User login
Interview with John Corboy, MD, on discontinuing disease modifying therapy in elderly patients with MS

Discontinuing
How would you characterize the prevalence of MS in the elderly?
DR. CORBOY: A recent large demographic study put together by the National MS Society found that there’s almost a million individuals diagnosed with MS over the course of the last 40 to 50 years. The largest population segment was those aged 55 to 64 years. People with MS aged 55 or older constituted 46% of all those with MS.
What disease-modifying therapies (DMTs) are approved by the FDA for the elderly?
DR. CORBOY: Of the drugs that have received FDA approval, most are for individuals over the age of 18 and there’s no specific age cutoff. However, there’s no data supporting DMT use in people over the age of 55 because they were excluded from the studies.
There’s one DMT, fingolimod, that was approved for use in patients under the age of 18; all others are approved for 18 and above. However, none of them are explicitly approved for people over the age of 55, because there is no data to support it.
What is the goal of your study, the DISCO MS trial?
DR. CORBOY: The DISCO MS trial will be the first randomized, controlled, blinded discontinuation trial in the MS space. The objective is to assess the benefit of DMTs in patients over the age of 55.
Part of the rationale for the trial is that prior subgroup analyses have shown that the vast majority of the benefit that we’ve been able to measure with all of these DMTs is seen in those who are under age 45.
A number of studies have examined existing databases and individuals who were either randomly or deliberately taken off of their medication as they age, including people who were felt to be stable with no recent relapses and no recent changes on their MRI brain. These studies reinforced that when discontinuing medications, the individuals who were much more likely to have recurrence of disease activity were younger patients.
Pathological studies clearly show the number of acutely inflamed plaques in the white matter is dramatically lower in autopsies of older vs younger patients. There are different changes in older patients, with lymphocytic nodules in the meninges, gray matter plaques related to these meningeal nodules, microglial activation, and smaller numbers of active, or mostly, inactive, white matter plaques. It’s been difficult to show any substantial benefit in slowing disability progression, much of which is felt to not be associated with acute inflammatory disease in the aging patient. All of these medicines, which can be thought of as anti-inflammatory medicines, are very beneficial when patients are young but less so as they age.
Would you describe the DISCO MS study design?
DR. CORBOY: Our study looks at individuals who are 55 and older who have not had a relapse for at least 5 years, and who’ve not had a change on their brain scan for at least 3 years.
Individuals will be randomized to either stay on the medication that they’re currently taking or discontinue that medicine. They will be followed then for 2 years. The primary outcome will be either a new relapse or a new scan change. The examining investigators are blinded to whether the patient is currently taking a MS disease modifying therapy.
Secondary outcomes include progression of disability as measured by confirmed change on the Extended Disability Status Scale (EDSS).
The enrollment goal is about 300 patients. There are presently 15 sites. The goal is to have the study completed in about 3 years. We’re presently over halfway through enrollment.
We also have a number of patient-reported outcomes because we’re particularly interested in the patient’s view of what’s going on in terms of how they feel. Understanding that dynamic will be extremely important.
We are including both patients with relapsing MS and progressive forms of MS, noting that they should have no relapse and no scan change at study entry.
What are the challenges with this study?
DR. CORBOY: One challenge is interpreting the information with the assumption that the hypothesis is validated. The hypothesis is that in a stable population of older patients that we can safely discontinue DMTs.
If that is found to be true, the question is how many people will be affected? We know that about 46% of people with MS are 55 and older, but there are not really good estimates of the number of individuals 55 and older who remain on a DMT and who are stable by the definition I just described.
It can be safely said, I think, that a substantial number of the individuals 55 and older are still on DMTs. If there’s almost a million people with MS and 46% are 55 and older, that means around 400,000 people with MS in the United States are aged 55 and older. If only half of those are on a DMT, that leaves 200,000. If only half of those are stable and could go off therapy, that would mean perhaps 100,000 people could discontinue DMTs in the United States. If all those assumptions are true, that would be a substantial savings in the health care burden of the United States from a relatively small population of individuals.
Beyond the cost, there are adverse events associated with using these medications. Older patients are more likely to be at risk of complications of MS DMTs. There also are doctor visits, blood monitoring, and other things that are done over time, and the inconvenience of taking a medicine on a routine basis if, indeed, it’s really not necessary because there is no benefit. Moreover, older individuals have other conditions (eg diabetes, hypertension, arrhythmias, cancer, etc) that may limit their ability to use medications due to risk. We’re very interested to see the outcome.
Discontinuing
How would you characterize the prevalence of MS in the elderly?
DR. CORBOY: A recent large demographic study put together by the National MS Society found that there’s almost a million individuals diagnosed with MS over the course of the last 40 to 50 years. The largest population segment was those aged 55 to 64 years. People with MS aged 55 or older constituted 46% of all those with MS.
What disease-modifying therapies (DMTs) are approved by the FDA for the elderly?
DR. CORBOY: Of the drugs that have received FDA approval, most are for individuals over the age of 18 and there’s no specific age cutoff. However, there’s no data supporting DMT use in people over the age of 55 because they were excluded from the studies.
There’s one DMT, fingolimod, that was approved for use in patients under the age of 18; all others are approved for 18 and above. However, none of them are explicitly approved for people over the age of 55, because there is no data to support it.
What is the goal of your study, the DISCO MS trial?
DR. CORBOY: The DISCO MS trial will be the first randomized, controlled, blinded discontinuation trial in the MS space. The objective is to assess the benefit of DMTs in patients over the age of 55.
Part of the rationale for the trial is that prior subgroup analyses have shown that the vast majority of the benefit that we’ve been able to measure with all of these DMTs is seen in those who are under age 45.
A number of studies have examined existing databases and individuals who were either randomly or deliberately taken off of their medication as they age, including people who were felt to be stable with no recent relapses and no recent changes on their MRI brain. These studies reinforced that when discontinuing medications, the individuals who were much more likely to have recurrence of disease activity were younger patients.
Pathological studies clearly show the number of acutely inflamed plaques in the white matter is dramatically lower in autopsies of older vs younger patients. There are different changes in older patients, with lymphocytic nodules in the meninges, gray matter plaques related to these meningeal nodules, microglial activation, and smaller numbers of active, or mostly, inactive, white matter plaques. It’s been difficult to show any substantial benefit in slowing disability progression, much of which is felt to not be associated with acute inflammatory disease in the aging patient. All of these medicines, which can be thought of as anti-inflammatory medicines, are very beneficial when patients are young but less so as they age.
Would you describe the DISCO MS study design?
DR. CORBOY: Our study looks at individuals who are 55 and older who have not had a relapse for at least 5 years, and who’ve not had a change on their brain scan for at least 3 years.
Individuals will be randomized to either stay on the medication that they’re currently taking or discontinue that medicine. They will be followed then for 2 years. The primary outcome will be either a new relapse or a new scan change. The examining investigators are blinded to whether the patient is currently taking a MS disease modifying therapy.
Secondary outcomes include progression of disability as measured by confirmed change on the Extended Disability Status Scale (EDSS).
The enrollment goal is about 300 patients. There are presently 15 sites. The goal is to have the study completed in about 3 years. We’re presently over halfway through enrollment.
We also have a number of patient-reported outcomes because we’re particularly interested in the patient’s view of what’s going on in terms of how they feel. Understanding that dynamic will be extremely important.
We are including both patients with relapsing MS and progressive forms of MS, noting that they should have no relapse and no scan change at study entry.
What are the challenges with this study?
DR. CORBOY: One challenge is interpreting the information with the assumption that the hypothesis is validated. The hypothesis is that in a stable population of older patients that we can safely discontinue DMTs.
If that is found to be true, the question is how many people will be affected? We know that about 46% of people with MS are 55 and older, but there are not really good estimates of the number of individuals 55 and older who remain on a DMT and who are stable by the definition I just described.
It can be safely said, I think, that a substantial number of the individuals 55 and older are still on DMTs. If there’s almost a million people with MS and 46% are 55 and older, that means around 400,000 people with MS in the United States are aged 55 and older. If only half of those are on a DMT, that leaves 200,000. If only half of those are stable and could go off therapy, that would mean perhaps 100,000 people could discontinue DMTs in the United States. If all those assumptions are true, that would be a substantial savings in the health care burden of the United States from a relatively small population of individuals.
Beyond the cost, there are adverse events associated with using these medications. Older patients are more likely to be at risk of complications of MS DMTs. There also are doctor visits, blood monitoring, and other things that are done over time, and the inconvenience of taking a medicine on a routine basis if, indeed, it’s really not necessary because there is no benefit. Moreover, older individuals have other conditions (eg diabetes, hypertension, arrhythmias, cancer, etc) that may limit their ability to use medications due to risk. We’re very interested to see the outcome.
Discontinuing
How would you characterize the prevalence of MS in the elderly?
DR. CORBOY: A recent large demographic study put together by the National MS Society found that there’s almost a million individuals diagnosed with MS over the course of the last 40 to 50 years. The largest population segment was those aged 55 to 64 years. People with MS aged 55 or older constituted 46% of all those with MS.
What disease-modifying therapies (DMTs) are approved by the FDA for the elderly?
DR. CORBOY: Of the drugs that have received FDA approval, most are for individuals over the age of 18 and there’s no specific age cutoff. However, there’s no data supporting DMT use in people over the age of 55 because they were excluded from the studies.
There’s one DMT, fingolimod, that was approved for use in patients under the age of 18; all others are approved for 18 and above. However, none of them are explicitly approved for people over the age of 55, because there is no data to support it.
What is the goal of your study, the DISCO MS trial?
DR. CORBOY: The DISCO MS trial will be the first randomized, controlled, blinded discontinuation trial in the MS space. The objective is to assess the benefit of DMTs in patients over the age of 55.
Part of the rationale for the trial is that prior subgroup analyses have shown that the vast majority of the benefit that we’ve been able to measure with all of these DMTs is seen in those who are under age 45.
A number of studies have examined existing databases and individuals who were either randomly or deliberately taken off of their medication as they age, including people who were felt to be stable with no recent relapses and no recent changes on their MRI brain. These studies reinforced that when discontinuing medications, the individuals who were much more likely to have recurrence of disease activity were younger patients.
Pathological studies clearly show the number of acutely inflamed plaques in the white matter is dramatically lower in autopsies of older vs younger patients. There are different changes in older patients, with lymphocytic nodules in the meninges, gray matter plaques related to these meningeal nodules, microglial activation, and smaller numbers of active, or mostly, inactive, white matter plaques. It’s been difficult to show any substantial benefit in slowing disability progression, much of which is felt to not be associated with acute inflammatory disease in the aging patient. All of these medicines, which can be thought of as anti-inflammatory medicines, are very beneficial when patients are young but less so as they age.
Would you describe the DISCO MS study design?
DR. CORBOY: Our study looks at individuals who are 55 and older who have not had a relapse for at least 5 years, and who’ve not had a change on their brain scan for at least 3 years.
Individuals will be randomized to either stay on the medication that they’re currently taking or discontinue that medicine. They will be followed then for 2 years. The primary outcome will be either a new relapse or a new scan change. The examining investigators are blinded to whether the patient is currently taking a MS disease modifying therapy.
Secondary outcomes include progression of disability as measured by confirmed change on the Extended Disability Status Scale (EDSS).
The enrollment goal is about 300 patients. There are presently 15 sites. The goal is to have the study completed in about 3 years. We’re presently over halfway through enrollment.
We also have a number of patient-reported outcomes because we’re particularly interested in the patient’s view of what’s going on in terms of how they feel. Understanding that dynamic will be extremely important.
We are including both patients with relapsing MS and progressive forms of MS, noting that they should have no relapse and no scan change at study entry.
What are the challenges with this study?
DR. CORBOY: One challenge is interpreting the information with the assumption that the hypothesis is validated. The hypothesis is that in a stable population of older patients that we can safely discontinue DMTs.
If that is found to be true, the question is how many people will be affected? We know that about 46% of people with MS are 55 and older, but there are not really good estimates of the number of individuals 55 and older who remain on a DMT and who are stable by the definition I just described.
It can be safely said, I think, that a substantial number of the individuals 55 and older are still on DMTs. If there’s almost a million people with MS and 46% are 55 and older, that means around 400,000 people with MS in the United States are aged 55 and older. If only half of those are on a DMT, that leaves 200,000. If only half of those are stable and could go off therapy, that would mean perhaps 100,000 people could discontinue DMTs in the United States. If all those assumptions are true, that would be a substantial savings in the health care burden of the United States from a relatively small population of individuals.
Beyond the cost, there are adverse events associated with using these medications. Older patients are more likely to be at risk of complications of MS DMTs. There also are doctor visits, blood monitoring, and other things that are done over time, and the inconvenience of taking a medicine on a routine basis if, indeed, it’s really not necessary because there is no benefit. Moreover, older individuals have other conditions (eg diabetes, hypertension, arrhythmias, cancer, etc) that may limit their ability to use medications due to risk. We’re very interested to see the outcome.

Treating persistent catatonia when benzodiazepines fail
Many catatonia cases respond to benzodiazepines—especially lorazepam—but up to 30% do not. Electroconvulsive therapy (ECT) can be effective, but what’s the next step when ECT is unavailable or inappropriate for your patient?
To help you solve this dilemma, we describe our diagnosis and treatment decisions for a patient we call Mr. C. We explain how our process was guided by recent understandings of an abnormal neural circuit that appears to cause catatonia’s complex motor and behavioral symptoms.
This article describes that neurologic pathology and answers common questions about the clinical workup and treatment of catatonia.
CASE: TROUBLE IN TV LAND
Mr. C, age 69, caused a disturbance at a local TV station, demanding that they broadcast a manuscript he had written. Police took him to a local hospital, where he was stabilized and then transferred to a neuropsychiatric hospital for evaluation.
The psychiatric interview revealed that he had developed insomnia, excessive activity, and delusional thinking 2 weeks before admission. His medical history included coronary artery disease (CAD), hypertension, and hypothyroidism. Medications included thyroid hormone replacement therapy, furosemide, potassium, ranitidine, simvastatin, metoprolol, and lisinopril. CAD treatment included stent placement and nitroglycerin as needed.
He had been hospitalized in his 30s and treated with ECT for what he called “bad thoughts.” He said he improved after 1 month and had no subsequent psychiatric history. He denied drug or alcohol abuse.
Shortly after admission, he refused to eat or drink and after 1 week became dehydrated. He also showed mutism, immobility, and stupor. He was transferred to the medical service for IV rehydration.
MANY SCENARIOS AND SIGNS
Mr. C’s symptoms suggest possible catatonia, a neuropsychiatric syndrome of motor dysregulation found in up to 10% of acutely ill psychiatric inpatients.1,2 A movement disorder,1,2 catatonia occurs with general medical conditions and psychiatric disorders (Table 1).
Pathophysiology. Catatonic signs develop when aberrant signals from neurochemical abnormalities trigger a neural circuit that affects the medial gyrus of the orbital frontal lobe, the lateral gyrus, caudate nucleus, globus pallidus, and thalamus (Box).3-5
Presentation. A focused exam is required because patients with catatonia often do not provide a comprehensive or reliable history.2 They show mutism, characteristic postures, rigidity, aberrant speech, negativism, and stereotyped behaviors.1,2 They may present in an excited or retarded state:
- Excited patients may injure themselves or others and develop hyperthermia, tachycardia, and elevated blood pressure from excessive motor activity.
- Patients in a retarded state may present with bradykinesia and poor self-care. They may be unresponsive to external stimuli, develop catatonic stupor, and refuse to eat or drink.
Mr. C’s earlier insomnia, excessive activity, and delusional thinking (such as the TV station incident) may have signaled an excited catatonia. On admission to the medical service, however, he presented in a retarded state.
Signs. Part of the challenge with detecting catatonia’s signs is that there are so many; some rating scales list more than 20. Not all signs need to be present to make the diagnosis, however, and if you find one, others usually turn up in the examination.
A mnemonic from the Bush-Francis Catatonia Screening Instrument (Table 2) represents diagnostic signs in patients with the excited or retarded forms.2 We recommend that you review an authoritative text (see Related resources) to understand catatonia’s psychopathology.2
Table 1
Common diagnoses of patients with catatonia
| Psychiatric |
|
| Organic |
|
Catatonia is caused by neurochemical abnormalities including low GABA activity in the frontal cortex, low dopamine (D2) activity in the basal ganglia, high glutamate—N-methyl-D-aspartate (NMDA)—activity in the parietal cortex, or a combination of these.3-5 Catatonic signs occur when these neurochemical changes cause aberrant signals and trigger a neural circuit affecting the medial gyrus of the orbital frontal lobe, the lateral gyrus, caudate nucleus, globus pallidus, and thalamus (Figure).
Posturing occurs when the aberrant signal reaches the posterior parietal lobe. Patients’ bizarre and mundane postures in catatonia are maintained by “anosognosia of position.” For example, an individual does not know the position of rest for his arm, and it remains in an unusual position as if at rest.3
The PP goes on to influence the supplemental motor area (SMA), causing bradykinesia, rigidity, and other motor phenomena that catatonia shares with Parkinson’s disease. The SMA feeds back to the medial orbital gyrus, completing the neural circuit.3
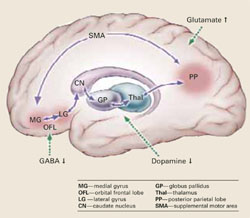
Regions such as the anterior cingulate area (ACA) and amygdala (1AMG) — also may be recruited into the expanded circuit. ACA recruitment may cause akinetic mutism, and fear is a symptom of AMG recruitment. If the anterior hypothalamus is affected, malignant catatonia or neuroleptic malignant syndrome may occur.3,5
This neural loop demonstrates an integrated model of psychosis. It may help explain why catatonia responds to treatment with lorazepam, ECT, and other agents such as antipsychotics and NMDA antagonists.
Illustration for CURRENT PSYCHIATRY by Marcia Hartsock, CMI
Table 2
WIRED `N MIRED: Mnemonic for detecting catatonia
| Waxy flexibility/catalepsy |
| Immobility/stupor |
| Refusal to eat or drink |
| Excitement |
| Deadpan staring |
| Negativism/negative symptoms |
| Mutism |
| Impulsivity |
| Rigidity |
| Echolalia/echopraxia |
| Direct observation |
CASE CONTINUED: MAKING THE DIAGNOSIS
In the medical unit, Mr. C was found to be in a catatonic stupor, with immobility, mutism (monosyllabic speech), catalepsy, intermittent waxy flexibility, withdrawal (refusal to eat and drink), automatic obedience, and mitgehen (exaggerated movements in response to light finger pressure, despite instructions to stay still). ECT workup was started, along with a trial of lorazepam, 1 mg tid.
Laboratory studies revealed high BUN/creatinine (80/2.0) that returned to normal range (BUN 7 to 21 mg/dL; creatinine 0.5 to 1.2 mg/dL) after 3 days of hydration. Because of Mr. C’s earlier excited symptoms and delusional thinking, we considered a diagnosis of bipolar disorder with catatonia. However, his symptoms did not improve with a trial of valproic acid (serum level 64 mcg/mL).
Head CT showed generalized atrophy and EEG showed delta slowing. Single-photo emission computer tomography (SPECT) showed areas of decreased perfusion in the cortex, with no perfusion in the left posterior parietal area (PP).
Mental status exam found Mr. C disoriented with poor short-term memory and unable to complete the Mini Mental State Examination (MMSE). His Bush-Francis Catatonia Rating Scale score was 28 and included many catatonic signs that would not be seen a patient with simple dehydration.
The workup supported a diagnosis of catatonia due to general medical condition (vascular dementia) and ruled out schizophrenia with catatonic features, bipolar disorder, or major depression with catatonia.
EVALUATION AND DIAGNOSIS
Medical causes. A careful history and thorough physical examination are essential for making an accurate diagnosis and ruling out medical conditions that could present with or mimic catatonia (Table 3). Medications that can induce catatonia include antipsychotics, corticosteroids, and disulfiram at therapeutic doses. Drug abuse (such as with phencyclidine), use of the general anesthetic ketamine, and benzodiazepine withdrawal may also lead to catatonia.
Head CT or MRI is indicated for patients being considered for ECT or for localizing neurologic findings. EEG can be useful when patients present with features of seizure activity—such as tongue biting, incontinence, or stupor—or with catatonia as a manifestation of delirium or dementia.
A history of head injury or neurologic disease warrants further neurologic investigation. Also consider a neurology consult when the patient has prolonged stupor or does not respond to initial drug therapy.
Psychiatric causes. The clinical setting may suggest the most likely primary psychiatric disorders to consider, such as:
- bipolar or major depression in acute inpatient psychiatric units
- autism and pervasive developmental disorders (PDD) in pediatric or PDD units
- catatonic schizophrenia in chronic psychotic patients
- somatoform or factitious disorders in forensic settings.
These generalizations are not clinically exclusive, of course, but may provide a starting point for the treatment team confronted with limited history and exam information.
Table 3
Catatonia workup: Recommended lab tests
| Test | Recommendation |
|---|---|
| Complete blood count with WBC differential | Look for leukocytosis |
| Serum chemistries | Look for electrolyte imbalances |
| Serum iron | May be low in NMS |
| Serum creatine kinase | If NMS is suspected |
| Brain MRI or CT | If structural lesion is suspected |
| Electroencephalography | If seizure disorder or brain abnormality is suspected |
| Lumbar puncture | If encephalitis or meningits is suspected |
| NMS: neuroleptic malignant syndrome | |
Initial treatment. Catatonia related to medical and psychiatric causes has been shown to respond to lorazepam and to ECT.6,7 Lorazepam is preferred because of its specificity for the GABAa receptor and ease of administration (oral, IM, or IV). Other agents that act on GABA—including amobarbital and zolpidem—have also been used. Catatonia’s hallmark features such as mutism and immobility have been shown to respond to lorazepam.8,9
ECT is a first-line treatment for catatonia with life-threatening conditions and should be considered for refractory cases.
Lorazepam. The starting dosage is usually 1 mg tid for healthy adults; 0.5 mg tid can be used for children and the elderly. Observe the patient for improvement in catatonic signs after the first dose and before giving the second. Dosages of up to 16 mg/d have been used.
In many cases, lorazepam can be tapered off after adequate treatment of the primary psychiatric condition. In severe cases, however—such as when patients refuse to eat or drink—lorazepam may be continued for as long as 1 year. Weigh the risk of benzodiazepine tolerance, dependence, and misuse versus the possibility of relapse and rehospitalization.
Medical catatonias and neuroleptic malignant syndrome (NMS) have responded favorably to ECT.8 Addressing the medical cause itself usually does not resolve catatonia, with the possible exception of seizure-induced (“ictal”) catatonia, which may respond to anticonvulsants and lorazepam.6,7
ECT. An ECT workup can begin as soon as a patient presents with catatonia. If lorazepam produces no response within 24 hours, consider ECT.
CASE CONTINUED: PERSISTENT SYMPTOMS
After three 1-mg doses of lorazepam, Mr. C became more alert and oriented but his catatonia symptoms persisted, as indicated by a Bush-Francis score of 23, significant grasp reflex, and gegenhalten (automatic rather than willful resistance to passive limb movement in proportion to the strength of the stimulus). An attempt to gradually increase lorazepam to 2 mg tid produced delirium. He remained confused even when lorazepam was reduced to 0.5 mg tid, so the drug was discontinued.
Mr. C’s neurologist added amantadine, 100 mg tid, and carbidopa/levodopa, 10/100 mg tid, to treat his parkinsonian rigidity.
WHAT NEXT? OTHER OPTIONS
Antipsychotics have been investigated as a possible treatment for catatonia. The literature suggests that conventional antipsychotics may cause catatonia and atypical antipsychotics may improve it. Conventional antipsychotics are best avoided in catatonia because they:
- appear less effective than other treatments in resolving catatonic symptoms8,10
- are associated with catatonic-like side effects, such as rigidity, akinesia, and staring10
- appear to increase NMS risk in patients with catatonic symptoms.11,12
Atypicals appear more effective in treating catatonia and less likely to cause NMS. Case reports13,14 indicate many of these agents can be effective and well tolerated in treating catatonic symptoms, although this was not the case for Mr. C.
Anticonvulsants such as valproate15 and carbamazepine, 600 to 1200 mg/d,16 may take longer to work than lorazepam but may be options for patients who do not respond to benzodiazepines.8,9
Amantadine, an N-methyl-D-aspartate (NMDA) antagonist, has been used with some success for catatonia that does not unrespond to lorazepam.17 However, amantidine’s dopamine agonist activity could worsen underlying psychosis.
Memantine—another NMDA antagonist—differs from amantadine despite having a similar chemical structure. Memantine is a noncompetitive antagonist at the NMDA receptor, without affinity for dopamine, norepinephrine, serotonin, or muscarinic receptors.18
Although no published data support using memantine in patients with catatonia, it might be considered for those who are not candidates for lorazepam or ECT. For instance, a double-blind, placebo-controlled study found that lorazepam was not effective for catatonic schizophrenia.19 We have found memantine to help in some patients with catatonic schizophrenia.
CASE CONTINUED: TRIAL OF MEMANTINE
Mr. C remained in a catatonic stupor, but we decided against ECT because he resumed eating and drinking and was not medically at risk. Quetiapine, 100 to 300 mg/d, was tried to address his dementia symptoms, confusion, and poor mentation. This trial was discontinued after Mr. C fell and was readmitted to the medical unit. We then added memantine, 5 mg bid.
In the first week after beginning memantine, Mr. C’s MMSE score was 21, consistent with vascular dementia, but he remained immobile and staring. Motor signs also persisted, including automatic obedience, ambitendency, and a grasp reflex.
The next week, we increased memantine to 10 mg bid. Mr. C was oriented to person, place, and time, and his affect was blunted. His MMSE score increased to 25, showing improved cognition and memory. His Bush-Francis scale score was 6, showing reduced catatonic signs, with remaining mild immobility, bradykinesia, speech-prompt mutism, staring, and grasp reflex.
He maintained this improvement on carbidopa/levodopa, 10/100 tid; amantadine, 100 mg tid; and memantine, 10 mg bid, and was discharged from the nursing home unit.
IMPROVEMENT WITH MEMANTINE
Memantine may reduce excess glutamate at the NMDA receptor in the parietal-SMA-frontal cortical circuit. It may help to increase GABA and dopamine, which are deficient in catatonia. Our patient with vascular dementia had a severe ischemic deficit in the posterior parietal area, as seen on SPECT.
Amantadine, another NMDA receptor antagonist, acts on dopamine neurons and may have anticholinergic-like side effects, whereas memantine does not. Although both drugs share antagonism at the NMDA glutamate receptor, noncompetitive binding is weak for amantadine and moderate for memantine. Memantine has some serotonin (5-HT3) antagonism, but neither agent has direct GABA activity.
Memantine can improve function in vascular dementia.20 Thus, Mr. C’s improvement may have been caused by the drug’s effect on his vascular dementia, the primary neuropsychiatric illness. However, his catatonic signs improved without antipsychotics, cholinesterase inhibitors, benzodiazepines, or ECT. No anticoagulation treatment or cerebral perfusion procedures account for his improved mental status.
CASE CONCLUSION
Mr. C went to live with his son’s family. Although he has problems with calculation, he shows good selfcare. When asked why he did not respond during his catatonic stupor, Mr. C stated that he believed the physician was an Internal Revenue Service agent asking him about serious tax problems. Upon reflection, he said he no longer believes this.
- Fink M, Taylor MA. Catatonia: a clinician’s guide to diagnosis and treatment. Cambridge, UK: Cambridge University Press, 2003.
- Caroff SN, Mann SC, Francis A, Fricchione GE. Catatonia: from psychopathology to neurobiology. Washington, DC: American Psychiatric Publishing, 2004.
- Mann SC, Caroff SN, Keck PE Jr, Lazarus A. Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003.
- Neuroleptic Malignant Syndrome Information Service. www.NMSIS.org.
Drug brand names
- Amantadine • Symmetrel
- Amobarbital • Amytal sodium
- Carbamazepine • Carbatrol, Equetro
- Carbidopa/levodopa • Sinemet
- Disulfiram • Antabuse
- Divalproex • Depakote
- Furosemide • Lasix
- Lisinopril • Prinivil, Zestril
- Lorazepam • Ativan
- Memantine • Namenda
- Metoprolol • Lopressor
- Ranitidine • Zantac
- Simvastatin • Zocor
- Valproic acid • Depakene
- Zolpidem • Ambien
Disclosure
Dr. Carroll and Dr. Hawkins are speakers for Forest Laboratories. The other authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
The authors thank Dr. Niraj Ahuja, consultant psychiatrist and honorary clinical lecturer (psychiatry), Newcastle, North Tyneside and Northumberland Mental Health Trust, UK, for assistance with the figure.
1. Taylor MA, Fink M. Catatonia in psychiatric classification: a home of its own. Am J Psychiatry 2003;160:1233-41.
2. Bush G, Fink M, Petrides G, et al. Catatonia I: Rating scale and standardized examination. Acta Psychiatr Scand 1996;93:129-36.
3. Northoff G. What catatonia can tell us about “top-down” modulation:” a neuropsychiatric hypothesis. Brain Behav Sci 2002;25:555-604.
4. Carroll BT. The universal field hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectrums 2000;5(7):26-33.
5. Carroll BT. Catatonia is the rosetta stone of psychosis (poster presentation). New York: American Psychiatric Association annual meeting, 2004.
6. Barnes MP, Saunders M, Walls TJ, et al. The syndrome of Karl Ludwig Kahlbaum. J Neurol Neurosurg Psychiatry 1986;49:991-6.
7. Carroll BT, Anfinson TJ, Kennedy JC, et al. Catatonic disorder due to general medical conditions. J Neuropsychiatry Clin Neurosci 1994;6:122-33.
8. Hawkins JM, Archer KJ, Strakowski SM, Keck PE. Somatic treatments of catatonia. Int J Psychiatry Med 1995;25:345-69.
9. Rosebush PI, Hildebrand AM, Furlong BG, Mazurek MF. Catatonic syndrome in a general psychiatric inpatient population: frequency, clinical presentation, and response to lorazepam. J Clin Psychiatry 1990;51:357-62.
10. Dose M. Neuroleptic-induced pseudo-catatonia. Pharmacopsychiatry 2001;34:262-4.
11. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. Arch Gen Psychiatry 1989;46:914-18.
12. White DAC. 17 catatonic patients diagnosed as neuroleptic malignant syndrome. CNS Spectrums 2000;5:58-65.
13. Levy WO, Nunez CY. Use of ziprasidone to treat bipolar-associated catatonia. Bipolar Disord 2004;6:166-7.
14. Hesslinger B, Walden J, Normann C. Acute and long-term treatment of catatonia with risperidone. Pharmacopsychiatry 2001;34:25-6.
15. Kruger S, Braunig P. Intravenous valproic acid in the treatment of severe catatonia. J Neuropsychiatry Clin Neurosci 2001;13:303-4.
16. Kritzinger PR, Jordaan GP. Catatonia: an open prospective series with carbamazepine. Int J Neuropsychopharmacol 2001;4:251-7.
17. Northoff G, Eckert J, Fritze J. Glutamatergic dysfunction in catatonia? Successful treatment of three acute akinetic catatonic patients with the NMDA antagonist amantadine. J Neurol Neurosurg Psychiatry 1997;62:404-6.
18. Namenda (memantine) Package labeling. Forest Laboratories, 2004.
19. Ungvari GS, Chie HFK, Chow LY, et al. Lorazepam for chronic catatonia: a random, double-blind, placebo-controlled, cross-over study. Psychopharmacol 1999;142:393-8.
20. Mobius HJ. Pharmacologic rationale for memantine in chronic cerebral hypoperfusion, especially vascular dementia. Alz Dis Assoc Disord 1999;13(suppl 3):172-8.
Many catatonia cases respond to benzodiazepines—especially lorazepam—but up to 30% do not. Electroconvulsive therapy (ECT) can be effective, but what’s the next step when ECT is unavailable or inappropriate for your patient?
To help you solve this dilemma, we describe our diagnosis and treatment decisions for a patient we call Mr. C. We explain how our process was guided by recent understandings of an abnormal neural circuit that appears to cause catatonia’s complex motor and behavioral symptoms.
This article describes that neurologic pathology and answers common questions about the clinical workup and treatment of catatonia.
CASE: TROUBLE IN TV LAND
Mr. C, age 69, caused a disturbance at a local TV station, demanding that they broadcast a manuscript he had written. Police took him to a local hospital, where he was stabilized and then transferred to a neuropsychiatric hospital for evaluation.
The psychiatric interview revealed that he had developed insomnia, excessive activity, and delusional thinking 2 weeks before admission. His medical history included coronary artery disease (CAD), hypertension, and hypothyroidism. Medications included thyroid hormone replacement therapy, furosemide, potassium, ranitidine, simvastatin, metoprolol, and lisinopril. CAD treatment included stent placement and nitroglycerin as needed.
He had been hospitalized in his 30s and treated with ECT for what he called “bad thoughts.” He said he improved after 1 month and had no subsequent psychiatric history. He denied drug or alcohol abuse.
Shortly after admission, he refused to eat or drink and after 1 week became dehydrated. He also showed mutism, immobility, and stupor. He was transferred to the medical service for IV rehydration.
MANY SCENARIOS AND SIGNS
Mr. C’s symptoms suggest possible catatonia, a neuropsychiatric syndrome of motor dysregulation found in up to 10% of acutely ill psychiatric inpatients.1,2 A movement disorder,1,2 catatonia occurs with general medical conditions and psychiatric disorders (Table 1).
Pathophysiology. Catatonic signs develop when aberrant signals from neurochemical abnormalities trigger a neural circuit that affects the medial gyrus of the orbital frontal lobe, the lateral gyrus, caudate nucleus, globus pallidus, and thalamus (Box).3-5
Presentation. A focused exam is required because patients with catatonia often do not provide a comprehensive or reliable history.2 They show mutism, characteristic postures, rigidity, aberrant speech, negativism, and stereotyped behaviors.1,2 They may present in an excited or retarded state:
- Excited patients may injure themselves or others and develop hyperthermia, tachycardia, and elevated blood pressure from excessive motor activity.
- Patients in a retarded state may present with bradykinesia and poor self-care. They may be unresponsive to external stimuli, develop catatonic stupor, and refuse to eat or drink.
Mr. C’s earlier insomnia, excessive activity, and delusional thinking (such as the TV station incident) may have signaled an excited catatonia. On admission to the medical service, however, he presented in a retarded state.
Signs. Part of the challenge with detecting catatonia’s signs is that there are so many; some rating scales list more than 20. Not all signs need to be present to make the diagnosis, however, and if you find one, others usually turn up in the examination.
A mnemonic from the Bush-Francis Catatonia Screening Instrument (Table 2) represents diagnostic signs in patients with the excited or retarded forms.2 We recommend that you review an authoritative text (see Related resources) to understand catatonia’s psychopathology.2
Table 1
Common diagnoses of patients with catatonia
| Psychiatric |
|
| Organic |
|
Catatonia is caused by neurochemical abnormalities including low GABA activity in the frontal cortex, low dopamine (D2) activity in the basal ganglia, high glutamate—N-methyl-D-aspartate (NMDA)—activity in the parietal cortex, or a combination of these.3-5 Catatonic signs occur when these neurochemical changes cause aberrant signals and trigger a neural circuit affecting the medial gyrus of the orbital frontal lobe, the lateral gyrus, caudate nucleus, globus pallidus, and thalamus (Figure).
Posturing occurs when the aberrant signal reaches the posterior parietal lobe. Patients’ bizarre and mundane postures in catatonia are maintained by “anosognosia of position.” For example, an individual does not know the position of rest for his arm, and it remains in an unusual position as if at rest.3
The PP goes on to influence the supplemental motor area (SMA), causing bradykinesia, rigidity, and other motor phenomena that catatonia shares with Parkinson’s disease. The SMA feeds back to the medial orbital gyrus, completing the neural circuit.3
Regions such as the anterior cingulate area (ACA) and amygdala (1AMG) — also may be recruited into the expanded circuit. ACA recruitment may cause akinetic mutism, and fear is a symptom of AMG recruitment. If the anterior hypothalamus is affected, malignant catatonia or neuroleptic malignant syndrome may occur.3,5
This neural loop demonstrates an integrated model of psychosis. It may help explain why catatonia responds to treatment with lorazepam, ECT, and other agents such as antipsychotics and NMDA antagonists.
Illustration for CURRENT PSYCHIATRY by Marcia Hartsock, CMI
Table 2
WIRED `N MIRED: Mnemonic for detecting catatonia
| Waxy flexibility/catalepsy |
| Immobility/stupor |
| Refusal to eat or drink |
| Excitement |
| Deadpan staring |
| Negativism/negative symptoms |
| Mutism |
| Impulsivity |
| Rigidity |
| Echolalia/echopraxia |
| Direct observation |
CASE CONTINUED: MAKING THE DIAGNOSIS
In the medical unit, Mr. C was found to be in a catatonic stupor, with immobility, mutism (monosyllabic speech), catalepsy, intermittent waxy flexibility, withdrawal (refusal to eat and drink), automatic obedience, and mitgehen (exaggerated movements in response to light finger pressure, despite instructions to stay still). ECT workup was started, along with a trial of lorazepam, 1 mg tid.
Laboratory studies revealed high BUN/creatinine (80/2.0) that returned to normal range (BUN 7 to 21 mg/dL; creatinine 0.5 to 1.2 mg/dL) after 3 days of hydration. Because of Mr. C’s earlier excited symptoms and delusional thinking, we considered a diagnosis of bipolar disorder with catatonia. However, his symptoms did not improve with a trial of valproic acid (serum level 64 mcg/mL).
Head CT showed generalized atrophy and EEG showed delta slowing. Single-photo emission computer tomography (SPECT) showed areas of decreased perfusion in the cortex, with no perfusion in the left posterior parietal area (PP).
Mental status exam found Mr. C disoriented with poor short-term memory and unable to complete the Mini Mental State Examination (MMSE). His Bush-Francis Catatonia Rating Scale score was 28 and included many catatonic signs that would not be seen a patient with simple dehydration.
The workup supported a diagnosis of catatonia due to general medical condition (vascular dementia) and ruled out schizophrenia with catatonic features, bipolar disorder, or major depression with catatonia.
EVALUATION AND DIAGNOSIS
Medical causes. A careful history and thorough physical examination are essential for making an accurate diagnosis and ruling out medical conditions that could present with or mimic catatonia (Table 3). Medications that can induce catatonia include antipsychotics, corticosteroids, and disulfiram at therapeutic doses. Drug abuse (such as with phencyclidine), use of the general anesthetic ketamine, and benzodiazepine withdrawal may also lead to catatonia.
Head CT or MRI is indicated for patients being considered for ECT or for localizing neurologic findings. EEG can be useful when patients present with features of seizure activity—such as tongue biting, incontinence, or stupor—or with catatonia as a manifestation of delirium or dementia.
A history of head injury or neurologic disease warrants further neurologic investigation. Also consider a neurology consult when the patient has prolonged stupor or does not respond to initial drug therapy.
Psychiatric causes. The clinical setting may suggest the most likely primary psychiatric disorders to consider, such as:
- bipolar or major depression in acute inpatient psychiatric units
- autism and pervasive developmental disorders (PDD) in pediatric or PDD units
- catatonic schizophrenia in chronic psychotic patients
- somatoform or factitious disorders in forensic settings.
These generalizations are not clinically exclusive, of course, but may provide a starting point for the treatment team confronted with limited history and exam information.
Table 3
Catatonia workup: Recommended lab tests
| Test | Recommendation |
|---|---|
| Complete blood count with WBC differential | Look for leukocytosis |
| Serum chemistries | Look for electrolyte imbalances |
| Serum iron | May be low in NMS |
| Serum creatine kinase | If NMS is suspected |
| Brain MRI or CT | If structural lesion is suspected |
| Electroencephalography | If seizure disorder or brain abnormality is suspected |
| Lumbar puncture | If encephalitis or meningits is suspected |
| NMS: neuroleptic malignant syndrome | |
Initial treatment. Catatonia related to medical and psychiatric causes has been shown to respond to lorazepam and to ECT.6,7 Lorazepam is preferred because of its specificity for the GABAa receptor and ease of administration (oral, IM, or IV). Other agents that act on GABA—including amobarbital and zolpidem—have also been used. Catatonia’s hallmark features such as mutism and immobility have been shown to respond to lorazepam.8,9
ECT is a first-line treatment for catatonia with life-threatening conditions and should be considered for refractory cases.
Lorazepam. The starting dosage is usually 1 mg tid for healthy adults; 0.5 mg tid can be used for children and the elderly. Observe the patient for improvement in catatonic signs after the first dose and before giving the second. Dosages of up to 16 mg/d have been used.
In many cases, lorazepam can be tapered off after adequate treatment of the primary psychiatric condition. In severe cases, however—such as when patients refuse to eat or drink—lorazepam may be continued for as long as 1 year. Weigh the risk of benzodiazepine tolerance, dependence, and misuse versus the possibility of relapse and rehospitalization.
Medical catatonias and neuroleptic malignant syndrome (NMS) have responded favorably to ECT.8 Addressing the medical cause itself usually does not resolve catatonia, with the possible exception of seizure-induced (“ictal”) catatonia, which may respond to anticonvulsants and lorazepam.6,7
ECT. An ECT workup can begin as soon as a patient presents with catatonia. If lorazepam produces no response within 24 hours, consider ECT.
CASE CONTINUED: PERSISTENT SYMPTOMS
After three 1-mg doses of lorazepam, Mr. C became more alert and oriented but his catatonia symptoms persisted, as indicated by a Bush-Francis score of 23, significant grasp reflex, and gegenhalten (automatic rather than willful resistance to passive limb movement in proportion to the strength of the stimulus). An attempt to gradually increase lorazepam to 2 mg tid produced delirium. He remained confused even when lorazepam was reduced to 0.5 mg tid, so the drug was discontinued.
Mr. C’s neurologist added amantadine, 100 mg tid, and carbidopa/levodopa, 10/100 mg tid, to treat his parkinsonian rigidity.
WHAT NEXT? OTHER OPTIONS
Antipsychotics have been investigated as a possible treatment for catatonia. The literature suggests that conventional antipsychotics may cause catatonia and atypical antipsychotics may improve it. Conventional antipsychotics are best avoided in catatonia because they:
- appear less effective than other treatments in resolving catatonic symptoms8,10
- are associated with catatonic-like side effects, such as rigidity, akinesia, and staring10
- appear to increase NMS risk in patients with catatonic symptoms.11,12
Atypicals appear more effective in treating catatonia and less likely to cause NMS. Case reports13,14 indicate many of these agents can be effective and well tolerated in treating catatonic symptoms, although this was not the case for Mr. C.
Anticonvulsants such as valproate15 and carbamazepine, 600 to 1200 mg/d,16 may take longer to work than lorazepam but may be options for patients who do not respond to benzodiazepines.8,9
Amantadine, an N-methyl-D-aspartate (NMDA) antagonist, has been used with some success for catatonia that does not unrespond to lorazepam.17 However, amantidine’s dopamine agonist activity could worsen underlying psychosis.
Memantine—another NMDA antagonist—differs from amantadine despite having a similar chemical structure. Memantine is a noncompetitive antagonist at the NMDA receptor, without affinity for dopamine, norepinephrine, serotonin, or muscarinic receptors.18
Although no published data support using memantine in patients with catatonia, it might be considered for those who are not candidates for lorazepam or ECT. For instance, a double-blind, placebo-controlled study found that lorazepam was not effective for catatonic schizophrenia.19 We have found memantine to help in some patients with catatonic schizophrenia.
CASE CONTINUED: TRIAL OF MEMANTINE
Mr. C remained in a catatonic stupor, but we decided against ECT because he resumed eating and drinking and was not medically at risk. Quetiapine, 100 to 300 mg/d, was tried to address his dementia symptoms, confusion, and poor mentation. This trial was discontinued after Mr. C fell and was readmitted to the medical unit. We then added memantine, 5 mg bid.
In the first week after beginning memantine, Mr. C’s MMSE score was 21, consistent with vascular dementia, but he remained immobile and staring. Motor signs also persisted, including automatic obedience, ambitendency, and a grasp reflex.
The next week, we increased memantine to 10 mg bid. Mr. C was oriented to person, place, and time, and his affect was blunted. His MMSE score increased to 25, showing improved cognition and memory. His Bush-Francis scale score was 6, showing reduced catatonic signs, with remaining mild immobility, bradykinesia, speech-prompt mutism, staring, and grasp reflex.
He maintained this improvement on carbidopa/levodopa, 10/100 tid; amantadine, 100 mg tid; and memantine, 10 mg bid, and was discharged from the nursing home unit.
IMPROVEMENT WITH MEMANTINE
Memantine may reduce excess glutamate at the NMDA receptor in the parietal-SMA-frontal cortical circuit. It may help to increase GABA and dopamine, which are deficient in catatonia. Our patient with vascular dementia had a severe ischemic deficit in the posterior parietal area, as seen on SPECT.
Amantadine, another NMDA receptor antagonist, acts on dopamine neurons and may have anticholinergic-like side effects, whereas memantine does not. Although both drugs share antagonism at the NMDA glutamate receptor, noncompetitive binding is weak for amantadine and moderate for memantine. Memantine has some serotonin (5-HT3) antagonism, but neither agent has direct GABA activity.
Memantine can improve function in vascular dementia.20 Thus, Mr. C’s improvement may have been caused by the drug’s effect on his vascular dementia, the primary neuropsychiatric illness. However, his catatonic signs improved without antipsychotics, cholinesterase inhibitors, benzodiazepines, or ECT. No anticoagulation treatment or cerebral perfusion procedures account for his improved mental status.
CASE CONCLUSION
Mr. C went to live with his son’s family. Although he has problems with calculation, he shows good selfcare. When asked why he did not respond during his catatonic stupor, Mr. C stated that he believed the physician was an Internal Revenue Service agent asking him about serious tax problems. Upon reflection, he said he no longer believes this.
- Fink M, Taylor MA. Catatonia: a clinician’s guide to diagnosis and treatment. Cambridge, UK: Cambridge University Press, 2003.
- Caroff SN, Mann SC, Francis A, Fricchione GE. Catatonia: from psychopathology to neurobiology. Washington, DC: American Psychiatric Publishing, 2004.
- Mann SC, Caroff SN, Keck PE Jr, Lazarus A. Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003.
- Neuroleptic Malignant Syndrome Information Service. www.NMSIS.org.
Drug brand names
- Amantadine • Symmetrel
- Amobarbital • Amytal sodium
- Carbamazepine • Carbatrol, Equetro
- Carbidopa/levodopa • Sinemet
- Disulfiram • Antabuse
- Divalproex • Depakote
- Furosemide • Lasix
- Lisinopril • Prinivil, Zestril
- Lorazepam • Ativan
- Memantine • Namenda
- Metoprolol • Lopressor
- Ranitidine • Zantac
- Simvastatin • Zocor
- Valproic acid • Depakene
- Zolpidem • Ambien
Disclosure
Dr. Carroll and Dr. Hawkins are speakers for Forest Laboratories. The other authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
The authors thank Dr. Niraj Ahuja, consultant psychiatrist and honorary clinical lecturer (psychiatry), Newcastle, North Tyneside and Northumberland Mental Health Trust, UK, for assistance with the figure.
Many catatonia cases respond to benzodiazepines—especially lorazepam—but up to 30% do not. Electroconvulsive therapy (ECT) can be effective, but what’s the next step when ECT is unavailable or inappropriate for your patient?
To help you solve this dilemma, we describe our diagnosis and treatment decisions for a patient we call Mr. C. We explain how our process was guided by recent understandings of an abnormal neural circuit that appears to cause catatonia’s complex motor and behavioral symptoms.
This article describes that neurologic pathology and answers common questions about the clinical workup and treatment of catatonia.
CASE: TROUBLE IN TV LAND
Mr. C, age 69, caused a disturbance at a local TV station, demanding that they broadcast a manuscript he had written. Police took him to a local hospital, where he was stabilized and then transferred to a neuropsychiatric hospital for evaluation.
The psychiatric interview revealed that he had developed insomnia, excessive activity, and delusional thinking 2 weeks before admission. His medical history included coronary artery disease (CAD), hypertension, and hypothyroidism. Medications included thyroid hormone replacement therapy, furosemide, potassium, ranitidine, simvastatin, metoprolol, and lisinopril. CAD treatment included stent placement and nitroglycerin as needed.
He had been hospitalized in his 30s and treated with ECT for what he called “bad thoughts.” He said he improved after 1 month and had no subsequent psychiatric history. He denied drug or alcohol abuse.
Shortly after admission, he refused to eat or drink and after 1 week became dehydrated. He also showed mutism, immobility, and stupor. He was transferred to the medical service for IV rehydration.
MANY SCENARIOS AND SIGNS
Mr. C’s symptoms suggest possible catatonia, a neuropsychiatric syndrome of motor dysregulation found in up to 10% of acutely ill psychiatric inpatients.1,2 A movement disorder,1,2 catatonia occurs with general medical conditions and psychiatric disorders (Table 1).
Pathophysiology. Catatonic signs develop when aberrant signals from neurochemical abnormalities trigger a neural circuit that affects the medial gyrus of the orbital frontal lobe, the lateral gyrus, caudate nucleus, globus pallidus, and thalamus (Box).3-5
Presentation. A focused exam is required because patients with catatonia often do not provide a comprehensive or reliable history.2 They show mutism, characteristic postures, rigidity, aberrant speech, negativism, and stereotyped behaviors.1,2 They may present in an excited or retarded state:
- Excited patients may injure themselves or others and develop hyperthermia, tachycardia, and elevated blood pressure from excessive motor activity.
- Patients in a retarded state may present with bradykinesia and poor self-care. They may be unresponsive to external stimuli, develop catatonic stupor, and refuse to eat or drink.
Mr. C’s earlier insomnia, excessive activity, and delusional thinking (such as the TV station incident) may have signaled an excited catatonia. On admission to the medical service, however, he presented in a retarded state.
Signs. Part of the challenge with detecting catatonia’s signs is that there are so many; some rating scales list more than 20. Not all signs need to be present to make the diagnosis, however, and if you find one, others usually turn up in the examination.
A mnemonic from the Bush-Francis Catatonia Screening Instrument (Table 2) represents diagnostic signs in patients with the excited or retarded forms.2 We recommend that you review an authoritative text (see Related resources) to understand catatonia’s psychopathology.2
Table 1
Common diagnoses of patients with catatonia
| Psychiatric |
|
| Organic |
|
Catatonia is caused by neurochemical abnormalities including low GABA activity in the frontal cortex, low dopamine (D2) activity in the basal ganglia, high glutamate—N-methyl-D-aspartate (NMDA)—activity in the parietal cortex, or a combination of these.3-5 Catatonic signs occur when these neurochemical changes cause aberrant signals and trigger a neural circuit affecting the medial gyrus of the orbital frontal lobe, the lateral gyrus, caudate nucleus, globus pallidus, and thalamus (Figure).
Posturing occurs when the aberrant signal reaches the posterior parietal lobe. Patients’ bizarre and mundane postures in catatonia are maintained by “anosognosia of position.” For example, an individual does not know the position of rest for his arm, and it remains in an unusual position as if at rest.3
The PP goes on to influence the supplemental motor area (SMA), causing bradykinesia, rigidity, and other motor phenomena that catatonia shares with Parkinson’s disease. The SMA feeds back to the medial orbital gyrus, completing the neural circuit.3
Regions such as the anterior cingulate area (ACA) and amygdala (1AMG) — also may be recruited into the expanded circuit. ACA recruitment may cause akinetic mutism, and fear is a symptom of AMG recruitment. If the anterior hypothalamus is affected, malignant catatonia or neuroleptic malignant syndrome may occur.3,5
This neural loop demonstrates an integrated model of psychosis. It may help explain why catatonia responds to treatment with lorazepam, ECT, and other agents such as antipsychotics and NMDA antagonists.
Illustration for CURRENT PSYCHIATRY by Marcia Hartsock, CMI
Table 2
WIRED `N MIRED: Mnemonic for detecting catatonia
| Waxy flexibility/catalepsy |
| Immobility/stupor |
| Refusal to eat or drink |
| Excitement |
| Deadpan staring |
| Negativism/negative symptoms |
| Mutism |
| Impulsivity |
| Rigidity |
| Echolalia/echopraxia |
| Direct observation |
CASE CONTINUED: MAKING THE DIAGNOSIS
In the medical unit, Mr. C was found to be in a catatonic stupor, with immobility, mutism (monosyllabic speech), catalepsy, intermittent waxy flexibility, withdrawal (refusal to eat and drink), automatic obedience, and mitgehen (exaggerated movements in response to light finger pressure, despite instructions to stay still). ECT workup was started, along with a trial of lorazepam, 1 mg tid.
Laboratory studies revealed high BUN/creatinine (80/2.0) that returned to normal range (BUN 7 to 21 mg/dL; creatinine 0.5 to 1.2 mg/dL) after 3 days of hydration. Because of Mr. C’s earlier excited symptoms and delusional thinking, we considered a diagnosis of bipolar disorder with catatonia. However, his symptoms did not improve with a trial of valproic acid (serum level 64 mcg/mL).
Head CT showed generalized atrophy and EEG showed delta slowing. Single-photo emission computer tomography (SPECT) showed areas of decreased perfusion in the cortex, with no perfusion in the left posterior parietal area (PP).
Mental status exam found Mr. C disoriented with poor short-term memory and unable to complete the Mini Mental State Examination (MMSE). His Bush-Francis Catatonia Rating Scale score was 28 and included many catatonic signs that would not be seen a patient with simple dehydration.
The workup supported a diagnosis of catatonia due to general medical condition (vascular dementia) and ruled out schizophrenia with catatonic features, bipolar disorder, or major depression with catatonia.
EVALUATION AND DIAGNOSIS
Medical causes. A careful history and thorough physical examination are essential for making an accurate diagnosis and ruling out medical conditions that could present with or mimic catatonia (Table 3). Medications that can induce catatonia include antipsychotics, corticosteroids, and disulfiram at therapeutic doses. Drug abuse (such as with phencyclidine), use of the general anesthetic ketamine, and benzodiazepine withdrawal may also lead to catatonia.
Head CT or MRI is indicated for patients being considered for ECT or for localizing neurologic findings. EEG can be useful when patients present with features of seizure activity—such as tongue biting, incontinence, or stupor—or with catatonia as a manifestation of delirium or dementia.
A history of head injury or neurologic disease warrants further neurologic investigation. Also consider a neurology consult when the patient has prolonged stupor or does not respond to initial drug therapy.
Psychiatric causes. The clinical setting may suggest the most likely primary psychiatric disorders to consider, such as:
- bipolar or major depression in acute inpatient psychiatric units
- autism and pervasive developmental disorders (PDD) in pediatric or PDD units
- catatonic schizophrenia in chronic psychotic patients
- somatoform or factitious disorders in forensic settings.
These generalizations are not clinically exclusive, of course, but may provide a starting point for the treatment team confronted with limited history and exam information.
Table 3
Catatonia workup: Recommended lab tests
| Test | Recommendation |
|---|---|
| Complete blood count with WBC differential | Look for leukocytosis |
| Serum chemistries | Look for electrolyte imbalances |
| Serum iron | May be low in NMS |
| Serum creatine kinase | If NMS is suspected |
| Brain MRI or CT | If structural lesion is suspected |
| Electroencephalography | If seizure disorder or brain abnormality is suspected |
| Lumbar puncture | If encephalitis or meningits is suspected |
| NMS: neuroleptic malignant syndrome | |
Initial treatment. Catatonia related to medical and psychiatric causes has been shown to respond to lorazepam and to ECT.6,7 Lorazepam is preferred because of its specificity for the GABAa receptor and ease of administration (oral, IM, or IV). Other agents that act on GABA—including amobarbital and zolpidem—have also been used. Catatonia’s hallmark features such as mutism and immobility have been shown to respond to lorazepam.8,9
ECT is a first-line treatment for catatonia with life-threatening conditions and should be considered for refractory cases.
Lorazepam. The starting dosage is usually 1 mg tid for healthy adults; 0.5 mg tid can be used for children and the elderly. Observe the patient for improvement in catatonic signs after the first dose and before giving the second. Dosages of up to 16 mg/d have been used.
In many cases, lorazepam can be tapered off after adequate treatment of the primary psychiatric condition. In severe cases, however—such as when patients refuse to eat or drink—lorazepam may be continued for as long as 1 year. Weigh the risk of benzodiazepine tolerance, dependence, and misuse versus the possibility of relapse and rehospitalization.
Medical catatonias and neuroleptic malignant syndrome (NMS) have responded favorably to ECT.8 Addressing the medical cause itself usually does not resolve catatonia, with the possible exception of seizure-induced (“ictal”) catatonia, which may respond to anticonvulsants and lorazepam.6,7
ECT. An ECT workup can begin as soon as a patient presents with catatonia. If lorazepam produces no response within 24 hours, consider ECT.
CASE CONTINUED: PERSISTENT SYMPTOMS
After three 1-mg doses of lorazepam, Mr. C became more alert and oriented but his catatonia symptoms persisted, as indicated by a Bush-Francis score of 23, significant grasp reflex, and gegenhalten (automatic rather than willful resistance to passive limb movement in proportion to the strength of the stimulus). An attempt to gradually increase lorazepam to 2 mg tid produced delirium. He remained confused even when lorazepam was reduced to 0.5 mg tid, so the drug was discontinued.
Mr. C’s neurologist added amantadine, 100 mg tid, and carbidopa/levodopa, 10/100 mg tid, to treat his parkinsonian rigidity.
WHAT NEXT? OTHER OPTIONS
Antipsychotics have been investigated as a possible treatment for catatonia. The literature suggests that conventional antipsychotics may cause catatonia and atypical antipsychotics may improve it. Conventional antipsychotics are best avoided in catatonia because they:
- appear less effective than other treatments in resolving catatonic symptoms8,10
- are associated with catatonic-like side effects, such as rigidity, akinesia, and staring10
- appear to increase NMS risk in patients with catatonic symptoms.11,12
Atypicals appear more effective in treating catatonia and less likely to cause NMS. Case reports13,14 indicate many of these agents can be effective and well tolerated in treating catatonic symptoms, although this was not the case for Mr. C.
Anticonvulsants such as valproate15 and carbamazepine, 600 to 1200 mg/d,16 may take longer to work than lorazepam but may be options for patients who do not respond to benzodiazepines.8,9
Amantadine, an N-methyl-D-aspartate (NMDA) antagonist, has been used with some success for catatonia that does not unrespond to lorazepam.17 However, amantidine’s dopamine agonist activity could worsen underlying psychosis.
Memantine—another NMDA antagonist—differs from amantadine despite having a similar chemical structure. Memantine is a noncompetitive antagonist at the NMDA receptor, without affinity for dopamine, norepinephrine, serotonin, or muscarinic receptors.18
Although no published data support using memantine in patients with catatonia, it might be considered for those who are not candidates for lorazepam or ECT. For instance, a double-blind, placebo-controlled study found that lorazepam was not effective for catatonic schizophrenia.19 We have found memantine to help in some patients with catatonic schizophrenia.
CASE CONTINUED: TRIAL OF MEMANTINE
Mr. C remained in a catatonic stupor, but we decided against ECT because he resumed eating and drinking and was not medically at risk. Quetiapine, 100 to 300 mg/d, was tried to address his dementia symptoms, confusion, and poor mentation. This trial was discontinued after Mr. C fell and was readmitted to the medical unit. We then added memantine, 5 mg bid.
In the first week after beginning memantine, Mr. C’s MMSE score was 21, consistent with vascular dementia, but he remained immobile and staring. Motor signs also persisted, including automatic obedience, ambitendency, and a grasp reflex.
The next week, we increased memantine to 10 mg bid. Mr. C was oriented to person, place, and time, and his affect was blunted. His MMSE score increased to 25, showing improved cognition and memory. His Bush-Francis scale score was 6, showing reduced catatonic signs, with remaining mild immobility, bradykinesia, speech-prompt mutism, staring, and grasp reflex.
He maintained this improvement on carbidopa/levodopa, 10/100 tid; amantadine, 100 mg tid; and memantine, 10 mg bid, and was discharged from the nursing home unit.
IMPROVEMENT WITH MEMANTINE
Memantine may reduce excess glutamate at the NMDA receptor in the parietal-SMA-frontal cortical circuit. It may help to increase GABA and dopamine, which are deficient in catatonia. Our patient with vascular dementia had a severe ischemic deficit in the posterior parietal area, as seen on SPECT.
Amantadine, another NMDA receptor antagonist, acts on dopamine neurons and may have anticholinergic-like side effects, whereas memantine does not. Although both drugs share antagonism at the NMDA glutamate receptor, noncompetitive binding is weak for amantadine and moderate for memantine. Memantine has some serotonin (5-HT3) antagonism, but neither agent has direct GABA activity.
Memantine can improve function in vascular dementia.20 Thus, Mr. C’s improvement may have been caused by the drug’s effect on his vascular dementia, the primary neuropsychiatric illness. However, his catatonic signs improved without antipsychotics, cholinesterase inhibitors, benzodiazepines, or ECT. No anticoagulation treatment or cerebral perfusion procedures account for his improved mental status.
CASE CONCLUSION
Mr. C went to live with his son’s family. Although he has problems with calculation, he shows good selfcare. When asked why he did not respond during his catatonic stupor, Mr. C stated that he believed the physician was an Internal Revenue Service agent asking him about serious tax problems. Upon reflection, he said he no longer believes this.
- Fink M, Taylor MA. Catatonia: a clinician’s guide to diagnosis and treatment. Cambridge, UK: Cambridge University Press, 2003.
- Caroff SN, Mann SC, Francis A, Fricchione GE. Catatonia: from psychopathology to neurobiology. Washington, DC: American Psychiatric Publishing, 2004.
- Mann SC, Caroff SN, Keck PE Jr, Lazarus A. Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003.
- Neuroleptic Malignant Syndrome Information Service. www.NMSIS.org.
Drug brand names
- Amantadine • Symmetrel
- Amobarbital • Amytal sodium
- Carbamazepine • Carbatrol, Equetro
- Carbidopa/levodopa • Sinemet
- Disulfiram • Antabuse
- Divalproex • Depakote
- Furosemide • Lasix
- Lisinopril • Prinivil, Zestril
- Lorazepam • Ativan
- Memantine • Namenda
- Metoprolol • Lopressor
- Ranitidine • Zantac
- Simvastatin • Zocor
- Valproic acid • Depakene
- Zolpidem • Ambien
Disclosure
Dr. Carroll and Dr. Hawkins are speakers for Forest Laboratories. The other authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
The authors thank Dr. Niraj Ahuja, consultant psychiatrist and honorary clinical lecturer (psychiatry), Newcastle, North Tyneside and Northumberland Mental Health Trust, UK, for assistance with the figure.
1. Taylor MA, Fink M. Catatonia in psychiatric classification: a home of its own. Am J Psychiatry 2003;160:1233-41.
2. Bush G, Fink M, Petrides G, et al. Catatonia I: Rating scale and standardized examination. Acta Psychiatr Scand 1996;93:129-36.
3. Northoff G. What catatonia can tell us about “top-down” modulation:” a neuropsychiatric hypothesis. Brain Behav Sci 2002;25:555-604.
4. Carroll BT. The universal field hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectrums 2000;5(7):26-33.
5. Carroll BT. Catatonia is the rosetta stone of psychosis (poster presentation). New York: American Psychiatric Association annual meeting, 2004.
6. Barnes MP, Saunders M, Walls TJ, et al. The syndrome of Karl Ludwig Kahlbaum. J Neurol Neurosurg Psychiatry 1986;49:991-6.
7. Carroll BT, Anfinson TJ, Kennedy JC, et al. Catatonic disorder due to general medical conditions. J Neuropsychiatry Clin Neurosci 1994;6:122-33.
8. Hawkins JM, Archer KJ, Strakowski SM, Keck PE. Somatic treatments of catatonia. Int J Psychiatry Med 1995;25:345-69.
9. Rosebush PI, Hildebrand AM, Furlong BG, Mazurek MF. Catatonic syndrome in a general psychiatric inpatient population: frequency, clinical presentation, and response to lorazepam. J Clin Psychiatry 1990;51:357-62.
10. Dose M. Neuroleptic-induced pseudo-catatonia. Pharmacopsychiatry 2001;34:262-4.
11. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. Arch Gen Psychiatry 1989;46:914-18.
12. White DAC. 17 catatonic patients diagnosed as neuroleptic malignant syndrome. CNS Spectrums 2000;5:58-65.
13. Levy WO, Nunez CY. Use of ziprasidone to treat bipolar-associated catatonia. Bipolar Disord 2004;6:166-7.
14. Hesslinger B, Walden J, Normann C. Acute and long-term treatment of catatonia with risperidone. Pharmacopsychiatry 2001;34:25-6.
15. Kruger S, Braunig P. Intravenous valproic acid in the treatment of severe catatonia. J Neuropsychiatry Clin Neurosci 2001;13:303-4.
16. Kritzinger PR, Jordaan GP. Catatonia: an open prospective series with carbamazepine. Int J Neuropsychopharmacol 2001;4:251-7.
17. Northoff G, Eckert J, Fritze J. Glutamatergic dysfunction in catatonia? Successful treatment of three acute akinetic catatonic patients with the NMDA antagonist amantadine. J Neurol Neurosurg Psychiatry 1997;62:404-6.
18. Namenda (memantine) Package labeling. Forest Laboratories, 2004.
19. Ungvari GS, Chie HFK, Chow LY, et al. Lorazepam for chronic catatonia: a random, double-blind, placebo-controlled, cross-over study. Psychopharmacol 1999;142:393-8.
20. Mobius HJ. Pharmacologic rationale for memantine in chronic cerebral hypoperfusion, especially vascular dementia. Alz Dis Assoc Disord 1999;13(suppl 3):172-8.
1. Taylor MA, Fink M. Catatonia in psychiatric classification: a home of its own. Am J Psychiatry 2003;160:1233-41.
2. Bush G, Fink M, Petrides G, et al. Catatonia I: Rating scale and standardized examination. Acta Psychiatr Scand 1996;93:129-36.
3. Northoff G. What catatonia can tell us about “top-down” modulation:” a neuropsychiatric hypothesis. Brain Behav Sci 2002;25:555-604.
4. Carroll BT. The universal field hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectrums 2000;5(7):26-33.
5. Carroll BT. Catatonia is the rosetta stone of psychosis (poster presentation). New York: American Psychiatric Association annual meeting, 2004.
6. Barnes MP, Saunders M, Walls TJ, et al. The syndrome of Karl Ludwig Kahlbaum. J Neurol Neurosurg Psychiatry 1986;49:991-6.
7. Carroll BT, Anfinson TJ, Kennedy JC, et al. Catatonic disorder due to general medical conditions. J Neuropsychiatry Clin Neurosci 1994;6:122-33.
8. Hawkins JM, Archer KJ, Strakowski SM, Keck PE. Somatic treatments of catatonia. Int J Psychiatry Med 1995;25:345-69.
9. Rosebush PI, Hildebrand AM, Furlong BG, Mazurek MF. Catatonic syndrome in a general psychiatric inpatient population: frequency, clinical presentation, and response to lorazepam. J Clin Psychiatry 1990;51:357-62.
10. Dose M. Neuroleptic-induced pseudo-catatonia. Pharmacopsychiatry 2001;34:262-4.
11. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. Arch Gen Psychiatry 1989;46:914-18.
12. White DAC. 17 catatonic patients diagnosed as neuroleptic malignant syndrome. CNS Spectrums 2000;5:58-65.
13. Levy WO, Nunez CY. Use of ziprasidone to treat bipolar-associated catatonia. Bipolar Disord 2004;6:166-7.
14. Hesslinger B, Walden J, Normann C. Acute and long-term treatment of catatonia with risperidone. Pharmacopsychiatry 2001;34:25-6.
15. Kruger S, Braunig P. Intravenous valproic acid in the treatment of severe catatonia. J Neuropsychiatry Clin Neurosci 2001;13:303-4.
16. Kritzinger PR, Jordaan GP. Catatonia: an open prospective series with carbamazepine. Int J Neuropsychopharmacol 2001;4:251-7.
17. Northoff G, Eckert J, Fritze J. Glutamatergic dysfunction in catatonia? Successful treatment of three acute akinetic catatonic patients with the NMDA antagonist amantadine. J Neurol Neurosurg Psychiatry 1997;62:404-6.
18. Namenda (memantine) Package labeling. Forest Laboratories, 2004.
19. Ungvari GS, Chie HFK, Chow LY, et al. Lorazepam for chronic catatonia: a random, double-blind, placebo-controlled, cross-over study. Psychopharmacol 1999;142:393-8.
20. Mobius HJ. Pharmacologic rationale for memantine in chronic cerebral hypoperfusion, especially vascular dementia. Alz Dis Assoc Disord 1999;13(suppl 3):172-8.