User login
Ibrutinib tops chlorambucil against CLL
AMSTERDAM – After 5 years, a large majority of patients with chronic lymphocytic leukemia treated with front-line ibrutinib (Imbruvica) have not experienced disease progression, and the median progression-free survival has still not been reached, long-term follow-up from the RESONATE-2 shows.

The 5-year estimated progression-free survival (PFS) rates were 70% for patients who had been randomized to receive ibrutinib monotherapy, compared with 12% for patients randomized to chlorambucil, reported Alessandra Tedeschi, MD, from Azienda Ospedaliera Niguarda Ca’ Granda in Milan.
Ibrutinib was also associated with a halving of risk for death, compared with chlorambucil, she said at the annual congress of the European Hematology Association.
“Importantly, the rate of progression during ibrutinib treatment was very low; only 8 – that is, 6% of patients” – experienced disease progression while receiving ibrutinib, she noted.
In the RESONATE-2 (PCYC-1115) trial, investigators enrolled 269 adults aged 65 years and older with previously untreated CLL/small lymphocytic lymphoma (SLL). Patients at the younger end of the age range (65-69 years) had to have comorbidities that would have made them ineligible for the FCR chemotherapy regimen (fludarabine, cyclophosphamide, and rituximab). Additionally, patients with the deleterious 17p deletion were excluded.
Patients were stratified by performance status and Rai stage and then randomized to receive either ibrutinib 420 mg once daily until disease progression or unacceptable toxicity (136 patients) or chlorambucil 0.5 mg/kg to a maximum of 0.8 mg/kg for up to 12 cycles (133 patients). The trial also had an extension study for patients who had disease progression as confirmed by an independent review committee or who had completed the RESONATE-2 trial. Of the 133 patients in the chlorambucil arm, 76 (57% of the intention-to-treat population) were crossed over to ibrutinib following disease progression.
The median duration of ibrutinib treatment was 57.1 months, with 73% of patients being on it for more than 3 years, 65% for more than 4 years, and 27% for more than 5 years. As of the data cutoff, 79 patients (58%) were continuing with ibrutinib on study.
At 5 years, 70% of ibrutinib-treated patients and 12% of chlorambucil-treated patients were estimated to be progression-free and alive (hazard ratio for PFS with ibrutinib 0.146 (95% confidence interval, 0.10-0.22). The benefit of ibrutinib was consistent for patients with high-risk genomic features, including the 11q deletion and unmutated immunoglobulin heavy-chain variable genes.
Estimated 5-year overall survival was also better with ibrutinib, at 83% vs. 68% (hazard ratio, 0.45; 95% CI, 0.266-0.761).
The most common grade 3 or greater adverse events occurring with ibrutinib were neutropenia (13%), pneumonia (12%), hypertension (8%), anemia (7%), hyponatremia (6%), atrial fibrillation (5%), and cataract (5%). The rates of most adverse events decreased over time, and dose reductions because of adverse events also diminished over time, from 5% of patients in the first year down to zero in years 4 through 5.
Patients responded to subsequent CLL therapies following ibrutinib discontinuation, including chemoimmunotherapy and other kinase inhibitors, Dr. Tedeschi said.
The trial was sponsored by Pharmacyclics with collaboration from Janssen Research & Development. Dr. Tedeschi reported advisory board activities with Janssen, AbbVie, and BeiGene.
SOURCE: Tedeschi A et al. EHA Congress, Abstract S107.
AMSTERDAM – After 5 years, a large majority of patients with chronic lymphocytic leukemia treated with front-line ibrutinib (Imbruvica) have not experienced disease progression, and the median progression-free survival has still not been reached, long-term follow-up from the RESONATE-2 shows.
The 5-year estimated progression-free survival (PFS) rates were 70% for patients who had been randomized to receive ibrutinib monotherapy, compared with 12% for patients randomized to chlorambucil, reported Alessandra Tedeschi, MD, from Azienda Ospedaliera Niguarda Ca’ Granda in Milan.
Ibrutinib was also associated with a halving of risk for death, compared with chlorambucil, she said at the annual congress of the European Hematology Association.
“Importantly, the rate of progression during ibrutinib treatment was very low; only 8 – that is, 6% of patients” – experienced disease progression while receiving ibrutinib, she noted.
In the RESONATE-2 (PCYC-1115) trial, investigators enrolled 269 adults aged 65 years and older with previously untreated CLL/small lymphocytic lymphoma (SLL). Patients at the younger end of the age range (65-69 years) had to have comorbidities that would have made them ineligible for the FCR chemotherapy regimen (fludarabine, cyclophosphamide, and rituximab). Additionally, patients with the deleterious 17p deletion were excluded.
Patients were stratified by performance status and Rai stage and then randomized to receive either ibrutinib 420 mg once daily until disease progression or unacceptable toxicity (136 patients) or chlorambucil 0.5 mg/kg to a maximum of 0.8 mg/kg for up to 12 cycles (133 patients). The trial also had an extension study for patients who had disease progression as confirmed by an independent review committee or who had completed the RESONATE-2 trial. Of the 133 patients in the chlorambucil arm, 76 (57% of the intention-to-treat population) were crossed over to ibrutinib following disease progression.
The median duration of ibrutinib treatment was 57.1 months, with 73% of patients being on it for more than 3 years, 65% for more than 4 years, and 27% for more than 5 years. As of the data cutoff, 79 patients (58%) were continuing with ibrutinib on study.
At 5 years, 70% of ibrutinib-treated patients and 12% of chlorambucil-treated patients were estimated to be progression-free and alive (hazard ratio for PFS with ibrutinib 0.146 (95% confidence interval, 0.10-0.22). The benefit of ibrutinib was consistent for patients with high-risk genomic features, including the 11q deletion and unmutated immunoglobulin heavy-chain variable genes.
Estimated 5-year overall survival was also better with ibrutinib, at 83% vs. 68% (hazard ratio, 0.45; 95% CI, 0.266-0.761).
The most common grade 3 or greater adverse events occurring with ibrutinib were neutropenia (13%), pneumonia (12%), hypertension (8%), anemia (7%), hyponatremia (6%), atrial fibrillation (5%), and cataract (5%). The rates of most adverse events decreased over time, and dose reductions because of adverse events also diminished over time, from 5% of patients in the first year down to zero in years 4 through 5.
Patients responded to subsequent CLL therapies following ibrutinib discontinuation, including chemoimmunotherapy and other kinase inhibitors, Dr. Tedeschi said.
The trial was sponsored by Pharmacyclics with collaboration from Janssen Research & Development. Dr. Tedeschi reported advisory board activities with Janssen, AbbVie, and BeiGene.
SOURCE: Tedeschi A et al. EHA Congress, Abstract S107.
AMSTERDAM – After 5 years, a large majority of patients with chronic lymphocytic leukemia treated with front-line ibrutinib (Imbruvica) have not experienced disease progression, and the median progression-free survival has still not been reached, long-term follow-up from the RESONATE-2 shows.
The 5-year estimated progression-free survival (PFS) rates were 70% for patients who had been randomized to receive ibrutinib monotherapy, compared with 12% for patients randomized to chlorambucil, reported Alessandra Tedeschi, MD, from Azienda Ospedaliera Niguarda Ca’ Granda in Milan.
Ibrutinib was also associated with a halving of risk for death, compared with chlorambucil, she said at the annual congress of the European Hematology Association.
“Importantly, the rate of progression during ibrutinib treatment was very low; only 8 – that is, 6% of patients” – experienced disease progression while receiving ibrutinib, she noted.
In the RESONATE-2 (PCYC-1115) trial, investigators enrolled 269 adults aged 65 years and older with previously untreated CLL/small lymphocytic lymphoma (SLL). Patients at the younger end of the age range (65-69 years) had to have comorbidities that would have made them ineligible for the FCR chemotherapy regimen (fludarabine, cyclophosphamide, and rituximab). Additionally, patients with the deleterious 17p deletion were excluded.
Patients were stratified by performance status and Rai stage and then randomized to receive either ibrutinib 420 mg once daily until disease progression or unacceptable toxicity (136 patients) or chlorambucil 0.5 mg/kg to a maximum of 0.8 mg/kg for up to 12 cycles (133 patients). The trial also had an extension study for patients who had disease progression as confirmed by an independent review committee or who had completed the RESONATE-2 trial. Of the 133 patients in the chlorambucil arm, 76 (57% of the intention-to-treat population) were crossed over to ibrutinib following disease progression.
The median duration of ibrutinib treatment was 57.1 months, with 73% of patients being on it for more than 3 years, 65% for more than 4 years, and 27% for more than 5 years. As of the data cutoff, 79 patients (58%) were continuing with ibrutinib on study.
At 5 years, 70% of ibrutinib-treated patients and 12% of chlorambucil-treated patients were estimated to be progression-free and alive (hazard ratio for PFS with ibrutinib 0.146 (95% confidence interval, 0.10-0.22). The benefit of ibrutinib was consistent for patients with high-risk genomic features, including the 11q deletion and unmutated immunoglobulin heavy-chain variable genes.
Estimated 5-year overall survival was also better with ibrutinib, at 83% vs. 68% (hazard ratio, 0.45; 95% CI, 0.266-0.761).
The most common grade 3 or greater adverse events occurring with ibrutinib were neutropenia (13%), pneumonia (12%), hypertension (8%), anemia (7%), hyponatremia (6%), atrial fibrillation (5%), and cataract (5%). The rates of most adverse events decreased over time, and dose reductions because of adverse events also diminished over time, from 5% of patients in the first year down to zero in years 4 through 5.
Patients responded to subsequent CLL therapies following ibrutinib discontinuation, including chemoimmunotherapy and other kinase inhibitors, Dr. Tedeschi said.
The trial was sponsored by Pharmacyclics with collaboration from Janssen Research & Development. Dr. Tedeschi reported advisory board activities with Janssen, AbbVie, and BeiGene.
SOURCE: Tedeschi A et al. EHA Congress, Abstract S107.
REPORTING FROM EHA CONGRESS
Predicting outcomes in acute leukemia, NSCLC
In this edition of “How I will treat my next patient,” I take a look at recent studies that examined ways to predict important outcomes in two very different settings, acute leukemia and advanced non–small cell lung cancer (NSCLC). They share the virtue of helping cancer specialists to increase their vigilance for clinically relevant complications and situations and to educate patients and families.

VTE risk in acute leukemia
The risk of venous thromboembolism (VTE) in cancer patients depends upon multiple patient-, tumor-, anatomic-, and treatment-related factors. The Khorana score has become an accepted standard for predicting the risks of VTE and assessing the relative value of various anticoagulants in cancer patients. However, the only hematologic malignancy that is specifically listed among the primary cancer sites in the Khorana score is “lymphoma.” VTE can develop during treatment for acute leukemia, especially among patients with acute lymphoblastic leukemia (ALL).
At the 2019 annual congress of the European Hematology Association, Alejandro Lazo-Langer, MD, and his colleagues proposed a scoring system to quantify the risks of VTE based on a retrospective cohort study of more than 500 acute leukemia patients, diagnosed from 2006-2017. They identified 77 patients with a VTE event, with a median time from diagnosis to VTE of 64 days. Among 20 possible predictive factors, 3 emerged in the final multivariate model – platelet count greater than 50,000 (1 point), ALL (2 points), and prior history of VTE (3 points).
Over a period of 12 months, patients with a score of more than 3 points had a cumulative incidence of VTE of 44%, in comparison with 10.5% among patients with lower scores. They were unable to discern whether particular antineoplastic regimens or drugs enhanced the risk.
The authors proposed that, if verified in a validation cohort study, the scoring system could lead to better patient education about signs and symptoms, more intensive surveillance for high-risk patients, and preventive interventions.
What this means in practice
Although a large number of patient records were reviewed for Dr. Lazo-Langer’s study, there were just 74 ALL patients, and it is unclear whether particular treatment regimens or drugs (such as L-asparaginase in ALL) enhance risk. Further study with a validation cohort (as was performed for the Khorana score for patients with other malignancies), is warranted. The study is thought provoking, but for now, in my opinion, standard clinical vigilance, surveillance, and education regarding VTE in leukemia patients remain appropriate.
Steroid impact in NSCLC with ICI therapy
Patients with autoimmune disease and individuals requiring active treatment with steroids (prednisone at 10 mg/day or more or the equivalent) were excluded from clinical trials that led to Food and Drug Administration approval of immune checkpoint inhibitor (ICI) agents. Recently published data indicate that treatment with 10 mg or more of daily prednisone correlates with poor outcome in NSCLC patients receiving ICI therapy (J Clin Oncol. 2018;36:2872-8; J Thoracic Oncol. 2018;13:1771-5). However, at the 2019 annual meeting of the American Society of Clinical Oncology, analyses of the CancerLinQ database showed that, among NSCLC patients, autoimmune disease and treatment for autoimmune disease are surprisingly prevalent. Should oncologists refuse to treat these patients with ICI agents, alone and in combination with chemotherapy or CTLA4 inhibitors?
Biagio Ricciuti, MD, and colleagues published a retrospective, single-institution record review of 650 advanced NSCLC patients who were treated with ICI plus or minus CTLA-4 inhibition on a correlative intramural research study. Patients who received ICI with concurrent cytotoxic chemotherapy were excluded. They gathered clinical-pathologic information about whether patients received concurrent corticosteroids (10 mg/day or more vs. less than 10 mg/day of prednisone or the equivalent) and the reason for steroid use (oncologic vs. cancer-unrelated indications).
Importantly, they gathered information about programmed death-ligand 1 (PD-L1) tumor proportion scores and tumor mutational burden.
Among the 14.3% patients receiving prednisone 10 mg/day or more at the start of ICI therapy, progression-free survival and overall survival were significantly worse – but only among the 66 patients who needed steroids for oncologic reasons (pain, brain metastases, anorexia, cancer-associated dyspnea). Among the 27 patients who received steroids for cancer-unrelated reasons (autoimmune disease, chronic obstructive pulmonary disease, hypersensitivity pneumonitis), progression-free and overall survival were no different than for patients on prednisone 0-9 mg/day. Imbalances in PD-L1 tumor proportion scores among the groups analyzed did not clearly account for the differences in survival.
What this means in practice
The potential for great treatment outcomes with single-agent ICIs in a subset of advanced NSCLC patients, coupled with the lack of an air-tight biomarker for benefit, has changed the timing of discussions between oncologists and patients about stopping antineoplastic treatment. Since we cannot identify the patients for whom ICI use is futile, the default position has been lenient on using these expensive and potentially toxic therapies.
If verified in a multi-institutional setting, with larger numbers of NSCLC patients receiving steroids for cancer-unrelated reasons, the observations of Dr. Ricciuti and colleagues could help clinicians confidently identify the time to focus discussions on supportive care only. In patients with short survival and strong rationale for maximizing supportive care, analyses like this one could help us deliver more appropriate treatment, instead of more treatment, thereby furthering the goals of personalized cancer patient management.
Dr. Lyss has been a community-based medical oncologist and clinical researcher for more than 35 years, practicing in St. Louis. His clinical and research interests are in the prevention, diagnosis, and treatment of breast and lung cancers and in expanding access to clinical trials to medically underserved populations.
In this edition of “How I will treat my next patient,” I take a look at recent studies that examined ways to predict important outcomes in two very different settings, acute leukemia and advanced non–small cell lung cancer (NSCLC). They share the virtue of helping cancer specialists to increase their vigilance for clinically relevant complications and situations and to educate patients and families.
VTE risk in acute leukemia
The risk of venous thromboembolism (VTE) in cancer patients depends upon multiple patient-, tumor-, anatomic-, and treatment-related factors. The Khorana score has become an accepted standard for predicting the risks of VTE and assessing the relative value of various anticoagulants in cancer patients. However, the only hematologic malignancy that is specifically listed among the primary cancer sites in the Khorana score is “lymphoma.” VTE can develop during treatment for acute leukemia, especially among patients with acute lymphoblastic leukemia (ALL).
At the 2019 annual congress of the European Hematology Association, Alejandro Lazo-Langer, MD, and his colleagues proposed a scoring system to quantify the risks of VTE based on a retrospective cohort study of more than 500 acute leukemia patients, diagnosed from 2006-2017. They identified 77 patients with a VTE event, with a median time from diagnosis to VTE of 64 days. Among 20 possible predictive factors, 3 emerged in the final multivariate model – platelet count greater than 50,000 (1 point), ALL (2 points), and prior history of VTE (3 points).
Over a period of 12 months, patients with a score of more than 3 points had a cumulative incidence of VTE of 44%, in comparison with 10.5% among patients with lower scores. They were unable to discern whether particular antineoplastic regimens or drugs enhanced the risk.
The authors proposed that, if verified in a validation cohort study, the scoring system could lead to better patient education about signs and symptoms, more intensive surveillance for high-risk patients, and preventive interventions.
What this means in practice
Although a large number of patient records were reviewed for Dr. Lazo-Langer’s study, there were just 74 ALL patients, and it is unclear whether particular treatment regimens or drugs (such as L-asparaginase in ALL) enhance risk. Further study with a validation cohort (as was performed for the Khorana score for patients with other malignancies), is warranted. The study is thought provoking, but for now, in my opinion, standard clinical vigilance, surveillance, and education regarding VTE in leukemia patients remain appropriate.
Steroid impact in NSCLC with ICI therapy
Patients with autoimmune disease and individuals requiring active treatment with steroids (prednisone at 10 mg/day or more or the equivalent) were excluded from clinical trials that led to Food and Drug Administration approval of immune checkpoint inhibitor (ICI) agents. Recently published data indicate that treatment with 10 mg or more of daily prednisone correlates with poor outcome in NSCLC patients receiving ICI therapy (J Clin Oncol. 2018;36:2872-8; J Thoracic Oncol. 2018;13:1771-5). However, at the 2019 annual meeting of the American Society of Clinical Oncology, analyses of the CancerLinQ database showed that, among NSCLC patients, autoimmune disease and treatment for autoimmune disease are surprisingly prevalent. Should oncologists refuse to treat these patients with ICI agents, alone and in combination with chemotherapy or CTLA4 inhibitors?
Biagio Ricciuti, MD, and colleagues published a retrospective, single-institution record review of 650 advanced NSCLC patients who were treated with ICI plus or minus CTLA-4 inhibition on a correlative intramural research study. Patients who received ICI with concurrent cytotoxic chemotherapy were excluded. They gathered clinical-pathologic information about whether patients received concurrent corticosteroids (10 mg/day or more vs. less than 10 mg/day of prednisone or the equivalent) and the reason for steroid use (oncologic vs. cancer-unrelated indications).
Importantly, they gathered information about programmed death-ligand 1 (PD-L1) tumor proportion scores and tumor mutational burden.
Among the 14.3% patients receiving prednisone 10 mg/day or more at the start of ICI therapy, progression-free survival and overall survival were significantly worse – but only among the 66 patients who needed steroids for oncologic reasons (pain, brain metastases, anorexia, cancer-associated dyspnea). Among the 27 patients who received steroids for cancer-unrelated reasons (autoimmune disease, chronic obstructive pulmonary disease, hypersensitivity pneumonitis), progression-free and overall survival were no different than for patients on prednisone 0-9 mg/day. Imbalances in PD-L1 tumor proportion scores among the groups analyzed did not clearly account for the differences in survival.
What this means in practice
The potential for great treatment outcomes with single-agent ICIs in a subset of advanced NSCLC patients, coupled with the lack of an air-tight biomarker for benefit, has changed the timing of discussions between oncologists and patients about stopping antineoplastic treatment. Since we cannot identify the patients for whom ICI use is futile, the default position has been lenient on using these expensive and potentially toxic therapies.
If verified in a multi-institutional setting, with larger numbers of NSCLC patients receiving steroids for cancer-unrelated reasons, the observations of Dr. Ricciuti and colleagues could help clinicians confidently identify the time to focus discussions on supportive care only. In patients with short survival and strong rationale for maximizing supportive care, analyses like this one could help us deliver more appropriate treatment, instead of more treatment, thereby furthering the goals of personalized cancer patient management.
Dr. Lyss has been a community-based medical oncologist and clinical researcher for more than 35 years, practicing in St. Louis. His clinical and research interests are in the prevention, diagnosis, and treatment of breast and lung cancers and in expanding access to clinical trials to medically underserved populations.
In this edition of “How I will treat my next patient,” I take a look at recent studies that examined ways to predict important outcomes in two very different settings, acute leukemia and advanced non–small cell lung cancer (NSCLC). They share the virtue of helping cancer specialists to increase their vigilance for clinically relevant complications and situations and to educate patients and families.
VTE risk in acute leukemia
The risk of venous thromboembolism (VTE) in cancer patients depends upon multiple patient-, tumor-, anatomic-, and treatment-related factors. The Khorana score has become an accepted standard for predicting the risks of VTE and assessing the relative value of various anticoagulants in cancer patients. However, the only hematologic malignancy that is specifically listed among the primary cancer sites in the Khorana score is “lymphoma.” VTE can develop during treatment for acute leukemia, especially among patients with acute lymphoblastic leukemia (ALL).
At the 2019 annual congress of the European Hematology Association, Alejandro Lazo-Langer, MD, and his colleagues proposed a scoring system to quantify the risks of VTE based on a retrospective cohort study of more than 500 acute leukemia patients, diagnosed from 2006-2017. They identified 77 patients with a VTE event, with a median time from diagnosis to VTE of 64 days. Among 20 possible predictive factors, 3 emerged in the final multivariate model – platelet count greater than 50,000 (1 point), ALL (2 points), and prior history of VTE (3 points).
Over a period of 12 months, patients with a score of more than 3 points had a cumulative incidence of VTE of 44%, in comparison with 10.5% among patients with lower scores. They were unable to discern whether particular antineoplastic regimens or drugs enhanced the risk.
The authors proposed that, if verified in a validation cohort study, the scoring system could lead to better patient education about signs and symptoms, more intensive surveillance for high-risk patients, and preventive interventions.
What this means in practice
Although a large number of patient records were reviewed for Dr. Lazo-Langer’s study, there were just 74 ALL patients, and it is unclear whether particular treatment regimens or drugs (such as L-asparaginase in ALL) enhance risk. Further study with a validation cohort (as was performed for the Khorana score for patients with other malignancies), is warranted. The study is thought provoking, but for now, in my opinion, standard clinical vigilance, surveillance, and education regarding VTE in leukemia patients remain appropriate.
Steroid impact in NSCLC with ICI therapy
Patients with autoimmune disease and individuals requiring active treatment with steroids (prednisone at 10 mg/day or more or the equivalent) were excluded from clinical trials that led to Food and Drug Administration approval of immune checkpoint inhibitor (ICI) agents. Recently published data indicate that treatment with 10 mg or more of daily prednisone correlates with poor outcome in NSCLC patients receiving ICI therapy (J Clin Oncol. 2018;36:2872-8; J Thoracic Oncol. 2018;13:1771-5). However, at the 2019 annual meeting of the American Society of Clinical Oncology, analyses of the CancerLinQ database showed that, among NSCLC patients, autoimmune disease and treatment for autoimmune disease are surprisingly prevalent. Should oncologists refuse to treat these patients with ICI agents, alone and in combination with chemotherapy or CTLA4 inhibitors?
Biagio Ricciuti, MD, and colleagues published a retrospective, single-institution record review of 650 advanced NSCLC patients who were treated with ICI plus or minus CTLA-4 inhibition on a correlative intramural research study. Patients who received ICI with concurrent cytotoxic chemotherapy were excluded. They gathered clinical-pathologic information about whether patients received concurrent corticosteroids (10 mg/day or more vs. less than 10 mg/day of prednisone or the equivalent) and the reason for steroid use (oncologic vs. cancer-unrelated indications).
Importantly, they gathered information about programmed death-ligand 1 (PD-L1) tumor proportion scores and tumor mutational burden.
Among the 14.3% patients receiving prednisone 10 mg/day or more at the start of ICI therapy, progression-free survival and overall survival were significantly worse – but only among the 66 patients who needed steroids for oncologic reasons (pain, brain metastases, anorexia, cancer-associated dyspnea). Among the 27 patients who received steroids for cancer-unrelated reasons (autoimmune disease, chronic obstructive pulmonary disease, hypersensitivity pneumonitis), progression-free and overall survival were no different than for patients on prednisone 0-9 mg/day. Imbalances in PD-L1 tumor proportion scores among the groups analyzed did not clearly account for the differences in survival.
What this means in practice
The potential for great treatment outcomes with single-agent ICIs in a subset of advanced NSCLC patients, coupled with the lack of an air-tight biomarker for benefit, has changed the timing of discussions between oncologists and patients about stopping antineoplastic treatment. Since we cannot identify the patients for whom ICI use is futile, the default position has been lenient on using these expensive and potentially toxic therapies.
If verified in a multi-institutional setting, with larger numbers of NSCLC patients receiving steroids for cancer-unrelated reasons, the observations of Dr. Ricciuti and colleagues could help clinicians confidently identify the time to focus discussions on supportive care only. In patients with short survival and strong rationale for maximizing supportive care, analyses like this one could help us deliver more appropriate treatment, instead of more treatment, thereby furthering the goals of personalized cancer patient management.
Dr. Lyss has been a community-based medical oncologist and clinical researcher for more than 35 years, practicing in St. Louis. His clinical and research interests are in the prevention, diagnosis, and treatment of breast and lung cancers and in expanding access to clinical trials to medically underserved populations.
ADMIRAL results solidify gilteritinib as new standard for FLT3-mutated AML
AMSTERDAM – For patients with FLT3-mutated, relapsed or refractory acute myeloid leukemia (AML), gilteritinib (Xospata) offers better median overall survival than salvage chemotherapy, according to results from the phase 3 ADMIRAL trial.

Patients treated with gilteritinib also more often responded to therapy and entered remission, reported lead author Alexander Perl, MD, of the University of Pennsylvania, Philadelphia.
To overcome resistance mechanisms to existing FLT3 inhibitors, drug developers have been seeking agents with activity against both FLT3-ITD and FLT3-TKD mutations, Dr. Perl explained during his presentation at the annual congress of the European Hematology Association. “Gilteritinib is one of these agents,” he said, noting a unique mechanism of action that also may limit toxicity concerns associated with existing FLT3 inhibitors.
The international ADMIRAL trial involved 371 patients with FLT3-mutated AML who had not responded to induction therapy or were untreated after first relapse.
The population was randomized in a 2:1 ratio to receive either gilteritinib 120 mg/day or one of four salvage chemotherapy regimens: azacitidine (AZA), low-dose cytarabine (LoDAC), mitoxantrone/etoposide/cytarabine (MEC), or fludarabine/cytarabine/granulocyte colony-stimulating factor/idarubicin (FLAG-IDA).
Coprimary endpoints were overall survival and the combined rate of complete remission and complete remission with partial hematologic recovery (CR/CRh). Secondary endpoints were complete remission rate and event-free survival.
Demographic data showed that the median patient age was 62 years with a broad range (19-85 years). Most patients were positive for FLT3-ITD (88.4%), while fewer tested positive for FLT3-TKD (8.4%) or both mutations (1.9%). Relapsed AML was more common than refractory disease (60.6% vs. 39.4%).
The efficacy analysis revealed that patients treated with gilteritinib had a median overall survival of 9.3 months, significantly longer than the 5.6 months among those treated with salvage chemotherapy (hazard ratio for death = 0.637; P = .0007). The 1-year survival rate was 37.1% for the gilteritinib group, compared with 16.7% among those who received chemotherapy.
The superiority of gilteritinib was further supported by twofold higher rates of CR/CRh (34.0% vs. 15.3%) and complete remission (21.1% vs. 10.5%). Similarly, median event-free survival was significantly longer in the gilteritinib group (2.8 vs. 0.7 months). Most subgroups, such as age and sex, showed consistent benefit.
Overall, gilteritinib demonstrated a favorable safety profile. After adjusting for exposure duration, serious treatment related adverse events were more common in the chemotherapy group than the gilteritinib group (9.2% vs. 7.1%). Common grade 3 or higher adverse events related to gilteritinib were anemia (19.5%), febrile neutropenia (15.4%), thrombocytopenia (12.2%), and decreased platelet count (12.2%).
“We were able to give [gilteritinib] in an outpatient setting,” Dr. Perl said.
Although comparisons between responses based on mutation type were not possible, owing to small sample sizes, Dr. Perl highlighted that gilteritinib showed activity against both FLT3 mutation subtypes.
“This drug has been approved on the results of this study,” Dr. Perl said. “Because of this, we have a new standard of care for this population.”
The study was funded by Astellas. The investigators reported financial relationships with AbbVie, Bayer, Takeda, and other companies.
SOURCE: Perl A et al. EHA Congress, Abstract S876.
AMSTERDAM – For patients with FLT3-mutated, relapsed or refractory acute myeloid leukemia (AML), gilteritinib (Xospata) offers better median overall survival than salvage chemotherapy, according to results from the phase 3 ADMIRAL trial.
Patients treated with gilteritinib also more often responded to therapy and entered remission, reported lead author Alexander Perl, MD, of the University of Pennsylvania, Philadelphia.
To overcome resistance mechanisms to existing FLT3 inhibitors, drug developers have been seeking agents with activity against both FLT3-ITD and FLT3-TKD mutations, Dr. Perl explained during his presentation at the annual congress of the European Hematology Association. “Gilteritinib is one of these agents,” he said, noting a unique mechanism of action that also may limit toxicity concerns associated with existing FLT3 inhibitors.
The international ADMIRAL trial involved 371 patients with FLT3-mutated AML who had not responded to induction therapy or were untreated after first relapse.
The population was randomized in a 2:1 ratio to receive either gilteritinib 120 mg/day or one of four salvage chemotherapy regimens: azacitidine (AZA), low-dose cytarabine (LoDAC), mitoxantrone/etoposide/cytarabine (MEC), or fludarabine/cytarabine/granulocyte colony-stimulating factor/idarubicin (FLAG-IDA).
Coprimary endpoints were overall survival and the combined rate of complete remission and complete remission with partial hematologic recovery (CR/CRh). Secondary endpoints were complete remission rate and event-free survival.
Demographic data showed that the median patient age was 62 years with a broad range (19-85 years). Most patients were positive for FLT3-ITD (88.4%), while fewer tested positive for FLT3-TKD (8.4%) or both mutations (1.9%). Relapsed AML was more common than refractory disease (60.6% vs. 39.4%).
The efficacy analysis revealed that patients treated with gilteritinib had a median overall survival of 9.3 months, significantly longer than the 5.6 months among those treated with salvage chemotherapy (hazard ratio for death = 0.637; P = .0007). The 1-year survival rate was 37.1% for the gilteritinib group, compared with 16.7% among those who received chemotherapy.
The superiority of gilteritinib was further supported by twofold higher rates of CR/CRh (34.0% vs. 15.3%) and complete remission (21.1% vs. 10.5%). Similarly, median event-free survival was significantly longer in the gilteritinib group (2.8 vs. 0.7 months). Most subgroups, such as age and sex, showed consistent benefit.
Overall, gilteritinib demonstrated a favorable safety profile. After adjusting for exposure duration, serious treatment related adverse events were more common in the chemotherapy group than the gilteritinib group (9.2% vs. 7.1%). Common grade 3 or higher adverse events related to gilteritinib were anemia (19.5%), febrile neutropenia (15.4%), thrombocytopenia (12.2%), and decreased platelet count (12.2%).
“We were able to give [gilteritinib] in an outpatient setting,” Dr. Perl said.
Although comparisons between responses based on mutation type were not possible, owing to small sample sizes, Dr. Perl highlighted that gilteritinib showed activity against both FLT3 mutation subtypes.
“This drug has been approved on the results of this study,” Dr. Perl said. “Because of this, we have a new standard of care for this population.”
The study was funded by Astellas. The investigators reported financial relationships with AbbVie, Bayer, Takeda, and other companies.
SOURCE: Perl A et al. EHA Congress, Abstract S876.
AMSTERDAM – For patients with FLT3-mutated, relapsed or refractory acute myeloid leukemia (AML), gilteritinib (Xospata) offers better median overall survival than salvage chemotherapy, according to results from the phase 3 ADMIRAL trial.
Patients treated with gilteritinib also more often responded to therapy and entered remission, reported lead author Alexander Perl, MD, of the University of Pennsylvania, Philadelphia.
To overcome resistance mechanisms to existing FLT3 inhibitors, drug developers have been seeking agents with activity against both FLT3-ITD and FLT3-TKD mutations, Dr. Perl explained during his presentation at the annual congress of the European Hematology Association. “Gilteritinib is one of these agents,” he said, noting a unique mechanism of action that also may limit toxicity concerns associated with existing FLT3 inhibitors.
The international ADMIRAL trial involved 371 patients with FLT3-mutated AML who had not responded to induction therapy or were untreated after first relapse.
The population was randomized in a 2:1 ratio to receive either gilteritinib 120 mg/day or one of four salvage chemotherapy regimens: azacitidine (AZA), low-dose cytarabine (LoDAC), mitoxantrone/etoposide/cytarabine (MEC), or fludarabine/cytarabine/granulocyte colony-stimulating factor/idarubicin (FLAG-IDA).
Coprimary endpoints were overall survival and the combined rate of complete remission and complete remission with partial hematologic recovery (CR/CRh). Secondary endpoints were complete remission rate and event-free survival.
Demographic data showed that the median patient age was 62 years with a broad range (19-85 years). Most patients were positive for FLT3-ITD (88.4%), while fewer tested positive for FLT3-TKD (8.4%) or both mutations (1.9%). Relapsed AML was more common than refractory disease (60.6% vs. 39.4%).
The efficacy analysis revealed that patients treated with gilteritinib had a median overall survival of 9.3 months, significantly longer than the 5.6 months among those treated with salvage chemotherapy (hazard ratio for death = 0.637; P = .0007). The 1-year survival rate was 37.1% for the gilteritinib group, compared with 16.7% among those who received chemotherapy.
The superiority of gilteritinib was further supported by twofold higher rates of CR/CRh (34.0% vs. 15.3%) and complete remission (21.1% vs. 10.5%). Similarly, median event-free survival was significantly longer in the gilteritinib group (2.8 vs. 0.7 months). Most subgroups, such as age and sex, showed consistent benefit.
Overall, gilteritinib demonstrated a favorable safety profile. After adjusting for exposure duration, serious treatment related adverse events were more common in the chemotherapy group than the gilteritinib group (9.2% vs. 7.1%). Common grade 3 or higher adverse events related to gilteritinib were anemia (19.5%), febrile neutropenia (15.4%), thrombocytopenia (12.2%), and decreased platelet count (12.2%).
“We were able to give [gilteritinib] in an outpatient setting,” Dr. Perl said.
Although comparisons between responses based on mutation type were not possible, owing to small sample sizes, Dr. Perl highlighted that gilteritinib showed activity against both FLT3 mutation subtypes.
“This drug has been approved on the results of this study,” Dr. Perl said. “Because of this, we have a new standard of care for this population.”
The study was funded by Astellas. The investigators reported financial relationships with AbbVie, Bayer, Takeda, and other companies.
SOURCE: Perl A et al. EHA Congress, Abstract S876.
REPORTING FROM EHA CONGRESS
Guadecitabine offers limited advantage over other standards for high-risk AML
AMSTERDAM – For treatment-naive patients with acute myeloid leukemia (AML) who are ineligible for chemotherapy, guadecitabine offers similar efficacy to other standard treatment options until four cycles are administered, after which guadecitabine offers a slight survival advantage, based on results from the phase 3 ASTRAL-1 trial.

Complete responders also derived greater benefit from guadecitabine, a new hypomethylating agent, reported lead author Pierre Fenaux, MD, PhD, of the Hôpital Saint Louis, Paris.
With 815 patients, ASTRAL-1 was the largest global, randomized trial to compare low-intensity therapy options in this elderly, unfit population – specifically, patients who were at least 75 years old or had an Eastern Cooperative Oncology Group (ECOG) performance status of 3 or more, Dr. Fenaux said at the annual congress of the European Hematology Association.
They were randomized in a 1:1 ratio to receive guadecitabine or one of three other treatment options: azacitidine, decitabine, or low-dose cytarabine. The coprimary endpoints of the trial were complete response rate and median overall survival. Safety measures were also investigated.
A demographic analysis showed that almost two-thirds of patients were at least 75 years old (62%), and about half had an ECOG status of 2 or 3, or bone marrow blasts. Approximately one-third of patients had poor-risk cytogenetics and a slightly higher proportion had secondary AML.
After a median follow-up of 25.5 months, patients had received, on average, five cycles of therapy. However, many patients (42%) received three or fewer cycles because of early death or disease progression. This therapy cessation rate was similar between the guadecitabine group (42.4%) and the other treatment group (40.8%).
The study failed to meet either coprimary endpoint across the entire patient population. Median overall survival was 7.10 months for guadecitabine versus 8.47 months for the other treatments, but this difference was not statistically significant (P = .73). Similarly, the complete response rate was slightly higher for guadecitabine (19.4% vs. 17.4%), but again, this finding carried a nonsignificant P value (P = .48).
The benefit offered by guadecitabine was realized only with extended treatment and in complete responders.
Patients who received a minimum of four cycles of guadecitabine had a median overall survival of 15.6 months, compared with 13.0 months for other treatments (P = .02). This benefit became more pronounced in those who received at least six cycles, which was associated with median overall survival of 19.5 months versus 14.9 months (P = .002). Complete responders also had extended survival when treated with guadecitabine, although this benefit was of a lesser magnitude (22.6 vs. 20.6 months; P = .07).
Most subgroup analyses, accounting for various clinical and genetic factors, showed no significant differences in primary outcomes between treatment arms, with one exception: TP53 mutations were associated with poor responses to guadecitabine, and a lack of the TP53 mutation predicted better responses to guadecitabine.
Adverse events were common, although most measures were not significantly different between treatment arms. For example, serious adverse events occurred in 81% and 75.5% of patients treated with guadecitabine and other options, respectively, while grade 3 or higher adverse events occurred in 91.5% of guadecitabine patients and 87.5% of patients treated with other options, but neither difference was statistically significant.
Adverse events leading to death occurred in 28.7% of patients treated with guadecitabine versus 29.8% of other patients, a nonsignificant difference. In contrast, Dr. Fenaux noted that patients treated with guadecitabine were significantly more likely to develop febrile neutropenia (33.9% vs. 26.5%), neutropenia (27.4% vs. 20.7%), and pneumonia (29.4% vs. 19.6%).
“In those patients [that received at least four cycles], there seemed to be some advantage of guadecitabine, which needs to be further explored,” Dr. Fenaux said. “But at least [this finding] suggests once more that for a hypomethylating agent to be efficacious, it requires a certain number of cycles, and whenever possible, at least 6 cycles to have full efficacy.”
The study was funded by Astex and Otsuka. The investigators reported additional relationships with Celgene, Janssen, and other companies.
SOURCE: Fenaux P et al. EHA Congress, Abstract S879.
AMSTERDAM – For treatment-naive patients with acute myeloid leukemia (AML) who are ineligible for chemotherapy, guadecitabine offers similar efficacy to other standard treatment options until four cycles are administered, after which guadecitabine offers a slight survival advantage, based on results from the phase 3 ASTRAL-1 trial.
Complete responders also derived greater benefit from guadecitabine, a new hypomethylating agent, reported lead author Pierre Fenaux, MD, PhD, of the Hôpital Saint Louis, Paris.
With 815 patients, ASTRAL-1 was the largest global, randomized trial to compare low-intensity therapy options in this elderly, unfit population – specifically, patients who were at least 75 years old or had an Eastern Cooperative Oncology Group (ECOG) performance status of 3 or more, Dr. Fenaux said at the annual congress of the European Hematology Association.
They were randomized in a 1:1 ratio to receive guadecitabine or one of three other treatment options: azacitidine, decitabine, or low-dose cytarabine. The coprimary endpoints of the trial were complete response rate and median overall survival. Safety measures were also investigated.
A demographic analysis showed that almost two-thirds of patients were at least 75 years old (62%), and about half had an ECOG status of 2 or 3, or bone marrow blasts. Approximately one-third of patients had poor-risk cytogenetics and a slightly higher proportion had secondary AML.
After a median follow-up of 25.5 months, patients had received, on average, five cycles of therapy. However, many patients (42%) received three or fewer cycles because of early death or disease progression. This therapy cessation rate was similar between the guadecitabine group (42.4%) and the other treatment group (40.8%).
The study failed to meet either coprimary endpoint across the entire patient population. Median overall survival was 7.10 months for guadecitabine versus 8.47 months for the other treatments, but this difference was not statistically significant (P = .73). Similarly, the complete response rate was slightly higher for guadecitabine (19.4% vs. 17.4%), but again, this finding carried a nonsignificant P value (P = .48).
The benefit offered by guadecitabine was realized only with extended treatment and in complete responders.
Patients who received a minimum of four cycles of guadecitabine had a median overall survival of 15.6 months, compared with 13.0 months for other treatments (P = .02). This benefit became more pronounced in those who received at least six cycles, which was associated with median overall survival of 19.5 months versus 14.9 months (P = .002). Complete responders also had extended survival when treated with guadecitabine, although this benefit was of a lesser magnitude (22.6 vs. 20.6 months; P = .07).
Most subgroup analyses, accounting for various clinical and genetic factors, showed no significant differences in primary outcomes between treatment arms, with one exception: TP53 mutations were associated with poor responses to guadecitabine, and a lack of the TP53 mutation predicted better responses to guadecitabine.
Adverse events were common, although most measures were not significantly different between treatment arms. For example, serious adverse events occurred in 81% and 75.5% of patients treated with guadecitabine and other options, respectively, while grade 3 or higher adverse events occurred in 91.5% of guadecitabine patients and 87.5% of patients treated with other options, but neither difference was statistically significant.
Adverse events leading to death occurred in 28.7% of patients treated with guadecitabine versus 29.8% of other patients, a nonsignificant difference. In contrast, Dr. Fenaux noted that patients treated with guadecitabine were significantly more likely to develop febrile neutropenia (33.9% vs. 26.5%), neutropenia (27.4% vs. 20.7%), and pneumonia (29.4% vs. 19.6%).
“In those patients [that received at least four cycles], there seemed to be some advantage of guadecitabine, which needs to be further explored,” Dr. Fenaux said. “But at least [this finding] suggests once more that for a hypomethylating agent to be efficacious, it requires a certain number of cycles, and whenever possible, at least 6 cycles to have full efficacy.”
The study was funded by Astex and Otsuka. The investigators reported additional relationships with Celgene, Janssen, and other companies.
SOURCE: Fenaux P et al. EHA Congress, Abstract S879.
AMSTERDAM – For treatment-naive patients with acute myeloid leukemia (AML) who are ineligible for chemotherapy, guadecitabine offers similar efficacy to other standard treatment options until four cycles are administered, after which guadecitabine offers a slight survival advantage, based on results from the phase 3 ASTRAL-1 trial.
Complete responders also derived greater benefit from guadecitabine, a new hypomethylating agent, reported lead author Pierre Fenaux, MD, PhD, of the Hôpital Saint Louis, Paris.
With 815 patients, ASTRAL-1 was the largest global, randomized trial to compare low-intensity therapy options in this elderly, unfit population – specifically, patients who were at least 75 years old or had an Eastern Cooperative Oncology Group (ECOG) performance status of 3 or more, Dr. Fenaux said at the annual congress of the European Hematology Association.
They were randomized in a 1:1 ratio to receive guadecitabine or one of three other treatment options: azacitidine, decitabine, or low-dose cytarabine. The coprimary endpoints of the trial were complete response rate and median overall survival. Safety measures were also investigated.
A demographic analysis showed that almost two-thirds of patients were at least 75 years old (62%), and about half had an ECOG status of 2 or 3, or bone marrow blasts. Approximately one-third of patients had poor-risk cytogenetics and a slightly higher proportion had secondary AML.
After a median follow-up of 25.5 months, patients had received, on average, five cycles of therapy. However, many patients (42%) received three or fewer cycles because of early death or disease progression. This therapy cessation rate was similar between the guadecitabine group (42.4%) and the other treatment group (40.8%).
The study failed to meet either coprimary endpoint across the entire patient population. Median overall survival was 7.10 months for guadecitabine versus 8.47 months for the other treatments, but this difference was not statistically significant (P = .73). Similarly, the complete response rate was slightly higher for guadecitabine (19.4% vs. 17.4%), but again, this finding carried a nonsignificant P value (P = .48).
The benefit offered by guadecitabine was realized only with extended treatment and in complete responders.
Patients who received a minimum of four cycles of guadecitabine had a median overall survival of 15.6 months, compared with 13.0 months for other treatments (P = .02). This benefit became more pronounced in those who received at least six cycles, which was associated with median overall survival of 19.5 months versus 14.9 months (P = .002). Complete responders also had extended survival when treated with guadecitabine, although this benefit was of a lesser magnitude (22.6 vs. 20.6 months; P = .07).
Most subgroup analyses, accounting for various clinical and genetic factors, showed no significant differences in primary outcomes between treatment arms, with one exception: TP53 mutations were associated with poor responses to guadecitabine, and a lack of the TP53 mutation predicted better responses to guadecitabine.
Adverse events were common, although most measures were not significantly different between treatment arms. For example, serious adverse events occurred in 81% and 75.5% of patients treated with guadecitabine and other options, respectively, while grade 3 or higher adverse events occurred in 91.5% of guadecitabine patients and 87.5% of patients treated with other options, but neither difference was statistically significant.
Adverse events leading to death occurred in 28.7% of patients treated with guadecitabine versus 29.8% of other patients, a nonsignificant difference. In contrast, Dr. Fenaux noted that patients treated with guadecitabine were significantly more likely to develop febrile neutropenia (33.9% vs. 26.5%), neutropenia (27.4% vs. 20.7%), and pneumonia (29.4% vs. 19.6%).
“In those patients [that received at least four cycles], there seemed to be some advantage of guadecitabine, which needs to be further explored,” Dr. Fenaux said. “But at least [this finding] suggests once more that for a hypomethylating agent to be efficacious, it requires a certain number of cycles, and whenever possible, at least 6 cycles to have full efficacy.”
The study was funded by Astex and Otsuka. The investigators reported additional relationships with Celgene, Janssen, and other companies.
SOURCE: Fenaux P et al. EHA Congress, Abstract S879.
REPORTING FROM EHA CONGRESS
Rituximab and vemurafenib could challenge frontline chemotherapy for HCL
AMSTERDAM – A combination of rituximab and the BRAF inhibitor vemurafenib could be the one-two punch needed for relapsed or refractory hairy cell leukemia (HCL), according to investigators.

Among evaluable patients treated with this combination, 96% achieved complete remission, reported lead author, Enrico Tiacci, MD, of the University and Hospital of Perugia, Italy.
This level of efficacy is “clearly superior to historical results with either agent alone,” Dr. Tiacci said during a presentation at the annual congress of the European Hematology Association, citing previous complete response rates with vemurafenib alone of 35%-40%. “[This combination] has potential for challenging chemotherapy in the frontline setting,” he said.
The phase 2 trial involved 31 patients with relapsed or refractory HCL who had received a median of three previous therapies. Eight of the patients (26%) had primary refractory disease. Patients received vemurafenib 960 mg, twice daily for 8 weeks and rituximab 375 mg/m2, every 2 weeks. After finishing vemurafenib, patients received rituximab four more times, keeping the interval of 2 weeks. Complete remission was defined as a normal blood count, no leukemic cells in bone marrow biopsies and blood smears, and no palpable splenomegaly.
Out of 31 patients, 27 were evaluable at data cutoff. Of these, 26 (96%) achieved complete remission. The investigators noted that two complete responders had incomplete platelet recovery at the end of treatment that resolved soon after, and two patients had persistent splenomegaly, but were considered to be in complete remission at 22.5 and 25 months after finishing therapy.
All of the complete responders had previously received purine analogs, while a few had been refractory to a prior BRAF inhibitor (n = 7) and/or rituximab (n = 5).
The investigators also pointed out that 15 out of 24 evaluable patients (63%) achieved complete remission just 4 weeks after starting the trial regimen. Almost two-thirds of patients (65%) were negative for minimal residual disease (MRD). The rate of progression-free survival at a median follow-up of 29.5 months was 83%. Disease progression occurred exclusively in patients who were MRD positive.
The combination was well tolerated; most adverse events were of grade 1 or 2, overlapping with the safety profile of each agent alone.
Reflecting on the study findings, Dr. Tiacci suggested that the combination could be most effective if delivered immediately, instead of after BRAF failure.
“Interestingly,” he said, “the relapse-free survival in patients naive to a BRAF inhibitor remained significantly longer than the relapse-free interval that patients previously exposed to a BRAF inhibitor enjoyed, both following monotherapy with a BRAF inhibitor and following subsequent combination with rituximab, potentially suggesting that vemurafenib should be used directly in combination with rituximab rather than being delivered first as a monotherapy and then added to rituximab at relapse.”
Randomized testing of the combination against the chemotherapy-based standard of care in the frontline setting is warranted, the investigators concluded.
Dr. Tiacci reported financial relationships with Roche, AbbVie, and Shire.
SOURCE: Tiacci E et al. EHA Congress, Abstract S104.
AMSTERDAM – A combination of rituximab and the BRAF inhibitor vemurafenib could be the one-two punch needed for relapsed or refractory hairy cell leukemia (HCL), according to investigators.
Among evaluable patients treated with this combination, 96% achieved complete remission, reported lead author, Enrico Tiacci, MD, of the University and Hospital of Perugia, Italy.
This level of efficacy is “clearly superior to historical results with either agent alone,” Dr. Tiacci said during a presentation at the annual congress of the European Hematology Association, citing previous complete response rates with vemurafenib alone of 35%-40%. “[This combination] has potential for challenging chemotherapy in the frontline setting,” he said.
The phase 2 trial involved 31 patients with relapsed or refractory HCL who had received a median of three previous therapies. Eight of the patients (26%) had primary refractory disease. Patients received vemurafenib 960 mg, twice daily for 8 weeks and rituximab 375 mg/m2, every 2 weeks. After finishing vemurafenib, patients received rituximab four more times, keeping the interval of 2 weeks. Complete remission was defined as a normal blood count, no leukemic cells in bone marrow biopsies and blood smears, and no palpable splenomegaly.
Out of 31 patients, 27 were evaluable at data cutoff. Of these, 26 (96%) achieved complete remission. The investigators noted that two complete responders had incomplete platelet recovery at the end of treatment that resolved soon after, and two patients had persistent splenomegaly, but were considered to be in complete remission at 22.5 and 25 months after finishing therapy.
All of the complete responders had previously received purine analogs, while a few had been refractory to a prior BRAF inhibitor (n = 7) and/or rituximab (n = 5).
The investigators also pointed out that 15 out of 24 evaluable patients (63%) achieved complete remission just 4 weeks after starting the trial regimen. Almost two-thirds of patients (65%) were negative for minimal residual disease (MRD). The rate of progression-free survival at a median follow-up of 29.5 months was 83%. Disease progression occurred exclusively in patients who were MRD positive.
The combination was well tolerated; most adverse events were of grade 1 or 2, overlapping with the safety profile of each agent alone.
Reflecting on the study findings, Dr. Tiacci suggested that the combination could be most effective if delivered immediately, instead of after BRAF failure.
“Interestingly,” he said, “the relapse-free survival in patients naive to a BRAF inhibitor remained significantly longer than the relapse-free interval that patients previously exposed to a BRAF inhibitor enjoyed, both following monotherapy with a BRAF inhibitor and following subsequent combination with rituximab, potentially suggesting that vemurafenib should be used directly in combination with rituximab rather than being delivered first as a monotherapy and then added to rituximab at relapse.”
Randomized testing of the combination against the chemotherapy-based standard of care in the frontline setting is warranted, the investigators concluded.
Dr. Tiacci reported financial relationships with Roche, AbbVie, and Shire.
SOURCE: Tiacci E et al. EHA Congress, Abstract S104.
AMSTERDAM – A combination of rituximab and the BRAF inhibitor vemurafenib could be the one-two punch needed for relapsed or refractory hairy cell leukemia (HCL), according to investigators.
Among evaluable patients treated with this combination, 96% achieved complete remission, reported lead author, Enrico Tiacci, MD, of the University and Hospital of Perugia, Italy.
This level of efficacy is “clearly superior to historical results with either agent alone,” Dr. Tiacci said during a presentation at the annual congress of the European Hematology Association, citing previous complete response rates with vemurafenib alone of 35%-40%. “[This combination] has potential for challenging chemotherapy in the frontline setting,” he said.
The phase 2 trial involved 31 patients with relapsed or refractory HCL who had received a median of three previous therapies. Eight of the patients (26%) had primary refractory disease. Patients received vemurafenib 960 mg, twice daily for 8 weeks and rituximab 375 mg/m2, every 2 weeks. After finishing vemurafenib, patients received rituximab four more times, keeping the interval of 2 weeks. Complete remission was defined as a normal blood count, no leukemic cells in bone marrow biopsies and blood smears, and no palpable splenomegaly.
Out of 31 patients, 27 were evaluable at data cutoff. Of these, 26 (96%) achieved complete remission. The investigators noted that two complete responders had incomplete platelet recovery at the end of treatment that resolved soon after, and two patients had persistent splenomegaly, but were considered to be in complete remission at 22.5 and 25 months after finishing therapy.
All of the complete responders had previously received purine analogs, while a few had been refractory to a prior BRAF inhibitor (n = 7) and/or rituximab (n = 5).
The investigators also pointed out that 15 out of 24 evaluable patients (63%) achieved complete remission just 4 weeks after starting the trial regimen. Almost two-thirds of patients (65%) were negative for minimal residual disease (MRD). The rate of progression-free survival at a median follow-up of 29.5 months was 83%. Disease progression occurred exclusively in patients who were MRD positive.
The combination was well tolerated; most adverse events were of grade 1 or 2, overlapping with the safety profile of each agent alone.
Reflecting on the study findings, Dr. Tiacci suggested that the combination could be most effective if delivered immediately, instead of after BRAF failure.
“Interestingly,” he said, “the relapse-free survival in patients naive to a BRAF inhibitor remained significantly longer than the relapse-free interval that patients previously exposed to a BRAF inhibitor enjoyed, both following monotherapy with a BRAF inhibitor and following subsequent combination with rituximab, potentially suggesting that vemurafenib should be used directly in combination with rituximab rather than being delivered first as a monotherapy and then added to rituximab at relapse.”
Randomized testing of the combination against the chemotherapy-based standard of care in the frontline setting is warranted, the investigators concluded.
Dr. Tiacci reported financial relationships with Roche, AbbVie, and Shire.
SOURCE: Tiacci E et al. EHA Congress, Abstract S104.
REPORTING FROM EHA CONGRESS
‘Robust antitumor immune responses’ observed in pediatric ALL
Pediatric acute lymphoblastic leukemia (ALL) may be more vulnerable to immunotherapies than previously thought, according to researchers.
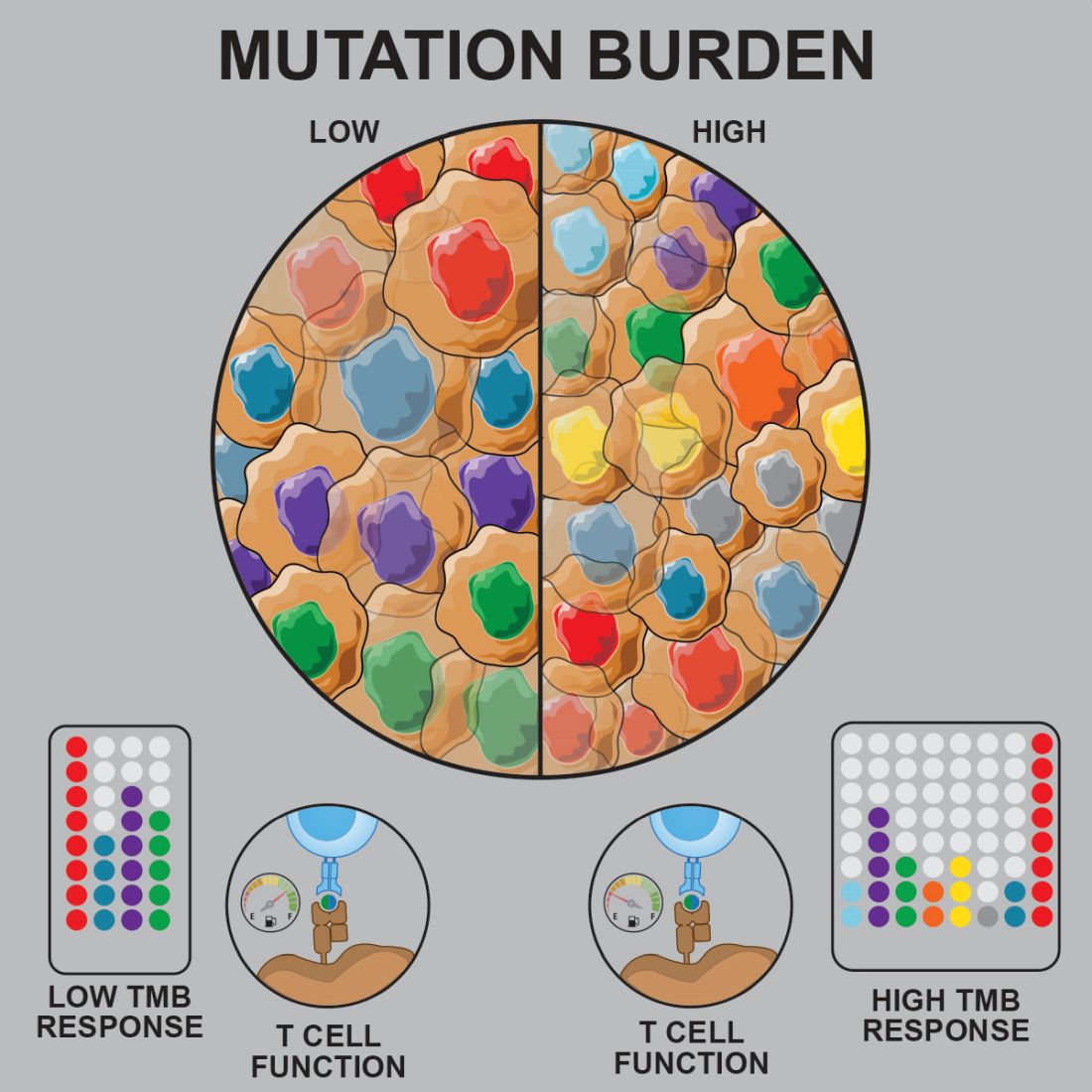
Prior studies suggested that tumors with a low mutational burden don’t elicit strong antitumor responses and therefore aren’t very susceptible to immunotherapy.
Now, researchers have found evidence to suggest that pediatric ALL induces “robust antitumor immune responses” despite a low mutational burden. The investigators identified tumor-associated CD8+ T cells that responded to 86% of neoantigens tested and recognized 68% of neoepitopes tested.
Anthony E. Zamora, PhD, of St. Jude Children’s Research Hospital in Memphis, Tenn., and colleagues recounted these findings in Science Translational Medicine.
The researchers analyzed samples from pediatric patients with ETV-associated ALL (n = 9) or ERG-associated ALL (n = 2) to determine how endogenous CD8+ T cells respond to patient-specific cancer neoantigens.
The investigators first assessed the ability of tumor-specific mutations and gene fusions to generate neoepitopes, or neoantigens predicted to bind patient-specific human leukocyte antigen (HLA) proteins. The team identified 5-28 neoepitopes per patient, including epitopes that spanned the fusion junction in patients with ETV6-RUNX1 fusions.
The researchers then tested whether CD8+ tumor infiltrating lymphocytes (TILs) were directly responsive to mutated neoepitopes. They observed cytokine responses across patient samples, noting that 31 of the 36 putative neoantigens tested (86%) were “immunogenic and capable of inducing robust cytokine responses.”
Next, the investigators mapped TIL responses to specific epitopes using patient-specific tetramers that corresponded to the previously identified neoepitopes. Seventeen of the 25 patient-specific tetramers (68%) bound to TILs above the background set by irrelevant HLA-matched tetramers.
“Within those responses, we observed immunodominance hierarchies among the distinct TIL populations, with a majority of tetramer-bound CD8+ T cells restricted to one or two putative neoepitopes,” the researchers noted.
The team also pointed out that seven of nine patients tested had CD8+ T cells that responded to ETV6-RUNX1.
Finally, the investigators performed transcriptional profiling of ALL-specific CD8+ TILs to assess inter- and intrapatient heterogeneity. The team identified three hierarchical clusters, which were characterized by transcriptional factors and regulators associated with:
- Functional effector CD8+ T cells (TBX21 and EOMES).
- Dysfunctional CD8+ T cells (STAT1/3/4, NR4A2/3, and BCL6).
- Exhausted CD8+ T cells (EOMES, MAF, PRDM1, and BATF).
Considering these findings together, the researchers concluded that “pediatric ALL elicits a potent neoepitope-specific CD8+ T-cell response.” Therefore, adoptive T-cell, monoclonal antibody, and targeted T-cell receptor therapies “should be explored” in pediatric ALL.
This research was supported by the National Institutes of Health, National Cancer Institute, National Institute of General Medical Sciences, Key for a Cure Foundation, and American Lebanese Syrian Associated Charities. The researchers disclosed patent applications and relationships with Pfizer, Amgen, and other companies.
SOURCE: Zamora AE et al. Sci. Transl. Med. 2019 Jun 26. doi: 10.1126/scitranslmed.aat8549.
Pediatric acute lymphoblastic leukemia (ALL) may be more vulnerable to immunotherapies than previously thought, according to researchers.
Prior studies suggested that tumors with a low mutational burden don’t elicit strong antitumor responses and therefore aren’t very susceptible to immunotherapy.
Now, researchers have found evidence to suggest that pediatric ALL induces “robust antitumor immune responses” despite a low mutational burden. The investigators identified tumor-associated CD8+ T cells that responded to 86% of neoantigens tested and recognized 68% of neoepitopes tested.
Anthony E. Zamora, PhD, of St. Jude Children’s Research Hospital in Memphis, Tenn., and colleagues recounted these findings in Science Translational Medicine.
The researchers analyzed samples from pediatric patients with ETV-associated ALL (n = 9) or ERG-associated ALL (n = 2) to determine how endogenous CD8+ T cells respond to patient-specific cancer neoantigens.
The investigators first assessed the ability of tumor-specific mutations and gene fusions to generate neoepitopes, or neoantigens predicted to bind patient-specific human leukocyte antigen (HLA) proteins. The team identified 5-28 neoepitopes per patient, including epitopes that spanned the fusion junction in patients with ETV6-RUNX1 fusions.
The researchers then tested whether CD8+ tumor infiltrating lymphocytes (TILs) were directly responsive to mutated neoepitopes. They observed cytokine responses across patient samples, noting that 31 of the 36 putative neoantigens tested (86%) were “immunogenic and capable of inducing robust cytokine responses.”
Next, the investigators mapped TIL responses to specific epitopes using patient-specific tetramers that corresponded to the previously identified neoepitopes. Seventeen of the 25 patient-specific tetramers (68%) bound to TILs above the background set by irrelevant HLA-matched tetramers.
“Within those responses, we observed immunodominance hierarchies among the distinct TIL populations, with a majority of tetramer-bound CD8+ T cells restricted to one or two putative neoepitopes,” the researchers noted.
The team also pointed out that seven of nine patients tested had CD8+ T cells that responded to ETV6-RUNX1.
Finally, the investigators performed transcriptional profiling of ALL-specific CD8+ TILs to assess inter- and intrapatient heterogeneity. The team identified three hierarchical clusters, which were characterized by transcriptional factors and regulators associated with:
- Functional effector CD8+ T cells (TBX21 and EOMES).
- Dysfunctional CD8+ T cells (STAT1/3/4, NR4A2/3, and BCL6).
- Exhausted CD8+ T cells (EOMES, MAF, PRDM1, and BATF).
Considering these findings together, the researchers concluded that “pediatric ALL elicits a potent neoepitope-specific CD8+ T-cell response.” Therefore, adoptive T-cell, monoclonal antibody, and targeted T-cell receptor therapies “should be explored” in pediatric ALL.
This research was supported by the National Institutes of Health, National Cancer Institute, National Institute of General Medical Sciences, Key for a Cure Foundation, and American Lebanese Syrian Associated Charities. The researchers disclosed patent applications and relationships with Pfizer, Amgen, and other companies.
SOURCE: Zamora AE et al. Sci. Transl. Med. 2019 Jun 26. doi: 10.1126/scitranslmed.aat8549.
Pediatric acute lymphoblastic leukemia (ALL) may be more vulnerable to immunotherapies than previously thought, according to researchers.
Prior studies suggested that tumors with a low mutational burden don’t elicit strong antitumor responses and therefore aren’t very susceptible to immunotherapy.
Now, researchers have found evidence to suggest that pediatric ALL induces “robust antitumor immune responses” despite a low mutational burden. The investigators identified tumor-associated CD8+ T cells that responded to 86% of neoantigens tested and recognized 68% of neoepitopes tested.
Anthony E. Zamora, PhD, of St. Jude Children’s Research Hospital in Memphis, Tenn., and colleagues recounted these findings in Science Translational Medicine.
The researchers analyzed samples from pediatric patients with ETV-associated ALL (n = 9) or ERG-associated ALL (n = 2) to determine how endogenous CD8+ T cells respond to patient-specific cancer neoantigens.
The investigators first assessed the ability of tumor-specific mutations and gene fusions to generate neoepitopes, or neoantigens predicted to bind patient-specific human leukocyte antigen (HLA) proteins. The team identified 5-28 neoepitopes per patient, including epitopes that spanned the fusion junction in patients with ETV6-RUNX1 fusions.
The researchers then tested whether CD8+ tumor infiltrating lymphocytes (TILs) were directly responsive to mutated neoepitopes. They observed cytokine responses across patient samples, noting that 31 of the 36 putative neoantigens tested (86%) were “immunogenic and capable of inducing robust cytokine responses.”
Next, the investigators mapped TIL responses to specific epitopes using patient-specific tetramers that corresponded to the previously identified neoepitopes. Seventeen of the 25 patient-specific tetramers (68%) bound to TILs above the background set by irrelevant HLA-matched tetramers.
“Within those responses, we observed immunodominance hierarchies among the distinct TIL populations, with a majority of tetramer-bound CD8+ T cells restricted to one or two putative neoepitopes,” the researchers noted.
The team also pointed out that seven of nine patients tested had CD8+ T cells that responded to ETV6-RUNX1.
Finally, the investigators performed transcriptional profiling of ALL-specific CD8+ TILs to assess inter- and intrapatient heterogeneity. The team identified three hierarchical clusters, which were characterized by transcriptional factors and regulators associated with:
- Functional effector CD8+ T cells (TBX21 and EOMES).
- Dysfunctional CD8+ T cells (STAT1/3/4, NR4A2/3, and BCL6).
- Exhausted CD8+ T cells (EOMES, MAF, PRDM1, and BATF).
Considering these findings together, the researchers concluded that “pediatric ALL elicits a potent neoepitope-specific CD8+ T-cell response.” Therefore, adoptive T-cell, monoclonal antibody, and targeted T-cell receptor therapies “should be explored” in pediatric ALL.
This research was supported by the National Institutes of Health, National Cancer Institute, National Institute of General Medical Sciences, Key for a Cure Foundation, and American Lebanese Syrian Associated Charities. The researchers disclosed patent applications and relationships with Pfizer, Amgen, and other companies.
SOURCE: Zamora AE et al. Sci. Transl. Med. 2019 Jun 26. doi: 10.1126/scitranslmed.aat8549.
FROM SCIENCE TRANSLATIONAL MEDICINE
Key clinical point: Preclinical research suggests pediatric acute lymphoblastic leukemia (ALL) induces “robust antitumor immune responses” despite a low mutational burden.
Major finding: Investigators identified tumor-associated CD8+ T cells that responded to 86% of neoantigens tested and recognized 68% of neoepitopes tested.
Study details: Analysis of samples from pediatric patients with ETV-associated ALL (n = 9) or ERG-associated ALL (n = 2).
Disclosures: The research was supported by the National Institutes of Health, National Cancer Institute, National Institute of General Medical Sciences, Key for a Cure Foundation, and American Lebanese Syrian Associated Charities. The researchers disclosed patent applications and relationships with Pfizer, Amgen, and other companies.
Source: Zamora AE et al. Sci. Transl. Med. 2019 Jun 26. doi: 10.1126/scitranslmed.aat8549.
Risk model could help predict VTE in acute leukemia
AMSTERDAM – A new clinical prediction model can determine the risk of venous thromboembolism in patients with leukemia, according to investigators.

The scoring system, which incorporates historical, morphological, and cytologic factors, was internally validated at multiple time points over the course of a year, reported lead author, Alejandro Lazo-Langner, MD, of the University of Western Ontario, London.
“It is important that we can predict or anticipate which patients [with acute leukemia] will develop venous thrombosis so that we can develop preventions and aim for better surveillance strategies,” Dr. Lazo-Langner said at the annual congress of the European Hematology Association. Venous thromboembolism (VTE) risk modeling is available for patients with solid tumors, but a similar prognostic tool for leukemia patients has been missing.
To fill this practice gap, Dr. Lazo-Langner and colleagues conducted a retrospective cohort study involving 501 patients with acute leukemia who were diagnosed between 2006 and 2017. Of these patients, 427 (85.2%) had myeloid lineage and 74 (14.8%) had lymphoblastic disease. VTE outcomes of interest included proximal lower- and upper-extremity deep vein thrombosis; pulmonary embolism; and thrombosis of unusual sites, such as splanchnic and cerebral. Patients were followed until last follow-up, VTE, or death. Single variable and multiple variable logistic regression were used sequentially to evaluate and confirm potential predictive factors, with nonparametric bootstrapping for internal validation.
After last follow-up, 77 patients (15.3%) had developed VTE; specifically, 44 patients had upper-extremity deep vein thrombosis, 28 had lower-extremity deep vein thrombosis or pulmonary embolism, and 5 had cerebral vein thrombosis. The median time from leukemia diagnosis to VTE was approximately 2 months (64 days). Out of 20 possible predictive factors, 7 were included in the multivariable model, and 3 constitute the final model. These three factors are platelet count greater than 50 x 109/L at time of diagnosis (1 point), lymphoblastic leukemia (2 points), and previous history of venous thromboembolism (3 points).
Dr. Lazo-Langner explained that leukemia patients at high risk of VTE are those with a score of 3 or more points. Using this risk threshold, the investigators found that the overall cumulative incidence of VTE in the high-risk group was 44.0%, compared with 10.5% in the low-risk group. Temporal analysis showed a widening disparity between the two groups, from 3 months (28.8% vs. 6.3%), to 6 months (41.1% vs. 7.9%), and 12 months (42.5% vs. 9.3%).
When asked if treatment type was evaluated, Dr. Lazo-Langner said that treatment type was evaluated but proved unfruitful for the model, which is designed for universal use in leukemia.
“We did include a number of different chemotherapy regimens,” he said. “The problem is, because we included both AML [acute myeloid leukemia] and ALL [acute lymphoblastic leukemia] lineage, and the cornerstone of treatment is different for both lineages. It’s difficult to actually include what kind of chemotherapy [patients had]. For instance, it is known that anthracyclines increase risk of thrombosis, but in both lineages, you use anthracyclines, so you really cannot use that as a predictor.”
Looking to the future, the next step will be validation in other cohorts. If this is successful, then Dr. Lazo-Langner speculated that clinicians could use the scoring system to direct monitoring and treatment. For example, patients with high scores and low platelet counts could receive earlier transfusional support, while all high-risk patients could be placed under more intensive surveillance and given additional education about thrombosis.
“I think recognizing symptoms early is important,” Dr. Lazo-Langner said, “and that would be training not only clinicians, but also nursing personnel and the patients themselves to be aware of the symptoms, so they can actually recognize them sooner.”
The study was funded by the Canadian Institutes of Health Research. Dr. Lazo-Langner is an investigator with the Canadian Venous Thromboembolism Clinical Trials and Outcomes Research (CanVECTOR) Network.
SOURCE: Lazo-Langner A et al. EHA 2019, Abstract S1642.
AMSTERDAM – A new clinical prediction model can determine the risk of venous thromboembolism in patients with leukemia, according to investigators.
The scoring system, which incorporates historical, morphological, and cytologic factors, was internally validated at multiple time points over the course of a year, reported lead author, Alejandro Lazo-Langner, MD, of the University of Western Ontario, London.
“It is important that we can predict or anticipate which patients [with acute leukemia] will develop venous thrombosis so that we can develop preventions and aim for better surveillance strategies,” Dr. Lazo-Langner said at the annual congress of the European Hematology Association. Venous thromboembolism (VTE) risk modeling is available for patients with solid tumors, but a similar prognostic tool for leukemia patients has been missing.
To fill this practice gap, Dr. Lazo-Langner and colleagues conducted a retrospective cohort study involving 501 patients with acute leukemia who were diagnosed between 2006 and 2017. Of these patients, 427 (85.2%) had myeloid lineage and 74 (14.8%) had lymphoblastic disease. VTE outcomes of interest included proximal lower- and upper-extremity deep vein thrombosis; pulmonary embolism; and thrombosis of unusual sites, such as splanchnic and cerebral. Patients were followed until last follow-up, VTE, or death. Single variable and multiple variable logistic regression were used sequentially to evaluate and confirm potential predictive factors, with nonparametric bootstrapping for internal validation.
After last follow-up, 77 patients (15.3%) had developed VTE; specifically, 44 patients had upper-extremity deep vein thrombosis, 28 had lower-extremity deep vein thrombosis or pulmonary embolism, and 5 had cerebral vein thrombosis. The median time from leukemia diagnosis to VTE was approximately 2 months (64 days). Out of 20 possible predictive factors, 7 were included in the multivariable model, and 3 constitute the final model. These three factors are platelet count greater than 50 x 109/L at time of diagnosis (1 point), lymphoblastic leukemia (2 points), and previous history of venous thromboembolism (3 points).
Dr. Lazo-Langner explained that leukemia patients at high risk of VTE are those with a score of 3 or more points. Using this risk threshold, the investigators found that the overall cumulative incidence of VTE in the high-risk group was 44.0%, compared with 10.5% in the low-risk group. Temporal analysis showed a widening disparity between the two groups, from 3 months (28.8% vs. 6.3%), to 6 months (41.1% vs. 7.9%), and 12 months (42.5% vs. 9.3%).
When asked if treatment type was evaluated, Dr. Lazo-Langner said that treatment type was evaluated but proved unfruitful for the model, which is designed for universal use in leukemia.
“We did include a number of different chemotherapy regimens,” he said. “The problem is, because we included both AML [acute myeloid leukemia] and ALL [acute lymphoblastic leukemia] lineage, and the cornerstone of treatment is different for both lineages. It’s difficult to actually include what kind of chemotherapy [patients had]. For instance, it is known that anthracyclines increase risk of thrombosis, but in both lineages, you use anthracyclines, so you really cannot use that as a predictor.”
Looking to the future, the next step will be validation in other cohorts. If this is successful, then Dr. Lazo-Langner speculated that clinicians could use the scoring system to direct monitoring and treatment. For example, patients with high scores and low platelet counts could receive earlier transfusional support, while all high-risk patients could be placed under more intensive surveillance and given additional education about thrombosis.
“I think recognizing symptoms early is important,” Dr. Lazo-Langner said, “and that would be training not only clinicians, but also nursing personnel and the patients themselves to be aware of the symptoms, so they can actually recognize them sooner.”
The study was funded by the Canadian Institutes of Health Research. Dr. Lazo-Langner is an investigator with the Canadian Venous Thromboembolism Clinical Trials and Outcomes Research (CanVECTOR) Network.
SOURCE: Lazo-Langner A et al. EHA 2019, Abstract S1642.
AMSTERDAM – A new clinical prediction model can determine the risk of venous thromboembolism in patients with leukemia, according to investigators.
The scoring system, which incorporates historical, morphological, and cytologic factors, was internally validated at multiple time points over the course of a year, reported lead author, Alejandro Lazo-Langner, MD, of the University of Western Ontario, London.
“It is important that we can predict or anticipate which patients [with acute leukemia] will develop venous thrombosis so that we can develop preventions and aim for better surveillance strategies,” Dr. Lazo-Langner said at the annual congress of the European Hematology Association. Venous thromboembolism (VTE) risk modeling is available for patients with solid tumors, but a similar prognostic tool for leukemia patients has been missing.
To fill this practice gap, Dr. Lazo-Langner and colleagues conducted a retrospective cohort study involving 501 patients with acute leukemia who were diagnosed between 2006 and 2017. Of these patients, 427 (85.2%) had myeloid lineage and 74 (14.8%) had lymphoblastic disease. VTE outcomes of interest included proximal lower- and upper-extremity deep vein thrombosis; pulmonary embolism; and thrombosis of unusual sites, such as splanchnic and cerebral. Patients were followed until last follow-up, VTE, or death. Single variable and multiple variable logistic regression were used sequentially to evaluate and confirm potential predictive factors, with nonparametric bootstrapping for internal validation.
After last follow-up, 77 patients (15.3%) had developed VTE; specifically, 44 patients had upper-extremity deep vein thrombosis, 28 had lower-extremity deep vein thrombosis or pulmonary embolism, and 5 had cerebral vein thrombosis. The median time from leukemia diagnosis to VTE was approximately 2 months (64 days). Out of 20 possible predictive factors, 7 were included in the multivariable model, and 3 constitute the final model. These three factors are platelet count greater than 50 x 109/L at time of diagnosis (1 point), lymphoblastic leukemia (2 points), and previous history of venous thromboembolism (3 points).
Dr. Lazo-Langner explained that leukemia patients at high risk of VTE are those with a score of 3 or more points. Using this risk threshold, the investigators found that the overall cumulative incidence of VTE in the high-risk group was 44.0%, compared with 10.5% in the low-risk group. Temporal analysis showed a widening disparity between the two groups, from 3 months (28.8% vs. 6.3%), to 6 months (41.1% vs. 7.9%), and 12 months (42.5% vs. 9.3%).
When asked if treatment type was evaluated, Dr. Lazo-Langner said that treatment type was evaluated but proved unfruitful for the model, which is designed for universal use in leukemia.
“We did include a number of different chemotherapy regimens,” he said. “The problem is, because we included both AML [acute myeloid leukemia] and ALL [acute lymphoblastic leukemia] lineage, and the cornerstone of treatment is different for both lineages. It’s difficult to actually include what kind of chemotherapy [patients had]. For instance, it is known that anthracyclines increase risk of thrombosis, but in both lineages, you use anthracyclines, so you really cannot use that as a predictor.”
Looking to the future, the next step will be validation in other cohorts. If this is successful, then Dr. Lazo-Langner speculated that clinicians could use the scoring system to direct monitoring and treatment. For example, patients with high scores and low platelet counts could receive earlier transfusional support, while all high-risk patients could be placed under more intensive surveillance and given additional education about thrombosis.
“I think recognizing symptoms early is important,” Dr. Lazo-Langner said, “and that would be training not only clinicians, but also nursing personnel and the patients themselves to be aware of the symptoms, so they can actually recognize them sooner.”
The study was funded by the Canadian Institutes of Health Research. Dr. Lazo-Langner is an investigator with the Canadian Venous Thromboembolism Clinical Trials and Outcomes Research (CanVECTOR) Network.
SOURCE: Lazo-Langner A et al. EHA 2019, Abstract S1642.
REPORTING FROM EHA CONGRESS
AML variants before transplant signal need for aggressive therapy
AMSTERDAM – Patients with acute myeloid leukemia who were in morphological complete remission prior to allogeneic hematopoietic cell transplant but had genomic evidence of a lingering AML variant had worse posttransplant outcomes when they underwent reduced-intensity conditioning, rather than myeloablative conditioning, investigators reported.

Among adults with AML in remission after induction therapy who were randomized in a clinical trial to either reduced-intensity conditioning (RIC) or myeloablative conditioning prior to transplant, those with known AML variants detected with ultra-deep genomic sequencing who underwent RIC had significantly greater risk for relapse, decreased disease-free survival (DFS), and worse overall survival (OS), compared with similar patients who underwent myeloablative conditioning (MAC), reported Christopher S. Hourigan, DM, DPhil, of the Laboratory of Myeloid Malignancies at the National Heart, Lung, and Blood Institute in Bethesda, Md.
The findings suggest that those patients with pretransplant AML variants who can tolerate MAC should get it, and that investigators need to find new options for patients who can’t, he said in an interview at the annual congress of the European Hematology Association.
“If I wasn’t a lab investigator and was a clinical trialist, I would be very excited about doing some randomized trials now to try see about novel targeted agents. For example, we have FLT3 inhibitors, we have IDH1 and IDH2 inhibitors, and I would be looking to try to combine reduced-intensity conditioning with additional therapy to try to lower the relapse rate for that group at the highest risk,” he said.
Previous studies have shown that, regardless of the method used – flow cytometry, quantitative polymerase chain reaction, or next-generation sequencing – minimal residual disease (MRD) detected in patients with AML in complete remission prior to transplant is associated with both cumulative incidence of relapse and worse overall survival.
Measurable, not minimal
Dr. Hourigan contends that the word “minimal” – the “M” in “MRD” – is a misnomer and should be replaced by the word “measurable,” because MRD really reflects the limitations of disease-detection technology.
“If you tell patients ‘you have minimal residual disease, and you have a huge chance of dying over the next few years,’ there’s nothing minimal about that,” he said.
The fundamental question that Dr. Hourigan and colleagues asked is, “is MRD just useful for predicting prognosis? Is this fate, or can we as doctors do something about it?”
To get answers, they examined whole-blood samples from patients enrolled in the BMT CTN 0901 trial, which compared survival and other outcomes following allogeneic hematopoietic stem cell transplants (allo-HSCT) with either RIC or MAC for pretransplant conditioning in patients with AML or the myelodysplastic syndrome.
The trial was halted early after just 272 of a planned 356 patients were enrolled, following evidence of a significantly higher relapse rate among patients who had undergone RIC.
“Strikingly, over half the AML patients receiving RIC relapsed within 18 months after getting transplants,” Dr. Hourigan said.
Relapse, survival differences
For this substudy, the National Institutes of Health investigators developed a custom 13-gene panel that would detect at least one AML variant in approximately 80% of patients who were included in a previous study of genomic classification and prognosis in AML.
They used ultra-deep genomic sequencing to look for variants in blood samples from 188 patients in BMT CTN 0901. There were no variants detected in the blood of 31% of patients who had undergone MAC or in 33% of those who had undergone RIC.
Among patients who did have detectable variants, the average number of variants per patient was 2.5.
In this cohort, transplant-related mortality (TRM) was higher with MAC at 27% vs. 20% with RIC at 3 years, but there were no differences in TRM within conditioning arms for patients, with or without AML variants.
Relapse rates in the cohort studied by Dr. Hourigan and his colleagues were virtually identical to those seen in the full study set, with an 18-month relapse rate of 16% for patients treated with MAC vs. 51% for those treated with RIC.
Among patients randomized to RIC, 3-year relapse rates were 57% for patients with detectable pretransplant AML variants, compared with 32% for those without variants (P less than .001).
Although there were no significant differences in 3-year OS by variant status among patients assigned to MAC, variant-positive patients assigned to RIC had significantly worse 3-year OS than those without variants (P = .04).
Among patients with no detectable variants, there were no significant differences in OS between the MAC or RIC arms. However, among patients with variants, survival was significantly worse with RIC (P = .02).
In multivariate analysis controlling for disease risk and donor group among patients who tested positive for an AML variant pretransplant, RIC was significantly associated with an increased risk for relapse (hazard ratio, 5.98; P less than .001); decreased DFS (HR, 2.80; P less than .001), and worse OS (HR, 2.16; P = .003).
“This study provides evidence that intervention for AML patients with evidence of MRD can result in improved survival,” Dr. Hourigan said.
Questions that still need to be addressed include whether variants in different genes confer different degrees of relapse risk, whether next-generation sequencing positivity is equivalent to MRD positivity, and whether the 13-gene panel could be improved upon to lower the chance for false negatives, he said.
The study was supported by the NIH. Dr. Hourigan reported research funding from Merck and Sellas Life Sciences AG, research collaboration with Qiagen and Archer, advisory board participation as an NIH official duty for Janssen and Novartis, and part-time employment with the Johns Hopkins School of Medicine.
SOURCE: Hourigan CS et al. EHA Congress, Abstract LB2600.
AMSTERDAM – Patients with acute myeloid leukemia who were in morphological complete remission prior to allogeneic hematopoietic cell transplant but had genomic evidence of a lingering AML variant had worse posttransplant outcomes when they underwent reduced-intensity conditioning, rather than myeloablative conditioning, investigators reported.
Among adults with AML in remission after induction therapy who were randomized in a clinical trial to either reduced-intensity conditioning (RIC) or myeloablative conditioning prior to transplant, those with known AML variants detected with ultra-deep genomic sequencing who underwent RIC had significantly greater risk for relapse, decreased disease-free survival (DFS), and worse overall survival (OS), compared with similar patients who underwent myeloablative conditioning (MAC), reported Christopher S. Hourigan, DM, DPhil, of the Laboratory of Myeloid Malignancies at the National Heart, Lung, and Blood Institute in Bethesda, Md.
The findings suggest that those patients with pretransplant AML variants who can tolerate MAC should get it, and that investigators need to find new options for patients who can’t, he said in an interview at the annual congress of the European Hematology Association.
“If I wasn’t a lab investigator and was a clinical trialist, I would be very excited about doing some randomized trials now to try see about novel targeted agents. For example, we have FLT3 inhibitors, we have IDH1 and IDH2 inhibitors, and I would be looking to try to combine reduced-intensity conditioning with additional therapy to try to lower the relapse rate for that group at the highest risk,” he said.
Previous studies have shown that, regardless of the method used – flow cytometry, quantitative polymerase chain reaction, or next-generation sequencing – minimal residual disease (MRD) detected in patients with AML in complete remission prior to transplant is associated with both cumulative incidence of relapse and worse overall survival.
Measurable, not minimal
Dr. Hourigan contends that the word “minimal” – the “M” in “MRD” – is a misnomer and should be replaced by the word “measurable,” because MRD really reflects the limitations of disease-detection technology.
“If you tell patients ‘you have minimal residual disease, and you have a huge chance of dying over the next few years,’ there’s nothing minimal about that,” he said.
The fundamental question that Dr. Hourigan and colleagues asked is, “is MRD just useful for predicting prognosis? Is this fate, or can we as doctors do something about it?”
To get answers, they examined whole-blood samples from patients enrolled in the BMT CTN 0901 trial, which compared survival and other outcomes following allogeneic hematopoietic stem cell transplants (allo-HSCT) with either RIC or MAC for pretransplant conditioning in patients with AML or the myelodysplastic syndrome.
The trial was halted early after just 272 of a planned 356 patients were enrolled, following evidence of a significantly higher relapse rate among patients who had undergone RIC.
“Strikingly, over half the AML patients receiving RIC relapsed within 18 months after getting transplants,” Dr. Hourigan said.
Relapse, survival differences
For this substudy, the National Institutes of Health investigators developed a custom 13-gene panel that would detect at least one AML variant in approximately 80% of patients who were included in a previous study of genomic classification and prognosis in AML.
They used ultra-deep genomic sequencing to look for variants in blood samples from 188 patients in BMT CTN 0901. There were no variants detected in the blood of 31% of patients who had undergone MAC or in 33% of those who had undergone RIC.
Among patients who did have detectable variants, the average number of variants per patient was 2.5.
In this cohort, transplant-related mortality (TRM) was higher with MAC at 27% vs. 20% with RIC at 3 years, but there were no differences in TRM within conditioning arms for patients, with or without AML variants.
Relapse rates in the cohort studied by Dr. Hourigan and his colleagues were virtually identical to those seen in the full study set, with an 18-month relapse rate of 16% for patients treated with MAC vs. 51% for those treated with RIC.
Among patients randomized to RIC, 3-year relapse rates were 57% for patients with detectable pretransplant AML variants, compared with 32% for those without variants (P less than .001).
Although there were no significant differences in 3-year OS by variant status among patients assigned to MAC, variant-positive patients assigned to RIC had significantly worse 3-year OS than those without variants (P = .04).
Among patients with no detectable variants, there were no significant differences in OS between the MAC or RIC arms. However, among patients with variants, survival was significantly worse with RIC (P = .02).
In multivariate analysis controlling for disease risk and donor group among patients who tested positive for an AML variant pretransplant, RIC was significantly associated with an increased risk for relapse (hazard ratio, 5.98; P less than .001); decreased DFS (HR, 2.80; P less than .001), and worse OS (HR, 2.16; P = .003).
“This study provides evidence that intervention for AML patients with evidence of MRD can result in improved survival,” Dr. Hourigan said.
Questions that still need to be addressed include whether variants in different genes confer different degrees of relapse risk, whether next-generation sequencing positivity is equivalent to MRD positivity, and whether the 13-gene panel could be improved upon to lower the chance for false negatives, he said.
The study was supported by the NIH. Dr. Hourigan reported research funding from Merck and Sellas Life Sciences AG, research collaboration with Qiagen and Archer, advisory board participation as an NIH official duty for Janssen and Novartis, and part-time employment with the Johns Hopkins School of Medicine.
SOURCE: Hourigan CS et al. EHA Congress, Abstract LB2600.
AMSTERDAM – Patients with acute myeloid leukemia who were in morphological complete remission prior to allogeneic hematopoietic cell transplant but had genomic evidence of a lingering AML variant had worse posttransplant outcomes when they underwent reduced-intensity conditioning, rather than myeloablative conditioning, investigators reported.
Among adults with AML in remission after induction therapy who were randomized in a clinical trial to either reduced-intensity conditioning (RIC) or myeloablative conditioning prior to transplant, those with known AML variants detected with ultra-deep genomic sequencing who underwent RIC had significantly greater risk for relapse, decreased disease-free survival (DFS), and worse overall survival (OS), compared with similar patients who underwent myeloablative conditioning (MAC), reported Christopher S. Hourigan, DM, DPhil, of the Laboratory of Myeloid Malignancies at the National Heart, Lung, and Blood Institute in Bethesda, Md.
The findings suggest that those patients with pretransplant AML variants who can tolerate MAC should get it, and that investigators need to find new options for patients who can’t, he said in an interview at the annual congress of the European Hematology Association.
“If I wasn’t a lab investigator and was a clinical trialist, I would be very excited about doing some randomized trials now to try see about novel targeted agents. For example, we have FLT3 inhibitors, we have IDH1 and IDH2 inhibitors, and I would be looking to try to combine reduced-intensity conditioning with additional therapy to try to lower the relapse rate for that group at the highest risk,” he said.
Previous studies have shown that, regardless of the method used – flow cytometry, quantitative polymerase chain reaction, or next-generation sequencing – minimal residual disease (MRD) detected in patients with AML in complete remission prior to transplant is associated with both cumulative incidence of relapse and worse overall survival.
Measurable, not minimal
Dr. Hourigan contends that the word “minimal” – the “M” in “MRD” – is a misnomer and should be replaced by the word “measurable,” because MRD really reflects the limitations of disease-detection technology.
“If you tell patients ‘you have minimal residual disease, and you have a huge chance of dying over the next few years,’ there’s nothing minimal about that,” he said.
The fundamental question that Dr. Hourigan and colleagues asked is, “is MRD just useful for predicting prognosis? Is this fate, or can we as doctors do something about it?”
To get answers, they examined whole-blood samples from patients enrolled in the BMT CTN 0901 trial, which compared survival and other outcomes following allogeneic hematopoietic stem cell transplants (allo-HSCT) with either RIC or MAC for pretransplant conditioning in patients with AML or the myelodysplastic syndrome.
The trial was halted early after just 272 of a planned 356 patients were enrolled, following evidence of a significantly higher relapse rate among patients who had undergone RIC.
“Strikingly, over half the AML patients receiving RIC relapsed within 18 months after getting transplants,” Dr. Hourigan said.
Relapse, survival differences
For this substudy, the National Institutes of Health investigators developed a custom 13-gene panel that would detect at least one AML variant in approximately 80% of patients who were included in a previous study of genomic classification and prognosis in AML.
They used ultra-deep genomic sequencing to look for variants in blood samples from 188 patients in BMT CTN 0901. There were no variants detected in the blood of 31% of patients who had undergone MAC or in 33% of those who had undergone RIC.
Among patients who did have detectable variants, the average number of variants per patient was 2.5.
In this cohort, transplant-related mortality (TRM) was higher with MAC at 27% vs. 20% with RIC at 3 years, but there were no differences in TRM within conditioning arms for patients, with or without AML variants.
Relapse rates in the cohort studied by Dr. Hourigan and his colleagues were virtually identical to those seen in the full study set, with an 18-month relapse rate of 16% for patients treated with MAC vs. 51% for those treated with RIC.
Among patients randomized to RIC, 3-year relapse rates were 57% for patients with detectable pretransplant AML variants, compared with 32% for those without variants (P less than .001).
Although there were no significant differences in 3-year OS by variant status among patients assigned to MAC, variant-positive patients assigned to RIC had significantly worse 3-year OS than those without variants (P = .04).
Among patients with no detectable variants, there were no significant differences in OS between the MAC or RIC arms. However, among patients with variants, survival was significantly worse with RIC (P = .02).
In multivariate analysis controlling for disease risk and donor group among patients who tested positive for an AML variant pretransplant, RIC was significantly associated with an increased risk for relapse (hazard ratio, 5.98; P less than .001); decreased DFS (HR, 2.80; P less than .001), and worse OS (HR, 2.16; P = .003).
“This study provides evidence that intervention for AML patients with evidence of MRD can result in improved survival,” Dr. Hourigan said.
Questions that still need to be addressed include whether variants in different genes confer different degrees of relapse risk, whether next-generation sequencing positivity is equivalent to MRD positivity, and whether the 13-gene panel could be improved upon to lower the chance for false negatives, he said.
The study was supported by the NIH. Dr. Hourigan reported research funding from Merck and Sellas Life Sciences AG, research collaboration with Qiagen and Archer, advisory board participation as an NIH official duty for Janssen and Novartis, and part-time employment with the Johns Hopkins School of Medicine.
SOURCE: Hourigan CS et al. EHA Congress, Abstract LB2600.
REPORTING FROM EHA CONGRESS
For tough AML, half respond to selinexor plus chemotherapy
AMSTERDAM – Patients with relapsed or refractory acute myeloid leukemia (AML) may be more likely to respond when selinexor is added to standard chemotherapy, according to investigators.

In a recent phase 2 trial, selinexor given with cytarabine and idarubicin led to a 50% overall response rate, reported lead author Walter Fiedler, MD, of University Medical Center Hamburg-Eppendorf (Germany). This response rate is at the upper end of what has been seen in published studies, Dr. Fiedler said at the annual congress of the European Hematology Association.
He also noted that giving a flat dose of selinexor improved tolerability in the trial, a significant finding in light of common adverse events and recent concerns from the Food and Drug Administration about the safety of selinexor for patients with multiple myeloma.
“The rationale to employ selinexor in this study is that there is a synergy between anthracyclines and selinexor,” Dr. Fiedler said, which may restore anthracycline sensitivity in relapsed or refractory patients. “Secondly, there is a c-myc reduction pathway that leads to a reduction of DNA damage repair genes such as Rad51 and Chk1, and this might result in inhibition of homologous recombination.”
The study involved 44 patients with relapsed or refractory AML, of whom 17 (39%) had previously received stem cell transplantation and 11 (25%) exhibited therapy-induced or secondary disease. The median patient age was 59.5 years.
Patients were given idarubicin 10 mg/m2 on days 1, 3, and 5, and cytarabine 100 mg/m2 on days 1-7. Initially, selinexor was given at a dose of 40 mg/m2 twice per week for 4 weeks, but this led to high rates of febrile neutropenia and grade 3 or higher diarrhea, along with prolonged aplasia. In response to this issue, after the first 27 patients, the dose was reduced to a flat amount of 60 mg, given twice weekly for 3 weeks.
For patients not undergoing transplantation after the first or second induction cycle, selinexor maintenance monotherapy was offered for up to 1 year.
The primary endpoint was overall remission rate, reported as complete remission, complete remission with incomplete blood count recovery, and morphological leukemia-free status. Secondary endpoints included the rate of partial remissions, percentage of patients being transplanted after induction, early death rate, overall survival, event-free survival, and relapse-free survival.
The efficacy analysis revealed an overall response rate of 50%. A total of 9 patients had complete remission (21.4%), 11 achieved complete remission with incomplete blood count recovery (26.2%), and 1 exhibited morphological leukemia-free status (2.4%). Of note, almost half of the patients (47%) who had relapsed after previous stem cell transplantation responded, as did three-quarters who tested positive for an NPM1 mutation. After a median follow-up of 8.2 months, the median overall survival was 8.2 months, relapse-free survival was 17.7 months, and event-free survival was 4.9 months.
Adverse events occurred frequently, with a majority of patients experiencing nausea (86%), diarrhea (83%), vomiting (74%), decreased appetite (71%), febrile neutropenia (67%), fatigue (64%), leukopenia (62%), thrombocytopenia (62%), or anemia (60%).
Grade 3 or higher adverse events were almost as common, and included febrile neutropenia (67%), leukopenia (62%), thrombocytopenia (62%), anemia (57%), and diarrhea (50%). Reducing the dose did improve tolerability, with notable drops in the rate of severe diarrhea (56% vs. 40%) and febrile neutropenia (85% vs. 33%). In total, 19% of patients discontinued treatment because of adverse events.
A total of 25 patients (60%) died during the study, with about half dying from disease progression (n = 12), and fewer succumbing to infectious complications, graft-versus-host disease, multiorgan failure, multiple brain infarct, or asystole. Two deaths, one from suspected hemophagocytosis and another from systemic inflammatory response syndrome, were considered possibly related to selinexor.
“The results should be further evaluated in a phase 3 study,” Dr. Fiedler said. However, plans for this are not yet underway, he said, adding that Karyopharm Therapeutics will be focusing its efforts on selinexor for myeloma first.
The study was funded by Karyopharm. Dr. Fielder reported financial relationships with Amgen, Pfizer, Jazz Pharmaceuticals, and other companies.
SOURCE: Fiedler W et al. EHA Congress, Abstract S880.
AMSTERDAM – Patients with relapsed or refractory acute myeloid leukemia (AML) may be more likely to respond when selinexor is added to standard chemotherapy, according to investigators.
In a recent phase 2 trial, selinexor given with cytarabine and idarubicin led to a 50% overall response rate, reported lead author Walter Fiedler, MD, of University Medical Center Hamburg-Eppendorf (Germany). This response rate is at the upper end of what has been seen in published studies, Dr. Fiedler said at the annual congress of the European Hematology Association.
He also noted that giving a flat dose of selinexor improved tolerability in the trial, a significant finding in light of common adverse events and recent concerns from the Food and Drug Administration about the safety of selinexor for patients with multiple myeloma.
“The rationale to employ selinexor in this study is that there is a synergy between anthracyclines and selinexor,” Dr. Fiedler said, which may restore anthracycline sensitivity in relapsed or refractory patients. “Secondly, there is a c-myc reduction pathway that leads to a reduction of DNA damage repair genes such as Rad51 and Chk1, and this might result in inhibition of homologous recombination.”
The study involved 44 patients with relapsed or refractory AML, of whom 17 (39%) had previously received stem cell transplantation and 11 (25%) exhibited therapy-induced or secondary disease. The median patient age was 59.5 years.
Patients were given idarubicin 10 mg/m2 on days 1, 3, and 5, and cytarabine 100 mg/m2 on days 1-7. Initially, selinexor was given at a dose of 40 mg/m2 twice per week for 4 weeks, but this led to high rates of febrile neutropenia and grade 3 or higher diarrhea, along with prolonged aplasia. In response to this issue, after the first 27 patients, the dose was reduced to a flat amount of 60 mg, given twice weekly for 3 weeks.
For patients not undergoing transplantation after the first or second induction cycle, selinexor maintenance monotherapy was offered for up to 1 year.
The primary endpoint was overall remission rate, reported as complete remission, complete remission with incomplete blood count recovery, and morphological leukemia-free status. Secondary endpoints included the rate of partial remissions, percentage of patients being transplanted after induction, early death rate, overall survival, event-free survival, and relapse-free survival.
The efficacy analysis revealed an overall response rate of 50%. A total of 9 patients had complete remission (21.4%), 11 achieved complete remission with incomplete blood count recovery (26.2%), and 1 exhibited morphological leukemia-free status (2.4%). Of note, almost half of the patients (47%) who had relapsed after previous stem cell transplantation responded, as did three-quarters who tested positive for an NPM1 mutation. After a median follow-up of 8.2 months, the median overall survival was 8.2 months, relapse-free survival was 17.7 months, and event-free survival was 4.9 months.
Adverse events occurred frequently, with a majority of patients experiencing nausea (86%), diarrhea (83%), vomiting (74%), decreased appetite (71%), febrile neutropenia (67%), fatigue (64%), leukopenia (62%), thrombocytopenia (62%), or anemia (60%).
Grade 3 or higher adverse events were almost as common, and included febrile neutropenia (67%), leukopenia (62%), thrombocytopenia (62%), anemia (57%), and diarrhea (50%). Reducing the dose did improve tolerability, with notable drops in the rate of severe diarrhea (56% vs. 40%) and febrile neutropenia (85% vs. 33%). In total, 19% of patients discontinued treatment because of adverse events.
A total of 25 patients (60%) died during the study, with about half dying from disease progression (n = 12), and fewer succumbing to infectious complications, graft-versus-host disease, multiorgan failure, multiple brain infarct, or asystole. Two deaths, one from suspected hemophagocytosis and another from systemic inflammatory response syndrome, were considered possibly related to selinexor.
“The results should be further evaluated in a phase 3 study,” Dr. Fiedler said. However, plans for this are not yet underway, he said, adding that Karyopharm Therapeutics will be focusing its efforts on selinexor for myeloma first.
The study was funded by Karyopharm. Dr. Fielder reported financial relationships with Amgen, Pfizer, Jazz Pharmaceuticals, and other companies.
SOURCE: Fiedler W et al. EHA Congress, Abstract S880.
AMSTERDAM – Patients with relapsed or refractory acute myeloid leukemia (AML) may be more likely to respond when selinexor is added to standard chemotherapy, according to investigators.
In a recent phase 2 trial, selinexor given with cytarabine and idarubicin led to a 50% overall response rate, reported lead author Walter Fiedler, MD, of University Medical Center Hamburg-Eppendorf (Germany). This response rate is at the upper end of what has been seen in published studies, Dr. Fiedler said at the annual congress of the European Hematology Association.
He also noted that giving a flat dose of selinexor improved tolerability in the trial, a significant finding in light of common adverse events and recent concerns from the Food and Drug Administration about the safety of selinexor for patients with multiple myeloma.
“The rationale to employ selinexor in this study is that there is a synergy between anthracyclines and selinexor,” Dr. Fiedler said, which may restore anthracycline sensitivity in relapsed or refractory patients. “Secondly, there is a c-myc reduction pathway that leads to a reduction of DNA damage repair genes such as Rad51 and Chk1, and this might result in inhibition of homologous recombination.”
The study involved 44 patients with relapsed or refractory AML, of whom 17 (39%) had previously received stem cell transplantation and 11 (25%) exhibited therapy-induced or secondary disease. The median patient age was 59.5 years.
Patients were given idarubicin 10 mg/m2 on days 1, 3, and 5, and cytarabine 100 mg/m2 on days 1-7. Initially, selinexor was given at a dose of 40 mg/m2 twice per week for 4 weeks, but this led to high rates of febrile neutropenia and grade 3 or higher diarrhea, along with prolonged aplasia. In response to this issue, after the first 27 patients, the dose was reduced to a flat amount of 60 mg, given twice weekly for 3 weeks.
For patients not undergoing transplantation after the first or second induction cycle, selinexor maintenance monotherapy was offered for up to 1 year.
The primary endpoint was overall remission rate, reported as complete remission, complete remission with incomplete blood count recovery, and morphological leukemia-free status. Secondary endpoints included the rate of partial remissions, percentage of patients being transplanted after induction, early death rate, overall survival, event-free survival, and relapse-free survival.
The efficacy analysis revealed an overall response rate of 50%. A total of 9 patients had complete remission (21.4%), 11 achieved complete remission with incomplete blood count recovery (26.2%), and 1 exhibited morphological leukemia-free status (2.4%). Of note, almost half of the patients (47%) who had relapsed after previous stem cell transplantation responded, as did three-quarters who tested positive for an NPM1 mutation. After a median follow-up of 8.2 months, the median overall survival was 8.2 months, relapse-free survival was 17.7 months, and event-free survival was 4.9 months.
Adverse events occurred frequently, with a majority of patients experiencing nausea (86%), diarrhea (83%), vomiting (74%), decreased appetite (71%), febrile neutropenia (67%), fatigue (64%), leukopenia (62%), thrombocytopenia (62%), or anemia (60%).
Grade 3 or higher adverse events were almost as common, and included febrile neutropenia (67%), leukopenia (62%), thrombocytopenia (62%), anemia (57%), and diarrhea (50%). Reducing the dose did improve tolerability, with notable drops in the rate of severe diarrhea (56% vs. 40%) and febrile neutropenia (85% vs. 33%). In total, 19% of patients discontinued treatment because of adverse events.
A total of 25 patients (60%) died during the study, with about half dying from disease progression (n = 12), and fewer succumbing to infectious complications, graft-versus-host disease, multiorgan failure, multiple brain infarct, or asystole. Two deaths, one from suspected hemophagocytosis and another from systemic inflammatory response syndrome, were considered possibly related to selinexor.
“The results should be further evaluated in a phase 3 study,” Dr. Fiedler said. However, plans for this are not yet underway, he said, adding that Karyopharm Therapeutics will be focusing its efforts on selinexor for myeloma first.
The study was funded by Karyopharm. Dr. Fielder reported financial relationships with Amgen, Pfizer, Jazz Pharmaceuticals, and other companies.
SOURCE: Fiedler W et al. EHA Congress, Abstract S880.
REPORTING FROM EHA CONGRESS
Hedgehog signaling offers prognostic, therapeutic potential in CLL
Activation of hedgehog (Hh) signaling could predict early disease progression in chronic lymphocytic leukemia (CLL) and offer a new therapeutic target, according to investigators.
Approximately 11% of treatment-naive patients had mutations associated with Hh signaling, reported lead author Emanuela M. Ghia, PhD, of the University of California, San Diego, and colleagues. In addition to early progression, Hh signaling was associated with expression of GLI1, which could be a future therapeutic target. In support of this possibility, in vitro experimentation with a GLI1 inhibitor showed high cytotoxicity for GLI1-positive CLL cells.
“Targeting GLI1 could block ligand-independent and ligand-dependent Hh pathway activation and perhaps overcome the apparent resistance to SMO inhibitors,” the investigators wrote in Blood.
Using the HALT Pan-Leukemia Gene Panel, which includes 103 genes, the investigators tested for mutations in cell samples from 841 patients with treatment-naive CLL. Specifically, the investigators focused on mutations that did not map to seven well-known signaling/metabolic pathways, such as Wnt and Notch.
This strategy revealed that 89 patients (11%) had nonsynonymous mutations in genes that drive Hh signaling. These mutations were highly associated with GLI1 expression (x2 test; P less than .0001), which stands to reason, as GLI1 is the main effector of the Hh signaling pathway. Of note, 62 of the 161 patients (38%) who did not test positive for an Hh pathway mutation still tested positive for GLI1, suggesting that they had Hh pathway activation, albeit without an identifiable mutational cause.
These findings are clinically significant, as GLI1 overexpression has been linked to numerous types of cancer and is an adverse prognostic indicator for acute myeloid leukemia and several carcinomas, the investigators wrote.
Considering these associations with other cancer types, the investigators looked for a relationship between GLI1 expression and outcomes in CLL. Comparing 103 patients with GLI1-positive disease with 107 GLI-negative patients revealed that GLI1 positivity was significantly associated with shorter median treatment-free survival (4.7 vs. 6.4 years; P = .002). Additional analysis showed that this prognostic relationship was present regardless of IgVH mutational status.
Based on these findings, the investigators concluded that “[a]ctivation of the Hh pathway can strongly influence disease progression in CLL.”
Two additional tests showed that GLI1-positivity was associated with cell survival. First, silencing GLI1 with a GLI1-specific small interfering RNA led to decreased cell viability. Second, GANT61, a small molecule inhibitor of GLI1, was highly cytotoxic for GLI1-positive cells. According to the investigators, these findings suggest that GLI1 could be a future therapeutic target.
“[T]his report shows that the Hh pathway frequently is activated in CLL and associated with relatively rapid disease progression, while identifying a new avenue for therapeutic intervention for patients with this disease,” they concluded.
The study was funded by the National Institutes of Health, the Cancer Stem Cell Consortium, the Canadian Institutes of Health Research, the Ontario Institute for Cancer Research, and other groups. The investigators reported having no competing financial interests.
SOURCE: Ghia EM et al. Blood. 2019 Jun 20;133(25):2651-63.
Activation of hedgehog (Hh) signaling could predict early disease progression in chronic lymphocytic leukemia (CLL) and offer a new therapeutic target, according to investigators.
Approximately 11% of treatment-naive patients had mutations associated with Hh signaling, reported lead author Emanuela M. Ghia, PhD, of the University of California, San Diego, and colleagues. In addition to early progression, Hh signaling was associated with expression of GLI1, which could be a future therapeutic target. In support of this possibility, in vitro experimentation with a GLI1 inhibitor showed high cytotoxicity for GLI1-positive CLL cells.
“Targeting GLI1 could block ligand-independent and ligand-dependent Hh pathway activation and perhaps overcome the apparent resistance to SMO inhibitors,” the investigators wrote in Blood.
Using the HALT Pan-Leukemia Gene Panel, which includes 103 genes, the investigators tested for mutations in cell samples from 841 patients with treatment-naive CLL. Specifically, the investigators focused on mutations that did not map to seven well-known signaling/metabolic pathways, such as Wnt and Notch.
This strategy revealed that 89 patients (11%) had nonsynonymous mutations in genes that drive Hh signaling. These mutations were highly associated with GLI1 expression (x2 test; P less than .0001), which stands to reason, as GLI1 is the main effector of the Hh signaling pathway. Of note, 62 of the 161 patients (38%) who did not test positive for an Hh pathway mutation still tested positive for GLI1, suggesting that they had Hh pathway activation, albeit without an identifiable mutational cause.
These findings are clinically significant, as GLI1 overexpression has been linked to numerous types of cancer and is an adverse prognostic indicator for acute myeloid leukemia and several carcinomas, the investigators wrote.
Considering these associations with other cancer types, the investigators looked for a relationship between GLI1 expression and outcomes in CLL. Comparing 103 patients with GLI1-positive disease with 107 GLI-negative patients revealed that GLI1 positivity was significantly associated with shorter median treatment-free survival (4.7 vs. 6.4 years; P = .002). Additional analysis showed that this prognostic relationship was present regardless of IgVH mutational status.
Based on these findings, the investigators concluded that “[a]ctivation of the Hh pathway can strongly influence disease progression in CLL.”
Two additional tests showed that GLI1-positivity was associated with cell survival. First, silencing GLI1 with a GLI1-specific small interfering RNA led to decreased cell viability. Second, GANT61, a small molecule inhibitor of GLI1, was highly cytotoxic for GLI1-positive cells. According to the investigators, these findings suggest that GLI1 could be a future therapeutic target.
“[T]his report shows that the Hh pathway frequently is activated in CLL and associated with relatively rapid disease progression, while identifying a new avenue for therapeutic intervention for patients with this disease,” they concluded.
The study was funded by the National Institutes of Health, the Cancer Stem Cell Consortium, the Canadian Institutes of Health Research, the Ontario Institute for Cancer Research, and other groups. The investigators reported having no competing financial interests.
SOURCE: Ghia EM et al. Blood. 2019 Jun 20;133(25):2651-63.
Activation of hedgehog (Hh) signaling could predict early disease progression in chronic lymphocytic leukemia (CLL) and offer a new therapeutic target, according to investigators.
Approximately 11% of treatment-naive patients had mutations associated with Hh signaling, reported lead author Emanuela M. Ghia, PhD, of the University of California, San Diego, and colleagues. In addition to early progression, Hh signaling was associated with expression of GLI1, which could be a future therapeutic target. In support of this possibility, in vitro experimentation with a GLI1 inhibitor showed high cytotoxicity for GLI1-positive CLL cells.
“Targeting GLI1 could block ligand-independent and ligand-dependent Hh pathway activation and perhaps overcome the apparent resistance to SMO inhibitors,” the investigators wrote in Blood.
Using the HALT Pan-Leukemia Gene Panel, which includes 103 genes, the investigators tested for mutations in cell samples from 841 patients with treatment-naive CLL. Specifically, the investigators focused on mutations that did not map to seven well-known signaling/metabolic pathways, such as Wnt and Notch.
This strategy revealed that 89 patients (11%) had nonsynonymous mutations in genes that drive Hh signaling. These mutations were highly associated with GLI1 expression (x2 test; P less than .0001), which stands to reason, as GLI1 is the main effector of the Hh signaling pathway. Of note, 62 of the 161 patients (38%) who did not test positive for an Hh pathway mutation still tested positive for GLI1, suggesting that they had Hh pathway activation, albeit without an identifiable mutational cause.
These findings are clinically significant, as GLI1 overexpression has been linked to numerous types of cancer and is an adverse prognostic indicator for acute myeloid leukemia and several carcinomas, the investigators wrote.
Considering these associations with other cancer types, the investigators looked for a relationship between GLI1 expression and outcomes in CLL. Comparing 103 patients with GLI1-positive disease with 107 GLI-negative patients revealed that GLI1 positivity was significantly associated with shorter median treatment-free survival (4.7 vs. 6.4 years; P = .002). Additional analysis showed that this prognostic relationship was present regardless of IgVH mutational status.
Based on these findings, the investigators concluded that “[a]ctivation of the Hh pathway can strongly influence disease progression in CLL.”
Two additional tests showed that GLI1-positivity was associated with cell survival. First, silencing GLI1 with a GLI1-specific small interfering RNA led to decreased cell viability. Second, GANT61, a small molecule inhibitor of GLI1, was highly cytotoxic for GLI1-positive cells. According to the investigators, these findings suggest that GLI1 could be a future therapeutic target.
“[T]his report shows that the Hh pathway frequently is activated in CLL and associated with relatively rapid disease progression, while identifying a new avenue for therapeutic intervention for patients with this disease,” they concluded.
The study was funded by the National Institutes of Health, the Cancer Stem Cell Consortium, the Canadian Institutes of Health Research, the Ontario Institute for Cancer Research, and other groups. The investigators reported having no competing financial interests.
SOURCE: Ghia EM et al. Blood. 2019 Jun 20;133(25):2651-63.
FROM BLOOD