User login
Anti-CGRP monoclonal antibody offers relief from migraine and comorbid depression
AUSTIN, TEX. – , new research shows.
Patients with both conditions who were randomly assigned to receive fremanezumab showed a statistically significant reduction in both the 17-item Hamilton Depression Rating Scale (HAMD-17) and the nine-criteria Patient Health Questionnaire (PHQ-9) scores, compared with matched controls who received placebo.
The results from the UNITE trial were presented at the annual meeting of the American Headache Society.
Long-standing questions
“It’s been well known for a long time that migraine is comorbid with a number of illnesses, and one of the most common is depression,” said study investigator Richard B. Lipton, a professor of neurology at Albert Einstein College of Medicine and the director of the Montefiore Headache Center, New York.
“Do you treat the depression? Do you treat the migraine? Do you independently treat both? Those have been long-standing questions for clinicians,” Dr. Lipton said.
Investigators randomly assigned 330 adults with migraine who were diagnosed with moderate-to-severe MDD (defined as a PHQ-9 score of 10 or greater) to receive 225 mg subcutaneous monthly fremanezumab (n = 164) or placebo (n = 166) for 12 weeks.
The trial continued as an open-label trial for another 12 weeks.
During the double-blind phase of the study, the mean change from baseline in the HAMD-17 score with placebo was –4.6 at week 8 and –5.4 at week 12, compared with –6.0 with fremanezumab at week 8 (P = .0205) and –6.7 at week 12 (P = .0228).
The change from baseline in PHQ-9 total score at week 8 was –5.8 for placebo and –7.1 for fremanezumab (P = .0283). At week 12, the change was –6.3 for placebo versus –7.8 for fremanezumab (P = .0108). These reductions were maintained throughout the open-label period of the trial.
The beneficial effect on depression and migraine demonstrated in the study is interesting on several levels, Dr. Lipton said.
“One, it tells us that if the patient has migraine and depression and you treat with fremanezumab, both disorders get better to a statistically significant degree. That’s critically important,” he said.
“The other thing, and this is actually what I find most interesting about this study, is that fremanezumab doesn’t get into the brain. There are many antimigraine therapies that do, so you can treat a patient with migraine and depression with a tricyclic antidepressant.”
“It may make the migraine better and the depression better, but you don’t know if the benefit in depression comes from the improvement in migraine, because of course the antidepressant works for both conditions. Maybe there are people who would disagree with this, but my interpretation [of the trial results] is that the depression got better because the migraine got better,” he added.
The link between migraine and depression is well established, Dr. Lipton added. Longitudinal studies have shown that people with depression but without migraine develop migraine at increased rates, compared with people with no depression. Conversely, people with migraine but no depression develop depression at increased rates.
“Both disorders may have a common substrate, but I also think many forms of chronic pain lead to depression, and that’s the part we’re making better,” he said.
If fremanezumab has this dual effect on migraine and depression, it is possible that other anti-CGRP drugs will have a similar effect, Dr. Lipton said.
“Honestly, my hope is that other companies that make effective drugs will do similar studies to see if other monoclonal antibodies that target CGRP have the same effect. My guess is that all of them work but until the studies are done, I’m going to use fremanezumab, the one that has been studied, in my patients.”
He added that depression is an important comorbidity of migraine and represents a huge challenge for clinicians. “A lot of headache patients want to know what to do about comorbid anxiety or comorbid depression. I run a headache center in a specialty practice, and when people come in with migraine, they almost always come in with migraine and depression or anxiety or another pain disorder, or something else, and one of the great challenges in the practice is managing these comorbidities,” he said.
A bidirectional relationship
The overlap between migraine and depression and anxiety has been known for quite a while, agreed Elizabeth W. Loder, MD, MPH, vice chair of academic affairs, department of neurology, Brigham and Women’s Hospital, and professor of neurology at Harvard Medical School, both in Boston.
“I think the relationship is generally viewed as bidirectional and causality is uncertain. I still do not think I would assume that any drug that reduces migraine would reduce depression,” said Dr. Loder.
However, she added, the fremanezumab study data are interesting. “The effects of any drug on depression could be due to improvement of migraine or it could be due to some other effect of the treatment on depression. That is what makes these results so intriguing. If the findings are borne out by other studies, it could mean that these treatments would be preferred to those older ones in patients with depression,” Dr. Loder said.
Also commenting on the findings, Huma Sheikh, MD, CEO of NY Neurology Medicine PC, said the study is important because it confirms the strong association between migraine and depression. “Both conditions have similar underlying neurobiological pathophysiologies, and if you are impacting one area in the brain with the CGRP inhibitors, you might also be targeting some of the receptors or pathways that are involved in depression,” Dr. Sheikh said.
The study was funded by Teva Pharmaceuticals. Dr. Lipton reported financial relationships with Teva and multiple other pharmaceutical companies. Dr. Loder and Dr. Sheikh have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
AUSTIN, TEX. – , new research shows.
Patients with both conditions who were randomly assigned to receive fremanezumab showed a statistically significant reduction in both the 17-item Hamilton Depression Rating Scale (HAMD-17) and the nine-criteria Patient Health Questionnaire (PHQ-9) scores, compared with matched controls who received placebo.
The results from the UNITE trial were presented at the annual meeting of the American Headache Society.
Long-standing questions
“It’s been well known for a long time that migraine is comorbid with a number of illnesses, and one of the most common is depression,” said study investigator Richard B. Lipton, a professor of neurology at Albert Einstein College of Medicine and the director of the Montefiore Headache Center, New York.
“Do you treat the depression? Do you treat the migraine? Do you independently treat both? Those have been long-standing questions for clinicians,” Dr. Lipton said.
Investigators randomly assigned 330 adults with migraine who were diagnosed with moderate-to-severe MDD (defined as a PHQ-9 score of 10 or greater) to receive 225 mg subcutaneous monthly fremanezumab (n = 164) or placebo (n = 166) for 12 weeks.
The trial continued as an open-label trial for another 12 weeks.
During the double-blind phase of the study, the mean change from baseline in the HAMD-17 score with placebo was –4.6 at week 8 and –5.4 at week 12, compared with –6.0 with fremanezumab at week 8 (P = .0205) and –6.7 at week 12 (P = .0228).
The change from baseline in PHQ-9 total score at week 8 was –5.8 for placebo and –7.1 for fremanezumab (P = .0283). At week 12, the change was –6.3 for placebo versus –7.8 for fremanezumab (P = .0108). These reductions were maintained throughout the open-label period of the trial.
The beneficial effect on depression and migraine demonstrated in the study is interesting on several levels, Dr. Lipton said.
“One, it tells us that if the patient has migraine and depression and you treat with fremanezumab, both disorders get better to a statistically significant degree. That’s critically important,” he said.
“The other thing, and this is actually what I find most interesting about this study, is that fremanezumab doesn’t get into the brain. There are many antimigraine therapies that do, so you can treat a patient with migraine and depression with a tricyclic antidepressant.”
“It may make the migraine better and the depression better, but you don’t know if the benefit in depression comes from the improvement in migraine, because of course the antidepressant works for both conditions. Maybe there are people who would disagree with this, but my interpretation [of the trial results] is that the depression got better because the migraine got better,” he added.
The link between migraine and depression is well established, Dr. Lipton added. Longitudinal studies have shown that people with depression but without migraine develop migraine at increased rates, compared with people with no depression. Conversely, people with migraine but no depression develop depression at increased rates.
“Both disorders may have a common substrate, but I also think many forms of chronic pain lead to depression, and that’s the part we’re making better,” he said.
If fremanezumab has this dual effect on migraine and depression, it is possible that other anti-CGRP drugs will have a similar effect, Dr. Lipton said.
“Honestly, my hope is that other companies that make effective drugs will do similar studies to see if other monoclonal antibodies that target CGRP have the same effect. My guess is that all of them work but until the studies are done, I’m going to use fremanezumab, the one that has been studied, in my patients.”
He added that depression is an important comorbidity of migraine and represents a huge challenge for clinicians. “A lot of headache patients want to know what to do about comorbid anxiety or comorbid depression. I run a headache center in a specialty practice, and when people come in with migraine, they almost always come in with migraine and depression or anxiety or another pain disorder, or something else, and one of the great challenges in the practice is managing these comorbidities,” he said.
A bidirectional relationship
The overlap between migraine and depression and anxiety has been known for quite a while, agreed Elizabeth W. Loder, MD, MPH, vice chair of academic affairs, department of neurology, Brigham and Women’s Hospital, and professor of neurology at Harvard Medical School, both in Boston.
“I think the relationship is generally viewed as bidirectional and causality is uncertain. I still do not think I would assume that any drug that reduces migraine would reduce depression,” said Dr. Loder.
However, she added, the fremanezumab study data are interesting. “The effects of any drug on depression could be due to improvement of migraine or it could be due to some other effect of the treatment on depression. That is what makes these results so intriguing. If the findings are borne out by other studies, it could mean that these treatments would be preferred to those older ones in patients with depression,” Dr. Loder said.
Also commenting on the findings, Huma Sheikh, MD, CEO of NY Neurology Medicine PC, said the study is important because it confirms the strong association between migraine and depression. “Both conditions have similar underlying neurobiological pathophysiologies, and if you are impacting one area in the brain with the CGRP inhibitors, you might also be targeting some of the receptors or pathways that are involved in depression,” Dr. Sheikh said.
The study was funded by Teva Pharmaceuticals. Dr. Lipton reported financial relationships with Teva and multiple other pharmaceutical companies. Dr. Loder and Dr. Sheikh have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
AUSTIN, TEX. – , new research shows.
Patients with both conditions who were randomly assigned to receive fremanezumab showed a statistically significant reduction in both the 17-item Hamilton Depression Rating Scale (HAMD-17) and the nine-criteria Patient Health Questionnaire (PHQ-9) scores, compared with matched controls who received placebo.
The results from the UNITE trial were presented at the annual meeting of the American Headache Society.
Long-standing questions
“It’s been well known for a long time that migraine is comorbid with a number of illnesses, and one of the most common is depression,” said study investigator Richard B. Lipton, a professor of neurology at Albert Einstein College of Medicine and the director of the Montefiore Headache Center, New York.
“Do you treat the depression? Do you treat the migraine? Do you independently treat both? Those have been long-standing questions for clinicians,” Dr. Lipton said.
Investigators randomly assigned 330 adults with migraine who were diagnosed with moderate-to-severe MDD (defined as a PHQ-9 score of 10 or greater) to receive 225 mg subcutaneous monthly fremanezumab (n = 164) or placebo (n = 166) for 12 weeks.
The trial continued as an open-label trial for another 12 weeks.
During the double-blind phase of the study, the mean change from baseline in the HAMD-17 score with placebo was –4.6 at week 8 and –5.4 at week 12, compared with –6.0 with fremanezumab at week 8 (P = .0205) and –6.7 at week 12 (P = .0228).
The change from baseline in PHQ-9 total score at week 8 was –5.8 for placebo and –7.1 for fremanezumab (P = .0283). At week 12, the change was –6.3 for placebo versus –7.8 for fremanezumab (P = .0108). These reductions were maintained throughout the open-label period of the trial.
The beneficial effect on depression and migraine demonstrated in the study is interesting on several levels, Dr. Lipton said.
“One, it tells us that if the patient has migraine and depression and you treat with fremanezumab, both disorders get better to a statistically significant degree. That’s critically important,” he said.
“The other thing, and this is actually what I find most interesting about this study, is that fremanezumab doesn’t get into the brain. There are many antimigraine therapies that do, so you can treat a patient with migraine and depression with a tricyclic antidepressant.”
“It may make the migraine better and the depression better, but you don’t know if the benefit in depression comes from the improvement in migraine, because of course the antidepressant works for both conditions. Maybe there are people who would disagree with this, but my interpretation [of the trial results] is that the depression got better because the migraine got better,” he added.
The link between migraine and depression is well established, Dr. Lipton added. Longitudinal studies have shown that people with depression but without migraine develop migraine at increased rates, compared with people with no depression. Conversely, people with migraine but no depression develop depression at increased rates.
“Both disorders may have a common substrate, but I also think many forms of chronic pain lead to depression, and that’s the part we’re making better,” he said.
If fremanezumab has this dual effect on migraine and depression, it is possible that other anti-CGRP drugs will have a similar effect, Dr. Lipton said.
“Honestly, my hope is that other companies that make effective drugs will do similar studies to see if other monoclonal antibodies that target CGRP have the same effect. My guess is that all of them work but until the studies are done, I’m going to use fremanezumab, the one that has been studied, in my patients.”
He added that depression is an important comorbidity of migraine and represents a huge challenge for clinicians. “A lot of headache patients want to know what to do about comorbid anxiety or comorbid depression. I run a headache center in a specialty practice, and when people come in with migraine, they almost always come in with migraine and depression or anxiety or another pain disorder, or something else, and one of the great challenges in the practice is managing these comorbidities,” he said.
A bidirectional relationship
The overlap between migraine and depression and anxiety has been known for quite a while, agreed Elizabeth W. Loder, MD, MPH, vice chair of academic affairs, department of neurology, Brigham and Women’s Hospital, and professor of neurology at Harvard Medical School, both in Boston.
“I think the relationship is generally viewed as bidirectional and causality is uncertain. I still do not think I would assume that any drug that reduces migraine would reduce depression,” said Dr. Loder.
However, she added, the fremanezumab study data are interesting. “The effects of any drug on depression could be due to improvement of migraine or it could be due to some other effect of the treatment on depression. That is what makes these results so intriguing. If the findings are borne out by other studies, it could mean that these treatments would be preferred to those older ones in patients with depression,” Dr. Loder said.
Also commenting on the findings, Huma Sheikh, MD, CEO of NY Neurology Medicine PC, said the study is important because it confirms the strong association between migraine and depression. “Both conditions have similar underlying neurobiological pathophysiologies, and if you are impacting one area in the brain with the CGRP inhibitors, you might also be targeting some of the receptors or pathways that are involved in depression,” Dr. Sheikh said.
The study was funded by Teva Pharmaceuticals. Dr. Lipton reported financial relationships with Teva and multiple other pharmaceutical companies. Dr. Loder and Dr. Sheikh have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
AT AHS 2023
Myasthenia gravis drug gets FDA nod
, the drug’s manufacturer, UCB, has announced.

Rozanolixizumab is a subcutaneous-infused humanized IgG4 monoclonal antibody that binds to the neonatal Fc receptor (FcRn), reducing the concentration of pathogenic IgG autoantibodies.
U.S. approval is based on results of the phase 3 MycarinG study involving 200 patients with AChR or MuSK autoantibody-positive gMG. Patients were randomly assigned to one of two rozanolixizumab groups (7 mg/kg or 10 mg/kg) or placebo for 6 weeks.
As reported last month in Lancet Neurology, rozanolixizumab led to statistically significant improvements in gMG-specific outcomes, including everyday activities such as breathing, talking, swallowing, and being able to rise from a chair.
“There is a significant need for new, innovative treatment options to reduce the day-to-day burden of gMG,” lead investigator Vera Bril, MD, professor of medicine (neurology), University of Toronto, said in a news release.
Rozanolixizumab is “a new treatment option, targeting one of the mechanisms of disease to provide symptom improvement in patient- and physician-reported outcomes at day 43,” Dr. Bril added.
The most common adverse reactions (reported in at least 10% of patients treated with rozanolixizumab) were headache, infections, diarrhea, pyrexia, hypersensitivity reactions, and nausea.
The company expects rozanolixizumab to be available in the United States during the third quarter of 2023.
The FDA granted the application for rozanolixizumab in gMG priority review.
A version of this article first appeared on Medscape.com.
, the drug’s manufacturer, UCB, has announced.
Rozanolixizumab is a subcutaneous-infused humanized IgG4 monoclonal antibody that binds to the neonatal Fc receptor (FcRn), reducing the concentration of pathogenic IgG autoantibodies.
U.S. approval is based on results of the phase 3 MycarinG study involving 200 patients with AChR or MuSK autoantibody-positive gMG. Patients were randomly assigned to one of two rozanolixizumab groups (7 mg/kg or 10 mg/kg) or placebo for 6 weeks.
As reported last month in Lancet Neurology, rozanolixizumab led to statistically significant improvements in gMG-specific outcomes, including everyday activities such as breathing, talking, swallowing, and being able to rise from a chair.
“There is a significant need for new, innovative treatment options to reduce the day-to-day burden of gMG,” lead investigator Vera Bril, MD, professor of medicine (neurology), University of Toronto, said in a news release.
Rozanolixizumab is “a new treatment option, targeting one of the mechanisms of disease to provide symptom improvement in patient- and physician-reported outcomes at day 43,” Dr. Bril added.
The most common adverse reactions (reported in at least 10% of patients treated with rozanolixizumab) were headache, infections, diarrhea, pyrexia, hypersensitivity reactions, and nausea.
The company expects rozanolixizumab to be available in the United States during the third quarter of 2023.
The FDA granted the application for rozanolixizumab in gMG priority review.
A version of this article first appeared on Medscape.com.
, the drug’s manufacturer, UCB, has announced.
Rozanolixizumab is a subcutaneous-infused humanized IgG4 monoclonal antibody that binds to the neonatal Fc receptor (FcRn), reducing the concentration of pathogenic IgG autoantibodies.
U.S. approval is based on results of the phase 3 MycarinG study involving 200 patients with AChR or MuSK autoantibody-positive gMG. Patients were randomly assigned to one of two rozanolixizumab groups (7 mg/kg or 10 mg/kg) or placebo for 6 weeks.
As reported last month in Lancet Neurology, rozanolixizumab led to statistically significant improvements in gMG-specific outcomes, including everyday activities such as breathing, talking, swallowing, and being able to rise from a chair.
“There is a significant need for new, innovative treatment options to reduce the day-to-day burden of gMG,” lead investigator Vera Bril, MD, professor of medicine (neurology), University of Toronto, said in a news release.
Rozanolixizumab is “a new treatment option, targeting one of the mechanisms of disease to provide symptom improvement in patient- and physician-reported outcomes at day 43,” Dr. Bril added.
The most common adverse reactions (reported in at least 10% of patients treated with rozanolixizumab) were headache, infections, diarrhea, pyrexia, hypersensitivity reactions, and nausea.
The company expects rozanolixizumab to be available in the United States during the third quarter of 2023.
The FDA granted the application for rozanolixizumab in gMG priority review.
A version of this article first appeared on Medscape.com.
FDA approves ritlecitinib for ages 12 and up for alopecia areata
Taken as a once-daily pill, ritlecitinib is a dual inhibitor of the TEC family of tyrosine kinases and of Janus kinase 3 (JAK3). The recommended dose of ritlecitinib, which will be marketed as Litfulo, is 50 mg once a day, according to the statement announcing the approval from Pfizer.
It is the second JAK inhibitor approved for treating alopecia areata, following approval of baricitinib (Olumiant) in June 2022 for AA in adults. Ritlecitinib is the first JAK inhibitor approved for children ages 12 and older with AA.
The European Medicines Agency has also accepted the Marketing Authorization Application for ritlecitinib in the same population and a decision is expected in the fourth quarter of this year.
Approval based on ALLEGRO trials
Approval was based on previously announced results from trials, including the phase 2b/3 ALLEGRO study of ritlecitinib in 718 patients aged 12 years and older with alopecia areata, with 50% of more scalp hair loss, as measured by the Severity of Alopecia Tool (SALT), including patients with alopecia totalis (complete scalp hair loss) and alopecia universalis (complete scalp, face, and body hair loss).
Patients in the trial were experiencing a current episode of alopecia areata that had lasted between 6 months and 10 years. They were randomized to receive once-daily ritlecitinib at doses of 30 mg or 50 mg (with or without 1 month of initial treatment with once-daily ritlecitinib 200 mg), ritlecitinib 10 mg, or placebo.
Statistically significantly higher proportions of patients treated with ritlecitinib 30 mg and 50 mg (with or without the loading dose) had 80% or more scalp hair coverage, as measured by a SALT score of 20 or less after 6 months of treatment versus placebo. After 6 months of treatment, among those on the 50-mg dose, 23% had achieved a SALT score of 20 or less, compared with 2% of those on placebo. The results were published in The Lancet.
According to the company release, efficacy and safety of ritlecitinib was consistent between those ages 12-17 and adults, and the most common adverse events reported in the study, in at least 4% of patients treated with ritlecitinib, were headache (10.8%), diarrhea (10%), acne (6.2%), rash (5.4%), and urticaria (4.6%).
Ritlecitinib labeling includes the boxed warning about the risk for serious infections, mortality, malignancy, major adverse cardiovascular events, and thrombosis, which is included in the labels for other JAK inhibitors.
Ritlecitinib evaluated for other diseases
In addition to alopecia areata, ritlecitinib has shown efficacy and acceptable safety in treating ulcerative colitis and is being evaluated for treating vitiligo, Crohn’s disease, and rheumatoid arthritis.
In the statement, the company says that ritlecitinib will be available “in the coming weeks.” The manufacturer says it also has completed regulatory submissions for ritlecitinib in the United Kingdom, China, and Japan, and expects decisions this year.
Alopecia areata affects about 6.8 million people in the United States and 147 million globally.
In a statement, Nicole Friedland, president and CEO of the National Alopecia Areata Foundation, said that NAAF “is thrilled to have a second FDA-approved treatment for alopecia areata, which is the first approved for adolescents.”
A version of this article first appeared on Medscape.com.
Taken as a once-daily pill, ritlecitinib is a dual inhibitor of the TEC family of tyrosine kinases and of Janus kinase 3 (JAK3). The recommended dose of ritlecitinib, which will be marketed as Litfulo, is 50 mg once a day, according to the statement announcing the approval from Pfizer.
It is the second JAK inhibitor approved for treating alopecia areata, following approval of baricitinib (Olumiant) in June 2022 for AA in adults. Ritlecitinib is the first JAK inhibitor approved for children ages 12 and older with AA.
The European Medicines Agency has also accepted the Marketing Authorization Application for ritlecitinib in the same population and a decision is expected in the fourth quarter of this year.
Approval based on ALLEGRO trials
Approval was based on previously announced results from trials, including the phase 2b/3 ALLEGRO study of ritlecitinib in 718 patients aged 12 years and older with alopecia areata, with 50% of more scalp hair loss, as measured by the Severity of Alopecia Tool (SALT), including patients with alopecia totalis (complete scalp hair loss) and alopecia universalis (complete scalp, face, and body hair loss).
Patients in the trial were experiencing a current episode of alopecia areata that had lasted between 6 months and 10 years. They were randomized to receive once-daily ritlecitinib at doses of 30 mg or 50 mg (with or without 1 month of initial treatment with once-daily ritlecitinib 200 mg), ritlecitinib 10 mg, or placebo.
Statistically significantly higher proportions of patients treated with ritlecitinib 30 mg and 50 mg (with or without the loading dose) had 80% or more scalp hair coverage, as measured by a SALT score of 20 or less after 6 months of treatment versus placebo. After 6 months of treatment, among those on the 50-mg dose, 23% had achieved a SALT score of 20 or less, compared with 2% of those on placebo. The results were published in The Lancet.
According to the company release, efficacy and safety of ritlecitinib was consistent between those ages 12-17 and adults, and the most common adverse events reported in the study, in at least 4% of patients treated with ritlecitinib, were headache (10.8%), diarrhea (10%), acne (6.2%), rash (5.4%), and urticaria (4.6%).
Ritlecitinib labeling includes the boxed warning about the risk for serious infections, mortality, malignancy, major adverse cardiovascular events, and thrombosis, which is included in the labels for other JAK inhibitors.
Ritlecitinib evaluated for other diseases
In addition to alopecia areata, ritlecitinib has shown efficacy and acceptable safety in treating ulcerative colitis and is being evaluated for treating vitiligo, Crohn’s disease, and rheumatoid arthritis.
In the statement, the company says that ritlecitinib will be available “in the coming weeks.” The manufacturer says it also has completed regulatory submissions for ritlecitinib in the United Kingdom, China, and Japan, and expects decisions this year.
Alopecia areata affects about 6.8 million people in the United States and 147 million globally.
In a statement, Nicole Friedland, president and CEO of the National Alopecia Areata Foundation, said that NAAF “is thrilled to have a second FDA-approved treatment for alopecia areata, which is the first approved for adolescents.”
A version of this article first appeared on Medscape.com.
Taken as a once-daily pill, ritlecitinib is a dual inhibitor of the TEC family of tyrosine kinases and of Janus kinase 3 (JAK3). The recommended dose of ritlecitinib, which will be marketed as Litfulo, is 50 mg once a day, according to the statement announcing the approval from Pfizer.
It is the second JAK inhibitor approved for treating alopecia areata, following approval of baricitinib (Olumiant) in June 2022 for AA in adults. Ritlecitinib is the first JAK inhibitor approved for children ages 12 and older with AA.
The European Medicines Agency has also accepted the Marketing Authorization Application for ritlecitinib in the same population and a decision is expected in the fourth quarter of this year.
Approval based on ALLEGRO trials
Approval was based on previously announced results from trials, including the phase 2b/3 ALLEGRO study of ritlecitinib in 718 patients aged 12 years and older with alopecia areata, with 50% of more scalp hair loss, as measured by the Severity of Alopecia Tool (SALT), including patients with alopecia totalis (complete scalp hair loss) and alopecia universalis (complete scalp, face, and body hair loss).
Patients in the trial were experiencing a current episode of alopecia areata that had lasted between 6 months and 10 years. They were randomized to receive once-daily ritlecitinib at doses of 30 mg or 50 mg (with or without 1 month of initial treatment with once-daily ritlecitinib 200 mg), ritlecitinib 10 mg, or placebo.
Statistically significantly higher proportions of patients treated with ritlecitinib 30 mg and 50 mg (with or without the loading dose) had 80% or more scalp hair coverage, as measured by a SALT score of 20 or less after 6 months of treatment versus placebo. After 6 months of treatment, among those on the 50-mg dose, 23% had achieved a SALT score of 20 or less, compared with 2% of those on placebo. The results were published in The Lancet.
According to the company release, efficacy and safety of ritlecitinib was consistent between those ages 12-17 and adults, and the most common adverse events reported in the study, in at least 4% of patients treated with ritlecitinib, were headache (10.8%), diarrhea (10%), acne (6.2%), rash (5.4%), and urticaria (4.6%).
Ritlecitinib labeling includes the boxed warning about the risk for serious infections, mortality, malignancy, major adverse cardiovascular events, and thrombosis, which is included in the labels for other JAK inhibitors.
Ritlecitinib evaluated for other diseases
In addition to alopecia areata, ritlecitinib has shown efficacy and acceptable safety in treating ulcerative colitis and is being evaluated for treating vitiligo, Crohn’s disease, and rheumatoid arthritis.
In the statement, the company says that ritlecitinib will be available “in the coming weeks.” The manufacturer says it also has completed regulatory submissions for ritlecitinib in the United Kingdom, China, and Japan, and expects decisions this year.
Alopecia areata affects about 6.8 million people in the United States and 147 million globally.
In a statement, Nicole Friedland, president and CEO of the National Alopecia Areata Foundation, said that NAAF “is thrilled to have a second FDA-approved treatment for alopecia areata, which is the first approved for adolescents.”
A version of this article first appeared on Medscape.com.
FDA approves talazoparib for metastatic prostate cancer
Talazoparib is already approved for adults with deleterious or suspected deleterious germline BRCA-mutated HER2-negative locally advanced or metastatic breast cancer. The new approval, granted following priority review, is based on findings from the randomized, placebo-controlled, phase 3 TALAPRO-2 trial, published in The Lancet.
The 399 patients in the study were randomly assigned in a 1:1 ratio to receive either enzalutamide 160 mg daily plus either talazoparib 0.5 mg or placebo daily. Median radiographic progression-free survival (PFS) was not reached in the treatment group; it was 13.8 months in the placebo group (hazard ratio, 0.45). In an exploratory analysis by BRCA mutation status, patients with BRCA-mutated disease who received talazoparib exhibited an even stronger median radiographic PFS (HR, 0.20; not reached vs. 11 months) in comparison with those without BRCA-mutated disease (HR, 0.72; 24.7 vs. 16.7 months).
Serious adverse reactions occurred in 30% of patients who received talazoparib plus enzalutamide. The most common serious adverse reactions, reported in more than 2% of patients, included anemia (9%) and fracture (3%). Discontinuation of talazoparib occurred in 10% of patients, according to a Pfizer statement.
Pfizer also noted that a marketing authorization application for the drug combination has been accepted for review by the European Medicines Agency.
“Despite treatment advancement in metastatic castration-resistant prostate cancer, the disease can progress quickly, and many patients may only receive one line of therapy,” lead investigator Neeraj Agarwal, MD, of the Huntsman Cancer Institute, University of Utah, Salt Lake City, said in a statement. Patients with metastatic castration-resistant prostate cancer harboring HRR genetic alterations have even worse outcomes, and thus the FDA’s approval of the talazoparib and enzalutamide combination “represents a treatment option deserving of excitement and attention.”
A version of this article originally appeared on Medscape.com.
Talazoparib is already approved for adults with deleterious or suspected deleterious germline BRCA-mutated HER2-negative locally advanced or metastatic breast cancer. The new approval, granted following priority review, is based on findings from the randomized, placebo-controlled, phase 3 TALAPRO-2 trial, published in The Lancet.
The 399 patients in the study were randomly assigned in a 1:1 ratio to receive either enzalutamide 160 mg daily plus either talazoparib 0.5 mg or placebo daily. Median radiographic progression-free survival (PFS) was not reached in the treatment group; it was 13.8 months in the placebo group (hazard ratio, 0.45). In an exploratory analysis by BRCA mutation status, patients with BRCA-mutated disease who received talazoparib exhibited an even stronger median radiographic PFS (HR, 0.20; not reached vs. 11 months) in comparison with those without BRCA-mutated disease (HR, 0.72; 24.7 vs. 16.7 months).
Serious adverse reactions occurred in 30% of patients who received talazoparib plus enzalutamide. The most common serious adverse reactions, reported in more than 2% of patients, included anemia (9%) and fracture (3%). Discontinuation of talazoparib occurred in 10% of patients, according to a Pfizer statement.
Pfizer also noted that a marketing authorization application for the drug combination has been accepted for review by the European Medicines Agency.
“Despite treatment advancement in metastatic castration-resistant prostate cancer, the disease can progress quickly, and many patients may only receive one line of therapy,” lead investigator Neeraj Agarwal, MD, of the Huntsman Cancer Institute, University of Utah, Salt Lake City, said in a statement. Patients with metastatic castration-resistant prostate cancer harboring HRR genetic alterations have even worse outcomes, and thus the FDA’s approval of the talazoparib and enzalutamide combination “represents a treatment option deserving of excitement and attention.”
A version of this article originally appeared on Medscape.com.
Talazoparib is already approved for adults with deleterious or suspected deleterious germline BRCA-mutated HER2-negative locally advanced or metastatic breast cancer. The new approval, granted following priority review, is based on findings from the randomized, placebo-controlled, phase 3 TALAPRO-2 trial, published in The Lancet.
The 399 patients in the study were randomly assigned in a 1:1 ratio to receive either enzalutamide 160 mg daily plus either talazoparib 0.5 mg or placebo daily. Median radiographic progression-free survival (PFS) was not reached in the treatment group; it was 13.8 months in the placebo group (hazard ratio, 0.45). In an exploratory analysis by BRCA mutation status, patients with BRCA-mutated disease who received talazoparib exhibited an even stronger median radiographic PFS (HR, 0.20; not reached vs. 11 months) in comparison with those without BRCA-mutated disease (HR, 0.72; 24.7 vs. 16.7 months).
Serious adverse reactions occurred in 30% of patients who received talazoparib plus enzalutamide. The most common serious adverse reactions, reported in more than 2% of patients, included anemia (9%) and fracture (3%). Discontinuation of talazoparib occurred in 10% of patients, according to a Pfizer statement.
Pfizer also noted that a marketing authorization application for the drug combination has been accepted for review by the European Medicines Agency.
“Despite treatment advancement in metastatic castration-resistant prostate cancer, the disease can progress quickly, and many patients may only receive one line of therapy,” lead investigator Neeraj Agarwal, MD, of the Huntsman Cancer Institute, University of Utah, Salt Lake City, said in a statement. Patients with metastatic castration-resistant prostate cancer harboring HRR genetic alterations have even worse outcomes, and thus the FDA’s approval of the talazoparib and enzalutamide combination “represents a treatment option deserving of excitement and attention.”
A version of this article originally appeared on Medscape.com.
Can a repurposed Parkinson’s drug slow ALS progression?
However, at least one expert believes the study has “significant flaws.”
Investigators randomly assigned 20 individuals with sporadic ALS to receive either ropinirole or placebo for 24 weeks. During the double-blind period, there was no difference between the groups in terms of decline in functional status.
However, during a further open-label extension period, the ropinirole group showed significant suppression of functional decline and an average of an additional 7 months of progression-free survival.
The researchers were able to predict clinical responsiveness to ropinirole in vitro by analyzing motor neurons derived from participants’ stem cells.
“We found that ropinirole is safe and tolerable for ALS patients and shows therapeutic promise at helping them sustain daily activity and muscle strength,” first author Satoru Morimoto, MD, of the department of physiology, Keio University School of Medicine, Tokyo, said in a news release.
The study was published online in Cell Stem Cell.
Feasibility study
“ALS is totally incurable and it’s a very difficult disease to treat,” senior author Hideyuki Okano, MD, PhD, professor, department of physiology, Keio University, said in the news release.
Preclinical animal models have “limited translational potential” for identifying drug candidates, but induced pluripotent stem cell (iPSC)–derived motor neurons (MNs) from ALS patients can “overcome these limitations for drug screening,” the authors write.
“We previously identified ropinirole [a dopamine D2 receptor agonist] as a potential anti-ALS drug in vitro by iPSC drug discovery,” Dr. Okano said.
The current trial was a randomized, placebo-controlled phase 1/2a feasibility trial that evaluated the safety, tolerability, and efficacy of ropinirole in patients with ALS, using several parameters:
- The revised ALS functional rating scale (ALSFRS-R) score.
- Composite functional endpoints.
- Event-free survival.
- Time to ≤ 50% forced vital capacity (FVC).
The trial consisted of a 12-week run-in period, a 24-week double-blind period, an open-label extension period that lasted from 4 to 24 weeks, and a 4-week follow-up period after administration.
Thirteen patients were assigned to receive ropinirole (23.1% women; mean age, 65.2 ± 12.6 years; 7.7% with clinically definite and 76.9% with clinically probable ALS); seven were assigned to receive placebo (57.1% women; mean age, 66.3 ± 7.5 years; 14.3% with clinically definite and 85.7% with clinically probable ALS).
Of the treatment group, 30.8% had a bulbar onset lesion vs. 57.1% in the placebo group. At baseline, the mean FVC was 94.4% ± 14.9 and 81.5% ± 23.2 in the ropinirole and placebo groups, respectively. The mean body mass index (BMI) was 22.91 ± 3.82 and 19.69 ± 2.63, respectively.
Of the participants,12 in the ropinirole and six in the control group completed the full 24-week treatment protocol; 12 in the ropinirole and five in the placebo group completed the open-label extension (participants who had received placebo were switched to the active drug).
However only seven participants in the ropinirole group and one participant in the placebo group completed the full 1-year trial.
‘Striking correlation’
“During the double-blind period, muscle strength and daily activity were maintained, but a decline in the ALSFRS-R … was not different from that in the placebo group,” the researchers write.
In the open-label extension period, the ropinirole group showed “significant suppression of ALSFRS-R decline,” with an ALSFRS-R score change of only 7.75 (95% confidence interval, 10.66-4.63) for the treatment group vs. 17.51 (95% CI, 22.46-12.56) for the placebo group.
The researchers used the assessment of function and survival (CAFS) score, which adjusts the ALSFRS-R score against mortality, to see whether functional benefits translated into improved survival.
The score “favored ropinirole” in the open-extension period and the entire treatment period but not in the double-blind period.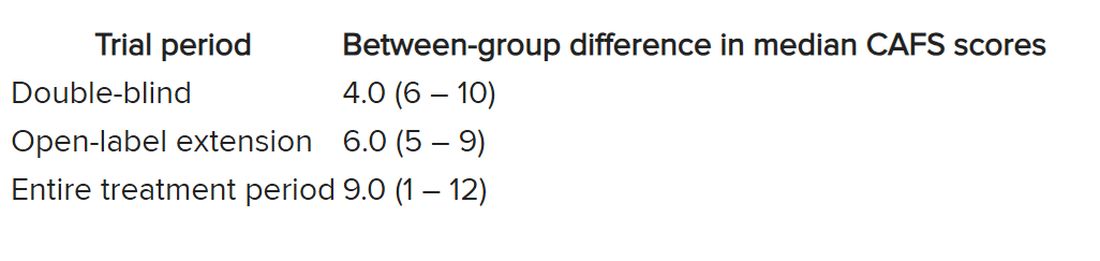
Disease progression events occurred in 7 of 7 (100%) participants in the placebo group and 7 of 13 (54%) in the ropinirole group, “suggesting a twofold decrease in disease progression” in the treatment group.
The ropinirole group experienced an additional 27.9 weeks of disease progression–free survival, compared with the placebo group.
“No participant discontinued treatment because of adverse experiences in either treatment group,” the authors report.
The analysis of iPSC-derived motor neurons from participants showed dopamine D2 receptor expression, as well as the potential involvement of the cholesterol pathway SREBP2 in the therapeutic effects of ropinirole. Lipid peroxide was also identified as a good “surrogate clinical marker to assess disease progression and drug efficacy.”
“We found a very striking correlation between a patient’s clinical response and the response of their motor neurons in vitro,” said Dr. Morimoto. “Patients whose motor neurons responded robustly to ropinirole in vitro had a much slower clinical disease progression with ropinirole treatment, while suboptimal responders showed much more rapid disease progression, despite taking ropinirole.”
Limitations include “small sample sizes and high attrition rates in the open-label extension period,” so “further validation” is required, the authors state.
Significant flaws
Commenting for this article, Carmel Armon, MD, MHS, professor of neurology, Loma Linda (Calif.) University, said the study “falls short of being a credible 1/2a clinical trial.”
Although the “intentions were good and the design not unusual,” the two groups were not “balanced on risk factors for faster progressing disease.” Rather, the placebo group was “tilted towards faster progressing disease” because there were more clinically definite and probable ALS patients in the placebo group than the treatment group, and there were more patients with bulbar onset.
Participants in the placebo group also had shorter median disease duration, lower BMI, and lower FVC, noted Dr. Armon, who was not involved with the study.
And only 1 in 7 control patients completed the open-label extension, compared with 7 of 13 patients in the intervention group.
“With these limitations, I would be disinclined to rely on the findings to justify a larger clinical trial,” Dr. Armon concluded.
The trial was sponsored by K Pharma. The study drug, active drugs, and placebo were supplied free of charge by GlaxoSmithKline K.K. Dr. Okano received grants from JSPS and AMED and grants and personal fees from K Pharma during the conduct of the study and personal fees from Sanbio, outside the submitted work. Dr. Okano has a patent on a therapeutic agent for ALS and composition for treatment licensed to K Pharma. The other authors’ disclosures and additional information are available in the original article. Dr. Armon reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
However, at least one expert believes the study has “significant flaws.”
Investigators randomly assigned 20 individuals with sporadic ALS to receive either ropinirole or placebo for 24 weeks. During the double-blind period, there was no difference between the groups in terms of decline in functional status.
However, during a further open-label extension period, the ropinirole group showed significant suppression of functional decline and an average of an additional 7 months of progression-free survival.
The researchers were able to predict clinical responsiveness to ropinirole in vitro by analyzing motor neurons derived from participants’ stem cells.
“We found that ropinirole is safe and tolerable for ALS patients and shows therapeutic promise at helping them sustain daily activity and muscle strength,” first author Satoru Morimoto, MD, of the department of physiology, Keio University School of Medicine, Tokyo, said in a news release.
The study was published online in Cell Stem Cell.
Feasibility study
“ALS is totally incurable and it’s a very difficult disease to treat,” senior author Hideyuki Okano, MD, PhD, professor, department of physiology, Keio University, said in the news release.
Preclinical animal models have “limited translational potential” for identifying drug candidates, but induced pluripotent stem cell (iPSC)–derived motor neurons (MNs) from ALS patients can “overcome these limitations for drug screening,” the authors write.
“We previously identified ropinirole [a dopamine D2 receptor agonist] as a potential anti-ALS drug in vitro by iPSC drug discovery,” Dr. Okano said.
The current trial was a randomized, placebo-controlled phase 1/2a feasibility trial that evaluated the safety, tolerability, and efficacy of ropinirole in patients with ALS, using several parameters:
- The revised ALS functional rating scale (ALSFRS-R) score.
- Composite functional endpoints.
- Event-free survival.
- Time to ≤ 50% forced vital capacity (FVC).
The trial consisted of a 12-week run-in period, a 24-week double-blind period, an open-label extension period that lasted from 4 to 24 weeks, and a 4-week follow-up period after administration.
Thirteen patients were assigned to receive ropinirole (23.1% women; mean age, 65.2 ± 12.6 years; 7.7% with clinically definite and 76.9% with clinically probable ALS); seven were assigned to receive placebo (57.1% women; mean age, 66.3 ± 7.5 years; 14.3% with clinically definite and 85.7% with clinically probable ALS).
Of the treatment group, 30.8% had a bulbar onset lesion vs. 57.1% in the placebo group. At baseline, the mean FVC was 94.4% ± 14.9 and 81.5% ± 23.2 in the ropinirole and placebo groups, respectively. The mean body mass index (BMI) was 22.91 ± 3.82 and 19.69 ± 2.63, respectively.
Of the participants,12 in the ropinirole and six in the control group completed the full 24-week treatment protocol; 12 in the ropinirole and five in the placebo group completed the open-label extension (participants who had received placebo were switched to the active drug).
However only seven participants in the ropinirole group and one participant in the placebo group completed the full 1-year trial.
‘Striking correlation’
“During the double-blind period, muscle strength and daily activity were maintained, but a decline in the ALSFRS-R … was not different from that in the placebo group,” the researchers write.
In the open-label extension period, the ropinirole group showed “significant suppression of ALSFRS-R decline,” with an ALSFRS-R score change of only 7.75 (95% confidence interval, 10.66-4.63) for the treatment group vs. 17.51 (95% CI, 22.46-12.56) for the placebo group.
The researchers used the assessment of function and survival (CAFS) score, which adjusts the ALSFRS-R score against mortality, to see whether functional benefits translated into improved survival.
The score “favored ropinirole” in the open-extension period and the entire treatment period but not in the double-blind period.
Disease progression events occurred in 7 of 7 (100%) participants in the placebo group and 7 of 13 (54%) in the ropinirole group, “suggesting a twofold decrease in disease progression” in the treatment group.
The ropinirole group experienced an additional 27.9 weeks of disease progression–free survival, compared with the placebo group.
“No participant discontinued treatment because of adverse experiences in either treatment group,” the authors report.
The analysis of iPSC-derived motor neurons from participants showed dopamine D2 receptor expression, as well as the potential involvement of the cholesterol pathway SREBP2 in the therapeutic effects of ropinirole. Lipid peroxide was also identified as a good “surrogate clinical marker to assess disease progression and drug efficacy.”
“We found a very striking correlation between a patient’s clinical response and the response of their motor neurons in vitro,” said Dr. Morimoto. “Patients whose motor neurons responded robustly to ropinirole in vitro had a much slower clinical disease progression with ropinirole treatment, while suboptimal responders showed much more rapid disease progression, despite taking ropinirole.”
Limitations include “small sample sizes and high attrition rates in the open-label extension period,” so “further validation” is required, the authors state.
Significant flaws
Commenting for this article, Carmel Armon, MD, MHS, professor of neurology, Loma Linda (Calif.) University, said the study “falls short of being a credible 1/2a clinical trial.”
Although the “intentions were good and the design not unusual,” the two groups were not “balanced on risk factors for faster progressing disease.” Rather, the placebo group was “tilted towards faster progressing disease” because there were more clinically definite and probable ALS patients in the placebo group than the treatment group, and there were more patients with bulbar onset.
Participants in the placebo group also had shorter median disease duration, lower BMI, and lower FVC, noted Dr. Armon, who was not involved with the study.
And only 1 in 7 control patients completed the open-label extension, compared with 7 of 13 patients in the intervention group.
“With these limitations, I would be disinclined to rely on the findings to justify a larger clinical trial,” Dr. Armon concluded.
The trial was sponsored by K Pharma. The study drug, active drugs, and placebo were supplied free of charge by GlaxoSmithKline K.K. Dr. Okano received grants from JSPS and AMED and grants and personal fees from K Pharma during the conduct of the study and personal fees from Sanbio, outside the submitted work. Dr. Okano has a patent on a therapeutic agent for ALS and composition for treatment licensed to K Pharma. The other authors’ disclosures and additional information are available in the original article. Dr. Armon reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
However, at least one expert believes the study has “significant flaws.”
Investigators randomly assigned 20 individuals with sporadic ALS to receive either ropinirole or placebo for 24 weeks. During the double-blind period, there was no difference between the groups in terms of decline in functional status.
However, during a further open-label extension period, the ropinirole group showed significant suppression of functional decline and an average of an additional 7 months of progression-free survival.
The researchers were able to predict clinical responsiveness to ropinirole in vitro by analyzing motor neurons derived from participants’ stem cells.
“We found that ropinirole is safe and tolerable for ALS patients and shows therapeutic promise at helping them sustain daily activity and muscle strength,” first author Satoru Morimoto, MD, of the department of physiology, Keio University School of Medicine, Tokyo, said in a news release.
The study was published online in Cell Stem Cell.
Feasibility study
“ALS is totally incurable and it’s a very difficult disease to treat,” senior author Hideyuki Okano, MD, PhD, professor, department of physiology, Keio University, said in the news release.
Preclinical animal models have “limited translational potential” for identifying drug candidates, but induced pluripotent stem cell (iPSC)–derived motor neurons (MNs) from ALS patients can “overcome these limitations for drug screening,” the authors write.
“We previously identified ropinirole [a dopamine D2 receptor agonist] as a potential anti-ALS drug in vitro by iPSC drug discovery,” Dr. Okano said.
The current trial was a randomized, placebo-controlled phase 1/2a feasibility trial that evaluated the safety, tolerability, and efficacy of ropinirole in patients with ALS, using several parameters:
- The revised ALS functional rating scale (ALSFRS-R) score.
- Composite functional endpoints.
- Event-free survival.
- Time to ≤ 50% forced vital capacity (FVC).
The trial consisted of a 12-week run-in period, a 24-week double-blind period, an open-label extension period that lasted from 4 to 24 weeks, and a 4-week follow-up period after administration.
Thirteen patients were assigned to receive ropinirole (23.1% women; mean age, 65.2 ± 12.6 years; 7.7% with clinically definite and 76.9% with clinically probable ALS); seven were assigned to receive placebo (57.1% women; mean age, 66.3 ± 7.5 years; 14.3% with clinically definite and 85.7% with clinically probable ALS).
Of the treatment group, 30.8% had a bulbar onset lesion vs. 57.1% in the placebo group. At baseline, the mean FVC was 94.4% ± 14.9 and 81.5% ± 23.2 in the ropinirole and placebo groups, respectively. The mean body mass index (BMI) was 22.91 ± 3.82 and 19.69 ± 2.63, respectively.
Of the participants,12 in the ropinirole and six in the control group completed the full 24-week treatment protocol; 12 in the ropinirole and five in the placebo group completed the open-label extension (participants who had received placebo were switched to the active drug).
However only seven participants in the ropinirole group and one participant in the placebo group completed the full 1-year trial.
‘Striking correlation’
“During the double-blind period, muscle strength and daily activity were maintained, but a decline in the ALSFRS-R … was not different from that in the placebo group,” the researchers write.
In the open-label extension period, the ropinirole group showed “significant suppression of ALSFRS-R decline,” with an ALSFRS-R score change of only 7.75 (95% confidence interval, 10.66-4.63) for the treatment group vs. 17.51 (95% CI, 22.46-12.56) for the placebo group.
The researchers used the assessment of function and survival (CAFS) score, which adjusts the ALSFRS-R score against mortality, to see whether functional benefits translated into improved survival.
The score “favored ropinirole” in the open-extension period and the entire treatment period but not in the double-blind period.
Disease progression events occurred in 7 of 7 (100%) participants in the placebo group and 7 of 13 (54%) in the ropinirole group, “suggesting a twofold decrease in disease progression” in the treatment group.
The ropinirole group experienced an additional 27.9 weeks of disease progression–free survival, compared with the placebo group.
“No participant discontinued treatment because of adverse experiences in either treatment group,” the authors report.
The analysis of iPSC-derived motor neurons from participants showed dopamine D2 receptor expression, as well as the potential involvement of the cholesterol pathway SREBP2 in the therapeutic effects of ropinirole. Lipid peroxide was also identified as a good “surrogate clinical marker to assess disease progression and drug efficacy.”
“We found a very striking correlation between a patient’s clinical response and the response of their motor neurons in vitro,” said Dr. Morimoto. “Patients whose motor neurons responded robustly to ropinirole in vitro had a much slower clinical disease progression with ropinirole treatment, while suboptimal responders showed much more rapid disease progression, despite taking ropinirole.”
Limitations include “small sample sizes and high attrition rates in the open-label extension period,” so “further validation” is required, the authors state.
Significant flaws
Commenting for this article, Carmel Armon, MD, MHS, professor of neurology, Loma Linda (Calif.) University, said the study “falls short of being a credible 1/2a clinical trial.”
Although the “intentions were good and the design not unusual,” the two groups were not “balanced on risk factors for faster progressing disease.” Rather, the placebo group was “tilted towards faster progressing disease” because there were more clinically definite and probable ALS patients in the placebo group than the treatment group, and there were more patients with bulbar onset.
Participants in the placebo group also had shorter median disease duration, lower BMI, and lower FVC, noted Dr. Armon, who was not involved with the study.
And only 1 in 7 control patients completed the open-label extension, compared with 7 of 13 patients in the intervention group.
“With these limitations, I would be disinclined to rely on the findings to justify a larger clinical trial,” Dr. Armon concluded.
The trial was sponsored by K Pharma. The study drug, active drugs, and placebo were supplied free of charge by GlaxoSmithKline K.K. Dr. Okano received grants from JSPS and AMED and grants and personal fees from K Pharma during the conduct of the study and personal fees from Sanbio, outside the submitted work. Dr. Okano has a patent on a therapeutic agent for ALS and composition for treatment licensed to K Pharma. The other authors’ disclosures and additional information are available in the original article. Dr. Armon reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM CELL STEM CELL
Starting indicated heart failure meds in-hospital: Progress, opportunities
Most patients aren’t receiving all the medications they should based on guidelines, nor are they getting them at the most effective time in their disease course, suggests a registry study of patients in the United States hospitalized with heart failure with reduced ejection fraction (HFrEF).
On average, one such medication was initiated per patient for every 6 days in the hospital.
Shortfalls in predischarge GDMT initiation disproportionately landed on women, patients at rural centers, and those with renal failure or other comorbidities. But they didn’t seem related to patient race or ethnicity in the study reported in JACC: Heart Failure.
The analysis covers the 3 years preceding the May 2020 first-time approval of a sodium-glucose cotransporter 2 (SGLT2) inhibitor for nondiabetic patients with HFrEF, and therefore doesn’t cover such drugs for that indication. The SGLT2 inhibitors would later join beta-blockers, renin-angiotensin system (RAS) inhibitors, and mineralocorticoid receptor antagonists (MRAs) in the quartet of core GDMT medications broadly indicated for HFrEF.
In-hospital initiation of GDMT for HFrEF is considered a predictor of being on those medications after discharge and is itself guideline recommended. There’s clear evidence that treatment with the four core medications boosts survival and cuts rehospitalization risk, and that “getting those on board as soon as possible will eventually benefit many patients,” Paul L. Hess, MD, MHS, said in an interview.
Dr. Hess, University of Colorado Anschutz Medical Campus, Aurora, is senior author on the report from the Get With The Guidelines–Heart Failure (GWTG-HF) quality improvement program of the American Heart Association.
Broad uptake of new medical therapies into practice may sometimes take 15 or more years from first publication, Dr. Hess said, so, “I find it encouraging in the study that over a shorter time period, 2017-2020, there was improvement.”
Indeed, the odds of in-hospital initiation of an indicated med during that period on average climbed a significant 8% every 3 months, the report states.
The finding suggests that “heart failure hospitalization is, in and of itself, an important intervention for getting folks on the appropriate medications,” Dr. Hess said. It also means “we’re getting better at it,” at least at the study’s 160 GWTG-HF participating hospitals nationwide.
Those centers, the report acknowledges, varied in size, geography, and teaching status but were not necessarily representative of all U.S. hospitals. In another potential limitation, the study couldn’t account for patients who weren’t prescribed all indicated medications for clinically valid reasons. It excluded patients with “clear contraindications,” Dr. Hess said. But there could have been “legitimate reasons” some indicated medications weren’t always prescribed, including patient frailty, hemodynamic intolerance, renal dysfunction, or polypharmacy concerns.
“Positive takeaways” from the analysis, notes an accompanying editorial, include improved prescription rates for key GDMT categories across more than 3 years of data, and evidence that in-hospital initiation “was feasible and, at least for some medications, reliably undertaken.”
Of note, new GDMT prescriptions from admission to discharge went from 70% to almost 98% for beta-blockers, from 59% to about 91% for RAS inhibitors, from about 26% to 56% for MRAs, and from 15.5% to 27.4% for hydralazine/nitrates, wrote Karen E. Joynt Maddox, MD, MPH, and Daniel K. Fox, MD, PhD, both of Washington University in St. Louis.
“Key areas for improvement,” they noted, include prescriptions for women, who were 12% less likely than men to have appropriate GDMT initiated during hospitalization (P < .001); and practice at rural hospitals, which were 40% less likely than urban centers to have patients on full GDMT by discharge (P = .017).
Although only 2.6% of the GWTG-HF centers were in rural locations, “rural hospitals make up approximately one-third of general acute care hospitals in this country,” the editorial states. They therefore “represent a key source of health disparity” in the United States in need of further study.
The analysis of 50,170 patients hospitalized with HFrEF compared the number of GDMT medications for which they were eligible, on at-hospital admission, and by discharge.
The drug categories included “evidence based beta blockers,” that is, bisoprolol, carvedilol, or sustained-release metoprolol; RAS inhibitors, specifically ACE inhibitors, angiotensin receptor blockers, or sacubitril/valsartan (Entresto); MRAs; SGLT2 inhibitors in patients with diabetes; diuretics for congestion; oral anticoagulants for atrial fibrillation; and hydralazine/nitrates in African Americans.
About 15% of the patients at hospital admission were on all indicated HFrEF medications for which they were eligible. The proportion more than doubled to 32.8% by discharge.
Factors significantly associated with reduced odds for in-hospital GDMT initiation include older age (odds ratio, 0.94 per 5-year increment), being female versus male (OR, 0.88), rural location (OR, 0.60), Medicaid versus Medicare or private insurance (OR, 0.93), stroke history (OR, 0.91), peripheral artery disease (OR, 0.93), chronic obstructive pulmonary disease or asthma (OR, 0.86), and renal insufficiency (OR, 0.77).
The findings suggest that there has been at least some progress in getting hospitalized patients “on the right meds” by discharge, Dr. Hess observed. To help address shortfalls in some patient groups, “there is interest in engaging pharmacists in helping us encourage providers on the front lines to initiate and titrate medications.”
The GWTG-HF program is sponsored, in part, by Novartis, Boehringer Ingelheim, Novo Nordisk, AstraZeneca, Bayer, Tylenol, and Alnylam Pharmaceuticals. Dr. Hess disclosed no relevant financial relationships. Dr. Maddox disclosed serving on the Health Policy Advisory Council for Centene. Dr. Fox reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Most patients aren’t receiving all the medications they should based on guidelines, nor are they getting them at the most effective time in their disease course, suggests a registry study of patients in the United States hospitalized with heart failure with reduced ejection fraction (HFrEF).
On average, one such medication was initiated per patient for every 6 days in the hospital.
Shortfalls in predischarge GDMT initiation disproportionately landed on women, patients at rural centers, and those with renal failure or other comorbidities. But they didn’t seem related to patient race or ethnicity in the study reported in JACC: Heart Failure.
The analysis covers the 3 years preceding the May 2020 first-time approval of a sodium-glucose cotransporter 2 (SGLT2) inhibitor for nondiabetic patients with HFrEF, and therefore doesn’t cover such drugs for that indication. The SGLT2 inhibitors would later join beta-blockers, renin-angiotensin system (RAS) inhibitors, and mineralocorticoid receptor antagonists (MRAs) in the quartet of core GDMT medications broadly indicated for HFrEF.
In-hospital initiation of GDMT for HFrEF is considered a predictor of being on those medications after discharge and is itself guideline recommended. There’s clear evidence that treatment with the four core medications boosts survival and cuts rehospitalization risk, and that “getting those on board as soon as possible will eventually benefit many patients,” Paul L. Hess, MD, MHS, said in an interview.
Dr. Hess, University of Colorado Anschutz Medical Campus, Aurora, is senior author on the report from the Get With The Guidelines–Heart Failure (GWTG-HF) quality improvement program of the American Heart Association.
Broad uptake of new medical therapies into practice may sometimes take 15 or more years from first publication, Dr. Hess said, so, “I find it encouraging in the study that over a shorter time period, 2017-2020, there was improvement.”
Indeed, the odds of in-hospital initiation of an indicated med during that period on average climbed a significant 8% every 3 months, the report states.
The finding suggests that “heart failure hospitalization is, in and of itself, an important intervention for getting folks on the appropriate medications,” Dr. Hess said. It also means “we’re getting better at it,” at least at the study’s 160 GWTG-HF participating hospitals nationwide.
Those centers, the report acknowledges, varied in size, geography, and teaching status but were not necessarily representative of all U.S. hospitals. In another potential limitation, the study couldn’t account for patients who weren’t prescribed all indicated medications for clinically valid reasons. It excluded patients with “clear contraindications,” Dr. Hess said. But there could have been “legitimate reasons” some indicated medications weren’t always prescribed, including patient frailty, hemodynamic intolerance, renal dysfunction, or polypharmacy concerns.
“Positive takeaways” from the analysis, notes an accompanying editorial, include improved prescription rates for key GDMT categories across more than 3 years of data, and evidence that in-hospital initiation “was feasible and, at least for some medications, reliably undertaken.”
Of note, new GDMT prescriptions from admission to discharge went from 70% to almost 98% for beta-blockers, from 59% to about 91% for RAS inhibitors, from about 26% to 56% for MRAs, and from 15.5% to 27.4% for hydralazine/nitrates, wrote Karen E. Joynt Maddox, MD, MPH, and Daniel K. Fox, MD, PhD, both of Washington University in St. Louis.
“Key areas for improvement,” they noted, include prescriptions for women, who were 12% less likely than men to have appropriate GDMT initiated during hospitalization (P < .001); and practice at rural hospitals, which were 40% less likely than urban centers to have patients on full GDMT by discharge (P = .017).
Although only 2.6% of the GWTG-HF centers were in rural locations, “rural hospitals make up approximately one-third of general acute care hospitals in this country,” the editorial states. They therefore “represent a key source of health disparity” in the United States in need of further study.
The analysis of 50,170 patients hospitalized with HFrEF compared the number of GDMT medications for which they were eligible, on at-hospital admission, and by discharge.
The drug categories included “evidence based beta blockers,” that is, bisoprolol, carvedilol, or sustained-release metoprolol; RAS inhibitors, specifically ACE inhibitors, angiotensin receptor blockers, or sacubitril/valsartan (Entresto); MRAs; SGLT2 inhibitors in patients with diabetes; diuretics for congestion; oral anticoagulants for atrial fibrillation; and hydralazine/nitrates in African Americans.
About 15% of the patients at hospital admission were on all indicated HFrEF medications for which they were eligible. The proportion more than doubled to 32.8% by discharge.
Factors significantly associated with reduced odds for in-hospital GDMT initiation include older age (odds ratio, 0.94 per 5-year increment), being female versus male (OR, 0.88), rural location (OR, 0.60), Medicaid versus Medicare or private insurance (OR, 0.93), stroke history (OR, 0.91), peripheral artery disease (OR, 0.93), chronic obstructive pulmonary disease or asthma (OR, 0.86), and renal insufficiency (OR, 0.77).
The findings suggest that there has been at least some progress in getting hospitalized patients “on the right meds” by discharge, Dr. Hess observed. To help address shortfalls in some patient groups, “there is interest in engaging pharmacists in helping us encourage providers on the front lines to initiate and titrate medications.”
The GWTG-HF program is sponsored, in part, by Novartis, Boehringer Ingelheim, Novo Nordisk, AstraZeneca, Bayer, Tylenol, and Alnylam Pharmaceuticals. Dr. Hess disclosed no relevant financial relationships. Dr. Maddox disclosed serving on the Health Policy Advisory Council for Centene. Dr. Fox reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Most patients aren’t receiving all the medications they should based on guidelines, nor are they getting them at the most effective time in their disease course, suggests a registry study of patients in the United States hospitalized with heart failure with reduced ejection fraction (HFrEF).
On average, one such medication was initiated per patient for every 6 days in the hospital.
Shortfalls in predischarge GDMT initiation disproportionately landed on women, patients at rural centers, and those with renal failure or other comorbidities. But they didn’t seem related to patient race or ethnicity in the study reported in JACC: Heart Failure.
The analysis covers the 3 years preceding the May 2020 first-time approval of a sodium-glucose cotransporter 2 (SGLT2) inhibitor for nondiabetic patients with HFrEF, and therefore doesn’t cover such drugs for that indication. The SGLT2 inhibitors would later join beta-blockers, renin-angiotensin system (RAS) inhibitors, and mineralocorticoid receptor antagonists (MRAs) in the quartet of core GDMT medications broadly indicated for HFrEF.
In-hospital initiation of GDMT for HFrEF is considered a predictor of being on those medications after discharge and is itself guideline recommended. There’s clear evidence that treatment with the four core medications boosts survival and cuts rehospitalization risk, and that “getting those on board as soon as possible will eventually benefit many patients,” Paul L. Hess, MD, MHS, said in an interview.
Dr. Hess, University of Colorado Anschutz Medical Campus, Aurora, is senior author on the report from the Get With The Guidelines–Heart Failure (GWTG-HF) quality improvement program of the American Heart Association.
Broad uptake of new medical therapies into practice may sometimes take 15 or more years from first publication, Dr. Hess said, so, “I find it encouraging in the study that over a shorter time period, 2017-2020, there was improvement.”
Indeed, the odds of in-hospital initiation of an indicated med during that period on average climbed a significant 8% every 3 months, the report states.
The finding suggests that “heart failure hospitalization is, in and of itself, an important intervention for getting folks on the appropriate medications,” Dr. Hess said. It also means “we’re getting better at it,” at least at the study’s 160 GWTG-HF participating hospitals nationwide.
Those centers, the report acknowledges, varied in size, geography, and teaching status but were not necessarily representative of all U.S. hospitals. In another potential limitation, the study couldn’t account for patients who weren’t prescribed all indicated medications for clinically valid reasons. It excluded patients with “clear contraindications,” Dr. Hess said. But there could have been “legitimate reasons” some indicated medications weren’t always prescribed, including patient frailty, hemodynamic intolerance, renal dysfunction, or polypharmacy concerns.
“Positive takeaways” from the analysis, notes an accompanying editorial, include improved prescription rates for key GDMT categories across more than 3 years of data, and evidence that in-hospital initiation “was feasible and, at least for some medications, reliably undertaken.”
Of note, new GDMT prescriptions from admission to discharge went from 70% to almost 98% for beta-blockers, from 59% to about 91% for RAS inhibitors, from about 26% to 56% for MRAs, and from 15.5% to 27.4% for hydralazine/nitrates, wrote Karen E. Joynt Maddox, MD, MPH, and Daniel K. Fox, MD, PhD, both of Washington University in St. Louis.
“Key areas for improvement,” they noted, include prescriptions for women, who were 12% less likely than men to have appropriate GDMT initiated during hospitalization (P < .001); and practice at rural hospitals, which were 40% less likely than urban centers to have patients on full GDMT by discharge (P = .017).
Although only 2.6% of the GWTG-HF centers were in rural locations, “rural hospitals make up approximately one-third of general acute care hospitals in this country,” the editorial states. They therefore “represent a key source of health disparity” in the United States in need of further study.
The analysis of 50,170 patients hospitalized with HFrEF compared the number of GDMT medications for which they were eligible, on at-hospital admission, and by discharge.
The drug categories included “evidence based beta blockers,” that is, bisoprolol, carvedilol, or sustained-release metoprolol; RAS inhibitors, specifically ACE inhibitors, angiotensin receptor blockers, or sacubitril/valsartan (Entresto); MRAs; SGLT2 inhibitors in patients with diabetes; diuretics for congestion; oral anticoagulants for atrial fibrillation; and hydralazine/nitrates in African Americans.
About 15% of the patients at hospital admission were on all indicated HFrEF medications for which they were eligible. The proportion more than doubled to 32.8% by discharge.
Factors significantly associated with reduced odds for in-hospital GDMT initiation include older age (odds ratio, 0.94 per 5-year increment), being female versus male (OR, 0.88), rural location (OR, 0.60), Medicaid versus Medicare or private insurance (OR, 0.93), stroke history (OR, 0.91), peripheral artery disease (OR, 0.93), chronic obstructive pulmonary disease or asthma (OR, 0.86), and renal insufficiency (OR, 0.77).
The findings suggest that there has been at least some progress in getting hospitalized patients “on the right meds” by discharge, Dr. Hess observed. To help address shortfalls in some patient groups, “there is interest in engaging pharmacists in helping us encourage providers on the front lines to initiate and titrate medications.”
The GWTG-HF program is sponsored, in part, by Novartis, Boehringer Ingelheim, Novo Nordisk, AstraZeneca, Bayer, Tylenol, and Alnylam Pharmaceuticals. Dr. Hess disclosed no relevant financial relationships. Dr. Maddox disclosed serving on the Health Policy Advisory Council for Centene. Dr. Fox reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JACC: HEART FAILURE
SSRI improves cognition, major depression in early dementia
TOPLINE:
METHODOLOGY:
- The multicenter MEMORY study included 82 subjects with MDD and early-stage dementia, mean age 70.3 years, mostly female (66%) and White (95%).
- Vortioxetine, a modulator of 5-hydroxytryptamine receptor activity and an inhibitor of the 5-HT transporter, initiated at 5 mg/day (recommended starting dose in older adults) with the dose up-titrated to 10 mg/day after a week and flexible dosing thereafter.
- Depression was assessed using the Montgomery-Åsberg Depression Rating Scale (MADRS), and cognition with the Digit Symbol Substitution Test (DSST) and Rey Auditory Verbal Learning Test.
TAKEAWAY:
- There was significant and clinically meaningful improvement in the severity of depressive symptoms, as measured by MADRS total score (the primary outcome), at all assessment time points (P < .0001).
- Improvements in depressive symptoms were irrespective of dementia type.
- There were also significant improvements in DSST total score (P < .0001) and in daily functioning and health-related quality of life (HRQoL).
- Vortioxetine was well tolerated; side effects, including nausea and abdominal pain, were mostly mild to moderate.
IN PRACTICE:
“Vortioxetine demonstrated effectiveness in clinically significantly improving depressive symptoms, cognitive performance, daily and global functioning, and HRQoL in patients with MDD and comorbid early-stage dementia treated for 12 weeks” the researchers noted.
STUDY DETAILS:
The study was conducted by Michael Cronquist Christensen from pharmaceutical company H. Lundbeck, Valby, Denmark, and colleagues. It was published online in the Journal of Affective Disorders.
LIMITATIONS:
The study is open label and lacked a control group. Learning effects were possible, which could contribute to improved cognitive performance, although significant improvement on the RAVLT was not observed until week 4, suggesting earning effects were minimal.
DISCLOSURES:
The study was funded by H. Lundbeck. Mr. Christensen is an employee of H. Lundbeck.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- The multicenter MEMORY study included 82 subjects with MDD and early-stage dementia, mean age 70.3 years, mostly female (66%) and White (95%).
- Vortioxetine, a modulator of 5-hydroxytryptamine receptor activity and an inhibitor of the 5-HT transporter, initiated at 5 mg/day (recommended starting dose in older adults) with the dose up-titrated to 10 mg/day after a week and flexible dosing thereafter.
- Depression was assessed using the Montgomery-Åsberg Depression Rating Scale (MADRS), and cognition with the Digit Symbol Substitution Test (DSST) and Rey Auditory Verbal Learning Test.
TAKEAWAY:
- There was significant and clinically meaningful improvement in the severity of depressive symptoms, as measured by MADRS total score (the primary outcome), at all assessment time points (P < .0001).
- Improvements in depressive symptoms were irrespective of dementia type.
- There were also significant improvements in DSST total score (P < .0001) and in daily functioning and health-related quality of life (HRQoL).
- Vortioxetine was well tolerated; side effects, including nausea and abdominal pain, were mostly mild to moderate.
IN PRACTICE:
“Vortioxetine demonstrated effectiveness in clinically significantly improving depressive symptoms, cognitive performance, daily and global functioning, and HRQoL in patients with MDD and comorbid early-stage dementia treated for 12 weeks” the researchers noted.
STUDY DETAILS:
The study was conducted by Michael Cronquist Christensen from pharmaceutical company H. Lundbeck, Valby, Denmark, and colleagues. It was published online in the Journal of Affective Disorders.
LIMITATIONS:
The study is open label and lacked a control group. Learning effects were possible, which could contribute to improved cognitive performance, although significant improvement on the RAVLT was not observed until week 4, suggesting earning effects were minimal.
DISCLOSURES:
The study was funded by H. Lundbeck. Mr. Christensen is an employee of H. Lundbeck.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- The multicenter MEMORY study included 82 subjects with MDD and early-stage dementia, mean age 70.3 years, mostly female (66%) and White (95%).
- Vortioxetine, a modulator of 5-hydroxytryptamine receptor activity and an inhibitor of the 5-HT transporter, initiated at 5 mg/day (recommended starting dose in older adults) with the dose up-titrated to 10 mg/day after a week and flexible dosing thereafter.
- Depression was assessed using the Montgomery-Åsberg Depression Rating Scale (MADRS), and cognition with the Digit Symbol Substitution Test (DSST) and Rey Auditory Verbal Learning Test.
TAKEAWAY:
- There was significant and clinically meaningful improvement in the severity of depressive symptoms, as measured by MADRS total score (the primary outcome), at all assessment time points (P < .0001).
- Improvements in depressive symptoms were irrespective of dementia type.
- There were also significant improvements in DSST total score (P < .0001) and in daily functioning and health-related quality of life (HRQoL).
- Vortioxetine was well tolerated; side effects, including nausea and abdominal pain, were mostly mild to moderate.
IN PRACTICE:
“Vortioxetine demonstrated effectiveness in clinically significantly improving depressive symptoms, cognitive performance, daily and global functioning, and HRQoL in patients with MDD and comorbid early-stage dementia treated for 12 weeks” the researchers noted.
STUDY DETAILS:
The study was conducted by Michael Cronquist Christensen from pharmaceutical company H. Lundbeck, Valby, Denmark, and colleagues. It was published online in the Journal of Affective Disorders.
LIMITATIONS:
The study is open label and lacked a control group. Learning effects were possible, which could contribute to improved cognitive performance, although significant improvement on the RAVLT was not observed until week 4, suggesting earning effects were minimal.
DISCLOSURES:
The study was funded by H. Lundbeck. Mr. Christensen is an employee of H. Lundbeck.
A version of this article first appeared on Medscape.com.
FDA OKs empagliflozin for children with type 2 diabetes
aged 10 years and older.
This approval represents only the second oral treatment option for children and adolescents with type 2 diabetes after metformin; the latter appears to be less effective for pediatric patients than for adults.
Injectable glucagonlike peptide–1 (GLP-1) agonists are also available for youth with type 2 diabetes. These include daily liraglutide (Victoza) and once-weekly extended-release exenatide (Bydureon/Bydureon BCise).
Jardiance has been approved for adults with type 2 diabetes since 2014, and Synjardy has been approved since 2015.
“Compared to adults, children with type 2 diabetes have limited treatment options, even though the disease and symptom onset generally progress more rapidly in children,” said Michelle Carey, MD, MPH.
“Today’s approvals provide much-needed additional treatment options for children with type 2 diabetes,” added Dr. Carey, associate director for therapeutic review for the division of diabetes, lipid disorders, and obesity in the FDA’s Center for Drug Evaluation and Research.
Type 2 diabetes rising exponentially in children, mainly non-Whites
Type 2 diabetes is rising exponentially in children and adolescents in the United States.
Data from the SEARCH for Diabetes in Youth study show that the incidence of type 2 diabetes among youth rose by about 5% per year between 2002 and 2015, and it continues to rise.
A more recent study found that a doubling of cases occurred during the pandemic, with youth often presenting with more severe disease. The majority of cases are among non-White racial groups.
Safety and efficacy data for empagliflozin for children came from the Diabetes Study of Linagliptin and Empagliflozin in Children and Adolescents (DINAMO) trial. That trial included 157 patients aged 10-17 years with A1c of 7% or above. Patients were randomly assigned to receive empagliflozin 10 mg or 25 mg daily, linagliptin (a DPP-4 inhibitor) 5 mg, or placebo for 26 weeks. Over 90% were also taking metformin, 40% in combination with insulin. All patients were given diet and exercise advice.
At week 26, the children treated with empagliflozin showed an average 0.2 percentage point decrease in A1c, compared with a 0.7-point increase among those taking placebo. Use of empagliflozin was also associated with lower fasting plasma glucose levels compared with placebo.
Side effects were similar to those seen in adults except for a higher risk of hypoglycemia, regardless of other glucose-lowering therapies that were being taken.
Reduction in A1c for participants treated with linagliptin was not statistically significant in comparison with placebo. There was a numerical reduction of 0.34% (P = .2935).
“Across the lifespan, we know that people living with type 2 diabetes have a high risk for many diabetes complications, so it’s important to recognize and treat diabetes early in its course,” Lori Laffel, MD, lead investigator of the DINAMO study, said in a press release from BI.
“These findings are particularly important given the need for more therapeutic options, especially oral agents, to manage type 2 diabetes in young people as, to date, metformin [has been] the only globally available oral treatment for youth,” added Dr. Laffel, chief of the pediatric, adolescent, and young adult section at the Joslin Diabetes Center and professor of pediatrics at Harvard Medical School, Boston.
A version of this article first appeared on Medscape.com.
aged 10 years and older.
This approval represents only the second oral treatment option for children and adolescents with type 2 diabetes after metformin; the latter appears to be less effective for pediatric patients than for adults.
Injectable glucagonlike peptide–1 (GLP-1) agonists are also available for youth with type 2 diabetes. These include daily liraglutide (Victoza) and once-weekly extended-release exenatide (Bydureon/Bydureon BCise).
Jardiance has been approved for adults with type 2 diabetes since 2014, and Synjardy has been approved since 2015.
“Compared to adults, children with type 2 diabetes have limited treatment options, even though the disease and symptom onset generally progress more rapidly in children,” said Michelle Carey, MD, MPH.
“Today’s approvals provide much-needed additional treatment options for children with type 2 diabetes,” added Dr. Carey, associate director for therapeutic review for the division of diabetes, lipid disorders, and obesity in the FDA’s Center for Drug Evaluation and Research.
Type 2 diabetes rising exponentially in children, mainly non-Whites
Type 2 diabetes is rising exponentially in children and adolescents in the United States.
Data from the SEARCH for Diabetes in Youth study show that the incidence of type 2 diabetes among youth rose by about 5% per year between 2002 and 2015, and it continues to rise.
A more recent study found that a doubling of cases occurred during the pandemic, with youth often presenting with more severe disease. The majority of cases are among non-White racial groups.
Safety and efficacy data for empagliflozin for children came from the Diabetes Study of Linagliptin and Empagliflozin in Children and Adolescents (DINAMO) trial. That trial included 157 patients aged 10-17 years with A1c of 7% or above. Patients were randomly assigned to receive empagliflozin 10 mg or 25 mg daily, linagliptin (a DPP-4 inhibitor) 5 mg, or placebo for 26 weeks. Over 90% were also taking metformin, 40% in combination with insulin. All patients were given diet and exercise advice.
At week 26, the children treated with empagliflozin showed an average 0.2 percentage point decrease in A1c, compared with a 0.7-point increase among those taking placebo. Use of empagliflozin was also associated with lower fasting plasma glucose levels compared with placebo.
Side effects were similar to those seen in adults except for a higher risk of hypoglycemia, regardless of other glucose-lowering therapies that were being taken.
Reduction in A1c for participants treated with linagliptin was not statistically significant in comparison with placebo. There was a numerical reduction of 0.34% (P = .2935).
“Across the lifespan, we know that people living with type 2 diabetes have a high risk for many diabetes complications, so it’s important to recognize and treat diabetes early in its course,” Lori Laffel, MD, lead investigator of the DINAMO study, said in a press release from BI.
“These findings are particularly important given the need for more therapeutic options, especially oral agents, to manage type 2 diabetes in young people as, to date, metformin [has been] the only globally available oral treatment for youth,” added Dr. Laffel, chief of the pediatric, adolescent, and young adult section at the Joslin Diabetes Center and professor of pediatrics at Harvard Medical School, Boston.
A version of this article first appeared on Medscape.com.
aged 10 years and older.
This approval represents only the second oral treatment option for children and adolescents with type 2 diabetes after metformin; the latter appears to be less effective for pediatric patients than for adults.
Injectable glucagonlike peptide–1 (GLP-1) agonists are also available for youth with type 2 diabetes. These include daily liraglutide (Victoza) and once-weekly extended-release exenatide (Bydureon/Bydureon BCise).
Jardiance has been approved for adults with type 2 diabetes since 2014, and Synjardy has been approved since 2015.
“Compared to adults, children with type 2 diabetes have limited treatment options, even though the disease and symptom onset generally progress more rapidly in children,” said Michelle Carey, MD, MPH.
“Today’s approvals provide much-needed additional treatment options for children with type 2 diabetes,” added Dr. Carey, associate director for therapeutic review for the division of diabetes, lipid disorders, and obesity in the FDA’s Center for Drug Evaluation and Research.
Type 2 diabetes rising exponentially in children, mainly non-Whites
Type 2 diabetes is rising exponentially in children and adolescents in the United States.
Data from the SEARCH for Diabetes in Youth study show that the incidence of type 2 diabetes among youth rose by about 5% per year between 2002 and 2015, and it continues to rise.
A more recent study found that a doubling of cases occurred during the pandemic, with youth often presenting with more severe disease. The majority of cases are among non-White racial groups.
Safety and efficacy data for empagliflozin for children came from the Diabetes Study of Linagliptin and Empagliflozin in Children and Adolescents (DINAMO) trial. That trial included 157 patients aged 10-17 years with A1c of 7% or above. Patients were randomly assigned to receive empagliflozin 10 mg or 25 mg daily, linagliptin (a DPP-4 inhibitor) 5 mg, or placebo for 26 weeks. Over 90% were also taking metformin, 40% in combination with insulin. All patients were given diet and exercise advice.
At week 26, the children treated with empagliflozin showed an average 0.2 percentage point decrease in A1c, compared with a 0.7-point increase among those taking placebo. Use of empagliflozin was also associated with lower fasting plasma glucose levels compared with placebo.
Side effects were similar to those seen in adults except for a higher risk of hypoglycemia, regardless of other glucose-lowering therapies that were being taken.
Reduction in A1c for participants treated with linagliptin was not statistically significant in comparison with placebo. There was a numerical reduction of 0.34% (P = .2935).
“Across the lifespan, we know that people living with type 2 diabetes have a high risk for many diabetes complications, so it’s important to recognize and treat diabetes early in its course,” Lori Laffel, MD, lead investigator of the DINAMO study, said in a press release from BI.
“These findings are particularly important given the need for more therapeutic options, especially oral agents, to manage type 2 diabetes in young people as, to date, metformin [has been] the only globally available oral treatment for youth,” added Dr. Laffel, chief of the pediatric, adolescent, and young adult section at the Joslin Diabetes Center and professor of pediatrics at Harvard Medical School, Boston.
A version of this article first appeared on Medscape.com.
Gilteritinib maintenance reduces relapse in MRD+ AML
The research was presented at the European Hematology Association Hybrid Congress 2023.
For the study, AML patients with the most common form of mutation in the proto-oncogene fms-like tyrosine kinase 3 (FLT3), known as the internal tandem duplication (ITD), were randomized to 24 months of maintenance therapy with either the FLT3 inhibitor gilteritinib or placebo.
The trial did not meet its primary endpoint, as there was no significant difference in relapse-free survival (RFS) between those assigned to the active drug and those given placebo, and there was no difference in overall survival rates.
However, subgroup analysis revealed that FLT3/ITD AML patients who were MRD+ after transplant, which represented approximately half of the participants, experienced a significant 48% improvement in RFS with gilteritinib versus placebo, while no benefit was seen in MRD– patients.
While acknowledging that the trial did not meet its primary endpoint, presenter Mark J. Levis, MD, PhD, program leader, hematologic malignancies and bone marrow transplant program, Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, said it was nevertheless “a successful study.”
“We learned how to use these drugs and in whom,” he continued, adding: “No, not everybody needs and should get a FLT3 inhibitor post-transplant, but we can use this [MRD] assay to identify who.”
Consequently, Dr. Levis believes that gilteritinib “should be a standard of care for those who are MRD positive,” although the decision to use it “should be balanced against the potential for toxicity,” compared with not adding an additional treatment after HCT.
He told a press conference that “we’re going to certainly make sure that patients who are MRD positive get [gilteritinib],” although the MRD negative patients “are going to be more questionable,” especially because the assay that they used in the study is not “perfect.”
Dr. Levis also suggested that the trial did not meet its endpoint because of regional differences in the clinical practice, such as in the number of treatment cycles prior to HCT, the time to transplant, and the previous use of a FLT3 inhibitor, all of which may have skewed the findings.
“Everybody in the world is convinced that they’re the best transplanter,” he said, and yet “they all do it differently, and the heterogeneity is astounding.”
He added: “If we’d restricted everybody [to a] pretransplant regimen, I suspect we would have had a different result than what we’re getting here, but this is releasing the drug into the world and saying: ‘Here, transplant however you want, however it’s practiced in the real world. Tell us how this works.’ ”
Approached for comment, Claudio Brunstein, MD, PhD, vice-chair of the department of hematology and oncology in the Cleveland Clinic Taussig Cancer Institute, said that while there was “some disappointment” with the results, he was “not surprised” that the trial did not meet its primary endpoint.
He said in an interview that the patient population was not of “high enough risk” to demonstrate an overall difference between gilteritinib and placebo, although he conceded that it is “hard to get to high-risk patients in a timely way” and so conduct a trial with them.
As to the notion that variations in clinical practice could have been responsible, Dr. Brunstein pointed out that it was a randomized trial, so the issue would have applied equally to both sides.
He nevertheless believes that it is “a very important study,” and “just the fact that it was done in the context of a number of drugs coming and being approved by the [U.S. Food and Drug Administration] in AML is quite remarkable.”
This is especially the case given that “many centers are already using [gilteritinib] as off-label maintenance therapy.”
Dr. Brunstein added that it is “good news” that the drug was effective in MRD+ patients, as it shows “you can overcome that with maintenance therapy rather than keeping giving more and more chemotherapy, especially as there are patients you’re worried about giving more intensive chemotherapy to make them MRD negative.”
He pointed out, however, that the assay used in the trial was “research grade” and very sensitive to MRD and “is not available everywhere, so there is an adjustment that the community will have to do to in order to apply this data.”
“But for those who are more obviously MRD positive with less sensitive assays, gilteritinib is already something that can be used,” Dr. Brunstein said.
Presenting the findings, Dr. Levis stated: “We all know that patients with FLT3/ITD AML have a high risk of relapse and are routinely referred for transplant. And we know that the detection of measurable residual disease pretransplant is highly predictive of outcome post-transplant.”
He continued that FLT3 inhibitors are “routinely given as post-transplant maintenance ... based on some prior trials, mostly with sorafenib.”
“But uncertainty exists as to the broad applicability of these trials,” Dr. Levis said. Moreover, the use of sorafenib in this context is “off label and can be difficult to tolerate,” and “we know that most patients are cured with allogeneic transplant alone.”
Gilteritinib is already known to be well tolerated as a monotherapy, and was approved by the FDA for the treatment of adult patients with FLT3 mutation–positive relapsed or refractory AML in 2018.
The investigators therefore examined whether it would be beneficial as a post-HCT maintenance therapy in FLT3-ITD AML. Patients were required to be in morphologic remission after one or two courses of induction therapy, with Dr. Levis underlining: “We did not allow patients who had been salvaged onto the study.”
They subsequently had a marrow aspirate sample taken for MRD analysis before undergoing allogeneic transplant, with any conditioning regimen, donor, or graft-versus-host disease (GVHD) prophylaxis allowed.
Between 30 and 90 days later, patients with successful engraftment who were able to take oral medication were then randomized to 24 months of maintenance therapy with either gilteritinib or placebo.
Dr. Levis showed that, among 620 patients screened at 110 centers in 16 countries, 356 were randomized between Aug. 15, 2017, and July 8, 2020. The median age was 53 years, and 49% of gilteritinib patients and 48% of those given placebo were female.
He noted that there was a “fairly even global distribution” of patients from North America, Europe, and the Asia/Pacific region, and that 60% of patients underwent a myeloablative conditioning regimen. Approximately the same proportion had received an FLT3 inhibitor prior to HCT.
MRD positivity, assessed at a cell count of ≥ 10-6, was observed pre-HCT in 47% of patients in both treatment groups, and in 50% of gilteritinib patients and 51% of placebo patients at both pre- and post-transplant assessments.
The treatment regimen was completed by 52.8% of patients assigned to gilteritinib and 53.9% in the placebo arm. Dr. Levis said that 18.5% and 20.3% of patients, respectively, experienced a grade 3/4 treatment emergent acute GVHD event, while 32.6% and 21.5%, respectively, had a grade ≥ 3 treatment emergent infection.
He noted that “adverse events were clearly more common in the gilteritinib arm and often led to either dose reduction or interruption, or withdrawal of treatment.”
The most common grade ≥ 3 treatment emergent adverse event was a decrease in neutrophil count, seen in 24.7% of gilteritinib patients and 7.9% of those given placebo, followed by reduced platelet count, in 15.2% and 5.6%, respectively, and anemia, in 6.2% and 1.7%, respectively.
Turning to the efficacy outcomes, Dr. Levis reported that the trial did not meet its primary endpoint, with no significant difference in RFS between the gilteritinib and placebo arms, at a hazard ratio of 0.679 (P = .0518). There was also no significant difference in the key secondary objective of overall survival, at a hazard ratio of 0.846 (P = .4394).
However, Dr. Levis noted that there was a “clear difference in the benefit of gilteritinib by region,” and, “at every level,” MRD predicted a benefit from gilteritinib, which he said was a “big surprise” and “really leapt out in the subgroup analysis.”
He explained that the researchers used a modified version of a two-step assay that has been used in previous studies, and was able to detect MRD at a sensitivity of approximately 1x10-6. “In our study, 98% of participants had samples pre- and post-[transplant].”
Regardless of treatment arm, MRD positivity measured at that sensitivity was associated with a significant reduction in overall survival, at a hazard ratio versus MRD– status of 0.514 (P = .0025).
When stratifying the patients by MRD status, the researchers found that, among MRD+ participants, gilteritinib was associated with a significant improvement in RFS, at a hazard ratio versus placebo of 0.515 (P = .0065), while there was no significant difference in MRD– patients.
Stratifying the patients by their conditioning regimen prior to HCT also revealed differences, with those undergoing myeloablative conditioning having significantly greater overall survival than those who underwent reduced-intensity conditioning, at a hazard ratio for death of 0.529 (P = .0027).
Dr. Levis said there is “no surprise there,” and the result could reflect the selection of fitter, younger patients to undergo the more intensive regimen.
He then showed that MRD+ patients who had undergone myeloablative conditioning had better overall survival with gilteritinib than placebo, at a hazard ratio for death of 0.418 (P = .0087). Again, the difference disappeared when looking at MRD– patients.
“So conditioning doesn’t help you in the setting of MRD,” Dr. Levis said.
Finally, he took a deeper dive into the regional differences in outcomes, noting that patients in the Asia/Pacific region, where gilteritinib showed no benefit over placebo, “were 10 years younger” than those in other regions, “tended to get myeloablative conditioning, and hardly ever used FLT3 inhibitors.”
In contrast, North American patients, who experienced a significant gilteritinib benefit in terms of RFS, underwent HCT an average of 26 days earlier than those elsewhere, and received fewer courses of chemotherapy pre-HCT. Moreover, 93.5% received an FLT3 inhibitor pretransplant.
The study was funded by Astellas Pharma Global Development. Dr. Levis declares relationships with Abbvie, Amgen, Astellas, Bristol-Myers-Squibb, Daiichi-Sankyo, GlaxoSmithKline, Jazz, Menarini, Pfizer, Sumitomo-Dainippon, Syndax, Takeda. Dr. Brunstein declares no relevant relationships.
The research was presented at the European Hematology Association Hybrid Congress 2023.
For the study, AML patients with the most common form of mutation in the proto-oncogene fms-like tyrosine kinase 3 (FLT3), known as the internal tandem duplication (ITD), were randomized to 24 months of maintenance therapy with either the FLT3 inhibitor gilteritinib or placebo.
The trial did not meet its primary endpoint, as there was no significant difference in relapse-free survival (RFS) between those assigned to the active drug and those given placebo, and there was no difference in overall survival rates.
However, subgroup analysis revealed that FLT3/ITD AML patients who were MRD+ after transplant, which represented approximately half of the participants, experienced a significant 48% improvement in RFS with gilteritinib versus placebo, while no benefit was seen in MRD– patients.
While acknowledging that the trial did not meet its primary endpoint, presenter Mark J. Levis, MD, PhD, program leader, hematologic malignancies and bone marrow transplant program, Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, said it was nevertheless “a successful study.”
“We learned how to use these drugs and in whom,” he continued, adding: “No, not everybody needs and should get a FLT3 inhibitor post-transplant, but we can use this [MRD] assay to identify who.”
Consequently, Dr. Levis believes that gilteritinib “should be a standard of care for those who are MRD positive,” although the decision to use it “should be balanced against the potential for toxicity,” compared with not adding an additional treatment after HCT.
He told a press conference that “we’re going to certainly make sure that patients who are MRD positive get [gilteritinib],” although the MRD negative patients “are going to be more questionable,” especially because the assay that they used in the study is not “perfect.”
Dr. Levis also suggested that the trial did not meet its endpoint because of regional differences in the clinical practice, such as in the number of treatment cycles prior to HCT, the time to transplant, and the previous use of a FLT3 inhibitor, all of which may have skewed the findings.
“Everybody in the world is convinced that they’re the best transplanter,” he said, and yet “they all do it differently, and the heterogeneity is astounding.”
He added: “If we’d restricted everybody [to a] pretransplant regimen, I suspect we would have had a different result than what we’re getting here, but this is releasing the drug into the world and saying: ‘Here, transplant however you want, however it’s practiced in the real world. Tell us how this works.’ ”
Approached for comment, Claudio Brunstein, MD, PhD, vice-chair of the department of hematology and oncology in the Cleveland Clinic Taussig Cancer Institute, said that while there was “some disappointment” with the results, he was “not surprised” that the trial did not meet its primary endpoint.
He said in an interview that the patient population was not of “high enough risk” to demonstrate an overall difference between gilteritinib and placebo, although he conceded that it is “hard to get to high-risk patients in a timely way” and so conduct a trial with them.
As to the notion that variations in clinical practice could have been responsible, Dr. Brunstein pointed out that it was a randomized trial, so the issue would have applied equally to both sides.
He nevertheless believes that it is “a very important study,” and “just the fact that it was done in the context of a number of drugs coming and being approved by the [U.S. Food and Drug Administration] in AML is quite remarkable.”
This is especially the case given that “many centers are already using [gilteritinib] as off-label maintenance therapy.”
Dr. Brunstein added that it is “good news” that the drug was effective in MRD+ patients, as it shows “you can overcome that with maintenance therapy rather than keeping giving more and more chemotherapy, especially as there are patients you’re worried about giving more intensive chemotherapy to make them MRD negative.”
He pointed out, however, that the assay used in the trial was “research grade” and very sensitive to MRD and “is not available everywhere, so there is an adjustment that the community will have to do to in order to apply this data.”
“But for those who are more obviously MRD positive with less sensitive assays, gilteritinib is already something that can be used,” Dr. Brunstein said.
Presenting the findings, Dr. Levis stated: “We all know that patients with FLT3/ITD AML have a high risk of relapse and are routinely referred for transplant. And we know that the detection of measurable residual disease pretransplant is highly predictive of outcome post-transplant.”
He continued that FLT3 inhibitors are “routinely given as post-transplant maintenance ... based on some prior trials, mostly with sorafenib.”
“But uncertainty exists as to the broad applicability of these trials,” Dr. Levis said. Moreover, the use of sorafenib in this context is “off label and can be difficult to tolerate,” and “we know that most patients are cured with allogeneic transplant alone.”
Gilteritinib is already known to be well tolerated as a monotherapy, and was approved by the FDA for the treatment of adult patients with FLT3 mutation–positive relapsed or refractory AML in 2018.
The investigators therefore examined whether it would be beneficial as a post-HCT maintenance therapy in FLT3-ITD AML. Patients were required to be in morphologic remission after one or two courses of induction therapy, with Dr. Levis underlining: “We did not allow patients who had been salvaged onto the study.”
They subsequently had a marrow aspirate sample taken for MRD analysis before undergoing allogeneic transplant, with any conditioning regimen, donor, or graft-versus-host disease (GVHD) prophylaxis allowed.
Between 30 and 90 days later, patients with successful engraftment who were able to take oral medication were then randomized to 24 months of maintenance therapy with either gilteritinib or placebo.
Dr. Levis showed that, among 620 patients screened at 110 centers in 16 countries, 356 were randomized between Aug. 15, 2017, and July 8, 2020. The median age was 53 years, and 49% of gilteritinib patients and 48% of those given placebo were female.
He noted that there was a “fairly even global distribution” of patients from North America, Europe, and the Asia/Pacific region, and that 60% of patients underwent a myeloablative conditioning regimen. Approximately the same proportion had received an FLT3 inhibitor prior to HCT.
MRD positivity, assessed at a cell count of ≥ 10-6, was observed pre-HCT in 47% of patients in both treatment groups, and in 50% of gilteritinib patients and 51% of placebo patients at both pre- and post-transplant assessments.
The treatment regimen was completed by 52.8% of patients assigned to gilteritinib and 53.9% in the placebo arm. Dr. Levis said that 18.5% and 20.3% of patients, respectively, experienced a grade 3/4 treatment emergent acute GVHD event, while 32.6% and 21.5%, respectively, had a grade ≥ 3 treatment emergent infection.
He noted that “adverse events were clearly more common in the gilteritinib arm and often led to either dose reduction or interruption, or withdrawal of treatment.”
The most common grade ≥ 3 treatment emergent adverse event was a decrease in neutrophil count, seen in 24.7% of gilteritinib patients and 7.9% of those given placebo, followed by reduced platelet count, in 15.2% and 5.6%, respectively, and anemia, in 6.2% and 1.7%, respectively.
Turning to the efficacy outcomes, Dr. Levis reported that the trial did not meet its primary endpoint, with no significant difference in RFS between the gilteritinib and placebo arms, at a hazard ratio of 0.679 (P = .0518). There was also no significant difference in the key secondary objective of overall survival, at a hazard ratio of 0.846 (P = .4394).
However, Dr. Levis noted that there was a “clear difference in the benefit of gilteritinib by region,” and, “at every level,” MRD predicted a benefit from gilteritinib, which he said was a “big surprise” and “really leapt out in the subgroup analysis.”
He explained that the researchers used a modified version of a two-step assay that has been used in previous studies, and was able to detect MRD at a sensitivity of approximately 1x10-6. “In our study, 98% of participants had samples pre- and post-[transplant].”
Regardless of treatment arm, MRD positivity measured at that sensitivity was associated with a significant reduction in overall survival, at a hazard ratio versus MRD– status of 0.514 (P = .0025).
When stratifying the patients by MRD status, the researchers found that, among MRD+ participants, gilteritinib was associated with a significant improvement in RFS, at a hazard ratio versus placebo of 0.515 (P = .0065), while there was no significant difference in MRD– patients.
Stratifying the patients by their conditioning regimen prior to HCT also revealed differences, with those undergoing myeloablative conditioning having significantly greater overall survival than those who underwent reduced-intensity conditioning, at a hazard ratio for death of 0.529 (P = .0027).
Dr. Levis said there is “no surprise there,” and the result could reflect the selection of fitter, younger patients to undergo the more intensive regimen.
He then showed that MRD+ patients who had undergone myeloablative conditioning had better overall survival with gilteritinib than placebo, at a hazard ratio for death of 0.418 (P = .0087). Again, the difference disappeared when looking at MRD– patients.
“So conditioning doesn’t help you in the setting of MRD,” Dr. Levis said.
Finally, he took a deeper dive into the regional differences in outcomes, noting that patients in the Asia/Pacific region, where gilteritinib showed no benefit over placebo, “were 10 years younger” than those in other regions, “tended to get myeloablative conditioning, and hardly ever used FLT3 inhibitors.”
In contrast, North American patients, who experienced a significant gilteritinib benefit in terms of RFS, underwent HCT an average of 26 days earlier than those elsewhere, and received fewer courses of chemotherapy pre-HCT. Moreover, 93.5% received an FLT3 inhibitor pretransplant.
The study was funded by Astellas Pharma Global Development. Dr. Levis declares relationships with Abbvie, Amgen, Astellas, Bristol-Myers-Squibb, Daiichi-Sankyo, GlaxoSmithKline, Jazz, Menarini, Pfizer, Sumitomo-Dainippon, Syndax, Takeda. Dr. Brunstein declares no relevant relationships.
The research was presented at the European Hematology Association Hybrid Congress 2023.
For the study, AML patients with the most common form of mutation in the proto-oncogene fms-like tyrosine kinase 3 (FLT3), known as the internal tandem duplication (ITD), were randomized to 24 months of maintenance therapy with either the FLT3 inhibitor gilteritinib or placebo.
The trial did not meet its primary endpoint, as there was no significant difference in relapse-free survival (RFS) between those assigned to the active drug and those given placebo, and there was no difference in overall survival rates.
However, subgroup analysis revealed that FLT3/ITD AML patients who were MRD+ after transplant, which represented approximately half of the participants, experienced a significant 48% improvement in RFS with gilteritinib versus placebo, while no benefit was seen in MRD– patients.
While acknowledging that the trial did not meet its primary endpoint, presenter Mark J. Levis, MD, PhD, program leader, hematologic malignancies and bone marrow transplant program, Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, said it was nevertheless “a successful study.”
“We learned how to use these drugs and in whom,” he continued, adding: “No, not everybody needs and should get a FLT3 inhibitor post-transplant, but we can use this [MRD] assay to identify who.”
Consequently, Dr. Levis believes that gilteritinib “should be a standard of care for those who are MRD positive,” although the decision to use it “should be balanced against the potential for toxicity,” compared with not adding an additional treatment after HCT.
He told a press conference that “we’re going to certainly make sure that patients who are MRD positive get [gilteritinib],” although the MRD negative patients “are going to be more questionable,” especially because the assay that they used in the study is not “perfect.”
Dr. Levis also suggested that the trial did not meet its endpoint because of regional differences in the clinical practice, such as in the number of treatment cycles prior to HCT, the time to transplant, and the previous use of a FLT3 inhibitor, all of which may have skewed the findings.
“Everybody in the world is convinced that they’re the best transplanter,” he said, and yet “they all do it differently, and the heterogeneity is astounding.”
He added: “If we’d restricted everybody [to a] pretransplant regimen, I suspect we would have had a different result than what we’re getting here, but this is releasing the drug into the world and saying: ‘Here, transplant however you want, however it’s practiced in the real world. Tell us how this works.’ ”
Approached for comment, Claudio Brunstein, MD, PhD, vice-chair of the department of hematology and oncology in the Cleveland Clinic Taussig Cancer Institute, said that while there was “some disappointment” with the results, he was “not surprised” that the trial did not meet its primary endpoint.
He said in an interview that the patient population was not of “high enough risk” to demonstrate an overall difference between gilteritinib and placebo, although he conceded that it is “hard to get to high-risk patients in a timely way” and so conduct a trial with them.
As to the notion that variations in clinical practice could have been responsible, Dr. Brunstein pointed out that it was a randomized trial, so the issue would have applied equally to both sides.
He nevertheless believes that it is “a very important study,” and “just the fact that it was done in the context of a number of drugs coming and being approved by the [U.S. Food and Drug Administration] in AML is quite remarkable.”
This is especially the case given that “many centers are already using [gilteritinib] as off-label maintenance therapy.”
Dr. Brunstein added that it is “good news” that the drug was effective in MRD+ patients, as it shows “you can overcome that with maintenance therapy rather than keeping giving more and more chemotherapy, especially as there are patients you’re worried about giving more intensive chemotherapy to make them MRD negative.”
He pointed out, however, that the assay used in the trial was “research grade” and very sensitive to MRD and “is not available everywhere, so there is an adjustment that the community will have to do to in order to apply this data.”
“But for those who are more obviously MRD positive with less sensitive assays, gilteritinib is already something that can be used,” Dr. Brunstein said.
Presenting the findings, Dr. Levis stated: “We all know that patients with FLT3/ITD AML have a high risk of relapse and are routinely referred for transplant. And we know that the detection of measurable residual disease pretransplant is highly predictive of outcome post-transplant.”
He continued that FLT3 inhibitors are “routinely given as post-transplant maintenance ... based on some prior trials, mostly with sorafenib.”
“But uncertainty exists as to the broad applicability of these trials,” Dr. Levis said. Moreover, the use of sorafenib in this context is “off label and can be difficult to tolerate,” and “we know that most patients are cured with allogeneic transplant alone.”
Gilteritinib is already known to be well tolerated as a monotherapy, and was approved by the FDA for the treatment of adult patients with FLT3 mutation–positive relapsed or refractory AML in 2018.
The investigators therefore examined whether it would be beneficial as a post-HCT maintenance therapy in FLT3-ITD AML. Patients were required to be in morphologic remission after one or two courses of induction therapy, with Dr. Levis underlining: “We did not allow patients who had been salvaged onto the study.”
They subsequently had a marrow aspirate sample taken for MRD analysis before undergoing allogeneic transplant, with any conditioning regimen, donor, or graft-versus-host disease (GVHD) prophylaxis allowed.
Between 30 and 90 days later, patients with successful engraftment who were able to take oral medication were then randomized to 24 months of maintenance therapy with either gilteritinib or placebo.
Dr. Levis showed that, among 620 patients screened at 110 centers in 16 countries, 356 were randomized between Aug. 15, 2017, and July 8, 2020. The median age was 53 years, and 49% of gilteritinib patients and 48% of those given placebo were female.
He noted that there was a “fairly even global distribution” of patients from North America, Europe, and the Asia/Pacific region, and that 60% of patients underwent a myeloablative conditioning regimen. Approximately the same proportion had received an FLT3 inhibitor prior to HCT.
MRD positivity, assessed at a cell count of ≥ 10-6, was observed pre-HCT in 47% of patients in both treatment groups, and in 50% of gilteritinib patients and 51% of placebo patients at both pre- and post-transplant assessments.
The treatment regimen was completed by 52.8% of patients assigned to gilteritinib and 53.9% in the placebo arm. Dr. Levis said that 18.5% and 20.3% of patients, respectively, experienced a grade 3/4 treatment emergent acute GVHD event, while 32.6% and 21.5%, respectively, had a grade ≥ 3 treatment emergent infection.
He noted that “adverse events were clearly more common in the gilteritinib arm and often led to either dose reduction or interruption, or withdrawal of treatment.”
The most common grade ≥ 3 treatment emergent adverse event was a decrease in neutrophil count, seen in 24.7% of gilteritinib patients and 7.9% of those given placebo, followed by reduced platelet count, in 15.2% and 5.6%, respectively, and anemia, in 6.2% and 1.7%, respectively.
Turning to the efficacy outcomes, Dr. Levis reported that the trial did not meet its primary endpoint, with no significant difference in RFS between the gilteritinib and placebo arms, at a hazard ratio of 0.679 (P = .0518). There was also no significant difference in the key secondary objective of overall survival, at a hazard ratio of 0.846 (P = .4394).
However, Dr. Levis noted that there was a “clear difference in the benefit of gilteritinib by region,” and, “at every level,” MRD predicted a benefit from gilteritinib, which he said was a “big surprise” and “really leapt out in the subgroup analysis.”
He explained that the researchers used a modified version of a two-step assay that has been used in previous studies, and was able to detect MRD at a sensitivity of approximately 1x10-6. “In our study, 98% of participants had samples pre- and post-[transplant].”
Regardless of treatment arm, MRD positivity measured at that sensitivity was associated with a significant reduction in overall survival, at a hazard ratio versus MRD– status of 0.514 (P = .0025).
When stratifying the patients by MRD status, the researchers found that, among MRD+ participants, gilteritinib was associated with a significant improvement in RFS, at a hazard ratio versus placebo of 0.515 (P = .0065), while there was no significant difference in MRD– patients.
Stratifying the patients by their conditioning regimen prior to HCT also revealed differences, with those undergoing myeloablative conditioning having significantly greater overall survival than those who underwent reduced-intensity conditioning, at a hazard ratio for death of 0.529 (P = .0027).
Dr. Levis said there is “no surprise there,” and the result could reflect the selection of fitter, younger patients to undergo the more intensive regimen.
He then showed that MRD+ patients who had undergone myeloablative conditioning had better overall survival with gilteritinib than placebo, at a hazard ratio for death of 0.418 (P = .0087). Again, the difference disappeared when looking at MRD– patients.
“So conditioning doesn’t help you in the setting of MRD,” Dr. Levis said.
Finally, he took a deeper dive into the regional differences in outcomes, noting that patients in the Asia/Pacific region, where gilteritinib showed no benefit over placebo, “were 10 years younger” than those in other regions, “tended to get myeloablative conditioning, and hardly ever used FLT3 inhibitors.”
In contrast, North American patients, who experienced a significant gilteritinib benefit in terms of RFS, underwent HCT an average of 26 days earlier than those elsewhere, and received fewer courses of chemotherapy pre-HCT. Moreover, 93.5% received an FLT3 inhibitor pretransplant.
The study was funded by Astellas Pharma Global Development. Dr. Levis declares relationships with Abbvie, Amgen, Astellas, Bristol-Myers-Squibb, Daiichi-Sankyo, GlaxoSmithKline, Jazz, Menarini, Pfizer, Sumitomo-Dainippon, Syndax, Takeda. Dr. Brunstein declares no relevant relationships.
AT EHA 2023
FDA OKs low-dose colchicine for broad CV indication
The Food and Drug Administration has approved the anti-inflammatory drug colchicine 0.5 mg tablets (Lodoco) as the first specific anti-inflammatory drug demonstrated to reduce the risk for myocardial infarction, stroke, coronary revascularization, and cardiovascular death in adult patients with established atherosclerotic disease or with multiple risk factors for cardiovascular disease.
The drug, which targets residual inflammation as an underlying cause of atherosclerotic cardiovascular disease, has a dosage of 0.5 mg once daily, and can be used alone or in combination with cholesterol-lowering medications. 
The drug’s manufacturer, Agepha Pharma, said it anticipates that Lodoco will be available for prescription in the second half of 2023.
Colchicine has been available for many years and used at higher doses for the acute treatment of gout and pericarditis, but the current formulation is a much lower dose for long-term use in patients with atherosclerotic heart disease.
Data supporting the approval has come from two major randomized trials, LoDoCo-2 and COLCOT.
In the LoDoCo-2 trial, the anti-inflammatory drug cut the risk of cardiovascular events by one third when added to standard prevention therapies in patients with chronic coronary disease. And in the COLCOT study, use of colchicine reduced cardiovascular events by 23% compared with placebo in patients with a recent MI.
Paul Ridker, MD, director of the Center for Cardiovascular Disease Prevention at Brigham and Women’s Hospital in Boston, who has been a pioneer in establishing inflammation as an underlying cause of atherosclerotic cardiovascular disease, welcomed the Lodoco approval.
‘A very big day for cardiology’
“This is a very big day for cardiology,” Dr. Ridker said in an interview.
“The FDA approval of colchicine for patients with atherosclerotic disease is a huge signal that physicians need to be aware of inflammation as a key player in cardiovascular disease,” he said.
Dr. Ridker was the lead author of a recent study showing that among patients receiving contemporary statins, inflammation assessed by high-sensitivity C-reactive protein (hsCRP) was a stronger predictor for risk of future cardiovascular events and death than LDL cholesterol.
He pointed out that
“That is virtually identical to the indication approved for statin therapy. That shows just how important the FDA thinks this is,” he commented.
But Dr. Ridker added that, while the label does not specify that Lodoco has to be used in addition to statin therapy, he believes that it will be used as additional therapy to statins in the vast majority of patients.
“This is not an alternative to statin therapy. In the randomized trials, the benefits were seen on top of statins,” he stressed.
Dr. Ridker believes that physicians will need time to feel comfortable with this new approach.
“Initially, I think, it will be used mainly by cardiologists who know about inflammation, but I believe over time it will be widely prescribed by internists, in much the same way as statins are used today,” he commented.
Dr. Ridker said he already uses low dose colchicine in his high-risk patients who have high levels of inflammation as seen on hsCRP testing. He believes this is where the drug will mostly be used initially, as this is where it is likely to be most effective.
The prescribing information states that Lodoco is contraindicated in patients who are taking strong CYP3A4 inhibitors or P-glycoprotein inhibitors, such as ketoconazole, fluconazole, and clarithromycin, and in patients with preexisting blood dyscrasias, renal failure, and severe hepatic impairment.
Common side effects reported in published clinical studies and literature with the use of colchicine are gastrointestinal symptoms (diarrhea, vomiting, abdominal cramping) and myalgia.
More serious adverse effects are listed as blood dyscrasias such as myelosuppression, leukopenia, granulocytopenia, thrombocytopenia, pancytopenia, and aplastic anemia; and neuromuscular toxicity in the form of myotoxicity including rhabdomyolysis, which may occur, especially in combination with other drugs known to cause this effect. If these adverse effects occur, it is recommended that the drug be stopped.
The prescribing information also notes that Lodoco may rarely and transiently impair fertility in males; and that patients with renal or hepatic impairment should be monitored closely for adverse effects of colchicine.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved the anti-inflammatory drug colchicine 0.5 mg tablets (Lodoco) as the first specific anti-inflammatory drug demonstrated to reduce the risk for myocardial infarction, stroke, coronary revascularization, and cardiovascular death in adult patients with established atherosclerotic disease or with multiple risk factors for cardiovascular disease.
The drug, which targets residual inflammation as an underlying cause of atherosclerotic cardiovascular disease, has a dosage of 0.5 mg once daily, and can be used alone or in combination with cholesterol-lowering medications.
The drug’s manufacturer, Agepha Pharma, said it anticipates that Lodoco will be available for prescription in the second half of 2023.
Colchicine has been available for many years and used at higher doses for the acute treatment of gout and pericarditis, but the current formulation is a much lower dose for long-term use in patients with atherosclerotic heart disease.
Data supporting the approval has come from two major randomized trials, LoDoCo-2 and COLCOT.
In the LoDoCo-2 trial, the anti-inflammatory drug cut the risk of cardiovascular events by one third when added to standard prevention therapies in patients with chronic coronary disease. And in the COLCOT study, use of colchicine reduced cardiovascular events by 23% compared with placebo in patients with a recent MI.
Paul Ridker, MD, director of the Center for Cardiovascular Disease Prevention at Brigham and Women’s Hospital in Boston, who has been a pioneer in establishing inflammation as an underlying cause of atherosclerotic cardiovascular disease, welcomed the Lodoco approval.
‘A very big day for cardiology’
“This is a very big day for cardiology,” Dr. Ridker said in an interview.
“The FDA approval of colchicine for patients with atherosclerotic disease is a huge signal that physicians need to be aware of inflammation as a key player in cardiovascular disease,” he said.
Dr. Ridker was the lead author of a recent study showing that among patients receiving contemporary statins, inflammation assessed by high-sensitivity C-reactive protein (hsCRP) was a stronger predictor for risk of future cardiovascular events and death than LDL cholesterol.
He pointed out that
“That is virtually identical to the indication approved for statin therapy. That shows just how important the FDA thinks this is,” he commented.
But Dr. Ridker added that, while the label does not specify that Lodoco has to be used in addition to statin therapy, he believes that it will be used as additional therapy to statins in the vast majority of patients.
“This is not an alternative to statin therapy. In the randomized trials, the benefits were seen on top of statins,” he stressed.
Dr. Ridker believes that physicians will need time to feel comfortable with this new approach.
“Initially, I think, it will be used mainly by cardiologists who know about inflammation, but I believe over time it will be widely prescribed by internists, in much the same way as statins are used today,” he commented.
Dr. Ridker said he already uses low dose colchicine in his high-risk patients who have high levels of inflammation as seen on hsCRP testing. He believes this is where the drug will mostly be used initially, as this is where it is likely to be most effective.
The prescribing information states that Lodoco is contraindicated in patients who are taking strong CYP3A4 inhibitors or P-glycoprotein inhibitors, such as ketoconazole, fluconazole, and clarithromycin, and in patients with preexisting blood dyscrasias, renal failure, and severe hepatic impairment.
Common side effects reported in published clinical studies and literature with the use of colchicine are gastrointestinal symptoms (diarrhea, vomiting, abdominal cramping) and myalgia.
More serious adverse effects are listed as blood dyscrasias such as myelosuppression, leukopenia, granulocytopenia, thrombocytopenia, pancytopenia, and aplastic anemia; and neuromuscular toxicity in the form of myotoxicity including rhabdomyolysis, which may occur, especially in combination with other drugs known to cause this effect. If these adverse effects occur, it is recommended that the drug be stopped.
The prescribing information also notes that Lodoco may rarely and transiently impair fertility in males; and that patients with renal or hepatic impairment should be monitored closely for adverse effects of colchicine.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved the anti-inflammatory drug colchicine 0.5 mg tablets (Lodoco) as the first specific anti-inflammatory drug demonstrated to reduce the risk for myocardial infarction, stroke, coronary revascularization, and cardiovascular death in adult patients with established atherosclerotic disease or with multiple risk factors for cardiovascular disease.
The drug, which targets residual inflammation as an underlying cause of atherosclerotic cardiovascular disease, has a dosage of 0.5 mg once daily, and can be used alone or in combination with cholesterol-lowering medications.
The drug’s manufacturer, Agepha Pharma, said it anticipates that Lodoco will be available for prescription in the second half of 2023.
Colchicine has been available for many years and used at higher doses for the acute treatment of gout and pericarditis, but the current formulation is a much lower dose for long-term use in patients with atherosclerotic heart disease.
Data supporting the approval has come from two major randomized trials, LoDoCo-2 and COLCOT.
In the LoDoCo-2 trial, the anti-inflammatory drug cut the risk of cardiovascular events by one third when added to standard prevention therapies in patients with chronic coronary disease. And in the COLCOT study, use of colchicine reduced cardiovascular events by 23% compared with placebo in patients with a recent MI.
Paul Ridker, MD, director of the Center for Cardiovascular Disease Prevention at Brigham and Women’s Hospital in Boston, who has been a pioneer in establishing inflammation as an underlying cause of atherosclerotic cardiovascular disease, welcomed the Lodoco approval.
‘A very big day for cardiology’
“This is a very big day for cardiology,” Dr. Ridker said in an interview.
“The FDA approval of colchicine for patients with atherosclerotic disease is a huge signal that physicians need to be aware of inflammation as a key player in cardiovascular disease,” he said.
Dr. Ridker was the lead author of a recent study showing that among patients receiving contemporary statins, inflammation assessed by high-sensitivity C-reactive protein (hsCRP) was a stronger predictor for risk of future cardiovascular events and death than LDL cholesterol.
He pointed out that
“That is virtually identical to the indication approved for statin therapy. That shows just how important the FDA thinks this is,” he commented.
But Dr. Ridker added that, while the label does not specify that Lodoco has to be used in addition to statin therapy, he believes that it will be used as additional therapy to statins in the vast majority of patients.
“This is not an alternative to statin therapy. In the randomized trials, the benefits were seen on top of statins,” he stressed.
Dr. Ridker believes that physicians will need time to feel comfortable with this new approach.
“Initially, I think, it will be used mainly by cardiologists who know about inflammation, but I believe over time it will be widely prescribed by internists, in much the same way as statins are used today,” he commented.
Dr. Ridker said he already uses low dose colchicine in his high-risk patients who have high levels of inflammation as seen on hsCRP testing. He believes this is where the drug will mostly be used initially, as this is where it is likely to be most effective.
The prescribing information states that Lodoco is contraindicated in patients who are taking strong CYP3A4 inhibitors or P-glycoprotein inhibitors, such as ketoconazole, fluconazole, and clarithromycin, and in patients with preexisting blood dyscrasias, renal failure, and severe hepatic impairment.
Common side effects reported in published clinical studies and literature with the use of colchicine are gastrointestinal symptoms (diarrhea, vomiting, abdominal cramping) and myalgia.
More serious adverse effects are listed as blood dyscrasias such as myelosuppression, leukopenia, granulocytopenia, thrombocytopenia, pancytopenia, and aplastic anemia; and neuromuscular toxicity in the form of myotoxicity including rhabdomyolysis, which may occur, especially in combination with other drugs known to cause this effect. If these adverse effects occur, it is recommended that the drug be stopped.
The prescribing information also notes that Lodoco may rarely and transiently impair fertility in males; and that patients with renal or hepatic impairment should be monitored closely for adverse effects of colchicine.
A version of this article first appeared on Medscape.com.