User login
Q Does unopposed estrogen increase the risk of breast cancer?
Expert Commentary
Although the US Food and Drug Administration requires drug treatment trials to include evidence of both benefit and risk, most clinical trials study adverse effects only over the short term, typically less than 2 years, with notable exceptions such as breast cancer adjuvant treatment trials.1 The assessment of long-term effects has largely fallen to the field of pharmacoepidemiology, and the most common research tool has been the observational cohort study.
Details of the studies
The WHI offered a unique opportunity to determine long-term benefits and risks from the 2 most commonly prescribed hormone regimens in the United States at the time the study began.2,3 In the estrogen-only arm, hysterectomized women were randomized to 0.625 mg daily of conjugated equine estrogen (CEE) or placebo, and were to be followed for 8 to 12 years to observe any major diseases that occurred, including breast cancer.
In 2004, the trial was stopped early after a mean 6.8 years of follow-up, because of a persistent elevated risk for stroke and no evidence of protection against coronary disease in the women randomized to estrogen.
Stefanick et al. In their closer look at WHI breast cancer data for the estrogen-only arm, Stefanick and colleagues found that women taking CEE had a nonstatistically significant 20% reduced risk of developing invasive breast cancer after a mean 7.1 years of follow-up. Examination over time did not suggest an increasing risk of breast cancer with CEE for up to 9 years of follow-up; rather, risk in CEE-treated women remained diminished, compared with placebo, throughout follow-up. Among women who had used estrogen alone for 5 or more years prior to enrollment in the WHI trial, the risk of invasive breast cancer increased nonsignificantly by 28% in CEE-treated women compared with placebo.
Chen et al. In their reanalysis of Nurses’ Health Study data from 28,835 postmenopausal women without a uterus, Chen et al observed comparable results after 5 to 9.9 years of CEE use—ie, a non-significant 13% reduced risk of breast cancer. However, among women who used CEE for 15 to 19.9 years, a nonstatistically significant 19% increase in the risk of invasive breast cancer was observed, and among women who used CEE for 20 or more years, a statistically significant 41% increased risk was seen.
Findings agree with earlier data
These 2 studies are in accord with previous observational studies of exogenous estrogen and the risk of breast cancer.4 A combined dataset representing more than 52,000 breast cancer cases and more than twice as many controls found that current or recent (past 1–4 years) use of a daily dose of unopposed CEE of 0.625 mg or less, for less than 5 years, was associated with a 23% reduced risk of breast cancer, compared with nonusers.4 Use of this formulation for 5 or more years was associated with a 64% increase in risk.
Similarly, the UK-based Million Women Study found that use of unopposed estrogen for less than 1 year reduced the risk for breast cancer by 19%, compared with never-users, but longer use increased risk by 25% to 37%.5
Bottom line: No heightened risk in the short term
Women choosing to take unopposed estrogen to control menopausal symptoms do not appear to face an increased risk of breast cancer if they use it for less than 5 years. Observational studies suggest they may increase their risk of breast cancer by using estrogen for 5 or more years, but no data from clinical trials are available past 7 years of follow-up.
The lower risk of breast cancer for women using unopposed estrogen for only short periods of time, seen in both the WHI clinical trial and the large observational studies, remains unexplained.
Dr. McTiernan is a consultant to Novartis Canada, Procter & Gamble, and Zymogenetics.
1. Mamounas EP. NSABP breast cancer clinical trials: recent results and future directions. Clin Med Res. 2003;1:309-326.
2. Anderson GL, Limacher M, Assaf AR, et al, for the Women’s Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized, controlled trial. JAMA. 2004;291:1701-1712.
3. Prentice R, Rossouw JR, Johnson SR, Freedman LS, McTiernan A. The role of randomized controlled trial in assessing the benefits and risks of long-term hormone replacement therapy: example of the Women’s Health Initiative. Menopause. 1996;3:71-76.
4. Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Lancet. 1997;350:1047-1059.
5. Million Women Study Collaborators. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003;362:419-427.
Expert Commentary
Although the US Food and Drug Administration requires drug treatment trials to include evidence of both benefit and risk, most clinical trials study adverse effects only over the short term, typically less than 2 years, with notable exceptions such as breast cancer adjuvant treatment trials.1 The assessment of long-term effects has largely fallen to the field of pharmacoepidemiology, and the most common research tool has been the observational cohort study.
Details of the studies
The WHI offered a unique opportunity to determine long-term benefits and risks from the 2 most commonly prescribed hormone regimens in the United States at the time the study began.2,3 In the estrogen-only arm, hysterectomized women were randomized to 0.625 mg daily of conjugated equine estrogen (CEE) or placebo, and were to be followed for 8 to 12 years to observe any major diseases that occurred, including breast cancer.
In 2004, the trial was stopped early after a mean 6.8 years of follow-up, because of a persistent elevated risk for stroke and no evidence of protection against coronary disease in the women randomized to estrogen.
Stefanick et al. In their closer look at WHI breast cancer data for the estrogen-only arm, Stefanick and colleagues found that women taking CEE had a nonstatistically significant 20% reduced risk of developing invasive breast cancer after a mean 7.1 years of follow-up. Examination over time did not suggest an increasing risk of breast cancer with CEE for up to 9 years of follow-up; rather, risk in CEE-treated women remained diminished, compared with placebo, throughout follow-up. Among women who had used estrogen alone for 5 or more years prior to enrollment in the WHI trial, the risk of invasive breast cancer increased nonsignificantly by 28% in CEE-treated women compared with placebo.
Chen et al. In their reanalysis of Nurses’ Health Study data from 28,835 postmenopausal women without a uterus, Chen et al observed comparable results after 5 to 9.9 years of CEE use—ie, a non-significant 13% reduced risk of breast cancer. However, among women who used CEE for 15 to 19.9 years, a nonstatistically significant 19% increase in the risk of invasive breast cancer was observed, and among women who used CEE for 20 or more years, a statistically significant 41% increased risk was seen.
Findings agree with earlier data
These 2 studies are in accord with previous observational studies of exogenous estrogen and the risk of breast cancer.4 A combined dataset representing more than 52,000 breast cancer cases and more than twice as many controls found that current or recent (past 1–4 years) use of a daily dose of unopposed CEE of 0.625 mg or less, for less than 5 years, was associated with a 23% reduced risk of breast cancer, compared with nonusers.4 Use of this formulation for 5 or more years was associated with a 64% increase in risk.
Similarly, the UK-based Million Women Study found that use of unopposed estrogen for less than 1 year reduced the risk for breast cancer by 19%, compared with never-users, but longer use increased risk by 25% to 37%.5
Bottom line: No heightened risk in the short term
Women choosing to take unopposed estrogen to control menopausal symptoms do not appear to face an increased risk of breast cancer if they use it for less than 5 years. Observational studies suggest they may increase their risk of breast cancer by using estrogen for 5 or more years, but no data from clinical trials are available past 7 years of follow-up.
The lower risk of breast cancer for women using unopposed estrogen for only short periods of time, seen in both the WHI clinical trial and the large observational studies, remains unexplained.
Dr. McTiernan is a consultant to Novartis Canada, Procter & Gamble, and Zymogenetics.
Expert Commentary
Although the US Food and Drug Administration requires drug treatment trials to include evidence of both benefit and risk, most clinical trials study adverse effects only over the short term, typically less than 2 years, with notable exceptions such as breast cancer adjuvant treatment trials.1 The assessment of long-term effects has largely fallen to the field of pharmacoepidemiology, and the most common research tool has been the observational cohort study.
Details of the studies
The WHI offered a unique opportunity to determine long-term benefits and risks from the 2 most commonly prescribed hormone regimens in the United States at the time the study began.2,3 In the estrogen-only arm, hysterectomized women were randomized to 0.625 mg daily of conjugated equine estrogen (CEE) or placebo, and were to be followed for 8 to 12 years to observe any major diseases that occurred, including breast cancer.
In 2004, the trial was stopped early after a mean 6.8 years of follow-up, because of a persistent elevated risk for stroke and no evidence of protection against coronary disease in the women randomized to estrogen.
Stefanick et al. In their closer look at WHI breast cancer data for the estrogen-only arm, Stefanick and colleagues found that women taking CEE had a nonstatistically significant 20% reduced risk of developing invasive breast cancer after a mean 7.1 years of follow-up. Examination over time did not suggest an increasing risk of breast cancer with CEE for up to 9 years of follow-up; rather, risk in CEE-treated women remained diminished, compared with placebo, throughout follow-up. Among women who had used estrogen alone for 5 or more years prior to enrollment in the WHI trial, the risk of invasive breast cancer increased nonsignificantly by 28% in CEE-treated women compared with placebo.
Chen et al. In their reanalysis of Nurses’ Health Study data from 28,835 postmenopausal women without a uterus, Chen et al observed comparable results after 5 to 9.9 years of CEE use—ie, a non-significant 13% reduced risk of breast cancer. However, among women who used CEE for 15 to 19.9 years, a nonstatistically significant 19% increase in the risk of invasive breast cancer was observed, and among women who used CEE for 20 or more years, a statistically significant 41% increased risk was seen.
Findings agree with earlier data
These 2 studies are in accord with previous observational studies of exogenous estrogen and the risk of breast cancer.4 A combined dataset representing more than 52,000 breast cancer cases and more than twice as many controls found that current or recent (past 1–4 years) use of a daily dose of unopposed CEE of 0.625 mg or less, for less than 5 years, was associated with a 23% reduced risk of breast cancer, compared with nonusers.4 Use of this formulation for 5 or more years was associated with a 64% increase in risk.
Similarly, the UK-based Million Women Study found that use of unopposed estrogen for less than 1 year reduced the risk for breast cancer by 19%, compared with never-users, but longer use increased risk by 25% to 37%.5
Bottom line: No heightened risk in the short term
Women choosing to take unopposed estrogen to control menopausal symptoms do not appear to face an increased risk of breast cancer if they use it for less than 5 years. Observational studies suggest they may increase their risk of breast cancer by using estrogen for 5 or more years, but no data from clinical trials are available past 7 years of follow-up.
The lower risk of breast cancer for women using unopposed estrogen for only short periods of time, seen in both the WHI clinical trial and the large observational studies, remains unexplained.
Dr. McTiernan is a consultant to Novartis Canada, Procter & Gamble, and Zymogenetics.
1. Mamounas EP. NSABP breast cancer clinical trials: recent results and future directions. Clin Med Res. 2003;1:309-326.
2. Anderson GL, Limacher M, Assaf AR, et al, for the Women’s Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized, controlled trial. JAMA. 2004;291:1701-1712.
3. Prentice R, Rossouw JR, Johnson SR, Freedman LS, McTiernan A. The role of randomized controlled trial in assessing the benefits and risks of long-term hormone replacement therapy: example of the Women’s Health Initiative. Menopause. 1996;3:71-76.
4. Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Lancet. 1997;350:1047-1059.
5. Million Women Study Collaborators. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003;362:419-427.
1. Mamounas EP. NSABP breast cancer clinical trials: recent results and future directions. Clin Med Res. 2003;1:309-326.
2. Anderson GL, Limacher M, Assaf AR, et al, for the Women’s Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized, controlled trial. JAMA. 2004;291:1701-1712.
3. Prentice R, Rossouw JR, Johnson SR, Freedman LS, McTiernan A. The role of randomized controlled trial in assessing the benefits and risks of long-term hormone replacement therapy: example of the Women’s Health Initiative. Menopause. 1996;3:71-76.
4. Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and hormone replacement therapy: collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Lancet. 1997;350:1047-1059.
5. Million Women Study Collaborators. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003;362:419-427.
Q Does acute infection raise the risk of venous thromboembolism?
In this study, the incidence ratio for deep venous thrombosis (DVT) following urinary tract infection was 2.10 (95% confidence interval [CI] 1.56–2.82), and for respiratory tract infection, it was 2.86 (95% CI 2.05–3.97).
The incidence ratio for pulmonary embolism (PE) following urinary tract infection was 2.11 (95% CI 1.38–3.23). Although the risk of PE following respiratory tract infection was 11-fold higher, possible misdiagnosis of PE as a respiratory infection precluded reliable estimates of the precise risk.
Details of the study
The study assessed the risk of a first-ever DVT or PE after acute urinary tract infection or acute systemic respiratory infection, excluding pharyngitis and coryza.
Data were from the United Kingdom’s Health Improvement Network, which has complete diagnostic and prescribing information, and covered the years 1987 to 2004, or approximately 20 million person-years.
One strength was use of a self-controlled case series method, which allowed patients to serve as their own controls, thus eliminating variation among individuals in risk factors for venous thromboembolism.
Patients were observed for 12 months after an acute urinary or respiratory tract infection to determine whether a thromboembolic event had occurred. Incidence ratios and confidence intervals were calculated, and the study had adequate power at 5% significance to detect a 4-fold difference during the first 2 weeks after acute infections.
Expert Commentary
The exact mechanism of thrombosis is still unknown, and the possibility of a common pathway not linked to a specific infection is intriguing. Uncovering the mechanism could help us direct therapy to a particular biochemical process.
Virchow proposed his triad of precipitating factors 150 years ago: venous stasis, increased coagulability of the blood, and vessel wall damage.1 It now seems entirely plausible that damage to the vessel wall need not be physical damage, but could include factors, such as inflammation, that affect endothelial function. As the authors noted, “Inflammation is a key determinant of endothelial function in both arteries and veins, and a link between infection and venous thrombosis via endothelial activation has been suggested.” In fact, earlier studies already identified infection as a potential risk factor for venous thromboembolism.2,3
Thromboembolic events occur at a rate of about 0.5 cases per 1,000 person-years and cause considerable morbidity and mortality.
How long to continue prophylaxis?
The study by Smeeth and colleagues should help ObGyns determine the level of prophylaxis appropriate for hospitalized patients. Less clear is whether thromboprophylaxis should be offered to women who have acute infections in an ambulatory setting. Although earlier studies suggested that thromboprophylaxis may be appropriate, I believe the question of whether every patient should receive preventive therapy remains unanswered.
Another unresolved issue: If prophylaxis is initiated, how long should it continue? Because the risk of a thromboembolic event does not return to baseline levels for 1 year, the duration of therapy could be lengthy. At the same time, the risks of anticoagulation are not inconsequential and may increase with extended therapy. As the greatest risk occurs during the first 8 weeks after infection, prophylaxis is most beneficial during this time.
Routine prophylaxis?
Given the data thus far, I do not believe therapy is warranted for every patient with an acute infection. Selective therapy may be justified.
1. Virchow RLK. Thrombosis and Emboli. Matzdorff AC, Bell WR, trans. Canton, Mass: Science History Publications; 1998.
2. Samama MM. An epidemiologic study of risk factors for deep vein thrombosis in medical outpatients: the Sirius study. Arch Intern Med. 2000;160:3415-3420.
3. Alikhan R, Cohen AT, Combe S, et al. Risk factors for venous thromboembolism in hospitalized patients with acute medical illness: analysis of the MEDENOX Study. Arch Intern Med. 2004;164:963-968.
In this study, the incidence ratio for deep venous thrombosis (DVT) following urinary tract infection was 2.10 (95% confidence interval [CI] 1.56–2.82), and for respiratory tract infection, it was 2.86 (95% CI 2.05–3.97).
The incidence ratio for pulmonary embolism (PE) following urinary tract infection was 2.11 (95% CI 1.38–3.23). Although the risk of PE following respiratory tract infection was 11-fold higher, possible misdiagnosis of PE as a respiratory infection precluded reliable estimates of the precise risk.
Details of the study
The study assessed the risk of a first-ever DVT or PE after acute urinary tract infection or acute systemic respiratory infection, excluding pharyngitis and coryza.
Data were from the United Kingdom’s Health Improvement Network, which has complete diagnostic and prescribing information, and covered the years 1987 to 2004, or approximately 20 million person-years.
One strength was use of a self-controlled case series method, which allowed patients to serve as their own controls, thus eliminating variation among individuals in risk factors for venous thromboembolism.
Patients were observed for 12 months after an acute urinary or respiratory tract infection to determine whether a thromboembolic event had occurred. Incidence ratios and confidence intervals were calculated, and the study had adequate power at 5% significance to detect a 4-fold difference during the first 2 weeks after acute infections.
Expert Commentary
The exact mechanism of thrombosis is still unknown, and the possibility of a common pathway not linked to a specific infection is intriguing. Uncovering the mechanism could help us direct therapy to a particular biochemical process.
Virchow proposed his triad of precipitating factors 150 years ago: venous stasis, increased coagulability of the blood, and vessel wall damage.1 It now seems entirely plausible that damage to the vessel wall need not be physical damage, but could include factors, such as inflammation, that affect endothelial function. As the authors noted, “Inflammation is a key determinant of endothelial function in both arteries and veins, and a link between infection and venous thrombosis via endothelial activation has been suggested.” In fact, earlier studies already identified infection as a potential risk factor for venous thromboembolism.2,3
Thromboembolic events occur at a rate of about 0.5 cases per 1,000 person-years and cause considerable morbidity and mortality.
How long to continue prophylaxis?
The study by Smeeth and colleagues should help ObGyns determine the level of prophylaxis appropriate for hospitalized patients. Less clear is whether thromboprophylaxis should be offered to women who have acute infections in an ambulatory setting. Although earlier studies suggested that thromboprophylaxis may be appropriate, I believe the question of whether every patient should receive preventive therapy remains unanswered.
Another unresolved issue: If prophylaxis is initiated, how long should it continue? Because the risk of a thromboembolic event does not return to baseline levels for 1 year, the duration of therapy could be lengthy. At the same time, the risks of anticoagulation are not inconsequential and may increase with extended therapy. As the greatest risk occurs during the first 8 weeks after infection, prophylaxis is most beneficial during this time.
Routine prophylaxis?
Given the data thus far, I do not believe therapy is warranted for every patient with an acute infection. Selective therapy may be justified.
In this study, the incidence ratio for deep venous thrombosis (DVT) following urinary tract infection was 2.10 (95% confidence interval [CI] 1.56–2.82), and for respiratory tract infection, it was 2.86 (95% CI 2.05–3.97).
The incidence ratio for pulmonary embolism (PE) following urinary tract infection was 2.11 (95% CI 1.38–3.23). Although the risk of PE following respiratory tract infection was 11-fold higher, possible misdiagnosis of PE as a respiratory infection precluded reliable estimates of the precise risk.
Details of the study
The study assessed the risk of a first-ever DVT or PE after acute urinary tract infection or acute systemic respiratory infection, excluding pharyngitis and coryza.
Data were from the United Kingdom’s Health Improvement Network, which has complete diagnostic and prescribing information, and covered the years 1987 to 2004, or approximately 20 million person-years.
One strength was use of a self-controlled case series method, which allowed patients to serve as their own controls, thus eliminating variation among individuals in risk factors for venous thromboembolism.
Patients were observed for 12 months after an acute urinary or respiratory tract infection to determine whether a thromboembolic event had occurred. Incidence ratios and confidence intervals were calculated, and the study had adequate power at 5% significance to detect a 4-fold difference during the first 2 weeks after acute infections.
Expert Commentary
The exact mechanism of thrombosis is still unknown, and the possibility of a common pathway not linked to a specific infection is intriguing. Uncovering the mechanism could help us direct therapy to a particular biochemical process.
Virchow proposed his triad of precipitating factors 150 years ago: venous stasis, increased coagulability of the blood, and vessel wall damage.1 It now seems entirely plausible that damage to the vessel wall need not be physical damage, but could include factors, such as inflammation, that affect endothelial function. As the authors noted, “Inflammation is a key determinant of endothelial function in both arteries and veins, and a link between infection and venous thrombosis via endothelial activation has been suggested.” In fact, earlier studies already identified infection as a potential risk factor for venous thromboembolism.2,3
Thromboembolic events occur at a rate of about 0.5 cases per 1,000 person-years and cause considerable morbidity and mortality.
How long to continue prophylaxis?
The study by Smeeth and colleagues should help ObGyns determine the level of prophylaxis appropriate for hospitalized patients. Less clear is whether thromboprophylaxis should be offered to women who have acute infections in an ambulatory setting. Although earlier studies suggested that thromboprophylaxis may be appropriate, I believe the question of whether every patient should receive preventive therapy remains unanswered.
Another unresolved issue: If prophylaxis is initiated, how long should it continue? Because the risk of a thromboembolic event does not return to baseline levels for 1 year, the duration of therapy could be lengthy. At the same time, the risks of anticoagulation are not inconsequential and may increase with extended therapy. As the greatest risk occurs during the first 8 weeks after infection, prophylaxis is most beneficial during this time.
Routine prophylaxis?
Given the data thus far, I do not believe therapy is warranted for every patient with an acute infection. Selective therapy may be justified.
1. Virchow RLK. Thrombosis and Emboli. Matzdorff AC, Bell WR, trans. Canton, Mass: Science History Publications; 1998.
2. Samama MM. An epidemiologic study of risk factors for deep vein thrombosis in medical outpatients: the Sirius study. Arch Intern Med. 2000;160:3415-3420.
3. Alikhan R, Cohen AT, Combe S, et al. Risk factors for venous thromboembolism in hospitalized patients with acute medical illness: analysis of the MEDENOX Study. Arch Intern Med. 2004;164:963-968.
1. Virchow RLK. Thrombosis and Emboli. Matzdorff AC, Bell WR, trans. Canton, Mass: Science History Publications; 1998.
2. Samama MM. An epidemiologic study of risk factors for deep vein thrombosis in medical outpatients: the Sirius study. Arch Intern Med. 2000;160:3415-3420.
3. Alikhan R, Cohen AT, Combe S, et al. Risk factors for venous thromboembolism in hospitalized patients with acute medical illness: analysis of the MEDENOX Study. Arch Intern Med. 2004;164:963-968.
Q Can a questionnaire differentiate urge and stress incontinence?
The 3IQ is therefore acceptable for use by primary care physicians, especially when treatment will be noninvasive.
Expert Commentary
When a woman complains of urinary incontinence, the clinician’s first objective is determining which type of incontinence she has so therapy can be appropriately targeted. This is not as straightforward as might be expected. The symptoms of urge and stress incontinence overlap significantly, necessitating simple or complex urodynamics.
Simple conservative treatments such as timed voiding and Kegel exercises may benefit all incontinent women. However, other treatments, such as anticholinergic drugs for urge incontinence or the new transobturator sling procedures for stress incontinence, require a more precise diagnosis. Thus was born the 3IQ.
1. During the last 3 months, have you leaked urine?
□ Yes
□ No
If no, no need to proceed
2. During the last 3 months, did you leak urine: (Check all that apply)
□ a. When you were performing a physical activity such as coughing, sneezing, lifting, or exercise?
□ b. When you had the urge or need to empty your bladder, but could not get to the toilet fast enough?
□ c. Without physical activity and without a sense of urgency?
3. During the last 3 months, did you leak urine most often: (Check only one)
□ a. When you were performing a physical activity?
□ b. When you had the urge or the feeling that you needed to empty your bladder?
□ c. Without physical activity and without a sense of urgency?
□ d. About equally as often with physical activity as with a sense of urgency?
Score by response to question 3:
a=stress or stress-dominant
b=urge or urge-dominant
c=other causes
d=mixed
Adapted from Brown JS, et al
How the 3IQ assesses symptoms
The 3IQ was completed by 301 women with untreated incontinence and was compared with a “gold standard” evaluation that included a history, physical examination (including neurologic evaluation), pelvic exam, cough stress test, measurement of postvoid residual, and review of a 3-day voiding diary.
Investigators did not evaluate each question individually, which is unfortunate, as it would be interesting to know whether a single question has similar value in distinguishing stress and urge incontinence.
All the women attended tertiary continence centers. It would be helpful to repeat this study in a primary care setting where women may have less severe symptoms and be less likely to undergo further evaluation.
Despite limited utility to ObGyns, the study is good news
The 3IQ appears to be particularly useful for selecting noninvasive therapy for women with urge incontinence. For surgeons who perform minimally invasive sling procedures for stress incontinence, the low specificity of this test renders it inappropriate. Thus, the 3IQ may be more useful for a family practitioner or internist than for the ObGyn who is also a surgeon.
The fact that this study was published in Annals of Internal Medicine makes it clear that primary care physicians are interested in the care of women with urinary incontinence. ObGyns should be happy about this, as studies like this one will increase awareness of the frequency of urinary incontinence and lead to further referrals to ObGyns for more extensive management.
The 3IQ is therefore acceptable for use by primary care physicians, especially when treatment will be noninvasive.
Expert Commentary
When a woman complains of urinary incontinence, the clinician’s first objective is determining which type of incontinence she has so therapy can be appropriately targeted. This is not as straightforward as might be expected. The symptoms of urge and stress incontinence overlap significantly, necessitating simple or complex urodynamics.
Simple conservative treatments such as timed voiding and Kegel exercises may benefit all incontinent women. However, other treatments, such as anticholinergic drugs for urge incontinence or the new transobturator sling procedures for stress incontinence, require a more precise diagnosis. Thus was born the 3IQ.
1. During the last 3 months, have you leaked urine?
□ Yes
□ No
If no, no need to proceed
2. During the last 3 months, did you leak urine: (Check all that apply)
□ a. When you were performing a physical activity such as coughing, sneezing, lifting, or exercise?
□ b. When you had the urge or need to empty your bladder, but could not get to the toilet fast enough?
□ c. Without physical activity and without a sense of urgency?
3. During the last 3 months, did you leak urine most often: (Check only one)
□ a. When you were performing a physical activity?
□ b. When you had the urge or the feeling that you needed to empty your bladder?
□ c. Without physical activity and without a sense of urgency?
□ d. About equally as often with physical activity as with a sense of urgency?
Score by response to question 3:
a=stress or stress-dominant
b=urge or urge-dominant
c=other causes
d=mixed
Adapted from Brown JS, et al
How the 3IQ assesses symptoms
The 3IQ was completed by 301 women with untreated incontinence and was compared with a “gold standard” evaluation that included a history, physical examination (including neurologic evaluation), pelvic exam, cough stress test, measurement of postvoid residual, and review of a 3-day voiding diary.
Investigators did not evaluate each question individually, which is unfortunate, as it would be interesting to know whether a single question has similar value in distinguishing stress and urge incontinence.
All the women attended tertiary continence centers. It would be helpful to repeat this study in a primary care setting where women may have less severe symptoms and be less likely to undergo further evaluation.
Despite limited utility to ObGyns, the study is good news
The 3IQ appears to be particularly useful for selecting noninvasive therapy for women with urge incontinence. For surgeons who perform minimally invasive sling procedures for stress incontinence, the low specificity of this test renders it inappropriate. Thus, the 3IQ may be more useful for a family practitioner or internist than for the ObGyn who is also a surgeon.
The fact that this study was published in Annals of Internal Medicine makes it clear that primary care physicians are interested in the care of women with urinary incontinence. ObGyns should be happy about this, as studies like this one will increase awareness of the frequency of urinary incontinence and lead to further referrals to ObGyns for more extensive management.
The 3IQ is therefore acceptable for use by primary care physicians, especially when treatment will be noninvasive.
Expert Commentary
When a woman complains of urinary incontinence, the clinician’s first objective is determining which type of incontinence she has so therapy can be appropriately targeted. This is not as straightforward as might be expected. The symptoms of urge and stress incontinence overlap significantly, necessitating simple or complex urodynamics.
Simple conservative treatments such as timed voiding and Kegel exercises may benefit all incontinent women. However, other treatments, such as anticholinergic drugs for urge incontinence or the new transobturator sling procedures for stress incontinence, require a more precise diagnosis. Thus was born the 3IQ.
1. During the last 3 months, have you leaked urine?
□ Yes
□ No
If no, no need to proceed
2. During the last 3 months, did you leak urine: (Check all that apply)
□ a. When you were performing a physical activity such as coughing, sneezing, lifting, or exercise?
□ b. When you had the urge or need to empty your bladder, but could not get to the toilet fast enough?
□ c. Without physical activity and without a sense of urgency?
3. During the last 3 months, did you leak urine most often: (Check only one)
□ a. When you were performing a physical activity?
□ b. When you had the urge or the feeling that you needed to empty your bladder?
□ c. Without physical activity and without a sense of urgency?
□ d. About equally as often with physical activity as with a sense of urgency?
Score by response to question 3:
a=stress or stress-dominant
b=urge or urge-dominant
c=other causes
d=mixed
Adapted from Brown JS, et al
How the 3IQ assesses symptoms
The 3IQ was completed by 301 women with untreated incontinence and was compared with a “gold standard” evaluation that included a history, physical examination (including neurologic evaluation), pelvic exam, cough stress test, measurement of postvoid residual, and review of a 3-day voiding diary.
Investigators did not evaluate each question individually, which is unfortunate, as it would be interesting to know whether a single question has similar value in distinguishing stress and urge incontinence.
All the women attended tertiary continence centers. It would be helpful to repeat this study in a primary care setting where women may have less severe symptoms and be less likely to undergo further evaluation.
Despite limited utility to ObGyns, the study is good news
The 3IQ appears to be particularly useful for selecting noninvasive therapy for women with urge incontinence. For surgeons who perform minimally invasive sling procedures for stress incontinence, the low specificity of this test renders it inappropriate. Thus, the 3IQ may be more useful for a family practitioner or internist than for the ObGyn who is also a surgeon.
The fact that this study was published in Annals of Internal Medicine makes it clear that primary care physicians are interested in the care of women with urinary incontinence. ObGyns should be happy about this, as studies like this one will increase awareness of the frequency of urinary incontinence and lead to further referrals to ObGyns for more extensive management.
Q Can twice-weekly metronidazole keep recurrent BV in check?
During the entire 28-week follow-up of this study, 26 women (51%) in the group receiving twice-weekly 0.75% metronidazole, 5 g intravaginally, had recurrent infection, compared with 33 women (75%) on placebo.
Expert Commentary
Not only is BV the most common vaginal infection in women of reproductive age, affecting 15% to 30% of nonpregnant women and roughly half of all gravidas, recurrence is a widespread problem: 33% of women have another infection within 3 months of treatment.
BV also can cause discomfort and serious reproductive tract complications, including postoperative upper genital tract infections and spontaneous preterm birth.
Until now, the usual approach to frequent recurrences of BV has been repeated short courses of treatment—often several per year. This is problematic because the drugs are expensive and “not entirely benign.”
BV diagnosis was rigorous
In this trial, BV was diagnosed using both clinical and microbiologic criteria. Clinical criteria for BV—better known as Amsel criteria—are the presence of at least 3 of the following: homogenous vaginal discharge, vaginal pH greater than 4.5, amine odor, and clue cells. The microbiologic definition of BV is based on Nugent criteria, which involves the assessment of a Gram-stained vaginal smear.
Antifungal may be needed
Women on suppressive therapy had a substantially increased frequency of vaginal candidiasis: 43.1% developed the infection, more than twice the frequency among controls. For this reason, physicians should be vigilant for candidiasis and consider suppressive therapy for it as well as BV.
Findings do not apply to gravidas
The findings of this study should not be extrapolated to a pregnant population.
Suppressive therapy is better than repeat courses
Although suppressive therapy did not eliminate recurrence, many women experienced major improvement. The increased frequency of candidiasis may warrant suppressive antifungal therapy.
In the “real world,” some providers base “diagnoses” on the symptoms reported rather than a rigorous physical exam. However, use of Amsel criteria is strongly encouraged, unless the lab is experienced in Nugent scoring of Gram-stained smears.
During the entire 28-week follow-up of this study, 26 women (51%) in the group receiving twice-weekly 0.75% metronidazole, 5 g intravaginally, had recurrent infection, compared with 33 women (75%) on placebo.
Expert Commentary
Not only is BV the most common vaginal infection in women of reproductive age, affecting 15% to 30% of nonpregnant women and roughly half of all gravidas, recurrence is a widespread problem: 33% of women have another infection within 3 months of treatment.
BV also can cause discomfort and serious reproductive tract complications, including postoperative upper genital tract infections and spontaneous preterm birth.
Until now, the usual approach to frequent recurrences of BV has been repeated short courses of treatment—often several per year. This is problematic because the drugs are expensive and “not entirely benign.”
BV diagnosis was rigorous
In this trial, BV was diagnosed using both clinical and microbiologic criteria. Clinical criteria for BV—better known as Amsel criteria—are the presence of at least 3 of the following: homogenous vaginal discharge, vaginal pH greater than 4.5, amine odor, and clue cells. The microbiologic definition of BV is based on Nugent criteria, which involves the assessment of a Gram-stained vaginal smear.
Antifungal may be needed
Women on suppressive therapy had a substantially increased frequency of vaginal candidiasis: 43.1% developed the infection, more than twice the frequency among controls. For this reason, physicians should be vigilant for candidiasis and consider suppressive therapy for it as well as BV.
Findings do not apply to gravidas
The findings of this study should not be extrapolated to a pregnant population.
Suppressive therapy is better than repeat courses
Although suppressive therapy did not eliminate recurrence, many women experienced major improvement. The increased frequency of candidiasis may warrant suppressive antifungal therapy.
In the “real world,” some providers base “diagnoses” on the symptoms reported rather than a rigorous physical exam. However, use of Amsel criteria is strongly encouraged, unless the lab is experienced in Nugent scoring of Gram-stained smears.
During the entire 28-week follow-up of this study, 26 women (51%) in the group receiving twice-weekly 0.75% metronidazole, 5 g intravaginally, had recurrent infection, compared with 33 women (75%) on placebo.
Expert Commentary
Not only is BV the most common vaginal infection in women of reproductive age, affecting 15% to 30% of nonpregnant women and roughly half of all gravidas, recurrence is a widespread problem: 33% of women have another infection within 3 months of treatment.
BV also can cause discomfort and serious reproductive tract complications, including postoperative upper genital tract infections and spontaneous preterm birth.
Until now, the usual approach to frequent recurrences of BV has been repeated short courses of treatment—often several per year. This is problematic because the drugs are expensive and “not entirely benign.”
BV diagnosis was rigorous
In this trial, BV was diagnosed using both clinical and microbiologic criteria. Clinical criteria for BV—better known as Amsel criteria—are the presence of at least 3 of the following: homogenous vaginal discharge, vaginal pH greater than 4.5, amine odor, and clue cells. The microbiologic definition of BV is based on Nugent criteria, which involves the assessment of a Gram-stained vaginal smear.
Antifungal may be needed
Women on suppressive therapy had a substantially increased frequency of vaginal candidiasis: 43.1% developed the infection, more than twice the frequency among controls. For this reason, physicians should be vigilant for candidiasis and consider suppressive therapy for it as well as BV.
Findings do not apply to gravidas
The findings of this study should not be extrapolated to a pregnant population.
Suppressive therapy is better than repeat courses
Although suppressive therapy did not eliminate recurrence, many women experienced major improvement. The increased frequency of candidiasis may warrant suppressive antifungal therapy.
In the “real world,” some providers base “diagnoses” on the symptoms reported rather than a rigorous physical exam. However, use of Amsel criteria is strongly encouraged, unless the lab is experienced in Nugent scoring of Gram-stained smears.
The HPV vaccine: A good start
Last month the Food and Drug Administration approved the first vaccine (Gardisil) to prevent infection with specific types of human papillomavirus (HPV). The approval is the culmination of 2 decades of work in a variety of disciplines that have clearly demonstrated that infection with specific types of high-risk HPV is required for the development of cervical cancer—and that the development of HPV-associated disease can be prevented through vaccination.
How and when will HPV vaccine be put to use?
It is important to recognize that FDA approval does not mean the HPV vaccine will be widely used. Five components are necessary for successful introduction:
1. FDA approval
FDA approval is based solely on the safety and effectiveness of the vaccine.
Indications. The FDA determined that the quadrivalent HPV vaccine is indicated for prevention of HPV 6, 11, 16, and 18 associated cervical cancer, genital warts, CIN, AIS, and VIN 2,3 and VAIN 2,3.
The indication applies to girls and women 9 to 26 years of age. The indication in girls 9 to 15 years of age, who were not included in the Phase II and III trials, was based on immunological “bridging” studies that demonstrated better immune responses to vaccination in this group than in the older group enrolled in the trials.
2. Federal-level recommendations
The second component of a successful vaccine introduction is the support of the Advisory Committee on Immunization Practices (ACIP),2 a congressionally mandated federal advisory committee coordinated by the Centers for Disease Control and Prevention (CDC). The ACIP advises the Secretary of Health and Human Services, as well as the Director of the CDC, on appropriate immunization practices for the United States.
Within several months of the June 2006 FDA approval, the ACIP will make a recommendation on the vaccine’s use. When making its recommendation, the ACIP takes into account the burden of disease in the population, safety and efficacy, and health economics of vaccination—will it be cost-effective?
Funding and insurance coverage at stake. The ACIP also determines whether the vaccine will be acceptable to physicians and consumers and what programmatic issues are associated with its introduction.
- Public funding. The ACIP recommendation strongly influences standard of practice, and is relied upon by the federal government and states for determining whether to include a vaccine in public funding programs.
- Insurers likewise look to the ACIP for guidance when setting reimbursement policy.
3. Achieving adequate funding
The third component of a successful vaccine introduction is achieving adequate funding. Federal and state programs such as the Vaccines for Children (VFC) program currently pay for more than half of all childhood vaccinations. Therefore it is important that these programs include the HPV vaccine in order to ensure it is widely available, especially among lower socio-economic groups.
4. Recommendations by professional societies
Societies such as the American Academy of Pediatrics, the American College of Obstetricians and Gynecologists, and the American Academy of Family Physicians, whose members provide most of the vaccinations in the US, have representatives who work directly with the ACIP. Many societies make their own vaccination recommendations, as well.
5. Advocacy and education
Other groups increase awareness of the need for vaccination among consumers, public health and government officials, and clinicians.
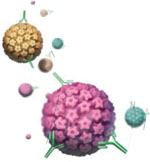
The quadrivalent vaccine consists of recombinant viral-like particles (VLPs) derived from the 4 different types of HPV (6, 11, 16, and 18) which, combined, account for approximately 70% of all invasive cervical cancers, 60% of high-grade CIN lesions, and more than 90% of genital warts, both in the United States and globally.
The biotechnology. The VLPs used in the HPV vaccines are produced by cloning the major viral capsid genes (L1) from the 4 different HPV types, inserting them into plasmids, and then producing large amounts of the L1 proteins of each of the HPV types separately in yeast. The recombinant L1 proteins are subsequently self-assembled into VLPs that structurally appear identical to HPV virions, but which lack both DNA and RNA.
Therefore, the VLPs are completely non-infectious and non-oncogenic. The purified VLPs are then mixed with an aluminum-containing adjuvant to produce the final vaccine.
Administration. It is administered intramuscularly as 3 injections over a 6-month period: at enrollment and at 2 and 6 months.
A bivalent vaccine for HPV 16 and 18 produced by GlaxoSmithKline is based on similar technology, and will most likely become available in 2007.
Clinical trials: 100% effective, well-accepted, safe
FDA approval of the quadrivalent HPV vaccine came approximately 18 months earlier than expected because, in the Phase III trials, the vaccine proved much more effective than originally predicted, thus shortening the length of follow-up needed to reach the endpoints.
Efficacy and safety studies. Generally, the vaccine appears to be well-accepted and safe. To date, the efficacy and safety of the quadrivalent vaccine have been evaluated in 4 Phase II and III studies that included 20,541 females aged 16 to 26 who were followed for a median of 2 to 4 years in the different studies.1 The endpoint for these trials was biopsy-confirmed high-grade cervical neoplasia, including CIN 2,3 and adenocarcinoma in-situ (AIS). The primary analyses of efficacy were conducted in the “per protocol population,” which consisted of women who received all 3 vaccinations and who were both DNA and serologically negative for the relevant HPV types during the 6-month vaccination period.
In each of the 4 trials, the quadrivalent vaccine was found to be 100% efficacious against HPV 16 and 18 associated CIN 2,3 and AIS. Similarly, the vaccine was also highly efficacious against HPV 6, 11, 16, or 18 associated genital warts.
Adverse events. Although injection site pain, swelling, erythema, and pruritus were increased in vaccine recipients compared to placebo, few subjects (0.1%) discontinued the study as a result of adverse experiences.
Varicella vaccine success story. Varicella vaccine was approved in the 1990s, but both physicians and the public were slow to accept it, in part because it was relatively expensive for physicians to buy, and insurance companies were slow to pay for vaccination. What brought about widespread adoption of varicella vaccination was education on the dangers of chicken pox, directed to clinicians, patients, and government officials. This led to appreciation of the benefits of vaccination. As a result, states changed their school entry requirements, and today, almost all children receive the varicella vaccine.
The unknowns
It is important to recognize that with all new vaccines there are unknowns when they are first introduced.
What will the ACIP recommend?
One of the biggest concerns about the HPV vaccines is that we do not yet know who the ACIP will recommend for vaccination. In the clinical trials, the HPV vaccine did not appear effective in women already exposed to HPV. Since the cumulative incidence of HPV infection in young women is approximately 40% within 2 years of initiating intercourse, we will need to target adolescents prior to onset of sexual activity.3 In the US, 7.4% of adolescents report having begun having sexual intercourse by 13 years of age, and about one third by the ninth grade.4 Therefore the most likely primary target population will be young girls 11 to 12 years of age. It is also likely that there will be a recommendation for “catch-up” vaccination of older girls and young women.
What is the duration of protection afforded by HPV vaccination?
We know that the vaccine appears to provide protection for at least 4 years, but if we vaccinate 11- to 12-year-old girls, will they require a booster later in life?5
How will HPV vaccination affect cervical screening?
Once vaccination is widespread, a significant reduction in CIN 2,3 will occur, necessitating changes in the age to start screening and screening frequency. However, it is unclear what percentage of the population will need to be vaccinated before we change screening policy.
Will HPV vaccine be efficacious in women over the age of 26 years?
Vaccine trials are currently underway in older women, but until these trials are completed, all we can tell our older, sexually active patients is that we simply don’t know if the vaccine will benefit them.
Will parents, adolescents, and the public at large accept the vaccine?
As with all issues relating to sexual behavior, it is likely that opinions will differ as to the acceptability of vaccinating young adolescents against a sexually transmitted disease.
1. Gardasil package insert. Released June 2006.
2. Orenstein S, Rodewald L, Hinman A. Immunization in the United States. In: Plotkin SA, Orenstein S, editors. Vaccines. 4th ed. Philadelphia: WB Saunders Company; 2004:1357-1386.
3. Winer RL, Lee SK, Hughes JP, Adam DE, Kiviat NB, Koutsky LA. Genital human papillomavirus infection: incidence and risk factors in a cohort of female university students. Am J Epidemiol. 2003;157:218-226.
4. Whalen L, Grunbaum J, Kinchen S, McManus T, Shanklin S, Kann L. Middle School: Youth Risk Behavior Survey 2003. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention; 2005.
5. Mao C, Koutsky LA, Ault KA, Wheeler CM, Brown DR, Wiley DJ, et al. Efficacy of human papillomavirus-16 vaccine to prevent cervical intraepithelial neoplasia: a randomized controlled trial. Obstet Gynecol. 2006;107:18-27.
Dr. Wright is the chairman of the upcoming ASCCP (American Society for Colposcopy and Cervical Pathology) 2006 Consensus Conference. He is an author of the 2001 Consensus Guidelines on Managing Women with Cytological and Histological Abnormalities, the 2004 Interim Guidance for Use of HPV DNA testing for Primary Screening, and the 2001 Bethesda System. He is a Professor of Pathology, Columbia University, New York City.
Dr. Wright serves as a consultant to GlaxoSmithKline.

Thomas C. Wright, MD
Thomas C. Wright, MD
Thomas C. Wright, MD
Last month the Food and Drug Administration approved the first vaccine (Gardisil) to prevent infection with specific types of human papillomavirus (HPV). The approval is the culmination of 2 decades of work in a variety of disciplines that have clearly demonstrated that infection with specific types of high-risk HPV is required for the development of cervical cancer—and that the development of HPV-associated disease can be prevented through vaccination.
How and when will HPV vaccine be put to use?
It is important to recognize that FDA approval does not mean the HPV vaccine will be widely used. Five components are necessary for successful introduction:
1. FDA approval
FDA approval is based solely on the safety and effectiveness of the vaccine.
Indications. The FDA determined that the quadrivalent HPV vaccine is indicated for prevention of HPV 6, 11, 16, and 18 associated cervical cancer, genital warts, CIN, AIS, and VIN 2,3 and VAIN 2,3.
The indication applies to girls and women 9 to 26 years of age. The indication in girls 9 to 15 years of age, who were not included in the Phase II and III trials, was based on immunological “bridging” studies that demonstrated better immune responses to vaccination in this group than in the older group enrolled in the trials.
2. Federal-level recommendations
The second component of a successful vaccine introduction is the support of the Advisory Committee on Immunization Practices (ACIP),2 a congressionally mandated federal advisory committee coordinated by the Centers for Disease Control and Prevention (CDC). The ACIP advises the Secretary of Health and Human Services, as well as the Director of the CDC, on appropriate immunization practices for the United States.
Within several months of the June 2006 FDA approval, the ACIP will make a recommendation on the vaccine’s use. When making its recommendation, the ACIP takes into account the burden of disease in the population, safety and efficacy, and health economics of vaccination—will it be cost-effective?
Funding and insurance coverage at stake. The ACIP also determines whether the vaccine will be acceptable to physicians and consumers and what programmatic issues are associated with its introduction.
- Public funding. The ACIP recommendation strongly influences standard of practice, and is relied upon by the federal government and states for determining whether to include a vaccine in public funding programs.
- Insurers likewise look to the ACIP for guidance when setting reimbursement policy.
3. Achieving adequate funding
The third component of a successful vaccine introduction is achieving adequate funding. Federal and state programs such as the Vaccines for Children (VFC) program currently pay for more than half of all childhood vaccinations. Therefore it is important that these programs include the HPV vaccine in order to ensure it is widely available, especially among lower socio-economic groups.
4. Recommendations by professional societies
Societies such as the American Academy of Pediatrics, the American College of Obstetricians and Gynecologists, and the American Academy of Family Physicians, whose members provide most of the vaccinations in the US, have representatives who work directly with the ACIP. Many societies make their own vaccination recommendations, as well.
5. Advocacy and education
Other groups increase awareness of the need for vaccination among consumers, public health and government officials, and clinicians.
The quadrivalent vaccine consists of recombinant viral-like particles (VLPs) derived from the 4 different types of HPV (6, 11, 16, and 18) which, combined, account for approximately 70% of all invasive cervical cancers, 60% of high-grade CIN lesions, and more than 90% of genital warts, both in the United States and globally.
The biotechnology. The VLPs used in the HPV vaccines are produced by cloning the major viral capsid genes (L1) from the 4 different HPV types, inserting them into plasmids, and then producing large amounts of the L1 proteins of each of the HPV types separately in yeast. The recombinant L1 proteins are subsequently self-assembled into VLPs that structurally appear identical to HPV virions, but which lack both DNA and RNA.
Therefore, the VLPs are completely non-infectious and non-oncogenic. The purified VLPs are then mixed with an aluminum-containing adjuvant to produce the final vaccine.
Administration. It is administered intramuscularly as 3 injections over a 6-month period: at enrollment and at 2 and 6 months.
A bivalent vaccine for HPV 16 and 18 produced by GlaxoSmithKline is based on similar technology, and will most likely become available in 2007.
Clinical trials: 100% effective, well-accepted, safe
FDA approval of the quadrivalent HPV vaccine came approximately 18 months earlier than expected because, in the Phase III trials, the vaccine proved much more effective than originally predicted, thus shortening the length of follow-up needed to reach the endpoints.
Efficacy and safety studies. Generally, the vaccine appears to be well-accepted and safe. To date, the efficacy and safety of the quadrivalent vaccine have been evaluated in 4 Phase II and III studies that included 20,541 females aged 16 to 26 who were followed for a median of 2 to 4 years in the different studies.1 The endpoint for these trials was biopsy-confirmed high-grade cervical neoplasia, including CIN 2,3 and adenocarcinoma in-situ (AIS). The primary analyses of efficacy were conducted in the “per protocol population,” which consisted of women who received all 3 vaccinations and who were both DNA and serologically negative for the relevant HPV types during the 6-month vaccination period.
In each of the 4 trials, the quadrivalent vaccine was found to be 100% efficacious against HPV 16 and 18 associated CIN 2,3 and AIS. Similarly, the vaccine was also highly efficacious against HPV 6, 11, 16, or 18 associated genital warts.
Adverse events. Although injection site pain, swelling, erythema, and pruritus were increased in vaccine recipients compared to placebo, few subjects (0.1%) discontinued the study as a result of adverse experiences.
Varicella vaccine success story. Varicella vaccine was approved in the 1990s, but both physicians and the public were slow to accept it, in part because it was relatively expensive for physicians to buy, and insurance companies were slow to pay for vaccination. What brought about widespread adoption of varicella vaccination was education on the dangers of chicken pox, directed to clinicians, patients, and government officials. This led to appreciation of the benefits of vaccination. As a result, states changed their school entry requirements, and today, almost all children receive the varicella vaccine.
The unknowns
It is important to recognize that with all new vaccines there are unknowns when they are first introduced.
What will the ACIP recommend?
One of the biggest concerns about the HPV vaccines is that we do not yet know who the ACIP will recommend for vaccination. In the clinical trials, the HPV vaccine did not appear effective in women already exposed to HPV. Since the cumulative incidence of HPV infection in young women is approximately 40% within 2 years of initiating intercourse, we will need to target adolescents prior to onset of sexual activity.3 In the US, 7.4% of adolescents report having begun having sexual intercourse by 13 years of age, and about one third by the ninth grade.4 Therefore the most likely primary target population will be young girls 11 to 12 years of age. It is also likely that there will be a recommendation for “catch-up” vaccination of older girls and young women.
What is the duration of protection afforded by HPV vaccination?
We know that the vaccine appears to provide protection for at least 4 years, but if we vaccinate 11- to 12-year-old girls, will they require a booster later in life?5
How will HPV vaccination affect cervical screening?
Once vaccination is widespread, a significant reduction in CIN 2,3 will occur, necessitating changes in the age to start screening and screening frequency. However, it is unclear what percentage of the population will need to be vaccinated before we change screening policy.
Will HPV vaccine be efficacious in women over the age of 26 years?
Vaccine trials are currently underway in older women, but until these trials are completed, all we can tell our older, sexually active patients is that we simply don’t know if the vaccine will benefit them.
Will parents, adolescents, and the public at large accept the vaccine?
As with all issues relating to sexual behavior, it is likely that opinions will differ as to the acceptability of vaccinating young adolescents against a sexually transmitted disease.
Last month the Food and Drug Administration approved the first vaccine (Gardisil) to prevent infection with specific types of human papillomavirus (HPV). The approval is the culmination of 2 decades of work in a variety of disciplines that have clearly demonstrated that infection with specific types of high-risk HPV is required for the development of cervical cancer—and that the development of HPV-associated disease can be prevented through vaccination.
How and when will HPV vaccine be put to use?
It is important to recognize that FDA approval does not mean the HPV vaccine will be widely used. Five components are necessary for successful introduction:
1. FDA approval
FDA approval is based solely on the safety and effectiveness of the vaccine.
Indications. The FDA determined that the quadrivalent HPV vaccine is indicated for prevention of HPV 6, 11, 16, and 18 associated cervical cancer, genital warts, CIN, AIS, and VIN 2,3 and VAIN 2,3.
The indication applies to girls and women 9 to 26 years of age. The indication in girls 9 to 15 years of age, who were not included in the Phase II and III trials, was based on immunological “bridging” studies that demonstrated better immune responses to vaccination in this group than in the older group enrolled in the trials.
2. Federal-level recommendations
The second component of a successful vaccine introduction is the support of the Advisory Committee on Immunization Practices (ACIP),2 a congressionally mandated federal advisory committee coordinated by the Centers for Disease Control and Prevention (CDC). The ACIP advises the Secretary of Health and Human Services, as well as the Director of the CDC, on appropriate immunization practices for the United States.
Within several months of the June 2006 FDA approval, the ACIP will make a recommendation on the vaccine’s use. When making its recommendation, the ACIP takes into account the burden of disease in the population, safety and efficacy, and health economics of vaccination—will it be cost-effective?
Funding and insurance coverage at stake. The ACIP also determines whether the vaccine will be acceptable to physicians and consumers and what programmatic issues are associated with its introduction.
- Public funding. The ACIP recommendation strongly influences standard of practice, and is relied upon by the federal government and states for determining whether to include a vaccine in public funding programs.
- Insurers likewise look to the ACIP for guidance when setting reimbursement policy.
3. Achieving adequate funding
The third component of a successful vaccine introduction is achieving adequate funding. Federal and state programs such as the Vaccines for Children (VFC) program currently pay for more than half of all childhood vaccinations. Therefore it is important that these programs include the HPV vaccine in order to ensure it is widely available, especially among lower socio-economic groups.
4. Recommendations by professional societies
Societies such as the American Academy of Pediatrics, the American College of Obstetricians and Gynecologists, and the American Academy of Family Physicians, whose members provide most of the vaccinations in the US, have representatives who work directly with the ACIP. Many societies make their own vaccination recommendations, as well.
5. Advocacy and education
Other groups increase awareness of the need for vaccination among consumers, public health and government officials, and clinicians.
The quadrivalent vaccine consists of recombinant viral-like particles (VLPs) derived from the 4 different types of HPV (6, 11, 16, and 18) which, combined, account for approximately 70% of all invasive cervical cancers, 60% of high-grade CIN lesions, and more than 90% of genital warts, both in the United States and globally.
The biotechnology. The VLPs used in the HPV vaccines are produced by cloning the major viral capsid genes (L1) from the 4 different HPV types, inserting them into plasmids, and then producing large amounts of the L1 proteins of each of the HPV types separately in yeast. The recombinant L1 proteins are subsequently self-assembled into VLPs that structurally appear identical to HPV virions, but which lack both DNA and RNA.
Therefore, the VLPs are completely non-infectious and non-oncogenic. The purified VLPs are then mixed with an aluminum-containing adjuvant to produce the final vaccine.
Administration. It is administered intramuscularly as 3 injections over a 6-month period: at enrollment and at 2 and 6 months.
A bivalent vaccine for HPV 16 and 18 produced by GlaxoSmithKline is based on similar technology, and will most likely become available in 2007.
Clinical trials: 100% effective, well-accepted, safe
FDA approval of the quadrivalent HPV vaccine came approximately 18 months earlier than expected because, in the Phase III trials, the vaccine proved much more effective than originally predicted, thus shortening the length of follow-up needed to reach the endpoints.
Efficacy and safety studies. Generally, the vaccine appears to be well-accepted and safe. To date, the efficacy and safety of the quadrivalent vaccine have been evaluated in 4 Phase II and III studies that included 20,541 females aged 16 to 26 who were followed for a median of 2 to 4 years in the different studies.1 The endpoint for these trials was biopsy-confirmed high-grade cervical neoplasia, including CIN 2,3 and adenocarcinoma in-situ (AIS). The primary analyses of efficacy were conducted in the “per protocol population,” which consisted of women who received all 3 vaccinations and who were both DNA and serologically negative for the relevant HPV types during the 6-month vaccination period.
In each of the 4 trials, the quadrivalent vaccine was found to be 100% efficacious against HPV 16 and 18 associated CIN 2,3 and AIS. Similarly, the vaccine was also highly efficacious against HPV 6, 11, 16, or 18 associated genital warts.
Adverse events. Although injection site pain, swelling, erythema, and pruritus were increased in vaccine recipients compared to placebo, few subjects (0.1%) discontinued the study as a result of adverse experiences.
Varicella vaccine success story. Varicella vaccine was approved in the 1990s, but both physicians and the public were slow to accept it, in part because it was relatively expensive for physicians to buy, and insurance companies were slow to pay for vaccination. What brought about widespread adoption of varicella vaccination was education on the dangers of chicken pox, directed to clinicians, patients, and government officials. This led to appreciation of the benefits of vaccination. As a result, states changed their school entry requirements, and today, almost all children receive the varicella vaccine.
The unknowns
It is important to recognize that with all new vaccines there are unknowns when they are first introduced.
What will the ACIP recommend?
One of the biggest concerns about the HPV vaccines is that we do not yet know who the ACIP will recommend for vaccination. In the clinical trials, the HPV vaccine did not appear effective in women already exposed to HPV. Since the cumulative incidence of HPV infection in young women is approximately 40% within 2 years of initiating intercourse, we will need to target adolescents prior to onset of sexual activity.3 In the US, 7.4% of adolescents report having begun having sexual intercourse by 13 years of age, and about one third by the ninth grade.4 Therefore the most likely primary target population will be young girls 11 to 12 years of age. It is also likely that there will be a recommendation for “catch-up” vaccination of older girls and young women.
What is the duration of protection afforded by HPV vaccination?
We know that the vaccine appears to provide protection for at least 4 years, but if we vaccinate 11- to 12-year-old girls, will they require a booster later in life?5
How will HPV vaccination affect cervical screening?
Once vaccination is widespread, a significant reduction in CIN 2,3 will occur, necessitating changes in the age to start screening and screening frequency. However, it is unclear what percentage of the population will need to be vaccinated before we change screening policy.
Will HPV vaccine be efficacious in women over the age of 26 years?
Vaccine trials are currently underway in older women, but until these trials are completed, all we can tell our older, sexually active patients is that we simply don’t know if the vaccine will benefit them.
Will parents, adolescents, and the public at large accept the vaccine?
As with all issues relating to sexual behavior, it is likely that opinions will differ as to the acceptability of vaccinating young adolescents against a sexually transmitted disease.
1. Gardasil package insert. Released June 2006.
2. Orenstein S, Rodewald L, Hinman A. Immunization in the United States. In: Plotkin SA, Orenstein S, editors. Vaccines. 4th ed. Philadelphia: WB Saunders Company; 2004:1357-1386.
3. Winer RL, Lee SK, Hughes JP, Adam DE, Kiviat NB, Koutsky LA. Genital human papillomavirus infection: incidence and risk factors in a cohort of female university students. Am J Epidemiol. 2003;157:218-226.
4. Whalen L, Grunbaum J, Kinchen S, McManus T, Shanklin S, Kann L. Middle School: Youth Risk Behavior Survey 2003. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention; 2005.
5. Mao C, Koutsky LA, Ault KA, Wheeler CM, Brown DR, Wiley DJ, et al. Efficacy of human papillomavirus-16 vaccine to prevent cervical intraepithelial neoplasia: a randomized controlled trial. Obstet Gynecol. 2006;107:18-27.
Dr. Wright is the chairman of the upcoming ASCCP (American Society for Colposcopy and Cervical Pathology) 2006 Consensus Conference. He is an author of the 2001 Consensus Guidelines on Managing Women with Cytological and Histological Abnormalities, the 2004 Interim Guidance for Use of HPV DNA testing for Primary Screening, and the 2001 Bethesda System. He is a Professor of Pathology, Columbia University, New York City.
Dr. Wright serves as a consultant to GlaxoSmithKline.
1. Gardasil package insert. Released June 2006.
2. Orenstein S, Rodewald L, Hinman A. Immunization in the United States. In: Plotkin SA, Orenstein S, editors. Vaccines. 4th ed. Philadelphia: WB Saunders Company; 2004:1357-1386.
3. Winer RL, Lee SK, Hughes JP, Adam DE, Kiviat NB, Koutsky LA. Genital human papillomavirus infection: incidence and risk factors in a cohort of female university students. Am J Epidemiol. 2003;157:218-226.
4. Whalen L, Grunbaum J, Kinchen S, McManus T, Shanklin S, Kann L. Middle School: Youth Risk Behavior Survey 2003. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention; 2005.
5. Mao C, Koutsky LA, Ault KA, Wheeler CM, Brown DR, Wiley DJ, et al. Efficacy of human papillomavirus-16 vaccine to prevent cervical intraepithelial neoplasia: a randomized controlled trial. Obstet Gynecol. 2006;107:18-27.
Dr. Wright is the chairman of the upcoming ASCCP (American Society for Colposcopy and Cervical Pathology) 2006 Consensus Conference. He is an author of the 2001 Consensus Guidelines on Managing Women with Cytological and Histological Abnormalities, the 2004 Interim Guidance for Use of HPV DNA testing for Primary Screening, and the 2001 Bethesda System. He is a Professor of Pathology, Columbia University, New York City.
Dr. Wright serves as a consultant to GlaxoSmithKline.
IN THIS ARTICLE
Q Does epidural early in labor lead to C-section?
Details of the study
Nulliparous women in early labor were randomized to receive epidural analgesia at the first request (“early” group, about 2.4 cm cervical dilation) or “late” (group in which the epidural was initiated at a mean dilation of 4.6 cm). Analgesia in the late group was provided by parenteral meperidine (Demerol) until cervical dilation increased.
There were no differences in:
- Cesarean delivery rates, either overall or for failure to progress
- Use of oxytocin
- Incidence of maternal fever
- Neonatal outcome as measured by Apgar score
- Presence of meconium
Expert commentary
Why is this study important? There has always been, and continues to be, controversy about epidural analgesia during labor and alleged adverse effects on progress and outcome of labor. Ohel and colleagues have added to the growing body of evidence on these alleged effects—or lack thereof.
Although recent studies have virtually eliminated epidural analgesia per se as an important or causative factor for intrapartum cesarean delivery, there is still some concern that early initiation may have other adverse effects.
What about other adverse effects?
A recent study by Wong et al1 drew the same conclusions as Ohel et al. However, the Wong study was criticized (a criticism with which I do not agree) for its use of a combined spinal-epidural technique, which was not thought to be representative of standard labor practice.
Ohel et al used a protocol that, by any definition, would be considered a typical epidural analgesia “cocktail.” Thus, it should lay to rest any further concerns about alleged adverse effects of early regional analgesia.
Induced and spontaneous labors were included. One potential criticism of this trial is the inclusion of both induced and spontaneous labors. Ohel et al acknowledged and addressed this concern, and provided separate analysis, including power analysis, for these 2 groups. The results were consistent.
Comply with her request
This investigation report was accompanied by a superb editorial,2 which concluded:
“…it is difficult to argue that epidural analgesia should be withheld from a woman who requests pain relief in labor. While such decisions should always be individualized, there should no longer be an arbitrary degree of cervical dilation before such a decision is considered.
“No longer should a patient be made to feel guilty about her wish for pain relief early in labor, powerless in her choices, or conflicted about the consequences of such a choice. Women should receive adequate pain relief when needed, as determined by the patient herself. What a concept—pain relief of real pain when requested.”
Imagine that.
The author reports no financial relationships relevant to this article.
Details of the study
Nulliparous women in early labor were randomized to receive epidural analgesia at the first request (“early” group, about 2.4 cm cervical dilation) or “late” (group in which the epidural was initiated at a mean dilation of 4.6 cm). Analgesia in the late group was provided by parenteral meperidine (Demerol) until cervical dilation increased.
There were no differences in:
- Cesarean delivery rates, either overall or for failure to progress
- Use of oxytocin
- Incidence of maternal fever
- Neonatal outcome as measured by Apgar score
- Presence of meconium
Expert commentary
Why is this study important? There has always been, and continues to be, controversy about epidural analgesia during labor and alleged adverse effects on progress and outcome of labor. Ohel and colleagues have added to the growing body of evidence on these alleged effects—or lack thereof.
Although recent studies have virtually eliminated epidural analgesia per se as an important or causative factor for intrapartum cesarean delivery, there is still some concern that early initiation may have other adverse effects.
What about other adverse effects?
A recent study by Wong et al1 drew the same conclusions as Ohel et al. However, the Wong study was criticized (a criticism with which I do not agree) for its use of a combined spinal-epidural technique, which was not thought to be representative of standard labor practice.
Ohel et al used a protocol that, by any definition, would be considered a typical epidural analgesia “cocktail.” Thus, it should lay to rest any further concerns about alleged adverse effects of early regional analgesia.
Induced and spontaneous labors were included. One potential criticism of this trial is the inclusion of both induced and spontaneous labors. Ohel et al acknowledged and addressed this concern, and provided separate analysis, including power analysis, for these 2 groups. The results were consistent.
Comply with her request
This investigation report was accompanied by a superb editorial,2 which concluded:
“…it is difficult to argue that epidural analgesia should be withheld from a woman who requests pain relief in labor. While such decisions should always be individualized, there should no longer be an arbitrary degree of cervical dilation before such a decision is considered.
“No longer should a patient be made to feel guilty about her wish for pain relief early in labor, powerless in her choices, or conflicted about the consequences of such a choice. Women should receive adequate pain relief when needed, as determined by the patient herself. What a concept—pain relief of real pain when requested.”
Imagine that.
The author reports no financial relationships relevant to this article.
Details of the study
Nulliparous women in early labor were randomized to receive epidural analgesia at the first request (“early” group, about 2.4 cm cervical dilation) or “late” (group in which the epidural was initiated at a mean dilation of 4.6 cm). Analgesia in the late group was provided by parenteral meperidine (Demerol) until cervical dilation increased.
There were no differences in:
- Cesarean delivery rates, either overall or for failure to progress
- Use of oxytocin
- Incidence of maternal fever
- Neonatal outcome as measured by Apgar score
- Presence of meconium
Expert commentary
Why is this study important? There has always been, and continues to be, controversy about epidural analgesia during labor and alleged adverse effects on progress and outcome of labor. Ohel and colleagues have added to the growing body of evidence on these alleged effects—or lack thereof.
Although recent studies have virtually eliminated epidural analgesia per se as an important or causative factor for intrapartum cesarean delivery, there is still some concern that early initiation may have other adverse effects.
What about other adverse effects?
A recent study by Wong et al1 drew the same conclusions as Ohel et al. However, the Wong study was criticized (a criticism with which I do not agree) for its use of a combined spinal-epidural technique, which was not thought to be representative of standard labor practice.
Ohel et al used a protocol that, by any definition, would be considered a typical epidural analgesia “cocktail.” Thus, it should lay to rest any further concerns about alleged adverse effects of early regional analgesia.
Induced and spontaneous labors were included. One potential criticism of this trial is the inclusion of both induced and spontaneous labors. Ohel et al acknowledged and addressed this concern, and provided separate analysis, including power analysis, for these 2 groups. The results were consistent.
Comply with her request
This investigation report was accompanied by a superb editorial,2 which concluded:
“…it is difficult to argue that epidural analgesia should be withheld from a woman who requests pain relief in labor. While such decisions should always be individualized, there should no longer be an arbitrary degree of cervical dilation before such a decision is considered.
“No longer should a patient be made to feel guilty about her wish for pain relief early in labor, powerless in her choices, or conflicted about the consequences of such a choice. Women should receive adequate pain relief when needed, as determined by the patient herself. What a concept—pain relief of real pain when requested.”
Imagine that.
The author reports no financial relationships relevant to this article.
Q Do atypical glandular cells on Pap test require aggressive follow-up?
This analysis found that roughly 29% of women with atypical glandular cells had conditions that required follow-up or treatment, and 5.2% had a malignancy. Therefore, methodical follow-up is not only warranted, but necessary.
Details of the study
Schnatz and colleagues reviewed 3,890 Pap tests that had a finding of atypical glandular cells of undetermined significance (AGUS), for which follow-up details were available.
They found these rates of pathology:
- 8.5% low-grade squamous intraepithelial lesions (LSIL)
- 11.1% high-grade squamous intraepithelial lesions (HSIL)
- 2.9% adenocarcinoma in situ
- 1.4% endometrial hyperplasia
- 5.2% malignancy, The distribution of cancers was:
- 57.6% endometrial adenocarcinoma
- 23.6% cervical adenocarcinoma
- 6.4% ovarian and fallopian tube carcinoma
- 5.4% squamous cell carcinoma of the cervix
- 6.9% other cancers
Expert commentary
Several years ago I gave a talk entitled, “AGUS scares me.” It still does.
For almost 2 decades I have been preaching that, except for a finding of invasive squamous cancer, the single most important cervical cytology report is one that confirms atypical glandular cells (now abbreviated as AGC under the Bethesda System). Virtually every paper written on the subject has shown that AGC are markers for cancer in a high proportion of cases, yet I continue to see clinicians react merely by repeating the sample, or quitting after a negative colposcopy.
Schnatz and colleagues confirmed that such action is insufficient. In their compilation of published research on the subject, 5.2% of all women with AGC and followup had invasive cancer, usually in the pelvis. That rate is many times higher than the cancer risk of HSIL, yet AGC often elicits a far less aggressive evaluation.
Does age matter?
I do wish the authors had done more to explore age as a variable. Most individual series are too small to establish guidelines based on age, but this review of the literature was an opportunity for meaningful observations.
Most of us are not as worried about a report of AGC in a young woman, particularly if she is pregnant. However, that clinical impression is based on very scant data. This study would have been a good place to investigate whether we should pursue AGC differently in young women, and perhaps at what age we should begin to consider endometrial biopsy.
1 in 15 cancers nonpelvic
Of the 203 invasive cancers associated with atypical glandular cells, most originated in the pelvic organs. The authors present a reasonable sequence of evaluation, starting with colposcopy, endocervical evaluation,endometrial biopsy, conization, and pelvic ultrasound—more or less in that order. However, 14 (6.9%), or 1 in 15, of the cancers were located outside the reproductive organs. The authors did not emphasize this finding as much as I would have liked.
Consider colonoscopy, ultrasound, CT
If no source of the AGC is identified in the pelvis, particularly if the patient is older than 50, consider further tests (eg, colonoscopy, abdominal ultrasound, computed tomography). Although nonpelvic malignancies responsible for AGC are often already metastasized, a few cures have been achieved with careful, thorough investigation.
The author reports no financial relationships relevant to this article.
This analysis found that roughly 29% of women with atypical glandular cells had conditions that required follow-up or treatment, and 5.2% had a malignancy. Therefore, methodical follow-up is not only warranted, but necessary.
Details of the study
Schnatz and colleagues reviewed 3,890 Pap tests that had a finding of atypical glandular cells of undetermined significance (AGUS), for which follow-up details were available.
They found these rates of pathology:
- 8.5% low-grade squamous intraepithelial lesions (LSIL)
- 11.1% high-grade squamous intraepithelial lesions (HSIL)
- 2.9% adenocarcinoma in situ
- 1.4% endometrial hyperplasia
- 5.2% malignancy, The distribution of cancers was:
- 57.6% endometrial adenocarcinoma
- 23.6% cervical adenocarcinoma
- 6.4% ovarian and fallopian tube carcinoma
- 5.4% squamous cell carcinoma of the cervix
- 6.9% other cancers
Expert commentary
Several years ago I gave a talk entitled, “AGUS scares me.” It still does.
For almost 2 decades I have been preaching that, except for a finding of invasive squamous cancer, the single most important cervical cytology report is one that confirms atypical glandular cells (now abbreviated as AGC under the Bethesda System). Virtually every paper written on the subject has shown that AGC are markers for cancer in a high proportion of cases, yet I continue to see clinicians react merely by repeating the sample, or quitting after a negative colposcopy.
Schnatz and colleagues confirmed that such action is insufficient. In their compilation of published research on the subject, 5.2% of all women with AGC and followup had invasive cancer, usually in the pelvis. That rate is many times higher than the cancer risk of HSIL, yet AGC often elicits a far less aggressive evaluation.
Does age matter?
I do wish the authors had done more to explore age as a variable. Most individual series are too small to establish guidelines based on age, but this review of the literature was an opportunity for meaningful observations.
Most of us are not as worried about a report of AGC in a young woman, particularly if she is pregnant. However, that clinical impression is based on very scant data. This study would have been a good place to investigate whether we should pursue AGC differently in young women, and perhaps at what age we should begin to consider endometrial biopsy.
1 in 15 cancers nonpelvic
Of the 203 invasive cancers associated with atypical glandular cells, most originated in the pelvic organs. The authors present a reasonable sequence of evaluation, starting with colposcopy, endocervical evaluation,endometrial biopsy, conization, and pelvic ultrasound—more or less in that order. However, 14 (6.9%), or 1 in 15, of the cancers were located outside the reproductive organs. The authors did not emphasize this finding as much as I would have liked.
Consider colonoscopy, ultrasound, CT
If no source of the AGC is identified in the pelvis, particularly if the patient is older than 50, consider further tests (eg, colonoscopy, abdominal ultrasound, computed tomography). Although nonpelvic malignancies responsible for AGC are often already metastasized, a few cures have been achieved with careful, thorough investigation.
The author reports no financial relationships relevant to this article.
This analysis found that roughly 29% of women with atypical glandular cells had conditions that required follow-up or treatment, and 5.2% had a malignancy. Therefore, methodical follow-up is not only warranted, but necessary.
Details of the study
Schnatz and colleagues reviewed 3,890 Pap tests that had a finding of atypical glandular cells of undetermined significance (AGUS), for which follow-up details were available.
They found these rates of pathology:
- 8.5% low-grade squamous intraepithelial lesions (LSIL)
- 11.1% high-grade squamous intraepithelial lesions (HSIL)
- 2.9% adenocarcinoma in situ
- 1.4% endometrial hyperplasia
- 5.2% malignancy, The distribution of cancers was:
- 57.6% endometrial adenocarcinoma
- 23.6% cervical adenocarcinoma
- 6.4% ovarian and fallopian tube carcinoma
- 5.4% squamous cell carcinoma of the cervix
- 6.9% other cancers
Expert commentary
Several years ago I gave a talk entitled, “AGUS scares me.” It still does.
For almost 2 decades I have been preaching that, except for a finding of invasive squamous cancer, the single most important cervical cytology report is one that confirms atypical glandular cells (now abbreviated as AGC under the Bethesda System). Virtually every paper written on the subject has shown that AGC are markers for cancer in a high proportion of cases, yet I continue to see clinicians react merely by repeating the sample, or quitting after a negative colposcopy.
Schnatz and colleagues confirmed that such action is insufficient. In their compilation of published research on the subject, 5.2% of all women with AGC and followup had invasive cancer, usually in the pelvis. That rate is many times higher than the cancer risk of HSIL, yet AGC often elicits a far less aggressive evaluation.
Does age matter?
I do wish the authors had done more to explore age as a variable. Most individual series are too small to establish guidelines based on age, but this review of the literature was an opportunity for meaningful observations.
Most of us are not as worried about a report of AGC in a young woman, particularly if she is pregnant. However, that clinical impression is based on very scant data. This study would have been a good place to investigate whether we should pursue AGC differently in young women, and perhaps at what age we should begin to consider endometrial biopsy.
1 in 15 cancers nonpelvic
Of the 203 invasive cancers associated with atypical glandular cells, most originated in the pelvic organs. The authors present a reasonable sequence of evaluation, starting with colposcopy, endocervical evaluation,endometrial biopsy, conization, and pelvic ultrasound—more or less in that order. However, 14 (6.9%), or 1 in 15, of the cancers were located outside the reproductive organs. The authors did not emphasize this finding as much as I would have liked.
Consider colonoscopy, ultrasound, CT
If no source of the AGC is identified in the pelvis, particularly if the patient is older than 50, consider further tests (eg, colonoscopy, abdominal ultrasound, computed tomography). Although nonpelvic malignancies responsible for AGC are often already metastasized, a few cures have been achieved with careful, thorough investigation.
The author reports no financial relationships relevant to this article.
Q Can stillbirth be predicted?
Expert Commentary
Stillbirth is the cause of about half of all perinatal deaths in the United States (6.4 per 1,000 births). In the past 50 years, the stillbirth rate has decreased about 4-fold, but is still roughly 10 times more than the rate for sudden infant death. The World Health Organization defines stillbirth as fetal loss in pregnancy beyond 20 weeks.
Cause depends on gestational age
Over the past 4 decades, the causes of stillbirth have shifted away from Rh disease and intrapartum loss and toward “unexplained” abruption, intrauterine growth restriction, malformations, and infection.
Categorizing stillbirths by gestational age gives a different rank order of causes. For early stillbirths (24–27 weeks), leading causes are infection (19%), abruption (14%), and anomalies (14%). For late stillbirths (≥28 weeks), the leading causes are “unexplained” (26%–40%, depending on gestational age), fetal malformations (14%–19%), and abruption (12%–18%).
Common risk factors
- Black race—the risk doubles even with adequate prenatal care
- Advanced maternal age, even after accounting for medical conditions
- Obesity, even after controlling for gestational diabetes and hypertension
- Thrombophilia—odds ratios range from 1.8 to 12
- Infection and “immunologic exposure,” including parvovirus B19, toxoplasmosis, and listeriosis
- Advanced reproductive technology, even among singletons
- Multiple gestation
- Medical diseases, particularly systemic lupus erythematosus
Practice recommendations
Fretts proposes these practices:
- Encourage smoking cessation.
- Assess risk factors, including screening for and treatment of diabetes and hypertension, and screening for congenital anomalies and intrauterine growth restriction.
- Consider obese women and women older than 35 years at higher risk.
- Because many stillbirths occur in women with no apparent risk factors, screen even low-risk patients with fetal kick counting in late pregnancy.
- Be vigorous in screening for and management of pregnancies affected by intrauterine growth restriction.
- Use a liberal antepartum testing strategy even in women at moderately increased risk (such as age >35 years).
- When stillbirth occurs, perform “appropriate and comprehensive stillbirth assessment,” most importantly an autopsy. Other tests to use selectively: fetal fasting glucose, hemoglobin A1C, Kleihauer-Betke, urine toxicology, and thrombophilia evaluation. “TORCH titers” to detect infection almost never further diagnosis if there are no findings from autopsy or the placenta.
- Induce labor within 24 hours of fetal death, to decrease maternal anxiety.
The author reports no financial relationships relevant to this article.
Expert Commentary
Stillbirth is the cause of about half of all perinatal deaths in the United States (6.4 per 1,000 births). In the past 50 years, the stillbirth rate has decreased about 4-fold, but is still roughly 10 times more than the rate for sudden infant death. The World Health Organization defines stillbirth as fetal loss in pregnancy beyond 20 weeks.
Cause depends on gestational age
Over the past 4 decades, the causes of stillbirth have shifted away from Rh disease and intrapartum loss and toward “unexplained” abruption, intrauterine growth restriction, malformations, and infection.
Categorizing stillbirths by gestational age gives a different rank order of causes. For early stillbirths (24–27 weeks), leading causes are infection (19%), abruption (14%), and anomalies (14%). For late stillbirths (≥28 weeks), the leading causes are “unexplained” (26%–40%, depending on gestational age), fetal malformations (14%–19%), and abruption (12%–18%).
Common risk factors
- Black race—the risk doubles even with adequate prenatal care
- Advanced maternal age, even after accounting for medical conditions
- Obesity, even after controlling for gestational diabetes and hypertension
- Thrombophilia—odds ratios range from 1.8 to 12
- Infection and “immunologic exposure,” including parvovirus B19, toxoplasmosis, and listeriosis
- Advanced reproductive technology, even among singletons
- Multiple gestation
- Medical diseases, particularly systemic lupus erythematosus
Practice recommendations
Fretts proposes these practices:
- Encourage smoking cessation.
- Assess risk factors, including screening for and treatment of diabetes and hypertension, and screening for congenital anomalies and intrauterine growth restriction.
- Consider obese women and women older than 35 years at higher risk.
- Because many stillbirths occur in women with no apparent risk factors, screen even low-risk patients with fetal kick counting in late pregnancy.
- Be vigorous in screening for and management of pregnancies affected by intrauterine growth restriction.
- Use a liberal antepartum testing strategy even in women at moderately increased risk (such as age >35 years).
- When stillbirth occurs, perform “appropriate and comprehensive stillbirth assessment,” most importantly an autopsy. Other tests to use selectively: fetal fasting glucose, hemoglobin A1C, Kleihauer-Betke, urine toxicology, and thrombophilia evaluation. “TORCH titers” to detect infection almost never further diagnosis if there are no findings from autopsy or the placenta.
- Induce labor within 24 hours of fetal death, to decrease maternal anxiety.
Expert Commentary
Stillbirth is the cause of about half of all perinatal deaths in the United States (6.4 per 1,000 births). In the past 50 years, the stillbirth rate has decreased about 4-fold, but is still roughly 10 times more than the rate for sudden infant death. The World Health Organization defines stillbirth as fetal loss in pregnancy beyond 20 weeks.
Cause depends on gestational age
Over the past 4 decades, the causes of stillbirth have shifted away from Rh disease and intrapartum loss and toward “unexplained” abruption, intrauterine growth restriction, malformations, and infection.
Categorizing stillbirths by gestational age gives a different rank order of causes. For early stillbirths (24–27 weeks), leading causes are infection (19%), abruption (14%), and anomalies (14%). For late stillbirths (≥28 weeks), the leading causes are “unexplained” (26%–40%, depending on gestational age), fetal malformations (14%–19%), and abruption (12%–18%).
Common risk factors
- Black race—the risk doubles even with adequate prenatal care
- Advanced maternal age, even after accounting for medical conditions
- Obesity, even after controlling for gestational diabetes and hypertension
- Thrombophilia—odds ratios range from 1.8 to 12
- Infection and “immunologic exposure,” including parvovirus B19, toxoplasmosis, and listeriosis
- Advanced reproductive technology, even among singletons
- Multiple gestation
- Medical diseases, particularly systemic lupus erythematosus
Practice recommendations
Fretts proposes these practices:
- Encourage smoking cessation.
- Assess risk factors, including screening for and treatment of diabetes and hypertension, and screening for congenital anomalies and intrauterine growth restriction.
- Consider obese women and women older than 35 years at higher risk.
- Because many stillbirths occur in women with no apparent risk factors, screen even low-risk patients with fetal kick counting in late pregnancy.
- Be vigorous in screening for and management of pregnancies affected by intrauterine growth restriction.
- Use a liberal antepartum testing strategy even in women at moderately increased risk (such as age >35 years).
- When stillbirth occurs, perform “appropriate and comprehensive stillbirth assessment,” most importantly an autopsy. Other tests to use selectively: fetal fasting glucose, hemoglobin A1C, Kleihauer-Betke, urine toxicology, and thrombophilia evaluation. “TORCH titers” to detect infection almost never further diagnosis if there are no findings from autopsy or the placenta.
- Induce labor within 24 hours of fetal death, to decrease maternal anxiety.
The author reports no financial relationships relevant to this article.
The author reports no financial relationships relevant to this article.
Q Are we doing a good job of screening for postpartum type 2 diabetes?
Expert Commentary
Although the clinical impact of gestational diabetes on pregnancy outcomes is still under debate, we know the condition heightens risk of developing type 2 diabetes later in life. As many as one third of parous women with diabetes had gestational diabetes, and the incidence of both gestational diabetes and subsequent type 2 diabetes seems to be increasing.
While the ADA recommends that maternal glycemic status be reassessed at roughly 6 weeks postpartum in women with gestational diabetes, the American College of Obstetricians and Gynecologists (ACOG) makes no clear recommendation. The ACOG practice bulletin on gestational diabetes1 acknowledges that such testing may be performed, but points out that no studies have confirmed its benefit.
Only 37% had the right kind of test
In the Smirnakis study, two thirds of the population of 197 women had some form of postpartum glucose measurement—but only 37% were tested according to ADA recommendations, which call for measurement of fasting glucose levels or undergoing the oral glucose tolerance test. In many cases, postpartum assessment consisted of only random glucose testing, which can overlook serious carbohydrate intolerance.
The only factors that significantly increased the likelihood of postpartum screening were glucose values after the 1-hour glucose loading test (during pregnancy) at or above the geometric mean (≥171 mg/dL) and/or a fasting glucose level (from the pregnancy diagnostic test) at or above the geometric mean (≥98 mg/dL).
This study did not report whether the need for insulin or an oral agent (class A2) during pregnancy was independently associated with the frequency of postpartum testing for type 2 diabetes. Such an association seems likely, given the fasting cutoff of 98 mg/dL.
Why aren’t we doing a better job?
Possible explanations of the low screening rates include a lack of direction from ACOG, confusion over provider responsibility (obstetrician versus primary care provider), lack of patient education, and the difficulty of mothers pursuing such testing while caring for a newborn.
Although it is unclear whether the 2-hour oral glucose tolerance test is clinically superior to a fasting glucose level for postpartum screening, it is generally accepted that some form of postpartum glucose assessment is needed in this high-risk group of women.
Unfortunately, we lack studies of efficacy and timing of diabetes screening in women with gestational diabetes.
Gestational diabetes always calls for postpartum glucose tolerance testing
A formal recommendation from ACOG would be a good first step, until further data are available. Even without such a recommendation, however, the ADA and most of us agree that early diagnosis and treatment of diabetes can potentially decrease long-term complications. For this reason, obstetricians should conduct postpartum glucose assessment using the 2-hour oral glucose tolerance test or fasting glucose for all women with a history of gestational diabetes.
1. American College of Obstetricians and Gynecologists. Practice Bulletin #30: Gestational Diabetes. Washington, DC: ACOG; 2001.
The author reports no financial relationships relevant to this article.
Expert Commentary
Although the clinical impact of gestational diabetes on pregnancy outcomes is still under debate, we know the condition heightens risk of developing type 2 diabetes later in life. As many as one third of parous women with diabetes had gestational diabetes, and the incidence of both gestational diabetes and subsequent type 2 diabetes seems to be increasing.
While the ADA recommends that maternal glycemic status be reassessed at roughly 6 weeks postpartum in women with gestational diabetes, the American College of Obstetricians and Gynecologists (ACOG) makes no clear recommendation. The ACOG practice bulletin on gestational diabetes1 acknowledges that such testing may be performed, but points out that no studies have confirmed its benefit.
Only 37% had the right kind of test
In the Smirnakis study, two thirds of the population of 197 women had some form of postpartum glucose measurement—but only 37% were tested according to ADA recommendations, which call for measurement of fasting glucose levels or undergoing the oral glucose tolerance test. In many cases, postpartum assessment consisted of only random glucose testing, which can overlook serious carbohydrate intolerance.
The only factors that significantly increased the likelihood of postpartum screening were glucose values after the 1-hour glucose loading test (during pregnancy) at or above the geometric mean (≥171 mg/dL) and/or a fasting glucose level (from the pregnancy diagnostic test) at or above the geometric mean (≥98 mg/dL).
This study did not report whether the need for insulin or an oral agent (class A2) during pregnancy was independently associated with the frequency of postpartum testing for type 2 diabetes. Such an association seems likely, given the fasting cutoff of 98 mg/dL.
Why aren’t we doing a better job?
Possible explanations of the low screening rates include a lack of direction from ACOG, confusion over provider responsibility (obstetrician versus primary care provider), lack of patient education, and the difficulty of mothers pursuing such testing while caring for a newborn.
Although it is unclear whether the 2-hour oral glucose tolerance test is clinically superior to a fasting glucose level for postpartum screening, it is generally accepted that some form of postpartum glucose assessment is needed in this high-risk group of women.
Unfortunately, we lack studies of efficacy and timing of diabetes screening in women with gestational diabetes.
Gestational diabetes always calls for postpartum glucose tolerance testing
A formal recommendation from ACOG would be a good first step, until further data are available. Even without such a recommendation, however, the ADA and most of us agree that early diagnosis and treatment of diabetes can potentially decrease long-term complications. For this reason, obstetricians should conduct postpartum glucose assessment using the 2-hour oral glucose tolerance test or fasting glucose for all women with a history of gestational diabetes.
Expert Commentary
Although the clinical impact of gestational diabetes on pregnancy outcomes is still under debate, we know the condition heightens risk of developing type 2 diabetes later in life. As many as one third of parous women with diabetes had gestational diabetes, and the incidence of both gestational diabetes and subsequent type 2 diabetes seems to be increasing.
While the ADA recommends that maternal glycemic status be reassessed at roughly 6 weeks postpartum in women with gestational diabetes, the American College of Obstetricians and Gynecologists (ACOG) makes no clear recommendation. The ACOG practice bulletin on gestational diabetes1 acknowledges that such testing may be performed, but points out that no studies have confirmed its benefit.
Only 37% had the right kind of test
In the Smirnakis study, two thirds of the population of 197 women had some form of postpartum glucose measurement—but only 37% were tested according to ADA recommendations, which call for measurement of fasting glucose levels or undergoing the oral glucose tolerance test. In many cases, postpartum assessment consisted of only random glucose testing, which can overlook serious carbohydrate intolerance.
The only factors that significantly increased the likelihood of postpartum screening were glucose values after the 1-hour glucose loading test (during pregnancy) at or above the geometric mean (≥171 mg/dL) and/or a fasting glucose level (from the pregnancy diagnostic test) at or above the geometric mean (≥98 mg/dL).
This study did not report whether the need for insulin or an oral agent (class A2) during pregnancy was independently associated with the frequency of postpartum testing for type 2 diabetes. Such an association seems likely, given the fasting cutoff of 98 mg/dL.
Why aren’t we doing a better job?
Possible explanations of the low screening rates include a lack of direction from ACOG, confusion over provider responsibility (obstetrician versus primary care provider), lack of patient education, and the difficulty of mothers pursuing such testing while caring for a newborn.
Although it is unclear whether the 2-hour oral glucose tolerance test is clinically superior to a fasting glucose level for postpartum screening, it is generally accepted that some form of postpartum glucose assessment is needed in this high-risk group of women.
Unfortunately, we lack studies of efficacy and timing of diabetes screening in women with gestational diabetes.
Gestational diabetes always calls for postpartum glucose tolerance testing
A formal recommendation from ACOG would be a good first step, until further data are available. Even without such a recommendation, however, the ADA and most of us agree that early diagnosis and treatment of diabetes can potentially decrease long-term complications. For this reason, obstetricians should conduct postpartum glucose assessment using the 2-hour oral glucose tolerance test or fasting glucose for all women with a history of gestational diabetes.
1. American College of Obstetricians and Gynecologists. Practice Bulletin #30: Gestational Diabetes. Washington, DC: ACOG; 2001.
The author reports no financial relationships relevant to this article.
1. American College of Obstetricians and Gynecologists. Practice Bulletin #30: Gestational Diabetes. Washington, DC: ACOG; 2001.
The author reports no financial relationships relevant to this article.
Q Is it reasonable to continue SSRIs during pregnancy?
About 10% to 20% of infants with PPHN do not survive, even with treatment. In the general population, PPHN typically occurs in 1 or 2 infants per 1,000 live births, but in women taking selective serotonin reuptake inhibitors (SSRIs), it occurs in 6 to 12 infants per 1,000 births.
Expert Commentary
These 2 studies add a lot to what we have learned1,2 about the risks and benefits of treating depression during pregnancy. The New England Journal of Medicine investigation was a sophisticated case-control study that identified an increased risk of PPHN associated with the use of SSRIs after 20 weeks’ gestation. The study in The Journal of the American Medical Association was a prospective naturalistic investigation in which longitudinal psychiatric assessments were conducted in pregnant women who elected to maintain or discontinue antidepressant therapy near the time of conception. No randomization occurred because of ethical concerns.
In the case-control study, no increased risk of PPHN was found for maternal SSRI use before 20 weeks’ gestation or for nonserotonergic antidepressants at any point during pregnancy. However, the relative risk of PPHN with late-pregnancy SSRI exposure, compared with early or no exposure, was 6.1 (95% confidence interval, 2.2–16.8).
In the JAMA study, significantly more women who stopped taking antidepressants had recurrence of major depression than did women who continued taking antidepressants: relapse occurred in 44 of 65 women (68%) after discontinuing the drugs, versus 21 of 82 women (26%) who continued treatment. Most recurrences emerged rapidly: 50% in the first trimester and 90% by the end of the second trimester.
Interestingly, women older than 32 had a 60% reduction in the rate of relapse, compared with women younger than 32. A history of depressive illness (>5 years) and/or multiple recurrences (>4 episodes) significantly increased the risk of relapse.
Women are most vulnerable to depression during childbearing years
Major depressive disorder affects about 12% of women per year, with greatest prevalence during the childbearing years.3 Maintenance antidepressant therapy is recommended for women with recurrent major depressive disorder (3 or more life-time episodes) because another episode is essentially certain without prophylaxis.
For many years, pregnancy was thought to be protective against psychiatric illness, but new data have refuted this theory.
Link between SSRIs and pulmonary hypertension not necessarily causal
In the general population, PPHN typically occurs in 1 or 2 infants per 1,000 live births and involves substantial morbidity and even mortality.
One possible mechanism for a causal relationship between PPHN and maternal SSRI use is that the lungs serve as a “reservoir” for antidepressant agents, and accumulation of SSRIs in the lungs has been reported. According to Chambers and colleagues, “Serotonin not only has vasoconstrictive properties that increase pulmonary vascular resistance, but also has mitogenic and comitogenic effects on pulmonary smooth-muscle cells. Thus, higher circulating levels of serotonin in the fetus and accumulation of serotonin in the fetal lung might result in the proliferation of smooth-muscle cells that is characteristic of PPHN.”
The study by Chambers and colleagues was a case-control investigation, however, which although useful for evaluating associations between exposures and adverse outcomes (especially for rare diseases such as PPHN), cannot establish causality.
What to tell patients
A clearer way to discuss the finding about PPHN is to focus on the absolute risk, which was 6 to 12 cases of PPHN per 1,000 births (0.6%–1.2%). In other words, 99% of women treated with an SSRI delivered infants without pulmonary hypertension.
What about nonserotonergic antidepressants?
Although no association was found between nonserotonergic antidepressants and PPHN, these drugs have their own set of risks and benefits. Bupropion or nonserotonergic tricyclic antidepressants (such as nortriptyline) are reasonable choices to treat depression during pregnancy. Unlike other types of antidepressants, therapeutic serum levels have been established for tricyclic antidepressants. Serum level monitoring during pregnancy can be a useful, if burdensome, component of follow-up.3
However, the potential for fetal cardiac toxicity (particularly with overdose) poses a risk not shared by SSRIs. Similar to SSRIs,2 tricyclic use is associated with neonatal signs (irritability, restlessness, tremor) when taken near term.1 Venlafaxine is a serotonin-norepinephrine reuptake inhibitor that has not been associated with major congenital malformations4; however, neonatal signs also occur with this serotonergic agent.
We have fewer data on reproductive outcomes with bupropion and venlafaxine use than with SSRIs or tricyclic antidepressants, but this lack of data does not necessarily mean there is a lack of adverse outcomes.
Weighing the risks
Add PPHN to the list of other reported risks of SSRIs during pregnancy, such as minor physical anomalies (no functional or cosmetic significance), preterm birth, decreased birth weight, neurobehavioral effects, and neonatal syndrome.2,5 These risks have not included major malformations or long-term developmental deficits in childhood.1,5
Untreated depression itself poses risks: preterm birth, growth restriction, preeclampsia, neonatal neurobehavioral effects, and impaired maternal function.6
After weighing these risks with her physician, a woman could reasonably decide that the benefits of drug treatment outweigh the risks, or she could decide the opposite. Certainly evidence-based nondrug therapies such as psychotherapy (cognitive behavioral or interpersonal psychotherapy), electroconvulsive therapy, and bright light therapy should be considered.7 However, not all women elect, respond to, or have access to such treatments.
Bottom line: No absolutes
Although our ability to quantitatively weight treatment choices improves with the publication of studies such as these, the path to the most favorable outcome remains a highly individualized decision with no absolutes. Continuing use of antidepressants does not guarantee that remission will be sustained. Careful monitoring and dose adjustment are necessary in order to maintain adequate serum levels in women who take tricyclics,8 particularly during the third trimester. Higher dose requirements also have been described for SSRIs because of increased drug metabolism during pregnancy.9
If the gravida decides to continue antidepressant therapy, she should choose the drug (an SSRI, tricyclic, or bupropion) that is most effective for her.
1. Wisner KL, Zarin DA, Holmboe ES, et al. Risk-benefit decision-making for treatment of depression during pregnancy. Am J Psychiatry. 2000;157:1933-1940.
2. Moses-Kolko EL, Bogen D, Perel J, et al. Neonatal signs after late in utero exposure to serotonin reuptake inhibitors: literature review and implications for clinical applications. JAMA. 2005;293:2372-2383.
3. Regier DA, Narrow WE, Rae DS, et al. The de facto US mental and addictive disorders service system. Epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry. 1993;50:85-94.
4. Einarson A, Fatoye B, Sarkar M, et al. Pregnancy outcome following gestational exposure to venlafaxine: a multicenter prospective controlled trial. Am J Psych. 2001;158:1728-1730.
5. Wisner K, Gelenberg A, Leonard H, Zarin D, Frank E. Pharmacologic treatment of depression during pregnancy. JAMA. 1999;282:1264-1269.
6. Bonari L, Pinto N, Ahn C, Einarson A, Steiner M, Koren G. Perinatal risks of untreated depression during pregnancy. Can J Psychiatry. 2004;49:726-735.
7. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). Am J Psychiatry. 2000;157(suppl):1-45.
8. Wisner KL, Perel JM, Wheeler SB. Tricyclic dose requirements across pregnancy. Am J Psychiatry. 1993;150:1541-1542.
9. Hostetter A, Stowe ZN, Strader JR, Jr, McLaughlin E, Llewellyn A. Dose of selective serotonin uptake inhibitors across pregnancy: clinical implications. Depress Anxiety. 2000;11:51-57.
Dr. Wisner has received grant/research support from Pfizer and serves as a speaker for GlaxoSmithKline and Pfizer. Dr. Hanusa has no financial relationships relevant to this article.
About 10% to 20% of infants with PPHN do not survive, even with treatment. In the general population, PPHN typically occurs in 1 or 2 infants per 1,000 live births, but in women taking selective serotonin reuptake inhibitors (SSRIs), it occurs in 6 to 12 infants per 1,000 births.
Expert Commentary
These 2 studies add a lot to what we have learned1,2 about the risks and benefits of treating depression during pregnancy. The New England Journal of Medicine investigation was a sophisticated case-control study that identified an increased risk of PPHN associated with the use of SSRIs after 20 weeks’ gestation. The study in The Journal of the American Medical Association was a prospective naturalistic investigation in which longitudinal psychiatric assessments were conducted in pregnant women who elected to maintain or discontinue antidepressant therapy near the time of conception. No randomization occurred because of ethical concerns.
In the case-control study, no increased risk of PPHN was found for maternal SSRI use before 20 weeks’ gestation or for nonserotonergic antidepressants at any point during pregnancy. However, the relative risk of PPHN with late-pregnancy SSRI exposure, compared with early or no exposure, was 6.1 (95% confidence interval, 2.2–16.8).
In the JAMA study, significantly more women who stopped taking antidepressants had recurrence of major depression than did women who continued taking antidepressants: relapse occurred in 44 of 65 women (68%) after discontinuing the drugs, versus 21 of 82 women (26%) who continued treatment. Most recurrences emerged rapidly: 50% in the first trimester and 90% by the end of the second trimester.
Interestingly, women older than 32 had a 60% reduction in the rate of relapse, compared with women younger than 32. A history of depressive illness (>5 years) and/or multiple recurrences (>4 episodes) significantly increased the risk of relapse.
Women are most vulnerable to depression during childbearing years
Major depressive disorder affects about 12% of women per year, with greatest prevalence during the childbearing years.3 Maintenance antidepressant therapy is recommended for women with recurrent major depressive disorder (3 or more life-time episodes) because another episode is essentially certain without prophylaxis.
For many years, pregnancy was thought to be protective against psychiatric illness, but new data have refuted this theory.
Link between SSRIs and pulmonary hypertension not necessarily causal
In the general population, PPHN typically occurs in 1 or 2 infants per 1,000 live births and involves substantial morbidity and even mortality.
One possible mechanism for a causal relationship between PPHN and maternal SSRI use is that the lungs serve as a “reservoir” for antidepressant agents, and accumulation of SSRIs in the lungs has been reported. According to Chambers and colleagues, “Serotonin not only has vasoconstrictive properties that increase pulmonary vascular resistance, but also has mitogenic and comitogenic effects on pulmonary smooth-muscle cells. Thus, higher circulating levels of serotonin in the fetus and accumulation of serotonin in the fetal lung might result in the proliferation of smooth-muscle cells that is characteristic of PPHN.”
The study by Chambers and colleagues was a case-control investigation, however, which although useful for evaluating associations between exposures and adverse outcomes (especially for rare diseases such as PPHN), cannot establish causality.
What to tell patients
A clearer way to discuss the finding about PPHN is to focus on the absolute risk, which was 6 to 12 cases of PPHN per 1,000 births (0.6%–1.2%). In other words, 99% of women treated with an SSRI delivered infants without pulmonary hypertension.
What about nonserotonergic antidepressants?
Although no association was found between nonserotonergic antidepressants and PPHN, these drugs have their own set of risks and benefits. Bupropion or nonserotonergic tricyclic antidepressants (such as nortriptyline) are reasonable choices to treat depression during pregnancy. Unlike other types of antidepressants, therapeutic serum levels have been established for tricyclic antidepressants. Serum level monitoring during pregnancy can be a useful, if burdensome, component of follow-up.3
However, the potential for fetal cardiac toxicity (particularly with overdose) poses a risk not shared by SSRIs. Similar to SSRIs,2 tricyclic use is associated with neonatal signs (irritability, restlessness, tremor) when taken near term.1 Venlafaxine is a serotonin-norepinephrine reuptake inhibitor that has not been associated with major congenital malformations4; however, neonatal signs also occur with this serotonergic agent.
We have fewer data on reproductive outcomes with bupropion and venlafaxine use than with SSRIs or tricyclic antidepressants, but this lack of data does not necessarily mean there is a lack of adverse outcomes.
Weighing the risks
Add PPHN to the list of other reported risks of SSRIs during pregnancy, such as minor physical anomalies (no functional or cosmetic significance), preterm birth, decreased birth weight, neurobehavioral effects, and neonatal syndrome.2,5 These risks have not included major malformations or long-term developmental deficits in childhood.1,5
Untreated depression itself poses risks: preterm birth, growth restriction, preeclampsia, neonatal neurobehavioral effects, and impaired maternal function.6
After weighing these risks with her physician, a woman could reasonably decide that the benefits of drug treatment outweigh the risks, or she could decide the opposite. Certainly evidence-based nondrug therapies such as psychotherapy (cognitive behavioral or interpersonal psychotherapy), electroconvulsive therapy, and bright light therapy should be considered.7 However, not all women elect, respond to, or have access to such treatments.
Bottom line: No absolutes
Although our ability to quantitatively weight treatment choices improves with the publication of studies such as these, the path to the most favorable outcome remains a highly individualized decision with no absolutes. Continuing use of antidepressants does not guarantee that remission will be sustained. Careful monitoring and dose adjustment are necessary in order to maintain adequate serum levels in women who take tricyclics,8 particularly during the third trimester. Higher dose requirements also have been described for SSRIs because of increased drug metabolism during pregnancy.9
If the gravida decides to continue antidepressant therapy, she should choose the drug (an SSRI, tricyclic, or bupropion) that is most effective for her.
About 10% to 20% of infants with PPHN do not survive, even with treatment. In the general population, PPHN typically occurs in 1 or 2 infants per 1,000 live births, but in women taking selective serotonin reuptake inhibitors (SSRIs), it occurs in 6 to 12 infants per 1,000 births.
Expert Commentary
These 2 studies add a lot to what we have learned1,2 about the risks and benefits of treating depression during pregnancy. The New England Journal of Medicine investigation was a sophisticated case-control study that identified an increased risk of PPHN associated with the use of SSRIs after 20 weeks’ gestation. The study in The Journal of the American Medical Association was a prospective naturalistic investigation in which longitudinal psychiatric assessments were conducted in pregnant women who elected to maintain or discontinue antidepressant therapy near the time of conception. No randomization occurred because of ethical concerns.
In the case-control study, no increased risk of PPHN was found for maternal SSRI use before 20 weeks’ gestation or for nonserotonergic antidepressants at any point during pregnancy. However, the relative risk of PPHN with late-pregnancy SSRI exposure, compared with early or no exposure, was 6.1 (95% confidence interval, 2.2–16.8).
In the JAMA study, significantly more women who stopped taking antidepressants had recurrence of major depression than did women who continued taking antidepressants: relapse occurred in 44 of 65 women (68%) after discontinuing the drugs, versus 21 of 82 women (26%) who continued treatment. Most recurrences emerged rapidly: 50% in the first trimester and 90% by the end of the second trimester.
Interestingly, women older than 32 had a 60% reduction in the rate of relapse, compared with women younger than 32. A history of depressive illness (>5 years) and/or multiple recurrences (>4 episodes) significantly increased the risk of relapse.
Women are most vulnerable to depression during childbearing years
Major depressive disorder affects about 12% of women per year, with greatest prevalence during the childbearing years.3 Maintenance antidepressant therapy is recommended for women with recurrent major depressive disorder (3 or more life-time episodes) because another episode is essentially certain without prophylaxis.
For many years, pregnancy was thought to be protective against psychiatric illness, but new data have refuted this theory.
Link between SSRIs and pulmonary hypertension not necessarily causal
In the general population, PPHN typically occurs in 1 or 2 infants per 1,000 live births and involves substantial morbidity and even mortality.
One possible mechanism for a causal relationship between PPHN and maternal SSRI use is that the lungs serve as a “reservoir” for antidepressant agents, and accumulation of SSRIs in the lungs has been reported. According to Chambers and colleagues, “Serotonin not only has vasoconstrictive properties that increase pulmonary vascular resistance, but also has mitogenic and comitogenic effects on pulmonary smooth-muscle cells. Thus, higher circulating levels of serotonin in the fetus and accumulation of serotonin in the fetal lung might result in the proliferation of smooth-muscle cells that is characteristic of PPHN.”
The study by Chambers and colleagues was a case-control investigation, however, which although useful for evaluating associations between exposures and adverse outcomes (especially for rare diseases such as PPHN), cannot establish causality.
What to tell patients
A clearer way to discuss the finding about PPHN is to focus on the absolute risk, which was 6 to 12 cases of PPHN per 1,000 births (0.6%–1.2%). In other words, 99% of women treated with an SSRI delivered infants without pulmonary hypertension.
What about nonserotonergic antidepressants?
Although no association was found between nonserotonergic antidepressants and PPHN, these drugs have their own set of risks and benefits. Bupropion or nonserotonergic tricyclic antidepressants (such as nortriptyline) are reasonable choices to treat depression during pregnancy. Unlike other types of antidepressants, therapeutic serum levels have been established for tricyclic antidepressants. Serum level monitoring during pregnancy can be a useful, if burdensome, component of follow-up.3
However, the potential for fetal cardiac toxicity (particularly with overdose) poses a risk not shared by SSRIs. Similar to SSRIs,2 tricyclic use is associated with neonatal signs (irritability, restlessness, tremor) when taken near term.1 Venlafaxine is a serotonin-norepinephrine reuptake inhibitor that has not been associated with major congenital malformations4; however, neonatal signs also occur with this serotonergic agent.
We have fewer data on reproductive outcomes with bupropion and venlafaxine use than with SSRIs or tricyclic antidepressants, but this lack of data does not necessarily mean there is a lack of adverse outcomes.
Weighing the risks
Add PPHN to the list of other reported risks of SSRIs during pregnancy, such as minor physical anomalies (no functional or cosmetic significance), preterm birth, decreased birth weight, neurobehavioral effects, and neonatal syndrome.2,5 These risks have not included major malformations or long-term developmental deficits in childhood.1,5
Untreated depression itself poses risks: preterm birth, growth restriction, preeclampsia, neonatal neurobehavioral effects, and impaired maternal function.6
After weighing these risks with her physician, a woman could reasonably decide that the benefits of drug treatment outweigh the risks, or she could decide the opposite. Certainly evidence-based nondrug therapies such as psychotherapy (cognitive behavioral or interpersonal psychotherapy), electroconvulsive therapy, and bright light therapy should be considered.7 However, not all women elect, respond to, or have access to such treatments.
Bottom line: No absolutes
Although our ability to quantitatively weight treatment choices improves with the publication of studies such as these, the path to the most favorable outcome remains a highly individualized decision with no absolutes. Continuing use of antidepressants does not guarantee that remission will be sustained. Careful monitoring and dose adjustment are necessary in order to maintain adequate serum levels in women who take tricyclics,8 particularly during the third trimester. Higher dose requirements also have been described for SSRIs because of increased drug metabolism during pregnancy.9
If the gravida decides to continue antidepressant therapy, she should choose the drug (an SSRI, tricyclic, or bupropion) that is most effective for her.
1. Wisner KL, Zarin DA, Holmboe ES, et al. Risk-benefit decision-making for treatment of depression during pregnancy. Am J Psychiatry. 2000;157:1933-1940.
2. Moses-Kolko EL, Bogen D, Perel J, et al. Neonatal signs after late in utero exposure to serotonin reuptake inhibitors: literature review and implications for clinical applications. JAMA. 2005;293:2372-2383.
3. Regier DA, Narrow WE, Rae DS, et al. The de facto US mental and addictive disorders service system. Epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry. 1993;50:85-94.
4. Einarson A, Fatoye B, Sarkar M, et al. Pregnancy outcome following gestational exposure to venlafaxine: a multicenter prospective controlled trial. Am J Psych. 2001;158:1728-1730.
5. Wisner K, Gelenberg A, Leonard H, Zarin D, Frank E. Pharmacologic treatment of depression during pregnancy. JAMA. 1999;282:1264-1269.
6. Bonari L, Pinto N, Ahn C, Einarson A, Steiner M, Koren G. Perinatal risks of untreated depression during pregnancy. Can J Psychiatry. 2004;49:726-735.
7. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). Am J Psychiatry. 2000;157(suppl):1-45.
8. Wisner KL, Perel JM, Wheeler SB. Tricyclic dose requirements across pregnancy. Am J Psychiatry. 1993;150:1541-1542.
9. Hostetter A, Stowe ZN, Strader JR, Jr, McLaughlin E, Llewellyn A. Dose of selective serotonin uptake inhibitors across pregnancy: clinical implications. Depress Anxiety. 2000;11:51-57.
Dr. Wisner has received grant/research support from Pfizer and serves as a speaker for GlaxoSmithKline and Pfizer. Dr. Hanusa has no financial relationships relevant to this article.
1. Wisner KL, Zarin DA, Holmboe ES, et al. Risk-benefit decision-making for treatment of depression during pregnancy. Am J Psychiatry. 2000;157:1933-1940.
2. Moses-Kolko EL, Bogen D, Perel J, et al. Neonatal signs after late in utero exposure to serotonin reuptake inhibitors: literature review and implications for clinical applications. JAMA. 2005;293:2372-2383.
3. Regier DA, Narrow WE, Rae DS, et al. The de facto US mental and addictive disorders service system. Epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry. 1993;50:85-94.
4. Einarson A, Fatoye B, Sarkar M, et al. Pregnancy outcome following gestational exposure to venlafaxine: a multicenter prospective controlled trial. Am J Psych. 2001;158:1728-1730.
5. Wisner K, Gelenberg A, Leonard H, Zarin D, Frank E. Pharmacologic treatment of depression during pregnancy. JAMA. 1999;282:1264-1269.
6. Bonari L, Pinto N, Ahn C, Einarson A, Steiner M, Koren G. Perinatal risks of untreated depression during pregnancy. Can J Psychiatry. 2004;49:726-735.
7. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). Am J Psychiatry. 2000;157(suppl):1-45.
8. Wisner KL, Perel JM, Wheeler SB. Tricyclic dose requirements across pregnancy. Am J Psychiatry. 1993;150:1541-1542.
9. Hostetter A, Stowe ZN, Strader JR, Jr, McLaughlin E, Llewellyn A. Dose of selective serotonin uptake inhibitors across pregnancy: clinical implications. Depress Anxiety. 2000;11:51-57.
Dr. Wisner has received grant/research support from Pfizer and serves as a speaker for GlaxoSmithKline and Pfizer. Dr. Hanusa has no financial relationships relevant to this article.