User login
EULAR Issues Recommendations for Managing SLE's Neuropsychiatric Effects
Neuropsychiatric manifestations in patients with systemic lupus erythematosus should be evaluated and treated initially as they would in patients without the autoimmune disease before being attributed to the condition, according to new recommendations put forth by a European League Against Rheumatism task force.
Using an evidence-based approach followed by expert consensus, the task force gathered relevant information on neuropsychiatric systemic lupus erythematosus (NPSLE), including prevalence, risk factors, diagnosis, monitoring, therapy, and prognosis and developed management recommendations in an effort to alleviate the persistent diagnostic and therapeutic challenges associated with the condition.
[Check out our coverage from the American College of Rheumatology's annual meeting.]
Based on a systematic literature and subsequent categorization of the quality of the evidence, lead author Dr. George K. Bertsias of the University of Crete in Heraklion and his colleagues on the task force of the EULAR standing committee for clinical affairs, determined that most NPSLE events occur at disease onset or within the first year of onset, and that while manifestations such as headache, mood disorders, anxiety, and mild cognitive dysfunction are common, they "do not usually reflect overt central nervous system lupus activity."
After excluding the mild manifestations noted above, cerebrovascular disease and seizures are the most common neuropsychiatric complications of SLE, with a cumulative incidence of 5%-15% and severe cognitive dysfunction, major depression, acute confusional state, peripheral nervous system disorders, and psychosis are all relatively uncommon, occurring in approximately 1%-5% of all SLE patients, the authors wrote (Ann. Rheum. Dis. 2010.;69:2074-82 [doi:10.1136/ard.2010.130476]).
In all SLE patients with new or unexplained symptoms or signs suggestive of neuropsychiatric disease, "the initial diagnostic work-up should be similar to that in non-SLE patients presenting with the same manifestations," the guidelines state. This is because the complications are generally not distinctive and could occur in the absence of SLE, according to the authors. Depending on the manifestation, the work-up may include lumbar puncture and cerebrospinal fluid analysis, EEG, neuropsychological assessment of cognitive function, nerve conduction studies, and neuroimaging, they wrote.
Possible causative factors for NPSLE include vascular injury of intracranial vessels, antibodies to neuronal antigens, ribosomes, and phospholipids-associated proteins, and inflammatory mediators, and risk factors include general SLE activity or damage, past or concurrent NPSLE, and moderate to high titers of persistently positive antiphospholipid antibodies, the authors wrote, noting that "these observations provide the rationale for primary and secondary prevention strategies."
Such strategies include glucocorticoids and immunosuppressant therapy "when the underlying pathogenesis is considered primarily inflammatory (such as in optic neuritis, transverse myelitis, or psychosis) or there is evidence of generalized SLE activity;" antiplatelet/antithrombotic therapy for moderate to high antiphospholipid antibodies or other features of antiphospholipid antibody syndrome, particularly thrombotic cerebrovascular disease; and symptomatic interventions and treatment of non-SLE factors as indicated, according to the guidelines. The efficacy of all of the interventions should be further defined in randomized controlled trial, the authors stressed.
The EULAR recommendations on NPSLE will be updated every 3 years, according to the authors, "with the inclusion of patients and individuals from other relevant professions, and the development of tools that will facilitate the dissemination and implementation of the recommendations."
This study was funded by the European League Against Rheumatism. The authors did not provide financial disclosures.
Neuropsychiatric manifestations in patients with systemic lupus erythematosus should be evaluated and treated initially as they would in patients without the autoimmune disease before being attributed to the condition, according to new recommendations put forth by a European League Against Rheumatism task force.
Using an evidence-based approach followed by expert consensus, the task force gathered relevant information on neuropsychiatric systemic lupus erythematosus (NPSLE), including prevalence, risk factors, diagnosis, monitoring, therapy, and prognosis and developed management recommendations in an effort to alleviate the persistent diagnostic and therapeutic challenges associated with the condition.
[Check out our coverage from the American College of Rheumatology's annual meeting.]
Based on a systematic literature and subsequent categorization of the quality of the evidence, lead author Dr. George K. Bertsias of the University of Crete in Heraklion and his colleagues on the task force of the EULAR standing committee for clinical affairs, determined that most NPSLE events occur at disease onset or within the first year of onset, and that while manifestations such as headache, mood disorders, anxiety, and mild cognitive dysfunction are common, they "do not usually reflect overt central nervous system lupus activity."
After excluding the mild manifestations noted above, cerebrovascular disease and seizures are the most common neuropsychiatric complications of SLE, with a cumulative incidence of 5%-15% and severe cognitive dysfunction, major depression, acute confusional state, peripheral nervous system disorders, and psychosis are all relatively uncommon, occurring in approximately 1%-5% of all SLE patients, the authors wrote (Ann. Rheum. Dis. 2010.;69:2074-82 [doi:10.1136/ard.2010.130476]).
In all SLE patients with new or unexplained symptoms or signs suggestive of neuropsychiatric disease, "the initial diagnostic work-up should be similar to that in non-SLE patients presenting with the same manifestations," the guidelines state. This is because the complications are generally not distinctive and could occur in the absence of SLE, according to the authors. Depending on the manifestation, the work-up may include lumbar puncture and cerebrospinal fluid analysis, EEG, neuropsychological assessment of cognitive function, nerve conduction studies, and neuroimaging, they wrote.
Possible causative factors for NPSLE include vascular injury of intracranial vessels, antibodies to neuronal antigens, ribosomes, and phospholipids-associated proteins, and inflammatory mediators, and risk factors include general SLE activity or damage, past or concurrent NPSLE, and moderate to high titers of persistently positive antiphospholipid antibodies, the authors wrote, noting that "these observations provide the rationale for primary and secondary prevention strategies."
Such strategies include glucocorticoids and immunosuppressant therapy "when the underlying pathogenesis is considered primarily inflammatory (such as in optic neuritis, transverse myelitis, or psychosis) or there is evidence of generalized SLE activity;" antiplatelet/antithrombotic therapy for moderate to high antiphospholipid antibodies or other features of antiphospholipid antibody syndrome, particularly thrombotic cerebrovascular disease; and symptomatic interventions and treatment of non-SLE factors as indicated, according to the guidelines. The efficacy of all of the interventions should be further defined in randomized controlled trial, the authors stressed.
The EULAR recommendations on NPSLE will be updated every 3 years, according to the authors, "with the inclusion of patients and individuals from other relevant professions, and the development of tools that will facilitate the dissemination and implementation of the recommendations."
This study was funded by the European League Against Rheumatism. The authors did not provide financial disclosures.
Neuropsychiatric manifestations in patients with systemic lupus erythematosus should be evaluated and treated initially as they would in patients without the autoimmune disease before being attributed to the condition, according to new recommendations put forth by a European League Against Rheumatism task force.
Using an evidence-based approach followed by expert consensus, the task force gathered relevant information on neuropsychiatric systemic lupus erythematosus (NPSLE), including prevalence, risk factors, diagnosis, monitoring, therapy, and prognosis and developed management recommendations in an effort to alleviate the persistent diagnostic and therapeutic challenges associated with the condition.
[Check out our coverage from the American College of Rheumatology's annual meeting.]
Based on a systematic literature and subsequent categorization of the quality of the evidence, lead author Dr. George K. Bertsias of the University of Crete in Heraklion and his colleagues on the task force of the EULAR standing committee for clinical affairs, determined that most NPSLE events occur at disease onset or within the first year of onset, and that while manifestations such as headache, mood disorders, anxiety, and mild cognitive dysfunction are common, they "do not usually reflect overt central nervous system lupus activity."
After excluding the mild manifestations noted above, cerebrovascular disease and seizures are the most common neuropsychiatric complications of SLE, with a cumulative incidence of 5%-15% and severe cognitive dysfunction, major depression, acute confusional state, peripheral nervous system disorders, and psychosis are all relatively uncommon, occurring in approximately 1%-5% of all SLE patients, the authors wrote (Ann. Rheum. Dis. 2010.;69:2074-82 [doi:10.1136/ard.2010.130476]).
In all SLE patients with new or unexplained symptoms or signs suggestive of neuropsychiatric disease, "the initial diagnostic work-up should be similar to that in non-SLE patients presenting with the same manifestations," the guidelines state. This is because the complications are generally not distinctive and could occur in the absence of SLE, according to the authors. Depending on the manifestation, the work-up may include lumbar puncture and cerebrospinal fluid analysis, EEG, neuropsychological assessment of cognitive function, nerve conduction studies, and neuroimaging, they wrote.
Possible causative factors for NPSLE include vascular injury of intracranial vessels, antibodies to neuronal antigens, ribosomes, and phospholipids-associated proteins, and inflammatory mediators, and risk factors include general SLE activity or damage, past or concurrent NPSLE, and moderate to high titers of persistently positive antiphospholipid antibodies, the authors wrote, noting that "these observations provide the rationale for primary and secondary prevention strategies."
Such strategies include glucocorticoids and immunosuppressant therapy "when the underlying pathogenesis is considered primarily inflammatory (such as in optic neuritis, transverse myelitis, or psychosis) or there is evidence of generalized SLE activity;" antiplatelet/antithrombotic therapy for moderate to high antiphospholipid antibodies or other features of antiphospholipid antibody syndrome, particularly thrombotic cerebrovascular disease; and symptomatic interventions and treatment of non-SLE factors as indicated, according to the guidelines. The efficacy of all of the interventions should be further defined in randomized controlled trial, the authors stressed.
The EULAR recommendations on NPSLE will be updated every 3 years, according to the authors, "with the inclusion of patients and individuals from other relevant professions, and the development of tools that will facilitate the dissemination and implementation of the recommendations."
This study was funded by the European League Against Rheumatism. The authors did not provide financial disclosures.
FROM THE ANNALS OF RHEUMATIC DISEASES
Briakinumab, Ustekinumab Trials Point to Future Psoriasis Treatment
Increased insight into the pathogenesis of psoriasis has led to the development of new drugs with novel mechanisms of action, as well as safer and more effective approaches to treatment with conventional drugs
As a result, treatment options for the management of patients with moderate to severe psoriasis in particular – nearly 25% of all patients with psoriasis, according to the National Psoriasis Foundation – have broadened substantially, and research reported during the past year suggests they are on track for continued expansion.

Among the most notable trials of the year were establishing the "unsurpassed efficacy" of Abbott’s investigational monoclonal antibody briakinumab, according to Dr. Craig Leonardi, a dermatologist and psoriasis specialist at St. Louis University. Like the recently approved psoriatic drug ustekinumab (Stelara), briakinumab is an injectable biologic agent that targets the interleukin-12 and -23 (IL-12/23) proteins, which are believed to promote the inflammation associated with psoriasis.
IL-12/23 Inhibition
Four phase III clinical studies of briakinumab were presented at the annual meeting of the European Academy of Dermatology and Venereology (EADV) in Gothenburg, Sweden.
– M06-890. In this trial, comparing the efficacy and safety of briakinumab to placebo, 80.7% of the 981 patients randomized to receive briakinumab every 4 weeks following an induction phase experienced a 75% improvement in psoriasis symptoms (PASI 75) at week 12, compared with 4.5% of the 484 patients randomized to placebo. Additionally, 61.6% and 32.2% of patients, respectively, saw 90% and 100% symptom clearance. And at week 52, 82.4% of patients who had achieved PASI 75 maintained at least that level of clearance.
– M10-315 and M10-114. In each of two 12-week trials comparing briakinumab to etanercept, significantly more patients randomized to briakinumab achieved Physician Global Assessment (PGA) scores of 0 or 1 and PASI 75 clearance. In the 350-patient M10-315 and the 347-patient M10-114 studies, respectively, 72.7% and 71% of patients randomized to briakinumab treatment achieved PGA 0 or 1, compared with 29.5% and 39.7% of patients randomized to etanercept therapy and 4.2% and 2.9% of those randomized to placebo.
Additionally, in the respective studies, 80.6% and 81.9% of the briakinumab-treated patients achieved PASI 75, compared with 39.6% and 56% of the etanercept patents, and 6.9% and 7.4% of the placebo groups.
– M10-255. The fourth trial of 317 patients was a head-to-head comparison with methotrexate in which 81.8% of briakinumab patients achieved PASI 75 clearance at 24 weeks, compared with 39.9% of those taking methotrexate. At 52 weeks, the PASI 75 clearance rates were 66.2% in the briakinumab group and 23.9% in the methotrexate group.
The most common adverse events observed across all four studies and in an ongoing open-label extension were upper respiratory infection, nasopharyngitis, headache, arthralgia, hypertension, and back pain. The incidence of infection and malignancy with briakinumab were higher than those with placebo, but were similar to those in patients treated with etanercept or methotrexate, according to a press release issued by Abbott.
Of particular note is the incidence of major adverse cardiovascular events (MACE) associated with briakinumab, Dr. Leonardi said in an interview. In the M06-890 trial, for example, seven briakinumab patients – all of whom had identifiable underlying cardiovascular risk factors, according to Abbott – experienced a MACE. A similar number of major cardiovascular events were observed in phase III trials of ustekinumab (Stelara), the IL 12/23 antagonist that received Food and Drug Administration approval for psoriasis treatment, he said. "Both drugs share the same mechanism of action, so it’s important to be cautious," he said. "Briakinumab has been submitted for FDA approval. We are awaiting review and comment."
Ustekinumab
Positive ustekinumab results were also presented last year. A pooled analysis of safety data reported at the summer academy meeting of the American Academy of Dermatology in Chicago demonstrated a favorable risk/benefit profile for up to 3 years of treatment.
Based on integrated data for 3,117 patients from the pivotal phase III PHOENIX 1 and PHOENIX 2 trials and the phase III ACCEPT trial, the investigators reported that the overall rates of adverse and serious adverse events were comparable in the 45-mg and 90-mg ustekinumab patient groups.
For the 45-mg and 90-mg patient groups, respectively, the per-hundred-patient-year rates were 0.82 and 1.50 for serious infections, 0.69 and 0.46 for noncutaneous malignancies, and 0.41 and 0.35 for MACE.
These rates were consistent with expectations for both general and psoriasis populations, and they remained stable over time, according to Dr. Leonardi, one of the study investigators. The maintenance of the favorable safety profile in patients who have been treated for several years is "encouraging," he said, noting that ongoing 5-year follow-up studies will enable continued monitoring of the drug’s safety.
Other important developments that may bode well for the treatment of psoriasis include data on IL-17 inhibitors, oral and topical janus kinase (JAK) inhibitors, an oral phosphodiesterase inhibitor, several IL-23 blockers, and topical niacin/calcipotriene, according to Dr. Leonardi and Dr. Alice B. Gottlieb of Tufts Medical Center in Boston.
Targeting IL-17
Recent studies have suggested that IL-17A–producing T cells have a crucial role in the pathogenesis of psoriasis, making them a potential treatment target.
In a phase II trial, a fully human IL-17 antibody AIN457 (Novartis) produced clinically meaningful reductions of disease activity at 4 and 12 weeks in 13 of 18 psoriasis patients who were randomized to receive a single 3-mg/kg intravenous infusion of the drug. At study weeks 4 and 12, respectively, the mean PASI score decreased by 58% and 63% in the AIN457 patients, compared with 4% and 9% in placebo patients, the authors wrote (Sci. Transl. Med. 2010;52:52ra72).
The results of a phase I study of another IL-17 drug – Amgen’s AMG 827, which binds to and blocks signaling via the IL-17 receptor – were reported at the EADV meeting. Most of the 16 patients who received a single dose of the fully human monoclonal antibody experienced substantial improvements in psoriasis symptoms. Based on the favorable findings, a phase II study is currently underway.
JAK 3 Inhibitors
The JAK pathways are also believed to have roles in the psoriasis disease cascade and as such have emerged as a treatment target. The four known JAK enzymes – JAK1, JAK2, JAK3, and tyrosine kinase (TYK) 2 – are components of signaling mechanisms used by multiple cytokines and growth factors that trigger dysregulated inflammatory pathways, according to Dr. Gottlieb. Oral and topical inhibitors of JAK are in phase II and III trials and have had promising results so far, she said in an interview.

In a 197-patient, phase II efficacy and safety study, Pfizer’s investigational oral JAK3 inhibitor, tentatively named tasocitinib (CP-690550), produced statistically significant responses at 12 weeks, compared with placebo in adults with moderate to severe psoriasis. The PASI 75 responses for patients randomized to twice daily tasocitinib at 2-mg, 5-mg, and 15-mg doses, respectively, were 25%, 40.8%, and 66.7%, compared with 2.0% for placebo, according to a company-issued statement. Additionally, as early as study week 4, "treatment with 5 and 15 mg twice daily of tasocitinib significantly improved patient-reported health-related quality of life outcomes."
Regarding safety, three patients randomized to tasocitinib treatment experienced a total of five serious adverse events during the study. Additionally, the investigators observed dose dependent decreases in mean neutrophil counts and hemoglobin values and increases in mean LDL, HDL, and total cholesterol levels. A large-scale, phase III trial program, called Oral Psoriasis Treatment (OPT) trials, is currently underway.
Topical JAK inhibitors are also under development, which presumably will avoid some of the complications associated with systemic therapy, according to Dr. Gottlieb, who is involved with investigations of Incyte’s topical JAK1/JAK2 inhibitor, INCB18424, which has demonstrated robust activity and safety in placebo-controlled, multidose, phase IIb trials in patients with mild to moderate psoriasis.
In studies reported by Dr. Gottlieb at last year’s annual meeting of the American Academy of Dermatology, topical application of INCB18424 cream led to significant improvement in the psoriatic lesions of treated patients at day 28, with the total lesion scores decreasing twofold, compared with placebo. "The effects were seen as early as 2 weeks, and they extended through" the study period," she said, noting that immunohistochemical staining and microarray gene analysis data confirmed the improvement in skin histology and the reduction in the psoriatic molecular signature.
Apremilast May Break Ground
Poised to be "the first oral medication with a new mechanism of action for psoriasis in almost 20 years," the phosphodiesterase 4 (PDE4) enzyme inhibitor apremilast (Celgene) demonstrated favorable results in a phase IIb study reported in December 2009 and has recently begun phase III trials, according to Dr. Leonardi.
In the phase IIb trial, 352 patients with moderate to severe plaque-type psoriasis were randomized to receive 10 mg, 20 mg or 30 mg of apremilast twice per day or placebo. Of the apremilast patients, 41% of the 30-mg group, 29% of the 20-mg group, and 11% of the 10-mg group achieved a PASI 75 after 16 weeks, compared with 6% of the placebo group, according a company statement.
A comparison of infection rates showed that infections occurred in 48% of the 30 mg apremilast patients and 33% of the placebo patients. No serious adverse events related to apremilast were reported.
The positive results have led to the initiation of two pivotal phase III trials: the multicenter ESTEEM 1 and ESTEEM 2 safety and efficacy studies comprising, respectively, 825 patients and 405 patients with moderate to severe plaque psoriasis. Primary outcome data from both studies will be available in the summer of 2011.
Topical Niacin/Calcipotriene
The nonsteroidal topical options for psoriasis will likely be expanded further by the addition of topical niacin (nicotinamide) to standard treatment with the synthetic vitamin D derivative calcipotriene, according to Dr. Gottlieb.
A recent 168-patient multicenter, double-blind, randomized trial reported that 50% of patients randomized to combination therapy with 0.005% calcipotriene and 1.4% nicotinamide achieved symptom clearance or near clearance, compared with 18.8% of patients randomized to placebo, 25% of patients treated with nicotinamide alone, and 31.5% of patients treated with calcipotriene alone.
The findings suggest that the combination therapy "may prove effective as an alternative therapeutic option to calcipotriene monotherapy and may provide an attractive option for patients seeking an effective corticosteroid-sparing topical psoriatic agent," the investigators wrote (J. Am. Acad. Dermatol. 2010;63:775-81).
In the midst of all of the new developments, it is important to note that tumor necrosis factor (TNF) antagonists continue to perform well, Dr. Leonardi stressed. "Given that the class is now 12 years old and includes 2 million patients, we are unlikely to learn of major safety risks at this point, and registry data support the notion that TNF blockade is cardioprotective in [rheumatoid arthritis], in distinction to the IL 12/23 blockers, where the MACE issue is unresolved," he said. "At this point, it is important for dermatologists to remember that the IL-12/23 drugs, in particular, are new, and since they are psoriasis specific, there are no other disease states where they were previously used."
Until the new drugs are in widespread use, "they should be used with caution," Dr. Leonardi continued. "The last psoriasis-specific biologic we used was Raptiva, which was voluntarily removed from the market by Genentech after 3 patients were identified with a rare and fatal central nervous system infection. At the time these patients were identified, the drug had been on the market for 5 years and had been used in 23,000 patients."
What this means in clinical practice is that ustekinumab or briakinumab may be reasonable options for patients with a history of failure of TNF antagonists or a history of central or peripheral demyelination, but until longer-term safety data are available, it should not be the first choice in the majority of treatment-naive patients, said Dr. Leonardi.
Dr. Leonardi is a consultant, investigator, and/or on the speakers bureau of Amgen, Abbott, Celgene, Centocor, Genentech, Incyte, Novartis Pfizer, and several other pharmaceutical companies.
Dr. Gottlieb has current consulting/advisory board agreements with Abbott, Amgen, Celgene, Incyte, and numerous other pharmaceutical companies.
Increased insight into the pathogenesis of psoriasis has led to the development of new drugs with novel mechanisms of action, as well as safer and more effective approaches to treatment with conventional drugs
As a result, treatment options for the management of patients with moderate to severe psoriasis in particular – nearly 25% of all patients with psoriasis, according to the National Psoriasis Foundation – have broadened substantially, and research reported during the past year suggests they are on track for continued expansion.
Among the most notable trials of the year were establishing the "unsurpassed efficacy" of Abbott’s investigational monoclonal antibody briakinumab, according to Dr. Craig Leonardi, a dermatologist and psoriasis specialist at St. Louis University. Like the recently approved psoriatic drug ustekinumab (Stelara), briakinumab is an injectable biologic agent that targets the interleukin-12 and -23 (IL-12/23) proteins, which are believed to promote the inflammation associated with psoriasis.
IL-12/23 Inhibition
Four phase III clinical studies of briakinumab were presented at the annual meeting of the European Academy of Dermatology and Venereology (EADV) in Gothenburg, Sweden.
– M06-890. In this trial, comparing the efficacy and safety of briakinumab to placebo, 80.7% of the 981 patients randomized to receive briakinumab every 4 weeks following an induction phase experienced a 75% improvement in psoriasis symptoms (PASI 75) at week 12, compared with 4.5% of the 484 patients randomized to placebo. Additionally, 61.6% and 32.2% of patients, respectively, saw 90% and 100% symptom clearance. And at week 52, 82.4% of patients who had achieved PASI 75 maintained at least that level of clearance.
– M10-315 and M10-114. In each of two 12-week trials comparing briakinumab to etanercept, significantly more patients randomized to briakinumab achieved Physician Global Assessment (PGA) scores of 0 or 1 and PASI 75 clearance. In the 350-patient M10-315 and the 347-patient M10-114 studies, respectively, 72.7% and 71% of patients randomized to briakinumab treatment achieved PGA 0 or 1, compared with 29.5% and 39.7% of patients randomized to etanercept therapy and 4.2% and 2.9% of those randomized to placebo.
Additionally, in the respective studies, 80.6% and 81.9% of the briakinumab-treated patients achieved PASI 75, compared with 39.6% and 56% of the etanercept patents, and 6.9% and 7.4% of the placebo groups.
– M10-255. The fourth trial of 317 patients was a head-to-head comparison with methotrexate in which 81.8% of briakinumab patients achieved PASI 75 clearance at 24 weeks, compared with 39.9% of those taking methotrexate. At 52 weeks, the PASI 75 clearance rates were 66.2% in the briakinumab group and 23.9% in the methotrexate group.
The most common adverse events observed across all four studies and in an ongoing open-label extension were upper respiratory infection, nasopharyngitis, headache, arthralgia, hypertension, and back pain. The incidence of infection and malignancy with briakinumab were higher than those with placebo, but were similar to those in patients treated with etanercept or methotrexate, according to a press release issued by Abbott.
Of particular note is the incidence of major adverse cardiovascular events (MACE) associated with briakinumab, Dr. Leonardi said in an interview. In the M06-890 trial, for example, seven briakinumab patients – all of whom had identifiable underlying cardiovascular risk factors, according to Abbott – experienced a MACE. A similar number of major cardiovascular events were observed in phase III trials of ustekinumab (Stelara), the IL 12/23 antagonist that received Food and Drug Administration approval for psoriasis treatment, he said. "Both drugs share the same mechanism of action, so it’s important to be cautious," he said. "Briakinumab has been submitted for FDA approval. We are awaiting review and comment."
Ustekinumab
Positive ustekinumab results were also presented last year. A pooled analysis of safety data reported at the summer academy meeting of the American Academy of Dermatology in Chicago demonstrated a favorable risk/benefit profile for up to 3 years of treatment.
Based on integrated data for 3,117 patients from the pivotal phase III PHOENIX 1 and PHOENIX 2 trials and the phase III ACCEPT trial, the investigators reported that the overall rates of adverse and serious adverse events were comparable in the 45-mg and 90-mg ustekinumab patient groups.
For the 45-mg and 90-mg patient groups, respectively, the per-hundred-patient-year rates were 0.82 and 1.50 for serious infections, 0.69 and 0.46 for noncutaneous malignancies, and 0.41 and 0.35 for MACE.
These rates were consistent with expectations for both general and psoriasis populations, and they remained stable over time, according to Dr. Leonardi, one of the study investigators. The maintenance of the favorable safety profile in patients who have been treated for several years is "encouraging," he said, noting that ongoing 5-year follow-up studies will enable continued monitoring of the drug’s safety.
Other important developments that may bode well for the treatment of psoriasis include data on IL-17 inhibitors, oral and topical janus kinase (JAK) inhibitors, an oral phosphodiesterase inhibitor, several IL-23 blockers, and topical niacin/calcipotriene, according to Dr. Leonardi and Dr. Alice B. Gottlieb of Tufts Medical Center in Boston.
Targeting IL-17
Recent studies have suggested that IL-17A–producing T cells have a crucial role in the pathogenesis of psoriasis, making them a potential treatment target.
In a phase II trial, a fully human IL-17 antibody AIN457 (Novartis) produced clinically meaningful reductions of disease activity at 4 and 12 weeks in 13 of 18 psoriasis patients who were randomized to receive a single 3-mg/kg intravenous infusion of the drug. At study weeks 4 and 12, respectively, the mean PASI score decreased by 58% and 63% in the AIN457 patients, compared with 4% and 9% in placebo patients, the authors wrote (Sci. Transl. Med. 2010;52:52ra72).
The results of a phase I study of another IL-17 drug – Amgen’s AMG 827, which binds to and blocks signaling via the IL-17 receptor – were reported at the EADV meeting. Most of the 16 patients who received a single dose of the fully human monoclonal antibody experienced substantial improvements in psoriasis symptoms. Based on the favorable findings, a phase II study is currently underway.
JAK 3 Inhibitors
The JAK pathways are also believed to have roles in the psoriasis disease cascade and as such have emerged as a treatment target. The four known JAK enzymes – JAK1, JAK2, JAK3, and tyrosine kinase (TYK) 2 – are components of signaling mechanisms used by multiple cytokines and growth factors that trigger dysregulated inflammatory pathways, according to Dr. Gottlieb. Oral and topical inhibitors of JAK are in phase II and III trials and have had promising results so far, she said in an interview.
In a 197-patient, phase II efficacy and safety study, Pfizer’s investigational oral JAK3 inhibitor, tentatively named tasocitinib (CP-690550), produced statistically significant responses at 12 weeks, compared with placebo in adults with moderate to severe psoriasis. The PASI 75 responses for patients randomized to twice daily tasocitinib at 2-mg, 5-mg, and 15-mg doses, respectively, were 25%, 40.8%, and 66.7%, compared with 2.0% for placebo, according to a company-issued statement. Additionally, as early as study week 4, "treatment with 5 and 15 mg twice daily of tasocitinib significantly improved patient-reported health-related quality of life outcomes."
Regarding safety, three patients randomized to tasocitinib treatment experienced a total of five serious adverse events during the study. Additionally, the investigators observed dose dependent decreases in mean neutrophil counts and hemoglobin values and increases in mean LDL, HDL, and total cholesterol levels. A large-scale, phase III trial program, called Oral Psoriasis Treatment (OPT) trials, is currently underway.
Topical JAK inhibitors are also under development, which presumably will avoid some of the complications associated with systemic therapy, according to Dr. Gottlieb, who is involved with investigations of Incyte’s topical JAK1/JAK2 inhibitor, INCB18424, which has demonstrated robust activity and safety in placebo-controlled, multidose, phase IIb trials in patients with mild to moderate psoriasis.
In studies reported by Dr. Gottlieb at last year’s annual meeting of the American Academy of Dermatology, topical application of INCB18424 cream led to significant improvement in the psoriatic lesions of treated patients at day 28, with the total lesion scores decreasing twofold, compared with placebo. "The effects were seen as early as 2 weeks, and they extended through" the study period," she said, noting that immunohistochemical staining and microarray gene analysis data confirmed the improvement in skin histology and the reduction in the psoriatic molecular signature.
Apremilast May Break Ground
Poised to be "the first oral medication with a new mechanism of action for psoriasis in almost 20 years," the phosphodiesterase 4 (PDE4) enzyme inhibitor apremilast (Celgene) demonstrated favorable results in a phase IIb study reported in December 2009 and has recently begun phase III trials, according to Dr. Leonardi.
In the phase IIb trial, 352 patients with moderate to severe plaque-type psoriasis were randomized to receive 10 mg, 20 mg or 30 mg of apremilast twice per day or placebo. Of the apremilast patients, 41% of the 30-mg group, 29% of the 20-mg group, and 11% of the 10-mg group achieved a PASI 75 after 16 weeks, compared with 6% of the placebo group, according a company statement.
A comparison of infection rates showed that infections occurred in 48% of the 30 mg apremilast patients and 33% of the placebo patients. No serious adverse events related to apremilast were reported.
The positive results have led to the initiation of two pivotal phase III trials: the multicenter ESTEEM 1 and ESTEEM 2 safety and efficacy studies comprising, respectively, 825 patients and 405 patients with moderate to severe plaque psoriasis. Primary outcome data from both studies will be available in the summer of 2011.
Topical Niacin/Calcipotriene
The nonsteroidal topical options for psoriasis will likely be expanded further by the addition of topical niacin (nicotinamide) to standard treatment with the synthetic vitamin D derivative calcipotriene, according to Dr. Gottlieb.
A recent 168-patient multicenter, double-blind, randomized trial reported that 50% of patients randomized to combination therapy with 0.005% calcipotriene and 1.4% nicotinamide achieved symptom clearance or near clearance, compared with 18.8% of patients randomized to placebo, 25% of patients treated with nicotinamide alone, and 31.5% of patients treated with calcipotriene alone.
The findings suggest that the combination therapy "may prove effective as an alternative therapeutic option to calcipotriene monotherapy and may provide an attractive option for patients seeking an effective corticosteroid-sparing topical psoriatic agent," the investigators wrote (J. Am. Acad. Dermatol. 2010;63:775-81).
In the midst of all of the new developments, it is important to note that tumor necrosis factor (TNF) antagonists continue to perform well, Dr. Leonardi stressed. "Given that the class is now 12 years old and includes 2 million patients, we are unlikely to learn of major safety risks at this point, and registry data support the notion that TNF blockade is cardioprotective in [rheumatoid arthritis], in distinction to the IL 12/23 blockers, where the MACE issue is unresolved," he said. "At this point, it is important for dermatologists to remember that the IL-12/23 drugs, in particular, are new, and since they are psoriasis specific, there are no other disease states where they were previously used."
Until the new drugs are in widespread use, "they should be used with caution," Dr. Leonardi continued. "The last psoriasis-specific biologic we used was Raptiva, which was voluntarily removed from the market by Genentech after 3 patients were identified with a rare and fatal central nervous system infection. At the time these patients were identified, the drug had been on the market for 5 years and had been used in 23,000 patients."
What this means in clinical practice is that ustekinumab or briakinumab may be reasonable options for patients with a history of failure of TNF antagonists or a history of central or peripheral demyelination, but until longer-term safety data are available, it should not be the first choice in the majority of treatment-naive patients, said Dr. Leonardi.
Dr. Leonardi is a consultant, investigator, and/or on the speakers bureau of Amgen, Abbott, Celgene, Centocor, Genentech, Incyte, Novartis Pfizer, and several other pharmaceutical companies.
Dr. Gottlieb has current consulting/advisory board agreements with Abbott, Amgen, Celgene, Incyte, and numerous other pharmaceutical companies.
Increased insight into the pathogenesis of psoriasis has led to the development of new drugs with novel mechanisms of action, as well as safer and more effective approaches to treatment with conventional drugs
As a result, treatment options for the management of patients with moderate to severe psoriasis in particular – nearly 25% of all patients with psoriasis, according to the National Psoriasis Foundation – have broadened substantially, and research reported during the past year suggests they are on track for continued expansion.
Among the most notable trials of the year were establishing the "unsurpassed efficacy" of Abbott’s investigational monoclonal antibody briakinumab, according to Dr. Craig Leonardi, a dermatologist and psoriasis specialist at St. Louis University. Like the recently approved psoriatic drug ustekinumab (Stelara), briakinumab is an injectable biologic agent that targets the interleukin-12 and -23 (IL-12/23) proteins, which are believed to promote the inflammation associated with psoriasis.
IL-12/23 Inhibition
Four phase III clinical studies of briakinumab were presented at the annual meeting of the European Academy of Dermatology and Venereology (EADV) in Gothenburg, Sweden.
– M06-890. In this trial, comparing the efficacy and safety of briakinumab to placebo, 80.7% of the 981 patients randomized to receive briakinumab every 4 weeks following an induction phase experienced a 75% improvement in psoriasis symptoms (PASI 75) at week 12, compared with 4.5% of the 484 patients randomized to placebo. Additionally, 61.6% and 32.2% of patients, respectively, saw 90% and 100% symptom clearance. And at week 52, 82.4% of patients who had achieved PASI 75 maintained at least that level of clearance.
– M10-315 and M10-114. In each of two 12-week trials comparing briakinumab to etanercept, significantly more patients randomized to briakinumab achieved Physician Global Assessment (PGA) scores of 0 or 1 and PASI 75 clearance. In the 350-patient M10-315 and the 347-patient M10-114 studies, respectively, 72.7% and 71% of patients randomized to briakinumab treatment achieved PGA 0 or 1, compared with 29.5% and 39.7% of patients randomized to etanercept therapy and 4.2% and 2.9% of those randomized to placebo.
Additionally, in the respective studies, 80.6% and 81.9% of the briakinumab-treated patients achieved PASI 75, compared with 39.6% and 56% of the etanercept patents, and 6.9% and 7.4% of the placebo groups.
– M10-255. The fourth trial of 317 patients was a head-to-head comparison with methotrexate in which 81.8% of briakinumab patients achieved PASI 75 clearance at 24 weeks, compared with 39.9% of those taking methotrexate. At 52 weeks, the PASI 75 clearance rates were 66.2% in the briakinumab group and 23.9% in the methotrexate group.
The most common adverse events observed across all four studies and in an ongoing open-label extension were upper respiratory infection, nasopharyngitis, headache, arthralgia, hypertension, and back pain. The incidence of infection and malignancy with briakinumab were higher than those with placebo, but were similar to those in patients treated with etanercept or methotrexate, according to a press release issued by Abbott.
Of particular note is the incidence of major adverse cardiovascular events (MACE) associated with briakinumab, Dr. Leonardi said in an interview. In the M06-890 trial, for example, seven briakinumab patients – all of whom had identifiable underlying cardiovascular risk factors, according to Abbott – experienced a MACE. A similar number of major cardiovascular events were observed in phase III trials of ustekinumab (Stelara), the IL 12/23 antagonist that received Food and Drug Administration approval for psoriasis treatment, he said. "Both drugs share the same mechanism of action, so it’s important to be cautious," he said. "Briakinumab has been submitted for FDA approval. We are awaiting review and comment."
Ustekinumab
Positive ustekinumab results were also presented last year. A pooled analysis of safety data reported at the summer academy meeting of the American Academy of Dermatology in Chicago demonstrated a favorable risk/benefit profile for up to 3 years of treatment.
Based on integrated data for 3,117 patients from the pivotal phase III PHOENIX 1 and PHOENIX 2 trials and the phase III ACCEPT trial, the investigators reported that the overall rates of adverse and serious adverse events were comparable in the 45-mg and 90-mg ustekinumab patient groups.
For the 45-mg and 90-mg patient groups, respectively, the per-hundred-patient-year rates were 0.82 and 1.50 for serious infections, 0.69 and 0.46 for noncutaneous malignancies, and 0.41 and 0.35 for MACE.
These rates were consistent with expectations for both general and psoriasis populations, and they remained stable over time, according to Dr. Leonardi, one of the study investigators. The maintenance of the favorable safety profile in patients who have been treated for several years is "encouraging," he said, noting that ongoing 5-year follow-up studies will enable continued monitoring of the drug’s safety.
Other important developments that may bode well for the treatment of psoriasis include data on IL-17 inhibitors, oral and topical janus kinase (JAK) inhibitors, an oral phosphodiesterase inhibitor, several IL-23 blockers, and topical niacin/calcipotriene, according to Dr. Leonardi and Dr. Alice B. Gottlieb of Tufts Medical Center in Boston.
Targeting IL-17
Recent studies have suggested that IL-17A–producing T cells have a crucial role in the pathogenesis of psoriasis, making them a potential treatment target.
In a phase II trial, a fully human IL-17 antibody AIN457 (Novartis) produced clinically meaningful reductions of disease activity at 4 and 12 weeks in 13 of 18 psoriasis patients who were randomized to receive a single 3-mg/kg intravenous infusion of the drug. At study weeks 4 and 12, respectively, the mean PASI score decreased by 58% and 63% in the AIN457 patients, compared with 4% and 9% in placebo patients, the authors wrote (Sci. Transl. Med. 2010;52:52ra72).
The results of a phase I study of another IL-17 drug – Amgen’s AMG 827, which binds to and blocks signaling via the IL-17 receptor – were reported at the EADV meeting. Most of the 16 patients who received a single dose of the fully human monoclonal antibody experienced substantial improvements in psoriasis symptoms. Based on the favorable findings, a phase II study is currently underway.
JAK 3 Inhibitors
The JAK pathways are also believed to have roles in the psoriasis disease cascade and as such have emerged as a treatment target. The four known JAK enzymes – JAK1, JAK2, JAK3, and tyrosine kinase (TYK) 2 – are components of signaling mechanisms used by multiple cytokines and growth factors that trigger dysregulated inflammatory pathways, according to Dr. Gottlieb. Oral and topical inhibitors of JAK are in phase II and III trials and have had promising results so far, she said in an interview.
In a 197-patient, phase II efficacy and safety study, Pfizer’s investigational oral JAK3 inhibitor, tentatively named tasocitinib (CP-690550), produced statistically significant responses at 12 weeks, compared with placebo in adults with moderate to severe psoriasis. The PASI 75 responses for patients randomized to twice daily tasocitinib at 2-mg, 5-mg, and 15-mg doses, respectively, were 25%, 40.8%, and 66.7%, compared with 2.0% for placebo, according to a company-issued statement. Additionally, as early as study week 4, "treatment with 5 and 15 mg twice daily of tasocitinib significantly improved patient-reported health-related quality of life outcomes."
Regarding safety, three patients randomized to tasocitinib treatment experienced a total of five serious adverse events during the study. Additionally, the investigators observed dose dependent decreases in mean neutrophil counts and hemoglobin values and increases in mean LDL, HDL, and total cholesterol levels. A large-scale, phase III trial program, called Oral Psoriasis Treatment (OPT) trials, is currently underway.
Topical JAK inhibitors are also under development, which presumably will avoid some of the complications associated with systemic therapy, according to Dr. Gottlieb, who is involved with investigations of Incyte’s topical JAK1/JAK2 inhibitor, INCB18424, which has demonstrated robust activity and safety in placebo-controlled, multidose, phase IIb trials in patients with mild to moderate psoriasis.
In studies reported by Dr. Gottlieb at last year’s annual meeting of the American Academy of Dermatology, topical application of INCB18424 cream led to significant improvement in the psoriatic lesions of treated patients at day 28, with the total lesion scores decreasing twofold, compared with placebo. "The effects were seen as early as 2 weeks, and they extended through" the study period," she said, noting that immunohistochemical staining and microarray gene analysis data confirmed the improvement in skin histology and the reduction in the psoriatic molecular signature.
Apremilast May Break Ground
Poised to be "the first oral medication with a new mechanism of action for psoriasis in almost 20 years," the phosphodiesterase 4 (PDE4) enzyme inhibitor apremilast (Celgene) demonstrated favorable results in a phase IIb study reported in December 2009 and has recently begun phase III trials, according to Dr. Leonardi.
In the phase IIb trial, 352 patients with moderate to severe plaque-type psoriasis were randomized to receive 10 mg, 20 mg or 30 mg of apremilast twice per day or placebo. Of the apremilast patients, 41% of the 30-mg group, 29% of the 20-mg group, and 11% of the 10-mg group achieved a PASI 75 after 16 weeks, compared with 6% of the placebo group, according a company statement.
A comparison of infection rates showed that infections occurred in 48% of the 30 mg apremilast patients and 33% of the placebo patients. No serious adverse events related to apremilast were reported.
The positive results have led to the initiation of two pivotal phase III trials: the multicenter ESTEEM 1 and ESTEEM 2 safety and efficacy studies comprising, respectively, 825 patients and 405 patients with moderate to severe plaque psoriasis. Primary outcome data from both studies will be available in the summer of 2011.
Topical Niacin/Calcipotriene
The nonsteroidal topical options for psoriasis will likely be expanded further by the addition of topical niacin (nicotinamide) to standard treatment with the synthetic vitamin D derivative calcipotriene, according to Dr. Gottlieb.
A recent 168-patient multicenter, double-blind, randomized trial reported that 50% of patients randomized to combination therapy with 0.005% calcipotriene and 1.4% nicotinamide achieved symptom clearance or near clearance, compared with 18.8% of patients randomized to placebo, 25% of patients treated with nicotinamide alone, and 31.5% of patients treated with calcipotriene alone.
The findings suggest that the combination therapy "may prove effective as an alternative therapeutic option to calcipotriene monotherapy and may provide an attractive option for patients seeking an effective corticosteroid-sparing topical psoriatic agent," the investigators wrote (J. Am. Acad. Dermatol. 2010;63:775-81).
In the midst of all of the new developments, it is important to note that tumor necrosis factor (TNF) antagonists continue to perform well, Dr. Leonardi stressed. "Given that the class is now 12 years old and includes 2 million patients, we are unlikely to learn of major safety risks at this point, and registry data support the notion that TNF blockade is cardioprotective in [rheumatoid arthritis], in distinction to the IL 12/23 blockers, where the MACE issue is unresolved," he said. "At this point, it is important for dermatologists to remember that the IL-12/23 drugs, in particular, are new, and since they are psoriasis specific, there are no other disease states where they were previously used."
Until the new drugs are in widespread use, "they should be used with caution," Dr. Leonardi continued. "The last psoriasis-specific biologic we used was Raptiva, which was voluntarily removed from the market by Genentech after 3 patients were identified with a rare and fatal central nervous system infection. At the time these patients were identified, the drug had been on the market for 5 years and had been used in 23,000 patients."
What this means in clinical practice is that ustekinumab or briakinumab may be reasonable options for patients with a history of failure of TNF antagonists or a history of central or peripheral demyelination, but until longer-term safety data are available, it should not be the first choice in the majority of treatment-naive patients, said Dr. Leonardi.
Dr. Leonardi is a consultant, investigator, and/or on the speakers bureau of Amgen, Abbott, Celgene, Centocor, Genentech, Incyte, Novartis Pfizer, and several other pharmaceutical companies.
Dr. Gottlieb has current consulting/advisory board agreements with Abbott, Amgen, Celgene, Incyte, and numerous other pharmaceutical companies.
EXPERT ANALYSIS
Neurostimulation Poised to Take On Tough Seizures
SAN ANTONIO – It’s still too soon to know whether neurostimulation will be the therapeutic advance that treatment-refractory epilepsy patients have been waiting for, but the possibility that it might be has the epilepsy community buzzing.
For approximately 30% of epilepsy patients, seizures cannot be controlled with antiepileptic drugs or surgery. New long-term safety and efficacy data for vagus nerve simulation (VNS) and the results of recent pivotal trials of two approaches to direct brain stimulation offer beacons of hope to these patients, Dr. Gregory K. Bergey said in a plenary session on neurostimulation at the annual meeting of the American Epilepsy Society.
"One of the frustrating things for those of us treating patients with epilepsy has been the fact that, although a number of new antiepileptic drugs have been developed over the past 10-15 years and most are better tolerated and have better pharmacokinetic profiles than earlier drugs, the number of patients with seizures that don’t respond to medical therapy has not been significantly reduced," said Dr. Bergey, director of the Johns Hopkins Epilepsy Center, Baltimore.
"So we’re stepping back and saying, ‘Is there some other way we can treat these patients?’ That has been the impetus for looking at neurostimulation, which has been around for well over a decade, and what we’re seeing is exciting."
Although the 40%-50% response rates observed in direct brain stimulation trials do not appear to be overwhelming, "this is just the beginning," Dr. Bergey stressed in an interview. "As opposed to a drug trial, where you go up to a certain dose and it either works or it doesn’t work, in the case of neurostimulation we don’t know the optimal stimulus parameters, and I think that’s what you’re going to begin to see over the next several years," he said.
"There’s going to be a lot of investigation into neurostimulation of the brain structures to try to figure out who are the best candidates and what the best stimulus parameters are. It’s easy to say we’re stimulating the brain, but do we stimulate 100 times per second, 50 times per second, 25 times per second, and what should the stimulus intensities be?"
Vagus Nerve Stimulation. Currently, Cyberonics’ VNS Therapy System is the only Food and Drug Administration–approved form of neurostimulation for the treatment of epilepsy. The technology was approved in 1997 for the treatment of medically refractory partial-onset seizures in patients 12 years or older. It consists of a stimulator that sends electric impulses to the left vagus nerve in the neck via a lead wire that is implanted under the skin. Studies since 1997 have indicated efficacy in generalized seizure disorders and children as well, according to Dr. Elinor Ben-Menachem, professor of neurology and epilepsy at the Institute for Clinical Neurosciences and Physiology, Göteborg (Sweden) University.
To date, more than 60,000 patients worldwide have been treated with VNS, and studies suggest that approximately 50% of patients who undergo the procedure experience a long-term decrease in mean seizure frequency of 50% or more. But fewer than 10% become seizure-free, Dr. Ben-Menachem said during the neurostimulation plenary presentation.
"[VNS] has a long history now, and what we know is that it does not cure or affect seizures immediately. We actually don’t notice a change in seizure activity until about 18 months or 2 years after starting."
For example, a recent long-term follow-up study of VNS patients in the Czech Republic showed that at 1 year post implantation, 44.4% of patients achieved more than 50% seizure reduction. The percentage of patients who reached that level of seizure reduction then increased from 58.7% at 2 years after implant to 64.4% at 5 years. At the 5-year mark, 15.5% of the patients had achieved a minimum 90% seizure reduction, and 5.5% were seizure free (Seizure 2009;18: 269-74).
The mechanism of action of VNS remains uncertain, but a number of possibilities have been suggested, including arousal of the reticular formation; stimulation of locus coeruleus and noradrenaline pathways; changes in a neurotransmitter, amino acid, or neuropeptide; or indirect thalamus stimulation, according to Dr. Ben-Menachem. "It’s also possible that there is long-term learning through synaptic structural changes," she said. "The more I work with this, the more I think it is a learning paradigm. It’s like learning to play the piano. You can’t just sit down and play, you have to redo and redo until the brain is trained."
In a recent study of 144 patients who had undergone VNS implantation, 10 patients were seizure free for more than 1 year post implantation, 89 patients experienced seizure improvement, and no changes were observed in 45 patients. "Stepwise multivariate analysis showed that unilateral interictal epileptiform discharges [IEDs], cortical dysgenesis, and younger age at implantation were independent predictors of seizure freedom in the long-term follow-up," they wrote (Seizure 2010;19: 264-8).
Most of the adverse events associated with VNS therapy, such as hoarseness and cough, tend to be mild and are stimulation related, Dr. Ben-Menachem explained. "Typically, they occur only during stimulation and they generally diminish over time on their own, or they may be diminished or eliminated by adjustment of the parameter settings."
Programmed Deep Brain Stimulation. The programmed deep brain stimulation device manufactured by Medtronic, one of the two emerging neurostimulation treatments for intractable epilepsy that is under FDA review, demonstrated efficacy in a pivotal trial that involved stimulation in the anterior thalamus. This site has connections with the temporal lobe, which is a common site for the origin of partial seizures, Dr. Bergey explained.
The device, which is already approved for Parkinson's disease, comprises two leads* that are implanted bilaterally into the target structure with a pulse generator placed below the clavicle. Each lead contains four electrodes. The device delivers stimuli at scheduled intervals "to hopefully modulate and reduce the number of seizures the patient is having," he said.
In the Medtronic-funded Stimulation of the Anterior Nucleus of the Thalamus for Epilepsy (SANTE) study, 110 patients with medically refractory partial seizures were implanted with the device and randomized to intermittent bilateral stimulation (1 minute on/5 minutes off) or no-stimulation for a 3-month blinded stage, followed by unblinded stimulation for all of the patients (Epilepsia 2010;51:899-908). At the end of the blinded period, patients who received stimulation experienced a median seizure reduction of 40.4%, compared with 14.5% of patients with the stimulator off, reported study coauthor Dr. Vincenta Salanova of Indiana University, Indianapolis.
In the open-label follow-up, 56% of all the patients had greater than 50% seizure reduction at 2 years, and there was a median 68% reduction in seizures among the 42 patients for whom 3-year data were available. Over the course of the study, "14 [12.7%] of the patients were seizure free for at least 6 months," she reported in a press briefing at the meeting.
Although the mechanism of action is not fully understood, Dr. Salanova said that "the thalamus has connections between the limbic system and the frontal lobe, so it’s possible that high-frequency stimulation may prevent the propagation of seizures."
Five deaths occurred in the study population, but none were attributed to lead implantation or stimulation, Dr. Salanova stressed. There were no symptomatic or clinically significant hemorrhages associated with implantation, but 4.5% of patients experienced asymptomatic intracranial hemorrhages – detected via neuroimaging – that were not clinically significant. Additionally, two patients experienced seizures that were linked to the stimulus, which were resolved by lowering the voltage.
Direct stimulation of the hippocampus may also offer seizure relief in some patients, according to Dr. Richard Wennberg of the University of Toronto. "The hippocampus is clinically recognized as a region of high epileptogenicity, and animal studies have demonstrated antiepileptic properties of electrical fields applied to the region," he said in a presentation during the neurostimulation plenary session, noting that the goal of direct hippocampal stimulation is to prevent seizure generation and spread from the temporal limbic region.
To date, the experimental procedure has been evaluated in small series and has shown some efficacy, Dr. Wennberg said. For example, in a recent study designed to assess the effect of continuous electrical stimulation of the hippocampus bilaterally, two patients with seizures from both mesial temporal lobes who were not candidates for surgical resection were implanted bilaterally with two four-contact electrodes along the hippocampal axis. After randomization to either stimulation on or off conditions for 3-month intervals, seizure frequency decreased by 33% during stimulation, and stayed and remained lower by 25% for the 3 months after stimulation was turned off, after which the seizure frequency returned to baseline, the authors reported. Although seizure frequency was reduced both during and for a period after bilateral hippocampal stimulation, "the overall impact in this study is not as robust as has been previously reported," the authors stated (Epilepsia 2010;51:304-7).
Responsive Neurostimulation. Another direct brain neurostimulating technology under FDA review is the Responsive Neurostimulator System (RNS) by NeuroPace. The system detects and aborts spontaneously occurring abnormal discharges* to prevent seizures, explained Dr. Lawrence J. Hirsch of Columbia University in New York. "It is designed to respond within seconds to abnormal activity in the brain by delivering a series of up to five stimuli to terminate the abnormal discharge."
The RNS device is implanted in a recess of the skull, and is connected to up to two four-contact electrodes that are placed within the brain or on the brain surface, depending on where the seizures begin. The device collects and stores seizure information, which the patient subsequently downloads to a laptop using a wand. Physicians can access the stored electrocorticograms via a secure Web page through which they can adjust detection and stimulation parameters specific to the individual patient, Dr. Hirsch said during the neurostimulation plenary session.
In the pivotal clinical trial of the RNS system, 191 patients with medically intractable, partial-onset seizures localized to one or two foci received the cranial implant. During a blinded period, patients received active or sham stimulation, followed by an open-label phase in which all the patients received active stimulation. During the entire blinded evaluation period, active stimulation was associated with a mean 37.9% reduction in seizure frequency, compared with a mean 17% reduction during the sham activation, Dr. Hirsch said.
"In the final month of the blinded period – month 4 to 5 – the respective reduction in seizure frequency was 42% and 9%." During the last 3 months of the open-label period, "47% of the patients had a greater than 50% seizure reduction," he said. "And at 4 years post implant, more than 50% of the patients had at least a 50% reduction in seizure frequency."
A subset analysis showed that neither prior surgery nor the number of seizure foci had an effect on treatment response, Dr. Hirsch noted. "It also showed that [RNS] is possibly more effective with medial temporal onset."
With respect to adverse events, implant site infections were reported in 5% of the patients, and led to explantation in 2%. The combined rate of status epilepticus reported in all trials of the device (256 patients) was 3.5%, and included episodes occurring between 5 months and 5 years post implant. Intracranial hemorrhage was reported in 4% of the patients, and included only one patient with neurological sequelae, which was chronic headache, he said.
The chronic, intracranial EEG recordings provided by the RNS technology have other potentially valuable uses, including seizure prediction/warning; seizure awareness and counting as a way to assess treatment efficacy; identification of circadian, catamenial, and other ictal and interictal patterns; and the lateralization of bitemporal seizures, Dr. Hirsch said.
Dr. Bergey disclosed financial relationships with Pfizer, UCB, and Eli Lilly. Dr. Ben-Menachem disclosed financial relationships with UCB, Eisai, Janssen, Cilag, Cyberonics, Lundbeck, and Sunovion. Dr. Wennberg disclosed a financial relationship with Medtronic. Dr. Hirsch reported having no financial disclosures.
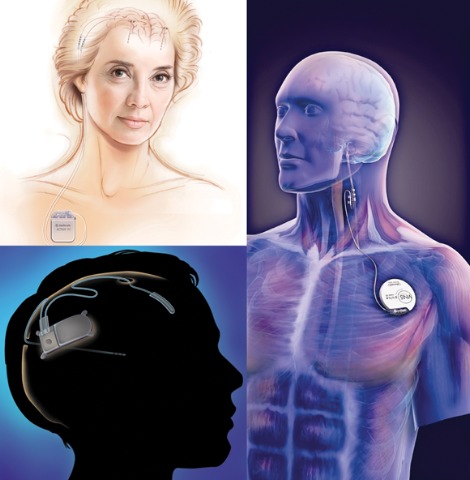
Medtronic's DBS system (top left) stimulates the anterior nucleus of the thalamus, whereas NeuroPace's RNS device (bottom left) responds to abnormal activity in targeted areas and Cyberonics' VNS Therapy System (right) periodically stimulates the left vagus nerve. (Photo Credit: top left: (c) Medtronic Inc., bottom left: (c) NeuroPace Inc., right: (c) Cyberonics Inc.)
* CORRECTION, 12/16/2010: The original version of this article misstated the function of the Responsive Neurostimulator System (RNS) by NeuroPace Inc. The system detects and aborts spontaneously occurring abnormal discharges. Also, the programmed deep brain stimulation device manufactured by Medtronic uses two leads that are placed bilaterally into the anterior thalamus. Each lead contains four electrodes. This version has been updated.
SAN ANTONIO – It’s still too soon to know whether neurostimulation will be the therapeutic advance that treatment-refractory epilepsy patients have been waiting for, but the possibility that it might be has the epilepsy community buzzing.
For approximately 30% of epilepsy patients, seizures cannot be controlled with antiepileptic drugs or surgery. New long-term safety and efficacy data for vagus nerve simulation (VNS) and the results of recent pivotal trials of two approaches to direct brain stimulation offer beacons of hope to these patients, Dr. Gregory K. Bergey said in a plenary session on neurostimulation at the annual meeting of the American Epilepsy Society.
"One of the frustrating things for those of us treating patients with epilepsy has been the fact that, although a number of new antiepileptic drugs have been developed over the past 10-15 years and most are better tolerated and have better pharmacokinetic profiles than earlier drugs, the number of patients with seizures that don’t respond to medical therapy has not been significantly reduced," said Dr. Bergey, director of the Johns Hopkins Epilepsy Center, Baltimore.
"So we’re stepping back and saying, ‘Is there some other way we can treat these patients?’ That has been the impetus for looking at neurostimulation, which has been around for well over a decade, and what we’re seeing is exciting."
Although the 40%-50% response rates observed in direct brain stimulation trials do not appear to be overwhelming, "this is just the beginning," Dr. Bergey stressed in an interview. "As opposed to a drug trial, where you go up to a certain dose and it either works or it doesn’t work, in the case of neurostimulation we don’t know the optimal stimulus parameters, and I think that’s what you’re going to begin to see over the next several years," he said.
"There’s going to be a lot of investigation into neurostimulation of the brain structures to try to figure out who are the best candidates and what the best stimulus parameters are. It’s easy to say we’re stimulating the brain, but do we stimulate 100 times per second, 50 times per second, 25 times per second, and what should the stimulus intensities be?"
Vagus Nerve Stimulation. Currently, Cyberonics’ VNS Therapy System is the only Food and Drug Administration–approved form of neurostimulation for the treatment of epilepsy. The technology was approved in 1997 for the treatment of medically refractory partial-onset seizures in patients 12 years or older. It consists of a stimulator that sends electric impulses to the left vagus nerve in the neck via a lead wire that is implanted under the skin. Studies since 1997 have indicated efficacy in generalized seizure disorders and children as well, according to Dr. Elinor Ben-Menachem, professor of neurology and epilepsy at the Institute for Clinical Neurosciences and Physiology, Göteborg (Sweden) University.
To date, more than 60,000 patients worldwide have been treated with VNS, and studies suggest that approximately 50% of patients who undergo the procedure experience a long-term decrease in mean seizure frequency of 50% or more. But fewer than 10% become seizure-free, Dr. Ben-Menachem said during the neurostimulation plenary presentation.
"[VNS] has a long history now, and what we know is that it does not cure or affect seizures immediately. We actually don’t notice a change in seizure activity until about 18 months or 2 years after starting."
For example, a recent long-term follow-up study of VNS patients in the Czech Republic showed that at 1 year post implantation, 44.4% of patients achieved more than 50% seizure reduction. The percentage of patients who reached that level of seizure reduction then increased from 58.7% at 2 years after implant to 64.4% at 5 years. At the 5-year mark, 15.5% of the patients had achieved a minimum 90% seizure reduction, and 5.5% were seizure free (Seizure 2009;18: 269-74).
The mechanism of action of VNS remains uncertain, but a number of possibilities have been suggested, including arousal of the reticular formation; stimulation of locus coeruleus and noradrenaline pathways; changes in a neurotransmitter, amino acid, or neuropeptide; or indirect thalamus stimulation, according to Dr. Ben-Menachem. "It’s also possible that there is long-term learning through synaptic structural changes," she said. "The more I work with this, the more I think it is a learning paradigm. It’s like learning to play the piano. You can’t just sit down and play, you have to redo and redo until the brain is trained."
In a recent study of 144 patients who had undergone VNS implantation, 10 patients were seizure free for more than 1 year post implantation, 89 patients experienced seizure improvement, and no changes were observed in 45 patients. "Stepwise multivariate analysis showed that unilateral interictal epileptiform discharges [IEDs], cortical dysgenesis, and younger age at implantation were independent predictors of seizure freedom in the long-term follow-up," they wrote (Seizure 2010;19: 264-8).
Most of the adverse events associated with VNS therapy, such as hoarseness and cough, tend to be mild and are stimulation related, Dr. Ben-Menachem explained. "Typically, they occur only during stimulation and they generally diminish over time on their own, or they may be diminished or eliminated by adjustment of the parameter settings."
Programmed Deep Brain Stimulation. The programmed deep brain stimulation device manufactured by Medtronic, one of the two emerging neurostimulation treatments for intractable epilepsy that is under FDA review, demonstrated efficacy in a pivotal trial that involved stimulation in the anterior thalamus. This site has connections with the temporal lobe, which is a common site for the origin of partial seizures, Dr. Bergey explained.
The device, which is already approved for Parkinson's disease, comprises two leads* that are implanted bilaterally into the target structure with a pulse generator placed below the clavicle. Each lead contains four electrodes. The device delivers stimuli at scheduled intervals "to hopefully modulate and reduce the number of seizures the patient is having," he said.
In the Medtronic-funded Stimulation of the Anterior Nucleus of the Thalamus for Epilepsy (SANTE) study, 110 patients with medically refractory partial seizures were implanted with the device and randomized to intermittent bilateral stimulation (1 minute on/5 minutes off) or no-stimulation for a 3-month blinded stage, followed by unblinded stimulation for all of the patients (Epilepsia 2010;51:899-908). At the end of the blinded period, patients who received stimulation experienced a median seizure reduction of 40.4%, compared with 14.5% of patients with the stimulator off, reported study coauthor Dr. Vincenta Salanova of Indiana University, Indianapolis.
In the open-label follow-up, 56% of all the patients had greater than 50% seizure reduction at 2 years, and there was a median 68% reduction in seizures among the 42 patients for whom 3-year data were available. Over the course of the study, "14 [12.7%] of the patients were seizure free for at least 6 months," she reported in a press briefing at the meeting.
Although the mechanism of action is not fully understood, Dr. Salanova said that "the thalamus has connections between the limbic system and the frontal lobe, so it’s possible that high-frequency stimulation may prevent the propagation of seizures."
Five deaths occurred in the study population, but none were attributed to lead implantation or stimulation, Dr. Salanova stressed. There were no symptomatic or clinically significant hemorrhages associated with implantation, but 4.5% of patients experienced asymptomatic intracranial hemorrhages – detected via neuroimaging – that were not clinically significant. Additionally, two patients experienced seizures that were linked to the stimulus, which were resolved by lowering the voltage.
Direct stimulation of the hippocampus may also offer seizure relief in some patients, according to Dr. Richard Wennberg of the University of Toronto. "The hippocampus is clinically recognized as a region of high epileptogenicity, and animal studies have demonstrated antiepileptic properties of electrical fields applied to the region," he said in a presentation during the neurostimulation plenary session, noting that the goal of direct hippocampal stimulation is to prevent seizure generation and spread from the temporal limbic region.
To date, the experimental procedure has been evaluated in small series and has shown some efficacy, Dr. Wennberg said. For example, in a recent study designed to assess the effect of continuous electrical stimulation of the hippocampus bilaterally, two patients with seizures from both mesial temporal lobes who were not candidates for surgical resection were implanted bilaterally with two four-contact electrodes along the hippocampal axis. After randomization to either stimulation on or off conditions for 3-month intervals, seizure frequency decreased by 33% during stimulation, and stayed and remained lower by 25% for the 3 months after stimulation was turned off, after which the seizure frequency returned to baseline, the authors reported. Although seizure frequency was reduced both during and for a period after bilateral hippocampal stimulation, "the overall impact in this study is not as robust as has been previously reported," the authors stated (Epilepsia 2010;51:304-7).
Responsive Neurostimulation. Another direct brain neurostimulating technology under FDA review is the Responsive Neurostimulator System (RNS) by NeuroPace. The system detects and aborts spontaneously occurring abnormal discharges* to prevent seizures, explained Dr. Lawrence J. Hirsch of Columbia University in New York. "It is designed to respond within seconds to abnormal activity in the brain by delivering a series of up to five stimuli to terminate the abnormal discharge."
The RNS device is implanted in a recess of the skull, and is connected to up to two four-contact electrodes that are placed within the brain or on the brain surface, depending on where the seizures begin. The device collects and stores seizure information, which the patient subsequently downloads to a laptop using a wand. Physicians can access the stored electrocorticograms via a secure Web page through which they can adjust detection and stimulation parameters specific to the individual patient, Dr. Hirsch said during the neurostimulation plenary session.
In the pivotal clinical trial of the RNS system, 191 patients with medically intractable, partial-onset seizures localized to one or two foci received the cranial implant. During a blinded period, patients received active or sham stimulation, followed by an open-label phase in which all the patients received active stimulation. During the entire blinded evaluation period, active stimulation was associated with a mean 37.9% reduction in seizure frequency, compared with a mean 17% reduction during the sham activation, Dr. Hirsch said.
"In the final month of the blinded period – month 4 to 5 – the respective reduction in seizure frequency was 42% and 9%." During the last 3 months of the open-label period, "47% of the patients had a greater than 50% seizure reduction," he said. "And at 4 years post implant, more than 50% of the patients had at least a 50% reduction in seizure frequency."
A subset analysis showed that neither prior surgery nor the number of seizure foci had an effect on treatment response, Dr. Hirsch noted. "It also showed that [RNS] is possibly more effective with medial temporal onset."
With respect to adverse events, implant site infections were reported in 5% of the patients, and led to explantation in 2%. The combined rate of status epilepticus reported in all trials of the device (256 patients) was 3.5%, and included episodes occurring between 5 months and 5 years post implant. Intracranial hemorrhage was reported in 4% of the patients, and included only one patient with neurological sequelae, which was chronic headache, he said.
The chronic, intracranial EEG recordings provided by the RNS technology have other potentially valuable uses, including seizure prediction/warning; seizure awareness and counting as a way to assess treatment efficacy; identification of circadian, catamenial, and other ictal and interictal patterns; and the lateralization of bitemporal seizures, Dr. Hirsch said.
Dr. Bergey disclosed financial relationships with Pfizer, UCB, and Eli Lilly. Dr. Ben-Menachem disclosed financial relationships with UCB, Eisai, Janssen, Cilag, Cyberonics, Lundbeck, and Sunovion. Dr. Wennberg disclosed a financial relationship with Medtronic. Dr. Hirsch reported having no financial disclosures.
Medtronic's DBS system (top left) stimulates the anterior nucleus of the thalamus, whereas NeuroPace's RNS device (bottom left) responds to abnormal activity in targeted areas and Cyberonics' VNS Therapy System (right) periodically stimulates the left vagus nerve. (Photo Credit: top left: (c) Medtronic Inc., bottom left: (c) NeuroPace Inc., right: (c) Cyberonics Inc.)
* CORRECTION, 12/16/2010: The original version of this article misstated the function of the Responsive Neurostimulator System (RNS) by NeuroPace Inc. The system detects and aborts spontaneously occurring abnormal discharges. Also, the programmed deep brain stimulation device manufactured by Medtronic uses two leads that are placed bilaterally into the anterior thalamus. Each lead contains four electrodes. This version has been updated.
SAN ANTONIO – It’s still too soon to know whether neurostimulation will be the therapeutic advance that treatment-refractory epilepsy patients have been waiting for, but the possibility that it might be has the epilepsy community buzzing.
For approximately 30% of epilepsy patients, seizures cannot be controlled with antiepileptic drugs or surgery. New long-term safety and efficacy data for vagus nerve simulation (VNS) and the results of recent pivotal trials of two approaches to direct brain stimulation offer beacons of hope to these patients, Dr. Gregory K. Bergey said in a plenary session on neurostimulation at the annual meeting of the American Epilepsy Society.
"One of the frustrating things for those of us treating patients with epilepsy has been the fact that, although a number of new antiepileptic drugs have been developed over the past 10-15 years and most are better tolerated and have better pharmacokinetic profiles than earlier drugs, the number of patients with seizures that don’t respond to medical therapy has not been significantly reduced," said Dr. Bergey, director of the Johns Hopkins Epilepsy Center, Baltimore.
"So we’re stepping back and saying, ‘Is there some other way we can treat these patients?’ That has been the impetus for looking at neurostimulation, which has been around for well over a decade, and what we’re seeing is exciting."
Although the 40%-50% response rates observed in direct brain stimulation trials do not appear to be overwhelming, "this is just the beginning," Dr. Bergey stressed in an interview. "As opposed to a drug trial, where you go up to a certain dose and it either works or it doesn’t work, in the case of neurostimulation we don’t know the optimal stimulus parameters, and I think that’s what you’re going to begin to see over the next several years," he said.
"There’s going to be a lot of investigation into neurostimulation of the brain structures to try to figure out who are the best candidates and what the best stimulus parameters are. It’s easy to say we’re stimulating the brain, but do we stimulate 100 times per second, 50 times per second, 25 times per second, and what should the stimulus intensities be?"
Vagus Nerve Stimulation. Currently, Cyberonics’ VNS Therapy System is the only Food and Drug Administration–approved form of neurostimulation for the treatment of epilepsy. The technology was approved in 1997 for the treatment of medically refractory partial-onset seizures in patients 12 years or older. It consists of a stimulator that sends electric impulses to the left vagus nerve in the neck via a lead wire that is implanted under the skin. Studies since 1997 have indicated efficacy in generalized seizure disorders and children as well, according to Dr. Elinor Ben-Menachem, professor of neurology and epilepsy at the Institute for Clinical Neurosciences and Physiology, Göteborg (Sweden) University.
To date, more than 60,000 patients worldwide have been treated with VNS, and studies suggest that approximately 50% of patients who undergo the procedure experience a long-term decrease in mean seizure frequency of 50% or more. But fewer than 10% become seizure-free, Dr. Ben-Menachem said during the neurostimulation plenary presentation.
"[VNS] has a long history now, and what we know is that it does not cure or affect seizures immediately. We actually don’t notice a change in seizure activity until about 18 months or 2 years after starting."
For example, a recent long-term follow-up study of VNS patients in the Czech Republic showed that at 1 year post implantation, 44.4% of patients achieved more than 50% seizure reduction. The percentage of patients who reached that level of seizure reduction then increased from 58.7% at 2 years after implant to 64.4% at 5 years. At the 5-year mark, 15.5% of the patients had achieved a minimum 90% seizure reduction, and 5.5% were seizure free (Seizure 2009;18: 269-74).
The mechanism of action of VNS remains uncertain, but a number of possibilities have been suggested, including arousal of the reticular formation; stimulation of locus coeruleus and noradrenaline pathways; changes in a neurotransmitter, amino acid, or neuropeptide; or indirect thalamus stimulation, according to Dr. Ben-Menachem. "It’s also possible that there is long-term learning through synaptic structural changes," she said. "The more I work with this, the more I think it is a learning paradigm. It’s like learning to play the piano. You can’t just sit down and play, you have to redo and redo until the brain is trained."
In a recent study of 144 patients who had undergone VNS implantation, 10 patients were seizure free for more than 1 year post implantation, 89 patients experienced seizure improvement, and no changes were observed in 45 patients. "Stepwise multivariate analysis showed that unilateral interictal epileptiform discharges [IEDs], cortical dysgenesis, and younger age at implantation were independent predictors of seizure freedom in the long-term follow-up," they wrote (Seizure 2010;19: 264-8).
Most of the adverse events associated with VNS therapy, such as hoarseness and cough, tend to be mild and are stimulation related, Dr. Ben-Menachem explained. "Typically, they occur only during stimulation and they generally diminish over time on their own, or they may be diminished or eliminated by adjustment of the parameter settings."
Programmed Deep Brain Stimulation. The programmed deep brain stimulation device manufactured by Medtronic, one of the two emerging neurostimulation treatments for intractable epilepsy that is under FDA review, demonstrated efficacy in a pivotal trial that involved stimulation in the anterior thalamus. This site has connections with the temporal lobe, which is a common site for the origin of partial seizures, Dr. Bergey explained.
The device, which is already approved for Parkinson's disease, comprises two leads* that are implanted bilaterally into the target structure with a pulse generator placed below the clavicle. Each lead contains four electrodes. The device delivers stimuli at scheduled intervals "to hopefully modulate and reduce the number of seizures the patient is having," he said.
In the Medtronic-funded Stimulation of the Anterior Nucleus of the Thalamus for Epilepsy (SANTE) study, 110 patients with medically refractory partial seizures were implanted with the device and randomized to intermittent bilateral stimulation (1 minute on/5 minutes off) or no-stimulation for a 3-month blinded stage, followed by unblinded stimulation for all of the patients (Epilepsia 2010;51:899-908). At the end of the blinded period, patients who received stimulation experienced a median seizure reduction of 40.4%, compared with 14.5% of patients with the stimulator off, reported study coauthor Dr. Vincenta Salanova of Indiana University, Indianapolis.
In the open-label follow-up, 56% of all the patients had greater than 50% seizure reduction at 2 years, and there was a median 68% reduction in seizures among the 42 patients for whom 3-year data were available. Over the course of the study, "14 [12.7%] of the patients were seizure free for at least 6 months," she reported in a press briefing at the meeting.
Although the mechanism of action is not fully understood, Dr. Salanova said that "the thalamus has connections between the limbic system and the frontal lobe, so it’s possible that high-frequency stimulation may prevent the propagation of seizures."
Five deaths occurred in the study population, but none were attributed to lead implantation or stimulation, Dr. Salanova stressed. There were no symptomatic or clinically significant hemorrhages associated with implantation, but 4.5% of patients experienced asymptomatic intracranial hemorrhages – detected via neuroimaging – that were not clinically significant. Additionally, two patients experienced seizures that were linked to the stimulus, which were resolved by lowering the voltage.
Direct stimulation of the hippocampus may also offer seizure relief in some patients, according to Dr. Richard Wennberg of the University of Toronto. "The hippocampus is clinically recognized as a region of high epileptogenicity, and animal studies have demonstrated antiepileptic properties of electrical fields applied to the region," he said in a presentation during the neurostimulation plenary session, noting that the goal of direct hippocampal stimulation is to prevent seizure generation and spread from the temporal limbic region.
To date, the experimental procedure has been evaluated in small series and has shown some efficacy, Dr. Wennberg said. For example, in a recent study designed to assess the effect of continuous electrical stimulation of the hippocampus bilaterally, two patients with seizures from both mesial temporal lobes who were not candidates for surgical resection were implanted bilaterally with two four-contact electrodes along the hippocampal axis. After randomization to either stimulation on or off conditions for 3-month intervals, seizure frequency decreased by 33% during stimulation, and stayed and remained lower by 25% for the 3 months after stimulation was turned off, after which the seizure frequency returned to baseline, the authors reported. Although seizure frequency was reduced both during and for a period after bilateral hippocampal stimulation, "the overall impact in this study is not as robust as has been previously reported," the authors stated (Epilepsia 2010;51:304-7).
Responsive Neurostimulation. Another direct brain neurostimulating technology under FDA review is the Responsive Neurostimulator System (RNS) by NeuroPace. The system detects and aborts spontaneously occurring abnormal discharges* to prevent seizures, explained Dr. Lawrence J. Hirsch of Columbia University in New York. "It is designed to respond within seconds to abnormal activity in the brain by delivering a series of up to five stimuli to terminate the abnormal discharge."
The RNS device is implanted in a recess of the skull, and is connected to up to two four-contact electrodes that are placed within the brain or on the brain surface, depending on where the seizures begin. The device collects and stores seizure information, which the patient subsequently downloads to a laptop using a wand. Physicians can access the stored electrocorticograms via a secure Web page through which they can adjust detection and stimulation parameters specific to the individual patient, Dr. Hirsch said during the neurostimulation plenary session.
In the pivotal clinical trial of the RNS system, 191 patients with medically intractable, partial-onset seizures localized to one or two foci received the cranial implant. During a blinded period, patients received active or sham stimulation, followed by an open-label phase in which all the patients received active stimulation. During the entire blinded evaluation period, active stimulation was associated with a mean 37.9% reduction in seizure frequency, compared with a mean 17% reduction during the sham activation, Dr. Hirsch said.
"In the final month of the blinded period – month 4 to 5 – the respective reduction in seizure frequency was 42% and 9%." During the last 3 months of the open-label period, "47% of the patients had a greater than 50% seizure reduction," he said. "And at 4 years post implant, more than 50% of the patients had at least a 50% reduction in seizure frequency."
A subset analysis showed that neither prior surgery nor the number of seizure foci had an effect on treatment response, Dr. Hirsch noted. "It also showed that [RNS] is possibly more effective with medial temporal onset."
With respect to adverse events, implant site infections were reported in 5% of the patients, and led to explantation in 2%. The combined rate of status epilepticus reported in all trials of the device (256 patients) was 3.5%, and included episodes occurring between 5 months and 5 years post implant. Intracranial hemorrhage was reported in 4% of the patients, and included only one patient with neurological sequelae, which was chronic headache, he said.
The chronic, intracranial EEG recordings provided by the RNS technology have other potentially valuable uses, including seizure prediction/warning; seizure awareness and counting as a way to assess treatment efficacy; identification of circadian, catamenial, and other ictal and interictal patterns; and the lateralization of bitemporal seizures, Dr. Hirsch said.
Dr. Bergey disclosed financial relationships with Pfizer, UCB, and Eli Lilly. Dr. Ben-Menachem disclosed financial relationships with UCB, Eisai, Janssen, Cilag, Cyberonics, Lundbeck, and Sunovion. Dr. Wennberg disclosed a financial relationship with Medtronic. Dr. Hirsch reported having no financial disclosures.
Medtronic's DBS system (top left) stimulates the anterior nucleus of the thalamus, whereas NeuroPace's RNS device (bottom left) responds to abnormal activity in targeted areas and Cyberonics' VNS Therapy System (right) periodically stimulates the left vagus nerve. (Photo Credit: top left: (c) Medtronic Inc., bottom left: (c) NeuroPace Inc., right: (c) Cyberonics Inc.)
* CORRECTION, 12/16/2010: The original version of this article misstated the function of the Responsive Neurostimulator System (RNS) by NeuroPace Inc. The system detects and aborts spontaneously occurring abnormal discharges. Also, the programmed deep brain stimulation device manufactured by Medtronic uses two leads that are placed bilaterally into the anterior thalamus. Each lead contains four electrodes. This version has been updated.
EXPERT ANALYSIS FROM THE ANNUAL MEETING OF THE AMERICAN EPILEPSY SOCIETY
New Psoriasis Treatments Offer Patients More Options
Increased insight into the pathogenesis of psoriasis has led to the development of new drugs with novel mechanisms of action, as well as safer and more effective approaches to treatment with conventional drugs
As a result, treatment options for the management of patients with moderate to severe psoriasis in particular – nearly 25% of all patients with psoriasis, according to the National Psoriasis Foundation – have broadened substantially, and research reported during the past year suggests they are on track for continued expansion.

Among the most notable trials of the year were those establishing the "unsurpassed efficacy" of Abbott’s investigational monoclonal antibody briakinumab, according to Dr. Craig Leonardi, a dermatologist and psoriasis specialist at St. Louis University. Like the recently approved psoriatic drug ustekinumab (Stelara), briakinumab is an injectable biologic agent that targets the interleukin-12 and -23 (IL-12/23) proteins, which are believed to promote the inflammation associated with psoriasis.
[Interrupting Biologic Therapy for Psoriasis: What to Expect]
IL-12/23 Inhibition
Four phase III clinical studies of briakinumab were presented at the annual meeting of the European Academy of Dermatology and Venereology (EADV) in Gothenburg, Sweden.
– M06-890. In this trial, comparing the efficacy and safety of briakinumab to placebo, 80.7% of the 981 patients randomized to receive briakinumab every 4 weeks following an induction phase experienced a 75% improvement in psoriasis symptoms (PASI 75) at week 12, compared with 4.5% of the 484 patients randomized to placebo. Additionally, 61.6% and 32.2% of patients, respectively, saw 90% and 100% symptom clearance. And at week 52, 82.4% of patients who had achieved PASI 75 maintained at least that level of clearance.
– M10-315 and M10-114. In each of two 12-week trials comparing briakinumab to etanercept, significantly more patients randomized to briakinumab achieved Physician Global Assessment (PGA) scores of 0 or 1 and PASI 75 clearance. In the 350-patient M10-315 and the 347-patient M10-114 studies, respectively, 72.7% and 71% of patients randomized to briakinumab treatment achieved PGA 0 or 1, compared with 29.5% and 39.7% of patients randomized to etanercept therapy and 4.2% and 2.9% of those randomized to placebo.
[Briakinumab Boosts Quality of Life for Psoriasis Patients]
Additionally, in the respective studies, 80.6% and 81.9% of the briakinumab-treated patients achieved PASI 75, compared with 39.6% and 56% of the etanercept patents, and 6.9% and 7.4% of the placebo groups.
– M10-255. The fourth trial of 317 patients was a head-to-head comparison with methotrexate in which 81.8% of briakinumab patients achieved PASI 75 clearance at 24 weeks, compared with 39.9% of those taking methotrexate. At 52 weeks, the PASI 75 clearance rates were 66.2% in the briakinumab group and 23.9% in the methotrexate group.
The most common adverse events observed across all four studies and in an ongoing open-label extension were upper respiratory infection, nasopharyngitis, headache, arthralgia, hypertension, and back pain. The incidence of infection and malignancy with briakinumab were higher than those with placebo, but were similar to those in patients treated with etanercept or methotrexate, according to a press release issued by Abbott.
Of particular note is the incidence of major adverse cardiovascular events (MACE) associated with briakinumab, Dr. Leonardi said in an interview. In the M06-890 trial, for example, seven briakinumab patients – all of whom had identifiable underlying cardiovascular risk factors, according to Abbott – experienced a MACE. A similar number of major cardiovascular events were observed in phase III trials of ustekinumab (Stelara), the IL 12/23 antagonist that received Food and Drug Administration approval for psoriasis treatment, he said. "Both drugs share the same mechanism of action, so it’s important to be cautious," he said. "Briakinumab has been submitted for FDA approval. We are awaiting review and comment."
Ustekinumab.
Positive ustekinumab results were also presented last year. A pooled analysis of safety data reported at the summer academy meeting of the American Academy of Dermatology in Chicago demonstrated a favorable risk/benefit profile for up to 3 years of treatment.
Based on integrated data for 3,117 patients from the pivotal phase III PHOENIX 1 and PHOENIX 2 trials and the phase III ACCEPT trial, the investigators reported that the overall rates of adverse and serious adverse events were comparable in the 45-mg and 90-mg ustekinumab patient groups.
For the 45-mg and 90-mg patient groups, respectively, the per-hundred-patient-year rates were 0.82 and 1.50 for serious infections, 0.69 and 0.46 for noncutaneous malignancies, and 0.41 and 0.35 for MACE.
These rates were consistent with expectations for both general and psoriasis populations, and they remained stable over time, according to Dr. Leonardi, one of the study investigators. The maintenance of the favorable safety profile in patients who have been treated for several years is "encouraging," he said, noting that ongoing 5-year follow-up studies will enable continued monitoring of the drug’s safety.

Other important developments that may bode well for the treatment of psoriasis include data on IL-17 inhibitors, oral and topical janus kinase (JAK) inhibitors, an oral phosphodiesterase inhibitor, several IL-23 blockers, and topical niacin/calcipotriene, according to Dr. Leonardi and Dr. Alice B. Gottlieb of Tufts Medical Center in Boston.
Targeting IL-17
Recent studies have suggested that IL-17A–producing T cells have a crucial role in the pathogenesis of psoriasis, making them a potential treatment target.
In a phase II trial, a fully human IL-17 antibody AIN457 (Novartis) produced clinically meaningful reductions of disease activity at 4 and 12 weeks in 13 of 18 psoriasis patients who were randomized to receive a single 3-mg/kg intravenous infusion of the drug. At study weeks 4 and 12, respectively, the mean PASI score decreased by 58% and 63% in the AIN457 patients, compared with 4% and 9% in placebo patients, the authors wrote (Sci. Transl. Med. 2010;52:52ra72).
The results of a phase I study of another IL-17 drug – Amgen’s AMG 827, which binds to and blocks signaling via the IL-17 receptor – were reported at the EADV meeting. Most of the 16 patients who received a single dose of the fully human monoclonal antibody experienced substantial improvements in psoriasis symptoms. Based on the favorable findings, a phase II study is currently underway.
JAK 3 Inhibitors
The JAK pathways are also believed to have roles in the psoriasis disease cascade and as such have emerged as a treatment target. The four known JAK enzymes – JAK1, JAK2, JAK3, and tyrosine kinase (TYK) 2 – are components of signaling mechanisms used by multiple cytokines and growth factors that trigger dysregulated inflammatory pathways, according to Dr. Gottlieb. Oral and topical inhibitors of JAK are in phase II and III trials and have had promising results so far, she said in an interview.
In a 197-patient, phase II efficacy and safety study, Pfizer’s investigational oral JAK3 inhibitor, tentatively named tasocitinib (CP-690550), produced statistically significant responses at 12 weeks, compared with placebo in adults with moderate to severe psoriasis. The PASI 75 responses for patients randomized to twice daily tasocitinib at 2-mg, 5-mg, and 15-mg doses, respectively, were 25%, 40.8%, and 66.7%, compared with 2.0% for placebo, according to a company-issued statement. Additionally, as early as study week 4, "treatment with 5 and 15 mg twice daily of tasocitinib significantly improved patient-reported health-related quality of life outcomes."
Regarding safety, three patients randomized to tasocitinib treatment experienced a total of five serious adverse events during the study. Additionally, the investigators observed dose dependent decreases in mean neutrophil counts and hemoglobin values and increases in mean LDL, HDL, and total cholesterol levels. A large-scale, phase III trial program, called Oral Psoriasis Treatment (OPT) trials, is currently underway.
Topical JAK inhibitors are also under development, which presumably will avoid some of the complications associated with systemic therapy, according to Dr. Gottlieb, who is involved with investigations of Incyte’s topical JAK1/JAK2 inhibitor, INCB18424, which has demonstrated robust activity and safety in placebo-controlled, multidose, phase IIb trials in patients with mild to moderate psoriasis.
In studies reported by Dr. Gottlieb at last year’s annual meeting of the American Academy of Dermatology, topical application of INCB18424 cream led to significant improvement in the psoriatic lesions of treated patients at day 28, with the total lesion scores decreasing twofold, compared with placebo. "The effects were seen as early as 2 weeks, and they extended through" the study period," she said, noting that immunohistochemical staining and microarray gene analysis data confirmed the improvement in skin histology and the reduction in the psoriatic molecular signature.
Apremilast May Break Ground
Poised to be "the first oral medication with a new mechanism of action for psoriasis in almost 20 years," the phosphodiesterase 4 (PDE4) enzyme inhibitor apremilast (Celgene) demonstrated favorable results in a phase IIb study reported in December 2009 and has recently begun phase III trials, according to Dr. Leonardi.
In the phase IIb trial, 352 patients with moderate to severe plaque-type psoriasis were randomized to receive 10 mg, 20 mg or 30 mg of apremilast twice per day or placebo. Of the apremilast patients, 41% of the 30-mg group, 29% of the 20-mg group, and 11% of the 10-mg group achieved a PASI 75 after 16 weeks, compared with 6% of the placebo group, according a company statement.
A comparison of infection rates showed that infections occurred in 48% of the 30 mg apremilast patients and 33% of the placebo patients. No serious adverse events related to apremilast were reported.
The positive results have led to the initiation of two pivotal phase III trials: the multicenter ESTEEM 1 and ESTEEM 2 safety and efficacy studies comprising, respectively, 825 patients and 405 patients with moderate to severe plaque psoriasis. Primary outcome data from both studies will be available in the summer of 2011.
Topical Niacin/Calcipotriene
The nonsteroidal topical options for psoriasis will likely be expanded further by the addition of topical niacin (nicotinamide) to standard treatment with the synthetic vitamin D derivative calcipotriene, according to Dr. Gottlieb.
A recent 168-patient multicenter, double-blind, randomized trial reported that 50% of patients randomized to combination therapy with 0.005% calcipotriene and 1.4% nicotinamide achieved symptom clearance or near clearance, compared with 18.8% of patients randomized to placebo, 25% of patients treated with nicotinamide alone, and 31.5% of patients treated with calcipotriene alone.
The findings suggest that the combination therapy "may prove effective as an alternative therapeutic option to calcipotriene monotherapy and may provide an attractive option for patients seeking an effective corticosteroid-sparing topical psoriatic agent," the investigators wrote (J. Am. Acad. Dermatol. 2010;63:775-81).
In the midst of all of the new developments, it is important to note that tumor necrosis factor (TNF) antagonists continue to perform well, Dr. Leonardi stressed. "Given that the class is now 12 years old and includes 2 million patients, we are unlikely to learn of major safety risks at this point, and registry data support the notion that TNF blockade is cardioprotective in [rheumatoid arthritis], in distinction to the IL 12/23 blockers, where the MACE issue is unresolved," he said. "At this point, it is important for dermatologists to remember that the IL-12/23 drugs, in particular, are new, and since they are psoriasis specific, there are no other disease states where they were previously used."
Until the new drugs are in widespread use, "they should be used with caution," Dr. Leonardi continued. "The last psoriasis-specific biologic we used was Raptiva, which was voluntarily removed from the market by Genentech after 3 patients were identified with a rare and fatal central nervous system infection. At the time these patients were identified, the drug had been on the market for 5 years and had been used in 23,000 patients."
[Topical Tar Making a Comeback in Psoriasis]
What this means in clinical practice is that ustekinumab or briakinumab may be reasonable options for patients with a history of failure of TNF antagonists or a history of central or peripheral demyelination, but until longer-term safety data are available, it should not be the first choice in the majority of treatment-naive patients, said Dr. Leonardi.
Dr. Leonardi is a consultant, investigator, and/or on the speakers bureau of Amgen, Abbott, Celgene, Centocor, Genentech, Incyte, Novartis Pfizer, and several other pharmaceutical companies.
Dr. Gottlieb has current consulting/advisory board agreements with Abbott, Amgen, Celgene, Incyte, and numerous other pharmaceutical companies.
Increased insight into the pathogenesis of psoriasis has led to the development of new drugs with novel mechanisms of action, as well as safer and more effective approaches to treatment with conventional drugs
As a result, treatment options for the management of patients with moderate to severe psoriasis in particular – nearly 25% of all patients with psoriasis, according to the National Psoriasis Foundation – have broadened substantially, and research reported during the past year suggests they are on track for continued expansion.
Among the most notable trials of the year were those establishing the "unsurpassed efficacy" of Abbott’s investigational monoclonal antibody briakinumab, according to Dr. Craig Leonardi, a dermatologist and psoriasis specialist at St. Louis University. Like the recently approved psoriatic drug ustekinumab (Stelara), briakinumab is an injectable biologic agent that targets the interleukin-12 and -23 (IL-12/23) proteins, which are believed to promote the inflammation associated with psoriasis.
[Interrupting Biologic Therapy for Psoriasis: What to Expect]
IL-12/23 Inhibition
Four phase III clinical studies of briakinumab were presented at the annual meeting of the European Academy of Dermatology and Venereology (EADV) in Gothenburg, Sweden.
– M06-890. In this trial, comparing the efficacy and safety of briakinumab to placebo, 80.7% of the 981 patients randomized to receive briakinumab every 4 weeks following an induction phase experienced a 75% improvement in psoriasis symptoms (PASI 75) at week 12, compared with 4.5% of the 484 patients randomized to placebo. Additionally, 61.6% and 32.2% of patients, respectively, saw 90% and 100% symptom clearance. And at week 52, 82.4% of patients who had achieved PASI 75 maintained at least that level of clearance.
– M10-315 and M10-114. In each of two 12-week trials comparing briakinumab to etanercept, significantly more patients randomized to briakinumab achieved Physician Global Assessment (PGA) scores of 0 or 1 and PASI 75 clearance. In the 350-patient M10-315 and the 347-patient M10-114 studies, respectively, 72.7% and 71% of patients randomized to briakinumab treatment achieved PGA 0 or 1, compared with 29.5% and 39.7% of patients randomized to etanercept therapy and 4.2% and 2.9% of those randomized to placebo.
[Briakinumab Boosts Quality of Life for Psoriasis Patients]
Additionally, in the respective studies, 80.6% and 81.9% of the briakinumab-treated patients achieved PASI 75, compared with 39.6% and 56% of the etanercept patents, and 6.9% and 7.4% of the placebo groups.
– M10-255. The fourth trial of 317 patients was a head-to-head comparison with methotrexate in which 81.8% of briakinumab patients achieved PASI 75 clearance at 24 weeks, compared with 39.9% of those taking methotrexate. At 52 weeks, the PASI 75 clearance rates were 66.2% in the briakinumab group and 23.9% in the methotrexate group.
The most common adverse events observed across all four studies and in an ongoing open-label extension were upper respiratory infection, nasopharyngitis, headache, arthralgia, hypertension, and back pain. The incidence of infection and malignancy with briakinumab were higher than those with placebo, but were similar to those in patients treated with etanercept or methotrexate, according to a press release issued by Abbott.
Of particular note is the incidence of major adverse cardiovascular events (MACE) associated with briakinumab, Dr. Leonardi said in an interview. In the M06-890 trial, for example, seven briakinumab patients – all of whom had identifiable underlying cardiovascular risk factors, according to Abbott – experienced a MACE. A similar number of major cardiovascular events were observed in phase III trials of ustekinumab (Stelara), the IL 12/23 antagonist that received Food and Drug Administration approval for psoriasis treatment, he said. "Both drugs share the same mechanism of action, so it’s important to be cautious," he said. "Briakinumab has been submitted for FDA approval. We are awaiting review and comment."
Ustekinumab.
Positive ustekinumab results were also presented last year. A pooled analysis of safety data reported at the summer academy meeting of the American Academy of Dermatology in Chicago demonstrated a favorable risk/benefit profile for up to 3 years of treatment.
Based on integrated data for 3,117 patients from the pivotal phase III PHOENIX 1 and PHOENIX 2 trials and the phase III ACCEPT trial, the investigators reported that the overall rates of adverse and serious adverse events were comparable in the 45-mg and 90-mg ustekinumab patient groups.
For the 45-mg and 90-mg patient groups, respectively, the per-hundred-patient-year rates were 0.82 and 1.50 for serious infections, 0.69 and 0.46 for noncutaneous malignancies, and 0.41 and 0.35 for MACE.
These rates were consistent with expectations for both general and psoriasis populations, and they remained stable over time, according to Dr. Leonardi, one of the study investigators. The maintenance of the favorable safety profile in patients who have been treated for several years is "encouraging," he said, noting that ongoing 5-year follow-up studies will enable continued monitoring of the drug’s safety.
Other important developments that may bode well for the treatment of psoriasis include data on IL-17 inhibitors, oral and topical janus kinase (JAK) inhibitors, an oral phosphodiesterase inhibitor, several IL-23 blockers, and topical niacin/calcipotriene, according to Dr. Leonardi and Dr. Alice B. Gottlieb of Tufts Medical Center in Boston.
Targeting IL-17
Recent studies have suggested that IL-17A–producing T cells have a crucial role in the pathogenesis of psoriasis, making them a potential treatment target.
In a phase II trial, a fully human IL-17 antibody AIN457 (Novartis) produced clinically meaningful reductions of disease activity at 4 and 12 weeks in 13 of 18 psoriasis patients who were randomized to receive a single 3-mg/kg intravenous infusion of the drug. At study weeks 4 and 12, respectively, the mean PASI score decreased by 58% and 63% in the AIN457 patients, compared with 4% and 9% in placebo patients, the authors wrote (Sci. Transl. Med. 2010;52:52ra72).
The results of a phase I study of another IL-17 drug – Amgen’s AMG 827, which binds to and blocks signaling via the IL-17 receptor – were reported at the EADV meeting. Most of the 16 patients who received a single dose of the fully human monoclonal antibody experienced substantial improvements in psoriasis symptoms. Based on the favorable findings, a phase II study is currently underway.
JAK 3 Inhibitors
The JAK pathways are also believed to have roles in the psoriasis disease cascade and as such have emerged as a treatment target. The four known JAK enzymes – JAK1, JAK2, JAK3, and tyrosine kinase (TYK) 2 – are components of signaling mechanisms used by multiple cytokines and growth factors that trigger dysregulated inflammatory pathways, according to Dr. Gottlieb. Oral and topical inhibitors of JAK are in phase II and III trials and have had promising results so far, she said in an interview.
In a 197-patient, phase II efficacy and safety study, Pfizer’s investigational oral JAK3 inhibitor, tentatively named tasocitinib (CP-690550), produced statistically significant responses at 12 weeks, compared with placebo in adults with moderate to severe psoriasis. The PASI 75 responses for patients randomized to twice daily tasocitinib at 2-mg, 5-mg, and 15-mg doses, respectively, were 25%, 40.8%, and 66.7%, compared with 2.0% for placebo, according to a company-issued statement. Additionally, as early as study week 4, "treatment with 5 and 15 mg twice daily of tasocitinib significantly improved patient-reported health-related quality of life outcomes."
Regarding safety, three patients randomized to tasocitinib treatment experienced a total of five serious adverse events during the study. Additionally, the investigators observed dose dependent decreases in mean neutrophil counts and hemoglobin values and increases in mean LDL, HDL, and total cholesterol levels. A large-scale, phase III trial program, called Oral Psoriasis Treatment (OPT) trials, is currently underway.
Topical JAK inhibitors are also under development, which presumably will avoid some of the complications associated with systemic therapy, according to Dr. Gottlieb, who is involved with investigations of Incyte’s topical JAK1/JAK2 inhibitor, INCB18424, which has demonstrated robust activity and safety in placebo-controlled, multidose, phase IIb trials in patients with mild to moderate psoriasis.
In studies reported by Dr. Gottlieb at last year’s annual meeting of the American Academy of Dermatology, topical application of INCB18424 cream led to significant improvement in the psoriatic lesions of treated patients at day 28, with the total lesion scores decreasing twofold, compared with placebo. "The effects were seen as early as 2 weeks, and they extended through" the study period," she said, noting that immunohistochemical staining and microarray gene analysis data confirmed the improvement in skin histology and the reduction in the psoriatic molecular signature.
Apremilast May Break Ground
Poised to be "the first oral medication with a new mechanism of action for psoriasis in almost 20 years," the phosphodiesterase 4 (PDE4) enzyme inhibitor apremilast (Celgene) demonstrated favorable results in a phase IIb study reported in December 2009 and has recently begun phase III trials, according to Dr. Leonardi.
In the phase IIb trial, 352 patients with moderate to severe plaque-type psoriasis were randomized to receive 10 mg, 20 mg or 30 mg of apremilast twice per day or placebo. Of the apremilast patients, 41% of the 30-mg group, 29% of the 20-mg group, and 11% of the 10-mg group achieved a PASI 75 after 16 weeks, compared with 6% of the placebo group, according a company statement.
A comparison of infection rates showed that infections occurred in 48% of the 30 mg apremilast patients and 33% of the placebo patients. No serious adverse events related to apremilast were reported.
The positive results have led to the initiation of two pivotal phase III trials: the multicenter ESTEEM 1 and ESTEEM 2 safety and efficacy studies comprising, respectively, 825 patients and 405 patients with moderate to severe plaque psoriasis. Primary outcome data from both studies will be available in the summer of 2011.
Topical Niacin/Calcipotriene
The nonsteroidal topical options for psoriasis will likely be expanded further by the addition of topical niacin (nicotinamide) to standard treatment with the synthetic vitamin D derivative calcipotriene, according to Dr. Gottlieb.
A recent 168-patient multicenter, double-blind, randomized trial reported that 50% of patients randomized to combination therapy with 0.005% calcipotriene and 1.4% nicotinamide achieved symptom clearance or near clearance, compared with 18.8% of patients randomized to placebo, 25% of patients treated with nicotinamide alone, and 31.5% of patients treated with calcipotriene alone.
The findings suggest that the combination therapy "may prove effective as an alternative therapeutic option to calcipotriene monotherapy and may provide an attractive option for patients seeking an effective corticosteroid-sparing topical psoriatic agent," the investigators wrote (J. Am. Acad. Dermatol. 2010;63:775-81).
In the midst of all of the new developments, it is important to note that tumor necrosis factor (TNF) antagonists continue to perform well, Dr. Leonardi stressed. "Given that the class is now 12 years old and includes 2 million patients, we are unlikely to learn of major safety risks at this point, and registry data support the notion that TNF blockade is cardioprotective in [rheumatoid arthritis], in distinction to the IL 12/23 blockers, where the MACE issue is unresolved," he said. "At this point, it is important for dermatologists to remember that the IL-12/23 drugs, in particular, are new, and since they are psoriasis specific, there are no other disease states where they were previously used."
Until the new drugs are in widespread use, "they should be used with caution," Dr. Leonardi continued. "The last psoriasis-specific biologic we used was Raptiva, which was voluntarily removed from the market by Genentech after 3 patients were identified with a rare and fatal central nervous system infection. At the time these patients were identified, the drug had been on the market for 5 years and had been used in 23,000 patients."
[Topical Tar Making a Comeback in Psoriasis]
What this means in clinical practice is that ustekinumab or briakinumab may be reasonable options for patients with a history of failure of TNF antagonists or a history of central or peripheral demyelination, but until longer-term safety data are available, it should not be the first choice in the majority of treatment-naive patients, said Dr. Leonardi.
Dr. Leonardi is a consultant, investigator, and/or on the speakers bureau of Amgen, Abbott, Celgene, Centocor, Genentech, Incyte, Novartis Pfizer, and several other pharmaceutical companies.
Dr. Gottlieb has current consulting/advisory board agreements with Abbott, Amgen, Celgene, Incyte, and numerous other pharmaceutical companies.
Increased insight into the pathogenesis of psoriasis has led to the development of new drugs with novel mechanisms of action, as well as safer and more effective approaches to treatment with conventional drugs
As a result, treatment options for the management of patients with moderate to severe psoriasis in particular – nearly 25% of all patients with psoriasis, according to the National Psoriasis Foundation – have broadened substantially, and research reported during the past year suggests they are on track for continued expansion.
Among the most notable trials of the year were those establishing the "unsurpassed efficacy" of Abbott’s investigational monoclonal antibody briakinumab, according to Dr. Craig Leonardi, a dermatologist and psoriasis specialist at St. Louis University. Like the recently approved psoriatic drug ustekinumab (Stelara), briakinumab is an injectable biologic agent that targets the interleukin-12 and -23 (IL-12/23) proteins, which are believed to promote the inflammation associated with psoriasis.
[Interrupting Biologic Therapy for Psoriasis: What to Expect]
IL-12/23 Inhibition
Four phase III clinical studies of briakinumab were presented at the annual meeting of the European Academy of Dermatology and Venereology (EADV) in Gothenburg, Sweden.
– M06-890. In this trial, comparing the efficacy and safety of briakinumab to placebo, 80.7% of the 981 patients randomized to receive briakinumab every 4 weeks following an induction phase experienced a 75% improvement in psoriasis symptoms (PASI 75) at week 12, compared with 4.5% of the 484 patients randomized to placebo. Additionally, 61.6% and 32.2% of patients, respectively, saw 90% and 100% symptom clearance. And at week 52, 82.4% of patients who had achieved PASI 75 maintained at least that level of clearance.
– M10-315 and M10-114. In each of two 12-week trials comparing briakinumab to etanercept, significantly more patients randomized to briakinumab achieved Physician Global Assessment (PGA) scores of 0 or 1 and PASI 75 clearance. In the 350-patient M10-315 and the 347-patient M10-114 studies, respectively, 72.7% and 71% of patients randomized to briakinumab treatment achieved PGA 0 or 1, compared with 29.5% and 39.7% of patients randomized to etanercept therapy and 4.2% and 2.9% of those randomized to placebo.
[Briakinumab Boosts Quality of Life for Psoriasis Patients]
Additionally, in the respective studies, 80.6% and 81.9% of the briakinumab-treated patients achieved PASI 75, compared with 39.6% and 56% of the etanercept patents, and 6.9% and 7.4% of the placebo groups.
– M10-255. The fourth trial of 317 patients was a head-to-head comparison with methotrexate in which 81.8% of briakinumab patients achieved PASI 75 clearance at 24 weeks, compared with 39.9% of those taking methotrexate. At 52 weeks, the PASI 75 clearance rates were 66.2% in the briakinumab group and 23.9% in the methotrexate group.
The most common adverse events observed across all four studies and in an ongoing open-label extension were upper respiratory infection, nasopharyngitis, headache, arthralgia, hypertension, and back pain. The incidence of infection and malignancy with briakinumab were higher than those with placebo, but were similar to those in patients treated with etanercept or methotrexate, according to a press release issued by Abbott.
Of particular note is the incidence of major adverse cardiovascular events (MACE) associated with briakinumab, Dr. Leonardi said in an interview. In the M06-890 trial, for example, seven briakinumab patients – all of whom had identifiable underlying cardiovascular risk factors, according to Abbott – experienced a MACE. A similar number of major cardiovascular events were observed in phase III trials of ustekinumab (Stelara), the IL 12/23 antagonist that received Food and Drug Administration approval for psoriasis treatment, he said. "Both drugs share the same mechanism of action, so it’s important to be cautious," he said. "Briakinumab has been submitted for FDA approval. We are awaiting review and comment."
Ustekinumab.
Positive ustekinumab results were also presented last year. A pooled analysis of safety data reported at the summer academy meeting of the American Academy of Dermatology in Chicago demonstrated a favorable risk/benefit profile for up to 3 years of treatment.
Based on integrated data for 3,117 patients from the pivotal phase III PHOENIX 1 and PHOENIX 2 trials and the phase III ACCEPT trial, the investigators reported that the overall rates of adverse and serious adverse events were comparable in the 45-mg and 90-mg ustekinumab patient groups.
For the 45-mg and 90-mg patient groups, respectively, the per-hundred-patient-year rates were 0.82 and 1.50 for serious infections, 0.69 and 0.46 for noncutaneous malignancies, and 0.41 and 0.35 for MACE.
These rates were consistent with expectations for both general and psoriasis populations, and they remained stable over time, according to Dr. Leonardi, one of the study investigators. The maintenance of the favorable safety profile in patients who have been treated for several years is "encouraging," he said, noting that ongoing 5-year follow-up studies will enable continued monitoring of the drug’s safety.
Other important developments that may bode well for the treatment of psoriasis include data on IL-17 inhibitors, oral and topical janus kinase (JAK) inhibitors, an oral phosphodiesterase inhibitor, several IL-23 blockers, and topical niacin/calcipotriene, according to Dr. Leonardi and Dr. Alice B. Gottlieb of Tufts Medical Center in Boston.
Targeting IL-17
Recent studies have suggested that IL-17A–producing T cells have a crucial role in the pathogenesis of psoriasis, making them a potential treatment target.
In a phase II trial, a fully human IL-17 antibody AIN457 (Novartis) produced clinically meaningful reductions of disease activity at 4 and 12 weeks in 13 of 18 psoriasis patients who were randomized to receive a single 3-mg/kg intravenous infusion of the drug. At study weeks 4 and 12, respectively, the mean PASI score decreased by 58% and 63% in the AIN457 patients, compared with 4% and 9% in placebo patients, the authors wrote (Sci. Transl. Med. 2010;52:52ra72).
The results of a phase I study of another IL-17 drug – Amgen’s AMG 827, which binds to and blocks signaling via the IL-17 receptor – were reported at the EADV meeting. Most of the 16 patients who received a single dose of the fully human monoclonal antibody experienced substantial improvements in psoriasis symptoms. Based on the favorable findings, a phase II study is currently underway.
JAK 3 Inhibitors
The JAK pathways are also believed to have roles in the psoriasis disease cascade and as such have emerged as a treatment target. The four known JAK enzymes – JAK1, JAK2, JAK3, and tyrosine kinase (TYK) 2 – are components of signaling mechanisms used by multiple cytokines and growth factors that trigger dysregulated inflammatory pathways, according to Dr. Gottlieb. Oral and topical inhibitors of JAK are in phase II and III trials and have had promising results so far, she said in an interview.
In a 197-patient, phase II efficacy and safety study, Pfizer’s investigational oral JAK3 inhibitor, tentatively named tasocitinib (CP-690550), produced statistically significant responses at 12 weeks, compared with placebo in adults with moderate to severe psoriasis. The PASI 75 responses for patients randomized to twice daily tasocitinib at 2-mg, 5-mg, and 15-mg doses, respectively, were 25%, 40.8%, and 66.7%, compared with 2.0% for placebo, according to a company-issued statement. Additionally, as early as study week 4, "treatment with 5 and 15 mg twice daily of tasocitinib significantly improved patient-reported health-related quality of life outcomes."
Regarding safety, three patients randomized to tasocitinib treatment experienced a total of five serious adverse events during the study. Additionally, the investigators observed dose dependent decreases in mean neutrophil counts and hemoglobin values and increases in mean LDL, HDL, and total cholesterol levels. A large-scale, phase III trial program, called Oral Psoriasis Treatment (OPT) trials, is currently underway.
Topical JAK inhibitors are also under development, which presumably will avoid some of the complications associated with systemic therapy, according to Dr. Gottlieb, who is involved with investigations of Incyte’s topical JAK1/JAK2 inhibitor, INCB18424, which has demonstrated robust activity and safety in placebo-controlled, multidose, phase IIb trials in patients with mild to moderate psoriasis.
In studies reported by Dr. Gottlieb at last year’s annual meeting of the American Academy of Dermatology, topical application of INCB18424 cream led to significant improvement in the psoriatic lesions of treated patients at day 28, with the total lesion scores decreasing twofold, compared with placebo. "The effects were seen as early as 2 weeks, and they extended through" the study period," she said, noting that immunohistochemical staining and microarray gene analysis data confirmed the improvement in skin histology and the reduction in the psoriatic molecular signature.
Apremilast May Break Ground
Poised to be "the first oral medication with a new mechanism of action for psoriasis in almost 20 years," the phosphodiesterase 4 (PDE4) enzyme inhibitor apremilast (Celgene) demonstrated favorable results in a phase IIb study reported in December 2009 and has recently begun phase III trials, according to Dr. Leonardi.
In the phase IIb trial, 352 patients with moderate to severe plaque-type psoriasis were randomized to receive 10 mg, 20 mg or 30 mg of apremilast twice per day or placebo. Of the apremilast patients, 41% of the 30-mg group, 29% of the 20-mg group, and 11% of the 10-mg group achieved a PASI 75 after 16 weeks, compared with 6% of the placebo group, according a company statement.
A comparison of infection rates showed that infections occurred in 48% of the 30 mg apremilast patients and 33% of the placebo patients. No serious adverse events related to apremilast were reported.
The positive results have led to the initiation of two pivotal phase III trials: the multicenter ESTEEM 1 and ESTEEM 2 safety and efficacy studies comprising, respectively, 825 patients and 405 patients with moderate to severe plaque psoriasis. Primary outcome data from both studies will be available in the summer of 2011.
Topical Niacin/Calcipotriene
The nonsteroidal topical options for psoriasis will likely be expanded further by the addition of topical niacin (nicotinamide) to standard treatment with the synthetic vitamin D derivative calcipotriene, according to Dr. Gottlieb.
A recent 168-patient multicenter, double-blind, randomized trial reported that 50% of patients randomized to combination therapy with 0.005% calcipotriene and 1.4% nicotinamide achieved symptom clearance or near clearance, compared with 18.8% of patients randomized to placebo, 25% of patients treated with nicotinamide alone, and 31.5% of patients treated with calcipotriene alone.
The findings suggest that the combination therapy "may prove effective as an alternative therapeutic option to calcipotriene monotherapy and may provide an attractive option for patients seeking an effective corticosteroid-sparing topical psoriatic agent," the investigators wrote (J. Am. Acad. Dermatol. 2010;63:775-81).
In the midst of all of the new developments, it is important to note that tumor necrosis factor (TNF) antagonists continue to perform well, Dr. Leonardi stressed. "Given that the class is now 12 years old and includes 2 million patients, we are unlikely to learn of major safety risks at this point, and registry data support the notion that TNF blockade is cardioprotective in [rheumatoid arthritis], in distinction to the IL 12/23 blockers, where the MACE issue is unresolved," he said. "At this point, it is important for dermatologists to remember that the IL-12/23 drugs, in particular, are new, and since they are psoriasis specific, there are no other disease states where they were previously used."
Until the new drugs are in widespread use, "they should be used with caution," Dr. Leonardi continued. "The last psoriasis-specific biologic we used was Raptiva, which was voluntarily removed from the market by Genentech after 3 patients were identified with a rare and fatal central nervous system infection. At the time these patients were identified, the drug had been on the market for 5 years and had been used in 23,000 patients."
[Topical Tar Making a Comeback in Psoriasis]
What this means in clinical practice is that ustekinumab or briakinumab may be reasonable options for patients with a history of failure of TNF antagonists or a history of central or peripheral demyelination, but until longer-term safety data are available, it should not be the first choice in the majority of treatment-naive patients, said Dr. Leonardi.
Dr. Leonardi is a consultant, investigator, and/or on the speakers bureau of Amgen, Abbott, Celgene, Centocor, Genentech, Incyte, Novartis Pfizer, and several other pharmaceutical companies.
Dr. Gottlieb has current consulting/advisory board agreements with Abbott, Amgen, Celgene, Incyte, and numerous other pharmaceutical companies.
Virologic Response Protects Against Death in Liver Transplant Patients With HCV
BOSTON – Achieving sustained virologic response to antiviral therapy was highly protective against liver-related death in liver transplant patients with chronic hepatitis C, according to an analysis of data collected over a 20-year period from 12 liver transplant centers in Italy.
Patients with recurrent HCV who were unable to achieve sustained virologic response (SVR) were at a significantly higher risk of liver-related mortality than were those for whom the virus remained suppressed 6 weeks after treatment, Dr. Maria Rendina reported at the annual meeting of the American Association for the Study of Liver Diseases.
For the study, the investigators retrospectively analyzed data on 464 patients who underwent liver transplantation between 1989 and 2008. All of the patients had viral and histologic confirmation of HCV recurrence and all were treated with recombinant or pegylated interferon plus ribavirin at standard doses for at least 1 year, Dr. Rendina said. Of the study population, 35% achieved SVR, defined as undetectable HCV RNA 6 weeks after the end of treatment, and approximately 30% died during the mean 6.8 years of posttreatment follow-up. "Liver-related mortality was the cause of death in 73% of the patients who did not survive, and all of the liver-related deaths except for one were in the group of patients who did not attain [SVR]," she said.
A comparison of the SVR and non-SVR patient characteristics showed no differences in immunosuppressive strategies between the two groups, according to Dr. Rendina of the University of Bari in Italy. Overall, the SVR patients received livers from younger donors. They also had longer treatment durations, higher cumulative doses of interferon, lower dropout rates, and lower diabetes incidence, she said.
In univariate analysis, older donor age, viral genotypes 1 or 4, diabetes, and lack of SVR to antiviral therapy were significant risk factors for death, Dr. Rendina reported. In a multivariate analysis, after adjustment for donor, viral, and recipient variables including age, gender, genotype, immunosuppression, creatinine, hepatocellular carcinoma, and diabetes, failure to achieve SVR remained "significantly and independently associated with a very high risk of liver-related death," she said, noting that the hazard ratio for liver-related death in the non-SVR group was 3.3.
"Antiviral therapy should be strongly pursued in liver transplant patients with recurrent hepatitis C infection because of the protective role of [SVR]," she said.
The hesitancy to pursue antiviral therapy for HCV recurrence after liver transplantation stems, in large part, from the absence of data regarding the relationship between viral eradication and long-term clinical outcome, said Dr. Rendina. It is further exacerbated by the frequency and severity of adverse events associated with the pegylated interferon/ribavirin standard of care – most notably flulike symptoms, hemolytic anemia, neutropenia, depression, and neurologic symptoms, she said, noting that more than 25% of the patients in the current study stopped treatment because of intolerable side effects.
Despite the drawbacks, antiviral therapy holds the most promise for improving HCV-infected patients’ long-term outcome post transplantation and should be used in clinical practice, Dr. Rendina stated. At the same time, "further randomized trials aimed at exploring various treatment options and the efficacy of new antiviral drugs should be undertaken," she said.
Dr. Rendina disclosed having no financial conflicts of interest with respect to her presentation.
BOSTON – Achieving sustained virologic response to antiviral therapy was highly protective against liver-related death in liver transplant patients with chronic hepatitis C, according to an analysis of data collected over a 20-year period from 12 liver transplant centers in Italy.
Patients with recurrent HCV who were unable to achieve sustained virologic response (SVR) were at a significantly higher risk of liver-related mortality than were those for whom the virus remained suppressed 6 weeks after treatment, Dr. Maria Rendina reported at the annual meeting of the American Association for the Study of Liver Diseases.
For the study, the investigators retrospectively analyzed data on 464 patients who underwent liver transplantation between 1989 and 2008. All of the patients had viral and histologic confirmation of HCV recurrence and all were treated with recombinant or pegylated interferon plus ribavirin at standard doses for at least 1 year, Dr. Rendina said. Of the study population, 35% achieved SVR, defined as undetectable HCV RNA 6 weeks after the end of treatment, and approximately 30% died during the mean 6.8 years of posttreatment follow-up. "Liver-related mortality was the cause of death in 73% of the patients who did not survive, and all of the liver-related deaths except for one were in the group of patients who did not attain [SVR]," she said.
A comparison of the SVR and non-SVR patient characteristics showed no differences in immunosuppressive strategies between the two groups, according to Dr. Rendina of the University of Bari in Italy. Overall, the SVR patients received livers from younger donors. They also had longer treatment durations, higher cumulative doses of interferon, lower dropout rates, and lower diabetes incidence, she said.
In univariate analysis, older donor age, viral genotypes 1 or 4, diabetes, and lack of SVR to antiviral therapy were significant risk factors for death, Dr. Rendina reported. In a multivariate analysis, after adjustment for donor, viral, and recipient variables including age, gender, genotype, immunosuppression, creatinine, hepatocellular carcinoma, and diabetes, failure to achieve SVR remained "significantly and independently associated with a very high risk of liver-related death," she said, noting that the hazard ratio for liver-related death in the non-SVR group was 3.3.
"Antiviral therapy should be strongly pursued in liver transplant patients with recurrent hepatitis C infection because of the protective role of [SVR]," she said.
The hesitancy to pursue antiviral therapy for HCV recurrence after liver transplantation stems, in large part, from the absence of data regarding the relationship between viral eradication and long-term clinical outcome, said Dr. Rendina. It is further exacerbated by the frequency and severity of adverse events associated with the pegylated interferon/ribavirin standard of care – most notably flulike symptoms, hemolytic anemia, neutropenia, depression, and neurologic symptoms, she said, noting that more than 25% of the patients in the current study stopped treatment because of intolerable side effects.
Despite the drawbacks, antiviral therapy holds the most promise for improving HCV-infected patients’ long-term outcome post transplantation and should be used in clinical practice, Dr. Rendina stated. At the same time, "further randomized trials aimed at exploring various treatment options and the efficacy of new antiviral drugs should be undertaken," she said.
Dr. Rendina disclosed having no financial conflicts of interest with respect to her presentation.
BOSTON – Achieving sustained virologic response to antiviral therapy was highly protective against liver-related death in liver transplant patients with chronic hepatitis C, according to an analysis of data collected over a 20-year period from 12 liver transplant centers in Italy.
Patients with recurrent HCV who were unable to achieve sustained virologic response (SVR) were at a significantly higher risk of liver-related mortality than were those for whom the virus remained suppressed 6 weeks after treatment, Dr. Maria Rendina reported at the annual meeting of the American Association for the Study of Liver Diseases.
For the study, the investigators retrospectively analyzed data on 464 patients who underwent liver transplantation between 1989 and 2008. All of the patients had viral and histologic confirmation of HCV recurrence and all were treated with recombinant or pegylated interferon plus ribavirin at standard doses for at least 1 year, Dr. Rendina said. Of the study population, 35% achieved SVR, defined as undetectable HCV RNA 6 weeks after the end of treatment, and approximately 30% died during the mean 6.8 years of posttreatment follow-up. "Liver-related mortality was the cause of death in 73% of the patients who did not survive, and all of the liver-related deaths except for one were in the group of patients who did not attain [SVR]," she said.
A comparison of the SVR and non-SVR patient characteristics showed no differences in immunosuppressive strategies between the two groups, according to Dr. Rendina of the University of Bari in Italy. Overall, the SVR patients received livers from younger donors. They also had longer treatment durations, higher cumulative doses of interferon, lower dropout rates, and lower diabetes incidence, she said.
In univariate analysis, older donor age, viral genotypes 1 or 4, diabetes, and lack of SVR to antiviral therapy were significant risk factors for death, Dr. Rendina reported. In a multivariate analysis, after adjustment for donor, viral, and recipient variables including age, gender, genotype, immunosuppression, creatinine, hepatocellular carcinoma, and diabetes, failure to achieve SVR remained "significantly and independently associated with a very high risk of liver-related death," she said, noting that the hazard ratio for liver-related death in the non-SVR group was 3.3.
"Antiviral therapy should be strongly pursued in liver transplant patients with recurrent hepatitis C infection because of the protective role of [SVR]," she said.
The hesitancy to pursue antiviral therapy for HCV recurrence after liver transplantation stems, in large part, from the absence of data regarding the relationship between viral eradication and long-term clinical outcome, said Dr. Rendina. It is further exacerbated by the frequency and severity of adverse events associated with the pegylated interferon/ribavirin standard of care – most notably flulike symptoms, hemolytic anemia, neutropenia, depression, and neurologic symptoms, she said, noting that more than 25% of the patients in the current study stopped treatment because of intolerable side effects.
Despite the drawbacks, antiviral therapy holds the most promise for improving HCV-infected patients’ long-term outcome post transplantation and should be used in clinical practice, Dr. Rendina stated. At the same time, "further randomized trials aimed at exploring various treatment options and the efficacy of new antiviral drugs should be undertaken," she said.
Dr. Rendina disclosed having no financial conflicts of interest with respect to her presentation.
FROM THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION FOR THE STUDY OF LIVER DISEASES
Virologic Response Protects Against Death in Liver Transplant Patients With HCV
BOSTON – Achieving sustained virologic response to antiviral therapy was highly protective against liver-related death in liver transplant patients with chronic hepatitis C, according to an analysis of data collected over a 20-year period from 12 liver transplant centers in Italy.
Patients with recurrent HCV who were unable to achieve sustained virologic response (SVR) were at a significantly higher risk of liver-related mortality than were those for whom the virus remained suppressed 6 weeks after treatment, Dr. Maria Rendina reported at the annual meeting of the American Association for the Study of Liver Diseases.
For the study, the investigators retrospectively analyzed data on 464 patients who underwent liver transplantation between 1989 and 2008. All of the patients had viral and histologic confirmation of HCV recurrence and all were treated with recombinant or pegylated interferon plus ribavirin at standard doses for at least 1 year, Dr. Rendina said. Of the study population, 35% achieved SVR, defined as undetectable HCV RNA 6 weeks after the end of treatment, and approximately 30% died during the mean 6.8 years of posttreatment follow-up. "Liver-related mortality was the cause of death in 73% of the patients who did not survive, and all of the liver-related deaths except for one were in the group of patients who did not attain [SVR]," she said.
A comparison of the SVR and non-SVR patient characteristics showed no differences in immunosuppressive strategies between the two groups, according to Dr. Rendina of the University of Bari in Italy. Overall, the SVR patients received livers from younger donors. They also had longer treatment durations, higher cumulative doses of interferon, lower dropout rates, and lower diabetes incidence, she said.
In univariate analysis, older donor age, viral genotypes 1 or 4, diabetes, and lack of SVR to antiviral therapy were significant risk factors for death, Dr. Rendina reported. In a multivariate analysis, after adjustment for donor, viral, and recipient variables including age, gender, genotype, immunosuppression, creatinine, hepatocellular carcinoma, and diabetes, failure to achieve SVR remained "significantly and independently associated with a very high risk of liver-related death," she said, noting that the hazard ratio for liver-related death in the non-SVR group was 3.3.
"Antiviral therapy should be strongly pursued in liver transplant patients with recurrent hepatitis C infection because of the protective role of [SVR]," she said.
The hesitancy to pursue antiviral therapy for HCV recurrence after liver transplantation stems, in large part, from the absence of data regarding the relationship between viral eradication and long-term clinical outcome, said Dr. Rendina. It is further exacerbated by the frequency and severity of adverse events associated with the pegylated interferon/ribavirin standard of care – most notably flulike symptoms, hemolytic anemia, neutropenia, depression, and neurologic symptoms, she said, noting that more than 25% of the patients in the current study stopped treatment because of intolerable side effects.
Despite the drawbacks, antiviral therapy holds the most promise for improving HCV-infected patients’ long-term outcome post transplantation and should be used in clinical practice, Dr. Rendina stated. At the same time, "further randomized trials aimed at exploring various treatment options and the efficacy of new antiviral drugs should be undertaken," she said.
Dr. Rendina disclosed having no financial conflicts of interest with respect to her presentation.
BOSTON – Achieving sustained virologic response to antiviral therapy was highly protective against liver-related death in liver transplant patients with chronic hepatitis C, according to an analysis of data collected over a 20-year period from 12 liver transplant centers in Italy.
Patients with recurrent HCV who were unable to achieve sustained virologic response (SVR) were at a significantly higher risk of liver-related mortality than were those for whom the virus remained suppressed 6 weeks after treatment, Dr. Maria Rendina reported at the annual meeting of the American Association for the Study of Liver Diseases.
For the study, the investigators retrospectively analyzed data on 464 patients who underwent liver transplantation between 1989 and 2008. All of the patients had viral and histologic confirmation of HCV recurrence and all were treated with recombinant or pegylated interferon plus ribavirin at standard doses for at least 1 year, Dr. Rendina said. Of the study population, 35% achieved SVR, defined as undetectable HCV RNA 6 weeks after the end of treatment, and approximately 30% died during the mean 6.8 years of posttreatment follow-up. "Liver-related mortality was the cause of death in 73% of the patients who did not survive, and all of the liver-related deaths except for one were in the group of patients who did not attain [SVR]," she said.
A comparison of the SVR and non-SVR patient characteristics showed no differences in immunosuppressive strategies between the two groups, according to Dr. Rendina of the University of Bari in Italy. Overall, the SVR patients received livers from younger donors. They also had longer treatment durations, higher cumulative doses of interferon, lower dropout rates, and lower diabetes incidence, she said.
In univariate analysis, older donor age, viral genotypes 1 or 4, diabetes, and lack of SVR to antiviral therapy were significant risk factors for death, Dr. Rendina reported. In a multivariate analysis, after adjustment for donor, viral, and recipient variables including age, gender, genotype, immunosuppression, creatinine, hepatocellular carcinoma, and diabetes, failure to achieve SVR remained "significantly and independently associated with a very high risk of liver-related death," she said, noting that the hazard ratio for liver-related death in the non-SVR group was 3.3.
"Antiviral therapy should be strongly pursued in liver transplant patients with recurrent hepatitis C infection because of the protective role of [SVR]," she said.
The hesitancy to pursue antiviral therapy for HCV recurrence after liver transplantation stems, in large part, from the absence of data regarding the relationship between viral eradication and long-term clinical outcome, said Dr. Rendina. It is further exacerbated by the frequency and severity of adverse events associated with the pegylated interferon/ribavirin standard of care – most notably flulike symptoms, hemolytic anemia, neutropenia, depression, and neurologic symptoms, she said, noting that more than 25% of the patients in the current study stopped treatment because of intolerable side effects.
Despite the drawbacks, antiviral therapy holds the most promise for improving HCV-infected patients’ long-term outcome post transplantation and should be used in clinical practice, Dr. Rendina stated. At the same time, "further randomized trials aimed at exploring various treatment options and the efficacy of new antiviral drugs should be undertaken," she said.
Dr. Rendina disclosed having no financial conflicts of interest with respect to her presentation.
BOSTON – Achieving sustained virologic response to antiviral therapy was highly protective against liver-related death in liver transplant patients with chronic hepatitis C, according to an analysis of data collected over a 20-year period from 12 liver transplant centers in Italy.
Patients with recurrent HCV who were unable to achieve sustained virologic response (SVR) were at a significantly higher risk of liver-related mortality than were those for whom the virus remained suppressed 6 weeks after treatment, Dr. Maria Rendina reported at the annual meeting of the American Association for the Study of Liver Diseases.
For the study, the investigators retrospectively analyzed data on 464 patients who underwent liver transplantation between 1989 and 2008. All of the patients had viral and histologic confirmation of HCV recurrence and all were treated with recombinant or pegylated interferon plus ribavirin at standard doses for at least 1 year, Dr. Rendina said. Of the study population, 35% achieved SVR, defined as undetectable HCV RNA 6 weeks after the end of treatment, and approximately 30% died during the mean 6.8 years of posttreatment follow-up. "Liver-related mortality was the cause of death in 73% of the patients who did not survive, and all of the liver-related deaths except for one were in the group of patients who did not attain [SVR]," she said.
A comparison of the SVR and non-SVR patient characteristics showed no differences in immunosuppressive strategies between the two groups, according to Dr. Rendina of the University of Bari in Italy. Overall, the SVR patients received livers from younger donors. They also had longer treatment durations, higher cumulative doses of interferon, lower dropout rates, and lower diabetes incidence, she said.
In univariate analysis, older donor age, viral genotypes 1 or 4, diabetes, and lack of SVR to antiviral therapy were significant risk factors for death, Dr. Rendina reported. In a multivariate analysis, after adjustment for donor, viral, and recipient variables including age, gender, genotype, immunosuppression, creatinine, hepatocellular carcinoma, and diabetes, failure to achieve SVR remained "significantly and independently associated with a very high risk of liver-related death," she said, noting that the hazard ratio for liver-related death in the non-SVR group was 3.3.
"Antiviral therapy should be strongly pursued in liver transplant patients with recurrent hepatitis C infection because of the protective role of [SVR]," she said.
The hesitancy to pursue antiviral therapy for HCV recurrence after liver transplantation stems, in large part, from the absence of data regarding the relationship between viral eradication and long-term clinical outcome, said Dr. Rendina. It is further exacerbated by the frequency and severity of adverse events associated with the pegylated interferon/ribavirin standard of care – most notably flulike symptoms, hemolytic anemia, neutropenia, depression, and neurologic symptoms, she said, noting that more than 25% of the patients in the current study stopped treatment because of intolerable side effects.
Despite the drawbacks, antiviral therapy holds the most promise for improving HCV-infected patients’ long-term outcome post transplantation and should be used in clinical practice, Dr. Rendina stated. At the same time, "further randomized trials aimed at exploring various treatment options and the efficacy of new antiviral drugs should be undertaken," she said.
Dr. Rendina disclosed having no financial conflicts of interest with respect to her presentation.
FROM THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION FOR THE STUDY OF LIVER DISEASES
Major Finding: In liver transplant patients with recurrent hepatitis C, failure to achieve a sustained virologic response to antiviral therapy was associated with a hazard ratio for liver-related death of 3.3.
Data Source: A retrospective study of 464 patients from 12 transplant centers in Italy who underwent antiviral therapy for recurrent hepatitis C infection.
Disclosures: Dr. Rendina reported having no financial conflicts of interest.
Telbivudine for Hep B in Pregnancy Appears Safe, Improves Outcomes
BOSTON – Limited use of the antiviral drug telbivudine is safe and effective for pregnant women with active hepatitis B virus infection and reduces perinatal transmission of the virus to their infants, according to Dr. Calvin Pan.
In the first open-label case-control trial investigating the safety and efficacy of telbivudine during the second to third trimester of pregnancy, the synthetic nucleoside analogue led to significant increases in rates of complete virologic response and normalization of alanine aminotransferase (ALT) levels in treated vs. untreated pregnant women with hepatitis B virus (HBV) infection, reported Dr. Pan of Mount Sinai School of Medicine in New York.
Additionally, there were no congenital defects identified up to 28 weeks post partum in the infants born to treated mothers, he reported at the annual meeting of the American Association for the Study of Liver Diseases.
Dr. Pan and his colleagues at the Second Affiliated Hospital of the Southeast University in Nanjing, China, where the study was conducted, enrolled 88 pregnant women who screened positive for hepatitis B e antigen (HBeAg) between gestational weeks 12 and 23 and who had serum HBV DNA levels greater than 6 log10 copies/mL and elevated ALT levels up to 10 times the upper limit of normal (40 IU/mL). Of the 88 patients, 53 opted to take telbivudine, and the remaining 35 served as the study’s control arm, he said.
Women in the treatment arm received 600 mg/day of telbivudine beginning sometime between 20 and 32 weeks gestation and continuing until a minimum of 4 weeks post partum, and infants born to women in both groups received hepatitis B immunoglobulin within 24 hours of birth and the HBV vaccine at birth, 1 month, and 6 months, Dr. Pan said.
The mean duration of telbivudine treatment was 15.5 weeks, and all of the women who took the drug remained in the study through at least postpartum week 4, compared with 92% of the controls, said Dr. Pan. Physical and laboratory examinations were conducted at baseline, at the time of delivery, and at 4 weeks post partum, he said, noting that a second study looked at infant outcomes through 28 weeks.
At the time of delivery and 4 weeks post partum, a complete virologic response was observed in 53% and 62% of the treated patients, respectively, while no patients in the control arm achieved a complete virologic response at either time point, Dr. Pan reported. "Both the control arm and the telbivudine arm had HBV DNA of approximately 8 logs. At the time of delivery, this was reduced substantially [to 2.35 log10 copies/mL] in the mothers in the treatment arm," he said.
With respect to ALT levels, 77% of the treatment group achieved normalization, compared with 29% of the control group, Dr. Pan said. Also, the levels of HBeAg dropped by 98% in the treatment group, which was a significantly greater decrease than the 60% drop observed in the control group; the latter was likely the result of natural viral clearance, he noted.
An evaluation of newborn outcomes showed no congenital deformities nor any differences in gestational age, infant height and weight, or Apgar scores between the two groups, said Dr. Pan. Significantly fewer babies born to telbivudine-treated mothers vs. untreated mothers had hepatitis B surface antigens or detectable levels of HBV RNA at birth (4% vs. 23%), he said.
Telbivudine appeared to be well tolerated as there were no adverse event–related treatment discontinuations, said Dr. Pan. Also, none of the patients experienced virologic breakthrough.
Despite concerns regarding antiviral treatment during pregnancy because of the potential risks to the fetus, the findings from this study suggest that limited treatment with telbivudine can improve maternal and child outcomes, Dr. Pan concluded.
The study was funded by the Chinese Department of Health with no commercial support. Dr. Pan reported having no relevant financial disclosures.
BOSTON – Limited use of the antiviral drug telbivudine is safe and effective for pregnant women with active hepatitis B virus infection and reduces perinatal transmission of the virus to their infants, according to Dr. Calvin Pan.
In the first open-label case-control trial investigating the safety and efficacy of telbivudine during the second to third trimester of pregnancy, the synthetic nucleoside analogue led to significant increases in rates of complete virologic response and normalization of alanine aminotransferase (ALT) levels in treated vs. untreated pregnant women with hepatitis B virus (HBV) infection, reported Dr. Pan of Mount Sinai School of Medicine in New York.
Additionally, there were no congenital defects identified up to 28 weeks post partum in the infants born to treated mothers, he reported at the annual meeting of the American Association for the Study of Liver Diseases.
Dr. Pan and his colleagues at the Second Affiliated Hospital of the Southeast University in Nanjing, China, where the study was conducted, enrolled 88 pregnant women who screened positive for hepatitis B e antigen (HBeAg) between gestational weeks 12 and 23 and who had serum HBV DNA levels greater than 6 log10 copies/mL and elevated ALT levels up to 10 times the upper limit of normal (40 IU/mL). Of the 88 patients, 53 opted to take telbivudine, and the remaining 35 served as the study’s control arm, he said.
Women in the treatment arm received 600 mg/day of telbivudine beginning sometime between 20 and 32 weeks gestation and continuing until a minimum of 4 weeks post partum, and infants born to women in both groups received hepatitis B immunoglobulin within 24 hours of birth and the HBV vaccine at birth, 1 month, and 6 months, Dr. Pan said.
The mean duration of telbivudine treatment was 15.5 weeks, and all of the women who took the drug remained in the study through at least postpartum week 4, compared with 92% of the controls, said Dr. Pan. Physical and laboratory examinations were conducted at baseline, at the time of delivery, and at 4 weeks post partum, he said, noting that a second study looked at infant outcomes through 28 weeks.
At the time of delivery and 4 weeks post partum, a complete virologic response was observed in 53% and 62% of the treated patients, respectively, while no patients in the control arm achieved a complete virologic response at either time point, Dr. Pan reported. "Both the control arm and the telbivudine arm had HBV DNA of approximately 8 logs. At the time of delivery, this was reduced substantially [to 2.35 log10 copies/mL] in the mothers in the treatment arm," he said.
With respect to ALT levels, 77% of the treatment group achieved normalization, compared with 29% of the control group, Dr. Pan said. Also, the levels of HBeAg dropped by 98% in the treatment group, which was a significantly greater decrease than the 60% drop observed in the control group; the latter was likely the result of natural viral clearance, he noted.
An evaluation of newborn outcomes showed no congenital deformities nor any differences in gestational age, infant height and weight, or Apgar scores between the two groups, said Dr. Pan. Significantly fewer babies born to telbivudine-treated mothers vs. untreated mothers had hepatitis B surface antigens or detectable levels of HBV RNA at birth (4% vs. 23%), he said.
Telbivudine appeared to be well tolerated as there were no adverse event–related treatment discontinuations, said Dr. Pan. Also, none of the patients experienced virologic breakthrough.
Despite concerns regarding antiviral treatment during pregnancy because of the potential risks to the fetus, the findings from this study suggest that limited treatment with telbivudine can improve maternal and child outcomes, Dr. Pan concluded.
The study was funded by the Chinese Department of Health with no commercial support. Dr. Pan reported having no relevant financial disclosures.
BOSTON – Limited use of the antiviral drug telbivudine is safe and effective for pregnant women with active hepatitis B virus infection and reduces perinatal transmission of the virus to their infants, according to Dr. Calvin Pan.
In the first open-label case-control trial investigating the safety and efficacy of telbivudine during the second to third trimester of pregnancy, the synthetic nucleoside analogue led to significant increases in rates of complete virologic response and normalization of alanine aminotransferase (ALT) levels in treated vs. untreated pregnant women with hepatitis B virus (HBV) infection, reported Dr. Pan of Mount Sinai School of Medicine in New York.
Additionally, there were no congenital defects identified up to 28 weeks post partum in the infants born to treated mothers, he reported at the annual meeting of the American Association for the Study of Liver Diseases.
Dr. Pan and his colleagues at the Second Affiliated Hospital of the Southeast University in Nanjing, China, where the study was conducted, enrolled 88 pregnant women who screened positive for hepatitis B e antigen (HBeAg) between gestational weeks 12 and 23 and who had serum HBV DNA levels greater than 6 log10 copies/mL and elevated ALT levels up to 10 times the upper limit of normal (40 IU/mL). Of the 88 patients, 53 opted to take telbivudine, and the remaining 35 served as the study’s control arm, he said.
Women in the treatment arm received 600 mg/day of telbivudine beginning sometime between 20 and 32 weeks gestation and continuing until a minimum of 4 weeks post partum, and infants born to women in both groups received hepatitis B immunoglobulin within 24 hours of birth and the HBV vaccine at birth, 1 month, and 6 months, Dr. Pan said.
The mean duration of telbivudine treatment was 15.5 weeks, and all of the women who took the drug remained in the study through at least postpartum week 4, compared with 92% of the controls, said Dr. Pan. Physical and laboratory examinations were conducted at baseline, at the time of delivery, and at 4 weeks post partum, he said, noting that a second study looked at infant outcomes through 28 weeks.
At the time of delivery and 4 weeks post partum, a complete virologic response was observed in 53% and 62% of the treated patients, respectively, while no patients in the control arm achieved a complete virologic response at either time point, Dr. Pan reported. "Both the control arm and the telbivudine arm had HBV DNA of approximately 8 logs. At the time of delivery, this was reduced substantially [to 2.35 log10 copies/mL] in the mothers in the treatment arm," he said.
With respect to ALT levels, 77% of the treatment group achieved normalization, compared with 29% of the control group, Dr. Pan said. Also, the levels of HBeAg dropped by 98% in the treatment group, which was a significantly greater decrease than the 60% drop observed in the control group; the latter was likely the result of natural viral clearance, he noted.
An evaluation of newborn outcomes showed no congenital deformities nor any differences in gestational age, infant height and weight, or Apgar scores between the two groups, said Dr. Pan. Significantly fewer babies born to telbivudine-treated mothers vs. untreated mothers had hepatitis B surface antigens or detectable levels of HBV RNA at birth (4% vs. 23%), he said.
Telbivudine appeared to be well tolerated as there were no adverse event–related treatment discontinuations, said Dr. Pan. Also, none of the patients experienced virologic breakthrough.
Despite concerns regarding antiviral treatment during pregnancy because of the potential risks to the fetus, the findings from this study suggest that limited treatment with telbivudine can improve maternal and child outcomes, Dr. Pan concluded.
The study was funded by the Chinese Department of Health with no commercial support. Dr. Pan reported having no relevant financial disclosures.
FROM THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION FOR THE STUDY OF LIVER DISEASES
Major Finding: A complete virologic response occurred in 53% of treated women when assessed at delivery and in 62% of treated women at 4 weeks post partum, while no patients in the control arm achieved a complete virologic response at either time point.
Data Source: An open-label, case-control trial of telbivudine in 88 pregnant women with active hepatitis B virus infection.
Disclosures: Dr. Pan reported that he had no relevant financial disclosures.
Immunoglobulin Doesn't Boost HBV Vaccine Prophylaxis in Newborns of Infected Mothers
BOSTON – The recombinant hepatitis B vaccine confers as much protection when given alone as it does when given together with hepatitis B immunoglobulin to newborns of chronically infected mothers, but neither regimen is optimally effective, a study has shown.
The randomized controlled trial assessed the hepatitis B virus (HBV) status of 222 infants born to mothers who tested positive for hepatitis B surface antigen (HBsAg). The rate of protection observed in infants who received only the vaccine was statistically similar to that of infants who received the vaccine plus hepatitis B immune globulin (HBIG).
A total of 39% of the vaccine-only group and 41% of the combination group remained infection free at a minimum of 18 weeks after birth, Dr. Shiv K. Sarin reported at the annual meeting of the American Association for the Study of Liver Diseases, noting that nearly half of the babies in both groups developed occult HBV infections.
The current standard of care for preventing HBV infection in babies born to mothers who are HBsAg positive is the recombinant hepatitis B virus vaccine plus HBIG, however previous studies have suggested the possibility that the vaccine alone may be as effective as the combination therapy, said Dr. Sarin of the Institute of Liver and Biliary Sciences in New Delhi.
To test this hypothesis, Dr. Sarin, along with lead investigator Dr. Chandana Pande, a research associate at G.B. Pant Hospital in New Delhi, and colleagues randomized the newborns of 222 women who screened positive for HBsAg during their prenatal care to receive the 0.5-mL recombinant HBV vaccine at birth, 6 weeks, 10 weeks, and 14 weeks, either alone (116 infants) or with 0.5 mL intramuscular HBIG (106 infants). Mothers on antiviral therapy and those with coinfections were excluded from the investigation, he said.
All of the babies were assessed at a minimum of 18 weeks for HBsAg, HBV-DNA, and antibodies to HBsAg (anti-HBs). The study’s primary end point was freedom from overt or occult HBV infection with adequate immune response, defined as anti-HBs titers of at least 10 IU/mL, Dr. Sarin said in a poster presentation.
Babies with overt HBV infection were those whose blood specimens tested positive for HBsAg by enzyme-linked immunosorbent assay, whereas babies with occult infection were negative for HBsAg but positive for HBV-DNA by polymerase chain reaction testing, he said. Babies with no infection but whose anti-HBs titers were less than 10 IU/mL were categorized as having a poor immune response.
At 18 weeks after birth, there were no significant differences between the combination therapy group and monotherapy group with respect to the number of babies meeting the study’s primary end point, Dr. Sarin reported. Specifically, 43 babies in the combination group and 45 in the vaccine-only group remained free of overt or occult HBV infection with adequate immune response, he said.
Of the babies not meeting the primary end point, 9 had overt HBV infection, including 2 in the combination group and 7 in the vaccine-only group, and 106 developed occult HBV infection, including 52 in the combination group and 54 in the vaccine-only group, Dr. Sarin said. Neither of these differences attained statistical significance, nor did the between-group difference in the number of infants demonstrating a poor immune response, he said. Nineteen babies in the combination group and 10 in the vaccine-only group had anti-HBs titers lower than 10 IU/mL.
The large number of babies in both groups who developed occult HBV infection "may be due to intrauterine transmission of the infection," Dr. Sarin suggested. The findings highlight the need for trials of antiviral agents during pregnancy in order to prevent cases in which immunoprophylaxis strategies need to be used, but then fail, he stressed.
Dr. Sarin and Dr. Pande said they had no relevant financial disclosures.
BOSTON – The recombinant hepatitis B vaccine confers as much protection when given alone as it does when given together with hepatitis B immunoglobulin to newborns of chronically infected mothers, but neither regimen is optimally effective, a study has shown.
The randomized controlled trial assessed the hepatitis B virus (HBV) status of 222 infants born to mothers who tested positive for hepatitis B surface antigen (HBsAg). The rate of protection observed in infants who received only the vaccine was statistically similar to that of infants who received the vaccine plus hepatitis B immune globulin (HBIG).
A total of 39% of the vaccine-only group and 41% of the combination group remained infection free at a minimum of 18 weeks after birth, Dr. Shiv K. Sarin reported at the annual meeting of the American Association for the Study of Liver Diseases, noting that nearly half of the babies in both groups developed occult HBV infections.
The current standard of care for preventing HBV infection in babies born to mothers who are HBsAg positive is the recombinant hepatitis B virus vaccine plus HBIG, however previous studies have suggested the possibility that the vaccine alone may be as effective as the combination therapy, said Dr. Sarin of the Institute of Liver and Biliary Sciences in New Delhi.
To test this hypothesis, Dr. Sarin, along with lead investigator Dr. Chandana Pande, a research associate at G.B. Pant Hospital in New Delhi, and colleagues randomized the newborns of 222 women who screened positive for HBsAg during their prenatal care to receive the 0.5-mL recombinant HBV vaccine at birth, 6 weeks, 10 weeks, and 14 weeks, either alone (116 infants) or with 0.5 mL intramuscular HBIG (106 infants). Mothers on antiviral therapy and those with coinfections were excluded from the investigation, he said.
All of the babies were assessed at a minimum of 18 weeks for HBsAg, HBV-DNA, and antibodies to HBsAg (anti-HBs). The study’s primary end point was freedom from overt or occult HBV infection with adequate immune response, defined as anti-HBs titers of at least 10 IU/mL, Dr. Sarin said in a poster presentation.
Babies with overt HBV infection were those whose blood specimens tested positive for HBsAg by enzyme-linked immunosorbent assay, whereas babies with occult infection were negative for HBsAg but positive for HBV-DNA by polymerase chain reaction testing, he said. Babies with no infection but whose anti-HBs titers were less than 10 IU/mL were categorized as having a poor immune response.
At 18 weeks after birth, there were no significant differences between the combination therapy group and monotherapy group with respect to the number of babies meeting the study’s primary end point, Dr. Sarin reported. Specifically, 43 babies in the combination group and 45 in the vaccine-only group remained free of overt or occult HBV infection with adequate immune response, he said.
Of the babies not meeting the primary end point, 9 had overt HBV infection, including 2 in the combination group and 7 in the vaccine-only group, and 106 developed occult HBV infection, including 52 in the combination group and 54 in the vaccine-only group, Dr. Sarin said. Neither of these differences attained statistical significance, nor did the between-group difference in the number of infants demonstrating a poor immune response, he said. Nineteen babies in the combination group and 10 in the vaccine-only group had anti-HBs titers lower than 10 IU/mL.
The large number of babies in both groups who developed occult HBV infection "may be due to intrauterine transmission of the infection," Dr. Sarin suggested. The findings highlight the need for trials of antiviral agents during pregnancy in order to prevent cases in which immunoprophylaxis strategies need to be used, but then fail, he stressed.
Dr. Sarin and Dr. Pande said they had no relevant financial disclosures.
BOSTON – The recombinant hepatitis B vaccine confers as much protection when given alone as it does when given together with hepatitis B immunoglobulin to newborns of chronically infected mothers, but neither regimen is optimally effective, a study has shown.
The randomized controlled trial assessed the hepatitis B virus (HBV) status of 222 infants born to mothers who tested positive for hepatitis B surface antigen (HBsAg). The rate of protection observed in infants who received only the vaccine was statistically similar to that of infants who received the vaccine plus hepatitis B immune globulin (HBIG).
A total of 39% of the vaccine-only group and 41% of the combination group remained infection free at a minimum of 18 weeks after birth, Dr. Shiv K. Sarin reported at the annual meeting of the American Association for the Study of Liver Diseases, noting that nearly half of the babies in both groups developed occult HBV infections.
The current standard of care for preventing HBV infection in babies born to mothers who are HBsAg positive is the recombinant hepatitis B virus vaccine plus HBIG, however previous studies have suggested the possibility that the vaccine alone may be as effective as the combination therapy, said Dr. Sarin of the Institute of Liver and Biliary Sciences in New Delhi.
To test this hypothesis, Dr. Sarin, along with lead investigator Dr. Chandana Pande, a research associate at G.B. Pant Hospital in New Delhi, and colleagues randomized the newborns of 222 women who screened positive for HBsAg during their prenatal care to receive the 0.5-mL recombinant HBV vaccine at birth, 6 weeks, 10 weeks, and 14 weeks, either alone (116 infants) or with 0.5 mL intramuscular HBIG (106 infants). Mothers on antiviral therapy and those with coinfections were excluded from the investigation, he said.
All of the babies were assessed at a minimum of 18 weeks for HBsAg, HBV-DNA, and antibodies to HBsAg (anti-HBs). The study’s primary end point was freedom from overt or occult HBV infection with adequate immune response, defined as anti-HBs titers of at least 10 IU/mL, Dr. Sarin said in a poster presentation.
Babies with overt HBV infection were those whose blood specimens tested positive for HBsAg by enzyme-linked immunosorbent assay, whereas babies with occult infection were negative for HBsAg but positive for HBV-DNA by polymerase chain reaction testing, he said. Babies with no infection but whose anti-HBs titers were less than 10 IU/mL were categorized as having a poor immune response.
At 18 weeks after birth, there were no significant differences between the combination therapy group and monotherapy group with respect to the number of babies meeting the study’s primary end point, Dr. Sarin reported. Specifically, 43 babies in the combination group and 45 in the vaccine-only group remained free of overt or occult HBV infection with adequate immune response, he said.
Of the babies not meeting the primary end point, 9 had overt HBV infection, including 2 in the combination group and 7 in the vaccine-only group, and 106 developed occult HBV infection, including 52 in the combination group and 54 in the vaccine-only group, Dr. Sarin said. Neither of these differences attained statistical significance, nor did the between-group difference in the number of infants demonstrating a poor immune response, he said. Nineteen babies in the combination group and 10 in the vaccine-only group had anti-HBs titers lower than 10 IU/mL.
The large number of babies in both groups who developed occult HBV infection "may be due to intrauterine transmission of the infection," Dr. Sarin suggested. The findings highlight the need for trials of antiviral agents during pregnancy in order to prevent cases in which immunoprophylaxis strategies need to be used, but then fail, he stressed.
Dr. Sarin and Dr. Pande said they had no relevant financial disclosures.
FROM THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION FOR THE STUDY OF LIVER DISEASES
Immunoglobulin Doesn't Boost HBV Vaccine Prophylaxis in Newborns of Infected Mothers
BOSTON – The recombinant hepatitis B vaccine confers as much protection when given alone as it does when given together with hepatitis B immunoglobulin to newborns of chronically infected mothers, but neither regimen is optimally effective, a study has shown.
The randomized controlled trial assessed the hepatitis B virus (HBV) status of 222 infants born to mothers who tested positive for hepatitis B surface antigen (HBsAg). The rate of protection observed in infants who received only the vaccine was statistically similar to that of infants who received the vaccine plus hepatitis B immune globulin (HBIG).
A total of 39% of the vaccine-only group and 41% of the combination group remained infection free at a minimum of 18 weeks after birth, Dr. Shiv K. Sarin reported at the annual meeting of the American Association for the Study of Liver Diseases, noting that nearly half of the babies in both groups developed occult HBV infections.
The current standard of care for preventing HBV infection in babies born to mothers who are HBsAg positive is the recombinant hepatitis B virus vaccine plus HBIG, however previous studies have suggested the possibility that the vaccine alone may be as effective as the combination therapy, said Dr. Sarin of the Institute of Liver and Biliary Sciences in New Delhi.
To test this hypothesis, Dr. Sarin, along with lead investigator Dr. Chandana Pande, a research associate at G.B. Pant Hospital in New Delhi, and colleagues randomized the newborns of 222 women who screened positive for HBsAg during their prenatal care to receive the 0.5-mL recombinant HBV vaccine at birth, 6 weeks, 10 weeks, and 14 weeks, either alone (116 infants) or with 0.5 mL intramuscular HBIG (106 infants). Mothers on antiviral therapy and those with coinfections were excluded from the investigation, he said.
All of the babies were assessed at a minimum of 18 weeks for HBsAg, HBV-DNA, and antibodies to HBsAg (anti-HBs). The study’s primary end point was freedom from overt or occult HBV infection with adequate immune response, defined as anti-HBs titers of at least 10 IU/mL, Dr. Sarin said in a poster presentation.
Babies with overt HBV infection were those whose blood specimens tested positive for HBsAg by enzyme-linked immunosorbent assay, whereas babies with occult infection were negative for HBsAg but positive for HBV-DNA by polymerase chain reaction testing, he said. Babies with no infection but whose anti-HBs titers were less than 10 IU/mL were categorized as having a poor immune response.
At 18 weeks after birth, there were no significant differences between the combination therapy group and monotherapy group with respect to the number of babies meeting the study’s primary end point, Dr. Sarin reported. Specifically, 43 babies in the combination group and 45 in the vaccine-only group remained free of overt or occult HBV infection with adequate immune response, he said.
Of the babies not meeting the primary end point, 9 had overt HBV infection, including 2 in the combination group and 7 in the vaccine-only group, and 106 developed occult HBV infection, including 52 in the combination group and 54 in the vaccine-only group, Dr. Sarin said. Neither of these differences attained statistical significance, nor did the between-group difference in the number of infants demonstrating a poor immune response, he said. Nineteen babies in the combination group and 10 in the vaccine-only group had anti-HBs titers lower than 10 IU/mL.
The large number of babies in both groups who developed occult HBV infection "may be due to intrauterine transmission of the infection," Dr. Sarin suggested. The findings highlight the need for trials of antiviral agents during pregnancy in order to prevent cases in which immunoprophylaxis strategies need to be used, but then fail, he stressed.
Dr. Sarin and Dr. Pande said they had no relevant financial disclosures.
BOSTON – The recombinant hepatitis B vaccine confers as much protection when given alone as it does when given together with hepatitis B immunoglobulin to newborns of chronically infected mothers, but neither regimen is optimally effective, a study has shown.
The randomized controlled trial assessed the hepatitis B virus (HBV) status of 222 infants born to mothers who tested positive for hepatitis B surface antigen (HBsAg). The rate of protection observed in infants who received only the vaccine was statistically similar to that of infants who received the vaccine plus hepatitis B immune globulin (HBIG).
A total of 39% of the vaccine-only group and 41% of the combination group remained infection free at a minimum of 18 weeks after birth, Dr. Shiv K. Sarin reported at the annual meeting of the American Association for the Study of Liver Diseases, noting that nearly half of the babies in both groups developed occult HBV infections.
The current standard of care for preventing HBV infection in babies born to mothers who are HBsAg positive is the recombinant hepatitis B virus vaccine plus HBIG, however previous studies have suggested the possibility that the vaccine alone may be as effective as the combination therapy, said Dr. Sarin of the Institute of Liver and Biliary Sciences in New Delhi.
To test this hypothesis, Dr. Sarin, along with lead investigator Dr. Chandana Pande, a research associate at G.B. Pant Hospital in New Delhi, and colleagues randomized the newborns of 222 women who screened positive for HBsAg during their prenatal care to receive the 0.5-mL recombinant HBV vaccine at birth, 6 weeks, 10 weeks, and 14 weeks, either alone (116 infants) or with 0.5 mL intramuscular HBIG (106 infants). Mothers on antiviral therapy and those with coinfections were excluded from the investigation, he said.
All of the babies were assessed at a minimum of 18 weeks for HBsAg, HBV-DNA, and antibodies to HBsAg (anti-HBs). The study’s primary end point was freedom from overt or occult HBV infection with adequate immune response, defined as anti-HBs titers of at least 10 IU/mL, Dr. Sarin said in a poster presentation.
Babies with overt HBV infection were those whose blood specimens tested positive for HBsAg by enzyme-linked immunosorbent assay, whereas babies with occult infection were negative for HBsAg but positive for HBV-DNA by polymerase chain reaction testing, he said. Babies with no infection but whose anti-HBs titers were less than 10 IU/mL were categorized as having a poor immune response.
At 18 weeks after birth, there were no significant differences between the combination therapy group and monotherapy group with respect to the number of babies meeting the study’s primary end point, Dr. Sarin reported. Specifically, 43 babies in the combination group and 45 in the vaccine-only group remained free of overt or occult HBV infection with adequate immune response, he said.
Of the babies not meeting the primary end point, 9 had overt HBV infection, including 2 in the combination group and 7 in the vaccine-only group, and 106 developed occult HBV infection, including 52 in the combination group and 54 in the vaccine-only group, Dr. Sarin said. Neither of these differences attained statistical significance, nor did the between-group difference in the number of infants demonstrating a poor immune response, he said. Nineteen babies in the combination group and 10 in the vaccine-only group had anti-HBs titers lower than 10 IU/mL.
The large number of babies in both groups who developed occult HBV infection "may be due to intrauterine transmission of the infection," Dr. Sarin suggested. The findings highlight the need for trials of antiviral agents during pregnancy in order to prevent cases in which immunoprophylaxis strategies need to be used, but then fail, he stressed.
Dr. Sarin and Dr. Pande said they had no relevant financial disclosures.
BOSTON – The recombinant hepatitis B vaccine confers as much protection when given alone as it does when given together with hepatitis B immunoglobulin to newborns of chronically infected mothers, but neither regimen is optimally effective, a study has shown.
The randomized controlled trial assessed the hepatitis B virus (HBV) status of 222 infants born to mothers who tested positive for hepatitis B surface antigen (HBsAg). The rate of protection observed in infants who received only the vaccine was statistically similar to that of infants who received the vaccine plus hepatitis B immune globulin (HBIG).
A total of 39% of the vaccine-only group and 41% of the combination group remained infection free at a minimum of 18 weeks after birth, Dr. Shiv K. Sarin reported at the annual meeting of the American Association for the Study of Liver Diseases, noting that nearly half of the babies in both groups developed occult HBV infections.
The current standard of care for preventing HBV infection in babies born to mothers who are HBsAg positive is the recombinant hepatitis B virus vaccine plus HBIG, however previous studies have suggested the possibility that the vaccine alone may be as effective as the combination therapy, said Dr. Sarin of the Institute of Liver and Biliary Sciences in New Delhi.
To test this hypothesis, Dr. Sarin, along with lead investigator Dr. Chandana Pande, a research associate at G.B. Pant Hospital in New Delhi, and colleagues randomized the newborns of 222 women who screened positive for HBsAg during their prenatal care to receive the 0.5-mL recombinant HBV vaccine at birth, 6 weeks, 10 weeks, and 14 weeks, either alone (116 infants) or with 0.5 mL intramuscular HBIG (106 infants). Mothers on antiviral therapy and those with coinfections were excluded from the investigation, he said.
All of the babies were assessed at a minimum of 18 weeks for HBsAg, HBV-DNA, and antibodies to HBsAg (anti-HBs). The study’s primary end point was freedom from overt or occult HBV infection with adequate immune response, defined as anti-HBs titers of at least 10 IU/mL, Dr. Sarin said in a poster presentation.
Babies with overt HBV infection were those whose blood specimens tested positive for HBsAg by enzyme-linked immunosorbent assay, whereas babies with occult infection were negative for HBsAg but positive for HBV-DNA by polymerase chain reaction testing, he said. Babies with no infection but whose anti-HBs titers were less than 10 IU/mL were categorized as having a poor immune response.
At 18 weeks after birth, there were no significant differences between the combination therapy group and monotherapy group with respect to the number of babies meeting the study’s primary end point, Dr. Sarin reported. Specifically, 43 babies in the combination group and 45 in the vaccine-only group remained free of overt or occult HBV infection with adequate immune response, he said.
Of the babies not meeting the primary end point, 9 had overt HBV infection, including 2 in the combination group and 7 in the vaccine-only group, and 106 developed occult HBV infection, including 52 in the combination group and 54 in the vaccine-only group, Dr. Sarin said. Neither of these differences attained statistical significance, nor did the between-group difference in the number of infants demonstrating a poor immune response, he said. Nineteen babies in the combination group and 10 in the vaccine-only group had anti-HBs titers lower than 10 IU/mL.
The large number of babies in both groups who developed occult HBV infection "may be due to intrauterine transmission of the infection," Dr. Sarin suggested. The findings highlight the need for trials of antiviral agents during pregnancy in order to prevent cases in which immunoprophylaxis strategies need to be used, but then fail, he stressed.
Dr. Sarin and Dr. Pande said they had no relevant financial disclosures.
FROM THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION FOR THE STUDY OF LIVER DISEASES
Major Finding: The addition of hepatitis B immunoglobulin to hepatitis B prophylaxis with the recombinant HBV vaccine does not confer additional protection to newborns of chronically infected mothers.
Data Source: A randomized controlled trial comparing rates of HBV infection in infants who received the recombinant HBV vaccine alone or in combination with hepatitis B immunoglobulin.
Disclosures: The investigators reported having no relevant financial disclosures.
Hepatitis C Vaccine Elicits Immune Response in Some Patients
BOSTON – A therapeutic vaccine against the hepatitis C virus was associated with a significantly higher sustained virologic response rate when added to standard-of-care treatment, and the findings justify further development of the vaccine, Dr. Paul Pockros reported at the annual meeting of the American Association for the Study of Liver Diseases.

In the proof-of-concept trial, 133 patients infected with hepatitis C virus (HCV) genotype 1 were randomized to either triple therapy comprising the experimental GI-5005 vaccine, which is designed to elicit a T-cell response specific to HCV, along with pegylated interferon alfa-2b plus ribavirin (P/R), or the standard P/R therapy alone.
The primary outcome measure was sustained virologic response (SVR), defined as undetectable HCV RNA at 6 months after treatment, said Dr. Pockros of the Scripps Clinic in La Jolla, Calif.
The 68 patients in the experimental group initially received monotherapy with the vaccine, consisting of five weekly injections followed by two monthly injections for a 12-week lead-in period, before progressing to the standard-of-care P/R treatment and once-monthly injections. The 65 patients in the control group received standard-of-care P/R treatment alone.
In both groups, the treatment duration was 48 weeks for treatment-naive patients and 72 weeks for those who had a poor or partial response to prior P/R treatment, said Dr. Pockros. Patients in whom prior P/R therapy induced no notable decrease in HCV viral load, as well as those in whom prior treatment initially resulted in undetectable HCV RNA followed by a viral rebound, were excluded from the analysis.
Among the study’s treatment-naive patients, 58% of those who received the vaccine achieved SVR, compared with 48% of those who received P/R alone. Among the prior poor and partial responders, the respective SVR rates in the vaccine and control groups were 17% and 5%, Dr. Pockros reported.
"There was a slight benefit in the treatment-naive and nonresponders, [but] this was numerical only and was not statistically significant," he said. However, the overall benefit in the vaccine vs. control groups was statistically significant, with respective SVR rates of 47% and 35%, he noted.
The vaccine strategy appears to be safe. "The most common associated adverse events were mild, transient injection-site reactions," said Dr. Pockros. Additionally, he stated, "the discontinuation rates due to adverse events were comparable [13%] in both the triple-therapy and standard-of-care arms."
Of particular interest, Dr. Pockros noted, was the T-cell response in a subgroup of difficult-to-treat patients receiving the vaccine. Previous studies have suggested that patients carrying the T allele of the IL28 gene are at high risk of treatment failure with the standard interferon-based therapy. The T-cell response in vaccine-treated patients, as measured by interferon-gamma enzyme-linked ImmunoSpot assay, "mimicked what we saw with a virologic response," he said.
"Four of five T/T allele patients had a T-cell response – one did not actually get treated – but none of the patients who received standard of care had a T-cell response."
This finding is consistent with the hypothesis that the GI-5005 vaccine corrects a fundamental deficit in cellular immunity in IL28B T/T genotype patients, he said. Although the findings need to be confirmed in additional studies with larger patient populations, the vaccine will likely be genotype specific, he noted.
Because the immune response is similar to that observed in HCV-infected patients who are able to clear the virus without treatment, future studies will evaluate the efficacy of monotherapy with the vaccine in genotype-specific patients, Dr. Pockros said.
The proof-of-concept study was funded by GlobeImmune, manufacturer of the vaccine. Dr. Pockros disclosed financial relationships with GlobeImmune, Genentech, Vertex, Merck, Gilead, Abbott, Pfizer, Phenomix, Tibotec, Pharmasset, 3RT, Novartis, Johnson & Johnson, Achillon, Regulus, Debio, Zymogenetics, and Human Genome Sciences.
BOSTON – A therapeutic vaccine against the hepatitis C virus was associated with a significantly higher sustained virologic response rate when added to standard-of-care treatment, and the findings justify further development of the vaccine, Dr. Paul Pockros reported at the annual meeting of the American Association for the Study of Liver Diseases.
In the proof-of-concept trial, 133 patients infected with hepatitis C virus (HCV) genotype 1 were randomized to either triple therapy comprising the experimental GI-5005 vaccine, which is designed to elicit a T-cell response specific to HCV, along with pegylated interferon alfa-2b plus ribavirin (P/R), or the standard P/R therapy alone.
The primary outcome measure was sustained virologic response (SVR), defined as undetectable HCV RNA at 6 months after treatment, said Dr. Pockros of the Scripps Clinic in La Jolla, Calif.
The 68 patients in the experimental group initially received monotherapy with the vaccine, consisting of five weekly injections followed by two monthly injections for a 12-week lead-in period, before progressing to the standard-of-care P/R treatment and once-monthly injections. The 65 patients in the control group received standard-of-care P/R treatment alone.
In both groups, the treatment duration was 48 weeks for treatment-naive patients and 72 weeks for those who had a poor or partial response to prior P/R treatment, said Dr. Pockros. Patients in whom prior P/R therapy induced no notable decrease in HCV viral load, as well as those in whom prior treatment initially resulted in undetectable HCV RNA followed by a viral rebound, were excluded from the analysis.
Among the study’s treatment-naive patients, 58% of those who received the vaccine achieved SVR, compared with 48% of those who received P/R alone. Among the prior poor and partial responders, the respective SVR rates in the vaccine and control groups were 17% and 5%, Dr. Pockros reported.
"There was a slight benefit in the treatment-naive and nonresponders, [but] this was numerical only and was not statistically significant," he said. However, the overall benefit in the vaccine vs. control groups was statistically significant, with respective SVR rates of 47% and 35%, he noted.
The vaccine strategy appears to be safe. "The most common associated adverse events were mild, transient injection-site reactions," said Dr. Pockros. Additionally, he stated, "the discontinuation rates due to adverse events were comparable [13%] in both the triple-therapy and standard-of-care arms."
Of particular interest, Dr. Pockros noted, was the T-cell response in a subgroup of difficult-to-treat patients receiving the vaccine. Previous studies have suggested that patients carrying the T allele of the IL28 gene are at high risk of treatment failure with the standard interferon-based therapy. The T-cell response in vaccine-treated patients, as measured by interferon-gamma enzyme-linked ImmunoSpot assay, "mimicked what we saw with a virologic response," he said.
"Four of five T/T allele patients had a T-cell response – one did not actually get treated – but none of the patients who received standard of care had a T-cell response."
This finding is consistent with the hypothesis that the GI-5005 vaccine corrects a fundamental deficit in cellular immunity in IL28B T/T genotype patients, he said. Although the findings need to be confirmed in additional studies with larger patient populations, the vaccine will likely be genotype specific, he noted.
Because the immune response is similar to that observed in HCV-infected patients who are able to clear the virus without treatment, future studies will evaluate the efficacy of monotherapy with the vaccine in genotype-specific patients, Dr. Pockros said.
The proof-of-concept study was funded by GlobeImmune, manufacturer of the vaccine. Dr. Pockros disclosed financial relationships with GlobeImmune, Genentech, Vertex, Merck, Gilead, Abbott, Pfizer, Phenomix, Tibotec, Pharmasset, 3RT, Novartis, Johnson & Johnson, Achillon, Regulus, Debio, Zymogenetics, and Human Genome Sciences.
BOSTON – A therapeutic vaccine against the hepatitis C virus was associated with a significantly higher sustained virologic response rate when added to standard-of-care treatment, and the findings justify further development of the vaccine, Dr. Paul Pockros reported at the annual meeting of the American Association for the Study of Liver Diseases.
In the proof-of-concept trial, 133 patients infected with hepatitis C virus (HCV) genotype 1 were randomized to either triple therapy comprising the experimental GI-5005 vaccine, which is designed to elicit a T-cell response specific to HCV, along with pegylated interferon alfa-2b plus ribavirin (P/R), or the standard P/R therapy alone.
The primary outcome measure was sustained virologic response (SVR), defined as undetectable HCV RNA at 6 months after treatment, said Dr. Pockros of the Scripps Clinic in La Jolla, Calif.
The 68 patients in the experimental group initially received monotherapy with the vaccine, consisting of five weekly injections followed by two monthly injections for a 12-week lead-in period, before progressing to the standard-of-care P/R treatment and once-monthly injections. The 65 patients in the control group received standard-of-care P/R treatment alone.
In both groups, the treatment duration was 48 weeks for treatment-naive patients and 72 weeks for those who had a poor or partial response to prior P/R treatment, said Dr. Pockros. Patients in whom prior P/R therapy induced no notable decrease in HCV viral load, as well as those in whom prior treatment initially resulted in undetectable HCV RNA followed by a viral rebound, were excluded from the analysis.
Among the study’s treatment-naive patients, 58% of those who received the vaccine achieved SVR, compared with 48% of those who received P/R alone. Among the prior poor and partial responders, the respective SVR rates in the vaccine and control groups were 17% and 5%, Dr. Pockros reported.
"There was a slight benefit in the treatment-naive and nonresponders, [but] this was numerical only and was not statistically significant," he said. However, the overall benefit in the vaccine vs. control groups was statistically significant, with respective SVR rates of 47% and 35%, he noted.
The vaccine strategy appears to be safe. "The most common associated adverse events were mild, transient injection-site reactions," said Dr. Pockros. Additionally, he stated, "the discontinuation rates due to adverse events were comparable [13%] in both the triple-therapy and standard-of-care arms."
Of particular interest, Dr. Pockros noted, was the T-cell response in a subgroup of difficult-to-treat patients receiving the vaccine. Previous studies have suggested that patients carrying the T allele of the IL28 gene are at high risk of treatment failure with the standard interferon-based therapy. The T-cell response in vaccine-treated patients, as measured by interferon-gamma enzyme-linked ImmunoSpot assay, "mimicked what we saw with a virologic response," he said.
"Four of five T/T allele patients had a T-cell response – one did not actually get treated – but none of the patients who received standard of care had a T-cell response."
This finding is consistent with the hypothesis that the GI-5005 vaccine corrects a fundamental deficit in cellular immunity in IL28B T/T genotype patients, he said. Although the findings need to be confirmed in additional studies with larger patient populations, the vaccine will likely be genotype specific, he noted.
Because the immune response is similar to that observed in HCV-infected patients who are able to clear the virus without treatment, future studies will evaluate the efficacy of monotherapy with the vaccine in genotype-specific patients, Dr. Pockros said.
The proof-of-concept study was funded by GlobeImmune, manufacturer of the vaccine. Dr. Pockros disclosed financial relationships with GlobeImmune, Genentech, Vertex, Merck, Gilead, Abbott, Pfizer, Phenomix, Tibotec, Pharmasset, 3RT, Novartis, Johnson & Johnson, Achillon, Regulus, Debio, Zymogenetics, and Human Genome Sciences.
FROM THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION FOR THE STUDY OF LIVER DISEASES