User login
CAR T therapy to enter early testing in multiple myeloma
Janssen Biotech is launching a phase 1b/2 trial of an .
The trial, which was cleared by the Food and Drug Administration to begin in the second half of 2018, will evaluate the safety and efficacy of LCAR-B38M (JNJ-68284528). The CAR T therapy targets B-cell Maturation Antigen and expresses a CAR protein that is identical to a product that was developed by Legend Biotech and evaluated in a first-in-human clinical study in China.
The drug is being developed as part of a collaboration between Legend Biotech and Janssen Biotech.
Janssen Biotech is launching a phase 1b/2 trial of an .
The trial, which was cleared by the Food and Drug Administration to begin in the second half of 2018, will evaluate the safety and efficacy of LCAR-B38M (JNJ-68284528). The CAR T therapy targets B-cell Maturation Antigen and expresses a CAR protein that is identical to a product that was developed by Legend Biotech and evaluated in a first-in-human clinical study in China.
The drug is being developed as part of a collaboration between Legend Biotech and Janssen Biotech.
Janssen Biotech is launching a phase 1b/2 trial of an .
The trial, which was cleared by the Food and Drug Administration to begin in the second half of 2018, will evaluate the safety and efficacy of LCAR-B38M (JNJ-68284528). The CAR T therapy targets B-cell Maturation Antigen and expresses a CAR protein that is identical to a product that was developed by Legend Biotech and evaluated in a first-in-human clinical study in China.
The drug is being developed as part of a collaboration between Legend Biotech and Janssen Biotech.
Novel Neuroendocrine Tumor in Multiple Endocrine Neoplasia Type 1 (FULL)
Neuroendocrine tumors (NETs) are uncommon and can occur in the context of genetic conditions. Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant disorder of the tumor suppressor gene of the same name—MEN1, which encodes for the protein menin. Multiple endocrine neoplasia type 1 is characterized clinically by the presence of 2 or more of the following NETs: parathyroid, pituitary, and pancreaticoduodenal.1 Pancreaticoduodenal NETs occur in 30% to 80% of patients with MEN1 and have malignant potential. Although the majority of pancreaticoduodenal NETs are nonfunctioning, patients may present with symptoms secondary to mass effect.
Genetic testing exists for MEN1, but not all genetic mutations that cause MEN1 have been discovered. Therefore, because negative genetic testing does not rule out MEN1, a diagnosis is based on tumor type and location. Neuroendocrine tumors of the biliary tree are rare, and there
are no well-accepted guidelines on how to stage them.2-4 The following case demonstrates an unusual initial presentation of a NET in the context of MEN1.
Case Report
A 29-year-old, active-duty African-American man deployed in Kuwait presented with icterus, flank pain, and hematuria. His past medical history was significant for nephrolithiasis, and his family history was notable for hyperparathyroidism. Laboratory results showed primary hyperparathyroidism and evidence of biliary obstruction.
A sestamibi scan demonstrated uptake in a location corresponding with the right inferior parathyroid gland. A computed tomography (CT) scan showed nephrolithiasis and hepatic biliary ductal dilatation. Magnetic resonance cholangiopancreatography (MRCP) revealed both intra- and extrahepatic ductal dilatation, focal narrowing of the proximal common bile duct, and possible adenopathy that was concerning for cholangiocarcinoma. Endoscopic retrograde cholangiopancreatography (ERCP) demonstrated a 1 cm to 2 cm focal stricture within the mid-common bile duct with intra- and extrahepatic ductal dilatation (Figure 1). An endoscopy showed no masses in the duodenum, and anendoscopic ultrasound showed no masses in the pancreas. Endoscopic brushings and endoscopic, ultrasound-guided, fine-needle aspiration
cytology were nondiagnostic. Exploratory laparotomy revealed a dilated hepatic bile duct, an inflamed porta hepatis, and a mass involving the distal hepatic bile duct. 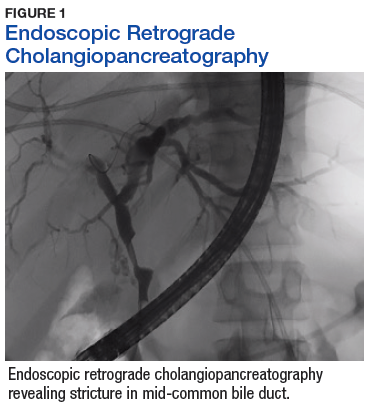
The patient underwent cholecystectomy, radical extra hepatic bile duct resection to the level of the hepatic bifurcation, and hepaticojejunostomy. Gross examination of the specimen showed a nodule centered in the distal common hepatic duct with an adjacent, 2-cm lymph node. The histologic examination revealed a neoplastic proliferation consisting of epithelioid cells with round nuclei and granular chromatin with amphophilic cytoplasm in a trabecular and nested architecture.
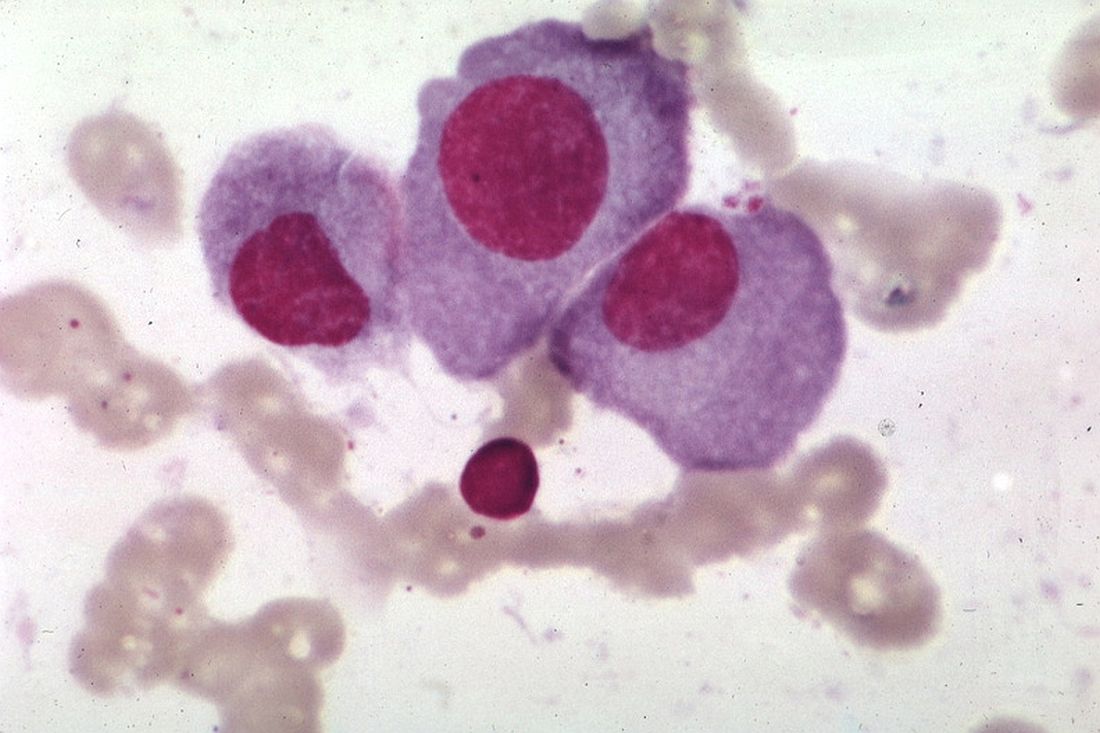
The tumor was centered in the submucosa, which is typical of gastrointestinal NETs (Figure 2). There was no evidence of direct tumor extension elsewhere. About 40% of the tumor cells contained eosinophilic, intracytoplasmic inclusions (Figure 3). The tumor did not involve the margins or lymph node. 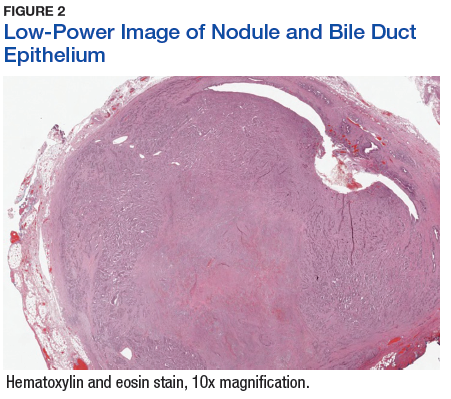
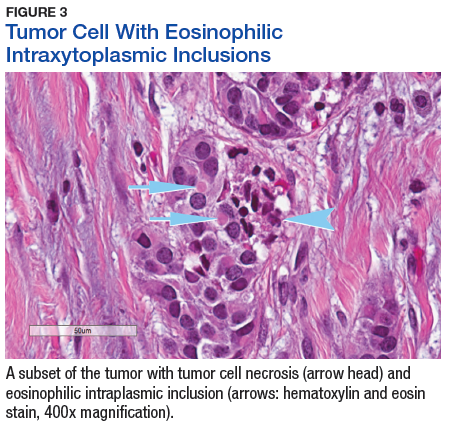
Positive staining with the neuroendocrine markers synaptophysin and chromagranin A confirmed a well-differentiated NET. The intracytoplasmic inclusions stained strongly positive for cytokeratin CAM 5.2. The tumor had higher-grade features, including tumor cell necrosis, a Ki-67 labeling index of 3%, and perineural invasion. The 2010 World Health Organization (WHO) criteria for NET of the digestive system classified this tumor as a grade 2, well-differentiated NET and as stage 1a (limited to the bile duct).4
Postoperatively, octreotide scan with single-photon emission computed tomography (SPECT)-CT did not show additional masses or lesions. Serum pancreatic polypeptide was elevated, with the remaining serum and plasma NET markers—including gastrin, glucagon, insulin, chromogranin A, and vasoactive intestinal polypeptide (VIP)—being within reference ranges. Genetic testing (GeneDx, Inc, Gaithersburg, MD) showed an E563X nonsense mutation in the MEN1 gene, confirming a MEN1 disorder. The patient then underwent a 4-gland parathyroidectomy with reimplantation; the parathyroid glands demonstrated hyperplasia in all 4 glands.
Biochemical follow-up at 14 months showed that the serum pancreatic polypeptide had normalized. There was no evidence of pituitary orpancreatic hypersecretion. The patient developed hypoparathyroidism, requiring calcium and calcitriol supplementation. Radiographic follow-up using abdominal magnetic resonance imaging at 16 months showed no evidence of disease.
Discussion
This case illustrates a genetic disease with an unusual initial presentation. Primary extrahepatic bile duct NETs are rare and have been reported previously in patients without MEN1.5-9 Neuroendocrine tumors in the hepatic bile duct in patients with MEN1 also have been reported but only after these tumors first appeared in the pancreas or duodenum.10 An extensive literature search revealed no prior reports extrahepatic bile duct NETs with MEN1 as the primary site or with biliary obstruction, which is why this patient’s presentation is particularly interesting.5,6,10-13 The table summarizes select reports of NETs. 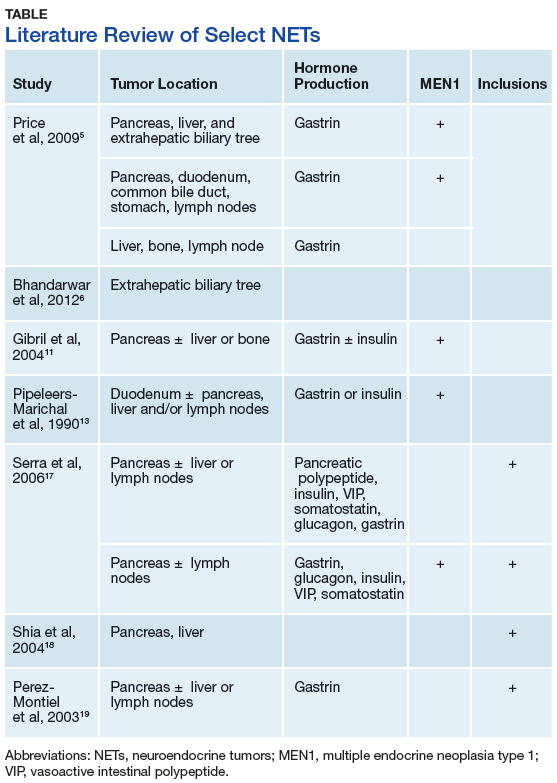
Tumor location in this patient was atypical, and genetic testing guided the management. Serum MEN1 genetic testing is indicated in patients with ≥ 2 tumors that are atypical but possibly associated with MEN1 (such as adrenal tumors, gastrinomas, and carcinoids) and in patients aged < 45 years with primary hyperparathyroidism.14,15 The patient in this study was aged 29 years and had hyperparathyroidism and an NET of the hepatic bile duct. This condition was sufficient to warrant genetic testing, the results of which affected the patient’s subsequent parathyroid surgery.15 Despite the suggestion of unifocal localization on the sestamibi scan, the patient underwent the more appropriate subtotal parathyroidectomy.14 The patient’s tumor most likely originated from a germline mutation of the MEN1 gene.
As a result of the patient’s genetic test results, his daughter also was tested. She was found to have the same mutation as her father and will undergo proper tumor surveillance for MEN1. There was no personal or family history of hemangioblastomas, renal cell carcinomas, or cystadenomas, which would have prompted testing for von Hippel-Lindau disease. Likewise, there was no personal or family history of café-au-lait macules and neurofibromas, which would have prompted testing for neurofibromatosis type 1.
Due to the paucity of cases, there are currently no well-accepted guidelines on how to stage extrahepatic biliary NETs.3-5,16 The WHO recommends staging according to adenocarcinomas of the gallbladder and bile duct.3 As such, the pathologic stage of this tumor would be stage 1a.
The significance of the intracytoplasmic inclusion in this case is unknown. Pancreatic NETs and neuroendocrine carcinomas have demonstrated intracytoplasmic inclusions that stain positively for keratin and may indicate more aggressive tumor behavior.17-19 In 1 report, electron microscopic examination demonstrated intermediate filaments with entrapped neurosecretory granules.18 In a series of 84 cases of pancreatic endocrine tumors, 14 had intracytoplasmic inclusions; of these, 5 had MEN1.17 In the present case, the patient continues to show no evidence of tumor recurrence at 16 months after resection.
Conclusion
Extrahepatic biliary neuroendocrine tumors are rare. Further investigation into biliary tree NET staging and future studies to determine the significance of intracytoplasmic inclusions may be beneficial. This case highlights the appropriate use of genetic testing and supports expanding the clinical diagnosis of MEN1 to include NETs of the extrahepatic bile duct.
Click here to read the digital edition.
1. Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: WB Saunders; 2011.
2. American Joint Committee on Cancer. Neuroendocrine Tumors. In: Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, eds. American Joint Committee on Cancer Staging Handbook. 7th ed. From the AJCC Cancer Staging Manual. New York, NY: Springer-Verlag; 2010:227-236.
3. Komminoth P, Arnold R, Capella C, et al. Neuroendocrine neoplasms of the gallbladder and extrahepatic bile ducts. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, et al, eds. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC Press; 2010:274-276.
4. Rindi G, Arnold R, Bosman FT. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, et al, eds. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC Press; 2010:13.
5. Price TN, Thompson GB, Lewis JT, Lloyd RV, Young WF. Zollinger-Ellison syndrome due to primary gastrinoma of the extrahepatic biliary tree: three case reports and review of literature. Endocr Pract. 2009;15(7):737-749.
6. Bhandarwar AH, Shaikh TA, Borisa AD, et al. Primary neuroendocrine tumor of the left hepatic duct: a case report with review of the literature. Case Rep Surg. 2012:786432.
7. Bhalla P, Powle V, Shah RC, Jagannath P. Neuroendocrine tumor of common hepatic duct. Indian J Gastroenterol. 2012;31(3):144-146.
8. Khan FA, Stevens-Chase A, Chaudhry R, Hashmi A, Edelman D, Weaver D. Extrahepatic biliary obstrution secondary to neuroendocrine tumor of the common hepatic duct. Int J Surg Case Rep. 2017;30:46-49.
9. Hong N, Kim HJ, Byun JH, et al. Neuroendocrine neoplasms of the extrahepatic bile duct: radiologic and clinical characteristics. Abdom Imaging. 2015;40(1):181-191.
10. Tonelli F, Giudici F, Nesi G, Batignani G, Brandi ML. Biliary tree gastrinomas in multiple endocrine neoplasia type 1 syndrome. World J Gastroenterol. 2013;19(45):8312-8320.
11. Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore). 2004;83(1):43-83.
12. Pieterman CRC, Conemans EB, Dreijerink KMA, et al. Thoracic and duodenopancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1: natural history and function of menin in tumorigenesis. Endocr Relat Cancer. 2014;21(3):R121-R142.
13. Pipeleers-Marichal M, Somers G, Willems G, et al. Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med. 1990;322(11):723-727.
14. Thakker RV, Newey PJ, Walls GV, et al; Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97(9):2990-3011.
15. Eastell R, Brandi ML, Costa AG, et al. Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99(10):3570-3579.
16. Michalopoulos N, Papavramidis TS, Karayannopoulou G, Pliakos I, Papavramidis ST, Kanellos I. Neuroendocrine tumors of extrahepatic biliary tract. Pathol Oncol Res. 2014;20(4):765-775.
17. Serra S, Asa SL, Chetty R. Intracytoplasmic inclusions (including the so-called “rhabdoid” phenotype) in pancreatic endocrine tumors. Endocr Pathol. 2006;17(1):75-81.
18. Shia J, Erlandson RA, Klimstra DS. Whorls of intermediate filaments with entrapped neurosecretory granules correspond to the “rhabdoid” inclusions seen in pancreatic endocrine
neoplasms. Am J Surg Pathol. 2004;28(2):271-273.
19. Perez-Montiel MD, Frankel WL, Suster S. Neuroendocrine carcinomas of the pancreas with ‘Rhabdoid’ features. Am J Surg Pathol. 2003;27(5):642-649.
Neuroendocrine tumors (NETs) are uncommon and can occur in the context of genetic conditions. Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant disorder of the tumor suppressor gene of the same name—MEN1, which encodes for the protein menin. Multiple endocrine neoplasia type 1 is characterized clinically by the presence of 2 or more of the following NETs: parathyroid, pituitary, and pancreaticoduodenal.1 Pancreaticoduodenal NETs occur in 30% to 80% of patients with MEN1 and have malignant potential. Although the majority of pancreaticoduodenal NETs are nonfunctioning, patients may present with symptoms secondary to mass effect.
Genetic testing exists for MEN1, but not all genetic mutations that cause MEN1 have been discovered. Therefore, because negative genetic testing does not rule out MEN1, a diagnosis is based on tumor type and location. Neuroendocrine tumors of the biliary tree are rare, and there
are no well-accepted guidelines on how to stage them.2-4 The following case demonstrates an unusual initial presentation of a NET in the context of MEN1.
Case Report
A 29-year-old, active-duty African-American man deployed in Kuwait presented with icterus, flank pain, and hematuria. His past medical history was significant for nephrolithiasis, and his family history was notable for hyperparathyroidism. Laboratory results showed primary hyperparathyroidism and evidence of biliary obstruction.
A sestamibi scan demonstrated uptake in a location corresponding with the right inferior parathyroid gland. A computed tomography (CT) scan showed nephrolithiasis and hepatic biliary ductal dilatation. Magnetic resonance cholangiopancreatography (MRCP) revealed both intra- and extrahepatic ductal dilatation, focal narrowing of the proximal common bile duct, and possible adenopathy that was concerning for cholangiocarcinoma. Endoscopic retrograde cholangiopancreatography (ERCP) demonstrated a 1 cm to 2 cm focal stricture within the mid-common bile duct with intra- and extrahepatic ductal dilatation (Figure 1). An endoscopy showed no masses in the duodenum, and anendoscopic ultrasound showed no masses in the pancreas. Endoscopic brushings and endoscopic, ultrasound-guided, fine-needle aspiration
cytology were nondiagnostic. Exploratory laparotomy revealed a dilated hepatic bile duct, an inflamed porta hepatis, and a mass involving the distal hepatic bile duct.
The patient underwent cholecystectomy, radical extra hepatic bile duct resection to the level of the hepatic bifurcation, and hepaticojejunostomy. Gross examination of the specimen showed a nodule centered in the distal common hepatic duct with an adjacent, 2-cm lymph node. The histologic examination revealed a neoplastic proliferation consisting of epithelioid cells with round nuclei and granular chromatin with amphophilic cytoplasm in a trabecular and nested architecture.
The tumor was centered in the submucosa, which is typical of gastrointestinal NETs (Figure 2). There was no evidence of direct tumor extension elsewhere. About 40% of the tumor cells contained eosinophilic, intracytoplasmic inclusions (Figure 3). The tumor did not involve the margins or lymph node.
Positive staining with the neuroendocrine markers synaptophysin and chromagranin A confirmed a well-differentiated NET. The intracytoplasmic inclusions stained strongly positive for cytokeratin CAM 5.2. The tumor had higher-grade features, including tumor cell necrosis, a Ki-67 labeling index of 3%, and perineural invasion. The 2010 World Health Organization (WHO) criteria for NET of the digestive system classified this tumor as a grade 2, well-differentiated NET and as stage 1a (limited to the bile duct).4
Postoperatively, octreotide scan with single-photon emission computed tomography (SPECT)-CT did not show additional masses or lesions. Serum pancreatic polypeptide was elevated, with the remaining serum and plasma NET markers—including gastrin, glucagon, insulin, chromogranin A, and vasoactive intestinal polypeptide (VIP)—being within reference ranges. Genetic testing (GeneDx, Inc, Gaithersburg, MD) showed an E563X nonsense mutation in the MEN1 gene, confirming a MEN1 disorder. The patient then underwent a 4-gland parathyroidectomy with reimplantation; the parathyroid glands demonstrated hyperplasia in all 4 glands.
Biochemical follow-up at 14 months showed that the serum pancreatic polypeptide had normalized. There was no evidence of pituitary orpancreatic hypersecretion. The patient developed hypoparathyroidism, requiring calcium and calcitriol supplementation. Radiographic follow-up using abdominal magnetic resonance imaging at 16 months showed no evidence of disease.
Discussion
This case illustrates a genetic disease with an unusual initial presentation. Primary extrahepatic bile duct NETs are rare and have been reported previously in patients without MEN1.5-9 Neuroendocrine tumors in the hepatic bile duct in patients with MEN1 also have been reported but only after these tumors first appeared in the pancreas or duodenum.10 An extensive literature search revealed no prior reports extrahepatic bile duct NETs with MEN1 as the primary site or with biliary obstruction, which is why this patient’s presentation is particularly interesting.5,6,10-13 The table summarizes select reports of NETs.
Tumor location in this patient was atypical, and genetic testing guided the management. Serum MEN1 genetic testing is indicated in patients with ≥ 2 tumors that are atypical but possibly associated with MEN1 (such as adrenal tumors, gastrinomas, and carcinoids) and in patients aged < 45 years with primary hyperparathyroidism.14,15 The patient in this study was aged 29 years and had hyperparathyroidism and an NET of the hepatic bile duct. This condition was sufficient to warrant genetic testing, the results of which affected the patient’s subsequent parathyroid surgery.15 Despite the suggestion of unifocal localization on the sestamibi scan, the patient underwent the more appropriate subtotal parathyroidectomy.14 The patient’s tumor most likely originated from a germline mutation of the MEN1 gene.
As a result of the patient’s genetic test results, his daughter also was tested. She was found to have the same mutation as her father and will undergo proper tumor surveillance for MEN1. There was no personal or family history of hemangioblastomas, renal cell carcinomas, or cystadenomas, which would have prompted testing for von Hippel-Lindau disease. Likewise, there was no personal or family history of café-au-lait macules and neurofibromas, which would have prompted testing for neurofibromatosis type 1.
Due to the paucity of cases, there are currently no well-accepted guidelines on how to stage extrahepatic biliary NETs.3-5,16 The WHO recommends staging according to adenocarcinomas of the gallbladder and bile duct.3 As such, the pathologic stage of this tumor would be stage 1a.
The significance of the intracytoplasmic inclusion in this case is unknown. Pancreatic NETs and neuroendocrine carcinomas have demonstrated intracytoplasmic inclusions that stain positively for keratin and may indicate more aggressive tumor behavior.17-19 In 1 report, electron microscopic examination demonstrated intermediate filaments with entrapped neurosecretory granules.18 In a series of 84 cases of pancreatic endocrine tumors, 14 had intracytoplasmic inclusions; of these, 5 had MEN1.17 In the present case, the patient continues to show no evidence of tumor recurrence at 16 months after resection.
Conclusion
Extrahepatic biliary neuroendocrine tumors are rare. Further investigation into biliary tree NET staging and future studies to determine the significance of intracytoplasmic inclusions may be beneficial. This case highlights the appropriate use of genetic testing and supports expanding the clinical diagnosis of MEN1 to include NETs of the extrahepatic bile duct.
Click here to read the digital edition.
Neuroendocrine tumors (NETs) are uncommon and can occur in the context of genetic conditions. Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant disorder of the tumor suppressor gene of the same name—MEN1, which encodes for the protein menin. Multiple endocrine neoplasia type 1 is characterized clinically by the presence of 2 or more of the following NETs: parathyroid, pituitary, and pancreaticoduodenal.1 Pancreaticoduodenal NETs occur in 30% to 80% of patients with MEN1 and have malignant potential. Although the majority of pancreaticoduodenal NETs are nonfunctioning, patients may present with symptoms secondary to mass effect.
Genetic testing exists for MEN1, but not all genetic mutations that cause MEN1 have been discovered. Therefore, because negative genetic testing does not rule out MEN1, a diagnosis is based on tumor type and location. Neuroendocrine tumors of the biliary tree are rare, and there
are no well-accepted guidelines on how to stage them.2-4 The following case demonstrates an unusual initial presentation of a NET in the context of MEN1.
Case Report
A 29-year-old, active-duty African-American man deployed in Kuwait presented with icterus, flank pain, and hematuria. His past medical history was significant for nephrolithiasis, and his family history was notable for hyperparathyroidism. Laboratory results showed primary hyperparathyroidism and evidence of biliary obstruction.
A sestamibi scan demonstrated uptake in a location corresponding with the right inferior parathyroid gland. A computed tomography (CT) scan showed nephrolithiasis and hepatic biliary ductal dilatation. Magnetic resonance cholangiopancreatography (MRCP) revealed both intra- and extrahepatic ductal dilatation, focal narrowing of the proximal common bile duct, and possible adenopathy that was concerning for cholangiocarcinoma. Endoscopic retrograde cholangiopancreatography (ERCP) demonstrated a 1 cm to 2 cm focal stricture within the mid-common bile duct with intra- and extrahepatic ductal dilatation (Figure 1). An endoscopy showed no masses in the duodenum, and anendoscopic ultrasound showed no masses in the pancreas. Endoscopic brushings and endoscopic, ultrasound-guided, fine-needle aspiration
cytology were nondiagnostic. Exploratory laparotomy revealed a dilated hepatic bile duct, an inflamed porta hepatis, and a mass involving the distal hepatic bile duct.
The patient underwent cholecystectomy, radical extra hepatic bile duct resection to the level of the hepatic bifurcation, and hepaticojejunostomy. Gross examination of the specimen showed a nodule centered in the distal common hepatic duct with an adjacent, 2-cm lymph node. The histologic examination revealed a neoplastic proliferation consisting of epithelioid cells with round nuclei and granular chromatin with amphophilic cytoplasm in a trabecular and nested architecture.
The tumor was centered in the submucosa, which is typical of gastrointestinal NETs (Figure 2). There was no evidence of direct tumor extension elsewhere. About 40% of the tumor cells contained eosinophilic, intracytoplasmic inclusions (Figure 3). The tumor did not involve the margins or lymph node.
Positive staining with the neuroendocrine markers synaptophysin and chromagranin A confirmed a well-differentiated NET. The intracytoplasmic inclusions stained strongly positive for cytokeratin CAM 5.2. The tumor had higher-grade features, including tumor cell necrosis, a Ki-67 labeling index of 3%, and perineural invasion. The 2010 World Health Organization (WHO) criteria for NET of the digestive system classified this tumor as a grade 2, well-differentiated NET and as stage 1a (limited to the bile duct).4
Postoperatively, octreotide scan with single-photon emission computed tomography (SPECT)-CT did not show additional masses or lesions. Serum pancreatic polypeptide was elevated, with the remaining serum and plasma NET markers—including gastrin, glucagon, insulin, chromogranin A, and vasoactive intestinal polypeptide (VIP)—being within reference ranges. Genetic testing (GeneDx, Inc, Gaithersburg, MD) showed an E563X nonsense mutation in the MEN1 gene, confirming a MEN1 disorder. The patient then underwent a 4-gland parathyroidectomy with reimplantation; the parathyroid glands demonstrated hyperplasia in all 4 glands.
Biochemical follow-up at 14 months showed that the serum pancreatic polypeptide had normalized. There was no evidence of pituitary orpancreatic hypersecretion. The patient developed hypoparathyroidism, requiring calcium and calcitriol supplementation. Radiographic follow-up using abdominal magnetic resonance imaging at 16 months showed no evidence of disease.
Discussion
This case illustrates a genetic disease with an unusual initial presentation. Primary extrahepatic bile duct NETs are rare and have been reported previously in patients without MEN1.5-9 Neuroendocrine tumors in the hepatic bile duct in patients with MEN1 also have been reported but only after these tumors first appeared in the pancreas or duodenum.10 An extensive literature search revealed no prior reports extrahepatic bile duct NETs with MEN1 as the primary site or with biliary obstruction, which is why this patient’s presentation is particularly interesting.5,6,10-13 The table summarizes select reports of NETs.
Tumor location in this patient was atypical, and genetic testing guided the management. Serum MEN1 genetic testing is indicated in patients with ≥ 2 tumors that are atypical but possibly associated with MEN1 (such as adrenal tumors, gastrinomas, and carcinoids) and in patients aged < 45 years with primary hyperparathyroidism.14,15 The patient in this study was aged 29 years and had hyperparathyroidism and an NET of the hepatic bile duct. This condition was sufficient to warrant genetic testing, the results of which affected the patient’s subsequent parathyroid surgery.15 Despite the suggestion of unifocal localization on the sestamibi scan, the patient underwent the more appropriate subtotal parathyroidectomy.14 The patient’s tumor most likely originated from a germline mutation of the MEN1 gene.
As a result of the patient’s genetic test results, his daughter also was tested. She was found to have the same mutation as her father and will undergo proper tumor surveillance for MEN1. There was no personal or family history of hemangioblastomas, renal cell carcinomas, or cystadenomas, which would have prompted testing for von Hippel-Lindau disease. Likewise, there was no personal or family history of café-au-lait macules and neurofibromas, which would have prompted testing for neurofibromatosis type 1.
Due to the paucity of cases, there are currently no well-accepted guidelines on how to stage extrahepatic biliary NETs.3-5,16 The WHO recommends staging according to adenocarcinomas of the gallbladder and bile duct.3 As such, the pathologic stage of this tumor would be stage 1a.
The significance of the intracytoplasmic inclusion in this case is unknown. Pancreatic NETs and neuroendocrine carcinomas have demonstrated intracytoplasmic inclusions that stain positively for keratin and may indicate more aggressive tumor behavior.17-19 In 1 report, electron microscopic examination demonstrated intermediate filaments with entrapped neurosecretory granules.18 In a series of 84 cases of pancreatic endocrine tumors, 14 had intracytoplasmic inclusions; of these, 5 had MEN1.17 In the present case, the patient continues to show no evidence of tumor recurrence at 16 months after resection.
Conclusion
Extrahepatic biliary neuroendocrine tumors are rare. Further investigation into biliary tree NET staging and future studies to determine the significance of intracytoplasmic inclusions may be beneficial. This case highlights the appropriate use of genetic testing and supports expanding the clinical diagnosis of MEN1 to include NETs of the extrahepatic bile duct.
Click here to read the digital edition.
1. Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: WB Saunders; 2011.
2. American Joint Committee on Cancer. Neuroendocrine Tumors. In: Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, eds. American Joint Committee on Cancer Staging Handbook. 7th ed. From the AJCC Cancer Staging Manual. New York, NY: Springer-Verlag; 2010:227-236.
3. Komminoth P, Arnold R, Capella C, et al. Neuroendocrine neoplasms of the gallbladder and extrahepatic bile ducts. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, et al, eds. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC Press; 2010:274-276.
4. Rindi G, Arnold R, Bosman FT. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, et al, eds. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC Press; 2010:13.
5. Price TN, Thompson GB, Lewis JT, Lloyd RV, Young WF. Zollinger-Ellison syndrome due to primary gastrinoma of the extrahepatic biliary tree: three case reports and review of literature. Endocr Pract. 2009;15(7):737-749.
6. Bhandarwar AH, Shaikh TA, Borisa AD, et al. Primary neuroendocrine tumor of the left hepatic duct: a case report with review of the literature. Case Rep Surg. 2012:786432.
7. Bhalla P, Powle V, Shah RC, Jagannath P. Neuroendocrine tumor of common hepatic duct. Indian J Gastroenterol. 2012;31(3):144-146.
8. Khan FA, Stevens-Chase A, Chaudhry R, Hashmi A, Edelman D, Weaver D. Extrahepatic biliary obstrution secondary to neuroendocrine tumor of the common hepatic duct. Int J Surg Case Rep. 2017;30:46-49.
9. Hong N, Kim HJ, Byun JH, et al. Neuroendocrine neoplasms of the extrahepatic bile duct: radiologic and clinical characteristics. Abdom Imaging. 2015;40(1):181-191.
10. Tonelli F, Giudici F, Nesi G, Batignani G, Brandi ML. Biliary tree gastrinomas in multiple endocrine neoplasia type 1 syndrome. World J Gastroenterol. 2013;19(45):8312-8320.
11. Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore). 2004;83(1):43-83.
12. Pieterman CRC, Conemans EB, Dreijerink KMA, et al. Thoracic and duodenopancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1: natural history and function of menin in tumorigenesis. Endocr Relat Cancer. 2014;21(3):R121-R142.
13. Pipeleers-Marichal M, Somers G, Willems G, et al. Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med. 1990;322(11):723-727.
14. Thakker RV, Newey PJ, Walls GV, et al; Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97(9):2990-3011.
15. Eastell R, Brandi ML, Costa AG, et al. Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99(10):3570-3579.
16. Michalopoulos N, Papavramidis TS, Karayannopoulou G, Pliakos I, Papavramidis ST, Kanellos I. Neuroendocrine tumors of extrahepatic biliary tract. Pathol Oncol Res. 2014;20(4):765-775.
17. Serra S, Asa SL, Chetty R. Intracytoplasmic inclusions (including the so-called “rhabdoid” phenotype) in pancreatic endocrine tumors. Endocr Pathol. 2006;17(1):75-81.
18. Shia J, Erlandson RA, Klimstra DS. Whorls of intermediate filaments with entrapped neurosecretory granules correspond to the “rhabdoid” inclusions seen in pancreatic endocrine
neoplasms. Am J Surg Pathol. 2004;28(2):271-273.
19. Perez-Montiel MD, Frankel WL, Suster S. Neuroendocrine carcinomas of the pancreas with ‘Rhabdoid’ features. Am J Surg Pathol. 2003;27(5):642-649.
1. Melmed S, Polonsky KS, Larsen PR, Kronenberg HM, eds. Williams Textbook of Endocrinology. 12th ed. Philadelphia, PA: WB Saunders; 2011.
2. American Joint Committee on Cancer. Neuroendocrine Tumors. In: Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A, eds. American Joint Committee on Cancer Staging Handbook. 7th ed. From the AJCC Cancer Staging Manual. New York, NY: Springer-Verlag; 2010:227-236.
3. Komminoth P, Arnold R, Capella C, et al. Neuroendocrine neoplasms of the gallbladder and extrahepatic bile ducts. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, et al, eds. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC Press; 2010:274-276.
4. Rindi G, Arnold R, Bosman FT. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, et al, eds. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: IARC Press; 2010:13.
5. Price TN, Thompson GB, Lewis JT, Lloyd RV, Young WF. Zollinger-Ellison syndrome due to primary gastrinoma of the extrahepatic biliary tree: three case reports and review of literature. Endocr Pract. 2009;15(7):737-749.
6. Bhandarwar AH, Shaikh TA, Borisa AD, et al. Primary neuroendocrine tumor of the left hepatic duct: a case report with review of the literature. Case Rep Surg. 2012:786432.
7. Bhalla P, Powle V, Shah RC, Jagannath P. Neuroendocrine tumor of common hepatic duct. Indian J Gastroenterol. 2012;31(3):144-146.
8. Khan FA, Stevens-Chase A, Chaudhry R, Hashmi A, Edelman D, Weaver D. Extrahepatic biliary obstrution secondary to neuroendocrine tumor of the common hepatic duct. Int J Surg Case Rep. 2017;30:46-49.
9. Hong N, Kim HJ, Byun JH, et al. Neuroendocrine neoplasms of the extrahepatic bile duct: radiologic and clinical characteristics. Abdom Imaging. 2015;40(1):181-191.
10. Tonelli F, Giudici F, Nesi G, Batignani G, Brandi ML. Biliary tree gastrinomas in multiple endocrine neoplasia type 1 syndrome. World J Gastroenterol. 2013;19(45):8312-8320.
11. Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore). 2004;83(1):43-83.
12. Pieterman CRC, Conemans EB, Dreijerink KMA, et al. Thoracic and duodenopancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1: natural history and function of menin in tumorigenesis. Endocr Relat Cancer. 2014;21(3):R121-R142.
13. Pipeleers-Marichal M, Somers G, Willems G, et al. Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med. 1990;322(11):723-727.
14. Thakker RV, Newey PJ, Walls GV, et al; Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97(9):2990-3011.
15. Eastell R, Brandi ML, Costa AG, et al. Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99(10):3570-3579.
16. Michalopoulos N, Papavramidis TS, Karayannopoulou G, Pliakos I, Papavramidis ST, Kanellos I. Neuroendocrine tumors of extrahepatic biliary tract. Pathol Oncol Res. 2014;20(4):765-775.
17. Serra S, Asa SL, Chetty R. Intracytoplasmic inclusions (including the so-called “rhabdoid” phenotype) in pancreatic endocrine tumors. Endocr Pathol. 2006;17(1):75-81.
18. Shia J, Erlandson RA, Klimstra DS. Whorls of intermediate filaments with entrapped neurosecretory granules correspond to the “rhabdoid” inclusions seen in pancreatic endocrine
neoplasms. Am J Surg Pathol. 2004;28(2):271-273.
19. Perez-Montiel MD, Frankel WL, Suster S. Neuroendocrine carcinomas of the pancreas with ‘Rhabdoid’ features. Am J Surg Pathol. 2003;27(5):642-649.
What Makes Squamous Cell Cancers Different? Genomics May Explain
Squamous cell carcinomas (SCCs) associated with smoking and human papillomavirus (HPV) have distinct genomic signatures, say researchers from a National Institutes of Health-supported study. That is one of the findings that may help distinguish SCCs from other cancers and point the way to new research and treatment.
The researchers used new analytic tools and data from the recently completed PanCancer Atlas to investigate similarities and differences among SCCs in the head and neck, lung, esophagus, cervix, and bladder. The PanCancer Atlas is a detailed analysis from a dataset containing molecular and clinical information on more than 10,000 tumors from 33 forms of cancer.
The researchers combined multiple platforms of genomic data from 1,400 SCC samples into integrated analyses, creating visual clusters of tumors based on genomic characteristics.
Squamous cell carcinomas had genomic features that set them apart from other cancers, the researchers found. The most common were gains or losses of the sections of certain chromosomes, making it likely that those regions harbor genes important to the development of SCCs.
The current study expands on research reported in 2014 and 2015, which compared genomic features of SCCs in head and neck cancer associated with smoking (a risk factor for head and neck cancer [HNC]) and HPV (a risk factor for cervical and some HNCs). Certain features were present in tumors associated with both, whereas others were exclusive to only 1 of the 2. The researchers also found similarities in the genomic characteristics of HNCs with lung cancers, some bladder cancers, and cervical cancer.
Squamous cell carcinomas (SCCs) associated with smoking and human papillomavirus (HPV) have distinct genomic signatures, say researchers from a National Institutes of Health-supported study. That is one of the findings that may help distinguish SCCs from other cancers and point the way to new research and treatment.
The researchers used new analytic tools and data from the recently completed PanCancer Atlas to investigate similarities and differences among SCCs in the head and neck, lung, esophagus, cervix, and bladder. The PanCancer Atlas is a detailed analysis from a dataset containing molecular and clinical information on more than 10,000 tumors from 33 forms of cancer.
The researchers combined multiple platforms of genomic data from 1,400 SCC samples into integrated analyses, creating visual clusters of tumors based on genomic characteristics.
Squamous cell carcinomas had genomic features that set them apart from other cancers, the researchers found. The most common were gains or losses of the sections of certain chromosomes, making it likely that those regions harbor genes important to the development of SCCs.
The current study expands on research reported in 2014 and 2015, which compared genomic features of SCCs in head and neck cancer associated with smoking (a risk factor for head and neck cancer [HNC]) and HPV (a risk factor for cervical and some HNCs). Certain features were present in tumors associated with both, whereas others were exclusive to only 1 of the 2. The researchers also found similarities in the genomic characteristics of HNCs with lung cancers, some bladder cancers, and cervical cancer.
Squamous cell carcinomas (SCCs) associated with smoking and human papillomavirus (HPV) have distinct genomic signatures, say researchers from a National Institutes of Health-supported study. That is one of the findings that may help distinguish SCCs from other cancers and point the way to new research and treatment.
The researchers used new analytic tools and data from the recently completed PanCancer Atlas to investigate similarities and differences among SCCs in the head and neck, lung, esophagus, cervix, and bladder. The PanCancer Atlas is a detailed analysis from a dataset containing molecular and clinical information on more than 10,000 tumors from 33 forms of cancer.
The researchers combined multiple platforms of genomic data from 1,400 SCC samples into integrated analyses, creating visual clusters of tumors based on genomic characteristics.
Squamous cell carcinomas had genomic features that set them apart from other cancers, the researchers found. The most common were gains or losses of the sections of certain chromosomes, making it likely that those regions harbor genes important to the development of SCCs.
The current study expands on research reported in 2014 and 2015, which compared genomic features of SCCs in head and neck cancer associated with smoking (a risk factor for head and neck cancer [HNC]) and HPV (a risk factor for cervical and some HNCs). Certain features were present in tumors associated with both, whereas others were exclusive to only 1 of the 2. The researchers also found similarities in the genomic characteristics of HNCs with lung cancers, some bladder cancers, and cervical cancer.
Giving Dexamethasone a New Lease on Life
Dexamethasone (Dex), a synthetic glucocorticoid, for years has been widely used both to treat adverse effects of antitumor agents and in direct chemotherapy regimens for hematologic malignancies, such as leukemia and lymphoma. But might it be modified to work against solid cancers as well? Researchers from Advanced Radiation Technology Institute, Medical Device Development Center, and University of Science and Technology in South Korea, suggest that ionizing radiation could produce new anticancer options from an old drug.
The researchers irradiated Dex with γ- rays to produce ionizing-radiation-irradiated.
Dex (Dex-IR), then investigated its effects on human lung cancer cells (cell lines H1650, A549, and H1299). The researchers used ionizing radiation because introducing energy into materials can produce favorable changes; irradiated materials with sufficiently high energy can decompose to yield very reactive intermediate molecules and form new ones. In this study, γ -irradiation produced “remarkable changes” in the chemical properties of dexamethasone; changes included degradation products, such as methanol vapor and carbon monoxide.
Original Dex inhibits the proliferation of non-small cell lung cancer (NSCLC) cells but has minimal cytotoxic effects, the researchers say. However, Dex-IR not only significantly inhibited the proliferation of NSCLC cells, but also induced apoptosis, arrested cell cycles of H1650 lung cancer cells, and significantly reduced cells’ invasiveness.
The researchers say their results “strongly suggest” a direct link between the chemical derivatives of Dex and inhibition of NSCLC cell growth. Their findings are the first evidence that γ -irradiated Dex represents a novel class of anticancer agents for lung cancer.
Lee EH, Park CH, Choi HJ, Kawala RA, Bai HW, Chung BY. PLoS One. 2018;13(4):e0194341.
doi: 10.1371/journal.pone.0194341.
Dexamethasone (Dex), a synthetic glucocorticoid, for years has been widely used both to treat adverse effects of antitumor agents and in direct chemotherapy regimens for hematologic malignancies, such as leukemia and lymphoma. But might it be modified to work against solid cancers as well? Researchers from Advanced Radiation Technology Institute, Medical Device Development Center, and University of Science and Technology in South Korea, suggest that ionizing radiation could produce new anticancer options from an old drug.
The researchers irradiated Dex with γ- rays to produce ionizing-radiation-irradiated.
Dex (Dex-IR), then investigated its effects on human lung cancer cells (cell lines H1650, A549, and H1299). The researchers used ionizing radiation because introducing energy into materials can produce favorable changes; irradiated materials with sufficiently high energy can decompose to yield very reactive intermediate molecules and form new ones. In this study, γ -irradiation produced “remarkable changes” in the chemical properties of dexamethasone; changes included degradation products, such as methanol vapor and carbon monoxide.
Original Dex inhibits the proliferation of non-small cell lung cancer (NSCLC) cells but has minimal cytotoxic effects, the researchers say. However, Dex-IR not only significantly inhibited the proliferation of NSCLC cells, but also induced apoptosis, arrested cell cycles of H1650 lung cancer cells, and significantly reduced cells’ invasiveness.
The researchers say their results “strongly suggest” a direct link between the chemical derivatives of Dex and inhibition of NSCLC cell growth. Their findings are the first evidence that γ -irradiated Dex represents a novel class of anticancer agents for lung cancer.
Lee EH, Park CH, Choi HJ, Kawala RA, Bai HW, Chung BY. PLoS One. 2018;13(4):e0194341.
doi: 10.1371/journal.pone.0194341.
Dexamethasone (Dex), a synthetic glucocorticoid, for years has been widely used both to treat adverse effects of antitumor agents and in direct chemotherapy regimens for hematologic malignancies, such as leukemia and lymphoma. But might it be modified to work against solid cancers as well? Researchers from Advanced Radiation Technology Institute, Medical Device Development Center, and University of Science and Technology in South Korea, suggest that ionizing radiation could produce new anticancer options from an old drug.
The researchers irradiated Dex with γ- rays to produce ionizing-radiation-irradiated.
Dex (Dex-IR), then investigated its effects on human lung cancer cells (cell lines H1650, A549, and H1299). The researchers used ionizing radiation because introducing energy into materials can produce favorable changes; irradiated materials with sufficiently high energy can decompose to yield very reactive intermediate molecules and form new ones. In this study, γ -irradiation produced “remarkable changes” in the chemical properties of dexamethasone; changes included degradation products, such as methanol vapor and carbon monoxide.
Original Dex inhibits the proliferation of non-small cell lung cancer (NSCLC) cells but has minimal cytotoxic effects, the researchers say. However, Dex-IR not only significantly inhibited the proliferation of NSCLC cells, but also induced apoptosis, arrested cell cycles of H1650 lung cancer cells, and significantly reduced cells’ invasiveness.
The researchers say their results “strongly suggest” a direct link between the chemical derivatives of Dex and inhibition of NSCLC cell growth. Their findings are the first evidence that γ -irradiated Dex represents a novel class of anticancer agents for lung cancer.
Lee EH, Park CH, Choi HJ, Kawala RA, Bai HW, Chung BY. PLoS One. 2018;13(4):e0194341.
doi: 10.1371/journal.pone.0194341.
FDA places partial hold on trials after secondary lymphoma
The drugmaker after a pediatric patient developed a secondary T-cell lymphoma.
The Food and Drug Administration had issued a partial clinical hold in April on new enrollment of any patients with genetically defined solid tumors and hematologic malignancies. Patients already enrolled who have not had disease progression can continue to receive tazemetostat.
Tazemetostat is a first-in-class EZH2 inhibitor being studied as monotherapy in phase 1 and 2 trials for certain molecularly defined solid tumors, follicular lymphoma and diffuse large B-cell lymphoma, mesothelioma, and in combination studies of DLBCL and non–small cell lung cancer.
Epizyme is currently working to update informed consent, the investigator’s brochure, and study protocols, the company said in a statement.
The drugmaker after a pediatric patient developed a secondary T-cell lymphoma.
The Food and Drug Administration had issued a partial clinical hold in April on new enrollment of any patients with genetically defined solid tumors and hematologic malignancies. Patients already enrolled who have not had disease progression can continue to receive tazemetostat.
Tazemetostat is a first-in-class EZH2 inhibitor being studied as monotherapy in phase 1 and 2 trials for certain molecularly defined solid tumors, follicular lymphoma and diffuse large B-cell lymphoma, mesothelioma, and in combination studies of DLBCL and non–small cell lung cancer.
Epizyme is currently working to update informed consent, the investigator’s brochure, and study protocols, the company said in a statement.
The drugmaker after a pediatric patient developed a secondary T-cell lymphoma.
The Food and Drug Administration had issued a partial clinical hold in April on new enrollment of any patients with genetically defined solid tumors and hematologic malignancies. Patients already enrolled who have not had disease progression can continue to receive tazemetostat.
Tazemetostat is a first-in-class EZH2 inhibitor being studied as monotherapy in phase 1 and 2 trials for certain molecularly defined solid tumors, follicular lymphoma and diffuse large B-cell lymphoma, mesothelioma, and in combination studies of DLBCL and non–small cell lung cancer.
Epizyme is currently working to update informed consent, the investigator’s brochure, and study protocols, the company said in a statement.
PDPK1 could be novel target in MCL
Researchers may have found a new therapeutic approach for treating mantle cell lymphoma (MCL) by targeting 3-phosphoinositide-dependent protein kinase 1 (PDPK1).
Saori Maegawa and colleagues at Kyoto Prefectural University of Medicine in Japan, evaluated PDPK1 activity in patient-derived primary B-cell lymphoma cells by immunohistochemical staining of p-PDPK1Ser241 (p-PDPK1) in tissue specimens from seven patients with MCL, six patients with diffuse large B-cell lymphoma, and five patients with follicular lymphoma. All specimens were biopsied at initial diagnosis, before starting treatment.

“Our study showed that PDPK1 inhibition caused inactivation of RSK2-NTKD, as well as the decrease of total RSK2 protein, but not of AKT, in MCL-derived cells,” the researchers wrote in Experimental Hematology. “This implies that RSK2 activity is mainly regulated by PDPK1 at both the transcriptional expression and post-translational levels, but AKT activity is regulated by a signaling pathway that does not interact with a PDPK1-mediated pathway in MCL.”
If a PDPK1 inhibitor is pursued as clinical target, the researchers said careful monitoring for hyperglycemia may be required since impaired glucose metabolism is commonly seen with AKT inhibitors. Future research in MCL could also be directed toward the targeting of RSK2-NTKD, the researchers wrote.
SOURCE: Maegawa S et al. Exp Hematol. 2018 Mar;59:72-81.e2.
Researchers may have found a new therapeutic approach for treating mantle cell lymphoma (MCL) by targeting 3-phosphoinositide-dependent protein kinase 1 (PDPK1).
Saori Maegawa and colleagues at Kyoto Prefectural University of Medicine in Japan, evaluated PDPK1 activity in patient-derived primary B-cell lymphoma cells by immunohistochemical staining of p-PDPK1Ser241 (p-PDPK1) in tissue specimens from seven patients with MCL, six patients with diffuse large B-cell lymphoma, and five patients with follicular lymphoma. All specimens were biopsied at initial diagnosis, before starting treatment.
“Our study showed that PDPK1 inhibition caused inactivation of RSK2-NTKD, as well as the decrease of total RSK2 protein, but not of AKT, in MCL-derived cells,” the researchers wrote in Experimental Hematology. “This implies that RSK2 activity is mainly regulated by PDPK1 at both the transcriptional expression and post-translational levels, but AKT activity is regulated by a signaling pathway that does not interact with a PDPK1-mediated pathway in MCL.”
If a PDPK1 inhibitor is pursued as clinical target, the researchers said careful monitoring for hyperglycemia may be required since impaired glucose metabolism is commonly seen with AKT inhibitors. Future research in MCL could also be directed toward the targeting of RSK2-NTKD, the researchers wrote.
SOURCE: Maegawa S et al. Exp Hematol. 2018 Mar;59:72-81.e2.
Researchers may have found a new therapeutic approach for treating mantle cell lymphoma (MCL) by targeting 3-phosphoinositide-dependent protein kinase 1 (PDPK1).
Saori Maegawa and colleagues at Kyoto Prefectural University of Medicine in Japan, evaluated PDPK1 activity in patient-derived primary B-cell lymphoma cells by immunohistochemical staining of p-PDPK1Ser241 (p-PDPK1) in tissue specimens from seven patients with MCL, six patients with diffuse large B-cell lymphoma, and five patients with follicular lymphoma. All specimens were biopsied at initial diagnosis, before starting treatment.
“Our study showed that PDPK1 inhibition caused inactivation of RSK2-NTKD, as well as the decrease of total RSK2 protein, but not of AKT, in MCL-derived cells,” the researchers wrote in Experimental Hematology. “This implies that RSK2 activity is mainly regulated by PDPK1 at both the transcriptional expression and post-translational levels, but AKT activity is regulated by a signaling pathway that does not interact with a PDPK1-mediated pathway in MCL.”
If a PDPK1 inhibitor is pursued as clinical target, the researchers said careful monitoring for hyperglycemia may be required since impaired glucose metabolism is commonly seen with AKT inhibitors. Future research in MCL could also be directed toward the targeting of RSK2-NTKD, the researchers wrote.
SOURCE: Maegawa S et al. Exp Hematol. 2018 Mar;59:72-81.e2.
FROM EXPERIMENTAL HEMATOLOGY
Updated CLL guidelines incorporate a decade of advances
include new and revised recommendations based on major advances in genomics, targeted therapies, and biomarkers that have occurred since the last iteration in 2008.
The guidelines are an update from a consensus document issued a decade ago by the International Workshop on CLL, focusing on the conduct of clinical trials in patients with CLL. The new guidelines are published in Blood.
Major changes or additions include:
Molecular genetics: The updated guidelines recognize the clinical importance of specific genomic alterations/mutations on response to standard chemotherapy or chemoimmunotherapy, including the 17p deletion and mutations in TP53.
“Therefore, the assessment of both del(17p) and TP53 mutation has prognostic and predictive value and should guide therapeutic decisions in routine practice. For clinical trials, it is recommended that molecular genetics be performed prior to treating a patient on protocol,” the guidelines state.
IGHV mutational status: The mutational status of immunoglobulin variable heavy chain (IGHV) genes has been demonstrated to offer important prognostic information, according to the guidelines authors led by Michael Hallek, MD of the University of Cologne, Germany.
Specifically, leukemia with IGHV genes without somatic mutations are associated with worse clinical outcomes, compared with leukemia with IGHV mutations. Patients with mutated IGHV and other prognostic factors such as favorable cytogenetics or minimal residual disease (MRD) negativity generally have excellent outcomes with a chemoimmunotherapy regimen consisting of fludarabine, cyclophosphamide, and rituximab, the authors noted.
Biomarkers: The guidelines call for standardization and use in prospective clinical trials of assays for serum markers such as soluble CD23, thymidine kinase, and beta-2-microglobulin. These markers have been shown in several studies to be associated with overall survival or progression-free survival, and of these markers, beta-2-microglobulin “has retained independent prognostic value in several multiparameter scores,” the guidelines state.
The authors also tip their hats to recently developed or improved prognostic scores, especially the CLL International Prognostic Index (CLL-IPI), which incorporates clinical stage, age, IGHV mutational status, beta-2-microglobulin, and del(17p) and/or TP53 mutations.
Organ function assessment: Not new, but improved in the current version of the guidelines, are recommendations for evaluation of splenomegaly, hepatomegaly, and lymphadenopathy in response assessment. These recommendations were harmonized with the relevant sections of the updated lymphoma response guidelines.
Continuous therapy: The guidelines panel recommends assessment of response duration during continuous therapy with oral agents and after the end of therapy, especially after chemotherapy or chemoimmunotherapy.
“Study protocols should provide detailed specifications of the planned time points for the assessment of the treatment response under continuous therapy. Response durations of less than six months are not considered clinically relevant,” the panel cautioned.
Response assessments for treatments with a maintenance phase should be performed at a minimum of 2 months after patients achieve their best responses.
MRD: The guidelines call for minimal residual disease (MRD) assessment in clinical trials aimed at maximizing remission depth, with emphasis on reporting the sensitivity of the MRD evaluation method used, and the type of tissue assessed.
Antiviral prophylaxis: The guidelines caution that because patients treated with anti-CD20 antibodies, such as rituximab or obinutuzumab, could have reactivation of hepatitis B virus (HBV) infections, patients should be tested for HBV serological status before starting on an anti-CD20 agent.
“Progressive multifocal leukoencephalopathy has been reported in a few CLL patients treated with anti-CD20 antibodies; therefore, infections with John Cunningham (JC) virus should be ruled out in situations of unclear neurological symptoms,” the panel recommended.
They note that patients younger than 65 treated with fludarabine-based therapy in the first line do not require routine monitoring or infection prophylaxis, due to the low reported incidence of infections in this group.
The authors reported having no financial disclosures related to the guidelines.
include new and revised recommendations based on major advances in genomics, targeted therapies, and biomarkers that have occurred since the last iteration in 2008.
The guidelines are an update from a consensus document issued a decade ago by the International Workshop on CLL, focusing on the conduct of clinical trials in patients with CLL. The new guidelines are published in Blood.
Major changes or additions include:
Molecular genetics: The updated guidelines recognize the clinical importance of specific genomic alterations/mutations on response to standard chemotherapy or chemoimmunotherapy, including the 17p deletion and mutations in TP53.
“Therefore, the assessment of both del(17p) and TP53 mutation has prognostic and predictive value and should guide therapeutic decisions in routine practice. For clinical trials, it is recommended that molecular genetics be performed prior to treating a patient on protocol,” the guidelines state.
IGHV mutational status: The mutational status of immunoglobulin variable heavy chain (IGHV) genes has been demonstrated to offer important prognostic information, according to the guidelines authors led by Michael Hallek, MD of the University of Cologne, Germany.
Specifically, leukemia with IGHV genes without somatic mutations are associated with worse clinical outcomes, compared with leukemia with IGHV mutations. Patients with mutated IGHV and other prognostic factors such as favorable cytogenetics or minimal residual disease (MRD) negativity generally have excellent outcomes with a chemoimmunotherapy regimen consisting of fludarabine, cyclophosphamide, and rituximab, the authors noted.
Biomarkers: The guidelines call for standardization and use in prospective clinical trials of assays for serum markers such as soluble CD23, thymidine kinase, and beta-2-microglobulin. These markers have been shown in several studies to be associated with overall survival or progression-free survival, and of these markers, beta-2-microglobulin “has retained independent prognostic value in several multiparameter scores,” the guidelines state.
The authors also tip their hats to recently developed or improved prognostic scores, especially the CLL International Prognostic Index (CLL-IPI), which incorporates clinical stage, age, IGHV mutational status, beta-2-microglobulin, and del(17p) and/or TP53 mutations.
Organ function assessment: Not new, but improved in the current version of the guidelines, are recommendations for evaluation of splenomegaly, hepatomegaly, and lymphadenopathy in response assessment. These recommendations were harmonized with the relevant sections of the updated lymphoma response guidelines.
Continuous therapy: The guidelines panel recommends assessment of response duration during continuous therapy with oral agents and after the end of therapy, especially after chemotherapy or chemoimmunotherapy.
“Study protocols should provide detailed specifications of the planned time points for the assessment of the treatment response under continuous therapy. Response durations of less than six months are not considered clinically relevant,” the panel cautioned.
Response assessments for treatments with a maintenance phase should be performed at a minimum of 2 months after patients achieve their best responses.
MRD: The guidelines call for minimal residual disease (MRD) assessment in clinical trials aimed at maximizing remission depth, with emphasis on reporting the sensitivity of the MRD evaluation method used, and the type of tissue assessed.
Antiviral prophylaxis: The guidelines caution that because patients treated with anti-CD20 antibodies, such as rituximab or obinutuzumab, could have reactivation of hepatitis B virus (HBV) infections, patients should be tested for HBV serological status before starting on an anti-CD20 agent.
“Progressive multifocal leukoencephalopathy has been reported in a few CLL patients treated with anti-CD20 antibodies; therefore, infections with John Cunningham (JC) virus should be ruled out in situations of unclear neurological symptoms,” the panel recommended.
They note that patients younger than 65 treated with fludarabine-based therapy in the first line do not require routine monitoring or infection prophylaxis, due to the low reported incidence of infections in this group.
The authors reported having no financial disclosures related to the guidelines.
include new and revised recommendations based on major advances in genomics, targeted therapies, and biomarkers that have occurred since the last iteration in 2008.
The guidelines are an update from a consensus document issued a decade ago by the International Workshop on CLL, focusing on the conduct of clinical trials in patients with CLL. The new guidelines are published in Blood.
Major changes or additions include:
Molecular genetics: The updated guidelines recognize the clinical importance of specific genomic alterations/mutations on response to standard chemotherapy or chemoimmunotherapy, including the 17p deletion and mutations in TP53.
“Therefore, the assessment of both del(17p) and TP53 mutation has prognostic and predictive value and should guide therapeutic decisions in routine practice. For clinical trials, it is recommended that molecular genetics be performed prior to treating a patient on protocol,” the guidelines state.
IGHV mutational status: The mutational status of immunoglobulin variable heavy chain (IGHV) genes has been demonstrated to offer important prognostic information, according to the guidelines authors led by Michael Hallek, MD of the University of Cologne, Germany.
Specifically, leukemia with IGHV genes without somatic mutations are associated with worse clinical outcomes, compared with leukemia with IGHV mutations. Patients with mutated IGHV and other prognostic factors such as favorable cytogenetics or minimal residual disease (MRD) negativity generally have excellent outcomes with a chemoimmunotherapy regimen consisting of fludarabine, cyclophosphamide, and rituximab, the authors noted.
Biomarkers: The guidelines call for standardization and use in prospective clinical trials of assays for serum markers such as soluble CD23, thymidine kinase, and beta-2-microglobulin. These markers have been shown in several studies to be associated with overall survival or progression-free survival, and of these markers, beta-2-microglobulin “has retained independent prognostic value in several multiparameter scores,” the guidelines state.
The authors also tip their hats to recently developed or improved prognostic scores, especially the CLL International Prognostic Index (CLL-IPI), which incorporates clinical stage, age, IGHV mutational status, beta-2-microglobulin, and del(17p) and/or TP53 mutations.
Organ function assessment: Not new, but improved in the current version of the guidelines, are recommendations for evaluation of splenomegaly, hepatomegaly, and lymphadenopathy in response assessment. These recommendations were harmonized with the relevant sections of the updated lymphoma response guidelines.
Continuous therapy: The guidelines panel recommends assessment of response duration during continuous therapy with oral agents and after the end of therapy, especially after chemotherapy or chemoimmunotherapy.
“Study protocols should provide detailed specifications of the planned time points for the assessment of the treatment response under continuous therapy. Response durations of less than six months are not considered clinically relevant,” the panel cautioned.
Response assessments for treatments with a maintenance phase should be performed at a minimum of 2 months after patients achieve their best responses.
MRD: The guidelines call for minimal residual disease (MRD) assessment in clinical trials aimed at maximizing remission depth, with emphasis on reporting the sensitivity of the MRD evaluation method used, and the type of tissue assessed.
Antiviral prophylaxis: The guidelines caution that because patients treated with anti-CD20 antibodies, such as rituximab or obinutuzumab, could have reactivation of hepatitis B virus (HBV) infections, patients should be tested for HBV serological status before starting on an anti-CD20 agent.
“Progressive multifocal leukoencephalopathy has been reported in a few CLL patients treated with anti-CD20 antibodies; therefore, infections with John Cunningham (JC) virus should be ruled out in situations of unclear neurological symptoms,” the panel recommended.
They note that patients younger than 65 treated with fludarabine-based therapy in the first line do not require routine monitoring or infection prophylaxis, due to the low reported incidence of infections in this group.
The authors reported having no financial disclosures related to the guidelines.
FROM BLOOD
Clinical Puzzle: Lung Cancer or Hodgkin Lymphoma?
Patients with Hodgkin lymphoma have a 15% to 40% likelihood of pulmonary involvement, such as a solitary lung mass or cavitary lung lesion. But clinicians at Bassett Healthcare in Cooperstown, New York, were faced with a rare case of another presentation: an endobronchial obstructing mass.
The patient, a 40-year-old man, reported having had cough, fatigue, and progressive weight loss (despite a good appetite) for 8 months. Because he had a history of smoking, he was treated for bronchitis, but the cough worsened. He had no fever, night sweats, dyspnea, or chest pain (common features of Hodgkin lymphoma).
Auscultation revealed clear lungs, with no crackles or wheeze, and no dullness to percussion. Blood work was negative except for eosinophilia. A subsequent chest radiograph showed an irregular left hilar lung opacity. A computer tomography scan showed a cavitary consolidation of the left upper lobe of the lung. Fiber-optic bronchoscopy with tissue from the endobronchial mass indicated an obstructing lesion in the left upper lobe bronchus. The clinicians suspected lung cancer.
However, they also found inflammatory cells, and immunohistochemistry revealed findings consistent with Hodgkin lymphoma. The clinicians started the patient on chemotherapy. After 6 cycles, his symptoms resolved. Follow-up at 8 months showed no clinical evidence of recurrence.
As the clinicians found out, radiologically, Hodgkin lymphoma can mimic lung cancer. They advise histopathologic diagnosis for a patient presenting with lung mass.
Source:
Abid H, Khan J, Lone N. BMJ Case Rep. 2018;2018. pii: bcr-2017-223809.
doi: 10.1136/bcr-2017-223809.
Patients with Hodgkin lymphoma have a 15% to 40% likelihood of pulmonary involvement, such as a solitary lung mass or cavitary lung lesion. But clinicians at Bassett Healthcare in Cooperstown, New York, were faced with a rare case of another presentation: an endobronchial obstructing mass.
The patient, a 40-year-old man, reported having had cough, fatigue, and progressive weight loss (despite a good appetite) for 8 months. Because he had a history of smoking, he was treated for bronchitis, but the cough worsened. He had no fever, night sweats, dyspnea, or chest pain (common features of Hodgkin lymphoma).
Auscultation revealed clear lungs, with no crackles or wheeze, and no dullness to percussion. Blood work was negative except for eosinophilia. A subsequent chest radiograph showed an irregular left hilar lung opacity. A computer tomography scan showed a cavitary consolidation of the left upper lobe of the lung. Fiber-optic bronchoscopy with tissue from the endobronchial mass indicated an obstructing lesion in the left upper lobe bronchus. The clinicians suspected lung cancer.
However, they also found inflammatory cells, and immunohistochemistry revealed findings consistent with Hodgkin lymphoma. The clinicians started the patient on chemotherapy. After 6 cycles, his symptoms resolved. Follow-up at 8 months showed no clinical evidence of recurrence.
As the clinicians found out, radiologically, Hodgkin lymphoma can mimic lung cancer. They advise histopathologic diagnosis for a patient presenting with lung mass.
Source:
Abid H, Khan J, Lone N. BMJ Case Rep. 2018;2018. pii: bcr-2017-223809.
doi: 10.1136/bcr-2017-223809.
Patients with Hodgkin lymphoma have a 15% to 40% likelihood of pulmonary involvement, such as a solitary lung mass or cavitary lung lesion. But clinicians at Bassett Healthcare in Cooperstown, New York, were faced with a rare case of another presentation: an endobronchial obstructing mass.
The patient, a 40-year-old man, reported having had cough, fatigue, and progressive weight loss (despite a good appetite) for 8 months. Because he had a history of smoking, he was treated for bronchitis, but the cough worsened. He had no fever, night sweats, dyspnea, or chest pain (common features of Hodgkin lymphoma).
Auscultation revealed clear lungs, with no crackles or wheeze, and no dullness to percussion. Blood work was negative except for eosinophilia. A subsequent chest radiograph showed an irregular left hilar lung opacity. A computer tomography scan showed a cavitary consolidation of the left upper lobe of the lung. Fiber-optic bronchoscopy with tissue from the endobronchial mass indicated an obstructing lesion in the left upper lobe bronchus. The clinicians suspected lung cancer.
However, they also found inflammatory cells, and immunohistochemistry revealed findings consistent with Hodgkin lymphoma. The clinicians started the patient on chemotherapy. After 6 cycles, his symptoms resolved. Follow-up at 8 months showed no clinical evidence of recurrence.
As the clinicians found out, radiologically, Hodgkin lymphoma can mimic lung cancer. They advise histopathologic diagnosis for a patient presenting with lung mass.
Source:
Abid H, Khan J, Lone N. BMJ Case Rep. 2018;2018. pii: bcr-2017-223809.
doi: 10.1136/bcr-2017-223809.
FDA approves new option in Hodgkin lymphoma treatment
The Food and Drug Administration has approved brentuximab vedotin, in combination with chemotherapy, for previously untreated adults with stage III or IV classical Hodgkin lymphoma.
The drug, which is marketed by Seattle Genetics as Adcetris, is already approved in classical Hodgkin lymphoma after relapse and after stem cell transplant when the patient is at risk of relapse or progression. The drug is also approved to treat both systemic anaplastic large cell lymphoma (ALCL) and primary cutaneous ALCL after failure on other treatments.
The modified 2-year progression-free survival in the trial was 82.1% for patients receiving brentuximab plus AVD versus 77.2% for ABVD (P = .03), a 23% relative risk reduction (N Engl J Med. 2018;378:331-44).
Common side effects of brentuximab vedotin include neutropenia, anemia, peripheral neuropathy, nausea, fatigue, constipation, diarrhea, vomiting, and pyrexia. The drug carries a boxed warning highlighting the risk of John Cunningham virus infection resulting in progressive multifocal leukoencephalopathy.
The Food and Drug Administration has approved brentuximab vedotin, in combination with chemotherapy, for previously untreated adults with stage III or IV classical Hodgkin lymphoma.
The drug, which is marketed by Seattle Genetics as Adcetris, is already approved in classical Hodgkin lymphoma after relapse and after stem cell transplant when the patient is at risk of relapse or progression. The drug is also approved to treat both systemic anaplastic large cell lymphoma (ALCL) and primary cutaneous ALCL after failure on other treatments.
The modified 2-year progression-free survival in the trial was 82.1% for patients receiving brentuximab plus AVD versus 77.2% for ABVD (P = .03), a 23% relative risk reduction (N Engl J Med. 2018;378:331-44).
Common side effects of brentuximab vedotin include neutropenia, anemia, peripheral neuropathy, nausea, fatigue, constipation, diarrhea, vomiting, and pyrexia. The drug carries a boxed warning highlighting the risk of John Cunningham virus infection resulting in progressive multifocal leukoencephalopathy.
The Food and Drug Administration has approved brentuximab vedotin, in combination with chemotherapy, for previously untreated adults with stage III or IV classical Hodgkin lymphoma.
The drug, which is marketed by Seattle Genetics as Adcetris, is already approved in classical Hodgkin lymphoma after relapse and after stem cell transplant when the patient is at risk of relapse or progression. The drug is also approved to treat both systemic anaplastic large cell lymphoma (ALCL) and primary cutaneous ALCL after failure on other treatments.
The modified 2-year progression-free survival in the trial was 82.1% for patients receiving brentuximab plus AVD versus 77.2% for ABVD (P = .03), a 23% relative risk reduction (N Engl J Med. 2018;378:331-44).
Common side effects of brentuximab vedotin include neutropenia, anemia, peripheral neuropathy, nausea, fatigue, constipation, diarrhea, vomiting, and pyrexia. The drug carries a boxed warning highlighting the risk of John Cunningham virus infection resulting in progressive multifocal leukoencephalopathy.
More evidence links increased BMI to higher multiple myeloma risk
A high body mass index in both early and later adulthood increases the risk for developing multiple myeloma (MM), according to a prospective analysis.
“This association did not significantly differ by gender but was nonetheless slightly stronger in men,” wrote Catherine R. Marinac, PhD, of the Dana-Farber Cancer Institute, Boston, and her colleagues. “MM risk was significantly positively associated with weight change and suggestive of a positive association for change in BMI since young adulthood. In contrast, we did not observe statistically significant associations of cumulative average physical activity or walking with MM risk.”
Dr. Marinac and her associates analyzed participants from the Nurses’ Health Study (NHS), the Health Professionals Follow-Up Study (HPFS), and the Women’s Health Study (WHS) with a pooled total of 575 MM cases and more than 5 million person-years of follow-up. From all of those databases, a combined baseline total of 49,374 men and 153,260 women were included in the analyses. Participants in all three of the cohorts were predominately white.
Each participant was required to report height and weight on a baseline questionnaire and updated weights on subsequent questionnaires. Using that height and weight information, the researchers calculated BMI. Physical activity also was reported using questionnaires, beginning in 1986 in the HPFS and NHS groups and at baseline for WHS, with all groups providing updates every 2-4 years. The researchers used the physical activity information to calculate the total metabolic equivalent (MET) hours of all activity and of walking per week.
Dr. Marinac and her team identified a total of 205 men from the HPFS cohort and 370 women (325 NHS, 45 WHS) with confirmed diagnoses of MM. The BMIs of those participants ranged from 23.8-25.8 kg/m2 at baseline and from 21.3-23.0 kg/m2 in young adulthood. Across all cohorts, each 5 kg/m2 increase in cumulative average adult BMI significantly increased the risk of MM by 17% (hazard ratio, 1.17; 95% confidence interval, 1.05-1.29).
In addition, the MM risk rose almost 30% for every 5 kg/m2 increase in young adult BMI (HR, 1.28; 95% CI, 1.12-1.47). Increased risk was not strictly related to changes in BMI but to incremental weight gain since young adulthood. (pooled HR, 1.04; 95% CI, 1.00-1.08; P = 0.03).
The study confirmed correlations between weight gain and increased MM risk, however, it also had certain limitations. For example, much of the data concerning weight, height, and physical activity were all self-reported. Another limitation is the sociodemographic heterogeneity of the study population.
Despite those limitations, Dr. Marinac emphasized that the study results add to evidence concerning weight gain and MM risk.
“Our findings support the growing body of literature demonstrating that a high BMI both early and later in adulthood is associated with the risk of MM, and suggest that maintaining a healthy body weight throughout life may be an important component to a much-needed MM prevention strategy,” wrote Dr. Marinac, who also is affiliated with the Harvard T.H. Chan School of Public Health, also in Boston.
“Further larger-scale studies aimed at clarifying the influence of obesity timing and duration and at directly evaluating the role of weight loss, ideally conducted in diverse prospective study populations and in [monoclonal gammopathy of undetermined significance] patients, will be important for elaborating the role of weight maintenance in MM prevention and for identifying high risk subgroups of patients that may benefit from weight loss.”
None of the researchers had competing financial interests to disclose.
SOURCE: Marinac CR et al. Br J Cancer. 2018 Mar 12. doi: 10.1038/s41416-018-0010-4.
A high body mass index in both early and later adulthood increases the risk for developing multiple myeloma (MM), according to a prospective analysis.
“This association did not significantly differ by gender but was nonetheless slightly stronger in men,” wrote Catherine R. Marinac, PhD, of the Dana-Farber Cancer Institute, Boston, and her colleagues. “MM risk was significantly positively associated with weight change and suggestive of a positive association for change in BMI since young adulthood. In contrast, we did not observe statistically significant associations of cumulative average physical activity or walking with MM risk.”
Dr. Marinac and her associates analyzed participants from the Nurses’ Health Study (NHS), the Health Professionals Follow-Up Study (HPFS), and the Women’s Health Study (WHS) with a pooled total of 575 MM cases and more than 5 million person-years of follow-up. From all of those databases, a combined baseline total of 49,374 men and 153,260 women were included in the analyses. Participants in all three of the cohorts were predominately white.
Each participant was required to report height and weight on a baseline questionnaire and updated weights on subsequent questionnaires. Using that height and weight information, the researchers calculated BMI. Physical activity also was reported using questionnaires, beginning in 1986 in the HPFS and NHS groups and at baseline for WHS, with all groups providing updates every 2-4 years. The researchers used the physical activity information to calculate the total metabolic equivalent (MET) hours of all activity and of walking per week.
Dr. Marinac and her team identified a total of 205 men from the HPFS cohort and 370 women (325 NHS, 45 WHS) with confirmed diagnoses of MM. The BMIs of those participants ranged from 23.8-25.8 kg/m2 at baseline and from 21.3-23.0 kg/m2 in young adulthood. Across all cohorts, each 5 kg/m2 increase in cumulative average adult BMI significantly increased the risk of MM by 17% (hazard ratio, 1.17; 95% confidence interval, 1.05-1.29).
In addition, the MM risk rose almost 30% for every 5 kg/m2 increase in young adult BMI (HR, 1.28; 95% CI, 1.12-1.47). Increased risk was not strictly related to changes in BMI but to incremental weight gain since young adulthood. (pooled HR, 1.04; 95% CI, 1.00-1.08; P = 0.03).
The study confirmed correlations between weight gain and increased MM risk, however, it also had certain limitations. For example, much of the data concerning weight, height, and physical activity were all self-reported. Another limitation is the sociodemographic heterogeneity of the study population.
Despite those limitations, Dr. Marinac emphasized that the study results add to evidence concerning weight gain and MM risk.
“Our findings support the growing body of literature demonstrating that a high BMI both early and later in adulthood is associated with the risk of MM, and suggest that maintaining a healthy body weight throughout life may be an important component to a much-needed MM prevention strategy,” wrote Dr. Marinac, who also is affiliated with the Harvard T.H. Chan School of Public Health, also in Boston.
“Further larger-scale studies aimed at clarifying the influence of obesity timing and duration and at directly evaluating the role of weight loss, ideally conducted in diverse prospective study populations and in [monoclonal gammopathy of undetermined significance] patients, will be important for elaborating the role of weight maintenance in MM prevention and for identifying high risk subgroups of patients that may benefit from weight loss.”
None of the researchers had competing financial interests to disclose.
SOURCE: Marinac CR et al. Br J Cancer. 2018 Mar 12. doi: 10.1038/s41416-018-0010-4.
A high body mass index in both early and later adulthood increases the risk for developing multiple myeloma (MM), according to a prospective analysis.
“This association did not significantly differ by gender but was nonetheless slightly stronger in men,” wrote Catherine R. Marinac, PhD, of the Dana-Farber Cancer Institute, Boston, and her colleagues. “MM risk was significantly positively associated with weight change and suggestive of a positive association for change in BMI since young adulthood. In contrast, we did not observe statistically significant associations of cumulative average physical activity or walking with MM risk.”
Dr. Marinac and her associates analyzed participants from the Nurses’ Health Study (NHS), the Health Professionals Follow-Up Study (HPFS), and the Women’s Health Study (WHS) with a pooled total of 575 MM cases and more than 5 million person-years of follow-up. From all of those databases, a combined baseline total of 49,374 men and 153,260 women were included in the analyses. Participants in all three of the cohorts were predominately white.
Each participant was required to report height and weight on a baseline questionnaire and updated weights on subsequent questionnaires. Using that height and weight information, the researchers calculated BMI. Physical activity also was reported using questionnaires, beginning in 1986 in the HPFS and NHS groups and at baseline for WHS, with all groups providing updates every 2-4 years. The researchers used the physical activity information to calculate the total metabolic equivalent (MET) hours of all activity and of walking per week.
Dr. Marinac and her team identified a total of 205 men from the HPFS cohort and 370 women (325 NHS, 45 WHS) with confirmed diagnoses of MM. The BMIs of those participants ranged from 23.8-25.8 kg/m2 at baseline and from 21.3-23.0 kg/m2 in young adulthood. Across all cohorts, each 5 kg/m2 increase in cumulative average adult BMI significantly increased the risk of MM by 17% (hazard ratio, 1.17; 95% confidence interval, 1.05-1.29).
In addition, the MM risk rose almost 30% for every 5 kg/m2 increase in young adult BMI (HR, 1.28; 95% CI, 1.12-1.47). Increased risk was not strictly related to changes in BMI but to incremental weight gain since young adulthood. (pooled HR, 1.04; 95% CI, 1.00-1.08; P = 0.03).
The study confirmed correlations between weight gain and increased MM risk, however, it also had certain limitations. For example, much of the data concerning weight, height, and physical activity were all self-reported. Another limitation is the sociodemographic heterogeneity of the study population.
Despite those limitations, Dr. Marinac emphasized that the study results add to evidence concerning weight gain and MM risk.
“Our findings support the growing body of literature demonstrating that a high BMI both early and later in adulthood is associated with the risk of MM, and suggest that maintaining a healthy body weight throughout life may be an important component to a much-needed MM prevention strategy,” wrote Dr. Marinac, who also is affiliated with the Harvard T.H. Chan School of Public Health, also in Boston.
“Further larger-scale studies aimed at clarifying the influence of obesity timing and duration and at directly evaluating the role of weight loss, ideally conducted in diverse prospective study populations and in [monoclonal gammopathy of undetermined significance] patients, will be important for elaborating the role of weight maintenance in MM prevention and for identifying high risk subgroups of patients that may benefit from weight loss.”
None of the researchers had competing financial interests to disclose.
SOURCE: Marinac CR et al. Br J Cancer. 2018 Mar 12. doi: 10.1038/s41416-018-0010-4.
FROM BRITISH JOURNAL OF CANCER
Key clinical point: Moderate increases in body mass index (BMI) can dramatically increase the risk of developing multiple myeloma (MM).
Major finding: Each 5 kg/m2 increase in cumulative average adult BMI significantly increased the risk of MM by 17%.
Study details: Prospective analysis of 49,374 men and 153,260 women from three databases.
Disclosures: None of the researchers had competing financial interests to disclose.
Source: Marinac CR et al. Br J Cancer. 2018 Mar 12. doi: 10.1038/s41416-018-0010-4.