User login
Happy Birthday, CATIE!

A decade after the CATIE study, the focus has shifted from effectiveness to neuroprotection
This past September, exactly 10 years after publication of the primary findings of the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study1—namely, that effectiveness (defined as all-cause discontinuation) was not different across first-generation antipsychotics (FGAs) and second generation antipsychotics (SGAs)— a new meta-analysis by Vita et al2 of differences in cortical gray-matter change between those 2 classes of antipsychotics offers a reminder: The clinical focus of the CATIE study overlooked important neurobiological and neuroprotective differences between FGAs and SGAs.
How drastically 1 decade can change the scientific perspective! Vita et al’s meta-analysis and meta-regression encompassed all 18 MRI studies of cortical gray matter in patients with schizophrenia.2 Earlier studies (published between 1983 and 2014) had lumped together patients who were receiving an FGA and those receiving an SGA, and authors reported overall reduction in cortical gray matter with prolonged antipsychotic treatment.
Remarkable findings emerge
When Vita et al2 analyzed FGA- and SGA-treated patients separately, however, they found a significant reduction in cortical gray matter in the FGA group but not in the SGA group. In fact, while higher daily dosages of FGAs were associated with greater reduction in cortical gray matter, higher dosages of SGAs were associated with lower cortical gray matter reduction and, in some samples, with an increase in volume of cortical gray matter.
The researchers hypothesized that the differential effects of FGAs and SGAs might be attributable to the neurotoxicity of typical FGAs and the neuroprotective effect of atypical SGAs.
Hindsight
The key neurobiological difference between FGAs and SGAs reported by Vita et al2 was not addressed in the CATIE study, leading, at that time, to a rush to judgment that all antipsychotics are the same. This conclusion emboldened managed-care organizations to mandate use of older (and cheaper) generic FGAs instead of newer (and more expensive) SGAs— most of which have become available as generic equivalents since the CATIE study was completed.
Investigators in the CATIE study— of which I was one—cannot be blamed for not focusing on neurotoxicity and neuroprotection; those data were not on the psychiatry’s radar when the CATIE study was designed in 1998. The major focus was on whether SGAs (new on the scene in the late 1990s) were more efficacious, safe, and tolerable (that is, more effective) than FGAs.
In fact, the first study reporting that SGAs stimulated neurogenesis (in animals) was published in 2002,3 when the CATIE study was more than half complete. Research into the neuroprotective properties of SGAs then grew rapidly. In fact, the principal investigator of the CATIE study conducted a head-to-head comparison of FGA haloperidol and SGA olanzapine in a sample of first-episode schizophrenia patients4; over 1 year of follow-up, it was determined that patients in the haloperidol-treated group exhibited significant brain volume loss on MRI but those in the olanzapine-treated group did not. This study was published in 2005—the same year the CATIE study was published!
SGAs offer neuroprotection
Over the past decade, the neuroprotective effects of SGAs5 and the neurotoxic effects of FGAs6 have been studied intensively, revealing that SGAs have multiple neuroprotective effects. These effects include:
• stimulation of the production of new brain cells (neurons and glia), known as neurogenesis5,7,8
• an increase in neurotrophic factors, such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF),9 which are found at a significantly low level in patients with psychosis10
• reversal of phencyclidine (PCP)-induced changes in gene expression11
• neuroprotection against ischemic stroke12-14
• reversal of PCP-induced loss of dendritic spines in the frontal cortex15
• prevention of oligodendrocyte damage caused by interferon gamma-stimulated microglia16,17
• reversal of loss of dendritic spines in the prefrontal cortex induced by dopamine depletion18
• an anti-inflammatory effect19,20
• protection against β-amyloid and hydrogen peroxide-induced cell death21
• protection against prefrontal cortical neuronal damage caused by dizocilpine (MK-801)22
• reversal of a PCP-induced decrease in the glutathione level and alteration of antioxidant defenses23
• protection of cortical neurons from glutamate neurotoxicity.24
One reason why SGAs are neuroprotective, but FGAs are not, can be attributed to their receptor profiles. FGAs block dopamine D2 receptors far more than serotonin 2A receptors, whereas SGAs do the opposite: They block 5-HT2A receptors 500% to 1,000% more than they block D2 receptors. This difference is associated in turn with a different neurobiological and neuroprotective profiles, such as a decrease or an increase in BDNF.25,26
Neither similar nor interchangeable
Since publication of the findings of the CATIE study, the primary investigator has proposed that neuroprotection can be a therapeutic strategy to prevent neurodegeneration and neurodeterioration associated with schizophrenia.27 Given the preponderance of data showing that SGAs have numerous neuroprotective properties but FGAs have many neurotoxic effects,6 the message to psychiatric practitioners, a decade after the CATIE study, is that the 2 generations of antipsychotic agents are not really similar or interchangeable. They might have similar clinical effectiveness but they exert very different neurobiological effects.
The proof of the pudding is in the eating: Despite the findings of the CATIE study, the vast majority of psychiatrists would prefer to treat their own family members with an SGA, not an FGA, if the need for antipsychotic medication arises.
1. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
2. Vita A, De Peri L, Deste G, et al. The effect of antipsychotic treatment on cortical gray matter changes in schizophrenia: does the class matter? A meta-analysis and meta-regression of longitudinal magnetic resonance imaging studies. Biol Psychiatry. 2015;78(6):403-412.
3. Wakade CG, Mahadik SP, Waller JL, et al. Atypical neuroleptics stimulate neurogenesis in adult rat brain. J Neurosci Res. 2002;69(1):72-79.
4. Lieberman JA, Tollefson GD, Charles C, et al; HGDH Study Group. Antipsychotic drug effects on brain morphology in first-episode psychosis. Arch Gen Psychiatry. 2005;62(4):361-370.
5. Nasrallah HA. Impaired neuroplasticity in schizophrenia and the neuro-regenerative effects of atypical antipsychotics. Medscape Psychiatry. http://www.medscape.org/viewarticle/569521. Published January 31, 2008. Accessed November 10, 2015.
6. Nasrallah HA. Haloperidol clearly is neurotoxic. Should it be banned? Current Psychiatry. 2012;12(7):7-8.
7. Nandra KS, Agius M. The differences between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99.
8. Nasrallah HA, Hopkins T, Pixley SK, et al. Differential effects of antipsychotic and antidepressant drugs on neurogenic region in rats. Brain Res. 2010;354:23-29.
9. Pillai A, Tery AV, Mahadik SP. Differential effects of long-term treatment with typical and atypical antipsychotics on NGF and BNDF levels in rat striatum and hippocampus. Schizophr Res. 2006;82(1):95-106.
10. Buckley PF, Pillai A, Evans D, et al. Brain derived neurotropic factor in first-episode psychosis. Schizophr Res. 2007;91(1-3):1-5.
11. Martin MV, Mimics K, Nisenbaum LK, et al. Olanzapine reversed brain gene expression changes induced by phencyclidines treatment in non-human primates. Mol Neuropsychiatry. 2015;1(2):82-93.
12. Yan BC, Park JH, Ahn JH, et al. Neuroprotection of posttreatment with risperidone, an atypical antipsychotic drug, in rat and gerbil models of ischemic stroke and the maintenance of antioxidants in a gerbil model of ischemic stroke. J Neurosci Res. 2014;92(6):795-807.
13. Yulug B, Yildiz A, Güzel O, et al. Risperidone attenuates brain damage after focal cerebral ischemia in vivo. Brain Res Bull. 2006;69(6):656-659.
14. Yulug B, Yildiz A, Hüdaoglu O, et al. Olanzapine attenuates brain damage after focal cerebral ischemia in vivo. Brain Res Bull. 2006;71(1-3):296-300.
15. Elsworth JD, Morrow BA. Hajszan T, et al. Phencyclidine-induced loss of asymmetric spine synapses in rodent prefrontal cortex is reversed by acute and chronic treatment with olanzapine. Neuropsychopharmacology. 2001;36(10):2054-2061.
16. Seki Y, Kato TA, Monji A, et al. Pretreatment of aripiprazole and minocycline, but not haloperidol, suppresses oligodendrocyte damage from interferon-y-stimulated microglia in co-culture model. Schizophr Res. 2013;151(1-3):20-28.
17. Bian Q, Kato T, Monji A, et al. The effect of atypical antipsychotics, perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-gamma. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):42-48.
18. Wang HD, Deutch AY. Dopamine depletion of the prefrontal cortex induces dendritic spine loss: reversal by atypical antipsychotic drug treatment. Neuropsychopharmacology. 2008;33(6):1276-1286.
19. Miller BJ, Buckley P, Seabolt W, et al. Meta-analysis of cytokine alternations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry. 2011;70(7):663-671.
20. Nasrallah HA. Beyond dopamine: The ‘other’ effects of antipsychotics. Current Psychiatry. 2013;12(6):8-9.
21. Yang MC, Lung FW. Neuroprotection of paliperidone on SH-SY5Y cells against β-amyloid peptide(25-35), N-methyl-4-phenylpyridinium ion, and hydrogen peroxide-induced cell death. Psychopharmacology (Berl). 2011;217(3):397-410.
22. Peng L, Zhu D, Feng X, et al. Paliperidone protects prefrontal cortical neurons from damages caused by MK-801 via Akt1/GSK3β _signaling pathway. Schizophr Res. 2013;147(1):14-23.23.
Stojkovic´ T, Radonjic´ NV, Velimirovic´ M, et al. Risperidone reverses phencyclidine induced decrease in glutathione levels and alternations of antioxidant defense in rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(1):192-199.
24. Koprivica V, Regardie K, Wolff C, et al. Aripiprazole protects cortical neurons from glutamate toxicity. Eur J Pharmacol. 2011;651(1-3):73-76.
25. Vaidya VA, Marek GJ, Aghajanian GK, et al. 5-HT2A receptor-mediated regulation of brain-derived neurotrophic factor mRNA in the hippocampus and the neocortex. J Neurosci. 1997;17(8):2785-2795.
26. Meridith GE, Switzer RC 3rd, Napier TC. Short-term, D2 receptor blockade induces synaptic degeneration, reduces levels of tyrosine hydroxylase and brain-derived neurotrophic factor, and enhances D2-mediated firing in the ventral pallidum. Brain Res. 2004;995(1):14-22.
27. Lieberman JA, Perkins DO, Jarskog LF. Neuroprotection: a therapeutic strategy to prevent deterioration associated with schizophrenia. CNS Spectr. 2007;12(suppl 4):1-13; quiz 14.
This past September, exactly 10 years after publication of the primary findings of the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study1—namely, that effectiveness (defined as all-cause discontinuation) was not different across first-generation antipsychotics (FGAs) and second generation antipsychotics (SGAs)— a new meta-analysis by Vita et al2 of differences in cortical gray-matter change between those 2 classes of antipsychotics offers a reminder: The clinical focus of the CATIE study overlooked important neurobiological and neuroprotective differences between FGAs and SGAs.
How drastically 1 decade can change the scientific perspective! Vita et al’s meta-analysis and meta-regression encompassed all 18 MRI studies of cortical gray matter in patients with schizophrenia.2 Earlier studies (published between 1983 and 2014) had lumped together patients who were receiving an FGA and those receiving an SGA, and authors reported overall reduction in cortical gray matter with prolonged antipsychotic treatment.
Remarkable findings emerge
When Vita et al2 analyzed FGA- and SGA-treated patients separately, however, they found a significant reduction in cortical gray matter in the FGA group but not in the SGA group. In fact, while higher daily dosages of FGAs were associated with greater reduction in cortical gray matter, higher dosages of SGAs were associated with lower cortical gray matter reduction and, in some samples, with an increase in volume of cortical gray matter.
The researchers hypothesized that the differential effects of FGAs and SGAs might be attributable to the neurotoxicity of typical FGAs and the neuroprotective effect of atypical SGAs.
Hindsight
The key neurobiological difference between FGAs and SGAs reported by Vita et al2 was not addressed in the CATIE study, leading, at that time, to a rush to judgment that all antipsychotics are the same. This conclusion emboldened managed-care organizations to mandate use of older (and cheaper) generic FGAs instead of newer (and more expensive) SGAs— most of which have become available as generic equivalents since the CATIE study was completed.
Investigators in the CATIE study— of which I was one—cannot be blamed for not focusing on neurotoxicity and neuroprotection; those data were not on the psychiatry’s radar when the CATIE study was designed in 1998. The major focus was on whether SGAs (new on the scene in the late 1990s) were more efficacious, safe, and tolerable (that is, more effective) than FGAs.
In fact, the first study reporting that SGAs stimulated neurogenesis (in animals) was published in 2002,3 when the CATIE study was more than half complete. Research into the neuroprotective properties of SGAs then grew rapidly. In fact, the principal investigator of the CATIE study conducted a head-to-head comparison of FGA haloperidol and SGA olanzapine in a sample of first-episode schizophrenia patients4; over 1 year of follow-up, it was determined that patients in the haloperidol-treated group exhibited significant brain volume loss on MRI but those in the olanzapine-treated group did not. This study was published in 2005—the same year the CATIE study was published!
SGAs offer neuroprotection
Over the past decade, the neuroprotective effects of SGAs5 and the neurotoxic effects of FGAs6 have been studied intensively, revealing that SGAs have multiple neuroprotective effects. These effects include:
• stimulation of the production of new brain cells (neurons and glia), known as neurogenesis5,7,8
• an increase in neurotrophic factors, such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF),9 which are found at a significantly low level in patients with psychosis10
• reversal of phencyclidine (PCP)-induced changes in gene expression11
• neuroprotection against ischemic stroke12-14
• reversal of PCP-induced loss of dendritic spines in the frontal cortex15
• prevention of oligodendrocyte damage caused by interferon gamma-stimulated microglia16,17
• reversal of loss of dendritic spines in the prefrontal cortex induced by dopamine depletion18
• an anti-inflammatory effect19,20
• protection against β-amyloid and hydrogen peroxide-induced cell death21
• protection against prefrontal cortical neuronal damage caused by dizocilpine (MK-801)22
• reversal of a PCP-induced decrease in the glutathione level and alteration of antioxidant defenses23
• protection of cortical neurons from glutamate neurotoxicity.24
One reason why SGAs are neuroprotective, but FGAs are not, can be attributed to their receptor profiles. FGAs block dopamine D2 receptors far more than serotonin 2A receptors, whereas SGAs do the opposite: They block 5-HT2A receptors 500% to 1,000% more than they block D2 receptors. This difference is associated in turn with a different neurobiological and neuroprotective profiles, such as a decrease or an increase in BDNF.25,26
Neither similar nor interchangeable
Since publication of the findings of the CATIE study, the primary investigator has proposed that neuroprotection can be a therapeutic strategy to prevent neurodegeneration and neurodeterioration associated with schizophrenia.27 Given the preponderance of data showing that SGAs have numerous neuroprotective properties but FGAs have many neurotoxic effects,6 the message to psychiatric practitioners, a decade after the CATIE study, is that the 2 generations of antipsychotic agents are not really similar or interchangeable. They might have similar clinical effectiveness but they exert very different neurobiological effects.
The proof of the pudding is in the eating: Despite the findings of the CATIE study, the vast majority of psychiatrists would prefer to treat their own family members with an SGA, not an FGA, if the need for antipsychotic medication arises.
This past September, exactly 10 years after publication of the primary findings of the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study1—namely, that effectiveness (defined as all-cause discontinuation) was not different across first-generation antipsychotics (FGAs) and second generation antipsychotics (SGAs)— a new meta-analysis by Vita et al2 of differences in cortical gray-matter change between those 2 classes of antipsychotics offers a reminder: The clinical focus of the CATIE study overlooked important neurobiological and neuroprotective differences between FGAs and SGAs.
How drastically 1 decade can change the scientific perspective! Vita et al’s meta-analysis and meta-regression encompassed all 18 MRI studies of cortical gray matter in patients with schizophrenia.2 Earlier studies (published between 1983 and 2014) had lumped together patients who were receiving an FGA and those receiving an SGA, and authors reported overall reduction in cortical gray matter with prolonged antipsychotic treatment.
Remarkable findings emerge
When Vita et al2 analyzed FGA- and SGA-treated patients separately, however, they found a significant reduction in cortical gray matter in the FGA group but not in the SGA group. In fact, while higher daily dosages of FGAs were associated with greater reduction in cortical gray matter, higher dosages of SGAs were associated with lower cortical gray matter reduction and, in some samples, with an increase in volume of cortical gray matter.
The researchers hypothesized that the differential effects of FGAs and SGAs might be attributable to the neurotoxicity of typical FGAs and the neuroprotective effect of atypical SGAs.
Hindsight
The key neurobiological difference between FGAs and SGAs reported by Vita et al2 was not addressed in the CATIE study, leading, at that time, to a rush to judgment that all antipsychotics are the same. This conclusion emboldened managed-care organizations to mandate use of older (and cheaper) generic FGAs instead of newer (and more expensive) SGAs— most of which have become available as generic equivalents since the CATIE study was completed.
Investigators in the CATIE study— of which I was one—cannot be blamed for not focusing on neurotoxicity and neuroprotection; those data were not on the psychiatry’s radar when the CATIE study was designed in 1998. The major focus was on whether SGAs (new on the scene in the late 1990s) were more efficacious, safe, and tolerable (that is, more effective) than FGAs.
In fact, the first study reporting that SGAs stimulated neurogenesis (in animals) was published in 2002,3 when the CATIE study was more than half complete. Research into the neuroprotective properties of SGAs then grew rapidly. In fact, the principal investigator of the CATIE study conducted a head-to-head comparison of FGA haloperidol and SGA olanzapine in a sample of first-episode schizophrenia patients4; over 1 year of follow-up, it was determined that patients in the haloperidol-treated group exhibited significant brain volume loss on MRI but those in the olanzapine-treated group did not. This study was published in 2005—the same year the CATIE study was published!
SGAs offer neuroprotection
Over the past decade, the neuroprotective effects of SGAs5 and the neurotoxic effects of FGAs6 have been studied intensively, revealing that SGAs have multiple neuroprotective effects. These effects include:
• stimulation of the production of new brain cells (neurons and glia), known as neurogenesis5,7,8
• an increase in neurotrophic factors, such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF),9 which are found at a significantly low level in patients with psychosis10
• reversal of phencyclidine (PCP)-induced changes in gene expression11
• neuroprotection against ischemic stroke12-14
• reversal of PCP-induced loss of dendritic spines in the frontal cortex15
• prevention of oligodendrocyte damage caused by interferon gamma-stimulated microglia16,17
• reversal of loss of dendritic spines in the prefrontal cortex induced by dopamine depletion18
• an anti-inflammatory effect19,20
• protection against β-amyloid and hydrogen peroxide-induced cell death21
• protection against prefrontal cortical neuronal damage caused by dizocilpine (MK-801)22
• reversal of a PCP-induced decrease in the glutathione level and alteration of antioxidant defenses23
• protection of cortical neurons from glutamate neurotoxicity.24
One reason why SGAs are neuroprotective, but FGAs are not, can be attributed to their receptor profiles. FGAs block dopamine D2 receptors far more than serotonin 2A receptors, whereas SGAs do the opposite: They block 5-HT2A receptors 500% to 1,000% more than they block D2 receptors. This difference is associated in turn with a different neurobiological and neuroprotective profiles, such as a decrease or an increase in BDNF.25,26
Neither similar nor interchangeable
Since publication of the findings of the CATIE study, the primary investigator has proposed that neuroprotection can be a therapeutic strategy to prevent neurodegeneration and neurodeterioration associated with schizophrenia.27 Given the preponderance of data showing that SGAs have numerous neuroprotective properties but FGAs have many neurotoxic effects,6 the message to psychiatric practitioners, a decade after the CATIE study, is that the 2 generations of antipsychotic agents are not really similar or interchangeable. They might have similar clinical effectiveness but they exert very different neurobiological effects.
The proof of the pudding is in the eating: Despite the findings of the CATIE study, the vast majority of psychiatrists would prefer to treat their own family members with an SGA, not an FGA, if the need for antipsychotic medication arises.
1. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
2. Vita A, De Peri L, Deste G, et al. The effect of antipsychotic treatment on cortical gray matter changes in schizophrenia: does the class matter? A meta-analysis and meta-regression of longitudinal magnetic resonance imaging studies. Biol Psychiatry. 2015;78(6):403-412.
3. Wakade CG, Mahadik SP, Waller JL, et al. Atypical neuroleptics stimulate neurogenesis in adult rat brain. J Neurosci Res. 2002;69(1):72-79.
4. Lieberman JA, Tollefson GD, Charles C, et al; HGDH Study Group. Antipsychotic drug effects on brain morphology in first-episode psychosis. Arch Gen Psychiatry. 2005;62(4):361-370.
5. Nasrallah HA. Impaired neuroplasticity in schizophrenia and the neuro-regenerative effects of atypical antipsychotics. Medscape Psychiatry. http://www.medscape.org/viewarticle/569521. Published January 31, 2008. Accessed November 10, 2015.
6. Nasrallah HA. Haloperidol clearly is neurotoxic. Should it be banned? Current Psychiatry. 2012;12(7):7-8.
7. Nandra KS, Agius M. The differences between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99.
8. Nasrallah HA, Hopkins T, Pixley SK, et al. Differential effects of antipsychotic and antidepressant drugs on neurogenic region in rats. Brain Res. 2010;354:23-29.
9. Pillai A, Tery AV, Mahadik SP. Differential effects of long-term treatment with typical and atypical antipsychotics on NGF and BNDF levels in rat striatum and hippocampus. Schizophr Res. 2006;82(1):95-106.
10. Buckley PF, Pillai A, Evans D, et al. Brain derived neurotropic factor in first-episode psychosis. Schizophr Res. 2007;91(1-3):1-5.
11. Martin MV, Mimics K, Nisenbaum LK, et al. Olanzapine reversed brain gene expression changes induced by phencyclidines treatment in non-human primates. Mol Neuropsychiatry. 2015;1(2):82-93.
12. Yan BC, Park JH, Ahn JH, et al. Neuroprotection of posttreatment with risperidone, an atypical antipsychotic drug, in rat and gerbil models of ischemic stroke and the maintenance of antioxidants in a gerbil model of ischemic stroke. J Neurosci Res. 2014;92(6):795-807.
13. Yulug B, Yildiz A, Güzel O, et al. Risperidone attenuates brain damage after focal cerebral ischemia in vivo. Brain Res Bull. 2006;69(6):656-659.
14. Yulug B, Yildiz A, Hüdaoglu O, et al. Olanzapine attenuates brain damage after focal cerebral ischemia in vivo. Brain Res Bull. 2006;71(1-3):296-300.
15. Elsworth JD, Morrow BA. Hajszan T, et al. Phencyclidine-induced loss of asymmetric spine synapses in rodent prefrontal cortex is reversed by acute and chronic treatment with olanzapine. Neuropsychopharmacology. 2001;36(10):2054-2061.
16. Seki Y, Kato TA, Monji A, et al. Pretreatment of aripiprazole and minocycline, but not haloperidol, suppresses oligodendrocyte damage from interferon-y-stimulated microglia in co-culture model. Schizophr Res. 2013;151(1-3):20-28.
17. Bian Q, Kato T, Monji A, et al. The effect of atypical antipsychotics, perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-gamma. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):42-48.
18. Wang HD, Deutch AY. Dopamine depletion of the prefrontal cortex induces dendritic spine loss: reversal by atypical antipsychotic drug treatment. Neuropsychopharmacology. 2008;33(6):1276-1286.
19. Miller BJ, Buckley P, Seabolt W, et al. Meta-analysis of cytokine alternations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry. 2011;70(7):663-671.
20. Nasrallah HA. Beyond dopamine: The ‘other’ effects of antipsychotics. Current Psychiatry. 2013;12(6):8-9.
21. Yang MC, Lung FW. Neuroprotection of paliperidone on SH-SY5Y cells against β-amyloid peptide(25-35), N-methyl-4-phenylpyridinium ion, and hydrogen peroxide-induced cell death. Psychopharmacology (Berl). 2011;217(3):397-410.
22. Peng L, Zhu D, Feng X, et al. Paliperidone protects prefrontal cortical neurons from damages caused by MK-801 via Akt1/GSK3β _signaling pathway. Schizophr Res. 2013;147(1):14-23.23.
Stojkovic´ T, Radonjic´ NV, Velimirovic´ M, et al. Risperidone reverses phencyclidine induced decrease in glutathione levels and alternations of antioxidant defense in rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(1):192-199.
24. Koprivica V, Regardie K, Wolff C, et al. Aripiprazole protects cortical neurons from glutamate toxicity. Eur J Pharmacol. 2011;651(1-3):73-76.
25. Vaidya VA, Marek GJ, Aghajanian GK, et al. 5-HT2A receptor-mediated regulation of brain-derived neurotrophic factor mRNA in the hippocampus and the neocortex. J Neurosci. 1997;17(8):2785-2795.
26. Meridith GE, Switzer RC 3rd, Napier TC. Short-term, D2 receptor blockade induces synaptic degeneration, reduces levels of tyrosine hydroxylase and brain-derived neurotrophic factor, and enhances D2-mediated firing in the ventral pallidum. Brain Res. 2004;995(1):14-22.
27. Lieberman JA, Perkins DO, Jarskog LF. Neuroprotection: a therapeutic strategy to prevent deterioration associated with schizophrenia. CNS Spectr. 2007;12(suppl 4):1-13; quiz 14.
1. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
2. Vita A, De Peri L, Deste G, et al. The effect of antipsychotic treatment on cortical gray matter changes in schizophrenia: does the class matter? A meta-analysis and meta-regression of longitudinal magnetic resonance imaging studies. Biol Psychiatry. 2015;78(6):403-412.
3. Wakade CG, Mahadik SP, Waller JL, et al. Atypical neuroleptics stimulate neurogenesis in adult rat brain. J Neurosci Res. 2002;69(1):72-79.
4. Lieberman JA, Tollefson GD, Charles C, et al; HGDH Study Group. Antipsychotic drug effects on brain morphology in first-episode psychosis. Arch Gen Psychiatry. 2005;62(4):361-370.
5. Nasrallah HA. Impaired neuroplasticity in schizophrenia and the neuro-regenerative effects of atypical antipsychotics. Medscape Psychiatry. http://www.medscape.org/viewarticle/569521. Published January 31, 2008. Accessed November 10, 2015.
6. Nasrallah HA. Haloperidol clearly is neurotoxic. Should it be banned? Current Psychiatry. 2012;12(7):7-8.
7. Nandra KS, Agius M. The differences between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99.
8. Nasrallah HA, Hopkins T, Pixley SK, et al. Differential effects of antipsychotic and antidepressant drugs on neurogenic region in rats. Brain Res. 2010;354:23-29.
9. Pillai A, Tery AV, Mahadik SP. Differential effects of long-term treatment with typical and atypical antipsychotics on NGF and BNDF levels in rat striatum and hippocampus. Schizophr Res. 2006;82(1):95-106.
10. Buckley PF, Pillai A, Evans D, et al. Brain derived neurotropic factor in first-episode psychosis. Schizophr Res. 2007;91(1-3):1-5.
11. Martin MV, Mimics K, Nisenbaum LK, et al. Olanzapine reversed brain gene expression changes induced by phencyclidines treatment in non-human primates. Mol Neuropsychiatry. 2015;1(2):82-93.
12. Yan BC, Park JH, Ahn JH, et al. Neuroprotection of posttreatment with risperidone, an atypical antipsychotic drug, in rat and gerbil models of ischemic stroke and the maintenance of antioxidants in a gerbil model of ischemic stroke. J Neurosci Res. 2014;92(6):795-807.
13. Yulug B, Yildiz A, Güzel O, et al. Risperidone attenuates brain damage after focal cerebral ischemia in vivo. Brain Res Bull. 2006;69(6):656-659.
14. Yulug B, Yildiz A, Hüdaoglu O, et al. Olanzapine attenuates brain damage after focal cerebral ischemia in vivo. Brain Res Bull. 2006;71(1-3):296-300.
15. Elsworth JD, Morrow BA. Hajszan T, et al. Phencyclidine-induced loss of asymmetric spine synapses in rodent prefrontal cortex is reversed by acute and chronic treatment with olanzapine. Neuropsychopharmacology. 2001;36(10):2054-2061.
16. Seki Y, Kato TA, Monji A, et al. Pretreatment of aripiprazole and minocycline, but not haloperidol, suppresses oligodendrocyte damage from interferon-y-stimulated microglia in co-culture model. Schizophr Res. 2013;151(1-3):20-28.
17. Bian Q, Kato T, Monji A, et al. The effect of atypical antipsychotics, perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-gamma. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):42-48.
18. Wang HD, Deutch AY. Dopamine depletion of the prefrontal cortex induces dendritic spine loss: reversal by atypical antipsychotic drug treatment. Neuropsychopharmacology. 2008;33(6):1276-1286.
19. Miller BJ, Buckley P, Seabolt W, et al. Meta-analysis of cytokine alternations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry. 2011;70(7):663-671.
20. Nasrallah HA. Beyond dopamine: The ‘other’ effects of antipsychotics. Current Psychiatry. 2013;12(6):8-9.
21. Yang MC, Lung FW. Neuroprotection of paliperidone on SH-SY5Y cells against β-amyloid peptide(25-35), N-methyl-4-phenylpyridinium ion, and hydrogen peroxide-induced cell death. Psychopharmacology (Berl). 2011;217(3):397-410.
22. Peng L, Zhu D, Feng X, et al. Paliperidone protects prefrontal cortical neurons from damages caused by MK-801 via Akt1/GSK3β _signaling pathway. Schizophr Res. 2013;147(1):14-23.23.
Stojkovic´ T, Radonjic´ NV, Velimirovic´ M, et al. Risperidone reverses phencyclidine induced decrease in glutathione levels and alternations of antioxidant defense in rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(1):192-199.
24. Koprivica V, Regardie K, Wolff C, et al. Aripiprazole protects cortical neurons from glutamate toxicity. Eur J Pharmacol. 2011;651(1-3):73-76.
25. Vaidya VA, Marek GJ, Aghajanian GK, et al. 5-HT2A receptor-mediated regulation of brain-derived neurotrophic factor mRNA in the hippocampus and the neocortex. J Neurosci. 1997;17(8):2785-2795.
26. Meridith GE, Switzer RC 3rd, Napier TC. Short-term, D2 receptor blockade induces synaptic degeneration, reduces levels of tyrosine hydroxylase and brain-derived neurotrophic factor, and enhances D2-mediated firing in the ventral pallidum. Brain Res. 2004;995(1):14-22.
27. Lieberman JA, Perkins DO, Jarskog LF. Neuroprotection: a therapeutic strategy to prevent deterioration associated with schizophrenia. CNS Spectr. 2007;12(suppl 4):1-13; quiz 14.
Non-drug therapies for refractory schizophrenia
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Needed: A biopsychosocial ‘therapeutic placenta’ for people with schizophrenia
Consider stroke. Guidelines for acute treatment, access, intervention, prevention of post-hospitalization relapse, and rehabilitation are extensively spelled out and implemented.1 (The Box outlines Mayo Clinic guidelines for stroke management, as a demonstration of the comprehensiveness of the approach.)

Schizophrenia and related severe mental illnesses (SMI) need a similar all-inclusive system that seamlessly provides the myriad components of care needed for this vulnerable population. I propose the term “therapeutic placenta” to describe what people with a disabling SMI brain disorder deserve, just as stroke patients do.
Closing asylums: Psychosocial abruptio placentae
In a past Editorial,2 I described the appalling consequences of eliminating the asylum, an entity that I believe must be a key component of the SMI therapeutic placenta. The asylum is to schizophrenia as the skilled nursing home is to stroke. SMI patients suffered extensively when asylums were shut down; they lost a medical refuge with psychiatric and primary care, nursing and social work support, occupational and recreational therapies, and work therapy (farming, carpentry shop, cafeteria, laundry, etc.). For SMI, these services are the psychosocial counterpart of various physical rehabilitation therapies for stroke patients that no one would ever dare to eliminate.
Persons with schizophrenia and other SMI have suffered tragically with rupture of the main components of the therapeutic placenta that existed for decades before the advent of medications. The massive homelessness, widespread incarceration, persistent poverty, rampant access to alcohol and drugs of abuse, early death due to lack of primary care, and absence of meaningful opportunities for vocational rehabilitation are all consequences of a neglectful society that refuses to fund a therapeutic placenta for the SMI population.
The public mental health system in charge of SMI patients is broken, disconnected, and failing to provide the necessary components of a therapeutic placenta. It should not be surprising to witness the terribly stressful life and premature mortality of SMI patients, who are modern-day les misérables.
The Table lists what I consider to be the necessary spectrum of health care services through the life of an SMI patient that an optimal therapeutic placenta must provide until an effective prevention or a cure for SMI is discovered.
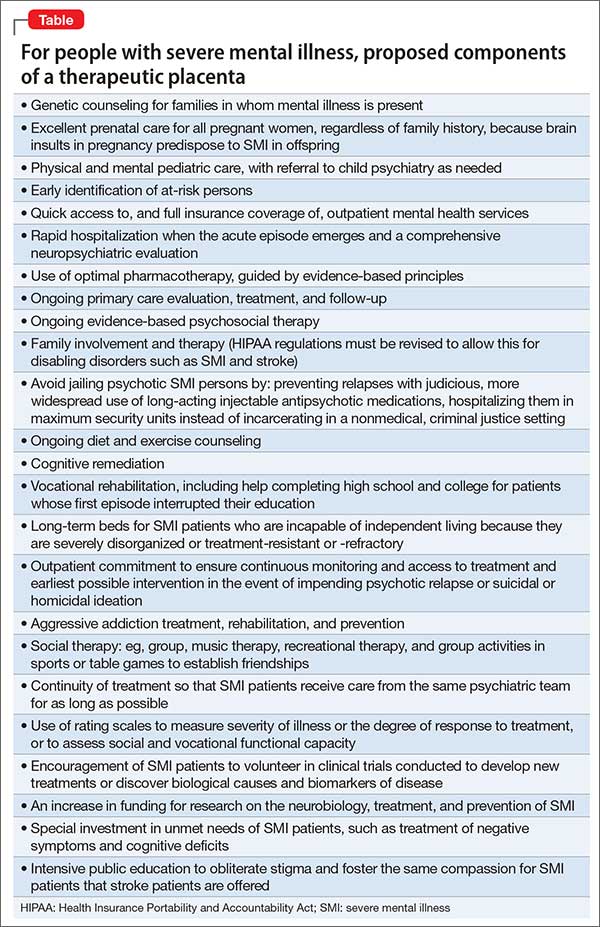
Reasons to be hopeful
Admittedly, encouraging steps are being made toward establishing a therapeutic placenta for SMI:
The RAISE Study3and Navigate Program4 demonstrate that implementing a comprehensive program of acute treatment and psychosocial interventions and rehabilitation yields better outcomes in SMI.
The Institute of Medicine released a landmark report on psychosocial interventions for mental illness and substance abuse disorders. It outlines a new model for establishing the effectiveness of intervention and the implementation of psychosocial strategies in clinical practice.5
The 21st Century Cures Act, if passed by Congress and signed by the President, will increase funding for the National Institutes of Health, which in turn will bolster the budgets of the National Institute of Mental Health, National Institute on Drug Abuse, and the National Institute on Alcohol Abuse and Alcoholism and enhance the chances of discovering better treatments and prevention of SMI.
The Helping Families in Mental Health Crisis Act, more directly relevant to mental health and psychiatry, proposes, if passed, to:
• enhance evidence-based and scientifically validated interventions in the public sector
• raise the profile of mental health within the federal government by creating a position of Assistant Secretary for Mental Health in the U.S. Department of Health and Human Services, who will have oversight of both research and mental health care within the federal government.
Unacceptable disparity must be remedied
Planning an effective therapeutic placenta is imperative if health care for SMI patients is to approach the comprehensive spectrum of treatment, rehabilitation, and prevention available to stroke patients. Although stroke is regarded as a sensory-motor brain disorder, it is also associated with mental symptoms, just as schizophrenia is associated with sensory-motor symptoms. Both are disabling brain disorders: one, physically and cognitively; the other, mentally and socially. Both require a therapeutic placenta: Stroke is supported by one; schizophrenia is not. This is an unacceptable disparity that must be addressed—soon.
1. Jauch EC, Saver JL, Adams HP Jr, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44(3):870-947.
2. Nasrallah HA. Bring back the asylums? Current Psychiatry. 2008;7(3):19-20.
3. Kane JM, Schooler NR, Marcy P, et al. The RAISE early treatment program for first-episode psychosis: background, rationale, and study design. J Clin Psychiatry. 2015;76(3):240-246.
4. Mueser KT, Penn DL, Addington J, et al. The NAVIGATE program for first-episode psychosis: rationale, overview, and description of psychosocial components. Psychiatr Serv. 2015;66(7):680-690.
5. The National Academy of Sciences. Psychosocial interventions for mental and substance use disorders: a framework for establishing evidence-based standards. Washington, DC. http:// iom.nationalacademies.org/Reports/2015/ Psychosocial-Interventions-Mental-Substance- Abuse-Disorders.aspx. Published July 14, 2015. Accessed September 3, 2015.
Consider stroke. Guidelines for acute treatment, access, intervention, prevention of post-hospitalization relapse, and rehabilitation are extensively spelled out and implemented.1 (The Box outlines Mayo Clinic guidelines for stroke management, as a demonstration of the comprehensiveness of the approach.)
Schizophrenia and related severe mental illnesses (SMI) need a similar all-inclusive system that seamlessly provides the myriad components of care needed for this vulnerable population. I propose the term “therapeutic placenta” to describe what people with a disabling SMI brain disorder deserve, just as stroke patients do.
Closing asylums: Psychosocial abruptio placentae
In a past Editorial,2 I described the appalling consequences of eliminating the asylum, an entity that I believe must be a key component of the SMI therapeutic placenta. The asylum is to schizophrenia as the skilled nursing home is to stroke. SMI patients suffered extensively when asylums were shut down; they lost a medical refuge with psychiatric and primary care, nursing and social work support, occupational and recreational therapies, and work therapy (farming, carpentry shop, cafeteria, laundry, etc.). For SMI, these services are the psychosocial counterpart of various physical rehabilitation therapies for stroke patients that no one would ever dare to eliminate.
Persons with schizophrenia and other SMI have suffered tragically with rupture of the main components of the therapeutic placenta that existed for decades before the advent of medications. The massive homelessness, widespread incarceration, persistent poverty, rampant access to alcohol and drugs of abuse, early death due to lack of primary care, and absence of meaningful opportunities for vocational rehabilitation are all consequences of a neglectful society that refuses to fund a therapeutic placenta for the SMI population.
The public mental health system in charge of SMI patients is broken, disconnected, and failing to provide the necessary components of a therapeutic placenta. It should not be surprising to witness the terribly stressful life and premature mortality of SMI patients, who are modern-day les misérables.
The Table lists what I consider to be the necessary spectrum of health care services through the life of an SMI patient that an optimal therapeutic placenta must provide until an effective prevention or a cure for SMI is discovered.
Reasons to be hopeful
Admittedly, encouraging steps are being made toward establishing a therapeutic placenta for SMI:
The RAISE Study3and Navigate Program4 demonstrate that implementing a comprehensive program of acute treatment and psychosocial interventions and rehabilitation yields better outcomes in SMI.
The Institute of Medicine released a landmark report on psychosocial interventions for mental illness and substance abuse disorders. It outlines a new model for establishing the effectiveness of intervention and the implementation of psychosocial strategies in clinical practice.5
The 21st Century Cures Act, if passed by Congress and signed by the President, will increase funding for the National Institutes of Health, which in turn will bolster the budgets of the National Institute of Mental Health, National Institute on Drug Abuse, and the National Institute on Alcohol Abuse and Alcoholism and enhance the chances of discovering better treatments and prevention of SMI.
The Helping Families in Mental Health Crisis Act, more directly relevant to mental health and psychiatry, proposes, if passed, to:
• enhance evidence-based and scientifically validated interventions in the public sector
• raise the profile of mental health within the federal government by creating a position of Assistant Secretary for Mental Health in the U.S. Department of Health and Human Services, who will have oversight of both research and mental health care within the federal government.
Unacceptable disparity must be remedied
Planning an effective therapeutic placenta is imperative if health care for SMI patients is to approach the comprehensive spectrum of treatment, rehabilitation, and prevention available to stroke patients. Although stroke is regarded as a sensory-motor brain disorder, it is also associated with mental symptoms, just as schizophrenia is associated with sensory-motor symptoms. Both are disabling brain disorders: one, physically and cognitively; the other, mentally and socially. Both require a therapeutic placenta: Stroke is supported by one; schizophrenia is not. This is an unacceptable disparity that must be addressed—soon.
Consider stroke. Guidelines for acute treatment, access, intervention, prevention of post-hospitalization relapse, and rehabilitation are extensively spelled out and implemented.1 (The Box outlines Mayo Clinic guidelines for stroke management, as a demonstration of the comprehensiveness of the approach.)
Schizophrenia and related severe mental illnesses (SMI) need a similar all-inclusive system that seamlessly provides the myriad components of care needed for this vulnerable population. I propose the term “therapeutic placenta” to describe what people with a disabling SMI brain disorder deserve, just as stroke patients do.
Closing asylums: Psychosocial abruptio placentae
In a past Editorial,2 I described the appalling consequences of eliminating the asylum, an entity that I believe must be a key component of the SMI therapeutic placenta. The asylum is to schizophrenia as the skilled nursing home is to stroke. SMI patients suffered extensively when asylums were shut down; they lost a medical refuge with psychiatric and primary care, nursing and social work support, occupational and recreational therapies, and work therapy (farming, carpentry shop, cafeteria, laundry, etc.). For SMI, these services are the psychosocial counterpart of various physical rehabilitation therapies for stroke patients that no one would ever dare to eliminate.
Persons with schizophrenia and other SMI have suffered tragically with rupture of the main components of the therapeutic placenta that existed for decades before the advent of medications. The massive homelessness, widespread incarceration, persistent poverty, rampant access to alcohol and drugs of abuse, early death due to lack of primary care, and absence of meaningful opportunities for vocational rehabilitation are all consequences of a neglectful society that refuses to fund a therapeutic placenta for the SMI population.
The public mental health system in charge of SMI patients is broken, disconnected, and failing to provide the necessary components of a therapeutic placenta. It should not be surprising to witness the terribly stressful life and premature mortality of SMI patients, who are modern-day les misérables.
The Table lists what I consider to be the necessary spectrum of health care services through the life of an SMI patient that an optimal therapeutic placenta must provide until an effective prevention or a cure for SMI is discovered.
Reasons to be hopeful
Admittedly, encouraging steps are being made toward establishing a therapeutic placenta for SMI:
The RAISE Study3and Navigate Program4 demonstrate that implementing a comprehensive program of acute treatment and psychosocial interventions and rehabilitation yields better outcomes in SMI.
The Institute of Medicine released a landmark report on psychosocial interventions for mental illness and substance abuse disorders. It outlines a new model for establishing the effectiveness of intervention and the implementation of psychosocial strategies in clinical practice.5
The 21st Century Cures Act, if passed by Congress and signed by the President, will increase funding for the National Institutes of Health, which in turn will bolster the budgets of the National Institute of Mental Health, National Institute on Drug Abuse, and the National Institute on Alcohol Abuse and Alcoholism and enhance the chances of discovering better treatments and prevention of SMI.
The Helping Families in Mental Health Crisis Act, more directly relevant to mental health and psychiatry, proposes, if passed, to:
• enhance evidence-based and scientifically validated interventions in the public sector
• raise the profile of mental health within the federal government by creating a position of Assistant Secretary for Mental Health in the U.S. Department of Health and Human Services, who will have oversight of both research and mental health care within the federal government.
Unacceptable disparity must be remedied
Planning an effective therapeutic placenta is imperative if health care for SMI patients is to approach the comprehensive spectrum of treatment, rehabilitation, and prevention available to stroke patients. Although stroke is regarded as a sensory-motor brain disorder, it is also associated with mental symptoms, just as schizophrenia is associated with sensory-motor symptoms. Both are disabling brain disorders: one, physically and cognitively; the other, mentally and socially. Both require a therapeutic placenta: Stroke is supported by one; schizophrenia is not. This is an unacceptable disparity that must be addressed—soon.
1. Jauch EC, Saver JL, Adams HP Jr, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44(3):870-947.
2. Nasrallah HA. Bring back the asylums? Current Psychiatry. 2008;7(3):19-20.
3. Kane JM, Schooler NR, Marcy P, et al. The RAISE early treatment program for first-episode psychosis: background, rationale, and study design. J Clin Psychiatry. 2015;76(3):240-246.
4. Mueser KT, Penn DL, Addington J, et al. The NAVIGATE program for first-episode psychosis: rationale, overview, and description of psychosocial components. Psychiatr Serv. 2015;66(7):680-690.
5. The National Academy of Sciences. Psychosocial interventions for mental and substance use disorders: a framework for establishing evidence-based standards. Washington, DC. http:// iom.nationalacademies.org/Reports/2015/ Psychosocial-Interventions-Mental-Substance- Abuse-Disorders.aspx. Published July 14, 2015. Accessed September 3, 2015.
1. Jauch EC, Saver JL, Adams HP Jr, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44(3):870-947.
2. Nasrallah HA. Bring back the asylums? Current Psychiatry. 2008;7(3):19-20.
3. Kane JM, Schooler NR, Marcy P, et al. The RAISE early treatment program for first-episode psychosis: background, rationale, and study design. J Clin Psychiatry. 2015;76(3):240-246.
4. Mueser KT, Penn DL, Addington J, et al. The NAVIGATE program for first-episode psychosis: rationale, overview, and description of psychosocial components. Psychiatr Serv. 2015;66(7):680-690.
5. The National Academy of Sciences. Psychosocial interventions for mental and substance use disorders: a framework for establishing evidence-based standards. Washington, DC. http:// iom.nationalacademies.org/Reports/2015/ Psychosocial-Interventions-Mental-Substance- Abuse-Disorders.aspx. Published July 14, 2015. Accessed September 3, 2015.
What do you do when clozapine fails?
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Do you practice sophisticated psychiatry? 10 Proposed foundations of advanced care
Some psychiatrists are rapid adopters of the latest discoveries. Others wait before they adopt new modalities and change their practice accordingly. Then, there are some—admittedly, a minority—who stubbornly persist in practicing exactly as they did 30 or 40 years ago when they completed residency.
What are the foundations of exemplary, advanced, brain-based psychiatric care?
Here are my 10 proposed tenets of excellence in psychiatric practice. They reflect superior assessment and management of patients as well as personal growth and contributions to the specialty.
Provide a complete medical assessment for every patient at the first lifetime psychiatric contact, whether inpatient or outpatient. This includes routine physical and neurologic examinations and a panel of basic laboratory tests (complete blood count, liver and kidney functions, urine screen, thyroid-stimulating hormone, electrolytes, fasting glucose, and fasting lipids). All vital signs are measured and recorded. Referrals to other medical specialists are made as needed.
This medical assessment must, of course, include a comprehensive psychiatric evaluation: personal history, social history, medical history, family history, and a complete neuropsychiatric mental status examination.
Create a thorough 3-generation pedigree of all relatives, indicating not only psychopathology, addiction, and legal problems but also medical (especially neurologic) disorders and cause of death.
Perform basic assessment of brain structure and function (a MRI scan, a neurocognitive battery, and tests of neurologic soft signs).
Measure biomarkers that reflect potential harm to the brain according to emerging research—eg, pro-inflammatory markers (such as C-reactive protein [CRP], interleukin-6, and tumor necrosis factor alpha [TNF-α]) and oxidative stress biomarkers of increased free radical activity (superoxide dismutase [SOD], glutathione, thiobarbituric acid [GSH] reactive substances [TBARS], and catalase).
Maintain measurement-based practice, in which:
• severity of illness is measured by a specific, appropriate rating scale (eg, Positive and Negative Syndrome Scale for schizophrenia [PANSS], Young Mania Rating Scale [YMRS], Montgomery-Åsberg Depression Rating Scale [MADRS] for depression, Hamilton Anxiety Rating Scale [HAM-A] for anxiety, Yale-Brown Obsessive Compulsive Scale [Y-BOCS] for obsessions and compulsions)
• degree of response to treatment is measured as a reflection of the extent of drop in the total score of those rating scales, which are administered at every visit
• severity of common side effects is measured by the Simpson-Angus Scale (SAS) for parkinsonism, the Barnes Akathisia Rating Scale (BARS), the Abnormal Involuntary Movement Scale (AIMS) for tardive dyskinesia, the Glasgow Antipsychotic Side-effect Scale (GASS), etc.
Use tier-1 evidence-based psychiatry (that is, findings from large, placebo-controlled, double-blind studies) to select best treatments. This includes being familiar with:
• principles of meta-analysis
• the meaning of low, medium, and large effect sizes
• for every medication used, the calculation and clinical implications of number needed to treat (NNT) and number needed to harm (NNH).
Always combine the dual management approaches of pharmacotherapy plus psychotherapy/psychosocial therapy.
Share knowledge and experience gleaned from practice with the community of psychiatrists, including:
• writing letters to the editor about a clinical matter
• submitting case reports or case series for publication
• teaching students or residents at the local medical school (after obtaining adjunct faculty status).
In addition, psychiatrists should educate the public to eliminate misperceptions and erase stigma about mental illness.
Participate in creating new psychiatric knowledge by developing skills to become a clinical trialist, so that you can participate as an investigator in multicenter clinical trials of new medications, or, at least, refer patients for possible participation in ongoing clinical trials conducted at local academic centers.
Engage in effective and continuous life-learning, by:
• attending weekly Grand Rounds at the nearest academic department of psychiatry
• attending national continuing medical education conferences annually
• scanning PubMed regularly (at least 3 times a week, if not daily) for the latest research related to one’s patients or to read about advances in one’s clinical subspecialty; read the abstracts and download several PDFs a week for subsequent reading.
Some readers will agree with part, but not all, of these proposed components of advanced psychiatric practice. That’s to be expected; I welcome your letters rebutting some tenets, or proposing additional ones, of a sophisticated psychiatric practice. After all, sophistication is a journey, not a destination.
Some psychiatrists are rapid adopters of the latest discoveries. Others wait before they adopt new modalities and change their practice accordingly. Then, there are some—admittedly, a minority—who stubbornly persist in practicing exactly as they did 30 or 40 years ago when they completed residency.
What are the foundations of exemplary, advanced, brain-based psychiatric care?
Here are my 10 proposed tenets of excellence in psychiatric practice. They reflect superior assessment and management of patients as well as personal growth and contributions to the specialty.
Provide a complete medical assessment for every patient at the first lifetime psychiatric contact, whether inpatient or outpatient. This includes routine physical and neurologic examinations and a panel of basic laboratory tests (complete blood count, liver and kidney functions, urine screen, thyroid-stimulating hormone, electrolytes, fasting glucose, and fasting lipids). All vital signs are measured and recorded. Referrals to other medical specialists are made as needed.
This medical assessment must, of course, include a comprehensive psychiatric evaluation: personal history, social history, medical history, family history, and a complete neuropsychiatric mental status examination.
Create a thorough 3-generation pedigree of all relatives, indicating not only psychopathology, addiction, and legal problems but also medical (especially neurologic) disorders and cause of death.
Perform basic assessment of brain structure and function (a MRI scan, a neurocognitive battery, and tests of neurologic soft signs).
Measure biomarkers that reflect potential harm to the brain according to emerging research—eg, pro-inflammatory markers (such as C-reactive protein [CRP], interleukin-6, and tumor necrosis factor alpha [TNF-α]) and oxidative stress biomarkers of increased free radical activity (superoxide dismutase [SOD], glutathione, thiobarbituric acid [GSH] reactive substances [TBARS], and catalase).
Maintain measurement-based practice, in which:
• severity of illness is measured by a specific, appropriate rating scale (eg, Positive and Negative Syndrome Scale for schizophrenia [PANSS], Young Mania Rating Scale [YMRS], Montgomery-Åsberg Depression Rating Scale [MADRS] for depression, Hamilton Anxiety Rating Scale [HAM-A] for anxiety, Yale-Brown Obsessive Compulsive Scale [Y-BOCS] for obsessions and compulsions)
• degree of response to treatment is measured as a reflection of the extent of drop in the total score of those rating scales, which are administered at every visit
• severity of common side effects is measured by the Simpson-Angus Scale (SAS) for parkinsonism, the Barnes Akathisia Rating Scale (BARS), the Abnormal Involuntary Movement Scale (AIMS) for tardive dyskinesia, the Glasgow Antipsychotic Side-effect Scale (GASS), etc.
Use tier-1 evidence-based psychiatry (that is, findings from large, placebo-controlled, double-blind studies) to select best treatments. This includes being familiar with:
• principles of meta-analysis
• the meaning of low, medium, and large effect sizes
• for every medication used, the calculation and clinical implications of number needed to treat (NNT) and number needed to harm (NNH).
Always combine the dual management approaches of pharmacotherapy plus psychotherapy/psychosocial therapy.
Share knowledge and experience gleaned from practice with the community of psychiatrists, including:
• writing letters to the editor about a clinical matter
• submitting case reports or case series for publication
• teaching students or residents at the local medical school (after obtaining adjunct faculty status).
In addition, psychiatrists should educate the public to eliminate misperceptions and erase stigma about mental illness.
Participate in creating new psychiatric knowledge by developing skills to become a clinical trialist, so that you can participate as an investigator in multicenter clinical trials of new medications, or, at least, refer patients for possible participation in ongoing clinical trials conducted at local academic centers.
Engage in effective and continuous life-learning, by:
• attending weekly Grand Rounds at the nearest academic department of psychiatry
• attending national continuing medical education conferences annually
• scanning PubMed regularly (at least 3 times a week, if not daily) for the latest research related to one’s patients or to read about advances in one’s clinical subspecialty; read the abstracts and download several PDFs a week for subsequent reading.
Some readers will agree with part, but not all, of these proposed components of advanced psychiatric practice. That’s to be expected; I welcome your letters rebutting some tenets, or proposing additional ones, of a sophisticated psychiatric practice. After all, sophistication is a journey, not a destination.
Some psychiatrists are rapid adopters of the latest discoveries. Others wait before they adopt new modalities and change their practice accordingly. Then, there are some—admittedly, a minority—who stubbornly persist in practicing exactly as they did 30 or 40 years ago when they completed residency.
What are the foundations of exemplary, advanced, brain-based psychiatric care?
Here are my 10 proposed tenets of excellence in psychiatric practice. They reflect superior assessment and management of patients as well as personal growth and contributions to the specialty.
Provide a complete medical assessment for every patient at the first lifetime psychiatric contact, whether inpatient or outpatient. This includes routine physical and neurologic examinations and a panel of basic laboratory tests (complete blood count, liver and kidney functions, urine screen, thyroid-stimulating hormone, electrolytes, fasting glucose, and fasting lipids). All vital signs are measured and recorded. Referrals to other medical specialists are made as needed.
This medical assessment must, of course, include a comprehensive psychiatric evaluation: personal history, social history, medical history, family history, and a complete neuropsychiatric mental status examination.
Create a thorough 3-generation pedigree of all relatives, indicating not only psychopathology, addiction, and legal problems but also medical (especially neurologic) disorders and cause of death.
Perform basic assessment of brain structure and function (a MRI scan, a neurocognitive battery, and tests of neurologic soft signs).
Measure biomarkers that reflect potential harm to the brain according to emerging research—eg, pro-inflammatory markers (such as C-reactive protein [CRP], interleukin-6, and tumor necrosis factor alpha [TNF-α]) and oxidative stress biomarkers of increased free radical activity (superoxide dismutase [SOD], glutathione, thiobarbituric acid [GSH] reactive substances [TBARS], and catalase).
Maintain measurement-based practice, in which:
• severity of illness is measured by a specific, appropriate rating scale (eg, Positive and Negative Syndrome Scale for schizophrenia [PANSS], Young Mania Rating Scale [YMRS], Montgomery-Åsberg Depression Rating Scale [MADRS] for depression, Hamilton Anxiety Rating Scale [HAM-A] for anxiety, Yale-Brown Obsessive Compulsive Scale [Y-BOCS] for obsessions and compulsions)
• degree of response to treatment is measured as a reflection of the extent of drop in the total score of those rating scales, which are administered at every visit
• severity of common side effects is measured by the Simpson-Angus Scale (SAS) for parkinsonism, the Barnes Akathisia Rating Scale (BARS), the Abnormal Involuntary Movement Scale (AIMS) for tardive dyskinesia, the Glasgow Antipsychotic Side-effect Scale (GASS), etc.
Use tier-1 evidence-based psychiatry (that is, findings from large, placebo-controlled, double-blind studies) to select best treatments. This includes being familiar with:
• principles of meta-analysis
• the meaning of low, medium, and large effect sizes
• for every medication used, the calculation and clinical implications of number needed to treat (NNT) and number needed to harm (NNH).
Always combine the dual management approaches of pharmacotherapy plus psychotherapy/psychosocial therapy.
Share knowledge and experience gleaned from practice with the community of psychiatrists, including:
• writing letters to the editor about a clinical matter
• submitting case reports or case series for publication
• teaching students or residents at the local medical school (after obtaining adjunct faculty status).
In addition, psychiatrists should educate the public to eliminate misperceptions and erase stigma about mental illness.
Participate in creating new psychiatric knowledge by developing skills to become a clinical trialist, so that you can participate as an investigator in multicenter clinical trials of new medications, or, at least, refer patients for possible participation in ongoing clinical trials conducted at local academic centers.
Engage in effective and continuous life-learning, by:
• attending weekly Grand Rounds at the nearest academic department of psychiatry
• attending national continuing medical education conferences annually
• scanning PubMed regularly (at least 3 times a week, if not daily) for the latest research related to one’s patients or to read about advances in one’s clinical subspecialty; read the abstracts and download several PDFs a week for subsequent reading.
Some readers will agree with part, but not all, of these proposed components of advanced psychiatric practice. That’s to be expected; I welcome your letters rebutting some tenets, or proposing additional ones, of a sophisticated psychiatric practice. After all, sophistication is a journey, not a destination.
Managing first-episode psychosis: Rationale and evidence for nonstandard first-line treatments for schizophrenia
First-episode psychosis (FEP) in schizophrenia is characterized by high response rates to antipsychotic therapy, followed by frequent antipsychotic discontinuation and elevated relapse rates soon after maintenance treatment begins.1,2 With subsequent episodes, time to response progressively increases and likelihood of response decreases.3,4
To address these issues, this article—the second of 2 parts5—describes the rationale and evidence for using nonstandard first-line antipsychotic therapies to manage FEP. Specifically, we discuss when clinicians might consider monotherapy exceeding FDA-approved maximum dosages, combination therapy, long-acting injectable antipsychotics (LAIA), or clozapine.
Monotherapy beyond FDA-approved dosages
Treatment guidelines for FEP recommend oral antipsychotic dosages in the lower half of the treatment range and lower than those that are required for multi-episode schizophrenia.6-16 Ultimately, clinicians prescribe individualized dosages for their patients based on symptom improvement and tolerability. The optimal dosage at which to achieve a favorable D2 receptor occupancy likely will vary from patient to patient.17
To control symptoms, higher dosages may be needed than those used in FEP clinical trials, recommended by guidelines for FEP or multi-episode patients, or approved by the FDA. Patients seen in everyday practice may be more complicated (eg, have a comorbid condition or history of nonresponse) than study populations. Higher dosages also may be reasonable to overcome drug−drug interactions (eg, cigarette smoking-mediated cytochrome P450 1A2 induction, resulting in increased olanzapine metabolism),18 or to establish antipsychotic failure if adequate trials at lower dosages have resulted in a suboptimal response and the patient is not experiencing tolerability or safety concerns.
In a study of low-, full-, and high-dosage antipsychotic therapy in FEP, an additional 15% of patients responded to higher dosages of olanzapine and risperidone after failing to respond to a standard dosage.19 A study of data from the Recovery After an Initial Schizophrenia Episode Project’s Early Treatment Program (RAISE-ETP) found that, of participants identified who may benefit from therapy modification, 8.8% were prescribed an antipsychotic (often, olanzapine, risperidone, and haloperidol) at a higher-than-recommended dosage.20 Of note, only olanzapine was prescribed at higher than FDA-approved dosages.
Antipsychotic combination therapy
Prescribing combinations of antipsychotics—antipsychotic polypharmacy (APP)— has a negative connotation because of limited efficacy and safety data,21 and limited endorsement in schizophrenia treatment guidelines.9,13 Caution with APP is warranted; a complex medication regimen may increase the potential for adverse effects, poorer adherence, and adverse drug-drug interactions.9 APP has been shown to independently predict both shorter treatment duration and discontinuation before 1 year.22
Nonetheless, the clinician and patient may share the decision to implement APP and observe whether benefits outweigh risks in situations such as:
• to optimize neuroreceptor occupancy and targets (eg, attempting to achieve adequate D2 receptor blockade while minimizing side effects secondary to binding other receptors)
• to manage co-existing symptom domains (eg, mood changes, aggression, negative symptoms, disorganization, and cognitive deficits)
• to mitigate antipsychotic-induced side effects (eg, initiating aripiprazole to treat hyperprolactinemia induced by another antipsychotic to which the patient has achieved a favorable response).23
Clinicians report using APP to treat as many as 50% of patients with a history of multiple psychotic episodes.23 For FEP patients, 23% of participants in the RAISE-ETP trial who were identified as possibly benefiting from therapy modification were prescribed APP.20 Regrettably, researchers have not found evidence to support a reported rationale for using APP—that lower dosages of individual antipsychotics when used in combination may avoid high-dosage prescriptions.24
Before implementing APP, thoroughly explore and manage reasons for a patient’s suboptimal response to monotherapy.25 An adequate trial with any antipsychotic should be at the highest tolerated dosage for 12 to 16 weeks. Be mindful that response to an APP trial may be the result of additional time on the original antipsychotic.
Long-acting injectable antipsychotics in FEP
Guideline recommendations. Most older guidelines for schizophrenia treatment suggest LAIA after multiple relapses related to medication nonadherence or when a patient prefers injected medication (Table 1).6-13 Expert consensus guidelines also recommend considering LAIA in patients who lack insight into their illness. The Texas Medication Algorithm Project (TMAP) guidelines7 state LAIA can be considered for inadequate adherence at any stage, whereas the 2010 British Association for Psychopharmacology (BAP) guidelines9 express uncertainty about their use in FEP, because of limited evidence. Both the BAP and National Institute for Health and Care Excellence guidelines13 urge clinicians to consider LAIA when avoiding nonadherence is a treatment priority.
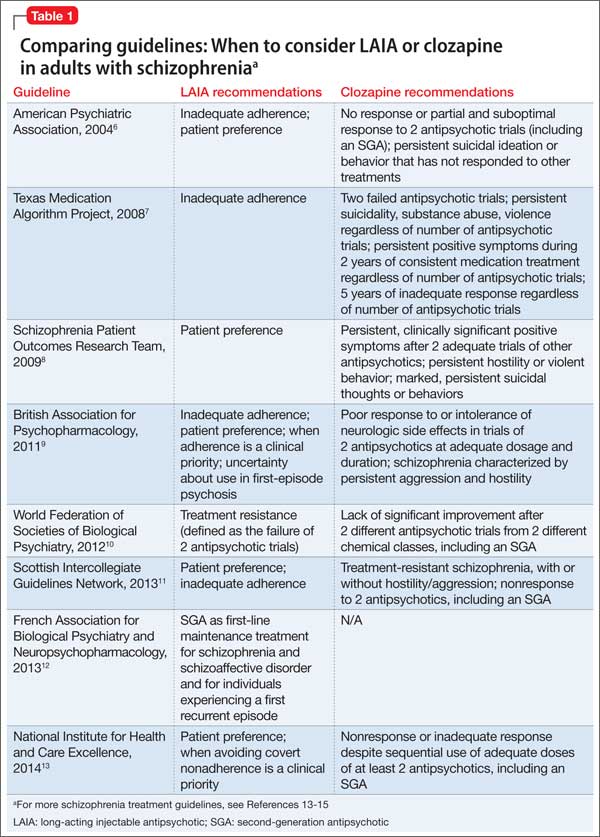
Recently, the French Association for Biological Psychiatry and Neuro-psychopharmacology (AFPBN) created expert consensus guidelines12 on using LAIA in practice. They recommend long-acting injectable second-generation antipsychotics (SGAs) as first-line maintenance treatment for schizophrenia and schizoaffective disorder and for individuals experiencing a first recurrent episode. The World Federation of Societies of Biological Psychiatry guidelines contain LAIA dosage recommendations for FEP (Table 2).10

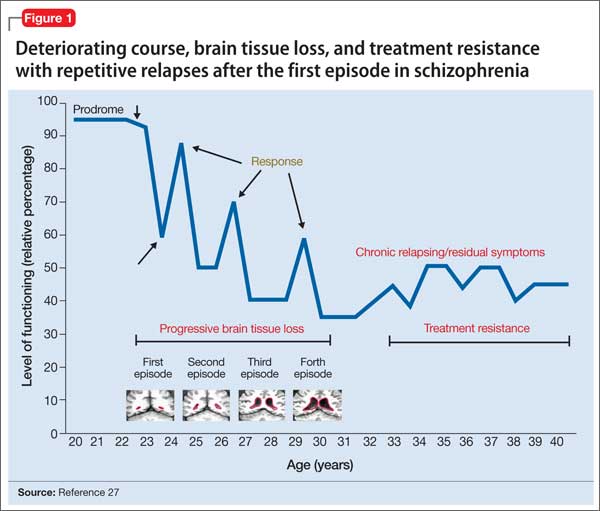
Advances have been made in understanding the serious neurobiological adverse effects of psychotic relapses, including neuroinflammation and oxidative stress, that may explain the atrophic changes observed with psychotic episodes starting with the FEP. Protecting the patient from a second episode has become a vital therapeutic management goal26 (Figure 127).
Concerns. Compared with oral antipsychotics, LAIA offers clinical advantages:
• improved pharmacokinetic profile (lower “peaks” and higher “valleys”)
• more consistent plasma concentrations (no variability related to administration timing or food effects)
• no first-pass metabolism, which can ease the process of finding the lowest effective and safe dosage
• reduced administration burden and objective tracking of adherence with typical dosing every 2 to 4 weeks
• less stigmatizing than oral medication for FEP patients, such as college students living in a dormitory.28,29
Barriers to LAIA use include:
• slow dosage titration and increased time to reach steady state drug level
• oral supplementation for some (eg, risperidone microspheres and aripiprazole long-acting injectable)
• logistical challenges for some (eg, 3-hour post-injection monitoring for delirium sedation syndrome with olanzapine pamoate)
• additional planning to coordinate care for scheduled injections
• higher expenses up front
• local injection site reactions
• dosage adjustment difficulties if adverse effects occur.28,29
Adoption rates of LAIA are low, especially for FEP.30 Most surveys indicate that (1) physicians believe LAIA treatment is ineffective for FEP31 and (2) patients do not prefer injectable to oral antipsychotics,32 despite evidence to the contrary.33,34 A survey of 198 psychiatrists identified 3 factors that influenced their decisions against using LAIA patients with FEP:
• limited availability of SGA depot formulations (4, to date, in the United States)
• frequent rejection by the patient when LAIA is offered without adequate explanation or encouragement
• skepticism of FEP patients (and their family) who lack experience with relapse.35
In reality, when SGA depots were introduced in the United Kingdom, prescribing rates of LAIA did not increase. As for patient rejection being a major reason for not prescribing LAIA, few patients (5% to 36%) are offered depot injections, particularly in FEP.29 Most patients using LAIA are chronic, multi-episode, violent people who are receiving medications involuntarily.29 Interestingly, this survey did not find 2 factors to be influential in psychiatrists’ decision not to use LAIA in FEP:
• guidelines do not explicitly recommend depot treatment in FEP
• treatment in FEP may be limited to 1 year, therefore depot administration is not worthwhile.35
Preliminary evidence. At least a dozen studies have explored LAIA treatment for FEP, with the use of fluphenazine decanoate,36 perphenazine enanthate37 (discontinued), and risperidone microspheres.37-48 The research demonstrates the efficacy and safety of LAIA in FEP as measured by these endpoints:
• improved symptom control38,40-43,46,48
• adherence43,44,48
• reduced relapse rates37,43 and rehospitalizations37,47
• lesser reductions in white matter brain volume45
• no differences in extrapyramidal side effects or prolactin-associated adverse effects.48
A few small studies demonstrate significant differences in outcomes between risperidone LAIA and oral comparator groups (Table 3).43-45 Ongoing studies of LAIA use in FEP are comparing paliperidone palmitate with risperidone microspheres and other oral antipsychotics.49-51 No studies are examining olanzapine pamoate in FEP, likely because several guidelines do not recommended its use. No studies have been published regarding aripiprazole long-acting injectable in FEP. This LAIA formulation was approved in February 2013, and robust studies of the oral formulation in FEP are limited.52

Discussion and recommendations. Psychiatrists relying on subjective measures of antipsychotic adherence may inaccurately assess whether patients meet this criterion for LAIA use.53 LAIA could combat the high relapse rate in FEP, yet depot antipsychotics are prescribed infrequently for FEP patients (eg, for only 9.5% of participants in the RAISE-ETP study).20 Most schizophrenia treatment guidelines do not discuss LAIA use specifically in FEP, although the AFPBN expert consensus guidelines published in 2013 do recommend SGA depot formulations in FEP.12 SGA LAIA may be preferable, given its neuroprotective effects, in contrast to the neurotoxicity concerns of FGA LAIA.54,55
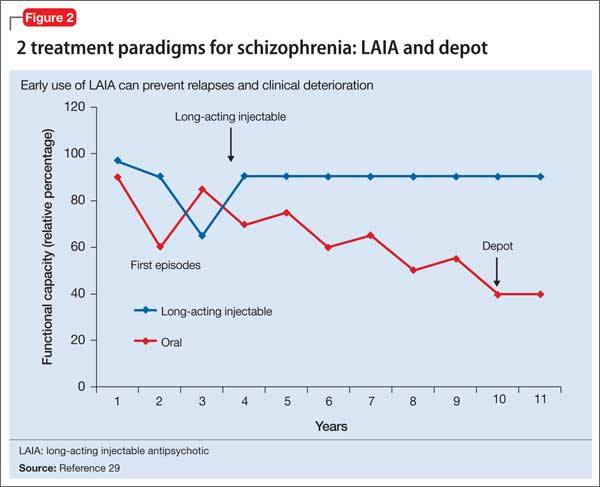
Relapses begin within a few months of illness stabilization after FEP, and >50% of patients relapse within 1 or 2 years2—the recommended minimum treatment duration for FEP.8,9,13 The use of LAIA is advisable in any patient with schizophrenia for whom long-term antipsychotic therapy is indicated.56 LAIA administration requirements objectively track medication adherence, which allows clinicians to be proactive in relapse prevention. Not using an intervention in FEP that improves adherence and decreases relapse rates contradicts our goal of instituting early, effective treatment to improve long-term functional outcomes (Figure 2).29
Considering clozapine in FEP
Guideline recommendations. Schizo-phrenia treatment guidelines and FDA labeling57 reserve clozapine for third-line treatment of refractory schizophrenia after 2 adequate antipsychotic trials have failed despite optimal dosing (Table 1).6-13 Some guidelines specify 1 of the 2 failed antipsychotic trials must include an SGA.6,7,10,11,13-16 Most say clozapine may be considered in patients with chronic aggression or hostility,7-9,14,16 or suicidal thoughts and behaviors.6-8,14,16 TMAP guidelines recommend a clozapine trial with concomitant substance abuse, persistent positive symptoms during 2 years of consistent medication treatment, and after 5 years of inadequate response (“treatment resistance”), regardless of the number of antipsychotic trials.7
Rationale and concerns. Clozapine is a superior choice for treatment-refractory delusions or hallucinations of schizophrenia, because it markedly enhances the response rate to antipsychotic therapy.58 Researchers therefore have investigated whether clozapine, compared with other antipsychotics, would yield more favorable initial and long-term outcomes when used first-line in FEP.
Preliminary evidence. Five studies have explored the use of clozapine as first-line therapy in FEP (Table 4).59-63 Interpreting the results is difficult because clozapine trials may be brief (mostly, 12 to 52 weeks); lack a comparator arm; suffer from a high attrition rate; enroll few patients; and lack potentially important outcome measures such as negative symptoms, suicidality, and functional assessment.
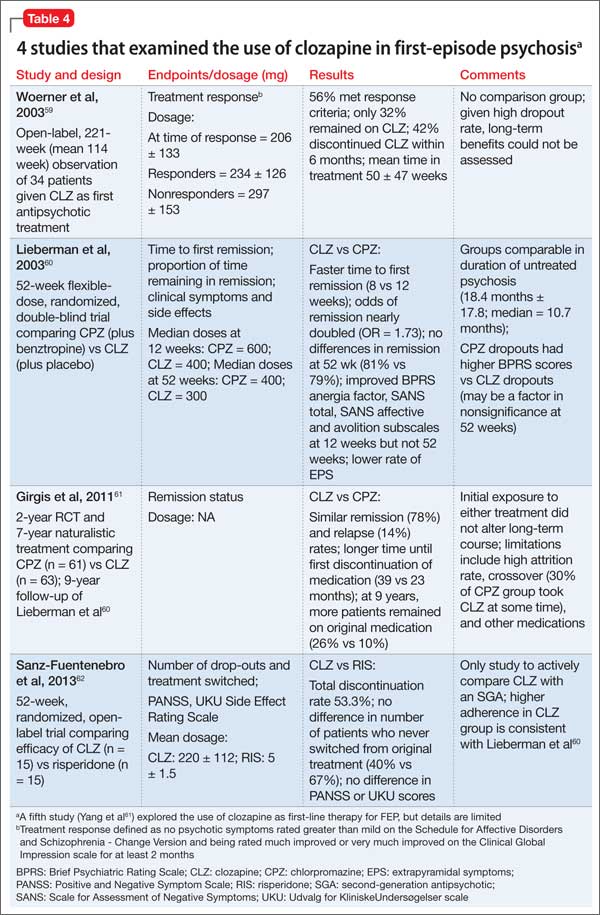
Overall, these studies demonstrate clozapine is as efficacious in this patient population as chlorpromazine (no difference in remission at 1-year, although clozapine-treated patients remitted faster and stayed in remission longer)60,61 or risperidone (no difference in Positive and Negative Syndrome Scale scores).62
At present, clozapine has not been shown superior to other antipsychotics as a first-line treatment for FEP. Research does underscore the importance of a clozapine trial as third-line treatment for FEP patients who have not responded well to 2 SGA trials.63 Many of these nonresponders (77%) have demonstrated a favorable response when promptly switched to clozapine.64
Discussion and recommendations. The limited evidence argues against using clozapine earlier than as third-line treatment in FEP. Perhaps the high treatment response that characterizes FEP creates a ceiling effect that obscures differences in antipsychotic efficacy at this stage.65 Clozapine use as first-line treatment should be re-evaluated with more robust methodology. One approach could be to assess its benefit in FEP by the duration of untreated psychosis.
The odds of achieving remission have been shown to decrease by 15% for each year that psychosis has not been treated.59 Studies exploring the use of clozapine as a second-line agent for FEP also are warranted, as antipsychotic response during subsequent trials is substantially reduced. In fact, the Scottish Intercollegiate Guidelines Network guidelines recommend this as an area for future research.11
For now, clozapine should continue to be reserved as second- or third-line treatment in a patient with FEP. The risks of clozapine’s potentially serious adverse effects (eg, agranulocytosis, seizures, obesity, diabetes, dyslipidemia, myocarditis, pancreatitis, hypotension, sialorrhea, severe sedation, ileus) can be justified only in the treatment of severe and persistent psychotic symptoms.57
Bottom Line
Nonstandard use of antipsychotic monotherapy dosages beyond the approved FDA limit and combination antipsychotic therapy may be reasonable for select first-episode psychosis (FEP) patients. Strongly consider long-acting injectable antipsychotics in FEP to proactively combat the high relapse rate and more easily identify antipsychotic failure. Continue to use clozapine as second- or third-line therapy in FEP: Studies have not found that it is more efficacious than other antipsychotics for first-line use.
Related Resource
• Recovery After an Initial Schizophrenia Episode (RAISE) Project Early Treatment Program. National Institute of Mental Health. http://raiseetp.org.
Drug Brand Names
Aripiprazole • Abilify, Abilify Maintena
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fluphenazine decanoate • Prolixin-D
Haloperidol • Haldol
Haloperidol decanoate • Haldol-D
Olanzapine • Zyprexa
Olanzapine pamoate • Zyprexa Relprevv
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone microspheres • Risperdal Consta
Disclosures
Dr. Gardner reports no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products. Dr. Nasrallah is a consultant to Acadia, Alkermes, Lundbeck, Janssen, Merck, Otsuka, and Sunovion, and is a speaker for Alkermes, Lundbeck, Janssen, Otsuka, and Sunovion.
1. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804.
2. Bradford DW, Perkins DO, Lieberman JA. Pharmacological management of first-episode schizophrenia and related nonaffective psychoses. Drugs. 2003;63(21):2265-2283.
3. Lieberman JA, Koreen AR, Chakos M, et al. Factors influencing treatment response and outcome of first-episode schizophrenia: implications for understanding the pathophysiology of schizophrenia. J Clin Psychiatry. 1996;57(suppl 9):5-9.
4. Agid O, Arenovich T, Sajeev G, et al. An algorithm-based approach to first-episode schizophrenia: response rates over 3 prospective antipsychotic trials with a retrospective data analysis. J Clin Psychiatry. 2011;72(11):1439-1444.
5. Gardner KN, Nasrallah HA. Managing first-episode psychosis. An early stage of schizophrenia with distinct treatment needs. Current Psychiatry. 2015;14(5):32-34,36-40,42.
6. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
7. Texas Department of State Health Services. Texas Medication Algorithm Project (TMAP) Procedural Manual. Schizophrenia Treatment Algorithms. http://www.jpshealthnet.org/sites/default/files/ tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed June 11, 2015.
8. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
9. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620.
10. Hasan A, Falkai P, Wobrok T, et al; WFSBP Task force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44.
11. Scottish Intercollegiate Guidelines Network. SIGN 131: Management of schizophrenia. http://www.sign.ac.uk/ pdf/sign131.pdf. Published March 2013. Accessed June 11, 2015.
12. Llorca PM, Abbar M, Courtet P, et al. Guidelines for the use and management of long-acting injectable antipsychotics in serous mental illness. BMC Psychiatry. 2013;13:340.
13. National Institute for Health and Care Excellence. NICE clinical guideline 178: Psychosis and schizophrenia in adults: treatment and management. https://www.nice.org. uk/guidance/cg178/resources/guidance-psychosis-and-schizophrenia-in-adults-treatment-and-management-pdf. Updated March 2014. Accessed June 16, 2015.
14. Canadian Psychiatric Association. Clinical practice guidelines. Treatment of schizophrenia. Can J Psychiatry. 2005;50(13 suppl 1):7S-57S.
15. McEvoy JP, Scheifler PL, Frances A. The expert consensus guideline series: treatment of schizophrenia. J Clin Psychiatry. 1999;60(suppl 11):3-80.
16. Marder SR, Essock SM, Miller AL, et al. The Mount Sinai conference on the pharmacotherapy of schizophrenia. Schizophr Bull. 2002;28(1):5-16.
17. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157(4):514-520.
18. Fankhauser MP. Drug interactions with tobacco smoke: implications for patient care. Current Psychiatry. 2013;12(1):12-16.
19. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022.
20. Robinson DG, Schooler NR, John M, et al. Prescription practices in the treatment of first-episode schizophrenia spectrum disorders: data from the national RAISE-ETP study. Am J Psychiatry. 2015;172(3):237-248.
21. Correll CU, Rummel-Kluge C, Corves C, et al. Antipsychotic combinations vs monotherapy in schizophrenia: a meta-analysis of randomized controlled trials. Schizophr Bull. 2009;35(2):443-457.
22. Fisher MD, Reilly K, Isenberg K, et al. Antipsychotic patterns of use in patients with schizophrenia: polypharmacy versus monotherapy. BMC Psychiatry. 2014;14(1):341.
23. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
24. John AP, Dragovic M. Antipsychotic polypharmacy is not associated with reduced dose of individual antipsychotics in schizophrenia. J Clin Psychopharmacol. 2015;35(2):193-195.
25. Nasrallah HA. Treatment-resistant schizophrenia. Current Psychiatry. http://www.currentpsychiatry.com/specialty-focus/schizophrenia-other-psychotic-disorders/article/ treatment-resistant-schizophrenia/9be7bba3713d4a4cd68aa 8c92b79e5b1.html. Accessed June 16, 2015.
26. Alvarez-Jiménez M, Parker AG, Hetrick SE, et al. Preventing the second episode: a systematic review and meta-analysis of psychosocial and pharmacological trials in first-episode psychosis. Schizophr Bull. 2011;37(3):619-630.
27. Nasrallah HA, Smeltzer DJ. Contemporary diagnosis and management of the patient with schizophrenia. 2nd ed. Newton, PA: Handbooks in Health Care Co; 2011.
28. McEvoy JP. Risks versus benefits of different types of long-acting injectable antipsychotics. J Clin Psychiatry. 2006;67(suppl 5):15-18.
29. Agid O, Foussias G, Remington G. Long-acting injectable antipsychotics in the treatment of schizophrenia: their role in relapse prevention. Expert Opin Pharmacother. 2010;11(14):2301-2317.
30. Kirschner M, Theodoridou A, Fusar-Poli P, et al. Patients’ and clinicians’ attitude towards long-acting depot antipsychotics in subjects with a first episode psychosis. Ther Adv Psychophamacol. 2013;3(2):89-99.
31. Heres S, Hamann J, Mendel R, et al. Identifying the profile of optimal candidates for antipsychotic depot therapy: A cluster analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1987-1993.
32. Heres S, Lambert M, Vauth R. Treatment of early episode in patents with schizophrenia: the role of long acting antipsychotics. Eur Psychiatry. 2014;29(suppl 2):1409-1413.
33. Heres S, Schmitz FS, Leucht S, et al. The attitude of patients towards antipsychotic depot treatment. Int Clin Psychopharmacol. 2007;22(5):275-282.
34. Weiden PJ, Schooler NR, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.
35. Heres S, Reichhart T, Hamann J, et al. Psychiatrists’ attitude to antipsychotic depot treatment in patients with first-episode schizophrenia. Eur Psychiatry. 2011;26(5):297-301.
36. Kane JM, Rifkin A, Quitkin F, et al. Fluphenazine vs placebo in patients with remitted, acute first-episode schizophrenia. Arch Gen Psychiatry. 1982;39(1):70-73.
37. Tiihonen J, Wahlbeck K, Lönnqvist J, et al. Effectiveness of antipsychotic treatments in a nationwide cohort of patients in a community care after first hospitalization due to schizophrenia and schizoaffective disorder: observational follow-up study. BMJ. 2006;333(7561):224.
38. Parellada E, Andrezina R, Milanova V, et al. Patients in the early phases of schizophrenia and schizoaffective disorders effectively treated with risperidone long-acting injectable. J Psychopharmacol. 2005;19(suppl 5):5-14.
39. Malla A, Binder C, Chue P. Comparison of long-acting injectable risperidone and oral novel antipsychotic drugs for treatment in early phase of schizophrenia spectrum psychosis. Proceedings of the 61st Annual Convention Society of Biological Psychiatry; Toronto, Canada; 2006.
40. Lasser RA, Bossie CA, Zhu Y, et al. Long-acting risperidone in young adults with early schizophrenia or schizoaffective illness. Ann Clin Psychiatry. 2007;19(2):65-71.
41. Emsley R, Oosthuizen P, Koen L, et al. Remission in patients with first-episode schizophrenia receiving assured antipsychotic medication: a study with risperidone long-acting injection. Int Clin Psychopharmacol. 2008;23(6):325-331.
42. Emsley R, Oosthuizen P, Koen L, et al. Oral versus injectable antipsychotic treatment in early psychosis: post hoc comparison of two studies. Clin Ther. 2008;30(12):2378-2386.
43. Kim B, Lee SH, Choi TK, et al. Effectiveness of risperidone long-acting injection in first-episode schizophrenia: in naturalistic setting. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1231-1235.
44. Weiden PJ, Schooler NJ, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.
45. Bartzokis G, Lu PH, Amar CP, et al. Long acting injection versus oral risperidone in first-episode schizophrenia: differential impact on white matter myelination trajectory. Schizophr Res. 2011;132(1):35-41.
46. Napryeyenko O, Burba B, Martinez G, et al. Risperidone long-acting injectable in recent-onset schizophrenia examined with clinician and patient self-report measures. J Clin Psychopharmacol. 2010;30(2):200-202.
47. Tiihonen J, Haukka J, Taylor M, et al. A nationwide cohort study of oral and depot antipsychotics after first hospitalization for schizophrenia. Am J Psychiatry. 2011;168(6):603-609.
48. Dubois V, Megens J, Mertens C, et al. Long-acting risperidone in early-episode schizophrenia. Acta Psychiatrica Belgica. 2011;111(1):9-21.
49. ClinicalTrials.gov. Oral risperidone versus injectable paliperidone palmitate for treating first-episode schizophrenia. https://clinicaltrials.gov/ct2/show/ NCT01451736. Accessed June 16, 2015.
50. ClinicalTrials.gov. Brain myelination effects of paliperidone palmitate versus oral risperidone in first episode schizophrenia. https://clinicaltrials.gov/ct2/ show/NCT01458379. Accessed June 16, 2015.
51. ClinicalTrials.gov. Effects of paliperidone palmitate versus oral antipsychotics on clinical outcomes and MRI measures. https://clinicaltrials.gov/ct2/show/NCT01359293. Accessed June 16, 2016.
52. U.S. Food and Drug Administration. Drugs@FDA. http:// www.accessdata.fda.gov/scripts/cder/drugsatfda. Accessed January 11, 2015.
53. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):1-46; quiz 47-48.
54. Nandra KS, Agius M. The difference between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99.
55. Nasrallah HA. Haloperidol is clearly neurotoxic. Should it be banned? Current Psychiatry. 2013;12(7):7-8.
56. Kane JM, Garcia-Ribora C. Clinical guideline recommendations for antipsychotic long-acting injections. Br J Psychiatry. 2009;52:S63-S67.
57. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
58. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
59. Woerner MG, Robinson DG, Alvir JMJ, et al. Clozapine as a first treatment for schizophrenia. Am J Psychiatry. 2003;160(8):1514-1516.
60. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naive first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003.
61. Girgis RR, Phillips MR, Li X, et al. Clozapine v. chlorpromazine in treatment-naive, first-episode schizophrenia: 9-year outcomes of a randomised clinical trial. Br J Psychiatry. 2011;199(4):281-288.
62. Sanz-Fuentenebro J, Taboada D, Palomo T, et al. Randomized trial of clozapine vs. risperidone in treatment-naïve first-episode schizophrenia: results after one year. Schizophr Res. 2013;149(1-3):156-161.
63. Yang PD, Ji Z. The efficacy and related factors of clozapine on first-episode schizophrenia. Chin J Nerv Ment Dis. 1997;23:155-158.
64. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022.
65. Remington G, Agid O, Foussias G, et al. Clozapine’s role in the treatment of first-episode schizophrenia. Am J Psychiatry. 2013;170(2):146-151.
First-episode psychosis (FEP) in schizophrenia is characterized by high response rates to antipsychotic therapy, followed by frequent antipsychotic discontinuation and elevated relapse rates soon after maintenance treatment begins.1,2 With subsequent episodes, time to response progressively increases and likelihood of response decreases.3,4
To address these issues, this article—the second of 2 parts5—describes the rationale and evidence for using nonstandard first-line antipsychotic therapies to manage FEP. Specifically, we discuss when clinicians might consider monotherapy exceeding FDA-approved maximum dosages, combination therapy, long-acting injectable antipsychotics (LAIA), or clozapine.
Monotherapy beyond FDA-approved dosages
Treatment guidelines for FEP recommend oral antipsychotic dosages in the lower half of the treatment range and lower than those that are required for multi-episode schizophrenia.6-16 Ultimately, clinicians prescribe individualized dosages for their patients based on symptom improvement and tolerability. The optimal dosage at which to achieve a favorable D2 receptor occupancy likely will vary from patient to patient.17
To control symptoms, higher dosages may be needed than those used in FEP clinical trials, recommended by guidelines for FEP or multi-episode patients, or approved by the FDA. Patients seen in everyday practice may be more complicated (eg, have a comorbid condition or history of nonresponse) than study populations. Higher dosages also may be reasonable to overcome drug−drug interactions (eg, cigarette smoking-mediated cytochrome P450 1A2 induction, resulting in increased olanzapine metabolism),18 or to establish antipsychotic failure if adequate trials at lower dosages have resulted in a suboptimal response and the patient is not experiencing tolerability or safety concerns.
In a study of low-, full-, and high-dosage antipsychotic therapy in FEP, an additional 15% of patients responded to higher dosages of olanzapine and risperidone after failing to respond to a standard dosage.19 A study of data from the Recovery After an Initial Schizophrenia Episode Project’s Early Treatment Program (RAISE-ETP) found that, of participants identified who may benefit from therapy modification, 8.8% were prescribed an antipsychotic (often, olanzapine, risperidone, and haloperidol) at a higher-than-recommended dosage.20 Of note, only olanzapine was prescribed at higher than FDA-approved dosages.
Antipsychotic combination therapy
Prescribing combinations of antipsychotics—antipsychotic polypharmacy (APP)— has a negative connotation because of limited efficacy and safety data,21 and limited endorsement in schizophrenia treatment guidelines.9,13 Caution with APP is warranted; a complex medication regimen may increase the potential for adverse effects, poorer adherence, and adverse drug-drug interactions.9 APP has been shown to independently predict both shorter treatment duration and discontinuation before 1 year.22
Nonetheless, the clinician and patient may share the decision to implement APP and observe whether benefits outweigh risks in situations such as:
• to optimize neuroreceptor occupancy and targets (eg, attempting to achieve adequate D2 receptor blockade while minimizing side effects secondary to binding other receptors)
• to manage co-existing symptom domains (eg, mood changes, aggression, negative symptoms, disorganization, and cognitive deficits)
• to mitigate antipsychotic-induced side effects (eg, initiating aripiprazole to treat hyperprolactinemia induced by another antipsychotic to which the patient has achieved a favorable response).23
Clinicians report using APP to treat as many as 50% of patients with a history of multiple psychotic episodes.23 For FEP patients, 23% of participants in the RAISE-ETP trial who were identified as possibly benefiting from therapy modification were prescribed APP.20 Regrettably, researchers have not found evidence to support a reported rationale for using APP—that lower dosages of individual antipsychotics when used in combination may avoid high-dosage prescriptions.24
Before implementing APP, thoroughly explore and manage reasons for a patient’s suboptimal response to monotherapy.25 An adequate trial with any antipsychotic should be at the highest tolerated dosage for 12 to 16 weeks. Be mindful that response to an APP trial may be the result of additional time on the original antipsychotic.
Long-acting injectable antipsychotics in FEP
Guideline recommendations. Most older guidelines for schizophrenia treatment suggest LAIA after multiple relapses related to medication nonadherence or when a patient prefers injected medication (Table 1).6-13 Expert consensus guidelines also recommend considering LAIA in patients who lack insight into their illness. The Texas Medication Algorithm Project (TMAP) guidelines7 state LAIA can be considered for inadequate adherence at any stage, whereas the 2010 British Association for Psychopharmacology (BAP) guidelines9 express uncertainty about their use in FEP, because of limited evidence. Both the BAP and National Institute for Health and Care Excellence guidelines13 urge clinicians to consider LAIA when avoiding nonadherence is a treatment priority.
Recently, the French Association for Biological Psychiatry and Neuro-psychopharmacology (AFPBN) created expert consensus guidelines12 on using LAIA in practice. They recommend long-acting injectable second-generation antipsychotics (SGAs) as first-line maintenance treatment for schizophrenia and schizoaffective disorder and for individuals experiencing a first recurrent episode. The World Federation of Societies of Biological Psychiatry guidelines contain LAIA dosage recommendations for FEP (Table 2).10
Advances have been made in understanding the serious neurobiological adverse effects of psychotic relapses, including neuroinflammation and oxidative stress, that may explain the atrophic changes observed with psychotic episodes starting with the FEP. Protecting the patient from a second episode has become a vital therapeutic management goal26 (Figure 127).
Concerns. Compared with oral antipsychotics, LAIA offers clinical advantages:
• improved pharmacokinetic profile (lower “peaks” and higher “valleys”)
• more consistent plasma concentrations (no variability related to administration timing or food effects)
• no first-pass metabolism, which can ease the process of finding the lowest effective and safe dosage
• reduced administration burden and objective tracking of adherence with typical dosing every 2 to 4 weeks
• less stigmatizing than oral medication for FEP patients, such as college students living in a dormitory.28,29
Barriers to LAIA use include:
• slow dosage titration and increased time to reach steady state drug level
• oral supplementation for some (eg, risperidone microspheres and aripiprazole long-acting injectable)
• logistical challenges for some (eg, 3-hour post-injection monitoring for delirium sedation syndrome with olanzapine pamoate)
• additional planning to coordinate care for scheduled injections
• higher expenses up front
• local injection site reactions
• dosage adjustment difficulties if adverse effects occur.28,29
Adoption rates of LAIA are low, especially for FEP.30 Most surveys indicate that (1) physicians believe LAIA treatment is ineffective for FEP31 and (2) patients do not prefer injectable to oral antipsychotics,32 despite evidence to the contrary.33,34 A survey of 198 psychiatrists identified 3 factors that influenced their decisions against using LAIA patients with FEP:
• limited availability of SGA depot formulations (4, to date, in the United States)
• frequent rejection by the patient when LAIA is offered without adequate explanation or encouragement
• skepticism of FEP patients (and their family) who lack experience with relapse.35
In reality, when SGA depots were introduced in the United Kingdom, prescribing rates of LAIA did not increase. As for patient rejection being a major reason for not prescribing LAIA, few patients (5% to 36%) are offered depot injections, particularly in FEP.29 Most patients using LAIA are chronic, multi-episode, violent people who are receiving medications involuntarily.29 Interestingly, this survey did not find 2 factors to be influential in psychiatrists’ decision not to use LAIA in FEP:
• guidelines do not explicitly recommend depot treatment in FEP
• treatment in FEP may be limited to 1 year, therefore depot administration is not worthwhile.35
Preliminary evidence. At least a dozen studies have explored LAIA treatment for FEP, with the use of fluphenazine decanoate,36 perphenazine enanthate37 (discontinued), and risperidone microspheres.37-48 The research demonstrates the efficacy and safety of LAIA in FEP as measured by these endpoints:
• improved symptom control38,40-43,46,48
• adherence43,44,48
• reduced relapse rates37,43 and rehospitalizations37,47
• lesser reductions in white matter brain volume45
• no differences in extrapyramidal side effects or prolactin-associated adverse effects.48
A few small studies demonstrate significant differences in outcomes between risperidone LAIA and oral comparator groups (Table 3).43-45 Ongoing studies of LAIA use in FEP are comparing paliperidone palmitate with risperidone microspheres and other oral antipsychotics.49-51 No studies are examining olanzapine pamoate in FEP, likely because several guidelines do not recommended its use. No studies have been published regarding aripiprazole long-acting injectable in FEP. This LAIA formulation was approved in February 2013, and robust studies of the oral formulation in FEP are limited.52
Discussion and recommendations. Psychiatrists relying on subjective measures of antipsychotic adherence may inaccurately assess whether patients meet this criterion for LAIA use.53 LAIA could combat the high relapse rate in FEP, yet depot antipsychotics are prescribed infrequently for FEP patients (eg, for only 9.5% of participants in the RAISE-ETP study).20 Most schizophrenia treatment guidelines do not discuss LAIA use specifically in FEP, although the AFPBN expert consensus guidelines published in 2013 do recommend SGA depot formulations in FEP.12 SGA LAIA may be preferable, given its neuroprotective effects, in contrast to the neurotoxicity concerns of FGA LAIA.54,55
Relapses begin within a few months of illness stabilization after FEP, and >50% of patients relapse within 1 or 2 years2—the recommended minimum treatment duration for FEP.8,9,13 The use of LAIA is advisable in any patient with schizophrenia for whom long-term antipsychotic therapy is indicated.56 LAIA administration requirements objectively track medication adherence, which allows clinicians to be proactive in relapse prevention. Not using an intervention in FEP that improves adherence and decreases relapse rates contradicts our goal of instituting early, effective treatment to improve long-term functional outcomes (Figure 2).29
Considering clozapine in FEP
Guideline recommendations. Schizo-phrenia treatment guidelines and FDA labeling57 reserve clozapine for third-line treatment of refractory schizophrenia after 2 adequate antipsychotic trials have failed despite optimal dosing (Table 1).6-13 Some guidelines specify 1 of the 2 failed antipsychotic trials must include an SGA.6,7,10,11,13-16 Most say clozapine may be considered in patients with chronic aggression or hostility,7-9,14,16 or suicidal thoughts and behaviors.6-8,14,16 TMAP guidelines recommend a clozapine trial with concomitant substance abuse, persistent positive symptoms during 2 years of consistent medication treatment, and after 5 years of inadequate response (“treatment resistance”), regardless of the number of antipsychotic trials.7
Rationale and concerns. Clozapine is a superior choice for treatment-refractory delusions or hallucinations of schizophrenia, because it markedly enhances the response rate to antipsychotic therapy.58 Researchers therefore have investigated whether clozapine, compared with other antipsychotics, would yield more favorable initial and long-term outcomes when used first-line in FEP.
Preliminary evidence. Five studies have explored the use of clozapine as first-line therapy in FEP (Table 4).59-63 Interpreting the results is difficult because clozapine trials may be brief (mostly, 12 to 52 weeks); lack a comparator arm; suffer from a high attrition rate; enroll few patients; and lack potentially important outcome measures such as negative symptoms, suicidality, and functional assessment.
Overall, these studies demonstrate clozapine is as efficacious in this patient population as chlorpromazine (no difference in remission at 1-year, although clozapine-treated patients remitted faster and stayed in remission longer)60,61 or risperidone (no difference in Positive and Negative Syndrome Scale scores).62
At present, clozapine has not been shown superior to other antipsychotics as a first-line treatment for FEP. Research does underscore the importance of a clozapine trial as third-line treatment for FEP patients who have not responded well to 2 SGA trials.63 Many of these nonresponders (77%) have demonstrated a favorable response when promptly switched to clozapine.64
Discussion and recommendations. The limited evidence argues against using clozapine earlier than as third-line treatment in FEP. Perhaps the high treatment response that characterizes FEP creates a ceiling effect that obscures differences in antipsychotic efficacy at this stage.65 Clozapine use as first-line treatment should be re-evaluated with more robust methodology. One approach could be to assess its benefit in FEP by the duration of untreated psychosis.
The odds of achieving remission have been shown to decrease by 15% for each year that psychosis has not been treated.59 Studies exploring the use of clozapine as a second-line agent for FEP also are warranted, as antipsychotic response during subsequent trials is substantially reduced. In fact, the Scottish Intercollegiate Guidelines Network guidelines recommend this as an area for future research.11
For now, clozapine should continue to be reserved as second- or third-line treatment in a patient with FEP. The risks of clozapine’s potentially serious adverse effects (eg, agranulocytosis, seizures, obesity, diabetes, dyslipidemia, myocarditis, pancreatitis, hypotension, sialorrhea, severe sedation, ileus) can be justified only in the treatment of severe and persistent psychotic symptoms.57
Bottom Line
Nonstandard use of antipsychotic monotherapy dosages beyond the approved FDA limit and combination antipsychotic therapy may be reasonable for select first-episode psychosis (FEP) patients. Strongly consider long-acting injectable antipsychotics in FEP to proactively combat the high relapse rate and more easily identify antipsychotic failure. Continue to use clozapine as second- or third-line therapy in FEP: Studies have not found that it is more efficacious than other antipsychotics for first-line use.
Related Resource
• Recovery After an Initial Schizophrenia Episode (RAISE) Project Early Treatment Program. National Institute of Mental Health. http://raiseetp.org.
Drug Brand Names
Aripiprazole • Abilify, Abilify Maintena
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fluphenazine decanoate • Prolixin-D
Haloperidol • Haldol
Haloperidol decanoate • Haldol-D
Olanzapine • Zyprexa
Olanzapine pamoate • Zyprexa Relprevv
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone microspheres • Risperdal Consta
Disclosures
Dr. Gardner reports no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products. Dr. Nasrallah is a consultant to Acadia, Alkermes, Lundbeck, Janssen, Merck, Otsuka, and Sunovion, and is a speaker for Alkermes, Lundbeck, Janssen, Otsuka, and Sunovion.
First-episode psychosis (FEP) in schizophrenia is characterized by high response rates to antipsychotic therapy, followed by frequent antipsychotic discontinuation and elevated relapse rates soon after maintenance treatment begins.1,2 With subsequent episodes, time to response progressively increases and likelihood of response decreases.3,4
To address these issues, this article—the second of 2 parts5—describes the rationale and evidence for using nonstandard first-line antipsychotic therapies to manage FEP. Specifically, we discuss when clinicians might consider monotherapy exceeding FDA-approved maximum dosages, combination therapy, long-acting injectable antipsychotics (LAIA), or clozapine.
Monotherapy beyond FDA-approved dosages
Treatment guidelines for FEP recommend oral antipsychotic dosages in the lower half of the treatment range and lower than those that are required for multi-episode schizophrenia.6-16 Ultimately, clinicians prescribe individualized dosages for their patients based on symptom improvement and tolerability. The optimal dosage at which to achieve a favorable D2 receptor occupancy likely will vary from patient to patient.17
To control symptoms, higher dosages may be needed than those used in FEP clinical trials, recommended by guidelines for FEP or multi-episode patients, or approved by the FDA. Patients seen in everyday practice may be more complicated (eg, have a comorbid condition or history of nonresponse) than study populations. Higher dosages also may be reasonable to overcome drug−drug interactions (eg, cigarette smoking-mediated cytochrome P450 1A2 induction, resulting in increased olanzapine metabolism),18 or to establish antipsychotic failure if adequate trials at lower dosages have resulted in a suboptimal response and the patient is not experiencing tolerability or safety concerns.
In a study of low-, full-, and high-dosage antipsychotic therapy in FEP, an additional 15% of patients responded to higher dosages of olanzapine and risperidone after failing to respond to a standard dosage.19 A study of data from the Recovery After an Initial Schizophrenia Episode Project’s Early Treatment Program (RAISE-ETP) found that, of participants identified who may benefit from therapy modification, 8.8% were prescribed an antipsychotic (often, olanzapine, risperidone, and haloperidol) at a higher-than-recommended dosage.20 Of note, only olanzapine was prescribed at higher than FDA-approved dosages.
Antipsychotic combination therapy
Prescribing combinations of antipsychotics—antipsychotic polypharmacy (APP)— has a negative connotation because of limited efficacy and safety data,21 and limited endorsement in schizophrenia treatment guidelines.9,13 Caution with APP is warranted; a complex medication regimen may increase the potential for adverse effects, poorer adherence, and adverse drug-drug interactions.9 APP has been shown to independently predict both shorter treatment duration and discontinuation before 1 year.22
Nonetheless, the clinician and patient may share the decision to implement APP and observe whether benefits outweigh risks in situations such as:
• to optimize neuroreceptor occupancy and targets (eg, attempting to achieve adequate D2 receptor blockade while minimizing side effects secondary to binding other receptors)
• to manage co-existing symptom domains (eg, mood changes, aggression, negative symptoms, disorganization, and cognitive deficits)
• to mitigate antipsychotic-induced side effects (eg, initiating aripiprazole to treat hyperprolactinemia induced by another antipsychotic to which the patient has achieved a favorable response).23
Clinicians report using APP to treat as many as 50% of patients with a history of multiple psychotic episodes.23 For FEP patients, 23% of participants in the RAISE-ETP trial who were identified as possibly benefiting from therapy modification were prescribed APP.20 Regrettably, researchers have not found evidence to support a reported rationale for using APP—that lower dosages of individual antipsychotics when used in combination may avoid high-dosage prescriptions.24
Before implementing APP, thoroughly explore and manage reasons for a patient’s suboptimal response to monotherapy.25 An adequate trial with any antipsychotic should be at the highest tolerated dosage for 12 to 16 weeks. Be mindful that response to an APP trial may be the result of additional time on the original antipsychotic.
Long-acting injectable antipsychotics in FEP
Guideline recommendations. Most older guidelines for schizophrenia treatment suggest LAIA after multiple relapses related to medication nonadherence or when a patient prefers injected medication (Table 1).6-13 Expert consensus guidelines also recommend considering LAIA in patients who lack insight into their illness. The Texas Medication Algorithm Project (TMAP) guidelines7 state LAIA can be considered for inadequate adherence at any stage, whereas the 2010 British Association for Psychopharmacology (BAP) guidelines9 express uncertainty about their use in FEP, because of limited evidence. Both the BAP and National Institute for Health and Care Excellence guidelines13 urge clinicians to consider LAIA when avoiding nonadherence is a treatment priority.
Recently, the French Association for Biological Psychiatry and Neuro-psychopharmacology (AFPBN) created expert consensus guidelines12 on using LAIA in practice. They recommend long-acting injectable second-generation antipsychotics (SGAs) as first-line maintenance treatment for schizophrenia and schizoaffective disorder and for individuals experiencing a first recurrent episode. The World Federation of Societies of Biological Psychiatry guidelines contain LAIA dosage recommendations for FEP (Table 2).10
Advances have been made in understanding the serious neurobiological adverse effects of psychotic relapses, including neuroinflammation and oxidative stress, that may explain the atrophic changes observed with psychotic episodes starting with the FEP. Protecting the patient from a second episode has become a vital therapeutic management goal26 (Figure 127).
Concerns. Compared with oral antipsychotics, LAIA offers clinical advantages:
• improved pharmacokinetic profile (lower “peaks” and higher “valleys”)
• more consistent plasma concentrations (no variability related to administration timing or food effects)
• no first-pass metabolism, which can ease the process of finding the lowest effective and safe dosage
• reduced administration burden and objective tracking of adherence with typical dosing every 2 to 4 weeks
• less stigmatizing than oral medication for FEP patients, such as college students living in a dormitory.28,29
Barriers to LAIA use include:
• slow dosage titration and increased time to reach steady state drug level
• oral supplementation for some (eg, risperidone microspheres and aripiprazole long-acting injectable)
• logistical challenges for some (eg, 3-hour post-injection monitoring for delirium sedation syndrome with olanzapine pamoate)
• additional planning to coordinate care for scheduled injections
• higher expenses up front
• local injection site reactions
• dosage adjustment difficulties if adverse effects occur.28,29
Adoption rates of LAIA are low, especially for FEP.30 Most surveys indicate that (1) physicians believe LAIA treatment is ineffective for FEP31 and (2) patients do not prefer injectable to oral antipsychotics,32 despite evidence to the contrary.33,34 A survey of 198 psychiatrists identified 3 factors that influenced their decisions against using LAIA patients with FEP:
• limited availability of SGA depot formulations (4, to date, in the United States)
• frequent rejection by the patient when LAIA is offered without adequate explanation or encouragement
• skepticism of FEP patients (and their family) who lack experience with relapse.35
In reality, when SGA depots were introduced in the United Kingdom, prescribing rates of LAIA did not increase. As for patient rejection being a major reason for not prescribing LAIA, few patients (5% to 36%) are offered depot injections, particularly in FEP.29 Most patients using LAIA are chronic, multi-episode, violent people who are receiving medications involuntarily.29 Interestingly, this survey did not find 2 factors to be influential in psychiatrists’ decision not to use LAIA in FEP:
• guidelines do not explicitly recommend depot treatment in FEP
• treatment in FEP may be limited to 1 year, therefore depot administration is not worthwhile.35
Preliminary evidence. At least a dozen studies have explored LAIA treatment for FEP, with the use of fluphenazine decanoate,36 perphenazine enanthate37 (discontinued), and risperidone microspheres.37-48 The research demonstrates the efficacy and safety of LAIA in FEP as measured by these endpoints:
• improved symptom control38,40-43,46,48
• adherence43,44,48
• reduced relapse rates37,43 and rehospitalizations37,47
• lesser reductions in white matter brain volume45
• no differences in extrapyramidal side effects or prolactin-associated adverse effects.48
A few small studies demonstrate significant differences in outcomes between risperidone LAIA and oral comparator groups (Table 3).43-45 Ongoing studies of LAIA use in FEP are comparing paliperidone palmitate with risperidone microspheres and other oral antipsychotics.49-51 No studies are examining olanzapine pamoate in FEP, likely because several guidelines do not recommended its use. No studies have been published regarding aripiprazole long-acting injectable in FEP. This LAIA formulation was approved in February 2013, and robust studies of the oral formulation in FEP are limited.52
Discussion and recommendations. Psychiatrists relying on subjective measures of antipsychotic adherence may inaccurately assess whether patients meet this criterion for LAIA use.53 LAIA could combat the high relapse rate in FEP, yet depot antipsychotics are prescribed infrequently for FEP patients (eg, for only 9.5% of participants in the RAISE-ETP study).20 Most schizophrenia treatment guidelines do not discuss LAIA use specifically in FEP, although the AFPBN expert consensus guidelines published in 2013 do recommend SGA depot formulations in FEP.12 SGA LAIA may be preferable, given its neuroprotective effects, in contrast to the neurotoxicity concerns of FGA LAIA.54,55
Relapses begin within a few months of illness stabilization after FEP, and >50% of patients relapse within 1 or 2 years2—the recommended minimum treatment duration for FEP.8,9,13 The use of LAIA is advisable in any patient with schizophrenia for whom long-term antipsychotic therapy is indicated.56 LAIA administration requirements objectively track medication adherence, which allows clinicians to be proactive in relapse prevention. Not using an intervention in FEP that improves adherence and decreases relapse rates contradicts our goal of instituting early, effective treatment to improve long-term functional outcomes (Figure 2).29
Considering clozapine in FEP
Guideline recommendations. Schizo-phrenia treatment guidelines and FDA labeling57 reserve clozapine for third-line treatment of refractory schizophrenia after 2 adequate antipsychotic trials have failed despite optimal dosing (Table 1).6-13 Some guidelines specify 1 of the 2 failed antipsychotic trials must include an SGA.6,7,10,11,13-16 Most say clozapine may be considered in patients with chronic aggression or hostility,7-9,14,16 or suicidal thoughts and behaviors.6-8,14,16 TMAP guidelines recommend a clozapine trial with concomitant substance abuse, persistent positive symptoms during 2 years of consistent medication treatment, and after 5 years of inadequate response (“treatment resistance”), regardless of the number of antipsychotic trials.7
Rationale and concerns. Clozapine is a superior choice for treatment-refractory delusions or hallucinations of schizophrenia, because it markedly enhances the response rate to antipsychotic therapy.58 Researchers therefore have investigated whether clozapine, compared with other antipsychotics, would yield more favorable initial and long-term outcomes when used first-line in FEP.
Preliminary evidence. Five studies have explored the use of clozapine as first-line therapy in FEP (Table 4).59-63 Interpreting the results is difficult because clozapine trials may be brief (mostly, 12 to 52 weeks); lack a comparator arm; suffer from a high attrition rate; enroll few patients; and lack potentially important outcome measures such as negative symptoms, suicidality, and functional assessment.
Overall, these studies demonstrate clozapine is as efficacious in this patient population as chlorpromazine (no difference in remission at 1-year, although clozapine-treated patients remitted faster and stayed in remission longer)60,61 or risperidone (no difference in Positive and Negative Syndrome Scale scores).62
At present, clozapine has not been shown superior to other antipsychotics as a first-line treatment for FEP. Research does underscore the importance of a clozapine trial as third-line treatment for FEP patients who have not responded well to 2 SGA trials.63 Many of these nonresponders (77%) have demonstrated a favorable response when promptly switched to clozapine.64
Discussion and recommendations. The limited evidence argues against using clozapine earlier than as third-line treatment in FEP. Perhaps the high treatment response that characterizes FEP creates a ceiling effect that obscures differences in antipsychotic efficacy at this stage.65 Clozapine use as first-line treatment should be re-evaluated with more robust methodology. One approach could be to assess its benefit in FEP by the duration of untreated psychosis.
The odds of achieving remission have been shown to decrease by 15% for each year that psychosis has not been treated.59 Studies exploring the use of clozapine as a second-line agent for FEP also are warranted, as antipsychotic response during subsequent trials is substantially reduced. In fact, the Scottish Intercollegiate Guidelines Network guidelines recommend this as an area for future research.11
For now, clozapine should continue to be reserved as second- or third-line treatment in a patient with FEP. The risks of clozapine’s potentially serious adverse effects (eg, agranulocytosis, seizures, obesity, diabetes, dyslipidemia, myocarditis, pancreatitis, hypotension, sialorrhea, severe sedation, ileus) can be justified only in the treatment of severe and persistent psychotic symptoms.57
Bottom Line
Nonstandard use of antipsychotic monotherapy dosages beyond the approved FDA limit and combination antipsychotic therapy may be reasonable for select first-episode psychosis (FEP) patients. Strongly consider long-acting injectable antipsychotics in FEP to proactively combat the high relapse rate and more easily identify antipsychotic failure. Continue to use clozapine as second- or third-line therapy in FEP: Studies have not found that it is more efficacious than other antipsychotics for first-line use.
Related Resource
• Recovery After an Initial Schizophrenia Episode (RAISE) Project Early Treatment Program. National Institute of Mental Health. http://raiseetp.org.
Drug Brand Names
Aripiprazole • Abilify, Abilify Maintena
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fluphenazine decanoate • Prolixin-D
Haloperidol • Haldol
Haloperidol decanoate • Haldol-D
Olanzapine • Zyprexa
Olanzapine pamoate • Zyprexa Relprevv
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone microspheres • Risperdal Consta
Disclosures
Dr. Gardner reports no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products. Dr. Nasrallah is a consultant to Acadia, Alkermes, Lundbeck, Janssen, Merck, Otsuka, and Sunovion, and is a speaker for Alkermes, Lundbeck, Janssen, Otsuka, and Sunovion.
1. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804.
2. Bradford DW, Perkins DO, Lieberman JA. Pharmacological management of first-episode schizophrenia and related nonaffective psychoses. Drugs. 2003;63(21):2265-2283.
3. Lieberman JA, Koreen AR, Chakos M, et al. Factors influencing treatment response and outcome of first-episode schizophrenia: implications for understanding the pathophysiology of schizophrenia. J Clin Psychiatry. 1996;57(suppl 9):5-9.
4. Agid O, Arenovich T, Sajeev G, et al. An algorithm-based approach to first-episode schizophrenia: response rates over 3 prospective antipsychotic trials with a retrospective data analysis. J Clin Psychiatry. 2011;72(11):1439-1444.
5. Gardner KN, Nasrallah HA. Managing first-episode psychosis. An early stage of schizophrenia with distinct treatment needs. Current Psychiatry. 2015;14(5):32-34,36-40,42.
6. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
7. Texas Department of State Health Services. Texas Medication Algorithm Project (TMAP) Procedural Manual. Schizophrenia Treatment Algorithms. http://www.jpshealthnet.org/sites/default/files/ tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed June 11, 2015.
8. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
9. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620.
10. Hasan A, Falkai P, Wobrok T, et al; WFSBP Task force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44.
11. Scottish Intercollegiate Guidelines Network. SIGN 131: Management of schizophrenia. http://www.sign.ac.uk/ pdf/sign131.pdf. Published March 2013. Accessed June 11, 2015.
12. Llorca PM, Abbar M, Courtet P, et al. Guidelines for the use and management of long-acting injectable antipsychotics in serous mental illness. BMC Psychiatry. 2013;13:340.
13. National Institute for Health and Care Excellence. NICE clinical guideline 178: Psychosis and schizophrenia in adults: treatment and management. https://www.nice.org. uk/guidance/cg178/resources/guidance-psychosis-and-schizophrenia-in-adults-treatment-and-management-pdf. Updated March 2014. Accessed June 16, 2015.
14. Canadian Psychiatric Association. Clinical practice guidelines. Treatment of schizophrenia. Can J Psychiatry. 2005;50(13 suppl 1):7S-57S.
15. McEvoy JP, Scheifler PL, Frances A. The expert consensus guideline series: treatment of schizophrenia. J Clin Psychiatry. 1999;60(suppl 11):3-80.
16. Marder SR, Essock SM, Miller AL, et al. The Mount Sinai conference on the pharmacotherapy of schizophrenia. Schizophr Bull. 2002;28(1):5-16.
17. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157(4):514-520.
18. Fankhauser MP. Drug interactions with tobacco smoke: implications for patient care. Current Psychiatry. 2013;12(1):12-16.
19. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022.
20. Robinson DG, Schooler NR, John M, et al. Prescription practices in the treatment of first-episode schizophrenia spectrum disorders: data from the national RAISE-ETP study. Am J Psychiatry. 2015;172(3):237-248.
21. Correll CU, Rummel-Kluge C, Corves C, et al. Antipsychotic combinations vs monotherapy in schizophrenia: a meta-analysis of randomized controlled trials. Schizophr Bull. 2009;35(2):443-457.
22. Fisher MD, Reilly K, Isenberg K, et al. Antipsychotic patterns of use in patients with schizophrenia: polypharmacy versus monotherapy. BMC Psychiatry. 2014;14(1):341.
23. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
24. John AP, Dragovic M. Antipsychotic polypharmacy is not associated with reduced dose of individual antipsychotics in schizophrenia. J Clin Psychopharmacol. 2015;35(2):193-195.
25. Nasrallah HA. Treatment-resistant schizophrenia. Current Psychiatry. http://www.currentpsychiatry.com/specialty-focus/schizophrenia-other-psychotic-disorders/article/ treatment-resistant-schizophrenia/9be7bba3713d4a4cd68aa 8c92b79e5b1.html. Accessed June 16, 2015.
26. Alvarez-Jiménez M, Parker AG, Hetrick SE, et al. Preventing the second episode: a systematic review and meta-analysis of psychosocial and pharmacological trials in first-episode psychosis. Schizophr Bull. 2011;37(3):619-630.
27. Nasrallah HA, Smeltzer DJ. Contemporary diagnosis and management of the patient with schizophrenia. 2nd ed. Newton, PA: Handbooks in Health Care Co; 2011.
28. McEvoy JP. Risks versus benefits of different types of long-acting injectable antipsychotics. J Clin Psychiatry. 2006;67(suppl 5):15-18.
29. Agid O, Foussias G, Remington G. Long-acting injectable antipsychotics in the treatment of schizophrenia: their role in relapse prevention. Expert Opin Pharmacother. 2010;11(14):2301-2317.
30. Kirschner M, Theodoridou A, Fusar-Poli P, et al. Patients’ and clinicians’ attitude towards long-acting depot antipsychotics in subjects with a first episode psychosis. Ther Adv Psychophamacol. 2013;3(2):89-99.
31. Heres S, Hamann J, Mendel R, et al. Identifying the profile of optimal candidates for antipsychotic depot therapy: A cluster analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1987-1993.
32. Heres S, Lambert M, Vauth R. Treatment of early episode in patents with schizophrenia: the role of long acting antipsychotics. Eur Psychiatry. 2014;29(suppl 2):1409-1413.
33. Heres S, Schmitz FS, Leucht S, et al. The attitude of patients towards antipsychotic depot treatment. Int Clin Psychopharmacol. 2007;22(5):275-282.
34. Weiden PJ, Schooler NR, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.
35. Heres S, Reichhart T, Hamann J, et al. Psychiatrists’ attitude to antipsychotic depot treatment in patients with first-episode schizophrenia. Eur Psychiatry. 2011;26(5):297-301.
36. Kane JM, Rifkin A, Quitkin F, et al. Fluphenazine vs placebo in patients with remitted, acute first-episode schizophrenia. Arch Gen Psychiatry. 1982;39(1):70-73.
37. Tiihonen J, Wahlbeck K, Lönnqvist J, et al. Effectiveness of antipsychotic treatments in a nationwide cohort of patients in a community care after first hospitalization due to schizophrenia and schizoaffective disorder: observational follow-up study. BMJ. 2006;333(7561):224.
38. Parellada E, Andrezina R, Milanova V, et al. Patients in the early phases of schizophrenia and schizoaffective disorders effectively treated with risperidone long-acting injectable. J Psychopharmacol. 2005;19(suppl 5):5-14.
39. Malla A, Binder C, Chue P. Comparison of long-acting injectable risperidone and oral novel antipsychotic drugs for treatment in early phase of schizophrenia spectrum psychosis. Proceedings of the 61st Annual Convention Society of Biological Psychiatry; Toronto, Canada; 2006.
40. Lasser RA, Bossie CA, Zhu Y, et al. Long-acting risperidone in young adults with early schizophrenia or schizoaffective illness. Ann Clin Psychiatry. 2007;19(2):65-71.
41. Emsley R, Oosthuizen P, Koen L, et al. Remission in patients with first-episode schizophrenia receiving assured antipsychotic medication: a study with risperidone long-acting injection. Int Clin Psychopharmacol. 2008;23(6):325-331.
42. Emsley R, Oosthuizen P, Koen L, et al. Oral versus injectable antipsychotic treatment in early psychosis: post hoc comparison of two studies. Clin Ther. 2008;30(12):2378-2386.
43. Kim B, Lee SH, Choi TK, et al. Effectiveness of risperidone long-acting injection in first-episode schizophrenia: in naturalistic setting. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1231-1235.
44. Weiden PJ, Schooler NJ, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.
45. Bartzokis G, Lu PH, Amar CP, et al. Long acting injection versus oral risperidone in first-episode schizophrenia: differential impact on white matter myelination trajectory. Schizophr Res. 2011;132(1):35-41.
46. Napryeyenko O, Burba B, Martinez G, et al. Risperidone long-acting injectable in recent-onset schizophrenia examined with clinician and patient self-report measures. J Clin Psychopharmacol. 2010;30(2):200-202.
47. Tiihonen J, Haukka J, Taylor M, et al. A nationwide cohort study of oral and depot antipsychotics after first hospitalization for schizophrenia. Am J Psychiatry. 2011;168(6):603-609.
48. Dubois V, Megens J, Mertens C, et al. Long-acting risperidone in early-episode schizophrenia. Acta Psychiatrica Belgica. 2011;111(1):9-21.
49. ClinicalTrials.gov. Oral risperidone versus injectable paliperidone palmitate for treating first-episode schizophrenia. https://clinicaltrials.gov/ct2/show/ NCT01451736. Accessed June 16, 2015.
50. ClinicalTrials.gov. Brain myelination effects of paliperidone palmitate versus oral risperidone in first episode schizophrenia. https://clinicaltrials.gov/ct2/ show/NCT01458379. Accessed June 16, 2015.
51. ClinicalTrials.gov. Effects of paliperidone palmitate versus oral antipsychotics on clinical outcomes and MRI measures. https://clinicaltrials.gov/ct2/show/NCT01359293. Accessed June 16, 2016.
52. U.S. Food and Drug Administration. Drugs@FDA. http:// www.accessdata.fda.gov/scripts/cder/drugsatfda. Accessed January 11, 2015.
53. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):1-46; quiz 47-48.
54. Nandra KS, Agius M. The difference between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99.
55. Nasrallah HA. Haloperidol is clearly neurotoxic. Should it be banned? Current Psychiatry. 2013;12(7):7-8.
56. Kane JM, Garcia-Ribora C. Clinical guideline recommendations for antipsychotic long-acting injections. Br J Psychiatry. 2009;52:S63-S67.
57. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
58. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
59. Woerner MG, Robinson DG, Alvir JMJ, et al. Clozapine as a first treatment for schizophrenia. Am J Psychiatry. 2003;160(8):1514-1516.
60. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naive first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003.
61. Girgis RR, Phillips MR, Li X, et al. Clozapine v. chlorpromazine in treatment-naive, first-episode schizophrenia: 9-year outcomes of a randomised clinical trial. Br J Psychiatry. 2011;199(4):281-288.
62. Sanz-Fuentenebro J, Taboada D, Palomo T, et al. Randomized trial of clozapine vs. risperidone in treatment-naïve first-episode schizophrenia: results after one year. Schizophr Res. 2013;149(1-3):156-161.
63. Yang PD, Ji Z. The efficacy and related factors of clozapine on first-episode schizophrenia. Chin J Nerv Ment Dis. 1997;23:155-158.
64. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022.
65. Remington G, Agid O, Foussias G, et al. Clozapine’s role in the treatment of first-episode schizophrenia. Am J Psychiatry. 2013;170(2):146-151.
1. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804.
2. Bradford DW, Perkins DO, Lieberman JA. Pharmacological management of first-episode schizophrenia and related nonaffective psychoses. Drugs. 2003;63(21):2265-2283.
3. Lieberman JA, Koreen AR, Chakos M, et al. Factors influencing treatment response and outcome of first-episode schizophrenia: implications for understanding the pathophysiology of schizophrenia. J Clin Psychiatry. 1996;57(suppl 9):5-9.
4. Agid O, Arenovich T, Sajeev G, et al. An algorithm-based approach to first-episode schizophrenia: response rates over 3 prospective antipsychotic trials with a retrospective data analysis. J Clin Psychiatry. 2011;72(11):1439-1444.
5. Gardner KN, Nasrallah HA. Managing first-episode psychosis. An early stage of schizophrenia with distinct treatment needs. Current Psychiatry. 2015;14(5):32-34,36-40,42.
6. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
7. Texas Department of State Health Services. Texas Medication Algorithm Project (TMAP) Procedural Manual. Schizophrenia Treatment Algorithms. http://www.jpshealthnet.org/sites/default/files/ tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed June 11, 2015.
8. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
9. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620.
10. Hasan A, Falkai P, Wobrok T, et al; WFSBP Task force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44.
11. Scottish Intercollegiate Guidelines Network. SIGN 131: Management of schizophrenia. http://www.sign.ac.uk/ pdf/sign131.pdf. Published March 2013. Accessed June 11, 2015.
12. Llorca PM, Abbar M, Courtet P, et al. Guidelines for the use and management of long-acting injectable antipsychotics in serous mental illness. BMC Psychiatry. 2013;13:340.
13. National Institute for Health and Care Excellence. NICE clinical guideline 178: Psychosis and schizophrenia in adults: treatment and management. https://www.nice.org. uk/guidance/cg178/resources/guidance-psychosis-and-schizophrenia-in-adults-treatment-and-management-pdf. Updated March 2014. Accessed June 16, 2015.
14. Canadian Psychiatric Association. Clinical practice guidelines. Treatment of schizophrenia. Can J Psychiatry. 2005;50(13 suppl 1):7S-57S.
15. McEvoy JP, Scheifler PL, Frances A. The expert consensus guideline series: treatment of schizophrenia. J Clin Psychiatry. 1999;60(suppl 11):3-80.
16. Marder SR, Essock SM, Miller AL, et al. The Mount Sinai conference on the pharmacotherapy of schizophrenia. Schizophr Bull. 2002;28(1):5-16.
17. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157(4):514-520.
18. Fankhauser MP. Drug interactions with tobacco smoke: implications for patient care. Current Psychiatry. 2013;12(1):12-16.
19. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022.
20. Robinson DG, Schooler NR, John M, et al. Prescription practices in the treatment of first-episode schizophrenia spectrum disorders: data from the national RAISE-ETP study. Am J Psychiatry. 2015;172(3):237-248.
21. Correll CU, Rummel-Kluge C, Corves C, et al. Antipsychotic combinations vs monotherapy in schizophrenia: a meta-analysis of randomized controlled trials. Schizophr Bull. 2009;35(2):443-457.
22. Fisher MD, Reilly K, Isenberg K, et al. Antipsychotic patterns of use in patients with schizophrenia: polypharmacy versus monotherapy. BMC Psychiatry. 2014;14(1):341.
23. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
24. John AP, Dragovic M. Antipsychotic polypharmacy is not associated with reduced dose of individual antipsychotics in schizophrenia. J Clin Psychopharmacol. 2015;35(2):193-195.
25. Nasrallah HA. Treatment-resistant schizophrenia. Current Psychiatry. http://www.currentpsychiatry.com/specialty-focus/schizophrenia-other-psychotic-disorders/article/ treatment-resistant-schizophrenia/9be7bba3713d4a4cd68aa 8c92b79e5b1.html. Accessed June 16, 2015.
26. Alvarez-Jiménez M, Parker AG, Hetrick SE, et al. Preventing the second episode: a systematic review and meta-analysis of psychosocial and pharmacological trials in first-episode psychosis. Schizophr Bull. 2011;37(3):619-630.
27. Nasrallah HA, Smeltzer DJ. Contemporary diagnosis and management of the patient with schizophrenia. 2nd ed. Newton, PA: Handbooks in Health Care Co; 2011.
28. McEvoy JP. Risks versus benefits of different types of long-acting injectable antipsychotics. J Clin Psychiatry. 2006;67(suppl 5):15-18.
29. Agid O, Foussias G, Remington G. Long-acting injectable antipsychotics in the treatment of schizophrenia: their role in relapse prevention. Expert Opin Pharmacother. 2010;11(14):2301-2317.
30. Kirschner M, Theodoridou A, Fusar-Poli P, et al. Patients’ and clinicians’ attitude towards long-acting depot antipsychotics in subjects with a first episode psychosis. Ther Adv Psychophamacol. 2013;3(2):89-99.
31. Heres S, Hamann J, Mendel R, et al. Identifying the profile of optimal candidates for antipsychotic depot therapy: A cluster analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1987-1993.
32. Heres S, Lambert M, Vauth R. Treatment of early episode in patents with schizophrenia: the role of long acting antipsychotics. Eur Psychiatry. 2014;29(suppl 2):1409-1413.
33. Heres S, Schmitz FS, Leucht S, et al. The attitude of patients towards antipsychotic depot treatment. Int Clin Psychopharmacol. 2007;22(5):275-282.
34. Weiden PJ, Schooler NR, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.
35. Heres S, Reichhart T, Hamann J, et al. Psychiatrists’ attitude to antipsychotic depot treatment in patients with first-episode schizophrenia. Eur Psychiatry. 2011;26(5):297-301.
36. Kane JM, Rifkin A, Quitkin F, et al. Fluphenazine vs placebo in patients with remitted, acute first-episode schizophrenia. Arch Gen Psychiatry. 1982;39(1):70-73.
37. Tiihonen J, Wahlbeck K, Lönnqvist J, et al. Effectiveness of antipsychotic treatments in a nationwide cohort of patients in a community care after first hospitalization due to schizophrenia and schizoaffective disorder: observational follow-up study. BMJ. 2006;333(7561):224.
38. Parellada E, Andrezina R, Milanova V, et al. Patients in the early phases of schizophrenia and schizoaffective disorders effectively treated with risperidone long-acting injectable. J Psychopharmacol. 2005;19(suppl 5):5-14.
39. Malla A, Binder C, Chue P. Comparison of long-acting injectable risperidone and oral novel antipsychotic drugs for treatment in early phase of schizophrenia spectrum psychosis. Proceedings of the 61st Annual Convention Society of Biological Psychiatry; Toronto, Canada; 2006.
40. Lasser RA, Bossie CA, Zhu Y, et al. Long-acting risperidone in young adults with early schizophrenia or schizoaffective illness. Ann Clin Psychiatry. 2007;19(2):65-71.
41. Emsley R, Oosthuizen P, Koen L, et al. Remission in patients with first-episode schizophrenia receiving assured antipsychotic medication: a study with risperidone long-acting injection. Int Clin Psychopharmacol. 2008;23(6):325-331.
42. Emsley R, Oosthuizen P, Koen L, et al. Oral versus injectable antipsychotic treatment in early psychosis: post hoc comparison of two studies. Clin Ther. 2008;30(12):2378-2386.
43. Kim B, Lee SH, Choi TK, et al. Effectiveness of risperidone long-acting injection in first-episode schizophrenia: in naturalistic setting. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1231-1235.
44. Weiden PJ, Schooler NJ, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.
45. Bartzokis G, Lu PH, Amar CP, et al. Long acting injection versus oral risperidone in first-episode schizophrenia: differential impact on white matter myelination trajectory. Schizophr Res. 2011;132(1):35-41.
46. Napryeyenko O, Burba B, Martinez G, et al. Risperidone long-acting injectable in recent-onset schizophrenia examined with clinician and patient self-report measures. J Clin Psychopharmacol. 2010;30(2):200-202.
47. Tiihonen J, Haukka J, Taylor M, et al. A nationwide cohort study of oral and depot antipsychotics after first hospitalization for schizophrenia. Am J Psychiatry. 2011;168(6):603-609.
48. Dubois V, Megens J, Mertens C, et al. Long-acting risperidone in early-episode schizophrenia. Acta Psychiatrica Belgica. 2011;111(1):9-21.
49. ClinicalTrials.gov. Oral risperidone versus injectable paliperidone palmitate for treating first-episode schizophrenia. https://clinicaltrials.gov/ct2/show/ NCT01451736. Accessed June 16, 2015.
50. ClinicalTrials.gov. Brain myelination effects of paliperidone palmitate versus oral risperidone in first episode schizophrenia. https://clinicaltrials.gov/ct2/ show/NCT01458379. Accessed June 16, 2015.
51. ClinicalTrials.gov. Effects of paliperidone palmitate versus oral antipsychotics on clinical outcomes and MRI measures. https://clinicaltrials.gov/ct2/show/NCT01359293. Accessed June 16, 2016.
52. U.S. Food and Drug Administration. Drugs@FDA. http:// www.accessdata.fda.gov/scripts/cder/drugsatfda. Accessed January 11, 2015.
53. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):1-46; quiz 47-48.
54. Nandra KS, Agius M. The difference between typical and atypical antipsychotics: the effects on neurogenesis. Psychiatr Danub. 2012;24(suppl 1):S95-S99.
55. Nasrallah HA. Haloperidol is clearly neurotoxic. Should it be banned? Current Psychiatry. 2013;12(7):7-8.
56. Kane JM, Garcia-Ribora C. Clinical guideline recommendations for antipsychotic long-acting injections. Br J Psychiatry. 2009;52:S63-S67.
57. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
58. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
59. Woerner MG, Robinson DG, Alvir JMJ, et al. Clozapine as a first treatment for schizophrenia. Am J Psychiatry. 2003;160(8):1514-1516.
60. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naive first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003.
61. Girgis RR, Phillips MR, Li X, et al. Clozapine v. chlorpromazine in treatment-naive, first-episode schizophrenia: 9-year outcomes of a randomised clinical trial. Br J Psychiatry. 2011;199(4):281-288.
62. Sanz-Fuentenebro J, Taboada D, Palomo T, et al. Randomized trial of clozapine vs. risperidone in treatment-naïve first-episode schizophrenia: results after one year. Schizophr Res. 2013;149(1-3):156-161.
63. Yang PD, Ji Z. The efficacy and related factors of clozapine on first-episode schizophrenia. Chin J Nerv Ment Dis. 1997;23:155-158.
64. Agid O, Schulze L, Arenovich T, et al. Antipsychotic response in first-episode schizophrenia: efficacy of high doses and switching. Eur Neuropsychopharmacol. 2013;23(9):1017-1022.
65. Remington G, Agid O, Foussias G, et al. Clozapine’s role in the treatment of first-episode schizophrenia. Am J Psychiatry. 2013;170(2):146-151.

Is there only 1 neurobiologic psychiatric disorder, with different clinical expressions?
In a report of a study that was published recently in a top-tier psychiatry journal,1 researchers describe a stunning finding that challenges the notion that there is a plethora of psychiatric brain disorders. They conducted a large meta-analysis of 193 published brain imaging studies of people with schizophrenia, bipolar disorder, major depression, obsessive-compulsive disorder (OCD), anxiety, and addiction. They found that those 6 supposedly discrete illnesses are all associated with a varying degree of shrinkage (atrophy or hypoplasia) of the same 3 brain regions:
• Dorsal anterior cingulate cortex. This region around the frontal part of the corpus callosum controls rational cognitive processes, reward anticipation, decision making, empathy, impulse control, and emotional response. Francis Crick, the Nobel laureate who first described the structure of DNA, hypothesized that the anterior cingulate sulcus might even be the center of what we call “free will.”
• Left insula and right insula. The insulae are the cortical regions deep inside the lateral sulcus, which is the fissure that separates the temporal lobe from the parietal and frontal lobes. The functions of the insulae include consciousness, emotions, perceptions, motor control, self-awareness, cognitive functioning, and interpersonal experience. (In addicts, the insular cortex is activated when they are exposed to environmental cues that trigger craving because the insulae are a target for the dopamine system. Notably, it has been reported that, when cigarette addicts suffer a stroke that damages the insulae, they stop smoking completely.)
The 3 regions of the brain, in which pathology extends across 6 DSM-5 diagnoses, work together to manage high-level executive functions, such as working memory, reasoning, and flexible thinking. The degree of dysfunction varies among the 6 clinical disorders, with schizophrenia having the highest severity.
Neurobiological commonality
The idea of shared neurobiological underpinnings among 6 distinct psychiatric disorders flies in the face of the entrenched DSM model, in which those 6 disorders are distinct disease entities. Other studies (including the Bipolar Schizophrenia Network on Intermediate Phenotypes) also found prominent biological similarities in varying degrees across schizophrenia, schizoaffective disorder, and bipolar disorder.2,3 The Research Domain Criteria of the National Institute of Mental Health also embraces the dimensional approach to neurobiological biomarkers across various psychiatric disorders.
A common genetic substrate also is emerging. A recently published genome-wide association study, conducted on 33,332 psychiatric patients and 27,888 controls,4 revealed that a number of genes are shared by 5 different psychiatric disorders: schizophrenia, autism, bipolar disorder, major depression, and attention-deficit/hyperactivity disorder. The genetic phenomenon of the same genes manifesting in different clinical phenotypes is called pleiotropy, and is consistent with shared neurobiological findings.
Genetic and brain structural commonalities among multiple DSM diagnostic categories might explain some well-known clinical observations:
• frequent comorbidity of certain psychiatric disorders, such as depression and addiction in schizophrenia; anxiety and OCD in bipolar disorder; depression with OCD and addictions; and so on
• the presence of intermediate phenotypes in unaffected family members, such as cognitive dysfunction in the parents of patients with schizophrenia, compared with parents of matched healthy controls5
• the much higher rate of psychopathology among family members of patients with a major psychiatric disorder, compared with the general population.6
Core inflexible thinking. So what about clinical features across those disorders that share genetic and neurobiologic similarities? Psychiatrists may agree that symptoms of schizophrenia, bipolar disorder, major depression, OCD, anxiety, and addiction appear very different. However, given that reasoning and flexible thinking are functions of the insulae, which are shrunken in all 6 disorders, one can postulate that inflexible thinking (fixed false beliefs also are called psychotic delusions) might be a common feature across all those disorders. Namely:
• schizophrenia is known for paranoid or implausible delusions
• bipolar disorder is characterized by grandiose delusions
• major depressive disorder is associated with a fixed false belief of worthlessness as well as hopelessness
• anxiety patients harbor the fixed false belief of impending doom or death (the plane will crash if they are a passenger on it)
• OCD manifests as ego-dystonic false beliefs (obsessions) that can progress into ego-syntonic delusions
• people with an alcohol or tobacco addiction are in delusional denial that they are not really addicted or that they will not be harmed by their drug of abuse. Pathologic gamblers harbor the false belief that they will soon reverse their fortunes and “win big.”
It seems that poor reality testing and impaired reasoning is a common feature of not only all 6 psychiatric disorders with shared neurobiology, but others, too, including anorexia nervosa, body dysmorphic disorder, delirium, and dementia.
From a thick volume to… a booklet?
Can you envision a day when psychiatric disorders are conceptualized as having a common genetic, neurobiological, and clinical core, with some variability in phenotype and behavior? If further brain research steers psychiatric nosology in that direction, we might end up with a DSM of 10 pages instead of almost 1,000, with an “Appendix” of genetic, neuroimaging, and other emerging biomarkers.
Bold scientific prophecies often sound delusional—until they come true….
1. Goodkind M, Eickhoff SB, Oathes DJ, et al. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry. 2015;72(4):305-315.
2. Hill SK, Reilly JL, Keefe RS, et al. Neuropsychological impairments in schizophrenia and psychotic bipolar disorder: findings from the Bipolar- Schizophrenia Network on Intermediate Phenotypes (B-SNIP) study. Am J Psychiatry. 2013;170(11):1275-1284.
3. Skudlarski P, Scretlen DJ, Thaker GK, et al. Diffusion tensor imaging white matter endophenotypes in patients with schizophrenia or psychotic bipolar disorder and their relatives. Am J Psychiatry. 2013;170(8):886-898.
4. Cross-Disorder Group of Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381(9875):1371-1379.
5. Appels MC, Sitskoorn MM, Westers P, et al. Cognitive dysfunction in parents of schizophrenia patients parallel the deficits found in patients. Schizophrenia Res. 2003;63(3):285-293.
6. Braff DL. The importance of endophenotypes in schizophrenia research. Schizophrenia Res. 2015;163(1-3):1-8.
In a report of a study that was published recently in a top-tier psychiatry journal,1 researchers describe a stunning finding that challenges the notion that there is a plethora of psychiatric brain disorders. They conducted a large meta-analysis of 193 published brain imaging studies of people with schizophrenia, bipolar disorder, major depression, obsessive-compulsive disorder (OCD), anxiety, and addiction. They found that those 6 supposedly discrete illnesses are all associated with a varying degree of shrinkage (atrophy or hypoplasia) of the same 3 brain regions:
• Dorsal anterior cingulate cortex. This region around the frontal part of the corpus callosum controls rational cognitive processes, reward anticipation, decision making, empathy, impulse control, and emotional response. Francis Crick, the Nobel laureate who first described the structure of DNA, hypothesized that the anterior cingulate sulcus might even be the center of what we call “free will.”
• Left insula and right insula. The insulae are the cortical regions deep inside the lateral sulcus, which is the fissure that separates the temporal lobe from the parietal and frontal lobes. The functions of the insulae include consciousness, emotions, perceptions, motor control, self-awareness, cognitive functioning, and interpersonal experience. (In addicts, the insular cortex is activated when they are exposed to environmental cues that trigger craving because the insulae are a target for the dopamine system. Notably, it has been reported that, when cigarette addicts suffer a stroke that damages the insulae, they stop smoking completely.)
The 3 regions of the brain, in which pathology extends across 6 DSM-5 diagnoses, work together to manage high-level executive functions, such as working memory, reasoning, and flexible thinking. The degree of dysfunction varies among the 6 clinical disorders, with schizophrenia having the highest severity.
Neurobiological commonality
The idea of shared neurobiological underpinnings among 6 distinct psychiatric disorders flies in the face of the entrenched DSM model, in which those 6 disorders are distinct disease entities. Other studies (including the Bipolar Schizophrenia Network on Intermediate Phenotypes) also found prominent biological similarities in varying degrees across schizophrenia, schizoaffective disorder, and bipolar disorder.2,3 The Research Domain Criteria of the National Institute of Mental Health also embraces the dimensional approach to neurobiological biomarkers across various psychiatric disorders.
A common genetic substrate also is emerging. A recently published genome-wide association study, conducted on 33,332 psychiatric patients and 27,888 controls,4 revealed that a number of genes are shared by 5 different psychiatric disorders: schizophrenia, autism, bipolar disorder, major depression, and attention-deficit/hyperactivity disorder. The genetic phenomenon of the same genes manifesting in different clinical phenotypes is called pleiotropy, and is consistent with shared neurobiological findings.
Genetic and brain structural commonalities among multiple DSM diagnostic categories might explain some well-known clinical observations:
• frequent comorbidity of certain psychiatric disorders, such as depression and addiction in schizophrenia; anxiety and OCD in bipolar disorder; depression with OCD and addictions; and so on
• the presence of intermediate phenotypes in unaffected family members, such as cognitive dysfunction in the parents of patients with schizophrenia, compared with parents of matched healthy controls5
• the much higher rate of psychopathology among family members of patients with a major psychiatric disorder, compared with the general population.6
Core inflexible thinking. So what about clinical features across those disorders that share genetic and neurobiologic similarities? Psychiatrists may agree that symptoms of schizophrenia, bipolar disorder, major depression, OCD, anxiety, and addiction appear very different. However, given that reasoning and flexible thinking are functions of the insulae, which are shrunken in all 6 disorders, one can postulate that inflexible thinking (fixed false beliefs also are called psychotic delusions) might be a common feature across all those disorders. Namely:
• schizophrenia is known for paranoid or implausible delusions
• bipolar disorder is characterized by grandiose delusions
• major depressive disorder is associated with a fixed false belief of worthlessness as well as hopelessness
• anxiety patients harbor the fixed false belief of impending doom or death (the plane will crash if they are a passenger on it)
• OCD manifests as ego-dystonic false beliefs (obsessions) that can progress into ego-syntonic delusions
• people with an alcohol or tobacco addiction are in delusional denial that they are not really addicted or that they will not be harmed by their drug of abuse. Pathologic gamblers harbor the false belief that they will soon reverse their fortunes and “win big.”
It seems that poor reality testing and impaired reasoning is a common feature of not only all 6 psychiatric disorders with shared neurobiology, but others, too, including anorexia nervosa, body dysmorphic disorder, delirium, and dementia.
From a thick volume to… a booklet?
Can you envision a day when psychiatric disorders are conceptualized as having a common genetic, neurobiological, and clinical core, with some variability in phenotype and behavior? If further brain research steers psychiatric nosology in that direction, we might end up with a DSM of 10 pages instead of almost 1,000, with an “Appendix” of genetic, neuroimaging, and other emerging biomarkers.
Bold scientific prophecies often sound delusional—until they come true….
In a report of a study that was published recently in a top-tier psychiatry journal,1 researchers describe a stunning finding that challenges the notion that there is a plethora of psychiatric brain disorders. They conducted a large meta-analysis of 193 published brain imaging studies of people with schizophrenia, bipolar disorder, major depression, obsessive-compulsive disorder (OCD), anxiety, and addiction. They found that those 6 supposedly discrete illnesses are all associated with a varying degree of shrinkage (atrophy or hypoplasia) of the same 3 brain regions:
• Dorsal anterior cingulate cortex. This region around the frontal part of the corpus callosum controls rational cognitive processes, reward anticipation, decision making, empathy, impulse control, and emotional response. Francis Crick, the Nobel laureate who first described the structure of DNA, hypothesized that the anterior cingulate sulcus might even be the center of what we call “free will.”
• Left insula and right insula. The insulae are the cortical regions deep inside the lateral sulcus, which is the fissure that separates the temporal lobe from the parietal and frontal lobes. The functions of the insulae include consciousness, emotions, perceptions, motor control, self-awareness, cognitive functioning, and interpersonal experience. (In addicts, the insular cortex is activated when they are exposed to environmental cues that trigger craving because the insulae are a target for the dopamine system. Notably, it has been reported that, when cigarette addicts suffer a stroke that damages the insulae, they stop smoking completely.)
The 3 regions of the brain, in which pathology extends across 6 DSM-5 diagnoses, work together to manage high-level executive functions, such as working memory, reasoning, and flexible thinking. The degree of dysfunction varies among the 6 clinical disorders, with schizophrenia having the highest severity.
Neurobiological commonality
The idea of shared neurobiological underpinnings among 6 distinct psychiatric disorders flies in the face of the entrenched DSM model, in which those 6 disorders are distinct disease entities. Other studies (including the Bipolar Schizophrenia Network on Intermediate Phenotypes) also found prominent biological similarities in varying degrees across schizophrenia, schizoaffective disorder, and bipolar disorder.2,3 The Research Domain Criteria of the National Institute of Mental Health also embraces the dimensional approach to neurobiological biomarkers across various psychiatric disorders.
A common genetic substrate also is emerging. A recently published genome-wide association study, conducted on 33,332 psychiatric patients and 27,888 controls,4 revealed that a number of genes are shared by 5 different psychiatric disorders: schizophrenia, autism, bipolar disorder, major depression, and attention-deficit/hyperactivity disorder. The genetic phenomenon of the same genes manifesting in different clinical phenotypes is called pleiotropy, and is consistent with shared neurobiological findings.
Genetic and brain structural commonalities among multiple DSM diagnostic categories might explain some well-known clinical observations:
• frequent comorbidity of certain psychiatric disorders, such as depression and addiction in schizophrenia; anxiety and OCD in bipolar disorder; depression with OCD and addictions; and so on
• the presence of intermediate phenotypes in unaffected family members, such as cognitive dysfunction in the parents of patients with schizophrenia, compared with parents of matched healthy controls5
• the much higher rate of psychopathology among family members of patients with a major psychiatric disorder, compared with the general population.6
Core inflexible thinking. So what about clinical features across those disorders that share genetic and neurobiologic similarities? Psychiatrists may agree that symptoms of schizophrenia, bipolar disorder, major depression, OCD, anxiety, and addiction appear very different. However, given that reasoning and flexible thinking are functions of the insulae, which are shrunken in all 6 disorders, one can postulate that inflexible thinking (fixed false beliefs also are called psychotic delusions) might be a common feature across all those disorders. Namely:
• schizophrenia is known for paranoid or implausible delusions
• bipolar disorder is characterized by grandiose delusions
• major depressive disorder is associated with a fixed false belief of worthlessness as well as hopelessness
• anxiety patients harbor the fixed false belief of impending doom or death (the plane will crash if they are a passenger on it)
• OCD manifests as ego-dystonic false beliefs (obsessions) that can progress into ego-syntonic delusions
• people with an alcohol or tobacco addiction are in delusional denial that they are not really addicted or that they will not be harmed by their drug of abuse. Pathologic gamblers harbor the false belief that they will soon reverse their fortunes and “win big.”
It seems that poor reality testing and impaired reasoning is a common feature of not only all 6 psychiatric disorders with shared neurobiology, but others, too, including anorexia nervosa, body dysmorphic disorder, delirium, and dementia.
From a thick volume to… a booklet?
Can you envision a day when psychiatric disorders are conceptualized as having a common genetic, neurobiological, and clinical core, with some variability in phenotype and behavior? If further brain research steers psychiatric nosology in that direction, we might end up with a DSM of 10 pages instead of almost 1,000, with an “Appendix” of genetic, neuroimaging, and other emerging biomarkers.
Bold scientific prophecies often sound delusional—until they come true….
1. Goodkind M, Eickhoff SB, Oathes DJ, et al. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry. 2015;72(4):305-315.
2. Hill SK, Reilly JL, Keefe RS, et al. Neuropsychological impairments in schizophrenia and psychotic bipolar disorder: findings from the Bipolar- Schizophrenia Network on Intermediate Phenotypes (B-SNIP) study. Am J Psychiatry. 2013;170(11):1275-1284.
3. Skudlarski P, Scretlen DJ, Thaker GK, et al. Diffusion tensor imaging white matter endophenotypes in patients with schizophrenia or psychotic bipolar disorder and their relatives. Am J Psychiatry. 2013;170(8):886-898.
4. Cross-Disorder Group of Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381(9875):1371-1379.
5. Appels MC, Sitskoorn MM, Westers P, et al. Cognitive dysfunction in parents of schizophrenia patients parallel the deficits found in patients. Schizophrenia Res. 2003;63(3):285-293.
6. Braff DL. The importance of endophenotypes in schizophrenia research. Schizophrenia Res. 2015;163(1-3):1-8.
1. Goodkind M, Eickhoff SB, Oathes DJ, et al. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry. 2015;72(4):305-315.
2. Hill SK, Reilly JL, Keefe RS, et al. Neuropsychological impairments in schizophrenia and psychotic bipolar disorder: findings from the Bipolar- Schizophrenia Network on Intermediate Phenotypes (B-SNIP) study. Am J Psychiatry. 2013;170(11):1275-1284.
3. Skudlarski P, Scretlen DJ, Thaker GK, et al. Diffusion tensor imaging white matter endophenotypes in patients with schizophrenia or psychotic bipolar disorder and their relatives. Am J Psychiatry. 2013;170(8):886-898.
4. Cross-Disorder Group of Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381(9875):1371-1379.
5. Appels MC, Sitskoorn MM, Westers P, et al. Cognitive dysfunction in parents of schizophrenia patients parallel the deficits found in patients. Schizophrenia Res. 2003;63(3):285-293.
6. Braff DL. The importance of endophenotypes in schizophrenia research. Schizophrenia Res. 2015;163(1-3):1-8.
Beyond dopamine: Brain repair tactics in schizophrenia
For the past 60 years, the standard of care has remained one-dimensional in this brain syndrome, even though the clinical and neurobiological complexities of schizophrenia are multidimensional. Dopamine D2 receptor antagonists, discovered serendipitously in the 1950s, have remained the mainstay of treatment, despite momentous insights about the neurodevelopmental and neurodegenerative processes of schizophrenia.
Why do we ignore abundant evidence that the brain in schizophrenia needs extensive structural repair, not simply a reduction in the activity of a single neurotransmitter in the mesolimbic dopamine tract? Perhaps the age-old dogmatic pessimism that neurodegeneration cannot be reversed has inhibited the field from attempting to escape the dopamine box, so to speak, and from developing innovative, even radical, approaches to repair of the brain of persons with schizophrenia.
But radical thinking is justified when dealing with a cruel brain syndrome that disables young adults in the prime of life.
We should exploit neuroprotective tactics
Several neuroprotective approaches to preventing or reversing the degenerative changes across brain regions in schizophrenia are now recognized. Indirect evidence exists for such interventions in animal models, but the results of few controlled human studies have been published.
Here are my proposals for using neuroprotective tactics to address the unmet need to repair the brain of patients ravaged by neurotoxic psychotic relapses.
Promote 100% adherence to antipsychotic therapy. The simplest tactic to protect the brain from atrophy in patients with schizophrenia is to use long-acting injectable antipsychotic agents immediately after the first psychotic episode. The risk of a psychotic relapse is far lower (7-fold lower, according to a study performed at the University of California, Los Angeles, that soon will be published) with an injectable medication than with oral medication in first-episode patients. Preventing psychotic episodes is, logically, the most important neuroprotective tactic.
Enhance neurogenesis. The brain has 2 neurogenic regions that produce progenitor cells (stem cells) that gradually mature and differentiate into neurons and glia. That is how the brain naturally replenishes itself throughout life. This adult neurogenesis process, carried out in the dentate gyrus of the hippocampus and in the subventricular zone, stops during psychosis but resumes when psychosis remits.
Second-generation antipsychotics (but not first-generation agents) stimulate neurogenesis in animals.1 Haloperidol, in fact, does the opposite—suppressing neurogenesis and causing neuronal death via 15 different molecular mechanisms (see my editorial, “Haloperidol clearly is neurotoxic. Should it be banned?,” in the July 2013 issue).
Other psychotropics also induce neurogenesis, including selective serotonin reuptake inhibitors (SSRIs), which increase hippocampal neurogenesis (atypical antipsychotics appear to increase neurogenesis in the subventricular zone).2 SSRIs often have been used in schizophrenia patients for 2 common comorbid conditions: depression and anxiety. These agents can help regenerate brain tissue, in addition to providing their approved therapeutic indications.
Lithium and valproate have been shown to be neuroprotective3 and to stimulate neurogenesis. Both are often used in schizoaffective disorder, bipolar type; they can exert a neuroprotective effect in addition to their clinical usefulness. The combination of an SSRI or lithium with a second-generation antipsychotic could be synergistic in turbocharging neurogenesis. This sounds like polypharmacy—but it is a rational approach that deserves to be put to the test.
Increase neurotrophins, such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF). When neurotrophin levels decline, the brain starts shrinking because of apoptosis. Psychosis lowers neurotrophins drastically—by approximately 60%. Atypical antipsychotics have been reported to increase the level of neurotrophins; haloperidol actually lowers those levels.4
Decrease inflammation. Psychosis has been shown to be associated with neuro-inflammation, as reflected in a surge of pro-inflammatory cytokines (released from activated microglia).5 A rise in interleukin-6, tumor necrosis factor-alpha, interferon-gamma, and other pro-inflammatory markers has been extensively documented in many studies.
With that observed rise in mind, several controlled studies have shown that adding an anti-inflammatory agent (aspirin, a nonsteroidal anti-inflammatory drug, a COX-2 inhibitor, or minocycline) to an antipsychotic can accentuate the therapeutic response, especially during a first episode of psychosis.6 Note also that second-generation antipsychotics have anti-inflammatory effects7 as well that might be part of their efficacy beyond blocking dopamine D2 receptors.
Decrease free radicals. Microglia are activated by psychosis to release free radicals, also known as reactive oxygen species; these include nitric oxide, superoxide, and peroxynitrate. All these species are destructive to brain tissue. Using an adjunctive strong antioxidant, such as N-acetyl cysteine,8 with an antipsychotic might help neutralize destructive effects of free radicals and protect the brain from tissue loss during a psychotic episode.
Avoid apoptosis inducers. Several substances can initiate programmed cell death (apoptosis), which is triggered during psychosis (believed to be caused by increased dopamine and, possibly, glutamate, activity) and which leads to brain atrophy. Patients with schizophrenia must be protected from these apoptosis inducers:
• amphetamine
• cocaine
• Cannabis
• lipid peroxidation products
• inflammatory cytokines.
Apoptosis can be inhibited by maintaining high levels of neurotrophic factors. Atypical, but not typical, antipsychotics increase levels of neurotrophins, such as NGF and BDNF.4 In addition, the Bcl-2 family of proteins inhibits apoptosis,9 and drugs such as lithium and valproate can induce Bcl-2 and protect against apoptosis and neuronal loss.3
Restore white-matter integrity. Numerous studies using diffusion tensor imaging have revealed that myelin is reduced or lacks integrity in schizophrenia. This results in loss of critical connectivity among brain regions, which might explain psychotic and cognitive symptoms. One possible way to repair white matter, which becomes more damaged after multiple psychotic episodes, is to use drugs indicated to treat the demyelinating disorder multiple sclerosis. Antagonists of LINGO-1, a negative regulator of axonal myelination, are a prominent possibility; a recent study reported altered signaling of LINGO-1 in schizophrenia.10
Decrease excessive glutamate. Because glutamate is neurotoxic and might contribute to brain-tissue loss during psychosis, it is important to reduce glutamate activity in schizophrenia. Lamotrigine and valproate are both known to do that.11 Several studies indicate that adjunctive lamotrigine might be helpful in schizophrenia.12
Inhibit caspase-3, also known as the “death cascade,” which is involved in brain-tissue loss. Eicosapentaenoic acid is an omega-3 fatty acid that inhibits caspase-3. Interestingly, omega-3 levels in patients with schizophrenia are significantly lower than in healthy subjects.13 Lithium also can inhibit caspase-3.
Do these proposals sound radical?
Most of the recommendations I’ve made here are not employed in the clinical practice of psychiatry. These ideas must be put to the test in controlled clinical trials.
The crux of my argument is that we need to think outside the “dopamine box” and focus on brain repair if we are to make progress in reversing, even preventing, neurodegeneration and clinical deterioration in this disabling brain syndrome. Just as cancer often is treated with rational polypharmacy, schizophrenia might need a similar approach. To vanquish schizophrenia—a goal that has eluded us—it is imperative to pursue radically novel and disruptive therapeutic strategies. The ideas I’ve listed here sound the call that the quest to repair the brain in schizophrenia must begin, and soon.
1. Agius N, Nandra, KS. Do atypical antipsychotics promote neurogenesis as a class effect? Psychiatr Danub. 2012;24(suppl 1):S191-S193.
2. Nasrallah HA, Hopkins T, Pixley SK. Differential effects of antipsychotic and antidepressant drugs on neurogenic regions in rats. Brain Res. 2010;1354:23-29.
3. Chiu CT, Wang Z, Hunsberger JG, et al. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol Rev. 2013;65(1):105-142.
4. Parikh V, Khan MM, Terry A, et al. Differential effects of typical and atypical antipsychotics on nerve growth factor and choline acetyltransferase expression in the cortex and nucleus basalis of rats. J Psychiatr Res. 2004;38(5):521-529.
5. Monji A, Kato TA, Mizoguchi Y, et al. Neuro-inflammation in schizophrenia especially focused on the role of microglia. Prog Neuropsychopharmacol Biol Psychiatry. 2013;42:115-121.
6. Sommer IE, deWitte L, Begemann M, et al. Nonsteriodal anti-inflammatory drugs in schizophrenia: ready for practice or a good start? A meta-analysis. J Clin Psychiatry. 2012;73(4):414-419.
7. Bian Q, Kato T, Monji A, et al. The effect of atypical anti-psychotics perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-gamma. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):42-48.
8. Berk M, Copolov D, Dean O, et al. N-acetyl cysteine as a glutathione precursor for schizophrenia—a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64(5):361-368.
9. Huang J, Fairbrother W, Reed JC, et al. Therapeutic targeting of Bcl-2 family for treatment of B-cell malignancies. Expert Rev Hematol. 2015;8(3):283-297.
10. Fernandez-Enright F, Andrews JL, Newell KA, et al. Novel implications of Lingo-1 and its signaling partners in schizophrenia. Transl Psychiatry. 2014; 4:e348.
11. Zink M, Correll CU. Glutamatergic agents for schizophrenia: current evidence and perspectives. Expert Rev Clin Pharmacol. 2015;8(3):335-352.
12. Kremer I, Vass A, Gorelik I, et al. Placebo-controlled trial of lamotrigine added to conventional and atypical antipsychotics in schizophrenia. Biol Psychiatry. 2004;56(6):444-446.
13. McEvoy J, Baillie RA, Zhu H, et al. Lipidomics reveals early metabolic changes in subjects with schizophrenia: effects of atypical antipsychotics. PLoS One. 2013;8(7):e68717.
For the past 60 years, the standard of care has remained one-dimensional in this brain syndrome, even though the clinical and neurobiological complexities of schizophrenia are multidimensional. Dopamine D2 receptor antagonists, discovered serendipitously in the 1950s, have remained the mainstay of treatment, despite momentous insights about the neurodevelopmental and neurodegenerative processes of schizophrenia.
Why do we ignore abundant evidence that the brain in schizophrenia needs extensive structural repair, not simply a reduction in the activity of a single neurotransmitter in the mesolimbic dopamine tract? Perhaps the age-old dogmatic pessimism that neurodegeneration cannot be reversed has inhibited the field from attempting to escape the dopamine box, so to speak, and from developing innovative, even radical, approaches to repair of the brain of persons with schizophrenia.
But radical thinking is justified when dealing with a cruel brain syndrome that disables young adults in the prime of life.
We should exploit neuroprotective tactics
Several neuroprotective approaches to preventing or reversing the degenerative changes across brain regions in schizophrenia are now recognized. Indirect evidence exists for such interventions in animal models, but the results of few controlled human studies have been published.
Here are my proposals for using neuroprotective tactics to address the unmet need to repair the brain of patients ravaged by neurotoxic psychotic relapses.
Promote 100% adherence to antipsychotic therapy. The simplest tactic to protect the brain from atrophy in patients with schizophrenia is to use long-acting injectable antipsychotic agents immediately after the first psychotic episode. The risk of a psychotic relapse is far lower (7-fold lower, according to a study performed at the University of California, Los Angeles, that soon will be published) with an injectable medication than with oral medication in first-episode patients. Preventing psychotic episodes is, logically, the most important neuroprotective tactic.
Enhance neurogenesis. The brain has 2 neurogenic regions that produce progenitor cells (stem cells) that gradually mature and differentiate into neurons and glia. That is how the brain naturally replenishes itself throughout life. This adult neurogenesis process, carried out in the dentate gyrus of the hippocampus and in the subventricular zone, stops during psychosis but resumes when psychosis remits.
Second-generation antipsychotics (but not first-generation agents) stimulate neurogenesis in animals.1 Haloperidol, in fact, does the opposite—suppressing neurogenesis and causing neuronal death via 15 different molecular mechanisms (see my editorial, “Haloperidol clearly is neurotoxic. Should it be banned?,” in the July 2013 issue).
Other psychotropics also induce neurogenesis, including selective serotonin reuptake inhibitors (SSRIs), which increase hippocampal neurogenesis (atypical antipsychotics appear to increase neurogenesis in the subventricular zone).2 SSRIs often have been used in schizophrenia patients for 2 common comorbid conditions: depression and anxiety. These agents can help regenerate brain tissue, in addition to providing their approved therapeutic indications.
Lithium and valproate have been shown to be neuroprotective3 and to stimulate neurogenesis. Both are often used in schizoaffective disorder, bipolar type; they can exert a neuroprotective effect in addition to their clinical usefulness. The combination of an SSRI or lithium with a second-generation antipsychotic could be synergistic in turbocharging neurogenesis. This sounds like polypharmacy—but it is a rational approach that deserves to be put to the test.
Increase neurotrophins, such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF). When neurotrophin levels decline, the brain starts shrinking because of apoptosis. Psychosis lowers neurotrophins drastically—by approximately 60%. Atypical antipsychotics have been reported to increase the level of neurotrophins; haloperidol actually lowers those levels.4
Decrease inflammation. Psychosis has been shown to be associated with neuro-inflammation, as reflected in a surge of pro-inflammatory cytokines (released from activated microglia).5 A rise in interleukin-6, tumor necrosis factor-alpha, interferon-gamma, and other pro-inflammatory markers has been extensively documented in many studies.
With that observed rise in mind, several controlled studies have shown that adding an anti-inflammatory agent (aspirin, a nonsteroidal anti-inflammatory drug, a COX-2 inhibitor, or minocycline) to an antipsychotic can accentuate the therapeutic response, especially during a first episode of psychosis.6 Note also that second-generation antipsychotics have anti-inflammatory effects7 as well that might be part of their efficacy beyond blocking dopamine D2 receptors.
Decrease free radicals. Microglia are activated by psychosis to release free radicals, also known as reactive oxygen species; these include nitric oxide, superoxide, and peroxynitrate. All these species are destructive to brain tissue. Using an adjunctive strong antioxidant, such as N-acetyl cysteine,8 with an antipsychotic might help neutralize destructive effects of free radicals and protect the brain from tissue loss during a psychotic episode.
Avoid apoptosis inducers. Several substances can initiate programmed cell death (apoptosis), which is triggered during psychosis (believed to be caused by increased dopamine and, possibly, glutamate, activity) and which leads to brain atrophy. Patients with schizophrenia must be protected from these apoptosis inducers:
• amphetamine
• cocaine
• Cannabis
• lipid peroxidation products
• inflammatory cytokines.
Apoptosis can be inhibited by maintaining high levels of neurotrophic factors. Atypical, but not typical, antipsychotics increase levels of neurotrophins, such as NGF and BDNF.4 In addition, the Bcl-2 family of proteins inhibits apoptosis,9 and drugs such as lithium and valproate can induce Bcl-2 and protect against apoptosis and neuronal loss.3
Restore white-matter integrity. Numerous studies using diffusion tensor imaging have revealed that myelin is reduced or lacks integrity in schizophrenia. This results in loss of critical connectivity among brain regions, which might explain psychotic and cognitive symptoms. One possible way to repair white matter, which becomes more damaged after multiple psychotic episodes, is to use drugs indicated to treat the demyelinating disorder multiple sclerosis. Antagonists of LINGO-1, a negative regulator of axonal myelination, are a prominent possibility; a recent study reported altered signaling of LINGO-1 in schizophrenia.10
Decrease excessive glutamate. Because glutamate is neurotoxic and might contribute to brain-tissue loss during psychosis, it is important to reduce glutamate activity in schizophrenia. Lamotrigine and valproate are both known to do that.11 Several studies indicate that adjunctive lamotrigine might be helpful in schizophrenia.12
Inhibit caspase-3, also known as the “death cascade,” which is involved in brain-tissue loss. Eicosapentaenoic acid is an omega-3 fatty acid that inhibits caspase-3. Interestingly, omega-3 levels in patients with schizophrenia are significantly lower than in healthy subjects.13 Lithium also can inhibit caspase-3.
Do these proposals sound radical?
Most of the recommendations I’ve made here are not employed in the clinical practice of psychiatry. These ideas must be put to the test in controlled clinical trials.
The crux of my argument is that we need to think outside the “dopamine box” and focus on brain repair if we are to make progress in reversing, even preventing, neurodegeneration and clinical deterioration in this disabling brain syndrome. Just as cancer often is treated with rational polypharmacy, schizophrenia might need a similar approach. To vanquish schizophrenia—a goal that has eluded us—it is imperative to pursue radically novel and disruptive therapeutic strategies. The ideas I’ve listed here sound the call that the quest to repair the brain in schizophrenia must begin, and soon.
For the past 60 years, the standard of care has remained one-dimensional in this brain syndrome, even though the clinical and neurobiological complexities of schizophrenia are multidimensional. Dopamine D2 receptor antagonists, discovered serendipitously in the 1950s, have remained the mainstay of treatment, despite momentous insights about the neurodevelopmental and neurodegenerative processes of schizophrenia.
Why do we ignore abundant evidence that the brain in schizophrenia needs extensive structural repair, not simply a reduction in the activity of a single neurotransmitter in the mesolimbic dopamine tract? Perhaps the age-old dogmatic pessimism that neurodegeneration cannot be reversed has inhibited the field from attempting to escape the dopamine box, so to speak, and from developing innovative, even radical, approaches to repair of the brain of persons with schizophrenia.
But radical thinking is justified when dealing with a cruel brain syndrome that disables young adults in the prime of life.
We should exploit neuroprotective tactics
Several neuroprotective approaches to preventing or reversing the degenerative changes across brain regions in schizophrenia are now recognized. Indirect evidence exists for such interventions in animal models, but the results of few controlled human studies have been published.
Here are my proposals for using neuroprotective tactics to address the unmet need to repair the brain of patients ravaged by neurotoxic psychotic relapses.
Promote 100% adherence to antipsychotic therapy. The simplest tactic to protect the brain from atrophy in patients with schizophrenia is to use long-acting injectable antipsychotic agents immediately after the first psychotic episode. The risk of a psychotic relapse is far lower (7-fold lower, according to a study performed at the University of California, Los Angeles, that soon will be published) with an injectable medication than with oral medication in first-episode patients. Preventing psychotic episodes is, logically, the most important neuroprotective tactic.
Enhance neurogenesis. The brain has 2 neurogenic regions that produce progenitor cells (stem cells) that gradually mature and differentiate into neurons and glia. That is how the brain naturally replenishes itself throughout life. This adult neurogenesis process, carried out in the dentate gyrus of the hippocampus and in the subventricular zone, stops during psychosis but resumes when psychosis remits.
Second-generation antipsychotics (but not first-generation agents) stimulate neurogenesis in animals.1 Haloperidol, in fact, does the opposite—suppressing neurogenesis and causing neuronal death via 15 different molecular mechanisms (see my editorial, “Haloperidol clearly is neurotoxic. Should it be banned?,” in the July 2013 issue).
Other psychotropics also induce neurogenesis, including selective serotonin reuptake inhibitors (SSRIs), which increase hippocampal neurogenesis (atypical antipsychotics appear to increase neurogenesis in the subventricular zone).2 SSRIs often have been used in schizophrenia patients for 2 common comorbid conditions: depression and anxiety. These agents can help regenerate brain tissue, in addition to providing their approved therapeutic indications.
Lithium and valproate have been shown to be neuroprotective3 and to stimulate neurogenesis. Both are often used in schizoaffective disorder, bipolar type; they can exert a neuroprotective effect in addition to their clinical usefulness. The combination of an SSRI or lithium with a second-generation antipsychotic could be synergistic in turbocharging neurogenesis. This sounds like polypharmacy—but it is a rational approach that deserves to be put to the test.
Increase neurotrophins, such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF). When neurotrophin levels decline, the brain starts shrinking because of apoptosis. Psychosis lowers neurotrophins drastically—by approximately 60%. Atypical antipsychotics have been reported to increase the level of neurotrophins; haloperidol actually lowers those levels.4
Decrease inflammation. Psychosis has been shown to be associated with neuro-inflammation, as reflected in a surge of pro-inflammatory cytokines (released from activated microglia).5 A rise in interleukin-6, tumor necrosis factor-alpha, interferon-gamma, and other pro-inflammatory markers has been extensively documented in many studies.
With that observed rise in mind, several controlled studies have shown that adding an anti-inflammatory agent (aspirin, a nonsteroidal anti-inflammatory drug, a COX-2 inhibitor, or minocycline) to an antipsychotic can accentuate the therapeutic response, especially during a first episode of psychosis.6 Note also that second-generation antipsychotics have anti-inflammatory effects7 as well that might be part of their efficacy beyond blocking dopamine D2 receptors.
Decrease free radicals. Microglia are activated by psychosis to release free radicals, also known as reactive oxygen species; these include nitric oxide, superoxide, and peroxynitrate. All these species are destructive to brain tissue. Using an adjunctive strong antioxidant, such as N-acetyl cysteine,8 with an antipsychotic might help neutralize destructive effects of free radicals and protect the brain from tissue loss during a psychotic episode.
Avoid apoptosis inducers. Several substances can initiate programmed cell death (apoptosis), which is triggered during psychosis (believed to be caused by increased dopamine and, possibly, glutamate, activity) and which leads to brain atrophy. Patients with schizophrenia must be protected from these apoptosis inducers:
• amphetamine
• cocaine
• Cannabis
• lipid peroxidation products
• inflammatory cytokines.
Apoptosis can be inhibited by maintaining high levels of neurotrophic factors. Atypical, but not typical, antipsychotics increase levels of neurotrophins, such as NGF and BDNF.4 In addition, the Bcl-2 family of proteins inhibits apoptosis,9 and drugs such as lithium and valproate can induce Bcl-2 and protect against apoptosis and neuronal loss.3
Restore white-matter integrity. Numerous studies using diffusion tensor imaging have revealed that myelin is reduced or lacks integrity in schizophrenia. This results in loss of critical connectivity among brain regions, which might explain psychotic and cognitive symptoms. One possible way to repair white matter, which becomes more damaged after multiple psychotic episodes, is to use drugs indicated to treat the demyelinating disorder multiple sclerosis. Antagonists of LINGO-1, a negative regulator of axonal myelination, are a prominent possibility; a recent study reported altered signaling of LINGO-1 in schizophrenia.10
Decrease excessive glutamate. Because glutamate is neurotoxic and might contribute to brain-tissue loss during psychosis, it is important to reduce glutamate activity in schizophrenia. Lamotrigine and valproate are both known to do that.11 Several studies indicate that adjunctive lamotrigine might be helpful in schizophrenia.12
Inhibit caspase-3, also known as the “death cascade,” which is involved in brain-tissue loss. Eicosapentaenoic acid is an omega-3 fatty acid that inhibits caspase-3. Interestingly, omega-3 levels in patients with schizophrenia are significantly lower than in healthy subjects.13 Lithium also can inhibit caspase-3.
Do these proposals sound radical?
Most of the recommendations I’ve made here are not employed in the clinical practice of psychiatry. These ideas must be put to the test in controlled clinical trials.
The crux of my argument is that we need to think outside the “dopamine box” and focus on brain repair if we are to make progress in reversing, even preventing, neurodegeneration and clinical deterioration in this disabling brain syndrome. Just as cancer often is treated with rational polypharmacy, schizophrenia might need a similar approach. To vanquish schizophrenia—a goal that has eluded us—it is imperative to pursue radically novel and disruptive therapeutic strategies. The ideas I’ve listed here sound the call that the quest to repair the brain in schizophrenia must begin, and soon.
1. Agius N, Nandra, KS. Do atypical antipsychotics promote neurogenesis as a class effect? Psychiatr Danub. 2012;24(suppl 1):S191-S193.
2. Nasrallah HA, Hopkins T, Pixley SK. Differential effects of antipsychotic and antidepressant drugs on neurogenic regions in rats. Brain Res. 2010;1354:23-29.
3. Chiu CT, Wang Z, Hunsberger JG, et al. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol Rev. 2013;65(1):105-142.
4. Parikh V, Khan MM, Terry A, et al. Differential effects of typical and atypical antipsychotics on nerve growth factor and choline acetyltransferase expression in the cortex and nucleus basalis of rats. J Psychiatr Res. 2004;38(5):521-529.
5. Monji A, Kato TA, Mizoguchi Y, et al. Neuro-inflammation in schizophrenia especially focused on the role of microglia. Prog Neuropsychopharmacol Biol Psychiatry. 2013;42:115-121.
6. Sommer IE, deWitte L, Begemann M, et al. Nonsteriodal anti-inflammatory drugs in schizophrenia: ready for practice or a good start? A meta-analysis. J Clin Psychiatry. 2012;73(4):414-419.
7. Bian Q, Kato T, Monji A, et al. The effect of atypical anti-psychotics perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-gamma. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):42-48.
8. Berk M, Copolov D, Dean O, et al. N-acetyl cysteine as a glutathione precursor for schizophrenia—a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64(5):361-368.
9. Huang J, Fairbrother W, Reed JC, et al. Therapeutic targeting of Bcl-2 family for treatment of B-cell malignancies. Expert Rev Hematol. 2015;8(3):283-297.
10. Fernandez-Enright F, Andrews JL, Newell KA, et al. Novel implications of Lingo-1 and its signaling partners in schizophrenia. Transl Psychiatry. 2014; 4:e348.
11. Zink M, Correll CU. Glutamatergic agents for schizophrenia: current evidence and perspectives. Expert Rev Clin Pharmacol. 2015;8(3):335-352.
12. Kremer I, Vass A, Gorelik I, et al. Placebo-controlled trial of lamotrigine added to conventional and atypical antipsychotics in schizophrenia. Biol Psychiatry. 2004;56(6):444-446.
13. McEvoy J, Baillie RA, Zhu H, et al. Lipidomics reveals early metabolic changes in subjects with schizophrenia: effects of atypical antipsychotics. PLoS One. 2013;8(7):e68717.
1. Agius N, Nandra, KS. Do atypical antipsychotics promote neurogenesis as a class effect? Psychiatr Danub. 2012;24(suppl 1):S191-S193.
2. Nasrallah HA, Hopkins T, Pixley SK. Differential effects of antipsychotic and antidepressant drugs on neurogenic regions in rats. Brain Res. 2010;1354:23-29.
3. Chiu CT, Wang Z, Hunsberger JG, et al. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol Rev. 2013;65(1):105-142.
4. Parikh V, Khan MM, Terry A, et al. Differential effects of typical and atypical antipsychotics on nerve growth factor and choline acetyltransferase expression in the cortex and nucleus basalis of rats. J Psychiatr Res. 2004;38(5):521-529.
5. Monji A, Kato TA, Mizoguchi Y, et al. Neuro-inflammation in schizophrenia especially focused on the role of microglia. Prog Neuropsychopharmacol Biol Psychiatry. 2013;42:115-121.
6. Sommer IE, deWitte L, Begemann M, et al. Nonsteriodal anti-inflammatory drugs in schizophrenia: ready for practice or a good start? A meta-analysis. J Clin Psychiatry. 2012;73(4):414-419.
7. Bian Q, Kato T, Monji A, et al. The effect of atypical anti-psychotics perospirone, ziprasidone and quetiapine on microglial activation induced by interferon-gamma. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):42-48.
8. Berk M, Copolov D, Dean O, et al. N-acetyl cysteine as a glutathione precursor for schizophrenia—a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64(5):361-368.
9. Huang J, Fairbrother W, Reed JC, et al. Therapeutic targeting of Bcl-2 family for treatment of B-cell malignancies. Expert Rev Hematol. 2015;8(3):283-297.
10. Fernandez-Enright F, Andrews JL, Newell KA, et al. Novel implications of Lingo-1 and its signaling partners in schizophrenia. Transl Psychiatry. 2014; 4:e348.
11. Zink M, Correll CU. Glutamatergic agents for schizophrenia: current evidence and perspectives. Expert Rev Clin Pharmacol. 2015;8(3):335-352.
12. Kremer I, Vass A, Gorelik I, et al. Placebo-controlled trial of lamotrigine added to conventional and atypical antipsychotics in schizophrenia. Biol Psychiatry. 2004;56(6):444-446.
13. McEvoy J, Baillie RA, Zhu H, et al. Lipidomics reveals early metabolic changes in subjects with schizophrenia: effects of atypical antipsychotics. PLoS One. 2013;8(7):e68717.
Managing first-episode psychosis: An early stage of schizophrenia with distinct treatment needs
The less time that passes between the onset of psychosis and initiation of appropriate treatment, the greater the patient’s odds of recovery.1 However, relapse prevention is a major clinical challenge because >80% of patients will relapse within 5 years, and, on average, 40% to 50% of patients with a first-episode schizophrenia will relapse within 2 years depending on the definition used and patient characteristics.2 Although there are several explanations and contributing factors to relapses, nonadherence—partial or complete discontinuation of antipsychotics—is a primary risk factor, contributing to a 5-fold increase in relapse risk.3
As such, optimal antipsychotic selection, dosing, and monitoring play an important role in managing this illness. Patients with first-episode psychosis (FEP) are unusual in some ways, compared with patients with multiple episodes of psychosis and represent a different stage of schizophrenia.
In this 2-part series, we will discuss pharmacotherapy for FEP. This article focuses on antipsychotic selection, dosage, and duration of treatment among these patients. The second article, in the July 2015 issue, reviews the rationale and evidence for non-standard, first-line therapies, including long-acting injectable antipsychotics and clozapine.
Defining FEP
FEP refers to a patient who has presented, been evaluated, and received treatment for the first psychotic episode associated with a schizophrenia spectrum diagnosis.4 FEP is part of a trajectory marked by tran sitional periods. The patient transitions from being “healthy” to a prodromal state characterized by: (1) nonpsychotic behavioral disturbances such as depression or obsessive-compulsive disorder, (2) attenuated psychotic symptoms not requiring treatment, then converting to (3) psychotic symptoms prompting initial presentation for antipsychotic pharmacotherapy, leading to (4) a formal diagnosis of schizophreniform disorder and, subsequently, schizophrenia, requiring treatment to stabilize symptoms.
There are 2 critical periods along this continuum: prodromal stage and the duration of untreated psychosis (DUP). The prodromal period is a retrospectively identified time where the patient shows initial nonpsychotic disturbances (eg, cognitive and behavioral symptoms) before exhibiting clinical diagnostic criteria for a schizophrenia spectrum disorder. Approximately one-third of patients exhibiting these symptoms convert to psychosis within 1 year, and early treatment engagement at this stage has been shown to improve outcomes.5 The DUP is the time from when a patient has noticeable psychotic symptoms to initiation of drug treatment. The DUP is a consistent predictor of clinical outcome in schizophrenia, including negative symptoms, quality of life, and functional capacity.1
Antipsychotic selection
Treatment goals for FEP patients include:
• minimizing the DUP
• rapidly stabilizing psychosis
• achieving full symptomatic remission
• preventing relapse.
Several treatment guidelines for managing schizophrenia offer variable recommendations for initial antipsychotic treatment in patients with first-episode schizophrenia (Table 1).6-15 Most recommend second-generation antipsychotics (SGAs) over first-generation antipsychotics (FGAs)6,8,9,13,15 with specific recommendations on minimizing neurologic and metabolic adverse effects—to which FEP patients are susceptible—by avoiding high-potency and neurotoxic FGAs (eg, haloperidol and fluphenazine),7 clozapine,11,14 olanzapine,11 or ziprasidone.14 Two guidelines—the National Institute for Health and Care Excellence and the Scottish Intercollegiate Guidelines Network—do not state a preference for antipsychotic selection.10,12
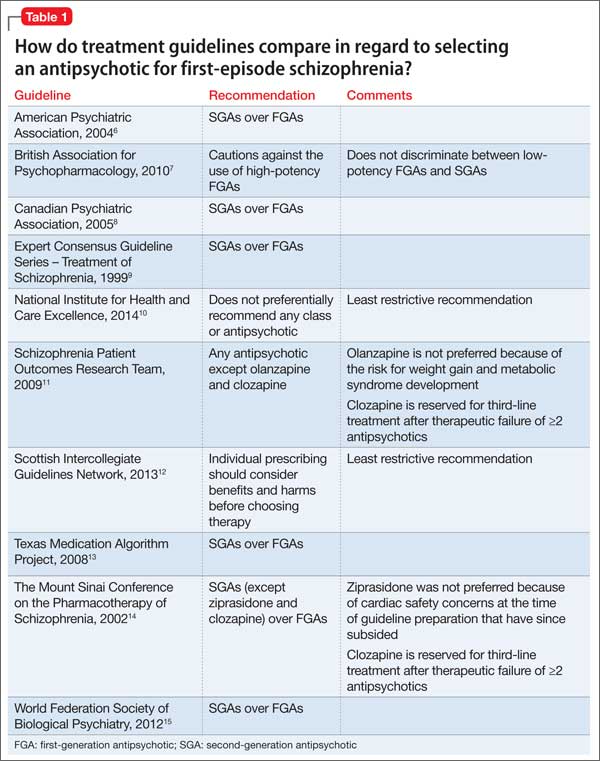
The rationale for these recommendations is based on efficacy data, tolerability differences, FDA-approved indications, and recent FDA approvals with sparse post-marketing data. Of note, there are a lack of robust data for newer antipsychotics (eg, aripiprazole, paliperidone, iloperidone, asenapine, and lurasidone) in effectively and safely treating FEP; however, given the results of other antipsychotics studies, it is likely the efficacy and tolerability of these drugs can be extrapolated from experience with multi-episode patients.
Study design and demographics. Research studies of FEP share some similarities in study design; however, there is enough variability to make it difficult to compare studies and generalize findings (Table 2).16 The variability of DUP is a limitation when comparing studies because it is a significant predictor of clinical outcome. Patients who abuse substances—and often are more challenging to treat17—typically are excluded from these trials, which could explain the high response rate documented in studies of first-episode schizophrenia.

In addition, some FEP patients included in clinical trials might not be truly antipsychotic naïve; an estimated 25% to 75% of patients in these studies are antipsychotic naïve. This is an important consideration when comparing data on adverse effects that occur early in treatment. Additionally, acknowledging the advantages and disadvantages of how to handle missing data is critical because of the high dropout rate observed in these studies.18
Efficacy. There is a high response rate to antipsychotic therapy—ranging from 46% to 96%, depending on the study—in patients with first-episode schizophrenia.3 The response mainly is seen in reduction of positive symptoms because typically negative and cognitive symptoms do not respond to antipsychotics. One study reported only 29% of patients achieved both positive and negative symptom remission.19 It is likely that secondary negative symptoms caused by social withdrawal, reduced speech, and avoidance improve when positive symptoms subside, but primary negative symptoms endure.In general, there is a lack of evidence suggesting that 1 antipsychotic class or agent is more effective than another. Studies mainly assess effectiveness using the primary outcome measure of all-cause discontinuation, such as the Clinical Antipsychotic Trials of Intervention Effectiveness study.20 This outcome measure is a mixture of patient preference, tolerability, and efficacy that provides a more generalizable gauge on how well the treatment works in the clinic rather than tightly regulated settings such as clinical trials. A recent meta-analysis supports no differences in efficacy among antipsychotics in early-episode psychosis.21
Tolerability. Because there are no significant differences among antipsychotic classes or agents in terms of efficacy in first-episode schizophrenia, drug selection is guided mainly by (1) the adverse effect profile and (2) what should be avoided depending on patient-specific variables. Evidence suggests first-episode patients are more sensitive to adverse effects of antipsychotics, particularly neurologic side effects (see this article at CurrentPsychiatry.com for a table comparing adverse effects of antipsychotics in first-episode psychosis).18,22-29 Overall adverse effect profiles remain similar across FEP or multi-episode patients, but tend to be more exaggerated in drug-naïve patients with FEP.
Regarding FGA side effects, McEvoy et al18 demonstrated the neuroleptic threshold occurs at 50% lower haloperidol dosages in patients with first-episode schizophrenia (2.1 mg/d) compared with multi-episode schizophrenia (4.3 mg/d). Other trials suggest SGAs are associated with a lower risk of extrapyramidal side effects (EPS) or use of adjunctive therapies such as anticholinergic drugs or benzodiazepines.23-27 An exception to this statement is that higher risperidone dosages (≥4 to 6 mg/d) have been found to have higher rates of EPS and use of adjunctive medications to treat these symptoms in FEP.26 This is important because studies report higher discontinuation rates with more severe adverse effects of antipsychotics.
Cardiometabolic effects are of particular concern in first-episode patients because most weight gain happens in the first 3 to 4 months of treatment and remains throughout the first year.18,24,29,30 Studies have shown that olanzapine, quetiapine, and risperidone are associated with more clinically significant weight gain compared with haloperidol and ziprasidone.23-25 Olanzapine-associated weight gain has been reported to be twice that of quetiapine and risperidone.18 Regardless, the EUFEST trial did not find a difference in clinically significant weight gain after 12 months among the antipsychotics studied, including haloperidol and ziprasidone.25
Weight gain associated with these antipsychotics is accompanied by changes in fasting triglycerides, glucose, total cholesterol,23 and high-density lipoprotein cholesterol as well as an increase in body mass index (BMI) categorization29 (eg, shift from normal to overweight).18,25 Patients with lower baseline BMI and in racial minority groups might experience more rapid weight gain regardless of antipsychotic selection.29,30
Hyperprolactinemia could be under-recognized and could contribute to early treatment discontinuation.31 Evidence in patients with first-episode schizophrenia suggests similar outcomes as those seen in multi-episode patients, in whom risperidone is associated with higher prolactin elevations and clinically significant hyperprolactinemia (eg, galactorrhea and gynecomastia) compared with olanzapine, quetiapine, and low-dose haloperidol.18,23,24 However, there is a lack of studies that assess whether long-term therapy with strong D2 receptor antagonists increases the risk of bone demineralization or pathological fractures when started before patients’ bones reach maximum density in their mid-20s.31
Antipsychotic dosing
Given the high rate of treatment response in FEP and patients’ higher sensitivity to antipsychotic adverse effects, particularly EPS, guidelines recommend antipsychotic dosages lower than those used for multi-episode schizophrenia,11 especially FGAs. Based on trial data, commonly used dosages include:
• haloperidol, ≤5 mg/d23-25,29
• olanzapine, 10 mg/d18,23,25,29
• risperidone, ≤4 to 6 mg/d.18,24,29,32
In general, haloperidol and risperidone, 2 to 3 mg/d, were well tolerated and effective in trials. Higher quetiapine dosages of 500 to 600 mg/d could be required.11,18,25
According to a survey on prescribing practices of antipsychotic selection and dosing in first-episode schizophrenia,4 clinical prescribing practices tend to use unnecessarily high initial antipsychotic dosing compared with trial data. There also is variability in the usual target antipsychotic dosage ranging from 50% lower dosages to normal dosages in chronic schizophrenia to above FDA-approved maximum dosages for olanzapine (which may be necessary to counteract tobacco-induced cytochrome P450 1A2 enzyme induction).
In addition, these clinicians reported prescribing aripiprazole, an antipsychotic with weaker evidence (eg, case reports, case series, open-label studies) supporting its efficacy and tolerability in FEP. These prescribing practices could reflect attempts to reduce the DUP and achieve symptom remission, so long as tolerability is not a concern.
Essentially, prescribed dosages should be based on symptom improvement and tolerability. This ideal dosage will vary as illustrated by Kapur et al,33 who reported that FEP patients (N = 20) given haloperidol, 1 mg or 2.5 mg/d, had D2 receptor occupancy rates of 38% to 87%, which was significantly dose-related (1 mg/d mean = 59%, 2.5 mg/d mean = 75%). Clinical response and EPS significantly increased as D2 receptor occupancy exceeded 65% and 78%, respectively.
Antipsychotic response
When should you expect to see symptom improvement in patients with first-episode schizophrenia?
Emsley et al34 reported a 77.6% response rate among first-episode patients (N = 522) treated with low dosages of risperidone (mean modal dosage [MMD] = 3.3 mg/d) and haloperidol (MMD = 2.9 mg/d). They found variable response times that were evenly dispersed over a 10-week period. Nearly one-quarter (22.5%) did not respond until after week 4 and 11.2% did not respond until after week 8. In a study of FEP patients (N = 112) treated with olanzapine (MMD = 11.8 mg/d) or risperidone (MMD = 3.9 mg/d), Gallego et al35 reported a cumulative response of 39.6% at week 8 and 65.1% at week 16.
Although there is evidence that, among multi-episode patients, early nonresponse to antipsychotic therapy could predict subsequent nonresponse,36 the evidence is mixed for first-episode schizophrenia. Studies by Emsley et al34 and Gallego et al35 did not find that early nonresponse at weeks 1 or 2 predicted subsequent nonresponse at week 4 or later. However, other studies support the idea that early nonresponse predicts subsequent nonresponse and early antipsychotic response predicts future response in first-episode patients, with good specificity and sensitivity.37,38
Overall, treatment response in first-episode schizophrenia is variable. An adequate antipsychotic trial may be longer, 8 to 16 weeks, compared with 4 to 8 weeks in multi-episode patients. Because research suggests that failure to respond to treatment may lead to medication nonadherence,39 it is reasonable to consider switching antipsychotics when a patient experiences minimal or no response to antipsychotic therapy at week 2; however, this should be a patient-specific decision.
How long should you continue therapy after symptom remission?
There is a lack of consensus on the duration of therapy for a patient treated for first-episode schizophrenia because a small percentage (10% to 20%) do not relapse after the first psychotic episode.3 In general, treatment guidelines and expert consensus statements recommend at least 1 to 2 years of treatment before considering a discontinuation trial.7,10-11 Discuss the benefits and risks of maintenance treatment with your patient and obtain informed consent. With patients with minimal insight, obtaining proper consent is not possible and the physician must exercise judgment unilaterally, if necessary, after educating the family.
After at least 12 months of treatment, antipsychotic therapy could continue indefinitely, depending on patient-specific factors. There are no predictors for identifying patients who do not require maintenance therapy beyond the first psychotic episode. The absence of negative and cognitive deficits could provide clues that a patient might be a candidate for antipsychotic tapering.
Predicting the treatment course
Research investigating clinical predictors or biomarkers that forecast whether a patient will respond to treatment is preliminary. Many characteristics have been identified (Table 31,3,4,23,25,40) and include shorter DUP,1 poorer premorbid function,3 antipsychotic discontinuation,3 a trusting patient-doctor relationship,41 and antipsychotic-related adverse effects,23,25 which are predictive of response, nonresponse, relapse, adherence, and nonadherence, respectively.
Bottom Line
The goals of pharmacological treatment of first-episode schizophrenia are to minimize the duration of untreated psychosis and target full remission of positive symptoms using the lowest possible antipsychotic dosages. Pharmacotherapy should continued for 1 to 2 years, with longer duration considered if it is discussed with the patient and with vigilant monitoring for adverse effects and suboptimal medication nonadherence to prevent relapse.
Editor’s note: The second article in this series in the July 2015 issue reviews the rationale and evidence for non-standard, first-line therapies, including long-acting injectable antipsychotics and clozapine.
Related Resources
• Recovery After an Initial Schizophrenia Episode (RAISE) Project Early Treatment Program. National Institute of Mental Health. http://raiseetp.org.
• Martens L, Baker S. Promoting recovery from first episode psychosis: a guide for families. Centre for Addiction and Mental Health. http://www.camh.ca/en/hospital/ Documents/www.camh.net/AboutCAMH/Guideto CAMH/MentalHealthPrograms/SchizophreniaProgram/ 3936PromotingRecoveryFirstEpisodePsychosisfinal.pdf.
Drug Brand Names
Aripiprazole • Abilify Lurasidone • Latuda
Asenapine • Saphris Olanzapine • Zyprexa
Clozapine • Clozaril Paliperidone • Invega
Fluphenazine • Prolixin Quetiapine • Seroquel
Iloperidone • Fanapt Risperidone • Risperdal
Haloperidol • Haldol Ziprasidone • Geodon
Disclosures
Dr. Gardner reports no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
Dr. Nasrallah is a consultant to Acadia, Alkermes, Lundbeck, Janssen, Merck, Otsuka, and Sunovion, and is a speaker for Alkermes, Lundbeck, Janssen, Otsuka, and Sunovion.
1. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804.
2. Bradford DW, Perkins DO, Lieberman JA. Pharmacological management of first-episode schizophrenia and related nonaffective psychoses. Drugs. 2003;63(21):2265-2283.
3. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following a response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
4. Weiden PJ, Buckley PF, Grody M. Understanding and treating “first-episode” schizophrenia. Psychiatr Clin North Am. 2007;30(3):481-510.
5. Madaan V, Bestha DP, Kolli V. Schizophrenia prodrome: an optimal approach. Current Psychiatry. 2014;13(3):16-20, 29-30.
6. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
7. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620.
8. Canadian Psychiatric Association. Clinical practice guideline. Treatment of schizophrenia. Can J Psychiatry. 2005;50(13 suppl 1):7S-57S.
9. McEvoy JP, Scheifler PL, Frances A. Treatment of schizophrenia 1999. Expert consensus guideline series. J Clin Psychiatry. 1999;60(suppl 11):4-80.
10. National Institute for Health and Care Excellence (NICE). Clinical guideline 178: Psychosis and schizophrenia in adults: treatment and management. London, United Kingdom: National Institute for Health and Care Excellence (NICE); 2014.
11. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
12. Scottish Intercollegiate Guidelines Network (SIGN). Management of schizophrenia. Edinburgh, Scotland: Scottish Intercollegiate Guidelines Network; 2013. SIGN publication no. 131.
13. Argo TR, Crismon ML, Miller AL, et al. Texas Medication Algorithm Project procedural manual. Schizophrenia treatment algorithms. Austin, Texas: Texas Department of State Health Services; 2008.
14. Marder SR, Essock SM, Miller Al, et al. The Mount Sinai conference on the pharmacotherapy of schizophrenia. Schizophr Bull. 2002;28(1):5-16.
15. Bandelow B, Zohar J, Hollander E, et al; WFSBP Task Force on Treatment Guidelines for Anxiety, Obsessive-Compulsive and Post-Traumatic Stress Disorders. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
16. Robinson DG, Woerner MG, Alvir JMJ, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psych. 1999;56(3):241-247.
17. Green AI, Tohen MF, Hamer RM, et al. First episode schizophrenia-related psychosis and substance use disorders: acute response to olanzapine and haloperidol. Schizophr Res. 2004;66(2-3):125-135.
18. McEvoy JP, Lieberman JA, Perkins DO, et al. Efficacy and tolerability of olanzapine, quetiapine, and risperidone in the treatment of early psychosis: a randomized, double-blind 52-week comparison. Am J Psychiatry. 2007;164(7): 1050-1060.
19. Henry LP, Amminger GP, Harris MG, et al. The EPPIC follow-up study of first-episode psychosis: longer-term clinical and functional outcome 7 years after index admission. J Clin Psychiatry. 2010;71(6):716-728.
20. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. New Engl J Med. 2005; 353(12):1209-1223.
21. Crossley NA, Constante M, McGuire P, et al. Efficacy of atypical v. typical antipsychotics in the treatment of early psychosis: meta-analysis. Br J Psychiatry. 2010;196(6):434-439.
22. McEvoy JP, Hogarty GE, Steingard S. Optimal dose of neuroleptic in acute schizophrenia: a controlled study of the neuroleptic threshold and higher haloperidol dose. Arch Gen Psych. 1991;48(8):739-745.
23. Lieberman JA, Tollefson G, Tohen M, et al; HGDH Study Group. Comparative efficacy and safety of atypical and conventional antipsychotic drugs in first-episode psychosis: a randomized, double-blind trial of olanzapine versus haloperidol. Am J Psychiatry. 2003;160(8):1396-1404.
24. Schooler N, Rabinowitz J, Davidson M, et al; Early Psychosis Global Working Group. Risperidone and haloperidol in first-episode psychosis: a long-term randomized trial. Am J Psychiatry. 2005;162(5):947-953.
25. Kahn RS, Fleischhacker WW, Boter H, et al; EUFEST study group. Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: an open randomised clinical trial. Lancet. 2008;371(9618):1085-1097.
26. Emsley RA; Risperidone Working Group. Risperidone in the treatment of first-episode psychotic patients: a double-blind multicenter study. Schizophr Bull. 1999;25(4):721-729.
27. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naïve first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003.
28. Girgis RR, Phillips MR, Li X, et al. Clozapine v. chlorpromazine in treatment-naive, first-episode schizophrenia: 9-year outcomes of a randomised clinical trial. Br J Psychiatry. 2011;199(4):281-288.
29. Robinson DG, Woerner MG, Napolitano B, et al. Randomized comparison of olanzapine versus risperidone for the treatment of first-episode schizophrenia: 4-month outcomes. Am J Psychiatry. 2006;163(12):2096-2102.
30. Zipursky RB, Gu H, Green AI, et al. Course and predictors of weight gain in people with first-episode psychosis treated with olanzapine or haloperidol. Br J Psychiatry. 2005;187:537-543.
31. Taylor M, Waight A, Leonard B. Advances in the understanding and challenges facing the management of first-episode schizophrenia. J Psychopharmacol. 2012; 26(suppl 5):3-5.
32. Merlo MC, Hofer H, Gekle W, et al. Risperidone, 2mg/day vs. 4mg/day, in first-episode, acutely psychotic patients: treatment efficacy and effects on fine motor functioning. J Clin Psychiatry. 2002;63(10):885-891.
33. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D2 occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157(4):514-520.
34. Emsley R, Rabinowitz J, Medori R. Time course for antipsychotic treatment response in first-episode schizophrenia. Am J Psychiatry. 2006;163(4):743-745.
35. Gallego JA, Robinson DG, Sevy SM, et al. Time to treatment response in first-episode schizophrenia: should acute treatment trials last several months? J Clin Psychiatry. 2011;72(12):1691-1696.
36. Gardner KN, Bostwick JR. Antipsychotic treatment response in schizophrenia. Am J Health Sys Pharm. 2012;69(21):1872-1879.
37. Stauffer VL, Case M, Kinon BJ, et al. Early response to antipsychotic therapy as a clinical marker of subsequent response in the treatment of patients with first-episode psychosis. Psychiatry Res. 2011;187(1-2):42-48.
38. Schennach-Wolff R, Seemüller FH, Mayr A, et al. An early improvement threshold to predict response and remission in first-episode schizophrenia. Br J Psychiatry. 2010;196(6):460-466.
39. Perkins DO, Gu H, Weiden PJ, et al; Comparison of Atypicals in First Episode study group. Predictors of treatment discontinuation and medication nonadherence in patients recovering from a first episode of schizophrenia, schizophreniform disorder, or schizoaffective disorder: a randomized, double-blind, flexible-dose, multicenter study. J Clin Psychiatry. 2008;69(1):106-113.
40. Garner B, Berger GE, Nicolo JP, et al. Pituitary volume and early treatment response in drug-naïve first-episode psychosis patients. Schizophr Res. 2009;113(1):65-71.
41. Sapra M, Weiden PJ, Schooler NR, et al. Reasons for adherence and nonadherence: a pilot study comparing first-and multi-episode schizophrenia patients. Clin Schizophr Relat Psychoses. 2014;7(4):199-206.
The less time that passes between the onset of psychosis and initiation of appropriate treatment, the greater the patient’s odds of recovery.1 However, relapse prevention is a major clinical challenge because >80% of patients will relapse within 5 years, and, on average, 40% to 50% of patients with a first-episode schizophrenia will relapse within 2 years depending on the definition used and patient characteristics.2 Although there are several explanations and contributing factors to relapses, nonadherence—partial or complete discontinuation of antipsychotics—is a primary risk factor, contributing to a 5-fold increase in relapse risk.3
As such, optimal antipsychotic selection, dosing, and monitoring play an important role in managing this illness. Patients with first-episode psychosis (FEP) are unusual in some ways, compared with patients with multiple episodes of psychosis and represent a different stage of schizophrenia.
In this 2-part series, we will discuss pharmacotherapy for FEP. This article focuses on antipsychotic selection, dosage, and duration of treatment among these patients. The second article, in the July 2015 issue, reviews the rationale and evidence for non-standard, first-line therapies, including long-acting injectable antipsychotics and clozapine.
Defining FEP
FEP refers to a patient who has presented, been evaluated, and received treatment for the first psychotic episode associated with a schizophrenia spectrum diagnosis.4 FEP is part of a trajectory marked by tran sitional periods. The patient transitions from being “healthy” to a prodromal state characterized by: (1) nonpsychotic behavioral disturbances such as depression or obsessive-compulsive disorder, (2) attenuated psychotic symptoms not requiring treatment, then converting to (3) psychotic symptoms prompting initial presentation for antipsychotic pharmacotherapy, leading to (4) a formal diagnosis of schizophreniform disorder and, subsequently, schizophrenia, requiring treatment to stabilize symptoms.
There are 2 critical periods along this continuum: prodromal stage and the duration of untreated psychosis (DUP). The prodromal period is a retrospectively identified time where the patient shows initial nonpsychotic disturbances (eg, cognitive and behavioral symptoms) before exhibiting clinical diagnostic criteria for a schizophrenia spectrum disorder. Approximately one-third of patients exhibiting these symptoms convert to psychosis within 1 year, and early treatment engagement at this stage has been shown to improve outcomes.5 The DUP is the time from when a patient has noticeable psychotic symptoms to initiation of drug treatment. The DUP is a consistent predictor of clinical outcome in schizophrenia, including negative symptoms, quality of life, and functional capacity.1
Antipsychotic selection
Treatment goals for FEP patients include:
• minimizing the DUP
• rapidly stabilizing psychosis
• achieving full symptomatic remission
• preventing relapse.
Several treatment guidelines for managing schizophrenia offer variable recommendations for initial antipsychotic treatment in patients with first-episode schizophrenia (Table 1).6-15 Most recommend second-generation antipsychotics (SGAs) over first-generation antipsychotics (FGAs)6,8,9,13,15 with specific recommendations on minimizing neurologic and metabolic adverse effects—to which FEP patients are susceptible—by avoiding high-potency and neurotoxic FGAs (eg, haloperidol and fluphenazine),7 clozapine,11,14 olanzapine,11 or ziprasidone.14 Two guidelines—the National Institute for Health and Care Excellence and the Scottish Intercollegiate Guidelines Network—do not state a preference for antipsychotic selection.10,12
The rationale for these recommendations is based on efficacy data, tolerability differences, FDA-approved indications, and recent FDA approvals with sparse post-marketing data. Of note, there are a lack of robust data for newer antipsychotics (eg, aripiprazole, paliperidone, iloperidone, asenapine, and lurasidone) in effectively and safely treating FEP; however, given the results of other antipsychotics studies, it is likely the efficacy and tolerability of these drugs can be extrapolated from experience with multi-episode patients.
Study design and demographics. Research studies of FEP share some similarities in study design; however, there is enough variability to make it difficult to compare studies and generalize findings (Table 2).16 The variability of DUP is a limitation when comparing studies because it is a significant predictor of clinical outcome. Patients who abuse substances—and often are more challenging to treat17—typically are excluded from these trials, which could explain the high response rate documented in studies of first-episode schizophrenia.
In addition, some FEP patients included in clinical trials might not be truly antipsychotic naïve; an estimated 25% to 75% of patients in these studies are antipsychotic naïve. This is an important consideration when comparing data on adverse effects that occur early in treatment. Additionally, acknowledging the advantages and disadvantages of how to handle missing data is critical because of the high dropout rate observed in these studies.18
Efficacy. There is a high response rate to antipsychotic therapy—ranging from 46% to 96%, depending on the study—in patients with first-episode schizophrenia.3 The response mainly is seen in reduction of positive symptoms because typically negative and cognitive symptoms do not respond to antipsychotics. One study reported only 29% of patients achieved both positive and negative symptom remission.19 It is likely that secondary negative symptoms caused by social withdrawal, reduced speech, and avoidance improve when positive symptoms subside, but primary negative symptoms endure.In general, there is a lack of evidence suggesting that 1 antipsychotic class or agent is more effective than another. Studies mainly assess effectiveness using the primary outcome measure of all-cause discontinuation, such as the Clinical Antipsychotic Trials of Intervention Effectiveness study.20 This outcome measure is a mixture of patient preference, tolerability, and efficacy that provides a more generalizable gauge on how well the treatment works in the clinic rather than tightly regulated settings such as clinical trials. A recent meta-analysis supports no differences in efficacy among antipsychotics in early-episode psychosis.21
Tolerability. Because there are no significant differences among antipsychotic classes or agents in terms of efficacy in first-episode schizophrenia, drug selection is guided mainly by (1) the adverse effect profile and (2) what should be avoided depending on patient-specific variables. Evidence suggests first-episode patients are more sensitive to adverse effects of antipsychotics, particularly neurologic side effects (see this article at CurrentPsychiatry.com for a table comparing adverse effects of antipsychotics in first-episode psychosis).18,22-29 Overall adverse effect profiles remain similar across FEP or multi-episode patients, but tend to be more exaggerated in drug-naïve patients with FEP.
Regarding FGA side effects, McEvoy et al18 demonstrated the neuroleptic threshold occurs at 50% lower haloperidol dosages in patients with first-episode schizophrenia (2.1 mg/d) compared with multi-episode schizophrenia (4.3 mg/d). Other trials suggest SGAs are associated with a lower risk of extrapyramidal side effects (EPS) or use of adjunctive therapies such as anticholinergic drugs or benzodiazepines.23-27 An exception to this statement is that higher risperidone dosages (≥4 to 6 mg/d) have been found to have higher rates of EPS and use of adjunctive medications to treat these symptoms in FEP.26 This is important because studies report higher discontinuation rates with more severe adverse effects of antipsychotics.
Cardiometabolic effects are of particular concern in first-episode patients because most weight gain happens in the first 3 to 4 months of treatment and remains throughout the first year.18,24,29,30 Studies have shown that olanzapine, quetiapine, and risperidone are associated with more clinically significant weight gain compared with haloperidol and ziprasidone.23-25 Olanzapine-associated weight gain has been reported to be twice that of quetiapine and risperidone.18 Regardless, the EUFEST trial did not find a difference in clinically significant weight gain after 12 months among the antipsychotics studied, including haloperidol and ziprasidone.25
Weight gain associated with these antipsychotics is accompanied by changes in fasting triglycerides, glucose, total cholesterol,23 and high-density lipoprotein cholesterol as well as an increase in body mass index (BMI) categorization29 (eg, shift from normal to overweight).18,25 Patients with lower baseline BMI and in racial minority groups might experience more rapid weight gain regardless of antipsychotic selection.29,30
Hyperprolactinemia could be under-recognized and could contribute to early treatment discontinuation.31 Evidence in patients with first-episode schizophrenia suggests similar outcomes as those seen in multi-episode patients, in whom risperidone is associated with higher prolactin elevations and clinically significant hyperprolactinemia (eg, galactorrhea and gynecomastia) compared with olanzapine, quetiapine, and low-dose haloperidol.18,23,24 However, there is a lack of studies that assess whether long-term therapy with strong D2 receptor antagonists increases the risk of bone demineralization or pathological fractures when started before patients’ bones reach maximum density in their mid-20s.31
Antipsychotic dosing
Given the high rate of treatment response in FEP and patients’ higher sensitivity to antipsychotic adverse effects, particularly EPS, guidelines recommend antipsychotic dosages lower than those used for multi-episode schizophrenia,11 especially FGAs. Based on trial data, commonly used dosages include:
• haloperidol, ≤5 mg/d23-25,29
• olanzapine, 10 mg/d18,23,25,29
• risperidone, ≤4 to 6 mg/d.18,24,29,32
In general, haloperidol and risperidone, 2 to 3 mg/d, were well tolerated and effective in trials. Higher quetiapine dosages of 500 to 600 mg/d could be required.11,18,25
According to a survey on prescribing practices of antipsychotic selection and dosing in first-episode schizophrenia,4 clinical prescribing practices tend to use unnecessarily high initial antipsychotic dosing compared with trial data. There also is variability in the usual target antipsychotic dosage ranging from 50% lower dosages to normal dosages in chronic schizophrenia to above FDA-approved maximum dosages for olanzapine (which may be necessary to counteract tobacco-induced cytochrome P450 1A2 enzyme induction).
In addition, these clinicians reported prescribing aripiprazole, an antipsychotic with weaker evidence (eg, case reports, case series, open-label studies) supporting its efficacy and tolerability in FEP. These prescribing practices could reflect attempts to reduce the DUP and achieve symptom remission, so long as tolerability is not a concern.
Essentially, prescribed dosages should be based on symptom improvement and tolerability. This ideal dosage will vary as illustrated by Kapur et al,33 who reported that FEP patients (N = 20) given haloperidol, 1 mg or 2.5 mg/d, had D2 receptor occupancy rates of 38% to 87%, which was significantly dose-related (1 mg/d mean = 59%, 2.5 mg/d mean = 75%). Clinical response and EPS significantly increased as D2 receptor occupancy exceeded 65% and 78%, respectively.
Antipsychotic response
When should you expect to see symptom improvement in patients with first-episode schizophrenia?
Emsley et al34 reported a 77.6% response rate among first-episode patients (N = 522) treated with low dosages of risperidone (mean modal dosage [MMD] = 3.3 mg/d) and haloperidol (MMD = 2.9 mg/d). They found variable response times that were evenly dispersed over a 10-week period. Nearly one-quarter (22.5%) did not respond until after week 4 and 11.2% did not respond until after week 8. In a study of FEP patients (N = 112) treated with olanzapine (MMD = 11.8 mg/d) or risperidone (MMD = 3.9 mg/d), Gallego et al35 reported a cumulative response of 39.6% at week 8 and 65.1% at week 16.
Although there is evidence that, among multi-episode patients, early nonresponse to antipsychotic therapy could predict subsequent nonresponse,36 the evidence is mixed for first-episode schizophrenia. Studies by Emsley et al34 and Gallego et al35 did not find that early nonresponse at weeks 1 or 2 predicted subsequent nonresponse at week 4 or later. However, other studies support the idea that early nonresponse predicts subsequent nonresponse and early antipsychotic response predicts future response in first-episode patients, with good specificity and sensitivity.37,38
Overall, treatment response in first-episode schizophrenia is variable. An adequate antipsychotic trial may be longer, 8 to 16 weeks, compared with 4 to 8 weeks in multi-episode patients. Because research suggests that failure to respond to treatment may lead to medication nonadherence,39 it is reasonable to consider switching antipsychotics when a patient experiences minimal or no response to antipsychotic therapy at week 2; however, this should be a patient-specific decision.
How long should you continue therapy after symptom remission?
There is a lack of consensus on the duration of therapy for a patient treated for first-episode schizophrenia because a small percentage (10% to 20%) do not relapse after the first psychotic episode.3 In general, treatment guidelines and expert consensus statements recommend at least 1 to 2 years of treatment before considering a discontinuation trial.7,10-11 Discuss the benefits and risks of maintenance treatment with your patient and obtain informed consent. With patients with minimal insight, obtaining proper consent is not possible and the physician must exercise judgment unilaterally, if necessary, after educating the family.
After at least 12 months of treatment, antipsychotic therapy could continue indefinitely, depending on patient-specific factors. There are no predictors for identifying patients who do not require maintenance therapy beyond the first psychotic episode. The absence of negative and cognitive deficits could provide clues that a patient might be a candidate for antipsychotic tapering.
Predicting the treatment course
Research investigating clinical predictors or biomarkers that forecast whether a patient will respond to treatment is preliminary. Many characteristics have been identified (Table 31,3,4,23,25,40) and include shorter DUP,1 poorer premorbid function,3 antipsychotic discontinuation,3 a trusting patient-doctor relationship,41 and antipsychotic-related adverse effects,23,25 which are predictive of response, nonresponse, relapse, adherence, and nonadherence, respectively.
Bottom Line
The goals of pharmacological treatment of first-episode schizophrenia are to minimize the duration of untreated psychosis and target full remission of positive symptoms using the lowest possible antipsychotic dosages. Pharmacotherapy should continued for 1 to 2 years, with longer duration considered if it is discussed with the patient and with vigilant monitoring for adverse effects and suboptimal medication nonadherence to prevent relapse.
Editor’s note: The second article in this series in the July 2015 issue reviews the rationale and evidence for non-standard, first-line therapies, including long-acting injectable antipsychotics and clozapine.
Related Resources
• Recovery After an Initial Schizophrenia Episode (RAISE) Project Early Treatment Program. National Institute of Mental Health. http://raiseetp.org.
• Martens L, Baker S. Promoting recovery from first episode psychosis: a guide for families. Centre for Addiction and Mental Health. http://www.camh.ca/en/hospital/ Documents/www.camh.net/AboutCAMH/Guideto CAMH/MentalHealthPrograms/SchizophreniaProgram/ 3936PromotingRecoveryFirstEpisodePsychosisfinal.pdf.
Drug Brand Names
Aripiprazole • Abilify Lurasidone • Latuda
Asenapine • Saphris Olanzapine • Zyprexa
Clozapine • Clozaril Paliperidone • Invega
Fluphenazine • Prolixin Quetiapine • Seroquel
Iloperidone • Fanapt Risperidone • Risperdal
Haloperidol • Haldol Ziprasidone • Geodon
Disclosures
Dr. Gardner reports no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
Dr. Nasrallah is a consultant to Acadia, Alkermes, Lundbeck, Janssen, Merck, Otsuka, and Sunovion, and is a speaker for Alkermes, Lundbeck, Janssen, Otsuka, and Sunovion.
The less time that passes between the onset of psychosis and initiation of appropriate treatment, the greater the patient’s odds of recovery.1 However, relapse prevention is a major clinical challenge because >80% of patients will relapse within 5 years, and, on average, 40% to 50% of patients with a first-episode schizophrenia will relapse within 2 years depending on the definition used and patient characteristics.2 Although there are several explanations and contributing factors to relapses, nonadherence—partial or complete discontinuation of antipsychotics—is a primary risk factor, contributing to a 5-fold increase in relapse risk.3
As such, optimal antipsychotic selection, dosing, and monitoring play an important role in managing this illness. Patients with first-episode psychosis (FEP) are unusual in some ways, compared with patients with multiple episodes of psychosis and represent a different stage of schizophrenia.
In this 2-part series, we will discuss pharmacotherapy for FEP. This article focuses on antipsychotic selection, dosage, and duration of treatment among these patients. The second article, in the July 2015 issue, reviews the rationale and evidence for non-standard, first-line therapies, including long-acting injectable antipsychotics and clozapine.
Defining FEP
FEP refers to a patient who has presented, been evaluated, and received treatment for the first psychotic episode associated with a schizophrenia spectrum diagnosis.4 FEP is part of a trajectory marked by tran sitional periods. The patient transitions from being “healthy” to a prodromal state characterized by: (1) nonpsychotic behavioral disturbances such as depression or obsessive-compulsive disorder, (2) attenuated psychotic symptoms not requiring treatment, then converting to (3) psychotic symptoms prompting initial presentation for antipsychotic pharmacotherapy, leading to (4) a formal diagnosis of schizophreniform disorder and, subsequently, schizophrenia, requiring treatment to stabilize symptoms.
There are 2 critical periods along this continuum: prodromal stage and the duration of untreated psychosis (DUP). The prodromal period is a retrospectively identified time where the patient shows initial nonpsychotic disturbances (eg, cognitive and behavioral symptoms) before exhibiting clinical diagnostic criteria for a schizophrenia spectrum disorder. Approximately one-third of patients exhibiting these symptoms convert to psychosis within 1 year, and early treatment engagement at this stage has been shown to improve outcomes.5 The DUP is the time from when a patient has noticeable psychotic symptoms to initiation of drug treatment. The DUP is a consistent predictor of clinical outcome in schizophrenia, including negative symptoms, quality of life, and functional capacity.1
Antipsychotic selection
Treatment goals for FEP patients include:
• minimizing the DUP
• rapidly stabilizing psychosis
• achieving full symptomatic remission
• preventing relapse.
Several treatment guidelines for managing schizophrenia offer variable recommendations for initial antipsychotic treatment in patients with first-episode schizophrenia (Table 1).6-15 Most recommend second-generation antipsychotics (SGAs) over first-generation antipsychotics (FGAs)6,8,9,13,15 with specific recommendations on minimizing neurologic and metabolic adverse effects—to which FEP patients are susceptible—by avoiding high-potency and neurotoxic FGAs (eg, haloperidol and fluphenazine),7 clozapine,11,14 olanzapine,11 or ziprasidone.14 Two guidelines—the National Institute for Health and Care Excellence and the Scottish Intercollegiate Guidelines Network—do not state a preference for antipsychotic selection.10,12
The rationale for these recommendations is based on efficacy data, tolerability differences, FDA-approved indications, and recent FDA approvals with sparse post-marketing data. Of note, there are a lack of robust data for newer antipsychotics (eg, aripiprazole, paliperidone, iloperidone, asenapine, and lurasidone) in effectively and safely treating FEP; however, given the results of other antipsychotics studies, it is likely the efficacy and tolerability of these drugs can be extrapolated from experience with multi-episode patients.
Study design and demographics. Research studies of FEP share some similarities in study design; however, there is enough variability to make it difficult to compare studies and generalize findings (Table 2).16 The variability of DUP is a limitation when comparing studies because it is a significant predictor of clinical outcome. Patients who abuse substances—and often are more challenging to treat17—typically are excluded from these trials, which could explain the high response rate documented in studies of first-episode schizophrenia.
In addition, some FEP patients included in clinical trials might not be truly antipsychotic naïve; an estimated 25% to 75% of patients in these studies are antipsychotic naïve. This is an important consideration when comparing data on adverse effects that occur early in treatment. Additionally, acknowledging the advantages and disadvantages of how to handle missing data is critical because of the high dropout rate observed in these studies.18
Efficacy. There is a high response rate to antipsychotic therapy—ranging from 46% to 96%, depending on the study—in patients with first-episode schizophrenia.3 The response mainly is seen in reduction of positive symptoms because typically negative and cognitive symptoms do not respond to antipsychotics. One study reported only 29% of patients achieved both positive and negative symptom remission.19 It is likely that secondary negative symptoms caused by social withdrawal, reduced speech, and avoidance improve when positive symptoms subside, but primary negative symptoms endure.In general, there is a lack of evidence suggesting that 1 antipsychotic class or agent is more effective than another. Studies mainly assess effectiveness using the primary outcome measure of all-cause discontinuation, such as the Clinical Antipsychotic Trials of Intervention Effectiveness study.20 This outcome measure is a mixture of patient preference, tolerability, and efficacy that provides a more generalizable gauge on how well the treatment works in the clinic rather than tightly regulated settings such as clinical trials. A recent meta-analysis supports no differences in efficacy among antipsychotics in early-episode psychosis.21
Tolerability. Because there are no significant differences among antipsychotic classes or agents in terms of efficacy in first-episode schizophrenia, drug selection is guided mainly by (1) the adverse effect profile and (2) what should be avoided depending on patient-specific variables. Evidence suggests first-episode patients are more sensitive to adverse effects of antipsychotics, particularly neurologic side effects (see this article at CurrentPsychiatry.com for a table comparing adverse effects of antipsychotics in first-episode psychosis).18,22-29 Overall adverse effect profiles remain similar across FEP or multi-episode patients, but tend to be more exaggerated in drug-naïve patients with FEP.
Regarding FGA side effects, McEvoy et al18 demonstrated the neuroleptic threshold occurs at 50% lower haloperidol dosages in patients with first-episode schizophrenia (2.1 mg/d) compared with multi-episode schizophrenia (4.3 mg/d). Other trials suggest SGAs are associated with a lower risk of extrapyramidal side effects (EPS) or use of adjunctive therapies such as anticholinergic drugs or benzodiazepines.23-27 An exception to this statement is that higher risperidone dosages (≥4 to 6 mg/d) have been found to have higher rates of EPS and use of adjunctive medications to treat these symptoms in FEP.26 This is important because studies report higher discontinuation rates with more severe adverse effects of antipsychotics.
Cardiometabolic effects are of particular concern in first-episode patients because most weight gain happens in the first 3 to 4 months of treatment and remains throughout the first year.18,24,29,30 Studies have shown that olanzapine, quetiapine, and risperidone are associated with more clinically significant weight gain compared with haloperidol and ziprasidone.23-25 Olanzapine-associated weight gain has been reported to be twice that of quetiapine and risperidone.18 Regardless, the EUFEST trial did not find a difference in clinically significant weight gain after 12 months among the antipsychotics studied, including haloperidol and ziprasidone.25
Weight gain associated with these antipsychotics is accompanied by changes in fasting triglycerides, glucose, total cholesterol,23 and high-density lipoprotein cholesterol as well as an increase in body mass index (BMI) categorization29 (eg, shift from normal to overweight).18,25 Patients with lower baseline BMI and in racial minority groups might experience more rapid weight gain regardless of antipsychotic selection.29,30
Hyperprolactinemia could be under-recognized and could contribute to early treatment discontinuation.31 Evidence in patients with first-episode schizophrenia suggests similar outcomes as those seen in multi-episode patients, in whom risperidone is associated with higher prolactin elevations and clinically significant hyperprolactinemia (eg, galactorrhea and gynecomastia) compared with olanzapine, quetiapine, and low-dose haloperidol.18,23,24 However, there is a lack of studies that assess whether long-term therapy with strong D2 receptor antagonists increases the risk of bone demineralization or pathological fractures when started before patients’ bones reach maximum density in their mid-20s.31
Antipsychotic dosing
Given the high rate of treatment response in FEP and patients’ higher sensitivity to antipsychotic adverse effects, particularly EPS, guidelines recommend antipsychotic dosages lower than those used for multi-episode schizophrenia,11 especially FGAs. Based on trial data, commonly used dosages include:
• haloperidol, ≤5 mg/d23-25,29
• olanzapine, 10 mg/d18,23,25,29
• risperidone, ≤4 to 6 mg/d.18,24,29,32
In general, haloperidol and risperidone, 2 to 3 mg/d, were well tolerated and effective in trials. Higher quetiapine dosages of 500 to 600 mg/d could be required.11,18,25
According to a survey on prescribing practices of antipsychotic selection and dosing in first-episode schizophrenia,4 clinical prescribing practices tend to use unnecessarily high initial antipsychotic dosing compared with trial data. There also is variability in the usual target antipsychotic dosage ranging from 50% lower dosages to normal dosages in chronic schizophrenia to above FDA-approved maximum dosages for olanzapine (which may be necessary to counteract tobacco-induced cytochrome P450 1A2 enzyme induction).
In addition, these clinicians reported prescribing aripiprazole, an antipsychotic with weaker evidence (eg, case reports, case series, open-label studies) supporting its efficacy and tolerability in FEP. These prescribing practices could reflect attempts to reduce the DUP and achieve symptom remission, so long as tolerability is not a concern.
Essentially, prescribed dosages should be based on symptom improvement and tolerability. This ideal dosage will vary as illustrated by Kapur et al,33 who reported that FEP patients (N = 20) given haloperidol, 1 mg or 2.5 mg/d, had D2 receptor occupancy rates of 38% to 87%, which was significantly dose-related (1 mg/d mean = 59%, 2.5 mg/d mean = 75%). Clinical response and EPS significantly increased as D2 receptor occupancy exceeded 65% and 78%, respectively.
Antipsychotic response
When should you expect to see symptom improvement in patients with first-episode schizophrenia?
Emsley et al34 reported a 77.6% response rate among first-episode patients (N = 522) treated with low dosages of risperidone (mean modal dosage [MMD] = 3.3 mg/d) and haloperidol (MMD = 2.9 mg/d). They found variable response times that were evenly dispersed over a 10-week period. Nearly one-quarter (22.5%) did not respond until after week 4 and 11.2% did not respond until after week 8. In a study of FEP patients (N = 112) treated with olanzapine (MMD = 11.8 mg/d) or risperidone (MMD = 3.9 mg/d), Gallego et al35 reported a cumulative response of 39.6% at week 8 and 65.1% at week 16.
Although there is evidence that, among multi-episode patients, early nonresponse to antipsychotic therapy could predict subsequent nonresponse,36 the evidence is mixed for first-episode schizophrenia. Studies by Emsley et al34 and Gallego et al35 did not find that early nonresponse at weeks 1 or 2 predicted subsequent nonresponse at week 4 or later. However, other studies support the idea that early nonresponse predicts subsequent nonresponse and early antipsychotic response predicts future response in first-episode patients, with good specificity and sensitivity.37,38
Overall, treatment response in first-episode schizophrenia is variable. An adequate antipsychotic trial may be longer, 8 to 16 weeks, compared with 4 to 8 weeks in multi-episode patients. Because research suggests that failure to respond to treatment may lead to medication nonadherence,39 it is reasonable to consider switching antipsychotics when a patient experiences minimal or no response to antipsychotic therapy at week 2; however, this should be a patient-specific decision.
How long should you continue therapy after symptom remission?
There is a lack of consensus on the duration of therapy for a patient treated for first-episode schizophrenia because a small percentage (10% to 20%) do not relapse after the first psychotic episode.3 In general, treatment guidelines and expert consensus statements recommend at least 1 to 2 years of treatment before considering a discontinuation trial.7,10-11 Discuss the benefits and risks of maintenance treatment with your patient and obtain informed consent. With patients with minimal insight, obtaining proper consent is not possible and the physician must exercise judgment unilaterally, if necessary, after educating the family.
After at least 12 months of treatment, antipsychotic therapy could continue indefinitely, depending on patient-specific factors. There are no predictors for identifying patients who do not require maintenance therapy beyond the first psychotic episode. The absence of negative and cognitive deficits could provide clues that a patient might be a candidate for antipsychotic tapering.
Predicting the treatment course
Research investigating clinical predictors or biomarkers that forecast whether a patient will respond to treatment is preliminary. Many characteristics have been identified (Table 31,3,4,23,25,40) and include shorter DUP,1 poorer premorbid function,3 antipsychotic discontinuation,3 a trusting patient-doctor relationship,41 and antipsychotic-related adverse effects,23,25 which are predictive of response, nonresponse, relapse, adherence, and nonadherence, respectively.
Bottom Line
The goals of pharmacological treatment of first-episode schizophrenia are to minimize the duration of untreated psychosis and target full remission of positive symptoms using the lowest possible antipsychotic dosages. Pharmacotherapy should continued for 1 to 2 years, with longer duration considered if it is discussed with the patient and with vigilant monitoring for adverse effects and suboptimal medication nonadherence to prevent relapse.
Editor’s note: The second article in this series in the July 2015 issue reviews the rationale and evidence for non-standard, first-line therapies, including long-acting injectable antipsychotics and clozapine.
Related Resources
• Recovery After an Initial Schizophrenia Episode (RAISE) Project Early Treatment Program. National Institute of Mental Health. http://raiseetp.org.
• Martens L, Baker S. Promoting recovery from first episode psychosis: a guide for families. Centre for Addiction and Mental Health. http://www.camh.ca/en/hospital/ Documents/www.camh.net/AboutCAMH/Guideto CAMH/MentalHealthPrograms/SchizophreniaProgram/ 3936PromotingRecoveryFirstEpisodePsychosisfinal.pdf.
Drug Brand Names
Aripiprazole • Abilify Lurasidone • Latuda
Asenapine • Saphris Olanzapine • Zyprexa
Clozapine • Clozaril Paliperidone • Invega
Fluphenazine • Prolixin Quetiapine • Seroquel
Iloperidone • Fanapt Risperidone • Risperdal
Haloperidol • Haldol Ziprasidone • Geodon
Disclosures
Dr. Gardner reports no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
Dr. Nasrallah is a consultant to Acadia, Alkermes, Lundbeck, Janssen, Merck, Otsuka, and Sunovion, and is a speaker for Alkermes, Lundbeck, Janssen, Otsuka, and Sunovion.
1. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804.
2. Bradford DW, Perkins DO, Lieberman JA. Pharmacological management of first-episode schizophrenia and related nonaffective psychoses. Drugs. 2003;63(21):2265-2283.
3. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following a response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
4. Weiden PJ, Buckley PF, Grody M. Understanding and treating “first-episode” schizophrenia. Psychiatr Clin North Am. 2007;30(3):481-510.
5. Madaan V, Bestha DP, Kolli V. Schizophrenia prodrome: an optimal approach. Current Psychiatry. 2014;13(3):16-20, 29-30.
6. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
7. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620.
8. Canadian Psychiatric Association. Clinical practice guideline. Treatment of schizophrenia. Can J Psychiatry. 2005;50(13 suppl 1):7S-57S.
9. McEvoy JP, Scheifler PL, Frances A. Treatment of schizophrenia 1999. Expert consensus guideline series. J Clin Psychiatry. 1999;60(suppl 11):4-80.
10. National Institute for Health and Care Excellence (NICE). Clinical guideline 178: Psychosis and schizophrenia in adults: treatment and management. London, United Kingdom: National Institute for Health and Care Excellence (NICE); 2014.
11. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
12. Scottish Intercollegiate Guidelines Network (SIGN). Management of schizophrenia. Edinburgh, Scotland: Scottish Intercollegiate Guidelines Network; 2013. SIGN publication no. 131.
13. Argo TR, Crismon ML, Miller AL, et al. Texas Medication Algorithm Project procedural manual. Schizophrenia treatment algorithms. Austin, Texas: Texas Department of State Health Services; 2008.
14. Marder SR, Essock SM, Miller Al, et al. The Mount Sinai conference on the pharmacotherapy of schizophrenia. Schizophr Bull. 2002;28(1):5-16.
15. Bandelow B, Zohar J, Hollander E, et al; WFSBP Task Force on Treatment Guidelines for Anxiety, Obsessive-Compulsive and Post-Traumatic Stress Disorders. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
16. Robinson DG, Woerner MG, Alvir JMJ, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psych. 1999;56(3):241-247.
17. Green AI, Tohen MF, Hamer RM, et al. First episode schizophrenia-related psychosis and substance use disorders: acute response to olanzapine and haloperidol. Schizophr Res. 2004;66(2-3):125-135.
18. McEvoy JP, Lieberman JA, Perkins DO, et al. Efficacy and tolerability of olanzapine, quetiapine, and risperidone in the treatment of early psychosis: a randomized, double-blind 52-week comparison. Am J Psychiatry. 2007;164(7): 1050-1060.
19. Henry LP, Amminger GP, Harris MG, et al. The EPPIC follow-up study of first-episode psychosis: longer-term clinical and functional outcome 7 years after index admission. J Clin Psychiatry. 2010;71(6):716-728.
20. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. New Engl J Med. 2005; 353(12):1209-1223.
21. Crossley NA, Constante M, McGuire P, et al. Efficacy of atypical v. typical antipsychotics in the treatment of early psychosis: meta-analysis. Br J Psychiatry. 2010;196(6):434-439.
22. McEvoy JP, Hogarty GE, Steingard S. Optimal dose of neuroleptic in acute schizophrenia: a controlled study of the neuroleptic threshold and higher haloperidol dose. Arch Gen Psych. 1991;48(8):739-745.
23. Lieberman JA, Tollefson G, Tohen M, et al; HGDH Study Group. Comparative efficacy and safety of atypical and conventional antipsychotic drugs in first-episode psychosis: a randomized, double-blind trial of olanzapine versus haloperidol. Am J Psychiatry. 2003;160(8):1396-1404.
24. Schooler N, Rabinowitz J, Davidson M, et al; Early Psychosis Global Working Group. Risperidone and haloperidol in first-episode psychosis: a long-term randomized trial. Am J Psychiatry. 2005;162(5):947-953.
25. Kahn RS, Fleischhacker WW, Boter H, et al; EUFEST study group. Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: an open randomised clinical trial. Lancet. 2008;371(9618):1085-1097.
26. Emsley RA; Risperidone Working Group. Risperidone in the treatment of first-episode psychotic patients: a double-blind multicenter study. Schizophr Bull. 1999;25(4):721-729.
27. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naïve first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003.
28. Girgis RR, Phillips MR, Li X, et al. Clozapine v. chlorpromazine in treatment-naive, first-episode schizophrenia: 9-year outcomes of a randomised clinical trial. Br J Psychiatry. 2011;199(4):281-288.
29. Robinson DG, Woerner MG, Napolitano B, et al. Randomized comparison of olanzapine versus risperidone for the treatment of first-episode schizophrenia: 4-month outcomes. Am J Psychiatry. 2006;163(12):2096-2102.
30. Zipursky RB, Gu H, Green AI, et al. Course and predictors of weight gain in people with first-episode psychosis treated with olanzapine or haloperidol. Br J Psychiatry. 2005;187:537-543.
31. Taylor M, Waight A, Leonard B. Advances in the understanding and challenges facing the management of first-episode schizophrenia. J Psychopharmacol. 2012; 26(suppl 5):3-5.
32. Merlo MC, Hofer H, Gekle W, et al. Risperidone, 2mg/day vs. 4mg/day, in first-episode, acutely psychotic patients: treatment efficacy and effects on fine motor functioning. J Clin Psychiatry. 2002;63(10):885-891.
33. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D2 occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157(4):514-520.
34. Emsley R, Rabinowitz J, Medori R. Time course for antipsychotic treatment response in first-episode schizophrenia. Am J Psychiatry. 2006;163(4):743-745.
35. Gallego JA, Robinson DG, Sevy SM, et al. Time to treatment response in first-episode schizophrenia: should acute treatment trials last several months? J Clin Psychiatry. 2011;72(12):1691-1696.
36. Gardner KN, Bostwick JR. Antipsychotic treatment response in schizophrenia. Am J Health Sys Pharm. 2012;69(21):1872-1879.
37. Stauffer VL, Case M, Kinon BJ, et al. Early response to antipsychotic therapy as a clinical marker of subsequent response in the treatment of patients with first-episode psychosis. Psychiatry Res. 2011;187(1-2):42-48.
38. Schennach-Wolff R, Seemüller FH, Mayr A, et al. An early improvement threshold to predict response and remission in first-episode schizophrenia. Br J Psychiatry. 2010;196(6):460-466.
39. Perkins DO, Gu H, Weiden PJ, et al; Comparison of Atypicals in First Episode study group. Predictors of treatment discontinuation and medication nonadherence in patients recovering from a first episode of schizophrenia, schizophreniform disorder, or schizoaffective disorder: a randomized, double-blind, flexible-dose, multicenter study. J Clin Psychiatry. 2008;69(1):106-113.
40. Garner B, Berger GE, Nicolo JP, et al. Pituitary volume and early treatment response in drug-naïve first-episode psychosis patients. Schizophr Res. 2009;113(1):65-71.
41. Sapra M, Weiden PJ, Schooler NR, et al. Reasons for adherence and nonadherence: a pilot study comparing first-and multi-episode schizophrenia patients. Clin Schizophr Relat Psychoses. 2014;7(4):199-206.
1. Perkins DO, Gu H, Boteva K, et al. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: a critical review and meta-analysis. Am J Psychiatry. 2005;162(10):1785-1804.
2. Bradford DW, Perkins DO, Lieberman JA. Pharmacological management of first-episode schizophrenia and related nonaffective psychoses. Drugs. 2003;63(21):2265-2283.
3. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following a response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
4. Weiden PJ, Buckley PF, Grody M. Understanding and treating “first-episode” schizophrenia. Psychiatr Clin North Am. 2007;30(3):481-510.
5. Madaan V, Bestha DP, Kolli V. Schizophrenia prodrome: an optimal approach. Current Psychiatry. 2014;13(3):16-20, 29-30.
6. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
7. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620.
8. Canadian Psychiatric Association. Clinical practice guideline. Treatment of schizophrenia. Can J Psychiatry. 2005;50(13 suppl 1):7S-57S.
9. McEvoy JP, Scheifler PL, Frances A. Treatment of schizophrenia 1999. Expert consensus guideline series. J Clin Psychiatry. 1999;60(suppl 11):4-80.
10. National Institute for Health and Care Excellence (NICE). Clinical guideline 178: Psychosis and schizophrenia in adults: treatment and management. London, United Kingdom: National Institute for Health and Care Excellence (NICE); 2014.
11. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
12. Scottish Intercollegiate Guidelines Network (SIGN). Management of schizophrenia. Edinburgh, Scotland: Scottish Intercollegiate Guidelines Network; 2013. SIGN publication no. 131.
13. Argo TR, Crismon ML, Miller AL, et al. Texas Medication Algorithm Project procedural manual. Schizophrenia treatment algorithms. Austin, Texas: Texas Department of State Health Services; 2008.
14. Marder SR, Essock SM, Miller Al, et al. The Mount Sinai conference on the pharmacotherapy of schizophrenia. Schizophr Bull. 2002;28(1):5-16.
15. Bandelow B, Zohar J, Hollander E, et al; WFSBP Task Force on Treatment Guidelines for Anxiety, Obsessive-Compulsive and Post-Traumatic Stress Disorders. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders - first revision. World J Biol Psychiatry. 2008;9(4):248-312.
16. Robinson DG, Woerner MG, Alvir JMJ, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psych. 1999;56(3):241-247.
17. Green AI, Tohen MF, Hamer RM, et al. First episode schizophrenia-related psychosis and substance use disorders: acute response to olanzapine and haloperidol. Schizophr Res. 2004;66(2-3):125-135.
18. McEvoy JP, Lieberman JA, Perkins DO, et al. Efficacy and tolerability of olanzapine, quetiapine, and risperidone in the treatment of early psychosis: a randomized, double-blind 52-week comparison. Am J Psychiatry. 2007;164(7): 1050-1060.
19. Henry LP, Amminger GP, Harris MG, et al. The EPPIC follow-up study of first-episode psychosis: longer-term clinical and functional outcome 7 years after index admission. J Clin Psychiatry. 2010;71(6):716-728.
20. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. New Engl J Med. 2005; 353(12):1209-1223.
21. Crossley NA, Constante M, McGuire P, et al. Efficacy of atypical v. typical antipsychotics in the treatment of early psychosis: meta-analysis. Br J Psychiatry. 2010;196(6):434-439.
22. McEvoy JP, Hogarty GE, Steingard S. Optimal dose of neuroleptic in acute schizophrenia: a controlled study of the neuroleptic threshold and higher haloperidol dose. Arch Gen Psych. 1991;48(8):739-745.
23. Lieberman JA, Tollefson G, Tohen M, et al; HGDH Study Group. Comparative efficacy and safety of atypical and conventional antipsychotic drugs in first-episode psychosis: a randomized, double-blind trial of olanzapine versus haloperidol. Am J Psychiatry. 2003;160(8):1396-1404.
24. Schooler N, Rabinowitz J, Davidson M, et al; Early Psychosis Global Working Group. Risperidone and haloperidol in first-episode psychosis: a long-term randomized trial. Am J Psychiatry. 2005;162(5):947-953.
25. Kahn RS, Fleischhacker WW, Boter H, et al; EUFEST study group. Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: an open randomised clinical trial. Lancet. 2008;371(9618):1085-1097.
26. Emsley RA; Risperidone Working Group. Risperidone in the treatment of first-episode psychotic patients: a double-blind multicenter study. Schizophr Bull. 1999;25(4):721-729.
27. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naïve first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003.
28. Girgis RR, Phillips MR, Li X, et al. Clozapine v. chlorpromazine in treatment-naive, first-episode schizophrenia: 9-year outcomes of a randomised clinical trial. Br J Psychiatry. 2011;199(4):281-288.
29. Robinson DG, Woerner MG, Napolitano B, et al. Randomized comparison of olanzapine versus risperidone for the treatment of first-episode schizophrenia: 4-month outcomes. Am J Psychiatry. 2006;163(12):2096-2102.
30. Zipursky RB, Gu H, Green AI, et al. Course and predictors of weight gain in people with first-episode psychosis treated with olanzapine or haloperidol. Br J Psychiatry. 2005;187:537-543.
31. Taylor M, Waight A, Leonard B. Advances in the understanding and challenges facing the management of first-episode schizophrenia. J Psychopharmacol. 2012; 26(suppl 5):3-5.
32. Merlo MC, Hofer H, Gekle W, et al. Risperidone, 2mg/day vs. 4mg/day, in first-episode, acutely psychotic patients: treatment efficacy and effects on fine motor functioning. J Clin Psychiatry. 2002;63(10):885-891.
33. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D2 occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157(4):514-520.
34. Emsley R, Rabinowitz J, Medori R. Time course for antipsychotic treatment response in first-episode schizophrenia. Am J Psychiatry. 2006;163(4):743-745.
35. Gallego JA, Robinson DG, Sevy SM, et al. Time to treatment response in first-episode schizophrenia: should acute treatment trials last several months? J Clin Psychiatry. 2011;72(12):1691-1696.
36. Gardner KN, Bostwick JR. Antipsychotic treatment response in schizophrenia. Am J Health Sys Pharm. 2012;69(21):1872-1879.
37. Stauffer VL, Case M, Kinon BJ, et al. Early response to antipsychotic therapy as a clinical marker of subsequent response in the treatment of patients with first-episode psychosis. Psychiatry Res. 2011;187(1-2):42-48.
38. Schennach-Wolff R, Seemüller FH, Mayr A, et al. An early improvement threshold to predict response and remission in first-episode schizophrenia. Br J Psychiatry. 2010;196(6):460-466.
39. Perkins DO, Gu H, Weiden PJ, et al; Comparison of Atypicals in First Episode study group. Predictors of treatment discontinuation and medication nonadherence in patients recovering from a first episode of schizophrenia, schizophreniform disorder, or schizoaffective disorder: a randomized, double-blind, flexible-dose, multicenter study. J Clin Psychiatry. 2008;69(1):106-113.
40. Garner B, Berger GE, Nicolo JP, et al. Pituitary volume and early treatment response in drug-naïve first-episode psychosis patients. Schizophr Res. 2009;113(1):65-71.
41. Sapra M, Weiden PJ, Schooler NR, et al. Reasons for adherence and nonadherence: a pilot study comparing first-and multi-episode schizophrenia patients. Clin Schizophr Relat Psychoses. 2014;7(4):199-206.
