User login
ACP: Average-risk women under 50 can postpone mammogram
(CBE) for screening in such women of any age, according to a new guideline from the American College of Physicians.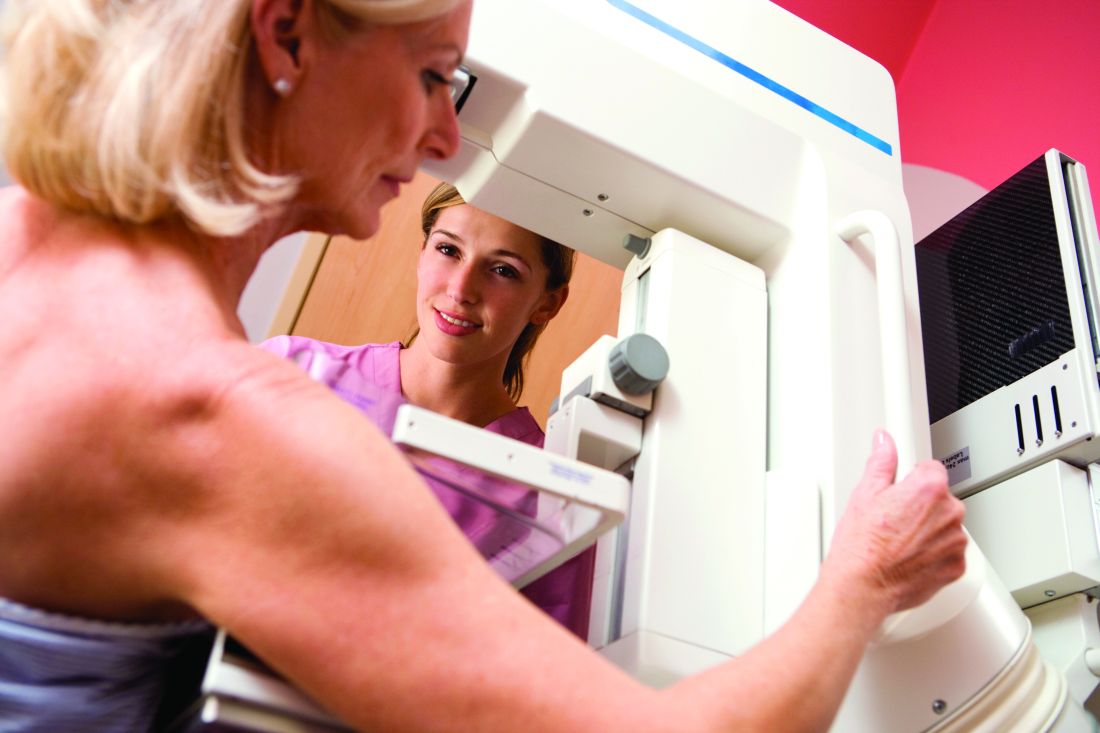
Further, clinicians should discuss whether to screen with mammography in average-risk women aged 40-49 years and consider potential harms and benefits, as well as patient preferences. Providers should discontinue screening average-risk women at age 75 years and women with a life expectancy of 10 years or less, Amir Qaseem, MD, PhD, of the ACP and colleagues wrote on behalf of the ACP Clinical Guidelines Committee.
The ACP guidance also addresses the varying recommendations from other organizations on the age at which to start and stop screening and on screening intervals, noting that “areas of disagreement include screening in women aged 40 to 49 years, screening in women aged 75 years or older, and recommended screening intervals,” and stresses the importance of patient input.
“Women should be informed participants in personalized decisions about breast cancer screening,” the authors wrote, adding that those under age 50 years without a clear preference for screening should not be screened.
However, the evidence shows that most average-risk women with no symptoms will benefit from mammography every other year beginning at age 50 years, they said.
The statement, published online April 8 in the Annals of Internal Medicine, was derived from a review of seven existing English-language breast cancer screening guidelines and the evidence cited in those guidelines. It’s intended to be a resource for all clinicians.
It differs from the 2017 American College of Obstetricians and Gynecologists (ACOG) guidelines in that ACOG recommends CBE and does not address screening in those with a life expectancy of less than 10 years. It also differs from the 2016 U.S. Preventive Services Task Force (USPSTF) guidelines, which make no recommendation on CBE and also do not address screening in those with a life expectancy of less than 10 years.
Other guidelines, such as those from the American College of Radiology, American Cancer Society (ACS), the Canadian Task Force on Preventive Health Care, and the National Comprehensive Cancer Network, recommend CBE, and the World Health Organization guidelines recommend CBE in low resource settings.
“Although CBE continues to be used as part of the examination of symptomatic women, data are sparse on screening asymptomatic women using CBE alone or combined with mammography,” the ACP guideline authors wrote. “The ACS recommends against CBE in average-risk women of any age because of the lack of demonstrated benefit and the potential for false-positive results.”
The guidance, which does not apply to patients with prior abnormal screening results or those at higher breast cancer risk, also includes an evidence-driven “talking points with patients” section based on frequently asked questions.
An important goal of the ACP Clinical Guidelines Committee in developing the guidance is to reduce overdiagnosis and overtreatment, which affects about 20% of women diagnosed over a 10-year period.
The committee reviewed all national guidelines published in English between January 1, 2013, and November 15, 2017, in the National Guideline Clearinghouse or Guidelines International Network library, and it also selected other guidelines commonly used in clinical practice. The committee evaluated the quality of each by using the Appraisal of Guidelines for Research and Evaluation II (AGREE II) instrument.
Alex Krist, MD, the USPSTF vice-chairperson, offered support for the “shift toward shared decision making that is emerging” and added it’s “part of a larger movement toward empowering people with information not only about the potential benefits but also the potential harms of screening tests.”
“In its 2016 recommendation, the Task Force found that the value of mammography increases with age, with women ages 50-74 benefiting most from screening. For women in their 40s, the Task Force also found that mammography screening every two years can be effective,” he told this publication. “We recommend that the decision to start screening should be an individual one, taking into account a woman’s health history, preferences, and how she values the different potential benefits and harms.”
Dr. Krist further noted that the USPSTF, ACP, and many others “have all affirmed that mammography is an important tool to reduce breast cancer mortality and that the benefits of mammography increase with age.”
Likewise, Robert Smith, PhD, vice president of cancer screening for the ACS, noted that the ACP guidance generally aligns with ACS and USPSTF guidelines because all “support informed decision making starting at age 40, and screening every two years starting at age 50 (USPSTF) or 55 (ACS).”
“The fact that all guidelines are not totally in sync is not unexpected. ... The most important thing to recognize is that all of these guidelines stress that regular mammography plays an important role in breast cancer early detection, and women should be aware of its benefits and limitations, and also remain vigilant and report any breast changes,” he said.
The guidance authors reported having no conflicts of interest.
SOURCE: Qaseem A et al., Ann Intern Med. 2019. doi: 10.7326/M18-2147.
The ACP guidance statements provide “clarity and simplicity amidst the chaos of diverging guidelines,” Joann G. Elmore, MD, and Christoph I. Lee, MD, wrote in an editorial that accompanied the guideline (Ann Intern Med. 2019. doi: 10.7326/M19-0726).
The four statements included in the guidance represent the convergence of differing recommendations, but they also highlight points for physicians to consider in shared decision making with patients, the editorial authors wrote.
Lacking, however, is advice on how clinicians should go about stopping screening in certain patients, they noted.
“We need reliable ways to determine life expectancy given comorbid conditions, as well as methods to appropriately manage the discussion about stopping screening. ... The cessation of routine screening is a highly uncomfortable situation for which we as clinicians currently have little guidance and few tools. At this crossroads of confusion, we need a clear path toward informed, tailored, risk-based screening for breast cancer,” they wrote adding that future guidance statements should “move beyond emphasizing variation across guidelines and instead provide more advice on how to implement high-value screening and deimplement low-value screening.”
Dr. Elmore is with the University of California, Los Angeles. Dr. Lee is with the University of Washington, Seattle.
The ACP guidance statements provide “clarity and simplicity amidst the chaos of diverging guidelines,” Joann G. Elmore, MD, and Christoph I. Lee, MD, wrote in an editorial that accompanied the guideline (Ann Intern Med. 2019. doi: 10.7326/M19-0726).
The four statements included in the guidance represent the convergence of differing recommendations, but they also highlight points for physicians to consider in shared decision making with patients, the editorial authors wrote.
Lacking, however, is advice on how clinicians should go about stopping screening in certain patients, they noted.
“We need reliable ways to determine life expectancy given comorbid conditions, as well as methods to appropriately manage the discussion about stopping screening. ... The cessation of routine screening is a highly uncomfortable situation for which we as clinicians currently have little guidance and few tools. At this crossroads of confusion, we need a clear path toward informed, tailored, risk-based screening for breast cancer,” they wrote adding that future guidance statements should “move beyond emphasizing variation across guidelines and instead provide more advice on how to implement high-value screening and deimplement low-value screening.”
Dr. Elmore is with the University of California, Los Angeles. Dr. Lee is with the University of Washington, Seattle.
The ACP guidance statements provide “clarity and simplicity amidst the chaos of diverging guidelines,” Joann G. Elmore, MD, and Christoph I. Lee, MD, wrote in an editorial that accompanied the guideline (Ann Intern Med. 2019. doi: 10.7326/M19-0726).
The four statements included in the guidance represent the convergence of differing recommendations, but they also highlight points for physicians to consider in shared decision making with patients, the editorial authors wrote.
Lacking, however, is advice on how clinicians should go about stopping screening in certain patients, they noted.
“We need reliable ways to determine life expectancy given comorbid conditions, as well as methods to appropriately manage the discussion about stopping screening. ... The cessation of routine screening is a highly uncomfortable situation for which we as clinicians currently have little guidance and few tools. At this crossroads of confusion, we need a clear path toward informed, tailored, risk-based screening for breast cancer,” they wrote adding that future guidance statements should “move beyond emphasizing variation across guidelines and instead provide more advice on how to implement high-value screening and deimplement low-value screening.”
Dr. Elmore is with the University of California, Los Angeles. Dr. Lee is with the University of Washington, Seattle.
(CBE) for screening in such women of any age, according to a new guideline from the American College of Physicians.
Further, clinicians should discuss whether to screen with mammography in average-risk women aged 40-49 years and consider potential harms and benefits, as well as patient preferences. Providers should discontinue screening average-risk women at age 75 years and women with a life expectancy of 10 years or less, Amir Qaseem, MD, PhD, of the ACP and colleagues wrote on behalf of the ACP Clinical Guidelines Committee.
The ACP guidance also addresses the varying recommendations from other organizations on the age at which to start and stop screening and on screening intervals, noting that “areas of disagreement include screening in women aged 40 to 49 years, screening in women aged 75 years or older, and recommended screening intervals,” and stresses the importance of patient input.
“Women should be informed participants in personalized decisions about breast cancer screening,” the authors wrote, adding that those under age 50 years without a clear preference for screening should not be screened.
However, the evidence shows that most average-risk women with no symptoms will benefit from mammography every other year beginning at age 50 years, they said.
The statement, published online April 8 in the Annals of Internal Medicine, was derived from a review of seven existing English-language breast cancer screening guidelines and the evidence cited in those guidelines. It’s intended to be a resource for all clinicians.
It differs from the 2017 American College of Obstetricians and Gynecologists (ACOG) guidelines in that ACOG recommends CBE and does not address screening in those with a life expectancy of less than 10 years. It also differs from the 2016 U.S. Preventive Services Task Force (USPSTF) guidelines, which make no recommendation on CBE and also do not address screening in those with a life expectancy of less than 10 years.
Other guidelines, such as those from the American College of Radiology, American Cancer Society (ACS), the Canadian Task Force on Preventive Health Care, and the National Comprehensive Cancer Network, recommend CBE, and the World Health Organization guidelines recommend CBE in low resource settings.
“Although CBE continues to be used as part of the examination of symptomatic women, data are sparse on screening asymptomatic women using CBE alone or combined with mammography,” the ACP guideline authors wrote. “The ACS recommends against CBE in average-risk women of any age because of the lack of demonstrated benefit and the potential for false-positive results.”
The guidance, which does not apply to patients with prior abnormal screening results or those at higher breast cancer risk, also includes an evidence-driven “talking points with patients” section based on frequently asked questions.
An important goal of the ACP Clinical Guidelines Committee in developing the guidance is to reduce overdiagnosis and overtreatment, which affects about 20% of women diagnosed over a 10-year period.
The committee reviewed all national guidelines published in English between January 1, 2013, and November 15, 2017, in the National Guideline Clearinghouse or Guidelines International Network library, and it also selected other guidelines commonly used in clinical practice. The committee evaluated the quality of each by using the Appraisal of Guidelines for Research and Evaluation II (AGREE II) instrument.
Alex Krist, MD, the USPSTF vice-chairperson, offered support for the “shift toward shared decision making that is emerging” and added it’s “part of a larger movement toward empowering people with information not only about the potential benefits but also the potential harms of screening tests.”
“In its 2016 recommendation, the Task Force found that the value of mammography increases with age, with women ages 50-74 benefiting most from screening. For women in their 40s, the Task Force also found that mammography screening every two years can be effective,” he told this publication. “We recommend that the decision to start screening should be an individual one, taking into account a woman’s health history, preferences, and how she values the different potential benefits and harms.”
Dr. Krist further noted that the USPSTF, ACP, and many others “have all affirmed that mammography is an important tool to reduce breast cancer mortality and that the benefits of mammography increase with age.”
Likewise, Robert Smith, PhD, vice president of cancer screening for the ACS, noted that the ACP guidance generally aligns with ACS and USPSTF guidelines because all “support informed decision making starting at age 40, and screening every two years starting at age 50 (USPSTF) or 55 (ACS).”
“The fact that all guidelines are not totally in sync is not unexpected. ... The most important thing to recognize is that all of these guidelines stress that regular mammography plays an important role in breast cancer early detection, and women should be aware of its benefits and limitations, and also remain vigilant and report any breast changes,” he said.
The guidance authors reported having no conflicts of interest.
SOURCE: Qaseem A et al., Ann Intern Med. 2019. doi: 10.7326/M18-2147.
(CBE) for screening in such women of any age, according to a new guideline from the American College of Physicians.
Further, clinicians should discuss whether to screen with mammography in average-risk women aged 40-49 years and consider potential harms and benefits, as well as patient preferences. Providers should discontinue screening average-risk women at age 75 years and women with a life expectancy of 10 years or less, Amir Qaseem, MD, PhD, of the ACP and colleagues wrote on behalf of the ACP Clinical Guidelines Committee.
The ACP guidance also addresses the varying recommendations from other organizations on the age at which to start and stop screening and on screening intervals, noting that “areas of disagreement include screening in women aged 40 to 49 years, screening in women aged 75 years or older, and recommended screening intervals,” and stresses the importance of patient input.
“Women should be informed participants in personalized decisions about breast cancer screening,” the authors wrote, adding that those under age 50 years without a clear preference for screening should not be screened.
However, the evidence shows that most average-risk women with no symptoms will benefit from mammography every other year beginning at age 50 years, they said.
The statement, published online April 8 in the Annals of Internal Medicine, was derived from a review of seven existing English-language breast cancer screening guidelines and the evidence cited in those guidelines. It’s intended to be a resource for all clinicians.
It differs from the 2017 American College of Obstetricians and Gynecologists (ACOG) guidelines in that ACOG recommends CBE and does not address screening in those with a life expectancy of less than 10 years. It also differs from the 2016 U.S. Preventive Services Task Force (USPSTF) guidelines, which make no recommendation on CBE and also do not address screening in those with a life expectancy of less than 10 years.
Other guidelines, such as those from the American College of Radiology, American Cancer Society (ACS), the Canadian Task Force on Preventive Health Care, and the National Comprehensive Cancer Network, recommend CBE, and the World Health Organization guidelines recommend CBE in low resource settings.
“Although CBE continues to be used as part of the examination of symptomatic women, data are sparse on screening asymptomatic women using CBE alone or combined with mammography,” the ACP guideline authors wrote. “The ACS recommends against CBE in average-risk women of any age because of the lack of demonstrated benefit and the potential for false-positive results.”
The guidance, which does not apply to patients with prior abnormal screening results or those at higher breast cancer risk, also includes an evidence-driven “talking points with patients” section based on frequently asked questions.
An important goal of the ACP Clinical Guidelines Committee in developing the guidance is to reduce overdiagnosis and overtreatment, which affects about 20% of women diagnosed over a 10-year period.
The committee reviewed all national guidelines published in English between January 1, 2013, and November 15, 2017, in the National Guideline Clearinghouse or Guidelines International Network library, and it also selected other guidelines commonly used in clinical practice. The committee evaluated the quality of each by using the Appraisal of Guidelines for Research and Evaluation II (AGREE II) instrument.
Alex Krist, MD, the USPSTF vice-chairperson, offered support for the “shift toward shared decision making that is emerging” and added it’s “part of a larger movement toward empowering people with information not only about the potential benefits but also the potential harms of screening tests.”
“In its 2016 recommendation, the Task Force found that the value of mammography increases with age, with women ages 50-74 benefiting most from screening. For women in their 40s, the Task Force also found that mammography screening every two years can be effective,” he told this publication. “We recommend that the decision to start screening should be an individual one, taking into account a woman’s health history, preferences, and how she values the different potential benefits and harms.”
Dr. Krist further noted that the USPSTF, ACP, and many others “have all affirmed that mammography is an important tool to reduce breast cancer mortality and that the benefits of mammography increase with age.”
Likewise, Robert Smith, PhD, vice president of cancer screening for the ACS, noted that the ACP guidance generally aligns with ACS and USPSTF guidelines because all “support informed decision making starting at age 40, and screening every two years starting at age 50 (USPSTF) or 55 (ACS).”
“The fact that all guidelines are not totally in sync is not unexpected. ... The most important thing to recognize is that all of these guidelines stress that regular mammography plays an important role in breast cancer early detection, and women should be aware of its benefits and limitations, and also remain vigilant and report any breast changes,” he said.
The guidance authors reported having no conflicts of interest.
SOURCE: Qaseem A et al., Ann Intern Med. 2019. doi: 10.7326/M18-2147.
REPORTING FROM THE ANNALS OF INTERNAL MEDICINE
FDA approves palbociclib for men with HR+/HER2- advanced breast cancer
The Food and Drug Administration has expanded the indication of palbociclib (Ibrance) in combination with specific endocrine therapies for hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative advanced or metastatic breast cancer in men.

Approval was based on postmarketing reports and electronic health records showing that the safety profile for men is consistent with that of women, the FDA said in a statement.
Less than 1% of all cases of breast cancer occur in men, but in the majority of those cases the tumors do express hormone receptors. Men are more likely to be diagnosed at a more advanced stage of disease. “According to the current clinical practice standards, male patients with breast cancer are treated similarly to women with breast cancer,” the FDA said.
The kinase inhibitor palbociclib was initially approved in 2015, in combination with an aromatase inhibitor, for postmenopausal women as first-line treatment of advanced disease.
The most common side effects in patients taking palbociclib are infections, leukopenia, fatigue, nausea, stomatitis, anemia, hair loss, diarrhea, and thrombocytopenia.
Because of the potential for genotoxicity, the FDA advised health care providers to tell male patients with female partners of reproductive potential to use effective contraception during treatment with palbociclib and for 3 months after the last dose.
The Food and Drug Administration has expanded the indication of palbociclib (Ibrance) in combination with specific endocrine therapies for hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative advanced or metastatic breast cancer in men.
Approval was based on postmarketing reports and electronic health records showing that the safety profile for men is consistent with that of women, the FDA said in a statement.
Less than 1% of all cases of breast cancer occur in men, but in the majority of those cases the tumors do express hormone receptors. Men are more likely to be diagnosed at a more advanced stage of disease. “According to the current clinical practice standards, male patients with breast cancer are treated similarly to women with breast cancer,” the FDA said.
The kinase inhibitor palbociclib was initially approved in 2015, in combination with an aromatase inhibitor, for postmenopausal women as first-line treatment of advanced disease.
The most common side effects in patients taking palbociclib are infections, leukopenia, fatigue, nausea, stomatitis, anemia, hair loss, diarrhea, and thrombocytopenia.
Because of the potential for genotoxicity, the FDA advised health care providers to tell male patients with female partners of reproductive potential to use effective contraception during treatment with palbociclib and for 3 months after the last dose.
The Food and Drug Administration has expanded the indication of palbociclib (Ibrance) in combination with specific endocrine therapies for hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative advanced or metastatic breast cancer in men.
Approval was based on postmarketing reports and electronic health records showing that the safety profile for men is consistent with that of women, the FDA said in a statement.
Less than 1% of all cases of breast cancer occur in men, but in the majority of those cases the tumors do express hormone receptors. Men are more likely to be diagnosed at a more advanced stage of disease. “According to the current clinical practice standards, male patients with breast cancer are treated similarly to women with breast cancer,” the FDA said.
The kinase inhibitor palbociclib was initially approved in 2015, in combination with an aromatase inhibitor, for postmenopausal women as first-line treatment of advanced disease.
The most common side effects in patients taking palbociclib are infections, leukopenia, fatigue, nausea, stomatitis, anemia, hair loss, diarrhea, and thrombocytopenia.
Because of the potential for genotoxicity, the FDA advised health care providers to tell male patients with female partners of reproductive potential to use effective contraception during treatment with palbociclib and for 3 months after the last dose.
Polyglutamine diseases are rare, but not the mutations that cause them
Polyglutamine diseases are a group of hereditary neurodegenerative disorders caused by mutations in which a trinucleotide repeat expands pathologically on a disease-associated gene. The diseases are rare, with the most common among them – Huntington disease – affecting between 10 and 14 per 100,000 people in Western countries, where prevalence is highest.
In polyglutamine diseases, which include the spinocerebellar ataxias, dentatorubral-pallidoluysian atrophy, and spinal bulbar muscular atrophy, higher CAG (cytosine-adenine-guanine) repeat numbers are associated with greater disease severity, faster progression, or earlier age at onset.
In research published April 1 in JAMA Neurology, investigators report that more than one-tenth of the European population carries CAG expansions that fall short of the repeats needed to cause any of 9 polyglutamine diseases – but are enough to put them at risk of having children who develop one. A smaller number of people – about 1% – carry enough CAG repeats to cause one of the diseases late in life.
For their research, Sarah L. Gardiner, MD, of Leiden (the Netherlands) University, and her colleagues looked at polyglutamine expansion variants for nine diseases in samples from 14,196 adults (56% of whom were women) from the Netherlands, Scotland, and Ireland. The samples were taken from five population-based cohort studies conducted between 1997 and 2012, and all subjects were without a history of polyglutamine disease or major depression.
Of these, 10.7% had a CAG repeat number on a disease-associated gene that was in the intermediate range, defined as a number of repeats that cannot cause disease but for which “expansion into the fully pathological range has been observed on intergenerational transmission,” Dr. Gardiner and her colleagues wrote. And some 1.3% of subjects were found to have CAG repeats within the disease-causing range, “mostly in the lower range associated with elderly onset.”
The investigators found no differences in sex, age, or body mass index between individuals with CAG repeat numbers within the pathological range and individuals with CAG repeat numbers within the normal or intermediate range.
Whether carriers of immediate or lower-range pathological CAG repeats went on to develop disease could not be measured, as follow-up data were not available. Another limitation of the study, the investigators acknowledged, was that the genotyping method used “did not allow us to determine the presence of trinucleotide interruptions,” which can affect disease penetrance.
“A late age at onset, a reduced penetrance, or the presence of interruptions could all explain the asymptomatic status of our carriers of intermediate and pathological polyglutamine disease–associated alleles at the time of assessment,” Dr. Gardiner and her colleagues wrote.
This study was funded by the European Union and Dutch government agencies; one of the population-based cohort studies from which the study sample was taken received some support from Bristol-Myers-Squibb. One of Dr. Gardiner’s coauthors, Raymund A. C. Roos, MD, PhD, disclosed being an adviser for UniQure, a gene-therapy firm, and no other conflicts of interest were reported.
SOURCE: Gardiner et al. JAMA Neurol. 2019 Apr 1. doi: 10.001/jamaneurol.2019.042.
Gardiner et al. describe the results of an appraisal of polyglutamine expansion variants in more than 14,000 individuals from the Netherlands, Scotland, and Ireland. Given the relative rarity of polyglutamine repeat disease, the first question that comes to mind is why were so many individuals identified with repeats in the pathogenic range? Based on our understanding of disease prevalence, it is unlikely that each of these individuals will become affected; therefore, this work suggests a reduced penetrance of these mutations. The findings are illustrative of a growing theme in human disease genetics: There are a very large number of apparently healthy individuals in the general population who carry mutations associated with various diseases. The phenomenon of reduced penetrance, where mutations cause disease in some but not all carriers, overlaps and arguably may be the same as that of variable expressivity, where the same mutation can lead to very different disease outcomes in different individuals. It is extremely likely that second-generation sequencing and population-scale screening will continue to reveal similar themes. We continue to appreciate the increasing complexity of the human genome and its relationship to disease, even those diseases we thought of previously as simple “single-gene” disorders.
Monia B. Hammer, PhD, and Andrew B. Singleton, PhD, are with the National Institute on Aging, National Institutes of Health, Bethesda, Md. Dr. Hammer and Dr. Singleton report no financial conflicts of interest related to their editorial.
Gardiner et al. describe the results of an appraisal of polyglutamine expansion variants in more than 14,000 individuals from the Netherlands, Scotland, and Ireland. Given the relative rarity of polyglutamine repeat disease, the first question that comes to mind is why were so many individuals identified with repeats in the pathogenic range? Based on our understanding of disease prevalence, it is unlikely that each of these individuals will become affected; therefore, this work suggests a reduced penetrance of these mutations. The findings are illustrative of a growing theme in human disease genetics: There are a very large number of apparently healthy individuals in the general population who carry mutations associated with various diseases. The phenomenon of reduced penetrance, where mutations cause disease in some but not all carriers, overlaps and arguably may be the same as that of variable expressivity, where the same mutation can lead to very different disease outcomes in different individuals. It is extremely likely that second-generation sequencing and population-scale screening will continue to reveal similar themes. We continue to appreciate the increasing complexity of the human genome and its relationship to disease, even those diseases we thought of previously as simple “single-gene” disorders.
Monia B. Hammer, PhD, and Andrew B. Singleton, PhD, are with the National Institute on Aging, National Institutes of Health, Bethesda, Md. Dr. Hammer and Dr. Singleton report no financial conflicts of interest related to their editorial.
Gardiner et al. describe the results of an appraisal of polyglutamine expansion variants in more than 14,000 individuals from the Netherlands, Scotland, and Ireland. Given the relative rarity of polyglutamine repeat disease, the first question that comes to mind is why were so many individuals identified with repeats in the pathogenic range? Based on our understanding of disease prevalence, it is unlikely that each of these individuals will become affected; therefore, this work suggests a reduced penetrance of these mutations. The findings are illustrative of a growing theme in human disease genetics: There are a very large number of apparently healthy individuals in the general population who carry mutations associated with various diseases. The phenomenon of reduced penetrance, where mutations cause disease in some but not all carriers, overlaps and arguably may be the same as that of variable expressivity, where the same mutation can lead to very different disease outcomes in different individuals. It is extremely likely that second-generation sequencing and population-scale screening will continue to reveal similar themes. We continue to appreciate the increasing complexity of the human genome and its relationship to disease, even those diseases we thought of previously as simple “single-gene” disorders.
Monia B. Hammer, PhD, and Andrew B. Singleton, PhD, are with the National Institute on Aging, National Institutes of Health, Bethesda, Md. Dr. Hammer and Dr. Singleton report no financial conflicts of interest related to their editorial.
Polyglutamine diseases are a group of hereditary neurodegenerative disorders caused by mutations in which a trinucleotide repeat expands pathologically on a disease-associated gene. The diseases are rare, with the most common among them – Huntington disease – affecting between 10 and 14 per 100,000 people in Western countries, where prevalence is highest.
In polyglutamine diseases, which include the spinocerebellar ataxias, dentatorubral-pallidoluysian atrophy, and spinal bulbar muscular atrophy, higher CAG (cytosine-adenine-guanine) repeat numbers are associated with greater disease severity, faster progression, or earlier age at onset.
In research published April 1 in JAMA Neurology, investigators report that more than one-tenth of the European population carries CAG expansions that fall short of the repeats needed to cause any of 9 polyglutamine diseases – but are enough to put them at risk of having children who develop one. A smaller number of people – about 1% – carry enough CAG repeats to cause one of the diseases late in life.
For their research, Sarah L. Gardiner, MD, of Leiden (the Netherlands) University, and her colleagues looked at polyglutamine expansion variants for nine diseases in samples from 14,196 adults (56% of whom were women) from the Netherlands, Scotland, and Ireland. The samples were taken from five population-based cohort studies conducted between 1997 and 2012, and all subjects were without a history of polyglutamine disease or major depression.
Of these, 10.7% had a CAG repeat number on a disease-associated gene that was in the intermediate range, defined as a number of repeats that cannot cause disease but for which “expansion into the fully pathological range has been observed on intergenerational transmission,” Dr. Gardiner and her colleagues wrote. And some 1.3% of subjects were found to have CAG repeats within the disease-causing range, “mostly in the lower range associated with elderly onset.”
The investigators found no differences in sex, age, or body mass index between individuals with CAG repeat numbers within the pathological range and individuals with CAG repeat numbers within the normal or intermediate range.
Whether carriers of immediate or lower-range pathological CAG repeats went on to develop disease could not be measured, as follow-up data were not available. Another limitation of the study, the investigators acknowledged, was that the genotyping method used “did not allow us to determine the presence of trinucleotide interruptions,” which can affect disease penetrance.
“A late age at onset, a reduced penetrance, or the presence of interruptions could all explain the asymptomatic status of our carriers of intermediate and pathological polyglutamine disease–associated alleles at the time of assessment,” Dr. Gardiner and her colleagues wrote.
This study was funded by the European Union and Dutch government agencies; one of the population-based cohort studies from which the study sample was taken received some support from Bristol-Myers-Squibb. One of Dr. Gardiner’s coauthors, Raymund A. C. Roos, MD, PhD, disclosed being an adviser for UniQure, a gene-therapy firm, and no other conflicts of interest were reported.
SOURCE: Gardiner et al. JAMA Neurol. 2019 Apr 1. doi: 10.001/jamaneurol.2019.042.
Polyglutamine diseases are a group of hereditary neurodegenerative disorders caused by mutations in which a trinucleotide repeat expands pathologically on a disease-associated gene. The diseases are rare, with the most common among them – Huntington disease – affecting between 10 and 14 per 100,000 people in Western countries, where prevalence is highest.
In polyglutamine diseases, which include the spinocerebellar ataxias, dentatorubral-pallidoluysian atrophy, and spinal bulbar muscular atrophy, higher CAG (cytosine-adenine-guanine) repeat numbers are associated with greater disease severity, faster progression, or earlier age at onset.
In research published April 1 in JAMA Neurology, investigators report that more than one-tenth of the European population carries CAG expansions that fall short of the repeats needed to cause any of 9 polyglutamine diseases – but are enough to put them at risk of having children who develop one. A smaller number of people – about 1% – carry enough CAG repeats to cause one of the diseases late in life.
For their research, Sarah L. Gardiner, MD, of Leiden (the Netherlands) University, and her colleagues looked at polyglutamine expansion variants for nine diseases in samples from 14,196 adults (56% of whom were women) from the Netherlands, Scotland, and Ireland. The samples were taken from five population-based cohort studies conducted between 1997 and 2012, and all subjects were without a history of polyglutamine disease or major depression.
Of these, 10.7% had a CAG repeat number on a disease-associated gene that was in the intermediate range, defined as a number of repeats that cannot cause disease but for which “expansion into the fully pathological range has been observed on intergenerational transmission,” Dr. Gardiner and her colleagues wrote. And some 1.3% of subjects were found to have CAG repeats within the disease-causing range, “mostly in the lower range associated with elderly onset.”
The investigators found no differences in sex, age, or body mass index between individuals with CAG repeat numbers within the pathological range and individuals with CAG repeat numbers within the normal or intermediate range.
Whether carriers of immediate or lower-range pathological CAG repeats went on to develop disease could not be measured, as follow-up data were not available. Another limitation of the study, the investigators acknowledged, was that the genotyping method used “did not allow us to determine the presence of trinucleotide interruptions,” which can affect disease penetrance.
“A late age at onset, a reduced penetrance, or the presence of interruptions could all explain the asymptomatic status of our carriers of intermediate and pathological polyglutamine disease–associated alleles at the time of assessment,” Dr. Gardiner and her colleagues wrote.
This study was funded by the European Union and Dutch government agencies; one of the population-based cohort studies from which the study sample was taken received some support from Bristol-Myers-Squibb. One of Dr. Gardiner’s coauthors, Raymund A. C. Roos, MD, PhD, disclosed being an adviser for UniQure, a gene-therapy firm, and no other conflicts of interest were reported.
SOURCE: Gardiner et al. JAMA Neurol. 2019 Apr 1. doi: 10.001/jamaneurol.2019.042.
FROM JAMA NEUROLOGY
Marcela Romero-Reyes, DDS, PhD, Comments on Peripheral and Central Headache Challenges

###
Dr. Rapoport: Do you commonly see patients who present with symptoms of both central and peripheral symptoms in practice?
Dr. Romero-Reyes: Yes, I see patients that present with temporomandibular disorders (TMD) and headache comorbidity, as well as patients with migraine, tension-type headache, and cervicogenic headache with myofascial pain.
Dr. Rapoport: Why do you think this condition is so challenging to treat?
Dr. Romero-Reyes: I think this is because of the lack of understanding and awareness that in addition to the multifactorial nature of headache disorders, other types of disorders that are not neurovascular in origin may influence trigeminovascular nociception, and these types of non-neurovascular disorders involve the skill and knowledge of other expertise.
Headaches receiving inputs from extracranial structures such as in TMD (temporomandibular joint [TMJ] and muscles of mastication) and/or cervical structures (cervical spine, cervical muscles) require multidisciplinary evaluation and management. In these cases, the management should involve a neurologist specialized in headache disorders, a dentist trained in TMD and orofacial pain disorders, and a physical therapist with special training in craniofacial and cervical Therapeutics. Multidisciplinary communication is key for successful management.
Another reason is that myofascial pain (MFP) is often overlooked in patients with headache disorders. In my experience, patients with episodic and chronic migraine, episodic and chronic tension-type headache, cervicogenic headache, and patients presenting TMD and headache comorbidity can present trigger points in the craniofacial and cervical muscles, an indication of MFP. It has been reported that these patients present a higher disability impact. The presence of MFP may be contributing to the activation of the trigeminovascular system and therefore facilitate, exacerbate, and perpetuate headache symptomatology and may accelerate the progression to a more chronic form of the disorder.
Dr. Rapoport: In your opinion, is this considered a controversial topic? Why or why not?
Dr. Romero-Reyes: Yes, I think it is necessary to clarify that tenderness in the back of the head or of neck muscles present in headache patients does not necessarily imply that it is due to a nerve compression. This could also be caused by local myalgia but more commonly, from latent or active myofascial trigger points present in the muscles of the area being palpated, or by referred pain beyond the area of the muscle being palpated. Suboccipital muscles (in the occiput area) are not the only muscle group that is associated with headache and neck pain symptomatology. For example, the trapezius muscle, which is an overlooked source of tension- type and cervicogenic headache, can present trigger points that can refer pain to the shoulder, neck, head, face and the eye. In addition, other craniofacial and cervical muscles such as the sternocleidomastoid (SCM) and temporalis muscles have been shown to be associated with headache symptomatology in the migraineur, as well as the chronic tension-type headache patient. Other muscles that also refer to the craniofacial area and can elicit headache and neck pain symptomatology include the masseter, occipitofrontalis, splenius capitis, splenius cervicis, semispinalis capitis, semispinalis cervicis and multifidi (cervical). The presence of trigger points in these muscles do not support or warrant the need to be removed or managed with non-conservative approaches.
Myofascial trigger points can result from muscle injury and overload, parafunctional activity, and poor head and neck posture. MFP is characterized by a regional pain and presence of localized tender areas (trigger points) in muscle, fascia or tendons that reproduce pain when palpated, and produce a pattern of regional pain spreading along the muscle palpated, or beyond the location boundary of the muscle palpated. It has been shown by microdyalisis that inflammatory mediators and neuropeptides are present in the area of an active trigger point. In addition, an increase of electromyography activity has been shown in trigger points in patients with chronic tension-type headache when compared with controls.
The importance of an evaluation by a skilled clinician in the craniofacial and cervical area to verify the source of pain is critical. The patient may be reporting pain in one area, but the source of the pain is in another area, and this is typical symptomatology present when there are active trigger points. In addition, an assessment of any contributing factors arising from the cervical spine (eg, poor posture) and craniofacial area (eg, TMD) that may exacerbate headache symptomatology is vital to proper diagnosis.
In my experience, patients with migraine, tension-type headache, cervicogenic headache, and TMD and headache comorbidity present MFP perpetuating headache symptomatology. MFP is not managed by surgical interventions. This perpetuating factor can be managed effectively with conservative measures. The plan is tailored for each patient’s needs. In general, the plan of management may include trigger point injections in the muscle with anesthetics, dry needling, and a physical therapy plan that may include education regarding habits and posture, exercises and physical therapy modalities, which are crucial to relieve pain and increase function. In cases of TMD and headache comorbidity, an occlusal appliance (stabilization appliance) can be included if necessary. We should also consider behavioral therapies (especially EMG biofeedback training) and some oral anti-inflammatories or muscle relaxants in the beginning of management, together with the plan of management mentioned above.
With these approaches to manage the MFP component in headache patients, I have been able to see that in migraineurs with MFP, the frequency and severity of the attacks decrease significantly. The patient may still experience migraine attacks, but feel happy to have the possibility to reduce medication intake and be in more control of their pain. In patients with tension-type headache, I have seen this even more dramatically.
This is telling us that headache pathophysiology involves a “conversation” between the peripheral and central nervous system, which influence each other. Peripheral nociceptive input coming from extracranial structures can induce trigeminovascular activation and therefore exacerbate a headache disorder and vice versa. Chronic myofascial pain may be the result of central sensitization due to the protracted peripheral nociceptive input (eg, poor posture, neck strain, parafunctional activity), therefore perpetuating the headache disorder even more.
Dr. Rapoport: Do you have any other comments about the article Treatment Challenges When Headache Has Central and Peripheral Involvement that you would like to share with our readers?
Dr. Romero-Reyes: It is simplistic to say migraine is either a peripheral or a central disorder, or that symptoms are either peripheral or central. Beyond thinking about migraine pain, migraine is fundamentally a brain (central) disorder. Its associated symptoms (nausea, phonophobia, photophobia) tell us this. Migraine headache is complex, and most likely the result of central mechanisms that can be influenced by peripheral inputs from the craniofacial and cervical region.
Embarking on surgical interventions for the management of headache disorders warrants a caution since it is still an experimental research question and the need of such therapies should be evaluated against conservative management. We are in a very exciting and hopeful time for migraine management. New evidence-based options from biological agents, such as anti-calcitonin gene-related peptide (CGRP) therapies, to non-pharmacological approaches, such as neuromodulation, can be offered to the patients. If the patient is experiencing pain in the neck area or other craniofacial area, it is recommended to have a thorough evaluation by a physical therapist with special training in cervical and craniofacial therapeutics and/or a dentist trained in TMD and orofacial pain disorders to work in consultation with a neurologist to elaborate a personalized management plan. Do not overlook the contribution of myofascial pain (trigger points) as well as TMD in the symptomatology of headache disorders. Few patients need to undergo surgical measures of peripheral nerves and muscles for improvement. An exhaustive evaluation must be undertaken first.
Resources for patients:
AHS
https://americanheadachesociety.org/
https://americanheadachesociety.org/wp-content/uploads/2018/06/Choosing-Wisely-Flyer.pdf
AAOP
https://aaop.clubexpress.com/content.aspx?sl=1152088466
PTBCTT
###
Dr. Rapoport: Do you commonly see patients who present with symptoms of both central and peripheral symptoms in practice?
Dr. Romero-Reyes: Yes, I see patients that present with temporomandibular disorders (TMD) and headache comorbidity, as well as patients with migraine, tension-type headache, and cervicogenic headache with myofascial pain.
Dr. Rapoport: Why do you think this condition is so challenging to treat?
Dr. Romero-Reyes: I think this is because of the lack of understanding and awareness that in addition to the multifactorial nature of headache disorders, other types of disorders that are not neurovascular in origin may influence trigeminovascular nociception, and these types of non-neurovascular disorders involve the skill and knowledge of other expertise.
Headaches receiving inputs from extracranial structures such as in TMD (temporomandibular joint [TMJ] and muscles of mastication) and/or cervical structures (cervical spine, cervical muscles) require multidisciplinary evaluation and management. In these cases, the management should involve a neurologist specialized in headache disorders, a dentist trained in TMD and orofacial pain disorders, and a physical therapist with special training in craniofacial and cervical Therapeutics. Multidisciplinary communication is key for successful management.
Another reason is that myofascial pain (MFP) is often overlooked in patients with headache disorders. In my experience, patients with episodic and chronic migraine, episodic and chronic tension-type headache, cervicogenic headache, and patients presenting TMD and headache comorbidity can present trigger points in the craniofacial and cervical muscles, an indication of MFP. It has been reported that these patients present a higher disability impact. The presence of MFP may be contributing to the activation of the trigeminovascular system and therefore facilitate, exacerbate, and perpetuate headache symptomatology and may accelerate the progression to a more chronic form of the disorder.
Dr. Rapoport: In your opinion, is this considered a controversial topic? Why or why not?
Dr. Romero-Reyes: Yes, I think it is necessary to clarify that tenderness in the back of the head or of neck muscles present in headache patients does not necessarily imply that it is due to a nerve compression. This could also be caused by local myalgia but more commonly, from latent or active myofascial trigger points present in the muscles of the area being palpated, or by referred pain beyond the area of the muscle being palpated. Suboccipital muscles (in the occiput area) are not the only muscle group that is associated with headache and neck pain symptomatology. For example, the trapezius muscle, which is an overlooked source of tension- type and cervicogenic headache, can present trigger points that can refer pain to the shoulder, neck, head, face and the eye. In addition, other craniofacial and cervical muscles such as the sternocleidomastoid (SCM) and temporalis muscles have been shown to be associated with headache symptomatology in the migraineur, as well as the chronic tension-type headache patient. Other muscles that also refer to the craniofacial area and can elicit headache and neck pain symptomatology include the masseter, occipitofrontalis, splenius capitis, splenius cervicis, semispinalis capitis, semispinalis cervicis and multifidi (cervical). The presence of trigger points in these muscles do not support or warrant the need to be removed or managed with non-conservative approaches.
Myofascial trigger points can result from muscle injury and overload, parafunctional activity, and poor head and neck posture. MFP is characterized by a regional pain and presence of localized tender areas (trigger points) in muscle, fascia or tendons that reproduce pain when palpated, and produce a pattern of regional pain spreading along the muscle palpated, or beyond the location boundary of the muscle palpated. It has been shown by microdyalisis that inflammatory mediators and neuropeptides are present in the area of an active trigger point. In addition, an increase of electromyography activity has been shown in trigger points in patients with chronic tension-type headache when compared with controls.
The importance of an evaluation by a skilled clinician in the craniofacial and cervical area to verify the source of pain is critical. The patient may be reporting pain in one area, but the source of the pain is in another area, and this is typical symptomatology present when there are active trigger points. In addition, an assessment of any contributing factors arising from the cervical spine (eg, poor posture) and craniofacial area (eg, TMD) that may exacerbate headache symptomatology is vital to proper diagnosis.
In my experience, patients with migraine, tension-type headache, cervicogenic headache, and TMD and headache comorbidity present MFP perpetuating headache symptomatology. MFP is not managed by surgical interventions. This perpetuating factor can be managed effectively with conservative measures. The plan is tailored for each patient’s needs. In general, the plan of management may include trigger point injections in the muscle with anesthetics, dry needling, and a physical therapy plan that may include education regarding habits and posture, exercises and physical therapy modalities, which are crucial to relieve pain and increase function. In cases of TMD and headache comorbidity, an occlusal appliance (stabilization appliance) can be included if necessary. We should also consider behavioral therapies (especially EMG biofeedback training) and some oral anti-inflammatories or muscle relaxants in the beginning of management, together with the plan of management mentioned above.
With these approaches to manage the MFP component in headache patients, I have been able to see that in migraineurs with MFP, the frequency and severity of the attacks decrease significantly. The patient may still experience migraine attacks, but feel happy to have the possibility to reduce medication intake and be in more control of their pain. In patients with tension-type headache, I have seen this even more dramatically.
This is telling us that headache pathophysiology involves a “conversation” between the peripheral and central nervous system, which influence each other. Peripheral nociceptive input coming from extracranial structures can induce trigeminovascular activation and therefore exacerbate a headache disorder and vice versa. Chronic myofascial pain may be the result of central sensitization due to the protracted peripheral nociceptive input (eg, poor posture, neck strain, parafunctional activity), therefore perpetuating the headache disorder even more.
Dr. Rapoport: Do you have any other comments about the article Treatment Challenges When Headache Has Central and Peripheral Involvement that you would like to share with our readers?
Dr. Romero-Reyes: It is simplistic to say migraine is either a peripheral or a central disorder, or that symptoms are either peripheral or central. Beyond thinking about migraine pain, migraine is fundamentally a brain (central) disorder. Its associated symptoms (nausea, phonophobia, photophobia) tell us this. Migraine headache is complex, and most likely the result of central mechanisms that can be influenced by peripheral inputs from the craniofacial and cervical region.
Embarking on surgical interventions for the management of headache disorders warrants a caution since it is still an experimental research question and the need of such therapies should be evaluated against conservative management. We are in a very exciting and hopeful time for migraine management. New evidence-based options from biological agents, such as anti-calcitonin gene-related peptide (CGRP) therapies, to non-pharmacological approaches, such as neuromodulation, can be offered to the patients. If the patient is experiencing pain in the neck area or other craniofacial area, it is recommended to have a thorough evaluation by a physical therapist with special training in cervical and craniofacial therapeutics and/or a dentist trained in TMD and orofacial pain disorders to work in consultation with a neurologist to elaborate a personalized management plan. Do not overlook the contribution of myofascial pain (trigger points) as well as TMD in the symptomatology of headache disorders. Few patients need to undergo surgical measures of peripheral nerves and muscles for improvement. An exhaustive evaluation must be undertaken first.
Resources for patients:
AHS
https://americanheadachesociety.org/
https://americanheadachesociety.org/wp-content/uploads/2018/06/Choosing-Wisely-Flyer.pdf
AAOP
https://aaop.clubexpress.com/content.aspx?sl=1152088466
PTBCTT
###
Dr. Rapoport: Do you commonly see patients who present with symptoms of both central and peripheral symptoms in practice?
Dr. Romero-Reyes: Yes, I see patients that present with temporomandibular disorders (TMD) and headache comorbidity, as well as patients with migraine, tension-type headache, and cervicogenic headache with myofascial pain.
Dr. Rapoport: Why do you think this condition is so challenging to treat?
Dr. Romero-Reyes: I think this is because of the lack of understanding and awareness that in addition to the multifactorial nature of headache disorders, other types of disorders that are not neurovascular in origin may influence trigeminovascular nociception, and these types of non-neurovascular disorders involve the skill and knowledge of other expertise.
Headaches receiving inputs from extracranial structures such as in TMD (temporomandibular joint [TMJ] and muscles of mastication) and/or cervical structures (cervical spine, cervical muscles) require multidisciplinary evaluation and management. In these cases, the management should involve a neurologist specialized in headache disorders, a dentist trained in TMD and orofacial pain disorders, and a physical therapist with special training in craniofacial and cervical Therapeutics. Multidisciplinary communication is key for successful management.
Another reason is that myofascial pain (MFP) is often overlooked in patients with headache disorders. In my experience, patients with episodic and chronic migraine, episodic and chronic tension-type headache, cervicogenic headache, and patients presenting TMD and headache comorbidity can present trigger points in the craniofacial and cervical muscles, an indication of MFP. It has been reported that these patients present a higher disability impact. The presence of MFP may be contributing to the activation of the trigeminovascular system and therefore facilitate, exacerbate, and perpetuate headache symptomatology and may accelerate the progression to a more chronic form of the disorder.
Dr. Rapoport: In your opinion, is this considered a controversial topic? Why or why not?
Dr. Romero-Reyes: Yes, I think it is necessary to clarify that tenderness in the back of the head or of neck muscles present in headache patients does not necessarily imply that it is due to a nerve compression. This could also be caused by local myalgia but more commonly, from latent or active myofascial trigger points present in the muscles of the area being palpated, or by referred pain beyond the area of the muscle being palpated. Suboccipital muscles (in the occiput area) are not the only muscle group that is associated with headache and neck pain symptomatology. For example, the trapezius muscle, which is an overlooked source of tension- type and cervicogenic headache, can present trigger points that can refer pain to the shoulder, neck, head, face and the eye. In addition, other craniofacial and cervical muscles such as the sternocleidomastoid (SCM) and temporalis muscles have been shown to be associated with headache symptomatology in the migraineur, as well as the chronic tension-type headache patient. Other muscles that also refer to the craniofacial area and can elicit headache and neck pain symptomatology include the masseter, occipitofrontalis, splenius capitis, splenius cervicis, semispinalis capitis, semispinalis cervicis and multifidi (cervical). The presence of trigger points in these muscles do not support or warrant the need to be removed or managed with non-conservative approaches.
Myofascial trigger points can result from muscle injury and overload, parafunctional activity, and poor head and neck posture. MFP is characterized by a regional pain and presence of localized tender areas (trigger points) in muscle, fascia or tendons that reproduce pain when palpated, and produce a pattern of regional pain spreading along the muscle palpated, or beyond the location boundary of the muscle palpated. It has been shown by microdyalisis that inflammatory mediators and neuropeptides are present in the area of an active trigger point. In addition, an increase of electromyography activity has been shown in trigger points in patients with chronic tension-type headache when compared with controls.
The importance of an evaluation by a skilled clinician in the craniofacial and cervical area to verify the source of pain is critical. The patient may be reporting pain in one area, but the source of the pain is in another area, and this is typical symptomatology present when there are active trigger points. In addition, an assessment of any contributing factors arising from the cervical spine (eg, poor posture) and craniofacial area (eg, TMD) that may exacerbate headache symptomatology is vital to proper diagnosis.
In my experience, patients with migraine, tension-type headache, cervicogenic headache, and TMD and headache comorbidity present MFP perpetuating headache symptomatology. MFP is not managed by surgical interventions. This perpetuating factor can be managed effectively with conservative measures. The plan is tailored for each patient’s needs. In general, the plan of management may include trigger point injections in the muscle with anesthetics, dry needling, and a physical therapy plan that may include education regarding habits and posture, exercises and physical therapy modalities, which are crucial to relieve pain and increase function. In cases of TMD and headache comorbidity, an occlusal appliance (stabilization appliance) can be included if necessary. We should also consider behavioral therapies (especially EMG biofeedback training) and some oral anti-inflammatories or muscle relaxants in the beginning of management, together with the plan of management mentioned above.
With these approaches to manage the MFP component in headache patients, I have been able to see that in migraineurs with MFP, the frequency and severity of the attacks decrease significantly. The patient may still experience migraine attacks, but feel happy to have the possibility to reduce medication intake and be in more control of their pain. In patients with tension-type headache, I have seen this even more dramatically.
This is telling us that headache pathophysiology involves a “conversation” between the peripheral and central nervous system, which influence each other. Peripheral nociceptive input coming from extracranial structures can induce trigeminovascular activation and therefore exacerbate a headache disorder and vice versa. Chronic myofascial pain may be the result of central sensitization due to the protracted peripheral nociceptive input (eg, poor posture, neck strain, parafunctional activity), therefore perpetuating the headache disorder even more.
Dr. Rapoport: Do you have any other comments about the article Treatment Challenges When Headache Has Central and Peripheral Involvement that you would like to share with our readers?
Dr. Romero-Reyes: It is simplistic to say migraine is either a peripheral or a central disorder, or that symptoms are either peripheral or central. Beyond thinking about migraine pain, migraine is fundamentally a brain (central) disorder. Its associated symptoms (nausea, phonophobia, photophobia) tell us this. Migraine headache is complex, and most likely the result of central mechanisms that can be influenced by peripheral inputs from the craniofacial and cervical region.
Embarking on surgical interventions for the management of headache disorders warrants a caution since it is still an experimental research question and the need of such therapies should be evaluated against conservative management. We are in a very exciting and hopeful time for migraine management. New evidence-based options from biological agents, such as anti-calcitonin gene-related peptide (CGRP) therapies, to non-pharmacological approaches, such as neuromodulation, can be offered to the patients. If the patient is experiencing pain in the neck area or other craniofacial area, it is recommended to have a thorough evaluation by a physical therapist with special training in cervical and craniofacial therapeutics and/or a dentist trained in TMD and orofacial pain disorders to work in consultation with a neurologist to elaborate a personalized management plan. Do not overlook the contribution of myofascial pain (trigger points) as well as TMD in the symptomatology of headache disorders. Few patients need to undergo surgical measures of peripheral nerves and muscles for improvement. An exhaustive evaluation must be undertaken first.
Resources for patients:
AHS
https://americanheadachesociety.org/
https://americanheadachesociety.org/wp-content/uploads/2018/06/Choosing-Wisely-Flyer.pdf
AAOP
https://aaop.clubexpress.com/content.aspx?sl=1152088466
PTBCTT
Amyloid brain imaging changed clinical management in 60% of MCI and dementia patients
.

Diagnoses changed from Alzheimer’s disease to non–Alzheimer’s disease in 25% of 11,409 patients and from non–Alzheimer’s disease to Alzheimer’s disease in 10.5%, reported Gil Rabinovici, MD, and his colleagues. The use of Alzheimer’s disease drugs doubled in amyloid-positive MCI patients, and increased by a third in amyloid-positive dementia patients. Physicians involved in the study said the scans provided key clinical information in 82% of cases with post-scan management changes.
Scans also benefited amyloid-negative patients. Before the scan, 71% of these carried an Alzheimer’s disease diagnosis; afterward, just 10% did, opening the way for an accurate diagnosis and more effective treatment.
The study was powered to detect a 30% or greater change in the MCI and dementia groups. The 60% change emphasize how useful amyloid PET scans could be in clinical practice, Dr. Rabinovici, the study’s lead author and principal investigator, said in a press statement.
“We are impressed by the magnitude of these results, which make it clear that amyloid PET imaging can have a major impact on how we diagnose and care for patients with Alzheimer’s disease and other forms of cognitive decline,” said Dr. Rabinovici of the University of California, San Francisco.
Alzheimer’s Association leaders were similarly pleased.
“These results present highly credible, large-scale evidence that amyloid PET imaging can be a powerful tool to improve the accuracy of Alzheimer’s diagnosis and lead to better medical management, especially in difficult-to-diagnose cases,” said Maria C. Carrillo, PhD, chief science officer of the Alzheimer’s Association and a coauthor of the study. “It is important that amyloid PET imaging be more broadly accessible to those who need it.”

Next steps
Ultimately, investigators hope the nationwide-wide, open-label study will prove the clinical value of amyloid PET scanning and convince the Centers for Medicare & Medicaid Services to make the test a fully covered service for those who meet the appropriate use criteria set forth by the Alzheimer’s Association and the Society of Nuclear Medicine and Molecular Imaging.
IDEAS’ second goal – showing that the scans improve health outcomes – is scheduled for 2020. These data are a key component of the CMS decision, but they might be a tough sell, Clifford R. Jack Jr., MD, and Ronald C. Petersen, MD, PhD, wrote in an accompanying editorial. Dr. Jack and Dr. Petersen are affiliated with the Mayo Clinic in Rochester, Minn.
“For CMS to cover the cost of amyloid PET, it must be demonstrated that the result of a scan has an effect on patient outcomes, not just patient care processes – and, without a disease-modifying therapy available, that might be a challenge,” they wrote.
IDEAS is a funding collaboration of the CMS, the Alzheimer’s Association, Avid Radiopharmaceuticals/Eli Lilly, General Electric Healthcare, Piramal Imaging, and the American College of Radiology. Dr. Rabinovici had no financial disclosures.
SOURCE: Rabinovici GD et al. JAMA. 2019 Apr 2. doi: 10.1001/jama.2019.2000.
Current clinical practice does not routinely include biomarkers, and if given a choice, most patients would prefer brain imaging to spinal fluid-based testing, so IDEAS may be making imaging-based biomarker characterization a real possibility in the future.

Richard J. Caselli, MD, is professor of neurology at the Mayo Clinic Arizona in Scottsdale and associate director and clinical core director of the Arizona Alzheimer’s Disease Center. He made these comments in an interview.
Current clinical practice does not routinely include biomarkers, and if given a choice, most patients would prefer brain imaging to spinal fluid-based testing, so IDEAS may be making imaging-based biomarker characterization a real possibility in the future.
Richard J. Caselli, MD, is professor of neurology at the Mayo Clinic Arizona in Scottsdale and associate director and clinical core director of the Arizona Alzheimer’s Disease Center. He made these comments in an interview.
Current clinical practice does not routinely include biomarkers, and if given a choice, most patients would prefer brain imaging to spinal fluid-based testing, so IDEAS may be making imaging-based biomarker characterization a real possibility in the future.
Richard J. Caselli, MD, is professor of neurology at the Mayo Clinic Arizona in Scottsdale and associate director and clinical core director of the Arizona Alzheimer’s Disease Center. He made these comments in an interview.
.
Diagnoses changed from Alzheimer’s disease to non–Alzheimer’s disease in 25% of 11,409 patients and from non–Alzheimer’s disease to Alzheimer’s disease in 10.5%, reported Gil Rabinovici, MD, and his colleagues. The use of Alzheimer’s disease drugs doubled in amyloid-positive MCI patients, and increased by a third in amyloid-positive dementia patients. Physicians involved in the study said the scans provided key clinical information in 82% of cases with post-scan management changes.
Scans also benefited amyloid-negative patients. Before the scan, 71% of these carried an Alzheimer’s disease diagnosis; afterward, just 10% did, opening the way for an accurate diagnosis and more effective treatment.
The study was powered to detect a 30% or greater change in the MCI and dementia groups. The 60% change emphasize how useful amyloid PET scans could be in clinical practice, Dr. Rabinovici, the study’s lead author and principal investigator, said in a press statement.
“We are impressed by the magnitude of these results, which make it clear that amyloid PET imaging can have a major impact on how we diagnose and care for patients with Alzheimer’s disease and other forms of cognitive decline,” said Dr. Rabinovici of the University of California, San Francisco.
Alzheimer’s Association leaders were similarly pleased.
“These results present highly credible, large-scale evidence that amyloid PET imaging can be a powerful tool to improve the accuracy of Alzheimer’s diagnosis and lead to better medical management, especially in difficult-to-diagnose cases,” said Maria C. Carrillo, PhD, chief science officer of the Alzheimer’s Association and a coauthor of the study. “It is important that amyloid PET imaging be more broadly accessible to those who need it.”
Next steps
Ultimately, investigators hope the nationwide-wide, open-label study will prove the clinical value of amyloid PET scanning and convince the Centers for Medicare & Medicaid Services to make the test a fully covered service for those who meet the appropriate use criteria set forth by the Alzheimer’s Association and the Society of Nuclear Medicine and Molecular Imaging.
IDEAS’ second goal – showing that the scans improve health outcomes – is scheduled for 2020. These data are a key component of the CMS decision, but they might be a tough sell, Clifford R. Jack Jr., MD, and Ronald C. Petersen, MD, PhD, wrote in an accompanying editorial. Dr. Jack and Dr. Petersen are affiliated with the Mayo Clinic in Rochester, Minn.
“For CMS to cover the cost of amyloid PET, it must be demonstrated that the result of a scan has an effect on patient outcomes, not just patient care processes – and, without a disease-modifying therapy available, that might be a challenge,” they wrote.
IDEAS is a funding collaboration of the CMS, the Alzheimer’s Association, Avid Radiopharmaceuticals/Eli Lilly, General Electric Healthcare, Piramal Imaging, and the American College of Radiology. Dr. Rabinovici had no financial disclosures.
SOURCE: Rabinovici GD et al. JAMA. 2019 Apr 2. doi: 10.1001/jama.2019.2000.
.
Diagnoses changed from Alzheimer’s disease to non–Alzheimer’s disease in 25% of 11,409 patients and from non–Alzheimer’s disease to Alzheimer’s disease in 10.5%, reported Gil Rabinovici, MD, and his colleagues. The use of Alzheimer’s disease drugs doubled in amyloid-positive MCI patients, and increased by a third in amyloid-positive dementia patients. Physicians involved in the study said the scans provided key clinical information in 82% of cases with post-scan management changes.
Scans also benefited amyloid-negative patients. Before the scan, 71% of these carried an Alzheimer’s disease diagnosis; afterward, just 10% did, opening the way for an accurate diagnosis and more effective treatment.
The study was powered to detect a 30% or greater change in the MCI and dementia groups. The 60% change emphasize how useful amyloid PET scans could be in clinical practice, Dr. Rabinovici, the study’s lead author and principal investigator, said in a press statement.
“We are impressed by the magnitude of these results, which make it clear that amyloid PET imaging can have a major impact on how we diagnose and care for patients with Alzheimer’s disease and other forms of cognitive decline,” said Dr. Rabinovici of the University of California, San Francisco.
Alzheimer’s Association leaders were similarly pleased.
“These results present highly credible, large-scale evidence that amyloid PET imaging can be a powerful tool to improve the accuracy of Alzheimer’s diagnosis and lead to better medical management, especially in difficult-to-diagnose cases,” said Maria C. Carrillo, PhD, chief science officer of the Alzheimer’s Association and a coauthor of the study. “It is important that amyloid PET imaging be more broadly accessible to those who need it.”
Next steps
Ultimately, investigators hope the nationwide-wide, open-label study will prove the clinical value of amyloid PET scanning and convince the Centers for Medicare & Medicaid Services to make the test a fully covered service for those who meet the appropriate use criteria set forth by the Alzheimer’s Association and the Society of Nuclear Medicine and Molecular Imaging.
IDEAS’ second goal – showing that the scans improve health outcomes – is scheduled for 2020. These data are a key component of the CMS decision, but they might be a tough sell, Clifford R. Jack Jr., MD, and Ronald C. Petersen, MD, PhD, wrote in an accompanying editorial. Dr. Jack and Dr. Petersen are affiliated with the Mayo Clinic in Rochester, Minn.
“For CMS to cover the cost of amyloid PET, it must be demonstrated that the result of a scan has an effect on patient outcomes, not just patient care processes – and, without a disease-modifying therapy available, that might be a challenge,” they wrote.
IDEAS is a funding collaboration of the CMS, the Alzheimer’s Association, Avid Radiopharmaceuticals/Eli Lilly, General Electric Healthcare, Piramal Imaging, and the American College of Radiology. Dr. Rabinovici had no financial disclosures.
SOURCE: Rabinovici GD et al. JAMA. 2019 Apr 2. doi: 10.1001/jama.2019.2000.
FROM THE JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION
FDA approves Mavenclad for treatment of relapsing MS
including relapsing/remitting and active secondary progressive disease.
The drug’s manufacturer, EMD Serono, said in a press release that cladribine is the first short-course oral therapy for such patients, and its use is generally recommended for patients who have had an inadequate response to, or are unable to tolerate, an alternate drug indicated for the treatment of MS. Cladribine is not recommended for use in patients with clinically isolated syndrome.
The agency’s decision is based on results from a clinical trial of 1,326 patients with relapsing MS who had experienced at least one relapse in the previous 12 months. Patients who received cladribine had significantly fewer relapses than did those who received placebo; the progression of disability was also significantly reduced in the cladribine group, compared with placebo, according to the FDA’s announcement.
The most common adverse events associated with cladribine include upper respiratory tract infections, headache, and decreased lymphocyte counts. In addition, the medication must be dispensed with a patient medication guide because the label includes a boxed warning for increased risk of malignancy and fetal harm. Other warnings include a risk for decreased lymphocyte count, hematologic toxicity and bone marrow suppression, and graft-versus-host-disease.
“We are committed to supporting the development of safe and effective treatments for patients with multiple sclerosis. The approval of Mavenclad represents an additional option for patients who have tried another treatment without success,” Billy Dunn, MD, director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research, said in the announcement.
The approved dose of cladribine is 3.5 mg/kg body weight over 2 years, administered as one treatment course of 1.75 mg/kg per year, each consisting of 2 treatment weeks. Additional courses of cladribine are not to be administered because retreatment with cladribine during years 3 and 4 may further increase the risk of malignancy. The safety and efficacy of reinitiating cladribine more than 2 years after completing two treatment courses has not been studied, according to EMD Serono.
Cladribine is approved in more than 50 other countries and was approved for use in the European Union in August 2017.
including relapsing/remitting and active secondary progressive disease.
The drug’s manufacturer, EMD Serono, said in a press release that cladribine is the first short-course oral therapy for such patients, and its use is generally recommended for patients who have had an inadequate response to, or are unable to tolerate, an alternate drug indicated for the treatment of MS. Cladribine is not recommended for use in patients with clinically isolated syndrome.
The agency’s decision is based on results from a clinical trial of 1,326 patients with relapsing MS who had experienced at least one relapse in the previous 12 months. Patients who received cladribine had significantly fewer relapses than did those who received placebo; the progression of disability was also significantly reduced in the cladribine group, compared with placebo, according to the FDA’s announcement.
The most common adverse events associated with cladribine include upper respiratory tract infections, headache, and decreased lymphocyte counts. In addition, the medication must be dispensed with a patient medication guide because the label includes a boxed warning for increased risk of malignancy and fetal harm. Other warnings include a risk for decreased lymphocyte count, hematologic toxicity and bone marrow suppression, and graft-versus-host-disease.
“We are committed to supporting the development of safe and effective treatments for patients with multiple sclerosis. The approval of Mavenclad represents an additional option for patients who have tried another treatment without success,” Billy Dunn, MD, director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research, said in the announcement.
The approved dose of cladribine is 3.5 mg/kg body weight over 2 years, administered as one treatment course of 1.75 mg/kg per year, each consisting of 2 treatment weeks. Additional courses of cladribine are not to be administered because retreatment with cladribine during years 3 and 4 may further increase the risk of malignancy. The safety and efficacy of reinitiating cladribine more than 2 years after completing two treatment courses has not been studied, according to EMD Serono.
Cladribine is approved in more than 50 other countries and was approved for use in the European Union in August 2017.
including relapsing/remitting and active secondary progressive disease.
The drug’s manufacturer, EMD Serono, said in a press release that cladribine is the first short-course oral therapy for such patients, and its use is generally recommended for patients who have had an inadequate response to, or are unable to tolerate, an alternate drug indicated for the treatment of MS. Cladribine is not recommended for use in patients with clinically isolated syndrome.
The agency’s decision is based on results from a clinical trial of 1,326 patients with relapsing MS who had experienced at least one relapse in the previous 12 months. Patients who received cladribine had significantly fewer relapses than did those who received placebo; the progression of disability was also significantly reduced in the cladribine group, compared with placebo, according to the FDA’s announcement.
The most common adverse events associated with cladribine include upper respiratory tract infections, headache, and decreased lymphocyte counts. In addition, the medication must be dispensed with a patient medication guide because the label includes a boxed warning for increased risk of malignancy and fetal harm. Other warnings include a risk for decreased lymphocyte count, hematologic toxicity and bone marrow suppression, and graft-versus-host-disease.
“We are committed to supporting the development of safe and effective treatments for patients with multiple sclerosis. The approval of Mavenclad represents an additional option for patients who have tried another treatment without success,” Billy Dunn, MD, director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research, said in the announcement.
The approved dose of cladribine is 3.5 mg/kg body weight over 2 years, administered as one treatment course of 1.75 mg/kg per year, each consisting of 2 treatment weeks. Additional courses of cladribine are not to be administered because retreatment with cladribine during years 3 and 4 may further increase the risk of malignancy. The safety and efficacy of reinitiating cladribine more than 2 years after completing two treatment courses has not been studied, according to EMD Serono.
Cladribine is approved in more than 50 other countries and was approved for use in the European Union in August 2017.
FDA proposes updates to mammography regulations
Proposed changes to federal mammography regulations aim to provide more information for doctors and patients, as well as standardize patient information on breast density’s impact on screening.

The Food and Drug Administration posted a new proposed rule online March 27 that would “expand the information mammography facilities must provide to patients and health care professionals, allowing for more informed medical decision making,” the agency said in a statement. “It would also modernize mammography quality standards and better position the FDA to enforce regulations that apply to the safety and quality of mammography services.”
Key among the proposed changes is the addition of breast density information to the summary letter provided to patients and to the medical report provided to referring health care professionals.
“The FDA is proposing specific language that would explain how breast density can influence the accuracy of mammography and would recommend patients with dense breasts talk to their health care provider about high breast density and how it relates to breast cancer risk and their individual situation,” the agency said in a statement.
Laurie Margolies, MD, section chief of breast imaging at Mount Sinai Health System in New York, said the regulations would bring some uniformity to the communication process.
“It builds on the experience of the 37 states and the District of Columbia, all of whom have passed dense breast notification laws, and can serve to unify those disparate regulations into one that would be uniform throughout the country to give one clear message to women and health care providers,” Dr. Margolies said in an interview.
She noted that dense breasts are very common and communicating issues that are related to them, including the potential need for supplemental screening, are important.
“Almost half of American women have dense breasts and why that is significant is because the dense breast issue not only increases one’s risk of getting breast cancer, but it also can hide small breast cancers on the mammogram,” she said.
If you take 1,000 women with dense breasts and then do a breast ultrasound as a supplemental screening measure, “you will find three more small node-negative breast cancers that would not have come to light if you didn’t do the extra supplemental screening,” underscoring the importance of communicating dense breast information, she continued.
FDA also is seeking to enhance information provided to health care professionals by codifying three additional categories for mammogram assessments, including the addition of the “known biopsy proven malignancy” category, which the agency says would help identify which scans were being used to evaluate treatment of already diagnosed cancers.
Both patients and health care professionals would receive more detailed information about the mammography facility under the proposed rule.
FDA is proposing modernization of quality standards to help the agency enforce regulations, including giving the agency the authority to notify patients and health care professionals directly if a mammography facility does not meet quality standards and that a reevaluation or repeat of the exam at a different facility may be needed. The proposed amendments also include requiring that only digital accessory components specifically FDA approved or cleared for mammography be used or that facilities use components that otherwise meet the requirements, and stronger record-keeping requirements.
Dr. Margolies did note one potential deficiency in the proposed rule – the lack of any information on health insurance coverage of supplemental screening for women with dense breasts, though she noted this may not fall under the FDA’s authority.
“It would do the most good if everybody could get the supplemental screening without regards to their ability to pay for it out of pocket,” she said.
The proposal amends regulations issued under the Mammography Quality Standards Act of 1992, which gives FDA oversight authority over mammography facilities, including accreditation, certification, annual inspection, and enforcement of standards.
Comments on the proposal are due 90 days after the proposed rule is published in the Federal Register, which is scheduled for March 28.
Proposed changes to federal mammography regulations aim to provide more information for doctors and patients, as well as standardize patient information on breast density’s impact on screening.
The Food and Drug Administration posted a new proposed rule online March 27 that would “expand the information mammography facilities must provide to patients and health care professionals, allowing for more informed medical decision making,” the agency said in a statement. “It would also modernize mammography quality standards and better position the FDA to enforce regulations that apply to the safety and quality of mammography services.”
Key among the proposed changes is the addition of breast density information to the summary letter provided to patients and to the medical report provided to referring health care professionals.
“The FDA is proposing specific language that would explain how breast density can influence the accuracy of mammography and would recommend patients with dense breasts talk to their health care provider about high breast density and how it relates to breast cancer risk and their individual situation,” the agency said in a statement.
Laurie Margolies, MD, section chief of breast imaging at Mount Sinai Health System in New York, said the regulations would bring some uniformity to the communication process.
“It builds on the experience of the 37 states and the District of Columbia, all of whom have passed dense breast notification laws, and can serve to unify those disparate regulations into one that would be uniform throughout the country to give one clear message to women and health care providers,” Dr. Margolies said in an interview.
She noted that dense breasts are very common and communicating issues that are related to them, including the potential need for supplemental screening, are important.
“Almost half of American women have dense breasts and why that is significant is because the dense breast issue not only increases one’s risk of getting breast cancer, but it also can hide small breast cancers on the mammogram,” she said.
If you take 1,000 women with dense breasts and then do a breast ultrasound as a supplemental screening measure, “you will find three more small node-negative breast cancers that would not have come to light if you didn’t do the extra supplemental screening,” underscoring the importance of communicating dense breast information, she continued.
FDA also is seeking to enhance information provided to health care professionals by codifying three additional categories for mammogram assessments, including the addition of the “known biopsy proven malignancy” category, which the agency says would help identify which scans were being used to evaluate treatment of already diagnosed cancers.
Both patients and health care professionals would receive more detailed information about the mammography facility under the proposed rule.
FDA is proposing modernization of quality standards to help the agency enforce regulations, including giving the agency the authority to notify patients and health care professionals directly if a mammography facility does not meet quality standards and that a reevaluation or repeat of the exam at a different facility may be needed. The proposed amendments also include requiring that only digital accessory components specifically FDA approved or cleared for mammography be used or that facilities use components that otherwise meet the requirements, and stronger record-keeping requirements.
Dr. Margolies did note one potential deficiency in the proposed rule – the lack of any information on health insurance coverage of supplemental screening for women with dense breasts, though she noted this may not fall under the FDA’s authority.
“It would do the most good if everybody could get the supplemental screening without regards to their ability to pay for it out of pocket,” she said.
The proposal amends regulations issued under the Mammography Quality Standards Act of 1992, which gives FDA oversight authority over mammography facilities, including accreditation, certification, annual inspection, and enforcement of standards.
Comments on the proposal are due 90 days after the proposed rule is published in the Federal Register, which is scheduled for March 28.
Proposed changes to federal mammography regulations aim to provide more information for doctors and patients, as well as standardize patient information on breast density’s impact on screening.
The Food and Drug Administration posted a new proposed rule online March 27 that would “expand the information mammography facilities must provide to patients and health care professionals, allowing for more informed medical decision making,” the agency said in a statement. “It would also modernize mammography quality standards and better position the FDA to enforce regulations that apply to the safety and quality of mammography services.”
Key among the proposed changes is the addition of breast density information to the summary letter provided to patients and to the medical report provided to referring health care professionals.
“The FDA is proposing specific language that would explain how breast density can influence the accuracy of mammography and would recommend patients with dense breasts talk to their health care provider about high breast density and how it relates to breast cancer risk and their individual situation,” the agency said in a statement.
Laurie Margolies, MD, section chief of breast imaging at Mount Sinai Health System in New York, said the regulations would bring some uniformity to the communication process.
“It builds on the experience of the 37 states and the District of Columbia, all of whom have passed dense breast notification laws, and can serve to unify those disparate regulations into one that would be uniform throughout the country to give one clear message to women and health care providers,” Dr. Margolies said in an interview.
She noted that dense breasts are very common and communicating issues that are related to them, including the potential need for supplemental screening, are important.
“Almost half of American women have dense breasts and why that is significant is because the dense breast issue not only increases one’s risk of getting breast cancer, but it also can hide small breast cancers on the mammogram,” she said.
If you take 1,000 women with dense breasts and then do a breast ultrasound as a supplemental screening measure, “you will find three more small node-negative breast cancers that would not have come to light if you didn’t do the extra supplemental screening,” underscoring the importance of communicating dense breast information, she continued.
FDA also is seeking to enhance information provided to health care professionals by codifying three additional categories for mammogram assessments, including the addition of the “known biopsy proven malignancy” category, which the agency says would help identify which scans were being used to evaluate treatment of already diagnosed cancers.
Both patients and health care professionals would receive more detailed information about the mammography facility under the proposed rule.
FDA is proposing modernization of quality standards to help the agency enforce regulations, including giving the agency the authority to notify patients and health care professionals directly if a mammography facility does not meet quality standards and that a reevaluation or repeat of the exam at a different facility may be needed. The proposed amendments also include requiring that only digital accessory components specifically FDA approved or cleared for mammography be used or that facilities use components that otherwise meet the requirements, and stronger record-keeping requirements.
Dr. Margolies did note one potential deficiency in the proposed rule – the lack of any information on health insurance coverage of supplemental screening for women with dense breasts, though she noted this may not fall under the FDA’s authority.
“It would do the most good if everybody could get the supplemental screening without regards to their ability to pay for it out of pocket,” she said.
The proposal amends regulations issued under the Mammography Quality Standards Act of 1992, which gives FDA oversight authority over mammography facilities, including accreditation, certification, annual inspection, and enforcement of standards.
Comments on the proposal are due 90 days after the proposed rule is published in the Federal Register, which is scheduled for March 28.
Anastrozole/fulvestrant prolongs OS in metastatic ER+ breast cancer
For women with metastatic hormone receptor–positive breast cancer, the addition of the selective estrogen receptor modifier fulvestrant (Faslodex) to the aromatase inhibitor anastrozole (Arimidex and generics) resulted in a small but significant improvement in overall survival, according to final results from a randomized phase 3 trial.
Among 694 patients randomized for whom data were available, the hazard ratio for death with the combination when compared with anastrozole alone was 0.82 (P = .03), reported Rita S. Mehta, MD, from the University of California (Irvine) Medical Center and her colleagues.
The benefit of the combination was highest for patients without prior exposure to adjuvant endocrine therapy.
“Furthermore, sequential therapy with anastrozole and fulvestrant (45% of patients crossed over to fulvestrant alone) did not negate the significance of the long-term overall survival benefit with the combination therapy as compared with anastrozole,” the investigators wrote in The New England Journal of Medicine.
The current report is the final survival analysis of the trial. The primary results were reported in 2012 (N Engl J Med. 2012; 367:435-44). A total of 707 postmenopausal women with previously untreated metastatic disease were randomly assigned to receive either 1 mg of anastrozole orally every day with crossover to fulvestrant alone strongly encouraged if the disease progressed or to anastrozole and fulvestrant in combination. Randomization was stratified according to prior adjuvant tamoxifen use. A total of 694 women had data available for analysis.
The primary analysis, conducted at a median follow-up of 35 months, showed a median progression-free survival (PFS) with anastrozole alone of 13.5 months, compared with 15.0 months for anastrozole/fulvestrant (HR for progression or death 0.80; P = .007). Respective median overall survival was 41.3 months and 47.7 months (HR for death 0.81; P = .05).
The current, final analysis, conducted at a median follow-up of 7 years in patients who did not have disease progression, showed 261 deaths among 345 women (76%) in the anastrozole-only group, compared with 247 deaths among 349 women (71%) in the combination group (HR for death 0.82; P = .03).
Overall survival was longer for those women who had not previously received tamoxifen who were treated with the combination, at a median of 52.2 months versus 40.3 months for women not previously treated with tamoxifen who received anastrozole alone (hazard ratio, 0.73; 95% confidence interval, 0.58-0.92). In contrast, there was no significant difference in OS between the two treatment groups in women who had previously received tamoxifen.
Approximately 45% of patients initially randomized to anastrozole alone were crossed over to fulvestrant.
The incidence of long-term toxic effects and treatment-related deaths was similar between the groups. Previously reported treatment-related deaths with the combination included pulmonary emboli in two patients and a cerebrovascular ischemic event in one patients.
At the time of data cutoff for the final report, 15% of patients in the combination-therapy group and 13% in the anastrozole-only group had experienced grade 3 toxicities.
The study was supported by National Cancer Institute grants and by AstraZeneca. Dr. Mehta reported institutional and personal grants from AstraZeneca and others. Multiple coauthors reported similar relationships.
SOURCE: Mehta RS et al. N Engl J Med. 2019;380:1226-34.
For women with metastatic hormone receptor–positive breast cancer, the addition of the selective estrogen receptor modifier fulvestrant (Faslodex) to the aromatase inhibitor anastrozole (Arimidex and generics) resulted in a small but significant improvement in overall survival, according to final results from a randomized phase 3 trial.
Among 694 patients randomized for whom data were available, the hazard ratio for death with the combination when compared with anastrozole alone was 0.82 (P = .03), reported Rita S. Mehta, MD, from the University of California (Irvine) Medical Center and her colleagues.
The benefit of the combination was highest for patients without prior exposure to adjuvant endocrine therapy.
“Furthermore, sequential therapy with anastrozole and fulvestrant (45% of patients crossed over to fulvestrant alone) did not negate the significance of the long-term overall survival benefit with the combination therapy as compared with anastrozole,” the investigators wrote in The New England Journal of Medicine.
The current report is the final survival analysis of the trial. The primary results were reported in 2012 (N Engl J Med. 2012; 367:435-44). A total of 707 postmenopausal women with previously untreated metastatic disease were randomly assigned to receive either 1 mg of anastrozole orally every day with crossover to fulvestrant alone strongly encouraged if the disease progressed or to anastrozole and fulvestrant in combination. Randomization was stratified according to prior adjuvant tamoxifen use. A total of 694 women had data available for analysis.
The primary analysis, conducted at a median follow-up of 35 months, showed a median progression-free survival (PFS) with anastrozole alone of 13.5 months, compared with 15.0 months for anastrozole/fulvestrant (HR for progression or death 0.80; P = .007). Respective median overall survival was 41.3 months and 47.7 months (HR for death 0.81; P = .05).
The current, final analysis, conducted at a median follow-up of 7 years in patients who did not have disease progression, showed 261 deaths among 345 women (76%) in the anastrozole-only group, compared with 247 deaths among 349 women (71%) in the combination group (HR for death 0.82; P = .03).
Overall survival was longer for those women who had not previously received tamoxifen who were treated with the combination, at a median of 52.2 months versus 40.3 months for women not previously treated with tamoxifen who received anastrozole alone (hazard ratio, 0.73; 95% confidence interval, 0.58-0.92). In contrast, there was no significant difference in OS between the two treatment groups in women who had previously received tamoxifen.
Approximately 45% of patients initially randomized to anastrozole alone were crossed over to fulvestrant.
The incidence of long-term toxic effects and treatment-related deaths was similar between the groups. Previously reported treatment-related deaths with the combination included pulmonary emboli in two patients and a cerebrovascular ischemic event in one patients.
At the time of data cutoff for the final report, 15% of patients in the combination-therapy group and 13% in the anastrozole-only group had experienced grade 3 toxicities.
The study was supported by National Cancer Institute grants and by AstraZeneca. Dr. Mehta reported institutional and personal grants from AstraZeneca and others. Multiple coauthors reported similar relationships.
SOURCE: Mehta RS et al. N Engl J Med. 2019;380:1226-34.
For women with metastatic hormone receptor–positive breast cancer, the addition of the selective estrogen receptor modifier fulvestrant (Faslodex) to the aromatase inhibitor anastrozole (Arimidex and generics) resulted in a small but significant improvement in overall survival, according to final results from a randomized phase 3 trial.
Among 694 patients randomized for whom data were available, the hazard ratio for death with the combination when compared with anastrozole alone was 0.82 (P = .03), reported Rita S. Mehta, MD, from the University of California (Irvine) Medical Center and her colleagues.
The benefit of the combination was highest for patients without prior exposure to adjuvant endocrine therapy.
“Furthermore, sequential therapy with anastrozole and fulvestrant (45% of patients crossed over to fulvestrant alone) did not negate the significance of the long-term overall survival benefit with the combination therapy as compared with anastrozole,” the investigators wrote in The New England Journal of Medicine.
The current report is the final survival analysis of the trial. The primary results were reported in 2012 (N Engl J Med. 2012; 367:435-44). A total of 707 postmenopausal women with previously untreated metastatic disease were randomly assigned to receive either 1 mg of anastrozole orally every day with crossover to fulvestrant alone strongly encouraged if the disease progressed or to anastrozole and fulvestrant in combination. Randomization was stratified according to prior adjuvant tamoxifen use. A total of 694 women had data available for analysis.
The primary analysis, conducted at a median follow-up of 35 months, showed a median progression-free survival (PFS) with anastrozole alone of 13.5 months, compared with 15.0 months for anastrozole/fulvestrant (HR for progression or death 0.80; P = .007). Respective median overall survival was 41.3 months and 47.7 months (HR for death 0.81; P = .05).
The current, final analysis, conducted at a median follow-up of 7 years in patients who did not have disease progression, showed 261 deaths among 345 women (76%) in the anastrozole-only group, compared with 247 deaths among 349 women (71%) in the combination group (HR for death 0.82; P = .03).
Overall survival was longer for those women who had not previously received tamoxifen who were treated with the combination, at a median of 52.2 months versus 40.3 months for women not previously treated with tamoxifen who received anastrozole alone (hazard ratio, 0.73; 95% confidence interval, 0.58-0.92). In contrast, there was no significant difference in OS between the two treatment groups in women who had previously received tamoxifen.
Approximately 45% of patients initially randomized to anastrozole alone were crossed over to fulvestrant.
The incidence of long-term toxic effects and treatment-related deaths was similar between the groups. Previously reported treatment-related deaths with the combination included pulmonary emboli in two patients and a cerebrovascular ischemic event in one patients.
At the time of data cutoff for the final report, 15% of patients in the combination-therapy group and 13% in the anastrozole-only group had experienced grade 3 toxicities.
The study was supported by National Cancer Institute grants and by AstraZeneca. Dr. Mehta reported institutional and personal grants from AstraZeneca and others. Multiple coauthors reported similar relationships.
SOURCE: Mehta RS et al. N Engl J Med. 2019;380:1226-34.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Anastrozole/fulvestrant improved survival when compared with anastrozole alone.
Major finding: The hazard ratio for death with the combination was 0.82 (P = .03).
Study details: Final survival analysis of a phase 3, randomized trial in 694 women with metastatic hormone receptor–positive breast cancer.
Disclosures: The study was supported by National Cancer Institute grants and by AstraZeneca. Dr. Mehta reported institutional and personal grants from AstraZeneca and others. Multiple coauthors reported similar relationships.
Source: Mehta RS et al. N Engl J Med. 2019;380:1226-34.
Many EMS protocols for status epilepticus do not follow evidence-based guidelines
“Many protocols did not follow evidence-based guidelines and did not accurately define generalized convulsive status epilepticus,” said John P. Betjemann, MD, associate professor of neurology at the University of California, San Francisco, and his colleagues. They reported their findings in the March 26 issue of JAMA.
Generalized convulsive status epilepticus is a neurologic emergency, and trials published in 2001 and 2012 found that benzodiazepines are effective prehospital treatments for patients with generalized convulsive status epilepticus. These trials informed a 2016 evidence-based guideline that cites level A evidence for intramuscular midazolam, IV lorazepam, and IV diazepam as initial treatment options for adults.
To determine whether EMS system protocols follow these recommendations, the investigators reviewed treatment protocols from 33 EMS systems that cover the 58 counties in California. The researchers reviewed EMS system protocols between May and June 2018 to determine when they were last updated and whether they defined generalized convulsive status epilepticus according to the guideline (namely, 5 or more minutes of continuous seizure or two or more discrete seizures between which a patient has incomplete recovery of consciousness). They also determined whether the protocols included any of the three benzodiazepines in the guideline and, if so, at what dose and using which route of administration.
Protocols’ most recent revision dates ranged between 2007 and 2018. Twenty-seven protocols (81.8%) were revised after the second clinical trial was published in 2012, and 17 (51.5%) were revised after the 2016 guideline. Seven EMS system protocols (21.2%) defined generalized convulsive status epilepticus according to the guideline. Thirty-two protocols (97.0%) included intramuscular midazolam, 2 (6.1%) included IV lorazepam, and 5 (15.2%) included IV diazepam.
Although the protocols “appropriately emphasized” intramuscular midazolam, the protocol doses often were lower than those used in the trials or recommended in the guideline. In addition, most protocols listed IV and intraosseous midazolam as options, although these treatments were not studied in the trials nor recommended in the guideline. In all, six of the protocols (18.2%) recommended at least one medication by the route and dose suggested in the trials or in the guideline.
“Why EMS system protocols deviate from the evidence and how this affects patient outcomes deserves further study,” the authors said.
The researchers noted that they examined EMS protocols in only one state and that “protocols may not necessarily reflect what emergency medical technicians actually do in practice.” In addition, the researchers accessed the most recent protocols by consulting EMS system websites rather than by contacting each EMS system for its most up-to-date protocol.
The authors reported personal compensation from JAMA Neurology and from Continuum Audio unrelated to the present study, as well as grants from the National Institutes of Health.
SOURCE: Betjemann JP et al. JAMA. 2019 Mar 26.
“Many protocols did not follow evidence-based guidelines and did not accurately define generalized convulsive status epilepticus,” said John P. Betjemann, MD, associate professor of neurology at the University of California, San Francisco, and his colleagues. They reported their findings in the March 26 issue of JAMA.
Generalized convulsive status epilepticus is a neurologic emergency, and trials published in 2001 and 2012 found that benzodiazepines are effective prehospital treatments for patients with generalized convulsive status epilepticus. These trials informed a 2016 evidence-based guideline that cites level A evidence for intramuscular midazolam, IV lorazepam, and IV diazepam as initial treatment options for adults.
To determine whether EMS system protocols follow these recommendations, the investigators reviewed treatment protocols from 33 EMS systems that cover the 58 counties in California. The researchers reviewed EMS system protocols between May and June 2018 to determine when they were last updated and whether they defined generalized convulsive status epilepticus according to the guideline (namely, 5 or more minutes of continuous seizure or two or more discrete seizures between which a patient has incomplete recovery of consciousness). They also determined whether the protocols included any of the three benzodiazepines in the guideline and, if so, at what dose and using which route of administration.
Protocols’ most recent revision dates ranged between 2007 and 2018. Twenty-seven protocols (81.8%) were revised after the second clinical trial was published in 2012, and 17 (51.5%) were revised after the 2016 guideline. Seven EMS system protocols (21.2%) defined generalized convulsive status epilepticus according to the guideline. Thirty-two protocols (97.0%) included intramuscular midazolam, 2 (6.1%) included IV lorazepam, and 5 (15.2%) included IV diazepam.
Although the protocols “appropriately emphasized” intramuscular midazolam, the protocol doses often were lower than those used in the trials or recommended in the guideline. In addition, most protocols listed IV and intraosseous midazolam as options, although these treatments were not studied in the trials nor recommended in the guideline. In all, six of the protocols (18.2%) recommended at least one medication by the route and dose suggested in the trials or in the guideline.
“Why EMS system protocols deviate from the evidence and how this affects patient outcomes deserves further study,” the authors said.
The researchers noted that they examined EMS protocols in only one state and that “protocols may not necessarily reflect what emergency medical technicians actually do in practice.” In addition, the researchers accessed the most recent protocols by consulting EMS system websites rather than by contacting each EMS system for its most up-to-date protocol.
The authors reported personal compensation from JAMA Neurology and from Continuum Audio unrelated to the present study, as well as grants from the National Institutes of Health.
SOURCE: Betjemann JP et al. JAMA. 2019 Mar 26.
“Many protocols did not follow evidence-based guidelines and did not accurately define generalized convulsive status epilepticus,” said John P. Betjemann, MD, associate professor of neurology at the University of California, San Francisco, and his colleagues. They reported their findings in the March 26 issue of JAMA.
Generalized convulsive status epilepticus is a neurologic emergency, and trials published in 2001 and 2012 found that benzodiazepines are effective prehospital treatments for patients with generalized convulsive status epilepticus. These trials informed a 2016 evidence-based guideline that cites level A evidence for intramuscular midazolam, IV lorazepam, and IV diazepam as initial treatment options for adults.
To determine whether EMS system protocols follow these recommendations, the investigators reviewed treatment protocols from 33 EMS systems that cover the 58 counties in California. The researchers reviewed EMS system protocols between May and June 2018 to determine when they were last updated and whether they defined generalized convulsive status epilepticus according to the guideline (namely, 5 or more minutes of continuous seizure or two or more discrete seizures between which a patient has incomplete recovery of consciousness). They also determined whether the protocols included any of the three benzodiazepines in the guideline and, if so, at what dose and using which route of administration.
Protocols’ most recent revision dates ranged between 2007 and 2018. Twenty-seven protocols (81.8%) were revised after the second clinical trial was published in 2012, and 17 (51.5%) were revised after the 2016 guideline. Seven EMS system protocols (21.2%) defined generalized convulsive status epilepticus according to the guideline. Thirty-two protocols (97.0%) included intramuscular midazolam, 2 (6.1%) included IV lorazepam, and 5 (15.2%) included IV diazepam.
Although the protocols “appropriately emphasized” intramuscular midazolam, the protocol doses often were lower than those used in the trials or recommended in the guideline. In addition, most protocols listed IV and intraosseous midazolam as options, although these treatments were not studied in the trials nor recommended in the guideline. In all, six of the protocols (18.2%) recommended at least one medication by the route and dose suggested in the trials or in the guideline.
“Why EMS system protocols deviate from the evidence and how this affects patient outcomes deserves further study,” the authors said.
The researchers noted that they examined EMS protocols in only one state and that “protocols may not necessarily reflect what emergency medical technicians actually do in practice.” In addition, the researchers accessed the most recent protocols by consulting EMS system websites rather than by contacting each EMS system for its most up-to-date protocol.
The authors reported personal compensation from JAMA Neurology and from Continuum Audio unrelated to the present study, as well as grants from the National Institutes of Health.
SOURCE: Betjemann JP et al. JAMA. 2019 Mar 26.
FROM JAMA
Key clinical point: Many emergency medical services (EMS) system protocols may not follow evidence-based guidelines or accurately define generalized convulsive status epilepticus.
Major finding: In all, 18.2% of the protocols recommended at least one medication by the route and at the dose suggested in clinical trials or in an evidence-based guideline.
Study details: A review of treatment protocols from 33 EMS systems that cover the 58 counties in California.
Disclosures: The authors reported personal compensation from JAMA Neurology and Continuum Audio unrelated to the present study and grants from the National Institutes of Health.
Source: Betjemann JP et al. JAMA. 2019 March 26.
Interview with John Corboy, MD, on discontinuing disease modifying therapy in elderly patients with MS

Discontinuing
How would you characterize the prevalence of MS in the elderly?
DR. CORBOY: A recent large demographic study put together by the National MS Society found that there’s almost a million individuals diagnosed with MS over the course of the last 40 to 50 years. The largest population segment was those aged 55 to 64 years. People with MS aged 55 or older constituted 46% of all those with MS.
What disease-modifying therapies (DMTs) are approved by the FDA for the elderly?
DR. CORBOY: Of the drugs that have received FDA approval, most are for individuals over the age of 18 and there’s no specific age cutoff. However, there’s no data supporting DMT use in people over the age of 55 because they were excluded from the studies.
There’s one DMT, fingolimod, that was approved for use in patients under the age of 18; all others are approved for 18 and above. However, none of them are explicitly approved for people over the age of 55, because there is no data to support it.
What is the goal of your study, the DISCO MS trial?
DR. CORBOY: The DISCO MS trial will be the first randomized, controlled, blinded discontinuation trial in the MS space. The objective is to assess the benefit of DMTs in patients over the age of 55.
Part of the rationale for the trial is that prior subgroup analyses have shown that the vast majority of the benefit that we’ve been able to measure with all of these DMTs is seen in those who are under age 45.
A number of studies have examined existing databases and individuals who were either randomly or deliberately taken off of their medication as they age, including people who were felt to be stable with no recent relapses and no recent changes on their MRI brain. These studies reinforced that when discontinuing medications, the individuals who were much more likely to have recurrence of disease activity were younger patients.
Pathological studies clearly show the number of acutely inflamed plaques in the white matter is dramatically lower in autopsies of older vs younger patients. There are different changes in older patients, with lymphocytic nodules in the meninges, gray matter plaques related to these meningeal nodules, microglial activation, and smaller numbers of active, or mostly, inactive, white matter plaques. It’s been difficult to show any substantial benefit in slowing disability progression, much of which is felt to not be associated with acute inflammatory disease in the aging patient. All of these medicines, which can be thought of as anti-inflammatory medicines, are very beneficial when patients are young but less so as they age.
Would you describe the DISCO MS study design?
DR. CORBOY: Our study looks at individuals who are 55 and older who have not had a relapse for at least 5 years, and who’ve not had a change on their brain scan for at least 3 years.
Individuals will be randomized to either stay on the medication that they’re currently taking or discontinue that medicine. They will be followed then for 2 years. The primary outcome will be either a new relapse or a new scan change. The examining investigators are blinded to whether the patient is currently taking a MS disease modifying therapy.
Secondary outcomes include progression of disability as measured by confirmed change on the Extended Disability Status Scale (EDSS).
The enrollment goal is about 300 patients. There are presently 15 sites. The goal is to have the study completed in about 3 years. We’re presently over halfway through enrollment.
We also have a number of patient-reported outcomes because we’re particularly interested in the patient’s view of what’s going on in terms of how they feel. Understanding that dynamic will be extremely important.
We are including both patients with relapsing MS and progressive forms of MS, noting that they should have no relapse and no scan change at study entry.
What are the challenges with this study?
DR. CORBOY: One challenge is interpreting the information with the assumption that the hypothesis is validated. The hypothesis is that in a stable population of older patients that we can safely discontinue DMTs.
If that is found to be true, the question is how many people will be affected? We know that about 46% of people with MS are 55 and older, but there are not really good estimates of the number of individuals 55 and older who remain on a DMT and who are stable by the definition I just described.
It can be safely said, I think, that a substantial number of the individuals 55 and older are still on DMTs. If there’s almost a million people with MS and 46% are 55 and older, that means around 400,000 people with MS in the United States are aged 55 and older. If only half of those are on a DMT, that leaves 200,000. If only half of those are stable and could go off therapy, that would mean perhaps 100,000 people could discontinue DMTs in the United States. If all those assumptions are true, that would be a substantial savings in the health care burden of the United States from a relatively small population of individuals.
Beyond the cost, there are adverse events associated with using these medications. Older patients are more likely to be at risk of complications of MS DMTs. There also are doctor visits, blood monitoring, and other things that are done over time, and the inconvenience of taking a medicine on a routine basis if, indeed, it’s really not necessary because there is no benefit. Moreover, older individuals have other conditions (eg diabetes, hypertension, arrhythmias, cancer, etc) that may limit their ability to use medications due to risk. We’re very interested to see the outcome.
Discontinuing
How would you characterize the prevalence of MS in the elderly?
DR. CORBOY: A recent large demographic study put together by the National MS Society found that there’s almost a million individuals diagnosed with MS over the course of the last 40 to 50 years. The largest population segment was those aged 55 to 64 years. People with MS aged 55 or older constituted 46% of all those with MS.
What disease-modifying therapies (DMTs) are approved by the FDA for the elderly?
DR. CORBOY: Of the drugs that have received FDA approval, most are for individuals over the age of 18 and there’s no specific age cutoff. However, there’s no data supporting DMT use in people over the age of 55 because they were excluded from the studies.
There’s one DMT, fingolimod, that was approved for use in patients under the age of 18; all others are approved for 18 and above. However, none of them are explicitly approved for people over the age of 55, because there is no data to support it.
What is the goal of your study, the DISCO MS trial?
DR. CORBOY: The DISCO MS trial will be the first randomized, controlled, blinded discontinuation trial in the MS space. The objective is to assess the benefit of DMTs in patients over the age of 55.
Part of the rationale for the trial is that prior subgroup analyses have shown that the vast majority of the benefit that we’ve been able to measure with all of these DMTs is seen in those who are under age 45.
A number of studies have examined existing databases and individuals who were either randomly or deliberately taken off of their medication as they age, including people who were felt to be stable with no recent relapses and no recent changes on their MRI brain. These studies reinforced that when discontinuing medications, the individuals who were much more likely to have recurrence of disease activity were younger patients.
Pathological studies clearly show the number of acutely inflamed plaques in the white matter is dramatically lower in autopsies of older vs younger patients. There are different changes in older patients, with lymphocytic nodules in the meninges, gray matter plaques related to these meningeal nodules, microglial activation, and smaller numbers of active, or mostly, inactive, white matter plaques. It’s been difficult to show any substantial benefit in slowing disability progression, much of which is felt to not be associated with acute inflammatory disease in the aging patient. All of these medicines, which can be thought of as anti-inflammatory medicines, are very beneficial when patients are young but less so as they age.
Would you describe the DISCO MS study design?
DR. CORBOY: Our study looks at individuals who are 55 and older who have not had a relapse for at least 5 years, and who’ve not had a change on their brain scan for at least 3 years.
Individuals will be randomized to either stay on the medication that they’re currently taking or discontinue that medicine. They will be followed then for 2 years. The primary outcome will be either a new relapse or a new scan change. The examining investigators are blinded to whether the patient is currently taking a MS disease modifying therapy.
Secondary outcomes include progression of disability as measured by confirmed change on the Extended Disability Status Scale (EDSS).
The enrollment goal is about 300 patients. There are presently 15 sites. The goal is to have the study completed in about 3 years. We’re presently over halfway through enrollment.
We also have a number of patient-reported outcomes because we’re particularly interested in the patient’s view of what’s going on in terms of how they feel. Understanding that dynamic will be extremely important.
We are including both patients with relapsing MS and progressive forms of MS, noting that they should have no relapse and no scan change at study entry.
What are the challenges with this study?
DR. CORBOY: One challenge is interpreting the information with the assumption that the hypothesis is validated. The hypothesis is that in a stable population of older patients that we can safely discontinue DMTs.
If that is found to be true, the question is how many people will be affected? We know that about 46% of people with MS are 55 and older, but there are not really good estimates of the number of individuals 55 and older who remain on a DMT and who are stable by the definition I just described.
It can be safely said, I think, that a substantial number of the individuals 55 and older are still on DMTs. If there’s almost a million people with MS and 46% are 55 and older, that means around 400,000 people with MS in the United States are aged 55 and older. If only half of those are on a DMT, that leaves 200,000. If only half of those are stable and could go off therapy, that would mean perhaps 100,000 people could discontinue DMTs in the United States. If all those assumptions are true, that would be a substantial savings in the health care burden of the United States from a relatively small population of individuals.
Beyond the cost, there are adverse events associated with using these medications. Older patients are more likely to be at risk of complications of MS DMTs. There also are doctor visits, blood monitoring, and other things that are done over time, and the inconvenience of taking a medicine on a routine basis if, indeed, it’s really not necessary because there is no benefit. Moreover, older individuals have other conditions (eg diabetes, hypertension, arrhythmias, cancer, etc) that may limit their ability to use medications due to risk. We’re very interested to see the outcome.
Discontinuing
How would you characterize the prevalence of MS in the elderly?
DR. CORBOY: A recent large demographic study put together by the National MS Society found that there’s almost a million individuals diagnosed with MS over the course of the last 40 to 50 years. The largest population segment was those aged 55 to 64 years. People with MS aged 55 or older constituted 46% of all those with MS.
What disease-modifying therapies (DMTs) are approved by the FDA for the elderly?
DR. CORBOY: Of the drugs that have received FDA approval, most are for individuals over the age of 18 and there’s no specific age cutoff. However, there’s no data supporting DMT use in people over the age of 55 because they were excluded from the studies.
There’s one DMT, fingolimod, that was approved for use in patients under the age of 18; all others are approved for 18 and above. However, none of them are explicitly approved for people over the age of 55, because there is no data to support it.
What is the goal of your study, the DISCO MS trial?
DR. CORBOY: The DISCO MS trial will be the first randomized, controlled, blinded discontinuation trial in the MS space. The objective is to assess the benefit of DMTs in patients over the age of 55.
Part of the rationale for the trial is that prior subgroup analyses have shown that the vast majority of the benefit that we’ve been able to measure with all of these DMTs is seen in those who are under age 45.
A number of studies have examined existing databases and individuals who were either randomly or deliberately taken off of their medication as they age, including people who were felt to be stable with no recent relapses and no recent changes on their MRI brain. These studies reinforced that when discontinuing medications, the individuals who were much more likely to have recurrence of disease activity were younger patients.
Pathological studies clearly show the number of acutely inflamed plaques in the white matter is dramatically lower in autopsies of older vs younger patients. There are different changes in older patients, with lymphocytic nodules in the meninges, gray matter plaques related to these meningeal nodules, microglial activation, and smaller numbers of active, or mostly, inactive, white matter plaques. It’s been difficult to show any substantial benefit in slowing disability progression, much of which is felt to not be associated with acute inflammatory disease in the aging patient. All of these medicines, which can be thought of as anti-inflammatory medicines, are very beneficial when patients are young but less so as they age.
Would you describe the DISCO MS study design?
DR. CORBOY: Our study looks at individuals who are 55 and older who have not had a relapse for at least 5 years, and who’ve not had a change on their brain scan for at least 3 years.
Individuals will be randomized to either stay on the medication that they’re currently taking or discontinue that medicine. They will be followed then for 2 years. The primary outcome will be either a new relapse or a new scan change. The examining investigators are blinded to whether the patient is currently taking a MS disease modifying therapy.
Secondary outcomes include progression of disability as measured by confirmed change on the Extended Disability Status Scale (EDSS).
The enrollment goal is about 300 patients. There are presently 15 sites. The goal is to have the study completed in about 3 years. We’re presently over halfway through enrollment.
We also have a number of patient-reported outcomes because we’re particularly interested in the patient’s view of what’s going on in terms of how they feel. Understanding that dynamic will be extremely important.
We are including both patients with relapsing MS and progressive forms of MS, noting that they should have no relapse and no scan change at study entry.
What are the challenges with this study?
DR. CORBOY: One challenge is interpreting the information with the assumption that the hypothesis is validated. The hypothesis is that in a stable population of older patients that we can safely discontinue DMTs.
If that is found to be true, the question is how many people will be affected? We know that about 46% of people with MS are 55 and older, but there are not really good estimates of the number of individuals 55 and older who remain on a DMT and who are stable by the definition I just described.
It can be safely said, I think, that a substantial number of the individuals 55 and older are still on DMTs. If there’s almost a million people with MS and 46% are 55 and older, that means around 400,000 people with MS in the United States are aged 55 and older. If only half of those are on a DMT, that leaves 200,000. If only half of those are stable and could go off therapy, that would mean perhaps 100,000 people could discontinue DMTs in the United States. If all those assumptions are true, that would be a substantial savings in the health care burden of the United States from a relatively small population of individuals.
Beyond the cost, there are adverse events associated with using these medications. Older patients are more likely to be at risk of complications of MS DMTs. There also are doctor visits, blood monitoring, and other things that are done over time, and the inconvenience of taking a medicine on a routine basis if, indeed, it’s really not necessary because there is no benefit. Moreover, older individuals have other conditions (eg diabetes, hypertension, arrhythmias, cancer, etc) that may limit their ability to use medications due to risk. We’re very interested to see the outcome.
