User login
Whether to anticoagulate: Toward a more reasoned approach
The article by Hagerty and Rich in this issue of the Cleveland Clinic Journal of Medicine1 covers an important topic—whether elderly patients with atrial fibrillation should receive anticoagulant therapy for it, or whether the risk of bleeding with this therapy outweighs the benefit of preventing stroke.
BETTER RISK PREDICTORS ARE NEEDED
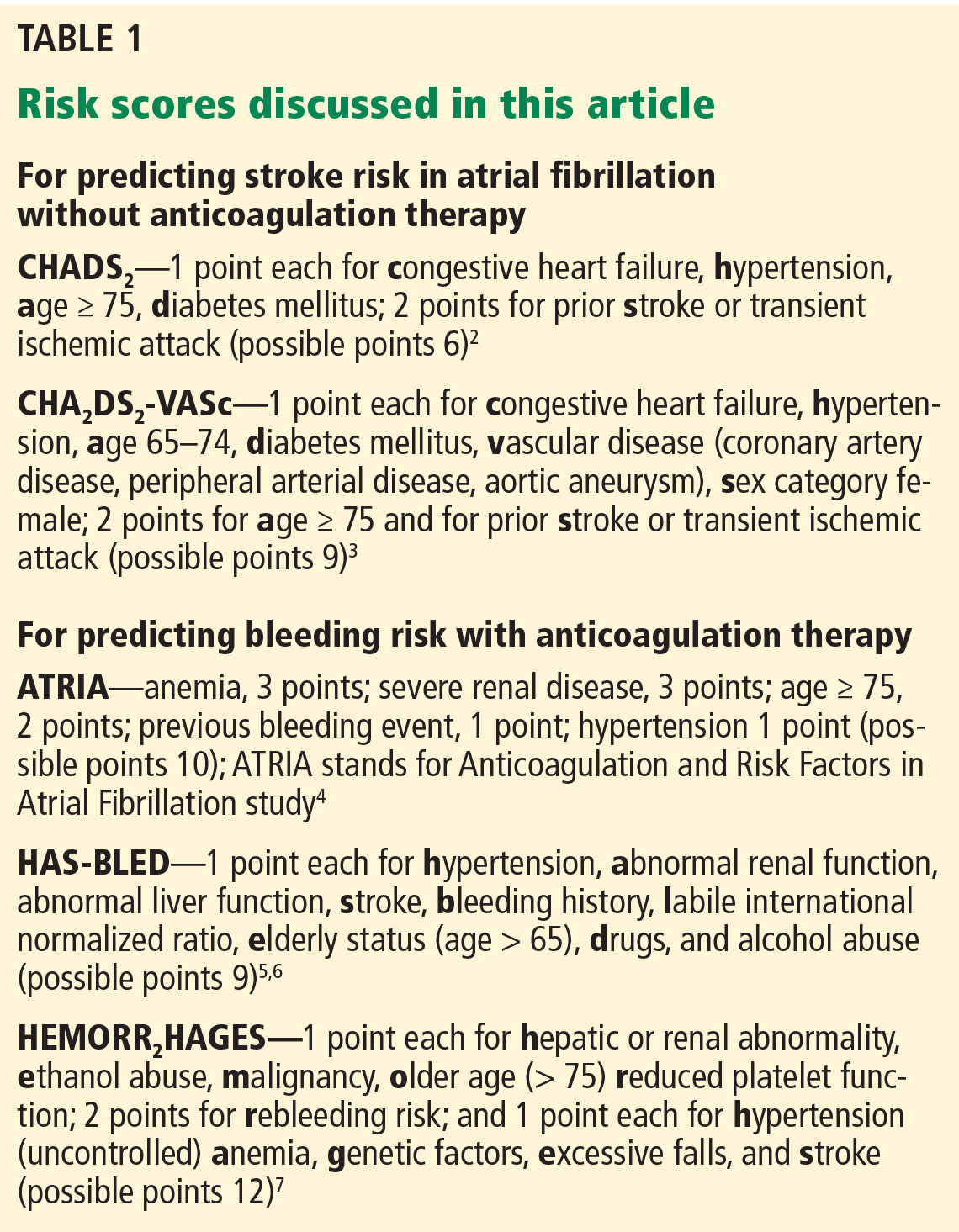
Prediction tools are available for estimating the risk of stroke in patients with atrial fibrillation without anticoagulation2,3 and to estimate bleeding risk from anticoagulation4–7 (Table 1). Both tools have limitations, but as Hagerty and Rich point out, the stroke risk scales are likely better than the bleeding risk scales.
For example, Fang et al8 note that the risk of intracranial hemorrhage increases significantly after age 85. The bleeding risk scales use lower age cutoffs than this, perhaps increasing their sensitivity but decreasing their specificity.
Although HAS-BLED5,6 includes antiplatelet drugs such as nonsteroidal anti-inflammatory drugs and aspirin as risk factors for bleeding, ATRIA4 and HEMORR2HAGES7 do not.
Other drugs such as macrolides, quinolones, and high-dose corticosteroids raise the international normalized ratio (INR). These are typically used short-term, but can cause major fluctuations in the INR that may not be detected by monthly INR checks. Incorporating the short-term use of such drugs into bleeding risk scales would be difficult if not impossible a priori. Yet clinicians should be aware that these drugs can affect bleeding risk.
As Hagerty and Rich note,1 the bleeding risk scores were developed for warfarin, and their applicability to patients treated with novel oral anticoagulants is uncertain.
All three of the available bleeding risk scales consider prior bleeding as a risk factor, but the severity of the prior bleeding varies. Although it is understandable to include major bleeding as a risk factor since it carries an increased risk of death, minor bleeding can affect morbidity and quality of life. Only the ATRIA score4 considers both major and minor bleeding, while HEMORR2HAGES7 does not specify bleeding severity, and HAS-BLED5,6 considers only major bleeding. Clearly, there is a need to update these existing bleeding risk scores so that they can apply to novel oral anticoagulants and consider both major and minor bleeding.
As the authors note, for predicting the risk of stroke, the CHA2DS2-VASc score3 provides more precision than the CHADS2 score2 at the lower end of the benefit spectrum. Unfortunately, there is no similar screening tool to predict bleeding risk from anticoagulation with greater precision in the middle to lower part of the risk spectrum.
THE PATIENT’S PREFERENCES MATTER
The patient’s life expectancy and personal preferences are important independent factors that affect the decision of whether to anticoagulate or not. It is the responsibility of clinicians who care for older adults to make sure that these two important considerations are included in any anticoagulation decision-making for this group of patients.
- Hagerty T, Rich MW. Fall risk and anticoagulation for atrial fibrillation in the elderly: a delicate balance. Cleve Clin J Med 2017; 84:35–40.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the Euro Heart Survey on atrial fibrillation. Chest 2010; 137:263–272.
- Fang MC, Go AS, Chang Y, et al. A new risk scheme to predict warfarin-associated hemorrhage: the ATRIA (Anticoagulation and Risk Factors in Atrial Fibrillation) study. J Am Coll Cardiol 2011; 58:395–401.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Lip GY, Frison L, Halperin JL, Lane DA. Comparative validation of a novel risk score for predicting bleeding risk in anticoagulated patients with atrial fibrillation: the HAS-BLED (Hypertension, Abnormal Renal/Liver Function, Stroke, Bleeding History or Predisposition, Labile INR, Elderly, Drugs/Alcohol Concomitantly) score. J Am Coll Cardiol 2011; 57:173–180.
- Gage BF, Yan Y, Milligan PE, et al. Clinical classification schemes for predicting hemorrhage: results from the National Registry of Atrial Fibrillation (NRAF). Am Heart J 2006; 151:713–719.
- Fang MC, Chang Y, Hylek EM, et al. Advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Ann Intern Med 2004; 141:745–752.
The article by Hagerty and Rich in this issue of the Cleveland Clinic Journal of Medicine1 covers an important topic—whether elderly patients with atrial fibrillation should receive anticoagulant therapy for it, or whether the risk of bleeding with this therapy outweighs the benefit of preventing stroke.
BETTER RISK PREDICTORS ARE NEEDED
Prediction tools are available for estimating the risk of stroke in patients with atrial fibrillation without anticoagulation2,3 and to estimate bleeding risk from anticoagulation4–7 (Table 1). Both tools have limitations, but as Hagerty and Rich point out, the stroke risk scales are likely better than the bleeding risk scales.
For example, Fang et al8 note that the risk of intracranial hemorrhage increases significantly after age 85. The bleeding risk scales use lower age cutoffs than this, perhaps increasing their sensitivity but decreasing their specificity.
Although HAS-BLED5,6 includes antiplatelet drugs such as nonsteroidal anti-inflammatory drugs and aspirin as risk factors for bleeding, ATRIA4 and HEMORR2HAGES7 do not.
Other drugs such as macrolides, quinolones, and high-dose corticosteroids raise the international normalized ratio (INR). These are typically used short-term, but can cause major fluctuations in the INR that may not be detected by monthly INR checks. Incorporating the short-term use of such drugs into bleeding risk scales would be difficult if not impossible a priori. Yet clinicians should be aware that these drugs can affect bleeding risk.
As Hagerty and Rich note,1 the bleeding risk scores were developed for warfarin, and their applicability to patients treated with novel oral anticoagulants is uncertain.
All three of the available bleeding risk scales consider prior bleeding as a risk factor, but the severity of the prior bleeding varies. Although it is understandable to include major bleeding as a risk factor since it carries an increased risk of death, minor bleeding can affect morbidity and quality of life. Only the ATRIA score4 considers both major and minor bleeding, while HEMORR2HAGES7 does not specify bleeding severity, and HAS-BLED5,6 considers only major bleeding. Clearly, there is a need to update these existing bleeding risk scores so that they can apply to novel oral anticoagulants and consider both major and minor bleeding.
As the authors note, for predicting the risk of stroke, the CHA2DS2-VASc score3 provides more precision than the CHADS2 score2 at the lower end of the benefit spectrum. Unfortunately, there is no similar screening tool to predict bleeding risk from anticoagulation with greater precision in the middle to lower part of the risk spectrum.
THE PATIENT’S PREFERENCES MATTER
The patient’s life expectancy and personal preferences are important independent factors that affect the decision of whether to anticoagulate or not. It is the responsibility of clinicians who care for older adults to make sure that these two important considerations are included in any anticoagulation decision-making for this group of patients.
The article by Hagerty and Rich in this issue of the Cleveland Clinic Journal of Medicine1 covers an important topic—whether elderly patients with atrial fibrillation should receive anticoagulant therapy for it, or whether the risk of bleeding with this therapy outweighs the benefit of preventing stroke.
BETTER RISK PREDICTORS ARE NEEDED
Prediction tools are available for estimating the risk of stroke in patients with atrial fibrillation without anticoagulation2,3 and to estimate bleeding risk from anticoagulation4–7 (Table 1). Both tools have limitations, but as Hagerty and Rich point out, the stroke risk scales are likely better than the bleeding risk scales.
For example, Fang et al8 note that the risk of intracranial hemorrhage increases significantly after age 85. The bleeding risk scales use lower age cutoffs than this, perhaps increasing their sensitivity but decreasing their specificity.
Although HAS-BLED5,6 includes antiplatelet drugs such as nonsteroidal anti-inflammatory drugs and aspirin as risk factors for bleeding, ATRIA4 and HEMORR2HAGES7 do not.
Other drugs such as macrolides, quinolones, and high-dose corticosteroids raise the international normalized ratio (INR). These are typically used short-term, but can cause major fluctuations in the INR that may not be detected by monthly INR checks. Incorporating the short-term use of such drugs into bleeding risk scales would be difficult if not impossible a priori. Yet clinicians should be aware that these drugs can affect bleeding risk.
As Hagerty and Rich note,1 the bleeding risk scores were developed for warfarin, and their applicability to patients treated with novel oral anticoagulants is uncertain.
All three of the available bleeding risk scales consider prior bleeding as a risk factor, but the severity of the prior bleeding varies. Although it is understandable to include major bleeding as a risk factor since it carries an increased risk of death, minor bleeding can affect morbidity and quality of life. Only the ATRIA score4 considers both major and minor bleeding, while HEMORR2HAGES7 does not specify bleeding severity, and HAS-BLED5,6 considers only major bleeding. Clearly, there is a need to update these existing bleeding risk scores so that they can apply to novel oral anticoagulants and consider both major and minor bleeding.
As the authors note, for predicting the risk of stroke, the CHA2DS2-VASc score3 provides more precision than the CHADS2 score2 at the lower end of the benefit spectrum. Unfortunately, there is no similar screening tool to predict bleeding risk from anticoagulation with greater precision in the middle to lower part of the risk spectrum.
THE PATIENT’S PREFERENCES MATTER
The patient’s life expectancy and personal preferences are important independent factors that affect the decision of whether to anticoagulate or not. It is the responsibility of clinicians who care for older adults to make sure that these two important considerations are included in any anticoagulation decision-making for this group of patients.
- Hagerty T, Rich MW. Fall risk and anticoagulation for atrial fibrillation in the elderly: a delicate balance. Cleve Clin J Med 2017; 84:35–40.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the Euro Heart Survey on atrial fibrillation. Chest 2010; 137:263–272.
- Fang MC, Go AS, Chang Y, et al. A new risk scheme to predict warfarin-associated hemorrhage: the ATRIA (Anticoagulation and Risk Factors in Atrial Fibrillation) study. J Am Coll Cardiol 2011; 58:395–401.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Lip GY, Frison L, Halperin JL, Lane DA. Comparative validation of a novel risk score for predicting bleeding risk in anticoagulated patients with atrial fibrillation: the HAS-BLED (Hypertension, Abnormal Renal/Liver Function, Stroke, Bleeding History or Predisposition, Labile INR, Elderly, Drugs/Alcohol Concomitantly) score. J Am Coll Cardiol 2011; 57:173–180.
- Gage BF, Yan Y, Milligan PE, et al. Clinical classification schemes for predicting hemorrhage: results from the National Registry of Atrial Fibrillation (NRAF). Am Heart J 2006; 151:713–719.
- Fang MC, Chang Y, Hylek EM, et al. Advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Ann Intern Med 2004; 141:745–752.
- Hagerty T, Rich MW. Fall risk and anticoagulation for atrial fibrillation in the elderly: a delicate balance. Cleve Clin J Med 2017; 84:35–40.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the Euro Heart Survey on atrial fibrillation. Chest 2010; 137:263–272.
- Fang MC, Go AS, Chang Y, et al. A new risk scheme to predict warfarin-associated hemorrhage: the ATRIA (Anticoagulation and Risk Factors in Atrial Fibrillation) study. J Am Coll Cardiol 2011; 58:395–401.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Lip GY, Frison L, Halperin JL, Lane DA. Comparative validation of a novel risk score for predicting bleeding risk in anticoagulated patients with atrial fibrillation: the HAS-BLED (Hypertension, Abnormal Renal/Liver Function, Stroke, Bleeding History or Predisposition, Labile INR, Elderly, Drugs/Alcohol Concomitantly) score. J Am Coll Cardiol 2011; 57:173–180.
- Gage BF, Yan Y, Milligan PE, et al. Clinical classification schemes for predicting hemorrhage: results from the National Registry of Atrial Fibrillation (NRAF). Am Heart J 2006; 151:713–719.
- Fang MC, Chang Y, Hylek EM, et al. Advanced age, anticoagulation intensity, and risk for intracranial hemorrhage among patients taking warfarin for atrial fibrillation. Ann Intern Med 2004; 141:745–752.

What is the role of roflumilast in chronic obstructive pulmonary disease?
Roflumilast has been shown to reduce rates of acute exacerbation in patients with severe chronic obstructive pulmonary disease (COPD), ie, forced expiratory volume in 1 second (FEV1) less than 50% with symptoms of chronic bronchitis and a history of exacerbations.
Roflumilast is a selective phosphodiesterase 4 (PDE4) inhibitor that acts on airway smooth muscle cells and various inflammatory cells. By blocking PDE4, roflumilast raises cyclic adenosine monophosphate levels within these cells, curtailing the inflammatory response.1,2
Roflumilast is not a bronchodilator, although modest improvements in FEV1 have been documented in clinical trials when it was used as maintenance therapy.
TRIALS OF ROFLUMILAST
Several trials have investigated the efficacy of roflumilast in COPD (Table 1).
The RECORD trial

The RECORD trial1 in 2005 was the first large randomized controlled trial of roflumilast in moderate to severe COPD. At a dose of 500 µg orally daily, there was a modest but statistically significant improvement in the postbronchodilator FEV1. There was also improvement in the St. George Respiratory Questionnaire score in the treatment arm, but this was not statistically significant. The study also found a reduction in acute exacerbations of COPD with roflumilast, which was a secondary end point.1
The results of this study spurred interest in roflumilast as well as criticism of the design of the study. First, COPD patients on inhaled maintenance therapy such as an inhaled corticosteroid and long-acting beta-agonist combination or a long-acting muscarinic antagonist had their medications held during the study. Second, the average FEV1 was 54% of predicted, indicative of a study population with less severe disease.1
The RATIO trial
Taking into account the results of the RECORD trial, the RATIO trial3 in 2007 recruited patients with more severe COPD—ie, Global Initiative for Chronic Obstructive Lung Disease (GOLD) class III and IV—and included the rate of acute exacerbations as a primary end point. Maintenance therapy with inhaled corticosteroids was continued in patients already taking them. However, long-acting beta-agonists and long-acting muscarinic antagonist therapies were held.3
Again, roflumilast improved postbronchodilator FEV1 compared with placebo. A reduction in acute exacerbations was seen but was not statistically significant except in subgroup analysis, where a statistically significant reduction in acute exacerbations was noted for patients with very severe (GOLD class IV) COPD.3
Post hoc analysis from the RATIO trial suggested that patients with chronic bronchitis and patients with a history of frequent exacerbations were more likely to respond to roflumilast.2
The EOS and HELIOS trials
In 2009, the results of the EOS and HELIOS trials of roflumilast in patients with severe COPD were published.4 These trials allowed continuation of long-acting beta-agonists and muscarinic antagonists. The prebronchodilator FEV1 improved modestly when roflumilast was added to a long-acting bronchodilator. These studies ran for only 24 weeks, and the rate of acute exacerbations was not a primary end point, although the results did show a trend toward reduction of exacerbations.4
The AURA and HERMES trials
Also in 2009 was the publication of the results of two 52-week placebo-controlled trials (AURA and HERMES) of roflumilast in patients with severe COPD with chronic bronchitis and a history of frequent exacerbations.5 Maintenance therapy with long-acting beta-agonists was continued, whereas inhaled corticosteroids and long-acting muscarinic antagonists were held. Statistically significant improvements in prebronchodilator FEV1 and reduction in the rate of exacerbations were observed in the roflumilast group (17% reduction, 95% confidence interval 8–25, P < .0003).5
The REACT trial
The REACT trial6 randomized 1,945 patients with severe COPD already on maximal recommended combination inhaled corticosteroid and long-acting beta-agonist therapy to receive either roflumilast or placebo. The patients’ ratio of FEV1 to forced vital capacity was less than 70%, their postbronchodilator FEV1 was less than 50%, and they had chronic bronchitis and a history of at least two acute exacerbations during the past year. They had also been on combination therapy for the previous year. Patients who were on long-acting muscarinic-antagonist therapy (70% of the cohort) were included, and continued with their medication.
Patients were followed for 52 weeks. There was a significant reduction in the rate of exacerbations in the roflumilast group vs placebo (0.823 vs 0.959; risk ratio 0.858; 95% confidence interval 0.740–0.995; P = .0424).6 As in previous trials, the roflumilast group showed an improvement in postbronchodilator FEV1. The study also showed a reduction in hospital admissions in the treatment group.6
ADVERSE EFFECTS OF ROFLUMILAST
Roflumilast is known to have adverse effects significant enough to reduce compliance, the most common being diarrhea, weight loss, and nausea.2,6,7 In the REACT trial,6 11% of patients in the roflumilast group vs 5% in the placebo group dropped out of the study because of adverse drug effects. Diarrhea was reported in 10% and weight loss in 9% of patients taking roflumilast. Weight loss has been shown to be reversible upon stopping roflumilast.2 There has been no evidence of increased risk of death or serious adverse events in studies of roflumilast in patients with COPD.2 However, the benefit-to-harm ratio suggests that roflumilast provides a net benefit only in patients at high risk of severe exacerbations.7
- Rabe KF, Bateman ED, O’Donnell DE, Witte S, Bredenbroker D, Bethke TD. Roflumilast—an oral anti-inflammatory treatment for chronic obstructive pulmonary disease: a randomized controlled trial. Lancet 2005; 366:63–71.
- Field SK. Roflumilast, a novel phosphodiesterase 4 inhibitor, for COPD patients with a history of exacerbations. Clin Med Insights Circ Respir Pulm Med 2011; 5:57–70.
- Calverley PM, Sanchez-Toril F, McIvor A, Teichmann P, Bredenbroeker D, Fabbri LM. Effect of 1-year treatment with roflumilast in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007; 176:154–161.
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al; M2-127 and M2-128 study groups. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomized clinical trials. Lancet 2009; 374:695–703.
- Calverley PM, Rabe KF, Goehring U-M, Kristiansen S, Fabbri LM, Martinez FJ. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomized clinical trials. Lancet 2009; 374:684–95.
- Martinez FJ, Calverley PM, Goehring UM, Brose M, Fabbri LM, Rabe KF. Effect of roflumilast on exacerbations in patients with severe chronic obstructive pulmonary disease uncontrolled by combination therapy (REACT): a multicentre randomised controlled trial. Lancet 2015; 385:857–866.
- Yu T, Fain K, Boyd CM, et al. Benefits and harms of roflumilast in moderate to severe COPD. Thorax 2014; 69:616–622.
Roflumilast has been shown to reduce rates of acute exacerbation in patients with severe chronic obstructive pulmonary disease (COPD), ie, forced expiratory volume in 1 second (FEV1) less than 50% with symptoms of chronic bronchitis and a history of exacerbations.
Roflumilast is a selective phosphodiesterase 4 (PDE4) inhibitor that acts on airway smooth muscle cells and various inflammatory cells. By blocking PDE4, roflumilast raises cyclic adenosine monophosphate levels within these cells, curtailing the inflammatory response.1,2
Roflumilast is not a bronchodilator, although modest improvements in FEV1 have been documented in clinical trials when it was used as maintenance therapy.
TRIALS OF ROFLUMILAST
Several trials have investigated the efficacy of roflumilast in COPD (Table 1).
The RECORD trial
The RECORD trial1 in 2005 was the first large randomized controlled trial of roflumilast in moderate to severe COPD. At a dose of 500 µg orally daily, there was a modest but statistically significant improvement in the postbronchodilator FEV1. There was also improvement in the St. George Respiratory Questionnaire score in the treatment arm, but this was not statistically significant. The study also found a reduction in acute exacerbations of COPD with roflumilast, which was a secondary end point.1
The results of this study spurred interest in roflumilast as well as criticism of the design of the study. First, COPD patients on inhaled maintenance therapy such as an inhaled corticosteroid and long-acting beta-agonist combination or a long-acting muscarinic antagonist had their medications held during the study. Second, the average FEV1 was 54% of predicted, indicative of a study population with less severe disease.1
The RATIO trial
Taking into account the results of the RECORD trial, the RATIO trial3 in 2007 recruited patients with more severe COPD—ie, Global Initiative for Chronic Obstructive Lung Disease (GOLD) class III and IV—and included the rate of acute exacerbations as a primary end point. Maintenance therapy with inhaled corticosteroids was continued in patients already taking them. However, long-acting beta-agonists and long-acting muscarinic antagonist therapies were held.3
Again, roflumilast improved postbronchodilator FEV1 compared with placebo. A reduction in acute exacerbations was seen but was not statistically significant except in subgroup analysis, where a statistically significant reduction in acute exacerbations was noted for patients with very severe (GOLD class IV) COPD.3
Post hoc analysis from the RATIO trial suggested that patients with chronic bronchitis and patients with a history of frequent exacerbations were more likely to respond to roflumilast.2
The EOS and HELIOS trials
In 2009, the results of the EOS and HELIOS trials of roflumilast in patients with severe COPD were published.4 These trials allowed continuation of long-acting beta-agonists and muscarinic antagonists. The prebronchodilator FEV1 improved modestly when roflumilast was added to a long-acting bronchodilator. These studies ran for only 24 weeks, and the rate of acute exacerbations was not a primary end point, although the results did show a trend toward reduction of exacerbations.4
The AURA and HERMES trials
Also in 2009 was the publication of the results of two 52-week placebo-controlled trials (AURA and HERMES) of roflumilast in patients with severe COPD with chronic bronchitis and a history of frequent exacerbations.5 Maintenance therapy with long-acting beta-agonists was continued, whereas inhaled corticosteroids and long-acting muscarinic antagonists were held. Statistically significant improvements in prebronchodilator FEV1 and reduction in the rate of exacerbations were observed in the roflumilast group (17% reduction, 95% confidence interval 8–25, P < .0003).5
The REACT trial
The REACT trial6 randomized 1,945 patients with severe COPD already on maximal recommended combination inhaled corticosteroid and long-acting beta-agonist therapy to receive either roflumilast or placebo. The patients’ ratio of FEV1 to forced vital capacity was less than 70%, their postbronchodilator FEV1 was less than 50%, and they had chronic bronchitis and a history of at least two acute exacerbations during the past year. They had also been on combination therapy for the previous year. Patients who were on long-acting muscarinic-antagonist therapy (70% of the cohort) were included, and continued with their medication.
Patients were followed for 52 weeks. There was a significant reduction in the rate of exacerbations in the roflumilast group vs placebo (0.823 vs 0.959; risk ratio 0.858; 95% confidence interval 0.740–0.995; P = .0424).6 As in previous trials, the roflumilast group showed an improvement in postbronchodilator FEV1. The study also showed a reduction in hospital admissions in the treatment group.6
ADVERSE EFFECTS OF ROFLUMILAST
Roflumilast is known to have adverse effects significant enough to reduce compliance, the most common being diarrhea, weight loss, and nausea.2,6,7 In the REACT trial,6 11% of patients in the roflumilast group vs 5% in the placebo group dropped out of the study because of adverse drug effects. Diarrhea was reported in 10% and weight loss in 9% of patients taking roflumilast. Weight loss has been shown to be reversible upon stopping roflumilast.2 There has been no evidence of increased risk of death or serious adverse events in studies of roflumilast in patients with COPD.2 However, the benefit-to-harm ratio suggests that roflumilast provides a net benefit only in patients at high risk of severe exacerbations.7
Roflumilast has been shown to reduce rates of acute exacerbation in patients with severe chronic obstructive pulmonary disease (COPD), ie, forced expiratory volume in 1 second (FEV1) less than 50% with symptoms of chronic bronchitis and a history of exacerbations.
Roflumilast is a selective phosphodiesterase 4 (PDE4) inhibitor that acts on airway smooth muscle cells and various inflammatory cells. By blocking PDE4, roflumilast raises cyclic adenosine monophosphate levels within these cells, curtailing the inflammatory response.1,2
Roflumilast is not a bronchodilator, although modest improvements in FEV1 have been documented in clinical trials when it was used as maintenance therapy.
TRIALS OF ROFLUMILAST
Several trials have investigated the efficacy of roflumilast in COPD (Table 1).
The RECORD trial
The RECORD trial1 in 2005 was the first large randomized controlled trial of roflumilast in moderate to severe COPD. At a dose of 500 µg orally daily, there was a modest but statistically significant improvement in the postbronchodilator FEV1. There was also improvement in the St. George Respiratory Questionnaire score in the treatment arm, but this was not statistically significant. The study also found a reduction in acute exacerbations of COPD with roflumilast, which was a secondary end point.1
The results of this study spurred interest in roflumilast as well as criticism of the design of the study. First, COPD patients on inhaled maintenance therapy such as an inhaled corticosteroid and long-acting beta-agonist combination or a long-acting muscarinic antagonist had their medications held during the study. Second, the average FEV1 was 54% of predicted, indicative of a study population with less severe disease.1
The RATIO trial
Taking into account the results of the RECORD trial, the RATIO trial3 in 2007 recruited patients with more severe COPD—ie, Global Initiative for Chronic Obstructive Lung Disease (GOLD) class III and IV—and included the rate of acute exacerbations as a primary end point. Maintenance therapy with inhaled corticosteroids was continued in patients already taking them. However, long-acting beta-agonists and long-acting muscarinic antagonist therapies were held.3
Again, roflumilast improved postbronchodilator FEV1 compared with placebo. A reduction in acute exacerbations was seen but was not statistically significant except in subgroup analysis, where a statistically significant reduction in acute exacerbations was noted for patients with very severe (GOLD class IV) COPD.3
Post hoc analysis from the RATIO trial suggested that patients with chronic bronchitis and patients with a history of frequent exacerbations were more likely to respond to roflumilast.2
The EOS and HELIOS trials
In 2009, the results of the EOS and HELIOS trials of roflumilast in patients with severe COPD were published.4 These trials allowed continuation of long-acting beta-agonists and muscarinic antagonists. The prebronchodilator FEV1 improved modestly when roflumilast was added to a long-acting bronchodilator. These studies ran for only 24 weeks, and the rate of acute exacerbations was not a primary end point, although the results did show a trend toward reduction of exacerbations.4
The AURA and HERMES trials
Also in 2009 was the publication of the results of two 52-week placebo-controlled trials (AURA and HERMES) of roflumilast in patients with severe COPD with chronic bronchitis and a history of frequent exacerbations.5 Maintenance therapy with long-acting beta-agonists was continued, whereas inhaled corticosteroids and long-acting muscarinic antagonists were held. Statistically significant improvements in prebronchodilator FEV1 and reduction in the rate of exacerbations were observed in the roflumilast group (17% reduction, 95% confidence interval 8–25, P < .0003).5
The REACT trial
The REACT trial6 randomized 1,945 patients with severe COPD already on maximal recommended combination inhaled corticosteroid and long-acting beta-agonist therapy to receive either roflumilast or placebo. The patients’ ratio of FEV1 to forced vital capacity was less than 70%, their postbronchodilator FEV1 was less than 50%, and they had chronic bronchitis and a history of at least two acute exacerbations during the past year. They had also been on combination therapy for the previous year. Patients who were on long-acting muscarinic-antagonist therapy (70% of the cohort) were included, and continued with their medication.
Patients were followed for 52 weeks. There was a significant reduction in the rate of exacerbations in the roflumilast group vs placebo (0.823 vs 0.959; risk ratio 0.858; 95% confidence interval 0.740–0.995; P = .0424).6 As in previous trials, the roflumilast group showed an improvement in postbronchodilator FEV1. The study also showed a reduction in hospital admissions in the treatment group.6
ADVERSE EFFECTS OF ROFLUMILAST
Roflumilast is known to have adverse effects significant enough to reduce compliance, the most common being diarrhea, weight loss, and nausea.2,6,7 In the REACT trial,6 11% of patients in the roflumilast group vs 5% in the placebo group dropped out of the study because of adverse drug effects. Diarrhea was reported in 10% and weight loss in 9% of patients taking roflumilast. Weight loss has been shown to be reversible upon stopping roflumilast.2 There has been no evidence of increased risk of death or serious adverse events in studies of roflumilast in patients with COPD.2 However, the benefit-to-harm ratio suggests that roflumilast provides a net benefit only in patients at high risk of severe exacerbations.7
- Rabe KF, Bateman ED, O’Donnell DE, Witte S, Bredenbroker D, Bethke TD. Roflumilast—an oral anti-inflammatory treatment for chronic obstructive pulmonary disease: a randomized controlled trial. Lancet 2005; 366:63–71.
- Field SK. Roflumilast, a novel phosphodiesterase 4 inhibitor, for COPD patients with a history of exacerbations. Clin Med Insights Circ Respir Pulm Med 2011; 5:57–70.
- Calverley PM, Sanchez-Toril F, McIvor A, Teichmann P, Bredenbroeker D, Fabbri LM. Effect of 1-year treatment with roflumilast in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007; 176:154–161.
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al; M2-127 and M2-128 study groups. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomized clinical trials. Lancet 2009; 374:695–703.
- Calverley PM, Rabe KF, Goehring U-M, Kristiansen S, Fabbri LM, Martinez FJ. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomized clinical trials. Lancet 2009; 374:684–95.
- Martinez FJ, Calverley PM, Goehring UM, Brose M, Fabbri LM, Rabe KF. Effect of roflumilast on exacerbations in patients with severe chronic obstructive pulmonary disease uncontrolled by combination therapy (REACT): a multicentre randomised controlled trial. Lancet 2015; 385:857–866.
- Yu T, Fain K, Boyd CM, et al. Benefits and harms of roflumilast in moderate to severe COPD. Thorax 2014; 69:616–622.
- Rabe KF, Bateman ED, O’Donnell DE, Witte S, Bredenbroker D, Bethke TD. Roflumilast—an oral anti-inflammatory treatment for chronic obstructive pulmonary disease: a randomized controlled trial. Lancet 2005; 366:63–71.
- Field SK. Roflumilast, a novel phosphodiesterase 4 inhibitor, for COPD patients with a history of exacerbations. Clin Med Insights Circ Respir Pulm Med 2011; 5:57–70.
- Calverley PM, Sanchez-Toril F, McIvor A, Teichmann P, Bredenbroeker D, Fabbri LM. Effect of 1-year treatment with roflumilast in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007; 176:154–161.
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al; M2-127 and M2-128 study groups. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomized clinical trials. Lancet 2009; 374:695–703.
- Calverley PM, Rabe KF, Goehring U-M, Kristiansen S, Fabbri LM, Martinez FJ. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomized clinical trials. Lancet 2009; 374:684–95.
- Martinez FJ, Calverley PM, Goehring UM, Brose M, Fabbri LM, Rabe KF. Effect of roflumilast on exacerbations in patients with severe chronic obstructive pulmonary disease uncontrolled by combination therapy (REACT): a multicentre randomised controlled trial. Lancet 2015; 385:857–866.
- Yu T, Fain K, Boyd CM, et al. Benefits and harms of roflumilast in moderate to severe COPD. Thorax 2014; 69:616–622.

Benign prostatic hyperplasia: Evaluation and medical management in primary care
Primary care physicians are uniquely positioned to screen for benign prostatic hyperplasia (BPH) and lower urinary tract symptoms, to perform the initial diagnostic workup, and to start medical therapy in uncomplicated cases. Effective medical therapy is available but underutilized in the primary care setting.1
This overview covers how to identify and evaluate patients with lower urinary tract symptoms, initiate therapy, and identify factors warranting timely urology referral.
TWO MECHANISMS: STATIC, DYNAMIC
BPH is a histologic diagnosis of proliferation of smooth muscle, epithelium, and stromal cells within the transition zone of the prostate,2 which surrounds the proximal urethra.
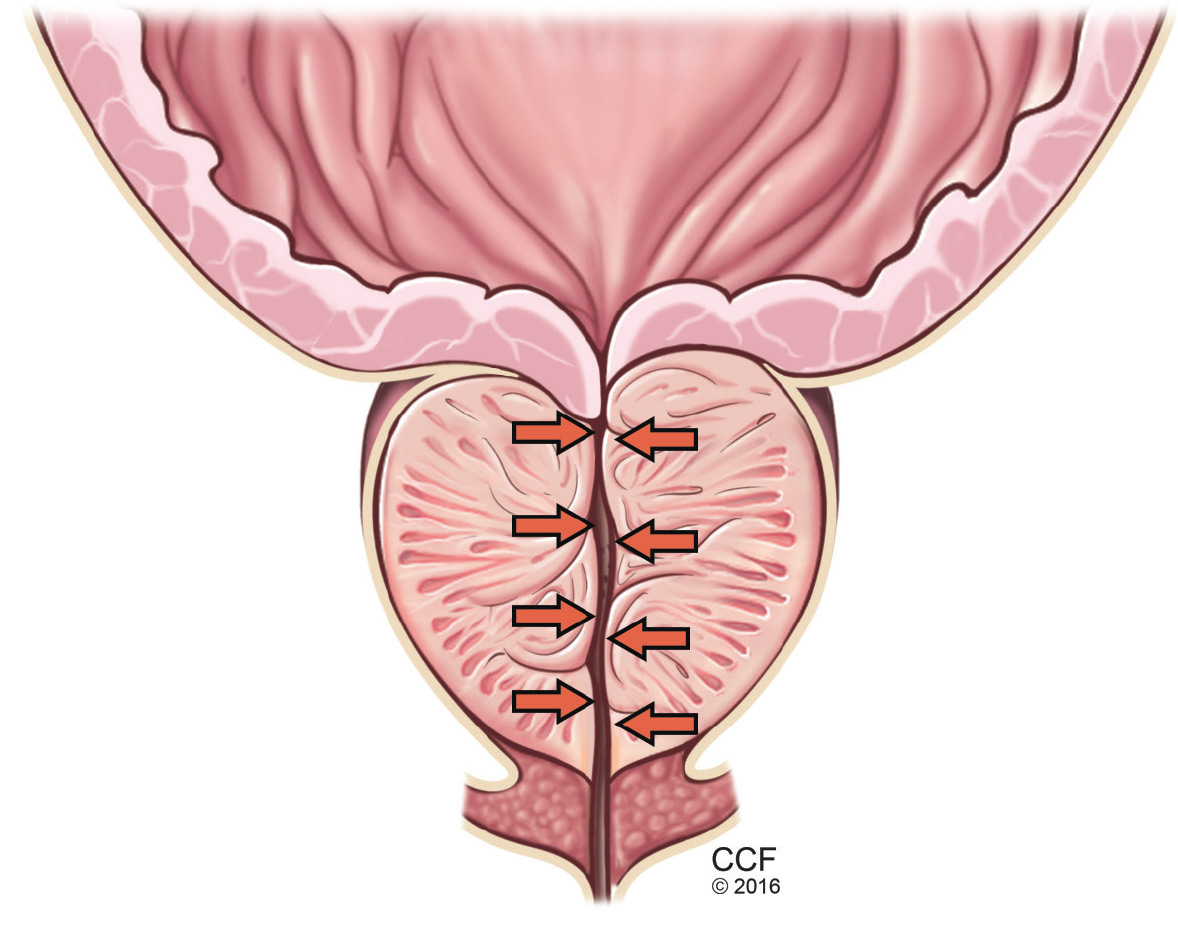
Symptoms arise through two mechanisms: static, in which the hyperplastic prostatic tissue compresses the urethra (Figure 1); and dynamic, with increased adrenergic nervous system and prostatic smooth muscle tone (Figure 2).3 Both mechanisms increase resistance to urinary flow at the level of the bladder outlet.
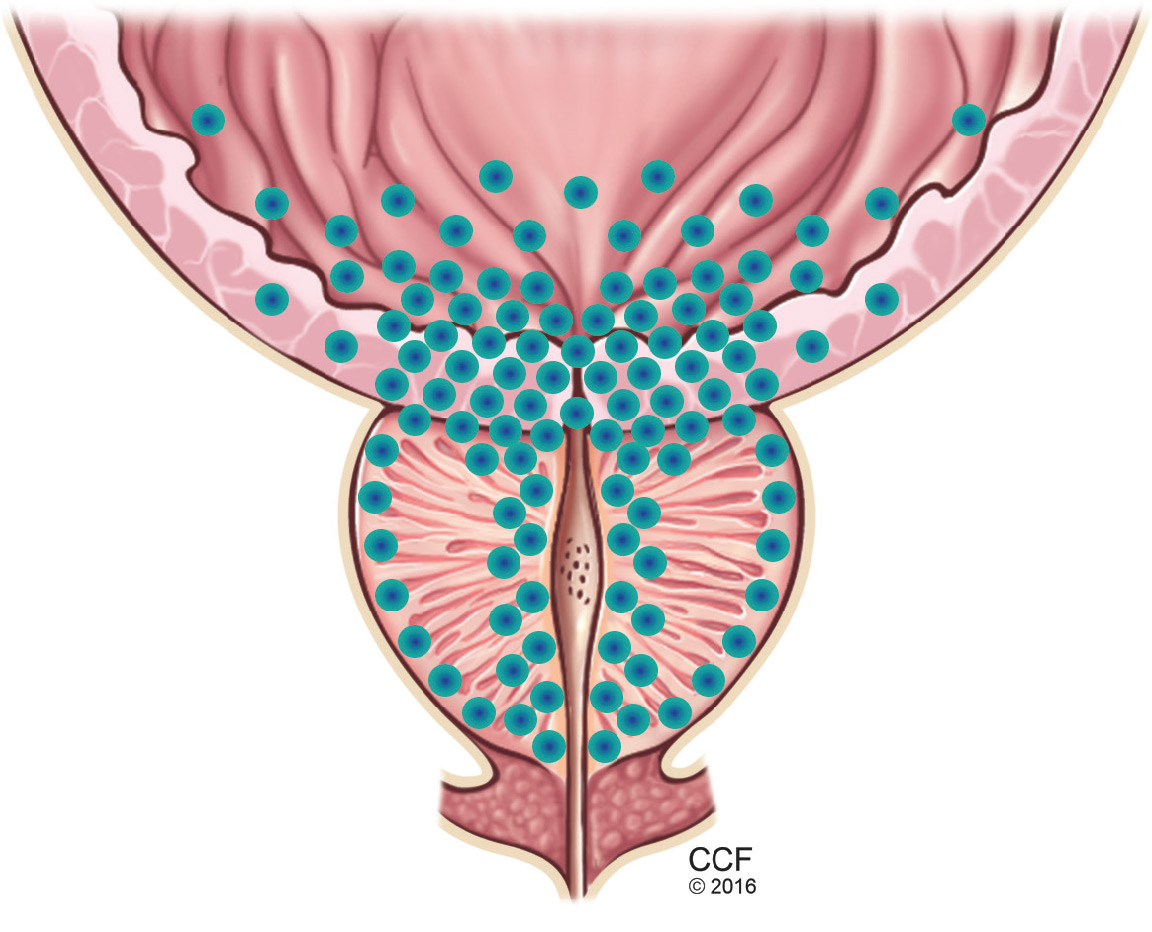
As an adaptive change to overcome outlet resistance and maintain urinary flow, the detrusor muscles undergo hypertrophy. However, over time the bladder may develop diminished compliance and increased detrusor activity, causing symptoms such as urinary frequency and urgency. Chronic bladder outlet obstruction can lead to bladder decompensation and detrusor underactivity, manifesting as incomplete emptying, urinary hesitancy, intermittency (starting and stopping while voiding), a weakened urinary stream, and urinary retention.
MOST MEN EVENTUALLY DEVELOP BPH
Autopsy studies have shown that BPH increases in prevalence with age beginning around age 30 and reaching a peak prevalence of 88% in men in their 80s.4 This trend parallels those of the incidence and severity of lower urinary tract symptoms.5
In the year 2000 alone, BPH was responsible for 4.5 million physician visits at an estimated direct cost of $1.1 billion, not including the cost of pharmacotherapy.6
OFFICE WORKUP
BPH can cause lower urinary tract symptoms that fall into two categories: storage and emptying. Storage symptoms include urinary frequency, urgency, and nocturia, whereas emptying symptoms include weak stream, hesitancy, intermittency, incomplete emptying, straining, and postvoid dribbling.
History and differential diagnosis
Assessment begins with characterizing the patient’s symptoms and determining those that are most bothersome. Because BPH is just one of many possible causes of lower urinary tract symptoms, a detailed medical history is necessary to evaluate for other conditions that may cause lower urinary tract dysfunction or complicate its treatment.
Obstructive urinary symptoms can arise from BPH or from other conditions, including urethral stricture disease and neurogenic voiding dysfunction.
Irritative voiding symptoms such as urinary urgency and frequency can result from detrusor overactivity secondary to BPH, but can also be caused by neurologic disease, malignancy, initiation of diuretic therapy, high fluid intake, or consumption of bladder irritants such as caffeine, alcohol, and spicy foods.
Urinary frequency is sometimes a presenting symptom of undiagnosed or poorly controlled diabetes mellitus resulting from glucosuria and polyuria. Iatrogenic causes of polyuria include the new hypoglycemic agents canagliflozin and dapagliflozin, which block renal glucose reabsorption, improving glycemic control by inducing urinary
glucose loss.7
Nocturia has many possible nonurologic causes including heart failure (in which excess extravascular fluid shifts to the intravascular space when the patient lies down, resulting in polyuria), obstructive sleep apnea, and behavioral factors such as high evening fluid intake. In these cases, patients usually have nocturnal polyuria (greater than one-third of 24-hour urine output at night) rather than only nocturia (waking at night to void). A fluid diary is a simple tool that can differentiate these two conditions.
Hematuria can develop in patients with BPH with bleeding from congested prostatic or bladder neck vessels; however, hematuria may indicate an underlying malignancy or urolithiasis, for which a urologic workup is indicated.
The broad differential diagnosis for the different lower urinary tract symptoms highlights the importance of obtaining a thorough history.
Physical examination
A general examination should include the following:
Body mass index. Obese patients are at risk of obstructive sleep apnea, which can cause nocturnal polyuria.
Gait. Abnormal gait may suggest a neurologic condition such as Parkinson disease or stroke that can also affect lower urinary tract function.
Lower abdomen. A palpable bladder suggests urinary retention.
External genitalia. Penile causes of urinary obstruction include urethral meatal stenosis or a palpable urethral mass.
Digital rectal examination can reveal benign prostatic enlargement or nodules or firmness, which suggest malignancy and warrant urologic referral.
Neurologic examination, including evaluation of anal sphincter tone and lower extremity sensorimotor function.
Feet. Bilateral lower-extremity edema may be due to heart failure or venous insufficiency.
The International Prostate Symptom Score
All men with lower urinary tract symptoms should complete the International Prostate Symptom Score (IPSS) survey, consisting of seven questions about urinary symptoms plus one about quality of life.8 Specifically, it asks the patient, “Over the past month, how often have you…”
- Had a sensation of not emptying your bladder completely after you finish urinating?
- Had to urinate again less than 2 hours after you finished urinating?
- Found you stopped and started again several times when you urinated?
- Found it difficult to postpone urination?
- Had a weak urinary stream?
- Had to push or strain to begin urination?
Each question above is scored as 0 (not at all), 1 (less than 1 time in 5), 2 (less than half the time), 3 (about half the time), 4 (more than half the time, or 5 (almost always).
- Over the past month, how many times did you most typically get up to urinate from the time you went to bed until the time you got up in the morning?
This question is scored from 0 (none) to 5 (5 times or more).
- If you were to spend the rest of your life with your urinary condition the way it is now, how would you feel about that?
This question is scored as 0 (delighted), 1 (pleased), 2 (mostly satisfied), 3 (mixed: equally satisfied and dissatisfied), 4 (mostly dissatisfied), 5 (unhappy), or 6 (terrible).
A total score of 1 to 7 is categorized as mild, 8 to 19 moderate, and 20 to 35 severe.
The questionnaire can also be used to evaluate for disease progression and response to treatment over time. A change of 3 points is clinically significant, as patients are unable to discern a difference below this threshold.9
Urinalysis
Urinalysis is recommended to assess for urinary tract infection, hematuria, proteinuria, or glucosuria.
Fluid diary
A fluid diary is useful for patients complaining of frequency or nocturia and can help quantify the volume of fluid intake, frequency of urination, and volumes voided. The patient should complete the diary over a 24-hour period, recording the time and volume of fluid intake and each void. This aids in diagnosing polyuria (> 3 L of urine output per 24 hours), nocturnal polyuria, and behavioral causes of symptoms, including excessive total fluid intake or high evening fluid intake contributing to nocturia.
Serum creatinine not recommended
Measuring serum creatinine is not recommended in the initial BPH workup, as men with lower urinary tract symptoms are not at higher risk of renal failure than those without these symptoms.10
Prostate-specific antigen
Prostate-specific antigen (PSA) is a glycoprotein primarily produced by prostatic luminal epithelial cells. It is most commonly discussed in the setting of prostate cancer screening, but its utility extends to guiding the management of BPH.
PSA levels correlate with prostate volume and subsequent growth.11 In addition, the risks of developing acute urinary retention or needing surgical intervention rise with increasing PSA.12 Among men in the Proscar Long-Term Efficacy and Safety Study, the risk of acute urinary retention or BPH-related surgery after 4 years in the watchful-waiting arm was 7.8% in men with a PSA of 1.3 ng/dL or less, compared with 19.9% in men with a PSA greater than 3.2 ng/dL.11 Therefore, men with BPH and an elevated PSA are at higher risk with watchful waiting and may be better served with medical therapy.
In addition, American Urological Association guidelines recommend measuring serum PSA levels in men with a life expectancy greater than 10 years in whom the diagnosis of prostate cancer would alter management.10
Urologic referral

If the initial evaluation reveals hematuria, recurrent urinary tract infection, a palpable bladder, abnormal findings on digital rectal examination suggesting prostate cancer, or a history of or risk factors for urethral stricture or neurologic disease, the patient should be referred to a urologist for further evaluation (Table 1).10 Other patients who should undergo urologic evaluation are those with persistent bothersome symptoms after basic management and those who desire referral.
Adjunctive tests
Patients referred for urologic evaluation may require additional tests for diagnosis and to guide management.
Postvoid residual volume is easily measured with either abdominal ultrasonography or catheterization and is often included in the urologic evaluation of BPH. Patients vary considerably in their residual volume, which correlates poorly with BPH, symptom severity, or surgical success. However, those with a residual volume of more than 100 mL have a slightly higher rate of failure with watchful waiting.13 Postvoid residual volume is not routinely monitored in patients with a low residual volume unless there is a significant change in urinary symptoms. Conversely, patients with a volume greater than 200 mL should be monitored closely for worsening urinary retention, especially if considering anticholinergic therapy.
There is no absolute threshold postvoid residual volume above which therapy is mandatory. Rather, the decision to intervene is based on symptom severity and whether sequelae of urinary retention (eg, incontinence, urinary tract infection, hematuria, hydronephrosis, renal dysfunction) are present.
Uroflowmetry is a noninvasive test measuring the urinary flow rate during voiding and is recommended during specialist evaluation of men with lower urinary tract symptoms and suspected BPH.10 Though a diminished urinary flow rate may be detected in men with bladder outlet obstruction from BPH, it cannot differentiate obstruction from detrusor underactivity, both of which may result in reduced urinary flow. Urodynamic studies can help differentiate between these two mechanisms of lower urinary tract symptoms. Uroflowmetry may be useful in selecting surgical candidates, as patients with a maximum urinary flow rate of 15 mL/second or greater have been shown to have lower rates of surgical success.14
Urodynamic studies. If the diagnosis of bladder outlet obstruction remains in doubt, urodynamic studies can differentiate obstruction from detrusor underactivity. Urodynamic studies allow simultaneous measurement of urinary flow and detrusor pressure, differentiating between obstruction (manifesting as diminished urinary flow with normal or elevated detrusor pressure) and detrusor underactivity (diminished urinary flow with diminished detrusor pressure). Nomograms15 and the easily calculated bladder outlet obstruction index16 are simple tools used to differentiate these two causes of diminished urinary flow.
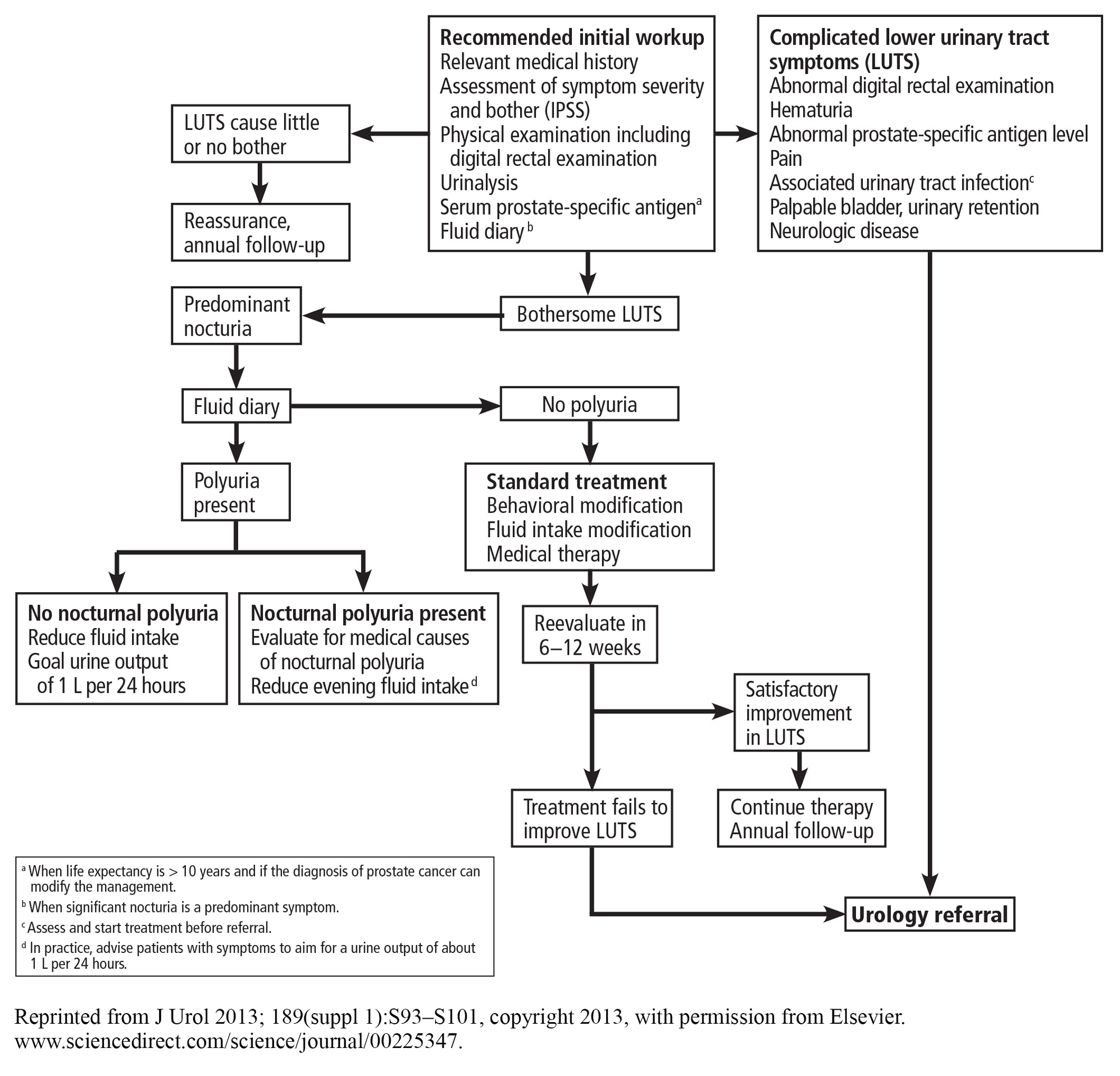
Cystourethroscopy is not recommended for routine evaluation of BPH. Indications for cystourethroscopy include hematuria and the presence of a risk factor for urethral stricture disease such as urethritis, prior urethral instrumentation, or perineal trauma. Cystourethroscopy can also aid in surgical planning when intervention is considered.
An algorithm for diagnostic workup and management of BPH and lower urinary tract symptoms is shown in Figure 3.17
MANAGEMENT STRATEGIES FOR BPH
While BPH is rarely life-threatening, it can significantly detract from a patient’s quality of life. The goal of treatment is not only to alleviate bothersome symptoms, but also to prevent disease progression and disease-related complications.
BPH tends to progress
Understanding the natural history of BPH is imperative to appropriately counsel patients on management options, which include watchful waiting, behavioral modification, pharmacologic therapy, and surgery.
In a randomized trial,18 men with moderately symptomatic BPH underwent either surgery or, in the control group, watchful waiting. At 5 years, the failure rate was 21% with watchful waiting vs 10% with surgery (P < .0004). (Failure was defined as a composite of death, repeated or intractable urinary retention, residual urine volume > 350 mL, development of bladder calculus, new persistent incontinence requiring use of a pad or other incontinence device, symptom score in the severe range [> 24 at 1 visit or score of 21 or higher at two consecutive visits, with 27 being the maximum score], or a doubling of baseline serum creatinine.) In the watchful-waiting group, 36% of the men crossed over to surgery. Men with more bothersome symptoms at enrollment were at higher risk of progressing to surgery.
In a longitudinal study of men with BPH and mild symptoms (IPSS < 8), the risk of progression to moderate or severe symptoms (IPPS ≥ 8) was 31% at 4 years.19
The Olmsted County Study of Urinary Symptoms and Health Status Among Men20 found that the peak urinary flow rate decreased by a mean of 2.1% per year, declining faster in older men who had a lower peak flow at baseline. In this cohort, the IPSS increased by a mean of 0.18 points per year, with a greater increase in older men.21
Though men managed with watchful waiting are at no higher risk of death or renal failure than men managed surgically,17 population-based studies have demonstrated an overall risk of acute urinary retention of 6.8/1,000 person-years with watchful waiting. Older men with a larger prostate, higher symptom score, and lower peak urinary flow rate are at higher risk of acute urinary retention and progression to needing BPH treatment.22,23
There is evidence that patients progressing to needing surgery after an initial period of watchful waiting have worse surgical outcomes than men managed surgically at the onset.18 This observation must be considered in counseling and selecting patients for watchful waiting. Ideal candidates include patients who have mild or moderate symptoms that cause little bother.10 Patients electing watchful waiting warrant annual follow-up including history, physical examination, and symptom assessment with the IPSS.
Behavioral modification
Behavioral modification should be incorporated into whichever management strategy a patient elects. Such modifications include:
- Reducing total or evening fluid intake for patients with urinary frequency or nocturia.
- Minimizing consumption of bladder irritants such as alcohol and caffeine, which exacerbate storage symptoms.
- Smoking cessation counseling.
- For patients with lower extremity edema who complain of nocturia, using compression stockings or elevating their legs in the afternoon to mobilize lower extremity edema and promote diuresis before going to sleep. If these measures fail, initiating or increasing the dose of a diuretic should be considered. Patients on diuretic therapy with nocturnal lower urinary tract symptoms should be instructed to take diuretics in the morning and early afternoon to avoid diuresis just before bed.
MEDICAL MANAGEMENT
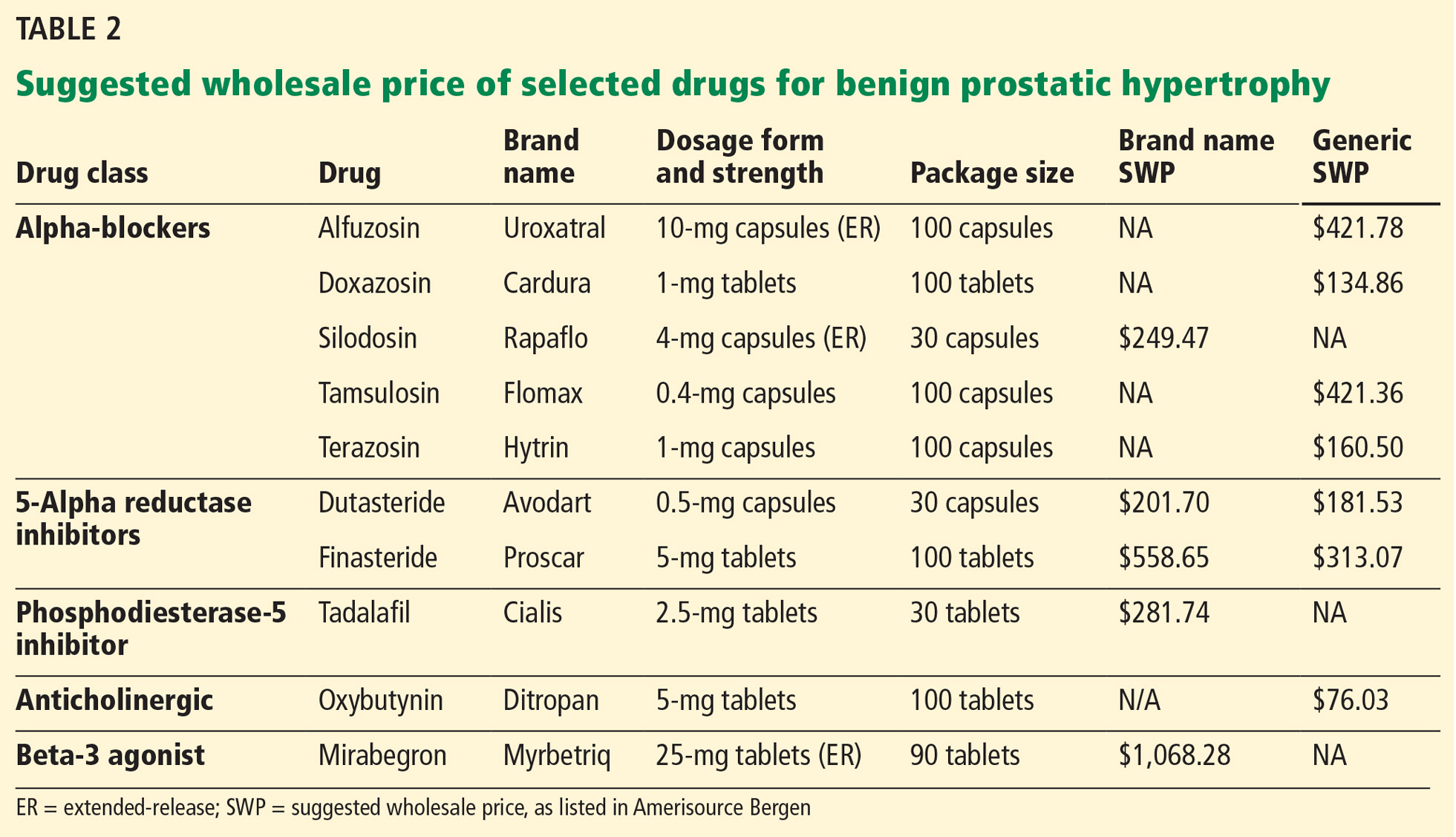
Drugs for BPH include alpha-adrenergic blockers, 5-alpha reductase inhibitors, anticholinergics, beta-3 agonists, and phosphodiesterase-5 inhibitors. Costs of selected agents in these classes are listed in Table 2.
Alpha-adrenergic receptor blockers
Alpha-adrenergic receptors are found throughout the body and modulate smooth muscle tone.24 The alpha-1a receptor is the predominant subtype found in the bladder neck and prostate25 (Figure 2) and is a target of therapy. By antagonizing the alpha-1a receptor, alpha-blockers relax the smooth muscle in the prostate and bladder neck, reduce bladder outlet resistance, and improve urinary flow.26
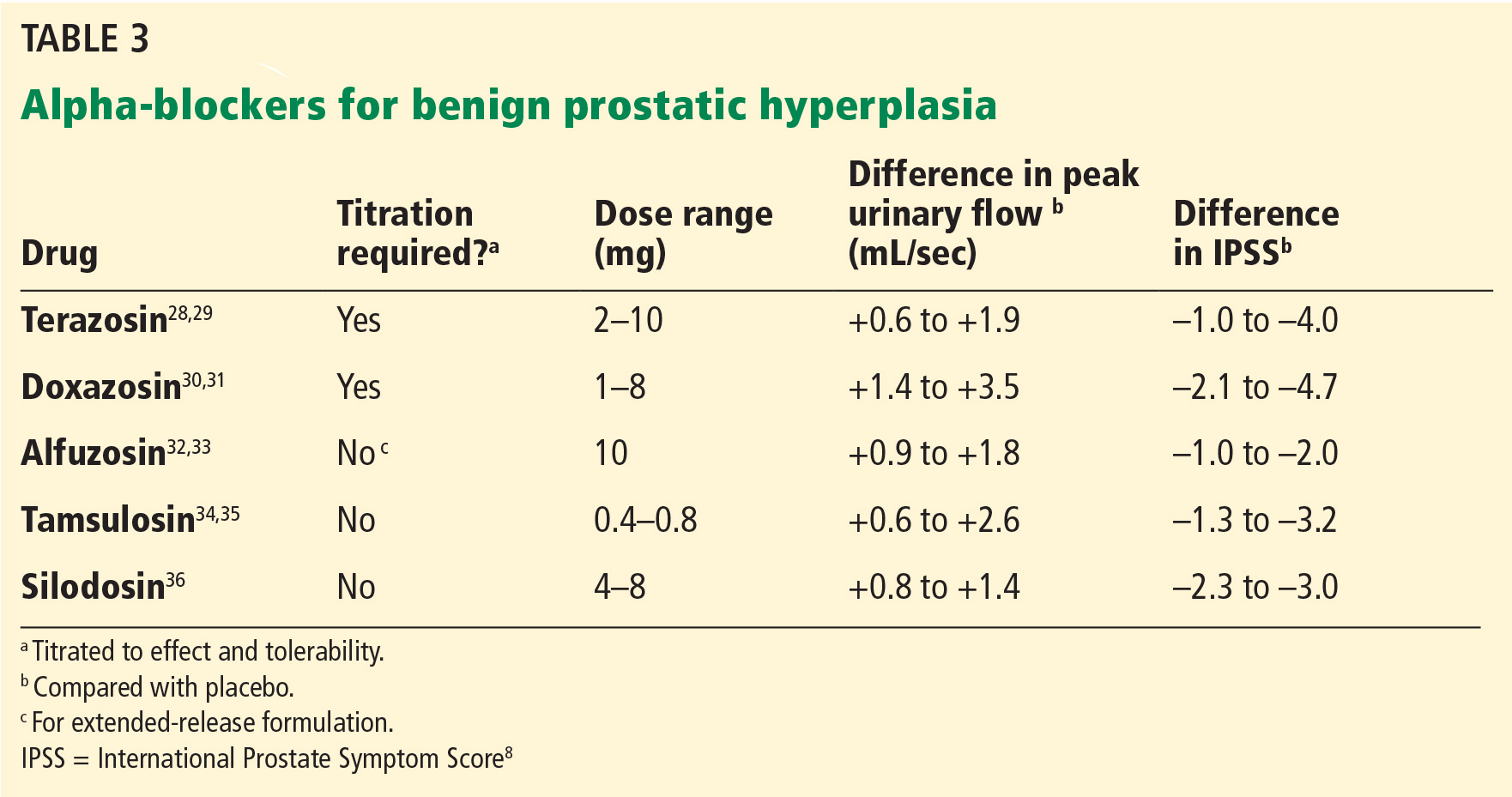
In clinical trials in BPH, alpha-blockers improved the symptom score by 30% to 45% and increased the peak urinary flow rate by 15% to 30% from baseline values.27 These agents have a rapid onset (within a few days) and result in significant symptom improvement. They are all about the same in efficacy (Table 3),28–36 with no strong evidence that any one of them is superior to another; thus, decisions about which agent to use must consider differences in receptor subtype specificity, adverse-effect profile, and tolerability.
In the Medical Therapy of Prostatic Symptoms (MTOPS) trial,37 men randomized to the alpha-blocker doxazosin had a 39% lower risk of BPH progression than with placebo, largely due to symptom score reduction. However, doxazosin failed to reduce the risk of progressing to acute urinary retention or surgical intervention. Though rapidly effective in reducing symptoms, alpha-blocker monotherapy may not be the best option in men at higher risk of BPH progression, as discussed below.
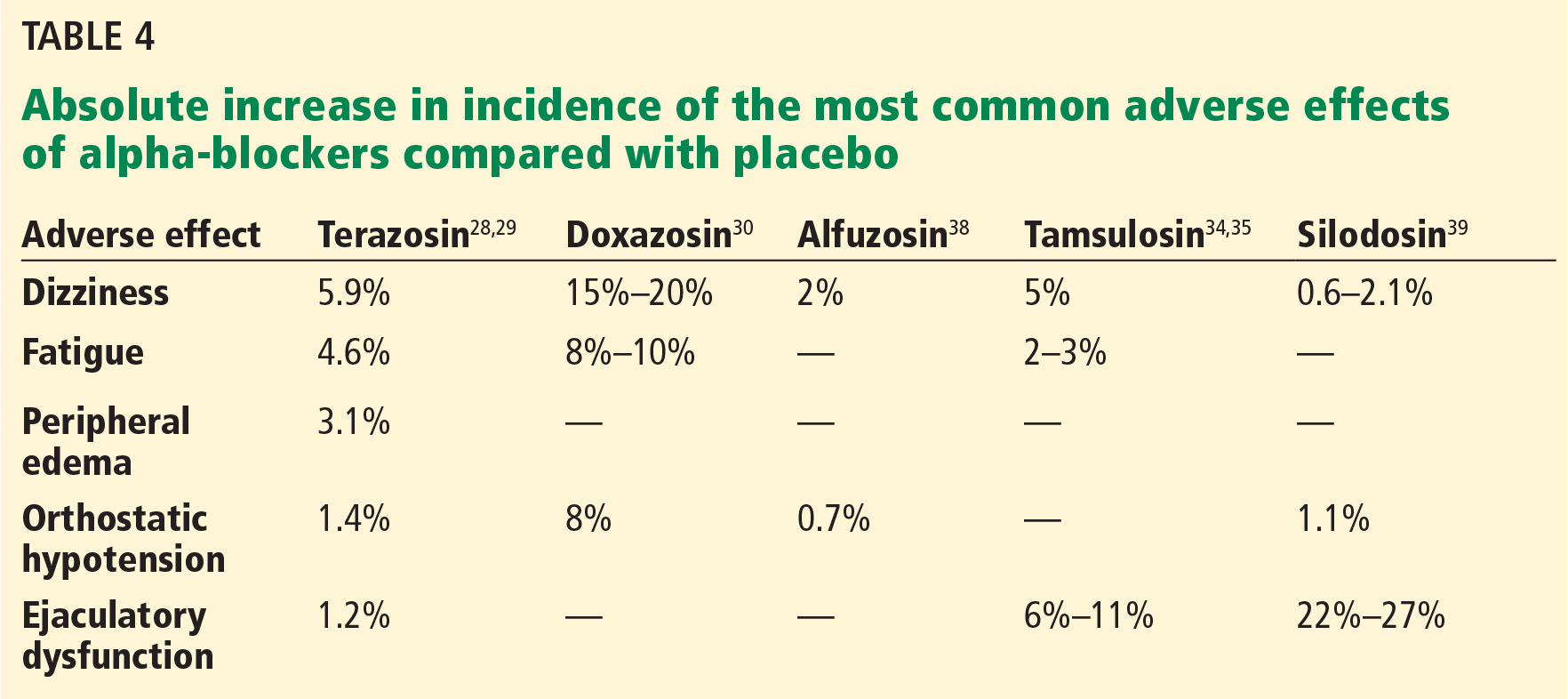
Before starting this therapy, patients must be counseled about common side effects such as dizziness, fatigue, peripheral edema, orthostatic hypotension, and ejaculatory dysfunction. The incidence of adverse effects varies among agents (Table 4).28–30,34,35,38,39
To maximize efficacy of alpha-blocker therapy, it is imperative to understand dosing variations among agents.
Alpha-blockers are classified as uroselective or non-uroselective based on alpha-1a receptor subtype specificity. The non-uroselective alpha-blockers doxazosin and terazosin need to be titrated because the higher the dose the greater the efficacy, but also the greater the blood pressure-lowering effect and other side effects.25 Though non-uroselective, alfuzosin does not affect blood pressure and does not require dose titration. Similarly, the uroselective alpha-blockers tamsulosin and silodosin can be initiated at a therapeutic dose.
Terazosin, a non-uroselective agent, can lower blood pressure and often causes dizziness. It should be started at 2 mg and titrated to side effects, efficacy, or maximum therapeutic dose (10 mg daily).28
Doxazosin has a high, dose-related incidence of dizziness (up to 20%) and must be titrated, starting at 1 mg to a maximum 8 mg.30
Alfuzosin, tamsulosin, and silodosin do not require titration and can be initiated at the therapeutic doses listed in Table 3. Of note, obese patients often require 0.8 mg tamsulosin for maximum efficacy due to a higher volume of distribution.
Before initiating an alpha-blocker, a physician must determine whether a patient plans to undergo cataract surgery, as the use of alpha-blockers is associated with intraoperative floppy iris syndrome. This condition is marked by poor intraoperative pupil dilation, increasing the risk of surgical complications.40 It is unclear whether discontinuing alpha-blockers before cataract surgery reduces the risk of intraoperative floppy iris syndrome. As such, alpha-blocker therapy should be delayed in patients planning to undergo cataract surgery.
5-Alpha reductase inhibitors
Prostate growth is androgen-dependent and mediated predominantly by dihydrotestosterone, which is generated from testosterone by the action of 5-alpha reductase. There are two 5-alpha reductase isoenzymes: type 1, expressed in the liver and skin, and type 2, expressed primarily in the prostate.
There are also two 5-alpha reductase inhibitors: dutasteride and finasteride. Dutasteride inhibits both isoenzymes, while finasteride is selective for type 2. By inhibiting both isoenzymes, dutasteride reduces the serum dihydrotestosterone concentration more than finasteride does (by 95% vs 70%), and also reduces the intraprostatic dihydrotestosterone concentration more (by 94% vs 80%).41–43 Both agents induce apoptosis of prostatic stroma, with a resultant 20% to 25% mean reduction in prostate volume.41,42
Finasteride and dutasteride are believed to mitigate the static obstructive component of BPH, with similar improvements in urinary flow rate (1.6–2.2 mL/sec) and symptom score (–2.7 to – 4.5 points) in men with an enlarged prostate.41,42 Indeed, data from the MTOPS trial showed that men with a prostate volume of 30 grams or greater or a PSA level of 1.5 ng/mL or greater are most likely to benefit from 5-alpha reductase inhibitors.37 Maximum symptomatic improvement is seen after 3 to 6 months of 5-alpha reductase inhibitor therapy.
In addition to improving urinary flow and lower urinary tract symptoms, finasteride has been shown to reduce the risk of disease progression in men with prostates greater than 30 grams.44 Compared with placebo, these drugs significantly reduce the risk of developing acute urinary retention or requiring BPH-related surgery, a benefit not seen with alpha-blockers.37 To estimate prostate volume, most practitioners rely on digital rectal examination. Though less precise than transrectal ultrasonography, digital rectal examination can identify men with significant prostatic enlargement likely to benefit from this therapy.
Before starting 5-alpha reductase inhibitor therapy, patients should be counseled about common adverse effects such as erectile dysfunction (occurring in 5%–8%), decreased libido (5%), ejaculatory dysfunction (1%–5%), and gynecomastia (1%).
Combination therapy
The MTOPS trial37 randomized patients to receive doxazosin, finasteride, both, or placebo. The combination of doxazosin (an alpha-blocker) and finasteride (a 5-alpha reductase inhibitor) reduced the risk of disease progression to a greater extent than doxazosin or finasteride alone. It also reduced the IPSS more and increased the peak urinary flow rate more. Similar results have been seen with the combination of dutasteride and tamsulosin.45
Given its superior efficacy and benefits in preventing disease progression, combination therapy should be considered for men with an enlarged prostate and moderate to severe lower urinary tract symptoms.
Anticholinergic agents
Anticholinergic agents block muscarinic receptors within the detrusor muscle, resulting in relaxation. They are used in the treatment of overactive bladder for symptoms of urinary urgency, frequency, and urge incontinence.
Anticholinergics were historically contraindicated in men with BPH because of concern about urinary retention. However, in men with a postvoid residual volume less than 200 mL, anticholinergics do not increase the risk of urinary retention.46 Further, greater symptom improvement has been demonstrated with the addition of anticholinergics to alpha-blocker therapy for men with BPH, irritative lower urinary tract symptoms, and a low postvoid residual volume.47
Beta-3 agonists
Anticholinergic side effects often limit the use of anticholinergic agents. An alternative in such instances is the beta-3 agonist mirabegron. By activating beta-3 adrenergic receptors in the bladder wall, mirabegron promotes detrusor relaxation and inhibits detrusor overactivity.48 Mirabegron does not have anticholinergic side effects and is generally well tolerated, though poorly controlled hypertension is a contraindication to its use.
Phosphodiesterase-5 inhibitors
Phosphodiesterase-5 (PDE5) inhibitors are a mainstay in the treatment of erectile dysfunction. These agents act within penile corporal smooth muscle cells and antagonize PDE5, resulting in cyclic guanosine monophosphate accumulation and smooth muscle relaxation. PDE5 is also found within the prostate and its inhibition is believed to reduce prostatic smooth muscle tone. Randomized studies have demonstrated significant improvement in lower urinary tract symptoms with PDE5 inhibitors, with an average 2-point IPSS improvement on a PDE5 inhibitor compared with placebo.49
Tadalafil is the only drug of this class approved by the FDA for the treatment of lower urinary tract symptoms, though other agents have demonstrated similar efficacy.
Dual therapy with a PDE5 inhibitor and an alpha-blocker has greater efficacy than either monotherapy alone; however, caution must be exercised as these agents are titrated to avoid symptomatic hypotension. Lower urinary tract symptoms and sexual dysfunction often coexist; PDE5 inhibitors are appropriate in the management of such cases.
SURGERY FOR BPH
Even with effective medical therapy, the disease will progress in some men. In the MTOPS trial,37 the 4-year incidence of disease progression was 10% for men on alpha-blocker or 5-alpha reductase inhibitor monotherapy and 5% for men on combination therapy; from 1% to 3% of those in the various treatment groups needed surgery. With this in mind, patients whose symptoms do not improve with medical therapy, whose symptoms progress, or who simply are interested in surgery should be referred for urologic evaluation.
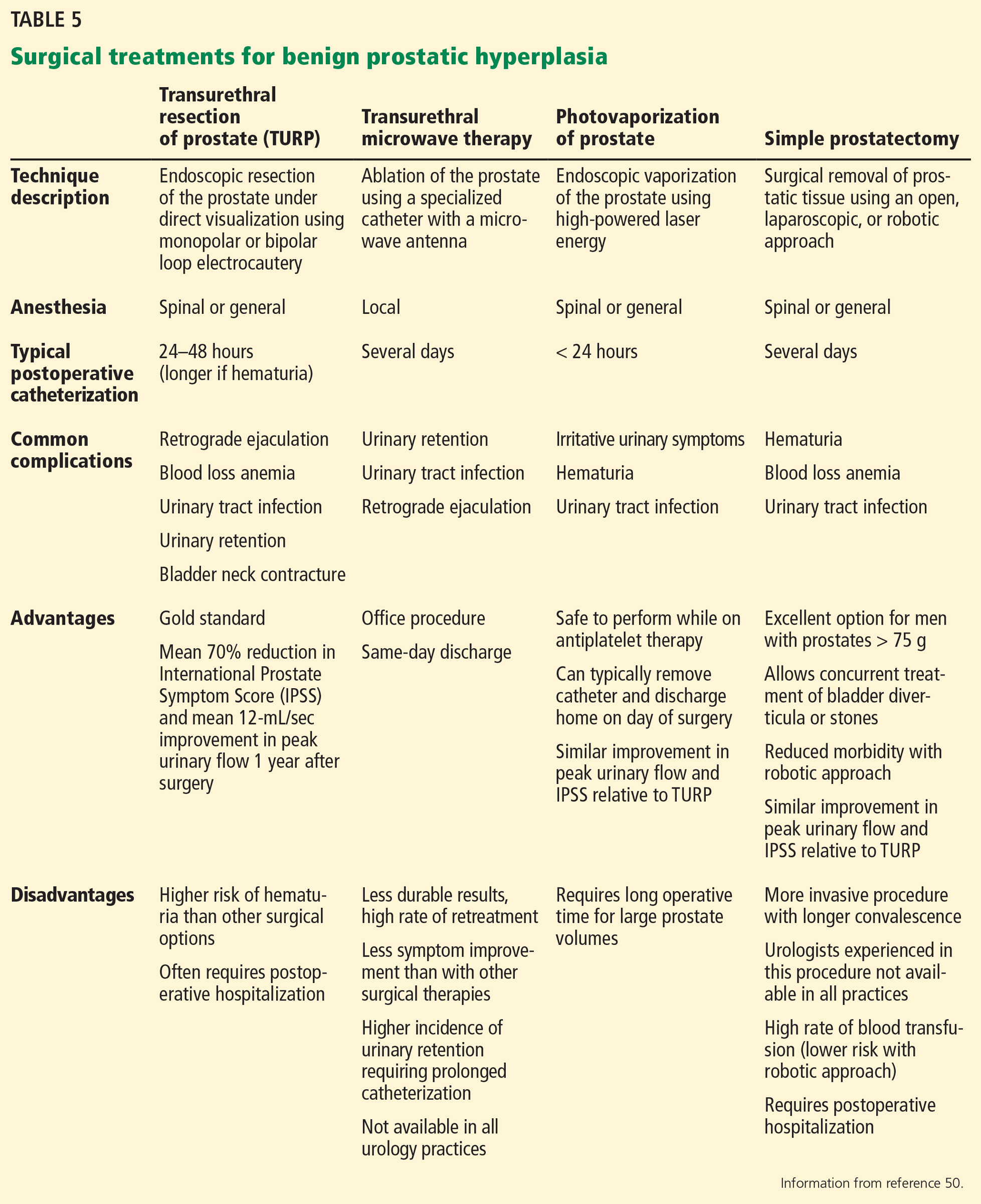
A number of effective surgical therapies are available for men with BPH (Table 5), providing excellent 1-year outcomes including a mean 70% reduction in IPSS and a mean 12 mL/sec improvement in peak urinary flow.50 Given the efficacy of surgical therapy, men who do not improve with medical therapy who demonstrate any of the findings outlined in Table 1 warrant urologic evaluation.
Acknowledgments: We would like to thank Mary Ellen Amos, PharmD, and Kara Sink, BS, RPh, for their assistance in obtaining the suggested wholesale pricing information included in Table 2.
- Wei JT, Miner MM, Steers WD, et al; BPH Registry Steering Committee. Benign prostatic hyperplasia evaluation and management by urologists and primary care physicians: practice patterns from the observational BPH registry. J Urol 2011; 186:971–976.
- McNeal J. Pathology of benign prostatic hyperplasia. insight into etiology. Urol Clin North Am 1990; 17:477–486.
- Roehrborn CG, Schwinn DA. Alpha1-adrenergic receptors and their inhibitors in lower urinary tract symptoms and benign prostatic hyperplasia. J Urol 2004; 171:1029–1035.
- Berry SJ, Coffey DS, Walsh PC, Ewing LL. The development of human benign prostatic hyperplasia with age. J Urol 1984; 132:474–479.
- Platz EA, Smit E, Curhan GC, Nyberg LM, Giovannucci E. Prevalence of and racial/ethnic variation in lower urinary tract symptoms and noncancer prostate surgery in US men. Urology 2002; 59:877–883.
- Wei JT, Calhoun E, Jacobsen SJ. Urologic diseases in America project: benign prostatic hyperplasia. J Urol 2008; 179(suppl):S75–S80.
- Scheen AJ, Paquot N. Metabolic effects of SGLT-2 inhibitors beyond increased glucosuria: a review of the clinical evidence. Diabetes Metab 2014; 40(suppl 1):S4–S11.
- Barry MJ, Fowler FJ Jr, O’Leary MP, et al. The American Urological Association symptom index for benign prostatic hyperplasia. The Measurement Committee of the American Urological Association. J Urol 1992; 148:1549–1564.
- Barry MJ, Williford WO, Chang Y, et al. Benign prostatic hyperplasia specific health status measures in clinical research: how much change in the American Urological Association symptom index and the benign prostatic hyperplasia impact index is perceptible to patients? J Urol 1995; 154:1770–1774.
- McVary KT, Roehrborn CG, Avins AL, et al. Update on AUA guideline on the management of benign prostatic hyperplasia. J Urol 2011; 185:1793–1803.
- Roehrborn CG, McConnell J, Bonilla J, et al. Serum prostate specific antigen is a strong predictor of future prostate growth in men with benign prostatic hyperplasia. PROSCAR long-term efficacy and safety study. J Urol 2000; 163:13–20.
- Roehrborn CG, McConnell JD, Lieber M, et al. Serum prostate-specific antigen concentration is a powerful predictor of acute urinary retention and need for surgery in men with clinical benign prostatic hyperplasia. PLESS Study Group. Urology 1999; 53:473–480.
- Wasson JH, Reda DJ, Bruskewitz RC, Elinson J, Keller AM, Henderson WG. A comparison of transurethral surgery with watchful waiting for moderate symptoms of benign prostatic hyperplasia. The Veterans Affairs Cooperative Study Group on Transurethral Resection of the Prostate. N Engl J Med 1995; 332:75–79.
- Jensen KM, Bruskewitz RC, Iversen P, Madsen PO. Spontaneous uroflowmetry in prostatism. Urology 1984; 24:403–409.
- Abrams PH, Griffiths DJ. The assessment of prostatic obstruction from urodynamic measurements and from residual urine. Br J Urol 1979; 51:129–134.
- Lim CS, Abrams P. The Abrams-Griffiths nomogram. World J Urol 1995; 13:34–39.
- Abrams P, Chapple C, Khoury S, Roehrborn C, de la Rosette J; International Consultation on New Developments in Prostate Cancer and Prostate Diseases. Evaluation and treatment of lower urinary tract symptoms in older men. J Urol 2013; 189(suppl 1):S93–S101.
- Flanigan RC, Reda DJ, Wasson JH, Anderson RJ, Abdellatif M, Bruskewitz RC. 5-year outcome of surgical resection and watchful waiting for men with moderately symptomatic benign prostatic hyperplasia: a Department of Veterans Affairs cooperative study. J Urol 1998; 160:12–17.
- Djavan B, Fong YK, Harik M, et al. Longitudinal study of men with mild symptoms of bladder outlet obstruction treated with watchful waiting for four years. Urology 2004; 64:1144–1148.
- Roberts RO, Jacobsen SJ, Jacobson DJ, Rhodes T, Girman CJ, Lieber MM. Longitudinal changes in peak urinary flow rates in a community based cohort. J Urol 2000; 163:107–113.
- Jacobsen SJ, Girman CJ, Guess HA, Rhodes T, Oesterling JE, Lieber MM. Natural history of prostatism: longitudinal changes in voiding symptoms in community dwelling men. J Urol 1996; 155:595–600.
- Jacobsen SJ, Jacobson DJ, Girman CJ, et al. Treatment for benign prostatic hyperplasia among community dwelling men: the Olmsted County study of urinary symptoms and health status. J Urol 1999; 162:1301–1306.
- Jacobsen SJ, Jacobson DJ, Girman CJ, et al. Natural history of prostatism: risk factors for acute urinary retention. J Urol 1997; 158:481–487.
- Kobayashi S, Tang R, Shapiro E, Lepor H. Characterization and localization of prostatic alpha 1 adrenoceptors using radioligand receptor binding on slide-mounted tissue section. J Urol 1993; 150:2002–2006.
- Kirby RS, Pool JL. Alpha adrenoceptor blockade in the treatment of benign prostatic hyperplasia: past, present and future. Br J Urol 1997; 80:521–532.
- Kirby RS, Pool JL. Alpha adrenoceptor blockade in the treatment of benign prostatic hyperplasia: past, present and future. Br J Urol 1997; 80:521–532.
- Milani S, Djavan B. Lower urinary tract symptoms suggestive of benign prostatic hyperplasia: latest update on alpha-adrenoceptor antagonists. BJU Int 2005; 95(suppl 4):29–36.
- Lepor H, Auerbach S, Puras-Baez A, et al. A randomized, placebo-controlled multicenter study of the efficacy and safety of terazosin in the treatment of benign prostatic hyperplasia. J Urol 1992; 148:1467–1474.
- Roehrborn CG, Oesterling JE, Auerbach S, et al. The Hytrin Community Assessment Trial study: a one-year study of terazosin versus placebo in the treatment of men with symptomatic benign prostatic hyperplasia. HYCAT Investigator Group. Urology 1996; 47:159–168.
- Gillenwater JY, Conn RL, Chrysant SG, et al. Doxazosin for the treatment of benign prostatic hyperplasia in patients with mild to moderate essential hypertension: a double-blind, placebo-controlled, dose-response multicenter study. J Urol 1995; 154:110–115.
- Chapple CR, Carter P, Christmas TJ, et al. A three month double-blind study of doxazosin as treatment for benign prostatic bladder outlet obstruction. Br J Urol 1994; 74:50–56.
- Buzelin JM, Roth S, Geffriaud-Ricouard C, Delauche-Cavallier MC. Efficacy and safety of sustained-release alfuzosin 5 mg in patients with benign prostatic hyperplasia. ALGEBI Study Group. Eur Urol 1997; 31:190–198.
- van Kerrebroeck P, Jardin A, Laval KU, van Cangh P. Efficacy and safety of a new prolonged release formulation of alfuzosin 10 mg once daily versus alfuzosin 2.5 mg thrice daily and placebo in patients with symptomatic benign prostatic hyperplasia. ALFORTI Study Group. Eur Urol 2000; 37:306–313.
- Narayan P, Tewari A. A second phase III multicenter placebo controlled study of 2 dosages of modified release tamsulosin in patients with symptoms of benign prostatic hyperplasia. United States 93-01 Study Group. J Urol 1998; 160:1701–1706.
- Lepor H. Phase III multicenter placebo-controlled study of tamsulosin in benign prostatic hyperplasia. Tamsulosin Investigator Group. Urology 1998; 51:892–900.
- Ding H, Du W, Hou ZZ, Wang HZ, Wang ZP. Silodosin is effective for treatment of LUTS in men with BPH: a systematic review. Asian J Androl 2013; 15:121–128.
- McConnell JD, Roehrborn CG, Bautista OM, et al; Medical Therapy of Prostatic Symptoms (MTOPS) Research Group. The long-term effect of doxazosin, finasteride, and combination therapy on the clinical progression of benign prostatic hyperplasia. N Engl J Med 2003; 349:2387–2398.
- Jardin A, Bensadoun H, Delauche-Cavallier MC, Attali P. Alfuzosin for treatment of benign prostatic hypertrophy. The BPH-ALF Group. Lancet 1991; 337:1457–1461.
- Marks LS, Gittelman MC, Hill LA, Volinn W, Hoel G. Rapid efficacy of the highly selective alpha1A-adrenoceptor antagonist silodosin in men with signs and symptoms of benign prostatic hyperplasia: pooled results of 2 phase 3 studies. J Urol 2009; 181:2634–2640.
- Chang DF, Campbell JR. Intraoperative floppy iris syndrome associated with tamsulosin. J Cataract Refract Surg 2005; 31:664–673.
- Gormley GJ, Stoner E, Bruskewitz RC, et al. The effect of finasteride in men with benign prostatic hyperplasia. The Finasteride Study Group. N Engl J Med 1992; 327:1185–1191.
- Roehrborn CG, Boyle P, Nickel JC, Hoefner K, Andriole G; ARIA3001 ARIA3002 and ARIA3003 Study Investigators. Efficacy and safety of a dual inhibitor of 5-alpha-reductase types 1 and 2 (dutasteride) in men with benign prostatic hyperplasia. Urology 2002; 60:434–441.
- Clark RV, Hermann DJ, Cunningham GR, Wilson TH, Morrill BB, Hobbs S. Marked suppression of dihydrotestosterone in men with benign prostatic hyperplasia by dutasteride, a dual 5alpha-reductase inhibitor. J Clin Endocrinol Metab 2004; 89:2179–2184.
- Kaplan SA, Lee JY, Meehan AG, Kusek JW; MTOPS Research Group. Long-term treatment with finasteride improves clinical progression of benign prostatic hyperplasia in men with an enlarged versus a smaller prostate: data from the MTOPS trial. J Urol 2011; 185:1369–1373.
- Roehrborn CG, Siami P, Barkin J, et al; CombAT Study Group. The effects of combination therapy with dutasteride and tamsulosin on clinical outcomes in men with symptomatic benign prostatic hyperplasia: 4-year results from the CombAT study. Eur Urol 2010; 57:123–131.
- Abrams P, Kaplan S, De Koning Gans HJ, Millard R. Safety and tolerability of tolterodine for the treatment of overactive bladder in men with bladder outlet obstruction. J Urol 2006; 175:999–1004.
- Kaplan SA, Roehrborn CG, Rovner ES, Carlsson M, Bavendam T, Guan Z. Tolterodine and tamsulosin for treatment of men with lower urinary tract symptoms and overactive bladder: a randomized controlled trial. JAMA 2006; 296:2319–2328.
- Suarez O, Osborn D, Kaufman M, Reynolds WS, Dmochowski R. Mirabegron for male lower urinary tract symptoms. Curr Urol Rep 2013; 14:580–584.
- Oelke M, Giuliano F, Mirone V, Xu L, Cox D, Viktrup L. Monotherapy with tadalafil or tamsulosin similarly improved lower urinary tract symptoms suggestive of benign prostatic hyperplasia in an international, randomised, parallel, placebo-controlled clinical trial. Eur Urol 2012; 61:917–925.
- Welliver C, McVary KT. Minimally invasive and endoscopic management of benign prostatic hyperplasia. In: Wein AJ, Kavoussi LR, Partin AW, Peters CA, eds. Campbell-Walsh Urology. 11th ed. Philadelphia, PA: Elsevier; 2016:2504–2534.
Primary care physicians are uniquely positioned to screen for benign prostatic hyperplasia (BPH) and lower urinary tract symptoms, to perform the initial diagnostic workup, and to start medical therapy in uncomplicated cases. Effective medical therapy is available but underutilized in the primary care setting.1
This overview covers how to identify and evaluate patients with lower urinary tract symptoms, initiate therapy, and identify factors warranting timely urology referral.
TWO MECHANISMS: STATIC, DYNAMIC
BPH is a histologic diagnosis of proliferation of smooth muscle, epithelium, and stromal cells within the transition zone of the prostate,2 which surrounds the proximal urethra.
Symptoms arise through two mechanisms: static, in which the hyperplastic prostatic tissue compresses the urethra (Figure 1); and dynamic, with increased adrenergic nervous system and prostatic smooth muscle tone (Figure 2).3 Both mechanisms increase resistance to urinary flow at the level of the bladder outlet.
As an adaptive change to overcome outlet resistance and maintain urinary flow, the detrusor muscles undergo hypertrophy. However, over time the bladder may develop diminished compliance and increased detrusor activity, causing symptoms such as urinary frequency and urgency. Chronic bladder outlet obstruction can lead to bladder decompensation and detrusor underactivity, manifesting as incomplete emptying, urinary hesitancy, intermittency (starting and stopping while voiding), a weakened urinary stream, and urinary retention.
MOST MEN EVENTUALLY DEVELOP BPH
Autopsy studies have shown that BPH increases in prevalence with age beginning around age 30 and reaching a peak prevalence of 88% in men in their 80s.4 This trend parallels those of the incidence and severity of lower urinary tract symptoms.5
In the year 2000 alone, BPH was responsible for 4.5 million physician visits at an estimated direct cost of $1.1 billion, not including the cost of pharmacotherapy.6
OFFICE WORKUP
BPH can cause lower urinary tract symptoms that fall into two categories: storage and emptying. Storage symptoms include urinary frequency, urgency, and nocturia, whereas emptying symptoms include weak stream, hesitancy, intermittency, incomplete emptying, straining, and postvoid dribbling.
History and differential diagnosis
Assessment begins with characterizing the patient’s symptoms and determining those that are most bothersome. Because BPH is just one of many possible causes of lower urinary tract symptoms, a detailed medical history is necessary to evaluate for other conditions that may cause lower urinary tract dysfunction or complicate its treatment.
Obstructive urinary symptoms can arise from BPH or from other conditions, including urethral stricture disease and neurogenic voiding dysfunction.
Irritative voiding symptoms such as urinary urgency and frequency can result from detrusor overactivity secondary to BPH, but can also be caused by neurologic disease, malignancy, initiation of diuretic therapy, high fluid intake, or consumption of bladder irritants such as caffeine, alcohol, and spicy foods.
Urinary frequency is sometimes a presenting symptom of undiagnosed or poorly controlled diabetes mellitus resulting from glucosuria and polyuria. Iatrogenic causes of polyuria include the new hypoglycemic agents canagliflozin and dapagliflozin, which block renal glucose reabsorption, improving glycemic control by inducing urinary
glucose loss.7
Nocturia has many possible nonurologic causes including heart failure (in which excess extravascular fluid shifts to the intravascular space when the patient lies down, resulting in polyuria), obstructive sleep apnea, and behavioral factors such as high evening fluid intake. In these cases, patients usually have nocturnal polyuria (greater than one-third of 24-hour urine output at night) rather than only nocturia (waking at night to void). A fluid diary is a simple tool that can differentiate these two conditions.
Hematuria can develop in patients with BPH with bleeding from congested prostatic or bladder neck vessels; however, hematuria may indicate an underlying malignancy or urolithiasis, for which a urologic workup is indicated.
The broad differential diagnosis for the different lower urinary tract symptoms highlights the importance of obtaining a thorough history.
Physical examination
A general examination should include the following:
Body mass index. Obese patients are at risk of obstructive sleep apnea, which can cause nocturnal polyuria.
Gait. Abnormal gait may suggest a neurologic condition such as Parkinson disease or stroke that can also affect lower urinary tract function.
Lower abdomen. A palpable bladder suggests urinary retention.
External genitalia. Penile causes of urinary obstruction include urethral meatal stenosis or a palpable urethral mass.
Digital rectal examination can reveal benign prostatic enlargement or nodules or firmness, which suggest malignancy and warrant urologic referral.
Neurologic examination, including evaluation of anal sphincter tone and lower extremity sensorimotor function.
Feet. Bilateral lower-extremity edema may be due to heart failure or venous insufficiency.
The International Prostate Symptom Score
All men with lower urinary tract symptoms should complete the International Prostate Symptom Score (IPSS) survey, consisting of seven questions about urinary symptoms plus one about quality of life.8 Specifically, it asks the patient, “Over the past month, how often have you…”
- Had a sensation of not emptying your bladder completely after you finish urinating?
- Had to urinate again less than 2 hours after you finished urinating?
- Found you stopped and started again several times when you urinated?
- Found it difficult to postpone urination?
- Had a weak urinary stream?
- Had to push or strain to begin urination?
Each question above is scored as 0 (not at all), 1 (less than 1 time in 5), 2 (less than half the time), 3 (about half the time), 4 (more than half the time, or 5 (almost always).
- Over the past month, how many times did you most typically get up to urinate from the time you went to bed until the time you got up in the morning?
This question is scored from 0 (none) to 5 (5 times or more).
- If you were to spend the rest of your life with your urinary condition the way it is now, how would you feel about that?
This question is scored as 0 (delighted), 1 (pleased), 2 (mostly satisfied), 3 (mixed: equally satisfied and dissatisfied), 4 (mostly dissatisfied), 5 (unhappy), or 6 (terrible).
A total score of 1 to 7 is categorized as mild, 8 to 19 moderate, and 20 to 35 severe.
The questionnaire can also be used to evaluate for disease progression and response to treatment over time. A change of 3 points is clinically significant, as patients are unable to discern a difference below this threshold.9
Urinalysis
Urinalysis is recommended to assess for urinary tract infection, hematuria, proteinuria, or glucosuria.
Fluid diary
A fluid diary is useful for patients complaining of frequency or nocturia and can help quantify the volume of fluid intake, frequency of urination, and volumes voided. The patient should complete the diary over a 24-hour period, recording the time and volume of fluid intake and each void. This aids in diagnosing polyuria (> 3 L of urine output per 24 hours), nocturnal polyuria, and behavioral causes of symptoms, including excessive total fluid intake or high evening fluid intake contributing to nocturia.
Serum creatinine not recommended
Measuring serum creatinine is not recommended in the initial BPH workup, as men with lower urinary tract symptoms are not at higher risk of renal failure than those without these symptoms.10
Prostate-specific antigen
Prostate-specific antigen (PSA) is a glycoprotein primarily produced by prostatic luminal epithelial cells. It is most commonly discussed in the setting of prostate cancer screening, but its utility extends to guiding the management of BPH.
PSA levels correlate with prostate volume and subsequent growth.11 In addition, the risks of developing acute urinary retention or needing surgical intervention rise with increasing PSA.12 Among men in the Proscar Long-Term Efficacy and Safety Study, the risk of acute urinary retention or BPH-related surgery after 4 years in the watchful-waiting arm was 7.8% in men with a PSA of 1.3 ng/dL or less, compared with 19.9% in men with a PSA greater than 3.2 ng/dL.11 Therefore, men with BPH and an elevated PSA are at higher risk with watchful waiting and may be better served with medical therapy.
In addition, American Urological Association guidelines recommend measuring serum PSA levels in men with a life expectancy greater than 10 years in whom the diagnosis of prostate cancer would alter management.10
Urologic referral
If the initial evaluation reveals hematuria, recurrent urinary tract infection, a palpable bladder, abnormal findings on digital rectal examination suggesting prostate cancer, or a history of or risk factors for urethral stricture or neurologic disease, the patient should be referred to a urologist for further evaluation (Table 1).10 Other patients who should undergo urologic evaluation are those with persistent bothersome symptoms after basic management and those who desire referral.
Adjunctive tests
Patients referred for urologic evaluation may require additional tests for diagnosis and to guide management.
Postvoid residual volume is easily measured with either abdominal ultrasonography or catheterization and is often included in the urologic evaluation of BPH. Patients vary considerably in their residual volume, which correlates poorly with BPH, symptom severity, or surgical success. However, those with a residual volume of more than 100 mL have a slightly higher rate of failure with watchful waiting.13 Postvoid residual volume is not routinely monitored in patients with a low residual volume unless there is a significant change in urinary symptoms. Conversely, patients with a volume greater than 200 mL should be monitored closely for worsening urinary retention, especially if considering anticholinergic therapy.
There is no absolute threshold postvoid residual volume above which therapy is mandatory. Rather, the decision to intervene is based on symptom severity and whether sequelae of urinary retention (eg, incontinence, urinary tract infection, hematuria, hydronephrosis, renal dysfunction) are present.
Uroflowmetry is a noninvasive test measuring the urinary flow rate during voiding and is recommended during specialist evaluation of men with lower urinary tract symptoms and suspected BPH.10 Though a diminished urinary flow rate may be detected in men with bladder outlet obstruction from BPH, it cannot differentiate obstruction from detrusor underactivity, both of which may result in reduced urinary flow. Urodynamic studies can help differentiate between these two mechanisms of lower urinary tract symptoms. Uroflowmetry may be useful in selecting surgical candidates, as patients with a maximum urinary flow rate of 15 mL/second or greater have been shown to have lower rates of surgical success.14
Urodynamic studies. If the diagnosis of bladder outlet obstruction remains in doubt, urodynamic studies can differentiate obstruction from detrusor underactivity. Urodynamic studies allow simultaneous measurement of urinary flow and detrusor pressure, differentiating between obstruction (manifesting as diminished urinary flow with normal or elevated detrusor pressure) and detrusor underactivity (diminished urinary flow with diminished detrusor pressure). Nomograms15 and the easily calculated bladder outlet obstruction index16 are simple tools used to differentiate these two causes of diminished urinary flow.
Cystourethroscopy is not recommended for routine evaluation of BPH. Indications for cystourethroscopy include hematuria and the presence of a risk factor for urethral stricture disease such as urethritis, prior urethral instrumentation, or perineal trauma. Cystourethroscopy can also aid in surgical planning when intervention is considered.
An algorithm for diagnostic workup and management of BPH and lower urinary tract symptoms is shown in Figure 3.17
MANAGEMENT STRATEGIES FOR BPH
While BPH is rarely life-threatening, it can significantly detract from a patient’s quality of life. The goal of treatment is not only to alleviate bothersome symptoms, but also to prevent disease progression and disease-related complications.
BPH tends to progress
Understanding the natural history of BPH is imperative to appropriately counsel patients on management options, which include watchful waiting, behavioral modification, pharmacologic therapy, and surgery.
In a randomized trial,18 men with moderately symptomatic BPH underwent either surgery or, in the control group, watchful waiting. At 5 years, the failure rate was 21% with watchful waiting vs 10% with surgery (P < .0004). (Failure was defined as a composite of death, repeated or intractable urinary retention, residual urine volume > 350 mL, development of bladder calculus, new persistent incontinence requiring use of a pad or other incontinence device, symptom score in the severe range [> 24 at 1 visit or score of 21 or higher at two consecutive visits, with 27 being the maximum score], or a doubling of baseline serum creatinine.) In the watchful-waiting group, 36% of the men crossed over to surgery. Men with more bothersome symptoms at enrollment were at higher risk of progressing to surgery.
In a longitudinal study of men with BPH and mild symptoms (IPSS < 8), the risk of progression to moderate or severe symptoms (IPPS ≥ 8) was 31% at 4 years.19
The Olmsted County Study of Urinary Symptoms and Health Status Among Men20 found that the peak urinary flow rate decreased by a mean of 2.1% per year, declining faster in older men who had a lower peak flow at baseline. In this cohort, the IPSS increased by a mean of 0.18 points per year, with a greater increase in older men.21
Though men managed with watchful waiting are at no higher risk of death or renal failure than men managed surgically,17 population-based studies have demonstrated an overall risk of acute urinary retention of 6.8/1,000 person-years with watchful waiting. Older men with a larger prostate, higher symptom score, and lower peak urinary flow rate are at higher risk of acute urinary retention and progression to needing BPH treatment.22,23
There is evidence that patients progressing to needing surgery after an initial period of watchful waiting have worse surgical outcomes than men managed surgically at the onset.18 This observation must be considered in counseling and selecting patients for watchful waiting. Ideal candidates include patients who have mild or moderate symptoms that cause little bother.10 Patients electing watchful waiting warrant annual follow-up including history, physical examination, and symptom assessment with the IPSS.
Behavioral modification
Behavioral modification should be incorporated into whichever management strategy a patient elects. Such modifications include:
- Reducing total or evening fluid intake for patients with urinary frequency or nocturia.
- Minimizing consumption of bladder irritants such as alcohol and caffeine, which exacerbate storage symptoms.
- Smoking cessation counseling.
- For patients with lower extremity edema who complain of nocturia, using compression stockings or elevating their legs in the afternoon to mobilize lower extremity edema and promote diuresis before going to sleep. If these measures fail, initiating or increasing the dose of a diuretic should be considered. Patients on diuretic therapy with nocturnal lower urinary tract symptoms should be instructed to take diuretics in the morning and early afternoon to avoid diuresis just before bed.
MEDICAL MANAGEMENT
Drugs for BPH include alpha-adrenergic blockers, 5-alpha reductase inhibitors, anticholinergics, beta-3 agonists, and phosphodiesterase-5 inhibitors. Costs of selected agents in these classes are listed in Table 2.
Alpha-adrenergic receptor blockers
Alpha-adrenergic receptors are found throughout the body and modulate smooth muscle tone.24 The alpha-1a receptor is the predominant subtype found in the bladder neck and prostate25 (Figure 2) and is a target of therapy. By antagonizing the alpha-1a receptor, alpha-blockers relax the smooth muscle in the prostate and bladder neck, reduce bladder outlet resistance, and improve urinary flow.26
In clinical trials in BPH, alpha-blockers improved the symptom score by 30% to 45% and increased the peak urinary flow rate by 15% to 30% from baseline values.27 These agents have a rapid onset (within a few days) and result in significant symptom improvement. They are all about the same in efficacy (Table 3),28–36 with no strong evidence that any one of them is superior to another; thus, decisions about which agent to use must consider differences in receptor subtype specificity, adverse-effect profile, and tolerability.
In the Medical Therapy of Prostatic Symptoms (MTOPS) trial,37 men randomized to the alpha-blocker doxazosin had a 39% lower risk of BPH progression than with placebo, largely due to symptom score reduction. However, doxazosin failed to reduce the risk of progressing to acute urinary retention or surgical intervention. Though rapidly effective in reducing symptoms, alpha-blocker monotherapy may not be the best option in men at higher risk of BPH progression, as discussed below.
Before starting this therapy, patients must be counseled about common side effects such as dizziness, fatigue, peripheral edema, orthostatic hypotension, and ejaculatory dysfunction. The incidence of adverse effects varies among agents (Table 4).28–30,34,35,38,39
To maximize efficacy of alpha-blocker therapy, it is imperative to understand dosing variations among agents.
Alpha-blockers are classified as uroselective or non-uroselective based on alpha-1a receptor subtype specificity. The non-uroselective alpha-blockers doxazosin and terazosin need to be titrated because the higher the dose the greater the efficacy, but also the greater the blood pressure-lowering effect and other side effects.25 Though non-uroselective, alfuzosin does not affect blood pressure and does not require dose titration. Similarly, the uroselective alpha-blockers tamsulosin and silodosin can be initiated at a therapeutic dose.
Terazosin, a non-uroselective agent, can lower blood pressure and often causes dizziness. It should be started at 2 mg and titrated to side effects, efficacy, or maximum therapeutic dose (10 mg daily).28
Doxazosin has a high, dose-related incidence of dizziness (up to 20%) and must be titrated, starting at 1 mg to a maximum 8 mg.30
Alfuzosin, tamsulosin, and silodosin do not require titration and can be initiated at the therapeutic doses listed in Table 3. Of note, obese patients often require 0.8 mg tamsulosin for maximum efficacy due to a higher volume of distribution.
Before initiating an alpha-blocker, a physician must determine whether a patient plans to undergo cataract surgery, as the use of alpha-blockers is associated with intraoperative floppy iris syndrome. This condition is marked by poor intraoperative pupil dilation, increasing the risk of surgical complications.40 It is unclear whether discontinuing alpha-blockers before cataract surgery reduces the risk of intraoperative floppy iris syndrome. As such, alpha-blocker therapy should be delayed in patients planning to undergo cataract surgery.
5-Alpha reductase inhibitors
Prostate growth is androgen-dependent and mediated predominantly by dihydrotestosterone, which is generated from testosterone by the action of 5-alpha reductase. There are two 5-alpha reductase isoenzymes: type 1, expressed in the liver and skin, and type 2, expressed primarily in the prostate.
There are also two 5-alpha reductase inhibitors: dutasteride and finasteride. Dutasteride inhibits both isoenzymes, while finasteride is selective for type 2. By inhibiting both isoenzymes, dutasteride reduces the serum dihydrotestosterone concentration more than finasteride does (by 95% vs 70%), and also reduces the intraprostatic dihydrotestosterone concentration more (by 94% vs 80%).41–43 Both agents induce apoptosis of prostatic stroma, with a resultant 20% to 25% mean reduction in prostate volume.41,42
Finasteride and dutasteride are believed to mitigate the static obstructive component of BPH, with similar improvements in urinary flow rate (1.6–2.2 mL/sec) and symptom score (–2.7 to – 4.5 points) in men with an enlarged prostate.41,42 Indeed, data from the MTOPS trial showed that men with a prostate volume of 30 grams or greater or a PSA level of 1.5 ng/mL or greater are most likely to benefit from 5-alpha reductase inhibitors.37 Maximum symptomatic improvement is seen after 3 to 6 months of 5-alpha reductase inhibitor therapy.
In addition to improving urinary flow and lower urinary tract symptoms, finasteride has been shown to reduce the risk of disease progression in men with prostates greater than 30 grams.44 Compared with placebo, these drugs significantly reduce the risk of developing acute urinary retention or requiring BPH-related surgery, a benefit not seen with alpha-blockers.37 To estimate prostate volume, most practitioners rely on digital rectal examination. Though less precise than transrectal ultrasonography, digital rectal examination can identify men with significant prostatic enlargement likely to benefit from this therapy.
Before starting 5-alpha reductase inhibitor therapy, patients should be counseled about common adverse effects such as erectile dysfunction (occurring in 5%–8%), decreased libido (5%), ejaculatory dysfunction (1%–5%), and gynecomastia (1%).
Combination therapy
The MTOPS trial37 randomized patients to receive doxazosin, finasteride, both, or placebo. The combination of doxazosin (an alpha-blocker) and finasteride (a 5-alpha reductase inhibitor) reduced the risk of disease progression to a greater extent than doxazosin or finasteride alone. It also reduced the IPSS more and increased the peak urinary flow rate more. Similar results have been seen with the combination of dutasteride and tamsulosin.45
Given its superior efficacy and benefits in preventing disease progression, combination therapy should be considered for men with an enlarged prostate and moderate to severe lower urinary tract symptoms.
Anticholinergic agents
Anticholinergic agents block muscarinic receptors within the detrusor muscle, resulting in relaxation. They are used in the treatment of overactive bladder for symptoms of urinary urgency, frequency, and urge incontinence.
Anticholinergics were historically contraindicated in men with BPH because of concern about urinary retention. However, in men with a postvoid residual volume less than 200 mL, anticholinergics do not increase the risk of urinary retention.46 Further, greater symptom improvement has been demonstrated with the addition of anticholinergics to alpha-blocker therapy for men with BPH, irritative lower urinary tract symptoms, and a low postvoid residual volume.47
Beta-3 agonists
Anticholinergic side effects often limit the use of anticholinergic agents. An alternative in such instances is the beta-3 agonist mirabegron. By activating beta-3 adrenergic receptors in the bladder wall, mirabegron promotes detrusor relaxation and inhibits detrusor overactivity.48 Mirabegron does not have anticholinergic side effects and is generally well tolerated, though poorly controlled hypertension is a contraindication to its use.
Phosphodiesterase-5 inhibitors
Phosphodiesterase-5 (PDE5) inhibitors are a mainstay in the treatment of erectile dysfunction. These agents act within penile corporal smooth muscle cells and antagonize PDE5, resulting in cyclic guanosine monophosphate accumulation and smooth muscle relaxation. PDE5 is also found within the prostate and its inhibition is believed to reduce prostatic smooth muscle tone. Randomized studies have demonstrated significant improvement in lower urinary tract symptoms with PDE5 inhibitors, with an average 2-point IPSS improvement on a PDE5 inhibitor compared with placebo.49
Tadalafil is the only drug of this class approved by the FDA for the treatment of lower urinary tract symptoms, though other agents have demonstrated similar efficacy.
Dual therapy with a PDE5 inhibitor and an alpha-blocker has greater efficacy than either monotherapy alone; however, caution must be exercised as these agents are titrated to avoid symptomatic hypotension. Lower urinary tract symptoms and sexual dysfunction often coexist; PDE5 inhibitors are appropriate in the management of such cases.
SURGERY FOR BPH
Even with effective medical therapy, the disease will progress in some men. In the MTOPS trial,37 the 4-year incidence of disease progression was 10% for men on alpha-blocker or 5-alpha reductase inhibitor monotherapy and 5% for men on combination therapy; from 1% to 3% of those in the various treatment groups needed surgery. With this in mind, patients whose symptoms do not improve with medical therapy, whose symptoms progress, or who simply are interested in surgery should be referred for urologic evaluation.
A number of effective surgical therapies are available for men with BPH (Table 5), providing excellent 1-year outcomes including a mean 70% reduction in IPSS and a mean 12 mL/sec improvement in peak urinary flow.50 Given the efficacy of surgical therapy, men who do not improve with medical therapy who demonstrate any of the findings outlined in Table 1 warrant urologic evaluation.
Acknowledgments: We would like to thank Mary Ellen Amos, PharmD, and Kara Sink, BS, RPh, for their assistance in obtaining the suggested wholesale pricing information included in Table 2.
Primary care physicians are uniquely positioned to screen for benign prostatic hyperplasia (BPH) and lower urinary tract symptoms, to perform the initial diagnostic workup, and to start medical therapy in uncomplicated cases. Effective medical therapy is available but underutilized in the primary care setting.1
This overview covers how to identify and evaluate patients with lower urinary tract symptoms, initiate therapy, and identify factors warranting timely urology referral.
TWO MECHANISMS: STATIC, DYNAMIC
BPH is a histologic diagnosis of proliferation of smooth muscle, epithelium, and stromal cells within the transition zone of the prostate,2 which surrounds the proximal urethra.
Symptoms arise through two mechanisms: static, in which the hyperplastic prostatic tissue compresses the urethra (Figure 1); and dynamic, with increased adrenergic nervous system and prostatic smooth muscle tone (Figure 2).3 Both mechanisms increase resistance to urinary flow at the level of the bladder outlet.
As an adaptive change to overcome outlet resistance and maintain urinary flow, the detrusor muscles undergo hypertrophy. However, over time the bladder may develop diminished compliance and increased detrusor activity, causing symptoms such as urinary frequency and urgency. Chronic bladder outlet obstruction can lead to bladder decompensation and detrusor underactivity, manifesting as incomplete emptying, urinary hesitancy, intermittency (starting and stopping while voiding), a weakened urinary stream, and urinary retention.
MOST MEN EVENTUALLY DEVELOP BPH
Autopsy studies have shown that BPH increases in prevalence with age beginning around age 30 and reaching a peak prevalence of 88% in men in their 80s.4 This trend parallels those of the incidence and severity of lower urinary tract symptoms.5
In the year 2000 alone, BPH was responsible for 4.5 million physician visits at an estimated direct cost of $1.1 billion, not including the cost of pharmacotherapy.6
OFFICE WORKUP
BPH can cause lower urinary tract symptoms that fall into two categories: storage and emptying. Storage symptoms include urinary frequency, urgency, and nocturia, whereas emptying symptoms include weak stream, hesitancy, intermittency, incomplete emptying, straining, and postvoid dribbling.
History and differential diagnosis
Assessment begins with characterizing the patient’s symptoms and determining those that are most bothersome. Because BPH is just one of many possible causes of lower urinary tract symptoms, a detailed medical history is necessary to evaluate for other conditions that may cause lower urinary tract dysfunction or complicate its treatment.
Obstructive urinary symptoms can arise from BPH or from other conditions, including urethral stricture disease and neurogenic voiding dysfunction.
Irritative voiding symptoms such as urinary urgency and frequency can result from detrusor overactivity secondary to BPH, but can also be caused by neurologic disease, malignancy, initiation of diuretic therapy, high fluid intake, or consumption of bladder irritants such as caffeine, alcohol, and spicy foods.
Urinary frequency is sometimes a presenting symptom of undiagnosed or poorly controlled diabetes mellitus resulting from glucosuria and polyuria. Iatrogenic causes of polyuria include the new hypoglycemic agents canagliflozin and dapagliflozin, which block renal glucose reabsorption, improving glycemic control by inducing urinary
glucose loss.7
Nocturia has many possible nonurologic causes including heart failure (in which excess extravascular fluid shifts to the intravascular space when the patient lies down, resulting in polyuria), obstructive sleep apnea, and behavioral factors such as high evening fluid intake. In these cases, patients usually have nocturnal polyuria (greater than one-third of 24-hour urine output at night) rather than only nocturia (waking at night to void). A fluid diary is a simple tool that can differentiate these two conditions.
Hematuria can develop in patients with BPH with bleeding from congested prostatic or bladder neck vessels; however, hematuria may indicate an underlying malignancy or urolithiasis, for which a urologic workup is indicated.
The broad differential diagnosis for the different lower urinary tract symptoms highlights the importance of obtaining a thorough history.
Physical examination
A general examination should include the following:
Body mass index. Obese patients are at risk of obstructive sleep apnea, which can cause nocturnal polyuria.
Gait. Abnormal gait may suggest a neurologic condition such as Parkinson disease or stroke that can also affect lower urinary tract function.
Lower abdomen. A palpable bladder suggests urinary retention.
External genitalia. Penile causes of urinary obstruction include urethral meatal stenosis or a palpable urethral mass.
Digital rectal examination can reveal benign prostatic enlargement or nodules or firmness, which suggest malignancy and warrant urologic referral.
Neurologic examination, including evaluation of anal sphincter tone and lower extremity sensorimotor function.
Feet. Bilateral lower-extremity edema may be due to heart failure or venous insufficiency.
The International Prostate Symptom Score
All men with lower urinary tract symptoms should complete the International Prostate Symptom Score (IPSS) survey, consisting of seven questions about urinary symptoms plus one about quality of life.8 Specifically, it asks the patient, “Over the past month, how often have you…”
- Had a sensation of not emptying your bladder completely after you finish urinating?
- Had to urinate again less than 2 hours after you finished urinating?
- Found you stopped and started again several times when you urinated?
- Found it difficult to postpone urination?
- Had a weak urinary stream?
- Had to push or strain to begin urination?
Each question above is scored as 0 (not at all), 1 (less than 1 time in 5), 2 (less than half the time), 3 (about half the time), 4 (more than half the time, or 5 (almost always).
- Over the past month, how many times did you most typically get up to urinate from the time you went to bed until the time you got up in the morning?
This question is scored from 0 (none) to 5 (5 times or more).
- If you were to spend the rest of your life with your urinary condition the way it is now, how would you feel about that?
This question is scored as 0 (delighted), 1 (pleased), 2 (mostly satisfied), 3 (mixed: equally satisfied and dissatisfied), 4 (mostly dissatisfied), 5 (unhappy), or 6 (terrible).
A total score of 1 to 7 is categorized as mild, 8 to 19 moderate, and 20 to 35 severe.
The questionnaire can also be used to evaluate for disease progression and response to treatment over time. A change of 3 points is clinically significant, as patients are unable to discern a difference below this threshold.9
Urinalysis
Urinalysis is recommended to assess for urinary tract infection, hematuria, proteinuria, or glucosuria.
Fluid diary
A fluid diary is useful for patients complaining of frequency or nocturia and can help quantify the volume of fluid intake, frequency of urination, and volumes voided. The patient should complete the diary over a 24-hour period, recording the time and volume of fluid intake and each void. This aids in diagnosing polyuria (> 3 L of urine output per 24 hours), nocturnal polyuria, and behavioral causes of symptoms, including excessive total fluid intake or high evening fluid intake contributing to nocturia.
Serum creatinine not recommended
Measuring serum creatinine is not recommended in the initial BPH workup, as men with lower urinary tract symptoms are not at higher risk of renal failure than those without these symptoms.10
Prostate-specific antigen
Prostate-specific antigen (PSA) is a glycoprotein primarily produced by prostatic luminal epithelial cells. It is most commonly discussed in the setting of prostate cancer screening, but its utility extends to guiding the management of BPH.
PSA levels correlate with prostate volume and subsequent growth.11 In addition, the risks of developing acute urinary retention or needing surgical intervention rise with increasing PSA.12 Among men in the Proscar Long-Term Efficacy and Safety Study, the risk of acute urinary retention or BPH-related surgery after 4 years in the watchful-waiting arm was 7.8% in men with a PSA of 1.3 ng/dL or less, compared with 19.9% in men with a PSA greater than 3.2 ng/dL.11 Therefore, men with BPH and an elevated PSA are at higher risk with watchful waiting and may be better served with medical therapy.
In addition, American Urological Association guidelines recommend measuring serum PSA levels in men with a life expectancy greater than 10 years in whom the diagnosis of prostate cancer would alter management.10
Urologic referral
If the initial evaluation reveals hematuria, recurrent urinary tract infection, a palpable bladder, abnormal findings on digital rectal examination suggesting prostate cancer, or a history of or risk factors for urethral stricture or neurologic disease, the patient should be referred to a urologist for further evaluation (Table 1).10 Other patients who should undergo urologic evaluation are those with persistent bothersome symptoms after basic management and those who desire referral.
Adjunctive tests
Patients referred for urologic evaluation may require additional tests for diagnosis and to guide management.
Postvoid residual volume is easily measured with either abdominal ultrasonography or catheterization and is often included in the urologic evaluation of BPH. Patients vary considerably in their residual volume, which correlates poorly with BPH, symptom severity, or surgical success. However, those with a residual volume of more than 100 mL have a slightly higher rate of failure with watchful waiting.13 Postvoid residual volume is not routinely monitored in patients with a low residual volume unless there is a significant change in urinary symptoms. Conversely, patients with a volume greater than 200 mL should be monitored closely for worsening urinary retention, especially if considering anticholinergic therapy.
There is no absolute threshold postvoid residual volume above which therapy is mandatory. Rather, the decision to intervene is based on symptom severity and whether sequelae of urinary retention (eg, incontinence, urinary tract infection, hematuria, hydronephrosis, renal dysfunction) are present.
Uroflowmetry is a noninvasive test measuring the urinary flow rate during voiding and is recommended during specialist evaluation of men with lower urinary tract symptoms and suspected BPH.10 Though a diminished urinary flow rate may be detected in men with bladder outlet obstruction from BPH, it cannot differentiate obstruction from detrusor underactivity, both of which may result in reduced urinary flow. Urodynamic studies can help differentiate between these two mechanisms of lower urinary tract symptoms. Uroflowmetry may be useful in selecting surgical candidates, as patients with a maximum urinary flow rate of 15 mL/second or greater have been shown to have lower rates of surgical success.14
Urodynamic studies. If the diagnosis of bladder outlet obstruction remains in doubt, urodynamic studies can differentiate obstruction from detrusor underactivity. Urodynamic studies allow simultaneous measurement of urinary flow and detrusor pressure, differentiating between obstruction (manifesting as diminished urinary flow with normal or elevated detrusor pressure) and detrusor underactivity (diminished urinary flow with diminished detrusor pressure). Nomograms15 and the easily calculated bladder outlet obstruction index16 are simple tools used to differentiate these two causes of diminished urinary flow.
Cystourethroscopy is not recommended for routine evaluation of BPH. Indications for cystourethroscopy include hematuria and the presence of a risk factor for urethral stricture disease such as urethritis, prior urethral instrumentation, or perineal trauma. Cystourethroscopy can also aid in surgical planning when intervention is considered.
An algorithm for diagnostic workup and management of BPH and lower urinary tract symptoms is shown in Figure 3.17
MANAGEMENT STRATEGIES FOR BPH
While BPH is rarely life-threatening, it can significantly detract from a patient’s quality of life. The goal of treatment is not only to alleviate bothersome symptoms, but also to prevent disease progression and disease-related complications.
BPH tends to progress
Understanding the natural history of BPH is imperative to appropriately counsel patients on management options, which include watchful waiting, behavioral modification, pharmacologic therapy, and surgery.
In a randomized trial,18 men with moderately symptomatic BPH underwent either surgery or, in the control group, watchful waiting. At 5 years, the failure rate was 21% with watchful waiting vs 10% with surgery (P < .0004). (Failure was defined as a composite of death, repeated or intractable urinary retention, residual urine volume > 350 mL, development of bladder calculus, new persistent incontinence requiring use of a pad or other incontinence device, symptom score in the severe range [> 24 at 1 visit or score of 21 or higher at two consecutive visits, with 27 being the maximum score], or a doubling of baseline serum creatinine.) In the watchful-waiting group, 36% of the men crossed over to surgery. Men with more bothersome symptoms at enrollment were at higher risk of progressing to surgery.
In a longitudinal study of men with BPH and mild symptoms (IPSS < 8), the risk of progression to moderate or severe symptoms (IPPS ≥ 8) was 31% at 4 years.19
The Olmsted County Study of Urinary Symptoms and Health Status Among Men20 found that the peak urinary flow rate decreased by a mean of 2.1% per year, declining faster in older men who had a lower peak flow at baseline. In this cohort, the IPSS increased by a mean of 0.18 points per year, with a greater increase in older men.21
Though men managed with watchful waiting are at no higher risk of death or renal failure than men managed surgically,17 population-based studies have demonstrated an overall risk of acute urinary retention of 6.8/1,000 person-years with watchful waiting. Older men with a larger prostate, higher symptom score, and lower peak urinary flow rate are at higher risk of acute urinary retention and progression to needing BPH treatment.22,23
There is evidence that patients progressing to needing surgery after an initial period of watchful waiting have worse surgical outcomes than men managed surgically at the onset.18 This observation must be considered in counseling and selecting patients for watchful waiting. Ideal candidates include patients who have mild or moderate symptoms that cause little bother.10 Patients electing watchful waiting warrant annual follow-up including history, physical examination, and symptom assessment with the IPSS.
Behavioral modification
Behavioral modification should be incorporated into whichever management strategy a patient elects. Such modifications include:
- Reducing total or evening fluid intake for patients with urinary frequency or nocturia.
- Minimizing consumption of bladder irritants such as alcohol and caffeine, which exacerbate storage symptoms.
- Smoking cessation counseling.
- For patients with lower extremity edema who complain of nocturia, using compression stockings or elevating their legs in the afternoon to mobilize lower extremity edema and promote diuresis before going to sleep. If these measures fail, initiating or increasing the dose of a diuretic should be considered. Patients on diuretic therapy with nocturnal lower urinary tract symptoms should be instructed to take diuretics in the morning and early afternoon to avoid diuresis just before bed.
MEDICAL MANAGEMENT
Drugs for BPH include alpha-adrenergic blockers, 5-alpha reductase inhibitors, anticholinergics, beta-3 agonists, and phosphodiesterase-5 inhibitors. Costs of selected agents in these classes are listed in Table 2.
Alpha-adrenergic receptor blockers
Alpha-adrenergic receptors are found throughout the body and modulate smooth muscle tone.24 The alpha-1a receptor is the predominant subtype found in the bladder neck and prostate25 (Figure 2) and is a target of therapy. By antagonizing the alpha-1a receptor, alpha-blockers relax the smooth muscle in the prostate and bladder neck, reduce bladder outlet resistance, and improve urinary flow.26
In clinical trials in BPH, alpha-blockers improved the symptom score by 30% to 45% and increased the peak urinary flow rate by 15% to 30% from baseline values.27 These agents have a rapid onset (within a few days) and result in significant symptom improvement. They are all about the same in efficacy (Table 3),28–36 with no strong evidence that any one of them is superior to another; thus, decisions about which agent to use must consider differences in receptor subtype specificity, adverse-effect profile, and tolerability.
In the Medical Therapy of Prostatic Symptoms (MTOPS) trial,37 men randomized to the alpha-blocker doxazosin had a 39% lower risk of BPH progression than with placebo, largely due to symptom score reduction. However, doxazosin failed to reduce the risk of progressing to acute urinary retention or surgical intervention. Though rapidly effective in reducing symptoms, alpha-blocker monotherapy may not be the best option in men at higher risk of BPH progression, as discussed below.
Before starting this therapy, patients must be counseled about common side effects such as dizziness, fatigue, peripheral edema, orthostatic hypotension, and ejaculatory dysfunction. The incidence of adverse effects varies among agents (Table 4).28–30,34,35,38,39
To maximize efficacy of alpha-blocker therapy, it is imperative to understand dosing variations among agents.
Alpha-blockers are classified as uroselective or non-uroselective based on alpha-1a receptor subtype specificity. The non-uroselective alpha-blockers doxazosin and terazosin need to be titrated because the higher the dose the greater the efficacy, but also the greater the blood pressure-lowering effect and other side effects.25 Though non-uroselective, alfuzosin does not affect blood pressure and does not require dose titration. Similarly, the uroselective alpha-blockers tamsulosin and silodosin can be initiated at a therapeutic dose.
Terazosin, a non-uroselective agent, can lower blood pressure and often causes dizziness. It should be started at 2 mg and titrated to side effects, efficacy, or maximum therapeutic dose (10 mg daily).28
Doxazosin has a high, dose-related incidence of dizziness (up to 20%) and must be titrated, starting at 1 mg to a maximum 8 mg.30
Alfuzosin, tamsulosin, and silodosin do not require titration and can be initiated at the therapeutic doses listed in Table 3. Of note, obese patients often require 0.8 mg tamsulosin for maximum efficacy due to a higher volume of distribution.
Before initiating an alpha-blocker, a physician must determine whether a patient plans to undergo cataract surgery, as the use of alpha-blockers is associated with intraoperative floppy iris syndrome. This condition is marked by poor intraoperative pupil dilation, increasing the risk of surgical complications.40 It is unclear whether discontinuing alpha-blockers before cataract surgery reduces the risk of intraoperative floppy iris syndrome. As such, alpha-blocker therapy should be delayed in patients planning to undergo cataract surgery.
5-Alpha reductase inhibitors
Prostate growth is androgen-dependent and mediated predominantly by dihydrotestosterone, which is generated from testosterone by the action of 5-alpha reductase. There are two 5-alpha reductase isoenzymes: type 1, expressed in the liver and skin, and type 2, expressed primarily in the prostate.
There are also two 5-alpha reductase inhibitors: dutasteride and finasteride. Dutasteride inhibits both isoenzymes, while finasteride is selective for type 2. By inhibiting both isoenzymes, dutasteride reduces the serum dihydrotestosterone concentration more than finasteride does (by 95% vs 70%), and also reduces the intraprostatic dihydrotestosterone concentration more (by 94% vs 80%).41–43 Both agents induce apoptosis of prostatic stroma, with a resultant 20% to 25% mean reduction in prostate volume.41,42
Finasteride and dutasteride are believed to mitigate the static obstructive component of BPH, with similar improvements in urinary flow rate (1.6–2.2 mL/sec) and symptom score (–2.7 to – 4.5 points) in men with an enlarged prostate.41,42 Indeed, data from the MTOPS trial showed that men with a prostate volume of 30 grams or greater or a PSA level of 1.5 ng/mL or greater are most likely to benefit from 5-alpha reductase inhibitors.37 Maximum symptomatic improvement is seen after 3 to 6 months of 5-alpha reductase inhibitor therapy.
In addition to improving urinary flow and lower urinary tract symptoms, finasteride has been shown to reduce the risk of disease progression in men with prostates greater than 30 grams.44 Compared with placebo, these drugs significantly reduce the risk of developing acute urinary retention or requiring BPH-related surgery, a benefit not seen with alpha-blockers.37 To estimate prostate volume, most practitioners rely on digital rectal examination. Though less precise than transrectal ultrasonography, digital rectal examination can identify men with significant prostatic enlargement likely to benefit from this therapy.
Before starting 5-alpha reductase inhibitor therapy, patients should be counseled about common adverse effects such as erectile dysfunction (occurring in 5%–8%), decreased libido (5%), ejaculatory dysfunction (1%–5%), and gynecomastia (1%).
Combination therapy
The MTOPS trial37 randomized patients to receive doxazosin, finasteride, both, or placebo. The combination of doxazosin (an alpha-blocker) and finasteride (a 5-alpha reductase inhibitor) reduced the risk of disease progression to a greater extent than doxazosin or finasteride alone. It also reduced the IPSS more and increased the peak urinary flow rate more. Similar results have been seen with the combination of dutasteride and tamsulosin.45
Given its superior efficacy and benefits in preventing disease progression, combination therapy should be considered for men with an enlarged prostate and moderate to severe lower urinary tract symptoms.
Anticholinergic agents
Anticholinergic agents block muscarinic receptors within the detrusor muscle, resulting in relaxation. They are used in the treatment of overactive bladder for symptoms of urinary urgency, frequency, and urge incontinence.
Anticholinergics were historically contraindicated in men with BPH because of concern about urinary retention. However, in men with a postvoid residual volume less than 200 mL, anticholinergics do not increase the risk of urinary retention.46 Further, greater symptom improvement has been demonstrated with the addition of anticholinergics to alpha-blocker therapy for men with BPH, irritative lower urinary tract symptoms, and a low postvoid residual volume.47
Beta-3 agonists
Anticholinergic side effects often limit the use of anticholinergic agents. An alternative in such instances is the beta-3 agonist mirabegron. By activating beta-3 adrenergic receptors in the bladder wall, mirabegron promotes detrusor relaxation and inhibits detrusor overactivity.48 Mirabegron does not have anticholinergic side effects and is generally well tolerated, though poorly controlled hypertension is a contraindication to its use.
Phosphodiesterase-5 inhibitors
Phosphodiesterase-5 (PDE5) inhibitors are a mainstay in the treatment of erectile dysfunction. These agents act within penile corporal smooth muscle cells and antagonize PDE5, resulting in cyclic guanosine monophosphate accumulation and smooth muscle relaxation. PDE5 is also found within the prostate and its inhibition is believed to reduce prostatic smooth muscle tone. Randomized studies have demonstrated significant improvement in lower urinary tract symptoms with PDE5 inhibitors, with an average 2-point IPSS improvement on a PDE5 inhibitor compared with placebo.49
Tadalafil is the only drug of this class approved by the FDA for the treatment of lower urinary tract symptoms, though other agents have demonstrated similar efficacy.
Dual therapy with a PDE5 inhibitor and an alpha-blocker has greater efficacy than either monotherapy alone; however, caution must be exercised as these agents are titrated to avoid symptomatic hypotension. Lower urinary tract symptoms and sexual dysfunction often coexist; PDE5 inhibitors are appropriate in the management of such cases.
SURGERY FOR BPH
Even with effective medical therapy, the disease will progress in some men. In the MTOPS trial,37 the 4-year incidence of disease progression was 10% for men on alpha-blocker or 5-alpha reductase inhibitor monotherapy and 5% for men on combination therapy; from 1% to 3% of those in the various treatment groups needed surgery. With this in mind, patients whose symptoms do not improve with medical therapy, whose symptoms progress, or who simply are interested in surgery should be referred for urologic evaluation.
A number of effective surgical therapies are available for men with BPH (Table 5), providing excellent 1-year outcomes including a mean 70% reduction in IPSS and a mean 12 mL/sec improvement in peak urinary flow.50 Given the efficacy of surgical therapy, men who do not improve with medical therapy who demonstrate any of the findings outlined in Table 1 warrant urologic evaluation.
Acknowledgments: We would like to thank Mary Ellen Amos, PharmD, and Kara Sink, BS, RPh, for their assistance in obtaining the suggested wholesale pricing information included in Table 2.
- Wei JT, Miner MM, Steers WD, et al; BPH Registry Steering Committee. Benign prostatic hyperplasia evaluation and management by urologists and primary care physicians: practice patterns from the observational BPH registry. J Urol 2011; 186:971–976.
- McNeal J. Pathology of benign prostatic hyperplasia. insight into etiology. Urol Clin North Am 1990; 17:477–486.
- Roehrborn CG, Schwinn DA. Alpha1-adrenergic receptors and their inhibitors in lower urinary tract symptoms and benign prostatic hyperplasia. J Urol 2004; 171:1029–1035.
- Berry SJ, Coffey DS, Walsh PC, Ewing LL. The development of human benign prostatic hyperplasia with age. J Urol 1984; 132:474–479.
- Platz EA, Smit E, Curhan GC, Nyberg LM, Giovannucci E. Prevalence of and racial/ethnic variation in lower urinary tract symptoms and noncancer prostate surgery in US men. Urology 2002; 59:877–883.
- Wei JT, Calhoun E, Jacobsen SJ. Urologic diseases in America project: benign prostatic hyperplasia. J Urol 2008; 179(suppl):S75–S80.
- Scheen AJ, Paquot N. Metabolic effects of SGLT-2 inhibitors beyond increased glucosuria: a review of the clinical evidence. Diabetes Metab 2014; 40(suppl 1):S4–S11.
- Barry MJ, Fowler FJ Jr, O’Leary MP, et al. The American Urological Association symptom index for benign prostatic hyperplasia. The Measurement Committee of the American Urological Association. J Urol 1992; 148:1549–1564.
- Barry MJ, Williford WO, Chang Y, et al. Benign prostatic hyperplasia specific health status measures in clinical research: how much change in the American Urological Association symptom index and the benign prostatic hyperplasia impact index is perceptible to patients? J Urol 1995; 154:1770–1774.
- McVary KT, Roehrborn CG, Avins AL, et al. Update on AUA guideline on the management of benign prostatic hyperplasia. J Urol 2011; 185:1793–1803.
- Roehrborn CG, McConnell J, Bonilla J, et al. Serum prostate specific antigen is a strong predictor of future prostate growth in men with benign prostatic hyperplasia. PROSCAR long-term efficacy and safety study. J Urol 2000; 163:13–20.
- Roehrborn CG, McConnell JD, Lieber M, et al. Serum prostate-specific antigen concentration is a powerful predictor of acute urinary retention and need for surgery in men with clinical benign prostatic hyperplasia. PLESS Study Group. Urology 1999; 53:473–480.
- Wasson JH, Reda DJ, Bruskewitz RC, Elinson J, Keller AM, Henderson WG. A comparison of transurethral surgery with watchful waiting for moderate symptoms of benign prostatic hyperplasia. The Veterans Affairs Cooperative Study Group on Transurethral Resection of the Prostate. N Engl J Med 1995; 332:75–79.
- Jensen KM, Bruskewitz RC, Iversen P, Madsen PO. Spontaneous uroflowmetry in prostatism. Urology 1984; 24:403–409.
- Abrams PH, Griffiths DJ. The assessment of prostatic obstruction from urodynamic measurements and from residual urine. Br J Urol 1979; 51:129–134.
- Lim CS, Abrams P. The Abrams-Griffiths nomogram. World J Urol 1995; 13:34–39.
- Abrams P, Chapple C, Khoury S, Roehrborn C, de la Rosette J; International Consultation on New Developments in Prostate Cancer and Prostate Diseases. Evaluation and treatment of lower urinary tract symptoms in older men. J Urol 2013; 189(suppl 1):S93–S101.
- Flanigan RC, Reda DJ, Wasson JH, Anderson RJ, Abdellatif M, Bruskewitz RC. 5-year outcome of surgical resection and watchful waiting for men with moderately symptomatic benign prostatic hyperplasia: a Department of Veterans Affairs cooperative study. J Urol 1998; 160:12–17.
- Djavan B, Fong YK, Harik M, et al. Longitudinal study of men with mild symptoms of bladder outlet obstruction treated with watchful waiting for four years. Urology 2004; 64:1144–1148.
- Roberts RO, Jacobsen SJ, Jacobson DJ, Rhodes T, Girman CJ, Lieber MM. Longitudinal changes in peak urinary flow rates in a community based cohort. J Urol 2000; 163:107–113.
- Jacobsen SJ, Girman CJ, Guess HA, Rhodes T, Oesterling JE, Lieber MM. Natural history of prostatism: longitudinal changes in voiding symptoms in community dwelling men. J Urol 1996; 155:595–600.
- Jacobsen SJ, Jacobson DJ, Girman CJ, et al. Treatment for benign prostatic hyperplasia among community dwelling men: the Olmsted County study of urinary symptoms and health status. J Urol 1999; 162:1301–1306.
- Jacobsen SJ, Jacobson DJ, Girman CJ, et al. Natural history of prostatism: risk factors for acute urinary retention. J Urol 1997; 158:481–487.
- Kobayashi S, Tang R, Shapiro E, Lepor H. Characterization and localization of prostatic alpha 1 adrenoceptors using radioligand receptor binding on slide-mounted tissue section. J Urol 1993; 150:2002–2006.
- Kirby RS, Pool JL. Alpha adrenoceptor blockade in the treatment of benign prostatic hyperplasia: past, present and future. Br J Urol 1997; 80:521–532.
- Kirby RS, Pool JL. Alpha adrenoceptor blockade in the treatment of benign prostatic hyperplasia: past, present and future. Br J Urol 1997; 80:521–532.
- Milani S, Djavan B. Lower urinary tract symptoms suggestive of benign prostatic hyperplasia: latest update on alpha-adrenoceptor antagonists. BJU Int 2005; 95(suppl 4):29–36.
- Lepor H, Auerbach S, Puras-Baez A, et al. A randomized, placebo-controlled multicenter study of the efficacy and safety of terazosin in the treatment of benign prostatic hyperplasia. J Urol 1992; 148:1467–1474.
- Roehrborn CG, Oesterling JE, Auerbach S, et al. The Hytrin Community Assessment Trial study: a one-year study of terazosin versus placebo in the treatment of men with symptomatic benign prostatic hyperplasia. HYCAT Investigator Group. Urology 1996; 47:159–168.
- Gillenwater JY, Conn RL, Chrysant SG, et al. Doxazosin for the treatment of benign prostatic hyperplasia in patients with mild to moderate essential hypertension: a double-blind, placebo-controlled, dose-response multicenter study. J Urol 1995; 154:110–115.
- Chapple CR, Carter P, Christmas TJ, et al. A three month double-blind study of doxazosin as treatment for benign prostatic bladder outlet obstruction. Br J Urol 1994; 74:50–56.
- Buzelin JM, Roth S, Geffriaud-Ricouard C, Delauche-Cavallier MC. Efficacy and safety of sustained-release alfuzosin 5 mg in patients with benign prostatic hyperplasia. ALGEBI Study Group. Eur Urol 1997; 31:190–198.
- van Kerrebroeck P, Jardin A, Laval KU, van Cangh P. Efficacy and safety of a new prolonged release formulation of alfuzosin 10 mg once daily versus alfuzosin 2.5 mg thrice daily and placebo in patients with symptomatic benign prostatic hyperplasia. ALFORTI Study Group. Eur Urol 2000; 37:306–313.
- Narayan P, Tewari A. A second phase III multicenter placebo controlled study of 2 dosages of modified release tamsulosin in patients with symptoms of benign prostatic hyperplasia. United States 93-01 Study Group. J Urol 1998; 160:1701–1706.
- Lepor H. Phase III multicenter placebo-controlled study of tamsulosin in benign prostatic hyperplasia. Tamsulosin Investigator Group. Urology 1998; 51:892–900.
- Ding H, Du W, Hou ZZ, Wang HZ, Wang ZP. Silodosin is effective for treatment of LUTS in men with BPH: a systematic review. Asian J Androl 2013; 15:121–128.
- McConnell JD, Roehrborn CG, Bautista OM, et al; Medical Therapy of Prostatic Symptoms (MTOPS) Research Group. The long-term effect of doxazosin, finasteride, and combination therapy on the clinical progression of benign prostatic hyperplasia. N Engl J Med 2003; 349:2387–2398.
- Jardin A, Bensadoun H, Delauche-Cavallier MC, Attali P. Alfuzosin for treatment of benign prostatic hypertrophy. The BPH-ALF Group. Lancet 1991; 337:1457–1461.
- Marks LS, Gittelman MC, Hill LA, Volinn W, Hoel G. Rapid efficacy of the highly selective alpha1A-adrenoceptor antagonist silodosin in men with signs and symptoms of benign prostatic hyperplasia: pooled results of 2 phase 3 studies. J Urol 2009; 181:2634–2640.
- Chang DF, Campbell JR. Intraoperative floppy iris syndrome associated with tamsulosin. J Cataract Refract Surg 2005; 31:664–673.
- Gormley GJ, Stoner E, Bruskewitz RC, et al. The effect of finasteride in men with benign prostatic hyperplasia. The Finasteride Study Group. N Engl J Med 1992; 327:1185–1191.
- Roehrborn CG, Boyle P, Nickel JC, Hoefner K, Andriole G; ARIA3001 ARIA3002 and ARIA3003 Study Investigators. Efficacy and safety of a dual inhibitor of 5-alpha-reductase types 1 and 2 (dutasteride) in men with benign prostatic hyperplasia. Urology 2002; 60:434–441.
- Clark RV, Hermann DJ, Cunningham GR, Wilson TH, Morrill BB, Hobbs S. Marked suppression of dihydrotestosterone in men with benign prostatic hyperplasia by dutasteride, a dual 5alpha-reductase inhibitor. J Clin Endocrinol Metab 2004; 89:2179–2184.
- Kaplan SA, Lee JY, Meehan AG, Kusek JW; MTOPS Research Group. Long-term treatment with finasteride improves clinical progression of benign prostatic hyperplasia in men with an enlarged versus a smaller prostate: data from the MTOPS trial. J Urol 2011; 185:1369–1373.
- Roehrborn CG, Siami P, Barkin J, et al; CombAT Study Group. The effects of combination therapy with dutasteride and tamsulosin on clinical outcomes in men with symptomatic benign prostatic hyperplasia: 4-year results from the CombAT study. Eur Urol 2010; 57:123–131.
- Abrams P, Kaplan S, De Koning Gans HJ, Millard R. Safety and tolerability of tolterodine for the treatment of overactive bladder in men with bladder outlet obstruction. J Urol 2006; 175:999–1004.
- Kaplan SA, Roehrborn CG, Rovner ES, Carlsson M, Bavendam T, Guan Z. Tolterodine and tamsulosin for treatment of men with lower urinary tract symptoms and overactive bladder: a randomized controlled trial. JAMA 2006; 296:2319–2328.
- Suarez O, Osborn D, Kaufman M, Reynolds WS, Dmochowski R. Mirabegron for male lower urinary tract symptoms. Curr Urol Rep 2013; 14:580–584.
- Oelke M, Giuliano F, Mirone V, Xu L, Cox D, Viktrup L. Monotherapy with tadalafil or tamsulosin similarly improved lower urinary tract symptoms suggestive of benign prostatic hyperplasia in an international, randomised, parallel, placebo-controlled clinical trial. Eur Urol 2012; 61:917–925.
- Welliver C, McVary KT. Minimally invasive and endoscopic management of benign prostatic hyperplasia. In: Wein AJ, Kavoussi LR, Partin AW, Peters CA, eds. Campbell-Walsh Urology. 11th ed. Philadelphia, PA: Elsevier; 2016:2504–2534.
- Wei JT, Miner MM, Steers WD, et al; BPH Registry Steering Committee. Benign prostatic hyperplasia evaluation and management by urologists and primary care physicians: practice patterns from the observational BPH registry. J Urol 2011; 186:971–976.
- McNeal J. Pathology of benign prostatic hyperplasia. insight into etiology. Urol Clin North Am 1990; 17:477–486.
- Roehrborn CG, Schwinn DA. Alpha1-adrenergic receptors and their inhibitors in lower urinary tract symptoms and benign prostatic hyperplasia. J Urol 2004; 171:1029–1035.
- Berry SJ, Coffey DS, Walsh PC, Ewing LL. The development of human benign prostatic hyperplasia with age. J Urol 1984; 132:474–479.
- Platz EA, Smit E, Curhan GC, Nyberg LM, Giovannucci E. Prevalence of and racial/ethnic variation in lower urinary tract symptoms and noncancer prostate surgery in US men. Urology 2002; 59:877–883.
- Wei JT, Calhoun E, Jacobsen SJ. Urologic diseases in America project: benign prostatic hyperplasia. J Urol 2008; 179(suppl):S75–S80.
- Scheen AJ, Paquot N. Metabolic effects of SGLT-2 inhibitors beyond increased glucosuria: a review of the clinical evidence. Diabetes Metab 2014; 40(suppl 1):S4–S11.
- Barry MJ, Fowler FJ Jr, O’Leary MP, et al. The American Urological Association symptom index for benign prostatic hyperplasia. The Measurement Committee of the American Urological Association. J Urol 1992; 148:1549–1564.
- Barry MJ, Williford WO, Chang Y, et al. Benign prostatic hyperplasia specific health status measures in clinical research: how much change in the American Urological Association symptom index and the benign prostatic hyperplasia impact index is perceptible to patients? J Urol 1995; 154:1770–1774.
- McVary KT, Roehrborn CG, Avins AL, et al. Update on AUA guideline on the management of benign prostatic hyperplasia. J Urol 2011; 185:1793–1803.
- Roehrborn CG, McConnell J, Bonilla J, et al. Serum prostate specific antigen is a strong predictor of future prostate growth in men with benign prostatic hyperplasia. PROSCAR long-term efficacy and safety study. J Urol 2000; 163:13–20.
- Roehrborn CG, McConnell JD, Lieber M, et al. Serum prostate-specific antigen concentration is a powerful predictor of acute urinary retention and need for surgery in men with clinical benign prostatic hyperplasia. PLESS Study Group. Urology 1999; 53:473–480.
- Wasson JH, Reda DJ, Bruskewitz RC, Elinson J, Keller AM, Henderson WG. A comparison of transurethral surgery with watchful waiting for moderate symptoms of benign prostatic hyperplasia. The Veterans Affairs Cooperative Study Group on Transurethral Resection of the Prostate. N Engl J Med 1995; 332:75–79.
- Jensen KM, Bruskewitz RC, Iversen P, Madsen PO. Spontaneous uroflowmetry in prostatism. Urology 1984; 24:403–409.
- Abrams PH, Griffiths DJ. The assessment of prostatic obstruction from urodynamic measurements and from residual urine. Br J Urol 1979; 51:129–134.
- Lim CS, Abrams P. The Abrams-Griffiths nomogram. World J Urol 1995; 13:34–39.
- Abrams P, Chapple C, Khoury S, Roehrborn C, de la Rosette J; International Consultation on New Developments in Prostate Cancer and Prostate Diseases. Evaluation and treatment of lower urinary tract symptoms in older men. J Urol 2013; 189(suppl 1):S93–S101.
- Flanigan RC, Reda DJ, Wasson JH, Anderson RJ, Abdellatif M, Bruskewitz RC. 5-year outcome of surgical resection and watchful waiting for men with moderately symptomatic benign prostatic hyperplasia: a Department of Veterans Affairs cooperative study. J Urol 1998; 160:12–17.
- Djavan B, Fong YK, Harik M, et al. Longitudinal study of men with mild symptoms of bladder outlet obstruction treated with watchful waiting for four years. Urology 2004; 64:1144–1148.
- Roberts RO, Jacobsen SJ, Jacobson DJ, Rhodes T, Girman CJ, Lieber MM. Longitudinal changes in peak urinary flow rates in a community based cohort. J Urol 2000; 163:107–113.
- Jacobsen SJ, Girman CJ, Guess HA, Rhodes T, Oesterling JE, Lieber MM. Natural history of prostatism: longitudinal changes in voiding symptoms in community dwelling men. J Urol 1996; 155:595–600.
- Jacobsen SJ, Jacobson DJ, Girman CJ, et al. Treatment for benign prostatic hyperplasia among community dwelling men: the Olmsted County study of urinary symptoms and health status. J Urol 1999; 162:1301–1306.
- Jacobsen SJ, Jacobson DJ, Girman CJ, et al. Natural history of prostatism: risk factors for acute urinary retention. J Urol 1997; 158:481–487.
- Kobayashi S, Tang R, Shapiro E, Lepor H. Characterization and localization of prostatic alpha 1 adrenoceptors using radioligand receptor binding on slide-mounted tissue section. J Urol 1993; 150:2002–2006.
- Kirby RS, Pool JL. Alpha adrenoceptor blockade in the treatment of benign prostatic hyperplasia: past, present and future. Br J Urol 1997; 80:521–532.
- Kirby RS, Pool JL. Alpha adrenoceptor blockade in the treatment of benign prostatic hyperplasia: past, present and future. Br J Urol 1997; 80:521–532.
- Milani S, Djavan B. Lower urinary tract symptoms suggestive of benign prostatic hyperplasia: latest update on alpha-adrenoceptor antagonists. BJU Int 2005; 95(suppl 4):29–36.
- Lepor H, Auerbach S, Puras-Baez A, et al. A randomized, placebo-controlled multicenter study of the efficacy and safety of terazosin in the treatment of benign prostatic hyperplasia. J Urol 1992; 148:1467–1474.
- Roehrborn CG, Oesterling JE, Auerbach S, et al. The Hytrin Community Assessment Trial study: a one-year study of terazosin versus placebo in the treatment of men with symptomatic benign prostatic hyperplasia. HYCAT Investigator Group. Urology 1996; 47:159–168.
- Gillenwater JY, Conn RL, Chrysant SG, et al. Doxazosin for the treatment of benign prostatic hyperplasia in patients with mild to moderate essential hypertension: a double-blind, placebo-controlled, dose-response multicenter study. J Urol 1995; 154:110–115.
- Chapple CR, Carter P, Christmas TJ, et al. A three month double-blind study of doxazosin as treatment for benign prostatic bladder outlet obstruction. Br J Urol 1994; 74:50–56.
- Buzelin JM, Roth S, Geffriaud-Ricouard C, Delauche-Cavallier MC. Efficacy and safety of sustained-release alfuzosin 5 mg in patients with benign prostatic hyperplasia. ALGEBI Study Group. Eur Urol 1997; 31:190–198.
- van Kerrebroeck P, Jardin A, Laval KU, van Cangh P. Efficacy and safety of a new prolonged release formulation of alfuzosin 10 mg once daily versus alfuzosin 2.5 mg thrice daily and placebo in patients with symptomatic benign prostatic hyperplasia. ALFORTI Study Group. Eur Urol 2000; 37:306–313.
- Narayan P, Tewari A. A second phase III multicenter placebo controlled study of 2 dosages of modified release tamsulosin in patients with symptoms of benign prostatic hyperplasia. United States 93-01 Study Group. J Urol 1998; 160:1701–1706.
- Lepor H. Phase III multicenter placebo-controlled study of tamsulosin in benign prostatic hyperplasia. Tamsulosin Investigator Group. Urology 1998; 51:892–900.
- Ding H, Du W, Hou ZZ, Wang HZ, Wang ZP. Silodosin is effective for treatment of LUTS in men with BPH: a systematic review. Asian J Androl 2013; 15:121–128.
- McConnell JD, Roehrborn CG, Bautista OM, et al; Medical Therapy of Prostatic Symptoms (MTOPS) Research Group. The long-term effect of doxazosin, finasteride, and combination therapy on the clinical progression of benign prostatic hyperplasia. N Engl J Med 2003; 349:2387–2398.
- Jardin A, Bensadoun H, Delauche-Cavallier MC, Attali P. Alfuzosin for treatment of benign prostatic hypertrophy. The BPH-ALF Group. Lancet 1991; 337:1457–1461.
- Marks LS, Gittelman MC, Hill LA, Volinn W, Hoel G. Rapid efficacy of the highly selective alpha1A-adrenoceptor antagonist silodosin in men with signs and symptoms of benign prostatic hyperplasia: pooled results of 2 phase 3 studies. J Urol 2009; 181:2634–2640.
- Chang DF, Campbell JR. Intraoperative floppy iris syndrome associated with tamsulosin. J Cataract Refract Surg 2005; 31:664–673.
- Gormley GJ, Stoner E, Bruskewitz RC, et al. The effect of finasteride in men with benign prostatic hyperplasia. The Finasteride Study Group. N Engl J Med 1992; 327:1185–1191.
- Roehrborn CG, Boyle P, Nickel JC, Hoefner K, Andriole G; ARIA3001 ARIA3002 and ARIA3003 Study Investigators. Efficacy and safety of a dual inhibitor of 5-alpha-reductase types 1 and 2 (dutasteride) in men with benign prostatic hyperplasia. Urology 2002; 60:434–441.
- Clark RV, Hermann DJ, Cunningham GR, Wilson TH, Morrill BB, Hobbs S. Marked suppression of dihydrotestosterone in men with benign prostatic hyperplasia by dutasteride, a dual 5alpha-reductase inhibitor. J Clin Endocrinol Metab 2004; 89:2179–2184.
- Kaplan SA, Lee JY, Meehan AG, Kusek JW; MTOPS Research Group. Long-term treatment with finasteride improves clinical progression of benign prostatic hyperplasia in men with an enlarged versus a smaller prostate: data from the MTOPS trial. J Urol 2011; 185:1369–1373.
- Roehrborn CG, Siami P, Barkin J, et al; CombAT Study Group. The effects of combination therapy with dutasteride and tamsulosin on clinical outcomes in men with symptomatic benign prostatic hyperplasia: 4-year results from the CombAT study. Eur Urol 2010; 57:123–131.
- Abrams P, Kaplan S, De Koning Gans HJ, Millard R. Safety and tolerability of tolterodine for the treatment of overactive bladder in men with bladder outlet obstruction. J Urol 2006; 175:999–1004.
- Kaplan SA, Roehrborn CG, Rovner ES, Carlsson M, Bavendam T, Guan Z. Tolterodine and tamsulosin for treatment of men with lower urinary tract symptoms and overactive bladder: a randomized controlled trial. JAMA 2006; 296:2319–2328.
- Suarez O, Osborn D, Kaufman M, Reynolds WS, Dmochowski R. Mirabegron for male lower urinary tract symptoms. Curr Urol Rep 2013; 14:580–584.
- Oelke M, Giuliano F, Mirone V, Xu L, Cox D, Viktrup L. Monotherapy with tadalafil or tamsulosin similarly improved lower urinary tract symptoms suggestive of benign prostatic hyperplasia in an international, randomised, parallel, placebo-controlled clinical trial. Eur Urol 2012; 61:917–925.
- Welliver C, McVary KT. Minimally invasive and endoscopic management of benign prostatic hyperplasia. In: Wein AJ, Kavoussi LR, Partin AW, Peters CA, eds. Campbell-Walsh Urology. 11th ed. Philadelphia, PA: Elsevier; 2016:2504–2534.
KEY POINTS
- Watchful waiting is appropriate for patients with mild to moderate symptoms that cause minimal bother.
- Patients with severe or bothersome symptoms should be offered pharmacotherapy, not only to improve symptoms but also to reduce the risk of disease progression.
- Several effective, minimally invasive surgical options are available for patients whose symptoms do not respond to medical therapy. These patients and those with abnormal findings on diagnostic evaluation warrant referral to a urologist for further evaluation.
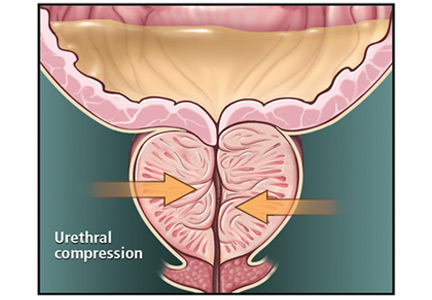
Fall risk and anticoagulation for atrial fibrillation in the elderly: A delicate balance
An 86-year-old woman with hypertension, osteoporosis, and mild cognitive impairment presents with episodes of palpitations and heart “fluttering.” These episodes occur 1 to 2 times per week, last for up to several hours, and are associated with mild shortness of breath and reduced activity tolerance. She is widowed and lives in a retirement facility, but she is independent in activities of daily living. She has fallen twice in the past year without significant injury.
Physical examination is unremarkable. An electrocardiogram demonstrates sinus rhythm with left ventricular hypertrophy. A 30-day event monitor reveals several episodes of paroxysmal atrial fibrillation that correspond with her symptoms. A subsequent echocardiogram shows normal left ventricular systolic function, mild diastolic dysfunction, and no significant valvular abnormalities. Laboratory studies, including thyroid-stimulating hormone, are normal.
What is this patient’s risk of stroke? What is her risk of major bleeding from anticoagulation? How should fall risk be addressed in the decision-making process? What other factors should be considered?
AGE, ATRIAL FIBRILLATION, AND STROKE RISK
The prevalence of atrial fibrillation increases with age, and nearly half of patients with atrial fibrillation in the United States are 75 or older.1 In addition, older age is an independent risk factor for stroke in patients with atrial fibrillation, and the proportion of strokes attributable to atrial fibrillation increases exponentially with age:
- 1.5% at age 50 to 59
- 2.8% at age 60 to 69
- 9.9% at age 70 to 79
- 23.5% at age 80 to 89.2
Numerous large randomized trials have shown that anticoagulation with warfarin reduces the risk of stroke by about two-thirds in patients with atrial fibrillation, and that this benefit extends to the elderly.
In the Birmingham Atrial Fibrillation Treatment of the Aged trial,3 973 patients at least 75 years old (mean age 81.5, 55% male) were randomized to receive either warfarin with a target international normalized ratio of 2.0 to 3.0 or aspirin 75 mg/day. Over an average follow-up of 2.7 years, the composite outcome of fatal or disabling stroke, arterial embolism, or intracranial hemorrhage occurred in 24 (4.9%) of the 488 patients in the warfarin group and 48 (9.9%) of the 485 patients in the aspirin group (absolute yearly risk reduction 2%, 95% confidence interval 0.7–3.2, number needed to treat 50 for 1 year). Importantly, the benefit of warfarin was similar in men and women, and in patients ages 75 to 79, 80 to 84, and 85 and older.
More recently, the oral anticoagulants dabigatran, rivaroxaban, apixaban, and edoxaban have been shown to be at least as effective as warfarin with respect to both stroke prevention and major bleeding complications, and subgroup analyses have confirmed similar outcomes in older and younger patients.4,5
But despite the proven value of anticoagulation for stroke prevention in older adults, only 40% to 60% of older patients who are suitable candidates for anticoagulation actually receive it.6 Moreover, the proportion of patients who are treated declines progressively with age. The most frequently cited reason for nontreatment is perception of a high risk of falls and associated concerns about bleeding, especially intracranial hemorrhage.7–10
BALANCING STROKE RISK VS BLEEDING RISK
Balancing the risk of stroke against the risk of bleeding related to falls is a commonly encountered conundrum in older patients with atrial fibrillation.
Stroke risk
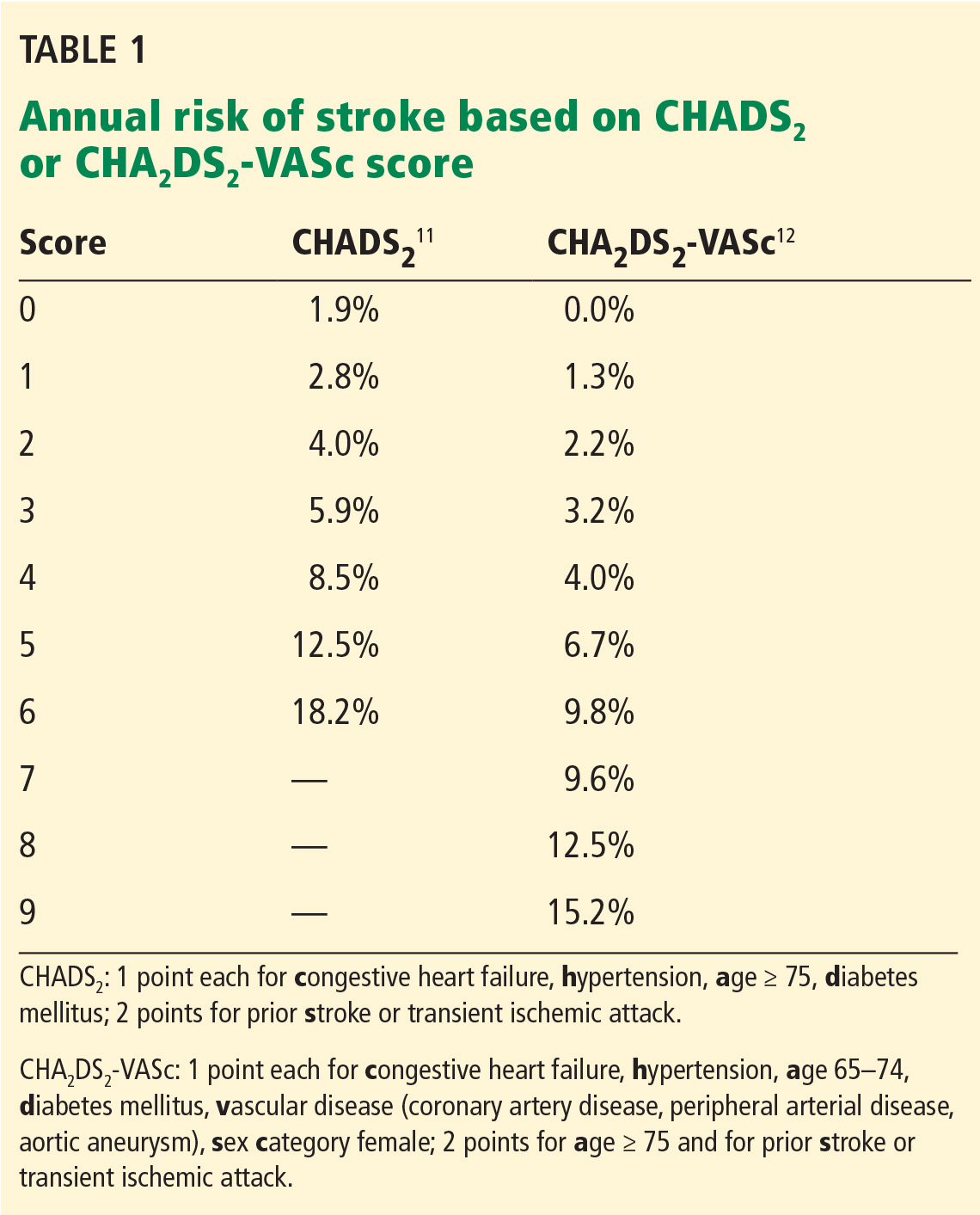
The CHADS2 score was, until recently, the most widely used method for assessing stroke risk in patients with nonvalvular atrial fibrillation. CHADS2 assigns 1 point each for congestive heart failure, hypertension, age ≥ 75, and diabetes, and 2 points for prior stroke or transient ischemic attack (range 0–6 points). Annual stroke risk based on the CHADS2 score ranges from about 2% to about 18%
(Table 1).11
The CHA2DS2-VASc score,12 a modification of CHADS2, appears to assess the risk of stroke more accurately, especially at the lower end of the scale, and recent guidelines for managing atrial fibrillation recommend using the CHA2DS2-VASc algorithm.13 CHA2DS2-VASc is similar to CHADS2, except that it assigns 1 point for ages 65 to 74, 2 points for ages 75 and older, 1 point for vascular disease (coronary artery disease, peripheral arterial disease, aortic aneurysm), and 1 point for female sex (Table 1).11,12
For both CHADS2 and CHA2DS2-VASc, systemic anticoagulation is recommended for patients who have a score of 2 or higher. Our patient’s CHADS2 score is 2, and her CHA2DS2-VASc score is 4, corresponding to an annual estimated stroke risk of 4% with both scores (Table 1). Note, however, that the CHA2DS2-VASc score provides more information at the lower end of the spectrum.
Bleeding risk
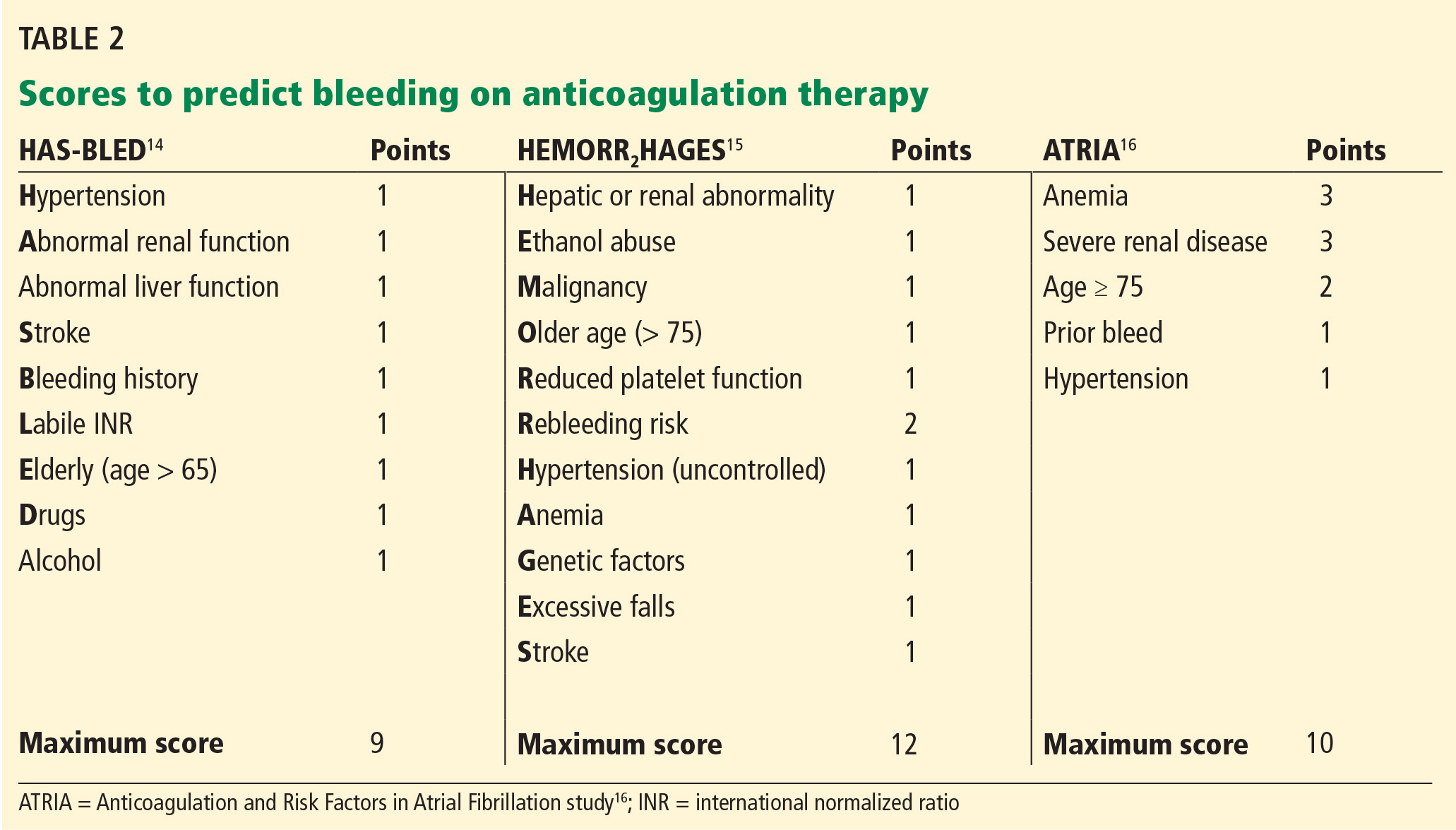
Several scoring systems for assessing bleeding risk have also been developed (Table 2).14–16 Of these, the HAS-BLED score has come to be used more widely in recent years.
Perhaps not surprisingly, some of the same factors associated with risk of stroke also predict increased risk of bleeding (eg, older age, hypertension, prior stroke).14 Note, however, that history of falling or high risk of falling is only included in one of the bleeding risk models (HEMORR2HAGES).15
These tools are somewhat limited by their lack of consideration of concomitant antiplatelet therapy (only included in HAS-BLED) or history of bleeding (only ATRIA16 considers major and minor bleeding, HEMORR2HAGES does not specify bleeding severity, and HAS-BLED only considers major bleeding). The models also fail to include medications such as antibiotics or antiarrhythmic agents, which are commonly used by older patients with atrial fibrillation and may increase bleeding risk. In addition, all bleeding risk scores were developed for warfarin, and their applicability to patients treated with the newer oral anticoagulants has not been established.
At the time of presentation, our patient has a HAS-BLED score of 2 (1 point each for age and hypertension), placing her at intermediate risk of bleeding.14
Fear the clot, not the bleed
So how does one balance the risk of stroke vs the risk of bleeding? An adage from the early days of thrombolytic therapy for acute myocardial infarction was “fear the clot, not the bleed.” In other words, in the present context the consequences of a thrombus embolizing from the heart to the brain are likely to be more devastating and more permanent than the consequences of anticoagulation-associated hemorrhage.
Support for this view is underscored by a 2015 study by Lip et al,17 who examined stroke and bleeding risks and outcomes in a large real-world population of patients age 75 and older. The analysis included 819 patients ages 85 to 89 and 386 patients age 90 and older. The key finding was that the oldest patients derived the greatest net benefit from anticoagulation.
Moreover, the Canadian stroke registry of 3,197 patients, mean age 79, showed that advanced age was a more potent risk factor for ischemic stroke than it was for hemorrhagic stroke.18
Thus, the benefit from anticoagulation in patients with atrial fibrillation does not appear to have an upper age limit.
FALLS AND ANTICOAGULATION
Falls are an important source of morbidity, disability, and activity curtailment in older adults and, like atrial fibrillation, the incidence and prevalence of falls increase with age. In community-dwelling adults age 65 and older, the overall proportion with at least 1 fall in the preceding year ranges from about 30% to 40%.19 However, the rate increases with age and exceeds 50% in nursing home residents.20
Although anticoagulation is associated with a higher risk of bleeding in patients who fall, the absolute risk is small.
In a study of older adults with nonvalvular atrial fibrillation, a history of falls or documented high risk of falling was associated with a risk of intracranial hemorrhage during follow-up that was 1.9 times higher.21 Importantly, however, this risk did not differ among patients treated with warfarin, aspirin, or no antithrombotic therapy. In this analysis, patients with a CHADS2 score of 2 or higher benefited from anticoagulation, whether or not they were considered to be at risk for falls.
In another study,22 it was estimated that an individual would have to fall 295 times in 1 year for the risk of fall-related major bleeding to outweigh the benefit of warfarin in reducing the risk of stroke.
Thus, based on available evidence, perception of a high risk of falling should not be construed as justification for withholding anticoagulation in older patients who are otherwise suitable candidates for such therapy.
AT WHAT POINT DOES BLEEDING RISK OUTWEIGH ANTICOAGULATION BENEFIT?
Absolute contraindications to anticoagulation include an intracranial hemorrhage or neurosurgical procedure with high risk for bleeding within the past 30 days, an intracranial neoplasm or vascular abnormality with high risk of bleeding, recurrent life-threatening gastrointestinal or other bleeding events, and severe bleeding disorders, including severe thrombocytopenia.
In patients with atrial fibrillation at high risk of bleeding as assessed by one of the bleeding risk scores and relatively low risk of ischemic stroke, the risk of anticoagulation may outweigh the benefit, although no studies have specifically addressed this issue.
In patients with frequent falls, including injurious falls, the benefits of anticoagulation usually outweigh the risks of bleeding, but management should incorporate interventions designed to mitigate fall risk.
Finally, in patients with a poor prognosis approaching the end of life, the risks and burdens of anticoagulation may exceed the perceived benefits, in which case discontinuation of anticoagulation may be appropriate.
SHOULD OUR PATIENT RECEIVE ANTICOAGULATION?
As noted above, our patient has a high risk of stroke and a moderate risk of bleeding, and multiple lines of evidence indicate that the benefits of anticoagulation (ie, prevention of stroke and systemic embolization) substantially outweigh the risks of bleeding. Although she has a history of falls, which may seem to muddy the waters, this factor should not play a major role in decision-making. Moreover, her advanced age should, if anything, be considered a point in favor of anticoagulation. So from the scientific standpoint, anticoagulation is the clear winner.
A shared decision
But that is not the end of the story. Since there is tension between benefits and risks with either approach (ie, anticoagulation or no anticoagulation), it is important to discuss the issues and options with the patient and relevant caregivers. Most older adults have witnessed the ravages of stroke in a friend or relative, and a recent study showed that most would be willing to accept a modest risk of bleeding to prevent a stroke.23
However, this is ultimately a personal decision for each patient, and in accordance with the principle of patient autonomy, the patient’s expressed wishes should be honored by using a process of shared decision-making.
Which anticoagulant?
Finally, what about the choice of anticoagulation? The complexities of using warfarin, including its narrow therapeutic range and myriad interactions with other medications and foods, can make it a less appealing option for both patient and provider.
We recommend a novel oral anticoagulant as first-line therapy in the absence of contraindications such as severe renal insufficiency, and prefer apixaban because it is the only agent shown to be superior to warfarin with respect to both stroke prevention and bleeding risk.24
Important disadvantages of the novel oral anticoagulants include their higher cost and lack of an effective antidote in the event of clinically significant bleeding (with the exception of idarucizumab, which was recently approved for reversal of serious bleeding associated with dabigatran), issues that may be of particular concern to older adults. While there is no therapeutic range to monitor for the newer agents, more frequent monitoring for occult anemia may be needed.
Thus, selection of an anticoagulant should also be individualized through shared decision-making.
Is aspirin alone an alternative?
And what if the patient chooses to forgo anticoagulation? In that case, aspirin 75 to 325 mg/day may seem reasonable, but there is scant evidence that aspirin is beneficial for stroke prevention in patients with atrial fibrillation in this age group, and aspirin, too, is associated with an increased risk of bleeding.25
As a result, current US and European guidelines recommend a very limited role for aspirin as a single agent in the management of atrial fibrillation.26 The joint 2014 guidelines of the American Heart Association, American College of Cardiology, and Heart Rhythm Society give aspirin a class IIB recommendation (ie, it “may” be considered), level of evidence C (ie, very limited) for use as an alternative to no antithrombotic therapy or systemic anticoagulation only in patients with a CHA2DS2-VASc score of 1, thereby excluding all patients age 75 and older.13
In most cases, aspirin as sole prophylaxis against stroke in atrial fibrillation should be avoided in the absence of another indication for its use, such as coexisting coronary artery disease or peripheral arterial disease.
A COMPLEX DECISION
In summary, the decisions surrounding anticoagulation of elderly patients with nonvalvular atrial fibrillation are complex. Accurate assessment of stroke risk is key, and although bleeding risk is also an essential consideration, it is important not to overemphasize bleeding and fall risks in the decision-making process.
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285:2370–2375.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991; 22:983–988.
- Mant J, Hobbs FD, Fletcher K, et al; BAFTA investigators; Midland Research Practices Network (MidReC). Warfarin versus aspirin for stroke prevention in an elderly community population with atrial fibrillation (the Birmingham Atrial Fibrillation Treatment of the Aged Study, BAFTA): a randomised controlled trial. Lancet 2007; 370:493–503.
- Chatterjee S, Sardar P, Biondi-Zoccai G, Kumbhani DJ. New oral anticoagulants and the risk of intracranial hemorrhage: traditional and Bayesian meta-analysis and mixed treatment comparison of randomized trials of new oral anticoagulants in atrial fibrillation. JAMA Neurology 2013; 70:1486–1490.
- Sardar P, Chatterjee S, Chaudhari S, Lip GY. New oral anticoagulants in elderly adults: evidence from a meta-analysis of randomized trials. J Am Geriatr Soc 2014; 62:857–864.
- Rich MW. Atrial fibrillation in long term care. J Am Med Dir Assoc 2012; 13:688–691.
- McCrory DC, Matchar DB, Samsa G, Sanders LL, Pritchett EL. Physician attitudes about anticoagulation for nonvalvular atrial fibrillation in the elderly. Arch Intern Med 1995; 155:277–281.
- Pugh D, Pugh J, Mead GE. Attitudes of physicians regarding anticoagulation for atrial fibrillation: a systematic review. Age Ageing 2011; 40:675–683.
- Sellers MB, Newby LK. Atrial fibrillation, anticoagulation, fall risk, and outcomes in elderly patients. Am Heart J 2011; 161:241–246.
- Bahri O, Roca F, Lechani T, et al. Underuse of oral anticoagulation for individuals with atrial fibrillation in a nursing home setting in France: comparisons of resident characteristics and physician attitude. J Am Geriatr Soc 2015; 63:71–76.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the Euro Heart Survey on Atrial Fibrillation. Chest 2010; 137:263–272.
- January CT, Wann LS, Alpert JS, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2014; 64:e1–e76.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Gage BF, Yan Y, Milligan PE, et al. Clinical classification schemes for predicting hemorrhage: results from the National Registry of Atrial Fibrillation (NRAF). Am Heart J 2006; 151:713–719.
- Fang MC, Go AS, Chang Y, et al. A new risk scheme to predict warfarin-associated hemorrhage: The ATRIA (Anticoagulation and Risk Factors in Atrial Fibrillation) Study. J Am Coll Cardiol 2011; 58:395–401.
- Lip GY, Clementy N, Pericart L, Banerjee A, Fauchier L. Stroke and major bleeding risk in elderly patients aged ≥ 75 years with atrial fibrillation: the Loire Valley atrial fibrillation project. Stroke 2015; 46:143–150.
- McGrath ER, Kapral MK, Fang J, et al; Investigators of the Registry of the Canadian Stroke Network. Which risk factors are more associated with ischemic stroke than intracerebral hemorrhage in patients with atrial fibrillation? Stroke 2012; 43:2048–2054.
- Phelan EA, Mahoney JE, Voit JC, Stevens JA. Assessment and management of fall risk in primary care settings. Med Clin North Am 2015; 99:281–293.
- Deandrea S, Bravi F, Turati F, Lucenteforte E, La Vecchia C, Negri E. Risk factors for falls in older people in nursing homes and hospitals. A systematic review and meta-analysis. Arch Gerontol Geriatr 2013; 56:407–415.
- Gage BF, Birman-Deych E, Kerzner R, Radford MJ, Nilasena DS, Rich MW. Incidence of intracranial hemorrhage in patients with atrial fibrillation who are prone to fall. Am J Med 2005; 118:612–617.
- Man-Son-Hing M, Nichol G, Lau A, Laupacis A. Choosing antithrombotic therapy for elderly patients with atrial fibrillation who are at risk for falls. Arch Intern Med 1999; 159:677–685.
- Riva N, Smith DE, Lip GY, Lane DA. Advancing age and bleeding risk are the strongest barriers to anticoagulant prescription in atrial fibrillation. Age Ageing 2011; 40:653–655.
- De Caterina R, Andersson U, Alexander JH, et al; ARISTOTLE Investigators. History of bleeding and outcomes with apixaban versus warfarin in patients with atrial fibrillation in the Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation trial. Am Heart J 2016; 175:175–183.
- Ben Freedman S, Gersh BJ, Lip GY. Misperceptions of aspirin efficacy and safety may perpetuate anticoagulant underutilization in atrial fibrillation. Eur Heart J 2015; 36:653–656.
- Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J 2012; 33:2719–2747.
An 86-year-old woman with hypertension, osteoporosis, and mild cognitive impairment presents with episodes of palpitations and heart “fluttering.” These episodes occur 1 to 2 times per week, last for up to several hours, and are associated with mild shortness of breath and reduced activity tolerance. She is widowed and lives in a retirement facility, but she is independent in activities of daily living. She has fallen twice in the past year without significant injury.
Physical examination is unremarkable. An electrocardiogram demonstrates sinus rhythm with left ventricular hypertrophy. A 30-day event monitor reveals several episodes of paroxysmal atrial fibrillation that correspond with her symptoms. A subsequent echocardiogram shows normal left ventricular systolic function, mild diastolic dysfunction, and no significant valvular abnormalities. Laboratory studies, including thyroid-stimulating hormone, are normal.
What is this patient’s risk of stroke? What is her risk of major bleeding from anticoagulation? How should fall risk be addressed in the decision-making process? What other factors should be considered?
AGE, ATRIAL FIBRILLATION, AND STROKE RISK
The prevalence of atrial fibrillation increases with age, and nearly half of patients with atrial fibrillation in the United States are 75 or older.1 In addition, older age is an independent risk factor for stroke in patients with atrial fibrillation, and the proportion of strokes attributable to atrial fibrillation increases exponentially with age:
- 1.5% at age 50 to 59
- 2.8% at age 60 to 69
- 9.9% at age 70 to 79
- 23.5% at age 80 to 89.2
Numerous large randomized trials have shown that anticoagulation with warfarin reduces the risk of stroke by about two-thirds in patients with atrial fibrillation, and that this benefit extends to the elderly.
In the Birmingham Atrial Fibrillation Treatment of the Aged trial,3 973 patients at least 75 years old (mean age 81.5, 55% male) were randomized to receive either warfarin with a target international normalized ratio of 2.0 to 3.0 or aspirin 75 mg/day. Over an average follow-up of 2.7 years, the composite outcome of fatal or disabling stroke, arterial embolism, or intracranial hemorrhage occurred in 24 (4.9%) of the 488 patients in the warfarin group and 48 (9.9%) of the 485 patients in the aspirin group (absolute yearly risk reduction 2%, 95% confidence interval 0.7–3.2, number needed to treat 50 for 1 year). Importantly, the benefit of warfarin was similar in men and women, and in patients ages 75 to 79, 80 to 84, and 85 and older.
More recently, the oral anticoagulants dabigatran, rivaroxaban, apixaban, and edoxaban have been shown to be at least as effective as warfarin with respect to both stroke prevention and major bleeding complications, and subgroup analyses have confirmed similar outcomes in older and younger patients.4,5
But despite the proven value of anticoagulation for stroke prevention in older adults, only 40% to 60% of older patients who are suitable candidates for anticoagulation actually receive it.6 Moreover, the proportion of patients who are treated declines progressively with age. The most frequently cited reason for nontreatment is perception of a high risk of falls and associated concerns about bleeding, especially intracranial hemorrhage.7–10
BALANCING STROKE RISK VS BLEEDING RISK
Balancing the risk of stroke against the risk of bleeding related to falls is a commonly encountered conundrum in older patients with atrial fibrillation.
Stroke risk
The CHADS2 score was, until recently, the most widely used method for assessing stroke risk in patients with nonvalvular atrial fibrillation. CHADS2 assigns 1 point each for congestive heart failure, hypertension, age ≥ 75, and diabetes, and 2 points for prior stroke or transient ischemic attack (range 0–6 points). Annual stroke risk based on the CHADS2 score ranges from about 2% to about 18%
(Table 1).11
The CHA2DS2-VASc score,12 a modification of CHADS2, appears to assess the risk of stroke more accurately, especially at the lower end of the scale, and recent guidelines for managing atrial fibrillation recommend using the CHA2DS2-VASc algorithm.13 CHA2DS2-VASc is similar to CHADS2, except that it assigns 1 point for ages 65 to 74, 2 points for ages 75 and older, 1 point for vascular disease (coronary artery disease, peripheral arterial disease, aortic aneurysm), and 1 point for female sex (Table 1).11,12
For both CHADS2 and CHA2DS2-VASc, systemic anticoagulation is recommended for patients who have a score of 2 or higher. Our patient’s CHADS2 score is 2, and her CHA2DS2-VASc score is 4, corresponding to an annual estimated stroke risk of 4% with both scores (Table 1). Note, however, that the CHA2DS2-VASc score provides more information at the lower end of the spectrum.
Bleeding risk
Several scoring systems for assessing bleeding risk have also been developed (Table 2).14–16 Of these, the HAS-BLED score has come to be used more widely in recent years.
Perhaps not surprisingly, some of the same factors associated with risk of stroke also predict increased risk of bleeding (eg, older age, hypertension, prior stroke).14 Note, however, that history of falling or high risk of falling is only included in one of the bleeding risk models (HEMORR2HAGES).15
These tools are somewhat limited by their lack of consideration of concomitant antiplatelet therapy (only included in HAS-BLED) or history of bleeding (only ATRIA16 considers major and minor bleeding, HEMORR2HAGES does not specify bleeding severity, and HAS-BLED only considers major bleeding). The models also fail to include medications such as antibiotics or antiarrhythmic agents, which are commonly used by older patients with atrial fibrillation and may increase bleeding risk. In addition, all bleeding risk scores were developed for warfarin, and their applicability to patients treated with the newer oral anticoagulants has not been established.
At the time of presentation, our patient has a HAS-BLED score of 2 (1 point each for age and hypertension), placing her at intermediate risk of bleeding.14
Fear the clot, not the bleed
So how does one balance the risk of stroke vs the risk of bleeding? An adage from the early days of thrombolytic therapy for acute myocardial infarction was “fear the clot, not the bleed.” In other words, in the present context the consequences of a thrombus embolizing from the heart to the brain are likely to be more devastating and more permanent than the consequences of anticoagulation-associated hemorrhage.
Support for this view is underscored by a 2015 study by Lip et al,17 who examined stroke and bleeding risks and outcomes in a large real-world population of patients age 75 and older. The analysis included 819 patients ages 85 to 89 and 386 patients age 90 and older. The key finding was that the oldest patients derived the greatest net benefit from anticoagulation.
Moreover, the Canadian stroke registry of 3,197 patients, mean age 79, showed that advanced age was a more potent risk factor for ischemic stroke than it was for hemorrhagic stroke.18
Thus, the benefit from anticoagulation in patients with atrial fibrillation does not appear to have an upper age limit.
FALLS AND ANTICOAGULATION
Falls are an important source of morbidity, disability, and activity curtailment in older adults and, like atrial fibrillation, the incidence and prevalence of falls increase with age. In community-dwelling adults age 65 and older, the overall proportion with at least 1 fall in the preceding year ranges from about 30% to 40%.19 However, the rate increases with age and exceeds 50% in nursing home residents.20
Although anticoagulation is associated with a higher risk of bleeding in patients who fall, the absolute risk is small.
In a study of older adults with nonvalvular atrial fibrillation, a history of falls or documented high risk of falling was associated with a risk of intracranial hemorrhage during follow-up that was 1.9 times higher.21 Importantly, however, this risk did not differ among patients treated with warfarin, aspirin, or no antithrombotic therapy. In this analysis, patients with a CHADS2 score of 2 or higher benefited from anticoagulation, whether or not they were considered to be at risk for falls.
In another study,22 it was estimated that an individual would have to fall 295 times in 1 year for the risk of fall-related major bleeding to outweigh the benefit of warfarin in reducing the risk of stroke.
Thus, based on available evidence, perception of a high risk of falling should not be construed as justification for withholding anticoagulation in older patients who are otherwise suitable candidates for such therapy.
AT WHAT POINT DOES BLEEDING RISK OUTWEIGH ANTICOAGULATION BENEFIT?
Absolute contraindications to anticoagulation include an intracranial hemorrhage or neurosurgical procedure with high risk for bleeding within the past 30 days, an intracranial neoplasm or vascular abnormality with high risk of bleeding, recurrent life-threatening gastrointestinal or other bleeding events, and severe bleeding disorders, including severe thrombocytopenia.
In patients with atrial fibrillation at high risk of bleeding as assessed by one of the bleeding risk scores and relatively low risk of ischemic stroke, the risk of anticoagulation may outweigh the benefit, although no studies have specifically addressed this issue.
In patients with frequent falls, including injurious falls, the benefits of anticoagulation usually outweigh the risks of bleeding, but management should incorporate interventions designed to mitigate fall risk.
Finally, in patients with a poor prognosis approaching the end of life, the risks and burdens of anticoagulation may exceed the perceived benefits, in which case discontinuation of anticoagulation may be appropriate.
SHOULD OUR PATIENT RECEIVE ANTICOAGULATION?
As noted above, our patient has a high risk of stroke and a moderate risk of bleeding, and multiple lines of evidence indicate that the benefits of anticoagulation (ie, prevention of stroke and systemic embolization) substantially outweigh the risks of bleeding. Although she has a history of falls, which may seem to muddy the waters, this factor should not play a major role in decision-making. Moreover, her advanced age should, if anything, be considered a point in favor of anticoagulation. So from the scientific standpoint, anticoagulation is the clear winner.
A shared decision
But that is not the end of the story. Since there is tension between benefits and risks with either approach (ie, anticoagulation or no anticoagulation), it is important to discuss the issues and options with the patient and relevant caregivers. Most older adults have witnessed the ravages of stroke in a friend or relative, and a recent study showed that most would be willing to accept a modest risk of bleeding to prevent a stroke.23
However, this is ultimately a personal decision for each patient, and in accordance with the principle of patient autonomy, the patient’s expressed wishes should be honored by using a process of shared decision-making.
Which anticoagulant?
Finally, what about the choice of anticoagulation? The complexities of using warfarin, including its narrow therapeutic range and myriad interactions with other medications and foods, can make it a less appealing option for both patient and provider.
We recommend a novel oral anticoagulant as first-line therapy in the absence of contraindications such as severe renal insufficiency, and prefer apixaban because it is the only agent shown to be superior to warfarin with respect to both stroke prevention and bleeding risk.24
Important disadvantages of the novel oral anticoagulants include their higher cost and lack of an effective antidote in the event of clinically significant bleeding (with the exception of idarucizumab, which was recently approved for reversal of serious bleeding associated with dabigatran), issues that may be of particular concern to older adults. While there is no therapeutic range to monitor for the newer agents, more frequent monitoring for occult anemia may be needed.
Thus, selection of an anticoagulant should also be individualized through shared decision-making.
Is aspirin alone an alternative?
And what if the patient chooses to forgo anticoagulation? In that case, aspirin 75 to 325 mg/day may seem reasonable, but there is scant evidence that aspirin is beneficial for stroke prevention in patients with atrial fibrillation in this age group, and aspirin, too, is associated with an increased risk of bleeding.25
As a result, current US and European guidelines recommend a very limited role for aspirin as a single agent in the management of atrial fibrillation.26 The joint 2014 guidelines of the American Heart Association, American College of Cardiology, and Heart Rhythm Society give aspirin a class IIB recommendation (ie, it “may” be considered), level of evidence C (ie, very limited) for use as an alternative to no antithrombotic therapy or systemic anticoagulation only in patients with a CHA2DS2-VASc score of 1, thereby excluding all patients age 75 and older.13
In most cases, aspirin as sole prophylaxis against stroke in atrial fibrillation should be avoided in the absence of another indication for its use, such as coexisting coronary artery disease or peripheral arterial disease.
A COMPLEX DECISION
In summary, the decisions surrounding anticoagulation of elderly patients with nonvalvular atrial fibrillation are complex. Accurate assessment of stroke risk is key, and although bleeding risk is also an essential consideration, it is important not to overemphasize bleeding and fall risks in the decision-making process.
An 86-year-old woman with hypertension, osteoporosis, and mild cognitive impairment presents with episodes of palpitations and heart “fluttering.” These episodes occur 1 to 2 times per week, last for up to several hours, and are associated with mild shortness of breath and reduced activity tolerance. She is widowed and lives in a retirement facility, but she is independent in activities of daily living. She has fallen twice in the past year without significant injury.
Physical examination is unremarkable. An electrocardiogram demonstrates sinus rhythm with left ventricular hypertrophy. A 30-day event monitor reveals several episodes of paroxysmal atrial fibrillation that correspond with her symptoms. A subsequent echocardiogram shows normal left ventricular systolic function, mild diastolic dysfunction, and no significant valvular abnormalities. Laboratory studies, including thyroid-stimulating hormone, are normal.
What is this patient’s risk of stroke? What is her risk of major bleeding from anticoagulation? How should fall risk be addressed in the decision-making process? What other factors should be considered?
AGE, ATRIAL FIBRILLATION, AND STROKE RISK
The prevalence of atrial fibrillation increases with age, and nearly half of patients with atrial fibrillation in the United States are 75 or older.1 In addition, older age is an independent risk factor for stroke in patients with atrial fibrillation, and the proportion of strokes attributable to atrial fibrillation increases exponentially with age:
- 1.5% at age 50 to 59
- 2.8% at age 60 to 69
- 9.9% at age 70 to 79
- 23.5% at age 80 to 89.2
Numerous large randomized trials have shown that anticoagulation with warfarin reduces the risk of stroke by about two-thirds in patients with atrial fibrillation, and that this benefit extends to the elderly.
In the Birmingham Atrial Fibrillation Treatment of the Aged trial,3 973 patients at least 75 years old (mean age 81.5, 55% male) were randomized to receive either warfarin with a target international normalized ratio of 2.0 to 3.0 or aspirin 75 mg/day. Over an average follow-up of 2.7 years, the composite outcome of fatal or disabling stroke, arterial embolism, or intracranial hemorrhage occurred in 24 (4.9%) of the 488 patients in the warfarin group and 48 (9.9%) of the 485 patients in the aspirin group (absolute yearly risk reduction 2%, 95% confidence interval 0.7–3.2, number needed to treat 50 for 1 year). Importantly, the benefit of warfarin was similar in men and women, and in patients ages 75 to 79, 80 to 84, and 85 and older.
More recently, the oral anticoagulants dabigatran, rivaroxaban, apixaban, and edoxaban have been shown to be at least as effective as warfarin with respect to both stroke prevention and major bleeding complications, and subgroup analyses have confirmed similar outcomes in older and younger patients.4,5
But despite the proven value of anticoagulation for stroke prevention in older adults, only 40% to 60% of older patients who are suitable candidates for anticoagulation actually receive it.6 Moreover, the proportion of patients who are treated declines progressively with age. The most frequently cited reason for nontreatment is perception of a high risk of falls and associated concerns about bleeding, especially intracranial hemorrhage.7–10
BALANCING STROKE RISK VS BLEEDING RISK
Balancing the risk of stroke against the risk of bleeding related to falls is a commonly encountered conundrum in older patients with atrial fibrillation.
Stroke risk
The CHADS2 score was, until recently, the most widely used method for assessing stroke risk in patients with nonvalvular atrial fibrillation. CHADS2 assigns 1 point each for congestive heart failure, hypertension, age ≥ 75, and diabetes, and 2 points for prior stroke or transient ischemic attack (range 0–6 points). Annual stroke risk based on the CHADS2 score ranges from about 2% to about 18%
(Table 1).11
The CHA2DS2-VASc score,12 a modification of CHADS2, appears to assess the risk of stroke more accurately, especially at the lower end of the scale, and recent guidelines for managing atrial fibrillation recommend using the CHA2DS2-VASc algorithm.13 CHA2DS2-VASc is similar to CHADS2, except that it assigns 1 point for ages 65 to 74, 2 points for ages 75 and older, 1 point for vascular disease (coronary artery disease, peripheral arterial disease, aortic aneurysm), and 1 point for female sex (Table 1).11,12
For both CHADS2 and CHA2DS2-VASc, systemic anticoagulation is recommended for patients who have a score of 2 or higher. Our patient’s CHADS2 score is 2, and her CHA2DS2-VASc score is 4, corresponding to an annual estimated stroke risk of 4% with both scores (Table 1). Note, however, that the CHA2DS2-VASc score provides more information at the lower end of the spectrum.
Bleeding risk
Several scoring systems for assessing bleeding risk have also been developed (Table 2).14–16 Of these, the HAS-BLED score has come to be used more widely in recent years.
Perhaps not surprisingly, some of the same factors associated with risk of stroke also predict increased risk of bleeding (eg, older age, hypertension, prior stroke).14 Note, however, that history of falling or high risk of falling is only included in one of the bleeding risk models (HEMORR2HAGES).15
These tools are somewhat limited by their lack of consideration of concomitant antiplatelet therapy (only included in HAS-BLED) or history of bleeding (only ATRIA16 considers major and minor bleeding, HEMORR2HAGES does not specify bleeding severity, and HAS-BLED only considers major bleeding). The models also fail to include medications such as antibiotics or antiarrhythmic agents, which are commonly used by older patients with atrial fibrillation and may increase bleeding risk. In addition, all bleeding risk scores were developed for warfarin, and their applicability to patients treated with the newer oral anticoagulants has not been established.
At the time of presentation, our patient has a HAS-BLED score of 2 (1 point each for age and hypertension), placing her at intermediate risk of bleeding.14
Fear the clot, not the bleed
So how does one balance the risk of stroke vs the risk of bleeding? An adage from the early days of thrombolytic therapy for acute myocardial infarction was “fear the clot, not the bleed.” In other words, in the present context the consequences of a thrombus embolizing from the heart to the brain are likely to be more devastating and more permanent than the consequences of anticoagulation-associated hemorrhage.
Support for this view is underscored by a 2015 study by Lip et al,17 who examined stroke and bleeding risks and outcomes in a large real-world population of patients age 75 and older. The analysis included 819 patients ages 85 to 89 and 386 patients age 90 and older. The key finding was that the oldest patients derived the greatest net benefit from anticoagulation.
Moreover, the Canadian stroke registry of 3,197 patients, mean age 79, showed that advanced age was a more potent risk factor for ischemic stroke than it was for hemorrhagic stroke.18
Thus, the benefit from anticoagulation in patients with atrial fibrillation does not appear to have an upper age limit.
FALLS AND ANTICOAGULATION
Falls are an important source of morbidity, disability, and activity curtailment in older adults and, like atrial fibrillation, the incidence and prevalence of falls increase with age. In community-dwelling adults age 65 and older, the overall proportion with at least 1 fall in the preceding year ranges from about 30% to 40%.19 However, the rate increases with age and exceeds 50% in nursing home residents.20
Although anticoagulation is associated with a higher risk of bleeding in patients who fall, the absolute risk is small.
In a study of older adults with nonvalvular atrial fibrillation, a history of falls or documented high risk of falling was associated with a risk of intracranial hemorrhage during follow-up that was 1.9 times higher.21 Importantly, however, this risk did not differ among patients treated with warfarin, aspirin, or no antithrombotic therapy. In this analysis, patients with a CHADS2 score of 2 or higher benefited from anticoagulation, whether or not they were considered to be at risk for falls.
In another study,22 it was estimated that an individual would have to fall 295 times in 1 year for the risk of fall-related major bleeding to outweigh the benefit of warfarin in reducing the risk of stroke.
Thus, based on available evidence, perception of a high risk of falling should not be construed as justification for withholding anticoagulation in older patients who are otherwise suitable candidates for such therapy.
AT WHAT POINT DOES BLEEDING RISK OUTWEIGH ANTICOAGULATION BENEFIT?
Absolute contraindications to anticoagulation include an intracranial hemorrhage or neurosurgical procedure with high risk for bleeding within the past 30 days, an intracranial neoplasm or vascular abnormality with high risk of bleeding, recurrent life-threatening gastrointestinal or other bleeding events, and severe bleeding disorders, including severe thrombocytopenia.
In patients with atrial fibrillation at high risk of bleeding as assessed by one of the bleeding risk scores and relatively low risk of ischemic stroke, the risk of anticoagulation may outweigh the benefit, although no studies have specifically addressed this issue.
In patients with frequent falls, including injurious falls, the benefits of anticoagulation usually outweigh the risks of bleeding, but management should incorporate interventions designed to mitigate fall risk.
Finally, in patients with a poor prognosis approaching the end of life, the risks and burdens of anticoagulation may exceed the perceived benefits, in which case discontinuation of anticoagulation may be appropriate.
SHOULD OUR PATIENT RECEIVE ANTICOAGULATION?
As noted above, our patient has a high risk of stroke and a moderate risk of bleeding, and multiple lines of evidence indicate that the benefits of anticoagulation (ie, prevention of stroke and systemic embolization) substantially outweigh the risks of bleeding. Although she has a history of falls, which may seem to muddy the waters, this factor should not play a major role in decision-making. Moreover, her advanced age should, if anything, be considered a point in favor of anticoagulation. So from the scientific standpoint, anticoagulation is the clear winner.
A shared decision
But that is not the end of the story. Since there is tension between benefits and risks with either approach (ie, anticoagulation or no anticoagulation), it is important to discuss the issues and options with the patient and relevant caregivers. Most older adults have witnessed the ravages of stroke in a friend or relative, and a recent study showed that most would be willing to accept a modest risk of bleeding to prevent a stroke.23
However, this is ultimately a personal decision for each patient, and in accordance with the principle of patient autonomy, the patient’s expressed wishes should be honored by using a process of shared decision-making.
Which anticoagulant?
Finally, what about the choice of anticoagulation? The complexities of using warfarin, including its narrow therapeutic range and myriad interactions with other medications and foods, can make it a less appealing option for both patient and provider.
We recommend a novel oral anticoagulant as first-line therapy in the absence of contraindications such as severe renal insufficiency, and prefer apixaban because it is the only agent shown to be superior to warfarin with respect to both stroke prevention and bleeding risk.24
Important disadvantages of the novel oral anticoagulants include their higher cost and lack of an effective antidote in the event of clinically significant bleeding (with the exception of idarucizumab, which was recently approved for reversal of serious bleeding associated with dabigatran), issues that may be of particular concern to older adults. While there is no therapeutic range to monitor for the newer agents, more frequent monitoring for occult anemia may be needed.
Thus, selection of an anticoagulant should also be individualized through shared decision-making.
Is aspirin alone an alternative?
And what if the patient chooses to forgo anticoagulation? In that case, aspirin 75 to 325 mg/day may seem reasonable, but there is scant evidence that aspirin is beneficial for stroke prevention in patients with atrial fibrillation in this age group, and aspirin, too, is associated with an increased risk of bleeding.25
As a result, current US and European guidelines recommend a very limited role for aspirin as a single agent in the management of atrial fibrillation.26 The joint 2014 guidelines of the American Heart Association, American College of Cardiology, and Heart Rhythm Society give aspirin a class IIB recommendation (ie, it “may” be considered), level of evidence C (ie, very limited) for use as an alternative to no antithrombotic therapy or systemic anticoagulation only in patients with a CHA2DS2-VASc score of 1, thereby excluding all patients age 75 and older.13
In most cases, aspirin as sole prophylaxis against stroke in atrial fibrillation should be avoided in the absence of another indication for its use, such as coexisting coronary artery disease or peripheral arterial disease.
A COMPLEX DECISION
In summary, the decisions surrounding anticoagulation of elderly patients with nonvalvular atrial fibrillation are complex. Accurate assessment of stroke risk is key, and although bleeding risk is also an essential consideration, it is important not to overemphasize bleeding and fall risks in the decision-making process.
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285:2370–2375.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991; 22:983–988.
- Mant J, Hobbs FD, Fletcher K, et al; BAFTA investigators; Midland Research Practices Network (MidReC). Warfarin versus aspirin for stroke prevention in an elderly community population with atrial fibrillation (the Birmingham Atrial Fibrillation Treatment of the Aged Study, BAFTA): a randomised controlled trial. Lancet 2007; 370:493–503.
- Chatterjee S, Sardar P, Biondi-Zoccai G, Kumbhani DJ. New oral anticoagulants and the risk of intracranial hemorrhage: traditional and Bayesian meta-analysis and mixed treatment comparison of randomized trials of new oral anticoagulants in atrial fibrillation. JAMA Neurology 2013; 70:1486–1490.
- Sardar P, Chatterjee S, Chaudhari S, Lip GY. New oral anticoagulants in elderly adults: evidence from a meta-analysis of randomized trials. J Am Geriatr Soc 2014; 62:857–864.
- Rich MW. Atrial fibrillation in long term care. J Am Med Dir Assoc 2012; 13:688–691.
- McCrory DC, Matchar DB, Samsa G, Sanders LL, Pritchett EL. Physician attitudes about anticoagulation for nonvalvular atrial fibrillation in the elderly. Arch Intern Med 1995; 155:277–281.
- Pugh D, Pugh J, Mead GE. Attitudes of physicians regarding anticoagulation for atrial fibrillation: a systematic review. Age Ageing 2011; 40:675–683.
- Sellers MB, Newby LK. Atrial fibrillation, anticoagulation, fall risk, and outcomes in elderly patients. Am Heart J 2011; 161:241–246.
- Bahri O, Roca F, Lechani T, et al. Underuse of oral anticoagulation for individuals with atrial fibrillation in a nursing home setting in France: comparisons of resident characteristics and physician attitude. J Am Geriatr Soc 2015; 63:71–76.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the Euro Heart Survey on Atrial Fibrillation. Chest 2010; 137:263–272.
- January CT, Wann LS, Alpert JS, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2014; 64:e1–e76.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Gage BF, Yan Y, Milligan PE, et al. Clinical classification schemes for predicting hemorrhage: results from the National Registry of Atrial Fibrillation (NRAF). Am Heart J 2006; 151:713–719.
- Fang MC, Go AS, Chang Y, et al. A new risk scheme to predict warfarin-associated hemorrhage: The ATRIA (Anticoagulation and Risk Factors in Atrial Fibrillation) Study. J Am Coll Cardiol 2011; 58:395–401.
- Lip GY, Clementy N, Pericart L, Banerjee A, Fauchier L. Stroke and major bleeding risk in elderly patients aged ≥ 75 years with atrial fibrillation: the Loire Valley atrial fibrillation project. Stroke 2015; 46:143–150.
- McGrath ER, Kapral MK, Fang J, et al; Investigators of the Registry of the Canadian Stroke Network. Which risk factors are more associated with ischemic stroke than intracerebral hemorrhage in patients with atrial fibrillation? Stroke 2012; 43:2048–2054.
- Phelan EA, Mahoney JE, Voit JC, Stevens JA. Assessment and management of fall risk in primary care settings. Med Clin North Am 2015; 99:281–293.
- Deandrea S, Bravi F, Turati F, Lucenteforte E, La Vecchia C, Negri E. Risk factors for falls in older people in nursing homes and hospitals. A systematic review and meta-analysis. Arch Gerontol Geriatr 2013; 56:407–415.
- Gage BF, Birman-Deych E, Kerzner R, Radford MJ, Nilasena DS, Rich MW. Incidence of intracranial hemorrhage in patients with atrial fibrillation who are prone to fall. Am J Med 2005; 118:612–617.
- Man-Son-Hing M, Nichol G, Lau A, Laupacis A. Choosing antithrombotic therapy for elderly patients with atrial fibrillation who are at risk for falls. Arch Intern Med 1999; 159:677–685.
- Riva N, Smith DE, Lip GY, Lane DA. Advancing age and bleeding risk are the strongest barriers to anticoagulant prescription in atrial fibrillation. Age Ageing 2011; 40:653–655.
- De Caterina R, Andersson U, Alexander JH, et al; ARISTOTLE Investigators. History of bleeding and outcomes with apixaban versus warfarin in patients with atrial fibrillation in the Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation trial. Am Heart J 2016; 175:175–183.
- Ben Freedman S, Gersh BJ, Lip GY. Misperceptions of aspirin efficacy and safety may perpetuate anticoagulant underutilization in atrial fibrillation. Eur Heart J 2015; 36:653–656.
- Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J 2012; 33:2719–2747.
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285:2370–2375.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991; 22:983–988.
- Mant J, Hobbs FD, Fletcher K, et al; BAFTA investigators; Midland Research Practices Network (MidReC). Warfarin versus aspirin for stroke prevention in an elderly community population with atrial fibrillation (the Birmingham Atrial Fibrillation Treatment of the Aged Study, BAFTA): a randomised controlled trial. Lancet 2007; 370:493–503.
- Chatterjee S, Sardar P, Biondi-Zoccai G, Kumbhani DJ. New oral anticoagulants and the risk of intracranial hemorrhage: traditional and Bayesian meta-analysis and mixed treatment comparison of randomized trials of new oral anticoagulants in atrial fibrillation. JAMA Neurology 2013; 70:1486–1490.
- Sardar P, Chatterjee S, Chaudhari S, Lip GY. New oral anticoagulants in elderly adults: evidence from a meta-analysis of randomized trials. J Am Geriatr Soc 2014; 62:857–864.
- Rich MW. Atrial fibrillation in long term care. J Am Med Dir Assoc 2012; 13:688–691.
- McCrory DC, Matchar DB, Samsa G, Sanders LL, Pritchett EL. Physician attitudes about anticoagulation for nonvalvular atrial fibrillation in the elderly. Arch Intern Med 1995; 155:277–281.
- Pugh D, Pugh J, Mead GE. Attitudes of physicians regarding anticoagulation for atrial fibrillation: a systematic review. Age Ageing 2011; 40:675–683.
- Sellers MB, Newby LK. Atrial fibrillation, anticoagulation, fall risk, and outcomes in elderly patients. Am Heart J 2011; 161:241–246.
- Bahri O, Roca F, Lechani T, et al. Underuse of oral anticoagulation for individuals with atrial fibrillation in a nursing home setting in France: comparisons of resident characteristics and physician attitude. J Am Geriatr Soc 2015; 63:71–76.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA 2001; 285:2864–2870.
- Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: the Euro Heart Survey on Atrial Fibrillation. Chest 2010; 137:263–272.
- January CT, Wann LS, Alpert JS, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2014; 64:e1–e76.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Gage BF, Yan Y, Milligan PE, et al. Clinical classification schemes for predicting hemorrhage: results from the National Registry of Atrial Fibrillation (NRAF). Am Heart J 2006; 151:713–719.
- Fang MC, Go AS, Chang Y, et al. A new risk scheme to predict warfarin-associated hemorrhage: The ATRIA (Anticoagulation and Risk Factors in Atrial Fibrillation) Study. J Am Coll Cardiol 2011; 58:395–401.
- Lip GY, Clementy N, Pericart L, Banerjee A, Fauchier L. Stroke and major bleeding risk in elderly patients aged ≥ 75 years with atrial fibrillation: the Loire Valley atrial fibrillation project. Stroke 2015; 46:143–150.
- McGrath ER, Kapral MK, Fang J, et al; Investigators of the Registry of the Canadian Stroke Network. Which risk factors are more associated with ischemic stroke than intracerebral hemorrhage in patients with atrial fibrillation? Stroke 2012; 43:2048–2054.
- Phelan EA, Mahoney JE, Voit JC, Stevens JA. Assessment and management of fall risk in primary care settings. Med Clin North Am 2015; 99:281–293.
- Deandrea S, Bravi F, Turati F, Lucenteforte E, La Vecchia C, Negri E. Risk factors for falls in older people in nursing homes and hospitals. A systematic review and meta-analysis. Arch Gerontol Geriatr 2013; 56:407–415.
- Gage BF, Birman-Deych E, Kerzner R, Radford MJ, Nilasena DS, Rich MW. Incidence of intracranial hemorrhage in patients with atrial fibrillation who are prone to fall. Am J Med 2005; 118:612–617.
- Man-Son-Hing M, Nichol G, Lau A, Laupacis A. Choosing antithrombotic therapy for elderly patients with atrial fibrillation who are at risk for falls. Arch Intern Med 1999; 159:677–685.
- Riva N, Smith DE, Lip GY, Lane DA. Advancing age and bleeding risk are the strongest barriers to anticoagulant prescription in atrial fibrillation. Age Ageing 2011; 40:653–655.
- De Caterina R, Andersson U, Alexander JH, et al; ARISTOTLE Investigators. History of bleeding and outcomes with apixaban versus warfarin in patients with atrial fibrillation in the Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation trial. Am Heart J 2016; 175:175–183.
- Ben Freedman S, Gersh BJ, Lip GY. Misperceptions of aspirin efficacy and safety may perpetuate anticoagulant underutilization in atrial fibrillation. Eur Heart J 2015; 36:653–656.
- Camm AJ, Lip GY, De Caterina R, et al; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation. Developed with the special contribution of the European Heart Rhythm Association. Eur Heart J 2012; 33:2719–2747.
KEY POINTS
- For most patients in this category, the benefits of anticoagulation outweigh the risks.
- Although they are not perfect, scoring systems have been developed to predict the risk of stroke without anticoagulation and the risk of bleeding with anticoagulation.
- The decision-making process is complex and should be shared with the patient and the patient’s family and caregivers.
Non-TNF-Targeted Therapy in Unresponsive RA More Effective than a Second Anti-TNF Drug
Study Overview
Objective. To determine whether a non–tumor necrosis factor (TNF)-targeted drug is more effective than a second anti-TNF drug in rheumatoid arthritis (RA) patients who have had an inadequate response to a first anti-TNF drug.
Design. 52-week pragmatic, multicenter, open-label, parallel-group, randomized clinical trial (the “Rotation or Change” trial).
Setting and participants. 300 patients who were at least 18 years old were recruited from December 2009 to August 2012 from 47 French clinical centers. These patients had to have a diagnosis of RA according to the 1987 American College of Rheumatology criteria, presence of erosions, a DAS28-ESR (a measure of disease burden using patient global health, tender and swollen joint counts, and the erythrocyte sedimentation rate) of 3.2 or more, and insufficient response to an anti-TNF according to the physician (based on 1 or more of: persistent tender and swollen joints, persistent disease activity according to patient global assessment, elevated levels of acute-phase reactants, and dependence on analgesics, nonsteroidal anti-inflammatory drugs, or corticosteroids). In addition, patients had to have a stable dose of oral corticosteroids of 15 mg/d or less of equivalent prednisone within 4 weeks before enrollment, a stable dose of synthetic disease-modifying antirheumatic drugs (DMARDs) within 4 weeks of enrollment, and informed written consent. Exclusion criteria included cessation of the first anti-TNF agent due only to an adverse event, previous treatment with 2 or more anti-TNF agents, previous treatment with abatacept, rituximab, or tocilizumab, a contraindication to all anti-TNF agents and other biologics such as an infection or cancer, pregnancy and breastfeeding.
Intervention. Patients were randomly assigned in equal proportions to receive either a non-TNF biologic (abatacept, rituximab, or tocilizumab) or a second anti-TNF agent (adalimumab, certolizumab, etanercept, infliximab, or golimumab); the choice of agent after randomization was decided by the physician. The starting dose and frequency of treatment was predetermined. Golimumab was not available for use at the time of this study. The choice of future dosing and frequency of the treatment was left up to the treating physician in both groups. The assigned drug treatments continued for 12 months but were allowed to be discontinued for adverse events, patient choice, or inefficacy. Treatment and dose adjustments for oral corticosteroids and glucocorticoid intra-articular injections were allowed for both treatment groups.
Main outcome measures. The primary outcome was the proportion of patients at week 24 with a good or moderate European League Against Rheumatism (EULAR) response. A good EULAR response is defined as a decrease in DAS28-ESR of more than 1.2 points leading to a score of 3.2 or lower while a moderate EULAR response is defined as a decrease of more than 0.6 and resulting in a score of 5.1 points or lower. Secondary end points were EULAR response at weeks 12 and 52, DAS28-ESR at weeks 12, 24, and 52, low disease activity (DAS28-ESR < 3.2) and remission (DAS28-ESR < 2.6) at weeks 12, 24, and 52, mean oral corticosteroid use at weeks 24 and 52, therapeutic maintenance (defined as the proportion of patients who did not discontinue the assigned biologic treatment) at weeks 24 and 52, and health assessment questionnaire (HAQ) score (range, 0–3 with 0 representing the best and 3 the worst outcomes) at weeks 12, 24, and 52. Safety including serious adverse events as well as serious infections was also evaluated throughout the study.
Main results. 300 patients were randomized. The 2 groups were not different with regard to demographic and disease characteristics. In the non-TNF group of 150 patients, 33 of 146 patients (23%) received abatacept, 41 (28%) rituximab, and 70 (48%) tocilizumab; 2 patients (1%) did not receive the intervention as planned, 1 patient received adalimumab and 1 patient received no treatment. For the anti-TNF group, 57 of 146 patients (39%) received adalimumab, 23 (16%) certolizumab, 53 (36%) etanercept, and 8 (5%) infliximab. Five patients (3%) did not receive the intervention assigned as 2 patients received rituximab, 1 patient received tocilizumab, and 2 patients received no treatment. About two-thirds of patients in each group received concomitant methotrexate and about half in each group received oral corticosteroids.
With regard to the primary outcome, at week 24 101 of 146 patients (69%) in the non-TNF group and 76 (52%) in the second anti-TNF group achieved a good or moderate EULAR response, with 39% with a good response and 30% with a moderate response in the non-TNF group and 21% with a good response and 31% with a moderate response in the second anti-TNF group (odds ratio [OR], 2.06; 95% confidence interval [CI], 1.27 to 3.37; P = 0.004, with imputation of missing data; absolute difference, 17.2%; 95% CI, 6.2% to 28.2%). The DAS28-ESR was lower in the non-TNF group (mean difference adjusted for baseline differences, −0.43; 95% CI, −0.72 to −0.14; P = 0.004). More patients in the non-TNF group vs the second anti-TNF group showed low disease activity at week 24 (45% vs 28%; OR, 2.09; 95% CI, 1.27 to 3.43; P = 0.004) and at week 52 (41% vs 23%; OR, 2.26; 95% CI, 1.33 to 3.86; P = 0.003).
The mean DAS28-ESR change from baseline was greater for patients in the non-TNF group than for patients in the second anti-TNF group with a 24-week mean difference of −0.43 (95% CI,−0.72 to −0.14; P = 0.004) and 52-week mean difference of −0.38 (95% CI, −0.69 to −0.08; P = 0.01).
The proportion of EULAR good and moderate responders at week 24 did not significantly differ with abatacept, rituximab, and tocilizumab treatment. The therapeutic maintenance rate, defined as the proportion of patients who continued the biologic treatment, was found to be significantly higher at weeks 24 and 52 in the non-TNF group than in the second anti-TNF group. The mean change from baseline to weeks 24 and 52 in the level of prednisone doses was not significantly different between patients between treatment groups.
With respect to safety, 16 patients (11%) in the non-TNF group experienced 18 serious adverse events and 8 patients (5%) in the second anti-TNF group experienced 13 events (P = 0.10) with 7 patients (5%) in each group developing serious infections.
Conclusion. In patients with RA previously treated with an anti-TNF drug with an inadequate response, the use of a non-TNF biologic agent was found to be more effective in achieving a good or moderate disease activity response at 24 weeks compared with a second anti-TNF medication.
Commentary
In patients with RA who have shown an inadequate response to methotrexate, TNF-α inhibitors have been shown to improve quality of life. However, it has been shown that almost one-third of patients have an insufficient and inadequate response to anti-TNF agents and continue to have persistent disease activity [1–3].Alternative treatments are therefore needed, but there is currently little guidance available for choosing the next treatment.
There are 3 placebo-controlled trials that have shown that switching to a non–TNF-targeted therapy may be appropriate [4–6]. The most commonly used non-TNF agents are abatacept, rituximab, and tocilizumab. However, there is evidence that switching to another anti-TNF agent after failure of a first can also be a good choice, as the molecular structure of TNF-inhibitors and their affinity for membrane and TNF-α vary. There were 2 randomized placebo-controlled trials that reported that approximately half of patients with RA with insufficient response to a TNF-α inhibitor responded to a second anti-TNF drug [7,8].
Although there have been observational studies addressing this question, this is the first randomized controlled trial to evaluate the efficacy of a non-TNF-targeted biologic compared to a second anti-TNF drug to treat RA in patients with an insufficient response to a first anti-TNF drug. Data showed that at week 24, 69% in the non-TNF group and 52% in the anti-TNF group achieved a good or moderate EULAR response. The non-TNF treatment was also associated with a better EULAR response than a second anti-TNF drug at weeks 12 and 52. The DAS28-ESR and the number of patients achieving low disease activity status were found to be greater at months 6 and 12 in the non-TNF group than in the second anti-TNF group. One strength of the study is its pragmatic design—the study evaluated the effectiveness of interventions under real-life, routine practice conditions where physicians often choose one drug over another for reasons based on the habits or characteristics of the patient. The comparison of strategies and not individual drugs more appropriately addresses the questions that physicians face in daily practice. However, there were some limitations including the lack of blinding by participants, the exclusion of some biologic agents such as golimunab, the lack of assessment of individual drug efficacy, and the fact that approximately 40% of patients in each group did not have concomitant treatment with methotrexate, an agent known to improve the efficacy of most biologic agents.
Applications for Clinical Practice
This is the first randomized controlled trial to evaluate the efficacy of a non-TNF-targeted biologic vs. a second anti-TNF in patients with RA who have an insufficient response to a first anti-TNF drug. Further studies addressing the limitations identified in this study are needed before physicians can employ these findings in clinical practice.
—Anita Laloo, MD
1. Hyrich KL, Lunt M, Watson KD, et al; British Society for Rheumatology Biologics Register. Outcomes after switching from one antitumor necrosis factor alpha agent to a second anti-tumor necrosis factor alpha agent in patients with rheumatoid arthritis: results from a large UK national cohort study. Arthritis Rheum 2007;56:13–20.
2. Hetland ML, Christensen IJ, Tarp U, et al; All Departments of Rheumatology in Denmark. Direct comparison of treatment responses, remission rates, and drug adherence in patients with rheumatoid arthritis treated with adalimumab, etanercept, or infliximab: results from eight years of surveillance of clinical practice in the nationwide Danish DANBIO registry. Arthritis Rheum 2010;62:22–32.
3. Smolen JS, Landewé R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann Rheum Dis 2010;69:964–75.
4. Cohen SB, Emery P, Greenwald MW, et al; REFLEX Trial Group. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase 3 trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum 2006;54:2793–806.
5. Emery P, Keystone E, Tony HP, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis 2008;67:1516–23.
6. Genovese MC, Becker JC, Schiff M, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N Engl J Med 2005;353:1114–23.
7. Smolen JS, Kay J, Doyle MK, et al; GO-AFTER study investigators. Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor alpha inhibitors (GO-AFTER study): a multicentre, randomised, double-blind, placebo-controlled, phase III trial. Lancet 2009;374:210–21.
8. Schiff MH, von Kempis J, Goldblum R, et al. Rheumatoid arthritis secondary non-responders to TNF can attain an efficacious and safe response by switching to certolizumab pegol: a phase IV, randomised, multicentre, double-blind, 12-week study, followed by a 12-week open-label phase. Ann Rheum Dis 2014;73:2174–7.
Study Overview
Objective. To determine whether a non–tumor necrosis factor (TNF)-targeted drug is more effective than a second anti-TNF drug in rheumatoid arthritis (RA) patients who have had an inadequate response to a first anti-TNF drug.
Design. 52-week pragmatic, multicenter, open-label, parallel-group, randomized clinical trial (the “Rotation or Change” trial).
Setting and participants. 300 patients who were at least 18 years old were recruited from December 2009 to August 2012 from 47 French clinical centers. These patients had to have a diagnosis of RA according to the 1987 American College of Rheumatology criteria, presence of erosions, a DAS28-ESR (a measure of disease burden using patient global health, tender and swollen joint counts, and the erythrocyte sedimentation rate) of 3.2 or more, and insufficient response to an anti-TNF according to the physician (based on 1 or more of: persistent tender and swollen joints, persistent disease activity according to patient global assessment, elevated levels of acute-phase reactants, and dependence on analgesics, nonsteroidal anti-inflammatory drugs, or corticosteroids). In addition, patients had to have a stable dose of oral corticosteroids of 15 mg/d or less of equivalent prednisone within 4 weeks before enrollment, a stable dose of synthetic disease-modifying antirheumatic drugs (DMARDs) within 4 weeks of enrollment, and informed written consent. Exclusion criteria included cessation of the first anti-TNF agent due only to an adverse event, previous treatment with 2 or more anti-TNF agents, previous treatment with abatacept, rituximab, or tocilizumab, a contraindication to all anti-TNF agents and other biologics such as an infection or cancer, pregnancy and breastfeeding.
Intervention. Patients were randomly assigned in equal proportions to receive either a non-TNF biologic (abatacept, rituximab, or tocilizumab) or a second anti-TNF agent (adalimumab, certolizumab, etanercept, infliximab, or golimumab); the choice of agent after randomization was decided by the physician. The starting dose and frequency of treatment was predetermined. Golimumab was not available for use at the time of this study. The choice of future dosing and frequency of the treatment was left up to the treating physician in both groups. The assigned drug treatments continued for 12 months but were allowed to be discontinued for adverse events, patient choice, or inefficacy. Treatment and dose adjustments for oral corticosteroids and glucocorticoid intra-articular injections were allowed for both treatment groups.
Main outcome measures. The primary outcome was the proportion of patients at week 24 with a good or moderate European League Against Rheumatism (EULAR) response. A good EULAR response is defined as a decrease in DAS28-ESR of more than 1.2 points leading to a score of 3.2 or lower while a moderate EULAR response is defined as a decrease of more than 0.6 and resulting in a score of 5.1 points or lower. Secondary end points were EULAR response at weeks 12 and 52, DAS28-ESR at weeks 12, 24, and 52, low disease activity (DAS28-ESR < 3.2) and remission (DAS28-ESR < 2.6) at weeks 12, 24, and 52, mean oral corticosteroid use at weeks 24 and 52, therapeutic maintenance (defined as the proportion of patients who did not discontinue the assigned biologic treatment) at weeks 24 and 52, and health assessment questionnaire (HAQ) score (range, 0–3 with 0 representing the best and 3 the worst outcomes) at weeks 12, 24, and 52. Safety including serious adverse events as well as serious infections was also evaluated throughout the study.
Main results. 300 patients were randomized. The 2 groups were not different with regard to demographic and disease characteristics. In the non-TNF group of 150 patients, 33 of 146 patients (23%) received abatacept, 41 (28%) rituximab, and 70 (48%) tocilizumab; 2 patients (1%) did not receive the intervention as planned, 1 patient received adalimumab and 1 patient received no treatment. For the anti-TNF group, 57 of 146 patients (39%) received adalimumab, 23 (16%) certolizumab, 53 (36%) etanercept, and 8 (5%) infliximab. Five patients (3%) did not receive the intervention assigned as 2 patients received rituximab, 1 patient received tocilizumab, and 2 patients received no treatment. About two-thirds of patients in each group received concomitant methotrexate and about half in each group received oral corticosteroids.
With regard to the primary outcome, at week 24 101 of 146 patients (69%) in the non-TNF group and 76 (52%) in the second anti-TNF group achieved a good or moderate EULAR response, with 39% with a good response and 30% with a moderate response in the non-TNF group and 21% with a good response and 31% with a moderate response in the second anti-TNF group (odds ratio [OR], 2.06; 95% confidence interval [CI], 1.27 to 3.37; P = 0.004, with imputation of missing data; absolute difference, 17.2%; 95% CI, 6.2% to 28.2%). The DAS28-ESR was lower in the non-TNF group (mean difference adjusted for baseline differences, −0.43; 95% CI, −0.72 to −0.14; P = 0.004). More patients in the non-TNF group vs the second anti-TNF group showed low disease activity at week 24 (45% vs 28%; OR, 2.09; 95% CI, 1.27 to 3.43; P = 0.004) and at week 52 (41% vs 23%; OR, 2.26; 95% CI, 1.33 to 3.86; P = 0.003).
The mean DAS28-ESR change from baseline was greater for patients in the non-TNF group than for patients in the second anti-TNF group with a 24-week mean difference of −0.43 (95% CI,−0.72 to −0.14; P = 0.004) and 52-week mean difference of −0.38 (95% CI, −0.69 to −0.08; P = 0.01).
The proportion of EULAR good and moderate responders at week 24 did not significantly differ with abatacept, rituximab, and tocilizumab treatment. The therapeutic maintenance rate, defined as the proportion of patients who continued the biologic treatment, was found to be significantly higher at weeks 24 and 52 in the non-TNF group than in the second anti-TNF group. The mean change from baseline to weeks 24 and 52 in the level of prednisone doses was not significantly different between patients between treatment groups.
With respect to safety, 16 patients (11%) in the non-TNF group experienced 18 serious adverse events and 8 patients (5%) in the second anti-TNF group experienced 13 events (P = 0.10) with 7 patients (5%) in each group developing serious infections.
Conclusion. In patients with RA previously treated with an anti-TNF drug with an inadequate response, the use of a non-TNF biologic agent was found to be more effective in achieving a good or moderate disease activity response at 24 weeks compared with a second anti-TNF medication.
Commentary
In patients with RA who have shown an inadequate response to methotrexate, TNF-α inhibitors have been shown to improve quality of life. However, it has been shown that almost one-third of patients have an insufficient and inadequate response to anti-TNF agents and continue to have persistent disease activity [1–3].Alternative treatments are therefore needed, but there is currently little guidance available for choosing the next treatment.
There are 3 placebo-controlled trials that have shown that switching to a non–TNF-targeted therapy may be appropriate [4–6]. The most commonly used non-TNF agents are abatacept, rituximab, and tocilizumab. However, there is evidence that switching to another anti-TNF agent after failure of a first can also be a good choice, as the molecular structure of TNF-inhibitors and their affinity for membrane and TNF-α vary. There were 2 randomized placebo-controlled trials that reported that approximately half of patients with RA with insufficient response to a TNF-α inhibitor responded to a second anti-TNF drug [7,8].
Although there have been observational studies addressing this question, this is the first randomized controlled trial to evaluate the efficacy of a non-TNF-targeted biologic compared to a second anti-TNF drug to treat RA in patients with an insufficient response to a first anti-TNF drug. Data showed that at week 24, 69% in the non-TNF group and 52% in the anti-TNF group achieved a good or moderate EULAR response. The non-TNF treatment was also associated with a better EULAR response than a second anti-TNF drug at weeks 12 and 52. The DAS28-ESR and the number of patients achieving low disease activity status were found to be greater at months 6 and 12 in the non-TNF group than in the second anti-TNF group. One strength of the study is its pragmatic design—the study evaluated the effectiveness of interventions under real-life, routine practice conditions where physicians often choose one drug over another for reasons based on the habits or characteristics of the patient. The comparison of strategies and not individual drugs more appropriately addresses the questions that physicians face in daily practice. However, there were some limitations including the lack of blinding by participants, the exclusion of some biologic agents such as golimunab, the lack of assessment of individual drug efficacy, and the fact that approximately 40% of patients in each group did not have concomitant treatment with methotrexate, an agent known to improve the efficacy of most biologic agents.
Applications for Clinical Practice
This is the first randomized controlled trial to evaluate the efficacy of a non-TNF-targeted biologic vs. a second anti-TNF in patients with RA who have an insufficient response to a first anti-TNF drug. Further studies addressing the limitations identified in this study are needed before physicians can employ these findings in clinical practice.
—Anita Laloo, MD
Study Overview
Objective. To determine whether a non–tumor necrosis factor (TNF)-targeted drug is more effective than a second anti-TNF drug in rheumatoid arthritis (RA) patients who have had an inadequate response to a first anti-TNF drug.
Design. 52-week pragmatic, multicenter, open-label, parallel-group, randomized clinical trial (the “Rotation or Change” trial).
Setting and participants. 300 patients who were at least 18 years old were recruited from December 2009 to August 2012 from 47 French clinical centers. These patients had to have a diagnosis of RA according to the 1987 American College of Rheumatology criteria, presence of erosions, a DAS28-ESR (a measure of disease burden using patient global health, tender and swollen joint counts, and the erythrocyte sedimentation rate) of 3.2 or more, and insufficient response to an anti-TNF according to the physician (based on 1 or more of: persistent tender and swollen joints, persistent disease activity according to patient global assessment, elevated levels of acute-phase reactants, and dependence on analgesics, nonsteroidal anti-inflammatory drugs, or corticosteroids). In addition, patients had to have a stable dose of oral corticosteroids of 15 mg/d or less of equivalent prednisone within 4 weeks before enrollment, a stable dose of synthetic disease-modifying antirheumatic drugs (DMARDs) within 4 weeks of enrollment, and informed written consent. Exclusion criteria included cessation of the first anti-TNF agent due only to an adverse event, previous treatment with 2 or more anti-TNF agents, previous treatment with abatacept, rituximab, or tocilizumab, a contraindication to all anti-TNF agents and other biologics such as an infection or cancer, pregnancy and breastfeeding.
Intervention. Patients were randomly assigned in equal proportions to receive either a non-TNF biologic (abatacept, rituximab, or tocilizumab) or a second anti-TNF agent (adalimumab, certolizumab, etanercept, infliximab, or golimumab); the choice of agent after randomization was decided by the physician. The starting dose and frequency of treatment was predetermined. Golimumab was not available for use at the time of this study. The choice of future dosing and frequency of the treatment was left up to the treating physician in both groups. The assigned drug treatments continued for 12 months but were allowed to be discontinued for adverse events, patient choice, or inefficacy. Treatment and dose adjustments for oral corticosteroids and glucocorticoid intra-articular injections were allowed for both treatment groups.
Main outcome measures. The primary outcome was the proportion of patients at week 24 with a good or moderate European League Against Rheumatism (EULAR) response. A good EULAR response is defined as a decrease in DAS28-ESR of more than 1.2 points leading to a score of 3.2 or lower while a moderate EULAR response is defined as a decrease of more than 0.6 and resulting in a score of 5.1 points or lower. Secondary end points were EULAR response at weeks 12 and 52, DAS28-ESR at weeks 12, 24, and 52, low disease activity (DAS28-ESR < 3.2) and remission (DAS28-ESR < 2.6) at weeks 12, 24, and 52, mean oral corticosteroid use at weeks 24 and 52, therapeutic maintenance (defined as the proportion of patients who did not discontinue the assigned biologic treatment) at weeks 24 and 52, and health assessment questionnaire (HAQ) score (range, 0–3 with 0 representing the best and 3 the worst outcomes) at weeks 12, 24, and 52. Safety including serious adverse events as well as serious infections was also evaluated throughout the study.
Main results. 300 patients were randomized. The 2 groups were not different with regard to demographic and disease characteristics. In the non-TNF group of 150 patients, 33 of 146 patients (23%) received abatacept, 41 (28%) rituximab, and 70 (48%) tocilizumab; 2 patients (1%) did not receive the intervention as planned, 1 patient received adalimumab and 1 patient received no treatment. For the anti-TNF group, 57 of 146 patients (39%) received adalimumab, 23 (16%) certolizumab, 53 (36%) etanercept, and 8 (5%) infliximab. Five patients (3%) did not receive the intervention assigned as 2 patients received rituximab, 1 patient received tocilizumab, and 2 patients received no treatment. About two-thirds of patients in each group received concomitant methotrexate and about half in each group received oral corticosteroids.
With regard to the primary outcome, at week 24 101 of 146 patients (69%) in the non-TNF group and 76 (52%) in the second anti-TNF group achieved a good or moderate EULAR response, with 39% with a good response and 30% with a moderate response in the non-TNF group and 21% with a good response and 31% with a moderate response in the second anti-TNF group (odds ratio [OR], 2.06; 95% confidence interval [CI], 1.27 to 3.37; P = 0.004, with imputation of missing data; absolute difference, 17.2%; 95% CI, 6.2% to 28.2%). The DAS28-ESR was lower in the non-TNF group (mean difference adjusted for baseline differences, −0.43; 95% CI, −0.72 to −0.14; P = 0.004). More patients in the non-TNF group vs the second anti-TNF group showed low disease activity at week 24 (45% vs 28%; OR, 2.09; 95% CI, 1.27 to 3.43; P = 0.004) and at week 52 (41% vs 23%; OR, 2.26; 95% CI, 1.33 to 3.86; P = 0.003).
The mean DAS28-ESR change from baseline was greater for patients in the non-TNF group than for patients in the second anti-TNF group with a 24-week mean difference of −0.43 (95% CI,−0.72 to −0.14; P = 0.004) and 52-week mean difference of −0.38 (95% CI, −0.69 to −0.08; P = 0.01).
The proportion of EULAR good and moderate responders at week 24 did not significantly differ with abatacept, rituximab, and tocilizumab treatment. The therapeutic maintenance rate, defined as the proportion of patients who continued the biologic treatment, was found to be significantly higher at weeks 24 and 52 in the non-TNF group than in the second anti-TNF group. The mean change from baseline to weeks 24 and 52 in the level of prednisone doses was not significantly different between patients between treatment groups.
With respect to safety, 16 patients (11%) in the non-TNF group experienced 18 serious adverse events and 8 patients (5%) in the second anti-TNF group experienced 13 events (P = 0.10) with 7 patients (5%) in each group developing serious infections.
Conclusion. In patients with RA previously treated with an anti-TNF drug with an inadequate response, the use of a non-TNF biologic agent was found to be more effective in achieving a good or moderate disease activity response at 24 weeks compared with a second anti-TNF medication.
Commentary
In patients with RA who have shown an inadequate response to methotrexate, TNF-α inhibitors have been shown to improve quality of life. However, it has been shown that almost one-third of patients have an insufficient and inadequate response to anti-TNF agents and continue to have persistent disease activity [1–3].Alternative treatments are therefore needed, but there is currently little guidance available for choosing the next treatment.
There are 3 placebo-controlled trials that have shown that switching to a non–TNF-targeted therapy may be appropriate [4–6]. The most commonly used non-TNF agents are abatacept, rituximab, and tocilizumab. However, there is evidence that switching to another anti-TNF agent after failure of a first can also be a good choice, as the molecular structure of TNF-inhibitors and their affinity for membrane and TNF-α vary. There were 2 randomized placebo-controlled trials that reported that approximately half of patients with RA with insufficient response to a TNF-α inhibitor responded to a second anti-TNF drug [7,8].
Although there have been observational studies addressing this question, this is the first randomized controlled trial to evaluate the efficacy of a non-TNF-targeted biologic compared to a second anti-TNF drug to treat RA in patients with an insufficient response to a first anti-TNF drug. Data showed that at week 24, 69% in the non-TNF group and 52% in the anti-TNF group achieved a good or moderate EULAR response. The non-TNF treatment was also associated with a better EULAR response than a second anti-TNF drug at weeks 12 and 52. The DAS28-ESR and the number of patients achieving low disease activity status were found to be greater at months 6 and 12 in the non-TNF group than in the second anti-TNF group. One strength of the study is its pragmatic design—the study evaluated the effectiveness of interventions under real-life, routine practice conditions where physicians often choose one drug over another for reasons based on the habits or characteristics of the patient. The comparison of strategies and not individual drugs more appropriately addresses the questions that physicians face in daily practice. However, there were some limitations including the lack of blinding by participants, the exclusion of some biologic agents such as golimunab, the lack of assessment of individual drug efficacy, and the fact that approximately 40% of patients in each group did not have concomitant treatment with methotrexate, an agent known to improve the efficacy of most biologic agents.
Applications for Clinical Practice
This is the first randomized controlled trial to evaluate the efficacy of a non-TNF-targeted biologic vs. a second anti-TNF in patients with RA who have an insufficient response to a first anti-TNF drug. Further studies addressing the limitations identified in this study are needed before physicians can employ these findings in clinical practice.
—Anita Laloo, MD
1. Hyrich KL, Lunt M, Watson KD, et al; British Society for Rheumatology Biologics Register. Outcomes after switching from one antitumor necrosis factor alpha agent to a second anti-tumor necrosis factor alpha agent in patients with rheumatoid arthritis: results from a large UK national cohort study. Arthritis Rheum 2007;56:13–20.
2. Hetland ML, Christensen IJ, Tarp U, et al; All Departments of Rheumatology in Denmark. Direct comparison of treatment responses, remission rates, and drug adherence in patients with rheumatoid arthritis treated with adalimumab, etanercept, or infliximab: results from eight years of surveillance of clinical practice in the nationwide Danish DANBIO registry. Arthritis Rheum 2010;62:22–32.
3. Smolen JS, Landewé R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann Rheum Dis 2010;69:964–75.
4. Cohen SB, Emery P, Greenwald MW, et al; REFLEX Trial Group. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase 3 trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum 2006;54:2793–806.
5. Emery P, Keystone E, Tony HP, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis 2008;67:1516–23.
6. Genovese MC, Becker JC, Schiff M, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N Engl J Med 2005;353:1114–23.
7. Smolen JS, Kay J, Doyle MK, et al; GO-AFTER study investigators. Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor alpha inhibitors (GO-AFTER study): a multicentre, randomised, double-blind, placebo-controlled, phase III trial. Lancet 2009;374:210–21.
8. Schiff MH, von Kempis J, Goldblum R, et al. Rheumatoid arthritis secondary non-responders to TNF can attain an efficacious and safe response by switching to certolizumab pegol: a phase IV, randomised, multicentre, double-blind, 12-week study, followed by a 12-week open-label phase. Ann Rheum Dis 2014;73:2174–7.
1. Hyrich KL, Lunt M, Watson KD, et al; British Society for Rheumatology Biologics Register. Outcomes after switching from one antitumor necrosis factor alpha agent to a second anti-tumor necrosis factor alpha agent in patients with rheumatoid arthritis: results from a large UK national cohort study. Arthritis Rheum 2007;56:13–20.
2. Hetland ML, Christensen IJ, Tarp U, et al; All Departments of Rheumatology in Denmark. Direct comparison of treatment responses, remission rates, and drug adherence in patients with rheumatoid arthritis treated with adalimumab, etanercept, or infliximab: results from eight years of surveillance of clinical practice in the nationwide Danish DANBIO registry. Arthritis Rheum 2010;62:22–32.
3. Smolen JS, Landewé R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann Rheum Dis 2010;69:964–75.
4. Cohen SB, Emery P, Greenwald MW, et al; REFLEX Trial Group. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase 3 trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum 2006;54:2793–806.
5. Emery P, Keystone E, Tony HP, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis 2008;67:1516–23.
6. Genovese MC, Becker JC, Schiff M, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N Engl J Med 2005;353:1114–23.
7. Smolen JS, Kay J, Doyle MK, et al; GO-AFTER study investigators. Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor alpha inhibitors (GO-AFTER study): a multicentre, randomised, double-blind, placebo-controlled, phase III trial. Lancet 2009;374:210–21.
8. Schiff MH, von Kempis J, Goldblum R, et al. Rheumatoid arthritis secondary non-responders to TNF can attain an efficacious and safe response by switching to certolizumab pegol: a phase IV, randomised, multicentre, double-blind, 12-week study, followed by a 12-week open-label phase. Ann Rheum Dis 2014;73:2174–7.
Left ventricular thrombosis can still complicate acute myocardial infarction
A 62-year-old man with hypertension, type 2 diabetes mellitus, and hypercholesterolemia presented to the emergency department with substernal chest pain that started about 15 hours earlier while he was at rest watching television.
On examination, his pulse was 92 beats per minute and regular, his blood pressure was 160/88 mm Hg, and he had no evidence of jugular venous distention or pedal edema. Lung examination was positive for bibasilar crackles.
Electrocardiography revealed Q waves with ST elevation in leads I, aVL, V4, V5, and V6 with reciprocal ST depression in leads II, III, and aVF.
His troponin T level on presentation was markedly elevated.

He underwent heart catheterization and was found to have 100% occlusion of the proximal left anterior descending artery. He underwent successful percutaneous coronary intervention with placement of a drug-eluting stent, and afterward had grade 3 flow on the Thrombolysis in Myocardial Infarction (TIMI) scale.
Echocardiography the next day revealed a mobile echo-dense mass in the left ventricular apex (Figure 1) and a left ventricular ejection fraction of 35%.
THE INCIDENCE OF LEFT VENTRICULAR THROMBOSIS IN ACUTE MI
1. What is the incidence of left ventricular thrombosis after acute myocardial infarction (MI), now that primary percutaneous coronary intervention is common?
- 0.1%
- 2%
- 20%
- 40%
Left ventricular thrombosis is a serious complication of acute MI that can cause systemic thromboembolism, including stroke.1 Before thrombolytic therapy was available, this complication occurred in 20% to 60% of patients with acute MI.2,3 But early reperfusion strategies, anticoagulation for the first 48 hours, and dual antiplatelet therapy have reduced the incidence of this complication significantly.
In the thrombolytic era, the incidence of left ventricular thrombosis was 5.1% in the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI) 3 study, which had 8,326 patients. A subset of patients who had an anterior MI had almost double the incidence (11.5%).3

The incidence has further declined with the advent of primary percutaneous coronary intervention, likely thanks to enhanced myocardial salvage, and now ranges from 2.5% to 15% (Table 1).4–11 The largest observational study, with 2,911 patients undergoing percutaneous coronary intervention, reported an incidence of 2.5% within 3 to 5 days of the MI.7 At our center, the incidence was found to be even lower, 1.8% in 1,700 patients presenting with ST-elevation MI undergoing primary percutaneous coronary intervention. Hence, of the answers to the question above, 2% would be closest.
Large infarct size with a low left ventricular ejection fraction (< 40%), anterior wall MI, hypertension, and delay in time from symptom onset to intervention were independent predictors of left ventricular thrombus formation in most studies.7,12 The risk is highest during the first 2 weeks after MI, and thrombosis almost never occurs more than 3 months after the index event.5,13–16
WHAT IS THE PATHOGENESIS OF LEFT VENTRICULAR THROMBOSIS?
A large transmural infarct results in loss of contractile function, which causes stagnation and pooling of blood adjacent to the infarcted ventricular segment. In addition, endocardial injury exposes tissue factor, which then initiates the coagulation cascade. To make matters worse, MI results in a hypercoagulable state through unclear mechanisms, which completes the Virchow triad for thrombus formation. Elevations of D-dimer, fibrinogen, anticardiolipin antibodies (IgM and IgG), and tissue factor have also been reported after acute MI.17
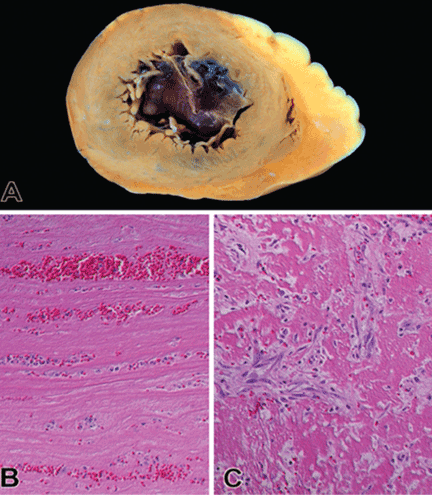
Thrombus formation begins with platelet aggregation at the site of endocardial damage, forming a platelet plug, followed by activation of clotting factors. These thrombi are referred to as “mural,” as they adhere to the chamber wall (endocardium). They are composed of fibrin and entrapped red and white blood cells (Figure 2).
The natural course of thrombus evolution is established but variable. A left ventricular thrombus may dislodge and embolize, resulting in stroke or other thromboembolic complications. Alternately, it can dissolve over time, aided by intrinsic fibrinolytic mechanisms. On other occasions, the thrombus may organize, a process characterized by ingrowth of smooth muscle cells, fibroblasts, and endothelium.
HOW IS LEFT VENTRICULAR THROMBOSIS DIAGNOSED?
2. What is the best imaging test for detecting a thrombus?
- Transesophageal echocardiography
- Transthoracic echocardiography
- Cardiac magnetic resonance imaging (MRI) without gadolinium contrast
- Cardiac MRI with gadolinium contrast
Evaluation of left ventricular function after acute MI carries a class I indication (ie, it should be performed).18
Echocardiography is commonly used, and it has a 60% sensitivity to detect a thrombus.19 In patients with poorer transthoracic echocardiographic windows, contrast can be used to better delineate the left ventricular cavity and show the thrombus. Transesophageal echocardiography is seldom useful, as the left ventricular apex is foreshortened and in the far field.
A left ventricular thrombus is confirmed if an echo-dense mass with well-demarcated margins distinct from the endocardium is seen throughout the cardiac cycle. It should be evident in at least two different views (apical and short-axis) and should be adjacent to a hypokinetic or akinetic left ventricular wall. False-positive findings can occur due to misidentified false tendons, papillary muscles, and trabeculae.
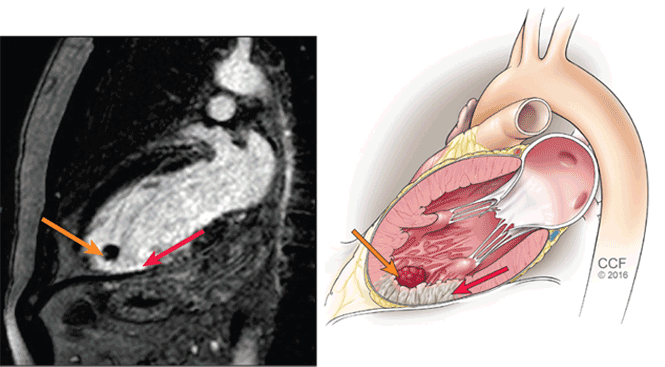
Cardiac MRI with late gadolinium enhancement is now the gold standard for diagnostic imaging, as it accurately characterizes the shape, size, and location of the thrombus (Figure 3). Gadolinium contrast increases the enhancement of the ventricular cavity, thus allowing easy detection of thrombus, which appears dark. Cardiac MRI with delayed enhancement has 88% to 91% sensitivity and 99% specificity to detect left ventricular thrombosis.20,21 However, compared with echocardiography, routine cardiac MRI is time-intensive, costly, and not routinely available. As a result, it should be performed only in patients with poor acoustic windows and a high clinical suspicion of left ventricular thrombosis.
Delayed-contrast cardiac computed tomography can be used to identify left ventricular thrombosis, using absence of contrast uptake. The need to use contrast is a disadvantage, but computed tomography can be an alternative in patients with contraindications to cardiac MRI.
WHAT COMPLICATIONS ARISE FROM LEFT VENTRICULAR THROMBOSIS?
The most feared complication of left ventricular thrombosis is thromboembolism. Cardioembolic stroke is generally severe, prone to early and long-term recurrence, and associated with a higher death rate than noncardioembolic ischemic stroke.22,23 Thrombi associated with thromboembolism are often acute and mobile rather than organized and immobile.24 They may embolize to the brain, spleen, kidneys, and bowel.25 In a meta-analysis of 11 studies, the pooled odds ratio for risk of embolization was 5.45 (95% confidence interval [CI] 3.02–9.83) with left ventricular thrombi vs without.26 Before systemic thrombolysis and antiplatelet therapy became available, stroke rates ranged from 1.5% to 10%.27–29
In a meta-analysis of 22 studies from 1978 to 2004, the incidence of ischemic stroke after MI during hospitalization was around 11.1 per 1,000 MIs.30 This study found that anterior MI was associated with a higher risk of stroke, but reported no difference in the incidence of stroke with percutaneous coronary intervention, systemic thrombolysis, or no reperfusion.
In a large prospective cohort study of 2,160 patients,31 259 (12%) had a stroke after MI. In multivariable analysis, age, diabetes, and previous stroke were predictors of stroke after MI. This study reported significantly fewer strokes in patients who underwent percutaneous coronary intervention than with other or no reperfusion therapies.31
ANTICOAGULATION TREATMENT
3. How would you treat a patient who has a drug-eluting stent in the left anterior descending artery and a new diagnosis of left ventricular thrombosis?
- Warfarin
- Aspirin and clopidogrel
- Aspirin, clopidogrel, and warfarin
- Aspirin and warfarin
The management of left ventricular thrombosis has been summarized in guidelines from the American College of Chest Physicians (ACCP) in 2012,32 and from the American College of Cardiology/American Heart Association in 2013,18 which recommend anticoagulation for at least 3 months, or indefinitely if bleeding risk is low, for all patients developing a left ventricular thrombus.
For patients with acute MI and left ventricular thrombosis, the ACCP guidelines recommend warfarin with a target international normalized ratio of 2.0 to 3.0 plus dual antiplatelet therapy (eg, aspirin plus clopidogrel) for 3 months, after which warfarin is discontinued but dual antiplatelet therapy is continued for up to 12 months.32
The European Society of Cardiology guidelines33 recommend 6 months of anticoagulation. However, if the patient is receiving dual antiplatelet therapy, they recommend repeated imaging of the left ventricle after 3 months of anticoagulation, which may allow for earlier discontinuation of anticoagulation if the thrombus has resolved and apical wall motion has recovered. Therefore, most experts recommend 3 months of anticoagulation when used in combination with dual antiplatelet therapy and repeating echocardiography at 3 months to safely discontinue anticoagulation. The best answer to the question posed here is aspirin, clopidogrel, and warfarin.
Decisions about antithrombotic therapy may also depend on stent type and the patient’s bleeding risk. With bare-metal stents, dual antiplatelet therapy along with anticoagulation should be used for 1 month, after which anticoagulation should be used with a single antiplatelet agent for another 2 months; after this, the anticoagulant can be discontinued and dual antiplatelet therapy can be resumed for a total of 12 months. Newer anticoagulants such as rivaroxaban, dabigatran, edoxaban, and apixaban may also have a role, but they have not yet been studied for this indication.
Surgical thrombectomy is rarely considered now, given the known efficacy of anticoagulants in dissolving the thrombus. It was done in the past for large, mobile, or protruding left ventricular thrombi, which have a higher potential for embolization.34 Currently, it can be done under very special circumstances, such as before placement of a left ventricular assist device or if the thrombus is large, to prevent embolism.35,36
BLEEDING COMPLICATIONS WITH TRIPLE ANTITHROMBOTIC THERAPY
After stent placement, almost all patients need to be on dual antiplatelet therapy for a specified duration depending on the type and generation of stent used. Such patients end up on “triple” antithrombotic therapy (two antiplatelet drugs plus an anticoagulant), which poses a high risk of bleeding.37 Consideration needs to be given to the risks of stroke, stent thrombosis, and major bleeding when selecting the antithrombotic regimen.38 Triple antithrombotic therapy has been associated with a risk of fatal and nonfatal bleeding of 4% to 16% when used for indications such as atrial fibrillation.39–41
Risks of triple antithrombotic therapy (aspirin 80–100 mg, clopidogrel 75 mg, and warfarin) were compared with those of clopidogrel plus warfarin in the What Is the Optimal Antiplatelet and Anticoagulant therapy in Patients With Oral Anticoagulation and Coronary Stenting Trial,37 which reported a significantly lower risk of major and minor bleeding with clopidogrel-plus-warfarin therapy than with triple antithrombotic therapy, 14.3% vs 31.7% (hazard ratio 0.40, 95% CI 0.28–0.58, P < .0001).
Additionally, the increased risk of major and minor bleeding associated with triple antithrombotic therapy has been confirmed in many observational studies; other studies found a trend toward lower risk with triple therapy, but this was not statistically significant (Table 2).38,40,42–55 A large multicenter European trial is being conducted to compare dual antiplatelet therapy vs triple antithrombotic therapy in patients with left ventricular thrombosis.
CASE FOLLOW-UP
Our patient was started on warfarin, clopidogrel 75 mg, and aspirin 75 mg at the time of discharge. He was continued on warfarin for 3 months, at which time a follow-up echocardiogram showed no thrombus in the left ventricle. Warfarin was discontinued, and he had no thromboembolic complications.
TAKE-HOME POINTS
Left ventricular thrombosis after an acute MI is very important to detect, as it can lead to serious complications through arterial embolism.
The incidence of left ventricular thrombosis has declined significantly with the use of percutaneous coronary intervention. However, it may still occur in a small number of patients with larger infarcts owing to delay in revascularization or proximal (left main or left anterior descending) occlusions with larger infarct size.
Echocardiography, which is routinely performed after acute MI to assess myocardial function, uncovers most left ventricular thrombi. In high-risk cases, MRI with late gadolinium enhancement can increase the diagnostic yield.
Anticoagulation with warfarin is recommended for at least 3 months. Post-MI patients undergoing stent implantation may need triple antithrombotic therapy, which, however, increases the bleeding risk significantly. Large randomized trials are needed to guide physicians in risk stratification of such patients.
- Lip GY, Piotrponikowski P, Andreotti F, et al; Heart Failure Association (EHFA) of the European Society of Cardiology (ESC) and the ESC Working Group on Thrombosis. Thromboembolism and antithrombotic therapy for heart failure in sinus rhythm: an executive summary of a joint consensus document from the ESC Heart Failure Association and the ESC Working Group on Thrombosis. Thromb Haemost 2012; 108:1009–1022.
- Turpie AG, Robinson JG, Doyle DJ, et al. Comparison of high-dose with low-dose subcutaneous heparin to prevent left ventricular mural thrombosis in patients with acute transmural anterior myocardial infarction. N Engl J Med 1989; 320:352–357.
- Chiarella F, Santoro E, Domenicucci S, Maggioni A, Vecchio C. Predischarge two-dimensional echocardiographic evaluation of left ventricular thrombosis after acute myocardial infarction in the GISSI-3 study. Am J Cardiol 1998; 81:822–827.
- Kalra A, Jang IK. Prevalence of early left ventricular thrombus after primary coronary intervention for acute myocardial infarction. J Thromb Thrombolysis 2000; 10:133–136.
- Nayak D, Aronow WS, Sukhija R, McClung JA, Monsen CE, Belkin RN. Comparison of frequency of left ventricular thrombi in patients with anterior wall versus non-anterior wall acute myocardial infarction treated with antithrombotic and antiplatelet therapy with or without coronary revascularization. Am J Cardiol 2004; 93:1529–1530.
- Rehan A, Kanwar M, Rosman H, et al. Incidence of post myocardial infarction left ventricular thrombus formation in the era of primary percutaneous intervention and glycoprotein IIb/IIIa inhibitors. A prospective observational study. Cardiovasc Ultrasound 2006;4:20.
- Zielinska M, Kaczmarek K, Tylkowski M. Predictors of left ventricular thrombus formation in acute myocardial infarction treated with successful primary angioplasty with stenting. Am J Med Sci 2008; 335:171–176.
- Osherov AB, Borovik-Raz M, Aronson D, et al. Incidence of early left ventricular thrombus after acute anterior wall myocardial infarction in the primary coronary intervention era. Am Heart J 2009; 157:1074–1080.
- Solheim S, Seljeflot I, Lunde K, et al. Frequency of left ventricular thrombus in patients with anterior wall acute myocardial infarction treated with percutaneous coronary intervention and dual antiplatelet therapy. Am J Cardiol 2010; 106:1197–1200.
- Shacham Y, Leshem-Rubinow E, Ben Assa E, et al. Comparison of C-reactive protein and fibrinogen levels in patients having anterior wall ST-segment elevation myocardial infarction with versus without left ventricular thrombus (from a primary percutaneous coronary intervention cohort). Am J Cardiol 2013; 112:57–60.
- Gianstefani S, Douiri A, Delithanasis I, et al. Incidence and predictors of early left ventricular thrombus after ST-elevation myocardial infarction in the contemporary era of primary percutaneous coronary intervention. Am J Cardiol 2014; 113:1111–1116.
- Shacham Y, Birati EY, Rogovski O, Cogan Y, Keren G, Roth A. Left ventricular thrombus formation and bleeding complications during continuous in-hospital anticoagulation for acute anterior myocardial infarction. Isr Med Assoc J 2012; 14:742–746.
- Asinger RW, Mikell FL, Elsperger J, Hodges M. Incidence of left-ventricular thrombosis after acute transmural myocardial infarction. Serial evaluation by two-dimensional echocardiography. N Engl J Med 1981; 305:297–302.
- Nihoyannopoulos P, Smith GC, Maseri A, Foale RA. The natural history of left ventricular thrombus in myocardial infarction: a rationale in support of masterly inactivity. J Am Coll Cardiol 1989; 14:903–911.
- Weinreich DJ, Burke JF, Pauletto FJ. Left ventricular mural thrombi complicating acute myocardial infarction. Long-term follow-up with serial echocardiography. Ann Intern Med 1984; 100:789–794.
- Greaves SC, Zhi G, Lee RT, et al. Incidence and natural history of left ventricular thrombus following anterior wall acute myocardial infarction. Am J Cardiol 1997; 80:442–448.
- Solheim S, Seljeflot I, Lunde K, et al. Prothrombotic markers in patients with acute myocardial infarction and left ventricular thrombus formation treated with pci and dual antiplatelet therapy. Thromb J 2013; 11:1.
- O’Gara PT, Kushner FG, Ascheim DD, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013; 127:e362–e425.
- Weinsaft JW, Kim HW, Crowley AL, et al. LV thrombus detection by routine echocardiography: insights into performance characteristics using delayed enhancement CMR. JACC Cardiovasc Imaging 2011; 4:702–712.
- Mollet NR, Dymarkowski S, Volders W, et al. Visualization of ventricular thrombi with contrast-enhanced magnetic resonance imaging in patients with ischemic heart disease. Circulation 2002; 106:2873–2876.
- Srichai MB, Junor C, Rodriguez LL, et al. Clinical, imaging, and pathological characteristics of left ventricular thrombus: a comparison of contrast-enhanced magnetic resonance imaging, transthoracic echocardiography, and transesophageal echocardiography with surgical or pathological validation. Am Heart J 2006; 152:75–84.
- Eriksson SE, Olsson JE. Survival and recurrent strokes in patients with different subtypes of stroke: a fourteen-year follow-up study. Cerebrovasc Dis 2001; 12:171–180.
- Grau AJ, Weimar C, Buggle F, et al. Risk factors, outcome, and treatment in subtypes of ischemic stroke: the German Stroke Data Bank. Stroke 2001; 32:2559–2566.
- Keren A, Goldberg S, Gottlieb S, et al. Natural history of left ventricular thrombi: their appearance and resolution in the posthospitalization period of acute myocardial infarction. J Am Coll Cardiol 1990; 15:790–800.
- Jordan RA, Miller RD, Edwards JE, Parker RL. Thrombo-embolism in acute and in healed myocardial infarction. I. Intracardiac mural thrombosis. Circulation 1952; 6:1–6.
- Vaitkus PT, Barnathan ES. Embolic potential, prevention and management of mural thrombus complicating anterior myocardial infarction: a meta-analysis. J Am Coll Cardiol 1993; 22:1004–1009.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- Cabin HS, Roberts WC. Left ventricular aneurysm, intraaneurysmal thrombus and systemic embolus in coronary heart disease. Chest 1980; 77:586–590.
- Keating EC, Gross SA, Schlamowitz RA, et al. Mural thrombi in myocardial infarctions. Prospective evaluation by two-dimensional echocardiography. Am J Med 1983; 74:989–995.
- Witt BJ, Ballman KV, Brown RD Jr, Meverden RA, Jacobsen SJ, Roger VL. The incidence of stroke after myocardial infarction: a meta-analysis. Am J Med 2006; 119:354.e1–354.e9.
- Witt BJ, Brown RD Jr, Jacobsen SJ, Weston SA, Yawn BP, Roger VL. A community-based study of stroke incidence after myocardial infarction. Ann Intern Med 2005; 143:785–792.
- Vandvik PO, Lincoff AM, Gore JM, et al; American College of Chest Physicians. Primary and secondary prevention of cardiovascular disease: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl):e637S–e68S.
- Steg G, James SK, Atar D, et al. ESC guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J 2012; 33:2569–2619.
- Nili M, Deviri E, Jortner R, Strasberg B, Levy MJ. Surgical removal of a mobile, pedunculated left ventricular thrombus: report of 4 cases. Ann Thorac Surg 1988; 46:396–400.
- Kanemitsu S, Miyake Y, Okabe M. Surgical removal of a left ventricular thrombus associated with cardiac sarcoidosis. Interact Cardiovasc Thorac Surg 2008; 7:333–335.
- Engin C, Yagdi T, Balcioglu O, et al. Left ventricular assist device implantation in heart failure patients with a left ventricular thrombus. Transplant Proc 2013; 45:1017–1019.
- Dewilde WJ, Oirbans T, Verheugt FW, et al; WOEST study investigators. Use of clopidogrel with or without aspirin in patients taking oral anticoagulant therapy and undergoing percutaneous coronary intervention: an open-label, randomised, controlled trial. Lancet 2013; 381:1107–1115.
- Faxon DP, Eikelboom JW, Berger PB, et al. Antithrombotic therapy in patients with atrial fibrillation undergoing coronary stenting: a North American perspective: executive summary. Circ Cardiovasc Interv 2011; 4:522–534.
- Hansen ML, Sorensen R, Clausen MT, et al. Risk of bleeding with single, dual, or triple therapy with warfarin, aspirin, and clopidogrel in patients with atrial fibrillation. Arch Intern Med 2010; 170:1433–1441.
- Karjalainen PP, Porela P, Ylitalo A, et al. Safety and efficacy of combined antiplatelet-warfarin therapy after coronary stenting. Eur Heart J 2007; 28:726–732.
- Doyle BJ, Rihal CS, Gastineau DA, Holmes DR Jr. Bleeding, blood transfusion, and increased mortality after percutaneous coronary intervention: implications for contemporary practice. J Am Coll Cardiol 2009; 53:2019–2027.
- Azoulay L, Dell’Aniello S, Simon T, Renoux C, Suissa S. The concurrent use of antithrombotic therapies and the risk of bleeding in patients with atrial fibrillation. Thromb Haemost 2013; 109:431–439.
- Deshmukh A, Hilleman DE, Del Core M, Nair CK. Antithrombotic regimens in patients with indication for long-term anticoagulation undergoing coronary interventions-systematic analysis, review of literature, and implications on management. Am J Ther 2013; 20:654–663.
- Fosbol EL, Wang TY, Li S, et al. Warfarin use among older atrial fibrillation patients with non-ST-segment elevation myocardial infarction managed with coronary stenting and dual antiplatelet therapy. Am Heart J 2013; 166:864–870.
- Gao F, Zhou YJ, Wang ZJ, et al. Meta-analysis of the combination of warfarin and dual antiplatelet therapy after coronary stenting in patients with indications for chronic oral anticoagulation. Int J Cardiol 2011; 148:96–101.
- Hansen ML, Sorensen R, Clausen MT, et al. Risk of bleeding with single, dual, or triple therapy with warfarin, aspirin, and clopidogrel in patients with atrial fibrillation. Arch Intern Med 2010; 170:1433–1441.
- Hermosillo AJ, Spinler SA. Aspirin, clopidogrel, and warfarin: is the combination appropriate and effective or inappropriate and too dangerous? Ann Pharmacother 2008; 42:790–805.
- Holmes DR Jr, Kereiakes DJ, Kleiman NS, Moliterno DJ, Patti G, Grines CL. Combining antiplatelet and anticoagulant therapies. J Am Coll Cardiol 2009; 54:95–109.
- Khurram Z, Chou E, Minutello R, et al. Combination therapy with aspirin, clopidogrel and warfarin following coronary stenting is associated with a significant risk of bleeding. J Invasive Cardiol 2006; 18:162–164.
- Orford JL, Fasseas P, Melby S, et al. Safety and efficacy of aspirin, clopidogrel, and warfarin after coronary stent placement in patients with an indication for anticoagulation. Am Heart J 2004; 147:463–467.
- Porter A, Konstantino Y, Iakobishvili Z, Shachar L, Battler A, Hasdai D. Short-term triple therapy with aspirin, warfarin, and a thienopyridine among patients undergoing percutaneous coronary intervention. Catheter Cardiovasc Interv 2006; 68:56–61.
- DeEugenio D, Kolman L, DeCaro M, et al. Risk of major bleeding with concomitant dual antiplatelet therapy after percutaneous coronary intervention in patients receiving long-term warfarin therapy. Pharmacotherapy 2007; 27:691–696.
- Ruiz-Nodar JM, Marin F, Hurtado JA, et al. Anticoagulant and antiplatelet therapy use in 426 patients with atrial fibrillation undergoing percutaneous coronary intervention and stent implantation implications for bleeding risk and prognosis. J Am Coll Cardiol 2008; 51:818–825.
- Sarafoff N, Ndrepepa G, Mehilli J, et al. Aspirin and clopidogrel with or without phenprocoumon after drug eluting coronary stent placement in patients on chronic oral anticoagulation. J Intern Med 2008; 264:472–480.
- Rossini R, Musumeci GF, Lettieri CF, et al. Long-term outcomes in patients undergoing coronary stenting on dual oral antiplatelet treatment requiring oral anticoagulant therapy. Am J Cardiol 2008; 102:1618–1623.
A 62-year-old man with hypertension, type 2 diabetes mellitus, and hypercholesterolemia presented to the emergency department with substernal chest pain that started about 15 hours earlier while he was at rest watching television.
On examination, his pulse was 92 beats per minute and regular, his blood pressure was 160/88 mm Hg, and he had no evidence of jugular venous distention or pedal edema. Lung examination was positive for bibasilar crackles.
Electrocardiography revealed Q waves with ST elevation in leads I, aVL, V4, V5, and V6 with reciprocal ST depression in leads II, III, and aVF.
His troponin T level on presentation was markedly elevated.
He underwent heart catheterization and was found to have 100% occlusion of the proximal left anterior descending artery. He underwent successful percutaneous coronary intervention with placement of a drug-eluting stent, and afterward had grade 3 flow on the Thrombolysis in Myocardial Infarction (TIMI) scale.
Echocardiography the next day revealed a mobile echo-dense mass in the left ventricular apex (Figure 1) and a left ventricular ejection fraction of 35%.
THE INCIDENCE OF LEFT VENTRICULAR THROMBOSIS IN ACUTE MI
1. What is the incidence of left ventricular thrombosis after acute myocardial infarction (MI), now that primary percutaneous coronary intervention is common?
- 0.1%
- 2%
- 20%
- 40%
Left ventricular thrombosis is a serious complication of acute MI that can cause systemic thromboembolism, including stroke.1 Before thrombolytic therapy was available, this complication occurred in 20% to 60% of patients with acute MI.2,3 But early reperfusion strategies, anticoagulation for the first 48 hours, and dual antiplatelet therapy have reduced the incidence of this complication significantly.
In the thrombolytic era, the incidence of left ventricular thrombosis was 5.1% in the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI) 3 study, which had 8,326 patients. A subset of patients who had an anterior MI had almost double the incidence (11.5%).3
The incidence has further declined with the advent of primary percutaneous coronary intervention, likely thanks to enhanced myocardial salvage, and now ranges from 2.5% to 15% (Table 1).4–11 The largest observational study, with 2,911 patients undergoing percutaneous coronary intervention, reported an incidence of 2.5% within 3 to 5 days of the MI.7 At our center, the incidence was found to be even lower, 1.8% in 1,700 patients presenting with ST-elevation MI undergoing primary percutaneous coronary intervention. Hence, of the answers to the question above, 2% would be closest.
Large infarct size with a low left ventricular ejection fraction (< 40%), anterior wall MI, hypertension, and delay in time from symptom onset to intervention were independent predictors of left ventricular thrombus formation in most studies.7,12 The risk is highest during the first 2 weeks after MI, and thrombosis almost never occurs more than 3 months after the index event.5,13–16
WHAT IS THE PATHOGENESIS OF LEFT VENTRICULAR THROMBOSIS?
A large transmural infarct results in loss of contractile function, which causes stagnation and pooling of blood adjacent to the infarcted ventricular segment. In addition, endocardial injury exposes tissue factor, which then initiates the coagulation cascade. To make matters worse, MI results in a hypercoagulable state through unclear mechanisms, which completes the Virchow triad for thrombus formation. Elevations of D-dimer, fibrinogen, anticardiolipin antibodies (IgM and IgG), and tissue factor have also been reported after acute MI.17
Thrombus formation begins with platelet aggregation at the site of endocardial damage, forming a platelet plug, followed by activation of clotting factors. These thrombi are referred to as “mural,” as they adhere to the chamber wall (endocardium). They are composed of fibrin and entrapped red and white blood cells (Figure 2).
The natural course of thrombus evolution is established but variable. A left ventricular thrombus may dislodge and embolize, resulting in stroke or other thromboembolic complications. Alternately, it can dissolve over time, aided by intrinsic fibrinolytic mechanisms. On other occasions, the thrombus may organize, a process characterized by ingrowth of smooth muscle cells, fibroblasts, and endothelium.
HOW IS LEFT VENTRICULAR THROMBOSIS DIAGNOSED?
2. What is the best imaging test for detecting a thrombus?
- Transesophageal echocardiography
- Transthoracic echocardiography
- Cardiac magnetic resonance imaging (MRI) without gadolinium contrast
- Cardiac MRI with gadolinium contrast
Evaluation of left ventricular function after acute MI carries a class I indication (ie, it should be performed).18
Echocardiography is commonly used, and it has a 60% sensitivity to detect a thrombus.19 In patients with poorer transthoracic echocardiographic windows, contrast can be used to better delineate the left ventricular cavity and show the thrombus. Transesophageal echocardiography is seldom useful, as the left ventricular apex is foreshortened and in the far field.
A left ventricular thrombus is confirmed if an echo-dense mass with well-demarcated margins distinct from the endocardium is seen throughout the cardiac cycle. It should be evident in at least two different views (apical and short-axis) and should be adjacent to a hypokinetic or akinetic left ventricular wall. False-positive findings can occur due to misidentified false tendons, papillary muscles, and trabeculae.
Cardiac MRI with late gadolinium enhancement is now the gold standard for diagnostic imaging, as it accurately characterizes the shape, size, and location of the thrombus (Figure 3). Gadolinium contrast increases the enhancement of the ventricular cavity, thus allowing easy detection of thrombus, which appears dark. Cardiac MRI with delayed enhancement has 88% to 91% sensitivity and 99% specificity to detect left ventricular thrombosis.20,21 However, compared with echocardiography, routine cardiac MRI is time-intensive, costly, and not routinely available. As a result, it should be performed only in patients with poor acoustic windows and a high clinical suspicion of left ventricular thrombosis.
Delayed-contrast cardiac computed tomography can be used to identify left ventricular thrombosis, using absence of contrast uptake. The need to use contrast is a disadvantage, but computed tomography can be an alternative in patients with contraindications to cardiac MRI.
WHAT COMPLICATIONS ARISE FROM LEFT VENTRICULAR THROMBOSIS?
The most feared complication of left ventricular thrombosis is thromboembolism. Cardioembolic stroke is generally severe, prone to early and long-term recurrence, and associated with a higher death rate than noncardioembolic ischemic stroke.22,23 Thrombi associated with thromboembolism are often acute and mobile rather than organized and immobile.24 They may embolize to the brain, spleen, kidneys, and bowel.25 In a meta-analysis of 11 studies, the pooled odds ratio for risk of embolization was 5.45 (95% confidence interval [CI] 3.02–9.83) with left ventricular thrombi vs without.26 Before systemic thrombolysis and antiplatelet therapy became available, stroke rates ranged from 1.5% to 10%.27–29
In a meta-analysis of 22 studies from 1978 to 2004, the incidence of ischemic stroke after MI during hospitalization was around 11.1 per 1,000 MIs.30 This study found that anterior MI was associated with a higher risk of stroke, but reported no difference in the incidence of stroke with percutaneous coronary intervention, systemic thrombolysis, or no reperfusion.
In a large prospective cohort study of 2,160 patients,31 259 (12%) had a stroke after MI. In multivariable analysis, age, diabetes, and previous stroke were predictors of stroke after MI. This study reported significantly fewer strokes in patients who underwent percutaneous coronary intervention than with other or no reperfusion therapies.31
ANTICOAGULATION TREATMENT
3. How would you treat a patient who has a drug-eluting stent in the left anterior descending artery and a new diagnosis of left ventricular thrombosis?
- Warfarin
- Aspirin and clopidogrel
- Aspirin, clopidogrel, and warfarin
- Aspirin and warfarin
The management of left ventricular thrombosis has been summarized in guidelines from the American College of Chest Physicians (ACCP) in 2012,32 and from the American College of Cardiology/American Heart Association in 2013,18 which recommend anticoagulation for at least 3 months, or indefinitely if bleeding risk is low, for all patients developing a left ventricular thrombus.
For patients with acute MI and left ventricular thrombosis, the ACCP guidelines recommend warfarin with a target international normalized ratio of 2.0 to 3.0 plus dual antiplatelet therapy (eg, aspirin plus clopidogrel) for 3 months, after which warfarin is discontinued but dual antiplatelet therapy is continued for up to 12 months.32
The European Society of Cardiology guidelines33 recommend 6 months of anticoagulation. However, if the patient is receiving dual antiplatelet therapy, they recommend repeated imaging of the left ventricle after 3 months of anticoagulation, which may allow for earlier discontinuation of anticoagulation if the thrombus has resolved and apical wall motion has recovered. Therefore, most experts recommend 3 months of anticoagulation when used in combination with dual antiplatelet therapy and repeating echocardiography at 3 months to safely discontinue anticoagulation. The best answer to the question posed here is aspirin, clopidogrel, and warfarin.
Decisions about antithrombotic therapy may also depend on stent type and the patient’s bleeding risk. With bare-metal stents, dual antiplatelet therapy along with anticoagulation should be used for 1 month, after which anticoagulation should be used with a single antiplatelet agent for another 2 months; after this, the anticoagulant can be discontinued and dual antiplatelet therapy can be resumed for a total of 12 months. Newer anticoagulants such as rivaroxaban, dabigatran, edoxaban, and apixaban may also have a role, but they have not yet been studied for this indication.
Surgical thrombectomy is rarely considered now, given the known efficacy of anticoagulants in dissolving the thrombus. It was done in the past for large, mobile, or protruding left ventricular thrombi, which have a higher potential for embolization.34 Currently, it can be done under very special circumstances, such as before placement of a left ventricular assist device or if the thrombus is large, to prevent embolism.35,36
BLEEDING COMPLICATIONS WITH TRIPLE ANTITHROMBOTIC THERAPY
After stent placement, almost all patients need to be on dual antiplatelet therapy for a specified duration depending on the type and generation of stent used. Such patients end up on “triple” antithrombotic therapy (two antiplatelet drugs plus an anticoagulant), which poses a high risk of bleeding.37 Consideration needs to be given to the risks of stroke, stent thrombosis, and major bleeding when selecting the antithrombotic regimen.38 Triple antithrombotic therapy has been associated with a risk of fatal and nonfatal bleeding of 4% to 16% when used for indications such as atrial fibrillation.39–41
Risks of triple antithrombotic therapy (aspirin 80–100 mg, clopidogrel 75 mg, and warfarin) were compared with those of clopidogrel plus warfarin in the What Is the Optimal Antiplatelet and Anticoagulant therapy in Patients With Oral Anticoagulation and Coronary Stenting Trial,37 which reported a significantly lower risk of major and minor bleeding with clopidogrel-plus-warfarin therapy than with triple antithrombotic therapy, 14.3% vs 31.7% (hazard ratio 0.40, 95% CI 0.28–0.58, P < .0001).
Additionally, the increased risk of major and minor bleeding associated with triple antithrombotic therapy has been confirmed in many observational studies; other studies found a trend toward lower risk with triple therapy, but this was not statistically significant (Table 2).38,40,42–55 A large multicenter European trial is being conducted to compare dual antiplatelet therapy vs triple antithrombotic therapy in patients with left ventricular thrombosis.
CASE FOLLOW-UP
Our patient was started on warfarin, clopidogrel 75 mg, and aspirin 75 mg at the time of discharge. He was continued on warfarin for 3 months, at which time a follow-up echocardiogram showed no thrombus in the left ventricle. Warfarin was discontinued, and he had no thromboembolic complications.
TAKE-HOME POINTS
Left ventricular thrombosis after an acute MI is very important to detect, as it can lead to serious complications through arterial embolism.
The incidence of left ventricular thrombosis has declined significantly with the use of percutaneous coronary intervention. However, it may still occur in a small number of patients with larger infarcts owing to delay in revascularization or proximal (left main or left anterior descending) occlusions with larger infarct size.
Echocardiography, which is routinely performed after acute MI to assess myocardial function, uncovers most left ventricular thrombi. In high-risk cases, MRI with late gadolinium enhancement can increase the diagnostic yield.
Anticoagulation with warfarin is recommended for at least 3 months. Post-MI patients undergoing stent implantation may need triple antithrombotic therapy, which, however, increases the bleeding risk significantly. Large randomized trials are needed to guide physicians in risk stratification of such patients.
A 62-year-old man with hypertension, type 2 diabetes mellitus, and hypercholesterolemia presented to the emergency department with substernal chest pain that started about 15 hours earlier while he was at rest watching television.
On examination, his pulse was 92 beats per minute and regular, his blood pressure was 160/88 mm Hg, and he had no evidence of jugular venous distention or pedal edema. Lung examination was positive for bibasilar crackles.
Electrocardiography revealed Q waves with ST elevation in leads I, aVL, V4, V5, and V6 with reciprocal ST depression in leads II, III, and aVF.
His troponin T level on presentation was markedly elevated.
He underwent heart catheterization and was found to have 100% occlusion of the proximal left anterior descending artery. He underwent successful percutaneous coronary intervention with placement of a drug-eluting stent, and afterward had grade 3 flow on the Thrombolysis in Myocardial Infarction (TIMI) scale.
Echocardiography the next day revealed a mobile echo-dense mass in the left ventricular apex (Figure 1) and a left ventricular ejection fraction of 35%.
THE INCIDENCE OF LEFT VENTRICULAR THROMBOSIS IN ACUTE MI
1. What is the incidence of left ventricular thrombosis after acute myocardial infarction (MI), now that primary percutaneous coronary intervention is common?
- 0.1%
- 2%
- 20%
- 40%
Left ventricular thrombosis is a serious complication of acute MI that can cause systemic thromboembolism, including stroke.1 Before thrombolytic therapy was available, this complication occurred in 20% to 60% of patients with acute MI.2,3 But early reperfusion strategies, anticoagulation for the first 48 hours, and dual antiplatelet therapy have reduced the incidence of this complication significantly.
In the thrombolytic era, the incidence of left ventricular thrombosis was 5.1% in the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI) 3 study, which had 8,326 patients. A subset of patients who had an anterior MI had almost double the incidence (11.5%).3
The incidence has further declined with the advent of primary percutaneous coronary intervention, likely thanks to enhanced myocardial salvage, and now ranges from 2.5% to 15% (Table 1).4–11 The largest observational study, with 2,911 patients undergoing percutaneous coronary intervention, reported an incidence of 2.5% within 3 to 5 days of the MI.7 At our center, the incidence was found to be even lower, 1.8% in 1,700 patients presenting with ST-elevation MI undergoing primary percutaneous coronary intervention. Hence, of the answers to the question above, 2% would be closest.
Large infarct size with a low left ventricular ejection fraction (< 40%), anterior wall MI, hypertension, and delay in time from symptom onset to intervention were independent predictors of left ventricular thrombus formation in most studies.7,12 The risk is highest during the first 2 weeks after MI, and thrombosis almost never occurs more than 3 months after the index event.5,13–16
WHAT IS THE PATHOGENESIS OF LEFT VENTRICULAR THROMBOSIS?
A large transmural infarct results in loss of contractile function, which causes stagnation and pooling of blood adjacent to the infarcted ventricular segment. In addition, endocardial injury exposes tissue factor, which then initiates the coagulation cascade. To make matters worse, MI results in a hypercoagulable state through unclear mechanisms, which completes the Virchow triad for thrombus formation. Elevations of D-dimer, fibrinogen, anticardiolipin antibodies (IgM and IgG), and tissue factor have also been reported after acute MI.17
Thrombus formation begins with platelet aggregation at the site of endocardial damage, forming a platelet plug, followed by activation of clotting factors. These thrombi are referred to as “mural,” as they adhere to the chamber wall (endocardium). They are composed of fibrin and entrapped red and white blood cells (Figure 2).
The natural course of thrombus evolution is established but variable. A left ventricular thrombus may dislodge and embolize, resulting in stroke or other thromboembolic complications. Alternately, it can dissolve over time, aided by intrinsic fibrinolytic mechanisms. On other occasions, the thrombus may organize, a process characterized by ingrowth of smooth muscle cells, fibroblasts, and endothelium.
HOW IS LEFT VENTRICULAR THROMBOSIS DIAGNOSED?
2. What is the best imaging test for detecting a thrombus?
- Transesophageal echocardiography
- Transthoracic echocardiography
- Cardiac magnetic resonance imaging (MRI) without gadolinium contrast
- Cardiac MRI with gadolinium contrast
Evaluation of left ventricular function after acute MI carries a class I indication (ie, it should be performed).18
Echocardiography is commonly used, and it has a 60% sensitivity to detect a thrombus.19 In patients with poorer transthoracic echocardiographic windows, contrast can be used to better delineate the left ventricular cavity and show the thrombus. Transesophageal echocardiography is seldom useful, as the left ventricular apex is foreshortened and in the far field.
A left ventricular thrombus is confirmed if an echo-dense mass with well-demarcated margins distinct from the endocardium is seen throughout the cardiac cycle. It should be evident in at least two different views (apical and short-axis) and should be adjacent to a hypokinetic or akinetic left ventricular wall. False-positive findings can occur due to misidentified false tendons, papillary muscles, and trabeculae.
Cardiac MRI with late gadolinium enhancement is now the gold standard for diagnostic imaging, as it accurately characterizes the shape, size, and location of the thrombus (Figure 3). Gadolinium contrast increases the enhancement of the ventricular cavity, thus allowing easy detection of thrombus, which appears dark. Cardiac MRI with delayed enhancement has 88% to 91% sensitivity and 99% specificity to detect left ventricular thrombosis.20,21 However, compared with echocardiography, routine cardiac MRI is time-intensive, costly, and not routinely available. As a result, it should be performed only in patients with poor acoustic windows and a high clinical suspicion of left ventricular thrombosis.
Delayed-contrast cardiac computed tomography can be used to identify left ventricular thrombosis, using absence of contrast uptake. The need to use contrast is a disadvantage, but computed tomography can be an alternative in patients with contraindications to cardiac MRI.
WHAT COMPLICATIONS ARISE FROM LEFT VENTRICULAR THROMBOSIS?
The most feared complication of left ventricular thrombosis is thromboembolism. Cardioembolic stroke is generally severe, prone to early and long-term recurrence, and associated with a higher death rate than noncardioembolic ischemic stroke.22,23 Thrombi associated with thromboembolism are often acute and mobile rather than organized and immobile.24 They may embolize to the brain, spleen, kidneys, and bowel.25 In a meta-analysis of 11 studies, the pooled odds ratio for risk of embolization was 5.45 (95% confidence interval [CI] 3.02–9.83) with left ventricular thrombi vs without.26 Before systemic thrombolysis and antiplatelet therapy became available, stroke rates ranged from 1.5% to 10%.27–29
In a meta-analysis of 22 studies from 1978 to 2004, the incidence of ischemic stroke after MI during hospitalization was around 11.1 per 1,000 MIs.30 This study found that anterior MI was associated with a higher risk of stroke, but reported no difference in the incidence of stroke with percutaneous coronary intervention, systemic thrombolysis, or no reperfusion.
In a large prospective cohort study of 2,160 patients,31 259 (12%) had a stroke after MI. In multivariable analysis, age, diabetes, and previous stroke were predictors of stroke after MI. This study reported significantly fewer strokes in patients who underwent percutaneous coronary intervention than with other or no reperfusion therapies.31
ANTICOAGULATION TREATMENT
3. How would you treat a patient who has a drug-eluting stent in the left anterior descending artery and a new diagnosis of left ventricular thrombosis?
- Warfarin
- Aspirin and clopidogrel
- Aspirin, clopidogrel, and warfarin
- Aspirin and warfarin
The management of left ventricular thrombosis has been summarized in guidelines from the American College of Chest Physicians (ACCP) in 2012,32 and from the American College of Cardiology/American Heart Association in 2013,18 which recommend anticoagulation for at least 3 months, or indefinitely if bleeding risk is low, for all patients developing a left ventricular thrombus.
For patients with acute MI and left ventricular thrombosis, the ACCP guidelines recommend warfarin with a target international normalized ratio of 2.0 to 3.0 plus dual antiplatelet therapy (eg, aspirin plus clopidogrel) for 3 months, after which warfarin is discontinued but dual antiplatelet therapy is continued for up to 12 months.32
The European Society of Cardiology guidelines33 recommend 6 months of anticoagulation. However, if the patient is receiving dual antiplatelet therapy, they recommend repeated imaging of the left ventricle after 3 months of anticoagulation, which may allow for earlier discontinuation of anticoagulation if the thrombus has resolved and apical wall motion has recovered. Therefore, most experts recommend 3 months of anticoagulation when used in combination with dual antiplatelet therapy and repeating echocardiography at 3 months to safely discontinue anticoagulation. The best answer to the question posed here is aspirin, clopidogrel, and warfarin.
Decisions about antithrombotic therapy may also depend on stent type and the patient’s bleeding risk. With bare-metal stents, dual antiplatelet therapy along with anticoagulation should be used for 1 month, after which anticoagulation should be used with a single antiplatelet agent for another 2 months; after this, the anticoagulant can be discontinued and dual antiplatelet therapy can be resumed for a total of 12 months. Newer anticoagulants such as rivaroxaban, dabigatran, edoxaban, and apixaban may also have a role, but they have not yet been studied for this indication.
Surgical thrombectomy is rarely considered now, given the known efficacy of anticoagulants in dissolving the thrombus. It was done in the past for large, mobile, or protruding left ventricular thrombi, which have a higher potential for embolization.34 Currently, it can be done under very special circumstances, such as before placement of a left ventricular assist device or if the thrombus is large, to prevent embolism.35,36
BLEEDING COMPLICATIONS WITH TRIPLE ANTITHROMBOTIC THERAPY
After stent placement, almost all patients need to be on dual antiplatelet therapy for a specified duration depending on the type and generation of stent used. Such patients end up on “triple” antithrombotic therapy (two antiplatelet drugs plus an anticoagulant), which poses a high risk of bleeding.37 Consideration needs to be given to the risks of stroke, stent thrombosis, and major bleeding when selecting the antithrombotic regimen.38 Triple antithrombotic therapy has been associated with a risk of fatal and nonfatal bleeding of 4% to 16% when used for indications such as atrial fibrillation.39–41
Risks of triple antithrombotic therapy (aspirin 80–100 mg, clopidogrel 75 mg, and warfarin) were compared with those of clopidogrel plus warfarin in the What Is the Optimal Antiplatelet and Anticoagulant therapy in Patients With Oral Anticoagulation and Coronary Stenting Trial,37 which reported a significantly lower risk of major and minor bleeding with clopidogrel-plus-warfarin therapy than with triple antithrombotic therapy, 14.3% vs 31.7% (hazard ratio 0.40, 95% CI 0.28–0.58, P < .0001).
Additionally, the increased risk of major and minor bleeding associated with triple antithrombotic therapy has been confirmed in many observational studies; other studies found a trend toward lower risk with triple therapy, but this was not statistically significant (Table 2).38,40,42–55 A large multicenter European trial is being conducted to compare dual antiplatelet therapy vs triple antithrombotic therapy in patients with left ventricular thrombosis.
CASE FOLLOW-UP
Our patient was started on warfarin, clopidogrel 75 mg, and aspirin 75 mg at the time of discharge. He was continued on warfarin for 3 months, at which time a follow-up echocardiogram showed no thrombus in the left ventricle. Warfarin was discontinued, and he had no thromboembolic complications.
TAKE-HOME POINTS
Left ventricular thrombosis after an acute MI is very important to detect, as it can lead to serious complications through arterial embolism.
The incidence of left ventricular thrombosis has declined significantly with the use of percutaneous coronary intervention. However, it may still occur in a small number of patients with larger infarcts owing to delay in revascularization or proximal (left main or left anterior descending) occlusions with larger infarct size.
Echocardiography, which is routinely performed after acute MI to assess myocardial function, uncovers most left ventricular thrombi. In high-risk cases, MRI with late gadolinium enhancement can increase the diagnostic yield.
Anticoagulation with warfarin is recommended for at least 3 months. Post-MI patients undergoing stent implantation may need triple antithrombotic therapy, which, however, increases the bleeding risk significantly. Large randomized trials are needed to guide physicians in risk stratification of such patients.
- Lip GY, Piotrponikowski P, Andreotti F, et al; Heart Failure Association (EHFA) of the European Society of Cardiology (ESC) and the ESC Working Group on Thrombosis. Thromboembolism and antithrombotic therapy for heart failure in sinus rhythm: an executive summary of a joint consensus document from the ESC Heart Failure Association and the ESC Working Group on Thrombosis. Thromb Haemost 2012; 108:1009–1022.
- Turpie AG, Robinson JG, Doyle DJ, et al. Comparison of high-dose with low-dose subcutaneous heparin to prevent left ventricular mural thrombosis in patients with acute transmural anterior myocardial infarction. N Engl J Med 1989; 320:352–357.
- Chiarella F, Santoro E, Domenicucci S, Maggioni A, Vecchio C. Predischarge two-dimensional echocardiographic evaluation of left ventricular thrombosis after acute myocardial infarction in the GISSI-3 study. Am J Cardiol 1998; 81:822–827.
- Kalra A, Jang IK. Prevalence of early left ventricular thrombus after primary coronary intervention for acute myocardial infarction. J Thromb Thrombolysis 2000; 10:133–136.
- Nayak D, Aronow WS, Sukhija R, McClung JA, Monsen CE, Belkin RN. Comparison of frequency of left ventricular thrombi in patients with anterior wall versus non-anterior wall acute myocardial infarction treated with antithrombotic and antiplatelet therapy with or without coronary revascularization. Am J Cardiol 2004; 93:1529–1530.
- Rehan A, Kanwar M, Rosman H, et al. Incidence of post myocardial infarction left ventricular thrombus formation in the era of primary percutaneous intervention and glycoprotein IIb/IIIa inhibitors. A prospective observational study. Cardiovasc Ultrasound 2006;4:20.
- Zielinska M, Kaczmarek K, Tylkowski M. Predictors of left ventricular thrombus formation in acute myocardial infarction treated with successful primary angioplasty with stenting. Am J Med Sci 2008; 335:171–176.
- Osherov AB, Borovik-Raz M, Aronson D, et al. Incidence of early left ventricular thrombus after acute anterior wall myocardial infarction in the primary coronary intervention era. Am Heart J 2009; 157:1074–1080.
- Solheim S, Seljeflot I, Lunde K, et al. Frequency of left ventricular thrombus in patients with anterior wall acute myocardial infarction treated with percutaneous coronary intervention and dual antiplatelet therapy. Am J Cardiol 2010; 106:1197–1200.
- Shacham Y, Leshem-Rubinow E, Ben Assa E, et al. Comparison of C-reactive protein and fibrinogen levels in patients having anterior wall ST-segment elevation myocardial infarction with versus without left ventricular thrombus (from a primary percutaneous coronary intervention cohort). Am J Cardiol 2013; 112:57–60.
- Gianstefani S, Douiri A, Delithanasis I, et al. Incidence and predictors of early left ventricular thrombus after ST-elevation myocardial infarction in the contemporary era of primary percutaneous coronary intervention. Am J Cardiol 2014; 113:1111–1116.
- Shacham Y, Birati EY, Rogovski O, Cogan Y, Keren G, Roth A. Left ventricular thrombus formation and bleeding complications during continuous in-hospital anticoagulation for acute anterior myocardial infarction. Isr Med Assoc J 2012; 14:742–746.
- Asinger RW, Mikell FL, Elsperger J, Hodges M. Incidence of left-ventricular thrombosis after acute transmural myocardial infarction. Serial evaluation by two-dimensional echocardiography. N Engl J Med 1981; 305:297–302.
- Nihoyannopoulos P, Smith GC, Maseri A, Foale RA. The natural history of left ventricular thrombus in myocardial infarction: a rationale in support of masterly inactivity. J Am Coll Cardiol 1989; 14:903–911.
- Weinreich DJ, Burke JF, Pauletto FJ. Left ventricular mural thrombi complicating acute myocardial infarction. Long-term follow-up with serial echocardiography. Ann Intern Med 1984; 100:789–794.
- Greaves SC, Zhi G, Lee RT, et al. Incidence and natural history of left ventricular thrombus following anterior wall acute myocardial infarction. Am J Cardiol 1997; 80:442–448.
- Solheim S, Seljeflot I, Lunde K, et al. Prothrombotic markers in patients with acute myocardial infarction and left ventricular thrombus formation treated with pci and dual antiplatelet therapy. Thromb J 2013; 11:1.
- O’Gara PT, Kushner FG, Ascheim DD, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013; 127:e362–e425.
- Weinsaft JW, Kim HW, Crowley AL, et al. LV thrombus detection by routine echocardiography: insights into performance characteristics using delayed enhancement CMR. JACC Cardiovasc Imaging 2011; 4:702–712.
- Mollet NR, Dymarkowski S, Volders W, et al. Visualization of ventricular thrombi with contrast-enhanced magnetic resonance imaging in patients with ischemic heart disease. Circulation 2002; 106:2873–2876.
- Srichai MB, Junor C, Rodriguez LL, et al. Clinical, imaging, and pathological characteristics of left ventricular thrombus: a comparison of contrast-enhanced magnetic resonance imaging, transthoracic echocardiography, and transesophageal echocardiography with surgical or pathological validation. Am Heart J 2006; 152:75–84.
- Eriksson SE, Olsson JE. Survival and recurrent strokes in patients with different subtypes of stroke: a fourteen-year follow-up study. Cerebrovasc Dis 2001; 12:171–180.
- Grau AJ, Weimar C, Buggle F, et al. Risk factors, outcome, and treatment in subtypes of ischemic stroke: the German Stroke Data Bank. Stroke 2001; 32:2559–2566.
- Keren A, Goldberg S, Gottlieb S, et al. Natural history of left ventricular thrombi: their appearance and resolution in the posthospitalization period of acute myocardial infarction. J Am Coll Cardiol 1990; 15:790–800.
- Jordan RA, Miller RD, Edwards JE, Parker RL. Thrombo-embolism in acute and in healed myocardial infarction. I. Intracardiac mural thrombosis. Circulation 1952; 6:1–6.
- Vaitkus PT, Barnathan ES. Embolic potential, prevention and management of mural thrombus complicating anterior myocardial infarction: a meta-analysis. J Am Coll Cardiol 1993; 22:1004–1009.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- Cabin HS, Roberts WC. Left ventricular aneurysm, intraaneurysmal thrombus and systemic embolus in coronary heart disease. Chest 1980; 77:586–590.
- Keating EC, Gross SA, Schlamowitz RA, et al. Mural thrombi in myocardial infarctions. Prospective evaluation by two-dimensional echocardiography. Am J Med 1983; 74:989–995.
- Witt BJ, Ballman KV, Brown RD Jr, Meverden RA, Jacobsen SJ, Roger VL. The incidence of stroke after myocardial infarction: a meta-analysis. Am J Med 2006; 119:354.e1–354.e9.
- Witt BJ, Brown RD Jr, Jacobsen SJ, Weston SA, Yawn BP, Roger VL. A community-based study of stroke incidence after myocardial infarction. Ann Intern Med 2005; 143:785–792.
- Vandvik PO, Lincoff AM, Gore JM, et al; American College of Chest Physicians. Primary and secondary prevention of cardiovascular disease: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl):e637S–e68S.
- Steg G, James SK, Atar D, et al. ESC guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J 2012; 33:2569–2619.
- Nili M, Deviri E, Jortner R, Strasberg B, Levy MJ. Surgical removal of a mobile, pedunculated left ventricular thrombus: report of 4 cases. Ann Thorac Surg 1988; 46:396–400.
- Kanemitsu S, Miyake Y, Okabe M. Surgical removal of a left ventricular thrombus associated with cardiac sarcoidosis. Interact Cardiovasc Thorac Surg 2008; 7:333–335.
- Engin C, Yagdi T, Balcioglu O, et al. Left ventricular assist device implantation in heart failure patients with a left ventricular thrombus. Transplant Proc 2013; 45:1017–1019.
- Dewilde WJ, Oirbans T, Verheugt FW, et al; WOEST study investigators. Use of clopidogrel with or without aspirin in patients taking oral anticoagulant therapy and undergoing percutaneous coronary intervention: an open-label, randomised, controlled trial. Lancet 2013; 381:1107–1115.
- Faxon DP, Eikelboom JW, Berger PB, et al. Antithrombotic therapy in patients with atrial fibrillation undergoing coronary stenting: a North American perspective: executive summary. Circ Cardiovasc Interv 2011; 4:522–534.
- Hansen ML, Sorensen R, Clausen MT, et al. Risk of bleeding with single, dual, or triple therapy with warfarin, aspirin, and clopidogrel in patients with atrial fibrillation. Arch Intern Med 2010; 170:1433–1441.
- Karjalainen PP, Porela P, Ylitalo A, et al. Safety and efficacy of combined antiplatelet-warfarin therapy after coronary stenting. Eur Heart J 2007; 28:726–732.
- Doyle BJ, Rihal CS, Gastineau DA, Holmes DR Jr. Bleeding, blood transfusion, and increased mortality after percutaneous coronary intervention: implications for contemporary practice. J Am Coll Cardiol 2009; 53:2019–2027.
- Azoulay L, Dell’Aniello S, Simon T, Renoux C, Suissa S. The concurrent use of antithrombotic therapies and the risk of bleeding in patients with atrial fibrillation. Thromb Haemost 2013; 109:431–439.
- Deshmukh A, Hilleman DE, Del Core M, Nair CK. Antithrombotic regimens in patients with indication for long-term anticoagulation undergoing coronary interventions-systematic analysis, review of literature, and implications on management. Am J Ther 2013; 20:654–663.
- Fosbol EL, Wang TY, Li S, et al. Warfarin use among older atrial fibrillation patients with non-ST-segment elevation myocardial infarction managed with coronary stenting and dual antiplatelet therapy. Am Heart J 2013; 166:864–870.
- Gao F, Zhou YJ, Wang ZJ, et al. Meta-analysis of the combination of warfarin and dual antiplatelet therapy after coronary stenting in patients with indications for chronic oral anticoagulation. Int J Cardiol 2011; 148:96–101.
- Hansen ML, Sorensen R, Clausen MT, et al. Risk of bleeding with single, dual, or triple therapy with warfarin, aspirin, and clopidogrel in patients with atrial fibrillation. Arch Intern Med 2010; 170:1433–1441.
- Hermosillo AJ, Spinler SA. Aspirin, clopidogrel, and warfarin: is the combination appropriate and effective or inappropriate and too dangerous? Ann Pharmacother 2008; 42:790–805.
- Holmes DR Jr, Kereiakes DJ, Kleiman NS, Moliterno DJ, Patti G, Grines CL. Combining antiplatelet and anticoagulant therapies. J Am Coll Cardiol 2009; 54:95–109.
- Khurram Z, Chou E, Minutello R, et al. Combination therapy with aspirin, clopidogrel and warfarin following coronary stenting is associated with a significant risk of bleeding. J Invasive Cardiol 2006; 18:162–164.
- Orford JL, Fasseas P, Melby S, et al. Safety and efficacy of aspirin, clopidogrel, and warfarin after coronary stent placement in patients with an indication for anticoagulation. Am Heart J 2004; 147:463–467.
- Porter A, Konstantino Y, Iakobishvili Z, Shachar L, Battler A, Hasdai D. Short-term triple therapy with aspirin, warfarin, and a thienopyridine among patients undergoing percutaneous coronary intervention. Catheter Cardiovasc Interv 2006; 68:56–61.
- DeEugenio D, Kolman L, DeCaro M, et al. Risk of major bleeding with concomitant dual antiplatelet therapy after percutaneous coronary intervention in patients receiving long-term warfarin therapy. Pharmacotherapy 2007; 27:691–696.
- Ruiz-Nodar JM, Marin F, Hurtado JA, et al. Anticoagulant and antiplatelet therapy use in 426 patients with atrial fibrillation undergoing percutaneous coronary intervention and stent implantation implications for bleeding risk and prognosis. J Am Coll Cardiol 2008; 51:818–825.
- Sarafoff N, Ndrepepa G, Mehilli J, et al. Aspirin and clopidogrel with or without phenprocoumon after drug eluting coronary stent placement in patients on chronic oral anticoagulation. J Intern Med 2008; 264:472–480.
- Rossini R, Musumeci GF, Lettieri CF, et al. Long-term outcomes in patients undergoing coronary stenting on dual oral antiplatelet treatment requiring oral anticoagulant therapy. Am J Cardiol 2008; 102:1618–1623.
- Lip GY, Piotrponikowski P, Andreotti F, et al; Heart Failure Association (EHFA) of the European Society of Cardiology (ESC) and the ESC Working Group on Thrombosis. Thromboembolism and antithrombotic therapy for heart failure in sinus rhythm: an executive summary of a joint consensus document from the ESC Heart Failure Association and the ESC Working Group on Thrombosis. Thromb Haemost 2012; 108:1009–1022.
- Turpie AG, Robinson JG, Doyle DJ, et al. Comparison of high-dose with low-dose subcutaneous heparin to prevent left ventricular mural thrombosis in patients with acute transmural anterior myocardial infarction. N Engl J Med 1989; 320:352–357.
- Chiarella F, Santoro E, Domenicucci S, Maggioni A, Vecchio C. Predischarge two-dimensional echocardiographic evaluation of left ventricular thrombosis after acute myocardial infarction in the GISSI-3 study. Am J Cardiol 1998; 81:822–827.
- Kalra A, Jang IK. Prevalence of early left ventricular thrombus after primary coronary intervention for acute myocardial infarction. J Thromb Thrombolysis 2000; 10:133–136.
- Nayak D, Aronow WS, Sukhija R, McClung JA, Monsen CE, Belkin RN. Comparison of frequency of left ventricular thrombi in patients with anterior wall versus non-anterior wall acute myocardial infarction treated with antithrombotic and antiplatelet therapy with or without coronary revascularization. Am J Cardiol 2004; 93:1529–1530.
- Rehan A, Kanwar M, Rosman H, et al. Incidence of post myocardial infarction left ventricular thrombus formation in the era of primary percutaneous intervention and glycoprotein IIb/IIIa inhibitors. A prospective observational study. Cardiovasc Ultrasound 2006;4:20.
- Zielinska M, Kaczmarek K, Tylkowski M. Predictors of left ventricular thrombus formation in acute myocardial infarction treated with successful primary angioplasty with stenting. Am J Med Sci 2008; 335:171–176.
- Osherov AB, Borovik-Raz M, Aronson D, et al. Incidence of early left ventricular thrombus after acute anterior wall myocardial infarction in the primary coronary intervention era. Am Heart J 2009; 157:1074–1080.
- Solheim S, Seljeflot I, Lunde K, et al. Frequency of left ventricular thrombus in patients with anterior wall acute myocardial infarction treated with percutaneous coronary intervention and dual antiplatelet therapy. Am J Cardiol 2010; 106:1197–1200.
- Shacham Y, Leshem-Rubinow E, Ben Assa E, et al. Comparison of C-reactive protein and fibrinogen levels in patients having anterior wall ST-segment elevation myocardial infarction with versus without left ventricular thrombus (from a primary percutaneous coronary intervention cohort). Am J Cardiol 2013; 112:57–60.
- Gianstefani S, Douiri A, Delithanasis I, et al. Incidence and predictors of early left ventricular thrombus after ST-elevation myocardial infarction in the contemporary era of primary percutaneous coronary intervention. Am J Cardiol 2014; 113:1111–1116.
- Shacham Y, Birati EY, Rogovski O, Cogan Y, Keren G, Roth A. Left ventricular thrombus formation and bleeding complications during continuous in-hospital anticoagulation for acute anterior myocardial infarction. Isr Med Assoc J 2012; 14:742–746.
- Asinger RW, Mikell FL, Elsperger J, Hodges M. Incidence of left-ventricular thrombosis after acute transmural myocardial infarction. Serial evaluation by two-dimensional echocardiography. N Engl J Med 1981; 305:297–302.
- Nihoyannopoulos P, Smith GC, Maseri A, Foale RA. The natural history of left ventricular thrombus in myocardial infarction: a rationale in support of masterly inactivity. J Am Coll Cardiol 1989; 14:903–911.
- Weinreich DJ, Burke JF, Pauletto FJ. Left ventricular mural thrombi complicating acute myocardial infarction. Long-term follow-up with serial echocardiography. Ann Intern Med 1984; 100:789–794.
- Greaves SC, Zhi G, Lee RT, et al. Incidence and natural history of left ventricular thrombus following anterior wall acute myocardial infarction. Am J Cardiol 1997; 80:442–448.
- Solheim S, Seljeflot I, Lunde K, et al. Prothrombotic markers in patients with acute myocardial infarction and left ventricular thrombus formation treated with pci and dual antiplatelet therapy. Thromb J 2013; 11:1.
- O’Gara PT, Kushner FG, Ascheim DD, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013; 127:e362–e425.
- Weinsaft JW, Kim HW, Crowley AL, et al. LV thrombus detection by routine echocardiography: insights into performance characteristics using delayed enhancement CMR. JACC Cardiovasc Imaging 2011; 4:702–712.
- Mollet NR, Dymarkowski S, Volders W, et al. Visualization of ventricular thrombi with contrast-enhanced magnetic resonance imaging in patients with ischemic heart disease. Circulation 2002; 106:2873–2876.
- Srichai MB, Junor C, Rodriguez LL, et al. Clinical, imaging, and pathological characteristics of left ventricular thrombus: a comparison of contrast-enhanced magnetic resonance imaging, transthoracic echocardiography, and transesophageal echocardiography with surgical or pathological validation. Am Heart J 2006; 152:75–84.
- Eriksson SE, Olsson JE. Survival and recurrent strokes in patients with different subtypes of stroke: a fourteen-year follow-up study. Cerebrovasc Dis 2001; 12:171–180.
- Grau AJ, Weimar C, Buggle F, et al. Risk factors, outcome, and treatment in subtypes of ischemic stroke: the German Stroke Data Bank. Stroke 2001; 32:2559–2566.
- Keren A, Goldberg S, Gottlieb S, et al. Natural history of left ventricular thrombi: their appearance and resolution in the posthospitalization period of acute myocardial infarction. J Am Coll Cardiol 1990; 15:790–800.
- Jordan RA, Miller RD, Edwards JE, Parker RL. Thrombo-embolism in acute and in healed myocardial infarction. I. Intracardiac mural thrombosis. Circulation 1952; 6:1–6.
- Vaitkus PT, Barnathan ES. Embolic potential, prevention and management of mural thrombus complicating anterior myocardial infarction: a meta-analysis. J Am Coll Cardiol 1993; 22:1004–1009.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- Cabin HS, Roberts WC. Left ventricular aneurysm, intraaneurysmal thrombus and systemic embolus in coronary heart disease. Chest 1980; 77:586–590.
- Keating EC, Gross SA, Schlamowitz RA, et al. Mural thrombi in myocardial infarctions. Prospective evaluation by two-dimensional echocardiography. Am J Med 1983; 74:989–995.
- Witt BJ, Ballman KV, Brown RD Jr, Meverden RA, Jacobsen SJ, Roger VL. The incidence of stroke after myocardial infarction: a meta-analysis. Am J Med 2006; 119:354.e1–354.e9.
- Witt BJ, Brown RD Jr, Jacobsen SJ, Weston SA, Yawn BP, Roger VL. A community-based study of stroke incidence after myocardial infarction. Ann Intern Med 2005; 143:785–792.
- Vandvik PO, Lincoff AM, Gore JM, et al; American College of Chest Physicians. Primary and secondary prevention of cardiovascular disease: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl):e637S–e68S.
- Steg G, James SK, Atar D, et al. ESC guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J 2012; 33:2569–2619.
- Nili M, Deviri E, Jortner R, Strasberg B, Levy MJ. Surgical removal of a mobile, pedunculated left ventricular thrombus: report of 4 cases. Ann Thorac Surg 1988; 46:396–400.
- Kanemitsu S, Miyake Y, Okabe M. Surgical removal of a left ventricular thrombus associated with cardiac sarcoidosis. Interact Cardiovasc Thorac Surg 2008; 7:333–335.
- Engin C, Yagdi T, Balcioglu O, et al. Left ventricular assist device implantation in heart failure patients with a left ventricular thrombus. Transplant Proc 2013; 45:1017–1019.
- Dewilde WJ, Oirbans T, Verheugt FW, et al; WOEST study investigators. Use of clopidogrel with or without aspirin in patients taking oral anticoagulant therapy and undergoing percutaneous coronary intervention: an open-label, randomised, controlled trial. Lancet 2013; 381:1107–1115.
- Faxon DP, Eikelboom JW, Berger PB, et al. Antithrombotic therapy in patients with atrial fibrillation undergoing coronary stenting: a North American perspective: executive summary. Circ Cardiovasc Interv 2011; 4:522–534.
- Hansen ML, Sorensen R, Clausen MT, et al. Risk of bleeding with single, dual, or triple therapy with warfarin, aspirin, and clopidogrel in patients with atrial fibrillation. Arch Intern Med 2010; 170:1433–1441.
- Karjalainen PP, Porela P, Ylitalo A, et al. Safety and efficacy of combined antiplatelet-warfarin therapy after coronary stenting. Eur Heart J 2007; 28:726–732.
- Doyle BJ, Rihal CS, Gastineau DA, Holmes DR Jr. Bleeding, blood transfusion, and increased mortality after percutaneous coronary intervention: implications for contemporary practice. J Am Coll Cardiol 2009; 53:2019–2027.
- Azoulay L, Dell’Aniello S, Simon T, Renoux C, Suissa S. The concurrent use of antithrombotic therapies and the risk of bleeding in patients with atrial fibrillation. Thromb Haemost 2013; 109:431–439.
- Deshmukh A, Hilleman DE, Del Core M, Nair CK. Antithrombotic regimens in patients with indication for long-term anticoagulation undergoing coronary interventions-systematic analysis, review of literature, and implications on management. Am J Ther 2013; 20:654–663.
- Fosbol EL, Wang TY, Li S, et al. Warfarin use among older atrial fibrillation patients with non-ST-segment elevation myocardial infarction managed with coronary stenting and dual antiplatelet therapy. Am Heart J 2013; 166:864–870.
- Gao F, Zhou YJ, Wang ZJ, et al. Meta-analysis of the combination of warfarin and dual antiplatelet therapy after coronary stenting in patients with indications for chronic oral anticoagulation. Int J Cardiol 2011; 148:96–101.
- Hansen ML, Sorensen R, Clausen MT, et al. Risk of bleeding with single, dual, or triple therapy with warfarin, aspirin, and clopidogrel in patients with atrial fibrillation. Arch Intern Med 2010; 170:1433–1441.
- Hermosillo AJ, Spinler SA. Aspirin, clopidogrel, and warfarin: is the combination appropriate and effective or inappropriate and too dangerous? Ann Pharmacother 2008; 42:790–805.
- Holmes DR Jr, Kereiakes DJ, Kleiman NS, Moliterno DJ, Patti G, Grines CL. Combining antiplatelet and anticoagulant therapies. J Am Coll Cardiol 2009; 54:95–109.
- Khurram Z, Chou E, Minutello R, et al. Combination therapy with aspirin, clopidogrel and warfarin following coronary stenting is associated with a significant risk of bleeding. J Invasive Cardiol 2006; 18:162–164.
- Orford JL, Fasseas P, Melby S, et al. Safety and efficacy of aspirin, clopidogrel, and warfarin after coronary stent placement in patients with an indication for anticoagulation. Am Heart J 2004; 147:463–467.
- Porter A, Konstantino Y, Iakobishvili Z, Shachar L, Battler A, Hasdai D. Short-term triple therapy with aspirin, warfarin, and a thienopyridine among patients undergoing percutaneous coronary intervention. Catheter Cardiovasc Interv 2006; 68:56–61.
- DeEugenio D, Kolman L, DeCaro M, et al. Risk of major bleeding with concomitant dual antiplatelet therapy after percutaneous coronary intervention in patients receiving long-term warfarin therapy. Pharmacotherapy 2007; 27:691–696.
- Ruiz-Nodar JM, Marin F, Hurtado JA, et al. Anticoagulant and antiplatelet therapy use in 426 patients with atrial fibrillation undergoing percutaneous coronary intervention and stent implantation implications for bleeding risk and prognosis. J Am Coll Cardiol 2008; 51:818–825.
- Sarafoff N, Ndrepepa G, Mehilli J, et al. Aspirin and clopidogrel with or without phenprocoumon after drug eluting coronary stent placement in patients on chronic oral anticoagulation. J Intern Med 2008; 264:472–480.
- Rossini R, Musumeci GF, Lettieri CF, et al. Long-term outcomes in patients undergoing coronary stenting on dual oral antiplatelet treatment requiring oral anticoagulant therapy. Am J Cardiol 2008; 102:1618–1623.
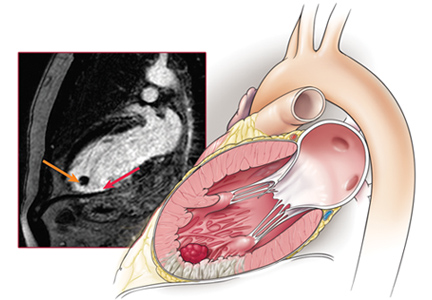
Correction: Anemia of chronic kidney disease
The article “Anemia of chronic kidney disease: Treat it, but not too aggressively” by Drs. Georges Nakhoul and James F. Simon (Cleve Clin J Med 2016; 83:613–624) contained a typographical error. In Table 2, the target ferritin level in chronic kidney disease is given as greater than 100 ng/dL, and for end-stage renal disease 200 to 1,200 ng/dL. Ferritin levels are measured in ng/mL, not ng/dL.
The article “Anemia of chronic kidney disease: Treat it, but not too aggressively” by Drs. Georges Nakhoul and James F. Simon (Cleve Clin J Med 2016; 83:613–624) contained a typographical error. In Table 2, the target ferritin level in chronic kidney disease is given as greater than 100 ng/dL, and for end-stage renal disease 200 to 1,200 ng/dL. Ferritin levels are measured in ng/mL, not ng/dL.
The article “Anemia of chronic kidney disease: Treat it, but not too aggressively” by Drs. Georges Nakhoul and James F. Simon (Cleve Clin J Med 2016; 83:613–624) contained a typographical error. In Table 2, the target ferritin level in chronic kidney disease is given as greater than 100 ng/dL, and for end-stage renal disease 200 to 1,200 ng/dL. Ferritin levels are measured in ng/mL, not ng/dL.
Evolution of heart failure management: Miles to go
The woods are lovely, dark and deep,
But I have promises to keep,
And miles to go before I sleep,
And miles to go before I sleep.
—Robert Frost, “Stopping by Woods on a Snowy Evening”1
Frost's words are simple yet elegant. They can be interpreted many ways. I see the allegory of life as a journey in this poem. The passage, like the woods, is beautiful, but there is a long, long way to go.
And so it is with the treatment of heart failure. There is beauty in our understanding of the syndrome’s physiologic complexities and natural history, and of effective treatments uncovered. Still, we’ve a monstrous climb ahead to get to the summit of this clinical challenge in order to start a real descent.
THE PAST, PRESENT, AND FUTURE OF HEART FAILURE THERAPY
Okwuosa et al,2 in this issue of the Journal, have capably summarized the ABCs of treating heart failure with reduced ejection fraction (also called systolic heart failure), approaching the subject from a perspective on past, present, and future therapies. They summarize heart failure interventions with a guideline-based philosophy, pointing out that these care paths are supposed to be evidence-based. They observe that in the 1960s the standard of care was digitalis, diuretics (furosemide first became available in 1967), and rest. That was about all we had for this problem.
There are now many drugs, devices, and operations that help patients with heart failure. But they never really cure the disease or, more aptly, the syndrome—and therapies are supposed to cure. This limitation of present therapies is important, given the disturbing epidemiology of heart failure, its economic cost, and the suffering of patients. That burden is well detailed.
In addition to curing, the overarching goals of treatment generally are to ameliorate distressing symptoms and to prevent comorbidities. In heart failure with reduced ejection fraction, we want to prevent premature death, stroke, myocardial infarction, congestive states, hospitalization, renal insufficiency, renal failure, cachexia, inanition, feebleness, and respiratory distress, among others.
The ABC mnemonic of Okwuosa et al will help caregivers remember the basics. It is important, however, to put algorithms into proper perspective and to look toward the future.
PROBLEMS WITH EVIDENCE-BASED MEDICINE
Several problems with our current heart failure treatments are rooted in how we perform clinical trials, arguably the premier method of determining truth in clinical practice and the foundation of evidence-based medicine.3,4
Do the trials represent real-world practice?
Were the clinical trials that led to regulatory approval and professional society endorsement of the therapies that we prescribe in our offices done in the same sorts of patients as those in our waiting rooms asking for help? Perhaps, for the most part, they have been. And thus, Okwuosa et al have crafted a work relevant to all of us and every patient.
But I believe there are major gaps in the types of participants enrolled in trials, eg, underrepresentation of certain racial and ethnic groups, not to mention the relative paucity of women. The very elderly (a rapidly growing population) have largely been ignored as well, and participants with significant renal insufficiency, anemia, and diabetes mellitus seem far fewer than what we deal with in a busy clinic.
In addition, Okwuosa et al focus only on patients with reduced left ventricular ejection fraction, a group that makes up only about half of the heart failure crowd.
What about quality of life and other important outcomes?
Clinical trials in heart failure with reduced ejection fraction have generally focused on major clinical end points (primarily, but not exclusively, mortality), to the exclusion of quality of life. Though sometimes included in trials, quality-of-life metrics generally get relegated to second-class seats or ‘tween-deck steerage. Perhaps that is because measuring quality of life can be time-consuming and difficult.
Yet, in the words of sociologist William Bruce Cameron, not everything that counts can be counted, and not everything that can be counted counts. That goes for quality of life.
Lies, damned lies, and P values
Quandaries in data management and analysis include what to do about trial dropouts, study power, precision of statistical analysis, intention-to-treat principles, and choice of the P value that defines significance (or not) for any end point observation. Of course, there are myriad sophisticated mathematical and statistical reasons to justify why we don’t simply count on-treatment participants or allow imputation of results when patients or results drop out, forcing us to worship at the altar of P < .05.
A review of the P value concept5 recently appeared with an accompanying editorial by Kyriacou6 that concluded that “the automatic application of dichotomized hypothesis testing based on prearranged levels of statistical significance should be substituted with a more complex process using effect estimates, confidence intervals, and even P values, thereby permitting scientists, statisticians, and clinicians to use their own inferential capabilities to assign scientific significance.”6
How many great treatments have we tossed out because of rigid reliance on old-fashioned approaches to determining therapeutic evidence? Many treatments studied have had great results in a minority of patients in clinical trials but did not have a major positive (or negative) impact on the overall cohort (with lack of primary end point statistical significance). And what to do when the primary end point is a neutral or negative one but secondary end points are positive? Why not focus more attention on those patients benefiting from an intervention despite the overall results of any trial?
Dilemmas of trials
Other issues are that clinical trials cost too much, and that recruitment and follow-up take too long. Intercurrent therapies (and guidelines) can emerge that jeopardize the trial itself or make observations untimely. The dilemma of stacking therapies one on top of another, often making patient compliance impossible, is another problem with clinical trials. Yet this is how we get to the ABCs.
A NEW WAY TO DO TRIALS
The information provided by Okwuosa et al is useful and encouraging, but too many gaps exist in our heart failure therapies to permit us to celebrate with exuberance. Too many patients still suffer, too many die too young, and the costs are still too great.
Perhaps the future of therapeutic development should embrace different and better ways to demonstrate real value (relying on the equation of value equals outcomes meaningful to patients, divided by cost) of therapies, including the old, the new, the trashed and the underdeveloped. More creative data analysis to reexamine the current tools on the shelf and the ones tried but discarded is essential.
A position paper from the Cardiovascular Round Table of the European Society of Cardiology concluded that “a coordinated effort involving academia, regulators, industry and payors will help to foster better and more effective conduct of clinical cardiovascular trials, supporting earlier availability of innovative therapies and better management of cardiovascular diseases.”7
Lauer and D’Agostino,8 also in an editorial, argued for innovative methods of doing clinical trials and discovering truth about therapies that are applicable to the future of developing treatments for heart failure with reduced ejection fraction. They noted that “the randomized registry trial represents a disruptive technology” and wondered if it will be “given serious consideration as a way to resolve the recognized limitations of current clinical-trial design.”8
Indeed, conducting megatrials with existing megadatabases using a registry format could help. Registries emerging from early adaptive trial design efforts, particularly when Bayesian analysis theory is applied, might help inform clinical experience faster and more efficiently. Bayesian analysis is a statistical approach that attempts to estimate parameters of an underlying distribution of events in an ongoing fashion based on the observed distribution. A clinical trial of stem cell therapies could, at the end of the trial, be turned into a multicenter registry that would continue to inform us about the more real-world application of newer treatment approaches.
Though the therapeutic cupboard for heart failure is certainly not bare, as Okwuosa et al point out, it is wanting. Let’s look for new therapeutic ABCs differently. We should be attacking the real challenge—curing the disease processes that cause the syndrome. Yes, there are miles to go before we sleep.
- Frost R. Stopping by Woods on a Snowy Evening. In: New Hampshire. New York, Henry Holt, 1923.
- Okwuoso IS, Ojeifo O, Nwabueze C, et al. The ABCs of managing systolic heart failure: the past, present and future. Cleve Clin J Med 2016; 83:753–765.
- Samman Tahhan A, Vaduganathan M, Kelkar A, et al. Trends in heart failure clinical trials from 2001–2012. J Card Fail 2016; 22:171–179.
- Cohn JN. Trials and tribulations. J Card Fail 2016; 22:180–181.
- Chavalarias D, Wallach JD, Li AH, Ioannidis JP. Evolution of reporting P values in the biomedical literature, 1990–2015. JAMA 2016; 315:1141–1148.
- Kyriacou DN. The enduring evolution of the P value. JAMA 2016; 315:1113–1115.
- Jackson N, Atar D, Borentain M, et al. Improving clinical trials for cardiovascular diseases: a position paper from the Cardiovascular Round Table of the European Society of Cardiology. Eur Heart J 2016; 37:747–754.
- Lauer MS, D’Agostino RB Sr. The randomized registry trial—the next disruptive technology in clinical research? N Engl J Med 2013; 369:1579–1581.
The woods are lovely, dark and deep,
But I have promises to keep,
And miles to go before I sleep,
And miles to go before I sleep.
—Robert Frost, “Stopping by Woods on a Snowy Evening”1
Frost's words are simple yet elegant. They can be interpreted many ways. I see the allegory of life as a journey in this poem. The passage, like the woods, is beautiful, but there is a long, long way to go.
And so it is with the treatment of heart failure. There is beauty in our understanding of the syndrome’s physiologic complexities and natural history, and of effective treatments uncovered. Still, we’ve a monstrous climb ahead to get to the summit of this clinical challenge in order to start a real descent.
THE PAST, PRESENT, AND FUTURE OF HEART FAILURE THERAPY
Okwuosa et al,2 in this issue of the Journal, have capably summarized the ABCs of treating heart failure with reduced ejection fraction (also called systolic heart failure), approaching the subject from a perspective on past, present, and future therapies. They summarize heart failure interventions with a guideline-based philosophy, pointing out that these care paths are supposed to be evidence-based. They observe that in the 1960s the standard of care was digitalis, diuretics (furosemide first became available in 1967), and rest. That was about all we had for this problem.
There are now many drugs, devices, and operations that help patients with heart failure. But they never really cure the disease or, more aptly, the syndrome—and therapies are supposed to cure. This limitation of present therapies is important, given the disturbing epidemiology of heart failure, its economic cost, and the suffering of patients. That burden is well detailed.
In addition to curing, the overarching goals of treatment generally are to ameliorate distressing symptoms and to prevent comorbidities. In heart failure with reduced ejection fraction, we want to prevent premature death, stroke, myocardial infarction, congestive states, hospitalization, renal insufficiency, renal failure, cachexia, inanition, feebleness, and respiratory distress, among others.
The ABC mnemonic of Okwuosa et al will help caregivers remember the basics. It is important, however, to put algorithms into proper perspective and to look toward the future.
PROBLEMS WITH EVIDENCE-BASED MEDICINE
Several problems with our current heart failure treatments are rooted in how we perform clinical trials, arguably the premier method of determining truth in clinical practice and the foundation of evidence-based medicine.3,4
Do the trials represent real-world practice?
Were the clinical trials that led to regulatory approval and professional society endorsement of the therapies that we prescribe in our offices done in the same sorts of patients as those in our waiting rooms asking for help? Perhaps, for the most part, they have been. And thus, Okwuosa et al have crafted a work relevant to all of us and every patient.
But I believe there are major gaps in the types of participants enrolled in trials, eg, underrepresentation of certain racial and ethnic groups, not to mention the relative paucity of women. The very elderly (a rapidly growing population) have largely been ignored as well, and participants with significant renal insufficiency, anemia, and diabetes mellitus seem far fewer than what we deal with in a busy clinic.
In addition, Okwuosa et al focus only on patients with reduced left ventricular ejection fraction, a group that makes up only about half of the heart failure crowd.
What about quality of life and other important outcomes?
Clinical trials in heart failure with reduced ejection fraction have generally focused on major clinical end points (primarily, but not exclusively, mortality), to the exclusion of quality of life. Though sometimes included in trials, quality-of-life metrics generally get relegated to second-class seats or ‘tween-deck steerage. Perhaps that is because measuring quality of life can be time-consuming and difficult.
Yet, in the words of sociologist William Bruce Cameron, not everything that counts can be counted, and not everything that can be counted counts. That goes for quality of life.
Lies, damned lies, and P values
Quandaries in data management and analysis include what to do about trial dropouts, study power, precision of statistical analysis, intention-to-treat principles, and choice of the P value that defines significance (or not) for any end point observation. Of course, there are myriad sophisticated mathematical and statistical reasons to justify why we don’t simply count on-treatment participants or allow imputation of results when patients or results drop out, forcing us to worship at the altar of P < .05.
A review of the P value concept5 recently appeared with an accompanying editorial by Kyriacou6 that concluded that “the automatic application of dichotomized hypothesis testing based on prearranged levels of statistical significance should be substituted with a more complex process using effect estimates, confidence intervals, and even P values, thereby permitting scientists, statisticians, and clinicians to use their own inferential capabilities to assign scientific significance.”6
How many great treatments have we tossed out because of rigid reliance on old-fashioned approaches to determining therapeutic evidence? Many treatments studied have had great results in a minority of patients in clinical trials but did not have a major positive (or negative) impact on the overall cohort (with lack of primary end point statistical significance). And what to do when the primary end point is a neutral or negative one but secondary end points are positive? Why not focus more attention on those patients benefiting from an intervention despite the overall results of any trial?
Dilemmas of trials
Other issues are that clinical trials cost too much, and that recruitment and follow-up take too long. Intercurrent therapies (and guidelines) can emerge that jeopardize the trial itself or make observations untimely. The dilemma of stacking therapies one on top of another, often making patient compliance impossible, is another problem with clinical trials. Yet this is how we get to the ABCs.
A NEW WAY TO DO TRIALS
The information provided by Okwuosa et al is useful and encouraging, but too many gaps exist in our heart failure therapies to permit us to celebrate with exuberance. Too many patients still suffer, too many die too young, and the costs are still too great.
Perhaps the future of therapeutic development should embrace different and better ways to demonstrate real value (relying on the equation of value equals outcomes meaningful to patients, divided by cost) of therapies, including the old, the new, the trashed and the underdeveloped. More creative data analysis to reexamine the current tools on the shelf and the ones tried but discarded is essential.
A position paper from the Cardiovascular Round Table of the European Society of Cardiology concluded that “a coordinated effort involving academia, regulators, industry and payors will help to foster better and more effective conduct of clinical cardiovascular trials, supporting earlier availability of innovative therapies and better management of cardiovascular diseases.”7
Lauer and D’Agostino,8 also in an editorial, argued for innovative methods of doing clinical trials and discovering truth about therapies that are applicable to the future of developing treatments for heart failure with reduced ejection fraction. They noted that “the randomized registry trial represents a disruptive technology” and wondered if it will be “given serious consideration as a way to resolve the recognized limitations of current clinical-trial design.”8
Indeed, conducting megatrials with existing megadatabases using a registry format could help. Registries emerging from early adaptive trial design efforts, particularly when Bayesian analysis theory is applied, might help inform clinical experience faster and more efficiently. Bayesian analysis is a statistical approach that attempts to estimate parameters of an underlying distribution of events in an ongoing fashion based on the observed distribution. A clinical trial of stem cell therapies could, at the end of the trial, be turned into a multicenter registry that would continue to inform us about the more real-world application of newer treatment approaches.
Though the therapeutic cupboard for heart failure is certainly not bare, as Okwuosa et al point out, it is wanting. Let’s look for new therapeutic ABCs differently. We should be attacking the real challenge—curing the disease processes that cause the syndrome. Yes, there are miles to go before we sleep.
The woods are lovely, dark and deep,
But I have promises to keep,
And miles to go before I sleep,
And miles to go before I sleep.
—Robert Frost, “Stopping by Woods on a Snowy Evening”1
Frost's words are simple yet elegant. They can be interpreted many ways. I see the allegory of life as a journey in this poem. The passage, like the woods, is beautiful, but there is a long, long way to go.
And so it is with the treatment of heart failure. There is beauty in our understanding of the syndrome’s physiologic complexities and natural history, and of effective treatments uncovered. Still, we’ve a monstrous climb ahead to get to the summit of this clinical challenge in order to start a real descent.
THE PAST, PRESENT, AND FUTURE OF HEART FAILURE THERAPY
Okwuosa et al,2 in this issue of the Journal, have capably summarized the ABCs of treating heart failure with reduced ejection fraction (also called systolic heart failure), approaching the subject from a perspective on past, present, and future therapies. They summarize heart failure interventions with a guideline-based philosophy, pointing out that these care paths are supposed to be evidence-based. They observe that in the 1960s the standard of care was digitalis, diuretics (furosemide first became available in 1967), and rest. That was about all we had for this problem.
There are now many drugs, devices, and operations that help patients with heart failure. But they never really cure the disease or, more aptly, the syndrome—and therapies are supposed to cure. This limitation of present therapies is important, given the disturbing epidemiology of heart failure, its economic cost, and the suffering of patients. That burden is well detailed.
In addition to curing, the overarching goals of treatment generally are to ameliorate distressing symptoms and to prevent comorbidities. In heart failure with reduced ejection fraction, we want to prevent premature death, stroke, myocardial infarction, congestive states, hospitalization, renal insufficiency, renal failure, cachexia, inanition, feebleness, and respiratory distress, among others.
The ABC mnemonic of Okwuosa et al will help caregivers remember the basics. It is important, however, to put algorithms into proper perspective and to look toward the future.
PROBLEMS WITH EVIDENCE-BASED MEDICINE
Several problems with our current heart failure treatments are rooted in how we perform clinical trials, arguably the premier method of determining truth in clinical practice and the foundation of evidence-based medicine.3,4
Do the trials represent real-world practice?
Were the clinical trials that led to regulatory approval and professional society endorsement of the therapies that we prescribe in our offices done in the same sorts of patients as those in our waiting rooms asking for help? Perhaps, for the most part, they have been. And thus, Okwuosa et al have crafted a work relevant to all of us and every patient.
But I believe there are major gaps in the types of participants enrolled in trials, eg, underrepresentation of certain racial and ethnic groups, not to mention the relative paucity of women. The very elderly (a rapidly growing population) have largely been ignored as well, and participants with significant renal insufficiency, anemia, and diabetes mellitus seem far fewer than what we deal with in a busy clinic.
In addition, Okwuosa et al focus only on patients with reduced left ventricular ejection fraction, a group that makes up only about half of the heart failure crowd.
What about quality of life and other important outcomes?
Clinical trials in heart failure with reduced ejection fraction have generally focused on major clinical end points (primarily, but not exclusively, mortality), to the exclusion of quality of life. Though sometimes included in trials, quality-of-life metrics generally get relegated to second-class seats or ‘tween-deck steerage. Perhaps that is because measuring quality of life can be time-consuming and difficult.
Yet, in the words of sociologist William Bruce Cameron, not everything that counts can be counted, and not everything that can be counted counts. That goes for quality of life.
Lies, damned lies, and P values
Quandaries in data management and analysis include what to do about trial dropouts, study power, precision of statistical analysis, intention-to-treat principles, and choice of the P value that defines significance (or not) for any end point observation. Of course, there are myriad sophisticated mathematical and statistical reasons to justify why we don’t simply count on-treatment participants or allow imputation of results when patients or results drop out, forcing us to worship at the altar of P < .05.
A review of the P value concept5 recently appeared with an accompanying editorial by Kyriacou6 that concluded that “the automatic application of dichotomized hypothesis testing based on prearranged levels of statistical significance should be substituted with a more complex process using effect estimates, confidence intervals, and even P values, thereby permitting scientists, statisticians, and clinicians to use their own inferential capabilities to assign scientific significance.”6
How many great treatments have we tossed out because of rigid reliance on old-fashioned approaches to determining therapeutic evidence? Many treatments studied have had great results in a minority of patients in clinical trials but did not have a major positive (or negative) impact on the overall cohort (with lack of primary end point statistical significance). And what to do when the primary end point is a neutral or negative one but secondary end points are positive? Why not focus more attention on those patients benefiting from an intervention despite the overall results of any trial?
Dilemmas of trials
Other issues are that clinical trials cost too much, and that recruitment and follow-up take too long. Intercurrent therapies (and guidelines) can emerge that jeopardize the trial itself or make observations untimely. The dilemma of stacking therapies one on top of another, often making patient compliance impossible, is another problem with clinical trials. Yet this is how we get to the ABCs.
A NEW WAY TO DO TRIALS
The information provided by Okwuosa et al is useful and encouraging, but too many gaps exist in our heart failure therapies to permit us to celebrate with exuberance. Too many patients still suffer, too many die too young, and the costs are still too great.
Perhaps the future of therapeutic development should embrace different and better ways to demonstrate real value (relying on the equation of value equals outcomes meaningful to patients, divided by cost) of therapies, including the old, the new, the trashed and the underdeveloped. More creative data analysis to reexamine the current tools on the shelf and the ones tried but discarded is essential.
A position paper from the Cardiovascular Round Table of the European Society of Cardiology concluded that “a coordinated effort involving academia, regulators, industry and payors will help to foster better and more effective conduct of clinical cardiovascular trials, supporting earlier availability of innovative therapies and better management of cardiovascular diseases.”7
Lauer and D’Agostino,8 also in an editorial, argued for innovative methods of doing clinical trials and discovering truth about therapies that are applicable to the future of developing treatments for heart failure with reduced ejection fraction. They noted that “the randomized registry trial represents a disruptive technology” and wondered if it will be “given serious consideration as a way to resolve the recognized limitations of current clinical-trial design.”8
Indeed, conducting megatrials with existing megadatabases using a registry format could help. Registries emerging from early adaptive trial design efforts, particularly when Bayesian analysis theory is applied, might help inform clinical experience faster and more efficiently. Bayesian analysis is a statistical approach that attempts to estimate parameters of an underlying distribution of events in an ongoing fashion based on the observed distribution. A clinical trial of stem cell therapies could, at the end of the trial, be turned into a multicenter registry that would continue to inform us about the more real-world application of newer treatment approaches.
Though the therapeutic cupboard for heart failure is certainly not bare, as Okwuosa et al point out, it is wanting. Let’s look for new therapeutic ABCs differently. We should be attacking the real challenge—curing the disease processes that cause the syndrome. Yes, there are miles to go before we sleep.
- Frost R. Stopping by Woods on a Snowy Evening. In: New Hampshire. New York, Henry Holt, 1923.
- Okwuoso IS, Ojeifo O, Nwabueze C, et al. The ABCs of managing systolic heart failure: the past, present and future. Cleve Clin J Med 2016; 83:753–765.
- Samman Tahhan A, Vaduganathan M, Kelkar A, et al. Trends in heart failure clinical trials from 2001–2012. J Card Fail 2016; 22:171–179.
- Cohn JN. Trials and tribulations. J Card Fail 2016; 22:180–181.
- Chavalarias D, Wallach JD, Li AH, Ioannidis JP. Evolution of reporting P values in the biomedical literature, 1990–2015. JAMA 2016; 315:1141–1148.
- Kyriacou DN. The enduring evolution of the P value. JAMA 2016; 315:1113–1115.
- Jackson N, Atar D, Borentain M, et al. Improving clinical trials for cardiovascular diseases: a position paper from the Cardiovascular Round Table of the European Society of Cardiology. Eur Heart J 2016; 37:747–754.
- Lauer MS, D’Agostino RB Sr. The randomized registry trial—the next disruptive technology in clinical research? N Engl J Med 2013; 369:1579–1581.
- Frost R. Stopping by Woods on a Snowy Evening. In: New Hampshire. New York, Henry Holt, 1923.
- Okwuoso IS, Ojeifo O, Nwabueze C, et al. The ABCs of managing systolic heart failure: the past, present and future. Cleve Clin J Med 2016; 83:753–765.
- Samman Tahhan A, Vaduganathan M, Kelkar A, et al. Trends in heart failure clinical trials from 2001–2012. J Card Fail 2016; 22:171–179.
- Cohn JN. Trials and tribulations. J Card Fail 2016; 22:180–181.
- Chavalarias D, Wallach JD, Li AH, Ioannidis JP. Evolution of reporting P values in the biomedical literature, 1990–2015. JAMA 2016; 315:1141–1148.
- Kyriacou DN. The enduring evolution of the P value. JAMA 2016; 315:1113–1115.
- Jackson N, Atar D, Borentain M, et al. Improving clinical trials for cardiovascular diseases: a position paper from the Cardiovascular Round Table of the European Society of Cardiology. Eur Heart J 2016; 37:747–754.
- Lauer MS, D’Agostino RB Sr. The randomized registry trial—the next disruptive technology in clinical research? N Engl J Med 2013; 369:1579–1581.

Dual antiplatelet therapy for acute coronary syndromes: How long to continue?
Percutaneous coronary intervention for acute coronary syndromes has evolved, and so, hand in hand, has antiplatelet therapy. With the advent of clopidogrel and newer agents, several studies demonstrated the benefits of dual antiplatelet therapy in preventing major vascular ischemic complications. The findings culminated in a guideline recommendation for at least 12 months of dual antiplatelet therapy after placement of a drug-eluting stent, when feasible—a class I recommendation (treatment should be given), level of evidence B (limited populations evaluated).1,2 But extending dual antiplatelet therapy beyond 12 months had no strong favorable evidence until the recent Dual Antiplatelet Therapy (DAPT) study3 shed light on this topic.
Here, we review the evidence thus far on the optimal duration of dual antiplatelet therapy in the secondary prevention of coronary artery disease.
PLATELETS IN ACUTE CORONARY SYNDROMES AND STENT THROMBOSIS
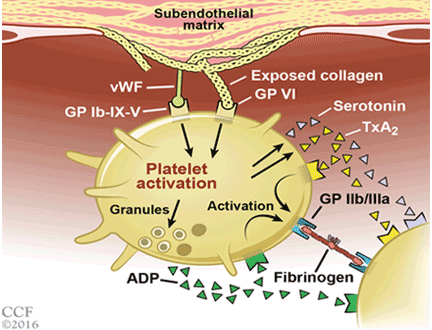
Acute coronary syndromes begin with fissuring or ulceration of a vulnerable atherosclerotic plaque, followed by thrombosis and occlusion, mediated by platelet adhesion, activation, and aggregation (Figure 1). Transient occlusion results in unstable angina or non-ST-elevation myocardial infarction, while total occlusion usually results in ST-elevation myocardial infarction.
Platelet aggregation is prominent among the mechanisms leading to stent thrombosis and vaso-occlusive ischemic complications after percutaneous coronary intervention. Thus, antiplatelet agents play a vital role in both primary and secondary prevention of cardiovascular events.4–6
Adhesion, activation, and aggregation
Adhesion. Disruption of the vascular endothelium as a result of vulnerable plaque fissuring or ulceration exposes subendothelial thrombogenic collagen and von Willebrand factor to blood. Collagen engages platelets through their glycoprotein (GP) Ia, IIa, and VI receptors, and von Willebrand factor binds platelets through the GP Ib-IX-V receptor.
Activation. Once platelets adhere to the subendothelium, they undergo a conformational change and become activated. Simultaneous release of various autocrine and paracrine mediators including adenosine diphosphate, serotonin, epinephrine, thromboxane, and various ligand-receptor interactions all contribute to the activation cascade. Adenosine diphosphate binds to the platelet receptor P2Y1, leading to an increase in intracellular calcium, and it binds to P2Y12, leading to a decrease in cyclic adenosine monophosphate, both of which cause GP IIb/IIIa receptor activation. Thromboxane A2 released by platelets by cyclo-oxygenase 1 binds to alpha or beta variant receptors and contributes to GP IIb/IIIa activation through elevation of intracellular calcium levels.
Aggregation and thrombosis. Exposure of tissue factor to plasma following plaque rupture activates the coagulation cascade via the extrinsic pathway, which generates thrombin, a powerful platelet activator that causes thrombus formation via fibrin. Thrombin binds to protease-activated receptors PAR-1 and PAR-4 on platelets, causing an increase in intracellular calcium and a decrease in cyclic adenosine monophosphate with subsequent GP IIb/IIIa activation. GP IIb/IIIa facilitates platelet aggregation by binding to fibrinogen and forming a stable platelet thrombus.
In the early stages of thrombus formation, platelets predominate (“white” thrombi); further organization with fibrin results in older “red” thrombi. The stages of thrombi vary in non-ST-elevation and ST-elevation myocardial infarction and are prognostic markers of death.4–8
PERCUTANEOUS INTERVENTION, RESTENOSIS, AND STENT THROMBOSIS
Percutaneous coronary intervention, the preferred means of revascularization for many patients, is performed emergently in patients with ST-elevation myocardial infarction, urgently in those with acute coronary syndromes without ST elevation, and electively in those with stable ischemic symptoms.
Percutaneous revascularization techniques have evolved from balloon angioplasty to bare-metal stents to drug-eluting stents, but each of these procedures has been associated with a periprocedural and postprocedural risk of thrombosis.

Balloon angioplasty was associated with vascular intimal injury, inciting elastic vascular recoil and smooth muscle cell proliferation leading to restenosis.
Bare-metal stents reduced the restenosis rate by eliminating vascular recoil, although restenosis still occurred within the stent because of neointimal proliferation of vascular smooth muscle cells. This was an important limitation, as both acute and subacute stent thrombosis were refractory to aggressive anticoagulation regimens that were associated with major bleeding complications and longer hospital length of stay. Stenting became mainstream practice only after the ISAR9 and STARS10 trials showed that dual antiplatelet therapy controlled stent thrombosis.
Drug-eluting stents coated with anti-proliferative and anti-inflammatory polymers markedly reduced in-stent restenosis rates by suppressing the initial vascular smooth-muscle proliferative response. However, they were still associated with late and very late stent thrombosis with incomplete endothelialization, even up to 40 months after implantation. Proposed mechanisms include incomplete stent apposition and inflammatory hypersensitivity reactions to the polymer coating. Incomplete stent apposition associated with low-velocity blood flow at the junction of the stent strut and vessel wall, together with delayed endothelialization, promotes platelet adhesion and aggregation, followed by thrombus formation.11
Second-generation drug-eluting stents have thinner struts and more biocompatible polymers and are thought to favor more complete re-endothelialization, reducing the rates of stent thrombosis.8,12,13
Predictors of early stent thrombosis
The Dutch Stent Thrombosis Registry and other studies looked at risk factors for stent thrombosis.14,15
Procedure-related factors included:
- Stent undersizing
- Residual uncovered dissections after angioplasty
- Longer stents
- Low flow after angioplasty (< 3 on the 0–3 Thrombolysis in Myocardial Infarction [TIMI] scale).
Lesion-related factors included:
- Intermediate coronary artery disease both proximal and distal to the culprit lesions
- Bifurcation lesions.
Patient-related factors included:
- Low left ventricular ejection fraction
- Diabetes mellitus
- Peripheral arterial disease Premature discontinuation of clopidogrel.
ANTIPLATELET AGENTS: MECHANISM OF ACTION
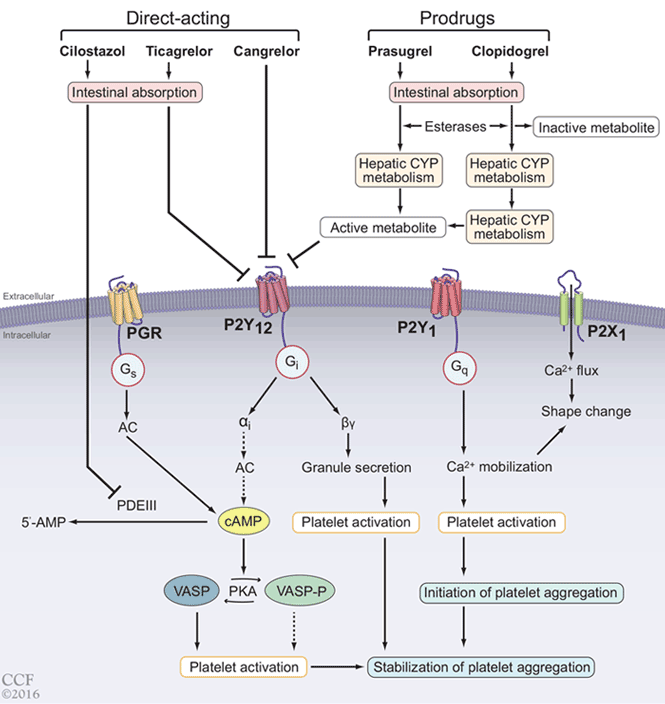
Various pathways play synergistic roles in platelet activation and aggregation and thrombus formation, and different antiplatelet agents inhibit these specific pathways, thus complementing each other and having additive effects (Figure 2, Table 1).5,16–21
Aspirin inhibits cyclo-oxygenase 1
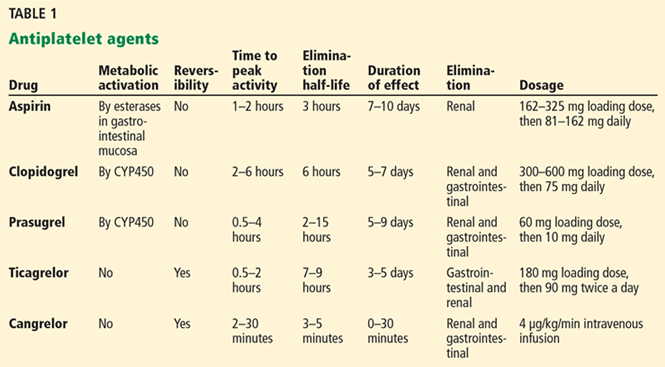
Cyclo-oxygenase 1, found in platelets, endothelial cells, and other cells, catalyzes the conversion of arachidonic acid to thromboxane A2. Aspirin irreversibly inhibits cyclo-oxygenase 1 by acetylating its serine residue, preventing formation of thromboxane A2 and preventing platelet activation and aggregation.
P2Y12 ADP receptor antagonists
Clopidogrel and prasugrel are thienopyridine agents that irreversibly inhibit the P2Y12 receptor, thereby preventing binding of adenosine diphosphate and the subsequent platelet activation-aggregation cascade. They are both prodrugs and require conversion by cytochrome P450 enzymes to active metabolites. Prasugrel is 10 times more potent than clopidogrel due to more efficient formation of its active metabolite, and it achieves a comparable effect on platelet inhibition 30 minutes faster than the peak effect of clopidogrel at 6 hours. The overall peak inhibitory effect of prasugrel is twice that of clopidogrel.22
Ticagrelor, a cyclopentyl-triazolo-pyrimidine, directly and reversibly inhibits the P2Y12 ADP receptor. Unlike clopidogrel and prasugrel, it does not need to be converted to an active metabolite, and it noncompetitively inhibits P2Y12 at a site different from the adenosine diphosphate binding site.23 Like prasugrel, ticagrelor inhibits platelet function more rapidly and more completely than clopidogrel.
Cangrelor, an intravenously administered analogue of adenosine triphosphate, reversibly inhibits the P2Y12 receptor. It has undergone phase 3 trials but is not yet approved for clinical use.24
WHY DUAL ANTIPLATELET THERAPY?
Aspirin is good, clopidogrel is better
Aspirin has a well-validated role in both primary and secondary prevention of coronary and noncoronary atherosclerotic vascular disease.
The CAPRIE trial found clopidogrel monotherapy to be superior to aspirin monotherapy in patients with established atherosclerotic vascular disease.25
After stenting, short-term dual therapy is better than short-term warfarin
Thrombotic complications in the early postprocedural period were a major limitation of stenting, and existing anticoagulation regimens were ineffective in preventing them.26,27
The ISAR trial studied the benefit of combined antiplatelet vs anticoagulant therapy after stent placement. Patients randomized to receive combined aspirin plus ticlopidine (an early P2Y12 inhibitor) had significantly lower rates of primary cardiac, hemorrhagic, and vascular events at 30 days.9 Two other trials confirmed this finding.28,29
STARS10 also confirmed the benefit of aspirin and ticlopidine after stenting. Patients were randomly assigned to aspirin alone, aspirin plus warfarin, or aspirin plus ticlopidine after stent placement. The rate of stent thrombosis at 30 days was significantly lower in the dual antiplatelet group than in the other two groups. The dual antiplatelet group had a higher rate of bleeding than the aspirin-alone group, but the rate was similar to that of the aspirin-plus-warfarin group.
Long-term dual antiplatelet therapy is beneficial in several situations
ISAR and STARS were landmark trials that showed stent thrombosis could be reduced by dual antiplatelet therapy for a 30-day period. However, the long-term role of dual antiplatelet therapy was still unknown.
The CURE trial30–32 randomized patients presenting with acute coronary syndromes without ST elevation to receive clopidogrel plus aspirin or placebo plus aspirin for 3 to 12 months. The rate of the primary end point (cardiac death, nonfatal myocardial infarction, or stroke) was significantly lower in the clopidogrel-plus-aspirin group. A similar benefit of dual antiplatelet therapy was seen in the subgroup of patients who underwent percutaneous coronary intervention. Both pretreatment with clopidogrel plus aspirin for a median of 10 days prior to percutaneous intervention and continuing it for a mean of 9 months reduced major adverse cardiovascular events.
The CREDO trial20 found that the combination of clopidogrel and aspirin significantly reduced the incidence of death, myocardial infarction, or stroke at 1 year after percutaneous coronary intervention. A subgroup of patients in this trial who had a longer pretreatment interval with a loading clopidogrel dose showed a benefit at 28 days, which was not as evident with a shorter loading dose interval.
The CLARITY-TIMI 28 trial33,34 showed the advantage of adding clopidogrel to aspirin in patients receiving fibrinolytic therapy for ST-elevation myocardial infarction. Adding clopidogrel both improved the patency of the infarct-related artery and reduced ischemic complications. In patients who subsequently underwent percutaneous coronary intervention and stenting, clopidogrel pretreatment was associated with a significant decrease in ischemic complications before and after the procedure. There was no significant increase in bleeding complications in either group.
COMMIT/CCS 235 also showed the benefit of dual antiplatelet therapy in patients with ST-elevation myocardial infarction. Clopidogrel added to aspirin during the short-term in-hospital or postdischarge treatment period significantly reduced a composite end point of reinfarction, death, or stroke as well as death from any cause.
The CHARISMA trial36–38 aimed to determine if patients who were more stable (ie, no recent acute coronary syndrome event or percutaneous coronary intervention) would benefit. Overall, CHARISMA showed no benefit of adding clopidogrel to aspirin compared with aspirin alone in a broad population of patients with established vascular disease (secondary prevention) or risk factors for vascular disease (primary prevention).
But importantly, though no benefit was seen in the primary prevention group, the large subgroup of patients with established atherosclerotic vascular disease (12,153 of the 15,603 patients in the trial) did benefit from dual antiplatelet therapy.36,37 This subgroup showed an overall reduction in absolute risk of 1.5% (relative risk 0.88, P = .046) over a median follow-up of 27.6 months. This benefit was even more apparent in the 9,478 patients with prior myocardial infarction, stroke, or peripheral artery disease, for whom the relative risk reduction was 17.1% (P = .01) and the reduction in absolute risk 1.5%.38
These results are comparable to the 2% absolute risk reduction in the CURE trial for similar end points over 9 months. In both studies, there was no significant increase in the risk of major bleeding or intracranial bleeding in the clopidogrel-plus-aspirin groups, although minor bleeding was increased by dual antiplatelet therapy.
The rate of severe bleeding, which was the primary safety end point in CHARISMA, was not significantly different in the clopidogrel-plus-aspirin group compared with the placebo-plus-aspirin group (relative risk 1.25, 95% CI 0.97–1.61, P = .09).
Thus, although the CHARISMA findings were negative overall, the positive finding observed in the predominant subgroup of patients with established vascular disease can therefore be considered supportive of the results of the subsequent trials discussed below.
The PEGASUS-TIMI 54 trial39 studied the benefit of adding ticagrelor (60 or 90 mg) to low-dose aspirin in patients with stable coronary artery disease who had had a myocardial infarction 1 to 3 years earlier.
Confirming the results of the CHARISMA subgroup analysis, the incidence of the ischemic primary efficacy end point (a composite of cardiovascular death, myocardial infarction, and stroke) was significantly lower in both groups receiving ticagrelor plus aspirin compared with those receiving placebo plus aspirin. The Kaplan-Meier rate at 3 years for the ticagrelor 90 mg-plus-aspirin group was 7.85% vs 9.04% for the placebo-plus-aspirin group (hazard ratio 0.85, 95% confidence interval [CI] 0.75–0.96, P = .008). The rate for the ticagrelor 60 mg-plus-aspirin group was 7.77% vs 9.04% for the placebo-plus-aspirin group (hazard ratio 0.84, 95% CI 0.74–0.95, P = .004).
The rates of all TIMI major and minor bleeding, as well as bleeding requiring transfusion or discontinuation of the study drug, were significantly higher in both ticagrelor dosing groups than in the placebo group (P < .01 for both groups vs placebo). The rates of fatal bleeding and nonfatal intracranial hemorrhage were not significantly higher. Although there was an overall reduction in ischemic end points with the addition of ticagrelor, there was also a significantly higher incidence of bleeding in this group.
Comment. Thus, with or without percutaneous coronary intervention in acute coronary syndrome as well as in stable coronary artery disease, dual antiplatelet therapy was shown to improve outcomes and decrease ischemic complications compared with aspirin alone. It provided benefit in the setting of acute coronary syndrome (in the CURE trial) and percutaneous coronary intervention (in the CREDO trial) for up to 1 year.
Major questions remained to be addressed:
- Do the results of CREDO, which was performed before the current interventional era and the use of drug-eluting stents, reflect outcomes after current interventional practice?
- Could shorter periods of dual antiplatelet therapy be sufficient, especially with newer stents with less risk of late thrombosis?
- Does the benefit of dual antiplatelet therapy extend beyond the 1-year time period tested in those trials to date?
RECOMMENDATIONS FOR DOSING
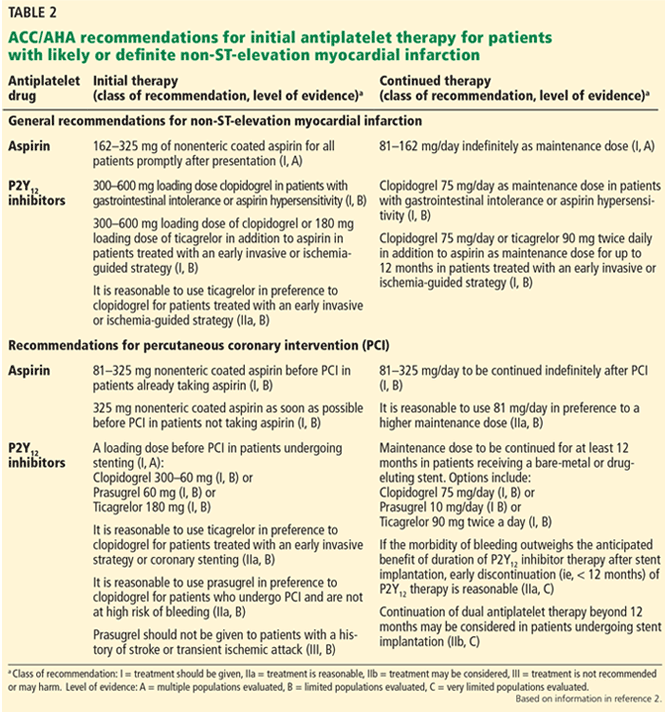
The American College of Cardiology Foundation/American Heart Association guidelines for dosing of antiplatelet agents for non-ST-elevation myocardial infarction are summarized in Table 2, and those for ST-elevation myocardial infarction are summarized in Table 3.1,2
WOULD SHORTER THERAPY AFTER STENTING WORK AS WELL?
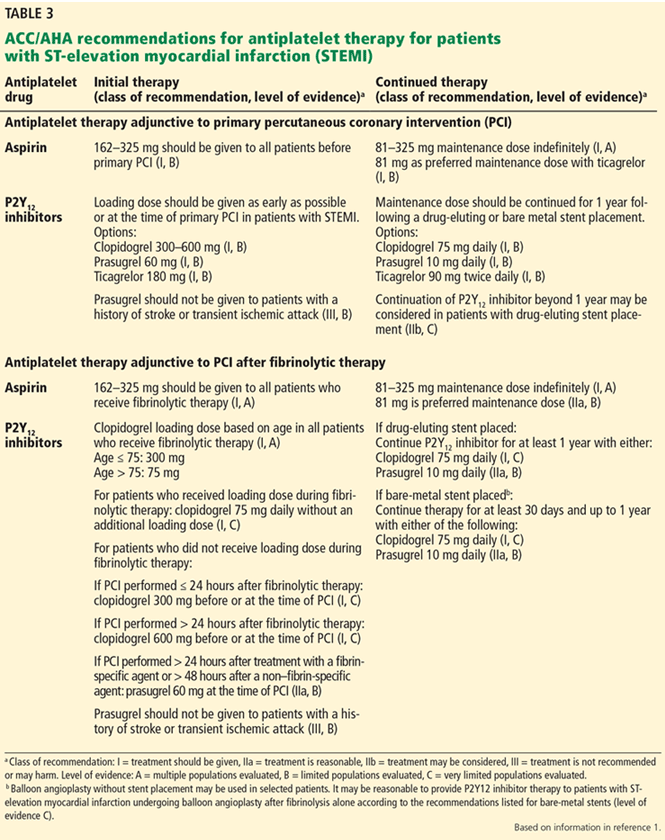
The American College of Cardiology Foundation/American Heart Association currently recommend dual antiplatelet therapy for at least 12 months after drug-eluting stent placement, with shorter courses appropriate for patients who develop excessive bleeding complications or who are at high risk of bleeding.
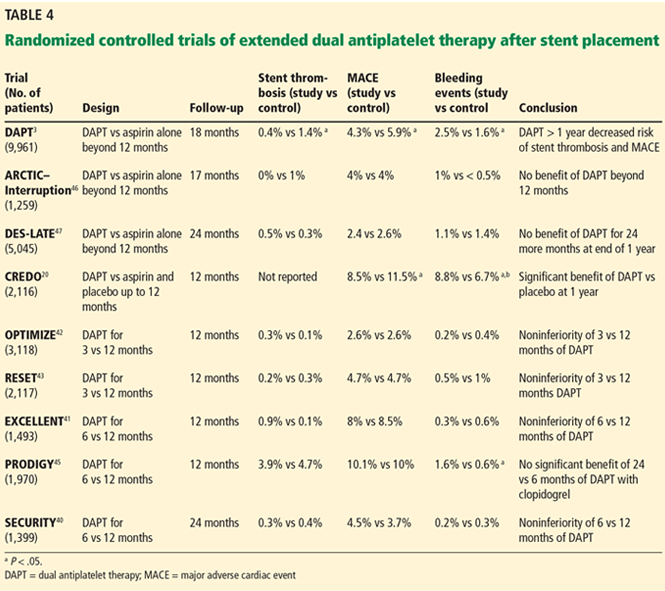
Four trials (Table 4) evaluated whether shorter durations of dual antiplatelet therapy would suffice: SECURITY,40 EXCELLENT,41 OPTIMIZE,42 and RESET.43 All of them showed that short-duration therapy was not inferior to standard-duration therapy.44 These studies were comparable in that:
- Patients were randomized at the time of percutaneous coronary intervention or within 24 hours of it.
- Most patients received a second-generation drug-eluting stent, with the following exceptions: in EXCELLENT,41 one-fourth of patients received a Cypher first-generation drug-eluting stent, and in RESET,43 approximately one-fourth of the patients received a sirolimus-eluting stent in the standard-duration group for short lesions. Those patients with longer lesions in the RESET standard-duration group received an everolimus drug-eluting stent.
- The second antiplatelet added to aspirin in all studies was clopidogrel, with the exception of the SECURITY trial, in which fewer than 2% of patients received ticagrelor or prasugrel.40
- All the trials except RESET excluded patients who had had a myocardial infarction within 72 hours, and thus most patients studied had a lower risk profile.
- All of the trials sought to study noninferiority of short- vs standard-duration dual antiplatelet therapy, defined as the occurrence of a primary end point at 1 year (a composite of cardiovascular death, myocardial infarction, stroke, stent thrombosis, target vessel failure or revascularization, or bleeding).
Their low-risk patient populations and infrequent end points rendered these studies underpowered to make definitive conclusions about the relative efficacy of 6-months vs 12-months of dual antiplatelet therapy.
WOULD LONGER THERAPY BE BETTER?
The PRODIGY trial45 assessed durations of dual antiplatelet therapy both shorter and longer than the conventional 1 year, randomizing patients undergoing placement of a bare-metal stent, first-generation drug-eluting stent, or second-generation drug-eluting stent to receive aspirin and clopidogrel for either 6 months or 24 months. The study showed no significant difference in primary outcomes in the short- or long-duration groups.
Other trials that compared the standard 12 months of dual antiplatelet therapy with extended duration beyond 12 months were DAPT,3 ARCTIC-Interruption,46 and DES-LATE.47 The trials were comparable in that:
- All patients were randomized after completing 12 months of dual antiplatelet therapy following drug-eluting stent placement.
- All patients who were included had been free of major cardiac ischemic events or bleeding during the 12 months following stent placement.
- The primary aim of all three studies was to compare primary end points in groups receiving aspirin alone vs extended dual antiplatelet therapy. The primary end point was a composite of death due to a cardiovascular cause, nonfatal myocardial infarction, stroke, or stent thrombosis.
- The principal safety end point was bleeding.
Although the two earlier studies (ARCTIC-Interruption and DES-LATE) did not show any benefit of extended dual antiplatelet therapy compared with the standard 12-month duration, the recent DAPT study did.
The DAPT study
The DAPT study3 was an international, multicenter, placebo-controlled, double-blind randomized trial designed to examine the benefit of dual antiplatelet therapy beyond 1 year in a patient population large enough to provide definitive assessment of benefit and risk.
A total of 9,961 patients who received drug-eluting stents were randomized after 12 months of dual antiplatelet therapy to receive either a thienopyridine (clopidogrel or prasugrel) plus aspirin or placebo plus aspirin. They were followed for an additional 18 months. The coprimary efficacy end points were stent thrombosis and a composite of death, myocardial infarction, or stroke, while the primary safety end point was moderate or severe bleeding. The patients were also observed from months 30 to 33 on aspirin alone after stopping the thienopyridine.
Results. Longer therapy substantially reduced the risks of stent thrombosis (hazard ratio [HR] 0.29, 95% confidence interval [CI] 0.17–0.48) and the composite ischemic end point (HR 0.71, 95% CI 0.59–0.85). Follow-up during the 3-month thienopyridine discontinuation phase starting at 30 months revealed convergence of the ischemic event-rate curves in the two groups, which suggested that continuing dual antiplatelet therapy beyond 30 months might have been beneficial. Myocardial infarction unrelated to stent thrombosis accounted for 55% of the treatment benefit of dual antiplatelet therapy.
The risk of bleeding was higher in the thienopyridine group during the treatment period (2.5% vs 1.6%, P = .001). There was also a higher rate of noncardiovascular mortality in the thienopyridine group, although this difference may have been due to chance.3,48
Why were the results different?
All three trials included first- and second-generation drug-eluting stents, with different proportions in different trials. In ARCTIC-Interruption,46 43% of the patients in the continuation group had a first-generation stent, as did 64% of the patients in the dual antiplatelet group of DES-LATE.47 In the DAPT trial,3 38% of the patients in the longer-duration arm had a first-generation stent, and in 26% of cases it was a paclitaxel-eluting stent.
Only clopidogrel was used as the second antiplatelet agent in DES-LATE, whereas prasugrel was used in 10% of patients in ARCTIC-Interruption and 35% in DAPT.
Yet none of these differences seem to explain the differences in outcome among the studies. ARCTIC-Interruption and DES-LATE did not show any benefit of continued dual antiplatelet therapy beyond 12 months. DAPT showed benefit of extended therapy with prasugrel or with clopidogrel, and with first-generation or second-generation drug-eluting stents. The most likely explanation for the different results was that DAPT was the only trial sufficiently powered to definitively assess the end points, including stent thrombosis.
A balance between ischemic efficacy and bleeding risk is the major consideration with any antithrombotic and antiplatelet therapy. In the three largest trials we discussed (the vascular disease subgroups of CHARISMA,38 PEGASUS,39 and DAPT3), comparison of the prespecified efficacy and safety end points of each trial suggests that dual antiplatelet therapy has a net benefit, particularly given the irreversible nature of ischemic end points.
In CHARISMA,38 60 cardiovascular deaths, myocardial infarctions, or strokes were prevented per year per 10,000 patients treated, at the cost of 28 excess moderate bleeding events.
In PEGASUS,39 42 cardiovascular deaths, myocardial infarctions, or strokes were prevented, at the cost of 79 excess bleeding events requiring transfusion.
In DAPT (a selected population who had tolerated dual antiplatelet therapy for 1 year), 106 deaths, myocardial infarctions, or stroke events were prevented, at the cost of 47 excess moderate bleeding events.3
Indirect comparisons between trials are problematic, given different end point definitions, populations, and background therapies. But their results suggest that less-intensive inhibition with clopidogrel as the second antiplatelet long-term (as in CHARISMA) may provide the best balance of benefit vs risk.
BALANCING RISK AND BENEFIT
The evidence is unequivocal that dual antiplatelet therapy suppresses coronary ischemic complications resulting from thrombosis at sites of spontaneous plaque rupture following acute coronary syndromes or mechanical plaque disruption and foreign body implantation associated with percutaneous coronary intervention.
Three large-scale trials (DAPT,3 PEGASUS,39 and the secondary prevention subgroup of CHARISMA38) showed that the protective effect of dual antiplatelet therapy continues with prolonged therapy in patients who have experienced an acute coronary syndrome event or have received a drug-eluting stent. That benefit seems to be due to the action of these therapies on the culprit vessel (the one that caused the acute coronary syndrome or the site of stenting), as well as nonculprit arteries, emphasizing that dual antiplatelet therapy protects against atherosclerosis progression and future plaque rupture events.
For the durations studied in the longest trials thus far, 30 months (DAPT3) and 36 months (PEGASUS39), event curves continue to diverge, indicating that the advantage of dual antiplatelet therapy may persist for an indefinite period of time. Thus, indefinite therapy with dual antiplatelet agents can be supported, particularly in patients with advanced coronary artery disease or those who have had multiple coronary events.
We believe that the balance of evidence suggests that smaller studies that failed to show a benefit of longer-term therapy were underpowered to do so.
The ischemic protection is associated with the adverse effect of increased bleeding risk. Unfortunately, there has been little success in guiding dual antiplatelet therapy based on ischemic vs bleeding risk, in part because the same factors that predict risk of ischemic complications seem to predict increased susceptibility to bleeding. Nevertheless, indirect comparisons between studies suggest that for longer-term therapy clopidogrel may be superior to ticagrelor or prasugrel: the absolute excess bleeding risk with dual antiplatelet therapy vs aspirin in the CHARISMA secondary prevention subgroup was less than that in PEGASUS, with similar absolute reductions in ischemic events. So while the TRITON-TIMI 3822 and PLATO23 trials support the superiority of prasugrel or ticagrelor over clopidogrel for the first year after acute coronary syndrome, subsequent years of therapy may best be provided with clopidogrel.
Some patients may have identifiable factors that place them at very high risk of bleeding—need for surgical procedures, need for anticoagulation, or occurrence of bleeding complications or excessive “nuisance bleeding.” In those patients, the data suggest that dual antiplatelet therapy could be discontinued after 6 months, or perhaps even 3 months in the highest bleeding risk circumstances after second-generation drug-eluting stent placement.
WOEST49 was an open-label randomized controlled trial that studied the safety of antiplatelet regimens in patients on anticoagulation requiring percutaneous coronary interventions. Patients were randomized to double therapy with anticoagulant and clopidogrel vs triple therapy with additional aspirin following percutaneous coronary intervention. The primary end point was bleeding events within 1 year. Clopidogrel without aspirin was associated with significantly fewer bleeding events compared with triple therapy, with no increase in adverse ischemic events. The strategy tested in the WOEST trial seems reasonable in the specific group of patients who require ongoing anticoagulant therapy after drug-eluting stent placement, recognizing that the trial was somewhat underpowered to make definitive conclusions, particularly in patients at high risk for stent thrombosis.
Based on the results of PEGASUS and the CHARISMA subgroup with established ischemic burden, in which dual antiplatelet therapy was started after an interruption following the index coronary event, it is also reasonable to restart long-term dual antiplatelet therapy in patients who require interruption for short-term indications such as a surgical procedure.
- American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:e78–e140.
- Amsterdam EA, Wenger NK, Brindis RG, et al; ACC/AHA Task Force Members. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 130:e344–e426.
- Mauri L, Kereiakes DJ, Yeh RW, et al; DAPT Study Investigators. Twelve or 30 months of dual antiplatelet therapy after drug-eluting stents. N Engl J Med 2014; 371:2155–2166.
- Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J 2010; 74:597–607.
- Papp J, Kenyeres P, Toth K. Clinical importance of antiplatelet drugs in cardiovascular diseases. Clin Hemorheol Microcirc 2013; 53:81–96.
- Showkathali R, Natarajan A. Antiplatelet and antithrombin strategies in acute coronary syndrome: state-of-the-art review. Curr Cardiol Rev 2012; 8:239–249.
- Angiolillo DJ. The evolution of antiplatelet therapy in the treatment of acute coronary syndromes: from aspirin to the present day. Drugs 2012; 72:2087–2116.
- Claessen BE, Henriques JP, Jaffer FA, Mehran R, Piek JJ, Dangas GD. Stent thrombosis: a clinical perspective. JACC Cardiovasc Interv 2014; 7:1081–1092.
- Schomig A, Neumann FJ, Kastrati A, et al. A randomized comparison of antiplatelet and anticoagulant therapy after the placement of coronary-artery stents. N Engl J Med 1996; 334:1084–1089.
- Leon MB, Baim DS, Popma JJ, et al. A clinical trial comparing three antithrombotic-drug regimens after coronary-artery stenting. Stent Anticoagulation Restenosis Study Investigators. N Engl J Med 1998; 339:1665–1671.
- Joner M, Finn AV, Farb A, et al. Pathology of drug-eluting stents in humans: delayed healing and late thrombotic risk. J Am Coll Cardiol 2006; 48:193–202.
- Nikam N, Steinberg TB, Steinberg DH. Advances in stent technologies and their effect on clinical efficacy and safety. Med Devices (Auckl) 2014; 7:165–178.
- Simard T, Hibbert B, Ramirez FD, Froeschl M, Chen YX, O’Brien ER. The evolution of coronary stents: a brief review. Can J Cardiol 2014; 30:35–45.
- Byrne RA, Joner M, Kastrati A. Stent thrombosis and restenosis: what have we learned and where are we going? The Andreas Gruntzig Lecture ESC 2014. Eur Heart J 2015; 36:3320–3331.
- van Werkum JW, Heestermans AA, Zomer AC, et al. Predictors of coronary stent thrombosis: the Dutch Stent Thrombosis Registry. J Am Coll Cardiol 2009; 53:1399–1409.
- Berger JS. Aspirin, clopidogrel, and ticagrelor in acute coronary syndromes. Am J Cardiol 2013; 112:737–745.
- Franchi F, Angiolillo DJ. Novel antiplatelet agents in acute coronary syndrome. Nat Rev Cardiol 2015; 12:30–47.
- Patrono C, Rocca B. The future of antiplatelet therapy in cardiovascular disease. Annu Rev Med 2010; 61:49–61.
- Park SJ, Kang SM, Park DW. Dual antiplatelet therapy after drug-eluting stents: defining the proper duration. Coron Artery Dis 2014; 25:83–89.
- Steinhubl SR, Berger PB, Mann JT 3rd, et al; CREDO Investigators. Clopidogrel for the reduction of events during observation. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Nusca A, Patti G. Platelet function and inhibition in ischemic heart disease. Curr Cardiol Rep 2012; 14:457–467.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Genereux P, Stone GW, Harrington RA, et al; CHAMPION PHOENIX Investigators. Impact of intraprocedural stent thrombosis during percutaneous coronary intervention: Insights from the CHAMPION PHOENIX Trial (Clinical Trial Comparing Cangrelor to Clopidogrel Standard of Care Therapy in Subjects Who Require Percutaneous Coronary Intervention). J Am Coll Cardiol 2014; 63:619–629.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Brilakis ES, Patel VG, Banerjee S. Medical management after coronary stent implantation: a review. JAMA 2013; 310:189–198.
- Warren J, Baber U, Mehran R. Antiplatelet therapy after drug-eluting stent implantation. J Cardiol 2015; 65:98–104.
- Urban P, Macaya C, Rupprecht HJ, et al. Randomized evaluation of anticoagulation versus antiplatelet therapy after coronary stent implantation in high-risk patients: the Multicenter Aspirin and Ticlopidine Trial After Intracoronary Stenting (MATTIS). Circulation 1998; 98:2126–2132.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: The PCI-CURE study. Lancet 2001; 358:527–533.
- Morais J. Insights from CURE: using clopidogrel on top of standard therapy. Cerebrovasc Dis 2002; 13(suppl 1):17–21.
- Ferguson JJ. Clopidogrel plus aspirin in patients with acute myocardial infarction treated with fibrinolytic therapy—CLARITY-TIMI 28. Future Cardiol 2005; 1:605–610.
- Sabatine MS, Cannon CP, Gibson CM, et al; Clopidogrel as Adjunctive Reperfusion Therapy (CLARITY)-Thrombolysis in Myocardial Infarction (TIMI) 28 Investigators. Effect of clopidogrel pretreatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005; 294:1224–1232.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bonaca MP, Bhatt DL, Cohen M, et al; PEGASUS-TIMI 54 Steering Committee and Investigators. Long-term use of ticagrelor in patients with prior myocardial infarction. N Engl J Med 2015; 372:1791–1800.
- Colombo A, Chieffo A, Frasheri A, et al. Second-generation drug-eluting stent implantation followed by 6- versus 12-month dual antiplatelet therapy: the SECURITY randomized clinical trial. J Am Coll Cardiol 2014; 64:2086–2097.
- Gwon HC, Hahn JY, Park KW, et al. Six-month versus 12-month dual antiplatelet therapy after implantation of drug-eluting stents: the Efficacy of Xience/Promus versus Cypher to Reduce Late Loss After Stenting (EXCELLENT) randomized, multicenter study. Circulation 2012; 125:505–513.
- Feres F, Costa RA, Abizaid A, et al; OPTIMIZE Trial Investigators. Three vs twelve months of dual antiplatelet therapy after zotarolimus-eluting stents: the OPTIMIZE randomized trial. JAMA 2013; 310:2510–2522.
- Kim BK, Hong MK, Shin DH, et al; RESET Investigators. A new strategy for discontinuation of dual antiplatelet therapy: the RESET Trial (REal Safety and Efficacy of 3-month dual antiplatelet Therapy following endeavor zotarolimus-eluting stent implantation). J Am Coll Cardiol 2012; 60:1340–1348.
- El-Hayek G, Messerli F, Bangalore S, et al. Meta-analysis of randomized clinical trials comparing short-term versus long-term dual antiplatelet therapy following drug-eluting stents. Am J Cardiol 2014; 114:236–242.
- Valgimigli M, Campo G, Monti M, et al; Prolonging Dual Antiplatelet Treatment After Grading Stent-Induced Intimal Hyperplasia Study (PRODIGY) Investigators. Short- versus long-term duration of dual-antiplatelet therapy after coronary stenting: a randomized multicenter trial. Circulation 2012; 125:2015–2026.
- Collet JP, Silvain J, Barthelemy O, et al; ARCTIC investigators. Dual-antiplatelet treatment beyond 1 year after drug-eluting stent implantation (ARCTIC-Interruption): a randomised trial. Lancet 2014; 384:1577–1585.
- Lee CW, Ahn JM, Park DW, et al. Optimal duration of dual antiplatelet therapy after drug-eluting stent implantation: a randomized, controlled trial. Circulation 2014; 129:304–312.
- Kwok CS, Bulluck H, Ryding AD, Loke YK. Benefits and harms of extending the duration of dual antiplatelet therapy after percutaneous coronary intervention with drug-eluting stents: a meta-analysis. ScientificWorldJournal 2014; 2014:794078.
- Dewilde WJ, Oirbans T, Verheugt FW, et al; WOEST study investigators. Use of clopidogrel with or without aspirin in patients taking oral anticoagulant therapy and undergoing percutaneous coronary intervention: an open-label, randomised, controlled trial. Lancet 2013; 381:1107–1115.
Percutaneous coronary intervention for acute coronary syndromes has evolved, and so, hand in hand, has antiplatelet therapy. With the advent of clopidogrel and newer agents, several studies demonstrated the benefits of dual antiplatelet therapy in preventing major vascular ischemic complications. The findings culminated in a guideline recommendation for at least 12 months of dual antiplatelet therapy after placement of a drug-eluting stent, when feasible—a class I recommendation (treatment should be given), level of evidence B (limited populations evaluated).1,2 But extending dual antiplatelet therapy beyond 12 months had no strong favorable evidence until the recent Dual Antiplatelet Therapy (DAPT) study3 shed light on this topic.
Here, we review the evidence thus far on the optimal duration of dual antiplatelet therapy in the secondary prevention of coronary artery disease.
PLATELETS IN ACUTE CORONARY SYNDROMES AND STENT THROMBOSIS
Acute coronary syndromes begin with fissuring or ulceration of a vulnerable atherosclerotic plaque, followed by thrombosis and occlusion, mediated by platelet adhesion, activation, and aggregation (Figure 1). Transient occlusion results in unstable angina or non-ST-elevation myocardial infarction, while total occlusion usually results in ST-elevation myocardial infarction.
Platelet aggregation is prominent among the mechanisms leading to stent thrombosis and vaso-occlusive ischemic complications after percutaneous coronary intervention. Thus, antiplatelet agents play a vital role in both primary and secondary prevention of cardiovascular events.4–6
Adhesion, activation, and aggregation
Adhesion. Disruption of the vascular endothelium as a result of vulnerable plaque fissuring or ulceration exposes subendothelial thrombogenic collagen and von Willebrand factor to blood. Collagen engages platelets through their glycoprotein (GP) Ia, IIa, and VI receptors, and von Willebrand factor binds platelets through the GP Ib-IX-V receptor.
Activation. Once platelets adhere to the subendothelium, they undergo a conformational change and become activated. Simultaneous release of various autocrine and paracrine mediators including adenosine diphosphate, serotonin, epinephrine, thromboxane, and various ligand-receptor interactions all contribute to the activation cascade. Adenosine diphosphate binds to the platelet receptor P2Y1, leading to an increase in intracellular calcium, and it binds to P2Y12, leading to a decrease in cyclic adenosine monophosphate, both of which cause GP IIb/IIIa receptor activation. Thromboxane A2 released by platelets by cyclo-oxygenase 1 binds to alpha or beta variant receptors and contributes to GP IIb/IIIa activation through elevation of intracellular calcium levels.
Aggregation and thrombosis. Exposure of tissue factor to plasma following plaque rupture activates the coagulation cascade via the extrinsic pathway, which generates thrombin, a powerful platelet activator that causes thrombus formation via fibrin. Thrombin binds to protease-activated receptors PAR-1 and PAR-4 on platelets, causing an increase in intracellular calcium and a decrease in cyclic adenosine monophosphate with subsequent GP IIb/IIIa activation. GP IIb/IIIa facilitates platelet aggregation by binding to fibrinogen and forming a stable platelet thrombus.
In the early stages of thrombus formation, platelets predominate (“white” thrombi); further organization with fibrin results in older “red” thrombi. The stages of thrombi vary in non-ST-elevation and ST-elevation myocardial infarction and are prognostic markers of death.4–8
PERCUTANEOUS INTERVENTION, RESTENOSIS, AND STENT THROMBOSIS
Percutaneous coronary intervention, the preferred means of revascularization for many patients, is performed emergently in patients with ST-elevation myocardial infarction, urgently in those with acute coronary syndromes without ST elevation, and electively in those with stable ischemic symptoms.
Percutaneous revascularization techniques have evolved from balloon angioplasty to bare-metal stents to drug-eluting stents, but each of these procedures has been associated with a periprocedural and postprocedural risk of thrombosis.
Balloon angioplasty was associated with vascular intimal injury, inciting elastic vascular recoil and smooth muscle cell proliferation leading to restenosis.
Bare-metal stents reduced the restenosis rate by eliminating vascular recoil, although restenosis still occurred within the stent because of neointimal proliferation of vascular smooth muscle cells. This was an important limitation, as both acute and subacute stent thrombosis were refractory to aggressive anticoagulation regimens that were associated with major bleeding complications and longer hospital length of stay. Stenting became mainstream practice only after the ISAR9 and STARS10 trials showed that dual antiplatelet therapy controlled stent thrombosis.
Drug-eluting stents coated with anti-proliferative and anti-inflammatory polymers markedly reduced in-stent restenosis rates by suppressing the initial vascular smooth-muscle proliferative response. However, they were still associated with late and very late stent thrombosis with incomplete endothelialization, even up to 40 months after implantation. Proposed mechanisms include incomplete stent apposition and inflammatory hypersensitivity reactions to the polymer coating. Incomplete stent apposition associated with low-velocity blood flow at the junction of the stent strut and vessel wall, together with delayed endothelialization, promotes platelet adhesion and aggregation, followed by thrombus formation.11
Second-generation drug-eluting stents have thinner struts and more biocompatible polymers and are thought to favor more complete re-endothelialization, reducing the rates of stent thrombosis.8,12,13
Predictors of early stent thrombosis
The Dutch Stent Thrombosis Registry and other studies looked at risk factors for stent thrombosis.14,15
Procedure-related factors included:
- Stent undersizing
- Residual uncovered dissections after angioplasty
- Longer stents
- Low flow after angioplasty (< 3 on the 0–3 Thrombolysis in Myocardial Infarction [TIMI] scale).
Lesion-related factors included:
- Intermediate coronary artery disease both proximal and distal to the culprit lesions
- Bifurcation lesions.
Patient-related factors included:
- Low left ventricular ejection fraction
- Diabetes mellitus
- Peripheral arterial disease Premature discontinuation of clopidogrel.
ANTIPLATELET AGENTS: MECHANISM OF ACTION
Various pathways play synergistic roles in platelet activation and aggregation and thrombus formation, and different antiplatelet agents inhibit these specific pathways, thus complementing each other and having additive effects (Figure 2, Table 1).5,16–21
Aspirin inhibits cyclo-oxygenase 1
Cyclo-oxygenase 1, found in platelets, endothelial cells, and other cells, catalyzes the conversion of arachidonic acid to thromboxane A2. Aspirin irreversibly inhibits cyclo-oxygenase 1 by acetylating its serine residue, preventing formation of thromboxane A2 and preventing platelet activation and aggregation.
P2Y12 ADP receptor antagonists
Clopidogrel and prasugrel are thienopyridine agents that irreversibly inhibit the P2Y12 receptor, thereby preventing binding of adenosine diphosphate and the subsequent platelet activation-aggregation cascade. They are both prodrugs and require conversion by cytochrome P450 enzymes to active metabolites. Prasugrel is 10 times more potent than clopidogrel due to more efficient formation of its active metabolite, and it achieves a comparable effect on platelet inhibition 30 minutes faster than the peak effect of clopidogrel at 6 hours. The overall peak inhibitory effect of prasugrel is twice that of clopidogrel.22
Ticagrelor, a cyclopentyl-triazolo-pyrimidine, directly and reversibly inhibits the P2Y12 ADP receptor. Unlike clopidogrel and prasugrel, it does not need to be converted to an active metabolite, and it noncompetitively inhibits P2Y12 at a site different from the adenosine diphosphate binding site.23 Like prasugrel, ticagrelor inhibits platelet function more rapidly and more completely than clopidogrel.
Cangrelor, an intravenously administered analogue of adenosine triphosphate, reversibly inhibits the P2Y12 receptor. It has undergone phase 3 trials but is not yet approved for clinical use.24
WHY DUAL ANTIPLATELET THERAPY?
Aspirin is good, clopidogrel is better
Aspirin has a well-validated role in both primary and secondary prevention of coronary and noncoronary atherosclerotic vascular disease.
The CAPRIE trial found clopidogrel monotherapy to be superior to aspirin monotherapy in patients with established atherosclerotic vascular disease.25
After stenting, short-term dual therapy is better than short-term warfarin
Thrombotic complications in the early postprocedural period were a major limitation of stenting, and existing anticoagulation regimens were ineffective in preventing them.26,27
The ISAR trial studied the benefit of combined antiplatelet vs anticoagulant therapy after stent placement. Patients randomized to receive combined aspirin plus ticlopidine (an early P2Y12 inhibitor) had significantly lower rates of primary cardiac, hemorrhagic, and vascular events at 30 days.9 Two other trials confirmed this finding.28,29
STARS10 also confirmed the benefit of aspirin and ticlopidine after stenting. Patients were randomly assigned to aspirin alone, aspirin plus warfarin, or aspirin plus ticlopidine after stent placement. The rate of stent thrombosis at 30 days was significantly lower in the dual antiplatelet group than in the other two groups. The dual antiplatelet group had a higher rate of bleeding than the aspirin-alone group, but the rate was similar to that of the aspirin-plus-warfarin group.
Long-term dual antiplatelet therapy is beneficial in several situations
ISAR and STARS were landmark trials that showed stent thrombosis could be reduced by dual antiplatelet therapy for a 30-day period. However, the long-term role of dual antiplatelet therapy was still unknown.
The CURE trial30–32 randomized patients presenting with acute coronary syndromes without ST elevation to receive clopidogrel plus aspirin or placebo plus aspirin for 3 to 12 months. The rate of the primary end point (cardiac death, nonfatal myocardial infarction, or stroke) was significantly lower in the clopidogrel-plus-aspirin group. A similar benefit of dual antiplatelet therapy was seen in the subgroup of patients who underwent percutaneous coronary intervention. Both pretreatment with clopidogrel plus aspirin for a median of 10 days prior to percutaneous intervention and continuing it for a mean of 9 months reduced major adverse cardiovascular events.
The CREDO trial20 found that the combination of clopidogrel and aspirin significantly reduced the incidence of death, myocardial infarction, or stroke at 1 year after percutaneous coronary intervention. A subgroup of patients in this trial who had a longer pretreatment interval with a loading clopidogrel dose showed a benefit at 28 days, which was not as evident with a shorter loading dose interval.
The CLARITY-TIMI 28 trial33,34 showed the advantage of adding clopidogrel to aspirin in patients receiving fibrinolytic therapy for ST-elevation myocardial infarction. Adding clopidogrel both improved the patency of the infarct-related artery and reduced ischemic complications. In patients who subsequently underwent percutaneous coronary intervention and stenting, clopidogrel pretreatment was associated with a significant decrease in ischemic complications before and after the procedure. There was no significant increase in bleeding complications in either group.
COMMIT/CCS 235 also showed the benefit of dual antiplatelet therapy in patients with ST-elevation myocardial infarction. Clopidogrel added to aspirin during the short-term in-hospital or postdischarge treatment period significantly reduced a composite end point of reinfarction, death, or stroke as well as death from any cause.
The CHARISMA trial36–38 aimed to determine if patients who were more stable (ie, no recent acute coronary syndrome event or percutaneous coronary intervention) would benefit. Overall, CHARISMA showed no benefit of adding clopidogrel to aspirin compared with aspirin alone in a broad population of patients with established vascular disease (secondary prevention) or risk factors for vascular disease (primary prevention).
But importantly, though no benefit was seen in the primary prevention group, the large subgroup of patients with established atherosclerotic vascular disease (12,153 of the 15,603 patients in the trial) did benefit from dual antiplatelet therapy.36,37 This subgroup showed an overall reduction in absolute risk of 1.5% (relative risk 0.88, P = .046) over a median follow-up of 27.6 months. This benefit was even more apparent in the 9,478 patients with prior myocardial infarction, stroke, or peripheral artery disease, for whom the relative risk reduction was 17.1% (P = .01) and the reduction in absolute risk 1.5%.38
These results are comparable to the 2% absolute risk reduction in the CURE trial for similar end points over 9 months. In both studies, there was no significant increase in the risk of major bleeding or intracranial bleeding in the clopidogrel-plus-aspirin groups, although minor bleeding was increased by dual antiplatelet therapy.
The rate of severe bleeding, which was the primary safety end point in CHARISMA, was not significantly different in the clopidogrel-plus-aspirin group compared with the placebo-plus-aspirin group (relative risk 1.25, 95% CI 0.97–1.61, P = .09).
Thus, although the CHARISMA findings were negative overall, the positive finding observed in the predominant subgroup of patients with established vascular disease can therefore be considered supportive of the results of the subsequent trials discussed below.
The PEGASUS-TIMI 54 trial39 studied the benefit of adding ticagrelor (60 or 90 mg) to low-dose aspirin in patients with stable coronary artery disease who had had a myocardial infarction 1 to 3 years earlier.
Confirming the results of the CHARISMA subgroup analysis, the incidence of the ischemic primary efficacy end point (a composite of cardiovascular death, myocardial infarction, and stroke) was significantly lower in both groups receiving ticagrelor plus aspirin compared with those receiving placebo plus aspirin. The Kaplan-Meier rate at 3 years for the ticagrelor 90 mg-plus-aspirin group was 7.85% vs 9.04% for the placebo-plus-aspirin group (hazard ratio 0.85, 95% confidence interval [CI] 0.75–0.96, P = .008). The rate for the ticagrelor 60 mg-plus-aspirin group was 7.77% vs 9.04% for the placebo-plus-aspirin group (hazard ratio 0.84, 95% CI 0.74–0.95, P = .004).
The rates of all TIMI major and minor bleeding, as well as bleeding requiring transfusion or discontinuation of the study drug, were significantly higher in both ticagrelor dosing groups than in the placebo group (P < .01 for both groups vs placebo). The rates of fatal bleeding and nonfatal intracranial hemorrhage were not significantly higher. Although there was an overall reduction in ischemic end points with the addition of ticagrelor, there was also a significantly higher incidence of bleeding in this group.
Comment. Thus, with or without percutaneous coronary intervention in acute coronary syndrome as well as in stable coronary artery disease, dual antiplatelet therapy was shown to improve outcomes and decrease ischemic complications compared with aspirin alone. It provided benefit in the setting of acute coronary syndrome (in the CURE trial) and percutaneous coronary intervention (in the CREDO trial) for up to 1 year.
Major questions remained to be addressed:
- Do the results of CREDO, which was performed before the current interventional era and the use of drug-eluting stents, reflect outcomes after current interventional practice?
- Could shorter periods of dual antiplatelet therapy be sufficient, especially with newer stents with less risk of late thrombosis?
- Does the benefit of dual antiplatelet therapy extend beyond the 1-year time period tested in those trials to date?
RECOMMENDATIONS FOR DOSING
The American College of Cardiology Foundation/American Heart Association guidelines for dosing of antiplatelet agents for non-ST-elevation myocardial infarction are summarized in Table 2, and those for ST-elevation myocardial infarction are summarized in Table 3.1,2
WOULD SHORTER THERAPY AFTER STENTING WORK AS WELL?
The American College of Cardiology Foundation/American Heart Association currently recommend dual antiplatelet therapy for at least 12 months after drug-eluting stent placement, with shorter courses appropriate for patients who develop excessive bleeding complications or who are at high risk of bleeding.
Four trials (Table 4) evaluated whether shorter durations of dual antiplatelet therapy would suffice: SECURITY,40 EXCELLENT,41 OPTIMIZE,42 and RESET.43 All of them showed that short-duration therapy was not inferior to standard-duration therapy.44 These studies were comparable in that:
- Patients were randomized at the time of percutaneous coronary intervention or within 24 hours of it.
- Most patients received a second-generation drug-eluting stent, with the following exceptions: in EXCELLENT,41 one-fourth of patients received a Cypher first-generation drug-eluting stent, and in RESET,43 approximately one-fourth of the patients received a sirolimus-eluting stent in the standard-duration group for short lesions. Those patients with longer lesions in the RESET standard-duration group received an everolimus drug-eluting stent.
- The second antiplatelet added to aspirin in all studies was clopidogrel, with the exception of the SECURITY trial, in which fewer than 2% of patients received ticagrelor or prasugrel.40
- All the trials except RESET excluded patients who had had a myocardial infarction within 72 hours, and thus most patients studied had a lower risk profile.
- All of the trials sought to study noninferiority of short- vs standard-duration dual antiplatelet therapy, defined as the occurrence of a primary end point at 1 year (a composite of cardiovascular death, myocardial infarction, stroke, stent thrombosis, target vessel failure or revascularization, or bleeding).
Their low-risk patient populations and infrequent end points rendered these studies underpowered to make definitive conclusions about the relative efficacy of 6-months vs 12-months of dual antiplatelet therapy.
WOULD LONGER THERAPY BE BETTER?
The PRODIGY trial45 assessed durations of dual antiplatelet therapy both shorter and longer than the conventional 1 year, randomizing patients undergoing placement of a bare-metal stent, first-generation drug-eluting stent, or second-generation drug-eluting stent to receive aspirin and clopidogrel for either 6 months or 24 months. The study showed no significant difference in primary outcomes in the short- or long-duration groups.
Other trials that compared the standard 12 months of dual antiplatelet therapy with extended duration beyond 12 months were DAPT,3 ARCTIC-Interruption,46 and DES-LATE.47 The trials were comparable in that:
- All patients were randomized after completing 12 months of dual antiplatelet therapy following drug-eluting stent placement.
- All patients who were included had been free of major cardiac ischemic events or bleeding during the 12 months following stent placement.
- The primary aim of all three studies was to compare primary end points in groups receiving aspirin alone vs extended dual antiplatelet therapy. The primary end point was a composite of death due to a cardiovascular cause, nonfatal myocardial infarction, stroke, or stent thrombosis.
- The principal safety end point was bleeding.
Although the two earlier studies (ARCTIC-Interruption and DES-LATE) did not show any benefit of extended dual antiplatelet therapy compared with the standard 12-month duration, the recent DAPT study did.
The DAPT study
The DAPT study3 was an international, multicenter, placebo-controlled, double-blind randomized trial designed to examine the benefit of dual antiplatelet therapy beyond 1 year in a patient population large enough to provide definitive assessment of benefit and risk.
A total of 9,961 patients who received drug-eluting stents were randomized after 12 months of dual antiplatelet therapy to receive either a thienopyridine (clopidogrel or prasugrel) plus aspirin or placebo plus aspirin. They were followed for an additional 18 months. The coprimary efficacy end points were stent thrombosis and a composite of death, myocardial infarction, or stroke, while the primary safety end point was moderate or severe bleeding. The patients were also observed from months 30 to 33 on aspirin alone after stopping the thienopyridine.
Results. Longer therapy substantially reduced the risks of stent thrombosis (hazard ratio [HR] 0.29, 95% confidence interval [CI] 0.17–0.48) and the composite ischemic end point (HR 0.71, 95% CI 0.59–0.85). Follow-up during the 3-month thienopyridine discontinuation phase starting at 30 months revealed convergence of the ischemic event-rate curves in the two groups, which suggested that continuing dual antiplatelet therapy beyond 30 months might have been beneficial. Myocardial infarction unrelated to stent thrombosis accounted for 55% of the treatment benefit of dual antiplatelet therapy.
The risk of bleeding was higher in the thienopyridine group during the treatment period (2.5% vs 1.6%, P = .001). There was also a higher rate of noncardiovascular mortality in the thienopyridine group, although this difference may have been due to chance.3,48
Why were the results different?
All three trials included first- and second-generation drug-eluting stents, with different proportions in different trials. In ARCTIC-Interruption,46 43% of the patients in the continuation group had a first-generation stent, as did 64% of the patients in the dual antiplatelet group of DES-LATE.47 In the DAPT trial,3 38% of the patients in the longer-duration arm had a first-generation stent, and in 26% of cases it was a paclitaxel-eluting stent.
Only clopidogrel was used as the second antiplatelet agent in DES-LATE, whereas prasugrel was used in 10% of patients in ARCTIC-Interruption and 35% in DAPT.
Yet none of these differences seem to explain the differences in outcome among the studies. ARCTIC-Interruption and DES-LATE did not show any benefit of continued dual antiplatelet therapy beyond 12 months. DAPT showed benefit of extended therapy with prasugrel or with clopidogrel, and with first-generation or second-generation drug-eluting stents. The most likely explanation for the different results was that DAPT was the only trial sufficiently powered to definitively assess the end points, including stent thrombosis.
A balance between ischemic efficacy and bleeding risk is the major consideration with any antithrombotic and antiplatelet therapy. In the three largest trials we discussed (the vascular disease subgroups of CHARISMA,38 PEGASUS,39 and DAPT3), comparison of the prespecified efficacy and safety end points of each trial suggests that dual antiplatelet therapy has a net benefit, particularly given the irreversible nature of ischemic end points.
In CHARISMA,38 60 cardiovascular deaths, myocardial infarctions, or strokes were prevented per year per 10,000 patients treated, at the cost of 28 excess moderate bleeding events.
In PEGASUS,39 42 cardiovascular deaths, myocardial infarctions, or strokes were prevented, at the cost of 79 excess bleeding events requiring transfusion.
In DAPT (a selected population who had tolerated dual antiplatelet therapy for 1 year), 106 deaths, myocardial infarctions, or stroke events were prevented, at the cost of 47 excess moderate bleeding events.3
Indirect comparisons between trials are problematic, given different end point definitions, populations, and background therapies. But their results suggest that less-intensive inhibition with clopidogrel as the second antiplatelet long-term (as in CHARISMA) may provide the best balance of benefit vs risk.
BALANCING RISK AND BENEFIT
The evidence is unequivocal that dual antiplatelet therapy suppresses coronary ischemic complications resulting from thrombosis at sites of spontaneous plaque rupture following acute coronary syndromes or mechanical plaque disruption and foreign body implantation associated with percutaneous coronary intervention.
Three large-scale trials (DAPT,3 PEGASUS,39 and the secondary prevention subgroup of CHARISMA38) showed that the protective effect of dual antiplatelet therapy continues with prolonged therapy in patients who have experienced an acute coronary syndrome event or have received a drug-eluting stent. That benefit seems to be due to the action of these therapies on the culprit vessel (the one that caused the acute coronary syndrome or the site of stenting), as well as nonculprit arteries, emphasizing that dual antiplatelet therapy protects against atherosclerosis progression and future plaque rupture events.
For the durations studied in the longest trials thus far, 30 months (DAPT3) and 36 months (PEGASUS39), event curves continue to diverge, indicating that the advantage of dual antiplatelet therapy may persist for an indefinite period of time. Thus, indefinite therapy with dual antiplatelet agents can be supported, particularly in patients with advanced coronary artery disease or those who have had multiple coronary events.
We believe that the balance of evidence suggests that smaller studies that failed to show a benefit of longer-term therapy were underpowered to do so.
The ischemic protection is associated with the adverse effect of increased bleeding risk. Unfortunately, there has been little success in guiding dual antiplatelet therapy based on ischemic vs bleeding risk, in part because the same factors that predict risk of ischemic complications seem to predict increased susceptibility to bleeding. Nevertheless, indirect comparisons between studies suggest that for longer-term therapy clopidogrel may be superior to ticagrelor or prasugrel: the absolute excess bleeding risk with dual antiplatelet therapy vs aspirin in the CHARISMA secondary prevention subgroup was less than that in PEGASUS, with similar absolute reductions in ischemic events. So while the TRITON-TIMI 3822 and PLATO23 trials support the superiority of prasugrel or ticagrelor over clopidogrel for the first year after acute coronary syndrome, subsequent years of therapy may best be provided with clopidogrel.
Some patients may have identifiable factors that place them at very high risk of bleeding—need for surgical procedures, need for anticoagulation, or occurrence of bleeding complications or excessive “nuisance bleeding.” In those patients, the data suggest that dual antiplatelet therapy could be discontinued after 6 months, or perhaps even 3 months in the highest bleeding risk circumstances after second-generation drug-eluting stent placement.
WOEST49 was an open-label randomized controlled trial that studied the safety of antiplatelet regimens in patients on anticoagulation requiring percutaneous coronary interventions. Patients were randomized to double therapy with anticoagulant and clopidogrel vs triple therapy with additional aspirin following percutaneous coronary intervention. The primary end point was bleeding events within 1 year. Clopidogrel without aspirin was associated with significantly fewer bleeding events compared with triple therapy, with no increase in adverse ischemic events. The strategy tested in the WOEST trial seems reasonable in the specific group of patients who require ongoing anticoagulant therapy after drug-eluting stent placement, recognizing that the trial was somewhat underpowered to make definitive conclusions, particularly in patients at high risk for stent thrombosis.
Based on the results of PEGASUS and the CHARISMA subgroup with established ischemic burden, in which dual antiplatelet therapy was started after an interruption following the index coronary event, it is also reasonable to restart long-term dual antiplatelet therapy in patients who require interruption for short-term indications such as a surgical procedure.
Percutaneous coronary intervention for acute coronary syndromes has evolved, and so, hand in hand, has antiplatelet therapy. With the advent of clopidogrel and newer agents, several studies demonstrated the benefits of dual antiplatelet therapy in preventing major vascular ischemic complications. The findings culminated in a guideline recommendation for at least 12 months of dual antiplatelet therapy after placement of a drug-eluting stent, when feasible—a class I recommendation (treatment should be given), level of evidence B (limited populations evaluated).1,2 But extending dual antiplatelet therapy beyond 12 months had no strong favorable evidence until the recent Dual Antiplatelet Therapy (DAPT) study3 shed light on this topic.
Here, we review the evidence thus far on the optimal duration of dual antiplatelet therapy in the secondary prevention of coronary artery disease.
PLATELETS IN ACUTE CORONARY SYNDROMES AND STENT THROMBOSIS
Acute coronary syndromes begin with fissuring or ulceration of a vulnerable atherosclerotic plaque, followed by thrombosis and occlusion, mediated by platelet adhesion, activation, and aggregation (Figure 1). Transient occlusion results in unstable angina or non-ST-elevation myocardial infarction, while total occlusion usually results in ST-elevation myocardial infarction.
Platelet aggregation is prominent among the mechanisms leading to stent thrombosis and vaso-occlusive ischemic complications after percutaneous coronary intervention. Thus, antiplatelet agents play a vital role in both primary and secondary prevention of cardiovascular events.4–6
Adhesion, activation, and aggregation
Adhesion. Disruption of the vascular endothelium as a result of vulnerable plaque fissuring or ulceration exposes subendothelial thrombogenic collagen and von Willebrand factor to blood. Collagen engages platelets through their glycoprotein (GP) Ia, IIa, and VI receptors, and von Willebrand factor binds platelets through the GP Ib-IX-V receptor.
Activation. Once platelets adhere to the subendothelium, they undergo a conformational change and become activated. Simultaneous release of various autocrine and paracrine mediators including adenosine diphosphate, serotonin, epinephrine, thromboxane, and various ligand-receptor interactions all contribute to the activation cascade. Adenosine diphosphate binds to the platelet receptor P2Y1, leading to an increase in intracellular calcium, and it binds to P2Y12, leading to a decrease in cyclic adenosine monophosphate, both of which cause GP IIb/IIIa receptor activation. Thromboxane A2 released by platelets by cyclo-oxygenase 1 binds to alpha or beta variant receptors and contributes to GP IIb/IIIa activation through elevation of intracellular calcium levels.
Aggregation and thrombosis. Exposure of tissue factor to plasma following plaque rupture activates the coagulation cascade via the extrinsic pathway, which generates thrombin, a powerful platelet activator that causes thrombus formation via fibrin. Thrombin binds to protease-activated receptors PAR-1 and PAR-4 on platelets, causing an increase in intracellular calcium and a decrease in cyclic adenosine monophosphate with subsequent GP IIb/IIIa activation. GP IIb/IIIa facilitates platelet aggregation by binding to fibrinogen and forming a stable platelet thrombus.
In the early stages of thrombus formation, platelets predominate (“white” thrombi); further organization with fibrin results in older “red” thrombi. The stages of thrombi vary in non-ST-elevation and ST-elevation myocardial infarction and are prognostic markers of death.4–8
PERCUTANEOUS INTERVENTION, RESTENOSIS, AND STENT THROMBOSIS
Percutaneous coronary intervention, the preferred means of revascularization for many patients, is performed emergently in patients with ST-elevation myocardial infarction, urgently in those with acute coronary syndromes without ST elevation, and electively in those with stable ischemic symptoms.
Percutaneous revascularization techniques have evolved from balloon angioplasty to bare-metal stents to drug-eluting stents, but each of these procedures has been associated with a periprocedural and postprocedural risk of thrombosis.
Balloon angioplasty was associated with vascular intimal injury, inciting elastic vascular recoil and smooth muscle cell proliferation leading to restenosis.
Bare-metal stents reduced the restenosis rate by eliminating vascular recoil, although restenosis still occurred within the stent because of neointimal proliferation of vascular smooth muscle cells. This was an important limitation, as both acute and subacute stent thrombosis were refractory to aggressive anticoagulation regimens that were associated with major bleeding complications and longer hospital length of stay. Stenting became mainstream practice only after the ISAR9 and STARS10 trials showed that dual antiplatelet therapy controlled stent thrombosis.
Drug-eluting stents coated with anti-proliferative and anti-inflammatory polymers markedly reduced in-stent restenosis rates by suppressing the initial vascular smooth-muscle proliferative response. However, they were still associated with late and very late stent thrombosis with incomplete endothelialization, even up to 40 months after implantation. Proposed mechanisms include incomplete stent apposition and inflammatory hypersensitivity reactions to the polymer coating. Incomplete stent apposition associated with low-velocity blood flow at the junction of the stent strut and vessel wall, together with delayed endothelialization, promotes platelet adhesion and aggregation, followed by thrombus formation.11
Second-generation drug-eluting stents have thinner struts and more biocompatible polymers and are thought to favor more complete re-endothelialization, reducing the rates of stent thrombosis.8,12,13
Predictors of early stent thrombosis
The Dutch Stent Thrombosis Registry and other studies looked at risk factors for stent thrombosis.14,15
Procedure-related factors included:
- Stent undersizing
- Residual uncovered dissections after angioplasty
- Longer stents
- Low flow after angioplasty (< 3 on the 0–3 Thrombolysis in Myocardial Infarction [TIMI] scale).
Lesion-related factors included:
- Intermediate coronary artery disease both proximal and distal to the culprit lesions
- Bifurcation lesions.
Patient-related factors included:
- Low left ventricular ejection fraction
- Diabetes mellitus
- Peripheral arterial disease Premature discontinuation of clopidogrel.
ANTIPLATELET AGENTS: MECHANISM OF ACTION
Various pathways play synergistic roles in platelet activation and aggregation and thrombus formation, and different antiplatelet agents inhibit these specific pathways, thus complementing each other and having additive effects (Figure 2, Table 1).5,16–21
Aspirin inhibits cyclo-oxygenase 1
Cyclo-oxygenase 1, found in platelets, endothelial cells, and other cells, catalyzes the conversion of arachidonic acid to thromboxane A2. Aspirin irreversibly inhibits cyclo-oxygenase 1 by acetylating its serine residue, preventing formation of thromboxane A2 and preventing platelet activation and aggregation.
P2Y12 ADP receptor antagonists
Clopidogrel and prasugrel are thienopyridine agents that irreversibly inhibit the P2Y12 receptor, thereby preventing binding of adenosine diphosphate and the subsequent platelet activation-aggregation cascade. They are both prodrugs and require conversion by cytochrome P450 enzymes to active metabolites. Prasugrel is 10 times more potent than clopidogrel due to more efficient formation of its active metabolite, and it achieves a comparable effect on platelet inhibition 30 minutes faster than the peak effect of clopidogrel at 6 hours. The overall peak inhibitory effect of prasugrel is twice that of clopidogrel.22
Ticagrelor, a cyclopentyl-triazolo-pyrimidine, directly and reversibly inhibits the P2Y12 ADP receptor. Unlike clopidogrel and prasugrel, it does not need to be converted to an active metabolite, and it noncompetitively inhibits P2Y12 at a site different from the adenosine diphosphate binding site.23 Like prasugrel, ticagrelor inhibits platelet function more rapidly and more completely than clopidogrel.
Cangrelor, an intravenously administered analogue of adenosine triphosphate, reversibly inhibits the P2Y12 receptor. It has undergone phase 3 trials but is not yet approved for clinical use.24
WHY DUAL ANTIPLATELET THERAPY?
Aspirin is good, clopidogrel is better
Aspirin has a well-validated role in both primary and secondary prevention of coronary and noncoronary atherosclerotic vascular disease.
The CAPRIE trial found clopidogrel monotherapy to be superior to aspirin monotherapy in patients with established atherosclerotic vascular disease.25
After stenting, short-term dual therapy is better than short-term warfarin
Thrombotic complications in the early postprocedural period were a major limitation of stenting, and existing anticoagulation regimens were ineffective in preventing them.26,27
The ISAR trial studied the benefit of combined antiplatelet vs anticoagulant therapy after stent placement. Patients randomized to receive combined aspirin plus ticlopidine (an early P2Y12 inhibitor) had significantly lower rates of primary cardiac, hemorrhagic, and vascular events at 30 days.9 Two other trials confirmed this finding.28,29
STARS10 also confirmed the benefit of aspirin and ticlopidine after stenting. Patients were randomly assigned to aspirin alone, aspirin plus warfarin, or aspirin plus ticlopidine after stent placement. The rate of stent thrombosis at 30 days was significantly lower in the dual antiplatelet group than in the other two groups. The dual antiplatelet group had a higher rate of bleeding than the aspirin-alone group, but the rate was similar to that of the aspirin-plus-warfarin group.
Long-term dual antiplatelet therapy is beneficial in several situations
ISAR and STARS were landmark trials that showed stent thrombosis could be reduced by dual antiplatelet therapy for a 30-day period. However, the long-term role of dual antiplatelet therapy was still unknown.
The CURE trial30–32 randomized patients presenting with acute coronary syndromes without ST elevation to receive clopidogrel plus aspirin or placebo plus aspirin for 3 to 12 months. The rate of the primary end point (cardiac death, nonfatal myocardial infarction, or stroke) was significantly lower in the clopidogrel-plus-aspirin group. A similar benefit of dual antiplatelet therapy was seen in the subgroup of patients who underwent percutaneous coronary intervention. Both pretreatment with clopidogrel plus aspirin for a median of 10 days prior to percutaneous intervention and continuing it for a mean of 9 months reduced major adverse cardiovascular events.
The CREDO trial20 found that the combination of clopidogrel and aspirin significantly reduced the incidence of death, myocardial infarction, or stroke at 1 year after percutaneous coronary intervention. A subgroup of patients in this trial who had a longer pretreatment interval with a loading clopidogrel dose showed a benefit at 28 days, which was not as evident with a shorter loading dose interval.
The CLARITY-TIMI 28 trial33,34 showed the advantage of adding clopidogrel to aspirin in patients receiving fibrinolytic therapy for ST-elevation myocardial infarction. Adding clopidogrel both improved the patency of the infarct-related artery and reduced ischemic complications. In patients who subsequently underwent percutaneous coronary intervention and stenting, clopidogrel pretreatment was associated with a significant decrease in ischemic complications before and after the procedure. There was no significant increase in bleeding complications in either group.
COMMIT/CCS 235 also showed the benefit of dual antiplatelet therapy in patients with ST-elevation myocardial infarction. Clopidogrel added to aspirin during the short-term in-hospital or postdischarge treatment period significantly reduced a composite end point of reinfarction, death, or stroke as well as death from any cause.
The CHARISMA trial36–38 aimed to determine if patients who were more stable (ie, no recent acute coronary syndrome event or percutaneous coronary intervention) would benefit. Overall, CHARISMA showed no benefit of adding clopidogrel to aspirin compared with aspirin alone in a broad population of patients with established vascular disease (secondary prevention) or risk factors for vascular disease (primary prevention).
But importantly, though no benefit was seen in the primary prevention group, the large subgroup of patients with established atherosclerotic vascular disease (12,153 of the 15,603 patients in the trial) did benefit from dual antiplatelet therapy.36,37 This subgroup showed an overall reduction in absolute risk of 1.5% (relative risk 0.88, P = .046) over a median follow-up of 27.6 months. This benefit was even more apparent in the 9,478 patients with prior myocardial infarction, stroke, or peripheral artery disease, for whom the relative risk reduction was 17.1% (P = .01) and the reduction in absolute risk 1.5%.38
These results are comparable to the 2% absolute risk reduction in the CURE trial for similar end points over 9 months. In both studies, there was no significant increase in the risk of major bleeding or intracranial bleeding in the clopidogrel-plus-aspirin groups, although minor bleeding was increased by dual antiplatelet therapy.
The rate of severe bleeding, which was the primary safety end point in CHARISMA, was not significantly different in the clopidogrel-plus-aspirin group compared with the placebo-plus-aspirin group (relative risk 1.25, 95% CI 0.97–1.61, P = .09).
Thus, although the CHARISMA findings were negative overall, the positive finding observed in the predominant subgroup of patients with established vascular disease can therefore be considered supportive of the results of the subsequent trials discussed below.
The PEGASUS-TIMI 54 trial39 studied the benefit of adding ticagrelor (60 or 90 mg) to low-dose aspirin in patients with stable coronary artery disease who had had a myocardial infarction 1 to 3 years earlier.
Confirming the results of the CHARISMA subgroup analysis, the incidence of the ischemic primary efficacy end point (a composite of cardiovascular death, myocardial infarction, and stroke) was significantly lower in both groups receiving ticagrelor plus aspirin compared with those receiving placebo plus aspirin. The Kaplan-Meier rate at 3 years for the ticagrelor 90 mg-plus-aspirin group was 7.85% vs 9.04% for the placebo-plus-aspirin group (hazard ratio 0.85, 95% confidence interval [CI] 0.75–0.96, P = .008). The rate for the ticagrelor 60 mg-plus-aspirin group was 7.77% vs 9.04% for the placebo-plus-aspirin group (hazard ratio 0.84, 95% CI 0.74–0.95, P = .004).
The rates of all TIMI major and minor bleeding, as well as bleeding requiring transfusion or discontinuation of the study drug, were significantly higher in both ticagrelor dosing groups than in the placebo group (P < .01 for both groups vs placebo). The rates of fatal bleeding and nonfatal intracranial hemorrhage were not significantly higher. Although there was an overall reduction in ischemic end points with the addition of ticagrelor, there was also a significantly higher incidence of bleeding in this group.
Comment. Thus, with or without percutaneous coronary intervention in acute coronary syndrome as well as in stable coronary artery disease, dual antiplatelet therapy was shown to improve outcomes and decrease ischemic complications compared with aspirin alone. It provided benefit in the setting of acute coronary syndrome (in the CURE trial) and percutaneous coronary intervention (in the CREDO trial) for up to 1 year.
Major questions remained to be addressed:
- Do the results of CREDO, which was performed before the current interventional era and the use of drug-eluting stents, reflect outcomes after current interventional practice?
- Could shorter periods of dual antiplatelet therapy be sufficient, especially with newer stents with less risk of late thrombosis?
- Does the benefit of dual antiplatelet therapy extend beyond the 1-year time period tested in those trials to date?
RECOMMENDATIONS FOR DOSING
The American College of Cardiology Foundation/American Heart Association guidelines for dosing of antiplatelet agents for non-ST-elevation myocardial infarction are summarized in Table 2, and those for ST-elevation myocardial infarction are summarized in Table 3.1,2
WOULD SHORTER THERAPY AFTER STENTING WORK AS WELL?
The American College of Cardiology Foundation/American Heart Association currently recommend dual antiplatelet therapy for at least 12 months after drug-eluting stent placement, with shorter courses appropriate for patients who develop excessive bleeding complications or who are at high risk of bleeding.
Four trials (Table 4) evaluated whether shorter durations of dual antiplatelet therapy would suffice: SECURITY,40 EXCELLENT,41 OPTIMIZE,42 and RESET.43 All of them showed that short-duration therapy was not inferior to standard-duration therapy.44 These studies were comparable in that:
- Patients were randomized at the time of percutaneous coronary intervention or within 24 hours of it.
- Most patients received a second-generation drug-eluting stent, with the following exceptions: in EXCELLENT,41 one-fourth of patients received a Cypher first-generation drug-eluting stent, and in RESET,43 approximately one-fourth of the patients received a sirolimus-eluting stent in the standard-duration group for short lesions. Those patients with longer lesions in the RESET standard-duration group received an everolimus drug-eluting stent.
- The second antiplatelet added to aspirin in all studies was clopidogrel, with the exception of the SECURITY trial, in which fewer than 2% of patients received ticagrelor or prasugrel.40
- All the trials except RESET excluded patients who had had a myocardial infarction within 72 hours, and thus most patients studied had a lower risk profile.
- All of the trials sought to study noninferiority of short- vs standard-duration dual antiplatelet therapy, defined as the occurrence of a primary end point at 1 year (a composite of cardiovascular death, myocardial infarction, stroke, stent thrombosis, target vessel failure or revascularization, or bleeding).
Their low-risk patient populations and infrequent end points rendered these studies underpowered to make definitive conclusions about the relative efficacy of 6-months vs 12-months of dual antiplatelet therapy.
WOULD LONGER THERAPY BE BETTER?
The PRODIGY trial45 assessed durations of dual antiplatelet therapy both shorter and longer than the conventional 1 year, randomizing patients undergoing placement of a bare-metal stent, first-generation drug-eluting stent, or second-generation drug-eluting stent to receive aspirin and clopidogrel for either 6 months or 24 months. The study showed no significant difference in primary outcomes in the short- or long-duration groups.
Other trials that compared the standard 12 months of dual antiplatelet therapy with extended duration beyond 12 months were DAPT,3 ARCTIC-Interruption,46 and DES-LATE.47 The trials were comparable in that:
- All patients were randomized after completing 12 months of dual antiplatelet therapy following drug-eluting stent placement.
- All patients who were included had been free of major cardiac ischemic events or bleeding during the 12 months following stent placement.
- The primary aim of all three studies was to compare primary end points in groups receiving aspirin alone vs extended dual antiplatelet therapy. The primary end point was a composite of death due to a cardiovascular cause, nonfatal myocardial infarction, stroke, or stent thrombosis.
- The principal safety end point was bleeding.
Although the two earlier studies (ARCTIC-Interruption and DES-LATE) did not show any benefit of extended dual antiplatelet therapy compared with the standard 12-month duration, the recent DAPT study did.
The DAPT study
The DAPT study3 was an international, multicenter, placebo-controlled, double-blind randomized trial designed to examine the benefit of dual antiplatelet therapy beyond 1 year in a patient population large enough to provide definitive assessment of benefit and risk.
A total of 9,961 patients who received drug-eluting stents were randomized after 12 months of dual antiplatelet therapy to receive either a thienopyridine (clopidogrel or prasugrel) plus aspirin or placebo plus aspirin. They were followed for an additional 18 months. The coprimary efficacy end points were stent thrombosis and a composite of death, myocardial infarction, or stroke, while the primary safety end point was moderate or severe bleeding. The patients were also observed from months 30 to 33 on aspirin alone after stopping the thienopyridine.
Results. Longer therapy substantially reduced the risks of stent thrombosis (hazard ratio [HR] 0.29, 95% confidence interval [CI] 0.17–0.48) and the composite ischemic end point (HR 0.71, 95% CI 0.59–0.85). Follow-up during the 3-month thienopyridine discontinuation phase starting at 30 months revealed convergence of the ischemic event-rate curves in the two groups, which suggested that continuing dual antiplatelet therapy beyond 30 months might have been beneficial. Myocardial infarction unrelated to stent thrombosis accounted for 55% of the treatment benefit of dual antiplatelet therapy.
The risk of bleeding was higher in the thienopyridine group during the treatment period (2.5% vs 1.6%, P = .001). There was also a higher rate of noncardiovascular mortality in the thienopyridine group, although this difference may have been due to chance.3,48
Why were the results different?
All three trials included first- and second-generation drug-eluting stents, with different proportions in different trials. In ARCTIC-Interruption,46 43% of the patients in the continuation group had a first-generation stent, as did 64% of the patients in the dual antiplatelet group of DES-LATE.47 In the DAPT trial,3 38% of the patients in the longer-duration arm had a first-generation stent, and in 26% of cases it was a paclitaxel-eluting stent.
Only clopidogrel was used as the second antiplatelet agent in DES-LATE, whereas prasugrel was used in 10% of patients in ARCTIC-Interruption and 35% in DAPT.
Yet none of these differences seem to explain the differences in outcome among the studies. ARCTIC-Interruption and DES-LATE did not show any benefit of continued dual antiplatelet therapy beyond 12 months. DAPT showed benefit of extended therapy with prasugrel or with clopidogrel, and with first-generation or second-generation drug-eluting stents. The most likely explanation for the different results was that DAPT was the only trial sufficiently powered to definitively assess the end points, including stent thrombosis.
A balance between ischemic efficacy and bleeding risk is the major consideration with any antithrombotic and antiplatelet therapy. In the three largest trials we discussed (the vascular disease subgroups of CHARISMA,38 PEGASUS,39 and DAPT3), comparison of the prespecified efficacy and safety end points of each trial suggests that dual antiplatelet therapy has a net benefit, particularly given the irreversible nature of ischemic end points.
In CHARISMA,38 60 cardiovascular deaths, myocardial infarctions, or strokes were prevented per year per 10,000 patients treated, at the cost of 28 excess moderate bleeding events.
In PEGASUS,39 42 cardiovascular deaths, myocardial infarctions, or strokes were prevented, at the cost of 79 excess bleeding events requiring transfusion.
In DAPT (a selected population who had tolerated dual antiplatelet therapy for 1 year), 106 deaths, myocardial infarctions, or stroke events were prevented, at the cost of 47 excess moderate bleeding events.3
Indirect comparisons between trials are problematic, given different end point definitions, populations, and background therapies. But their results suggest that less-intensive inhibition with clopidogrel as the second antiplatelet long-term (as in CHARISMA) may provide the best balance of benefit vs risk.
BALANCING RISK AND BENEFIT
The evidence is unequivocal that dual antiplatelet therapy suppresses coronary ischemic complications resulting from thrombosis at sites of spontaneous plaque rupture following acute coronary syndromes or mechanical plaque disruption and foreign body implantation associated with percutaneous coronary intervention.
Three large-scale trials (DAPT,3 PEGASUS,39 and the secondary prevention subgroup of CHARISMA38) showed that the protective effect of dual antiplatelet therapy continues with prolonged therapy in patients who have experienced an acute coronary syndrome event or have received a drug-eluting stent. That benefit seems to be due to the action of these therapies on the culprit vessel (the one that caused the acute coronary syndrome or the site of stenting), as well as nonculprit arteries, emphasizing that dual antiplatelet therapy protects against atherosclerosis progression and future plaque rupture events.
For the durations studied in the longest trials thus far, 30 months (DAPT3) and 36 months (PEGASUS39), event curves continue to diverge, indicating that the advantage of dual antiplatelet therapy may persist for an indefinite period of time. Thus, indefinite therapy with dual antiplatelet agents can be supported, particularly in patients with advanced coronary artery disease or those who have had multiple coronary events.
We believe that the balance of evidence suggests that smaller studies that failed to show a benefit of longer-term therapy were underpowered to do so.
The ischemic protection is associated with the adverse effect of increased bleeding risk. Unfortunately, there has been little success in guiding dual antiplatelet therapy based on ischemic vs bleeding risk, in part because the same factors that predict risk of ischemic complications seem to predict increased susceptibility to bleeding. Nevertheless, indirect comparisons between studies suggest that for longer-term therapy clopidogrel may be superior to ticagrelor or prasugrel: the absolute excess bleeding risk with dual antiplatelet therapy vs aspirin in the CHARISMA secondary prevention subgroup was less than that in PEGASUS, with similar absolute reductions in ischemic events. So while the TRITON-TIMI 3822 and PLATO23 trials support the superiority of prasugrel or ticagrelor over clopidogrel for the first year after acute coronary syndrome, subsequent years of therapy may best be provided with clopidogrel.
Some patients may have identifiable factors that place them at very high risk of bleeding—need for surgical procedures, need for anticoagulation, or occurrence of bleeding complications or excessive “nuisance bleeding.” In those patients, the data suggest that dual antiplatelet therapy could be discontinued after 6 months, or perhaps even 3 months in the highest bleeding risk circumstances after second-generation drug-eluting stent placement.
WOEST49 was an open-label randomized controlled trial that studied the safety of antiplatelet regimens in patients on anticoagulation requiring percutaneous coronary interventions. Patients were randomized to double therapy with anticoagulant and clopidogrel vs triple therapy with additional aspirin following percutaneous coronary intervention. The primary end point was bleeding events within 1 year. Clopidogrel without aspirin was associated with significantly fewer bleeding events compared with triple therapy, with no increase in adverse ischemic events. The strategy tested in the WOEST trial seems reasonable in the specific group of patients who require ongoing anticoagulant therapy after drug-eluting stent placement, recognizing that the trial was somewhat underpowered to make definitive conclusions, particularly in patients at high risk for stent thrombosis.
Based on the results of PEGASUS and the CHARISMA subgroup with established ischemic burden, in which dual antiplatelet therapy was started after an interruption following the index coronary event, it is also reasonable to restart long-term dual antiplatelet therapy in patients who require interruption for short-term indications such as a surgical procedure.
- American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:e78–e140.
- Amsterdam EA, Wenger NK, Brindis RG, et al; ACC/AHA Task Force Members. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 130:e344–e426.
- Mauri L, Kereiakes DJ, Yeh RW, et al; DAPT Study Investigators. Twelve or 30 months of dual antiplatelet therapy after drug-eluting stents. N Engl J Med 2014; 371:2155–2166.
- Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J 2010; 74:597–607.
- Papp J, Kenyeres P, Toth K. Clinical importance of antiplatelet drugs in cardiovascular diseases. Clin Hemorheol Microcirc 2013; 53:81–96.
- Showkathali R, Natarajan A. Antiplatelet and antithrombin strategies in acute coronary syndrome: state-of-the-art review. Curr Cardiol Rev 2012; 8:239–249.
- Angiolillo DJ. The evolution of antiplatelet therapy in the treatment of acute coronary syndromes: from aspirin to the present day. Drugs 2012; 72:2087–2116.
- Claessen BE, Henriques JP, Jaffer FA, Mehran R, Piek JJ, Dangas GD. Stent thrombosis: a clinical perspective. JACC Cardiovasc Interv 2014; 7:1081–1092.
- Schomig A, Neumann FJ, Kastrati A, et al. A randomized comparison of antiplatelet and anticoagulant therapy after the placement of coronary-artery stents. N Engl J Med 1996; 334:1084–1089.
- Leon MB, Baim DS, Popma JJ, et al. A clinical trial comparing three antithrombotic-drug regimens after coronary-artery stenting. Stent Anticoagulation Restenosis Study Investigators. N Engl J Med 1998; 339:1665–1671.
- Joner M, Finn AV, Farb A, et al. Pathology of drug-eluting stents in humans: delayed healing and late thrombotic risk. J Am Coll Cardiol 2006; 48:193–202.
- Nikam N, Steinberg TB, Steinberg DH. Advances in stent technologies and their effect on clinical efficacy and safety. Med Devices (Auckl) 2014; 7:165–178.
- Simard T, Hibbert B, Ramirez FD, Froeschl M, Chen YX, O’Brien ER. The evolution of coronary stents: a brief review. Can J Cardiol 2014; 30:35–45.
- Byrne RA, Joner M, Kastrati A. Stent thrombosis and restenosis: what have we learned and where are we going? The Andreas Gruntzig Lecture ESC 2014. Eur Heart J 2015; 36:3320–3331.
- van Werkum JW, Heestermans AA, Zomer AC, et al. Predictors of coronary stent thrombosis: the Dutch Stent Thrombosis Registry. J Am Coll Cardiol 2009; 53:1399–1409.
- Berger JS. Aspirin, clopidogrel, and ticagrelor in acute coronary syndromes. Am J Cardiol 2013; 112:737–745.
- Franchi F, Angiolillo DJ. Novel antiplatelet agents in acute coronary syndrome. Nat Rev Cardiol 2015; 12:30–47.
- Patrono C, Rocca B. The future of antiplatelet therapy in cardiovascular disease. Annu Rev Med 2010; 61:49–61.
- Park SJ, Kang SM, Park DW. Dual antiplatelet therapy after drug-eluting stents: defining the proper duration. Coron Artery Dis 2014; 25:83–89.
- Steinhubl SR, Berger PB, Mann JT 3rd, et al; CREDO Investigators. Clopidogrel for the reduction of events during observation. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Nusca A, Patti G. Platelet function and inhibition in ischemic heart disease. Curr Cardiol Rep 2012; 14:457–467.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Genereux P, Stone GW, Harrington RA, et al; CHAMPION PHOENIX Investigators. Impact of intraprocedural stent thrombosis during percutaneous coronary intervention: Insights from the CHAMPION PHOENIX Trial (Clinical Trial Comparing Cangrelor to Clopidogrel Standard of Care Therapy in Subjects Who Require Percutaneous Coronary Intervention). J Am Coll Cardiol 2014; 63:619–629.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Brilakis ES, Patel VG, Banerjee S. Medical management after coronary stent implantation: a review. JAMA 2013; 310:189–198.
- Warren J, Baber U, Mehran R. Antiplatelet therapy after drug-eluting stent implantation. J Cardiol 2015; 65:98–104.
- Urban P, Macaya C, Rupprecht HJ, et al. Randomized evaluation of anticoagulation versus antiplatelet therapy after coronary stent implantation in high-risk patients: the Multicenter Aspirin and Ticlopidine Trial After Intracoronary Stenting (MATTIS). Circulation 1998; 98:2126–2132.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: The PCI-CURE study. Lancet 2001; 358:527–533.
- Morais J. Insights from CURE: using clopidogrel on top of standard therapy. Cerebrovasc Dis 2002; 13(suppl 1):17–21.
- Ferguson JJ. Clopidogrel plus aspirin in patients with acute myocardial infarction treated with fibrinolytic therapy—CLARITY-TIMI 28. Future Cardiol 2005; 1:605–610.
- Sabatine MS, Cannon CP, Gibson CM, et al; Clopidogrel as Adjunctive Reperfusion Therapy (CLARITY)-Thrombolysis in Myocardial Infarction (TIMI) 28 Investigators. Effect of clopidogrel pretreatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005; 294:1224–1232.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bonaca MP, Bhatt DL, Cohen M, et al; PEGASUS-TIMI 54 Steering Committee and Investigators. Long-term use of ticagrelor in patients with prior myocardial infarction. N Engl J Med 2015; 372:1791–1800.
- Colombo A, Chieffo A, Frasheri A, et al. Second-generation drug-eluting stent implantation followed by 6- versus 12-month dual antiplatelet therapy: the SECURITY randomized clinical trial. J Am Coll Cardiol 2014; 64:2086–2097.
- Gwon HC, Hahn JY, Park KW, et al. Six-month versus 12-month dual antiplatelet therapy after implantation of drug-eluting stents: the Efficacy of Xience/Promus versus Cypher to Reduce Late Loss After Stenting (EXCELLENT) randomized, multicenter study. Circulation 2012; 125:505–513.
- Feres F, Costa RA, Abizaid A, et al; OPTIMIZE Trial Investigators. Three vs twelve months of dual antiplatelet therapy after zotarolimus-eluting stents: the OPTIMIZE randomized trial. JAMA 2013; 310:2510–2522.
- Kim BK, Hong MK, Shin DH, et al; RESET Investigators. A new strategy for discontinuation of dual antiplatelet therapy: the RESET Trial (REal Safety and Efficacy of 3-month dual antiplatelet Therapy following endeavor zotarolimus-eluting stent implantation). J Am Coll Cardiol 2012; 60:1340–1348.
- El-Hayek G, Messerli F, Bangalore S, et al. Meta-analysis of randomized clinical trials comparing short-term versus long-term dual antiplatelet therapy following drug-eluting stents. Am J Cardiol 2014; 114:236–242.
- Valgimigli M, Campo G, Monti M, et al; Prolonging Dual Antiplatelet Treatment After Grading Stent-Induced Intimal Hyperplasia Study (PRODIGY) Investigators. Short- versus long-term duration of dual-antiplatelet therapy after coronary stenting: a randomized multicenter trial. Circulation 2012; 125:2015–2026.
- Collet JP, Silvain J, Barthelemy O, et al; ARCTIC investigators. Dual-antiplatelet treatment beyond 1 year after drug-eluting stent implantation (ARCTIC-Interruption): a randomised trial. Lancet 2014; 384:1577–1585.
- Lee CW, Ahn JM, Park DW, et al. Optimal duration of dual antiplatelet therapy after drug-eluting stent implantation: a randomized, controlled trial. Circulation 2014; 129:304–312.
- Kwok CS, Bulluck H, Ryding AD, Loke YK. Benefits and harms of extending the duration of dual antiplatelet therapy after percutaneous coronary intervention with drug-eluting stents: a meta-analysis. ScientificWorldJournal 2014; 2014:794078.
- Dewilde WJ, Oirbans T, Verheugt FW, et al; WOEST study investigators. Use of clopidogrel with or without aspirin in patients taking oral anticoagulant therapy and undergoing percutaneous coronary intervention: an open-label, randomised, controlled trial. Lancet 2013; 381:1107–1115.
- American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:e78–e140.
- Amsterdam EA, Wenger NK, Brindis RG, et al; ACC/AHA Task Force Members. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 130:e344–e426.
- Mauri L, Kereiakes DJ, Yeh RW, et al; DAPT Study Investigators. Twelve or 30 months of dual antiplatelet therapy after drug-eluting stents. N Engl J Med 2014; 371:2155–2166.
- Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J 2010; 74:597–607.
- Papp J, Kenyeres P, Toth K. Clinical importance of antiplatelet drugs in cardiovascular diseases. Clin Hemorheol Microcirc 2013; 53:81–96.
- Showkathali R, Natarajan A. Antiplatelet and antithrombin strategies in acute coronary syndrome: state-of-the-art review. Curr Cardiol Rev 2012; 8:239–249.
- Angiolillo DJ. The evolution of antiplatelet therapy in the treatment of acute coronary syndromes: from aspirin to the present day. Drugs 2012; 72:2087–2116.
- Claessen BE, Henriques JP, Jaffer FA, Mehran R, Piek JJ, Dangas GD. Stent thrombosis: a clinical perspective. JACC Cardiovasc Interv 2014; 7:1081–1092.
- Schomig A, Neumann FJ, Kastrati A, et al. A randomized comparison of antiplatelet and anticoagulant therapy after the placement of coronary-artery stents. N Engl J Med 1996; 334:1084–1089.
- Leon MB, Baim DS, Popma JJ, et al. A clinical trial comparing three antithrombotic-drug regimens after coronary-artery stenting. Stent Anticoagulation Restenosis Study Investigators. N Engl J Med 1998; 339:1665–1671.
- Joner M, Finn AV, Farb A, et al. Pathology of drug-eluting stents in humans: delayed healing and late thrombotic risk. J Am Coll Cardiol 2006; 48:193–202.
- Nikam N, Steinberg TB, Steinberg DH. Advances in stent technologies and their effect on clinical efficacy and safety. Med Devices (Auckl) 2014; 7:165–178.
- Simard T, Hibbert B, Ramirez FD, Froeschl M, Chen YX, O’Brien ER. The evolution of coronary stents: a brief review. Can J Cardiol 2014; 30:35–45.
- Byrne RA, Joner M, Kastrati A. Stent thrombosis and restenosis: what have we learned and where are we going? The Andreas Gruntzig Lecture ESC 2014. Eur Heart J 2015; 36:3320–3331.
- van Werkum JW, Heestermans AA, Zomer AC, et al. Predictors of coronary stent thrombosis: the Dutch Stent Thrombosis Registry. J Am Coll Cardiol 2009; 53:1399–1409.
- Berger JS. Aspirin, clopidogrel, and ticagrelor in acute coronary syndromes. Am J Cardiol 2013; 112:737–745.
- Franchi F, Angiolillo DJ. Novel antiplatelet agents in acute coronary syndrome. Nat Rev Cardiol 2015; 12:30–47.
- Patrono C, Rocca B. The future of antiplatelet therapy in cardiovascular disease. Annu Rev Med 2010; 61:49–61.
- Park SJ, Kang SM, Park DW. Dual antiplatelet therapy after drug-eluting stents: defining the proper duration. Coron Artery Dis 2014; 25:83–89.
- Steinhubl SR, Berger PB, Mann JT 3rd, et al; CREDO Investigators. Clopidogrel for the reduction of events during observation. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 288:2411–2420.
- Nusca A, Patti G. Platelet function and inhibition in ischemic heart disease. Curr Cardiol Rep 2012; 14:457–467.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Genereux P, Stone GW, Harrington RA, et al; CHAMPION PHOENIX Investigators. Impact of intraprocedural stent thrombosis during percutaneous coronary intervention: Insights from the CHAMPION PHOENIX Trial (Clinical Trial Comparing Cangrelor to Clopidogrel Standard of Care Therapy in Subjects Who Require Percutaneous Coronary Intervention). J Am Coll Cardiol 2014; 63:619–629.
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet 1996; 348:1329–1339.
- Brilakis ES, Patel VG, Banerjee S. Medical management after coronary stent implantation: a review. JAMA 2013; 310:189–198.
- Warren J, Baber U, Mehran R. Antiplatelet therapy after drug-eluting stent implantation. J Cardiol 2015; 65:98–104.
- Urban P, Macaya C, Rupprecht HJ, et al. Randomized evaluation of anticoagulation versus antiplatelet therapy after coronary stent implantation in high-risk patients: the Multicenter Aspirin and Ticlopidine Trial After Intracoronary Stenting (MATTIS). Circulation 1998; 98:2126–2132.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: The PCI-CURE study. Lancet 2001; 358:527–533.
- Morais J. Insights from CURE: using clopidogrel on top of standard therapy. Cerebrovasc Dis 2002; 13(suppl 1):17–21.
- Ferguson JJ. Clopidogrel plus aspirin in patients with acute myocardial infarction treated with fibrinolytic therapy—CLARITY-TIMI 28. Future Cardiol 2005; 1:605–610.
- Sabatine MS, Cannon CP, Gibson CM, et al; Clopidogrel as Adjunctive Reperfusion Therapy (CLARITY)-Thrombolysis in Myocardial Infarction (TIMI) 28 Investigators. Effect of clopidogrel pretreatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA 2005; 294:1224–1232.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bonaca MP, Bhatt DL, Cohen M, et al; PEGASUS-TIMI 54 Steering Committee and Investigators. Long-term use of ticagrelor in patients with prior myocardial infarction. N Engl J Med 2015; 372:1791–1800.
- Colombo A, Chieffo A, Frasheri A, et al. Second-generation drug-eluting stent implantation followed by 6- versus 12-month dual antiplatelet therapy: the SECURITY randomized clinical trial. J Am Coll Cardiol 2014; 64:2086–2097.
- Gwon HC, Hahn JY, Park KW, et al. Six-month versus 12-month dual antiplatelet therapy after implantation of drug-eluting stents: the Efficacy of Xience/Promus versus Cypher to Reduce Late Loss After Stenting (EXCELLENT) randomized, multicenter study. Circulation 2012; 125:505–513.
- Feres F, Costa RA, Abizaid A, et al; OPTIMIZE Trial Investigators. Three vs twelve months of dual antiplatelet therapy after zotarolimus-eluting stents: the OPTIMIZE randomized trial. JAMA 2013; 310:2510–2522.
- Kim BK, Hong MK, Shin DH, et al; RESET Investigators. A new strategy for discontinuation of dual antiplatelet therapy: the RESET Trial (REal Safety and Efficacy of 3-month dual antiplatelet Therapy following endeavor zotarolimus-eluting stent implantation). J Am Coll Cardiol 2012; 60:1340–1348.
- El-Hayek G, Messerli F, Bangalore S, et al. Meta-analysis of randomized clinical trials comparing short-term versus long-term dual antiplatelet therapy following drug-eluting stents. Am J Cardiol 2014; 114:236–242.
- Valgimigli M, Campo G, Monti M, et al; Prolonging Dual Antiplatelet Treatment After Grading Stent-Induced Intimal Hyperplasia Study (PRODIGY) Investigators. Short- versus long-term duration of dual-antiplatelet therapy after coronary stenting: a randomized multicenter trial. Circulation 2012; 125:2015–2026.
- Collet JP, Silvain J, Barthelemy O, et al; ARCTIC investigators. Dual-antiplatelet treatment beyond 1 year after drug-eluting stent implantation (ARCTIC-Interruption): a randomised trial. Lancet 2014; 384:1577–1585.
- Lee CW, Ahn JM, Park DW, et al. Optimal duration of dual antiplatelet therapy after drug-eluting stent implantation: a randomized, controlled trial. Circulation 2014; 129:304–312.
- Kwok CS, Bulluck H, Ryding AD, Loke YK. Benefits and harms of extending the duration of dual antiplatelet therapy after percutaneous coronary intervention with drug-eluting stents: a meta-analysis. ScientificWorldJournal 2014; 2014:794078.
- Dewilde WJ, Oirbans T, Verheugt FW, et al; WOEST study investigators. Use of clopidogrel with or without aspirin in patients taking oral anticoagulant therapy and undergoing percutaneous coronary intervention: an open-label, randomised, controlled trial. Lancet 2013; 381:1107–1115.
KEY POINTS
- The outcomes of patients with acute coronary syndrome events have been improving as percutaneous coronary intervention and its accompanying medical therapy have evolved.
- Newer, more potent antiplatelet agents are preferred over clopidogrel when possible.
- Two earlier studies showed no advantage of extended dual antiplatelet therapy over the standard 12-month duration, but the recent Dual Antiplatelet Therapy trial did.
- The protection against ischemia afforded by dual antiplatelet therapy comes at the price of increased risk of bleeding.
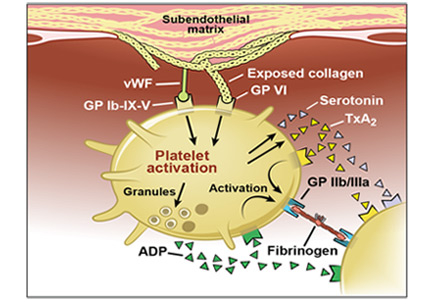
Evidence-Based Deprescribing: Reversing the Tide of Potentially Inappropriate Polypharmacy
From the Department of Internal Medicine and Clinical Epidemiology, Princess Alexandra Hospital, Ipswich Road, Woolloongabba, Queensland, Australia (Dr. Scott), School of Medicine, The University of Queensland, Herston Road, Brisbane, Australia (Dr. Scott), Centre of Research Excellence in Quality & Safety in Integrated Primary-Secondary Care, The University of Queensland, Herston Road, Brisbane, Australia (Ms. Anderson), and Charming Institute, Camp Hill, Brisbane, Queensland, Australia (Dr. Freeman).
Abstract
- Objective: To review the adverse drug events (ADEs) risk of polypharmacy; the process of deprescribing and evidence of efficacy in reducing inappropriate polypharmacy; the enablers and barriers to deprescribing; and patient and system of care level strategies that can be employed to enhance deprescribing.
- Methods: Literature review.
- Results: Inappropriate polypharmacy, especially in older people, imposes a significant burden of ADEs, ill health, disability, hospitalization and even death. The single most important predictor of inappropriate prescribing and risk of ADEs in older patients is the number of prescribed medicines. Deprescribing is the process of systematically reviewing, identifying, and discontinuing potentially inappropriate medicines (PIMs), aimed at minimizing polypharmacy and improving patient outcomes. Evidence of efficacy for deprescribing is emerging from randomized trials and observational studies, and deprescribing protocols have been developed and validated for clinical use. Barriers and enablers to deprescribing by individual prescribers center on 4 themes: (1) raising awareness of the prevalence and characteristics of PIMs; (2) overcoming clinical inertia whereby discontinuing medicines is seen as being a low value proposition compared to maintaining the status quo; (3) increasing skills and competence (self-efficacy) in deprescribing; and (4) countering external and logistical factors that impede the process.
- Conclusion: In optimizing the scale and effects of deprescribing in clinical practice, strategies that promote depresribing will need to be applied at both the level of individual patient–prescriber encounters and systems of care.
In developed countries in the modern era, about 30% of patients aged 65 years or older are prescribed 5 or more medicines [1]. Over the past decade, the prevalence of polypharmacy (use of > 5 prescription drugs) in the adult population of the United States has doubled from 8.2% in 1999–2000 to 15% in 2011–2012 [2]. While many patients may benefit from such polypharmacy [3] (defined here as 5 or more regularly prescribed medicines), it comes with increased risk of adverse drug events (ADEs) in older people [4] due to physiological changes of aging that alter pharmacokinetic and pharmacodynamic responses to medicines [5]. Approximately 1 in 5 medicines commonly used in older people may be inappropriate [6], rising to a third among those living in residential aged care facilities [7]. Among nursing home residents with advanced dementia, more than half receive at least 1 medicine with questionable benefit [8]. Approximately 50% of hospitalized nursing home or ambulatory care patients receive 1 or more unnecessary medicines [9]. Observational studies have documented ADEs in at least 15% of older patients, contributing to ill health [10], disability [11], hospitalization [12] and readmissions [13], increased length of stay, and, in some cases, death [14]. This high level of iatrogenic harm from potentially inappropriate medicines (PIMs) mandates a response from clinicians responsible for managing medicines.
In this narrative review, we aim to detail the ADE risk of polypharmacy, the process of deprescribing and evidence of its efficacy in reducing potentially inappropriate polypharmacy, the enablers and barriers to deprescribing, and patient and system of care level strategies that can be employed in enhancing deprescribing.
Polypharmacy As a Risk Factor for Medicine-Related Harm
The number of medicines a patient is taking is the single most important predictor of medicine-related harm [15]. One report estimated the risk of ADEs as a contributory cause of patients presenting acutely to hospital emergency departments to be 13% for 2 drugs, 38% for 4 drugs, and 82% for 7 drugs or more [16]. The more medicines an individual takes, the greater their risk of experiencing an adverse drug reaction, a drug-drug interaction, a drug-disease interaction, cascade prescribing (where more medicines are added to counteract side effects of existing medicines), nonadherence, and drug errors (wrong drug, wrong dose, missed doses, erroneous dosing frequency) [17–20]. Once the number of regular medicines rises above 5 (commonly regarded as the threshold for defining polypharmacy), observational data suggest that additional medicines independently increase the risk of frailty, falling, and hospital admission [21].
The benefits of many medicines in frail older people remain unquantified. As many as 50% of clinical trials have a specific upper age limit and approximately 80% of clinical trials exclude people with comorbidities [22,23]. Single-disease treatment guidelines based on such trials are often extrapolated to older people with multimorbidity despite an absence of evidence for benefit [24] and with little consideration of the potential burdens and harms of polypharmacy resulting from treating multiple diseases in the one patient [25]. By contrast, the risks from many medicines in older people are well known. Older people are at high risk of ADEs and toxicity due to reduced renal and liver function and age-related changes in physiological reserve, body composition, and cellular metabolism [26]. While the adverse effects of polypharmacy or of comorbidities targeted for treatment are difficult to separate, the burden of medicine-induced decline in function and quality of life is becoming better defined and appreciated [27].
Defining Evidence-Based Deprescribing
While many definitions have been proposed [28], we define evidence-based deprescribing as follows: the active process of systematically reviewing medicines being used by individual patients and, using best available evidence, identifying and discontinuing those associated with unfavorable risk–benefit trade-offs within the context of illness severity, advanced age, multi-morbidity, physical and emotional capacity, life expectancy, care goals, and personal preferences [29]. An enlarging body of research has demonstrated the feasibility, safety and patient benefit of deprescribing, as discussed further below. It employs evidence-based frameworks that assist the prescriber [30] and are patient-centered [31].
Importantly, deprescribing should be seen as part of the good prescribing continuum, which spans medicine initiation, titrating, changing, or adding medicines, and switching or ceasing medicines. Deprescribing is not about denying effective treatment to eligible patients. It is a positive, patient-centered intervention, with inherent uncertainties, and requires shared decision-making, informed patient consent and close monitoring of effects [32]. Deprescribing involves diagnosing a problem (use of a PIM), making a therapeutic decision (withdrawing it with close follow-up) and altering the natural history of the problem (reducing incidence of medicine-related adverse events).
Our definition of evidence-based deprescribing is a form of direct deprescribing applied at the level of the individual patient-prescriber/pharmacist encounter. Direct deprescribing uses explicit, systematic processes (such as using an algorithm or structured deprescribing framework or guide) applied by individual prescribers (or pharmacists) to the medicine regimens of individual patients (ie, at the patient level), and which targets either specific classes of medicines or all medicines that are potentially inappropriate. This is in contrast to indirect deprescribing, which uses more generic, programmatic strategies aimed at prescribers as a whole (ie, at the population or system level) and which seek to improve quality use of medicines in general, including both underuse and overuse of medicines. Indirect deprescribing entails a broader aim of medicines optimization in which deprescribing is a possible outcome but not necessarily the sole focus. Such strategies include pharmacist or physician medicine reviews, education programs for clinicians and/or patients, academic detailing, audit and feedback, geriatric assessment, multidisciplinary teams, prescribing restrictions, and government policies, all of which aim to reduce the overall burden of PIMs among broad groups of patients. While intuitively the 2 approaches in combination should exert synergistic effects superior to those of either by itself, this has not been studied.
Evidence For Deprescribing
Indirect Deprescribing
Overall, the research into indirect interventions has been highly heterogenous in terms of interventions and measures of medicine use. Research has often been of low to moderate quality, focused more on changes to prescribing patterns and less on clinical outcomes, been of short duration, and produced mixed results [33]. In a 2013 systematic review of 36 studies involving different interventions involving frail older patients in various settings, 22 of 26 quantitative studies reported statistically significant reductions in the proportions of medicines deemed unnecessary (defined using various criteria), ranging from 3 to 20 percentage points [34]. A more recent review of 20 trials of pharmacist-led reviews in both inpatient and outpatient settings reported a small reduction in the mean number of prescribed medicines (–0.48, 95% confidence interval [CI] –0.89 to –0.07) but no effects on mortality or readmissions, although unplanned hospitalizations were reduced in patients with heart failure [35]. A 2012 review of 10 controlled and 20 randomized studies revealed statistically significant reductions in the number of medicines in most of the controlled studies, although mixed results in the randomized studies [36]. Another 2012 review of 10 studies of different designs concluded that interventions were beneficial in reducing potentially inappropriate prescribing and medicine-related problems [37]. A 2013 review of 15 studies of academic detailing of family physicians showed a modest decline in the number of medications of certain classes such as benzodiazepines and nonsteroidal anti-inflammatory drugs [38]. Another 2013 review restricted to 8 randomized trials of various interventions involving nursing home patients suggested medicine-related problems were more frequently identified and resolved, together with improvement in medicine appropriateness [39]. In 2 randomized trials conducted in aged care facilities and centered on educational interventions, one aimed at prescribers [40] and the other at nursing staff [41],the number of potentially harmful medicines and days in hospital was significantly reduced [40,41], combined with slower declines in health-related quality of life [40]. In a randomized trial, patient education provided through community pharmacists led to a 77% reduction in benzodiazepine use among chronic users at 6 months with no withdrawal seizures or other ill effects [42].
Direct Deprescribing Targeting Specific Classes of Medicines
The evidence base for direct patient-level deprescribing is more rigorous as it pertains to specific classes of medicines. A 2008 systematic review of 31 trials (15 randomized, 16 observational) that withdrew a single class of medicine in older people demonstrated that, with appropriate patient selection and education coupled with careful withdrawal and close monitoring, antihypertensive agents, psychotropic medicines, and benzodiazepines could be discontinued without harm in 20% to 100% of patients, although psychotropics showed a high post-trial rate of recommencement [43]. Another review of 9 randomized trials demonstrated the safety of withdrawing antipsychotic agents that had been used continuously for behavioural and psychological symptoms in more than 80% of subjects with dementia [44]. In an observational study, cessation of inappropriate antihypertensives was associated with fewer cardiovascular events and deaths over a 5-year follow-up period [45]. A recent randomized trial of statin withdrawal in patients with advanced illness and of whom half had a prognosis of less than 12 months demonstrated improved quality of life and no increased risk of cardiovascular events over the following 60 days [46].
Direct Deprescribing Targeting All Medicines
The evidence base for direct patient-level deprescribing that assesses all medicines, not just specific medicine classes, features several high-quality observational studies and controlled trials, and subgroup findings from a recent comprehensive systematic review. In this review of 132 studies, which included 56 randomized controlled trials [47], mortality was shown in randomized trials to be decreased by 38% as a result of direct (ie, patient-level) deprescribing interventions. However, this effect was not seen in studies of indirect deprescribing comprising mainly generic educational interventions. While space prevents a detailed analysis of all relevant trials, some of the more commonly cited sentinel studies are mentioned here.
In a controlled trial involving 190 patients in aged care facilities, a structured approach to deprescribing (Good Palliative–Geriatric Practice algorithm) resulted in 63% of patients having, on average, 2.8 medicines per patient discontinued, and was associated with a halving in both annual mortality and referrals to acute care hospitals [48]. In another prospective uncontrolled study, the same approach applied to a cohort of 70 community-dwelling older patients resulted in an average of 4.4 medicines prescribed to 64 patients being recommended for discontinuation, of which 81% were successfully discontinued, with 88% of patients reporting global improvements in health [49]. In a prospective cohort study of 50 older hospitalized patients receiving a median of 10 regular medicines on admission, a formal deprescribing process led to the cessation of just over 1 in 3 medicines by discharge, representing 4 fewer medicines per patient [50]. During a median follow-up period of just over 2.5 months for 39 patients, less than 5% of ceased medicines were recommenced in 3 patients for relapsing symptoms, with no deaths or acute presentations to hospital attributable to cessation of medicines. A multidisciplinary hospital clinic for older patients over a 3-month period achieved cessation of 22% of medicines in 17 patients without ill effect [51].
Two randomized studies used the Screening Tool of Older People’s Prescriptions (STOPP) to reduce the use of PIMs in older hospital inpatients [52,53]. One reported significantly reduced PIMs use in the intervention group at discharge and 6 months post-discharge, no change in the rate of hospital readmission, and non-significant reductions in falls, all cause-mortality, and primary care visits during the 6-month follow-up period [52]. The second study reported reduced PIMs use in the intervention group of frail older patients on discharge, although the proportion of people prescribed at least 1 PIM was not altered [53].
Recently, a randomized trial of a deprescribing intervention applied to aged care residents resulted in successful discontinuation of 207 (59%) of 348 medicines targeted for deprescribing, and a mean reduction of 2 medicines per patient at 12 months compared to none in controls, with no differences in mortality or hospital admissions [54]. The evidence for direct deprescribing is limited by relatively few high-quality randomized trials, small patient samples, short duration of follow-up, selection of specific subsets of patients, and the absence of comprehensive re-prescribing data and clinical outcomes.
Methods Used for Direct Deprescribing
At the level of individual patient care, various instruments have been developed to assist the deprescribing process. Screening tools or criteria such as the Beers criteria and STOPP tool help identify medicines more likely than not to be inappropriate for a given set of circumstances and are widely used by research pharmacists. Deprescribing guidelines directed at particular medications (or drug classes) [55], or specific patient populations [56], can identify clinical scenarios where a particular drug is likely to be inappropriate, and how to safely wean or discontinue it.
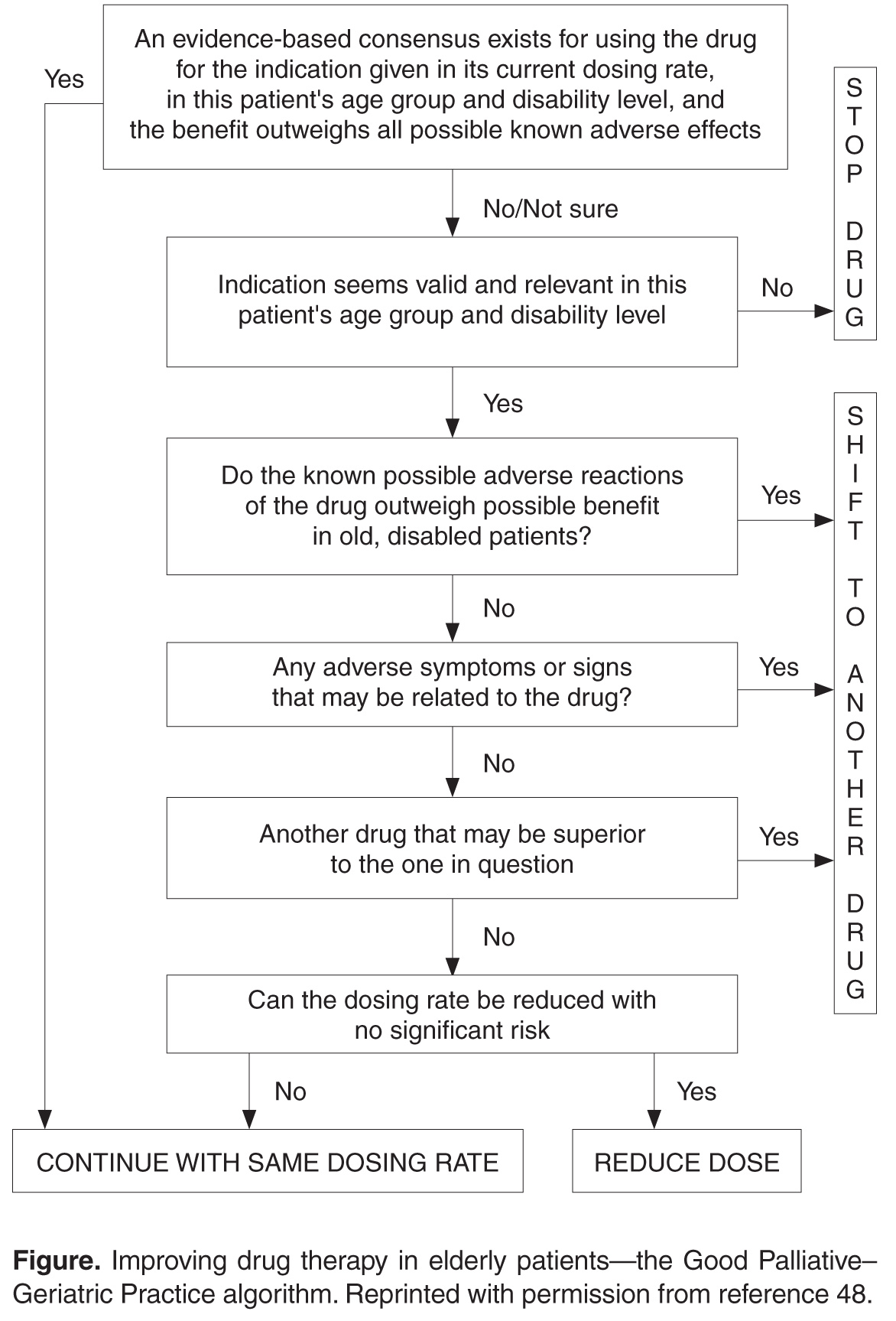
In applying a more nuanced, patient-centered approach to deprescribing, structured guides comprising algorithms, flowcharts, or tables describe sequential steps in deciding which medications used by an individual patient should be targeted for discontinuation after due attention to all relevant factors. Such guides prompt a more systematic appraisal of all medications being used. In a recent review of 7 structured guides that had undergone some form of efficacy testing [59], the strongest evidence of efficacy and clinician acceptability was seen for the Good Palliative–Geriatric Practice algorithm [48] (Figure) and the CEASE protocol [29,30,50,60] (Table). Both have been subject to a process of development and refinement over months to years involving multiple clinician prescribers and pharmacists. 
Clinical Circumstances Conducive to Deprescribing
Deprescribing should be especially considered in any older patient presenting with a new symptom or clinical syndrome suggestive of adverse medicine effects. The advent of advanced or end-stage disease, terminal illness, dementia, extreme frailty, or full dependence on others for all cares marks a stage of a person’s life when limited life expectancy and changed goals of care call for a re-appraisal of the benefits of current medicines. Lack of response in controlling symptoms despite optimal adherence and dosing or conversely the absence of symptoms for long periods of time should challenge the need for ongoing regular use of medicines. Similarly, the lack of verification, or indeed repudiation, of past diagnostic labels which gave rise to indications for medicines in the first place should prompt consideration of discontinuation. Patients receiving single medicines or combinations of medicines, both of which are high risk, should attract attention [63], as should use of preventive medicines for scenarios associated with no increased disease risk despite medicine cessation (eg, ceasing alendronate after 5 years of treatment results in no increase in osteoporotic fracture risk over the ensuing 5 years [64]; ceasing statins for primary prevention after a prolonged period results in no increase in cardiovascular events 8 years after discontinuation [65]). Evidence that has emerged that strongly contradicts previously held beliefs as to the indications for certain medicines (eg, aspirin as primary prevention of cardiovascular disease) should lead to a higher frequency of their discontinuation. Finally, medicines which impose demands on patients which they deem intolerable in terms of dietary and lifestyle restrictions, adverse side effects, medicine monitoring (such as warfarin), financial cost, or any other reason likely to result in nonadherence, should be considered candidates for deprescribing [25].
Barriers to Deprescribing
The most effective strategy to reducing potentially inappropriate polypharmacy is for doctors to prescribe and patients to consume fewer medicines. Unfortunately, both doctors and patients often lack confidence about when and how to cease medicines [66–69]. In a recent systematic review comprised mostly of studies involving general practitioners in primary care [66], 4 themes emerged. First, prescribers may be unaware of their own instances of inappropriate prescribing in older people until this is pointed out to them. Poor insight may be attributable in part to insufficient education in geriatric pharmacology. Second, clinical inertia manifesting as failure to act despite an awareness of PIMs may arise from deprescribing being viewed as a risky affair [70], with doctors fearful of provoking withdrawal syndromes or disease complications, and damaging their reputation and relationships with patients or colleagues in the process. Continuing inappropriate medicines is reinforced by prescriber beliefs that to do so is a safer or kinder course of action for the patient. Third, self-perceptions of being ill-equipped, in terms of the necessary knowledge and skills, to deprescribe appropriately (lack of self-efficacy) may be a barrier, even if one accepts the need for deprescribing. Information deficits around benefit-harm trade-offs of particular drugs and alternative treatments (both drug and non-drug), especially for older, frail, multi-morbid patients, contribute to the problem. Confidence to deprescribe is further undermined by the lack of clear documentation regarding reasons drugs were originally prescribed by other doctors, outcomes of past trials of discontinuation, and current patient care goals. Fourth, several external or logistical constraints may hamper deprescribing efforts such as perceived patient unwillingness to deprescribe certain medicines, lack of prescriber time, poor remuneration, and community and professional attitudes toward more rather than less use of medicines.
Deprescribing in hospital settings led by specialists appears to be no better than in general practice, although it has been less well studied. While an episode of acute inpatient care may afford an opportunity to review and reduce medicine lists, studies suggest the opposite occurs. In a New Zealand audit of 424 patients of mean age 80 years admitted acutely to a medical unit, chronically administered medications increased during hospital stay from a mean of 6.6 to 7.7 [71]. Similarly, in an Australian study investigating medication changes for 1220 patients of mean age 81 years admitted to general medical units of 11 acute care hospitals, the mean number of regularly administered medications rose from 7.1 on admission to 7.6 at discharge [72]. It is likely the same drivers behind failure to deprescribe in primary care also operate in secondary and tertiary care settings. Part of the problem is under-recognition of medicine-related geriatric syndromes on the part of hospital physicians and pharmacists [73].
Patients in both the community and residential aged care facilities frequently express a desire to have their medicines reduced in number, especially if advised by their treating clinician [74,75]. Having said this, many remain wary of discontinuing specific medicines [67], sharing the same fears of evoking withdrawal syndromes or disease relapse as do prescribers, and recounting the strong advice of past specialists to never withhold any medicines without first seeking their advice.
A challenge for all involved in deprescribing is gaining agreement on what are the most important factors that determine when, how, and in whom deprescribing should be conducted. Recent qualitative studies suggest that doctors, pharmacists, nursing staff, and patients and their families, while in broad agreement that deprescribing is worthwhile, often differ in their perspectives on what takes priority in selecting medicines for deprescribing in individual patients, and how it should be done and by whom [76,77].
Strategies That May Facilitate Deprescribing
While deprescribing presents some challenges, there are several strategies that can facilitate it at both the level of individual clinical encounters and at the level of whole populations and systems of care.
Individual Clinical Encounters
Within individual clinician–patient encounters, patients should be empowered to ask their doctors and pharmacists the following questions:
- What are my treatment options (including non-medicine options) for my condition?
- What are the possible benefits and harms of each medicine?
- What might be reasonable grounds for stopping a medicine?
In turn, doctors and pharmacists should ask in a nonjudgmental fashion, at every encounter, whether patients are experiencing any side effects, administration and monitoring problems, or other barriers to adherence associated with any of their medicines.
The issue of deprescribing should be framed as an attempt to alleviate symptoms (of drug toxicity), improve quality of life (from drug-induced disability), and lessen the risk of morbid events (especially ADEs) in the future. Compelling evidence that identifies circumstances in which medicines can be safely withdrawn while reducing the risk of ADEs needs to be emphasized. Specialists must play a sentinel leadership role in advising and authorizing other health professionals to deprescribe in situations where benefits of medications they have prescribed are no longer outweighed by the harms [60,78].
In language they can understand, patients should be informed of the benefit–harm trade-offs specific to them of continuing or discontinuing a particular medicine, as far as these can be specified. Patients often overestimate the benefits and underestimate the harms of treatments [79]. Providing such personalised information can substantially alter perceptions of risk and change attitudes towards discontinuation [80]. Eliciting patients’ beliefs about the necessity for each individual medicine and spending time, using an empathic manner, to dispel or qualify those at odds with evidence and clinical judgement renders deprescribing more acceptable to patients.
In estimating treatment benefit–harm trade-offs in individual patients, disease risk prediction tools (http://www.medal.org/), evidence tables [81,82], and decision aids are increasingly available. Prognostication tools (http://eprognosis.ucsf.edu) combined with trial-based time-to-event data can be used to determine if medicine-specific time until benefit exceeds remaining life span.
Deprescribing is best performed by reducing medicines one at a time over several encounters with the same overseeing generalist clinician with whom patients have established a trusting and collaborative relationship. This provides repeated opportunities to discuss and assuage any fears of discontinuing a medicine, and to adjust the deprescribing plan according to changes in clinical circumstances and revised treatment goals. Practice-based pharmacists can review patients’ medicine lists and apply screening criteria to identify medicines more likely to be unnecessary or harmful, which then helps initiate and guide deprescribing. Integrating a structured deprescribing protocol—and reminders to use it—into electronic health records, and providing decision support and data collection for future reference, reduce the cognitive burden on prescribers [83]. Practical guidance in how to safely wean and cease particular classes of medicines in older people can be accessed from various sources [84,85]. Seeking input from clinical pharmacologists, pharmacists, nurses, and other salient care providers on a case-by-case basis in the form of interactive case conferences provides support, seeks consensus, and shares the risk and responsibility for deprescribing recommendations [86].
System of Care
The success of deprescribing efforts in realizing better population health will be compromised unless all key stakeholders involved in quality use of medicines commit to operationalizing deprescribing strategies at the system of care level. Position statements on deprescribing in multi-morbid populations should be formulated and promulgated by all professional societies of prescribers (primary care, specialists, pharmacists, dentists, nurse practitioners). Professional development programs as well as undergraduate, graduate, and postgraduate courses in medicine, pharmacy, and nursing should include training in deprescribing as a core curricular element.
Researchers seeking funding and/or ethics approval for research projects involving medicines should be required to collect, analyze, and report data on the frequency of, and reasons for, withdrawal of drugs in trial subjects. This helps build the evidence base of medicine-related harm. In turn, government funders of research should require more researchers to design and conduct clinical trials that recruit multi-morbid patients, including specific subgroups (eg, patients with dementia), and aim to define medicine benefits and harms using patient risk stratification methods. Pharmaceutical companies should sponsor research on how to deprescribe their medicines within trials that also aim to assess efficacy and safety. Medicine regulatory authorities such as the Food and Drug Administration should mandate that this information be supplied at the time the company submits their application to have the medicine approved and listed for public subsidy. Trialists should adopt the word “deprescribing” in abstract titles for research on prescriber-initiated medicine discontinuation so that relevant articles can be more accurately indexed in, and retrieved from, bibliographic databases using recently formulated medical subject headings in Medline (“depresciptions”).
Editors of medical journals should promote a deprescribing agenda as a quality and safety issue for patient care, with the “Less is More” series in JAMA Internal Medicine and “Too much medicine” series in BMJ being good examples. Clinical guideline developers should formulate treatment recommendations specific to the needs of multi-morbid patients which acknowledge the limited evidence base for many medicines in such populations. These should take account of commonly encountered clinical scenarios where disease-specific medicines may engender greater risk of harm, and provide cautionary notes regarding initiation and discontinuation of medicines associated with high-risk.
Pharmacists need to instruct patients in how to identify medicine-induced harm and side effects, and how to collaborate with their prescribing clinicians in safely discontinuing high-risk medicines. Ideally, patients being admitted to residential aged care facilities should have their medicine lists reviewed by a pharmacist in flagging medicines eligible for deprescribing. Organizations and services responsible for providing quality use of medicines information (medicines handbooks, prescribing guidelines, drug safety bulletins) should describe when and how deprescribing should be performed in regards to specific medicines. This information should be cross-referenced to clinical guidelines and position statements dealing with the same medicine. Vendors of medicine prescribing software should be encouraged to incorporate flags and alerts which prompt prescribers to consider medicine cessation in high-risk patients.
Government and statutory bodies with responsibility for health care (health departments, quality and safety commissions, practice accreditation services, health care standard–setting bodies) should fund more research to develop and evaluate medicine safety standards aimed at reducing inappropriate use of medicines. Accreditation procedures for hospitals and primary care organizations should mandate the adoption of professional development and quality measurement systems that support and monitor patients receiving multiple medicines. Organizations responsible for conducting pharmacovigilance studies should issue medicine-specific deprescribing alerts whenever their data suggest higher than expected incidence of medicine-related adverse events in older populations receiving such medicines.
Conclusion
Inappropriate medicine use and polypharmacy is a growing issue among older and multi-morbid patients. The cumulative evidence of the safety and benefits of deprescribing argues for its adoption on the part of all prescribers, as well as its support by pharmacists and others responsible for optimizing use of medicines. Widespread implementation within routine care of an evidence-based approach to deprescribing in all patients receiving polypharmacy has its challenges, but also considerable potential to relieve unnecessary suffering and disability. More high quality research is needed in defining the circumstances under which deprescribing confers maximal benefit in terms of improved clinical outcomes.
Corresponding author: Ian A. Scott, Dept. of Internal Medicine and Clinical Epidemiology, Princess Alexandra Hospital, Brisbane, Australia 4102, [email protected].
Financial disclosures: None.
1. Qato DM, Alexander GC, Conti RM, et . Use of prescription and over-the-counter medications and dietary supplements among older adults in the United States. JAMA 2008;300:2867–78.
2. Kantor ED, Rehm CD, Haas JS, et al. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA 2015;314:1818–31.
3. Wise J. Polypharmacy: a necessary evil. BMJ 2013;347: f7033.
4. Gnjidic D, Hilmer SN, Blyth FM, et al. Polypharmacy cutoff and outcomes: five or more medicines were used to identify community-dwelling older men at risk of different adverse outcomes. J Clin Epidemiol 2012;65:989–95.
5. Atkin PA, Veitch PC, Veitch EM, Ogle SJ. The epidemiology of serious adverse drug reactions among the elderly. Medicines Aging 1999;14:141–52.
6. Roughead EE, Anderson B, Gilbert AL. Potentially inappropriate prescribing among Australian veterans and war widows/widowers. Intern Med J 2007;37:402–5.
7. Stafford AC, Alswayan MS, Tenni PC. Inappropriate prescribing in older residents of Australian care homes. Clin Pharmacol Therapeut 2011;36:33–44.
8. Tjia J, Briesacher BA, Peterson D, et al. Use of medications of questionable benefit in advanced dementia. JAMA Intern Med 2014;174:1763–71.
9. Tjia J, Velten SJ, Parsons C, et al. Studies to reduce unnecessary medication use in frail older adults: A systematic review. Drugs Aging 2013;30:285–307.
10. Anathhanam AS, Powis RA, Cracknell AL, Robson J. Impact of prescribed medicines on patient safety in older people. Ther Adv Drug Saf 2012;3:165–74.
11. Opondo D, Eslami S, Visscher S, et al. Inappropriateness of medication prescriptions to elderly patients in the primary care setting: a systematic review. PLoS One 2012;7(8):e43617.
12. Kalisch LM, Caughey GE, Barratt JD, et al. Prevalence of preventable medication-related hospitalizations in Australia: an opportunity to reduce harm. Int J Qual Health Care 2012;24:239–49.
13. Bero LA, Lipton HL, Bird JA. Characterisation of geriatric drug-related hospital readmissions. Med Care 1991;29:989–1003.
14. Jyrkkä J, Enlund H, Korhonen MJ, et al. Polypharmacy status as an indicator of mortality in an elderly population. Drugs Aging 2009;26:1039–48.
15. Steinman MA, Miao Y, Boscardin WJ, et al. Prescribing quality in older veterans: a multifocal approach. J Gen Intern Med 2014;29:1379–86.
16. Goldberg R, Mabee J, Chan L, Wong S. Drug-drug and drug-disease interactions in the ED: analysis of a high-risk population. Am J Emerg Med 1996;14:447–50.
17. Elliott RA, Booth JC. Problems with medicine use in older Australians: a review of recent literature. J Pharm Pract Res 2014;44:258–71.
18. Barat I, Andreasen F, Damsgaard EM. Drug therapy in the elderly: what doctors believe and patients actually do. Br J Clin Pharmacol 2001;51:615–22.
19. Chapman RH, Benner JS, Petrilla AA, et al. Predictors of adherence with antihypertensive and lipid-lowering therapy. Arch Intern Med 2005;165:1147–52.
20. Gnjidic D, Hilmer SN. Emergency hospitalizations for adverse drug events. N Engl J Med 2012;366:859.
21. Gnjidic D, Hilmer SN, Blyth FM, Naganathan V, Waite L, et al. Polypharmacy cutoff and outcomes: five or more medicines were used to identify community-dwelling older men at risk of different adverse outcomes. J Clin Epidemiol 2012;65:989–95.
22. Cherubini A, Oristrell J, Pla X, et al. The persistent exclusion of older patients from ongoing clinical trials regarding heart failure. Arch Intern Med 2011;171:550–6.
23. Bugeja G, Kumar A, Banerjee AK. Exclusion of elderly people from clinical research: a descriptive study of published reports. BMJ 1997;315:1059.
24. Mangin D, Heath I, Jamoulle M. Beyond diagnosis: rising to the multimorbidity challenge. BMJ 2012;344:e3526.
25. Boyd CM, Darer J, Boult C, et al. Clinical practice guidelines and quality of care for older patients with multiple comorbid diseases: implications for pay for performance. JAMA 2005;294:716–24.
26. McLean AJ, Le Couteur DG. Aging biology and geriatric clinical pharmacology. Pharmacol Rev 2004;56:163–84.
27. Hilmer SN, Mager DE, Simonsick EM, et al. Drug Burden Index score and functional decline in older people. Am J Med 2009;122:1142–9.
28. Reeve E, Gnjidic D, Long J, Hilmer S. A systematic review of the emerging definition of ‘deprescribing’ with network analysis: implications for future research and clinical practice. Br J Clin Pharmacol 2015;80:1254–68.
29. Scott IA, Hilmer SN, Reeve E, et al. Reducing inappropriate polypharmacy – the process of deprescribing. JAMA Intern Med 2015;175:827–34.
30. Scott IA, Gray LA, Martin JH, et al. Deciding when to stop: towards evidence-based deprescribing of drugs in older populations. Evidence-based Med 2013;18:121–4.
31. Reeve E, Shakib S, Hendrix I, et al. Review of deprescribing processes and development of an evidence-based, patient-centred deprescribing process. Br J Clin Pharmacol 2014;78:738–47.
32. Alldred D. Deprescribing: a brave new word? Int J Pharm Pract. 2014;22:2–3.
33. Kaur S, Mitchell G, Vitetta L, Roberts MS. Interventions that can reduce inappropriate prescribing in the elderly: a systematic review. Drugs Aging 2009;26:1013–28.
34. Tjia J, Velten SJ, Parsons C, et al. Studies to reduce unnecessary medication use in frail older adults: A systematic review. Drugs Aging 2013;30:285–307.
35. Thomas R, Huntley AL, Mann M, et al. Pharmacist-led interventions to reduce unplanned admissions for older people: a systematic review and meta-analysis of randomised controlled trials. Age Ageing 2014;43:174–87.
36. Gnjidic D, Le Couteur DG, Kouladjian L, Hilmer SN. Deprescribing trials: Methods to reduce polypharmacy and the impact on prescribing and clinical outcomes. Clin Geriatr Med 2012;28:237–53.
37. Patterson SM, Hughes C, Kerse N, et al. Interventions to improve use of polypharmacy for older people. Cochrane Database Syst Rev 2012;5:CD008165.
38. Chhina HK, Bhole VM, Goldsmith C, et al. Effectiveness of academic detailing to optimize medication prescribing behaviour of family physicians. J Pharm Pharm Sci 2013;16:511–29.
39. Alldred DP, Raynor DK, Hughes C, et al. Interventions to optimise prescribing for older people in care homes. Cochrane Database Syst Rev 2013;CD009095.
40. García-Gollarte F, Baleriola-Júlvez J, Ferrero-López I, et al. An educational intervention on drug use in nursing homes improves health outcomes and resource utilization and reduces inappropriate drug prescription. J Am Dir Assoc 2014;15:885–91.
41. Pitkälä KH, Juola A-L, Kautiainen H, Soini H, et al. Education to reduce potentially harmful medication use among residents of assisted living facilities: A randomized controlled trial. J Am Dir Assoc 2014;15:892–8.
42. Tannenbaum C, Martin P, Tamblyn R, et al. Reduction of inappropriate benzodiazepine prescriptions among older adults through direct patient education. The EMPOWER cluster randomized trial. JAMA Intern Med 2014;174:890–8.
43. Iyer S, Naganathan V, McLachlan AJ, Le Couteur DG. Medication withdrawal trials in people aged 65 years and older: a systematic review. Drugs Aging 2008;25:1021–31.
44. Declercq T, Petrovic M, Azermai M, et al. Withdrawal versus continuation of chronic antipsychotic medicines for behavioural and psychological symptoms in older people with dementia. Cochrane Database Syst Rev 2013;3:CD007726.
45. Ekbom T, Lindholm LH, Odén A, et al. A 5-year prospective, observational study of the withdrawal of antihypertensive treatment in elderly people. J Intern Med 1994;235:581–588.
46. Kutner JS, Blatchford PJ, Taylor DH Jr, et al. Safety and benefit of discontinuing statin therapy in the setting of advanced, life-limiting illness: a randomized clinical trial. JAMA Intern Med 2015;175:691–700.
47. Page AT, Clifford RM, Potter K, Schwartz D, Etherton-Beer CD. The feasibility and effect of deprescribing in older adults on mortality and health: a systematic review and meta-analysis. Br J Clin Pharmacol 2016 Apr 14. [Epub ahead of print]
48. Garfinkel D, Zur-Gil S, Ben-Israel J. The war against polypharmacy: a new cost-effective geriatric-palliative approach for improving drug therapy in disabled elderly people. Isr Med Assoc J 2007;9:430–4.
49. Garfinkel D, Mangin D. Feasibility study of a systematic approach for discontinuation of multiple medicines in older adults: addressing polypharmacy. Arch Intern Med 2010;170:1648–54.
50. McKean M, Pillans P, Scott IA. A medication review and deprescribing method for hospitalised older patients receiving multiple medications. Intern Med J 2016;46:35–42.
51. Mudge A, Radnedge K, Kasper K, et al. Effects of a pilot multidisciplinary clinic for frequent attending elderly patients on deprescribing. Aust Health Rev 2015; Jul 6. [Epub ahead of print]
52. Gallagher PF, O’Connor MN, O’Mahony D. Prevention of potentially inappropriate prescribing for elderly Patients: A randomized controlled trial using STOPP/START criteria. Clin Pharmacol Therap 2011;89:845–54.
53. Dalleur O, Boland B, Losseau C, et al. Reduction of potentially inappropriate medications using the STOPP criteria in frail older inpatients: a randomised controlled study. Drugs Aging 2014;31:291–8.
54. Potter K, Flicker L, Page A, Etherton-Beer C. Deprescribing in frail older people: A randomised controlled trial. PLoS One 2016;11(3):e0149984.
55. Conklin J, Farrell B, Ward N, et al. Developmental evaluation as a strategy to enhance the uptake and use of deprescribing guidelines: protocol for a multiple case study. Implement Sci 2015;10:91–101.
56. Lindsay J, Dooley M, Martin J, et al. The development and evaluation of an oncological palliative care deprescribing guideline: the ‘OncPal deprescribing guideline’ Support Care Cancer 2015;23:71–8.
57. Miller GC, Valenti L, Britt H, Bayram C. Drugs causing adverse events in patients aged 45 or older: a randomised survey of Australian general practice patients. BMJ Open 2013;3:e003701.
58. Budnitz DS, Lovegrove MC, Shebab N, Richards CL. Emergency hospitalisations for adverse drug events in older Americans. N Engl J Med 2011;365:2002–12.
59. Scott IA, Andersen K, Freeman C. Review of structured guides for deprescribing. Eur J Hosp Pharm 2016. In press.
60. Scott IA, Le Couteur D. Physicians need to take the lead in deprescribing. Intern Med J 2015;45:352–6.
61. Poudel A, Ballokova A, Hubbard RE, et al. An algorithm of medication review in residential aged care facilities: focus on minimizing use of high risk medications. Geriatr Gerontol Int Sep 3. [Epub ahead of print]
62. Scott IA, Martin JH, Gray LA, Mitchell CA. Effects of a drug minimisation guide on prescribing intentions in elderly persons with polypharmacy. Drugs Ageing 2012;29:659–67.
63. Bennett A, Gnjidic D, Gillett M, et al. Prevalence and impact of fall-risk-increasing drugs, polypharmacy, and drug-drug interactions in robust versus frail hospitalised falls patients: a prospective cohort study. Drugs Aging 2014;31:225–32.
64. Black DM, Schwartz AV, Ensrud KE, et al. FLEX Research Group. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomised trial. JAMA 2006;296:2927–38.
65. Sever PS, Chang CL, Gupta AK, et al. The Anglo-Scandinavian Cardiac Outcomes Trial: 11-year mortality follow-up of the lipid lowering arm in the UK. Eur Heart J 2011;32:2525–32.
66. Anderson K, Stowasser D, Freeman C, Scott I. Prescriber barriers and enablers to minimising potentially inappropriate medications in adults: a systematic review and thematic synthesis. BMJ Open 2014;4.
67. Reeve E, To J, Hendrix I, et al. Patient barriers to and enablers of deprescribing: a systematic review. Drugs Aging 2013;30:793–807.
68. Palagyi A, Keay L, Harper J, et al. Barricades and brickwalls—a qualitative study exploring perceptions of medication use and deprescribing in long-term care. BMC Geriatr 2016;16:15.
69. Garfinkel D, Ilhan B, Bahat G. Routine deprescribing of chronic medications to combat polypharmacy. Ther Adv Drug Saf 2015;6:212–33.
70. Reeve E, Shakib S, Hendrix I, et al. The benefits and harms of deprescribing. Med J Aust 2014;201:386–9.
71. Betteridge TM, Frampton CM, Jardine DL. Polypharmacy – we make it worse! A cross-sectional study from an acute admissions unit. Intern Med J 2012;42:208–11.
72. Hubbard RE, Peel NM, Scott IA, et al. Polypharmacy among inpatients aged 70 years or older in Australia. Med J Aust 2015;202:373–7.
73. Klopotowska JE, Wierenga PC, Smorenburg SM, et al. Recognition of adverse drug events in older hospitalized medical patients. Eur J Clin Pharmacol 2013;69:75–85.
74. Reeve E, Wiese MD, Hendrix I, et al. People’s attitudes, beliefs, and experiences regarding polypharmacy and willingness to deprescribe. J Am Geriatr Soc 2013;61:1508–14.
75. Kalogianis MJ, Wimmer BC, Turner JP, et al. Are residents of aged care facilities willing to have their medications deprescribed? Res Social Adm Pharm 2015. Published online 18 Dec 2015.
76. Turner JP, Edwards S, Stanners M, et al. What factors are important for deprescribing in Australian long-term care facilities? Perspectives of residents and health professionals. BMJ Open 2016;6:e009781.
77. Page AT, Etherton-Beer CD, Clifford RM, et al. Deprescribing in frail older people - Do doctors and pharmacists agree? Res Social Adm Pharm 2015;12:438–49.
78. Luymes CH, van der Kleij RM, Poortvliet RK, et al. Deprescribing potentially inappropriate preventive cardiovascular medication: Barriers and enablers for patients and general practitioners. Ann Pharmacother 2016 Mar 3. [Epub ahead of print]
79. Hoffmann TC, Del Mar C. Patients’ expectations of the benefits and harms of treatments, screening, and tests: a systematic review. JAMA Intern Med 2015;175:274–86.
80. Martin P, Tamblyn R, Ahmed S, Tannenbaum C. A drug education tool developed for older adults changes knowledge, beliefs and risk perceptions about inappropriate benzodiazepine prescriptions in the elderly. Patient Educ Couns 2013;92:81–7.
81. Hamilton H, Gallagher P, Ryan C, et al. Potentially inappropriate medicines defined by STOPP criteria and the risk of adverse drug events in older hospitalized patients. Arch Intern Med 2011;171:1013–7.
82. NHS Highland. Polypharmacy: guidance for prescribing in frail adults. Accessed at: www.nhshighland.scot.nhs.uk/publications/documents/guidelines/polypharmacy guidance for prescribing in frail adults.pdf.
83. Anderson K, Foster MM, Freeman CR, Scott IA. A multifaceted intervention to reduce inappropriate polypharmacy in primary care: research co-creation opportunities in a pilot study. Med J Aust 2016;204:S41–4.
84. A practical guide to stopping medicines in older people. Accessed at: www.bpac.org.nz/magazine/2010/april/stopGuide.asp.
85. www.cpsedu.com.au/posts/view/46/Deprescribing-Documents-now-Available-for-Download.
86. Bregnhøj L, Thirstrup S, Kristensen MB, et al. Combined intervention programme reduces inappropriate prescribing in elderly patients exposed to polypharmacy in primary care. Eur J Clin Pharmacol 2009;65:199–207.
From the Department of Internal Medicine and Clinical Epidemiology, Princess Alexandra Hospital, Ipswich Road, Woolloongabba, Queensland, Australia (Dr. Scott), School of Medicine, The University of Queensland, Herston Road, Brisbane, Australia (Dr. Scott), Centre of Research Excellence in Quality & Safety in Integrated Primary-Secondary Care, The University of Queensland, Herston Road, Brisbane, Australia (Ms. Anderson), and Charming Institute, Camp Hill, Brisbane, Queensland, Australia (Dr. Freeman).
Abstract
- Objective: To review the adverse drug events (ADEs) risk of polypharmacy; the process of deprescribing and evidence of efficacy in reducing inappropriate polypharmacy; the enablers and barriers to deprescribing; and patient and system of care level strategies that can be employed to enhance deprescribing.
- Methods: Literature review.
- Results: Inappropriate polypharmacy, especially in older people, imposes a significant burden of ADEs, ill health, disability, hospitalization and even death. The single most important predictor of inappropriate prescribing and risk of ADEs in older patients is the number of prescribed medicines. Deprescribing is the process of systematically reviewing, identifying, and discontinuing potentially inappropriate medicines (PIMs), aimed at minimizing polypharmacy and improving patient outcomes. Evidence of efficacy for deprescribing is emerging from randomized trials and observational studies, and deprescribing protocols have been developed and validated for clinical use. Barriers and enablers to deprescribing by individual prescribers center on 4 themes: (1) raising awareness of the prevalence and characteristics of PIMs; (2) overcoming clinical inertia whereby discontinuing medicines is seen as being a low value proposition compared to maintaining the status quo; (3) increasing skills and competence (self-efficacy) in deprescribing; and (4) countering external and logistical factors that impede the process.
- Conclusion: In optimizing the scale and effects of deprescribing in clinical practice, strategies that promote depresribing will need to be applied at both the level of individual patient–prescriber encounters and systems of care.
In developed countries in the modern era, about 30% of patients aged 65 years or older are prescribed 5 or more medicines [1]. Over the past decade, the prevalence of polypharmacy (use of > 5 prescription drugs) in the adult population of the United States has doubled from 8.2% in 1999–2000 to 15% in 2011–2012 [2]. While many patients may benefit from such polypharmacy [3] (defined here as 5 or more regularly prescribed medicines), it comes with increased risk of adverse drug events (ADEs) in older people [4] due to physiological changes of aging that alter pharmacokinetic and pharmacodynamic responses to medicines [5]. Approximately 1 in 5 medicines commonly used in older people may be inappropriate [6], rising to a third among those living in residential aged care facilities [7]. Among nursing home residents with advanced dementia, more than half receive at least 1 medicine with questionable benefit [8]. Approximately 50% of hospitalized nursing home or ambulatory care patients receive 1 or more unnecessary medicines [9]. Observational studies have documented ADEs in at least 15% of older patients, contributing to ill health [10], disability [11], hospitalization [12] and readmissions [13], increased length of stay, and, in some cases, death [14]. This high level of iatrogenic harm from potentially inappropriate medicines (PIMs) mandates a response from clinicians responsible for managing medicines.
In this narrative review, we aim to detail the ADE risk of polypharmacy, the process of deprescribing and evidence of its efficacy in reducing potentially inappropriate polypharmacy, the enablers and barriers to deprescribing, and patient and system of care level strategies that can be employed in enhancing deprescribing.
Polypharmacy As a Risk Factor for Medicine-Related Harm
The number of medicines a patient is taking is the single most important predictor of medicine-related harm [15]. One report estimated the risk of ADEs as a contributory cause of patients presenting acutely to hospital emergency departments to be 13% for 2 drugs, 38% for 4 drugs, and 82% for 7 drugs or more [16]. The more medicines an individual takes, the greater their risk of experiencing an adverse drug reaction, a drug-drug interaction, a drug-disease interaction, cascade prescribing (where more medicines are added to counteract side effects of existing medicines), nonadherence, and drug errors (wrong drug, wrong dose, missed doses, erroneous dosing frequency) [17–20]. Once the number of regular medicines rises above 5 (commonly regarded as the threshold for defining polypharmacy), observational data suggest that additional medicines independently increase the risk of frailty, falling, and hospital admission [21].
The benefits of many medicines in frail older people remain unquantified. As many as 50% of clinical trials have a specific upper age limit and approximately 80% of clinical trials exclude people with comorbidities [22,23]. Single-disease treatment guidelines based on such trials are often extrapolated to older people with multimorbidity despite an absence of evidence for benefit [24] and with little consideration of the potential burdens and harms of polypharmacy resulting from treating multiple diseases in the one patient [25]. By contrast, the risks from many medicines in older people are well known. Older people are at high risk of ADEs and toxicity due to reduced renal and liver function and age-related changes in physiological reserve, body composition, and cellular metabolism [26]. While the adverse effects of polypharmacy or of comorbidities targeted for treatment are difficult to separate, the burden of medicine-induced decline in function and quality of life is becoming better defined and appreciated [27].
Defining Evidence-Based Deprescribing
While many definitions have been proposed [28], we define evidence-based deprescribing as follows: the active process of systematically reviewing medicines being used by individual patients and, using best available evidence, identifying and discontinuing those associated with unfavorable risk–benefit trade-offs within the context of illness severity, advanced age, multi-morbidity, physical and emotional capacity, life expectancy, care goals, and personal preferences [29]. An enlarging body of research has demonstrated the feasibility, safety and patient benefit of deprescribing, as discussed further below. It employs evidence-based frameworks that assist the prescriber [30] and are patient-centered [31].
Importantly, deprescribing should be seen as part of the good prescribing continuum, which spans medicine initiation, titrating, changing, or adding medicines, and switching or ceasing medicines. Deprescribing is not about denying effective treatment to eligible patients. It is a positive, patient-centered intervention, with inherent uncertainties, and requires shared decision-making, informed patient consent and close monitoring of effects [32]. Deprescribing involves diagnosing a problem (use of a PIM), making a therapeutic decision (withdrawing it with close follow-up) and altering the natural history of the problem (reducing incidence of medicine-related adverse events).
Our definition of evidence-based deprescribing is a form of direct deprescribing applied at the level of the individual patient-prescriber/pharmacist encounter. Direct deprescribing uses explicit, systematic processes (such as using an algorithm or structured deprescribing framework or guide) applied by individual prescribers (or pharmacists) to the medicine regimens of individual patients (ie, at the patient level), and which targets either specific classes of medicines or all medicines that are potentially inappropriate. This is in contrast to indirect deprescribing, which uses more generic, programmatic strategies aimed at prescribers as a whole (ie, at the population or system level) and which seek to improve quality use of medicines in general, including both underuse and overuse of medicines. Indirect deprescribing entails a broader aim of medicines optimization in which deprescribing is a possible outcome but not necessarily the sole focus. Such strategies include pharmacist or physician medicine reviews, education programs for clinicians and/or patients, academic detailing, audit and feedback, geriatric assessment, multidisciplinary teams, prescribing restrictions, and government policies, all of which aim to reduce the overall burden of PIMs among broad groups of patients. While intuitively the 2 approaches in combination should exert synergistic effects superior to those of either by itself, this has not been studied.
Evidence For Deprescribing
Indirect Deprescribing
Overall, the research into indirect interventions has been highly heterogenous in terms of interventions and measures of medicine use. Research has often been of low to moderate quality, focused more on changes to prescribing patterns and less on clinical outcomes, been of short duration, and produced mixed results [33]. In a 2013 systematic review of 36 studies involving different interventions involving frail older patients in various settings, 22 of 26 quantitative studies reported statistically significant reductions in the proportions of medicines deemed unnecessary (defined using various criteria), ranging from 3 to 20 percentage points [34]. A more recent review of 20 trials of pharmacist-led reviews in both inpatient and outpatient settings reported a small reduction in the mean number of prescribed medicines (–0.48, 95% confidence interval [CI] –0.89 to –0.07) but no effects on mortality or readmissions, although unplanned hospitalizations were reduced in patients with heart failure [35]. A 2012 review of 10 controlled and 20 randomized studies revealed statistically significant reductions in the number of medicines in most of the controlled studies, although mixed results in the randomized studies [36]. Another 2012 review of 10 studies of different designs concluded that interventions were beneficial in reducing potentially inappropriate prescribing and medicine-related problems [37]. A 2013 review of 15 studies of academic detailing of family physicians showed a modest decline in the number of medications of certain classes such as benzodiazepines and nonsteroidal anti-inflammatory drugs [38]. Another 2013 review restricted to 8 randomized trials of various interventions involving nursing home patients suggested medicine-related problems were more frequently identified and resolved, together with improvement in medicine appropriateness [39]. In 2 randomized trials conducted in aged care facilities and centered on educational interventions, one aimed at prescribers [40] and the other at nursing staff [41],the number of potentially harmful medicines and days in hospital was significantly reduced [40,41], combined with slower declines in health-related quality of life [40]. In a randomized trial, patient education provided through community pharmacists led to a 77% reduction in benzodiazepine use among chronic users at 6 months with no withdrawal seizures or other ill effects [42].
Direct Deprescribing Targeting Specific Classes of Medicines
The evidence base for direct patient-level deprescribing is more rigorous as it pertains to specific classes of medicines. A 2008 systematic review of 31 trials (15 randomized, 16 observational) that withdrew a single class of medicine in older people demonstrated that, with appropriate patient selection and education coupled with careful withdrawal and close monitoring, antihypertensive agents, psychotropic medicines, and benzodiazepines could be discontinued without harm in 20% to 100% of patients, although psychotropics showed a high post-trial rate of recommencement [43]. Another review of 9 randomized trials demonstrated the safety of withdrawing antipsychotic agents that had been used continuously for behavioural and psychological symptoms in more than 80% of subjects with dementia [44]. In an observational study, cessation of inappropriate antihypertensives was associated with fewer cardiovascular events and deaths over a 5-year follow-up period [45]. A recent randomized trial of statin withdrawal in patients with advanced illness and of whom half had a prognosis of less than 12 months demonstrated improved quality of life and no increased risk of cardiovascular events over the following 60 days [46].
Direct Deprescribing Targeting All Medicines
The evidence base for direct patient-level deprescribing that assesses all medicines, not just specific medicine classes, features several high-quality observational studies and controlled trials, and subgroup findings from a recent comprehensive systematic review. In this review of 132 studies, which included 56 randomized controlled trials [47], mortality was shown in randomized trials to be decreased by 38% as a result of direct (ie, patient-level) deprescribing interventions. However, this effect was not seen in studies of indirect deprescribing comprising mainly generic educational interventions. While space prevents a detailed analysis of all relevant trials, some of the more commonly cited sentinel studies are mentioned here.
In a controlled trial involving 190 patients in aged care facilities, a structured approach to deprescribing (Good Palliative–Geriatric Practice algorithm) resulted in 63% of patients having, on average, 2.8 medicines per patient discontinued, and was associated with a halving in both annual mortality and referrals to acute care hospitals [48]. In another prospective uncontrolled study, the same approach applied to a cohort of 70 community-dwelling older patients resulted in an average of 4.4 medicines prescribed to 64 patients being recommended for discontinuation, of which 81% were successfully discontinued, with 88% of patients reporting global improvements in health [49]. In a prospective cohort study of 50 older hospitalized patients receiving a median of 10 regular medicines on admission, a formal deprescribing process led to the cessation of just over 1 in 3 medicines by discharge, representing 4 fewer medicines per patient [50]. During a median follow-up period of just over 2.5 months for 39 patients, less than 5% of ceased medicines were recommenced in 3 patients for relapsing symptoms, with no deaths or acute presentations to hospital attributable to cessation of medicines. A multidisciplinary hospital clinic for older patients over a 3-month period achieved cessation of 22% of medicines in 17 patients without ill effect [51].
Two randomized studies used the Screening Tool of Older People’s Prescriptions (STOPP) to reduce the use of PIMs in older hospital inpatients [52,53]. One reported significantly reduced PIMs use in the intervention group at discharge and 6 months post-discharge, no change in the rate of hospital readmission, and non-significant reductions in falls, all cause-mortality, and primary care visits during the 6-month follow-up period [52]. The second study reported reduced PIMs use in the intervention group of frail older patients on discharge, although the proportion of people prescribed at least 1 PIM was not altered [53].
Recently, a randomized trial of a deprescribing intervention applied to aged care residents resulted in successful discontinuation of 207 (59%) of 348 medicines targeted for deprescribing, and a mean reduction of 2 medicines per patient at 12 months compared to none in controls, with no differences in mortality or hospital admissions [54]. The evidence for direct deprescribing is limited by relatively few high-quality randomized trials, small patient samples, short duration of follow-up, selection of specific subsets of patients, and the absence of comprehensive re-prescribing data and clinical outcomes.
Methods Used for Direct Deprescribing
At the level of individual patient care, various instruments have been developed to assist the deprescribing process. Screening tools or criteria such as the Beers criteria and STOPP tool help identify medicines more likely than not to be inappropriate for a given set of circumstances and are widely used by research pharmacists. Deprescribing guidelines directed at particular medications (or drug classes) [55], or specific patient populations [56], can identify clinical scenarios where a particular drug is likely to be inappropriate, and how to safely wean or discontinue it.
In applying a more nuanced, patient-centered approach to deprescribing, structured guides comprising algorithms, flowcharts, or tables describe sequential steps in deciding which medications used by an individual patient should be targeted for discontinuation after due attention to all relevant factors. Such guides prompt a more systematic appraisal of all medications being used. In a recent review of 7 structured guides that had undergone some form of efficacy testing [59], the strongest evidence of efficacy and clinician acceptability was seen for the Good Palliative–Geriatric Practice algorithm [48] (Figure) and the CEASE protocol [29,30,50,60] (Table). Both have been subject to a process of development and refinement over months to years involving multiple clinician prescribers and pharmacists.
Clinical Circumstances Conducive to Deprescribing
Deprescribing should be especially considered in any older patient presenting with a new symptom or clinical syndrome suggestive of adverse medicine effects. The advent of advanced or end-stage disease, terminal illness, dementia, extreme frailty, or full dependence on others for all cares marks a stage of a person’s life when limited life expectancy and changed goals of care call for a re-appraisal of the benefits of current medicines. Lack of response in controlling symptoms despite optimal adherence and dosing or conversely the absence of symptoms for long periods of time should challenge the need for ongoing regular use of medicines. Similarly, the lack of verification, or indeed repudiation, of past diagnostic labels which gave rise to indications for medicines in the first place should prompt consideration of discontinuation. Patients receiving single medicines or combinations of medicines, both of which are high risk, should attract attention [63], as should use of preventive medicines for scenarios associated with no increased disease risk despite medicine cessation (eg, ceasing alendronate after 5 years of treatment results in no increase in osteoporotic fracture risk over the ensuing 5 years [64]; ceasing statins for primary prevention after a prolonged period results in no increase in cardiovascular events 8 years after discontinuation [65]). Evidence that has emerged that strongly contradicts previously held beliefs as to the indications for certain medicines (eg, aspirin as primary prevention of cardiovascular disease) should lead to a higher frequency of their discontinuation. Finally, medicines which impose demands on patients which they deem intolerable in terms of dietary and lifestyle restrictions, adverse side effects, medicine monitoring (such as warfarin), financial cost, or any other reason likely to result in nonadherence, should be considered candidates for deprescribing [25].
Barriers to Deprescribing
The most effective strategy to reducing potentially inappropriate polypharmacy is for doctors to prescribe and patients to consume fewer medicines. Unfortunately, both doctors and patients often lack confidence about when and how to cease medicines [66–69]. In a recent systematic review comprised mostly of studies involving general practitioners in primary care [66], 4 themes emerged. First, prescribers may be unaware of their own instances of inappropriate prescribing in older people until this is pointed out to them. Poor insight may be attributable in part to insufficient education in geriatric pharmacology. Second, clinical inertia manifesting as failure to act despite an awareness of PIMs may arise from deprescribing being viewed as a risky affair [70], with doctors fearful of provoking withdrawal syndromes or disease complications, and damaging their reputation and relationships with patients or colleagues in the process. Continuing inappropriate medicines is reinforced by prescriber beliefs that to do so is a safer or kinder course of action for the patient. Third, self-perceptions of being ill-equipped, in terms of the necessary knowledge and skills, to deprescribe appropriately (lack of self-efficacy) may be a barrier, even if one accepts the need for deprescribing. Information deficits around benefit-harm trade-offs of particular drugs and alternative treatments (both drug and non-drug), especially for older, frail, multi-morbid patients, contribute to the problem. Confidence to deprescribe is further undermined by the lack of clear documentation regarding reasons drugs were originally prescribed by other doctors, outcomes of past trials of discontinuation, and current patient care goals. Fourth, several external or logistical constraints may hamper deprescribing efforts such as perceived patient unwillingness to deprescribe certain medicines, lack of prescriber time, poor remuneration, and community and professional attitudes toward more rather than less use of medicines.
Deprescribing in hospital settings led by specialists appears to be no better than in general practice, although it has been less well studied. While an episode of acute inpatient care may afford an opportunity to review and reduce medicine lists, studies suggest the opposite occurs. In a New Zealand audit of 424 patients of mean age 80 years admitted acutely to a medical unit, chronically administered medications increased during hospital stay from a mean of 6.6 to 7.7 [71]. Similarly, in an Australian study investigating medication changes for 1220 patients of mean age 81 years admitted to general medical units of 11 acute care hospitals, the mean number of regularly administered medications rose from 7.1 on admission to 7.6 at discharge [72]. It is likely the same drivers behind failure to deprescribe in primary care also operate in secondary and tertiary care settings. Part of the problem is under-recognition of medicine-related geriatric syndromes on the part of hospital physicians and pharmacists [73].
Patients in both the community and residential aged care facilities frequently express a desire to have their medicines reduced in number, especially if advised by their treating clinician [74,75]. Having said this, many remain wary of discontinuing specific medicines [67], sharing the same fears of evoking withdrawal syndromes or disease relapse as do prescribers, and recounting the strong advice of past specialists to never withhold any medicines without first seeking their advice.
A challenge for all involved in deprescribing is gaining agreement on what are the most important factors that determine when, how, and in whom deprescribing should be conducted. Recent qualitative studies suggest that doctors, pharmacists, nursing staff, and patients and their families, while in broad agreement that deprescribing is worthwhile, often differ in their perspectives on what takes priority in selecting medicines for deprescribing in individual patients, and how it should be done and by whom [76,77].
Strategies That May Facilitate Deprescribing
While deprescribing presents some challenges, there are several strategies that can facilitate it at both the level of individual clinical encounters and at the level of whole populations and systems of care.
Individual Clinical Encounters
Within individual clinician–patient encounters, patients should be empowered to ask their doctors and pharmacists the following questions:
- What are my treatment options (including non-medicine options) for my condition?
- What are the possible benefits and harms of each medicine?
- What might be reasonable grounds for stopping a medicine?
In turn, doctors and pharmacists should ask in a nonjudgmental fashion, at every encounter, whether patients are experiencing any side effects, administration and monitoring problems, or other barriers to adherence associated with any of their medicines.
The issue of deprescribing should be framed as an attempt to alleviate symptoms (of drug toxicity), improve quality of life (from drug-induced disability), and lessen the risk of morbid events (especially ADEs) in the future. Compelling evidence that identifies circumstances in which medicines can be safely withdrawn while reducing the risk of ADEs needs to be emphasized. Specialists must play a sentinel leadership role in advising and authorizing other health professionals to deprescribe in situations where benefits of medications they have prescribed are no longer outweighed by the harms [60,78].
In language they can understand, patients should be informed of the benefit–harm trade-offs specific to them of continuing or discontinuing a particular medicine, as far as these can be specified. Patients often overestimate the benefits and underestimate the harms of treatments [79]. Providing such personalised information can substantially alter perceptions of risk and change attitudes towards discontinuation [80]. Eliciting patients’ beliefs about the necessity for each individual medicine and spending time, using an empathic manner, to dispel or qualify those at odds with evidence and clinical judgement renders deprescribing more acceptable to patients.
In estimating treatment benefit–harm trade-offs in individual patients, disease risk prediction tools (http://www.medal.org/), evidence tables [81,82], and decision aids are increasingly available. Prognostication tools (http://eprognosis.ucsf.edu) combined with trial-based time-to-event data can be used to determine if medicine-specific time until benefit exceeds remaining life span.
Deprescribing is best performed by reducing medicines one at a time over several encounters with the same overseeing generalist clinician with whom patients have established a trusting and collaborative relationship. This provides repeated opportunities to discuss and assuage any fears of discontinuing a medicine, and to adjust the deprescribing plan according to changes in clinical circumstances and revised treatment goals. Practice-based pharmacists can review patients’ medicine lists and apply screening criteria to identify medicines more likely to be unnecessary or harmful, which then helps initiate and guide deprescribing. Integrating a structured deprescribing protocol—and reminders to use it—into electronic health records, and providing decision support and data collection for future reference, reduce the cognitive burden on prescribers [83]. Practical guidance in how to safely wean and cease particular classes of medicines in older people can be accessed from various sources [84,85]. Seeking input from clinical pharmacologists, pharmacists, nurses, and other salient care providers on a case-by-case basis in the form of interactive case conferences provides support, seeks consensus, and shares the risk and responsibility for deprescribing recommendations [86].
System of Care
The success of deprescribing efforts in realizing better population health will be compromised unless all key stakeholders involved in quality use of medicines commit to operationalizing deprescribing strategies at the system of care level. Position statements on deprescribing in multi-morbid populations should be formulated and promulgated by all professional societies of prescribers (primary care, specialists, pharmacists, dentists, nurse practitioners). Professional development programs as well as undergraduate, graduate, and postgraduate courses in medicine, pharmacy, and nursing should include training in deprescribing as a core curricular element.
Researchers seeking funding and/or ethics approval for research projects involving medicines should be required to collect, analyze, and report data on the frequency of, and reasons for, withdrawal of drugs in trial subjects. This helps build the evidence base of medicine-related harm. In turn, government funders of research should require more researchers to design and conduct clinical trials that recruit multi-morbid patients, including specific subgroups (eg, patients with dementia), and aim to define medicine benefits and harms using patient risk stratification methods. Pharmaceutical companies should sponsor research on how to deprescribe their medicines within trials that also aim to assess efficacy and safety. Medicine regulatory authorities such as the Food and Drug Administration should mandate that this information be supplied at the time the company submits their application to have the medicine approved and listed for public subsidy. Trialists should adopt the word “deprescribing” in abstract titles for research on prescriber-initiated medicine discontinuation so that relevant articles can be more accurately indexed in, and retrieved from, bibliographic databases using recently formulated medical subject headings in Medline (“depresciptions”).
Editors of medical journals should promote a deprescribing agenda as a quality and safety issue for patient care, with the “Less is More” series in JAMA Internal Medicine and “Too much medicine” series in BMJ being good examples. Clinical guideline developers should formulate treatment recommendations specific to the needs of multi-morbid patients which acknowledge the limited evidence base for many medicines in such populations. These should take account of commonly encountered clinical scenarios where disease-specific medicines may engender greater risk of harm, and provide cautionary notes regarding initiation and discontinuation of medicines associated with high-risk.
Pharmacists need to instruct patients in how to identify medicine-induced harm and side effects, and how to collaborate with their prescribing clinicians in safely discontinuing high-risk medicines. Ideally, patients being admitted to residential aged care facilities should have their medicine lists reviewed by a pharmacist in flagging medicines eligible for deprescribing. Organizations and services responsible for providing quality use of medicines information (medicines handbooks, prescribing guidelines, drug safety bulletins) should describe when and how deprescribing should be performed in regards to specific medicines. This information should be cross-referenced to clinical guidelines and position statements dealing with the same medicine. Vendors of medicine prescribing software should be encouraged to incorporate flags and alerts which prompt prescribers to consider medicine cessation in high-risk patients.
Government and statutory bodies with responsibility for health care (health departments, quality and safety commissions, practice accreditation services, health care standard–setting bodies) should fund more research to develop and evaluate medicine safety standards aimed at reducing inappropriate use of medicines. Accreditation procedures for hospitals and primary care organizations should mandate the adoption of professional development and quality measurement systems that support and monitor patients receiving multiple medicines. Organizations responsible for conducting pharmacovigilance studies should issue medicine-specific deprescribing alerts whenever their data suggest higher than expected incidence of medicine-related adverse events in older populations receiving such medicines.
Conclusion
Inappropriate medicine use and polypharmacy is a growing issue among older and multi-morbid patients. The cumulative evidence of the safety and benefits of deprescribing argues for its adoption on the part of all prescribers, as well as its support by pharmacists and others responsible for optimizing use of medicines. Widespread implementation within routine care of an evidence-based approach to deprescribing in all patients receiving polypharmacy has its challenges, but also considerable potential to relieve unnecessary suffering and disability. More high quality research is needed in defining the circumstances under which deprescribing confers maximal benefit in terms of improved clinical outcomes.
Corresponding author: Ian A. Scott, Dept. of Internal Medicine and Clinical Epidemiology, Princess Alexandra Hospital, Brisbane, Australia 4102, [email protected].
Financial disclosures: None.
From the Department of Internal Medicine and Clinical Epidemiology, Princess Alexandra Hospital, Ipswich Road, Woolloongabba, Queensland, Australia (Dr. Scott), School of Medicine, The University of Queensland, Herston Road, Brisbane, Australia (Dr. Scott), Centre of Research Excellence in Quality & Safety in Integrated Primary-Secondary Care, The University of Queensland, Herston Road, Brisbane, Australia (Ms. Anderson), and Charming Institute, Camp Hill, Brisbane, Queensland, Australia (Dr. Freeman).
Abstract
- Objective: To review the adverse drug events (ADEs) risk of polypharmacy; the process of deprescribing and evidence of efficacy in reducing inappropriate polypharmacy; the enablers and barriers to deprescribing; and patient and system of care level strategies that can be employed to enhance deprescribing.
- Methods: Literature review.
- Results: Inappropriate polypharmacy, especially in older people, imposes a significant burden of ADEs, ill health, disability, hospitalization and even death. The single most important predictor of inappropriate prescribing and risk of ADEs in older patients is the number of prescribed medicines. Deprescribing is the process of systematically reviewing, identifying, and discontinuing potentially inappropriate medicines (PIMs), aimed at minimizing polypharmacy and improving patient outcomes. Evidence of efficacy for deprescribing is emerging from randomized trials and observational studies, and deprescribing protocols have been developed and validated for clinical use. Barriers and enablers to deprescribing by individual prescribers center on 4 themes: (1) raising awareness of the prevalence and characteristics of PIMs; (2) overcoming clinical inertia whereby discontinuing medicines is seen as being a low value proposition compared to maintaining the status quo; (3) increasing skills and competence (self-efficacy) in deprescribing; and (4) countering external and logistical factors that impede the process.
- Conclusion: In optimizing the scale and effects of deprescribing in clinical practice, strategies that promote depresribing will need to be applied at both the level of individual patient–prescriber encounters and systems of care.
In developed countries in the modern era, about 30% of patients aged 65 years or older are prescribed 5 or more medicines [1]. Over the past decade, the prevalence of polypharmacy (use of > 5 prescription drugs) in the adult population of the United States has doubled from 8.2% in 1999–2000 to 15% in 2011–2012 [2]. While many patients may benefit from such polypharmacy [3] (defined here as 5 or more regularly prescribed medicines), it comes with increased risk of adverse drug events (ADEs) in older people [4] due to physiological changes of aging that alter pharmacokinetic and pharmacodynamic responses to medicines [5]. Approximately 1 in 5 medicines commonly used in older people may be inappropriate [6], rising to a third among those living in residential aged care facilities [7]. Among nursing home residents with advanced dementia, more than half receive at least 1 medicine with questionable benefit [8]. Approximately 50% of hospitalized nursing home or ambulatory care patients receive 1 or more unnecessary medicines [9]. Observational studies have documented ADEs in at least 15% of older patients, contributing to ill health [10], disability [11], hospitalization [12] and readmissions [13], increased length of stay, and, in some cases, death [14]. This high level of iatrogenic harm from potentially inappropriate medicines (PIMs) mandates a response from clinicians responsible for managing medicines.
In this narrative review, we aim to detail the ADE risk of polypharmacy, the process of deprescribing and evidence of its efficacy in reducing potentially inappropriate polypharmacy, the enablers and barriers to deprescribing, and patient and system of care level strategies that can be employed in enhancing deprescribing.
Polypharmacy As a Risk Factor for Medicine-Related Harm
The number of medicines a patient is taking is the single most important predictor of medicine-related harm [15]. One report estimated the risk of ADEs as a contributory cause of patients presenting acutely to hospital emergency departments to be 13% for 2 drugs, 38% for 4 drugs, and 82% for 7 drugs or more [16]. The more medicines an individual takes, the greater their risk of experiencing an adverse drug reaction, a drug-drug interaction, a drug-disease interaction, cascade prescribing (where more medicines are added to counteract side effects of existing medicines), nonadherence, and drug errors (wrong drug, wrong dose, missed doses, erroneous dosing frequency) [17–20]. Once the number of regular medicines rises above 5 (commonly regarded as the threshold for defining polypharmacy), observational data suggest that additional medicines independently increase the risk of frailty, falling, and hospital admission [21].
The benefits of many medicines in frail older people remain unquantified. As many as 50% of clinical trials have a specific upper age limit and approximately 80% of clinical trials exclude people with comorbidities [22,23]. Single-disease treatment guidelines based on such trials are often extrapolated to older people with multimorbidity despite an absence of evidence for benefit [24] and with little consideration of the potential burdens and harms of polypharmacy resulting from treating multiple diseases in the one patient [25]. By contrast, the risks from many medicines in older people are well known. Older people are at high risk of ADEs and toxicity due to reduced renal and liver function and age-related changes in physiological reserve, body composition, and cellular metabolism [26]. While the adverse effects of polypharmacy or of comorbidities targeted for treatment are difficult to separate, the burden of medicine-induced decline in function and quality of life is becoming better defined and appreciated [27].
Defining Evidence-Based Deprescribing
While many definitions have been proposed [28], we define evidence-based deprescribing as follows: the active process of systematically reviewing medicines being used by individual patients and, using best available evidence, identifying and discontinuing those associated with unfavorable risk–benefit trade-offs within the context of illness severity, advanced age, multi-morbidity, physical and emotional capacity, life expectancy, care goals, and personal preferences [29]. An enlarging body of research has demonstrated the feasibility, safety and patient benefit of deprescribing, as discussed further below. It employs evidence-based frameworks that assist the prescriber [30] and are patient-centered [31].
Importantly, deprescribing should be seen as part of the good prescribing continuum, which spans medicine initiation, titrating, changing, or adding medicines, and switching or ceasing medicines. Deprescribing is not about denying effective treatment to eligible patients. It is a positive, patient-centered intervention, with inherent uncertainties, and requires shared decision-making, informed patient consent and close monitoring of effects [32]. Deprescribing involves diagnosing a problem (use of a PIM), making a therapeutic decision (withdrawing it with close follow-up) and altering the natural history of the problem (reducing incidence of medicine-related adverse events).
Our definition of evidence-based deprescribing is a form of direct deprescribing applied at the level of the individual patient-prescriber/pharmacist encounter. Direct deprescribing uses explicit, systematic processes (such as using an algorithm or structured deprescribing framework or guide) applied by individual prescribers (or pharmacists) to the medicine regimens of individual patients (ie, at the patient level), and which targets either specific classes of medicines or all medicines that are potentially inappropriate. This is in contrast to indirect deprescribing, which uses more generic, programmatic strategies aimed at prescribers as a whole (ie, at the population or system level) and which seek to improve quality use of medicines in general, including both underuse and overuse of medicines. Indirect deprescribing entails a broader aim of medicines optimization in which deprescribing is a possible outcome but not necessarily the sole focus. Such strategies include pharmacist or physician medicine reviews, education programs for clinicians and/or patients, academic detailing, audit and feedback, geriatric assessment, multidisciplinary teams, prescribing restrictions, and government policies, all of which aim to reduce the overall burden of PIMs among broad groups of patients. While intuitively the 2 approaches in combination should exert synergistic effects superior to those of either by itself, this has not been studied.
Evidence For Deprescribing
Indirect Deprescribing
Overall, the research into indirect interventions has been highly heterogenous in terms of interventions and measures of medicine use. Research has often been of low to moderate quality, focused more on changes to prescribing patterns and less on clinical outcomes, been of short duration, and produced mixed results [33]. In a 2013 systematic review of 36 studies involving different interventions involving frail older patients in various settings, 22 of 26 quantitative studies reported statistically significant reductions in the proportions of medicines deemed unnecessary (defined using various criteria), ranging from 3 to 20 percentage points [34]. A more recent review of 20 trials of pharmacist-led reviews in both inpatient and outpatient settings reported a small reduction in the mean number of prescribed medicines (–0.48, 95% confidence interval [CI] –0.89 to –0.07) but no effects on mortality or readmissions, although unplanned hospitalizations were reduced in patients with heart failure [35]. A 2012 review of 10 controlled and 20 randomized studies revealed statistically significant reductions in the number of medicines in most of the controlled studies, although mixed results in the randomized studies [36]. Another 2012 review of 10 studies of different designs concluded that interventions were beneficial in reducing potentially inappropriate prescribing and medicine-related problems [37]. A 2013 review of 15 studies of academic detailing of family physicians showed a modest decline in the number of medications of certain classes such as benzodiazepines and nonsteroidal anti-inflammatory drugs [38]. Another 2013 review restricted to 8 randomized trials of various interventions involving nursing home patients suggested medicine-related problems were more frequently identified and resolved, together with improvement in medicine appropriateness [39]. In 2 randomized trials conducted in aged care facilities and centered on educational interventions, one aimed at prescribers [40] and the other at nursing staff [41],the number of potentially harmful medicines and days in hospital was significantly reduced [40,41], combined with slower declines in health-related quality of life [40]. In a randomized trial, patient education provided through community pharmacists led to a 77% reduction in benzodiazepine use among chronic users at 6 months with no withdrawal seizures or other ill effects [42].
Direct Deprescribing Targeting Specific Classes of Medicines
The evidence base for direct patient-level deprescribing is more rigorous as it pertains to specific classes of medicines. A 2008 systematic review of 31 trials (15 randomized, 16 observational) that withdrew a single class of medicine in older people demonstrated that, with appropriate patient selection and education coupled with careful withdrawal and close monitoring, antihypertensive agents, psychotropic medicines, and benzodiazepines could be discontinued without harm in 20% to 100% of patients, although psychotropics showed a high post-trial rate of recommencement [43]. Another review of 9 randomized trials demonstrated the safety of withdrawing antipsychotic agents that had been used continuously for behavioural and psychological symptoms in more than 80% of subjects with dementia [44]. In an observational study, cessation of inappropriate antihypertensives was associated with fewer cardiovascular events and deaths over a 5-year follow-up period [45]. A recent randomized trial of statin withdrawal in patients with advanced illness and of whom half had a prognosis of less than 12 months demonstrated improved quality of life and no increased risk of cardiovascular events over the following 60 days [46].
Direct Deprescribing Targeting All Medicines
The evidence base for direct patient-level deprescribing that assesses all medicines, not just specific medicine classes, features several high-quality observational studies and controlled trials, and subgroup findings from a recent comprehensive systematic review. In this review of 132 studies, which included 56 randomized controlled trials [47], mortality was shown in randomized trials to be decreased by 38% as a result of direct (ie, patient-level) deprescribing interventions. However, this effect was not seen in studies of indirect deprescribing comprising mainly generic educational interventions. While space prevents a detailed analysis of all relevant trials, some of the more commonly cited sentinel studies are mentioned here.
In a controlled trial involving 190 patients in aged care facilities, a structured approach to deprescribing (Good Palliative–Geriatric Practice algorithm) resulted in 63% of patients having, on average, 2.8 medicines per patient discontinued, and was associated with a halving in both annual mortality and referrals to acute care hospitals [48]. In another prospective uncontrolled study, the same approach applied to a cohort of 70 community-dwelling older patients resulted in an average of 4.4 medicines prescribed to 64 patients being recommended for discontinuation, of which 81% were successfully discontinued, with 88% of patients reporting global improvements in health [49]. In a prospective cohort study of 50 older hospitalized patients receiving a median of 10 regular medicines on admission, a formal deprescribing process led to the cessation of just over 1 in 3 medicines by discharge, representing 4 fewer medicines per patient [50]. During a median follow-up period of just over 2.5 months for 39 patients, less than 5% of ceased medicines were recommenced in 3 patients for relapsing symptoms, with no deaths or acute presentations to hospital attributable to cessation of medicines. A multidisciplinary hospital clinic for older patients over a 3-month period achieved cessation of 22% of medicines in 17 patients without ill effect [51].
Two randomized studies used the Screening Tool of Older People’s Prescriptions (STOPP) to reduce the use of PIMs in older hospital inpatients [52,53]. One reported significantly reduced PIMs use in the intervention group at discharge and 6 months post-discharge, no change in the rate of hospital readmission, and non-significant reductions in falls, all cause-mortality, and primary care visits during the 6-month follow-up period [52]. The second study reported reduced PIMs use in the intervention group of frail older patients on discharge, although the proportion of people prescribed at least 1 PIM was not altered [53].
Recently, a randomized trial of a deprescribing intervention applied to aged care residents resulted in successful discontinuation of 207 (59%) of 348 medicines targeted for deprescribing, and a mean reduction of 2 medicines per patient at 12 months compared to none in controls, with no differences in mortality or hospital admissions [54]. The evidence for direct deprescribing is limited by relatively few high-quality randomized trials, small patient samples, short duration of follow-up, selection of specific subsets of patients, and the absence of comprehensive re-prescribing data and clinical outcomes.
Methods Used for Direct Deprescribing
At the level of individual patient care, various instruments have been developed to assist the deprescribing process. Screening tools or criteria such as the Beers criteria and STOPP tool help identify medicines more likely than not to be inappropriate for a given set of circumstances and are widely used by research pharmacists. Deprescribing guidelines directed at particular medications (or drug classes) [55], or specific patient populations [56], can identify clinical scenarios where a particular drug is likely to be inappropriate, and how to safely wean or discontinue it.
In applying a more nuanced, patient-centered approach to deprescribing, structured guides comprising algorithms, flowcharts, or tables describe sequential steps in deciding which medications used by an individual patient should be targeted for discontinuation after due attention to all relevant factors. Such guides prompt a more systematic appraisal of all medications being used. In a recent review of 7 structured guides that had undergone some form of efficacy testing [59], the strongest evidence of efficacy and clinician acceptability was seen for the Good Palliative–Geriatric Practice algorithm [48] (Figure) and the CEASE protocol [29,30,50,60] (Table). Both have been subject to a process of development and refinement over months to years involving multiple clinician prescribers and pharmacists.
Clinical Circumstances Conducive to Deprescribing
Deprescribing should be especially considered in any older patient presenting with a new symptom or clinical syndrome suggestive of adverse medicine effects. The advent of advanced or end-stage disease, terminal illness, dementia, extreme frailty, or full dependence on others for all cares marks a stage of a person’s life when limited life expectancy and changed goals of care call for a re-appraisal of the benefits of current medicines. Lack of response in controlling symptoms despite optimal adherence and dosing or conversely the absence of symptoms for long periods of time should challenge the need for ongoing regular use of medicines. Similarly, the lack of verification, or indeed repudiation, of past diagnostic labels which gave rise to indications for medicines in the first place should prompt consideration of discontinuation. Patients receiving single medicines or combinations of medicines, both of which are high risk, should attract attention [63], as should use of preventive medicines for scenarios associated with no increased disease risk despite medicine cessation (eg, ceasing alendronate after 5 years of treatment results in no increase in osteoporotic fracture risk over the ensuing 5 years [64]; ceasing statins for primary prevention after a prolonged period results in no increase in cardiovascular events 8 years after discontinuation [65]). Evidence that has emerged that strongly contradicts previously held beliefs as to the indications for certain medicines (eg, aspirin as primary prevention of cardiovascular disease) should lead to a higher frequency of their discontinuation. Finally, medicines which impose demands on patients which they deem intolerable in terms of dietary and lifestyle restrictions, adverse side effects, medicine monitoring (such as warfarin), financial cost, or any other reason likely to result in nonadherence, should be considered candidates for deprescribing [25].
Barriers to Deprescribing
The most effective strategy to reducing potentially inappropriate polypharmacy is for doctors to prescribe and patients to consume fewer medicines. Unfortunately, both doctors and patients often lack confidence about when and how to cease medicines [66–69]. In a recent systematic review comprised mostly of studies involving general practitioners in primary care [66], 4 themes emerged. First, prescribers may be unaware of their own instances of inappropriate prescribing in older people until this is pointed out to them. Poor insight may be attributable in part to insufficient education in geriatric pharmacology. Second, clinical inertia manifesting as failure to act despite an awareness of PIMs may arise from deprescribing being viewed as a risky affair [70], with doctors fearful of provoking withdrawal syndromes or disease complications, and damaging their reputation and relationships with patients or colleagues in the process. Continuing inappropriate medicines is reinforced by prescriber beliefs that to do so is a safer or kinder course of action for the patient. Third, self-perceptions of being ill-equipped, in terms of the necessary knowledge and skills, to deprescribe appropriately (lack of self-efficacy) may be a barrier, even if one accepts the need for deprescribing. Information deficits around benefit-harm trade-offs of particular drugs and alternative treatments (both drug and non-drug), especially for older, frail, multi-morbid patients, contribute to the problem. Confidence to deprescribe is further undermined by the lack of clear documentation regarding reasons drugs were originally prescribed by other doctors, outcomes of past trials of discontinuation, and current patient care goals. Fourth, several external or logistical constraints may hamper deprescribing efforts such as perceived patient unwillingness to deprescribe certain medicines, lack of prescriber time, poor remuneration, and community and professional attitudes toward more rather than less use of medicines.
Deprescribing in hospital settings led by specialists appears to be no better than in general practice, although it has been less well studied. While an episode of acute inpatient care may afford an opportunity to review and reduce medicine lists, studies suggest the opposite occurs. In a New Zealand audit of 424 patients of mean age 80 years admitted acutely to a medical unit, chronically administered medications increased during hospital stay from a mean of 6.6 to 7.7 [71]. Similarly, in an Australian study investigating medication changes for 1220 patients of mean age 81 years admitted to general medical units of 11 acute care hospitals, the mean number of regularly administered medications rose from 7.1 on admission to 7.6 at discharge [72]. It is likely the same drivers behind failure to deprescribe in primary care also operate in secondary and tertiary care settings. Part of the problem is under-recognition of medicine-related geriatric syndromes on the part of hospital physicians and pharmacists [73].
Patients in both the community and residential aged care facilities frequently express a desire to have their medicines reduced in number, especially if advised by their treating clinician [74,75]. Having said this, many remain wary of discontinuing specific medicines [67], sharing the same fears of evoking withdrawal syndromes or disease relapse as do prescribers, and recounting the strong advice of past specialists to never withhold any medicines without first seeking their advice.
A challenge for all involved in deprescribing is gaining agreement on what are the most important factors that determine when, how, and in whom deprescribing should be conducted. Recent qualitative studies suggest that doctors, pharmacists, nursing staff, and patients and their families, while in broad agreement that deprescribing is worthwhile, often differ in their perspectives on what takes priority in selecting medicines for deprescribing in individual patients, and how it should be done and by whom [76,77].
Strategies That May Facilitate Deprescribing
While deprescribing presents some challenges, there are several strategies that can facilitate it at both the level of individual clinical encounters and at the level of whole populations and systems of care.
Individual Clinical Encounters
Within individual clinician–patient encounters, patients should be empowered to ask their doctors and pharmacists the following questions:
- What are my treatment options (including non-medicine options) for my condition?
- What are the possible benefits and harms of each medicine?
- What might be reasonable grounds for stopping a medicine?
In turn, doctors and pharmacists should ask in a nonjudgmental fashion, at every encounter, whether patients are experiencing any side effects, administration and monitoring problems, or other barriers to adherence associated with any of their medicines.
The issue of deprescribing should be framed as an attempt to alleviate symptoms (of drug toxicity), improve quality of life (from drug-induced disability), and lessen the risk of morbid events (especially ADEs) in the future. Compelling evidence that identifies circumstances in which medicines can be safely withdrawn while reducing the risk of ADEs needs to be emphasized. Specialists must play a sentinel leadership role in advising and authorizing other health professionals to deprescribe in situations where benefits of medications they have prescribed are no longer outweighed by the harms [60,78].
In language they can understand, patients should be informed of the benefit–harm trade-offs specific to them of continuing or discontinuing a particular medicine, as far as these can be specified. Patients often overestimate the benefits and underestimate the harms of treatments [79]. Providing such personalised information can substantially alter perceptions of risk and change attitudes towards discontinuation [80]. Eliciting patients’ beliefs about the necessity for each individual medicine and spending time, using an empathic manner, to dispel or qualify those at odds with evidence and clinical judgement renders deprescribing more acceptable to patients.
In estimating treatment benefit–harm trade-offs in individual patients, disease risk prediction tools (http://www.medal.org/), evidence tables [81,82], and decision aids are increasingly available. Prognostication tools (http://eprognosis.ucsf.edu) combined with trial-based time-to-event data can be used to determine if medicine-specific time until benefit exceeds remaining life span.
Deprescribing is best performed by reducing medicines one at a time over several encounters with the same overseeing generalist clinician with whom patients have established a trusting and collaborative relationship. This provides repeated opportunities to discuss and assuage any fears of discontinuing a medicine, and to adjust the deprescribing plan according to changes in clinical circumstances and revised treatment goals. Practice-based pharmacists can review patients’ medicine lists and apply screening criteria to identify medicines more likely to be unnecessary or harmful, which then helps initiate and guide deprescribing. Integrating a structured deprescribing protocol—and reminders to use it—into electronic health records, and providing decision support and data collection for future reference, reduce the cognitive burden on prescribers [83]. Practical guidance in how to safely wean and cease particular classes of medicines in older people can be accessed from various sources [84,85]. Seeking input from clinical pharmacologists, pharmacists, nurses, and other salient care providers on a case-by-case basis in the form of interactive case conferences provides support, seeks consensus, and shares the risk and responsibility for deprescribing recommendations [86].
System of Care
The success of deprescribing efforts in realizing better population health will be compromised unless all key stakeholders involved in quality use of medicines commit to operationalizing deprescribing strategies at the system of care level. Position statements on deprescribing in multi-morbid populations should be formulated and promulgated by all professional societies of prescribers (primary care, specialists, pharmacists, dentists, nurse practitioners). Professional development programs as well as undergraduate, graduate, and postgraduate courses in medicine, pharmacy, and nursing should include training in deprescribing as a core curricular element.
Researchers seeking funding and/or ethics approval for research projects involving medicines should be required to collect, analyze, and report data on the frequency of, and reasons for, withdrawal of drugs in trial subjects. This helps build the evidence base of medicine-related harm. In turn, government funders of research should require more researchers to design and conduct clinical trials that recruit multi-morbid patients, including specific subgroups (eg, patients with dementia), and aim to define medicine benefits and harms using patient risk stratification methods. Pharmaceutical companies should sponsor research on how to deprescribe their medicines within trials that also aim to assess efficacy and safety. Medicine regulatory authorities such as the Food and Drug Administration should mandate that this information be supplied at the time the company submits their application to have the medicine approved and listed for public subsidy. Trialists should adopt the word “deprescribing” in abstract titles for research on prescriber-initiated medicine discontinuation so that relevant articles can be more accurately indexed in, and retrieved from, bibliographic databases using recently formulated medical subject headings in Medline (“depresciptions”).
Editors of medical journals should promote a deprescribing agenda as a quality and safety issue for patient care, with the “Less is More” series in JAMA Internal Medicine and “Too much medicine” series in BMJ being good examples. Clinical guideline developers should formulate treatment recommendations specific to the needs of multi-morbid patients which acknowledge the limited evidence base for many medicines in such populations. These should take account of commonly encountered clinical scenarios where disease-specific medicines may engender greater risk of harm, and provide cautionary notes regarding initiation and discontinuation of medicines associated with high-risk.
Pharmacists need to instruct patients in how to identify medicine-induced harm and side effects, and how to collaborate with their prescribing clinicians in safely discontinuing high-risk medicines. Ideally, patients being admitted to residential aged care facilities should have their medicine lists reviewed by a pharmacist in flagging medicines eligible for deprescribing. Organizations and services responsible for providing quality use of medicines information (medicines handbooks, prescribing guidelines, drug safety bulletins) should describe when and how deprescribing should be performed in regards to specific medicines. This information should be cross-referenced to clinical guidelines and position statements dealing with the same medicine. Vendors of medicine prescribing software should be encouraged to incorporate flags and alerts which prompt prescribers to consider medicine cessation in high-risk patients.
Government and statutory bodies with responsibility for health care (health departments, quality and safety commissions, practice accreditation services, health care standard–setting bodies) should fund more research to develop and evaluate medicine safety standards aimed at reducing inappropriate use of medicines. Accreditation procedures for hospitals and primary care organizations should mandate the adoption of professional development and quality measurement systems that support and monitor patients receiving multiple medicines. Organizations responsible for conducting pharmacovigilance studies should issue medicine-specific deprescribing alerts whenever their data suggest higher than expected incidence of medicine-related adverse events in older populations receiving such medicines.
Conclusion
Inappropriate medicine use and polypharmacy is a growing issue among older and multi-morbid patients. The cumulative evidence of the safety and benefits of deprescribing argues for its adoption on the part of all prescribers, as well as its support by pharmacists and others responsible for optimizing use of medicines. Widespread implementation within routine care of an evidence-based approach to deprescribing in all patients receiving polypharmacy has its challenges, but also considerable potential to relieve unnecessary suffering and disability. More high quality research is needed in defining the circumstances under which deprescribing confers maximal benefit in terms of improved clinical outcomes.
Corresponding author: Ian A. Scott, Dept. of Internal Medicine and Clinical Epidemiology, Princess Alexandra Hospital, Brisbane, Australia 4102, [email protected].
Financial disclosures: None.
1. Qato DM, Alexander GC, Conti RM, et . Use of prescription and over-the-counter medications and dietary supplements among older adults in the United States. JAMA 2008;300:2867–78.
2. Kantor ED, Rehm CD, Haas JS, et al. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA 2015;314:1818–31.
3. Wise J. Polypharmacy: a necessary evil. BMJ 2013;347: f7033.
4. Gnjidic D, Hilmer SN, Blyth FM, et al. Polypharmacy cutoff and outcomes: five or more medicines were used to identify community-dwelling older men at risk of different adverse outcomes. J Clin Epidemiol 2012;65:989–95.
5. Atkin PA, Veitch PC, Veitch EM, Ogle SJ. The epidemiology of serious adverse drug reactions among the elderly. Medicines Aging 1999;14:141–52.
6. Roughead EE, Anderson B, Gilbert AL. Potentially inappropriate prescribing among Australian veterans and war widows/widowers. Intern Med J 2007;37:402–5.
7. Stafford AC, Alswayan MS, Tenni PC. Inappropriate prescribing in older residents of Australian care homes. Clin Pharmacol Therapeut 2011;36:33–44.
8. Tjia J, Briesacher BA, Peterson D, et al. Use of medications of questionable benefit in advanced dementia. JAMA Intern Med 2014;174:1763–71.
9. Tjia J, Velten SJ, Parsons C, et al. Studies to reduce unnecessary medication use in frail older adults: A systematic review. Drugs Aging 2013;30:285–307.
10. Anathhanam AS, Powis RA, Cracknell AL, Robson J. Impact of prescribed medicines on patient safety in older people. Ther Adv Drug Saf 2012;3:165–74.
11. Opondo D, Eslami S, Visscher S, et al. Inappropriateness of medication prescriptions to elderly patients in the primary care setting: a systematic review. PLoS One 2012;7(8):e43617.
12. Kalisch LM, Caughey GE, Barratt JD, et al. Prevalence of preventable medication-related hospitalizations in Australia: an opportunity to reduce harm. Int J Qual Health Care 2012;24:239–49.
13. Bero LA, Lipton HL, Bird JA. Characterisation of geriatric drug-related hospital readmissions. Med Care 1991;29:989–1003.
14. Jyrkkä J, Enlund H, Korhonen MJ, et al. Polypharmacy status as an indicator of mortality in an elderly population. Drugs Aging 2009;26:1039–48.
15. Steinman MA, Miao Y, Boscardin WJ, et al. Prescribing quality in older veterans: a multifocal approach. J Gen Intern Med 2014;29:1379–86.
16. Goldberg R, Mabee J, Chan L, Wong S. Drug-drug and drug-disease interactions in the ED: analysis of a high-risk population. Am J Emerg Med 1996;14:447–50.
17. Elliott RA, Booth JC. Problems with medicine use in older Australians: a review of recent literature. J Pharm Pract Res 2014;44:258–71.
18. Barat I, Andreasen F, Damsgaard EM. Drug therapy in the elderly: what doctors believe and patients actually do. Br J Clin Pharmacol 2001;51:615–22.
19. Chapman RH, Benner JS, Petrilla AA, et al. Predictors of adherence with antihypertensive and lipid-lowering therapy. Arch Intern Med 2005;165:1147–52.
20. Gnjidic D, Hilmer SN. Emergency hospitalizations for adverse drug events. N Engl J Med 2012;366:859.
21. Gnjidic D, Hilmer SN, Blyth FM, Naganathan V, Waite L, et al. Polypharmacy cutoff and outcomes: five or more medicines were used to identify community-dwelling older men at risk of different adverse outcomes. J Clin Epidemiol 2012;65:989–95.
22. Cherubini A, Oristrell J, Pla X, et al. The persistent exclusion of older patients from ongoing clinical trials regarding heart failure. Arch Intern Med 2011;171:550–6.
23. Bugeja G, Kumar A, Banerjee AK. Exclusion of elderly people from clinical research: a descriptive study of published reports. BMJ 1997;315:1059.
24. Mangin D, Heath I, Jamoulle M. Beyond diagnosis: rising to the multimorbidity challenge. BMJ 2012;344:e3526.
25. Boyd CM, Darer J, Boult C, et al. Clinical practice guidelines and quality of care for older patients with multiple comorbid diseases: implications for pay for performance. JAMA 2005;294:716–24.
26. McLean AJ, Le Couteur DG. Aging biology and geriatric clinical pharmacology. Pharmacol Rev 2004;56:163–84.
27. Hilmer SN, Mager DE, Simonsick EM, et al. Drug Burden Index score and functional decline in older people. Am J Med 2009;122:1142–9.
28. Reeve E, Gnjidic D, Long J, Hilmer S. A systematic review of the emerging definition of ‘deprescribing’ with network analysis: implications for future research and clinical practice. Br J Clin Pharmacol 2015;80:1254–68.
29. Scott IA, Hilmer SN, Reeve E, et al. Reducing inappropriate polypharmacy – the process of deprescribing. JAMA Intern Med 2015;175:827–34.
30. Scott IA, Gray LA, Martin JH, et al. Deciding when to stop: towards evidence-based deprescribing of drugs in older populations. Evidence-based Med 2013;18:121–4.
31. Reeve E, Shakib S, Hendrix I, et al. Review of deprescribing processes and development of an evidence-based, patient-centred deprescribing process. Br J Clin Pharmacol 2014;78:738–47.
32. Alldred D. Deprescribing: a brave new word? Int J Pharm Pract. 2014;22:2–3.
33. Kaur S, Mitchell G, Vitetta L, Roberts MS. Interventions that can reduce inappropriate prescribing in the elderly: a systematic review. Drugs Aging 2009;26:1013–28.
34. Tjia J, Velten SJ, Parsons C, et al. Studies to reduce unnecessary medication use in frail older adults: A systematic review. Drugs Aging 2013;30:285–307.
35. Thomas R, Huntley AL, Mann M, et al. Pharmacist-led interventions to reduce unplanned admissions for older people: a systematic review and meta-analysis of randomised controlled trials. Age Ageing 2014;43:174–87.
36. Gnjidic D, Le Couteur DG, Kouladjian L, Hilmer SN. Deprescribing trials: Methods to reduce polypharmacy and the impact on prescribing and clinical outcomes. Clin Geriatr Med 2012;28:237–53.
37. Patterson SM, Hughes C, Kerse N, et al. Interventions to improve use of polypharmacy for older people. Cochrane Database Syst Rev 2012;5:CD008165.
38. Chhina HK, Bhole VM, Goldsmith C, et al. Effectiveness of academic detailing to optimize medication prescribing behaviour of family physicians. J Pharm Pharm Sci 2013;16:511–29.
39. Alldred DP, Raynor DK, Hughes C, et al. Interventions to optimise prescribing for older people in care homes. Cochrane Database Syst Rev 2013;CD009095.
40. García-Gollarte F, Baleriola-Júlvez J, Ferrero-López I, et al. An educational intervention on drug use in nursing homes improves health outcomes and resource utilization and reduces inappropriate drug prescription. J Am Dir Assoc 2014;15:885–91.
41. Pitkälä KH, Juola A-L, Kautiainen H, Soini H, et al. Education to reduce potentially harmful medication use among residents of assisted living facilities: A randomized controlled trial. J Am Dir Assoc 2014;15:892–8.
42. Tannenbaum C, Martin P, Tamblyn R, et al. Reduction of inappropriate benzodiazepine prescriptions among older adults through direct patient education. The EMPOWER cluster randomized trial. JAMA Intern Med 2014;174:890–8.
43. Iyer S, Naganathan V, McLachlan AJ, Le Couteur DG. Medication withdrawal trials in people aged 65 years and older: a systematic review. Drugs Aging 2008;25:1021–31.
44. Declercq T, Petrovic M, Azermai M, et al. Withdrawal versus continuation of chronic antipsychotic medicines for behavioural and psychological symptoms in older people with dementia. Cochrane Database Syst Rev 2013;3:CD007726.
45. Ekbom T, Lindholm LH, Odén A, et al. A 5-year prospective, observational study of the withdrawal of antihypertensive treatment in elderly people. J Intern Med 1994;235:581–588.
46. Kutner JS, Blatchford PJ, Taylor DH Jr, et al. Safety and benefit of discontinuing statin therapy in the setting of advanced, life-limiting illness: a randomized clinical trial. JAMA Intern Med 2015;175:691–700.
47. Page AT, Clifford RM, Potter K, Schwartz D, Etherton-Beer CD. The feasibility and effect of deprescribing in older adults on mortality and health: a systematic review and meta-analysis. Br J Clin Pharmacol 2016 Apr 14. [Epub ahead of print]
48. Garfinkel D, Zur-Gil S, Ben-Israel J. The war against polypharmacy: a new cost-effective geriatric-palliative approach for improving drug therapy in disabled elderly people. Isr Med Assoc J 2007;9:430–4.
49. Garfinkel D, Mangin D. Feasibility study of a systematic approach for discontinuation of multiple medicines in older adults: addressing polypharmacy. Arch Intern Med 2010;170:1648–54.
50. McKean M, Pillans P, Scott IA. A medication review and deprescribing method for hospitalised older patients receiving multiple medications. Intern Med J 2016;46:35–42.
51. Mudge A, Radnedge K, Kasper K, et al. Effects of a pilot multidisciplinary clinic for frequent attending elderly patients on deprescribing. Aust Health Rev 2015; Jul 6. [Epub ahead of print]
52. Gallagher PF, O’Connor MN, O’Mahony D. Prevention of potentially inappropriate prescribing for elderly Patients: A randomized controlled trial using STOPP/START criteria. Clin Pharmacol Therap 2011;89:845–54.
53. Dalleur O, Boland B, Losseau C, et al. Reduction of potentially inappropriate medications using the STOPP criteria in frail older inpatients: a randomised controlled study. Drugs Aging 2014;31:291–8.
54. Potter K, Flicker L, Page A, Etherton-Beer C. Deprescribing in frail older people: A randomised controlled trial. PLoS One 2016;11(3):e0149984.
55. Conklin J, Farrell B, Ward N, et al. Developmental evaluation as a strategy to enhance the uptake and use of deprescribing guidelines: protocol for a multiple case study. Implement Sci 2015;10:91–101.
56. Lindsay J, Dooley M, Martin J, et al. The development and evaluation of an oncological palliative care deprescribing guideline: the ‘OncPal deprescribing guideline’ Support Care Cancer 2015;23:71–8.
57. Miller GC, Valenti L, Britt H, Bayram C. Drugs causing adverse events in patients aged 45 or older: a randomised survey of Australian general practice patients. BMJ Open 2013;3:e003701.
58. Budnitz DS, Lovegrove MC, Shebab N, Richards CL. Emergency hospitalisations for adverse drug events in older Americans. N Engl J Med 2011;365:2002–12.
59. Scott IA, Andersen K, Freeman C. Review of structured guides for deprescribing. Eur J Hosp Pharm 2016. In press.
60. Scott IA, Le Couteur D. Physicians need to take the lead in deprescribing. Intern Med J 2015;45:352–6.
61. Poudel A, Ballokova A, Hubbard RE, et al. An algorithm of medication review in residential aged care facilities: focus on minimizing use of high risk medications. Geriatr Gerontol Int Sep 3. [Epub ahead of print]
62. Scott IA, Martin JH, Gray LA, Mitchell CA. Effects of a drug minimisation guide on prescribing intentions in elderly persons with polypharmacy. Drugs Ageing 2012;29:659–67.
63. Bennett A, Gnjidic D, Gillett M, et al. Prevalence and impact of fall-risk-increasing drugs, polypharmacy, and drug-drug interactions in robust versus frail hospitalised falls patients: a prospective cohort study. Drugs Aging 2014;31:225–32.
64. Black DM, Schwartz AV, Ensrud KE, et al. FLEX Research Group. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomised trial. JAMA 2006;296:2927–38.
65. Sever PS, Chang CL, Gupta AK, et al. The Anglo-Scandinavian Cardiac Outcomes Trial: 11-year mortality follow-up of the lipid lowering arm in the UK. Eur Heart J 2011;32:2525–32.
66. Anderson K, Stowasser D, Freeman C, Scott I. Prescriber barriers and enablers to minimising potentially inappropriate medications in adults: a systematic review and thematic synthesis. BMJ Open 2014;4.
67. Reeve E, To J, Hendrix I, et al. Patient barriers to and enablers of deprescribing: a systematic review. Drugs Aging 2013;30:793–807.
68. Palagyi A, Keay L, Harper J, et al. Barricades and brickwalls—a qualitative study exploring perceptions of medication use and deprescribing in long-term care. BMC Geriatr 2016;16:15.
69. Garfinkel D, Ilhan B, Bahat G. Routine deprescribing of chronic medications to combat polypharmacy. Ther Adv Drug Saf 2015;6:212–33.
70. Reeve E, Shakib S, Hendrix I, et al. The benefits and harms of deprescribing. Med J Aust 2014;201:386–9.
71. Betteridge TM, Frampton CM, Jardine DL. Polypharmacy – we make it worse! A cross-sectional study from an acute admissions unit. Intern Med J 2012;42:208–11.
72. Hubbard RE, Peel NM, Scott IA, et al. Polypharmacy among inpatients aged 70 years or older in Australia. Med J Aust 2015;202:373–7.
73. Klopotowska JE, Wierenga PC, Smorenburg SM, et al. Recognition of adverse drug events in older hospitalized medical patients. Eur J Clin Pharmacol 2013;69:75–85.
74. Reeve E, Wiese MD, Hendrix I, et al. People’s attitudes, beliefs, and experiences regarding polypharmacy and willingness to deprescribe. J Am Geriatr Soc 2013;61:1508–14.
75. Kalogianis MJ, Wimmer BC, Turner JP, et al. Are residents of aged care facilities willing to have their medications deprescribed? Res Social Adm Pharm 2015. Published online 18 Dec 2015.
76. Turner JP, Edwards S, Stanners M, et al. What factors are important for deprescribing in Australian long-term care facilities? Perspectives of residents and health professionals. BMJ Open 2016;6:e009781.
77. Page AT, Etherton-Beer CD, Clifford RM, et al. Deprescribing in frail older people - Do doctors and pharmacists agree? Res Social Adm Pharm 2015;12:438–49.
78. Luymes CH, van der Kleij RM, Poortvliet RK, et al. Deprescribing potentially inappropriate preventive cardiovascular medication: Barriers and enablers for patients and general practitioners. Ann Pharmacother 2016 Mar 3. [Epub ahead of print]
79. Hoffmann TC, Del Mar C. Patients’ expectations of the benefits and harms of treatments, screening, and tests: a systematic review. JAMA Intern Med 2015;175:274–86.
80. Martin P, Tamblyn R, Ahmed S, Tannenbaum C. A drug education tool developed for older adults changes knowledge, beliefs and risk perceptions about inappropriate benzodiazepine prescriptions in the elderly. Patient Educ Couns 2013;92:81–7.
81. Hamilton H, Gallagher P, Ryan C, et al. Potentially inappropriate medicines defined by STOPP criteria and the risk of adverse drug events in older hospitalized patients. Arch Intern Med 2011;171:1013–7.
82. NHS Highland. Polypharmacy: guidance for prescribing in frail adults. Accessed at: www.nhshighland.scot.nhs.uk/publications/documents/guidelines/polypharmacy guidance for prescribing in frail adults.pdf.
83. Anderson K, Foster MM, Freeman CR, Scott IA. A multifaceted intervention to reduce inappropriate polypharmacy in primary care: research co-creation opportunities in a pilot study. Med J Aust 2016;204:S41–4.
84. A practical guide to stopping medicines in older people. Accessed at: www.bpac.org.nz/magazine/2010/april/stopGuide.asp.
85. www.cpsedu.com.au/posts/view/46/Deprescribing-Documents-now-Available-for-Download.
86. Bregnhøj L, Thirstrup S, Kristensen MB, et al. Combined intervention programme reduces inappropriate prescribing in elderly patients exposed to polypharmacy in primary care. Eur J Clin Pharmacol 2009;65:199–207.
1. Qato DM, Alexander GC, Conti RM, et . Use of prescription and over-the-counter medications and dietary supplements among older adults in the United States. JAMA 2008;300:2867–78.
2. Kantor ED, Rehm CD, Haas JS, et al. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA 2015;314:1818–31.
3. Wise J. Polypharmacy: a necessary evil. BMJ 2013;347: f7033.
4. Gnjidic D, Hilmer SN, Blyth FM, et al. Polypharmacy cutoff and outcomes: five or more medicines were used to identify community-dwelling older men at risk of different adverse outcomes. J Clin Epidemiol 2012;65:989–95.
5. Atkin PA, Veitch PC, Veitch EM, Ogle SJ. The epidemiology of serious adverse drug reactions among the elderly. Medicines Aging 1999;14:141–52.
6. Roughead EE, Anderson B, Gilbert AL. Potentially inappropriate prescribing among Australian veterans and war widows/widowers. Intern Med J 2007;37:402–5.
7. Stafford AC, Alswayan MS, Tenni PC. Inappropriate prescribing in older residents of Australian care homes. Clin Pharmacol Therapeut 2011;36:33–44.
8. Tjia J, Briesacher BA, Peterson D, et al. Use of medications of questionable benefit in advanced dementia. JAMA Intern Med 2014;174:1763–71.
9. Tjia J, Velten SJ, Parsons C, et al. Studies to reduce unnecessary medication use in frail older adults: A systematic review. Drugs Aging 2013;30:285–307.
10. Anathhanam AS, Powis RA, Cracknell AL, Robson J. Impact of prescribed medicines on patient safety in older people. Ther Adv Drug Saf 2012;3:165–74.
11. Opondo D, Eslami S, Visscher S, et al. Inappropriateness of medication prescriptions to elderly patients in the primary care setting: a systematic review. PLoS One 2012;7(8):e43617.
12. Kalisch LM, Caughey GE, Barratt JD, et al. Prevalence of preventable medication-related hospitalizations in Australia: an opportunity to reduce harm. Int J Qual Health Care 2012;24:239–49.
13. Bero LA, Lipton HL, Bird JA. Characterisation of geriatric drug-related hospital readmissions. Med Care 1991;29:989–1003.
14. Jyrkkä J, Enlund H, Korhonen MJ, et al. Polypharmacy status as an indicator of mortality in an elderly population. Drugs Aging 2009;26:1039–48.
15. Steinman MA, Miao Y, Boscardin WJ, et al. Prescribing quality in older veterans: a multifocal approach. J Gen Intern Med 2014;29:1379–86.
16. Goldberg R, Mabee J, Chan L, Wong S. Drug-drug and drug-disease interactions in the ED: analysis of a high-risk population. Am J Emerg Med 1996;14:447–50.
17. Elliott RA, Booth JC. Problems with medicine use in older Australians: a review of recent literature. J Pharm Pract Res 2014;44:258–71.
18. Barat I, Andreasen F, Damsgaard EM. Drug therapy in the elderly: what doctors believe and patients actually do. Br J Clin Pharmacol 2001;51:615–22.
19. Chapman RH, Benner JS, Petrilla AA, et al. Predictors of adherence with antihypertensive and lipid-lowering therapy. Arch Intern Med 2005;165:1147–52.
20. Gnjidic D, Hilmer SN. Emergency hospitalizations for adverse drug events. N Engl J Med 2012;366:859.
21. Gnjidic D, Hilmer SN, Blyth FM, Naganathan V, Waite L, et al. Polypharmacy cutoff and outcomes: five or more medicines were used to identify community-dwelling older men at risk of different adverse outcomes. J Clin Epidemiol 2012;65:989–95.
22. Cherubini A, Oristrell J, Pla X, et al. The persistent exclusion of older patients from ongoing clinical trials regarding heart failure. Arch Intern Med 2011;171:550–6.
23. Bugeja G, Kumar A, Banerjee AK. Exclusion of elderly people from clinical research: a descriptive study of published reports. BMJ 1997;315:1059.
24. Mangin D, Heath I, Jamoulle M. Beyond diagnosis: rising to the multimorbidity challenge. BMJ 2012;344:e3526.
25. Boyd CM, Darer J, Boult C, et al. Clinical practice guidelines and quality of care for older patients with multiple comorbid diseases: implications for pay for performance. JAMA 2005;294:716–24.
26. McLean AJ, Le Couteur DG. Aging biology and geriatric clinical pharmacology. Pharmacol Rev 2004;56:163–84.
27. Hilmer SN, Mager DE, Simonsick EM, et al. Drug Burden Index score and functional decline in older people. Am J Med 2009;122:1142–9.
28. Reeve E, Gnjidic D, Long J, Hilmer S. A systematic review of the emerging definition of ‘deprescribing’ with network analysis: implications for future research and clinical practice. Br J Clin Pharmacol 2015;80:1254–68.
29. Scott IA, Hilmer SN, Reeve E, et al. Reducing inappropriate polypharmacy – the process of deprescribing. JAMA Intern Med 2015;175:827–34.
30. Scott IA, Gray LA, Martin JH, et al. Deciding when to stop: towards evidence-based deprescribing of drugs in older populations. Evidence-based Med 2013;18:121–4.
31. Reeve E, Shakib S, Hendrix I, et al. Review of deprescribing processes and development of an evidence-based, patient-centred deprescribing process. Br J Clin Pharmacol 2014;78:738–47.
32. Alldred D. Deprescribing: a brave new word? Int J Pharm Pract. 2014;22:2–3.
33. Kaur S, Mitchell G, Vitetta L, Roberts MS. Interventions that can reduce inappropriate prescribing in the elderly: a systematic review. Drugs Aging 2009;26:1013–28.
34. Tjia J, Velten SJ, Parsons C, et al. Studies to reduce unnecessary medication use in frail older adults: A systematic review. Drugs Aging 2013;30:285–307.
35. Thomas R, Huntley AL, Mann M, et al. Pharmacist-led interventions to reduce unplanned admissions for older people: a systematic review and meta-analysis of randomised controlled trials. Age Ageing 2014;43:174–87.
36. Gnjidic D, Le Couteur DG, Kouladjian L, Hilmer SN. Deprescribing trials: Methods to reduce polypharmacy and the impact on prescribing and clinical outcomes. Clin Geriatr Med 2012;28:237–53.
37. Patterson SM, Hughes C, Kerse N, et al. Interventions to improve use of polypharmacy for older people. Cochrane Database Syst Rev 2012;5:CD008165.
38. Chhina HK, Bhole VM, Goldsmith C, et al. Effectiveness of academic detailing to optimize medication prescribing behaviour of family physicians. J Pharm Pharm Sci 2013;16:511–29.
39. Alldred DP, Raynor DK, Hughes C, et al. Interventions to optimise prescribing for older people in care homes. Cochrane Database Syst Rev 2013;CD009095.
40. García-Gollarte F, Baleriola-Júlvez J, Ferrero-López I, et al. An educational intervention on drug use in nursing homes improves health outcomes and resource utilization and reduces inappropriate drug prescription. J Am Dir Assoc 2014;15:885–91.
41. Pitkälä KH, Juola A-L, Kautiainen H, Soini H, et al. Education to reduce potentially harmful medication use among residents of assisted living facilities: A randomized controlled trial. J Am Dir Assoc 2014;15:892–8.
42. Tannenbaum C, Martin P, Tamblyn R, et al. Reduction of inappropriate benzodiazepine prescriptions among older adults through direct patient education. The EMPOWER cluster randomized trial. JAMA Intern Med 2014;174:890–8.
43. Iyer S, Naganathan V, McLachlan AJ, Le Couteur DG. Medication withdrawal trials in people aged 65 years and older: a systematic review. Drugs Aging 2008;25:1021–31.
44. Declercq T, Petrovic M, Azermai M, et al. Withdrawal versus continuation of chronic antipsychotic medicines for behavioural and psychological symptoms in older people with dementia. Cochrane Database Syst Rev 2013;3:CD007726.
45. Ekbom T, Lindholm LH, Odén A, et al. A 5-year prospective, observational study of the withdrawal of antihypertensive treatment in elderly people. J Intern Med 1994;235:581–588.
46. Kutner JS, Blatchford PJ, Taylor DH Jr, et al. Safety and benefit of discontinuing statin therapy in the setting of advanced, life-limiting illness: a randomized clinical trial. JAMA Intern Med 2015;175:691–700.
47. Page AT, Clifford RM, Potter K, Schwartz D, Etherton-Beer CD. The feasibility and effect of deprescribing in older adults on mortality and health: a systematic review and meta-analysis. Br J Clin Pharmacol 2016 Apr 14. [Epub ahead of print]
48. Garfinkel D, Zur-Gil S, Ben-Israel J. The war against polypharmacy: a new cost-effective geriatric-palliative approach for improving drug therapy in disabled elderly people. Isr Med Assoc J 2007;9:430–4.
49. Garfinkel D, Mangin D. Feasibility study of a systematic approach for discontinuation of multiple medicines in older adults: addressing polypharmacy. Arch Intern Med 2010;170:1648–54.
50. McKean M, Pillans P, Scott IA. A medication review and deprescribing method for hospitalised older patients receiving multiple medications. Intern Med J 2016;46:35–42.
51. Mudge A, Radnedge K, Kasper K, et al. Effects of a pilot multidisciplinary clinic for frequent attending elderly patients on deprescribing. Aust Health Rev 2015; Jul 6. [Epub ahead of print]
52. Gallagher PF, O’Connor MN, O’Mahony D. Prevention of potentially inappropriate prescribing for elderly Patients: A randomized controlled trial using STOPP/START criteria. Clin Pharmacol Therap 2011;89:845–54.
53. Dalleur O, Boland B, Losseau C, et al. Reduction of potentially inappropriate medications using the STOPP criteria in frail older inpatients: a randomised controlled study. Drugs Aging 2014;31:291–8.
54. Potter K, Flicker L, Page A, Etherton-Beer C. Deprescribing in frail older people: A randomised controlled trial. PLoS One 2016;11(3):e0149984.
55. Conklin J, Farrell B, Ward N, et al. Developmental evaluation as a strategy to enhance the uptake and use of deprescribing guidelines: protocol for a multiple case study. Implement Sci 2015;10:91–101.
56. Lindsay J, Dooley M, Martin J, et al. The development and evaluation of an oncological palliative care deprescribing guideline: the ‘OncPal deprescribing guideline’ Support Care Cancer 2015;23:71–8.
57. Miller GC, Valenti L, Britt H, Bayram C. Drugs causing adverse events in patients aged 45 or older: a randomised survey of Australian general practice patients. BMJ Open 2013;3:e003701.
58. Budnitz DS, Lovegrove MC, Shebab N, Richards CL. Emergency hospitalisations for adverse drug events in older Americans. N Engl J Med 2011;365:2002–12.
59. Scott IA, Andersen K, Freeman C. Review of structured guides for deprescribing. Eur J Hosp Pharm 2016. In press.
60. Scott IA, Le Couteur D. Physicians need to take the lead in deprescribing. Intern Med J 2015;45:352–6.
61. Poudel A, Ballokova A, Hubbard RE, et al. An algorithm of medication review in residential aged care facilities: focus on minimizing use of high risk medications. Geriatr Gerontol Int Sep 3. [Epub ahead of print]
62. Scott IA, Martin JH, Gray LA, Mitchell CA. Effects of a drug minimisation guide on prescribing intentions in elderly persons with polypharmacy. Drugs Ageing 2012;29:659–67.
63. Bennett A, Gnjidic D, Gillett M, et al. Prevalence and impact of fall-risk-increasing drugs, polypharmacy, and drug-drug interactions in robust versus frail hospitalised falls patients: a prospective cohort study. Drugs Aging 2014;31:225–32.
64. Black DM, Schwartz AV, Ensrud KE, et al. FLEX Research Group. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomised trial. JAMA 2006;296:2927–38.
65. Sever PS, Chang CL, Gupta AK, et al. The Anglo-Scandinavian Cardiac Outcomes Trial: 11-year mortality follow-up of the lipid lowering arm in the UK. Eur Heart J 2011;32:2525–32.
66. Anderson K, Stowasser D, Freeman C, Scott I. Prescriber barriers and enablers to minimising potentially inappropriate medications in adults: a systematic review and thematic synthesis. BMJ Open 2014;4.
67. Reeve E, To J, Hendrix I, et al. Patient barriers to and enablers of deprescribing: a systematic review. Drugs Aging 2013;30:793–807.
68. Palagyi A, Keay L, Harper J, et al. Barricades and brickwalls—a qualitative study exploring perceptions of medication use and deprescribing in long-term care. BMC Geriatr 2016;16:15.
69. Garfinkel D, Ilhan B, Bahat G. Routine deprescribing of chronic medications to combat polypharmacy. Ther Adv Drug Saf 2015;6:212–33.
70. Reeve E, Shakib S, Hendrix I, et al. The benefits and harms of deprescribing. Med J Aust 2014;201:386–9.
71. Betteridge TM, Frampton CM, Jardine DL. Polypharmacy – we make it worse! A cross-sectional study from an acute admissions unit. Intern Med J 2012;42:208–11.
72. Hubbard RE, Peel NM, Scott IA, et al. Polypharmacy among inpatients aged 70 years or older in Australia. Med J Aust 2015;202:373–7.
73. Klopotowska JE, Wierenga PC, Smorenburg SM, et al. Recognition of adverse drug events in older hospitalized medical patients. Eur J Clin Pharmacol 2013;69:75–85.
74. Reeve E, Wiese MD, Hendrix I, et al. People’s attitudes, beliefs, and experiences regarding polypharmacy and willingness to deprescribe. J Am Geriatr Soc 2013;61:1508–14.
75. Kalogianis MJ, Wimmer BC, Turner JP, et al. Are residents of aged care facilities willing to have their medications deprescribed? Res Social Adm Pharm 2015. Published online 18 Dec 2015.
76. Turner JP, Edwards S, Stanners M, et al. What factors are important for deprescribing in Australian long-term care facilities? Perspectives of residents and health professionals. BMJ Open 2016;6:e009781.
77. Page AT, Etherton-Beer CD, Clifford RM, et al. Deprescribing in frail older people - Do doctors and pharmacists agree? Res Social Adm Pharm 2015;12:438–49.
78. Luymes CH, van der Kleij RM, Poortvliet RK, et al. Deprescribing potentially inappropriate preventive cardiovascular medication: Barriers and enablers for patients and general practitioners. Ann Pharmacother 2016 Mar 3. [Epub ahead of print]
79. Hoffmann TC, Del Mar C. Patients’ expectations of the benefits and harms of treatments, screening, and tests: a systematic review. JAMA Intern Med 2015;175:274–86.
80. Martin P, Tamblyn R, Ahmed S, Tannenbaum C. A drug education tool developed for older adults changes knowledge, beliefs and risk perceptions about inappropriate benzodiazepine prescriptions in the elderly. Patient Educ Couns 2013;92:81–7.
81. Hamilton H, Gallagher P, Ryan C, et al. Potentially inappropriate medicines defined by STOPP criteria and the risk of adverse drug events in older hospitalized patients. Arch Intern Med 2011;171:1013–7.
82. NHS Highland. Polypharmacy: guidance for prescribing in frail adults. Accessed at: www.nhshighland.scot.nhs.uk/publications/documents/guidelines/polypharmacy guidance for prescribing in frail adults.pdf.
83. Anderson K, Foster MM, Freeman CR, Scott IA. A multifaceted intervention to reduce inappropriate polypharmacy in primary care: research co-creation opportunities in a pilot study. Med J Aust 2016;204:S41–4.
84. A practical guide to stopping medicines in older people. Accessed at: www.bpac.org.nz/magazine/2010/april/stopGuide.asp.
85. www.cpsedu.com.au/posts/view/46/Deprescribing-Documents-now-Available-for-Download.
86. Bregnhøj L, Thirstrup S, Kristensen MB, et al. Combined intervention programme reduces inappropriate prescribing in elderly patients exposed to polypharmacy in primary care. Eur J Clin Pharmacol 2009;65:199–207.