User login
EC grants immunotherapy orphan designation
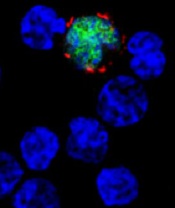
among uninfected cells (blue)
Image courtesy of
Benjamin Chaigne-Delalande
The European Commission (EC) has granted orphan drug designation for CMD-003 (baltaleucel-T) as a treatment for nasal type extranodal NK/T-cell lymphoma and post-transplant lymphoproliferative disorder.
CMD-003 consists of patient-derived T cells that have been activated to kill malignant cells expressing antigens associated with Epstein-Barr virus (EBV).
The T cells specifically target 4 EBV epitopes—LMP1, LMP2, EBNA, and BARF1.
CMD-003 is being developed by Cell Medica and the Baylor College of Medicine, with funding provided, in part, by the Cancer Prevention and Research Institute of Texas.
About orphan designation
Orphan designation from the EC provides regulatory and financial incentives for companies to develop and market therapies that treat a life-threatening or chronically debilitating condition affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity in the European Union if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure.
CMD-003 also has orphan designation from the US Food and Drug Administration to treat all EBV-associated non-Hodgkin lymphomas.
CMD-003-related research
CMD-003 is currently under investigation in a phase 2 trial, CITADEL, for the treatment of advanced NK/T-cell lymphoma.
Researchers have not published results from any trials of CMD-003, but they have published results with EBV-specific T-cell products related to CMD-003.
In one study, published in the Journal of Clinical Oncology, researchers administered cytotoxic T lymphocytes (CTLs) in 50 patients with EBV-associated Hodgkin or non-Hodgkin lymphoma.
Twenty-nine of the patients were in remission when they received CTL infusions, but they were at a high risk of relapse. The remaining 21 patients had relapsed or refractory disease at the time of CTL infusion.
Twenty-seven of the patients who received CTLs as an adjuvant treatment remained in remission at 3.1 years after treatment.
Their 2-year event-free survival rate was 82%. None of the patients died of lymphoma, but 9 died from complications associated with the chemotherapy and radiation they had received.
Of the 21 patients with relapsed or refractory disease, 13 responded to CTL infusions, and 11 patients achieved a complete response. In this group, the 2-year event-free survival rate was about 50%.
The researchers said there were no toxicities that were definitively related to CTL infusion.
One patient had central nervous system deterioration 2 weeks after infusion. This was attributed to disease progression but could possibly have been treatment-related.
Another patient developed respiratory complications about 4 weeks after a second CTL infusion that may have been treatment-related. However, the researchers attributed it to an intercurrent infection, and the patient made a complete recovery. ![]()
among uninfected cells (blue)
Image courtesy of
Benjamin Chaigne-Delalande
The European Commission (EC) has granted orphan drug designation for CMD-003 (baltaleucel-T) as a treatment for nasal type extranodal NK/T-cell lymphoma and post-transplant lymphoproliferative disorder.
CMD-003 consists of patient-derived T cells that have been activated to kill malignant cells expressing antigens associated with Epstein-Barr virus (EBV).
The T cells specifically target 4 EBV epitopes—LMP1, LMP2, EBNA, and BARF1.
CMD-003 is being developed by Cell Medica and the Baylor College of Medicine, with funding provided, in part, by the Cancer Prevention and Research Institute of Texas.
About orphan designation
Orphan designation from the EC provides regulatory and financial incentives for companies to develop and market therapies that treat a life-threatening or chronically debilitating condition affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity in the European Union if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure.
CMD-003 also has orphan designation from the US Food and Drug Administration to treat all EBV-associated non-Hodgkin lymphomas.
CMD-003-related research
CMD-003 is currently under investigation in a phase 2 trial, CITADEL, for the treatment of advanced NK/T-cell lymphoma.
Researchers have not published results from any trials of CMD-003, but they have published results with EBV-specific T-cell products related to CMD-003.
In one study, published in the Journal of Clinical Oncology, researchers administered cytotoxic T lymphocytes (CTLs) in 50 patients with EBV-associated Hodgkin or non-Hodgkin lymphoma.
Twenty-nine of the patients were in remission when they received CTL infusions, but they were at a high risk of relapse. The remaining 21 patients had relapsed or refractory disease at the time of CTL infusion.
Twenty-seven of the patients who received CTLs as an adjuvant treatment remained in remission at 3.1 years after treatment.
Their 2-year event-free survival rate was 82%. None of the patients died of lymphoma, but 9 died from complications associated with the chemotherapy and radiation they had received.
Of the 21 patients with relapsed or refractory disease, 13 responded to CTL infusions, and 11 patients achieved a complete response. In this group, the 2-year event-free survival rate was about 50%.
The researchers said there were no toxicities that were definitively related to CTL infusion.
One patient had central nervous system deterioration 2 weeks after infusion. This was attributed to disease progression but could possibly have been treatment-related.
Another patient developed respiratory complications about 4 weeks after a second CTL infusion that may have been treatment-related. However, the researchers attributed it to an intercurrent infection, and the patient made a complete recovery. ![]()
among uninfected cells (blue)
Image courtesy of
Benjamin Chaigne-Delalande
The European Commission (EC) has granted orphan drug designation for CMD-003 (baltaleucel-T) as a treatment for nasal type extranodal NK/T-cell lymphoma and post-transplant lymphoproliferative disorder.
CMD-003 consists of patient-derived T cells that have been activated to kill malignant cells expressing antigens associated with Epstein-Barr virus (EBV).
The T cells specifically target 4 EBV epitopes—LMP1, LMP2, EBNA, and BARF1.
CMD-003 is being developed by Cell Medica and the Baylor College of Medicine, with funding provided, in part, by the Cancer Prevention and Research Institute of Texas.
About orphan designation
Orphan designation from the EC provides regulatory and financial incentives for companies to develop and market therapies that treat a life-threatening or chronically debilitating condition affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity in the European Union if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure.
CMD-003 also has orphan designation from the US Food and Drug Administration to treat all EBV-associated non-Hodgkin lymphomas.
CMD-003-related research
CMD-003 is currently under investigation in a phase 2 trial, CITADEL, for the treatment of advanced NK/T-cell lymphoma.
Researchers have not published results from any trials of CMD-003, but they have published results with EBV-specific T-cell products related to CMD-003.
In one study, published in the Journal of Clinical Oncology, researchers administered cytotoxic T lymphocytes (CTLs) in 50 patients with EBV-associated Hodgkin or non-Hodgkin lymphoma.
Twenty-nine of the patients were in remission when they received CTL infusions, but they were at a high risk of relapse. The remaining 21 patients had relapsed or refractory disease at the time of CTL infusion.
Twenty-seven of the patients who received CTLs as an adjuvant treatment remained in remission at 3.1 years after treatment.
Their 2-year event-free survival rate was 82%. None of the patients died of lymphoma, but 9 died from complications associated with the chemotherapy and radiation they had received.
Of the 21 patients with relapsed or refractory disease, 13 responded to CTL infusions, and 11 patients achieved a complete response. In this group, the 2-year event-free survival rate was about 50%.
The researchers said there were no toxicities that were definitively related to CTL infusion.
One patient had central nervous system deterioration 2 weeks after infusion. This was attributed to disease progression but could possibly have been treatment-related.
Another patient developed respiratory complications about 4 weeks after a second CTL infusion that may have been treatment-related. However, the researchers attributed it to an intercurrent infection, and the patient made a complete recovery. ![]()
Switch may increase supply of HSCs from cord blood

Photo courtesy of NHS
Researchers have discovered a genetic switch that could increase the supply of hematopoietic stem cells (HSCs) derived from cord blood, according to a paper published in Cell Stem Cell.
However, experiments in mice suggest that flipping the switch may also spur the development of myeloproliferative disease.
The researchers explained that, when an HSC divides, it produces multipotent progenitor (MPP) cells immediately downstream that retain the ability to differentiate but have lost the ability to self-renew.
By analyzing murine and human models of hematopoiesis, the team discovered that a microRNA, miR-125a, controls self-renewal and is normally switched on in HSCs but turned off in MPPs.
“Our work shows that if we artificially throw the switch on in those downstream cells [MPPs], we can endow them with stemness, and they basically become stem cells and can be maintained over the long term,” said study author John Dick, PhD, of Princess Margaret Cancer Centre at University Health Network in Toronto, Ontario, Canada.
Specifically, Dr Dick and his colleagues found that overexpression of miR-125a induced stem cell potential in both human MPPs and an as-yet-unidentified population within the CD34+38+ committed progenitor compartment.
Overexpression of miR-125a had a similar effect in murine MPPs. When the researchers transplanted these MPPs into recipient mice, the cells exhibited increased self-renewal and generated high levels of multi-lineage reconstitution for up to 16 weeks after transplant.
Unfortunately, when the miR-125a-overexpressed MPPs were transplanted into secondary and tertiary recipient mice, the animals developed symptoms of a myeloproliferative disease.
The researchers said this result is in line with findings from previous studies and has been shown to be dependent upon sustained expression of miR-125a and dosage.
The current study suggested that the miR-125a-induced myeloproliferative disease occurs, in part, as a function of replicative stress.
The researchers therefore speculated that, with careful titration of miR-125a levels, it may be possible to reap the beneficial self-renewal effects of miR-125a without inducing myeloproliferative disease.
In fact, the team hopes that, in the future, approaches combining miR-125a-modified MPPs with existing small-molecule compounds will enable the expansion of cord blood units for transplant. ![]()
Photo courtesy of NHS
Researchers have discovered a genetic switch that could increase the supply of hematopoietic stem cells (HSCs) derived from cord blood, according to a paper published in Cell Stem Cell.
However, experiments in mice suggest that flipping the switch may also spur the development of myeloproliferative disease.
The researchers explained that, when an HSC divides, it produces multipotent progenitor (MPP) cells immediately downstream that retain the ability to differentiate but have lost the ability to self-renew.
By analyzing murine and human models of hematopoiesis, the team discovered that a microRNA, miR-125a, controls self-renewal and is normally switched on in HSCs but turned off in MPPs.
“Our work shows that if we artificially throw the switch on in those downstream cells [MPPs], we can endow them with stemness, and they basically become stem cells and can be maintained over the long term,” said study author John Dick, PhD, of Princess Margaret Cancer Centre at University Health Network in Toronto, Ontario, Canada.
Specifically, Dr Dick and his colleagues found that overexpression of miR-125a induced stem cell potential in both human MPPs and an as-yet-unidentified population within the CD34+38+ committed progenitor compartment.
Overexpression of miR-125a had a similar effect in murine MPPs. When the researchers transplanted these MPPs into recipient mice, the cells exhibited increased self-renewal and generated high levels of multi-lineage reconstitution for up to 16 weeks after transplant.
Unfortunately, when the miR-125a-overexpressed MPPs were transplanted into secondary and tertiary recipient mice, the animals developed symptoms of a myeloproliferative disease.
The researchers said this result is in line with findings from previous studies and has been shown to be dependent upon sustained expression of miR-125a and dosage.
The current study suggested that the miR-125a-induced myeloproliferative disease occurs, in part, as a function of replicative stress.
The researchers therefore speculated that, with careful titration of miR-125a levels, it may be possible to reap the beneficial self-renewal effects of miR-125a without inducing myeloproliferative disease.
In fact, the team hopes that, in the future, approaches combining miR-125a-modified MPPs with existing small-molecule compounds will enable the expansion of cord blood units for transplant. ![]()
Photo courtesy of NHS
Researchers have discovered a genetic switch that could increase the supply of hematopoietic stem cells (HSCs) derived from cord blood, according to a paper published in Cell Stem Cell.
However, experiments in mice suggest that flipping the switch may also spur the development of myeloproliferative disease.
The researchers explained that, when an HSC divides, it produces multipotent progenitor (MPP) cells immediately downstream that retain the ability to differentiate but have lost the ability to self-renew.
By analyzing murine and human models of hematopoiesis, the team discovered that a microRNA, miR-125a, controls self-renewal and is normally switched on in HSCs but turned off in MPPs.
“Our work shows that if we artificially throw the switch on in those downstream cells [MPPs], we can endow them with stemness, and they basically become stem cells and can be maintained over the long term,” said study author John Dick, PhD, of Princess Margaret Cancer Centre at University Health Network in Toronto, Ontario, Canada.
Specifically, Dr Dick and his colleagues found that overexpression of miR-125a induced stem cell potential in both human MPPs and an as-yet-unidentified population within the CD34+38+ committed progenitor compartment.
Overexpression of miR-125a had a similar effect in murine MPPs. When the researchers transplanted these MPPs into recipient mice, the cells exhibited increased self-renewal and generated high levels of multi-lineage reconstitution for up to 16 weeks after transplant.
Unfortunately, when the miR-125a-overexpressed MPPs were transplanted into secondary and tertiary recipient mice, the animals developed symptoms of a myeloproliferative disease.
The researchers said this result is in line with findings from previous studies and has been shown to be dependent upon sustained expression of miR-125a and dosage.
The current study suggested that the miR-125a-induced myeloproliferative disease occurs, in part, as a function of replicative stress.
The researchers therefore speculated that, with careful titration of miR-125a levels, it may be possible to reap the beneficial self-renewal effects of miR-125a without inducing myeloproliferative disease.
In fact, the team hopes that, in the future, approaches combining miR-125a-modified MPPs with existing small-molecule compounds will enable the expansion of cord blood units for transplant. ![]()
Immunotherapy may benefit relapsed HSCT recipients
Photo from Business Wire
Results of a phase 1 study suggest that repeated doses of the immunotherapy drug ipilimumab is a feasible treatment option for patients with hematologic diseases who relapse after allogeneic hematopoietic stem cell transplant (HSCT).
Seven of the 28 patients studied responded to the treatment, but immune-mediated toxic effects and graft-vs-host disease (GVHD) occurred as well.
These results were published in NEJM.
Ipilimumab, which is already approved to treat unresectable or metastatic melanoma, works by blocking the immune checkpoint CTLA-4. Blockade of CTLA-4 has been shown to augment T-cell activation and proliferation.
“We believe [,in the case of relapse after HSCT,] the donor immune cells are present but can’t recognize the tumor cells because of inhibitory signals that disguise them,” said study author Matthew Davids, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“By blocking the checkpoint, you allow the donor cells to see the cancer cells.”
Dr Davids and his colleagues tested this theory in 28 patients who had relapsed after allogeneic HSCT. The patients had acute myeloid leukemia (AML, n=12), Hodgkin lymphoma (n=7), non-Hodgkin lymphoma (n=4), myelodysplastic syndromes (MDS, n=2), multiple myeloma (n=1), myeloproliferative neoplasm (n=1), or acute lymphoblastic leukemia (n=1).
Patients had received a median of 3 prior treatment regimens, excluding HSCT (range, 1 to 14), and 20 patients (71%) had received treatment for relapse after transplant. Eight patients (29%) previously had grade 1/2 acute GVHD, and 16 (57%) previously had chronic GVHD.
The median time from transplant to initial treatment with ipilimumab was 675 days (range, 198 to 1830), and the median time from relapse to initial treatment with ipilimumab was 97 days (range, 0 to
1415).
Patients received induction therapy with ipilimumab at a dose of 3 mg/kg or 10 mg/kg every 3 weeks for a total of 4 doses. Those who had a clinical benefit received additional doses every 12 weeks for up to 60 weeks.
Safety
Five patients discontinued ipilimumab due to dose-limiting toxic effects. Four of these patients had GVHD, and 1 had severe immune-related adverse events.
Dose-limiting GVHD presented as chronic GVHD of the liver in 3 patients and acute GVHD of the gut in 1 patient.
Immune-related adverse events included death (n=1), pneumonitis (2 grade 2 events, 1 grade 4 event), colitis (1 grade 3 event), immune thrombocytopenia (1 grade 2 event), and diarrhea (1 grade 2 event).
Efficacy
There were no responses in patients who received ipilimumab at 3 mg/kg. Among the 22 patients who received ipilimumab at 10 mg/kg, 5 had a complete response, and 2 had a partial response.
Six other patients did not qualify as having responses but had a decrease in their tumor burden. Altogether, ipilimumab reduced tumor burden in 59% of patients.
The complete responses occurred in 4 patients with extramedullary AML and 1 patient with MDS developing into AML. Two of the AML patients remained in complete response at 12 and 15 months, and the patient with MDS remained in complete response at 16 months.
At a median follow-up of 15 months (range, 8 to 27), the median duration of response had not been reached. Responses were associated with in situ infiltration of cytotoxic CD8+ T cells, decreased activation of regulatory T cells, and expansion of subpopulations of effector T cells.
The 1-year overall survival rate was 49%.
The investigators said these encouraging results have set the stage for larger trials of checkpoint blockade in this patient population. Further research is planned to determine whether immunotherapy drugs could be given to high-risk patients to prevent relapse. ![]()
Photo from Business Wire
Results of a phase 1 study suggest that repeated doses of the immunotherapy drug ipilimumab is a feasible treatment option for patients with hematologic diseases who relapse after allogeneic hematopoietic stem cell transplant (HSCT).
Seven of the 28 patients studied responded to the treatment, but immune-mediated toxic effects and graft-vs-host disease (GVHD) occurred as well.
These results were published in NEJM.
Ipilimumab, which is already approved to treat unresectable or metastatic melanoma, works by blocking the immune checkpoint CTLA-4. Blockade of CTLA-4 has been shown to augment T-cell activation and proliferation.
“We believe [,in the case of relapse after HSCT,] the donor immune cells are present but can’t recognize the tumor cells because of inhibitory signals that disguise them,” said study author Matthew Davids, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“By blocking the checkpoint, you allow the donor cells to see the cancer cells.”
Dr Davids and his colleagues tested this theory in 28 patients who had relapsed after allogeneic HSCT. The patients had acute myeloid leukemia (AML, n=12), Hodgkin lymphoma (n=7), non-Hodgkin lymphoma (n=4), myelodysplastic syndromes (MDS, n=2), multiple myeloma (n=1), myeloproliferative neoplasm (n=1), or acute lymphoblastic leukemia (n=1).
Patients had received a median of 3 prior treatment regimens, excluding HSCT (range, 1 to 14), and 20 patients (71%) had received treatment for relapse after transplant. Eight patients (29%) previously had grade 1/2 acute GVHD, and 16 (57%) previously had chronic GVHD.
The median time from transplant to initial treatment with ipilimumab was 675 days (range, 198 to 1830), and the median time from relapse to initial treatment with ipilimumab was 97 days (range, 0 to
1415).
Patients received induction therapy with ipilimumab at a dose of 3 mg/kg or 10 mg/kg every 3 weeks for a total of 4 doses. Those who had a clinical benefit received additional doses every 12 weeks for up to 60 weeks.
Safety
Five patients discontinued ipilimumab due to dose-limiting toxic effects. Four of these patients had GVHD, and 1 had severe immune-related adverse events.
Dose-limiting GVHD presented as chronic GVHD of the liver in 3 patients and acute GVHD of the gut in 1 patient.
Immune-related adverse events included death (n=1), pneumonitis (2 grade 2 events, 1 grade 4 event), colitis (1 grade 3 event), immune thrombocytopenia (1 grade 2 event), and diarrhea (1 grade 2 event).
Efficacy
There were no responses in patients who received ipilimumab at 3 mg/kg. Among the 22 patients who received ipilimumab at 10 mg/kg, 5 had a complete response, and 2 had a partial response.
Six other patients did not qualify as having responses but had a decrease in their tumor burden. Altogether, ipilimumab reduced tumor burden in 59% of patients.
The complete responses occurred in 4 patients with extramedullary AML and 1 patient with MDS developing into AML. Two of the AML patients remained in complete response at 12 and 15 months, and the patient with MDS remained in complete response at 16 months.
At a median follow-up of 15 months (range, 8 to 27), the median duration of response had not been reached. Responses were associated with in situ infiltration of cytotoxic CD8+ T cells, decreased activation of regulatory T cells, and expansion of subpopulations of effector T cells.
The 1-year overall survival rate was 49%.
The investigators said these encouraging results have set the stage for larger trials of checkpoint blockade in this patient population. Further research is planned to determine whether immunotherapy drugs could be given to high-risk patients to prevent relapse. ![]()
Photo from Business Wire
Results of a phase 1 study suggest that repeated doses of the immunotherapy drug ipilimumab is a feasible treatment option for patients with hematologic diseases who relapse after allogeneic hematopoietic stem cell transplant (HSCT).
Seven of the 28 patients studied responded to the treatment, but immune-mediated toxic effects and graft-vs-host disease (GVHD) occurred as well.
These results were published in NEJM.
Ipilimumab, which is already approved to treat unresectable or metastatic melanoma, works by blocking the immune checkpoint CTLA-4. Blockade of CTLA-4 has been shown to augment T-cell activation and proliferation.
“We believe [,in the case of relapse after HSCT,] the donor immune cells are present but can’t recognize the tumor cells because of inhibitory signals that disguise them,” said study author Matthew Davids, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“By blocking the checkpoint, you allow the donor cells to see the cancer cells.”
Dr Davids and his colleagues tested this theory in 28 patients who had relapsed after allogeneic HSCT. The patients had acute myeloid leukemia (AML, n=12), Hodgkin lymphoma (n=7), non-Hodgkin lymphoma (n=4), myelodysplastic syndromes (MDS, n=2), multiple myeloma (n=1), myeloproliferative neoplasm (n=1), or acute lymphoblastic leukemia (n=1).
Patients had received a median of 3 prior treatment regimens, excluding HSCT (range, 1 to 14), and 20 patients (71%) had received treatment for relapse after transplant. Eight patients (29%) previously had grade 1/2 acute GVHD, and 16 (57%) previously had chronic GVHD.
The median time from transplant to initial treatment with ipilimumab was 675 days (range, 198 to 1830), and the median time from relapse to initial treatment with ipilimumab was 97 days (range, 0 to
1415).
Patients received induction therapy with ipilimumab at a dose of 3 mg/kg or 10 mg/kg every 3 weeks for a total of 4 doses. Those who had a clinical benefit received additional doses every 12 weeks for up to 60 weeks.
Safety
Five patients discontinued ipilimumab due to dose-limiting toxic effects. Four of these patients had GVHD, and 1 had severe immune-related adverse events.
Dose-limiting GVHD presented as chronic GVHD of the liver in 3 patients and acute GVHD of the gut in 1 patient.
Immune-related adverse events included death (n=1), pneumonitis (2 grade 2 events, 1 grade 4 event), colitis (1 grade 3 event), immune thrombocytopenia (1 grade 2 event), and diarrhea (1 grade 2 event).
Efficacy
There were no responses in patients who received ipilimumab at 3 mg/kg. Among the 22 patients who received ipilimumab at 10 mg/kg, 5 had a complete response, and 2 had a partial response.
Six other patients did not qualify as having responses but had a decrease in their tumor burden. Altogether, ipilimumab reduced tumor burden in 59% of patients.
The complete responses occurred in 4 patients with extramedullary AML and 1 patient with MDS developing into AML. Two of the AML patients remained in complete response at 12 and 15 months, and the patient with MDS remained in complete response at 16 months.
At a median follow-up of 15 months (range, 8 to 27), the median duration of response had not been reached. Responses were associated with in situ infiltration of cytotoxic CD8+ T cells, decreased activation of regulatory T cells, and expansion of subpopulations of effector T cells.
The 1-year overall survival rate was 49%.
The investigators said these encouraging results have set the stage for larger trials of checkpoint blockade in this patient population. Further research is planned to determine whether immunotherapy drugs could be given to high-risk patients to prevent relapse. ![]()
Old drug could treat CMV

Image by Ed Uthman
Research published in PLOS Pathogens suggests that emetine, a drug once used to treat amebiasis and induce vomiting in cases of poisoning, may also halt replication of cytomegalovirus (CMV).
Although there are drugs available to treat CMV, they can cause serious toxicity.
Furthermore, resistant strains of CMV can emerge, creating “a desperate need for other ways to control this virus,” according to Ravit Boger, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
Dr Boger and her colleagues decided to search for drugs to treat CMV by screening 1280 pharmacologically active compounds already approved by the US Food and Drug Administration.
The researchers found a “hit” with emetine, a drug that was used in the past to treat amebiasis before other drugs took its place decades ago.
Results showed that emetine can inhibit CMV at much lower doses than those used for amebiasis, and less frequent doses might be effective for CMV inhibition as well.
In vitro and in vivo tests showed that very low doses of emetine significantly reduced viral replication—75 nm in vitro and 0.1 mg/kg in mice.
Furthermore, with a half-life of 35 hours, the drug exerted its effects over a sustained period, effectively inhibiting virus replication at 14 days after 3 doses.
Additional investigation revealed that emetine’s action against CMV was due to its effects on proteins that control the cell cycle.
Dr Boger cautioned that there is a long way to go before emetine or similar agents can be considered for the treatment of CMV.
“But if further research affirms its potential value, emetine might eventually be used in patients who don’t respond to approved anti-CMV drugs, alone or in combination with these,” she said.
Dr Boger and her colleagues also believe that gaining a better understanding of emetine’s activity in cells could lead to the discovery of new drugs that take advantage of the same or similar pathways. ![]()
Image by Ed Uthman
Research published in PLOS Pathogens suggests that emetine, a drug once used to treat amebiasis and induce vomiting in cases of poisoning, may also halt replication of cytomegalovirus (CMV).
Although there are drugs available to treat CMV, they can cause serious toxicity.
Furthermore, resistant strains of CMV can emerge, creating “a desperate need for other ways to control this virus,” according to Ravit Boger, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
Dr Boger and her colleagues decided to search for drugs to treat CMV by screening 1280 pharmacologically active compounds already approved by the US Food and Drug Administration.
The researchers found a “hit” with emetine, a drug that was used in the past to treat amebiasis before other drugs took its place decades ago.
Results showed that emetine can inhibit CMV at much lower doses than those used for amebiasis, and less frequent doses might be effective for CMV inhibition as well.
In vitro and in vivo tests showed that very low doses of emetine significantly reduced viral replication—75 nm in vitro and 0.1 mg/kg in mice.
Furthermore, with a half-life of 35 hours, the drug exerted its effects over a sustained period, effectively inhibiting virus replication at 14 days after 3 doses.
Additional investigation revealed that emetine’s action against CMV was due to its effects on proteins that control the cell cycle.
Dr Boger cautioned that there is a long way to go before emetine or similar agents can be considered for the treatment of CMV.
“But if further research affirms its potential value, emetine might eventually be used in patients who don’t respond to approved anti-CMV drugs, alone or in combination with these,” she said.
Dr Boger and her colleagues also believe that gaining a better understanding of emetine’s activity in cells could lead to the discovery of new drugs that take advantage of the same or similar pathways. ![]()
Image by Ed Uthman
Research published in PLOS Pathogens suggests that emetine, a drug once used to treat amebiasis and induce vomiting in cases of poisoning, may also halt replication of cytomegalovirus (CMV).
Although there are drugs available to treat CMV, they can cause serious toxicity.
Furthermore, resistant strains of CMV can emerge, creating “a desperate need for other ways to control this virus,” according to Ravit Boger, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
Dr Boger and her colleagues decided to search for drugs to treat CMV by screening 1280 pharmacologically active compounds already approved by the US Food and Drug Administration.
The researchers found a “hit” with emetine, a drug that was used in the past to treat amebiasis before other drugs took its place decades ago.
Results showed that emetine can inhibit CMV at much lower doses than those used for amebiasis, and less frequent doses might be effective for CMV inhibition as well.
In vitro and in vivo tests showed that very low doses of emetine significantly reduced viral replication—75 nm in vitro and 0.1 mg/kg in mice.
Furthermore, with a half-life of 35 hours, the drug exerted its effects over a sustained period, effectively inhibiting virus replication at 14 days after 3 doses.
Additional investigation revealed that emetine’s action against CMV was due to its effects on proteins that control the cell cycle.
Dr Boger cautioned that there is a long way to go before emetine or similar agents can be considered for the treatment of CMV.
“But if further research affirms its potential value, emetine might eventually be used in patients who don’t respond to approved anti-CMV drugs, alone or in combination with these,” she said.
Dr Boger and her colleagues also believe that gaining a better understanding of emetine’s activity in cells could lead to the discovery of new drugs that take advantage of the same or similar pathways. ![]()
Drug granted breakthrough, orphan designation for cGVHD

Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ibrutinib (Imbruvica), a Bruton’s tyrosine kinase inhibitor, as a potential treatment for chronic graft-versus-host-disease (cGVHD) in patients who have failed 1 or more lines of systemic therapy.
The FDA has also granted ibrutinib orphan drug designation for this indication.
The request for breakthrough therapy designation and orphan designation for ibrutinib in patients with cGVHD was based on preliminary data from a phase 1b/2 study of patients with steroid-dependent or refractory cGVHD.
Results from this trial were presented at the 2015 ASCO Annual Meeting (abstract 7024) and the 2016 EBMT meeting (abstract P124).
About ibrutinib
Ibrutinib is an oral, once-daily therapy that inhibits Bruton’s tyrosine kinase, a signaling molecule in the B-cell receptor signaling complex that plays an important role in the survival and spread of malignant B cells.
Ibrutinib is FDA-approved to treat patients with chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL), including those with 17p deletion, patients with mantle cell lymphoma (MCL) who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia.
Accelerated approval was granted for the MCL indication based on overall response rate. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted ibrutinib breakthrough designation for the treatment of relapsed or refractory MCL, Waldenström’s macroglobulinemia, and CLL/SLL patients with 17p deletion. The FDA also granted ibrutinib orphan designation for all 3 indications.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
About breakthrough designation
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the drug is approved. ![]()
Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ibrutinib (Imbruvica), a Bruton’s tyrosine kinase inhibitor, as a potential treatment for chronic graft-versus-host-disease (cGVHD) in patients who have failed 1 or more lines of systemic therapy.
The FDA has also granted ibrutinib orphan drug designation for this indication.
The request for breakthrough therapy designation and orphan designation for ibrutinib in patients with cGVHD was based on preliminary data from a phase 1b/2 study of patients with steroid-dependent or refractory cGVHD.
Results from this trial were presented at the 2015 ASCO Annual Meeting (abstract 7024) and the 2016 EBMT meeting (abstract P124).
About ibrutinib
Ibrutinib is an oral, once-daily therapy that inhibits Bruton’s tyrosine kinase, a signaling molecule in the B-cell receptor signaling complex that plays an important role in the survival and spread of malignant B cells.
Ibrutinib is FDA-approved to treat patients with chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL), including those with 17p deletion, patients with mantle cell lymphoma (MCL) who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia.
Accelerated approval was granted for the MCL indication based on overall response rate. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted ibrutinib breakthrough designation for the treatment of relapsed or refractory MCL, Waldenström’s macroglobulinemia, and CLL/SLL patients with 17p deletion. The FDA also granted ibrutinib orphan designation for all 3 indications.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
About breakthrough designation
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the drug is approved. ![]()
Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ibrutinib (Imbruvica), a Bruton’s tyrosine kinase inhibitor, as a potential treatment for chronic graft-versus-host-disease (cGVHD) in patients who have failed 1 or more lines of systemic therapy.
The FDA has also granted ibrutinib orphan drug designation for this indication.
The request for breakthrough therapy designation and orphan designation for ibrutinib in patients with cGVHD was based on preliminary data from a phase 1b/2 study of patients with steroid-dependent or refractory cGVHD.
Results from this trial were presented at the 2015 ASCO Annual Meeting (abstract 7024) and the 2016 EBMT meeting (abstract P124).
About ibrutinib
Ibrutinib is an oral, once-daily therapy that inhibits Bruton’s tyrosine kinase, a signaling molecule in the B-cell receptor signaling complex that plays an important role in the survival and spread of malignant B cells.
Ibrutinib is FDA-approved to treat patients with chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL), including those with 17p deletion, patients with mantle cell lymphoma (MCL) who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia.
Accelerated approval was granted for the MCL indication based on overall response rate. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted ibrutinib breakthrough designation for the treatment of relapsed or refractory MCL, Waldenström’s macroglobulinemia, and CLL/SLL patients with 17p deletion. The FDA also granted ibrutinib orphan designation for all 3 indications.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
About breakthrough designation
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the drug is approved. ![]()
Study: CMV doesn’t lower risk of relapse, death
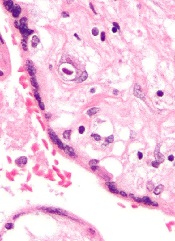
Small studies have suggested that early cytomegalovirus (CMV) reactivation may protect against leukemia relapse and even death after hematopoietic stem cell transplant.
However, a new study, based on data from about 9500 patients, suggests otherwise.
Results showed no association between CMV reactivation and relapse but suggested CMV reactivation increases the risk of non-relapse mortality.
Researchers reported these findings in Blood.
“The original purpose of the study was to confirm that CMV infection may prevent leukemia relapse, prevent death, and become a major therapeutic tool for improving patient survival rates,” said study author Pierre Teira, MD, of the University of Montreal in Quebec, Canada.
“However, we found the exact opposite. Our results clearly show that . . . the virus not only does not prevent leukemia relapse [it] also remains a major factor associated with the risk of death. Monitoring of CMV after transplantation remains a priority for patients.”
For this study, Dr Teira and his colleagues analyzed data from 9469 patients who received a transplant between 2003 and 2010.
The patients had acute myeloid leukemia (AML, n=5310), acute lymphoblastic leukemia (ALL, n=1883), chronic myeloid leukemia (CML, n=1079), or myelodysplastic syndromes (MDS, n=1197).
The median time to initial CMV reactivation was 41 days (range, 1-362 days).
The researchers found no significant association between CMV reactivation and disease relapse for AML (P=0.60), ALL (P=0.08), CML (P=0.94), or MDS (P=0.58).
However, CMV reactivation was associated with a significantly higher risk of nonrelapse mortality for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0004), and MDS (P=0.0002).
Therefore, CMV reactivation was associated with significantly lower overall survival for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0005), and MDS (P=0.003).
“Deaths due to uncontrolled CMV reactivation are virtually zero in this study, so uncontrolled CMV reactivation is not what reduces survival rates after transplantation,” Dr Teira noted. “The link between this common virus and increased risk of death remains a biological mystery.”
One possible explanation is that CMV decreases the ability of the patient’s immune system to fight against other types of infection. This is supported by the fact that death rates from infections other than CMV are higher in patients infected with CMV or patients whose donors were.
For researchers, the next step is therefore to verify whether the latest generation of anti-CMV treatments can prevent both reactivation of the virus and weakening of the patient’s immune system against other types of infection in the presence of CMV infection.
“CMV has a complex impact on the outcomes for transplant patients, and, each year, more than 30,000 patients around the world receive bone marrow transplants from donors,” Dr Teira said.
“It is therefore essential for future research to better understand the role played by CMV after bone marrow transplantation and improve the chances of success of the transplant. This will help to better choose the right donor for the right patient.” ![]()
Small studies have suggested that early cytomegalovirus (CMV) reactivation may protect against leukemia relapse and even death after hematopoietic stem cell transplant.
However, a new study, based on data from about 9500 patients, suggests otherwise.
Results showed no association between CMV reactivation and relapse but suggested CMV reactivation increases the risk of non-relapse mortality.
Researchers reported these findings in Blood.
“The original purpose of the study was to confirm that CMV infection may prevent leukemia relapse, prevent death, and become a major therapeutic tool for improving patient survival rates,” said study author Pierre Teira, MD, of the University of Montreal in Quebec, Canada.
“However, we found the exact opposite. Our results clearly show that . . . the virus not only does not prevent leukemia relapse [it] also remains a major factor associated with the risk of death. Monitoring of CMV after transplantation remains a priority for patients.”
For this study, Dr Teira and his colleagues analyzed data from 9469 patients who received a transplant between 2003 and 2010.
The patients had acute myeloid leukemia (AML, n=5310), acute lymphoblastic leukemia (ALL, n=1883), chronic myeloid leukemia (CML, n=1079), or myelodysplastic syndromes (MDS, n=1197).
The median time to initial CMV reactivation was 41 days (range, 1-362 days).
The researchers found no significant association between CMV reactivation and disease relapse for AML (P=0.60), ALL (P=0.08), CML (P=0.94), or MDS (P=0.58).
However, CMV reactivation was associated with a significantly higher risk of nonrelapse mortality for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0004), and MDS (P=0.0002).
Therefore, CMV reactivation was associated with significantly lower overall survival for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0005), and MDS (P=0.003).
“Deaths due to uncontrolled CMV reactivation are virtually zero in this study, so uncontrolled CMV reactivation is not what reduces survival rates after transplantation,” Dr Teira noted. “The link between this common virus and increased risk of death remains a biological mystery.”
One possible explanation is that CMV decreases the ability of the patient’s immune system to fight against other types of infection. This is supported by the fact that death rates from infections other than CMV are higher in patients infected with CMV or patients whose donors were.
For researchers, the next step is therefore to verify whether the latest generation of anti-CMV treatments can prevent both reactivation of the virus and weakening of the patient’s immune system against other types of infection in the presence of CMV infection.
“CMV has a complex impact on the outcomes for transplant patients, and, each year, more than 30,000 patients around the world receive bone marrow transplants from donors,” Dr Teira said.
“It is therefore essential for future research to better understand the role played by CMV after bone marrow transplantation and improve the chances of success of the transplant. This will help to better choose the right donor for the right patient.” ![]()
Small studies have suggested that early cytomegalovirus (CMV) reactivation may protect against leukemia relapse and even death after hematopoietic stem cell transplant.
However, a new study, based on data from about 9500 patients, suggests otherwise.
Results showed no association between CMV reactivation and relapse but suggested CMV reactivation increases the risk of non-relapse mortality.
Researchers reported these findings in Blood.
“The original purpose of the study was to confirm that CMV infection may prevent leukemia relapse, prevent death, and become a major therapeutic tool for improving patient survival rates,” said study author Pierre Teira, MD, of the University of Montreal in Quebec, Canada.
“However, we found the exact opposite. Our results clearly show that . . . the virus not only does not prevent leukemia relapse [it] also remains a major factor associated with the risk of death. Monitoring of CMV after transplantation remains a priority for patients.”
For this study, Dr Teira and his colleagues analyzed data from 9469 patients who received a transplant between 2003 and 2010.
The patients had acute myeloid leukemia (AML, n=5310), acute lymphoblastic leukemia (ALL, n=1883), chronic myeloid leukemia (CML, n=1079), or myelodysplastic syndromes (MDS, n=1197).
The median time to initial CMV reactivation was 41 days (range, 1-362 days).
The researchers found no significant association between CMV reactivation and disease relapse for AML (P=0.60), ALL (P=0.08), CML (P=0.94), or MDS (P=0.58).
However, CMV reactivation was associated with a significantly higher risk of nonrelapse mortality for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0004), and MDS (P=0.0002).
Therefore, CMV reactivation was associated with significantly lower overall survival for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0005), and MDS (P=0.003).
“Deaths due to uncontrolled CMV reactivation are virtually zero in this study, so uncontrolled CMV reactivation is not what reduces survival rates after transplantation,” Dr Teira noted. “The link between this common virus and increased risk of death remains a biological mystery.”
One possible explanation is that CMV decreases the ability of the patient’s immune system to fight against other types of infection. This is supported by the fact that death rates from infections other than CMV are higher in patients infected with CMV or patients whose donors were.
For researchers, the next step is therefore to verify whether the latest generation of anti-CMV treatments can prevent both reactivation of the virus and weakening of the patient’s immune system against other types of infection in the presence of CMV infection.
“CMV has a complex impact on the outcomes for transplant patients, and, each year, more than 30,000 patients around the world receive bone marrow transplants from donors,” Dr Teira said.
“It is therefore essential for future research to better understand the role played by CMV after bone marrow transplantation and improve the chances of success of the transplant. This will help to better choose the right donor for the right patient.” ![]()
EBV-CTL product classified as ATMP
among uninfected cells (blue)
Image courtesy of Benjamin
Chaigne-Delalande
A cytotoxic T-lymphocyte product that targets Epstein-Barr virus (EBV-CTLs) has been classified as an advanced therapy medicinal product (ATMP) by the European Medicines Agency (EMA).
The EBV-CTLs are being developed by Atara Biotherapeutics, Inc., to treat patients with EBV post-transplant lymphoproliferative disorder (EBV-PTLD).
ATMP classification was established to regulate cell and gene therapy and tissue-engineered medicinal products, support the development of these products, and provide a benchmark for the level of quality compliance for pharmaceutical practices.
ATMP classification can provide developers with scientific regulatory guidance, help clarify the applicable regulatory framework and development path, and provide access to all relevant services and incentives offered by the EMA. It can also be advantageous when submitting clinical trial dossiers to national regulatory authorities within the European Union.
About EBV-CTLs
Atara Bio’s EBV-CTL product utilizes a technology in which T cells are collected from the blood of third-party donors and then exposed to EBV antigens. The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
In the context of EBV-PTLD, the EBV-CTLs find the cancer cells expressing EBV and kill them.
Atara Bio’s EBV-CTL product is currently being studied in phase 2 trials of patients with EBV-associated cancers, including PTLD and nasopharyngeal carcinoma.
Results of a phase 1/2 study of EBV-CTLs were presented at the APHON 37th Annual Conference and Exhibit and the 2015 ASCO Annual Meeting.
Atara Bio’s EBV-CTL product has orphan designation in the European Union and the US, as well as breakthrough designation in the US. ![]()
among uninfected cells (blue)
Image courtesy of Benjamin
Chaigne-Delalande
A cytotoxic T-lymphocyte product that targets Epstein-Barr virus (EBV-CTLs) has been classified as an advanced therapy medicinal product (ATMP) by the European Medicines Agency (EMA).
The EBV-CTLs are being developed by Atara Biotherapeutics, Inc., to treat patients with EBV post-transplant lymphoproliferative disorder (EBV-PTLD).
ATMP classification was established to regulate cell and gene therapy and tissue-engineered medicinal products, support the development of these products, and provide a benchmark for the level of quality compliance for pharmaceutical practices.
ATMP classification can provide developers with scientific regulatory guidance, help clarify the applicable regulatory framework and development path, and provide access to all relevant services and incentives offered by the EMA. It can also be advantageous when submitting clinical trial dossiers to national regulatory authorities within the European Union.
About EBV-CTLs
Atara Bio’s EBV-CTL product utilizes a technology in which T cells are collected from the blood of third-party donors and then exposed to EBV antigens. The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
In the context of EBV-PTLD, the EBV-CTLs find the cancer cells expressing EBV and kill them.
Atara Bio’s EBV-CTL product is currently being studied in phase 2 trials of patients with EBV-associated cancers, including PTLD and nasopharyngeal carcinoma.
Results of a phase 1/2 study of EBV-CTLs were presented at the APHON 37th Annual Conference and Exhibit and the 2015 ASCO Annual Meeting.
Atara Bio’s EBV-CTL product has orphan designation in the European Union and the US, as well as breakthrough designation in the US. ![]()
among uninfected cells (blue)
Image courtesy of Benjamin
Chaigne-Delalande
A cytotoxic T-lymphocyte product that targets Epstein-Barr virus (EBV-CTLs) has been classified as an advanced therapy medicinal product (ATMP) by the European Medicines Agency (EMA).
The EBV-CTLs are being developed by Atara Biotherapeutics, Inc., to treat patients with EBV post-transplant lymphoproliferative disorder (EBV-PTLD).
ATMP classification was established to regulate cell and gene therapy and tissue-engineered medicinal products, support the development of these products, and provide a benchmark for the level of quality compliance for pharmaceutical practices.
ATMP classification can provide developers with scientific regulatory guidance, help clarify the applicable regulatory framework and development path, and provide access to all relevant services and incentives offered by the EMA. It can also be advantageous when submitting clinical trial dossiers to national regulatory authorities within the European Union.
About EBV-CTLs
Atara Bio’s EBV-CTL product utilizes a technology in which T cells are collected from the blood of third-party donors and then exposed to EBV antigens. The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
In the context of EBV-PTLD, the EBV-CTLs find the cancer cells expressing EBV and kill them.
Atara Bio’s EBV-CTL product is currently being studied in phase 2 trials of patients with EBV-associated cancers, including PTLD and nasopharyngeal carcinoma.
Results of a phase 1/2 study of EBV-CTLs were presented at the APHON 37th Annual Conference and Exhibit and the 2015 ASCO Annual Meeting.
Atara Bio’s EBV-CTL product has orphan designation in the European Union and the US, as well as breakthrough designation in the US.
EMA recommends authorization of T-cell product

Image courtesy of NIAID
The European Medicines Agency (EMA) has recommended granting conditional marketing authorization for a T-cell product known as Zalmoxis.
Zalmoxis is intended for use as an adjunctive therapy to aid immune reconstitution and help treat graft-versus-host disease (GVHD) in adults receiving a haploidentical hematopoietic stem cell transplant to treat hematologic malignancy.
Zalmoxis consists of allogeneic T cells genetically modified with a retroviral vector encoding for a truncated form of the human low affinity nerve growth factor receptor (ΔLNGFR) and the herpes simplex I virus thymidine kinase (HSV-TK Mut2).
This modification makes the T cells susceptible to treatment with the drug ganciclovir. So if a patient develops GVHD, he can be treated with ganciclovir, which should kill the modified T cells and prevent further development of the disease.
Zalmoxis is being developed by MolMed S.p.A.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) recommended conditional approval for Zalmoxis. Conditional approval is one of the agency’s main mechanisms to facilitate earlier access to medicines that fulfill unmet medical needs.
Conditional approval allows the EMA to recommend a medicine for marketing authorization before the availability of confirmatory clinical trial data, if the benefits of making this medicine available to patients immediately outweigh the risks inherent in the lack of comprehensive data.
Zalmoxis was also assessed by the Committee on Advanced Therapies (CAT), the EMA’s specialized scientific committee for advanced therapy medicinal products, such as gene or cell therapies.
At its June 2016 meeting, the CAT recommended a conditional marketing authorization for Zalmoxis. The CHMP then considered the CAT’s recommendation and agreed with it.
The recommendation has been sent to the European Commission, which will adopt a decision on marketing authorization that will apply to the European Economic Area.
If Zalmoxis is granted conditional marketing authorization, MolMed S.p.A. must provide the EMA with results from an ongoing phase 3 trial (TK008; NCT00914628).
Until the complete data from this trial are available, the CAT and the CHMP will review the benefits and risks of Zalmoxis annually to determine whether the conditional marketing authorization can be maintained.
Zalmoxis was designated as an orphan medicinal product in 2003. Orphan designation gives drug developers access to incentives such as fee reductions for scientific advice and the opportunity to obtain 10 years of market exclusivity for an authorized orphan-designated medicine.
Image courtesy of NIAID
The European Medicines Agency (EMA) has recommended granting conditional marketing authorization for a T-cell product known as Zalmoxis.
Zalmoxis is intended for use as an adjunctive therapy to aid immune reconstitution and help treat graft-versus-host disease (GVHD) in adults receiving a haploidentical hematopoietic stem cell transplant to treat hematologic malignancy.
Zalmoxis consists of allogeneic T cells genetically modified with a retroviral vector encoding for a truncated form of the human low affinity nerve growth factor receptor (ΔLNGFR) and the herpes simplex I virus thymidine kinase (HSV-TK Mut2).
This modification makes the T cells susceptible to treatment with the drug ganciclovir. So if a patient develops GVHD, he can be treated with ganciclovir, which should kill the modified T cells and prevent further development of the disease.
Zalmoxis is being developed by MolMed S.p.A.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) recommended conditional approval for Zalmoxis. Conditional approval is one of the agency’s main mechanisms to facilitate earlier access to medicines that fulfill unmet medical needs.
Conditional approval allows the EMA to recommend a medicine for marketing authorization before the availability of confirmatory clinical trial data, if the benefits of making this medicine available to patients immediately outweigh the risks inherent in the lack of comprehensive data.
Zalmoxis was also assessed by the Committee on Advanced Therapies (CAT), the EMA’s specialized scientific committee for advanced therapy medicinal products, such as gene or cell therapies.
At its June 2016 meeting, the CAT recommended a conditional marketing authorization for Zalmoxis. The CHMP then considered the CAT’s recommendation and agreed with it.
The recommendation has been sent to the European Commission, which will adopt a decision on marketing authorization that will apply to the European Economic Area.
If Zalmoxis is granted conditional marketing authorization, MolMed S.p.A. must provide the EMA with results from an ongoing phase 3 trial (TK008; NCT00914628).
Until the complete data from this trial are available, the CAT and the CHMP will review the benefits and risks of Zalmoxis annually to determine whether the conditional marketing authorization can be maintained.
Zalmoxis was designated as an orphan medicinal product in 2003. Orphan designation gives drug developers access to incentives such as fee reductions for scientific advice and the opportunity to obtain 10 years of market exclusivity for an authorized orphan-designated medicine.
Image courtesy of NIAID
The European Medicines Agency (EMA) has recommended granting conditional marketing authorization for a T-cell product known as Zalmoxis.
Zalmoxis is intended for use as an adjunctive therapy to aid immune reconstitution and help treat graft-versus-host disease (GVHD) in adults receiving a haploidentical hematopoietic stem cell transplant to treat hematologic malignancy.
Zalmoxis consists of allogeneic T cells genetically modified with a retroviral vector encoding for a truncated form of the human low affinity nerve growth factor receptor (ΔLNGFR) and the herpes simplex I virus thymidine kinase (HSV-TK Mut2).
This modification makes the T cells susceptible to treatment with the drug ganciclovir. So if a patient develops GVHD, he can be treated with ganciclovir, which should kill the modified T cells and prevent further development of the disease.
Zalmoxis is being developed by MolMed S.p.A.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) recommended conditional approval for Zalmoxis. Conditional approval is one of the agency’s main mechanisms to facilitate earlier access to medicines that fulfill unmet medical needs.
Conditional approval allows the EMA to recommend a medicine for marketing authorization before the availability of confirmatory clinical trial data, if the benefits of making this medicine available to patients immediately outweigh the risks inherent in the lack of comprehensive data.
Zalmoxis was also assessed by the Committee on Advanced Therapies (CAT), the EMA’s specialized scientific committee for advanced therapy medicinal products, such as gene or cell therapies.
At its June 2016 meeting, the CAT recommended a conditional marketing authorization for Zalmoxis. The CHMP then considered the CAT’s recommendation and agreed with it.
The recommendation has been sent to the European Commission, which will adopt a decision on marketing authorization that will apply to the European Economic Area.
If Zalmoxis is granted conditional marketing authorization, MolMed S.p.A. must provide the EMA with results from an ongoing phase 3 trial (TK008; NCT00914628).
Until the complete data from this trial are available, the CAT and the CHMP will review the benefits and risks of Zalmoxis annually to determine whether the conditional marketing authorization can be maintained.
Zalmoxis was designated as an orphan medicinal product in 2003. Orphan designation gives drug developers access to incentives such as fee reductions for scientific advice and the opportunity to obtain 10 years of market exclusivity for an authorized orphan-designated medicine.
FDA grants drug breakthrough designation to treat GVHD

Photo from Business Wire
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ruxolitinib (Jakafi) for the treatment of patients with acute graft-versus-host disease (GVHD).
Ruxolitinib is a JAK1/2 inhibitor that is already FDA-approved to treat patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea.
The drug is also approved to treat patients with intermediate or high-risk myelofibrosis (MF), including primary MF, post-polycythemia vera MF, and post-essential thrombocythemia MF.
Ruxolitinib is marketed as Jakafi by Incyte in the US and as Jakavi by Novartis outside the US.
“Receiving breakthrough therapy designation from the FDA recognizes the severe nature of acute GVHD, the clear unmet medical need of these patients, and the potential, based on clinical evidence to date, for ruxolitinib to address the urgent needs of patients with this life-threatening disease,” said Steven Stein, MD, Incyte’s chief medical officer.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Photo from Business Wire
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ruxolitinib (Jakafi) for the treatment of patients with acute graft-versus-host disease (GVHD).
Ruxolitinib is a JAK1/2 inhibitor that is already FDA-approved to treat patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea.
The drug is also approved to treat patients with intermediate or high-risk myelofibrosis (MF), including primary MF, post-polycythemia vera MF, and post-essential thrombocythemia MF.
Ruxolitinib is marketed as Jakafi by Incyte in the US and as Jakavi by Novartis outside the US.
“Receiving breakthrough therapy designation from the FDA recognizes the severe nature of acute GVHD, the clear unmet medical need of these patients, and the potential, based on clinical evidence to date, for ruxolitinib to address the urgent needs of patients with this life-threatening disease,” said Steven Stein, MD, Incyte’s chief medical officer.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Photo from Business Wire
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ruxolitinib (Jakafi) for the treatment of patients with acute graft-versus-host disease (GVHD).
Ruxolitinib is a JAK1/2 inhibitor that is already FDA-approved to treat patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea.
The drug is also approved to treat patients with intermediate or high-risk myelofibrosis (MF), including primary MF, post-polycythemia vera MF, and post-essential thrombocythemia MF.
Ruxolitinib is marketed as Jakafi by Incyte in the US and as Jakavi by Novartis outside the US.
“Receiving breakthrough therapy designation from the FDA recognizes the severe nature of acute GVHD, the clear unmet medical need of these patients, and the potential, based on clinical evidence to date, for ruxolitinib to address the urgent needs of patients with this life-threatening disease,” said Steven Stein, MD, Incyte’s chief medical officer.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Therapy granted fast track designation to treat GVHD
![]()
Photo by Chad McNeeley
The US Food and Drug Administration (FDA) has granted fast track designation for ProTmune™, a programmed cellular immunotherapy, to reduce the incidence and severity of acute graft-versus-host disease (GVHD) in patients undergoing allogeneic hematopoietic stem cell transplant (HSCT).
ProTmune is produced by modulating a donor-sourced, mobilized peripheral blood (mPB) graft ex vivo with 2 small molecules (FT1050 and FT4145) to enhance the biological properties and therapeutic function of the graft’s immune cells.
The programmed mPB graft is adoptively transferred and administered to a patient as a one-time intravenous infusion.
ProTmune is being developed by Fate Therapeutics, Inc.
The company is conducting a phase 1/2 trial testing ProTmune for the prevention of acute GVHD and cytomegalovirus infection in adults with hematologic malignancies who are undergoing HSCT.
The trial design consists of an initial 10-subject, phase 1 stage, during which all subjects undergoing allogeneic mPB HSCT will receive ProTmune.
Following an independent data monitoring committee safety review, a 60-subject, randomized, controlled phase 2 stage is expected to begin. In this stage, subjects undergoing allogeneic mPB HSCT will be assigned to receive either ProTmune or a conventional mPB cell graft in a 1:1 ratio.
About fast track designation
The FDA’s fast track program is designed to facilitate and expedite the development and review of new drugs intended to treat serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the drug may be allowed to submit sections of the biologic license application or new drug application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings with the FDA to discuss the drug’s development plan and ensure collection of the appropriate data needed to support drug approval. And the designation allows for more frequent written communication from the FDA about things such as the design of proposed clinical trials and the use of biomarkers.
![]()
Photo by Chad McNeeley
The US Food and Drug Administration (FDA) has granted fast track designation for ProTmune™, a programmed cellular immunotherapy, to reduce the incidence and severity of acute graft-versus-host disease (GVHD) in patients undergoing allogeneic hematopoietic stem cell transplant (HSCT).
ProTmune is produced by modulating a donor-sourced, mobilized peripheral blood (mPB) graft ex vivo with 2 small molecules (FT1050 and FT4145) to enhance the biological properties and therapeutic function of the graft’s immune cells.
The programmed mPB graft is adoptively transferred and administered to a patient as a one-time intravenous infusion.
ProTmune is being developed by Fate Therapeutics, Inc.
The company is conducting a phase 1/2 trial testing ProTmune for the prevention of acute GVHD and cytomegalovirus infection in adults with hematologic malignancies who are undergoing HSCT.
The trial design consists of an initial 10-subject, phase 1 stage, during which all subjects undergoing allogeneic mPB HSCT will receive ProTmune.
Following an independent data monitoring committee safety review, a 60-subject, randomized, controlled phase 2 stage is expected to begin. In this stage, subjects undergoing allogeneic mPB HSCT will be assigned to receive either ProTmune or a conventional mPB cell graft in a 1:1 ratio.
About fast track designation
The FDA’s fast track program is designed to facilitate and expedite the development and review of new drugs intended to treat serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the drug may be allowed to submit sections of the biologic license application or new drug application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings with the FDA to discuss the drug’s development plan and ensure collection of the appropriate data needed to support drug approval. And the designation allows for more frequent written communication from the FDA about things such as the design of proposed clinical trials and the use of biomarkers.
![]()
Photo by Chad McNeeley
The US Food and Drug Administration (FDA) has granted fast track designation for ProTmune™, a programmed cellular immunotherapy, to reduce the incidence and severity of acute graft-versus-host disease (GVHD) in patients undergoing allogeneic hematopoietic stem cell transplant (HSCT).
ProTmune is produced by modulating a donor-sourced, mobilized peripheral blood (mPB) graft ex vivo with 2 small molecules (FT1050 and FT4145) to enhance the biological properties and therapeutic function of the graft’s immune cells.
The programmed mPB graft is adoptively transferred and administered to a patient as a one-time intravenous infusion.
ProTmune is being developed by Fate Therapeutics, Inc.
The company is conducting a phase 1/2 trial testing ProTmune for the prevention of acute GVHD and cytomegalovirus infection in adults with hematologic malignancies who are undergoing HSCT.
The trial design consists of an initial 10-subject, phase 1 stage, during which all subjects undergoing allogeneic mPB HSCT will receive ProTmune.
Following an independent data monitoring committee safety review, a 60-subject, randomized, controlled phase 2 stage is expected to begin. In this stage, subjects undergoing allogeneic mPB HSCT will be assigned to receive either ProTmune or a conventional mPB cell graft in a 1:1 ratio.
About fast track designation
The FDA’s fast track program is designed to facilitate and expedite the development and review of new drugs intended to treat serious or life-threatening conditions and address unmet medical need.
Through the fast track program, a product may be eligible for priority review. In addition, the company developing the drug may be allowed to submit sections of the biologic license application or new drug application on a rolling basis as data become available.
Fast track designation also provides the company with opportunities for more frequent meetings with the FDA to discuss the drug’s development plan and ensure collection of the appropriate data needed to support drug approval. And the designation allows for more frequent written communication from the FDA about things such as the design of proposed clinical trials and the use of biomarkers.