User login
HIV-associated Kaposi sarcoma responds to checkpoint inhibitors
Checkpoint inhibitor therapy is effective for patients with HIV-associated Kaposi sarcoma (KS), a recent study has found.
Partial or complete remission was achieved by a majority of patients; others currently have stable disease lasting longer than 6 months, reported Natalie Galanina, MD, of Rebecca and John Moores Cancer Center at the University of California, San Diego, and her colleagues. Earlier this year, investigators reported similar responses to checkpoint inhibitors in two patients with KS that wasn’t associated with HIV.
“An association has been demonstrated between chronic viral infection, malignancy, and up-regulation of programmed death receptor 1 (PD-1) on CD8+ cytotoxic T-lymphocytes,” the authors wrote in Cancer Immunology Research. In particular, “HIV-specific CD8+ T cells have increased PD-1 expression, which … promotes a cellular milieu conducive to oncogenesis.” These factors, together with the results from the previous study, have suggested that checkpoint inhibitors may be effective for patients with HIV-associated KS.
The retrospective study involved 320 patients treated with immunotherapy at Moores Cancer Center from August 2013 through December 2017. From this group, nine cases of HIV-associated KS were found. Median CD4 count was 256 cells/mcL and median viral load was 20 copies/mL. Eight patients were treated with nivolumab and one was treated with pembrolizumab. Median age was 44 years. All patients were male and receiving antiretroviral therapy.
Six patients (67%) achieved remission, with five attaining partial remission and one attaining complete remission (gastrointestinal disease). Of the remaining three patients, two currently have stable disease lasting longer than 6 months, and one has stable disease lasting longer than 3 months.
Muscle aches, pruritus, and low-grade fever were the most common adverse events. No grade 3 or higher drug-related adverse events occurred.
“Most of our patients received one to four prior lines of therapy but still responded to checkpoint blockade,” the authors wrote. “Our observations suggest that patients with HIV-associated KS have high [response rates] to PD-1 checkpoint blockade, without significant toxicity, even in the presence of low [tumor mutational burden] and/or lack of PD-L1 expression.”
Authors reported compensation from Incyte, Genentech, Merck, Pfizer, and others.
SOURCE: Galanina et al. Cancer Immunol Res. doi: 10.1158/2326-6066.CIR-18-0121.
Checkpoint inhibitor therapy is effective for patients with HIV-associated Kaposi sarcoma (KS), a recent study has found.
Partial or complete remission was achieved by a majority of patients; others currently have stable disease lasting longer than 6 months, reported Natalie Galanina, MD, of Rebecca and John Moores Cancer Center at the University of California, San Diego, and her colleagues. Earlier this year, investigators reported similar responses to checkpoint inhibitors in two patients with KS that wasn’t associated with HIV.
“An association has been demonstrated between chronic viral infection, malignancy, and up-regulation of programmed death receptor 1 (PD-1) on CD8+ cytotoxic T-lymphocytes,” the authors wrote in Cancer Immunology Research. In particular, “HIV-specific CD8+ T cells have increased PD-1 expression, which … promotes a cellular milieu conducive to oncogenesis.” These factors, together with the results from the previous study, have suggested that checkpoint inhibitors may be effective for patients with HIV-associated KS.
The retrospective study involved 320 patients treated with immunotherapy at Moores Cancer Center from August 2013 through December 2017. From this group, nine cases of HIV-associated KS were found. Median CD4 count was 256 cells/mcL and median viral load was 20 copies/mL. Eight patients were treated with nivolumab and one was treated with pembrolizumab. Median age was 44 years. All patients were male and receiving antiretroviral therapy.
Six patients (67%) achieved remission, with five attaining partial remission and one attaining complete remission (gastrointestinal disease). Of the remaining three patients, two currently have stable disease lasting longer than 6 months, and one has stable disease lasting longer than 3 months.
Muscle aches, pruritus, and low-grade fever were the most common adverse events. No grade 3 or higher drug-related adverse events occurred.
“Most of our patients received one to four prior lines of therapy but still responded to checkpoint blockade,” the authors wrote. “Our observations suggest that patients with HIV-associated KS have high [response rates] to PD-1 checkpoint blockade, without significant toxicity, even in the presence of low [tumor mutational burden] and/or lack of PD-L1 expression.”
Authors reported compensation from Incyte, Genentech, Merck, Pfizer, and others.
SOURCE: Galanina et al. Cancer Immunol Res. doi: 10.1158/2326-6066.CIR-18-0121.
Checkpoint inhibitor therapy is effective for patients with HIV-associated Kaposi sarcoma (KS), a recent study has found.
Partial or complete remission was achieved by a majority of patients; others currently have stable disease lasting longer than 6 months, reported Natalie Galanina, MD, of Rebecca and John Moores Cancer Center at the University of California, San Diego, and her colleagues. Earlier this year, investigators reported similar responses to checkpoint inhibitors in two patients with KS that wasn’t associated with HIV.
“An association has been demonstrated between chronic viral infection, malignancy, and up-regulation of programmed death receptor 1 (PD-1) on CD8+ cytotoxic T-lymphocytes,” the authors wrote in Cancer Immunology Research. In particular, “HIV-specific CD8+ T cells have increased PD-1 expression, which … promotes a cellular milieu conducive to oncogenesis.” These factors, together with the results from the previous study, have suggested that checkpoint inhibitors may be effective for patients with HIV-associated KS.
The retrospective study involved 320 patients treated with immunotherapy at Moores Cancer Center from August 2013 through December 2017. From this group, nine cases of HIV-associated KS were found. Median CD4 count was 256 cells/mcL and median viral load was 20 copies/mL. Eight patients were treated with nivolumab and one was treated with pembrolizumab. Median age was 44 years. All patients were male and receiving antiretroviral therapy.
Six patients (67%) achieved remission, with five attaining partial remission and one attaining complete remission (gastrointestinal disease). Of the remaining three patients, two currently have stable disease lasting longer than 6 months, and one has stable disease lasting longer than 3 months.
Muscle aches, pruritus, and low-grade fever were the most common adverse events. No grade 3 or higher drug-related adverse events occurred.
“Most of our patients received one to four prior lines of therapy but still responded to checkpoint blockade,” the authors wrote. “Our observations suggest that patients with HIV-associated KS have high [response rates] to PD-1 checkpoint blockade, without significant toxicity, even in the presence of low [tumor mutational burden] and/or lack of PD-L1 expression.”
Authors reported compensation from Incyte, Genentech, Merck, Pfizer, and others.
SOURCE: Galanina et al. Cancer Immunol Res. doi: 10.1158/2326-6066.CIR-18-0121.
FROM CANCER IMMUNOLOGY RESEARCH
Key clinical point: Checkpoint inhibitor therapy is effective for patients with HIV-associated Kaposi sarcoma.
Major finding: Two-thirds of patients (67%) with HIV-associated Kaposi sarcoma achieved partial or complete remission when treated with immune checkpoint blockade.
Study details: A retrospective study involving nine patients with Kaposi sarcoma treated with either nivolumab or pembrolizumab at the Rebecca and John Moores Cancer Center at the University of California, San Diego, (UCSD) from August 2013 through December 2017.
Disclosures: Authors reported compensation from Incyte, Genentech, Merck, Pfizer, and others.
Source: Galanina et al. Cancer Immunol Res. 2018 Sept 7. doi: 10.1158/2326-6066.CIR-18-0121.
Novel molecular assay: Promising results in bone and soft tissue tumor evaluation
, according to researchers.
The technique of anchored multiplex polymerase chain reaction (AMP)–based targeted next-generation sequencing (NGS) had a failure rate of 14% but, nonetheless, worked favorably when compared with conventional techniques, which were associated with several false positives in this study, the researchers reported in the Journal of Molecular Diagnostics.
Two new fusion partners for the USP6 gene were found using AMP-based targeted NGS in this study, which thus contributed to the “further unraveling of the molecular landscape” for these tumors, added corresponding author Judith V.M.G. Bovée, MD, PhD, of the department of pathology at Leiden (the Netherlands) University Medical Center and her colleagues.
While the genetics of bone and soft tissue tumors have diagnostic value in clinical practice, standard fluorescence in situ hybridization (FISH) and reverse transcriptase PCR are associated with several drawbacks, such as a high false negative rate in the case of FISH, Dr. Bovée and her coauthors wrote.
Accordingly, the researchers evaluated the applicability of a targeted sequencing assay (Archer FusionPlex Sarcoma kit, which was developed by ArcherDX) aimed at 26 genes relevant to bone and soft tissue tumor diagnostics.
Besides allowing for assessment of multiple target genes in a single assay, this technique circumvents the need to know both fusion partners for translocation detection, which opens up the possibility of identifying novel or rare fusion partners, investigators noted.
AMP-based targeted NGS was used to evaluate 81 bone and soft tissue tumor samples, and of those, 48 cases showed a fusion. For the remaining 33 cases in which no fusion was detected, 22 were considered truly negative because samples met all criteria for good quality, while the remaining 11 (14%) were considered not reliable because of insufficient quality, investigators reported.
The samples were also evaluated through use of FISH, reverse transcriptase PCR, or both in 58 cases and use of immunohistochemistry in 16 cases; for the remaining seven cases, no assay or immunohistochemistry could be applied because of a lack of availability, according to investigators.
Among the 48 entities that were fusion-positive according to AMP-based targeted NGS, 29 were validated using standard molecular assays, and of those, 25 had concordant results. Further analysis of the four discordant cases with a third independent technique confirmed the AMP-based targeted NGS findings, according to the published report.
Among the 22 fusion-negative high-quality samples, 19 were validated using FISH, and one case was found to be discordant; however, despite use of a third independent technique, this discrepancy could not be resolved, investigators said.
The AMP-based targeted NGS technique identified COL1A1 and SEC31A as novel fusion partners for USP6 in two cases of nodular fasciitis. Those fusion partners had been previously described in aneurysmal bone cysts, according to investigators.
Despite the promising results for the novel assay, conventional methods were sufficient in this study to confirm translocations in straightforward cases and ordinary rearrangements, according to the investigators.
“Both reverse transcription PCR and FISH are not only quick and easy to conduct but are also of low cost and high analytical validity and accuracy, which make them attractive methods,” they wrote.
The work by Dr. Bovée and her colleagues was supported by Leiden University Medical Center. The department of pathology and the department of cell and chemical biology at the medical center receive royalty payments from Kreatech/Leica, which provided a COL1A1/PDGFB fusion probe used in the research.
SOURCE: Lam SW et al. J Mol Diagn. 2018 Aug 20;20(5):653-63.
, according to researchers.
The technique of anchored multiplex polymerase chain reaction (AMP)–based targeted next-generation sequencing (NGS) had a failure rate of 14% but, nonetheless, worked favorably when compared with conventional techniques, which were associated with several false positives in this study, the researchers reported in the Journal of Molecular Diagnostics.
Two new fusion partners for the USP6 gene were found using AMP-based targeted NGS in this study, which thus contributed to the “further unraveling of the molecular landscape” for these tumors, added corresponding author Judith V.M.G. Bovée, MD, PhD, of the department of pathology at Leiden (the Netherlands) University Medical Center and her colleagues.
While the genetics of bone and soft tissue tumors have diagnostic value in clinical practice, standard fluorescence in situ hybridization (FISH) and reverse transcriptase PCR are associated with several drawbacks, such as a high false negative rate in the case of FISH, Dr. Bovée and her coauthors wrote.
Accordingly, the researchers evaluated the applicability of a targeted sequencing assay (Archer FusionPlex Sarcoma kit, which was developed by ArcherDX) aimed at 26 genes relevant to bone and soft tissue tumor diagnostics.
Besides allowing for assessment of multiple target genes in a single assay, this technique circumvents the need to know both fusion partners for translocation detection, which opens up the possibility of identifying novel or rare fusion partners, investigators noted.
AMP-based targeted NGS was used to evaluate 81 bone and soft tissue tumor samples, and of those, 48 cases showed a fusion. For the remaining 33 cases in which no fusion was detected, 22 were considered truly negative because samples met all criteria for good quality, while the remaining 11 (14%) were considered not reliable because of insufficient quality, investigators reported.
The samples were also evaluated through use of FISH, reverse transcriptase PCR, or both in 58 cases and use of immunohistochemistry in 16 cases; for the remaining seven cases, no assay or immunohistochemistry could be applied because of a lack of availability, according to investigators.
Among the 48 entities that were fusion-positive according to AMP-based targeted NGS, 29 were validated using standard molecular assays, and of those, 25 had concordant results. Further analysis of the four discordant cases with a third independent technique confirmed the AMP-based targeted NGS findings, according to the published report.
Among the 22 fusion-negative high-quality samples, 19 were validated using FISH, and one case was found to be discordant; however, despite use of a third independent technique, this discrepancy could not be resolved, investigators said.
The AMP-based targeted NGS technique identified COL1A1 and SEC31A as novel fusion partners for USP6 in two cases of nodular fasciitis. Those fusion partners had been previously described in aneurysmal bone cysts, according to investigators.
Despite the promising results for the novel assay, conventional methods were sufficient in this study to confirm translocations in straightforward cases and ordinary rearrangements, according to the investigators.
“Both reverse transcription PCR and FISH are not only quick and easy to conduct but are also of low cost and high analytical validity and accuracy, which make them attractive methods,” they wrote.
The work by Dr. Bovée and her colleagues was supported by Leiden University Medical Center. The department of pathology and the department of cell and chemical biology at the medical center receive royalty payments from Kreatech/Leica, which provided a COL1A1/PDGFB fusion probe used in the research.
SOURCE: Lam SW et al. J Mol Diagn. 2018 Aug 20;20(5):653-63.
, according to researchers.
The technique of anchored multiplex polymerase chain reaction (AMP)–based targeted next-generation sequencing (NGS) had a failure rate of 14% but, nonetheless, worked favorably when compared with conventional techniques, which were associated with several false positives in this study, the researchers reported in the Journal of Molecular Diagnostics.
Two new fusion partners for the USP6 gene were found using AMP-based targeted NGS in this study, which thus contributed to the “further unraveling of the molecular landscape” for these tumors, added corresponding author Judith V.M.G. Bovée, MD, PhD, of the department of pathology at Leiden (the Netherlands) University Medical Center and her colleagues.
While the genetics of bone and soft tissue tumors have diagnostic value in clinical practice, standard fluorescence in situ hybridization (FISH) and reverse transcriptase PCR are associated with several drawbacks, such as a high false negative rate in the case of FISH, Dr. Bovée and her coauthors wrote.
Accordingly, the researchers evaluated the applicability of a targeted sequencing assay (Archer FusionPlex Sarcoma kit, which was developed by ArcherDX) aimed at 26 genes relevant to bone and soft tissue tumor diagnostics.
Besides allowing for assessment of multiple target genes in a single assay, this technique circumvents the need to know both fusion partners for translocation detection, which opens up the possibility of identifying novel or rare fusion partners, investigators noted.
AMP-based targeted NGS was used to evaluate 81 bone and soft tissue tumor samples, and of those, 48 cases showed a fusion. For the remaining 33 cases in which no fusion was detected, 22 were considered truly negative because samples met all criteria for good quality, while the remaining 11 (14%) were considered not reliable because of insufficient quality, investigators reported.
The samples were also evaluated through use of FISH, reverse transcriptase PCR, or both in 58 cases and use of immunohistochemistry in 16 cases; for the remaining seven cases, no assay or immunohistochemistry could be applied because of a lack of availability, according to investigators.
Among the 48 entities that were fusion-positive according to AMP-based targeted NGS, 29 were validated using standard molecular assays, and of those, 25 had concordant results. Further analysis of the four discordant cases with a third independent technique confirmed the AMP-based targeted NGS findings, according to the published report.
Among the 22 fusion-negative high-quality samples, 19 were validated using FISH, and one case was found to be discordant; however, despite use of a third independent technique, this discrepancy could not be resolved, investigators said.
The AMP-based targeted NGS technique identified COL1A1 and SEC31A as novel fusion partners for USP6 in two cases of nodular fasciitis. Those fusion partners had been previously described in aneurysmal bone cysts, according to investigators.
Despite the promising results for the novel assay, conventional methods were sufficient in this study to confirm translocations in straightforward cases and ordinary rearrangements, according to the investigators.
“Both reverse transcription PCR and FISH are not only quick and easy to conduct but are also of low cost and high analytical validity and accuracy, which make them attractive methods,” they wrote.
The work by Dr. Bovée and her colleagues was supported by Leiden University Medical Center. The department of pathology and the department of cell and chemical biology at the medical center receive royalty payments from Kreatech/Leica, which provided a COL1A1/PDGFB fusion probe used in the research.
SOURCE: Lam SW et al. J Mol Diagn. 2018 Aug 20;20(5):653-63.
FROM THE JOURNAL OF MOLECULAR DIAGNOSTICS
Key clinical point: Anchored multiplex PCR (AMP)-based targeted next-generation sequencing (NGS) may be superior to conventional molecular assays in the evaluation of bone and soft tissue tumor samples.
Major finding: Standard techniques yielded 4 false negatives out of 29 samples that were fusion-positive by AMP-based targeted NGS.
Study details: Analysis of 81 bone and soft tissue tumor samples evaluated by AMP-based targeted NGS and conventional techniques.
Disclosures: The research was supported by Leiden (the Netherlands) University Medical Center, which receives royalty payments from Kreatech/Leica.
Source: Lam SW et al. J Mol Diagn. 2018 Aug 20;20(5):653-63.
SEAL: Selinexor extends PFS in advanced dedifferentiated liposarcoma
The investigational drug selinexor appears to be improving progression-free survival in patients with advanced dedifferentiated liposarcoma, based on phase 2 results from the randomized, placebo-controlled SEAL study.
But the statistical significance of the improvements varied depending on whether progression-free survival (PFS) was assessed by the World Health Organization criteria, which looks at two-dimensional measurements of these irregular three-dimensional objects, or RECIST v1.1 criteria, which only looks at a unidimensional measure, reported Mrinal M. Gounder, MD, of Memorial Sloan Kettering Cancer Center, New York, at the annual meeting of the American Society of Clinical Oncology. When tumor response was based on WHO criteria, there was no difference in median PFS for the 24 patients on active therapy (1.4 months) and the 27 patients on placebo (1.8 months). By RECIST v1.1 criteria, however, median PFS was 5.6 months with selinexor.
Dedifferentiated liposarcoma is incurable, and palliative therapies are associated with an overall survival of 11-20 months in these patients. Selinexor is an oral selective inhibitor of exportin-1 which exports proteins from the nucleus into the cytoplasm. The drug appears to prevent p53 from leaving the nucleus, thereby protecting it from overexpressed MDM2, which is a negative regulator of p53, but the drug might have other potential mechanisms of action.
The double-blind study included 56 evaluable patients who had progressive dedifferentiated liposarcoma and had received at least one prior systemic therapy. Patients’ median age was 61 years and they had received a median of two prior therapies. Patients were randomized to get either 60 mg of selinexor (26 patients) or placebo (30 patients) twice weekly until their disease progressed or they were no longer able to tolerate therapy. Patients whose disease progressed on placebo (24 patients) were allowed to cross over to open-label selinexor therapy.
Treatments were unblinded for 51 of the patients, 24 on selinexor and 27 on placebo. Disease progression as confirmed by Independent Central Radiological Review using WHO criteria was the main reason for ending blinded treatment.
Grade 1/2 adverse events for selinexor versus placebo, respectively, were nausea (85% vs. 31%), anorexia (62% vs. 14%), and fatigue (58% vs. 45%). The comparable rates of grade 3/4 adverse events were hyponatremia (15% vs. 0%), anemia (15% vs. 7%), and thrombocytopenia (12% vs. 0%). Selinexor dose was reduced because of adverse events in 12 patients.
In a discussion of the study’s implications, Mark Andrew Dickson, MD, also of Memorial Sloan Kettering Cancer Center, called the adverse events profile “mostly manageable but predictable grade 1/2 adverse events ... and median progression-free survival of 5 and a half months is quite encouraging.
“Changing response assessment method midtrial in a study with progression-free survival as the primary endpoint is obviously problematic, but it also highlights how difficult it is to measure three-dimensional tumors like complex retroperitoneal liposarcomas, which move and change and grow and shrink over time,” he said. “And I would conclude that RECIST is probably the worst method of tumor assessment for sarcoma, except for all the other methods of tumor assessment.”
To illustrate the difficulty of measuring tumor response, Dr. Dickson presented examples of different tumor shapes and scenarios where one method would indicate tumor progression and the other would indicate stable disease.
“There can be differences between the two methods in how progression responds and is determined. And you can do this experiment with a number of different shapes and find scenarios where one method would call it progression at a different time than the other. So this is really critically important when we look at the results of the clinical trial, because it was designed to look at WHO PFS. And you can see that, based on that, there was no significant difference between the selinexor and placebo arm,” he said.
Additionally, he reviewed cases from the study where “either way you measure this, you can see that [the] tumor is getting smaller over time,” as well as cases where the tumor grew in patients on placebo first, but decreased in size after switching to the active therapy.
“The improvement in progression-free survival is promising and ... selinexor probably does have activity in dediff lipo compared to historical data,” said Dr. Dickson, adding that he looks forward to selinexor progressing to a randomized, phase 3 trial and “seeing those data perhaps next year.”
Dr. Gounder disclosed financial relationships with multiple drug companies including Karyopharm Therapeutics, the maker of selinexor. Dr. Dickson disclosed a consult or adviser role with Celgene and research funding from Eli Lilly.
SOURCE: Gounder M et al. ASCO 2018, Abstract 11512.
The investigational drug selinexor appears to be improving progression-free survival in patients with advanced dedifferentiated liposarcoma, based on phase 2 results from the randomized, placebo-controlled SEAL study.
But the statistical significance of the improvements varied depending on whether progression-free survival (PFS) was assessed by the World Health Organization criteria, which looks at two-dimensional measurements of these irregular three-dimensional objects, or RECIST v1.1 criteria, which only looks at a unidimensional measure, reported Mrinal M. Gounder, MD, of Memorial Sloan Kettering Cancer Center, New York, at the annual meeting of the American Society of Clinical Oncology. When tumor response was based on WHO criteria, there was no difference in median PFS for the 24 patients on active therapy (1.4 months) and the 27 patients on placebo (1.8 months). By RECIST v1.1 criteria, however, median PFS was 5.6 months with selinexor.
Dedifferentiated liposarcoma is incurable, and palliative therapies are associated with an overall survival of 11-20 months in these patients. Selinexor is an oral selective inhibitor of exportin-1 which exports proteins from the nucleus into the cytoplasm. The drug appears to prevent p53 from leaving the nucleus, thereby protecting it from overexpressed MDM2, which is a negative regulator of p53, but the drug might have other potential mechanisms of action.
The double-blind study included 56 evaluable patients who had progressive dedifferentiated liposarcoma and had received at least one prior systemic therapy. Patients’ median age was 61 years and they had received a median of two prior therapies. Patients were randomized to get either 60 mg of selinexor (26 patients) or placebo (30 patients) twice weekly until their disease progressed or they were no longer able to tolerate therapy. Patients whose disease progressed on placebo (24 patients) were allowed to cross over to open-label selinexor therapy.
Treatments were unblinded for 51 of the patients, 24 on selinexor and 27 on placebo. Disease progression as confirmed by Independent Central Radiological Review using WHO criteria was the main reason for ending blinded treatment.
Grade 1/2 adverse events for selinexor versus placebo, respectively, were nausea (85% vs. 31%), anorexia (62% vs. 14%), and fatigue (58% vs. 45%). The comparable rates of grade 3/4 adverse events were hyponatremia (15% vs. 0%), anemia (15% vs. 7%), and thrombocytopenia (12% vs. 0%). Selinexor dose was reduced because of adverse events in 12 patients.
In a discussion of the study’s implications, Mark Andrew Dickson, MD, also of Memorial Sloan Kettering Cancer Center, called the adverse events profile “mostly manageable but predictable grade 1/2 adverse events ... and median progression-free survival of 5 and a half months is quite encouraging.
“Changing response assessment method midtrial in a study with progression-free survival as the primary endpoint is obviously problematic, but it also highlights how difficult it is to measure three-dimensional tumors like complex retroperitoneal liposarcomas, which move and change and grow and shrink over time,” he said. “And I would conclude that RECIST is probably the worst method of tumor assessment for sarcoma, except for all the other methods of tumor assessment.”
To illustrate the difficulty of measuring tumor response, Dr. Dickson presented examples of different tumor shapes and scenarios where one method would indicate tumor progression and the other would indicate stable disease.
“There can be differences between the two methods in how progression responds and is determined. And you can do this experiment with a number of different shapes and find scenarios where one method would call it progression at a different time than the other. So this is really critically important when we look at the results of the clinical trial, because it was designed to look at WHO PFS. And you can see that, based on that, there was no significant difference between the selinexor and placebo arm,” he said.
Additionally, he reviewed cases from the study where “either way you measure this, you can see that [the] tumor is getting smaller over time,” as well as cases where the tumor grew in patients on placebo first, but decreased in size after switching to the active therapy.
“The improvement in progression-free survival is promising and ... selinexor probably does have activity in dediff lipo compared to historical data,” said Dr. Dickson, adding that he looks forward to selinexor progressing to a randomized, phase 3 trial and “seeing those data perhaps next year.”
Dr. Gounder disclosed financial relationships with multiple drug companies including Karyopharm Therapeutics, the maker of selinexor. Dr. Dickson disclosed a consult or adviser role with Celgene and research funding from Eli Lilly.
SOURCE: Gounder M et al. ASCO 2018, Abstract 11512.
The investigational drug selinexor appears to be improving progression-free survival in patients with advanced dedifferentiated liposarcoma, based on phase 2 results from the randomized, placebo-controlled SEAL study.
But the statistical significance of the improvements varied depending on whether progression-free survival (PFS) was assessed by the World Health Organization criteria, which looks at two-dimensional measurements of these irregular three-dimensional objects, or RECIST v1.1 criteria, which only looks at a unidimensional measure, reported Mrinal M. Gounder, MD, of Memorial Sloan Kettering Cancer Center, New York, at the annual meeting of the American Society of Clinical Oncology. When tumor response was based on WHO criteria, there was no difference in median PFS for the 24 patients on active therapy (1.4 months) and the 27 patients on placebo (1.8 months). By RECIST v1.1 criteria, however, median PFS was 5.6 months with selinexor.
Dedifferentiated liposarcoma is incurable, and palliative therapies are associated with an overall survival of 11-20 months in these patients. Selinexor is an oral selective inhibitor of exportin-1 which exports proteins from the nucleus into the cytoplasm. The drug appears to prevent p53 from leaving the nucleus, thereby protecting it from overexpressed MDM2, which is a negative regulator of p53, but the drug might have other potential mechanisms of action.
The double-blind study included 56 evaluable patients who had progressive dedifferentiated liposarcoma and had received at least one prior systemic therapy. Patients’ median age was 61 years and they had received a median of two prior therapies. Patients were randomized to get either 60 mg of selinexor (26 patients) or placebo (30 patients) twice weekly until their disease progressed or they were no longer able to tolerate therapy. Patients whose disease progressed on placebo (24 patients) were allowed to cross over to open-label selinexor therapy.
Treatments were unblinded for 51 of the patients, 24 on selinexor and 27 on placebo. Disease progression as confirmed by Independent Central Radiological Review using WHO criteria was the main reason for ending blinded treatment.
Grade 1/2 adverse events for selinexor versus placebo, respectively, were nausea (85% vs. 31%), anorexia (62% vs. 14%), and fatigue (58% vs. 45%). The comparable rates of grade 3/4 adverse events were hyponatremia (15% vs. 0%), anemia (15% vs. 7%), and thrombocytopenia (12% vs. 0%). Selinexor dose was reduced because of adverse events in 12 patients.
In a discussion of the study’s implications, Mark Andrew Dickson, MD, also of Memorial Sloan Kettering Cancer Center, called the adverse events profile “mostly manageable but predictable grade 1/2 adverse events ... and median progression-free survival of 5 and a half months is quite encouraging.
“Changing response assessment method midtrial in a study with progression-free survival as the primary endpoint is obviously problematic, but it also highlights how difficult it is to measure three-dimensional tumors like complex retroperitoneal liposarcomas, which move and change and grow and shrink over time,” he said. “And I would conclude that RECIST is probably the worst method of tumor assessment for sarcoma, except for all the other methods of tumor assessment.”
To illustrate the difficulty of measuring tumor response, Dr. Dickson presented examples of different tumor shapes and scenarios where one method would indicate tumor progression and the other would indicate stable disease.
“There can be differences between the two methods in how progression responds and is determined. And you can do this experiment with a number of different shapes and find scenarios where one method would call it progression at a different time than the other. So this is really critically important when we look at the results of the clinical trial, because it was designed to look at WHO PFS. And you can see that, based on that, there was no significant difference between the selinexor and placebo arm,” he said.
Additionally, he reviewed cases from the study where “either way you measure this, you can see that [the] tumor is getting smaller over time,” as well as cases where the tumor grew in patients on placebo first, but decreased in size after switching to the active therapy.
“The improvement in progression-free survival is promising and ... selinexor probably does have activity in dediff lipo compared to historical data,” said Dr. Dickson, adding that he looks forward to selinexor progressing to a randomized, phase 3 trial and “seeing those data perhaps next year.”
Dr. Gounder disclosed financial relationships with multiple drug companies including Karyopharm Therapeutics, the maker of selinexor. Dr. Dickson disclosed a consult or adviser role with Celgene and research funding from Eli Lilly.
SOURCE: Gounder M et al. ASCO 2018, Abstract 11512.
FROM ASCO 2018
Key clinical point: The investigational drug selinexor appears to be improving progression-free survival (PFS) in patients with advanced dedifferentiated liposarcoma.
Major finding: When tumor response was based on World Health Organization criteria, there was no difference in median PFS for the 24 patients on active therapy (1.4 months) and the 27 patients on placebo (1.8 months). By RECIST v1.1 criteria, however, median PFS was 5.6 months with selinexor.
Study details: Phase 2 results from 56 patients with dedifferentiated liposarcoma in the randomized, placebo-controlled SEAL study.
Disclosures: Dr. Gounder reported financial relationships with multiple drug companies including Karyopharm Therapeutics, the maker of selinexor. Dr. Dickson reported a consultant or adviser role with Celgene and research funding from Eli Lilly.
Source: Gounder M et al. ASCO 2018, Abstract 11512.
Chromoplexy linked to aggressive Ewing sarcomas
Chromoplexy, a sudden burst of complex, loop-like gene rearrangements that gives rise to a fusion gene, appears to be associated with aggressive Ewing sarcomas, based on a study of 124 tumors reported in Science.
Ewing sarcomas with complex karyotypes are associated with a poorer prognosis compared with those with simpler karyotypes. The new findings show that these complex karyotypes are the product of chromoplexy, and that chromoplexy-generated fusions arise early, giving rise to both primary and relapse Ewing sarcoma tumors, which can continue to evolve in parallel.
Analysis of the sequence context surrounding chromoplexy breaks may provide clues and potentially point to a therapeutic vulnerability that could be used to treat Ewing sarcomas. Further, given the preference of chromoplexy events for transcriptionally active regions, Ewing sarcomas arising from chromoplexy may be responsive to immune checkpoint inhibition.
In a study of the whole genomes of 124 Ewing sarcomas, chromoplexy rather than simple reciprocal translocations defined the gene fusions seen in 52 tumors (42%). Ewing sarcoma involves fusions between EWSR1, a gene encoding an RNA binding protein, and E26 transformation-specific (ETS) transcription factors.
“Our analyses reveal rearrangement bursts (chromoplectic loops) as a source of gene fusion in human bone and soft tissue tumors. Ewing sarcomas with complex karyotypes are associated with a poorer prognosis than those with simpler karyotypes, and here we show chromoplexy as the mechanism in 42% of tumors. It is possible that the chromoplectic tumor’s additional gene disruptions and fusions contribute to the difference in patient survival,” wrote Nathaniel D. Anderson of the Hospital for Sick Children, Toronto, and the University of Toronto, and his colleagues.
Standard reciprocal translocations involve DNA breaks in two fusion partners. Chromoplexy involves three or more breakpoints in the genome. A loop pattern emerges as these three or more broken chromosome ends are forced to find a new partner. The result is the formation of functional EWSR1-FLI1 or EWSR1-ERG fusions that, upon expression, provide a selective growth or survival advantage
The researchers found that the loop rearrangements always contained the disease-defining fusion at the center, but they disrupted multiple additional genes. The loops occurred preferentially in early replicating and transcriptionally active genomic regions.
They found similar loops forming canonical fusions in three other sarcoma types.
“Our whole-genome sequence data support a model in which there is an early clone of (Ewing sarcoma), containing EWSR1-ETS and chromoplexy, arising at least 1 year before diagnosis, which gives rise to both the primary and metastatic or relapse tumors. Whether the bursts ... are chance events or driven by specific mutational processes, akin to the RAG machinery operative in leukemia, remains to be established. As an increasing and diverse number of tumor genome sequences become available, we may be able to define further rearrangement processes that underlie fusion genes and thus unravel the causes of fusion-driven human cancers,” the researchers wrote.
The clinical features and demographics of the study patients were typical of Ewing sarcoma patients. Average patient age at diagnosis was 14.8 years (2.8 to 36.6 years); the male to female ratio was 1.38:1; and 14 patients had relapsed, with 13 having died from their disease.
About half of fusions between the EWS RNA binding protein 1 (EWSR1) gene on chromosome 22 and an E26 transformation-specific (ETS) family transcription factor gene, either FLI1 at 11q24 or ERG at 21q11 arose via chromoplexy.
SOURCE: Anderson et al. Science 2018 Aug 31. doi: 10.1126/science.aam8419.
The contribution of genetic analysis to the current standard of care for Ewing sarcoma is limited to confirmation of the diagnostic EWSR1-FLI1 or EWSR1-ERG fusions. The discovery of genomic patterns associated with subsets of Ewing sarcomas raises the question of whether additional molecular diagnostic modalities are warranted. If chromoplexy events are important clinical biomarkers for disease aggressiveness in this tumor, as the authors suggest, their findings may support a new indication for clinical whole genome sequencing.
Analysis of additional patient samples will be needed, however, to confirm that the presence of chromoplexy is an independent prognostic predictor in Ewing sarcoma. This is because the researchers find that chromoplexy-driven Ewing sarcoma more likely contains tumor protein 53 (TP53) mutations. Because TP53 and stromal antigen 2 (STAG2) mutations and genomic complexity have each been associated with more aggressive Ewing sarcoma, dissecting the contribution of these factors to poor clinical outcomes in chromoplexy-derived Ewing sarcoma will be an important area of future work.
More generally, the study has important clinical implications for the genomic diagnosis of these and other cancers, as well as the expanding biological role of complex rearrangements in cancer evolution.
Could chromoplexy events in Ewing sarcoma be linked, for example, to the activity of an aberrantly expressed endogenous transposase such as PiggyBac transposase 5 (PGBD5), which was recently implicated in the genesis of the pathogenic gene rearrangements in childhood malignant rhabdoid tumors? An alternative possibility is a constitutional or acquired DNA repair defect (Science 2018 Aug 31. doi: 10.1126/science.aau8231).
Marcin Imielinski is with the Meyer Cancer Center, Cornell University, and the New York Genome Center, New York. Marc Ladanyi is with Memorial Sloan Kettering Cancer Center, New York. They made their remarks in an editorial in Science that accompanied the study.
The contribution of genetic analysis to the current standard of care for Ewing sarcoma is limited to confirmation of the diagnostic EWSR1-FLI1 or EWSR1-ERG fusions. The discovery of genomic patterns associated with subsets of Ewing sarcomas raises the question of whether additional molecular diagnostic modalities are warranted. If chromoplexy events are important clinical biomarkers for disease aggressiveness in this tumor, as the authors suggest, their findings may support a new indication for clinical whole genome sequencing.
Analysis of additional patient samples will be needed, however, to confirm that the presence of chromoplexy is an independent prognostic predictor in Ewing sarcoma. This is because the researchers find that chromoplexy-driven Ewing sarcoma more likely contains tumor protein 53 (TP53) mutations. Because TP53 and stromal antigen 2 (STAG2) mutations and genomic complexity have each been associated with more aggressive Ewing sarcoma, dissecting the contribution of these factors to poor clinical outcomes in chromoplexy-derived Ewing sarcoma will be an important area of future work.
More generally, the study has important clinical implications for the genomic diagnosis of these and other cancers, as well as the expanding biological role of complex rearrangements in cancer evolution.
Could chromoplexy events in Ewing sarcoma be linked, for example, to the activity of an aberrantly expressed endogenous transposase such as PiggyBac transposase 5 (PGBD5), which was recently implicated in the genesis of the pathogenic gene rearrangements in childhood malignant rhabdoid tumors? An alternative possibility is a constitutional or acquired DNA repair defect (Science 2018 Aug 31. doi: 10.1126/science.aau8231).
Marcin Imielinski is with the Meyer Cancer Center, Cornell University, and the New York Genome Center, New York. Marc Ladanyi is with Memorial Sloan Kettering Cancer Center, New York. They made their remarks in an editorial in Science that accompanied the study.
The contribution of genetic analysis to the current standard of care for Ewing sarcoma is limited to confirmation of the diagnostic EWSR1-FLI1 or EWSR1-ERG fusions. The discovery of genomic patterns associated with subsets of Ewing sarcomas raises the question of whether additional molecular diagnostic modalities are warranted. If chromoplexy events are important clinical biomarkers for disease aggressiveness in this tumor, as the authors suggest, their findings may support a new indication for clinical whole genome sequencing.
Analysis of additional patient samples will be needed, however, to confirm that the presence of chromoplexy is an independent prognostic predictor in Ewing sarcoma. This is because the researchers find that chromoplexy-driven Ewing sarcoma more likely contains tumor protein 53 (TP53) mutations. Because TP53 and stromal antigen 2 (STAG2) mutations and genomic complexity have each been associated with more aggressive Ewing sarcoma, dissecting the contribution of these factors to poor clinical outcomes in chromoplexy-derived Ewing sarcoma will be an important area of future work.
More generally, the study has important clinical implications for the genomic diagnosis of these and other cancers, as well as the expanding biological role of complex rearrangements in cancer evolution.
Could chromoplexy events in Ewing sarcoma be linked, for example, to the activity of an aberrantly expressed endogenous transposase such as PiggyBac transposase 5 (PGBD5), which was recently implicated in the genesis of the pathogenic gene rearrangements in childhood malignant rhabdoid tumors? An alternative possibility is a constitutional or acquired DNA repair defect (Science 2018 Aug 31. doi: 10.1126/science.aau8231).
Marcin Imielinski is with the Meyer Cancer Center, Cornell University, and the New York Genome Center, New York. Marc Ladanyi is with Memorial Sloan Kettering Cancer Center, New York. They made their remarks in an editorial in Science that accompanied the study.
Chromoplexy, a sudden burst of complex, loop-like gene rearrangements that gives rise to a fusion gene, appears to be associated with aggressive Ewing sarcomas, based on a study of 124 tumors reported in Science.
Ewing sarcomas with complex karyotypes are associated with a poorer prognosis compared with those with simpler karyotypes. The new findings show that these complex karyotypes are the product of chromoplexy, and that chromoplexy-generated fusions arise early, giving rise to both primary and relapse Ewing sarcoma tumors, which can continue to evolve in parallel.
Analysis of the sequence context surrounding chromoplexy breaks may provide clues and potentially point to a therapeutic vulnerability that could be used to treat Ewing sarcomas. Further, given the preference of chromoplexy events for transcriptionally active regions, Ewing sarcomas arising from chromoplexy may be responsive to immune checkpoint inhibition.
In a study of the whole genomes of 124 Ewing sarcomas, chromoplexy rather than simple reciprocal translocations defined the gene fusions seen in 52 tumors (42%). Ewing sarcoma involves fusions between EWSR1, a gene encoding an RNA binding protein, and E26 transformation-specific (ETS) transcription factors.
“Our analyses reveal rearrangement bursts (chromoplectic loops) as a source of gene fusion in human bone and soft tissue tumors. Ewing sarcomas with complex karyotypes are associated with a poorer prognosis than those with simpler karyotypes, and here we show chromoplexy as the mechanism in 42% of tumors. It is possible that the chromoplectic tumor’s additional gene disruptions and fusions contribute to the difference in patient survival,” wrote Nathaniel D. Anderson of the Hospital for Sick Children, Toronto, and the University of Toronto, and his colleagues.
Standard reciprocal translocations involve DNA breaks in two fusion partners. Chromoplexy involves three or more breakpoints in the genome. A loop pattern emerges as these three or more broken chromosome ends are forced to find a new partner. The result is the formation of functional EWSR1-FLI1 or EWSR1-ERG fusions that, upon expression, provide a selective growth or survival advantage
The researchers found that the loop rearrangements always contained the disease-defining fusion at the center, but they disrupted multiple additional genes. The loops occurred preferentially in early replicating and transcriptionally active genomic regions.
They found similar loops forming canonical fusions in three other sarcoma types.
“Our whole-genome sequence data support a model in which there is an early clone of (Ewing sarcoma), containing EWSR1-ETS and chromoplexy, arising at least 1 year before diagnosis, which gives rise to both the primary and metastatic or relapse tumors. Whether the bursts ... are chance events or driven by specific mutational processes, akin to the RAG machinery operative in leukemia, remains to be established. As an increasing and diverse number of tumor genome sequences become available, we may be able to define further rearrangement processes that underlie fusion genes and thus unravel the causes of fusion-driven human cancers,” the researchers wrote.
The clinical features and demographics of the study patients were typical of Ewing sarcoma patients. Average patient age at diagnosis was 14.8 years (2.8 to 36.6 years); the male to female ratio was 1.38:1; and 14 patients had relapsed, with 13 having died from their disease.
About half of fusions between the EWS RNA binding protein 1 (EWSR1) gene on chromosome 22 and an E26 transformation-specific (ETS) family transcription factor gene, either FLI1 at 11q24 or ERG at 21q11 arose via chromoplexy.
SOURCE: Anderson et al. Science 2018 Aug 31. doi: 10.1126/science.aam8419.
Chromoplexy, a sudden burst of complex, loop-like gene rearrangements that gives rise to a fusion gene, appears to be associated with aggressive Ewing sarcomas, based on a study of 124 tumors reported in Science.
Ewing sarcomas with complex karyotypes are associated with a poorer prognosis compared with those with simpler karyotypes. The new findings show that these complex karyotypes are the product of chromoplexy, and that chromoplexy-generated fusions arise early, giving rise to both primary and relapse Ewing sarcoma tumors, which can continue to evolve in parallel.
Analysis of the sequence context surrounding chromoplexy breaks may provide clues and potentially point to a therapeutic vulnerability that could be used to treat Ewing sarcomas. Further, given the preference of chromoplexy events for transcriptionally active regions, Ewing sarcomas arising from chromoplexy may be responsive to immune checkpoint inhibition.
In a study of the whole genomes of 124 Ewing sarcomas, chromoplexy rather than simple reciprocal translocations defined the gene fusions seen in 52 tumors (42%). Ewing sarcoma involves fusions between EWSR1, a gene encoding an RNA binding protein, and E26 transformation-specific (ETS) transcription factors.
“Our analyses reveal rearrangement bursts (chromoplectic loops) as a source of gene fusion in human bone and soft tissue tumors. Ewing sarcomas with complex karyotypes are associated with a poorer prognosis than those with simpler karyotypes, and here we show chromoplexy as the mechanism in 42% of tumors. It is possible that the chromoplectic tumor’s additional gene disruptions and fusions contribute to the difference in patient survival,” wrote Nathaniel D. Anderson of the Hospital for Sick Children, Toronto, and the University of Toronto, and his colleagues.
Standard reciprocal translocations involve DNA breaks in two fusion partners. Chromoplexy involves three or more breakpoints in the genome. A loop pattern emerges as these three or more broken chromosome ends are forced to find a new partner. The result is the formation of functional EWSR1-FLI1 or EWSR1-ERG fusions that, upon expression, provide a selective growth or survival advantage
The researchers found that the loop rearrangements always contained the disease-defining fusion at the center, but they disrupted multiple additional genes. The loops occurred preferentially in early replicating and transcriptionally active genomic regions.
They found similar loops forming canonical fusions in three other sarcoma types.
“Our whole-genome sequence data support a model in which there is an early clone of (Ewing sarcoma), containing EWSR1-ETS and chromoplexy, arising at least 1 year before diagnosis, which gives rise to both the primary and metastatic or relapse tumors. Whether the bursts ... are chance events or driven by specific mutational processes, akin to the RAG machinery operative in leukemia, remains to be established. As an increasing and diverse number of tumor genome sequences become available, we may be able to define further rearrangement processes that underlie fusion genes and thus unravel the causes of fusion-driven human cancers,” the researchers wrote.
The clinical features and demographics of the study patients were typical of Ewing sarcoma patients. Average patient age at diagnosis was 14.8 years (2.8 to 36.6 years); the male to female ratio was 1.38:1; and 14 patients had relapsed, with 13 having died from their disease.
About half of fusions between the EWS RNA binding protein 1 (EWSR1) gene on chromosome 22 and an E26 transformation-specific (ETS) family transcription factor gene, either FLI1 at 11q24 or ERG at 21q11 arose via chromoplexy.
SOURCE: Anderson et al. Science 2018 Aug 31. doi: 10.1126/science.aam8419.
FROM SCIENCE
Key clinical point: Chromoplexy, a sudden burst of complex, loop-like gene rearrangements that gives rise to a fusion gene, appears to be associated with aggressive Ewing sarcomas.
Major finding: Chromoplexy rather than simple reciprocal translocations defined the gene fusions seen in 42% of Ewing sarcoma tumors.
Study details: A study of the whole genomes of 124 Ewing sarcomas.
Disclosures: This research project was conducted with support from C17 and partially funded by Ewings Cancer Foundation of Canada and Childhood Cancer Canada Foundation. The authors declared no competing interests.
Source: Anderson et al. Science 2018 Aug 31. doi: 10.1126/science.aam8419.
Addressing the rarity and complexities of sarcomas
The rarity and complexities of bone and soft tissue sarcomas pose a major challenge to effective treatment. Historically, there has been a blanket approach to treatment, but more recently that has begun to change thanks to genome profiling studies and novel clinical trial strategies. Here, we discuss the resulting enrichment of the therapeutic armamentarium with molecularly targeted and immune therapies.
A challenging tumor type
Sarcomas are a large group of histologically diverse cancers that arise in the mesenchymal cells. They can be broadly divided into bone and soft tissue sarcomas (STS) but are further subdivided according to the type of cell from which they derive; osteosarcomas in the bone, rhabdomyosarcomas in the skeletal muscle, liposarcomas in the fat tissues, leiomyosarcomas in the smooth muscle, and chondrosarcomas in the cartilaginous tissue, for example.
Each sarcoma subtype itself encompasses a range of different cancers with unique biology. Under the umbrella of liposarcoma, for example, are well/dedifferentiated liposarcomas and myxoid liposarcomas, which have very different pathologies and clinical courses.
As a whole, sarcomas are extremely rare tumors, accounting for less than 1% of all adult cancers, although they disproportionately affect children and young adults, with a prevalence closer to 15%.1,2 Certain sarcoma subtypes are exceptionally rare, with only a few cases diagnosed worldwide each year, whereas liposarcomas are at the other end of the spectrum, comprising the most common form of STS (Figure 1).3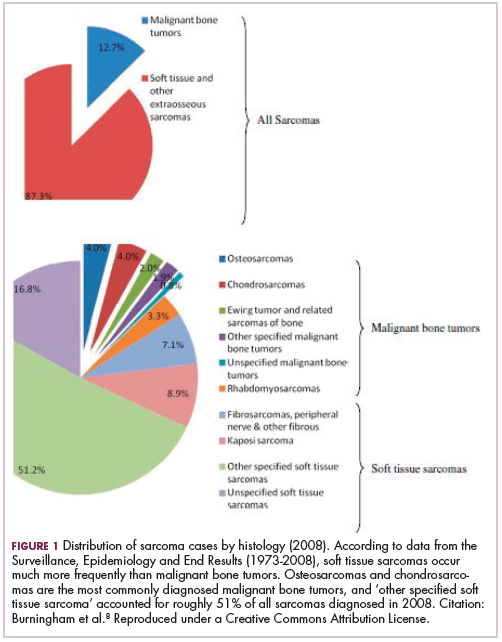
In the early stages, sarcomas are generally highly treatable with a combination of surgical resection, chemotherapy, and radiation therapy. However, many patients develop advanced, metastatic disease, which presents much more of a challenge.4,5
Magic bullet for GIST
Despite their clear heterogeneity and complexity, sarcomas have tended to be treated as a single entity. Chemotherapy has played a central role in the treatment of advanced sarcomas and continues to do so, with 2 newer drugs approved by the United States Food and Drug Administration (FDA) in the past several years.6,7
The development of targeted therapy, on the other hand, for the most part proved unsuccessful. In general, studies examining the somatic mutation landscape in sarcomas found very few that were highly recurrent. The exception was gastrointestinal stromal tumors (GIST), which represent around 8% of STS.8 Frequent mutations in several highly targetable tyrosine kinases, notably KIT, which is mutated in around 85% of cases,9 and platelet-derived growth factor receptor alpha (PDGFRα) were identified in these tumors.10This prompted the development of tyrosine kinase inhibitors (TKIs), targeting these and other kinases, for the treatment of patients with GIST, and culminated in the approval of imatinib for this indication in 2002. This revolutionized the treatment of GIST, which had a poor prognosis and were resistant to chemotherapy, extending median overall survival in patients with metastatic disease almost to 5 years.11-13
Imatinib was also shown to benefit patients with surgically resectable disease and was subsequently approved in the adjuvant setting in 2008. A recent trial demonstrated that 3-year continuation of adjuvant imatinib resulted in a significantly longer progression-free survival (PFS) compared with 1 year of adjuvant imatinib, and even longer time periods are now being evaluated.14,15 The TKIs sunitinib and regorafenib have also been approved for the treatment of patients who become resistant to imatinib.16,17 Avapritinib, a newer, more specific inhibitor of KIT is also being evaluated in patients with GIST (Table).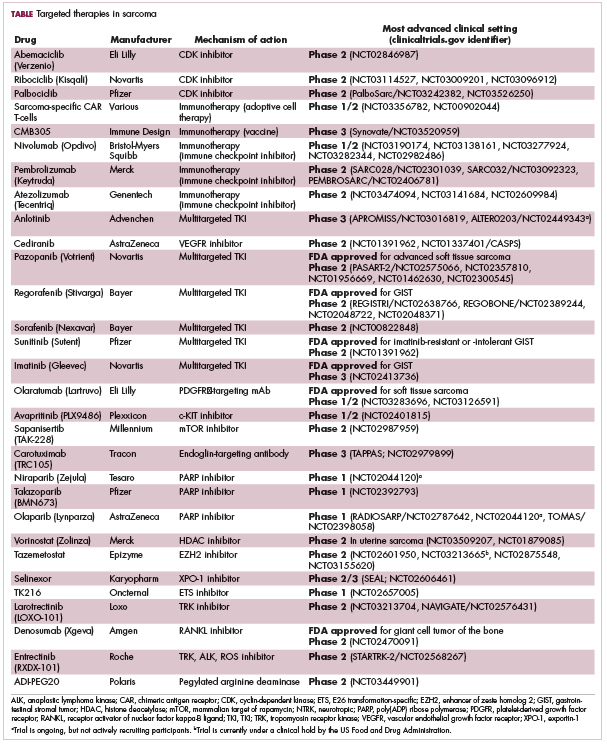
Long-sought success for STS
Sunitinib and regorafenib include PDGFRα and the vascular endothelial growth factor receptors (VEGFRs) among their targets, receptors that play crucial roles in the formation of new blood vessels (angiogenesis). Many types of non-GIST sarcomas have been shown to be highly vascularized and express high levels of both of those receptors and other angiogenic proteins, which sparked interest in the development of multitargeted TKIs and other anti-angiogenic drugs in patients with STS.18
In 2012, pazopanib became the first FDA-approved molecularly targeted therapy for the treatment of non-GIST sarcomas. Approval in the second-line setting was based on the demonstration of a 3-month improvement in PFS compared with placebo.19 Four years later, the monoclonal antibody olaratumab, a more specific inhibitor of PDGFRα, was approved in combination with doxorubicin, marking the first front-line approval for more than 4 decades.20Numerous other anti-angiogenic drugs continue to be evaluated for the treatment of advanced STS. Among them, anlotinib is being tested in phase 3 clinical trials, and results from the ALTER0203 trial were presented at the 2018 annual meeting of the American Society of Clinical Oncology (ASCO).21 After failure of chemotherapy, 223 patients were randomly assigned to receive either anlotinib or placebo. Anlotinib significantly improved median PFS across all patients, compared with placebo (6.27 vs 1.4 months, respectively; hazard ratio [HR], 0.33; P < .0001), but was especially effective in patients with alveolar soft part sarcoma (ASPS; mPFS: 18.2 vs 3 months) and was well tolerated.21
Sarcoma secrets revealed
Advancements in genome sequencing technologies have made it possible to interrogate the molecular underpinnings of sarcomas in greater detail. However, their rarity presents a significant technical challenge, with a dearth of samples available for genomic testing. Large-scale worldwide collaborative efforts have facilitated the collection of sufficiently large patient populations to provide statistically robust data in many cases. The Cancer Genome Atlas has established a rare tumor characterization project to facilitate the genomic sequencing of rare cancer types like sarcomas.
Genome sequencing studies have revealed 2 types of sarcomas: those with relatively stable genomes and few molecular alterations, exemplified by Ewing sarcoma, which has a mutational load of 0.15 mutations/Megabase (Mb); and those that are much more complex with frequent somatic mutations, the prime example being leiomyosarcoma. The latter are characterized by mutations in the TP53 gene, dubbed the “guardian of the genome” for its essential role in genome stability.
The 2 types are likely to require very different therapeutic strategies. Although genomically complex tumors offer up lots of potential targets for therapy, they also display significant heterogeneity and it can be challenging to find a shared target across different tumor samples. The p53 protein would make a logical target but, to date, tumor suppressor proteins are not readily druggable.
The most common type of molecular alterations in sarcomas are chromosomal translocations, where part of a chromosome breaks off and becomes reattached to another chromosome. This can result in the formation of a gene fusion when parts of 2 different genes are brought together in a way in which the genetic code can still be read, leading to the formation of a fusion protein with altered activity.22-25
In sarcomas, these chromosomal translocations predominantly involve genes encoding transcription factors and the gene fusion results in their aberrant expression and activation of the transcriptional programs that they regulate.
Ewing sarcoma is a prime example of a sarcoma that is defined by chromosomal translocations. Most often, the resulting gene fusions occur between members of theten-eleven translocation (TET) family of RNA-binding proteins and the E26 transformation-specific (ETS) family of transcription factors. The most common fusion is between the EWSR1 and FLI1 genes, observed in between 85% and 90% of cases.
Significant efforts have been made to target EWSR1-FLI1. Since direct targeting of transcription factors is challenging, those efforts focused on targeting the aberrant transcriptional programs that they initiate. A major downstream target is the insulin-like growth factor receptor 1 (IGF1R) and numerous IGF1R inhibitors were developed and tested in patients with Ewing sarcoma, but unfortunately success was limited. Attention turned to the mammalian target of rapamycin (mTOR) as a potential mechanism of resistance to IGF1R inhibitors and explanation for the limited responses. Clinical trials combining mTOR and IGF1R inhibitors also proved unsuccessful.26
Although overall these trials were deemed failures, they were notable for the dramatic responses that were seen in 1 or 2 patients. Researchers are probing these “exceptional responses” using novel N-of-1 clinical trial designs that focus on a single patient (Figure 2).27-30 More recently, the first drug to specifically target the EWSR1-FLI1 fusion protein was developed. TK216 binds to the fusion protein and prevents it from binding to RNA helicase A, thereby blocking its function.31
Another type of gene fusion, involving the neurotrophic tropomyosin receptor kinase (NTRK) genes, has recently come into the spotlight for the treatment of lung cancer. According to a recent study, NTRK fusions may also be common in sarcomas. They were observed in 8% of patients with breast sarcomas, 5% with fibrosarcomas, and 5% with stomach or small intestine sarcomas.32
The NTRK genes encode TRK proteins and several small molecule inhibitors of TRK have been developed to treat patients with NTRK fusion-positive cancers. Another novel clinical trial design – the basket trial – is being used to test these inhibitors. This type of trial uses a tumor-agnostic approach, recruiting patients with all different histological subtypes of cancer that are unified by the shared presence of a specific molecular alteration.33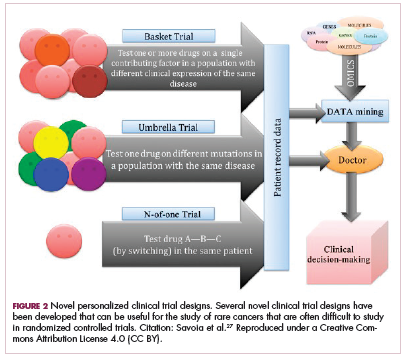
Repurposing gynecologic cancer drugs
More recently, a third group of sarcomas was categorized, with intermediate genomic complexity. These tumors, including well/dedifferentiated liposarcomas, were characterized by amplifications of chromosome 12, involving genes such as cyclin-dependent kinase 4 (CDK4). In fact, more than 90% of patients with well/dedifferentiated sarcomas display CDK4 amplification, making it a logical therapeutic target.36
CDK4 encodes CDK4 protein, a cell cycle-associated protein that regulates the transition from G1-S phase, known as the restriction point, beyond which the cell commits to undergoing mitosis. Aberrant expression of CDK4 in cancer drives the hallmark process of unchecked cellular proliferation.
Some small molecule CDK4/6 inhibitors have been developed and have shown significant promise in the treatment of breast cancer. They are also being evaluatedin patients with sarcoma whose tumors display CDK4 overexpression. In a recently published phase 2 trial of palbociclib in 60 patients with well/dedifferentiated liposarcomas, there was 1 CR.37
Another group of drugs that has advanced the treatment of gynecologic cancers comprises the poly (ADP-ribose) polymerase (PARP) inhibitors. In this context, PARP inhibitors are used in patients with mutations in the breast cancer susceptibility genes, BRCA1/2. The BRCA and PARP proteins are both involved in DNA repair pathways and the inhibition of PARP in patients who already have a defective BRCA pathway renders a lethal double blow to the cancer cell. According to the Broad Institute Cancer Cell Line Encyclopedia, Ewing sarcomas express high levels of the PARP1 enzyme, which could render them sensitive to PARP inhibition. Preclinical studies seemed to confirm that sensitivity, however, so far this has yet to translate into success in clinical trials, with no objective responses observed as yet.38
Expanding the field
Other treatment strategies being tested in patients with sarcoma are moving the field beyond conventional targeted therapies. There has been substantial focus in recent years on epigenetic alterations and their potential role in the development of cancer. Epigenetics is the secondary layer of regulation that acts on the genome and directs the spatial and temporal expression of genes.
Both DNA and the histone proteins they are packaged up with to form chromatin in nondividing cells can be modified by the attachment of chemical groups, such as acetyl and methyl groups, which can alter access to the DNA for transcription.
EZH2 is an enzyme that participates in histone methylation and thereby regulates transcriptional repression. Some types of sarcoma are characterized by a loss of expression of the INI1 gene, also known as SMARCB1. The INI1 protein is part of a chromatin remodeling complex that relieves transcriptional repression and when INI1 is lost, cells become dependent upon EZH2.39Clinical trials of the EZH2 inhibitor tazemetostat are ongoing in several types of sarcoma. Results from a phase 2 study in adults with INI1-negative tumors were presented at ASCO in 2017. Among 31 patients treated with 800 mg tazemetostat in continuous 28-day cycles, mPFS was 5.7 months, disease control rate was 10%, and confirmed overall response rate was 13%. The FDA has granted tazemetostat orphan drug designation in this indication.40A pediatric basket trial of tazemetostat is also ongoing, but the FDA recently placed it under a clinical hold as a result of a safety update from the trial in which a pediatric patient with advanced poorly differentiated chordoma developed a secondary T-cell lymphoma.41
Targeting the unique metabolism of sarcomas may offer a promising therapeutic strategy, although this is in the preliminary stages of evaluation. A recent study showed that the expression of the argininosuccinate synthase 1 enzyme, which is involved in the generation of arginine through the urea cycle, was lost in up to 90% of STS. A pegylated arginine deaminase (ADI-PEG20), is being evaluated in a phase 2 clinical trial.42
Finally, the concept of using immunotherapy to boost the anti-tumor immune response is also being examined in sarcomas. A significant number of cases of STS, osteosarcoma and GIST have been shown to express programmed cell death protein-ligand 1, therefore the use of immune checkpoint inhibitors that block this ligand or its receptor and help to reactive tumor-infiltrating T cells, could be a beneficial strategy.
Limited activity has been observed in studies conducted to date, however combination therapies, especially with inhibitors of the indoleamine 2,3-dioxygenase (IDO) enzyme, which plays a key role in immunosuppression, could help to harness the power of these drugs. Studies have suggested that sarcomas may be infiltrated by immunosuppressive macrophages that express IDO.43
It is generally believed that immunotherapy is most effective in tumors that are highly mutated because that allows a large number of cancer antigens to provoke an anti-tumor immune response. However, a single highly expressed antigen can also be strongly immunogenic. Synovial sarcomas have a relatively low mutational burden but they do express high levels of the cancer testis antigen NY-ESO-1.
NY-ESO-1 has provided a useful target for the development of adoptive cell therapies and vaccines for the treatment of sarcomas. CMB305 is an NY-ESO-1 vaccine that also incorporates a toll-like receptor 4 agonist. It is being evaluated in the phase 3 Synovate study as maintenance monotherapy in patients with locally advanced, unresectable or metastatic synovial sarcoma. In a phase 1 study, at a median follow-up of just under 18 months, the median OS for all 25 patients was 23.7 months.44
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
2. Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer. 2006;119(12):2922-2930.
3. Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012;2(1):14.
4. Italiano A, Mathoulin-Pelissier S, Cesne AL, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049-1054.
5. Savina M, Le Cesne A, Blay JY, et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: the METASARC observational study. BMC Med. 2017;15(1):78.
6. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34(8):786-793.
7. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387(10028):1629-1637.
8. Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg. 2014;260(3):416-421; discussion 421-412.
9. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342-4349.
10. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708-710.
11. Dagher R, Cohen M, Williams G, et al. Approval summary. Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034-3038.
12. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626-632.
13. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127-1134.
14. Zhao R, Wang Y, Huang Y, et al. Adjuvant imatinib for patients with high-risk gastrointestinal stromal tumors: a retrospective cohort study. Scientific Reports. 2017;7:16834.
15. Raut C, Espat N, Maki R, Araujo D, Williams T, Wolff J. Extended treatment with adjuvant imatinib (IM) for patients (pts) with high-risk primary gastrointestinal stromal tumor (GIST): The PERSIST-5 study. J Clin Oncol. 2017;35(15_suppl):11009.
16. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302.
17. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-1338.
18. Versleijen-Jonkers YM, Vlenterie M, van de Luijtgaarden AC, van der Graaf WT. Anti-angiogenic therapy, a new player in the field of sarcoma treatment. Crit Rev Oncol Hematol. 2014;91(2):172-185.
19. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879-1886.
20. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
21. Chi Y, Yao Y, Wang S, et al. Anlotinib for metastatic soft tissue sarcoma: A randomized, double-blind, placebo-controlled and multi-centered clinical trial. J Clin Oncol. 2018;36(suppl):abstr 11503.
22. Brohl AS, Shah HR, Wang Y-C, Kasarskis A, Maki RG. The somatic mutational landscape in soft tissue sarcoma: Early results from TCGA data. J Clin Oncol. 2015;33(15_suppl):10508-10508.
23. Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326-1341.
24. Jour G, Scarborough JD, Jones RL, et al. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Hum Pathol. 2014;45(8):1563-1571.
25. Yang J-L. Investigation of osteosarcoma genomics and its impact on targeted therapy: an international collaboration to conquer human osteosarcoma. Chin J Cancer. 2014;33(12):575-580.
26. Cidre-Aranaz F, Alonso J. EWS/FLI1 target genes and therapeutic opportunities in Ewing sarcoma. Front Oncol. 2015;5:162.
27. Savoia C, Volpe M, Grassi G, Borghi C, Agabiti Rosei E, Touyz RM. Personalized medicine-a modern approach for the diagnosis and management of hypertension. Clin Sci (Lond). 2017;131(22):2671-2685.
28. Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop. 2016;7(9):527-538.
29. van Maldegem AM, Bovée JVMG, Peterse EFP, Hogendoorn PCW, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171-180.
30. Subbiah V, Hess KR, Khawaja MR, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016;6:35448.
31. Jessen K, Moseley E, Chung EYL, et al. TK216, a novel, small molecule inhibitor of the ETS-family of transcription factors, displays anti-tumor activity in AML and DLBCL. Blood. 2016;128(22):4035-4035.
32. Sankhala K, Potts S, Christiansen J, et al. Immunohistochemistry screening to increase the efficacy of next-generation sequencing for detection of NTRK, ROS1, and ALK gene rearrangements (fusions) in sarcoma patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 9-12, 2016, 2016; Lisbon, Portugal.
33. Renfro LA, An MW, Mandrekar SJ. Precision oncology: a new era of cancer clinical trials. Cancer Lett. 2017;387:121-126.
34. DuBois S, Laetsch T, Federman N, et al. The use of larotrectinib in the management of locally advanced pediatric NTRK-fusion sarcoma. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
35. Multani P, Manavel E, Hornby Z. Preliminary evidence of clinical response to entrectinib in three sarcome patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
36. Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42(8):715-721.
37. Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol. 2016;2(7):937-940.
38. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603-607.
39. Kenichi K, Yoshinao O. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Science. 2017;108(4):547-552.
40. US Food and Drug Administration. Orphan drug designations and approvals. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=544416. Designated date September 28, 2017. Accessed July 4, 2018.
41. Press release. Epizyme provides update regarding tazemetostat clinical program. https://globenewswire.com/news-release/2018/04/23/1485765/0/en/Epizyme-Provides-Update-Regarding-Tazemetostat-Clinical-Program.html. Released April 23, 2018. Accessed July 4, 2018.
42. Bean GR, Kremer JC, Prudner BC, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death &Amp; Disease. 2016;7:e2406.
43. Bourcier K, Italiano A. Newer therapeutic strategies for soft-tissue sarcomas. Pharmacol Ther. 2018;188:118-123.
44. Somaiah N, Chawla SP, Block MS, et al. Immune response, safety, and survival impact from CMB305 in NY-ESO-1+ recurrent soft tissue sarcomas (STS). J Clin Oncol. 2017;35(15_suppl):11006-11006.
The rarity and complexities of bone and soft tissue sarcomas pose a major challenge to effective treatment. Historically, there has been a blanket approach to treatment, but more recently that has begun to change thanks to genome profiling studies and novel clinical trial strategies. Here, we discuss the resulting enrichment of the therapeutic armamentarium with molecularly targeted and immune therapies.
A challenging tumor type
Sarcomas are a large group of histologically diverse cancers that arise in the mesenchymal cells. They can be broadly divided into bone and soft tissue sarcomas (STS) but are further subdivided according to the type of cell from which they derive; osteosarcomas in the bone, rhabdomyosarcomas in the skeletal muscle, liposarcomas in the fat tissues, leiomyosarcomas in the smooth muscle, and chondrosarcomas in the cartilaginous tissue, for example.
Each sarcoma subtype itself encompasses a range of different cancers with unique biology. Under the umbrella of liposarcoma, for example, are well/dedifferentiated liposarcomas and myxoid liposarcomas, which have very different pathologies and clinical courses.
As a whole, sarcomas are extremely rare tumors, accounting for less than 1% of all adult cancers, although they disproportionately affect children and young adults, with a prevalence closer to 15%.1,2 Certain sarcoma subtypes are exceptionally rare, with only a few cases diagnosed worldwide each year, whereas liposarcomas are at the other end of the spectrum, comprising the most common form of STS (Figure 1).3
In the early stages, sarcomas are generally highly treatable with a combination of surgical resection, chemotherapy, and radiation therapy. However, many patients develop advanced, metastatic disease, which presents much more of a challenge.4,5
Magic bullet for GIST
Despite their clear heterogeneity and complexity, sarcomas have tended to be treated as a single entity. Chemotherapy has played a central role in the treatment of advanced sarcomas and continues to do so, with 2 newer drugs approved by the United States Food and Drug Administration (FDA) in the past several years.6,7
The development of targeted therapy, on the other hand, for the most part proved unsuccessful. In general, studies examining the somatic mutation landscape in sarcomas found very few that were highly recurrent. The exception was gastrointestinal stromal tumors (GIST), which represent around 8% of STS.8 Frequent mutations in several highly targetable tyrosine kinases, notably KIT, which is mutated in around 85% of cases,9 and platelet-derived growth factor receptor alpha (PDGFRα) were identified in these tumors.10This prompted the development of tyrosine kinase inhibitors (TKIs), targeting these and other kinases, for the treatment of patients with GIST, and culminated in the approval of imatinib for this indication in 2002. This revolutionized the treatment of GIST, which had a poor prognosis and were resistant to chemotherapy, extending median overall survival in patients with metastatic disease almost to 5 years.11-13
Imatinib was also shown to benefit patients with surgically resectable disease and was subsequently approved in the adjuvant setting in 2008. A recent trial demonstrated that 3-year continuation of adjuvant imatinib resulted in a significantly longer progression-free survival (PFS) compared with 1 year of adjuvant imatinib, and even longer time periods are now being evaluated.14,15 The TKIs sunitinib and regorafenib have also been approved for the treatment of patients who become resistant to imatinib.16,17 Avapritinib, a newer, more specific inhibitor of KIT is also being evaluated in patients with GIST (Table).
Long-sought success for STS
Sunitinib and regorafenib include PDGFRα and the vascular endothelial growth factor receptors (VEGFRs) among their targets, receptors that play crucial roles in the formation of new blood vessels (angiogenesis). Many types of non-GIST sarcomas have been shown to be highly vascularized and express high levels of both of those receptors and other angiogenic proteins, which sparked interest in the development of multitargeted TKIs and other anti-angiogenic drugs in patients with STS.18
In 2012, pazopanib became the first FDA-approved molecularly targeted therapy for the treatment of non-GIST sarcomas. Approval in the second-line setting was based on the demonstration of a 3-month improvement in PFS compared with placebo.19 Four years later, the monoclonal antibody olaratumab, a more specific inhibitor of PDGFRα, was approved in combination with doxorubicin, marking the first front-line approval for more than 4 decades.20Numerous other anti-angiogenic drugs continue to be evaluated for the treatment of advanced STS. Among them, anlotinib is being tested in phase 3 clinical trials, and results from the ALTER0203 trial were presented at the 2018 annual meeting of the American Society of Clinical Oncology (ASCO).21 After failure of chemotherapy, 223 patients were randomly assigned to receive either anlotinib or placebo. Anlotinib significantly improved median PFS across all patients, compared with placebo (6.27 vs 1.4 months, respectively; hazard ratio [HR], 0.33; P < .0001), but was especially effective in patients with alveolar soft part sarcoma (ASPS; mPFS: 18.2 vs 3 months) and was well tolerated.21
Sarcoma secrets revealed
Advancements in genome sequencing technologies have made it possible to interrogate the molecular underpinnings of sarcomas in greater detail. However, their rarity presents a significant technical challenge, with a dearth of samples available for genomic testing. Large-scale worldwide collaborative efforts have facilitated the collection of sufficiently large patient populations to provide statistically robust data in many cases. The Cancer Genome Atlas has established a rare tumor characterization project to facilitate the genomic sequencing of rare cancer types like sarcomas.
Genome sequencing studies have revealed 2 types of sarcomas: those with relatively stable genomes and few molecular alterations, exemplified by Ewing sarcoma, which has a mutational load of 0.15 mutations/Megabase (Mb); and those that are much more complex with frequent somatic mutations, the prime example being leiomyosarcoma. The latter are characterized by mutations in the TP53 gene, dubbed the “guardian of the genome” for its essential role in genome stability.
The 2 types are likely to require very different therapeutic strategies. Although genomically complex tumors offer up lots of potential targets for therapy, they also display significant heterogeneity and it can be challenging to find a shared target across different tumor samples. The p53 protein would make a logical target but, to date, tumor suppressor proteins are not readily druggable.
The most common type of molecular alterations in sarcomas are chromosomal translocations, where part of a chromosome breaks off and becomes reattached to another chromosome. This can result in the formation of a gene fusion when parts of 2 different genes are brought together in a way in which the genetic code can still be read, leading to the formation of a fusion protein with altered activity.22-25
In sarcomas, these chromosomal translocations predominantly involve genes encoding transcription factors and the gene fusion results in their aberrant expression and activation of the transcriptional programs that they regulate.
Ewing sarcoma is a prime example of a sarcoma that is defined by chromosomal translocations. Most often, the resulting gene fusions occur between members of theten-eleven translocation (TET) family of RNA-binding proteins and the E26 transformation-specific (ETS) family of transcription factors. The most common fusion is between the EWSR1 and FLI1 genes, observed in between 85% and 90% of cases.
Significant efforts have been made to target EWSR1-FLI1. Since direct targeting of transcription factors is challenging, those efforts focused on targeting the aberrant transcriptional programs that they initiate. A major downstream target is the insulin-like growth factor receptor 1 (IGF1R) and numerous IGF1R inhibitors were developed and tested in patients with Ewing sarcoma, but unfortunately success was limited. Attention turned to the mammalian target of rapamycin (mTOR) as a potential mechanism of resistance to IGF1R inhibitors and explanation for the limited responses. Clinical trials combining mTOR and IGF1R inhibitors also proved unsuccessful.26
Although overall these trials were deemed failures, they were notable for the dramatic responses that were seen in 1 or 2 patients. Researchers are probing these “exceptional responses” using novel N-of-1 clinical trial designs that focus on a single patient (Figure 2).27-30 More recently, the first drug to specifically target the EWSR1-FLI1 fusion protein was developed. TK216 binds to the fusion protein and prevents it from binding to RNA helicase A, thereby blocking its function.31
Another type of gene fusion, involving the neurotrophic tropomyosin receptor kinase (NTRK) genes, has recently come into the spotlight for the treatment of lung cancer. According to a recent study, NTRK fusions may also be common in sarcomas. They were observed in 8% of patients with breast sarcomas, 5% with fibrosarcomas, and 5% with stomach or small intestine sarcomas.32
The NTRK genes encode TRK proteins and several small molecule inhibitors of TRK have been developed to treat patients with NTRK fusion-positive cancers. Another novel clinical trial design – the basket trial – is being used to test these inhibitors. This type of trial uses a tumor-agnostic approach, recruiting patients with all different histological subtypes of cancer that are unified by the shared presence of a specific molecular alteration.33
Repurposing gynecologic cancer drugs
More recently, a third group of sarcomas was categorized, with intermediate genomic complexity. These tumors, including well/dedifferentiated liposarcomas, were characterized by amplifications of chromosome 12, involving genes such as cyclin-dependent kinase 4 (CDK4). In fact, more than 90% of patients with well/dedifferentiated sarcomas display CDK4 amplification, making it a logical therapeutic target.36
CDK4 encodes CDK4 protein, a cell cycle-associated protein that regulates the transition from G1-S phase, known as the restriction point, beyond which the cell commits to undergoing mitosis. Aberrant expression of CDK4 in cancer drives the hallmark process of unchecked cellular proliferation.
Some small molecule CDK4/6 inhibitors have been developed and have shown significant promise in the treatment of breast cancer. They are also being evaluatedin patients with sarcoma whose tumors display CDK4 overexpression. In a recently published phase 2 trial of palbociclib in 60 patients with well/dedifferentiated liposarcomas, there was 1 CR.37
Another group of drugs that has advanced the treatment of gynecologic cancers comprises the poly (ADP-ribose) polymerase (PARP) inhibitors. In this context, PARP inhibitors are used in patients with mutations in the breast cancer susceptibility genes, BRCA1/2. The BRCA and PARP proteins are both involved in DNA repair pathways and the inhibition of PARP in patients who already have a defective BRCA pathway renders a lethal double blow to the cancer cell. According to the Broad Institute Cancer Cell Line Encyclopedia, Ewing sarcomas express high levels of the PARP1 enzyme, which could render them sensitive to PARP inhibition. Preclinical studies seemed to confirm that sensitivity, however, so far this has yet to translate into success in clinical trials, with no objective responses observed as yet.38
Expanding the field
Other treatment strategies being tested in patients with sarcoma are moving the field beyond conventional targeted therapies. There has been substantial focus in recent years on epigenetic alterations and their potential role in the development of cancer. Epigenetics is the secondary layer of regulation that acts on the genome and directs the spatial and temporal expression of genes.
Both DNA and the histone proteins they are packaged up with to form chromatin in nondividing cells can be modified by the attachment of chemical groups, such as acetyl and methyl groups, which can alter access to the DNA for transcription.
EZH2 is an enzyme that participates in histone methylation and thereby regulates transcriptional repression. Some types of sarcoma are characterized by a loss of expression of the INI1 gene, also known as SMARCB1. The INI1 protein is part of a chromatin remodeling complex that relieves transcriptional repression and when INI1 is lost, cells become dependent upon EZH2.39Clinical trials of the EZH2 inhibitor tazemetostat are ongoing in several types of sarcoma. Results from a phase 2 study in adults with INI1-negative tumors were presented at ASCO in 2017. Among 31 patients treated with 800 mg tazemetostat in continuous 28-day cycles, mPFS was 5.7 months, disease control rate was 10%, and confirmed overall response rate was 13%. The FDA has granted tazemetostat orphan drug designation in this indication.40A pediatric basket trial of tazemetostat is also ongoing, but the FDA recently placed it under a clinical hold as a result of a safety update from the trial in which a pediatric patient with advanced poorly differentiated chordoma developed a secondary T-cell lymphoma.41
Targeting the unique metabolism of sarcomas may offer a promising therapeutic strategy, although this is in the preliminary stages of evaluation. A recent study showed that the expression of the argininosuccinate synthase 1 enzyme, which is involved in the generation of arginine through the urea cycle, was lost in up to 90% of STS. A pegylated arginine deaminase (ADI-PEG20), is being evaluated in a phase 2 clinical trial.42
Finally, the concept of using immunotherapy to boost the anti-tumor immune response is also being examined in sarcomas. A significant number of cases of STS, osteosarcoma and GIST have been shown to express programmed cell death protein-ligand 1, therefore the use of immune checkpoint inhibitors that block this ligand or its receptor and help to reactive tumor-infiltrating T cells, could be a beneficial strategy.
Limited activity has been observed in studies conducted to date, however combination therapies, especially with inhibitors of the indoleamine 2,3-dioxygenase (IDO) enzyme, which plays a key role in immunosuppression, could help to harness the power of these drugs. Studies have suggested that sarcomas may be infiltrated by immunosuppressive macrophages that express IDO.43
It is generally believed that immunotherapy is most effective in tumors that are highly mutated because that allows a large number of cancer antigens to provoke an anti-tumor immune response. However, a single highly expressed antigen can also be strongly immunogenic. Synovial sarcomas have a relatively low mutational burden but they do express high levels of the cancer testis antigen NY-ESO-1.
NY-ESO-1 has provided a useful target for the development of adoptive cell therapies and vaccines for the treatment of sarcomas. CMB305 is an NY-ESO-1 vaccine that also incorporates a toll-like receptor 4 agonist. It is being evaluated in the phase 3 Synovate study as maintenance monotherapy in patients with locally advanced, unresectable or metastatic synovial sarcoma. In a phase 1 study, at a median follow-up of just under 18 months, the median OS for all 25 patients was 23.7 months.44
The rarity and complexities of bone and soft tissue sarcomas pose a major challenge to effective treatment. Historically, there has been a blanket approach to treatment, but more recently that has begun to change thanks to genome profiling studies and novel clinical trial strategies. Here, we discuss the resulting enrichment of the therapeutic armamentarium with molecularly targeted and immune therapies.
A challenging tumor type
Sarcomas are a large group of histologically diverse cancers that arise in the mesenchymal cells. They can be broadly divided into bone and soft tissue sarcomas (STS) but are further subdivided according to the type of cell from which they derive; osteosarcomas in the bone, rhabdomyosarcomas in the skeletal muscle, liposarcomas in the fat tissues, leiomyosarcomas in the smooth muscle, and chondrosarcomas in the cartilaginous tissue, for example.
Each sarcoma subtype itself encompasses a range of different cancers with unique biology. Under the umbrella of liposarcoma, for example, are well/dedifferentiated liposarcomas and myxoid liposarcomas, which have very different pathologies and clinical courses.
As a whole, sarcomas are extremely rare tumors, accounting for less than 1% of all adult cancers, although they disproportionately affect children and young adults, with a prevalence closer to 15%.1,2 Certain sarcoma subtypes are exceptionally rare, with only a few cases diagnosed worldwide each year, whereas liposarcomas are at the other end of the spectrum, comprising the most common form of STS (Figure 1).3
In the early stages, sarcomas are generally highly treatable with a combination of surgical resection, chemotherapy, and radiation therapy. However, many patients develop advanced, metastatic disease, which presents much more of a challenge.4,5
Magic bullet for GIST
Despite their clear heterogeneity and complexity, sarcomas have tended to be treated as a single entity. Chemotherapy has played a central role in the treatment of advanced sarcomas and continues to do so, with 2 newer drugs approved by the United States Food and Drug Administration (FDA) in the past several years.6,7
The development of targeted therapy, on the other hand, for the most part proved unsuccessful. In general, studies examining the somatic mutation landscape in sarcomas found very few that were highly recurrent. The exception was gastrointestinal stromal tumors (GIST), which represent around 8% of STS.8 Frequent mutations in several highly targetable tyrosine kinases, notably KIT, which is mutated in around 85% of cases,9 and platelet-derived growth factor receptor alpha (PDGFRα) were identified in these tumors.10This prompted the development of tyrosine kinase inhibitors (TKIs), targeting these and other kinases, for the treatment of patients with GIST, and culminated in the approval of imatinib for this indication in 2002. This revolutionized the treatment of GIST, which had a poor prognosis and were resistant to chemotherapy, extending median overall survival in patients with metastatic disease almost to 5 years.11-13
Imatinib was also shown to benefit patients with surgically resectable disease and was subsequently approved in the adjuvant setting in 2008. A recent trial demonstrated that 3-year continuation of adjuvant imatinib resulted in a significantly longer progression-free survival (PFS) compared with 1 year of adjuvant imatinib, and even longer time periods are now being evaluated.14,15 The TKIs sunitinib and regorafenib have also been approved for the treatment of patients who become resistant to imatinib.16,17 Avapritinib, a newer, more specific inhibitor of KIT is also being evaluated in patients with GIST (Table).
Long-sought success for STS
Sunitinib and regorafenib include PDGFRα and the vascular endothelial growth factor receptors (VEGFRs) among their targets, receptors that play crucial roles in the formation of new blood vessels (angiogenesis). Many types of non-GIST sarcomas have been shown to be highly vascularized and express high levels of both of those receptors and other angiogenic proteins, which sparked interest in the development of multitargeted TKIs and other anti-angiogenic drugs in patients with STS.18
In 2012, pazopanib became the first FDA-approved molecularly targeted therapy for the treatment of non-GIST sarcomas. Approval in the second-line setting was based on the demonstration of a 3-month improvement in PFS compared with placebo.19 Four years later, the monoclonal antibody olaratumab, a more specific inhibitor of PDGFRα, was approved in combination with doxorubicin, marking the first front-line approval for more than 4 decades.20Numerous other anti-angiogenic drugs continue to be evaluated for the treatment of advanced STS. Among them, anlotinib is being tested in phase 3 clinical trials, and results from the ALTER0203 trial were presented at the 2018 annual meeting of the American Society of Clinical Oncology (ASCO).21 After failure of chemotherapy, 223 patients were randomly assigned to receive either anlotinib or placebo. Anlotinib significantly improved median PFS across all patients, compared with placebo (6.27 vs 1.4 months, respectively; hazard ratio [HR], 0.33; P < .0001), but was especially effective in patients with alveolar soft part sarcoma (ASPS; mPFS: 18.2 vs 3 months) and was well tolerated.21
Sarcoma secrets revealed
Advancements in genome sequencing technologies have made it possible to interrogate the molecular underpinnings of sarcomas in greater detail. However, their rarity presents a significant technical challenge, with a dearth of samples available for genomic testing. Large-scale worldwide collaborative efforts have facilitated the collection of sufficiently large patient populations to provide statistically robust data in many cases. The Cancer Genome Atlas has established a rare tumor characterization project to facilitate the genomic sequencing of rare cancer types like sarcomas.
Genome sequencing studies have revealed 2 types of sarcomas: those with relatively stable genomes and few molecular alterations, exemplified by Ewing sarcoma, which has a mutational load of 0.15 mutations/Megabase (Mb); and those that are much more complex with frequent somatic mutations, the prime example being leiomyosarcoma. The latter are characterized by mutations in the TP53 gene, dubbed the “guardian of the genome” for its essential role in genome stability.
The 2 types are likely to require very different therapeutic strategies. Although genomically complex tumors offer up lots of potential targets for therapy, they also display significant heterogeneity and it can be challenging to find a shared target across different tumor samples. The p53 protein would make a logical target but, to date, tumor suppressor proteins are not readily druggable.
The most common type of molecular alterations in sarcomas are chromosomal translocations, where part of a chromosome breaks off and becomes reattached to another chromosome. This can result in the formation of a gene fusion when parts of 2 different genes are brought together in a way in which the genetic code can still be read, leading to the formation of a fusion protein with altered activity.22-25
In sarcomas, these chromosomal translocations predominantly involve genes encoding transcription factors and the gene fusion results in their aberrant expression and activation of the transcriptional programs that they regulate.
Ewing sarcoma is a prime example of a sarcoma that is defined by chromosomal translocations. Most often, the resulting gene fusions occur between members of theten-eleven translocation (TET) family of RNA-binding proteins and the E26 transformation-specific (ETS) family of transcription factors. The most common fusion is between the EWSR1 and FLI1 genes, observed in between 85% and 90% of cases.
Significant efforts have been made to target EWSR1-FLI1. Since direct targeting of transcription factors is challenging, those efforts focused on targeting the aberrant transcriptional programs that they initiate. A major downstream target is the insulin-like growth factor receptor 1 (IGF1R) and numerous IGF1R inhibitors were developed and tested in patients with Ewing sarcoma, but unfortunately success was limited. Attention turned to the mammalian target of rapamycin (mTOR) as a potential mechanism of resistance to IGF1R inhibitors and explanation for the limited responses. Clinical trials combining mTOR and IGF1R inhibitors also proved unsuccessful.26
Although overall these trials were deemed failures, they were notable for the dramatic responses that were seen in 1 or 2 patients. Researchers are probing these “exceptional responses” using novel N-of-1 clinical trial designs that focus on a single patient (Figure 2).27-30 More recently, the first drug to specifically target the EWSR1-FLI1 fusion protein was developed. TK216 binds to the fusion protein and prevents it from binding to RNA helicase A, thereby blocking its function.31
Another type of gene fusion, involving the neurotrophic tropomyosin receptor kinase (NTRK) genes, has recently come into the spotlight for the treatment of lung cancer. According to a recent study, NTRK fusions may also be common in sarcomas. They were observed in 8% of patients with breast sarcomas, 5% with fibrosarcomas, and 5% with stomach or small intestine sarcomas.32
The NTRK genes encode TRK proteins and several small molecule inhibitors of TRK have been developed to treat patients with NTRK fusion-positive cancers. Another novel clinical trial design – the basket trial – is being used to test these inhibitors. This type of trial uses a tumor-agnostic approach, recruiting patients with all different histological subtypes of cancer that are unified by the shared presence of a specific molecular alteration.33
Repurposing gynecologic cancer drugs
More recently, a third group of sarcomas was categorized, with intermediate genomic complexity. These tumors, including well/dedifferentiated liposarcomas, were characterized by amplifications of chromosome 12, involving genes such as cyclin-dependent kinase 4 (CDK4). In fact, more than 90% of patients with well/dedifferentiated sarcomas display CDK4 amplification, making it a logical therapeutic target.36
CDK4 encodes CDK4 protein, a cell cycle-associated protein that regulates the transition from G1-S phase, known as the restriction point, beyond which the cell commits to undergoing mitosis. Aberrant expression of CDK4 in cancer drives the hallmark process of unchecked cellular proliferation.
Some small molecule CDK4/6 inhibitors have been developed and have shown significant promise in the treatment of breast cancer. They are also being evaluatedin patients with sarcoma whose tumors display CDK4 overexpression. In a recently published phase 2 trial of palbociclib in 60 patients with well/dedifferentiated liposarcomas, there was 1 CR.37
Another group of drugs that has advanced the treatment of gynecologic cancers comprises the poly (ADP-ribose) polymerase (PARP) inhibitors. In this context, PARP inhibitors are used in patients with mutations in the breast cancer susceptibility genes, BRCA1/2. The BRCA and PARP proteins are both involved in DNA repair pathways and the inhibition of PARP in patients who already have a defective BRCA pathway renders a lethal double blow to the cancer cell. According to the Broad Institute Cancer Cell Line Encyclopedia, Ewing sarcomas express high levels of the PARP1 enzyme, which could render them sensitive to PARP inhibition. Preclinical studies seemed to confirm that sensitivity, however, so far this has yet to translate into success in clinical trials, with no objective responses observed as yet.38
Expanding the field
Other treatment strategies being tested in patients with sarcoma are moving the field beyond conventional targeted therapies. There has been substantial focus in recent years on epigenetic alterations and their potential role in the development of cancer. Epigenetics is the secondary layer of regulation that acts on the genome and directs the spatial and temporal expression of genes.
Both DNA and the histone proteins they are packaged up with to form chromatin in nondividing cells can be modified by the attachment of chemical groups, such as acetyl and methyl groups, which can alter access to the DNA for transcription.
EZH2 is an enzyme that participates in histone methylation and thereby regulates transcriptional repression. Some types of sarcoma are characterized by a loss of expression of the INI1 gene, also known as SMARCB1. The INI1 protein is part of a chromatin remodeling complex that relieves transcriptional repression and when INI1 is lost, cells become dependent upon EZH2.39Clinical trials of the EZH2 inhibitor tazemetostat are ongoing in several types of sarcoma. Results from a phase 2 study in adults with INI1-negative tumors were presented at ASCO in 2017. Among 31 patients treated with 800 mg tazemetostat in continuous 28-day cycles, mPFS was 5.7 months, disease control rate was 10%, and confirmed overall response rate was 13%. The FDA has granted tazemetostat orphan drug designation in this indication.40A pediatric basket trial of tazemetostat is also ongoing, but the FDA recently placed it under a clinical hold as a result of a safety update from the trial in which a pediatric patient with advanced poorly differentiated chordoma developed a secondary T-cell lymphoma.41
Targeting the unique metabolism of sarcomas may offer a promising therapeutic strategy, although this is in the preliminary stages of evaluation. A recent study showed that the expression of the argininosuccinate synthase 1 enzyme, which is involved in the generation of arginine through the urea cycle, was lost in up to 90% of STS. A pegylated arginine deaminase (ADI-PEG20), is being evaluated in a phase 2 clinical trial.42
Finally, the concept of using immunotherapy to boost the anti-tumor immune response is also being examined in sarcomas. A significant number of cases of STS, osteosarcoma and GIST have been shown to express programmed cell death protein-ligand 1, therefore the use of immune checkpoint inhibitors that block this ligand or its receptor and help to reactive tumor-infiltrating T cells, could be a beneficial strategy.
Limited activity has been observed in studies conducted to date, however combination therapies, especially with inhibitors of the indoleamine 2,3-dioxygenase (IDO) enzyme, which plays a key role in immunosuppression, could help to harness the power of these drugs. Studies have suggested that sarcomas may be infiltrated by immunosuppressive macrophages that express IDO.43
It is generally believed that immunotherapy is most effective in tumors that are highly mutated because that allows a large number of cancer antigens to provoke an anti-tumor immune response. However, a single highly expressed antigen can also be strongly immunogenic. Synovial sarcomas have a relatively low mutational burden but they do express high levels of the cancer testis antigen NY-ESO-1.
NY-ESO-1 has provided a useful target for the development of adoptive cell therapies and vaccines for the treatment of sarcomas. CMB305 is an NY-ESO-1 vaccine that also incorporates a toll-like receptor 4 agonist. It is being evaluated in the phase 3 Synovate study as maintenance monotherapy in patients with locally advanced, unresectable or metastatic synovial sarcoma. In a phase 1 study, at a median follow-up of just under 18 months, the median OS for all 25 patients was 23.7 months.44
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
2. Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer. 2006;119(12):2922-2930.
3. Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012;2(1):14.
4. Italiano A, Mathoulin-Pelissier S, Cesne AL, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049-1054.
5. Savina M, Le Cesne A, Blay JY, et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: the METASARC observational study. BMC Med. 2017;15(1):78.
6. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34(8):786-793.
7. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387(10028):1629-1637.
8. Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg. 2014;260(3):416-421; discussion 421-412.
9. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342-4349.
10. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708-710.
11. Dagher R, Cohen M, Williams G, et al. Approval summary. Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034-3038.
12. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626-632.
13. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127-1134.
14. Zhao R, Wang Y, Huang Y, et al. Adjuvant imatinib for patients with high-risk gastrointestinal stromal tumors: a retrospective cohort study. Scientific Reports. 2017;7:16834.
15. Raut C, Espat N, Maki R, Araujo D, Williams T, Wolff J. Extended treatment with adjuvant imatinib (IM) for patients (pts) with high-risk primary gastrointestinal stromal tumor (GIST): The PERSIST-5 study. J Clin Oncol. 2017;35(15_suppl):11009.
16. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302.
17. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-1338.
18. Versleijen-Jonkers YM, Vlenterie M, van de Luijtgaarden AC, van der Graaf WT. Anti-angiogenic therapy, a new player in the field of sarcoma treatment. Crit Rev Oncol Hematol. 2014;91(2):172-185.
19. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879-1886.
20. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
21. Chi Y, Yao Y, Wang S, et al. Anlotinib for metastatic soft tissue sarcoma: A randomized, double-blind, placebo-controlled and multi-centered clinical trial. J Clin Oncol. 2018;36(suppl):abstr 11503.
22. Brohl AS, Shah HR, Wang Y-C, Kasarskis A, Maki RG. The somatic mutational landscape in soft tissue sarcoma: Early results from TCGA data. J Clin Oncol. 2015;33(15_suppl):10508-10508.
23. Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326-1341.
24. Jour G, Scarborough JD, Jones RL, et al. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Hum Pathol. 2014;45(8):1563-1571.
25. Yang J-L. Investigation of osteosarcoma genomics and its impact on targeted therapy: an international collaboration to conquer human osteosarcoma. Chin J Cancer. 2014;33(12):575-580.
26. Cidre-Aranaz F, Alonso J. EWS/FLI1 target genes and therapeutic opportunities in Ewing sarcoma. Front Oncol. 2015;5:162.
27. Savoia C, Volpe M, Grassi G, Borghi C, Agabiti Rosei E, Touyz RM. Personalized medicine-a modern approach for the diagnosis and management of hypertension. Clin Sci (Lond). 2017;131(22):2671-2685.
28. Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop. 2016;7(9):527-538.
29. van Maldegem AM, Bovée JVMG, Peterse EFP, Hogendoorn PCW, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171-180.
30. Subbiah V, Hess KR, Khawaja MR, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016;6:35448.
31. Jessen K, Moseley E, Chung EYL, et al. TK216, a novel, small molecule inhibitor of the ETS-family of transcription factors, displays anti-tumor activity in AML and DLBCL. Blood. 2016;128(22):4035-4035.
32. Sankhala K, Potts S, Christiansen J, et al. Immunohistochemistry screening to increase the efficacy of next-generation sequencing for detection of NTRK, ROS1, and ALK gene rearrangements (fusions) in sarcoma patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 9-12, 2016, 2016; Lisbon, Portugal.
33. Renfro LA, An MW, Mandrekar SJ. Precision oncology: a new era of cancer clinical trials. Cancer Lett. 2017;387:121-126.
34. DuBois S, Laetsch T, Federman N, et al. The use of larotrectinib in the management of locally advanced pediatric NTRK-fusion sarcoma. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
35. Multani P, Manavel E, Hornby Z. Preliminary evidence of clinical response to entrectinib in three sarcome patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
36. Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42(8):715-721.
37. Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol. 2016;2(7):937-940.
38. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603-607.
39. Kenichi K, Yoshinao O. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Science. 2017;108(4):547-552.
40. US Food and Drug Administration. Orphan drug designations and approvals. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=544416. Designated date September 28, 2017. Accessed July 4, 2018.
41. Press release. Epizyme provides update regarding tazemetostat clinical program. https://globenewswire.com/news-release/2018/04/23/1485765/0/en/Epizyme-Provides-Update-Regarding-Tazemetostat-Clinical-Program.html. Released April 23, 2018. Accessed July 4, 2018.
42. Bean GR, Kremer JC, Prudner BC, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death &Amp; Disease. 2016;7:e2406.
43. Bourcier K, Italiano A. Newer therapeutic strategies for soft-tissue sarcomas. Pharmacol Ther. 2018;188:118-123.
44. Somaiah N, Chawla SP, Block MS, et al. Immune response, safety, and survival impact from CMB305 in NY-ESO-1+ recurrent soft tissue sarcomas (STS). J Clin Oncol. 2017;35(15_suppl):11006-11006.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
2. Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer. 2006;119(12):2922-2930.
3. Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012;2(1):14.
4. Italiano A, Mathoulin-Pelissier S, Cesne AL, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049-1054.
5. Savina M, Le Cesne A, Blay JY, et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: the METASARC observational study. BMC Med. 2017;15(1):78.
6. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34(8):786-793.
7. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387(10028):1629-1637.
8. Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg. 2014;260(3):416-421; discussion 421-412.
9. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342-4349.
10. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708-710.
11. Dagher R, Cohen M, Williams G, et al. Approval summary. Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034-3038.
12. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626-632.
13. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127-1134.
14. Zhao R, Wang Y, Huang Y, et al. Adjuvant imatinib for patients with high-risk gastrointestinal stromal tumors: a retrospective cohort study. Scientific Reports. 2017;7:16834.
15. Raut C, Espat N, Maki R, Araujo D, Williams T, Wolff J. Extended treatment with adjuvant imatinib (IM) for patients (pts) with high-risk primary gastrointestinal stromal tumor (GIST): The PERSIST-5 study. J Clin Oncol. 2017;35(15_suppl):11009.
16. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302.
17. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-1338.
18. Versleijen-Jonkers YM, Vlenterie M, van de Luijtgaarden AC, van der Graaf WT. Anti-angiogenic therapy, a new player in the field of sarcoma treatment. Crit Rev Oncol Hematol. 2014;91(2):172-185.
19. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879-1886.
20. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
21. Chi Y, Yao Y, Wang S, et al. Anlotinib for metastatic soft tissue sarcoma: A randomized, double-blind, placebo-controlled and multi-centered clinical trial. J Clin Oncol. 2018;36(suppl):abstr 11503.
22. Brohl AS, Shah HR, Wang Y-C, Kasarskis A, Maki RG. The somatic mutational landscape in soft tissue sarcoma: Early results from TCGA data. J Clin Oncol. 2015;33(15_suppl):10508-10508.
23. Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326-1341.
24. Jour G, Scarborough JD, Jones RL, et al. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Hum Pathol. 2014;45(8):1563-1571.
25. Yang J-L. Investigation of osteosarcoma genomics and its impact on targeted therapy: an international collaboration to conquer human osteosarcoma. Chin J Cancer. 2014;33(12):575-580.
26. Cidre-Aranaz F, Alonso J. EWS/FLI1 target genes and therapeutic opportunities in Ewing sarcoma. Front Oncol. 2015;5:162.
27. Savoia C, Volpe M, Grassi G, Borghi C, Agabiti Rosei E, Touyz RM. Personalized medicine-a modern approach for the diagnosis and management of hypertension. Clin Sci (Lond). 2017;131(22):2671-2685.
28. Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop. 2016;7(9):527-538.
29. van Maldegem AM, Bovée JVMG, Peterse EFP, Hogendoorn PCW, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171-180.
30. Subbiah V, Hess KR, Khawaja MR, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016;6:35448.
31. Jessen K, Moseley E, Chung EYL, et al. TK216, a novel, small molecule inhibitor of the ETS-family of transcription factors, displays anti-tumor activity in AML and DLBCL. Blood. 2016;128(22):4035-4035.
32. Sankhala K, Potts S, Christiansen J, et al. Immunohistochemistry screening to increase the efficacy of next-generation sequencing for detection of NTRK, ROS1, and ALK gene rearrangements (fusions) in sarcoma patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 9-12, 2016, 2016; Lisbon, Portugal.
33. Renfro LA, An MW, Mandrekar SJ. Precision oncology: a new era of cancer clinical trials. Cancer Lett. 2017;387:121-126.
34. DuBois S, Laetsch T, Federman N, et al. The use of larotrectinib in the management of locally advanced pediatric NTRK-fusion sarcoma. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
35. Multani P, Manavel E, Hornby Z. Preliminary evidence of clinical response to entrectinib in three sarcome patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
36. Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42(8):715-721.
37. Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol. 2016;2(7):937-940.
38. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603-607.
39. Kenichi K, Yoshinao O. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Science. 2017;108(4):547-552.
40. US Food and Drug Administration. Orphan drug designations and approvals. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=544416. Designated date September 28, 2017. Accessed July 4, 2018.
41. Press release. Epizyme provides update regarding tazemetostat clinical program. https://globenewswire.com/news-release/2018/04/23/1485765/0/en/Epizyme-Provides-Update-Regarding-Tazemetostat-Clinical-Program.html. Released April 23, 2018. Accessed July 4, 2018.
42. Bean GR, Kremer JC, Prudner BC, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death &Amp; Disease. 2016;7:e2406.
43. Bourcier K, Italiano A. Newer therapeutic strategies for soft-tissue sarcomas. Pharmacol Ther. 2018;188:118-123.
44. Somaiah N, Chawla SP, Block MS, et al. Immune response, safety, and survival impact from CMB305 in NY-ESO-1+ recurrent soft tissue sarcomas (STS). J Clin Oncol. 2017;35(15_suppl):11006-11006.
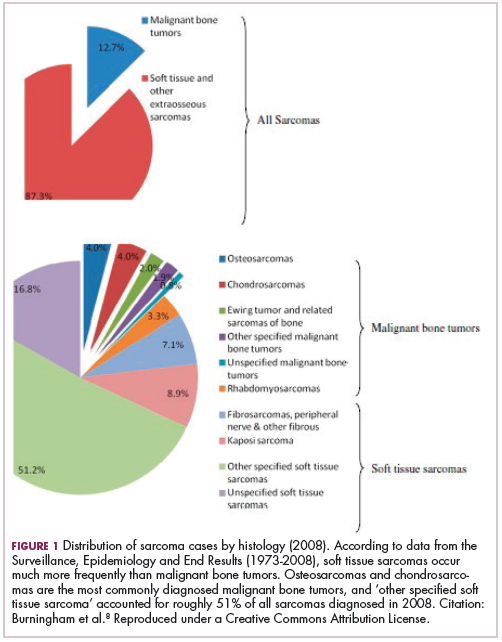
Lurbinectedin shows activity against relapsed Ewing’s
CHICAGO – Single-agent lurbinectedin (PM1183, Zepsyre) showed “encouraging” activity against advanced Ewing’s Sarcoma in previously treated adults, results of a phase 2 study indicated.
Among 28 adults with Ewing’s sarcoma (ES) that had relapsed after up to two prior lines of therapy, treatment with lurbinectedin was associated with five partial responses and six cases of stable disease, reported Vivek Subbiah, MD, from the University of Texas MD Anderson Cancer Center in Houston and his colleagues.
“Treatment in combination with other agents is warranted in this patient population,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
Lurbinectedin’s mechanism of action is through blocking DNA transcription and inducing DNA double-strand breaks, which leads to programmed cell death.
“Moreover, in sarcomas associated with translocations, such as ES, in which the translocation produces a fusion protein that acts as a deregulated transcription factor, lurbinectedin might interfere with the binding of this protein to specific DNA promoters and thus with the synthesis of downstream proteins,” the investigators wrote.
This agent is being investigated against ES as part of a phase 2 basket trial, which is testing the drug against a variety of malignancies. The ES cohort in this study included 15 adults who had received up to two prior chemotherapy regimens. The trial rules called for recruitment of a minimum of 10 more patients if at least one of the first 15 had a confirmed response. There were two responses among the 15 patients, leading to an expansion cohort with 13 patients, for a total of 28 in the current analysis.
The median patient age was 33 years (range, 18-74 years). The majority of patients had good performance status, with Eastern Cooperative Oncology Group scores of 0 (11 patients) or 1 (15 patients); one patient had an ECOG PS score of 2, and one had unknown status.
All but one patient had received a minimum of two prior lines of therapy.
The patients were treated with lurbinectedin 3.2 mg/m2 in a 1-hour infusion on day 1 of every 21-day cycle, with the longest duration of therapy out to 14 cycles.
Among 25 evaluable patients, eight had tumor shrinkage, ranging from less than 5% (two patients) to more than 45% (four patients).
Median progression-free survival (PFS) was 2.7 months. The 4-month PFS rate was 42.9%, and the 6-month rates was 21.4%.
A total of 11 patients had some clinical benefit, including five partial responses and six cases of stable disease.
Grade 3 or 4 treatment-related adverse events included febrile neutropenia (two events grade 3 and two grade 4), anemia (five events, all grade 3), neutropenia (five grade 3 and 10 grade 4), thrombocytopenia (four grade 3 events), and elevated alanine aminotransferase levels (two grade 3 events).
Myelosuppression was transient and manageable with granulocyte colony-stimulating factor, the investigators said.
The study was supported by PharmaMar. Dr. Subbiah disclosed travel, accommodations, and/or expenses from the company and from Bayer; a consulting or advisory role with MedImmune; and institutional research funding from PharmaMar and multiple other companies.
SOURCE: Subbiah V et al. ASCO 2018, Abstract 11519.
CHICAGO – Single-agent lurbinectedin (PM1183, Zepsyre) showed “encouraging” activity against advanced Ewing’s Sarcoma in previously treated adults, results of a phase 2 study indicated.
Among 28 adults with Ewing’s sarcoma (ES) that had relapsed after up to two prior lines of therapy, treatment with lurbinectedin was associated with five partial responses and six cases of stable disease, reported Vivek Subbiah, MD, from the University of Texas MD Anderson Cancer Center in Houston and his colleagues.
“Treatment in combination with other agents is warranted in this patient population,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
Lurbinectedin’s mechanism of action is through blocking DNA transcription and inducing DNA double-strand breaks, which leads to programmed cell death.
“Moreover, in sarcomas associated with translocations, such as ES, in which the translocation produces a fusion protein that acts as a deregulated transcription factor, lurbinectedin might interfere with the binding of this protein to specific DNA promoters and thus with the synthesis of downstream proteins,” the investigators wrote.
This agent is being investigated against ES as part of a phase 2 basket trial, which is testing the drug against a variety of malignancies. The ES cohort in this study included 15 adults who had received up to two prior chemotherapy regimens. The trial rules called for recruitment of a minimum of 10 more patients if at least one of the first 15 had a confirmed response. There were two responses among the 15 patients, leading to an expansion cohort with 13 patients, for a total of 28 in the current analysis.
The median patient age was 33 years (range, 18-74 years). The majority of patients had good performance status, with Eastern Cooperative Oncology Group scores of 0 (11 patients) or 1 (15 patients); one patient had an ECOG PS score of 2, and one had unknown status.
All but one patient had received a minimum of two prior lines of therapy.
The patients were treated with lurbinectedin 3.2 mg/m2 in a 1-hour infusion on day 1 of every 21-day cycle, with the longest duration of therapy out to 14 cycles.
Among 25 evaluable patients, eight had tumor shrinkage, ranging from less than 5% (two patients) to more than 45% (four patients).
Median progression-free survival (PFS) was 2.7 months. The 4-month PFS rate was 42.9%, and the 6-month rates was 21.4%.
A total of 11 patients had some clinical benefit, including five partial responses and six cases of stable disease.
Grade 3 or 4 treatment-related adverse events included febrile neutropenia (two events grade 3 and two grade 4), anemia (five events, all grade 3), neutropenia (five grade 3 and 10 grade 4), thrombocytopenia (four grade 3 events), and elevated alanine aminotransferase levels (two grade 3 events).
Myelosuppression was transient and manageable with granulocyte colony-stimulating factor, the investigators said.
The study was supported by PharmaMar. Dr. Subbiah disclosed travel, accommodations, and/or expenses from the company and from Bayer; a consulting or advisory role with MedImmune; and institutional research funding from PharmaMar and multiple other companies.
SOURCE: Subbiah V et al. ASCO 2018, Abstract 11519.
CHICAGO – Single-agent lurbinectedin (PM1183, Zepsyre) showed “encouraging” activity against advanced Ewing’s Sarcoma in previously treated adults, results of a phase 2 study indicated.
Among 28 adults with Ewing’s sarcoma (ES) that had relapsed after up to two prior lines of therapy, treatment with lurbinectedin was associated with five partial responses and six cases of stable disease, reported Vivek Subbiah, MD, from the University of Texas MD Anderson Cancer Center in Houston and his colleagues.
“Treatment in combination with other agents is warranted in this patient population,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
Lurbinectedin’s mechanism of action is through blocking DNA transcription and inducing DNA double-strand breaks, which leads to programmed cell death.
“Moreover, in sarcomas associated with translocations, such as ES, in which the translocation produces a fusion protein that acts as a deregulated transcription factor, lurbinectedin might interfere with the binding of this protein to specific DNA promoters and thus with the synthesis of downstream proteins,” the investigators wrote.
This agent is being investigated against ES as part of a phase 2 basket trial, which is testing the drug against a variety of malignancies. The ES cohort in this study included 15 adults who had received up to two prior chemotherapy regimens. The trial rules called for recruitment of a minimum of 10 more patients if at least one of the first 15 had a confirmed response. There were two responses among the 15 patients, leading to an expansion cohort with 13 patients, for a total of 28 in the current analysis.
The median patient age was 33 years (range, 18-74 years). The majority of patients had good performance status, with Eastern Cooperative Oncology Group scores of 0 (11 patients) or 1 (15 patients); one patient had an ECOG PS score of 2, and one had unknown status.
All but one patient had received a minimum of two prior lines of therapy.
The patients were treated with lurbinectedin 3.2 mg/m2 in a 1-hour infusion on day 1 of every 21-day cycle, with the longest duration of therapy out to 14 cycles.
Among 25 evaluable patients, eight had tumor shrinkage, ranging from less than 5% (two patients) to more than 45% (four patients).
Median progression-free survival (PFS) was 2.7 months. The 4-month PFS rate was 42.9%, and the 6-month rates was 21.4%.
A total of 11 patients had some clinical benefit, including five partial responses and six cases of stable disease.
Grade 3 or 4 treatment-related adverse events included febrile neutropenia (two events grade 3 and two grade 4), anemia (five events, all grade 3), neutropenia (five grade 3 and 10 grade 4), thrombocytopenia (four grade 3 events), and elevated alanine aminotransferase levels (two grade 3 events).
Myelosuppression was transient and manageable with granulocyte colony-stimulating factor, the investigators said.
The study was supported by PharmaMar. Dr. Subbiah disclosed travel, accommodations, and/or expenses from the company and from Bayer; a consulting or advisory role with MedImmune; and institutional research funding from PharmaMar and multiple other companies.
SOURCE: Subbiah V et al. ASCO 2018, Abstract 11519.
REPORTING FROM ASCO 2018
Key clinical point: Lurbinectedin showed single-agent activity against Ewing’s sarcoma.
Major finding: Out of 28 patients, 5 had partial responses, and 6 had stable disease.
Study details: Phase 2 basket trial of lurbinectedin including 15 patients with Ewing’s sarcoma in the primary cohort and 13 in an expansion cohort.
Disclosures: The study was supported by PharmaMar. Dr. Subbiah disclosed travel, accommodations, and/or expenses from the company and from Bayer; a consulting or advisory role with MedImmune; and institutional research funding from PharmaMar and multiple other companies.
Source: Subbiah V et al. ASCO 2018, Abstract 11519.
Adjuvant chemotherapy benefits high-risk sarcoma patients
CHICAGO – Patients with high-risk soft-tissue sarcomas identified by patient data and a risk calculator had significantly better overall and disease-free survival when they were treated with adjuvant doxorubicin and ifosfamide, a retrospective analysis from a randomized clinical trial showed.
The findings suggest that future clinical trials for sarcoma therapies may need to focus on specific risk categories, investigators said.
Among 290 patients with soft tissues sarcomas (STS) of the trunk wall or extremities, adjuvant chemotherapy with doxorubicin, ifosfamide, mesna, and lenograstim more than halved the risk of death for patients determined by the nomogram to have a low probability of 10-year overall survival, reported Sandro Pasquali, MD, from the Fondazione IRCCS Istituto Nazionale dei Tumori in Milan, and his colleagues.
“These findings interpret conflicting results of randomized controlled trials on perioperative chemotherapy in STS showing that inclusion of low-risk tumors have diluted the effect of chemotherapy leading to negative results and small study effect in meta-analysis,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
The clinical trial in question, EORTC-STBSG 62931, results of which were published in 2012 in The Lancet Oncology, was technically a failure, because it did not show a significant benefit of adjuvant chemotherapy vs. observation in patients with STS.
To see whether adjuvant chemotherapy may have benefited select patients, the investigators calculated 10-year probabilities of overall survival (P-OS) for 290 patients with STS of the trunk wall or extremities out of the total trial cohort of 351 patients. The P-OS for each of three categories – low (51% or less), intermediate (52%-66%), and high (67% or greater) – was calculated using individual patient data and the freely available smartphone-based nomogram Sarculator (available in the Apple App Store and Google Play).
They calculated disease-free survival at the study median follow-up of 8 years.
The tumor histologies included malignant fibrous histiocytoma/undifferentiated pleomorphic sarcoma, liposarcoma, leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, and others.
A total of 52 patients were in the low P-OS group, including 24 treated with observation, and 28 with adjuvant chemotherapy. Respective numbers for the intermediate and high P-OS categories were 34/34, and 90/80.
The investigators found that for patients in the low P-OS group, adjuvant chemotherapy cut the risk of death by slightly more than half, with a hazard ratio of 0.46 (P = .033). In contrast, there were no significant differences in the risk of death for patients at either intermediate or high probability of 10 year OS (HR, 1.00 and 1.08, respectively; P values not significant).
Similarly, adjuvant chemotherapy cut the risk of disease progression by the same amount, with an HR of 0.46 (P = .021), whereas there was no additional benefit among patients at either intermediate or high probability of 10-year OS (HR 0.74 and 0.90; P values were not significant).
The absolute risk reduction for adjuvant chemotherapy was 21% (8-yr disease-free survival of 34% for adjuvant chemotherapy vs. 13% for observation), with a number needed to treated of 4.76.
The study was supported by the European Organization for Research and Treatment of Cancer. Dr. Pasquali reported having no conflicts of interest.
SOURCE: Pasquali S et al. ASCO 2018, Abstract 115118.
CHICAGO – Patients with high-risk soft-tissue sarcomas identified by patient data and a risk calculator had significantly better overall and disease-free survival when they were treated with adjuvant doxorubicin and ifosfamide, a retrospective analysis from a randomized clinical trial showed.
The findings suggest that future clinical trials for sarcoma therapies may need to focus on specific risk categories, investigators said.
Among 290 patients with soft tissues sarcomas (STS) of the trunk wall or extremities, adjuvant chemotherapy with doxorubicin, ifosfamide, mesna, and lenograstim more than halved the risk of death for patients determined by the nomogram to have a low probability of 10-year overall survival, reported Sandro Pasquali, MD, from the Fondazione IRCCS Istituto Nazionale dei Tumori in Milan, and his colleagues.
“These findings interpret conflicting results of randomized controlled trials on perioperative chemotherapy in STS showing that inclusion of low-risk tumors have diluted the effect of chemotherapy leading to negative results and small study effect in meta-analysis,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
The clinical trial in question, EORTC-STBSG 62931, results of which were published in 2012 in The Lancet Oncology, was technically a failure, because it did not show a significant benefit of adjuvant chemotherapy vs. observation in patients with STS.
To see whether adjuvant chemotherapy may have benefited select patients, the investigators calculated 10-year probabilities of overall survival (P-OS) for 290 patients with STS of the trunk wall or extremities out of the total trial cohort of 351 patients. The P-OS for each of three categories – low (51% or less), intermediate (52%-66%), and high (67% or greater) – was calculated using individual patient data and the freely available smartphone-based nomogram Sarculator (available in the Apple App Store and Google Play).
They calculated disease-free survival at the study median follow-up of 8 years.
The tumor histologies included malignant fibrous histiocytoma/undifferentiated pleomorphic sarcoma, liposarcoma, leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, and others.
A total of 52 patients were in the low P-OS group, including 24 treated with observation, and 28 with adjuvant chemotherapy. Respective numbers for the intermediate and high P-OS categories were 34/34, and 90/80.
The investigators found that for patients in the low P-OS group, adjuvant chemotherapy cut the risk of death by slightly more than half, with a hazard ratio of 0.46 (P = .033). In contrast, there were no significant differences in the risk of death for patients at either intermediate or high probability of 10 year OS (HR, 1.00 and 1.08, respectively; P values not significant).
Similarly, adjuvant chemotherapy cut the risk of disease progression by the same amount, with an HR of 0.46 (P = .021), whereas there was no additional benefit among patients at either intermediate or high probability of 10-year OS (HR 0.74 and 0.90; P values were not significant).
The absolute risk reduction for adjuvant chemotherapy was 21% (8-yr disease-free survival of 34% for adjuvant chemotherapy vs. 13% for observation), with a number needed to treated of 4.76.
The study was supported by the European Organization for Research and Treatment of Cancer. Dr. Pasquali reported having no conflicts of interest.
SOURCE: Pasquali S et al. ASCO 2018, Abstract 115118.
CHICAGO – Patients with high-risk soft-tissue sarcomas identified by patient data and a risk calculator had significantly better overall and disease-free survival when they were treated with adjuvant doxorubicin and ifosfamide, a retrospective analysis from a randomized clinical trial showed.
The findings suggest that future clinical trials for sarcoma therapies may need to focus on specific risk categories, investigators said.
Among 290 patients with soft tissues sarcomas (STS) of the trunk wall or extremities, adjuvant chemotherapy with doxorubicin, ifosfamide, mesna, and lenograstim more than halved the risk of death for patients determined by the nomogram to have a low probability of 10-year overall survival, reported Sandro Pasquali, MD, from the Fondazione IRCCS Istituto Nazionale dei Tumori in Milan, and his colleagues.
“These findings interpret conflicting results of randomized controlled trials on perioperative chemotherapy in STS showing that inclusion of low-risk tumors have diluted the effect of chemotherapy leading to negative results and small study effect in meta-analysis,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
The clinical trial in question, EORTC-STBSG 62931, results of which were published in 2012 in The Lancet Oncology, was technically a failure, because it did not show a significant benefit of adjuvant chemotherapy vs. observation in patients with STS.
To see whether adjuvant chemotherapy may have benefited select patients, the investigators calculated 10-year probabilities of overall survival (P-OS) for 290 patients with STS of the trunk wall or extremities out of the total trial cohort of 351 patients. The P-OS for each of three categories – low (51% or less), intermediate (52%-66%), and high (67% or greater) – was calculated using individual patient data and the freely available smartphone-based nomogram Sarculator (available in the Apple App Store and Google Play).
They calculated disease-free survival at the study median follow-up of 8 years.
The tumor histologies included malignant fibrous histiocytoma/undifferentiated pleomorphic sarcoma, liposarcoma, leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor, and others.
A total of 52 patients were in the low P-OS group, including 24 treated with observation, and 28 with adjuvant chemotherapy. Respective numbers for the intermediate and high P-OS categories were 34/34, and 90/80.
The investigators found that for patients in the low P-OS group, adjuvant chemotherapy cut the risk of death by slightly more than half, with a hazard ratio of 0.46 (P = .033). In contrast, there were no significant differences in the risk of death for patients at either intermediate or high probability of 10 year OS (HR, 1.00 and 1.08, respectively; P values not significant).
Similarly, adjuvant chemotherapy cut the risk of disease progression by the same amount, with an HR of 0.46 (P = .021), whereas there was no additional benefit among patients at either intermediate or high probability of 10-year OS (HR 0.74 and 0.90; P values were not significant).
The absolute risk reduction for adjuvant chemotherapy was 21% (8-yr disease-free survival of 34% for adjuvant chemotherapy vs. 13% for observation), with a number needed to treated of 4.76.
The study was supported by the European Organization for Research and Treatment of Cancer. Dr. Pasquali reported having no conflicts of interest.
SOURCE: Pasquali S et al. ASCO 2018, Abstract 115118.
REPORTING FROM ASCO 2018
Key clinical point: Patients with high-risk soft-tissue sarcomas benefit from adjuvant chemotherapy.
Major finding: Hazard ratios for death and disease progression in patients with a low probability of 10-year overall survival were 0.46 for each.
Study details: Retrospective analysis of a randomized phase 3 trial comparing adjuvant chemotherapy with observation in 290 patients with soft tissue sarcomas of the trunk wall or extremities.
Disclosures: The study was supported by the European Organisation for Research and Treatment of Cancer. Dr. Pasquali reported having no conflicts of interest.
Source: Pasquali S et al. ASCO 2018, Abstract 115118.
Rapid drug alteration a bust in metastatic GIST
CHICAGO – For patients with gastrointestinal stromal tumor (GIST) with KIT mutations conferring resistance to imatinib, a strategy of rapid alteration of drugs with complementary activity against KIT mutations is feasible but has thus far failed to yield significant clinical benefits, investigators said.
There were no objective responses among 12 patients treated continuously with 3 days of sunitinib (Sutent) followed by 4 days of regorafenib (Stivarga), and although 4 patients had stable disease in the short term, in each case the disease progressed within 16 weeks, reported Cesar Serrano-Garcia, MD, from the Dana-Farber Cancer Institute and Brigham and Women’s Hospital in Boston, and his colleagues.
“Drug exposure is critical to effectively target specific resistant subpopulations and low exposure may have contributed to the lack of efficacy in this cohort,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
The investigators noted that the main mechanism of resistance to imatinib (Gleevec) in GIST is polyclonal emergence of KIT secondary mutations. They then theorized that rapid alteration of sunitinib with regorafenib, which both have complementary activity against different KIT resistance mutations, could be a novel therapeutic strategy for controlling imatinib-resistant disease.
Both agents are active against KIT and platelet-derived growth factor receptor alpha (PDGFR-alpha). Sunitinib has stronger activity against ATP-binding pocket mutations, and regorafenib is more effective against activation loop oncoproteins, the investigators explained.
They conducted a phase Ib trial to evaluate the safety and preliminary efficacy of the strategy in patients with metastatic GIST that had advanced on therapy with all established protocols. The trial had a standard 3+3 design to determine the recommended phase 2 dose; a total of 14 patients were enrolled, but only 12 received one or more complete cycles.
The median patient age was 63.5%. Nine patients had Eastern Cooperative Oncology Group performance status of 0, and five had an ECOG status of 1. The patients had received a median of four prior lines of therapy, and all had received at least three lines.
The primary mutations were at KIT exon 11 in eight patients, exon 9 in five patients, and a KIT/PDGFR-alpha wild type in one patient.
Of the 12 patients who received one or more complete cycles, 7 were treated with sunitinib 37.5 mg daily for 3 days, followed by regorafenib 120 mg daily for 4 days. There were no dose-limiting toxicities in this group. The median number of cycles delivered was 2 (range 1-4).
The other five patients were treated with sunitinib at the same 37.5 mg daily dose for 3 days, followed immediately by regorafenib 160 mg daily for 4 days. There were two dose-limiting toxicities in this group, both grade 3 hypophosphatemia, one of which was refractory to phosphorous replacement.
Antitumor activity according to Response Evaluation Criteria in Solid Tumors version 1.1 included four cases of stable disease at the time of the efficacy analysis, and eight cases of disease progression. The median progression-free survival was 1.9 months. As noted before, there were no complete or partial responses among the 12 patients.
A pharmacokinetic profile at cycle 1 showed that neither drug reached its reported active blood drug levels.
The patients appeared to tolerate the treatment well, with grade 1 or 2 fatigue in all patients being the most common adverse events. Grade 3 or 4 events included hand-foot syndrome, hypertension, and hypophosphatemia in two patients each.
As noted, the authors acknowledged that low drug exposure levels may explain the lack of any responses in this cohort.
“Therapeutic strategies based on KIT inhibition remain crucial in GIST patients progressing to multiple lines,” they wrote.
The study was supported by an ASCO Young Investigator Award, Pfizer, and Bayer. Dr. Serrano-Garcia disclosed honoraria from Bayer, a consulting or advisory role for Deciphera, research funding from Bayer and Deciphera, and travel accommodations and expenses from Pfizer.
SOURCE: Serrano-Garcia C et al. ASCO 2018, Abstract 11510.
CHICAGO – For patients with gastrointestinal stromal tumor (GIST) with KIT mutations conferring resistance to imatinib, a strategy of rapid alteration of drugs with complementary activity against KIT mutations is feasible but has thus far failed to yield significant clinical benefits, investigators said.
There were no objective responses among 12 patients treated continuously with 3 days of sunitinib (Sutent) followed by 4 days of regorafenib (Stivarga), and although 4 patients had stable disease in the short term, in each case the disease progressed within 16 weeks, reported Cesar Serrano-Garcia, MD, from the Dana-Farber Cancer Institute and Brigham and Women’s Hospital in Boston, and his colleagues.
“Drug exposure is critical to effectively target specific resistant subpopulations and low exposure may have contributed to the lack of efficacy in this cohort,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
The investigators noted that the main mechanism of resistance to imatinib (Gleevec) in GIST is polyclonal emergence of KIT secondary mutations. They then theorized that rapid alteration of sunitinib with regorafenib, which both have complementary activity against different KIT resistance mutations, could be a novel therapeutic strategy for controlling imatinib-resistant disease.
Both agents are active against KIT and platelet-derived growth factor receptor alpha (PDGFR-alpha). Sunitinib has stronger activity against ATP-binding pocket mutations, and regorafenib is more effective against activation loop oncoproteins, the investigators explained.
They conducted a phase Ib trial to evaluate the safety and preliminary efficacy of the strategy in patients with metastatic GIST that had advanced on therapy with all established protocols. The trial had a standard 3+3 design to determine the recommended phase 2 dose; a total of 14 patients were enrolled, but only 12 received one or more complete cycles.
The median patient age was 63.5%. Nine patients had Eastern Cooperative Oncology Group performance status of 0, and five had an ECOG status of 1. The patients had received a median of four prior lines of therapy, and all had received at least three lines.
The primary mutations were at KIT exon 11 in eight patients, exon 9 in five patients, and a KIT/PDGFR-alpha wild type in one patient.
Of the 12 patients who received one or more complete cycles, 7 were treated with sunitinib 37.5 mg daily for 3 days, followed by regorafenib 120 mg daily for 4 days. There were no dose-limiting toxicities in this group. The median number of cycles delivered was 2 (range 1-4).
The other five patients were treated with sunitinib at the same 37.5 mg daily dose for 3 days, followed immediately by regorafenib 160 mg daily for 4 days. There were two dose-limiting toxicities in this group, both grade 3 hypophosphatemia, one of which was refractory to phosphorous replacement.
Antitumor activity according to Response Evaluation Criteria in Solid Tumors version 1.1 included four cases of stable disease at the time of the efficacy analysis, and eight cases of disease progression. The median progression-free survival was 1.9 months. As noted before, there were no complete or partial responses among the 12 patients.
A pharmacokinetic profile at cycle 1 showed that neither drug reached its reported active blood drug levels.
The patients appeared to tolerate the treatment well, with grade 1 or 2 fatigue in all patients being the most common adverse events. Grade 3 or 4 events included hand-foot syndrome, hypertension, and hypophosphatemia in two patients each.
As noted, the authors acknowledged that low drug exposure levels may explain the lack of any responses in this cohort.
“Therapeutic strategies based on KIT inhibition remain crucial in GIST patients progressing to multiple lines,” they wrote.
The study was supported by an ASCO Young Investigator Award, Pfizer, and Bayer. Dr. Serrano-Garcia disclosed honoraria from Bayer, a consulting or advisory role for Deciphera, research funding from Bayer and Deciphera, and travel accommodations and expenses from Pfizer.
SOURCE: Serrano-Garcia C et al. ASCO 2018, Abstract 11510.
CHICAGO – For patients with gastrointestinal stromal tumor (GIST) with KIT mutations conferring resistance to imatinib, a strategy of rapid alteration of drugs with complementary activity against KIT mutations is feasible but has thus far failed to yield significant clinical benefits, investigators said.
There were no objective responses among 12 patients treated continuously with 3 days of sunitinib (Sutent) followed by 4 days of regorafenib (Stivarga), and although 4 patients had stable disease in the short term, in each case the disease progressed within 16 weeks, reported Cesar Serrano-Garcia, MD, from the Dana-Farber Cancer Institute and Brigham and Women’s Hospital in Boston, and his colleagues.
“Drug exposure is critical to effectively target specific resistant subpopulations and low exposure may have contributed to the lack of efficacy in this cohort,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
The investigators noted that the main mechanism of resistance to imatinib (Gleevec) in GIST is polyclonal emergence of KIT secondary mutations. They then theorized that rapid alteration of sunitinib with regorafenib, which both have complementary activity against different KIT resistance mutations, could be a novel therapeutic strategy for controlling imatinib-resistant disease.
Both agents are active against KIT and platelet-derived growth factor receptor alpha (PDGFR-alpha). Sunitinib has stronger activity against ATP-binding pocket mutations, and regorafenib is more effective against activation loop oncoproteins, the investigators explained.
They conducted a phase Ib trial to evaluate the safety and preliminary efficacy of the strategy in patients with metastatic GIST that had advanced on therapy with all established protocols. The trial had a standard 3+3 design to determine the recommended phase 2 dose; a total of 14 patients were enrolled, but only 12 received one or more complete cycles.
The median patient age was 63.5%. Nine patients had Eastern Cooperative Oncology Group performance status of 0, and five had an ECOG status of 1. The patients had received a median of four prior lines of therapy, and all had received at least three lines.
The primary mutations were at KIT exon 11 in eight patients, exon 9 in five patients, and a KIT/PDGFR-alpha wild type in one patient.
Of the 12 patients who received one or more complete cycles, 7 were treated with sunitinib 37.5 mg daily for 3 days, followed by regorafenib 120 mg daily for 4 days. There were no dose-limiting toxicities in this group. The median number of cycles delivered was 2 (range 1-4).
The other five patients were treated with sunitinib at the same 37.5 mg daily dose for 3 days, followed immediately by regorafenib 160 mg daily for 4 days. There were two dose-limiting toxicities in this group, both grade 3 hypophosphatemia, one of which was refractory to phosphorous replacement.
Antitumor activity according to Response Evaluation Criteria in Solid Tumors version 1.1 included four cases of stable disease at the time of the efficacy analysis, and eight cases of disease progression. The median progression-free survival was 1.9 months. As noted before, there were no complete or partial responses among the 12 patients.
A pharmacokinetic profile at cycle 1 showed that neither drug reached its reported active blood drug levels.
The patients appeared to tolerate the treatment well, with grade 1 or 2 fatigue in all patients being the most common adverse events. Grade 3 or 4 events included hand-foot syndrome, hypertension, and hypophosphatemia in two patients each.
As noted, the authors acknowledged that low drug exposure levels may explain the lack of any responses in this cohort.
“Therapeutic strategies based on KIT inhibition remain crucial in GIST patients progressing to multiple lines,” they wrote.
The study was supported by an ASCO Young Investigator Award, Pfizer, and Bayer. Dr. Serrano-Garcia disclosed honoraria from Bayer, a consulting or advisory role for Deciphera, research funding from Bayer and Deciphera, and travel accommodations and expenses from Pfizer.
SOURCE: Serrano-Garcia C et al. ASCO 2018, Abstract 11510.
REPORTING FROM ASCO 2018
Key clinical point: The strategy of rapid alteration of drugs to overcome mutations conferring imatinib resistance in gastrointestinal stromal tumor (GIST) was feasible but ineffective.
Major finding: There were no objective responses among 12 patients treated with the strategy.
Study details: A phase Ib clinical trial in 12 patients with heavily pretreated metastatic GIST.
Disclosures: The study was supported by an ASCO Young Investigator Award, Pfizer, and Bayer. Dr. Serrano-Garcia disclosed honoraria from Bayer, a consulting or advisory role for Deciphera, research funding from Bayer and Deciphera, and travel accommodations and expenses from Pfizer.
Source: Serrano-Garcia C et al. ASCO 2018, Abstract 11510.
ctDNA profiles pre- and posttreatment KIT mutations in GIST
CHICAGO – Detection of circulating tumor DNA (ctDNA), aka “liquid biopsy,” may serve as a noninvasive marker for disease heterogeneity and aid in the assessment of clinical responses to therapy for patients with gastrointestinal stromal tumors (GIST), according to investigators.
In a phase 1 trial of the investigational agent DCC-2618, a pan-KIT/platelet-derived growth factor receptor alpha (PDGFRA) switch control inhibitor, identification of ctDNA by next-generation sequencing (NGS) was accomplished in the majority of patients, with findings that support the need for a broad-spectrum KIT inhibitor for patients with GIST resistant to imatinib (Gleevec), reported Suzanne George, MD, of Dana-Farber Cancer Institute in Boston, and her colleagues.
“This data demonstrates for the first time that the distribution of resistance mutations in KIT across exons 13, 14, 17, and 18 or a combination thereof is similar in 2nd, 3rd, and 4th-line patients,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
The dose-finding and escalation trial included baseline evaluations of KIT/PDGFRA mutations with both ctDNA and fresh tumor biopsy, and ctDNA measurements during treatment.
Biopsy detected 68 KIT mutations at baseline in 81 patients, and ctDNA detected 75 mutations in 95 patients. Some patients had multiple mutations within one exon.
An analysis of mutations by response showed that of 73 patients with detectable KIT mutations by ctDNA at baseline, 35 became KIT ctDNA negative during at least one treatment time point. Of this group, 8 had a partial response (PR) and 27 had stable disease (SD). In all, 57 of the 73 patients had a more than 50% reduction in KIT mutation allele frequency (MAF).
Some patients with stable disease remained KIT negative out to 60 weeks following the first DCC-2618 dose.
The investigators also looked at ctDNA at baseline (21 patients) and post treatment (20 patients) in those who had a PR as their best response. Ten of these patients had KIT mutations detected at baseline, and of this group, eight became KIT negative after treatment; one had no detectable mutations in one exon and one exon with an MAF less than .1%. No posttreatment samples were available for the remaining patient.
There were preliminary data suggesting that DCC-2618 in the second line could be more efficacious than sunitinib (Sutent) in the same setting, and that in KIT-driven GIST DCC-2618 may provide more benefit in the second line compared with the fourth or subsequent lines of therapy, the authors stated.
“The mutational profile of KIT in tumors and plasma at baseline in GIST patients supports the need for a broad spectrum KIT inhibitor in all post-imatinib lines of therapy,” they wrote.
The trial is supported by Deciphera Pharmaceuticals. Dr. George disclosed stock or other ownership in Abbott Laboratories and Abbvie, consulting/advising for AstraZeneca, Blueprint Medicines, and Deciphera, and institutional research funding from Ariad, Bayer, Blueprint Medicine, Deciphera, Novartis, and Pfizer.
SOURCE: George S et al. ASCO 2018. Abstract 11511.
CHICAGO – Detection of circulating tumor DNA (ctDNA), aka “liquid biopsy,” may serve as a noninvasive marker for disease heterogeneity and aid in the assessment of clinical responses to therapy for patients with gastrointestinal stromal tumors (GIST), according to investigators.
In a phase 1 trial of the investigational agent DCC-2618, a pan-KIT/platelet-derived growth factor receptor alpha (PDGFRA) switch control inhibitor, identification of ctDNA by next-generation sequencing (NGS) was accomplished in the majority of patients, with findings that support the need for a broad-spectrum KIT inhibitor for patients with GIST resistant to imatinib (Gleevec), reported Suzanne George, MD, of Dana-Farber Cancer Institute in Boston, and her colleagues.
“This data demonstrates for the first time that the distribution of resistance mutations in KIT across exons 13, 14, 17, and 18 or a combination thereof is similar in 2nd, 3rd, and 4th-line patients,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
The dose-finding and escalation trial included baseline evaluations of KIT/PDGFRA mutations with both ctDNA and fresh tumor biopsy, and ctDNA measurements during treatment.
Biopsy detected 68 KIT mutations at baseline in 81 patients, and ctDNA detected 75 mutations in 95 patients. Some patients had multiple mutations within one exon.
An analysis of mutations by response showed that of 73 patients with detectable KIT mutations by ctDNA at baseline, 35 became KIT ctDNA negative during at least one treatment time point. Of this group, 8 had a partial response (PR) and 27 had stable disease (SD). In all, 57 of the 73 patients had a more than 50% reduction in KIT mutation allele frequency (MAF).
Some patients with stable disease remained KIT negative out to 60 weeks following the first DCC-2618 dose.
The investigators also looked at ctDNA at baseline (21 patients) and post treatment (20 patients) in those who had a PR as their best response. Ten of these patients had KIT mutations detected at baseline, and of this group, eight became KIT negative after treatment; one had no detectable mutations in one exon and one exon with an MAF less than .1%. No posttreatment samples were available for the remaining patient.
There were preliminary data suggesting that DCC-2618 in the second line could be more efficacious than sunitinib (Sutent) in the same setting, and that in KIT-driven GIST DCC-2618 may provide more benefit in the second line compared with the fourth or subsequent lines of therapy, the authors stated.
“The mutational profile of KIT in tumors and plasma at baseline in GIST patients supports the need for a broad spectrum KIT inhibitor in all post-imatinib lines of therapy,” they wrote.
The trial is supported by Deciphera Pharmaceuticals. Dr. George disclosed stock or other ownership in Abbott Laboratories and Abbvie, consulting/advising for AstraZeneca, Blueprint Medicines, and Deciphera, and institutional research funding from Ariad, Bayer, Blueprint Medicine, Deciphera, Novartis, and Pfizer.
SOURCE: George S et al. ASCO 2018. Abstract 11511.
CHICAGO – Detection of circulating tumor DNA (ctDNA), aka “liquid biopsy,” may serve as a noninvasive marker for disease heterogeneity and aid in the assessment of clinical responses to therapy for patients with gastrointestinal stromal tumors (GIST), according to investigators.
In a phase 1 trial of the investigational agent DCC-2618, a pan-KIT/platelet-derived growth factor receptor alpha (PDGFRA) switch control inhibitor, identification of ctDNA by next-generation sequencing (NGS) was accomplished in the majority of patients, with findings that support the need for a broad-spectrum KIT inhibitor for patients with GIST resistant to imatinib (Gleevec), reported Suzanne George, MD, of Dana-Farber Cancer Institute in Boston, and her colleagues.
“This data demonstrates for the first time that the distribution of resistance mutations in KIT across exons 13, 14, 17, and 18 or a combination thereof is similar in 2nd, 3rd, and 4th-line patients,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
The dose-finding and escalation trial included baseline evaluations of KIT/PDGFRA mutations with both ctDNA and fresh tumor biopsy, and ctDNA measurements during treatment.
Biopsy detected 68 KIT mutations at baseline in 81 patients, and ctDNA detected 75 mutations in 95 patients. Some patients had multiple mutations within one exon.
An analysis of mutations by response showed that of 73 patients with detectable KIT mutations by ctDNA at baseline, 35 became KIT ctDNA negative during at least one treatment time point. Of this group, 8 had a partial response (PR) and 27 had stable disease (SD). In all, 57 of the 73 patients had a more than 50% reduction in KIT mutation allele frequency (MAF).
Some patients with stable disease remained KIT negative out to 60 weeks following the first DCC-2618 dose.
The investigators also looked at ctDNA at baseline (21 patients) and post treatment (20 patients) in those who had a PR as their best response. Ten of these patients had KIT mutations detected at baseline, and of this group, eight became KIT negative after treatment; one had no detectable mutations in one exon and one exon with an MAF less than .1%. No posttreatment samples were available for the remaining patient.
There were preliminary data suggesting that DCC-2618 in the second line could be more efficacious than sunitinib (Sutent) in the same setting, and that in KIT-driven GIST DCC-2618 may provide more benefit in the second line compared with the fourth or subsequent lines of therapy, the authors stated.
“The mutational profile of KIT in tumors and plasma at baseline in GIST patients supports the need for a broad spectrum KIT inhibitor in all post-imatinib lines of therapy,” they wrote.
The trial is supported by Deciphera Pharmaceuticals. Dr. George disclosed stock or other ownership in Abbott Laboratories and Abbvie, consulting/advising for AstraZeneca, Blueprint Medicines, and Deciphera, and institutional research funding from Ariad, Bayer, Blueprint Medicine, Deciphera, Novartis, and Pfizer.
SOURCE: George S et al. ASCO 2018. Abstract 11511.
REPORTING FROM ASCO 2018
Key clinical point: Circulating tumor DNA can be used for mutational profiling and responses assessment in patients with advanced imatinib-resistant GIST.
Major finding: Of 73 patients with detectable KIT mutations by ctDNA at baseline, 35 became KIT ctDNA negative during at least one treatment time point.
Study details: Subanalyses from a phase 1 trial of DCC-2618.
Disclosures: The trial is supported by Deciphera Pharmaceuticals. Dr. George disclosed stock or other ownership in Abbott Laboratories and Abbvie, consulting/advising for AstraZeneca, Blueprint Medicines, and Deciphera, and institutional research funding from Ariad, Bayer, Blueprint Medicine, Deciphera, Novartis, and Pfizer.
Source: George S et al. ASCO 2018. Abstract 11511.
Novel TKI PLX9486 showed efficacy against KIT mutations in GIST
CHICAGO – A combination of the investigational agent PLX9486 with another novel tyrosine kinase inhibitor (TKI) showed some efficacy against a range of primary and secondary KIT mutations in patients with gastrointestinal stromal tumor (GIST), the results of a phase 1 dose escalation study have suggested.
Among 39 patients with GIST who had progressed on imatinib and other TKIs, the rates of clinical benefit at 16 weeks were 64% for 11 patients treated with PLX8486 monotherapy at a dose of 1,000 mg daily and 67% for 9 patients treated with PLX9486 and the investigational TKI pexidartinib.
One patient in the 1,000 mg monotherapy group had a partial response on interim analysis. The median progression-free survival in this dose group was 6 months, which was “significantly better than at lower doses,” reported Andrew J. Wagner, MD, PhD, from the Dana-Farber Cancer Institute in Boston and his colleagues.
“The combination of PLX9486 with either pexidartinib or sunitinib is generally well tolerated and toxicities are typically grade 1 or 2 in nature and reversible,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
PLX9486 is an inhibitor of KIT primary mutations in exons 9 and 11 and secondary resistance mutations in exons 17 and 18. Compared with other KIT-targeted TKIs, PLX9486 has complementary selectivity for mutant forms of KIT with a greater than 150-fold selectivity for mutant versus wild-type KIT, the investigators explained.
“Combinations of PLX9486 with either pexidartinib (PLX3397) or sunitinib potentially inhibit and address all common primary and secondary KIT mutations,” they wrote.
The investigators conducted a phase 1, open-label, dose-escalation study with two parts. The first part was designed to study the safety and pharmacokinetics of single-agent PLX9486 and established a maximum tolerated dose (MTD) for phase 2 studies. The second part was designed to study the drug as a single agent at the recommended phase 2 dose in GIST and other solid tumors with KIT mutations and also in combination with either pexidartinib or sunitinib in patients with GIST.
They found that single-agent PLX9486 was well tolerated at all doses tested (250, 300, 350, 500, and 1,000 mg daily) and that it selectively inhibited a spectrum of KIT mutations, “including difficult to treat exon 17/18 activation loop variants.”
The combination of PLX9486 at 500 mg and pexidartinib 600 mg was associated with three partial responses and a clinical benefit rate of 67%, with a PFS on interim analysis of 6 months.
The efficacy of single agent PLX9486 was suggested by circulating tumor DNA studies, which showed reductions in circulating tumor DNA levels of exons 11 and 17/18, which reflected the selectivity profile of the TKI.
In the PLX9486 dose escalation phase, there were three cases of grade 3 or 4 toxicities, including one case each of fatigue, creatinine phosphokinase increase, and hypophosphatemia.
The combination of PLX9486 and pexidartinib was associated with grade 1 or 2 adverse events, including hair color changes in five patients; fatigue and decreased appetite in four patients each; anemia, diarrhea, nausea, alanine aminotransferase increase, and aspartate aminotransferase increase in three patients each; and weight loss, maculopapular rash, and hypertension in two patients each.
At the time of the poster presentation, the sunitinib cohort was still accruing, and interim efficacy data were not available.
“Given these interim results, it is anticipated that the selectivity profile and potency of PLX9486 + sunitinib combination will achieve broader and more durable coverage of primary and secondary KIT mutations,” Dr. Wagner and his associates wrote.
SOURCE: Wagner AJ et al. ASCO 2018, Abstract 11509.
CHICAGO – A combination of the investigational agent PLX9486 with another novel tyrosine kinase inhibitor (TKI) showed some efficacy against a range of primary and secondary KIT mutations in patients with gastrointestinal stromal tumor (GIST), the results of a phase 1 dose escalation study have suggested.
Among 39 patients with GIST who had progressed on imatinib and other TKIs, the rates of clinical benefit at 16 weeks were 64% for 11 patients treated with PLX8486 monotherapy at a dose of 1,000 mg daily and 67% for 9 patients treated with PLX9486 and the investigational TKI pexidartinib.
One patient in the 1,000 mg monotherapy group had a partial response on interim analysis. The median progression-free survival in this dose group was 6 months, which was “significantly better than at lower doses,” reported Andrew J. Wagner, MD, PhD, from the Dana-Farber Cancer Institute in Boston and his colleagues.
“The combination of PLX9486 with either pexidartinib or sunitinib is generally well tolerated and toxicities are typically grade 1 or 2 in nature and reversible,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
PLX9486 is an inhibitor of KIT primary mutations in exons 9 and 11 and secondary resistance mutations in exons 17 and 18. Compared with other KIT-targeted TKIs, PLX9486 has complementary selectivity for mutant forms of KIT with a greater than 150-fold selectivity for mutant versus wild-type KIT, the investigators explained.
“Combinations of PLX9486 with either pexidartinib (PLX3397) or sunitinib potentially inhibit and address all common primary and secondary KIT mutations,” they wrote.
The investigators conducted a phase 1, open-label, dose-escalation study with two parts. The first part was designed to study the safety and pharmacokinetics of single-agent PLX9486 and established a maximum tolerated dose (MTD) for phase 2 studies. The second part was designed to study the drug as a single agent at the recommended phase 2 dose in GIST and other solid tumors with KIT mutations and also in combination with either pexidartinib or sunitinib in patients with GIST.
They found that single-agent PLX9486 was well tolerated at all doses tested (250, 300, 350, 500, and 1,000 mg daily) and that it selectively inhibited a spectrum of KIT mutations, “including difficult to treat exon 17/18 activation loop variants.”
The combination of PLX9486 at 500 mg and pexidartinib 600 mg was associated with three partial responses and a clinical benefit rate of 67%, with a PFS on interim analysis of 6 months.
The efficacy of single agent PLX9486 was suggested by circulating tumor DNA studies, which showed reductions in circulating tumor DNA levels of exons 11 and 17/18, which reflected the selectivity profile of the TKI.
In the PLX9486 dose escalation phase, there were three cases of grade 3 or 4 toxicities, including one case each of fatigue, creatinine phosphokinase increase, and hypophosphatemia.
The combination of PLX9486 and pexidartinib was associated with grade 1 or 2 adverse events, including hair color changes in five patients; fatigue and decreased appetite in four patients each; anemia, diarrhea, nausea, alanine aminotransferase increase, and aspartate aminotransferase increase in three patients each; and weight loss, maculopapular rash, and hypertension in two patients each.
At the time of the poster presentation, the sunitinib cohort was still accruing, and interim efficacy data were not available.
“Given these interim results, it is anticipated that the selectivity profile and potency of PLX9486 + sunitinib combination will achieve broader and more durable coverage of primary and secondary KIT mutations,” Dr. Wagner and his associates wrote.
SOURCE: Wagner AJ et al. ASCO 2018, Abstract 11509.
CHICAGO – A combination of the investigational agent PLX9486 with another novel tyrosine kinase inhibitor (TKI) showed some efficacy against a range of primary and secondary KIT mutations in patients with gastrointestinal stromal tumor (GIST), the results of a phase 1 dose escalation study have suggested.
Among 39 patients with GIST who had progressed on imatinib and other TKIs, the rates of clinical benefit at 16 weeks were 64% for 11 patients treated with PLX8486 monotherapy at a dose of 1,000 mg daily and 67% for 9 patients treated with PLX9486 and the investigational TKI pexidartinib.
One patient in the 1,000 mg monotherapy group had a partial response on interim analysis. The median progression-free survival in this dose group was 6 months, which was “significantly better than at lower doses,” reported Andrew J. Wagner, MD, PhD, from the Dana-Farber Cancer Institute in Boston and his colleagues.
“The combination of PLX9486 with either pexidartinib or sunitinib is generally well tolerated and toxicities are typically grade 1 or 2 in nature and reversible,” they wrote in a poster presented at the annual meeting of the American Society of Clinical Oncology.
PLX9486 is an inhibitor of KIT primary mutations in exons 9 and 11 and secondary resistance mutations in exons 17 and 18. Compared with other KIT-targeted TKIs, PLX9486 has complementary selectivity for mutant forms of KIT with a greater than 150-fold selectivity for mutant versus wild-type KIT, the investigators explained.
“Combinations of PLX9486 with either pexidartinib (PLX3397) or sunitinib potentially inhibit and address all common primary and secondary KIT mutations,” they wrote.
The investigators conducted a phase 1, open-label, dose-escalation study with two parts. The first part was designed to study the safety and pharmacokinetics of single-agent PLX9486 and established a maximum tolerated dose (MTD) for phase 2 studies. The second part was designed to study the drug as a single agent at the recommended phase 2 dose in GIST and other solid tumors with KIT mutations and also in combination with either pexidartinib or sunitinib in patients with GIST.
They found that single-agent PLX9486 was well tolerated at all doses tested (250, 300, 350, 500, and 1,000 mg daily) and that it selectively inhibited a spectrum of KIT mutations, “including difficult to treat exon 17/18 activation loop variants.”
The combination of PLX9486 at 500 mg and pexidartinib 600 mg was associated with three partial responses and a clinical benefit rate of 67%, with a PFS on interim analysis of 6 months.
The efficacy of single agent PLX9486 was suggested by circulating tumor DNA studies, which showed reductions in circulating tumor DNA levels of exons 11 and 17/18, which reflected the selectivity profile of the TKI.
In the PLX9486 dose escalation phase, there were three cases of grade 3 or 4 toxicities, including one case each of fatigue, creatinine phosphokinase increase, and hypophosphatemia.
The combination of PLX9486 and pexidartinib was associated with grade 1 or 2 adverse events, including hair color changes in five patients; fatigue and decreased appetite in four patients each; anemia, diarrhea, nausea, alanine aminotransferase increase, and aspartate aminotransferase increase in three patients each; and weight loss, maculopapular rash, and hypertension in two patients each.
At the time of the poster presentation, the sunitinib cohort was still accruing, and interim efficacy data were not available.
“Given these interim results, it is anticipated that the selectivity profile and potency of PLX9486 + sunitinib combination will achieve broader and more durable coverage of primary and secondary KIT mutations,” Dr. Wagner and his associates wrote.
SOURCE: Wagner AJ et al. ASCO 2018, Abstract 11509.
PRESENTED AT ASCO 2018
Key clinical point: The novel tyrosine kinase inhibitor PLX9486 showed activity against resistance mutations in gastrointestinal stromal tumors.
Major finding: The combination of PLX9486 at 500 mg and pexidartinib 600 mg was associated with three partial responses and a clinical benefit rate of 67% with a PFS on interim analysis of 6 months.
Study details: Phase 1 dose-escalation, safety and pharmacokinetics study in 39 patients with GIST, four with adenocarcinomas, and one with follicular lymphoma.
Disclosures: The study was sponsored by Plexxikon. Dr. Wagner disclosed consulting or advisory roles with Prime Therapeutics, Lilly, and Loxo Oncology, as well as having received institutional research funding from AADi, Celldex Therapeutics, Daiichi Sankyo, Karyopharm Therapeutics, Lilly, and Plexxikon.
Source: Wagner AJ et al. ASCO 2018, Abstract 11509.