diffusing capacity of the lungs for carbon monoxide (DLCO) on pulmonary function testing, is also common. Because pulmonary perfusion is higher in the lung bases, where IPF is also predominant, the DLCO is often reduced to a greater extent than the FVC.
PROGNOSTIC INDICATORS
Clinicians typically view IPF as a relentless and progressive process, but its course is variable and can be uncertain in an individual patient (Figure 1).15,16 Nevertheless, over time, most patients have a decline in lung function leading to respiratory failure. Respiratory failure, often preceded by a subacute deterioration (over weeks to months) or an acute deterioration (< 4 weeks), is the most common cause of death, but comorbid diseases such as lung cancer, infection, and heart failure are also common causes of death in these patients.17,18
Predictors of mortality include worsening FVC, DLCO, symptoms, and physiologic impairment, manifested by a decline in the 6-minute walking test or worsening exertional hypoxemia.19–22 Other common comorbidities linked with impaired quality of life and poor prognosis include obstructive sleep apnea, gastroesophageal reflux disease, and depression.16,23 Retrospective studies suggest that most IPF patients die 2 to 5 years after symptom onset. With the lag from symptom onset to final diagnosis, the average life expectancy is as little as 2 years from the time of diagnosis.9,18,24,25
Two staging systems have been developed to predict short-term and long-term mortality risk based on sex, age, and physiologic parameters.23,24 The GAP (gender, age, physiology) index provides an estimate of the risk of death for a cohort of patients: a score of 0 to 8 is calculated, and the score is then categorized as stage I, II, or III. Each stage is associated with 1-, 2-, and 3-year mortality rates, with stage III having the highest rates. The GAP calculator (www.acponline.org/journals/annals/extras/gap) provides an estimate of the risk of death for an individual patient. The application of these tools for the management of IPF is evolving; however, they may be helpful for counseling patients about disease prognosis.
CLUES TO DIAGNOSIS
Histologic patterns
UIP on histologic study is also seen in fibrotic lung diseases other than IPF, including connective tissue disease-associated interstitial lung disease, inhalational or occupational interstitial lung disease, and chronic hypersensitivity pneumonitis.26–29 Consequently, the diagnosis of IPF requires exclusion of other known causes of UIP.
According to the 2011 guidelines,16 the histology of interstitial lung disease can be categorized as definite UIP, probable UIP, or possible UIP, or as an atypical pattern suggesting another diagnosis. If no definite cause of the interstitial lung abnormality is found, the level of certainty of the histopathologic pattern of UIP helps formulate the clinical diagnosis and management plan.
Clues on computed tomography
The UIP nomenclature also describes patterns on high-resolution computed tomography (HRCT). HRCT is done without contrast and produces thin-sliced images (usually < 1.5 mm) in inspiratory, expiratory, and prone views; this allows detection of air trapping, which may indicate an airway-centric alternative diagnosis.
On HRCT, UIP appears as reticular opacities, often with traction bronchiectasis or bronchiolectasis, usually with a basilar and peripheral predominance. Honeycombing is a key feature and appears as clustered cystic spaces with well-defined walls in the periphery of the lung parenchyma. Ground-glass opacities are not a prominent feature of UIP, and although they do not exclude a UIP pattern, they should spur consideration of other diagnoses.16 Reactive mediastinal and hilar lymphadenopathy is another common feature of UIP.
When evaluating results of HRCT for UIP, the radiologist categorizes the pattern as definite UIP, possible UIP, or inconsistent. The definite pattern meets all the above features and has none of the features suggesting an alternative diagnosis (Figure 3). The possible pattern includes all the above features with the exception of honeycombing. If the predominant features on HRCT include any atypical finding listed above, then the study is considered inconsistent with UIP. If the pattern on HRCT is considered definite, evaluation of pathology is not necessary. If the pattern is categorized as possible or is inconsistent, then surgical lung biopsy-confirmed UIP is necessary for the definitive diagnosis of IPF.
However, evidence is emerging that in the correct clinical scenario, possible UIP behaves similarly to definite UIP and may be sufficient to make the clinical diagnosis of IPF even without surgical biopsy confirmation.30
A DIAGNOSTIC ALGORITHM FOR IPF
Given the multitude of interstitial lung diseases, their complexities, and the lack of a gold standard definitive diagnostic test, the diagnosis of IPF can be difficult, requiring the integration of clinical, radiologic, and, if necessary, pathologic findings.
Demographic features
The patient’s demographic features and risk factors dictate the initial clinical suspicion of IPF compared with other interstitial lung diseases. The incidence of IPF increases with age, and IPF is more common in men. A history of smoking is another risk factor.31 A 45-year-old never-smoker is much less likely to have IPF than a 70-year-old former smoker, and a 70-year-old man is more likely to have IPF than a woman of the same age. Thus, the finding of interstitial lung disease in a patient with a demographic profile that is not typical (ie, a younger woman who never smoked) should prompt an exhaustive investigation for another diagnosis such as hypersensitivity pneumonitis or connective tissue disease.
Key elements of the history
After considering the demographic profile and risk factors, the next step in the evaluation is a thorough and accurate medical history. This should include assessment of the severity of dyspnea and cough, signs and symptoms of connective tissue disease (eg, arthralgias, sicca symptoms, Raynaud phenomenon, difficulty swallowing), and gastroesophageal reflux disease, which can be associated with connective tissue disease and, independently, with IPF.
It is also important to identify any environmental exposures that suggest pneumoconiosis or chronic hypersensitivity pneumonitis. The most common risk factors for hypersensitivity pneumonitis are birds and bird feathers, molds, fungi, hot tub use, and some industrial chemicals.32
A medication history is important. Many medications are associated with interstitial lung disease, but amiodarone, bleomycin, methotrexate, and nitrofurantoin are among the common offenders.33
A thorough family history is necessary, as there are familial forms of IPF.
Focus of the physical examination
The physical examination must include careful auscultation for rales. While rales are not specific for IPF, they are the most common pulmonary abnormality. Detailed skin, musculoskeletal, and cardiovascular examinations are also important to evaluate for rheumatologic signs, clubbing, or evidence of heart failure or pulmonary hypertension.
Laboratory tests
Laboratory testing should include a serologic autoantibody panel to evaluate for connective tissue diseases that can manifest as interstitial lung disease, including rheumatoid arthritis, dermatopolymyositis, scleroderma, Sjögren syndrome, and undifferentiated or mixed connective tissue disease. Typical preliminary laboratory tests are antinuclear antibody, rheumatoid factor, erythrocyte sedimentation rate, and C-reactive protein. Others may include anticyclic citrullinated peptide (anti-CCP), anti-Scl-70, anti-RNP, anti-SS-A, anti-SS-B, and anti-Jo-1.16 The breadth of the panel should depend on patient demographics and findings in the history or physical examination that increase or decrease the likelihood of a connective tissue disease.
Lung function testing
Assessing the patient’s pulmonary physiology should include spirometry, DLCO, and body plethysmography (lung volumes). In most cases, IPF manifests with restrictive physiology. Once restrictive physiology is confirmed with a low total lung capacity, FVC testing can be used as a longitudinal surrogate, as it is less expensive and easier for the patient to perform. In general, a lower total lung capacity or FVC indicates more severe impairment.
The DLCO serves as another marker of severity but is less reliable due to baseline variability and difficulties performing the maneuver.
A 6-minute walk test is another crucial physiologic assessment tool that can quantify exertional hypoxemia and functional status (ie, distance walked), and can assist in prognosis.
Imaging
Most patients undergo chest radiography in the workup for undiagnosed dyspnea. However, chest radiography is not adequate to formulate an accurate diagnosis in suspected interstitial lung disease, and a normal radiograph cannot exclude changes that might reflect early phases of the disease. As the disease progresses, the plain radiograph can show reticulonodular opacities and honeycombing in the peripheral and lower lung zones (Figure 3).34
The decision whether to order HRCT in the workup for a patient who has dyspnea and a normal chest radiograph is challenging. We recommend cross-sectional imaging when physiologic testing shows restriction or low DLCO, or when there is a high index of suspicion for underlying lung disease as the cause of symptoms.
Expert consultation can aid with this decision, especially when the underlying cause of dyspnea remains unclear after initial studies have been completed. Otherwise, HRCT is an essential test in the evaluation of interstitial lung disease.
Bronchoscopy’s role controversial
If the pattern on HRCT is nondiagnostic, then surgical biopsy is necessary, and the diagnosis of IPF requires a histologic pattern of UIP as described above.16,35
Although bronchoscopy can be valuable if an alternative diagnosis such as sarcoidosis or chronic hypersensitivity pneumonitis is suspected, the role of bronchoscopic biopsy in the workup of IPF is controversial. The patchy nature of UIP does not lend itself to the relatively small biopsy samples obtained through bronchoscopy.36,37
Surgical biopsy options
The favored biopsy approach is surgical, using either an open or a video-assisted thoracoscopic technique. The latter is preferred as it is less invasive, requires a shorter length of hospital stay, and allows a faster recovery.38 Bronchoscopic cryobiopsy, currently under investigation, is a potentially valuable tool whose role in diagnosing IPF is evolving.
Frequently, neither HRCT nor surgical lung biopsy demonstrates UIP, making the definitive diagnosis of IPF difficult. Moreover, some patients with nondiagnostic HRCT results are unable to tolerate surgical lung biopsy because of severely impaired lung function or other comorbidities.
The role of multidisciplinary discussions
When surgical lung biopsy is not possible, current practice at leading centers uses a multidisciplinary approach to allow for a confident diagnosis.30,39 Discussions take place between pulmonologists, pathologists, radiologists, and other specialists to collectively consider all aspects of a case before rendering a consensus opinion on the diagnosis and the management plan. If the discussion leads to a consensus diagnosis of IPF, then the patient’s clinician can move forward with treatment options. If not, the group can recommend further workup or alternative diagnoses and treatment regimens. The multidisciplinary group is also well positioned to consider the relative risks and benefits of moving forward with surgical lung biopsy for individual patients.
This approach illustrates the importance of referring these patients to centers of excellence in diagnosing and managing complex cases of interstitial lung disease, including IPF.40
TREATMENT OF IPF
Antifibrotic therapy
Antifibrotic therapy is a choice between pirfenidone and nintedanib.
Pirfenidone, which has an undefined molecular target, was approved based on the results of 3 trials.41,42 Pooled analyses from these trials showed a reduction in the decline from baseline in FVC percent predicted and improved progression-free survival.43 Pooled and meta-analyses of pirfenidone clinical trials have shown a mortality benefit, although no individual study has shown such an effect on mortality rates.44
The major adverse effects of pirfenidone are gastrointestinal distress and photosensitivity rash.
Nintedanib is a triple tyrosine kinase inhibitor that broadly targets fibroblast growth factor, vascular endothelial growth factor, and platelet-derived growth factor receptors. Combined analysis of 2 concurrent trials45 showed that nintedanib reduced the decline in FVC, similarly to pirfenidone. The major adverse event associated with nintedanib was diarrhea. Since it inhibits vascular endothelial growth factor, there is a risk of hematologic complications such as bleeding or clotting events.
Because pirfenidone and nintedanib can increase aminotransferase levels, regular monitoring is recommended.
To date, no trial has compared pirfenidone and nintedanib in terms of their efficacy and tolerability. Therefore, the choice of agent is based on the patient’s preference after a discussion of potential risks and expected benefits, a review of each drug’s side effects, and consideration of comorbid conditions and physician experience.
Patients need to understand that these drugs slow the rate of decline in FVC but have not been shown to improve symptoms or functional status.
Corticosteroids are not routine
Corticosteroids should not be used routinely in the treatment of IPF. Although steroids, alone or in combination with other immunosuppressive medications, were commonly used for IPF in the past, such use was not based on results of randomized controlled trials.46 Retrospective controlled studies have failed to show that corticosteroids improve mortality rates in IPF; indeed, they have shown that corticosteroids confer substantial morbidity.47,48 In addition, a randomized controlled trial combining corticosteroids with N-acetylcysteine and azathioprine was stopped early due to an increased risk of death and hospitalization.49 Collectively, these data suggest that corticosteroids confer no benefit and are potentially harmful. Their use in IPF is discouraged, and the joint international guidelines recommend against immunosuppression to treat IPF.16
Other treatments
The guidelines offer additional suggestions for the management of IPF.
Preliminary evidence suggests that microaspiration associated with abnormal gastroesophageal acid reflux is a risk factor for IPF. As such, there is a weak recommendation for aggressive treatment of reflux disease.50 However, because evidence suggests that proton-pump inhibitor therapy may be associated with adverse renal or central nervous system effects, this recommendation bears caution. It is hoped that ongoing studies will provide further insight into the role of acid-suppression in the management of IPF.51,52
Further treatment recommendations include best supportive management such as supplemental oxygen, pulmonary rehabilitation, and vaccinations.
Prompt referral for lung transplant is imperative. IPF is now the most common indication for lung transplant, and given the poor overall prognosis of advanced IPF, transplant confers a survival benefit in appropriately selected patients.53,54 Table 2 provides an evidence-based checklist for the workup and management of IPF.
ACUTE EXACERBATIONS OF IPF
The unpredictable nature of IPF can manifest in the form of acute exacerbations without an identifiable cause. The loosely defined diagnostic criteria for the diagnosis of acute exacerbations are a previous or new diagnosis of IPF, worsening or development of dyspnea in the last 30 days, and new bilateral ground-glass or consolidative changes with a background of UIP on HRCT.16
A new definition has been proposed55 to facilitate research in the characterization and treatment of acute exacerbations of IPF. The new definition includes all causes of respiratory deterioration except for heart failure and volume overload. It is less strict about the 30-day time frame. This newer definition is based on the lack of evidence differentiating outcomes when an acute deterioration is associated with known or unknown etiologies.55
The incidence of acute exacerbations is variable, with a 1- and 3-year incidence ranging between 8.6% and 23.9% depending on the criteria used.56 In general, acute exacerbations carry a grim prognosis, with a median life expectancy of 2.2 months.57
There is no approved therapy for exacerbations of IPF. Rather, treatment is mainly supportive with supplemental oxygen and mechanical ventilation. Current guidelines have a weak recommendation for the use of corticosteroids, but there are no recommendations regarding dose, route, or duration of therapy. Other treatments, primarily immunomodulatory agents, have been suggested but lack evidence of benefit.
Acknowledgments: Pathology images were provided by Carol Farver, MD, Pathology Institute, Cleveland Clinic. Radiology images were provided by Ruchi Yadav, MD, Imaging Institute, Cleveland Clinic.
- Brown KK, Raghu G. Medical treatment for pulmonary fibrosis: current trends, concepts, and prospects. Clin Chest Med 2004; 25(4):759–772, vii. doi:10.1016/j.ccm.2004.08.003
- Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 2013; 19(5):453–459. doi:10.1097/MCP.0b013e328363f48d
- Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014; 2(7):566–572. doi:10.1016/S2213-2600(14)70101-8
- Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; 21(126):355–361. doi:10.1183/09059180.00002512
- Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46(3):795–806. doi:10.1183/09031936.00185114
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268(2):563–571. doi:10.1148/radiol.13120816
- Southern BD, Scheraga RG, Yadav R. Managing interstitial lung disease detected on CT during lung cancer screening. Cleve Clin J Med 2016; 83(1):55–65. doi:10.3949/ccjm.83a.14157
- King TE Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001; 164(5):1025–1032. doi:10.1164/ajrccm.164.6.2001056
- Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184(7):842–847. doi:10.1164/rccm.201104-0668OC
- Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med 2018; 18(1):19. doi:10.1186/s12890-018-0575-y
- Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res 2015; 165(1):48–60. doi:10.1016/j.trsl.2014.03.011
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378(9807):1949–1961. doi:10.1016/S0140-6736(11)60052-4
- Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis 2008; 3:8. doi:10.1186/1750-1172-3-8
- Raghu G. Idiopathic pulmonary fibrosis. A rational clinical approach. Chest 1987; 92(1):148–154. doi:10.1378/chest.92.1.148
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Panos RJ, Mortenson RL, Niccoli SA, King TE Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med 1990; 88(4):396–404. doi:10.1016/0002-9343(90)90495-Y
- Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183(4):431–440. doi:10.1164/rccm.201006-0894CI
- Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003; 168(5):538–542. doi:10.1164/rccm.200211-1311OC
- Flaherty KR, Andrei AC, Murray S, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174(7):803–809. doi:10.1164/rccm.200604-488OC
- Jegal Y, Kim DS, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171(6):639–644. doi:10.1164/rccm.200403-331OC
- Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003; 168(5):531–537. doi:10.1164/rccm.200210-1245OC
- King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med 2017; 5(1):72–84. doi:10.1016/S2213-2600(16)30222-3
- Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156(1):684–691. doi:10.7326/0003-4819-156-10-201205150-00004
- Rudd RM, Prescott RJ, Chalmers JC, Johnston ID; Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. British Thoracic Society study on cryptogenic fibrosing alveolitis: response to treatment and survival. Thorax 2007; 62(1):62–66. doi:10.1136/thx.2005.045591
- Gutsche M, Rosen GD, Swigris JJ. Connective tissue disease-associated interstitial lung disease: a review. Curr Respir Care Rep 2012; 1:224–232. doi:10.1007/s13665-012-0028-7
- Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007; 175(7):705–711. doi:10.1164/rccm.200607-912OC
- Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006; 3(4):293–298. doi:10.1513/pats.200512-131TK
- Vourlekis JS, Schwarz MI, Cherniack RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med 2004; 116(10):662–668. doi:10.1016/j.amjmed.2003.12.030
- Brownell R, Moua T, Henry TS, et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax 2017; 72(5):424–429. doi:10.1136/thoraxjnl-2016-209671
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997; 155(1):242–248. doi:10.1164/ajrccm.155.1.9001319
- Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012; 186(4):314–324. doi:10.1164/rccm.201203-0513CI
- Schwaiblmair M, Behr W, Haeckel T, Markl B, Foerg W, Berghaus T. Drug induced interstitial lung disease. Open Respir Med J 2012; 6:63–74. doi:10.2174/1874306401206010063
- Grenier P, Valeyre D, Cluzel P, Brauner MW, Lenoir S, Chastang C. Chronic diffuse interstitial lung disease: diagnostic value of chest radiography and high-resolution CT. Radiology 1991; 179(1):123–132. doi:10.1148/radiology.179.1.2006262
- Lynch JP 3rd, Huynh RH, Fishbein MC, Saggar R, Belperio JA, Weigt SS. Idiopathic pulmonary fibrosis: epidemiology, clinical features, prognosis, and management. Semin Respir Crit Care Med 2016; 37(3):331–357. doi:10.1055/s-0036-1582011
- Berbescu EA, Katzenstein AL, Snow JL, Zisman DA. Transbronchial biopsy in usual interstitial pneumonia. Chest 2006; 129(5):1126–1131. doi:10.1378/chest.129.5.1126
- Ohshimo S, Bonella F, Cui A, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179(11):1043–1047. doi:10.1164/rccm.200808-1313OC
- Oparka J, Yan TD, Ryan E, Dunning J. Does video-assisted thoracic surgery provide a safe alternative to conventional techniques in patients with limited pulmonary function who are otherwise suitable for lung resection? Interact Cardiovasc Thorac Surg 2013; 17(1):159–162. doi:10.1093/icvts/ivt097
- Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170(8):904–910. doi:10.1164/rccm.200402-147OC
- Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med 2016; 4(7):557–565. doi:10.1016/S2213-2600(16)30033-9
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779):1760–1769. doi:10.1016/S0140-6736(11)60405-4
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016; 47(1):243–253. doi:10.1183/13993003.00026-2015
- Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5(1):33–41. doi:10.1016/S2213-2600(16)30326-5
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Richeldi L, Davies HR, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2003: 3:CD002880. doi:10.1002/14651858.CD002880
- Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med 2000; 161(4 pt 1):1172–1178. doi:10.1164/ajrccm.161.4.9907002
- Gay SE, Kazerooni EA, Toews GB, et al. Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Crit Care Med 1998; 157(4 pt 1):1063–1072. doi:10.1164/ajrccm.157.4.9703022
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J 2006; 27(1):136–142. doi:10.1183/09031936.06.00037005
- Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia: a pharmacoepidemiological claims data analysis. JAMA Neurol 2016; 73(4):410–416. doi:10.1001/jamaneurol.2015.4791
- Xie Y, Bowe B, Li T, Xian H, Balasubramanian S, Al-Aly Z. Proton pump inhibitors and risk of incident CKD and progression to ESRD. J Am Soc Nephrol 2016; 27(10):3153–3163. doi:10.1681/ASN.2015121377
- Thabut G, Mal H, Castier Y, et al. Survival benefit of lung transplantation for patients with idiopathic pulmonary fibrosis. J Thorac Cardiovasc Surg 2003; 126(2):469–475. doi:10.1016/S0022-5223(03)00600-7
- Valapour M, Skeans MA, Smith JM, et al. Lung. Am J Transplant 2016; 16(suppl 2):141–168. doi:10.1111/ajt.13671
- Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med 2016; 194(3):265–275. doi:10.1164/rccm.201604-0801CI
- Kondoh Y, Taniguchi H, Katsuta T, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27(2):103–110. doi:10.1016/j.resinv.2015.04.005
- Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37(2):356–363. doi:10.1183/09031936.00159709
- Brown KK, Raghu G. Medical treatment for pulmonary fibrosis: current trends, concepts, and prospects. Clin Chest Med 2004; 25(4):759–772, vii. doi:10.1016/j.ccm.2004.08.003
- Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 2013; 19(5):453–459. doi:10.1097/MCP.0b013e328363f48d
- Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014; 2(7):566–572. doi:10.1016/S2213-2600(14)70101-8
- Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; 21(126):355–361. doi:10.1183/09059180.00002512
- Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46(3):795–806. doi:10.1183/09031936.00185114
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268(2):563–571. doi:10.1148/radiol.13120816
- Southern BD, Scheraga RG, Yadav R. Managing interstitial lung disease detected on CT during lung cancer screening. Cleve Clin J Med 2016; 83(1):55–65. doi:10.3949/ccjm.83a.14157
- King TE Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001; 164(5):1025–1032. doi:10.1164/ajrccm.164.6.2001056
- Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184(7):842–847. doi:10.1164/rccm.201104-0668OC
- Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med 2018; 18(1):19. doi:10.1186/s12890-018-0575-y
- Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res 2015; 165(1):48–60. doi:10.1016/j.trsl.2014.03.011
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378(9807):1949–1961. doi:10.1016/S0140-6736(11)60052-4
- Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis 2008; 3:8. doi:10.1186/1750-1172-3-8
- Raghu G. Idiopathic pulmonary fibrosis. A rational clinical approach. Chest 1987; 92(1):148–154. doi:10.1378/chest.92.1.148
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Panos RJ, Mortenson RL, Niccoli SA, King TE Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med 1990; 88(4):396–404. doi:10.1016/0002-9343(90)90495-Y
- Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183(4):431–440. doi:10.1164/rccm.201006-0894CI
- Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003; 168(5):538–542. doi:10.1164/rccm.200211-1311OC
- Flaherty KR, Andrei AC, Murray S, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174(7):803–809. doi:10.1164/rccm.200604-488OC
- Jegal Y, Kim DS, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171(6):639–644. doi:10.1164/rccm.200403-331OC
- Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003; 168(5):531–537. doi:10.1164/rccm.200210-1245OC
- King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med 2017; 5(1):72–84. doi:10.1016/S2213-2600(16)30222-3
- Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156(1):684–691. doi:10.7326/0003-4819-156-10-201205150-00004
- Rudd RM, Prescott RJ, Chalmers JC, Johnston ID; Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. British Thoracic Society study on cryptogenic fibrosing alveolitis: response to treatment and survival. Thorax 2007; 62(1):62–66. doi:10.1136/thx.2005.045591
- Gutsche M, Rosen GD, Swigris JJ. Connective tissue disease-associated interstitial lung disease: a review. Curr Respir Care Rep 2012; 1:224–232. doi:10.1007/s13665-012-0028-7
- Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007; 175(7):705–711. doi:10.1164/rccm.200607-912OC
- Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006; 3(4):293–298. doi:10.1513/pats.200512-131TK
- Vourlekis JS, Schwarz MI, Cherniack RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med 2004; 116(10):662–668. doi:10.1016/j.amjmed.2003.12.030
- Brownell R, Moua T, Henry TS, et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax 2017; 72(5):424–429. doi:10.1136/thoraxjnl-2016-209671
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997; 155(1):242–248. doi:10.1164/ajrccm.155.1.9001319
- Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012; 186(4):314–324. doi:10.1164/rccm.201203-0513CI
- Schwaiblmair M, Behr W, Haeckel T, Markl B, Foerg W, Berghaus T. Drug induced interstitial lung disease. Open Respir Med J 2012; 6:63–74. doi:10.2174/1874306401206010063
- Grenier P, Valeyre D, Cluzel P, Brauner MW, Lenoir S, Chastang C. Chronic diffuse interstitial lung disease: diagnostic value of chest radiography and high-resolution CT. Radiology 1991; 179(1):123–132. doi:10.1148/radiology.179.1.2006262
- Lynch JP 3rd, Huynh RH, Fishbein MC, Saggar R, Belperio JA, Weigt SS. Idiopathic pulmonary fibrosis: epidemiology, clinical features, prognosis, and management. Semin Respir Crit Care Med 2016; 37(3):331–357. doi:10.1055/s-0036-1582011
- Berbescu EA, Katzenstein AL, Snow JL, Zisman DA. Transbronchial biopsy in usual interstitial pneumonia. Chest 2006; 129(5):1126–1131. doi:10.1378/chest.129.5.1126
- Ohshimo S, Bonella F, Cui A, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179(11):1043–1047. doi:10.1164/rccm.200808-1313OC
- Oparka J, Yan TD, Ryan E, Dunning J. Does video-assisted thoracic surgery provide a safe alternative to conventional techniques in patients with limited pulmonary function who are otherwise suitable for lung resection? Interact Cardiovasc Thorac Surg 2013; 17(1):159–162. doi:10.1093/icvts/ivt097
- Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170(8):904–910. doi:10.1164/rccm.200402-147OC
- Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med 2016; 4(7):557–565. doi:10.1016/S2213-2600(16)30033-9
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779):1760–1769. doi:10.1016/S0140-6736(11)60405-4
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016; 47(1):243–253. doi:10.1183/13993003.00026-2015
- Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5(1):33–41. doi:10.1016/S2213-2600(16)30326-5
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Richeldi L, Davies HR, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2003: 3:CD002880. doi:10.1002/14651858.CD002880
- Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med 2000; 161(4 pt 1):1172–1178. doi:10.1164/ajrccm.161.4.9907002
- Gay SE, Kazerooni EA, Toews GB, et al. Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Crit Care Med 1998; 157(4 pt 1):1063–1072. doi:10.1164/ajrccm.157.4.9703022
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J 2006; 27(1):136–142. doi:10.1183/09031936.06.00037005
- Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia: a pharmacoepidemiological claims data analysis. JAMA Neurol 2016; 73(4):410–416. doi:10.1001/jamaneurol.2015.4791
- Xie Y, Bowe B, Li T, Xian H, Balasubramanian S, Al-Aly Z. Proton pump inhibitors and risk of incident CKD and progression to ESRD. J Am Soc Nephrol 2016; 27(10):3153–3163. doi:10.1681/ASN.2015121377
- Thabut G, Mal H, Castier Y, et al. Survival benefit of lung transplantation for patients with idiopathic pulmonary fibrosis. J Thorac Cardiovasc Surg 2003; 126(2):469–475. doi:10.1016/S0022-5223(03)00600-7
- Valapour M, Skeans MA, Smith JM, et al. Lung. Am J Transplant 2016; 16(suppl 2):141–168. doi:10.1111/ajt.13671
- Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med 2016; 194(3):265–275. doi:10.1164/rccm.201604-0801CI
- Kondoh Y, Taniguchi H, Katsuta T, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27(2):103–110. doi:10.1016/j.resinv.2015.04.005
- Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37(2):356–363. doi:10.1183/09031936.00159709
KEY POINTS
- IPF is characterized by a pattern of usual interstitial pneumonia on imaging and histopathology without another known etiology.
- We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis and to initiate appropriate therapy.
- Specialized centers offer advice on prognosis, enrollment in disease registries and clinical trials, and candidacy for lung transplant.
It takes a village to care for the patient with idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is a devastating progressive fibrosing interstitial lung disease associated with a high burden of morbidity and death.1 A clinical diagnosis of IPF is made only after careful interpretation of integrated clinical, radiologic, and often histopathologic data.
Interstitial lung disease encompasses a broad spectrum of parenchymal lung diseases, and a classification of IPF is restricted to a lung injury pattern of usual interstitial pneumonia (UIP) based on high-resolution computed tomography or surgical lung biopsy, after all known causes of UIP have been excluded.1
However, a lung injury pattern of UIP is not synonymous with IPF, as UIP can be seen with connective tissue disease, chronic hypersensitivity pneumonitis, drug toxicity, and sarcoidosis.1 As such, rendering a diagnosis of IPF requires a thorough evaluation to exclude such diverse potential etiologies.
In this issue of the Cleveland Clinic Journal of Medicine, Tolle and colleagues2 provide an up-to-date, broad overview of IPF focused on what the primary care provider needs to know about the disease. Their review is timely and serves as a useful primer for the practicing clinician.
The field of IPF is actively evolving, as this era has been witness to a recent paradigm shift in pharmacologic management. Immunosuppression is no longer recommended3 and may even be harmful.4 And the US Food and Drug Administration has approved 2 antifibrotic drugs—pirfenidone and nintedanib—that have been shown to delay progression of IPF.5,6
Primary care providers have a unique opportunity to play an integral role in the evaluation and care of patients with IPF, in particular with earlier disease recognition, initial disease assessment, and timely specialty consultative referral—as well as implementing a comprehensive longitudinal care plan.
EARLIER DISEASE RECOGNITION
IPF is a rare disease primarily affecting men over the age of 65.1 It is reasonable to presume that many or most of these individuals ultimately diagnosed with IPF are already seeking routine care for existing common medical conditions such as hypertension or dyslipidemia—or at least having periodic routine health maintenance assessments. Such evaluations may offer an opportunity for earlier recognition of an underlying fibrotic lung disease that may be subclinical in nature.
IPF has a lower-lung zone predominance. The importance of chest auscultation, particularly listening carefully to the lung bases, is poignantly highlighted in a recent editorial: “It is time that the stethoscope draped around the neck of physicians, which tends to be used for identification purposes rather than for medical diagnosis, be also the (presently only) genuine tool for an earlier diagnosis of IPF.”7
Advances in imaging also provide an opportunity for earlier diagnosis. Many patients undergo screening computed tomography for coronary calcium scoring or lung cancer surveillance, and these studies may incidentally identify subtle interstitial lung abnormalities. These incidental findings should lead to further investigation, as they have been shown to be functionally important and carry risk of progression to clinical interstitial lung disease.8
INITIAL ASSESSMENT, TIMELY REFERRAL
But whether evidence of interstitial lung disease is detected incidentally or during testing for respiratory symptoms, further evaluation is necessary. Primary care providers are uniquely positioned to initiate the assessment and to expedite and guide further evaluation and specialty referral consultation to ensure an accurate diagnosis. They can also help grade the severity of the disease with pulmonary function testing, oxygen assessments at rest and with ambulation, and ordering thoracic high-resolution computed tomography to provide valuable information about disease extent and interstitial lung disease pattern.
General practitioners may assess for features suggesting connective tissue disease that would warrant specific serologic testing and dedicated rheumatologic consultation.
Finally, given the rarity, complexity, and challenges of interstitial lung disease, an effective multidisciplinary team consisting of clinicians, radiologists, and pathologists enhances diagnostic accuracy.9 This may also help general practitioners deviate from normal patterns of referral to general pulmonary providers, and instead refer patients to specialized centers with dedicated clinical and research expertise in interstitial lung disease.
IMPLEMENTING A COMPREHENSIVE, LONGITUDINAL CARE PLAN
The primary care practitioner often has developed long-term relationships with patients ultimately diagnosed with IPF, and because of this is particularly well positioned to help implement a collaborative and comprehensive care plan. Logistical realities such as distance to a specialty center, limited insurance coverage for specialty visits, and limited specialty availability all reinforce the central role that primary care practitioners play in ensuring that patients adhere to a comprehensive treatment program.
Primary providers may be very experienced and more inclined to manage a number of the common and often important comorbid conditions seen in patients with IPF, such as gastroesophageal reflux disease, obstructive sleep apnea, and depression. Reinforcing to the patient the need to adhere to adjunctive therapies such as supplemental oxygen and pulmonary rehabilitation is another key opportunity to actively engage in the management of patients with IPF.
Primary providers may also play a central role in IPF care through prevention strategies such as smoking cessation and ensuring appropriate immunization against seasonal influenza, pneumococcal pneumonia, and pertussis, among other age-appropriate vaccinations.
With the introduction and expansion of use of nintedanib and pirfenidone for IPF over the past few years, general practitioners may be called on to help manage common gastrointestinal side effects associated with pirfenidone (primarily nausea) and nintedanib (primarily diarrhea), and to be aware of potential drug-drug interactions and other medication-related toxicities.
Finally, as IPF remains a progressive disease, primary care practitioners are often well positioned to help implement palliative care, hospice care, and end-of-life care.
Despite recent advances in treatment, IPF remains a devastating lung disease with a high degree of morbidity and mortality. It takes a village to help care for the IPF patient. And as key members of the healthcare team, primary care providers have unique and important opportunities to help in the early recognition, thorough assessment, and comprehensive management of patients with IPF.
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Tolle L, Southern BD, Culver D, Horowitz JC. Idiopathic pulmonary fibrosis: what primary care physicians need to know. Cleve Clin J Med 2018; 85(5):377–386. doi:10.3949/ccjm.85a.17018
- Raghu G, Richeldi L. Current approaches to the management of idiopathic pulmonary fibrosis. Respir Med 2017; 129:24–30. doi:10.1016/j.rmed.2017.05.017
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Cottin V, Cordier JF. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J 2012; 40(3):519–521. doi:10.1183/09031936.00001612
- Doyle TJ, Hunninghake GM, Rosas IO. Subclinical interstitial lung disease: why you should care. Am J Respir Crit Care Med 2012; 185(11):1147–1153. doi:10.1164/rccm.201108-1420PP
- Walsh SLF, Maher TM, Kolb M, et al; IPF Project Consortium. Diagnostic accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: an international case-cohort study. Eur Respir J 2017; 50(2):1700936. doi:10.1183/13993003.00936-2017
Idiopathic pulmonary fibrosis (IPF) is a devastating progressive fibrosing interstitial lung disease associated with a high burden of morbidity and death.1 A clinical diagnosis of IPF is made only after careful interpretation of integrated clinical, radiologic, and often histopathologic data.
Interstitial lung disease encompasses a broad spectrum of parenchymal lung diseases, and a classification of IPF is restricted to a lung injury pattern of usual interstitial pneumonia (UIP) based on high-resolution computed tomography or surgical lung biopsy, after all known causes of UIP have been excluded.1
However, a lung injury pattern of UIP is not synonymous with IPF, as UIP can be seen with connective tissue disease, chronic hypersensitivity pneumonitis, drug toxicity, and sarcoidosis.1 As such, rendering a diagnosis of IPF requires a thorough evaluation to exclude such diverse potential etiologies.
In this issue of the Cleveland Clinic Journal of Medicine, Tolle and colleagues2 provide an up-to-date, broad overview of IPF focused on what the primary care provider needs to know about the disease. Their review is timely and serves as a useful primer for the practicing clinician.
The field of IPF is actively evolving, as this era has been witness to a recent paradigm shift in pharmacologic management. Immunosuppression is no longer recommended3 and may even be harmful.4 And the US Food and Drug Administration has approved 2 antifibrotic drugs—pirfenidone and nintedanib—that have been shown to delay progression of IPF.5,6
Primary care providers have a unique opportunity to play an integral role in the evaluation and care of patients with IPF, in particular with earlier disease recognition, initial disease assessment, and timely specialty consultative referral—as well as implementing a comprehensive longitudinal care plan.
EARLIER DISEASE RECOGNITION
IPF is a rare disease primarily affecting men over the age of 65.1 It is reasonable to presume that many or most of these individuals ultimately diagnosed with IPF are already seeking routine care for existing common medical conditions such as hypertension or dyslipidemia—or at least having periodic routine health maintenance assessments. Such evaluations may offer an opportunity for earlier recognition of an underlying fibrotic lung disease that may be subclinical in nature.
IPF has a lower-lung zone predominance. The importance of chest auscultation, particularly listening carefully to the lung bases, is poignantly highlighted in a recent editorial: “It is time that the stethoscope draped around the neck of physicians, which tends to be used for identification purposes rather than for medical diagnosis, be also the (presently only) genuine tool for an earlier diagnosis of IPF.”7
Advances in imaging also provide an opportunity for earlier diagnosis. Many patients undergo screening computed tomography for coronary calcium scoring or lung cancer surveillance, and these studies may incidentally identify subtle interstitial lung abnormalities. These incidental findings should lead to further investigation, as they have been shown to be functionally important and carry risk of progression to clinical interstitial lung disease.8
INITIAL ASSESSMENT, TIMELY REFERRAL
But whether evidence of interstitial lung disease is detected incidentally or during testing for respiratory symptoms, further evaluation is necessary. Primary care providers are uniquely positioned to initiate the assessment and to expedite and guide further evaluation and specialty referral consultation to ensure an accurate diagnosis. They can also help grade the severity of the disease with pulmonary function testing, oxygen assessments at rest and with ambulation, and ordering thoracic high-resolution computed tomography to provide valuable information about disease extent and interstitial lung disease pattern.
General practitioners may assess for features suggesting connective tissue disease that would warrant specific serologic testing and dedicated rheumatologic consultation.
Finally, given the rarity, complexity, and challenges of interstitial lung disease, an effective multidisciplinary team consisting of clinicians, radiologists, and pathologists enhances diagnostic accuracy.9 This may also help general practitioners deviate from normal patterns of referral to general pulmonary providers, and instead refer patients to specialized centers with dedicated clinical and research expertise in interstitial lung disease.
IMPLEMENTING A COMPREHENSIVE, LONGITUDINAL CARE PLAN
The primary care practitioner often has developed long-term relationships with patients ultimately diagnosed with IPF, and because of this is particularly well positioned to help implement a collaborative and comprehensive care plan. Logistical realities such as distance to a specialty center, limited insurance coverage for specialty visits, and limited specialty availability all reinforce the central role that primary care practitioners play in ensuring that patients adhere to a comprehensive treatment program.
Primary providers may be very experienced and more inclined to manage a number of the common and often important comorbid conditions seen in patients with IPF, such as gastroesophageal reflux disease, obstructive sleep apnea, and depression. Reinforcing to the patient the need to adhere to adjunctive therapies such as supplemental oxygen and pulmonary rehabilitation is another key opportunity to actively engage in the management of patients with IPF.
Primary providers may also play a central role in IPF care through prevention strategies such as smoking cessation and ensuring appropriate immunization against seasonal influenza, pneumococcal pneumonia, and pertussis, among other age-appropriate vaccinations.
With the introduction and expansion of use of nintedanib and pirfenidone for IPF over the past few years, general practitioners may be called on to help manage common gastrointestinal side effects associated with pirfenidone (primarily nausea) and nintedanib (primarily diarrhea), and to be aware of potential drug-drug interactions and other medication-related toxicities.
Finally, as IPF remains a progressive disease, primary care practitioners are often well positioned to help implement palliative care, hospice care, and end-of-life care.
Despite recent advances in treatment, IPF remains a devastating lung disease with a high degree of morbidity and mortality. It takes a village to help care for the IPF patient. And as key members of the healthcare team, primary care providers have unique and important opportunities to help in the early recognition, thorough assessment, and comprehensive management of patients with IPF.
Idiopathic pulmonary fibrosis (IPF) is a devastating progressive fibrosing interstitial lung disease associated with a high burden of morbidity and death.1 A clinical diagnosis of IPF is made only after careful interpretation of integrated clinical, radiologic, and often histopathologic data.
Interstitial lung disease encompasses a broad spectrum of parenchymal lung diseases, and a classification of IPF is restricted to a lung injury pattern of usual interstitial pneumonia (UIP) based on high-resolution computed tomography or surgical lung biopsy, after all known causes of UIP have been excluded.1
However, a lung injury pattern of UIP is not synonymous with IPF, as UIP can be seen with connective tissue disease, chronic hypersensitivity pneumonitis, drug toxicity, and sarcoidosis.1 As such, rendering a diagnosis of IPF requires a thorough evaluation to exclude such diverse potential etiologies.
In this issue of the Cleveland Clinic Journal of Medicine, Tolle and colleagues2 provide an up-to-date, broad overview of IPF focused on what the primary care provider needs to know about the disease. Their review is timely and serves as a useful primer for the practicing clinician.
The field of IPF is actively evolving, as this era has been witness to a recent paradigm shift in pharmacologic management. Immunosuppression is no longer recommended3 and may even be harmful.4 And the US Food and Drug Administration has approved 2 antifibrotic drugs—pirfenidone and nintedanib—that have been shown to delay progression of IPF.5,6
Primary care providers have a unique opportunity to play an integral role in the evaluation and care of patients with IPF, in particular with earlier disease recognition, initial disease assessment, and timely specialty consultative referral—as well as implementing a comprehensive longitudinal care plan.
EARLIER DISEASE RECOGNITION
IPF is a rare disease primarily affecting men over the age of 65.1 It is reasonable to presume that many or most of these individuals ultimately diagnosed with IPF are already seeking routine care for existing common medical conditions such as hypertension or dyslipidemia—or at least having periodic routine health maintenance assessments. Such evaluations may offer an opportunity for earlier recognition of an underlying fibrotic lung disease that may be subclinical in nature.
IPF has a lower-lung zone predominance. The importance of chest auscultation, particularly listening carefully to the lung bases, is poignantly highlighted in a recent editorial: “It is time that the stethoscope draped around the neck of physicians, which tends to be used for identification purposes rather than for medical diagnosis, be also the (presently only) genuine tool for an earlier diagnosis of IPF.”7
Advances in imaging also provide an opportunity for earlier diagnosis. Many patients undergo screening computed tomography for coronary calcium scoring or lung cancer surveillance, and these studies may incidentally identify subtle interstitial lung abnormalities. These incidental findings should lead to further investigation, as they have been shown to be functionally important and carry risk of progression to clinical interstitial lung disease.8
INITIAL ASSESSMENT, TIMELY REFERRAL
But whether evidence of interstitial lung disease is detected incidentally or during testing for respiratory symptoms, further evaluation is necessary. Primary care providers are uniquely positioned to initiate the assessment and to expedite and guide further evaluation and specialty referral consultation to ensure an accurate diagnosis. They can also help grade the severity of the disease with pulmonary function testing, oxygen assessments at rest and with ambulation, and ordering thoracic high-resolution computed tomography to provide valuable information about disease extent and interstitial lung disease pattern.
General practitioners may assess for features suggesting connective tissue disease that would warrant specific serologic testing and dedicated rheumatologic consultation.
Finally, given the rarity, complexity, and challenges of interstitial lung disease, an effective multidisciplinary team consisting of clinicians, radiologists, and pathologists enhances diagnostic accuracy.9 This may also help general practitioners deviate from normal patterns of referral to general pulmonary providers, and instead refer patients to specialized centers with dedicated clinical and research expertise in interstitial lung disease.
IMPLEMENTING A COMPREHENSIVE, LONGITUDINAL CARE PLAN
The primary care practitioner often has developed long-term relationships with patients ultimately diagnosed with IPF, and because of this is particularly well positioned to help implement a collaborative and comprehensive care plan. Logistical realities such as distance to a specialty center, limited insurance coverage for specialty visits, and limited specialty availability all reinforce the central role that primary care practitioners play in ensuring that patients adhere to a comprehensive treatment program.
Primary providers may be very experienced and more inclined to manage a number of the common and often important comorbid conditions seen in patients with IPF, such as gastroesophageal reflux disease, obstructive sleep apnea, and depression. Reinforcing to the patient the need to adhere to adjunctive therapies such as supplemental oxygen and pulmonary rehabilitation is another key opportunity to actively engage in the management of patients with IPF.
Primary providers may also play a central role in IPF care through prevention strategies such as smoking cessation and ensuring appropriate immunization against seasonal influenza, pneumococcal pneumonia, and pertussis, among other age-appropriate vaccinations.
With the introduction and expansion of use of nintedanib and pirfenidone for IPF over the past few years, general practitioners may be called on to help manage common gastrointestinal side effects associated with pirfenidone (primarily nausea) and nintedanib (primarily diarrhea), and to be aware of potential drug-drug interactions and other medication-related toxicities.
Finally, as IPF remains a progressive disease, primary care practitioners are often well positioned to help implement palliative care, hospice care, and end-of-life care.
Despite recent advances in treatment, IPF remains a devastating lung disease with a high degree of morbidity and mortality. It takes a village to help care for the IPF patient. And as key members of the healthcare team, primary care providers have unique and important opportunities to help in the early recognition, thorough assessment, and comprehensive management of patients with IPF.
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Tolle L, Southern BD, Culver D, Horowitz JC. Idiopathic pulmonary fibrosis: what primary care physicians need to know. Cleve Clin J Med 2018; 85(5):377–386. doi:10.3949/ccjm.85a.17018
- Raghu G, Richeldi L. Current approaches to the management of idiopathic pulmonary fibrosis. Respir Med 2017; 129:24–30. doi:10.1016/j.rmed.2017.05.017
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Cottin V, Cordier JF. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J 2012; 40(3):519–521. doi:10.1183/09031936.00001612
- Doyle TJ, Hunninghake GM, Rosas IO. Subclinical interstitial lung disease: why you should care. Am J Respir Crit Care Med 2012; 185(11):1147–1153. doi:10.1164/rccm.201108-1420PP
- Walsh SLF, Maher TM, Kolb M, et al; IPF Project Consortium. Diagnostic accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: an international case-cohort study. Eur Respir J 2017; 50(2):1700936. doi:10.1183/13993003.00936-2017
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Tolle L, Southern BD, Culver D, Horowitz JC. Idiopathic pulmonary fibrosis: what primary care physicians need to know. Cleve Clin J Med 2018; 85(5):377–386. doi:10.3949/ccjm.85a.17018
- Raghu G, Richeldi L. Current approaches to the management of idiopathic pulmonary fibrosis. Respir Med 2017; 129:24–30. doi:10.1016/j.rmed.2017.05.017
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Cottin V, Cordier JF. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J 2012; 40(3):519–521. doi:10.1183/09031936.00001612
- Doyle TJ, Hunninghake GM, Rosas IO. Subclinical interstitial lung disease: why you should care. Am J Respir Crit Care Med 2012; 185(11):1147–1153. doi:10.1164/rccm.201108-1420PP
- Walsh SLF, Maher TM, Kolb M, et al; IPF Project Consortium. Diagnostic accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: an international case-cohort study. Eur Respir J 2017; 50(2):1700936. doi:10.1183/13993003.00936-2017

Eyes of the mimicker
Lumbar puncture study revealed 34 nucleated cells/µL (94% lymphocytes), protein 58 mg/dL, and glucose 62 mg/dL. Cerebrospinal fluid Venereal Disease Research Laboratory and fluorescent treponemal antibody absorption tests were reactive, confirming a diagnosis of ocular syphilis.
The patient was admitted to the hospital for treatment with intravenous penicillin G. After 5 days, he was discharged with instructions to complete a 10-day course of intravenous ceftriaxone (chosen for its ease of administration), for a total of 14 days of antibiotic therapy. His vision improved with treatment.
He continued to follow up with ophthalmology and infectious disease. Subsequent dilated fundus examinations showed resolution of pathology in the left eye, resolution of Roth spots in the right eye, and resolution of the subhyaloid hemorrhage. Repeat cerebrospinal fluid study examination was planned if the serum rapid plasma reagin had not become nonreactive 24 months after treatment.
RECOGNIZING AND MANAGING OCULAR SYPHILIS AND NEUROSYPHILIS
In addition to ocular syphilis and neurosyphilis, the differential diagnosis for Roth spots and disc edema on dilated funduscopy includes endocarditis, viral retinitis, and autoimmune or inflammatory conditions such as sarcoidosis and vasculitis.
In our patient, infectious endocarditis was considered, given his history of intermittent fevers and rigors, but it was ultimately ruled out by negative blood cultures and the absence of valvular vegetations on echocardiography.
The large subhyaloid hemorrhage raised suspicion of leukemia, but this was ruled out by the normal total white blood cell count and differential. HIV, herpetic retinitis, and toxoplasmosis were also considered, but laboratory tests for these infections were negative.
Typically, retinal precipitates are more characteristic of syphilitic retinitis and distinguish it from other infectious causes such as herpetic retinitis and toxoplasmosis.1 Additionally, ocular syphilis more commonly manifests as uveitis or panuveitis.1,2 Our patient’s ocular syphilis presented with white-centered retinal hemorrhages, subhyaloid hemorrhage, and optic disc edema.
Who is at highest risk?
About 90% of syphilis cases occur in men, and 81% occur in men who have sex with men. The US Centers for Disease Control and Prevention (CDC) thus recommends annual syphilis testing for men who have sex with men.3
Classically, syphilis was called “the great imitator” because it mimicked manifestations of other diseases. Patients with ocular manifestations of syphilis may not have other neurologic symptoms.4,5 Nevertheless, cerebrospinal fluid examination should be done in all instances of ocular syphilis, as many patients with ocular syphilis have evidence of neurosyphilis on testing.2 The CDC also recommends follow-up cerebrospinal fluid analysis to assess treatment response.2 This was planned in our patient.
- Fu EX, Geraets RL, Dodds EM, et al. Superficial retinal precipitates in patients with syphilitic retinitis. Retina 2010; 30(7):1135–1143. doi:10.1097/IAE.0b013e3181cdf3ae
- US Centers for Disease Control and Prevention. Sexually Transmitted Diseases. Clinical Advisory: Ocular Syphilis in the United States, March 24, 2016. www.cdc.gov/std/syphilis/clinicaladvisoryos2015.htm. Accessed March 28, 2018.
- US Centers for Disease Control and Prevention. Sexually Transmitted Disease Surveillance, 2015. www.cdc.gov/std/stats15/std-surveillance-2015-print.pdf. Accessed March 28, 2018.
- Rishi E, Govindarajan MV, Biswas J, Agarwal M, Sudharshan S, Rishi P. Syphilitic uveitis as the presenting feature of HIV. Indian J Ophthalmol 2016; 64(2):149–150. doi:10.4103/0301-4738.179714
- Zhang R, Qian J, Guo J, et al. Clinical manifestations and treatment outcomes of syphilitic uveitis in a Chinese population. J Ophthalmol 2016; 2016:2797028. doi:10.1155/2016/2797028
Lumbar puncture study revealed 34 nucleated cells/µL (94% lymphocytes), protein 58 mg/dL, and glucose 62 mg/dL. Cerebrospinal fluid Venereal Disease Research Laboratory and fluorescent treponemal antibody absorption tests were reactive, confirming a diagnosis of ocular syphilis.
The patient was admitted to the hospital for treatment with intravenous penicillin G. After 5 days, he was discharged with instructions to complete a 10-day course of intravenous ceftriaxone (chosen for its ease of administration), for a total of 14 days of antibiotic therapy. His vision improved with treatment.
He continued to follow up with ophthalmology and infectious disease. Subsequent dilated fundus examinations showed resolution of pathology in the left eye, resolution of Roth spots in the right eye, and resolution of the subhyaloid hemorrhage. Repeat cerebrospinal fluid study examination was planned if the serum rapid plasma reagin had not become nonreactive 24 months after treatment.
RECOGNIZING AND MANAGING OCULAR SYPHILIS AND NEUROSYPHILIS
In addition to ocular syphilis and neurosyphilis, the differential diagnosis for Roth spots and disc edema on dilated funduscopy includes endocarditis, viral retinitis, and autoimmune or inflammatory conditions such as sarcoidosis and vasculitis.
In our patient, infectious endocarditis was considered, given his history of intermittent fevers and rigors, but it was ultimately ruled out by negative blood cultures and the absence of valvular vegetations on echocardiography.
The large subhyaloid hemorrhage raised suspicion of leukemia, but this was ruled out by the normal total white blood cell count and differential. HIV, herpetic retinitis, and toxoplasmosis were also considered, but laboratory tests for these infections were negative.
Typically, retinal precipitates are more characteristic of syphilitic retinitis and distinguish it from other infectious causes such as herpetic retinitis and toxoplasmosis.1 Additionally, ocular syphilis more commonly manifests as uveitis or panuveitis.1,2 Our patient’s ocular syphilis presented with white-centered retinal hemorrhages, subhyaloid hemorrhage, and optic disc edema.
Who is at highest risk?
About 90% of syphilis cases occur in men, and 81% occur in men who have sex with men. The US Centers for Disease Control and Prevention (CDC) thus recommends annual syphilis testing for men who have sex with men.3
Classically, syphilis was called “the great imitator” because it mimicked manifestations of other diseases. Patients with ocular manifestations of syphilis may not have other neurologic symptoms.4,5 Nevertheless, cerebrospinal fluid examination should be done in all instances of ocular syphilis, as many patients with ocular syphilis have evidence of neurosyphilis on testing.2 The CDC also recommends follow-up cerebrospinal fluid analysis to assess treatment response.2 This was planned in our patient.
Lumbar puncture study revealed 34 nucleated cells/µL (94% lymphocytes), protein 58 mg/dL, and glucose 62 mg/dL. Cerebrospinal fluid Venereal Disease Research Laboratory and fluorescent treponemal antibody absorption tests were reactive, confirming a diagnosis of ocular syphilis.
The patient was admitted to the hospital for treatment with intravenous penicillin G. After 5 days, he was discharged with instructions to complete a 10-day course of intravenous ceftriaxone (chosen for its ease of administration), for a total of 14 days of antibiotic therapy. His vision improved with treatment.
He continued to follow up with ophthalmology and infectious disease. Subsequent dilated fundus examinations showed resolution of pathology in the left eye, resolution of Roth spots in the right eye, and resolution of the subhyaloid hemorrhage. Repeat cerebrospinal fluid study examination was planned if the serum rapid plasma reagin had not become nonreactive 24 months after treatment.
RECOGNIZING AND MANAGING OCULAR SYPHILIS AND NEUROSYPHILIS
In addition to ocular syphilis and neurosyphilis, the differential diagnosis for Roth spots and disc edema on dilated funduscopy includes endocarditis, viral retinitis, and autoimmune or inflammatory conditions such as sarcoidosis and vasculitis.
In our patient, infectious endocarditis was considered, given his history of intermittent fevers and rigors, but it was ultimately ruled out by negative blood cultures and the absence of valvular vegetations on echocardiography.
The large subhyaloid hemorrhage raised suspicion of leukemia, but this was ruled out by the normal total white blood cell count and differential. HIV, herpetic retinitis, and toxoplasmosis were also considered, but laboratory tests for these infections were negative.
Typically, retinal precipitates are more characteristic of syphilitic retinitis and distinguish it from other infectious causes such as herpetic retinitis and toxoplasmosis.1 Additionally, ocular syphilis more commonly manifests as uveitis or panuveitis.1,2 Our patient’s ocular syphilis presented with white-centered retinal hemorrhages, subhyaloid hemorrhage, and optic disc edema.
Who is at highest risk?
About 90% of syphilis cases occur in men, and 81% occur in men who have sex with men. The US Centers for Disease Control and Prevention (CDC) thus recommends annual syphilis testing for men who have sex with men.3
Classically, syphilis was called “the great imitator” because it mimicked manifestations of other diseases. Patients with ocular manifestations of syphilis may not have other neurologic symptoms.4,5 Nevertheless, cerebrospinal fluid examination should be done in all instances of ocular syphilis, as many patients with ocular syphilis have evidence of neurosyphilis on testing.2 The CDC also recommends follow-up cerebrospinal fluid analysis to assess treatment response.2 This was planned in our patient.
- Fu EX, Geraets RL, Dodds EM, et al. Superficial retinal precipitates in patients with syphilitic retinitis. Retina 2010; 30(7):1135–1143. doi:10.1097/IAE.0b013e3181cdf3ae
- US Centers for Disease Control and Prevention. Sexually Transmitted Diseases. Clinical Advisory: Ocular Syphilis in the United States, March 24, 2016. www.cdc.gov/std/syphilis/clinicaladvisoryos2015.htm. Accessed March 28, 2018.
- US Centers for Disease Control and Prevention. Sexually Transmitted Disease Surveillance, 2015. www.cdc.gov/std/stats15/std-surveillance-2015-print.pdf. Accessed March 28, 2018.
- Rishi E, Govindarajan MV, Biswas J, Agarwal M, Sudharshan S, Rishi P. Syphilitic uveitis as the presenting feature of HIV. Indian J Ophthalmol 2016; 64(2):149–150. doi:10.4103/0301-4738.179714
- Zhang R, Qian J, Guo J, et al. Clinical manifestations and treatment outcomes of syphilitic uveitis in a Chinese population. J Ophthalmol 2016; 2016:2797028. doi:10.1155/2016/2797028
- Fu EX, Geraets RL, Dodds EM, et al. Superficial retinal precipitates in patients with syphilitic retinitis. Retina 2010; 30(7):1135–1143. doi:10.1097/IAE.0b013e3181cdf3ae
- US Centers for Disease Control and Prevention. Sexually Transmitted Diseases. Clinical Advisory: Ocular Syphilis in the United States, March 24, 2016. www.cdc.gov/std/syphilis/clinicaladvisoryos2015.htm. Accessed March 28, 2018.
- US Centers for Disease Control and Prevention. Sexually Transmitted Disease Surveillance, 2015. www.cdc.gov/std/stats15/std-surveillance-2015-print.pdf. Accessed March 28, 2018.
- Rishi E, Govindarajan MV, Biswas J, Agarwal M, Sudharshan S, Rishi P. Syphilitic uveitis as the presenting feature of HIV. Indian J Ophthalmol 2016; 64(2):149–150. doi:10.4103/0301-4738.179714
- Zhang R, Qian J, Guo J, et al. Clinical manifestations and treatment outcomes of syphilitic uveitis in a Chinese population. J Ophthalmol 2016; 2016:2797028. doi:10.1155/2016/2797028