User login
FDA approves Oxbryta for sickle cell disease treatment
The Food and Drug Administration has approved voxelotor (Oxbryta) for adults and pediatric patients aged 12 years and older with sickle cell disease.

Approval was based on results from HOPE, a randomized, double-blind, placebo-controlled, multicenter trial of 274 patients with sickle cell disease (median age, 24 years) with a baseline hemoglobin level between 5.5 and 10.5 g/dL. Just over half of patients (51.1%) who received voxelotor at 1,500 mg had a hemoglobin increase of at least 1 g/dL over the 24-week study period, compared with 6.5% of patients who received placebo.
Patients in the 1,500-mg group also had reduced indirect bilirubin and percent reticulocyte count at –29.1% and –19.9%, respectively, compared with placebo, where the change was –3.2% and 4.5%, respectively.
The most common adverse events associated with voxelotor are headache, diarrhea, abdominal pain, nausea, rash, fatigue and pyrexia. The recommended voxelotor dose is 1,500 mg orally once daily with or without food, according to the FDA.
The Food and Drug Administration has approved voxelotor (Oxbryta) for adults and pediatric patients aged 12 years and older with sickle cell disease.
Approval was based on results from HOPE, a randomized, double-blind, placebo-controlled, multicenter trial of 274 patients with sickle cell disease (median age, 24 years) with a baseline hemoglobin level between 5.5 and 10.5 g/dL. Just over half of patients (51.1%) who received voxelotor at 1,500 mg had a hemoglobin increase of at least 1 g/dL over the 24-week study period, compared with 6.5% of patients who received placebo.
Patients in the 1,500-mg group also had reduced indirect bilirubin and percent reticulocyte count at –29.1% and –19.9%, respectively, compared with placebo, where the change was –3.2% and 4.5%, respectively.
The most common adverse events associated with voxelotor are headache, diarrhea, abdominal pain, nausea, rash, fatigue and pyrexia. The recommended voxelotor dose is 1,500 mg orally once daily with or without food, according to the FDA.
The Food and Drug Administration has approved voxelotor (Oxbryta) for adults and pediatric patients aged 12 years and older with sickle cell disease.
Approval was based on results from HOPE, a randomized, double-blind, placebo-controlled, multicenter trial of 274 patients with sickle cell disease (median age, 24 years) with a baseline hemoglobin level between 5.5 and 10.5 g/dL. Just over half of patients (51.1%) who received voxelotor at 1,500 mg had a hemoglobin increase of at least 1 g/dL over the 24-week study period, compared with 6.5% of patients who received placebo.
Patients in the 1,500-mg group also had reduced indirect bilirubin and percent reticulocyte count at –29.1% and –19.9%, respectively, compared with placebo, where the change was –3.2% and 4.5%, respectively.
The most common adverse events associated with voxelotor are headache, diarrhea, abdominal pain, nausea, rash, fatigue and pyrexia. The recommended voxelotor dose is 1,500 mg orally once daily with or without food, according to the FDA.
FDA approves Givlaari for treatment of acute hepatic porphyria
The Food and Drug Administration has approved givosiran (Givlaari) for the treatment of adult patients with acute hepatic porphyria, a genetic disorder that causes buildup of porphyrin molecules.

“This buildup can cause acute attacks, known as porphyria attacks, which can lead to severe pain and paralysis, respiratory failure, seizures, and mental status changes. These attacks occur suddenly and can produce permanent neurological damage and death. Prior to today’s approval, treatment options have only provided partial relief from the intense unremitting pain that characterizes these attacks,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
Approval for givosiran is based on results from a clinical trial of 94 patients with acute hepatic porphyria. Patients who received givosiran experienced 70% fewer porphyria attacks that required hospitalization, urgent health care visits, or home intravenous hemin injections compared with patients who received a placebo.
The most common adverse events associated with givosiran were nausea and injection site reactions. Patients receiving the medication should be monitored for anaphylactic reaction and renal function, and liver function should be tested before and periodically during treatment.
“The drug approved today can treat this disease by helping to reduce the number of attacks that disrupt the lives of patients,” said Dr. Pazdur, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
The Food and Drug Administration has approved givosiran (Givlaari) for the treatment of adult patients with acute hepatic porphyria, a genetic disorder that causes buildup of porphyrin molecules.
“This buildup can cause acute attacks, known as porphyria attacks, which can lead to severe pain and paralysis, respiratory failure, seizures, and mental status changes. These attacks occur suddenly and can produce permanent neurological damage and death. Prior to today’s approval, treatment options have only provided partial relief from the intense unremitting pain that characterizes these attacks,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
Approval for givosiran is based on results from a clinical trial of 94 patients with acute hepatic porphyria. Patients who received givosiran experienced 70% fewer porphyria attacks that required hospitalization, urgent health care visits, or home intravenous hemin injections compared with patients who received a placebo.
The most common adverse events associated with givosiran were nausea and injection site reactions. Patients receiving the medication should be monitored for anaphylactic reaction and renal function, and liver function should be tested before and periodically during treatment.
“The drug approved today can treat this disease by helping to reduce the number of attacks that disrupt the lives of patients,” said Dr. Pazdur, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
The Food and Drug Administration has approved givosiran (Givlaari) for the treatment of adult patients with acute hepatic porphyria, a genetic disorder that causes buildup of porphyrin molecules.
“This buildup can cause acute attacks, known as porphyria attacks, which can lead to severe pain and paralysis, respiratory failure, seizures, and mental status changes. These attacks occur suddenly and can produce permanent neurological damage and death. Prior to today’s approval, treatment options have only provided partial relief from the intense unremitting pain that characterizes these attacks,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
Approval for givosiran is based on results from a clinical trial of 94 patients with acute hepatic porphyria. Patients who received givosiran experienced 70% fewer porphyria attacks that required hospitalization, urgent health care visits, or home intravenous hemin injections compared with patients who received a placebo.
The most common adverse events associated with givosiran were nausea and injection site reactions. Patients receiving the medication should be monitored for anaphylactic reaction and renal function, and liver function should be tested before and periodically during treatment.
“The drug approved today can treat this disease by helping to reduce the number of attacks that disrupt the lives of patients,” said Dr. Pazdur, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
FDA approves treatment for sickle cell pain crises
The Food and Drug Administration has approved crizanlizumab-tmca (Adakveo) to reduce the frequency of vaso-occlusive crisis, a common complication of sickle cell disease.

The drug is approved for patients aged 16 years and older. It was approved on the strength of the SUSTAIN trial, which randomized 198 patients with sickle cell disease and a history of vaso-occlusive crisis to crizanlizumab or placebo. Patients who received crizanlizumab had a median annual rate of 1.63 health care visits for vaso-occlusive crises, compared with patients who received placebo and had a median annual rate of 2.98 visits. The drug also delayed the first vaso-occlusive crisis after starting treatment from 1.4 months to 4.1 months, according to the FDA.
“Adakveo is the first targeted therapy approved for sickle cell disease, specifically inhibiting selectin, a substance that contributes to cells sticking together and leads to vaso-occlusive crisis,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement. “Vaso-occlusive crisis can be extremely painful and is a frequent reason for emergency department visits and hospitalization for patients with sickle cell disease.”
Common adverse events associated with crizanlizumab included back pain, nausea, pyrexia, and arthralgia. The FDA advised physicians to monitor patients for infusion-related reactions.
The Food and Drug Administration has approved crizanlizumab-tmca (Adakveo) to reduce the frequency of vaso-occlusive crisis, a common complication of sickle cell disease.
The drug is approved for patients aged 16 years and older. It was approved on the strength of the SUSTAIN trial, which randomized 198 patients with sickle cell disease and a history of vaso-occlusive crisis to crizanlizumab or placebo. Patients who received crizanlizumab had a median annual rate of 1.63 health care visits for vaso-occlusive crises, compared with patients who received placebo and had a median annual rate of 2.98 visits. The drug also delayed the first vaso-occlusive crisis after starting treatment from 1.4 months to 4.1 months, according to the FDA.
“Adakveo is the first targeted therapy approved for sickle cell disease, specifically inhibiting selectin, a substance that contributes to cells sticking together and leads to vaso-occlusive crisis,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement. “Vaso-occlusive crisis can be extremely painful and is a frequent reason for emergency department visits and hospitalization for patients with sickle cell disease.”
Common adverse events associated with crizanlizumab included back pain, nausea, pyrexia, and arthralgia. The FDA advised physicians to monitor patients for infusion-related reactions.
The Food and Drug Administration has approved crizanlizumab-tmca (Adakveo) to reduce the frequency of vaso-occlusive crisis, a common complication of sickle cell disease.
The drug is approved for patients aged 16 years and older. It was approved on the strength of the SUSTAIN trial, which randomized 198 patients with sickle cell disease and a history of vaso-occlusive crisis to crizanlizumab or placebo. Patients who received crizanlizumab had a median annual rate of 1.63 health care visits for vaso-occlusive crises, compared with patients who received placebo and had a median annual rate of 2.98 visits. The drug also delayed the first vaso-occlusive crisis after starting treatment from 1.4 months to 4.1 months, according to the FDA.
“Adakveo is the first targeted therapy approved for sickle cell disease, specifically inhibiting selectin, a substance that contributes to cells sticking together and leads to vaso-occlusive crisis,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement. “Vaso-occlusive crisis can be extremely painful and is a frequent reason for emergency department visits and hospitalization for patients with sickle cell disease.”
Common adverse events associated with crizanlizumab included back pain, nausea, pyrexia, and arthralgia. The FDA advised physicians to monitor patients for infusion-related reactions.
FDA approves anemia treatment for transfusion-dependent beta thalassemia patients
The Food and Drug Administration has approved the first treatment for anemia in adults with transfusion-dependent beta thalassemia.
Luspatercept-aamt (Reblozyl) is an erythroid maturation agent that reduced the transfusion burden for patients with beta thalassemia in the BELIEVE trial of 336 patients. In total, 21% of patients who received luspatercept-aamt achieved at least a 33% reduction in red blood cell transfusions, compared with 4.5% of patients who received placebo, according to the FDA.
Common side effects associated with luspatercept-aamt were headache, bone pain, arthralgia, fatigue, cough, abdominal pain, diarrhea, and dizziness. Patients taking the agent should be monitored for thrombosis, the FDA advised.
Celgene, which makes luspatercept-aamt, said the agent would be available about 1 week following the FDA approval.
The FDA is also evaluating luspatercept-aamt as an anemia treatment in adults with very-low– to intermediate-risk myelodysplastic syndromes who have ring sideroblasts and require red blood cell transfusions. The agency is expected to take action on that application in April 2020.
The Food and Drug Administration has approved the first treatment for anemia in adults with transfusion-dependent beta thalassemia.
Luspatercept-aamt (Reblozyl) is an erythroid maturation agent that reduced the transfusion burden for patients with beta thalassemia in the BELIEVE trial of 336 patients. In total, 21% of patients who received luspatercept-aamt achieved at least a 33% reduction in red blood cell transfusions, compared with 4.5% of patients who received placebo, according to the FDA.
Common side effects associated with luspatercept-aamt were headache, bone pain, arthralgia, fatigue, cough, abdominal pain, diarrhea, and dizziness. Patients taking the agent should be monitored for thrombosis, the FDA advised.
Celgene, which makes luspatercept-aamt, said the agent would be available about 1 week following the FDA approval.
The FDA is also evaluating luspatercept-aamt as an anemia treatment in adults with very-low– to intermediate-risk myelodysplastic syndromes who have ring sideroblasts and require red blood cell transfusions. The agency is expected to take action on that application in April 2020.
The Food and Drug Administration has approved the first treatment for anemia in adults with transfusion-dependent beta thalassemia.
Luspatercept-aamt (Reblozyl) is an erythroid maturation agent that reduced the transfusion burden for patients with beta thalassemia in the BELIEVE trial of 336 patients. In total, 21% of patients who received luspatercept-aamt achieved at least a 33% reduction in red blood cell transfusions, compared with 4.5% of patients who received placebo, according to the FDA.
Common side effects associated with luspatercept-aamt were headache, bone pain, arthralgia, fatigue, cough, abdominal pain, diarrhea, and dizziness. Patients taking the agent should be monitored for thrombosis, the FDA advised.
Celgene, which makes luspatercept-aamt, said the agent would be available about 1 week following the FDA approval.
The FDA is also evaluating luspatercept-aamt as an anemia treatment in adults with very-low– to intermediate-risk myelodysplastic syndromes who have ring sideroblasts and require red blood cell transfusions. The agency is expected to take action on that application in April 2020.
Best practice alerts really can work
SAN ANTONIO – Clinicians don’t appear to mind too much when their red blood cell orders are flagged for review by a best practice alert system, and alert fatigue doesn’t seem to hamper patient blood management efforts, investigators in a single-center study reported.
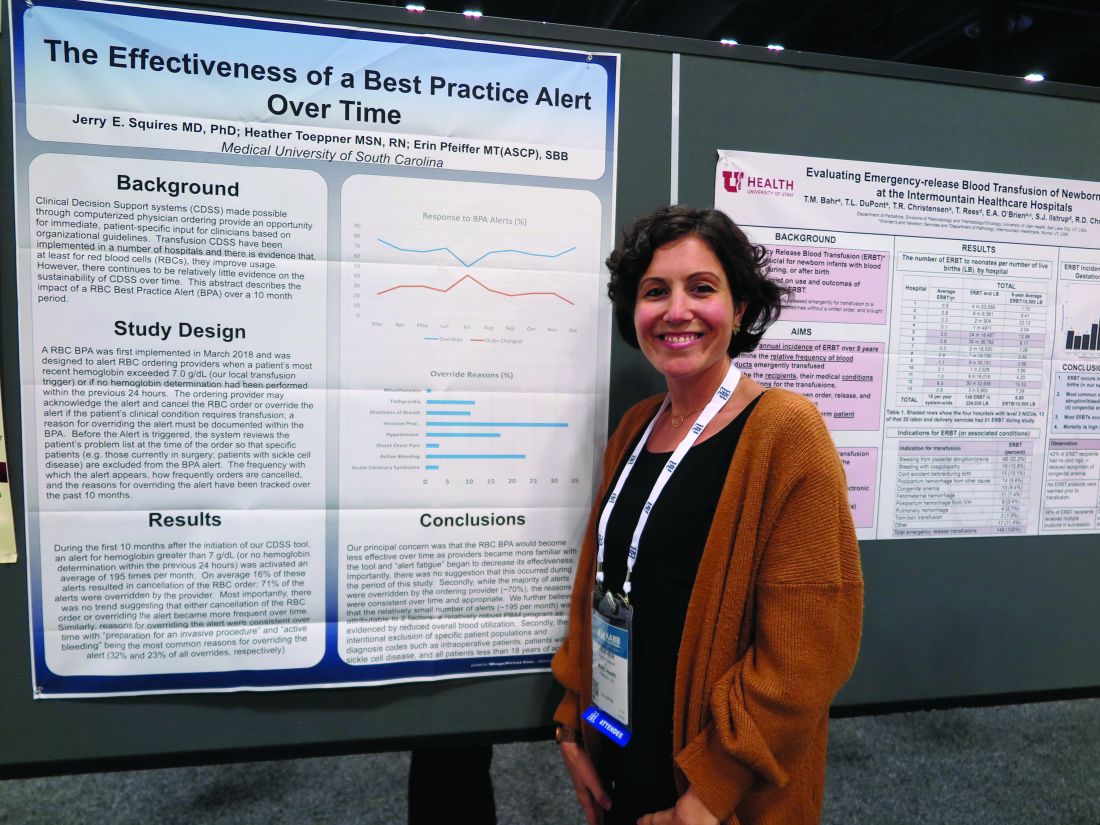
At the Medical University of South Carolina, Charleston (MUSC), if clinicians order RBC transfusions for patients with hemoglobin levels over 7.0 g/dL or for patients who did not have a hemoglobin determination over the past 24 hours, they receive a best practice alert. They must acknowledge it and cancel the order, or override it and document a reason in the medical record.
Although approximately 70% of alerts were overridden, the reasons for the overrides “were consistent over time and appropriate,” reported Jerry E. Squires, MD, PhD, and colleagues from MUSC in a poster presentation at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The goal of the study was to find out if the effectiveness of the alert was wearing out after months of active use by clinicians. “Is it true that they’re clicking too much and they’re inundated with other [best practice alerts], and are they even paying attention?” said coauthor Heather Toeppner, RN, also from MUSC, in an interview. “All in all, we found that the alert is making a lasting impression in our institution,” she said.
Transfusion clinical decision support systems that produce automated alerts for clinicians can improve usage and reduce waste of RBCs, but whether the effect is sustained over time was unknown, Ms. Toeppner said, prompting the investigators to study the effect of the RBC best practice alert over 10 months.
As noted, the alert is triggered when providers order RBCs for patients with hemoglobin levels over 7.0 g/dL or when there is no record of a hemoglobin test in the chart within the past 24 hours. Before the alert is triggered, however, the system reviews the record and excludes alerts for patients with specific conditions, such as concurrent surgery or sickle cell disease.
The authors found that the alert was triggered an average of 195 times per month over the 10 months studied. On average, 16% of the alerts resulted in a cancellation of the RBC order, and 71% of alerts were overridden.
“Most importantly, there was no trend suggesting that either cancellation of the RBC order or overriding the alert became more frequent over time,” the investigators wrote. “Similarly, reasons for overriding the alert were consistent over time, with ‘preparation for an invasive procedure’ and ‘active bleeding’ being the most common reasons for overriding the alert (32% and 23% of all overrides, respectively).”
Other common reasons for overrides included tachycardia, shortness of breath, hypotension, onset of chest pain, and acute coronary syndrome.
Interestingly, but perhaps not surprisingly, they found that overrides dropped sharply and changed orders rose by the same magnitude in July, when new residents started their rotations.
The investigators wrote that the relatively small number of alerts may be attributable to their institution’s robust patient blood management program and the intentional exclusion of orders for patients with specific diagnostic codes, including intraoperative patients, those with sickle cell disease, and all patients aged younger than 18 years.
The study was internally funded. The authors reported having no conflicts of interest.
SAN ANTONIO – Clinicians don’t appear to mind too much when their red blood cell orders are flagged for review by a best practice alert system, and alert fatigue doesn’t seem to hamper patient blood management efforts, investigators in a single-center study reported.
At the Medical University of South Carolina, Charleston (MUSC), if clinicians order RBC transfusions for patients with hemoglobin levels over 7.0 g/dL or for patients who did not have a hemoglobin determination over the past 24 hours, they receive a best practice alert. They must acknowledge it and cancel the order, or override it and document a reason in the medical record.
Although approximately 70% of alerts were overridden, the reasons for the overrides “were consistent over time and appropriate,” reported Jerry E. Squires, MD, PhD, and colleagues from MUSC in a poster presentation at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The goal of the study was to find out if the effectiveness of the alert was wearing out after months of active use by clinicians. “Is it true that they’re clicking too much and they’re inundated with other [best practice alerts], and are they even paying attention?” said coauthor Heather Toeppner, RN, also from MUSC, in an interview. “All in all, we found that the alert is making a lasting impression in our institution,” she said.
Transfusion clinical decision support systems that produce automated alerts for clinicians can improve usage and reduce waste of RBCs, but whether the effect is sustained over time was unknown, Ms. Toeppner said, prompting the investigators to study the effect of the RBC best practice alert over 10 months.
As noted, the alert is triggered when providers order RBCs for patients with hemoglobin levels over 7.0 g/dL or when there is no record of a hemoglobin test in the chart within the past 24 hours. Before the alert is triggered, however, the system reviews the record and excludes alerts for patients with specific conditions, such as concurrent surgery or sickle cell disease.
The authors found that the alert was triggered an average of 195 times per month over the 10 months studied. On average, 16% of the alerts resulted in a cancellation of the RBC order, and 71% of alerts were overridden.
“Most importantly, there was no trend suggesting that either cancellation of the RBC order or overriding the alert became more frequent over time,” the investigators wrote. “Similarly, reasons for overriding the alert were consistent over time, with ‘preparation for an invasive procedure’ and ‘active bleeding’ being the most common reasons for overriding the alert (32% and 23% of all overrides, respectively).”
Other common reasons for overrides included tachycardia, shortness of breath, hypotension, onset of chest pain, and acute coronary syndrome.
Interestingly, but perhaps not surprisingly, they found that overrides dropped sharply and changed orders rose by the same magnitude in July, when new residents started their rotations.
The investigators wrote that the relatively small number of alerts may be attributable to their institution’s robust patient blood management program and the intentional exclusion of orders for patients with specific diagnostic codes, including intraoperative patients, those with sickle cell disease, and all patients aged younger than 18 years.
The study was internally funded. The authors reported having no conflicts of interest.
SAN ANTONIO – Clinicians don’t appear to mind too much when their red blood cell orders are flagged for review by a best practice alert system, and alert fatigue doesn’t seem to hamper patient blood management efforts, investigators in a single-center study reported.
At the Medical University of South Carolina, Charleston (MUSC), if clinicians order RBC transfusions for patients with hemoglobin levels over 7.0 g/dL or for patients who did not have a hemoglobin determination over the past 24 hours, they receive a best practice alert. They must acknowledge it and cancel the order, or override it and document a reason in the medical record.
Although approximately 70% of alerts were overridden, the reasons for the overrides “were consistent over time and appropriate,” reported Jerry E. Squires, MD, PhD, and colleagues from MUSC in a poster presentation at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The goal of the study was to find out if the effectiveness of the alert was wearing out after months of active use by clinicians. “Is it true that they’re clicking too much and they’re inundated with other [best practice alerts], and are they even paying attention?” said coauthor Heather Toeppner, RN, also from MUSC, in an interview. “All in all, we found that the alert is making a lasting impression in our institution,” she said.
Transfusion clinical decision support systems that produce automated alerts for clinicians can improve usage and reduce waste of RBCs, but whether the effect is sustained over time was unknown, Ms. Toeppner said, prompting the investigators to study the effect of the RBC best practice alert over 10 months.
As noted, the alert is triggered when providers order RBCs for patients with hemoglobin levels over 7.0 g/dL or when there is no record of a hemoglobin test in the chart within the past 24 hours. Before the alert is triggered, however, the system reviews the record and excludes alerts for patients with specific conditions, such as concurrent surgery or sickle cell disease.
The authors found that the alert was triggered an average of 195 times per month over the 10 months studied. On average, 16% of the alerts resulted in a cancellation of the RBC order, and 71% of alerts were overridden.
“Most importantly, there was no trend suggesting that either cancellation of the RBC order or overriding the alert became more frequent over time,” the investigators wrote. “Similarly, reasons for overriding the alert were consistent over time, with ‘preparation for an invasive procedure’ and ‘active bleeding’ being the most common reasons for overriding the alert (32% and 23% of all overrides, respectively).”
Other common reasons for overrides included tachycardia, shortness of breath, hypotension, onset of chest pain, and acute coronary syndrome.
Interestingly, but perhaps not surprisingly, they found that overrides dropped sharply and changed orders rose by the same magnitude in July, when new residents started their rotations.
The investigators wrote that the relatively small number of alerts may be attributable to their institution’s robust patient blood management program and the intentional exclusion of orders for patients with specific diagnostic codes, including intraoperative patients, those with sickle cell disease, and all patients aged younger than 18 years.
The study was internally funded. The authors reported having no conflicts of interest.
REPORTING FROM AABB 2019
NIH seeks gene-based cures for HIV, sickle cell disease
The National Institutes of Health and the Bill & Melinda Gates Foundation have announced that they plan to invest $100 million each over the next 4 years to develop affordable, gene-based cures for sickle cell disease (SCD) and HIV.
The initiative follows an announcement from President Trump that set a goal of ending the HIV epidemic in the United States in the next 10 years, seeking to reduce the number of diagnoses by 90% by 2030. The Trump administration has also identified SCD as an “intractable health challenge with the potential for dramatic advances in the coming years,” the NIH said in a statement.
Gene-based therapy has become a reality in recent years thanks to dramatic advances, but the cost is prohibitive in many parts of the world. “The collaboration between the NIH and the Gates Foundation sets out a bold goal of advancing safe, effective, and durable gene-based cures to clinical trials in the United States and relevant countries in sub-Saharan Africa within the next 7-10 years. The ultimate goal is to scale and implement these treatments globally in areas hardest hit by these diseases,” the NIH said.
Both diseases are a significant burden on low- and middle-income countries, as 95% of the 38 million people living with HIV globally are in the developing world, with 67% living in sub-Saharan Africa; about half of the HIV-infected population receives no treatment for the disease. An estimated 15 million children will be born with SCD over the next 30 years, with three-quarters of those births occurring in sub-Saharan Africa. About 50%-90% of children born with SCD will die before age 5 years.
The collaboration will focus on coordination in two areas: identifying potential candidate cures for SCD and HIV for preclinical and clinical evaluation, and defining long-term opportunities to work together and with African partners on advancing promising candidates to late-phase clinical trials, with funding to be determined as candidates progress.
“In recent years, gene-based treatments have been groundbreaking for rare genetic disorders and infectious diseases. While these treatments are exciting, people in low- and middle-income countries do not have access to these breakthroughs. By working with the NIH and scientists across Africa, we aim to ensure these approaches will improve the lives of those most in need and bring the incredible promise of gene-based treatments to the world of public health,” said Trevor Mundel, MD, PhD, president of the global health program at the Bill & Melinda Gates Foundation.
The National Institutes of Health and the Bill & Melinda Gates Foundation have announced that they plan to invest $100 million each over the next 4 years to develop affordable, gene-based cures for sickle cell disease (SCD) and HIV.
The initiative follows an announcement from President Trump that set a goal of ending the HIV epidemic in the United States in the next 10 years, seeking to reduce the number of diagnoses by 90% by 2030. The Trump administration has also identified SCD as an “intractable health challenge with the potential for dramatic advances in the coming years,” the NIH said in a statement.
Gene-based therapy has become a reality in recent years thanks to dramatic advances, but the cost is prohibitive in many parts of the world. “The collaboration between the NIH and the Gates Foundation sets out a bold goal of advancing safe, effective, and durable gene-based cures to clinical trials in the United States and relevant countries in sub-Saharan Africa within the next 7-10 years. The ultimate goal is to scale and implement these treatments globally in areas hardest hit by these diseases,” the NIH said.
Both diseases are a significant burden on low- and middle-income countries, as 95% of the 38 million people living with HIV globally are in the developing world, with 67% living in sub-Saharan Africa; about half of the HIV-infected population receives no treatment for the disease. An estimated 15 million children will be born with SCD over the next 30 years, with three-quarters of those births occurring in sub-Saharan Africa. About 50%-90% of children born with SCD will die before age 5 years.
The collaboration will focus on coordination in two areas: identifying potential candidate cures for SCD and HIV for preclinical and clinical evaluation, and defining long-term opportunities to work together and with African partners on advancing promising candidates to late-phase clinical trials, with funding to be determined as candidates progress.
“In recent years, gene-based treatments have been groundbreaking for rare genetic disorders and infectious diseases. While these treatments are exciting, people in low- and middle-income countries do not have access to these breakthroughs. By working with the NIH and scientists across Africa, we aim to ensure these approaches will improve the lives of those most in need and bring the incredible promise of gene-based treatments to the world of public health,” said Trevor Mundel, MD, PhD, president of the global health program at the Bill & Melinda Gates Foundation.
The National Institutes of Health and the Bill & Melinda Gates Foundation have announced that they plan to invest $100 million each over the next 4 years to develop affordable, gene-based cures for sickle cell disease (SCD) and HIV.
The initiative follows an announcement from President Trump that set a goal of ending the HIV epidemic in the United States in the next 10 years, seeking to reduce the number of diagnoses by 90% by 2030. The Trump administration has also identified SCD as an “intractable health challenge with the potential for dramatic advances in the coming years,” the NIH said in a statement.
Gene-based therapy has become a reality in recent years thanks to dramatic advances, but the cost is prohibitive in many parts of the world. “The collaboration between the NIH and the Gates Foundation sets out a bold goal of advancing safe, effective, and durable gene-based cures to clinical trials in the United States and relevant countries in sub-Saharan Africa within the next 7-10 years. The ultimate goal is to scale and implement these treatments globally in areas hardest hit by these diseases,” the NIH said.
Both diseases are a significant burden on low- and middle-income countries, as 95% of the 38 million people living with HIV globally are in the developing world, with 67% living in sub-Saharan Africa; about half of the HIV-infected population receives no treatment for the disease. An estimated 15 million children will be born with SCD over the next 30 years, with three-quarters of those births occurring in sub-Saharan Africa. About 50%-90% of children born with SCD will die before age 5 years.
The collaboration will focus on coordination in two areas: identifying potential candidate cures for SCD and HIV for preclinical and clinical evaluation, and defining long-term opportunities to work together and with African partners on advancing promising candidates to late-phase clinical trials, with funding to be determined as candidates progress.
“In recent years, gene-based treatments have been groundbreaking for rare genetic disorders and infectious diseases. While these treatments are exciting, people in low- and middle-income countries do not have access to these breakthroughs. By working with the NIH and scientists across Africa, we aim to ensure these approaches will improve the lives of those most in need and bring the incredible promise of gene-based treatments to the world of public health,” said Trevor Mundel, MD, PhD, president of the global health program at the Bill & Melinda Gates Foundation.
Autoimmune Hemolytic Anemia: Treatment of Common Types
Autoimmune hemolytic anemia (AIHA) is mediated by antibodies, and in most cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. AIHA caused by a drug reaction is rare, with an estimated annual incidence of 1:1,000,000 for severe drug-related AIHA.1 This article reviews the management of the more common types of AIHA, with a focus on warm, cold, and drug-induced AIHA; the evaluation and diagnosis of AIHA is reviewed in a separate article.
Warm Autoimmune Hemolytic Anemia
In AIHA, hemolysis is mediated by antibodies that bind to the surface of red blood cells. AIHA in which IgG antibodies are the offending antibodies is referred to as warm AIHA. “Warm” refers to the fact that the antibody binds best at body temperature (37°C). In warm AIHA, testing will show IgG molecules attached to the surface of the red cells, with 50% of patients also showing C3. Between 50% and 90% of AIHA cases are due to warm antibodies.2,3 The incidence of warm AIHA varies by series but is approximately 1 case per 100,000 patients per year; this form of hemolysis affects women more frequently than men.4,5
Therapeutic Options
First Line
Steroids. The goal of therapy in warm AIHA can be hard to define. However, most would agree that a hematocrit above 30% (or higher to prevent symptoms) with a minimal increase in the reticulocyte count—reflective of a significantly slowed hemolytic process—is a reasonable goal. Initial management of warm AIHA is prednisone at a standard dose of 1 mg/kg daily (Table 1).6,7 Patients should be also started on proton-pump inhibitors to prevent ulcers. It can take up to 3 weeks for patients to respond to prednisone therapy. Once the patient’s hematocrit is above 30%, the prednisone is slowly tapered. Although approximately 80% of patients will respond to steroids, only 30% can be fully tapered off steroids. For patients who can be maintained on a daily steroid dose of 10 mg or less, steroids may be the most reasonable long-term therapy. In addition, because active hemolysis leads to an increased demand for folic acid, patients with warm AIHA are often prescribed folic acid 1 mg daily to prevent megaloblastic anemia due to folic acid deficiency.

Rituximab. Increasingly, rituximab (anti-CD20) therapy is added to the initial steroids. Two clinical trials showed both increased long-term and short-term responses with the use of rituximab.8,9 An important consideration is that most patients respond gradually to rituximab over weeks, so a rapid response should not be expected. Most studies have used the traditional dosing of 375 mg/m2 weekly for 4 weeks. These responses appear to be durable, but as in immune thrombocytopenia (ITP), repeat treatment with rituximab is effective.
The major side effects of rituximab are infusion reactions, which are often worse with the first dose. These reactions can be controlled with antihistamines, steroids, and, for severe rigors, meperidine. Rarely, patients can develop neutropenia (approximately 1:500) that appears to be autoimmune in nature. Infections appear to be only minimally increased with the use of rituximab.10 One group at risk is chronic carriers of hepatitis B virus, who may experience a reactivation of the virus that can be fatal. Thus, patients being considered for rituximab need to be screened for hepatitis B virus carrier state.11 Patients receiving rituximab are at very slight risk for progressive multifocal leukoencephalopathy, which is more common in patients with cancer and in heavily immunosuppressed patients. The overall risk is unknown but is less than 1:50,000.
Second Line
Splenectomy. For patients who cannot be weaned from steroids or in whom steroid therapy fails, there is no standard therapy. Currently, the 2 main choices are splenectomy or rituximab (anti-CD20) therapy if the patient did not receive it first line. Splenectomy is the classic therapy for warm AIHA. Reported response rates in the literature range from 50% to 80%, with 50% to 60% remaining in remission.12-16 Timing of the procedure is a balance between allowing time for the steroids to work and the risk of toxicity of steroids. In a patient who has low presurgical risk and has either refractory disease or cannot be weaned from high doses of steroids, splenectomy should be done sooner. Splenectomy can be delayed or other therapy tried first in patients who require lower doses of steroids or have medical risk factors for surgery. Most splenectomies are performed via laparoscopy. The small incisions allow for quicker healing, and the laparoscopic approach provides better visualization of the abdomen to find and remove accessory spleens. When splenectomy is performed by experienced surgeons, the mortality rate is low (< 0.5%).17
The most concerning complication of splenectomy is overwhelming post-splenectomy infection (OPSI).18 In adults, the spleen appears to play a minimal role in immunity except for protecting against certain encap-sulated organisms. Splenectomized patients infected with an encapsulated organism (eg, Pneumococcus) will develop overwhelming sepsis within hours. These patients will often have disseminated intravascular coagulation and will rapidly progress to purpura fulminans. Approximately 40% to 50% of patients will die of sepsis even when the infection is detected early. The overall lifetime risk of sepsis may be as high as 1:500. The organism that most commonly causes sepsis in splenectomized patients is Streptococcus pneumoniae, reported in over 50% of cases. Neisseria meningitidis and Haemophilus influenzae have also been implicated in many cases.19 Overwhelming sepsis after dog bites has been reported due to Capnocytophaga canimorsus infections. Patients are also at increased risk of developing severe malaria and severe babesiosis.18
Patients who have undergone splenectomy need to be warned about the risk of OPSI and instructed to report to the emergency department readily if they develop a fever greater than 101°F (38.3°C) or shaking chills. Once in the emergency department, blood cultures should be obtained rapidly and the patient started on antibiotic coverage with vancomycin and ceftriaxone (or levofloxacin if allergic to beta-lactams).20 For patients in remote areas, some physicians will prescribe prophylactic antibiotics to take while they are traveling to a health care provider or even recommend a “standby” antibiotic dose to take while traveling to health care.5 This usually consists of amoxicillin or a macrolide for penicillin-allergic patients.
Patients in whom splenectomy is being planned or considered should be vaccinated for pneumococcal, meningococcal, and influenza infections (Table 2).18 If there is a plan to treat with rituximab, patients should first be vaccinated since they will not be able to mount an immune response after being treated with rituximab.

Third Line
The therapeutic options for patients who do not respond to either splenectomy or rituximab are much less certain.5,6 Although intravenous immune globulin is a standard therapy for ITP, response rates are low in warm AIHA.17 Numerous therapies have been reported in small series, but no clear approach has emerged. Options include azathioprine, cyclophosphamide, mycophenolate, cyclosporine, danazol, and alemtuzumab. Our approach has been to use mycophenolate for patients requiring high doses of steroids or transfusions. Patients who respond to lower doses of steroids may be good candidates for danazol to help wean them off steroids.
Treatment of Warm AIHA with Associated Diseases
Warm AIHA can complicate several diseases. Patients with systemic lupus erythematosus (SLE) can develop warm AIHA as part of their disease complex. The initial treatment approach is the same, but data suggest that splenectomy may not be as effective.13,17 Also, many SLE patients have complex medical conditions, making surgeries riskier. For SLE patients who are refractory or cannot be weaned from steroids, rituximab may be the better choice. Babesiosis, particularly in asplenic patients, has been associated with the development of AIHA.21,22
Of the malignances associated with AIHA, chronic lymphocytic leukemia (CLL) has the strongest association.4,23 Series show that 5% to 10% of patients with CLL will have warm AIHA. AIHA can appear concurrent with CLL or develop during the course of the disease. The introduction of purine analogs such as fludarabine led to a dramatic increase in the incidence of warm AIHA in treated patients.24 It is speculated that these powerful agents reduce the number and effectiveness of T cells that hold in check the autoantibody response, leading to warm AIHA.25 However, when these purine analogs are used in com-bination with agents such as cyclophosphamide or rituximab (with their immunosuppressive effects), the rates of warm AIHA have been lower.23
The approach to patients with CLL and warm AIHA depends on the state of their CLL.23 For patients who have low-stage CLL that does not need treatment, the standard approach to warm AIHA should be steroids, splenectomy, and rituximab.24 For patients with higher-stage CLL, the treatment for the leukemia will often provide therapy for the warm AIHA. The combination of rituximab-cyclophosphamide-dexamethasone has been reported to be effective for both the AIHA and CLL components.26 The use of ibrutinib has also been reported to be effective.27
A rare but important variant of warm AIHA is Evans syndrome.28,29 This is the combination of AIHA and ITP. Approximately 1% to 3% of AIHA cases are the Evans variant. The ITP can precede, be concurrent with, or develop after the AIHA. The diagnosis of Evans syndrome should raise concern for underlying disorders. In young adults, immunodeficiency disorders such as autoimmune lymphoproliferative disease (ALPS) need to be considered. In older patients, Evans syndrome is often associated with T cell lymphomas. The sparse literature on Evans syndrome suggests that it can be more refractory to standard therapy, with response rates to splenectomy in the 50% range.28,30 In patients with lymphoma, antineoplastic therapy is crucial. There is increasing data showing that mycophenolate or sirolimus may be effective for patients with ALPS in whom splenectomy or rituximab therapy is unsuccessful.31
Warm AIHA with IgA or IgM Antibodies
In rare patients with warm AIHA, IgA or IgM is the implicated antibody. The literature suggests that patients with IgA AIHA may have more severe hemolysis.32 Patients with IgM AIHA often have a severe course with a fatal outcome.33 In such cases, the patient’s plasma may show spontaneous hemolysis and agglutination. The DAT may not be strongly positive or may show C3 reactivity only. The clinical clues are C3 reactivity with no cold agglutinins and severe hemolysis, sometimes with an intravascular component. Treatment is the same as for warm AIHA, including the use of rituximab.34
Cold Autoimmune Hemolytic Anemia
In cold AIHA, the hemolysis is mediated by IgM antibodies directed against red cells.35 As discussed earlier, the term “cold” refers to the fact that the antibody binds maximally at temperatures below 37°C. The most efficient temperature for binding is called the “thermal amplitude,” and, in theory, the higher the thermal amplitude, the more virulent the antibody. An antibody titer can be calculated at each reaction temperature from 4°C to 37°C by serial dilutions of the patient’s serum prior to incubating with reagent red cells. Rarely, the IgM can fix complement rapidly, leading to intravascular hemolysis. In most patients, complement is fixed through C3, and the C3-coated red cells are taken up by macrophages in the mononuclear phagocyte system, primarily in the liver.3
The DAT in patients with cold AIHA will show cells coated with C3. The blood smear will often show ag-glutination of the blood, and if the blood cools before being analyzed, the agglutination will interfere with the analysis. Titers of cold agglutinin can range from 1:1000 to over 1 million. The IgM autoantibodies are most often directed against the I/i antigens on the red blood cell membrane, with 90% against I antigen.35 The I antigen specificity is typical with primary cold agglutinin disease and after Mycoplasma infection. The i antigen specificity is most typical of Epstein-Barr virus and cytomegalovirus infections in children. In young patients, cold AIHA often occurs following an infection, including viral and Mycoplasma infections, and the course is self-limited.35,36 The hemolysis usually starts 2 to 3 weeks after the illness and will last for 4 to 6 weeks. In older patients, the etiology in over 90% of cases is a B-cell lymphoproliferative disorder, usually with monoclonal kappa B-cells.37 The most common disorders are marginal zone lymphoma, small lymphocytic lymphoma, and lymphoplasmacytic lymphoma.3
Therapeutic Options
It is important to diagnose cold AIHA because the standard therapy for warm AIHA (steroids) is ineffective in cold AIHA. Because C3-coated red cells are taken up primarily in the liver, removing the spleen is also an ineffective therapy. Simple measures to help with cold AIHA should be employed.37 Patients should be kept in a warm environment and should try to avoid the cold. If transfusions are needed, they should be infused via blood warmers to prevent hemolysis. In rare patients with severe hemolysis, therapeutic plasma exchange (TPE) can be considered.38 Given that the culprit antibody is IgM—mostly intravascular—use of TPE may slow the hemolysis to give time for other therapies to take effect.
Treatment of cold AIHA remains difficult (Table 3). Because most patients with primary AIHA have underlying lymphoproliferative diseases, chlorambucil has been used in the past to treat cold AIHA. However, responses were rare and the drug could worsen the anemia.38 Currently, the drug of choice is rituximab. Response is seen in 45% to 75% of patients, but is almost always a partial response and retreatment is often necessary.17,37,39 As with other autoimmune hematologic diseases, there can be a delay in response that ranges from 2 weeks to 4 months (median time, 1.5 months).37 Given the lack of robust response (complete and durable) with rituximab, the Berentsen group has explored adding bendamustine to rituximab.40 In a prospective trial, 71% of patients responded with a 40% complete response rate. Therapy was well tolerated, with only 29% of patients needing dose reduction Although more toxic, this combination can be considered in patients with aggressive disease. A small study of the use of bortezomib showed a good response in one-third of patients.41 There is increasing use of the C5 complement inhibitor eculizumab to halt the hemolysis, but further study of this agent is also required.36,42,43 Blockers of C1s complement, which block hemolysis by preventing complement activation, are currently being studied in clinical trials.44

Since most patients with cold AIHA are older, a frequent issue that must be considered is cardiac surgery. The concern is that the hypothermia involved with most heart bypass procedures will lead to agglutination and hemolysis. The development of normothermic bypass has expanded treatment options. A recommended approach in patients who have known cold agglutinins is to measure the thermal amplitude of the antibody preoperatively. If the thermal amplitude is above 18°C, normothermic bypass should be done, if feasible.45 If not feasible, preoperative TPE should be considered.
Paroxysmal Cold Hemoglobinuria
A unique cold AIHA is paroxysmal cold hemoglobinuria (PCH).3,46 This cold hemolytic syndrome most often occurs in children following a viral infection, but in the past it complicated any stage of syphilis.47 The mediating antibody in PCH is an IgG antibody directed against the P antigen on the red blood cell. This antibody binds best at temperatures below 37°C, fixing complement at cold temperatures, but then activates the complement cascade at body temperature.48 Because this antibody can fix complement, hemolysis can be rapid and severe, leading to extreme anemia. The DAT is often weakly positive but can be negative. The diagnostic test for PCH is the Donath–Landsteiner test. This complex test is performed by incubating 3 blood samples, 1 at 0° to 4°C, another at 37°C, and a third at 0° to 4°C and then at 37°C. The diagnosis of PCH is made if only the third tube shows hemolysis.35 PCH can persist for 1 to 3 months but is almost always self-limiting. In severe case, steroids may be of benefit.
Drug-Induced Hemolytic Anemia
AIHA caused by a drug reaction is rare, with a lower incidence than drug-related ITP. The rate of severe drug-related AIHA is estimated at 1:1,000,000, but less severe cases may be missed.1 Most patients will have a positive DAT without signs of hemolysis, but in rare cases patients will have relentless hemolysis resulting in death.
Mechanisms
Multiple mechanisms for drug-induced immune hemolysis have been proposed, including drug-absorption (hapten-induced) and immune complex mechanisms.1,49 The hapten mechanism is most often associated with the use of high-dose penicillin.50 High doses of penicillin or similar drugs such as piperacillin lead to incorporation of the drug into the red cell membrane by binding to proteins. Patients will manifest a positive DAT with IgG antibody but not complement.51 The patient’s plasma will be reactive only with penicillin-coated red cells and not with normal cells. As mentioned, very few patients will have hemolysis, and if they have hemolysis, it will resolve in a few days after discontinuation of the offending drug.52
Binding of a drug-antibody complex to the red cell membrane may cause hemolysis via the immune com-plex mechanism.53 Again, most often the patient will have just a positive DAT, but rarely patients will have life-threatening hemolysis upon exposure or reexposure to the drug. The onset of hemolysis is rapid, with signs of acute illness and intravascular hemolysis. The paradigm drug is quinine, but many other drugs have been implicated. Testing shows a positive Coombs test with anti-complement but not anti-IgG.50 This pattern is due to the effectiveness of the tertiary complex at fixing complement. The patient’s plasma reacts with red cells only when the drug is added.
A form of immune complex hemolysis associated with both disseminated intravascular coagulation (DIC) and brisk hemolysis has been recognized. Patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone.53,54) have developed this syndrome.50,55-59 The clinical symptoms start 7 to 10 days after the drug is administered; often the patient has only received the antibiotic for surgical prophylaxis. Immune hemolysis with acute hematocrit drop, hypotension, and DIC ensues. The patients are often believed to have sepsis and are often reexposed to the cephalosporin, resulting in worsening of the clinical status. The outcome is often fatal due to massive hemolysis and thrombosis.56,60,62
Finally, 8% to 36% of patients taking methyldopa will develop a positive DAT after 6 months of therapy, with less than 1% showing hemolysis.52,63 The hemolysis in these patients is indistinguishable from warm AIHA, consistent with the notion that methyldopa induces an AIHA. The hemolysis often resolves rapidly after the methyldopa is stopped, but the Coombs test may remain positive for months.63 This type of drug-induced hemolytic anemia has been reported with levodopa, procainamide, and chlorpromazine, but fludarabine is the most common cause currently. This form of AIHA is now being seen with increased use of checkpoint inhibitors.64
Diagnosis
In many patients, the first clue to the presence of drug AIHA is the finding of a positive DAT. Rarely, patients will have severe hemolysis, but in many patients the hemolytic process is mild and may be wrongly assumed to be part of the underlying illness. Finding the offending drug can be a challenge, unless a patient has recently started a new drug; in a hospitalized patient on multiple agents, identifying the problem drug is difficult. Patients recently started on “suspect drugs,” especially the most common agents cefotetan, ceftriaxone, and piperacillin, should have these agents stopped (Table 4).1,49,65 Specialty laboratories such as the Blood Center of Wisconsin or the Los Angeles Red Cross can perform in vitro studies of drug interactions that can confirm the clinical diagnosis of drug-induced AIHA.

Treatment
Therapy for patients with positive DAT without signs of hemolysis is uncertain. If the drug is essential, then the patient can be observed. If the patient has hemolysis, the drug needs to be stopped and the patient observed for signs of end-organ damage. It is doubtful that steroids or other autoimmune-directed therapy is effective. For patients with the DIC-hemolysis syndrome, there are anecdotal reports that TPE may be helpful.1
Summary
AIHA can range from an abnormal laboratory test (positive DAT and signs of hemolysis) to an acute, life-threatening illness. Treatment is guided by the laboratory work-up and evaluation of the patient’s clinical status. While rituximab is promising for many patients, the lack of robust clinical trials hinders the treatment of patients who fail standard therapies.
1. Garratty G. Immune hemolytic anemia associated with drug therapy. Blood Rev. 2010;24:143-150.
2. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
3. Berentsen S. Cold agglutinin disease. Hematology Am Soc Hematol Educ Program. 2016;2016:226-231.
4. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
5. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
6. Kyrle PA, Rosendaal FR, Eichinger S. Risk assessment for re¬current venous thrombosis. Lancet. 2010;376:2032-2039.
7. Go RS, Winters JL, Kay NE. How I treat autoimmune hemolytic anemia. Blood. 2017;129:2971-2979.
8. Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013;163:393-399
9. Michel M, Terriou L, Roudot-Thoraval F, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study). Am J Hematol. 2017;92:23-27.
10. Gea-Banacloche JC. Rituximab-associated infections. Semin Hematol. 2010;47:187-198.
11. Loomba R, Liang TJ. Hepatitis B reactivation associated with immune suppressive and biological modifier therapies: current concepts, management strategies, and future directions. Gastroenterology. 2017;152:1297-1309.
12. Coon WW. Splenectomy in the treatment of hemolytic anemia. Arch Surg. 1985;120:625-628.
13. Akpek G, McAneny D, Weintraub L. Comparative response to splenectomy in coombs-positive autoimmune hemolytic anemia with or without associated disease. Am J Hematol. 1999;61:98-102.
14. Patel NY, Chilsen AM, Mathiason MA, et al. Outcomes and complications after splenectomy for hematologic disorders. Am J Surg. 2012;204:1014-1020.
15. Crowther M, Chan YL, Garbett IK, et al. Evidence-based focused review of the treatment of idiopathic warm immune hemolytic anemia in adults. Blood. 2011;118:4036-4040.
16. Giudice V, Rosamilio R, Ferrara I, et al. Efficacy and safety of splenectomy in adult autoimmune hemolytic anemia. Open Med (Wars). 2016;11:374-380.
17. Lechner K, Jager U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010;116:1831-1838.
18. Rodeghiero F, Ruggeri M. Short- and long-term risks of splenectomy for benign haematological disorders: should we revisit the indications? Br J Haematol. 2012;158:16-29.
19. Ahmed N, Bialowas C, Kuo YH, Zawodniak L. Impact of preinjury anticoagulation in patients with traumatic brain injury. South Med J. 2009;102:476-480.
20. Morgan TL, Tomich EB. Overwhelming post-splenectomy infection (OPSI): a case report and review of the literature. J Emerg Med. 2012;43:758-763.
21. Woolley AE, Montgomery MW, Savage WJ, et al. Post-babesiosis warm autoimmune hemolytic anemia. N Engl J Med. 2017;376:939-946.
22. Shatzel JJ, Donohoe K, Chu NQ, et al. Profound autoimmune hemolysis and Evans syndrome in two asplenic patients with babesiosis. Transfusion. 2015;55:661-665.
23. Hodgson K, Ferrer G, Montserrat E, Moreno C. Chronic lymphocytic leukemia and autoimmunity: a systematic review. Haematologica. 2011;96:752-761.
24. Hamblin TJ. Autoimmune complications of chronic lymphocytic leukemia. Semin Oncol. 2006;33:230-239.
25. Tertian G, Cartron J, Bayle C, et al. Fatal intravascular au¬toimmune hemolytic anemia after fludarabine treatment for chronic lymphocytic leukemia. Hematol Cell Ther. 1996;38:359-360.
26. Rossignol J, Michallet AS, Oberic L, et al. Rituximab-cyclophosphamide-dexamethasone combination in the management of autoimmune cytopenias associated with chronic lymphocytic leukemia. Leukemia. 2011;25:473-478.
27. Hampel PJ, Larson MC, Kabat B, et al. Autoimmune cytopenias in patients with chronic lymphocytic leukaemia treated with ibrutinib in routine clinical practice at an academic medical centre. Br J Haematol. 2018;183:421-427.
28. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114:3167-3172.
29. Jaime-Pérez JC, Aguilar-Calderón PE, Salazar-Cavazos L, Gómez-Almaguer D. Evans syndrome: clinical perspectives, biological insights and treatment modalities. J Blood Med. 2018;9:171-184.
30. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology. 2008;13:356-360.
31. Bride KL, Vincent T, Smith-Whitley K, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016;127:17-28.
32. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell au¬toantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
33. Garratty G, Arndt P, Domen R, et al. Severe autoimmune hemolytic anemia associated with IgM warm autoantibodies directed against determinants on or associated with glycophorin A. Vox Sang. 1997;72:124-130.
34. Wakim M, Shah A, Arndt PA, et al. Successful anti-CD20 monoclonal antibody treatment of severe autoimmune hemolytic anemia due to warm reactive IgM autoantibody in a child with common variable immunodeficiency. Am J Hematol. 2004;76:152-155.
35. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
36. Berentsen S, Tjonnfjord GE. Diagnosis and treatment of cold agglutinin mediated autoimmune hemolytic anemia. Blood Rev. 2012;26:107-115.
37. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
38. King KE, Ness PM. Treatment of autoimmune hemolytic anemia. Semin Hematol. 2005;42:131136.
39. Barcellini W, Zaja F, Zaninoni A, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. 2012;119:3691-3697.
40. Berentsen S, Randen U, Oksman M, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. 2017;130:537-541.
41. Rossi G, Gramegna D, Paoloni F, et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: a phase 2 prospective GIMEMA study. Blood. 2018;132:547-550.
42. Makishima K, Obara N, Ishitsuka K, et al. High efficacy of eculizumab treatment for fulminant hemolytic anemia in primary cold agglutinin disease. Ann Hematol. 2019;98:1031-1032.
43. Roth A, Huttmann A, Rother RP, et al. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood. 2009;113:38853886.
44. Jäger U, D'Sa S, Schörgenhofer C, et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: a first-in-human trial. Blood. 2019;133:893-901.
45. Agarwal SK, Ghosh PK, Gupta D. Cardiac surgery and cold-reactive proteins. Ann Thorac Surg. 1995;60:1143-1150.
46. Shanbhag S, Spivak J. Paroxysmal cold hemoglobinuria. Hematol Oncol Clin North Am. 2015;29:473-478.
47. Kumar ND, Sethi S, Pandhi RK. Paroxysmal cold haemoglobinuria in syphilis patients. Genitourin Med. 1993;69:76.
48. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
49. Pierce A, Nester T. Pathology consultation on drug-induced hemolytic anemia. Am J Clin Pathol. 2011;136:7-12.
50. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Med Rev. 1993;7:255-267.
51. Petz LD, Mueller-Eckhardt C. Drug-induced immune hemolytic anemia. Transfusion. 1992;32:202-204.
52. Packman CH, Leddy JP. Drug-related immune hemolytic anemia. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ, eds. William’s Hematology. 5th ed. New York: McGraw-Hill; 1995:691-704.
53. Garratty G. Drug-induced immune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2009;73-79.
54. Leicht HB, Weinig E, Mayer B, et al. Ceftriaxone-induced hemolytic anemia with severe renal failure: a case report and review of literature. BMC Pharmacol Toxicol. 2018;19:67.
55. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis. 1992;15:863-865.
56. Garratty G, Nance S, Lloyd M, Domen R. Fatal im¬mune hemolytic anemia due to cefotetan. Transfusion. 1992;32:269-271.
57. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes: case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion. 1999;39:306-309.
58. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion. 1999;39:1239-1246.
59. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol. 2006;81:186-188.
60. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr. 1995;126(5 Pt 1):813-815.
61. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1995;14:1116-1117.
62. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone [see comments]. J Pediatr. 1995;126(5 Pt 1):816-817.
63. Petz LD. Drug-induced autoimmune hemolytic anemia. Transfusion Med Rev. 1993;7:242-254.
64. Leaf RK, Ferreri C, Rangachari D, et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am J Hematol. 2019;94:563-574.
65. DeLoughery T. Drug induced immune hematological disease. Allerg Immunol Clin. 1998;18:829-841.
Autoimmune hemolytic anemia (AIHA) is mediated by antibodies, and in most cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. AIHA caused by a drug reaction is rare, with an estimated annual incidence of 1:1,000,000 for severe drug-related AIHA.1 This article reviews the management of the more common types of AIHA, with a focus on warm, cold, and drug-induced AIHA; the evaluation and diagnosis of AIHA is reviewed in a separate article.
Warm Autoimmune Hemolytic Anemia
In AIHA, hemolysis is mediated by antibodies that bind to the surface of red blood cells. AIHA in which IgG antibodies are the offending antibodies is referred to as warm AIHA. “Warm” refers to the fact that the antibody binds best at body temperature (37°C). In warm AIHA, testing will show IgG molecules attached to the surface of the red cells, with 50% of patients also showing C3. Between 50% and 90% of AIHA cases are due to warm antibodies.2,3 The incidence of warm AIHA varies by series but is approximately 1 case per 100,000 patients per year; this form of hemolysis affects women more frequently than men.4,5
Therapeutic Options
First Line
Steroids. The goal of therapy in warm AIHA can be hard to define. However, most would agree that a hematocrit above 30% (or higher to prevent symptoms) with a minimal increase in the reticulocyte count—reflective of a significantly slowed hemolytic process—is a reasonable goal. Initial management of warm AIHA is prednisone at a standard dose of 1 mg/kg daily (Table 1).6,7 Patients should be also started on proton-pump inhibitors to prevent ulcers. It can take up to 3 weeks for patients to respond to prednisone therapy. Once the patient’s hematocrit is above 30%, the prednisone is slowly tapered. Although approximately 80% of patients will respond to steroids, only 30% can be fully tapered off steroids. For patients who can be maintained on a daily steroid dose of 10 mg or less, steroids may be the most reasonable long-term therapy. In addition, because active hemolysis leads to an increased demand for folic acid, patients with warm AIHA are often prescribed folic acid 1 mg daily to prevent megaloblastic anemia due to folic acid deficiency.
Rituximab. Increasingly, rituximab (anti-CD20) therapy is added to the initial steroids. Two clinical trials showed both increased long-term and short-term responses with the use of rituximab.8,9 An important consideration is that most patients respond gradually to rituximab over weeks, so a rapid response should not be expected. Most studies have used the traditional dosing of 375 mg/m2 weekly for 4 weeks. These responses appear to be durable, but as in immune thrombocytopenia (ITP), repeat treatment with rituximab is effective.
The major side effects of rituximab are infusion reactions, which are often worse with the first dose. These reactions can be controlled with antihistamines, steroids, and, for severe rigors, meperidine. Rarely, patients can develop neutropenia (approximately 1:500) that appears to be autoimmune in nature. Infections appear to be only minimally increased with the use of rituximab.10 One group at risk is chronic carriers of hepatitis B virus, who may experience a reactivation of the virus that can be fatal. Thus, patients being considered for rituximab need to be screened for hepatitis B virus carrier state.11 Patients receiving rituximab are at very slight risk for progressive multifocal leukoencephalopathy, which is more common in patients with cancer and in heavily immunosuppressed patients. The overall risk is unknown but is less than 1:50,000.
Second Line
Splenectomy. For patients who cannot be weaned from steroids or in whom steroid therapy fails, there is no standard therapy. Currently, the 2 main choices are splenectomy or rituximab (anti-CD20) therapy if the patient did not receive it first line. Splenectomy is the classic therapy for warm AIHA. Reported response rates in the literature range from 50% to 80%, with 50% to 60% remaining in remission.12-16 Timing of the procedure is a balance between allowing time for the steroids to work and the risk of toxicity of steroids. In a patient who has low presurgical risk and has either refractory disease or cannot be weaned from high doses of steroids, splenectomy should be done sooner. Splenectomy can be delayed or other therapy tried first in patients who require lower doses of steroids or have medical risk factors for surgery. Most splenectomies are performed via laparoscopy. The small incisions allow for quicker healing, and the laparoscopic approach provides better visualization of the abdomen to find and remove accessory spleens. When splenectomy is performed by experienced surgeons, the mortality rate is low (< 0.5%).17
The most concerning complication of splenectomy is overwhelming post-splenectomy infection (OPSI).18 In adults, the spleen appears to play a minimal role in immunity except for protecting against certain encap-sulated organisms. Splenectomized patients infected with an encapsulated organism (eg, Pneumococcus) will develop overwhelming sepsis within hours. These patients will often have disseminated intravascular coagulation and will rapidly progress to purpura fulminans. Approximately 40% to 50% of patients will die of sepsis even when the infection is detected early. The overall lifetime risk of sepsis may be as high as 1:500. The organism that most commonly causes sepsis in splenectomized patients is Streptococcus pneumoniae, reported in over 50% of cases. Neisseria meningitidis and Haemophilus influenzae have also been implicated in many cases.19 Overwhelming sepsis after dog bites has been reported due to Capnocytophaga canimorsus infections. Patients are also at increased risk of developing severe malaria and severe babesiosis.18
Patients who have undergone splenectomy need to be warned about the risk of OPSI and instructed to report to the emergency department readily if they develop a fever greater than 101°F (38.3°C) or shaking chills. Once in the emergency department, blood cultures should be obtained rapidly and the patient started on antibiotic coverage with vancomycin and ceftriaxone (or levofloxacin if allergic to beta-lactams).20 For patients in remote areas, some physicians will prescribe prophylactic antibiotics to take while they are traveling to a health care provider or even recommend a “standby” antibiotic dose to take while traveling to health care.5 This usually consists of amoxicillin or a macrolide for penicillin-allergic patients.
Patients in whom splenectomy is being planned or considered should be vaccinated for pneumococcal, meningococcal, and influenza infections (Table 2).18 If there is a plan to treat with rituximab, patients should first be vaccinated since they will not be able to mount an immune response after being treated with rituximab.
Third Line
The therapeutic options for patients who do not respond to either splenectomy or rituximab are much less certain.5,6 Although intravenous immune globulin is a standard therapy for ITP, response rates are low in warm AIHA.17 Numerous therapies have been reported in small series, but no clear approach has emerged. Options include azathioprine, cyclophosphamide, mycophenolate, cyclosporine, danazol, and alemtuzumab. Our approach has been to use mycophenolate for patients requiring high doses of steroids or transfusions. Patients who respond to lower doses of steroids may be good candidates for danazol to help wean them off steroids.
Treatment of Warm AIHA with Associated Diseases
Warm AIHA can complicate several diseases. Patients with systemic lupus erythematosus (SLE) can develop warm AIHA as part of their disease complex. The initial treatment approach is the same, but data suggest that splenectomy may not be as effective.13,17 Also, many SLE patients have complex medical conditions, making surgeries riskier. For SLE patients who are refractory or cannot be weaned from steroids, rituximab may be the better choice. Babesiosis, particularly in asplenic patients, has been associated with the development of AIHA.21,22
Of the malignances associated with AIHA, chronic lymphocytic leukemia (CLL) has the strongest association.4,23 Series show that 5% to 10% of patients with CLL will have warm AIHA. AIHA can appear concurrent with CLL or develop during the course of the disease. The introduction of purine analogs such as fludarabine led to a dramatic increase in the incidence of warm AIHA in treated patients.24 It is speculated that these powerful agents reduce the number and effectiveness of T cells that hold in check the autoantibody response, leading to warm AIHA.25 However, when these purine analogs are used in com-bination with agents such as cyclophosphamide or rituximab (with their immunosuppressive effects), the rates of warm AIHA have been lower.23
The approach to patients with CLL and warm AIHA depends on the state of their CLL.23 For patients who have low-stage CLL that does not need treatment, the standard approach to warm AIHA should be steroids, splenectomy, and rituximab.24 For patients with higher-stage CLL, the treatment for the leukemia will often provide therapy for the warm AIHA. The combination of rituximab-cyclophosphamide-dexamethasone has been reported to be effective for both the AIHA and CLL components.26 The use of ibrutinib has also been reported to be effective.27
A rare but important variant of warm AIHA is Evans syndrome.28,29 This is the combination of AIHA and ITP. Approximately 1% to 3% of AIHA cases are the Evans variant. The ITP can precede, be concurrent with, or develop after the AIHA. The diagnosis of Evans syndrome should raise concern for underlying disorders. In young adults, immunodeficiency disorders such as autoimmune lymphoproliferative disease (ALPS) need to be considered. In older patients, Evans syndrome is often associated with T cell lymphomas. The sparse literature on Evans syndrome suggests that it can be more refractory to standard therapy, with response rates to splenectomy in the 50% range.28,30 In patients with lymphoma, antineoplastic therapy is crucial. There is increasing data showing that mycophenolate or sirolimus may be effective for patients with ALPS in whom splenectomy or rituximab therapy is unsuccessful.31
Warm AIHA with IgA or IgM Antibodies
In rare patients with warm AIHA, IgA or IgM is the implicated antibody. The literature suggests that patients with IgA AIHA may have more severe hemolysis.32 Patients with IgM AIHA often have a severe course with a fatal outcome.33 In such cases, the patient’s plasma may show spontaneous hemolysis and agglutination. The DAT may not be strongly positive or may show C3 reactivity only. The clinical clues are C3 reactivity with no cold agglutinins and severe hemolysis, sometimes with an intravascular component. Treatment is the same as for warm AIHA, including the use of rituximab.34
Cold Autoimmune Hemolytic Anemia
In cold AIHA, the hemolysis is mediated by IgM antibodies directed against red cells.35 As discussed earlier, the term “cold” refers to the fact that the antibody binds maximally at temperatures below 37°C. The most efficient temperature for binding is called the “thermal amplitude,” and, in theory, the higher the thermal amplitude, the more virulent the antibody. An antibody titer can be calculated at each reaction temperature from 4°C to 37°C by serial dilutions of the patient’s serum prior to incubating with reagent red cells. Rarely, the IgM can fix complement rapidly, leading to intravascular hemolysis. In most patients, complement is fixed through C3, and the C3-coated red cells are taken up by macrophages in the mononuclear phagocyte system, primarily in the liver.3
The DAT in patients with cold AIHA will show cells coated with C3. The blood smear will often show ag-glutination of the blood, and if the blood cools before being analyzed, the agglutination will interfere with the analysis. Titers of cold agglutinin can range from 1:1000 to over 1 million. The IgM autoantibodies are most often directed against the I/i antigens on the red blood cell membrane, with 90% against I antigen.35 The I antigen specificity is typical with primary cold agglutinin disease and after Mycoplasma infection. The i antigen specificity is most typical of Epstein-Barr virus and cytomegalovirus infections in children. In young patients, cold AIHA often occurs following an infection, including viral and Mycoplasma infections, and the course is self-limited.35,36 The hemolysis usually starts 2 to 3 weeks after the illness and will last for 4 to 6 weeks. In older patients, the etiology in over 90% of cases is a B-cell lymphoproliferative disorder, usually with monoclonal kappa B-cells.37 The most common disorders are marginal zone lymphoma, small lymphocytic lymphoma, and lymphoplasmacytic lymphoma.3
Therapeutic Options
It is important to diagnose cold AIHA because the standard therapy for warm AIHA (steroids) is ineffective in cold AIHA. Because C3-coated red cells are taken up primarily in the liver, removing the spleen is also an ineffective therapy. Simple measures to help with cold AIHA should be employed.37 Patients should be kept in a warm environment and should try to avoid the cold. If transfusions are needed, they should be infused via blood warmers to prevent hemolysis. In rare patients with severe hemolysis, therapeutic plasma exchange (TPE) can be considered.38 Given that the culprit antibody is IgM—mostly intravascular—use of TPE may slow the hemolysis to give time for other therapies to take effect.
Treatment of cold AIHA remains difficult (Table 3). Because most patients with primary AIHA have underlying lymphoproliferative diseases, chlorambucil has been used in the past to treat cold AIHA. However, responses were rare and the drug could worsen the anemia.38 Currently, the drug of choice is rituximab. Response is seen in 45% to 75% of patients, but is almost always a partial response and retreatment is often necessary.17,37,39 As with other autoimmune hematologic diseases, there can be a delay in response that ranges from 2 weeks to 4 months (median time, 1.5 months).37 Given the lack of robust response (complete and durable) with rituximab, the Berentsen group has explored adding bendamustine to rituximab.40 In a prospective trial, 71% of patients responded with a 40% complete response rate. Therapy was well tolerated, with only 29% of patients needing dose reduction Although more toxic, this combination can be considered in patients with aggressive disease. A small study of the use of bortezomib showed a good response in one-third of patients.41 There is increasing use of the C5 complement inhibitor eculizumab to halt the hemolysis, but further study of this agent is also required.36,42,43 Blockers of C1s complement, which block hemolysis by preventing complement activation, are currently being studied in clinical trials.44
Since most patients with cold AIHA are older, a frequent issue that must be considered is cardiac surgery. The concern is that the hypothermia involved with most heart bypass procedures will lead to agglutination and hemolysis. The development of normothermic bypass has expanded treatment options. A recommended approach in patients who have known cold agglutinins is to measure the thermal amplitude of the antibody preoperatively. If the thermal amplitude is above 18°C, normothermic bypass should be done, if feasible.45 If not feasible, preoperative TPE should be considered.
Paroxysmal Cold Hemoglobinuria
A unique cold AIHA is paroxysmal cold hemoglobinuria (PCH).3,46 This cold hemolytic syndrome most often occurs in children following a viral infection, but in the past it complicated any stage of syphilis.47 The mediating antibody in PCH is an IgG antibody directed against the P antigen on the red blood cell. This antibody binds best at temperatures below 37°C, fixing complement at cold temperatures, but then activates the complement cascade at body temperature.48 Because this antibody can fix complement, hemolysis can be rapid and severe, leading to extreme anemia. The DAT is often weakly positive but can be negative. The diagnostic test for PCH is the Donath–Landsteiner test. This complex test is performed by incubating 3 blood samples, 1 at 0° to 4°C, another at 37°C, and a third at 0° to 4°C and then at 37°C. The diagnosis of PCH is made if only the third tube shows hemolysis.35 PCH can persist for 1 to 3 months but is almost always self-limiting. In severe case, steroids may be of benefit.
Drug-Induced Hemolytic Anemia
AIHA caused by a drug reaction is rare, with a lower incidence than drug-related ITP. The rate of severe drug-related AIHA is estimated at 1:1,000,000, but less severe cases may be missed.1 Most patients will have a positive DAT without signs of hemolysis, but in rare cases patients will have relentless hemolysis resulting in death.
Mechanisms
Multiple mechanisms for drug-induced immune hemolysis have been proposed, including drug-absorption (hapten-induced) and immune complex mechanisms.1,49 The hapten mechanism is most often associated with the use of high-dose penicillin.50 High doses of penicillin or similar drugs such as piperacillin lead to incorporation of the drug into the red cell membrane by binding to proteins. Patients will manifest a positive DAT with IgG antibody but not complement.51 The patient’s plasma will be reactive only with penicillin-coated red cells and not with normal cells. As mentioned, very few patients will have hemolysis, and if they have hemolysis, it will resolve in a few days after discontinuation of the offending drug.52
Binding of a drug-antibody complex to the red cell membrane may cause hemolysis via the immune com-plex mechanism.53 Again, most often the patient will have just a positive DAT, but rarely patients will have life-threatening hemolysis upon exposure or reexposure to the drug. The onset of hemolysis is rapid, with signs of acute illness and intravascular hemolysis. The paradigm drug is quinine, but many other drugs have been implicated. Testing shows a positive Coombs test with anti-complement but not anti-IgG.50 This pattern is due to the effectiveness of the tertiary complex at fixing complement. The patient’s plasma reacts with red cells only when the drug is added.
A form of immune complex hemolysis associated with both disseminated intravascular coagulation (DIC) and brisk hemolysis has been recognized. Patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone.53,54) have developed this syndrome.50,55-59 The clinical symptoms start 7 to 10 days after the drug is administered; often the patient has only received the antibiotic for surgical prophylaxis. Immune hemolysis with acute hematocrit drop, hypotension, and DIC ensues. The patients are often believed to have sepsis and are often reexposed to the cephalosporin, resulting in worsening of the clinical status. The outcome is often fatal due to massive hemolysis and thrombosis.56,60,62
Finally, 8% to 36% of patients taking methyldopa will develop a positive DAT after 6 months of therapy, with less than 1% showing hemolysis.52,63 The hemolysis in these patients is indistinguishable from warm AIHA, consistent with the notion that methyldopa induces an AIHA. The hemolysis often resolves rapidly after the methyldopa is stopped, but the Coombs test may remain positive for months.63 This type of drug-induced hemolytic anemia has been reported with levodopa, procainamide, and chlorpromazine, but fludarabine is the most common cause currently. This form of AIHA is now being seen with increased use of checkpoint inhibitors.64
Diagnosis
In many patients, the first clue to the presence of drug AIHA is the finding of a positive DAT. Rarely, patients will have severe hemolysis, but in many patients the hemolytic process is mild and may be wrongly assumed to be part of the underlying illness. Finding the offending drug can be a challenge, unless a patient has recently started a new drug; in a hospitalized patient on multiple agents, identifying the problem drug is difficult. Patients recently started on “suspect drugs,” especially the most common agents cefotetan, ceftriaxone, and piperacillin, should have these agents stopped (Table 4).1,49,65 Specialty laboratories such as the Blood Center of Wisconsin or the Los Angeles Red Cross can perform in vitro studies of drug interactions that can confirm the clinical diagnosis of drug-induced AIHA.
Treatment
Therapy for patients with positive DAT without signs of hemolysis is uncertain. If the drug is essential, then the patient can be observed. If the patient has hemolysis, the drug needs to be stopped and the patient observed for signs of end-organ damage. It is doubtful that steroids or other autoimmune-directed therapy is effective. For patients with the DIC-hemolysis syndrome, there are anecdotal reports that TPE may be helpful.1
Summary
AIHA can range from an abnormal laboratory test (positive DAT and signs of hemolysis) to an acute, life-threatening illness. Treatment is guided by the laboratory work-up and evaluation of the patient’s clinical status. While rituximab is promising for many patients, the lack of robust clinical trials hinders the treatment of patients who fail standard therapies.
Autoimmune hemolytic anemia (AIHA) is mediated by antibodies, and in most cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. AIHA caused by a drug reaction is rare, with an estimated annual incidence of 1:1,000,000 for severe drug-related AIHA.1 This article reviews the management of the more common types of AIHA, with a focus on warm, cold, and drug-induced AIHA; the evaluation and diagnosis of AIHA is reviewed in a separate article.
Warm Autoimmune Hemolytic Anemia
In AIHA, hemolysis is mediated by antibodies that bind to the surface of red blood cells. AIHA in which IgG antibodies are the offending antibodies is referred to as warm AIHA. “Warm” refers to the fact that the antibody binds best at body temperature (37°C). In warm AIHA, testing will show IgG molecules attached to the surface of the red cells, with 50% of patients also showing C3. Between 50% and 90% of AIHA cases are due to warm antibodies.2,3 The incidence of warm AIHA varies by series but is approximately 1 case per 100,000 patients per year; this form of hemolysis affects women more frequently than men.4,5
Therapeutic Options
First Line
Steroids. The goal of therapy in warm AIHA can be hard to define. However, most would agree that a hematocrit above 30% (or higher to prevent symptoms) with a minimal increase in the reticulocyte count—reflective of a significantly slowed hemolytic process—is a reasonable goal. Initial management of warm AIHA is prednisone at a standard dose of 1 mg/kg daily (Table 1).6,7 Patients should be also started on proton-pump inhibitors to prevent ulcers. It can take up to 3 weeks for patients to respond to prednisone therapy. Once the patient’s hematocrit is above 30%, the prednisone is slowly tapered. Although approximately 80% of patients will respond to steroids, only 30% can be fully tapered off steroids. For patients who can be maintained on a daily steroid dose of 10 mg or less, steroids may be the most reasonable long-term therapy. In addition, because active hemolysis leads to an increased demand for folic acid, patients with warm AIHA are often prescribed folic acid 1 mg daily to prevent megaloblastic anemia due to folic acid deficiency.
Rituximab. Increasingly, rituximab (anti-CD20) therapy is added to the initial steroids. Two clinical trials showed both increased long-term and short-term responses with the use of rituximab.8,9 An important consideration is that most patients respond gradually to rituximab over weeks, so a rapid response should not be expected. Most studies have used the traditional dosing of 375 mg/m2 weekly for 4 weeks. These responses appear to be durable, but as in immune thrombocytopenia (ITP), repeat treatment with rituximab is effective.
The major side effects of rituximab are infusion reactions, which are often worse with the first dose. These reactions can be controlled with antihistamines, steroids, and, for severe rigors, meperidine. Rarely, patients can develop neutropenia (approximately 1:500) that appears to be autoimmune in nature. Infections appear to be only minimally increased with the use of rituximab.10 One group at risk is chronic carriers of hepatitis B virus, who may experience a reactivation of the virus that can be fatal. Thus, patients being considered for rituximab need to be screened for hepatitis B virus carrier state.11 Patients receiving rituximab are at very slight risk for progressive multifocal leukoencephalopathy, which is more common in patients with cancer and in heavily immunosuppressed patients. The overall risk is unknown but is less than 1:50,000.
Second Line
Splenectomy. For patients who cannot be weaned from steroids or in whom steroid therapy fails, there is no standard therapy. Currently, the 2 main choices are splenectomy or rituximab (anti-CD20) therapy if the patient did not receive it first line. Splenectomy is the classic therapy for warm AIHA. Reported response rates in the literature range from 50% to 80%, with 50% to 60% remaining in remission.12-16 Timing of the procedure is a balance between allowing time for the steroids to work and the risk of toxicity of steroids. In a patient who has low presurgical risk and has either refractory disease or cannot be weaned from high doses of steroids, splenectomy should be done sooner. Splenectomy can be delayed or other therapy tried first in patients who require lower doses of steroids or have medical risk factors for surgery. Most splenectomies are performed via laparoscopy. The small incisions allow for quicker healing, and the laparoscopic approach provides better visualization of the abdomen to find and remove accessory spleens. When splenectomy is performed by experienced surgeons, the mortality rate is low (< 0.5%).17
The most concerning complication of splenectomy is overwhelming post-splenectomy infection (OPSI).18 In adults, the spleen appears to play a minimal role in immunity except for protecting against certain encap-sulated organisms. Splenectomized patients infected with an encapsulated organism (eg, Pneumococcus) will develop overwhelming sepsis within hours. These patients will often have disseminated intravascular coagulation and will rapidly progress to purpura fulminans. Approximately 40% to 50% of patients will die of sepsis even when the infection is detected early. The overall lifetime risk of sepsis may be as high as 1:500. The organism that most commonly causes sepsis in splenectomized patients is Streptococcus pneumoniae, reported in over 50% of cases. Neisseria meningitidis and Haemophilus influenzae have also been implicated in many cases.19 Overwhelming sepsis after dog bites has been reported due to Capnocytophaga canimorsus infections. Patients are also at increased risk of developing severe malaria and severe babesiosis.18
Patients who have undergone splenectomy need to be warned about the risk of OPSI and instructed to report to the emergency department readily if they develop a fever greater than 101°F (38.3°C) or shaking chills. Once in the emergency department, blood cultures should be obtained rapidly and the patient started on antibiotic coverage with vancomycin and ceftriaxone (or levofloxacin if allergic to beta-lactams).20 For patients in remote areas, some physicians will prescribe prophylactic antibiotics to take while they are traveling to a health care provider or even recommend a “standby” antibiotic dose to take while traveling to health care.5 This usually consists of amoxicillin or a macrolide for penicillin-allergic patients.
Patients in whom splenectomy is being planned or considered should be vaccinated for pneumococcal, meningococcal, and influenza infections (Table 2).18 If there is a plan to treat with rituximab, patients should first be vaccinated since they will not be able to mount an immune response after being treated with rituximab.
Third Line
The therapeutic options for patients who do not respond to either splenectomy or rituximab are much less certain.5,6 Although intravenous immune globulin is a standard therapy for ITP, response rates are low in warm AIHA.17 Numerous therapies have been reported in small series, but no clear approach has emerged. Options include azathioprine, cyclophosphamide, mycophenolate, cyclosporine, danazol, and alemtuzumab. Our approach has been to use mycophenolate for patients requiring high doses of steroids or transfusions. Patients who respond to lower doses of steroids may be good candidates for danazol to help wean them off steroids.
Treatment of Warm AIHA with Associated Diseases
Warm AIHA can complicate several diseases. Patients with systemic lupus erythematosus (SLE) can develop warm AIHA as part of their disease complex. The initial treatment approach is the same, but data suggest that splenectomy may not be as effective.13,17 Also, many SLE patients have complex medical conditions, making surgeries riskier. For SLE patients who are refractory or cannot be weaned from steroids, rituximab may be the better choice. Babesiosis, particularly in asplenic patients, has been associated with the development of AIHA.21,22
Of the malignances associated with AIHA, chronic lymphocytic leukemia (CLL) has the strongest association.4,23 Series show that 5% to 10% of patients with CLL will have warm AIHA. AIHA can appear concurrent with CLL or develop during the course of the disease. The introduction of purine analogs such as fludarabine led to a dramatic increase in the incidence of warm AIHA in treated patients.24 It is speculated that these powerful agents reduce the number and effectiveness of T cells that hold in check the autoantibody response, leading to warm AIHA.25 However, when these purine analogs are used in com-bination with agents such as cyclophosphamide or rituximab (with their immunosuppressive effects), the rates of warm AIHA have been lower.23
The approach to patients with CLL and warm AIHA depends on the state of their CLL.23 For patients who have low-stage CLL that does not need treatment, the standard approach to warm AIHA should be steroids, splenectomy, and rituximab.24 For patients with higher-stage CLL, the treatment for the leukemia will often provide therapy for the warm AIHA. The combination of rituximab-cyclophosphamide-dexamethasone has been reported to be effective for both the AIHA and CLL components.26 The use of ibrutinib has also been reported to be effective.27
A rare but important variant of warm AIHA is Evans syndrome.28,29 This is the combination of AIHA and ITP. Approximately 1% to 3% of AIHA cases are the Evans variant. The ITP can precede, be concurrent with, or develop after the AIHA. The diagnosis of Evans syndrome should raise concern for underlying disorders. In young adults, immunodeficiency disorders such as autoimmune lymphoproliferative disease (ALPS) need to be considered. In older patients, Evans syndrome is often associated with T cell lymphomas. The sparse literature on Evans syndrome suggests that it can be more refractory to standard therapy, with response rates to splenectomy in the 50% range.28,30 In patients with lymphoma, antineoplastic therapy is crucial. There is increasing data showing that mycophenolate or sirolimus may be effective for patients with ALPS in whom splenectomy or rituximab therapy is unsuccessful.31
Warm AIHA with IgA or IgM Antibodies
In rare patients with warm AIHA, IgA or IgM is the implicated antibody. The literature suggests that patients with IgA AIHA may have more severe hemolysis.32 Patients with IgM AIHA often have a severe course with a fatal outcome.33 In such cases, the patient’s plasma may show spontaneous hemolysis and agglutination. The DAT may not be strongly positive or may show C3 reactivity only. The clinical clues are C3 reactivity with no cold agglutinins and severe hemolysis, sometimes with an intravascular component. Treatment is the same as for warm AIHA, including the use of rituximab.34
Cold Autoimmune Hemolytic Anemia
In cold AIHA, the hemolysis is mediated by IgM antibodies directed against red cells.35 As discussed earlier, the term “cold” refers to the fact that the antibody binds maximally at temperatures below 37°C. The most efficient temperature for binding is called the “thermal amplitude,” and, in theory, the higher the thermal amplitude, the more virulent the antibody. An antibody titer can be calculated at each reaction temperature from 4°C to 37°C by serial dilutions of the patient’s serum prior to incubating with reagent red cells. Rarely, the IgM can fix complement rapidly, leading to intravascular hemolysis. In most patients, complement is fixed through C3, and the C3-coated red cells are taken up by macrophages in the mononuclear phagocyte system, primarily in the liver.3
The DAT in patients with cold AIHA will show cells coated with C3. The blood smear will often show ag-glutination of the blood, and if the blood cools before being analyzed, the agglutination will interfere with the analysis. Titers of cold agglutinin can range from 1:1000 to over 1 million. The IgM autoantibodies are most often directed against the I/i antigens on the red blood cell membrane, with 90% against I antigen.35 The I antigen specificity is typical with primary cold agglutinin disease and after Mycoplasma infection. The i antigen specificity is most typical of Epstein-Barr virus and cytomegalovirus infections in children. In young patients, cold AIHA often occurs following an infection, including viral and Mycoplasma infections, and the course is self-limited.35,36 The hemolysis usually starts 2 to 3 weeks after the illness and will last for 4 to 6 weeks. In older patients, the etiology in over 90% of cases is a B-cell lymphoproliferative disorder, usually with monoclonal kappa B-cells.37 The most common disorders are marginal zone lymphoma, small lymphocytic lymphoma, and lymphoplasmacytic lymphoma.3
Therapeutic Options
It is important to diagnose cold AIHA because the standard therapy for warm AIHA (steroids) is ineffective in cold AIHA. Because C3-coated red cells are taken up primarily in the liver, removing the spleen is also an ineffective therapy. Simple measures to help with cold AIHA should be employed.37 Patients should be kept in a warm environment and should try to avoid the cold. If transfusions are needed, they should be infused via blood warmers to prevent hemolysis. In rare patients with severe hemolysis, therapeutic plasma exchange (TPE) can be considered.38 Given that the culprit antibody is IgM—mostly intravascular—use of TPE may slow the hemolysis to give time for other therapies to take effect.
Treatment of cold AIHA remains difficult (Table 3). Because most patients with primary AIHA have underlying lymphoproliferative diseases, chlorambucil has been used in the past to treat cold AIHA. However, responses were rare and the drug could worsen the anemia.38 Currently, the drug of choice is rituximab. Response is seen in 45% to 75% of patients, but is almost always a partial response and retreatment is often necessary.17,37,39 As with other autoimmune hematologic diseases, there can be a delay in response that ranges from 2 weeks to 4 months (median time, 1.5 months).37 Given the lack of robust response (complete and durable) with rituximab, the Berentsen group has explored adding bendamustine to rituximab.40 In a prospective trial, 71% of patients responded with a 40% complete response rate. Therapy was well tolerated, with only 29% of patients needing dose reduction Although more toxic, this combination can be considered in patients with aggressive disease. A small study of the use of bortezomib showed a good response in one-third of patients.41 There is increasing use of the C5 complement inhibitor eculizumab to halt the hemolysis, but further study of this agent is also required.36,42,43 Blockers of C1s complement, which block hemolysis by preventing complement activation, are currently being studied in clinical trials.44
Since most patients with cold AIHA are older, a frequent issue that must be considered is cardiac surgery. The concern is that the hypothermia involved with most heart bypass procedures will lead to agglutination and hemolysis. The development of normothermic bypass has expanded treatment options. A recommended approach in patients who have known cold agglutinins is to measure the thermal amplitude of the antibody preoperatively. If the thermal amplitude is above 18°C, normothermic bypass should be done, if feasible.45 If not feasible, preoperative TPE should be considered.
Paroxysmal Cold Hemoglobinuria
A unique cold AIHA is paroxysmal cold hemoglobinuria (PCH).3,46 This cold hemolytic syndrome most often occurs in children following a viral infection, but in the past it complicated any stage of syphilis.47 The mediating antibody in PCH is an IgG antibody directed against the P antigen on the red blood cell. This antibody binds best at temperatures below 37°C, fixing complement at cold temperatures, but then activates the complement cascade at body temperature.48 Because this antibody can fix complement, hemolysis can be rapid and severe, leading to extreme anemia. The DAT is often weakly positive but can be negative. The diagnostic test for PCH is the Donath–Landsteiner test. This complex test is performed by incubating 3 blood samples, 1 at 0° to 4°C, another at 37°C, and a third at 0° to 4°C and then at 37°C. The diagnosis of PCH is made if only the third tube shows hemolysis.35 PCH can persist for 1 to 3 months but is almost always self-limiting. In severe case, steroids may be of benefit.
Drug-Induced Hemolytic Anemia
AIHA caused by a drug reaction is rare, with a lower incidence than drug-related ITP. The rate of severe drug-related AIHA is estimated at 1:1,000,000, but less severe cases may be missed.1 Most patients will have a positive DAT without signs of hemolysis, but in rare cases patients will have relentless hemolysis resulting in death.
Mechanisms
Multiple mechanisms for drug-induced immune hemolysis have been proposed, including drug-absorption (hapten-induced) and immune complex mechanisms.1,49 The hapten mechanism is most often associated with the use of high-dose penicillin.50 High doses of penicillin or similar drugs such as piperacillin lead to incorporation of the drug into the red cell membrane by binding to proteins. Patients will manifest a positive DAT with IgG antibody but not complement.51 The patient’s plasma will be reactive only with penicillin-coated red cells and not with normal cells. As mentioned, very few patients will have hemolysis, and if they have hemolysis, it will resolve in a few days after discontinuation of the offending drug.52
Binding of a drug-antibody complex to the red cell membrane may cause hemolysis via the immune com-plex mechanism.53 Again, most often the patient will have just a positive DAT, but rarely patients will have life-threatening hemolysis upon exposure or reexposure to the drug. The onset of hemolysis is rapid, with signs of acute illness and intravascular hemolysis. The paradigm drug is quinine, but many other drugs have been implicated. Testing shows a positive Coombs test with anti-complement but not anti-IgG.50 This pattern is due to the effectiveness of the tertiary complex at fixing complement. The patient’s plasma reacts with red cells only when the drug is added.
A form of immune complex hemolysis associated with both disseminated intravascular coagulation (DIC) and brisk hemolysis has been recognized. Patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone.53,54) have developed this syndrome.50,55-59 The clinical symptoms start 7 to 10 days after the drug is administered; often the patient has only received the antibiotic for surgical prophylaxis. Immune hemolysis with acute hematocrit drop, hypotension, and DIC ensues. The patients are often believed to have sepsis and are often reexposed to the cephalosporin, resulting in worsening of the clinical status. The outcome is often fatal due to massive hemolysis and thrombosis.56,60,62
Finally, 8% to 36% of patients taking methyldopa will develop a positive DAT after 6 months of therapy, with less than 1% showing hemolysis.52,63 The hemolysis in these patients is indistinguishable from warm AIHA, consistent with the notion that methyldopa induces an AIHA. The hemolysis often resolves rapidly after the methyldopa is stopped, but the Coombs test may remain positive for months.63 This type of drug-induced hemolytic anemia has been reported with levodopa, procainamide, and chlorpromazine, but fludarabine is the most common cause currently. This form of AIHA is now being seen with increased use of checkpoint inhibitors.64
Diagnosis
In many patients, the first clue to the presence of drug AIHA is the finding of a positive DAT. Rarely, patients will have severe hemolysis, but in many patients the hemolytic process is mild and may be wrongly assumed to be part of the underlying illness. Finding the offending drug can be a challenge, unless a patient has recently started a new drug; in a hospitalized patient on multiple agents, identifying the problem drug is difficult. Patients recently started on “suspect drugs,” especially the most common agents cefotetan, ceftriaxone, and piperacillin, should have these agents stopped (Table 4).1,49,65 Specialty laboratories such as the Blood Center of Wisconsin or the Los Angeles Red Cross can perform in vitro studies of drug interactions that can confirm the clinical diagnosis of drug-induced AIHA.
Treatment
Therapy for patients with positive DAT without signs of hemolysis is uncertain. If the drug is essential, then the patient can be observed. If the patient has hemolysis, the drug needs to be stopped and the patient observed for signs of end-organ damage. It is doubtful that steroids or other autoimmune-directed therapy is effective. For patients with the DIC-hemolysis syndrome, there are anecdotal reports that TPE may be helpful.1
Summary
AIHA can range from an abnormal laboratory test (positive DAT and signs of hemolysis) to an acute, life-threatening illness. Treatment is guided by the laboratory work-up and evaluation of the patient’s clinical status. While rituximab is promising for many patients, the lack of robust clinical trials hinders the treatment of patients who fail standard therapies.
1. Garratty G. Immune hemolytic anemia associated with drug therapy. Blood Rev. 2010;24:143-150.
2. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
3. Berentsen S. Cold agglutinin disease. Hematology Am Soc Hematol Educ Program. 2016;2016:226-231.
4. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
5. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
6. Kyrle PA, Rosendaal FR, Eichinger S. Risk assessment for re¬current venous thrombosis. Lancet. 2010;376:2032-2039.
7. Go RS, Winters JL, Kay NE. How I treat autoimmune hemolytic anemia. Blood. 2017;129:2971-2979.
8. Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013;163:393-399
9. Michel M, Terriou L, Roudot-Thoraval F, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study). Am J Hematol. 2017;92:23-27.
10. Gea-Banacloche JC. Rituximab-associated infections. Semin Hematol. 2010;47:187-198.
11. Loomba R, Liang TJ. Hepatitis B reactivation associated with immune suppressive and biological modifier therapies: current concepts, management strategies, and future directions. Gastroenterology. 2017;152:1297-1309.
12. Coon WW. Splenectomy in the treatment of hemolytic anemia. Arch Surg. 1985;120:625-628.
13. Akpek G, McAneny D, Weintraub L. Comparative response to splenectomy in coombs-positive autoimmune hemolytic anemia with or without associated disease. Am J Hematol. 1999;61:98-102.
14. Patel NY, Chilsen AM, Mathiason MA, et al. Outcomes and complications after splenectomy for hematologic disorders. Am J Surg. 2012;204:1014-1020.
15. Crowther M, Chan YL, Garbett IK, et al. Evidence-based focused review of the treatment of idiopathic warm immune hemolytic anemia in adults. Blood. 2011;118:4036-4040.
16. Giudice V, Rosamilio R, Ferrara I, et al. Efficacy and safety of splenectomy in adult autoimmune hemolytic anemia. Open Med (Wars). 2016;11:374-380.
17. Lechner K, Jager U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010;116:1831-1838.
18. Rodeghiero F, Ruggeri M. Short- and long-term risks of splenectomy for benign haematological disorders: should we revisit the indications? Br J Haematol. 2012;158:16-29.
19. Ahmed N, Bialowas C, Kuo YH, Zawodniak L. Impact of preinjury anticoagulation in patients with traumatic brain injury. South Med J. 2009;102:476-480.
20. Morgan TL, Tomich EB. Overwhelming post-splenectomy infection (OPSI): a case report and review of the literature. J Emerg Med. 2012;43:758-763.
21. Woolley AE, Montgomery MW, Savage WJ, et al. Post-babesiosis warm autoimmune hemolytic anemia. N Engl J Med. 2017;376:939-946.
22. Shatzel JJ, Donohoe K, Chu NQ, et al. Profound autoimmune hemolysis and Evans syndrome in two asplenic patients with babesiosis. Transfusion. 2015;55:661-665.
23. Hodgson K, Ferrer G, Montserrat E, Moreno C. Chronic lymphocytic leukemia and autoimmunity: a systematic review. Haematologica. 2011;96:752-761.
24. Hamblin TJ. Autoimmune complications of chronic lymphocytic leukemia. Semin Oncol. 2006;33:230-239.
25. Tertian G, Cartron J, Bayle C, et al. Fatal intravascular au¬toimmune hemolytic anemia after fludarabine treatment for chronic lymphocytic leukemia. Hematol Cell Ther. 1996;38:359-360.
26. Rossignol J, Michallet AS, Oberic L, et al. Rituximab-cyclophosphamide-dexamethasone combination in the management of autoimmune cytopenias associated with chronic lymphocytic leukemia. Leukemia. 2011;25:473-478.
27. Hampel PJ, Larson MC, Kabat B, et al. Autoimmune cytopenias in patients with chronic lymphocytic leukaemia treated with ibrutinib in routine clinical practice at an academic medical centre. Br J Haematol. 2018;183:421-427.
28. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114:3167-3172.
29. Jaime-Pérez JC, Aguilar-Calderón PE, Salazar-Cavazos L, Gómez-Almaguer D. Evans syndrome: clinical perspectives, biological insights and treatment modalities. J Blood Med. 2018;9:171-184.
30. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology. 2008;13:356-360.
31. Bride KL, Vincent T, Smith-Whitley K, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016;127:17-28.
32. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell au¬toantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
33. Garratty G, Arndt P, Domen R, et al. Severe autoimmune hemolytic anemia associated with IgM warm autoantibodies directed against determinants on or associated with glycophorin A. Vox Sang. 1997;72:124-130.
34. Wakim M, Shah A, Arndt PA, et al. Successful anti-CD20 monoclonal antibody treatment of severe autoimmune hemolytic anemia due to warm reactive IgM autoantibody in a child with common variable immunodeficiency. Am J Hematol. 2004;76:152-155.
35. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
36. Berentsen S, Tjonnfjord GE. Diagnosis and treatment of cold agglutinin mediated autoimmune hemolytic anemia. Blood Rev. 2012;26:107-115.
37. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
38. King KE, Ness PM. Treatment of autoimmune hemolytic anemia. Semin Hematol. 2005;42:131136.
39. Barcellini W, Zaja F, Zaninoni A, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. 2012;119:3691-3697.
40. Berentsen S, Randen U, Oksman M, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. 2017;130:537-541.
41. Rossi G, Gramegna D, Paoloni F, et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: a phase 2 prospective GIMEMA study. Blood. 2018;132:547-550.
42. Makishima K, Obara N, Ishitsuka K, et al. High efficacy of eculizumab treatment for fulminant hemolytic anemia in primary cold agglutinin disease. Ann Hematol. 2019;98:1031-1032.
43. Roth A, Huttmann A, Rother RP, et al. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood. 2009;113:38853886.
44. Jäger U, D'Sa S, Schörgenhofer C, et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: a first-in-human trial. Blood. 2019;133:893-901.
45. Agarwal SK, Ghosh PK, Gupta D. Cardiac surgery and cold-reactive proteins. Ann Thorac Surg. 1995;60:1143-1150.
46. Shanbhag S, Spivak J. Paroxysmal cold hemoglobinuria. Hematol Oncol Clin North Am. 2015;29:473-478.
47. Kumar ND, Sethi S, Pandhi RK. Paroxysmal cold haemoglobinuria in syphilis patients. Genitourin Med. 1993;69:76.
48. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
49. Pierce A, Nester T. Pathology consultation on drug-induced hemolytic anemia. Am J Clin Pathol. 2011;136:7-12.
50. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Med Rev. 1993;7:255-267.
51. Petz LD, Mueller-Eckhardt C. Drug-induced immune hemolytic anemia. Transfusion. 1992;32:202-204.
52. Packman CH, Leddy JP. Drug-related immune hemolytic anemia. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ, eds. William’s Hematology. 5th ed. New York: McGraw-Hill; 1995:691-704.
53. Garratty G. Drug-induced immune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2009;73-79.
54. Leicht HB, Weinig E, Mayer B, et al. Ceftriaxone-induced hemolytic anemia with severe renal failure: a case report and review of literature. BMC Pharmacol Toxicol. 2018;19:67.
55. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis. 1992;15:863-865.
56. Garratty G, Nance S, Lloyd M, Domen R. Fatal im¬mune hemolytic anemia due to cefotetan. Transfusion. 1992;32:269-271.
57. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes: case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion. 1999;39:306-309.
58. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion. 1999;39:1239-1246.
59. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol. 2006;81:186-188.
60. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr. 1995;126(5 Pt 1):813-815.
61. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1995;14:1116-1117.
62. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone [see comments]. J Pediatr. 1995;126(5 Pt 1):816-817.
63. Petz LD. Drug-induced autoimmune hemolytic anemia. Transfusion Med Rev. 1993;7:242-254.
64. Leaf RK, Ferreri C, Rangachari D, et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am J Hematol. 2019;94:563-574.
65. DeLoughery T. Drug induced immune hematological disease. Allerg Immunol Clin. 1998;18:829-841.
1. Garratty G. Immune hemolytic anemia associated with drug therapy. Blood Rev. 2010;24:143-150.
2. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
3. Berentsen S. Cold agglutinin disease. Hematology Am Soc Hematol Educ Program. 2016;2016:226-231.
4. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
5. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
6. Kyrle PA, Rosendaal FR, Eichinger S. Risk assessment for re¬current venous thrombosis. Lancet. 2010;376:2032-2039.
7. Go RS, Winters JL, Kay NE. How I treat autoimmune hemolytic anemia. Blood. 2017;129:2971-2979.
8. Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013;163:393-399
9. Michel M, Terriou L, Roudot-Thoraval F, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study). Am J Hematol. 2017;92:23-27.
10. Gea-Banacloche JC. Rituximab-associated infections. Semin Hematol. 2010;47:187-198.
11. Loomba R, Liang TJ. Hepatitis B reactivation associated with immune suppressive and biological modifier therapies: current concepts, management strategies, and future directions. Gastroenterology. 2017;152:1297-1309.
12. Coon WW. Splenectomy in the treatment of hemolytic anemia. Arch Surg. 1985;120:625-628.
13. Akpek G, McAneny D, Weintraub L. Comparative response to splenectomy in coombs-positive autoimmune hemolytic anemia with or without associated disease. Am J Hematol. 1999;61:98-102.
14. Patel NY, Chilsen AM, Mathiason MA, et al. Outcomes and complications after splenectomy for hematologic disorders. Am J Surg. 2012;204:1014-1020.
15. Crowther M, Chan YL, Garbett IK, et al. Evidence-based focused review of the treatment of idiopathic warm immune hemolytic anemia in adults. Blood. 2011;118:4036-4040.
16. Giudice V, Rosamilio R, Ferrara I, et al. Efficacy and safety of splenectomy in adult autoimmune hemolytic anemia. Open Med (Wars). 2016;11:374-380.
17. Lechner K, Jager U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010;116:1831-1838.
18. Rodeghiero F, Ruggeri M. Short- and long-term risks of splenectomy for benign haematological disorders: should we revisit the indications? Br J Haematol. 2012;158:16-29.
19. Ahmed N, Bialowas C, Kuo YH, Zawodniak L. Impact of preinjury anticoagulation in patients with traumatic brain injury. South Med J. 2009;102:476-480.
20. Morgan TL, Tomich EB. Overwhelming post-splenectomy infection (OPSI): a case report and review of the literature. J Emerg Med. 2012;43:758-763.
21. Woolley AE, Montgomery MW, Savage WJ, et al. Post-babesiosis warm autoimmune hemolytic anemia. N Engl J Med. 2017;376:939-946.
22. Shatzel JJ, Donohoe K, Chu NQ, et al. Profound autoimmune hemolysis and Evans syndrome in two asplenic patients with babesiosis. Transfusion. 2015;55:661-665.
23. Hodgson K, Ferrer G, Montserrat E, Moreno C. Chronic lymphocytic leukemia and autoimmunity: a systematic review. Haematologica. 2011;96:752-761.
24. Hamblin TJ. Autoimmune complications of chronic lymphocytic leukemia. Semin Oncol. 2006;33:230-239.
25. Tertian G, Cartron J, Bayle C, et al. Fatal intravascular au¬toimmune hemolytic anemia after fludarabine treatment for chronic lymphocytic leukemia. Hematol Cell Ther. 1996;38:359-360.
26. Rossignol J, Michallet AS, Oberic L, et al. Rituximab-cyclophosphamide-dexamethasone combination in the management of autoimmune cytopenias associated with chronic lymphocytic leukemia. Leukemia. 2011;25:473-478.
27. Hampel PJ, Larson MC, Kabat B, et al. Autoimmune cytopenias in patients with chronic lymphocytic leukaemia treated with ibrutinib in routine clinical practice at an academic medical centre. Br J Haematol. 2018;183:421-427.
28. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114:3167-3172.
29. Jaime-Pérez JC, Aguilar-Calderón PE, Salazar-Cavazos L, Gómez-Almaguer D. Evans syndrome: clinical perspectives, biological insights and treatment modalities. J Blood Med. 2018;9:171-184.
30. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology. 2008;13:356-360.
31. Bride KL, Vincent T, Smith-Whitley K, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016;127:17-28.
32. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell au¬toantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
33. Garratty G, Arndt P, Domen R, et al. Severe autoimmune hemolytic anemia associated with IgM warm autoantibodies directed against determinants on or associated with glycophorin A. Vox Sang. 1997;72:124-130.
34. Wakim M, Shah A, Arndt PA, et al. Successful anti-CD20 monoclonal antibody treatment of severe autoimmune hemolytic anemia due to warm reactive IgM autoantibody in a child with common variable immunodeficiency. Am J Hematol. 2004;76:152-155.
35. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
36. Berentsen S, Tjonnfjord GE. Diagnosis and treatment of cold agglutinin mediated autoimmune hemolytic anemia. Blood Rev. 2012;26:107-115.
37. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
38. King KE, Ness PM. Treatment of autoimmune hemolytic anemia. Semin Hematol. 2005;42:131136.
39. Barcellini W, Zaja F, Zaninoni A, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. 2012;119:3691-3697.
40. Berentsen S, Randen U, Oksman M, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. 2017;130:537-541.
41. Rossi G, Gramegna D, Paoloni F, et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: a phase 2 prospective GIMEMA study. Blood. 2018;132:547-550.
42. Makishima K, Obara N, Ishitsuka K, et al. High efficacy of eculizumab treatment for fulminant hemolytic anemia in primary cold agglutinin disease. Ann Hematol. 2019;98:1031-1032.
43. Roth A, Huttmann A, Rother RP, et al. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood. 2009;113:38853886.
44. Jäger U, D'Sa S, Schörgenhofer C, et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: a first-in-human trial. Blood. 2019;133:893-901.
45. Agarwal SK, Ghosh PK, Gupta D. Cardiac surgery and cold-reactive proteins. Ann Thorac Surg. 1995;60:1143-1150.
46. Shanbhag S, Spivak J. Paroxysmal cold hemoglobinuria. Hematol Oncol Clin North Am. 2015;29:473-478.
47. Kumar ND, Sethi S, Pandhi RK. Paroxysmal cold haemoglobinuria in syphilis patients. Genitourin Med. 1993;69:76.
48. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
49. Pierce A, Nester T. Pathology consultation on drug-induced hemolytic anemia. Am J Clin Pathol. 2011;136:7-12.
50. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Med Rev. 1993;7:255-267.
51. Petz LD, Mueller-Eckhardt C. Drug-induced immune hemolytic anemia. Transfusion. 1992;32:202-204.
52. Packman CH, Leddy JP. Drug-related immune hemolytic anemia. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ, eds. William’s Hematology. 5th ed. New York: McGraw-Hill; 1995:691-704.
53. Garratty G. Drug-induced immune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2009;73-79.
54. Leicht HB, Weinig E, Mayer B, et al. Ceftriaxone-induced hemolytic anemia with severe renal failure: a case report and review of literature. BMC Pharmacol Toxicol. 2018;19:67.
55. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis. 1992;15:863-865.
56. Garratty G, Nance S, Lloyd M, Domen R. Fatal im¬mune hemolytic anemia due to cefotetan. Transfusion. 1992;32:269-271.
57. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes: case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion. 1999;39:306-309.
58. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion. 1999;39:1239-1246.
59. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol. 2006;81:186-188.
60. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr. 1995;126(5 Pt 1):813-815.
61. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1995;14:1116-1117.
62. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone [see comments]. J Pediatr. 1995;126(5 Pt 1):816-817.
63. Petz LD. Drug-induced autoimmune hemolytic anemia. Transfusion Med Rev. 1993;7:242-254.
64. Leaf RK, Ferreri C, Rangachari D, et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am J Hematol. 2019;94:563-574.
65. DeLoughery T. Drug induced immune hematological disease. Allerg Immunol Clin. 1998;18:829-841.
Autoimmune Hemolytic Anemia: Evaluation and Diagnosis
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons.1,2 AIHA is mediated by antibodies, and in the majority of cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C (Table 1). This article series reviews the most common types of AIHA, with an overview of evaluation and diagnosis presented in this article and management of warm, cold, and drug-induced AIHA reviewed in a separate article.

Pathogenesis
In most cases, the ultimate etiology of AIHA is unknown. In warm AIHA, the target epitopes in most cases are Rh proteins.2 What leads the immune system to target these proteins is unidentified, but one theory is that an initial immune response to a foreign antigen starts to cross-react with the Rh proteins and the immune system fails to suppress this autoreactive response, leading to hemolysis. In IgG-mediated (warm) hemolysis, the red cells become coated with IgG molecules, which mark the cells for uptake and destruction by splenic macrophages.3 In "cold" AIHA, IgM molecules fix complement to the surface of red blood cells. Rarely, this can lead to activation of the full complement cascade, resulting in red cell lysis, but more often it is stopped at the C3 stage, leading to C3-coated red cells which are then taken up by hepatic macrophages.4
Suspecting the Diagnosis
In many patients, it is the symptoms and signs of anemia that lead to suspicion of hemolysis. Older patients often present earlier in the course of the disease due to lack of tolerance of anemia, especially if there is a sudden drop in the red blood cell count. Dark, cola-colored urine resulting from the presence of free hemoglobin may be noted by some patients. Patients with rapid-onset hemolysis may note lumbar back pain, and those with cold agglutinins often note symptoms related to agglutination of red cells in the peripheral circulation, such as the development of acrocyanosis in cold weather.5 In rare cases, patients will have abdominal pain when eating cold food due to ischemia related to agglutination of red cells in the viscera. Some patients with cold agglutinins can have an exacerbation of their hemolysis with cold exposure.
Unlike patients with immune thrombocytopenia, those with AIHA may have mild splenomegaly on exam. The presence of enlarged lymph nodes or massive splenomegaly should raise concern about concomitant lymphoma or chronic lymphocytic leukemia.
Making the Diagnosis
The 2 key steps in diagnosis are (1) demonstrating hemolysis and (2) demonstrating the autoimmune component.
Laboratory Evaluation for Hemolysis
Hemolysis is proven by finding evidence of both red cell breakdown and the compensatory increase in red cell production this stimulates (Table 2). The following sections discuss the laboratory tests that are performed to investigate hemolysis.

Lactate Dehydrogenase
When red cells undergo hemolysis, they release their contents, which are mostly comprised of hemoglobin but also include lactate dehydrogenase (LDH), an enzyme found in high concentration in red cells. Most patients with hemolysis will have an elevated LDH level, making this a sensitive test. However, because many other processes, including liver disease and pneumonia, also raise the serum LDH level, this finding is not specific for hemolysis.
Serum Bilirubin
Hemoglobin is salvaged by haptoglobin, and the heme moiety is broken down first to bilirubin and then to urobilinogen, which is excreted in the urine.2 Bilirubin produced from the breakdown of heme is not conjugated, but rather is delivered to the liver, where it is conjugated and excreted into the bile. In hemolysis, the concentration of unconjugated bilirubin (indirect bilirubin) is increased, while in liver disease the level of conjugated bilirubin (direct bilirubin) is increased. However, if the patient has concomitant liver disease with an increased direct bilirubin level, the serum bilirubin test is not reliable.
Serum Haptoglobin
Haptoglobin binds free serum hemoglobin and is taken up by the liver. Haptoglobin usually falls to very low levels in hemolysis. A confounder is that haptoglobin is an acute phase reactant and can rise with systemic disease or inflammation. However, patients with advanced liver disease will have low haptoglobin levels due to lack of synthesis, and up to 2% of the population may congenitally lack haptoglobin.1
Serum Hemoglobin
If the hemolysis is very rapid, the amount of free hemoglobin released will overwhelm the binding capacity of haptoglobin and lead to free hemoglobin in the plasma. This can be crudely quantified by examining the plasma color. Even minute amounts of free hemoglobin will turn the plasma pink. In fulminant hemolysis, the plasma will be cola-colored.
Reticulocyte Count
In most patients with hemolysis, the destruction of red cells is accompanied by an increase in the reticulocyte count. Reticulocytes are red cells that still contain RNA and are a marker of red cells that are approximately 24 hours old or less. Traditionally, reticulocytes were measured manually by staining the blood smear with vital blue and counting the percentage of cells that absorb the stain; this percentage needs to be adjusted for the hematocrit. Usually a percentage above 1.5% is considered indicative of an elevated reticulocyte count. Recently, automated complete blood count machines have taken advantage of the fact that reticulocytes will absorb certain stains; these machines can directly measure the reticulocyte count via flow cytometry, which results in an “absolute” reticulocyte count. The reticulocyte count obtained using this method does not have to be corrected for hematocrit, and levels of approximately 90,000/μL are considered raised. However, the reticulocyte count can also be raised in blood loss or in patients who have other causes of anemia (eg, iron deficiency) under treatment. In addition, as many as 25% of patients with AIHA will never have raised counts for various reasons, such as nutritional deficiency, autoimmune destruction of red cell precursors, or lack of erythropoietin.
Blood Smear
The blood smear provides vital information. The hallmark laboratory parameter of AIHA is spherocytes seen on the blood smear. In AIHA, antibodies and/or complement attach to the red cells, and when the antibodies or complement are taken up by macrophages in the spleen some of the red blood cell mem-brane is removed as well, decreasing the surface area of the cell. As the surface area of the red cell decreases with each pass through the spleen, the cell's shape changes from a biconcave disk to a sphere before the cell is destroyed, reflecting the fact that a sphere has the smallest surface area for a given volume. The vast majority of patients with AIHA will have spherocytes on the blood smear. However, spherocytes are not specific to AIHA, as they can be seen in hereditary spherocytosis, Wilson’s disease, clostridial sepsis, and severe burns.
Patients with cold agglutinins will often have red cell agglutination on the blood smear. In addition, patients with AIHA will often have a raised mean corpuscular volume (MCV) for 2 reasons. In patients with brisk reticulocytosis, the MCV will be raised due to the large size of the reticulocyte. In patients with cold agglutinin disease, the MCV may be falsely raised due to clumping of the red blood cells.
Urinary Hemosiderin
When hemoglobin is excreted by the kidney, the iron is deposited in the tubules. When the tubule cells are sloughed off, they appear in the urine. The urine can be stained for iron, and a positive result is another sign of hemolysis. Hemosiderinuria is a later sign of hemolysis, as it takes 1 week for iron-laden tubule cells to be excreted in sufficient quantities to be detected in the urine.
Urinary Hemoglobin
One other sign of hemolysis is the presence of hemoglobin in the urine. A quick way to demonstrate hemoglobinuria is to check the urine with a dipstick followed by a microscopic exam. In hemolysis, the dipstick will detect “blood,” while the microscopic exam will be negative for red cells.
Laboratory Evaluation for Autoimmune Component
The autoimmune component is shown by demonstrating the presence of either IgG molecules or complement on the surface of red blood cells.4,6 This can be done by performing the direct antiglobulin test (DAT) or Coombs test. IgG bound to red cells will not agglutinate them, but if IgM that is directed against IgG or C3 is added, the red cells will agglutinate, proving that there is IgG and/or C3 on the red cell membrane. The finding of a positive DAT in the setting of a hemolytic anemia is diagnostic of AIHA. Beware of individuals with concomitant weak positive DAT and other causes of hemolysis. The strength of the DAT result and the degree of hemolysis must match in order to conclude that the hemolysis is immune-mediated.
There are several pitfalls to the DAT. One is that a positive DAT is found in 1:1000 patients in the normal population and in up to several percent of ill patients, especially those with elevated gamma globulin, such as patients with liver disease or HIV infection.6 Administration of intravenous immunoglobulin (IVIG) can also create a positive DAT. Conversely, patients can have AIHA with a negative DAT.7-9 For some patients, the number of IgG molecules bound to the red cell is below the detection limit of the DAT reagents. Other patients can have IgA or “warm” IgM as the cause of the AIHA.10 Specialty laboratories can test for these possibilities. The diagnosis of DAT-negative AIHA should be made with caution, and other causes of hemolysis, such as hereditary spherocytosis or paroxysmal hematuria, should be excluded.
Transfusion Therapy
Performing transfusions can be very difficult in patients with AIHA.2 The presence of the autoantibody can interfere with typing of the blood and almost always interferes with the crossmatch, since this final step consists of mixing the patient’s serum or plasma with donor red cells. In most patients with AIHA, the autoantibodies will react with any donor cells, rendering a negative crossmatch impossible. Without the crossmatch, the concern is that underlying alloantibodies can be missed. Studies indicate that 15% to 32% of patients will have underlying alloantibodies, which can lead to transfusion reactions.2 However, there are 2 considerations that may mitigate these concerns.11,12 First, patients who have never been transfused or pregnant will rarely have alloantibodies. Second, a patient who has been transfused in the remote past may have an anamnestic antibody response but not an immediate hemolytic reaction.
The transfusion service can take several steps to identify alloantibodies. Occasionally, if the autoantibody is weakly reacting when the patient’s serum is tested against a panel of reagent red cells, the alloantibodies can be identified by their stronger reactions as compared with the weakly reactive autoantibody. The most common technique for identifying alloantibodies is the autoadsorption technique.4,13 This involves incu-bating the patient’s red cells with the patient’s serum to adsorb the autoantibody. After a period of incubation, the cells are pelleted and the serum is collected as the supernatant. The adsorbed serum may be incubated with another sample of the patient’s cells for a second adsorption if the initial agglutination reactions of the patient’s serum with the reagent cells were strong. After 1 to 3 adsorptions, the adsorbed serum is tested with a red cell panel in order to check for “leftover” alloantibodies.
When a patient is first suspected of having AIHA, a generous sample of blood should be given to the transfusion service to allow for adequate testing. Many centers will test the blood not only for blood groups ABO and D but also perform full Rh typing plus check for Kidd, Duffy and Kell status.14 Increasingly, this is performed by direct genetic sequencing for the appropriate genotypes. This can allow transfusion of phenotypically matched red blood cells to lessen the risk of alloantibody formation.
One difficult issue is timing of transfusion. Clinicians are often hesitant to transfuse patients with AIHA due to fear of reactions, but in patients with severe anemia, especially elderly patients or those with heart disease, transfusion can be lifesaving. Since in some cases it may take hours to screen for alloantibodies, it is often preferable to transfuse patients with severe anemia and observe carefully for reaction.
1. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
2. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
3. Seve P, Philippe P, Dufour JF, et al. Autoimmune hemolytic anemia: classification and therapeutic approaches. Expert Rev Hematol. 2008;1:189-204.
4. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
5. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
6. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
7. Michel M. Classification and therapeutic approaches in autoimmune hemolytic anemia: an update. Expert Rev Hematol. 2011;4:607-618.
8. Garratty G. Immune hemolytic anemia associated with negative routine serology. Semin Hematol. 2005;42:156-164.
9. Sachs UJ, Roder L, Santoso S, Bein G. Does a negative direct antiglobulin test exclude warm autoimmune haemolytic anaemia? A prospective study of 504 cases. Br J Haematol. 2006;132:655-656.
10. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell autoantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
11. Petz LD. “Least incompatible” units for transfusion in autoimmune hemolytic anemia: should we eliminate this meaningless term? A commentary for clinicians and transfusion medicine professionals. Transfusion. 2003;43:1503-1507.
12. Blackall DP. How do I approach patients with warm-reactive autoantibodies? Transfusion. 2011;51:14-17.
13. Winkelmayer WC, Liu J, Setoguchi S, Choudhry NK. Effectiveness and safety of warfarin initiation in older hemodialysis patients with incident atrial fibrillation. Clin J Am Soc Nephrol. 2011;6:2662-2668.
14. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons.1,2 AIHA is mediated by antibodies, and in the majority of cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C (Table 1). This article series reviews the most common types of AIHA, with an overview of evaluation and diagnosis presented in this article and management of warm, cold, and drug-induced AIHA reviewed in a separate article.
Pathogenesis
In most cases, the ultimate etiology of AIHA is unknown. In warm AIHA, the target epitopes in most cases are Rh proteins.2 What leads the immune system to target these proteins is unidentified, but one theory is that an initial immune response to a foreign antigen starts to cross-react with the Rh proteins and the immune system fails to suppress this autoreactive response, leading to hemolysis. In IgG-mediated (warm) hemolysis, the red cells become coated with IgG molecules, which mark the cells for uptake and destruction by splenic macrophages.3 In "cold" AIHA, IgM molecules fix complement to the surface of red blood cells. Rarely, this can lead to activation of the full complement cascade, resulting in red cell lysis, but more often it is stopped at the C3 stage, leading to C3-coated red cells which are then taken up by hepatic macrophages.4
Suspecting the Diagnosis
In many patients, it is the symptoms and signs of anemia that lead to suspicion of hemolysis. Older patients often present earlier in the course of the disease due to lack of tolerance of anemia, especially if there is a sudden drop in the red blood cell count. Dark, cola-colored urine resulting from the presence of free hemoglobin may be noted by some patients. Patients with rapid-onset hemolysis may note lumbar back pain, and those with cold agglutinins often note symptoms related to agglutination of red cells in the peripheral circulation, such as the development of acrocyanosis in cold weather.5 In rare cases, patients will have abdominal pain when eating cold food due to ischemia related to agglutination of red cells in the viscera. Some patients with cold agglutinins can have an exacerbation of their hemolysis with cold exposure.
Unlike patients with immune thrombocytopenia, those with AIHA may have mild splenomegaly on exam. The presence of enlarged lymph nodes or massive splenomegaly should raise concern about concomitant lymphoma or chronic lymphocytic leukemia.
Making the Diagnosis
The 2 key steps in diagnosis are (1) demonstrating hemolysis and (2) demonstrating the autoimmune component.
Laboratory Evaluation for Hemolysis
Hemolysis is proven by finding evidence of both red cell breakdown and the compensatory increase in red cell production this stimulates (Table 2). The following sections discuss the laboratory tests that are performed to investigate hemolysis.
Lactate Dehydrogenase
When red cells undergo hemolysis, they release their contents, which are mostly comprised of hemoglobin but also include lactate dehydrogenase (LDH), an enzyme found in high concentration in red cells. Most patients with hemolysis will have an elevated LDH level, making this a sensitive test. However, because many other processes, including liver disease and pneumonia, also raise the serum LDH level, this finding is not specific for hemolysis.
Serum Bilirubin
Hemoglobin is salvaged by haptoglobin, and the heme moiety is broken down first to bilirubin and then to urobilinogen, which is excreted in the urine.2 Bilirubin produced from the breakdown of heme is not conjugated, but rather is delivered to the liver, where it is conjugated and excreted into the bile. In hemolysis, the concentration of unconjugated bilirubin (indirect bilirubin) is increased, while in liver disease the level of conjugated bilirubin (direct bilirubin) is increased. However, if the patient has concomitant liver disease with an increased direct bilirubin level, the serum bilirubin test is not reliable.
Serum Haptoglobin
Haptoglobin binds free serum hemoglobin and is taken up by the liver. Haptoglobin usually falls to very low levels in hemolysis. A confounder is that haptoglobin is an acute phase reactant and can rise with systemic disease or inflammation. However, patients with advanced liver disease will have low haptoglobin levels due to lack of synthesis, and up to 2% of the population may congenitally lack haptoglobin.1
Serum Hemoglobin
If the hemolysis is very rapid, the amount of free hemoglobin released will overwhelm the binding capacity of haptoglobin and lead to free hemoglobin in the plasma. This can be crudely quantified by examining the plasma color. Even minute amounts of free hemoglobin will turn the plasma pink. In fulminant hemolysis, the plasma will be cola-colored.
Reticulocyte Count
In most patients with hemolysis, the destruction of red cells is accompanied by an increase in the reticulocyte count. Reticulocytes are red cells that still contain RNA and are a marker of red cells that are approximately 24 hours old or less. Traditionally, reticulocytes were measured manually by staining the blood smear with vital blue and counting the percentage of cells that absorb the stain; this percentage needs to be adjusted for the hematocrit. Usually a percentage above 1.5% is considered indicative of an elevated reticulocyte count. Recently, automated complete blood count machines have taken advantage of the fact that reticulocytes will absorb certain stains; these machines can directly measure the reticulocyte count via flow cytometry, which results in an “absolute” reticulocyte count. The reticulocyte count obtained using this method does not have to be corrected for hematocrit, and levels of approximately 90,000/μL are considered raised. However, the reticulocyte count can also be raised in blood loss or in patients who have other causes of anemia (eg, iron deficiency) under treatment. In addition, as many as 25% of patients with AIHA will never have raised counts for various reasons, such as nutritional deficiency, autoimmune destruction of red cell precursors, or lack of erythropoietin.
Blood Smear
The blood smear provides vital information. The hallmark laboratory parameter of AIHA is spherocytes seen on the blood smear. In AIHA, antibodies and/or complement attach to the red cells, and when the antibodies or complement are taken up by macrophages in the spleen some of the red blood cell mem-brane is removed as well, decreasing the surface area of the cell. As the surface area of the red cell decreases with each pass through the spleen, the cell's shape changes from a biconcave disk to a sphere before the cell is destroyed, reflecting the fact that a sphere has the smallest surface area for a given volume. The vast majority of patients with AIHA will have spherocytes on the blood smear. However, spherocytes are not specific to AIHA, as they can be seen in hereditary spherocytosis, Wilson’s disease, clostridial sepsis, and severe burns.
Patients with cold agglutinins will often have red cell agglutination on the blood smear. In addition, patients with AIHA will often have a raised mean corpuscular volume (MCV) for 2 reasons. In patients with brisk reticulocytosis, the MCV will be raised due to the large size of the reticulocyte. In patients with cold agglutinin disease, the MCV may be falsely raised due to clumping of the red blood cells.
Urinary Hemosiderin
When hemoglobin is excreted by the kidney, the iron is deposited in the tubules. When the tubule cells are sloughed off, they appear in the urine. The urine can be stained for iron, and a positive result is another sign of hemolysis. Hemosiderinuria is a later sign of hemolysis, as it takes 1 week for iron-laden tubule cells to be excreted in sufficient quantities to be detected in the urine.
Urinary Hemoglobin
One other sign of hemolysis is the presence of hemoglobin in the urine. A quick way to demonstrate hemoglobinuria is to check the urine with a dipstick followed by a microscopic exam. In hemolysis, the dipstick will detect “blood,” while the microscopic exam will be negative for red cells.
Laboratory Evaluation for Autoimmune Component
The autoimmune component is shown by demonstrating the presence of either IgG molecules or complement on the surface of red blood cells.4,6 This can be done by performing the direct antiglobulin test (DAT) or Coombs test. IgG bound to red cells will not agglutinate them, but if IgM that is directed against IgG or C3 is added, the red cells will agglutinate, proving that there is IgG and/or C3 on the red cell membrane. The finding of a positive DAT in the setting of a hemolytic anemia is diagnostic of AIHA. Beware of individuals with concomitant weak positive DAT and other causes of hemolysis. The strength of the DAT result and the degree of hemolysis must match in order to conclude that the hemolysis is immune-mediated.
There are several pitfalls to the DAT. One is that a positive DAT is found in 1:1000 patients in the normal population and in up to several percent of ill patients, especially those with elevated gamma globulin, such as patients with liver disease or HIV infection.6 Administration of intravenous immunoglobulin (IVIG) can also create a positive DAT. Conversely, patients can have AIHA with a negative DAT.7-9 For some patients, the number of IgG molecules bound to the red cell is below the detection limit of the DAT reagents. Other patients can have IgA or “warm” IgM as the cause of the AIHA.10 Specialty laboratories can test for these possibilities. The diagnosis of DAT-negative AIHA should be made with caution, and other causes of hemolysis, such as hereditary spherocytosis or paroxysmal hematuria, should be excluded.
Transfusion Therapy
Performing transfusions can be very difficult in patients with AIHA.2 The presence of the autoantibody can interfere with typing of the blood and almost always interferes with the crossmatch, since this final step consists of mixing the patient’s serum or plasma with donor red cells. In most patients with AIHA, the autoantibodies will react with any donor cells, rendering a negative crossmatch impossible. Without the crossmatch, the concern is that underlying alloantibodies can be missed. Studies indicate that 15% to 32% of patients will have underlying alloantibodies, which can lead to transfusion reactions.2 However, there are 2 considerations that may mitigate these concerns.11,12 First, patients who have never been transfused or pregnant will rarely have alloantibodies. Second, a patient who has been transfused in the remote past may have an anamnestic antibody response but not an immediate hemolytic reaction.
The transfusion service can take several steps to identify alloantibodies. Occasionally, if the autoantibody is weakly reacting when the patient’s serum is tested against a panel of reagent red cells, the alloantibodies can be identified by their stronger reactions as compared with the weakly reactive autoantibody. The most common technique for identifying alloantibodies is the autoadsorption technique.4,13 This involves incu-bating the patient’s red cells with the patient’s serum to adsorb the autoantibody. After a period of incubation, the cells are pelleted and the serum is collected as the supernatant. The adsorbed serum may be incubated with another sample of the patient’s cells for a second adsorption if the initial agglutination reactions of the patient’s serum with the reagent cells were strong. After 1 to 3 adsorptions, the adsorbed serum is tested with a red cell panel in order to check for “leftover” alloantibodies.
When a patient is first suspected of having AIHA, a generous sample of blood should be given to the transfusion service to allow for adequate testing. Many centers will test the blood not only for blood groups ABO and D but also perform full Rh typing plus check for Kidd, Duffy and Kell status.14 Increasingly, this is performed by direct genetic sequencing for the appropriate genotypes. This can allow transfusion of phenotypically matched red blood cells to lessen the risk of alloantibody formation.
One difficult issue is timing of transfusion. Clinicians are often hesitant to transfuse patients with AIHA due to fear of reactions, but in patients with severe anemia, especially elderly patients or those with heart disease, transfusion can be lifesaving. Since in some cases it may take hours to screen for alloantibodies, it is often preferable to transfuse patients with severe anemia and observe carefully for reaction.
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons.1,2 AIHA is mediated by antibodies, and in the majority of cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C (Table 1). This article series reviews the most common types of AIHA, with an overview of evaluation and diagnosis presented in this article and management of warm, cold, and drug-induced AIHA reviewed in a separate article.
Pathogenesis
In most cases, the ultimate etiology of AIHA is unknown. In warm AIHA, the target epitopes in most cases are Rh proteins.2 What leads the immune system to target these proteins is unidentified, but one theory is that an initial immune response to a foreign antigen starts to cross-react with the Rh proteins and the immune system fails to suppress this autoreactive response, leading to hemolysis. In IgG-mediated (warm) hemolysis, the red cells become coated with IgG molecules, which mark the cells for uptake and destruction by splenic macrophages.3 In "cold" AIHA, IgM molecules fix complement to the surface of red blood cells. Rarely, this can lead to activation of the full complement cascade, resulting in red cell lysis, but more often it is stopped at the C3 stage, leading to C3-coated red cells which are then taken up by hepatic macrophages.4
Suspecting the Diagnosis
In many patients, it is the symptoms and signs of anemia that lead to suspicion of hemolysis. Older patients often present earlier in the course of the disease due to lack of tolerance of anemia, especially if there is a sudden drop in the red blood cell count. Dark, cola-colored urine resulting from the presence of free hemoglobin may be noted by some patients. Patients with rapid-onset hemolysis may note lumbar back pain, and those with cold agglutinins often note symptoms related to agglutination of red cells in the peripheral circulation, such as the development of acrocyanosis in cold weather.5 In rare cases, patients will have abdominal pain when eating cold food due to ischemia related to agglutination of red cells in the viscera. Some patients with cold agglutinins can have an exacerbation of their hemolysis with cold exposure.
Unlike patients with immune thrombocytopenia, those with AIHA may have mild splenomegaly on exam. The presence of enlarged lymph nodes or massive splenomegaly should raise concern about concomitant lymphoma or chronic lymphocytic leukemia.
Making the Diagnosis
The 2 key steps in diagnosis are (1) demonstrating hemolysis and (2) demonstrating the autoimmune component.
Laboratory Evaluation for Hemolysis
Hemolysis is proven by finding evidence of both red cell breakdown and the compensatory increase in red cell production this stimulates (Table 2). The following sections discuss the laboratory tests that are performed to investigate hemolysis.
Lactate Dehydrogenase
When red cells undergo hemolysis, they release their contents, which are mostly comprised of hemoglobin but also include lactate dehydrogenase (LDH), an enzyme found in high concentration in red cells. Most patients with hemolysis will have an elevated LDH level, making this a sensitive test. However, because many other processes, including liver disease and pneumonia, also raise the serum LDH level, this finding is not specific for hemolysis.
Serum Bilirubin
Hemoglobin is salvaged by haptoglobin, and the heme moiety is broken down first to bilirubin and then to urobilinogen, which is excreted in the urine.2 Bilirubin produced from the breakdown of heme is not conjugated, but rather is delivered to the liver, where it is conjugated and excreted into the bile. In hemolysis, the concentration of unconjugated bilirubin (indirect bilirubin) is increased, while in liver disease the level of conjugated bilirubin (direct bilirubin) is increased. However, if the patient has concomitant liver disease with an increased direct bilirubin level, the serum bilirubin test is not reliable.
Serum Haptoglobin
Haptoglobin binds free serum hemoglobin and is taken up by the liver. Haptoglobin usually falls to very low levels in hemolysis. A confounder is that haptoglobin is an acute phase reactant and can rise with systemic disease or inflammation. However, patients with advanced liver disease will have low haptoglobin levels due to lack of synthesis, and up to 2% of the population may congenitally lack haptoglobin.1
Serum Hemoglobin
If the hemolysis is very rapid, the amount of free hemoglobin released will overwhelm the binding capacity of haptoglobin and lead to free hemoglobin in the plasma. This can be crudely quantified by examining the plasma color. Even minute amounts of free hemoglobin will turn the plasma pink. In fulminant hemolysis, the plasma will be cola-colored.
Reticulocyte Count
In most patients with hemolysis, the destruction of red cells is accompanied by an increase in the reticulocyte count. Reticulocytes are red cells that still contain RNA and are a marker of red cells that are approximately 24 hours old or less. Traditionally, reticulocytes were measured manually by staining the blood smear with vital blue and counting the percentage of cells that absorb the stain; this percentage needs to be adjusted for the hematocrit. Usually a percentage above 1.5% is considered indicative of an elevated reticulocyte count. Recently, automated complete blood count machines have taken advantage of the fact that reticulocytes will absorb certain stains; these machines can directly measure the reticulocyte count via flow cytometry, which results in an “absolute” reticulocyte count. The reticulocyte count obtained using this method does not have to be corrected for hematocrit, and levels of approximately 90,000/μL are considered raised. However, the reticulocyte count can also be raised in blood loss or in patients who have other causes of anemia (eg, iron deficiency) under treatment. In addition, as many as 25% of patients with AIHA will never have raised counts for various reasons, such as nutritional deficiency, autoimmune destruction of red cell precursors, or lack of erythropoietin.
Blood Smear
The blood smear provides vital information. The hallmark laboratory parameter of AIHA is spherocytes seen on the blood smear. In AIHA, antibodies and/or complement attach to the red cells, and when the antibodies or complement are taken up by macrophages in the spleen some of the red blood cell mem-brane is removed as well, decreasing the surface area of the cell. As the surface area of the red cell decreases with each pass through the spleen, the cell's shape changes from a biconcave disk to a sphere before the cell is destroyed, reflecting the fact that a sphere has the smallest surface area for a given volume. The vast majority of patients with AIHA will have spherocytes on the blood smear. However, spherocytes are not specific to AIHA, as they can be seen in hereditary spherocytosis, Wilson’s disease, clostridial sepsis, and severe burns.
Patients with cold agglutinins will often have red cell agglutination on the blood smear. In addition, patients with AIHA will often have a raised mean corpuscular volume (MCV) for 2 reasons. In patients with brisk reticulocytosis, the MCV will be raised due to the large size of the reticulocyte. In patients with cold agglutinin disease, the MCV may be falsely raised due to clumping of the red blood cells.
Urinary Hemosiderin
When hemoglobin is excreted by the kidney, the iron is deposited in the tubules. When the tubule cells are sloughed off, they appear in the urine. The urine can be stained for iron, and a positive result is another sign of hemolysis. Hemosiderinuria is a later sign of hemolysis, as it takes 1 week for iron-laden tubule cells to be excreted in sufficient quantities to be detected in the urine.
Urinary Hemoglobin
One other sign of hemolysis is the presence of hemoglobin in the urine. A quick way to demonstrate hemoglobinuria is to check the urine with a dipstick followed by a microscopic exam. In hemolysis, the dipstick will detect “blood,” while the microscopic exam will be negative for red cells.
Laboratory Evaluation for Autoimmune Component
The autoimmune component is shown by demonstrating the presence of either IgG molecules or complement on the surface of red blood cells.4,6 This can be done by performing the direct antiglobulin test (DAT) or Coombs test. IgG bound to red cells will not agglutinate them, but if IgM that is directed against IgG or C3 is added, the red cells will agglutinate, proving that there is IgG and/or C3 on the red cell membrane. The finding of a positive DAT in the setting of a hemolytic anemia is diagnostic of AIHA. Beware of individuals with concomitant weak positive DAT and other causes of hemolysis. The strength of the DAT result and the degree of hemolysis must match in order to conclude that the hemolysis is immune-mediated.
There are several pitfalls to the DAT. One is that a positive DAT is found in 1:1000 patients in the normal population and in up to several percent of ill patients, especially those with elevated gamma globulin, such as patients with liver disease or HIV infection.6 Administration of intravenous immunoglobulin (IVIG) can also create a positive DAT. Conversely, patients can have AIHA with a negative DAT.7-9 For some patients, the number of IgG molecules bound to the red cell is below the detection limit of the DAT reagents. Other patients can have IgA or “warm” IgM as the cause of the AIHA.10 Specialty laboratories can test for these possibilities. The diagnosis of DAT-negative AIHA should be made with caution, and other causes of hemolysis, such as hereditary spherocytosis or paroxysmal hematuria, should be excluded.
Transfusion Therapy
Performing transfusions can be very difficult in patients with AIHA.2 The presence of the autoantibody can interfere with typing of the blood and almost always interferes with the crossmatch, since this final step consists of mixing the patient’s serum or plasma with donor red cells. In most patients with AIHA, the autoantibodies will react with any donor cells, rendering a negative crossmatch impossible. Without the crossmatch, the concern is that underlying alloantibodies can be missed. Studies indicate that 15% to 32% of patients will have underlying alloantibodies, which can lead to transfusion reactions.2 However, there are 2 considerations that may mitigate these concerns.11,12 First, patients who have never been transfused or pregnant will rarely have alloantibodies. Second, a patient who has been transfused in the remote past may have an anamnestic antibody response but not an immediate hemolytic reaction.
The transfusion service can take several steps to identify alloantibodies. Occasionally, if the autoantibody is weakly reacting when the patient’s serum is tested against a panel of reagent red cells, the alloantibodies can be identified by their stronger reactions as compared with the weakly reactive autoantibody. The most common technique for identifying alloantibodies is the autoadsorption technique.4,13 This involves incu-bating the patient’s red cells with the patient’s serum to adsorb the autoantibody. After a period of incubation, the cells are pelleted and the serum is collected as the supernatant. The adsorbed serum may be incubated with another sample of the patient’s cells for a second adsorption if the initial agglutination reactions of the patient’s serum with the reagent cells were strong. After 1 to 3 adsorptions, the adsorbed serum is tested with a red cell panel in order to check for “leftover” alloantibodies.
When a patient is first suspected of having AIHA, a generous sample of blood should be given to the transfusion service to allow for adequate testing. Many centers will test the blood not only for blood groups ABO and D but also perform full Rh typing plus check for Kidd, Duffy and Kell status.14 Increasingly, this is performed by direct genetic sequencing for the appropriate genotypes. This can allow transfusion of phenotypically matched red blood cells to lessen the risk of alloantibody formation.
One difficult issue is timing of transfusion. Clinicians are often hesitant to transfuse patients with AIHA due to fear of reactions, but in patients with severe anemia, especially elderly patients or those with heart disease, transfusion can be lifesaving. Since in some cases it may take hours to screen for alloantibodies, it is often preferable to transfuse patients with severe anemia and observe carefully for reaction.
1. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
2. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
3. Seve P, Philippe P, Dufour JF, et al. Autoimmune hemolytic anemia: classification and therapeutic approaches. Expert Rev Hematol. 2008;1:189-204.
4. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
5. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
6. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
7. Michel M. Classification and therapeutic approaches in autoimmune hemolytic anemia: an update. Expert Rev Hematol. 2011;4:607-618.
8. Garratty G. Immune hemolytic anemia associated with negative routine serology. Semin Hematol. 2005;42:156-164.
9. Sachs UJ, Roder L, Santoso S, Bein G. Does a negative direct antiglobulin test exclude warm autoimmune haemolytic anaemia? A prospective study of 504 cases. Br J Haematol. 2006;132:655-656.
10. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell autoantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
11. Petz LD. “Least incompatible” units for transfusion in autoimmune hemolytic anemia: should we eliminate this meaningless term? A commentary for clinicians and transfusion medicine professionals. Transfusion. 2003;43:1503-1507.
12. Blackall DP. How do I approach patients with warm-reactive autoantibodies? Transfusion. 2011;51:14-17.
13. Winkelmayer WC, Liu J, Setoguchi S, Choudhry NK. Effectiveness and safety of warfarin initiation in older hemodialysis patients with incident atrial fibrillation. Clin J Am Soc Nephrol. 2011;6:2662-2668.
14. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
1. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
2. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
3. Seve P, Philippe P, Dufour JF, et al. Autoimmune hemolytic anemia: classification and therapeutic approaches. Expert Rev Hematol. 2008;1:189-204.
4. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
5. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
6. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
7. Michel M. Classification and therapeutic approaches in autoimmune hemolytic anemia: an update. Expert Rev Hematol. 2011;4:607-618.
8. Garratty G. Immune hemolytic anemia associated with negative routine serology. Semin Hematol. 2005;42:156-164.
9. Sachs UJ, Roder L, Santoso S, Bein G. Does a negative direct antiglobulin test exclude warm autoimmune haemolytic anaemia? A prospective study of 504 cases. Br J Haematol. 2006;132:655-656.
10. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell autoantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
11. Petz LD. “Least incompatible” units for transfusion in autoimmune hemolytic anemia: should we eliminate this meaningless term? A commentary for clinicians and transfusion medicine professionals. Transfusion. 2003;43:1503-1507.
12. Blackall DP. How do I approach patients with warm-reactive autoantibodies? Transfusion. 2011;51:14-17.
13. Winkelmayer WC, Liu J, Setoguchi S, Choudhry NK. Effectiveness and safety of warfarin initiation in older hemodialysis patients with incident atrial fibrillation. Clin J Am Soc Nephrol. 2011;6:2662-2668.
14. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
Readmission burden high for those with sickle cell disease
, according to the Agency for Healthcare Research and Quality.
The 30-day all-cause readmission rate for index stays with a principal diagnosis of SCD was 33.5% in 2016, compared with 12.5% for non-SCD hospital stays. Patients with a secondary diagnosis of SCD had readmission rates of 32.9% with a pain crisis and 21.0% without one, and the overall readmission rate for an index stay with any SCD diagnosis was 31.1%, Kathryn R. Fingar, PhD, MPH, of IBM Watson Health, Sacramento, Calif., and associates wrote in an AHRQ statistical brief.
When age is factored in, the readmission gap between a principal SCD diagnosis and non-SCD becomes even greater – and smaller. The difference was greatest for patients aged 18-34 years – 39.4% with a principal diagnosis of SCD versus 7.8% without any SCD – and then narrowed as patients got older. For those aged 65 years and older, the rates were 22.6% with a principal diagnosis of SCD and 15.9% without, the investigators reported.
The approximately 100,000 Americans with SCD accounted for 134,000 admissions in 2016, and more than three-quarters of those stays involved a pain crisis. A principal diagnosis of SCD was recorded for almost 96,000 of those visits, and nearly all (96%) of those stays involved a pain crisis. For those with a secondary diagnosis of SCD, the most common reasons for hospitalization were diseases of the respiratory system (14.3% of those stays) and infectious and parasitic diseases (13.2%).
Patients with SCD were more likely than non-SCD patients to be admitted from the ED (79.6% vs. 51.3%), and they were more likely to discharged against medical advice (4.1% vs. 1.2%). Among those who left the hospital against medical advice, patients with SCD were much more likely to be readmitted than those without SCD (46.6% vs. 26.5%), based on data from the AHRQ’s Nationwide Readmissions Database.
Improved treatment of complications has “reduced mortality rates so that nearly 95% of individuals born with SCD in the United States reach 18 years of age [but] limited knowledge of SCD treatment guidelines among healthcare professionals continues to pose a barrier to effective patient-provider relationships, and this barrier contributes to lower quality of life,” Dr. Fingar and associates wrote.
, according to the Agency for Healthcare Research and Quality.
The 30-day all-cause readmission rate for index stays with a principal diagnosis of SCD was 33.5% in 2016, compared with 12.5% for non-SCD hospital stays. Patients with a secondary diagnosis of SCD had readmission rates of 32.9% with a pain crisis and 21.0% without one, and the overall readmission rate for an index stay with any SCD diagnosis was 31.1%, Kathryn R. Fingar, PhD, MPH, of IBM Watson Health, Sacramento, Calif., and associates wrote in an AHRQ statistical brief.
When age is factored in, the readmission gap between a principal SCD diagnosis and non-SCD becomes even greater – and smaller. The difference was greatest for patients aged 18-34 years – 39.4% with a principal diagnosis of SCD versus 7.8% without any SCD – and then narrowed as patients got older. For those aged 65 years and older, the rates were 22.6% with a principal diagnosis of SCD and 15.9% without, the investigators reported.
The approximately 100,000 Americans with SCD accounted for 134,000 admissions in 2016, and more than three-quarters of those stays involved a pain crisis. A principal diagnosis of SCD was recorded for almost 96,000 of those visits, and nearly all (96%) of those stays involved a pain crisis. For those with a secondary diagnosis of SCD, the most common reasons for hospitalization were diseases of the respiratory system (14.3% of those stays) and infectious and parasitic diseases (13.2%).
Patients with SCD were more likely than non-SCD patients to be admitted from the ED (79.6% vs. 51.3%), and they were more likely to discharged against medical advice (4.1% vs. 1.2%). Among those who left the hospital against medical advice, patients with SCD were much more likely to be readmitted than those without SCD (46.6% vs. 26.5%), based on data from the AHRQ’s Nationwide Readmissions Database.
Improved treatment of complications has “reduced mortality rates so that nearly 95% of individuals born with SCD in the United States reach 18 years of age [but] limited knowledge of SCD treatment guidelines among healthcare professionals continues to pose a barrier to effective patient-provider relationships, and this barrier contributes to lower quality of life,” Dr. Fingar and associates wrote.
, according to the Agency for Healthcare Research and Quality.
The 30-day all-cause readmission rate for index stays with a principal diagnosis of SCD was 33.5% in 2016, compared with 12.5% for non-SCD hospital stays. Patients with a secondary diagnosis of SCD had readmission rates of 32.9% with a pain crisis and 21.0% without one, and the overall readmission rate for an index stay with any SCD diagnosis was 31.1%, Kathryn R. Fingar, PhD, MPH, of IBM Watson Health, Sacramento, Calif., and associates wrote in an AHRQ statistical brief.
When age is factored in, the readmission gap between a principal SCD diagnosis and non-SCD becomes even greater – and smaller. The difference was greatest for patients aged 18-34 years – 39.4% with a principal diagnosis of SCD versus 7.8% without any SCD – and then narrowed as patients got older. For those aged 65 years and older, the rates were 22.6% with a principal diagnosis of SCD and 15.9% without, the investigators reported.
The approximately 100,000 Americans with SCD accounted for 134,000 admissions in 2016, and more than three-quarters of those stays involved a pain crisis. A principal diagnosis of SCD was recorded for almost 96,000 of those visits, and nearly all (96%) of those stays involved a pain crisis. For those with a secondary diagnosis of SCD, the most common reasons for hospitalization were diseases of the respiratory system (14.3% of those stays) and infectious and parasitic diseases (13.2%).
Patients with SCD were more likely than non-SCD patients to be admitted from the ED (79.6% vs. 51.3%), and they were more likely to discharged against medical advice (4.1% vs. 1.2%). Among those who left the hospital against medical advice, patients with SCD were much more likely to be readmitted than those without SCD (46.6% vs. 26.5%), based on data from the AHRQ’s Nationwide Readmissions Database.
Improved treatment of complications has “reduced mortality rates so that nearly 95% of individuals born with SCD in the United States reach 18 years of age [but] limited knowledge of SCD treatment guidelines among healthcare professionals continues to pose a barrier to effective patient-provider relationships, and this barrier contributes to lower quality of life,” Dr. Fingar and associates wrote.
In older patients with immune-mediated TTP, atypical features may delay diagnosis
Older patients with immune thrombotic thrombocytopenic purpura (iTTP) more often have an atypical neurological presentation, which could result in a delayed diagnosis, according to authors of a recent retrospective analysis.
“Practitioners should be aware of this in order to shorten the time to treatment, which could improve the prognosis in older iTTP patients,” Paul Coppo, MD, PhD, of Hôpital Saint-Antoine, Paris, and coauthors wrote in Blood.
The older patients also had increased 1-month and 1-year mortality compared with younger patients, and had more than a threefold risk of long-term mortality compared with elderly patients without iTTP, according to the study report.
The analysis included 411 patients with iTTP entered into a national registry in France between 2000 and 2016. Seventy-one patients were 60 years of age or older.
Time from hospital admission to diagnosis was 3 days for those older patients, versus just 1 day for patients under 60 years of age (P = .0001), Dr. Coppo and colleagues reported.
Clinical records were available for 67 of the older iTTP patients, of whom 17 had no evidence of delayed diagnosis. The remainder had a “possible diagnostic delay,” according to the report; among those, the iTTP diagnosis was preceded by neurological manifestations in 26 cases, and transient ischemic stroke that usually led to focal deficiency or aphasia in 14 cases. Other features preceding the diagnosis included malaise, behavioral abnormalities, seizure, and dizziness.
Many of these findings are “not specific to a disease, and they are less alarming than in young patients,” the researchers wrote. “In this context, the presence of a thrombocytopenia with anemia should alert physicians to this possible rare diagnosis.”
Older patients also presented with less pronounced cytopenias compared with younger patients, which could have contributed to a late diagnosis, they added.
Older age is a known risk factor for mortality related to iTTP. In the present study, rates of 1-month mortality were 37% for patients aged 60 years and older, and 9% for those younger than age 60 (P less than .0001). The 1-year mortality rates were 49% and 11% for older and younger patients, respectively (P less than .0001).
Compared with older individuals without iTTP from a different study, older iTTP patients had a lower long-term survival rate. iTTP remained an independent risk factor for death even after adjustment for age, sex, and some comorbidities (hazard ratio, 3.44; 95% confidence interval, 2.02-5.87).
The study was partly funded by a grant from the French Ministry of Health. Dr. Coppo reported that he is a clinical advisory board member for Alexion, Ablynx (now part of Sanofi), Shire, and Octapharma. Two other co-authors reported participating in advisory boards for Ablynx.
SOURCE: Prevel R et al. Blood. 2019 Sep 17. doi: 10.1182/blood.2019000748.
Older patients with immune thrombotic thrombocytopenic purpura (iTTP) more often have an atypical neurological presentation, which could result in a delayed diagnosis, according to authors of a recent retrospective analysis.
“Practitioners should be aware of this in order to shorten the time to treatment, which could improve the prognosis in older iTTP patients,” Paul Coppo, MD, PhD, of Hôpital Saint-Antoine, Paris, and coauthors wrote in Blood.
The older patients also had increased 1-month and 1-year mortality compared with younger patients, and had more than a threefold risk of long-term mortality compared with elderly patients without iTTP, according to the study report.
The analysis included 411 patients with iTTP entered into a national registry in France between 2000 and 2016. Seventy-one patients were 60 years of age or older.
Time from hospital admission to diagnosis was 3 days for those older patients, versus just 1 day for patients under 60 years of age (P = .0001), Dr. Coppo and colleagues reported.
Clinical records were available for 67 of the older iTTP patients, of whom 17 had no evidence of delayed diagnosis. The remainder had a “possible diagnostic delay,” according to the report; among those, the iTTP diagnosis was preceded by neurological manifestations in 26 cases, and transient ischemic stroke that usually led to focal deficiency or aphasia in 14 cases. Other features preceding the diagnosis included malaise, behavioral abnormalities, seizure, and dizziness.
Many of these findings are “not specific to a disease, and they are less alarming than in young patients,” the researchers wrote. “In this context, the presence of a thrombocytopenia with anemia should alert physicians to this possible rare diagnosis.”
Older patients also presented with less pronounced cytopenias compared with younger patients, which could have contributed to a late diagnosis, they added.
Older age is a known risk factor for mortality related to iTTP. In the present study, rates of 1-month mortality were 37% for patients aged 60 years and older, and 9% for those younger than age 60 (P less than .0001). The 1-year mortality rates were 49% and 11% for older and younger patients, respectively (P less than .0001).
Compared with older individuals without iTTP from a different study, older iTTP patients had a lower long-term survival rate. iTTP remained an independent risk factor for death even after adjustment for age, sex, and some comorbidities (hazard ratio, 3.44; 95% confidence interval, 2.02-5.87).
The study was partly funded by a grant from the French Ministry of Health. Dr. Coppo reported that he is a clinical advisory board member for Alexion, Ablynx (now part of Sanofi), Shire, and Octapharma. Two other co-authors reported participating in advisory boards for Ablynx.
SOURCE: Prevel R et al. Blood. 2019 Sep 17. doi: 10.1182/blood.2019000748.
Older patients with immune thrombotic thrombocytopenic purpura (iTTP) more often have an atypical neurological presentation, which could result in a delayed diagnosis, according to authors of a recent retrospective analysis.
“Practitioners should be aware of this in order to shorten the time to treatment, which could improve the prognosis in older iTTP patients,” Paul Coppo, MD, PhD, of Hôpital Saint-Antoine, Paris, and coauthors wrote in Blood.
The older patients also had increased 1-month and 1-year mortality compared with younger patients, and had more than a threefold risk of long-term mortality compared with elderly patients without iTTP, according to the study report.
The analysis included 411 patients with iTTP entered into a national registry in France between 2000 and 2016. Seventy-one patients were 60 years of age or older.
Time from hospital admission to diagnosis was 3 days for those older patients, versus just 1 day for patients under 60 years of age (P = .0001), Dr. Coppo and colleagues reported.
Clinical records were available for 67 of the older iTTP patients, of whom 17 had no evidence of delayed diagnosis. The remainder had a “possible diagnostic delay,” according to the report; among those, the iTTP diagnosis was preceded by neurological manifestations in 26 cases, and transient ischemic stroke that usually led to focal deficiency or aphasia in 14 cases. Other features preceding the diagnosis included malaise, behavioral abnormalities, seizure, and dizziness.
Many of these findings are “not specific to a disease, and they are less alarming than in young patients,” the researchers wrote. “In this context, the presence of a thrombocytopenia with anemia should alert physicians to this possible rare diagnosis.”
Older patients also presented with less pronounced cytopenias compared with younger patients, which could have contributed to a late diagnosis, they added.
Older age is a known risk factor for mortality related to iTTP. In the present study, rates of 1-month mortality were 37% for patients aged 60 years and older, and 9% for those younger than age 60 (P less than .0001). The 1-year mortality rates were 49% and 11% for older and younger patients, respectively (P less than .0001).
Compared with older individuals without iTTP from a different study, older iTTP patients had a lower long-term survival rate. iTTP remained an independent risk factor for death even after adjustment for age, sex, and some comorbidities (hazard ratio, 3.44; 95% confidence interval, 2.02-5.87).
The study was partly funded by a grant from the French Ministry of Health. Dr. Coppo reported that he is a clinical advisory board member for Alexion, Ablynx (now part of Sanofi), Shire, and Octapharma. Two other co-authors reported participating in advisory boards for Ablynx.
SOURCE: Prevel R et al. Blood. 2019 Sep 17. doi: 10.1182/blood.2019000748.
FROM BLOOD