User login
Can intraputamenal infusions of GDNF treat Parkinson’s disease?
researchers reported. The investigational therapy, delivered through a skull-mounted port, was well tolerated in a 40-week, randomized, controlled trial and a 40-week, open-label extension.
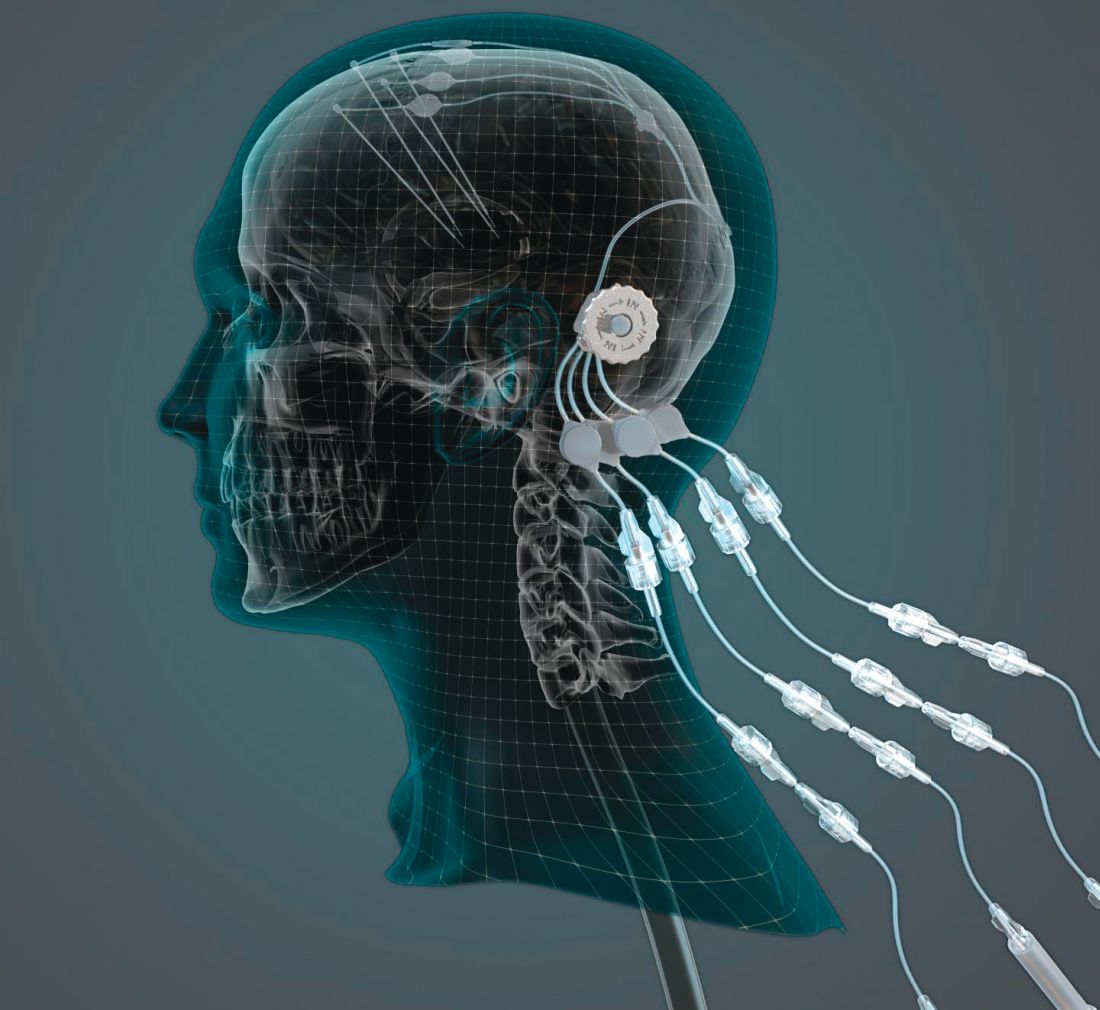
Neither study met its primary endpoint, but post hoc analyses suggest possible clinical benefits. In addition, PET imaging after the 40-week, randomized trial found significantly increased 18F-DOPA uptake in patients who received GDNF. The randomized trial was published in the March 2019 issue of Brain; data from the open-label extension were published online ahead of print Feb. 26, 2019, in the Journal of Parkinson’s Disease.
“The spatial and relative magnitude of the improvement in the brain scans is beyond anything seen previously in trials of surgically delivered growth-factor treatments for Parkinson’s [disease],” said principal investigator Alan L. Whone, MBChB, PhD, of the University of Bristol (England) and North Bristol National Health Service Trust. “This represents some of the most compelling evidence yet that we may have a means to possibly reawaken and restore the dopamine brain cells that are gradually destroyed in Parkinson’s [disease].”
Nevertheless, the trial did not confirm clinical benefits. The hypothesis that growth factors can benefit patients with Parkinson’s disease may be incorrect, the researchers acknowledged. It also is possible that the hypothesis is valid and that a trial with a higher GDNF dose, longer treatment duration, patients with an earlier disease stage, or different outcome measures would yield positive results. GDNF warrants further study, they wrote.
The findings could have implications for other neurologic disorders as well.
“This trial has shown that we can safely and repeatedly infuse drugs directly into patients’ brains over months or years. This is a significant breakthrough in our ability to treat neurologic conditions ... because most drugs that might work cannot cross from the bloodstream into the brain,” said Steven Gill, MB, MS. Mr. Gill, of the North Bristol NHS Trust and the U.K.-based engineering firm Renishaw, designed the convection-enhanced delivery system used in the studies.
A neurotrophic protein
GDNF has neurorestorative and neuroprotective effects in animal models of Parkinson’s disease. In open-label studies, continuous, low-rate intraputamenal administration of GDNF has shown signs of potential efficacy, but a placebo-controlled trial did not replicate clinical benefits. In the present studies, the researchers assessed intermittent GDNF administration using convection-enhanced delivery, which can achieve wider and more even distribution of GDNF, compared with the previous approach.
The researchers conducted a single-center, randomized, double-blind, placebo-controlled trial to study this novel administration approach. Patients were aged 35-75 years, had motor symptoms for at least 5 years, and had moderate disease severity in the off state (that is, Hoehn and Yahr stage 2-3 and Unified Parkinson’s Disease Rating Scale motor score–part III [UPDRS-III] of 25-45).
In a pilot stage of the trial, six patients were randomized 2:1 to receive GDNF (120 mcg per putamen) or placebo. In the primary stage, another 35 patients were randomized 1:1 to GDNF or placebo. The primary outcome was the percentage change from baseline to week 40 in the off-state UPDRS-III among patients from the primary stage of the trial. Further analyses included all 41 patients from the pilot and primary stages.
Patients in the primary analysis had a mean age of 56.4 years and mean disease duration of 10.9 years. About half were female.
Results on primary and secondary clinical endpoints did not significantly differ between the groups. Average off state UPDRS motor score decreased by 17.3 in the active treatment group, compared with 11.8 in the placebo group.
A post hoc analysis, however, found that nine patients (43%) in the active-treatment group had a large, clinically important motor improvement of 10 or more points in the off state, whereas no placebo patients did. These “10-point responders in the GDNF group are a potential focus of interest; however, as this is a post hoc finding we would not wish to overinterpret its meaning,” Dr. Whone and his colleagues wrote. Among patients who received GDNF, PET imaging demonstrated significantly increased 18F-DOPA uptake throughout the putamen, ranging from a 25% increase in the left anterior putamen to a 100% increase in both posterior putamena, whereas patients who received placebo did not have significantly increased uptake.
No drug-related serious adverse events were reported. “The majority of device-related adverse events were port site associated, most commonly local hypertrophic scarring or infections, amenable to antibiotics,” the investigators wrote. “The frequency of these declined during the trial as surgical and device handling experience improved.”
Open-label extension
By week 80, when all participants had received GDNF, both groups showed moderate to large improvement in symptoms, compared with baseline. From baseline to week 80, percentage change in UPDRS motor score in the off state did not significantly differ between patients who received GDNF for 80 weeks and patients who received placebo followed by GDNF (26.7% vs. 27.6%). Secondary endpoints also did not differ between the groups. Treatment compliance was 97.8%; no patients discontinued the study.
The trials were funded by Parkinson’s UK with support from the Cure Parkinson’s Trust and in association with the North Bristol NHS Trust. GDNF and additional resources and funding were provided by MedGenesis Therapeutix, which owns the license for GDNF and received funding from the Michael J. Fox Foundation for Parkinson’s Research. Renishaw manufactured the convection-enhanced delivery device on behalf of North Bristol NHS Trust. The Gatsby Foundation provided a 3T MRI scanner. Some study authors are employed by and have shares or share options with MedGenesis Therapeutix. Other authors are employees of Renishaw. Dr. Gill is Renishaw’s medical director and may have a future royalty share from the drug delivery system that he invented.
SOURCES: Whone AL et al. Brain. 2019 Feb 26. doi: 10.1093/brain/awz023; Whone AL et al. J Parkinsons Dis. 2019 Feb 26. doi: 10.3233/JPD-191576.
researchers reported. The investigational therapy, delivered through a skull-mounted port, was well tolerated in a 40-week, randomized, controlled trial and a 40-week, open-label extension.
Neither study met its primary endpoint, but post hoc analyses suggest possible clinical benefits. In addition, PET imaging after the 40-week, randomized trial found significantly increased 18F-DOPA uptake in patients who received GDNF. The randomized trial was published in the March 2019 issue of Brain; data from the open-label extension were published online ahead of print Feb. 26, 2019, in the Journal of Parkinson’s Disease.
“The spatial and relative magnitude of the improvement in the brain scans is beyond anything seen previously in trials of surgically delivered growth-factor treatments for Parkinson’s [disease],” said principal investigator Alan L. Whone, MBChB, PhD, of the University of Bristol (England) and North Bristol National Health Service Trust. “This represents some of the most compelling evidence yet that we may have a means to possibly reawaken and restore the dopamine brain cells that are gradually destroyed in Parkinson’s [disease].”
Nevertheless, the trial did not confirm clinical benefits. The hypothesis that growth factors can benefit patients with Parkinson’s disease may be incorrect, the researchers acknowledged. It also is possible that the hypothesis is valid and that a trial with a higher GDNF dose, longer treatment duration, patients with an earlier disease stage, or different outcome measures would yield positive results. GDNF warrants further study, they wrote.
The findings could have implications for other neurologic disorders as well.
“This trial has shown that we can safely and repeatedly infuse drugs directly into patients’ brains over months or years. This is a significant breakthrough in our ability to treat neurologic conditions ... because most drugs that might work cannot cross from the bloodstream into the brain,” said Steven Gill, MB, MS. Mr. Gill, of the North Bristol NHS Trust and the U.K.-based engineering firm Renishaw, designed the convection-enhanced delivery system used in the studies.
A neurotrophic protein
GDNF has neurorestorative and neuroprotective effects in animal models of Parkinson’s disease. In open-label studies, continuous, low-rate intraputamenal administration of GDNF has shown signs of potential efficacy, but a placebo-controlled trial did not replicate clinical benefits. In the present studies, the researchers assessed intermittent GDNF administration using convection-enhanced delivery, which can achieve wider and more even distribution of GDNF, compared with the previous approach.
The researchers conducted a single-center, randomized, double-blind, placebo-controlled trial to study this novel administration approach. Patients were aged 35-75 years, had motor symptoms for at least 5 years, and had moderate disease severity in the off state (that is, Hoehn and Yahr stage 2-3 and Unified Parkinson’s Disease Rating Scale motor score–part III [UPDRS-III] of 25-45).
In a pilot stage of the trial, six patients were randomized 2:1 to receive GDNF (120 mcg per putamen) or placebo. In the primary stage, another 35 patients were randomized 1:1 to GDNF or placebo. The primary outcome was the percentage change from baseline to week 40 in the off-state UPDRS-III among patients from the primary stage of the trial. Further analyses included all 41 patients from the pilot and primary stages.
Patients in the primary analysis had a mean age of 56.4 years and mean disease duration of 10.9 years. About half were female.
Results on primary and secondary clinical endpoints did not significantly differ between the groups. Average off state UPDRS motor score decreased by 17.3 in the active treatment group, compared with 11.8 in the placebo group.
A post hoc analysis, however, found that nine patients (43%) in the active-treatment group had a large, clinically important motor improvement of 10 or more points in the off state, whereas no placebo patients did. These “10-point responders in the GDNF group are a potential focus of interest; however, as this is a post hoc finding we would not wish to overinterpret its meaning,” Dr. Whone and his colleagues wrote. Among patients who received GDNF, PET imaging demonstrated significantly increased 18F-DOPA uptake throughout the putamen, ranging from a 25% increase in the left anterior putamen to a 100% increase in both posterior putamena, whereas patients who received placebo did not have significantly increased uptake.
No drug-related serious adverse events were reported. “The majority of device-related adverse events were port site associated, most commonly local hypertrophic scarring or infections, amenable to antibiotics,” the investigators wrote. “The frequency of these declined during the trial as surgical and device handling experience improved.”
Open-label extension
By week 80, when all participants had received GDNF, both groups showed moderate to large improvement in symptoms, compared with baseline. From baseline to week 80, percentage change in UPDRS motor score in the off state did not significantly differ between patients who received GDNF for 80 weeks and patients who received placebo followed by GDNF (26.7% vs. 27.6%). Secondary endpoints also did not differ between the groups. Treatment compliance was 97.8%; no patients discontinued the study.
The trials were funded by Parkinson’s UK with support from the Cure Parkinson’s Trust and in association with the North Bristol NHS Trust. GDNF and additional resources and funding were provided by MedGenesis Therapeutix, which owns the license for GDNF and received funding from the Michael J. Fox Foundation for Parkinson’s Research. Renishaw manufactured the convection-enhanced delivery device on behalf of North Bristol NHS Trust. The Gatsby Foundation provided a 3T MRI scanner. Some study authors are employed by and have shares or share options with MedGenesis Therapeutix. Other authors are employees of Renishaw. Dr. Gill is Renishaw’s medical director and may have a future royalty share from the drug delivery system that he invented.
SOURCES: Whone AL et al. Brain. 2019 Feb 26. doi: 10.1093/brain/awz023; Whone AL et al. J Parkinsons Dis. 2019 Feb 26. doi: 10.3233/JPD-191576.
researchers reported. The investigational therapy, delivered through a skull-mounted port, was well tolerated in a 40-week, randomized, controlled trial and a 40-week, open-label extension.
Neither study met its primary endpoint, but post hoc analyses suggest possible clinical benefits. In addition, PET imaging after the 40-week, randomized trial found significantly increased 18F-DOPA uptake in patients who received GDNF. The randomized trial was published in the March 2019 issue of Brain; data from the open-label extension were published online ahead of print Feb. 26, 2019, in the Journal of Parkinson’s Disease.
“The spatial and relative magnitude of the improvement in the brain scans is beyond anything seen previously in trials of surgically delivered growth-factor treatments for Parkinson’s [disease],” said principal investigator Alan L. Whone, MBChB, PhD, of the University of Bristol (England) and North Bristol National Health Service Trust. “This represents some of the most compelling evidence yet that we may have a means to possibly reawaken and restore the dopamine brain cells that are gradually destroyed in Parkinson’s [disease].”
Nevertheless, the trial did not confirm clinical benefits. The hypothesis that growth factors can benefit patients with Parkinson’s disease may be incorrect, the researchers acknowledged. It also is possible that the hypothesis is valid and that a trial with a higher GDNF dose, longer treatment duration, patients with an earlier disease stage, or different outcome measures would yield positive results. GDNF warrants further study, they wrote.
The findings could have implications for other neurologic disorders as well.
“This trial has shown that we can safely and repeatedly infuse drugs directly into patients’ brains over months or years. This is a significant breakthrough in our ability to treat neurologic conditions ... because most drugs that might work cannot cross from the bloodstream into the brain,” said Steven Gill, MB, MS. Mr. Gill, of the North Bristol NHS Trust and the U.K.-based engineering firm Renishaw, designed the convection-enhanced delivery system used in the studies.
A neurotrophic protein
GDNF has neurorestorative and neuroprotective effects in animal models of Parkinson’s disease. In open-label studies, continuous, low-rate intraputamenal administration of GDNF has shown signs of potential efficacy, but a placebo-controlled trial did not replicate clinical benefits. In the present studies, the researchers assessed intermittent GDNF administration using convection-enhanced delivery, which can achieve wider and more even distribution of GDNF, compared with the previous approach.
The researchers conducted a single-center, randomized, double-blind, placebo-controlled trial to study this novel administration approach. Patients were aged 35-75 years, had motor symptoms for at least 5 years, and had moderate disease severity in the off state (that is, Hoehn and Yahr stage 2-3 and Unified Parkinson’s Disease Rating Scale motor score–part III [UPDRS-III] of 25-45).
In a pilot stage of the trial, six patients were randomized 2:1 to receive GDNF (120 mcg per putamen) or placebo. In the primary stage, another 35 patients were randomized 1:1 to GDNF or placebo. The primary outcome was the percentage change from baseline to week 40 in the off-state UPDRS-III among patients from the primary stage of the trial. Further analyses included all 41 patients from the pilot and primary stages.
Patients in the primary analysis had a mean age of 56.4 years and mean disease duration of 10.9 years. About half were female.
Results on primary and secondary clinical endpoints did not significantly differ between the groups. Average off state UPDRS motor score decreased by 17.3 in the active treatment group, compared with 11.8 in the placebo group.
A post hoc analysis, however, found that nine patients (43%) in the active-treatment group had a large, clinically important motor improvement of 10 or more points in the off state, whereas no placebo patients did. These “10-point responders in the GDNF group are a potential focus of interest; however, as this is a post hoc finding we would not wish to overinterpret its meaning,” Dr. Whone and his colleagues wrote. Among patients who received GDNF, PET imaging demonstrated significantly increased 18F-DOPA uptake throughout the putamen, ranging from a 25% increase in the left anterior putamen to a 100% increase in both posterior putamena, whereas patients who received placebo did not have significantly increased uptake.
No drug-related serious adverse events were reported. “The majority of device-related adverse events were port site associated, most commonly local hypertrophic scarring or infections, amenable to antibiotics,” the investigators wrote. “The frequency of these declined during the trial as surgical and device handling experience improved.”
Open-label extension
By week 80, when all participants had received GDNF, both groups showed moderate to large improvement in symptoms, compared with baseline. From baseline to week 80, percentage change in UPDRS motor score in the off state did not significantly differ between patients who received GDNF for 80 weeks and patients who received placebo followed by GDNF (26.7% vs. 27.6%). Secondary endpoints also did not differ between the groups. Treatment compliance was 97.8%; no patients discontinued the study.
The trials were funded by Parkinson’s UK with support from the Cure Parkinson’s Trust and in association with the North Bristol NHS Trust. GDNF and additional resources and funding were provided by MedGenesis Therapeutix, which owns the license for GDNF and received funding from the Michael J. Fox Foundation for Parkinson’s Research. Renishaw manufactured the convection-enhanced delivery device on behalf of North Bristol NHS Trust. The Gatsby Foundation provided a 3T MRI scanner. Some study authors are employed by and have shares or share options with MedGenesis Therapeutix. Other authors are employees of Renishaw. Dr. Gill is Renishaw’s medical director and may have a future royalty share from the drug delivery system that he invented.
SOURCES: Whone AL et al. Brain. 2019 Feb 26. doi: 10.1093/brain/awz023; Whone AL et al. J Parkinsons Dis. 2019 Feb 26. doi: 10.3233/JPD-191576.
Breast cancer survivors offer realistic strategies for easing cost burden
A qualitative study representing the patient perspective provides insight on reducing economic burden after breast cancer, including specific recommendations for changes to insurance, supportive services, financial assistance, and protective policies.
As part of a 6-month observational study conducted in 2015, Lorraine T. Dean, ScD, of Johns Hopkins Schools of Public Health and Medicine, Baltimore, and her associates, interviewed 40 women diagnosed with invasive stage I-III breast cancer who had completed active cancer treatment. All patients, who reported having more than one lymph node removed resided in Pennsylvania or New Jersey. The mean age of the women was 64 years.
Of those interviewed, 53% were white; 42.5% were black. More than half of participants (53%) were college graduates or had received a graduate degree. Annual income for 58% of the patients ranged from $30,000 to $70,000; 11% earned under $30,000. All participants included in the study were insured, including 82.5% who had private insurance. The patients had been diagnosed a mean of 12 years prior. Breast cancer–related lymphedema was reported in 60% of patients, Dr. Dean and her associates reported in a report published in Cancer.
Among the 40 participants, 27 made recommendations for easing economic burden, including nine key recommendations across four significant areas: insurance, supportive services and care, financial assistance, and protective policies. These findings are consistent with previous studies that examined patient recommendations, but they address additional areas where cost-saving services and policies could be offered or improved upon, the investigators noted.
Insurance-related recommendations included offering more complementary and integrative treatments as well as helping patients understand what insurance plans cover and how to adjust to changes under new insurance plans. Providing high-quality plans with low copays, premiums, and deductibles that cover required as well as elective cancer-related services, and covering lymphedema-related materials and treatments also were flagged as important.
Supportive service recommendations included addressing psychosocial costs through expansion of support groups and buddy services, offering extended home health services following cancer treatment, and providing domestic assistance with household chores, child care, and transportation.
Financial assistance that broadens financial aid and social services eligibility to those not classified as being in poverty was considered important.
Protective policy recommendations focused on expanding employment and medical leave policies concerning the amount of time offered off from work.
Patient recommendations offer just one viewpoint concerning potential challenges to the overall system, but “their thoughts on how it can be improved add value to decision-making processes,” noted Dr. Dean and her associates.
They were careful to acknowledge the benefits of the Patient Protection and Affordable Care Act, but they noted that it does not include provisions to address the adverse treatment effects of conditions such as cancer. While some states already have successfully passed legislation requiring private insurance carriers to cover lymphedema treatment, similar legislation should be adopted at a national level through joint efforts of Congress and the Department of Labor, they advised.
Any such efforts to make sweeping changes within the insurance industry would take considerable effort on the part of patients, providers, insurers, and state and federal policy makers, as well as the pharmaceutical industry. Yet, such “top-down and bottom-up strategies that involve all parties are warranted,” they urged.
Several important limitations of the study are worth noting. All participants were from the East Coast, had insurance coverage, and reported an overall low level of economic burden. Responses may have differed had the study been conducted in other regions of the country. The study was voluntary, so it is important to consider that patients with greater financial challenges may not have had time to enroll and participate, which suggests that the level of economic burden affecting this population actually could be understated.
SOURCE: Dean LT et al. Cancer 2019 Mar 6. doi: 10.1002/cncr.32012.
A qualitative study representing the patient perspective provides insight on reducing economic burden after breast cancer, including specific recommendations for changes to insurance, supportive services, financial assistance, and protective policies.
As part of a 6-month observational study conducted in 2015, Lorraine T. Dean, ScD, of Johns Hopkins Schools of Public Health and Medicine, Baltimore, and her associates, interviewed 40 women diagnosed with invasive stage I-III breast cancer who had completed active cancer treatment. All patients, who reported having more than one lymph node removed resided in Pennsylvania or New Jersey. The mean age of the women was 64 years.
Of those interviewed, 53% were white; 42.5% were black. More than half of participants (53%) were college graduates or had received a graduate degree. Annual income for 58% of the patients ranged from $30,000 to $70,000; 11% earned under $30,000. All participants included in the study were insured, including 82.5% who had private insurance. The patients had been diagnosed a mean of 12 years prior. Breast cancer–related lymphedema was reported in 60% of patients, Dr. Dean and her associates reported in a report published in Cancer.
Among the 40 participants, 27 made recommendations for easing economic burden, including nine key recommendations across four significant areas: insurance, supportive services and care, financial assistance, and protective policies. These findings are consistent with previous studies that examined patient recommendations, but they address additional areas where cost-saving services and policies could be offered or improved upon, the investigators noted.
Insurance-related recommendations included offering more complementary and integrative treatments as well as helping patients understand what insurance plans cover and how to adjust to changes under new insurance plans. Providing high-quality plans with low copays, premiums, and deductibles that cover required as well as elective cancer-related services, and covering lymphedema-related materials and treatments also were flagged as important.
Supportive service recommendations included addressing psychosocial costs through expansion of support groups and buddy services, offering extended home health services following cancer treatment, and providing domestic assistance with household chores, child care, and transportation.
Financial assistance that broadens financial aid and social services eligibility to those not classified as being in poverty was considered important.
Protective policy recommendations focused on expanding employment and medical leave policies concerning the amount of time offered off from work.
Patient recommendations offer just one viewpoint concerning potential challenges to the overall system, but “their thoughts on how it can be improved add value to decision-making processes,” noted Dr. Dean and her associates.
They were careful to acknowledge the benefits of the Patient Protection and Affordable Care Act, but they noted that it does not include provisions to address the adverse treatment effects of conditions such as cancer. While some states already have successfully passed legislation requiring private insurance carriers to cover lymphedema treatment, similar legislation should be adopted at a national level through joint efforts of Congress and the Department of Labor, they advised.
Any such efforts to make sweeping changes within the insurance industry would take considerable effort on the part of patients, providers, insurers, and state and federal policy makers, as well as the pharmaceutical industry. Yet, such “top-down and bottom-up strategies that involve all parties are warranted,” they urged.
Several important limitations of the study are worth noting. All participants were from the East Coast, had insurance coverage, and reported an overall low level of economic burden. Responses may have differed had the study been conducted in other regions of the country. The study was voluntary, so it is important to consider that patients with greater financial challenges may not have had time to enroll and participate, which suggests that the level of economic burden affecting this population actually could be understated.
SOURCE: Dean LT et al. Cancer 2019 Mar 6. doi: 10.1002/cncr.32012.
A qualitative study representing the patient perspective provides insight on reducing economic burden after breast cancer, including specific recommendations for changes to insurance, supportive services, financial assistance, and protective policies.
As part of a 6-month observational study conducted in 2015, Lorraine T. Dean, ScD, of Johns Hopkins Schools of Public Health and Medicine, Baltimore, and her associates, interviewed 40 women diagnosed with invasive stage I-III breast cancer who had completed active cancer treatment. All patients, who reported having more than one lymph node removed resided in Pennsylvania or New Jersey. The mean age of the women was 64 years.
Of those interviewed, 53% were white; 42.5% were black. More than half of participants (53%) were college graduates or had received a graduate degree. Annual income for 58% of the patients ranged from $30,000 to $70,000; 11% earned under $30,000. All participants included in the study were insured, including 82.5% who had private insurance. The patients had been diagnosed a mean of 12 years prior. Breast cancer–related lymphedema was reported in 60% of patients, Dr. Dean and her associates reported in a report published in Cancer.
Among the 40 participants, 27 made recommendations for easing economic burden, including nine key recommendations across four significant areas: insurance, supportive services and care, financial assistance, and protective policies. These findings are consistent with previous studies that examined patient recommendations, but they address additional areas where cost-saving services and policies could be offered or improved upon, the investigators noted.
Insurance-related recommendations included offering more complementary and integrative treatments as well as helping patients understand what insurance plans cover and how to adjust to changes under new insurance plans. Providing high-quality plans with low copays, premiums, and deductibles that cover required as well as elective cancer-related services, and covering lymphedema-related materials and treatments also were flagged as important.
Supportive service recommendations included addressing psychosocial costs through expansion of support groups and buddy services, offering extended home health services following cancer treatment, and providing domestic assistance with household chores, child care, and transportation.
Financial assistance that broadens financial aid and social services eligibility to those not classified as being in poverty was considered important.
Protective policy recommendations focused on expanding employment and medical leave policies concerning the amount of time offered off from work.
Patient recommendations offer just one viewpoint concerning potential challenges to the overall system, but “their thoughts on how it can be improved add value to decision-making processes,” noted Dr. Dean and her associates.
They were careful to acknowledge the benefits of the Patient Protection and Affordable Care Act, but they noted that it does not include provisions to address the adverse treatment effects of conditions such as cancer. While some states already have successfully passed legislation requiring private insurance carriers to cover lymphedema treatment, similar legislation should be adopted at a national level through joint efforts of Congress and the Department of Labor, they advised.
Any such efforts to make sweeping changes within the insurance industry would take considerable effort on the part of patients, providers, insurers, and state and federal policy makers, as well as the pharmaceutical industry. Yet, such “top-down and bottom-up strategies that involve all parties are warranted,” they urged.
Several important limitations of the study are worth noting. All participants were from the East Coast, had insurance coverage, and reported an overall low level of economic burden. Responses may have differed had the study been conducted in other regions of the country. The study was voluntary, so it is important to consider that patients with greater financial challenges may not have had time to enroll and participate, which suggests that the level of economic burden affecting this population actually could be understated.
SOURCE: Dean LT et al. Cancer 2019 Mar 6. doi: 10.1002/cncr.32012.
FROM CANCER
Combo shows promise in HER2-positive breast cancer with brain mets
The combination of neratinib and capecitabine showed positive efficacy outcomes, but also a high degree of toxicity, in patients with progressive HER2-positive breast cancer and brain metastases, according to results from a phase 2 study.
“Neratinib [is] an irreversible pan-HER tyrosine kinase inhibitor that inhibits signal transduction,” wrote Rachel A Freedman, MD, MPH, of the Dana-Farber Cancer Institute in Boston, and her colleagues. Their report is in the Journal of Clinical Oncology. The researchers grouped patients into two cohorts, those without previous lapatinib (n = 37) and those with previous lapatinib exposure (n = 12), which were termed cohort 3A and 3B, respectively. All study participants were given neratinib 240 mg once daily in combination with capecitabine 750 mg/m2 twice daily for a total of 14 days, followed by 7 days without capecitabine.
The primary outcome measured was the composite central nervous system (CNS) objective response rate (ORR) of each individual cohort, which was defined as a decrease of at least 50% in the total target CNS lesion volumes, in the absence of other markers of further progression.
After analysis, Dr. Freedman and her colleagues found that the CNS ORR was 49% (95% confidence interval, 32%-66%) and 33% (95% CI, 10%-65%) in cohorts 3A and 3B, respectively. In addition, the team reported that the median progression-free survival was 5.5 and 3.1 months in the same respective cohorts.
With respect to safety, the most frequently seen adverse event was diarrhea, with 29% of study participants having grade 3 toxicity. The researchers reported that no grade 4 adverse events were seen.
“It is possible that selection bias affected toxicity events, because no patients in cohort 3B stopped treatment [due to] toxicity,” they said.
A key limitation of the study was the lack of a comparison group, which could be added in future trials.
“Future studies could examine local therapy versus systemic therapy in CNS disease and additionally explore the role of other neratinib-based combination regimens,” they concluded.
The study was supported by grant funding from Puma Biotechnology, the Translational Breast Cancer Research Consortium’s foundation partners, the American Cancer Society, Susan G. Komen for the Cure, the Breast Cancer Research Foundation, and the Dana-Farber/Harvard Cancer Center. The authors reported financial affiliations with Puma Biotechnology, Genentech, Eli Lilly, Novartis, Pfizer, and others.
SOURCE: Freedman RA et al. J Clin Oncol. 2019 Mar 12. doi: 10.1200/JCO.18.01511.
The combination of neratinib and capecitabine showed positive efficacy outcomes, but also a high degree of toxicity, in patients with progressive HER2-positive breast cancer and brain metastases, according to results from a phase 2 study.
“Neratinib [is] an irreversible pan-HER tyrosine kinase inhibitor that inhibits signal transduction,” wrote Rachel A Freedman, MD, MPH, of the Dana-Farber Cancer Institute in Boston, and her colleagues. Their report is in the Journal of Clinical Oncology. The researchers grouped patients into two cohorts, those without previous lapatinib (n = 37) and those with previous lapatinib exposure (n = 12), which were termed cohort 3A and 3B, respectively. All study participants were given neratinib 240 mg once daily in combination with capecitabine 750 mg/m2 twice daily for a total of 14 days, followed by 7 days without capecitabine.
The primary outcome measured was the composite central nervous system (CNS) objective response rate (ORR) of each individual cohort, which was defined as a decrease of at least 50% in the total target CNS lesion volumes, in the absence of other markers of further progression.
After analysis, Dr. Freedman and her colleagues found that the CNS ORR was 49% (95% confidence interval, 32%-66%) and 33% (95% CI, 10%-65%) in cohorts 3A and 3B, respectively. In addition, the team reported that the median progression-free survival was 5.5 and 3.1 months in the same respective cohorts.
With respect to safety, the most frequently seen adverse event was diarrhea, with 29% of study participants having grade 3 toxicity. The researchers reported that no grade 4 adverse events were seen.
“It is possible that selection bias affected toxicity events, because no patients in cohort 3B stopped treatment [due to] toxicity,” they said.
A key limitation of the study was the lack of a comparison group, which could be added in future trials.
“Future studies could examine local therapy versus systemic therapy in CNS disease and additionally explore the role of other neratinib-based combination regimens,” they concluded.
The study was supported by grant funding from Puma Biotechnology, the Translational Breast Cancer Research Consortium’s foundation partners, the American Cancer Society, Susan G. Komen for the Cure, the Breast Cancer Research Foundation, and the Dana-Farber/Harvard Cancer Center. The authors reported financial affiliations with Puma Biotechnology, Genentech, Eli Lilly, Novartis, Pfizer, and others.
SOURCE: Freedman RA et al. J Clin Oncol. 2019 Mar 12. doi: 10.1200/JCO.18.01511.
The combination of neratinib and capecitabine showed positive efficacy outcomes, but also a high degree of toxicity, in patients with progressive HER2-positive breast cancer and brain metastases, according to results from a phase 2 study.
“Neratinib [is] an irreversible pan-HER tyrosine kinase inhibitor that inhibits signal transduction,” wrote Rachel A Freedman, MD, MPH, of the Dana-Farber Cancer Institute in Boston, and her colleagues. Their report is in the Journal of Clinical Oncology. The researchers grouped patients into two cohorts, those without previous lapatinib (n = 37) and those with previous lapatinib exposure (n = 12), which were termed cohort 3A and 3B, respectively. All study participants were given neratinib 240 mg once daily in combination with capecitabine 750 mg/m2 twice daily for a total of 14 days, followed by 7 days without capecitabine.
The primary outcome measured was the composite central nervous system (CNS) objective response rate (ORR) of each individual cohort, which was defined as a decrease of at least 50% in the total target CNS lesion volumes, in the absence of other markers of further progression.
After analysis, Dr. Freedman and her colleagues found that the CNS ORR was 49% (95% confidence interval, 32%-66%) and 33% (95% CI, 10%-65%) in cohorts 3A and 3B, respectively. In addition, the team reported that the median progression-free survival was 5.5 and 3.1 months in the same respective cohorts.
With respect to safety, the most frequently seen adverse event was diarrhea, with 29% of study participants having grade 3 toxicity. The researchers reported that no grade 4 adverse events were seen.
“It is possible that selection bias affected toxicity events, because no patients in cohort 3B stopped treatment [due to] toxicity,” they said.
A key limitation of the study was the lack of a comparison group, which could be added in future trials.
“Future studies could examine local therapy versus systemic therapy in CNS disease and additionally explore the role of other neratinib-based combination regimens,” they concluded.
The study was supported by grant funding from Puma Biotechnology, the Translational Breast Cancer Research Consortium’s foundation partners, the American Cancer Society, Susan G. Komen for the Cure, the Breast Cancer Research Foundation, and the Dana-Farber/Harvard Cancer Center. The authors reported financial affiliations with Puma Biotechnology, Genentech, Eli Lilly, Novartis, Pfizer, and others.
SOURCE: Freedman RA et al. J Clin Oncol. 2019 Mar 12. doi: 10.1200/JCO.18.01511.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
FDA approves another trastuzumab biosimilar for HER2-positive breast cancer, gastric cancer
The Food and Drug Administration has approved Trazimera (trastuzumab-qyyp), a biosimilar of Herceptin (trastuzumab), for the treatment of HER2-positive breast cancer and HER2-positive metastatic gastric or gastroesophageal junction adenocarcinoma.

FDA approval was based on a review of a comprehensive data package, which included results from the REFLECTIONS B327-02 trial. In this trial, Trazimera was found to have clinical equivalence with trastuzumab in the first-line treatment setting in patients with HER2-positive metastatic breast cancer.
The most common adverse events associated with Trazimera in patients with breast cancer include fever, nausea, vomiting, infusion reactions, diarrhea, infections, increased cough, headache, fatigue, shortness of breath, rash, low white and red blood cell counts, and muscle pain. For patients with metastatic adenocarcinoma, the most common adverse events include low white and red blood cell counts; diarrhea; fatigue; swelling of the mouth lining, mucous membranes, nose, or throat; weight loss; upper respiratory tract infections; fever; low platelet counts; and change in taste.
“Approximately 15-30% of breast cancers and 10-30% of gastric cancers are HER2-positive, which is associated with aggressive disease and poor prognoses for patients. With the availability of biosimilars like Trazimera in the U.S., oncologists will have additional treatment options to choose from, which may help provide patients with greater access to the medicines they need,” Mark Pegram, MD, director of the breast oncology program at the Stanford Women’s Cancer Center at Stanford (Calif.) University, said in the press release.
Find the full press release on the Pfizer website.
The Food and Drug Administration has approved Trazimera (trastuzumab-qyyp), a biosimilar of Herceptin (trastuzumab), for the treatment of HER2-positive breast cancer and HER2-positive metastatic gastric or gastroesophageal junction adenocarcinoma.
FDA approval was based on a review of a comprehensive data package, which included results from the REFLECTIONS B327-02 trial. In this trial, Trazimera was found to have clinical equivalence with trastuzumab in the first-line treatment setting in patients with HER2-positive metastatic breast cancer.
The most common adverse events associated with Trazimera in patients with breast cancer include fever, nausea, vomiting, infusion reactions, diarrhea, infections, increased cough, headache, fatigue, shortness of breath, rash, low white and red blood cell counts, and muscle pain. For patients with metastatic adenocarcinoma, the most common adverse events include low white and red blood cell counts; diarrhea; fatigue; swelling of the mouth lining, mucous membranes, nose, or throat; weight loss; upper respiratory tract infections; fever; low platelet counts; and change in taste.
“Approximately 15-30% of breast cancers and 10-30% of gastric cancers are HER2-positive, which is associated with aggressive disease and poor prognoses for patients. With the availability of biosimilars like Trazimera in the U.S., oncologists will have additional treatment options to choose from, which may help provide patients with greater access to the medicines they need,” Mark Pegram, MD, director of the breast oncology program at the Stanford Women’s Cancer Center at Stanford (Calif.) University, said in the press release.
Find the full press release on the Pfizer website.
The Food and Drug Administration has approved Trazimera (trastuzumab-qyyp), a biosimilar of Herceptin (trastuzumab), for the treatment of HER2-positive breast cancer and HER2-positive metastatic gastric or gastroesophageal junction adenocarcinoma.
FDA approval was based on a review of a comprehensive data package, which included results from the REFLECTIONS B327-02 trial. In this trial, Trazimera was found to have clinical equivalence with trastuzumab in the first-line treatment setting in patients with HER2-positive metastatic breast cancer.
The most common adverse events associated with Trazimera in patients with breast cancer include fever, nausea, vomiting, infusion reactions, diarrhea, infections, increased cough, headache, fatigue, shortness of breath, rash, low white and red blood cell counts, and muscle pain. For patients with metastatic adenocarcinoma, the most common adverse events include low white and red blood cell counts; diarrhea; fatigue; swelling of the mouth lining, mucous membranes, nose, or throat; weight loss; upper respiratory tract infections; fever; low platelet counts; and change in taste.
“Approximately 15-30% of breast cancers and 10-30% of gastric cancers are HER2-positive, which is associated with aggressive disease and poor prognoses for patients. With the availability of biosimilars like Trazimera in the U.S., oncologists will have additional treatment options to choose from, which may help provide patients with greater access to the medicines they need,” Mark Pegram, MD, director of the breast oncology program at the Stanford Women’s Cancer Center at Stanford (Calif.) University, said in the press release.
Find the full press release on the Pfizer website.
Novel immunostimulant combo shows early efficacy
SAN FRANCISCO – A combination of two novel immune-stimulating agents has shown early evidence of efficacy against malignant melanoma, leiomyosarcoma, and triple-negative breast cancer in a phase 1b, dose-escalating study.

Among 11 evaluable patients enrolled in a trial of NKTR-262, a small molecule agonist of toll-like receptors (TLR) 7/8, and bempegaldesleukin, an interleukin-2 pathway agonist, 2 had a partial response and 3 had stable disease, reported Adi Diab, MD, from the University of Texas MD Anderson Cancer Center, Houston, and his colleagues.
Patients tolerated the combination well, and there have been no serious adverse events or dose-limiting toxicities.
“Pharmacodynamic data demonstrate both activation of the systemic adaptive and the local innate immune system, and we have seen early evidence of clinical activity in patients who are refractory to checkpoint inhibitors with immunotherapy regimens,” Dr. Diab said at the American Society of Clinical Oncology (ASCO) – Society for Immunotherapy of Cancer (SITC): Clinical Immuno-Oncology Symposium.
NKTR-262 is injected into tumors and is designed to be retained in the tumor microenvironment where it helps to activate antigen-presenting cells, such as dendritic cells, and primes development of new, antigen-specific cytotoxic T cells. Bempegaldesleukin is a cytokine that works within the IL-2 pathway to increase CD8-positive T cells and natural killer (NK) cells in the tumor microenvironment.
The rationale for the combination is that NKTR-262 can activate innate immunity in cells surrounding the tumor microenvironment and activate the machinery of antigen-presenting cells, and bempegaldesleukin can prime and boost a systemic tumor immune response that can ultimately mediate antitumor activity in distant lesions, Dr. Adib said.
In preclinical models, the combination of these agents led to a robust antitumor effect that also involved distant lesions through mediation of the abscopal effect, in which treatment of a tumor activates an immune response against distant tumor cells as well, Dr. Diab said.
The REVEAL study is an ongoing, phase 1b/2 trial looking at the combination in melanoma, Merkel cell carcinoma, triple-negative breast cancer (TNBC), ovarian cancer, renal cell carcinoma, colorectal cancer, urothelial carcinoma, and sarcoma.
The primary goal of the study is to evaluate safety and determine the optimal phase 2 dose of the combination, evaluate biomarkers of response, and assess antitumor activity. As of Jan. 23, 2019, 13 patients were enrolled and evaluable for safety, and 11 were evaluable for the preliminary efficacy analysis.
The most common treatment-related adverse events (TRAEs) with the combination were transient grade 1 or 2 flu-like symptoms, rash, fatigue, pruritus, and nausea. One patients developed grade 3 maculopapular rash and leukocytosis.
Most of the TRAEs are attributable to bempegaldesleukin. There were no immune-mediated AEs and no TRAEs resulted in study discontinuation.
Tumor biopsies obtained 24 hours after injection of NKTR-262 confirmed the activation of TLR 7/8 and robust induction of type 1 interferon, interferon-alpha, and interferon-beta gene-related signatures necessary for optimal antigen presentation.
Dr. Diab noted that in a different trial of bempegaldesleukin monotherapy there was no significant increase in the type 1 interferon gene signature, but the agent did promote activation of the adaptive immune system.
The complementary nature of the two novel agents could also be demonstrated in evaluation of peripheral blood samples, which showed that, although there was no proliferation of T or NK cells following NKTR-262 injection, the addition of bempegaldesleukin resulted in the proliferation of both effector T cells and NK cells to enhance the systemic immune response.
The preliminary efficacy analysis showed that two of five patients with stage IV melanoma who experienced disease progression on prior immune checkpoint inhibitors had partial responses, including one who had a 100% reduction in target lesions and the other with a 50% reduction. In addition, two patients with heavily pretreated leiomyosarcoma had stable disease as the best response, as did the single patient with TNBC.
The maximum tolerated dose of the combination has not been identified, and the investigators are continuing to enroll patients.
The REVEAL study is supported by Nektar Therapeutics. Dr. Diab reported institutional research funding, consulting fees, and advisory board participation from Nektar, Bristol-Myers Squib, Idera Pharmaceuticals, Jounce Therapeutics, and Array BioPharma.
SOURCE: Diab A et al. ASCO-SITC, Abstract 26.
SAN FRANCISCO – A combination of two novel immune-stimulating agents has shown early evidence of efficacy against malignant melanoma, leiomyosarcoma, and triple-negative breast cancer in a phase 1b, dose-escalating study.
Among 11 evaluable patients enrolled in a trial of NKTR-262, a small molecule agonist of toll-like receptors (TLR) 7/8, and bempegaldesleukin, an interleukin-2 pathway agonist, 2 had a partial response and 3 had stable disease, reported Adi Diab, MD, from the University of Texas MD Anderson Cancer Center, Houston, and his colleagues.
Patients tolerated the combination well, and there have been no serious adverse events or dose-limiting toxicities.
“Pharmacodynamic data demonstrate both activation of the systemic adaptive and the local innate immune system, and we have seen early evidence of clinical activity in patients who are refractory to checkpoint inhibitors with immunotherapy regimens,” Dr. Diab said at the American Society of Clinical Oncology (ASCO) – Society for Immunotherapy of Cancer (SITC): Clinical Immuno-Oncology Symposium.
NKTR-262 is injected into tumors and is designed to be retained in the tumor microenvironment where it helps to activate antigen-presenting cells, such as dendritic cells, and primes development of new, antigen-specific cytotoxic T cells. Bempegaldesleukin is a cytokine that works within the IL-2 pathway to increase CD8-positive T cells and natural killer (NK) cells in the tumor microenvironment.
The rationale for the combination is that NKTR-262 can activate innate immunity in cells surrounding the tumor microenvironment and activate the machinery of antigen-presenting cells, and bempegaldesleukin can prime and boost a systemic tumor immune response that can ultimately mediate antitumor activity in distant lesions, Dr. Adib said.
In preclinical models, the combination of these agents led to a robust antitumor effect that also involved distant lesions through mediation of the abscopal effect, in which treatment of a tumor activates an immune response against distant tumor cells as well, Dr. Diab said.
The REVEAL study is an ongoing, phase 1b/2 trial looking at the combination in melanoma, Merkel cell carcinoma, triple-negative breast cancer (TNBC), ovarian cancer, renal cell carcinoma, colorectal cancer, urothelial carcinoma, and sarcoma.
The primary goal of the study is to evaluate safety and determine the optimal phase 2 dose of the combination, evaluate biomarkers of response, and assess antitumor activity. As of Jan. 23, 2019, 13 patients were enrolled and evaluable for safety, and 11 were evaluable for the preliminary efficacy analysis.
The most common treatment-related adverse events (TRAEs) with the combination were transient grade 1 or 2 flu-like symptoms, rash, fatigue, pruritus, and nausea. One patients developed grade 3 maculopapular rash and leukocytosis.
Most of the TRAEs are attributable to bempegaldesleukin. There were no immune-mediated AEs and no TRAEs resulted in study discontinuation.
Tumor biopsies obtained 24 hours after injection of NKTR-262 confirmed the activation of TLR 7/8 and robust induction of type 1 interferon, interferon-alpha, and interferon-beta gene-related signatures necessary for optimal antigen presentation.
Dr. Diab noted that in a different trial of bempegaldesleukin monotherapy there was no significant increase in the type 1 interferon gene signature, but the agent did promote activation of the adaptive immune system.
The complementary nature of the two novel agents could also be demonstrated in evaluation of peripheral blood samples, which showed that, although there was no proliferation of T or NK cells following NKTR-262 injection, the addition of bempegaldesleukin resulted in the proliferation of both effector T cells and NK cells to enhance the systemic immune response.
The preliminary efficacy analysis showed that two of five patients with stage IV melanoma who experienced disease progression on prior immune checkpoint inhibitors had partial responses, including one who had a 100% reduction in target lesions and the other with a 50% reduction. In addition, two patients with heavily pretreated leiomyosarcoma had stable disease as the best response, as did the single patient with TNBC.
The maximum tolerated dose of the combination has not been identified, and the investigators are continuing to enroll patients.
The REVEAL study is supported by Nektar Therapeutics. Dr. Diab reported institutional research funding, consulting fees, and advisory board participation from Nektar, Bristol-Myers Squib, Idera Pharmaceuticals, Jounce Therapeutics, and Array BioPharma.
SOURCE: Diab A et al. ASCO-SITC, Abstract 26.
SAN FRANCISCO – A combination of two novel immune-stimulating agents has shown early evidence of efficacy against malignant melanoma, leiomyosarcoma, and triple-negative breast cancer in a phase 1b, dose-escalating study.
Among 11 evaluable patients enrolled in a trial of NKTR-262, a small molecule agonist of toll-like receptors (TLR) 7/8, and bempegaldesleukin, an interleukin-2 pathway agonist, 2 had a partial response and 3 had stable disease, reported Adi Diab, MD, from the University of Texas MD Anderson Cancer Center, Houston, and his colleagues.
Patients tolerated the combination well, and there have been no serious adverse events or dose-limiting toxicities.
“Pharmacodynamic data demonstrate both activation of the systemic adaptive and the local innate immune system, and we have seen early evidence of clinical activity in patients who are refractory to checkpoint inhibitors with immunotherapy regimens,” Dr. Diab said at the American Society of Clinical Oncology (ASCO) – Society for Immunotherapy of Cancer (SITC): Clinical Immuno-Oncology Symposium.
NKTR-262 is injected into tumors and is designed to be retained in the tumor microenvironment where it helps to activate antigen-presenting cells, such as dendritic cells, and primes development of new, antigen-specific cytotoxic T cells. Bempegaldesleukin is a cytokine that works within the IL-2 pathway to increase CD8-positive T cells and natural killer (NK) cells in the tumor microenvironment.
The rationale for the combination is that NKTR-262 can activate innate immunity in cells surrounding the tumor microenvironment and activate the machinery of antigen-presenting cells, and bempegaldesleukin can prime and boost a systemic tumor immune response that can ultimately mediate antitumor activity in distant lesions, Dr. Adib said.
In preclinical models, the combination of these agents led to a robust antitumor effect that also involved distant lesions through mediation of the abscopal effect, in which treatment of a tumor activates an immune response against distant tumor cells as well, Dr. Diab said.
The REVEAL study is an ongoing, phase 1b/2 trial looking at the combination in melanoma, Merkel cell carcinoma, triple-negative breast cancer (TNBC), ovarian cancer, renal cell carcinoma, colorectal cancer, urothelial carcinoma, and sarcoma.
The primary goal of the study is to evaluate safety and determine the optimal phase 2 dose of the combination, evaluate biomarkers of response, and assess antitumor activity. As of Jan. 23, 2019, 13 patients were enrolled and evaluable for safety, and 11 were evaluable for the preliminary efficacy analysis.
The most common treatment-related adverse events (TRAEs) with the combination were transient grade 1 or 2 flu-like symptoms, rash, fatigue, pruritus, and nausea. One patients developed grade 3 maculopapular rash and leukocytosis.
Most of the TRAEs are attributable to bempegaldesleukin. There were no immune-mediated AEs and no TRAEs resulted in study discontinuation.
Tumor biopsies obtained 24 hours after injection of NKTR-262 confirmed the activation of TLR 7/8 and robust induction of type 1 interferon, interferon-alpha, and interferon-beta gene-related signatures necessary for optimal antigen presentation.
Dr. Diab noted that in a different trial of bempegaldesleukin monotherapy there was no significant increase in the type 1 interferon gene signature, but the agent did promote activation of the adaptive immune system.
The complementary nature of the two novel agents could also be demonstrated in evaluation of peripheral blood samples, which showed that, although there was no proliferation of T or NK cells following NKTR-262 injection, the addition of bempegaldesleukin resulted in the proliferation of both effector T cells and NK cells to enhance the systemic immune response.
The preliminary efficacy analysis showed that two of five patients with stage IV melanoma who experienced disease progression on prior immune checkpoint inhibitors had partial responses, including one who had a 100% reduction in target lesions and the other with a 50% reduction. In addition, two patients with heavily pretreated leiomyosarcoma had stable disease as the best response, as did the single patient with TNBC.
The maximum tolerated dose of the combination has not been identified, and the investigators are continuing to enroll patients.
The REVEAL study is supported by Nektar Therapeutics. Dr. Diab reported institutional research funding, consulting fees, and advisory board participation from Nektar, Bristol-Myers Squib, Idera Pharmaceuticals, Jounce Therapeutics, and Array BioPharma.
SOURCE: Diab A et al. ASCO-SITC, Abstract 26.
REPORTING FROM ASCO-SITC
Neurologists grappling with patients who embrace ‘stem cell tourism’
DALLAS – Stem cell tourism – the unethical practice of offering unproven cellular preparations to patients for a variety of conditions – is increasingly sought by patients with incurable conditions such as multiple sclerosis and amyotrophic lateral sclerosis, results from a novel survey suggest.

In fact, most academic neurologists have been approached by patients with incurable conditions who ask them about stem cell therapy, while about two-thirds have had at least one patient who has undergone stem cell therapy.
“It’s really scary,” Wijdan Rai, MBBS, the study’s first author, said in an interview at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “This is a more prevalent issue than we think, and the complication rates are higher than we think.”
According to the study’s senior author, Jaime Imitola, MD, who directs the Progressive Multiple Sclerosis Multidisciplinary Clinic and Translational Research Program at the Ohio State University Wexner Medical Center, Columbus, the results “call for the creation of a nationwide registry where neurologists can document adverse reactions to stem cell procedures and further support dedicated patient and neurologist education as we have proposed before” (See Semin Neurol. 2018; 38[2]:176-81 and JAMA Neurol. 2015;72[11]:1342-5).
In an effort to understand the experiences and attitudes of academic neurologists regarding stem cell tourism and patient-reported complications, the researchers developed a 25-question survey disseminated via Synapse, a web tool from the American Academy of Neurology. Respondents were asked about demographic information, frequency of patient questioning, perception of physician competence, patient complications and experiences, and attitudes toward increased physician education.
Dr. Rai, who is a senior neurology resident at the medical center, presented findings from 204 neurologist respondents, of whom 31% identified themselves as MS specialists. Nearly all respondents (91%) said they have been approached by patients with incurable conditions seeking information about stem cells (37% of whom had diagnosis of MS). In addition, 65% have had at least one patient that has undergone “stem cell therapy,” and 73% said it would be “helpful” or “very helpful” to have an evidence-based patient education tool on the topic. “Patients most often wanted general information,” Dr. Rai said. “However, 50% requested permission to undergo a stem cell procedure, and 31% approached their neurologist after the procedure.”
Survey respondents reported that 33% of the stem cell interventions were performed in the United States and 22% abroad, while 37% reported both in the U.S. and abroad. Patients underwent the procedures in China, Germany, the Bahamas, Mexico, Russia, and Costa Rica. Three-quarters of respondents (75%) indicated no patient experiencing complications from the stem cell interventions. However, 25% reported patients experiencing a variety of complications from the procedures, including strokes, meningoencephalitis, quadriparesis, MS deterioration, sepsis, hepatitis C, seizures, meningitis from intrathecal cell injections, infections, and spinal cord tumors. “At least three respondents had a patient who died as a direct complication from stem cell therapy,” Dr. Rai said.
In their poster, the researchers recommended a “multipronged approach to improve education of MS patients from exploitation and engaging multiple stakeholders in the field, including MS academic societies, licensing boards, and legislative bodies. Specifically, we call for creation of evidence-based education for both neurologists and patients, including physical resources that neurologists can use when discussing stem cell interventions with patients and videos on proper counseling during these visits.”
Colleagues from OSU’s Laboratory for Neural Stem Cells and Functional Neurogenetics contributed to this work. The researchers reported having no financial disclosures.
SOURCE: Rai W et al. ACTRIMS Forum 2019, Poster 237.
DALLAS – Stem cell tourism – the unethical practice of offering unproven cellular preparations to patients for a variety of conditions – is increasingly sought by patients with incurable conditions such as multiple sclerosis and amyotrophic lateral sclerosis, results from a novel survey suggest.
In fact, most academic neurologists have been approached by patients with incurable conditions who ask them about stem cell therapy, while about two-thirds have had at least one patient who has undergone stem cell therapy.
“It’s really scary,” Wijdan Rai, MBBS, the study’s first author, said in an interview at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “This is a more prevalent issue than we think, and the complication rates are higher than we think.”
According to the study’s senior author, Jaime Imitola, MD, who directs the Progressive Multiple Sclerosis Multidisciplinary Clinic and Translational Research Program at the Ohio State University Wexner Medical Center, Columbus, the results “call for the creation of a nationwide registry where neurologists can document adverse reactions to stem cell procedures and further support dedicated patient and neurologist education as we have proposed before” (See Semin Neurol. 2018; 38[2]:176-81 and JAMA Neurol. 2015;72[11]:1342-5).
In an effort to understand the experiences and attitudes of academic neurologists regarding stem cell tourism and patient-reported complications, the researchers developed a 25-question survey disseminated via Synapse, a web tool from the American Academy of Neurology. Respondents were asked about demographic information, frequency of patient questioning, perception of physician competence, patient complications and experiences, and attitudes toward increased physician education.
Dr. Rai, who is a senior neurology resident at the medical center, presented findings from 204 neurologist respondents, of whom 31% identified themselves as MS specialists. Nearly all respondents (91%) said they have been approached by patients with incurable conditions seeking information about stem cells (37% of whom had diagnosis of MS). In addition, 65% have had at least one patient that has undergone “stem cell therapy,” and 73% said it would be “helpful” or “very helpful” to have an evidence-based patient education tool on the topic. “Patients most often wanted general information,” Dr. Rai said. “However, 50% requested permission to undergo a stem cell procedure, and 31% approached their neurologist after the procedure.”
Survey respondents reported that 33% of the stem cell interventions were performed in the United States and 22% abroad, while 37% reported both in the U.S. and abroad. Patients underwent the procedures in China, Germany, the Bahamas, Mexico, Russia, and Costa Rica. Three-quarters of respondents (75%) indicated no patient experiencing complications from the stem cell interventions. However, 25% reported patients experiencing a variety of complications from the procedures, including strokes, meningoencephalitis, quadriparesis, MS deterioration, sepsis, hepatitis C, seizures, meningitis from intrathecal cell injections, infections, and spinal cord tumors. “At least three respondents had a patient who died as a direct complication from stem cell therapy,” Dr. Rai said.
In their poster, the researchers recommended a “multipronged approach to improve education of MS patients from exploitation and engaging multiple stakeholders in the field, including MS academic societies, licensing boards, and legislative bodies. Specifically, we call for creation of evidence-based education for both neurologists and patients, including physical resources that neurologists can use when discussing stem cell interventions with patients and videos on proper counseling during these visits.”
Colleagues from OSU’s Laboratory for Neural Stem Cells and Functional Neurogenetics contributed to this work. The researchers reported having no financial disclosures.
SOURCE: Rai W et al. ACTRIMS Forum 2019, Poster 237.
DALLAS – Stem cell tourism – the unethical practice of offering unproven cellular preparations to patients for a variety of conditions – is increasingly sought by patients with incurable conditions such as multiple sclerosis and amyotrophic lateral sclerosis, results from a novel survey suggest.
In fact, most academic neurologists have been approached by patients with incurable conditions who ask them about stem cell therapy, while about two-thirds have had at least one patient who has undergone stem cell therapy.
“It’s really scary,” Wijdan Rai, MBBS, the study’s first author, said in an interview at the meeting held by the Americas Committee for Treatment and Research in Multiple Sclerosis. “This is a more prevalent issue than we think, and the complication rates are higher than we think.”
According to the study’s senior author, Jaime Imitola, MD, who directs the Progressive Multiple Sclerosis Multidisciplinary Clinic and Translational Research Program at the Ohio State University Wexner Medical Center, Columbus, the results “call for the creation of a nationwide registry where neurologists can document adverse reactions to stem cell procedures and further support dedicated patient and neurologist education as we have proposed before” (See Semin Neurol. 2018; 38[2]:176-81 and JAMA Neurol. 2015;72[11]:1342-5).
In an effort to understand the experiences and attitudes of academic neurologists regarding stem cell tourism and patient-reported complications, the researchers developed a 25-question survey disseminated via Synapse, a web tool from the American Academy of Neurology. Respondents were asked about demographic information, frequency of patient questioning, perception of physician competence, patient complications and experiences, and attitudes toward increased physician education.
Dr. Rai, who is a senior neurology resident at the medical center, presented findings from 204 neurologist respondents, of whom 31% identified themselves as MS specialists. Nearly all respondents (91%) said they have been approached by patients with incurable conditions seeking information about stem cells (37% of whom had diagnosis of MS). In addition, 65% have had at least one patient that has undergone “stem cell therapy,” and 73% said it would be “helpful” or “very helpful” to have an evidence-based patient education tool on the topic. “Patients most often wanted general information,” Dr. Rai said. “However, 50% requested permission to undergo a stem cell procedure, and 31% approached their neurologist after the procedure.”
Survey respondents reported that 33% of the stem cell interventions were performed in the United States and 22% abroad, while 37% reported both in the U.S. and abroad. Patients underwent the procedures in China, Germany, the Bahamas, Mexico, Russia, and Costa Rica. Three-quarters of respondents (75%) indicated no patient experiencing complications from the stem cell interventions. However, 25% reported patients experiencing a variety of complications from the procedures, including strokes, meningoencephalitis, quadriparesis, MS deterioration, sepsis, hepatitis C, seizures, meningitis from intrathecal cell injections, infections, and spinal cord tumors. “At least three respondents had a patient who died as a direct complication from stem cell therapy,” Dr. Rai said.
In their poster, the researchers recommended a “multipronged approach to improve education of MS patients from exploitation and engaging multiple stakeholders in the field, including MS academic societies, licensing boards, and legislative bodies. Specifically, we call for creation of evidence-based education for both neurologists and patients, including physical resources that neurologists can use when discussing stem cell interventions with patients and videos on proper counseling during these visits.”
Colleagues from OSU’s Laboratory for Neural Stem Cells and Functional Neurogenetics contributed to this work. The researchers reported having no financial disclosures.
SOURCE: Rai W et al. ACTRIMS Forum 2019, Poster 237.
REPORTING FROM ACTRIMS FORUM 2019
FDA approves Tecentriq plus Abraxane in breast cancer
The Food and Drug Administration has granted accelerated approval for the combination of atezolizumab (Tecentriq) plus nanoparticle albumin–bound paclitaxel (nab-paclitaxel; Abraxane) for the treatment of adults with unresectable locally advanced or metastatic programmed death-ligand 1 (PD-L1)–positive triple-negative breast cancer (TNBC).
This conditional approval is granted to medicines that fill an unmet medical need for serious or life-threatening diseases or conditions, but the FDA may require confirmatory trials to provide verification and description of clinical benefit to allow continued approval.
The approval is based on the phase 3 IMpassion130 trial (NCT02425891), which enrolled 902 patients with unresectable, locally advanced or metastatic TNBC who had not received prior lines of chemo for metastatic disease, according to Genentech.
The multicenter, randomized, double-blind study has been evaluating the drug combination’s efficacy, safety, and pharmacokinetics. Compared with placebo plus nab-paclitaxel, atezolizumab/nab-paclitaxel demonstrated significantly superior progression-free survival (median PFS, 7.4 months vs. 4.8 months; hazard ratio, 0.60; 95% confidence interval, 0.48-0.77; P less than .0001).
The overall survival data for the intention-to-treat population remains immature, but further data will be shared with the FDA in the future, according to Genentech.
No new safety signals were seen in the atezolizumab/nab-paclitaxel arm, and the combination’s safety appeared consistent with the known safety profiles of each medicine individually.
The most common grade 3-4 events (occurring in more than 2% of patients) in the combination arm included low red blood cells, low white blood cells, feeling tired, low blood potassium level, and pneumonia.
The most common side effects (occurring in more than 20% of patients) in the combination arm included hair loss, tingling, nausea, diarrhea, headache, low red blood cells, low white blood cells, and decreased appetite.
Atezolizumab is a monoclonal antibody that binds to the PD-L1 receptor, which could possibly lead to the reactivation of T cells; however, atezolizumab also may interact with other cells in the body. Nab-paclitaxel is an injectable suspension of the common chemotherapy drug.
The Food and Drug Administration has granted accelerated approval for the combination of atezolizumab (Tecentriq) plus nanoparticle albumin–bound paclitaxel (nab-paclitaxel; Abraxane) for the treatment of adults with unresectable locally advanced or metastatic programmed death-ligand 1 (PD-L1)–positive triple-negative breast cancer (TNBC).
This conditional approval is granted to medicines that fill an unmet medical need for serious or life-threatening diseases or conditions, but the FDA may require confirmatory trials to provide verification and description of clinical benefit to allow continued approval.
The approval is based on the phase 3 IMpassion130 trial (NCT02425891), which enrolled 902 patients with unresectable, locally advanced or metastatic TNBC who had not received prior lines of chemo for metastatic disease, according to Genentech.
The multicenter, randomized, double-blind study has been evaluating the drug combination’s efficacy, safety, and pharmacokinetics. Compared with placebo plus nab-paclitaxel, atezolizumab/nab-paclitaxel demonstrated significantly superior progression-free survival (median PFS, 7.4 months vs. 4.8 months; hazard ratio, 0.60; 95% confidence interval, 0.48-0.77; P less than .0001).
The overall survival data for the intention-to-treat population remains immature, but further data will be shared with the FDA in the future, according to Genentech.
No new safety signals were seen in the atezolizumab/nab-paclitaxel arm, and the combination’s safety appeared consistent with the known safety profiles of each medicine individually.
The most common grade 3-4 events (occurring in more than 2% of patients) in the combination arm included low red blood cells, low white blood cells, feeling tired, low blood potassium level, and pneumonia.
The most common side effects (occurring in more than 20% of patients) in the combination arm included hair loss, tingling, nausea, diarrhea, headache, low red blood cells, low white blood cells, and decreased appetite.
Atezolizumab is a monoclonal antibody that binds to the PD-L1 receptor, which could possibly lead to the reactivation of T cells; however, atezolizumab also may interact with other cells in the body. Nab-paclitaxel is an injectable suspension of the common chemotherapy drug.
The Food and Drug Administration has granted accelerated approval for the combination of atezolizumab (Tecentriq) plus nanoparticle albumin–bound paclitaxel (nab-paclitaxel; Abraxane) for the treatment of adults with unresectable locally advanced or metastatic programmed death-ligand 1 (PD-L1)–positive triple-negative breast cancer (TNBC).
This conditional approval is granted to medicines that fill an unmet medical need for serious or life-threatening diseases or conditions, but the FDA may require confirmatory trials to provide verification and description of clinical benefit to allow continued approval.
The approval is based on the phase 3 IMpassion130 trial (NCT02425891), which enrolled 902 patients with unresectable, locally advanced or metastatic TNBC who had not received prior lines of chemo for metastatic disease, according to Genentech.
The multicenter, randomized, double-blind study has been evaluating the drug combination’s efficacy, safety, and pharmacokinetics. Compared with placebo plus nab-paclitaxel, atezolizumab/nab-paclitaxel demonstrated significantly superior progression-free survival (median PFS, 7.4 months vs. 4.8 months; hazard ratio, 0.60; 95% confidence interval, 0.48-0.77; P less than .0001).
The overall survival data for the intention-to-treat population remains immature, but further data will be shared with the FDA in the future, according to Genentech.
No new safety signals were seen in the atezolizumab/nab-paclitaxel arm, and the combination’s safety appeared consistent with the known safety profiles of each medicine individually.
The most common grade 3-4 events (occurring in more than 2% of patients) in the combination arm included low red blood cells, low white blood cells, feeling tired, low blood potassium level, and pneumonia.
The most common side effects (occurring in more than 20% of patients) in the combination arm included hair loss, tingling, nausea, diarrhea, headache, low red blood cells, low white blood cells, and decreased appetite.
Atezolizumab is a monoclonal antibody that binds to the PD-L1 receptor, which could possibly lead to the reactivation of T cells; however, atezolizumab also may interact with other cells in the body. Nab-paclitaxel is an injectable suspension of the common chemotherapy drug.
Novel biomarker could predict resistance to palbociclib
, according to a gene expression analysis.
Nicholas C. Turner, MD, PhD, of Royal Marsden Hospital and Institute of Cancer Research in London, and his colleagues used a gene expression panel to detect biomarkers related to the efficacy of palbociclib plus fulvestrant in patients with endocrine-pretreated metastatic breast cancer.
“No predictive biomarkers have been identified in randomized trials of CDK4/6 inhibitors,” the researchers wrote in the Journal of Clinical Oncology.
Study participants were randomly assigned to receive either combination palbociclib and fulvestrant (n = 194) or placebo and fulvestrant (n = 108). The primary analysis was completed using data from the PALOMA-3 trial, which included 10 genes selected from a panel search of 2,534 genes.
The association between level of gene expression and efficacy of palbociclib combination therapy was evaluated by way of advanced statistical analysis.
After analysis, the efficacy of palbociclib was found to be reduced with high levels of cyclin E1 mRNA expression compared with low levels (median PFS palbociclib arm, 7.6 vs. 14.1 months; placebo arm, 4.0 vs. 4.8 months; P = .00238).
“These data suggest that CCNE1 mRNA expression may be associated with the benefit from palbociclib in early-stage breast cancer,” they wrote.
The authors acknowledged that one key limitation of the study was that the biomarkers identified may not be relevant to other CDK4/6 inhibitor combinations.
“Additional methodologic and clinical validations are warranted to elucidate the role of CCNE1 mRNA expression as a biomarker of CDK4/6 inhibitor therapy,” they concluded.
The study was supported by Pfizer. The authors reported financial interests with AbbVie, AstraZeneca, Genentech, Novartis, Pfizer, and others.
SOURCE: Turner NC et al. J Clin Oncol. 2019 Feb 26. doi: 10.1200/JCO.18.00925.
, according to a gene expression analysis.
Nicholas C. Turner, MD, PhD, of Royal Marsden Hospital and Institute of Cancer Research in London, and his colleagues used a gene expression panel to detect biomarkers related to the efficacy of palbociclib plus fulvestrant in patients with endocrine-pretreated metastatic breast cancer.
“No predictive biomarkers have been identified in randomized trials of CDK4/6 inhibitors,” the researchers wrote in the Journal of Clinical Oncology.
Study participants were randomly assigned to receive either combination palbociclib and fulvestrant (n = 194) or placebo and fulvestrant (n = 108). The primary analysis was completed using data from the PALOMA-3 trial, which included 10 genes selected from a panel search of 2,534 genes.
The association between level of gene expression and efficacy of palbociclib combination therapy was evaluated by way of advanced statistical analysis.
After analysis, the efficacy of palbociclib was found to be reduced with high levels of cyclin E1 mRNA expression compared with low levels (median PFS palbociclib arm, 7.6 vs. 14.1 months; placebo arm, 4.0 vs. 4.8 months; P = .00238).
“These data suggest that CCNE1 mRNA expression may be associated with the benefit from palbociclib in early-stage breast cancer,” they wrote.
The authors acknowledged that one key limitation of the study was that the biomarkers identified may not be relevant to other CDK4/6 inhibitor combinations.
“Additional methodologic and clinical validations are warranted to elucidate the role of CCNE1 mRNA expression as a biomarker of CDK4/6 inhibitor therapy,” they concluded.
The study was supported by Pfizer. The authors reported financial interests with AbbVie, AstraZeneca, Genentech, Novartis, Pfizer, and others.
SOURCE: Turner NC et al. J Clin Oncol. 2019 Feb 26. doi: 10.1200/JCO.18.00925.
, according to a gene expression analysis.
Nicholas C. Turner, MD, PhD, of Royal Marsden Hospital and Institute of Cancer Research in London, and his colleagues used a gene expression panel to detect biomarkers related to the efficacy of palbociclib plus fulvestrant in patients with endocrine-pretreated metastatic breast cancer.
“No predictive biomarkers have been identified in randomized trials of CDK4/6 inhibitors,” the researchers wrote in the Journal of Clinical Oncology.
Study participants were randomly assigned to receive either combination palbociclib and fulvestrant (n = 194) or placebo and fulvestrant (n = 108). The primary analysis was completed using data from the PALOMA-3 trial, which included 10 genes selected from a panel search of 2,534 genes.
The association between level of gene expression and efficacy of palbociclib combination therapy was evaluated by way of advanced statistical analysis.
After analysis, the efficacy of palbociclib was found to be reduced with high levels of cyclin E1 mRNA expression compared with low levels (median PFS palbociclib arm, 7.6 vs. 14.1 months; placebo arm, 4.0 vs. 4.8 months; P = .00238).
“These data suggest that CCNE1 mRNA expression may be associated with the benefit from palbociclib in early-stage breast cancer,” they wrote.
The authors acknowledged that one key limitation of the study was that the biomarkers identified may not be relevant to other CDK4/6 inhibitor combinations.
“Additional methodologic and clinical validations are warranted to elucidate the role of CCNE1 mRNA expression as a biomarker of CDK4/6 inhibitor therapy,” they concluded.
The study was supported by Pfizer. The authors reported financial interests with AbbVie, AstraZeneca, Genentech, Novartis, Pfizer, and others.
SOURCE: Turner NC et al. J Clin Oncol. 2019 Feb 26. doi: 10.1200/JCO.18.00925.
FROM JOURNAL OF CLINICAL ONCOLOGY
FDA urges caution with robotic devices in cancer surgery
A new safety communication from the Food and Drug Administration on the use of robotically assisted surgical devices for mastectomy and other cancer-related surgeries in women encourages physician-patient dialogue and suggests that, moving forward, data on specific oncologic outcomes – not only perioperative and short-term outcomes – are key.
The FDA is “warning patients and providers that the use of robotically assisted surgical devices for any cancer-related surgery has not been granted marketing authorization by the agency, and therefore the survival benefits to patients when compared to traditional surgery have not been established,” Terri Cornelison, MD, PhD, assistant director for the health of women in the FDA’s Center for Devices and Radiological Health, said in a statement.
The safety communication focuses on women and calls attention specifically to robotically-assisted mastectomy and hysterectomy for early cervical cancers. It says there is “limited, preliminary evidence that the use of robotically-assisted surgical devices for treatment or prevention of cancers that primarily (breast) or exclusively (cervical) affect women may be associated with diminished long-term survival.”
The FDA cited a multicenter randomized trial that found that minimally invasive radical hysterectomy in women with cervical cancer (laparoscopic and robotically assisted) was associated with a lower rate of long-term survival compared with open surgery (N Engl J Med. 2018;379:1895-1904).
The communication does not refer to any other specific studies. Regarding current evidence on robotically-assisted mastectomies, the FDA safety communication says simply that safety and effectiveness have not been established and that the agency is “aware of scientific literature and media publications describing surgeons and hospital systems that use robotically-assisted surgical devices for mastectomy.”
Robotically-assisted mastectomy
Walton Taylor, MD, president of the American Society of Breast Surgeons and a surgeon with Texas Health Physicians Group in Dallas, said that the FDA’s concern is valid. “I really hope that robotic surgery turns out to be good [for mastectomy]. It’s awesome technology that can be great for patients,” he said. “But we have to gather real data that shows that long-term and short-term outcomes – from a cancer standpoint – are as good as with the open procedure ... that there aren’t negative unintended consequences.”
Right now, Dr. Taylor said, robotic mastectomy “is not commonplace by any means.”
The technique for robotic nipple-sparing mastectomy (NSM) was first described by Antonio Toesca, MD, of the European Institute of Oncology in Milan (Ann Surg. 2017;266[2]:e28-e30).
In an editorial published recently in Annals of Surgical Oncology, Jesse C. Selber, MD, MPH, of the department of plastic surgery at the University of Texas MD Anderson Cancer Center in Houston, described the technique as a “natural next step in the evolution of minimally invasive breast surgery that has the potential to mitigate the challenges associated with traditional NSM” (Ann Surg Oncol. 2019;26[1]:10-11). Robotic nipple-sparing mastectomy is catching on in Europe” with very promising early results, he wrote.
At least a couple of practices promoted their performance of robotic mastectomy last year. Northwell Health, a large network of hospitals, outpatient facilities, and physicians in New York, announced in March 2018 that Neil Tanna, MD, and Alan Kadison, MD, of the divisions of plastic and reconstructive surgery and surgical oncology, respectively, had performed the first robotic nipple-sparing mastectomy and breast reconstructive surgery in the United States. Their patient carried the BRCA gene and had a preventive mastectomy at Northwell Health’s Long Island Jewish Medical Center.
In October 2018, a surgeon in Tinton Falls, N.J., Stephen Chagares, MD, announced that he had performed the first robotic nipple-sparing mastectomy with reconstruction in a patient with breast cancer at Monmouth Medical Center. His press release described a 3-cm incision “to the side of the breast, tucked neatly behind the armpit.” Both Dr. Chagares and Dr. Tanna had traveled to Milan to train with Dr. Toesca, according to the press releases.
Both of these cases – as well as a decision by Monmouth Medical Center in December 2018 to suspend the surgery pending further review – were mentioned in a letter submitted to the FDA in mid-December by Hooman Noorchashm, MD, PhD, a Philadelphia cardiothoracic surgeon-turned-patient-advocate whose wife Amy Josephine Reed, MD, PhD, died of uterine cancer in May 2017 following a laparoscopic hysterectomy performed with a power morcellator.
In his complaint, Dr. Noorchashm urged the agency to issue a warning about the “potentially dangerous/premature application” of robotic mastectomy for the treatment of breast cancer or BRCA carrier status outside the setting of randomized controlled trials with primary cancer–related outcomes metrics or an investigational device exemption from the FDA. (Receipt of the letter was acknowledged by the Allegation of Regulatory Misconduct Branch of the FDA several days later.)
In an interview, Dr. Noorchashm said he wants to see a regulatory framework that doesn’t allow 510(k) devices (devices requiring a premarket notification to the FDA) to modify an existing standard of care without having been shown to have noninferior primary outcomes. When devices are used in the diagnosis or treatment of cancerous or potentially cancerous tissue, he said, this means primary oncologic outcomes must be shown to be noninferior.
“When you have 510(k) devices able to inject themselves and affect existing standards of care without any sort of clinical trial requirement, you get the standard of care changing without any outcomes data to back it up,” he said. “That’s what happened with the power morcellator. Physicians started using it without any sort of prospective data, level 1 outcomes data, and it dramatically changed the conduct of hysterectomies.”
In its safety communication, the FDA encourages the establishment of patient registries to gather data on robotically-assisted surgical devices for all uses, including the prevention and treatment of cancer. It also says that while the agency’s evaluation of the devices has generally focused on complication rates at 30 days, the FDA “anticipates” that their use in the prevention or treatment of cancer “would be supported by specific clinical outcomes, such as local cancer recurrence, disease-free survival, or overall survival at time periods much longer than 30 days.”
The American Society of Breast Surgeons has a Nipple Sparing Mastectomy Registry that is collecting oncologic outcomes as well as aesthetic outcomes and other metrics on 2,000 patients. “In the last year or two, we’ve seen nipple-sparing mastectomy become much more commonplace,” said Dr. Taylor. Thus far, the registry does not include robotic procedures, but “if there were interest in a registry specifically for robotic nipple-sparing mastectomy, we would do it in a heartbeat.”
Gynecologic oncology surgery
The randomized controlled study on radical hysterectomy for cervical cancer that caught the FDA’s attention reported lower rates of disease-free survival at 4.5 years with minimally invasive surgery than with open abdominal surgery (86% versus 96.5%) and lower rates of overall survival at 3 years.
The phase 3 multicenter Laparoscopic Approach to Cervical Cancer trial recruited more than 600 women with stage IA1, IA2, or IB1 cervical cancer. Most (91.9%) had IB1 disease and either squamous-cell carcinoma, adenocarcinoma, or adenosquamous carcinoma. Differences in the outcomes remained after adjustment for age, body mass index, disease stage, lymphovascular invasion, and lymph-node involvement. The findings led to early termination of the study.
The study did not single out robotically-assisted surgery. It was a two-arm study and was “not powered to analyze laparoscopy versus robotics,” lead author Pedro T. Ramirez, MD, of the University of Texas MD Anderson Cancer Center, said in an interview. “But based on our numbers, we saw no difference [in outcomes] between the two groups.” Of the patients who underwent minimally invasive surgery, 84.4% underwent laparoscopy and 15.6% underwent robot-assisted surgery.
The study, funded by MD Anderson and Medtronic, has been criticized for potential design and conduct issues. Outside experts pointed out that the study involved extremely small numbers of patients at each of the 33 participating centers, and that cancer recurrences were clustered at 14 of these centers. It’s important to appreciate, Dr. Ramirez said in the interview, that the majority of patients were accrued in these 14 centers.
In its safety communication, the FDA noted that other researchers have reported no statistically significant difference in long-term survival when open and minimally invasive approaches to radical hysterectomy for cervical cancer have been compared.
Asked to comment on the FDA’s safety communication, Dwight D. Im, MD, who leads the National Institute of Robotic Surgery at Mercy in Baltimore, said in an e-mail that “while robotic surgery may advance into new areas, such as mastectomy and cancer prevention, more research must be done and this should be part of any conversation between gyn-surgeons who are experienced in the realm of robotic surgery, and their patients.”
Regarding the treatment of cervical cancer, “I think it is safe to say that most gynecologic oncologists now offer only open laparotomies until we have more data comparing open to minimally invasive (laparoscopic and robotic) approaches,” he said.
The FDA said in a briefing document accompanying the safety communication that it has received a “small number of medical device reports of patient injury when [robotically-assisted surgical devices] are used in cancer-related procedures.”
According to the FDA spokesperson, 5 of 32 medical device reports received between January 2016 and December 2018 describe patients who underwent hysterectomy and experienced metastases afterward. It does not appear that any of the 5 cases were a direct result of a system error or device malfunction, and the complications described in the reports are not unique to robotically-assisted surgical devices, the spokesperson said.
The safety communication “reflects the agency’s commitment to enhancing the oversight of device safety as part of our Medical Device Action Plan, as well as the agency’s ongoing commitment to advancing women’s health.”
Dr. Taylor reported that he has no current financial disclosures. Dr. Ramirez reported to the New England Journal of Medicine that he had no relevant disclosures. Dr. Im reported that he is a speaker for Intuitive Surgical, which manufacturers the da Vinci Surgical System, as well as for Conmed and Ethicon.
A new safety communication from the Food and Drug Administration on the use of robotically assisted surgical devices for mastectomy and other cancer-related surgeries in women encourages physician-patient dialogue and suggests that, moving forward, data on specific oncologic outcomes – not only perioperative and short-term outcomes – are key.
The FDA is “warning patients and providers that the use of robotically assisted surgical devices for any cancer-related surgery has not been granted marketing authorization by the agency, and therefore the survival benefits to patients when compared to traditional surgery have not been established,” Terri Cornelison, MD, PhD, assistant director for the health of women in the FDA’s Center for Devices and Radiological Health, said in a statement.
The safety communication focuses on women and calls attention specifically to robotically-assisted mastectomy and hysterectomy for early cervical cancers. It says there is “limited, preliminary evidence that the use of robotically-assisted surgical devices for treatment or prevention of cancers that primarily (breast) or exclusively (cervical) affect women may be associated with diminished long-term survival.”
The FDA cited a multicenter randomized trial that found that minimally invasive radical hysterectomy in women with cervical cancer (laparoscopic and robotically assisted) was associated with a lower rate of long-term survival compared with open surgery (N Engl J Med. 2018;379:1895-1904).
The communication does not refer to any other specific studies. Regarding current evidence on robotically-assisted mastectomies, the FDA safety communication says simply that safety and effectiveness have not been established and that the agency is “aware of scientific literature and media publications describing surgeons and hospital systems that use robotically-assisted surgical devices for mastectomy.”
Robotically-assisted mastectomy
Walton Taylor, MD, president of the American Society of Breast Surgeons and a surgeon with Texas Health Physicians Group in Dallas, said that the FDA’s concern is valid. “I really hope that robotic surgery turns out to be good [for mastectomy]. It’s awesome technology that can be great for patients,” he said. “But we have to gather real data that shows that long-term and short-term outcomes – from a cancer standpoint – are as good as with the open procedure ... that there aren’t negative unintended consequences.”
Right now, Dr. Taylor said, robotic mastectomy “is not commonplace by any means.”
The technique for robotic nipple-sparing mastectomy (NSM) was first described by Antonio Toesca, MD, of the European Institute of Oncology in Milan (Ann Surg. 2017;266[2]:e28-e30).
In an editorial published recently in Annals of Surgical Oncology, Jesse C. Selber, MD, MPH, of the department of plastic surgery at the University of Texas MD Anderson Cancer Center in Houston, described the technique as a “natural next step in the evolution of minimally invasive breast surgery that has the potential to mitigate the challenges associated with traditional NSM” (Ann Surg Oncol. 2019;26[1]:10-11). Robotic nipple-sparing mastectomy is catching on in Europe” with very promising early results, he wrote.
At least a couple of practices promoted their performance of robotic mastectomy last year. Northwell Health, a large network of hospitals, outpatient facilities, and physicians in New York, announced in March 2018 that Neil Tanna, MD, and Alan Kadison, MD, of the divisions of plastic and reconstructive surgery and surgical oncology, respectively, had performed the first robotic nipple-sparing mastectomy and breast reconstructive surgery in the United States. Their patient carried the BRCA gene and had a preventive mastectomy at Northwell Health’s Long Island Jewish Medical Center.
In October 2018, a surgeon in Tinton Falls, N.J., Stephen Chagares, MD, announced that he had performed the first robotic nipple-sparing mastectomy with reconstruction in a patient with breast cancer at Monmouth Medical Center. His press release described a 3-cm incision “to the side of the breast, tucked neatly behind the armpit.” Both Dr. Chagares and Dr. Tanna had traveled to Milan to train with Dr. Toesca, according to the press releases.
Both of these cases – as well as a decision by Monmouth Medical Center in December 2018 to suspend the surgery pending further review – were mentioned in a letter submitted to the FDA in mid-December by Hooman Noorchashm, MD, PhD, a Philadelphia cardiothoracic surgeon-turned-patient-advocate whose wife Amy Josephine Reed, MD, PhD, died of uterine cancer in May 2017 following a laparoscopic hysterectomy performed with a power morcellator.
In his complaint, Dr. Noorchashm urged the agency to issue a warning about the “potentially dangerous/premature application” of robotic mastectomy for the treatment of breast cancer or BRCA carrier status outside the setting of randomized controlled trials with primary cancer–related outcomes metrics or an investigational device exemption from the FDA. (Receipt of the letter was acknowledged by the Allegation of Regulatory Misconduct Branch of the FDA several days later.)
In an interview, Dr. Noorchashm said he wants to see a regulatory framework that doesn’t allow 510(k) devices (devices requiring a premarket notification to the FDA) to modify an existing standard of care without having been shown to have noninferior primary outcomes. When devices are used in the diagnosis or treatment of cancerous or potentially cancerous tissue, he said, this means primary oncologic outcomes must be shown to be noninferior.
“When you have 510(k) devices able to inject themselves and affect existing standards of care without any sort of clinical trial requirement, you get the standard of care changing without any outcomes data to back it up,” he said. “That’s what happened with the power morcellator. Physicians started using it without any sort of prospective data, level 1 outcomes data, and it dramatically changed the conduct of hysterectomies.”
In its safety communication, the FDA encourages the establishment of patient registries to gather data on robotically-assisted surgical devices for all uses, including the prevention and treatment of cancer. It also says that while the agency’s evaluation of the devices has generally focused on complication rates at 30 days, the FDA “anticipates” that their use in the prevention or treatment of cancer “would be supported by specific clinical outcomes, such as local cancer recurrence, disease-free survival, or overall survival at time periods much longer than 30 days.”
The American Society of Breast Surgeons has a Nipple Sparing Mastectomy Registry that is collecting oncologic outcomes as well as aesthetic outcomes and other metrics on 2,000 patients. “In the last year or two, we’ve seen nipple-sparing mastectomy become much more commonplace,” said Dr. Taylor. Thus far, the registry does not include robotic procedures, but “if there were interest in a registry specifically for robotic nipple-sparing mastectomy, we would do it in a heartbeat.”
Gynecologic oncology surgery
The randomized controlled study on radical hysterectomy for cervical cancer that caught the FDA’s attention reported lower rates of disease-free survival at 4.5 years with minimally invasive surgery than with open abdominal surgery (86% versus 96.5%) and lower rates of overall survival at 3 years.
The phase 3 multicenter Laparoscopic Approach to Cervical Cancer trial recruited more than 600 women with stage IA1, IA2, or IB1 cervical cancer. Most (91.9%) had IB1 disease and either squamous-cell carcinoma, adenocarcinoma, or adenosquamous carcinoma. Differences in the outcomes remained after adjustment for age, body mass index, disease stage, lymphovascular invasion, and lymph-node involvement. The findings led to early termination of the study.
The study did not single out robotically-assisted surgery. It was a two-arm study and was “not powered to analyze laparoscopy versus robotics,” lead author Pedro T. Ramirez, MD, of the University of Texas MD Anderson Cancer Center, said in an interview. “But based on our numbers, we saw no difference [in outcomes] between the two groups.” Of the patients who underwent minimally invasive surgery, 84.4% underwent laparoscopy and 15.6% underwent robot-assisted surgery.
The study, funded by MD Anderson and Medtronic, has been criticized for potential design and conduct issues. Outside experts pointed out that the study involved extremely small numbers of patients at each of the 33 participating centers, and that cancer recurrences were clustered at 14 of these centers. It’s important to appreciate, Dr. Ramirez said in the interview, that the majority of patients were accrued in these 14 centers.
In its safety communication, the FDA noted that other researchers have reported no statistically significant difference in long-term survival when open and minimally invasive approaches to radical hysterectomy for cervical cancer have been compared.
Asked to comment on the FDA’s safety communication, Dwight D. Im, MD, who leads the National Institute of Robotic Surgery at Mercy in Baltimore, said in an e-mail that “while robotic surgery may advance into new areas, such as mastectomy and cancer prevention, more research must be done and this should be part of any conversation between gyn-surgeons who are experienced in the realm of robotic surgery, and their patients.”
Regarding the treatment of cervical cancer, “I think it is safe to say that most gynecologic oncologists now offer only open laparotomies until we have more data comparing open to minimally invasive (laparoscopic and robotic) approaches,” he said.
The FDA said in a briefing document accompanying the safety communication that it has received a “small number of medical device reports of patient injury when [robotically-assisted surgical devices] are used in cancer-related procedures.”
According to the FDA spokesperson, 5 of 32 medical device reports received between January 2016 and December 2018 describe patients who underwent hysterectomy and experienced metastases afterward. It does not appear that any of the 5 cases were a direct result of a system error or device malfunction, and the complications described in the reports are not unique to robotically-assisted surgical devices, the spokesperson said.
The safety communication “reflects the agency’s commitment to enhancing the oversight of device safety as part of our Medical Device Action Plan, as well as the agency’s ongoing commitment to advancing women’s health.”
Dr. Taylor reported that he has no current financial disclosures. Dr. Ramirez reported to the New England Journal of Medicine that he had no relevant disclosures. Dr. Im reported that he is a speaker for Intuitive Surgical, which manufacturers the da Vinci Surgical System, as well as for Conmed and Ethicon.
A new safety communication from the Food and Drug Administration on the use of robotically assisted surgical devices for mastectomy and other cancer-related surgeries in women encourages physician-patient dialogue and suggests that, moving forward, data on specific oncologic outcomes – not only perioperative and short-term outcomes – are key.
The FDA is “warning patients and providers that the use of robotically assisted surgical devices for any cancer-related surgery has not been granted marketing authorization by the agency, and therefore the survival benefits to patients when compared to traditional surgery have not been established,” Terri Cornelison, MD, PhD, assistant director for the health of women in the FDA’s Center for Devices and Radiological Health, said in a statement.
The safety communication focuses on women and calls attention specifically to robotically-assisted mastectomy and hysterectomy for early cervical cancers. It says there is “limited, preliminary evidence that the use of robotically-assisted surgical devices for treatment or prevention of cancers that primarily (breast) or exclusively (cervical) affect women may be associated with diminished long-term survival.”
The FDA cited a multicenter randomized trial that found that minimally invasive radical hysterectomy in women with cervical cancer (laparoscopic and robotically assisted) was associated with a lower rate of long-term survival compared with open surgery (N Engl J Med. 2018;379:1895-1904).
The communication does not refer to any other specific studies. Regarding current evidence on robotically-assisted mastectomies, the FDA safety communication says simply that safety and effectiveness have not been established and that the agency is “aware of scientific literature and media publications describing surgeons and hospital systems that use robotically-assisted surgical devices for mastectomy.”
Robotically-assisted mastectomy
Walton Taylor, MD, president of the American Society of Breast Surgeons and a surgeon with Texas Health Physicians Group in Dallas, said that the FDA’s concern is valid. “I really hope that robotic surgery turns out to be good [for mastectomy]. It’s awesome technology that can be great for patients,” he said. “But we have to gather real data that shows that long-term and short-term outcomes – from a cancer standpoint – are as good as with the open procedure ... that there aren’t negative unintended consequences.”
Right now, Dr. Taylor said, robotic mastectomy “is not commonplace by any means.”
The technique for robotic nipple-sparing mastectomy (NSM) was first described by Antonio Toesca, MD, of the European Institute of Oncology in Milan (Ann Surg. 2017;266[2]:e28-e30).
In an editorial published recently in Annals of Surgical Oncology, Jesse C. Selber, MD, MPH, of the department of plastic surgery at the University of Texas MD Anderson Cancer Center in Houston, described the technique as a “natural next step in the evolution of minimally invasive breast surgery that has the potential to mitigate the challenges associated with traditional NSM” (Ann Surg Oncol. 2019;26[1]:10-11). Robotic nipple-sparing mastectomy is catching on in Europe” with very promising early results, he wrote.
At least a couple of practices promoted their performance of robotic mastectomy last year. Northwell Health, a large network of hospitals, outpatient facilities, and physicians in New York, announced in March 2018 that Neil Tanna, MD, and Alan Kadison, MD, of the divisions of plastic and reconstructive surgery and surgical oncology, respectively, had performed the first robotic nipple-sparing mastectomy and breast reconstructive surgery in the United States. Their patient carried the BRCA gene and had a preventive mastectomy at Northwell Health’s Long Island Jewish Medical Center.
In October 2018, a surgeon in Tinton Falls, N.J., Stephen Chagares, MD, announced that he had performed the first robotic nipple-sparing mastectomy with reconstruction in a patient with breast cancer at Monmouth Medical Center. His press release described a 3-cm incision “to the side of the breast, tucked neatly behind the armpit.” Both Dr. Chagares and Dr. Tanna had traveled to Milan to train with Dr. Toesca, according to the press releases.
Both of these cases – as well as a decision by Monmouth Medical Center in December 2018 to suspend the surgery pending further review – were mentioned in a letter submitted to the FDA in mid-December by Hooman Noorchashm, MD, PhD, a Philadelphia cardiothoracic surgeon-turned-patient-advocate whose wife Amy Josephine Reed, MD, PhD, died of uterine cancer in May 2017 following a laparoscopic hysterectomy performed with a power morcellator.
In his complaint, Dr. Noorchashm urged the agency to issue a warning about the “potentially dangerous/premature application” of robotic mastectomy for the treatment of breast cancer or BRCA carrier status outside the setting of randomized controlled trials with primary cancer–related outcomes metrics or an investigational device exemption from the FDA. (Receipt of the letter was acknowledged by the Allegation of Regulatory Misconduct Branch of the FDA several days later.)
In an interview, Dr. Noorchashm said he wants to see a regulatory framework that doesn’t allow 510(k) devices (devices requiring a premarket notification to the FDA) to modify an existing standard of care without having been shown to have noninferior primary outcomes. When devices are used in the diagnosis or treatment of cancerous or potentially cancerous tissue, he said, this means primary oncologic outcomes must be shown to be noninferior.
“When you have 510(k) devices able to inject themselves and affect existing standards of care without any sort of clinical trial requirement, you get the standard of care changing without any outcomes data to back it up,” he said. “That’s what happened with the power morcellator. Physicians started using it without any sort of prospective data, level 1 outcomes data, and it dramatically changed the conduct of hysterectomies.”
In its safety communication, the FDA encourages the establishment of patient registries to gather data on robotically-assisted surgical devices for all uses, including the prevention and treatment of cancer. It also says that while the agency’s evaluation of the devices has generally focused on complication rates at 30 days, the FDA “anticipates” that their use in the prevention or treatment of cancer “would be supported by specific clinical outcomes, such as local cancer recurrence, disease-free survival, or overall survival at time periods much longer than 30 days.”
The American Society of Breast Surgeons has a Nipple Sparing Mastectomy Registry that is collecting oncologic outcomes as well as aesthetic outcomes and other metrics on 2,000 patients. “In the last year or two, we’ve seen nipple-sparing mastectomy become much more commonplace,” said Dr. Taylor. Thus far, the registry does not include robotic procedures, but “if there were interest in a registry specifically for robotic nipple-sparing mastectomy, we would do it in a heartbeat.”
Gynecologic oncology surgery
The randomized controlled study on radical hysterectomy for cervical cancer that caught the FDA’s attention reported lower rates of disease-free survival at 4.5 years with minimally invasive surgery than with open abdominal surgery (86% versus 96.5%) and lower rates of overall survival at 3 years.
The phase 3 multicenter Laparoscopic Approach to Cervical Cancer trial recruited more than 600 women with stage IA1, IA2, or IB1 cervical cancer. Most (91.9%) had IB1 disease and either squamous-cell carcinoma, adenocarcinoma, or adenosquamous carcinoma. Differences in the outcomes remained after adjustment for age, body mass index, disease stage, lymphovascular invasion, and lymph-node involvement. The findings led to early termination of the study.
The study did not single out robotically-assisted surgery. It was a two-arm study and was “not powered to analyze laparoscopy versus robotics,” lead author Pedro T. Ramirez, MD, of the University of Texas MD Anderson Cancer Center, said in an interview. “But based on our numbers, we saw no difference [in outcomes] between the two groups.” Of the patients who underwent minimally invasive surgery, 84.4% underwent laparoscopy and 15.6% underwent robot-assisted surgery.
The study, funded by MD Anderson and Medtronic, has been criticized for potential design and conduct issues. Outside experts pointed out that the study involved extremely small numbers of patients at each of the 33 participating centers, and that cancer recurrences were clustered at 14 of these centers. It’s important to appreciate, Dr. Ramirez said in the interview, that the majority of patients were accrued in these 14 centers.
In its safety communication, the FDA noted that other researchers have reported no statistically significant difference in long-term survival when open and minimally invasive approaches to radical hysterectomy for cervical cancer have been compared.
Asked to comment on the FDA’s safety communication, Dwight D. Im, MD, who leads the National Institute of Robotic Surgery at Mercy in Baltimore, said in an e-mail that “while robotic surgery may advance into new areas, such as mastectomy and cancer prevention, more research must be done and this should be part of any conversation between gyn-surgeons who are experienced in the realm of robotic surgery, and their patients.”
Regarding the treatment of cervical cancer, “I think it is safe to say that most gynecologic oncologists now offer only open laparotomies until we have more data comparing open to minimally invasive (laparoscopic and robotic) approaches,” he said.
The FDA said in a briefing document accompanying the safety communication that it has received a “small number of medical device reports of patient injury when [robotically-assisted surgical devices] are used in cancer-related procedures.”
According to the FDA spokesperson, 5 of 32 medical device reports received between January 2016 and December 2018 describe patients who underwent hysterectomy and experienced metastases afterward. It does not appear that any of the 5 cases were a direct result of a system error or device malfunction, and the complications described in the reports are not unique to robotically-assisted surgical devices, the spokesperson said.
The safety communication “reflects the agency’s commitment to enhancing the oversight of device safety as part of our Medical Device Action Plan, as well as the agency’s ongoing commitment to advancing women’s health.”
Dr. Taylor reported that he has no current financial disclosures. Dr. Ramirez reported to the New England Journal of Medicine that he had no relevant disclosures. Dr. Im reported that he is a speaker for Intuitive Surgical, which manufacturers the da Vinci Surgical System, as well as for Conmed and Ethicon.
Anthracycline-free regimen OK in HER2-negative early breast cancer
It appears to be safe to hold the anthracycline in patients with HER2-negative early breast cancer who are at intermediate-to-high genomic risk, results of a large randomized trial suggest.
Among both pre- and postmenopausal women with pathologic stage T1 to T4c with positive nodes or node-negative but high-risk early breast cancer, there were no significant differences in 5-year outcomes for patients treated with six cycles of docetaxel and cyclophosphamide (TC) or four cycles of epirubicin and cyclophosphamide followed by four cycles of docetaxel (EC-T), reported Ulrike Nitz, MD, from the West German Study Group in Mönchengladbach, Germany, and her colleagues in the West German Study PlanB Trial.
Disease-free survival (DFS, the primary endpoint), distant recurrence-free interval (dRFI), and overall survival (OS) “were excellent and virtually identical in patients who received the anthracycline-containing or the anthracycline-free regimen. Subgroups that benefited from the anthracycline-containing regimen were not identified by interaction analysis, although a potentially clinically relevant benefit in particular (e.g., high-risk) subgroups cannot be ruled out,” they wrote. The report is in Journal of Clinical Oncology.
The investigators noted that anthracyclines are associated with increased risk for cardiac disease and hematologic malignancies, prompting investigators in other trials to consider anthracycline-free regimens.
“As the number of long-term survivors, elderly patients, and patients with preexisting cardiac risk factors increases, the toxicity profile becomes a more important discriminator in adjuvant treatment selection,” they wrote.
The investigators enrolled and randomized 2,449 women (median age 55, range 25-77 years) who had histologically confirmed, unilateral primary invasive breast cancer, adequate surgical treatment, and no evidence of metastatic disease. The patients all had HER2-negative disease, pT1 to pT4c, known hormone receptor status, and either pN+ or pN0 with one or more risk factors.
The intention-to-treat analysis included 1,227 patients assigned to EC-T and 1,222 assigned to TC in the efficacy population, and 1,167 and 1,178 patients, respectively, in the safety population.
After a median follow-up of 60 months, the 5-year DFS rate in the TC-treated group was 89.6%, compared with 89.9% in the EC-T–treated group. The estimated 5-year dRFI rates were 94.1% vs. 93.4%, and the estimated 5-year OS rates were 94.7% vs. 94.5%, respectively. None of the comparisons were statistically significant.
There were five treatment-related deaths in the TC arm, (one each from urosepsis, Streptococcus septicemia, peritonitis/diverticulitis, Staphylococcus epidermidis septicemia, and pulmonary embolism), and one in the EC-T arm (from septicemia).
In an interim safety analysis, the rate of febrile neutropenia was 6.1% in the TC arm and 3.9% in the EC-T arm, leading to a recommendation for “generous” prophylaxis with granulocyte-colony stimulating factor, and ciprofloxacine for patients with a history of diverticulitis or chronic infectious GI disease, or expected duration of neutropenia greater than 1 week.
Rates of grade 3 or 4 leukopenia, neutropenia, nausea, vomiting, peripheral polyneuropathy, hand-foot syndrome, mucositis/stomatitis, arthralgia, myalgia, and fatigue were significantly higher among patients treated with EC-T. There were numerically more grade 3-4 infections and febrile neutropenia within the TC arm, but this trend did not reach statistical significance.
The investigators noted that the results of their trial provide the strongest evidence for patients with pathologic NO or N1 disease, and that the trial did not examine the question of dose-dense chemotherapy in patients with high-risk early breast cancer.
Genomic Health, Sanofi, and Amgen supported the study. Dr. Nitz and multiple coauthors disclosed financial relationships with these companies and others.
SOURCE: Nitz U et al. J Clin Oncol. 2019 Feb 20. doi: 10.1200/JCO.18.00028.
It appears to be safe to hold the anthracycline in patients with HER2-negative early breast cancer who are at intermediate-to-high genomic risk, results of a large randomized trial suggest.
Among both pre- and postmenopausal women with pathologic stage T1 to T4c with positive nodes or node-negative but high-risk early breast cancer, there were no significant differences in 5-year outcomes for patients treated with six cycles of docetaxel and cyclophosphamide (TC) or four cycles of epirubicin and cyclophosphamide followed by four cycles of docetaxel (EC-T), reported Ulrike Nitz, MD, from the West German Study Group in Mönchengladbach, Germany, and her colleagues in the West German Study PlanB Trial.
Disease-free survival (DFS, the primary endpoint), distant recurrence-free interval (dRFI), and overall survival (OS) “were excellent and virtually identical in patients who received the anthracycline-containing or the anthracycline-free regimen. Subgroups that benefited from the anthracycline-containing regimen were not identified by interaction analysis, although a potentially clinically relevant benefit in particular (e.g., high-risk) subgroups cannot be ruled out,” they wrote. The report is in Journal of Clinical Oncology.
The investigators noted that anthracyclines are associated with increased risk for cardiac disease and hematologic malignancies, prompting investigators in other trials to consider anthracycline-free regimens.
“As the number of long-term survivors, elderly patients, and patients with preexisting cardiac risk factors increases, the toxicity profile becomes a more important discriminator in adjuvant treatment selection,” they wrote.
The investigators enrolled and randomized 2,449 women (median age 55, range 25-77 years) who had histologically confirmed, unilateral primary invasive breast cancer, adequate surgical treatment, and no evidence of metastatic disease. The patients all had HER2-negative disease, pT1 to pT4c, known hormone receptor status, and either pN+ or pN0 with one or more risk factors.
The intention-to-treat analysis included 1,227 patients assigned to EC-T and 1,222 assigned to TC in the efficacy population, and 1,167 and 1,178 patients, respectively, in the safety population.
After a median follow-up of 60 months, the 5-year DFS rate in the TC-treated group was 89.6%, compared with 89.9% in the EC-T–treated group. The estimated 5-year dRFI rates were 94.1% vs. 93.4%, and the estimated 5-year OS rates were 94.7% vs. 94.5%, respectively. None of the comparisons were statistically significant.
There were five treatment-related deaths in the TC arm, (one each from urosepsis, Streptococcus septicemia, peritonitis/diverticulitis, Staphylococcus epidermidis septicemia, and pulmonary embolism), and one in the EC-T arm (from septicemia).
In an interim safety analysis, the rate of febrile neutropenia was 6.1% in the TC arm and 3.9% in the EC-T arm, leading to a recommendation for “generous” prophylaxis with granulocyte-colony stimulating factor, and ciprofloxacine for patients with a history of diverticulitis or chronic infectious GI disease, or expected duration of neutropenia greater than 1 week.
Rates of grade 3 or 4 leukopenia, neutropenia, nausea, vomiting, peripheral polyneuropathy, hand-foot syndrome, mucositis/stomatitis, arthralgia, myalgia, and fatigue were significantly higher among patients treated with EC-T. There were numerically more grade 3-4 infections and febrile neutropenia within the TC arm, but this trend did not reach statistical significance.
The investigators noted that the results of their trial provide the strongest evidence for patients with pathologic NO or N1 disease, and that the trial did not examine the question of dose-dense chemotherapy in patients with high-risk early breast cancer.
Genomic Health, Sanofi, and Amgen supported the study. Dr. Nitz and multiple coauthors disclosed financial relationships with these companies and others.
SOURCE: Nitz U et al. J Clin Oncol. 2019 Feb 20. doi: 10.1200/JCO.18.00028.
It appears to be safe to hold the anthracycline in patients with HER2-negative early breast cancer who are at intermediate-to-high genomic risk, results of a large randomized trial suggest.
Among both pre- and postmenopausal women with pathologic stage T1 to T4c with positive nodes or node-negative but high-risk early breast cancer, there were no significant differences in 5-year outcomes for patients treated with six cycles of docetaxel and cyclophosphamide (TC) or four cycles of epirubicin and cyclophosphamide followed by four cycles of docetaxel (EC-T), reported Ulrike Nitz, MD, from the West German Study Group in Mönchengladbach, Germany, and her colleagues in the West German Study PlanB Trial.
Disease-free survival (DFS, the primary endpoint), distant recurrence-free interval (dRFI), and overall survival (OS) “were excellent and virtually identical in patients who received the anthracycline-containing or the anthracycline-free regimen. Subgroups that benefited from the anthracycline-containing regimen were not identified by interaction analysis, although a potentially clinically relevant benefit in particular (e.g., high-risk) subgroups cannot be ruled out,” they wrote. The report is in Journal of Clinical Oncology.
The investigators noted that anthracyclines are associated with increased risk for cardiac disease and hematologic malignancies, prompting investigators in other trials to consider anthracycline-free regimens.
“As the number of long-term survivors, elderly patients, and patients with preexisting cardiac risk factors increases, the toxicity profile becomes a more important discriminator in adjuvant treatment selection,” they wrote.
The investigators enrolled and randomized 2,449 women (median age 55, range 25-77 years) who had histologically confirmed, unilateral primary invasive breast cancer, adequate surgical treatment, and no evidence of metastatic disease. The patients all had HER2-negative disease, pT1 to pT4c, known hormone receptor status, and either pN+ or pN0 with one or more risk factors.
The intention-to-treat analysis included 1,227 patients assigned to EC-T and 1,222 assigned to TC in the efficacy population, and 1,167 and 1,178 patients, respectively, in the safety population.
After a median follow-up of 60 months, the 5-year DFS rate in the TC-treated group was 89.6%, compared with 89.9% in the EC-T–treated group. The estimated 5-year dRFI rates were 94.1% vs. 93.4%, and the estimated 5-year OS rates were 94.7% vs. 94.5%, respectively. None of the comparisons were statistically significant.
There were five treatment-related deaths in the TC arm, (one each from urosepsis, Streptococcus septicemia, peritonitis/diverticulitis, Staphylococcus epidermidis septicemia, and pulmonary embolism), and one in the EC-T arm (from septicemia).
In an interim safety analysis, the rate of febrile neutropenia was 6.1% in the TC arm and 3.9% in the EC-T arm, leading to a recommendation for “generous” prophylaxis with granulocyte-colony stimulating factor, and ciprofloxacine for patients with a history of diverticulitis or chronic infectious GI disease, or expected duration of neutropenia greater than 1 week.
Rates of grade 3 or 4 leukopenia, neutropenia, nausea, vomiting, peripheral polyneuropathy, hand-foot syndrome, mucositis/stomatitis, arthralgia, myalgia, and fatigue were significantly higher among patients treated with EC-T. There were numerically more grade 3-4 infections and febrile neutropenia within the TC arm, but this trend did not reach statistical significance.
The investigators noted that the results of their trial provide the strongest evidence for patients with pathologic NO or N1 disease, and that the trial did not examine the question of dose-dense chemotherapy in patients with high-risk early breast cancer.
Genomic Health, Sanofi, and Amgen supported the study. Dr. Nitz and multiple coauthors disclosed financial relationships with these companies and others.
SOURCE: Nitz U et al. J Clin Oncol. 2019 Feb 20. doi: 10.1200/JCO.18.00028.
FROM JOURNAL of CLINICAL ONCOLOGY