User login
Complaints of foot pain
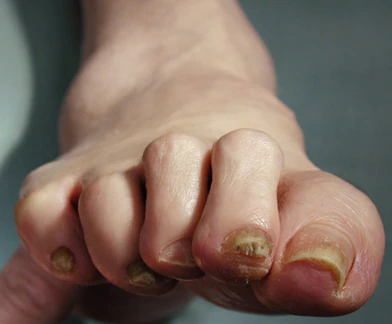
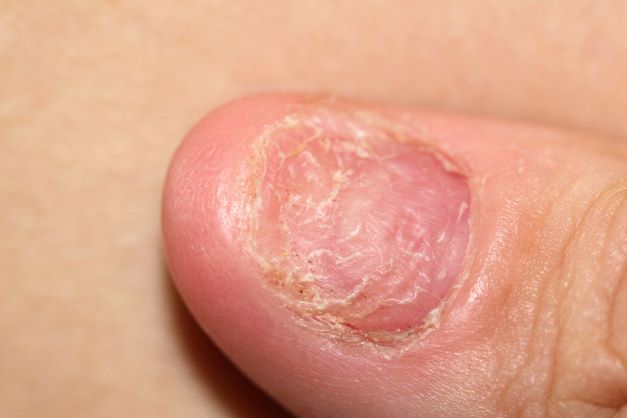
This patient's physical findings are consistent with a diagnosis of claw toe, which can be caused by diabetes-related peripheral neuropathy.
According to the International Diabetes Federation, diabetes currently affects approximately 537 million adults worldwide. The number of individuals living with diabetes is expected to exceed 640 million by 2030 and 780 million by 2045. In the United States, more than 37 million people are living with diabetes.
Foot complications related to diabetes represent a significant economic and social burden and can profoundly affect a patient's quality of life and medical outcomes. Common diabetes-related foot complications include foot deformity and peripheral neuropathy, both of which increase the risk for ulceration and amputation. The most common deformity is at the metatarsophalangeal joint (MTPJ). As many as 85% of patients with a history of ulcers and amputation have an MTPJ deformity such as claw toe or hammertoe.
Although they are often grouped together, claw toe and hammertoe have distinct features. Extended MTPJ, flexed proximal interphalangeal joint (PIPJ), and flexed distal interphalangeal joint (DIPJ) are characteristic of claw toe. While hammertoe also has extended MTPJ and flexed PIPJ, the DIPJ is extended rather than flexed. In both cases, the area of high pressure at risk for skin breakdown and ulceration is at the metatarsal head as a result of MTPJ hyperextension deformity.
Prompt detection and care of diabetes-related foot complications can minimize progression and negative consequences on patients' health and quality of life. According to the American Diabetes Association, all patients with diabetes should undergo a comprehensive foot evaluation at least annually to identify risk factors for ulceration and amputation, which include foot deformities, poor glycemic control, peripheral neuropathy, cigarette smoking, preulcerative callus or corn, peripheral artery disease, chronic kidney disease, visual impairment, and a history of ulceration or amputation. When patients present with a history of ulceration or amputation, a foot inspection should be conducted at each visit.
A comprehensive foot evaluation should include inspection of the skin, evaluation of any foot deformities, a neurologic assessment (10-g monofilament testing with at least one other assessment: pinprick, temperature, vibration), and a vascular assessment, including pulses in the legs and feet.
Patients should be educated on risk factors and appropriate management of foot-related complications, including the importance of effective glycemic control and daily monitoring of feet. Treatment may be medical, surgical, or both, as indicated by the individual patient's presentation. Conservative treatment approaches include footwear that is extra wide or deep, avoiding high-heeled and narrow-toed shoes, use of a metatarsal bar or pad, cushioning sleeves or stocking caps with silicon linings, and a longitudinal pad beneath the toes.
Complete recommendations on achieving glycemic control in T2D can be found in the 2022 American Diabetes Association Standards of Medical Care. Guidelines on foot care are also available.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's physical findings are consistent with a diagnosis of claw toe, which can be caused by diabetes-related peripheral neuropathy.
According to the International Diabetes Federation, diabetes currently affects approximately 537 million adults worldwide. The number of individuals living with diabetes is expected to exceed 640 million by 2030 and 780 million by 2045. In the United States, more than 37 million people are living with diabetes.
Foot complications related to diabetes represent a significant economic and social burden and can profoundly affect a patient's quality of life and medical outcomes. Common diabetes-related foot complications include foot deformity and peripheral neuropathy, both of which increase the risk for ulceration and amputation. The most common deformity is at the metatarsophalangeal joint (MTPJ). As many as 85% of patients with a history of ulcers and amputation have an MTPJ deformity such as claw toe or hammertoe.
Although they are often grouped together, claw toe and hammertoe have distinct features. Extended MTPJ, flexed proximal interphalangeal joint (PIPJ), and flexed distal interphalangeal joint (DIPJ) are characteristic of claw toe. While hammertoe also has extended MTPJ and flexed PIPJ, the DIPJ is extended rather than flexed. In both cases, the area of high pressure at risk for skin breakdown and ulceration is at the metatarsal head as a result of MTPJ hyperextension deformity.
Prompt detection and care of diabetes-related foot complications can minimize progression and negative consequences on patients' health and quality of life. According to the American Diabetes Association, all patients with diabetes should undergo a comprehensive foot evaluation at least annually to identify risk factors for ulceration and amputation, which include foot deformities, poor glycemic control, peripheral neuropathy, cigarette smoking, preulcerative callus or corn, peripheral artery disease, chronic kidney disease, visual impairment, and a history of ulceration or amputation. When patients present with a history of ulceration or amputation, a foot inspection should be conducted at each visit.
A comprehensive foot evaluation should include inspection of the skin, evaluation of any foot deformities, a neurologic assessment (10-g monofilament testing with at least one other assessment: pinprick, temperature, vibration), and a vascular assessment, including pulses in the legs and feet.
Patients should be educated on risk factors and appropriate management of foot-related complications, including the importance of effective glycemic control and daily monitoring of feet. Treatment may be medical, surgical, or both, as indicated by the individual patient's presentation. Conservative treatment approaches include footwear that is extra wide or deep, avoiding high-heeled and narrow-toed shoes, use of a metatarsal bar or pad, cushioning sleeves or stocking caps with silicon linings, and a longitudinal pad beneath the toes.
Complete recommendations on achieving glycemic control in T2D can be found in the 2022 American Diabetes Association Standards of Medical Care. Guidelines on foot care are also available.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's physical findings are consistent with a diagnosis of claw toe, which can be caused by diabetes-related peripheral neuropathy.
According to the International Diabetes Federation, diabetes currently affects approximately 537 million adults worldwide. The number of individuals living with diabetes is expected to exceed 640 million by 2030 and 780 million by 2045. In the United States, more than 37 million people are living with diabetes.
Foot complications related to diabetes represent a significant economic and social burden and can profoundly affect a patient's quality of life and medical outcomes. Common diabetes-related foot complications include foot deformity and peripheral neuropathy, both of which increase the risk for ulceration and amputation. The most common deformity is at the metatarsophalangeal joint (MTPJ). As many as 85% of patients with a history of ulcers and amputation have an MTPJ deformity such as claw toe or hammertoe.
Although they are often grouped together, claw toe and hammertoe have distinct features. Extended MTPJ, flexed proximal interphalangeal joint (PIPJ), and flexed distal interphalangeal joint (DIPJ) are characteristic of claw toe. While hammertoe also has extended MTPJ and flexed PIPJ, the DIPJ is extended rather than flexed. In both cases, the area of high pressure at risk for skin breakdown and ulceration is at the metatarsal head as a result of MTPJ hyperextension deformity.
Prompt detection and care of diabetes-related foot complications can minimize progression and negative consequences on patients' health and quality of life. According to the American Diabetes Association, all patients with diabetes should undergo a comprehensive foot evaluation at least annually to identify risk factors for ulceration and amputation, which include foot deformities, poor glycemic control, peripheral neuropathy, cigarette smoking, preulcerative callus or corn, peripheral artery disease, chronic kidney disease, visual impairment, and a history of ulceration or amputation. When patients present with a history of ulceration or amputation, a foot inspection should be conducted at each visit.
A comprehensive foot evaluation should include inspection of the skin, evaluation of any foot deformities, a neurologic assessment (10-g monofilament testing with at least one other assessment: pinprick, temperature, vibration), and a vascular assessment, including pulses in the legs and feet.
Patients should be educated on risk factors and appropriate management of foot-related complications, including the importance of effective glycemic control and daily monitoring of feet. Treatment may be medical, surgical, or both, as indicated by the individual patient's presentation. Conservative treatment approaches include footwear that is extra wide or deep, avoiding high-heeled and narrow-toed shoes, use of a metatarsal bar or pad, cushioning sleeves or stocking caps with silicon linings, and a longitudinal pad beneath the toes.
Complete recommendations on achieving glycemic control in T2D can be found in the 2022 American Diabetes Association Standards of Medical Care. Guidelines on foot care are also available.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
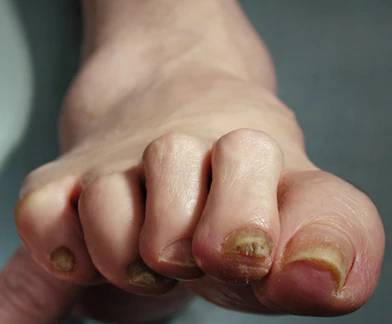
A 59-year-old woman newly diagnosed with type 2 diabetes (T2D) and hypercholesterolemia presents with complaints of foot pain, particularly while wearing shoes. Physical examination reveals an extended metatarsophalangeal joint, a flexed proximal interphalangeal joint, and flexed distal interphalangeal joint. Her toenails are discolored with a yellowish hue, and callus formation is noted over the metatarsal area. The patient reports pain at the tip of the toe from pressure against the point of the distal phalanx. She states that she has not experienced any numbness, tingling, or muscle weakness. Before her recent diabetes diagnosis, the patient had not been receiving regular medical care. The patient's current medications include metformin 500 mg/d, empagliflozin 10 mg/d, and rosuvastatin 10 mg/d.
Severe abdominal pain and vomiting
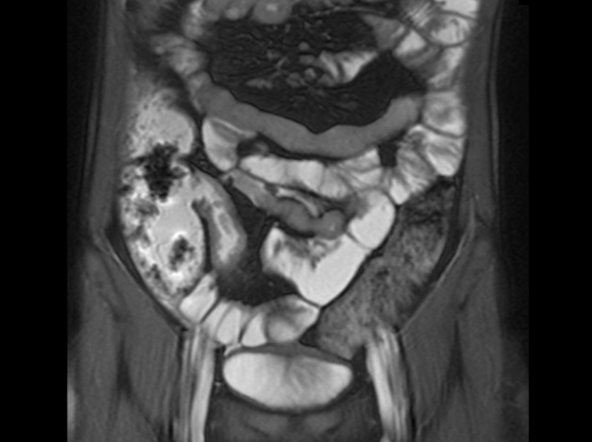
On the basis of the patient’s family history, personal history, and presentation, the likely diagnosis is Crohn disease. Although the disease may be diagnosed at any age, onset shows a bimodal distribution, with the first, more predominant wave occurring in adolescence and early adulthood. Peak global onset is between the ages of 15 and 30. Compared with adult-onset disease, pediatric Crohn disease is associated with a more serious disease course. Patients of Ashkenazi Jewish descent are at higher risk of developing this autoimmune disease than any other ethnic group.
Colonoscopy is the first-line approach for diagnosing and monitoring inflammatory bowel disease. Typical findings in patients with Crohn disease include histologic changes, such as focal crypt irregularity, transmural lymphoid aggregates, fissures and fistulas, and perianal disorders. In the differential diagnosis, ulcerative colitis (UC) must be carefully ruled out. UC involves only the large bowel, rarely causes fistulas, and is frequently seen with bleeding. Crohn disease is characteristically noncontiguous, with linear ulcerations of a cobblestone appearance. In addition, noncaseating granulomas are specific for Crohn disease. Micronutrient and vitamin levels are usually low, as seen in the present case. During workup, fecal calprotectin can help differentiate inflammatory bowel disease from irritable bowel syndrome.
The patient in this case may be a candidate for 5-aminosalicylic acid, together with a nutritional plan, used in mild or moderate cases of pediatric Crohn disease. Clinical improvement plus a decrease of fecal calprotectin would be an indication of positive treatment response. Being newly diagnosed, if the patient does not achieve remission after the induction period, he may be at risk for a more complicated disease course. Treatment for Crohn disease in the pediatric setting, as in the adult setting, should be implemented through a step-up approach. Other treatment options for pediatric disease include antibiotics; immunomodulators; and, in moderate-to severe cases, corticosteroids and biologics.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the patient’s family history, personal history, and presentation, the likely diagnosis is Crohn disease. Although the disease may be diagnosed at any age, onset shows a bimodal distribution, with the first, more predominant wave occurring in adolescence and early adulthood. Peak global onset is between the ages of 15 and 30. Compared with adult-onset disease, pediatric Crohn disease is associated with a more serious disease course. Patients of Ashkenazi Jewish descent are at higher risk of developing this autoimmune disease than any other ethnic group.
Colonoscopy is the first-line approach for diagnosing and monitoring inflammatory bowel disease. Typical findings in patients with Crohn disease include histologic changes, such as focal crypt irregularity, transmural lymphoid aggregates, fissures and fistulas, and perianal disorders. In the differential diagnosis, ulcerative colitis (UC) must be carefully ruled out. UC involves only the large bowel, rarely causes fistulas, and is frequently seen with bleeding. Crohn disease is characteristically noncontiguous, with linear ulcerations of a cobblestone appearance. In addition, noncaseating granulomas are specific for Crohn disease. Micronutrient and vitamin levels are usually low, as seen in the present case. During workup, fecal calprotectin can help differentiate inflammatory bowel disease from irritable bowel syndrome.
The patient in this case may be a candidate for 5-aminosalicylic acid, together with a nutritional plan, used in mild or moderate cases of pediatric Crohn disease. Clinical improvement plus a decrease of fecal calprotectin would be an indication of positive treatment response. Being newly diagnosed, if the patient does not achieve remission after the induction period, he may be at risk for a more complicated disease course. Treatment for Crohn disease in the pediatric setting, as in the adult setting, should be implemented through a step-up approach. Other treatment options for pediatric disease include antibiotics; immunomodulators; and, in moderate-to severe cases, corticosteroids and biologics.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the patient’s family history, personal history, and presentation, the likely diagnosis is Crohn disease. Although the disease may be diagnosed at any age, onset shows a bimodal distribution, with the first, more predominant wave occurring in adolescence and early adulthood. Peak global onset is between the ages of 15 and 30. Compared with adult-onset disease, pediatric Crohn disease is associated with a more serious disease course. Patients of Ashkenazi Jewish descent are at higher risk of developing this autoimmune disease than any other ethnic group.
Colonoscopy is the first-line approach for diagnosing and monitoring inflammatory bowel disease. Typical findings in patients with Crohn disease include histologic changes, such as focal crypt irregularity, transmural lymphoid aggregates, fissures and fistulas, and perianal disorders. In the differential diagnosis, ulcerative colitis (UC) must be carefully ruled out. UC involves only the large bowel, rarely causes fistulas, and is frequently seen with bleeding. Crohn disease is characteristically noncontiguous, with linear ulcerations of a cobblestone appearance. In addition, noncaseating granulomas are specific for Crohn disease. Micronutrient and vitamin levels are usually low, as seen in the present case. During workup, fecal calprotectin can help differentiate inflammatory bowel disease from irritable bowel syndrome.
The patient in this case may be a candidate for 5-aminosalicylic acid, together with a nutritional plan, used in mild or moderate cases of pediatric Crohn disease. Clinical improvement plus a decrease of fecal calprotectin would be an indication of positive treatment response. Being newly diagnosed, if the patient does not achieve remission after the induction period, he may be at risk for a more complicated disease course. Treatment for Crohn disease in the pediatric setting, as in the adult setting, should be implemented through a step-up approach. Other treatment options for pediatric disease include antibiotics; immunomodulators; and, in moderate-to severe cases, corticosteroids and biologics.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
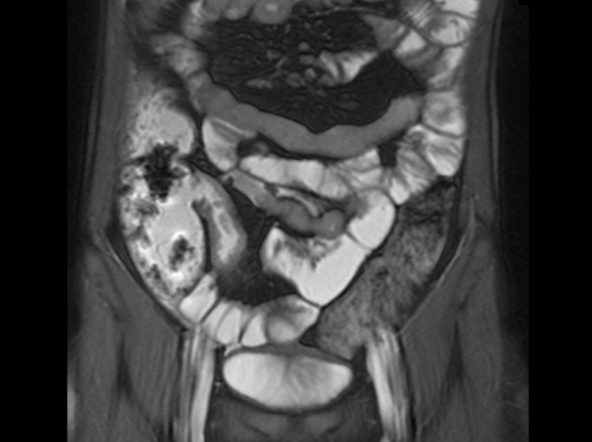
A 12-year-old boy presents to an urgent care center with severe abdominal pain, nausea, and vomiting. His height is 5 ft 3 in and weight is 99 lb (BMI 18.7). The patient has a history of chronic diarrhea and reports intermittent abdominal pain that began about 6 months ago. During this time, he has lost about 12 lb, as many foods exacerbate his symptoms, though his mother notes that even plain foods can bother his stomach. Further questioning reveals that his father has moderate Crohn disease (age of onset, 29 years), his sister has celiac disease, and the patient is of Ashkenazi Jewish descent. His body temperature is 100.2 °F. Vitamin B12, serum iron, total iron binding capacity, calcium, and magnesium are low. Stool cultures are negative. Ileocolonoscopy shows small aphthous erosions in the large intestine and in the terminal ileum.
Severe pulsing headache
On the basis of the patient's presentation and described history, the likely diagnosis is migraine. By adolescence, migraine is much more common among female patients and can be connected to the menstrual cycle. The early symptoms before onset of head pain reported by this patient characterize the prodromal phase, which can occur 1-2 days before the headache, followed by the aura phase. Approximately one third of patients with migraine experience episodes with aura, like the visual disturbance described in this case.
Migraine can be diagnosed on a clinical basis, but certain neurologic symptoms with headache should be considered red flags and prompt further workup (ie, stiff neck or fever, or history of head injury or major trauma). Spontaneous internal carotid artery dissection, for example, should be investigated in the differential of younger patients who have severe headache before onset of neurologic symptoms. Patients who present with migraine are very frequently misdiagnosed as having sinus headaches or sinusitis. Relevant clinical findings of acute sinusitis are sinus tenderness or pressure; pain over the cheek which radiates to the frontal region or teeth; redness of nose, cheeks, or eyelids; pain to the vertex, temple, or occiput; postnasal discharge; a blocked nose; coughing or pharyngeal irritation; facial pain; and hyposmia. Tension-type headaches usually are associated with mild or moderate bilateral pain, causing a steady ache as opposed to the throbbing of migraines. Basilar migraine, common among female patients, is marked by vertebrobasilar insufficiency.
The American Headache Society defines migraine by the occurrence of at least five episodes. These attacks must last 4-72 hours and have at least two of these four characteristics: unilateral location, pulsating quality, moderate or severe pain intensity, and aggravation by or causing avoidance of routine physical activity. During these episodes, the patient must experience either photophobia and phonophobia or nausea and/or vomiting. If these signs and symptoms cannot be explained by another diagnosis, the patient is very likely presenting with migraine.
Identifying an effective treatment for migraines is often associated with a trial-and-error period, with an average 4-year gap between diagnosis and initiation of preventive medications. Because the patient's migraines do not seem to respond to non-steroidal anti inflammatory drugs, she may be a candidate for other treatments of mild-to-moderate migraines: nonopioid analgesics, acetaminophen, or caffeinated analgesic combinations. If attacks are moderate or severe, or even mild to moderate but do not respond well to therapy, migraine-specific agents are recommended: triptans, dihydroergotamine (DHE), small-molecule calcitonin gene-related peptide (CGRP) receptor antagonists (gepants), and selective serotonin (5-HT1F) receptor agonists (ditans).
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the patient's presentation and described history, the likely diagnosis is migraine. By adolescence, migraine is much more common among female patients and can be connected to the menstrual cycle. The early symptoms before onset of head pain reported by this patient characterize the prodromal phase, which can occur 1-2 days before the headache, followed by the aura phase. Approximately one third of patients with migraine experience episodes with aura, like the visual disturbance described in this case.
Migraine can be diagnosed on a clinical basis, but certain neurologic symptoms with headache should be considered red flags and prompt further workup (ie, stiff neck or fever, or history of head injury or major trauma). Spontaneous internal carotid artery dissection, for example, should be investigated in the differential of younger patients who have severe headache before onset of neurologic symptoms. Patients who present with migraine are very frequently misdiagnosed as having sinus headaches or sinusitis. Relevant clinical findings of acute sinusitis are sinus tenderness or pressure; pain over the cheek which radiates to the frontal region or teeth; redness of nose, cheeks, or eyelids; pain to the vertex, temple, or occiput; postnasal discharge; a blocked nose; coughing or pharyngeal irritation; facial pain; and hyposmia. Tension-type headaches usually are associated with mild or moderate bilateral pain, causing a steady ache as opposed to the throbbing of migraines. Basilar migraine, common among female patients, is marked by vertebrobasilar insufficiency.
The American Headache Society defines migraine by the occurrence of at least five episodes. These attacks must last 4-72 hours and have at least two of these four characteristics: unilateral location, pulsating quality, moderate or severe pain intensity, and aggravation by or causing avoidance of routine physical activity. During these episodes, the patient must experience either photophobia and phonophobia or nausea and/or vomiting. If these signs and symptoms cannot be explained by another diagnosis, the patient is very likely presenting with migraine.
Identifying an effective treatment for migraines is often associated with a trial-and-error period, with an average 4-year gap between diagnosis and initiation of preventive medications. Because the patient's migraines do not seem to respond to non-steroidal anti inflammatory drugs, she may be a candidate for other treatments of mild-to-moderate migraines: nonopioid analgesics, acetaminophen, or caffeinated analgesic combinations. If attacks are moderate or severe, or even mild to moderate but do not respond well to therapy, migraine-specific agents are recommended: triptans, dihydroergotamine (DHE), small-molecule calcitonin gene-related peptide (CGRP) receptor antagonists (gepants), and selective serotonin (5-HT1F) receptor agonists (ditans).
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the patient's presentation and described history, the likely diagnosis is migraine. By adolescence, migraine is much more common among female patients and can be connected to the menstrual cycle. The early symptoms before onset of head pain reported by this patient characterize the prodromal phase, which can occur 1-2 days before the headache, followed by the aura phase. Approximately one third of patients with migraine experience episodes with aura, like the visual disturbance described in this case.
Migraine can be diagnosed on a clinical basis, but certain neurologic symptoms with headache should be considered red flags and prompt further workup (ie, stiff neck or fever, or history of head injury or major trauma). Spontaneous internal carotid artery dissection, for example, should be investigated in the differential of younger patients who have severe headache before onset of neurologic symptoms. Patients who present with migraine are very frequently misdiagnosed as having sinus headaches or sinusitis. Relevant clinical findings of acute sinusitis are sinus tenderness or pressure; pain over the cheek which radiates to the frontal region or teeth; redness of nose, cheeks, or eyelids; pain to the vertex, temple, or occiput; postnasal discharge; a blocked nose; coughing or pharyngeal irritation; facial pain; and hyposmia. Tension-type headaches usually are associated with mild or moderate bilateral pain, causing a steady ache as opposed to the throbbing of migraines. Basilar migraine, common among female patients, is marked by vertebrobasilar insufficiency.
The American Headache Society defines migraine by the occurrence of at least five episodes. These attacks must last 4-72 hours and have at least two of these four characteristics: unilateral location, pulsating quality, moderate or severe pain intensity, and aggravation by or causing avoidance of routine physical activity. During these episodes, the patient must experience either photophobia and phonophobia or nausea and/or vomiting. If these signs and symptoms cannot be explained by another diagnosis, the patient is very likely presenting with migraine.
Identifying an effective treatment for migraines is often associated with a trial-and-error period, with an average 4-year gap between diagnosis and initiation of preventive medications. Because the patient's migraines do not seem to respond to non-steroidal anti inflammatory drugs, she may be a candidate for other treatments of mild-to-moderate migraines: nonopioid analgesics, acetaminophen, or caffeinated analgesic combinations. If attacks are moderate or severe, or even mild to moderate but do not respond well to therapy, migraine-specific agents are recommended: triptans, dihydroergotamine (DHE), small-molecule calcitonin gene-related peptide (CGRP) receptor antagonists (gepants), and selective serotonin (5-HT1F) receptor agonists (ditans).
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.

An 18-year-old female patient presents with severe pulsing headache that began about 6 hours earlier. She describes feeling tired and irritable for the past 2 days and that she has had difficulty concentrating. Earlier in the day, before headache onset, she became extremely fatigued. Describing a "blinding light" in her vision, she is currently highly photophobic. The patient took four ibuprofen 2 hours ago. There is no significant medical history. She is on a regimen of estrogen-progestin and spironolactone for acne. Following advice from her primary care practitioner, she takes magnesium and vitamin B for headache prevention. The patient reports that she does not believe that she has migraines because she has never vomited during an episode. The patient explains that she has always had frequent headaches but that this is the sixth or seventh episode of this type and severity that she has had in the past year. The headaches do not seem to align with her menstrual cycle.

Paresthesias along forearm
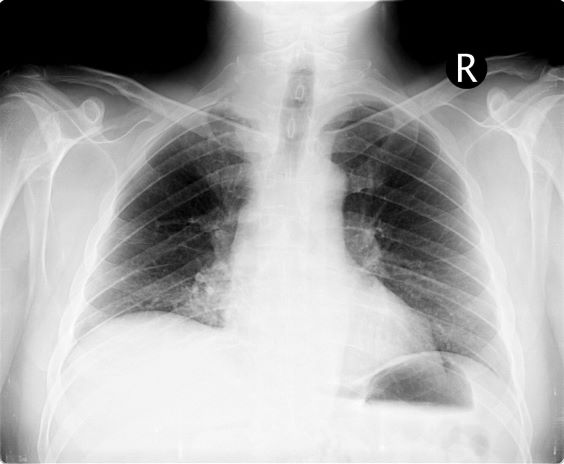
On the basis of this presentation, and the findings from the chest x-ray (as shown), the likely diagnosis is non–small cell lung cancer (NSCLC), Pancoast tumor, also known as superior sulcus tumor. Pancoast tumors are rare, representing about 3%-5% of all lung cancers, and invade the structures in the apex of the chest, including the first thoracic ribs or periosteum, the lower nerve roots of the bronchial plexus, the sympathetic chain and stellate ganglion, or the subclavian vessels. The majority of Pancoast tumors are non–small cell carcinomas.
Because of their pulmonary location, Pancoast tumors are characterized by several distinct symptoms. As seen in this case, patients often present with shoulder pain that worsens over time, especially with invasion of the chest wall and brachial plexus. The pain may radiate to the neck; axilla; anterior chest wall; and medial aspect of the arm, forearm, and wrist. If Pancoast tumors infiltrate the ulnar nerve, patients may present with weakness and muscle atrophy of the intrinsic muscles of the hand. In addition, invasion of the sympathetic chain and of the inferior cervical ganglion can cause Horner syndrome (ptosis, miosis, enophthalmos, and anhidrosis). Lastly, upper-arm edema may develop, signaling invasion and potentially occlusion of the subclavian vein.
During workup, CT-guided core biopsy is the first-line diagnostic test for Pancoast tumors. CT of the chest can confirm the presence of an apical mass and its position in relation to other structures of the thoracic inlet. MRI can further assess suspected brachial plexus, subclavian vessels, spine, and neural foramina invasion, specifying the extent of the disease and of the amount of nerve-root involvement.
For resectable Pancoast tumors, the National Comprehensive Cancer Network recommends chemoradiation, followed by surgical resection and chemotherapy. Preoperative chemoradiation together with surgical resection has shown a 2-year survival between 50% and 70%. Depending on biomarker status (certain EGFR mutations or programmed death ligand 1 levels ≥ 1%), the addition of either atezolizumab or osimertinib is advised. However, the positioning of Pancoast tumors can pose a surgical challenge, and if the lesion remains unresectable after preoperative concurrent chemoradiation, then consolidation immunotherapy with durvalumab is recommended.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of this presentation, and the findings from the chest x-ray (as shown), the likely diagnosis is non–small cell lung cancer (NSCLC), Pancoast tumor, also known as superior sulcus tumor. Pancoast tumors are rare, representing about 3%-5% of all lung cancers, and invade the structures in the apex of the chest, including the first thoracic ribs or periosteum, the lower nerve roots of the bronchial plexus, the sympathetic chain and stellate ganglion, or the subclavian vessels. The majority of Pancoast tumors are non–small cell carcinomas.
Because of their pulmonary location, Pancoast tumors are characterized by several distinct symptoms. As seen in this case, patients often present with shoulder pain that worsens over time, especially with invasion of the chest wall and brachial plexus. The pain may radiate to the neck; axilla; anterior chest wall; and medial aspect of the arm, forearm, and wrist. If Pancoast tumors infiltrate the ulnar nerve, patients may present with weakness and muscle atrophy of the intrinsic muscles of the hand. In addition, invasion of the sympathetic chain and of the inferior cervical ganglion can cause Horner syndrome (ptosis, miosis, enophthalmos, and anhidrosis). Lastly, upper-arm edema may develop, signaling invasion and potentially occlusion of the subclavian vein.
During workup, CT-guided core biopsy is the first-line diagnostic test for Pancoast tumors. CT of the chest can confirm the presence of an apical mass and its position in relation to other structures of the thoracic inlet. MRI can further assess suspected brachial plexus, subclavian vessels, spine, and neural foramina invasion, specifying the extent of the disease and of the amount of nerve-root involvement.
For resectable Pancoast tumors, the National Comprehensive Cancer Network recommends chemoradiation, followed by surgical resection and chemotherapy. Preoperative chemoradiation together with surgical resection has shown a 2-year survival between 50% and 70%. Depending on biomarker status (certain EGFR mutations or programmed death ligand 1 levels ≥ 1%), the addition of either atezolizumab or osimertinib is advised. However, the positioning of Pancoast tumors can pose a surgical challenge, and if the lesion remains unresectable after preoperative concurrent chemoradiation, then consolidation immunotherapy with durvalumab is recommended.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of this presentation, and the findings from the chest x-ray (as shown), the likely diagnosis is non–small cell lung cancer (NSCLC), Pancoast tumor, also known as superior sulcus tumor. Pancoast tumors are rare, representing about 3%-5% of all lung cancers, and invade the structures in the apex of the chest, including the first thoracic ribs or periosteum, the lower nerve roots of the bronchial plexus, the sympathetic chain and stellate ganglion, or the subclavian vessels. The majority of Pancoast tumors are non–small cell carcinomas.
Because of their pulmonary location, Pancoast tumors are characterized by several distinct symptoms. As seen in this case, patients often present with shoulder pain that worsens over time, especially with invasion of the chest wall and brachial plexus. The pain may radiate to the neck; axilla; anterior chest wall; and medial aspect of the arm, forearm, and wrist. If Pancoast tumors infiltrate the ulnar nerve, patients may present with weakness and muscle atrophy of the intrinsic muscles of the hand. In addition, invasion of the sympathetic chain and of the inferior cervical ganglion can cause Horner syndrome (ptosis, miosis, enophthalmos, and anhidrosis). Lastly, upper-arm edema may develop, signaling invasion and potentially occlusion of the subclavian vein.
During workup, CT-guided core biopsy is the first-line diagnostic test for Pancoast tumors. CT of the chest can confirm the presence of an apical mass and its position in relation to other structures of the thoracic inlet. MRI can further assess suspected brachial plexus, subclavian vessels, spine, and neural foramina invasion, specifying the extent of the disease and of the amount of nerve-root involvement.
For resectable Pancoast tumors, the National Comprehensive Cancer Network recommends chemoradiation, followed by surgical resection and chemotherapy. Preoperative chemoradiation together with surgical resection has shown a 2-year survival between 50% and 70%. Depending on biomarker status (certain EGFR mutations or programmed death ligand 1 levels ≥ 1%), the addition of either atezolizumab or osimertinib is advised. However, the positioning of Pancoast tumors can pose a surgical challenge, and if the lesion remains unresectable after preoperative concurrent chemoradiation, then consolidation immunotherapy with durvalumab is recommended.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
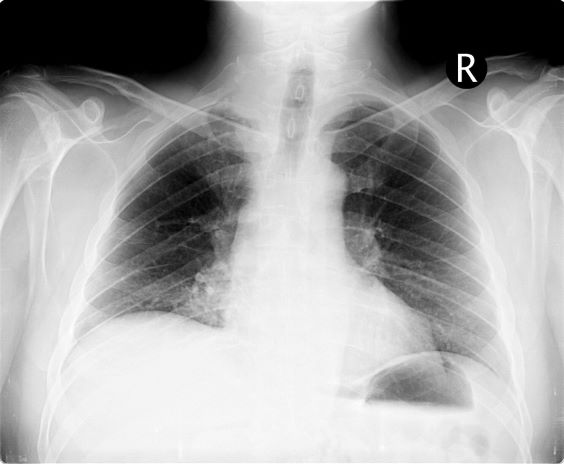
A 54-year-old man presents with shoulder pain and paresthesias along the medial side of the forearm. The patient has a 50–pack-year history of smoking. He reports that the pain began about 6 weeks ago, at which point he scheduled an orthopedic consultation. Physical examination is also notable for facial flushing. Breathing is normal, with no shortness of breath. Chest radiography reveals asymmetry of the apices (right apex is more opaque than the left). Invasion of the ribs is also seen.
Elbow tenderness and swollen joints
The diagnosis for this case is psoriatic arthritis (PsA). The 2018 American College of Rheumatology/National Psoriasis Foundation guidelines offer current treatment recommendations for this condition. For patients with active PsA who are treatment-naive, treatment recommendations are:
- Tumor necrosis factor (TNF) inhibitors are preferred over oral small molecules (OSMs), interleukin (IL)–17 inhibitors, or IL-12/23 inhibitors
- OSMs are recommended over IL-17 inhibitors or IL-12/23 inhibitors
- Methotrexate is recommended over nonsteroidal anti-inflammatory drugs
- IL-17 inhibitors are recommended over IL-12/23 inhibitors
Treatment recommendations for patients with active PsA despite the use of OSMs are:
- Switching to a TNF inhibitor over another OSM, IL-17 or IL-12/23 inhibitors, abatacept, or tofacitinib
- Switching to an IL-17 inhibitor over another OSM, IL-12/23 inhibitor, abatacept, or tofacitinib
- Switching to an IL-12/23 inhibitor over another OSM, abatacept, or tofacitinib.
International groups, such as the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) and the European League Against Rheumatism (EULAR), have published treatment recommendations as well. These address both PsA and, to a lesser extent, psoriasis. The GRAPPA recommendations consider six domains of involvement (peripheral arthritis, axial disease, enthesitis, dactylitis, skin psoriasis, and nail psoriasis) and use a grid approach to account for various levels of disease activity and severity. The EULAR recommendations use an algorithmic approach that focuses mainly on musculoskeletal manifestations, specifically peripheral arthritis; manifestations, such as dactylitis, enthesitis, and skin and nail involvement, are considered separately.
Psoriasis precedes the onset of PsA in 60%-80% of patients (sometimes by up to 20 years but usually by less than 10 years); but in as many as 15%-20% of patients, arthritis appears before psoriasis. On occasion, arthritis and psoriasis appear simultaneously.
Patients with PsA are typically seronegative for rheumatoid factor and antinuclear antibody; antinuclear antibody titers in persons with PsA do not differ from those of age- and sex-matched controls. C-reactive protein may be elevated but is often normal. Lack of C-reactive protein elevation, however, does not mean that systemic inflammation is absent but rather indicates that a different type of systemic inflammation may be at play for those patients.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The diagnosis for this case is psoriatic arthritis (PsA). The 2018 American College of Rheumatology/National Psoriasis Foundation guidelines offer current treatment recommendations for this condition. For patients with active PsA who are treatment-naive, treatment recommendations are:
- Tumor necrosis factor (TNF) inhibitors are preferred over oral small molecules (OSMs), interleukin (IL)–17 inhibitors, or IL-12/23 inhibitors
- OSMs are recommended over IL-17 inhibitors or IL-12/23 inhibitors
- Methotrexate is recommended over nonsteroidal anti-inflammatory drugs
- IL-17 inhibitors are recommended over IL-12/23 inhibitors
Treatment recommendations for patients with active PsA despite the use of OSMs are:
- Switching to a TNF inhibitor over another OSM, IL-17 or IL-12/23 inhibitors, abatacept, or tofacitinib
- Switching to an IL-17 inhibitor over another OSM, IL-12/23 inhibitor, abatacept, or tofacitinib
- Switching to an IL-12/23 inhibitor over another OSM, abatacept, or tofacitinib.
International groups, such as the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) and the European League Against Rheumatism (EULAR), have published treatment recommendations as well. These address both PsA and, to a lesser extent, psoriasis. The GRAPPA recommendations consider six domains of involvement (peripheral arthritis, axial disease, enthesitis, dactylitis, skin psoriasis, and nail psoriasis) and use a grid approach to account for various levels of disease activity and severity. The EULAR recommendations use an algorithmic approach that focuses mainly on musculoskeletal manifestations, specifically peripheral arthritis; manifestations, such as dactylitis, enthesitis, and skin and nail involvement, are considered separately.
Psoriasis precedes the onset of PsA in 60%-80% of patients (sometimes by up to 20 years but usually by less than 10 years); but in as many as 15%-20% of patients, arthritis appears before psoriasis. On occasion, arthritis and psoriasis appear simultaneously.
Patients with PsA are typically seronegative for rheumatoid factor and antinuclear antibody; antinuclear antibody titers in persons with PsA do not differ from those of age- and sex-matched controls. C-reactive protein may be elevated but is often normal. Lack of C-reactive protein elevation, however, does not mean that systemic inflammation is absent but rather indicates that a different type of systemic inflammation may be at play for those patients.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The diagnosis for this case is psoriatic arthritis (PsA). The 2018 American College of Rheumatology/National Psoriasis Foundation guidelines offer current treatment recommendations for this condition. For patients with active PsA who are treatment-naive, treatment recommendations are:
- Tumor necrosis factor (TNF) inhibitors are preferred over oral small molecules (OSMs), interleukin (IL)–17 inhibitors, or IL-12/23 inhibitors
- OSMs are recommended over IL-17 inhibitors or IL-12/23 inhibitors
- Methotrexate is recommended over nonsteroidal anti-inflammatory drugs
- IL-17 inhibitors are recommended over IL-12/23 inhibitors
Treatment recommendations for patients with active PsA despite the use of OSMs are:
- Switching to a TNF inhibitor over another OSM, IL-17 or IL-12/23 inhibitors, abatacept, or tofacitinib
- Switching to an IL-17 inhibitor over another OSM, IL-12/23 inhibitor, abatacept, or tofacitinib
- Switching to an IL-12/23 inhibitor over another OSM, abatacept, or tofacitinib.
International groups, such as the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) and the European League Against Rheumatism (EULAR), have published treatment recommendations as well. These address both PsA and, to a lesser extent, psoriasis. The GRAPPA recommendations consider six domains of involvement (peripheral arthritis, axial disease, enthesitis, dactylitis, skin psoriasis, and nail psoriasis) and use a grid approach to account for various levels of disease activity and severity. The EULAR recommendations use an algorithmic approach that focuses mainly on musculoskeletal manifestations, specifically peripheral arthritis; manifestations, such as dactylitis, enthesitis, and skin and nail involvement, are considered separately.
Psoriasis precedes the onset of PsA in 60%-80% of patients (sometimes by up to 20 years but usually by less than 10 years); but in as many as 15%-20% of patients, arthritis appears before psoriasis. On occasion, arthritis and psoriasis appear simultaneously.
Patients with PsA are typically seronegative for rheumatoid factor and antinuclear antibody; antinuclear antibody titers in persons with PsA do not differ from those of age- and sex-matched controls. C-reactive protein may be elevated but is often normal. Lack of C-reactive protein elevation, however, does not mean that systemic inflammation is absent but rather indicates that a different type of systemic inflammation may be at play for those patients.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
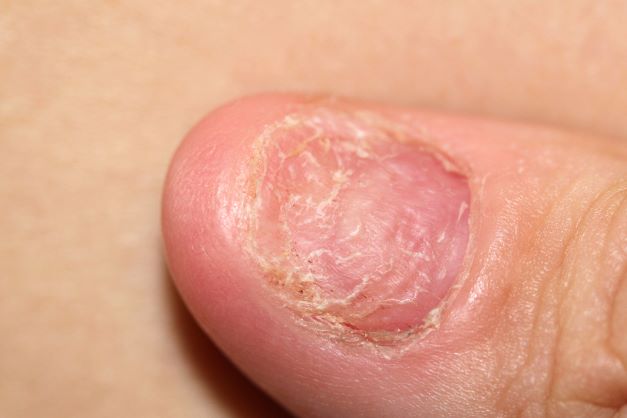
A 45-year-old man presents with complaints of intermittent joint aches to the point that he can no longer golf and has trouble with his handwriting. He has a 6-year history of scalp psoriasis that he has controlled with a salicylic acid shampoo. On physical examination, he has tenderness over both elbows and in his metacarpophalangeal and proximal interphalangeal joints on both hands. Swollen joints are noted in the proximal and distal joints of the right hand. His fingernails show uniform pitting. Neurologic examination shows no sensory deficits or hyperesthesia. Motor examination is unremarkable, as are chest and abdominal findings. Blood pressure is 128/80 mm Hg. Radiographic findings showed periarticular soft-tissue swelling of the distal interphalangeal joints of the right second and fourth fingers and left thumb, although no significant bony abnormalities were observed. There is asymmetric narrowing of the joint space in the interphalangeal joints. Laboratory findings reveal an erythrocyte sedimentation rate of 35 mm/h, negative for rheumatoid factor, negative for antinuclear antibody, and C-reactive protein level of 9 mg/dL.
Lower abdominal pain and dehydration
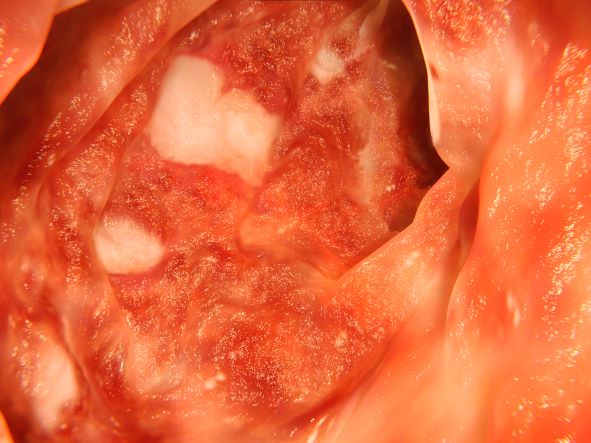
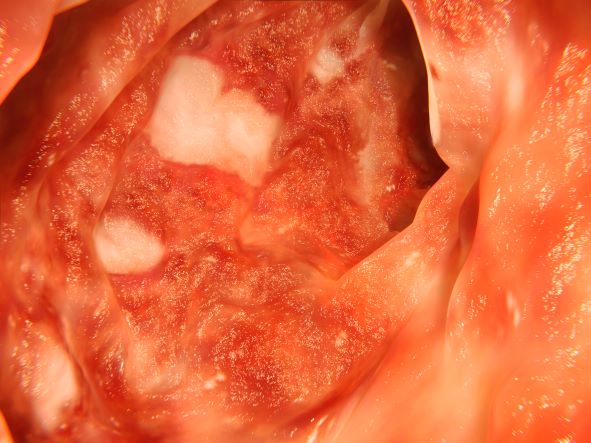
The diagnosis in this patient is ulcerative colitis (UC) on the basis of physical examination, laboratory values, and endoscopy. However, this patient has the most extensive form, pancolitis, which means that inflammation and damage extend the entire length of the colon.
The diagnosis of UC is best made with endoscopy and mucosal biopsy for histopathology. Characteristic findings are abnormal erythematous mucosa, with or without ulceration, extending from the rectum to a part or all of the colon and uniform inflammation, without intervening areas of normal mucosa (skip lesions tend to characterize Crohn disease). Contact bleeding may also be observed, with mucus identified in the lumen of the bowel.
The bowel wall is thin or of normal thickness, but edema, accumulation of fat, and hypertrophy of the muscle layer may give it the appearance of a thickened bowel wall. The disease is largely confined to the mucosa and, to a lesser extent, the submucosa.
Laboratory studies are helpful to exclude other diagnoses and assess the patient's nutritional status, and serologic markers aid in the differential diagnosis of inflammatory bowel disease. Radiographic imaging has an important role in differentiation of UC from Crohn disease. Fistulas or the presence of small bowel disease are seen only in Crohn disease.
According the American Gastroenterological Association, drug classes for the long-term management of moderate to severe UC include tumor necrosis factor–alpha antagonists, anti-integrin agent (vedolizumab), Janus kinase inhibitor (tofacitinib), interleukin 12/23 antagonist (ustekinumab), and immunomodulators (thiopurines, methotrexate). Most drugs that are initiated for the induction of remission are continued as maintenance therapy if they are effective. This is not the case, however, if corticosteroids or cyclosporine are necessary to induce remission.
This patient's pancolitis presentation is also acute and severe, defined as more than 6 bloody bowel movements per day plus one of the following: fever > 100.4 °F, hemoglobin level < 10.5 g/dL, heart rate > 90 beats/min, erythrocyte sedimentation rate > 30 mm/h, or C-reactive protein level > 30 mg/dL). This requires hospitalization and treatment with intravenous corticosteroids (hydrocortisone 400 mg/d or methylprednisolone 60 mg/d). Considered a medical emergency, the situation requires prompt recognition and multidisciplinary management. In patients who fail therapy with 3-5 days of intravenous corticosteroids, medical rescue therapy is indicated with either infliximab or cyclosporine. If all measures fail, the patient may need emergency surgery.
Hospitalized patients with acute severe UC have short-term colectomy rates of 25%-30%.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The diagnosis in this patient is ulcerative colitis (UC) on the basis of physical examination, laboratory values, and endoscopy. However, this patient has the most extensive form, pancolitis, which means that inflammation and damage extend the entire length of the colon.
The diagnosis of UC is best made with endoscopy and mucosal biopsy for histopathology. Characteristic findings are abnormal erythematous mucosa, with or without ulceration, extending from the rectum to a part or all of the colon and uniform inflammation, without intervening areas of normal mucosa (skip lesions tend to characterize Crohn disease). Contact bleeding may also be observed, with mucus identified in the lumen of the bowel.
The bowel wall is thin or of normal thickness, but edema, accumulation of fat, and hypertrophy of the muscle layer may give it the appearance of a thickened bowel wall. The disease is largely confined to the mucosa and, to a lesser extent, the submucosa.
Laboratory studies are helpful to exclude other diagnoses and assess the patient's nutritional status, and serologic markers aid in the differential diagnosis of inflammatory bowel disease. Radiographic imaging has an important role in differentiation of UC from Crohn disease. Fistulas or the presence of small bowel disease are seen only in Crohn disease.
According the American Gastroenterological Association, drug classes for the long-term management of moderate to severe UC include tumor necrosis factor–alpha antagonists, anti-integrin agent (vedolizumab), Janus kinase inhibitor (tofacitinib), interleukin 12/23 antagonist (ustekinumab), and immunomodulators (thiopurines, methotrexate). Most drugs that are initiated for the induction of remission are continued as maintenance therapy if they are effective. This is not the case, however, if corticosteroids or cyclosporine are necessary to induce remission.
This patient's pancolitis presentation is also acute and severe, defined as more than 6 bloody bowel movements per day plus one of the following: fever > 100.4 °F, hemoglobin level < 10.5 g/dL, heart rate > 90 beats/min, erythrocyte sedimentation rate > 30 mm/h, or C-reactive protein level > 30 mg/dL). This requires hospitalization and treatment with intravenous corticosteroids (hydrocortisone 400 mg/d or methylprednisolone 60 mg/d). Considered a medical emergency, the situation requires prompt recognition and multidisciplinary management. In patients who fail therapy with 3-5 days of intravenous corticosteroids, medical rescue therapy is indicated with either infliximab or cyclosporine. If all measures fail, the patient may need emergency surgery.
Hospitalized patients with acute severe UC have short-term colectomy rates of 25%-30%.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The diagnosis in this patient is ulcerative colitis (UC) on the basis of physical examination, laboratory values, and endoscopy. However, this patient has the most extensive form, pancolitis, which means that inflammation and damage extend the entire length of the colon.
The diagnosis of UC is best made with endoscopy and mucosal biopsy for histopathology. Characteristic findings are abnormal erythematous mucosa, with or without ulceration, extending from the rectum to a part or all of the colon and uniform inflammation, without intervening areas of normal mucosa (skip lesions tend to characterize Crohn disease). Contact bleeding may also be observed, with mucus identified in the lumen of the bowel.
The bowel wall is thin or of normal thickness, but edema, accumulation of fat, and hypertrophy of the muscle layer may give it the appearance of a thickened bowel wall. The disease is largely confined to the mucosa and, to a lesser extent, the submucosa.
Laboratory studies are helpful to exclude other diagnoses and assess the patient's nutritional status, and serologic markers aid in the differential diagnosis of inflammatory bowel disease. Radiographic imaging has an important role in differentiation of UC from Crohn disease. Fistulas or the presence of small bowel disease are seen only in Crohn disease.
According the American Gastroenterological Association, drug classes for the long-term management of moderate to severe UC include tumor necrosis factor–alpha antagonists, anti-integrin agent (vedolizumab), Janus kinase inhibitor (tofacitinib), interleukin 12/23 antagonist (ustekinumab), and immunomodulators (thiopurines, methotrexate). Most drugs that are initiated for the induction of remission are continued as maintenance therapy if they are effective. This is not the case, however, if corticosteroids or cyclosporine are necessary to induce remission.
This patient's pancolitis presentation is also acute and severe, defined as more than 6 bloody bowel movements per day plus one of the following: fever > 100.4 °F, hemoglobin level < 10.5 g/dL, heart rate > 90 beats/min, erythrocyte sedimentation rate > 30 mm/h, or C-reactive protein level > 30 mg/dL). This requires hospitalization and treatment with intravenous corticosteroids (hydrocortisone 400 mg/d or methylprednisolone 60 mg/d). Considered a medical emergency, the situation requires prompt recognition and multidisciplinary management. In patients who fail therapy with 3-5 days of intravenous corticosteroids, medical rescue therapy is indicated with either infliximab or cyclosporine. If all measures fail, the patient may need emergency surgery.
Hospitalized patients with acute severe UC have short-term colectomy rates of 25%-30%.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
A 76-year-old man presents with complaints of severe lower abdominal pain and dehydration. He reports bloody diarrhea of 2 weeks' duration and an unintentional 12-lb weight loss. Dietary alterations and loperamide have not helped. He has a fever of 102.1 °F. Medications include naproxen 440 mg/d for osteoarthritis, losartan 50 mg/d and amlodipine 5 mg/d for hypertension, and simvastatin 20 mg/d for dyslipidemia.
Physical examination reveals tenderness, particularly at the left lower quadrant of the abdomen, without rebound tenderness or guarding. Bowel sounds are active. He has a purulent rectal discharge. Stool cultures for the pathogens are negative. He has hypoalbuminemia (2.5 g/dL). He is positive for perinuclear antineutrophil cytoplasmic antibodies. Serum carcinoembryonic antigen test is negative. C-reactive protein is 32 mg/dL.
The patient is admitted to the hospital and receives intravenous fluids. Colonoscopy reveals inflammation and visible ulcers in the mucosa throughout the entire length of the colon.
Episodes of visual disturbance
On the basis of the history, examination, and investigations, retinal migraine was diagnosed according to the International Classification of Headache Disorders, third edition (1.2 migraine with aura; 1.2.4 retinal migraine). This classification system describes retinal migraine as a subtype of migraine with aura.
Retinal migraine (also called ophthalmic or ocular migraine) is relatively rare but is sometimes a cause of transient monocular blindness in young adults. It manifests as recurrent attacks of unilateral visual disturbance (positive symptoms) or blindness (negative symptoms) lasting from minutes to 1 hour, associated with minimal or no headache.
Some patients describe a positive visual symptom/disturbance in a mosaic pattern of scotomata that gradually enlarge, producing total or near-total unilateral visual loss. Precipitating factors may include emotional stress, hypertension, and hormonal contraceptive pills, as well as exercise, high altitude, dehydration, smoking, hypoglycemia, and hyperthermia.
Retinal migraine is believed to result from transient vasospasm of the choroidal or retinal arteries. A history of recurrent attacks of transient monocular visual disturbance or blindness, with or without a headache and without other neurologic symptoms, can suggest retinal migraine. A personal or family history of migraine can confirm the diagnosis.
Ruling out eye disease or vascular causes, especially when risk factors for arteriosclerosis exist, is important; that is, the condition must be differentiated from ocular or vascular causes of transient monocular blindness, mainly carotid artery disease.
Carotid duplex ultrasonography, transcranial Doppler ultrasonography, magnetic resonance angiography, or CT angiography of the brain may be helpful. Fluorescein or cerebral angiography is rarely necessary. A hypercoagulability workup and evaluation of the erythrocyte sedimentation rate may be useful in excluding other coagulation disorders associated with retinal vasculopathy.
Regarding management, calcium-channel blockers have shown some efficacy. Even in patients with low blood pressure, nifedipine 10-20 mg/d is generally tolerated. From the available literature on treatment of this condition, it is recommended that triptans, ergots, and beta-blockers be used with caution or avoided in patients with retinal migraine owing to the potential for exacerbating vasoconstriction of the retinal artery. Transient vision loss in retinal migraine has been associated with future onset of permanent vision loss from occlusive conditions such as central retinal artery occlusion and branch retinal artery occlusion.
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the history, examination, and investigations, retinal migraine was diagnosed according to the International Classification of Headache Disorders, third edition (1.2 migraine with aura; 1.2.4 retinal migraine). This classification system describes retinal migraine as a subtype of migraine with aura.
Retinal migraine (also called ophthalmic or ocular migraine) is relatively rare but is sometimes a cause of transient monocular blindness in young adults. It manifests as recurrent attacks of unilateral visual disturbance (positive symptoms) or blindness (negative symptoms) lasting from minutes to 1 hour, associated with minimal or no headache.
Some patients describe a positive visual symptom/disturbance in a mosaic pattern of scotomata that gradually enlarge, producing total or near-total unilateral visual loss. Precipitating factors may include emotional stress, hypertension, and hormonal contraceptive pills, as well as exercise, high altitude, dehydration, smoking, hypoglycemia, and hyperthermia.
Retinal migraine is believed to result from transient vasospasm of the choroidal or retinal arteries. A history of recurrent attacks of transient monocular visual disturbance or blindness, with or without a headache and without other neurologic symptoms, can suggest retinal migraine. A personal or family history of migraine can confirm the diagnosis.
Ruling out eye disease or vascular causes, especially when risk factors for arteriosclerosis exist, is important; that is, the condition must be differentiated from ocular or vascular causes of transient monocular blindness, mainly carotid artery disease.
Carotid duplex ultrasonography, transcranial Doppler ultrasonography, magnetic resonance angiography, or CT angiography of the brain may be helpful. Fluorescein or cerebral angiography is rarely necessary. A hypercoagulability workup and evaluation of the erythrocyte sedimentation rate may be useful in excluding other coagulation disorders associated with retinal vasculopathy.
Regarding management, calcium-channel blockers have shown some efficacy. Even in patients with low blood pressure, nifedipine 10-20 mg/d is generally tolerated. From the available literature on treatment of this condition, it is recommended that triptans, ergots, and beta-blockers be used with caution or avoided in patients with retinal migraine owing to the potential for exacerbating vasoconstriction of the retinal artery. Transient vision loss in retinal migraine has been associated with future onset of permanent vision loss from occlusive conditions such as central retinal artery occlusion and branch retinal artery occlusion.
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the history, examination, and investigations, retinal migraine was diagnosed according to the International Classification of Headache Disorders, third edition (1.2 migraine with aura; 1.2.4 retinal migraine). This classification system describes retinal migraine as a subtype of migraine with aura.
Retinal migraine (also called ophthalmic or ocular migraine) is relatively rare but is sometimes a cause of transient monocular blindness in young adults. It manifests as recurrent attacks of unilateral visual disturbance (positive symptoms) or blindness (negative symptoms) lasting from minutes to 1 hour, associated with minimal or no headache.
Some patients describe a positive visual symptom/disturbance in a mosaic pattern of scotomata that gradually enlarge, producing total or near-total unilateral visual loss. Precipitating factors may include emotional stress, hypertension, and hormonal contraceptive pills, as well as exercise, high altitude, dehydration, smoking, hypoglycemia, and hyperthermia.
Retinal migraine is believed to result from transient vasospasm of the choroidal or retinal arteries. A history of recurrent attacks of transient monocular visual disturbance or blindness, with or without a headache and without other neurologic symptoms, can suggest retinal migraine. A personal or family history of migraine can confirm the diagnosis.
Ruling out eye disease or vascular causes, especially when risk factors for arteriosclerosis exist, is important; that is, the condition must be differentiated from ocular or vascular causes of transient monocular blindness, mainly carotid artery disease.
Carotid duplex ultrasonography, transcranial Doppler ultrasonography, magnetic resonance angiography, or CT angiography of the brain may be helpful. Fluorescein or cerebral angiography is rarely necessary. A hypercoagulability workup and evaluation of the erythrocyte sedimentation rate may be useful in excluding other coagulation disorders associated with retinal vasculopathy.
Regarding management, calcium-channel blockers have shown some efficacy. Even in patients with low blood pressure, nifedipine 10-20 mg/d is generally tolerated. From the available literature on treatment of this condition, it is recommended that triptans, ergots, and beta-blockers be used with caution or avoided in patients with retinal migraine owing to the potential for exacerbating vasoconstriction of the retinal artery. Transient vision loss in retinal migraine has been associated with future onset of permanent vision loss from occlusive conditions such as central retinal artery occlusion and branch retinal artery occlusion.
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.

A 23-year-old woman presents with sudden recurrent episodes of visual disturbance (extreme blurriness and partial blindness) in her right eye. She had seven or eight episodes over 30 hours; each episode lasted for 5-7 minutes, with spontaneous and full recovery. These were not associated with flashes of light, tingling, numbness, fever, or headache. She was asymptomatic between episodes.
She had normal vision in her left eye during these episodes, which she checked by covering both eyes alternately with her hands. The only significant history was four episodes of migraine with aura 3 years ago, which resolved spontaneously and did not recur. Family history was noncontributory. She had no history of illicit drug use or alcohol use.
On examination, her vital signs were normal. Blood pressure was 110/80 mm Hg, pulse 85 beats/min, and respiratory rate 16 breaths/min. There was no lymphadenopathy, and jugular venous pressure was not elevated. Visual acuity was 6/6, with normal visual fields and perimetry. Fundoscopy was normal. Complete blood count, liver function tests, renal function tests, erythrocyte sedimentation rate, antineutrophil antibodies, electrocardiography, transthoracic echocardiography, carotid Doppler, and MRI of the brain with contrast were all normal. She is taking no medications.

Flickering sensation in eyes
The American Diabetes Association (ADA) position statement on diabetic retinopathy states that hyperglycemia has been the most consistently associated risk factor for retinopathy. A large and consistent set of observational studies and clinical trials confirms the association of poor glucose control and retinopathy.
The Diabetes Control and Complications Trial (DCCT), a randomized controlled clinical trial of intensive glycemic control vs conventional glycemic control in people with type 1 diabetes (T1D), demonstrated that intensive therapy reduced the development or progression of diabetic retinopathy by 34%-76%. The DCCT also demonstrated a definitive relationship between hyperglycemia and diabetic microvascular complications, including retinopathy. Early treatment with intensive therapy was effective.
The UK Prospective Diabetes Study (UKPDS) of patients with newly diagnosed T2D conclusively demonstrated that improved blood glucose control reduced the risk for retinopathy and nephropathy and, possibly, neuropathy. The overall microvascular complication rate was decreased by 25% in patients receiving intensive therapy vs conventional therapy. Epidemiologic analysis of the UKPDS data showed a continuous relationship between the risk for microvascular complications and glycemia, such that every percentage-point decrease in A1c (eg, 9% to 8%) was associated with a 35% reduction in the risk for microvascular complications.
More recently, the ACCORD trial of medical therapies demonstrated that intensive glycemic control reduced the risk for progression of diabetic retinopathy in people with T2D of 10 years' duration. This study included 2856 ACCORD participants enrolled in the ACCORD Eye Study and followed for 4 years.
The ADA recommends screening by an ophthalmologist for diabetic retinopathy within 5 years of the diagnosis of T1D and at the time of diagnosis of T2D. Women with preexisting diabetes who are planning pregnancy or who have become pregnant should be screened before pregnancy or in the first trimester.
While optimization of blood glucose, blood pressure, and serum lipid levels in conjunction with appropriately scheduled dilated eye examinations can substantially decrease the risk for vision loss from diabetic retinopathy, a significant proportion of those affected with diabetes develop diabetic macular edema or proliferative changes that require intervention. ADA treatment recommendations are:
• Refer patients with any level of macular edema, severe nonproliferative diabetic retinopathy (a precursor of proliferative diabetic retinopathy), or proliferative diabetic retinopathy to an ophthalmologist knowledgeable and experienced in the management and treatment of diabetic retinopathy.
• Laser photocoagulation therapy reduces the risk for vision loss in patients with high-risk proliferative diabetic retinopathy and, in some cases, severe nonproliferative diabetic retinopathy.
• Intravitreous injections of anti–vascular endothelial growth factor are indicated for central-involved diabetic macular edema, which occurs beneath the foveal center and may threaten reading vision.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The American Diabetes Association (ADA) position statement on diabetic retinopathy states that hyperglycemia has been the most consistently associated risk factor for retinopathy. A large and consistent set of observational studies and clinical trials confirms the association of poor glucose control and retinopathy.
The Diabetes Control and Complications Trial (DCCT), a randomized controlled clinical trial of intensive glycemic control vs conventional glycemic control in people with type 1 diabetes (T1D), demonstrated that intensive therapy reduced the development or progression of diabetic retinopathy by 34%-76%. The DCCT also demonstrated a definitive relationship between hyperglycemia and diabetic microvascular complications, including retinopathy. Early treatment with intensive therapy was effective.
The UK Prospective Diabetes Study (UKPDS) of patients with newly diagnosed T2D conclusively demonstrated that improved blood glucose control reduced the risk for retinopathy and nephropathy and, possibly, neuropathy. The overall microvascular complication rate was decreased by 25% in patients receiving intensive therapy vs conventional therapy. Epidemiologic analysis of the UKPDS data showed a continuous relationship between the risk for microvascular complications and glycemia, such that every percentage-point decrease in A1c (eg, 9% to 8%) was associated with a 35% reduction in the risk for microvascular complications.
More recently, the ACCORD trial of medical therapies demonstrated that intensive glycemic control reduced the risk for progression of diabetic retinopathy in people with T2D of 10 years' duration. This study included 2856 ACCORD participants enrolled in the ACCORD Eye Study and followed for 4 years.
The ADA recommends screening by an ophthalmologist for diabetic retinopathy within 5 years of the diagnosis of T1D and at the time of diagnosis of T2D. Women with preexisting diabetes who are planning pregnancy or who have become pregnant should be screened before pregnancy or in the first trimester.
While optimization of blood glucose, blood pressure, and serum lipid levels in conjunction with appropriately scheduled dilated eye examinations can substantially decrease the risk for vision loss from diabetic retinopathy, a significant proportion of those affected with diabetes develop diabetic macular edema or proliferative changes that require intervention. ADA treatment recommendations are:
• Refer patients with any level of macular edema, severe nonproliferative diabetic retinopathy (a precursor of proliferative diabetic retinopathy), or proliferative diabetic retinopathy to an ophthalmologist knowledgeable and experienced in the management and treatment of diabetic retinopathy.
• Laser photocoagulation therapy reduces the risk for vision loss in patients with high-risk proliferative diabetic retinopathy and, in some cases, severe nonproliferative diabetic retinopathy.
• Intravitreous injections of anti–vascular endothelial growth factor are indicated for central-involved diabetic macular edema, which occurs beneath the foveal center and may threaten reading vision.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The American Diabetes Association (ADA) position statement on diabetic retinopathy states that hyperglycemia has been the most consistently associated risk factor for retinopathy. A large and consistent set of observational studies and clinical trials confirms the association of poor glucose control and retinopathy.
The Diabetes Control and Complications Trial (DCCT), a randomized controlled clinical trial of intensive glycemic control vs conventional glycemic control in people with type 1 diabetes (T1D), demonstrated that intensive therapy reduced the development or progression of diabetic retinopathy by 34%-76%. The DCCT also demonstrated a definitive relationship between hyperglycemia and diabetic microvascular complications, including retinopathy. Early treatment with intensive therapy was effective.
The UK Prospective Diabetes Study (UKPDS) of patients with newly diagnosed T2D conclusively demonstrated that improved blood glucose control reduced the risk for retinopathy and nephropathy and, possibly, neuropathy. The overall microvascular complication rate was decreased by 25% in patients receiving intensive therapy vs conventional therapy. Epidemiologic analysis of the UKPDS data showed a continuous relationship between the risk for microvascular complications and glycemia, such that every percentage-point decrease in A1c (eg, 9% to 8%) was associated with a 35% reduction in the risk for microvascular complications.
More recently, the ACCORD trial of medical therapies demonstrated that intensive glycemic control reduced the risk for progression of diabetic retinopathy in people with T2D of 10 years' duration. This study included 2856 ACCORD participants enrolled in the ACCORD Eye Study and followed for 4 years.
The ADA recommends screening by an ophthalmologist for diabetic retinopathy within 5 years of the diagnosis of T1D and at the time of diagnosis of T2D. Women with preexisting diabetes who are planning pregnancy or who have become pregnant should be screened before pregnancy or in the first trimester.
While optimization of blood glucose, blood pressure, and serum lipid levels in conjunction with appropriately scheduled dilated eye examinations can substantially decrease the risk for vision loss from diabetic retinopathy, a significant proportion of those affected with diabetes develop diabetic macular edema or proliferative changes that require intervention. ADA treatment recommendations are:
• Refer patients with any level of macular edema, severe nonproliferative diabetic retinopathy (a precursor of proliferative diabetic retinopathy), or proliferative diabetic retinopathy to an ophthalmologist knowledgeable and experienced in the management and treatment of diabetic retinopathy.
• Laser photocoagulation therapy reduces the risk for vision loss in patients with high-risk proliferative diabetic retinopathy and, in some cases, severe nonproliferative diabetic retinopathy.
• Intravitreous injections of anti–vascular endothelial growth factor are indicated for central-involved diabetic macular edema, which occurs beneath the foveal center and may threaten reading vision.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
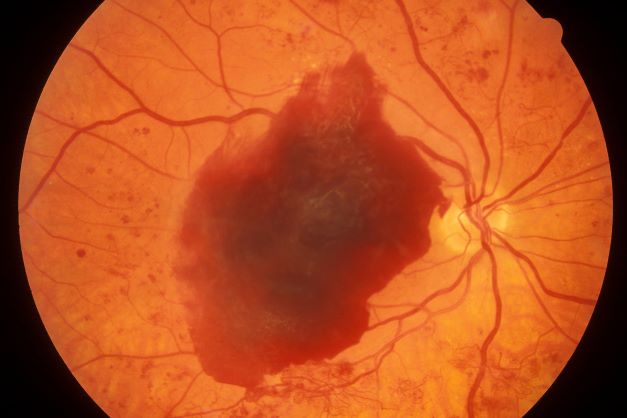
A 48-year-old Black man with type 2 diabetes (T2D) presented with complaints of a "flickering" sensation and a decrease in brightness of colors in both eyes as well as floaters in his left eye for several weeks. He reported that his symptoms fluctuate with changes in his blood glucose levels. His last eye examination was 2 years ago and his ocular history was unremarkable. His medical history was significant with a history of hypertension and T2D requiring insulin. His most recent glycated hemoglobin (A1c), 2 months ago, was 8.4%. His BMI was 31.2. The patient's medications were dulaglutide 0.75 mg injection pen, glargine insulin 42 units, losartan 100 mg, and amlodipine 10 mg.
On examination, his best-corrected visual acuity was 20/20 in the right eye and 20/30 in the left eye. Confrontation fields were intact, extraocular movements were full and extensive, and both pupils were equal, round, and reactive to light without afferent pupillary defects. Anterior segment examination was unremarkable in both eyes, without iris neovascularization. Intraocular pressures were 17 mm Hg in the right eye and 16 mm Hg in the left eye. On dilated fundus examination, the cup-to-disc ratio was 0.45 horizontally and vertically, with the presence of 1/4 disc diameters of neovascularization of the disc in the right eye and 2/3 disc diameters of neovascularization of the disc in the left eye.
Posterior segment findings were significant for scattered microaneurysms and dot/blot hemorrhages in the maculae. In the periphery of both eyes, there were tortuous vessels, scatter microaneurysms with dot/blot hemorrhages, and multiple areas of neovascularization elsewhere, with several foci of vitreous traction. There was no vitreous hemorrhage or tractional retinal detachment of either eye.
Spectral domain optical coherence tomography revealed an epiretinal membrane in the right eye and a blunted foveal contour with parafoveal cystic spaces, probably secondary to vitreomacular contraction. The left eye also had an epiretinal membrane and blunted foveal contour secondary to vitreomacular adhesion. The patient was diagnosed with bilateral high-risk proliferative diabetic retinopathy.
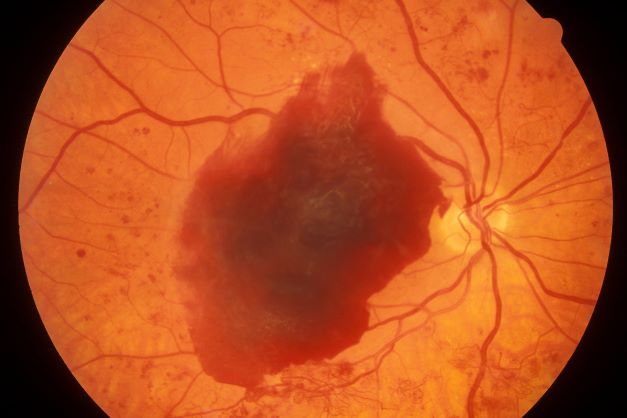
Pain and photophobia
On the basis of the patient's medical history and presentation, this is probably a case of uveitis, a common extra-articular manifestation of psoriatic disease. In fact, the presence of uveitis can help distinguish PsA from osteoarthritis. Uveitis is characterized by inflammation of the uvea tract, with the retina, optic nerve, vitreous body, and sclera potentially becoming inflamed as well. Among patients with PsA, the prevalence of uveitis rises with ongoing disease duration, though the condition may also precede the development of PsA in patients with psoriasis, and is common among patients with severe psoriatic disease in Western and Asian populations. Overall, the prevalence of uveitis has been estimated to be 6%-9%. HLA-B27 genotype is strongly associated with uveitis in patients with concomitant PsA.
Symptoms of uveitis, as seen in the present case, include blurred vision, photophobia, pain, and ciliary flush. The condition is classified as anterior, intermediate, posterior, or panuveitis, with the majority of cases diagnosed as anterior. In anterior uveitis, the inflamed pupil may become constricted or take on an irregular shape caused by iris adhesions to the anterior lens capsule. Uveitis in PsA is bilateral and has a chronic relapsing course. Onset is typically insidious.
Workup for uveitis should comprise visual acuity testing, slit lamp biomicroscopy, measurement of intraocular pressures, and a dilated eye exam. Conditions in the differential which threaten a patient's sight include retinal vasculitis, vitritis, cystoid macular edema, Behçet disease, and tubulo-interstitial nephritis. Other autoimmune diseases which can cause uveitis with systemic manifestations (multiple sclerosis, sarcoidosis, lupus) should be investigated. Infectious causes must also be eliminated. However, considering this patient's history of psoriatic disease, uveitis should be highly suspected.
Uveitis demands urgent treatment to control ocular inflammation. Tumor necrosis factor (TNF) inhibitors are the recommended first-line and second-line treatment for PsA, including in patients with complications such as uveitis. However, etanercept should not be used as it is less effective than adalimumab or other TNF inhibitors for uveitis. Because uveitis may sometimes respond to MTX therapy, patients with severe PsA may use a biologic agent in combination with MTX if they have had a partial response to current MTX therapy, as recommended by the American College of Rheumatology.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the patient's medical history and presentation, this is probably a case of uveitis, a common extra-articular manifestation of psoriatic disease. In fact, the presence of uveitis can help distinguish PsA from osteoarthritis. Uveitis is characterized by inflammation of the uvea tract, with the retina, optic nerve, vitreous body, and sclera potentially becoming inflamed as well. Among patients with PsA, the prevalence of uveitis rises with ongoing disease duration, though the condition may also precede the development of PsA in patients with psoriasis, and is common among patients with severe psoriatic disease in Western and Asian populations. Overall, the prevalence of uveitis has been estimated to be 6%-9%. HLA-B27 genotype is strongly associated with uveitis in patients with concomitant PsA.
Symptoms of uveitis, as seen in the present case, include blurred vision, photophobia, pain, and ciliary flush. The condition is classified as anterior, intermediate, posterior, or panuveitis, with the majority of cases diagnosed as anterior. In anterior uveitis, the inflamed pupil may become constricted or take on an irregular shape caused by iris adhesions to the anterior lens capsule. Uveitis in PsA is bilateral and has a chronic relapsing course. Onset is typically insidious.
Workup for uveitis should comprise visual acuity testing, slit lamp biomicroscopy, measurement of intraocular pressures, and a dilated eye exam. Conditions in the differential which threaten a patient's sight include retinal vasculitis, vitritis, cystoid macular edema, Behçet disease, and tubulo-interstitial nephritis. Other autoimmune diseases which can cause uveitis with systemic manifestations (multiple sclerosis, sarcoidosis, lupus) should be investigated. Infectious causes must also be eliminated. However, considering this patient's history of psoriatic disease, uveitis should be highly suspected.
Uveitis demands urgent treatment to control ocular inflammation. Tumor necrosis factor (TNF) inhibitors are the recommended first-line and second-line treatment for PsA, including in patients with complications such as uveitis. However, etanercept should not be used as it is less effective than adalimumab or other TNF inhibitors for uveitis. Because uveitis may sometimes respond to MTX therapy, patients with severe PsA may use a biologic agent in combination with MTX if they have had a partial response to current MTX therapy, as recommended by the American College of Rheumatology.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the patient's medical history and presentation, this is probably a case of uveitis, a common extra-articular manifestation of psoriatic disease. In fact, the presence of uveitis can help distinguish PsA from osteoarthritis. Uveitis is characterized by inflammation of the uvea tract, with the retina, optic nerve, vitreous body, and sclera potentially becoming inflamed as well. Among patients with PsA, the prevalence of uveitis rises with ongoing disease duration, though the condition may also precede the development of PsA in patients with psoriasis, and is common among patients with severe psoriatic disease in Western and Asian populations. Overall, the prevalence of uveitis has been estimated to be 6%-9%. HLA-B27 genotype is strongly associated with uveitis in patients with concomitant PsA.
Symptoms of uveitis, as seen in the present case, include blurred vision, photophobia, pain, and ciliary flush. The condition is classified as anterior, intermediate, posterior, or panuveitis, with the majority of cases diagnosed as anterior. In anterior uveitis, the inflamed pupil may become constricted or take on an irregular shape caused by iris adhesions to the anterior lens capsule. Uveitis in PsA is bilateral and has a chronic relapsing course. Onset is typically insidious.
Workup for uveitis should comprise visual acuity testing, slit lamp biomicroscopy, measurement of intraocular pressures, and a dilated eye exam. Conditions in the differential which threaten a patient's sight include retinal vasculitis, vitritis, cystoid macular edema, Behçet disease, and tubulo-interstitial nephritis. Other autoimmune diseases which can cause uveitis with systemic manifestations (multiple sclerosis, sarcoidosis, lupus) should be investigated. Infectious causes must also be eliminated. However, considering this patient's history of psoriatic disease, uveitis should be highly suspected.
Uveitis demands urgent treatment to control ocular inflammation. Tumor necrosis factor (TNF) inhibitors are the recommended first-line and second-line treatment for PsA, including in patients with complications such as uveitis. However, etanercept should not be used as it is less effective than adalimumab or other TNF inhibitors for uveitis. Because uveitis may sometimes respond to MTX therapy, patients with severe PsA may use a biologic agent in combination with MTX if they have had a partial response to current MTX therapy, as recommended by the American College of Rheumatology.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
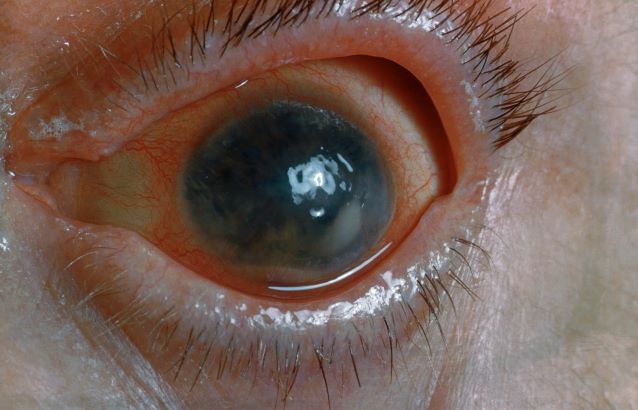
A 48-year-old male patient presents with blurred vision, pain, and photophobia. He recently had a routine visit with an ophthalmologist, which was normal. The affected pupil appears irregular in shape. The anterior chamber appears foggy. Local ciliary flush is observed on slit lamp exam. The physical examination is also notable for axial arthropathy. The patient has an 11-year history of moderate to severe psoriatic arthritis (PsA) which he typically manages with methotrexate (MTX) therapy, to which he has had a partial response. He was initially diagnosed when he presented with worsening psoriasis and enthesitis on the insertion sites of the plantar fascia, as well as dactylitis.
Dry cough and dyspnea
Based on the patient's presentation and workup, the likely diagnosis is adenosquamous carcinoma of the lung, a relatively rare subtype of non–small cell lung cancer (NSCLC). Adenosquamous carcinoma displays qualities of both squamous cell carcinoma and adenocarcinoma; for definitive diagnosis, the cancer must contain 10% of each of these major NSCLC subtypes. Maeda and colleagues concluded that adenosquamous carcinoma occurs more frequently among men and that the age at the time of diagnosis is higher among such cancers compared with adenocarcinoma. Several studies have confirmed that adenosquamous carcinoma of the lung is also more prevalent among smokers.
Though a diagnosis of adenosquamous carcinoma may be suspected after small biopsies, cytology, or excisional biopsies, definitive diagnosis necessitates a resection specimen. If any adenocarcinoma component is observed in a biopsy specimen that is otherwise squamous, as in the present case, this finding is an indication for molecular testing. Epidermal growth factor receptor (EGFR) mutations may be present in adenosquamous carcinoma cancers, despite a majority of cancers with EGFR mutations being among nonsmokers or former light smokers with adenocarcinoma histology. In addition, even for patients diagnosed with squamous cell carcinoma, adenosquamous carcinoma should be considered if genetic testing suggests EGFR mutations.
Relative to adenocarcinoma and squamous cell carcinoma, adenosquamous carcinoma has higher grade malignancy, more advanced postoperative stage, and stronger lymph nodal invasiveness. In terms of treatment, surgical resection is the curative option for adenosquamous carcinoma of the lung, with lobectomy with lymphadenectomy considered for first-line treatment. Though the most beneficial chemotherapy regimen for patients with adenosquamous carcinoma of the lung remains the subject of investigation, platinum-based doublet chemotherapy is the current standard treatment option. EGFR tyrosine kinase inhibitors may be an effective option for EGFR-positive patients.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
Based on the patient's presentation and workup, the likely diagnosis is adenosquamous carcinoma of the lung, a relatively rare subtype of non–small cell lung cancer (NSCLC). Adenosquamous carcinoma displays qualities of both squamous cell carcinoma and adenocarcinoma; for definitive diagnosis, the cancer must contain 10% of each of these major NSCLC subtypes. Maeda and colleagues concluded that adenosquamous carcinoma occurs more frequently among men and that the age at the time of diagnosis is higher among such cancers compared with adenocarcinoma. Several studies have confirmed that adenosquamous carcinoma of the lung is also more prevalent among smokers.
Though a diagnosis of adenosquamous carcinoma may be suspected after small biopsies, cytology, or excisional biopsies, definitive diagnosis necessitates a resection specimen. If any adenocarcinoma component is observed in a biopsy specimen that is otherwise squamous, as in the present case, this finding is an indication for molecular testing. Epidermal growth factor receptor (EGFR) mutations may be present in adenosquamous carcinoma cancers, despite a majority of cancers with EGFR mutations being among nonsmokers or former light smokers with adenocarcinoma histology. In addition, even for patients diagnosed with squamous cell carcinoma, adenosquamous carcinoma should be considered if genetic testing suggests EGFR mutations.
Relative to adenocarcinoma and squamous cell carcinoma, adenosquamous carcinoma has higher grade malignancy, more advanced postoperative stage, and stronger lymph nodal invasiveness. In terms of treatment, surgical resection is the curative option for adenosquamous carcinoma of the lung, with lobectomy with lymphadenectomy considered for first-line treatment. Though the most beneficial chemotherapy regimen for patients with adenosquamous carcinoma of the lung remains the subject of investigation, platinum-based doublet chemotherapy is the current standard treatment option. EGFR tyrosine kinase inhibitors may be an effective option for EGFR-positive patients.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
Based on the patient's presentation and workup, the likely diagnosis is adenosquamous carcinoma of the lung, a relatively rare subtype of non–small cell lung cancer (NSCLC). Adenosquamous carcinoma displays qualities of both squamous cell carcinoma and adenocarcinoma; for definitive diagnosis, the cancer must contain 10% of each of these major NSCLC subtypes. Maeda and colleagues concluded that adenosquamous carcinoma occurs more frequently among men and that the age at the time of diagnosis is higher among such cancers compared with adenocarcinoma. Several studies have confirmed that adenosquamous carcinoma of the lung is also more prevalent among smokers.
Though a diagnosis of adenosquamous carcinoma may be suspected after small biopsies, cytology, or excisional biopsies, definitive diagnosis necessitates a resection specimen. If any adenocarcinoma component is observed in a biopsy specimen that is otherwise squamous, as in the present case, this finding is an indication for molecular testing. Epidermal growth factor receptor (EGFR) mutations may be present in adenosquamous carcinoma cancers, despite a majority of cancers with EGFR mutations being among nonsmokers or former light smokers with adenocarcinoma histology. In addition, even for patients diagnosed with squamous cell carcinoma, adenosquamous carcinoma should be considered if genetic testing suggests EGFR mutations.
Relative to adenocarcinoma and squamous cell carcinoma, adenosquamous carcinoma has higher grade malignancy, more advanced postoperative stage, and stronger lymph nodal invasiveness. In terms of treatment, surgical resection is the curative option for adenosquamous carcinoma of the lung, with lobectomy with lymphadenectomy considered for first-line treatment. Though the most beneficial chemotherapy regimen for patients with adenosquamous carcinoma of the lung remains the subject of investigation, platinum-based doublet chemotherapy is the current standard treatment option. EGFR tyrosine kinase inhibitors may be an effective option for EGFR-positive patients.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
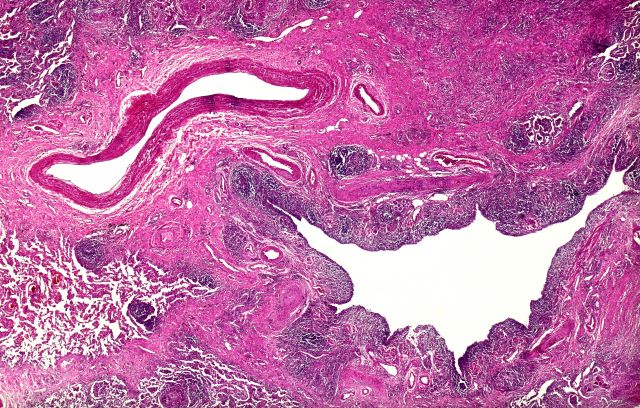
A 58-year-old man with a 20-year–pack history of smoking initially presented with a persistent dry cough and dyspnea. Clubbing was noted on physical examination and breath sounds in the right upper lung were weak. Other than hypertension, which the patient manages with angiotensin-converting enzyme (ACE) inhibitors, medical history is unremarkable. The patient notes that this medication has always made him cough, but dyspnea has only developed over the past 6 weeks. Respiratory symptoms prompted a chest radiograph which revealed a mass in the upper lobe of the right lung. Transbronchial lung biopsy of the right lung reveals components of adenocarcinoma; the specimen is otherwise squamous.